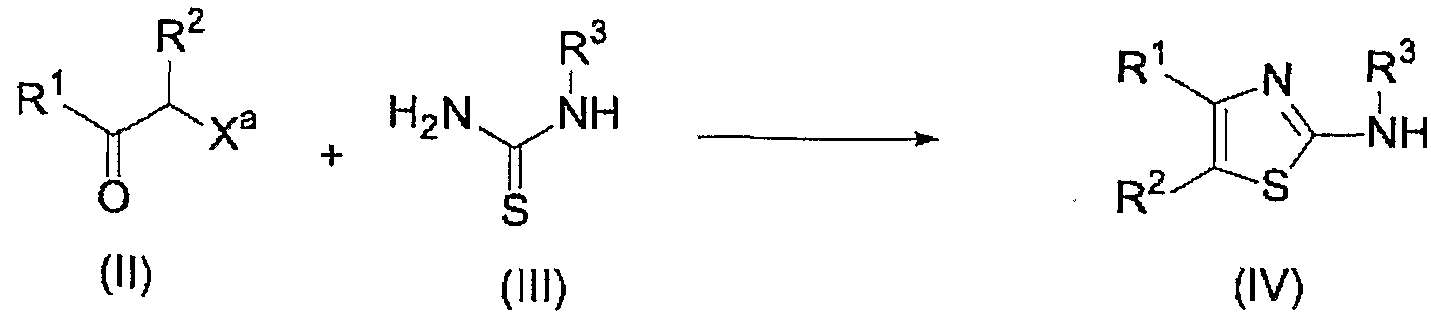明細書 Specification
チアゾール誘導体 Thiazole derivatives
技術分野 Technical field
本発明は、 II - HSD1 (11)8—ヒドロキシステロイド デヒドロゲナ一ゼタイ プ 1 )阻害剤として有用な新規チアゾール誘導体、 その医薬上許容される塩、 及びその溶媒和物などに関する。 背景技術 The present invention relates to a novel thiazole derivative useful as an II-HSD1 (11) 8-hydroxysteroid dehydrogenase type 1) inhibitor, a pharmaceutically acceptable salt thereof, a solvate thereof and the like. Background art
11)8-HSD1 コルチゾンをコルチゾ一ルへ変換する酵素である。 113-HSD1 は、 肝臓、 内臓脂肪などで発現し、 細胞内のコルチゾール濃度を各臓器レベル で増幅するファクタ一として機能すると考えられている。 11)8-HSD1は、 肝臓 において糖の新生に関与することが示唆されている。 このため、 11j8- HSD1の 活性を阻害すると、 肝臓における糖の新生を抑制できると期待される。 更に 11 - HSD1は、 内臓脂肪の蓄積に関係していることが示唆されている。 このた め、 11i8- HSD1の活性を阻害すると、 内臓脂肪の蓄積を抑制できると期待され る。 11)8- HSD1の阻害剤として、 Barf Tらの報告 (J Med Chem. 2002 :3813- 5) 、 WO 01/090090、 WO 01/090091、 WO 01/090092 WO 01/090093、 W0 11) 8-HSD1 An enzyme that converts cortisone to cortisone. 113-HSD1 is expressed in the liver, visceral fat, etc., and is thought to function as a factor that amplifies intracellular cortisol levels at the level of each organ. 11) 8-HSD1 has been suggested to be involved in gluconeogenesis in the liver. Therefore, inhibition of 11j8-HSD1 activity is expected to be able to suppress hepatic gluconeogenesis. Furthermore, 11-HSD1 has been suggested to be involved in visceral fat accumulation. Therefore, inhibition of 11i8-HSD1 activity is expected to suppress visceral fat accumulation. 11) As inhibitors of 8-HSD1, reports by Barf T et al. (J Med Chem. 2002: 3813-5), WO 01/090090, WO 01/090091, WO 01/090092 WO 01/090093, W0
01/090094、 W0 03/043999 に記載されているチアゾール誘導体が知られている。 Thiazole derivatives described in WO 01/090094, WO 03/043999 are known.
W001/090090 (特許文献 1 ) には、 以下の式で表されるチアゾール誘導体を 含む 11/S- HSD1 阻害剤が開示されている。 下記式において、 Xは、 メチレン基、 又はカルボニル基であり ;ー Yは、 メチレン基、 カルボニル基、 又は単結合で あり ;— R2は、 〜 アルキルなどである (同公報第 6頁第 9行目〜第 8頁 第 11行目) 。
W001 / 090090 (Patent Document 1) discloses an 11 / S-HSD1 inhibitor containing a thiazole derivative represented by the following formula. In the following formulas, X is is methylene group, or carbonyl group; over Y is methylene group, a carbonyl group, or a single bond; - R 2 is like-alkyl (same publication page 6, 9 Lines-page 8 line 11).
WO 01/090091 (特許文献 2) には、 以下の式で表されるチアゾ一ル誘導体を 含む 11;8- HSD〗 阻害剤が開示されている。 下記式において、 Aは、 アルキル基、 ビニル基、 又は 3— (ェチルー 3—メチルブタネ一卜) である。 WO 01/090091 (Patent Document 2) discloses an 11; 8-HSD 阻 害 inhibitor containing a thiazol derivative represented by the following formula. In the following formula, A is an alkyl group, a vinyl group, or 3- (ethyl-3-methylbutane).
W001/090092 (特許文献 3) には、 以下:の式で表されるチアゾール誘導体を 含む 11^-HSD1阻害剤が開示されている。 下記式において、 Aは、 置換基を有 してもよいァリール基、 又はへテロアリール環である (同公報第 6頁第 26行 目〜第 8頁第 7行目) 。 W001 / 090092 (Patent Document 3) discloses an 11 ^ -HSD1 inhibitor containing a thiazole derivative represented by the following formula: In the following formula, A is an aryl group which may have a substituent, or a heteroaryl ring (page 6, line 26 to page 8, line 7).
WO 01/090093 (特許文献 4) には、 以下の式で表されるチアゾ一ル誘導体を 含む 113- HSD1阻害剤が開示されている。 下記式からわかるとおり、 この文献 に開示されるチアゾール環の 4位は無置換である。 WO 01/090093 (Patent Document 4) discloses a 113-HSD1 inhibitor containing a thiazol derivative represented by the following formula. As can be seen from the following formula, the 4-position of the thiazole ring disclosed in this document is unsubstituted.
W001/090094 (特許文献 5) には、 以下の式で表されるチアソール誘導体を 含む 11 ;8- HSD1阻害剤が開示されている。 W001 / 090094 (Patent Document 5) discloses an 11; 8-HSD1 inhibitor containing a thiazole derivative represented by the following formula.
W003/043999 (特許文献 6) には、 以下の式で表されるチアゾール誘導体を 含む 11)8- HSD1阻害剤が開示されている。 下記式において、 Bは、 水素原子、 C1〜C6アルキル、 又はジメチルァミノメチル基である (同公報第 6頁第 25行 目〜第 8頁第 22行目) 。
W003 / 043999 (Patent Document 6) discloses an 11) 8-HSD1 inhibitor containing a thiazole derivative represented by the following formula. In the following formula, B is a hydrogen atom, a C 1 -C 6 alkyl, or a dimethylaminomethyl group (page 6, line 25 to page 8, line 22 of the same publication).
2ノ Y— X 2 no Y—X
すなわち、 ァダマンチル基を有するチアゾール誘導体を含有する 11)8-HSD1 阻害剤は知られていない。 That is, an 11-HSD1 inhibitor containing a thiazole derivative having an adamantyl group is not known.
なお、 IliS-HSDI阻害剤ではないが、 ァダマンチル基を有するチアゾール誘 導体として、 以下の化合物が知られている (特許文献 7 (米国特許明細書 Mo. 5, 378, 706, 第 13及び第 14欄) 。 下記式からもわかるとおり、 この化合物に は、 スルホニル基 (一 SO?—) が存在しない。 Although not an IliS-HSDI inhibitor, the following compounds are known as thiazole derivatives having an adamantyl group (Patent Document 7 (U.S. Patent Specification Mo. 5, 378, 706, Nos. 13 and 14). Column) As can be seen from the following formula, this compound does not have a sulfonyl group (one SO ? —).
[特許文献 1 ] W001/090090 [Patent Document 1] W001 / 090090
[特許文献 2] W001/090091 [Patent Document 2] W001 / 090091
[特許文献 3] W001/090092 [Patent Document 3] W001 / 090092
[特許文献 4] W001/090093 [Patent Document 4] W001 / 090093
[特許文献 5] W001/090094 [Patent Document 5] W001 / 090094
[特許文献 6] W003/043999 [Patent Document 6] W003 / 043999
[特許文献 7 ] 米国特許明細書 No. 5, 378, 706 [Patent Document 7] U.S. Patent Specification No. 5, 378, 706
[非特許文献 1 ] J Med Chem. 2002 :3813-5
発明の開示 [Non-patent Document 1] J Med Chem. 2002: 3813-5 Disclosure of the invention
上記に説明した公知の 11 ;8 - HSD1阻害剤は、 阻害活性が十分とはいえず医薬 品として満足できるものではない。 このため、 11 i8 - HSD1阻害作用による十分 な治療効果を有し、 医薬品として満足できる化合物の開発が望まれている。 本発明者らは、 上記目的を達成するために鋭意検討を重ねた結果、 ァダマン チル基を有する特定のチアゾール誘導体が優れた 11 )8 - HSD1阻害活性を有する ことを見出し、 本発明を完成するに至った。 The known 11; 8-HSD1 inhibitors described above have insufficient inhibitory activity and are not satisfactory as pharmaceuticals. For this reason, there is a demand for the development of a compound that has a sufficient therapeutic effect by 11i8-HSD1 inhibitory action and is satisfactory as a pharmaceutical. The present inventors have conducted intensive studies to achieve the above object, and as a result, have found that a specific thiazole derivative having an adamantyl group has excellent 11) 8-HSD1 inhibitory activity, thereby completing the present invention. Reached.
すなわち、 本発明は、 以下の新規チアゾール誘導体、 その医薬上許容される 塩、 及びその溶媒和物などに関する。 That is, the present invention relates to the following novel thiazole derivatives, pharmaceutically acceptable salts thereof, and solvates thereof.
[式中、 , [Where,,
R1は、 ァダマンチル基、 又は 「水酸基、 ハロゲン原子、 (;广 アルキル基、 及び 〜 アルコキシ基」 から選ばれる 1〜3個の置換基を有するァダマンチ ル基を表し; R 1 represents an adamantyl group or an adamantyl group having 1 to 3 substituents selected from “hydroxyl group, halogen atom, (; broad alkyl group, and ~ alkoxy group”;
R2は、 水素原子、 〜 アルコキシカルボニル基、 〜( 5アルキル基、 又は 「水酸基、 ハロゲン原子、 及び 〜 アルコキシ基」 から選ばれる 1〜3個の 置換基を有する (^〜(:5アルキル基を表し; R 2 has 1 to 3 substituents selected from a hydrogen atom, an alkoxycarbonyl group, a ( 5 alkyl group, or a “hydroxyl group, a halogen atom, and an alkoxy group” (^ to (: 5 alkyl groups Represents;
R3は、 水素原子、 〜 アルキル基、 C2〜C6アルケニル基、 C2〜C6アルキニル 基;又は 「水酸基、 力ルバモイル基、 ハロゲン原子、 〜 アルコキシ—フエ ニル基、 アミノ基、 〜 アルキルアミノ基、 〜 ジァルキルアミノ基及び C广 C5アルコキシ基」 から選ばれる 1〜3個の置換基を有する 〜 アルキル 基を表し;
A環はァリール基またはへテロアリール基を表し; R 3 is a hydrogen atom, an alkyl group, a C 2 -C 6 alkenyl group, a C 2 -C 6 alkynyl group; or a “hydroxyl group, a carbamoyl group, a halogen atom, an alkoxy-phenyl group, an amino group, an alkyl group; an amino group, ~ ~ alkyl group having 1 to 3 substituents selected from Jiarukiruamino group and a C广C 5 alkoxy group "; Ring A represents an aryl or heteroaryl group;
R4 、 R5、 R6 、 R7 および R8はそれぞれ、 ハロゲン原子、 シァノ基、 ニトロ基、 式一 X1— X2— X3 (式中、 X1は、 単結合、 酸素原子、 硫黄原子、 スルフィニル 基、 スルホニル基、 カルボニル基、 式—NX4—、 式— CO— NX4—、 式一 N X4— CO—、 式— NX4— CO— NX4—、 式一 NX4— CO— O—、 式— S 02 — NX4—、 式一 NX4_S02—、 式一 CO— O—または式—O— CO—を表 し; X2は、 単結合、 〜 アルキレン基を表し; X3および X4は、 それぞれ、 水素原子、 〜 アルキル基、 C3〜C6シクロアルキル基、 C2~C6アルケニル基、 C2〜C6アルキニル基、 ァリール基、 ヘテロァリール基または脂環式へテロ環基 を表す。 ただし、 X2、 X3および X4は、 下記置換基 Y群より選ばれる 1 ~3個 の置換基を有していてもよい。 ) 、 又は、 式一 X1— X5— X6 (式中、 X1は、 前記と同義であり ; X5は、 C广 C5アルキレン基を表し; X6は、 式—ox7、 式 一 NX7X8、 式— CO— NX7X8、 式一 N X7— CO— X7、 式一 S02— NX7X8、 式—NX7— S02— X8、 式— CO— O— X7または式— 0— CO— X7を表し; X7および X8は、 それぞれ、 水素原子、 ァリール基または 〜 アルキル基を 表す。 ただし、 X5、 X6、 X7および X8は、 下記置換基 Y群より選ばれる 1〜 3個の置換基を有していてもよい。 ) を表すか、 又は!?4、 R R6 、 R7 および R8のうち隣接する 2個の基が一緒になつて式一 X -X5— X1— (式中、 X1およ び X5は、 前記と同義である) で表される環構造を形成し;置換基 Y群とは、 ハロゲン原子、 シァノ基、 ニトロ基、 水酸基、 メチル基、 トリフル才ロメチル 基、 ァセチル基、 メチルチオ基またはメ卜キシ基からなる群を表す。 ] で表さ れるチアゾール誘導体、 その薬学的に許容される塩、 又はその溶媒和物。 本発明によれば、 優れた 113-HSD1 阻害活性を有する新規チアゾール誘導体、 その医薬上許容される塩、 及びその溶媒和物などを提供できる。 発明を実施するための最良の形態 R 4 , R 5 , R 6 , R 7 and R 8 each represent a halogen atom, a cyano group, a nitro group, a formula X 1 — X 2 — X 3 (wherein X 1 is a single bond, an oxygen atom, Sulfur atom, sulfinyl group, sulfonyl group, carbonyl group, formula —NX 4 —, formula — CO— NX 4 —, formula — NX 4 — CO—, formula — NX 4 — CO— NX 4 —, formula — NX 4 — CO- O-, formula - S 0 2 - NX 4 - , wherein one NX 4 _S0 2 -, and Table formula one CO- O-or formula -O- CO-; X 2 is a single bond, - an alkylene group X 3 and X 4 are each a hydrogen atom, an alkyl group, a C 3 -C 6 cycloalkyl group, a C 2 -C 6 alkenyl group, a C 2 -C 6 alkynyl group, an aryl group, a heteroaryl group or to alicyclic represents a heterocyclic group. However, X 2, X 3 and X 4 may have 1 to 3 substituents selected from the following substituent group Y.), or the formula X 1 - X 5 - X 6 ( wherein, X 1 is as defined above; X 5 represents a C广C 5 alkylene group; X 6 has the formula -ox 7, wherein one NX 7 X 8 , Formula — CO— NX 7 X 8 , Formula NX 7 — CO— X 7 , Formula 1 S0 2 — NX 7 X 8 , Formula — NX 7 — S 0 2 — X 8 , Formula — CO— O— X 7 or . formula - 0- CO- represents X 7; X 7 and X 8 each represent a hydrogen atom, Ariru group or ~ alkyl group provided that, X 5, X 6, X 7 and X 8, following substituent group Y. 1 to may have three substituents selected from the group.) or represents, or!? 4, two neighboring groups of RR 6, R 7 and R 8 summer together X-X 5 —X 1 — (wherein X 1 and X 5 are the same as defined above); and the substituent Y group is a halogen atom, Cyano group, nitro group, hydroxyl group, methyl group, trimethyl group, acetyl Group, a group consisting of methylthio group or main Bok alkoxy group. ] The thiazole derivative represented by these, its pharmaceutically acceptable salt, or its solvate. According to the present invention, a novel thiazole derivative having an excellent 113-HSD1 inhibitory activity, a pharmaceutically acceptable salt thereof, a solvate thereof, and the like can be provided. BEST MODE FOR CARRYING OUT THE INVENTION
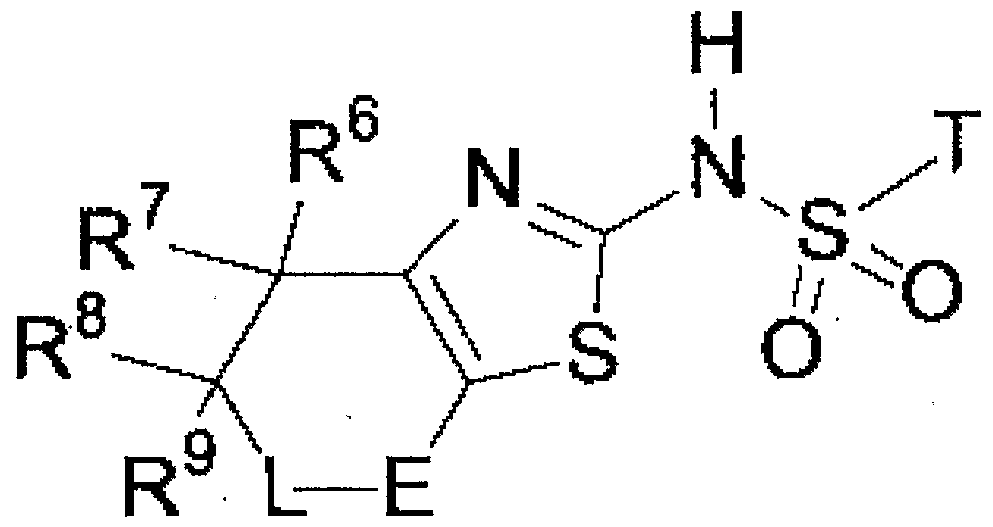
本発明のチアゾ一ル誘導体は、 下記式 (1 ) で表される化合物である。
The thiazole derivative of the present invention is a compound represented by the following formula (1).
式 (1 ) において、 R1は、 ァダマンチル基、 又は、 「水酸基、 ハロゲン原子.、 〜 アルキル基、 及び 〜じ 5アルコキシ基」 から選ばれる 1 ~3個の置 換基を有するァダマンチル基を表す。 好ましくは、. R1はァダマンチル基、 又は、 水酸基又は Ci Cgアルコキシ基で置換されたァダマンチル基である。 更に好 ましくは、 R1はァダマンチル基又は 3—ヒドロキシァダマンチル基である。 In the formula (1), R 1 represents an adamantyl group or an adamantyl group having 1 to 3 substituents selected from “hydroxyl group, halogen atom, 〜alkyl group, and じ5 alkoxy group”. . Preferably, R 1 is an adamantyl group or an adamantyl group substituted with a hydroxyl group or a CiCg alkoxy group. More preferably, R 1 is an adamantyl group or a 3-hydroxyadamantyl group.
式 (1 ) において、 R2は、 水素原子、 〜 アルコキシカルボニル基、 ~(5 アルキル基、 又は 「水酸基、 ハロゲン原子、 及び (〜 アルコキシ基 J から選 ばれる 1 ~ 3個の置換基を有する 〜 アルキル基を表す。 好ましくは、 は、 水素原子、 〜 アルキル基、 又は水酸基で置換された 〜 アルキル基であ る。 更に好ましくは、 R2は、 水素原子又はメチル基である。 In the formula (1), R 2 is a hydrogen atom, - an alkoxycarbonyl group, - (5 alkyl group, or "hydroxyl, ~ having a halogen atom, and (a barrel selected from-alkoxy group J ~ 3 substituents Represents an alkyl group, preferably is a hydrogen atom, an alkyl group, or an alkyl group substituted with a hydroxyl group, and more preferably, R 2 is a hydrogen atom or a methyl group.
式 (1 ) において、 R3は、 水素原子、 〜〇5アルキル基、 じ广 アルケニ ル基、 C Csアルキニル基;又は、 「水酸基、 力ルバモイル基、 ハロゲン原 子、 C广 C5アルコキシ-フエニル基、 アミノ基、 〜 アルキルアミノ基、 C2〜C7ジアルキルアミノ基及び 〜 アルコキシ基」 から選ばれる 1 ~3 個の置換基を有する C Csアルキル基を表す。 好ましくは、 R3は、 水素原子、 じ,〜じ アルキル基である。 更に好ましくは、 R2は、 水素原子又はメチル基で ある。 In the formula (1), R 3 is a hydrogen atom, ~〇 5 alkyl group, Ji广alkenyl Le group, C Cs alkynyl group; or "hydroxyl, force Rubamoiru group, halogen atom, C广C 5 alkoxy - phenyl It represents group, an amino group, - an alkylamino group, a C Cs alkyl groups having from 1 to 3 substituents selected from C 2 -C 7 dialkylamino group and ~ alkoxy group ". Preferably, R 3 is a hydrogen atom, a di- or di-alkyl group. More preferably, R 2 is a hydrogen atom or a methyl group.
式 (1 ) において、 A環は、 ァリール基又はへテロアリール基を表す。 好ま しい A環は、 フエニル基、 1 一ナフチル基、 又は 2—ナフチル基; ピロリル基、 フリル基、 チェニル基、 才キサゾリル基、 イソ才キサゾリル基、 イミダゾリル
基、 チアゾリル基、 イソチアゾリル基、 ピラゾリル基、 卜リアゾリル基、 テ卜 ラゾリル基、 1,3, 4-才キサジァゾリル基、 1,2, 4-才キサジァゾリル基、 1, 2,4- チアジアゾリル基、 ピリジル基、 ビラジニル基、 ピリミジニル基、 ピリダジニ ル基、 1, 2, 4-卜リアジニル基、 1, 2, 3-卜リアジニル基、 1, 3, 5-卜リアジニル基、 ベンズ才キサゾリル基、 ベンズイソキサゾリル基、 ベンゾチアゾリル基、 ベン ズイソチアゾリル基、 ベンズイミダゾリル基、 ベンゾ卜リアゾリル基、 ベンゾ チアジアゾリル基、 ベンゾフラザニル基、 ベンゾピラニル基、 チアナフテニル 基、 イソチアナフテニル基、 ベンゾフラニル基、 イソべンゾフラニル基、 ベン ゾチェ二ル基、 イソインドリル基、 インドリル基、 インダゾリル基、 イソキノ リル基、 キノリル基、 フタラジニル基、 キノキサリニル基、 キナゾリニル基、 シンノリニル基、 2, 1, 3-ベンズ才キサジァゾリル基、 ベンゾキサジニル基、 ク マリル基、 ナフチリジニル基、 プリニル基、 プテリジニル基、 チェノフラニル 基、 イミダゾチアゾリル基、 イミダゾピリジニル基、 ピロ口ピリジニル基、 ピ ロロピリミジニル基、 ピリドピリミジニル基である。 更に好ましい A環は、 フ ェニル基、 1 一ナフチル基、 又は 2—ナフチル基; ピリジル基、 イミダゾリル 基、 ピリミジニル基、 ピラゾリル基、 フリル基、 チェニル基、 イソ才キサゾー ル基、 チアゾリル基、 ピロリル基、 キノリニル基、 ベンゾチ才フエニル基、 ベ ンゾフラニル基、 又はべンゾ才キサゾリル基である。 また更に好ましい A環は、 フエニル基、 ベンゾチ才フエニル基、 チェニル基、 又はべンゾフラニル基であ る。 In the formula (1), the ring A represents an aryl group or a heteroaryl group. Preferred A ring is phenyl, 1-naphthyl, or 2-naphthyl; pyrrolyl, furyl, phenyl, thienyl, oxazolyl, isoxazolyl, imidazolyl Group, thiazolyl group, isothiazolyl group, pyrazolyl group, triazolyl group, tetrazolyl group, 1,3,4-year-old oxadiazolyl group, 1,2,4-year-old oxadiazolyl group, 1,2,4-thiadiazolyl group, pyridyl Group, birazinyl group, pyrimidinyl group, pyridazinyl group, 1,2,4-triazinyl group, 1,2,3-triazinyl group, 1,3,5-triazinyl group, benzixyloxazolyl group, benzisoxa Zolyl group, benzothiazolyl group, benzoisothiazolyl group, benzimidazolyl group, benzotriazolyl group, benzothiadiazolyl group, benzofurazanyl group, benzopyranyl group, thianaphthenyl group, isothianaphthenyl group, benzofuranyl group, isobenzofuranyl group, benzofuranzyl group Group, isoindolyl group, indolyl group, indazolyl group, isoquinolyl group, Ryl, phthalazinyl, quinoxalinyl, quinazolinyl, cinnolinyl, 2,1,3-benzoxadiazolyl, benzoxazinyl, cumaryl, naphthyridinyl, purinyl, pteridinyl, chenofuranyl, imidazothiazolyl , An imidazopyridinyl group, a pyrrolidone pyridinyl group, a pyrrolopyrimidinyl group, and a pyridopyrimidinyl group. More preferred ring A is phenyl, 1-naphthyl or 2-naphthyl; pyridyl, imidazolyl, pyrimidinyl, pyrazolyl, furyl, chenyl, isoxazole, thiazolyl, pyrrolyl. A quinolinyl group, a benzothylphenyl group, a benzofuranyl group, or a benzoxyloxazolyl group. Still more preferred ring A is a phenyl group, a benzothienylphenyl group, a phenyl group, or a benzofuranyl group.
式 (1 ) において、 R4 、 R5、 R6 、 R7 および R8はそれぞれ、 ハロゲン原子、 シァノ基、 ニトロ基、 式—X1— X2— X3 (式中、 X1は、 単結合、 酸素原子、 硫 黄原子、 スルフィニル基、 スルホニル基、 カルボニル基、 式— NX4—、 式一 C 0— NX4—、 式一 NX4— CO—、 式—N X4—CO— N X4—、 式一 NX4— C O— 0—、 式— S02— NX4—、 式—NX4— S02—、 式一 CO— 0—または式 一 0— CO—を表し; X2は、 単結合、 ~(:5アルキレン基を表し; X3および X4は、 それぞれ、 水素原子、 〜(:5アルキル基、 C3~C6シクロアルキル基、 C2 〜(; 6アルケニル基、 C2~C6アルキニル基、 ァリール基、 ヘテロァリール基また は脂環式へテロ環基を表す。 ただし、 X2、 X3および X4は、 下記置換基 Y群よ
り選ばれる 1〜3個の置換基を有していてもよい。 ) 、 又は、 式一 X1— X5 -X6 (式中、 X1は、 前記と同義であり ; X5は、 ^〜(:5アルキレン基を表し; X6は、 式一 OX7、 式一 NX7X8、 式一 CO— NX7X8、 式一 NX7— CO— X7、 式— S02— NX7X8、 式ーNX7—S02—X8、 式— CO— 0— X7または式一 0 一 CO— X7を表し; X7および X8は、 それぞれ、 水素原子、 ァリール基または 〜 アルキル基を表す。 ただし、 X5、 X6、 X7および X8は、 下記置換基 Y群 より選ばれる〗〜 3個の置換基を有していてもよい。 ) を表すか、 又は R4 、 R5、 R6 、 R7 および R8のうち隣接する 2個の基が一緒になつて式一 X1— X5— X1— (式中、 X1および X5は、 前記と同義である) で表される環構造を形成し;置 換基丫群とは、 ハロゲン原子、 シァノ基、 ニトロ基、 水酸基、 メチル基、 トリ フル才ロメチル基、 ァセチル基、 メチルチオ基またはメ卜キシ基からなる群を 表す。 好ましい R4 、 R5、 R6 、 R7 および R8は、 それぞれ、 ハロゲン原子、 シァ ノ基、 二卜口基、 式一 X1— X2— X3 (式中、 X1は、 単結合、 酸素原子、 スルホ ニル基、 カルポニル基、 式一 NX4—、 式一 NX4— CO—、 式一 NX4— CO— NX4—、 式一 NX4— CO— 0—、 式— S 02— NX4—又は式— CO— 0—を表 し; X2は、 単結合、 〜 アルキレン基を表し; X3および X4は、 それぞれ、 水素原子、 〜 アルキル基、 C3〜C6シクロアルキル基、 ァリール基、 ヘテロ ァリール基または脂環式へテロ環基を表す。 ただし、 X2、 X3および X4は、 下 記置換基 Y群より選ばれる〗〜 3個の置換基を有していてもよい。 ) 、 又は、 式一 X1— X5— X6 (式中、 X'は、 単結合、 酸素原子、 であリ ; X5は、 〜 アルキレン基を表し; X6は、 式一 OX7、 式一 NX7X8、 式— CO— ΝΧ7Χ8、 式一 NX7— CO— X.7又は式一 CO— 0— X7を表し; X7および X8は、 それぞ れ、 水素原子、 ァリール基または 〜 アルキル基を表す。 ただし、 X5、 X6、 X7および X8は、 下記置換基 Y群より選ばれる〗〜3個の置換基を有していて もよい。 ) を表すか、 又は R4 、 R5、 R6 、 R7および R8のうち隣接する 2個の基 が一緒になつて式— X1— XS-X1— (式中、 X1は、 単結合、 酸素原子、 であり、 X5は、 〜 アルキレン基である) で表される環構造を形成し;置換基 Y群と は、 ハロゲン原子、 シァノ基、 二卜口基、 水酸基、 メチル基、 トリフル才ロメ チル基、 ァセチル基、 メチルチオ基またはメ卜キシ基からなる群である。
更に好ましい R4 、 R5、 R6 、 R7 および R8は、 それぞれ、 ハロゲン原子、 シァ ノ基又は式一 X1— X2— X3 (式中、 X1は、 単結合、 酸素原子又はカルボニル基 を表し; X2は、 単結合、 〜 アルキレン基を表し; X3および X4は、 それぞ れ、 水素原子、 〜(:5アルキル基、 C3~C6シクロアルキル基、 ヘテロァリール 基または脂環式へテロ環基を表す。 ただし、 X2、 X3および X4は、 下記置換基 Y群より選ばれる 1〜3個の置換基を有していてもよい。 ) 又は 式—X1— X 5— X6 (式中、 X1は、 単結合、 酸素原子、 であり ; X5は、 〜 アルキレン基 を表し; X6は、 式一 OX7又は式一 NX7X8を表し; X7および X8は、 それぞ れ、 水素原子または ~(5アルキル基を表す。 ただし、 X5、 X6、 X7および X8 は、 下記置換基 Y群より選ばれる 1〜 3個の置換基を有していてもよい。 ) を 表すか、 又は R4 、 R R6 、 R7 および R8のうち隣接する 2個の基が一緒になつ て式一 X1— X5— X1— (式中、 X1は、 単結合、 酸素原子、 であり、 X5は、 〜C5アルキレン基である) で表される環構造を形成し;置換基 Y群とは、 ハロ ゲン原子、 シァノ基、 ニトロ基、 水酸基、 メチル基、 卜リフル才ロメチル基、 ァセチル基、 メチルチオ基またはメ卜キシ基からなる群である。 In the formula (1), R 4 , R 5 , R 6 , R 7 and R 8 are each a halogen atom, a cyano group, a nitro group, a formula —X 1 —X 2 —X 3 (wherein X 1 is single bond, an oxygen atom, sulfur atom, a sulfinyl group, a sulfonyl group, a carbonyl group, the formula - NX 4 -, wherein one C 0- NX 4 -, wherein one NX 4 - CO-, wherein -NX 4 -CO- NX 4 —, Formula 1 NX 4 — CO— 0—, Formula — S0 2 — NX 4 —, Formula — NX 4 — S0 2 —, Formula 1 CO— 0— or Formula 1 0—CO—; X 2 a single bond, - (: represents a 5 alkylene group; X 3 and X 4 are each a hydrogen atom, - (: 5 alkyl group, C 3 ~ C 6 cycloalkyl group, C 2 ~ (; 6 alkenyl group, Represents a C 2 to C 6 alkynyl group, an aryl group, a heteroaryl group or an alicyclic heterocyclic group, provided that X 2 , X 3 and X 4 are the same as those of the following substituent group Y It may have 1 to 3 substituents selected. X 1 —X 5 -X 6 (wherein X 1 has the same meaning as described above; X 5 represents ^ 〜 (: 5 alkylene group; X 6 is a group represented by the formula OX 7) , wherein one NX 7 X 8, wherein one CO- NX 7 X 8, wherein one NX 7 - CO- X 7, wherein - S0 2 - NX 7 X 8 , Shiki NX 7 -S0 2 -X 8, wherein - X— represents X— 7 or a formula—CO—X— 7 ; X 7 and X 8 each represent a hydrogen atom, an aryl group or an alkyl group, provided that X 5 , X 6 , X 7 and X 8 may have〗 to 3 substituents selected from the following substituent group Y.) or a group adjacent to R 4 , R 5 , R 6 , R 7 and R 8 The two groups together form a ring structure of the formula X 1 — X 5 — X 1 — (wherein X 1 and X 5 are as defined above); Groups include halogen atoms, cyano groups, nitro groups, hydroxyl groups, methyl groups, R 4 , R 5 , R 6 , R 7 and R 8 each represent a halogen atom, a cyano group or a nitro group, each of which represents a methyl group, an acetyl group, a methylthio group or a methoxy group; , Formula 1 X 1 — X 2 — X 3 (where X 1 is a single bond, an oxygen atom, a sulfonyl group, a carbonyl group, Formula 1 NX 4 —, Formula 1 NX 4 — CO—, Formula 1 NX 4 — CO— NX 4 —, formula — NX 4 — CO— 0—, formula — S 0 2 — NX 4 — or formula — CO— 0—; X 2 represents a single bond, an alkylene group; X 3 and X 4 each represent a hydrogen atom, an alkyl group, a C 3 -C 6 cycloalkyl group, an aryl group, a heteroaryl group or an alicyclic heterocyclic group, provided that X 2 , X 3 and X 4 may have〗 to 3 substituents selected from the following substituent group Y.) or a compound represented by the formula: X 1 — X 5 — X 6 , wherein X ′ is Single bond, an oxygen atom, der Li; X 5 represents a ~ alkylene group; X 6 has the formula one OX 7, wherein one NX 7 X 8, wherein - CO- ΝΧ 7 Χ 8, wherein one NX 7 - CO- represents X. 7 or formula one CO- 0- X 7; X 7 and X 8, which respectively represent a hydrogen atom, Ariru group or ~ alkyl group. However, X 5 , X 6 , X 7 and X 8 may have 1 to 3 substituents selected from the following substituent group Y. ) Or two adjacent groups of R 4 , R 5 , R 6 , R 7 and R 8 are joined together to form a formula — X 1 — XS-X 1 — (wherein X 1 is , A single bond, an oxygen atom, and X 5 is an alkylene group); and the substituent group Y is a halogen atom, a cyano group, a dihydroxy group, a hydroxyl group, A group consisting of a methyl group, a trifluoromethyl group, an acetyl group, a methylthio group and a methoxy group. More preferably, R 4 , R 5 , R 6 , R 7 and R 8 are each a halogen atom, a cyano group or a compound of the formula X 1 —X 2 —X 3 (wherein X 1 is a single bond, an oxygen atom X 2 represents a single bond, to an alkylene group; X 3 and X 4 each represent a hydrogen atom, to (: 5 alkyl group, C 3 to C 6 cycloalkyl group, heteroaryl) Represents a group or an alicyclic heterocyclic group, provided that X 2 , X 3 and X 4 may have 1 to 3 substituents selected from the following substituent group Y.) —X 1 —X 5 —X 6 (wherein, X 1 is a single bond or an oxygen atom; X 5 represents an alkylene group; X 6 is a formula OX 7 or a formula NX 7 X represents 8;. X 7 and X 8, which respectively represent a hydrogen atom or - (5-alkyl group provided that, X 5, X 6, X 7 and X 8 is selected from the following substituent group Y May have 1 to 3 substituents.) Or two adjacent groups among R 4 , RR 6 , R 7 and R 8 are taken together to form a group of formula X 1 — X 5 — X 1 — (wherein, X 1 is a single bond, an oxygen atom, and X 5 is a C 5 alkylene group); and a substituent group Y Is a group consisting of a halogen atom, a cyano group, a nitro group, a hydroxyl group, a methyl group, a trimethyl group, an acetyl group, a methylthio group or a methoxy group.
最も好ましい R4 、 R5、 R6 、 R7 および R8は、 それぞれ、 ハロゲン原子、 シァ ノ基、 メチ^基、 ェチル基、 卜リフル才ロメチル基、 メ卜キシ基である。 以下では、 本明細書において用いられる各置換基などについて説明する。 Most preferably, R 4 , R 5 , R 6 , R 7 and R 8 are a halogen atom, a cyano group, a methyl group, an ethyl group, a trimethyl group, and a methoxy group, respectively. Hereinafter, each substituent and the like used in the present specification will be described.
「ハロゲン原子」 として、 フッ素原子、 塩素原子、 臭素原子又はヨウ素原子が あげられる。 Examples of the “halogen atom” include a fluorine atom, a chlorine atom, a bromine atom and an iodine atom.
