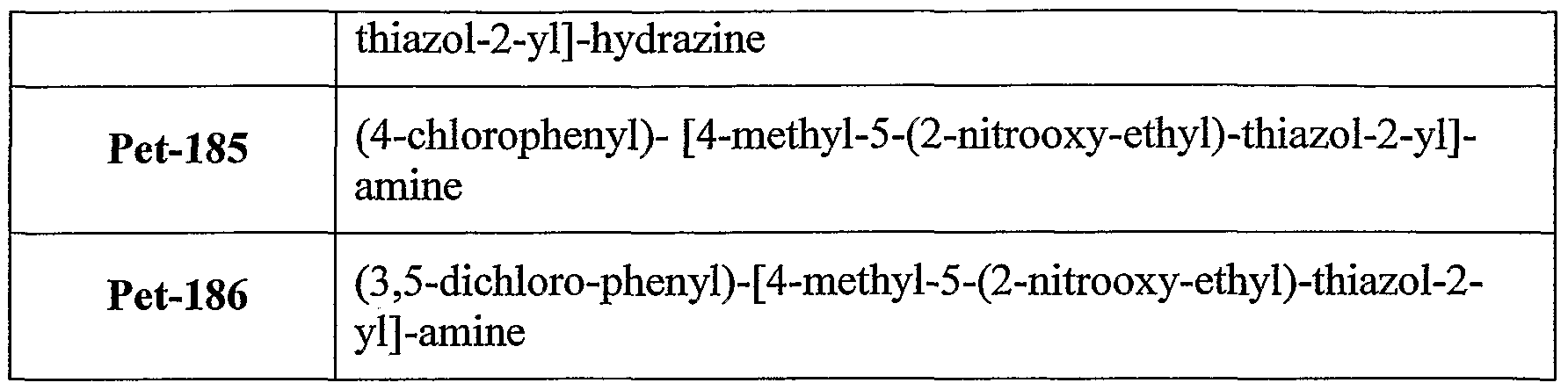NITRIC OXIDE DONORS FOR TREATING INFLAMMATORY BOWEL DISEASES
FIELD AND BACKGROUND OF THE INVENTION The present invention relates to a novel class of NO-donating compounds and their use in the treatment of inflammatory bowel diseases such as, for example, Crohn's disease and ulcerative colitis. Inflammatory bowel disease, or IBD, is a collective term encompassing related, but distinct, inflammatory disorders of the gastrointestinal tract, such as Crohn's disease (CD), ulcerative colitis (UC), indeterminate colitis, microscopic colitis and collagenous colitis, with Crohn's disease and ulcerative colitis being the most common diseases. Ulcerative colitis is confined to the large intestine (colon) and rectum, and involves only the inner lining of the intestinal wall. Crohn's disease may affect any section of the gastrointestinal tract (e.g., mouth, esophagus, stomach, small intestine, large intestine, rectum and anus) and may involve all layers of the intestinal wall. Both diseases, as well as other IBDs, are characterized by abdominal pain and cramping, diarrhea, rectal and/or intestinal bleeding, weight loss and fever. The symptoms of these diseases are usually progressive, and sufferers typically experience periods of remission followed by severe flare-ups. Less frequent, but also possible, IBD symptoms reflect mucosal inflammation of other sections of the GI tract, such as duodenitis, jejunitis and proctitis. Since patients oftentimes seek medical assistance at advanced stages of the disease, most of the IBDs are diagnosed only when the condition becomes chronic. A detailed description of IBD symptoms is found in, for example, Northfield, Drugs, Vol. 14, pages 198-206 (1977); Blaker et al, Eur. J. Pediatr., Vol. 139, pages 162-164 (1982); Singleton, The Gastroenterology Annual, pages 268-310 (1983); Saco et al, J. Amer. Acad. Dermatol, Vol. 4, pages 619-629 (1981); Prantera et al, Ital. J. Gastroenterol., Vol. 13, pages 24-27 (1981); Sales et al, Arch. Int. Med., Vol. 143, pages 294-299 (1983); and Ament, Inflammatory Bowel Diseases, Martinus Nijhoff Publ., Boston, Mass., pages 254-268 (1982). For most patients, IBD is a chronic condition with symptoms lasting for months to years. It is most common in young adults, but can occur at any age. It is found worldwide, but it is most common in industrialized countries such as the United
States, England, and northern Europe. In fact, IBD affects an estimated two million people in the United States alone. Protracted IBD is also known as a risk factor for colon cancer. Diagnosis of IBD is based on the clinical symptoms, the use of a barium enema for radioscopy, and/or direct visualization (sigmoidoscopy or colonoscopy), with the latter being the most accurate test. For the diagnosis of Crohn's disease, see, for example, U.S. Patents Nos. 6,348,452 and 6,297,015. The exact causes of IBD are not yet understood. Common hypotheses include, for example, disorders in the immune system and actions of pro-inflammatory cytokines and selective activation of lymphocyte subsets, which perpetuate unrestrained activation of an inflammatory response in the intestine. To date IBD has no cure. Patients afflicted with IBD are generally treated, currently with therapies that are directed at reducing the iriflammatory processes, and at reducing the effects of the inflammatory processes on the patients. The presently known medical treatment of IBD is intended to decrease the number, frequency and severity of acute exacerbations of inflammatory bowel disease and to prevent secondary complications, but at best, the results are disappointing. The presently known methods for treating IBD have involved anti- inflammatory drugs, immunosuppressive drugs and surgery. The most commonly used medications to treat IBD are non-steroidal anti- inflammatory drugs (NSAID) such as paracetamol and the family of salicylates. Preparations of salicylate are effective in treating mild to moderate disease and can also decrease the frequency of disease flares when the medications are taken on a prolonged basis. Examples of salicylates include sulfasalazine, olsalazine, and mesalamine. Particularly, sulfasalazine and related drugs having the bioactive 5- amino-salicylic acid (5-ASA) moiety are widely used to control moderate IBD symptoms and to maintain remission. All of these medications are given orally in high doses for maximal therapeutic benefit. Although a recent study questioned the link between non-steroidal anti-inflammatory drugs and the exacerbations of inflammatory bowel disease [Forrest, K. et al, Aliment Pharmacol Ther., 2004;20(10): 1035-43], treatments with these medications is typically accompanied with adverse side effects such as nausea, dizziness, changes in blood chemistry (including anemia and leukopenia), skin rashes and drug dependence. In addition,
recent findings, as well we novel regulations of the FDA have implicated these medications with elevation of blood pressure, cardiac arrest and death. Corticosteroids are more potent and faster-acting anti-inflammatory drugs in the treatment of IBD, as compared with salicylates. Prednisone, for example, is a corticosteroid commonly used in the treatment of severe cases of IBD. Nevertheless, potentially serious side effects limit the use of corticosteroids to patients with more severe disease. Side effects of corticosteroids usually occur upon long term use and include thinning of the bone and skin, infections, diabetes, muscle wasting, rounding of facial features, psychiatric disturbances, and, on rare occasions, destruction of hip joints. In cases where IBD patients do not respond to salicylates or corticosteroids, medications that suppress the immune system, namely immunosuppressants, are used. Examples of immunosuppressants include azathioprine and 6-mercaptopurine. However, as immunosuppressants may render the patient immuno-compromised and susceptible to infections and other diseases, the use thereof in the treatment of IBD is oftentimes not recommended. In more severe cases or when the drug therapy fails to relieve the symptoms of
IBD, surgical procedures are used. Typical surgical procedures include colectomy, proctocolectomy and ileostomy (See, Cecil Textbook of Medicine, 19th Edition, Wyngaarden et al, ed., 1992). These surgical treatments are radical procedures that often profoundly alter the everyday life of the patient. In addition to the presently common methods of treating IBD described above, other methods of treating gastrointestinal disorders are disclosed, for example, in U.S. Patents Nos. 5,110,795; 5,112,856; 5,216,002; 5,238,931; 5,292,771; 5,312,818; 5,324,738; 5,331,013; 5,340,801; 5,368,854; 5,391,555; 5,552,439; 5,569,680; 5,599,795; 5,604,231; 5,691,343; 5,693,645; 5,710,181; and 6,372,733, and in WO 95/16461, WO 97/35596, and U.S. Patent Applications having the Publication Nos. 20020006432 and 20040067223. The presently known methods for treating IBD fail to provide a solution for IBD sufferers as these methods (i) fail to provide a substantial cure for IBD, but rather provide treatment of the symptoms; and (ii) include either drug therapy that is accompanied by severe adverse side effects or invasive surgical treatments, both affecting the sufferer's quality of life and may further be life threatening.
There is thus a widely recognized need for new pharmaceuticals which are beneficial in the non-invasive treatment of IBD, which would be safe, effective and side effects-free. Nitric oxide (NO) is a pluripotent free radical with moderate reactivity, which gives rise to a multitude of organ-specific regulatory functions and mediates multiple physiological and pathophysiological processes in the cardiovascular and neurological systems [Evig, CB. et al, Nitric Oxide, 2004, 10(3), 119-29]. The beneficial effects of nitric oxide (NO) as a therapeutic agent in general, and as a blood vessel dilator (vasodilator) in particular, was first observed in 1857, and were demonstrated by the therapeutic activity of a family of compounds, known as nitrovasodilators, which have been used purposely for almost 150 years. The biological activity of NO can be separated into direct and indirect actions
[Wink D.A. and Mitchell J.B., Free Radic. Biol Med. 1998; 25:434-56]. Direct actions typically involve reactions in which the NO radical interacts directly with a biological molecule or target, whereby indirect reactions occur when the final effector molecule is generated by the interaction of NO with reactive oxygen species. A representative example of direct actions is the direct interaction of NO with metal-containing proteins or with organic free radicals. Direct interaction of NO with metals occurs in vivo primarily with iron-containing proteins via such moieties as a haem, leading to the formation of stable nitrosyl adducts [Wink D.A. et al., J. Biol. Chem. 1997; 272:11147-51]. The most notable is the reaction of NO and guanylate cyclase, which leads to the formation of cGMP from GTP [Moncada S. et al., Pharmacol. Rev. 1991;43:109-42]. cGMP has significant regulatory and anti- inflammatory effects, such as the regulation of vascular tone and the inhibition of platelet aggregation and leucocyte adhesion. Superoxide (O2 ~) scavenging is another direct action of NO, which serves to protect haem-containing enzymes involved in prostaglandin synthesis (e.g. cyclooxygenase) from reduction to their inactive forms [Rubbo H. et al., J. Biol. Chem. 1994; 269:26066-75]. Several studies suggest that NO may also modulate iron-catalyzed oxidation reactions by acting as an iron chelator. In vitro, NO can dramatically inhibit the O2 ~-driven Fenton reaction (a most important iron-catalyzed oxidation reaction that produces powerful oxidants such as the hydroxy radical OH-),
suggesting that it may have remarkable antioxidant capabilities [Rubbo, H. et al, J. Biol. Chem. 1994; 269: 26066-75]. Taken- together, the above observations suggest that the direct effects of NO would be involved primarily, but not exclusively, in regulatory, protective and/or anti- inflammatory processes in vivo. In contrast, the indirect actions of NO are mediated by intermediate reactive nitrogen oxide species derived from the interactions of NO with O2 or O2 ~, which give rise to two types of chemical stress: nitrosative and oxidative. Both types of chemical stress are generally thought to be associated with certain pathophysiological situations, such as inflammation, where de novo expression of inducible nitric oxide synthase (iNOS) occurs [Grisham, M.B. et al, Am. J. Physiol. Gastrointest. Liver Physiol. 1999; 39: G315-21]. Biological NO is synthesized by the enzyme nitric oxide synthase (NOS) that generates NO from L-arginine by oxidation of a terminal nitrogen in the amino acid, yielding NO and L-citrulline. This enzyme exists in three different forms (referred to as isoforms): NOS-1, NOS-2 and NOS-3. Each isoform generates NO under different conditions. NOS-1 is the neural isoform (also known as the brain isoform) and is a key component in synaptic transmission. NOS-2 (also known as inducible NOS (iNOS) is responsible for generating high concentrations of NO (100 to 1000 folds higher then the normal NO biological concentration), typically in response to the presence of bacteria. iNOS is produced by macrophages and is responsible for their effects to repair injury and warding off infections. iNOS is regulated at the transcriptional level and is sensitive to inhibitors of DNA transcription and protein synthesis, such as actinomycin-D and cycloheximide [Morris S.M. and Billiar T.R., Am. J. Physiol. 1994; 266.E829-39]. NO production by iNOS is delayed by several hours following stimulation, but once induced is active for periods as long as 5 days. The delay between stimulation and enzyme generation suggests the requirement of de novo synthesis of a cofactor, e.g. tetrahydrobiopterin [Stuehr D.J. and Griffith O.W., Adv. Enzymol Relat. Areas Mol. Biol. 1992; 65:287-346] for achieving maximal activity. NOS-3 (also known as endothelial NOS or eNOS) is found in endothelial cells lining the inner surface of all blood vessels and lymph ducts. eNOS is activated by the pulsatile flow of blood through vessels, which exerts "shear stress" on the membrane of the endothelial cells. The NO generated by eNOS is responsibly for
maintaining the diameter of blood vessels, to thereby maintain an optimal level of tissues perfusion, as well as for the growth of new blood vessels (angiogenesis). The role of NO in the emergence, progression and remission of IBD is still a subject for active research. At present there are many indications that NO is involved in IBD in a crucial way, yet even the most basic question whether NO is beneficial or harmful in IBD conditions remains a matter of boisterous debates, mostly due to perplexing results and inadequate animal and disease models [Kolios, G., V. Nalatas, et al. (2004), Immunology 113(4): 427-37]. The amounting evidences for NO association in IBD conditions may be summarize as follows: NO is not cytotoxic for intestinal tissue and may be an indispensable homeostatic regulator; NO production levels are heightened during IBD inflammation; and chronic overproduction of NO via sustained overexpression of iNOS may be detrimental in IBD conditions. These circumstantial empirical evidences leave the above key question open: are the heightened NO levels cause the inflammation or aggravate IBD conditions, or whether NO overproduction in the settings of local or systemic inflammatory responses that has been evolutionary selected to occur because it provides the host with an overall survival advantage. Studies conducted to examine the direct effects of NO on epithelial cell integrity have shown that NO per se is not cytotoxic for intestinal tissue [Kubes, P., et al, Am. J. Physiol. 1995; 269: G34-41]. On the contrary, eNOS-derived NO appears to be a homeostatic regulator of numerous essential functions of the gastrointestinal mucosa, such as maintenance of adequate perfusion [Moncada, S., Ada Physiol
Scand. 1992; 145: 201-27], and regulation of microvascular and epithelial permeability [Alican, I. and Kubes, P., Am. J. Physiol, 1996; 33: G225-37 and Kubes,
P., Am. J. Physiol. 1992; 262: GI 138-42]. The latter strongly reflects the functional integrity of the gastrointestinal mucosa barrier, and its disturbance is considered to be a quantitative index of injury or dysfunction. Inhibition of NO production has been found to increase the epithelial permeability to substances of low molecular weight, whereby this effect was reversed when NO donors were applied. This function of NO has been attributed to both an increase of cGMP content of intestinal epithelia and to the NO suppressive effects on platelet-activation factor (PAF) and histamine secretion by mucosal mast cells [Kanwar, S. et al., Am. J. Physiol. 1994; 266: G222-9].
Administration of exogenous NO by means of NO-donors has been reported to protect the gastrointestinal mucosa against damage induced by several irritants [Whittle, B. J. et al. (1990), Rr. J. Pharmacol. 99(3): 607-11 and Kitagawa, H. et al (1990), J. Pharmacol. Exp. Ther. 253(3): 1133-7], including maintenance of blood " flow, inhibition of platelet and leucocyte adhesion and/or aggregation within the vasculature, down-regulation of mast cell reactivity, and modulation of oxidative stress [Alican, I. and Kubes, P., Am. J. Physiol. 1996; 33: G225-37, Peng, H.B. et al, J. Biol. Chem. 1995; 270: 14214-9 and Payne, D. and Kubes, P., Am. J. Physiol. 1993; 265: GI 89-95]. Accordingly, NO donors have been found to double the plasma antioxidant capacity of animals subjected to reperfusion-induced mucosal injury. These findings suggest a potential application for these compounds in situations in which the gastrointestinal mucosa is exposed to noxious substances or in which mucosal defense is impaired. In addition, NO-donors have been shown to accelerate the healing of preexisting ulcers in the gastrointestinal tract [Elliott, S. N. et al. (1995), Gastroenterology 109(2): 524-30]. Furthermore, transdermal application of a nitroglycerin patch, a clinically used mode of continuous administration of an NO donor, largely used in clinical settings, has been shown to protect the integrity of the gastric mucosa in indomethacin-treated rats [Calatayud, S. et al. (1999), Br. J. Pharmacol. 127(5): 1111-8]. This preparation, when applied to patients who are non steroidal anti-inflammatory drugs (NSAIDs) users has been shown to exhibit a significantly lower risk of gastrointestinal bleeding [Lanas, A. et al. (1998), J. Int. Med. Res. 26(3): 120-8], suggesting that the nitroglycerin patch represents a rational clinical alternative for the prevention of gastric damage. Production of large quantities of NO via the up-regulation of iNOS can have a variety of effects, which may be detrimental or beneficial depending on the amount, duration and anatomical site of synthesis. Production of large quantities of NO can inhibit key enzymes in the mitochondrial electron transport chain and citric acid cycle by nitrosylation of reactive groups, which are essential for enzyme catalytic function [Forstermann, U. et al, Hypertension 199 ,' 23: 1121-31 and Kurose, I. et al, J. Gastroenterol. Hepatol. 1995; 10 (Suppl. 1):S68-71]. Since NO may inhibits DNA synthesis via inactivation of the ribonucleotide reductase enzyme, it may also exert anti-proliferative activity. The above mechanisms may account for the cytotoxic and cytostatic effects of macrophage-derived NO on tumor cells and micro-organisms
[Tepperman, B.L. et al, Am. J. Physiol. 1993; 265: G214-G218 and Fukuo, K. et al, J. Clin. Invest. 1995; 95: 669-16]. Indeed, iNOS-induced NO has been found to exert a direct antimicrobial effect [Fang, F.C., J Clin. Invest. 1997; 99: 2818-25]. Enteroinvasive bacteria (bacteria that can cause inflammation of the stomach and bowels), such as Escherichia coli, Salmonella and Shigella, can directly induce iNOS expression, suggesting an important role of iNOS in the intestinal antibacterial response [Witthoft, T. et al, Am. J. Physiol. 1998; 275: G564-71 and Kolios, G. et al, Gut 1998; 43: 56-63]. Thus, apart from being an important component of the host defense system, iNOS-mediated NO production may occasionally become part of a dysregulated immune response, resulting in chronic inflammatory disorders. One of the settings where this hypothesis has been most vigorously tested is in IBD, where NO produced following the up-regulation of iNOS in epithelial cells has been closely associated with the initiation and maintenance of intestinal inflammation. Several studies have shown that the inhibition of NO causes many of the hallmark features of intestinal inflammation, whereas the delivery of exogenous NO originating, e.g., from NO-donating compounds, reduces the sequelae of acute inflammation. On the other hand, the up-regulation of the NO producing iNOS has been shown to correlate well with prolonged colonic inflammation, especially within epithelial cells around inflammatory foci [Kolios, G. et al, Gut 1998; 43: 56-63]. Excess NO produced by the iNOS may theoretically exacerbate the clinicopathological features of ulcerative colitis (UC) by direct cytotoxicity, activation of neutrophils [Ribbons, K.A. et al, Gastroenterology 1995; 108: 705-11], vasodilatation, reduced smooth muscle tone [Middleton, S.J. et al, Gut 1993; 34: 814- 7], increased production of nitrosamines (to cause cancer) [Ohshima, H. and Bartsch, H., MutatRes. 1994; 305: 253-64], and interaction with superoxide to form the highly toxic peroxynitrite radical [Singer, I.I. etal, Gastroenterology 1996; 111: 871-85]. The link between up-regulation and activity of iNOS to the exacerbation of IBD may not be correlated to NO, but rather to a family of species, metabolites and co-products that react differently in other environmental conditions, such as citruUine, the co-product of iNOS. The concentrations of citruUine were found to be higher in rectal biopsy specimens from patients with active UC than in those from patients with quiescent disease or a normal histology, while incubation with Nω-monomethyl-L-
arginine (L-NMMA), an effective inhibitor of all types of NOS, significantly reduced the concentration of citruUine in colonic biopsies, suggesting that the increased biosynthesis of citruUine must be a consequence of NO synthase activity, which simultaneously produces NO [Middleton, S.J. et al, Lancet 1993; 341: 465-6]. Epithelial disruption in the small and large intestine following the expression of iNOS as a consequence of endotoxin challenge has also been well characterized [Tepperman, B. L. et al. (1994), J. Pharmacol. Exp. Ther. 271(3): 1477-82]. This again is a feature of the gut inflammatory reaction that may reflect an important involvement of iNOS. It has been demonstrated that the organism implicated in the pathogenesis of peptic ulceration, Helicobacter pylori, can elaborate a factor that induces iNOS in macrophage cell lines [Wilson, K. T. et al. (1996), Gastroenterology 111(6): 1524-33]. A few empirical conclusions stem from the contradictional studies described above: NO is an indispensable protector of intestinal lining mucosal cells; selective inhibition of NO producing iNOS improves IBD symptoms [Barrachina, M. D. et al. (2001), Curr. Pharm. Des. 7(1): 31-48 and Martinez-Cuesta, M. A. et al. (1997), J. Pharm. Pharmacol. 49(10): 988-90]; and exogenous NO, e.g., NO derived from NO- donors, reduces the sequelae of acute iriflammation. Pharmacological compounds that release NO (also known as NO-donors) have been useful tools for evaluating the pivotal role of NO in physiology and therapeutics. These agents constitute two broad classes of compounds, those that release NO or one of its redox congeners spontaneously, and those that require enzymatic metabolism to generate NO. Several commonly used cardiovascular drugs exert their beneficial action, in part, by modulating the NO pathway. While NO is a gas, it may be directly administered by inhalation. However, although this administration route is used in cases where improved patient oxygenation is required, as, for example, in pulmonary hypertension (high blood pressure in the lungs) and in patients with sickle cell anemia, such direct administration of the NO active form may not reach the target organ and/or biological system, and is oftentimes associated with both biochemical and medical complications, including, for example, methemoglobinemia and direct pulmonary injury.
