WO2004089960A2 - Process for preparation of cyclosporin “a” analogues having a terminal diene group - Google Patents
Process for preparation of cyclosporin “a” analogues having a terminal diene groupInfo
- Publication number
- WO2004089960A2 WO2004089960A2 PCT/EP2004/003504 EP2004003504W WO2004089960A2 WO 2004089960 A2 WO2004089960 A2 WO 2004089960A2 EP 2004003504 W EP2004003504 W EP 2004003504W WO 2004089960 A2 WO2004089960 A2 WO 2004089960A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- conducted
- process according
- dichloromethane
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 76
- 230000008569 process Effects 0.000 title claims abstract description 65
- 238000002360 preparation method Methods 0.000 title claims abstract description 35
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 title claims abstract description 18
- 229930105110 Cyclosporin A Natural products 0.000 title abstract description 21
- 108010036949 Cyclosporine Proteins 0.000 title abstract description 21
- 229960001265 ciclosporin Drugs 0.000 title abstract description 21
- 229930182912 cyclosporin Natural products 0.000 title 1
- 125000002897 diene group Chemical group 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 47
- 239000010936 titanium Substances 0.000 claims abstract description 22
- 229910052719 titanium Inorganic materials 0.000 claims abstract description 14
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 claims abstract description 13
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 220
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 128
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 120
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 96
- 229910001868 water Inorganic materials 0.000 claims description 85
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 82
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 82
- 238000006243 chemical reaction Methods 0.000 claims description 75
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 57
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 42
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 41
- 235000019253 formic acid Nutrition 0.000 claims description 41
- 239000011541 reaction mixture Substances 0.000 claims description 38
- 239000000203 mixture Substances 0.000 claims description 37
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 claims description 33
- 239000012074 organic phase Substances 0.000 claims description 33
- 238000010626 work up procedure Methods 0.000 claims description 23
- 239000002904 solvent Substances 0.000 claims description 22
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 claims description 20
- 125000006239 protecting group Chemical group 0.000 claims description 18
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 claims description 18
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 claims description 16
- 125000000217 alkyl group Chemical group 0.000 claims description 16
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 claims description 16
- 229910052739 hydrogen Inorganic materials 0.000 claims description 16
- HYWCXWRMUZYRPH-UHFFFAOYSA-N trimethyl(prop-2-enyl)silane Chemical compound C[Si](C)(C)CC=C HYWCXWRMUZYRPH-UHFFFAOYSA-N 0.000 claims description 16
- NHDIQVFFNDKAQU-UHFFFAOYSA-N tripropan-2-yl borate Chemical compound CC(C)OB(OC(C)C)OC(C)C NHDIQVFFNDKAQU-UHFFFAOYSA-N 0.000 claims description 14
- 239000001257 hydrogen Substances 0.000 claims description 12
- 230000002378 acidificating effect Effects 0.000 claims description 11
- 239000006184 cosolvent Substances 0.000 claims description 11
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 11
- YNLAOSYQHBDIKW-UHFFFAOYSA-M diethylaluminium chloride Chemical compound CC[Al](Cl)CC YNLAOSYQHBDIKW-UHFFFAOYSA-M 0.000 claims description 9
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 claims description 8
- 229940011051 isopropyl acetate Drugs 0.000 claims description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 8
- 239000013638 trimer Substances 0.000 claims description 8
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims description 7
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 7
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 7
- WRECIMRULFAWHA-UHFFFAOYSA-N trimethyl borate Chemical compound COB(OC)OC WRECIMRULFAWHA-UHFFFAOYSA-N 0.000 claims description 5
- VBKNTGMWIPUCRF-UHFFFAOYSA-M potassium;fluoride;hydrofluoride Chemical compound F.[F-].[K+] VBKNTGMWIPUCRF-UHFFFAOYSA-M 0.000 claims description 4
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 claims 2
- 150000003892 tartrate salts Chemical class 0.000 claims 2
- 239000003153 chemical reaction reagent Substances 0.000 abstract description 95
- -1 cyclosporin A aldehyde Chemical class 0.000 abstract description 66
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 abstract description 3
- 229910052782 aluminium Inorganic materials 0.000 abstract description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 abstract description 2
- 239000004411 aluminium Substances 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 117
- 229960005289 voclosporin Drugs 0.000 description 57
- 238000005937 allylation reaction Methods 0.000 description 46
- 230000008030 elimination Effects 0.000 description 46
- 238000003379 elimination reaction Methods 0.000 description 46
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 37
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 37
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 31
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 28
- 239000000725 suspension Substances 0.000 description 26
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 24
- 238000003756 stirring Methods 0.000 description 22
- 238000005481 NMR spectroscopy Methods 0.000 description 21
- 238000002425 crystallisation Methods 0.000 description 19
- 230000008025 crystallization Effects 0.000 description 19
- 238000003786 synthesis reaction Methods 0.000 description 19
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 18
- 229920006395 saturated elastomer Polymers 0.000 description 17
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 16
- 230000015572 biosynthetic process Effects 0.000 description 15
- 229910052796 boron Inorganic materials 0.000 description 15
- 239000000543 intermediate Substances 0.000 description 15
- 239000012071 phase Substances 0.000 description 15
- 239000011780 sodium chloride Substances 0.000 description 15
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 14
- 230000007062 hydrolysis Effects 0.000 description 14
- 238000006460 hydrolysis reaction Methods 0.000 description 14
- 229940093499 ethyl acetate Drugs 0.000 description 13
- 235000019439 ethyl acetate Nutrition 0.000 description 13
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 13
- 238000004128 high performance liquid chromatography Methods 0.000 description 13
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 13
- GMDGXJQUWIQOLE-UHFFFAOYSA-N prop-2-enylborane Chemical compound BCC=C GMDGXJQUWIQOLE-UHFFFAOYSA-N 0.000 description 13
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 12
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 12
- 238000005160 1H NMR spectroscopy Methods 0.000 description 11
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 11
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical compound FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 description 11
- 239000011734 sodium Substances 0.000 description 11
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 10
- 238000011065 in-situ storage Methods 0.000 description 10
- 239000007787 solid Substances 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 239000003960 organic solvent Substances 0.000 description 9
- 239000000843 powder Substances 0.000 description 9
- 229940095064 tartrate Drugs 0.000 description 9
- 229910015900 BF3 Inorganic materials 0.000 description 8
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 8
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 8
- 235000019341 magnesium sulphate Nutrition 0.000 description 8
- 229910052751 metal Inorganic materials 0.000 description 8
- 239000002184 metal Substances 0.000 description 8
- 239000000126 substance Substances 0.000 description 8
- 230000004913 activation Effects 0.000 description 7
- 239000008346 aqueous phase Substances 0.000 description 7
- 230000005595 deprotonation Effects 0.000 description 7
- 238000010537 deprotonation reaction Methods 0.000 description 7
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 7
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 6
- 239000002841 Lewis acid Substances 0.000 description 6
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- KUSICUWKCBCAHV-ZSINMPTNSA-N [(e,1r,2r)-1-[(2s,5s,11s,14s,17s,20s,23r,26s,29s,32s)-5-ethyl-1,7,10,16,20,23,25,28,31-nonamethyl-11,17,26,29-tetrakis(2-methylpropyl)-3,6,9,12,15,18,21,24,27,30,33-undecaoxo-14,32-di(propan-2-yl)-1,4,7,10,13,16,19,22,25,28,31-undecazacyclotritriacont-2-y Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](OC(C)=O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O KUSICUWKCBCAHV-ZSINMPTNSA-N 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 239000006260 foam Substances 0.000 description 6
- 235000011167 hydrochloric acid Nutrition 0.000 description 6
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 6
- GWYFCOCPABKNJV-UHFFFAOYSA-M isovalerate Chemical compound CC(C)CC([O-])=O GWYFCOCPABKNJV-UHFFFAOYSA-M 0.000 description 6
- 150000007517 lewis acids Chemical class 0.000 description 6
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 239000012455 biphasic mixture Substances 0.000 description 5
- 230000000875 corresponding effect Effects 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 239000002274 desiccant Substances 0.000 description 5
- YMWUJEATGCHHMB-DICFDUPASA-N dichloromethane-d2 Chemical compound [2H]C([2H])(Cl)Cl YMWUJEATGCHHMB-DICFDUPASA-N 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 238000005580 one pot reaction Methods 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- 229910052700 potassium Inorganic materials 0.000 description 5
- 229910052710 silicon Inorganic materials 0.000 description 5
- BICRTLVBTLFLRD-PTWUADNWSA-N voclosporin Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C=C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O BICRTLVBTLFLRD-PTWUADNWSA-N 0.000 description 5
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- ZMANZCXQSJIPKH-UHFFFAOYSA-N N,N-Diethylethanamine Substances CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 4
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 4
- 238000006293 aldehyde allylation reaction Methods 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 150000001450 anions Chemical class 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- 239000002808 molecular sieve Substances 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 238000012552 review Methods 0.000 description 4
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 4
