WO2004103272A2 - Improved cytotoxic agents comprising new maytansinoids - Google Patents
Improved cytotoxic agents comprising new maytansinoids Download PDFInfo
- Publication number
- WO2004103272A2 WO2004103272A2 PCT/US2004/013314 US2004013314W WO2004103272A2 WO 2004103272 A2 WO2004103272 A2 WO 2004103272A2 US 2004013314 W US2004013314 W US 2004013314W WO 2004103272 A2 WO2004103272 A2 WO 2004103272A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- maytansinoid
- carbon atoms
- alkenyl
- methyl
- Prior art date
Links
- 229940127089 cytotoxic agent Drugs 0.000 title description 5
- 239000002254 cytotoxic agent Substances 0.000 title description 5
- 231100000599 cytotoxic agent Toxicity 0.000 title description 5
- 238000000034 method Methods 0.000 claims abstract description 196
- 239000011230 binding agent Substances 0.000 claims abstract description 121
- 125000003396 thiol group Chemical class [H]S* 0.000 claims abstract description 62
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 42
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 39
- 125000004434 sulfur atom Chemical group 0.000 claims abstract description 39
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 claims abstract description 20
- 125000004432 carbon atom Chemical group C* 0.000 claims description 244
- 125000003342 alkenyl group Chemical group 0.000 claims description 216
- 150000001875 compounds Chemical class 0.000 claims description 190
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 159
- -1 heterocycloalkyl radical Chemical class 0.000 claims description 145
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 141
- 125000006615 aromatic heterocyclic group Chemical group 0.000 claims description 118
- 125000000217 alkyl group Chemical group 0.000 claims description 114
- GDFAOVXKHJXLEI-VKHMYHEASA-N N-methyl-L-alanine Chemical compound C[NH2+][C@@H](C)C([O-])=O GDFAOVXKHJXLEI-VKHMYHEASA-N 0.000 claims description 109
- 239000000203 mixture Substances 0.000 claims description 87
- QWPXBEHQFHACTK-KZVYIGENSA-N (10e,12e)-86-chloro-12,14,4-trihydroxy-85,14-dimethoxy-33,2,7,10-tetramethyl-15,16-dihydro-14h-7-aza-1(6,4)-oxazina-3(2,3)-oxirana-8(1,3)-benzenacyclotetradecaphane-10,12-dien-6-one Chemical compound CN1C(=O)CC(O)C2(C)OC2C(C)C(OC(=O)N2)CC2(O)C(OC)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 QWPXBEHQFHACTK-KZVYIGENSA-N 0.000 claims description 68
- QWPXBEHQFHACTK-UHFFFAOYSA-N Maytansinol Natural products CN1C(=O)CC(O)C2(C)OC2C(C)C(OC(=O)N2)CC2(O)C(OC)C=CC=C(C)CC2=CC(OC)=C(Cl)C1=C2 QWPXBEHQFHACTK-UHFFFAOYSA-N 0.000 claims description 58
- GDFAOVXKHJXLEI-UHFFFAOYSA-N L-N-Boc-N-methylalanine Natural products CNC(C)C(O)=O GDFAOVXKHJXLEI-UHFFFAOYSA-N 0.000 claims description 54
- 238000011282 treatment Methods 0.000 claims description 53
- NEAFWRKPYYJETG-UHFFFAOYSA-N 4-sulfanylpentanoic acid Chemical compound CC(S)CCC(O)=O NEAFWRKPYYJETG-UHFFFAOYSA-N 0.000 claims description 44
- 125000003107 substituted aryl group Chemical group 0.000 claims description 43
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 claims description 42
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 37
- XYONNSVDNIRXKZ-UHFFFAOYSA-N S-methyl methanethiosulfonate Chemical compound CSS(C)(=O)=O XYONNSVDNIRXKZ-UHFFFAOYSA-N 0.000 claims description 36
- 150000003839 salts Chemical class 0.000 claims description 36
- 239000012453 solvate Substances 0.000 claims description 36
- GDFAOVXKHJXLEI-GSVOUGTGSA-N (2r)-2-(methylamino)propanoic acid Chemical compound CN[C@H](C)C(O)=O GDFAOVXKHJXLEI-GSVOUGTGSA-N 0.000 claims description 33
- 238000006243 chemical reaction Methods 0.000 claims description 33
- NMHMNPHRMNGLLB-UHFFFAOYSA-N phloretic acid Chemical compound OC(=O)CCC1=CC=C(O)C=C1 NMHMNPHRMNGLLB-UHFFFAOYSA-N 0.000 claims description 33
- 230000032050 esterification Effects 0.000 claims description 32
- 238000005886 esterification reaction Methods 0.000 claims description 32
- 125000002252 acyl group Chemical group 0.000 claims description 31
- 238000004519 manufacturing process Methods 0.000 claims description 30
- 125000001424 substituent group Chemical group 0.000 claims description 30
- 229940125773 compound 10 Drugs 0.000 claims description 29
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 claims description 29
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 27
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 23
- 230000008569 process Effects 0.000 claims description 23
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 claims description 22
- 229940126543 compound 14 Drugs 0.000 claims description 22
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 21
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 claims description 21
- 229940125876 compound 15a Drugs 0.000 claims description 20
- CXYRUNPLKGGUJF-OZVSTBQFSA-M pamine Chemical compound [Br-].C1([C@@H](CO)C(=O)OC2C[C@@H]3[N+]([C@H](C2)[C@@H]2[C@H]3O2)(C)C)=CC=CC=C1 CXYRUNPLKGGUJF-OZVSTBQFSA-M 0.000 claims description 20
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 claims description 18
- 239000003085 diluting agent Substances 0.000 claims description 17
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 17
- SYIFSIGCMOMQRB-UHFFFAOYSA-N 4-methyl-4-sulfanylpentanoic acid Chemical compound CC(C)(S)CCC(O)=O SYIFSIGCMOMQRB-UHFFFAOYSA-N 0.000 claims description 16
- 150000002148 esters Chemical class 0.000 claims description 16
- 239000008194 pharmaceutical composition Substances 0.000 claims description 16
- TZSZZENYCISATO-WIOPSUGQSA-N rodatristat Chemical compound CCOC(=O)[C@@H]1CC2(CN1)CCN(CC2)c1cc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)nc(N)n1 TZSZZENYCISATO-WIOPSUGQSA-N 0.000 claims description 16
- 150000001413 amino acids Chemical group 0.000 claims description 15
- 239000003937 drug carrier Substances 0.000 claims description 15
- 230000030833 cell death Effects 0.000 claims description 14
- 125000002228 disulfide group Chemical group 0.000 claims description 14
- 238000004128 high performance liquid chromatography Methods 0.000 claims description 14
- 230000001939 inductive effect Effects 0.000 claims description 14
- 239000000377 silicon dioxide Substances 0.000 claims description 14
- 125000005414 dithiopyridyl group Chemical group 0.000 claims description 12
- 239000002253 acid Substances 0.000 claims description 11
- 125000003118 aryl group Chemical group 0.000 claims description 11
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 11
- 125000000392 cycloalkenyl group Chemical group 0.000 claims description 11
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 11
- 150000001721 carbon Chemical group 0.000 claims description 10
- 125000004119 disulfanediyl group Chemical group *SS* 0.000 claims description 10
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 claims description 8
- 150000001450 anions Chemical class 0.000 claims description 8
- 229940125846 compound 25 Drugs 0.000 claims description 8
- 229910052739 hydrogen Inorganic materials 0.000 claims description 8
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 claims description 7
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 claims description 7
- 229940125758 compound 15 Drugs 0.000 claims description 7
- 229940125810 compound 20 Drugs 0.000 claims description 7
- 230000003301 hydrolyzing effect Effects 0.000 claims description 7
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 claims description 6
- 229940125961 compound 24 Drugs 0.000 claims description 6
- 125000005179 haloacetyl group Chemical group 0.000 claims description 5
- 229940024606 amino acid Drugs 0.000 claims description 4
- 235000001014 amino acid Nutrition 0.000 claims description 4
- 150000003573 thiols Chemical group 0.000 claims 12
- MSSQOQPKGAMUSY-LEAFIULHSA-N 2-[1-[2-[(4r,6s)-8-chloro-6-(2,3-dimethoxyphenyl)-4,6-dihydropyrrolo[1,2-a][4,1]benzoxazepin-4-yl]acetyl]piperidin-4-yl]acetic acid Chemical compound COC1=CC=CC([C@@H]2C3=CC(Cl)=CC=C3N3C=CC=C3[C@@H](CC(=O)N3CCC(CC(O)=O)CC3)O2)=C1OC MSSQOQPKGAMUSY-LEAFIULHSA-N 0.000 claims 9
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 claims 3
- 206010028980 Neoplasm Diseases 0.000 abstract description 54
- 230000001472 cytotoxic effect Effects 0.000 abstract description 26
- 239000003814 drug Substances 0.000 abstract description 26
- 231100000433 cytotoxic Toxicity 0.000 abstract description 25
- 241001465754 Metazoa Species 0.000 abstract description 24
- 230000015572 biosynthetic process Effects 0.000 abstract description 22
- 238000003786 synthesis reaction Methods 0.000 abstract description 21
- 239000003795 chemical substances by application Substances 0.000 abstract description 9
- 238000006467 substitution reaction Methods 0.000 abstract description 3
- 229940124597 therapeutic agent Drugs 0.000 abstract description 3
- 230000001225 therapeutic effect Effects 0.000 abstract description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 146
- 210000004027 cell Anatomy 0.000 description 140
- 239000000562 conjugate Substances 0.000 description 122
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 81
- 239000000243 solution Substances 0.000 description 54
- 235000019439 ethyl acetate Nutrition 0.000 description 52
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 36
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 36
- 239000000047 product Substances 0.000 description 33
- 238000000338 in vitro Methods 0.000 description 25
- 229940079593 drug Drugs 0.000 description 23
- 239000002904 solvent Substances 0.000 description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- 239000012044 organic layer Substances 0.000 description 21
- JAMUSZRJCHASRL-UHFFFAOYSA-N 4-methylpentanedithioic acid Chemical compound CC(C)CCC(S)=S JAMUSZRJCHASRL-UHFFFAOYSA-N 0.000 description 20
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 20
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 20
- 239000011541 reaction mixture Substances 0.000 description 20
- LJFFDOBFKICLHN-IXWHRVGISA-N [(1S,2R,3S,5S,6S,16E,18E,20R,21S)-11-chloro-21-hydroxy-12,20-dimethoxy-2,5,9,16-tetramethyl-8,23-dioxo-4,24-dioxa-9,22-diazatetracyclo[19.3.1.110,14.03,5]hexacosa-10,12,14(26),16,18-pentaen-6-yl] (2S)-2-[methyl(4-sulfanylpentanoyl)amino]propanoate Chemical compound CO[C@@H]([C@@]1(O)C[C@H](OC(=O)N1)[C@@H](C)[C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(=O)CCC(C)S)CC(=O)N1C)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 LJFFDOBFKICLHN-IXWHRVGISA-N 0.000 description 19
- 238000003756 stirring Methods 0.000 description 18
- 230000003013 cytotoxicity Effects 0.000 description 16
