AN IMPROVED PROCESS FOR THE PREPARATION OF GEFITINIB
FIELD OF INVENTION
The present invention relates to an improved process for the preparation of Gefitinib. Gefitinib which is (4-(3'-chloro-4'-fluorophenylamino)-7-methoxy-6-[3-(4-morpho- linyl)propoxy]quinazoline) has the formula-I given below. It is an addition to the arsenal of anti-tumor agents, especially the epidermal growth factor receptor-tyrosine kinase inhibitors, that has shown promising results in pre-clinical and clinical trials. The anti- tumor activity of gefitinib has provoked interest in its potential for clinical application in the treatment of a variety of solid tumors, especially non-small cell lung cancer and hematological malignancies. The safety and oral bioavailability of this drug has encouraged many countries to launch this drug.
BACKGROUND OF INVENTION Gefitinib of the formula-I is reported for the first time by Gibson, KH (Zeneca) in EP 0823900. Equivalent patents for gefitinib are US 5770599 and WO 9633980. Antitumor activity of gefitinib is described in Clin. Cancer Res., 2000, 6, 2053-63. Pharmacokinetics and tolerability of gefitinib is described in Clin. Pharmacokin., 2001, 40, 297-306. Clinical studies of gefitinib related to lung cancer are described in Lung Cancer, 2000, 29 (Suppl. 1), page 72.
Process for the preparation of gefitinib disclosed in EP 0823900 starts with the quinazoline derivative of the formula-II (Scheme-I). Quinazoline derivative of the
formula-II is reacted with methanesulfonic acid and L-methionine to get the demethylated derivative of the formula-Ill. Protection of the hydroxy group present in compound of the formula-Ill as its acetate followed by reaction with thionyl chloride gave the chloro derivative of the formula- V. Reaction of the compound of the formula-V with 3-chloro-4-fluoroaniline gave the anilino derivative of the formula- VI. Deprotection of the acetate group followed by etherification of the resulting compound with 3- morpholinopropyl chloride gave crude gefitinib of the formula-I after column chromatography. This material was further purified by recrystallization from toluene.
The main draw back in this process is formation of impurities during the introduction of morpholinopropyl side chain. During this stage main impurity that can form is N- alkylated derivative of the formula- VIII. Due to its chemically related property, removal of this impurity needs column chromatography. Such a process is not commercially viable for bulk production and also will not be cost effective. Also the process involves
steps like protection and deprotection of phenolic hydroxy group during the synthesis of process. This leads to more number of steps in making gefitinib and therefore the process
VIII become lengthy and time consuming. Also, the basic raw material used in the synthesis is not readily available on commercial scale and it needs to be prepared from 2-amino-4,5- dimethoxybenzoic acid as oer the process given in EP 0566226.
SUMMARY OF INVENTION Keeping in view of the difficulties in the above described process for the preparation of gefitinib on a commercial scale, we aimed to develop a simple and economically viable and commercially applicable process for the preparation of gefitinib of the formula-I.
Accordingly, the main objective of the present invention is to provide an improved process for the preparation of gefitinib of the formula-I which is simple and economical •*' and commercially applicable.
Still another objective of the present invention is to provide an improved process for the preparation of gefitinib of the formula-I which involves employment of readily and cheaply available raw materials.
Yet another objective of the present invention is to avoid the formation of impurities such as compound of the formula- VIII.
Still another objective of the present invention is to provide an improved process for the preparation of gefitinib of the formula-I which has short in number of steps making it simple and economical.
According to another objective of the present invention there is provided novel compound of the formula-X, which is an intermediate for the preparation of compound of the formula-I and a process for its preparation.
According to still another objective of the present invention there is provided novel compound of the formula-XI, which is an intermediate for the preparation of compound of the formula-I and a process for its preparation.
According to yet another objective of the present invention there is provided novel compound of the formula-XII, which is an intermediate for the preparation of compound of the formula-I and a process for its preparation.
According to further objective of the present invention there is provided novel compound of the formula-XIII, which is an intermediate for the preparation of compound of the formula-I and a process for its preparation.
t1 V f # U U £ £ J
According to another objective of the present invention there is provided novel compound of the formula-XIV, which is an intermediate for the preparation of compound of the formula-I and a process for its preparation.
According to still another objective of the present invention there is provided novel compound of the formula-XV, which is an intermediate for the preparation of compound of the formula-I and a process for its preparation.
During our sustained research in developing an improved process for the preparation of gefitinib of the formula-I, we realized that if we introduce the morpholinopropyl side chain at an early stage of the synthesis, the process would be simpler, shorter and free from related impurities.