- 108010057559 voclosporin Proteins 0.000 description 4
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 150000001735 carboxylic acids Chemical class 0.000 description 3
- 235000001729 chan in Nutrition 0.000 description 3
- 239000003638 chemical reducing agent Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000005100 correlation spectroscopy Methods 0.000 description 3
- 150000001993 dienes Chemical group 0.000 description 3
- 238000006735 epoxidation reaction Methods 0.000 description 3
- 150000002170 ethers Chemical class 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 239000012634 fragment Substances 0.000 description 3
- 238000005570 heteronuclear single quantum coherence Methods 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 229910052744 lithium Inorganic materials 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 229940098779 methanesulfonic acid Drugs 0.000 description 3
- 239000007800 oxidant agent Substances 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 238000007248 oxidative elimination reaction Methods 0.000 description 3
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 230000009257 reactivity Effects 0.000 description 3
- 229910052707 ruthenium Inorganic materials 0.000 description 3
- 238000007086 side reaction Methods 0.000 description 3
- 239000011877 solvent mixture Substances 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 230000007704 transition Effects 0.000 description 3
- PBIMIGNDTBRRPI-UHFFFAOYSA-N trifluoro borate Chemical compound FOB(OF)OF PBIMIGNDTBRRPI-UHFFFAOYSA-N 0.000 description 3
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- DBERHVIZRVGDFO-UHFFFAOYSA-N Acetoxyacetone Chemical compound CC(=O)COC(C)=O DBERHVIZRVGDFO-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 0 CC(C)C[C@@](C(N[C@](C(C)C)C(N(C)[C@@](CC(C)C)C(N[C@@](C)C(N[C@](C)C(N(C)[C@@](CC(C)C)C(N(C)[C@@](CC(C)C)C(N(C)[C@@](C(C)C)C(N(C)[C@@]([C@@]([C@](C)CC=CC=C)O)C(N[C@@](*)C(N(C)C1)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)N(C)C1=O Chemical compound CC(C)C[C@@](C(N[C@](C(C)C)C(N(C)[C@@](CC(C)C)C(N[C@@](C)C(N[C@](C)C(N(C)[C@@](CC(C)C)C(N(C)[C@@](CC(C)C)C(N(C)[C@@](C(C)C)C(N(C)[C@@]([C@@]([C@](C)CC=CC=C)O)C(N[C@@](*)C(N(C)C1)=O)=O)=O)=O)=O)=O)=O)=O)=O)=O)N(C)C1=O 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- 238000003109 Karl Fischer titration Methods 0.000 description 2
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 2
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 150000001242 acetic acid derivatives Chemical class 0.000 description 2
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 2
- 239000012346 acetyl chloride Substances 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000001336 alkenes Chemical class 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 230000002051 biphasic effect Effects 0.000 description 2
- 125000005620 boronic acid group Chemical class 0.000 description 2
- BRTALTYTFFNPAC-UHFFFAOYSA-N boroxin Chemical compound B1OBOBO1 BRTALTYTFFNPAC-UHFFFAOYSA-N 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 238000010668 complexation reaction Methods 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- UAIZDWNSWGTKFZ-UHFFFAOYSA-L ethylaluminum(2+);dichloride Chemical compound CC[Al](Cl)Cl UAIZDWNSWGTKFZ-UHFFFAOYSA-L 0.000 description 2
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- WQYVRQLZKVEZGA-UHFFFAOYSA-N hypochlorite Chemical compound Cl[O-] WQYVRQLZKVEZGA-UHFFFAOYSA-N 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- QSOMFNQEXNFPNU-UHFFFAOYSA-L magnesium;hydrogen sulfate;hydroxide;hydrate Chemical compound O.O.[Mg+2].[O-]S([O-])(=O)=O QSOMFNQEXNFPNU-UHFFFAOYSA-L 0.000 description 2
- 238000004452 microanalysis Methods 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 108010007425 oligomycin sensitivity conferring protein Proteins 0.000 description 2
- 125000002524 organometallic group Chemical group 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 150000002924 oxiranes Chemical class 0.000 description 2
- 238000005949 ozonolysis reaction Methods 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- QGLVEAGMVUQOJP-UHFFFAOYSA-N prop-2-enylboronic acid Chemical compound OB(O)CC=C QGLVEAGMVUQOJP-UHFFFAOYSA-N 0.000 description 2
- 239000003586 protic polar solvent Substances 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 230000000707 stereoselective effect Effects 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 150000003899 tartaric acid esters Chemical class 0.000 description 2
- NYBWUHOMYZZKOR-UHFFFAOYSA-N tes-adt Chemical class C1=C2C(C#C[Si](CC)(CC)CC)=C(C=C3C(SC=C3)=C3)C3=C(C#C[Si](CC)(CC)CC)C2=CC2=C1SC=C2 NYBWUHOMYZZKOR-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 238000001551 total correlation spectroscopy Methods 0.000 description 2
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 2
- UKRDPEFKFJNXQM-UHFFFAOYSA-N vinylsilane Chemical compound [SiH3]C=C UKRDPEFKFJNXQM-UHFFFAOYSA-N 0.000 description 2
- UQCJFKIZCSWJEH-UHFFFAOYSA-N (1-acetyloxy-2-oxopropyl) acetate Chemical compound CC(=O)OC(C(C)=O)OC(C)=O UQCJFKIZCSWJEH-UHFFFAOYSA-N 0.000 description 1
- OGNVQLDIPUXYDH-ZPKKHLQPSA-N (2R,3R,4S)-3-(2-methylpropanoylamino)-4-(4-phenyltriazol-1-yl)-2-[(1R,2R)-1,2,3-trihydroxypropyl]-3,4-dihydro-2H-pyran-6-carboxylic acid Chemical compound CC(C)C(=O)N[C@H]1[C@H]([C@H](O)[C@H](O)CO)OC(C(O)=O)=C[C@@H]1N1N=NC(C=2C=CC=CC=2)=C1 OGNVQLDIPUXYDH-ZPKKHLQPSA-N 0.000 description 1
- YFTHTJAPODJVSL-UHFFFAOYSA-N 2-(1-benzothiophen-5-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(SC=C2)C2=C1 YFTHTJAPODJVSL-UHFFFAOYSA-N 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- AMCCZIJPRTYFAD-PWSUYJOCSA-N CC(C)NC([C@H](C[C@H](C)CC=O)N(C)C(C)=O)=O Chemical compound CC(C)NC([C@H](C[C@H](C)CC=O)N(C)C(C)=O)=O AMCCZIJPRTYFAD-PWSUYJOCSA-N 0.000 description 1
- QFPPFTSMTRWDFJ-QHZWATSCSA-N CC[C@H](CCC/C=C/[Si](C)(C)C)C(C(C=N)(C(C)=O)N(C)C(C)=O)=C Chemical compound CC[C@H](CCC/C=C/[Si](C)(C)C)C(C(C=N)(C(C)=O)N(C)C(C)=O)=C QFPPFTSMTRWDFJ-QHZWATSCSA-N 0.000 description 1
- ZIDSZJSOTYLEFW-UHFFFAOYSA-N COC(=O)C1BOOC1C(=O)OC Chemical compound COC(=O)C1BOOC1C(=O)OC ZIDSZJSOTYLEFW-UHFFFAOYSA-N 0.000 description 1
- DMGGGIHRKRZJHE-IMJQCBOHSA-N C[C@H](C/C=C/C=C)C([C@@H](C(NC)=O)N(C)C([I](C)C)=O)[I]=O Chemical compound C[C@H](C/C=C/C=C)C([C@@H](C(NC)=O)N(C)C([I](C)C)=O)[I]=O DMGGGIHRKRZJHE-IMJQCBOHSA-N 0.000 description 1
- XUAYINXDDXLJAN-VQSFLMFPSA-N C[C@H](C/C=C/C=C)[C@H]([C@@H](C(NC)=O)N(C)C=O)O Chemical compound C[C@H](C/C=C/C=C)[C@H]([C@@H](C(NC)=O)N(C)C=O)O XUAYINXDDXLJAN-VQSFLMFPSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 241000122205 Chamaeleonidae Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- GSNUFIFRDBKVIE-UHFFFAOYSA-N DMF Natural products CC1=CC=C(C)O1 GSNUFIFRDBKVIE-UHFFFAOYSA-N 0.000 description 1
- 238000005698 Diels-Alder reaction Methods 0.000 description 1
- ZGTMUACCHSMWAC-UHFFFAOYSA-L EDTA disodium salt (anhydrous) Chemical compound [Na+].[Na+].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O ZGTMUACCHSMWAC-UHFFFAOYSA-L 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 238000012565 NMR experiment Methods 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- 238000003527 Peterson olefination reaction Methods 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- VKMHCIUSNZRODM-GQCTYLIASA-N [(e)-3-trimethylsilylprop-2-enyl]boronic acid Chemical compound C[Si](C)(C)\C=C\CB(O)O VKMHCIUSNZRODM-GQCTYLIASA-N 0.000 description 1
- 238000007171 acid catalysis Methods 0.000 description 1
- 239000012445 acidic reagent Substances 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 125000005234 alkyl aluminium group Chemical group 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 150000001722 carbon compounds Chemical class 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 230000000536 complexating effect Effects 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000005934 crotylation reaction Methods 0.000 description 1
- 150000003997 cyclic ketones Chemical class 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- XEBCWEDRGPSHQH-UHFFFAOYSA-N diisopropyl tartrate Chemical compound CC(C)OC(=O)C(O)C(O)C(=O)OC(C)C XEBCWEDRGPSHQH-UHFFFAOYSA-N 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000012039 electrophile Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- CCGKOQOJPYTBIH-UHFFFAOYSA-N ethenone Chemical compound C=C=O CCGKOQOJPYTBIH-UHFFFAOYSA-N 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000000457 gamma-lactone group Chemical group 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 229910001385 heavy metal Inorganic materials 0.000 description 1
- DKAGJZJALZXOOV-UHFFFAOYSA-N hydrate;hydrochloride Chemical compound O.Cl DKAGJZJALZXOOV-UHFFFAOYSA-N 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- OJURWUUOVGOHJZ-UHFFFAOYSA-N methyl 2-[(2-acetyloxyphenyl)methyl-[2-[(2-acetyloxyphenyl)methyl-(2-methoxy-2-oxoethyl)amino]ethyl]amino]acetate Chemical compound C=1C=CC=C(OC(C)=O)C=1CN(CC(=O)OC)CCN(CC(=O)OC)CC1=CC=CC=C1OC(C)=O OJURWUUOVGOHJZ-UHFFFAOYSA-N 0.000 description 1
- 230000037230 mobility Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 238000005935 nucleophilic addition reaction Methods 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 238000006053 organic reaction Methods 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- ZEJYUSNKPNYKPO-UHFFFAOYSA-N prop-1-ene Chemical compound CC=[CH-] ZEJYUSNKPNYKPO-UHFFFAOYSA-N 0.000 description 1
- ZTEKSGWPGRYKHH-UHFFFAOYSA-N propan-2-ylboron Chemical class [B]C(C)C ZTEKSGWPGRYKHH-UHFFFAOYSA-N 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000010503 protodeborylation reaction Methods 0.000 description 1
- 230000005588 protonation Effects 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 229910001927 ruthenium tetroxide Inorganic materials 0.000 description 1
- SCPYDCQAZCOKTP-UHFFFAOYSA-N silanol Chemical compound [SiH3]O SCPYDCQAZCOKTP-UHFFFAOYSA-N 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 230000000153 supplemental effect Effects 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- DHCDFWKWKRSZHF-UHFFFAOYSA-L thiosulfate(2-) Chemical compound [O-]S([S-])(=O)=O DHCDFWKWKRSZHF-UHFFFAOYSA-L 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 238000006478 transmetalation reaction Methods 0.000 description 1
- 238000004104 two-dimensional total correlation spectroscopy Methods 0.000 description 1
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/64—Cyclic peptides containing only normal peptide links
- C07K7/645—Cyclosporins; Related peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/12—Preparation of carboxylic acid amides by reactions not involving the formation of carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0803—Compounds with Si-C or Si-Si linkages
Definitions
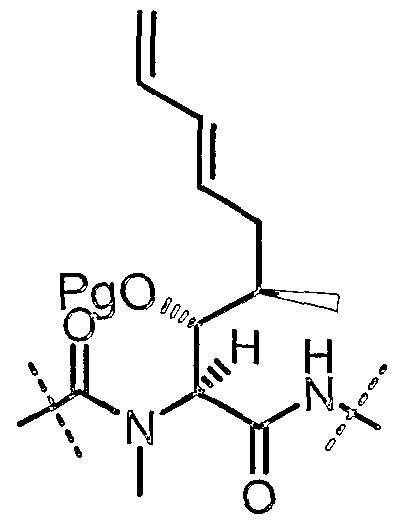
- This invention relates to a new process for the preparation of cyclosporin A analog of formula I:
- the cyclosporin A analog of formula I is structually identical to cyclosporin A except for modification at the 1 -amino acid residue.