- 231100000135 cytotoxicity Toxicity 0.000 description 16
- 239000011734 sodium Substances 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 15
- 238000001727 in vivo Methods 0.000 description 15
- 125000005647 linker group Chemical group 0.000 description 15
- 238000002360 preparation method Methods 0.000 description 15
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 14
- GYFCEVXCVFNSCL-UHFFFAOYSA-N 4-methyl-4-(methyldisulfanyl)pentanoic acid Chemical compound CSSC(C)(C)CCC(O)=O GYFCEVXCVFNSCL-UHFFFAOYSA-N 0.000 description 13
- 229930126263 Maytansine Natural products 0.000 description 13
- 241000699670 Mus sp. Species 0.000 description 13
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 13
- WKPWGQKGSOKKOO-RSFHAFMBSA-N maytansine Chemical compound CO[C@@H]([C@@]1(O)C[C@](OC(=O)N1)([C@H]([C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(C)=O)CC(=O)N1C)C)[H])\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 WKPWGQKGSOKKOO-RSFHAFMBSA-N 0.000 description 13
- 230000004614 tumor growth Effects 0.000 description 13
- 238000005160 1H NMR spectroscopy Methods 0.000 description 12
- 208000012766 Growth delay Diseases 0.000 description 12
- 238000002390 rotary evaporation Methods 0.000 description 12
- ZNSPHKJFQDEABI-NZQKXSOJSA-N Nc1nc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)cc(n1)N1CCC2(CN[C@@H](C2)C(O)=O)CC1 Chemical compound Nc1nc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)cc(n1)N1CCC2(CN[C@@H](C2)C(O)=O)CC1 ZNSPHKJFQDEABI-NZQKXSOJSA-N 0.000 description 11
- 201000011510 cancer Diseases 0.000 description 11
- 238000002523 gelfiltration Methods 0.000 description 11
- 229960005558 mertansine Drugs 0.000 description 11
- 229920005654 Sephadex Polymers 0.000 description 10
- 239000012507 Sephadex™ Substances 0.000 description 10
- 239000000427 antigen Substances 0.000 description 10
- 108091007433 antigens Proteins 0.000 description 10
- 102000036639 antigens Human genes 0.000 description 10
- 229910052786 argon Inorganic materials 0.000 description 10
- 239000012300 argon atmosphere Substances 0.000 description 10
- 238000004587 chromatography analysis Methods 0.000 description 10
- 239000008367 deionised water Substances 0.000 description 10
- 229910021641 deionized water Inorganic materials 0.000 description 10
- 239000010410 layer Substances 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 10
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 10
- 0 C[C@@](C(OC(CC(N(C)c(cc(CC(C)=CC=CC(C(C1)(N2)O)OC)cc3OC)c3Cl)=O)C3(C)OC3C(C)C1OC2=O)=O)N(C)C(*)=O Chemical compound C[C@@](C(OC(CC(N(C)c(cc(CC(C)=CC=CC(C(C1)(N2)O)OC)cc3OC)c3Cl)=O)C3(C)OC3C(C)C1OC2=O)=O)N(C)C(*)=O 0.000 description 9
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 9
- 238000005481 NMR spectroscopy Methods 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 230000008033 biological extinction Effects 0.000 description 9
- 229940127121 immunoconjugate Drugs 0.000 description 9
- 230000003389 potentiating effect Effects 0.000 description 9
- 239000000741 silica gel Substances 0.000 description 9
- 229910002027 silica gel Inorganic materials 0.000 description 9
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 8
- ANZJBCHSOXCCRQ-FKUXLPTCSA-N mertansine Chemical compound CO[C@@H]([C@@]1(O)C[C@H](OC(=O)N1)[C@@H](C)[C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(=O)CCS)CC(=O)N1C)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 ANZJBCHSOXCCRQ-FKUXLPTCSA-N 0.000 description 8
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 7
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 7
- 239000000611 antibody drug conjugate Substances 0.000 description 7
- 239000012062 aqueous buffer Substances 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 208000029742 colonic neoplasm Diseases 0.000 description 7
- 210000004881 tumor cell Anatomy 0.000 description 7
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- 241000699666 Mus <mouse, genus> Species 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 206010052779 Transplant rejections Diseases 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 229940049595 antibody-drug conjugate Drugs 0.000 description 6
- 210000000988 bone and bone Anatomy 0.000 description 6
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 6
- 229960000975 daunorubicin Drugs 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 208000024908 graft versus host disease Diseases 0.000 description 6
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 6
- 230000014759 maintenance of location Effects 0.000 description 6
- 239000002953 phosphate buffered saline Substances 0.000 description 6
- 230000001988 toxicity Effects 0.000 description 6
- 231100000419 toxicity Toxicity 0.000 description 6
- 239000011592 zinc chloride Substances 0.000 description 6
- 208000009329 Graft vs Host Disease Diseases 0.000 description 5
- 239000000637 Melanocyte-Stimulating Hormone Substances 0.000 description 5
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 5
- 238000011579 SCID mouse model Methods 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- 208000025113 myeloid leukemia Diseases 0.000 description 5
- 229910000160 potassium phosphate Inorganic materials 0.000 description 5
- 235000011009 potassium phosphates Nutrition 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- 238000007920 subcutaneous administration Methods 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 230000003442 weekly effect Effects 0.000 description 5
- JSHOVKSMJRQOGY-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-(pyridin-2-yldisulfanyl)butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCSSC1=CC=CC=N1 JSHOVKSMJRQOGY-UHFFFAOYSA-N 0.000 description 4
- ITOFPJRDSCGOSA-KZLRUDJFSA-N (2s)-2-[[(4r)-4-[(3r,5r,8r,9s,10s,13r,14s,17r)-3-hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H](CC[C@]13C)[C@@H]2[C@@H]3CC[C@@H]1[C@H](C)CCC(=O)N[C@H](C(O)=O)CC1=CNC2=CC=CC=C12 ITOFPJRDSCGOSA-KZLRUDJFSA-N 0.000 description 4
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 4
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 4
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 4
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 4
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 4
- 241001529936 Murinae Species 0.000 description 4
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 229960003767 alanine Drugs 0.000 description 4
- 239000003098 androgen Substances 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 239000001273 butane Substances 0.000 description 4
- 230000022534 cell killing Effects 0.000 description 4
- 230000021615 conjugation Effects 0.000 description 4
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 4
- 230000005917 in vivo anti-tumor Effects 0.000 description 4
- 239000008363 phosphate buffer Substances 0.000 description 4
- 239000008057 potassium phosphate buffer Substances 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 229910000162 sodium phosphate Inorganic materials 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 235000005074 zinc chloride Nutrition 0.000 description 4
- JWDFQMWEFLOOED-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(pyridin-2-yldisulfanyl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCSSC1=CC=CC=N1 JWDFQMWEFLOOED-UHFFFAOYSA-N 0.000 description 3
- BQWBEDSJTMWJAE-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[(2-iodoacetyl)amino]benzoate Chemical compound C1=CC(NC(=O)CI)=CC=C1C(=O)ON1C(=O)CCC1=O BQWBEDSJTMWJAE-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 3
- 206010009944 Colon cancer Diseases 0.000 description 3
- ZGTMUACCHSMWAC-UHFFFAOYSA-L EDTA disodium salt (anhydrous) Chemical compound [Na+].[Na+].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O ZGTMUACCHSMWAC-UHFFFAOYSA-L 0.000 description 3
- 229910004373 HOAc Inorganic materials 0.000 description 3
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 3
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 3
- 241000187747 Streptomyces Species 0.000 description 3
- 210000001744 T-lymphocyte Anatomy 0.000 description 3
- 241000700605 Viruses Species 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 231100000050 cytotoxic potential Toxicity 0.000 description 3
- 229940011871 estrogen Drugs 0.000 description 3
- 239000000262 estrogen Substances 0.000 description 3
- 235000019152 folic acid Nutrition 0.000 description 3
- 239000011724 folic acid Substances 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 230000002045 lasting effect Effects 0.000 description 3
- 208000032839 leukemia Diseases 0.000 description 3
- 238000001819 mass spectrum Methods 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- SVVGCFZPFZGWRG-OTKBOCOUSA-N maytansinoid dm4 Chemical compound CO[C@@H]([C@@]1(O)C[C@H](OC(=O)N1)C(C)(C)[C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(=O)CCC(C)(C)S)CC(=O)N1C)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 SVVGCFZPFZGWRG-OTKBOCOUSA-N 0.000 description 3
- 201000001441 melanoma Diseases 0.000 description 3
- 229960000485 methotrexate Drugs 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 239000012679 serum free medium Substances 0.000 description 3
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 3
- PUPZLCDOIYMWBV-BYPYZUCNSA-N (S)-butane-1,3-diol Chemical compound C[C@H](O)CCO PUPZLCDOIYMWBV-BYPYZUCNSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 208000023275 Autoimmune disease Diseases 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 102400001368 Epidermal growth factor Human genes 0.000 description 2
- 101800003838 Epidermal growth factor Proteins 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 2
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 2
- 101000945318 Homo sapiens Calponin-1 Proteins 0.000 description 2
- 101000652736 Homo sapiens Transgelin Proteins 0.000 description 2
- 108091006905 Human Serum Albumin Proteins 0.000 description 2
- 102000008100 Human Serum Albumin Human genes 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- 150000008575 L-amino acids Chemical class 0.000 description 2
- XNSAINXGIQZQOO-UHFFFAOYSA-N L-pyroglutamyl-L-histidyl-L-proline amide Natural products NC(=O)C1CCCN1C(=O)C(NC(=O)C1NC(=O)CC1)CC1=CN=CN1 XNSAINXGIQZQOO-UHFFFAOYSA-N 0.000 description 2
- 108010074338 Lymphokines Proteins 0.000 description 2
- 102000008072 Lymphokines Human genes 0.000 description 2
- 102000029749 Microtubule Human genes 0.000 description 2
- 108091022875 Microtubule Proteins 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 102000005157 Somatostatin Human genes 0.000 description 2
- 108010056088 Somatostatin Proteins 0.000 description 2
