WO2005030258A2 - Small molecule compositions and methods for increasing drug efficiency using compositions thereof - Google Patents
Small molecule compositions and methods for increasing drug efficiency using compositions thereof Download PDFInfo
- Publication number
- WO2005030258A2 WO2005030258A2 PCT/US2004/031147 US2004031147W WO2005030258A2 WO 2005030258 A2 WO2005030258 A2 WO 2005030258A2 US 2004031147 W US2004031147 W US 2004031147W WO 2005030258 A2 WO2005030258 A2 WO 2005030258A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- conjugate
- aryl
- arylureido
- alkylureido
- Prior art date
Links
- 0 C[C@](C(*)=C(*)[C@@](CO)C=C)N(C(N1*)O)C=C(*)C1=O Chemical compound C[C@](C(*)=C(*)[C@@](CO)C=C)N(C(N1*)O)C=C(*)C1=O 0.000 description 14
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/08—Linear peptides containing only normal peptide links having 12 to 20 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/337—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having four-membered rings, e.g. taxol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/475—Quinolines; Isoquinolines having an indole ring, e.g. yohimbine, reserpine, strychnine, vinblastine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/04—X-ray contrast preparations
- A61K49/0404—X-ray contrast preparations containing barium sulfate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- FIELD Conjugates, compositions and methods for improving drug efficiency are provided.
- the conjugates provided are for delivery of therapeutic agents for treating a variety of disorders, such as, proliferative diseases, autoimmune diseases, infectious diseases and inflammatory diseases, are provided.
- Th.e conjugates contain drug moieties and substrates for kinases other than hexokinases, protein kinases and lipid kinases non-releasably linked thereto, optionally by a non-releasing linker.
- BACKGROUND Many potent, but relatively non-specific drugs have been developed, for the treatment of cancer. Examples of drugs for the treatment of cancer include taxanes or taxoids, vinca alkaloids, alkylating agents, camptothecins, and anthracycliries.
- Another method for antimetabolite treatment of cancer uses a purine containing compound whose action depends upon HGRT, and the most common cause of resistance is the deficiency of this enzyme.
- Targeting is lacking with antimetabolites used for treatment of cancer as evidenced by the poor discrimination between normal cells and cancer cells.
- the antimetabolites used in cancer chemotherapy are prodrugs, since additional metabolic events are needed once cellular entry is gained for a therapeutic effect to be realized.
- the anti-metabolite 5-FdU a pyrimidine base, must be converted to a nucleotide for inhibition of thymidine synthetase, which is responsible for the therapeutic effect.
- Viral anti-metabolites target the viral thymidine kinase (TK), but trapping or accumulation is not responsible for their therapeutic effect.
- Viral anti-metabolites acted upon by viral TK include pyrimidine and purine containing compounds.
- Anti- metabolites used in anti- viral therapy are also prodrugs, and must be subject to downstream intracellular enzymes for conversion to the nucleoside triphosphate, and incorporation into the developing DNA strand to inhibit the DNA synthesizing machinery of a viral infected cell, which is the actual event responsible for the therapeutic effect.
- Viral diseases treated by anti-metabolites include the DNA viruses HSV-1, HSV-1, VZV, EBV, CMV and the RNA viruses HTLV-1 and HIV.
- RNA viral infections are especially problematic and despite the advances in treating AIDS caused by HJV infection, the disease caused by this virus is invariably fatal.
- Drug effectiveness can also be limited by resistance to the drug which develops during treatment. This resistance is exemplified by the treatment of cancer wherein drug is actively removed from the treated cell by a P-glycoprotein transporter or wherein the effectiveness of the drug is diminished by over-expression of the enzyme upon which the drug acts.
- the compounds are conjugates that contain a drug moiety and a substrate for a kinase other than a hexokinase, a protein kinase or a lipid kinase non-releasably linked thereto.
- the drug moieties include therapeutic agents, such as a cytotoxic agents, and diagnostic agents, such as labeled moieties and imaging agents other than compounds containing a carboranyl, hydroxyboryl and rare earth crypate moiety.
- the substrates are substrates for a kinase other than a hexokinase, a protein kinase or a lipid kinase.
- the drug moiety is a therapeutic agent other than a compound containing a carboranyl or hydroxyboryl moiety. In certain embodiments, the drug moiety is a label other than a compound containing a rare earth crypate moiety.
- the conjugates contain one or more substrates for one or a plurality of kinases other than a hexokinase, a protein kinase or a lipid kinase non- releasably linked thereto, either directly or via a non-releasing linker to a drug moiety, such as a cytotoxic agent.
- the conjugates provided herein contain the following components: (substrate) t , (linker) q , and (drug)a in which: at least one substrate for a kinase other than a hexokinase, a protein kinase or a lipid kinase is non-releasably linked, optinally via a linker, to a drug moiety, t is 1 to 6 and each substrate is the same or different, and is generally 1 or 2; q is 0 to 6; 0 to 4; 0 or 1 ; d is 1 to 6, in certain embodiment 1 or 2 and each drug moieties are the same or different; linker refers to any non-releasing linker; and the drug is any therapeutic agent, such as a cytotoxic agent, including an anti-cancer drug, a diagnostic agent, such as an imaging agent or labeled moiety other than compounds containing a carboranyl, hydroxyboryl and rare earth crypate moiety.
- the drug moiety of the drug conjugate may be derived from a naturally occurring or synthetic compound that may be obtained from a wide variety of sources, including libraries of synthetic or natural compounds.
- exemplary drug moieties can be cytotoxic agents, including, but not limited to, anti-infective agents, antihelminthic, antiprotozoal agents, antimalarial agents, antiamebic agents, antileiscmanial drugs, antitrichomonal agents, antitrypanosomal agents, sulfonamides, antimycobacterial drugs, or antiviral chemotherapeutics.
- the conjugates for use in the compositions and methods provided herein have formula (1): (D) d -(L) q -(S) t (1) or a pharmaceutically acceptable derivative thereof, wherein D is a drug moiety; d is 1-6, or is 1 or 2; L is a non- releasing linker; q is 0 to 6, or is 0 or 1; S is a substrate for a kinase other than a hexokinase, a protein kinase or a lipid kinase; and t is 1 to 6, or is 1 or 2, or is 1.
- the drug moiety is covalently attached, optionally via a non-releasing linker, to the substrate.
- the conjugation of the drug moiety(s) or non-releasing linker linked thereto can be at various positions of the substrate.
- conjugation to the drug moiety(s) or non-releasing linker linked thereto can be at various positions of the substrate.
- the kinase is overexpressed, overactive or exhibits undesired activity in a target system.
- the action of the kinase on the substrate results in a negative charge on the conjugate.
- the action of the kinase on the substrate may result in improved drug efficiency.
- the target system may be a cell, tissue or organ, hi particular embodiments, the cell is a tumor cell or a tumor-associated endothelial cell.
- the target system may also be associated with cancer, inflammation, angiogenesis, autoimmune syndromes, transplant rejection or osteoporosis.
- the substrate in certain embodiments, has a molecular weight of between about 50 amu and 1000 amu. In other embodiments, the substrate has a molecular weight of more than 1000 amu such as when the substrate exists as a dimer.
- the conjugates have formula (2) D - L- S' (2) wherein D and L are as defined in formula (1); and S' is a substrate for a phosphotransferase other than a hexokinase, a protein kinase or a lipid kinase.
- contemplated phosphotransferase are designated by the Enzyme Commission under the general category number EC 2.7.1 with the exceptions of the specific EC numbers 2.7.1.1 (hexokinase), 2.71.37 (protein kinase), 2.7 ' .1.91 (sphinganine kinae) and EC numbers designating other lipid kinases.
- the phospho group acceptor is a nucleoside.
- the substrate in the conjugates herein can be a substrate for a kinase including, but not limited to, thymidine kinase, viral thymidine kinase, TK-1, deoxycytidine kinase, deoxyguanosine kinase.
- the substrate in certain embodiments, is phosphorylated upon action of a kinase such as thymidine kinase, viral thymidine kinase, TK-1, deoxycytidine kinase, deoxyguanosine kinase.
- the substrate is nucleoside.
- nucleosides for use as substrates in the conjugates provided herein include, but are not limited to, cytosine, uridine, thymidine, guanosine, adenosine, or derivatives thereof.
- the drug moiety can be a hydrophobic drug, hi certain embodiments, D can be a detectable label.
- the drug is an anti- cancer drug.
- Pharmaceutical compositions containing a conjugate of formula 1 and a pharmaceutically acceptable carrier are provided herein. Also provided are methods for using the conjugates. The methods provided are methods for treating conditions caused by undesirable chronic or abenant cellular activation, migration, proliferation or survival (ACAMPS).
- the conjugates are for used in methods for treating cancer. Also provided are methods of improving drug efficiency by administering a therapeutically effective amount of a conjugate provided herein to a target system or organism, wherein the action of the kinase on the substrate results in improved drug efficiency. In one embodiment, methods for identifying kinase substrates capable of selectively accumulating in a target system are provided.
- the methods contain the steps of: a) contacting one or more conjugates with a kinase that is overexpressed, overactive or that exhibits undesired activity in a target system; and b) determining kinase activity on one or more conjugates.
- the method for identifying kinase substrates capable of selectively accumulating in a target system further contains the steps of: c) determining a first amount or a plurality of first amounts of one or more conjugates in the target system; and d) determining a second amount or a plurality of second amounts of one or more conjugates in a non-target system.
- one or more conjugates may contain a detectable label.
- the label may be radioactive or fluorescent.
- the radioactive lable is a radioactive compound other than a compound containing rare earth crypate moiety.
- the target system may be associated with cancer, inflammation, angiogenesis, autoimmune syndromes, transplant rejection or osteoporosis.
- the target system may be a cell, tissue or organ.
- the cell may be a tumor cell or a tumor- associated endothelial cell.
- methods for identifying conjugates capable of exhibiting selective toxicity against a target system are provided. The methods contain the steps of: a) contacting one or more conjugates containing a drug moiety with a target system; and b) determining the cytotoxicity of the one or more conjugates against the target system.
- drug conjugate or a “conjugate” refers to compounds having one or more drug moieties non-releasably linked, optionally via a non-releasable linker, to a substrate for one or more kinase other hexokinase, a protein kinase or a lipid kinase.
- the drug-substrate conjugates provided herein retain a significant fraction of drug activity within the conjugate and the desired therapeutic effect is elicited by the drug-substrate conjugate without having the need to cleave the drug from the substrate.
- the drug moiety or the substrate moiety in the conjugate can be present in a form of a pharmaceutically acceptable derivative that renders the conjugate biologically inactive.
- the inactive drug-substrate conjugate can be converted to the active drug-substrate conjugate under physiological conditions without having the need to cleave the drug-substrate conjugate.
- substrate is a molecule which is subject to phosphorylation by an enzyme, other than a hexokinase, a protein kinase or a lipid kinase, and encompasses species which can be converted by chemical and/or enzymatic reaction(s) to a substrate upon or after introduction of the molecule (in conjugate form) to a target system or organism.
- the substrates for use herein include, but are not limited to substrates for nucleoside kinases such as thymidine kinase, deoxycytidine kinase and deoxyguanosine kinase.
- the substrates for nuceoside kinases include, but are not limited to, natural and non-natural nucleosides and their analogs, and natural and non-natural bases for nucleosides, such as purines and pyrimidines and their analogs.
- drug or “drug moiety” is any drug or other agent that is intended for delivery to a targeted cell or tissue, such as cells or tissues associated with abe ⁇ ant cellular activation, migration, proliferation or survival, other than a compound containing a carboranyl, hydroxyboryl or rare earth cryptate containing moiety.
- Drug moiety for use herein include, but are not limited to, anti-cancer agents, anti-angiogenic agents, cytotoxic agents and labels other than compounds containing a carboranyl, hydroxyboryl or rare earth cryptate containing moieties, as described herein and known to those of skill in the art.
- an anti-cancer agent (used interchangeably with "anti-tumor or anti-neoplasm agent”) refers to any agents used in the treatment of cancer.
- anti-neoplasm agents include anti-angiogenic agents, alkylating agents, antimetabolite, certain natural products that are anti-neoplasm agents, platinum coordination complexes, anthracenediones, substituted ureas, methylhydrazine derivatives, adrenocortical suppressants, certain hormones, antagonists and anti-cancer polysaccharides.
- anti-angiogenic agent refers to any compound, that, when used alone or in combination with other treatment or compounds, can alleviate, reduce, ameliorate, prevent, or place or maintain in a state of remission, one or more clinical symptoms or diagnostic markers associated with undesired and/or uncontrolled angiogenesis.
- an anti-angiogenic agent refers to an agent that inhibits the establishment or maintenance of vasculature.
- agents include, but are not limited to, anti-tumor agents, and agents for treatments of other disorders associated with undesirable angiogenesis, such as diabetic retinopathies, hyperproliferative disorders and others.
- drug-linker construct refers to a chemical combination wherein a drug moiety and a linker moiety are covalently attached.
- a drug-substrate construct refers to a chemical combination wherein a drug moiety and a substrate moiety are covalently attached.
- linker-substrate construct refers to a chemical combination wherein a linker moiety and a substrate moiety are covalently attached.
- fraction of activity refers to an amount of the desired biological activity of a test compound, such as a drug-substrate conjugate provided herein, compared with the biological activity of the unconjugated drug or unconjugated substrate.
- the desired biological activity for the conjugates, the parent drugs or the substrates can be measured by any method known in the art, including, but not limited to, cytotoxicity assay, tubulin polymerisation assay and thymidine kinase activity assays described herein.
- the biological activity of the conjugates provided herein is greater than the activity of the parent drug moiety.
- a "significant fraction" referes to the activity of from about 5% up to about 100% of the biological activity, from about 5% up to about 95%, from about 5% up to about 90%, from about 5% up to about 80%, up to about 70%, up to about 60%, or up to about 50% of the biological activity. Significant fraction is also mean to include biological activity of 100% or more.
- subject is an animal, typically a mammal, including human, such as a patient.
- antiant refers to any biological process, cellular activation, migration, proliferation or survival, enzyme level or activity that is in excess of that associated with normal physiology.
- chronic refers to a biological process, cellular activation, migration, proliferation or survival, enzyme level or activity that is persistent or lasts longer than that associated with normal physiology.
- undesirable refers to normal physiological processes that occur at an undesirable time, such as but not limited to, immune responses associated with transplant rejection and/or graft versus host disease.
- ACAMPS refers to abe ⁇ ant cellular activation, migration, proliferation or survival.
- ACAMPS conditions are characterized by undesirable or abe ⁇ ant activation, migration, proliferation or survival of tumor cells, endothelial cells, B cells, T cells, macrophages, granulocytes including neutrophils, eosinophils and basopbils, monocytes, platelets, fibroblasts, other connective tissue cells, osteoblasts, osteoclasts and progenitors of many of these cell types.
- ACAMPS-related conditions include, but are not limited to, cancer, coronary restenosis, osteoporosis and syndromes characterized by chronic inflammation and/or autoimmunity.
- hydrophobic drug refers to any organic or inorganic compound or substance having biological or pharmaceutical activity with water solubility of less than 100 mg/ml, having a log P greater than 2, being lipid soluble or not adsorbing water.
- effective amount of therapeutic response refers to an amount which is effective in prolonging the survivability of the patient beyond the survivability in the absence of such treatment. Prolonging survivability also refers to improving the clinical disposition or physical well-being of the patient.
- therapeutically effective amount refers to an amount which is effective, upon single or multiple dose administration to the patient, in controlling tumor growth.
- controlling tumor growth refers to slowing, interrupting, a ⁇ esting or stopping the migration or proliferation of tumor or tumor-associated endothelial cells.
- the cytotoxic selectivity of the conjugates provided herein is assessed by comparing conjugate cytotoxicity against normal cells to the conjugate cytotoxicity in the tumor cells. Typically, the conjugates show highter cytotoxicity selectivity for tumor cells as compared to the normal cells.
- the term “cytotoxic selectivity index” refers to the ratio of EC 50 of the conjugate in tumor cells to the EC 50 of the conjugate in normal cell. In certain embodiments, the conjugates provided herein have higher cytotoxic selectivity for tumor cells than that of the parent drug.
- the conjugates provided herein show inproved cytotoxic selectivity index as compared to the parent drug.
- the cytotoxic selectivity index values for the conjugates provided herein are calculated by the methods provided herein.
- the term "improved drug efficiency" refers to a property of a drug within the conjugate which is improved relative to the drug in free form. Improved drug efficiency includes, but is not limited to, increased solubility, altered pharmacokinetics, including adsorption, distribution, metabolism and excretion, an increase in maximum tolerated dose, a reduction of side effects, an increase in cytotoxic selectivity index, an ability to surmount or avoid resistance mechanisms, or an ability to be administered chronically or more frequently.
- a more efficient drug may have an improved cytotoxic selectivity index as compared to a less efficient drug.
- the improvement in the cytotoxic selectivity index is at least 1.5 fold greater is the conjugate.
- non releasing linker moiety or “non releasable linker moiety” refers to a linker moiety that is attached to a drug moiety through a covalent bond or functionality which remains substantially intact under physiological conditions during a period of time required for eliciting a pharmacological response such that the pharmacological response is not due to free drug. Typically, the time is sufficient for uptake of the conjugate by the target system.
- linkage remains from about 10% up to about 100% intact under physiologic conditions in a period of about 0.1 hours up to about 3 hours.
- the linker is more than 50% intact, in another embodiment, more than 60%, more than 70%, 80% or 90% intact. Evaluation of the stability of such linkage can be made by one of skill in the art using methods known in the art.
- linker moiety refers to the intervening atoms between the drug moiety and substrate.
- a linker precursor, used interchangeably with linker precursor moity is a compound that is used in the synthesis of a drug linker construct or a substrate linker construct.
- linker and “linking moiety” herein refer to any moiety that non-releasably connects the substrate moiety and drug moiety of the conjugate to one another.
- the linking moiety can be a covalent bond or a chemical functional group that directly connects the drug moiety to the substrate.
- the linking moiety can contain a series of covalently bonded atoms and their substituents which are collectively refened to as a linking group.
- Linking moieties are characterized by a first covalent bond or a chemical functional group that connects the drug moiety to a first end of the linker group and a second covalent bond or chemical functional group that connects the second end of the linker group to the substrate.
- the first and second functionality, which independently may or may not be present, and the linker group are collectively refened to as the linker moiety.
- the linker moiety is defined by the linking group, the first functionality if present and the second functionality if present.
- the linker moiety contains atoms interposed between the drug moiety and substrate, independent of the source of these atoms and the reaction sequence used to synthesize the conjugate.
- non-releasably linked refers to linkage of a drug moiety through a covalent bond or functionality wherein the linkage remains substantially intact under physiological conditions during a period of time required for eliciting a pharmacological response such that the pharmacological response is not due to free drug.
- the linkage remains from about 10% up to about 100% intact under physiologic conditions in a period of about 0.1 hours up to about 3 hours.
- the linker is more than 50% intact, in another embodiment, more than 60%, more than 70%, 80% or 90% intact.
- L', L refers to linker groups or covalent bonds that connect the first and the second functionalities of the linker or the linking moiety.
- label or "labeling agen 'is a molecule that allows for the manipulation and/or detection of the conjugate which contains the label. Examples of labels include spectroscopic probes such as chromophores, fluorophores, and contrast agents.
- the label may also be a radioactive molecule or a molecule that is part of a specific binding pair well known in the art such as biotin and streptavidin.
- the radioactive lable for use herein is a radioactive compound other than a compound containing a rare earth crypate moiety.
- nucleoside refers to a molecule composed of a heterocyclic base and a carbohydrate. Typically, a nucleoside is composed of a heterocyclic nitrogenous base in N-glycosidic linkage with a sugar. Nucleosides are recognized in the art to include natural bases (standard), and non-natural bases well known in the art.
- the carbohydrates include the true sugars found in natural nucleosides or a species replacing the ribofuranosyl moiety or acyclic sugars.
- the heterocyclic nitrogenous bases are generally located at the 1' position of a nucleoside sugar moiety.
- Nucleosides generally contain a base and sugar group.
- the nucleosides can be unmodified or modified at the sugar, and or base moiety, (also refened to interchangeably as nucleoside analogs, modified nucleosides, non-natural nucleosides, non-standard nucleosides; see for example, Eckstein et al, International PCT Publication No. WO 92/07065 and Usman et al, International PCT Publication No. WO 93/15187).
- the heterocyclic base is typically thymine, uracil, cytosine, adenine or guanine.
- the carbohydrate shall be understood to mean the true sugar found in natural nucleosides or a species replacing the ribofuranosyl moiety or acyclic sugars, hi certain embodiments, acyclic sugars contain 3-6 carbon atoms and include, for example, the acyclic sugar moieties present in acyclovir (- CH2-O-CH2 CH2-OH), ganciclovir (-CH2-O-CH(CH2 OH)-CH2-OH), and the like. Natural nucleosides have the ⁇ -D-configuration.
- nucleoside shall be understood to encompass unnatural configurations and species replacing the true sugar that lack an anomeric carbon.
- heteocyclic base is attached to the carbohydrate through a carbon-nitrogen bond.
- nucleoside shall be understood to encompass species wherein the heterocyclic base and carbohydrate are attached through a carbon-carbon bond (C-nucleosides).
- target system is a cell, tissue or organ which is responsible for the genesis or maintenance of a disease state or is responsible for or associated with the condition being treated.
- biological activity refers to the in vivo activities of a compound or physiological responses that result upon in vivo administration of a compound, composition or other mixture.
- Biological activity thus, encompasses therapeutic effects and pharmacokinetic behaviour of such compounds, compositions and mixtures. Biological activities can be observed in in vitro systems designed to test for such activities.
- pharmaceutically acceptable derivatives of a compound include salts, esters, enol ethers, enol esters, acetals, ketals, orthoesters, hemiacetals, hemiketals, acids, bases, solvates, hydrates or prodrugs thereof. Such derivatives may be readily prepared by those of skill in this art using known methods for such derivatization.
- compositions produced may be administered to animals or humans without substantial toxic effects and either are pharmaceutically active or are prodrugs.
- Pharmaceutically acceptable salts include, but are not limited to, amine salts, such as but not limited to N,N'-dibenzylethylenediamine, chloroprocaine, choline, ammonia, diethanolamine and other hydroxyalkylamines, ethylenediamine, N-methylglucamine, procaine, N-benzylphenethylamine, l-para-chlorobenzyl-2- py ⁇ olidin- -ylmethylbenzimidazole, diethylamineand other alkylamines, piperazine and tris(hydroxymethyl)aminomethane; alkali metal salts, such as but not limited to lithium, potassium and sodium; alkali earth metal salts, such as but not limited to barium, calcium and magnesium; transition metal salts, such as but not limited to zinc; and inorganic salts, such as but not limited to, sodium hydrogen phosphat
- esters include, but are not limited to, alkyl, alkenyl, alkynyl, aryl, heteroaryl, aralkyl, heteroaralkyl, cycloalkyl and heterocyclyl esters of acidic groups, including, but not limited to, carboxylic acids, phosphoric acids, phosphinic acids, sulfonic acids, sulfinic acids and boronic acids.
- Pharmaceutically acceptable solvates and hydrates are complexes of a compound with one or more solvent or water molecules, or 1 to about 100, or 1 to about 10, or one to about 2, 3 or 4, solvent or water molecules.
- treatment means any manner in which one or more of the symptoms of a disease or disorder are ameliorated or otherwise beneficially altered. Treatment also encompasses any pharmaceutical use of the compositions herein, such as use for treating a cancer.
- amelioration of the symptoms of a particular disorder by administration of a particular compound or pharmaceutical composition refers to any lessening, whether permanent or temporary, lasting or transient that can be attributed to or associated with administration of the composition.
- EC 50 refers to a dosage, concentration or amount of a particular test compound that elicits a dose-dependent response at 50% of maximal expression of a particular response that is induced, provoked or potentiated by the particular test compound.
- the compounds provided herein may contain chiral centers. Such chiral centers may be of either the (R) or (S) configuration, or may be a mixture thereof.