アルキル基 J とは、 炭素数 1〜5のアルキル基を意味し、 例えば、 メチル基、 ェチル基、 プロピル基、 イソプロピル基、 ブチル基、 イソブチル基、 sec-ブチル基、 tert-プチル基、 ペンチル基、 イソペンチル基、 1, 1ージメ チルェチル基、 1 , 1ージメチルプロピル基、 又は 1 一ェチルプロピル基など があげられる。 Alkyl group J means an alkyl group having 1 to 5 carbon atoms, for example, methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group And an isopentyl group, a 1,1-dimethylethyl group, a 1,1-dimethylpropyl group, and a 1-ethylpropyl group.
「C3〜C6シクロアルキル基」 として、 例えばシクロプロピル基、 クロブ チル基、 シクロペンチル基、 シクロへキシル基があげられる。
「ァリール基」 とは、 芳香族炭化水素の環に結合する水素原子が 1個遊離し て生ずる基であり、 例えばフエニル基、 1 一ナフチル基、 2—ナフチル基があ げられる。 Examples of the “C 3 -C 6 cycloalkyl group” include a cyclopropyl group, a clobutyl group, a cyclopentyl group, and a cyclohexyl group. An “aryl group” is a group formed by liberating one hydrogen atom bonded to a ring of an aromatic hydrocarbon, and includes, for example, a phenyl group, a 1.1-naphthyl group and a 2-naphthyl group.
「ヘテロァリール基」 とは、 酸素原子、 硫黄原子、 窒素原子から任意に選ば れる 1〜4個の元素を環内に有する単環あるいは縮合芳香環であり、 例えばピ 口リル基、 フリル基、 チェニル基、 ォキサゾリル基、 イソ才キサゾリル基、 ィ ミダゾリル基、 チアゾリル基、 イソチアゾリル基、 ピラゾリル基、 卜リアゾリ ル基、 テ卜ラゾリル基、 1, 3, 4-才キサジァゾリル基、 1, 2, 4-才キサジァゾリル 基、 1, 2, 4-チアジアゾリル基、 ピリジル基、 ビラジニル基、 ピリミジニル基、 ピリダジニル基、 1, 2, 4-卜リアジニル基、 1, 2, 3-卜リアジニル基、 1, 3, 5-卜リ アジニル基、 ベンズォキサゾリル基、 ベンズイソキサゾリル基、 ベンゾチアゾ リル基、 ベンズイソチアゾリル基、 ベンズイミダゾリル基、 ベンゾ卜リアゾリ ル基、 ベンゾチアジアゾリル基、 ベンゾフラザニル基、 ベンゾピラニル基、 チ アナフテニル基、 イソチアナフテニル基、 ベンゾフラニル基、 イソベンゾフラ ニル基、 ベンゾチェ二ル基、 イソインドリル基、 インドリル基、 インダゾリル 基、 イソキノリル基、 キノリル基、 フタラジニル基、 キノキサリニル基、 キナ ゾリニル基、. シンノリニル基、 2, 1, 3-ベンズォキサジァゾリル基、 ベンゾキサ ジニル基、 クマリル基、 ナフチリジニル基、 プリニル基、 プテリジニル基、 チ エノフラニル基、 イミダゾチアゾリル基、 イミダゾピリジニル基、 ピロ口ピリ ジニル基、 ピロ口ピリミジニル基、 ピリドピリミジニル基があげられる。 The "heteroaryl group" is a monocyclic or condensed aromatic ring having 1 to 4 elements arbitrarily selected from an oxygen atom, a sulfur atom, and a nitrogen atom in the ring. Group, oxazolyl group, isoxazole group, imidazolyl group, thiazolyl group, isothiazolyl group, pyrazolyl group, triazolyl group, tetrazolyl group, 1,3,4-year-old Oxadiazolyl, 1,2,4-thiadiazolyl, pyridyl, birazinyl, pyrimidinyl, pyridazinyl, 1,2,4-triazinyl, 1,2,3-triazinyl, 1,3,5- Tri azinyl, benzoxazolyl, benzisoxazolyl, benzothiazolyl, benzisothiazolyl, benzimidazolyl, benzotriazolyl, benzothiazole Diazolyl group, benzofurazanyl group, benzopyranyl group, thianaphthenyl group, isothianaphthenyl group, benzofuranyl group, isobenzofuranyl group, benzocenyl group, isoindolyl group, indolyl group, indazolyl group, isoquinolyl group, quinolyl group, phthalazinyl group, quinoxalinyl group Quinazolinyl group, cinnolinyl group, 2,1,3-benzoxadiazolyl group, benzoxazinyl group, cumaryl group, naphthyridinyl group, purinyl group, pteridinyl group, thienofuranyl group, imidazothiazolyl group, Examples thereof include an imidazopyridinyl group, a pyridinyl group having a pyro opening, a pyrimidinyl group having a pyro opening, and a pyridopyrimidinyl group.
「C2~ C 6アルケニル基 J とは、 炭素数 2 ~ 6個の直鎖状、 分枝状又は環状 のアルケニル基を意味し、 例えばビニル基、 ァリル基、 1 一プロぺニル基、 2 一プロぺニル基、 イソプロぺニル基、 2—ブテニル基、 3—ブテニル基、 イソ ブテニル基、 4一ペンテニル基、 又は 5—へキセニル基があげられる。 `` C 2 -C 6 alkenyl group J means a straight-chain, branched or cyclic alkenyl group having 2 to 6 carbon atoms, for example, a vinyl group, an aryl group, a 1-propenyl group, Examples thereof include a monopropenyl group, an isopropenyl group, a 2-butenyl group, a 3-butenyl group, an isobutenyl group, a 4-pentenyl group, and a 5-hexenyl group.
「C 2~ C 6アルキニル基」 とは、 炭素数 2〜6個の直鎖状、 分枝状又は環状 のアルキニル基を意味し、 例えばェチニル基、 1 一プロピニル基、 2—プロピ ニル基、 3—プチ二ル基、 4一ペンチニル基、 又は 5—へキシニル基があげら れる。
「 〜 アルキレン基」 とは、 前記 「 〜^のアルキル基」 からさらに 任意の水素原子を 1個除いて誘導される二価の基を意味し、 例えばメチレン基、“C 2 -C 6 alkynyl group” means a linear, branched or cyclic alkynyl group having 2 to 6 carbon atoms, such as ethynyl group, 1-propynyl group, 2-propynyl group, Examples thereof include a 3-butynyl group, a 4-pentynyl group, and a 5-hexynyl group. "-Alkylene group" means a divalent group derived from the above-mentioned "alkyl group of ~" by removing one arbitrary hydrogen atom, for example, a methylene group,
1 , 1 一エチレン基、 1, 2—エチレン基、 1, 1 一プロピレン基、 1, 3— プロピレン基、 テ卜ラメチレン基、 又はペンタメチレン基があげられる。 Examples thereof include a 1,1-ethylene group, a 1,2-ethylene group, a 1,1-propylene group, a 1,3-propylene group, a tetramethylene group, and a pentamethylene group.
「脂環式へテロ環基」 として、 例えばァゼチジン環、 ピロリジン環、 ピペリ ジン環、 テ卜ラヒドロフラン環、 テ卜ラヒドロピラン環、 モルホリン環、 チ才 モルホリン環、 ピぺラジン環、 チアゾリジン環、 ジ才キサン環、 イミダゾリン 環、 又はチアゾリン環があげられ Examples of the “alicyclic heterocyclic group” include, for example, an azetidine ring, a pyrrolidine ring, a piperidine ring, a tetrahydrofuran ring, a tetrahydropyran ring, a morpholine ring, and a morpholine ring, a piperazine ring, a thiazolidine ring, Xan ring, imidazoline ring, or thiazoline ring
る。 The
「 〜〇5アルコキシ基」 とは、 炭素数 1〜5個の直鎖状、 分枝状又は環状 のアルコキシ基を意味し、 例えば、 メ卜キシ基、 ェ卜キシ基、 プロポキシ基、 イソプロポキシ基、 シクロプロポキシ基、 ブ卜キシ基、 又は tert—プ卜キシ基 である。 The "~〇 5 alkoxy group", the number 1-5 straight, means branched or cyclic alkoxy group, for example, main Bok alkoxy group, E Bok alkoxy group, a propoxy group, isopropoxy Group, cyclopropoxy group, butoxy group, or tert-butoxy group.
「 〜 アルコキシ一フエニル基」 とは、 前記 〜 アルコキシ基で置 換されたフエ二ル基を意味し、 例えば、 4—メ卜キシフエニル基、 又は 4—ェ 卜キシフエニル基があげられる。 The term "-alkoxy-phenyl group" means a phenyl group substituted with the above-mentioned -alkoxy group, and examples thereof include a 4-methoxyphenyl group and a 4-ethoxyphenyl group.
「 〜 アルキルアミノ基」 とは、 炭素数 1〜5個の直鎖状、 分枝状又は 環状のアルキル基で置換されたアミノ基を意味し、 例えば、 メチルァミノ基、 ェチルァミノ基、 プロピルアミノ基、 イソプロピルアミノ基、 シクロプロピル アミノ基、 プチルァミノ基、 tert—プチルァミノ基、 ペンチルァミノ基、 又は ネオペンチルァミノ基があげられる。 "-Alkylamino group" means an amino group substituted with a linear, branched or cyclic alkyl group having 1 to 5 carbon atoms, such as a methylamino group, an ethylamino group, a propylamino group, Examples include an isopropylamino group, a cyclopropylamino group, a butylamino group, a tert-butylamino group, a pentylamino group, and a neopentylamino group.
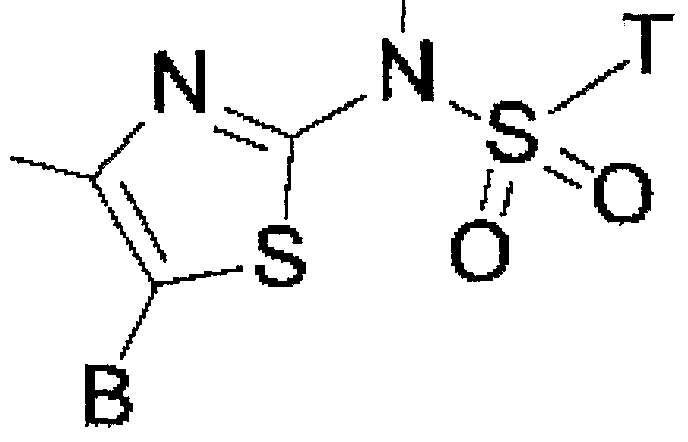
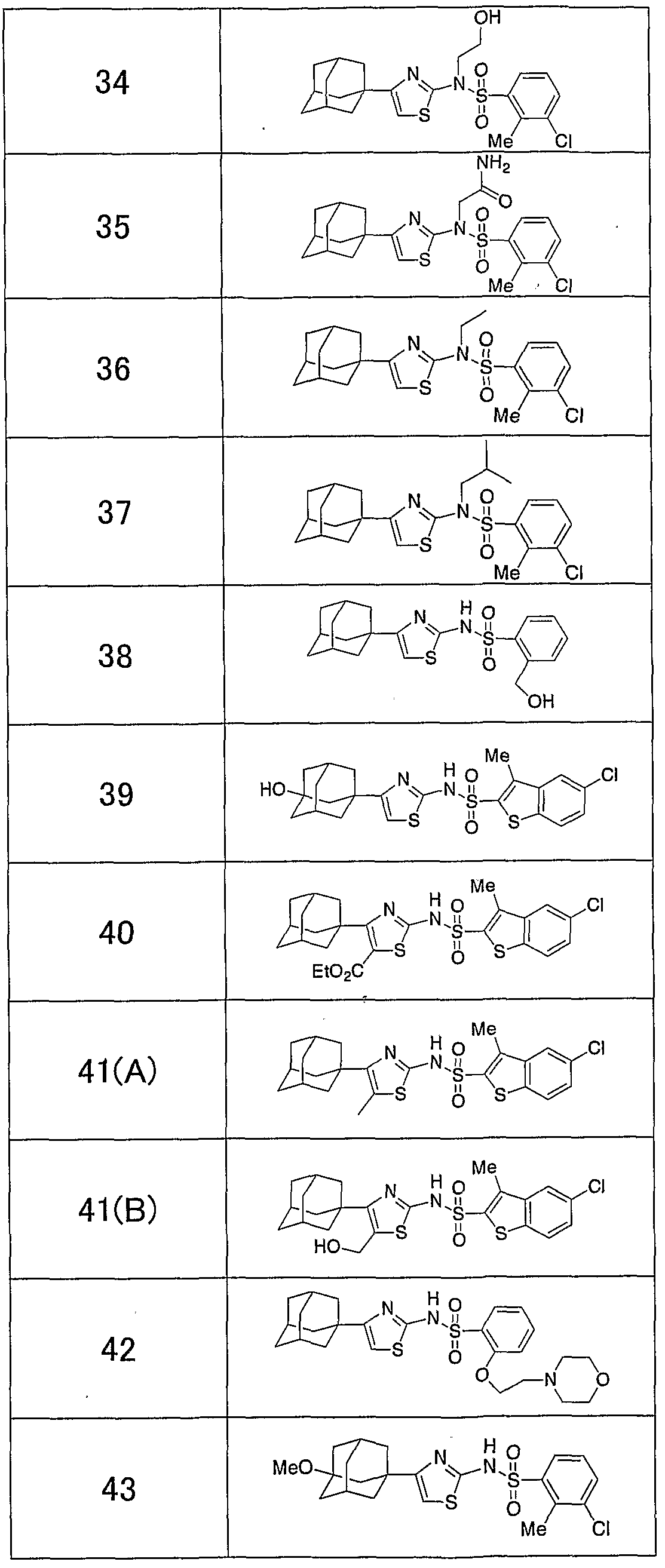
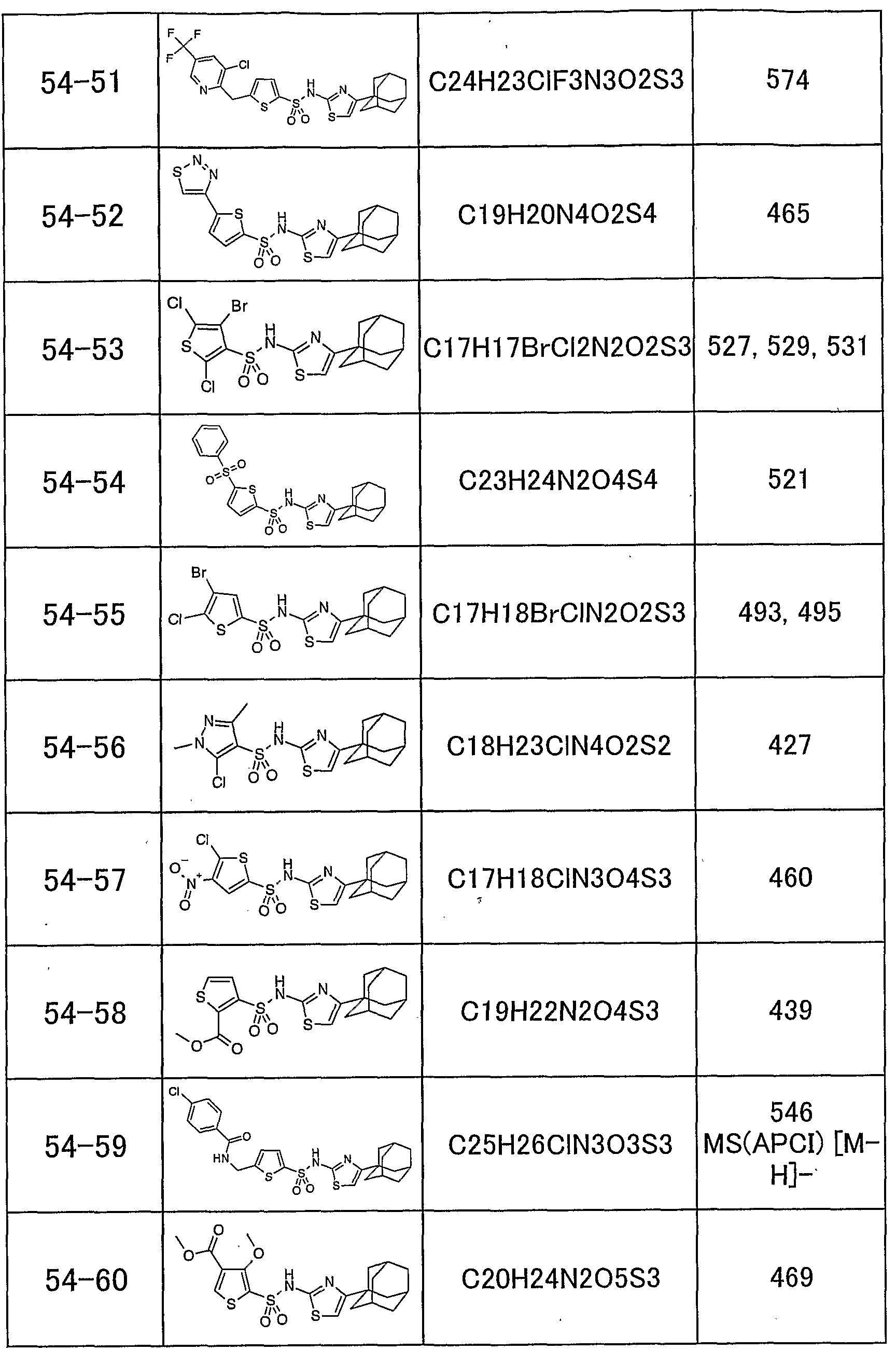
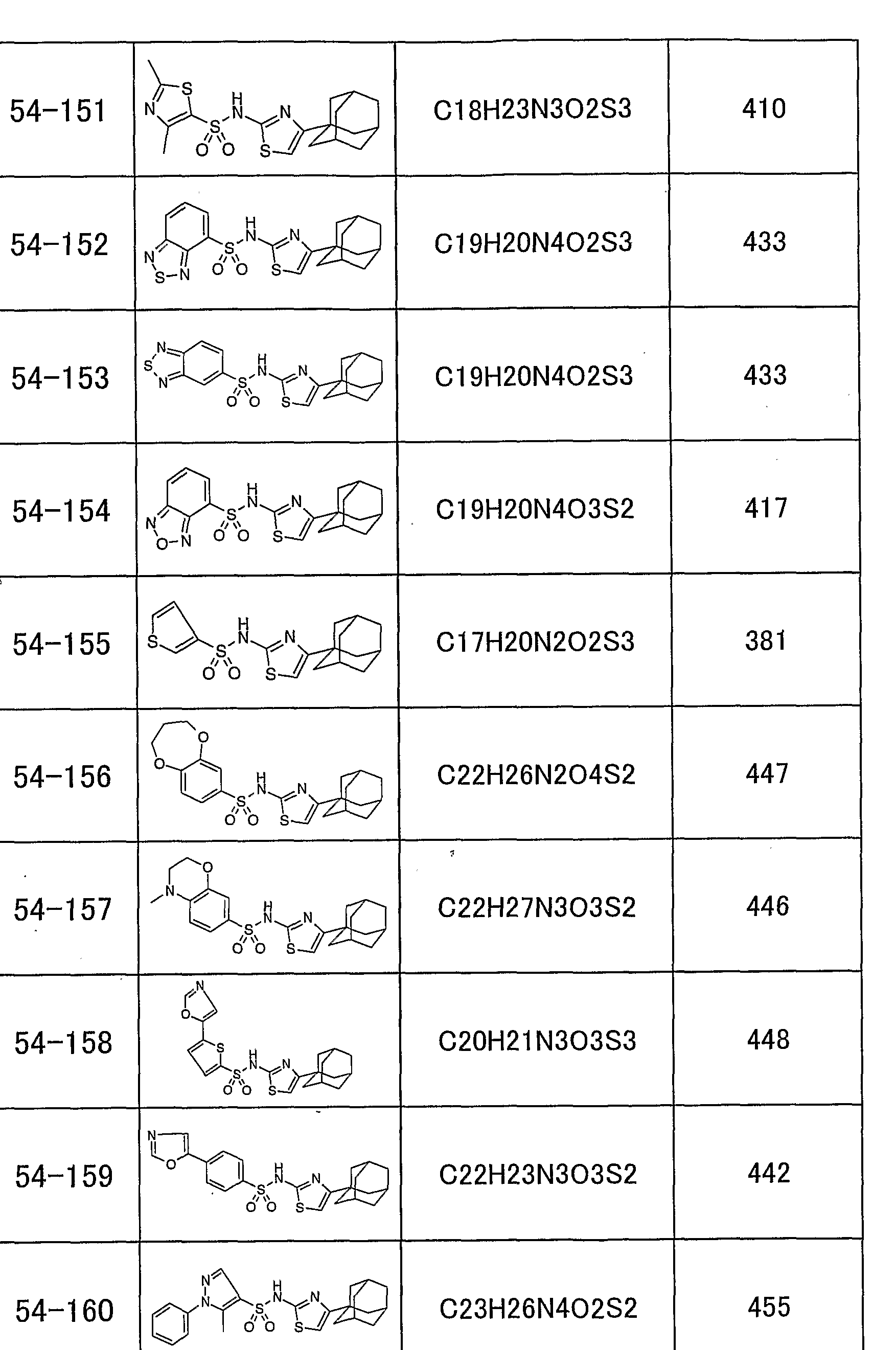
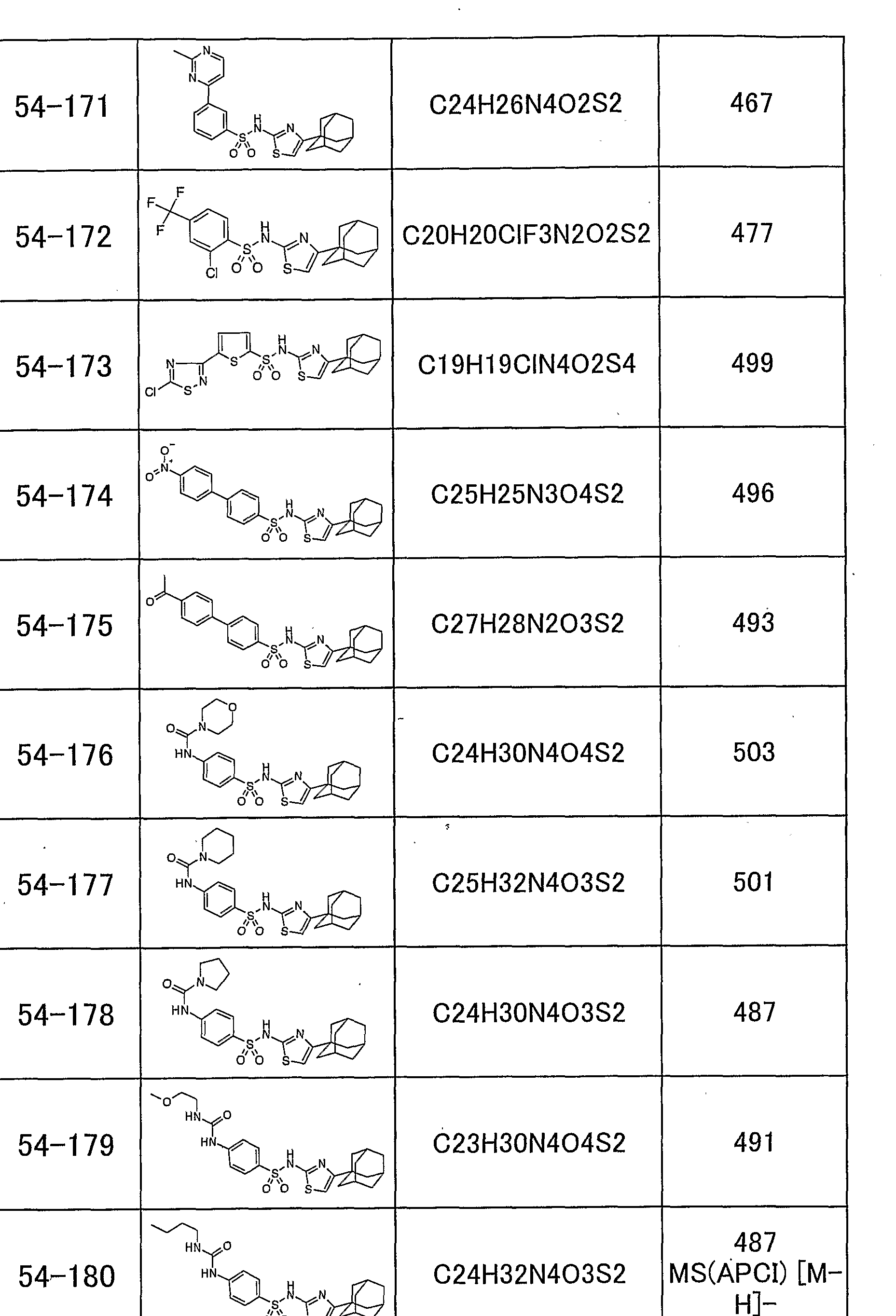
「 〜 ジアルキルアミノ基」 とは、 同一又は異なった炭素数 1 ~ 5個の 直鎖状、 分枝状又は環状の 2つのアルキル基で置換されたアミノ基を意味し、 例えば、 ジメチルァミノ基、 メチルェチルァミノ基、 ジェチルァミノ基、 又は ジプロピルアミノ基があげられる。 本発明のチアゾール誘導体の具体的な例として、 以下のものがあげられる。 すなわち、 化合物 1 : 4一 (1 -ァダマンチル) _ 2— (1 -ナフチルスルホニ ルァミノ) チアゾ一ル、 化合物 2 : 4一 (1 -ァダマンチル) — 2— (2, 4
5—トリクロ口フエニルスルホニルァミノ) チアゾール、 化合物 3 : 4 - (1 - ァダマンチル) 一 2— [ (2—チェニルスルホニル) ァミノ]チアゾール、 化合 物 4 : 4一 (1 -ァダマンチル) 一 2— (4一ブロモフエニルスルホニルアミ ノ) チアゾール、 化合物 5 : 4一 (1 -ァダマンチル) —2— (3—クロ口— 2—メチルフエニルスルホニルァミノ) チアゾール、 化合物 6 : 4- (1 -ァ ダマンチル) 一 2- ( ェニルスルホニルァミノ)チアゾール、 化合物 7 : 4— (1 -ァダマンチル) ー2— [ (5—プロモー 6—クロ口ピリジン一 3—ィル) ス ルホニルァミノ] チアゾール、 ィ匕合物 8 : 4— (1 -ァダマンチル) —2— [ (6 -モルホリノピリジン— 3—ィル) スルホニルァミノ] チアゾ一ル、 化 合物 9 : 4— (1 -ァダマンチル) 一 2— [ (6 -フエノキシピリジン一 3—ィ ル) スルホニルァミノ] チアゾール、 化合物 1 0 : 4— (1 -ァダマンチル) 一 2— [ (ピリジン一 2—ィル) スルホニルァミノ] チアゾール、 化合物 1 1 : 4- (1 -ァダマンチル) 一 2— [ (ピリジン一 3—ィル) スルホニルァ ミノ] チアゾール、 化合物 1 2 : 4— (1ーァダマンチル) —2— [N- (4 -ビフエニルスルホニル) 一 N—メチルァミノ] チアゾ一ル、 化合物 1 3 : 4 一 (1ーァダマンチル) 一 2— [N- [ (5 -クロ口 - 3 -メチルベンゾ [b]チ 才フェン- 2 -ィル) スルホ二ソレ]—N—メチルァミノ〕 チアゾール、 ィ匕合物 1 4 : 4一 (1—ァダマンチル) 一 2— [Ντメチルー N— (4 - η -プロピルフ ェニルスルホニル) ァミノ] チアゾ一ル、 化合物 1 5 : 4— (1ーァダマンチ ル) 一 2— [Ν— (4 -フルオロフェニルスルホニル) 一 Ν—メチルァミノ] チ ァゾ一ル、 化合物 1 6 : 4— ( 1 ーァダマンチル) 一 2— [Ν—メチルー Ν—The term "-dialkylamino group" means an amino group substituted by two linear or branched or cyclic alkyl groups having 1 to 5 carbon atoms, which are the same or different, such as a dimethylamino group and a methyl group. Examples thereof include an ethylamino group, a getylamino group, and a dipropylamino group. The following are specific examples of the thiazole derivative of the present invention. That is, compound 1: 41- (1-adamantyl) — 2— (1-naphthylsulfonylamino) thiazol, compound 2: 41- (1-adamantyl) — 2— (2, 4 5-Triclomethylphenylsulfonylamino) thiazole, compound 3: 4- (1-adamantyl) 1 2-[(2-Chenylsulfonyl) amino] thiazole, compound 4: 4-1 (1-adamantyl) 1 2 — (4-Bromophenylsulfonylamino) thiazole, compound 5: 4- (1-adamantyl) —2 -— (3-chloromethyl-2-methylphenylsulfonylamino) thiazole, compound 6: 4- (1 -A-damantyl) -1-2- (enylsulfonylamino) thiazole, compound 7: 4— (1-adamantyl) -2-[[(5-promo 6-chloropyridine-13-yl) sulfonylamino] thiazole, i 9: 4— (1 -adamantyl)-2-[(6-morpholinopyridine-3-yl) sulfonylamino] thiazolyl, compound 9: 4— (1-adamantyl) 1 2— [(6-Phenoxypyridine-1 3 — (Yl) sulfonylamino] thiazole, compound 10: 4— (1-adamantyl) 1-2 — [(Pyridine-12-2-yl) sulfonylamino] thiazole, compound 11 1: 4- (1-adamantyl) I 2 — [(Pyridine-13-yl) sulfonylamino] thiazole, compound 12: 4— (1-adamantyl) —2 -— [N- (4-biphenylsulfonyl) -1-N-methylamino] thiazol, compound 1 3: 4 1 (1-adamantyl) 1 2-[N-[(5-chloro-3 -methylbenzo [b] thiaphen-2-yl) sulfonisole] -N-methylamino] thiazole, Compound 14: 4-1- (1-adamantyl) 1-2- [Ντ-methyl-N— (4-η-propylphenylsulfonyl) amino] thiazol, compound 15: 4— (1-adamantyl) 1-2- [ Ν— (4-Fluorophenylsulfonyl) monomethylamino Sol, Compound 16: 4— (1-adamantyl) 1-2— [Ν-methyl-Ν—
(4—モルホリノフエニルスルホニル) ァミノ] チアゾ一ル、 化合物 1 7 : 4 一 (1ーァダマンチル) 一2— [Ν—メチルー Ν— [4- (4 -メチルビペラジ ニル) フエニルスルホニル] ァミノ] チアゾ一ル、 化合物 1 8 : 4— (1ーァ ダマンチル) 一 2— [Ν— [4 - [ビス (2 -ヒドロキシェチル) ァミノ] フエ ニルスルホニル] 一 Ν—メチルァミノ]チアゾ一ル、 化合物 1 9 : 4— (1ーァ ダマンチル) 一 2— [Ν— [4 - [ (2 -ヒドロキシェチル) ァミノ] フエニル スルホニル] —Ν—メチルァミノ]チアゾール、 化合物 20 : 4一 (1—ァダマ ンチル) 一 2— [Ν— [4 - (2 -ヒドロキシェ卜キシ) フエニルスルホニル]
一 N—メチルァミノ]チアゾール、 ィ匕合物 2 1 : 4- (1—ァダマンチル) 一 2 - [N- (4 -フルオロフェニルスルホニル) 一 N— (4 -メ卜キシベンジル) ァミノ]チアゾール、 化合物 22 : 4一 (1 —ァダマンチル) — 2— [N— (4 -メ卜キシベンジル) — N— (4 -モルホリノフエニルスルホニル) ァミノ]チ ァゾール、 化合物 23 : 4- (1—ァダマンチル) —2— (4 -モルホリノフ ェニルスルホニルァミノ) チアゾール、 化合物 24 4 - (1ーァダマンチ ル) - 2 - [N- (4 -メ卜キシベンジル) 一 N— [4 - (2 -モルホリノエ卜 キシ) フエニルスルホニル] ァミノ]チアゾール、 化合物 25 : 4一 (1—ァ ダマンチル) 一 2— [4 - (2 -モルホリノエ卜キシ) フエニルスルホニルァ ミノ]チアゾ一ル、 化合物 26 : 4- (1ーァダマンチル) 一 2— [N— [4 - [2 - (ジメチルァミノ) エトキシ] フエニルスルホニル] —N— (4 -メ卜 キシベンジル) ァミノ]チアゾ一ル、 化合物 27 : 4— (1—ァダマンチル) 一 2_[4 - [2 - (ジメチルァミノ) エトキシ] フエニルスルホニルァミノ] チアゾール、 化合物 28 : 4一 ( 1ーァダマンチル) - 2 - [4 - [2 - (4 - ピペラジニル)ェ卜キシ]フエニルスルホニルァミノ]チアゾール、 化合物 2(4-Morpholinophenylsulfonyl) amino] thiazol, compound 17: 4-1 (1-adamantyl) 1-2- [Ν-methyl-Ν— [4- (4-methylbiperazinyl) phenylsulfonyl] amino] thiazo-1 Compound 18: 4— (1-Damantyl) 12— [一 — [4- [bis (2-hydroxyethyl) amino] phenylsulfonyl] -1-methylamino] thiazole, Compound 19 : 4— (1-Adamantyl) 1 2— [Ν— [4-[(2-Hydroxyethyl) amino] phenylsulfonyl] —Ν—Methylamino] thiazole, Compound 20: 4-1- (1-adamantyl) 2— [Ν— [4- (2-hydroxyethoxy) phenylsulfonyl] 1-N-Methylamino] thiazole, diamide 21 1: 4- (1-adamantyl) 1-2- [N- (4-fluorophenylsulfonyl) -1-N— (4-methoxybenzyl) amino] thiazole, compound 22 : 4- (1-adamantyl) — 2 -— [N— (4-methoxybenzyl) — N— (4-morpholinophenylsulfonyl) amino] thiazole, compound 23: 4- (1-adamantyl) —2— (4-morpholinophenylsulfonylamino) thiazole, compound 24 4- (1-adamantyl) -2- (N- (4-methoxybenzyl) -1-N— [4- (2-morpholinoethoxy) phenylsulfonyl] [Amino] thiazole, Compound 25: 4- (1-Adamantyl) 1-2— [4- (2-Morpholinoethoxy) phenylsulfonylamino] thiazol, Compound 26: 4- (1-Adamantyl) 1-2— [N— [4-[2-(dimethylamino) Toxi] phenylsulfonyl] —N— (4-methoxybenzyl) amino, thiazol, compound 27: 4- (1-adamantyl) 1-2_ [4- [2- (dimethylamino) ethoxy] phenylsulfonylamino ] Thiazole, compound 28: 4- (1-adamantyl) -2- [4- [2- (4-piperazinyl) ethoxy] phenylsulfonylamino] thiazole, compound 2
9 : 4- (1ーァダマンチル) 一 2— [N— (4 -ヒドロキシフエニルスルホニ ル) 一 N— (4 -メ卜キシベンジル) ァミノ]チアゾール、 化合物 30 : 4-9: 4- (1-adamantyl) -1-2 -— [N— (4-hydroxyphenylsulfonyl) -1-N— (4-methoxybenzyl) amino] thiazole, compound 30: 4-
(1ーァダマンチル) 一 2— [4 - (カルボキシメ卜キシ) フエニルスルホニ ルァミノ]チアゾール、 化合物 3 1 : 4- (1ーァダマンチル) 一 2— [N— [4 - [ (ァミノカルボニル) メ卜キシ]フエニルスルホニル]一 N— (4 -メ卜キシ ベンジル) ァミノ]チアゾ一ル、 化合物 32 : 4一 (1—ァダマンチル) 一 2 -[4 - [ (ァミノカルボニル) メ卜キシ]フエニルスルホニルァミノ]チアゾー ル、 化合物 33 : 4— (1ーァダマンチル) 一 2— [N- (3—クロロー 2— メチルフエニルスルホニル) 一 N—メチルァミノ] チアゾール、 化合物 34 : 4一 (1ーァダマンチル) 一 2— [N- (3—クロロー 2—メチルフエニルス ルホニル) ー[\|一 (2 -ヒドロキシェチル) ァミノ] チアゾール、 化合物 3 5 : 4- (1ーァダマンチル) —2— [N— (ァミノカルボニルメチル) 一 |\|— (3—クロ口 _ 2—メチルフエニルスルホニル) ァミノ] チアゾール、 ィ匕合物 36 : 4- (1ーァダマンチル) 一 2— [N— (3—クロロー 2—メチルフエ
ニルスルホニル) 一 N—ェチルァミノ] チアゾール、 ィ匕合物 37 : 4— (1 - ァダマンチル) —2— [N- (3—クロロー 2—メチルフエニルスルホニル) —N—イソプチルァミノ] チアゾール、 ィ匕合物 38 : 4- (1ーァダマンチ ル) -2- [2— (ヒドロキシメチル) フエニルスルホニルァミノ] チアゾー ル、 化合物 39 (3) : 4 - (3—ヒドロキシァダマンタン一 1 一ィル) - 2 一 [N- [ (5 -クロロ - 3 -メチルベンゾ [b]チ才フェン- 2 -ィル) スルホ 二ル]— N—メチルァミノ] チアゾール、 化合物 40 (2) : 4一 (1 -ァダマ ンチル) -5- (ェ卜キシカルボニル) 一 2— (5-クロ口- 3-メチル -1-ベン ゾチェン -2-ィル)スルホニル]ァミノ Iチアゾール、 化合物 41 (A) : 4- (1 -ァダマンチル) — 5—メチル— 2— (5-クロ口- 3-メチル -1-ベンゾチ ェン- 2-ィル)スルホニル]ァミノ 1チアゾール、 化合物 41 (B) : 4- (1 - ァダマンチル) - 5 - (ヒドロキシメチル) 一 2— {[(5-クロ口- 3-メチル- 1 - ベンゾチェン- 2-ィル)スルホニル]ァミノ Iチアゾール、 化合物 42 (4) : 4 一 (1—ァダマンチル) 一 2— [2 - (2 -モルホリノエ卜キシ) フエニルス ルホニルァミノ]チアゾール、 化合物 43 (6) : 3—クロ口— N— [4— (3 ーメトキシ一 1—ァダマンチル) 一 1, 3—チアゾールー 2—ィル]一 2—メ チルベンゼンスルホンアミド、 化合物 44 : 5—クロロー N— [4— (3—メ卜 キシー 1ーァダマンチル) 一 1, 3—チアゾールー 2—ィル]一 3—メチル— 1 一ベンゾチォフェン一 2—スルホンアミド、 化合物 45 : 4- (1ーァダマ ンチル) 一 2— (2—ナフチルスルホニルァミノ) チアゾール、 化合物 46 : 4- (1 -ァダマンチル) 一 2— (2, 3, 4一卜リクロロフエニルスルホニ ルァミノ) チアゾ一ル、 化合物 47 : 4一 (1 -ァダマンチル) 一 2— (2, 4, 5—トリフルオロフェニルスルホニルァミノ) チアゾ一ル、 化合物 48 : 4 - (1 -ァダマンチル) ー2— (2, 6—ジクロ口フエニルスルホニルアミ ノ) チアゾール、 化合物 49 : 4- (1 -ァダマンチル) -2- [ (3—プロ モー 5—クロロチ才フェン一 2—ィル) スルホニルァミノ] チアゾール、 化合 物 50 : 4— (1 -ァダマンチル) 一 2— (2, 3—ジクロロフエニルスルホ ニルァミノ) チアゾール、 化合物 5 1 : 4一 (1 -ァダマンチル) — 2— (2, 4ージフル才ロフエニルスルホニルァミノ) チアゾ一ル、 化合物 52 : 4一
(1 -ァダマンチル) —2— [ (2, 5—ジクロロチ才フェン一 3—ィル) ス ルホニルァミノ] チアゾール、 ィ匕合物 53 : 4一 (1 -ァダマンチル) 一 2—(1-adamantyl) 1 2— [4- (carboxymethoxy) phenylsulfonylamino] thiazole, compound 31: 4- (1-adamantyl) 12— [N— [4 — [(aminocarbonyl) methoxy] Phenylsulfonyl] -1-N— (4-methoxybenzyl) amino] thiazole, compound 32: 4- (1-adamantyl) 1-2- [4-[(aminocarbonyl) methoxy] phenylsulfonyl [Amino] thiazole, compound 33: 4- (1-adamantyl) -12- [N- (3-chloro-2-methylphenylsulfonyl) -1-N-methylamino] thiazole, compound 34: 4-1 (1-adamantyl) 1-2 — [N- (3-chloro-2-methylphenylsulfonyl)-[\ |-(2-hydroxyethyl) amino] thiazole, compound 35: 4- (1-adamantyl) —2— [N— (amino Carbonylmethyl) 1 | \ | — (3—black mouth _ 2-Methylphenylsulfonyl) amino] thiazole, conjugated compound 36: 4- (1-adamantyl) -1-2- [N- (3-chloro-2-methylphen) Nylsulfonyl) 1-N-ethylamino] thiazole, diazole 37: 4- (1 -adamantyl) —2— [N- (3-chloro-2-methylphenylsulfonyl) -N-isobutylamino] thiazole, diamide Compound 38: 4- (1-adamantyl) -2- [2- (hydroxymethyl) phenylsulfonylamino] thiazole, compound 39 (3): 4- (3-hydroxyadamantane) -2- [N-[(5-Chloro-3-methylbenzo [b] thinphen-2-yl) sulfonyl] -N-methylamino] thiazole, compound 40 (2): 4- (1-adama) ) -5- (Ethoxycarbonyl) 1-2- (5-chloro-3-methyl-1-benzo-2-yn) sulfonyl] amino Ithiazole, compound 41 (A): 4- (1 -Adamantyl) — 5-methyl-2--2- (5-chloro-3-methyl-1-benzophen-2-yl) sulfonyl] amino 1 thia Compound 41 (B): 4- (1-adamantyl) -5- (hydroxymethyl) 1-2-{[(5-chloro-3-methyl-1-benzobenzo-2-yl) sulfonyl] amino I Thiazole, Compound 42 (4): 4 1- (1-adamantyl) 1 2— [2- (2-morpholinoethoxy) phenylsulfonylamino] thiazole, Compound 43 (6): 3—Cross— N— [4— (3-methoxy-11-adamantyl) -1,3-thiazol-2-yl] 1-2-methylbenzenesulfonamide, compound 44: 5-chloro-N— [4- (3-methoxy-1-adamantyl) 1-1 , 3-Thiazol-2-yl] -1-methyl-1-benzothiophene-2-sulfonamide, compound 45: 4- (1-adamantyl) -12- (2-naphthylsulfonylamino) thiazole, compound 46: 4 -(1 -adamantyl) 1 2— (2,3,4) Nylsulfonylamino) thiazole, compound 47: 4- (1-adamantyl) 1-2- (2,4,5-trifluorophenylsulfonylamino) thiazole, compound 48: 4- (1-adamantyl) -2 — (2,6-Dichloromouth phenylsulfonylamino) thiazole, compound 49: 4- (1-adamantyl) -2-[(3-promo 5-chlorothiophen-1-yl) sulfonylamino] Thiazole, Compound 50: 4- (1-adamantyl) 1-2- (2,3-dichlorophenylsulfonylamino) Thiazole, compound 51 1: 4-1 (1-adamantyl) — 2 -— (2,4-difuroflov) Enylsulfonylamino) thiazole, compound 52: 41 (1-adamantyl) —2— [(2,5-dichlorothienephen-1-yl) sulfonylamino] thiazole, iridani compound 53: 4-1 (1-adamantyl) one 2—
(3—クロ口— 5—フル才ロ— 2—メチルフエニルスルホニルァミノ) チアゾ ール、 化合物 4 _ 1 : 4- (1 -ァダマンチル) 一 2— (2, 4, 6—卜リメ チルフエニルスルホニルァミノ) チアゾール、 化合物 4一 2 : 4— (1 -ァダ マンチル) 一 2— (3—卜リフル才ロメチルフエニルスルホニルァミノ) チア ゾール、 化合物 4一 3 : 4一 (1 -ァダマンチル) 一 2— (4一プロピルフエ ニルスルホニルァミノ) チアゾール、 化合物 4— 4 : 4一 (1 -ァダマンチ ル) -2- (2—メチルフエニルスルホニルァミノ) チアゾール、 ィ匕合物 4一 5 : 4— (1 -ァダマンチル) 一 2— (4—フルオロフェニルスルホニルアミ ノ) チアゾール、 化合物 4— 6 : 4一 (1 -ァダマンチル) 一 2— (2, 5- ジメチルフエニルスルホニルァミノ) チアゾール、 化合物 4一 7 : 4— (1 - ァダマンチル) —2— (ビフエニルスルホニルァミノ) チアゾール、 化合物 4 一 8 : 4— (1 -ァダマンチル) 一 2— (4一クロ口, 2, 5—ジメチルフエ ニルスルホニルァミノ) チアゾール、 化合物 4一 9 : 4一 (1 -ァダマンチ ル) 一 2— (3, 5—ジメチルイソ才キサゾールスルホニルァミノ) チアゾー ル、 化合物 4—10 : 4— (1 -ァダマンチル) 一 2— (5—クロロー 3—メチ ルーベンゾ [b] チ才フェン一 2ィル—スルホニルァミノ) チアゾール、 化合 物 4— 11 : 4— (1 -ァダマンチル) 一 2— (2, 5—ジクロ口フエニルスル ホニルァミノ) チアゾ一ル、 化合物 4一 12 : 4— (1 -ァダマンチル) 一 2— (4一二卜口フエニルスルホニルァミノ) チアゾ一ル、 化合物 4—13 : 4一 (1 -ァダマンチル) 一 2— (4ーメ卜キシフエニルスルホニルァミノ) チア ゾール、 化合物 4一 14 : 4- (1 -ァダマンチル) 一 2— (2, 4—ジメトキ シフエニルスルホニルァミノ) チアゾ一ル、 化合物 4—15 : 4— (1 -ァダマ ンチル) 一 2— (3—ニトロフエニルスルホニルァミノ) チアゾ一ル、 化合物 4-16 : N— [4一 (4ーァダマンタン一 1ーィルーチアゾ一ルー 2—ィルー スルファモイル) 一フエニル]ーァセ卜アミド、 化合物 4—17 : 4— (1 -ァ ダマンチル) 一 2— (4一クロ口フエニルスルホニルァミノ) チアゾ一ル、 化 合物 4— 18 : 4— (1 -ァダマンチル) 一 2— (4一フエノキシフエニルスル