- 102000011923 Thyrotropin Human genes 0.000 description 2
- 108010061174 Thyrotropin Proteins 0.000 description 2
- 239000000627 Thyrotropin-Releasing Hormone Substances 0.000 description 2
- 102400000336 Thyrotropin-releasing hormone Human genes 0.000 description 2
- 101800004623 Thyrotropin-releasing hormone Proteins 0.000 description 2
- 102000004338 Transferrin Human genes 0.000 description 2
- 108090000901 Transferrin Proteins 0.000 description 2
- 102100031013 Transgelin Human genes 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 238000012742 biochemical analysis Methods 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- 244000309464 bull Species 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 231100000096 clonogenic assay Toxicity 0.000 description 2
- 238000009643 clonogenic assay Methods 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 125000004093 cyano group Chemical group *C#N 0.000 description 2
- 230000017858 demethylation Effects 0.000 description 2
- 238000010520 demethylation reaction Methods 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- WQOXQRCZOLPYPM-UHFFFAOYSA-N dimethyl disulfide Chemical compound CSSC WQOXQRCZOLPYPM-UHFFFAOYSA-N 0.000 description 2
- 230000003467 diminishing effect Effects 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 235000013601 eggs Nutrition 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 229940116977 epidermal growth factor Drugs 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 238000000684 flow cytometry Methods 0.000 description 2
- 229940014144 folate Drugs 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 229940051026 immunotoxin Drugs 0.000 description 2
- 239000002596 immunotoxin Substances 0.000 description 2
- 231100000608 immunotoxin Toxicity 0.000 description 2
- 230000002637 immunotoxin Effects 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 238000010829 isocratic elution Methods 0.000 description 2
- 230000002147 killing effect Effects 0.000 description 2
- 210000004698 lymphocyte Anatomy 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 2
- 230000011278 mitosis Effects 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 238000007747 plating Methods 0.000 description 2
- JCBJVAJGLKENNC-UHFFFAOYSA-M potassium ethyl xanthate Chemical compound [K+].CCOC([S-])=S JCBJVAJGLKENNC-UHFFFAOYSA-M 0.000 description 2
- 230000001376 precipitating effect Effects 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 235000019833 protease Nutrition 0.000 description 2
- XNSAINXGIQZQOO-SRVKXCTJSA-N protirelin Chemical compound NC(=O)[C@@H]1CCCN1C(=O)[C@@H](NC(=O)[C@H]1NC(=O)CC1)CC1=CN=CN1 XNSAINXGIQZQOO-SRVKXCTJSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- MNWBNISUBARLIT-UHFFFAOYSA-N sodium cyanide Chemical compound [Na+].N#[C-] MNWBNISUBARLIT-UHFFFAOYSA-N 0.000 description 2
- 239000011877 solvent mixture Substances 0.000 description 2
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 2
- 229960000553 somatostatin Drugs 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 229940034199 thyrotropin-releasing hormone Drugs 0.000 description 2
- 108700012359 toxins Proteins 0.000 description 2
- 239000012581 transferrin Substances 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- APJYDQYYACXCRM-UHFFFAOYSA-N tryptamine Chemical compound C1=CC=C2C(CCN)=CNC2=C1 APJYDQYYACXCRM-UHFFFAOYSA-N 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- 239000011782 vitamin Substances 0.000 description 2
- 235000013343 vitamin Nutrition 0.000 description 2
- 229940088594 vitamin Drugs 0.000 description 2
- 229930003231 vitamin Natural products 0.000 description 2
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (1R)-1,3-butanediol Natural products CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 1
- VIMMECPCYZXUCI-MIMFYIINSA-N (4s,6r)-6-[(1e)-4,4-bis(4-fluorophenyl)-3-(1-methyltetrazol-5-yl)buta-1,3-dienyl]-4-hydroxyoxan-2-one Chemical compound CN1N=NN=C1C(\C=C\[C@@H]1OC(=O)C[C@@H](O)C1)=C(C=1C=CC(F)=CC=1)C1=CC=C(F)C=C1 VIMMECPCYZXUCI-MIMFYIINSA-N 0.000 description 1
- WZIMSXIXZTUBSO-UHFFFAOYSA-N 2-[[bis(carboxymethyl)amino]methyl-(carboxymethyl)amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)CN(CC(O)=O)CC(O)=O WZIMSXIXZTUBSO-UHFFFAOYSA-N 0.000 description 1
- PUUZUVNGEQEYKF-UHFFFAOYSA-N 2-methylpentanedithioic acid Chemical compound CCCC(C)C(S)=S PUUZUVNGEQEYKF-UHFFFAOYSA-N 0.000 description 1
- IMGGANUNCHXAQF-UHFFFAOYSA-N 2-sulfanylpentanoic acid Chemical compound CCCC(S)C(O)=O IMGGANUNCHXAQF-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- DJBRKGZFUXKLKO-UHFFFAOYSA-N 3-(pyridin-2-yldisulfanyl)propanoic acid Chemical compound OC(=O)CCSSC1=CC=CC=N1 DJBRKGZFUXKLKO-UHFFFAOYSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 241000186046 Actinomyces Species 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000004881 Amebiasis Diseases 0.000 description 1
- 206010001980 Amoebiasis Diseases 0.000 description 1
- DGBBXVWXOHSLTG-UHFFFAOYSA-N Ansamitocin P2 Natural products CC1C2OC2(C)C(OC(=O)CC)CC(=O)N(C)C(C(=C(OC)C=2)Cl)=CC=2CC(C)=CC=CC(OC)C2(O)NC(=O)OC1C2 DGBBXVWXOHSLTG-UHFFFAOYSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 208000011691 Burkitt lymphomas Diseases 0.000 description 1
- 108090000565 Capsid Proteins Proteins 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 1
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 206010011831 Cytomegalovirus infection Diseases 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 241000257465 Echinoidea Species 0.000 description 1
- 101100001669 Emericella variicolor andD gene Proteins 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 101150021185 FGF gene Proteins 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- 208000006552 Lewis Lung Carcinoma Diseases 0.000 description 1
- 206010024715 Liver transplant rejection Diseases 0.000 description 1
- 206010051604 Lung transplant rejection Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 208000028018 Lymphocytic leukaemia Diseases 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 240000001427 Mallotus nudiflorus Species 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241001441512 Maytenus serrata Species 0.000 description 1
- 229940121849 Mitotic inhibitor Drugs 0.000 description 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 1
- 125000000199 N-methyl-L-alanine group Chemical group 0.000 description 1
- 208000002454 Nasopharyngeal Carcinoma Diseases 0.000 description 1
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 241000187654 Nocardia Species 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- 108010020346 Polyglutamic Acid Proteins 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 208000009052 Precursor T-Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010029180 Sialic Acid Binding Ig-like Lectin 3 Proteins 0.000 description 1
- 102000001555 Sialic Acid Binding Ig-like Lectin 3 Human genes 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 208000029052 T-cell acute lymphoblastic leukemia Diseases 0.000 description 1
- 102400001320 Transforming growth factor alpha Human genes 0.000 description 1
- 101800004564 Transforming growth factor alpha Proteins 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 108010067390 Viral Proteins Proteins 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- UGAPHEBNTGUMBB-UHFFFAOYSA-N acetic acid;ethyl acetate Chemical compound CC(O)=O.CCOC(C)=O UGAPHEBNTGUMBB-UHFFFAOYSA-N 0.000 description 1
- XURZGOTTZHKXTQ-UHFFFAOYSA-N acetonitrile;lithium Chemical compound [Li].CC#N XURZGOTTZHKXTQ-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 201000011186 acute T cell leukemia Diseases 0.000 description 1
- 150000001263 acyl chlorides Chemical class 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 125000005042 acyloxymethyl group Chemical group 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 229940064734 aminobenzoate Drugs 0.000 description 1
- 229940094957 androgens and estrogen Drugs 0.000 description 1
- DGBBXVWXOHSLTG-UMDRASRXSA-N ansamitocin p 2 Chemical compound C([C@@H]([C@@]1(O[C@H]1[C@@H]1C)C)OC(=O)CC)C(=O)N(C)C(C(=C(OC)C=2)Cl)=CC=2C\C(C)=C\C=C\[C@@H](OC)[C@]2(O)NC(=O)O[C@H]1C2 DGBBXVWXOHSLTG-UMDRASRXSA-N 0.000 description 1
- 230000003127 anti-melanomic effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 238000007068 beta-elimination reaction Methods 0.000 description 1
- 238000012925 biological evaluation Methods 0.000 description 1
- 210000002798 bone marrow cell Anatomy 0.000 description 1
- 201000008274 breast adenocarcinoma Diseases 0.000 description 1
- 201000008275 breast carcinoma Diseases 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229930195731 calicheamicin Natural products 0.000 description 1
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- GTZCVFVGUGFEME-HNQUOIGGSA-N cis-Aconitic acid Natural products OC(=O)C\C(C(O)=O)=C/C(O)=O GTZCVFVGUGFEME-HNQUOIGGSA-N 0.000 description 1
- GTZCVFVGUGFEME-IWQZZHSRSA-N cis-aconitic acid Chemical compound OC(=O)C\C(C(O)=O)=C\C(O)=O GTZCVFVGUGFEME-IWQZZHSRSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 229940047120 colony stimulating factors Drugs 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 150000001923 cyclic compounds Chemical class 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- UFULAYFCSOUIOV-UHFFFAOYSA-N cysteamine Chemical class NCCS UFULAYFCSOUIOV-UHFFFAOYSA-N 0.000 description 1
- 231100000263 cytotoxicity test Toxicity 0.000 description 1
- 238000006298 dechlorination reaction Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 210000001163 endosome Anatomy 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 239000003687 estradiol congener Substances 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 102000006815 folate receptor Human genes 0.000 description 1
- 108020005243 folate receptor Proteins 0.000 description 1
- 229960000304 folic acid Drugs 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 201000006592 giardiasis Diseases 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 208000003747 lymphoid leukemia Diseases 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 230000002132 lysosomal effect Effects 0.000 description 1
- 210000003712 lysosome Anatomy 0.000 description 1
- 230000001868 lysosomic effect Effects 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 210000002752 melanocyte Anatomy 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 230000000394 mitotic effect Effects 0.000 description 1
- 229940125645 monoclonal antibody drug Drugs 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 210000000066 myeloid cell Anatomy 0.000 description 1
