WO2004083263A1 - Polymerisation and oligomerisation catalysts - Google Patents
Polymerisation and oligomerisation catalysts Download PDFInfo
- Publication number
- WO2004083263A1 WO2004083263A1 PCT/GB2004/001184 GB2004001184W WO2004083263A1 WO 2004083263 A1 WO2004083263 A1 WO 2004083263A1 GB 2004001184 W GB2004001184 W GB 2004001184W WO 2004083263 A1 WO2004083263 A1 WO 2004083263A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- groups
- formula
- group
- polymerisation
- catalyst
- Prior art date
Links
- 0 CC1=C(N=CN=C)N=C(C)*1 Chemical compound CC1=C(N=CN=C)N=C(C)*1 0.000 description 9
- VPYXFGGWQNGBRD-YRRMYEEHSA-N C(C/N=C/c([nH]c1c(cc2)-c3ccccc3)nc1c2-c1ccccc1)C(c1ccccc1)c1ccccc1 Chemical compound C(C/N=C/c([nH]c1c(cc2)-c3ccccc3)nc1c2-c1ccccc1)C(c1ccccc1)c1ccccc1 VPYXFGGWQNGBRD-YRRMYEEHSA-N 0.000 description 1
- UMODNIZMTKYGNV-LFIBNONCSA-N C(c1ccccc1)/N=C/c1nc2ccccc2[nH]1 Chemical compound C(c1ccccc1)/N=C/c1nc2ccccc2[nH]1 UMODNIZMTKYGNV-LFIBNONCSA-N 0.000 description 1
- PXYBTSOOEVCYFY-UHFFFAOYSA-N C(c1nc(cccc2)c2[nH]1)=N/c(cccc1)c1-c1ccccc1 Chemical compound C(c1nc(cccc2)c2[nH]1)=N/c(cccc1)c1-c1ccccc1 PXYBTSOOEVCYFY-UHFFFAOYSA-N 0.000 description 1
- YHJRRXPBPGGVNP-WGOQTCKBSA-N CC(C)C(C)/N=C/c([nH]c1c(cc2)-c3ccccc3)nc1c2-c1ccccc1 Chemical compound CC(C)C(C)/N=C/c([nH]c1c(cc2)-c3ccccc3)nc1c2-c1ccccc1 YHJRRXPBPGGVNP-WGOQTCKBSA-N 0.000 description 1
- SKDFOEZWYUIRBW-WPWMEQJKSA-N CC(C)C(C)/N=C/c1nc(c(-c2ccccc2)ccc2-c3ccccc3)c2[n]1C Chemical compound CC(C)C(C)/N=C/c1nc(c(-c2ccccc2)ccc2-c3ccccc3)c2[n]1C SKDFOEZWYUIRBW-WPWMEQJKSA-N 0.000 description 1
- BEGRGLDCTSOOSF-PCLIKHOPSA-N CC(C)C/N=C/c1nc(c(-c2ccccc2)ccc2-c3ccccc3)c2[nH]1 Chemical compound CC(C)C/N=C/c1nc(c(-c2ccccc2)ccc2-c3ccccc3)c2[nH]1 BEGRGLDCTSOOSF-PCLIKHOPSA-N 0.000 description 1
- SPOIMFLPLONJPQ-MTDXEUNCSA-N CC(CCCC1)C1/N=C/c([nH]c1c(cc2)-c3ccccc3)nc1c2-c1ccccc1 Chemical compound CC(CCCC1)C1/N=C/c([nH]c1c(cc2)-c3ccccc3)nc1c2-c1ccccc1 SPOIMFLPLONJPQ-MTDXEUNCSA-N 0.000 description 1
- OFUNZXHOEUGOOZ-MTDXEUNCSA-N COc(cccc1)c1/N=C/c([nH]c1c(cc2)-c3ccccc3)nc1c2-c1ccccc1 Chemical compound COc(cccc1)c1/N=C/c([nH]c1c(cc2)-c3ccccc3)nc1c2-c1ccccc1 OFUNZXHOEUGOOZ-MTDXEUNCSA-N 0.000 description 1
- GSMSPZSPXSKKGT-XIEYBQDHSA-N C[n]1c2ccccc2nc1/C=N/CCC(c1ccccc1)c1ccccc1 Chemical compound C[n]1c2ccccc2nc1/C=N/CCC(c1ccccc1)c1ccccc1 GSMSPZSPXSKKGT-XIEYBQDHSA-N 0.000 description 1
- RBFTUXPNWAUJJG-PXLXIMEGSA-N C[n]1c2ccccc2nc1/C=N/c1ccccc1-c1ccccc1 Chemical compound C[n]1c2ccccc2nc1/C=N/c1ccccc1-c1ccccc1 RBFTUXPNWAUJJG-PXLXIMEGSA-N 0.000 description 1
- QXYGYSONSTWNFE-MHWRWJLKSA-N Cc(cccc1)c1/N=C/c1nc2ccccc2[nH]1 Chemical compound Cc(cccc1)c1/N=C/c1nc2ccccc2[nH]1 QXYGYSONSTWNFE-MHWRWJLKSA-N 0.000 description 1