In a search for alternative routes for administering NO, it was found that NO may be delivered and generated in situ by means of prodrugs. These prodrugs are known as NO-donors, which are metabolized by means of an enzymatic mechanism so as to generate or release active NO. NO-donors, which are also referred to interchangeably, herein and in the art, as NO prodrugs or NO-donating agents) are pharmacologically active substances that spontaneously release, or are metabolized to, NO or its redox congeners. However, while the beneficial effects of administering NO-donors have been widely recognized, treatment with conventional nitrate preparations is typically limited by their therapeutic bioavailability half-life, lack of selectivity, systemic absorption accompanied by potentially adverse hemodynamic effects, and drug tolerance (namely, reduced medicinal response which develops during prolonged use), with the latter being with the presently most limiting feature associated with administration of NO-donors [Ignarro LJ. et al, J Cardiovasc Pharmacol. 1999; 34: 879-886, Kojda G. et al, Cardiovasc Res. 1999; 43: 562-571, Loscalzo J. et al, Humana Press; 2000, Loscalzo J. et al, Circ Res. 2001; 88: 756-762, Loscalzo J., Circulation. 2000; 101: 2126-2129 and Napoli C. et al, Nitric Oxide. 2001; 5: 88- 97]. The inadequacies in current NO-donor prodrugs have limited their use to only short-term management of angina pectoris and acute heart failure. As an alternative treatment, novel NO-donating drugs which may offer selective effects, a prolonged half-life, and/or a reduced incidence of drug tolerance are currently in various developmental stages. Among these are diazeniumdiolates, known as "NONOates" (1 -substituted diazen-l-ium-l,2-diolates, e.g., DETA NONOate) [Keefer LK. et al, Methods Enzymol. 1996, 268, pp. 281-93], S- nitrosothiols (e.g., SNAP) [Ng ES, Kubes P, Can J Physiol Pharmacol. 2003, 81(8), pp. 759-64] and mesoionic oxatriazoles (e.g., GEA3162 or l,2,3,4-oxatriazolium-5- amino-3-(3,4-dichlorophenyl)-chloride) [Karup G. et al, Pol J Pharmacol. 1994, 46(6), pp. 541-52]. However, heretofore these compounds are still in pre-clinical phases and are mostly used as biochemical and pharmacological tools Some conjugates of NSAIDs and a moiety that generates NO have been recently synthesized, and their properties have been characterized in several species [Wallace, J. L. et al. (1994), Gastroenterology 107(1): 173-9]. Since it has been shown that some of the adverse side effects associated with NSAIDs involve
decreased NO levels, these conjugates were aimed at maintaining the therapeutic properties of the NSAIDs while minimizing their side effects [Muscara, M. N. et al. (1998), Life Sci. 62(15): PL235-40 and Wallace, J. L. et al. (1998), J. Clin. Gastroenterol. 27 Suppl 1: S28-34]. In view of the limitations associated with the presently know drugs and methods for the treatment of IBD, and the limitations associated with utilizing the presently known NO-donors, there is a widely recognized need for, and it would be highly advantageous to have NO-donating compounds which are devoid of the above limitations and can be used in the treatment of IBD.
SUMMARY OF THE INVENTION According to one aspect of the present invention there is provided a method of treating an inflammatory bowel disease, which comprises administering to a subject in need thereof a therapeutically effective amount of an NO-donating compound comprising an NO-releasing group and a chemical moiety being covalently attached to the NO-releasing group, such that when NO is released from the compound a residue which is a naturally occurring metabolite is formed. According to further features in preferred embodiments of the invention described below, the administration of the NO-donors is effected orally, rectally, intravenously, topically, intranasally, intradermally, transdermally, subcutaneously, intramuscularly, intrperitoneally, intraperitoneally, by inhalation or by intrathecal catheter. According to still further features in the described preferred embodiments the method further includes administering to the subject an additional active ingredient, the additional active ingredient being capable of treating the inflammatory bowel disease. According to another aspect of the present invention there is provided a pharmaceutical composition identified for use in the treatment of an inflammatory bowel disease comprising, as an active ingredient, a NO-donating compound as described herein and a pharmaceutically acceptable carrier. According to further features in preferred embodiments of the invention described below, the pharmaceutical composition further includes an additional active ingredient being capable of treating the inflammatory bowel disease.
According to still further features in the described preferred embodiments the pharmaceutical composition is packaged in a packaging material and identified in print, in or on the packaging material, for use in the treatment of the inflammatory bowel disease. According to another aspect of the present invention there is provided a use of an NO-donating compound as described herein in the treatment of an inflammatory bowel disease. According to another aspect of the present invention there is provided a use of an NO-donating compound as described herein for the preparation of a medicament for treating an inflammatory bowel disease. According to further features in preferred embodiments of the invention described below, the formation of the naturally occurring metabolite substantially prevents or decreases a development of tolerance to the NO-donating compound. According to still further features in the described preferred embodiments the naturally occurring metabolite residue that is capable of inhibiting an activity of inducible nitric oxide synthase. According to still further features in preferred embodiments of the invention described below, the NO-releasing group is selected from the group consisting of an - ONO2 group, a -SNO group, a diazeniumdiolate and a mesoionic oxatriazole. According to still further features in preferred embodiments of the invention described below, the naturally occurring metabolite is a thiamine metabolite. According to still further features in the described preferred embodiments the chemical moiety includes a substituted or unsubstituted tbiazole ring. According to still further features in the described preferred embodiments the NO-donating compound further includes a bioactive agent residue covalently attached to the chemical moiety. According to still further features in the described preferred embodiments the bioactive agent residue is attached to the chemical moiety via a biocleavable moiety. According to still further features in the described preferred embodiments the bioactive agent residue is selected from the group consisting of an inducible nitric oxide synthase inhibitor residue, an inflammatory bowel disease drug residue, a fatty acid residue, a metabolite residue, a carbohydrate residue, an amino acid residue, a peptide residue, a protein residue, a hydroxamic acid residue, a nicotinic acid residue,
a nicotinamide residue, a carnitine residue, a co-enzyme residue, a beta carotene residue, a bromelain residue, a steroidal anti-inflammatory agent residue, a non- steroidal anti-inflammatory drug residue, an anti-psychotic agent residue, an anti- thrombogenic agent residue, an anti-platelet agent residue, an anti-coagulant residue, an anti-diabetic agent residue, a growth factor residue, a statin residue, a toxin residue, an antimicrobial agent residue, an analgesic residue, an anti-metabolic agent residue, a vasoactive agent residue, a vasodilator agent residue, a prostaglandin residue, a hormone residue, a thrombin inhibitor residue, an enzyme residue, an oligonucleotide residue, a nucleic acid residue, an antisense residue, a protein residue, an antibody residue, an antigen residue, a vitamin residue, an immunoglobulin residue, a cytokine residue, a cardiovascular agent residue, a chemotherapeutic agent residue, an antioxidant residue, a phospholipid residue, an anti-proliferative agent residue, a heparin residue, and any combination thereof. According to still further features in the described preferred embodiments the biocleavable moiety is selected from the group consisting of amide, carboxylate, carbonate, carbamate, phosphate, hydrazide, thiohydrazide, disulfide, epoxide, peroxo and methyleneamine. According to still further features in the described preferred embodiments, the NO-donating compound utilized in the present invention has the general formula I:
Formula I wherein: A is selected from the group consisting of alkenyl, alkoxy, alkyl, alkynyl, amine, amine-oxide, aryl, aryloxy, azo, borate, C-amide, carbonyl, C-carboxylate, C-thiocarboxylate, cycloalkyl, diazo, disulfide, guanidine, guanyl, haloalkyl, heteroalicyclic, heteroaryl, hydrazine, N-amide, N-carbamate, N-dithiocarbamate, nitro, N-sulfonamide, N-thiocarbamate, O-carbamate, O-carboxylate, O-thiocarbamate, O-thiocarboxylate, oxime, oxygen, sulfur, peroxo, phosphate, phosphine-oxide, phosphine-sulfide, phosphinyl, phosphite, phosphonate, pyrophosphate, S-dithiocarbamate, silaza, silicate, siloxy, silyl, S-sulfonamide,
sulfate, sulfite, sulfonate, sulfoxide, sulfur, thioalkoxy, thioaryloxy, thiocarbonyl, thiophosphate, thiosulfate, thiosulfite, thiourea, triphosphate, urea, a biocleavable moiety and any combination thereof, or absent; X is selected from the group consisting of acyl-halide, alkenyl, alkoxy, alkyl, alkynyl, amine, amine-oxide, aryl, aryloxy, azo, borate, C-amide, carbonyl,
C-carboxylate, C-thiocarboxylate, cyano, cycloalkyl, diazo, disulfide, guanidine, guanyl, halide, haloalkyl, heteroalicyclic, heteroaryl, hydrazine, hydrogen, hydroxy,
N-amide, N-carbamate, N-dithiocarbamate, nitro, N-sulfonamide, N-thiocarbamate,
O-carbamate, O-carboxylate, O-thiocarbamate, O-thiocarboxylate, oxime, peroxo, phosphate, phosphine-oxide, phosphine-sulfide, phosphinyl, phosphite, phosphonate, pyrophosphate, S-dithiocarbamate, silaza, silicate, siloxy, silyl, S-sulfonamide, sulfate, sulfite, sulfonate, sulfoxide, thioalkoxy, thioaryloxy, thiocarbonyl, thiohydroxy, thiophosphate, thiosulfate, thiosulfite, thiourea, triphosphate, urea, a bioactive agent residue as described herein, a moiety containing one or more NO- releasing group as described herein, a substituted or unsubstituted tliiazole and any combination thereof; B is selected from the group consisting of a saturated or unsaturated, substituted or unsubstituted alkylene chain having 1-20 carbon atoms, and a saturated or unsaturated, substituted or unsubstituted alkylene chain having 1-20 carbon atoms interrupted by one or more heteroatom, whereby the heteroatom or heteroatoms include oxygen, sulfur, nitrogen, phosphor, silicon and any combination thereof; Y is the NO-releasing group; and Z is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, amine, cycloalkyl, heteroalicyclic, aryl, heteroaryl, halide, haloalkyl, hydroxy, thiohydroxy, alkoxy, thioalkoxy, aryloxy and thioaryloxy. According to still further features in the described preferred embodiments the NO-releasing group denoted as Y in formula I is selected from the group consisting of a -ONO2 group, a -SNO group, a diazeniumdiolate and a mesoionic oxatriazole. Preferably Y is an -ONO group. According to still further features in the described preferred embodiments the group denoted Z in formula I is alkyl. Preferably the alkyl is methyl. According to still further features in the described preferred embodiments the group denoted B in formula I is an ethylene chain.
According to still further features in the described preferred embodiments the group denoted B in formula I is selected from the group consisting of -CH2-CH2-O- CH2- , -CH2-CH2-NH-CH2- and -CH2-CH2-S-CH2-. According to still further features in the described preferred embodiments the group denoted X in formula I is aryl. Preferably the aryl is selected from the group consisting of a substituted phenyl and an unsubstituted phenyl. According to still further features in the described preferred embodiments the group denoted X in formula I is heteroaryl. Preferably the heteroaryl is selected from the group consisting of pyridin-3-yl and N-methylpyrazin-2-amine-6-yl. According to still further features in the described preferred embodiments the group denoted X in formula I is amine. Preferably the amine is selected from the group consisting of-NH , prop-2-en-l -amine and naphthalen-1 -amine. According to still further features in the described preferred embodiments the group denoted X in formula I is a bioactive agent as described above. According to still further features in the described preferred embodiments the bioactive agent is a non-steroidal anti-inflammatory drug residue. According to still further features in the described preferred embodiments the non-steroidal anti- inflammatory drug is selected from the group consisting of aspirin, celecoxib, diclofenac, diflunisal, etodolac, fenoprofen, flurbiprofen, ibuprofen, indomethacin, ketoprofen, ketorolac, meclofenamate, mefenamic acid, nabumetone, naproxen, oxaprozin, oxyphenbutazone, phenylbutazone, piroxicam, rofecoxib sulindac and tolmetin. Preferably the non-steroidal anti-inflammatory drug residue is selected from the group consisting of an aspirin residue, an ibuprofen residue and a naproxen residue. According to still further features in the described preferred embodiments the bioactive agent residue is an inflammatory bowel disease drug residue. According to still further features in the described preferred embodiments the inflammatory bowel disease drug is selected from the group consisting of 5- aminosalicylic acid, 4-aminophenylacetic acid, sulphasalazine, olsalazine, mesalazine, rifaximin, rifampin, hydrocortisone, prednisolone, budesonide, azathioprine, 6- mercaptopurine, cyclosporin, methotrexate, metronidazole, tinidazole, loperamide, diphenoxylate, atropine, cholestylamine, colestipol and paracetamol.
According to still further features in the described preferred embodiments the bioactive agent residue is an inducible nitric oxide synthase inhibitor residue. According to still further features in the described preferred embodiments the inducible nitric oxide synthase inhibitor is selected from the group consisting of (-)- noformycin, (1 S,5S,6R,7R)-7-chloro-5-methyl-2-aza-bicyclo[4.1.0]heptan-3-imine,
(S,E)-3-(4-chlorophenyl)-N-(l-oxo-l-(2-oxo-2-(4-(6-(trifluoromethyl)pyrimidin-4- yloxy)piperidin- 1 -yl)ethylamino)-3 -(pyridin-2-yl)propan-2-yl)acrylamide, 1 -amino-2- hydroxy-guanidine, 2-aminoethyl-isothiourea, 2-benzyl-2-thio-pseudourea, 2- iminobiotin, 3-hydroxy-4-methyl-5-pentyl-2-iminopyrrolidine, 4-methyl-5- propyloxazolidin-2-imine, 4-methyl-5-propylthiazol-2-amine, 5-tert-butyl-4- methylthiazol-2-amine, 8-(3-chlorostyryl)caffeine, alloxazine, aminoguanidine, deltoin, dexamethasone, geldanamycin, Gingivex®, guanidinoethyldisulphide, imperatorin, L-canavanine, L-N6-(l-iminoethyl)lysine 5-tetrazole amide,, mercaptoethylguanidine, methyl 4-(2-(lH-imidazol- 1 -yl)pyrimidin-4-yl)-3-(2- (benzo [d] [ 1 ,3] dioxol-5-ylmethylamino)-2-oxoethyl)piperazine- 1 -carboxylate, N-(3 -
(aminomethyl)benzyl) acetamidine, N-(5(S)-amino-6,7- dihydroxyheptyl)ethanimidamide, . NG-monomemyl-L-arginine, N-iminoethyl-L- lysine, N-iminoethyl-L-ornithine, Nω-nitro-L-arginine methyl ester, S-(4-nitrobenzyl)-
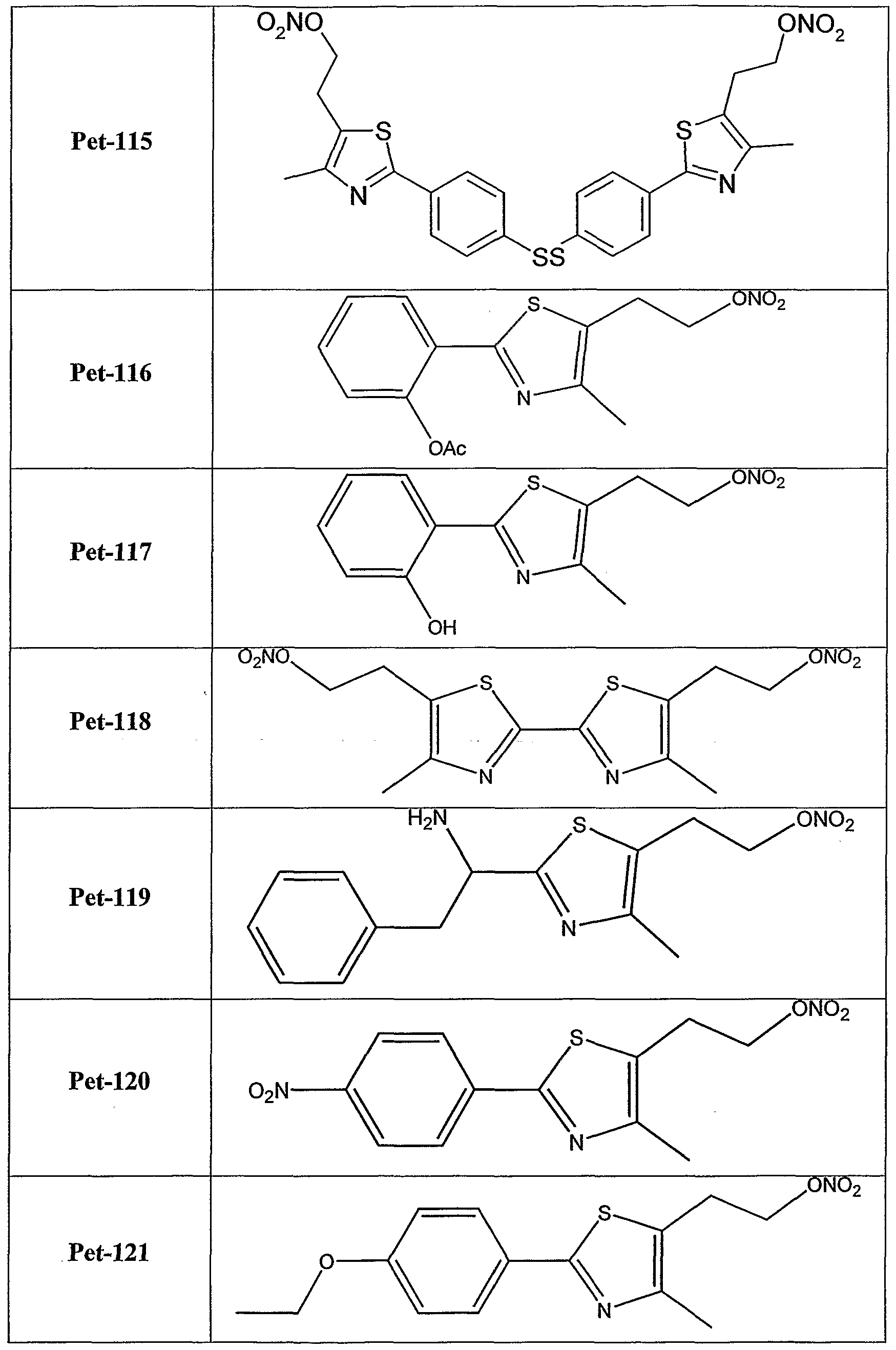
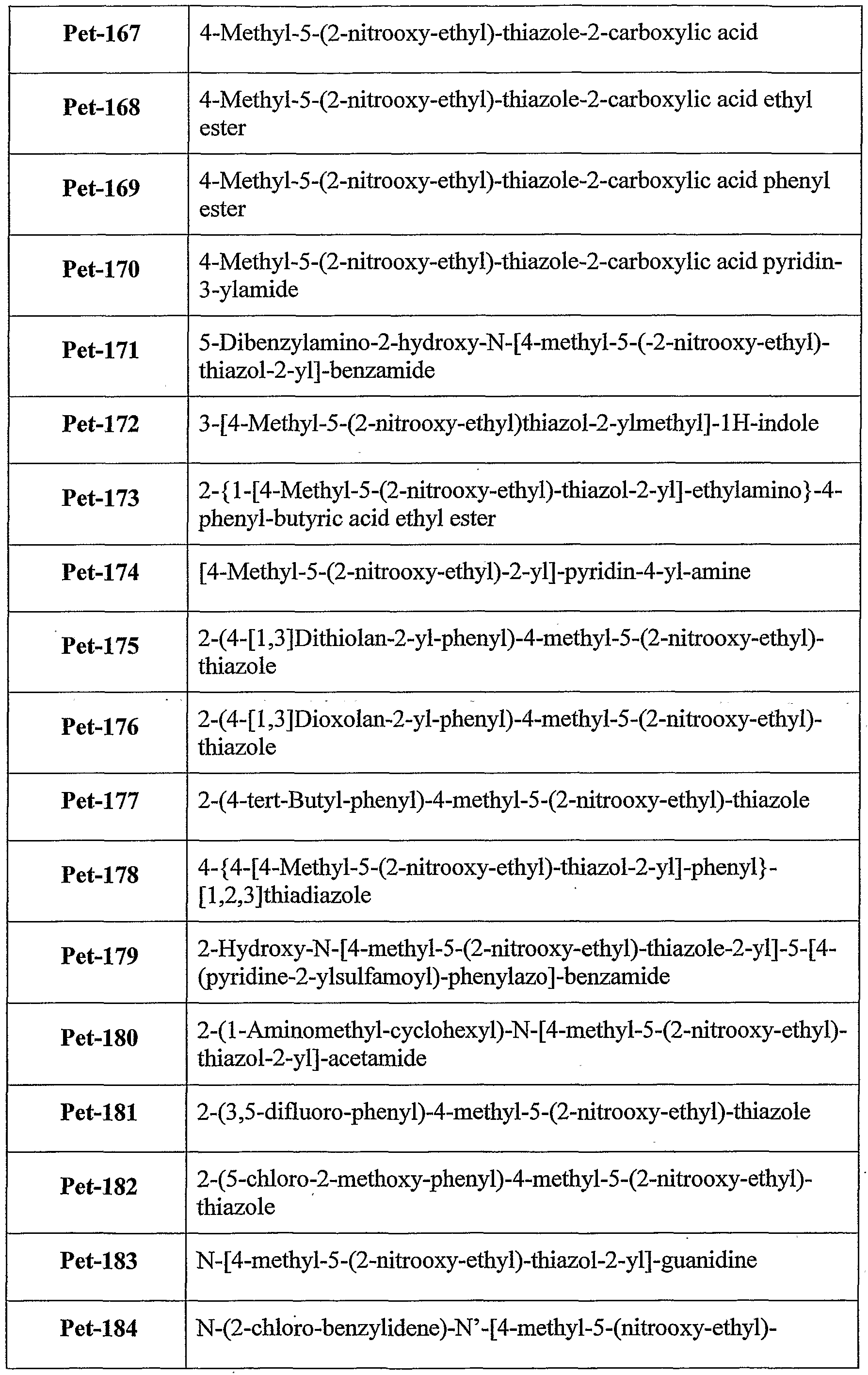
6-thioinosine, S,S'-l,4-phenylene-bis(l,2-ethanediyl)bis-isotbiourea, salicylate, S- ethylisothiourea and S-methylisothiourea. According to still further features in the described preferred embodiments the bioactive agent residue is a metabolite residue. According to still further features in the described preferred embodiments the metabolite residue is a nicotinic acid residue. According to still further features in the described preferred embodiments the group denoted A in formula I is a biocleavable moiety. Preferably, the biocleavable moiety is selected from the group consisting of amide, carboxylate, carbonate, carbamate, phosphate, hydrazide, thiohydrazide, disulfide, epoxide, peroxo and methyleneamine. According to still further features in the described preferred embodiments A is a biocleavable moiety and X is a bioactive agent residue as described hereinabove. Exemplary NO-donating compounds utilized in the various aspects of the present invention are set forth in Tables 1 and 2 hereinbelow.