ホニルァミノ) チアゾール、 化合物 4— 1 9 : 4— (1 -ァダマンチル) 一 2 _ ( 4—プチルフエニルスルホニルァミノ) チアゾール、 ィ匕合物 4一 2 0 : 4一 (1 -ァダマンチル) 一 2— ( 1, 1—ジメチルプロピルフエニルスルホニル ァミノ) チアゾール、 化合物 4一 2 1 : 4— (1 -ァダマンチル) —2— (ベン ゾ [ b ] チ才フェン— 2ィル—スルホニルァミノ) チアゾール、 ィ匕合物 4— 2 2 : 4 - (1 -ァダマンチル) 一 2— (ベンゾ [ b ] チ才フェン— 3ィル—ス ルホニルァミノ) チアゾール及び化合物 4一 2 3 : 2 - ( 4一 (ァダマンタン - 1—ィル—チアゾール— 2ィルースルファモイル) 一ベンゼン酸メチルエス テル、 化合物 4一 2 4 : 4一 ( 4 - (ァダマンタン一 1ーィルーチアゾールー 2ィルースルファモイル) 一ベンゼン酸、 化合物 4一 2 5 : 4一 (1 -ァダマ ンチル) ー2— (ベンゾフラン一 3—ィルースルホニルァミノ) チアゾール等 である。 (3-Mouth—5-methyl-2-methylphenylsulfonylamino) thiazole, compound 4 _ 1: 4- (1-adamantyl) 1 2— (2,4,6-trimethyl) Phenylsulfonylamino) thiazole, compound 4-2: 4- (1-adamantyl) 1-2- (3-trimethyl romethylphenylsulfonylamino) thiazole, compound 4-1 3: 4-1 ( 1-adamantyl) 1- 2- (4-propylphenylsulfonylamino) thiazole, compound 4-4: 4- (1-adamantyl) -2- (2-methylphenylsulfonylamino) thiazole, diamide 4-5: 4- (1-adamantyl) 1-2- (4-fluorophenylsulfonylamino) thiazole, compound 4-6: 4-1 (1-adamantyl) -12- (2,5-dimethylphenylsulfonyl) Mino) thiazole, compound 4 7: 4— (1- Adamantyl) —2— (Biphenylsulfonylamino) thiazole, compound 418: 4— (1-adamantyl) 12— (4,1-, 2,5-dimethylphenylsulfonylamino) thiazole, compound 41 9: 4-1 (1-adamantyl) -12- (3,5-dimethylisoxoxazolesulfonylamino) thiazole, compound 4-10: 4- (1-adamantyl) 1-2 (5-chloro-3-) Methyl benzo [b] thylphen-2-yl-sulfonylamino) thiazole, compound 4-11: 4- (1-adamantyl) -12- (2,5-dichloromouth phenylsulfonylamino) thiazol, compound 4-12: 4— (1-adamantyl) 1 2— (412 phenylsulfonylamino) thiazole, compound 4-13: 4—1 (1-adamantyl) 1 2— (4-meth Xyphenylsulfonylamino Thiazole, compound 4- 14: 4- (1-adamantyl) 1-2- (2,4-dimethoxyphenylsulfonylamino) thiazol, compound 4-15: 4- (1-adamantyl) 1-2- (3-Nitrophenylsulfonylamino) thiazole, compound 4-16: N— [4- (4-adamantane-1-1-yruthiazo-1-ru-2-ylsulfamoyl) -phenyl] acetamide, compound 4-17: 4 — (1-adamantyl) 1 2-(4-phenylsulfonylamino) thiazole, compound 4-18: 4-(1-adamantyl) 1 2-(4-phenoxyphenylsulfur) Honylamino) thiazole, compound 4-1 9: 4- (1-adamantyl) 1 2 _ (4-butylphenylsulfonylamino) thiazole, i-conjugated compound 4 1 0: 4 1 (1-adamantyl) 1 2 — (1,1-Dimethylpropylphenylsulfonylamino) thiazole, compound 4-1 21: 4- (1-adamantyl) —2— (Benzo [b] thifenphen-2-yl-sulfonylamino) thiazole匕 匕 合 4 — — 物 4 4 4 4 合 合 4 合 匕 合 合 物 合 物 合 合 物 合 合 合 合 合 合 合 物 合 合 合 合 合 合 合. Adamantane-1-yl-thiazole-2-ylsulfamoyl) methyl ester monobenzene, compound 4 2 4: 4-1 (4--(adamantane-1-ylutiazole-2-ylsulfamoyl) monobenzeneic acid The compound 4 1 2 5: 4 1 1 - Adama pentyl) over 2- (benzofuran one 3- I Lou sulfonyl § amino) thiazole and the like.
本発明のチアゾール誘導体は、 その医薬上許容される塩、 又はその溶媒和物 であっても良い。 以下、 本発明のチアゾール誘導体、 その医薬上許容される塩、 及びその溶媒和物を含めて、 「本発明の化合物」 ともいう。 The thiazole derivative of the present invention may be a pharmaceutically acceptable salt thereof, or a solvate thereof. Hereinafter, the thiazole derivative of the present invention, its pharmaceutically acceptable salt, and its solvate are also referred to as “the compound of the present invention”.
本明細書において、 溶媒和物としては、 水和物など医薬上許容される溶媒和 物があげられる。 本発明のチアゾール誘導体は、 大気にさらされ、 又は再結晶 することなどにより、 水分を吸収し、 吸着水がつく場合や、 水和物となる場合 がある。 本発明のチアゾール誘導体の医薬上許容される溶媒和物は、 そのよう な水和物をも含む。 In the present specification, examples of the solvate include pharmaceutically acceptable solvates such as hydrates. The thiazole derivative of the present invention may absorb water by being exposed to the air or recrystallized, and may form adsorbed water or may be a hydrate. Pharmaceutically acceptable solvates of the thiazole derivatives of the present invention also include such hydrates.
本明細書において、 医薬上許容される塩とは、 例えば、 塩酸塩、 臭化水素酸 塩、 ヨウ化水素酸塩、 リン酸塩、 硫酸塩、 硝酸塩のような鉱酸塩;メタンスル ホン酸塩、 エタンスルホン酸塩、 ベンゼンスルホン酸塩、 p—トルエンスルホ ン酸塩のようなスルホン酸塩;シユウ酸塩、 酒石酸塩、 クェン酸塩、 マレイン 酸塩、 コハク酸塩、 酢酸塩、 安息香酸塩、 マンデル酸塩、 ァスコルビン酸塩、 乳酸塩、 ダルコン酸塩、 リンゴ酸塩のような有機酸塩等の酸付加塩、 グリシン 塩、 リジン塩、 アルギニン塩、 オル二チン塩、 グルタミン酸塩、 ァスパラギン 酸塩のようなアミノ酸塩、 あるいはリチウム塩、 ナトリウム塩、 カリウム塩、 カルシウム塩、 マグネシウム塩のような無機塩又はアンモニゥ厶塩、 卜リエチ
ルァミン塩、 ジイソプロピルアミン塩、 シクロへキシルァミン塩のような有機 塩基との塩であり得、 好適には塩酸塩、 臭化水素酸塩、 リン酸塩、 硫酸塩、 メ タンスルホン酸塩、 P—卜ルエンスルホン酸塩、 シユウ酸塩、 酒石酸塩、 クェ ン酸塩、 酢酸塩、 乳酸塩、 グルタミン酸塩、 ァスパラギン酸塩、 ナトリウム塩、 カリウム塩、 アンモニゥ厶塩又は卜リエチルァミン塩があげられ、 好ましくは ナトリウム塩、 塩酸塩又は硫酸塩であり、 より好ましくは塩酸塩である。 なお、 本明細書において、 本発明の化合物には、 生体内において代謝されて本発明の 本発明の化合物に変換される化合物、 いわゆるプロドラッグも含まれる。 In the present specification, pharmaceutically acceptable salts include, for example, mineral salts such as hydrochloride, hydrobromide, hydroiodide, phosphate, sulfate, and nitrate; methanesulfonate Sulfonates such as ethanesulfonate, benzenesulfonate, p-toluenesulfonate; oxalate, tartrate, citrate, maleate, succinate, acetate, benzoate Acid addition salts such as organic acid salts such as mandelate, ascorbate, lactate, dalconate, malate, glycine, lysine, arginine, orditin, glutamate, aspartate Amino acid salts such as salts, or inorganic salts such as lithium salts, sodium salts, potassium salts, calcium salts, and magnesium salts or ammonium salts; It may be a salt with an organic base such as a luminamine salt, a diisopropylamine salt, a cyclohexylamine salt, and is preferably a hydrochloride, a hydrobromide, a phosphate, a sulfate, a methanesulfonate, or a Toluene sulfonate, oxalate, tartrate, citrate, acetate, lactate, glutamate, aspartate, sodium, potassium, ammonium, or triethylamine salts, preferably It is a sodium salt, a hydrochloride or a sulfate, and more preferably a hydrochloride. In the present specification, the compound of the present invention also includes a compound which is metabolized in a living body and converted into the compound of the present invention, so-called prodrug.
本発明のチアゾール誘導体は、 不斉中心を持つことがあり、 その場合種々の 光学異性体又は配置のものが存在する。 したがって、 本発明の化合物は、 The thiazole derivative of the present invention may have an asymmetric center, in which case, there are various optical isomers or configurations. Thus, the compounds of the present invention
( + ) および (一) の別々の光学活性体として、 およびラセミ体又は (土) 混 合物として存在し得る。 また、 不斉中心を 2個以上持つ化合物の場合には、 さ らにそれぞれの光学異性によるジァステレオマーも存在する。 本発明のチアゾ ール誘導体は、 これらすベての型を、 任意の割合で含む。 たとえば、 ジァステ レオマーは当業者によく知られた方法、 たとえば分別結晶法等によって分離す ることができ、 また、 光学活性体はこの目的のためによく知られた有機化学的 手法によって得ることができる。 また、 本発明のチアゾ一ル誘導体は、 シス体、 卜ランス体などの異性体が存在することがある。 本発明のチアゾ一ル誘導体は、 それらの異性体、 及びそれらの異性体を任意の割合で含んだものも含む。 以下、 本発明の化合物の製造方法に用いられる反応スキームの例を説明する。 本発明の化合物は、 上記特許文献、 非特許文献を含めた公知文献に従い、 公知 の有機合成の手法を採用して製造できる。 また、 下記の反応式で表される各製 造工程においても、 公知の手法を採用することができる。 なお、 各工程におけ る原材料となる化合物は、 公知化合物であるか、 又は公知化合物から容易に合 成できる化合物である。
[反応式 1 ] It can exist as separate (+) and (1) optically active forms and as a racemic or (soil) mixture. In the case of a compound having two or more asymmetric centers, diastereomers due to each optical isomer also exist. The thiazole derivative of the present invention contains all of these types in an arbitrary ratio. For example, diastereomers can be separated by methods well known to those skilled in the art, such as fractional crystallization, and optically active forms can be obtained by well-known organic chemistry techniques for this purpose. it can. In addition, the thiazole derivative of the present invention may have isomers such as cis- and trans-isomers. The thiazole derivatives of the present invention include their isomers and those containing these isomers in any proportion. Hereinafter, examples of the reaction scheme used in the method for producing the compound of the present invention will be described. The compound of the present invention can be produced according to known literature including the above-mentioned patent literature and non-patent literature and employing a known organic synthesis technique. In each of the production steps represented by the following reaction formulas, known methods can be employed. The compound serving as a raw material in each step is a known compound or a compound that can be easily synthesized from a known compound. [Reaction formula 1]
(式中、 R R2および R3 はそれぞれ前記と同意義である。 Xaは塩素原子、 臭素原子、 ヨウ素原子、 メタンスルホニル才キシ基または卜シル才キシ基を示 す。 ) (Wherein, RR 2 and R 3 have the same meanings as described above. X a represents a chlorine atom, a bromine atom, an iodine atom, a methanesulfonyl group or a trisyl group.)
この工程は、 化合物 (I I ) と化合物 (I I I ) を縮合してァミノチアゾール誘 導体 (I V) を得る工程である。 この反応に使用する溶媒としては、 エタノール、 メタノール、 N, N—ジメチルホルムアミド、 クロ口ホルム等が挙げられ、 反応 は 0〜1 00°Cで行うことが出来る。 この際、 生成する塩酸、 または臭化水素酸を 捕捉するために塩基を添加してもよい。 添加する塩基として卜リェチルァミン、 ジィソプロピルェチルァミン等のアミン類または炭酸力リゥ厶ゃ炭酸水素ナ卜 リウ厶等の無機塩基等が挙げられる。 また得られた化合物 (I V) を酸との塩と 'して得た場合は、 酢酸ェチル等の有機溶媒に溶解し、 無機塩基の水溶液で洗浄 を行うことにより、 フリーのァミンとして単離することも出来る。 In this step, the compound (II) is condensed with the compound (III) to obtain an aminothiazole derivative (IV). Examples of the solvent used for this reaction include ethanol, methanol, N, N-dimethylformamide, and chloroform. The reaction can be carried out at 0 to 100 ° C. At this time, a base may be added to capture the generated hydrochloric acid or hydrobromic acid. Examples of the base to be added include amines such as triethylamine and disopropylethylamine, and inorganic bases such as carbon dioxide and sodium hydrogencarbonate. When the obtained compound (IV) is obtained as a salt with an acid, the compound is dissolved in an organic solvent such as ethyl acetate and washed with an aqueous solution of an inorganic base to be isolated as free amine. You can do it.
[反応式 2 ][Reaction formula 2]
(V) (VI) (V) (VI)
(式中、 R1 R2および R3 はそれぞれ前記と同意義である。 ) (Wherein, R 1 R 2 and R 3 are each as defined above.)
この工程は、 化合物 (V) のァミノ基に R3を導入し化合物 (V I ) を得る工程 である。 In this step, R 3 is introduced into the amino group of compound (V) to obtain compound (VI).
例えば、 脱離基で置換された R3、 即ち R3— Xa (Xaは前記と同意義である) を 用い化合物 (V) と反応を行う場合は、 反応に使用する溶媒としてはジクロ口 メタン、 クロ口ホルム、 N, N—ジメチルホルムアミド、 エタノール等が挙げら
れ、 反応は 0〜100°Cで行うことが出来る。 この際、 反応は適当な塩基を用いて 行うことが出来、 塩基としてとして卜リエチルァミン、 ジイソプロピルェチル ァミン等のアミン類または炭酸力リウ厶ゃ炭酸水素ナ卜リゥ厶等の無機塩基等 が挙げられる。 For example, when reacting with compound (V) using R 3 substituted with a leaving group, that is, R 3 —X a (X a has the same meaning as described above), the solvent used for the reaction is dichloromethane. Methane, chloroform, N, N-dimethylformamide, ethanol, etc. The reaction can be carried out at 0 to 100 ° C. In this case, the reaction can be carried out using an appropriate base, and examples of the base include amines such as triethylamine and diisopropylethylamine, and inorganic bases such as lithium carbonate and sodium hydrogencarbonate. .
また例えば、 別の例としては、 アルデヒド誘導体と還元剤を用いる還元的ァ ミノ化反応を挙げることが出来る。 還元剤には卜リアセ卜キシ水素化ホウ素ナ 卜リゥ厶ゃ水素化シァノホウ素ナ卜リウ厶、 水素化ホウ素ナ卜リゥ厶等が挙げ られ、 必要に応じて氷酢酸や卜シル酸等の酸を添加することが出来る。 この反 応に使用する溶媒としてはジクロロメタンやクロ口ホルム、 テ卜ラヒドロフラ ン、 ジォキサン、 1, 2-ジクロロェタン等が挙げられ、 反応は- 20〜100°Cで行う ことが出来る。 Another example is a reductive amination reaction using an aldehyde derivative and a reducing agent. Examples of the reducing agent include triacetoxy sodium borohydride, sodium cyanoborohydride, sodium borohydride and the like, and if necessary, an acid such as glacial acetic acid or tosylic acid. Can be added. Solvents used for this reaction include dichloromethane, chloroform, tetrahydrofuran, dioxane, 1,2-dichloroethane and the like, and the reaction can be carried out at −20 to 100 ° C.
[ [
(式中、 R\ R2、 R3、 R4、 R5、 R6、 R7および R8は前記と同意義である。 ) この工程は、 化合物 (IV) のァミノ基をスルホニル化することによりスルホ ンアミド誘導体 (VI I ) を得る工程である。 (Wherein R \ R 2 , R 3 , R 4 , R 5 , R 6 , R 7 and R 8 are as defined above.) This step sulfonylates the amino group of compound (IV). This is a step of obtaining a sulfonamide derivative (VII).
例えば、 スルホニルクロリドゃスルホニルブロミド等のスルホニルハライド を用いることでスルホンアミド誘導体を得ることが出来る。 この反応に使用す る溶媒としては、 ピリジン、 ジクロロメタン、 クロ口ホルム、 1, 2-ジクロロェ タン、 テ卜ラヒドロフラン、 ジ才キサン、 トルエン、 酢酸ェチル等が挙げられ、 反応は- 50〜100°Cで行うことが出来る。 この際反応は、 適当な塩基を用いて行 うことが出来、 塩基の例としては、 卜リエチルァミン、 ジイソプロピルァミン、 4一ジメチルァミノピリジン等のァミン類ゃ炭酸力リゥ厶等の無機塩基等が挙 げられる。 また、 この際、 R1、 R2および R3にスルホニルハライドと反応しやす
い基がある場合には、 これを保護して行うことができる。 例えば、 水酸基が存 在した場合に卜リメチルシリル化を先行することで、 選択的に目的物を得るこ とができる。 For example, a sulfonamide derivative can be obtained by using a sulfonyl halide such as sulfonyl chloride-sulfonyl bromide. Examples of the solvent used in this reaction include pyridine, dichloromethane, chloroform, 1,2-dichloroethane, tetrahydrofuran, dioxane, toluene, and ethyl acetate. Can be done. In this case, the reaction can be carried out using an appropriate base. Examples of the base include amines such as triethylamine, diisopropylamine, and 4-dimethylaminopyridine; and inorganic bases such as carbon dioxide. Are listed. At this time, R 1 , R 2 and R 3 are liable to react with sulfonyl halide. If there are any groups, this can be done with protection. For example, when a hydroxyl group is present, the desired product can be selectively obtained by preceding trimethylsilylation.
また例えば、 スルホンアミド化反応の別の例として 1一べンゾ卜リアゾリル エステルゃスクシンィミジルエステル等の活性エステルを用いて行うことが出 来る。 反応溶媒としてはジクロロメタン、 クロ口ホルム、 1 , 2—ジクロロェ タン、 N, N—ジメチルホルムアミド、 テトラヒドロフラン、 ジ才キサン、 トル ェン、 酢酸ェチル等が挙げられる。 この反応は— 50〜50°Cで行うことが出来る。 Further, for example, another example of the sulfonamidation reaction can be carried out using an active ester such as 1-benzotriazolyl ester / succinimidyl ester. Examples of the reaction solvent include dichloromethane, chloroform, 1,2-dichloroethane, N, N-dimethylformamide, tetrahydrofuran, dioxane, toluene, and ethyl acetate. This reaction can be carried out at -50 to 50 ° C.
[反応式 4 ] [Reaction formula 4]
(式中、 R1、 R2、 R3、 R4、 R5、 R6、 R7および R8は前記と同意義である。 Xbは ァミノ基の保護基を示す。 ) (Wherein, R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 and R 8 are as defined above. X b represents a protecting group for an amino group.)
この工程は、 化合物 (V I I ) の保護基 Xbを脱保護して化合物 (V I I I ) を得る 工程である。 例えば、 Xbが 4ーメ卜キシベンジル基等の酸で脱保護される基の 場合は、 塩酸、 硫酸、 トリフル才ロ酢酸、 p—卜ルエンスルホン酸、 メタンス ルホン酸等の酸を用い、 脱保護することができる。 この際、 有機溶媒又は水で 希釈又は溶解して行うことができ、 反応温度は— 5 0 °Cから 5 0 °Cで行うこと ができる。 有機溶媒としては、 例えばエタノール、 メタノール、 テ卜ラヒドロ フラン、 N , N—ジメチルホルムアミド、 ジクロロメタン、 クロ口ホルム、 Ί , 2—ジクロ C!エタン等があげられる。
[反応式 5] In this step, the protecting group Xb of the compound (VII) is deprotected to obtain the compound (VIII). For example, when X b is a group that can be deprotected with an acid such as a 4-methoxybenzyl group, the deprotection is performed using an acid such as hydrochloric acid, sulfuric acid, trifluroacetic acid, p-toluenesulfonic acid, and methanesulfonic acid. Can be protected. At this time, the reaction can be performed by diluting or dissolving with an organic solvent or water, and the reaction can be performed at a reaction temperature of −50 ° C. to 50 ° C. Examples of the organic solvent include ethanol, methanol, tetrahydrofuran, N, N-dimethylformamide, dichloromethane, chloroform, Ί, 2-dichloro C! Ethane, and the like. [Reaction formula 5]
(式中、 R2、 R3、 R5、 R6、 R7、 R8および Xaは前記と同意義である。 Ra は、 式一 X1— X2_X3または式— X1— X5— X6で表される基のうち X1が、 酸 素原子、 硫黄原子または式 _ NX4—で表されるものを示す。 X1、 X2、 X3、 X 4、 X5および X6は、 前記と同意義である。 ) (Wherein, R 2 , R 3 , R 5 , R 6 , R 7 , R 8 and X a are as defined above. Ra is a group represented by the formula X 1 —X 2 _X 3 or the formula —X 1 — X 5 — Of the groups represented by X 6 , X 1 represents an oxygen atom, a sulfur atom, or a compound represented by the formula _ NX 4 — X 1 , X 2 , X 3 , X 4 , X 5 and X 6 are as defined above.
この工程は、 脱離基を置換基に有する化合物 (X) を用い置換反応を行って R4を導入した化合物 (XI) を得る工程である。 In this step, the compound (X) having a leaving group as a substituent is subjected to a substitution reaction to obtain a compound (XI) into which R 4 has been introduced.
例えば、 Xaがフッ素の場合、 求核置換反応を用いてァミン、 ァニリン、 アル コール及びチオールを導入することが出来る。 また例えば、 2—メチルスルホ ニルエタノールなどを用いて同様の置換反応を行うと生成物としてハロゲン原 子が水酸基に変換された化合物 (XI) を得ることができる。 これら反応に使用 する溶媒としてジメチルスルホキシド、 N, N—ジメチルホルムアミド、 テ卜ラ ヒドロフラン、 トルエン等が挙げられ、 反応温度は 0〜200°Cで行うことが 出来る。 またこの反応は適当な塩基を添加して反応を行うことが出来、 塩基と して水素化ナトリウム、 tert -ブトキシカリウム、 n -ブチルリチウム、 リチ ゥ厶ジイソプロピルアミド、 リチウムへキサメチルジシラジド等ゃ炭酸力リゥ 厶等の無機塩基が挙げられる。 また必要に応じて金属触媒を添加することも出 来る。 金属触媒としては卜リス (ジベンジリデンアセトン) ジパラジウムや酢 酸パラジウム等が挙げられる。 For example, when X a is fluorine, Amin using nucleophilic substitution reaction, Anirin, can be introduced alcohol and thiol. Further, for example, when a similar substitution reaction is performed using 2-methylsulfonylethanol or the like, a compound (XI) in which a halogen atom is converted to a hydroxyl group can be obtained as a product. Examples of the solvent used in these reactions include dimethyl sulfoxide, N, N-dimethylformamide, tetrahydrofuran, toluene and the like, and the reaction can be carried out at a temperature of 0 to 200 ° C. In addition, this reaction can be carried out by adding an appropriate base. Examples of the base include sodium hydride, potassium tert-butoxide, n-butyllithium, lithium diisopropylamide, lithium hexamethyldisilazide, and the like. And inorganic bases such as carbon dioxide realm. In addition, a metal catalyst can be added as needed. Examples of the metal catalyst include tris (dibenzylideneacetone) dipalladium and palladium acetate.
また例えば、 金属触媒と有機金属化合物を用いたクロスカップリング法によ り、 ビアリール等の炭素一炭素結合を構築することも出来る。 この反応に使用 する溶媒としてテ卜ラヒドロフラン、 トルエン、 1 , 2 -ジメ卜キシェタン等 が挙げられ、 反応温度は 0〜200°Cで行うことが出来る。 金属触媒としては テ卜ラキス (卜リフエニルホスフィン) パラジウムや卜リス (ジベンジリデン
アセトン) ジパラジウムやヒス (ァセチルァセ卜ナ卜) ニッケル等が挙げられ る。 有機金属化合物にはグリニャール試薬、 有機アルミニウム化合物、 ァリー ルボロン酸が挙げられる。 またこの反応は適当な塩基を添加して反応を行うこ とが出来、 塩基としては卜リエチルァミン、 ジイソプロピルアミン等のアミン や水酸化ナトリウム、 炭酸カリウム、 炭酸ナトリウム等の無機塩基が挙げられ る。 Also, for example, a carbon-carbon bond such as biaryl can be constructed by a cross-coupling method using a metal catalyst and an organometallic compound. Examples of the solvent used in this reaction include tetrahydrofuran, toluene, 1,2-dimethyloxetane and the like, and the reaction can be carried out at a temperature of 0 to 200 ° C. Metal catalysts include tetrakis (triphenylphosphine) palladium and tris (dibenzylidene) Acetone) dipalladium and hiss (acetyl acetate) nickel. Organometallic compounds include Grignard reagents, organoaluminum compounds and arylboronic acids. This reaction can be carried out by adding an appropriate base, and examples of the base include amines such as triethylamine and diisopropylamine and inorganic bases such as sodium hydroxide, potassium carbonate and sodium carbonate.
[反応式 6 ] [Reaction formula 6]
(式中、 R1 R2 R3 R5 R6 R7および R8は前記と同意義である。 R4bは、 式- X1— X X3または式— X1— X5— X6で表される基のうち X1が、 酸素原 子で表されるものを示す。 X1 X2 X3 X4 X5および X6は、 前記と同意義 である。 ) (Wherein, R 1 R 2 R 3 R 5 R 6 R 7 and R 8 are as defined above. R 4b is a group represented by the formula —X 1 —XX 3 or the formula —X 1 —X 5 —X 6 Among the groups represented, X 1 represents a group represented by an oxygen atom, and X 1 X 2 X 3 X 4 X 5 and X 6 are as defined above.
この工程は、 水酸基を置換基に有する化合物 (XI I) を用いて化合物 In this step, the compound (XI I) having a hydroxyl group as a substituent is
(XI I I) を得る工程である。 , (XI I I). ,
例えば、 ハロゲン化アルキルを用いた反応で、 水酸基のアルキル化を行うこ とができる。 この反応は適当な塩基を添加して反応を行うことが出来、 塩基と して水素化ナトリウム、 tert -プ卜キシカリウム、 n -プチルリチウ厶、 リチ ゥ厶ジイソプロピルアミド、 リチウムへキサメチルジシラジド等ゃ炭酸力リウ 厶等の無機塩基が挙げられる。 これら反応に使用する溶媒としてジメチルスル ホキシド、 N N—ジメチルホルムアミド、 テ卜ラヒドロフラン、 トルエン等が 挙げられ、 反応温度は 0 200°Cで行うことが出来る。
[反応式 7 ] For example, a hydroxyl group can be alkylated by a reaction using an alkyl halide. This reaction can be carried out by adding an appropriate base, and sodium hydride, potassium tert-butoxide, n-butyllithium, lithium diisopropylamide, lithium hexamethyldisilazide can be used as the base. Inorganic bases such as carbon dioxide are included. Solvents used for these reactions include dimethyl sulfoxide, NN-dimethylformamide, tetrahydrofuran, toluene and the like, and the reaction can be carried out at 0 200 ° C. [Reaction formula 7]
(式中、 に R1, R2、 R3、 R5、 R6、 R7、 R8、 X1、 X5および X7は前記と同意義であ る。 Xbは、 ァミノ基の保護基を表す。 ) (Wherein, R 1 , R 2 , R 3 , R 5 , R 6 , R 7 , R 8 , X 1 , X 5 and X 7 have the same meanings as described above. X b is an amino group Represents a protecting group.)