Accordingly, the invention provides an improved process for the preparation of compound of formula-I,
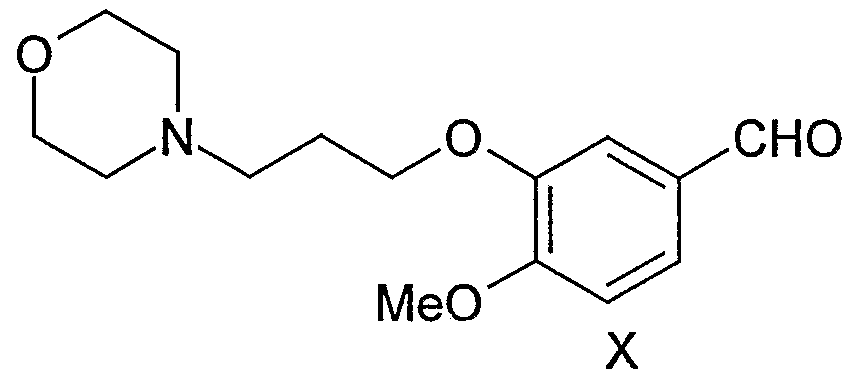
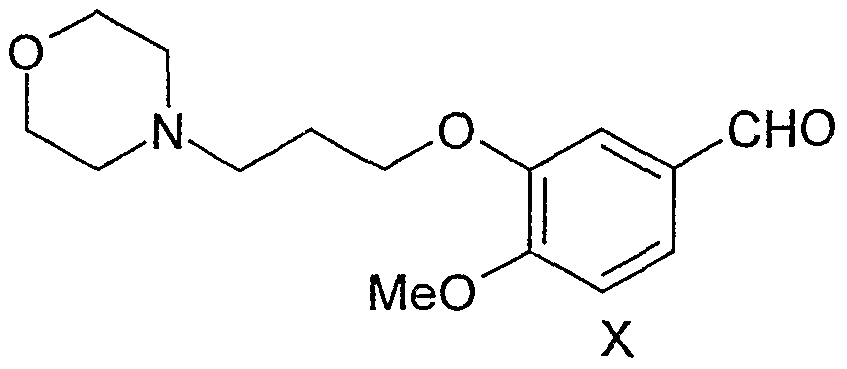
I which comprises, (i) Reacting iso-vanillin of the formula-IX,
IX with 3-morpholinopropyl halide wherein the term halide represents CI, Br, or I in the presence of a base to get the novel compound of the, formula-X,
(ϋ) Nitrating the compound of the formula-X using a nitrating reagent at a temperature in the range of 5 to 60 °C, to get the novel nitro derivative of the formula-XI,
(iii) Reacting the compound of the formula-XI with hydroxylamine salt in a polar solvent medium at a temperature in the range of 20 to 60 °C in the presence of a base to get the novel oxime derivative of the formula-XII,
O 2005/070 W W W • » — »* . fc, tf
(iv) Reacting the compound of the formula-XII with a dehydrating agent at a temperature in the range of 20 to 140 °C in the presence or absence of a solvent to get the novel compound of the formula-XIII,
(v) Hydrolyzing the compound of the formula-XIII using a base or an acid in a solvent medium and at a temperature in the range of 20 to 80 °C to get the novel amide compound of the formula-XIV,
(vi) Hydrogenating the compound of the formula-XIV in the presence of a metal catalyst and a polar solvent at a temperature in the range of 20 to 60 °C, and pressure in the range of 10 to 60psi to get the novel compound of the formula- XV,
(vii) Cyclizing the compound of the formula-XV using foπnic acid at a temperature in the range of 80 to 110 °C to get the quinazolone derivative of the formula-XVI,
XVI (viii) Converting the keto group present in the compound of the formula-XVI into a leaving group 'X' by conventional methods to get the compound of the formula-XVII,
XVII (X = CI, OAc, OMs, OBs, OTs) (ix) Condensing the compound of the formula-XVII with 3-chloro-4-fluoroaniline by conventional methods to get the compound of the formula-I
(x) Purifying the crude compound of the formula-I by acid / base treatment or by recrystallization from solvents to get pharmaceutically acceptable grade gefitinib of the formula-I.
The process is shown in Scheme-II.
XVII (X = CI, OAc, OMs, OBs, OTs)
Scheme-II
Accordingly, the basic raw material selected for the synthesis of gefitinib is iso-vanillin. Earlier reported process (WO 01/04102 within Example (25) for the preparation of compound of the formula-XVI consists of hydrolysis of gefitinib of formula-I with 6N
hydrochloric acid. Since gefitinib itself is a target compound, preparation of compound of formula-XVI from gefitinib is of no commercial value as such.
DETAILED DESCRIPTION OF INVENTION In a preferred embodiment of the present invention, iso-vanillin of the formula-IX is reacted with 3-morpholinopropyl halide in the presence of a base to get the O-alkylated compound of the formula-X. The 3-morpholinopropyl halide used in the alkylation step can be selected from chloro, bromo, or iodo derivative, preferably chloro or bromo compound. The base used in the reaction can be an inorganic base such as sodium or potassium hydroxide, hydrogen carbonate, carbonate, preferably sodium or potassium carbonate. Alternatively the base used in the alkylation step can be organic base such as diisopropyl ethylamine, triethylamine, pyridine, DBU (l,8-diazabicyclo[5.4.0]undec-7- ene), DABCO (l,4-diazabicyclo[2.2.2]octane, etc.
In the second step, nitration is done by using dilute nitric acid or a nitration mixture such as sulfuric acid/nitric acid. The percentage of nitric acid can be between 10-70%, preferably 30-60%. Temperature of the reaction is preferably, between 20-50 °C. The nitration product of the formula-XI can be isolated by neutralization of reaction mass with a base to basic pH followed by extraction into a solvent. The base used in neutralization can be an inorganic base or organic base. The inorganic base is selected from sodium or potassium hydroxide, carbonate, hydrogen carbonate. The organic base is selected from aqueous ammonia, ethylamine, propylamine, diethylamine, triethylamine, pyridine, etc., preferably ammonia. The neutralization pH is in the range of 7-12, preferably, 8-9. The solvent used for extraction of compound of formula-XI is selected from ethyl acetate, isopropyl acetate, methylene chloride, chloroform, ethylene dichloride, diisopropyl ether, toluene, xylene, etc, preferably ethyl acetate or methylene chloride. The isolated product of the formula-XI, having a purity of more than 90% can be purified further to more than 98% by doing recrystallization from solvents such as isopropanol, methanol, ethanol, acetone, methyl ethyl ketone, ethyl acetate or a mixture thereof.
Conversion of the aldehyde group present in compound of the formula-XI into its oxime derivative of the formula-XII can be done by using hydroxylamine salt and a polar solvent at ambient or elevated temperature in the presence of base. The hydroxylamine salt used is selected from hydroxylamine sulfate, chloride, nitrate, phosphate, etc, preferably sulfate or chloride. The solvent used during oxime preparation is selected from water, methanol, ethanol, isopropanol, or a mixture thereof. The temperature of reaction is between 20-60 °C, preferably between 25-45 °C. The base used in the reaction is selected from sodium or potassium hydroxide, carbonate or bicarbonate. The oxime of the formula-XII thus obtained is isolated by neutralization of reaction mass with a weak organic acid such as acetic acid to a pH of 7-8 and filtration of the resulting solid. The crude product of the formula-XII thus obtained can be recrystallized from a solvent such as ethyl acetate, methanol, ethanol, isopropanol, acetone, acetonitrile, methyl ethyl ketone, preferably ethyl acetate, acetonitrile or methyl ethyl ketone. Purity of the product after crystallization is more than 99%.
