WO2005063247A1 - Aryl sulfonamide compounds and uses related thereto - Google Patents
Aryl sulfonamide compounds and uses related thereto Download PDFInfo
- Publication number
- WO2005063247A1 WO2005063247A1 PCT/US2004/042842 US2004042842W WO2005063247A1 WO 2005063247 A1 WO2005063247 A1 WO 2005063247A1 US 2004042842 W US2004042842 W US 2004042842W WO 2005063247 A1 WO2005063247 A1 WO 2005063247A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- aryl
- heteroaryl
- disorder
- Prior art date
Links
- 0 C[C@@](CN(CC1(CC1)c1ccncc1)CC1)*1S(c1ccc(C(C)(C(F)(F)F)O)cc1)(=O)=O Chemical compound C[C@@](CN(CC1(CC1)c1ccncc1)CC1)*1S(c1ccc(C(C)(C(F)(F)F)O)cc1)(=O)=O 0.000 description 6
- CNLBEQKBEZCTGU-FWJOYPJLSA-N C[C@H](CN(CC1(CC1)C(O)=O)CC1)N1S(c1ccc(C(C)(C(F)(F)F)O)cc1)(=O)=O Chemical compound C[C@H](CN(CC1(CC1)C(O)=O)CC1)N1S(c1ccc(C(C)(C(F)(F)F)O)cc1)(=O)=O CNLBEQKBEZCTGU-FWJOYPJLSA-N 0.000 description 1
- BFGFWFKNUIQYPQ-UJONTBEJSA-N C[C@H](CN(CC1(CC1)c(cc1)cnc1Cl)CC1)N1S(c1ccc(C(C)(C(F)(F)F)O)cc1)(=O)=O Chemical compound C[C@H](CN(CC1(CC1)c(cc1)cnc1Cl)CC1)N1S(c1ccc(C(C)(C(F)(F)F)O)cc1)(=O)=O BFGFWFKNUIQYPQ-UJONTBEJSA-N 0.000 description 1
- YYJQXYCFCBXRDT-OQHSHRKDSA-N C[C@H](CN(CC1(CC1)c1cnccc1)CC1)N1S(c1ccc(C(C)(C(F)(F)F)O)cc1)(=O)=O Chemical compound C[C@H](CN(CC1(CC1)c1cnccc1)CC1)N1S(c1ccc(C(C)(C(F)(F)F)O)cc1)(=O)=O YYJQXYCFCBXRDT-OQHSHRKDSA-N 0.000 description 1
- QYPQMTIQUXFCGG-QGZVFWFLSA-N C[C@H](CN(CC1(Cc2cnccc2)CC1)CC1)N1S(c1ccc(C(C(F)(F)F)(C(F)(F)F)O)cc1)(=O)=O Chemical compound C[C@H](CN(CC1(Cc2cnccc2)CC1)CC1)N1S(c1ccc(C(C(F)(F)F)(C(F)(F)F)O)cc1)(=O)=O QYPQMTIQUXFCGG-QGZVFWFLSA-N 0.000 description 1
- XGQRCMUZWUGSAX-LLVKDONJSA-N C[C@H]1NCCN(CC2(CC2)c(cc2)cnc2Cl)C1 Chemical compound C[C@H]1NCCN(CC2(CC2)c(cc2)cnc2Cl)C1 XGQRCMUZWUGSAX-LLVKDONJSA-N 0.000 description 1
- JOMNTHCQHJPVAZ-RXMQYKEDSA-N C[C@H]1NCCNC1 Chemical compound C[C@H]1NCCNC1 JOMNTHCQHJPVAZ-RXMQYKEDSA-N 0.000 description 1
- JTVIGCDEICVAGC-UHFFFAOYSA-N O=CC1(CC1)c(cc1)cnc1Cl Chemical compound O=CC1(CC1)c(cc1)cnc1Cl JTVIGCDEICVAGC-UHFFFAOYSA-N 0.000 description 1
- BTAKPMQKLSAHFC-UHFFFAOYSA-N OCC1(CC1)c(cc1)cnc1Cl Chemical compound OCC1(CC1)c(cc1)cnc1Cl BTAKPMQKLSAHFC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
- C07D213/38—Radicals substituted by singly-bound nitrogen atoms having only hydrogen or hydrocarbon radicals attached to the substituent nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/61—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/04—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/22—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with hetero atoms directly attached to ring nitrogen atoms
- C07D295/26—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Definitions
- This invention is generally directed to novel compounds, compositions, and the use of either in methods for modulating hydroxysteroid dehydrogenases, such as 11/3-HSD1, and for treating or preventing diseases associated with the modulation of hydroxysteroid dehydrogenases, such as diabetes and obesity.
- the methods comprise the administration, to a patient in need thereof, of a therapeutically effective amount of an Aryl Sulfonamide Compound.
- Novel Aryl Sulfonamide Compounds or pharmaceutically acceptable salts, solvates, stereoisomers, or prodrugs thereof are presented herein.
- HSDs Hydroxysteroid dehydrogenases
- the 11-beta-hydroxysteroid dehydrogenases (11 ⁇ -HSDs) catalyze the interconversion of active glucocorticoids (such as cortisol and corticosterone), and their inert forms (such as cortisone and 11- dehydrocorticosterone).
- the isoform 11-beta-hydroxysteroid dehydrogenase type 1 (11/3- HSD1) is expressed in liver, adipose tissue, brain, lung and other glucocorticoid tissue and is a potential target for therapy directed at numerous disorders that may be ameliorated by reduction of glucocorticoid action, such as diabetes, obesity and age-related cognitive dysfunction. Seckl, et al., Endocrinology, 2001, 142:1371-1376.
- HSDs are also believed to play a role in obesity.
- Obesity is an important factor in Syndrome X as well as type II (non-insulin dependent) diabetes, and omental fat appears to be of central importance in the development of both of these disease, as abdominal obesity has been linked with glucose intolerance, hyperinsulinemia, hypertriglyceridemia, and other factors of Syndrome X (e.g., raised blood pressure, decreased levels of HDL and increased levels of VLDL).
- stromal cells pre-adipocytes
- resultsed in a decreased rate of differentiation into adipocytes This is predicted to result in diminished expansion (possibly reduction) ofthe omental fat depot, which may lead to reduced central obesity. Bujalska et al., Lancet 1997, 349:1210-1213.
- HSDs have also been implicated in the process of appetite control and therefore is believed to play an additional role in weight-related disorders. It is known that adrenalectomy attenuates the effect of fasting to increase both food intake and hypothalamic neuropeptide Y expression. This suggests that glucocorticoids play a role in promoting food intake and that inhibition of 11/3-HSD1 in the brain may increase satiety, thus resulting in a decreased food intake. Woods et al., Sct ' ercce 1998, 280:1378-1383.
- Another possible therapeutic effect associated with modulation of HSDs is that which is related to various pancreatic aliments. It is reported that inhibition of 11/3-HSD1 in murine pancreatic /3-cells results in increased insulin secretion. Davani et al., J. Biol. Chem. 2000, 275:34841-34844. This follows from the preceding discovery that glucocorticoids were previously found to be responsible for reduced pancreatic insulin release in vivo Billaudel et al., Horm. Metab. Res. 1979, 11:555-560. Thus, it is suggested that inhibition of 11/3-HSD1 would yield other beneficial effects in the treatment of diabetes other than the predicted effects on the liver and of fat reduction.
- 11 / 3-HSD1 also regulates glucocorticoid activity in the brain and thus contributes to neurotoxicity.
- Stress and/or glucocorticoids are known to influence cognitive function (de Quervain et al, Nature 1998, 394:787-790), and unpublished results indicate significant memory improvement in rats treated with a non-specific 11/3-HSD inhibitors.
- 11/3-HSD1 reactivates 11-DHC to corticosterone in hippocampal cells and can potentiate kinase neurotoxicity, resulting in age- related learning impairments. Therefore, selective inhibitors of 1 l -HSDl are believed to protect against hippocampal function decline with age. Yau et al., Proc Natl. Acad. Sci. USA 2001, 98:4716-4721. Thus, it has been hypothesized that inhibition of 11/3-HSD1 in the human brain would protect against deleterious glucocorticoid-mediated effects on neuronal function, such as cognitive impairment, depression, and increased appetite.
- HSDs are believed to play a role in immunomodulation based on the general perception that glucocorticoids suppress the immune system.
- HPA hypothalamopituitary- adrenal
- glucocorticoids help balance between cell-mediated responses and humoral responses.
- Increased glucocorticoid activity which may be induced by stress, is associated with a humoral response and as such, the inhibition of 11/3-HSD1 may result in shifting the response towards a cell-based reaction.
- 11 / 3-HSD1 is expressed exclusively in the basal cells ofthe corneal epithelium, the non-pigmented epithelialium ofthe cornea (the site of aqueous production), ciliary muscle, and the sphincter and dilator muscles ofthe iris.
- the distant isoenzyme 1 l -hydroxysteroid dehydrogenase type 2 (“11/3-HSD2") is highly expressed in the non-pigmented ciliary epithelium and corneal endothelium.
- No HSDs have been found at the trabecular meshwork, which is the site of drainage. Therefore, 1 l -HSDl is suggested to have a role in aqueous production.
- Glucocorticoids also play an essential role in skeletal development and function but are detrimental to such development and function when present in excess.
- Glucocorticoid- induced bone loss is partially derived from suppression of osteoblast proliferation and collagen synthesis, as reported in Kim et al., J. Endocrinol. 1999, 162:371 379. It has been reported that the detrimental effects of glucocorticoids on bone nodule formation can be lessened by adminisfration of carbenoxolone, which is a non-specific 11 / 3-HSDl inhibitor. Bellows et al., Bone 1998, 23:119-125.
- 11/3-HSD1 maybe responsible for providing increased levels of active glucocorticoid in osteoclasts, and thus in augmenting bone resorption.
- Cooper et al. Bone 2000, 27:375-381.
- This data suggests that inhibition of 11/3-HSDl may have beneficial effects against osteoporosis via one or more mechanisms which may act in parallel.
- the various isozymes ofthe 17-beta-hydroxysteroid dehydrogenases (17/3-HSDs) bind to androgen receptors or estrogen receptors and catalyze the interconversion of various sex hormones including esfradiol/estrone and testosterone/androstenedione.
- six isozymes have been identifed in humans and are expressed in various human tissues including endometrial tissue, breast tissue, colon tissue, and in the testes.
- 17-beta- hydroxysteroid dehydrogenase type 2 (17/3-HSD2) is expressed in human endometrium and it's activity has been reported to be linked to cervical cancer. Kitawaki et al., J Clin. Endocrin.
- 17-beta-hydroxysteroid dehydrogenase type 3 (17/3-HSD3) is expressed in the testes and it's modulation may be useful for the freatment of androgen-related disorders.
- Androgens and estrogens are active in their 17/3-hydroxy configurations, whereas their 17-keto derivatives do not bind to androgen and estrogen receptors and are thus inactive.
- the conversion between the active and inactive forms (esfradiol/estrone and testosterone/androstenedione) of sex hormones is catlyzed by members ofthe 17/3-HSD family.
- 17/3-HSD 1 catalyzes the formation of estradiol in breast tissue, which is important for the growth of malignant breast tumors. Labrie et al., Mol. Cell. Endocrinol. 1991, 78:C113- CI 18.
- a similar role has been suggested for 17/3-HSD4 in colon cancer. English et al., J.
- 17/3-HSD3 is almost exclusively expressed in the testes and converts androstenedione into testosterone. Deficiency of this enzyme during fetal develoment leads to male pseudohermaphroditism. Geissler et al., Nat. Genet. 1994, 7:34-39. Both 17/3-HSD3 and various 3 ⁇ -HSD isozymes are involved in complex metabolic pathways which lead to androgen shuffles between inactive and active forms. Penning et al., Biochem. J. 2000, 351:67-77. Thus, modulation of certain HSDs can have potentially beneficial effects in the freatment of androgen- and estrogen-related disorders.
- the 20-alpha-hydroxysteroid dehydrogenases (20 ⁇ -HSDs) catalyze the interconversion of progestins (such as between progesterone and 20 ⁇ -hydroxy progesterone).
- Other substrates for 20 ⁇ -HSDs include 17o;-hydroxypregnenolone or 17 ⁇ - hydroxyprogesterone, leading to 20 ⁇ -OH steroids.
- 20o!-HSD isoforms have been identified and 20 ⁇ -HSDs are expressed in various tissues, including the placenta, ovaries, testes and adrenals. Peltoketo, et al., J. Mol. Endocrinol. 1999, 23:1-11.
- the 3-alpha-hydroxysteroid dehydrogenases catalyze the interconversion ofthe androgens dihydrotestosterone (DHT) and 5 ⁇ -androstane-3o 17/3-diol and the interconversion ofthe androgens DHEA and androstenedione and therefore play an important role in androgen metabolism.
- DHT dihydrotestosterone
- DHEA dihydrotestosterone
- DHEA androstenedione and therefore play an important role in androgen metabolism.
- Aryl sulfonamide compounds and methods for their synthesis are disclosed in Klioze et al., J. Med. Chem. 1980, 23:677-679, and International Publication No. WO 01/02371. The disclosure of these publications, however, does not however encompass the Aryl Sulfonamide Compounds ofthe present invention nor the use ofthe disclosed compounds as HSD modulators.
- the present invention relates to novel compounds, compositions thereof and methods for modulating the activity of hydroxysteroid dehydrogenases (HSDs), such as 11/3- hydroxysteroid dehydrogenases, 17/3-hydroxysteroid dehydrogenases, 20 ⁇ -hydroxysteroid dehydrogenases, and 3 ⁇ -hydroxysteroid dehydrogenases, including all isoforms thereof, including but not limted to 11/3-hydroxysteroid dehydrogenase type 1 (hereinafter "11 / 3- HSDl”), 11/3-hydroxysteroid dehydrogenase type 2 (hereinafter "11/3-HSD2”), and 17/3- hydroxysteroid dehydrogenase type 3 (hereinafter "17/3-HSD3").
- HSDs hydroxysteroid dehydrogenases
- the components ofthe invention inhibit HSD activity.
- the present invention also relates to methods for treating or preventing diseases or disorders associated with the action of hydroxysteroid dehydrogenases, comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound or a pharmaceutically acceptable salt, solvate, stereoisomer, or prodrug thereof.
- the invention encompasses both selective and non-selective inhibitors of hydroxysteroid dehydrogenases.
- selective and non-selective inhibitors of hydroxysteroid dehydrogenases each have benefits in the treatment or prevention of diseases associated with, for example, abnormal glucose levels or hypothalamic function.
- Two types of selectivity are contemplated, that with respect to selectivity for HSDs as a class over other types of receptors or gene targets related to glucose metabolism, or those which are selective for various HSDs or specific isoforms thereof compared to other HSDs or specific isoforms thereof.
- the Aryl Sulfonamide Compounds can act as selective or non- selective 11/3-HSD inhibitors.
- the compounds may inhibit the interconversion of inactive 11-keto steroids with their active hydroxy equivalents.
- the present invention provides methods by which the conversion ofthe inactive form to the active form may be controlled, and useful therapeutic effects maybe obtained as a result of such control. More specifically, but not exclusively, the invention is concerned with interconversion between cortisone and cortisol in humans.
- the Aryl Sulfonamide Compounds can act as 11/3-HSD inhibitors in vivo.
- the Aryl Sulfonamide Compounds ofthe present invention may be orally active.
- the Aryl Sulfonamide Compounds are also useful for modulation of numerous metabolic functions including, but not limited to, one or more of: (i) regulation of carbohydrate metabolism, (ii) regulation of protein metabolism, (iii) regulation of lipid metabolism, (iv) regulation of normal growth and/or development, (v) influence on cognitive function, (vi) resistance to stress and mineralocorticoid activity.
- the Aryl Sulfonamide Compounds may also be useful for inhibiting hepatic gluconeogenesis, and may also be effective to relieve the effects of endogenous glucocorticoids in diabetes mellitus, obesity (including entripetal obesity), neuronal loss and/or the cognitive impairment of old age.
- the invention provides the use of an inhibitor of HSDs in methods directed to producing one or more therapeutic effects in a patient to whom the Aryl Sulfonamide Compound is administered, said therapeutic effects selected from inhibition of hepatic gluconeogenesis, an increase in insulin sensitivity in adipose tissue and muscle, and the prevention of or reduction in neuronal loss/cognitive impairment due to glucocorticoid-potentiated neurotoxicity or neural dysfunction or damage.
- the invention further provides methods for treating treatment a condition selected from the group consisting of: hepatic insulin resistance, adipose tissue insulin resistance, muscle insulin resistance, neuronal loss or dysfunction due to glucocorticoid potentiated neurotoxicity, and any combination ofthe aforementioned conditions, the methods comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound.
- the Aryl Sulfonamide Compounds ofthe invention include compounds having Formula (I): (I) or pharmaceutically acceptable salts, solvates, stereoisomers, or prodrugs thereof, wherein R 1 , R 2 and R 3 are independently selected from -H, -halo, -OH, -CN, -NO 2 , -d-C 8 alkyl, -C 2 -C 8 alkenyl, -C 2 -C 8 alkynyl, -alkoxy, -haloalkyl, -hydroxyalkyl, -cycloalkyl, - heterocycloalkyl, -heteroaryl and -aryl, and at least one of R 1 , R 2 and R 3 is other than -H; R 4 is selected from -H, -halo, -CN, -NO 2 , - -Cs alkyl, -C 2 -C 8 alkenyl, -C 2 -C 8 alky
- R 5 is selected from -H, -halo, -CN, -NO 2 , -d-C 8 alkyl, -C 2 -C 8 alkenyl, -C 2 -C 8 alkynyl, - alkoxy, -haloalkyl, -hydroxyalkyl, -cycloalkyl, -heterocycloalkyl, -heteroaryl, -aryl,
- R 6 is selected from -H, -halo, -CN, -NO 2 , -C ⁇ -C 8 alkyl, -C 2 -C 8 alkenyl, -C 2 -C 8 alkynyl, - alkoxy, -haloalkyl, -hydroxyalkyl, -cycloalkyl, -heterocycloalkyl, -heteroaryl, -aryl,
- R 7 is selected from -H, -halo, -CN, -N0 2 , amino and -d-Cs alkyl; and in some embodiments is in a position ortho to the sulfonamide moiety of formula I;
- Q is selected from the group consisting of -H, -halo, -CN, -NO , -Q-Cs alkyl, -C 2 -C 8 alkenyl, -C 2 -C 8 alkynyl, -alkoxy, -haloalkyl, -hydroxyalkyl, -cycloalkyl, -heterocycloalkyl, - heteroaryl, -aryl, -cycloalkyl-(C 1 -C 6 alkyl),
- R a is selected from 4-methoxyphenyl, 4-chlorophenyl and 4-bromophenyl and R b is 4-fluorophenyl or 4-bromophenyl.
- the invention provides pharmaceutical compositions comprising an Aryl Sulfonamide Compound and a pharmaceutically acceptable vehicle, carrier, excipient or diluent.
- the invention provides methods for freating insulin-dependent diabetes mellitus comprising admimstering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for freating non-insulin- dependent diabetes mellitus comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for treating insulin resistance comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for freating obesity comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for modulating cortisol production comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for modulating hepatic glucose production comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for modulating hypothalamic function comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for freating a hydroxysteroid dehydrogenase-mediated condition or disorder comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides method for modulating the function of a hydroxysteroid dehydrogenase in a cell comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for modulating a hydroxysteroid dehydrogenase, comprising admimstering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for treating an 11 /3-HSD 1 - mediated condition or disorder comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides method for modulating the function of 11/3-HSDl in a cell comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for modulating 11 /3-HSD 1 , comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for freating an 1 l/3-HSD2-mediated condition or disorder comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I)..
- the invention provides method for modulating the function of 11/3-HSD2 in a cell comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for modulating 11/3-HSD2, comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for treating an 17/3-HSD3 -mediated condition or disorder comprising admimstering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides method for modulating the function of 17/3-HSD3 in a cell comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention provides methods for modulating 17/3-HSD3, comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- d-C 6 alkyl refers to a straight or branched chain, saturated hydrocarbon having from 1 to 6 carbon atoms.