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 201000011216 nasopharynx carcinoma Diseases 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 125000006501 nitrophenyl group Chemical group 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- WYMSBXTXOHUIGT-UHFFFAOYSA-N paraoxon Chemical compound CCOP(=O)(OCC)OC1=CC=C([N+]([O-])=O)C=C1 WYMSBXTXOHUIGT-UHFFFAOYSA-N 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 230000036281 parasite infection Effects 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920002643 polyglutamic acid Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 201000001514 prostate carcinoma Diseases 0.000 description 1
- 231100000654 protein toxin Toxicity 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 230000010837 receptor-mediated endocytosis Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- RWWYLEGWBNMMLJ-YSOARWBDSA-N remdesivir Chemical compound NC1=NC=NN2C1=CC=C2[C@]1([C@@H]([C@@H]([C@H](O1)CO[P@](=O)(OC1=CC=CC=C1)N[C@H](C(=O)OCC(CC)CC)C)O)O)C#N RWWYLEGWBNMMLJ-YSOARWBDSA-N 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- QEVHRUUCFGRFIF-MDEJGZGSSA-N reserpine Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C(C5=CC=C(OC)C=C5N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 QEVHRUUCFGRFIF-MDEJGZGSSA-N 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 201000004409 schistosomiasis Diseases 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000003270 steroid hormone Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- CNHYKKNIIGEXAY-UHFFFAOYSA-N thiolan-2-imine Chemical compound N=C1CCCS1 CNHYKKNIIGEXAY-UHFFFAOYSA-N 0.000 description 1
- YONPGGFAJWQGJC-UHFFFAOYSA-K titanium(iii) chloride Chemical compound Cl[Ti](Cl)Cl YONPGGFAJWQGJC-UHFFFAOYSA-K 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- GTZCVFVGUGFEME-UHFFFAOYSA-N trans-aconitic acid Natural products OC(=O)CC(C(O)=O)=CC(O)=O GTZCVFVGUGFEME-UHFFFAOYSA-N 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/16—Peri-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/14—Ortho-condensed systems
- C07D491/147—Ortho-condensed systems the condensed system containing one ring with oxygen as ring hetero atom and two rings with nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6807—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug or compound being a sugar, nucleoside, nucleotide, nucleic acid, e.g. RNA antisense
- A61K47/6809—Antibiotics, e.g. antitumor antibiotics anthracyclins, adriamycin, doxorubicin or daunomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/04—Amoebicides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6867—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from a cell of a blood cancer
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Oncology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Virology (AREA)
- Cell Biology (AREA)
- Hematology (AREA)
- Rheumatology (AREA)
- Molecular Biology (AREA)
- Physical Education & Sports Medicine (AREA)
- AIDS & HIV (AREA)
- Pain & Pain Management (AREA)
- Neurology (AREA)
- Dermatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Transplantation (AREA)
- Biochemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description























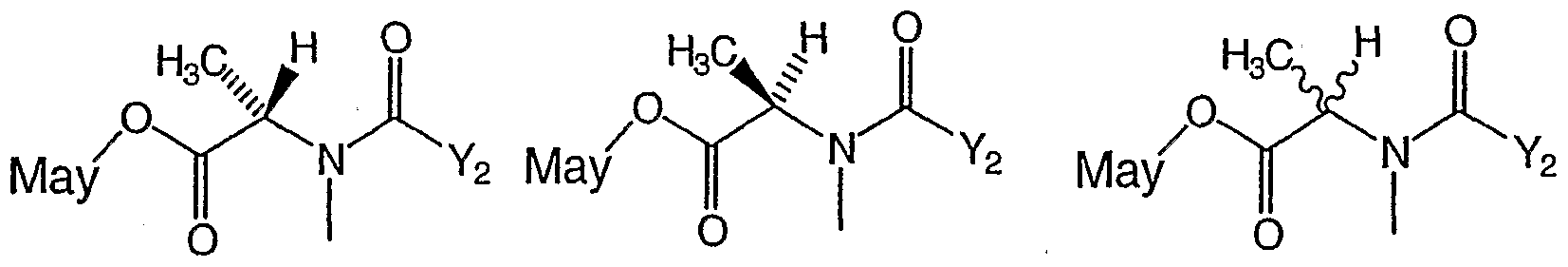
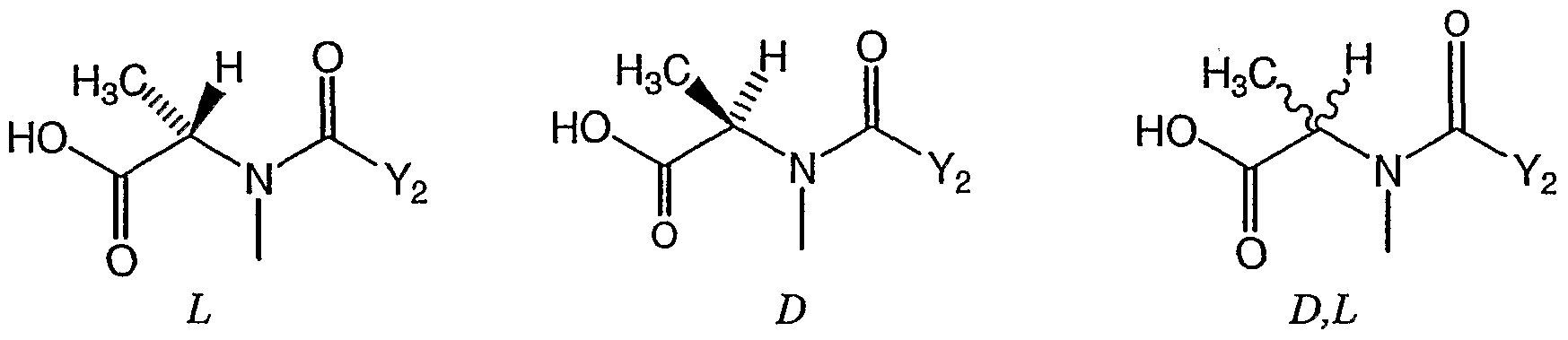
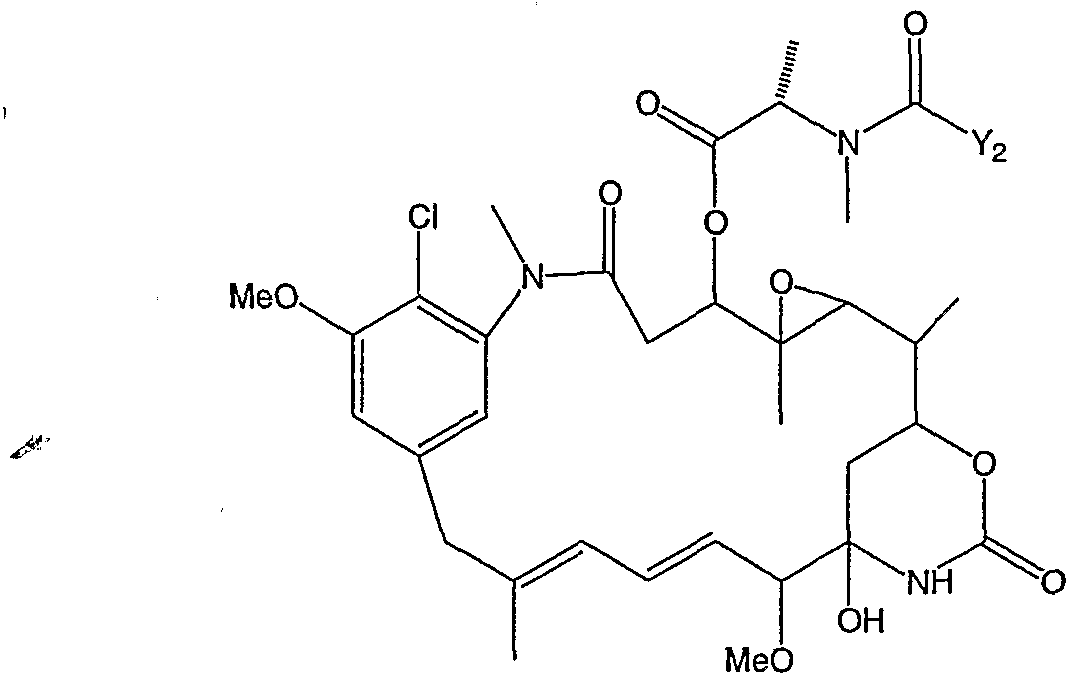





Claims















Priority Applications (30)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MXPA05011811A MXPA05011811A (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids. |
| PL04750945T PL1651162T3 (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids |
| CR20170291A CR20170291A (en) | 2003-05-20 | 2004-05-20 | IMPROVED CYTOTOXIC AGENTS UNDERSTANDING NEW MAITANSINÓIDES |
| ES04750945.0T ES2559670T3 (en) | 2003-05-20 | 2004-05-20 | Enhanced cytotoxic agents comprising new maitansinoids |
| JP2006532511A JP5208420B2 (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic drugs containing novel maytansinoids |
| EP04750945.0A EP1651162B1 (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids |
| MX2016009879A MX370281B (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids. |
| PL19160750T PL3524611T3 (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids |
| EP15190436.4A EP3031810B1 (en) | 2003-05-20 | 2004-05-20 | Maytansinoid-cell-binding agent conjugates |
| CA2525130A CA2525130C (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids |
| SI200432295T SI1651162T1 (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids |
| NZ542695A NZ542695A (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids |
| EP19160750.6A EP3524611B1 (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids |
| EA200501836A EA010909B1 (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids |
| DK04750945.0T DK1651162T3 (en) | 2003-05-20 | 2004-05-20 | IMPROVED CYTOTOXIC AGENTS WITH NEW MAYTANSINOIDS |
| AU2004240541A AU2004240541B2 (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids |
| KR1020057022166A KR101145506B1 (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids |
| BRPI0410748A BRPI0410748B8 (en) | 2003-05-20 | 2004-05-20 | maytansinoid compounds, their pharmaceutical compositions, methods of esterification of maytansinoids, as well as methods for their production, and maytansinoid-cell binding agent conjugate |
| EP20216572.6A EP3851126A1 (en) | 2003-05-20 | 2004-05-20 | Maytansinoid-cell-binding agent conjugates |
| MX2013008224A MX340862B (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids. |
| BRPI0419348A BRPI0419348B8 (en) | 2003-05-20 | 2004-05-20 | methods for making the maytansinoid-cell binding agent conjugate |
| IL171170A IL171170A (en) | 2003-05-20 | 2005-09-29 | Maytansinoid derivatives and pharmaceutical compositions containing the same, methods of producing the same and methods of use thereof |
| NO20056039A NO339597B1 (en) | 2003-05-20 | 2005-12-19 | Enhanced cytotoxic agents which include novel maytansinoids; process for their preparation; intermediates thereof and process for their preparation; conjugate between the maytansinoids and a cell binding agent; process for its preparation and its use in a therapeutic method or for the manufacture of a medicament, and a pharmaceutical preparation thereof. |
| IL213876A IL213876A (en) | 2003-05-20 | 2011-06-30 | Maytansinoid derivatives and pharmaceutical compositions containing the same |
| IL223297A IL223297A (en) | 2003-05-20 | 2012-11-27 | Maytansinoid derivatives and pharmaceutical compositions containing the same and methods of producing the same |
| IL231810A IL231810A (en) | 2003-05-20 | 2014-03-30 | Maytansinoid derivatives and pharmaceutical compositions containing the same, methods of producing the same and methods of use thereof for use in treating cancer |
| IL238894A IL238894A (en) | 2003-05-20 | 2015-05-19 | Maytansinoid derivatives and methods of producing the same |
| IL241211A IL241211B (en) | 2003-05-20 | 2015-09-06 | Maytansinoid derivatives and pharmaceutical compositions containing the same |
| HRP20160046TT HRP20160046T1 (en) | 2003-05-20 | 2016-01-14 | Improved cytotoxic agents comprising new maytansinoids |
| CY20211100265T CY1124278T1 (en) | 2003-05-20 | 2021-03-26 | IMPROVED CYTOTOXIC AGENTS CONTAINING NOVEL MAYTANSINOIDS |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US47173903P | 2003-05-20 | 2003-05-20 | |
| US60/471,739 | 2003-05-20 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004103272A2 true WO2004103272A2 (en) | 2004-12-02 |
| WO2004103272A3 WO2004103272A3 (en) | 2006-11-23 |
Family
ID=33476883
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/013314 WO2004103272A2 (en) | 2003-05-20 | 2004-05-20 | Improved cytotoxic agents comprising new maytansinoids |
Country Status (28)
| Country | Link |
|---|---|
| EP (4) | EP3851126A1 (en) |
| JP (2) | JP5208420B2 (en) |
| KR (1) | KR101145506B1 (en) |
| CN (2) | CN101186613B (en) |
| AU (1) | AU2004240541B2 (en) |
| BR (2) | BRPI0410748B8 (en) |
| CA (1) | CA2525130C (en) |
| CL (1) | CL2012000651A1 (en) |
| CO (1) | CO5660276A2 (en) |
| CR (1) | CR20170291A (en) |
| CY (2) | CY1117161T1 (en) |
| DK (2) | DK1651162T3 (en) |
| EA (1) | EA010909B1 (en) |
| EC (1) | ECSP056149A (en) |
| ES (2) | ES2863498T3 (en) |
| HK (1) | HK1116777A1 (en) |
| HR (2) | HRP20160046T1 (en) |
| HU (2) | HUE028314T2 (en) |
| IL (6) | IL171170A (en) |
| LT (1) | LT3524611T (en) |
| MX (3) | MX370281B (en) |
| NO (1) | NO339597B1 (en) |
| NZ (1) | NZ542695A (en) |
| PL (2) | PL1651162T3 (en) |
| PT (2) | PT1651162E (en) |
| SI (2) | SI1651162T1 (en) |
| WO (1) | WO2004103272A2 (en) |
| ZA (1) | ZA200507845B (en) |
Cited By (90)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006086733A3 (en) * | 2005-02-11 | 2007-06-07 | Immunogen Inc | Process for preparing maytansinoid antibody conjugates |
| WO2007024536A3 (en) * | 2005-08-24 | 2007-07-05 | Immunogen Inc | Process for preparing maytansinoid antibody conjugates |
| EP1813614A1 (en) | 2006-01-25 | 2007-08-01 | Sanofi-Aventis | Cytotoxic agents comprising new tomaymycin derivatives |
| EP1868649A2 (en) * | 2005-04-15 | 2007-12-26 | Immunogen, Inc. | Elimination of heterogeneous or mixed cell population in tumors |
| EP2062591A1 (en) | 2005-04-07 | 2009-05-27 | Novartis Vaccines and Diagnostics, Inc. | CACNA1E in cancer diagnosis detection and treatment |
| EP2083088A2 (en) | 2005-04-07 | 2009-07-29 | Novartis Vaccines and Diagnostics, Inc. | Cancer-related genes |
| WO2011001052A1 (en) | 2009-06-29 | 2011-01-06 | Sanofi-Aventis | Novel conjugates, preparation thereof, and therapeutic use thereof |
| WO2011023883A1 (en) | 2009-08-25 | 2011-03-03 | Sanofi-Aventis | Conjugates of pyrrolo[1,4]benzodiazepine dimers as anticancer agents |
| WO2011039721A1 (en) | 2009-10-02 | 2011-04-07 | Sanofi-Aventis | New maytansinoids and the use of said maytansinoids to prepare conjugates with an antibody |
| WO2011045396A2 (en) | 2009-10-16 | 2011-04-21 | Novartis Ag | Biomarkers of tumor pharmacodynamic response |
| EP2389946A1 (en) | 2006-03-23 | 2011-11-30 | Novartis AG | Anti-tumor cell antigen antibody therapeutics |
| EP2407483A1 (en) | 2006-04-13 | 2012-01-18 | Novartis Vaccines and Diagnostics, Inc. | Methods of treating, diagnosing or detecting cancers |
| WO2012016227A2 (en) | 2010-07-29 | 2012-02-02 | Xencor, Inc. | Antibodies with modified isoelectric points |
| WO2012014147A1 (en) | 2010-07-26 | 2012-02-02 | Sanofi | Anticancer derivatives, preparation thereof and therapeutic use thereof |
| WO2012045085A1 (en) | 2010-10-01 | 2012-04-05 | Oxford Biotherapeutics Ltd. | Anti-rori antibodies |