- DWPPPLHLYRWKBQ-GZTJUZNOSA-N Cc1ccccc1/N=C/c1nc2ccccc2[n]1C Chemical compound Cc1ccccc1/N=C/c1nc2ccccc2[n]1C DWPPPLHLYRWKBQ-GZTJUZNOSA-N 0.000 description 1
- LBBKWEDRPDGXPM-UHFFFAOYSA-N Cc1ccn[s]1 Chemical compound Cc1ccn[s]1 LBBKWEDRPDGXPM-UHFFFAOYSA-N 0.000 description 1
- CTANASBIJLHKQH-UHFFFAOYSA-N Cc1nc2ncncc2[o]1 Chemical compound Cc1nc2ncncc2[o]1 CTANASBIJLHKQH-UHFFFAOYSA-N 0.000 description 1
- NAVKNJCPYYUREB-UHFFFAOYSA-N Cc1nc2ncncc2[s]1 Chemical compound Cc1nc2ncncc2[s]1 NAVKNJCPYYUREB-UHFFFAOYSA-N 0.000 description 1
- XZGLNCKSNVGDNX-UHFFFAOYSA-N Cc1nnn[nH]1 Chemical compound Cc1nnn[nH]1 XZGLNCKSNVGDNX-UHFFFAOYSA-N 0.000 description 1
- IFBUREDCXVEQFT-UHFFFAOYSA-N Cc1nnn[o]1 Chemical compound Cc1nnn[o]1 IFBUREDCXVEQFT-UHFFFAOYSA-N 0.000 description 1
- PNMKAKOKTOWKAY-UHFFFAOYSA-N Cc1nnn[s]1 Chemical compound Cc1nnn[s]1 PNMKAKOKTOWKAY-UHFFFAOYSA-N 0.000 description 1
- KJXCQCQRWZMAPT-FYJGNVAPSA-N IC[n]1c2ccccc2nc1/C=N/c(cccc1)c1N1CCCC1 Chemical compound IC[n]1c2ccccc2nc1/C=N/c(cccc1)c1N1CCCC1 KJXCQCQRWZMAPT-FYJGNVAPSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/02—Carriers therefor
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F136/00—Homopolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds
- C08F136/02—Homopolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds
- C08F136/04—Homopolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds conjugated
- C08F136/06—Butadiene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F10/02—Ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/70—Iron group metals, platinum group metals or compounds thereof
- C08F4/7001—Iron group metals, platinum group metals or compounds thereof the metallic compound containing a multidentate ligand, i.e. a ligand capable of donating two or more pairs of electrons to form a coordinate or ionic bond
- C08F4/7003—Bidentate ligand
- C08F4/7004—Neutral ligand
- C08F4/7006—NN
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F110/00—Homopolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F110/02—Ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/02—Ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/16—Copolymers of ethene with alpha-alkenes, e.g. EP rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/639—Component covered by group C08F4/62 containing a transition metal-carbon bond
- C08F4/63912—Component covered by group C08F4/62 containing a transition metal-carbon bond in combination with an organoaluminium compound
Definitions
- the present invention relates to transition metal-based polymerisation and oligomerisation catalysts and to their use in the polymerisation, copolymerisation and oligomerisation of olefins.