- Representative d-C 6 alkyl groups include, but are not limited to methyl, ethyl, propyl, isopropyl, butyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, hexyl, isohexyl, and neohexyl.
- a -C ⁇ alkyl group can be unsubstituted or optionally substituted with one or more substituents as described herein below.
- d-C 8 alkyl refers to a straight or branched chain, saturated hydrocarbon having from 1 to 8 carbon atoms.
- Representative d-C 8 alkyl groups include, but are not limited to methyl, ethyl, propyl, isopropyl, butyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, hexyl, isohexyl, neohexyl, heptyl, isoheptyl, neoheptyl, octyl, isooctyl, and neooctyl.
- a d-C 8 alkyl group can be unsubstituted or optionally substituted with one or more substituents as described herein below.
- C 2 -C 8 alkenyl refers to a straight or branched chain unsaturated hydrocarbon containing 2-8 carbon atoms and at least one double bond.
- Examples of a C 2 -C 8 alkenyl group include, but are not limited to, ethylene, propylene, 1- butylene, 2-butylene, isobutylene, sec-butylene, 1-pentene, 2-pentene, isopentene, 1-hexene, 2-hexene, 3-hexene, isohexene, 1-heptene, 2-heptene, 3-heptene, isoheptene, 1-octene, 2- octene, 3-octene, 4-octene, and isooctene.
- C 2 -C 8 alkynyl refers to a straight or branched chain unsaturated hydrocarbon containing 2-8 carbon atoms and at least one triple bond.
- Examples of a C 2 -C 8 alkynyl group include, but are not limited to, acetylene, propyne, 1-butyne, 2- butyne, isobutyne, sec-butyne, 1-pentyne, 2-pentyne, isopentyne, 1-hexyne, 2-hexyne, 3- hexyne, isohexyne, 1-heptyne, 2-heptyne, 3-heptyne, isoheptyne, 1-octyne, 2-octyne, 3- octyne, 4-octyne, and isooctyne.
- d-C alkylene refers to a d-C 7 alkyl group in which one of the d-C 7 alkyl group's hydrogen atoms has been replaced with a bond.
- a d- C 7 alkylene include -CH 2 -, -CH 2 CH 2 -, -CH 2 CH 2 CH 2 -, -CH 2 CH 2 CH 2 CH 2 -, -CH 2 CH 2 CH 2 CH 2 CH 2 -, -CH 2 CH 2 CH 2 CH 2 CH 2 -, and -CH 2 CH 2 CH 2 CH 2 CH 2 CH 2 CH 2 -.
- d-C 6 alkoxy refers to a group having the formula -O- (d-C 6 alkyl).
- a d-C 6 alkoxy group include -O-methyl, -O-ethyl, -O-propyl, -O- isopropyl, -O-butyl, -O-sec-butyl, -O-tert-butyl, -O-pentyl, -O-isopentyl, -O-neopentyl, -O- hexyl, -O-isohexyl, and -O-neohexyl.
- aminoalkyl refers to a d-C 6 alkyl group wherein from one or more ofthe d-C 6 alkyl group's hydrogen atom is replaced with an amine of formula -N(Ra)2, wherein each occurrence of Ra is independently -H or d-C 6 alkyl.
- aminoalkyl groups include, but are not limited to, -CH 2 NH , -CH CH NH , -CH 2 CH 2 CH 2 NH 2 , -CH 2 CH 2 CH 2 CH 2 NH 2 , -CH 2 CH 2 CH 2 CH 2 CH 2 NH 2 , -CH 2 CH 2 CH 2 CH 2 CH 2 NH 2 , -CH 2 CH 2 CH 2 CH 2 CH 2 CH NH , t-butylamine and isopropylamine.
- aryl refers to a 6- to 14-membered monocyclic, bicyclic or tricyclic aromatic hydrocarbon ring system.
- Examples of an aryl group include phenyl and naphthyl.
- An aryl group can be unsubstituted or optionally substituted with one or more substituents as described herein below.
- cycloalkyl and “cycloalkane” are used interchangeably and refer to a 3- to 15-membered saturated or unsaturated non-aromatic monocyclic, bicyclic or tricyclic hydrocarbon ring system. Included in this class are cycloalkyl groups which are fused to a benzene ring and cycloalkyl groups which are spirocyclic, as well as spirocyclic and fused to a benzene ring.
- Representative cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclopentadienyl, cyclohexyl, cyclohexenyl, 1,3-cyclohexadienyl, cycloheptyl, cycloheptenyl, 1,3-cycloheptadienyl, 1,4- cycloheptadienyl, -1,3,5-cycloheptatrienyl, cyclooctyl, cyclooctenyl, 1,3-cyclooctadienyl, 1,4-cyclooctadienyl, -1,3,5-cyclooctatrienyl, decahydronaphthalene, octahydronaphthalene, hexahydronaphthalene, octahydroinden
- halo refers to -F, -CI, -Br or -I.
- haloalkyl refers to a d-C 6 alkyl group wherein from one or more ofthe d-C 6 alkyl group's hydrogen atom is replaced with a halogen atom, which can be the same or different.
- haloalkyl groups include, but are not limited to, trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, pentachloroethyl, and 10 1,1,1 -trifluoro-2-bromo-2-chloroethyl.
- heteroalkyl by itself or in combination with another term, means, unless otherwise stated, a stable straight or branched chain, or cyclic hydrocarbon radical, or combinations thereof, consisting ofthe stated number of carbon atoms and from one to three heteroatoms selected from the group consisting of O, N, Si and S, wherein the nitrogen and
- [ 5 sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quaternized.
- the heteroatom(s) O, N and S may be placed at any interior position ofthe heteroalkyl group.
- the heteroatom Si may be placed at any position ofthe heteroalkyl group, including the position at which the alkyl group is attached to the remainder ofthe molecule. Examples include -CH 2 -CH 2 -O-CH 3 , -CH 2 -CH 2 -NH-CH 3 , -CH 2 -CH 2 -N(CH 3 )-CH 3 , -CH 2 -S-
- d-C 7 heteroalkylene refers to a d-C alkylene in which one to three ofthe C 1 -C alkylene's -CH 2 - groups has been replaced by a sulfur atom, an 5 oxygen atom, or -NH-.
- a C ⁇ -C 7 heteroalkylene group can have a heteroatom at either or both of its termini.
- heteroaryl refers to an aromatic heterocycle ring of 5 to 14 members and having at least one heteroatom selected from nitrogen, oxygen and sulfur, and containing at least 1 carbon atom, including monocyclic, bicyclic, and tricyclic ring 0 systems.
- heteroaryls are triazolyl, tefrazolyl, oxadiazolyl, pyridyl, furyl, benzofuranyl, thiophenyl, benzothiophenyl, quinolinyl, pyrrolyl, indolyl, oxazolyl, benzoxazolyl, imidazolyl, benzimidazolyl, thiazolyl, benzothiazolyl, isoxazolyl, pyrazolyl, isothiazolyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, cinnolinyl, phthalazinyl, quinazolinyl, pyrimidyl, oxetanyl, azepinyl, piperazinyl, morpholinyl, dioxanyl, thietanyl and oxazolyl.
- a heteroaryl group can be unsub
- heteroatom is meant to include oxygen (O), nifrogen (N), and sulfur (S).
- heterocycle refers to 5- to 14-membered ring systems which are either saturated, unsaturated, or aromatic, and which contains from 1 to 4 heteroatoms independently selected from nifrogen, oxygen and sulfur, and wherein the nifrogen and sulfur heteroatoms may be optionally oxidized, and the nitrogen heteroatom may be optionally quaternized, including, including monocyclic, bicyclic, and tricyclic ring systems.
- the bicyclic and tricyclic ring systems may encompass a heterocycle or heteroaryl fused to a benzene ring.
- the heterocycle may be attached via any heteroatom or carbon atom.
- Heterocycles include heteroaryls as defined above.
- heterocycles include, but are not limited to, aziridinyl, oxiranyl, thiiranyl, friazolyl, tefrazolyl, azirinyl, diaziridinyl, diazirinyl, oxaziridinyl, azetidinyl, azetidinonyl, oxetanyl, thietanyl, piperidinyl, piperazinyl, morpholinyl, pyrrolyl, oxazinyl, thiazinyl, diazinyl, triazinyl, tetrazinyl, imidazolyl, tefrazolyl, pyrrolidinyl, isoxazolyl, furanyl, furazanyl, pyridinyl, oxazolyl, benzoxazolyl, benzisoxazolyl, thiazolyl, benzthiazolyl, azir
- hydroxyalkyl refers to a C ⁇ -C 6 alkyl group wherein from one or more ofthe C ⁇ -C 6 alkyl group's hydrogen atom is replaced with an -OH group.
- hydroxyalkyl groups include, but are not limited to, -CH 2 OH, -CH 2 CH 2 OH, -CH 2 CH 2 CH 2 OH, -CH 2 CH 2 CH 2 CH 2 OH, -CH 2 CH 2 CH 2 CH 2 OH, CH 2 CH 2 CH 2 CH 2 CH 2 OH, t-butanol and isopropanol.
- C 3 -C 8 hydroxyalkyl refers to a hydroxyalkyl group having from three to eight carbon atoms.
- R', R" and R'" each independently refer to hydrogen, unsubstituted (d-C 8 )alkyl or (d-C 8 )alkyl substituted with hydroxy, cyano or amino, unsubstituted hetero(C 1 -C 8 )alkyl, unsubstituted aryl and aryl substituted with one to three substituents selected from -halo, unsubstituted alkyl, unsubstituted alkoxy, unsubstituted thioalkoxy and unsubstituted aryl(d-C 4 )alkyl.
- R' and R" When R' and R" are attached to the same nitrogen atom, they can be combined with the nifrogen atom to form a 5-, 6- or 7- membered ring.
- -NR'R is meant to include 1-pyrrolidinyl and 4-morpholinyl.
- an alkyl or heteroalkyl group will have from zero to three substituents, with those groups having two or fewer substituents being preferred in the present invention. More preferably, an alkyl or heteroalkyl radical will be unsubstituted or monosubstituted. Most preferably, an alkyl or heteroalkyl radical will be unsubstituted. From the above discussion of substituents, one of skill in the art will understand that the term "alkyl” is meant to include groups such as trihaloalkyl (e.g., -CF and -CH 2 CF 3 ).
- Preferred substituents for the alkyl and heteroalkyl radicals are selected from: -OR', -NR'R", -SR', -halo, -SiR'R"R'", -OC(O)R', -C(0)R', -CO 2 R', -C(O)NR'R",
- substituents are selected from: -OR', -NR'R", -halo, -OC(O)R', -CO 2 R', -C(O)NR'R", -OC(O)NR'R", -NR"C(O)R', -NR"CO 2 R', -NR'"SO 2 NR'R", -SO 2 R', -SO 2 NR'R", -NR"SO 2 R' -CN and -NO 2 .
- an aryl or heteroaryl group will have from zero to three substituents, with those groups having two or fewer substituents being preferred in the present invention.
- an aryl or heteroaryl group will be unsubstituted or monosubstituted.
- an aryl or heteroaryl group will be unsubstituted.
- Preferred substituents for aryl and heteroaryl groups are selected from: -halo, -OR', -OC(O)R', -NR'R", -SR', -R', -CN, -N0 2 , -CO 2 R', -CONR'R", -C(O)R', -OC(0)NR'R", -NR"C(O)R', -S(O)R', -SO 2 R', -SO 2 NR'R", -NR"SO 2 R', -N 3 , -CH(Ph) 2 , perfluoroalkoxy and perfluoro(d-C 4 )alkyl, where R' and R" are as defined above.
- substituents are selected from: -halo, -OR', -OC(0)R', -NR'R", -R', -CN, -NO 2 , -CO 2 R', -CONR'R", -NR"C(O)R', -SO 2 R', -SO 2 NR'R", -NR"SO 2 R', perfluoroalkoxy and perfluoro(d-C 4 )alkyl.
- Two ofthe substituents on adjacent atoms ofthe aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -T-C(O)-(CH 2 ) q -U-, wherein T and U are independently -NH-, -O-, -CH 2 - or a single bond, and q is an integer of from 0 to 2.
- two ofthe substituents on adjacent atoms ofthe aryl or heteroaryl ring may optionally be replaced with a substituent ofthe formula -A-(CH 2 ) r -B-, wherein A and B are independently -CH 2 -, -O-, -NH-, -S-, -S(O)-, -S(O) 2 -, -S(O) 2 NR'- or a single bond, and r is an integer of from 1 to 3.
- One ofthe single bonds ofthe new ring so formed may optionally be replaced with a double bond.
- two ofthe substituents on adjacent atoms ofthe aryl or heteroaryl ring may optionally be replaced with a substituent ofthe formula -(CH 2 ) S - X-(CH )r, where s and t are independently integers of from 0 to 3, and X is -O-, -NR'-, -S-, - S(O)-, -S(O) 2 -, or -S(O) 2 NR'-.
- the substituent R' in -NR'- and -S(O) 2 NR'- is selected from hydrogen or unsubstituted (d-C 6 )alkyl.
- substituent -CO 2 H may be optionally replaced with bioisosteric replacements such as: 0 II P ⁇ OH , % OH and the like. See, e.g., The Practice of Medicinal Chemistry; Wermuth, C.G., Ed.; Academic Press: New York, 1996; p. 203.
- the Aryl Sulfonamide Compound can also exist in various isomeric forms, including configurational, geometric and conformational isomers, as well as existing in various tautomeric forms, particularly those that differ in the point of attachment of a hydrogen atom.
- the term "isomer" is intended to encompass all isomeric forms of an Aryl Sulfonamide Compound, including tautomeric forms ofthe compound.
- Certain Aryl Sulfonamide Compounds may have asymmetric centers and therefore exist in different enantiomeric and diastereomeric forms.
- An Aryl Sulfonamide Compound can be in the form of an optical isomer or a diastereomer. Accordingly, the invention encompasses Aryl Sulfonamide Compounds and their uses as described herein in the form of their optical isomers, diasteriomers and mixtures thereof, including a racemic mixture.
- Optical isomers ofthe Aryl Sulfonamide Compounds can be obtained by known techniques such as asymmetric synthesis, chiral chromatography, simulated moving bed technology or via chemical separation of stereoisomers through the employment of optically active resolving agents.
- stereomerically pure compound means one stereoisomer of a compound that is substantially free of other stereoisomers of that compound.
- a stereomerically pure compound having one chiral center will be substantially free ofthe opposite enantiomer ofthe compound.
- a stereomerically pure a compound having two chiral centers will be substantially free of other diastereomers ofthe compound.
- a typical stereomerically pure compound comprises greater than about 80% by weight of one stereoisomer ofthe compound and less than about 20% by weight of other stereoisomers ofthe compound, more preferably greater than about 90% by weight of one stereoisomer of the compound and less than about 10% by weight of the other stereoisomers ofthe compound, even more preferably greater than about 95% by weight of one stereoisomer ofthe compound and less than about 5% by weight ofthe other stereoisomers ofthe compound, and most preferably greater than about 97% by weight of one stereoisomer ofthe compound and less than about 3% by weight ofthe other stereoisomers of the compound.
- An Aryl Sulfonamide Compound can be in the form of a pharmaceutically acceptable salt.
- pharmaceutically acceptable salt refers to a pharmaceutically acceptable organic or inorganic acid or base salt of an Aryl Sulfonamide Compound.
- Representative pharmaceutically acceptable salts include, e.g., alkali metal salts, alkali earth salts, ammonium salts, water- soluble and water-insoluble salts, such as the acetate, amsonate (4,4-diaminostilbene-2, 2 - disulfonate), benzenesulfonate, benzonate, bicarbonate, bisulfate, bitarfrate, borate, bromide, butyrate, calcium, calcium edetate, camsylate, carbonate, chloride, citrate, clavulariate, dihydrochloride, edetate, edisylate, estolate, esylate, fiunarate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexafluorophosphate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, hydroxynaphthoate, iodide, is
- the term "isolated and purified form” means that when isolated (e.g., from other components of a synthetic organic chemical reaction mixture), the isolate contains at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 55%, at least 60%, at least 65%, at least 70%, at least 75%, at least 80%, at least 85%, at least 90%, at least 95% or at least 98% of an Aryl Sulfonamide Compound by weight of the isolate. In one embodiment, the isolate contains at least 95% of an Aryl Sulfonamide Compound by weight ofthe isolate.
- prodrug means a derivative of a compound that can hydrolyze, oxidize, or otherwise react under biological conditions (in vitro or in vivo) to provide an active compound, particularly an Aryl Sulfonamide Compound.
- prodrugs include, but are not limited to, derivatives and metabolites of an Aryl Sulfonamide Compound that include biohydrolyzable groups such as biohydrolyzable amides, biohydrolyzable esters, biohydrolyzable carbamates, biohydrolyzable carbonates, biohydrolyzable ureides, and biohydrolyzable phosphate analogues (e.g., monophosphate, diphosphate or triphosphate).
- biohydrolyzable groups such as biohydrolyzable amides, biohydrolyzable esters, biohydrolyzable carbamates, biohydrolyzable carbonates, biohydrolyzable ureides, and biohydrolyzable phosphate analogues (e.g.
- prodrugs of compounds with carboxyl functional groups are the lower alkyl esters ofthe carboxylic acid.
- the carboxylate esters are conveniently formed by esterifying any ofthe carboxylic acid moieties present on the molecule.
- Prodrugs can typically be prepared using well-known methods, such as those described by Burger 's Medicinal Chemistry and Drug Discovery 6 ed. (Donald J. Abraham ed., 2001, Wiley) and Design and Application of Prodrugs (H. Bundgaard ed., 1985, Harwood Academic Publishers Gmfh).
- the terms “treat”, “treating” and “treatment” refer to the eradication or amelioration of a disease or symptoms associated with a disease. In certain embodiments, such terms refer to minimizing the spread or worsening ofthe disease resulting from the administration of one or more prophylactic or therapeutic agents to a patient with such a disease.
- the terms “prevent”, “preventing” and “prevention” refer to the prevention ofthe onset, recurrence or spread ofthe disease in a patient resulting from the adminisfration of a prophylactic or therapeutic agent.
- an effective amount refers to an amount of an Aryl Sulfonamide Compound or other active ingredient sufficient to provide a therapeutic or prophylactic benefit in the freatment or prevention of a disease or to delay or minimize symptoms associated with a disease.
- a therapeutically effective amount with respect to an Aryl Sulfonamide Compound means that amount of therapeutic agent alone, or in combination with other therapies, that provides a therapeutic benefit in the treatment or prevention of a disease. Used in connection with an Aryl Sulfonamide Compound, the term can encompass an amount that improves overall therapy, reduces or avoids symptoms or causes of disease, or enhances the therapeutic efficacy of or synergies with another therapeutic agent,
- Syndrome X refers to a collection of abnormalities including hyperinsulinemia, obesity, elevated levels of triglycerides, uric acid, fibrinogen, small dense LDL particles and plasminogen activator inhibitor 1 (PAI-1), and decreased levels of HDL cholesterol. Syndrome X is further meant to include metabolic syndrome.
- modulate refers to the ability of a compound to increase or decrease the function, or activity of a hydroxysteroid dehydrogenase, for example, 11 (3-HSD 1.
- Module as used herein in its various forms, is intended to encompass inhibition, antagonism, partial antagonism, activation, agonism and/or partial agonism ofthe activity associated with a hydroxysteroid dehydrogenase.
- Hydroxysteroid dehydrogenase inhibitors are compounds that, e.g., bind to, partially or totally block stimulation, decrease, prevent, delay activation, inactivate, desensitize, or down regulate signal transduction.
- Hydroxysteroid dehydrogenase activators are compounds that, e.g., bind to, stimulate, increase, open, activate, facilitate, enhance activation, sensitize or up regulate signal transduction.