- This analog is disclosed in WO 99/18120 and U.S. Provisional Patent Application No. 60/346,201. Hereinafter this analog is mentioned as (E)-ISA247.
- Tetrahedron Letters, Vol.22, No.29, p2751-2752, 1981 discloses one of the intermediates of the process of this invention, namely pinacol (E)-l-trimethylsilyl- l-propene-3-boronate, and the allylation process using it.
- Tetrahedron Letters, Vol.36, No.10, pl583, 1995 discloses allylation process using tartrate modified (E)- ⁇ -(trimethylsilyl)allylboronate.
- this invention provides a process for the preparation of a cyclosporin A analog of formula I
- R 1 is hydrogen, C ⁇ _ 8 alkyl or C 3 _ 8 cycloalkyl and/or, when R 1 is hydrogen, a trimer thereof;
- R 2 is C ⁇ _s alkyl or C 3 _ 8 cycloalkyl
- this invention provides intermediates for the process mentioned above.
- this invention provides processes for the preparation of these intermediates.
- step b) The process of (i), wherein step b) is conducted by
- step a-i), a-ii) or a-vi) is conducted in dichloromethane or toluene.
- step a-iii) is conducted in the presence of BF 3 .Et 2 O, formic acid, acetic acid or tartrate esters.
- step a-iii) is conducted in water/ dichloromethane or water/toluene.
- step a-iii) and b-i) are conducted in dichloromethane or tetrahydrofuran and in the presence of BF 3 .Et 2 0.
- step a-iii) is conducted in acetic acid and/or formic acid; or in a mixture of acetic acid and /or formic acid and one or two cosolvents selected from a group consisting of dichloromethane and tetrahydrofuran.
- step a-iii) is conducted in acetic acid and step b-i) is conducted by addition of formic acid to the reaction mixture.
- step a-iii) and b-i) are conducted in formic acid or acetic acid/ formic acid.
- step a-iv The process of (i) or (ii), wherein step a-iv) is conducted in water/dichloromethane or water/toluene.
- step a-v The process of (iii), wherein step a-v) is conducted in the presence of formic acid or acetic acid.
- step a-v) and b-i) are conducted in formic acid or acetic acid/formic acid.
- step a-v) and b-i) are conducted in a mixture of acetic acid/formic acid and co-solvent selected from dichloromethane, toluene, ethyl acetate and isopropyl acetate.
- step a-v) is conducted in acetic acid and step b-i) is conducted by addition of formic acid to the reaction mixture.
- step a-vii The process of (iii), wherein step a-vii) is conducted by allylating the compound of formula II with a reaction mixture prepared by reaction of the trimethylsilylallyllithium with diethylaluminum chloride.
- step a-viii The process of (iii), wherein step a-viii) is conducted by allylating the compound of formula II with a reaction mixture prepared by reaction of trimethylsilylallyllithium with titanium tetraisopropoxide or titanium chlorotriisopropoxide.
- C a _b alkyl denotes straight chain or branched alkyl residues containing a to b carbon atoms. Therefore, for example, " -s alkyl” means straight chain or branched alkyl residues containing 1 to 8 carbon atoms, such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl or tert-butyl.
- C3. 8 cycloalkyl refers to a saturated monovalent cyclic hydrocarbon radical of three to eight ring carbons e.g., cyclopropyl, cyclobutyl, cyclohexyl.
- Protecting group refers to a grouping of atoms that when attached to a reactive group in a molecule masks, reduces or prevents that reactivity. Examples of protecting groups can be found in T.W. Green and P.G. Futs, Protective Groups in Organic Chemistry, (Wiley, 2 nd ed. 1991) and Harrison and Harrison et al., Compendium of Synthetic Organic Methods, Vols. 1-8 (John Wiley and Sons,
- the starting materials and reagents used in the process of the present invention are either available from commercial suppliers such as Aldrich Chemical
- the starting materials and the intermediates of the reaction maybe isolated and purified if desired using conventional techniques, including but not limited to filtration, distillation, crystallization, chromatography. Such materials may be characterized using conventional means, including physical constants and spectral data.
- the all lmetal reagent also referred herein as allylating reagent is to be taken in a general sense and may comprise reagents where the metal part is based on boron although it is not per se a metal.
- step a) protected cyclosporin A aldehyde of formula II is allylated by ⁇ - silylated allylmetal reagent of formula III, IV, V, VI, VTI, VIII etc. to form a mixture of ⁇ -silylhomoallylic alcohol diastereomers of formula XI (For a general discussion about allylmetals and allylation of aldehydes see : W. R. Roush in "Allyl
- the aldehyde side chain preferably adopts a pseudo equatorial position in order to minimize 1,3-diaxial steric interactions.
- the relative configuration of the ⁇ -silylalcohol fragment will therefore be determined by the configuration of C-C double bond of the allylmetal reagent.
- trans- or cis- ⁇ -silylated allylmetals reagents should lead predominantly to the anti- or syn- ⁇ -silylalcohol isomer respectively. This holds in general, for example, for the allyl-boron, -titanium and -aluminum reagents.
- step b) the ⁇ -silylalcohol of formula XI is converted to (E)-ISA247 of formula I.
- Step b) can be carried out as illustrated in Scheme C.
- step b-i the ⁇ -silylalcohol of formula XI undergoes a Peterson elimination (For a general discussion about Peterson eliminations, see : D. J. Ager in "The Peterson Reaction", Synthesis 1984, p384-397 as well as references cited therein.) and the internal double bond is generated, i.e. the elimination of silanol from the ⁇ - silylalcohol moiety occurs.
- the success of the allylation-Peterson elimination sequence relies on the selective introduction of a relative anti or syn configuration of the ⁇ -silylalcohol moiety.
- Anti isomers should give the trans double bond under acidic Peterson elimination conditions whereas syn isomers would provide the cis double bond.
- the reaction proceeds via a mechanism where the hydroxyl and the silyl groups are in an anti conformation prior to elimination.
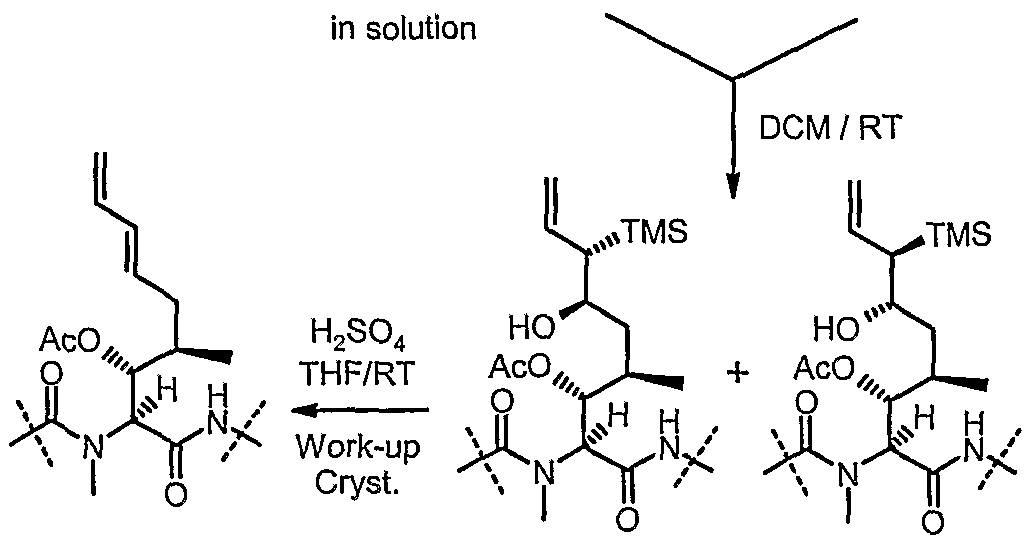
- trans- ⁇ -silylated allylmetals reagents are used for allylation of protected cyclosporin A aldehyde of formula II to form a mixture of anti- ⁇ -silylalcohol diastereomers of formula XL Therefore, a Peterson elimination is performed under acidic condition to form a trans double bond.