- the compounds provided herein may be enantiomerically pure, or be stereoisomeric or diastereomeric mixtures.
- administration of a compound in its (R) form is equivalent, for compounds that undergo epimerization in vivo, to administration of the compound in its (S) form.
- substantially pure means sufficiently homogeneous to appear free of readily detectable impurities as determined by standard methods of analysis, such as thin layer chromatography (TLC), gel electrophoresis, high performance liquid chromatography (HPLC) and mass spectrometry (MS), used by those of skill in the art to assess such purity, or sufficiently pure such that further purification would not detectably alter the physical and chemical properties, such as enzymatic and biological activities, of the substance.
- TLC thin layer chromatography
- HPLC high performance liquid chromatography
- MS mass spectrometry
- Optically active (+) and (-), (R)- and (S)-, or (D)- and (L)-isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques, such as reverse phase HPLC.
- the compounds described herein contain olefinic double bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended that the compounds include both E and Z geometric isomers.
- all tautomeric forms are also intended to be included.
- the nomenclature alkyl, alkoxy, carbonyl, etc. is used as is generally understood by those of skill in this art.
- alkyl, alkenyl and alkynyl carbon chains contain from 1 to 20 carbons, or 1 to 16 carbons, and are straight or branched.
- Alkenyl carbon chains of from 2 to 20 carbons in certain embodiments, contain 1 to 8 double bonds, and the alkenyl carbon chains of 2 to 16 carbons, in certain embodiments, contain 1 to 5 double bonds.
- Alkynyl carbon chains of from 2 to 20 carbons in certain embodiments, contain 1 to 8 triple bonds, and the alkynyl carbon chains of 2 to 16 carbons, in certain embodiments, contain 1 to 5 triple bonds.
- alkyl, alkenyl and alkynyl groups herein include, but are not limited to, methyl, ethyl, propyl, isopropyl, isobutyl, n-butyl, sec-butyl, tert-butyl, isopentyl, neopentyl, tert-pentyl, isohexyl, ethene, propene, butene, pentene, acetylene and hexyne.
- lower alkyl, lower alkenyl, and lower alkynyl refer to carbon chains having from about 1 or about 2 carbons up to about 6 carbons.
- alk(en)(yn)yl refers to an alkyl group containing at least one double bond and at least one triple bond.
- cycloalkyl refers to a saturated mono- or multicyclic ring system, in certain embodiments of 3 to 10 carbon atoms, in other embodiments of 3 to 6 carbon atoms; cycloalkenyl and cycloalkynyl refer to mono- or multicyclic ring systems that respectively include at least one double bond and at least one triple bond.
- Cycloalkenyl and cycloalkynyl groups may, in certain embodiments, contain 3 to 10 carbon atoms, with cycloalkenyl groups, in further embodiments, containing 4 to 7 carbon atoms and cycloalkynyl groups, in further embodiments, containing 8 to 10 carbon atoms.
- the ring systems of the cycloalkyl, cycloalkenyl and cycloalkynyl groups may be composed of one ring or two or more rings which may be joined together in a fused, bridged or spiro-connected fashion.
- Cycloalk(en)(yn)yl refers to a cycloalkyl group containing at least one double bond and at least one triple bond.
- substituted alkyl refers to alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl and cycloalkynyl groups, respectively, that are substituted with one or more substituents, in certain embodiments one to three or four substituents, where the substituents are as defined herein, generally selected from Q 1 .
- aryl refers to aromatic monocyclic or multicyclic groups containing from 6 to 19 carbon atoms.
- Aryl groups include, but are not limited to groups such as fluorenyl, substituted fluorenyl, phenyl, substituted phenyl, naphthyl and substituted naphthyl.
- heteroaryl refers to a monocyclic or multicyclic aromatic ring system, in certain embodiments, of about 5 to about 15 members where one or more, in one embodiment 1 to 3, of the atoms in the ring system is a heteroatom, that is, an element other than carbon, including but not limited to, nitrogen, oxygen or sulfur.
- heteroaryl group may be optionally fused to a benzene ring.
- Heteroaryl groups include, but are not limited to, furyl, imidazolyl, py ⁇ olidinyl, pyrimidinyl, tetrazolyl, thienyl, pyridyl, py ⁇ olyl, N-methylpy ⁇ olyl, quinolinyl and isoquinolinyl.
- a "heteroarylium” group is a heteroaryl group that is positively charged on one or more of the heteroatoms.
- heterocyclyl refers to a monocyclic or multicyclic non- aromatic ring system, in one embodiment of 3 to 10 members, in another embodiment of 4 to 7 members, in a further embodiment of 5 to 6 members, where one or more, in certain embodiments, 1 to 3, of the atoms in the ring system is a heteroatom, that is, an element other than carbon, including but not limited to, nitrogen, oxygen or sulfur.
- the nitrogen is optionally substituted with alkyl, alkenyl, alkynyl, aryl, heteroaryl, aralkyl, heteroaralkyl, cycloalkyl, heterocyclyl, cycloalkylalkyl, heterocyclylalkyl, acyl, guanidino, or the nitrogen may be quaternized to form an ammonium group where the substituents are selected as above.
- substituted aryl refers to aryl, heteroaryl and heterocyclyl groups, respectively, that are substituted with one or more substituents, in certain embodiments one to three or four substituents, where the substituents are as defined herein, generally selected from Q 1 .
- aralkyl refers to an alkyl group in which one of the hydrogen atoms of the alkyl is replaced by an aryl group.
- heterooaralkyl refers to an alkyl group in which one of the hydrogen atoms of the alkyl is replaced by a heteroaryl group.
- halo refers to F, CI, Br or I.
- pseudohalides or pseudohalo groups are groups that behave substantially similar to halides. Such compounds can be used in the same manner and treated in the same manner as halides. Pseudohalides include, but are not limited to, cyano, thiocyanate, selenocyanate, trifluoromethoxy, and azide.
- haloalkyl refers to an alkyl group in which one or more of the hydrogen atoms are replaced by halogen. Such groups include, but are not limited to, chloromethyl, trifluoromethyl and l-chloro-2-fluoroethyl.
- haloalkoxy refers to RO- in which R is a haloalkyl group.
- sulfinyl or “thionyl” refers to -S(O)-.
- sulfonyl or “sulfuryl” refers to -S(O) 2 -.
- sulfo refers to -S(O) 2 O-.
- carbboxy refers to a divalent radical, -C(O)O-.
- aminocarbonyl refers to -C(O)NH 2 .
- alkylaminocarbonyl refers to -C(O)NHR in which R is alkyl, including lower alkyl.
- dialkylaminocarbonyl refers to -C(O)NRR in which R and R are independently alkyl, including lower alkyl;
- carboxamide refers to groups of formula -NRCOR in which R and R are independently alkyl, including lower alkyl.
- diarylaminocarbonyl refers to -C(O)NRR' in which R and R' are independently selected from aryl, including lower aryl, such as phenyl.
- arylalkylaminocarbonyl refers to -C(O)NRR' in which one of R and R' is aryl, including lower aryl, such as phenyl, and the other of R and R' is alkyl, including lower alkyl.
- arylaminocarbonyl refers to -C(O)NHR in which R is aryl, including lower aryl, such as phenyl.
- hydroxycarbonyl refers to -COOH.
- alkoxycarbonyl refers to -C(O)OR in which R is alkyl, including lower alkyl.
- aryloxycarbonyl refers to -C(O)OR in which R is aryl, including lower aryl, such as phenyl.
- alkoxy and alkylthio refer to RO- and RS-, in which R is alkyl, including lower alkyl.
- aryloxy and arylthio refer to RO- and RS-, in which R is aryl, including lower aryl, such as phenyl.
- alkylene refers to a straight, branched or cyclic, in certain embodiments straight or branched, divalent aliphatic hydrocarbon group, in one embodiment having from 1 to about 20 carbon atoms, in another embodiment having from 1 to 12 carbons. In a further embodiment alkylene includes lower alkylene.
- Alkylene groups include, but are not limited to, methylene (-CH 2 -), ethylene (-CH 2 CH -), propylene (-(CH 2 ) 3 -), methylenedioxy (-O-CH 2 -O-) and ethylenedioxy (-O-(CH 2 ) 2 -O-).
- lower alkylene refers to alkylene groups having 1 to 6 carbons. In certain embodiments, alkylene groups are lower alkylene, including alkylene of 1 to 3 carbon atoms.
- “azaalkylene” refers to -(CRR) n -NR-(CRR) m -, where n and m are each independently an integer from 0 to 4.
- oxaalkylene refers to -(CRR) n -O-(CRR) m -, where n and m are each independently an integer from 0 to 4.
- the "R" groups in the definitions of azaalkylene, oxaalkylene and thiaalkylene are each independently selected from hydrogen and Q 1 , as defined herein.
- alkenylene refers to a straight, branched or cyclic, in one embodiment straight or branched, divalent aliphatic hydrocarbon group, in certain embodiments having from 2 to about 20 carbon atoms and at least one double bond, in other embodiments 1 to 12 carbons.
- alkenylene groups include lower alkenylene. There may be optionally inserted along the alkenylene group one or more oxygen, sulfur or substituted or unsubstituted nitrogen atoms, where the nitrogen substituent is alkyl.
- the term "lower alkenylene” refers to alkenylene groups having 2 to 6 carbons. In certain embodiments, alkenylene groups are lower alkenylene, including alkenylene of 3 to 4 carbon atoms.
- alkynylene refers to a straight, branched or cyclic, in certain embodiments straight or branched, divalent aliphatic hydrocarbon group, in one embodiment having from 2 to about 20 carbon atoms and at least one triple bond, in another embodiment 1 to 12 carbons. In a further embodiment, alkynylene includes lower alkynylene.
- Alkynylene groups include, but are not limited to, — C ⁇ C— C ⁇ C— , -C ⁇ C- and -C ⁇ C-CH 2 -.
- the term "lower alkynylene” refers to alkynylene groups having 2 to 6 carbons. In certain embodiments, alkynylene groups are lower alkynylene, including alkynylene of 3 to 4 carbon atoms.
- alk(en)(yn)ylene refers to a straight, branched or cyclic, in certain embodiments straight or branched, divalent aliphatic hydrocarbon group, in one embodiment having from 2 to about 20 carbon atoms and at least one triple bond, and at least one double bond; in another embodiment 1 to 12 carbons.
- alk(en)(yn)ylene includes lower alk(en)(yn)ylene. There may be optionally inserted along the alkynylene group one or more oxygen, sulfur or substituted or unsubstituted nitrogen atoms, where the nitrogen substituent is alkyl.
- the term "lower alk(en)(yn)ylene” refers to alk(en)(yn)ylene groups having up to 6 carbons, hi certain embodiments, alk(en)(yn)ylene groups have about 4 carbon atoms.
- cycloalkylene refers to a divalent saturated mono- or multicyclic ring system, in certain embodiments of 3 to 10 carbon atoms, in other embodiments 3 to 6 carbon atoms; cycloalkenylene and cycloalkynylene refer to divalent mono- or multicyclic ring systems that respectively include at least one double bond and at least one triple bond. Cycloalkenylene and cycloalkynylene groups may, in certain embodiments, contain 3 to 10 carbon atoms, with cycloalkenylene groups in certain embodiments containing 4 to 7 carbon atoms and cycloalkynylene groups in certain embodiments containing 8 to 10 carbon atoms.
- ring systems of the cycloalkylene, cycloalkenylene and cycloalkynylene groups may be composed of one ring or two or more rings which may be joined together in a fused, bridged or spiro-connected fashion.
- Cycloalk(en)(yn)ylene refers to a cycloalkylene group containing at least one double bond and at least one triple bond.
- substituted alkylene "substituted alkenylene,” “substituted alkynylene,” “substituted cycloalkylene,” “substituted cycloalkenylene,” and
- substituted cycloalkynylene refer to alkylene, alkenylene, alkynylene, cycloalkylene, cycloalkenylene and cycloalkynylene groups, respectively, that are substituted with one or more substituents, in certain embodiments one to three or four substituents, where the substituents are as defined herein, generally selected from Q 1 .
- arylene refers to a monocyclic or polycyclic, in certain embodiments monocyclic, divalent aromatic group, in one embodiment having from 5 to about 20 carbon atoms and at least one aromatic ring, in another embodiment 5 to 12 carbons. In further embodiments, arylene includes lower arylene.
- Arylene groups include, but are not limited to, 1,2-, 1,3- and 1,4-phenylene.
- the term “lower arylene” refers to arylene groups having 5 or 6 carbons.
- heteroarylene refers to a divalent monocyclic or multicyclic aromatic ring system, in one embodiment of about 5 to about 15 members where one or more, in certain embodiments 1 to 3, of the atoms in the ring system is a heteroatom, that is, an element other than carbon, including but not limited to, nitrogen, oxygen or sulfur.
- heterocyclylene refers to a divalent monocyclic or multicyclic non-aromatic ring system, in certain embodiments of 3 to 10 members, in one embodiment 4 to 7 members, in another embodiment 5 to 6 members, where one or more, including 1 to 3, of the atoms in the ring system is a heteroatom, that is, an element other than carbon, including but not limited to, nitrogen, oxygen or sulfur.
- substituted arylene refers to arylene, heteroarylene and heterocyclylene groups, respectively, that are substituted with one or more substituents, in certain embodiments one to three of four substituents, where the substituents are as defined herein, generally selected from Q 1 .
- arylalkylidene refers to an alkylidene group in which either R' or R" is an aryl group.
- Cycloalkylidene groups are those where R' and R" are linked to form a carbocyclic ring.
- Heterocyclylidene groups are those where at least one of R' and R" contain a heteroatom in the chain, and R' and R" are linked to form a heterocyclic ring.
- amido refers to the divalent group -C(O)NH-.
- Thioamido refers to the divalent group -C(S)NH-.
- Oxyamido refers to the divalent group - OC(O)NH-.
- Thiaamido refers to the divalent group -SC(O)NH-.
- Dithiaamido refers to the divalent group -SC(S)NH-.
- Ureido refers to the divalent group - HNC(O)NH-.
- Thioureido refers to the divalent group -HNC(S)NH-.
- aminocarbonyl refers to - NHC(O) group.
- aminocarbonyloxy refers to - NHC(O) O- group.
- “semicarbazide” refers to -NHC(O)NHNH
- “thiosemicarbizide refers to - NHC(S)NHNH
- “Carbazate” refers to the divalent group -OC(O)NHNH-.
- “Isothiocarbazate” refers to the divalent group -SC(O)NHNH-.
- “Thiocarbazate” refers to the divalent group -OC(S)NHNH-.
- “Sulfonylhydrazide” refers to the group - SO 2 NHNH-.
- “Hydrazide” refers to the divalent group -C(O)NHNH-.
- Hydrazinyl refers to the divalent group -NH-NH-.
- substituents there may be one or more substituents present.
- haloalkyl may include one or more of the same or different halogens.
- C 1-3 alkoxyphenyl may include one or more of the same or different alkoxy groups containing one, two or three carbons.
- PEG linker represents a polyethylene glycol chain containing the designated number of atoms in the chain between the drug moiety and the substrate, conjugated to the drug moiety at the first end and to the substrate at the second end.
- alkane linker represents an alkylene group having the designated number of atoms in the chain between the drug moiety and the substrate, conjugated to the drug moiety at the first end and to the substrate at the second end.
- thymidine as the enzyme substrate is attached to the linker at N3 of the nucleoside base.
- conjugate PXL-7Ca- ALK(6)-N3-THY is a paclitaxel thymidine conjugate, wherein N3 of thymidine is conjugated to paclitaxel at C7 with a C6 alkane unit via a carbamate functionality.
- Table 1 provides examples of various drug moieties with possible points of attachments and linking functionalities.
- Table 2 herein provides examples of various linker groups and the names thereof.
- the drug-substrate conjugates provided herein retain a significant fraction of parent drug activity within the conjugate and the desired therapeutic effect is elicited by the drug-substrate conjugate without having the need to cleave the drug from the substrate.
- the conjugates provided herein are not limited to specific drug, linker and substrate moieties. Various combinations of the drug, linker and substrate moieties can be prepared using synthetic methodologies known in the art and described herein. As discussed above, the conjugates can contain a plurality of substrates, a plurality of linkers and a plurality of drug moieties.
- the conjugates provided herein retain a significant fraction of biological activity of parent drug within the conjugate. In certain embodiments, the conjugates retain from about 5 % up to about 100% of the biological activity, from 5% up to about 95%, from about 5% up to about 90%, from about 5%> up to about 80%, up to about 70%, up to about 60% or up to about 50% of the biological activity of parent drug, hi certain embodiment the biological activity of the drug in the conjugate exceeds that of parent drug.
- the drug-substrate conjugates are selectively trapped or accumulated in target cells.
- the conjugates are selectively trapped or accumulated in target cells due to phosphorylation of the substrate in the conjugates by a kinase whose activity is involved in the condition being treated.
- doses of the drug-substrate conjugate required to elicit the same effective amount of therapeutic response as the parent drug can be reduced thereby resulting in a reduction of undesirable side effects.
- This allows for an increase in the duration of therapy, which is highly desirable in chronic disease settings.
- the standard drug dose in conjugate form can be increased without exceeding the tolerability of undesirable side effects to allow for more aggressive treatment.
- molecules capable of eliciting a desired pharmacological response but which elicit unacceptable side effects at doses below that required for an effective amount of therapeutic response may be transformed by conjugation into a molecule useful in the treatment of an ACAMPS condition.
- trapping or accumulation of drug conjugates by phosphorylation may prevent the efflux of cancer drugs such as vinca alkaloids, epipodophyllotoxins, taxanes/taxoids, and anthracyclines, by the membrane transporter P-glycoprotein, thus, preventing a major form of MDR.
- the substrate moiety in the conjugate may be any substrate for a kinase other than a hexokinase, a protein kinase or a lipid kinase that is overexpressed, overactive or that exhibits undesired activity in a target system.
- the action of the kinase on the substrate results in a modified conjugate wherein significant fraction of the activity of the drug moiety as well as the substrate moiety is retained.
- a target system e.g. cell, tissue or organ
- the drug- substrate conjugate is less able to exit the cell in comparison to the unmodified drug.
- the drug-substrate conjugates exhibit improved cytotoxic selectivity index over the parent drug.
- the drug- substrate conjugates exhibit improved solubility over the parent drug.
- the conjugates exhibit better serum stability than the parent drug. In certain embodiments, the conjugates exhibit better shelf life than the parent drug.
- the conjugates for use in the methods and compositions provided herein have the formula (1): (D) d -(L) q -(S) t (1) or a pharmaceutically acceptable derivative thereof, wherein D is a drug moiety; d is 1-6, or is 1 or 2; L is a non- releasing linker; q is 0 to 6, or is 0 or 1; S is a substrate for a kinase other than a hexokinase, a protein kinase or a lipid kinase; and t is 1 to 6, or is 1 or 2, or is 1.
- the drug moiety is covalently attached, optionally via a non-releasing linker, to the substrate.
- conjugates that contain two drug moieties, which are the same or different conjugated to the substrate moiety(s) or non-releasing linked thereto can be at various positions of the substrate.
- the conjugates have formula (2): D-L-S, (2) or a pharmaceutically acceptable derivative thereof, where the variables are as defined elsewhere herein.
- Exemplary substrates, drug moieties, linkers and exemplary conjugates are described in further detail below. It is intended herein that conjugates resulting from all combinations and/or permutations of the groups recited below for the variables of formulae (1) and (2) are encompassed within the instant disclosure.
- Drug Moiety The conjugates provided herein are intended for modifying a variety of biological responses.
- the drug moiety may be any molecule, as well as a binding portion, fragment or derivative thereof that is capable of modulating a biological process other than compounds containing a carboranyl, hydroxyboryl or rare earth cryptate containing moiety.
- the drug moiety encompasses any molecule that elicits a pharmacological response that may be used for the treatment or prevention of a disease.
- the drug moities are any moities, including proteins and polypeptides, small molecules and other molecules that possess or potentiate a desired biological activity.
- Such molecules include cytotoxic agents, such as, but are not limited to, a toxin such as abrin, ricin A, pseudomonas exotoxin, shiga toxin, diphtheria toxin and other such toxins and toxic portions and/or subunits or chains thereof; proteins such as, but not limited to, tumor necrosis factor, ⁇ -interferon, ⁇ - interferon, nerve growth factor, platelet derived growth factor, tissue plasminogen activator; or, biological response modifiers such as, for example, lymphokines, interleukin- 1 (IL-1), interleukin-2 (IL-2), interleukin-6 (IL-6), granulocyte macrophage colony stimulating factor (GMCSF), granulocyte colony stimulating factor (G-CSF), erythropoietin (EPO), pro-coagulants such as tissue factor and tissue factor variants, pro-apoptotic agents such FAS-ligand, fibroblast growth factors (FGF), nerve growth factor and other growth factors.
- the drug moiety of the drug conjugate may be derived from a naturally occurring or synthetic compound that may be obtained from a wide variety of sources, including libraries of synthetic or natural compounds. For example, numerous means are available for random and directed synthesis of a wide variety of organic compounds and biomolecules. Alternatively, libraries of natural compounds in the form of bacterial, fungal, plant and animal extracts are available or readily produced. Additionally, natural or synthetically produced libraries and compounds are readily modified through conventional chemical, physical and biochemical means, and may be used to produce combinatorial libraries. Known pharmacological agents may be subj ected to directed or random chemical modifications, such as acylation, alkylation, esterification, amidification, etc., to produce structural analogs.
- the drug moiety may be obtained from a library of naturally occurring or synthetic molecules, including a library of compounds produced through combinatorial means (i.e., a compound diversity combinatorial library). When obtained from such libraries, the drug moiety employed will have demonstrated some desirable activity in an appropriate screening assay for the activity. Combinatorial libraries, as well as methods for the production and screening, are known in the art.
- the drug moiety is a chemotherapeutic agent.
- chemotherapeutic agents include but are not limited to anti-infective agents, antihelminthic, antiprotozoal agents, antimalarial agents, antiamebic agents, antileiscmanial drugs, antitrichomonal agents, antitrypanosomal agents, sulfonamides, antimycobacterial drugs, or antiviral chemotherapeutics.
- Chemotherapeutic agents may also be antineoplastic agents or cytotoxic drugs, such as alkylating agents, plant alkaloids, antimetabolites, antibiotics, tubulin or microtubule binding agents and other anticellular proliferative agents.
- drugs of interest include but are not limited to central nervous system depressants and stimulants, respiratory tract drugs, pharmacodynamic agents, such as histamines and antihistamines, cardiovascular drugs, blood and hemopoietic system drugs, gastrointestinal tract drugs, and locally acting drugs including chemotherapeutic agents.
- Drug compounds of interest from which drug moieties may be derived are also listed in: Goodman & Gilman's, The Pharmacological Basis of Therapeutics (9th Ed) (Goodman, et al, eds.) (McGraw-Hill) (1996); and 1999 Physician's Desk Reference (1998) and Chu, E.; DeVita, V.T. Physicians' Cancer Chemotherapy Drug Manual 2003, Jones and Bartlett Publishers.
- Classes of cytotoxic agents for use herein include, for example, the a) anthracycline family of drugs, b) vinca alkaloid drugs, c) mitomycins, d) bleomycins, e) cytotoxic nucleosides, f) pteridine family of drugs, g) diynenes, h) estramustine, i) cyclophosphamide, j) taxanes, k) podophyllotoxins, 1) maytansanoids, m) epothilones, and n) combretastatin and analogs.
- the drug moiety is selected from a) doxorubicin, b) carminomycin, c) daunorubicin, d) aminopterin, e) methotrexate, f) methopterin, g) dichloromethotrexate, h) mitomycin C, i) porfiromycin, j) 5-fluorouracil, k) 6- mercaptopurine, 1) cytosine arabinoside, m) podophyllotoxin, n) etoposide, o) etoposide phosphate, p) melphalan, q) vinblastine, r) vincristine, s) leurosidine, t) vindesine, u) estramustine, v) cisplatin, w) cyclophosphamide, x) paclitaxel y) leurositte, z) 4-desacetylvinblastine,
- linking moiety is used to attach the drug covalently to the substrate.