- the ability of a compound to modulate a hydroxysteroid dehydrogenase can be demonstrated in an enzymatic assay or a cell-based assay.
- the inhibition of 11/3-HSDl may decrease cortisol levels in a patient and/or increase cortisone levels in a patient by blocking the conversion of cortisone to cortisol.
- the inhibition of 11/3-HSD2 can increase cortisol levels in a patient and/or decrease cortisone levels in a patient by blocking the conversion of cortisol to cortisone.
- a "patient” includes an animal (e.g., cow, horse, sheep, pig, chicken, turkey, quail, cat, dog, mouse, rat, rabbit or guinea pig), in one embodiment a mammal such as a non- primate and a primate (e.g., monkey and human), and in another embodiment a human.
- a patient is a human.
- the patient is a human infant, child, adolescent or adult.
- HSD hydroxysteroid dehydrogenase enzymes in general, including, but not limited to, 11-beta-hydroxysteroid dehydrogenases (1 l ⁇ -HSDs), 17-beta-hydroxysteroid dehydrogenases (17 ⁇ -HSDs), 20-alpha-hydroxysteroid dehydrogenases (20 ⁇ -HSDs), 3-alpha-hydroxysteroid dehydrogenases (3 ⁇ -HSDs), and all isoforms thereof.
- 11/3-HSDl refers to the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme, variant, or isoform thereof.
- 11/3-HSDl variants include proteins substantially homologous to native 11/3-HSDl, i.e., proteins having one or more naturally or non-naturally occurring amino acid deletions, insertions or substitutions (e.g., 11/3-HSDl derivatives, homologs and fragments).
- the amino acid sequence of a 11/3-HSDl variant preferably is at least about 80% identical to a native 11/3-HSDl, more preferably at least about 90% identical, and most preferably at least about 95% identical.
- 11 /3-HSD2 refers to the 11 -beta-hydroxysteroid dehydrogenase type 2 enzyme, variant, or isoform thereof.
- 11 3-HSD2 variants include proteins substantially homologous to native 11 / 3-HSD2, i.e., proteins having one or more naturally or non-naturally occurring amino acid deletions, insertions or substitutions (e.g., 11/3-HSD2 derivatives, homologs and fragments).
- the amino acid sequence of a 11/3-HSD2 variant preferably is at least about 80% identical to a native 11/3-HSD2, more preferably at least about 90% identical, and most preferably at least about 95% identical, (see Bart et al, J. Med. Chem., 2002, 45:3813-3815).
- 17/3-HSD3 refers to the 17-beta-hydroxysteroid dehydrogenase type 3 enzyme, variant, or isoform thereof.
- 173-HSD3 variants include proteins substantially homologous to native 17/3-HSD3, i.e., proteins having one or more naturally or non-naturally occurring amino acid deletions, insertions or substitutions (e.g., 17/3-HSD3 derivatives, homologs and fragments).
- the amino acid sequence of a 17/3-HSD3 variant preferably is at least about 80% identical to a native 17/3-HSD3, more preferably at least about 90% identical, and most preferably at least about 95% identical.
- HSD-responsive condition or disorder refers to a condition or disorder that responds favorably to modulation of a hydroxysteroid dehydrogenase enzyme (HSD).
- HSD hydroxysteroid dehydrogenase enzyme
- Favorable responses to HSD modulation include alleviation or abrogation ofthe disease and/or its .attendant symptoms, inhibition of the disease, i.e., arrest or reduction ofthe development ofthe disease, or its clinical symptoms, and regression ofthe disease or its clinical symptoms.
- An HSD-responsive condition or disease may be completely or partially responsive to HSD modulation.
- HSD-responsive condition or disorder may be associated with inappropriate, e.g., less than or greater than normal, HSD activity and at least partially responsive to or affected by HSD modulation (e.g., an HSD inhibitor results in some improvement in patient well-being in at least some patients). Inappropriate HSD functional activity might arise as the result of HSD expression in cells which normally do not express HSD, decreased HSD expression or increased HSD expression.
- An HSD-responsive condition or disorder may include condition or disorder mediated by any HSD or isoform thereof.
- 11/3-HSDl -responsive condition or disorder refers to a condition or disorder that responds favorably to modulation of 11/3-HSDl activity.
- Favorable responses to 11 / 3-HSDl modulation include alleviation or abrogation ofthe disease and/or its attendant symptoms, inhibition ofthe disease, i.e., arrest or reduction ofthe development ofthe disease, or its clinical symptoms, and regression ofthe disease or its clinical symptoms.
- An 11/3-HSDl -responsive condition or disease maybe completely or partially responsive to 11 / 3-HSDl modulation.
- An 11 / 3-HSDl -responsive condition or disorder may be associated with inappropriate, e.g., less than or greater than normal, 11 / 3-HSDl activity and at least partially responsive to or affected by 11 / 3-HSDl modulation (e.g., a 11/3-HSDl inhibitor results in some improvement in patient well-being in at least some patients).
- Inappropriate 11 / 3-HSDl functional activity might arise as the result of 11 / 3-HSDl expression in cells which normally do not express 11 / 3-HSDl, decreased 11/3- HSDl expression or increased 11/3-HSDl expression.
- a 11 / 3-HSDl -responsive condition or disorder may include a 11 (3-HSD 1 -mediated condition or disorder.
- 1 l/3-HSD2-responsive condition or disorder refers to a condition or disorder that responds favorably to modulation of 11/3-HSD2 activity.
- Favorable responses to 11/3-HSD2 modulation include alleviation or abrogation ofthe disease and/or its attendant symptoms, inhibition ofthe disease, i.e., arrest or reduction ofthe development ofthe disease, or its clinical symptoms, and regression ofthe disease or its clinical symptoms.
- An 1 l/3-HSD2-responsive condition or disease may be completely or partially responsive to 11/3-HSD2 modulation.
- An 1 l/3-HSD2-responsive condition or disorder may be associated with inappropriate, e.g., less than or greater than normal, 11 / 3-HSD2 activity and at least partially responsive to or affected by 11/3-HSD2 modulation (e.g., a 11/3-HSD2 inhibitor results in some improvement in patient well-being in at least some patients).
- 17/3-HSD3 -responsive condition or disorder refers to a condition or disorder that responds favorably to modulation of 17/3-HSD3 activity.
- Favorable responses to 17/3-HSD3 modulation include alleviation or abrogation ofthe disease and/or its attendant symptoms, inhibition ofthe disease, i.e., arrest or reduction ofthe development ofthe disease, or its clinical symptoms, and regression ofthe disease or its clinical symptoms.
- An 17/3-HSD3-responsive condition or disease may be completely or partially responsive to 17/3-HSD3 modulation.
- An 17/3-HSD3-responsive condition or disorder may be associated with inappropriate, e.g., less than or greater than normal, 17/3-HSD3 activity and at least partially responsive to or affected by 17/3-HSD3 modulation (e.g., a 17 / 3-HSD3 inhibitor results in some improvement in patient well-being in at least some patients).
- Inappropriate 17/3-HSD3 functional activity might arise as the result of 17/3-HSD3 expression in cells which normally do not express 17/3-HSD3, decreased 17/3- HSD3 expression or increased 17/3-HSD3 expression.
- a 17/3-HSD3 -responsive condition or disorder may include a 17/3-HSD3-mediated condition or disorder.
- HSD-mediated condition or disorder refers to a condition or disorder characterized by inappropriate, e.g., less than or greater than normal, activity of a hydroxysteroid dehydrogenase (HSD).
- HSD-mediated condition or disorder may be completely or partially characterized by inappropriate HSD activity.
- an HSD-mediated condition or disorder is one in which modulation of an HSD results in some effect on the underlying condition or disease (e.g., an HSD inhibitor results in some improvement in patient well-being in at least some patients).
- 11/3-HSDl -mediated condition or disorder refers to a condition or disorder characterized by inappropriate, e.g., less than or greater than normal, 11/3-HSDl activity.
- a 11/3-HSDl -mediated condition or disorder may be completely or partially characterized by inappropriate 11/3-HSDl activity.
- a 11/3-HSDl -mediated condition or disorder is one in which modulation of 11 / 3- HSDl results in some effect on the underlying condition or disease (e.g., a 11/3-HSDl inhibitor results in some improvement in patient well-being in at least some patients).
- 1 l/3-HSD2-mediated condition or disorder refers to a condition or disorder characterized by inappropriate, e.g., less than or greater than normal, 11/3-HSD2 activity.
- a 1 l/3-HSD2-mediated condition or disorder may be completely or partially characterized by inappropriate 11/3-HSD2 activity.
- a 1 l/3-HSD2-mediated condition or disorder is one in which modulation of 11/3- HSD2 results in some effect on the underlying condition or disease (e.g., a 11/3-HSD2 inhibitor results in some improvement in patient well-being in at least some patients).
- 17/3-HSD3 -mediated condition or disorder refers to a condition or disorder characterized by inappropriate, e.g., less than or greater than normal, 17/3-HSD3 activity.
- a 17/3-HSD3-mediated condition or disorder may be completely or partially characterized by inappropriate 17/3-HSD3 activity.
- a 17/3-HSD3-mediated condition or disorder is one in which modulation of 17/3- HSD3 results in some effect on the underlying condition or disease (e.g., a 17/3-HSD3 inhibitor results in some improvement in patient well-being in at least some patients).
- ATP is adenosine triphosphate
- t-BuOH is tert-butyl alcohol
- CHO is Chinese hamster ovary
- Dess- Martin Periodinane is l,l,l,-triacetoxy-l,l-dihyro-l,2-benziodoxol-3(lH)-one
- DIBAL- ⁇ is diisobutyl aluminum hydride
- DMEM Dulbecco's Modified Eagle Medium
- DMF is N,N- dimethylformamide
- Et 3 N is triethylamine
- Et 4 NCN is tefraethylammonium cyanide
- EtOAc is ethyl acetate
- EtO ⁇ is ethanol
- LA ⁇ is lithium aluminum hydride
- LDA is lithium diisopropylamide
- LiA ⁇ O'Bu ⁇ is lithium tri-tert-butoxyaluminohydride
- MeO ⁇ is ethanol
- LA ⁇
- R 1 , R 2 and R 3 are independently selected from -H, -halo, -OH, -CN, -NO 2 , -C C 8 alkyl, -C 2 -C 8 alkenyl, -C 2 -C 8 alkynyl, -alkoxy, -haloalkyl, -hydroxyalkyl, -cycloalkyl, - heterocycloalkyl, -heteroaryl and -aryl, and at least one of R 1 , R 2 and R 3 is other than -H;
- R 4 is -H, -halo, -CN, -NO 2 , -d-C 8 alkyl, -C 2 -C 8 alkenyl, -C 2 -C 8 alkynyl, - alkoxy, - haloalkyl, -C 2 -C 8 hydroxyalkyl
- a first subclass ofthe Aryl Sulfonamide compounds of Formula (I) is that where L 1 is -C 1 -C 7 alkylene-, L 2 is a direct bond, and R 5 and R 6 , together with the carbon atom to which they are attached, join to form a cycloalkane ring.
- a second subclass ofthe Aryl Sulfonamide compounds of Formula (I) is that where L 1 is -d-C 7 alkylene-, L 2 is -C 1 -C 7 alkylene-, and R 5 and R 6 , together with the carbon atom to which they are attached, join to form a cycloalkane ring.
- a third subclass ofthe Aryl Sulfonamide compounds of Formula (I) is that where L 1 is -d-C 7 alkylene-, L 2 is a direct bond, and R 5 and R 6 , together with the carbon atom to which they are attached, join to form a cycloalkane ring, and Q is -aryl or -heteroaryl.
- a fourth subclass ofthe Aryl Sulfonamide compounds of Formula (I) is that where L 1 is -C1-C7 alkylene-, L 2 is -C 1 -C7 alkylene-, and R 5 and R 6 , together with the carbon atom to which they are attached, join to form a cycloalkane ring, and Q is -aryl or -heteroaryl.
- a fifth subclass ofthe Aryl Sulfonamide compounds of Formula (I) is that where L 1 is -d-d alkylene-, L is a direct bond, and R and R , together with the carbon atom to which they are attached, join to form a cycloalkane ring, and Q is -COOH or -C(O)NH .
- a sixth subclass ofthe Aryl Sulfonamide compounds of Formula (I) is that where 1 "7 1 f
- L is -d-d alkylene-
- L is -d-d alkylene-
- R and R together with the carbon atom to which they are attached, join to form a cycloalkane ring
- Q is -COOH or -C(O)NH 2 .
- a seventh subclass ofthe Aryl Sulfonamide compounds of Formula (I) is that where R 4 is -H , -alkyl or -C 3 -C 6 hydroxyalkyl.
- An eighth subclass ofthe Aryl Sulfonamide compounds of Formula (I) is that where R ⁇ s -OH or -d-Cs alkyl.
- a ninth subclass ofthe Aryl Sulfonamide compounds of Formula (I) is that where R 1 is -OH and R 2 and R 3 are independently -Ci-Cs alkyl, or -haloalkyl.
- a tenth subclass ofthe Aryl Sulfonamide compounds of Formula (I) is that where R 1 , R 2 and R 3 are each -d-C 8 alkyl.
- An eleventh subclass ofthe Aryl Sulfonamide compounds of Formula (I) is that where L and L are each a direct bond, R and R together form a cycloalkane ring, and Q is -H. [0121] For each ofthe subclasses above wherein R 5 and R 6 together form a cycloalkane ring, the ring is optionally substituted with from one to three members selected from the substituents described above for "alkyl".
- aryl e.g., optionally substituted phenyl
- heteroaryl e.g., optionally substituted imidazolyl, triazolyl or pyridyl
- heterocycloalkyl e.g., morpholinyl, pyrrolidinyl and piperidinyl.
- R', R" and R'" each independently refer to hydrogen, unsubstituted (d-C 8 )alkyl or (d-C 8 )alkyl substituted with hydroxy, cyano or amino, unsubstituted hetero(C 1 -C 8 )alkyl, unsubstituted aryl and aryl substituted with one to three substituents selected from -halo, unsubstituted alkyl, unsubstituted alkoxy, unsubstituted thioalkoxy and unsubstituted aryl(C ⁇ -C 4 )alkyl.
- Aryl, heteroaryl and heterocyclyl groups directly attached to the cycloalkane ring are optionally substituted.
- L 1 is -CH 2 - and L 2 is a direct bond.
- L 1 is a direct bond and L 2 is -CH 2 -.
- L 1 and L 2 are each -CH 2 -.
- L and L are each a direct bond.
- Q is -aryl or -heteroaryl, optionally substituted with up to four groups independently chosen from -d-C 8 alkyl, -halo, -C0 2 R', C(O)N(R') and -CN.
- Q is pyridyl
- Q is imidazolyl
- Q is -COOH
- Q is -C(O)NH 2 .
- Q is -H.
- R 5 and R 6 together with the carbon atom to which they are attached, join to form a cyclopropane ring.
- R 5 and R 6 together with the carbon atom to which they are attached, join to form a cyclobutane ring.
- R 5 and R 6 together with the carbon atom to which they are attached, join to form a cyclopentane ring.
- R 4 is -H.
- R 4 is -CH 3 .
- R 4 is -CH 2 CH 2 OH.
- R 1 is -OH
- R 2 is -CH 3
- R 3 is CF 3 .
- R 1 is -OH
- R 2 is -CF 3
- R 3 is CF 3 .
- the Aryl Sulfonamide Compounds of Formula (I) have the formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , L 1 , L 2 and Q are as defined above for the compounds of Formula (I).
- the Aryl Sulfonamide Compounds of Formula (I) have the formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , L 1 , L 2 and Q are as defined above for the compounds of Formula (I).
- Aryl Sulfonamide Compounds of Formula (I) have the formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , L 1 , L 2 and Q are as defined above for the compounds of Formula (I).
- Aryl Sulfonamide Compounds of Formula (I) have the formula:
- R , R , R , R , R , L , L and Q are as defined above for the compounds of Formula (I).
- the Aryl Sulfonamide Compounds of Formula (I) have the formula: wherein R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , L 1 , L 2 and Q are as defined above for the compounds of Formula (I).
- Aryl Sulfonamide Compounds of Formula (I) have the formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , L 1 , L 2 and Q are as defined above for the compounds of Formula (I).
- Aryl Sulfonamide Compounds of Formula (I) have the formula:
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , L 1 , L 2 and Q are as defined above for the compounds of Formula (I).
- the Aryl sulfonyl portion has the formula:
- the Aryl sulfonyl portion has the formula:
- the Aryl Sulfonamide Compounds of Formula (I) have a substituted piperazine ring with the following stereochemistry:
- the Aryl Sulfonamide compounds of Formula (I) comprise an aryl sulfonyl piperazine component having the formula and stereochemistry below:
- Illustrative Aryl Sulfonamide compounds of Formula (I) include the compounds listed below:
- the Aryl Sulfonamide Compounds can have asymmetric centers and therefore exist in different enantiomeric and diastereomeric forms.
- This invention relates to the use of all optical isomers and stereoisomers ofthe Aryl Sulfonamide Compounds, and mixtures thereof, and to all pharmaceutical compositions and methods of freatment that may employ or contain them.
- racemates, racemic mixtures, and stereoisomers, particularly diastereomeric mixtures or diastereomerically pure compounds and enantiomers or enantiomerically pure compounds ofthe above are all encompassed.
- compositions comprising a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I) and a pharmaceutically acceptable vehicle, carrier, diluent or excipient.
- the invention further provides Aryl Sulfonamide Compounds of Formula (I) that are in isolated and purified form.
- the invention provides methods for freating diabetes comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I). [0158] The invention also provides methods for treating obesity comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention further provides methods for freating an HSD-mediated condition or disorder comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention further provides methods for freating an 11 / 3-HSD 1 -mediated condition or disorder comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention further provides methods for freating an 11 /3-HSD2-mediated condition or disorder comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention further provides methods for freating an 17/3-HSD3 -mediated condition or disorder comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention further provides methods for freating an HSD-responsive condition or disorder comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention further provides methods for freating an 11 /3-HSD 1 -responsive condition or disorder comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention further provides methods for freating an 1 l / 3-HSD2-responsive condition or disorder comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- the invention further provides methods for treating an 17/3-HSD3 -responsive condition or disorder comprising administering to a patient in need thereof a therapeutically effective amount of an Aryl Sulfonamide Compound of Formula (I).
- Aryl Sulfonamide Compounds can be made using synthetic methods well- known to one of ordinary skill in the art of organic synthesis or by using the synthetic procedures outlined below in Schemes 1-2.
- substituted sulfonamide compounds of formula A can be alkylated using elecfrophile compounds of formula B (wherein LG is an aldehyde or a good leaving group such as a halide, mesylate, or triflate) to provide compounds of Formula (I) using methods well-known to those of skill in the relevant art.
- the substituent(s) on the sulfonamide aryl ring can be further modified using known procedures to provide the desired compounds of Formula (I). Stereochemistry in the substituent may be set by substrate control, control via an auxiliary, or control via a chiral catalyst.
- substituted phenylsulfonyl chloride compounds of formula C can be alkylated using piperazine compounds of formula D to provide compounds of Formula (I) using methods well-known to those of skill in the relevant art.
- the substituent(s) on the sulfonamide aryl ring can be further modified using known procedures to provide the desired compounds of Formula (I).
- Stereochemistry in the substituent may be set by substrate control, control via an auxiliary, or control via a chiral catalyst.
- the Aryl Sulfonamide Compounds of Formula (I) can have one or more asymmetric centers and therefore exist in different enantiomeric and diastereomeric forms.
- An Aryl Sulfonamide Compound can be in the form of an optical isomer, an enantiomer, a racemate, or a diastereomer. Accordingly, the invention encompasses Aryl Sulfonamide Compounds and their uses as described herein in the form of their optical isomers, racemates, diastereomers, enantiomers, and mixtures thereof, including a racemic mixture.
- the synthetic routes provided above can be modified to use different starting materials and alternate reagents to accomplish the desired transformations.
- the compounds ofthe invention maybe synthesized via bond forming reactions which disconnect any torsional bond present in the compound.