- Typical acids for the acid-promoted reaction may include sulfuric acid, formic acid, chlorhydric acid, methanesulfonic acid tetrafluoroboric acid, perchloric acid, trifluoroacetic acid and various Lewis acids.
- Preferred acids are sulfuric acid, formic acid, methanesulfonic acid and BF 3 .Et 2 O, especially sulfuric acid, formic acid and BF 3 .Et 2 0.
- This step can be conducted at a reaction temperature from -70 °C to 50 °C.
- Preferred temperature range is 0 ° C to 50 °C, more preferably 20 °C to 40 °C for formic acid.
- Preferred temperature range is 0 °C to 40 °C, more preferably 20 °C to 30 °C for sulfuric and methanesulfonic acid.
- Preferred temperature range is -80 °C to 50 °C, preferably -80 °C to 25 °C, especially -80 °C to 0 °C for BF 3 .Et 2 O.
- E-acetyl-ISA247 can be purified by crystallization in MTBE (for example via solvent exchange from dichloromethane to MTBE) or in MeOH/water mixtures.
- step b-ii) the protecting group is removed, returning the functional group on that carbon to an alcohol.
- the conditions and reagents to be employed depend on the protecting group used, which are known to those skilled in the art.
- Acyl group (R'C(O)-; wherein R' is a linear saturated monovalent hydrocarbon radical of one to six carbon atoms or a branched saturated monovalent hydrocarbon radical of three to six carbon atoms) such as acetyl, propionyl, butyryl, isobutyryl, valeryl can preferably be used as a protecting group.
- the protecting group is an acetyl group, it can be removed, for example, by the treatment with K 2 C0 3 in methanol and water.
- Bases other than potassium carbonate that may be used to remove the protecting group include sodium hydroxide, sodium carbonate, sodium alkoxide and potassium alkoxide.
- the reagent of formula III or IV is needed to complete the allylation of acetyl-cyclosporin A aldehyde (II') within an acceptable timeframe.
- an activating agent such as a tartrate ester and/or dichloromethane as (co)-solvent.
- the reagent of formula Ilia can potentially exist in the form of cyclic trimer (boroxine) or oligomers (For an example of such behavior of a boronic acid, see: K. Ishihara, H. Kurihara, M. Matsumoto and H.
- a solution of reagent of formula Ilia can be generated by hydrolysis of complex of formula V in organic solvent/water mixture such as a dichloromethane/water, toluene/water, ethyl acetate/water, THF/water, chloroform/water mixture, preferably a dichloromethane/water mixture, preferably in the presence of an acid such as sulfuric acid, chlorhydric acid, acetic acid, preferably acetic acid. Allylation of acetyl-cyclosporin A aldehyde with a dichloromethane solution of reagent of formula Ilia prepared as just described can reach high conversions using as low as 2 equivalents of the reagent. In this case, isopropyl derivatives are of course absent.
- organic solvent/water mixture such as a dichloromethane/water, toluene/water, ethyl acetate/water, THF/water, chloroform/water mixture, preferably a dichloromethane/water mixture
- Toluene can be used as solvent for these reactions, however, marked solvent effects have been observed in these reactions.
- the allylation is best performed in polar non-coordinating solvents, preferably dichloromethane.
- allylation is preferably performed in dichloromethane using a concentrated solution of the crude boronic acid(>10%, preferably ca 50% concentration).
- Preferred reagent of formula III wherein R is hydrogen, C ⁇ _ 8 alkyl or C 3 . 8 cycloalkyl and/ or, when R 1 is hydrogen, a trimer thereof are those wherein R 1 is hydrogen, methyl, ethyl, propyl, isopropyl, butyl or benzyl, more preferably R 1 is hydrogen, methyl, ethyl, propyl, isopropyl or butyl, further preferably hydrogen, methyl, ethyl, propyl or butyl, especially preferably hydrogen.
- Allylation is performed in organic solvent such as ethyl acetate, THF, toluene, chloroform or dichloromethane, preferably in ethyl acetate, toluene or dichloromethane, more preferably in toluene or dichloromethane, especially in dichloromethane.
- organic solvent such as ethyl acetate, THF, toluene, chloroform or dichloromethane, preferably in ethyl acetate, toluene or dichloromethane, more preferably in toluene or dichloromethane, especially in dichloromethane.
- the Peterson elimination can take place directly on the allylation reaction mixture by addition of THF and sulfuric acid. Aqueous work-up and crystallization yields the (E)-acetyI-ISA247 (XIT).
- a tartrate ester such as, for example, L-(+)-dimethyltartrate
- a drying agent activates the boronic acid of formula Ilia by generating in-situ the corresponding boronate ester, a reagent class known in the literature to exhibit very high allylation reactivity.
- the reaction then proceeds partially or mainly through the generated boronate ester increasing the rate of the allylation.
- R is - 8 alkyl, preferably C ⁇ . 6 alkyl, more preferably methyl, ethyl or isopropyl, especially methyl.
- Allylation with reagent of formula IV is performed in organic solvent such as ethyl acetate, THF, toluene, chloroform or dichloromethane, preferably in ethyl acetate, toluene or dichloromethane, more preferably in toluene or dichloromethane, especially in dichloromethane.
- organic solvent such as ethyl acetate, THF, toluene, chloroform or dichloromethane, preferably in ethyl acetate, toluene or dichloromethane, more preferably in toluene or dichloromethane, especially in dichloromethane.
- reaction involving the use of crude boronic acid solution with or without tartrate activation should be performed at neutral or acidic pH (between 3 and 7, preferably between 5 and 6). Indeed when the pH is over 7, substantial amount of a side-product identified as the vinylsilane of formula XV are formed.
- a test reaction (performed without tartrate activation) where Et 3 N amine was added to reach a pH of 9-10 led to the almost exclusive formation of the vinylsilane product XV (as evidenced by MS, 1H NMR, COSY, TOCSY and HSQC NMR experiments). Such an effect was totally unexpected.
- the diethanolamine complex of formula V does not react at RT with acetyl-cyclosporin A aldehyde in non protic solvents like dichloromethane or THF.
- the complex of formula V represent a stable source of the corresponding boronic acid.
- a water/ organic solvent such as ethyl acetate, THF, dichloromethane or toluene, preferably ethyl acetate, dichloromethane or toluene, more preferably dichloromethane
- the diethanolamine complex V is hydrolyzed and liberates the reactive boronic acid as shown, for example, in Scheme H, which can then reacts with the acetyl- cyclosporin A aldehyde (II'), preferably at RT.
- Allylmetalation of acetyl-cyclosporin A aldehyde (IF) can also take place under non-aqueous conditions directly with complex of formula V. Indeed, protic solvents such as carboxylic acids are particularly effective. Solvent mixture could be acetic acid and/ or formic acid or a combination of acetic acid and/or formic acid and a co-solvent such as dichloromethane and THF. The allylation is best performed in acetic acid between RT and 35 °C.
- Another alternative consists in performing the addition of complex of formula V to acetyl-cyclosporin A aldehyde (IF) in the presence of a Lewis acid such as BF 3 .Et 2 0.
- a Lewis acid such as BF 3 .Et 2 0.
- the reaction with BF 3 .Et 2 O can be performed in a solvent such as dichloromethane or THF at a temperature ranging from -40 °C to RT. Under these conditions, the allylation can directly be followed by the Peterson elimination, yielding the expected (E)-acetyl-ISA247 (XIF).
- Reacting the allyltrifluoroborate VI and acetyl-cyclosporin A aldehyde (IF) in a biphasic water/ organic solvent, preferably water/dichloromethane mixture or water/toluene mixture, more preferably water/dichloromethane mixture at RT provides a mixture of anti ⁇ -trimethylsilylalcohol diastereomers (XF).
- a biphasic water/ organic solvent preferably water/dichloromethane mixture or water/toluene mixture, more preferably water/dichloromethane mixture at RT provides a mixture of anti ⁇ -trimethylsilylalcohol diastereomers (XF).
- the Peterson elimination is performed by addition of THF and sulfuric acid at a temperature of 0 °C to RT providing (E)-acetyl-ISA247 (XIF).
- Peterson elimination can also be performed under standard conditions (sulfuric acid in THF) after isolation of the anti ⁇ -trimethylsilylalcohol diastereomers to give E-acetyl-ISA247.
- the allylation can also be promoted by a Lewis acid.
- the allylation and the Peterson elimination can take place in-situ.
- addition of excess BF 3 .Et 2 0 to a suspension of allyltrifluoroborate VI (2 equiv.) in a solution of acetyl-cyclosporin A aldehyde (XIF) in dichloromethane at -70 °C provides after 60 min. reaction and aqueous work-up, (E)-acetyl-ISA247 (I).
- Solvents for the reaction are organic solvent such as dichloromethane, THF or toluene, preferably dichloromethane.