The present invention successfully addresses the shortcomings of the presently known configurations by providing a method of treating IBDs which utilizes a novel class of NO-donating agents that exhibit high therapeutic effect while being non- tolerance inducing. Unless otherwise defined, all technical and scientific terms used herein have the same meaning as commonly understood by one of ordinary skill in the art to which this invention belongs. Although methods and materials similar or equivalent to those described herein can be used in the practice or testing of the present invention, suitable methods and materials are described below. In case of conflict, the patent specification, including definitions, will control. In addition, the materials, methods, and examples are illustrative only and not intended to be limiting. As used herein, the term "treating" includes abrogating, substantially inhibiting, slowing or reversing the progression of a condition, substantially ameliorating clinical or aesthetical symptoms of a condition or substantially preventing the appearance of clinical or aesthetical symptoms of a condition. The term "comprising" means that other steps and ingredients that do not affect the final result can be added. This term encompasses the terms "consisting of and "consisting essentially of. The phrase "consisting essentially of means that the composition or method may include additional ingredients and/or steps, but only if the additional ingredients and/or steps do not materially alter the basic and novel characteristics of the claimed composition or method. The term "method" refers to manners, means, techniques and procedures for accomplishing a given task including, but not limited to, those manners, means, techniques and procedures either known to, or readily developed from known manners, means, techniques and procedures by practitioners of the chemical, pharmacological, biological, biochemical and medical arts. The term "active ingredient" refers to a pharmaceutical agent including any natural or synthetic chemical substance that subsequent to its application has, at the very least, at least one desired pharmaceutical or therapeutic effect. As used herein, the singular form "a," "an," and "the" include plural references unless the context clearly dictates otherwise. For example, the term "a compound" or
"at least one compound" may include a plurality of compounds, including mixtures thereof. Throughout this disclosure, various aspects of this invention can be presented in a range format. It should be understood that the description in range format is merely for convenience and brevity and should not be construed as an inflexible limitation on the scope of the invention. Accordingly, the description of a range should be considered to have specifically disclosed all the possible subranges as well as individual numerical values within that range. For example, description of a range such as from 1 to 6 should be considered to have specifically disclosed subranges such as from 1 to 3, from 1 to 4, from 1 to 5, from 2 to 4, from 2 to 6, from 3 to 6 etc., as well as individual numbers within that range, for example, 1, 2, 3, 4, 5, and 6. This applies regardless of the breadth of the range. Whenever a numerical range is indicated herein, it is meant to include any cited numeral (fractional or integral) within the indicated range. The phrases "ranging/ranges between" a first indicate number and a second indicate number and "ranging/ranges from" a first indicate number "to" a second indicate number are used herein interchangeably and are meant to include the first and second indicated numbers and all the fractional and integral numerals therebetween.
BRIEF DESCRIPTION OF THE DRAWINGS The invention is herein described, by way of example only, with reference to the accompanying drawings. With specific reference now to the drawings in detail, it is stressed that the particulars shown are by way of example and for purposes of illustrative discussion of the preferred embodiments of the present invention only, and are presented in the cause of providing what is believed to be the most useful and readily understood description of the principles and conceptual aspects of the invention. In this regard, no attempt is made to show structural details of the invention in more detail than is necessary for a fundamental understanding of the invention, the description taken with the drawings making apparent to those skilled in the art how the several forms of the invention may be embodied in practice. In the drawings: FIG. 1 is a bar graph demonstrating the in-vivo anti-inflammatory effect of Pet-152, Pet-154, Pet-155 and Pet-10, exemplary NO-donors according to the
present invention, as compared with the effect of 5-ASA, in colitis-induced rats, reflected by the effect on the activity of myeloperoxidase measured spectrophotometrically by following the decomposition of hydrogen peroxide in the presence of o-dianisidine in washed rat colon tissue (error bars represent the mean ±standard errors, n=4); FIG. 2 is a bar graph demonstrating the in-vivo anti-inflammatory effect of Pet-8 and Pet-8-OH, an exemplary NO-donor and NO-donor intermediate compound according to the present invention, as compared with the effect of 5-ASA, in colitis- induced rats, reflected by the effect on the activity of myeloperoxidase measured spectrophotometrically by following the decomposition of hydrogen peroxide in the presence of o-dianisidine in washed rat colon tissue (error bars represent the mean ±standard errors, n=4); FIG. 3 is a bar graph demonstrating the in-vivo anti-inflammatory effect of Pet-8, Pet-8-OH, Pet-l-OH and Pet-1, exemplary NO-donors and NO-donor intermediate compounds (Pet-8-OH and Pet-l-OH) according to the present invention, as compared with the effect of 5-ASA, in colitis-induced rats, reflected by the effect on the activity of myeloperoxidase measured spectrophotometrically by following the decomposition of hydrogen peroxide in the presence of o-dianisidine in washed rat colon tissue (error bars represent the mean ±standard errors, n=4); FIG. 4 is a bar graph demonstrating the in-vivo anti-inflammatory effect of
TBA (the thioamide starting material used in the synthesis of Pet-8), Pet-8-OH (the alcohol intermediate in the synthesis of Pet-8), Pet-8, Pet-1, exemplary NO-donors according to the present invention, as compared with the effect of 5-ASA, in colitis- induced rats, reflected by the effect on the activity of myeloperoxidase measured spectrophotometrically by following the decomposition of hydrogen peroxide in the presence of o-dianisidine in washed rat colon tissue (error bars represent the mean ±standard errors, n=4); and FIG. 5 is a bar graph demonstrating the in-vivo anti-inflammatory effect of Pet-8, Pet-12, Pet-149 and Pet-24, exemplary NO-donors according to the present invention, as compared with the effect of 5-ASA, in colitis-induced rats, reflected by the effect on the activity of myeloperoxidase measured spectrophotometrically by following the decomposition of hydrogen peroxide in the presence of o-dianisidine in washed rat colon tissue (error bars represent the mean ±standard errors, n=4).
DESCRIPTION OF THE PREFERRED EMBODIMENTS The present invention is of the use of a novel family of NO-donating compounds (NO-donors) in the treatment of inflammatory bowel diseases (IBDs). The NO-donating compounds utilized according to the present invention comprise one or more NO-releasing group(s) covalently attached to a chemical moiety, and are designed such that when NO is released from the compound, a residue which is a naturally occurring metabolite is formed. The NO-donating compounds may further include a bioactive moiety (e.g., a drug) and therefore may exhibit dual/synergistic therapeutic effects. The principles and operation of the present invention may be better understood with reference to the drawings and accompanying descriptions. Before explaining at least one embodiment of the invention in detail, it is to be understood that the invention is not limited in its application to the details set forth in the following description or exemplified by the Examples. The invention is capable of other embodiments or of being practiced or carried out in various ways. Also, it is to be understood that the phraseology and terminology employed herein is for the purpose of description and should not be regarded as limiting. As discussed hereinabove, inflammatory bowel diseases present a scientific and pharmaceutical challenge as to the exact causes and methods for treating these diseases. The amounting results of numerous studies point towards the involvement of nitric oxide (NO) in the disease, yet the question of its beneficial versus harmful effect remains unanswered. As further discussed above, partial evidence suggest that NO by itself is beneficial for the cells lining the digestive tract during inflammation, while its over-production by inducible nitric oxide synthase (iNOS), which is up- regulated in IBD conditions, may be responsible for some of its adverse effects. In U.S. Provisional Patent Application No. 60/651,619, by the present inventors, which is incorporated by reference as if fully set forth herein, a novel class of NO-donating compounds is disclosed. These NO-donating compounds comprise an NO-releasing group that is covalently attached to a chemical moiety and are designed such that upon releasing a bioactive NO, a residue of a naturally occurring metabolite is formed. While designing these novel NO-donating compounds, it was envisioned that such NO donors, when entering a biological system, would be subjected to enzymatic reactions which would result in the release of a bioactive NO
and the formation of a residue of a metabolite, whereby this residue, by being derived from a naturally occurring metabolite, would be characterized by inherent biocompatibility, non-toxicity, and efficient absorption, distribution, excretion, metabolism and other biocompatibility related advantages. Above all, it was envisioned that the cleavage of such compounds into a residue that is characterized by such an inherent biocompatibility, would result in preventing or at least in substantially decreasing the development of tolerance to such compounds upon repetitive administration thereof such that a major drawback of prevalent NO-donors would be circumvented. As is demonstrated in U.S. Provisional Patent Application No. 60/651,619, such NO-donors, which were based on thiazole, a residue of the metabolite vitamin B (thiamine), were found to be highly efficacious as vasodilators and in reducing hypertension, and were further found to be non-tolerance inducing upon repetitive administration thereof. As is further described in U.S. Provisional Patent Application No. 60/651,619, some of these NO-donors were designed to further include, in addition to the selected chemical moiety and the NO releasing group, one or more bioactive agent residue(s) and thus exhibit synergistic therapeutic effects, resulting from the dual therapeutic effect of the bioactive agent and the bioactive NO. In view of the abovementioned studies and the findings described in U.S. Provisional Patent Application No. 60/651,619, the present inventors have now envisioned that due to the recognized effect of the NO-donors in treating IBD and due to the unique features of the NO-donors described hereinabove, this new class of NO- donating compounds (also referred to herein interchangeably as NO-donors) could be beneficially used in the treatment of IBDs, while avoiding the adverse phenomena associated with the presently known NO-donors. While reducing the present invention to practice it was indeed found that the NO-donating compounds described in U.S. Provisional Patent Application No. 60/651,619 act as beneficial therapeutic agents in treating IBDs. As is demonstrated in the Examples section that follows, it was found, in in-vivo studies, that treatment with various compounds of this family resulted in a substantial reduction of IBD manifestations. The therapeutic activity of these compounds was found superior to the activity of widely used IBD drugs such as 5-ASA.
As is delineated hereinabove, inadequate somatic NO levels are associated with various biological dysfunctions, which typically result from or lead to adverse decrease in the somatic NO levels. Administering an NO-donating compound to subjects that suffer from such inadequate somatic NO levels therefore ameliorate the biological dysfunction itself or its symptoms. Hence, according to one aspect of the present invention there is provided a method of treating an inflammatory bowel disease, which is effected by administering to a subject in need thereof a therapeutically effective amount of an NO-donating compound. The NO-donating compound utilized in this and other aspects of the present invention comprises an NO-releasing group, as is defined and detailed hereinunder and a chemical moiety being covalently attached to the NO-releasing group. The chemical moiety and the NO-releasing group are selected and attached one to the other such that upon release of NO from the compound, a residue of a naturally occurring metabolite is formed. As is detailed hereinbelow, the NO-donating compounds utilized in this and other aspects of the present invention optionally and preferably further comprise a bioactive agent residue. The bioactive agent residue is preferably attached, either directly or indirectly, preferably via a biocleavable moiety, to the chemical moiety in the compound. As used herein, the phrase "chemical moiety" describes a residue, as this term is defined hereinbelow, of an organic substance. The term "residue", as used herein, refers herein to a major portion of a molecule, which is covalently linked to another molecule, herein the chemical moiety (e.g., a thiamine-derived thiazole), or alternatively, is formed upon cleavage of another molecule. As used herein, the term "metabolite" describes a substance that is typically associated with one or more metabolic processes, that is, a substance produced by a metabolic process, required for a metabolic process and/or participating in a metabolic process. As is discussed hereinabove and is further detailed in U.S. Provisional Patent
Application No. 60/651,619, due to the formation of a residue of a naturally occurring metabolite, the development of tolerance to the NO-donating compounds upon repetitive administration thereof is prevented or at least substantially decreased.
Hence, according to a preferred embodiment of the present invention the NO- donating compounds utilized in this and other aspects of the present invention are characterized as being non-tolerance inducing. As used herein, the phrase "non-tolerance inducing compound(s)" is meant to describe compounds which upon repetitive, administration thereof do not induce tolerance thereto. As is well known in the art, the term "tolerance" described a reduced medicinal response to an administered compound. As is further discussed in detail hereinabove, one of the major adverse effects associated with NO production as a response to inflammatory-damaged tissues in the up-regulation and intensified activity of iNOS. It was found that such an increased activity of iNOS oftentimes cause aggravation of the inflammatory process. Administering an NO-donating compound which can elevate the NO level in a subject while at the same time inhibits the iNOS activity is therefore highly beneficial. Hence, according to a preferred embodiment of the present invention, the chemical moiety and the NO-releasing group are selected and attached one to the other such that upon release of NO from the compound, a residue of a naturally occurring metabolite which is capable of inhibiting iNOS activity is formed. As is described in detail in U.S. Provisional Patent Application No. 60/651,619, a myriad of NO-donating compounds that were designed to form a residue of the metabolite vitamin B (thiamine) upon releasing bioactive NO were successfully prepared. More specifically, these NO-donating compounds were designed such that upon release of NO a thiazole residue is formed. These compounds were found highly active as vasodilators and in reducing hypertension while not inducing tolerance thereto. Vitamin Bi, a water soluble vitamin having the chemical name 3-[(4-amino-2- methyl-5-pyrimidinyl)methyl]-5-(2-hydroxyethyl)-4-methylthiazolium, is also known as thiamin, thiamine and aneurin. Thiamine is required by every cell of the body to process carbohydrates, fat, and protein and to form the fuel compound adenosine triphosphate (ATP).
Vitamin Bi Thiamin consists of a pyrimidine ring and a thiazole ring connected by a single-carbon bridging moiety, whereby the nitrogen in the thiazole ring being positively charged. It serves as a coenzyme for the decarboxylation of pyruvate and the oxidation of alpha keto-glutamic acid. The enzyme thiamin pyrophosphatekinase and adenosine triphosphate (ATP) convert thiamin into its metabolically active coenzyme form, thiamin pyrophosphate (TPP), which is also referred to in the art as thiamine diphosphate (TDP) and cocarboxylase. The reaction center of TPP is the relatively acidic proton on carbon 2 of the thiazole ring, which has the capacity to form a carbanion, whereby the latter readily undergoes nucleophilic addition to carbonyl groups. In the form of TPP, thiamin functions in the oxidative decarboxylation of alpha-keto acids, such as pyruvate and alpha-ketoglutarate, as a coenzyme for α//?/.α-ketoacid dehydrogenases. In addition TPP functions in the transketolase reaction of the pentose phosphate pathway as a coenzyme for transketolases. Both types of enzymes, alpha-ketoacid dehydrogenases and transketolases, cleave a carbon-carbon bond adjacent to a carbonyl group, releasing either carbon dioxide or an aldehyde. In the case of alpha- ketoacid dehydrogenases, the decarboxylation product is transferred to coenzyme A (CoA). Transketolases cleaves the carbon-carbon bond adjacent to the carbonyl group of an α/p/tø-ketosugar to give an activated glycoaldehyde. The glycoaldehyde is then combined with an aldose molecule to yield a new ketose. All known TPP dependent enzymes also require a divalent cation, commonly Mg
2+. Thiamine thus plays an important role in glucose metabolism and further appears to be involved in nerve transmission and/or excitation. As thiamine is involved in numerous biological pathways, it was assumed that any residue thereof would be characterized by the inherent biocompatibility described above. It was further assumed that the metabolic pathways of vitamin B} described hereinabove could participate in releasing a bioactive NO from an NO-releasing
group that is attached to a thiamine residue, similarly to the release of a phosphate group of TPP. As is demonstrated in the Examples section that follows, NO-donating compounds, which allow the formation a thiamine-derived thiazole residue upon release of NO, were found highly active in treating an IBD such as colitis. A precise understanding of the mechanism by which such NO-donating compounds exhibit the required therapeutic effect for treating IBDs is not required in order to practice the present invention, however, while not being bound to any particular mechanism or theory, it is assumed that the biocompatibility characteristics of thiamine-derived NO-donating compounds, detailed hereinabove, render these compounds highly efficacious in this respect. In addition, it is assumed that the thiamine-derived thiazole skeleton of such
NO-donating compounds may provide these compounds with a beneficial iNOS inhibitory activity. In a recent publication (Ueda. et al. 2004 Chem. Pharm. Bull. (Tokyo) 52(5), pp. 634-7) it was reported that 5-(l-methyl)ethyl-4-methylthiazol-2- ylamine was found to be a potent and selective candidate iNOS inhibitor.