この工程は、 A環置換基に保護されたアミノ基を有する化合物 (X IV) を用い て脱保護を行い、 ァミン誘導体 (XV) を得る工程である。 この脱保護について は PROTECT IVE GROUPS I N ORGA I C SYNTHES I S, THEODORA W. GREENE and PETER G. M WUTS著に記載の方法を用いることが出来る。 例えば Xbが tert -ブ 卜キシカルボニル基、 卜リチル基、 0—二卜口ベンゼンスルフエニル基等の酸 で脱保護される基の場合は、 塩酸、 硫酸、 トリフル才ロ酢酸、 p—トルエンス ルホン酸、 メタンスルホン酸等の酸を用い、 脱保護することができる。 この際、 有機溶媒又は水で希釈又は溶解して行うことができ、 反応温度は一 5 0 °Cから 5 0 °Cで行うことができる。 有機溶媒としては、 例えばエタノール、 メタノー ル、 テ卜ラヒドロフラン、 N , N—ジメ^ルホルムアミド、 ジクロロメタン、 クロ口ホルム、 1 , 2—ジクロロェタン等があげられる。 更に例えば、 X9がべ ンジル才キシカルボニル基等の加水素分解反応により脱保護される基の場合は、 パラジウム等の金属触媒を用いた加水素分解反応により脱保護することができ る。 溶媒としては、 エタノール、 メタノール、 テ卜ラヒドロフラン、 酢酸ェチ ル等の反応に関与しない溶媒を用いることができる。 反応温度は 0〜 1 0 0 °C で行うことができる。 また、 この反応に水素ガスを用いることもできるし、 ぎ 酸—ぎ酸アンモニゥ厶を例とする試薬の組み合わせで行うこともできる。 更に 例えば、 R bが塩基で脱保護されるフル才レニル才キシカルボニル基等の保護 基の場合は、 ジェチルァミン、 ピぺリジン、 アンモニア、 水酸化ナトリウム、 炭酸カリウム等の塩基を用いて脱保護することができる。 これらの塩基は、 単 独で、 あるいは溶媒に希釈又は懸濁して用いることができる。 この際、 溶媒と
しては水、 エタノール、 メタノール、 テ卜ラヒドロフラン、 N, N—ジメチル ホルムアミド、 ジクロロメタン、 クロ口ホルム、 1, 2—ジクロロエタン等を 用いることができる。 反応温度は 0〜1 0 0 °Cで行うことができる。 更に例え ば、 X9がァリル才キ カルボニル基等の金属触媒により脱保護される基の場合 は、 テトラキス (卜リフエニルホスフィン) パラジウム等を触媒又は試薬とし て用いることにより脱保護することができる。 この際、 ジクロロメタン、 クロ 口ホルム、 テ卜ラヒドロフラン等の反応に関与しない溶媒中で行うことができ る。 反応温度は 0〜 1 0 0 °Cで行うことができる。 In this step, deprotection is performed using a compound (XIV) having an amino group protected by a ring A substituent to obtain an amine derivative (XV). For this deprotection, a method described in PROTECTIVE IVE GROUPS IN ORGA IC SYNTHES IS, THEODORA W. GREENE and PETER G. M WUTS can be used. For example, when X b is a group that can be deprotected with an acid such as a tert-butoxycarbonyl group, a trityl group, or a 0-2-nitrobenzenesulfenyl group, hydrochloric acid, sulfuric acid, trifluroacetic acid, p- Deprotection can be performed using an acid such as toluenesulfonic acid or methanesulfonic acid. At this time, the reaction can be carried out by diluting or dissolving with an organic solvent or water, and the reaction can be carried out at a temperature of 150 ° C to 50 ° C. Examples of the organic solvent include ethanol, methanol, tetrahydrofuran, N, N-dimethylformamide, dichloromethane, chloroform, 1,2-dichloroethane, and the like. Further, for example, when X 9 is a group that can be deprotected by a hydrogenolysis reaction such as a benzyloxycarbonyl group, it can be deprotected by a hydrogenolysis reaction using a metal catalyst such as palladium. As the solvent, a solvent that does not participate in the reaction, such as ethanol, methanol, tetrahydrofuran, and ethyl acetate can be used. The reaction can be carried out at a temperature of 0 to 100 ° C. In addition, hydrogen gas can be used for this reaction, or a combination of reagents such as formic acid-ammonium formate can be used. Further, for example, in the case where R b is a protecting group such as a fluorinated benzyl group which is deprotected with a base, deprotection is performed using a base such as getylamine, piperidine, ammonia, sodium hydroxide, potassium carbonate, etc. be able to. These bases can be used alone or diluted or suspended in a solvent. At this time, the solvent and For example, water, ethanol, methanol, tetrahydrofuran, N, N-dimethylformamide, dichloromethane, chloroform, 1,2-dichloroethane, and the like can be used. The reaction can be carried out at a temperature of 0 to 100 ° C. Further, for example, when X 9 is a group which can be deprotected by a metal catalyst such as an arylcarbonyl group, it can be deprotected by using tetrakis (triphenylphosphine) palladium as a catalyst or a reagent. . At this time, the reaction can be carried out in a solvent which does not participate in the reaction, such as dichloromethane, chloroform, tetrahydrofuran and the like. The reaction can be carried out at a temperature of 0 to 100 ° C.
[反応式 8 ] [Reaction formula 8]
(式中、 R\ R2、 R3、 R5、 R6、 R7および R8は前記と同意義である。 Xeは、 水素原子または炭素数 1〜5のアルキル基を表す。 ) (Wherein, R \ R 2 , R 3 , R 5 , R 6 , R 7 and R 8 are as defined above. X e represents a hydrogen atom or an alkyl group having 1 to 5 carbon atoms.)
この工程は、 化合物 (XV I ) を用いて還元反応を行い、 ヒドロキシメチル体 (XV I I ) を得る工程である。 この反応は、 適当な還元法を用いた条件下で反応 を行うことができる。 用いる還元法としては、 例えば水素化リチウムアルミ二 ゥ厶を用いる方法があげられる。 用いる溶媒としては、 テ卜ラヒドロフラン、 ジ才キサン等の反応に関与しない溶媒を用いることができる。 反応は一 20〜 1 00°Cで行うことができる。 In this step, a reduction reaction is performed using the compound (XVI) to obtain a hydroxymethyl form (XVII). This reaction can be performed under conditions using an appropriate reduction method. Examples of the reduction method used include a method using lithium aluminum hydride. As a solvent to be used, a solvent that does not participate in the reaction, such as tetrahydrofuran or dioxane, can be used. The reaction can be carried out at between 120 and 100 ° C.
(式中、 R1, R2、 R3、 R4、 R5、 R6'、 R7および R8は前記と同意義である。 Xdは、 ,〜 アルコキシカルボ二ル基を表す。 ) (Wherein, R 1 , R 2 , R 3 , R 4 , R 5 , R 6 ′, R 7 and R 8 have the same meanings as described above. X d represents a, ア ル コ キ シ alkoxycarbonyl group. )
この工程は、 化合物 (XVI I I ) を用いて還元反応を行い、 直接 (X I X) を得る 工程、 及び還元されて得られた化合物を用いて更に変換反応を行い (XI X) を 得る工程である。 この還元反応は、 適当な還元法を用いた条件下で反応を行う ことができる。 用いる還元法としては、 例えば水素化リチウムアルミニウムを 用いる方法があげられる。 用いる溶媒としては、 テ卜ラヒドロフラン、 ジ才キ サン等の反応に関与しない溶媒を用いることができる'。 反応は— 20〜100°Cで 行うことができる。 本発明の化合物は、 後述の実施例により示されるとおり、 11 i8 - HSD1活性阻 害作用を有する。 したがって、 本発明の化合物は、 11 i8 -HSD1 の関与する疾患、 例えば、 肝臓における糖新生抑制、 又は内臓脂肪の蓄積抑制に有効に使用でき る。 すなわち、 本発明の化合物は、 11 )8 -HSD1の阻害剤;肝臓における糖新生 抑制、 又は内臓脂肪の蓄積抑制などの医薬として利用できる。 本発明の化合物 は、 単独又は薬学的あるいは薬剤学的に許容される担体又は希釈剤と共に投与 することができる。 本発明の化合物を 11 3 - HSD1阻害剤などとして使用する場 合は、 本発明の化合物をそのまま経口投与、 又は非経口投与してもよい。 また、 本発明の化合物を有効成分として含む剤として経口投与、 又は非経口投与して もよい。 非経口投与としては、 注射による静脈内投与があげられる。 This step is a step of performing a reduction reaction using the compound (XVI II) to directly obtain (XIX), and a step of further performing a conversion reaction using the compound obtained by reduction to obtain (XI X). . This reduction reaction can be performed under conditions using an appropriate reduction method. Examples of the reduction method used include a method using lithium aluminum hydride. As a solvent to be used, a solvent which does not participate in the reaction such as tetrahydrofuran and dioxane can be used. The reaction can be carried out at —20-100 ° C. The compounds of the present invention have an activity of inhibiting 11 i8 -HSD1 activity, as shown in the Examples below. Therefore, the compound of the present invention can be effectively used for diseases associated with 11 i8 -HSD1, for example, suppression of gluconeogenesis in the liver or suppression of accumulation of visceral fat. That is, the compound of the present invention can be used as a drug for 11) an inhibitor of 8-HSD1; suppression of gluconeogenesis in the liver, or suppression of accumulation of visceral fat. The compounds of the present invention can be administered alone or with a pharmaceutically or pharmaceutically acceptable carrier or diluent. When the compound of the present invention is used as a 113-HSD1 inhibitor or the like, the compound of the present invention may be directly administered orally or parenterally. Further, the compound of the present invention may be orally or parenterally administered as an agent containing the compound as an active ingredient. Parenteral administration includes intravenous administration by injection.
上記の剤を経口投与する場合は、 希釈剤、 賦形剤、 崩壊剤、 結合剤、 滑沢剤、 抗酸化剤、 コーティング剤、 界面活性剤、 可塑剤、 着色剤、 矯味矯臭剤などを 混合して、 本発明の化合物を有効成分として含む顆粒剤、 カプセル剤、 錠剤、 薬用ドロップ、 卜ローチ、 硬質キャンディ、 粉末剤、 噴霧剤、 などの製剤とし て投与されてもよい。 また、 適宜に甘味付け、 又は香味付けを行っても良い。 上記の剤を非経口投与する場合は、 本発明の化合物を有効成分として含む注射 剤、 点滴剤、 点眼剤、 クリーム、 膏薬、 坐薬、 ゼリー、 ジエル、 ペース卜、 口 ーシヨン、 軟膏、 水性懸濁液などの製剤として投与されてもよい。 製剤化する 際には、 通常の製剤化の方法を使用できる。
本発明の化合物は経口投与又は非経口投与でき、 例えば 1回につき 1mg〜 1000mg、 好ましくは 10mg〜200mg投与でき、 例えば 1 日当り 1回〜 3回投与す ればよい。 本発明の化合物の投与量は、 患者の年齢、 体重及び症状によって適 宜調整することができる。 本発明の化合物の 11 )8 -HSD1活性阻害を評価するには、 例えば、 実施例に記 載した方法など、 公知の手法に従つて行なうことができる。 以下に、 参考例、 実施例、 及び試験例を示して本発明を具体的に説明する。 しかしながら、 本発明は以下の実施例などに限られるものではない。 When the above agents are administered orally, diluents, excipients, disintegrants, binders, lubricants, antioxidants, coating agents, surfactants, plasticizers, coloring agents, flavoring agents, etc. are mixed. Then, they may be administered as preparations such as granules, capsules, tablets, medicinal drops, troches, hard candy, powders, sprays, etc., containing the compound of the present invention as an active ingredient. In addition, sweetening or flavoring may be performed as appropriate. When the above-mentioned preparations are to be administered parenterally, injections, drops, eye drops, creams, salves, suppositories, jellies, jewels, pastes, mouth lotions, ointments, aqueous suspensions containing the compound of the present invention as an active ingredient It may be administered as a formulation such as a liquid. In formulating, a usual formulation method can be used. The compound of the present invention can be administered orally or parenterally, for example, 1 mg to 1000 mg, preferably 10 mg to 200 mg at a time, for example, once to three times a day. The dose of the compound of the present invention can be appropriately adjusted according to the age, weight and condition of the patient. In order to evaluate the 11) 8-HSD1 activity inhibition of the compound of the present invention, for example, a known method such as the method described in Examples can be used. Hereinafter, the present invention will be specifically described with reference to Reference Examples, Examples, and Test Examples. However, the present invention is not limited to the following examples.
(参考例 1 ) 4 - (1 -ァダマンチル) 一 2—ァミノチアゾ一ル臭化水素 酸塩の合成 Reference Example 1 Synthesis of 4- (1-adamantyl) -12-aminothiazol hydrobromide
1ーァダマンチルプロモメチルケトン (4. 9 3g) をエタノール (1 1 0 ml) に溶解し、 チ才ゥレア (1 . 4 6g) を加え、 室温で 2時間攪拌した。 反 応液を減圧下濃縮し、 得られた残渣にィソプロピルエーテルとエタノールの 1 0 : 1混合溶媒 (60tn l ) を加え、 懸濁させて 2時間攪拌した。 無色粉末と して表題化合物 (6. 0 6 g) を得た。 ^MRの結果は、 以下のとおりであつ た。 1-adamantyl bromomethyl ketone (4.93 g) was dissolved in ethanol (110 ml), to which was added, and stirred at room temperature for 2 hours. The reaction solution was concentrated under reduced pressure, and a 10: 1 mixed solvent (60 tnl) of isopropyl ether and ethanol was added to the obtained residue, suspended, and stirred for 2 hours. The title compound (6.06 g) was obtained as a colorless powder. ^ MR results were as follows.
1H-隱 (300MHz, 画 - d6) δ: 8.74 (2Η, brs), 6.43(1 H, s), 2.08- 1.98 (3H, rn), 1.88-1.60 (12H, m) 1H-Oki (300MHz, drawing-d6) δ: 8.74 (2Η, brs), 6.43 (1 H, s), 2.08-1.98 (3H, rn), 1.88-1.60 (12H, m)
(参考例 2 ) 4 - (1 -ァダマンチル) — 2—ァミノチアゾールの合成 参考例 1と同様の方法で合成した 4一 (1 -ァダマンチル) 一 2—アミノチ ァゾール臭化水素酸塩 (700mg) を 1M水酸化ナトリウム水溶液 (80 ml) に懸濁させ、 酢酸ェチル (200m l X 3) で抽出した。 集めた有機相を 無水硫酸マグネシウムで乾燥し、 乾燥剤を濾去し、 溶媒を減圧留去した。 無色 アモルファスとして表題化合物 (484mg) を得た。 - Rの結果は、 以下 のとおりであった。
,H-匿 ( 300 MHz, 隨 - d6 ) δ : 6.78 (2H, brs), 6.00 ( 1H, s), 2.10-1.98 (3H, m), 1.83-1.58 (12H, m) (Reference Example 2) Synthesis of 4- (1-adamantyl) — 2-aminothiazole 41- (1-adamantyl) -12-aminothiazole hydrobromide (700 mg) synthesized in the same manner as in Reference Example 1. Was suspended in 1M aqueous sodium hydroxide solution (80 ml) and extracted with ethyl acetate (200 ml × 3). The collected organic phase was dried over anhydrous magnesium sulfate, the desiccant was removed by filtration, and the solvent was distilled off under reduced pressure. The title compound (484 mg) was obtained as a colorless amorphous. -The results of R were as follows. , H-concealed (300 MHz, optional-d6) δ: 6.78 (2H, brs), 6.00 (1H, s), 2.10-1.98 (3H, m), 1.83-1.58 (12H, m)
(参考例 3 ) 4一 (1 -ァダマンチル) 一 2— (メチルァミノ) チアゾー ルの合成 (Reference Example 3) Synthesis of 4- (1-adamantyl) 1-2- (methylamino) thiazole
1—ァダマンチルプロモメチルケトン (4. 93g) をエタノール (58ml) に懸濁し、 N—メチルチオウレァ (Ί . 73g) を加え、 室温で 0. 5時間攪 拌後、 イソプロピルエーテル(6 Oml)を加えて室温で更に 1. 5時間攪拌した。 析出した固体を吸引濾取して回収し、 無色粉末として表題化合物の臭化水素酸 塩 (3. 95 g) を得た。 母液から同様に 0. 5 1 g得た。 続いて、 これらの 固体を合わせて、 水 (1 70ml) に懸濁させ、 炭酸水素ナ卜リウ厶 (1. 6 1 g) を加えて室温で 2時間攪拌した。 固体を吸引濾取し、 水 (200ml) で洗 浄し、 無色粉末として表題化合物 (3. 04 g) を得た。 -NMRの結果は、 以 下のとおりであった。 1-adamantyl bromomethyl ketone (4.93 g) is suspended in ethanol (58 ml), N-methylthiourea (Ί.73 g) is added, and the mixture is stirred at room temperature for 0.5 hour, and then isopropyl ether (6 Oml) is added. In addition, the mixture was further stirred at room temperature for 1.5 hours. The precipitated solid was collected by suction filtration to give the title compound hydrobromide (3.95 g) as a colorless powder. Similarly, 0.51 g was obtained from the mother liquor. Subsequently, these solids were combined, suspended in water (170 ml), added with sodium hydrogen carbonate (1.61 g), and stirred at room temperature for 2 hours. The solid was collected by suction filtration and washed with water (200 ml) to give the title compound (3.04 g) as a colorless powder. The results of -NMR were as follows.
】Η-隱 ( 300 MHz, DMS0 ― d6) δ: 7, 34-7.24 (1 H, m), 6.08 (1H, ) Η-Oki (300 MHz, DMS0-d6) δ: 7, 34-7.24 (1 H, m), 6.08 (1H,
s) , 2.75 (3H, d, J=4.8Hz), 2.02-1.95 (3H, m), 1.85-1.79 (6H, m), 1.76-1.62 (6H, m) 実施例 1 , s), 2.75 (3H, d, J = 4.8Hz), 2.02-1.95 (3H, m), 1.85-1.79 (6H, m), 1.76-1.62 (6H, m)
4- (1 -ァダマンチル) —2— (1 -ナフチルスルホニルァミノ) チアゾ ールの合成 Synthesis of 4- (1 -adamantyl) —2— (1-naphthylsulfonylamino) thiazole
4— (1 -ァダマンチル) —2—ァミノチアゾ一ル臭化水素酸塩 (200m g) をピリジン (1. 2ml) に溶解し、 氷冷下で 1一ナフタレンスルホニル クロリド (1 44mg) を加え、 その後、 室温に戻して一晩攪拌した。 反応液 を氷冷した 6 M塩酸水溶液 (1 Om l ) に滴下して、 2時間攪拌後、 析出した 固体を濾取し、 6 M塩酸水溶液 (1 Om I ) 、 水 (1 Om I ) で洗浄し、 乾燥 して薄茶色固体として表題化合物 (225 mg) を得た。 MMRの結果は、 以 下のとぉリであった。
〕H-腫 ( 300 MHz, D SO - d6 ) δ: 12.66 (1H, brs), 8.75 (1H, dd, J =8.3, 0.8Hz) , 8.22-8.14 ( 2H, m), 8.08-8.03 (1H, m), 7.73-7.59 (3H, m), 6.30 (1H, s), 2.04-1.88 (3H, m) , 1.82-1.54 (12H, m) 4- (1-Adamantyl) -2-aminothiazol hydrobromide (200 mg) was dissolved in pyridine (1.2 ml), and under ice-cooling, 1-naphthalenesulfonyl chloride (144 mg) was added. The mixture was returned to room temperature and stirred overnight. The reaction mixture was added dropwise to an ice-cooled 6 M aqueous hydrochloric acid solution (1 OmI), and after stirring for 2 hours, the precipitated solid was collected by filtration and washed with a 6 M aqueous hydrochloric acid solution (1 OmI) and water (1 OmI). After washing and drying, the title compound (225 mg) was obtained as a light brown solid. The results of the MMR were as follows. H-tumor (300 MHz, DSO-d6) δ: 12.66 (1H, brs), 8.75 (1H, dd, J = 8.3, 0.8Hz), 8.22-8.14 (2H, m), 8.08-8.03 (1H , m), 7.73-7.59 (3H, m), 6.30 (1H, s), 2.04-1.88 (3H, m), 1.82-1.54 (12H, m)
実施例 2 Example 2
4一 (1 -ァダマンチル) —2— (2, 4, 5—トリクロ口フエニルスルホニ ルァミノ) チアゾールの合成 Synthesis of 4- (1-adamantyl) -2- (2,4,5-trichloromethylphenylsulfonylamino) thiazole
実施例 1と同様の方法で 4— (1 -ァダマンチル) ー2—ァミノチアゾール 臭化水素酸塩 (20 Omg) と 2, 4, 5 - 卜リクロロベンゼンスルホニルク ロリド (1 78mg) から、 薄桃色固体として表題化合物 (Ί 1 mg) を得た。 In the same manner as in Example 1, 4- (1-adamantyl) -2-aminothiazole hydrobromide (20 Omg) and 2,4,5-trichlorobenzenesulfonyl chloride (178 mg) were thinly diluted. The title compound (Ί1 mg) was obtained as a pink solid.
Rの結果は、 以下のとおりであった。 The results of R were as follows.
1H-隱 ( 300 MHz, 睡 - d6 ) δ 12.99 (1Η, brs), 8.14 (1H, s), 8.07 (1H, s), 6.41 (1H, s), 2.04-1.95 (3H, m), 1.84-1.76 (6H, tn) , 1.74-1.58 (6H, m) 1 H-Oki (300 MHz, sleep-d6) δ 12.99 (1Η, brs), 8.14 (1H, s), 8.07 (1H, s), 6.41 (1H, s), 2.04-1.95 (3H, m), 1.84-1.76 (6H, tn), 1.74-1.58 (6H, m)
実施例 3 Example 3
4一 (1 -ァダマンチル) 一 2— [ (2—チェニルスルホニル) ァミノ]チア ゾールの合成 Synthesis of 4- (1-adamantyl) -1-2-[(2-Chenylsulfonyl) amino] thiazole
実施例 1 と同様の方法で 4一 (1 -ァダマンチル) 一 2—ァミノチアゾール 臭化水素酸塩 (20 Omg) と 2 -チ才スエンスルホニルクロリド (1 1 6m g) から、 薄茶色粉末として表題化合物 (9 Omg) を得た。 NMRの結果は、 以下のとおりであった。 In the same manner as in Example 1, 4- (1-adamantyl) -12-aminothiazole hydrobromide (20 Omg) and 2-thiensuensulfonyl chloride (116 mg) were obtained as a light brown powder. The title compound (9 Omg) was obtained. The result of NMR was as follows.
1H -隱 ( 300 MHz, DMS0 - d6 ) δ 12.84 (1Η, brs), 7.82 (1H, dd, J =5.0, 1.4Hz), 7.55 ( 1H, dd, J = 3.7, 1.4 Hz), 7.11 (1H, dd, J = 5.0, 3.7 Hz), 6.40 (1H, s), 2.10-1.94 (3H, ml, 1.85-1.54 (12H, m) 1 H-Odd (300 MHz, DMS0-d6) δ 12.84 (1Η, brs), 7.82 (1H, dd, J = 5.0, 1.4 Hz), 7.55 (1H, dd, J = 3.7, 1.4 Hz), 7.11 ( 1H, dd, J = 5.0, 3.7 Hz), 6.40 (1H, s), 2.10-1.94 (3H, ml, 1.85-1.54 (12H, m)
実施例 4 Example 4
4一 (1 -ァダマンチル) 一 2— (4一ブロモフエニルスルホニルァミノ) チアゾールの合成 Synthesis of 4- (1-adamantyl) -1-2- (4-bromophenylsulfonylamino) thiazole
4— (1 -ァダマンチル) —2—ァミノチアゾ一ル臭化水素酸塩 (200m g) をピリジン (1. 2m l ) に溶解し、 4ージメチルァミノピリジン (7. 7mg) を加えた後、 氷冷下で 4一プロモベンゼンスルホニルクロリド (1 7
8mg) を加え、 その後、 室温に戻して一晚攪拌した。 反応液を氷冷した 6 M 塩酸水溶液 (1 Om l ) に滴下して、 2時間攪拌後、 析出した固体を濾取し、 6 M塩酸水溶液 ( 1 0 m I ) 、 水 ( 1 0 m I ) で洗浄し、 乾燥して無色粉末の 表題化合物 (239mg) を得た。 Rの結果は、 以下のとおりであった。 4- (1-adamantyl) -2-aminothiazol hydrobromide (200 mg) was dissolved in pyridine (1.2 ml), and 4-dimethylaminopyridine (7.7 mg) was added. Under ice-cooling 4-promobenzenesulfonyl chloride (17 8 mg), and then returned to room temperature and stirred for a while. The reaction mixture was added dropwise to an ice-cooled 6 M aqueous hydrochloric acid solution (1 Oml). After stirring for 2 hours, the precipitated solid was collected by filtration, and a 6 M aqueous hydrochloric acid solution (10 mI) and water (10 mI) were added. ) And dried to give the title compound (239 mg) as a colorless powder. The results of R were as follows.
1H-隱 ( 300 MHz, DMS0 - d6 ) δ 12.78ΠΗ, brs), 7.79-7.70 (4H, m) , 6.36 (ΙΗ' s), 2.02-1.94 (3Η, m), 1.84-1.58 (12Η, m) 1H-Oki (300 MHz, DMS0-d6) δ 12.78ΠΗ, brs), 7.79-7.70 (4H, m), 6.36 (ΙΗ's), 2.02-1.94 (3Η, m), 1.84-1.58 (12Η, m )
(実施例 4 _ 1〜実施例 4一 23) (Example 4_1 to Example 4-1 23)
以下、 実施例 4と同様の方法で、 化合物 4一 1〜化合物 4—23を得た。 これ らの化合物の構造式と、 得られた1 H- NMRの結果を表 1に示す。 化合物 4— 1〜 化合物 4一 23は、 以下のとおりである。 化合物 4一 1は、 4_ (1 -ァダマン チル) 一 2— (2, 4, 6—卜リメチルフエニルスルホニルァミノ) チアゾー ルであり、 化合物 4一 2は、 4 - (1 -ァダマンチル) - 2 - (3—トリフル 才ロメチルフエニルスルホニルァミノ) チアゾールであり、 化合物 4一 3は、 4一 (1 -ァダマンチル) 一 2— (4—プロピルフエニルスルホニルァミノ) チアゾールであり、 化合物 4— 4は、 4一 (1 -ァダマンチル) 一 2— (2— メチルフエニルスルホニルァミノ) チアゾールであり、 化合物 4一 5は、 4一 (1 -ァダマンチル) 一 2— (4一フル才ロフエニルスルホニルァミノ) チア ゾ一ルであり、 化合物 4一 6は、 4一 (1 ァダマンチル) 一 2— (2, 5— ジメチルフエニルスルホニルァミノ) チアゾ一ルであり、 化合物 4一 7は、 4 - (1 -ァダマンチル) 一 2— (ビフエニルスルホニルァミノ) チアゾールで あり、 化合物 4— 8は、 4— (1 -ァダマンチル) 一 2— (4一クロ口, 2, 5—ジメチルフエニルスルホニルァミノ) チアゾールであり、 化合物 4— 9は、 4- (1 -ァダマンチル) 一 2— (3, 5—ジメチルイソ才キサゾールスルホ ニルァミノ) チアゾールであり、 化合物 4— 10は、 4_ (1 -ァダマンチル) 一 2— (5—クロロー 3—メチル—ベンゾ [b] チ才フェン一 2ィルースルホ ニルァミノ) チアゾールであり、 化合物 4一 11は、 4一 (1 -ァダマンチル) —2— (2, 5—ジクロロフエニルスルホニルァミノ) チアゾ一ルであり、 ィ匕 合物 4一 ί2は、 4一 (1 -ァダマンチル) —2— (4—ニトロフエニルスルホ ニルァミノ) チアゾールであり、 ィ匕合物 4一 13は、 4— (1 -ァダマンチル)
—2— (4ーメ卜キシフエニルスルホニルァミノ) チアゾールであり、 化合物 4一〗 4は、 4 - (1 -ァダマンチル) -2- (2, 4ージメ卜キシフエニルス ルホニルァミノ) チアゾールであり、 化合物 4一 15は、 4一 (1 -ァダマンチ ル) -2- (3—二卜口フエニルスルホニルァミノ) チアゾ一ルであり、 化合 物 4— 16は、 N— [4— (4—ァダマンタン— 1 —ィル—チアゾ一ルー 2—ィ ルースルファモイル) 一フエニル]一ァセ卜アミドであり、 化合物 4—17は、 4一 (1 -ァダマンチル) 一 2— (4一クロ口フエニルスルホニルァミノ) チ ァゾールであり、 化合物 4一 18は、 4一 (1 -ァダマンチル) —2— (4ーフ エノキシフエニルスルホニルァミノ) チアゾールであり、 化合物 4一 19は、 4一 (1 -ァダマンチル) —2— (4一ブチルフエニルスルホニルァミノ) チ ァゾールであり、 化合物 4— 20は、 4— (1 -ァダマンチル) - 2 - (1 , 1ージメチルプロピルフエニルスルホニルァミノ) チアゾールであり、 化合物 4一 21は、 4 - (1 -ァダマンチル) - 2 - (ベンゾ [b] チ才フェン一 2ィ ル—スルホニルァミノ) チアゾールであり、 ィ匕合物 4— 22は、 4_ (1 -ァ ダマンチル) 一 2— (ベンゾ [b] チ才フェン一 3ィルースルホニルァミノ) チアゾールであり、 化合物 4— 23は、 2— (ァダマンタン一 1ーィルーチア ゾールー 2ィルースルファモイル) 一ベンゼン酸メチルエステルであり、 化合 物 4— 24は、 4— (ァダマンタン一 1一,ィル—チアゾール— 2ィルースルフ ァモイル) —ベンゼン酸であり、 そして化合物 4— 25は、 4一 (1 -ァダマ ンチル) —2— (ベンゾ [b] フラン— 2—ィルースルホニルァミノ) チアゾ ールである。 Thereafter, Compound 41-11 to Compound 4-23 were obtained in the same manner as in Example 4. Table 1 shows the structural formulas of these compounds and the obtained 1 H-NMR results. Compounds 4-1 to 4-1-23 are as follows. Compound 4-1 is 4_ (1-adamantyl) 1-2- (2,4,6-trimethylphenylsulfonylamino) thiazole, and compound 4-2 is 4- (1-adamantyl) -2--(3-trifluoromethylphenylsulfonylamino) thiazole; compound 4-1-3 is 4- (1-adamantyl) -12- (4-propylphenylsulfonylamino) thiazole; Compound 4-4 is 4- (1-adamantyl) 1-2- (2-methylphenylsulfonylamino) thiazole, and compound 4-1-5 is 4- (1-adamantyl) 1-2- (4-full Compound 41 is 6- (1 adamantyl) 12- (2,5-dimethylphenylsulfonylamino) thiazole, and Compound 41 is Compound 41. 7 is 4-(1-adamantyl) 1 2-(bihue Compound 4-8 is 4- (1-adamantyl) -12- (4 mono-, 2,5-dimethylphenylsulfonylamino) thiazole and compound 4-9 Is 4- (1-adamantyl) 1-2- (3,5-dimethylisoxazolesulfonylamino) thiazole, and compound 4-10 is 4_ (1-adamantyl) 1-2- (5-chloro-3-methyl —Benzo [b] thylphen-2-ylsulfonylamino) thiazole, compound 411 is 4- (1-adamantyl) —2— (2,5-dichlorophenylsulfonylamino) thiazole , 匕 合 物 4 ί 、 、 、 4 4 、 4 4 4 物 4 物 4 物 物 物 4 4 4 4 4 物 物 4 4 物 物 物 4 4 4 物 物 物-— 物 物 物 物 4 4 4 4 4-物 — 物 物 4 4 4 4 4. —2— (4-Methoxyoxysulfonylamino) thiazole, Compound 414 is 4- (1-adamantyl) -2- (2,4 dimethyloxyphenylsulfonylamino) thiazole, compound 4-15 is 4- (1-adamantyl) -2- (3-nitrophenylphenylsulfonylamino) thiazole, and compound 4-16 is N- [4- (4-adamantane). — 1 —yl-thiazo-l-2-l-l-sulfamoyl) -phenyl] -l-acetamide; compound 4-17 is 4-l- (1-adamantyl) -l 2- Sulfonylamino) thiazole, Compound 418 is 4-1 (1-adamantyl) —2— (4-phenoxyphenylsulfonylamino) thiazole, and Compound 419 is 4-1 (1 -Adamantly) —2— (4-butylphenylsulfonylamino) Compound 4-20 is 4- (1-adamantyl) -2- (1,1-dimethylpropylphenylsulfonylamino) thiazole; compound 4-1 21 is 4- (1-adamantyl) -2-(Benzo [b] thylphen-2yl-sulfonylamino) is a thiazole. Phen-1-ylsulfonylamino) thiazole, compound 4-23 is 2- (adamantane-1-ylutiazol-2-ylsulfamoyl) monobenzene methyl ester, and compound 4-24 is 4- (Adamantane-11, yl-thiazole-2-ylsulfamoyl) is a benzene acid, and compound 4-25 is 4- (1-adamantyl) -2- (benzo [b] furan-2-yl Sulfonylamino) thiazo Is Le.
表 1. 化合物 4一 1〜 4一 25の化合物の構造式と、 得られた Rの結果
Table 1. Structural formulas of compounds 411 to 425 and the obtained R results
l.00/S00idf/X3d f9Z.Z.60/S00Z OAV
12.98(1 H,brs),8.05— l.00 / S00idf / X3d f9Z.Z.60 / S00Z OAV 12.98 (1H, brs), 8.05-
7.97(2H,m),7.96(1H,s),7.53- 7.97 (2H, m), 7.96 (1H, s), 7.53-
4-21 7.43(2H,m),6.43(1H,s),2.00-4-21 7.43 (2H, m), 6.43 (1H, s), 2.00-
1.92(3H,m),1.80-1.73(6H,m),1.72-1.92 (3H, m), 1.80-1.73 (6H, m), 1.72-
1.57(6H,m). 1.57 (6H, m).
12.74(1 H,brs),8.45(1H,s),8.18- 8.13(1 H,m),8.11-8.06(1 H,m),7.55- 12.74 (1H, brs), 8.45 (1H, s), 8.18-8.13 (1H, m), 8.11-8.06 (1H, m), 7.55-
4-22 oo二: zェ 7.43(2H,m),6.36(1H,s),2.00- - 1.92(3H,m),1.80-1 J3(6H,m),1.72- 1.57(6H,m). 4-22 oo2: z 7.43 (2H, m), 6.36 (1H, s), 2.00--1.92 (3H, m), 1.80-1 J3 (6H, m), 1.72-1.57 (6H, m) .
12.69 (1H, br s), 7.95― 7.91 (1H, m), 7.69 - 7.62 (2H, m), 7.54― 7.51 (1H, m), 6.36 (1H, 12.69 (1H, br s), 7.95-7.91 (1H, m), 7.69-7.62 (2H, m), 7.54-7.51 (1H, m), 6.36 (1H,
4-23 4-23
s), 3.73 (3H, s), 2.10一 1.90 (3H, m), 1.85 - 1.75 (6H, m), 1.72一 1.60 (6H, m). s), 3.73 (3H, s), 2.10-1.90 (3H, m), 1.85-1.75 (6H, m), 1.72-1-1.60 (6H, m).
8.03 (2H, d, J=8.5Hz), 7.70 (2H, d, 8.03 (2H, d, J = 8.5Hz), 7.70 (2H, d,
4-24 J=8.5Hz), 6.78 (1H, s), 2.09 - 1.89 (9H, m), 4-24 J = 8.5Hz), 6.78 (1H, s), 2.09-1.89 (9H, m),
1.81-1.62 (6H, m). 1.81-1.62 (6H, m).
13.06 (1H, br s), 7.81-7.74 (1H, m), 7.71- 7.64 (2H, m), 7.52-7.44 (2H, m), 7.40-7.32 13.06 (1H, br s), 7.81-7.74 (1H, m), 7.71- 7.64 (2H, m), 7.52-7.44 (2H, m), 7.40-7.32
4-25 rsH° (1H, m), 6.48-6.45 (1H, m), 2.05 - 1.56 (15H, m). 4-25 r s H ° (1H, m), 6.48-6.45 (1H, m), 2.05-1.56 (15H, m).