- Carboxylic acids such as formic acid or acetic acid were found to dramatically enhance the rate of allylation.
- allylation of acetyl cyclosporin A aldehyde (IF) can reach conversion of over 95% within 5 hours at RT with 2 equivalents of reagent of formula VI, providing the ⁇ - silylalcohols (XI').
- formic acid promotes the Peterson elimination.
- (E)-acetyl-ISA247 (XIF) is obtained after extractive work-up ascertaining the relative anti stereochemistry of the intermediate ⁇ -silylalcohols.
- Peterson elimination can also be performed under standard conditions (sulfuric acid in THF) after isolation of the anti ⁇ -trimethylsilylalcohol diastereomers to give (E)-acetyl-ISA247.
- the allylmetalation and the following Peterson elimination can take place in one-pot.
- a combination of acetic acid and formic acid ca 1:1 v/v
- the allylation of acetyl-cyclosporin A aldehyde and the following Peterson elimination reach over 90% conversion within 60 min. at RT with 1.5 equivalent of reagent.
- Aqueous extractive work-up and crystallization furnishes (E)-acetyl-ISA247 (XIF).
- acetic acid formic acid and a suitable co-solvent
- Dichloromethane, toluene, ethyl acetate or isopropyl acetate, preferably isopropyl acetate could be used as co-solvent. Decrease in reactivity can be observed when using a co-solvent but this could be compensated by increasing the reaction temperature.
- reagent of formula VII to acetyl cyclosporin A aldehyde (IF) can be promoted by a Lewis acid such as BF 3 .Et 2 O at a temperature of -70 °C to
- the ⁇ -silylated allylmetal reagents required for the allylmetalation step are best generated from the corresponding allylsilanes via deprotonation, trapping with an adequate metal reagent and optionally by further complexation of the metal rest by a suitable ligand.
- the resulting reagents can, depending on their stability and the process, be used in situ or be isolated and stored.
- the allyltrimethylsilane is deprotonated by n- butyllithium in THF at a temperature ranging from 0 °C to 35 °C, preferably bewteen 0 °C and 25 °C for 30 min. up to 3 hours.
- This generates a trimethylsilylallyllithium intermediate.
- This intermediate most probably exists in solution as a ⁇ -allyl complex of lithium in a trans configuration (T. H. Chan in "Silylallyl Anions in Organic Synthesis: A Study in Regio- and Stereoselectivity",
- M and M' are a metallic fragment comprising the metal and its ligands.
- a solution of crude boronic acid of formula Ilia is obtained after deprotonation of allyltrimethylsilane, trapping with an electrophilic boron reagent and aqueous work- up.
- the deprotonation of allyltrimethylsilane is performed in THF with butyllithium, between 0 °C and 35 °C, preferably between 0 °C and 25 °C for 30min. to 3 hours.
- the electrophilic boron reagent is a trialkylborate such as triisopropyl borate or trimethyl borate, preferably triisopropyl borate.
- the trapping of the trimethylsilylallyllithium intermediate with triispropyl borate is performed between -80 °C and -20 °C, preferably below -60 °C for 30 min. to 2 hours.
- the trapping of the trimethylsilylallyllithium intermediate with trimethyl borate is performed between -80 °C and -60 °C for 30 min. to 2 hours.
- Crotylboronates Preparation of the Chiral Crotylboronates and Reactions with Achiral Aldehydes", Journal of the American Chemical Society 1990, 112, 6339 (see reference 17)).
- Concentrated solutions (>10% concentration, for example ca 50% concentration) are not stable at RT and decompose. They should be rapidly used. If needed they should be stored at 5 °C maximum.
- the boronic acid of formula Ilia might in principle also exists in the form of a cyclic trimer (also called a boroxine) or oligomers.
- a solution of reagent of formula Ilia can be prepared by hydrolysis of complex of formula V in an organic solvent/water mixture such as a dichloromethane/water, toluene/water, ethyl acetate/water, THF/water, chloroform/water mixture, preferably a dichloromethane/water mixture preferably in the presence of an acid such as sulfuric acid, chlorhydric acid, acetic acid, preferably acetic acid.
- an organic solvent/water mixture such as a dichloromethane/water, toluene/water, ethyl acetate/water, THF/water, chloroform/water mixture, preferably a dichloromethane/water mixture preferably in the presence of an acid such as sulfuric acid, chlorhydric acid, acetic acid, preferably acetic acid.
- Boronate reagent of formula IV where R is .s alkyl, preferably C ⁇ - 6 alkyl, more preferably methyl, ethyl or isopropyl, especially methyl is prepared by treating boron reagent of formula Ilia with the required tartrate ester in the presence of a drying agent such molecular sieves or magnesium sulfate, preferably magnesium sulfate.
- Reagent of formula IVa' is prepared by mixing a solution of boronic acid of formula Ilia with L-(+)-dimethyltartrate, in the presence of a drying agent such molecular sieves or magnesium sulfate, preferably magnesium sulfate , as evidenced by n B and 1H NMR analyses.
- ⁇ -silylated allylboron reagent of formula III This reagent can be prepared by treating boron reagent of formula Ilia with the required alcohol in the presence of a drying agent such molecular sieves or magnesium sulfate, preferably magnesium sulfate.
- a drying agent such molecular sieves or magnesium sulfate, preferably magnesium sulfate.
- R 8 cycloalkyl are those wherein R 1 is methyl, ethyl, propyl, isopropyl, butyl or benzyl, more preferably R 1 is methyl, ethyl, propyl, isopropyl or butyl, further preferably R 1 is methyl, ethyl, propyl or butyl, especially preferably R 1 is methyl.
- Ilia VI as a methanol solution
- allyltrifluoroborate potassium salts are prepared by treating the corresponding boronic acid with 3 equivalents of KHF 2 in a water/methanol solvent mixture (see for example : R. A. Batey in "Diastereoselective Allylation and Crotylation Reactions of Aldehydes with Potassium Allyl- and CiOtyltrifluoroborates under Lewis acid Catalysis", Synthesis 2000, pp 990-998).
- the required boronic acid solution is prepared by hydrolyzing the diethanolamine complex of formula V in a water/dichloromethane mixture in the presence of an acid such as acetic acid.
- the aqueous phase is discarded and the solvent is exchanged from dichloromethane to methanol.
- the diethanolamine complex V can be used directly as starting material.
- the trifluoroborate salts VI can be prepared, however, crystallization does not occur.
- pinacol complex VII can then be distilled under low pressure or used directly in the allylmetalation step.
- the preparation of the pinacol complex of formula VII is, for example, performed as illustrated in Scheme O.
- the trapping of the 1-trimethylsilylallyl lithium can be performed with 2-isopropoxy-4,4,5,5-tetramethyl-l,3,2-dioxaborolane, leading directly after aqueous work-up to the pinacol boronate of formula VII.
- the reagent can be prepared by deprotonation of allytrimethylsilane at room temperature with butyllithium, quench of the allylithium intermediate with tri-ispropylborate (between -80 °C to -20 °C, preferably between -80 °C and -30 °C), addition of pinacol and then aqueous work- up.
- the (trimethylsilyl) allyltitanium reagents are prepared in-situ via deprotonation of allyltrimethylsilane to form trimethylsilylallylithium, as described above, and reaction of this intermediate with titanium dichlorodiisopropoxide, titanium tetraisopropoxide or titanium chlorotriisopropoxide, preferably titanium tetraisopropoxide or titanium chlorotriisopropoxide at a temperature of-80 °C to 0 °C, preferably -80 °C to -30 °C, more preferably -80 °C to -50 °C, especially - 80 °C to -60 °C.
- the resulting titanium reagents are used in situ for the allylation of protected cyclosporin A aldehydes. Putative structures for theses reagents are presented below :
- the (trimethylsilyl) allylaluminum reagents are prepared in-situ via deprotonation of allyltrimethylsilane to form trimethylsilylallylithium, as described above, and reaction of this intermediate with a dialkylaluminum chloride such as diethylaluminum chloride or with an alkylaluminum dichloride such as ethylaluminum dichloride, preferably with diethylaluminum chloride, at a temperature of -80 °C to 0 °C, preferably -80 °C to -30 °C, more preferably -80 °C to -50 °C, especially -80 °C to -60 °C.
- the resulting aluminum reagents are used in situ for the allylation of protected cyclosporin A aldehydes.
- a protecting group is introduced in cyclosporin A of formula XIII, to protect hydroxyl group at the ⁇ -position of the side chain of the 1 -amino acid residue.
- Protecting groups are well known in organic synthesis, and have been discussed by J. R. Hanson in Chapter 2,"The Protection of Alcohols,” of the publication Protecting Groups in Organic Synthesis (Sheffield Academic Press, Sheffield, England, 1999), pp. 24-25. Hanson teaches how to protect hydroxyl groups by converting them to either esters or ethers. Acetate esters are perhaps the most frequently used type of chemistry for protecting hydroxyl groups. There are a wide range of conditions that maybe used to introduce the acetate group.
- reagents and solvents include acetic anhydride and pyridine; acetic anhydride, pyridine and dimethylaminopyridine (DMAP); acetic anhydride and sodium acetate; acetic anhydride and toluene-p-sulphonic acid, acetyl chloride, pyridine and DMAP; and ketene.
- DMAP is a useful acylation catalyst because of the formation of a highly reactive N-acylpyridium salt from the anhydride.
- the ⁇ -alcohol of cyclosporin A is protected as an acetate by reacting cyclosporin A (XIII) with acetyl chloride, ethyl acetate, or combinations thereof, forming the compound, acetyl cyclosporin A.
- the ⁇ - alcohol undergoes a nucleophilic addition to acetic anhydride, forming acetyl cyclosporin A and acetic acid.