4-methyl-5 -propylthiazol-2-amine
As can be seen from the structure presented above, this iNOS inhibitor has structural resembles to the thiazole portion of thiamine. As is discussed hereinabove, the beneficial effect of NO in inflammatory-damaged tissues is oftentimes adversely affected by the iNOS up-regulation and intensified activity as a result of the NO production. It is therefore postulated that thiamine-derived NO-donors can release the required bioactive NO while at the same time attenuating the adverse activity of iNOS and thus provide a synergistic effect. Hence, according to a preferred embodiment of the present invention, each of the NO-donating compounds utilized in this and other aspects of the present invention is designed such that upon release of NO, a residue of thiamine is formed. Each of
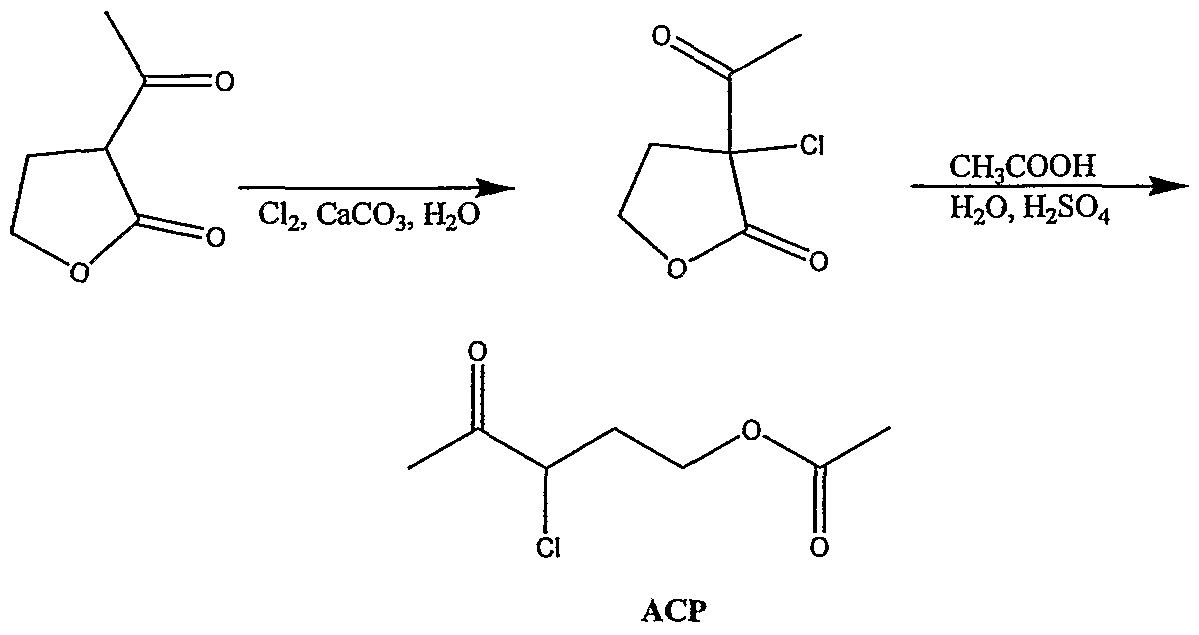
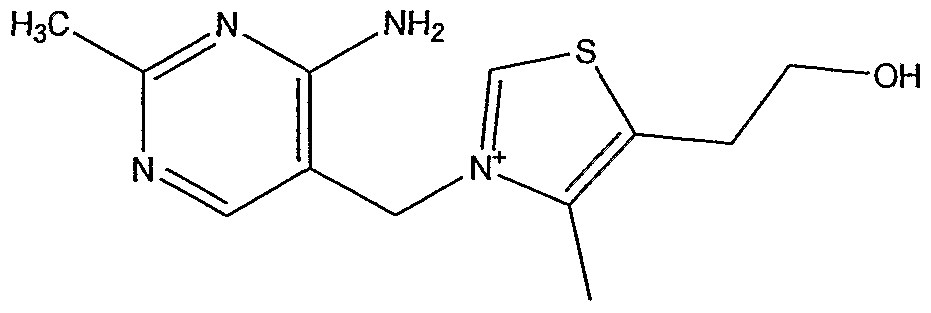
the NO-donating compounds according to this embodiment of the present invention therefore includes an NO-releasing group, as is detailed hereinunder, being covalently attached to a thiamine-derived thiazole ring. Derivatives of tliiazole are well-known in the art and are readily synthesized by well-established procedures. As is exemplified in the Examples section that follows, by selecting a suitable synthesis of a thiamine-derived thiazole, a variety of chemical parameters can be easily tailored, thus enabling the design and preparation of versatile thiazole-derived NO-donating compounds. The NO-donating compounds utilized in the present invention include, for example, a thiamine nitrated derivative, in which the hydroxyl end group at position 5
(see, formula I below) has been replaced by a -ONO2 group (see, for example, Pet-68 in Tables 1 and 2 below), and/or a pharmaceutically acceptable salt, prodrug, solvate and/or hydrate thereof, as these terms are defined hereinbelow. Alternatively, the NO-donating compounds utilized in the present invention, include a thiamine analog, which has a thiamine-derived thiazole ring as its basic structural unit and/or a pharmaceutically acceptable salt, prodrug, solvate and/or hydrate thereof. Such NO-donating- compounds utilized in the present invention are referred to herein interchangeably as thiazole-derived or thiazole-based compounds and are collectively represented by the general formula I:

Formula I wherein: A is selected from the group consisting of alkenyl, alkoxy, alkyl, alkynyl, amine, amine-oxide, aryl, aryloxy, azo, borate, C-amide, carbonyl, C-carboxylate, C-thiocarboxylate, cycloalkyl, diazo, disulfide, guanidine, guanyl, haloalkyl, heteroalicyclic, heteroaryl, hydrazine, N-amide, N-carbamate, N-dithiocarbamate, nitro, N-sulfonamide, N-thiocarbamate, O-carbamate, O-carboxylate, O-thiocarbamate, O-thiocarboxylate, oxime, oxygen, sulfur, peroxo, phosphate, phosphine-oxide, phosphine-sulfide, phosphinyl, phosphite, phosphonate, pyrophosphate, S-dithiocarbamate, silaza, silicate, siloxy, silyl, S-sulfonamide,
sulfate, sulfite, sulfonate, sulfoxide, sulfur, thioalkoxy, thioaryloxy, thiocarbonyl, thiophosphate, thiosulfate, thiosulfite, thiourea, triphosphate, urea, a biocleavable moiety and any combination thereof, or absent; X is selected from the group consisting of acyl halide, alkenyl, alkoxy, alkyl, alkynyl, amine, amine-oxide, aryl, aryloxy, azo, borate, C-amide, carbonyl,
C-carboxylate, C-thiocarboxylate, cyano, cycloalkyl, diazo, disulfide, guanidine, guanyl, halide, haloalkyl, heteroalicyclic, heteroaryl, hydrazine, hydrogen, hydroxy,
N-amide, N-carbamate, N-dithiocarbamate, nitro, N-sulfonamide, N-thiocarbamate,
O-carbamate, O-carboxylate, O-thiocarbamate, O-thiocarboxylate, oxime, peroxo, phosphate, phosphine-oxide, phosphine-sulfide, phosphinyl, phosphite, phosphonate, pyrophosphate, S-dithiocarbamate, silaza, silicate, siloxy, silyl, S-sulfonamide, sulfate, sulfite, sulfonate, sulfoxide, thioalkoxy, thioaryloxy, thiocarbonyl, thiohydroxy, thiophosphate, thiosulfate, thiosulfite, thiourea, triphosphate, urea, a bioactive agent residue, a moiety containing at least one NO-releasing group, a substituted or unsubstituted thiazole and any combination thereof; B is selected from the group consisting of a saturated or unsaturated, substituted or unsubstituted alkylene chain having 1-20 carbon atoms, and a saturated or unsaturated, substituted or unsubstituted alkylene chain having 1-20 carbon atoms interrupted by at least one heteroatom, whereby the at least one heteroatom comprises oxygen, sulfur, nitrogen, phosphor, silicon and any combination thereof; Y is an NO-releasing group; and Z is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, amine, cycloalkyl, heteroalicyclic, aryl, heteroaryl, halide, haloalkyl, hydroxy, thiohydroxy, alkoxy, thioalkoxy, aryloxy and thioaryloxy. As used herein, the term "amine" describes both a -NR'R" group and a -NR'- group, wherein R' and R" are each independently hydrogen, alkyl, cycloalkyl, aryl, as these terms are defined hereinbelow. The amine group can therefore be a primary amine, where both R' and R" are hydrogen, a secondary amine, where R' is hydrogen and R" is alkyl, cycloalkyl or aryl, or a tertiary amine, where each of R' and R" is independently alkyl, cycloalkyl or aryl. Alternatively, R' and R' ' can each independently be hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, amine, halide, sulfonate,
sulfoxide, phosphonate, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, nitro, azo, sulfonamide, carbonyl, C-carboxylate, O-carboxylate, N-thiocarbamate, O-thiocarbamate, urea, thiourea, N-carbamate, O-carbamate, C- amide, N-amide, guanyl, guanidine and hydrazine. The term "amine" is used herein to describe a -NR'R" group in cases where the amine is an end group, as defined hereunder, and is used herein to describe a - NR'- group in cases where the amine is a linking group. Herein throughout, the phrase "end group" describes a group (a substituent) that is attached to another moiety in the compound via one atom thereof. The phrase "linking group" describes a group (a substituent) that is attached to another moiety in the compound via two or more atoms thereof. The term "alkyl" describes a saturated aliphatic hydrocarbon including straight chain and branched chain groups. Preferably, the alkyl group has 1 to 20 carbon atoms. Whenever a numerical range; e.g., "1-20", is stated herein, it implies that the group, in this case the alkyl group, may contain 1 carbon atom, 2 carbon atoms, 3 carbon atoms, etc., up to and including 20 carbon atoms. More preferably, the alkyl is a medium size alkyl having 1 to 10 carbon atoms. Most preferably, unless otherwise indicated, the alkyl is a lower alkyl having 1 to 4 carbon atoms. The alkyl group may be substituted or unsubstituted. Substituted alkyl may have one or more substituents, whereby each substituent group can independently be, for example, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, amine, halide, sulfonate, sulfoxide, phosphonate, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, nitro, azo, sulfonamide, C-carboxylate, O- carboxylate, N-thiocarbamate, O-thiocarbamate, urea, thiourea, N-carbamate, O-carbamate, C-amide, N-amide, guanyl, guanidine and hydrazine. The alkyl group can be an end group, as this phrase is defined hereinabove, wherein it is attached to a single adjacent atom, or a liriking group, as this phrase is defined hereinabove, which connects two or more moieties via at least two carbons in its chain. The term "cycloalkyl" describes an all-carbon monocyclic or fused ring (i.e., rings which share an adjacent pair of carbon atoms) group where one or more of the rings does not have a completely conjugated pi-electron system. The cycloalkyl group may be substituted or unsubstituted. Substituted cycloalkyl may have one or
more substituents, whereby each substituent group can independently be, for example, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, amine, halide, sulfonate, sulfoxide, phosphonate, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, nitro, azo, sulfonamide, C- carboxylate, O-carboxylate, N-thiocarbamate, O-thiocarbamate, urea, thiourea, N-carbamate, O-carbamate, C-amide, N-amide, guanyl, guanidine and hydrazine. The cycloalkyl group can be an end group, as this phrase is defined hereinabove, wherein it is attached to a single adjacent atom, or a linking group, as this phrase is defined hereinabove, connecting two or more moieties at two or more positions thereof. The term "aryl" describes an all-carbon monocyclic or fused-ring polycyclic (i.e., rings which share adjacent pairs of carbon atoms) groups having a completely conjugated pi-electron system. The aryl group may be substituted or unsubstituted. Substituted aryl may have one or more substituents, whereby each substituent group can independently be, for example, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, amine, halide, sulfonate, sulfoxide, phosphonate, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, nitro, azo, sulfonamide, C-carboxylate, O-carboxylate, N-thiocarbamate, O-thiocarbamate, urea, thiourea, N-carbamate, O-carbamate, C-amide, N-amide, guanyl, guanidine and hydrazine. The aryl group can be an end group, as this term is defined hereinabove, wherein it is attached to a single adjacent atom, or a linking group, as this term is defined hereinabove, connecting two or more moieties at two or more positions thereof. The term "amine-oxide" describes a -N(OR')(R") or a -N(OR')- group, where R' and R" are as defined herein. This term refers to a -N(OR')(R") group in cases where the amine-oxide is an end group, as this phrase is defined hereinabove, and to a -N(OR')- group in cases where the amine-oxime is an end group, as this phrase is defined hereinabove. The term "halide" and "halo" describes fluorine, chlorine, bromine or iodine. The term "haloalkyl" describes an alkyl group as defined above, further substituted by one or more halide.
The term "sulfate" describes a -O-S(= )2-OR' end group, as this term is defined hereinabove, or an -O-S(=O)2-O- linking group, as these phrases are defined hereinabove, where R' is as defined hereinabove. The term "thiosulfate" describes a -O-S(=S)(=O)-OR' end group or a -O- S(=S)(:=:O)-O- linking group, as these phrases are defined hereinabove, where R' is as defined hereinabove. The term "sulfite" describes an -O-S(=O)-O-R' end group or a -O-S(=O)-O- group linking group, as these phrases are defined hereinabove, where R' is as defined hereinabove. The term "thiosulfite" describes a -O-S(=S)-O-R' end group or an -O-
S(=S)-O- group linking group, as these phrases are defined hereinabove, where R' is as defined hereinabove. The term "sulfinate" describes a -S(=O)-OR' end group or an -S(=O)-O- group linking group, as these phrases are defined hereinabove, where R' is as defined hereinabove. The term "sulfoxide" or "sulflnyl" describes a -S(=O)R' end group or an - S(=O)- linking group, as these phrases are defined hereinabove, where R' is as defined hereinabove. The term "sulfonate" describes a -S(=O)2-R' end group or an -S(=O) - linking group, as these phrases are defined hereinabove, where R' is as defined herein. The term "S-sulfonamide" describes a -S(=O) -NR'R" end group or a - S(=O)2-NR'- linking group, as these phrases are defined hereinabove, with R' and R" as defined herein. The term "N-sulfonamide" describes an R'S(=O)2-NR"- end group or a -S(=O)2-NR'- linking group, as these phrases are defined hereinabove, where R' and R" are as defined herein. The term "disulfide" refers to a -S-SR' end group or a -S-S- Unking group, as these phrases are defined hereinabove, where R' is as defined herein. The term "phosphonate" describes a -P(=O)(OR')(OR") end group or a -P(=O)(OR')(O)- linking group, as these phrases are defined hereinabove, with R' and R" as defined herein.
The term "phosphinyl" describes a -PR'R" end group or a -PR'- linking group, as these phrases are defined hereinabove, with R' and R" as defined hereinabove. The term' "phosphine oxide" describes a -P(=O)(R')(R") end group or a -P(=O)(R')- linking group, as these phrases are defined hereinabove, with R' and R" as defined herein. The term "phosphine sulfide" describes a -P(=S)(R')(R") end group or a -P(=S)(R')- linking group, as these phrases are defined hereinabove, with R' and R" as defined herein. The term "phosphate" describes an -O-P(=O)(OR')(OR") end group or an
-O-P(=O)(OR')( O)- linking group, as these phrases are defined hereinabove, with R', R" as defined herein. The term "phosphite" describes an -O-PR'(=O)(OR") end group or an -O- PR'(=O)(O)- linking group, as these phrases are defined hereinabove, with R' and R" as defined herein. The term "thiophosphate" describes an -O-P(=S)(OR')(OR") end group or an -O-P(=S)(OR')( O)- linking group, as these phrases are defined hereinabove, with R', R" as defined herein. The term "carbonyl" or "carbonate" as used herein, describes a -C(=O)-R' end group or a -C(=O)- linking group, as these phrases are defined hereinabove, with R' as defined herein. The term "thiocarbonyl " as used herein, describes a -C^S^R' end group or a -C(=S)- linking group, as these phrases are defined hereinabove, with R' as defined herein. The term "oxime" describes a =N-OH end group or a =N-O- linking group, as these phrases are defined hereinabove. The term "hydroxyl" describes a -OH group. The term "alkoxy" describes both an -O-alkyl and an -O-cycloalkyl group, as defined herein. The term "aryloxy" describes both an -O-aryl and an -O-heteroaryl group, as defined herein. The term "thiohydroxy" describes a -SH group.
The term "thioalkoxy" describes both a -S-alkyl group, and a -S-cycloalkyl group, as defined herein. The term "thioaryloxy" describes both a -S-aryl and a -S-heteroaryl group, as defined herein. The term "cyano" describes a -C≡N group. The term "isocyanate" describes an -N=C=O group. The term "nitro" describes an -NO2 group. The term "acyl halide" describes a -(C=O)R"" group wherein R"" is halide, as defined hereinabove. The term "azo" or "diazo" describes an -N=NR' end group or an -N=N- linking group, as these phrases are defined hereinabove, with R' as defined hereinabove. The term "peroxo" describes an -O-OR' end group or an -O-O- linking group, as these phrases are defined hereinabove, with R' as defined hereinabove. The term "C-carboxylate" describes a -C(=O)-OR' end group or a -C(=O)-O- linking group, as these phrases are defined hereinabove, where R' is as defined herein. The term "O-carboxylate" describes a -OC(=O)R' end group or a -OC(=O)- linking group, as these phrases are defined hereinabove, where R' is as defined herein. The term "C-thiocarboxylate" describes a -C(=S)-OR' end group or a -C(=S)- O- linking group, as these phrases are defined hereinabove, where R' is as defined herein. The term "O-thiocarboxylate" describes a -OC(=S)R' end group or a -OC(=S)- linking group, as these phrases are defined hereinabove, where R' is as defined herein. The term "N-carbamate" describes an R"OC(=O)-NR'- end group or a -OC(=O)-NR'- linking group, as these phrases are defined hereinabove, with R' and R" as defined herein. The term "O-carbamate" describes an -OC(=O)-NR'R" end group or an - OC(=O)-NR'- linking group, as these phrases are defined hereinabove, with R' and R" as defined herein. The term "O-thiocarbamate" describes a -OC(=S)-NR'R" end group or a
-OC(=S)-NR'- linking group, as these phrases are defined hereinabove, with R' and R" as defined herein.