Dehydration of the oxime compound of the formula-XII to the nitrile compound of the formula-XIII, can be done by using a dehydrating agent at elevated temperature in the presence or absence of a solvent. The dehydrating agent used is selected from acetic anhydride, propionic anhydride, phosphorous pentoxide, and preferably acetic anhydride. The temperature of reaction is between 20-140 °C, preferably 40-130 °C. In case of acetic or propionic anhydride the amount can be 5-10 fold excess. Isolation of the product of the formula-XIII can be done by distilling off the excess reagent and subsequently diluting the reaction mass with water and neutralization of the mass pH to basic. The base used for neutralization is selected from sodium or potassium hydroxide; carbonate, bicarbonate, ammonia, triethylamine, etc., preferably potassium carbonate or ammonia.
The amide compound of the formula-XV can be obtained in one-step process from the compound of the formula-XIII by reduction-cum-hydrolysis or in a step-wise manner. In the direct conversion process the suitable reagents are palladium or Raney nickel in the presence of hydrazine hydrate. The medium of reaction is aqueous alcoholic solvent, such
as methanol, ethanol, isopropanol. The temperature of reaction is preferably 30-80 °C. The crude solid product of the formula-XV is sufficiently pure enough for further conversion. r The stepwise process for the preparation of compound of the formula-XV from the compound of the formula-XIII consists of (i) hydrolysis of compound of the formula-XIII to compound of the formula-XIV and (ii) reduction of nitro group present in compound of the formula-XIV to compound of the formula-XV. Hydrolysis of the nitrile derivative of the formula-XIII can be done with acids such as 50-70% sulfuric acid, concentrated hydrochloric acid or poly phosphoric acid or with bases such as aqueous sodium or ' potassium hydroxide containing hydrogen peroxide. During acid hydrolysis the temperature of the reaction can be in the range of 20-80 °C, preferably 40-60 °C. During base hydrolysis the temperature of reaction can be in the range of 30-50 °C, preferably 40-45 °C.
Reduction of nitro group present in compound of the formula-XIV to amino compound of the formula-XV can be done by hydrogenation in the presence of a metal catalyst such as 2-10% palladium-on-carbon 5% rhodium-on-alumina, 2-5% platinum-on-carbon, or Raney nickel, preferably 5-10% palladium-on-carbon or Raney nickel. The hydrogen pressure required during reduction can be in the range of 10-60psi. The medium of reduction is alcoholic solvent such as methanol, ethanol, or isopropanol.
Treatment of compound of the formula-XV with formic acid at elevated temperature gives the quinazolone derivative of the formula-XVI. The formic acid used in the reaction can be of aqueous (1-20%) or anhydrous type. The temperature of reaction can be the boiling point of formic acid used in the reaction. The product of the formula-XVI can be isolated by neutralization of reaction mass with a base followed by filtration of the solid compound. The base used for neutralization can be selected from sodium or potassium hydroxide, carbonate, bicarbonate; ammonia, triethylamine, etc., preferably sodium or potassium bicarbonate or ammonia.
Conversion of the keto group present in compound of the formula-XVI into a leaving group 'X' present in compound of the formula-XVII can be done by treating the compound of the formula-XVI with reagents such as thionyl chloride, phosphorous pentachloride, phosphorous oxychloride, oxalyl chloride, acetic anhydride, methanesulfonyl cloride, benzenesulfonyl chloride, p-toluenesulfonyl chloride, etc., preferably, thionyl chloride, oxalyl chloride, phosphorous oxychloride or methanesulfonyl cloride in the absence or presence of solvent. The solvent employed in the reaction is selected from methylene chloride, chloroform, toluene, tetrahydrofuran, dioxane, acetonitrile, cyclohexane, preferably chloroform or cyclohexane. The crude product thus obtained can be directly used in the next step without any purification.
Condensation of 3-chloro-4-fluoroaniline with the compound of the formula-XVII can be done by following a similar process as given in WO 03/072108. Accordingly, compound of formula-XVII can be reacted with excess 3-chloro-4-fluoroaniline in isopropanol medium to get the crude gefitinib of formula-I. The crude product thus obtained can be recrystallized from solvents such as toluene, actonitrile, ethanol, isopropanol, ethyl acetate, ethyl methyl ketone or a mixture thereof. Alternatively, the crude product can be dissolved in aqueous acid, treated with carbon and neutralized with a base to get pure compound of formula-I. The acid used during purification is selected from organic acids such as acetic acid, propionic acid, oxalic acid, succinic acid, toluic acid, mandelic acid, tartaric acid, preferably acetic acid or oxalic acid; mineral acid such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, preferably hydrochloric acid or sulfuric acid. The base used during purification is selected from organic base such as ammonia, triethylamine, ethylamine, propylamine, preferably ammonia; the inorganic base such as sodium or potassium hydroxide, carbonate, bicarbonate, preferably sodium or potassium hydroxide.
According to another embodiment of the present invention there is provided novel compound of the formula-X, which is useful as an intermediate for the preparation of the compound of the formula-I,
Accordingly the invention also provides a process for the preparation of novel compound of the formula-X,
which comprises, (i) Reacting iso-vanillin of the formula-IX,
IX with 3-morpholinopropyl halide where halide represents CI, Br, I in the presence of a base to get the compound of the formula-X.
The base used in step (i) is selected from inorganic base such as sodium or potassium hydroxide, hydrogen carbonate, carbonate, preferably sodium or potassium carbonate. Alternatively the base used in the alkylation step can be organic base such as diisopropyl ethylamine, triethylamine, pyridine, DBU (l,8-diazabicyclo[5.4.0]undec-7-ene), DABCO (l,4-diazabicyclo[2.2.2]octane, preferably pyridine or DBU.