- linker and “linking moiety” herein refer to any moiety that non-releasably connects the substrate moiety and drug moiety of the conjugate to one another.
- the linking moiety can be a covalent bond or a chemical functional group that directly connects the drug moiety to the substrate.
- the linking moiety can contain a series of covalently bonded atoms and their substituents which are collectively refened to as a linking group.
- Linking moieties are characterized by a first covalent bond or a chemical functional group that connects the drug moiety to a first end of the linker group and a second covalent bond or chemical functional group that connects the second end of the linker group to the substrate.
- the first and second functionality which independently may or may not be present, and the linker group are collectively refened to as the linker moiety.
- the linker moiety is defined by the linking group, the first functionality if present and the second functionality if present.
- the linker moiety contains atoms interposed between the drug moiety and substrate, independent of the source of these atoms and the reaction sequence used to synthesize the conjugate.
- the linker moiety is chosen to serve as a spacer between the drug and the substrate, to remove or relieve steric hindrance that may interfere with substrate activity and/or the pharmacological effect of the drug.
- the linker moiety can also be chosen based on its effect on the hydrophobicity of the drug-substrate conjugate, to improve passive diffusion into the target cells or tissue or to improve pharmacokinetic or pharmacodynamic properties.
- linking moieties of interest can vary widely depending on the nature of the drug and substrate moieties.
- the linking moiety is biologically inert.
- Precursors for a variety of linkers are known to those of skill in the art, which may be used in the synthesis of conjugates provided herein.
- Linker precursors are desirably synthetically accessible and provide shelf-stable products; and do not add any intrinsic biological activity that interferes with the conjugates activity. When incorporated into the conjugates, they can add desirable properties such as increasing solubility or stability to the conjugate.
- Any bifunctional linker precursor in certain embodiments, heterobifunctional linking precursors that can form a non-releasable bond between the drug moiety and the substrate moiety, when incorporated into the conjugates, can be used in the synthesis of conjugates provided herein.
- a linker precusor can be homobifunctional.
- one or more of substrate moieties are linked to one or more drug moieties via a multifunctional linking moiety.
- Each of these functional groups can form a covalent linkage to a suitable functional group on the substrate or the drug to get a drug-linker or a substrate-linker construct.
- amino, hydroxy and hydrazino groups can each form a covalent bond with a reactive carboxyl group (e.g., a carboxyhc acid chloride or activated ester such as an N-hydroxysuccinimide ester (NHS)).
- a reactive carboxyl group e.g., a carboxyhc acid chloride or activated ester such as an N-hydroxysuccinimide ester (NHS)
- Other suitable bond forming groups are well-known in the art.
- the linking moiety, L can include linear or acyclic portions, cyclic portions, aromatic rings or combinations thereof.
- the linking moiety can have f om 1 to 100 main chain atoms other than hydrogen atoms, selected from C, N, O, S, P and Si. hi certain embodiments the linking moiety contains up to 50 main chain atoms other than hydrogen, up to 40, up to 30, up to 20, up to 15, up to 10, up to 5, up to 2 main chain atoms other than hydrogen. In certain embodiments the linking moiety is acyclic. In certain embodiments, the linking moieties contain oligomers of ethylene glycol or alkylene chains or mixtures thereof.
- linking moieties are, in certain embodiments, attached to the substrate via either an alkyl or amide connection, hi certain embodiments, the drug moiety is attached to the first end of the linker via an amide, sulfonamide, or ether connection. Illustrative synthetic schemes for forming such conjugates are discussed elsewhere herein for exemplary linkers for the conjugates provided herein.
- the linking moiety is a covalent bond between the drug moiety and the substrate moiety. Typically, this attachment is accomplished via coupling of a functional group on the drug with a compatible functional group on the substrate.
- the drug has an isocyanate, isothiocyanate or carboxyhc acid functional group that is used to attach the drug to a hydroxy or amino group present on the substrate moiety to form a carbamate, thiocarbamate, urea or thiourea linkage between the components.
- a variety of linking moieties depending on the nature of the drug and substrate moieties can be used in the conjugates provided herein. Suitable linking moieties can be selected by one of skill in the art based on the criteria set forth herein.
- the linking moiety can be selected by the following procedure: A first end of a linker precursor is used in synthesizing a linker-nucleoside construct according to the procedures illustrated by Schemess 2, 3 and 6 and described herein. It is subjected to a first test which determines nucleoside kinase activity. In one embodiment, the method of the first test is by observing ADP formation which is an obligatory product of phospho group transfer from ATP using a coupled enzyme assay. ADP, formed from substrate phosphorylation (in conjugate form), is used by pyruvate kinase to generate pyruvate from phospoenolpyruvate which in turn is converted to lactate by lactate dehydrogenase.
- the lactate results in the consumption of NADH which is followed spectrophotometrically.
- the rate of substrate phosphorylation (in conjugate form) is then directly related to the rate of decrease in the observed NADH signal.
- a linker of appropriate length and a nucleoside or nucleoside analog is found with an effective amount of kinase activity which may be expected to be retained in the drug conjugate.
- the linker found in the first test is subjected to a second test in certain embodiments, to determine suitability of the linker by connecting a second end of the linker precursor to a drug moiety.
- the site on the drug wherein the second end of the linker is attached is known to tolerate modification or may be shown to tolerate modification through a suitable functional group either pre-existing on the drug or on an analog thereof that is known to have an effective amount of the pharmacological activity of the parent drug.
- paclitaxel modifications at C7, CIO and C3 '-N are known to be tolerated as described in Springfield, Fortschr. Chem. Org. Naturst. 2002:84, 53-225, the disclosure of which is incorporated by reference.
- camptothecin analogs with suitable functionalities for linker attachment are described in Wall, et. al, J. Med. Chem. 1993: 36, 2689-2700 whose disclosure is incorporated by reference.
- conjugates based upon Vinblastine are prepared by use of the natural product O -deacetyl Vinblastine or Vindesine prepared according to Barnett, et. al. J. Med. Chem. 1978: 21, 88-96, whose disclosure is incorporated by reference.
- Vindesine and O 4 -deacetyl Vinblastine are characterized by a free hydroxyl group at C-4.
- vinblastine conjugates are prepared from O 4 - deacetyl-3-de-(methoxycarbonyl)-vinblastin-3-yl carbonyl azide through condensation with amines as described in Lavielle, et.al. J. Med. Chem. 1991: 34, 1998-2003, the disclosure of which is incorporated by reference.
- a second test of a drug-linker construct may then be determined by a functional assay which is predictive of pharmacological activity.
- a functional assay which is predictive of pharmacological activity.
- microtubule stabilization for paclitaxel drug linker constructs or microtubule disruption by vinblastine drug-linker constructs is determined with a tubulin polymerization assay as described in Barron et. al. Anal. Biochem. 2003:315, 49-56 the disclosure of which is incorporated by reference.
- Tubulin assembly or inhibition thereof can be monitored by fluorescence using the CytoDYNAMIX ScreenTM 10 kit available from Cytoskeleton (1830 S. Acoma St., Denver, CO).
- the kit is based upon an increase in quantum yield of florescence upon binding of a fluorophore to tubulin and microtubules and a 10X difference in affinity for microtubules compared to tubulin.
- Compounds such as paclitaxel which enhance tubulin assembly will therefore give an increase in emission whereas compounds such as vinblastine which inhibit tubulin assembly will give a decrease in emission.
- Tubulin assembly or inhibition thereof can also be monitored by light scattering which is approximated by the apparent absorption at 350 nm.
- doxorubicin-linker constructs can be screened by monitoring alteration in the ability of Topoisomerase II to catalyze the formation of relaxed conformation DNA from a super-coiled plasmid.
- a functional assay for camptothecin drug-linker constructs depends on Topoisomerase I binding to DNA an example of which is given in E>emarquay, Anti-Cancer Drugs 2001:12, 9-19 the disclosure of which is incorporated by reference. It should be appreciated that an appropriate linker may also be found by interchanging the order of the first and second tests.
- the linking moiety in the conjugates provided herein contains an alkylene chain containing from 1 up to 50 main chain atoms other than hydrogen. In certain embodiments, the alkylene chain contains 2, 3, 4, 5, 6, 7, 8, 9, 10 or 15 main chain atoms other than hydrogen. In other embodiments, the alkylene chain contains 3, 4, 5, 6, 7, 8 or 9 main chain atoms other than hydrogen.
- the linking moiety in the conjugates provided herein contains a polyethylene glycol (PEG) chain.
- PEG polyethylene glycol
- the PEGs for use herein can contain up to 50 main chain atoms other than hydrogen, hi certain embodiments, the PEG contains 5, 11, 13, 14, 22 or 29 main chain atoms other than hydrogen. In certain embodiments, the PEG contains 5, 11, 13 or 29 main chain atoms other than hydrogen.
- the linker moiety contains a combination of alkylene, PEG and maleimide units in the chain.
- the substrate moiety may be any substrate for a kinase that is overexpressed, overactive or that exhibits undesired activity in a target system, wherein the kinase is other than a hexokinase, a protein kinase or a lipid kinase.
- the substrate has a molecular weight between about 50 amu and 1000 amu.
- the kinase is present at a higher concentration or operates at a higher activity, or the activity is undesired or persistent in a cell type that contributes to the genesis or maintenance of the condition being treated in the target cell in comparison to other cells.
- Addition of a phosphate group by action of the kinase on the substrate confers a negative charge to the conjugate, thus trapping or accumulating the conjugate inside the targeted cells at concentrations higher than will be achieved in other cells not involved with the condition being treated.
- the action of the kinase on the substrate results in a modified conjugate in the target system (e.g.
- the kinase is associated with an ACAMPS-related condition.
- the substrate is a substrate for a kinase such as a nucleoside kinase.
- the substrate is a substrate for a kinase such as thymidine kinase, viral thymidine kinase, human thymidine kinase TK-1, deoxycytidine kinase, deoxyguanosine kinase.
- the substrate is a substrate for viral thymidine kinase, human thymidine kinase TK-1.
- the substrate is selected from nucleosides and their natural and non-natural analogs. Examples of nucleosides for use as substrates in the conjugates herein, but are not limited to, cytidine, uridine, thymidine, guanosine, adenosine, or derivatives thereof.
- the substrate is a nucleoside or nucleoside analog for thymidine kinase, viral thymidine kinase, TK-1, deoxycytidine kinase or deoxyguanosine kinase known or found to be activated in cells associated with ACAMPS-related conditions.
- Natural and non-natural nucleoside analogs are contemplated herein.
- the substrate is a nucleoside or nucleoside base which is converted to a substrate of thymidine kinase, viral thymidine kinase, TK-1 or deoxycytidine kinase by the action of thymidine phosphorylase or cytidine deaminase.
- Table 3 shows illustrative examples of known pyrimidine nucleoside analogs which are substrates for thymidine kinase, and deoxycytidine kinase; and purine analogs which are substrates for deoxycytidine kinase and deoxyguanosine kinase, for use in the conjugates and methods provided herein.
- D- and L-pyrimidine and purine nucleoside analogs are contemplated substrates.
- Other contemplated substrates include pyrimidine analogs covalently linked to a non-deoxy-ribose sugar having an anomeric carbon ( ⁇ and ⁇ anomers).
- Such substrates include but are not limited to ⁇ -L-2',3'-dideoxy-3'-thiacytidine (3TC), ⁇ -L- 1,3-dioxolane-cytidine (L-OddC), (North)-methanocarba-thymidine and analogs thereof.
- Still other contemplated substrates are acyclic and carbocyclic analogs of guanosine which are known in the art as substrates for viral thymidine kinase. (For a review see De Clerq, D.E. et al., Nucleosides, Nucleotides & Nucleic Acids 20:271- 285 (2001)).
- the substrate is a nucleoside or nucleoside analog substrate for a thymidine kinase (TK).
- TK thymidine kinase
- the TK is active within a cell type that contributes to the genesis or maintenance of a disease. Phosphorylation of the nucleoside or nucleoside analog by the TK leads to trapping or accumulation of the conjugate within the targeted cell type due to the drug-conjugate acquiring a negative charge. Due to failure to introduce the foreign gene into every cancer cell, previous efforts with gene therapy to effect targeting of a nucleoside prodrug to tumors by introducing a foreign TK into the cancer cell has not lead to clinical success (for review see Fillat, et. al. Cu ⁇ . Gene Ther. 2003: 3:13-26).
- the substrate is a nucleoside or nucleoside analog substrate for human thymidine TK-1 or a viral TK.
- the drug- nucleoside or drug- nucleoside analog conjugate in one embodiment, is effective in treating cancer through phosphorylation of the drug- nucleoside or drug-nucleoside analog conjugate by TK-1, leading in certain embodiment, to trapping or accumulation of the conjugate and hence the anti-cancer agent within the cancer cell. Therefore, trapping or accumulation is responsible for the therapeutic effect of these conjugates in the treatment of cancer.
- the therapeutic effect is due to the accumulated anti-cancer drug which is active in the conjugate and is not due to the nucleoside or nucleoside analog, which simply serves as a substrate for the targeting enzyme.
- the therapeutic effect of the drug conjugate is not dependent on release of free drug.
- no further intervention of intracellular proteins is required for activation of the drug within the conjugate. Further action by thymidylate kinase and incorporation into DNA is not precluded as an additional enhancement of the therapeutic effect of the drug conjugate in the treatment of cancer.
- the drug moiety and/or the substrate moiety in the conjugate can be present in a form of a pharmaceutically acceptable derivative that renders the conjugate biologically inactive.
- the inactive drug-substrate conjugate can be converted to the active drug-substrate conjugate under physiological conditions or by intracellular proteins without having the need to cleave the drug-substrate conjugate.
- the anti-cancer drug-nucleoside conjugates are effective in treating viral infections, such as DNA or RNA viral infections, by using a viral TK which results in trapping or accumulation of a drug which is responsible for the therapeutic effect of these conjugates.
- viral infections such as DNA or RNA viral infections
- a cell infected with a RNA or DNA virus is distinguished by a TK activity introduced by the virus into the cell.
- the viruses include, but are not limited to, HSV-1, HSV-2, VZV, EBV, CMV, HTLV-1 and HIV.
- a thymidine kinase that is abe ⁇ ant in a cell type involved in the genesis or maintenance of the condition is used to target the cell types with a conjugate of a drug to a nucleoside or nucleoside analog that is a substrate of the kinase.
- Addition of a phosphate group by action of the thymidine kinase on the nucleoside or nucleoside analog confers a negative charge to the conjugate thus trapping or accumulating the drug inside the targeted cells at concentrations higher than will be achieved in other cells not involved with the condition being treated.
- the therapeutic effect is due to the accumulated drug which is active in the conjugate and is not due to the nucleoside which simply serves as a substrate for the targeted enzyme or release of free drug. Therefore, as discussed above, no further intervention of intracellular proteins is required for activation of the drug within the conjugate though cleavage of the linker to give free drug.
- the intracellular proteins may activate the conjugate by converting the conjugate in the active form.
- the nucleoside conferring an additional therapeutic effect is not precluded. Further action by thymidylate kinase and incorporation into DNA is not precluded as an additional enhancement of the therapeutic effect of the drug conjugate in the treatment of viral infections.
- Thymidylate synthase (TS) and thymidine kinase (TK) are the key enzymes in the synthesis of pyrimidine nucleotides required for cell division.
- TS catalyzes the reductive methylation of dUMP to dTMP.
- TK directly catalyzes the phosphorylation of thymidine released from cells by DNA catabolism.
- Both enzymes are highly expressed in breast, gastric, ovarian, colorectal and bladder carcinomas to name a few.
- anti-metabolite drugs that target TS notably 5-fluorouracil (5-FU).
- the substrate is a substrate for deoxycytidine kinase, including, but not limited to, cytidine and uridine derivatives.
- Deoxycytidine kinase is known to phosphorylate cytostatic drugs (e.g., gemcitabine) for activation. It is contemplated that dCK may have greater tolerance towards uridine derivatives, in comparison to TK. For example, 5-amino-uridine which has been shown to have a 17 % activity towards dCK may be used as a substrate, utilizing the amino group as a potential site for linkage to the conjugate. Furthermore, dCK is known to have greater tolerance towards carbohydrate modifications than thymidine kinase. For example, dCK shows only modest discrimination between the natural nucleoside ⁇ -D-cytidine and its enantiomer ⁇ -L-cytidine.
- Contemplated substrates include, but are not limited to, cytostatic nucleosides known in the art to be substrates for TK and/or dCK and anti-viral nucleosides known in the art to be substrates for viral thymidine kinase. Additional contemplated substrates include but are not limited to ⁇ -D-, ⁇ -L-, ⁇ -D-, and ⁇ -L-nucleoside analogs and acyclic carbohydrate analogs, which may also utilize the acyclic carbohydrate as a potential site for linkage to the conjugate. 4.
- the conjugates provided herein contain a substrate that is a substrate for a nucleoside kinase and the conjugates have formula: N-L-D or a pharmaceutically acceptable derivative thereof, wherein N is a natural or non- natural nucleoside; L, which may or may not be present, is a non-releasing linker and D is a drug moiety.
- the drug is non-releasably linked to a carbohydrate or a base moiety of the nucleoside.
- the drug is linked to the carbohydrate moiety of the nucleoside.
- the drug is linked to the base of the nucleoside moiety of the nucleoside.
- the conjugates have a formula: S c -P ⁇ L-D, or a pharmaceutically acceptable derivative thereof, wherein S c is ribose, deoxyribose or analog thereof; P 1 is a purine, pyrimidine or analog thereof and other variables are as defined herein.
- the conjugates have a formula: P ⁇ S c -L-D, or a pharmaceutically acceptable derivative thereof, wherein S c is ribose, deoxyribose or analog thereof; P 1 is a purine, pyrimidine or analog thereof and other variables are as defined herein.
- the conjugates provided herein have formula:
- R 1 , R 3 , R 4 and R 5 are each independently Y, H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl; R 2 is Y, H, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl,
- R is Y, H or Cl-6 alkyl, C2-6 alkenyl or C2-6 alkynyl; W is CR e R f or O; R e and R f are each independently H or Cl-6 alkyl; Y is a linker-drug construct L-D, wherein L, which may or may not be present, is a non-releasing linker and D a drug moiety; R, R 1 and R 3 -R 5 are selected such that at least one of R, R 1 and R 3 -R 5 is Y and at least one of R, R 1 and R 3 -R 5 is OH; R -R 5 and R are unsubstituted or substituted with one or more substituents, in one embodiment 1-4 substituents, in another embodiment 1 or 2 substituents, each independently selected from Q 1 .
- Q 1 is halo, pseudohalo, hydroxy, oxo, thia, nitrile, nitro, formyl, mercapto, hydroxycarbonyl, hydroxycarbonylalkyl, alkyl, haloalkyl, polyhaloalkyl, aminoalkyl, diaminoalkyl, alkenyl containing 1 to 2 double bonds, alkynyl containing 1 to 2 triple bonds, cycloalkyl, cycloalkylalkyl, heterocyclyl, heterocyclylalk yl, aryl, heteroaryl, aralkyl, aralkenyl, aralkynyl, heteroarylalkyl, trialkylsilyl, dialkylarylsilyl, alkyldiarylsilyl, triarylsilyl, alkylidene, arylalkylidene, alkylcarbonyl, arylcarbonyl, heteroarylcarbonyl, alk
- dialkylphosphonyl alkylarylphosphonyl, diarylphosphonyl, hydroxyphosphonyl, alkylthio, arylthio, perfluoroalkylthio, hydroxycarbonylalkylthio, thiocyano, isothiocyano, alkylsulfinyloxy, alkylsulfonyloxy, arylsulfinyloxy, arylsulfonyloxy, hydroxysulfonyloxy, alkoxysulfonyloxy, aminosulfonyloxy, alkylaminosulfonyloxy, dialkylaminosulfonyloxy, arylaminosulfonyloxy, diary
- R 1 is, H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl.
- R 1 is OH.
- R 3 is Y, H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl.
- R 3 is hydroxy.
- R 3 is Y.
- R 4 is Y, H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl.
- R 4 is hydroxy.
- R 4 is H.
- R 4 is Y.
- R 5 is Y, H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl.
- R 5 is H.
- R 5 is Y.
- R 2 is Y, H, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, aryl or heteroaryl.
- R is H.
- R is Cl-6 alkyl.
- R 2 is methyl.
- R 2 is Y.
- R is Y, H or Cl-6 alkyl.
- R is H. hi another embodiment, R is Cl-6 alkyl.
- R is Y.
- W is CR e R f or O. In certain embodiments, W is O. hi certain embodiments, W is CR ⁇ .
- R e and R f are each H.
- Y is -L-D, where L is a non-releasing linker and D is a drug moiety.
- Y is D.
- -L- is -O- i-, where Li is non-releasing linker.
- -Li- is selected from a mono or bifunctional alkelene chain or mono or bifunctional polyethylene glycol chain.
- the conjugates have formula:
- the conjugates have formula:
- conjugates have formula: 2005/030258
- R la , R 3a , R 4a and R 5a are each independently Y; H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl;
- R 2a is Y, H, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, hydroxy, aryl or heteroaryl or halo ;
- ⁇ is a linker-drug construct L-D, wherein L, which may or may not be present, is a non-releasing linker and D is a drug moiety;
- R a and R b are each independently Y, H, or Cl-6 alkyl;
- R d is H or Cl-6 alkyl;
- a is CR ⁇ or O;
- R e and R f are each independently H or Cl-6 alkyl;
- R a and R b are selected such that at least one of R la -R 5a , R a and R b is Y and at least one of R la , R 3a - R 5a is OH; R la -R 5a , R a , R b and R d are unsubstituted or substituted with one or more substituents, in one embodiment 1-4 substituents, in another embodiment 1 or 2 substituents, selected from Q 1 .
- R la , R 3a , R 4a and R 5a are each independently Y, H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl.
- R la is Y, H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl.
- R la is Y or hydroxy.
- R is Y.
- R la is hydroxy.
- R 3a is Y or hydroxy, In certain embodiments, R 3a is Y. In certain embodiments, R 3a is hydroxy.
- R 4a is Y or hydroxy. In certain embodiments, R 4a is Y. In certain embodiments, R 4a hydroxy.
- R 5a is Y or hydroxy.
- R 5a is Y. In certain embodiments, R 5a is H. In one embodiment, R 2a is Y, H or Cl-6 alkyl. hi other embodiments, R 2a is Y ⁇ In other embodiments, R 2a is H. In other embodiment, R a and R are each independently Y, H or Cl-6 alkyl. In other embodiment, R a and R b are each independently H. In other embodiment, W a is O. In certain embodiments, Y is -L-D, where L is a non-releasing linker and D is a drug moiety, hi other embodiments, Y is D.
- -L- is — 0-(L ⁇ )q— , where Li is non-releasing linker and q is 0-2.
- -L ⁇ - is selected from a mono or bifunctional alkelene chain or mono or bifunctional polyethylene glycol chain.
- the conjugates have formula:
- R 3 and R 4b are each independently Y, H, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl;
- R is Y, H, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, aryl, heteroaryl or halo;
- Y is a linker-drug construct L-D, wherein L, which may or may not be present, is a non-releasing linker and D is a drug moiety;
- W b is CR e R f or O;
- R e and R f are each independently H or C 1 -6 alkyl;
- R a and R are independently Y, H, or Cl-6 alkyl;
- R d is H or Cl-6 alkyl;
- R l -R 4b , R a and R b are selected such that at least one of
- R l -R 4b , R a , R b and R d are unsubstituted or substituted with one or more substituents, in one embodiment 1-4 substituents, in another embodiment 1 or 2 substituents, selected from Q 1 .
- R 2b is Y or H.
- R 2b is Y.