- DMAP dimethylaminopyridine
- protecting groups other than acetate esters may be used to protect the ⁇ -alcohol of the 1 -amino acid residue of cyclosporin A.
- These protecting groups may include benzoate esters, substituted benzoate esters, ethers, and silyl ethers. Under certain reaction conditions, the acetate protecting group is prone to undesirable side reactions such as elimination and hydrolysis. Since benzoate esters, ethers and silyl ethers are often more resistant to such side reactions under those same reaction conditions, it is often advantageous to employ such protecting groups in place of acetate.
- step c-ii) the protected cyclosporin A of formula XIV is converted to a protected cyclosporin A aldehyde of formula II.
- This step can be carried out, for example, by using ozone as an oxidizing agent followed by work-up with a reducing agent to form a protected cyclosporin A aldehyde (II).
- Ozonolysis step is conducted at a temperature range from about -80 °C to 0 °C.
- the solvent used during the ozonolysis maybe a lower alcohol such as methanol.
- the reducing agent may be a trialkylphosphine such as tributylphosphine, a triarylphosphine, a trialykylamine such as triethylamine, an alkylaminosulfide, a thiosulfate or a dialkylsulfide such as dimethylsulfide.
- a protected cyclosporin A aldehyde (II) can be prepared by converting the protected cyclosporinA XIV, such as acetyl cyclosporin A, to the protected cyclosporin A epoxide with a monopersulfate, preferably oxone, in the presence of a ketone, such as acetoxyacetone or diacetoxyacetone. This step is performed in an organic solvent which is inert under these reaction conditions such as acetonitrile and water. Ethylenediamintetra-acetic acid disodium salt is added to capture any heavy metal ions which might be present. The epoxidation reaction is carried out preferably at a pH over 7.
- This epoxidation reaction is followed by oxidative cleavage of the epoxide with periodic acid or periodate salt under acidic conditions.
- the oxidation and the oxidative cleavage can be combined in a work-up procedure.
- Carlsen et al. in "A Greatly Improved Procedure for Ruthenium Tetroxide Catalyzed Oxidations of Organic Compounds," J. Org. Chem., Vol. 46, No. 19, pp 3736-3738 (1981). Carlsen et al. teach that, historically, the expense of ruthenium metal provided an incentive for the development of catalytic procedures, the most popular of which used periodate or hypochlorite as stoichiometric oxidants. These investigators found a loss of catalytic activity during the course of the reaction with the conventional use of ruthenium which they postulated to be due to the presence of carboxylic acids.
- nitriles to the reaction mixture, especially acetonitrile, was found to significantly enhance the rate and extent of the oxidative cleavage of alkenes in a CCl 4 /H 2 0/I ⁇ 4 _ system.
- protected cyclosporin A aldehyde (II) can be produced from protected cyclosporin A (XIV), such as acetyl cyclosporin A, by dissolving it in a mixture of acetonitrile and water, and then adding first sodium periodate and then ruthenium ' chloride hydrate. The aldehyde (II) maybe extracted with ethyl acetate.
- the organic phases were washed sequentially with 150 ml of a saturated aqueous NaCl solution, combined, dried over Na 2 SO 4 , filtered and concentrated under reduced pressure to ca 40 ml.
- the weight of the solution was adjusted to 53.6 g by addition of dichloromethane in order to obtain a ca 50% solution of boronic acid (based on the starting allyltrimethylsilane).
- (E)-acetyl-ISA247 can be recrystallized by dissolving the solid in dichloromethane at room temperature and exchanging the solvent to MTBE (by adding MTBE, concentrating the solution to half its volume under reduced pressure at 40°C and repeating these operation 2 to three times). The solution is cooled to room temperature and the crystallization then starts within a few minutes. The suspension is stirred at room temperature for 2h and 30min at 0°C. The crystals of (E)-acetyl-ISA247 are isolated after filtration, washing with MTBE and drying under reduced pressure at 40°C.
- the crude ⁇ -trimethylsilyalcohol diastereomers mixture (11 g, maximum 4.056 mmol) was dissolved in 25 ml THF. 0.679 ml (12.16 mmol, 3 equiv.) concentrated sulfuric were added dropwise maintaining the temperature between 20 °C and 25 °C. After 2 hours at RT, 50 ml half saturated aqueous NaCl solution were added. The resulting mixture was extracted twice with 50 ml MTBE. The organic phases were washed with 50ml of a half saturated aqueous NaCl solution, combined, dried over Na 2 SO and concentrated under reduce pressure at 40°C.
- the aqueous layer was separated and discarded.
- the organic phase was washed with 40 ml NaHC0 3 aq , 20 ml saturated NaCl aq , 40 ml saturated NaCl aq , dried over Na 2 SO 4 , filtered and concentrated at 40 °C under reduced pressure.
- the crude E-acetyl-ISA247 was re-dissolved in 200 ml MTBE and crystallization started within a few minutes. After 15 min.
- the organic phase was washed with 120 ml water, 204 ml 2M aqueous NaOH solution and 60 ml water. The organic phase was concentrated at 30 °C until the crystallization started. 200 ml MTBE were added and the suspension was concentrated to ca 220 ml. After stirring at RT for 2 hours and for 1 hour at 0-2 °C, the suspension was filtered. The solid was washed with 30 ml MTBE and dried at 50 °C under reduced pressure to provide 18 g of (E)-acetyl- ISA247 as a white powder in >98% double bond isomeric purity (by NMR).
- the filtrate was concentrated under reduced pressure at 40 °C to ca 25 ml.
- the solution was cooled to 0-2 °C and the a white suspension was obtained. After 30 min. at 0-2 °C, the suspension was filtered and the solid was washed with cold methanol (-20 °C) and dried under reduced pressure at 40 °C to give 3.4 g of a white powder.
- Ci 5 H ⁇ 3 F 3 BKSi (contains 1.08 equiv. H 2 O by Karl-Fischer titration and 0.5 equiv. KF) :
- reaction mixture was stirred for 2 hours at -76 °C. The temperature was slowly raised to -40 °C and the stirring was continued for a further 2 hours at -40 °C. The reaction mixture was poured onto a mixture consisting of 32.5 ml of a 1M aqueous HC1 solution and 20 ml MTBE. 16.2 ml of a
- the crude product was dissolved in 11.15 ml THF and 268 ⁇ l concentrated sulfuric acid were added. The reaction mixture was heated at 33 °C for 1.5 hour and then cooled to RT. 22 ml water were added and the reaction mixture was extracted with 22 ml MTBE. The aqueous phase was re-extracted with 11 ml MTBE. The organic layer were washed with 11 ml water, combined, dried over Na 2 S0 4 , filtered and concentrated at 40 °C under reduced pressure to give 1.89 g of crude (E)-acetyl-ISA247 as a beige powder. The crude product was re-dissolved in
- the crude product was dissolved in 8 ml THF at RT. The solution was cooled to 0-5 °C and 200 ⁇ l of concentrated sulfuric acid were added dropwise. The temperature was raised to RT and the reaction mixture was stirred 10 hours. 40 ml MTBE and 15 ml of water were added. The water phase was separated and discarded. The organic phase was washed 15 ml of a 5% aqueous NaHC0 3 solution, 15 ml of a half saturated aqueous NaCl solution, dried over Na 2 S0 4 , filtered and concentrated under reduced pressure to give 1.8 g of crude E-acetyl- ISA247. The crude diene was dissolved in 20 ml dichloromethane.
- the organic phase was washed with 35 ml of 1M aqueous HCl solution, 25 ml of water, 25 ml of a saturated aqueous NaCl solution, dried over Na 2 S0 4 , filtered and concentrated under reduced pressure to provide 2 g of the crude mixture of anti ⁇ -trimethylsilylalcohol diastereomers as a solidifying oil.
- the crude product was dissolved in 10 ml THF at RT. The solution was cooled to 0-5 °C and 200 ⁇ l of concentrated sulfuric acid were added dropwise. The temperature was raised to RT and the reaction mixture was stirred overnight. 40 ml MTBE and 15 ml of water were added. The water phase was separated and discarded. The organic phase was washed with 15 ml water, 15 ml of a 5% aqueous NaHC0 3 solution, 15 ml of a half saturated aqueous NaCl solution, filtered and concentrated under reduced pressure to give 1.8 g of crude E-acetyl-ISA247. The crude diene was redissolved in 35 ml of MTBE.
- the organic phase was concentrated to ca 280 ml. 130 ml water were added over ca 60 min. at 20-25 °C.