| WO2012061590A1 (en) * | 2010-11-03 | 2012-05-10 | Immunogen, Inc. | Cytotoxic agents comprising new ansamitocin derivatives |
| EP2474556A2 (en) | 2007-03-14 | 2012-07-11 | Novartis AG | APCDD1 inhibitors for treating, diagnosing or detecting cancer |
| EP2524929A1 (en) * | 2011-05-17 | 2012-11-21 | Sanofi | Use of anti-CD19 maytansinoid immunoconjugate antibody for the treatment of CD19+ B-cell malignancies syptoms |
| EP2550975A1 (en) * | 2011-07-29 | 2013-01-30 | Sanofi | Combination therapy for the treatment of CD19+ B-cell malignancies symptoms comprising an anti-CD19 maytansinoid immunoconjugate and rituximab |
| US8603483B2 (en) | 2004-12-09 | 2013-12-10 | Janssen Biotech, Inc. | Anti-integrin immunoconjugates, methods and uses |
| WO2014114207A1 (en) | 2013-01-23 | 2014-07-31 | 上海新理念生物医药科技有限公司 | Tridentate connexon and use thereof |
| US8795673B2 (en) | 2011-03-29 | 2014-08-05 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| WO2015009740A2 (en) | 2013-07-15 | 2015-01-22 | Cell Signaling Technology, Inc. | Anti-mucin 1 binding agents and uses thereof |
| WO2015149077A1 (en) | 2014-03-28 | 2015-10-01 | Xencor, Inc. | Bispecific antibodies that bind to cd38 and cd3 |
| US9150649B2 (en) | 2008-04-30 | 2015-10-06 | Immunogen, Inc. | Potent conjugates and hydrophilic linkers |
| WO2016014984A1 (en) | 2014-07-24 | 2016-01-28 | Xencor, Inc. | Rapid clearance of antigen complexes using novel antibodies |
| WO2016036804A1 (en) | 2014-09-03 | 2016-03-10 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives |
| US9376500B2 (en) | 2009-06-03 | 2016-06-28 | Immunogen, Inc. | Conjugation methods |
| WO2016141387A1 (en) | 2015-03-05 | 2016-09-09 | Xencor, Inc. | Modulation of t cells with bispecific antibodies and fc fusions |
| WO2016192527A1 (en) | 2015-05-29 | 2016-12-08 | Newbio Therapeutics, Inc. | Derivatives of dolastatin 10 and uses thereof |
| US9605084B2 (en) | 2013-03-15 | 2017-03-28 | Xencor, Inc. | Heterodimeric proteins |
| US9650446B2 (en) | 2013-01-14 | 2017-05-16 | Xencor, Inc. | Heterodimeric proteins |
| WO2017088734A1 (en) | 2015-11-23 | 2017-06-01 | 四川科伦博泰生物医药股份有限公司 | Anti-erbb2 antibody-drug conjugate and composition thereof, preparation method therefor, and application thereof |
| US9701759B2 (en) | 2013-01-14 | 2017-07-11 | Xencor, Inc. | Heterodimeric proteins |
| US9738722B2 (en) | 2013-01-15 | 2017-08-22 | Xencor, Inc. | Rapid clearance of antigen complexes using novel antibodies |
| US9850320B2 (en) | 2014-11-26 | 2017-12-26 | Xencor, Inc. | Heterodimeric antibodies to CD3 X CD20 |
| US9856327B2 (en) | 2014-11-26 | 2018-01-02 | Xencor, Inc. | Heterodimeric antibodies to CD3 X CD123 |
| EP3284755A1 (en) | 2010-12-30 | 2018-02-21 | Takeda Pharmaceutical Company Limited | Conjugated anti-cd38 antibodies |
| EP3311846A1 (en) | 2014-09-02 | 2018-04-25 | ImmunoGen, Inc. | Methods for formulating antibody drug conjugate compositions |
| EP3160518A4 (en) * | 2014-06-30 | 2018-05-23 | Tarveda Therapeutics, Inc. | Targeted conjugates and particles and formulations thereof |
| US10035817B2 (en) | 2012-10-04 | 2018-07-31 | Immunogen, Inc. | Method of purifying cell-binding agent-cytotoxic agent conjugates with a PVDF membrane |
| WO2018160539A1 (en) | 2017-02-28 | 2018-09-07 | Immunogen, Inc. | Maytansinoid derivatives with self-immolative peptide linkers and conjugates thereof |
| US10106624B2 (en) | 2013-03-15 | 2018-10-23 | Xencor, Inc. | Heterodimeric proteins |
| WO2018195243A1 (en) | 2017-04-20 | 2018-10-25 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives and conjugates thereof |
| US10131710B2 (en) | 2013-01-14 | 2018-11-20 | Xencor, Inc. | Optimized antibody variable regions |
| EP3421495A2 (en) | 2013-03-15 | 2019-01-02 | Xencor, Inc. | Modulation of t cells with bispecific antibodies and fc fusions |
| US10227410B2 (en) | 2015-12-07 | 2019-03-12 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and PSMA |
| WO2019105835A1 (en) | 2017-11-29 | 2019-06-06 | Bayer Consumer Care Ag | Combinations of copanlisib and anetumab ravtansine |
| US10316088B2 (en) | 2016-06-28 | 2019-06-11 | Xencor, Inc. | Heterodimeric antibodies that bind somatostatin receptor 2 |
| WO2019133652A1 (en) | 2017-12-28 | 2019-07-04 | Immunogen, Inc. | Benzodiazepine derivatives |
| US10428155B2 (en) | 2014-12-22 | 2019-10-01 | Xencor, Inc. | Trispecific antibodies |
| US10449258B2 (en) | 2015-06-09 | 2019-10-22 | Xdcexplorer (Shanghai) Co., Ltd. | Antibody drug conjugate, intermediate, preparation method, pharmaceutical composition and uses thereof |
| US10487155B2 (en) | 2013-01-14 | 2019-11-26 | Xencor, Inc. | Heterodimeric proteins |
| US10501543B2 (en) | 2016-10-14 | 2019-12-10 | Xencor, Inc. | IL15/IL15Rα heterodimeric Fc-fusion proteins |
| WO2019238843A1 (en) | 2018-06-14 | 2019-12-19 | Berlin-Chemie Ag | Pharmaceutical combinations |
| US10519242B2 (en) | 2013-03-15 | 2019-12-31 | Xencor, Inc. | Targeting regulatory T cells with heterodimeric proteins |
| EP3587448A1 (en) | 2013-03-15 | 2020-01-01 | Xencor, Inc. | Heterodimeric proteins |
| US10526417B2 (en) | 2014-11-26 | 2020-01-07 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CD38 |
| WO2020010079A2 (en) | 2018-07-02 | 2020-01-09 | Amgen Inc. | Anti-steap1 antigen-binding protein |
| US10532019B2 (en) | 2005-12-01 | 2020-01-14 | University Of Massachusetts Lowell | Botulinum nanoemulsions |
| WO2020023871A1 (en) | 2018-07-27 | 2020-01-30 | Promega Corporation | Quinone-containing conjugates |
| EP3611187A1 (en) | 2011-10-10 | 2020-02-19 | Xencor, Inc. | A method for purifying antibodies |
| US10654873B2 (en) | 2016-09-15 | 2020-05-19 | Polytherics Limited | Cytotoxic agents and conjugates thereof |
| US10787518B2 (en) | 2016-06-14 | 2020-09-29 | Xencor, Inc. | Bispecific checkpoint inhibitor antibodies |
| US10793632B2 (en) | 2016-08-30 | 2020-10-06 | Xencor, Inc. | Bispecific immunomodulatory antibodies that bind costimulatory and checkpoint receptors |
| WO2020234114A1 (en) | 2019-05-21 | 2020-11-26 | Bayer Aktiengesellschaft | A novel stable high concentration formulation for anetumab ravtansine |
| US10851178B2 (en) | 2011-10-10 | 2020-12-01 | Xencor, Inc. | Heterodimeric human IgG1 polypeptides with isoelectric point modifications |
| US10858417B2 (en) | 2013-03-15 | 2020-12-08 | Xencor, Inc. | Heterodimeric proteins |
| EP3778601A1 (en) | 2014-09-03 | 2021-02-17 | ImmunoGen, Inc. | Cytotoxic benzodiazepine derivatives |
| US10944190B2 (en) | 2012-09-26 | 2021-03-09 | Immunogen, Inc. | Methods for the acylation of maytansinol |
| US10968276B2 (en) | 2013-03-12 | 2021-04-06 | Xencor, Inc. | Optimized anti-CD3 variable regions |
| US10982006B2 (en) | 2018-04-04 | 2021-04-20 | Xencor, Inc. | Heterodimeric antibodies that bind fibroblast activation protein |
| US10981992B2 (en) | 2017-11-08 | 2021-04-20 | Xencor, Inc. | Bispecific immunomodulatory antibodies that bind costimulatory and checkpoint receptors |
| US11053316B2 (en) | 2013-01-14 | 2021-07-06 | Xencor, Inc. | Optimized antibody variable regions |
| US11084863B2 (en) | 2017-06-30 | 2021-08-10 | Xencor, Inc. | Targeted heterodimeric Fc fusion proteins containing IL-15 IL-15alpha and antigen binding domains |
| US11160871B2 (en) | 2015-10-28 | 2021-11-02 | Tarveda Therapeutics, Inc. | SSTR-targeted conjugates and particles and formulations thereof |
| US11311496B2 (en) | 2016-11-21 | 2022-04-26 | Eirion Therapeutics, Inc. | Transdermal delivery of large agents |
| US11312770B2 (en) | 2017-11-08 | 2022-04-26 | Xencor, Inc. | Bispecific and monospecific antibodies using novel anti-PD-1 sequences |
| US11319355B2 (en) | 2017-12-19 | 2022-05-03 | Xencor, Inc. | Engineered IL-2 Fc fusion proteins |
| US11358999B2 (en) | 2018-10-03 | 2022-06-14 | Xencor, Inc. | IL-12 heterodimeric Fc-fusion proteins |
| US11365258B2 (en) | 2017-03-10 | 2022-06-21 | Berlin-Chemie Ag | Pharmaceutical combinations comprising an anti-LY75 antibody |
| US11472890B2 (en) | 2019-03-01 | 2022-10-18 | Xencor, Inc. | Heterodimeric antibodies that bind ENPP3 and CD3 |
| US11505595B2 (en) | 2018-04-18 | 2022-11-22 | Xencor, Inc. | TIM-3 targeted heterodimeric fusion proteins containing IL-15/IL-15RA Fc-fusion proteins and TIM-3 antigen binding domains |
| US11524991B2 (en) | 2018-04-18 | 2022-12-13 | Xencor, Inc. | PD-1 targeted heterodimeric fusion proteins containing IL-15/IL-15Ra Fc-fusion proteins and PD-1 antigen binding domains and uses thereof |
| US11591401B2 (en) | 2020-08-19 | 2023-02-28 | Xencor, Inc. | Anti-CD28 compositions |
| US11634508B2 (en) | 2019-07-10 | 2023-04-25 | Cybrexa 2, Inc. | Peptide conjugates of cytotoxins as therapeutics |
| WO2023089314A1 (en) | 2021-11-18 | 2023-05-25 | Oxford Biotherapeutics Limited | Pharmaceutical combinations |
| US11739144B2 (en) | 2021-03-09 | 2023-08-29 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CLDN6 |
| US11859012B2 (en) | 2021-03-10 | 2024-01-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and GPC3 |
| US11919956B2 (en) | 2020-05-14 | 2024-03-05 | Xencor, Inc. | Heterodimeric antibodies that bind prostate specific membrane antigen (PSMA) and CD3 |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2525130C (en) * | 2003-05-20 | 2014-04-15 | Immunogen, Inc. | Improved cytotoxic agents comprising new maytansinoids |
| US9493578B2 (en) | 2009-09-02 | 2016-11-15 | Xencor, Inc. | Compositions and methods for simultaneous bivalent and monovalent co-engagement of antigens |
| EP2486023A4 (en) * | 2009-10-06 | 2014-05-07 | Immunogen Inc | Potent conjugates and hydrophilic linkers |
| RU2451010C1 (en) * | 2011-01-11 | 2012-05-20 | Закрытое Акционерное Общество "Ива Фарм" | Palladium-copper catalysts for homogeneous selective oxidation of thiol groups, combination and composition based on said catalysts and therapeutic treatment method |
| CN104974252B (en) * | 2014-04-01 | 2020-04-24 | 三生国健药业(上海)股份有限公司 | Antibody-small molecule drug conjugate for inhibiting tumor growth and preparation method and application thereof |
| SI3956332T1 (en) * | 2019-04-18 | 2023-05-31 | Indena S.P.A. | Diasteroselective process for the preparation of thiol- or disulfide-containing maytansinoid esters and intermediates thereof |
| CR20220057A (en) | 2019-07-10 | 2022-07-19 | Cybrexa 3 Inc | Peptide conjugates of microtubule-targeting agents as therapeutics |
Family Cites Families (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3896111A (en) | 1973-02-20 | 1975-07-22 | Research Corp | Ansa macrolides |
| US4151042A (en) | 1977-03-31 | 1979-04-24 | Takeda Chemical Industries, Ltd. | Method for producing maytansinol and its derivatives |