- the conjugates have formula:
- R 3c and R 4c are each independently Y, H, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl; R 2 is Y, H, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl,
- R q is Y, H or Cl-6 alkyl
- Y is a linker-drug construct L-D, wherein L, which may or may not be present, is a non-releasing linker and D is a drug moiety
- W c is CR e R f or O
- R e and R f are each independently H or Cl-6 alkyl
- R lc -R 4c and R q are selected such that at least one of R lc -R 4 ° or R q is Y;
- R lc -R 4c and R q are unsubstituted or substituted with one or more substituents, in one embodiment 1-4 substituents, in another embodiment 1 or 2 substituents, selected from Q 1 .
- R lc is Y. In certain embodiments, R 2c is Y. In certain embodiments, R q is Y. In another embodiment, the conjugates have formula: or a pharmaceutically acceptable derivative thereof, wherein R ld , R 3d , R 4d and R 5d are each independently Y, H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl; R 7d is Y, H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl, C2-6 alkynyl, thioalkyl orNR a R ; R.
- R sd is Y, H, alkyl, halo, SR d or NR a R ;
- R. 9d is Y, H, or Cl-6 alkyl;
- V is a linker-drug construct L-D, wherein L, which may or may not be present, is a non-releasing linker and D is a drug moiety;
- R ld -R 9d are selected such that at least one of R ld , R 3d , R 4d , R 5d or R 7d is Y and at least one of R ld , R 3d , R 4d , R 5d or R 7d is OH;
- R_ a and R are each independently Y, H, or Cl-6 alkyl;
- R d is H or Cl-6 alkyl;
- W d is CR e R f or O;
- R e and R f are each independently H or Cl-6 alkyl;
- R »5e a re each independently Y, H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl or C2-6 alkynyl;
- R 2e is Y, H, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl, C2-6 alkynyl or C3-6 cycloalkyl,;
- R ,8 o e e is Y, H, alkyl, halo, SR ⁇ or NR a a ⁇ Rj°.;
- R 9e is Y, H, or Cl-6 alkyl;
- Y is a linker-drug construct L-D, wherein L, which may or may not be present, is a non-releasing linker and D is a drug moiety;
- R le -R 5e , R 8e and R 9e are selected such that at least one of R le - R 5e ,
- R 6f is Cl-10 alkyl and optionally containing a heteroatom, C2-10 alkenyl or C2-10 alkynyl; 1 f * R is Y or hydroxy; R is Y, H, hydroxy, halo, azido, Cl-6 alkyl and optionally containing a heteroatom, C2-6 alkenyl, C2-6 alkynyl, SR d orNR a R c ; R 8f is Y, H, alkyl, SR d , halo or NR a R b ; R 9e is Y, H, or Cl-6 alkyl; Y is a linker-drug construct L-D, wherein L, which may or may not be present, is a non-releasing linker and D is a drug moiety; R 7f , R 8f and R 9f are selected such that at least one of R lf , R 7f , R 8f and R 9f is selected such that at least one of R lf
- R 3 , R 4 and R 5 are each independently H, optionally substituted hydroxy, halo, azido, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C2-6 alkenyl or alkynyl; wherein R 2 is H, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C2-6 alkenyl or alkynyl, C3-6 cycloalkyl, optionally substituted hydroxy, aryl or heteroaryl; formula (4): wherein R , R and R are independently H, optionally substituted hydroxy, halo, azido, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C2-6 alkenyl or alkynyl; wherein R is H or Cl-6 alkyl; formula (5):
- R 4 is H, optionally substituted hydroxy, halo, azido, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C2-6 alkenyl or alkynyl; wherein R is H, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C2-6 alkenyl or alkynyl, C3-6 cycloalkyl, optionally substituted hydroxy, aryl or heteroaryl; wherein R is H or Cl-6 alkyl; wherein X is O or is absent; formula (6):
- R 3 , R 4 and R 5 are independently H, optionally substituted hydroxy, halo, azido, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C2-6 alkenyl or alkynyl; wherein each R' and R" are independently H or lower alkyl; formula (7):
- R 3 and R 4 are independently H, optionally substituted, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C2-6 alkenyl or alkynyl; wherein R' and R' ' are independently H or lower alkyl; formula (8):
- R and R are independently H, optionally substituted hydroxy, halo, azido, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C2-6 alkenyl or alkynyl; wherein R 2 is H, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C2-6 alkenyl or alkynyl, C3-6 cycloalkyl, optionally substituted hydroxy, aryl or heteroaryl; wherein R' and R' ' are independently H or lower alkyl; wherein X is O or is absent; formula (9): wherein R 3 , R 4 and R 5 are independently H, optionally substituted hydroxy, halo, azido, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C2-6 alkenyl or alkynyl; wherein R 7 is H, optionally substituted hydroxy, halo, azido, Cl-6 optionally substituted alkyl and optionally containing a heteroatom
- R 3 , R and R 5 are independently H, optionally substituted hydroxy, halo, azido, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C2-6 alkenyl or alkynyl; wherein R 8 is H, halo, NR'R", wherein R' and R" are independently H or lower alkyl, or SR"", wherein R'" is H or lower alkyl; wherein R is H or lower alkyl; wherein W is C or O; wherein Z 1 , Z 2 and Z 3 are independently C or N; or formula (11): or a pharmaceutically acceptable derivative thereof, wherein R is an acyclic Cl-10 alkyl optionally substituted alkyl and optionally containing a heteroatom, or C2-10 alkenyl or alkynyl; wherein R 7 is H, optionally substituted hydroxy, halo, azido, Cl-6 optionally substituted alkyl and optionally containing a heteroatom, C3-6 cycloal
- the linker-drug moiety may be attached at other positions of the pyrimidine or purine nucleoside analog.
- the conjugates are paclitaxel-thymidine conjugates.
- the paclitaxel-thymidine conjugates contain a non-releasing linker between paclitaxel and thymidine.
- the linker contains an alkylene chain or PEG chain.
- the linker is bonded to thymidine via a covalent bond.
- the linker is bonded to paclitaxel via a first functionality.
- the first functionality is a carbamate.
- the substrate is conjugated to paclitaxel at C7 position, hi one embodiment, the paclitaxel-thymidine conjugates have formula:
- the conjugates are paclitaxel-thymidine conjugates in which the linker contains an alkylene chain or PEG chain and is bonded to thymidine via a covalent bond and to paclitaxel via a carbamate group at CIO.
- the paclitaxel-thymidine conjugates have formula:
- the paclitaxel-thymidine conjugates have formula:
- the conjugates are vinblastine-thymidine conjugates.
- the conjugates are desacetyl vinblastine-thymidine conjugates.
- the vinblastine-thymidine conjugates contain a non-releasing linker between vinblastine and thymidine.
- the linker contains an alkylene chain or PEG chain.
- the linker is bonded to thymidine via a covalent bond.
- the linker is bonded to C3 of vinblastine via an amide group.
- the vinblastine- thymidine conjugates have formula:
- the conjugates are doxorubicin-thymidine conjugates.
- the doxorubicin-thymidine conjugates contain a non- releasing linker between doxorubicin and thymidine.
- the linker contains one or more groups containing maleimide, an alkylene chain and PEG chain.
- the linker is bonded to thymidine via a covalent bond.
- the linker is bonded to doxorubicin via an amino group.
- the doxorubicin-thymidine conjugates have formula:
- conjugates are each independently selected from alkylene or PEG or a pharmaceutically acceptable derivative thereof. More examples of conjugates provided herein are provided in Table ? C. Preparation of the conjugates
- the conjugates provided herein can be prepared using any convenient methodology. In one approach, the conjugates are produced using a rational approach. In a rational approach, the conjugates are constructed from their individual components (e.g., drug, linker precursor and substrate). The components can be covalently bonded to one another through functional groups known in the art. Furthermore, the particular portion of the different components modified to provide for covalent linkage will be chosen so as not to substantially adversely interfere with that component' s desired binding activity.
- the functional groups can be present on the components or introduced onto the components using one or more steps, such as oxidation, reduction, cleavage reactions and the like.
- Examples of functional groups that can be used in covalently bonding the components to produce the conjugate include but are not limited to hydroxy, sulfhydryl, amino, carbonyl, and the like.
- certain moieties on the components may be protected using blocking groups, as is known in the art, see, e.g., Green & Wuts, Protective Groups in Organic Synthesis (John Wiley & Sons) (1991).
- Scheme 1 illustrates the general syntheses of linker nucleoside constructs with linker attachment at N-3 of a pyrimidine base of a nucleoside, followed by attachment of a drug to the lihker-nucleoside construct.
- Examples of functional groups on the drug for attaching to the linker-nucleoside construct include, but are not limited to, COOH, CHO, halogen, NHR, or OH, wherein m is a positive integer preferably between 1 and 20.
- the drug, linker precuser, and nucleoside used in the illustrated methods of conjugation are suitably protected in a manner consistent with the conditions required to affect conjugation, and the appropriate choice of protecting groups are within the ordinary skill of one in the art of chemical synthesis.
- the methylenes interposed between the two ends of the linker are for illustrative purposes only, and should not be construed as a limitation to the conjugates provided herein.
- the methylene subunits may be switched to ethyleneoxy subunits to give a polyethylene glycol based linker.
- the linker nucleoside construct is prepared by the condensation of thymidine or a suitable nucleoside analog with a linker precusor having a first end and a second end wherein the first end contains an appropriate leaving group and the second end contains a suitably protected functional group. Condensation is effected with a base with either the hydroxyl groups of the nucleoside carbohydrate protected or in free form. The intermediate with carbohydrate hydroxyl groups suitably protected is then subjected to deprotection to selectively remove a protecting group on the functional group on the second end of the linker.
- linker-nucleoside construct attachment of the linker-nucleoside construct to the drug uses amide bond coupling procedures well known in the art of peptide chemistry.
- the functional group on the drug is CHO
- reductive amination is performed with NaBH 4 , NaCNBH 4 NaB(OAc) 3 H or other suitable reducing groups to provide the drug conjugate.
- the functional group on the drug is OH
- coupling is affected by activation of the linker COOH group with DCC, or with any other acid activation agent well known in the art for ester bond formation.
- a functional group on the drug is halogen, alkylsulfonyloxy, arylsulfonyloxy, or any other suitable leaving group for nucleophilic displacement
- conjugation is through nucleophilic displacement by the free amine of the linker-nucleoside construct in the presence of Et 3 N or any other appropriate acid scavenger.
- the free functional group on the linker of the linker-nucleoside construct is a thiol
- condensation with the drug is effected by nucleophilic displacement of a leaving group on the drug, as given for the aforementioned nucleophilic displacement by free amine, after conversion of the thiol to a thiolate.
- the procedure for drug conjugation to a linker-nucleoside construct bearing a free thiol groups is also applicable to a free hydroxyl group on the linker to give a drug-linker-nucleoside conjugate with ether attachment of the linker to the drug.
- the conjugates may also be constructed using the same aforementioned chemical transformations for synthesis of drug-linker-nucleophile conjugates by first attaching the linker to the drug followed by attachment to N-3 of the nucleoside base.
- the purpose of the linker is to serve as a spacer between the drug and the nucleoside in order to remove or relieve steric interactions that may interfere with the kinase substrate activity of the nucleoside and/or the pharmacological effect of the drug so effective amounts of kinase and pharmacological activities remain.
- the linker may also be chosen based on its effect on the hydrophobicity of the drug conjugate to improve passive diffusion into the target cells or tissue. It should also be understood that cleavage of a bond within a linker to generate free drug is not required for the pharmacological effect of the drug that is incorporated into a conjugate.
- Example syntheses of nucleoside-linker constructs with linker attachment to the nucleoside C3 '-O is generally illustrated in Scheme 2a.
- nucleoside constructs may be synthesized from known 3'-O- (2-hydroxyethyl)-2'-deoxy-thymidine suitably protected at C5'-O and N3.
- nucleoside constructs may be synthesized from a suitably protected C5'-O, N3 uridine or a suitably protected C5'-O, C4-N cytidine wherein the hydroxyethyl group is introduced as reported for 2'-deoxy-thymidine.
- the hydroxyethyl group is transformed by S ern oxidation to provide an aldehyde functional group.
- a carboxyhc acid functional group may be introduced by reaction of a suitably protected 2'-deoxy-ribonucleoside with ClCH 2 COONa (Edge, M.D., et al. J. Chem. Soc. Perkin Trans. 1, 290-4 (1973)).
- the aldehyde or carboxyhc acid functional groups may be further elaborated to extend the linker, or may be used directly for attachment to a drug bearing an amino group.
- the hydroxyethyl group may be activated for nucleophilic displacement, for example, by treatment with triflic anhydride.
- Nucleophilic displacement may be from a drug nucleophile to give a drug-linker-nucleoside construct, or with the anion derived from a protected amine including phthalimido or bis-(t-butyloxycarbonyl)amine, to introduce a protected amine. After selective removal of the amine protecting group or groups, the free amine may be used to further extend the linker or may be used to attach the linker to the drug as described for Scheme 1.
- An alternative method to introduce the amino functional group and a method to introduce a thiol functional group are given in Teng, K., et al. US Pat. No. 6,087,482 the disclosure of which is incorporated by reference.
- C3'-O may be synthesized by selectively condensing thymidine or 2'-deoxyuridine with suitable protection at C5'-OH, or 2'-deoxy-cytosine with suitable protection at
- the second end is a functional group such as a selectively protected OH, N3, ester or a terminal alkene which may be converted once incorporated into the nucleoside-linker construct into an electrophile by for example selective deprotection to give a free OH which is transformed to an aldehyde by Swern oxidation or is sulfonated to provide a leaving group, Staudinger reduction of the azide to give an amine, selective hydrolysis of the ester to give a carboxyhc acid, or ozonolysis of the terminal alkene to provide an aldehyde or hydroboration to give a terminal OH which is further manipulated as described.
- Example syntheses of nucleoside-linker constructs with direct linker attachment to the nucleoside C3' carbon is generally illustrated in Scheme 2c.
- the alkyne Prior to selective deprotection of the functional group, or after formation of the drug conjugate the alkyne may be reduced to the alkene or alkane to proved further drug conjugates.
- Alternative methods to synthesize nucleosides having a C3'-C bond wherein the substituent to C3' contains a suitable functional group for drug attachment as described for Scheme 1 are given in Teng, K., et al. US Pat. No. 6,087,482 the disclosure of which is incorporated by reference.
- Scheme 3 teaches the synthesis of linker-nucleoside constructs at C-5.
- An alternative route for linker attachment to C5 through a carbon-carbon bond employs the Heck reaction using an alkene on the first end of the linker.
- An example of an alkene based linker with an appropriate second end is ethyl acrylate.
- hydrolysis with optional reduction of the double bond would provide a COOH group for further linker elaboration or attachment to a drug a described for Scheme 1 to give a drug-linker-nucleoside conjugate either with alkene or alkane functionality within the linker.
- Reduction of the alkyne in either the aforementioned linker-nucleoside construct or in a final drug conjugate would also provide linkers having alkene and alkane functionality within the linker.
- the nucleoside linker construct is then formed by silylation of the pyrimidine base with hexamethyldisilazane and a catalytic amount of trimethylsilylchloride followed by condensation with an appropriately protected ⁇ -D- ribosylchloride using ZnC12 in CC1 4 .
- 5-thio-2'deoxy-urudine maybe S-alkylated to directly give the nucleoside-linker construct.
- the fourth end may be a functionality that may be converted to a electrophile or nucleophile once incorporated into a nucleoside-linker construct and includes but is not limited to a selectively protected OH, N3, ester or a terminal alkene .
- thymidine or uracil nucleoside constructs or drug conjugates so described may be converted to their conesponding 2'deoxy cytidine analogs by treatment with a chlorinating agent such as POC13 to form a 4-C1 pyrimidine intermediate which is condensed with ammonia or a substituted amine.
- paclitaxel appropriately protected at the more reactive C2' hydroxyl, is condensed with a linker-nucleoside construct wherein n is 0 or a positive integer, preferably where n is between O and 20, wherein the second end of the linker bears a carboxyhc acid group.
- Coupling agent Condensation with a benzoic acid derivative containing a suitably protected amine wherein is preferentially zero, one or two using standard amide bond forming conditions then provides a paclitaxel derivative wherein a functionality amenable to conjugation is introduced into the C3'-N benzamido group.
- Deprotection of the amine followed by amide bond formation to a linker-nucleoside conjugate wherein the second end of the linker bears a COOH group gives after final deprotection the desired paclitaxel-linker-nucleoside conjugate with attachment at the C3'-N benzamido group.
- Drug-linker-purine nucleoside conjugates conesponding to Formulas 9, 10 and 11 are prepared according to the procedures illustrated in Schemes 1, 4 and 5 using a purine nucleoside linker construct wherein the linker is attached to C8 of the purine.
- Appropriate starting materials are exemplified by known compounds 8- bromo-2'deoxy-adenosine and 8-bromo-2'-deoxy-guanosine.
- Y is a nucleophile, preferably a thiolate, which displaces the C8-Br and becomes incorporated into the purine nucleoside-linker construct
- Z is an appropriately protected nucleophile or elecfrophile which may be used in the reaction sequences exemplified by Schemes 4 and 5 after selective deprotection.
- paclitaxel C-10 carbamates prepared directly from paclitaxel include some simple analogs derived from 10-O-deacetyl-7-O,10-O-bis-[N- (2,2,2-trichloroethyloxy)-aminocarbonyl] -paclitaxel as reported in Bourzat, J.Det al.; EPO Application 524,093 (1993).
- This synthetic methodology is not versatile since selective reaction of the amine input at C-10 is possible only in dichloromethane.
- a more general approach for the synthesis of C-10 carbamates starts from 10-deacetyl-baccatin-III.
- compound 5a can be converted in nearly quantitative yield into its CIO carbonylimidazole 6a by reaction with carbonyl- diimidazole (CDI) in dichloromethane at room temperature.
- CDI carbonyl- diimidazole
- Compound 6a can be reacted with amines in suitable solvents to yield the conesponding carbamate 8a, which can be deprotected to give 9a.
- the reaction can be carried out in non-polar solvents, such as dichloromethane or in protic solvents such as LPA or t-BuOH at elevated temperatures.
- the CIO- carbonylimidazole 6a can be activated with an alkylating agent such as an alkyl halide, alkyl sulfonate or di-alkyl-sulfate to give a N ⁇ alkyl-N ⁇ acyl imidazolium species represented by 7a.of Scheme 8.
- the alkylating agent is selected from dimetylsulfate and methyl iodide.
- the imidazolium species can then be reacted with various amines either in free or salt forms in protic solvents or aprotic solvents such as DMF, DMSO or dioxane.
- amine salts condensation with 7a is conducted in the presence of a hindered base such as DLEA.
- a hindered base such as DLEA.
- less reactive amines, such as arylamines or heteroarylamines may be condensed with 7a to obtain paclitaxel CIO carbamates with N-aryl or N- heteroaryl linker attachment.
- nucleophiles can be used in the reactions provided herein to prepare CIO paclitaxel carbamates.
- nucleophiles include, but are not limited to, primary and secondary amines, amine containing acids, such as ⁇ -amino acids, amino-sugars, such as glucosamine, arylamines, heteroarylamines, and ⁇ , - disubstituted alcohols.
- the thymidine-linker intermediate so obtained is subjected to catalytic hydrogenation using methanol with 10 wt% palladium on carbon and stirring under an atmosphere of H 2 for 16 h. Filtration of the reaction mixture on Celite, removal of volatiles in vacuo and lyophilization provides the thymidine-linker-amine intermediate of general structure 4.
- compositions provided herein contain therapeutically effective amounts of one or more of conjugates provided herein that are useful in the prevention, treatment, or amelioration of one or more of the symptoms of ACAMPS conditions.
- Such conditions include, but are not limited to, cancer, coronary restenosis, osteoporosis and syndromes characterized by chronic inflammation and/or autoimmunity.
- Chronic inflammation and/or autoimmune diseases include but are not limited to rheumatoid arthritis and other forms of arthritis, asthma, psoriasis, inflammatory bowel disease, systemic lupus erythematosus, systemic dermatomyositis, inflammatory ophthalmic diseases, autoimmune hematologic disorders, multiple sclerosis, vasculitis, idiopathic nephrotic syndrome, transplant rejection and graft versus host disease.
- the compositions contain one or more conjugates provided herein.
- the conjugates are preferably formulated into suitable pharmaceutical preparations such as solutions, suspensions, tablets, dispersible tablets, pills, capsules, powders, sustained release formulations or elixirs, for oral administration or in sterile solutions or suspensions for parenteral administration, as well as transdermal patch preparation and dry powder inhalers.
- suitable pharmaceutical preparations such as solutions, suspensions, tablets, dispersible tablets, pills, capsules, powders, sustained release formulations or elixirs, for oral administration or in sterile solutions or suspensions for parenteral administration, as well as transdermal patch preparation and dry powder inhalers.
- suitable pharmaceutical preparations such as solutions, suspensions, tablets, dispersible tablets, pills, capsules, powders, sustained release formulations or elixirs, for oral administration or in sterile solutions or suspensions for parenteral administration, as well as transdermal patch preparation and dry powder inhalers.
- the conjugates described above are formulated into pharmaceutical compositions using techniques and procedures well known in the art (see, e
- the conjugates may be derivatized as the conesponding salts, esters, enol ethers or esters, acids, bases, solvates, hydrates or prodrugs prior to formulation, as described above.
- concentrations of the conjugates in the compositions are effective for delivery of an amount, upon administration, that treats, prevents, or ameliorates one or more of the symptoms of conditions associated with ACAMPS.
- Such conditions include, but are not limited to, cancer, coronary restenosis, osteoporosis and syndromes characterized by chronic inflammation and/or autoimmunity.
- the compositions are formulated for single dosage administration.
- the weight fraction of conjugate is dissolved, suspended, dispersed or otherwise mixed in a selected vehicle at an effective concentration such that the treated condition is relieved or ameliorated.
- Pharmaceutical carriers or vehicles suitable for administration of the conjugates provided herein include any such carriers known to those skilled in the art to be suitable for the particular mode of administration.
- the conjugates may be formulated as the sole pharmaceutically active ingredient in the composition or may be combined with other active ingredients.
- Liposomal suspensions, including tissue-targeted liposomes, such as tumor-targeted liposomes may also be suitable as pharmaceutically acceptable carriers. These may be prepared according to methods known to those skilled in the art. For example, liposome formulations may be prepared as described in U.S. Patent No. 4,522,811.
- liposomes such as multilamellar vesicles (MLV's) maybe formed by drying down egg phosphatidyl choline and brain phosphatidyl serine (7:3 molar ratio) on the inside of a flask. A solution of a compound provided herein in phosphate buffered saline lacking divalent cations (PBS) is added and the flask shaken until the lipid film is dispersed. The resulting vesicles are washed to remove unencapsulated compound, pelleted by centrifugation, and then resuspended in PBS.
- PBS phosphate buffered saline lacking divalent cations
- the active conjugate is included in the pharmaceutically acceptable carrier in an amount sufficient to exert a therapeutically useful effect in the absence of undesirable side effects on the patient treated.
- the therapeutically effective concentration may be determined empirically by testing the conjugates in in vitro and in vivo systems described herein and then extrapolated therefrom for dosages for humans.
- concentration of active conjugate in the pharmaceutical composition will depend on absorption, inactivation and excretion rates of the active conjugate, the physicochemical characteristics of the conjugate, the dosage schedule, and amount administered as well as other factors known to those of skill in the art.
- the amount that is delivered is sufficient to ameliorate one or more of the symptoms of diseases or disorders associated with ACAMPS condition as described herein.
- a therapeutically effective dosage should produce a serum concentration of active ingredient of from about 0.1 ng/ml to about 50-100 ⁇ g/ml.
- the pharmaceutical compositions typically should provide a dosage of from about 0.001 mg to about 20O0 mg of conjugate per kilogram of body weight per day.
- Pharmaceutical dosage unit forms are prepared to provide from about 1 mg to about 1000 mg and preferably from about 10 to about 500 mg of the essential active ingredient or a combination of essential ingredients per dosage unit form.
- the active ingredient may be administered at once, or may be divided into a number of smaller doses to be administered at intervals of time.
- compositions include acids, bases, enol ethers and esters, salts, esters, hydrates, solvates and prodrug forms.