- the resulting white suspension was stirred 60 min. at room temperature.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Immunology (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Pharmacology & Pharmacy (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Transplantation (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description































Claims









Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002521116A CA2521116A1 (en) | 2003-04-08 | 2004-04-02 | Process for preparation of cyclosporin "a" analogues having a terminal diene group |
| MXPA05010794A MXPA05010794A (en) | 2003-04-08 | 2004-04-02 | PROCESS FOR PREPARATION OF CYCLOSPORIN aCúAaC° ANALOGUES HAVING A TERMINAL DIENE GROUP. |
| BRPI0409734-3A BRPI0409734A (en) | 2003-04-08 | 2004-04-02 | process for the preparation of closporin analog to |
| JP2006504961A JP2006525961A (en) | 2003-04-08 | 2004-04-02 | Process for the preparation of cyclosporine "A" analogs with terminal diene groups |
| EP04725333A EP1613646A2 (en) | 2003-04-08 | 2004-04-02 | Process for preparation of cyclosporin "a" analogues having a terminal diene group |
| AU2004228159A AU2004228159A1 (en) | 2003-04-08 | 2004-04-02 | Process for preparation of cyclosporin "A" analogues having a terminal diene group |
| NO20054478A NO20054478L (en) | 2003-04-08 | 2005-09-28 | Procedure for Preparation of Cyclosporin A Analog |
| TNP2005000255A TNSN05255A1 (en) | 2003-04-08 | 2005-10-07 | Process for preparation of cyclosporin "a" analogues having a terminal diene group. |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03007921 | 2003-04-08 | ||
| EP03007921.4 | 2003-04-08 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004089960A2 true WO2004089960A2 (en) | 2004-10-21 |
| WO2004089960A3 WO2004089960A3 (en) | 2005-01-06 |
Family
ID=33155122
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2004/003504 WO2004089960A2 (en) | 2003-04-08 | 2004-04-02 | Process for preparation of cyclosporin “a” analogues having a terminal diene group |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US20040220091A1 (en) |
| EP (1) | EP1613646A2 (en) |
| JP (1) | JP2006525961A (en) |
| KR (1) | KR20050119200A (en) |
| CN (1) | CN1798759A (en) |
| AU (1) | AU2004228159A1 (en) |
| BR (1) | BRPI0409734A (en) |
| CA (1) | CA2521116A1 (en) |
| CO (1) | CO5680450A2 (en) |
| EC (1) | ECSP056091A (en) |
| MA (1) | MA27831A1 (en) |
| MX (1) | MXPA05010794A (en) |
| NO (1) | NO20054478L (en) |
| RU (1) | RU2005130508A (en) |
| TN (1) | TNSN05255A1 (en) |
| TW (1) | TW200505946A (en) |
| WO (1) | WO2004089960A2 (en) |
| ZA (1) | ZA200507980B (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7449069B2 (en) | 2004-12-28 | 2008-11-11 | Lg Display Co., Ltd. | Slit coater having apparatus for supplying a coating solution |
| WO2010018211A1 (en) * | 2008-08-14 | 2010-02-18 | Minakem | Cyclopropyl- and cyclobutyl-dioxazaborocane or dioxazaborecane derivatives |
| US7696165B2 (en) | 2006-03-28 | 2010-04-13 | Albany Molecular Research, Inc. | Use of cyclosporin alkyne analogues for preventing or treating viral-induced disorders |
| US7696166B2 (en) | 2006-03-28 | 2010-04-13 | Albany Molecular Research, Inc. | Use of cyclosporin alkyne/alkene analogues for preventing or treating viral-induced disorders |
| US10265375B2 (en) | 2007-10-08 | 2019-04-23 | Aurinia Pharmaceuticals Inc. | Ophthalmic compositions |
| US11622991B2 (en) | 2017-05-12 | 2023-04-11 | Aurinia Pharmaceuticals Inc. | Protocol for treatment of lupus nephritis |
| EP4201952A1 (en) | 2021-12-21 | 2023-06-28 | Curia Spain, S.A.U. | Process for the controlled synthesis of voclosporin |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6859799B1 (en) | 1998-11-30 | 2005-02-22 | Gemstar Development Corporation | Search engine for video and graphics |
| US7103906B1 (en) | 2000-09-29 | 2006-09-05 | International Business Machines Corporation | User controlled multi-device media-on-demand system |
| CN101715109A (en) | 2000-10-11 | 2010-05-26 | 联合视频制品公司 | Systems and methods for providing storage of data on servers in an on-demand media delivery system |
| US7493646B2 (en) | 2003-01-30 | 2009-02-17 | United Video Properties, Inc. | Interactive television systems with digital video recording and adjustable reminders |
| WO2004082629A2 (en) * | 2003-03-17 | 2004-09-30 | Albany Molecular Research, Inc. | Novel cyclosporins |
| CA2575280C (en) * | 2004-07-29 | 2012-08-14 | Amr Technology, Inc. | Novel processes for stereoselective synthesis of trans isatx 247 |
| US8086575B2 (en) | 2004-09-23 | 2011-12-27 | Rovi Solutions Corporation | Methods and apparatus for integrating disparate media formats in a networked media system |
| US7378391B2 (en) * | 2004-09-29 | 2008-05-27 | Amr Technology, Inc. | Cyclosporin alkyne analogues and their pharmaceutical uses |
| WO2006039164A2 (en) * | 2004-09-29 | 2006-04-13 | Amr Technology, Inc. | Novel cyclosporin analogues and their pharmaceutical uses |
| WO2006041631A2 (en) * | 2004-10-06 | 2006-04-20 | Amr Technology, Inc. | Novel cyclosporin alkynes and their utility as pharmaceutical agents |
| US9681105B2 (en) | 2005-12-29 | 2017-06-13 | Rovi Guides, Inc. | Interactive media guidance system having multiple devices |
| US8607287B2 (en) | 2005-12-29 | 2013-12-10 | United Video Properties, Inc. | Interactive media guidance system having multiple devices |
| US20090019492A1 (en) | 2007-07-11 | 2009-01-15 | United Video Properties, Inc. | Systems and methods for mirroring and transcoding media content |
| US8601526B2 (en) | 2008-06-13 | 2013-12-03 | United Video Properties, Inc. | Systems and methods for displaying media content and media guidance information |
| CN102458370A (en) | 2009-06-09 | 2012-05-16 | 卢克斯生物科技公司 | Topical drug delivery systems for ophthalmic use |
| US9014546B2 (en) | 2009-09-23 | 2015-04-21 | Rovi Guides, Inc. | Systems and methods for automatically detecting users within detection regions of media devices |
| RU2630690C9 (en) | 2010-12-15 | 2017-12-28 | КонтраВир Фармасьютикалз, Инк. | Cyclosporin analogs molecules modified by 1 and 3 amino acids |
| US8805418B2 (en) | 2011-12-23 | 2014-08-12 | United Video Properties, Inc. | Methods and systems for performing actions based on location-based rules |
| AR090964A1 (en) | 2012-05-09 | 2014-12-17 | Novartis Ag | PROCESS FOR THE PREPARATION OF CYCLE UNDECAPEPTIDES |
| US9674563B2 (en) | 2013-11-04 | 2017-06-06 | Rovi Guides, Inc. | Systems and methods for recommending content |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0296122A2 (en) * | 1987-06-17 | 1988-12-21 | Sandoz Ag | Cyclosporins and their use as pharmaceuticals |
| WO1998048791A1 (en) * | 1997-04-30 | 1998-11-05 | The Regents Of The University Of California | Synthesis of discodermolide and analogs |
| WO1999018120A1 (en) * | 1997-10-08 | 1999-04-15 | Isotechnika Inc. | Deuterated cyclosporine analogs and their use as immunomodulating agents |
| EP1291347A2 (en) * | 2001-09-07 | 2003-03-12 | Clariant GmbH | Process for preparing bisallylboranes and non-aromatic boronic acids |
| WO2003033526A2 (en) * | 2001-10-19 | 2003-04-24 | Isotechnika Inc. | Synthesis of cyclosporin analogs |
| WO2003033527A2 (en) * | 2001-10-19 | 2003-04-24 | Isotechnika Inc. | Cyclosporine analogue mixtures and their use as immunomodulating agents |
-
2004
- 2004-03-31 TW TW093108938A patent/TW200505946A/en unknown
- 2004-04-02 AU AU2004228159A patent/AU2004228159A1/en not_active Abandoned
- 2004-04-02 CN CNA2004800154235A patent/CN1798759A/en active Pending
- 2004-04-02 CA CA002521116A patent/CA2521116A1/en not_active Abandoned
- 2004-04-02 RU RU2005130508/04A patent/RU2005130508A/en not_active Application Discontinuation
- 2004-04-02 BR BRPI0409734-3A patent/BRPI0409734A/en not_active Application Discontinuation
- 2004-04-02 ZA ZA200507980A patent/ZA200507980B/en unknown
- 2004-04-02 MX MXPA05010794A patent/MXPA05010794A/en unknown
- 2004-04-02 WO PCT/EP2004/003504 patent/WO2004089960A2/en not_active Application Discontinuation
- 2004-04-02 JP JP2006504961A patent/JP2006525961A/en not_active Withdrawn
- 2004-04-02 KR KR1020057019162A patent/KR20050119200A/en not_active Application Discontinuation
- 2004-04-02 EP EP04725333A patent/EP1613646A2/en not_active Withdrawn
- 2004-04-05 US US10/817,991 patent/US20040220091A1/en not_active Abandoned
-
2005
- 2005-09-28 NO NO20054478A patent/NO20054478L/en not_active Application Discontinuation
- 2005-10-06 CO CO05101444A patent/CO5680450A2/en not_active Application Discontinuation
- 2005-10-07 EC EC2005006091A patent/ECSP056091A/en unknown
- 2005-10-07 TN TNP2005000255A patent/TNSN05255A1/en unknown
- 2005-10-07 MA MA28540A patent/MA27831A1/en unknown
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0296122A2 (en) * | 1987-06-17 | 1988-12-21 | Sandoz Ag | Cyclosporins and their use as pharmaceuticals |
| WO1998048791A1 (en) * | 1997-04-30 | 1998-11-05 | The Regents Of The University Of California | Synthesis of discodermolide and analogs |
| WO1999018120A1 (en) * | 1997-10-08 | 1999-04-15 | Isotechnika Inc. | Deuterated cyclosporine analogs and their use as immunomodulating agents |