Accordingly the invention also provides a process for the preparation of novel compound of the formula-XI, which is useful as an intermediate for the preparation of the compound of the formula-I
which comprises, (i) Reacting iso-vanillin of the formula-IX,
IX with 3-morpholinopropyl halide where halide represents CI, Br, I in the presence of a base to get the compound of formula-X,
(ii) Nitrating the compound of the formula-X using nitrating reagent to get the nitro derivative of formula-XI.
Accordingly the invention also provides a process for the preparation of novel compound of the formula-XII, which is useful as an intermediate for the preparation of the compound of the formula-I
which comprises, (i) Reacting iso-vanillin of the formula-IX,
IX with 3-morpholinopropyl halide where halide represents CI, Br, I in the presence of a base to get the compound of formula-X,
(ϋ) Nitrating the compound of the formula-X using a nitrating reagent to get the nitro derivative of formula-XI,
(iii) Reacting the compound of the formula-XI with hydroxylamine salt in a polar solvent medium at a temperature in the range of 20 to 60°C in the presence of a base to get the oxime derivative of the formula-XII.
Accordingly the invention also provides a process for the preparation of novel compound of the formula-XIII, which is useful as an intermediate for the preparation of the compound of the formula-I
which comprises,
(i) Reacting iso-vanillin of the formula-IX,
IX with 3-morpholinopropyl halide where halide represents CI, Br, I in the presence of a base to get the compound of formula-X,
(ϋ) Nitrating the compound of the formula-X using a nitrating reagent to get the nitro derivative of formula-XI,
(iii) Reacting the compound of the formula-XI with hydroxylamine salt in a polar solvent medium at a temperature in the range of 20 to 60°C in the presence of a base to get the oxime derivative of the formula-XII,
(iv) Reacting the compound of the formula-XII with a dehydrating agent at a temperature in the range of 20 to 140°C to get the compound of the formula- XIII.
Accordingly the invention also provides a process for the preparation of novel compound of the formula-XIV,
XIV which is useful as an intermediate for the preparation of the compound of the formula-I. which comprises, (i) Reacting iso-vanillin of the formula-IX,
IX with 3-morpholinopropyl halide where halide represents CI, Br, I in the presence of a base to get the compound of formula-X,
(ii) Nitrating the compound of the formula-X using a nitrating reagent to get the nitro derivative of the formula-XI,
(iii) Reacting the compound of the formula-XI with hydroxylamine salt in a polar solvent medium at a temperature in the range of 20 to 60°C in the presence of a base to get the oxime derivative of the formula-XII,
(iv) Reacting the compound of the formula-XII with a dehydrating agent at a temperature in the range of 20 to 140°C to get the compound of fόrmula-XIII,
(v) Hydrolyzing the compound of the formula-XIII using a base or an acid in a solvent medium and at a temperature in the range of 20 to 80°C to get the amide compound of the formula-XIV.
Accordingly the invention also provides a process for the preparation of novel compound of the formula-XV, which is useful as an intermediate for the preparation of the compound of the formula-I
which comprises, (i) Reacting iso-vanillin of the formula-IX,
IX with 3-morpholinopropyl halide where halide represents CI, Br, I in the presence of a base to get the compound of the formula-X,
(ii) Nitrating the compound of the formula-X using a nitrating reagent to get the nitro derivative of the formula-XI,
(iii) Reacting the compound of the formula-XI with hydroxylamine salt in a polar solvent medium at a temperature in the range of 20 to 60°C in the presence of a base to get the oxime derivative of the formula-XII,
(iv) Reacting the compound of the formula-XII with a dehydrating agent at a temperature in the range of 20 to 140°C t to get the compound of the formula- XIII,
(V) Hydrolyzing the compound of the formula-XIII using a base or an acid in a solvent medium and a temperature in the range of 20 to 80°C to get the amide compound of the formula-XIV,
XIV (Vi) Hydrogenating the compound of the formula-XIV in the presence of a metal catalyst and a polar solvent at a temperature in the range of 20 to 60°C and a pressure in the range of 10 to 60psi to get the compound of the formula-XV.
In the above mentioned processes the various reagents and reaction conditions, which may be preferably employed, are described below.
The base used in step (i) is selected from inorganic base such as sodium or potassium hydroxide, hydrogen carbonate, carbonate, preferably sodium or potassium carbonate. Alternatively the base used in the alkylation step can be organic base such as diisopropyl ethylamine, triethylamine, pyridine, DBU, DABCO, preferably pyridine or DBU.
Nitrating reagent used in step (ii) is selected from nitric acid or a nitration mixture such as sulfuric acid/nitric acid. The percentage of nitric acid can be between 10-70%, preferably 30-60%. Temperature of the reaction is between 5-60°C, preferably, 20-50 °C.
Hydroxylamine salt used step (iii) is selected from hydroxylamine sulfate, chloride, nitrate, phosphate, etc, preferably sulfate or chloride. The solvent used during oxime
cpreparation is selected from water, methanol, ethanol, isopropanol, or a mixture thereof. The temperature of reaction is between 20-60°C, preferably 25-45 °C. The base used in the reaction is selected from sodium or potassium hydroxide, carbonate or bicarbonate. The dehydrating agent used in step (iv) is selected from acetic anhydride, propionic anhydride, phosphorous pentoxide, and preferably acetic anhydride. The temperature of reaction is between 20-140 °C, preferably 40-130 °C. Amount of dehydrating agent used is 2-15 fold excess, preferably 5-10 fold excess. The base used in hydrolysis step (v) is selected from aqueous sodium or potassium hydroxide containing hydrogen peroxide. During base hydrolysis the temperature of reaction can be in the range of 30-50 °C, preferably 40-45 °C: The acid used in step (v) is selected from 50-70% sulfuric acid, concentrated hydrochloric acid or poly phosphoric acid. During acid hydrolysis the temperature of the reaction can be in the range of 20-80 °C, preferably 40-60 °C.