The term "N-thiocarbamate" describes an R"OC(=S)NR'- end group or a -OC(=S)NR'- linking group, as these phrases are defined hereinabove, with R' and R" as defined herein. The term "S-dithiocarbamate" describes a -SC(=:S)-NR'R" end group or a -SC(=S)NR'- linking group, as these phrases are defined hereinabove, with R' and R" as defined herein. The term "N-dithiocarbamate" describes an R"SC(=S)NR'- end group or a -SC(=S)NR'- linking group, as these phrases are defined hereinabove, with R' and R" as defined herein. The term "urea", which is also referred to herein as "ureido", describes a
-NR'C(=O)-NR"R"' end group or a -NR'C(=O)-NR"- linking group, as these phrases are defined hereinabove, where R' and R" are as defined herein and R'" is as defined herein for R' and R". The term "thiourea", which is also referred to herein as "thioureido", describes a -NR'-C(=S)-NR"R'" end group or a -NR'-C(=S)-NR"- linking group, with R', R" and R'" as defined herein. The term "C-amide" describes a -C(=O)-NR'R" end group or a„-C(=O)-NR'- linking group, as these phrases are defined hereinabove, where R' and R" are as defined herein. The term "N-amide" describes a R'C(=O)-NR"- end group or a R'C(=O)-N- linking group, as these phrases are defined hereinabove, where R' and R" are as defined herein. The term "guanyl" describes a R'R"NC(=N)- end group or a -R'NC(=N)- linking group, as these phrases are defined hereinabove, where R' and R" are as defined herein. The term "guanidine" describes a -R'NC(=N)-NR"R'" end group or a - R'NC(=N)- NR"- linking group, as these phrases are defined hereinabove, where R', R" and R'" are as defined herein. The term "hydrazine" describes a -NR'-NR"R'" end group or a -NR'-NR"- linking group, as these phrases are defined hereinabove, with R\ R", and R"' as defined herein.
The term "silyl" describes a -SiR'R"R'" end group or a -SiR'R"- linking group, as these phrases are defined hereinabove, whereby each of R', R" and R'" are as defined herein. The term "siloxy" describes a -Si(OR')R"R'" end group or a -Si(OR')R"- linking group, as these phrases are defined hereinabove, whereby each of R', R" and R'" are as defined herein. The term "silaza" describes a -Si(NR'R")R"' end group or a -Si(NR'R")- linking group, as these phrases are defined hereinabove, whereby each of R', R" and R'" is as defined herein. The term "silicate" describes a -O-Si(OR')(OR")(OR'") end group or a
-O-Si(OR')(OR")- linking group, as these phrases are defined hereinabove, with R', R" and R'" as defined herein. The term "boryl" describes a -BR'R" end group or a -BR'- linking group, as these phrases are defined hereinabove, with R' and R" are as defined herein. The term "borate" describes a -O-B(OR')(OR") end group or a -O-B(OR')(O-) liriking group, as these phrases are defined hereinabove, with R' and R" are as defined herein. The term "heteroaryl" describes a monocyclic or fused ring (i.e., rings which share an adjacent pair of atoms) group having in the ring(s) one or more atoms, such as, for example, nitrogen, oxygen and sulfur and, in addition, having a completely conjugated pi-electron system. Examples, without limitation, of heteroaryl groups include pyrrole, furane, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrimidine, quinoline, isoquinoline and purine. The heteroaryl group may be substituted or unsubstituted. Substituted heteroaryl may have one or more substituents, whereby each substituent group can independently be, for example, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, amine, halide, sulfonate, sulfoxide, phosphonate, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, nitro, azo, sulfonamide, C- carboxylate, O-carboxylate, N-thiocarbamate, O-thiocarbamate, urea, thiourea, O- carbamate, N-carbamate, C-amide, N-amide, guanyl, guanidine and hydrazine. The heteroaryl group can be an end group, as this phrase is defined hereinabove, where it is attached to a single adjacent atom, or a linking group, as this phrase is defined
hereinabove, connecting two or more moieties at two or more positions thereof. Representative examples are pyridine, pyrrole, oxazole, indole, purine and the like. The term "heteroalicyclic" describes a monocyclic or fused ring group having in the ring(s) one or more atoms such as nitrogen, oxygen and sulfur. The rings may also have one or more double bonds. However, the rings do not have a completely conjugated pi-electron system. The heteroalicyclic may be substituted or unsubstituted. Substituted heteroalicyclic may have one or more substituents, whereby each substituent group can independently be, for example, hydroxyalkyl, trihaloalkyl, cycloalkyl, alkenyl, alkynyl, aryl, heteroaryl, heteroalicyclic, amine, halide, sulfonate, sulfoxide, phosphonate, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, nitro, azo, sulfonamide, C-carboxylate, O- carboxylate, N-thiocarbamate, O-thiocarbamate, urea, thiourea, O-carbamate, N- carbamate, C-amide, N-amide, guanyl, guanidine and hydrazine. The heteroalicyclic group can be an end group, as this phrase is defined hereinabove, where it is attached to a single adjacent atom, or a linking group, as this phrase is defined hereinabove, connecting two or more moieties at two or more positions thereof. Representative examples are piperidine, piperazine, tetrahydrofurane, tetrahydropyrane, morpholino and the like. As used herein, the phrase "NO-releasing group" describes a chemical moiety, which is capable of generating NO either spontaneously or by means of chemical or enzymatic reactions. Representative examples of suitable NO-releasing groups include, without limitation, nitrate esters such as, for example, -ONO2, S-nitrosothiol such as, for example, -SNO, diazeniumdiolates , also known as "NONOates" such as, for example, -N(NONO")- and -N(NONOH)-, and mesoionic oxatriazoles such as for example, 5-amino-[l,2,3,4]oxatriazol-2-ium and ,2,3,4-oxatriazolium-5-amino-3-(3,4- dichlorophenyl)-chloride. Preferably, the NO-releasing group, denoted as Y in the general Formula I as presented and discussed in detail below, is -ONO2. Thus, each of the compounds utilized in this and other aspects of the present invention has a thiazole ring, to which an NO-releasing group is attached, preferably at position 5 of the ring. The NO-releasing group can be attached directly to the thiazole ring, or, preferably via a spacer. The spacer, denoted as B in the general Formula I above, can be a saturated or unsaturated, substituted or unsubstituted hydrocarbon chain, and may optionally be
interrupted by one or more heteroatom(s) such as oxygen, sulfur, nitrogen, phosphor, silicon and any combination thereof. When the heteroatom is nitrogen, phosphor or silicon, the heteroatom is preferably substituted by e.g., hydrogen, alkyl, halide, cycloalkyl or aryl, as these terms are defined hereinabove. The chemical structure and length of the spacer may affect the biocompatibility, bioavailability, target specificity, and NO-releasing sensitivity of the compound. Preferably, B is a non-substituted, saturated alkylene chain. Thus, B is preferably a non-substituted alkylene chain and, more preferably, a short alkylene chain such as, for example, methylene, ethylene and propylene. Since in the thiazole ring of Thiamine position 5 is substituted by a hydroxyethlene, more preferably, B is ethylene. Alternatively, B can be a non-substituted, saturated alkylene chain interrupted by one heteroatom and can therefore be, for example, -CH2-CH2-O-CH - (methoxy ethylene), -CH2-CH2-NH-CH2- (ethyl-methyl-amine) and -CH2-CH2-S-CH2- (ethyl- methylsulfanyl). The thiazole ring may be further substituted at position 4, by variable substituents, denoted as Z in the general Formula I above, which may also be selected so as to affect the compound's pharmacokinetic properties such as biocompatibility, bioavailability, solubility and target specificity. Since in the thiazole ring of thiamine position 4 is substituted by a methyl, preferably Z is an alkyl, more preferably a lower alkyl such as methyl, ethyl and propyl, and more preferably, Z is methyl. Each of these NO-releasing compounds further includes a moiety that is covalently attached to position 2 of the thiazole ring. This moiety, denoted as X in the general Formula I above, can be a chemical moiety such as, for example, acyl- halide, alkenyl, alkoxy, alkyl, alkynyl, amine, amine-oxide, aryl, aryloxy, azo, borate, C-amide, carbonyl, C-carboxylate, C-thiocarboxylate, cyano, cycloalkyl, diazo, disulfide, guanidine, guanyl, halide, haloalkyl, heteroalicyclic, heteroaryl, hydrazine, hydrogen, hydroxy, N-amide, N-carbamate, N-dithiocarbamate, nitro, N-sulfonamide, N-thiocarbamate, O-carbamate, O-carboxylate, O-thiocarbamate, O-thiocarboxylate, oxime, peroxo, phosphate, phosphine-oxide, phosphine-sulfide, phosphinyl, phosphite, phosphonate, pyrophosphate, S-dithiocarbamate, silaza, silicate, siloxy,
silyl, S-sulfonamide, sulfate, sulfite, sulfonate, sulfoxide, thioalkoxy, thioaryloxy, thiocarbonyl, thiohydroxy, thiophosphate, thiosulfate, thiosulfite, thiourea, triphosphate, urea and any combination thereof. The attachment of such moieties may further affect the pharmacokinetic profile of the compound, as described hereinabove. Optionally, X can be a moiety containing one or more NO-releasing group(s).
Compounds in which X contains one or more NO-releasing group, in addition to the NO-releasing group in B (see, Formula I above), may exert enhanced capacity to elevate bioactive NO levels. Furthermore, the presence of more than one NO- releasing group in the same compound enables to incorporate therein different NO- releasing groups, which may be susceptible to more than one NO-releasing bio- and/or chemo-mechanism and thus further enhance the capacity of the compound to elevate NO levels and thus the therapeutic effect in treating IBD . Representative examples of compounds in which X is a moiety containing an NO-releasing group have been successfully prepared, and include l,4-bis-[4-methyl- 5-(2-nitrooxy)-ethyl)-thiazol-2-yl]-butane (Pet-13), bis-[4-methyl-5-(2-nitrooxy- ethyl)-thiazole-2-yl]-diazene (Pet-102) and 4,4'-dimethyl-5,5'-bis-(2-nitrooxy-ethyl)- [2,2']bithiazolyl (Pet-118) (see, Tables 1 and 2). Further optionally, X can be a thiazole, such that the compound contains two thiazole moieties attached therebetween. Such compounds may provide for additive advantageous effects of the thiazole residue, discussed hereinabove. When X is a thiazole ring, the thiazole can be substituted or non-substituted. When substituted, each substituent can be, for example, alkyl, haloalkyl, alkenyl, alkynyl, cycloalkyl, heteroalicyclic, aryl, heteroaryl, amine, C-amide, N-amide, halide, acyl-halide, haloalkyl, sulfonate, sulfoxide, phosphonate, hydroxy, alkoxy, aryloxy, thiohydroxy, thioalkoxy, thioaryloxy, cyano, nitro, azo, N-sulfonamide, S-sulfonamide, C-carboxylate, O-carboxylate, C-thiocarboxylate, O-thiocarboxylate, N-carbamate, O-carbamate, N-thiocarbamate, O-thiocarbamate, S-dithiocarbamate, N-dithiocarbamate, urea, thiourea, guanyl, guanidine and hydrazine, as these terms are defined hereinabove. Alternatively, one or more of the substituents on the thiazole ring can be a moiety containing an NO-releasing group, as described hereinabove. Such compounds may provide for additive advantageous effects of both the tliiazole residues and the NO-releasing groups.
As is further detailed hereinbelow, the NO-donating compounds utilized in the present invention optionally and preferably further comprise a bioactive agent residue. The bioactive residue is preferably attached, either directly or indirectly, preferably via a biocleavable moiety, to the chemical moiety in the compound. This capacity for dual therapeutic activity of these compounds extends their potential as unique remedies. Thus further optionally and preferably, X is a bioactive agent residue. The phrase "bioactive agent" is used herein to describe an agent capable of exerting a beneficial activity in a biological system (e.g., a living tissue or organ) of a subject. The beneficial activity includes, for example, a therapeutic activity per se, reduction of adverse side effects induced by another moiety or agent, and/or targeting and/or transportation of another moiety and/or agent towards a desired biological target. The conjugation of a bioactive agent to a chemical moiety having an NO- releasing group attached thereto is highly beneficial since it may provide for combined and even synergistic therapeutic effects of both the NO-releasing group and the bioactive agent. Representative examples of bioactive agents that can be beneficially incorporated in the NO-donating compounds utilized in the present invention include, without limitation, drugs, inhibitors, co-factors, co-enzymes, amino acids, peptides, proteins, inflammatory bowel disease drugs, inducible nitric oxide synthase inhibitors, fatty acids, metabolites, carbohydrates, hydroxamic acid, nicotinic acid, nicotinamides, carnitines, beta carotene, bromelain, steroidal anti-inflammatory agents, non-steroidal anti-inflammatory drugs (NSAIDs), anti-psychotic agents, anti- thrombogenic agents, anti-platelet agents, anti-coagulants, anti-diabetic agents, growth factors, statins, toxins, antimicrobial agents, analgesics, anti-metabolic agents, vasoactive agents, vasodilator agents, prostaglandins, hormones, thrombin, enzymes, oligonucleotides, nucleic acids, antisense, antibodies, antigens, vitamins, immunoglobulins, cytokines, cardiovascular agents, chemotherapeutic agents, antioxidants, phospholipids, anti-proliferative agents, and heparins. Particularly preferred bioactive agents that can be beneficially incorporated in the NO-donating compounds utilized in this and other aspects of the present invention include, without limitation, bioactive agents that may provide an added therapeutic value for treating an IBD, in addition to the release of the bioactive NO. These
include, for example, NSAIDs, which, as described hereinabove, are presently commonly used in treating IBD, other therapeutic agents for treating IBD (also referred to herein interchangeably as IBD drugs), iNOS inhibitors which, as discussed hereinabove, can beneficially affect IBD manifestations and therapeutically beneficial metabolites such as nicotinic acid. Additional preferred bioactive agents include agents that are effective in treating conditions that are associated with or exacerbated by IBD. Thus, according to a preferred embodiment of the present invention, the bioactive agent residue (X on Formula I above) is a non-steroidal anti-inflammatory drug residue. Non-limiting examples of non-steroidal anti-inflammatory drugs (NSAIDs) that can be beneficially incorporated in the NO-donors, according to the present invention include aspirin, celecoxib, diclofenac, diflunisal, etodolac, fenoprofen, flurbiprofen, ibuprofen, indomethacin, ketoprofen, ketorolac, meclofenamate, mefenamic acid, nabumetone, naproxen, oxaprozin, oxyphenbutazone, phenylbutazone, piroxicam, rofecoxib sulindac and tolmetin. As is discussed hereinabove, non-steroidal anti-inflammatory drugs (NSAIDs) are widely used as in treating IBDs. Chronic NSAID therapy effectively reduces the symptoms~bf IBDs7 but "is oftentimes associated with adverse gastrointestinal (GI) complications [Cash, J.M.; Klippel, J.H. New Engl J. Med., 1994, 330, 1368; Davies, N.M., Wallace, J.L. J. Gastroenterol, 1997, 32, 127; Wallace, J. Gastroenterol, 1997, 112, 1000], as well as high blood pressure and heart diseases. At the tissue level, the most common clinical manifestation of NSAID-related GI damage is a combination of gastroduodenal erosions and ulcerations often called NS AID-induced gastropathy, affecting at least 25 % of chronic NSAID patients. NSAID-induced gastropathy may limit long-term NSAID therapy and cause a significant financial burden to the healthcare system. In vivo NO generation has become the prime therapeutic target for reducing NSAID induced gastropathy associated with chronic NSAID use, common to IBD patients. A recently published data have shown that NO-donors effectively reduce gastric mucosal damage and may facilitate GI healing following chemical insult [Ko, J.K.; Cho, CH. Inflamm. Res., 1999, 48, 471]. As first conceptualized by Wallace and colleagues [Reuter, B., Wallace, J.L. A therapeutic application of nitric oxide: GI- sparing NSAIDs. In: Nitric Oxide: A Modulator of Cell-Cell Interactions in the
Microcirculation. (P. Kubes, ed.) R.G. Landes Company, 1995, pp. 157-168], modern drug discovery has focused on one general approach in an attempt to utilize the therapeutic potential of NO against NSAID-induced gastric damage: covalent modification of NSAIDs with NO-releasing moieties [Brzozowski T., et al, Dig Liver Dis. 200032(7), pp 583-94] . Due to the beneficial effect of compounds that can act as both NO-donors and anti-inflammatory agents, delineated above, the present inventors have designed and successfully prepared representative thiazole-based compounds, as described in U.S. Provisional Patent Application No. 60/651,619, which have a NSAID residue attached thereto (for example, as X in Formula I above). These include 2-[l-(6-methoxy- naphthalen-2-yl-ethyl]-4-methyl-5-(2-nitrooxy-ethyl)-thiazole (Pet 17), wherein X is a naproxen residue, 2-[l-(4-isobutyl-phenyl)-ethyl]-4-methyl-5-(2-nitrooxy-ethyl)- thiazole (Pet-66) wherein X is an ibuprofen residue and acetic acid 2-[4-methyl-5-(2- nitrooxy-ethyl)-thiazol-2-yl]-phenyl ester (Pet-116) wherein X is an aspirin residue (see, Tables 1 and 2). These compounds thus exert high NO-releasing efficacy, are non-tolerance inducing and therefore further exert the protective effect required to improve the safety and pharmacokinetic profile of the NSAID agent residue attached thereto, which is crucial in the treatment of IBDs. The combined therapeutic effects mentioned above are further advantageous when the bioactive agent is a substance used for the treatment of IBDs. Thus, according to another preferred embodiment of the present invention, the bioactive agent residue (X in Formula I above) is an inflammatory bowel disease drug residue, as is detail hereinabove. Non-limiting examples of inflammatory bowel disease drugs that can be beneficially incorporated in these NO-donors include 5-aminosalicylic acid, 4- aminophenylacetic acid, sulphasalazine, olsalazine, mesalazine, rifaximin, rifampin, hydrocortisone, prednisolone, budesonide, azathioprine, 6-mercaptopurine, cyclosporin, methotrexate, metronidazole, tinidazole, loperamide, diphenoxylate, atropine, cholestylamine, colestipol and paracetamol. Since recent research studies point to iNOS intensified activity as a possible cause of some of the adverse expressions of IBD, as discussed hereinabove, the combined therapeutic effects mentioned above may be further advantageous when the
bioactive agent is an inhibitor of inducible nitric oxide synthase (iNOS) which can be used for the treatment of IBDs. Thus, according to still another preferred embodiment of the present invention, the bioactive agent residue (X in Formula I above) is an inducible nitric oxide synthase inhibitor residue. Non-limiting examples of inducible nitric oxide synthase inhibitors, which can be beneficially incorporated in these NO-donors include (-)-noformycin, (1 S,5S,6R,7R)-7-chloro-5-methyl-2-aza-bicyclo[4.1.0]heptan-3-imine, (S,E)-3-(4- chlorophenyl)-N-( 1 -oxo- 1 -(2-oxo-2-(4-(6-(trifluoromethyl)pyrimidin-4-yloxy) piperidin- 1 -yl)ethylamino)-3 -(pyridin-2-yl)propan-2-yl)acrylamide, 1 -amino-2- hydroxy-guanidine, 2-aminoethyl-isothiourea, 2-benzyl-2-thio-pseudourea, 2- iminobiotin, 3-hydroxy-4-methyl-5-pentyl-2-iminopyrrolidine, 4-methyl-5- propyloxazolidin-2-imine, 4-methyl-5-propylthiazol-2-amine, 5-tert-butyl-4- methylthiazol-2-amine, 8-(3-chlorostyryl)caffeine, alloxazine, aminoguanidine, deltoin, dexamethasone, geldanamycin, Gingivex®, guamdinoethyldisulphide, imperatorin, L-canavanine, L-N6-(l-iminoethyl)lysine 5-tetrazole amide,, mercaptoethylguanidine, methyl 4-(2-(lH-imidazol-l-yl)pyrimidin-4-yl)-3-(2- (benzo [d] [ 1 ,3] dioxol-5 -ylmethylamino)-2-oxoethyl)piperazine- 1 -carboxylate, N-(3 - (aminomethyl)benzyl) acetamidine, N-(5(S)-amino-6,7- dihydroxyheptyl)ethanimidamide, NG-monomethyl-L-arginine, N-iminoethyl-L- lysine, N-iminoethyl-L-oπiithine, Nω-nitro-L-arginine methyl ester, S-(4-nitrobenzyl)- 6-thioinosine, S,S'-l,4-phenylene-bis(l,2-ethanediyl)bis-isothiourea, salicylate, S- ethylisothiourea and S-methylisothiourea. Each of the bioactive agent residues described above (for example, X in Formula I above) can be attached to the chemical moiety (e.g., the thiamine-derived thiazole ring) either directly or indirectly. When attached indirectly, ttte bioactive agent is attached to the chemical moiety (e.g., the thiazole ring) via a linking moiety, represented, for example, as A in Formula I above. The linking moiety (e.g., A in Formula I) can be, for example, alkenyl, alkoxy, alkyl, alkynyl, amine, amine-oxide, aryl, aryloxy, azo, borate, C-amide, carbonyl,
C-carboxylate, C-thiocarboxylate, cycloalkyl, diazo, disulfide, guanidine, guanyl, haloalkyl, heteroalicyclic, heteroaryl, hydrazine, N-amide, N-carbamate, N- dithiocarbamate, nitro, N-sulfonamide, N-thiocarbamate, O-carbamate,
O-carboxylate, O-thiocarbamate, O-thiocarboxylate, oxime, oxygen, peroxo, phosphate, phosphine-oxide, phosphine-sulfide, phosphinyl, phosphite, phosphonate, pyrophosphate, S-dithiocarbamate, silaza, silicate, siloxy, silyl, S-sulfonamide, sulfate, sulfite, sulfonate, sulfoxide, sulfur, thioalkoxy, thioaryloxy, thiocarbonyl, thiophosphate, thiosulfate, thiosulfite, thiourea, triphosphate, urea and any combination thereof, or absent. According to a preferred , embodiment of the present invention, the linking moiety (e.g., A in Formula I) is a biocleavable moiety. Representative examples of biocleavable moieties include, without limitation, amides, carboxylates, carbamates, phosphates, hydrazides, thiohydrazides, disulfides, epoxides, peroxo and methyleneamines. Such moieties are typically subjected to enzymatic cleavages in a biological system, by enzymes such as, for example, hydrolases, amidases, kinases, peptidases, phospholipases, Upases, proteases, esterases, epoxide hydrolases, nitrilases, glycosidases and the like. As used herein, the phrase "biocleavable moiety" describes a chemical moiety, which undergoes cleavage in a biological system such as, for example, the digestive system of an organism or an enzymatic system in a living cell. .Representative examples of biocleavable moieties are presented hereinbelow. As used herein, the term "hydrazide" describes a -C(=O)-NR'-NR"-R'" end group or a -C(=O)-NR'-NR"- linking group, as these phrases are defined hereinabove, where R', R" and R'" are as defined herein. As used herein, the term "thiohydrazide" describes a -C(=S)-NR'-NR"-R'" end group or a -C(=S)-NR'-NR"- linking group, as these phrases are defined hereinabove, where R', R" and R'" are as defined herein. Q As used herein, the term "epoxide" describes a R A' R" end group or a y R' R" Unking group, as these phrases are defined hereinabove, where R', R" and R'" are as defined herein. As used herein, the term "methyleneamine" describes an -NR'-CH2-CH=CR"R"' end group or a -NR'-CH2-CH=CR"- linking group, as these phrases are defined hereinabove, where R', R" and R'" are as defined herein.