実施例 5 , Example 5,
4一 (1 -ァダマンチル) 一 2— (3—クロロー 2—メチルフエニルスルホ ニルァミノ) チアゾールの合成 Synthesis of 4- (1-adamantyl) -2- (3-chloro-2-methylphenylsulfonylanamino) thiazole
4 - (1 -ァダマンチル) 一 2—ァミノチアゾール (20 Omg) をピリジ ン (1. 2ml) に溶解し、 氷冷下で 3—クロロー 2—メチルベンゼンスルホ ニルクロリド (2 1 3mg) を加え、 その後、 室温に戻して一晩攪拌した。 反 応液を氷冷した 6 M塩酸水溶液 (1 Om l ) に滴下して、 2時間攪拌後、 析出 した固体を濾取し、 6 M塩酸水溶液 (1 0m I ) 、 水 (1 0m I ) で洗浄した。 粗結晶をシリカゲルカラムクロマトグラフィー (展開溶媒 クロ口ホルム:メ 夕ノール = 1 00 : 1 ) で精製し、 薄茶色アモルファスとして表題化合物 (3 09mg) を得た。 1H- NMRの結果は、 以下のとおりであった。
1H-隱 ( 300 MHz, DMSO - d6 ) 6: 12.73 (1H, brs), 7.91 (1H, dd, J = 8.0, 1.2 Hz), 7.68 (1H, dd, J = 8.0, 1.2 Hz), 7.39 (1H, t, J = 8.0 Hz) , 6.33 (1H, s), 2.65 (3H, s), 2.0 4- (1-Adamantyl) -l- 2-Aminothiazole (20 Omg) is dissolved in pyridin (1.2 ml), and under ice-cooling, 3-chloro-2-methylbenzenesulfonyl chloride (213 mg) is added. Thereafter, the mixture was returned to room temperature and stirred overnight. The reaction solution was added dropwise to an ice-cooled 6 M aqueous hydrochloric acid solution (1 Oml). After stirring for 2 hours, the precipitated solid was collected by filtration, and a 6 M aqueous hydrochloric acid solution (10 mI) and water (10 mI) were added. And washed. The crude crystals were purified by silica gel column chromatography (developing solvent, solvent: chloroform: 100: 1) to give the title compound (309 mg) as a pale brown amorphous. The result of 1 H-NMR was as follows. 1 H-Oki (300 MHz, DMSO-d6) 6: 12.73 (1H, brs), 7.91 (1H, dd, J = 8.0, 1.2 Hz), 7.68 (1H, dd, J = 8.0, 1.2 Hz), 7.39 (1H, t, J = 8.0 Hz), 6.33 (1H, s), 2.65 (3H, s), 2.0
2-1.95 (3H, m), 1.83-1.74 (6H, m) , 1.73-1.58 (6H, m) 2-1.95 (3H, m), 1.83-1.74 (6H, m), 1.73-1.58 (6H, m)
実施例 6 Example 6
4 - (1 -ァダマンチル) 一 2—(フエニルスルホニルァミノ)チアゾールの 合成 Synthesis of 4- (1-adamantyl) 1-2- (phenylsulfonylamino) thiazole
実施例 5と同様の方法で 4一 (1 -ァダマンチル) 一 2—ァミノチアゾール (7 Omg) とベンゼンスルホニルクロリド (53mg) から、 薄黄色ァモル ファスとして表題化合物 (64mg) を得た。 NMRの結果は、 以下のとおり であった。 In the same manner as in Example 5, the title compound (64 mg) was obtained as a pale yellow amorphus from 41- (1-adamantyl) -12-aminothiazole (7 Omg) and benzenesulfonyl chloride (53 mg). The results of NMR were as follows.
】H -賺 ( 300 MHz, DMS0 - d6 ) δ :12.69 (1Η, brs), 7.84-7.78 (2H, m), 7.64-7.50 ( 3H, m), 6, 32 (1H, s), 2.02-1.95 (3H, m), 1.83-1.74 (6H, m) , 1.73-1.58 (6H, m) H-new (300 MHz, DMS0-d6) δ: 12.69 (1Η, brs), 7.84-7.78 (2H, m), 7.64-7.50 (3H, m), 6, 32 (1H, s), 2.02- 1.95 (3H, m), 1.83-1.74 (6H, m), 1.73-1.58 (6H, m)
実施例 7 Example 7
4一 (1 -ァダマンチル) — 2— [ (5—プロモ一6—クロ口ピリジン一 3—ィ ル) スルホニルァミノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) — 2 — [(5-promo-6-chloropyridine-13-yl) sulfonylamino] thiazole
4一 (1 -ァダマンチル) 一 2—ァミノチアゾール臭化水素酸塩 (200m g) をクロ口ホルム (1. 2m l ) に溶解し、 室温で 4—ジメチルァミノピリ ジン ( 7. 7 m g ) を加えた後、 氷冷下で卜リェチルァミン Π 77 μ I ) と 3 _ブロモ一2—クロ口ピリジン一 5—スルホニルクロリド (24 Omg) を 加え、 その後、 室温に戻して一晚攪拌した。 更に 3—プロモー 2—クロ口ピリ ジン— 5—スルホニルクロライド (74mg) を加えて、 室温で 2日間攪拌し た。 水で冷却しながら反応液にクロ口ホルム (1 Om l ) と 0. 5%炭酸水素 ナトリウム水溶液 (1 0m l ) を加えて、 析出した結晶を濾取し、 無色粉末と して表題化合物 (5 Omg) を得た。 - NMRの結果は、 以下のとおりであった。 4 Dissolve 2- (1-adamantyl) -1-2-aminothiazole hydrobromide (200 mg) in chloroform (1.2 ml) and add 4-dimethylaminopyridine (7.7 mg) at room temperature. ), And triethylamine (77 μI) and 3_bromo-12-chloropyridine-15-sulfonyl chloride (24 Omg) were added under ice-cooling, and the mixture was returned to room temperature and stirred for a while. Further, 3-promo 2-cyclomouth pyridine-5-sulfonyl chloride (74 mg) was added, and the mixture was stirred at room temperature for 2 days. While cooling with water, the reaction mixture was added with chloroform (1 Oml) and a 0.5% aqueous sodium hydrogen carbonate solution (10 ml), and the precipitated crystals were collected by filtration to give the title compound (colorless powder). 5 Omg). -The result of NMR was as follows.
】H -隨 ( 300 MHz, 隱 - d6 ) <5 :13.01 (1H, brs), 8.78 (1H, d, J = 2.2 Hz), 8.46 (1H, d, J = 2.2 Hz) , 6.45 (1H, s) , 2.04-1.95 (3H, m) , 1.83-1.76 (6H, m) , 1.74-1.58 (6H, m)
実施例 8 ) H-free (300 MHz, Oki-d6) <5: 13.01 (1H, brs), 8.78 (1H, d, J = 2.2 Hz), 8.46 (1H, d, J = 2.2 Hz), 6.45 (1H, s), 2.04-1.95 (3H, m), 1.83-1.76 (6H, m), 1.74-1.58 (6H, m) Example 8
4- (1 -ァダマンチル) 一 2— [ (6 -モルホリノピリジン一 3—ィル) ス ルホニルァミノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) -1-2-[(6-morpholinopyridine-13-yl) sulfonylamino] thiazole
実施例 7と同様の方法で 4一 (1 -ァダマンチル) 一 2—ァミノチアゾール 臭化水素酸塩 (20 Omg) と 6 -モルホリノー 3 -ピリジンスルホニルクロ リド (366 mg) から、 無色粉末として表題化合物 (1 34mg) を得た。 1H-剛 Rの結果は、 以下のとおりであった。 In the same manner as in Example 7, the title compound was obtained as a colorless powder from 4- (1-adamantyl) -12-aminothiazole hydrobromide (20 mg) and 6-morpholinol 3-pyridinesulfonyl chloride (366 mg). The compound (134 mg) was obtained. Results of 1 H- Tsuyoshi R were as follows.
】H -賺 ( 300 MHz, D S0 - d6 ), <5 :12.64 (1H, brs), 8.47 (1H, d, J=2.5Hz) , 7.81 (1 H, dd, J=9.0, 2.5Hz) , 6.89 (1 H, d, J=9.0Hz) , 6.29 (1 H, s) , 3.70- 3.63 (4H, m) , 3.60-3.53 (4H, m) , 2.02-1.93 (3H, m) , 1.82-1.74 (6H, m) , 1.73- 1.58(6H, m) H-new (300 MHz, D S0-d6), <5: 12.64 (1H, brs), 8.47 (1H, d, J = 2.5 Hz), 7.81 (1 H, dd, J = 9.0, 2.5 Hz) , 6.89 (1 H, d, J = 9.0 Hz), 6.29 (1 H, s), 3.70- 3.63 (4H, m), 3.60-3.53 (4H, m), 2.02-1.93 (3H, m), 1.82 -1.74 (6H, m), 1.73- 1.58 (6H, m)
実施例 9 Example 9
4一 (1 -ァダマンチル) 一 2— [ (6 -フエノキシピリジン一 3—ィル) ス ルホニルァミノ] チアゾールの合成 ' 4-Synthesis of 1- (1-adamantyl) 1-2-[(6-Phenoxypyridine-13-yl) sulfonylamino] thiazole ''
実施例 7と同様の方法で 4一 (1 -ァダマンチル) —2—ァミノチアゾ一ル 臭化水素酸塩 (20 Omg) と 6 -フエノキシ一 3 -ピリジンスルホニルクロ リド (376 mg) から、 薄黄色粉末として表題化合物 (1 99mg) を得た。 A light yellow powder was obtained from 4- (1-adamantyl) -2-aminothiazol hydrobromide (20 mg) and 6-phenoxy-13-pyridinesulfonyl chloride (376 mg) in the same manner as in Example 7. As a result, the title compound (199 mg) was obtained.
^ -剛 Rの結果は、 以下のとおりであった。, ^ -Go R results were as follows: ,
1H-N R ( 300 MHz, DMS0 - d6) (5 :12.83 (1H, brs), 1H-N R (300 MHz, DMS0-d6) (5: 12.83 (1H, brs),
8.52 (1H, dd, J=2.5, 0.6Hz), 8.17(1H, dd, J=8.6, 2.5Hz) , 7.49-7.41 (2H, m) ,8.52 (1H, dd, J = 2.5, 0.6Hz), 8.17 (1H, dd, J = 8.6, 2.5Hz), 7.49-7.41 (2H, m),
7.30-7.23 (1H, m) , 7.22-7.17 (2H, m), 7.15 (1H, dd, J=8.6, 0.6Hz) , 7.30-7.23 (1H, m), 7.22-7.17 (2H, m), 7.15 (1H, dd, J = 8.6, 0.6Hz),
6.37 (1H, s), 2.02-1.93 (3H, m) , 1.82-1.74 (6H, m), 1.73-1.58 (6H, m) 6.37 (1H, s), 2.02-1.93 (3H, m), 1.82-1.74 (6H, m), 1.73-1.58 (6H, m)
実施例 1 0 Example 1 0
4一 (1 -ァダマンチル) 一 2— [ (ピリジン一 2—ィル) スルホニルアミ ノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) 1-2-[(pyridine-12-yl) sulfonylamino] thiazole
実施例 7と同様の方法で 4— (1 -ァダマンチル) —2—ァミノチアゾール 臭化水素酸塩 (200mg) と 2 -ピリジンスルホニルクロリド (31 4 m g) から、 無色アモルファスとして表題化合物 (1 42mg) を得た。 NMR の結果は、 以下のとおりであった。
】H-隱 ( 300 MHz, DMSO - d6 ) δ :12.79ΠΗ, brs), 8.67-8.63 (1H, m), 8.03 (1H, td, J=7.7, 1.8Hz), 7.94 (1H, dt, J=7.7, 1.1Hz) , In the same manner as in Example 7, 4- (1-adamantyl) -2-aminothiazole hydrobromide (200 mg) and 2-pyridinesulfonyl chloride (314 mg) were used to give the title compound (142 mg) as a colorless amorphous. ). NMR results were as follows. H-Oki (300 MHz, DMSO-d6) δ: 12.79ΠΗ, brs), 8.67-8.63 (1H, m), 8.03 (1H, td, J = 7.7, 1.8Hz), 7.94 (1H, dt, J = 7.7, 1.1Hz),
7.60 (1H, ddd, J=7.5, 4.7, 1.3Hz), 6.36(1 l s), 2.02-1.94 (3H, m) , 1.83- 1.76 (6H, m), 1.73—1.58 (6H, m) 7.60 (1H, ddd, J = 7.5, 4.7, 1.3Hz), 6.36 (1 ls), 2.02-1.94 (3H, m), 1.83- 1.76 (6H, m), 1.73—1.58 (6H, m)
実施例 1 1 Example 11
4 - (1 -ァダマンチル) 一 2— [ (ピリジン一 3—ィル) スルホニルアミ ノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) 1-2-[(pyridine-13-yl) sulfonylamino] thiazole
実施例 7と同様の方法で 4一 (1 -ァダマンチル) 一 2—ァミノチアゾール 臭化水素酸塩 (200mg) と 3 -ピリジンスルホニルクロリド (299m g) から、 無色アモルファスとして表題化合物 (1 59mg) を得た。 1H- NMR の結果は、 以下のとおりであった。 In the same manner as in Example 7, 4- (1-adamantyl) -1-2-aminothiazole hydrobromide (200 mg) and 3-pyridinesulfonyl chloride (299 mg) were used to give the title compound (159 mg) as a colorless amorphous. Got. The result of 1 H-NMR was as follows.
1H-NMR ( 300 MHz, 固 -d6 ): 1H-NMR (300 MHz, solid -d6):
δ :12.87 (1Η, brs), 8.96 (1H, d, J=2.2Hz) , 8.78 (1H, dd, J=4.8, 1.6Hz) , 8.17 (1H, dd d, J=8.0, 2.2, 1.6Hz) , 7.60 (1H, dd, J=8.0, 4, 8Hz), 6, 39 (1H, s) , 2.02- 1.94 (3H, m) , 1.83-1.76 (6H, m) , 1.73-1.58 (6H, m) δ: 12.87 (1Η, brs), 8.96 (1H, d, J = 2.2Hz), 8.78 (1H, dd, J = 4.8, 1.6Hz), 8.17 (1H, dd d, J = 8.0, 2.2, 1.6Hz) ), 7.60 (1H, dd, J = 8.0, 4, 8Hz), 6, 39 (1H, s), 2.02- 1.94 (3H, m), 1.83-1.76 (6H, m), 1.73-1.58 (6H, m)
実施例 1 2 Example 1 2
4- (1ーァダマンチル) _2— [N— (4 -ビフエニルスルホニル) 一 N— メチルァミノ] チアゾ一ルの合成 , Synthesis of 4- (1-adamantyl) _2— [N— (4-biphenylsulfonyl) -1-N-methylamino] thiazolyl,
4一 (1 -ァダマンチル) 一 2— (メチルァミノ) チアゾール (200m g) をピリジン (3. 2m l ) に溶解し、 4—ジメチルァミノピリジン (98 mg) を加えた後、 氷冷下で 4 -ビフエニルスルホニルクロリド (407m g) を加え、 その後、 室温に戻して一晩攪拌した。 反応液を氷冷した 6 M塩酸 水溶液 (1 5m l ) に滴下して、 2時間攪拌後、 析出した固体を濾取し、 6M 塩酸水溶液 (1 Om I ) 、 水 (1 Om I ) で洗浄し、 乾燥して得られた固体を プレパラティブ T LC (展開溶媒 クロ口ホルム) で精製し、 無色ァモルファ スとして表題化合物 (1 1 6mg) を得た。 1H- NMRの結果は、 以下のとおりで あった。
-隱 (300MHz, D S0-d6) δ :7.96—7.86 (4H, m), 7.76-7.70 (2H, m) , 7.55 - 7.42 (3Η, m), 6.86 (1H, s), 3.39 (3H, s) , 2, 02-1.94 (3Η, m), 1.83— 4- (1-Adamantyl) -12- (methylamino) thiazole (200 mg) was dissolved in pyridine (3.2 ml), and 4-dimethylaminopyridine (98 mg) was added. -Biphenylsulfonyl chloride (407 mg) was added, and then the mixture was returned to room temperature and stirred overnight. The reaction solution was added dropwise to an ice-cooled 6 M aqueous hydrochloric acid solution (15 ml), and after stirring for 2 hours, the precipitated solid was collected by filtration and washed with a 6 M aqueous hydrochloric acid solution (1 Om I) and water (1 Om I). Then, the solid obtained by drying was purified by preparative TLC (developing solvent, chloroform) to obtain the title compound (116 mg) as a colorless amorphous. The result of 1 H-NMR was as follows. -Hidden (300MHz, D S0-d6) δ: 7.96-7.86 (4H, m), 7.76-7.70 (2H, m), 7.55-7.42 (3Η, m), 6.86 (1H, s), 3.39 (3H, s), 2, 02-1.94 (3Η, m), 1.83—
1.76 (6Η, m), 1.73- 1.58 (6H, m) 1.76 (6Η, m), 1.73- 1.58 (6H, m)
実施例 1 3 Example 1 3
4- (1—ァダマンチル) 一 2— [N— [ (5 -クロ口 - 3 -メチルベンゾ [b]チ才フェン- 2 -ィル) スルホニル]一 N—メチルァミノ] チアゾ一ルの合 実施例 12と同様の方法で 4一 (1 -ァダマンチル) —2— (メチルァミノ) チアゾール (20 Omg) と 5 -クロ口 - 3 -メチルベンゾ [b]チ才フェン - 2 -スルホニルクロリド (453mg) から、 無色アモルファスとして表題化 合物 (42mg) を得た。 NMRの結果は、 以下のとおりであった。 4- (1-Adamantyl) -1-2- [N-[(5-chloro-3-3-methylbenzo [b] thinphen-2-yl) sulfonyl] -1-N-methylamino] thiazol Example 12 Colorless amorphous from 4- (1-adamantyl) -2- (methylamino) thiazole (20 Omg) and 5-chloro-3,3-methylbenzo [b] thinphen-2-phenyl-2-sulfonyl chloride (453mg) in the same manner as As a result, the title compound (42 mg) was obtained. The result of NMR was as follows.
]H-N R (300MHz, DMS0-d6) ] HNR (300MHz, DMS0-d6)
δ :8.15 (1Η, dd, J=8.7, 0.5Hz) , 8.11 (1 H, dd, J=2.2, 0.5Hz) , 7.64 (1 H, dd, J=8.7, 2. 2Hz), 6.97 (1H, s), 3.41 (3H, s) , 2.46 (3H, s) , 2.02-1.94 (3H, m) , 1.77- 1.58 (12H, m) , δ: 8.15 (1Η, dd, J = 8.7, 0.5Hz), 8.11 (1H, dd, J = 2.2, 0.5Hz), 7.64 (1H, dd, J = 8.7, 2.2Hz), 6.97 (1H , s), 3.41 (3H, s), 2.46 (3H, s), 2.02-1.94 (3H, m), 1.77- 1.58 (12H, m),
実施例 1 4 Example 14
4- (1ーァダマンチル) 一 2— [ —メチル— N— (4 - n -プロピルフエ ニルスルホニル) ァミノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) -1-2 — [— methyl—N— (4-n-propylphenylsulfonyl) amino] thiazole
4一 (1 -ァダマンチル) ー2— (メチルァミノ) チアゾ一ル (200m g) をクロ口ホルム (1. 2m l ) に溶解し、 4ージメチルァミノピリジン (98mg) を加えた後、 水冷下で 4 - n -プロピルベンゼンスルホニルクロ ライド (352mg) を加え、 その後、 室温に戻して一晚攪拌した。 水で冷却 しながら、 反応液に 3M塩酸水溶液 (5rrH ) を加えて、 クロ口ホルム (1 8 m l ) で抽出し、 有機相を 3 M塩酸水溶液 (5m l ) 、 5%炭酸水素ナ卜リウ 厶水溶液 (5m l ) 、 水 (5m l ) 、 飽和食塩水 (5m l ) で順次洗浄した。 有機相を無水硫酸マグネシウムで乾燥後、 乾燥剤を濾去し、 溶媒を減圧留去し た。 得られた残渣をプレパラティブ TLC (展開溶媒 クロ口ホルム) で精製 し、 無色固体として表題化合物 (95mg) を得た。 Rの結果は、 以下の とおりであった。
^- MR (300MHz, D S0-d6) 4- (1-adamantyl) -2- (methylamino) thiazol (200 mg) was dissolved in chloroform (1.2 ml), and 4-dimethylaminopyridine (98 mg) was added. Then, 4-n-propylbenzenesulfonyl chloride (352 mg) was added, and then the mixture was returned to room temperature and stirred for a while. While cooling with water, a 3M aqueous solution of hydrochloric acid (5rrH) was added to the reaction solution, and the mixture was extracted with chloroform (18 ml). The organic phase was extracted with a 3M aqueous solution of hydrochloric acid (5ml) and 5% sodium bicarbonate. Washed sequentially with an aqueous solution (5 ml), water (5 ml), and a saturated saline solution (5 ml). After the organic phase was dried over anhydrous magnesium sulfate, the desiccant was removed by filtration, and the solvent was distilled off under reduced pressure. The obtained residue was purified by preparative TLC (developing solvent, black-mouthed form) to give the title compound (95 mg) as a colorless solid. The results of R were as follows. ^-MR (300MHz, D S0-d6)
δ :7.70 (2H, d, J=8.6Hz) , 7.44 (2H, d, J=8.6Hz), 6.84 (1H, s), 3.23 (3H, s) , 2.63 (2H, t, J=7.4 Hz) , 2.01-1.93 (3H, m), 1.78-1.52 (14H, m) , δ: 7.70 (2H, d, J = 8.6Hz), 7.44 (2H, d, J = 8.6Hz), 6.84 (1H, s), 3.23 (3H, s), 2.63 (2H, t, J = 7.4 Hz) ), 2.01-1.93 (3H, m), 1.78-1.52 (14H, m),
0.86 (3H, t, J=7.4 Hz) 0.86 (3H, t, J = 7.4 Hz)
実施例 1 5 Example 1 5
4 - ( 1ーァダマンチル) ー2— CN- (4 -フルオロフェニルスル木ニ ル) 一 N—メチルァミノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) -2-CN- (4-fluorophenylsulfonyl) -1-N-methylamino] thiazole
実施例 1 4と同様の方法で 4一 (1 -ァダマンチル) 一 2— (メチルアミ ノ) チアゾール (50 Omg) と 4 -フル才ロベンゼンスルホニルクロリド (588mg) から、 無色固体として表題化合物 (403mg) を得た。 ^- NMRの結果は、 以下のとおりであった。 In the same manner as in Example 14, the title compound (403 mg) was obtained as a colorless solid from 4- (1-adamantyl) -12- (methylamino) thiazole (50 Omg) and 4-furoserobenzenesulfonyl chloride (588 mg). Got. ^ -NMR results were as follows.
^- R (300MHz, DMSO- d6) ^ -R (300MHz, DMSO-d6)
δ :7.89 (2H, dd, J=8.9, 5.0Hz) , 7.49 (2H, t, J=8.9Hz) , 6.87 (1H, s) , 3.35 (3H, s) , 2. 02-1.93 (3H, m), 1.82-1.58 (12H, m) δ: 7.89 (2H, dd, J = 8.9, 5.0Hz), 7.49 (2H, t, J = 8.9Hz), 6.87 (1H, s), 3.35 (3H, s), 2.02.1.93 (3H, m), 1.82-1.58 (12H, m)
実施例 1 6 Example 16
4一 (1ーァダマンチル) 一 2— [N—メチルー N— (4—モルホリノフエ二 ルスルホニル) ァミノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) -1-2- [N-methyl-N- (4-morpholinophenylsulfonyl) amino] thiazole
実施例 15で得られた 4一 (1 ーァダマンチル) - 2 - [N- (4 -フルォロ フエニルスルホニル) —N—メチルァミノ] チアゾール (1 1 Omg) の DM S 0懸濁液 (1 m l ) にモルホリン ( 236 m g ) と炭酸力リウ厶 (4 1 m g) を加えて 1 60°Cで 2時間攪拌した。 水で冷却しながら、 反応液に水と飽 和食塩水の 1: 1混合液 (5m l ) を加えて攪拌し、 クロ口ホルム (1 5m I ) で抽出後、 有機相を水と飽和食塩水の 1 : 1混合液 (5 m I X 3) 、 飽和 食塩水 (5m l ) で順次洗浄した。 有機相を無水硫酸マグネシウムで乾燥後、 乾燥剤を濾去し、 溶媒を減圧留去した。 得られた残渣をシリカゲルカラムクロ マトグラフィー (展開溶媒 クロ口ホルム) で精製し、 無色固体として表題化 合物 (9 Omg) を得た。 NMRの結果は、 以下のとおりであった。 To a DMSO suspension (1 ml) of 4- (1-adamantyl) -2- [N- (4-fluorophenylsulfonyl) -N-methylamino] thiazole (11 mg) obtained in Example 15 was added. Morpholine (236 mg) and carbonated lithium (41 mg) were added, and the mixture was stirred at 160 ° C for 2 hours. While cooling with water, a 1: 1 mixture (5 ml) of water and saturated saline was added to the reaction mixture, and the mixture was stirred. After extraction with chloroform (15 ml), the organic phase was combined with water and saturated saline. The mixture was washed sequentially with a 1: 1 mixture (5 ml IX 3) and saturated saline (5 ml). After the organic phase was dried over anhydrous magnesium sulfate, the desiccant was removed by filtration, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent, black-mouthed form) to give the title compound (9 Omg) as a colorless solid. The result of NMR was as follows.
】H-瞧 (300MHz, D S0-d6) 】 H- 瞧 (300MHz, D S0-d6)
δ :7.60 (2Η, d, J=9.2Hz), 7, 05 (2H, d, J=9.2Hz) , 6.78 (1 H, s) , 3, 74-
3.67 (4H, m) , 3.32-3.25 (7H, m), 2.02-1.93 (3H, m), 1.82-1.74 (6H, m), 1.73— 1.58 (6H, m) δ: 7.60 (2Η, d, J = 9.2Hz), 7, 05 (2H, d, J = 9.2Hz), 6.78 (1 H, s), 3, 74- 3.67 (4H, m), 3.32-3.25 (7H, m), 2.02-1.93 (3H, m), 1.82-1.74 (6H, m), 1.73—1.58 (6H, m)
実施例 1 7 Example 17
4一 (1 —ァダマンチル) —2— [N—メチルー N— [4 - (4 -メチルピぺ ラジニル) フエニルスルホニル] ァミノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) -2- (N-methyl-N— [4- (4-methylpyrazinyl) phenylsulfonyl] amino] thiazole
実施例 1 6と同様の方法で 4一 (1ーァダマンチル) 一 2— [N- (4 -フ ル才ロフエニルスルホニル) 一 N—メチルァミノ] チアゾール (1 1 Omg) と 1 -メチルビペラジン (27 1 mg) から、 薄黄色アモルファスとして表題 化合物 (1 26mg) を得た。 NW1Rの結果は、 以下のとおりであった。 In the same manner as in Example 16, 4-1- (1-adamantyl) -12- [N- (4-fluorophenylsulfonyl) -1-N-methylamino] thiazole (11 Omg) and 1-methylbiperazine (27 1 mg ) To give the title compound (126 mg) as a pale-yellow amorphous. The results of NW1R were as follows.
^-N R (300MHz, DMS0-d6) ^ -NR (300MHz, DMS0-d6)
δ :7.56 (2Η, d, J=9.2Hz) , 7.03 (2H, d, J=9.2Hz) , 6.78 (1H, s) , 3.37- 3.26 (7H, m) , 2.41-2.38 (4H, m) , 2.20 (3H, s) , 2.02-1.94 (3H, m), 1.80- 1.75 (6H, m), 1.74-1.59 (6H, m) δ: 7.56 (2Η, d, J = 9.2Hz), 7.03 (2H, d, J = 9.2Hz), 6.78 (1H, s), 3.37-3.26 (7H, m), 2.41-2.38 (4H, m) , 2.20 (3H, s), 2.02-1.94 (3H, m), 1.80- 1.75 (6H, m), 1.74-1.59 (6H, m)
実施例 1 8 Example 18
4一 (1ーァダマンチル) 一 2— [N— [4 - [ビス (2 -ヒドロキシェチ ル) ァミノ] フエニルスルホニル] —N—メチルァミノ]チアゾールの合成 Synthesis of 4- (1-adamantyl) 1-2- [N- [4- [bis (2-hydroxyethyl) amino] phenylsulfonyl] -N-methylamino] thiazole
実施例 1 6と同様の方法で 4— (1ーァダマンチル) —2— [N— (4 -フ ル才ロフエニルスルホニル) 一 N—メチルァミノ] チアゾ一ル (1 2ひ mg) とジエタノールァミン (3 1 Omg) から、 無色粉末として表題化合物 (28 mg) を得た。 MMRの結果は、 以下のとおりであった。 In the same manner as in Example 16, 4- (1-adamantyl) -2-[[N- (4-fluorophenylsulfonyl) -N-methylamino] thiazolyl (12 mg) and diethanolamine ( From 31 Omg), the title compound (28 mg) was obtained as a colorless powder. The results of the MMR were as follows.
Ή-NMR (300MHz, DMS0-d6) Ή-NMR (300MHz, DMS0-d6)
(5 :7.50 (2H, d, J=9. OHz) , 6.79 (2H, d, J=9.0Hz) , 6.76 (1H, s) , 4.79 (2H, t, J=5.0Hz) , 3.59-3.44 (8H, m), 3.28 (3H, s), 2.04-1.93 (3H, m), 1.86-1.60 (12H, m) (5: 7.50 (2H, d, J = 9.OHz), 6.79 (2H, d, J = 9.0Hz), 6.76 (1H, s), 4.79 (2H, t, J = 5.0Hz), 3.59-3.44 (8H, m), 3.28 (3H, s), 2.04-1.93 (3H, m), 1.86-1.60 (12H, m)
実施例 1 9 Example 19
4 - (1ーァダマンチル) —2— [N— [4 - [ (2 -ヒドロキシェチル) ァ ミノ] フエニルスルホニル] —N—メチルァミノ]チアゾ一ルの合成 Synthesis of 4- (1-adamantyl) —2— [N— [4-[(2-hydroxyethyl) amino] phenylsulfonyl] —N-methylamino] thiazole
実施例 1 6と同様の方法で 4一 (1ーァダマンチル) —2— [N- (4 -フ ル才ロフエニルスルホニル) —メチルァミノ] チアゾール (1 2 Omg)
と 2 -アミノエ夕ノール (90mg) から、 薄黄色アモルファスとして表題化 合物 (1 O Omg) を得た。 1H- Rの結果は、 以下のとおりであった。 In the same manner as in Example 16, 4-1- (1-adamantyl) -2-[[N- (4-fluorophenylsulfonyl) -methylamino] thiazole (12 Omg) And 2-aminoenol (90 mg) to give the title compound (1 O Omg) as a pale yellow amorphous. The results of 1 H-R were as follows.
^-NM (300MHz, D S0-d6) ^ -NM (300MHz, D S0-d6)
δ :1.46 (2Η, d, J=9. OHz) , 6.81 (1H, t, J=5.5Hz) , 6.76 (1H, s), 6.65 (2H, d, J=9.0Hz) ,4.75 (1H, t, J=5.4Hz) , 3.53 (2H, td, J=5.8, 5.4Hz), 3.27 (3H, s) , 3.14 (2H, td, J=5. 8, 5.5Hz), 2.02-1.94 (3H, m), 1.82-1.76 (6H, m) , 1.75-1.60 (6H, m) δ: 1.46 (2Η, d, J = 9.OHz), 6.81 (1H, t, J = 5.5Hz), 6.76 (1H, s), 6.65 (2H, d, J = 9.0Hz), 4.75 (1H, t, J = 5.4Hz), 3.53 (2H, td, J = 5.8, 5.4Hz), 3.27 (3H, s), 3.14 (2H, td, J = 5.8, 5.5Hz), 2.02-1.94 (3H , m), 1.82-1.76 (6H, m), 1.75-1.60 (6H, m)
実施例 2 O Example 2 O
4 - (1ーァダマンチル) 一 2-[N— [4 - (2 -ヒドロキシェ卜キシ) フ ェニルスルホニル] 一 N—メチルァミノ]チアゾールの合成 Synthesis of 4- (1-adamantyl) -1-2- [N— [4- (2-hydroxyethoxy) phenylsulfonyl] -1-N-methylamino] thiazole
実施例 15で得られた 4— ( Ίーァダマンチル) 一 2— [N- (4 -フル才ロ フエニルスルホニル) 一N—メチルァミノ] チアゾール (1 20mg) とェチ レングリコール (0. 3m l ) の N, N—ジメチルホルムアミド (DM F) 溶 液 ( 0. 9m l ) を水で冷却しながら、 水素化ナ卜リウ厶 (60 %オイル 2 6mg) を加え、 反応温度を 90 °Cに昇温して 1. 5時間攪拌した。 水で冷却 しながら、 反応液に塩化アンモニゥ厶水溶液 (5m l ) を加えて攪拌し、 クロ 口ホルム (1 5m l ) で抽出し、 有機相を塩化アンモニゥ厶水溶液 (5m l ) 、 水と飽和食塩水の 1 : 1混合液 (5m l X 2) 、 飽和食塩水 (5m l ) で順次 洗浄した。 有機相を無水硫酸マグネシウムで乾燥後、 乾燥剤を濾去し、 溶媒を 減圧留去した。 得られた残渣をシリカゲルカラムクロマトグラフィー (展開溶 媒 クロ口ホルム:メタノール =1 60 : 1 ) で精製した。 更にプレパラティ ブ T LC (展開溶媒 クロ口ホルム) で精製し、 無色アモルファスとして表題 ィ匕合物 (1 O Omg) を得た。 NMRの結果は、 以下のとおりであった。 4- (Padamantyl) 1-2- [N- (4-fluorophenylsulfonyl) -1-N-methylamino] thiazole obtained in Example 15 (120 mg) and ethylene glycol (0.3 ml) While cooling the N, N-dimethylformamide (DMF) solution (0.9 ml) with water, sodium hydride (26 mg of 60% oil) was added, and the reaction temperature was raised to 90 ° C. Warmed and stirred for 1.5 hours. While cooling with water, add an aqueous ammonium chloride solution (5 ml) to the reaction solution, stir the mixture, extract with chloroform (15 ml), and saturate the organic phase with aqueous ammonium chloride solution (5 ml) and water. The mixture was washed successively with a 1: 1 mixture of saline (5 ml × 2) and saturated saline (5 ml). After the organic phase was dried over anhydrous magnesium sulfate, the desiccant was removed by filtration, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent, solvent: form: methanol = 160: 1). The product was further purified by preparative TLC (developing solvent, black-mouthed form) to give the title compound (1 O Omg) as a colorless amorphous. The result of NMR was as follows.
^ - NMR (300MHz, DMS0 - d6) ^-NMR (300MHz, DMS0-d6)
δ :7.73 (2Η, d, J=9. OHz) , 7.14 (2H, d, J=9. OHz) , 6.82 (1 H, s) , 4.92 (1 H, t, J=5.4Hz) , 4.07 (2H, t, J=4.8) , 3.72 (2H, td, J=5.4, 4.8Hz) , 3.32 (3H, s) , 2.02 - 1, 94 (3H, m) , 1.82-1.76鼠 m) , 1.75-1.60 (6H, m) δ: 7.73 (2Η, d, J = 9.OHz), 7.14 (2H, d, J = 9.OHz), 6.82 (1H, s), 4.92 (1H, t, J = 5.4Hz), 4.07 (2H, t, J = 4.8), 3.72 (2H, td, J = 5.4, 4.8Hz), 3.32 (3H, s), 2.02-1, 94 (3H, m), 1.82-1.76 mouse m), 1.75 -1.60 (6H, m)
実施例 2 1 Example 2 1
4一 (1ーァダマンチル) 一 2— [N— (4 -フルオロフェニルスルホニル) -N- (4 -メ卜キシベンジル) ァミノ]チアゾールの合成
実施例番号 4 - 5で得られた 4一 (1ーァダマンチル) 一 2— [ (4 -フル 才ロフエニルスルホニル) ァミノ] チアゾール (200mg) の DM F溶液Synthesis of 4- (1-adamantyl) -1-2- [N- (4-fluorophenylsulfonyl) -N- (4-methoxybenzyl) amino] thiazole DMF solution of 4- (1-adamantyl) 1-2-[(4-furofilophenylsulfonyl) amino] thiazole (200 mg) obtained in Example No. 4-5
(4. 0m l ) に水冷下で水素化ナ卜リウ厶 (60%オイル 22mg) を加 え、 続いて p -メ卜キシベンジルクロライド (0. 1 1 m l ) とヨウ化力リウ 厶 (1 27mg) を加え、 室温で 2時間攪拌した。 氷冷下で水 (1 Om I ) を 加えて攪拌し、 酢酸ェチル (20m l ) で抽出した後、 有機相を水 (1 0m l X4) 、 飽和食塩水 (1 Om l ) で順次洗浄した。 有機相を無水硫酸マグネシ ゥ厶で乾燥後、 乾燥剤を濾去し、 溶媒を減圧留去した。 得られた残渣をシリカ ゲルカラムクロマトグラフィー (展開溶媒 クロ口ホルム〜クロ口ホルム:メ 夕ノール =200 : 1 ) で精製し、 薄黄色油状物質として表題化合物 (21 1 mg) を得た。 の結果は、 以下のとおりであった。 (4.0 ml) was added with sodium hydride (22 mg of 60% oil) under water cooling, followed by p-methoxybenzyl chloride (0.1 ml) and potassium iodide (1 ml). 27 mg) and stirred at room temperature for 2 hours. After adding water (1 Om I) under ice cooling and stirring, extracting with ethyl acetate (20 ml), the organic phase was washed successively with water (10 ml X4) and saturated saline (1 Oml). . After the organic phase was dried over anhydrous magnesium sulfate, the desiccant was removed by filtration, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: black-mouthed form to black-mouthed form: methanol = 200: 1) to obtain the title compound (21 1 mg) as a pale yellow oily substance. The results were as follows.