- the derivative is selected such that its pharmacokinetic properties are superior to the conesponding neutral conjugate.
- effective concentrations or amounts of one or more of the conjugates described herein or pharmaceutically acceptable derivatives thereof are mixed with a suitable pharmaceutical carrier or vehicle for systemic, topical or local adminisfration to form pharmaceutical compositions.
- Conjugates are included in an amount effective for ameliorating one or more symptoms of, or for treating or preventing diseases or disorders associated with ACAMPS condition as described herein.
- concentration of active conjugate in the composition will depend on absorption, inactivation, excretion rates of the active conjugate, the dosage schedule, amount administered, particular formulation as well as other factors known to those of skill in the art.
- compositions are intended to be administered by a suitable route, including orally, parenterally, rectally, topically and locally.
- a suitable route including orally, parenterally, rectally, topically and locally.
- capsules and tablets are presently prefe ⁇ ed.
- the compositions are in liquid, semi- liquid or solid form and are formulated in a manner suitable for each route of administration.
- Prefe ⁇ ed modes of adminisfration include parenteral and oral modes of administration.
- Oral administration is presently most prefened.
- Solutions or suspensions used for parenteral, intradermal, subcutaneous, or topical application can include any of the following components: a sterile diluent, such as water for injection, saline solution, fixed oil, polyethylene glycol, glycerine, propylene glycol, dimethyl acetamide or other synthetic solvent; antimicrobial agents, such as benzyl alcohol and methyl parabens; antioxidants, such as ascorbic acid and sodium bisulfite; chelating agents, such as ethylenediaminetetraacetic acid (EDTA); buffers, such as acetates, citrates and phosphates; and agents for the adjustment of tonicity such as sodium chloride or dextrose.
- a sterile diluent such as water for injection, saline solution, fixed oil, polyethylene glycol, glycerine, propylene glycol, dimethyl acetamide or other synthetic solvent
- antimicrobial agents such as benzyl alcohol and methyl parabens
- parenteral preparations can be enclosed in ampules, disposable syringes or single or multiple dose vials made of glass, plastic or other suitable material.
- methods for solubilizing conjugates may be used. Such methods are known to those of skill in this art, and include, but are not limited to, using cosolvents, such as dimethylsulfoxide (DMSO), using surfactants, such as TWEEN®, or dissolution in aqueous sodium bicarbonate.
- cosolvents such as dimethylsulfoxide (DMSO)
- surfactants such as TWEEN®
- dissolution in aqueous sodium bicarbonate Upon mixing or addition of the conjugate(s), the resulting mixture may be a solution, suspension, emulsion or the like.
- the form of the resulting mixture depends upon a number of factors, including the intended mode of administration and the solubility of the conjugate in the selected carrier or vehicle.
- the effective concentration is sufficient for ameliorating the symptoms of the disease, disorder or condition treated and may be empirically determined.
- the pharmaceutical compositions are provided for adminisfration to humans and animals in unit dosage forms, such as tablets, capsules, pills, powders, granules, sterile parenteral solutions or suspensions, and oral solutions or suspensions, and oil- water emulsions containing suitable quantities of the conjugates or pharmaceutically acceptable derivatives thereof.
- the pharmaceutically therapeutically active conjugates and derivatives thereof are typically formulated and administered in unit-dosage forms or multiple-dosage forms.
- Unit-dose forms as used herein refers to physically discrete units suitable for human and animal subjects and packaged individually as is known in the art. Each unit-dose contains a predetermined quantity of the therapeutically active conjugate sufficient to produce the desired therapeutic effect, in association with the required pharmaceutical carrier, vehicle or diluent. Examples of unit-dose forms include ampules and syringes and individually packaged tablets or capsules. Unit-dose forms may be administered in fractions or multiples thereof.
- a multiple-dose form is a plurality of identical unit-dosage forms packaged in a single container to be administered in segregated unit-dose form. Examples of multiple-dose forms include vials, bottles of tablets or capsules or bottles of pints or gallons.
- the composition can contain along with the active ingredient: a diluent such as lactose, sucrose, dicalcium phosphate, or carboxymethylcellulose; a lubricant, such as magnesium stearate, calcium stearate and talc; and a binder such as starch, natural gums, such as gum acaciagelatin, glucose, molasses, polvinylpy ⁇ olidine, celluloses and derivatives thereof, povidone, crospovidones and other such binders known to those of skill in the art.
- a diluent such as lactose, sucrose, dicalcium phosphate, or carboxymethylcellulose
- a lubricant such as magnesium stearate, calcium stearate and talc
- a binder such as starch, natural gums, such as gum acaciagelatin, glucose, molasses, polvinylpy ⁇ olidine, celluloses and derivatives thereof, povidone,
- Liquid pharmaceutically administrable compositions can, for example, be prepared by dissolving, dispersing, or otherwise mixing an active conjugate as defined above and optional pharmaceutical adjuvants in a carrier, such as, for example, water, saline, aqueous dextrose, glycerol, glycols, ethanol, and the like, to thereby form a solution or suspension.
- a carrier such as, for example, water, saline, aqueous dextrose, glycerol, glycols, ethanol, and the like, to thereby form a solution or suspension.
- the pharmaceutical composition to be administered may also contain minor amounts of nontoxic auxiliary substances such as wetting agents, emulsifying agents, or solubilizing agents, pH buffering agents and the like, for example, acetate, sodium citrate, cyclodextrine derivatives, sorbitan monolaurate, triethanolamine sodium acetate, triethanolamine oleate, and other such agents.
- auxiliary substances such as wetting agents, emulsifying agents, or solubilizing agents, pH buffering agents and the like, for example, acetate, sodium citrate, cyclodextrine derivatives, sorbitan monolaurate, triethanolamine sodium acetate, triethanolamine oleate, and other such agents.
- auxiliary substances such as wetting agents, emulsifying agents, or solubilizing agents, pH buffering agents and the like, for example, acetate, sodium citrate, cyclodextrine derivatives, sorbitan monolaurate, triethanolamine sodium acetate, triethanolamine ole
- compositions containing active ingredient in the range of 0.005% to 100% with the balance made up from non-toxic carrier may be prepared.
- a pharmaceutically acceptable non-toxic composition is formed by the incorporation of any of the normally employed excipients, such as, for example pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, talcum, cellulose derivatives, sodium crosscarmellose, glucose, sucrose, magnesium carbonate or sodium saccharin.
- compositions include solutions, suspensions, tablets, capsules, powders and sustained release formulations, such as, but not limited to, implants and icroencapsulated delivery systems, and biodegradable, biocompatible polymers, such as collagen, ethylene vinyl acetate, polyanhydrides, polyglycolic acid, polyorthoesters, polylactic acid and others. Methods for preparation of these compositions are known to those skilled in the art.
- the contemplated compositions may contain 0.001%-100% active ingredient, preferably 0.1-85%, typically 75-95%.
- the active conjugates or pharmaceutically acceptable derivatives may be prepared with carriers that protect the conjugate against rapid elimination from the body, such as time release formulations or coatings.
- the compositions may include other active conjugates to obtain desired combinations of properties.
- compositions for oral administration Oral pharmaceutical dosage forms are either solid, gel or liquid.
- the solid dosage forms are tablets, capsules, granules, and bulk powders. Types of oral tablets include compressed, chewable lozenges and tablets which may be enteric-coated, sugar-coated or film-coated.
- Capsules may be hard or soft gelatin capsules, while granules and powders may be provided in non-effervescent or effervescent form with the combination of other ingredients known to those skilled in the art.
- the formulations are solid dosage forms, preferably capsules or tablets.
- the tablets, pills, capsules, troches and the like can contain any of the following ingredients, or conjugates of a similar nature: a binder; a diluent; a disintegrating agent; a lubricant; a glidant; a sweetening agent; and a flavoring agent.
- binders include microcrystalline cellulose, gum fragacanth, glucose solution, acacia mucilage, gelatin solution, sucrose and starch paste.
- Lubricants include talc, starch, magnesium or calcium stearate, lycopodium and stearic acid.
- Diluents include, for example, lactose, sucrose, starch, kaolin, salt, mannitol and dicalcium phosphate.
- Glidants include, but are not limited to, colloidal silicon dioxide.
- Disintegrating agents include crosscarmellose sodium, sodium starch glycolate, alginic acid, corn starch, potato starch, bentonite, methylcellulose, agar and carboxymethylcellulose.
- Coloring agents include, for example, any of the approved certified water soluble FD and C dyes, mixtures thereof; and water insoluble FD and C dyes suspended on alumina hydrate.
- Sweetening agents include sucrose, lactose, mannitol and artificial sweetening agents such as saccharin, and any number of spray dried flavors.
- Flavoring agents include natural flavors extracted from plants such as fruits and synthetic blends of compounds which produce a pleasant sensation, such as, but not limited to peppermint and methyl salicylate.
- Wetting agents include propylene glycol monostearate, sorbitan monooleate, diethylene glycol monolaurate and polyoxyethylene laural ether.
- Emetic-coatings include fatty acids, fats, waxes, shellac, ammoniated shellac and cellulose acetate phthalates.
- Film coatings include hydroxyethylcellulose, sodium carboxymethylcellulose, polyethylene glycol 4000 and cellulose acetate phthalate.
- the conjugate could be provided in a composition that protects it from the acidic environment of the stomach.
- the composition can be formulated in an enteric coating that maintains its integrity in the stomach and releases the active conjugate in the intestine.
- the composition may also be formulated in combination with an antacid or other such ingredient.
- the dosage unit form is a capsule, it can contain, in addition to material of the above type, a liquid carrier such as a fatty oil.
- dosage unit forms can contain various other materials which modify the physical form of the dosage unit, for example, coatings of sugar and other enteric agents.
- the conjugates can also be administered as a component of an elixir, suspension, syrup, wafer, sprinkle, chewing gum or the like.
- a syrup may contain, in addition to the active conjugates, sucrose as a sweetening agent and certain preservatives, dyes and colorings and flavors.
- the active materials can also be mixed with other active materials which do not impair the desired action, or with materials that supplement the desired action, such as antacids, H2 blockers, and diuretics.
- the active ingredient is a conjugate or pharmaceutically acceptable derivative thereof as described herein. Higher concentrations, up to about 98% by weight of the active ingredient maybe included.
- Pharmaceutically acceptable carriers included in tablets are binders, lubricants, diluents, disintegrating agents, coloring agents, flavoring agents, and wetting agents.
- Enteric-coated tablets because of the enteric-coating, resist the action of stomach acid and dissolve or disintegrate in the neutral or alkaline intestines.
- Sugar-coated tablets are compressed tablets to which different layers of pharmaceutically acceptable substances are applied.
- Film-coated tablets are compressed tablets which have been coated with a polymer or other suitable coating. Multiple compressed tablets are compressed tablets made by more than one compression cycle utilizing the pharmaceutically acceptable substances previously mentioned.
- Coloring agents may also be used in the above dosage forms. Flavoring and sweetening agents are used in compressed tablets, sugar-coated, multiple compressed and chewable tablets.
- Liquid oral dosage forms include aqueous solutions, emulsions, suspensions, solutions and/or suspensions reconstituted from non-effervescent granules and effervescent preparations reconstituted from effervescent granules.
- Aqueous solutions include, for example, elixirs and syrups. Emulsions are either oil-in- water or water-in-oil. Elixirs are clear, sweetened, hydroalcoholic preparations.
- Pharmaceutically acceptable carriers used in elixirs include solvents. Syrups are concentrated aqueous solutions of a sugar, for example, sucrose, and may contain a preservative.
- An emulsion is a two-phase system in which one liquid is dispersed in the form of small globules throughout another liquid.
- Pharmaceutically acceptable carriers used in emulsions are non-aqueous liquids, emulsifying agents and preservatives. Suspensions use pharmaceutically acceptable suspending agents and preservatives.
- Pharmaceutically acceptable substances used in non-effervescent granules, to be reconstituted into a liquid oral dosage form include diluents, sweeteners and wetting agents.
- Pharmaceutically acceptable substances used in effervescent granules, to be reconstituted into a liquid oral dosage form include organic acids and a source of carbon dioxide. Coloring and flavoring agents are used in all of the above dosage forms.
- Solvents include glycerin, sorbitol, ethyl alcohol and syrup.
- preservatives include glycerin, methyl and propylparaben, benzoic add, sodium benzoate and alcohol.
- non-aqueous liquids utilized in emulsions include mineral oil and cottonseed oil.
- emulsifying agents include gelatin, acacia, fragacanth, bentonite, and surfactants such as polyoxyethylene sorbitan monooleate.
- Suspending agents include sodium carboxymethylcellulose, pectin, fragacanth, Veegum and acacia.
- Diluents include lactose and sucrose.
- Sweetening agents include sucrose, syrups, glycerin and artificial sweetening agents such as saccharin.
- Wetting agents include propylene glycol monostearate, sorbitan monooleate, diethylene glycol monolaurate and polyoxyethylene lauryl ether.
- Organic adds include citric and tartaric acid.
- Sources of carbon dioxide include sodium bicarbonate and sodium carbonate.
- Coloring agents include any of the approved certified water soluble FD and C dyes, and mixtures thereof.
- Flavoring agents include natural flavors extracted from plants such fruits, and synthetic blends of compounds which produce a pleasant taste sensation.
- the solution or suspension in for example propylene carbonate, vegetable oils or triglycerides, is preferably encapsulated in a gelatin capsule.
- the solution e.g., for example, in a polyethylene glycol
- a pharmaceutically acceptable liquid carrier e.g., water
- liquid or semi-solid oral formulations may be prepared by dissolving or dispersing the active conjugate or salt in vegetable oils, glycols, triglycerides, propylene glycol esters (e.g., propylene carbonate) and other such carriers, and encapsulating these solutions or suspensions in hard or soft gelatin capsule shells.
- formulations include, but are not limited to, those containing a conjugate provided herein, a dialkylated mono- or poly-alkylene glycol, including, but not limited to, 1,2-dimethoxymethane, diglyme, triglyme, tetraglyme, polyethylene glycol-350-dimethyl ether, polyethylene glycol-550- dimethyl ether, polyethylene glycol-750-dimethyl ether wherein 350, 550 and 750 refer to the approximate average molecular weight of the polyethylene glycol, and one or more antioxidants, such as butylated hydroxytoluene (BHT), butylated hydroxyanisole (BHA), propyl gallate, vitamin E, hydroquinone, hydroxycoumarins, ethanolamine, lecithin, cephalin, ascorbic acid, malic acid, sorbitol, phosphoric acid,
- BHT butylated hydroxytoluene
- BHA butylated hydroxyanisole
- antioxidants
- formulations include, but are not limited to, aqueous alcoholic solutions including a pharmaceutically acceptable acetal.
- Alcohols used in these formulations are any pharmaceutically acceptable water-miscible solvents having one or more hydroxyl groups, including, but not limited to, propylene glycol and ethanol.
- Acetals include, but are not limited to, di(lower alkyl) acetals of lower alkyl aldehydes such as acetaldehyde diethyl acetal.
- tablets and capsules formulations may be coated as known by those of skill in the art in order to modify or sustain dissolution of the active ingredient.
- Injectables may be coated with a conventional enterically digestible coating, such as phenylsalicylate, waxes and cellulose acetate phthalate.
- enterically digestible coating such as phenylsalicylate, waxes and cellulose acetate phthalate.
- Parenteral administration generally characterized by injection, either subcutaneously, intramuscularly or intravenously is also contemplated herein.
- Injectables can be prepared in conventional forms, either as liquid solutions or suspensions, solid forms suitable for solution or suspension in liquid prior to injection, or as emulsions. Suitable excipients are, for example, water, saline, dextrose, glycerol or ethanol.
- compositions to be administered may also contain minor amounts of non-toxic auxiliary substances such as wetting or emulsifying agents, pH buffering agents, stabilizers, solubility enhancers, and other such agents, such as for example, sodium acetate, sorbitan monolaurate, triethanolamine oleate and cyclodextrins.
- auxiliary substances such as wetting or emulsifying agents, pH buffering agents, stabilizers, solubility enhancers, and other such agents, such as for example, sodium acetate, sorbitan monolaurate, triethanolamine oleate and cyclodextrins.
- implantation of a slow-release or sustained-release system such that a constant level of dosage is maintained (see, e.g., U.S. Patent No. 3,710,795) is also contemplated herein.
- a conjugate provided herein is dispersed in a solid inner matrix, e.g., polymethylmethacrylate, polybutylmethacrylate, plasticized or unplasticized polyvinylchloride, plasticized nylon, plasticized polyethyleneterephthalate, natural rubber, polyisoprene, polyisobutylene, polybutadiene, polyethylene, ethylene-vinylacetate copolymers, silicone rubbers, polydimethylsiloxanes, silicone carbonate copolymers, hydrophilic polymers such as hydrogels of esters of acrylic and methacrylic acid, collagen, cross- linked polyvinylalcohol and cross-linked partially hydrolyzed polyvinyl acetate, that is su ⁇ ounded by an outer polymeric membrane, e.g., polyethylene, polypropylene, ethylene/propylene copolymers, ethylene/ethyl acrylate copolymers, ethylene/vinylacetate copolymers, silicone rubbers, polydimethyl silox
- parenteral administration of the compositions includes intravenous, subcutaneous and intramuscular administrations.
- Preparations for parenteral administration include sterile solutions ready for injection, sterile dry soluble products, such as lyophilized powders, ready to be combined with a solvent just prior to use, including hypodermic tablets, sterile suspensions ready for injection, sterile dry insoluble products ready to be combined with a vehicle just prior to use and sterile emulsions.
- the solutions may be either aqueous or nonaqueous.
- suitable carriers include physiological saline or phosphate buffered saline (PBS), and solutions containing thickening and solubilizing agents, such as glucose, polyethylene glycol, and polypropylene glycol and mixtures thereof.
- Pharmaceutically acceptable carriers used in parenteral preparations include aqueous vehicles, nonaqueous vehicles, antimicrobial agents, isotonic agents, buffers, antioxidants, local anesthetics, suspending and dispersing agents, emulsifying agents, sequestering or chelating agents and other pharmaceutically acceptable substances.
- aqueous vehicles include Sodium Chloride Injection, Ringers Injection, Isotonic Dextrose injection, Sterile Water Injection, Dextrose and Lactated Ringers Injection.
- Nonaqueous parenteral vehicles include fixed oils of vegetable origin, cottonseed oil, corn oil, sesame oil and peanut oil.
- Antimicrobial agents in bacteriostatic or fungistatic concentrations must be added to parenteral preparations packaged in multiple-dose containers which include phenols or cresols, mercurials, benzyl alcohol, chlorobutanol, methyl and propyl p-hydroxybenzoic acid esters, thimerosal, benzalkonium chloride and benzethonium chloride.
- Isotonic agents include sodium chloride and dextrose. Buffers include phosphate and citrate.
- Antioxidants include sodium bisulfate.
- Local anesthetics include procaine hydrochloride.
- Suspending and dispersing agents include sodium carboxymethylcelluose, hydroxypropyl methylcellulose and polyvinylpy ⁇ olidone.
- Emulsifying agents include Polysorbate 80 (TWEEN® 80).
- a sequestering or chelating agent of metal ions include EDTA.
- Pharmaceutical carriers also include ethyl alcohol, polyethylene glycol and propylene glycol for water miscible vehicles and sodium hydroxide, hydrochloric acid, citric acid or lactic acid for pH adjustment. The concentration of the pharmaceutically active conjugate is adjusted so that an injection provides an effective amount to produce the desired pharmacological effect. The exact dose depends on the age, weight and condition of the patient or animal as is known in the art.
- the unit-dose parenteral preparations are packaged in an ampule, a vial or a syringe with a needle. All preparations for parenteral adminisfration must be sterile, as is known and practiced in the art. Illustratively, intravenous or intraarterial infusion of a sterile aqueous solution containing an active conjugate is an effective mode of adminisfration. Another embodiment is a sterile aqueous or oily solution or suspension containing an active material injected as necessary to produce the desired pharmacological effect. Injectables are designed for local and systemic administration.
- a therapeutically effective dosage is formulated to contain a concentration of at least about 0.1% w/w up to about 90% w/w or more, preferably more than 1% w/w of the active conjugate to the treated tissue(s).
- the active ingredient may be administered at once, or may be divided into a number of smaller doses to be administered at intervals of time. It is understood that the precise dosage and duration of treatment is a function of the tissue being treated and may be determined empirically using known testing protocols or by extrapolation from in vivo or in vitro test data. It is to be noted that concentrations and dosage values may also vary with the age of the individual treated.
- the conjugate may be suspended in micronized or other suitable form or may be derivatized to produce a more soluble active product or to produce a prodrug.
- the form of the resulting mixture depends upon a number of factors, including the intended mode of administration and the solubility of the conjugate in the selected carrier or vehicle.
- the effective concentration is sufficient for ameliorating the symptoms of the condition and may be empirically determined.
- Lyophilized powders of interest herein are also lyophilized powders, which can be reconstituted for administration as solutions, emulsions and other mixtures. They may also be reconstituted and formulated as solids or gels.
- the sterile, lyophilized powder is prepared by dissolving a conjugate provided herein, or a pharmaceutically acceptable derivative thereof, in a suitable solvent.
- the solvent may contain an excipient which improves the stability or other pharmacological component of the powder or reconstituted solution, prepared from the powder. Excipients that may be used include, but are not limited to, dextrose, sorbital, fructose, com syrup, xylitol, glycerin, glucose, sucrose or other suitable agent.
- the solvent may also contain a buffer, such as citrate, sodium or potassium phosphate or other such buffer known to those of skill in the art at, typically, about neutral pH.
- a buffer such as citrate, sodium or potassium phosphate or other such buffer known to those of skill in the art at, typically, about neutral pH.
- Subsequent sterile filtration of the solution followed by lyophilization under standard conditions known to those of skill in the art provides the desired formulation.
- the resulting solution will be apportioned into vials for lyophilization.
- Each vial will contain a single dosage (10-1000 mg, preferably 100- 500 mg) or multiple dosages of the conjugate.
- the lyophilized powder can be stored under appropriate conditions, such as at about 4 °C to room temperature. Reconstitution of this lyophilized powder with water for injection provides a formulation for use in parenteral adminisfration.
- Topical administration Topical mixtures are prepared as described for the local and systemic administration.
- the resulting mixture may be a solution, suspension, emulsions or the like and are formulated as creams, gels, ointments, emulsions, solutions, elixirs, lotions, suspensions, tinctures, pastes, foams, aerosols, irrigations, sprays, suppositories, bandages, dermal patches or any other formulations suitable for topical administration.
- the conjugates or pharmaceutically acceptable derivatives thereof may be formulated as aerosols for topical application, such as by inhalation (see, e.g., U.S. Patent Nos. 4,044,126, 4,414,209, and 4,364,923, which describe aerosols for delivery of a steroid useful for treatment of inflammatory diseases, particularly asthma).
- These formulations for adminisfration to the respiratory tract can be in the form of an aerosol or solution for a nebulizer, or as a microfine powder for insufflation, alone or in combination with an inert carrier such as lactose.
- the particles of the formulation will typically have diameters of less than 50 microns, preferably less than 10 microns.
- the conjugates may be formulated for local or topical application, such as for topical application to the skin and mucous membranes, such as in the eye, in the form of gels, creams, and lotions and for application to the eye or for intracisternal or intraspinal application.
- Topical adminisfration is contemplated for transdermal delivery and also for administration to the eyes or mucosa, or for inhalation therapies.
- Nasal solutions of the active conjugate alone or in combination with other pharmaceutically acceptable excipients can also be administered. These solutions, particularly those intended for ophthalmic use, may be formulated as 0.01% - 10% isotomc solutions, pH about 5-7, with appropriate salts. 5.
- compositions for other routes of administration are also contemplated herein.
- routes of administration such as topical application, transdermal patches, and rectal administration
- pharmaceutical dosage forms for rectal administration are rectal suppositories, capsules and tablets for systemic effect.
- Rectal suppositories are used herein mean solid bodies for insertion into the rectum which melt or soften at body temperature releasing one or more pharmacologically or therapeutically active ingredients.