| US4137230A (en) | 1977-11-14 | 1979-01-30 | Takeda Chemical Industries, Ltd. | Method for the production of maytansinoids |
| US4307016A (en) | 1978-03-24 | 1981-12-22 | Takeda Chemical Industries, Ltd. | Demethyl maytansinoids |
| US4265814A (en) | 1978-03-24 | 1981-05-05 | Takeda Chemical Industries | Matansinol 3-n-hexadecanoate |
| JPS5562090A (en) | 1978-10-27 | 1980-05-10 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| US4256746A (en) | 1978-11-14 | 1981-03-17 | Takeda Chemical Industries | Dechloromaytansinoids, their pharmaceutical compositions and method of use |
| JPS5566585A (en) | 1978-11-14 | 1980-05-20 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| JPS55164687A (en) | 1979-06-11 | 1980-12-22 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| JPS55102583A (en) | 1979-01-31 | 1980-08-05 | Takeda Chem Ind Ltd | 20-acyloxy-20-demethylmaytansinoid compound |
| JPS55162791A (en) | 1979-06-05 | 1980-12-18 | Takeda Chem Ind Ltd | Antibiotic c-15003pnd and its preparation |
| JPS55164685A (en) | 1979-06-08 | 1980-12-22 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| JPS55164686A (en) | 1979-06-11 | 1980-12-22 | Takeda Chem Ind Ltd | Novel maytansinoid compound and its preparation |
| US4309428A (en) | 1979-07-30 | 1982-01-05 | Takeda Chemical Industries, Ltd. | Maytansinoids |
| JPS5645483A (en) | 1979-09-19 | 1981-04-25 | Takeda Chem Ind Ltd | C-15003phm and its preparation |
| EP0028683A1 (en) | 1979-09-21 | 1981-05-20 | Takeda Chemical Industries, Ltd. | Antibiotic C-15003 PHO and production thereof |
| JPS5645485A (en) | 1979-09-21 | 1981-04-25 | Takeda Chem Ind Ltd | Production of c-15003pnd |
| WO1982001188A1 (en) | 1980-10-08 | 1982-04-15 | Takeda Chemical Industries Ltd | 4,5-deoxymaytansinoide compounds and process for preparing same |
| US4450254A (en) | 1980-11-03 | 1984-05-22 | Standard Oil Company | Impact improvement of high nitrile resins |
| US4315929A (en) | 1981-01-27 | 1982-02-16 | The United States Of America As Represented By The Secretary Of Agriculture | Method of controlling the European corn borer with trewiasine |
| US4313946A (en) | 1981-01-27 | 1982-02-02 | The United States Of America As Represented By The Secretary Of Agriculture | Chemotherapeutically active maytansinoids from Trewia nudiflora |
| JPS57192389A (en) | 1981-05-20 | 1982-11-26 | Takeda Chem Ind Ltd | Novel maytansinoid |
| US5208020A (en) * | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| CA2026147C (en) * | 1989-10-25 | 2006-02-07 | Ravi J. Chari | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| SE9102074D0 (en) | 1991-07-03 | 1991-07-03 | Kabi Pharmacia Ab | TOMOUR ANTIGEN SPECIFIC ANTIBODY |
| DK1024191T3 (en) | 1991-12-02 | 2008-12-08 | Medical Res Council | Preparation of autoantibodies displayed on phage surfaces from antibody segment libraries |
| US5639641A (en) | 1992-09-09 | 1997-06-17 | Immunogen Inc. | Resurfacing of rodent antibodies |
| CA2297070A1 (en) | 1997-08-01 | 1999-02-11 | Morphosys Ag | Novel method and phage for the identification of nucleic acid sequences encoding members of a multimeric (poly)peptide complex |
| CA2385528C (en) * | 1999-10-01 | 2013-12-10 | Immunogen, Inc. | Compositions and methods for treating cancer using immunoconjugates and chemotherapeutic agents |
| US6333410B1 (en) * | 2000-08-18 | 2001-12-25 | Immunogen, Inc. | Process for the preparation and purification of thiol-containing maytansinoids |
| EP1258255A1 (en) * | 2001-05-18 | 2002-11-20 | Boehringer Ingelheim International GmbH | Conjugates of an antibody to CD44 and a maytansinoid |
| US6441163B1 (en) * | 2001-05-31 | 2002-08-27 | Immunogen, Inc. | Methods for preparation of cytotoxic conjugates of maytansinoids and cell binding agents |
| PL374363A1 (en) * | 2001-09-05 | 2005-10-17 | Genentech, Inc. | Methods for the identification of polypeptide antigens associated with disorders involving aberrant cell proliferation and compositions useful for the treatment of such disorders |
| KR101424624B1 (en) * | 2003-05-14 | 2014-07-31 | 이뮤노젠 아이엔씨 | Drug Conjugate Composition |
| CA2525130C (en) * | 2003-05-20 | 2014-04-15 | Immunogen, Inc. | Improved cytotoxic agents comprising new maytansinoids |
| AU2004258955C1 (en) * | 2003-07-21 | 2012-07-26 | Immunogen, Inc. | A CA6 antigen-specific cytotoxic conjugate and methods of using the same |
-
2004
- 2004-05-20 CA CA2525130A patent/CA2525130C/en active Active
- 2004-05-20 EP EP20216572.6A patent/EP3851126A1/en active Pending
- 2004-05-20 JP JP2006532511A patent/JP5208420B2/en active Active
- 2004-05-20 EP EP04750945.0A patent/EP1651162B1/en active Active
- 2004-05-20 DK DK04750945.0T patent/DK1651162T3/en active
- 2004-05-20 CR CR20170291A patent/CR20170291A/en unknown
- 2004-05-20 BR BRPI0410748A patent/BRPI0410748B8/en active IP Right Grant
- 2004-05-20 SI SI200432295T patent/SI1651162T1/en unknown
- 2004-05-20 MX MX2016009879A patent/MX370281B/en unknown
- 2004-05-20 WO PCT/US2004/013314 patent/WO2004103272A2/en active Application Filing
- 2004-05-20 DK DK19160750.6T patent/DK3524611T3/en active
- 2004-05-20 NZ NZ542695A patent/NZ542695A/en unknown
- 2004-05-20 EA EA200501836A patent/EA010909B1/en unknown
- 2004-05-20 EP EP15190436.4A patent/EP3031810B1/en active Active
- 2004-05-20 PL PL04750945T patent/PL1651162T3/en unknown
- 2004-05-20 HU HUE04750945A patent/HUE028314T2/en unknown
- 2004-05-20 PT PT47509450T patent/PT1651162E/en unknown
- 2004-05-20 LT LTEP19160750.6T patent/LT3524611T/en unknown
- 2004-05-20 MX MX2013008224A patent/MX340862B/en unknown
- 2004-05-20 BR BRPI0419348A patent/BRPI0419348B8/en active IP Right Grant
- 2004-05-20 PL PL19160750T patent/PL3524611T3/en unknown
- 2004-05-20 CN CN200710194150.0A patent/CN101186613B/en active Active
- 2004-05-20 MX MXPA05011811A patent/MXPA05011811A/en active IP Right Grant
- 2004-05-20 EP EP19160750.6A patent/EP3524611B1/en active Active
- 2004-05-20 AU AU2004240541A patent/AU2004240541B2/en active Active
- 2004-05-20 KR KR1020057022166A patent/KR101145506B1/en active IP Right Grant
- 2004-05-20 ES ES19160750T patent/ES2863498T3/en active Active
- 2004-05-20 ES ES04750945.0T patent/ES2559670T3/en active Active
- 2004-05-20 CN CNA2004800134886A patent/CN1956722A/en active Pending
- 2004-05-20 HU HUE19160750A patent/HUE054074T2/en unknown
- 2004-05-20 PT PT191607506T patent/PT3524611T/en unknown
- 2004-05-20 SI SI200432508T patent/SI3524611T1/en unknown
-
2005
- 2005-09-28 ZA ZA200507845A patent/ZA200507845B/en unknown
- 2005-09-29 IL IL171170A patent/IL171170A/en active IP Right Grant
- 2005-11-03 CO CO05112351A patent/CO5660276A2/en unknown
- 2005-11-10 EC EC2005006149A patent/ECSP056149A/en unknown
- 2005-12-19 NO NO20056039A patent/NO339597B1/en unknown
-
2008
- 2008-07-02 HK HK08107275.3A patent/HK1116777A1/en unknown
-
2011
- 2011-06-30 IL IL213876A patent/IL213876A/en active IP Right Grant
-
2012
- 2012-03-14 CL CL2012000651A patent/CL2012000651A1/en unknown
- 2012-11-27 IL IL223297A patent/IL223297A/en active IP Right Grant
-
2013
- 2013-01-04 JP JP2013000052A patent/JP5563673B2/en active Active
-
2014
- 2014-03-30 IL IL231810A patent/IL231810A/en active IP Right Grant
-
2015
- 2015-05-19 IL IL238894A patent/IL238894A/en active IP Right Grant
- 2015-09-06 IL IL241211A patent/IL241211B/en active IP Right Grant
-
2016
- 2016-01-14 HR HRP20160046TT patent/HRP20160046T1/en unknown
- 2016-01-21 CY CY20161100067T patent/CY1117161T1/en unknown
-
2021
- 2021-03-19 HR HRP20210464TT patent/HRP20210464T1/en unknown
- 2021-03-26 CY CY20211100265T patent/CY1124278T1/en unknown
Non-Patent Citations (1)
| Title |
|---|
| See references of EP1651162A4 * |
Cited By (179)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8603483B2 (en) | 2004-12-09 | 2013-12-10 | Janssen Biotech, Inc. | Anti-integrin immunoconjugates, methods and uses |
| WO2006086733A3 (en) * | 2005-02-11 | 2007-06-07 | Immunogen Inc | Process for preparing maytansinoid antibody conjugates |
| JP2008531484A (en) * | 2005-02-11 | 2008-08-14 | イムノゲン インコーポレーティッド | Method for preparing stable drug conjugates |
| AU2006213662B2 (en) * | 2005-02-11 | 2010-08-05 | Immunogen, Inc. | Process for preparing stable drug conjugates |
| EP2062591A1 (en) | 2005-04-07 | 2009-05-27 | Novartis Vaccines and Diagnostics, Inc. | CACNA1E in cancer diagnosis detection and treatment |
| EP2083088A2 (en) | 2005-04-07 | 2009-07-29 | Novartis Vaccines and Diagnostics, Inc. | Cancer-related genes |
| US8388960B2 (en) | 2005-04-15 | 2013-03-05 | Immunogen Inc. | Elimination of heterogeneous or mixed cell population in tumors |
| EP1868649A2 (en) * | 2005-04-15 | 2007-12-26 | Immunogen, Inc. | Elimination of heterogeneous or mixed cell population in tumors |
| US8137669B2 (en) | 2005-04-15 | 2012-03-20 | Immunogen, Inc. | Elimination of heterogeneous or mixed cell population in tumors |
| EP1868649A4 (en) * | 2005-04-15 | 2011-06-29 | Immunogen Inc | Elimination of heterogeneous or mixed cell population in tumors |
| EP2662096A1 (en) * | 2005-08-24 | 2013-11-13 | ImmunoGen, Inc. | Process for preparing maytansinoid antibody conjugates |
| US11471536B2 (en) | 2005-08-24 | 2022-10-18 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| EP3539572A1 (en) * | 2005-08-24 | 2019-09-18 | ImmunoGen, Inc. | Process for preparing maytansinoid antibody conjugates |
| US9789204B2 (en) | 2005-08-24 | 2017-10-17 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| US7811572B2 (en) | 2005-08-24 | 2010-10-12 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| JP2013014604A (en) * | 2005-08-24 | 2013-01-24 | Immunogen Inc | Process for preparing maytansinoid antibody conjugate |
| US8933205B2 (en) | 2005-08-24 | 2015-01-13 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| US8383122B2 (en) | 2005-08-24 | 2013-02-26 | Immunogen, Inc. | Process for preparing purified drug conjugates |
| WO2007024536A3 (en) * | 2005-08-24 | 2007-07-05 | Immunogen Inc | Process for preparing maytansinoid antibody conjugates |
| JP2014196307A (en) * | 2005-08-24 | 2014-10-16 | イムノゲン インコーポレーティッド | Method for preparing maytansinoid-antibody conjugate |
| EA013327B1 (en) * | 2005-08-24 | 2010-04-30 | Иммуноджен, Инк. | Process for preparing purified drug conjugate |
| US10576034B2 (en) | 2005-12-01 | 2020-03-03 | University Of Massachusetts Lowell | Botulinum nanoemulsions |
| US10532019B2 (en) | 2005-12-01 | 2020-01-14 | University Of Massachusetts Lowell | Botulinum nanoemulsions |
| EP2386559A1 (en) | 2006-01-25 | 2011-11-16 | Sanofi | Cytotoxiv agents comprising new tomaymycin derivatives and their therapeutic use |