^ - NMR (300MHz, DMS0-d6) ^-NMR (300MHz, DMS0-d6)
δ :7.89 (2Η, dd, J=8.9, 5.1 Hz) , 7.47 (2H, t, J=8.9Hz) , 7.26 (2H, d, J=8.7Hz) , 6.89 - 6.83 (3H, m) , 4.98 (2H, s) , 3.71 (3H, s) , 2.02-1.94 (3H, m), 1.78—1.61 (12H, m) 実施例 22 δ: 7.89 (2Η, dd, J = 8.9, 5.1 Hz), 7.47 (2H, t, J = 8.9Hz), 7.26 (2H, d, J = 8.7Hz), 6.89-6.83 (3H, m), 4.98 (2H, s), 3.71 (3H, s), 2.02-1.94 (3H, m), 1.78-1.61 (12H, m) Example 22
4一 ( 1—ァダマンチル) —2-[N— (4 -メ卜キシベンジル) 一 N— (4 -モルホリノフエニルスルホニル) ァミノ]チアゾールの合成 Synthesis of 4- (1-adamantyl) -2- (N- (4-methoxybenzyl) -1-N- (4-morpholinophenylsulfonyl) amino] thiazole
実施例 1 6と同様の方法で 4一 (1ーァ:ダマンチル) 一 2— [N— (4 -フル オロフェニルスルホニル) 一 N— (4 -メ卜キシベンジル) ァミノ]チアゾール (20 Omg) とモルホリン (0. 3m l ) から、 無色アモルファスとして表 題化合物 (1 7 Omg) を得た。 -NMRの結果は、 以下のとおりであった。 In the same manner as in Example 16, 4-1- (1-α-damantyl) -12- [N— (4-fluorophenylsulfonyl) -1-N— (4-methoxybenzyl) amino] thiazole (20 Omg) The title compound (17 Omg) was obtained as a colorless amorphous from morpholine (0.3 ml). The result of -NMR was as follows.
^ -國 R (300MHz, DMS0-d6) ^ -Country R (300MHz, DMS0-d6)
δ :7.61 (2Η, d, J=9.2Hz) , 7.30 (2H, d, J=8.8Hz) , 7.02 (2H, d, J=9.2Hz) , 6.85 (2H, d, J=8.8Hz), 6.76 (1H, s), 4.94 (2H, s) , 3.74-3.67 (7H, m) , 3.32-3.26 (4H, m) , 2.02— 1.94 (3H, m), 1.78-1.61 (12H, m) δ: 7.61 (2Η, d, J = 9.2Hz), 7.30 (2H, d, J = 8.8Hz), 7.02 (2H, d, J = 9.2Hz), 6.85 (2H, d, J = 8.8Hz), 6.76 (1H, s), 4.94 (2H, s), 3.74-3.67 (7H, m), 3.32-3.26 (4H, m), 2.02--1.94 (3H, m), 1.78-1.61 (12H, m)
実施例 23 Example 23
4一 (1ーァダマンチル) 一2— (4 -モルホリノフエニルスルホニルアミ ノ) チアゾールの合成
実施例 22で得られた 4一 (Ίーァダマンチル) — 2— 一 (4 -メ卜キシ ベンジル) 一 N— (4 -モルホリノフエニルスルホニル) ァミノ]チアゾールSynthesis of 4- (1-adamantyl) -1- (4-morpholinophenylsulfonylamino) thiazole 4- (4-adamantyl) -2-1- (4-methoxybenzyl) -1-N- (4-morpholinophenylsulfonyl) amino] thiazole obtained in Example 22
(1 02mg) に水で冷却しながらトリフルォロ酢酸 (0. 5m l ) とァニソ ール (50 I ) を加え、 5分間攪拌した。 反応液を減圧下濃縮し、 得られた 残渣をシリ力ゲル力ラムクロマトグラフィー (展開溶媒 クロ口ホルム:メ夕 ノール =80 : 1 ) で精製し、 無色アモルファスとして表題化合物 (44m g) を得た。 隱 Rの結果は、 以下のとおりであった。 To the solution (102 mg) was added trifluoroacetic acid (0.5 ml) and anisol (50 I) while cooling with water, and the mixture was stirred for 5 minutes. The reaction solution was concentrated under reduced pressure, and the obtained residue was purified by silica gel gel column chromatography (developing solvent, chromate form: solvent = 80: 1) to give the title compound (44 mg) as a colorless amorphous. Was. The results of Oki R are as follows.
^-N R (300MHz, D S0-d6) ^ -N R (300MHz, D S0-d6)
a :12.52(1 H, brs) , 7.62 (2H, d J=9.0Hz) , 7.00 (2H, d, J=9.0Hz) , 6.26 (1 H, s) , 3.75 -3.68 (4H, m) , 3.25-3.18 (4H, m) , 2.02-1.94 (3H, m), 1.80-1.75 (6H, tn), 1 · 74- 1.59 (6H, m) a: 12.52 (1 H, brs), 7.62 (2H, d J = 9.0 Hz), 7.00 (2H, d, J = 9.0 Hz), 6.26 (1 H, s), 3.75 -3.68 (4H, m), 3.25-3.18 (4H, m), 2.02-1.94 (3H, m), 1.80-1.75 (6H, tn), 1 · 74-1.59 (6H, m)
実施例 24 Example 24
4 - (1ーァダマンチル) 一 2— [N— (4 -メ卜キシベンジル) — N— [4 - (2 -モルホリノエ卜キシ) フエニルスルホニル] ァミノ]チアゾールの合 実施例 2 1で得られた 4一 (1ーァダマンチル) — 2— [M— (4 -フル才ロ フエニルスルホニル) 一 N— (4 -メ卜キシベンジル) ァミノ]チアゾール (2 1 0 ) と1\1 - (2 -ヒドロキシェチル) モルホリン (0. 1 m l ) の DM F溶液 (2. Om I ) に、 水素化ナトリウム (60%オイル 36mg) を加 え、 室温で〗 5分間攪拌した。 氷冷下で水と飽和食塩水の 1 : 1混合液 (5 m I ) を加えて攪拌し、 クロ口ホルム (1 5m l ) で抽出し、 有機相を水と飽和 食塩水の 1 :〗 混合液 (5m I X 3) 、 飽和食塩水 (5m l ) で順次洗浄した。 有機相を無水硫酸マグネシウムで乾燥後、 乾燥剤を濾去し、 溶媒を減圧留去し た。 得られた残渣をシリカゲルカラムクロマトグラフィー (展開溶媒 n -へ キサン:酢酸ェチル =1 : 1 〜 酢酸ェチル) で精製し、 薄黄色油状物質とし て表題化合物 (1 49mg) を得た。 -NMRの結果は、 以下のとおりであった。 Synthesis of 4- (1-adamantyl) -1-2- [N— (4-methoxybenzyl) —N— [4- (2-morpholinoethoxy) phenylsulfonyl] amino] thiazole 4 obtained in Example 21 1- (1-adamantyl) — 2 -— [M— (4-fluorophenylsulfonyl) -1-N— (4-methoxybenzyl) amino] thiazole (2 10) and 1 \ 1- (2-hydroxyethyl) To a solution of morpholine (0.1 ml) in DMF (2. Om I), sodium hydride (36 mg of 60% oil) was added, and the mixture was stirred at room temperature for about 5 minutes. Under ice-cooling, a 1: 1 mixture of water and saturated saline (5 ml) was added, and the mixture was stirred and extracted with chloroform (15 ml). The organic phase was mixed with water and saturated saline 1:〗. The mixture was washed successively with a mixed solution (5 ml IX 3) and a saturated saline solution (5 ml). After the organic phase was dried over anhydrous magnesium sulfate, the desiccant was removed by filtration, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent n-hexane: ethyl acetate = 1: 1 to ethyl acetate) to obtain the title compound (149 mg) as a pale yellow oily substance. The result of -NMR was as follows.
】H-隱 (300MHz, D S0-d6) ] H-Oki (300MHz, D S0-d6)
δ :7.73 (2Η, d, J=9.0Hz), 7.28 (2H, d, J=8.9Hz) ,7.13 (2H, d, J=9.0Hz) , 6.85 (2H, d, J=8.9Hz), 6.80 (1H, s), 4.95 (2H, s) , 4.17 (2H, t, J=5.6Hz) , 3.70 (3H, s) , 3.59-
3.53(4H, m), 2.69 (2H, t, J=5.6Hz), 2.48-2.42 (4H, m) , 2.02 - 1.94 (3H, m) , 1.78- 1.61 (12H, m) δ: 7.73 (2Η, d, J = 9.0Hz), 7.28 (2H, d, J = 8.9Hz), 7.13 (2H, d, J = 9.0Hz), 6.85 (2H, d, J = 8.9Hz), 6.80 (1H, s), 4.95 (2H, s), 4.17 (2H, t, J = 5.6Hz), 3.70 (3H, s), 3.59- 3.53 (4H, m), 2.69 (2H, t, J = 5.6Hz), 2.48-2.42 (4H, m), 2.02-1.94 (3H, m), 1.78- 1.61 (12H, m)
実施例 25 Example 25
4- (1ーァダマンチル) 一 2— [4 - (2 -モルホリノエ卜キシ) フエ二 ルスルホニルァミノ〗チアゾ一ル · トリフル才ロ酢酸塩の合成 4- (1-adamantyl) -1-2- [4- (2-morpholinoethoxy) phenyl Synthesis of sulphonylaminoaminothiazol tritrifluoroacetate
実施例 23と同様の方法で 4一 (1 —ァダマンチル) — 2— [N— (4 -メ卜 キシベンジル) — N— [4 - (2 -モルホリノエ卜キシ) フエニルスルホニ ル] ァミノ] チアゾ一ル (1 43mg) から、 無色アモルファスとして表題化 合物 (1 44mg) を得た。 NMRの結果は、 以下のとおりであった。 In the same manner as in Example 23, 4- (1-adamantyl) —2- [N— (4-methoxybenzyl) —N— [4- (2-morpholinoethoxy) phenylsulfonyl] amino] thiazol ( 143 mg) to give the title compound (144 mg) as a colorless amorphous. The result of NMR was as follows.
^- MR (300MHz, DMS0-d6) ^-MR (300MHz, DMS0-d6)
δ :12.63 (1 Η, brs) , 7.77 (2Η, d, J=9. OHz) ,7.12 (2H, d, J=9. OHz) , 6.31 (1 H, s) , 4.40 (2H, brs) , 4.10-3.00 (m) , 2.02-1.94 (3H, m) , 1.80-1.75 (6H, m) , 1.74- 1.59鼠 m) δ: 12.63 (1Η, brs), 7.77 (2Η, d, J = 9.OHz), 7.12 (2H, d, J = 9.OHz), 6.31 (1H, s), 4.40 (2H, brs) , 4.10-3.00 (m), 2.02-1.94 (3H, m), 1.80-1.75 (6H, m), 1.74- 1.59 m
実施例 26 Example 26
4- (1ーァダマンチル) 一 2— [N— [4 - [2 - (ジメチルァミノ) エト キシ] フエニルスルホニル] — N— (4 -メ卜キシベンジル) ァミノ]チアゾー ルの合成 Synthesis of 4- (1-adamantyl) 1-2- [N— [4- [2- (dimethylamino) ethoxy] phenylsulfonyl] —N— (4-methoxybenzyl) amino] thiazole
実施例 24と同様の方法で 4一 (1ーァダマンチル) 一 2— [N— (4 -フル 才ロフエニルスルホニル) 一 N— (4 -メ卜キシベンジル) ァミノ]チアゾール (204mg) と N, N—ジメチルエタノールァミン ( 80 μ I ) から、 無色 粉末として表題化合物 (1 47mg) を得た。 - Rの結果は、 以下のとおり であった。 In the same manner as in Example 24, 4- (1-adamantyl) -12- [N- (4-fluorophenylsulfonyl) -1-N- (4-methoxybenzyl) amino] thiazole (204 mg) and N, N- The title compound (147 mg) was obtained as a colorless powder from dimethylethanolamine (80 μI). -The results of R were as follows.
】H -剛 R (300MHz, DMS0-d6) δ :7.73 (2Η, d, J=9. OHz) , 7.28 (2H, d, J=8.7Hz) , 7.13 (2H, d, J=9. OHz), 6.85 (2H, d, J=8.7Hz) , 6.79 (1H, s), 4.96 (2H, s) , H-rigid R (300MHz, DMS0-d6) δ: 7.73 (2Η, d, J = 9.OHz), 7.28 (2H, d, J = 8.7Hz), 7.13 (2H, d, J = 9.OHz) ), 6.85 (2H, d, J = 8.7Hz), 6.79 (1H, s), 4.96 (2H, s),
4.17 (2H, t, J=5.7Hz) , .3.71 (3H, s) , 2.63 (2H, t, J=5.7Hz) , 2.20 (6H, s) , 2.02- 1.94 (3H, tn), 1.78-1.60 (12H, m) 4.17 (2H, t, J = 5.7Hz), .3.71 (3H, s), 2.63 (2H, t, J = 5.7Hz), 2.20 (6H, s), 2.02- 1.94 (3H, tn), 1.78- 1.60 (12H, m)
実施例 27 Example 27
4一 — (1ーァダマンチル) 一 _2— [4 - [2 - (ジメチルァミノ) ェトキ シ] フエニルスルホニルァミノ]チアゾ一ル · トリフル才ロ酢酸塩の合成
05007106 4--1- (1-adamantyl) -1-_2— [4- [2- (Dimethylamino) ethoxy] phenylsulfonylamino] thiazol / trifluroacetate 05007106
45 実施例 23と同様の方法で 4一 (1—ァダマンチル) 一 2— [N_ [4 - [2 - (ジメチルァミノ) ェ卜キシ] フエニルスルホニル] — N— (4 -メ卜キシ ベンジル) ァミノ]チアゾール (1 33mg) から、 薄桃色アモルファスとし て表題化合物 (1 1 1 mg) を得た。 1H- NMR の結果は、 以下のとおりであった。 】H-隱 (300MHz, D S0-d6) δ :7.77 (2H, d, J=9. OHz), 7.12 (2H, d, J=9. OHz) , 6.31 (1H, s), 4.37 (2H, t, J=5. OHz) , 3.51 (2H, t, J=5. OHz) , 2.84 (6H, s) , 2.02- 1.94 (3H, m), 1.78— 1.60 (12H, m) 45 4- (1-adamantyl) -12- [N_ [4- [2- (dimethylamino) ethoxy] phenylsulfonyl] —N— (4-methoxybenzyl) amino in the same manner as in Example 23 The title compound (111 mg) was obtained as a pale pink amorphous from thiazole (133 mg). The result of 1 H-NMR was as follows. H-Oki (300MHz, D S0-d6) δ: 7.77 (2H, d, J = 9.OHz), 7.12 (2H, d, J = 9.OHz), 6.31 (1H, s), 4.37 (2H , t, J = 5. OHz), 3.51 (2H, t, J = 5. OHz), 2.84 (6H, s), 2.02- 1.94 (3H, m), 1.78- 1.60 (12H, m)
実施例 28 Example 28
4— (1ーァダマンチル) 一 2—〖4 - [2 - (4 -ピペラジニル)エトキシ] フエニルスルホニルァミノ]チアゾ一ルの合成 Synthesis of 4- (1-adamantyl) 1 2— 〖4- [2- (4-piperazinyl) ethoxy] phenylsulfonylamino] thiazol
(1 ) 4一 (1ーァダマンチル) 一 2— [M— [4 - [2 - [4 - (t -ブト キシカルボニル) ピペラジニル] ェ卜キシ] フエニルスルホニルー N_ (4 - メ卜キシベンジル) ァミノ]チアゾールの合成 (1) 4- (1-adamantyl) -1-2- (M— [4- [2- [4- (t-butoxycarbonyl) piperazinyl] ethoxy] phenylsulfonyl-N_ (4-methoxybenzyl) amino ] Synthesis of thiazole
実施例 24と同様の方法で 4一 (1ーァダマンチル) - 2 - [N- (4 -フル オロフェニルスルホニル) 一 N— (4 -メ卜キシベンジル) ァミノ]チアゾ一ル (25 Omg) と t -ブチル 4- (2 -ヒドロキシェチル) ピぺラジン - 1 - カルボキシレー卜 (225mg) から黄色油状物質として表題化合物 (1 28 mg) を得 。 1H- NMRの結果は、 以下のとぉリであった。 In the same manner as in Example 24, 4- (1-adamantyl) -2- [N- (4-fluorophenylsulfonyl) -1-N— (4-methoxybenzyl) amino] thiazol (25 Omg) and t- Butyl 4- (2-hydroxyethyl) pidazine-1-carboxylate (225 mg) gave the title compound (128 mg) as a yellow oil. The result of 1 H-NMR was as follows.
1H-NMR (300MHz, DMS0-d6) 1H-NMR (300MHz, DMS0-d6)
δ :7.73 (2Η, d, J=9, OHz) , 7.28 (2H, d, J=8.7Hz) , 7.13(2H, d, J=9. OHz) , 6.85 (2H, d, J=8.7Hz) , 6.79 (1H, s), 4.95 (2H, s) , 4.18(2H, t, J=5.4Hz) , 3.71 (3H, s), 3.34- 3, 27 (4H, m), 2.72 (2H, t, J=5.4Hz), 2.45-2.38 (4H, m) , 2.02-1.94 (3H, m), 1, 78- 1.60 (12H, m), 1.39 (9H, s) δ: 7.73 (2Η, d, J = 9, OHz), 7.28 (2H, d, J = 8.7Hz), 7.13 (2H, d, J = 9.OHz), 6.85 (2H, d, J = 8.7Hz) ), 6.79 (1H, s), 4.95 (2H, s), 4.18 (2H, t, J = 5.4Hz), 3.71 (3H, s), 3.34- 3, 27 (4H, m), 2.72 (2H, t, J = 5.4Hz), 2.45-2.38 (4H, m), 2.02-1.94 (3H, m), 1, 78-1.60 (12H, m), 1.39 (9H, s)
(2) 4- (1ーァダマンチル) 一 2— [4 - [2 - (4 -ピペラジニル)ェ 卜キシ]フエニルスルホニルァミノ ]チアゾールの合成 (2) Synthesis of 4- (1-adamantyl) -1-2- [4- [2- (4-piperazinyl) ethoxy] phenylsulfonylamino] thiazole
実施例 23と同様の方法で 4一 (1ーァダマンチル) — 2— [N— [4 - [2 - [4 - (t -ブ卜キシカルボニル) ピペラジニル] エトキシ] フエニルスルホ 二ルー N— (4 -メ卜キシベンジル) ァミノ]チアゾール (1 1 4mg) から粗 生成物を得た後、 シリカゲルカラムクロマトグラフィー (展開溶媒 クロロホ
ル厶:メタノール: 28%アンモニア水溶液 = 1 5 : 1 : 0. 1 〜 1 0 : 1 : 0. 1 ) で精製し、 無色粉末として表題化合物 (50mg) を得た。 - NMR の結果は、 以下のとおりであった。 In the same manner as in Example 23, 4- (1-adamantyl) —2-—N— [4- [2- [4- (t-butoxycarbonyl) piperazinyl] ethoxy] phenylsulfo 2-N— (4-methyl After obtaining the crude product from [methoxybenzyl) amino] thiazole (114 mg), silica gel column chromatography (developing solvent: chloropho Purification was performed using a solution of room temperature: room temperature: 28% aqueous ammonia solution = 15: 1: 0.1 to 10: 1: 0.1) to obtain the title compound (50 mg) as a colorless powder. -The result of NMR was as follows.
^- MR (300MHz, D S0-d6) ^-MR (300MHz, D S0-d6)
δ :7.68 (2Η, d, J=8.7Hz) , 6.97 (2H, d, J=8.7Hz) , 6.06 (1H, s), 4.10 (2H, t, J=5.5Hz) , 2.93-2.86 (4H, m), 2.73 (2H, t, J=5.5Hz), 1.58-2.47 (4H, m) , 2.01- 1.92 (3H, m), 1.79-1.58 (12H, m) δ: 7.68 (2Η, d, J = 8.7Hz), 6.97 (2H, d, J = 8.7Hz), 6.06 (1H, s), 4.10 (2H, t, J = 5.5Hz), 2.93-2.86 (4H , m), 2.73 (2H, t, J = 5.5Hz), 1.58-2.47 (4H, m), 2.01- 1.92 (3H, m), 1.79-1.58 (12H, m)
実施例 29 Example 29
4- (1ーァダマンチル) — 2— [N— (4 -ヒドロキシフエニルスルホニ ル) 一 N— (4 -メ卜キシベンジル) ァミノ]チアゾールの合成 Synthesis of 4- (1-adamantyl) — 2 -— [N— (4-hydroxyphenylsulfonyl) -N— (4-methoxybenzyl) amino] thiazole
4 - (1 —ァダマンチル) 一 2— [N— (4 -フルオロフェニルスルホニル) 一 N— (4 -メトキシベンジル) ァミノ]チアゾール (496mg) と 2 - (メ チルスルホニル) エタノールの DM F溶液 (1. 5m l ) に氷冷下で水素化ナ 卜リウ厶 (60%オイル 1 1 6mg) を加え、 氷冷下で 5分間攪拌した後、 室温に戻して 30分間攪拌した。 氷冷下、 1 M塩酸水溶液 (1 Om l ) を加え て発泡が止むまで攪拌し、 酢酸ェチル (20m l ) で抽出した。 有機相を水 DMF solution of 4- (1-adamantyl) -1-2- [N— (4-fluorophenylsulfonyl) -1-N— (4-methoxybenzyl) amino] thiazole (496 mg) and 2- (methylsulfonyl) ethanol (1 To 5 ml), sodium hydride (116 mg of 60% oil) was added under ice-cooling, and the mixture was stirred for 5 minutes under ice-cooling, then returned to room temperature and stirred for 30 minutes. Under ice-cooling, a 1 M aqueous hydrochloric acid solution (1 Oml) was added, the mixture was stirred until foaming ceased, and the mixture was extracted with ethyl acetate (20 ml). Organic phase with water
(1 Om I X3) 、 飽和食塩水 (1 0m l ) で順次洗浄した。 有機相を無水硫 酸マグネシウムで乾燥後、 乾燥剤を濾去し、 溶媒を減圧留去した。 得られた残 渣をシリカゲルカラムクロマトグラフィー (展開溶媒 クロ口ホル厶:メタノ ール = 1 00 : 1 ) で精製し、 薄黄色アモルファスとして表題化合物 (2 Ί 1 mg) を得た。 NWIRの結果は、 以下のとおりであった。 (1 Om I X3) and saturated saline (10 ml). After the organic phase was dried over anhydrous magnesium sulfate, the desiccant was removed by filtration, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent, solvent: methanol: 100: 1) to give the title compound (2.1 mg) as a pale yellow amorphous. The NWIR results were as follows.
Ή-NMR (300MHz, DMS0-d6) Ή-NMR (300MHz, DMS0-d6)
(5 :10.70 (1H, brs) , 7.64 (2Η, d, J=8.9Hz) , 7.28 (2H, d, J=8.9Hz) , 6.90 (2H, d, J=8.9 Hz) , 6.85 (2H, d, J=8.9Hz) , 6.78 (1H, s) , 4.94 (2H, s), 3.71 (3H, s) , 2.02 - 1.94 (3H, m), 1.78-1.61 (12H, m) (5: 10.70 (1H, brs), 7.64 (2Η, d, J = 8.9Hz), 7.28 (2H, d, J = 8.9Hz), 6.90 (2H, d, J = 8.9 Hz), 6.85 (2H, d, J = 8.9Hz), 6.78 (1H, s), 4.94 (2H, s), 3.71 (3H, s), 2.02-1.94 (3H, m), 1.78-1.61 (12H, m)
実施例 30 Example 30
4一 (1 —ァダマンチル) 一 2—— [4 - (カルボキシメ卜キシ) フエニルス ルホニルァミノ]チアゾールの合成
(1 ) 4 - (1ーァダマンチル) 一 2— [N— [4 - [ (t _プ卜キシカルボ ニル) メ卜キシ]フエニルスルホニノレ]— N— (4 -メ卜キシベンジル) ァミノ] チアゾールの合成 4-1- (1-adamantyl) -1-2- Synthesis of [4- (carboxymethoxy) phenylsulfonylamino] thiazole (1) 4- (1-adamantyl) -1-2- [N— [4-[(t_butoxycarbonyl) methoxy] phenylsulfoninole] —N— (4-methoxybenzyl) amino] thiazole Synthesis
実施例 2 9で得られた 4一 (1ーァダマンチル) — 2— [N— (4 -ヒドロキ シフエニルスルホニル) 一 N— (4 -メ卜キシベンジル) ァミノ]チアゾール (24 3mg) の DM F溶液 ( 2. 5m l ) に氷冷下で水素化ナ卜リウ厶 (6 0%オイル 1 1 6 mg) を加え、 5分間攪拌した後、 クロ口酢酸 t -プチル (1 3mg) を加えて室温に戻し、 2時間攪拌した。 氷冷下、 水 (1 0m I ) を加えて発泡が止むまで攪拌し、 酢酸ェチル (20m l ) で抽出した。 有 機相を水 (1 Om I X4) 、 飽和食塩水 (1 Om l ) で順次洗浄した。 有機相 を無水硫酸マグネシウムで乾燥後、 乾燥剤を濾去し、 溶媒を減圧留去した。 得 られた残渣をシリカゲルカラムクロマトグラフィー (展開溶媒 n -へキサ ン:酢酸ェチル =9: 1) で精製し、 無色アモルファスとして表題化合物 (2 5 4mg) を得た。 NMRの結果は、 以下のとおりであった。 A solution of 4- (1-adamantyl) —2- [N— (4-hydroxyphenylsulfonyl) -1-N— (4-methoxybenzyl) amino] thiazole (243 mg) obtained in Example 29 in DMF ( To 2.5 ml) was added sodium hydride (60 mg oil, 116 mg) under ice-cooling, and after stirring for 5 minutes, t-butyl acetate (13 mg) was added to the mixture, and the mixture was brought to room temperature. It was returned and stirred for 2 hours. Under ice-cooling, water (10 ml) was added, the mixture was stirred until foaming stopped, and the mixture was extracted with ethyl acetate (20 ml). The organic phase was washed successively with water (1 Om I X4) and saturated saline (1 Oml). After the organic phase was dried over anhydrous magnesium sulfate, the desiccant was removed by filtration, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent n-hexane: ethyl acetate = 9: 1) to give the title compound (254 mg) as a colorless amorphous. The result of NMR was as follows.
^-NWIR (300MHz, D S0-d6) ^ -NWIR (300MHz, DS0-d6)
δ :7.75 (2Η, d, J=9. OHz) , 7.28 (2H, d, J=8.8Hz) , 7.09 (2H, d, J=9. OHz), 6.85 (2H, d, J=8.8Hz) , 6.80 (1H, s) , 4.96 (2H, s) , 4.80 (2H, s) , 3.71 (3H, s), 2.02- 1.94 (3H, m), 1.78-1.60 (12H, m), 1.41 (9H, s), δ: 7.75 (2Η, d, J = 9.OHz), 7.28 (2H, d, J = 8.8Hz), 7.09 (2H, d, J = 9.OHz), 6.85 (2H, d, J = 8.8Hz) ), 6.80 (1H, s), 4.96 (2H, s), 4.80 (2H, s), 3.71 (3H, s), 2.02- 1.94 (3H, m), 1.78-1.60 (12H, m), 1.41 ( 9H, s),
(2) 4— (1ーァダマンチル) 一 2— [4 - (カルボキシメ卜キシ) フエ ニルスルホニルァミノ]チアゾールの合成 (2) Synthesis of 4- (1-adamantyl) -1-2- [4- (carboxymethoxy) phenylsulfonylamino] thiazole
実施例 2 3と同様の方法で 4一 ( 1ーァダマンチル) 一 2— [N— [4 In the same manner as in Example 23, 41 (1-adamantyl) 1 2— [N— [4
- [ (t -ブトキシカルボニル) メ卜キシ]フエニルスルホニル]一 N— (4 -メ 卜キシベンジル) ァミノ]チアゾール (20 9mg) から、 白色粉末として表 題化合物 (1 45mg) を得た。 - IIRの結果は、 以下のとおりであった。 -[(t-Butoxycarbonyl) methoxy] phenylsulfonyl] -N- (4-methoxybenzyl) amino] thiazole (209 mg) gave the title compound (145 mg) as a white powder. -The IIR results are as follows.
'W- R (300MHz, DMS0-d6) 'W-R (300MHz, DMS0-d6)
δ :12.61 (IH, brs) , 7.72 (2H, d, J=9. OHz) , 7.03 (2H, d, J=9. OHz), 6.30 (IH, s) , 4.76 (2H, s) , 2.02-1.94 (3H, tn) , 1.78—1.60 (12H, m) δ: 12.61 (IH, brs), 7.72 (2H, d, J = 9.OHz), 7.03 (2H, d, J = 9.OHz), 6.30 (IH, s), 4.76 (2H, s), 2.02 -1.94 (3H, tn), 1.78—1.60 (12H, m)
実施例 3 1
4一 (1 —ァダマンチル) _ 2— [|[一 [4 - [ (アミノカルボニル) メ卜キ シ]フエニルスルホニル]一 N— (4 -メ卜キシベンジル) ァミノ]チアゾールの 合成 Example 3 1 Synthesis of 4- (1-adamantyl) _2- [| [1- [4-[(aminocarbonyl) methoxy] phenylsulfonyl] -1-N— (4-methoxybenzyl) amino] thiazole
実施例 2 9で得られた 4一 (1—ァダマンチル) — 2— [N— (4 -ヒドロキ シフエニルスルホニル) —N— (4 -メ卜キシベンジル) ァミノ]チアゾ一ル (2 1 Omg) の DM F溶液 (2. Om l ) に氷冷下で水素化ナトリウム (6 0 %オイル 1 8mg) を加え、 5分間攪拌した後、 2 -クロロアセ卜アミド (7 7 mg) を加えて室温に戻し、 2時間攪拌した。 更にヨウ化カリウム (1 3 6mg) を加えて、 室温で 3時間攪拌した後、 80°Cに昇温して 3時間攪拌 した。 氷冷下、 水と飽和食塩水の 1 :〗混合液 (5m l ) を加えて発泡が止む まで攪拌し、 クロ口ホルム (2 0m で抽出した。 有機相を水と飽和食塩水 の 1 : 1混合液 ( 1 0 m I X 2 ) 、 飽和食塩水 (1 Om l ) で順次洗浄した。 有機相を無水硫酸マグネシウムで乾燥後、 乾燥剤を濾去し、 溶媒を減圧留去し た。 得られた残渣をシリカゲルカラムクロマトグラフィー (展開溶媒 クロ口 ホルム:メタノール =80 : 1 ) で精製し、 薄茶色油状物質として表題化合物 (1 6 Omg) を得た。 MMRの結果は、 以下のとおりであった。 Example 29 The 4- (1-adamantyl) —2- [N— (4-hydroxyphenylsulfonyl) —N— (4-methoxybenzyl) amino] thiazol (21 mg) obtained in Example 9 Sodium hydride (60% oil, 18mg) was added to the DMF solution (2. Oml) under ice-cooling, and the mixture was stirred for 5 minutes. Then, 2-chloroacetamide (77mg) was added, and the mixture was returned to room temperature. Stirred for 2 hours. Further, potassium iodide (136 mg) was added, and the mixture was stirred at room temperature for 3 hours, and then heated to 80 ° C and stirred for 3 hours. Under ice-cooling, a mixture of water and saturated saline 1: 5 (5 ml) was added, and the mixture was stirred until bubbling ceased, and the mixture was extracted with chloroform (20 m. The organic phase was mixed with water and saturated saline 1: 5). The mixture was washed successively with 1 mixture (10 m IX 2) and saturated saline (1 Oml) .The organic phase was dried over anhydrous magnesium sulfate, the desiccant was filtered off, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent, liquid: form: methanol = 80: 1) to give the title compound (16 Omg) as a pale brown oily substance. there were.
1H-NMR (300MHz, DMS0-d6) 1H-NMR (300MHz, DMS0-d6)
d :7.77 (2H, d, J=9.0Hz), 7.62 (1H, brs) , 7.43 OH, brs), 7.29 (2H, d, J=8.8Hz) , 7.1 2 (2H, d, J=9.0Hz) , 6.85 (2H, d, J=8.8Hz) , 6.79 (1H, s), 4.96 (2H, s), 4.55 (2H, s) , 3. 71 (3H, s), 2.02-1.94 (3H, m), 1.79-1.61 (12H, ra) d: 7.77 (2H, d, J = 9.0Hz), 7.62 (1H, brs), 7.43 OH, brs), 7.29 (2H, d, J = 8.8Hz), 7.12 (2H, d, J = 9.0Hz) ), 6.85 (2H, d, J = 8.8Hz), 6.79 (1H, s), 4.96 (2H, s), 4.55 (2H, s), 3.71 (3H, s), 2.02-1.94 (3H, m), 1.79-1.61 (12H, ra)
実施例 3 2 Example 3 2
4— ( 1—ァダマンチル) 一 2— [4 - [ (ァミノカルボニル) メ卜キシ]フ ェニルスルホニルァミノ]チアゾールの合成 Synthesis of 4- (1-adamantyl) -1-2- [4-[(Aminocarbonyl) methoxy] phenylsulfonylamino] thiazole
実施例 2 3と同様の方法で 4一 (1ーァダマンチル) 一 2— [N— [4 - [ (ァ ミノカルボニル) メトキシ]フエニルスルホニル]一 M— (4 -メ卜キシベンジ ル) ァミノ]チアゾ一ル (1 6 Omg) から、 無色粉末として表題化合物 (Ί 08mg) を得た。 I1Rの結果は、 以下のとおりであった。
1H-N R (300MHz, DMSO - d6) In the same manner as in Example 23, 4- (1-adamantyl) -12- [N- [4-[(aminocarbonyl) methoxy] phenylsulfonyl] -1-M- (4-methoxybenzyl) amino] thiazo The title compound (Ί08 mg) was obtained as a colorless powder from one liter (16 Omg). The I1R results were as follows. 1 HNR (300MHz, DMSO-d6)
δ :12.6.1 (1H, brs) , 7.77 (2H, d, J=9.0Hz) , 7.58 (1H, brs) , 7.41 (1H, brs) , 7.05 (2H, d, J=9. OHz), 6.30 (1H, s),4.51 (2H, s) , 2.02-1.92 (3H, m) , 1.84-1.56 (12H, m) 実施例 33 δ: 12.6.1 (1H, brs), 7.77 (2H, d, J = 9.0Hz), 7.58 (1H, brs), 7.41 (1H, brs), 7.05 (2H, d, J = 9.OHz), 6.30 (1H, s), 4.51 (2H, s), 2.02-1.92 (3H, m), 1.84-1.56 (12H, m) Example 33
4一 (1—ァダマンチル) —2— [N- (3—クロロー 2—メチルフエニル スルホニル) 一 N—メチルァミノ] チアゾ一ルの合成 Synthesis of 4- (1-adamantyl) -2- [N- (3-chloro-2-methylphenylsulfonyl) -1-N-methylamino] thiazol
実施例 5で得られた 4一 (1ーァダマンチル) —2— (3—クロ口— 2—メ チルフエニルスルホニルァミノ) チアゾール (1 O Omg) の DM F溶液 (1. A solution of 4- (1-adamantyl) -2- (3-chloro-2--2-methylphenylsulfonylamino) thiazole (1 O Omg) obtained in Example 5 in DMF (1.
Om l ) に氷冷下で水素化ナトリウム (60%オイル 1 1 mg) を加え、 3 - 0分間攪袢した後、 ヨウ化メチル (1 5 ^ 1 ) を加えて室温に戻し、 一晩攪拌 した。 氷冷下、 水 (5m l ) を加えて発泡が止むまで攪拌し、 酢酸ェチル (1Oml), add sodium hydride (11 mg of 60% oil) under ice-cooling, stir for 3-0 minutes, add methyl iodide (15 ^ 1), return to room temperature, and stir overnight did. Under ice-cooling, add water (5 ml), stir until foaming stops, and add ethyl acetate (1
Om l ) で抽出した。 有機相を水と飽和食塩水の 1 : 1 混合液 (5m I X 2) 、 飽和食塩水 (5m l ) で順次洗浄した。 有機相を無水硫酸マグネシウムで乾燥 後、 乾燥剤を濾去し、 溶媒を減圧留去した。 得られた残渣をシリカゲルカラム クロマトグラフィー (展開溶媒 n -へキサン:酢酸ェチル =20 : 1 ) で精 製し、 無色油状物質として表題化合物 (57mg) を得た。 1H- MMRの結果は、 以下のとおりであった。 Oml). The organic phase was washed successively with a 1: 1 mixture of water and saturated saline (5 ml IX 2) and saturated saline (5 ml). After the organic phase was dried over anhydrous magnesium sulfate, the desiccant was removed by filtration, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (eluent: n-hexane: ethyl acetate = 20: 1) to give the title compound (57 mg) as a colorless oil. The results of 1 H-MMR were as follows.