As discussed hereinabove, NO-donors which have a bioactive agent residue attached thereto offer exceptional advantages due to the dual functionality thereof (elevating the NO level by an NO-releasing group and exerting a beneficial activity by the bioactive agent). Incorporation of a biocleavable moiety which links between the bioactive agent residue and the chemical moiety attached to the NO-releasing group (e.g., a nitrated thiamine-derived thiazole ring) in such compounds provides for a release of the bioactive agent in a biological system and thus may improve the biological activity of both the NO-releasing part of the compound and the bioactive agent. Thus, according to another preferred embodiment of the present invention, the
NO-donating compounds utilized in this and other aspects of the present invention include a bioactive agent (e.g., X in Formula I), which is attached to the chemical moiety via a biocleavable moiety (e.g., A in formula I). As is described in the Examples section that follows and further in U.S. Provisional Patent Application No. 60/651,619, representative examples of thiamine- derived thiazole-based NO-donating compounds in which A is a biocleavable moiety and X is a bioactive residue have been successfully prepared and were shown higlily active in treating induced colitis These include 4-[l,2]dithiolan-3-yl-N-[4-methyl-5- (2-nitrooxy-ethyl)-thiazol-2-yl]-butyramide (Pet-151) wherein A is an amide and X is a residue of 5-[l,2]dithiolan-3-yl-pentanoic acid, also known as lipoic acid, which has been suggested as a therapeutic and prophylactic treatment of many age-related diseases, from heart disease and stroke to diabetes and IBD, 2-(4-Isobutyl-phenyl)-N- [4-methyl-5-(2-nitrooxy-ethyl)-thiazol-2-yl]-propionamide (Pet-152), wherein A is an amide and X is an ibuprofen residue, N-[4-Methyl-5-(2-nitrooxy-ethyl)-thiazol-2-yl]- nicotinamide (Pet-154) wherein A is an amide and X is a nicotinic acid residue, and allyl-[4-methyl-5-(2-nitrooxy-ethyl)-thiazol-2-yl]-amine (Pet-155) wherein A is an amine and X is aUyl (H2C=CH-CH2-). Other exemplary thiamine-derived thiazole-based NO-donating compounds in which A is a biocleavable moiety which have been successfully prepared include, for example, 4-memyl-5-(2-nitrooxy-ethyl)-thiazole-2-carboxylic acid N'-phthalazin-l-yl- hydrazide (Pet-153) wherein A is a hydrazide and X is phthalazine-1-yl, , N-[4- methyl-5-(2-nifrooxy-emyl)-thiazol-2-yl]-l-oxy-nicotinamide (Pet-156) wherein A is an amide and X is an oxidized nicotinic acid residue (pyridine l-oxide-3-yl), 4-
acetylamino-N-[4-methyl-5-(2-nitrooxy-ethyl)-thiazol-2-yl]-benzamide (Pet-157); 10,13-dimethyl-3-oxo-2,3,6,7,8,9,10,l l,12,13,14,15,16,17-tetradecahydro-lH- cyclopenta[a]phenanthren-17-yl ester (Pet-164) wherein A is an amide and X is a hormone residue; hexadecanoic acid [4-methyl-5-(2-nitrooxy-ethyl)-thiazol-2-yl]- amide (Pet-158) wherein A is an amide and X is 1-pentadecanyl (fatty acid),pyrrolidine-2-carboxylic acid [4-methyl-5-(2-nitrooxy-ethyl)-thiazol-2-yl]- amide (Pet-160); 2,6-difluoro-N-[4-methyl-5-(2-nitrooxy-ethyl)-thiazol-2-yl]- benzamide (Pet-161); 2-(2,4-dichloro-phenyl)-N-[4-methyl-5-(2-nitrooxy-ethyl)- thiazol-2-yl]-acetamide (Pet-162); and 2-(2,4-dichloro-phenoxy)-N-[4-methyl-5-(2- nitrooxy-ethyl)-thiazol-2-yl]-acetamide (Pet-163). As is further discussed hereinabove, NO-donors in which the bioactive agent residue is a NSAID residue are highly beneficial for use in the context of the present invention and hence attaching the NSAID residue to the thiazole ring that contains an NO-releasing moiety would be highly advantageous. Thus, according to a preferred embodiment of the present invention, A is a biocleavable moiety and X is a NSAID residue. As is further discussed hereinabove, NO-donors in which the bioactive agent residue is an IBD drug residue are highly beneficial for use in the context of the present invention and hence attaching the IBD drug residue to the thiazole ring that contains an NO-releasing moiety would be highly advantageous. Thus, according to another preferred embodiment of the present invention, A is a biocleavable moiety and X is a IBD drug residue. As is further discussed hereinabove, NO-donors in which the bioactive agent residue is an iNOS inhibitor residue are highly beneficial for use in the context of the present invention and hence attaching the iNOS inhibitor residue to the thiazole ring that contains an NO-releasing moiety would be highly advantageous. Thus, according to another preferred embodiment of the present invention, A is a biocleavable moiety and X is a iNOS inhibitor residue. As is further discussed hereinabove, NO-donors in which the bioactive agent residue is a metabolite residue such as nicotinic acid residue are highly beneficial for use in the context of the present invention and hence attaching the nicotinic acid residue to the thiazole ring that contains an NO-releasing moiety would be highly advantageous. Thus, according to still another preferred embodiment of the present
invention, A is a biocleavable moiety and X is a nicotinic acid residue, a derivative or an analog thereof. The chemical structures of exemplary compounds according to this embodiment of the present invention are set forth in Table 1 below. A list of preferred compounds according to the present invention is set forth in Table 2 below. As mentioned hereinabove, each of the NO-donating compounds described herein can be utilized in this and other aspects of the present invention in a form of a pharmaceutically acceptable salt, a prodrug, a solvate and/or a hydrate thereof. The phrase "pharmaceutically acceptable salt" refers to a charged species of the parent compound and its counter ion, which is typically used to modify the solubility characteristics of the parent compound and/or to reduce any significant irritation to an organism by the parent compound, while not abrogating the biological activity and properties of the administered compound. The term "prodrug" refers to an agent, which is converted into the active compound (the active parent drug) in vivo. Prodrugs are typically useful for facilitating the administration of the parent drug. They may, for instance, be bioavailable by oral administration whereas the parent drug is not. The prodrug may also have improved solubUity as compared with the parent drug in pharmaceutical compositions. Prodrugs are also often used to achieve a sustained release of the active compound in vivo. An example, without limitation, of a prodrug would be the NO-donating compound, having one or more carboxylic acid moieties, which is administered as an ester (the "prodrug"). Such a prodrug is hydrolysed in vivo, to thereby provide the free compound (the parent drug). The selected ester may affect both the solubility characteristics and the hydrolysis rate of the prodrug. The term "solvate" refers to a complex of variable stochiometry (e.g., di-, tri-, tetra-, penta-, hexa-, and so on), which is formed by a solute (the NO-donating compound) and a solvent, whereby the solvent does not interfere with the biological activity of the solute. Suitable solvents include, for example, ethanol, acetic acid and the like. The term "hydrate" refers to a solvate, as defined hereinabove, where the solvent is water. The beneficial characteristics of the NO-donating compounds described herein render such compounds highly suitable for use in the treatment of IBDs.
The NO-donating compounds described herein can thus be beneficially used in the treatment of various inflammatory bowel diseases and/or related conditions including, without limitation, gross and microscopic Crohn's disease, gross and microscopic ulcerative colitis, gross colonic adenocarcinoma, gross and microscopic pseudomembranous colitis, microscopic amebiasis, irritable bowel syndrome, celiac disease, diverticulosis, stomach and duodenal ulcers, gastro-esophageal reflux disease, Candida proliferation, candidiasis and interstitial cystitis of the bladder. As used herein, the phrase "therapeutically effective amount" describes an amount of the compound being admimstered which will relieve to some extent one or more of the symptoms of the disorder being treated, herein an inflammatory bowel disease as is detailed hereinabove. According to a preferred embodiment of the method according to this aspect of the present invention, a therapeutically effective amount of the NO-donating compounds described herein can range from about 1 mg/kg body to about 200 mg/kg body, more preferably from about 1 mg/kg body to about 100 mg/kg body and more preferably from about 1 mg/kg body to about 50 mg/kg body. As used herein the term "about" refers to ± 10 %. As is demonstrated in the Example section that follows, the NO-donating compounds described herein are at least as effective as 5-ASA in treating colitis- induced animals, and in most cases show superior efficacy as compared with 5-ASA. A typical treatment regime of 5-ASA, when used in the treatment of IBD such as ulcerative coUtis (either in onset or during remissions) includes administration of unit dosage of from about 5-10 mg/kg body to about 10 mg/kg body (estimating an 80 kilograms adult subject) taken orally three times daily for up to 6 weeks, or about 50 mg/kg body administered rectally once daily as a liquid (60 ml) for a time period that ranges from 3 weeks to 6 weeks. The NO-donating compounds described herein when utilized for treating IND according to the present invention can therefore be administered as a unit dosage form hat ranges from about 0.1 mg/kg body to about 50 mg/kg body, whereby such a unit dosage form can be administered from 1 to 6 times a day, more preferably from 1 to 3 times a day, or rectally once a day, for a period that ranges from 1 to 10 weeks.
The NO-donors described herein can be administered, for example, orally, rectally, intravenously, intraventricularly, topically, intranasally, intraperitoneally, intestinally, parenterally, intraocularly, intradermally, transdermally, subcutaneously, intramuscularly, transmucosally, by inhalation and/or by intrathecal catheter. Preferably, the NO-donors are administered orally or intravenously, and optionally rectally, transdermally or by intrathecal catheter, depending on the IBD condition and the subject being treated. Optionally, the NO-donors are administered by means of a medical device such as a catheter or a gastroscope that is designed for directly delivering the compounds to the afflicted site. The method of treating an IBD, according to this aspect of the present invention, can optionally be effected by co-administering to the subject, along with the NO-donating compound described herein, an additional active agent that may have an added therapeutic value in treating IBD and/or a condition exacerbated thereby. The additional active agent can be co-administered prior to, concomitantly or subsequent to administering the NO-releasing compound(s). The additional active agent can be, for example, an inflammatory bowel disease drug, an NSAID, an antimicrobial agent, an analgesic, a metabolite agent, an anti-metabolic agent, a chemotherapeutic agent, an antioxidants, an anti-proliferative agents and any other agent that may provide an added therapeutic value. By being highly beneficial in treating IBDs, the NO-donating compounds described herein can be efficiently used for the preparation of a medicament for treating an inflammatory bowel disease. In any of the aspects of the present invention, the NO-donors described herein, either alone or in combination with any other active agents, can be utilized either per se, or as a part of a pharmaceutical composition. Hence, according to another aspect of the present invention, there are provided pharmaceutical compositions identified for use in the treatment of an inflammatory bowel disease, which comprise, as an active ingredient, one or more of the NO-donors described above and a pharmaceutically acceptable carrier. The pharmaceutical composition may further comprise an additional active ingredient being capable of treating the inflammatory bowel disease, as detailed hereinabove.
As used herein a "pharmaceutical composition" or "medicament" refers to a preparation of one or more of the NO-donors described herein, with other chemical components such as pharmaceutically acceptable and suitable carriers and excipients.
The purpose of a pharmaceutical composition is to facilitate administration of a compound to an organism. Hereinafter, the term "pharmaceutically acceptable carrier" refers to a carrier or a diluent that does not cause significant irritation to an organism and does not abrogate the biological activity and properties of the administered compound. Examples, without limitations, of carriers are: propylene glycol, saline, emulsions and mixtures of organic solvents with water, as well as solid (e.g., powdered) and gaseous carriers. Herein the term "excipient" refers to an inert substance added to a pharmaceutical composition to further facilitate administration of a compound.
Examples, without limitation, of excipients include calcium carbonate, calcium phosphate, various sugars and types of starch, cellulose derivatives, gelatin, vegetable oils and polyethylene glycols. Techniques for formulation and administration of drugs may be found in "Remington's Pharmaceutical Sciences" Mack Publishing Co., Easton, PA, latest edition, which is incorporated herein by reference. Pharmaceutical compositions of the present invention may be manufactured by processes well known in the art, e.g., by means of conventional mixing, dissolving, granulating, dragee-making, levigating, emulsifying, encapsulating, entrapping or lyophilizing processes. Pharmaceutical compositions for use in accordance with the present invention thus may be formulated in conventional manner using one or more pharmaceutically acceptable carriers comprising excipients and auxiliaries, which facilitate processing of the NO-donors into preparations which, can be used pharmaceutically. Proper formulation is dependent upon the route of administration chosen. For injection, the NO-donors of the invention may be formulated in aqueous solutions, preferably in physiologically compatible buffers such as Hank's solution, Ringer's solution, or physiological saline buffer with or without organic solvents such as propylene glycol, polyethylene glycol.
For transmucosal administration, penetrants are used in the formulation. Such penetrants are generally known in the art. For oral administration, the NO-donors of the invention can be formulated readily by combining the NO-donors with pharmaceutically acceptable carriers well known in the art. Such carriers enable the NO-donors of the invention to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions, and the like, for oral ingestion by a patient. Pharmacological preparations for oral use can be made using a solid excipient, optionally grinding the resulting mixture, and processing the mixture of granules, after adding suitable auxiliaries if desired, to obtain tablets or dragee cores. Suitable excipients are, in particular, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl- cellulose, sodium carbomethylcellulose; and/or physiologically acceptable polymers such as polyvinylpyrrolidone (PNP). If desired, disintegrating agents may be added, such as cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate. Dragee cores are provided with suitable coatings. For this purpose, concentrated sugar solutions may be used which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, titanium dioxide, lacquer solutions and suitable organic solvents or solvent mixtures. Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active ΝO-donors doses. Pharmaceutical compositions, which can be used orally, include push-fit capsules made of gelatin as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol. The push-fit capsules may contain the active ingredients in admixture with filler such as lactose, binders such as starches, lubricants such as talc or magnesium stearate and, optionally, stabilizers. In soft capsules, the ΝO-donors may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols. In addition, stabilizers may be added. A formulations for oral administration should be in dosages suitable for the chosen route of administration. Preferably, formulations for oral administration
further include a protective coating, aimed at protecting or slowing enzymatic degradation of the preparation in the GI tract. For buccal administration, the compositions may take the form of tablets or lozenges formulated in conventional manner. For administration by inhalation, the NO-donors for use according to the present invention are conveniently delivered in the form of an aerosol spray presentation (which typically includes powdered, liquified and/or gaseous carriers) from a pressurized pack or a nebulizer, with the use of a suitable propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichloro-tetrafluoroethane or carbon dioxide. In the case of a pressurized aerosol, the dosage unit may be determined by providing a valve to deliver a metered amount. Capsules and cartridges of, e.g., gelatin for use in an inhaler or insufflator may be formulated containing a powder mix of the NO-donors and a suitable powder base such as, but not limited to, lactose or starch. The NO-donors described herein may be formulated for parenteral administration, e.g., by bolus injection or continuous infusion. Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multidose containers with optionally, an added preservative. The compositions may be suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents. Pharmaceutical compositions for parenteral administration include aqueous solutions of the NO-donors preparation in water-soluble form. Additionally, suspensions of the NO-donors may be prepared as appropriate oily injection suspensions and emulsions (e.g., water-in-oil, oil-in-water or water-in-oil in oil emulsions). Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acids esters such as ethyl oleate, triglycerides or liposomes. Aqueous injection suspensions may contain substances, which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol or dextran. Optionally, the suspension may also contain suitable stabilizers or agents, which increase the solubility of the NO-donors to allow for the preparation of highly concentrated solutions. Alternatively, the NO-donors may be in powder form for constitution with a suitable vehicle, e.g., sterile, pyrogen-free water, before use.
The NO-donors, described in U.S. Provisional Patent Application No. 60/651,619 and herein, may also be formulated in rectal compositions such as suppositories or retention enemas, using, e.g., conventional suppository bases such as cocoa butter or other glycerides. The pharmaceutical compositions herein described may also comprise suitable solid of gel phase carriers or excipients. Examples of such carriers or excipients include, but are not limited to, calcium carbonate, calcium phosphate, various sugars, starches, cellulose derivatives, gelatin and polymers such as polyethylene glycols. Pharmaceutical compositions suitable for use in context of the present invention include compositions wherein the active ingredients are contained in an amount effective to achieve the intended purpose. More specifically, a therapeutically effective amount means an amount of NO-donors effective to prevent, alleviate or ameliorate symptoms of disease or prolong the survival of the subject being treated. Determination of a therapeutically effective amount is well within the capability of those skilled in the art, especially in light of the detailed disclosure provided herein. For any NO-donors used in the methods of the invention, the therapeutically effective amount or dose can be estimated initially from activity assays in animals. For example, a dose can be formulated in animal models to achieve a circulating concentration range that includes the IC50 as determined by activity assays (e.g., the concentration of the test NO-donors, which achieves a half-maximal reduction of the mean arterial blood pressure). Such information can be used to more accurately determine useful doses in humans. As is demonstrated in the Examples section that follows, a therapeutically effective amount for the NO-donors, may range between about 1 mg/kg body to about 200 mg/kg body. Toxicity and therapeutic efficacy of the NO-donors described herein can be determined by standard pharmaceutical procedures in experimental animals, e.g., by determining the EC50, the IC50 and the LD50 (lethal dose causing death in 50 % of the tested animals) for a subject NO-donor. The data obtained from these activity assays and animal studies can be used in formulating a range of dosage 'for use in human. The dosage may vary depending upon the dosage form employed and the route of administration utilized. The exact formulation, route of administration and dosage
can be chosen by the individual physician in view of the patient's condition. (See e.g., Fingl et al., 1975, in "The Pharmacological Basis of Therapeutics", Ch. 1 p.l). Dosage amount and interval may be adjusted individually to provide plasma levels of the active moiety which are sufficient to maintain the desired effects, termed the minimal effective concentration (MEC). The MEC will vary for each preparation, but can be estimated from in vitro data; e.g., the concentration necessary to achieve 50-90 % vasorelaxation of contracted arteries. Dosages necessary to achieve the MEC will depend on individual characteristics and route of administration. HPLC assays or bioassays can be used to determine plasma concentrations. Dosage intervals can also be determined using the MEC value. Preparations should be administered using a regimen, which maintains plasma levels above the MEC for 10-90 % of the time, preferable between 30-90 % and most preferably 50-90 %. Depending on the severity and responsiveness of the condition to be treated, dosing can also be a single administration of a slow release composition described hereinabove, with course of treatment lasting from several days to several weeks or until cure is effected or diminution of the disease state is achieved. The amount of a composition to be administered will, of course, be dependent on the subject being treated, the severity of the affliction, the manner of administration, the judgment of the prescribing physician, etc. Compositions of the present invention may, if desired, be presented in a pack or dispenser device, such as an FDA (the U.S. Food and Drug Administration) approved kit, which may contain one or more unit dosage forms containing the active ingredient. The pack may, for example, comprise metal or plastic foil, such as, but not limited to a blister pack or a pressurized container (for inhalation). The pack or dispenser device may be accompanied by instructions for administration. The pack or dispenser may also be accompanied by a notice associated with the container in a form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals, which notice is reflective of approval by the agency of the form of the compositions for human or veterinary administration. Such notice, for example, may be of labeling approved by the U.S. Food and Drug Administration for prescription drugs or of an approved product insert. Compositions comprising a NO- donors of the invention formulated in a compatible pharmaceutical carrier may also be
prepared, placed in an appropriate container, and labeled for treatment of an indicated condition or diagnosis, as is detailed hereinabove. Thus, according to a preferred embodiment of the present invention, the pharmaceutical composition described herein is packaged in a packaging material and identified in print, in or on the packaging material, for use in the treatment of an inflammatory bowel disease.