| EP1291347A2 (en) * | 2001-09-07 | 2003-03-12 | Clariant GmbH | Process for preparing bisallylboranes and non-aromatic boronic acids |
| WO2003033526A2 (en) * | 2001-10-19 | 2003-04-24 | Isotechnika Inc. | Synthesis of cyclosporin analogs |
| WO2003033527A2 (en) * | 2001-10-19 | 2003-04-24 | Isotechnika Inc. | Cyclosporine analogue mixtures and their use as immunomodulating agents |
Non-Patent Citations (7)
| Title |
|---|
| BATEY R A ET AL: "Potassium Allyl- and Crotyltrifluoroborates: Stable and Efficient Agents for Allylation and Crotylation" TETRAHEDRON LETTERS, ELSEVIER SCIENCE PUBLISHERS, AMSTERDAM, NL, vol. 40, no. 23, 4 June 1999 (1999-06-04), pages 4289-4292, XP004164628 ISSN: 0040-4039 * |
| MANFRED T REETZ ET AL: "Stereoselective addition of organotitanium reagents to carbonyl compounds" CHEMISCHE BERICHTE, VERLAG CHEMIE GMBH. WEINHEIM, DE, vol. 118, no. 4, 1985, pages 1441-1454, XP002117771 ISSN: 0009-2940 * |
| MARRON T G ET AL: "Diastereoselective Synthesis of N-Benzoyl Mycalamine, the Fully Elaborated Trioxadecalin Nucleus of Mycalamide A. Control of the Key N-Acyl Aminal Stereocenter via Carbamate Acylation" TETRAHEDRON LETTERS, ELSEVIER SCIENCE PUBLISHERS, AMSTERDAM, NL, vol. 36, no. 10, 6 March 1995 (1995-03-06), pages 1581-1584, XP004028537 ISSN: 0040-4039 cited in the application * |
| RATHKE, M. R.; CHAO, E.; WU, G.: "The preparation and reactions of esters of dichloromethaneboronic acid" JOURNAL OF ORGANOMETALLIC CHEMISTRY, vol. 122, no. 2, 1976, pages 145-149, XP002296392 * |
| THADANI AVINASH N ET AL: "A mild protocol for allylation and highly diastereoselective syn or anti crotylation of aldehydes in biphasic and aqueous media utilizing potassium allyl- and crotyltrifluoroborates." ORGANIC LETTERS. 31 OCT 2002, vol. 4, no. 22, 31 October 2002 (2002-10-31), pages 3827-3830, XP002296391 ISSN: 1523-7060 * |
| TSAI D J S ET AL: "A STEREO CONTROLLED SYNTHESIS OF Z AND E TERMINAL DIENES FROM PINACOL E-1 TRI METHYLSILYL-1 PROPENE-3-BORONATE" TETRAHEDRON LETTERS, vol. 22, no. 29, 1981, pages 2751-2752, XP002288404 ISSN: 0040-4039 cited in the application * |
| YAMAMOTO, YOSHINORI ET AL: "Regioreversed reactions of the trimethylsilyl or phenylselenyl allylic carbanion with carbonyl compounds via allylic aluminum "ate" complexes" TETRAHEDRON LETTERS , 23(44), 4597-600 CODEN: TELEAY; ISSN: 0040-4039, 1982, XP002288405 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7449069B2 (en) | 2004-12-28 | 2008-11-11 | Lg Display Co., Ltd. | Slit coater having apparatus for supplying a coating solution |
| US7696165B2 (en) | 2006-03-28 | 2010-04-13 | Albany Molecular Research, Inc. | Use of cyclosporin alkyne analogues for preventing or treating viral-induced disorders |
| US7696166B2 (en) | 2006-03-28 | 2010-04-13 | Albany Molecular Research, Inc. | Use of cyclosporin alkyne/alkene analogues for preventing or treating viral-induced disorders |
| US10265375B2 (en) | 2007-10-08 | 2019-04-23 | Aurinia Pharmaceuticals Inc. | Ophthalmic compositions |
| US10973871B2 (en) | 2007-10-08 | 2021-04-13 | Aurinia Pharmaceuticals, Inc. | Ophthalmic compositions |
| WO2010018211A1 (en) * | 2008-08-14 | 2010-02-18 | Minakem | Cyclopropyl- and cyclobutyl-dioxazaborocane or dioxazaborecane derivatives |
| FR2934997A1 (en) * | 2008-08-14 | 2010-02-19 | Minakem | CYCLOPROPYL-AND CYCLOBUTYL-DIOXAZABOROCANES OR DIOXAZABORECANES DERIVATIVES |
| US11622991B2 (en) | 2017-05-12 | 2023-04-11 | Aurinia Pharmaceuticals Inc. | Protocol for treatment of lupus nephritis |
| EP4201952A1 (en) | 2021-12-21 | 2023-06-28 | Curia Spain, S.A.U. | Process for the controlled synthesis of voclosporin |
| WO2023118045A1 (en) | 2021-12-21 | 2023-06-29 | Curia Spain, S.A.U. | Process for the controlled synthesis of voclosporin |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2005130508A (en) | 2006-06-27 |
| WO2004089960A3 (en) | 2005-01-06 |
| CO5680450A2 (en) | 2006-09-29 |
| BRPI0409734A (en) | 2006-05-09 |
| CA2521116A1 (en) | 2004-10-21 |
| KR20050119200A (en) | 2005-12-20 |
| US20040220091A1 (en) | 2004-11-04 |
| AU2004228159A1 (en) | 2004-10-21 |
| NO20054478L (en) | 2005-10-17 |
| MXPA05010794A (en) | 2005-12-14 |
| NO20054478D0 (en) | 2005-09-28 |
| TW200505946A (en) | 2005-02-16 |
| TNSN05255A1 (en) | 2007-07-10 |
| ZA200507980B (en) | 2006-11-29 |
| ECSP056091A (en) | 2006-03-01 |
| MA27831A1 (en) | 2006-04-03 |
| EP1613646A2 (en) | 2006-01-11 |
| CN1798759A (en) | 2006-07-05 |
| JP2006525961A (en) | 2006-11-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2004089960A2 (en) | Process for preparation of cyclosporin “a” analogues having a terminal diene group | |
| Lachance et al. | Allylboration of carbonyl compounds | |
| Beruben et al. | Stereodefined substituted cyclopropyl zinc reagents from gem-bismetallics | |
| JPH0753508A (en) | Intermediate of astaxanthin, its production and production of astaxanthin | |
| Suzuki et al. | Novel type of carbozirconation reaction of alkynes | |
| Altendorfer et al. | Modular synthesis of polyene side chain analogues of the potent macrolide antibiotic etnangien by a flexible coupling strategy based on hetero-bis-metallated alkenes | |
| JP7377266B2 (en) | Method for producing mannose derivatives | |
| Matsuhashi et al. | The Palladium-Catalyzed Cross-Coupling Reaction of Organosilicon Compounds with Allylic Carbonates or Diene Monoxides. | |
| Lombardo et al. | The reaction of nitrones with organometallic compounds: Scope, limitations and synthetic applications | |
| KR100541590B1 (en) | Sulfurpentafluoride compounds and methods for making and using same | |
| D’yakonov et al. | Synthesis and transformations of metallacycles 38. The Cp 2 ZrCl 2-catalyzed cyclometallation of α, ω-diynes upon the action of RMgR’or R n AlCl 3− n | |
| US6444825B1 (en) | Process for the preparation of an oxazolidinecarboxylic acid which is useful for preparing therapeutically active taxoids | |
| Boucley et al. | Stereoselective addition of organomanganese reagents to chiral acylsilanes and aldehydes | |
| Prié et al. | Efficient preparation of β-trifluoromethyl acrylates and derivatives via palladium cross-coupling reactions | |
| RU2402564C2 (en) | Method of obtaining pyrazinoyl-(l)-phenylalanyl-(l)-leucine boronic acid or its anhydride | |
| Landais et al. | Studies on the Mercury‐Desilylation of Chiral Cyclopropylmethylsilanes‐A Stereocontrolled Access to Carba‐Sugars | |
| Amedio Jr et al. | A Practical Preparation of Methyl 4-(Trimethylsilyl)-benzoate: An Intermediate in the Synthesis of SDZ 63135 | |
| NZ247574A (en) | Preparation of 2,3-di(hydroxymethyl)cyclobutanol having both hydroxymethyl groups protected from the corresponding protected cyclobutanone | |
| Ibragimov et al. | Joint cycloalumination of ethylene and other unsaturated compounds with EtAlCl 2 in the presence of Cp 2 ZrCl 2. Synthesis of aluminacarbocycles | |
| Jankowski et al. | The reaction of trimethylsilyl ethylene oxide with α-sulfonyl anions and α, α-sulfonyl dianions. A method for stereocontrolled synthesis of (E)-and (Z)-allylic alcohols | |
| CN115521330B (en) | Alkynyl-containing alpha-silanol compound and preparation method thereof | |
| Hofman | Studies directed towards the total synthesis of (+)-himbacine | |
| JP2020511404A (en) | Method for preparing eribulin and intermediates thereof | |
| Cintrat et al. | An alternative route to enantioenriched α-alkoxyalkylstannanes by stereoselective opening of chiral α-stannylacetals with organometallic reagents | |
| Walkup et al. | Preparation of 2-(Tetrahydrofuran-2′-YL)-l, 4-Pentadienes From γ-Allenyl Alcohols via Oxymercuration Followed by Palladium (II)-Medlated all Ylation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 170912 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004725333 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004228159 Country of ref document: AU Ref document number: 542637 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2521116 Country of ref document: CA Ref document number: 4431/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200507980 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006504961 Country of ref document: JP Ref document number: 05101444 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2005/010794 Country of ref document: MX Ref document number: 1020057019162 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2005/0765 Country of ref document: YU |
|
| ENP | Entry into the national phase |
Ref document number: 2004228159 Country of ref document: AU Date of ref document: 20040402 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004228159 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005130508 Country of ref document: RU Ref document number: 1200501650 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20048154235 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057019162 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004725333 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0409734 Country of ref document: BR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2004725333 Country of ref document: EP |