The metal catalyst used in hydrogenation step (vi) is selected from 2-10% palladium-on- carbon 5% rhodium-on-alumina, 2-5% platinum-on-carbon, or Raney nickel, preferably 5-10% palladium-on-carbon or Raney nickel. The hydrogen pressure required during reduction can be in the range of 10-60psi. The medium of reduction is alcoholic solvent such as methanol, ethanol, or isopropanol.
The invention also provides a one step process for the preparation of compound of the formula-XV,
from the compound of formula-XIII,
which is useful as an intermediate for the preparation of the compound of the formula-I. which comprises, (i) Reacting the compound of the formula-XIII,
with hydrazine hydrate in the presence of metal catalyst such as palladium-on-carbon or Raney nickel in a polar solvent system at a temperature in the range of 30 to 80 °C to get the compound of the formula-XV.
The solvent used is selected from aqueous alcoholic solvent, such as methanol, ethanol, isopropanol. The temperature of reaction is preferably 40-60 °C.
The details of the invention are given in the Examples given below which are provided to illustrate the invention only and therefore should not be construed to limit the scope of the present invention.
Example 1 Preparation of 4-(3'-chloro-4'-fluoroanilino)-7-methoxy-6-(3-morphoIinopropoxy)- quinazoline (gefitinib) (i) Preparation of 4-Methoxy-3-[3-(4-morphoIinyl)propoxy]benzaldehyde
Into a IL, four necked, round-bottomed, flask equipped with a mechanical stirrer, reflux condenser and thermometer socket are charged dimethylformamide (500 mL), potassium carbonate (91 g), and iso-vanillin (50 g). After stirring the reaction mixture for 15min, 3-
. . , t ^
morpholinopropyl chloride (67.4 g) was added in 15min. The reaction mass was heated to 80 °C and maintained for 2 hr. TLC of the reaction mass indicated the absence of iso- vanillin. The reaction mass was cooled to 25-30 °C, filtered the inorganic material and washed the cake with 50 mL of dimethylformamide. Dimethylformamide was distilled of from the filtrate below 80 °C and the residue was poured into a wide petri dish while hot. After allowing the mass to cool to 25-30 °C, 91.5 g of 4-methoxy-3-£3-(4-morpholinyI)- propoxy]benzaldehyde was obtained as tan crystalline solid. M. P. is 52-54 °C. IR (KBr); 2954, 2844, 2813, 2761, 1675, 1586, 1512, 1436, 1402, 1356, 1270, 1240, 1161, 1137, 1116, 1071, 1022, 1002, 993, 946, 896, 862, 831, 730, and 641 cm'1. 1H-NMR (300MHz, CDC13): 9.85 (s, IH, -CHO); 7.44-7.48 (m, 2H, ar. H); 6.98 (d, J .= 8.4Hz, IH, ar. H); 4.16 (t, J = 6.6Hz, 2H, -OCHzCHz-); 3.95 (s, 3H, -OCH3); 3.73 (t, J = 4.4Hz, 4H, - CH2OCH2-); 2.38-2.62 (m, 6H, 3 x-NCH2-); 1.98-2.12 (m, 2H, -OCTfeCHjCHr).
(ii) Preparation of 4-methoxy-6-nitro-3-[3-(4-morphoIinyI)propoxy]benzaIdehyde Into a IL, four necked, round-bottomed, flask equipped with a mechanical stirrer, reflux condenser and thermometer socket are charged dil. Nitric acid (500 mL, 40% w/w). The reaction mass was heated to 40 °C and added 4-methoxy-3- [3 -(4-morpholiny ^-propoxyjbenzaldehyde (50 g) obtained by the process described in step (i) in lots over a period of 20-30min. After maintaining for 4hr at 40-45 °C reaction was found to be over by TLC. The reaction mass was cooled to 25-30 °C and poured into 500 mL of water at 25-30 °C. pH of the reaction mass was adjusted to 8.0 with aqueous ammonia (ca. 400 m.L) keeping the temperature below 30 °C. Product was extracted into ethyl acetate (3 x 400 mL and 1 x 200 mL). The combined ethyl acetate layer was dried over anhydrous sodium sulfate, distilled of solvent below 60 °C to get 4-methoxy-3-[3-(4-morpholinyl)- propoxyjbenzaldehyde quantitatively as light brown solid. A small sample was recrystallized from isoporpanol to get fine yellow needles. M. P. is 110-111 °C. IR (KBr): 3107, 2945, 2894, 2857, 2832, 1683, 1608, 1573, 1518, 1506, 1469, 1438, 1398, 1338, 1284, 1223, 1170, 1116, 1061, 1011, 906, 881, 862, 807, 738, and 643 cm"1. Η- NMR (300MHz, CDC13): 10.45 (s, IH, -CHO); 7.61 (s, IH, ar. H); 7.43 (s, IH, ar. H); 7.27 (s, IH, ar. H); 4.25 (t, J = 6.6Hz, 2H, -OCH2CH2-); 4.02 (s, 3H, -OCH3); 3.72 (t, J =
. <.