| US8163736B2 (en) | 2006-01-25 | 2012-04-24 | Sanofi-Aventis | Cytotoxic agents comprising new tomaymycin derivatives |
| EP1813614A1 (en) | 2006-01-25 | 2007-08-01 | Sanofi-Aventis | Cytotoxic agents comprising new tomaymycin derivatives |
| EP2371827A1 (en) | 2006-01-25 | 2011-10-05 | Sanofi | Cytotoxic agents comprising new tomaymycin derivatives and their therapeutic use |
| EP2389948A1 (en) | 2006-03-23 | 2011-11-30 | Novartis AG | Anti-tumor cell antigen antibody therapeutics |
| EP2389950A1 (en) | 2006-03-23 | 2011-11-30 | Novartis AG | Anti-tumor cell antigen antibody therapeutics |
| EP2389947A1 (en) | 2006-03-23 | 2011-11-30 | Novartis AG | Anti-tumor cell antigen antibody therapeutics |
| EP2389951A1 (en) | 2006-03-23 | 2011-11-30 | Novartis AG | Anti-tumor cell antigen antibody therapeutics |
| EP2389949A1 (en) | 2006-03-23 | 2011-11-30 | Novartis AG | Anti-tumor cell antigen antibody therapeutics |
| EP2389946A1 (en) | 2006-03-23 | 2011-11-30 | Novartis AG | Anti-tumor cell antigen antibody therapeutics |
| EP2407483A1 (en) | 2006-04-13 | 2012-01-18 | Novartis Vaccines and Diagnostics, Inc. | Methods of treating, diagnosing or detecting cancers |
| EP2474556A2 (en) | 2007-03-14 | 2012-07-11 | Novartis AG | APCDD1 inhibitors for treating, diagnosing or detecting cancer |
| US9150649B2 (en) | 2008-04-30 | 2015-10-06 | Immunogen, Inc. | Potent conjugates and hydrophilic linkers |
| US9376500B2 (en) | 2009-06-03 | 2016-06-28 | Immunogen, Inc. | Conjugation methods |
| US10815309B2 (en) | 2009-06-03 | 2020-10-27 | Immunogen, Inc. | Methods for preparing antibody-drug conjugates |
| US11498979B2 (en) | 2009-06-03 | 2022-11-15 | Immunogen, Inc. | Methods for preparing a purified maytansinoid conjugate in a solution |
| US9771432B2 (en) | 2009-06-03 | 2017-09-26 | Immunogen, Inc. | Conjugation methods |
| US10233257B2 (en) | 2009-06-03 | 2019-03-19 | Immunogen, Inc. | Methods for preparing antibody-drug conjugates |
| US8952147B2 (en) | 2009-06-29 | 2015-02-10 | Sanofi | Conjugates, preparation thereof, and therapeutic use thereof |
| WO2011001052A1 (en) | 2009-06-29 | 2011-01-06 | Sanofi-Aventis | Novel conjugates, preparation thereof, and therapeutic use thereof |
| US8481042B2 (en) | 2009-08-25 | 2013-07-09 | Sanofi | Conjugates of pyrrolo[1,4]benzodiazepine dimers as anticancer agents |
| WO2011023883A1 (en) | 2009-08-25 | 2011-03-03 | Sanofi-Aventis | Conjugates of pyrrolo[1,4]benzodiazepine dimers as anticancer agents |
| WO2011039721A1 (en) | 2009-10-02 | 2011-04-07 | Sanofi-Aventis | New maytansinoids and the use of said maytansinoids to prepare conjugates with an antibody |
| WO2011045396A2 (en) | 2009-10-16 | 2011-04-21 | Novartis Ag | Biomarkers of tumor pharmacodynamic response |
| US9056914B2 (en) | 2010-07-26 | 2015-06-16 | Sanofi | Anticancer derivatives, preparation thereof and therapeutic use thereof |
| WO2012014147A1 (en) | 2010-07-26 | 2012-02-02 | Sanofi | Anticancer derivatives, preparation thereof and therapeutic use thereof |
| WO2012016227A2 (en) | 2010-07-29 | 2012-02-02 | Xencor, Inc. | Antibodies with modified isoelectric points |
| EP3029066A2 (en) | 2010-07-29 | 2016-06-08 | Xencor, Inc. | Antibodies with modified isoelectric points |
| EP3828205A1 (en) | 2010-10-01 | 2021-06-02 | Oxford BioTherapeutics Ltd | Anti-ror1 antibodies |
| EP3219731A1 (en) | 2010-10-01 | 2017-09-20 | Oxford BioTherapeutics Ltd | Anti-ror1 antibodies |
| WO2012045085A1 (en) | 2010-10-01 | 2012-04-05 | Oxford Biotherapeutics Ltd. | Anti-rori antibodies |
| CN106349254A (en) * | 2010-11-03 | 2017-01-25 | 伊缪诺金公司 | Cytotoxic agents comprising new ansamitocin derivatives |
| AU2011323316A9 (en) * | 2010-11-03 | 2016-02-18 | Immunogen, Inc. | Cytotoxic agents comprising new ansamitocin derivatives |
| AU2011323316B2 (en) * | 2010-11-03 | 2016-02-25 | Immunogen, Inc. | Cytotoxic agents comprising new ansamitocin derivatives |
| US9090629B2 (en) | 2010-11-03 | 2015-07-28 | Immunogen, Inc. | Cytotoxic agents comprising new ansamitocin derivatives |
| CN103269712A (en) * | 2010-11-03 | 2013-08-28 | 伊缪诺金公司 | Cytotoxic agents comprising new ansamitocin derivatives |
| WO2012061590A1 (en) * | 2010-11-03 | 2012-05-10 | Immunogen, Inc. | Cytotoxic agents comprising new ansamitocin derivatives |
| CN103269712B (en) * | 2010-11-03 | 2016-09-21 | 伊缪诺金公司 | Comprise the cytotoxic agent of new ansamitocin derivant |
| EP3284755A1 (en) | 2010-12-30 | 2018-02-21 | Takeda Pharmaceutical Company Limited | Conjugated anti-cd38 antibodies |
| EP3798231A1 (en) | 2010-12-30 | 2021-03-31 | Takeda Pharmaceutical Company Limited | Conjugated anti-cd38 antibodies |
| US9428543B2 (en) | 2011-03-29 | 2016-08-30 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| US11090390B2 (en) | 2011-03-29 | 2021-08-17 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| US11744900B2 (en) | 2011-03-29 | 2023-09-05 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| US9914748B2 (en) | 2011-03-29 | 2018-03-13 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| US8795673B2 (en) | 2011-03-29 | 2014-08-05 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| US10435432B2 (en) | 2011-03-29 | 2019-10-08 | Immunogen, Inc. | Preparation of maytansinoid antibody conjugates by a one-step process |
| AU2012258254B2 (en) * | 2011-05-17 | 2016-04-21 | Sanofi | Use of anti-CD19 maytansinoid immunoconjugate antibody for the treatment of B-cell malignancies symptoms |
| EP2524929A1 (en) * | 2011-05-17 | 2012-11-21 | Sanofi | Use of anti-CD19 maytansinoid immunoconjugate antibody for the treatment of CD19+ B-cell malignancies syptoms |
| WO2012156455A1 (en) * | 2011-05-17 | 2012-11-22 | Sanofi | Use of anti-cd19 maytansinoid immunoconjugate antibody for the treatment of b-cell malignancies symptoms |
| EA028574B1 (en) * | 2011-05-17 | 2017-12-29 | Санофи | Method of treating b-cell malignancies expressing cd19 |
| CN103547596A (en) * | 2011-05-17 | 2014-01-29 | 赛诺菲 | Use of anti-CD19 maytansinoid immunoconjugate antibody for the treatment of b-cell malignancies symptoms |
| US9555126B2 (en) | 2011-05-17 | 2017-01-31 | Sanofi | Use of anti-CD19 maytansinoid immunoconjugate antibody for the treatment of B-cell malignancies symptoms |
| EP2550975A1 (en) * | 2011-07-29 | 2013-01-30 | Sanofi | Combination therapy for the treatment of CD19+ B-cell malignancies symptoms comprising an anti-CD19 maytansinoid immunoconjugate and rituximab |
| WO2013017540A1 (en) * | 2011-07-29 | 2013-02-07 | Sanofi | Combination therapy for the treatment of cd19+ b-cell malignancies symptoms comprising an anti-cd19 maytansinoid immunoconjugate and rituximab |
| EP3611187A1 (en) | 2011-10-10 | 2020-02-19 | Xencor, Inc. | A method for purifying antibodies |
| US10851178B2 (en) | 2011-10-10 | 2020-12-01 | Xencor, Inc. | Heterodimeric human IgG1 polypeptides with isoelectric point modifications |
| US10944190B2 (en) | 2012-09-26 | 2021-03-09 | Immunogen, Inc. | Methods for the acylation of maytansinol |
| US10035817B2 (en) | 2012-10-04 | 2018-07-31 | Immunogen, Inc. | Method of purifying cell-binding agent-cytotoxic agent conjugates with a PVDF membrane |
| US10131710B2 (en) | 2013-01-14 | 2018-11-20 | Xencor, Inc. | Optimized antibody variable regions |
| US11718667B2 (en) | 2013-01-14 | 2023-08-08 | Xencor, Inc. | Optimized antibody variable regions |
| US9701759B2 (en) | 2013-01-14 | 2017-07-11 | Xencor, Inc. | Heterodimeric proteins |
| US11634506B2 (en) | 2013-01-14 | 2023-04-25 | Xencor, Inc. | Heterodimeric proteins |
| US9650446B2 (en) | 2013-01-14 | 2017-05-16 | Xencor, Inc. | Heterodimeric proteins |
| US10472427B2 (en) | 2013-01-14 | 2019-11-12 | Xencor, Inc. | Heterodimeric proteins |
| US10487155B2 (en) | 2013-01-14 | 2019-11-26 | Xencor, Inc. | Heterodimeric proteins |
| US10738133B2 (en) | 2013-01-14 | 2020-08-11 | Xencor, Inc. | Heterodimeric proteins |
| US10738132B2 (en) | 2013-01-14 | 2020-08-11 | Xencor, Inc. | Heterodimeric proteins |
| US11053316B2 (en) | 2013-01-14 | 2021-07-06 | Xencor, Inc. | Optimized antibody variable regions |
| US9738722B2 (en) | 2013-01-15 | 2017-08-22 | Xencor, Inc. | Rapid clearance of antigen complexes using novel antibodies |
| WO2014114207A1 (en) | 2013-01-23 | 2014-07-31 | 上海新理念生物医药科技有限公司 | Tridentate connexon and use thereof |
| US10960082B2 (en) | 2013-01-23 | 2021-03-30 | Newbio Therapeutics, Inc. | Tridentate connexon and use thereof |
| US10968276B2 (en) | 2013-03-12 | 2021-04-06 | Xencor, Inc. | Optimized anti-CD3 variable regions |
| US9605084B2 (en) | 2013-03-15 | 2017-03-28 | Xencor, Inc. | Heterodimeric proteins |
| US11299554B2 (en) | 2013-03-15 | 2022-04-12 | Xencor, Inc. | Heterodimeric proteins |
| US10106624B2 (en) | 2013-03-15 | 2018-10-23 | Xencor, Inc. | Heterodimeric proteins |
| US10858417B2 (en) | 2013-03-15 | 2020-12-08 | Xencor, Inc. | Heterodimeric proteins |
| US11814423B2 (en) | 2013-03-15 | 2023-11-14 | Xencor, Inc. | Heterodimeric proteins |
| US10544187B2 (en) | 2013-03-15 | 2020-01-28 | Xencor, Inc. | Targeting regulatory T cells with heterodimeric proteins |
| EP3421495A2 (en) | 2013-03-15 | 2019-01-02 | Xencor, Inc. | Modulation of t cells with bispecific antibodies and fc fusions |
| US10287364B2 (en) | 2013-03-15 | 2019-05-14 | Xencor, Inc. | Heterodimeric proteins |
| EP3587448A1 (en) | 2013-03-15 | 2020-01-01 | Xencor, Inc. | Heterodimeric proteins |
| US10519242B2 (en) | 2013-03-15 | 2019-12-31 | Xencor, Inc. | Targeting regulatory T cells with heterodimeric proteins |
| WO2015009740A2 (en) | 2013-07-15 | 2015-01-22 | Cell Signaling Technology, Inc. | Anti-mucin 1 binding agents and uses thereof |
| EP3699200A1 (en) | 2013-07-15 | 2020-08-26 | Cell Signaling Technology, Inc. | Anti-mucin 1 binding agents and uses thereof |
| US9822186B2 (en) | 2014-03-28 | 2017-11-21 | Xencor, Inc. | Bispecific antibodies that bind to CD38 and CD3 |
| US11840579B2 (en) | 2014-03-28 | 2023-12-12 | Xencor, Inc. | Bispecific antibodies that bind to CD38 and CD3 |
| US10858451B2 (en) | 2014-03-28 | 2020-12-08 | Xencor, Inc. | Bispecific antibodies that bind to CD38 and CD3 |
| WO2015149077A1 (en) | 2014-03-28 | 2015-10-01 | Xencor, Inc. | Bispecific antibodies that bind to cd38 and cd3 |
| US10322191B2 (en) | 2014-06-30 | 2019-06-18 | Tarveda Therapeutics, Inc. | Targeted conjugates and particles and formulations thereof |
| US11458206B2 (en) | 2014-06-30 | 2022-10-04 | Tva (Abc), Llc | Targeted conjugates and particles and formulations thereof |
| US10624967B2 (en) | 2014-06-30 | 2020-04-21 | Tarveda Therapeutics, Inc. | Targeted conjugates and particles and formulations thereof |