- Commodity polyethylenes are commercially produced in a variety of different types and grades. Homopolymerisation of ethylene with transition metal based catalysts leads to the production of so-called "high density" grades of polyethylene. These polymers have relatively high stiffness and are useful for making articles where inherent rigidity is required. Copolymerisation of ethylene with higher 1 -olefins (eg butene, hexene or octene) is employed commercially to provide a wide variety of copolymers differing in density and in other important physical properties. Particularly important copolymers made by copolymerising ethylene with higher 1 -olefins using transition metal based catalysts are the copolymers having a density in the range of 0.91 to 0.93.
- 1 -olefins eg butene, hexene or octene
- linear low density polyethylene are in many respects similar to the so-called “low density” polyethylene produced by the high pressure free radical catalysed polymerisation of ethylene.
- Such polymers and copolymers are used extensively in the manufacture of flexible blown film.
- Polypropylenes are also commercially produced in a variety of different types and grades. Homopolymerisation of propylene with transition metal based catalysts leads to the production of grades with a wide variety of applications. Copolymers of propylene with ethylene or terpolymers with ethylene and higher 1 -olefins are also useful materials, often used in film applications.
- metallocene catalysts for example biscyclopentadienylzirconiumdichloride-activated with alumoxane
- metallocene catalysts for example biscyclopentadienylzirconiumdichloride-activated with alumoxane
- Other derivatives of metallocenes have been ' shown to be potentially useful for producing polypropylene with good activity, molecular weight and tacticity control.
- metallocene catalysts of this type suffer from a number of disadvantages, for example, high sensitivity to impurities when used with commercially available monomers, diluents and process gas streams, the need to use large quantities of expensive alumoxanes to achieve high activity, difficulties in putting the catalyst on to a suitable support and synthetic difficulties in the production of more complex catalyst structures suitable for polymerising propene in a tactic manner.
- Olefm oligomerisation is also a commercially important process, leading to the production of 1 -olefins (1-hexene, 1-octene, 1-decene, etc) that find utility in a wide range of applications, for example as comonomers for linear low densitypolyethylene, monomers for poly(l -olefins) and starting materials for surfactants.
- Catalysts based on a wide range of metal complexes may be used for this process and typically produce a so-called "Schulz-Flory" distribution of 1 -olefins. More recently catalysts have emerged that selectively produce only 1 -hexene by a distinctive trimerisation mechanism.
- An object of the present invention is to provide a catalyst suitable for polymerising or oligomerising monomers, for example, olefins, cycloolefins or diolefms, and especially for polymerising or oligomerising ethylene alone or propylene alone, or for copolymerising ethylene with higher 1 -olefins with high activity.
- a further object of the invention is to provide an improved process for the polymerisation of olefins.
- Yet another object of the present invention is to provide novel complexes based on certain transition metals.
- the catalysts described here show extremely high activity for polymerisation and oligomerisation which leads to many benefits including lower catalyst loadings in a commercial process and lower catalyst residues in the final product.
- the present invention provides a novel polymerisation catalyst comprising .
- Z comprises a five-membered heterocyclic group, the five membered heterocyclic group containing at least one carbon atom, at least one nitrogen atom and at least one other hetero atom selected from nitrogen, sulphur and oxygen, the remaining atoms in said ring being selected from nitrogen and carbon;
- M is a metal from Group 3 to 11 of the Periodic Table or a lanthanide metal;
- E 1 and E 2 are divalent groups independently selected from (i) aliphatic hydrocarbon, (ii) alicyclic hydrocarbon, (iii) aromatic hydrocarbon, (iv) alkyl substituted aromatic hydrocarbon (v) heterocyclic groups and (vi) heterosubstituted derivatives of said groups (i) to (v);
- D 1 and D 2 are donor atoms or groups;
- X is an anionic group, L is a neutral donor group;
- y and z are independently zero or integers such that the number of X and L groups satisfy the
- At least one of the atoms present in the ring of the five-membered heterocyclic group Z is preferably bonded directly to E 1 and preferably a second atom in the ring is bonded directly to M. Most preferably the atom in the five-membered ring bonded directly to E 1 is adjacent to a second atom in said ring, said second atom being bonded directly to M.
- the five-membered heterocyclic group Z preferably contains at least 2 carbon atoi ⁇ s in its ring and more preferably at least 3 carbon atoms in its ring.