^-NM (300MHz, DMS0-d6) , ^ -NM (300MHz, DMS0-d6),
6 :7.92 (1Η, d, J=8. OHz) ' 7.83 (1H, d, J=8. OHz) , 7.50 (1H, t, J=8. OHz) , 6.87 (1H, s) , 3.39 (3H, s) , 2.43 (3H, s) , 2.02-1.94 (3H, m) , 1.78-1.61 (12H, m) 6: 7.92 (1Η, d, J = 8.OHz) '7.83 (1H, d, J = 8.OHz), 7.50 (1H, t, J = 8.OHz), 6.87 (1H, s), 3.39 ( 3H, s), 2.43 (3H, s), 2.02-1.94 (3H, m), 1.78-1.61 (12H, m)
実施例 34 Example 34
4 - ( 1ーァダマンチル) 一 2— [N— (3—クロロー 2—メチルフエニル スルホニル) 一 N— (2 -ヒドロキシェチル) ァミノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) -1-2- [N- (3-chloro-2-methylphenylsulfonyl) -1-N- (2-hydroxyethyl) amino] thiazole
(1 ) 4一 (1—ァダマンチル) 一 2— [N- (3—クロ口— 2—メチル フエニルスルホニル) 一 W— [2 - (テ卜ラヒドロ - 2H—ピラン- 2 -ィル才 キシ) ェチル] ァミノ] チアゾールの合成 (1) 4 1 (1-adamantyl) 1 2— [N- (3-chloro- 2 -methylphenylsulfonyl) 1 W— [2-(tetrahydro-2H-pyran-2-ylyl) Synthesis of ethyl] amino] thiazole
実施例 33と同様の方法で 4一 (1 —ァダマンチル) 一 2— (3—クロ口— 2—メチルフエニルスルホニルァミノ) チアゾール (90mg) と 2 - (2 - プロモェ卜キシ)テ卜ラヒドロ - 2 H -ピラン (32 μ I ) から無色油状物質
/v:/ O 9s/-00s00ifcl£ 60sosAV In the same manner as in Example 33, 4- (1-adamantyl) -12- (3-chloro-2-methylphenylsulfonylamino) thiazole (90 mg) and 2- (2-promethoxy) tetrahydro -Colorless oil from 2H-pyran (32 μI) / v: / O 9s / -00s00ifcl £ 60sosAV
。 .
(s0¾9ρια憂 ¾-- , (s0¾9ρια ¾--,
9 9
( «cn^^)丄 ( v 6ϋ%ίιl09^-υEM..£
0分間攪拌した後、 2 -クロロアセ卜アミド (1 1 mg) とヨウ化カリウム (1 9mg) を加えて室温に戻し、 1. 5時間攪拌した。 反応温度を〗 00°C に昇温し、 4時間攪拌した。 2 -クロロアセ卜アミド (1 I mg) とヨウ化力 リウ厶 (1 9mg) を加えて、 1 00°Cで 3時間攪拌した後、 反応温度を 1 3 0°Cに昇温して、 6時間攪拌した。 更に 2 -クロロアセ卜アミド (1 I mg) とヨウ化カリウム (1 9mg) を加えて、 1 30°Cで 3時間攪拌した。 氷冷下、 水と飽和食塩水の 1 : 1混合液 (5m l ) を加えて、 酢酸ェチル (1 5m l ) で抽出した。 有機相を水と飽和食塩水の 1 : 1混合液 (5m I X2) 、 飽和食 塩水 (5m l ) で順次洗浄した。 有機相を無水硫酸マグネシウムで乾燥後、 乾 燥剤を濾去し、 溶媒を減圧留去した。 得られた残渣をシリカゲルカラムクロマ 卜グラフィー (展開溶媒 クロ口ホルム:メタノール =80 : 1 ) で精製し、 黄色油状物質として表題化合物 (27mg) を得た。 1H- NMRの結果は、 以下の とおりであった。 («Cn ^^) 丄 (v 6ϋ% ίιl09 ^ -υEM .. £ After stirring for 0 minutes, 2-chloroacetamide (11 mg) and potassium iodide (19 mg) were added, and the mixture was returned to room temperature and stirred for 1.5 hours. The reaction temperature was raised to about 00 ° C, and the mixture was stirred for 4 hours. After adding 2-chloroacetoamide (1 mg) and lithium iodide (19 mg) and stirring at 100 ° C for 3 hours, the reaction temperature was raised to 130 ° C and 6 Stirred for hours. Further, 2-chloroacetoamide (1 I mg) and potassium iodide (19 mg) were added, and the mixture was stirred at 130 ° C for 3 hours. Under ice-cooling, a 1: 1 mixture of water and saturated saline (5 ml) was added, and the mixture was extracted with ethyl acetate (15 ml). The organic phase was washed successively with a 1: 1 mixture of water and saturated saline (5 ml × 2) and saturated brine (5 ml). After the organic phase was dried over anhydrous magnesium sulfate, the drying agent was removed by filtration, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent, chromate form: methanol = 80: 1) to obtain the title compound (27 mg) as a yellow oily substance. The result of 1 H-NMR was as follows.
1H-NMR (300MHz, D S0-d6) 1H-NMR (300MHz, D S0-d6)
(5:8.02 (1H, d, J=8.0Hz), 7.82 (1H, d, J=8.0Hz) , 7.54 (1H, brs) , 7.48 (1H, t, J=8. OH z), 7.18 (1H, brs), 6.81 (1 H, s) 4.49 (2H, s) , 2.02-1 · 94 (3H, m), 1.78-1.61 (12H, m) 実施例 36 (5: 8.02 (1H, d, J = 8.0Hz), 7.82 (1H, d, J = 8.0Hz), 7.54 (1H, brs), 7.48 (1H, t, J = 8.OHz), 7.18 ( 1H, brs), 6.81 (1 H, s) 4.49 (2H, s), 2.02-194 (3H, m), 1.78-1.61 (12H, m) Example 36
4一 (1ーァダマンチル) 一 2— 一 (3—クロロー 2—メチルフエニル スルホニル) —N—ェチルァミノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) -2-1- (3-chloro-2-methylphenylsulfonyl) -N-ethylamino] thiazole
実施例 33と同様の方法で 4一 (1ーァダマンチル) 一 2— (3—クロ口— In the same manner as in Example 33, 4- (1-adamantyl) -1- (3-chloro-
2—メチルフエニルスルホニルァミノ) チアゾ一ル (88mg) とヨウ化工チ ル (23 I ) から黄色油状物質として表題化合物 (49mg) を得た。 ^ -The title compound (49 mg) was obtained as a yellow oil from 2-methylphenylsulfonylamino) thiazol (88 mg) and iodide tyl (23 I). ^-
NMRの結果は、 以下のとおりであった。 The result of NMR was as follows.
Ή-N R (300MHz, DMS0 - d6) Ή-N R (300MHz, DMS0-d6)
δ:7.91 (1Η, d, J=8. OHz) , 7.82 (1H, d, J=8.0Hz) , 7.49 (1H, t, J=8.0Hz) , 6.91 (1H, s)δ: 7.91 (1Η, d, J = 8.OHz), 7.82 (1H, d, J = 8.0Hz), 7.49 (1H, t, J = 8.0Hz), 6.91 (1H, s)
, 3.90 (2H, q, J=7. OHz) , 2.42 (3H, s) , 2.02-1.96 (3H, m), 1.78 -, 3.90 (2H, q, J = 7.OHz), 2.42 (3H, s), 2.02-1.96 (3H, m), 1.78-
1.61 (12H, m), 1.19 (3H, t, J=7. OHz) 1.61 (12H, m), 1.19 (3H, t, J = 7. OHz)
実施例 37
4 - (1ーァダマンチル) 一 2— CN- (3—クロロー 2—メチルフエニル スルホニル) 一 N—イソプチルァミノ] チアゾールの合成 Example 37 Synthesis of 4- (1-adamantyl) -1-2-CN- (3-chloro-2-methylphenylsulfonyl) -1-N-isobutylamino] thiazole
実施例 33と同様の方法で 4一 (1ーァダマンチル) 一 2— (3—クロロー 2—メチルフエニルスルホニルァミノ) チアゾール (90mg) とヨウ化イソ ブチル (1 22 μ I ) から黄色油状物質として表題化合物 (28mg) を得た。 In the same manner as in Example 33, 4- (1-adamantyl) -12- (3-chloro-2-methylphenylsulfonylamino) thiazole (90 mg) and isobutyl iodide (122 μI) were used as a yellow oily substance. The title compound (28 mg) was obtained.
1H -圖 Rの結果は、 以下のとおりであった。 The results of 1H-R are as follows.
-1H-N R (300MHz, DMS0-d6) - 1 HN R (300MHz, DMS0 -d6)
δ :7.88 (1Η, d, J=8. OHz), 7.80 (1H, d, J=8.0Hz) , 7.46 (1H, t, J=8.0Hz) , 6.94 (1H, s) , 3, 71 (ZH, d, J=7. OHz) , 2.38 (3H, s) , 2.02-1.92 (3H, m), 1, 91-1.80 (1H, m) , 1.78- 1.58(12H, m), 0.87 (6H, d J=7. OHz). δ: 7.88 (1Η, d, J = 8.OHz), 7.80 (1H, d, J = 8.0Hz), 7.46 (1H, t, J = 8.0Hz), 6.94 (1H, s), 3, 71 ( ZH, d, J = 7.OHz), 2.38 (3H, s), 2.02-1.92 (3H, m), 1, 91-1.80 (1H, m), 1.78-1.58 (12H, m), 0.87 (6H , d J = 7. OHz).
実施例 38 Example 38
4一 (1 —ァダマンチル) 一2— [2— (ヒドロキシメチル) フエニルスル ホニルァミノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) -1- [2- (hydroxymethyl) phenylsulfonylamino] thiazole
実施例 4一 23で得られた、 4— (1ーァダマンチル) 一 2— [2— (メ卜 キシカルボニル) フエニルスルホニルァミノ] チアゾール (250mg) をテ 卜ラヒドロフラン (1 OmL) に溶解し、 氷冷した。 この溶液に、 水素化リチ ゥ厶アルミニウム (44mg) を 5分間かけて加えた。 氷冷下で 1時間、 室温 で 3時間撹拌した後、 反応液を希塩酸に注,いだ。 得られた溶液を、 クロ口ホル 厶 (5 OmL) で抽出し、 飽和食塩水で洗浄し、 無水硫酸マグネシウムで乾燥 した。 乾燥剤を濾去し、 溶媒を減圧留去した。 得られた残渣をシリカゲルカラ 厶クロマトグラフィー (展開溶媒 酢酸ェチル: n—へキサン =1 : 1 ) で精 製して、 淡黄色アモルファスとして表題化合物 (1 80mg) を得た。 Example 4 4- (1-adamantyl) 1-2- [2- (methoxycarbonyl) phenylsulfonylamino] thiazole (250 mg) obtained in 23 was dissolved in tetrahydrofuran (1 OmL). Ice cooled. To this solution, lithium aluminum hydride (44 mg) was added over 5 minutes. After stirring for 1 hour under ice cooling and 3 hours at room temperature, the reaction solution was poured into diluted hydrochloric acid. The resulting solution was extracted with chloroform (5 OmL), washed with saturated saline, and dried over anhydrous magnesium sulfate. The drying agent was removed by filtration, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: ethyl acetate: n-hexane = 1: 1) to obtain the title compound (180 mg) as a pale yellow amorphous.
の結果は、 以下のとおりであった。 The results were as follows.
^-N R ( 300 MHz, DMS0 - d6 ). (5: 12.62 ( 1H, br s ), 7.86 ( 1H, d, J = 7.8 Hz ) , 7.76 ( 1H, d, J = 7.8 Hz ) , 7.59 ( 1H, t, J = 7.5 Hz ) , ^ -NR (300 MHz, DMS0-d6). (5: 12.62 (1H, br s), 7.86 (1H, d, J = 7.8 Hz), 7.76 (1H, d, J = 7.8 Hz), 7.59 (1H , t, J = 7.5 Hz),
7.39 ( 1H, t J = 7.5 Hz ) , 6.31 ( 1H, s ), 5.31 ( 1H, t, J = 5.5 Hz ), 4.95 ( 2H, d, J = 5.5 Hz ) , 2.04 - 1.94 ( 3H, m ), 1.86 - 1.76 ( 6H, m ), 1.74 - 1.60 ( 6H, m ) 7.39 (1H, t J = 7.5 Hz), 6.31 (1H, s), 5.31 (1H, t, J = 5.5 Hz), 4.95 (2H, d, J = 5.5 Hz), 2.04-1.94 (3H, m) , 1.86-1.76 (6H, m), 1.74-1.60 (6H, m)
実施例 39
(1 ) 1 - (3—ヒドロキシァダマンチル) エタノンの合成 Example 39 (1) Synthesis of 1- (3-hydroxyadamantyl) ethanone
3—ヒドロキシァダマンタン一 1—カルボン酸 (98 1 mg) のテ卜ラヒド 口フラン (20m I ) 溶液を氷冷し、 1 . 2 Mメチルリチウム溶液 (Ί 2. 5 m I、 ジェチルエーテル溶液) を 1 0分間で滴下した。 滴下終了後、 室温まで 温度を上げ、 6時間攪拌した。 反応液を水にあけ酢酸ェチル (50m l ) で抽 出し、 飽和食塩水 (5 0m l ) で洗浄した。 有機相を無水硫酸マグネシウムで 乾燥後、 乾燥剤を濾去し、 溶媒を減圧留去して、 無色固体として表題化合物 (40 Omg) を得た。 NMRの結果は、 以下のとおりであった。 A solution of 3-hydroxyadamantane-11-carboxylic acid (98 1 mg) in tetrahydrofuran (20 ml) was cooled on ice, and a 1.2 M methyllithium solution (Ί2.5 ml, getyl ether) was added. Solution) was added dropwise over 10 minutes. After the addition was completed, the temperature was raised to room temperature, and the mixture was stirred for 6 hours. The reaction solution was poured into water, extracted with ethyl acetate (50 ml), and washed with saturated saline (50 ml). After the organic phase was dried over anhydrous magnesium sulfate, the desiccant was removed by filtration, and the solvent was distilled off under reduced pressure to obtain the title compound (40 Omg) as a colorless solid. The result of NMR was as follows.
,[■!-隱 (300MHz, DMS0-d6) δ: 4.51 (1Η, s), 2.20- 2.10 (2H, m) , 2.06 (3Η, s) , 1.62-1. 7 (12Η, m) , [■! -Hidden (300MHz, DMS0-d6) δ: 4.51 (1Η, s), 2.20- 2.10 (2H, m), 2.06 (3Η, s), 1.62-1.7 (12Η, m)
(2) 4 - (3—ヒドロキシァダマンタン一 1 Jィル) 一 1, 3—チアゾール 一 2—ァミン合成 (2) Synthesis of 4- (3-hydroxyadamantane-1Jyl) 1-1,3-thiazole-12-amine
1 ― (3—ヒドロキシァダマンタン一 1—ィル) エタノン (3 9 Omg) の クロ口ホルム (1 Om l ) 溶液に、 室温で臭素 (0. 1 1 m l ) を加えた。 徐々に 60°Cまで加熱し、 同温で 1 0分間攪拌した。 溶媒を減圧留去し、 残渣 をエタノール (1 Om l ) に溶解し、 チォゥレア (1 5 2mg) を加え室温で 2日間攪拌した。 溶媒を減圧留去し、 残渣をシリカゲルカラムクロマ卜グラフ ィー (展開溶媒 クロ口ホルム:メタノ,ル: 28%アンモニア水 = 1 00 : 5 : 0. 5) で精製し、 無色固体として表題化合物 (2 5 Omg) を得た。 - NMRの結果は、 以下のとおりであった。 To a solution of 1- (3-hydroxyadamantane-11-yl) ethanone (39 Omg) in chloroform (1 Oml) was added bromine (0.11 ml) at room temperature. The mixture was gradually heated to 60 ° C and stirred at the same temperature for 10 minutes. The solvent was distilled off under reduced pressure, the residue was dissolved in ethanol (1 Oml), and thioprea (152 mg) was added, followed by stirring at room temperature for 2 days. The solvent was distilled off under reduced pressure, and the residue was purified by silica gel column chromatography (developing solvent: chloroform: methanol: 28% aqueous ammonia = 100: 5: 0.5) to give the title compound as a colorless solid. (25 Omg). -The result of NMR was as follows.
】Η-匿 (300MHz, DMS0-d6) δ: 6.77 (2Η, br ] Η-Hidden (300MHz, DMS0-d6) δ: 6.77 (2Η, br
s) , 6.00 (1 H, s) , 4.40 (1 H, s) , 2.14 (2H, br s) , 1.70-1.47 (12H, m) s), 6.00 (1 H, s), 4.40 (1 H, s), 2.14 (2H, br s), 1.70-1.47 (12H, m)
(3) 4一 (3—ヒドロキシァダマンタン一 1 一ィル) 一 2— [N_ [ (5 -ク ロロ - 3 -メチルベンゾ [b]チォフェン- 2 -ィル) スルホニル]一 N—メチル ァミノ] チアゾールの合成 (3) 4- (3-hydroxyadamantane-1) -1 2-[[N _ [(5-chloro-3-methylbenzo [b] thiophen-2-yl) sulfonyl] -1-N-methylamino ] Synthesis of thiazole
4— (3—ヒドロキシァダマンタン— 1 —ィル) — 1, 3—チアゾールー 2 —ァミン (22 Omg) をクロ口ホルム (5 m l ) に溶解し、 氷冷下卜リエチ ルァミン ( 0. 2 70m l ) と卜リメチルシリルクロリド ( 0. 1 2 3 m l ) を加え、 室温で 6時間攪拌した。 再び氷冷し 4—ジメチルァミノピリジン (1
08mg) と 5 -クロ口 - 3 -メチルベンゾ [b]チ才フェン - 2 -スルホニル クロリド (495mg) を加え、 室温に戻して一晩攪拌した。 28%アンモニ ァ水 (0. 2m l ) を加え室温で 2時間攪拌した後、 35%塩酸 (1 m l ) と メタノール (5m l ) を加えて 1 5分間攪拌した。 クロ口ホルム (50m l ) を加え、 飽和食塩水 (30m I X 3) で洗浄した。 有機相を無水硫酸マグネシ ゥ Aで乾燥後、 乾燥剤を濾去し、 溶媒を減圧留去した。 得られた残渣をシリカ ゲルカラムクロマトグラフィー (展開溶媒 クロ口ホルム:メタノール = 1 0 0 : 3) で精製し、 これに酢酸ェチルを加え、 析出した固体を濾取して淡黄色 固体として表題化合物 (1 53mg) を得た。 - MMRの結果は、 以下のとおり であった。 4- (3-Hydroxyadamantan-1-yl) -1,3-thiazol-2-amine (22 Omg) is dissolved in chloroform (5 ml), and triethylamine (0.2) is dissolved under ice-cooling. 70 ml) and trimethylsilyl chloride (0.123 ml) were added, and the mixture was stirred at room temperature for 6 hours. Cool again on ice and add 4-dimethylaminopyridine (1 08 mg) and 5-methyl-3 -methylbenzo [b] thiaphen-2-enesulfonyl chloride (495 mg) were added, and the mixture was returned to room temperature and stirred overnight. After adding 28% ammonia water (0.2 ml) and stirring at room temperature for 2 hours, 35% hydrochloric acid (1 ml) and methanol (5 ml) were added and stirred for 15 minutes. To the solution was added black-mouthed form (50 ml), and the mixture was washed with saturated saline (30 ml IX 3). After the organic phase was dried over anhydrous magnesium sulfate A, the desiccant was removed by filtration and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent, chromate form: methanol = 100: 3), ethyl acetate was added thereto, and the precipitated solid was collected by filtration to give the title compound as a pale yellow solid. (153 mg). -The results of the MMR are as follows.
1H-NMR (300MHz, D S0-d6) δ 12.98 (1Η, br 1 H-NMR (300MHz, D S0-d6) δ 12.98 (1Η, br
s) , 8.06 (1 H, d, J=8.6Hz) , 7.98 (1 H, d, J=2.1 Hz) , 7.54 (1 H, dd, J=8.6, s), 8.06 (1 H, d, J = 8.6 Hz), 7.98 (1 H, d, J = 2.1 Hz), 7.54 (1 H, dd, J = 8.6,
2.1Hz), 6.40 (1H, d, J=1.9Hz) , 2.61 (3H, s), 2.14 (2H, br s), 1.76-1.45 (12H, m) 実施例 40 2.1Hz), 6.40 (1H, d, J = 1.9Hz), 2.61 (3H, s), 2.14 (2H, br s), 1.76-1.45 (12H, m)
(1 ) 4- (1 -ァダマンチル) -5- (ェ卜キシカルボニル) -2-ァミノチアゾール 臭化水素酸塩の合成 (1) Synthesis of 4- (1-adamantyl) -5- (ethoxycarbonyl) -2-aminothiazole hydrobromide
ェチル 3- (1-ァダマンチル) -3-ォキソプロピオネー卜 (2. 0 Og) のェ夕 ノール溶液 (20ml) に臭素 (0. 4〗 ml,) を加え 80°Cで 30分攪拌した後、 室温に戻し、 チ才ゥレア (0. 49g) を 45°Cで 3時間攪拌した後、 室温に 戻して一晩攪拌した。 析出した固体を吸引濾取して回収し、 無色粉末として表 題化合物 (1. 31 g) を得た。 NMRの結果は、 以下のとおりであった。 Bromine (0.4 ml) was added to an ethanol solution (20 ml) of ethyl 3- (1-adamantyl) -3-oxopropionate (2.0 Og), and the mixture was stirred at 80 ° C for 30 minutes. Thereafter, the mixture was returned to room temperature, and stirred at 45 ° C. for 3 hours, and then returned to room temperature and stirred overnight. The precipitated solid was collected by suction filtration to give the title compound (1.31 g) as a colorless powder. The result of NMR was as follows.
1H-NMR (300MHz, D S0-d6) δ: 4.17 (2Η, q, J=7.1Hz), 2.15-2.08. (6H, m) , 2.04-1.95 (3H, m) , 1.78-1.64 (6H, m), 1.24 (3H, t, J=7.1 Hz) 1 H-NMR (300 MHz, DS0-d6) δ: 4.17 (2Η, q, J = 7.1 Hz), 2.15-2.08. (6H, m), 2.04-1.95 (3H, m), 1.78-1.64 (6H , m), 1.24 (3H, t, J = 7.1 Hz)
(2) 4一 (1 -ァダマンチル) —5— (ェ卜キシカルボニル) 一 2—([(5 - クロ口- 3-メチル -1-ベンゾチェン- 2-ィル)スルホニル]ァミノ 1チアゾールの合 実施例 4と同様の方法で 4- (1-ァダマンチル) -5- (エトキシカルボニル) - 2-ァミノチアゾール臭化水素酸塩 (500mg) と 5—クロロー 3—メチルベ ンゾ [b] チォフェン一 2—スルホニルクロリド (762 mg) から、 薄橙色
アモルファスとして表題化合物 (723mg) を得た。 】H- I\IMRの結果は、 以下 のとおりであった。 (2) 4- (1-adamantyl) -5- (ethoxycarbonyl) -12-([(5-chloro-3-methyl-1-benzophen-2-yl) sulfonyl] amino 1-thiazole In the same manner as in Example 4, 4- (1-adamantyl) -5- (ethoxycarbonyl) -2-aminothiazole hydrobromide (500 mg) and 5-chloro-3-methylbenzo [b] thiophene were used. From 2-sulfonyl chloride (762 mg), pale orange The title compound (723 mg) was obtained as amorphous. ] The results of H-I \ IMR are as follows.
!H- MR (300MHz, DMS0-d6) δ: 8.09 (1Η, d, J=8.6Hz) , 8.03 (1H, d, J=2.0Hz) , 7.57 (1H, dd, J=8.6, 2.0 Hz), 4.25 (2H, q, J=7.1 Hz), 2.63 (3H, s), 2.12-1.95 (9H, m) , 1.75-1.60 (6H, m), 1.27 (3H, t, J=7.1 Hz) 実施例 4 1 ! H-MR (300MHz, DMS0-d6) δ: 8.09 (1Η, d, J = 8.6Hz), 8.03 (1H, d, J = 2.0Hz), 7.57 (1H, dd, J = 8.6, 2.0 Hz) , 4.25 (2H, q, J = 7.1 Hz), 2.63 (3H, s), 2.12-1.95 (9H, m), 1.75-1.60 (6H, m), 1.27 (3H, t, J = 7.1 Hz) Example 4 1
4一 (1 -ァダマンチル) 一 5—メチル— 2— (5-クロ口- 3-メチル -1-ベン ゾチェン- 2-ィル)スルホニル]ァミノ iチアゾ一ルの合成 (A) 及び 4一 (1 - ァダマンチル) —5— (ヒドロキシメチル) 一 2— {[(5-クロ口- 3-メチル- 1- ベンゾチェン- 2-ィル)スルホニル]ァミノ)チアゾールの合成 (B) Synthesis of 4- (1-adamantyl) -1-5-methyl-2- (5-chloro-3-methyl-1-benzo-2-en) sulfonyl] amino ithiazolyl (A) and 4- ( Synthesis of 1-adamantyl) -5- (hydroxymethyl) 1-2-{[(5-chloro-3-methyl-1-benzocen-2-yl) sulfonyl] amino) thiazole (B)
実施例 40 (2) で得た 4一 (1 -ァダマンチル) —5— (エトキシカルボ ニル) 一 2— (5-ク口口 -3-メチル -1-ベンゾチェン- 2-ィル)スルホニル]ァミ ノ)チアゾール (600mg) のテ卜ラヒドロフラン溶液 (24ml) に氷冷下で 水素化リチウムアルミニウム (83mg) を加え、 室温に戻して 3時間攪拌し た後、 更に水素化リチウムアルミニウム (4 1 mg) を加えて、 室温でー晚攪 拌した。 反応液を氷冷下で 0. 5 M塩酸水溶液 (30m l ) に滴下した後、 ク ロロホルム (50m で抽出し飽和食塩水 (30tnl) で洗浄した。 有機相を無 水硫酸マグネシウムで乾燥後、 溶媒を減压留去した。 得られた残渣をシリカゲ ルクロマ卜グラフィー (展開溶媒 クロ口ホルム:メタノール = 1 00 : 1 ) で精製した後に、 更にプレパラティブ T LC (展開溶媒 クロ口ホルム:メタ ノール =80 : 1 ) で精製し、 無色アモルファスとして表題化合物 (A) (2 36mg) 及び薄黄色粉末として表題化合物 (B) (1 1 8mg) を得た。 ^- NMRの結果は、 以下のとおりであった。 Example 40 4- (1-Adamantyl) -5- (ethoxycarbonyl) 1-2- (5-co- mouth-3-methyl-1-benzocen-2-yl) sulfonyl] a obtained in (2) Lithium aluminum hydride (83 mg) was added to a tetrahydrofuran solution (24 ml) of (amino) thiazole (600 mg) under ice-cooling, and the mixture was returned to room temperature, stirred for 3 hours, and further mixed with lithium aluminum hydride (41 mg). ) And stirred at room temperature. The reaction solution was added dropwise to a 0.5 M aqueous hydrochloric acid solution (30 ml) under ice-cooling, then extracted with chloroform (50 m and washed with saturated saline (30 tnl). The organic phase was dried over anhydrous magnesium sulfate. The obtained residue was purified by silica gel chromatography (developing solvent: chloroform: methanol = 100: 1), and further purified by preparative TLC (developing solvent: chloroform: methanol). = 80: 1) to give the title compound (A) (236 mg) as a colorless amorphous and the title compound (B) (118 mg) as a pale yellow powder. Met.
表題化合物 (A) Title compound (A)
1H-NMR (300MHz, DMS0~d6) δ 12.40 (1Η, br s), 8.05 (1H, d, J=8.6Hz) , 7.97 (1H, d, J=2.1Hz), 7.54 (1H, dd, J=8.6, 2.1 Hz), 2.60 (3H, s), 2.27 (3H, s), 1.92-2.02 (9H, ml, 1.75-1.60 (6H, m) 1H-NMR (300MHz, DMS0 ~ d6) δ 12.40 (1Η, br s), 8.05 (1H, d, J = 8.6Hz), 7.97 (1H, d, J = 2.1Hz), 7.54 (1H, dd, J = 8.6, 2.1 Hz), 2.60 (3H, s), 2.27 (3H, s), 1.92-2.02 (9H, ml, 1.75-1.60 (6H, m)
表題化合物 (B)
^- MR (300MHz, DIWS0-d6) δ: 13.06 (1H, s), 8.06 (1H, d, J=8.6Hz) , 7.95 (1H, d, J=2.1Hz), 7.54 (1H, dd, J=8.6, 2.1 Hz), 5.79 (1H, t, J=5.5Hz) , 4.60 (2H, d, J=5.5Hz) , 2.60 (3H, s) , 2.00-1.89 (9H, m) , 1.75—1.59 (6H, m) Title compound (B) ^-MR (300MHz, DIWS0-d6) δ: 13.06 (1H, s), 8.06 (1H, d, J = 8.6Hz), 7.95 (1H, d, J = 2.1Hz), 7.54 (1H, dd, J) = 8.6, 2.1 Hz), 5.79 (1H, t, J = 5.5Hz), 4.60 (2H, d, J = 5.5Hz), 2.60 (3H, s), 2.00-1.89 (9H, m), 1.75—1.59 (6H, m)
実施例 42 Example 42
(1 ) 4- (1ーァダマンチル) 一 2— [ (2 -フル才ロフエニルスルホニ ル) ァミノ] チアゾールの合成 (1) Synthesis of 4- (1-adamantyl) -1-2-([2-furofilophenylsulfonyl) amino] thiazole
実施例 4と同様の方法で 4- (1-ァダマンチル) -2-ァミノチアゾール臭化水 素酸塩 (600mg) と 2—フル才ロベンゼンスルホニルクロリド (74 1 m g) から、 薄茶色粉末として表題化合物 (882mg) を得た。 1H-剛 Rの結果' は、 以下のとおりであった。 In the same manner as in Example 4, 4- (1-adamantyl) -2-aminothiazole hydrobromide (600 mg) and 2-furoserobenzenesulfonyl chloride (74 1 mg) were used as a light brown powder. The title compound (882 mg) was obtained. The results of 1 H-go R 'were as follows.
1H-NMR (300MHz, DMS0-d6) <5: 12.84 (1H, br s) , 7, 99 (1Η, m) , 7.84 (1Η, m), 7.20-7.45 (2Η, m) , 6.36 (1H, s), 1.60-2.06 (15H, m) 1 H-NMR (300MHz, DMS0 -d6) <5: 12.84 (1H, br s), 7, 99 (1Η, m), 7.84 (1Η, m), 7.20-7.45 (2Η, m), 6.36 (1H , s), 1.60-2.06 (15H, m)
(2) 4- (1ーァダマンチル) _2— [N— (2 -フルオロフェニルスルホニ ル) — N— (4 (2) 4- (1-adamantyl) _2— [N— (2-fluorophenylsulfonyl) — N— (4
-メドキシベンジル) ァミノ]チアゾールの合成 -Medoxybenzyl) amino] thiazole
実施例 2 1と同様の方法で 4一 (1ーァダマンチル) 一 2— [ (2 -フル才 口フエニルスルホニル) ァミノ] チアゾ"ル (800mg) と P -メ卜キシべ ンジルクロライド (444 M I ) から、 茶色油状物質として表題化合物 (20 Omg) を得た。 ^-NMRの結果は、 以下のとおりであった。 Example 21 In the same manner as in Example 1, 4- (1-adamantyl) 1-2-[(2-full-age phenylsulfonyl) amino] thiazo "(800 mg) and P-methoxybenzyl chloride (444 MI ) To give the title compound (20 Omg) as a brown oily substance.The results of ^ -NMR were as follows.
^- MR (300MHz, DMS0-d6) 6: 7.92 (1H, td, J=7.5, 1.7Hz), 7.80 (1H, m) , 7.55-7.40 (2H, m) , 7.28 (2H, d, J=8.8Hz) , 6.87 (2H, d, J=8.8 Hz) , 6.80 (1H, s,), 5.06 (2H, s), 3.71 (3H, s), 2.02-1.94 (3H, m), 1.75-1.60 (12H, m) ^-MR (300MHz, DMS0-d6) 6: 7.92 (1H, td, J = 7.5, 1.7Hz), 7.80 (1H, m), 7.55-7.40 (2H, m), 7.28 (2H, d, J = 8.8Hz), 6.87 (2H, d, J = 8.8 Hz), 6.80 (1H, s,), 5.06 (2H, s), 3.71 (3H, s), 2.02-1.94 (3H, m), 1.75-1.60 (12H, m)
(3) 4— (1ーァダマンチル) — 2— [N— (4 -メ卜キシベンジル) 一N— C2 - (2 -モルホリノエトキシ) フエニルスルホニル] ァミノ]チアゾール の合成 (3) Synthesis of 4 -— (1-adamantyl) — 2 -— [N— (4-Methoxybenzyl) 1N—C2- (2-morpholinoethoxy) phenylsulfonyl] aminoaminothiazole
実施例 24と同様の方法で 4一 (Ίーァダマンチル) 一 2— [N— (2 -フル オロフェニルスルホニル) — N— (4 -メ卜キシベンジル) ァミノ]チアゾール
(1 88mg) と N - (2 -ヒドロキシェチル) モルホリン (96mg) から、 薄茶色油状物質として表題化合物 (79mg) を得た。 1H- Rの結果は、 以下 のとおりであった。 In the same manner as in Example 24, 41- (adamantyl) -12- [N- (2-fluorophenylsulfonyl) -N- (4-methoxybenzyl) amino] thiazole (188 mg) and N- (2-hydroxyethyl) morpholine (96 mg) to give the title compound (79 mg) as a pale brown oil. The results of 1 H-R were as follows.
^- MR (300MHz, DMS0-d6) δ 7.93 (1Η, dd, J=7.9, 1.7Hz), 7.65 (1H, ml, 7.36 (2H, d, J=8.7Hz) , 7.29 (1H, d, J=8.4Hz), 7.12 (1H, t, J=7.9 Hz) , 6.87 (2H, d, J=8.7 Hz) , 6.65 (1H, s) , 5.24 (2H, s), 4.20 (2H, t, ^-MR (300MHz, DMS0-d6) δ 7.93 (1Η, dd, J = 7.9, 1.7Hz), 7.65 (1H, ml, 7.36 (2H, d, J = 8.7Hz), 7.29 (1H, d, J = 8.4Hz), 7.12 (1H, t, J = 7.9 Hz), 6.87 (2H, d, J = 8.7 Hz), 6.65 (1H, s), 5.24 (2H, s), 4.20 (2H, t,
J=6.0Hz) , 3.71 (3H, s) , 3.52 (4H, m), 2.58 (2H, t, J=6.0Hz) , 2.40 (4H, m), 2.02-1.94 (3H, m) , 1.80-1.60 (12H, m) J = 6.0Hz), 3.71 (3H, s), 3.52 (4H, m), 2.58 (2H, t, J = 6.0Hz), 2.40 (4H, m), 2.02-1.94 (3H, m), 1.80- 1.60 (12H, m)
(4) 4- (1 —ァダマンチル) 一 2— [2 - (2 -モルホリノエ卜キシ) フ ェニルスルホニルァミノ]チアゾールの合成 (4) Synthesis of 4- (1-adamantyl) -1-2- [2- (2-morpholinoethoxy) phenylsulfonylamino] thiazole
実施例 23と同様の方法で 4一 (1ーァダマンチル) 一 2— [N— (4 -メ卜 キシベンジル) —N— [2 - (2 -モルホリノエ卜キシ) フエニルスルホニ ル] ァミノ]チアゾール (67mg) から無色アモルファスとして表題化合物 (48mg) を得た。 1H- NMRの結果は、 以下のとおりであった。 In the same manner as in Example 23, 4- (1-adamantyl) 1-2- [N— (4-methoxybenzyl) —N— [2- (2-morpholinoethoxy) phenylsulfonyl] amino] thiazole (67 mg) The title compound (48 mg) was obtained as a colorless amorphous. The result of 1 H-NMR was as follows.