| EP3160518A4 (en) * | 2014-06-30 | 2018-05-23 | Tarveda Therapeutics, Inc. | Targeted conjugates and particles and formulations thereof |
| WO2016014984A1 (en) | 2014-07-24 | 2016-01-28 | Xencor, Inc. | Rapid clearance of antigen complexes using novel antibodies |
| EP3311846A1 (en) | 2014-09-02 | 2018-04-25 | ImmunoGen, Inc. | Methods for formulating antibody drug conjugate compositions |
| US10603388B2 (en) | 2014-09-02 | 2020-03-31 | Immunogen, Inc. | Methods for formulating antibody drug conjugate compositions |
| EP3778601A1 (en) | 2014-09-03 | 2021-02-17 | ImmunoGen, Inc. | Cytotoxic benzodiazepine derivatives |
| WO2016036804A1 (en) | 2014-09-03 | 2016-03-10 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives |
| US10526417B2 (en) | 2014-11-26 | 2020-01-07 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CD38 |
| US11859011B2 (en) | 2014-11-26 | 2024-01-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US11225528B2 (en) | 2014-11-26 | 2022-01-18 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US11673972B2 (en) | 2014-11-26 | 2023-06-13 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US11111315B2 (en) | 2014-11-26 | 2021-09-07 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US9850320B2 (en) | 2014-11-26 | 2017-12-26 | Xencor, Inc. | Heterodimeric antibodies to CD3 X CD20 |
| US9856327B2 (en) | 2014-11-26 | 2018-01-02 | Xencor, Inc. | Heterodimeric antibodies to CD3 X CD123 |
| US11945880B2 (en) | 2014-11-26 | 2024-04-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US10889653B2 (en) | 2014-11-26 | 2021-01-12 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US10913803B2 (en) | 2014-11-26 | 2021-02-09 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US11352442B2 (en) | 2014-11-26 | 2022-06-07 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CD38 |
| US10259887B2 (en) | 2014-11-26 | 2019-04-16 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and tumor antigens |
| US10428155B2 (en) | 2014-12-22 | 2019-10-01 | Xencor, Inc. | Trispecific antibodies |
| WO2016141387A1 (en) | 2015-03-05 | 2016-09-09 | Xencor, Inc. | Modulation of t cells with bispecific antibodies and fc fusions |
| US10227411B2 (en) | 2015-03-05 | 2019-03-12 | Xencor, Inc. | Modulation of T cells with bispecific antibodies and FC fusions |
| US11091548B2 (en) | 2015-03-05 | 2021-08-17 | Xencor, Inc. | Modulation of T cells with bispecific antibodies and Fc fusions |
| WO2016192527A1 (en) | 2015-05-29 | 2016-12-08 | Newbio Therapeutics, Inc. | Derivatives of dolastatin 10 and uses thereof |
| US10449258B2 (en) | 2015-06-09 | 2019-10-22 | Xdcexplorer (Shanghai) Co., Ltd. | Antibody drug conjugate, intermediate, preparation method, pharmaceutical composition and uses thereof |
| US11160871B2 (en) | 2015-10-28 | 2021-11-02 | Tarveda Therapeutics, Inc. | SSTR-targeted conjugates and particles and formulations thereof |
| WO2017088734A1 (en) | 2015-11-23 | 2017-06-01 | 四川科伦博泰生物医药股份有限公司 | Anti-erbb2 antibody-drug conjugate and composition thereof, preparation method therefor, and application thereof |
| US10227410B2 (en) | 2015-12-07 | 2019-03-12 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and PSMA |
| US11623957B2 (en) | 2015-12-07 | 2023-04-11 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and PSMA |
| US11492407B2 (en) | 2016-06-14 | 2022-11-08 | Xencor, Inc. | Bispecific checkpoint inhibitor antibodies |
| US11236170B2 (en) | 2016-06-14 | 2022-02-01 | Xencor, Inc. | Bispecific checkpoint inhibitor antibodies |
| US10787518B2 (en) | 2016-06-14 | 2020-09-29 | Xencor, Inc. | Bispecific checkpoint inhibitor antibodies |
| US10316088B2 (en) | 2016-06-28 | 2019-06-11 | Xencor, Inc. | Heterodimeric antibodies that bind somatostatin receptor 2 |
| US11225521B2 (en) | 2016-06-28 | 2022-01-18 | Xencor, Inc. | Heterodimeric antibodies that bind somatostatin receptor 2 |
| US10793632B2 (en) | 2016-08-30 | 2020-10-06 | Xencor, Inc. | Bispecific immunomodulatory antibodies that bind costimulatory and checkpoint receptors |
| US10654873B2 (en) | 2016-09-15 | 2020-05-19 | Polytherics Limited | Cytotoxic agents and conjugates thereof |
| US10501543B2 (en) | 2016-10-14 | 2019-12-10 | Xencor, Inc. | IL15/IL15Rα heterodimeric Fc-fusion proteins |
| US10550185B2 (en) | 2016-10-14 | 2020-02-04 | Xencor, Inc. | Bispecific heterodimeric fusion proteins containing IL-15-IL-15Rα Fc-fusion proteins and PD-1 antibody fragments |
| US11311496B2 (en) | 2016-11-21 | 2022-04-26 | Eirion Therapeutics, Inc. | Transdermal delivery of large agents |
| WO2018160539A1 (en) | 2017-02-28 | 2018-09-07 | Immunogen, Inc. | Maytansinoid derivatives with self-immolative peptide linkers and conjugates thereof |
| US11365258B2 (en) | 2017-03-10 | 2022-06-21 | Berlin-Chemie Ag | Pharmaceutical combinations comprising an anti-LY75 antibody |
| EP4257614A2 (en) | 2017-03-10 | 2023-10-11 | Berlin-Chemie AG | Pharmaceutical combinations comprising an anti-ly75 antibody |
| WO2018195243A1 (en) | 2017-04-20 | 2018-10-25 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives and conjugates thereof |
| US11084863B2 (en) | 2017-06-30 | 2021-08-10 | Xencor, Inc. | Targeted heterodimeric Fc fusion proteins containing IL-15 IL-15alpha and antigen binding domains |
| US10981992B2 (en) | 2017-11-08 | 2021-04-20 | Xencor, Inc. | Bispecific immunomodulatory antibodies that bind costimulatory and checkpoint receptors |
| US11312770B2 (en) | 2017-11-08 | 2022-04-26 | Xencor, Inc. | Bispecific and monospecific antibodies using novel anti-PD-1 sequences |
| WO2019105835A1 (en) | 2017-11-29 | 2019-06-06 | Bayer Consumer Care Ag | Combinations of copanlisib and anetumab ravtansine |
| US11319355B2 (en) | 2017-12-19 | 2022-05-03 | Xencor, Inc. | Engineered IL-2 Fc fusion proteins |
| WO2019133652A1 (en) | 2017-12-28 | 2019-07-04 | Immunogen, Inc. | Benzodiazepine derivatives |
| US10982006B2 (en) | 2018-04-04 | 2021-04-20 | Xencor, Inc. | Heterodimeric antibodies that bind fibroblast activation protein |
| US11505595B2 (en) | 2018-04-18 | 2022-11-22 | Xencor, Inc. | TIM-3 targeted heterodimeric fusion proteins containing IL-15/IL-15RA Fc-fusion proteins and TIM-3 antigen binding domains |
| US11524991B2 (en) | 2018-04-18 | 2022-12-13 | Xencor, Inc. | PD-1 targeted heterodimeric fusion proteins containing IL-15/IL-15Ra Fc-fusion proteins and PD-1 antigen binding domains and uses thereof |
| WO2019238843A1 (en) | 2018-06-14 | 2019-12-19 | Berlin-Chemie Ag | Pharmaceutical combinations |
| WO2020010079A2 (en) | 2018-07-02 | 2020-01-09 | Amgen Inc. | Anti-steap1 antigen-binding protein |
| US11857638B2 (en) | 2018-07-27 | 2024-01-02 | Promega Corporation | Quinone-containing conjugates |
| WO2020023871A1 (en) | 2018-07-27 | 2020-01-30 | Promega Corporation | Quinone-containing conjugates |
| US11358999B2 (en) | 2018-10-03 | 2022-06-14 | Xencor, Inc. | IL-12 heterodimeric Fc-fusion proteins |
| US11472890B2 (en) | 2019-03-01 | 2022-10-18 | Xencor, Inc. | Heterodimeric antibodies that bind ENPP3 and CD3 |
| WO2020234114A1 (en) | 2019-05-21 | 2020-11-26 | Bayer Aktiengesellschaft | A novel stable high concentration formulation for anetumab ravtansine |
| US11634508B2 (en) | 2019-07-10 | 2023-04-25 | Cybrexa 2, Inc. | Peptide conjugates of cytotoxins as therapeutics |
| US11919956B2 (en) | 2020-05-14 | 2024-03-05 | Xencor, Inc. | Heterodimeric antibodies that bind prostate specific membrane antigen (PSMA) and CD3 |
| US11919958B2 (en) | 2020-08-19 | 2024-03-05 | Xencor, Inc. | Anti-CD28 compositions |
| US11591401B2 (en) | 2020-08-19 | 2023-02-28 | Xencor, Inc. | Anti-CD28 compositions |
| US11739144B2 (en) | 2021-03-09 | 2023-08-29 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CLDN6 |
| US11859012B2 (en) | 2021-03-10 | 2024-01-02 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and GPC3 |
| WO2023089314A1 (en) | 2021-11-18 | 2023-05-25 | Oxford Biotherapeutics Limited | Pharmaceutical combinations |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20180043013A1 (en) | Cytotoxic agents comprising new maytansinoids (dm4) | |
| EP3524611B1 (en) | Improved cytotoxic agents comprising new maytansinoids | |
| EP0425235B1 (en) | Cytotoxic agents comprising maytansinoids and their therapeutic use | |
| US5208020A (en) | Cytotoxic agents comprising maytansinoids and their therapeutic use | |
| US8198417B2 (en) | Method of targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via a non-cleavable linker, said conjugates and methods of making said conjugates | |
| JP2013542958A (en) | Cytotoxic agents containing ansamitocin derivatives | |
| JP2024059792A (en) | Methods for targeting specific cell populations using cell-binding agent maytansinoid conjugates linked via non-cleavable linkers, said conjugates, and methods for making said conjugates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005/07845 Country of ref document: ZA Ref document number: 542695 Country of ref document: NZ Ref document number: 200507845 Country of ref document: ZA Ref document number: 2004240541 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 171170 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 542695 Country of ref document: NZ |
|
| ENP | Entry into the national phase |
Ref document number: 2004240541 Country of ref document: AU Date of ref document: 20040520 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004240541 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004750945 Country of ref document: EP Ref document number: 4915/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 05112351A Country of ref document: CO Ref document number: PA/a/2005/011811 Country of ref document: MX Ref document number: 05112351 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2525130 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20048134886 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2005-008100 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006532511 Country of ref document: JP Ref document number: 1020057022166 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200501836 Country of ref document: EA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057022166 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004750945 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0410748 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 223297 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 231810 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 238894 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 241211 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017291 Country of ref document: CR Ref document number: CR2017-000291 Country of ref document: CR |