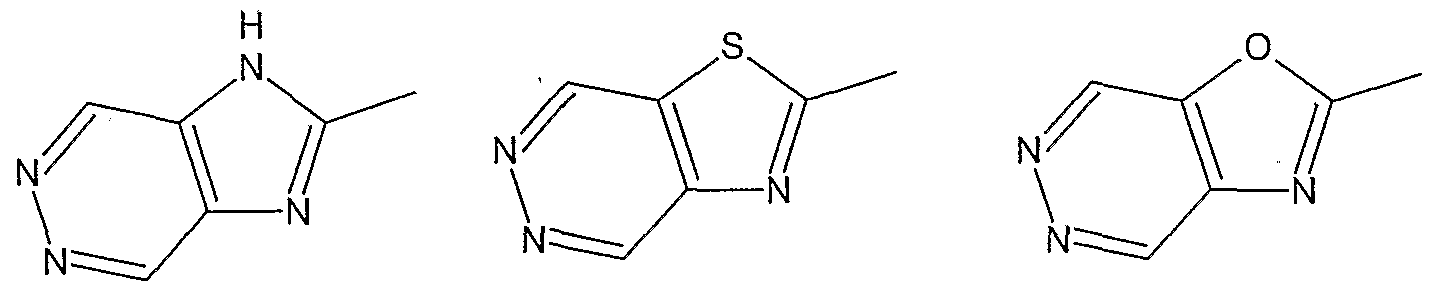
- suitable 5-membered heterocyclic groups are (but are not restricted to):
- Z, in Formula A is specifically an imidazole-containing group
- the present invention further provides a novel polymerisation catalyst comprising
- a fransition metal compound having the following Formula A, and optionally
- Z is specifically an imidazole-containing group
- M is a metal from Group 3 to 11 of the Periodic Table or a lanthanide metal
- E 1 and E 2 are divalent groups independently selected from (i) aliphatic hydrocarbon, (ii) alicyclic hydrocarbon, (iii) aromatic hydrocarbon, (iv) alkyl substituted aromatic hydrocarbon (v) heterocyclic groups and (vi) heterosubstituted derivatives of said groups (i) to (v);
- D 1 and D 2 are donor groups;
- X is an anionic group, L is a neutral donor group;
- y and z are independently zero or integers such that the number of X and L groups satisfy the valency and oxidation state of the metal M.
- D 1 and / or D 2 are donor atoms or " groups containing at least one donor atom.
- D 1 and / or D 2 can be, for example, groups having the same formula as recited for group Z.
- D and / or D can be groups comprising a five-membered heterocyclic group containing at least 2 carbon atoms in its ring and more preferably at least 3 carbon atoms in its ring.
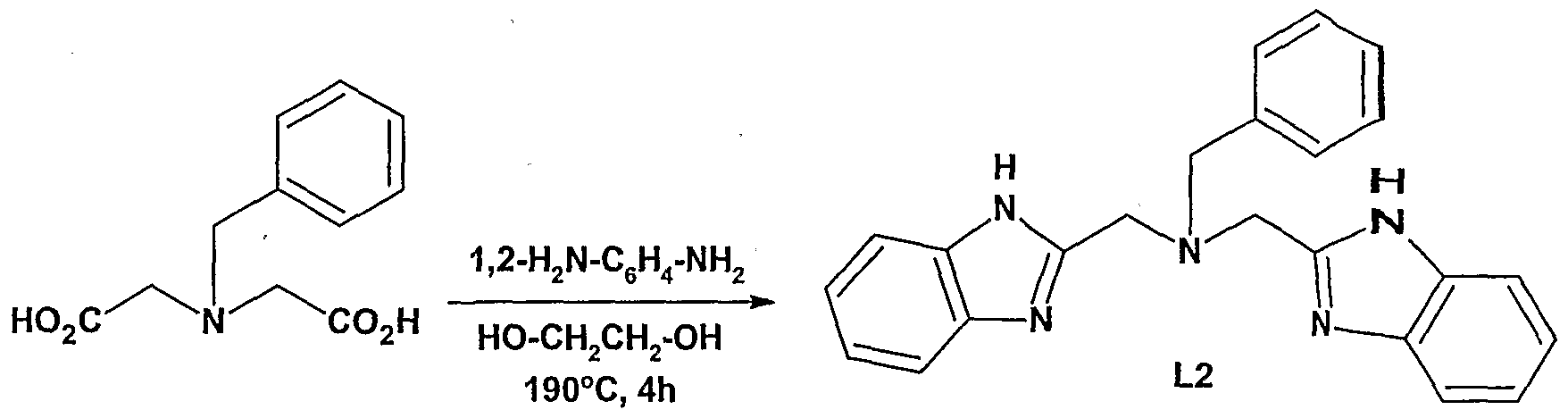
- D 1 and / or D 2 can be imidazole-containing groups if desired. When D 1 and / or D 2 are an imidazole-containing group this or these can be identical with Z. In a prefened embodiment D 2 and Z are identical imidazole containing groups.