- Particularly facile synthesis of compounds ofthe invention occurs when the synthesis proceeds via the connection of fragments at the disconnection points a, b, c and d, as shown below for an Aryl Sulfonamide Compound of Formula (I):
- fragments may be assembled in any order to synthesize compounds ofthe invention.
- compositions and single unit dosage forms comprising an Aryl Sulfonamide Compound, or a pharmaceutically acceptable stereoisomer, prodrug, salt, solvate, hydrate, or clathrate thereof, are also encompassed by the invention.
- Individual dosage forms ofthe invention may be suitable for oral, mucosal (including sub lingual, buccal, rectal, nasal, or vaginal), parenteral (including subcutaneous, intramuscular, bolus injection, intraarterial, or intravenous), fransdermal, or topical administration.
- Single unit dosage forms ofthe invention are suitable for oral, mucosal (e.g., nasal, sublingual, vaginal, buccal, or rectal), parenteral (e.g., subcutaneous, intravenous, bolus injection, intramuscular, or intraarterial), or fransdermal adminisfration to a patient.
- mucosal e.g., nasal, sublingual, vaginal, buccal, or rectal
- parenteral e.g., subcutaneous, intravenous, bolus injection, intramuscular, or intraarterial
- fransdermal adminisfration e.g., fransdermal adminisfration
- dosage forms include, but are not limited to: tablets; caplets; capsules, such as soft elastic gelatin capsules; cachets; troches; lozenges; dispersions; suppositories; ointments; cataplasms (poultices); pastes; powders; dressings; creams; plasters; solutions; patches; aerosols (e.g., nasal sprays or inhalers); gels; liquid dosage forms suitable for oral or mucosal adminisfration to a patient, including suspensions (e.g., aqueous or non-aqueous liquid suspensions, oil-in-water emulsions, or a water-in-oil liquid emulsions), solutions, and elixirs; liquid dosage forms suitable for parenteral adminisfration to a patient; and sterile solids (e.g., crystalline or amorphous solids) that can be reconstituted to provide liquid dosage forms suitable for parenteral adminisfration to a patient.
- suspensions
- composition, shape, and type of dosage forms ofthe invention will typically vary depending on their use.
- a dosage form used in the acute freatment of diabetes or a related disease may contain larger amounts of one or more ofthe active ingredients it comprises than a dosage form used in the chronic treatment ofthe same disease.
- a parenteral dosage form may contain smaller amounts of one or more ofthe active ingredients it comprises than an oral dosage form used to treat the same disease or disorder.
- Typical pharmaceutical compositions and dosage forms comprise one or more carriers, excipients or diluents.
- Suitable excipients are well known to those skilled in the art of pharmacy, and non-limiting examples of suitable excipients are provided herein. Whether a particular excipient is suitable for incorporation into a pharmaceutical composition or dosage form depends on a variety of factors well known in the art including, but not limited to, the way in which the dosage form will be administered to a patient.
- oral dosage forms such as tablets may contain excipients not suited for use in parenteral dosage forms. The suitability of a particular excipient may also depend on the specific active ingredients in the dosage form.
- This invention further encompasses anhydrous (e.g., ⁇ 1% water) pharmaceutical compositions and dosage forms comprising active ingredients, since water can facilitate the degradation of some compounds.
- water e.g., 5%
- water is widely accepted in the pharmaceutical arts as a means of simulating long-term storage in order to determine characteristics such as shelf-life or the stability of formulations over time. See, e.g., Jens T. Carstensen, Drug Stability: Principles & Practice, 2d. Ed., Marcel Dekker, NY, NY, 1995, pp. 379-80.
- water and heat accelerate the decomposition of some compounds.
- the effect of water on a formulation can be of great significance since moisture and/or humidity are commonly encountered during manufacture, handling, packaging, storage, shipment, and use of formulations.
- Anhydrous pharmaceutical compositions and dosage forms ofthe invention can be prepared using anhydrous or low moisture containing ingredients and low moisture or low humidity conditions.
- Pharmaceutical compositions and dosage forms that comprise lactose and at least one active ingredient that comprises a primary or secondary amine are preferably anhydrous if substantial contact with moisture and/or humidity during manufacturing, packaging, and/or storage is expected.
- anhydrous pharmaceutical composition should be prepared and stored such that its anhydrous nature is maintained. Accordingly, anhydrous compositions are preferably packaged using materials known to prevent exposure to water such that they can be included in suitable formulary kits. Examples of suitable packaging include, but are not limited to, hermetically sealed foils, plastics, unit dose containers (e.g., vials), blister packs, and strip packs.
- compositions and dosage forms that comprise one or more compounds that reduce the rate by which an active ingredient will decompose.
- compounds which are referred to herein as “stabilizers,” include, but are not limited to, antioxidants such as ascorbic acid, pH buffers, or salt buffers.
- the Aryl Sulfonamide Compound can be administered to a mammal (human, mouse, rat, rabbit, dog, cat, bovine, pig, monkey etc.) as an 11/3-HSDl modulator, a prophylactic or therapeutic drug of diabetes, a prophylactic or therapeutic drug of diabetic complication (retinopathy, nephropathy, neuropathy, cardiac infarction and cerebral infarction based on arteriosclerosis etc.), a prophylactic or therapeutic drug of hyperlipemia, a prophylactic or therapeutic drug of obesity, neurodegenerative disease and the like, or a prophylactic or therapeutic drug of diseases mediated by 11/3-HSDl.
- a mammal human, mouse, rat, rabbit, dog, cat, bovine, pig, monkey etc.
- an 11/3-HSDl modulator a prophylactic or therapeutic drug of diabetes
- a prophylactic or therapeutic drug of diabetic complication retinopathy, nephropathy, neuropathy, cardiac infarction and cerebral infarction
- the Aryl Sulfonamide Compound can be administered to a mammal concurrently with an additional therapeutic agent for the treatment of a disease, such as diabetes or obesity, with the aim ofthe prophylaxis or freatment of a disease.
- a disease such as diabetes or obesity
- the Aryl Sulfonamide Compounds ofthe present invention can be administered in combination with other therapeutic agents for the freatment or prevention of numerous diseases, including, but not limited to, diabetes and obesity.
- the compounds of the invention may be administered by oral, parenteral (e.g., inframuscular, intraperitoneal, intravenous, ICV, infracisternal injection or infusion, subcutaneous injection or implant), inhalation, nasal, vaginal, rectal, sublingual, or topical (e.g., fransdermal, local) routes of administration and may be formulated, alone or together, in suitable dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for each route of adminisfration.
- the invention also contemplates adminisfration ofthe compounds ofthe invention in a depot formulation, in which the active ingredient is released over a defined time period.
- the Aryl Sulfonamide Compound may be administered simultaneously with other another therapeutic agent that is useful for the freatment or prevention of diabetes, obesity or other disease or may be administered at a time prior to or subsequent to another therapeutic agent.
- a pharmaceutical composition containing the Aryl Sulfonamide Compound and an additional therapeutic agent can be administered.
- a pharmaceutical composition containing the Aryl Sulfonamide Compound and a pharmaceutical composition containing an additional therapeutic agent may be administered separately.
- the adminisfration routes of respective pharmaceutical compositions may be the same or different.
- the Aryl Sulfonamide Compound may be administered at a dose of 50 mg to 800 mg per adminisfration, which is given once to several times a day. In addition, the compound may be administered at a smaller dose.
- the combined pharmaceutical agent can be administered at a dose generally employed for the prophylaxis or freatment of diabetes or obesity or at a smaller dose than that.
- the amounts and specific types of active ingredients in a dosage form may differ depending on factors such as, but not limited to, the route by which it is to be administered to patients.
- typical dosage forms ofthe invention comprise an Aryl Sulfonamide Compound, or a pharmaceutically acceptable salt, solvate, clathrate, hydrate, polymorph or prodrug thereof.
- an appropriate dosage level will generally be from about 0.001 to about 100 mg per kg patient body weight per day which can be administered in single or multiple doses.
- the dosage level will be from about 0.01 to about 25 mg/kg per day; more preferably from about 0.05 to about 10 mg/kg per day.
- a suitable dosage level may be from about 0.01 to about 25 mg/kg per day, about 0.05 to about 10 mg/kg per day, or about 0.1 to about 5 mg/kg per day. Within this range the dosage may be from about 0.005 to about 0.05, about 0.05 to about 0.5 or about 0.5 to about 5.0 mg/kg per day.
- the dosage levels lie within the range of from about 0.1 mg to about 2000 mg per day, given as a single once-a-day dose in the morning but preferably as divided doses throughout the day taken with food. More preferably, the daily dose is administered twice daily in equally divided doses.
- a daily dose range should be from about 5 mg to about 500 mg per day, more preferably, between about 10 mg and about 200 mg per day. In managing the patient, the therapy should be initiated at a lower dose, perhaps from about 1 mg to about 25 mg, and increased if necessary up to from about 200 mg to about 2000 mg per day as either a single dose or divided doses, depending on the patient's global response.
- the weight ratio ofthe compound ofthe invention to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound ofthe invention is combined with an NSAID, the weight ratio ofthe compound of the invention to the NSATD will generally range from about 1000:1 to about 1:1000, preferably about 200:1 to about 1:200. Combinations of a compound ofthe invention and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used.
- compositions ofthe invention that are suitable for oral adminisfration can be presented as discrete dosage forms, including, but are not limited to, tablets (e.g., chewable tablets), caplets, capsules, and liquids (e.g., flavored syrups).
- dosage forms contain predetermined amounts of active ingredients, and may be prepared by methods of pharmacy well known to those skilled in the art. See generally, Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing, Easton PA (1990).
- Typical oral dosage forms ofthe invention are prepared by combining the active ingredient(s) in an intimate admixture with at least one excipient according to conventional pharmaceutical compounding techniques.
- Excipients can take a wide variety of forms depending on the form of preparation desired for admimsfration.
- excipients suitable for use in oral liquid or aerosol dosage forms include, but are not limited to, water, glycols, oils, alcohols, flavoring agents, preservatives, and coloring agents.
- excipients suitable for use in solid oral dosage forms include, but are not limited to, starches, sugars, micro-crystalline cellulose, diluents, granulating agents, lubricants, binders, and disintegrating agents.
- tablets and capsules represent the most advantageous oral dosage unit forms, in which case solid excipients are employed. If desired, tablets can be coated by standard aqueous or nonaqueous techniques. Such dosage forms can be prepared by any ofthe methods of pharmacy. In general, pharmaceutical compositions and dosage forms are prepared by uniformly and intimately admixing the active ingredients with liquid carriers, finely divided solid carriers, or both, and then shaping the product into the desired presentation if necessary.
- a tablet can be prepared by compression or molding.
- Compressed tablets can be prepared by compressing in a suitable machine the active ingredients in a free- flowing form such as powder or granules, optionally mixed with an excipient.
- Molded tablets can be made by molding in a suitable machine a mixture ofthe powdered compound moistened with an inert liquid diluent.
- excipients that can be used in oral dosage forms of the invention include, but are not limited to, binders, fillers, disintegrants, and lubricants.
- Binders suitable for use in pharmaceutical compositions and dosage forms include, but are not limited to, corn starch, potato starch, or other starches, gelatin, natural and synthetic gums such as acacia, sodium alginate, alginic acid, other alginates, powdered tragacanth, guar gum, cellulose and its derivatives (e.g., ethyl cellulose, cellulose acetate, carboxymethyl cellulose calcium, sodium carboxymethyl cellulose), polyvinyl pyrrolidone, methyl cellulose, pre-gelatinized starch, hydroxypropyl methyl cellulose, (e.g., Nos. 2208, 2906, 2910), microcrystalline cellulose, and mixtures thereof.
- fillers suitable for use in the pharmaceutical compositions and dosage forms disclosed herein include, but are not limited to, talc, calcium carbonate (e.g., granules or powder), microcrystalline cellulose, powdered cellulose, dexfrates, kaolin, mannitol, silicic acid, sorbitol, starch, pre-gelatinized starch, and mixtures thereof.
- the binder or filler in pharmaceutical compositions ofthe invention is typically present in from about 50 to about 99 weight percent ofthe pharmaceutical composition or dosage form.
- Suitable forms of microcrystalline cellulose include, but are not limited to, the materials sold as AVICEL-PH-101, AVICEL-PH-103 AVICEL RC-581, AVICEL-PH-105 (available from FMC Corporation, American Viscose Division, Avicel Sales, Marcus Hook, PA), and mixtures thereof.
- An specific binder is a mixture of microcrystalline cellulose and sodium carboxymethyl cellulose sold as AVICEL RC-581.
- Suitable anhydrous or low moisture excipients or additives include AVICEL-PH-103TM and Starch 1500 LM.
- Disintegrants are used in the compositions ofthe invention to provide tablets that disintegrate when exposed to an aqueous environment. Tablets that contain too much disintegrant may disintegrate in storage, while those that contain too little may not disintegrate at a desired rate or under the desired conditions. Thus, a sufficient amount of disintegrant that is neither too much nor too little to detrimentally alter the release ofthe active ingredients should be used to form solid oral dosage forms ofthe invention.
- the amount of disintegrant used varies based upon the type of formulation, and is readily discernible to those of ordinary skill in the art. Typical pharmaceutical compositions comprise from about 0.5 to about 15 weight percent of disintegrant, specifically from about 1 to about 5 weight percent of disintegrant.
- Disintegrants that can be used in pharmaceutical compositions and dosage forms of the invention include, but are not limited to, agar-agar, alginic acid, calcium carbonate, microcrystalline cellulose, croscarmellose sodium, crospovidone, polacrilin potassium, sodium starch glycolate, potato or tapioca starch, pre-gelatinized starch, other starches, clays, other algins, other celluloses, gums, and mixtures thereof.
- Lubricants that can be used in pharmaceutical compositions and dosage forms ofthe invention include, but are not limited to, calcium stearate, magnesium stearate, mineral oil, light mineral oil, glycerin, sorbitol, mannitol, polyethylene glycol, other glycols, stearic acid, sodium lauryl sulfate, talc, hydrogenated vegetable oil (e.g., peanut oil, cottonseed oil, sunflower oil, sesame oil, olive oil, corn oil, and soybean oil), zinc stearate, ethyl oleate, ethyl laureate, agar, and mixtures thereof.
- calcium stearate e.g., magnesium stearate, mineral oil, light mineral oil, glycerin, sorbitol, mannitol, polyethylene glycol, other glycols, stearic acid, sodium lauryl sulfate, talc
- hydrogenated vegetable oil e.g., peanut oil, cottonseed oil
- Additional lubricants include, for example, a syloid silica gel (AEROSIL 200, manufactured by W.R. Grace Co. of Baltimore, MD), a coagulated aerosol of synthetic silica (marketed by Degussa Co. of Piano, TX), CAB-0-SIL (a pyrogenic silicon dioxide product sold by Cabot Co. of Boston, MA), and mixtures thereof. If used at all, lubricants are typically used in an amount of less than about 1 weight percent ofthe pharmaceutical compositions or dosage forms into which they are incorporated.
- AEROSIL 200 manufactured by W.R. Grace Co. of Baltimore, MD
- a coagulated aerosol of synthetic silica marketed by Degussa Co. of Piano, TX
- CAB-0-SIL a pyrogenic silicon dioxide product sold by Cabot Co. of Boston, MA
- lubricants are typically used in an amount of less than about 1 weight percent ofthe pharmaceutical compositions or dosage forms into which they are incorporated.
- the compositions are preferably provided in the form of tablets containing about 1 to about 1000 milligrams ofthe active ingredient, hi other embodiments, the compositions are provided in provided in the form of tablets containing about 1.0, about 5.0, about 10.0, about 15.0. about 20.0, about 25.0, about 50.0, about 75.0, about 100.0, about 150.0, about 200.0, about 250.0, about 300.0, about 400.0, about 500.0, about 600.0, about 750.0, about 800.0, about 900.0, or about 1000.0 milligrams ofthe active ingredient for the symptomatic adjustment ofthe dosage to the patient to be treated.
- the compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
- Active ingredients ofthe invention can be administered by controlled release means or by delivery devices that are well known to those of ordinary skill in the art. Examples include, but are not limited to, those described in U.S. Patent Nos.: 3,845,770; 3,916,899; 3,536,809; 3,598,123; and 4,008,719, 5,674,533, 5,059,595, 5,591,767, 5,120,548, 5,073,543, 5,639,476, 5,354,556, and 5,733,566, each of which is incorporated herein by reference.
- Such dosage forms can be used to provide slow or confrolled-release of one or more active ingredients using, for example, hydropropylmethyl cellulose, other polymer matrices, gels, permeable membranes, osmotic systems, multilayer coatings, microp articles, liposomes, microspheres, or a combination thereof to provide the desired release profile in varying proportions.
- Suitable confrolled-release formulations known to those of ordinary skill in the art, including those described herein, can be readily selected for use with the active ingredients ofthe invention.
- the invention thus encompasses single unit dosage forms suitable for oral administration such as, but not limited to, tablets, capsules, gelcaps, and caplets that are adapted for confrolled-release.
- Confrolled-release pharmaceutical products can improve drug therapy over that achieved by their non-controlled counterparts.
- the use of an optimally designed confrolled-release preparation in medical treatment is characterized by a minimum of drug substance being employed to cure or control the condition in a minimum amount of time.
- confrolled-release formulations include extended activity ofthe drug, reduced dosage frequency, and increased patient compliance, hi addition, confrolled-release formulations can be used to affect the time of onset of action or other characteristics, such as blood levels ofthe drug, and can thus affect the occurrence of side (e.g., adverse) effects.
- confrolled-release formulations are designed to initially release an amount of drug (active ingredient) that promptly produces the desired therapeutic effect, and gradually and continually release of other amounts of drug to maintain this level of therapeutic or prophylactic effect over an extended period of time.
- drug active ingredient
- Confrolled-release of an active ingredient can be stimulated by various conditions including, but not limited to, pH, temperature, enzymes, water, or other physiological conditions or compounds.
- Parenteral dosage forms can be administered to patients by various routes including, but not limited to, subcutaneous, intravenous (including bolus injection), inframuscular, and infra-arterial. Because their adminisfration typically bypasses patients' natural defenses against contaminants, parenteral dosage forms are preferably sterile or capable of being sterilized prior to administration to a patient. Examples of parenteral dosage forms include, but are not limited to, solutions ready for injection, dry products ready to be dissolved or suspended in a pharmaceutically acceptable vehicle for injection, suspensions ready for injection, and emulsions. For example, lyophilized sterile compositions suitable for reconstitution into particulate-free dosage forms suitable for adminisfration to humans.
- Suitable vehicles that can be used to provide parenteral dosage forms ofthe invention are well known to those skilled in the art. Examples include, but are not limited to: Water for Injection USP; aqueous vehicles such as, but not limited to, Sodium Chloride Injection, Ringer's Injection, Dexfrose Injection, Dexfrose and Sodium Chloride Injection, and Lactated Ringer's Injection; water-miscible vehicles such as, but not limited to, ethyl alcohol, polyethylene glycol, and polypropylene glycol; and non-aqueous vehicles such as, but not limited to, corn oil, cottonseed oil, peanut oil, sesame oil, ethyl oleate, isopropyl myristate, and benzyl benzoate.
- water for Injection USP Water for Injection USP
- aqueous vehicles such as, but not limited to, Sodium Chloride Injection, Ringer's Injection, Dexfrose Injection, Dexfrose and
- Parenteral dosage forms are preferred for the methods of preventing, treating or managing disease in a cancer patient.
- Transdermal and topical dosage forms ofthe invention include, but are not limited to, creams, lotions, ointments, gels, solutions, emulsions, suspensions, or other forms known to one of skill in the art. See, e.g., Remington's Pharmaceutical Sciences, 18th eds., Mack Publishing, Easton PA (1990); and Introduction to Pharmaceutical Dosage Forms, 4th ed., Lea & Febiger, Philadelphia (1985).