Additional objects, advantages, and novel features of the present invention will become apparent to one ordinarily skilled in the art upon examination of the foUowing examples, which are not intended to be limiting. Additionally, each of the various embodiments and aspects of the present invention as delineated hereinabove and as claimed in the claims section below finds experimental support in the following examples. EXAMPLES Reference is now made to the following examples, which together with the above descriptions, illustrate the invention in a non limiting fashion.
CHEMICAL SYNTHESES Materials and Methods: Proton NMR spectra were recorded using Varian 300 MHz and Brucker 200 MHz spectrometer. Spectra were obtained in deutro-chloroform (CDC1
3), unless indicated otherwise. Chemical shifts (δ) are reported in parts per million (ppm) downfield from tetramethylsilane. Gas chromatography was carried out on a Hewelett-Packard (HP-6890) gas chromatograph having a 5% phenylsiloxan column and a FID detector. Ultraviolet (UV) spectra were run as solutions in ethanol on a Beckman MVl spectrophotometer. 5-Amino-2-hydroxybenzoic acid (also referred to herein as5-ASA and also known as 5-aminosalicylic acid, mesalazine and mesalmine) was purchased from Sigma-Aldrich, Israel.
5-ASA
2,4,6-Trinitrobenzene sulfonic acid (TNBS) was purchased from Sigma- Aldrich, Israel.
4-Methyl-5-thiazoleetlιanol and propionamide were purchased from Aldrich chemical Company, USA. Phosphorus pentasulfide was purchased from Merck, Darmstadt, Germany. Tetrahydrofuran (THF) was dried and was freshly distilled over sodium/benzophenone ketyl prior to use. Other reagents were purchased from different suppliers and were used without purification, unless otherwise indicated. 5-Acetoxy-3-chloro-2-pentanone (ACP) was prepared according to the synthetic pathway described in Scheme 1 below and the corresponding procedure published by Buchman [J Am. Chem. Soc. 58, 1803, 1936].
Scheme 1
Syntheses of 2-substituted-4-methyl-5-(2-nitrooxy-ethyl)-thiazole derivatives
— General Procedure: The general synthetic pathway for preparing 2-substituted-4-methyl-5-(2- nitrooxy-ethyl)-thiazole derivatives, which serve as thiazole-based NO-donor compounds according to a preferred embodiment of the present invention, is presented in Schemes 2-4 below. In general, a desired thioamide is first prepared from a corresponding amide (Scheme 2), and is thereafter reacted, via a condensation reaction, with an alpha-chloroketone such as ACP, so as to form a 4-substituted-5- thiazoleethanol derivative (a 4-methyl-5-thiazoleethanol derivative in case of ACP) (Scheme 3). The alcohol moiety of the latter is then reacted with nitric acid, so as to produce the desired NO-donor according to this preferred embodiment of the present invention (Scheme 4). Thus, according to a representative synthetic pathway, a desired thioamide (general Compound II) is typically prepared according to the present invention by placing a corresponding amide (general Compound I) in a dry solvent such as THF or toluene, slowly adding thereto phosphorus pentasulfide (P2S5), while stirring, for a time period of 20-30 minutes under controlled temperature, and heating the resulting mixture at reflux temperature for additional 2-3 hours.
Scheme 2
Compound I Compound II
The respective 4-methyl-5-thiazoleethanol (general Compound III) is prepared from the thioamide (Compound II) according to the Hantzsch procedure [Hantzsch and Trauman, 1888, Rer 21, 938], as is shown in Scheme 3 below, by adding to the thioamide reaction mixture 5-acetoxy-3-chloro-2-pentanone (ACP), over a time period of 15-20 minutes. The reaction mixture is then heated at reflux temperature for about 20 hours, and the solvent is thereafter removed by distillation at atmospheric pressure. The reaction mixture is then cooled to 25 °C, hydrochloric acid (10 %) is added, and the mixture is heated at reflux temperature for one additional hour. Extraction of the reaction mixture with dichloromethane, drying over sodium sulfate, and evaporation to dryness, results in the desired 4-methyl-5-thiazoleethanol derivative.
Scheme 3
Compound II ACP Compound III
Nitration of the 4-methyl-5-thiazoleethanol derivative (Compound III) is then carried out as is illustrated in Scheme 4 below, by drop wise addition of nitric acid
(85-90%) to sulfuric acid (95-98%) over a time period of 20 minutes while keeping
the temperature at 0-5 °C, followed by addition of the 4-methyl-5-thiazoleethanol derivative. After stirring the reaction mixture for additional 2-3 hours at 0-5 °C, it is poured carefully onto cold water, washed with sodium hydroxide 20 %, and extracted with dichloromethane. The extracts are combined, dried and evaporated to dryness, to thereby produce the respective 2-substituted-4-methyl-5-(2-nitrooxy-ethyl)-thiazole (general Compound IV, also denoted as Pet, whereby each specific derivative is identified by a numeral).
Scheme 4
Compound III Compound IV
Using the general procedure described above, a variety of NO-donor compounds according to an preferred embodiment of the present invention were prepared, as is detailed hereinbelow.
Preparation of2-(4-methylthiazol-5-yl)ethyl nitrate (Pet-1):
2-(4-Methyl-thiazol-5-yl)-ethanol (Pet-l-OH) was purchased from Aldrich chemical Company, USA in 98% purity and was used without further purification. Nitration of 2-(4-methylthiazol-5-yl)ethanol, was carried out according to the procedure described hereinabove, to give 7 grams (53 % yield) of 4-Methyl-5-(2-
nitrooxy-ethyl)-thiazole (Pet-1) as pale yellow liquid having a purity of 99 % as determined by gas chromatography. 1H-NMR (CDC13): δ = 2.41 (s, 3H, CH3), 3.17 (t, 2Η, CH2), 4.58 (t, 2Η, CH2), 8.60 (s, 1Η, Aromatic) ppm. Preparation of4-methyl-5-(2-nitrooxy-ethyl)-2-phenyl-thiazole (Pet-8):
2-(4-Methyl-2-phenyl-thiazol-5-yl)-ethanol (Pet-8-OΗ) was prepared, according to general procedures presented hereinabove, by adding 7.3 grams (0.053 moles) of thiobenzamide (TB A, obtained from Merck, Germany) to 200 ml dry THF, followed by addition of 9.5 grams (0.053 moles) 5-acetoxy-3-chloro-2-pentanone over a time period of 20 minutes. The reaction mixture was then refluxed at 80 °C for 24 hours, after which the THF was removed by evaporation. 100 ml of water and 15 ml of HCl solution (32 %) were added and the reaction mixture was refluxed for 1 hour at 90 °C. After cooling, the mixture was washed with two portions of 100 ml of dichloromethane to remove excess starting materials. The aqueous phase was turned basic (pH 8-9) using an aqueous solution of 5 N sodium hydroxide. The 2-(4-methyl- 2-phenyl-thiazol-5-yl)-ethanol was extracted with three portions of 100 ml of dichloromethane and the combined extracts were dried over sodium sulfate. After removal of the dichloromethane 8 grams (45 % yield) of the 2-(4-methyl-2-phenyl- thiazol-5-yl)-ethanol product were obtained as a reddish-brown liquid. The nitration of 2-(4-methyl-2-phenyl-thiazol-5-yl)-ethanol (Pet-8-OH) was carried out subsequently according to the procedure described hereinabove, to give 4- methyl-5-(2-nitrooxy-ethyl)-2-phenyl-thiazole) in 40 % yield. The product was purified by column chromatography using a mixture of 1:1:1 ethyl acetate:hexane:dichloromethane as eluent, to purity of 99.5 % as determined by HPLC and by thin-layer chromatography stained with diphenylamine as a marker for the nitrate ester moiety.
NMR (CDCI3): δ = 2.37 (s, 3H, CH3), 3.18 (t, 3Η, CH2), 4.57 (t, 2Η, CH2ONO2), 7.32-8.19 (m, 5Η, Aromatic) ppm. Preparation of 4-methyl-5-(2-nitrooxy-ethyl)-thiazole-2-ylamine (Pet-10):
2-(2-Amino-4-methyl-thiazol-5-yl)-ethanol was prepared according to general procedures presented hereinabove, by adding 20 grams (0.263 moles) of thiourea to 200 ml of dry toluene, followed by addition of 47 grams (0.263 moles) of 5-acetoxy- 3-chloro-2-pentanone over a time period of 20 minutes. The reaction mixture was heated at 80 °C for 24 hours and thereafter approximately 180 ml of toluene were removed by evaporation. 180 ml of water and 20 ml of HCL solution (32 %) were then added and the reaction mixture was refluxed for 1 hour at 90 °C. The organic phase was then removed by washing with chloroform, and the aqueous phase was turned basic (pH 8-9) using a 5 N solution of sodium hydroxide. The product was extracted once with 70 ml chloroform and once with 70 ml ethyl acetate. The combined organic extracts were dried over sodium sulfate and the solvents were removed by evaporation to give 30 grams (72 % yield) of violet-brown crystals. Subsequently, 4-methyl-5-(2-nitrooxy-ethyl)-thiazole-2-ylamine was prepared by drop-wise addition of 1.59 grams of nitric acid (70 %) to 2.48 grams of cooled sulfuric acid (95-98 %) at 0-5 °C over a time period of 20 minutes. Following, 4 grams (0.025 moles) of 2-(2-Amino-4-methyl-thiazol-5-yl)-ethanol were added over a time period of 45 minutes at 0-5 °C. After the addition was completed, the reaction mixture was stirred for 3 hours at 25 °C, and was then added carefully to 50 ml of cold water. The water solution was turned basic with an aqueous solution of 20 % sodium hydroxide and the aqueous phase was extracted with three portions of 75 grams of ethyl acetate. The organic extracts were combined, dried over sodium sulfate, filtered and evaporated to dryness under vacuum. Chromatography of the crude product on silica gel (using a mixture of 8:2 ethyl acetate:hexane as eluent)
gave 1.8 grams (35 % yield) of the product as orange oil having a purity of 98.5 % as determined by gas chromatography. 1H-NMR (CDC13): δ = 2.10 (s, 3H, CH3), 2.79 (t, 2Η, CH2), 3.37 (t, 2Η, CH2OH,), 4.87 (s, broad, NH2) ppm. Preparation of 3-[4-methyl-5-(2-nitrooxy-ethyl)-thiazole-2-yl]-pyridine
(Pet-12):
2-(4-Methyl-2-pyridin-3-yl-thiazole-5-yl)-ethanol was prepared according to the general procedure presented hereinabove, by adding 20 grams (0.145 moles) of thionicotinamide (purchased from Acros, Belgium) to 200 ml of dry toluene, followed by addition of 26 grams (0.145 moles) of ACP over a time period of 20 minutes. The reaction mixture heated for 24 hours at 80 °C and thereafter about 180 ml of toluene were removed by evaporation. 100 ml of water and 20 ml of HCl solution (32 %) were added and reflux was continued for 1 hour at 90 °C. The organic phase was then removed by washing with chloroform and the aqueous phase was turned basic (pH 8- 9) using a 5 N solution of NaOH. The 2-(4-methyl-2-pyridin-3-yl-thiazole-5-yl)- ethanol was extracted with three portions of 100 ml of chloroform and the combined extracts were dried over sodium sulfate. The chloroform was thereafter removed and the residue was purified by liquid chromatography, using a mixture of 9:1 ethyl acetate:methanol as eluent, to give 10 grams (31 % yield) of the 2-(4-methyl-2- pyridin-3-yl-thiazole-5-yl)-ethanol product as a violet-brown powder. 3-[4-Methyl-5-(2-nitrooxy-ethyl)-thiazole-2-yl]-pyridine was prepared by drop-wise addition of 1.145 grams of nitric acid (70 %) to 1.78 grams of cooled sulfuric acid (95-98 %) at 0-5 °C, followed by addition of 4 grams (0.0181 moles) 2- (4-methyl-2-pyridin-3-yl-thiazole-5-yl)-ethanol over a time period of 30 minutes at 0- 5 °C. After the addition was completed the reaction mixture was stirred for 30 minutes at 0-5 °C, and for one additional hour at room temperature. The reaction mixture was then added carefuUy to 25 ml of cold water. The water solution was turned basic with an aqueous 20 % solution of sodium hydroxide and the aqueous
phase was extracted with three portions of 75 grams of dichloromethane. The extracts were combined, dried over sodium sulfate, filtered and evaporated to dryness under vacuum. The nitration step was confirmed initially by thin-layer chromatography stained with diphenylamine as a marker for the nitrate ester moiety (Figure 1). Chromatography of the crude product on silica gel (using ethyl acetate as eluent) gave 1.5 grams (41 % yield) of Pet-12 as a pale yellow liquid having a purity of 99 % as determined by HPLC. 1H-NMR (CDC13): δ = 2,47 (s, 3H, CH3), 2.77 (t, 2Η, CH2, benzylic), 3.88 (t, 2Η, CH2ONO2), 7.44-8.56 (m, 4Η, Aromatic) ppm. Preparation of 2-(4-methyl-2-(6-(methylamino)pyrazin-2-yl)thiazol-5- yl)ethyl nitrate (Pet-24):

2-(4-methyl-2-(6-(methylamino)pyrazin-2-yl)thiazol-5-yl)ethanol was prepared according to the general procedure presented hereinabove, by adding 10 grams (0.059 moles) of 6-(methylamino)pyrazine-2-carbothioamide (purchased from Avocado, UK) to 150 ml of dry toluene, followed by addition of 10.62 grams (0.0.59 moles) of ACP over a time period of 20 minutes. The reaction mixture heated for 24 hours at 80 °C and thereafter about 140 ml of toluene were removed by evaporation. 100 ml of water and 15 ml of HCl solution (32 %) were added and reflux was continued for 1 hour at 90 °C. The organic phase was then removed by washing with chloroform and the aqueous phase was turned basic (pH 8-9) using a 5 N solution of NaOH. The 2-(4-methyl-2-(6-(methylammo)pyrazin-2-yl)thiazol-5-yl)ethanol was extracted with three portions of 100 ml of chloroform and the combined extracts were dried over sodium sulfate. The chloroform was thereafter removed and the residue was purified by liquid chromatography, using a mixture of 9:1 ethyl acetate:methanol as eluent, to give 11 grams (74 % yield) of the 2-(4-methyl-2-(6- (methylamino)pyrazin-2-yl)thiazol-5-yl)ethanol product as a brown solid.
2-(4-methyl-2-(6-(methylamino)pyrazin-2-yl)thiazol-5 -yl)ethyl nitrate was prepared by drop-wise addition of 2 grams of nitric acid (70 %) to 2 grams of cooled sulfuric acid (95-98 %) at 0-5 °C, followed by addition of 2 grams (6.78 mmoles) 2-
(4-methyl-2-(6-(methylamino)pyrazin-2-yl)thiazol-5-yl)ethanol over a time period of 20 minutes at 0-5 °C. After the addition was completed the reaction mixture was stirred for 30 minutes at 0-5 °C, and for one additional hour at room temperature. The reaction mixture was then added carefully to 25 ml of cold water. The water solution was turned basic with an aqueous 20 % solution of sodium hydroxide and the aqueous phase was extracted with three portions of 45 grams of dichloromethane. The extracts were combined, dried over sodium sulfate, filtered and evaporated to dryness under vacuum. The nitration step was confirmed initially by thin-layer chromatography stained with diphenylamine as a marker for the nitrate ester moiety. Chromatography of the crude product on silica gel (using ethyl acetate as eluent) gave 1.1 grams (47 % yield) of Pet-24 as a pale green liquid having a purity of 96 % as determined by HPLC. 1H-NMR (CDC13): δ = 2.44 (s, 3H, CH3), 2.51 (s, 3Η, NH-CH3), 3.70 (t, 2H, CH2), 4.77 (t, 2Η, CH2-ONO2), 7.67-7.98 (m, 2Η, Aromatic) ppm. Preparation of2-(2-(4-chlorophenyl)-4-methylthiazol-5-yl)ethyl nitrate (Pet- 56):
Pet-56 was prepared according to general procedure presented hereinabove and the procedure described above for the preparation of Pet-8, using 4-chlorothiobenzamide (obtained from Avocado, UK) as the starting material. The respective alcohol was obtained in 66 % yield as brown crystals whereby Pet-56 was obtained as pale yellow crystals (85 % yield) having a purity of 99 % as determined by thin-layer chromatography and gas chromatography. 1H-NMR (CDCI3) of the alcohol intermediate: δ = 2.38 (s, 3H, CH3), 2.96 (t, 2Η, CH2, J=6 Ηz), 3.82 (t, 2Η, CH2OH, J=6.3 Hz), 7.36 (d, 2H, Aromatic, J=2.1 Hz), 7.79 (d, 2H, Aromatic, J=2.1 Hz) ppm.
1H-NMR of Pet-56 (CDC13): δ = 2.41 (s, 3H, CH3), 3.17 (t, 2Η, CH2, J-6.6 Ηz), 4.60 (t, 2Η, CH2ONO2, J=6.6Ηz), 7.37 (d, 2H, Aromatic, J=6.6 Hz), 7.79 (d, 2H, Aromatic, J=6.9 Hz) ppm. Preparation of 4-methyl-5-(2-nitrooxy-ethyl)-2-(4-trifluoromethyl-phenyl)- thiazole (Pet-59):
Pet-59 was prepared according to general procedure presented hereinabove and the procedure described above for the preparation of Pet-8, using 4-(trifluoromethyl)-thiobenzamide (obtained from Avocado, UK) as the starting material. The respective alcohol was obtained in 73 % yield as brown crystals whereby Pet-59 was obtained as pale brown crystals (74 % yield) having a purity of 99 % as determined by thin-layer chromatography and gas chromatography. 1H-NMR (CDCI3) of the alcohol intermediate: δ = 2.42 (s, 3H, CH3), 3.01 (t,
2Η, CH2, J=6 Ηz), 3.85 (t, 2Η, CH2OH, J=6 Hz) ppm. 1H-NMR of Pet-59 (CDCI3): δ = 2.43 (s, 3H, CH3), 3.18 (t, 2Η, CH2, J=6.6 Ηz), 4.60 (t, 2Η, CH2ONO2, J=6.6Ηz) ppm. Preparation of Bh-[4-methyl-5-(2-nitrooxy-ethyl)-thiazol-2-yl]-diazene (Pet- 102):
Pet-102 was prepared according to general procedure presented hereinabove and the procedure described above for the preparation of Pet-3, using azodicarbonamide (Aldrich-Sigma) as the starting material, in an overall yield of 55%.