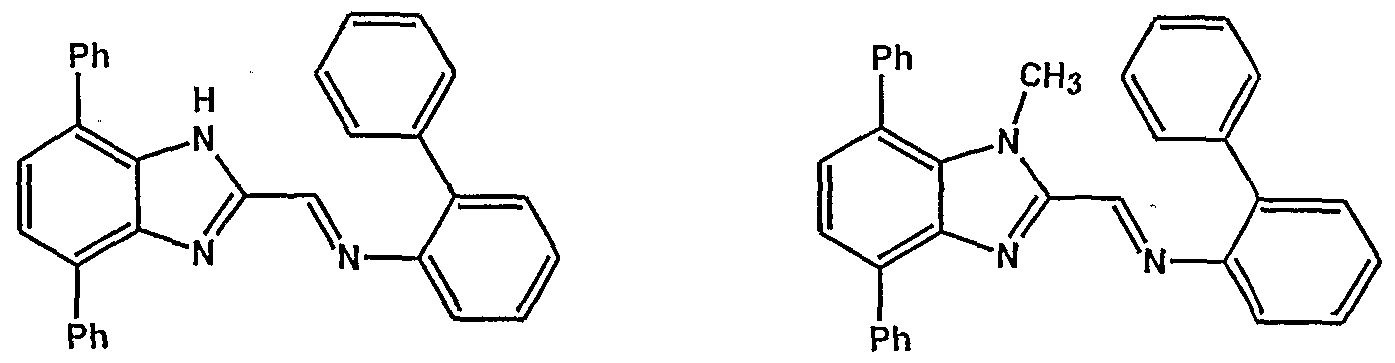
- the imidazole-containing group Z is preferably a group of formula I, II or in
- R 1 to R 11 are independently hydrogen or a monovalent (i) aliphatic hydrocarbon, (ii) alicyclic hydrocarbon, (iii) aromatic hydrocarbon, (iv) alkyl substituted aromatic hydrocarbon (v) heterocyclic groups, (vi) heterosubstituted derivatives of said groups (i) to (v), and (vii) hydrocarbyl-substituted heteroatom groups.
- the "free" valence bond on the left of Formulae I, II and HI provides at least one of the links of E into the rest of Formula A.
- the other link or links are preferably provided by at least one of the nitrogen atoms in the imidazole-containing group.
- R 1 to R 11 preferably contain 1 to 30, more preferably 2 to 20, most preferably 2 to 12 carbon atoms.
- suitable aliphatic hydrocarbon groups are methyl, ethyl, ethyleny], butyl, hexyl, isopropyl and tert-butyl.
- suitable alicyclic hydrocarbon groups are adamantyl, norbornyl, cyclopentyl and cyclohexyl.
- suitable aromatic hydrocarbon groups are phenyl, biphenyl, naphthyl, phenanthryl and anthryl.
- alkyl substituted aromatic hydrocarbon groups examples include benzyl, tolyl, mesityl, 2,6-diisopropylphenyl and 2,4,6-triisopropyl.
- suitable heterocyclic groups are 2-pyridinyl, 3-pyridinyl, 2-thiophenyl, 2-furanyl, 2-pyrrolyl, 2-quinolinyl.
- Suitable substituents for forming heterosubstituted derivatives of said groups R 1 to R n are, for example, chloro, bromo, fluoro, iodo, nitro, amino, cyano, ether, hydroxyl and silyl, methoxy, ethoxy, phenoxy (i.e.
- -OC 6 H 5 tolyloxy (i.e. -OC 6 H (CH 3 )), xylyloxy, mesityloxy, dimethylamino, diethylamino, methylethylamino, thiomethyl, thiophenyl and trimethylsilyl.
- suitable heterosubstituted derivatives of said groups (i) to (v) are 2-chloroethyl, 2-bromocyclohexyl, 2-nitrophenyl, 4-ethoxyphenyl, 4-chloro-2- pyridinyl, 4-dimethylaminophenyl and 4-methylaminophenyl.