- Transdermal dosage forms include "reservoir type” or "matrix type” patches, which can be applied to the skin and worn for a specific period of time to permit the penetration of a desired amount of active ingredients.
- Suitable excipients e.g., carriers and diluents
- other materials that can be used to provide transdermal and topical dosage forms encompassed by this invention are well known to those skilled in the pharmaceutical arts, and depend on the particular tissue to which a given pharmaceutical composition or dosage form will be applied.
- excipients include, but are not limited to, water, acetone, ethanol, ethylene glycol, propylene glycol, butane- 1, 3 -diol, isopropyl myristate, isopropyl palmitate, mineral oil, and mixtures thereof to form lotions, tinctures, creams, emulsions, gels or ointments, which are non-toxic and pharmaceutically acceptable.
- Moisturizers or humectants can also be added to pharmaceutical compositions and dosage forms if desired. Examples of such additional ingredients are well known in the art. See, e.g., Remington's Pharmaceutical Sciences, 18th eds., Mack Publishing, Easton PA (1990).
- penetration enhancers can be used to assist in delivering the active ingredients to the tissue.
- Suitable penetration enhancers include, but are not limited to: acetone; various alcohols such as ethanol, oleyl, and tefrahydrofuryl; alkyl sulfoxides such as dimethyl sulfoxide; dimethyl acetamide; dimethyl formamide; polyethylene glycol; pyrrolidones such as polyvinylpyrrolidone; Kollidon grades (Povidone, Polyvidone); urea; and various water-soluble or insoluble sugar esters such as Tween 80 (polysorbate 80) and Span 60 (sorbitan monostearate).
- the pH of a pharmaceutical composition or dosage form, or ofthe tissue to which the pharmaceutical composition or dosage form is applied may also be adjusted to improve delivery of one or more active ingredients.
- the polarity of a solvent carrier, its ionic strength, or tonicity can be adjusted to improve delivery.
- Compounds such as stearates can also be added to pharmaceutical compositions or dosage forms to advantageously alter the hydrophilicity or lipophilicity of one or more active ingredients so as to improve delivery.
- stearates can serve as a lipid vehicle for the formulation, as an emulsifying agent or surfactant, and as a delivery-enhancing or penetration-enhancing agent.
- Different salts, hydrates or solvates ofthe active ingredients can be used to further adjust the properties ofthe resulting composition.
- Mucosal dosage forms ofthe invention include, but are not limited to, ophthalmic solutions, sprays and aerosols, or other forms known to one of skill in the art. See, e.g., Remington's Pharmaceutical Sciences, 18th eds., Mack Publishing, Easton PA (1990); and Introduction to Pharmaceutical Dosage Forms, 4th ed., Lea & Febiger, Philadelphia (1985). Dosage forms suitable for treating mucosal tissues within the oral cavity can be formulated as mouthwashes or as oral gels.
- the aerosol comprises a carrier. In another embodiment, the aerosol is carrier free.
- a compound ofthe invention can also be administered directly to the lung by inhalation (see e.g., Tong et al., PCT Application, WO 97/39745; Clark et al, PCT Application, WO 99/47196, which are herein incorporated by reference).
- inhalation see e.g., Tong et al., PCT Application, WO 97/39745; Clark et al, PCT Application, WO 99/47196, which are herein incorporated by reference.
- an Aryl Sulfonamide Compound can be conveniently delivered to the lung by a number of different devices.
- a Metered Dose Inhaler which utilizes canisters that contain a suitable low boiling propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas can be used to deliver an Aryl Sulfonamide Compound directly to the lung.
- MDI devices are available from a number of suppliers such as 3M Corporation, Aventis, Boehringer Ingleheim, Forest Laboratories, Glaxo- Wellcome, Schering Plough and Vectura.
- a Dry Powder Inhaler (DPI) device can be used to administer an Aryl Sulfonamide Compound to the lung (See, e.g., Raleigh et al, Proc. Amer. Assoc. Cancer Research Annual Meeting, 1999, 40, 397, which is herein incorporated by reference).
- DPI devices typically use a mechanism such as a burst of gas to create a cloud of dry powder inside a container, which can then be inhaled by the patient.
- DPI devices are also well known in the art and can be purchased from a number of vendors which include, for example, Fisons, Glaxo- Wellcome, Inhale Therapeutic Systems, ML Laboratories, Qdose and Vectura.
- MDDPI multiple dose DPI
- a popular variation is the multiple dose DPI (“MDDPI”) system, which allows for the delivery of more than one therapeutic dose.
- MDDPI devices are available from companies such as AsfraZeneca, Glaxo Wellcome, IV AX, Schering Plough, SkyePharma and Vectura.
- capsules and cartridges of gelatin for use in an inhaler or insufflator can be formulated containing a powder mix ofthe compound and a suitable powder base such as lactose or starch for these systems.
- Aryl Sulfonamide Compound Another type of device that can be used to deliver an Aryl Sulfonamide Compound to the lung is a liquid spray device supplied, for example, by Aradigm Corporation.
- Liquid spray systems use extremely small nozzle holes to aerosolize liquid drug formulations that can then be directly inhaled into the lung.
- a nebulizer device is used to deliver an Aryl Sulfonamide Compound to the lung.
- Nebulizers create aerosols from liquid drug formulations by using, for example, ultrasonic energy to form fine particles that can be readily inhaled (See e.g., Verschoyle et al, British J Cancer, 1999, 80, Suppl 2, 96, which is herein incorporated by reference).
- Examples of nebulizers include devices supplied by Sheffield/Systemic Pulmonary Delivery Ltd. (See, Armer et al., U.S. Pat. No. 5,954,047; van der Linden et al., U.S. Pat. No.
- Inhaled compound ofthe invention delivered by nebulizer devices, is currently under investigation as a freatment for aerodigestive cancer (Engelke et al., Poster 342 at American Association of Cancer Research, San Francisco, Calif, Apr. 1-5, 2000) and lung cancer (Dahl et al., Poster 524 at American Association of Cancer Research, San Francisco, Calif., April 1- 5, 2000).
- an electrohydrodynamic (“EHD”) aerosol device is used to deliver an Aryl Sulfonamide Compound to the lung.
- EHD aerosol devices use electrical energy to aerosolize liquid drug solutions or suspensions (see e.g., Noakes et al., U.S. Pat. No. 4,765,539; Coffee, U.S. Pat. No., 4,962,885; Coffee, PCT Application, WO 94/12285; Coffee, PCT Application, WO 94/14543; Coffee, PCT Application, WO 95/26234, Coffee, PCT Application, WO 95/26235, Coffee, PCT Application, WO 95/32807, which are herein incorporated by reference).
- the electrochemical properties ofthe compound ofthe invention formulation may be important parameters to optimize when delivering this drug to the lung with an EHD aerosol device and such optimization is routinely performed by one of skill in the art.
- EHD aerosol devices may more efficiently delivery drugs to the lung than existing pulmonary delivery technologies.
- Other methods of infra-pulmonary delivery of an Aryl Sulfonamide Compound will be known to the skilled artisan and are within the scope of the invention.
- Liquid drug formulations suitable for use with nebulizers and liquid spray devices and EHD aerosol devices will typically include an Aryl Sulfonamide Compound with a pharmaceutically acceptable carrier.
- the pharmaceutically acceptable carrier is a liquid such as alcohol, water, polyethylene glycol or a perfluorocarbon.
- another material may be added to alter the aerosol properties ofthe solution or suspension of an Aryl Sulfonamide Compound.
- this material is liquid such as an alcohol, glycol, polyglycol or a fatty acid.
- Other methods of formulating liquid drug solutions or suspension suitable for use in aerosol devices are known to those of skill in the art (See, e.g., Biesalski, U.S. Pat. Nos.
- a compound ofthe invention can also be formulated in rectal or vaginal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter or other glycerides.
- an Aryl Sulfonamide Compound can also be formulated as a depot preparation.
- Such long acting formulations can be administered by implantation (for example subcutaneously or intramuscularly) or by inframuscular injection.
- the compounds can be formulated with suitable polymeric or hydrophobic materials (for example, as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- Liposomes and emulsions are well known examples of delivery vehicles that can be used to deliver an Aryl Sulfonamide Compound.
- Certain organic solvents such as dimethylsulfoxide can also be employed, although usually at the cost of greater toxicity.
- a compound ofthe invention can also be delivered in a confrolled release system.
- a pump can be used (Sefton, CRC Grit. Ref Biomed Eng., 1987, 14, 201; Buchwald et al., Surgery, 1980, 88, 507; Saudek et al., N. Engl. J Med, 1989, 321, 574).
- polymeric materials can be used (see Medical Applications of Confrolled Release, Langer and Wise (eds.), CRC Pres., Boca Raton, Fla. (1974); Confrolled Drug Bioavailability, Drug Product Design and Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger and Peppas, J Macromol. Sci. Rev. Macromol. Chem., 1983, 23, 61; see also Levy et al., Science 1985, 228, 190; During et al., Ann. Neurol., 1989,25,351; Howard et al., 1989, J. Neurosurg. 71, 105).
- a confrolled-release system can be placed in proximity of the target ofthe compounds ofthe invention, e.g., the lung, thus requiring only a fraction of the systemic dose (see, e.g., Goodson, in Medical Applications of Confrolled Release, supra, vol. 2, pp. 115 (1984)).
- Other confrolled-release system can be used (see e.g., Langer, Science, 1990, 249, 1527).
- Suitable excipients e.g., carriers and diluents
- excipients include, but are not limited to, water, ethanol, ethylene glycol, propylene glycol, butane- 1, 3 -diol, isopropyl myristate, isopropyl palmitate, mineral oil, and mixtures thereof, which are non-toxic and pharmaceutically acceptable.
- additional ingredients are well known in the art. See, e.g., Remington's Pharmaceutical Sciences, 18th eds., Mack Publishing, Easton PA (1990).
- the pH of a pharmaceutical composition or dosage form, or of the tissue to which the pharmaceutical composition or dosage form is applied can also be adjusted to improve delivery of one or more active ingredients.
- the polarity of a solvent carrier, its ionic strength, or tonicity can be adjusted to improve delivery.
- Compounds such as stearates can also be added to pharmaceutical compositions or dosage forms to advantageously alter the hydrophihcity or lipophilicity of one or more active ingredients so as to improve delivery.
- stearates can serve as a lipid vehicle for the formulation, as an emulsifying agent or surfactant, and as a delivery-enhancing or penetration-enhancing agent.
- Different salts, hydrates or solvates ofthe active ingredients can be used to further adjust the properties ofthe resulting composition.
- the invention provides methods of freating or preventing a condition or disorder associated with the modulation of hydroxysteroid dehydrogenases by administering to a patient having such a condition or disorder a therapeutically effective amount of a compound or composition of the invention.
- conditions and disorders including chronic diseases of humans or other species, can be freated with modulators, stimulators, or inhibitors of hydroxysteroid dehydrogenases, such as 11/3-HSDl. TREATMENT OR PREVENTION OF DIABETES
- Diabetes and diabetic conditions can be freated or prevented by adminisfration of a therapeutically effective amount of an Aryl Sulfonamide Compound.
- Types of diabetes that can be freated or prevented by administering a therapeutically effective amount of an Aryl Sulfonamide Compound include type I diabetes mellitus (juvenile onset diabetes, insulin dependent-diabetes mellitus or IDDM), type II diabetes mellitus (non-insulin-dependent diabetes mellitus or NIDDM), insulinopathies, diabetes associated with pancreatic disorders, diabetes associated with other disorders (such as Cushing's Syndrome, acromegaly, pheochromocytoma, glucagonoma, primary aldosteronism, and somatostatinoma), type A and type B insulin resistance syndromes, lipafrophic diabetes, and diabetes induced by ⁇ -cell toxins.
- IDDM insulin dependent-diabetes mellitus
- NIDDM non-insulin-dependent diabetes mellitus
- insulinopathies diabetes associated with pancreatic disorders, diabetes associated with other disorders (such as Cushing's Syndrome, acromegaly, phe
- the type of diabetes being freated is type II diabetes.
- Obesity can be freated or prevented by adminisfration of a therapeutically effective amount of an Aryl Sulfonamide Compound.
- Obesity may have genetic, environmental (e.g., expending less energy than is consumed) and regulatory determinants.
- Obesity includes exogenous, hyperinsulinar, hyperplasmic, hypothyroid, hypothalamic, symptomatic, infantile, upper body, alimentary, hypogonadal, simple and central obesity, hypophyseal adiposity and hyperphagia.
- Metabolic disorders such as hyperlidemia and diabetes, and cardiovascular disorders, such as hypertension and coronary artery disease, are commonly associated with obesity.
- Complications due to obesity may also be freated or prevented by administering a therapeutically effective amount of an Aryl Sulfonamide Compound.
- Such complications include, but are not limited to, sleep apnea, Pickwickian syndrome, orthopedic disturbances of weight-bearing and non-weight-bearing joints, and skin disorders resulting from increased sweat or skin secretions.
- Conditions that can be treated or prevented by administering a therapeutically effective amount of an Aryl Sulfonamide Compound include, but are not limited to any condition which is responsive to the modulation, preferably inhibition, of hydroxysteroid dehydrogenases or specific isoforms thereof, and thereby benefit from adminisfration of such a modulator.
- Representative conditions in this regard include, but are not limited to, These conditions and disorders include, but are not limited to, metabolic disorders and related cardiovascular risk factors such as syndrome X, polycystic ovarian disease, eating disorders (e.g., anorexia and bulimia), craniopharyngioma, Prader-Willi syndrome, Frohlich's syndrome, hyperlipidemia, dyslipidemia, hypercholesterolemia, hypertriglyceridemia, low HDL levels, high HDL levels, hyperglycemia, insulin resistance, hyperinsulinemia and Cushing's syndrome; diseases associated therewith such as hypertension, atherosclerosis, vascular restenosis, retinopathy and nephropathy; neurologic disorders such as neurodegenerative disease, neuropathy and muscle wasting; cognitive disorders, such as age- related learning disorders; androgen and/or estrogen-related disorders such as prostate cancer, colon cancer, breast cancer, benign prostatic hyperplasia, ovarian cancer, uterine cancer, and male pseudohermaphrodism; endo
- the present methods for treating or preventing further comprise the adminisfration of a therapeutically effective amoimt of another therapeutic agent useful for freating or preventing the diseases or disorders disclosed herein.
- the time in which the therapeutic effect ofthe other therapeutic agent is exerted overlaps with the time in which the therapeutic effect ofthe Aryl Sulfonamide Compound is exerted.
- the compounds ofthe invention can be combined or used in combination with other agents useful in the treatment, prevention, suppression or amelioration ofthe conditions or disorders for which compounds ofthe invention are useful, including diabetes, obesity, glaucoma, osteoporosis, cognitive disorders, immune disorders, depression and those pathologies noted above.
- Such other agents, or drugs may be administered, by a route and in an amount commonly used therefor, simultaneously or sequentially with an Aryl Sulfonamide
- compositions of the invention include those that also contain one or more other active ingredients or therapeutic agents, in addition to an Aryl Sulfonamide Compound.
- an Aryl Sulfonamide Compound can be administered with another therapeutic agent, including, but not limited to, anti-diabetic agents such as insulin, inhaled insulin (Exubera®), insulin mimetics, insulin secretogues, sulfonylureas (e.g., glyburide, meglinatide, glimepiride, gliclazide, glipizide, gliquidone, chloropropresponsivemide, tolbutamide, acetohexamide, glycopyramide, carbutamide, glibonuride, glisoxepid, glybuthiazole, glibuzole, glyhexamide, glymidine, glypinamide, phenbutamide, tolcylamide and tolazamide), biguanides (e.g., metformin (Glucophage ® )), -glucosidase inhibitors (e.g., a), metformin (Glucophage ® )),
- an Aryl for the treatment or prevention of obesity, an Aryl
- Sulfonamide Compound can be administered with another therapeutic agent, including, but not limited to, /33 adrenergic receptor agonists, leptin or derivatives thereof, neuropeptide Y (e.g., NPY5) antagonists, and mazindol.
- another therapeutic agent including, but not limited to, /33 adrenergic receptor agonists, leptin or derivatives thereof, neuropeptide Y (e.g., NPY5) antagonists, and mazindol.
- Examples of other therapeutic agents that may be combined with an Aryl Sulfonamide Compound, either administered separately or in the same pharmaceutical compositions, include, but are not limited to: (i) cholesterol lowering agents such as HMG- CoA reductase inhibitors (e.g., lovastatin, simvastatin (Zocor®), pravastatin, fluvastatin, atorvastatin (Lipitor®) and other statins), bile acid sequesvers (e.g., cholestyramine and colestipol), vitamin B 3 (also known as nicotinic acid, or niacin), vitamin B 6 (pyridoxine), vitamin B 12 (cyanocobalamin), fibric acid derivatives (e.g., gemfibrozil, clofibrate, fenofibrate and benzafibrate), probucol, nitroglycerin, and inhibitors of cholesterol absorption (e.g., beta-sitosterol and
- the weight ratio ofthe compound ofthe invention to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when an Aryl Sulfonamide
- the weight ratio ofthe compound ofthe invention to the NSAID will generally range from about 1000:1 to about 1:1000, preferably about 200:1 to about 1:200.
- Combinations of an Aryl Sulfonamide Compound and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used.
- the invention encompasses kits that can simplify the adminisfration of the Aryl Sulfonamide Compounds or composition ofthe invention to a patient.
- a typical kit of the invention comprises a unit dosage of an Aryl Sulfonamide Compound, hi one embodiment, the unit dosage form is in a container, which can be sterile, containing a therapeutically effective amount of an Aryl Sulfonamide Compound and a pharmaceutically acceptable vehicle. In another embodiment, the unit dosage form is in a container containing a therapeutically effective amount of an Aryl Sulfonamide Compound as a lyophilate or pharmaceutically acceptable salt, hi this instance, the kit can further comprise another container that contains a solution useful for the reconstitution ofthe lyophilate or dissolution ofthe salt. The kit can also comprise a label or printed instructions for use ofthe Aryl Sulfonamide Compounds.
- the kit comprises a unit dosage form of a composition of the invention.
- Kits ofthe invention can further comprise one or more devices that are useful for administering the unit dosage forms ofthe Aryl Sulfonamide Compounds or a composition of the invention.
- devices include, but are not limited to, a syringe, a drip bag, a patch or an enema, which optionally contain the unit dosage forms.
- Step a l-Pyridm-4-yl-cyclopropanecarboxylic acid ethyl ester.
- a 500 mL flask was charged with 2.5 g ethyl 4-pyridylacetate (15.15 mmol, 1.0 equiv.), 45 mL THF and 45 mL DMF, followed by the addition of 1.8 g sodium hydride (75.0 mmol, 5.0 equiv.). The resulting suspension was stirred at room temperature for 15 min, and then 2 mL 1,2- dibromoethane (46.38 mmol, 3.0 equiv.) was introduced via an addition funnel.
- Step b (l-Pyridin-4-yl-cyclopropyl) methanol.
- a 100 mL flask containing 165 mg l-pyridin-4-yl-cyclopropanecarboxylic acid ethyl ester (1.0 mmol, 1.0 equiv.) and 10 mL THF was carefully added 3.0 mL of 1.0 M DIBAL-H in toluene.
- the reaction was allowed to stir for 1 h at which it was diluted with 5 mL of 1 N HC1.
- the solution was then extracted (2 x MeOH/C ⁇ kCfe), washed (1 x brine), dried (Na 2 SO 4 ) and concentrated under reduced pressure.
- Step c 4-(l-ChloromethyIcyclopropyl) pyridine.
- a 250 mL flask containing 333 mg of l-pyridin-4-yl-cyclopropylmethanol 2.23 mmol, 1.0 equiv.) and 10 mL CH 2 C1 2 under N 2 was added 0.18 mL thionyl chloride (2.46 mmol, 1.1 equiv.). After stirring for 2 h, the solution was concentrated under reduced pressure to provide the product as a white solid which was sufficiently pure to continue to the next step.
- Step d (R)-3-Methyl-l-(l-pyridin-4-yl-cyclopropylmethyl) piperazine.