- Pharmaceutically acceptable substances utilized in rectal suppositories are bases or vehicles and agents to raise the melting point.
- bases examples include cocoa butter (theobroma oil), glycerin-gelatin, carbowax (polyoxyethylene glycol) and appropriate mixtures of mono-, di- and triglycerides of fatty acids. Combinations of the various bases may be used.
- Agents to raise the melting point of suppositories include spermaceti and wax. Rectal suppositories may be prepared either by the compressed method or by molding. The typical weight of a rectal suppository is about 2 to 3 gm. Tablets and capsules for rectal adminisfration are manufactured using the same pharmaceutically acceptable substance and by the same methods as for formulations for oral administration. 6.
- the conjugates or pharmaceutically acceptable derivatives can be packaged as articles of manufacture containing packaging material, a conjugate or pharmaceutically acceptable derivative thereof provided herein, which is used for treatment, prevention or amelioration of one or more symptoms associated with
- ACAMPS condition and a label that indicates that the conjugate or pharmaceutically acceptable derivative thereof is used for treatment, prevention or amelioration of one or more symptoms associated with ACAMPS condition.
- the articles of manufacture provided herein contain packaging materials.
- Packaging materials for use in packaging pharmaceutical products are well known to those of skill in the art. See, e.g., U.S. Patent Nos. 5,323,907, 5,052,558 and 5,033,252.
- Examples of pharmaceutical packaging materials include, but are not limited to, blister packs, bottles, tubes, inhalers, pumps, bags, vials, containers, syringes, bottles, and any packaging material suitable for a selected formulation and intended mode of administration and treatment.
- a wide array of formulations of the conjugates and compositions provided herein are contemplated as are a variety of treatments for any disorder associated with ACAMPS conditions.
- E. Evaluation of the Activity of the Conjugates Standard physiological, pharmacological and biochemical procedures are available for testing the conjugates to identify those that possess biological activity, including kinase activity. In vitro and in vivo assays that can be used to evaluate biological activity, such as cytotoxicity, which will depend upon the therapeutic agent being used in the conjugate. Exemplary assays are discussed briefly below with reference to cytotoxic conjugates (see, also, Examples). It is understood that the particular activity assayed will depend upon the conjugated therapeutic agent. 1.
- Thymidine kinase, viral thymidine kinase, TK-1 deoxycytidine kinase, and deoxyguanosine kinase activities are determined by subjecting a first end of a linker used in synthesizing linker-substrate constructs to a first test.
- the first test may involve observing ADP formation, an obligatory co-product of phospho group transfer from ATP which is catalyzed by the kinase to the C5' hydroxyl group or its equivalent in the nucleoside or nucleoside analog. Formation of ADP is followed by a coupled enzyme assay well known in the art.
- ADP formed from kinase phosphorylation
- pyruvate kinase is used by pyruvate kinase to generate pyruvate from phosphoenolpyruvate which in turn is converted to lactate by lactate dehydrogenase.
- lactate results in the consumption of NADH which is followed specfrophotometrically.
- the rate of nucleoside phosphorylation is then directly related to the rate of decrease in the observed NADH signal.
- Another test may involve monitoring the consumption of ATP.
- ATP concentrations at time 0 or after 4 hour incubation may be monitored by luciferase reaction (PKLightTM kit obtained from Cambrex Corporation, One Meadowlands Plaza, East Rutherford, NJ 07073), which generate a luminescence readout in the presence of ATP. Assays are initiated by mixing a kinase and a drug conjugate in the presence of 40 ⁇ M ATP. After 4 hour of incubation at 30°C, PKLightTM reagent is added and mixed well, and luminescence readout measured. The rate of drug conjugate phosphorylation is then directly related to the rate of decrease in the observed luminescence.
- PLLightTM kit obtained from Cambrex Corporation, One Meadowlands Plaza, East Rutherford, NJ 07073
- linkers of appropriate lengths and substrate with an effective amount of kinase activity which may be expected to be retained in the drug conjugate may be found.
- BSA is employed in the first test to prevent drug conjugate aggregation.
- Tubulin polymerization assay Drug-linker constructs may further be screened using functional assays predictive of biological activity. In one example, microtubule stabilization for paclitaxel drug linker constructs or microtubule disruption by vinblastine drug-linker constructs is determined with a tubulin polymerization assay (Ba ⁇ on, et al, Anal. Biochem. (2003) 315:49-56).
- Tubulin assembly or inhibition thereof may be monitored by fluorescence using the CytoDYNAMIX ScreenTM 10 kit available from Cytoskeleton (1830 S. Acoma St., Denver, CO).
- the kit is based upon an increase in quantum yield of florescence upon binding of a fluorophore to tubulin and microtubules and a 1 OX difference in affinity for microtubules compared to tubulin. Emission is monitored at 405 nm with excitation at 360 nm.
- the compounds such as paclitaxel which enhance tubulin assembly will therefore give an increase in emission whereas compounds such as vinblastine which inhibit tubulin assembly will give a decrease in emission.
- Tubulin assembly or inhibition may also be monitored by light scattering which is approximated by the apparent absorption at 350 nm.
- BSA is employed to prevent aggregation
- glycerol which is a tubulin polymerization enhancer, is omitted from the kit to increase the signal to noise ratio.
- activity of doxorubicin conjugates was assayed by monitoring alteration in the ability of Topoisomerase II, by electrophoresis, to catalyze the formation of relaxed conformation DNA from a super-coiled plasmid. The more active a conjugate is at a particular concentration the less relaxed conformation DNA is produced by the action of Topoisomerase II.
- a functional assay for camptothecin drug-linker constructs depends on inhibition of Topoisomerase I binding to DNA.
- a functional assay for camptothecin drug-linker constructs depends on inhibition of Topoisomerase I binding to DNA (Demarquay, Anti-Cancer Drugs (2001) 12:9-19).
- the enzyme (kinase) and biochemical microtubule polymerization results for all synthetic lots of each compound were combined and analyzed using GraphPad Prism® software to generate the mean ⁇ SD.
- results from all assays carried out with all synthetic lots of each compound were combined and analyzed using Graph Pad Prism software® to generate the mean ⁇ SD.
- the conjugates show increase in the cytotoxic selectivity index of the conjugate for tumor cells as compared to the cytotoxic selectivity index of the parent drug:
- PXL-7Ca-ALK(6)-N3-Thy conjugate as compared to the cytotoxic selectivity index of paclitaxel in exemplary cell lines, as illustrated by improved cytotoxic selectivity index index, is shown below: Cytotoxic Selectivity Index HFF/MCF7 HFF/HT29 Paclitaxel 1.4 0.6 PXL-7Ca- ALK(6)-N3-Thy 11.4 3.8
- the conjugates show better serum stability as compared to the parent drag as demonstrated by an exemplary conjugate below:
- PXL-7Ca- 10 100 84 90 80 38 55 ALK(6)-N3-Thy
- the assays described here may also be used to screen for direct subsfrate-drug conjugates (i.e., conjugates which contain no linker).
- F. Methods of use of the conjugates and compositions Methods of use of the conjugates and compositions provided herein are also provided. The methods involve both in vitro and in vivo uses of the conjugates and compositions. The methods provided herein can be used for increasing drug efficiency. In certain embodiments, methods for treating conditions caused by undesirable chronic or abenant cellular activation, migration, proliferation or survival (ACAMPS) are provided.
- ACAMPS conditions are characterized by undesirable or abe ⁇ ant activation, migration, proliferation or survival of tumor cells, endothelial cells, B cells, T cells, macrophages, granulocytes including neutrophils, eosinophils and basophils, monocytes, platelets, fibroblasts, other connective tissue cells, osteoblasts, osteoclasts and progenitors of many of these cell types.
- ACAMPS-related conditions include, but are not limited to, cancer, coronary restenosis, osteoporosis and syndromes characterized by chronic inflammation and/or autoimmunity.
- Chronic inflammation and/or autoimmune diseases include but are not limited to rheumatoid arthritis and other forms of arthritis, asthma, psoriasis, inflammatory bowel disease, systemic lupus erythematosus, systemic dermatomyositis, inflammatory ophthalmic diseases, autoimmune hematologic disorders, multiple sclerosis, vasculitis, idiopathic nephrotic syndrome, transplant rejection and graft versus host disease.
- cancers include, but are not limited to, non-small cell lung cancer, small cell lung cancer, head and neck squamous cancers, colorectal cancer, prostate cancer, and breast cancer, acute lymphocytic leukemia, adult acute myeloid leukemia, adult non-Hodgkin's lymphoma, brain tumors, cervical cancers, childhood cancers, childhood sarcoma, chronic lymphocytic leukemia, chronic myeloid leukemia, esophageal cancer, hairy cell leukemia, kidney cancer, liver cancer, multiple myeloma, neuroblastoma, oral cancer, pancreatic cancer, primary central nervous system lymphoma, skin cancer, and small-cell lung cancer.
- Childhood cancers amenable to treatment by the methods and with the compositions provided herein include, but are not limited to, brain stem glioma, cerebellar astrocytoma, cerebral astrocytoma, ependymoma, Ewing's sarcoma and family of tumors, germ cell tumor, Hodgkin's disease, ALL, AML, liver cancer, medulloblastoma, neuroblastoma, non-Hodgkin's lymphoma, osteosarcoma, malignant fibrous histiocytoma of bone, retinoblastoma, rhabdomyosarcoma, soft tissue sarcoma, supratentorial primitive neuroectodermal and pineal tumors, unusual childhood cancers, visual pathway and hypothalamic glioma, Wilms' tumor, and other childhood kidney tumors.
- the methods and compositions provided can also be used to treat cancers that originated from or have metastasized to the bone, brain, breast, digestive and gastrointestinal systems, endocrine system, blood, lung, respiratory system, thorax, musculoskeletal system, and skin.
- the methods are generally applicable to all cancers but have particularly significant therapeutic benefit in the freatment of solid tumors.
- the solid tumors are characterized by extensive regions of hypoxic tissue.
- the drug moieties provided in Table 7 are used in the conjugates, which are used in treating particular types of cancer.
- Paclitaxel Texane susceptible to MDR • Breast, Lung, Prostate, Ovarian, Head & Neck, Esophageal, Bladder ⁇ Doxorubicin (Anthracycline / MDR) • Breast, Lung, Ovarian, Bladder, Hepatoma, Neuroblastoma, Lymphoma Vinblastine (Vinca Alkaloid / MDR) • Breast, Lung, Prostate, Testicular, Renal, Lymphoma ⁇ Methotrexate (Antimetabolite) • Breast, Colorectal, Head & Neck, Leukemia, Lymphoma A" Cisplatin (DNA Crosslinking Agent) • Lung, Ovarian, Head & Neck, Esophageal, Bladder, Lymphoma
- the conjugates provided herein are produced using combinatorial methods to produce large libraries of potential conjugates. Methods for producing and screening combinatorial libraries of molecules are known in the art. The libraries of potential conjugates may then be screened for identification of a conjugate with the desired characteristics. Any convenient screening assay may be employed, where the particular screening assay may be known to those of skill in the art or developed in view of the specific molecule and property being studied. For example, the libraries of potential conjugates may be screened for selectivity by comparing the conjugate activity in the target cell or tissue type to conjugate activity in cells or tissues in which drag activity is not desired.
- a selective conjugate will affect the target in the desired cells (e.g., cells involved in a disease process), but affect the target in undesired cells to a lesser extent or not at all.
- the libraries of potential conjugates maybe screened for conjugates that exhibit enhanced drag efficiency as compared to the pharmacological activity of the unconjugated drug. For example, a more efficient drag will result in a desirable pharmacological response at a lower effective dose than a less efficient drag. In another example, a more efficient drug will have an improved cytotoxic selectivity index compared to a less efficient drag.
- the screening assay will involve observing the accumulation of the conjugate in the target system, in comparison to that of the unconjugated drug.
- the conjugates provided are distinguished by retention of drug activity or a significant fraction thereof within the conjugate and therefore do not rely on release of free drag or activation of the drag by an infra-cellular protein.
- the drag moiety and/or the substrate moiety in the conjugate can be present in a form of a pharmaceutically acceptable derivative that renders the conjugate biologically inactive.
- the inactive drag-substrate conjugate can be converted to the active drag-substrate conjugate under physiological conditions or by intracellular proteins without having the need to cleave the drag-substrate conjugate.
- the conjugates are selectively targeted or trapped by cancer or viral infected cells due to phosphorylation of the substrate (e.g. , nucleoside or nucleoside analog by a TK) whose activity is involved in the condition being treated. Accumulation of the drag conjugate into the cancer or viral infected cell types will occur by pushing the equilibrium of passive diffusion towards the cancer or viral infected cells as a result of preferential trapping due to the higher kinase activity within these cell types.
- Drugs such as paclitaxel and vinblastine can be prepared with a biotin moiety or fluorescent tag using procedures known in the art.
- Guillemard et al Anticancer Res . 1999 Nov-Dec; 19(6B):5127-30; Dubois et al, BioorgMed Chem. 1995 Oct; 3(10):1357-68; Chatterjee et al, Biochemistry. 2002 Nov 26; 41(47): 14010-8; Baloglu et al, BioorgMed Chem Lett. 2001 Sep 3; ll(17):2249-52; Li et al, Biochemistry. 2000 Jan 25; 39(3):616-23; Rao et al, BioorgMed Chem.
- Substrate libraries can be conjugated to drags (such as paclitaxel or vinblastine) which contain a biotin moiety or a fluorescent tag.
- drags such as paclitaxel or vinblastine
- a fluorescent drug such as doxorubicin
- the libraries need not be purified.
- conjugates can be incubated with various target cells (ACAMPS disease or normal), followed by removal of the extracellular medium, cell washing and isolation of phosphorylated (trapped or accumilated) conjugates from cell lysates using streptavidin or avidin affinity chromatography. Determination of the trapped or accumulated substrate by standard methods will identify a substrate of an overexpressed or activated kinase expressed in the diseased cell type (or disease-associated normal cell type). This provides a trapping or accumilation of the substrate candidate, which can then be used with the original drag or linked to other drags and optimized. Fluorescently tagged conjugates can be used with drag conjugate libraries that are produced in a "one conjugate per well" format.
- the libraries are incubated with tumor cells, endothelial cells or cells derived from any (ACAMPS) disease tissue, in a multi-well format, followed by washing and determination of well-associated fluorescence.
- Fluorescent drug conjugates that are retained to a high extent by diseased or other target cells represent novel drag candidates. Additionally, specificity can be assessed by comparing fluorescence uptake in the target cell to that in a normal cell type or one not associated with the disease of interest.
- the above methods are not limited to biotinylated or fluorescently tagged conjugates, but can be carried out with any tag or inherent property that facilitates purification or specfrophotometric visualization of conjugates specifically trapped or accumulated in target cells.
- the methods can be used to identify an overexpressed or abe ⁇ antly activated kinase that has not previously been associated with a particular disease.
- a biotinylated drag-substrate conjugate it could also be used to isolate the kinase in question from cell extracts via affinity chromatography.
- the kinase may be a previously identified or novel enzyme.
- the library and screening methods can be applied to small molecule or metabolic kinase substrates.
- conjugates provided herein may be administered as the sole active ingredient or in combination with other active ingredients.
- Other active ingredients that may be used in combination with the conjugates provided herein include but are not limited to, compounds known to treat ACAMPS conditions, anti-angiogenesis agents, anti-tumor agents, other cancer treatments and autoimmune agents.
- Such compounds include, in general, but are not limited to, alkylating agents, toxins, antiproliferative agents and tubulin binding agents.
- Classes of cytotoxic agents for use herein include, for example, the anthracycline family of drags, the vinca drugs, the mitomycins, the bleomycins, the cytotoxic nucleosides, the pteridine family of drags, diynenes, the maytansinoids, the epothilones, the taxanes and the podophyllotoxins.
- Example 6 provides a synthesis of a representative vinblastine drug-linker construct with amide linker to C3.
- Cbz benzyloxycarbonyl
- CDI 1,1' -carbonyldiimidazole
- DCM dichloromethane
- DIEA NN-diisopropylethylamine
- DMAP 4- (dimethylamino)pyridine
- DMF NN-dimethylformamide
- IP A isopropyl alcohol
- MeOH methanol
- MS mass spectroscopy
- RP-HPLC reversed phase high performance liquid chromatography
- RT room temperature
- TEA triethylamine
- TFA trifluoroacetic acid
- Ts Tosyl.
- the imidazolium salt 21 is then dissolved in DMSO (0.5 mL) and 3-(4-amino-phenyl)-propionic acid (500 mol%) is added. The reaction mixture is sti ⁇ ed for 30 minutes at room temperature, diluted with pyridine (0.5 mL). The resulting mixture is cooled to 0°C and HF/Py (233 ⁇ l) is added. Stirring is continued for 3 hours at room temperature. The reaction mixture is then diluted with EtOAc (5 mL) and extracted with saturated aqueous solution of CuSO 4 (3 x 1 mL) followed by water (2 x 2 mL).
- the reaction mixture was sti ⁇ ed at room temperature for 3 hours then concentrated in vacuo to give a residue that was purified by silica gel column chromatography eluting with 95:5 chloroform:methanol.
- the intermediate, 4-deacetyl-3-de- (methoxycarbonyI)-vinblastin-3-yl-N- ⁇ N'-Boc-2-[2-[2-[2- ethoxy]ethoxy]ethoxy]ethylamino ⁇ -aminocarbonyl) was dissolved into 120 mL 1:1 of DCM:TFA and the mixture was stirred at room temperature for 10 minutes.
- Exponentially growing cells (5,000 MCF-7 or HT-29; 1,500 HFF) were plated in 100 ⁇ l medium and incubated overnight (5% CO 2 , 37°C). Compounds (20 pM to 20 ⁇ M final concentration, 6-8 doses) and vehicle (DMSO) controls were added and the incubation was continued for an additional 72 hours. Final cell density was determined by incubating cultures with 25 ⁇ l AlamarBlue reagent (BioSource, Camarillo, CA) for 4 hours, followed by determination of fluorescence at excitation of 544 nm and emission of 590 nm with a SpectroMax Gemini EM fluorescence plate reader (Molecular Devices, Sunnyvale, CA).
- EC 50 values were generated from dose-response curves by a 4-parameter method using Softmax PRO software.
- Mean EC50s ( ⁇ SD) represent the average of all tests carried out for all lots of a given compound.
- Outlier EC 50 values ( ⁇ 7%) were identified and removed prior to analysis using the method of Hoaglin et al., J. Amer. Statistical Assoc, 81, 991-999 (1986).
- Cytotoxicity Assay (soft agar) Assays were carried out in 24-well plates with 0.5 ml bottom layers (0.8% agar) and 0.5 ml top layers (0.38% agar) in RPMI1640 medium containing 5% fetal calf serum.
- Top layers were plated with 1,250 MCF-7 or 5,000 HT-29 cells per well and drugs, compounds or vehicle controls in triplicate as described above. Plates were incubated as above for 10-14 days and then colony formation was assessed by adding 50 ⁇ l AlamarBlue to each well and determining EC50s as described above for monolayer assays. The EC 5 0 values for exemplary conjugates and patent drugs for normal and tumor cells different cell lines are provided in Figure 1-3.
- TKl was amplified using PCR with forward primer 5'-CAATCCATATGAGCTGCATTAACCTGC-3' and reverse primer 5'-TATTAAGCTTCTAGTTGGCAGGGCTGCAT-3 ⁇
- the PCR product was digested with Nde I and Hind EH.
- An N-terminal His-tagged TKl construct, TKl/pET28b(+) was generated by subcloning TKl into the Nde I and Hind III sites of prokaryotic expression vector pET28b (+) (Novagen, San Diego, CA).
- TKl was expressed from TKl/pET28b(+) using E. coli strain BL21-codon plus (Stratagene, San Diego, C A) and purified by Ni column chromatography.
- a coupled (kinetic) ATP depletion assay was developed to measure thymidine kinase activity.
- the reaction contained 100 mM Tris HCI, pH 7.5, 20 mM MgCl 2 , 100 mM KC1, 0.4 mM PEP, 0.2 mM NADH, Pyruvate Kinase (0.7 units) / Lactate Dehydrogenase (1.0 unit) (Sigma #P0294), 2.5-10 ⁇ g Thymidine Kinase, 100 ⁇ M thymidine or thymidine-drag conjugate in a volume of 75 ⁇ l.
- EXAMPLE 18 Fluorescence-based assays for enhancement (paclitaxel) and inhibition (vinblastine) of tubulin polymerization
- the assay kit (#BK011) was purchased from Cytoskeleton (Denver, CO). The assays were carried out according to the manufacturer's instructions, except that 1 mg ml BSA. (Sigma #A3059) was included in all assays.
- Paclitaxel assays were carried out in the absence of glycerol and vinblastine assays were carried out in the presence of 20% glycerol.
- Parent drugs and conjugates were tested at 0.75, 1.5, 3 and 10 micromolar final concentration and results represent a comparison of conjugate and parent drug curves obtained from the linear range of the dose responses.
- Mean percentages of paclitaxel or vinblastine activity ( ⁇ SD) represent the average of all
- Test articles prepared in DMSO as 5 mM stocks, were added to the cell culture media to a final concenfration of 10 mM. Aliquots (150 ml) were withdrawn in triplicate at 0, 4, 8, 24 and 72 hours and combined with the same volume of ice-cold acetonitrile to terminate the reaction. The mixture was centrifuged at 2,000 x g for 10 minutes. One part of the supernatant was mixed with four parts of deionized water to bring down the percentage of organic solvent. The diluted samples were then assayed by LC/MS for the test article. The natural log of the percent remaining was plotted versus time. A linear fit was used to determine the rate constant.
- the fit was truncated after the percent of remaining test article was less than 10%.
- the elimination half-lives associated with the disappearance of test articles were determined to compare their relative stability.
- the assays were carried out by Absorption Systems (Exton, PA).
- EXAMPLE 21 Liver microsome metabolic stability Human and mouse liver microsomes were obtained from Absorption Systems (Exton, PA) and Xenotech (Lenexa, KS), respectively.
- the reaction mixture contained microsomes (human or mouse) 1.0 mg/ml, potassium phosphate, pH 7.4 100 mM, magnesium chloride 10 mM, test article 10 mM, and was equilibrated at 37°C for 3 min.
- the reaction was initiated by adding NADPH (1 mM final), and the system was then incubated in a shaking water bath at 37°C. Aliquots (100 ml) were withdrawn in triplicate at 0, 15, 30, and 60 minutes and combined with 900 ml of ice- cold 50/50 acetonitrile/dH2O to terminate the reaction. Two controls (testosterone and propranolol) were run simultaneously with the test articles in separate reactions. The samples were assayed by LC MS for the test article. The natural log of the percent remaining was plotted versus time. A linear fit was used to determine the rate constant. The fit was truncated when percent remaining of the test article was less than 10%.
- the paclitaxel-thymidine conjugate exhibits cytotoxity against breast carcinoma, colon carcinoma and leukemia.
- EC50 values for paclitaxel and paclitaxel-thymidine conjugate were determined to be 5-8 nM and 75-170 nM, respectively.
- the vinblastine-thymidine conjugate shows a TK substrate activity conesponding to 27 % of thymidine, and a vinblastine activity conesponding to 76 % of vinblastine.
- the vinblastine-thymidine conjugate exhibits cytotoxicity against breast carcinoma, colon carcinoma and leukemia.
- EC50 values for vinblastine and vinblastine-thymidine conjugate were determined to be 1-2 nM and 11 -43 nM, respectively.