- hydrocarbyl-substituted heteroatom groups are chloro, bromo, fluoro, iodo, nitro, amino, cyano, ether, hydroxyl and silyl, methoxy, ethoxy, phenoxy (i.e. -OC 6 H 5 ), tolyloxy (i.e. -OC 6 H (CH 3 )), xylyloxy, mesityloxy, dimethylamino, diethylamino, methylethylamino, thiomethyl, thiophenyl and trimethylsilyl.
- Any of the substituents R 1 to R 11 may be linked to form cyclic structures.
- Substituents R to R may also suitably be inorganic groups such as fluoro, chloro, bromo, iodo, nitro, amino, cyano and hydroxyl.
- imidazole-containing groups may be obtained by removal of substituent Ri, for example by deprotonation when R 1 is hydrogen, to give formally monoanionic imidazole-containing groups.
- the imidazole-containing group has a structure described in formula m (a "benzimidazole”).
- R 1 is preferably hydrogen, an aliphatic hydrocarbon group, an aromatic hydrocarbon group or is removed to give a formally monoanionic
- R to R are preferably hydrogen, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- E 1 and E 2 can be the same or different.
- E is independently selected from divalent (i) aliphatic hydrocarbon, (ii) alicyclic hydrocarbon, (iii) aromatic hydrocarbon, (iv) alkyl substituted aromatic hydrocarbon (v) heterocyclic groups, (vi) heterosubstituted derivatives of said groups (i) to (v), and (vii) hydrocarbyl-substituted heteroatom groups.
- divalent groups E are -CH 2 -, -CH 2 CH 2 -, -CH 2 CH 2 CH 2 -, 1,2-phenylene, trans- 1,2-cyclopentane, trans-1,2- cyclohexane, 2,3-butane, 1,1 '-biphenyl, l,l '-binaphthyl, and -Si(Me) 2 -.
- E is an aliphatic or aromatic hydrocarbon group. More preferably the divalent group E is -CH 2 -.
- D 1 and D 2 can be the same or different donor groups, for example oxygen, sulfur, an amine, an imine or a phosphine.
- D 1 and D 2 are selected from oxygen, sulfur, an amine of formula -N(R 12 )- or a phosphine of formula -P(R 13 )- wherein R 12 and R 13 are hydrogen or (i) aliphatic hydrocarbon, (ii) alicyclic hydrocarbon, (iii) aromatic hydrocarbon, (iv) alkyl substituted aromatic hydrocarbon (v) heterocyclic groups, (vi) heterosubstituted derivatives of said groups (i) to (v), (vii) hydrocarbyl-substituted heteroatom groups and (viii) further imidazole-containing groups.
- R 12 or R 13 may be removed, for example by deprotonation when they are hydrogen, to give a formally monoanionic fragment; or if both R 12 or R 13 are removed they provide a formally dianionic fragment.
- D 2 is an amine of formula -N(R 12 )- as defined above.
- R 12 is preferably hydrogen, an aliphatic hydrocarbon, an aromatic hydrocarbon or a further imidazole containing group.
- D 2 is an imidazole-containing group.
- M is preferably a metal selected from Groups 3 to 11 of the periodic table, preferably from Groups 3 to 7, more preferably selected from Sc, Ti, Zr, Hfi V, Nb, Ta, Cr, Mo, W, Mn and most preferably V, Cr, Ti, Zr and Hf
- the anionic group X can be, for example, a halide, preferably chloride or bromide; or a hydrocarbyl group, for example, methyl, benzyl or phenyl; a carboxylate, for example, acetate or an acetylacetonate; an oxide; an amide, for example diethyl amide; an alkoxide, for example, methoxide, ethoxide or phenoxide; or a hydroxyl.
- X can be a non-coordinating or weakly-coordinating anion, for example, tefrafluoroborate, a fluorinated aryl borate or a triflate.
- the anionic groups X may be the same or different and may independently be monoanionic, dianionic or trianionic.
- the neutral donor group L can be, for example, a solvate molecule, for example diethyl ether or THF; an amine, for example, diethyl amine, trimethylamine or pyridine; a phosphine, for example trimethyl phosphine or triphenyl phosphine; or water; or an olefm or a tortral, conjugated or nonconjugated diene, optionally substituted with one or more groups selected from hydrocarbyl or trimethylsilyl groups, said group having up to 40 carbon atoms and forming a pi-complex with M.