4.8Hz, 4H, -CH
2OCH
2-); 2.44-2.57 (m, 6H, 3 x -NCH
2-); 2.03-2.14 (m, 2H, -
(iii) Preparation of 4-methoxy-6-nitro-3-[3-(4-morpholinyl)propoxy]benzaldoxime Into a 500-mL, four necked, round-bottomed, flask equipped with a mechanical stirrer, reflux condenser and thermometer socket are charged 150 mL of 95% methanol and 50 g of 4-methoxy-6-nitro-3-[3-(4-morpholinyl)propoxy]benzaldehyde obtained by the process described in step (ii). A solution of aqueous hydroxylamine sulfate (17.5 g dissolved in 50 mL of water) was added to the reaction mixture in 15 min. A solution of aqueous sodium hydroxide (15 g of sodium hydroxide dissolved in 75 mL of water) was added to the reaction mass slowly keeping the temperature between 30-35 °C. After maintaining the reaction mass at 30-35 °C for 4 hr reaction was found to be over by TLC. The reaction mass was quenched into 200 g of crushed ice under stirring and adjusted the pH of reaction mass to 7.0-8.0 with aqueous acetic acid (50% v/v). After the pH adjustment reaction mass was stirred for 30 min and filtered the product under vaccum. The wet cake was washed with 200 mL of water and dried 75-80 °C to get 4-methoxy-6- nitro-3-[3-(4-morphoϊinyl)propoxy]benzaldoxime (38 g) as bright yellow solid. A small sample was recrystallized from ethyl acetate to get analytically pure sample. M. P. is 166-168 °C. IR (KBr): 3479, 3105, 3015, 2960, 2859, 2837, 2733, 1615, 1577, 1523, 1500, 1412, 1355, 1328, 1270, 1215, 1117, 1069, 1010, 985, 946, 922, 861, 798, 753, 685, and 646 cm-1. 1H-NMR (300MHz, CDC13): 10.13 (br. s. exch. with D2O, IH, =NOH); 8.74 (s, IH, -CH=N-); 7.61 (s, IH, ar. H); 7.33 (s, IH, ar. H); 7.27 (s, IH, ar. H); 4.21 (t, J = 5.9Hz, 2H, -OCHjCHz); 3.95 (s, 3H, -OCH3); 3.77 (t, J = 4.8Hz, 4H, - CH2OCH2-); 2.54-2.68 (m, 6H, 3 x -NCH2-); 2.12-2.25 (m, 2H, -CH2CH2CH2-).
(iv) Preparation of 4-methoxy-6-nitro-3-[3-(4-morpholinyl)propoxy]benzonitriIe Into a 500-mL, four necked, round-bottomed, flask equipped with a mechanical stirrer, reflux condenser and thermometer socket are charged with acetic anhydride (108 g) and 20 g of 4-methoxy-6-nitro-3-[3-(4-morpholinyl)propoxy]benzaldoxime prepared by the process described in step (iii) . The reaction mass was slowly heated to 120-125 °C and maintained for 4 hr. The reaction mass was cooled to 80 °C and excess acetic anhydride
was distilled of under vaccum. The residue was cooled to 30-35 °C and added 200 L of water. After stirring for 15 min reaction mass became a clear solution. The reaction mass pH was adjusted to 7.0-8.0 using aqueous ammonia. The resultant precipitate was collected by filtration and washing with 200 mL of water. After drying the wet material in drier at 70-75 °C (17 g) of 4-methoxy-6-nitro-3-[3-(4-morpholinyl)-propoxy]- benzonitrile was obtained as a dark greenish yellow solid. M. P. is 138-140 °C. IR (KBr): 3109, 3063, 2926, 2830, 2226, 1609, 1574, 1535, 1459, 1443, 1400, 1343, 1299, 1254, 1231, 1181, 1147, 1116, 1060, 1006, 980, 916, 882, 860, 799, 754, 635, and 615 cm"1. 1H-NMR (300MHz, CDC13): 7.79 (s, IH, ar. H); 7.27 (s, IH, ar. H); 4.23 (t, J = 6.6Hz, 2H, -OCH2CH2-); 4.02 (s, 3H, -OCH3); 3.73 (t, J = 4.4Hz, 4H, -CH2OCH2-); 2.45- 2.57 (m, 6H, 3 x -NCH2); 2.01-2.15 (m, 2H, -CHz ^CH ).
(v) Preparation of 4-methoxy-6-nitro-3-[3-(4-morp olinyl)propoxy]-benzamide
Into a 500-mL, four necked, round-bottomed, flask equipped with a mechanical stirrer, reflux condenser and thermometer socket are charged 3% hydrogen peroxide (160 mL), aqueous potassium hydroxide (10 mL, 25%w/w), and 4-methoxy-6-nitro-3-[3-(4- morpholinyl)propoxy]benzonitrile (10 g). The reaction mixture was warmed to 45 °C and maintained for 1 hr. TLC of the reaction mass indicated the absence of starting nitrile compound. The reaction mass was cooled to 30-35 °C and filtered. After washing the wet cake with water it was dried in oven at 75-80 °C to get 4-methoxy-6-nitro-3-[3-(4- morpholinyl)propoxy]-benzamidε (8 g) as off white solid. M.P. is 187-188 °C. IR
(KBr): 3409, 3308, 3207, 2944, 2856, 1670, 1616, 1578, 1522, 1427, 1362, 1345, 1272,
1216, 1117, 1050, 906, 884, 863, 805, and 649 cm-1. 1H-NMR (300MHz, DMSO-d6):
7.97 (s, exch. with D2O, IH, -CONH2); 7.55 (s, IH exch. with D2O, 2H, CONH2 and ar. H); 7.09 (s, IH, ar. H); 4.15 (t, J = 4.6Hz, 2H, -OCH2CH2CH2-); 3.86 (s, 3H, -OCH3);
3.56 (t, J = 4.0Hz, 4H, -CH2OCH2-); 2.35-2.50 (m, 6H, 3 x -NCH2); 1.86-1.93 (m, 2H, -
OCH2CH2CH2-).