- EXAMPLE 23 A comparison of cytotoxic selectivity index for an exemplary conjugate and parent drag in tumors and normal cells shows the increase in the cytotoxic selectivity index of the conjugate for tumor cells as compared to the cytotoxic selectivity index of the parent drag: HFF Monolay MCF-7 MCF-7 Soft HT-29 HT-29 Soft er Monolayer Agar Monolayer Agar EC50 (nM) EC50(nM) EC50 (nM) EC50 (nM) EC50 (nM) EC50 (nM) EC50 (nM) EC50 (nM)
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Epidemiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Molecular Biology (AREA)
- Oncology (AREA)
- Pulmonology (AREA)
- Urology & Nephrology (AREA)
- Dermatology (AREA)
- Biomedical Technology (AREA)
- Genetics & Genomics (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Transplantation (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Ophthalmology & Optometry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Vascular Medicine (AREA)
- Pain & Pain Management (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description






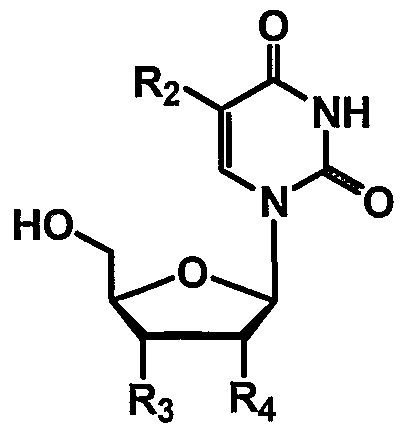






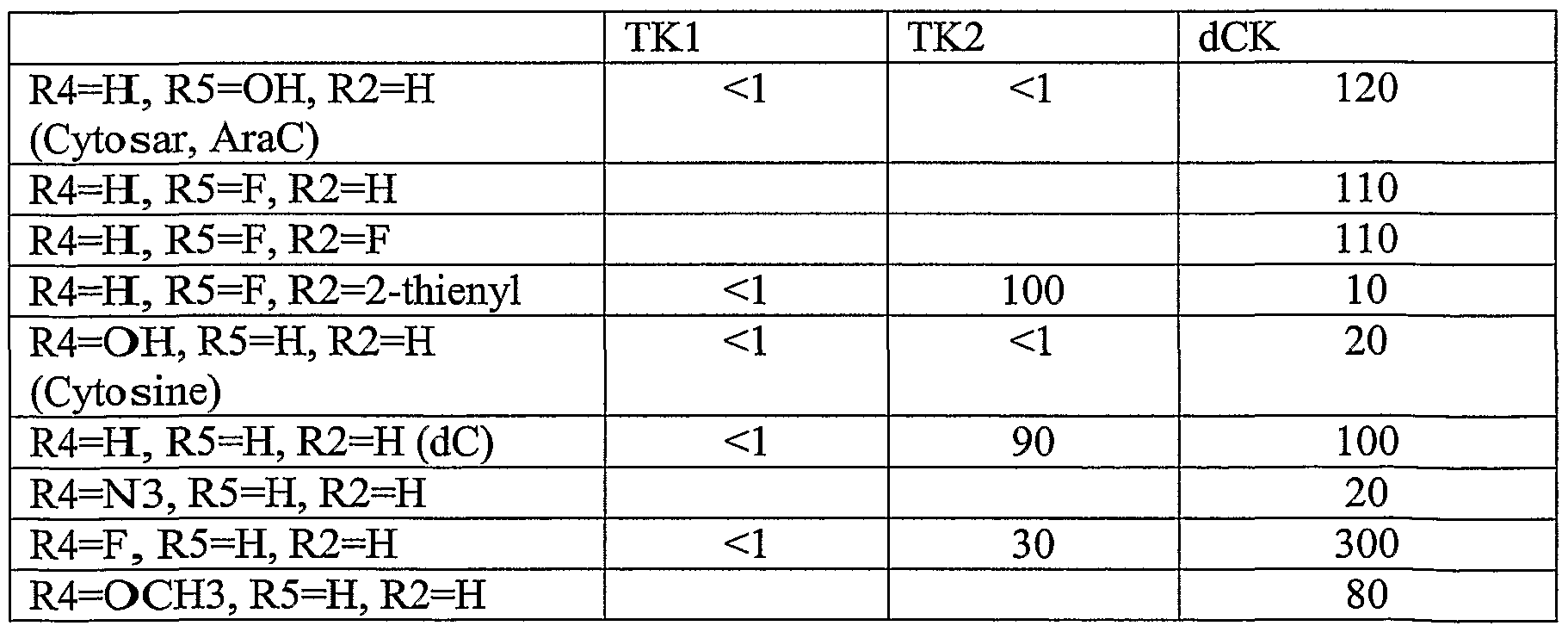






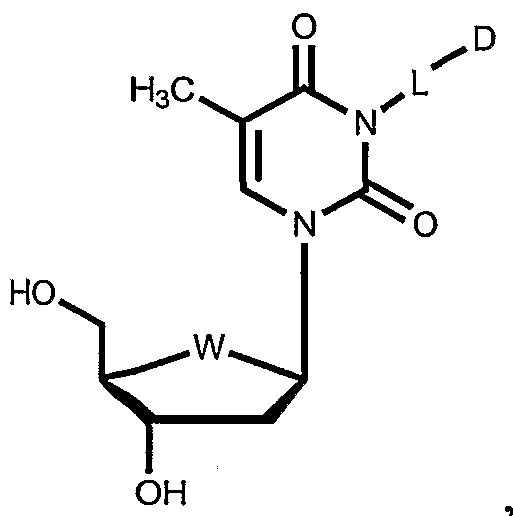




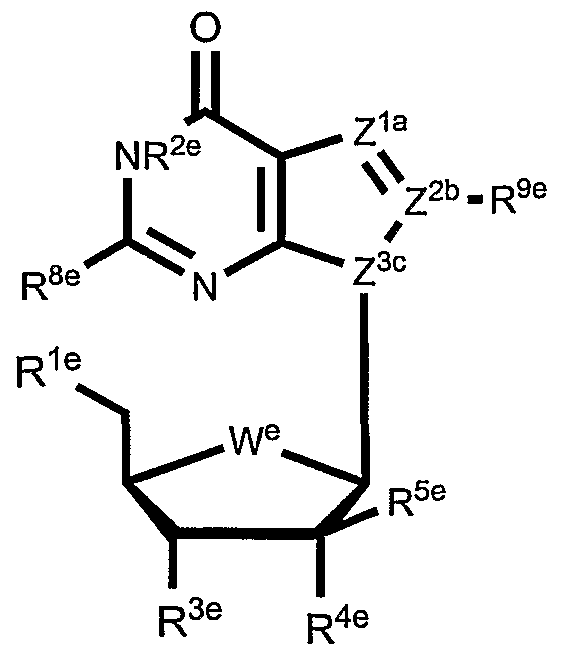












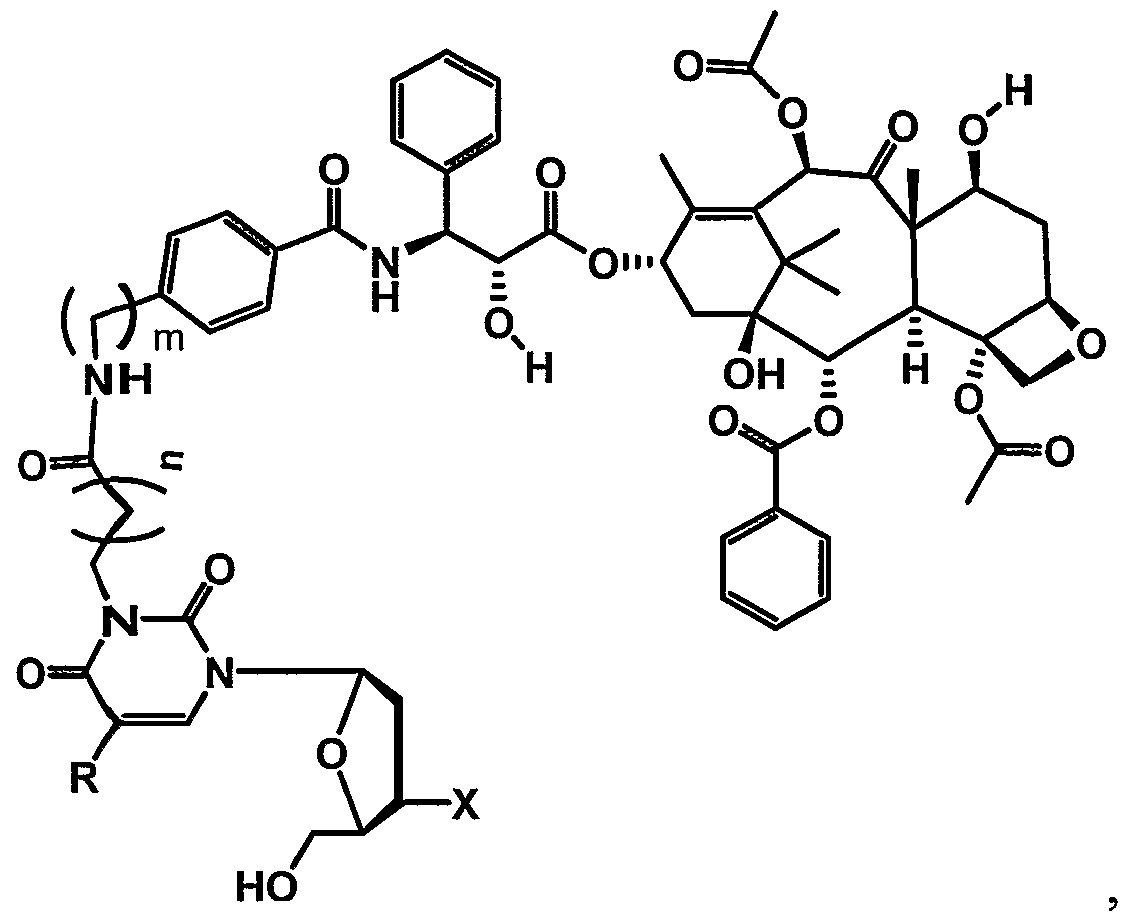














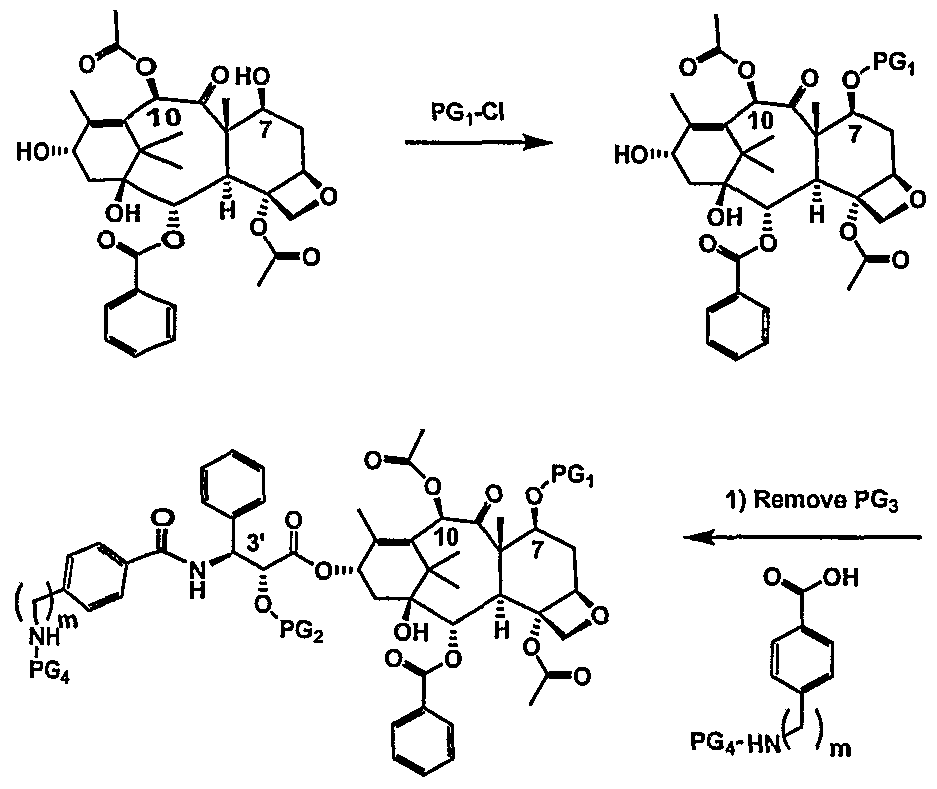





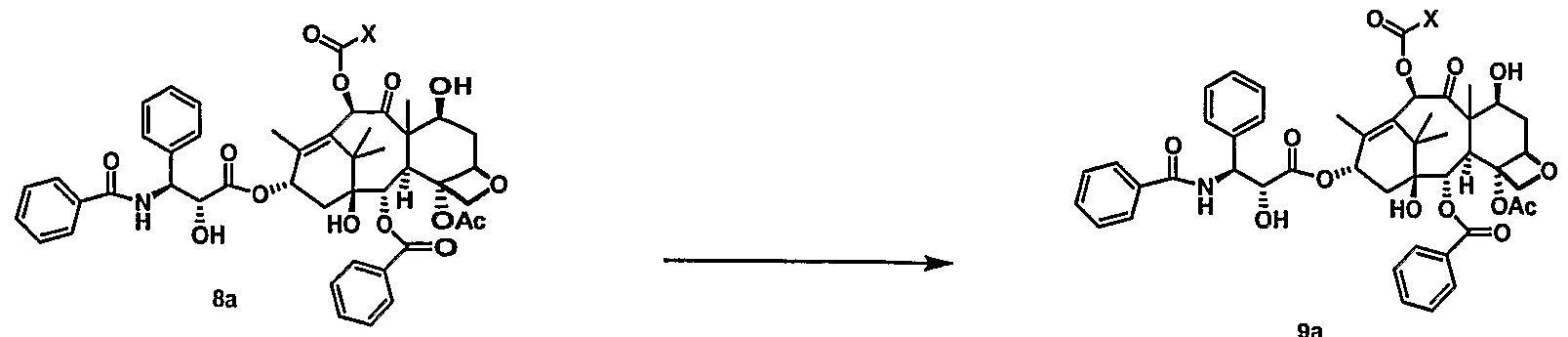

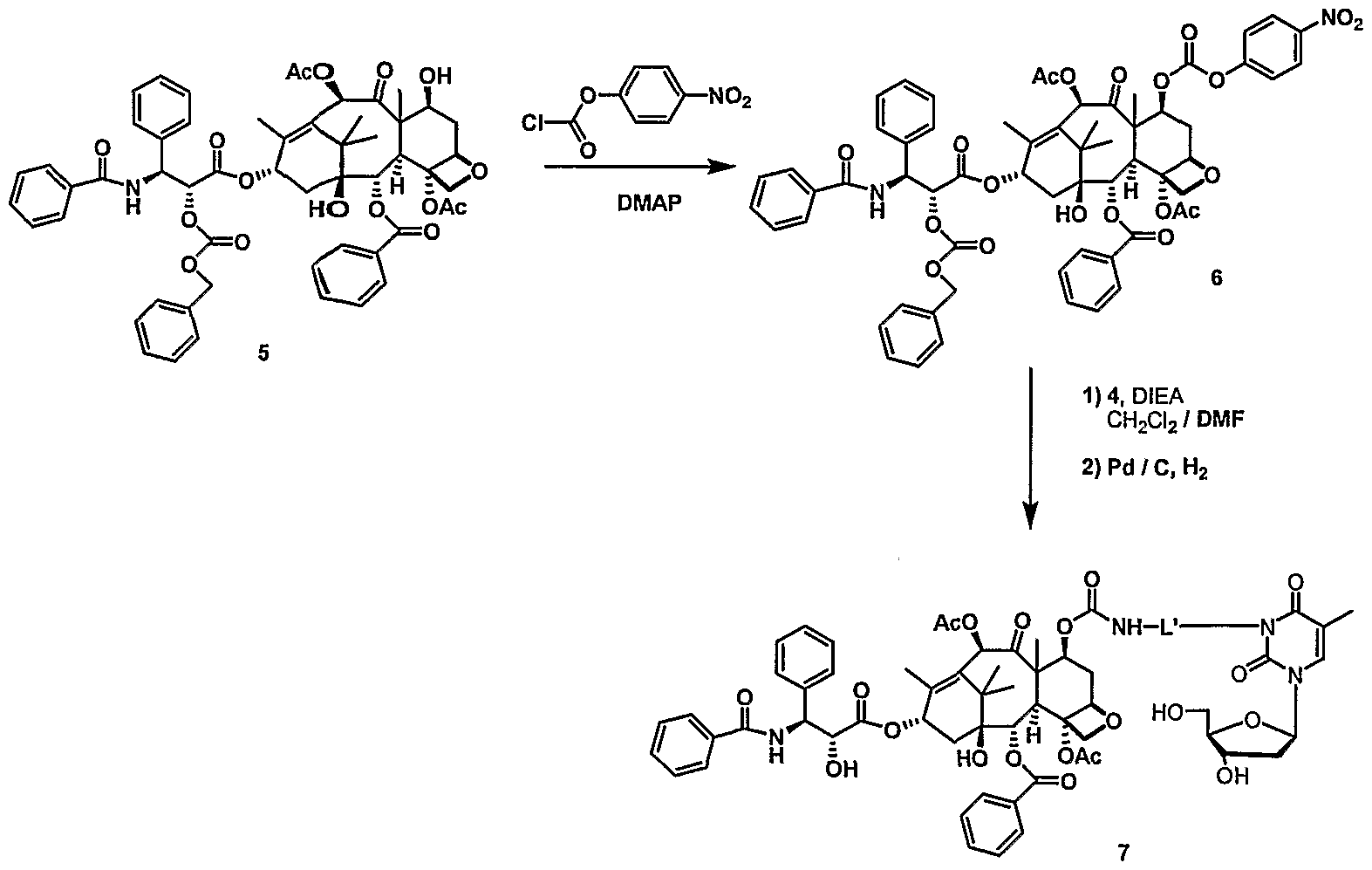




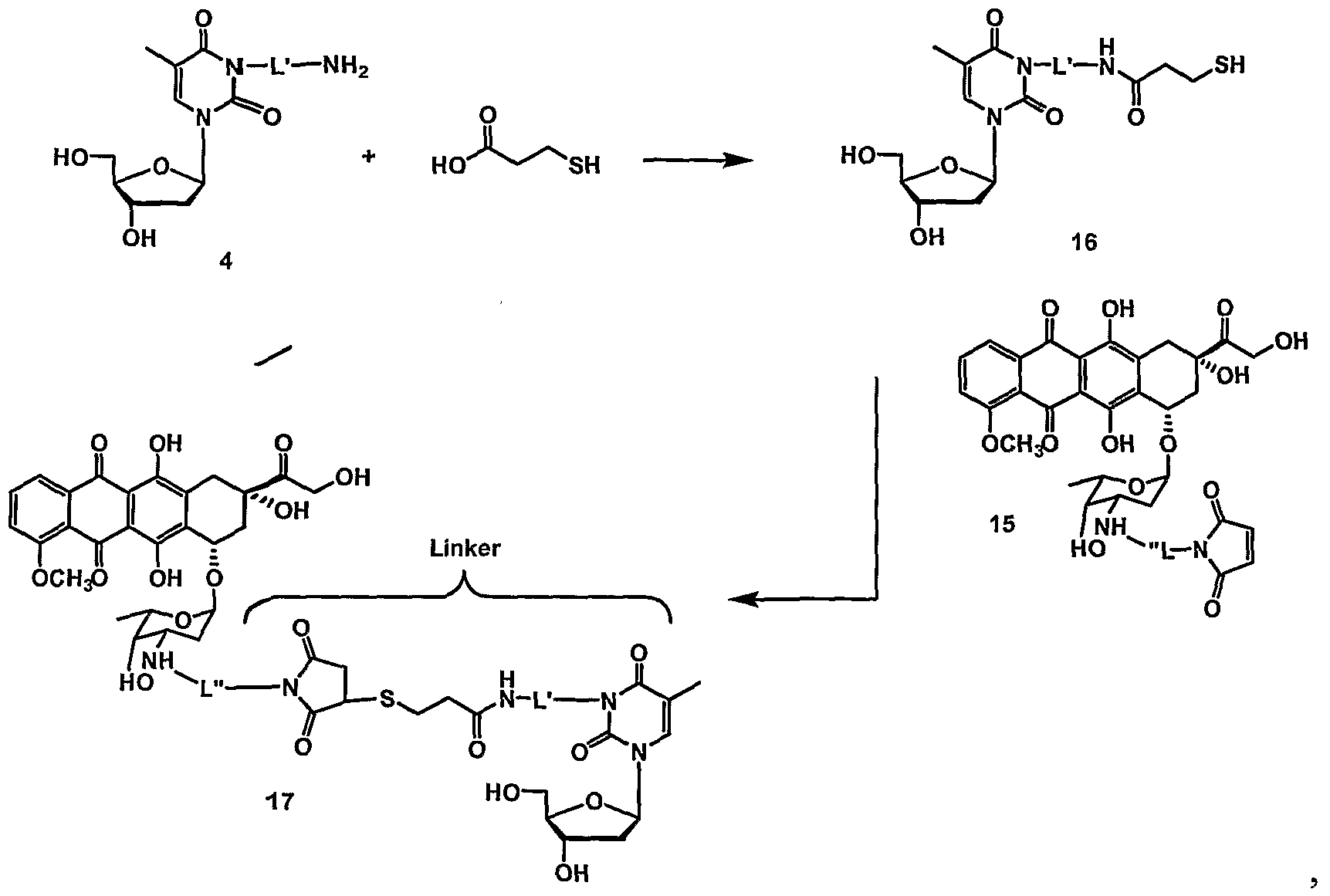












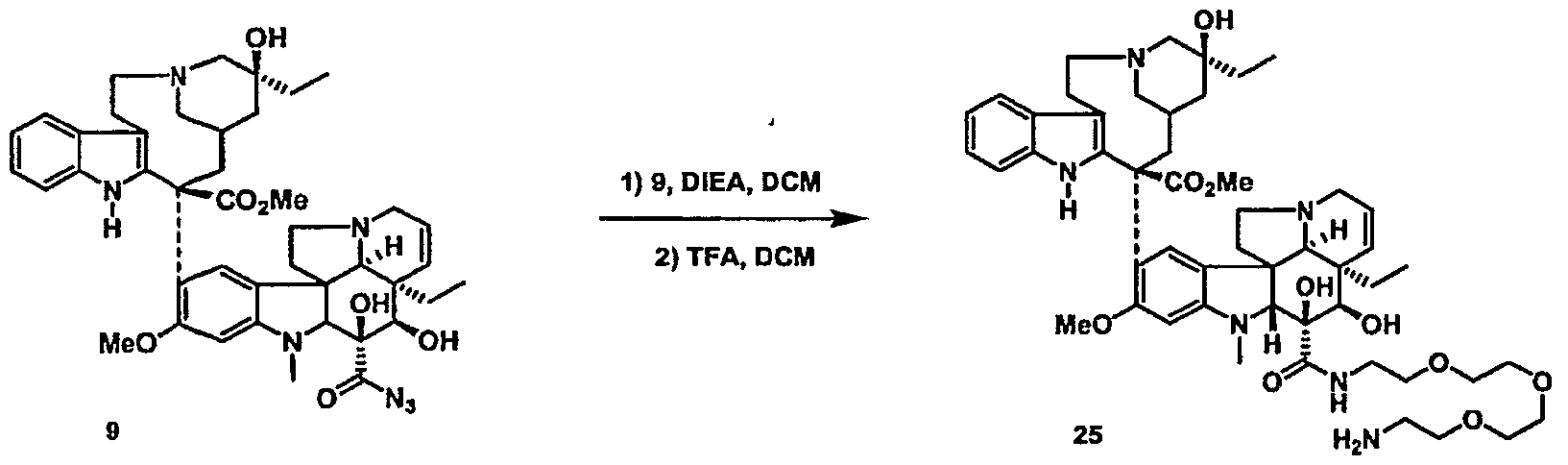







Claims





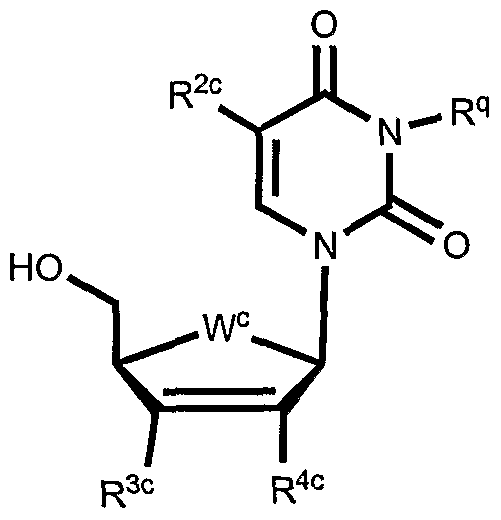

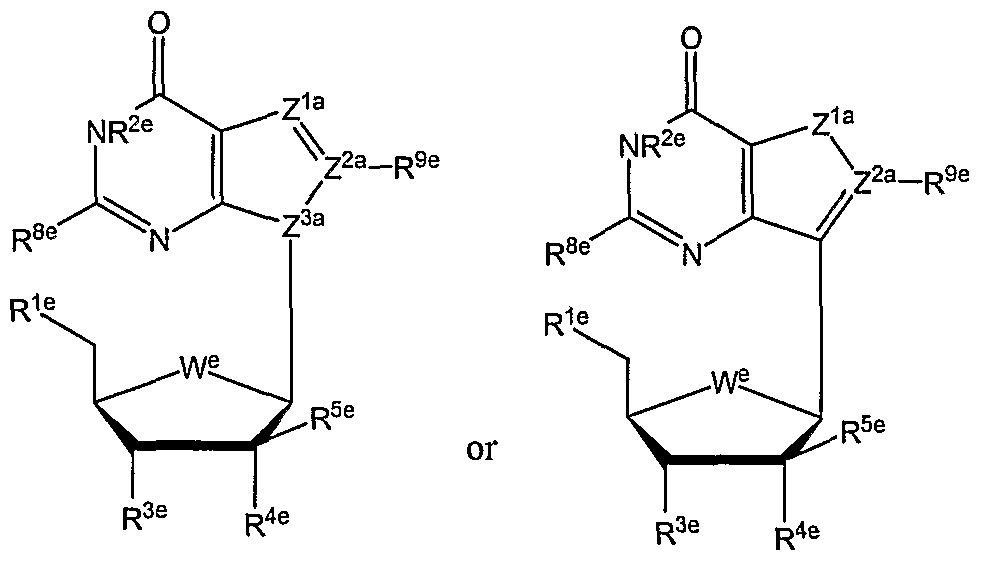
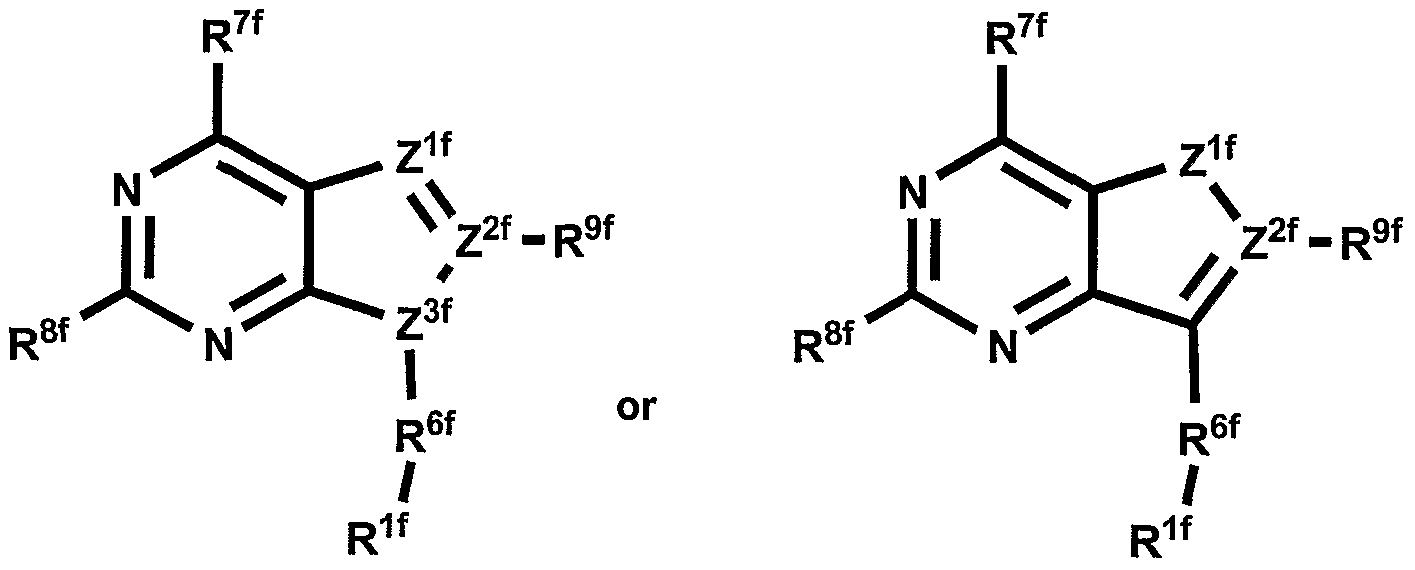






Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2004275770A AU2004275770A1 (en) | 2003-09-22 | 2004-09-22 | Small molecule compositions and methods for increasing drug efficiency using compositions thereof |
| EP04784836A EP1689439A2 (en) | 2003-09-22 | 2004-09-22 | Small molecule compositions and methods for increasing drug efficiency using compositions thereof |
| CA002539914A CA2539914A1 (en) | 2003-09-22 | 2004-09-22 | Small molecule compositions and methods for increasing drug efficiency using compositions thereof |
| JP2006527154A JP2007522094A (en) | 2003-09-22 | 2004-09-22 | Small molecule composition and method for increasing drug efficiency using the composition |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US50532503P | 2003-09-22 | 2003-09-22 | |
| US60/505,325 | 2003-09-22 | ||
| US58183504P | 2004-06-22 | 2004-06-22 | |
| US60/581,835 | 2004-06-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005030258A2 true WO2005030258A2 (en) | 2005-04-07 |
| WO2005030258A3 WO2005030258A3 (en) | 2005-09-22 |
Family
ID=34396244
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/031147 WO2005030258A2 (en) | 2003-09-22 | 2004-09-22 | Small molecule compositions and methods for increasing drug efficiency using compositions thereof |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1689439A2 (en) |
| JP (1) | JP2007522094A (en) |
| AU (1) | AU2004275770A1 (en) |
| CA (1) | CA2539914A1 (en) |
| WO (1) | WO2005030258A2 (en) |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007076160A2 (en) * | 2005-12-28 | 2007-07-05 | Acidophil Llc | C-10 carbamates of taxanes |
| EP2282724A2 (en) * | 2008-05-23 | 2011-02-16 | The University Of British Columbia | Modified drugs for use in liposomal nanoparticles |
| WO2014210564A1 (en) | 2013-06-27 | 2014-12-31 | Academia Sinica | Glycan conjugates and use thereof |
| US9556216B2 (en) | 2012-08-31 | 2017-01-31 | Novartis Ag | 2′-Ethynyl nucleoside derivatives for treatment of viral infections |
| US9732111B2 (en) | 2012-11-19 | 2017-08-15 | Merck Sharp & Dohme Corp. | 2′-alkynyl substituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| US9968628B2 (en) | 2000-05-26 | 2018-05-15 | Idenix Pharmaceuticals Llc | Methods and compositions for treating flaviviruses and pestiviruses |
| US9975965B2 (en) | 2015-01-16 | 2018-05-22 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US9982041B2 (en) | 2014-01-16 | 2018-05-29 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US10005847B2 (en) | 2014-05-27 | 2018-06-26 | Academia Sinica | Anti-HER2 glycoantibodies and uses thereof |
| US10023892B2 (en) | 2014-05-27 | 2018-07-17 | Academia Sinica | Compositions and methods relating to universal glycoforms for enhanced antibody efficacy |
| US10086054B2 (en) | 2013-06-26 | 2018-10-02 | Academia Sinica | RM2 antigens and use thereof |
| US10087236B2 (en) | 2009-12-02 | 2018-10-02 | Academia Sinica | Methods for modifying human antibodies by glycan engineering |
| US10111951B2 (en) | 2013-09-06 | 2018-10-30 | Academia Sinica | Human iNKT cell activation using glycolipids with altered glycosyl groups |
| US10118969B2 (en) | 2014-05-27 | 2018-11-06 | Academia Sinica | Compositions and methods relating to universal glycoforms for enhanced antibody efficacy |
| US10119972B2 (en) | 2014-03-27 | 2018-11-06 | Academia Sinica | Reactive labelling compounds and uses thereof |
| US10130714B2 (en) | 2012-04-14 | 2018-11-20 | Academia Sinica | Enhanced anti-influenza agents conjugated with anti-inflammatory activity |
| US10150818B2 (en) | 2014-01-16 | 2018-12-11 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US10214765B2 (en) | 2012-08-18 | 2019-02-26 | Academia Sinica | Cell-permeable probes for identification and imaging of sialidases |
| US10274488B2 (en) | 2008-07-15 | 2019-04-30 | Academia Sinica | Glycan arrays on PTFE-like aluminum coated glass slides and related methods |
| US10317393B2 (en) | 2007-03-23 | 2019-06-11 | Academia Sinica | Alkynyl sugar analogs for labeling and visualization of glycoconjugates in cells |
| US10336784B2 (en) | 2016-03-08 | 2019-07-02 | Academia Sinica | Methods for modular synthesis of N-glycans and arrays thereof |
| US10338069B2 (en) | 2010-04-12 | 2019-07-02 | Academia Sinica | Glycan arrays for high throughput screening of viruses |
| US10342858B2 (en) | 2015-01-24 | 2019-07-09 | Academia Sinica | Glycan conjugates and methods of use thereof |
| US10363265B2 (en) | 2000-05-23 | 2019-07-30 | Idenix Pharmaceuticals Llc | Methods and compositions for treating hepatitis C virus |
| US10495645B2 (en) | 2015-01-16 | 2019-12-03 | Academia Sinica | Cancer markers and methods of use thereof |
| US10525072B2 (en) | 2002-11-15 | 2020-01-07 | Idenix Pharmaceuticals Llc | 2′-branched nucleosides and flaviviridae mutation |
| US10533034B2 (en) | 2014-09-08 | 2020-01-14 | Academia Sinica | Human iNKT cell activation using glycolipids |
| US10538592B2 (en) | 2016-08-22 | 2020-01-21 | Cho Pharma, Inc. | Antibodies, binding fragments, and methods of use |
| US11332523B2 (en) | 2014-05-28 | 2022-05-17 | Academia Sinica | Anti-TNF-alpha glycoantibodies and uses thereof |
| US11377485B2 (en) | 2009-12-02 | 2022-07-05 | Academia Sinica | Methods for modifying human antibodies by glycan engineering |
| US11884739B2 (en) | 2014-05-27 | 2024-01-30 | Academia Sinica | Anti-CD20 glycoantibodies and uses thereof |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2019268412A1 (en) * | 2018-05-15 | 2020-12-03 | Interk Peptide Therapeutics Limited | Peptide activating agent |
| AU2019268411B2 (en) * | 2018-05-15 | 2024-03-14 | Interk Peptide Therapeutics Limited | Activating agents |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4368138A (en) * | 1979-01-10 | 1983-01-11 | Israel Marcus | Water soluble thymidine derivative |
| EP0735044A1 (en) * | 1995-03-10 | 1996-10-02 | Bristol-Myers Squibb Company | Preparation of D4T from 5-methyluridine |
| US5723589A (en) * | 1995-12-21 | 1998-03-03 | Icn Pharmaceuticals | Carbohydrate conjugated bio-active compounds |
-
2004
- 2004-09-22 AU AU2004275770A patent/AU2004275770A1/en not_active Abandoned
- 2004-09-22 CA CA002539914A patent/CA2539914A1/en not_active Abandoned
- 2004-09-22 JP JP2006527154A patent/JP2007522094A/en not_active Abandoned
- 2004-09-22 WO PCT/US2004/031147 patent/WO2005030258A2/en not_active Application Discontinuation
- 2004-09-22 EP EP04784836A patent/EP1689439A2/en not_active Withdrawn
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4368138A (en) * | 1979-01-10 | 1983-01-11 | Israel Marcus | Water soluble thymidine derivative |
| EP0735044A1 (en) * | 1995-03-10 | 1996-10-02 | Bristol-Myers Squibb Company | Preparation of D4T from 5-methyluridine |
| US5723589A (en) * | 1995-12-21 | 1998-03-03 | Icn Pharmaceuticals | Carbohydrate conjugated bio-active compounds |
Non-Patent Citations (5)
| Title |
|---|
| AKANNI ABRAHAM ET AL: "Estrogen-nucleic acid adducts: Dissection of the reaction of 3,4-estrone quinone and its radical anion and radical cation with deoxynucleosides and DNA" CHEMICAL RESEARCH IN TOXICOLOGY, vol. 12, no. 12, December 1999 (1999-12), pages 1247-1253, XP002315588 ISSN: 0893-228X * |
| CHENG Q Q ET AL: "Synthesis and biological evaluation of novel 9-functional heterocyclic coupled 7-deoxy-9-Dihydropaclitaxel analogue" BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 10, no. 5, March 2000 (2000-03), pages 517-521, XP004202451 ISSN: 0960-894X * |
| MOOSAVI-MOVAHEDI ALI A ET AL: "Design, synthesis, and anticancer activity of phosphonic acid diphosphate derivative of adenine-containing butenolide and its water-soluble derivatives of paclitaxel with high antitumor activity." BIOORGANIC & MEDICINAL CHEMISTRY, vol. 11, no. 20, 3 September 2003 (2003-09-03), pages 4303-4313, XP002315844 ISSN: 0968-0896 [retrieved on 2003-09-03] * |