- a 250 mL flask was charged with 250 mg (R)-(-)-2-methylpiperazine (10.0 mmol, 2.5 equiv.), 334 mg 4-(l-chloromethylcyclopropyl) pyridine (2.0 mmol, 1.0 equiv.) and 15 mL acetonitrile.
- the flask was equipped with a reflux condenser, and then placed into a preheated 100 °C bath.
- a portion ofthe product obtained above (462 mg, 2.0 mmol, 1.0 equiv.) in 10 mL CH 2 C1 2 was combined in a flask with 436 mg 4-acetylbenzenesulfonyl chloride (1.0 mmol, 1.0 equiv.) and 0.34 mL triethylamine (2.0 mmol, 1.2 equiv.).
- Step f l,l,l-Trifluoro-2- ⁇ 4-[2-(R)-methyl-4-(l-pyridm-4-yl-cyclopylmethyl) piperazine-l-sulfonyl]phenyl ⁇ propan-2-ol (1).
- Step a 4-(l,l,l,3,3,3-hexafluoropropan-2-ol-2-yl)benzenesulfonyl chloride.
- 4-(hexafluoro-2-hydroxylisopropyl)aniline (15.0g, 58 mmol)
- HC1 37% in water, 30 mL
- CH 3 COOH 9 mL
- NaNO 2 4.4g, 64 mmol
- Step b Using the same methods as Example 1, and substituting 4-(l, 1,1,3,3,3- hexafluoropropan-2-ol-2-yl)benzenesulfonyl chloride for 4-acetylbenzenesulfonyl chloride in step e of Example 1, l,l,l,3,3,3-hexafluoro-2- ⁇ 4-[2-(R)-methyl-4-(l-pyridin-4-yl- cyclopropylmethyl) piperazine- 1 -sulfonyl]phenyl ⁇ -propan-2-ol (4) was prepared.
- Step a 2-Pyridin-3-yl-methylmalonic acid monoethyl ester.
- a stirred solution of 2.72 mL diisopropylamine (19.55 mmol, 2.3 equiv.) in THF under N 2 was cooled to -10 °C and freated with 7.5 mL of n-BuLi in hexane. After 10 min, the mixture was cooled to -78 °C and 3-pyridin-3-yl-propionic acid ethyl ester (1.4 g, 8.5 mmol, 1.0 equiv.) was added. After stirring for an additional 20 min.
- Step b 2-Pyridin-3-yl-methylacrylic acid ethyl ester.
- 2-pyridin-3-yl- methylmalonic acid monoethyl ester (8.5 mmol, 1.0 equiv.) was combined in a flask with 73 mg piperazine (0.85 mmol, 0.1 equiv.), 255 mg paraformaldehyde (8.5 mmol, 1.0 equiv.) and 15 mL pyridine.
- the mixture was refluxed for 2 h, cooled to room temperature and then diluted with saturated NaHCO 3 .
- the solution was exfracted (2x 10% MeOH/CH 2 Cl 2 ), washed (1 x brine), dried (Na 2 SO 4 ), concentrated under reduced pressure to provide 0.92 g of the product as a white solid (4.82 mmol).
- Step c l-Pyridin-3-yl-methylcyclopropanecarboxylic acid ethyl ester.
- 2-pyridin-3-ylmethylacrylic acid monoethyl ester (4.07 mmol, 1.0 equiv.) and 25 mL CH 2 C1 2 under N 2 was added a solution of diazomethane (16.60 mmol, 4.08 equiv.) in ether. After stirring for 24 h, the solution was quenched with acetic acid and followed by saturated aqueous NaHCO 3 , and then extracted with 10% MeOH/CH 2 Cl 2 .
- Step d l,l,l,3,3,3-hexafluoro-2- ⁇ 4-[2-(R)-methyl-4-(l-pyridin-3-yl- methylcyclopropylmethyl) piperazine-l-sulfonyl]phenyl ⁇ propan-2-ol (5).
- Step a l-(6-Chloro-pyridin-3-yl) cyclopropanecarbaldehyde. 1.52 g Dess- Martin periodinane (3.6 mmol, 1.2 equiv.) was added to a solution of 549 mg [l-(6-chloro- ⁇ yridin-3-yl)cyclopropyl] methanol (3.0 mmol, 1.0 equiv.) in 30 mL THF. After stirring for 3 h, the solution was diluted with saturated NaHCO 3 , exfracted (2 x 10% MeOH/CH 2 Cl 2 ), washed (1 x brine), dried (Na 2 SO 4 ) and concenfrated under reduced pressure. Purification of the residue by flash chromatography (SiO 2 , 5% MeOH/CH 2 Cl 2 ) provided the product as a yellow solid (0.5 g, 2.75 mmol).
- Step b (R)-l-[l-(6 ⁇ Chloro-pyridin-3-yl) cyclopropylmethyl]-3- methylpiperazine.
- (R)-(-)-2-methylpiperiazine 13.81 mmol, 5.0 equiv.
- 500 mg l-(6-chloro-pyridin-3-yl) cyclopropanecarbaldehyde 2.76 "mmol, 1.0 equiv.) in 40 mL 1
- 2-dichloroethane was added 1.17 g NaBH(OAc) 3 (5.52 mmol, 2 equiv.).
- Step c 2-(4- ⁇ 4-[l-(6-chloro-pyridin-3-yl)cyclopropylmethyl]-2-(R)-methyl- piperazine-l-sulfonyl ⁇ phenyl)-l,l,l,l-trifluoro-propan-2-ol (6).
- Step a [l-(tert-Butyldimethylsilanyloxymethyl) cyclopropyl] methanol.
- tert-Butyldimethylsilyl chloride (4.5 g, 30.0 mmol, 1.0 equiv.) was added to a suspension of 3 g l,l-bis(hydroxymethyl) cyclopropane (30.0 mmol, 1.0 equiv.) and 4.08 g imidazole (60 mmol, 2 equiv.) in THF at 0 °C. The mixture was stirred at 0 °C for 30 min, and water was added.
- Step b Methanesulfonic acid l-(f ⁇ r ⁇ butyldimethylsilanyloxymethyl) cyclopropylmethyl ester.
- Methanesulfonyl chloride (0.92 mL, 2.0 mmol, 1.2 equiv.) was added to a solution of 2.16 g [l-(tert-butyldimethylsilanyloxymethyl) cyclopropyl] methanol (10.0 mmol, 1.0 equiv.) and 2.5 mL triethylamine (20 mmol, 2 equiv.) in 20 mL CH 2 C1 2 at 0 °C.
- Step c [l-(tert-ButyldimethyIsilanyloxymethyl)cyclopropylmethyl]-3-(R)- methylpiperazine.
- Methanesulfonic acid[ 1 -(tert-butyldimethylsilanyloxymethyl) cyclopropylmethyl ester (1.5 g, 5.1 mmol, 1.0 equiv.) was combined in a sealed tube with 1.28 g (R)-2-methylpiperazine (12.76 mmol, 2.5 equiv.).
- the mixture was heated at 130 °C for 24 h, cooled to room temperature and diluted with saturated NaHCO 3 .
- the solution was exfracted (2 x 10%) MeOH/CH 2 Cl 2 ), washed (1 x brine), dried (Na 2 SO 4 ) and concenfrated under reduced pressure to provide a colorless liquid which was used in the next step.
- Step d l,l,l-trifluoro-2- ⁇ 4-[4-(l-hydroxymethyl-l-yl-cyclopropyImethyl]-2-(R)- methylpiperazine-l-sulfonyl]phenyl ⁇ propan-2-ol (7).
- steps e and fin Example 1 substituting (R)-l-[l-(tert-butyldimethylsilanyloxymethyl) cyclopropylmethyl] -3- methylpiperazine for (R)-3 -methyl- l-(l-pyridin-4-yl-cyclopropylmethyl) piperazine in step e, Compound 7 was prepared.
- Step a (1 -Hydroxy methylcyclopropyl) acetonitrile. 5,7-Dioxa-spiro[2.5] octan- 6-one (1.48 g, 10.0 mmol, 1.0 equiv.) was combined in a flask with 3.12 g tetraethylammoniumcyanide (20.0 mmol, 2.0 equiv.) in 30 mL DMF. The mixture was heated at 70 °C for 24 h, cooled to room temperature and diluted with saturated NaHCO 3 .
- the solution was exfracted (2 x 10% MeOH/CH 2 Cl 2 ), washed (1 x brine), dried (Na 2 SO 4 ) and concenfrated under reduced pressure to provide 360 mg ofthe product as a colorless liquid which was used in the next step.
- Step a l-Pyridin-4-yl-cyclobutanecarboxylic acid ethyl ester.
- pyridin-4-yl-acetic acid ethyl ester 20.0 mmol, 1.0 equiv.
- THF 30.0 mmol
- DMF 30.0 mmol
- DMF 30.0 mmol
- 1,3-Dibromo-propane 30.0 mmol, 1.5 equiv.
- Step b (l-Pyridin-4-yI-cyclobutyl)-methanol.
- To a 250 mL flask was charged with the product obtained above (2.0 g, 9.7 mmol, 1.0 equiv.) and 10 mL THF.
- 30 mL DIBAL-H (1.0 mL in Hexanes, 30 mmol, 3.0 equiv.) was added in a flask.
- the resulting solution was allowed to stir for 2 h and then was diluted with saturated NaHCO and extracted (4 x 10% MeOH/CH 2 Cl 2 ).
- the organics were dried (MgSO ) and concenfrated under reduced pressure. Purification by flash chromatography (SiO 2 , 3% MeOH/CH 2 Ci 2 ) provided mthe product as a yellow oil (0.78 g, 4.8mmol).
- Step c Methanesulfonic acid l-pyridin-4-yl-cyclobutylmethyl ester.
- a portion ofthe product obtained above (750 mg, 4.6 mmol, 1.0 equiv.) in 50 mL CH 2 C1 2 was combined in a flask with 700 mg triethylamine (6.9 mmol, 1.5 equiv.) and 580 mg methanesulfonic acid chloride (5.1 mmol, 1.1 equiv.). The solution was allowed to stir for 1/2 h followed by dilution with saturated NaHC0 3 and extracted (3 x 10% MeOH/CH 2 Cl 2 ). The organics were dried (MgSO 4 ) and concentrated under reduced pressure.
- Step d l-(l-Pyridin-4-yl-cyclobutylmethyl)piperazine.
- a portion ofthe product obtained above (200 mg, 0.83 mmol, 1.0 equiv.) was combined in a pressure tube with 500 mg Piperazine. The resulting mixture was then placed into a preheated 100 °C bath. After stirring for 4 h, the mixture was diluted with 50mL CH 2 CI 2 and saturated NaHC0 3 , exfracted (3 x 10% MeOH/CH 2 Ci 2 ). The organics were dried (MgSO 4 ) and concentrated under reduced pressure. Purification by flash chromatography (Si0 2 , 4% MeOH/CH 2 Cl 2 , 8%
- Step d l- ⁇ 4-[4-(l-Pyridin-4-yl-cyclobutylmethyl)-piperazine-l-sulfonyl]- phenyl ⁇ -ethanone.
- the product obtained above 160 mg, 0.69 mmol, 1.0 equiv.
- 5 mL CH 2 C1 2 was combined in a flask with 152 mg 4-acetylbenzenesulfonyl chloride (0.69 mmol, 1.0 equiv.) and 142 mg triethylamine (1.40 mmol, 2.0 equiv.).
- the solution was allowed to stir for 1 h followed by dilution with 20 mL CH 2 C1 2 .
- Step a 1-Hydroxymethylcyclobutanecarboxylic acid ethyl ester.
- cyclobutane-l,l-dicarboxylic acid diethyl ester 25.0 mmol, 1.0 equiv.
- THF 5.0 mmol, 1.0 equiv.
- lithium tri-tert-butoxyaluminohydride 1.0M in THF, 55 mmol, 2.2 equiv.
- the solution was heated at reflux for 5h before cooled to room temperature.
- the suspension was diluted with Saturated NH C1. After stirring for lh, the suspension was filtered through Buckner funnel.
- Step b 1-Formylcyclobutanecarboxylic acid ethyl ester.
- Step c l-[3-(R )-Methyl-piperazin-l-ylmethyl]-cyclobutanecarboxylic acid ethyl ester.
- the product obtained above (470 mg, 3.0mmol, 1.0 equiv.) in 25 mL 1,2- dichloro-ethane was combined in a flask with 750 mg (R)-(-)-2-methylpiperazine (7.5mmol, 2.5 equiv.) and several drops of acetic acid.
- 2.54 g of sodium triacetoxyborohydride (12.0 mmol, 4.0 equiv.) was added and the suspension was allowed to stir overnight.
- the mixture was diluted with saturated NaHCO 3 , exfracted (3 x 10% MeOH/CH 2 Cl 2 ), dried (MgSO 4 ) and concenfrated under reduced pressure.
- Step d l ⁇ [4-(4-Acetyl-benzenesulfonyl)-3-( R )-methyl-piperazin ⁇ l-ylmethyI]- cyclobutanecarboxylic acid ethyl ester.
- the residue obtained above in 6 mL CH 2 CI 2 was combined in a flask with 272 mg 4-acetylbenzenesulfonyl chloride (1.25mmol) and 250 mg triethylamine (2.5 mmol). The solution was allowed to stir for 1 h followed by dilution with 20 mL CH 2 ⁇ 2 and saturated NaHCO 3 .
- Step e l- ⁇ 3-(R )-Methyl-4-[4-(2,2,2-trifluoro-l-hydroxy-l-methyl-ethyl)- benzenesulfonyl]-piperazin-l-ylmethyl ⁇ -cyclobutanecarboxylic acid ethyl ester.
- Step f l- ⁇ 3-(R )-Methyl-4-[4-(2,2,2-trifluoro-l-hydroxy-l-methyl-ethyl)- benzenesuIfonyl]-piperazin-l-ylmethyl ⁇ -cyclobutanecarboxylic acid.
- Step g. l- ⁇ 3-(R)-MethyI-4-[4-(2,2,2-trifluoro-l-hydroxy-l-methyl-ethyl)- benzenesulfonyl]-piperazin-l-ylmethyl ⁇ -cyclobutanecarboxylic acid amide (13).
- Step a 1-Hydroxymethyl-cyclopropanecarbonitrile.
- 1-cyano-cyclopropanecarboxylic acid ethyl ester 2.0 g, 14.4 mmol, 1.0 equiv.
- ethylene glycol dimethyl ether 100 mL
- MeOH 10 mL
- NaBH 4 4.4 g, 115.0 mmol, 8.0 equiv.
- the suspension was diluted with Saturated NaHCO 3 slowly, exfracted (3 x 10% MeOH/CH 2 Cl 2 ), dried (MgSO 4 ), and concenfrated under reduced pressure. Purification by flash chromatography (SiO 2 , CH 2 C1 2 , 5% MeOH/CH 2 Cl 2 ) provided the product as a colorless oil (1.25 g, 12.9 mmol).
- Step b Methanesulfonic acid 1-cyano-cyclopropylmethyl ester.
- the product obtained above (1.25 mg, 12.9 mmol, 1.0 equiv.) in 30 mL CH 2 C1 2 was combined in a flask with 2.6 g triethylamine (25.8 mmol, 2.0 equiv.) and 1.92 g methanesulfonic acid chloride (16.8. mmol, 1.3 equiv.) at 0 °C.
- the solution was allowed to stir for 1 h followed by dilution with saturated NaHCO 3 and exfracted (3 x 10% MeOH/CH 2 Cl 2 ).
- Step c l-(3-(R )-Methyl-piperazin-l-ylmethyl)-cyclopropanecarbonitrile.
- the product obtained above (1.9 g, 10.8 mmol, 1.0 equiv.) was combined in a pressure tube with 2.7 g (R)-(-)-2-methylpiperazine( 27.1 mmol, 2.5equiv.). The resulting mixture was then placed into a preheated 100 °C bath. After stirring for 12h, the mixture was cooled to room temperature, diluted with 50mL CH 2 C1 2 and saturated NaHCO 3 , exfracted (3 x 10%
- Step d l-[4-(4-Acetyl-benzenesulfonyl)-3-(R )-methyl-piperazin-l-ylmethyl]- cyclopropanecarbonitrile.
- the residue obtained above in 20 mL CH 2 C1 2 was combined in a flask with 1.88 g 4-acetylbenzenesulfonyl chloride (8.64 mmol) and 1.75 g triethylamine (17.3 mmol).
- the solution was allowed to stir for 1 h followed by dilution with 20 mL CH 2 C1 2 and saturated NaHCO 3 .
- the aqueous solution was exfracted (2 x 10% MeOH/CH 2 Ci2).
- the organics were dried (MgSO 4 ) and concentrated under reduced pressure. Purification by flash chromatography (Si ⁇ 2 , 1% MeOH/CH 2 Cl2) provided the product as a colorless oil (1.98 g, 5.48mmol).
- Step e l- ⁇ 3-(J? )-Methyl-4-[4-(2,2,2-trifluoro-l-hydroxy-l-methyl-ethyl)- benzenesulfonyl]-piperazin-l-ylmethyl ⁇ -cyclopropanecarbonitrile.
- a 100 mL flask containing product obtained above (1.98 g, 5.48 mmol, 1.0 equiv.) was charged with 22 mL TMS-CF 3 (0.5M in THF, 11 mmol, 2.0 equiv.).
- Step f l- ⁇ 3-(R )-Methyl-4-[4-(2,2,2-trifluoro-l-hydroxy-l-methyl-ethyl)- benzenesulfonyl]-piperazin-l-ylmethyl ⁇ -cyclopropanecarboxylic acid amide (15).
- Step g. (S)-2-(4-((R)-4-((l-Cyanocyclopropyl) methyl)-2-methyIpiperazin-l- ylsulfonyI)phenyl)-l,l,l-trifluoropropan-2-yl (S)-l-hydroxybutan-2-ylcarbamate.
- Step h l-(((R)-3-Methyl-4-(4-((S)-l,l,l-rifluoro-2-hydroxypropan-2- yl)phenylsulfonyl)piperazin-l-yl)methyl)cyclopropanecarboxamide (15b).
- Step i The diastereoisomers were resolved by HPLC. Flow rate 22 mL/min on a Chiralpak AD 20 mm i.d. x 250 mm 10 micron column (Daciel Chemical Industries Ltd), using isopropyl alcohol/hexanes (30/70) as the eluent. The first peak was collected to yield 1 -(((R)-3-methyl-4-(4-((S)- 1,1,1 -trifluoro-2-hydroxypropan-2-yl)phenylsulfonyl)piperazin-l - yl)methyl)cyclopro ⁇ anecarboxamide (15b).
- Step a 4-(l-Chloromethyl-cyclopropyl)-pyridine.
- a 50 mL flask containing 200 mg of (l-Pyridin-4-yl-cyclopropyl)-methanol (1.34mmol, 1.0 equiv.) in 10 mL CH 2 C1 2 was added 174 mg thionyl chloride (1.48mmol, 1.1 equiv.). After stirring for 1 h, the solution was concentrated under reduced pressure to provide the product as a brown solid.
- Step b 2-[l-Benzyl-4-(l-pyridin-4-yl-cyclopropylmethyl)-piperazin-2-yl]- ethanol.
- a 250 mL flask was charged with the product obtained above (1.34 mmol, 1.0 equiv.), 2-(l-benzyl-piperazin-2-yl)-ethanol (1.18 g, 5.36 mmol, 4.0 equiv.), and 10 mL acetonitrile.
- the flask was equipped with a reflux condenser, and then placed into a preheated 100 °C bath.
- Step c 2-[4-(l-Pyridin-4-yl-cyclopropylmethyl)-piperazin-2-yl]-ethanol.
- To a 100 mL flask was charged with 500 mg 5% Pd/C and 30mL EtOH under N 2 .
- the product obtained above (0.88 mmol) was then added followed by 10 mL cyclohexene.
- the flask was equipped with a reflux condenser, and then placed into a preheated 80 °C bath. After stirring for 2 h, the solution was hot filtered through a plug of celite.