(vi) Preparation of 2-Amino-4-methoxy-5-[3-(morpholinyI)propoxy]benzamide Into a IL, stainless steel hydrogenation kettle are charged methanol (150 mL), Raney nickel (2 g5 50% wet), and 4-methoxy-6-nitro-3-[3-(4-mo.φholinyl)propoxy]-benzamide
(5 g). The kettle was twice evacuated and filled with hydrogen. Hydrogenation was conducted at 25-30 °C and at a pressure of 20-25 psi for 2 hr. TLC of the reaction mass indicated the absence of starting nitro compound. The reaction mass was filtered on hiflow bed and the filtrate treated with carbon (2 g), distilled of solvent under reduced pressure to get 4.1 g of 2-amino-4-methoxy-5-[3-(morpholinyl)propoxy]benzamide as light greenish solid. M. P. is 153-154 °C. IR (KBr): 3447, 3308, 3191, 2947, 2863, 2820, 1661, 1632, 1582, 1542, 1517, 1466, 1444, 1391, 1265, 1230, 1204, 1116, 1048, 1004, 936, 906, 868, 842, and 583 cm-1. 1H-NMR (300MHz, DMSO-d6): 7.40 (br. s, exch. with D2O, IH, -CONH2); 7.11 (s, IH, ar. H); 6.80 (br. s, exch. with D2O, IH, -CONH2); 6.43 (br. s, exch. with D2O, 2H, ArNH2-); 6.23 (s, IH, ar. H); 3.85 (t, J = 6.6Hz, 2H, - OCH2CH2CH2-); 3.69 (s, 3H, -OCH3); 3.55 (t, J = 4.7Hz, 4H, -CH2OCH2-); 2.31-2.42 (m, 6H, 3 x -NCH2); 1.75-1.85 (m, 2H, -OCH2CU2 U2- .
(vii) Preparation of 7-methoxy-6-(3-morpholinopropoxy)-3,4-dihydroquinazoIin-4- one
Into a 500-mL, four necked, round-bottomed, flask equipped with a mechanical stirrer, reflux condenser and thermometer socket are charged 2-Amino-4-methoxy-5-[3- (morpholinyl)propoxy]benzamide (24 g) and formic acid (122 g, 98%). After stirring for 15-20 min at 25-30 °C reaction mass became a clear solution. The reaction mass was heated to 90-95 °C and maintained for 4 hr. TLC of reaction mass indicated the absence of starting compound. Excess formic acid was distilled of from the reaction mass keeping the temperature below 80 °C. The residue was cooled to 30-35 °C and added water (250 mL). After stirring for 10 min reaction mass became a clear solution. pH of the reaction mass was adjusted to 8.0-9.0 using aqueous ammonia. After the pH adjustment reaction mass was maintained for 30 min and filtered. The wet cake was washed with water, dried at 75-80 °C to get 7-methoxy-6-(3-morpholinopropoxy)-3,4-dihydroquinazoIin-4-one (15 g) as off white solid. M. P. is 244.5 °C.
(viii) Preparation of 4-chloro-6-(3-mopholinopropoxy)-7-methoxy-quinazoline Into a 500-mL, four necked, round-bottomed, flask equipped with a mechanical stirrer, reflux condenser, pressure equalizing addition funnel, and thermometer socket are
charged 7-methoxy-6-(3-morpholinopropoxy)-3,4-dihydroquinazolin-4-one (10 g), thionyl chloride (245 g or 150 mL), and dimethylformamide (2 g). The reaction mass was slowly heated to reflux temperature and maintained at reflux for lhr. Reaction was found to be over by TLC. Excess thionyl chloride was distilled off from reaction mass keeping the temperature below 50 °C. Toluene (50 mL) was added to the residue and distilled off under vaccum keeping the temperature below 50 °C. Addition and distillation of toluene was repeated twice and the residue cooled to 10 °C. Isopropanol ((50 mL) was added to the reaction mass and kept under stirring for 30 min without allowing the mass temperature to more than 25 °C. The resultant reaction mass was filtered and the solid washed with 50 mL of isoporanol (50 mL). The wet cake (17 g) thus obtained was directly used in next step without any delay.
(ix) Preparation of 4-(3'-chloro-4'-fluoroanilino)-7-methoxy-6-(3-morpholino- propoxy)-quinazoline Into a 500-mL, four necked, round-bottomed, flask equipped with a mechanical stirrer, reflux condenser, and thermometer socket are charged isopropanol (100 mL), 4-chloro-6- (3-mopholinopropoxy)-7-methoxy-quinazoline (17 g), and 3-chloro-4-fluoroaniline (10 g). The reaction mass was stirred and heated to reflux temperature. After maintaining at reflux temperature for 1 hr the reaction was found to be over by TLC. The reaction mass was cooled to 30-35 °C and filtered the reaction mass. The wet cake was washed with isopropanol (50 mL) and dried in the oven at 70-75 °C to get 13.7 g of the crude compound.
The above crude compound was taken into a 250-mL beaker and added 150 mL of water. After stirring and heating to 60 °C, pH of the reaction mass was adjusted to 9.5-10-0 with dilute sodium hydroxide solution. The reaction mass was cooled to 30-35 °C and filtered.
The wet cake was washed with water (50 mL) and dried at 75-80 °C to get 11 g of 4-(3'- chloro-4'-fluoroaniIino)-7-methoxy-6-(3-morphoIinopropoxy)-quinazoline as off white solid.
Example 2
Preparation of 4-(3'-chIoro-4'-fluoroanilino)-7-methoxy-6-(3-morpholinopropoxy)- quinazoline (gefitinib)
(i) Preparation of 2-amino-4-methoxy-5-[3-(morpholinyl)propoxy]benzamide Into a 500-mL, four necked, round-bottomed, flask equipped with a mechanical stirrer, reflux condenser and thermometer socket are charged with methanol (250 mL), 4- methoxy-6-nitro-3-[3-(4-moφholinyl)propoxy]benzonitrile (25 g), obtained according to the procedure given in Example 1 above and Raney nickel (10 g, 50% wet). A solution of hydrazine hydrate (17.5 g, 80 %) in methanol (25 mL) was slowly added to the reaction mass over a period of 2 hr maintaining the mass temperature at 40-45 °C. After the addition, reaction mass was maintained for another 2 hr at 40-45 °C and found to be over by TLC. The reaction mass was cooled to 30-35 °C and filtered off Raney nickel. Methanol was distilled of from the filtrate below 40 °C to get the title compound (25 g) as light greenish solid.