1H-NMR (CDC13): δ = 2.62 (s, 6H, 2 x CH3), 2.82(q, 4Η, 2 x CH2), 3.86(t, 4Η, 2x CH2ONO2) ppm. Preparation of 2-(4-methyl-2-(naphthalen-l-ylamino)thiazol-5-yl)ethyl nitrate (Pet-149):
According to the general procedure presented hereinabove, 200 ml of dry toluene and 10 grams (0.049 moles) of l-(naphthalen-l-yl)thiourea (obtained from Avocado, UK) were placed in a 500 ml round-bottomed flask fitted with a reflux condenser. 8.82 grams (0.049 moles) of 5-acetoxy-3-chloro-2-pentanone (ACP) were added to the solution over a time period of 20 minutes. The reaction mixture was refluxed at 80 °C for 24 hours, after which the toluene was removed by evaporation. 100 ml of water and 15 ml of ΗC1 solution (32 %) were added and the resulting mixture was refluxed for 1 hour at 90 °C. After cooling, the mixture was washed with two portions of 100 ml of dichloromethane to remove excess of starting materials. The aqueous phase was turned basic (pΗ 8-9) using 5 N solution of sodium hydroxide. The 2-(4-methyl-2-(naphthalen-l-ylamino)thiazol-5-yl)ethanol was extracted with three portions of 100 ml of dichloromethane and the combined extracts were dried over sodium sulfate. The dichloromethane was removed under vacuum to yield 3.7 grams (27 %) of 2-(4-methyl-2-(naphthalen-l-ylamino)thiazol-5-yl)ethanol, which was used in the subsequent nitration step without further purification. Nitration of 2-(4-methyl-2-(naphthalen-l-ylamino)thiazol-5-yl)ethanol was carried out by cooling 0.2 grams (0.002 moles) of sulfuric acid (95-98 %) to 0-5 °C and addition thereto of 0.13 grams (0.002 moles) nitric acid (70 %) drop-wise over a time period of 20 minutes while keeping the temperature between 0-5 °C. Following, 0.6 grams of 2-(4-methyl-2-(naphthalen-l-ylamino)thiazol-5-yl)ethanol (0.002 moles) were added over a time period of 10 minutes at 0-5 °C. After the addition was completed, the reaction mixture was stirred for 30 minutes at 0-5 °C. The reaction
mixture was then added carefully to 25 ml of cold water. The water solution was turned basic with an aqueous solution of sodium hydroxide and the aqueous phase was extracted with three 25 grams portions of a mixture of 1 : 1 ethyl acetate:ether. The organic extracts were combined, dried over sodium sulfate, filtered and evaporated to dryness. Chromatography of the crude product on silica gel (using a mixture of 2:1 ethyl acetate:hexane as eluent) gave 0.5 grams (76 % yield) of the product as a dark green solid having a purity of 97 % as determined by TLC. 1H-NMR (CDC13): δ = 2.52 (s, 3H, CH3), 3.42 (s, 2Η, CH2), 4.46 (t, 2Η, CH2ONO2), 7.21-8.09 (m, 7Η, Aromatic) ppm.
Using the general procedure and the exemplary procedures described above, Pet-2, Pet-3, Pet-4, Pet-5, Pet-6, Pet-7, Pet-9, Pet-11, Pet-13, Pet-17, Pet-44, Pet- 55, Pet-66, Pet-97, Pet-116, Pet-118, Pet-181, Pet-182, Pet-183, Pet-184, Pet-185 and Pet-186 presented in Tables 1 and 2, were prepared and analyzed. Other 2-substituted-4-methyl-5-(2-nitrooxy-ethyl)-thiazoles, as presented, for example, in Tables 1 and 2, are similarly prepared.
Preparation of NO-donors having a biocleavable moiety - General Procedure: The procedure presented hereinbelow is general procedure for the preparation of thiazole-based NO-donor according to a preferred embodiment of the present invention, having a biocleavable moiety between the thiazole residue and an additional moiety that is linked thereto. This procedure relies on the general synthetic pathway for preparing the desired 2-(2-substituted-4-methyl-thiazol-5-yl)-ethanol derivative (Compound IH) as presented hereinabove and described in Schemes 2 and 3, which serve as thiazole-based NO-donor compounds according to a preferred embodiment of the present invention. The general procedure is presented in Scheme 5 below. In general, a reactive derivative of a 2-(2-substituted-4-methyl-thiazol-5-yl)-ethanol (Compound V, Scheme 5), is first prepared, and is thereafter reacted with a desired compound having a second reactive group (K-X, Scheme 5). The first and the second reactive groups are selected capable of reacting therebetween, to thereby form a biocleavable moiety (A,
Scheme 5). The resulting compound (Compound VI) thus includes a thiazole moiety and a residue of the desired compound covalently linked therebetween by a biocleavable moiety. Scheme 5
Compound V Compound VI Thus, according to a representative synthetic pathway, the first reactive group on the thiazole derivative (Q in Compound V, Scheme 5) is, for example, an amine, the corresponding second reactive group on the desired compound (K) is, for example, a carboxylic acid, and the formed biocleavable moiety is, for example, an amide. The synthesis in this case is effected by adding dicyclohexylcarbodiimide (DCC) to an equal molar amount of the carboxylic acid derivative in dichloromethane. The mixture is stirred for 2 hours, followed by the addition of an equal molar amount of 4-methyl-5-(2-nitrooxy-ethyl)-thiazole-2-ylamine (Pet-10, prepared as described hereinabove). The reaction mixture is stirred for 8 hours, after which the organic layer is removed and washed with 5 % NaOH solution followed by 5 % HCl solution and finally with two portions of water to remove excess starting materials. The dichloromethane is dried using sodium sulfate and removed by evaporation to afford the corresponding [5-(2-hydroxy-ethyl)-4-methyl-thiazol-2-yl]- amide. In accordance with the representative general synthetic pathway presented hereinabove, the nitration of the [5-(2-hydroxy-ethyl)-4-methyl-thiazol-2-yl]-amide is afforded by the addition of 70 % nitric acid to acetic anhydride while stirring and maintaining the temperature between 20-30 °C by external cooling. The mixture is then cooled to -5 °C while stirring, followed by the addition of the [5-(2-hydroxy- ethyl)-4-methyl-thiazol-2-yl]-amide. The mixture is kept for 30 minutes at -5 °C and then heated to 10 °C and stirred for one hour. The resulting mixture is poured
thereafter into ice water and stirred for 1 hour. Aliquots of NaHCO3 are added until CO2 evolution ceases. The aqueous phase is extracted with three portions of ethyl acetate, and the combined extracts are dried over sodium sulfate and concentrated by evaporation (Scheme 6).
Scheme 6
In a second example, the first reactive group on the thiazole derivative (Q in
Compound V, Scheme 5) is, for example, hydroxyl, the corresponding second reactive group on the desired compound (K) is, for example, a carboxylic acid, and the formed biocleavable moiety is, for example, an ester. The synthesis in this case is executed by reacting an equal molar amount of the carboxylic acid derivative and an equal molar amount of the thiazole derivative in the presence of a catalytic amount of an acid or an equal molar amount of a base. In another example, the first reactive group on the thiazole derivative (Q in Compound V, Scheme 5) is, for example, a carboxylic acid, and the corresponding second reactive group on the desired compound (K) is, for example, hydrazine, and the formed biocleavable moiety is, for example, hydrazide. The synthesis in this case is effected by an equal molar amount of the carboxylic acid derivative and an equal molar amount of the thiazole derivative in the presence of a catalytic amount of an acid. The nitration step for the general examples above is carried out in accordance with the representative general synthetic pathway for the biocleavable amide described hereinabove. Using the general procedure described hereinabove, a variety of NO-donor compounds having a biocleavable moiety according to the present invention were prepared, as is detailed hereinbelow.
Preparation of N-[4-Methyl-5-(2-nitrooxy-ethyl)-thiazol-2-yl]-nicotinamide (Pet-154):
Pet-154 was prepared according to the general procedure presented hereinabove, and as described in Scheme 7 below. 16.7 grams (0.081 mol) of dicyclohexylcarbodiimide (DCC) were added to 10 grams (0.081 mol) of nicotinic acid in dichloromethane. The mixture was stirred for 2 hour, and 12.8 grams (0.081 mol) of 2-(2-amino-4-methyl-thiazol-5-yl)-ethanol were added thereto. The reaction mixture was stirred for additional 8 hours, and the organic layer was thereafter separated, washed with 5 % NaOH solution, 5 % HCl solution and two portions of water, to remove excess starting materials, and was dried using sodium sulfate. Removal of the solvent by evaporation gave 13 grams (61 % yield) of N-[5-(2- hydroxy-ethyl)-4-methyl-thiazol-2-yl]-nicotinamide as a pale yellow powder. 1.2 ml of 70 % nitric acid were added to 5 ml of acetic anhydride while stirring and maintaining the temperature between 20-30 °C by means of external cooling. The mixture was then cooled to —5 °C while stirring, 1 gram of N-[5-(2- hydroxy-ethyl)-4-methyl-thiazol-2-yl]-nicotinamide was added thereto and the resulting mixture was stirred for 30 minutes at -5 °C and for an additional hour at 10 °C. The mixture was then poured onto an ice-water mixture and was stirred for 1 hour. Aliquots of NaHCO3 were thereafter added to the reaction mixture until CO2 evolution ceased. The resulting orange-yellow aqueous solution was extracted with three portions of 15 ml ethyl acetate. The combined extracts were dried over sodium sulfate and concentrated by evaporation, to give the yellow solid product in 75 % yield. 1H-NMR (CDC13): δ = 2.48 (s, 3H, CH3), 3.39 (q, 2Η, CH2 benzylic), 4.08 (t, 2Η, CH2ONO2), 7.24-8.87 (m, 4Η, Pyridinic) ppm.
Scheme 7
Preparation of5-[l,2]Dithiolan-3-yl-pentanoic acid [4-methyl-5-(2-nitrooxy- ethyl)-thiazol-2-yl]-amide (Pet-151):
Pet-151 was prepared according to the general procedure described hereinabove and the procedure described above for the preparation of Pet-154, using 5-[l,2]dithiolan-3-yl-pentanoic acid (DL-Lipoic acid) and 4-methyl-5-(2-nitrooxy- ethyl)-thiazol-2-ylamine (Pet-10) as the starting materials. Pet-151 was obtained as a pale yellow liquid in an overall yield of 51 %. 1H-NMR (CDC1
3): δ = 1.61 (m, 2H, CH
2, alpha-S), 1.66 (m, 2Η, CH
2, beta-
S), 1.90 (m, 2Η, CH2, beta-C=(O)N), 2.25 (s, 3H, CH3), 2.36 (m, 2Η, CH2), 2.89 (t, 2Η, CH2, 3.54 (m, 1Η, CH-S-CΗ2), 3.80 (t, 2H, CH2-ONO2), 6.91 (s, 1Η, NH-amide) ppm.
Preparation of 2-(4-Isobutyl-phenyl)-N-[4-methyl-5-(2-nitrooxy-ethyl)- thiazol-2-ylJ-propionamide (Pet-152):
Pet-152 was prepared according to the general procedure described hereinabove and the procedure described above for the preparation of Pet-154, using 2-(4-isobutyl-phenyl)-propionic acid (Ibuprofen) and 4-methyl-5-(2-nitrooxy-ethyl)- thiazol-2-ylamine (Pet-10) as the starting materials, in an overall yield of 63 %. 1H-NMR (CDC13): δ = 0.86 (m, 6H, CH-(CH3)2, 1.18 (t, 2Η, CH2), 1.57 (d, 1Η, CH-CΗ3), 2.21 (s, 3H, CH3), 2.70 (m, 2Η, CH2), 3.06 (t, 2Η, CH2), 3.41 (q, 3Η, CH3-CH,), 3.81 (q, IH, CH3-CH), 4.52 (t, 2Η, CH2-ONO2), 7.12-7.83 (m, 4Η, aromatic) ppm. In addition, Pet-150, Pet-153, Pet-155, Pet-156, Pet-157, Pet-158, Pet-159,
Pet-160, Pet-161, Pet-162, Pet-163 and Pet-164, having a biocleavable moiety and presented in Tables 1 and 2, were prepared and analyzed according to similar procedures. Other 2-substituted-4-methyl-5-(2-nitrooxy-ethyl)-thiazole derivatives having a biocleavable moiety, as shown in Tables 1 and 2, have been similarly prepared.
Table 1
Table 2
IN VIVO ACTVIITY ASSAYS The anti-inflammatory effect of the NO-donors of the present invention was evaluated by treating colitis-induced rats with exemplary NO-donors according to the present invention and measuring thereafter the myeloperoxidase (MPO) activity in isolated colon tissues of the tested rats, according to the protocol described below. The effect of the NO-donors of the present invention was compared to that of 5-ASA, a presently used drug for treating ulcerative colitis. Myeloperoxidase (MPO) is a lysosomal enzyme that is found predominantly in the azurophilic granules of neutrophils (white blood cells). MPO utilizes hydrogen peroxidase to convert chloride to hypochlorous acid, whereby the produced hypochlorous acid may act as an anti-bacterial agent. MPO is known as an excellent inflammatory biomarker for autoimmune diseases, inflammatory diseases and cancer, and has been used as a quantitative index of inflammation in several tissues, including the intestine (Krawisz et al., 1984, Gastroenterology 87:1344-1350). Measuring the MPO activity in isolated colon tissues of the treated rats therefore served to record and score the level of the inflammation and for determining the effect of the tested compounds of the extent of the induced colitis. Protocol: Colitis induction in rats: Sprague Dawley rats were lightly anesthetized with ether and a rubber catheter (3 mm diameter) was inserted through the anal canal for a distance of 7 cm into the colon just proximal to the splenic flexure. Colitis was induced by rectal administration of 0.2 ml of 2,4,6-trinitrobenzene sulfonic acid (TNBS, 100 g/L dissolved in 50 % ethanol). This procedure was repeated once daily for one week. Administration of NO-donors and colon tissue sample preparation: One week after colitis induction, a daily dose of 20 mg/Kg of Pet-l-OH (the alcohol precursor used in the synthesis of Pet-1, vide supra), Pet-1, TBA
(thiobenzamide, the thioamide used in the synthesis of Pet-8, vide supra) Pet-8-OH (the alcohol precursor used in the synthesis of Pet-8, vide supra), Pet-8, Pet-10, Pet- 12, PET-24, Pet-56, Pet-149, Pet-151, Pet-152, Pet-154, Pet-155 and 5- aminosalicylic acid (5-ASA) was administered rectally to rats for a duration of four days. Rats were grouped as follows:
Following the treatment period, specimens of colon tissue were collected from the rats in each group, rinsed with buffered ice-cold saline to remove contaminating blood and were stored at -70 °C. Detection of myeloperoxidase activity: MPO activity in rats colon tissue was then assayed by determining the decomposition of hydrogen peroxide in the presence of o-dianisidine, as follows: Finely ground colon tissue (200 mg) was homogenized three times for 3 seconds at 4 °C with a Polytron™ homogenization device (Glen Mills Inc.) in 1 ml ice-cold homogenization solution containing 0.5 % hexadecyltrimethylammonium bromide (HTAB; Sigma) and 50 mmol/liter phosphate buffer set to pH 6. The homogenizer probe was rinsed twice with 1.0 ml homogenization solution, and the washings were combined with the homogenate. The homogenate was subjected to intensive sonication by ultrasound for 10 second, flash-frozen and thawed three times, and centrifuged at 13,000 rpm for 5 minutes at 4 °C. The supernatant was collected for analysis. 2.9 ml of a solution containing 50 mmol/liter phosphate buffer set to pH 6.0,
0.167 mg/ml O-dianizidine hydrochloride and 0.005 % hydrogen peroxide, were added to 0.1 ml of the obtained supernatant. Absorbance differences at 460 nm
(ΔOD460) were measured spectrophotometerically. One unit of MPO activity was defined as that degrading 1 μmol peroxide per unit at 25 °C.
EXPERIMENTAL RESULTS: The anti-inflammatory effect of exemplary NO-donors according to the present invention, compared with the effect of the known anti-inflammatory drug 5-ASA was measured by determining the MPO activity in colon tissues isolated from colitis- induced rats treated as described in the protocol above. The obtained results are summarized in Table 3 below and are further presented in Figures 1-5.
Table 3
Figure 1 presents the results obtained foUowing treatment with Pet-10, Pet-152, Pet-154 and Pet-155 and 5-ASA, and clearly shows the superior efficacy of
the NO-donors of the present invention in treating induced colitis compared with that of the commonly used drug, 5-ASA, and particularly the superior activity of Pet-155, Pet-152 and Pet-154 which contain a biological moiety that is attached to the thiazole ring via a biocleavable bond that can readily cleave in the body and release a bioactive moiety. Thus, Pet-152 can be cleaved so as to release an ibuprofen residue, a know NSAID agent and Pet-154 can be cleaved so as to release a nicotinic acid residue, a prevalent metabolite. The obtained results thus demonstrate the potent activity of such dual-active NO-donors according to the present invention. Figure 2 presents the results obtained following treatment with Pet-8-OH and Pet-8 (the first is the alcohol intermediate obtained in the synthesis of Pet-8) and 5- ASA, and clearly shows the superior efficacy of the NO-donors of the present invention in treating colitis as compared with that of the commonly used drug, 5-ASA. Furthermore, the results clearly show the superior activity of Pet-8 as compared with that of its alcohol intermediate Pet-8-OH and thus indicate that the presence of an NO releasing group is required for exerting such an activity. Figure 3 presents the results obtained following treatment with Pet-l-OH (the alcohol intermediate of Pet-1), Pet-1, Pet-8-OH (the alcohol intermediate of Pet-8) and Pet-8 and 5-ASA, and clearly shows the superior efficacy of the NO-donors of the present invention in treating induced colitis, and particularly that of Pet-8 as compared with that of the commonly used drug, 5-ASA. Again, the results show the superior activity of Pet-1 and Pet-8 as compared with that of its alcohol intermediates Pet-l- OH and Pet-8-OH and thus indicate that the presence of an NO releasing group is required for exerting such an activity. Figure 4 presents the results obtained following treatment with TBA (the thioamide used in the synthesis of Pet-8), Pet-8-OH (the alcohol intermediate in the synthesis of Pet-8), Pet-8, Pet-1 and 5-ASA, and clearly shows the superior efficacy of the NO-donors of the present invention in treating induced colitis as compared with that of the synthetic intermediate species and commonly used drug, 5-ASA. In addition to showing the superior activity of Pet-8 as compared with that of its alcohol intermediate Pet-8-OH, the results show the superior activity of the alcohol intermediate over that of the thioamide starting material, thus indicating that the presence of a thiazole ring is required for exerting anti-colitis activity.
Figure 5 presents the results obtained following treatment with Pet-8, Pet-12, Pet-24 and Pet-149 and 5-ASA, and clearly shows the superior efficacy of the NO- donors of the present invention in treating induced colitis, and particularly that of Pet- 8 and Pet-12 as compared with that of the commonly used drug, 5-ASA. Since Pet- 12 and Pet-8 both have an aromatic moiety (aryl or heteroaryl) attached to the thiazole ring, these results may suggest that such as an aryl/heteroaryl group attached to the thiazole ring may be required for effective activity of the NO-donors as anti-colitis agents. It is appreciated that certain features of the invention, which are, for clarity, described in the context of separate embodiments, may also be provided in combination in a single embodiment. Conversely, various features of the invention, which are, for brevity, described in the context of a single embodiment, may also be provided separately or in any suitable subcombination.
Although the invention has been described in conjunction with specific embodiments thereof, it is evident that many alternatives, modifications and variations will be apparent to those skilled in the art. Accordingly, it is intended to embrace all such alternatives, modifications and variations that fall within the spirit and broad scope of the appended claims.
AU publications, patents and patent applications mentioned in this specification are herein incorporated in their entirety by reference into the specification, to the same extent as if each individual publication, patent or patent application was specifically and individually indicated to be incorporated herein by reference. In addition, citation or identification of any reference in this application shall not be construed as an admission that such reference is available as prior art to the present invention.