- the celite plug was washed (3 x EtOH), and the combined EtOH fractions were concenfrated under reduced pressure to provide the product as a colorless oil.
- Step d l- ⁇ 4-[2-(2-HydroxyethyI)-4-(l-pyridin-4-yI-cyclopropylmethyl)- piperazine-l-sulfonyl] ⁇ phenyl ⁇ -ethanone.
- the product obtained above 0.88 mmol, 1.0 equiv.) in 5 mL CH 2 C1 2 was combined in a flask with 193 mg 4-acetylbenzenesulfonyl chloride (0.88mmol, 1.0 equiv.) and 178 mg triethylamine (1.76 mmol, 2.0 equiv.).
- a 100 mL flask containing a portion of product obtained above 26 mg, 0.059 mmol, 1.0 equiv.
- TMS-CF 3 0.5M in THF
- the solution was allowed to stir for 0.5 h followed by addition of 0.5 mL tetrabutylammonium fluoride(l .0M in THF).
- Step a A mixture of 1-aminocyclopropane-l-carboxylic acid methyl ester hydrochloride (1.0 g, 6.60 mmol) in H 2 O (4 mL), phosphoric acid (85 wt. % in water, 0.2 mL), glyoxal (40 wt.% in water, 0.76 mL, 6.60 mmol) and formaldehyde (37 wt.% in water, 0.50 mL, 6.60 mmol) was warmed and stirred in a 90 °C oil bath. To the mixture NH 4 C1 (354 mg, 6.60 mmol) in H 2 O (3 mL) was added dropwise. The stirring was continued for lh.
- the viscous solution was allowed to cool to room temperature and stirred for lh.
- the mixture was cooled to 0 °C and KOH (3N) was added dropwise to neutralize the solution to pH 7.
- the mixture was concenfrated and dried in vacuo. The obtained dry mixture was patiented to a reduction with LAH.
- Step b To the product obtained from the above reaction in THF (20 mL) LAH (1.0 M in THF, 13.2 mL, 13.2 mmol) was added dropwise at 0 °C. The ice- water bath was removed and stirring was continued at room temperature for 4 h. The solution was allowed to cool to 0 °C and H 2 O (0.4 mL), NaOH (15 wt. % in water, 0.4 mL) and H 2 O (1.2 mL) were added sequentially. The cold bath was removed, stirring was continued for 15min. The mixture was then filtered through a pad of Celite, using THF as a rinse. Evaporation ofthe combined filtrates in vacuo, and flash chromatography ofthe residue, using 1.5:8.5:0.05 MeOH-CH 2 Cl 2 -NH OH, provided 0.23 g product.
- Step c To a solution ofthe alcohol (0.23 g, 1.66 mmol) in CH 2 C1 2 , SOCl 2 ( 0.24 mL, 3.32 mmol) was added dropwise. Stirring at room temperature was continued for 14 h. Evaporation ofthe solvent and the remaining SOCl 2 in vacuo and trituration in EtOAc provided the chloride (195mg, HC1 salt).
- Step d A mixture of (R)-(-)-2-methyl ⁇ iperazine (348 mg, 3.48 mmol) and the chloride (195 mg, 0.99 mmol) was heated and stirred in a 100 °C oil bath for 5 h. The mixture was cooled to room temperature and dissolved in CH 2 C1 2 (3mL). Flash chromatography ofthe solution, using 1.5:8.5:0.05 MeOH-CH 2 Cl 2 -NH 4 OH, provided the coupling product (123 mg).
- Step e A mixture ofthe coupling product from step d (105.4mg, 0.48 mmol), acetylbenzenesulfonyl chloride (105mg, 0.53 mmol), NEt 3 (O.lmL, 0.58 mmol) in CH 2 C1 2 was stirred at room temperature for 14 h. Evaporation ofthe solvent in v ⁇ cuo, and flash chromatography ofthe residue, using 1 :9:0.05 MeOH-CH 2 Cl 2 -NH 4 OH, provided the sulfonamide (0.17 g).
- Step f To a mixture ofthe sulfonamide (0.17g, 0.42 mmol) and CF 3 SiMe 3 (0.5M in THF, 2.6 mL, 1.3 mmol) TBAF (1.0 M, 1.3 mL, 1.3 mmol) was added at room temperature. The mixture was stirred for 20 h, diluted with Et 2 O (20 mL). The solution was washed with saturated aqueous NaHCO 3 , and brine, dried, and concenfrated. Flash chromatography ofthe residue, using 0.5:9.5 MeOH-CH 2 Cl 2 , provided Compound 17 (125mg).
- the flow rate was 20mL/min on a Chiralcel AD- H 20mm I.D.x250mm, 5 micron column (Daciel chemical Industries LTD), using isopropyl alcohol/hexanes (8:92) as an eluent.
- the first peak was collected as 28b.
- the 11/3-HSDl inhibitory activity was examined by quantitative determination by an SPA (scintillation proximity assay) system ofthe suppressive action on the conversion from cortisone to cortisol using human 11/3-HSDl (hereinafter recombinant 11/3-HSDl) expressed using a baculo-virus system as an enzyme source.
- SPA sintillation proximity assay
- a reagent was added to a 96 well plate (96 well Opti-platesTM-96 (Packard)) to the following final concenfration and a volume of 100 ⁇ l was reacted at room temperature for 90 min.
- reaction solution used was 0.1 ⁇ g/ml recombinant 11/3-HSDl, 500 ⁇ M NADPH, 16 nM 3 H cortisone (Amersham Biosciences, 1.78 Tbq/mol) dissolved in 0.1% BSA (Sigma)-containing PBS and the test drug was 2 ⁇ l of a compound solution (dissolved in DMSO).
- the reaction was stopped by adding PBS (40 ⁇ l, containing 0.1% BSA (Sigma)) containing 0.08 ⁇ g of anti-cortisol mouse monoclonal antibody (East Coast Biologies), 365 ⁇ g SPA PVT mouse antibody-binding beads (Amersham Biosciences) and 175 ⁇ M carbenoxolone (Sigma) to the reaction solution. After the completion ofthe reaction, the plate was incubated overnight at room temperature and the radioactivity was measured by Topcount (Packard).
- the value (0% inhibition) ofthe well containing 2 ⁇ l of DMSO instead ofthe test drug was used, and for positive control, the value (100% inhibition) ofthe well containing carbenoxolone instead ofthe test drug at the final concenfration of 50 ⁇ M was used.
- the inhibition (%) ofthe test drug was calculated by ((value of control - value of test drug)/(value of control - value of positive control)) x 100 (%).
- the IC 5 o value was analyzed using a computer-based curve fitting soft.
- Recombinant human, mouse and rat 11/3-HSDl were expressed in baculovirus expression system, isolated by affinity purification and used as the enzyme sources for cortisone to cortisol conversion in vitro.
- H-Cortisone (Amersham Bioscience, 1.78 Tbq/mol. 49 Ci/mmol) was used as the substrate, and a monoclonal anti-cortisol antibody and the scintillation proximity assay (SPA) system were used to detect the product ofthe 11/3-HSD1- catalyzed reaction, 3 H-cortisol. Reactions took place at room temperature for 90 min.
- SPA scintillation proximity assay
- Opti-platesTM-96 (Packard) in 100 ⁇ L volume with 2 ⁇ L test compounds or control in DMSO, 0.1 ⁇ g/mL 11/3-HSDl protein, 500 ⁇ M NADPH and 16 nM radioactive cortisone, in PBS buffer supplemented with 0.1% BSA (Sigma). Reaction was stopped with the addition of 40 ⁇ L buffer containing 0.08 ⁇ g anti-cortisol monoclonal antibody (East Coast Biologies), 365 ⁇ g SPA PVT antibody-binding beads (Amersham Biosciences) and 175 ⁇ M carbenoxolone (Sigma).
- This cell-based assay measures the conversion of 3 H-cortisone to 3 H-cortisol in a HEK-293 cell line stably overexpressing human recombinant 11/3-HSDl .
- HEK-293 cells were grown in DMEM/F12 supplemented with 10% fetal bovine serum, and plated onto poly- D-lysine-coated 96-well assay plates (Costar 3903), 100,000 cells per well in 50 ⁇ L assay media (phenol free DMEM/F12 (frivifrogen) + 0.2% BSA + 1% antibiotic-antimycotic solutions).
- the solution was incubated at 37 °C for 24 h, and the reaction was initiated by the addition of 25 ⁇ L of assay media containing compounds of desired concentration and 25 ⁇ L of assay media containing 40 nM of 3 H-cortisone to each well.
- the reaction mixture was incubated at 37 °C for 90 min. and the reaction terminated by the addition of 25 ⁇ L of assay media containing 0.2 ⁇ g of anti-cortisol monoclonal antibody (East Coast Biologies), 500 ⁇ g SPA PVT antibody-binding beads (Amersham Biosciences) and 500 ⁇ M carbenoxolone (Sigma).
- Plates were incubated at room temperature for at least 2 h before being read on Topcount (Packard). The point of 50% inhibition of 11 / 3-HSDl enzyme activity (IC 50 ) was determined by computer-based curve fitting.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Endocrinology (AREA)
- Ophthalmology & Optometry (AREA)
- Urology & Nephrology (AREA)
- Cardiology (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Emergency Medicine (AREA)
- Hospice & Palliative Care (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Child & Adolescent Psychology (AREA)
- Immunology (AREA)
- Vascular Medicine (AREA)
- Dermatology (AREA)
Abstract
Description
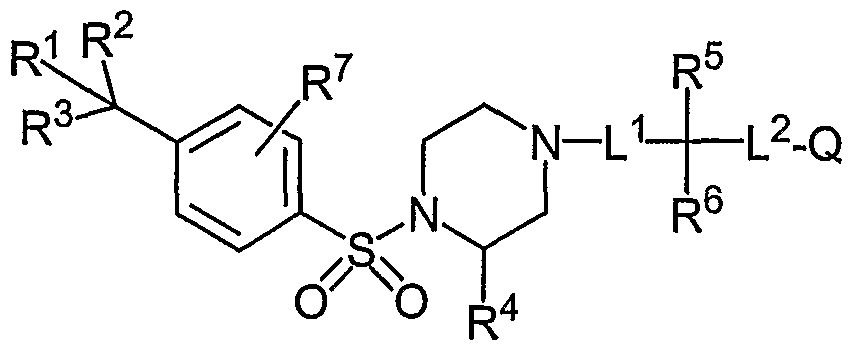


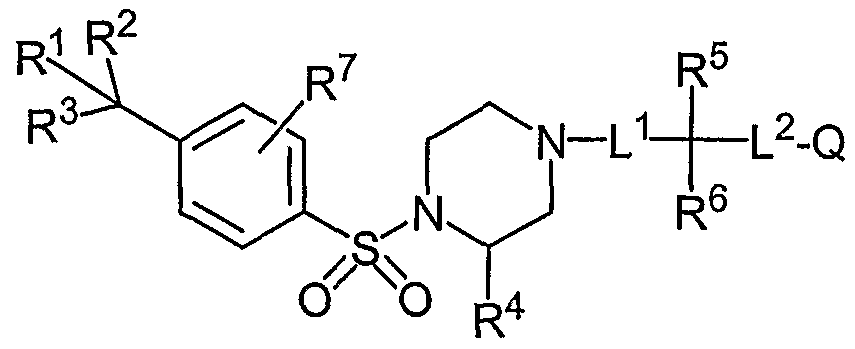



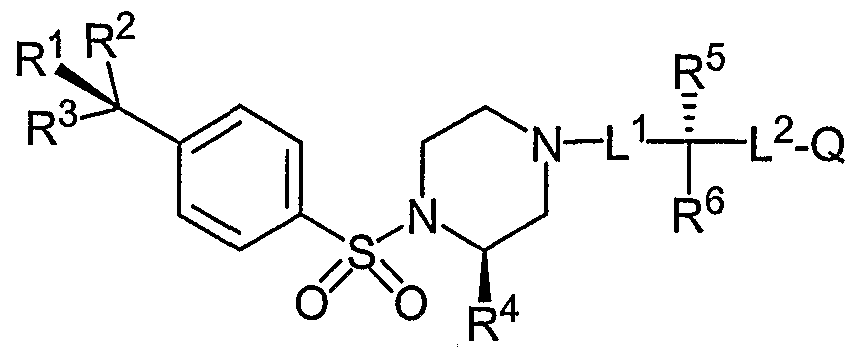

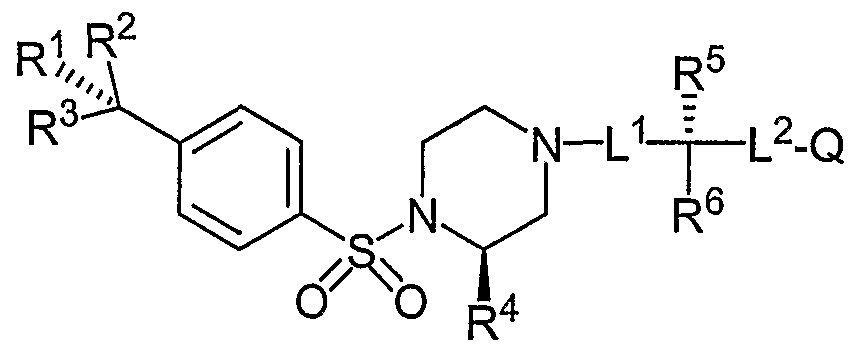




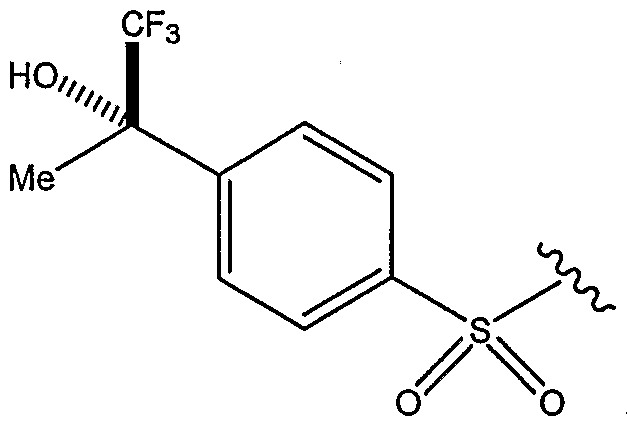














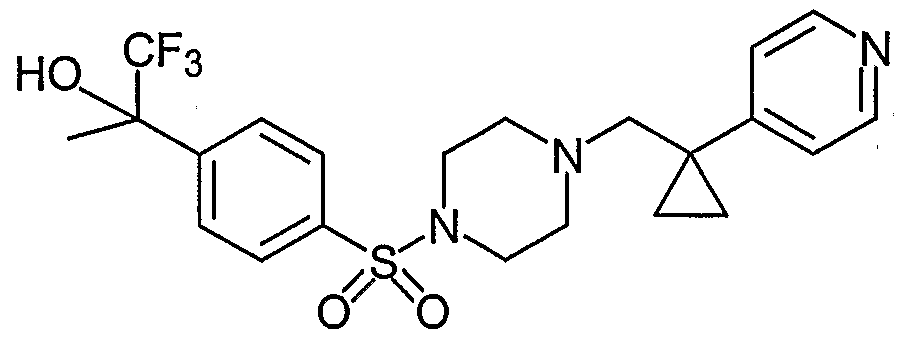



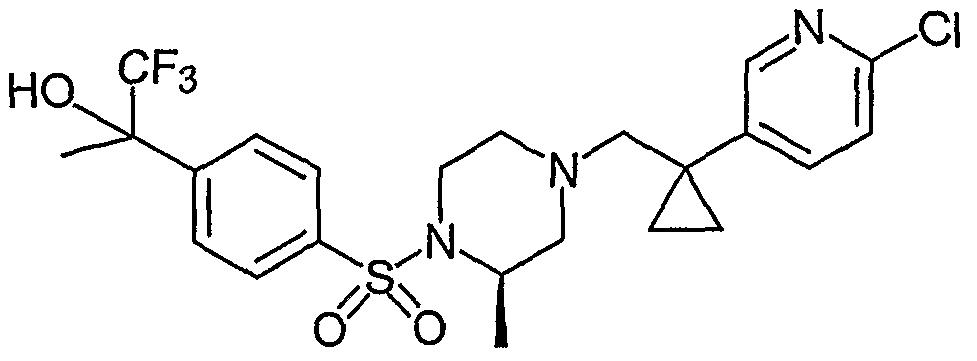









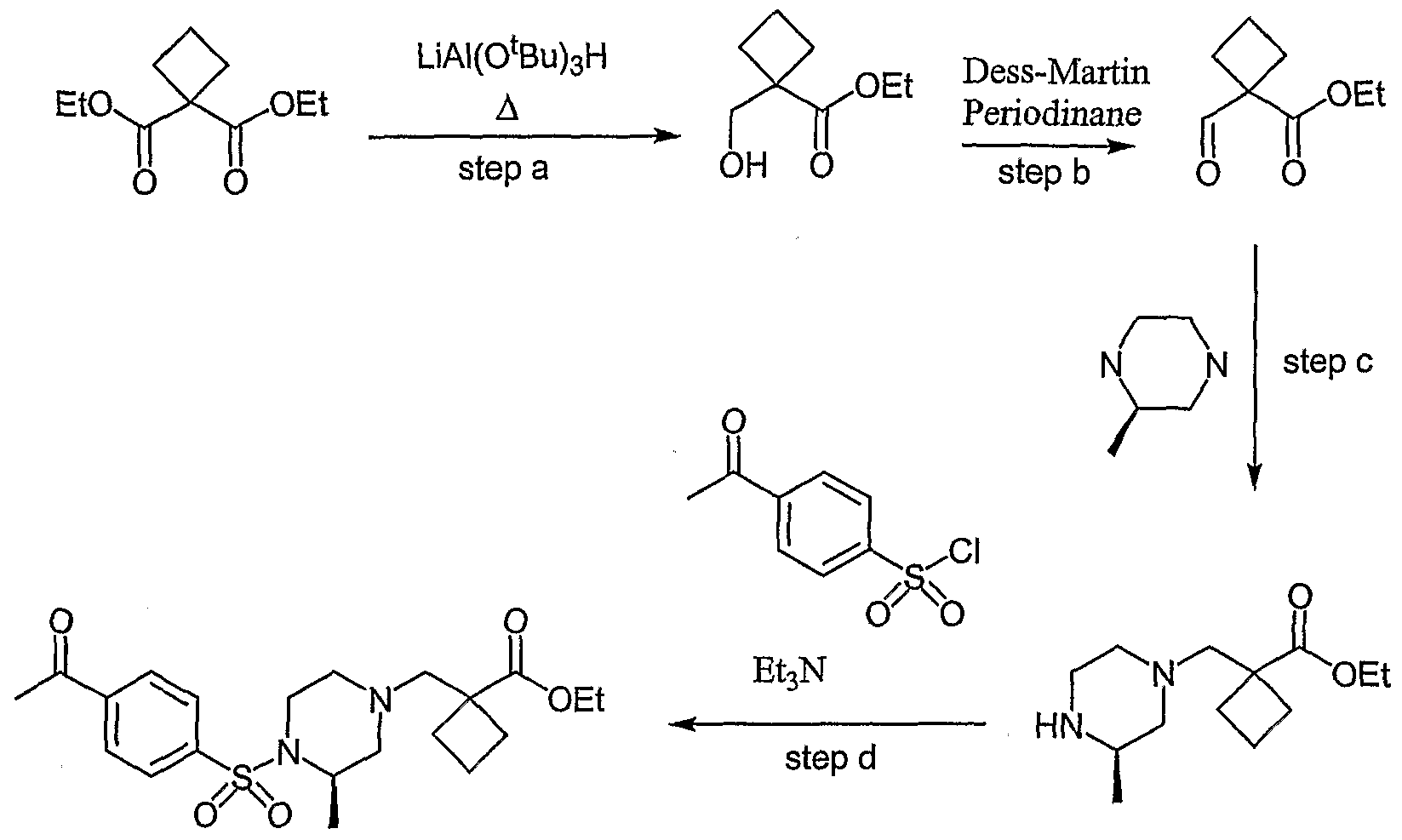














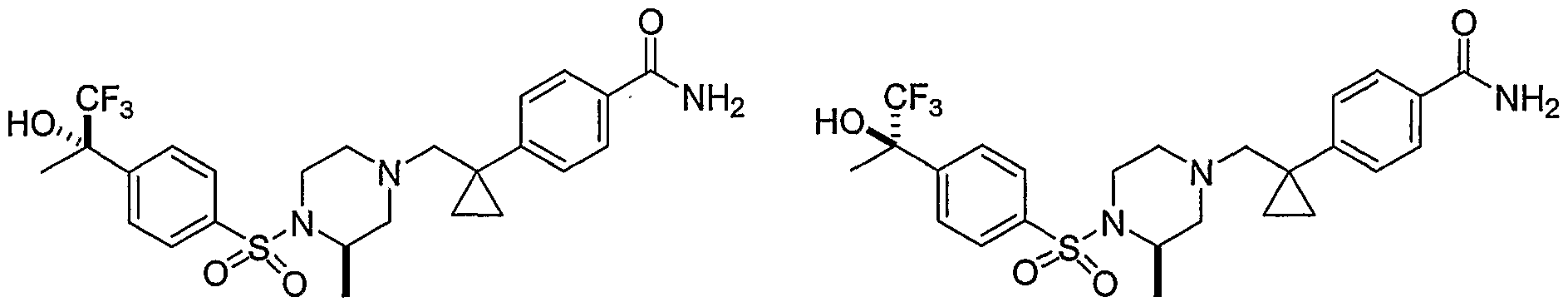




Claims





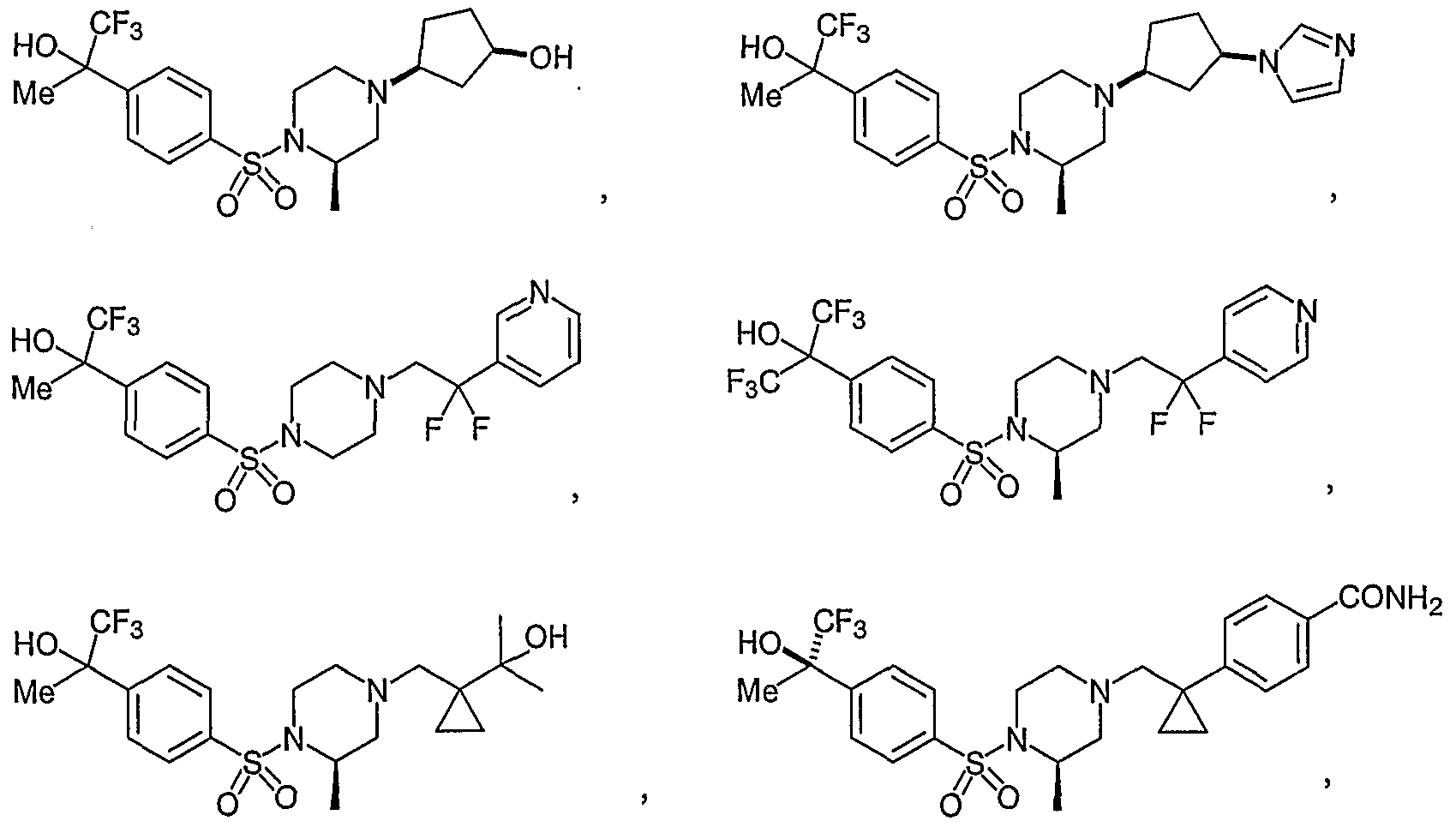




Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002548309A CA2548309A1 (en) | 2003-12-22 | 2004-12-20 | Aryl sulfonamide compounds and uses related thereto |
| JP2006547244A JP2007515490A (en) | 2003-12-22 | 2004-12-20 | Arylsulfonamide compounds and related uses |
| AU2004308932A AU2004308932A1 (en) | 2003-12-22 | 2004-12-20 | Aryl sulfonamide compounds and uses related thereto |
| EP04814971A EP1703908A4 (en) | 2003-12-22 | 2004-12-20 | Aryl sulfonamide compounds and uses related thereto |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US53192403P | 2003-12-22 | 2003-12-22 | |
| US60/531,924 | 2003-12-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005063247A1 true WO2005063247A1 (en) | 2005-07-14 |
Family
ID=34738718
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/042842 WO2005063247A1 (en) | 2003-12-22 | 2004-12-20 | Aryl sulfonamide compounds and uses related thereto |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US7365075B2 (en) |
| EP (1) | EP1703908A4 (en) |
| JP (1) | JP2007515490A (en) |
| AR (1) | AR047165A1 (en) |
| AU (1) | AU2004308932A1 (en) |
| CA (1) | CA2548309A1 (en) |
| TW (1) | TW200533335A (en) |
| WO (1) | WO2005063247A1 (en) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005110980A3 (en) * | 2004-04-14 | 2006-02-23 | Amgen Inc | Aryl sulfones and uses related thereto |
| WO2006105127A2 (en) * | 2005-03-31 | 2006-10-05 | Takeda San Diego, Inc. | Hydroxysteroid dehydrogenase inhibitors |
| WO2007070506A2 (en) * | 2005-12-14 | 2007-06-21 | Amgen Inc. | Diaza heterocyclic sulfonamide derivatives and their uses |
| WO2007073935A1 (en) * | 2005-12-29 | 2007-07-05 | Lek Pharmaceuticals D.D. | Heterocyclic compounds |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| EP1907357A1 (en) * | 2005-07-22 | 2008-04-09 | Amgen Inc. | Aniline sulfonamide derivatives and their uses |
| EP1918285A1 (en) * | 2006-11-03 | 2008-05-07 | Merck Sante | Diazepane-acetamide derivatives as selective 11beta-HSD1 inhibitors |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US7632838B2 (en) | 2006-02-07 | 2009-12-15 | Wyeth | 11-beta HSD1 inhibitors |