| NEWMAN M ET AL: "611 Introduction of specificity into cytotoxic drugs and improvement of therapeutic index by kinase-mediated trapping" EUROPEAN JOURNAL OF CANCER. SUPPLEMENT, PERGAMON, OXFORD, GB, vol. 2, no. 8, September 2004 (2004-09), page 185, XP004640055 ISSN: 1359-6349 * |
| VELAZQUEZ S ET AL: "Novel series of (ddN)-(TSAO-T) heterodimers as potential bi-functional inhibitors of HIV-1 RT. Studies in the linker and ddN region" NUCLEOSIDES AND NUCLEOTIDES, vol. 18, no. 4-5, April 1999 (1999-04), pages 1029-1030, XP009043245 ISSN: 0732-8311 * |
Cited By (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10363265B2 (en) | 2000-05-23 | 2019-07-30 | Idenix Pharmaceuticals Llc | Methods and compositions for treating hepatitis C virus |
| US10758557B2 (en) | 2000-05-23 | 2020-09-01 | Idenix Pharmaceuticals Llc | Methods and compositions for treating hepatitis C virus |
| US9968628B2 (en) | 2000-05-26 | 2018-05-15 | Idenix Pharmaceuticals Llc | Methods and compositions for treating flaviviruses and pestiviruses |
| US10525072B2 (en) | 2002-11-15 | 2020-01-07 | Idenix Pharmaceuticals Llc | 2′-branched nucleosides and flaviviridae mutation |
| US8138361B2 (en) | 2005-12-28 | 2012-03-20 | The Trustees Of The University Of Pennsylvania | C-10 carbamates of taxanes |
| WO2007076160A2 (en) * | 2005-12-28 | 2007-07-05 | Acidophil Llc | C-10 carbamates of taxanes |
| WO2007076160A3 (en) * | 2005-12-28 | 2007-11-15 | Acidophil Llc | C-10 carbamates of taxanes |
| US10317393B2 (en) | 2007-03-23 | 2019-06-11 | Academia Sinica | Alkynyl sugar analogs for labeling and visualization of glycoconjugates in cells |
| EP2282724A4 (en) * | 2008-05-23 | 2015-01-21 | Univ British Columbia | Modified drugs for use in liposomal nanoparticles |
| EP2282724A2 (en) * | 2008-05-23 | 2011-02-16 | The University Of British Columbia | Modified drugs for use in liposomal nanoparticles |
| US10274488B2 (en) | 2008-07-15 | 2019-04-30 | Academia Sinica | Glycan arrays on PTFE-like aluminum coated glass slides and related methods |
| US10087236B2 (en) | 2009-12-02 | 2018-10-02 | Academia Sinica | Methods for modifying human antibodies by glycan engineering |
| US11377485B2 (en) | 2009-12-02 | 2022-07-05 | Academia Sinica | Methods for modifying human antibodies by glycan engineering |
| US11267870B2 (en) | 2009-12-02 | 2022-03-08 | Academia Sinica | Methods for modifying human antibodies by glycan engineering |
| US10338069B2 (en) | 2010-04-12 | 2019-07-02 | Academia Sinica | Glycan arrays for high throughput screening of viruses |
| US10130714B2 (en) | 2012-04-14 | 2018-11-20 | Academia Sinica | Enhanced anti-influenza agents conjugated with anti-inflammatory activity |
| US10214765B2 (en) | 2012-08-18 | 2019-02-26 | Academia Sinica | Cell-permeable probes for identification and imaging of sialidases |
| US9814739B2 (en) | 2012-08-31 | 2017-11-14 | Novartis Ag | 2′-ethynyl nucleoside derivatives for treatment of viral infections |
| US9556216B2 (en) | 2012-08-31 | 2017-01-31 | Novartis Ag | 2′-Ethynyl nucleoside derivatives for treatment of viral infections |
| US9732111B2 (en) | 2012-11-19 | 2017-08-15 | Merck Sharp & Dohme Corp. | 2′-alkynyl substituted nucleoside derivatives and methods of use thereof for the treatment of viral diseases |
| US10086054B2 (en) | 2013-06-26 | 2018-10-02 | Academia Sinica | RM2 antigens and use thereof |
| US9981030B2 (en) | 2013-06-27 | 2018-05-29 | Academia Sinica | Glycan conjugates and use thereof |
| EP3013347A4 (en) * | 2013-06-27 | 2017-01-18 | Academia Sinica | Glycan conjugates and use thereof |
| WO2014210564A1 (en) | 2013-06-27 | 2014-12-31 | Academia Sinica | Glycan conjugates and use thereof |
| US10918714B2 (en) | 2013-09-06 | 2021-02-16 | Academia Sinica | Human iNKT cell activation using glycolipids with altered glycosyl groups |
| US10111951B2 (en) | 2013-09-06 | 2018-10-30 | Academia Sinica | Human iNKT cell activation using glycolipids with altered glycosyl groups |
| US9982041B2 (en) | 2014-01-16 | 2018-05-29 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US10150818B2 (en) | 2014-01-16 | 2018-12-11 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US10119972B2 (en) | 2014-03-27 | 2018-11-06 | Academia Sinica | Reactive labelling compounds and uses thereof |
| US11319567B2 (en) | 2014-05-27 | 2022-05-03 | Academia Sinica | Fucosidase from bacteroides and methods using the same |
| US10618973B2 (en) | 2014-05-27 | 2020-04-14 | Academia Sinica | Anti-HER2 glycoantibodies and uses thereof |
| US10118969B2 (en) | 2014-05-27 | 2018-11-06 | Academia Sinica | Compositions and methods relating to universal glycoforms for enhanced antibody efficacy |
| US10023892B2 (en) | 2014-05-27 | 2018-07-17 | Academia Sinica | Compositions and methods relating to universal glycoforms for enhanced antibody efficacy |
| US10005847B2 (en) | 2014-05-27 | 2018-06-26 | Academia Sinica | Anti-HER2 glycoantibodies and uses thereof |
| US11884739B2 (en) | 2014-05-27 | 2024-01-30 | Academia Sinica | Anti-CD20 glycoantibodies and uses thereof |
| US11332523B2 (en) | 2014-05-28 | 2022-05-17 | Academia Sinica | Anti-TNF-alpha glycoantibodies and uses thereof |
| US10533034B2 (en) | 2014-09-08 | 2020-01-14 | Academia Sinica | Human iNKT cell activation using glycolipids |
| US10495645B2 (en) | 2015-01-16 | 2019-12-03 | Academia Sinica | Cancer markers and methods of use thereof |
| US9975965B2 (en) | 2015-01-16 | 2018-05-22 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US10342858B2 (en) | 2015-01-24 | 2019-07-09 | Academia Sinica | Glycan conjugates and methods of use thereof |
| US10336784B2 (en) | 2016-03-08 | 2019-07-02 | Academia Sinica | Methods for modular synthesis of N-glycans and arrays thereof |
| US10538592B2 (en) | 2016-08-22 | 2020-01-21 | Cho Pharma, Inc. | Antibodies, binding fragments, and methods of use |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2005030258A3 (en) | 2005-09-22 |
| AU2004275770A1 (en) | 2005-04-07 |
| EP1689439A2 (en) | 2006-08-16 |
| JP2007522094A (en) | 2007-08-09 |
| CA2539914A1 (en) | 2005-04-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2005030258A2 (en) | Small molecule compositions and methods for increasing drug efficiency using compositions thereof | |
| US20050148534A1 (en) | Small molecule compositions and methods for increasing drug efficiency using compositions thereof | |
| JP2675749B2 (en) | Desazapurine-nucleoside-derivative | |
| US6573247B1 (en) | Anti-viral pyrimidine nucleoside analogues | |
| US20080132525A1 (en) | Inhibitors of DNA Methyltransferase | |
| US7019135B2 (en) | Anti-viral pyrimidine nucleoside analogues | |
| WO2005035003A2 (en) | Compositions and methods for increasing drug efficiency | |
| WO2003062257A1 (en) | Deazapurine nucleoside analogs and their use as therapeutic agents | |
| KR20070087687A (en) | Pyrimido compounds having antiproliferative activity | |
| JPH04505932A (en) | Three novel non-polyglutamate deazaaminopterins and their synthesis methods | |
| WO2003051898A1 (en) | Unusual nucleoside libraries, compounds, and preferred uses as antiviral and anticancer agents | |
| US6372725B1 (en) | Specific lipid conjugates to nucleoside diphosphates and their use as drugs | |
| US11453695B2 (en) | Nucleoside analogues and methods of use thereof | |
| WO1990009177A1 (en) | Cyclopentenyl pyrimidines and methods of using same | |
| KR20050109939A (en) | Nucleotide lipid ester derivatives | |
| Silamkoti et al. | Synthesis and biological activity of 2-fluoro adenine and 6-methyl purine nucleoside analogs as prodrugs for suicide gene therapy of cancer | |
| WO2020013712A1 (en) | Inhibitors of dnmt1 as anticancer therapeutic agents | |
| US7217815B2 (en) | 2-beta -modified-6-substituted adenosine analogs and their use as antiviral agents | |
| NZ569140A (en) | Dioxolane derivatives for the treatment of cancer | |
| Mugnaini et al. | Research on L-nucleosides. Synthesis and biological evaluation of a series of L-and D-2′, 3′-dideoxy-3′-[tris (methylthio) methyl]-β-pentofuranosyl nucleosides | |
| Bhattacharya et al. | Studies on the synthesis of furo [3, 2-d] pyrimidine C-nucleosides: new inosine analogues with antiprotozoan activity | |
| Janeba et al. | Synthesis of 8-amino-and N-substituted 8-aminoadenine derivatives of acyclic nucleoside and nucleotide analogs | |
| Suresh et al. | Convergent synthesis of 2-thioether-substituted (N)-methanocarba-adenosines as purine receptor agonists | |
| Mashelkar | Design, Synthesis and SARs of 7-Deaza-4'-thioadenosine Derivatives as Multi-Kinase Inhibitors targeting Cancer Using Polypharmacology Approach | |
| JP2022535386A (en) | carbocyclic nucleoside analogues |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006527154 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2539914 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004784836 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004275770 Country of ref document: AU |
|
| ENP | Entry into the national phase in: |
Ref document number: 2004275770 Country of ref document: AU Date of ref document: 20040922 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004275770 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004784836 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2004784836 Country of ref document: EP |