1H-NMR (300MHz, D S0-d6) 6: 7.82 (1H, dd, J=7.7, 1.7Hz) , 7.52 (1H, m), 7.19 (1H, d, J=8.6Hz) , 7.04 (1H, m) , 6.32 (1H, s), 4.10 (2H, t, J=6.6 Hz) , 3.55 (2H, m) , 2.55-2.37 (6H, m), 2.04-1.58 (15H, m) 1H-NMR (300MHz, DS0-d6) 6: 7.82 (1H, dd, J = 7.7, 1.7Hz), 7.52 (1H, m), 7.19 (1H, d, J = 8.6Hz), 7.04 (1H, m), 6.32 (1H, s), 4.10 (2H, t, J = 6.6 Hz), 3.55 (2H, m), 2.55-2.37 (6H, m), 2.04-1.58 (15H, m)
実施例 43 ' Example 43 '
( 1 ) 3—メ卜キシァダマンタン一 1一力ルボン酸メチルエステルの合成 水素化ナトリウム (60%オイル、 40 Omg) を n—へキサン (2 Om I X 3) で洗浄し、 DM F (30m l ) を加え、 更に 3—ヒドロキシァダマン夕 ンー 1一力ルボン酸 (98 Omg) を加えて室温で 1時間攪拌した。 この溶液 に、 ヨウ化メチル (0. 93m l ) を加え室温で一晩攪拌した。 反応液をジェ チルエーテル (1 50m l ) で希釈し、 水 (1 0 Om I X 3) 次いで飽和食塩 水 O 0 Om I ) で洗浄した。 有機相を無水硫酸マグネシウムで乾燥後、 乾燥 剤を濾去し、 溶媒を減圧留去した。 得られた残渣をシリカゲルカラムクロマ卜 グラフィー (展開溶媒 酢酸ェチル: n—へキサン: = 1 : 4) で精製し、 無色 油状物質として表題化合物 (80 Omg) を得た。 1H- NMRの結果は、 以下のと おりであった。
1H-賺 (300MHz, DMS0-d6) δ: 3.67 (3H, s), 3.25 (3H, s) , 2.36- 2.14 (2H, m), 1.92-1.53 (12H, m) (1) Synthesis of 3-Methoxy-adamantane-l-rubinic acid methyl ester Sodium hydride (60% oil, 40 Omg) was washed with n-hexane (2 Om IX 3) and DMF (30 ml) Was added, and 3-hydroxyadaman-1-yl rubonic acid (98 Omg) was further added, followed by stirring at room temperature for 1 hour. Methyl iodide (0.93 ml) was added to this solution, and the mixture was stirred at room temperature overnight. The reaction solution was diluted with ethyl ether (150 ml) and washed with water (10 Om IX 3) and then with saturated saline O 0 Om I). After the organic phase was dried over anhydrous magnesium sulfate, the desiccant was removed by filtration, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (developing solvent: ethyl acetate: n- hexane: = 1: 4) to give the title compound (80 Omg) as a colorless oily substance. The result of 1 H-NMR was as follows. 1 H-note (300MHz, DMS0-d6) δ: 3.67 (3H, s), 3.25 (3H, s), 2.36- 2.14 (2H, m), 1.92-1.53 (12H, m)
(2) 3—メ卜キシァダマンタン一 1一力ルボン酸の合成 (2) Synthesis of 3-methoxydamantane
3—メ卜キシァダマンタン一 1—カルボン酸メチルエステル (71 Omg) をメタノール (5 m I ) に溶解し、 5 M N aOH水溶液 (0. 95m l ) を 加えて、 一晚攪拌した。 更に 70°Cで 2時間攪拌した。 溶媒を減圧留去し、 残 渣に 0 · 5 M塩酸水溶液 (20m l ) を加え、 クロロホルム (50m l ) で抽 出した。 クロ口ホルム相を飽和食塩水で洗浄後、 無水硫酸マグネシウムで乾燥 した。 乾燥剤を濾去し、 溶媒を減圧留去し無色固体として表題化合物 (650 mg) を得た。 1H- の結果は、 以下のとおりであった。 3-Methoxyadamantane-1-carboxylic acid methyl ester (71 Omg) was dissolved in methanol (5 ml), a 5 M NaOH aqueous solution (0.95 ml) was added, and the mixture was stirred for a while. The mixture was further stirred at 70 ° C for 2 hours. The solvent was distilled off under reduced pressure, a 0.5 M aqueous hydrochloric acid solution (20 ml) was added to the residue, and the mixture was extracted with chloroform (50 ml). The porthole form phase was washed with saturated saline and dried over anhydrous magnesium sulfate. The drying agent was removed by filtration, and the solvent was distilled off under reduced pressure to obtain the title compound (650 mg) as a colorless solid. The result of 1 H- was as follows.
】H -匿 (300MHz, DMS0-d6) δ 12.11 (1Η, s) ,.3. Π (3Η, s), 2.23— ] H-hidden (300MHz, DMS0-d6) δ 12.11 (1Η, s), .3. Π (3Η, s), 2.23—
2.13 (2Η, m), 1.74-1.48 (12Η, m) 2.13 (2Η, m), 1.74-1.48 (12Η, m)
(3) Ν, 3—ジメ卜キシー Ν—メチルァダマンタン一 1—カルボン酸アミドの 合成 (3) Synthesis of Ν, 3-dimethyloxyΝ-methyladamantane-1-carboxamide
3—メ卜キシァダマンタン一 1—カルボン酸 (6 Ί Omg) と , 0-ジメチル ヒドロキシルァミン塩酸塩 (284mg) 、 1-ヒドロキシベンゾ卜リアゾール 1水和物 (426mg) 、 卜リエチルァミン (0. 44ml) の DMF (6ml) 溶液に、 氷冷下 1-ェチル -3- (3-ジメチルァミノプロピル)カルポジイミド塩酸 塩 (604mg) を加え 1 時間攪拌した後、 室温で一晩攪拌した。 反応終了後、 水を加え、 酢酸ェチルで抽出し、 飽和炭酸水素ナトリウム水溶液、 塩酸水 溶液、 飽和食塩水で順次洗浄した。 有機相を無水硫酸マグネシウムで乾燥後、 溶媒を減圧留去した。 残渣をシリカゲルカラムクロマトグラフィー (へキサ ン:酢酸ェチル == 1 : 1 ) で精製し、 無色油状物質として表題化合物 (540 mg) を得た。 -NMRの結果は、 以下のとおりであった。 3-Methoxy adamantane 1-carboxylic acid (6 Ί Omg), 0-dimethylhydroxylamine hydrochloride (284 mg), 1-hydroxybenzotriazole monohydrate (426 mg), triethylamine (0.44 ml) To a DMF (6 ml) solution of the above was added 1-ethyl-3- (3-dimethylaminopropyl) carboimide hydrochloride (604 mg) under ice-cooling, and the mixture was stirred for 1 hour and then at room temperature overnight. After completion of the reaction, water was added, and the mixture was extracted with ethyl acetate. After the organic phase was dried over anhydrous magnesium sulfate, the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (hexane: ethyl acetate == 1: 1) to give the title compound (540 mg) as a colorless oil. The result of -NMR was as follows.
^-NMR (300MHz, DMS0-d6) «5: 3.68 (3H, s), 3.26 (3H, s), 3.17 (3H, s), 2.16 — 2.36 (2H, m), 2.01-1.96 (2H, m), 1.94-1.87 (3H, m), 1.78-1.68 (4H, m), 1.64 - 1.54 (3H, m) ^ -NMR (300MHz, DMS0-d6) «5: 3.68 (3H, s), 3.26 (3H, s), 3.17 (3H, s), 2.16 — 2.36 (2H, m), 2.01-1.96 (2H, m ), 1.94-1.87 (3H, m), 1.78-1.68 (4H, m), 1.64-1.54 (3H, m)
(4) 1— (3—メ卜キシ一 1ーァダマンチル) エタノンの合成
窒素置換下、 N, 3—ジメ卜キシー N—メチルァダマンタン一 1—カルボン酸 アミド (540mg) のテ卜ラヒドロフラン (8ml) 溶液に氷冷下、 3Mメチ ルマグネシウムブロミドのジェチルエーテル溶液 (2. 82ml) を滴下し、 同 温で 3時間攪拌した。 反応終了後、 反応液を飽和塩化アンモニゥム水に (40 ml) にあけ、 クロ口ホルムで抽出し、 飽和食塩水で洗浄した。 有機相を無水硫 酸マグネシウムで乾燥後、 溶媒を 圧留去し、 無色油状物質として表題化合物 (40 Omg) を得た。 Rの結果は、 以下のとおりであった。 (4) Synthesis of 1- (3-methoxy-1-1-adamantyl) ethanone Under a nitrogen atmosphere, a 3M methylmagnesium bromide getyl ether solution (3M methylmagnesium bromide) was added to a tetrahydrofuran (8ml) solution of N, 3-dimethyloxyn-methyladamantane-11-carboxylic acid amide (540mg) under ice cooling. 2.82 ml) was added dropwise, and the mixture was stirred at the same temperature for 3 hours. After the completion of the reaction, the reaction solution was poured into saturated aqueous ammonium chloride (40 ml), extracted with chloroform, and washed with saturated saline. After the organic phase was dried over anhydrous magnesium sulfate, the solvent was distilled off under reduced pressure to obtain the title compound (40 Omg) as a colorless oily substance. The results of R were as follows.
'W- (300MHz, DMS0-d6) δ: 3.25 (3Η, s) , 2.41-2.24 (2Η, m), 2.12 (3Η, s), 1.83-1.70 (9H, m), 1.66-1.51 (3H, m) 'W- (300MHz, DMS0-d6) δ: 3.25 (3Η, s), 2.41-2.24 (2Η, m), 2.12 (3Η, s), 1.83-1.70 (9H, m), 1.66-1.51 (3H, m)
(5) 4— (3—メ卜キシー 1—ァダマンチル) — 1, 3—チアゾールー 2— アミン臭化水素酸塩の合成 (5) Synthesis of 4- (3-Methoxy-1-adamantyl) —1,3-thiazol-2-amine hydrobromide
実施例 39 (2) と同様の方法で 1 — (3—メ卜キシー 1—ァダマンチル) エタノン (400mg) と臭素 (0. 1 1 m l ) とチ才ゥレア (1 46mg) から、 無色粉末として表題化合物 (49 Omg) を得た。 - NMRの結果は、 以 下のとおりであった。 Example 39 In the same manner as in (2), 1- (3-methoxy-1-adamantyl) ethanone (400 mg), bromine (0.1 ml) and thiaurea (146 mg) were obtained as a colorless powder. The compound (49 Omg) was obtained. -The result of NMR was as follows.
^-NMR (300MHz, DWIS0-d6) δ: 6.50 (1Η, s), 3.14 (3H, s) , 2.39-2.12 (2H, m), 1.87-1.43 (12H, m) ^ -NMR (300MHz, DWIS0-d6) δ: 6.50 (1Η, s), 3.14 (3H, s), 2.39-2.12 (2H, m), 1.87-1.43 (12H, m)
(6) 3—クロロー N— [4— (3—メ卜キシー 1ーァダマンチル) 一 1, 3— チアゾ一ルー 2—ィル]— 2—メチルベンゼンスルホンアミドの合成 (6) Synthesis of 3-chloro-N- [4- (3-methoxy-1-adamantyl) -1-1,3-thiazo-1-yl-2-yl]-2-methylbenzenesulfonamide
実施例 4と同様の方法で 4一 (3—メ卜キシ— 1ーァダマンチル) — 1, 3 一チアゾール—2—アミン臭化水素酸塩 (1 03mg) と 3—クロロー 2—メ チルベンゼンスルホニルクロリド (1 35mg) から、 無色粉末として表題ィ匕 合物 (8 Omg) を得た。 NMRの結果は、 以下のとおりであった。 In the same manner as in Example 4, 4- (3-methoxy-1-adamantyl) -1,3-thiazole-2-amine hydrobromide (103 mg) and 3-chloro-2-methylbenzenesulfonyl chloride (135 mg) to give the title compound (8 Omg) as a colorless powder. The result of NMR was as follows.
^- MR (300MHz, D S0-d6) δ: 12.75 (1Η, br s) 7.91 (1H, dd, J=7.9, 1.4 Hz), 7.68 (1H, d, J=7.9 Hz) , 7.39 (1H, t like, J=7.9) , 6.39 (1H, s), 3.11 (3H, s), 2.65 (3H, s), 2.26-2.10 (2H, m), 1.80-1.73 (2H, m), 1.73-1.68 (4H, m), 1.67—1.59 (4H, m) , 1.58-1.47 (2H, m) ^-MR (300MHz, D S0-d6) δ: 12.75 (1Η, br s) 7.91 (1H, dd, J = 7.9, 1.4 Hz), 7.68 (1H, d, J = 7.9 Hz), 7.39 (1H, t like, J = 7.9), 6.39 (1H, s), 3.11 (3H, s), 2.65 (3H, s), 2.26-2.10 (2H, m), 1.80-1.73 (2H, m), 1.73-1.68 (4H, m), 1.67--1.59 (4H, m), 1.58-1.47 (2H, m)
実施例 44
5—クロ口一 N— [4— (3—メ卜キシー 1ーァダマンチル) — 1, 3—チアゾ 一ルー 2—ィル]—3—メチルー 1一ベンゾチ才フェン一 2—スルホンアミド の合成 Example 44 Synthesis of N- [4- (3-Methoxy-1-adamantyl) —1,3-thiazo-1-yl-2-yl] -3-methyl-1-benzothienephen-1--2-sulfonamide
実施例 4と同様の方法で 4— (3—メトキシ— 1—ァダマンチル) — 1 , 3 —チアゾ一ル—2—アミン臭化水素酸塩 (1 03 mg) と 5—クロロー 1 —ベ ンゾチォフェン一 2—スルホニルクロリド (1 68mg) から、 無色粉末とし て表題化合物 (6 1 mg) を得た。 ^-NMRの結果は、 以下のとおりであった。 In the same manner as in Example 4, 4- (3-methoxy-1-adamantyl) -1,3, thiazol-2-aminehydrobromide (103 mg) and 5-chloro-1-benzothiophene were used. The title compound (61 mg) was obtained as colorless powder from 2-sulfonyl chloride (168 mg). ^ -NMR results were as follows.
^- MR (300MHz, D S0-d6) <5: 12.96 (1H, brs), 8.06 (1H, d, J=8.7 Hz) , 7.98 ΠΗ' d, J=2.0 Hz) , , 7.54 OH, dd, J=8.7, 2.0 Hz), 6.45 (1H, s), 3.11 (3H, s), 2.61 (3H, s), 2.29-2.09 (2H, m), 1.77-1.73 (2H, m), 1.73-1.67 (4H, m), 1.66-1.59 (4H, m) , 1.57-1.46 (2H, m) ^-MR (300MHz, D S0-d6) <5: 12.96 (1H, brs), 8.06 (1H, d, J = 8.7 Hz), 7.98 d 'd, J = 2.0 Hz),, 7.54 OH, dd, J = 8.7, 2.0 Hz), 6.45 (1H, s), 3.11 (3H, s), 2.61 (3H, s), 2.29-2.09 (2H, m), 1.77-1.73 (2H, m), 1.73-1.67 (4H, m), 1.66-1.59 (4H, m), 1.57-1.46 (2H, m)
実施例 45 Example 45
4一 ( 1ーァダマンチル) 一 2— (2—ナフチルスルホニルァミノ) チアゾ一 ルの合成 4-Synthesis of 1- (1-adamantyl) 1-2- (2-naphthylsulfonylamino) thiazole
参考例 2で得た 4一 ( 1ーァダマンチル) 一 2—ァミノチアゾール (50m g) のクロ口ホルム (3m l ) 溶液に、 ピリジン (68. 3 μ. \ ) を加えた後、 2—ナフタレンスルホニルクロリド (1 9 1. 9mg) を加え、 マイクロゥェ ープ反応装置を用い、 1 00°Cで〗 5分攪拌した。 反応液にクロ口ホルムを加 え、 飽和塩化アンモニゥ厶水溶液洗浄し、 有機相を減圧で濃縮した。 得られた 残渣を T LCプレー卜 (富士シリシァ化学社製 T LCプレート NH) を用いて 精製したところ、 無色固体として表題化合物 (40. 1 mg) を得た。 - NMR の結果は、 以下のとおりであった。 To a solution of 4- (1-adamantyl) -12-aminothiazole (50 mg) in chloroform (3 ml) obtained in Reference Example 2 was added pyridine (68.3 μ. \), And then 2-naphthalene. Sulfonyl chloride (191.19 mg) was added, and the mixture was stirred at 100 ° C. for about 5 minutes using a microbeak reactor. To the reaction mixture was added chloroform, washed with a saturated aqueous ammonium chloride solution, and the organic phase was concentrated under reduced pressure. The resulting residue was purified using a TLC plate (TLC plate NH, manufactured by Fuji Silicon Chemicals Ltd.) to give the title compound (40.1 mg) as a colorless solid. -The result of NMR was as follows.
^- MR (300MHz, DMS0-d6) δ : 9.45 ΠΗ, s), 8.52 (1Η, s), 8.06-7.79 (4Η, m), 7.76-7.45 (2H, tn) , 5.93 (1H, s) , 2.20-1.96 (3H, m), 1.95-1.46 (12H, m) ^-MR (300MHz, DMS0-d6) δ: 9.45ΠΗ, s), 8.52 (1Η, s), 8.06-7.79 (4Η, m), 7.76-7.45 (2H, tn), 5.93 (1H, s), 2.20-1.96 (3H, m), 1.95-1.46 (12H, m)
実施例 46 Example 46
4— (1 -ァダマンチル) 一 2— (2, 3, 4—トリクロ口フエニルスルホニ ルァミノ) チアゾールの合成
実施例 45と同様の方法で、 表題化合物を得た。 - Rの結果は、 以下のと おりであった。 Synthesis of 4- (1-adamantyl) -1-2- (2,3,4-trichloromethylphenylsulfonylamino) thiazole The title compound was obtained in the same manner as in Example 45. -The results of R were as follows.
^-NMR (300MHz, DMS0~d6) 6: 8.10 (1H, d, J=8.6 Hz) , 7.51 (1H, d, J=8.6 Hz) , 6.00 (1H, s) , 2.21-2.01 (3H, m) , 1.93-1.62 (12H, m) ^ -NMR (300MHz, DMS0 ~ d6) 6: 8.10 (1H, d, J = 8.6 Hz), 7.51 (1H, d, J = 8.6 Hz), 6.00 (1H, s), 2.21-2.01 (3H, m ), 1.93-1.62 (12H, m)
実施例 47 Example 47
4一 (1 -ァダマンチル) 一 2— (2, 4, 5—トリフルオロフェニルスルホ ニルァミノ) チアゾールの合成 Synthesis of 4- (1-adamantyl) -1-2- (2,4,5-trifluorophenylsulfonylanamino) thiazole
実施例 45と同様の方法で、 表題化合物を得た。 1H- Rの結果は、 以下のと おりであった。 The title compound was obtained in the same manner as in Example 45. The results of 1 H-R were as follows.
(300MHz, DMS0-06) δ: 9.93 (1H, br s), 7.93-7.77 (1H, m), 7.09- (300MHz, DMS0-06) δ: 9.93 (1H, br s), 7.93-7.77 (1H, m), 7.09-
6.93 (1H, m), 6.02 (1H, s), 2.22-2.02 (3H, m), 1.92-1.66 (12H, m) 実施例 48 6.93 (1H, m), 6.02 (1H, s), 2.22-2.02 (3H, m), 1.92-1.66 (12H, m)
4一 (1 -ァダマンチル) 一 2— (2, 6—ジクロ口フエニルスルホニルアミ ノ) チアゾールの合成 Synthesis of 4- (1-adamantyl) -1-2- (2,6-dichroic phenylsulfonylamino) thiazole
実施例 45と同様の方法で、 表題化合物を得た。 刚 Rの結果は、 以下のと おりであった。 The title compound was obtained in the same manner as in Example 45.刚 The results of R were as follows.
1H-NMR (300MHz, DMS0-d6) δ: 9.85 (1Η, br s), 7.49-7.37 (2H, m), 7.33- 1 H-NMR (300 MHz, DMS0-d6) δ: 9.85 (1Η, br s), 7.49-7.37 (2H, m), 7.33-
7.20 (1H, m), 5.98 (1H, s), 2.18 - 2.04,(3H, m), 1.92-1.66 (9H, m), 1.65-1.51 (3H, m) 7.20 (1H, m), 5.98 (1H, s), 2.18-2.04, (3H, m), 1.92-1.66 (9H, m), 1.65-1.51 (3H, m)
実施例 49 Example 49
4一 (1 -ァダマンチル) 一 2— [ (3—プロモー 5—クロロチ才フェン— 2 一ィル) スルホニルァミノ] チアゾールの合成 Synthesis of 4- (1-adamantyl) -1-2-([3-promo 5-chlorothiophene-2-yl) sulfonylamino] thiazole
実施例 45と同様の方法で、 表題化合物を得た。 Rの結果は、 以下のと おりであった。 The title compound was obtained in the same manner as in Example 45. The results of R were as follows.
^- MR (300MHz, D S0-d6) δ: 9.83 (1Η, br s), 6.90 (1H, s), 6.03 (1H, s), 2.20-2.05 (3H, m), 1.95—1.54 (12H, m) ^-MR (300MHz, D S0-d6) δ: 9.83 (1Η, br s), 6.90 (1H, s), 6.03 (1H, s), 2.20-2.05 (3H, m), 1.95-1.54 (12H, m)
実施例 50 Example 50
4一 (1 -ァダマンチル) 一 2— (2, 3—ジクロ口フエニルスルホニルアミ ノ) チアゾ一ルの合成
実施例 45と同様の方法で、 表題化合物を得た。 MMRの結果は、 以下のと おりであった。 Synthesis of 4- (1-adamantyl) -1-2- (2,3-dichroic phenylsulfonylamino) thiazole The title compound was obtained in the same manner as in Example 45. The results of the MMR are as follows.
'W-MR (300MHz, DMS0-d6) δ: 9.85 (IH, br s), 8.16 (1H, dd, J=7.9, 1.55 Hz), 7.61 (1H, dd, J=8.1, 1.55 Hz), 7.37-7.29 (1H, m) , 5.99 (1H, s), 2.18-2.0.4 (3H, m) , 1.92-1.51 (12H, m) 'W-MR (300MHz, DMS0-d6) δ: 9.85 (IH, br s), 8.16 (1H, dd, J = 7.9, 1.55 Hz), 7.61 (1H, dd, J = 8.1, 1.55 Hz), 7.37 -7.29 (1H, m), 5.99 (1H, s), 2.18-2.0.4 (3H, m), 1.92-1.51 (12H, m)
実施例 5 1 Example 5 1
4— (1 -ァダマンチル) 一 2— (2, 4—ジフルオロフェニルスルホニルァ ミノ) チアゾールの合成 Synthesis of 4- (1-adamantyl) -1-2- (2,4-difluorophenylsulfonylamino) thiazole
実施例 45と同様の方法で、 表題化合物 ¾得た。 MMRの結果は、 以下のと おりであった。 The title compound was obtained in the same manner as in Example 45. The results of the MMR are as follows.
^-NMR (300MHz, DMS0-d6) δ: 9.87 (ΙΗ' br s), 8.17-7.89. (1H, m) , 7.04- 6.81 (2H, m), 5.99 (1H, s), 2.22-2.00 ' (3H, m), 1.97-1.57 (12H, m) 実施例 52 ^ -NMR (300MHz, DMS0-d6) δ: 9.87 (ΙΗ 'br s), 8.17-7.89. (1H, m), 7.04-6.81 (2H, m), 5.99 (1H, s), 2.22-2.00' (3H, m), 1.97-1.57 (12H, m) Example 52
4— (1 -ァダマンチル) —2— [ (2, 5—ジクロロチ才フェン— 3—ィ ル) スルホニルァミノ] チアゾールの合成 Synthesis of 4- (1 -adamantyl) —2— [(2,5-dichlorothienephen-3--3-yl) sulfonylamino] thiazole
実施例 45と同様の方法で、 表題化合物を得た。 Rの結果は、 以下のと おりであった。 The title compound was obtained in the same manner as in Example 45. The results of R were as follows.
^- MR (300MHz, DMS0-d6) δ: 9.81 (ΙΗ,, br s), 7.19 (1H, s), 6.00 (1H, s), 2.23-1.97 (3H, m) , 1.95-1.57 (12H, m) ^-MR (300MHz, DMS0-d6) δ: 9.81 (ΙΗ ,, br s), 7.19 (1H, s), 6.00 (1H, s), 2.23-1.97 (3H, m), 1.95-1.57 (12H, m)
実施例 53 Example 53
4 - (1 -ァダマンチル) 一 2— (3—クロ口— 5—フル才ロー 2—メチルフ ェニルスルホニルァミノ) チアゾールの合成 Synthesis of 4- (1 -adamantyl) -1-2- (3-chloro- mouth-5-full-rough 2-methylphenylsulfonylamino) thiazole
実施例 45と同様の方法で、 表題化合物を得た。 NMRの結果は、 以下のと おりであった。 The title compound was obtained in the same manner as in Example 45. The NMR results were as follows.
^- R (300MHz, DMS0-d6) δ: 9.84 (IH, br s), 7.86 (IH, d, J=7.5 Hz), 7.16 (IH, d, J=9.3 Hz) , 5.98 (IH, s), 2.38 (3H, s), 2.19-2.03 (3H, m), 1.93-1.61 (12H, m) ^ -R (300MHz, DMS0-d6) δ: 9.84 (IH, br s), 7.86 (IH, d, J = 7.5 Hz), 7.16 (IH, d, J = 9.3 Hz), 5.98 (IH, s) , 2.38 (3H, s), 2.19-2.03 (3H, m), 1.93-1.61 (12H, m)
上記実施例で得られた化合物の構造を表 2にまとめて表す。 Table 2 summarizes the structures of the compounds obtained in the above Examples.
表 2.. 実施例〗〜 53で得られた化合物の構造を表す。
Table 2. Represents the structures of the compounds obtained in Examples I to 53.
, OH rX OH , OH rX OH
1 OH ^rN 1 OH ^ r N
9999
l.00/S00Zdf/X3d 179..60/S00Z OAV
ϋ l.00 / S00Zdf / X3d 179..60 / S00Z OAV ϋ
I I
実施例 5 4 Example 5 4
化合物 5 4— 1〜化合物 5 4— 2 1 7の製造 Preparation of Compound 54-1-1 to Compound 54-217
N— (メチルポリスチレン) 一4— (メチルァミノ) ピリジン (1 5 0 I ) のクロ口ホルム (7 0 0 M I ) 溶液に、 参考例 2で得た 4— ( 1 ーァダマ
ンチル) — 2—ァミノチアゾール (30 Mmo I ) を加えた後、 それぞれ対応 するスルホニルクロリド (〗 20 mo I ) を加え、 50°Cでニ晚攪拌した。 反応液をろ過した後に、 溶媒を減圧で濃縮した。 得られた残渣を NHシリ ゲ ルカラムクロマトグラフィー (展開溶媒 クロ口ホルム:メタノール =20 : 1 ) で精製し、 下記表に示した目的物を得た。 構造式及び質量分析データを下 記表に記載した。
To a solution of N- (methylpolystyrene) 14- (methylamino) pyridine (150I) in chloroform (700 MI) was added 4- (1-adama) obtained in Reference Example 2. After addition of 2-aminothiazole (30 MmoI), the corresponding sulfonyl chloride (〗 20 moI) was added, and the mixture was stirred at 50 ° C. under nitrogen. After filtering the reaction solution, the solvent was concentrated under reduced pressure. The obtained residue was purified by NH silica gel column chromatography (developing solvent, chromate form: methanol = 20: 1) to obtain the desired compound shown in the following table. The structural formula and mass spectrometry data are shown in the table below.
MS(APCI) 化合物番号 構造式 分子式 MS (APCI) Compound number Structural formula Molecular formula
[M+H]+ [M + H] +
54-1 C21 H24N203S2 417 54-1 C21 H24N203S2 417
54-2 C23H30N2O2S2 431 54-2 C23H30N2O2S2 431
54-3 C19H21 BrN202S2 453, 455 54-3 C19H21 BrN202S2 453, 455
54-4 C23H30N2O3S2 447 54-4 C23H30N2O3S2 447
54-5 C19H21 BrN202S2 453, 455 54-5 C19H21 BrN202S2 453, 455
54-6 C17H19BrN202S3 459, 461 54-6 C17H19BrN202S3 459, 461
0 o s 0 o s
54-7 C17H19CIN202S3 415 54-7 C17H19CIN202S3 415
0 o s 0 o s
54-8 C20H21 N3O2S2 400 54-8 C20H21 N3O2S2 400
N N
54-9 C22H23N304S2 458 54-9 C22H23N304S2 458
54-10 z C25H29N302S2 468
54-10 z C25H29N302S2 468
OAVv:/s00ifcl£ 9s/-00 OAVv: / s00ifcl £ 9s / -00
cn cn αι cn cn αι
1 1 \ 1 ' 1 1 \ 1 '
1 i I 1 I 1 1 i I 1 I 1
<J3 σ) σ> Oi CD σ> σ¾ O) o CD 00 Jl CO ro <J3 σ) σ> Oi CD σ> σ¾ O) o CD 00 Jl CO ro
O O
。 ¾一 . ¾ 一
〇 〇 O 〇 〇 〇 〇 〇 O 〇 〇 〇
CDCD
O 〇 . 〇 〇 O 〇.
ェ ΓΟ ο ) ro CD ェ CO O CD ο) ro CD CO CO O
CD o ro I ro ェ ro CD o ro I ro e ro
0 P CO CO ro CD CD 0 P CO CO ro CD CD
〇 CT) 0 〇 a> (〇 CT) 0 〇 a>
σ 〇 〇 ) Z z ro ro Z z: σ 〇 〇) Z z ro ro Z z:
ro— 〇 O O 〇 ro— 〇 O O 〇
〇 〇 CO 0 n 〇 〇 〇 〇 CO 0 n 〇 〇
〇 ro ω ) ω ω (/) n CO 〇 ro ω) ω ω (/) n CO
GO GO ro GO ro > GO GO ω ro ro ro ro ^ ^ GO GO ro GO ro> GO GO ω ro ro ro ro ^ ^
00 4^ 00 4 ^
CD C cn cn CD C cn cn
ro CO O ro CO O
^ CD CO
^ CD CO
LLLL
l.00/S00Zdf/X3d 179..60/S00Z: OAV
l.00 / S00Zdf / X3d 179..60 / S00Z: OAV
/iz/3d/i1oso卜 o / iz / 3d / i1oso o
enen
^ 1 ^ ^ ^ 1 ^ ^
1 1 1 1 1 1 1 1 1 1
Ο CD O cn CO ro Ο CD O cn CO ro
ro ro
。 。 、ェ . . ,
O O
〇 o o O 〇 〇 o o O 〇
〇 to ro 〇 〇 o 〇 to ro 〇 〇 o
CD cr> O ro CO ェ o O ro CD cr> O ro CO e o O ro
O o O o
ェ ェ ェ ェ
PO CD ェ PO CD
CD o o N5 ro CD o o N5 ro
CD o ro O T| o -n CO "Π T| σ> o ro CD o ro O T | o -n CO "Π T | σ> o ro
CO "Π Γ z ro ro CO "Π Γ z ro ro
z CO ro Z z ro ro O z CO ro Z z ro ro O
〇 z 〇 〇 z 〇
〇 〇 〇 〇 〇 〇 ro 〇 〇 〇 〇 〇 〇 ro
r ω 〇 O CO if) 〇 r> in ω ro r ω 〇 O CO if) 〇 r> in ω ro
, CO 00 ro CO , CO 00 ro CO
CO ro GO ro CO ro GO ro
ΟΊ CO ΟΊ CO
-!≥>. cn ϋι o CO CD O 4^ CO 00 CO -! ≥> .cn ϋι o CO CD O 4 ^ CO 00 CO
O O
ai ai
CO CO
cn
cn
/v: O/-00s00ifcl£AV / v: O / -00s00ifcl £ AV
38 38
90l.00/S00Zdf/X3d 1"9んん 60/S00Z OAV
90l.00 / S00Zdf / X3d 1 "9 60 / S00Z OAV
S8 S8
90l.00/S00Zdf/X3d 1-9..60/S00Z ΟΛ\
90l.00 / S00Zdf / X3d 1-9..60 / S00Z ΟΛ \
fr8 fr8
90l.00/S00Zdf/X3d ^9..60/δ00Γ OAV
OV/osA 60s9uvld/ooifc0 oos 9. 90l.00 / S00Zdf / X3d ^ 9..60 / δ00Γ OAV OV / osA 60s9uvld / ooifc0 oos 9.
寸 LO (S3 LO 寸 00 CO Dimension LO (S3 LO Dimension 00 CO
CD CO o O LO CSI o LO 寸 寸 ir> in 寸 寸 寸 io LO 寸 LO O CD CO o O LO CSI o LO Dimension ir> in Dimension Dimension io LO Dimension LO O
CSJ ω 寸 CO CNJ CM CO CO CO 寸 CO CM 00 00 C ω ω C CSJ ω dimension CO CNJ CM CO CO CO dimension CO CM 00 00 C ω ω C
寸 〇 CM CM CO 寸 \r> 寸 寸 CO 〇 寸 〇 〇 〇 〇 O 〇 〇 〇 CO z 寸 CNJ 寸 csj CO CO CM CM Dimension 〇 CM CM CO Dimension \ r> Dimension Dimension CO 寸 Dimension 〇 〇 〇 〇 O 〇 〇 〇 CO z Dimension CNJ Dimension csj CO CO CM CM
CO Z z: Z : z CO Z z: Z: z
U- 寸 寸 寸 O σ> σ 寸 O U-dimensions dimension O σ> σ dimension O
CM CO CM CM CM CM Csi CNJ csi Csj CM CO CM CM CM CM Csi CNJ csi Csj
CM I X ェ ェ CM I X
X CM OJ CO CO o CSJ X CM OJ CO CO o CSJ
CM CM CM Csi CM M CNJ Csi Csi 〇 C 〇 〇 ϋ 〇 ϋ 〇' 〇 〇 〇 CM CM CM Csi CM M CNJ Csi Csi 〇 C 〇 〇 ϋ 〇 ϋ 〇 '〇 〇 〇
ェ2 o E 2 o
エ o IZ o D o IZ o
/ ェ z、 ,Ο こ . ェ/ E z,, Ο this. E
o (li '一。 o (li 'I.
1 ω / 、o 1 ω /, o
o / o /
—o —O
Csj CO 寸 LO O 00 〇 Csj CO size LO O 00 〇
CO CD CO CD CO CD o CO O CO CD CO CD CO CD o CO O
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 寸 寸 寸 寸 寸 寸 寸 寸 寸 寸 LO LO LO LO ι
1 1 1 1 Dimensions Dimensions Dimensions Dimensions Dimensions Dimensions Dimensions LO LO LO LO ι
9898
l.00/S00Zdf/X3d 179..60/S00Z OAV
β/ 60iz/3/i1diso i l.00 / S00Zdf / X3d 179..60 / S00Z OAV β / 60iz / 3 / i1diso i
OAVvud/fcl oosooz OAVvud / fcl oosooz
OM麵卜3diiミ卜 09 OM 麵 3dii Mi09
06 06
90l.00/£00ZdT/X3d t"9厶ん 60細 Z OAV
(試験例) 11i8HSD1阻害試験 90l.00 / £ 00ZdT / X3d t "9mm 60 fine Z OAV (Test example) 11i8HSD1 inhibition test
試験化合物の 11 β HSD1阻害活性を以下のようにして評価した。 The 11βHSD1 inhibitory activity of the test compound was evaluated as follows.
30mM TrisHCI (pH7.4)/1mM EDTA緩衝液中に 200nM NADPHを加えた反応液に酵 素源であるヒ卜肝臓ミクロソー厶 (Tissue Transformation Technologies社) を lO zg/mlとして添加し、 さらに試験化合物を加えた。 その後、 終濃度 100nM となるように基質であるコルチゾン溶液を加え、 反応を開始させた。 37°Cで 80 分間インキュベーションした後、 非特異的な阻害剤である 18)3グリチルレチ ン酸を終濃度 ΙΟΟ ΙΜとして加えることで反応を停止させた。 生成したコルチ ゾール量を HTRF8 (Homogeneous Time-Resolved Fluorescence) 法による検出 キット (日本シエーリング株式会社) を用いて定量した。 本系は、 ユーロピウ 厶で標識された抗コルチゾール抗体と XL665が標識されたコルチゾールの間で 生じる蛍光共鳴エネルギー移動を検出する系であり、 未標識のコルチゾールを 加えると競合反応により、 結合のシグナルが減弱する。 この時、 キット付属の 濃度既知のコルチゾールにより標準曲線を作製し、 反応によって生成するコル チゾール量を評価した。 酵素を含まないゥエルのコルチゾール生成量をバック グラウンド、 化合物を含まないゥエルのコルチゾール生成量を 100%の酵素活性 として、 それぞれの化合物について 50μ から公比 3の希釈系列につき評価し、 IC50値を算出した。 ' To the reaction solution containing 200 nM NADPH in 30 mM TrisHCI (pH 7.4) / 1 mM EDTA buffer, human liver microsomal enzyme (Tissue Transformation Technologies) as an enzyme source was added as lO zg / ml, and the test compound was added. Was added. Thereafter, a cortisone solution as a substrate was added to a final concentration of 100 nM, and the reaction was started. After incubation at 37 ° C for 80 minutes, the reaction was stopped by adding a non-specific inhibitor, 18) 3 glycyrrhetinic acid, as a final concentration of ΙΜ ΙΜ. The amount of generated cortisol was quantified using a detection kit (Nippon Schering Co., Ltd.) by the HTRF8 (Homogeneous Time-Resolved Fluorescence) method. This system detects the fluorescence resonance energy transfer that occurs between the anticortisol antibody labeled with europium and the cortisol labeled with XL665.When unlabeled cortisol is added, the binding signal is generated by a competitive reaction. Attenuate. At this time, a standard curve was prepared using cortisol of a known concentration included in the kit, and the amount of cortisol generated by the reaction was evaluated. Background cortisol production of Ueru without enzyme, cortisol production of Ueru without compound as 100% of enzyme activity was assessed per dilution series of common ratio 3 from 50μ for each compound, IC 50 values Calculated. '
実施例 4-10の化合物 IC5()値 16nM Compound IC 5 () value of Example 4-10 16 nM
実施例 4- 21の化合物 IC5。値 42nM 産業上の利用可能性 Compound IC 5 Example 4 21. Value 42nM Industrial availability
本発明のチアゾール誘導体は、 新規化合物として、 医療を含めた様々な分野 で利用され得る。 The thiazole derivative of the present invention can be used as a novel compound in various fields including medical treatment.
本発明のチアゾール誘導体、 その医薬上許容される塩、 及びその溶媒和物は、 11/8-HSD1阻害剤及び医薬組成物の有効成分などとして利用され得る。 The thiazole derivative of the present invention, a pharmaceutically acceptable salt thereof, and a solvate thereof can be used as an 11 / 8-HSD1 inhibitor and an active ingredient of a pharmaceutical composition.
本発明の HSD1阻害剤及び医薬組成物は、 医薬として利用され得る。
The HSD1 inhibitor and the pharmaceutical composition of the present invention can be used as a medicine.