| US7645773B2 (en) | 2006-01-18 | 2010-01-12 | Hoffmann-La Roche Inc. | Thiazoles as inhibitors of 11β-hydroxysteroid dehydrogenase |
| WO2010141550A3 (en) * | 2009-06-04 | 2011-09-01 | Wyeth Llc | 11beta-hydroxysteroid dehydrogenase type 1 (11-beta hsd1) inhibitors |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8524894B2 (en) | 2009-06-04 | 2013-09-03 | Laboratorios Salvat, S.A. | Inhibitor compounds of 11-beta-hydroxysteroid dehydrogenase type 1 |
| WO2022066277A1 (en) * | 2020-09-24 | 2022-03-31 | Gongwin Biopharm Co., Ltd | Method for reducing fat by administering benzenesulfonamide compositions |
| WO2023215205A1 (en) * | 2022-05-06 | 2023-11-09 | Merck Sharp & Dohme Llc | Orexin receptor agonists |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7879840B2 (en) | 2005-08-25 | 2011-02-01 | The Trustees Of Columbia University In The City Of New York | Agents for preventing and treating disorders involving modulation of the RyR receptors |
| US7718644B2 (en) | 2004-01-22 | 2010-05-18 | The Trustees Of Columbia University In The City Of New York | Anti-arrhythmic and heart failure drugs that target the leak in the ryanodine receptor (RyR2) and uses thereof |
| US8022058B2 (en) | 2000-05-10 | 2011-09-20 | The Trustees Of Columbia University In The City Of New York | Agents for preventing and treating disorders involving modulation of the RyR receptors |
| US8710045B2 (en) | 2004-01-22 | 2014-04-29 | The Trustees Of Columbia University In The City Of New York | Agents for preventing and treating disorders involving modulation of the ryanodine receptors |
| US7704990B2 (en) | 2005-08-25 | 2010-04-27 | The Trustees Of Columbia University In The City Of New York | Agents for preventing and treating disorders involving modulation of the RyR receptors |
| CN101417987B (en) * | 2007-10-24 | 2011-07-13 | 山东轩竹医药科技有限公司 | DPP-IV inhibitor with sulfonamide formyl piperazine structure |
| SG186885A1 (en) | 2010-06-04 | 2013-02-28 | Albany Molecular Res Inc | Glycine transporter-1 inhibitors, methods of making them, and uses thereof |
| MX2021009515A (en) * | 2019-02-08 | 2021-10-26 | Sanofi Sa | Biotechnological optimization of microorganisms for the 1,2-dehydrogenation of steroids. |
| CN114605282B (en) * | 2022-03-22 | 2024-10-08 | 江苏阿尔法集团盛基药业(宿迁)有限公司 | Preparation method of Montelukast sodium side chain intermediate |
| CN114605283A (en) * | 2022-03-22 | 2022-06-10 | 宿迁盛基医药科技有限公司 | Method for efficiently preparing montelukast sodium side chain intermediate |
| WO2024107615A1 (en) * | 2022-11-17 | 2024-05-23 | Merck Sharp & Dohme Llc | Orexin receptor agonists |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003033484A1 (en) * | 2001-10-11 | 2003-04-24 | Bayer Healthcare Ag | Substituted piperazine cyclohexane carboxilic acid amides and the use thereof |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63101849A (en) * | 1986-10-17 | 1988-05-06 | Konica Corp | Silver halide photographic sensitive material |
| US5656644A (en) * | 1994-07-20 | 1997-08-12 | Smithkline Beecham Corporation | Pyridyl imidazoles |
| IL104369A0 (en) * | 1992-01-13 | 1993-05-13 | Smithkline Beecham Corp | Novel compounds and compositions |
| WO2001002371A1 (en) | 1999-03-05 | 2001-01-11 | Nippon Soda Co., Ltd. | Optically active piperazine compounds, intermediates for the preparation of the same and processes for the preparation of both |
| DE19838300A1 (en) * | 1998-08-24 | 2000-03-02 | Bayer Ag | 9-dialkylaminopurinone derivatives |
| DE19838705A1 (en) * | 1998-08-26 | 2000-03-02 | Bayer Ag | New dihydro- (1,2,3) -triazolo- [4,5-d] pyrimidin-7-ones |
| WO2001024786A1 (en) * | 1999-05-13 | 2001-04-12 | Shionogi & Co., Ltd. | Preventive or therapeutic drugs for diabetes |
| DE10000739A1 (en) * | 2000-01-11 | 2001-07-12 | Merck Patent Gmbh | New piperidine and piperazine derivatives which are antagonists of certain serotonin receptors, are useful in treatment of, e.g. eating disorders, stroke, anxiety or Parkinson's disease |
| SE0001899D0 (en) | 2000-05-22 | 2000-05-22 | Pharmacia & Upjohn Ab | New compounds |
| EP1436281B1 (en) * | 2001-10-17 | 2010-06-16 | Schering Corporation | Piperidine- and piperazineacetamides as 17beta hydroxysteroid dehydrogenase type 3 inhibitors for the treatment of androgen dependent diseases |
| CN1633428A (en) | 2001-11-22 | 2005-06-29 | 比奥维特罗姆股份公司 | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
-
2004
- 2004-12-20 EP EP04814971A patent/EP1703908A4/en not_active Ceased
- 2004-12-20 JP JP2006547244A patent/JP2007515490A/en active Pending
- 2004-12-20 CA CA002548309A patent/CA2548309A1/en not_active Abandoned
- 2004-12-20 US US11/018,401 patent/US7365075B2/en not_active Expired - Fee Related
- 2004-12-20 WO PCT/US2004/042842 patent/WO2005063247A1/en active Application Filing
- 2004-12-20 AU AU2004308932A patent/AU2004308932A1/en not_active Abandoned
- 2004-12-22 TW TW093140141A patent/TW200533335A/en unknown
- 2004-12-22 AR ARP040104871A patent/AR047165A1/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003033484A1 (en) * | 2001-10-11 | 2003-04-24 | Bayer Healthcare Ag | Substituted piperazine cyclohexane carboxilic acid amides and the use thereof |
Cited By (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005110980A3 (en) * | 2004-04-14 | 2006-02-23 | Amgen Inc | Aryl sulfones and uses related thereto |
| US7402704B2 (en) | 2004-04-14 | 2008-07-22 | Amgen Inc. | Arylsulfones and uses related thereto |
| US7754890B2 (en) | 2004-04-14 | 2010-07-13 | Amgen Inc. | Arylsulfones and uses related thereto |
| WO2006105127A2 (en) * | 2005-03-31 | 2006-10-05 | Takeda San Diego, Inc. | Hydroxysteroid dehydrogenase inhibitors |
| WO2006105127A3 (en) * | 2005-03-31 | 2007-03-22 | Takeda San Diego Inc | Hydroxysteroid dehydrogenase inhibitors |
| US7759339B2 (en) | 2005-03-31 | 2010-07-20 | Takeda San Diego, Inc. | Hydroxysteroid dehydrogenase inhibitors |
| EP1907357A1 (en) * | 2005-07-22 | 2008-04-09 | Amgen Inc. | Aniline sulfonamide derivatives and their uses |
| WO2007070506A2 (en) * | 2005-12-14 | 2007-06-21 | Amgen Inc. | Diaza heterocyclic sulfonamide derivatives and their uses |
| WO2007070506A3 (en) * | 2005-12-14 | 2007-12-13 | Amgen Inc | Diaza heterocyclic sulfonamide derivatives and their uses |
| US7825122B2 (en) | 2005-12-14 | 2010-11-02 | Amgen Inc. | Diaza heterocyclic sulfonamide derivatives and their uses |
| WO2007073935A1 (en) * | 2005-12-29 | 2007-07-05 | Lek Pharmaceuticals D.D. | Heterocyclic compounds |
| US7645773B2 (en) | 2006-01-18 | 2010-01-12 | Hoffmann-La Roche Inc. | Thiazoles as inhibitors of 11β-hydroxysteroid dehydrogenase |
| US7632838B2 (en) | 2006-02-07 | 2009-12-15 | Wyeth | 11-beta HSD1 inhibitors |
| EP2351568A2 (en) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Uses of dpp-iv inhibitors |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| US8242107B2 (en) | 2006-11-03 | 2012-08-14 | Merck Patent Gesellschaft Mit Beschrankter Haftung | Diazepane-acetamide derivatives as selective 11β-HSD1 inhibitors |
| JP2010508312A (en) * | 2006-11-03 | 2010-03-18 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Diazepane-acetamide derivatives as selective 11β-HSD1 inhibitors |
| WO2008052638A1 (en) * | 2006-11-03 | 2008-05-08 | Merk Patent Gmbh | DIAZEPANE-ACETAMIDE DERIVATIVES AS SELECTIVE 11β-HSD1 INHIBITORS |
| EP1918285A1 (en) * | 2006-11-03 | 2008-05-07 | Merck Sante | Diazepane-acetamide derivatives as selective 11beta-HSD1 inhibitors |
| US8586577B2 (en) | 2006-11-03 | 2013-11-19 | Merck Patent Gmbh | Diazepane acetamide derivatives as selective 11B-HSD1 inhibitors |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010141550A3 (en) * | 2009-06-04 | 2011-09-01 | Wyeth Llc | 11beta-hydroxysteroid dehydrogenase type 1 (11-beta hsd1) inhibitors |
| US8822452B2 (en) | 2009-06-04 | 2014-09-02 | Laboratorios Salvat, S.A. | Inhibitor compounds of 11-beta-hydroxysteroid dehydrogenase type 1 |
| US8524894B2 (en) | 2009-06-04 | 2013-09-03 | Laboratorios Salvat, S.A. | Inhibitor compounds of 11-beta-hydroxysteroid dehydrogenase type 1 |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2022066277A1 (en) * | 2020-09-24 | 2022-03-31 | Gongwin Biopharm Co., Ltd | Method for reducing fat by administering benzenesulfonamide compositions |
| TWI788000B (en) * | 2020-09-24 | 2022-12-21 | 共信醫藥科技股份有限公司 | Use of benzenesulfonamide compositions in reducing fat |
| US11752160B2 (en) | 2020-09-24 | 2023-09-12 | Gongwin Biopharm Co., Ltd | Method for reducing fat by administering benzenesulfonamide compositions |
| WO2023215205A1 (en) * | 2022-05-06 | 2023-11-09 | Merck Sharp & Dohme Llc | Orexin receptor agonists |
Also Published As
| Publication number | Publication date |
|---|---|
| US20050277649A1 (en) | 2005-12-15 |
| AU2004308932A1 (en) | 2005-07-14 |
| EP1703908A4 (en) | 2009-07-08 |
| TW200533335A (en) | 2005-10-16 |
| US7365075B2 (en) | 2008-04-29 |
| AR047165A1 (en) | 2006-01-11 |
| CA2548309A1 (en) | 2005-07-14 |
| JP2007515490A (en) | 2007-06-14 |
| EP1703908A1 (en) | 2006-09-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7365075B2 (en) | Aryl sulfonamide compounds and uses related thereto | |
| US7825122B2 (en) | Diaza heterocyclic sulfonamide derivatives and their uses | |
| US7524848B2 (en) | Diaza heterocyclic amide compounds and their uses | |
| EP2029540B1 (en) | Benzamide derivatives and uses related thereto | |
| EP1904435B1 (en) | Benzamide derivatives and uses related thereto | |
| EP2044004B1 (en) | Benzamide derivatives as modulators of 11beta-hsd1 for treating diabetes and obesity | |
| EP1753421B1 (en) | Arylsulfonamides and uses as hydroxysteroid dehydrogenase | |
| EP1735275B1 (en) | Aryl sulfones and uses related thereto | |
| US8765788B2 (en) | Tricyclic inhibitors of hydroxysteroid dehydrogenases | |
| US8076376B2 (en) | Aniline sulfonamide derivatives and their uses | |
| MX2008007725A (en) | Diaza heterocyclic sulfonamide derivatives and their uses |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2548309 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/007141 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006547244 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004308932 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004814971 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2004308932 Country of ref document: AU Date of ref document: 20041220 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004308932 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004814971 Country of ref document: EP |