(ii) Preparation of 7-methoxy-6-(3-morpholinopropoxy)-3,4-dihydroquinazolin-4- one
Into a 500-mL, four necked, round-bottomed, flask equipped with a mechanical stirrer, reflux condenser and thermometer socket are charged 2-amino-4-methoxy-5-[3- (moφholinyl)propoxy]benzamide (10 g) and formic acid (60 g, 90%). After stirring for 15-20 min at 25-30 °C reaction mass became a clear solution. The reaction mass was heated to 90-95 °C and maintained for 4 hr. TLC of reaction mass indicated the absence of starting compound. Excess formic acid was distilled off from the reaction mass keeping the temperature below 80 °C. The residue was cooled to 30-35 °C and added water (150 mL). After stirring for 10 min reaction mass became a clear solution. pH of the reaction mass was adjusted to 8.0-9.0 using aqueous ammonia. After the pH adjustment reaction mass was maintained for 30 min and filtered. The wet cake was washed with water, dried at 75-80 °C to get 7-methoxy-6-(3-morpholinopropoxy)-3,4- dihydroquinazoIin-4-one (6 g) as off white solid.
(iii) Preparation of 4-chloro-6-(3-mopholinopropoxy)-7-methoxy-quinazoUne
Into a 500-mL, four necked, round-bottomed, flask equipped with a mechanical stirrer, o reflux condenser, pressure equalizing addition funnel, and thermometer socket are charged 7-methoxy-6-(3-moφholinopropoxy)-3,4-dihydroquinazolin-4-one (10 g), chloroform (100 mL), thionyl chloride (20 g), and dimethylformamide (1 g). The reaction mass was slowly heated to reflux temperature and maintained at reflux for lhr. Reaction was found to be over by TLC. Chloroform and excess thionyl chloride were distilled of from reaction mass keeping the temperature below 50 °C. Toluene (50 mL) was added to the residue and distilled of under vaccum keeping the temperature below 50 °C. Addition and distillation of toluene was repeated twice and the residue cooled to 10 °C. Isopropanol ((50 mL) was added to the reaction mass and kept under stirring for 30 min without allowing the mass temperature to more than 25 °C. The resultant reaction mass was filtered and the solid washed with 50 mL of isoporanol (50 mL). The wet cake (12 g) thus obtained was directly used in next step without any delay. ,
(iv) Preparation of 4-(3'-chloro-4'-fluoroanilino)-7-methoxy-6-(3-morpholino- propoxy)-quinazoline
Into a 500-mL, four necked, round-bottomed, flask equipped with a mechanical stirrer, reflux condenser, and thermometer socket are charged isopropanol (100 mL), 4-chloro-6- (3-mopholinopropoxy)-7-methoxy-quinazoline (12 g), triethylamine (5 mL), and 3- chloro-4-fluoroaniline (6 g). The reaction mass was stirred and heated to reflux temperature. After maintaining at reflux temperature reaction was found to be over by TLC. The reaction mass was cooled to 30-35 °C and filtered the reaction mass. The wet cake was washed with isopropanol (50 mL) and dried in the oven at 70-75 °C to get 10 g of the title compound.
Example 3
Recrystallization of gefitinib of formula-I from Acetonitrile
Into a 500 mL, three-necked RB flask was charged 5 g of gefitinib prepared by the process described in Example 1 and 350 mL of acetonitrile. The reaction mixture was heated to reflux temperature to get a clear solution. Carbon (1 g) was added to the
reaction mass and filtered under vaccum. The filtrate was cooled to 25-30 °C, maintained for 1 hr and filtered to get 4.1 g of pure gefitinib as white crystalline solid. M. P. is 197.4°C. Purity by HPLC is 99.83%.
Example 4
Recrystallization of gefitinib of formula-I from Isopropanol
Into a 500 mL, three-necked RB flask was charged 5 g of gefitinib prepared by the process described in Example 2 and 400 mL of isopropanol. The reaction mixture was heated to reflux temperature to get a clear solution. Carbon (1 g) was added to the reaction mass and filtered under vaccum. The filtrate was cooled to 0-5 °C, maintained for 1 hr and filtered to get 4.0 g of pure gefitinib as white crystalline solid. M. P. is 197.1 °C. Purity by HPLC is 99.82%.
Example 5 Recrystallization of gefitinib of formula-I from Ethyl acetate
Into a 500 mL, three-necked RB flask was charged 5 g of gefitinib prepared by the process described in Example 1 and 400 mL of ethyl acetate. The reaction mixture was heated to reflux temperature to get a clear solution. Carbon (1 g) was added to the reaction mass and filtered under vaccum. The filtrate was cooled to 25-30 °C, maintained for 1 hr and filtered to get 4 g of pure gefitinib as white crystalline solid. M. P. is 197.2°C. Purity by HPLC is 99.78%.
Example 6
Recrystallization of gefitinib of formula-I from Methyl ethyl ketone Into a 500 mL, three-necked RB flask was charged 5 g of gefitinib prepared by the process described in Example 1 and 350 mL of methyl ethyl ketone. The reaction mixture was heated to reflux temperature to get a clear solution. Carbon (1 g) was added to the reaction mass and filtered under vaccum. The filtrate was cooled to 10-15 °C, maintained for 1 hr and filtered to get 4.2 g of pure gefitinib as white crystalline solid. M. P. is 197.2°C. Purity by HPLC is 99.74%.
Example 7
Purification of gefitinib of formula-I by acid/base treatment
Into a 250 mL, three-necked RB flask was charged 5 g of crude gefitinib prepared by the process described in Example 1 and 100 mL of 5% aqueous acetic acid. The reaction mass was heated to 70-80 °C to get a clear solution. Carbon (1 g) was added to the reaction and filtered under vaccum. The filtrate was neutralized with aqueous ammonia to pH 8-9 and stirred for 30 min. The resulting precipitate was filtered and washed with 20 mL of water and 5 mL of isopropanol to get 4.25 g of pure gefitinib as white crystalline solid. M.P. is 197.0°C. HPLC purity is 99.76%.
• Advantages of Present Invention:
1. The gefitinib prepared by the process is of high purity (>99.5%).
2. Process does not require any chromatographic purification therefore simple and commercially applicable.
3. Process is free from impurity of the formula-XVIII.
4. Process involves novel compounds of the formulae-X to XV.