- a solvate molecule for example diethyl ether or THF
- an amine for example, diethyl amine, trimethylamine or pyridine
- a phosphine for example trimethyl phosphine or triphenyl phosphine
- water or an olefm or a damral, conjugated or nonconjugated diene, optionally
- L is a diene ligand
- it can be, for example s-trans- ⁇ 4 -l,4-diphenyl-l,3-butadiene; s-trans- ⁇ 4 -3-methyl-l,3- pentadiene; s-trans- ⁇ 4 -l,4-dibenzyl-l,3-butadiene; s-trans- ⁇ 4 -2,4-hexadiene; s-trans- ⁇ 4 - 1 ,3-pentadiene; s-trans- ⁇ 4 - 1 ,4-ditolyl- 1 ,3-butadiene; s-frans- ⁇ 4 - 1 ,4-bis(trimethylsilyl)- 1,3-butadiene; s-trans- ⁇ 4 -l,4-diphenyl-l,3-butadiene; s-cis- ⁇ 4 -3 -methyl- 1,3-pentadiene; s-cis- ⁇ 4
- the optional activator (2) for the catalyst of the present invention is suitably selected from organoaluminium compounds and organoboron compounds or mixtures thereof.
- organoaluminium compounds include trialkyaluminium compounds, for example, trimethylaluminium, triethylaluminium, tributylaluminium, toi-n-octylaluminium, ethylaluminium dichloride, diethylaluminium chloride, tris(pentafluorophenyl)aluminium and alumoxanes.
- Alumoxanes are well known in the art as typically the oligomeric compounds which can be prepared by the controlled addition of water to an alkylaluminium compound, for example trimethylaluminium.
- organoboron compounds are dimethylphenylammoniumtefra(phenyl)borate, frityltefra(phenyl)borate, triphenylboron, dimethylphenylammonium tetra(pentafluorophenyl)borate, sodium tetrakis[(bis-3,5-frifluoromethyl)phenyl]borate, H + (OEt 2 )[(bis-3,5- trifiuoromethyl)phenyl]borate, trityltetra(pentafluorophenyl)borate and tris(pentafluorophenyl) boron.
- organoaluminium compounds and organoboron compounds may be used.
- the quantity of activating compound selected from organoaluminium compounds and organoboron compounds to be employed is easily determined by simple testing, for example, by the preparation of small test samples which can be used to polymerise small quantities of the monomer(s) and thus to determine the activity of the produced catalyst. It is generally found that the quantity employed is sufficient to provide 0.1 to 20,000 atoms, preferably 1 to 2000 atoms of aluminium or boron per atom of M present in the compound of Formula A. Mixtures t»f different activating compounds may be used.
- EP1238989 discloses the use of activators (Lewis acids) selected from (b-1) ionic-bonding compounds having a CdCl 2 type or a Cdl 2 type of layered crystal structure;
- the activator employed in the present invention may be of the type disclosed in EP1238989 if desired.
- Such Lewis acids are those compounds which capable of receiving at least one electron pair and is capable of forming an ion pair by reaction with the transition metal complex.
- the Lewis acid includes the afore-mentioned (b-1) ionic- bonding compounds having a layered crystal structure of a CdCl 2 type or Cdl 2 type (b-2) clay . clay minerals, or ion-exchange layered compounds, (b-3) heteropoly compounds, and (b-4) halogenated lanthanoid compounds.
- the Lewis acid further includes SiO 2 , Al 2 O 3 , natural and synthetic zeolites which have Lewis acid points formed by heating or a like treatment, and complexes and mixtures thereof.
- US Patent 6399535 discloses a coordinating catalyst system capable of polymerizing olefins comprising: (I) as a pre-catalyst, at least one non-metallocene, non-constrained geometry, bidentate ligand containing transition metal compound or fridentate ligand containing transition metal compound capable of (A) being activated upon contact with the catalyst support- activator agglomerate of (II) or (B) being converted, upon contact with an organometallic compound, to an intermediate capable of being activated upon contact with the catalyst support-activator agglomerate of (II), wherein the transition metal is at least one member selected from Groups 3 to 10 of the Periodic table; in intimate contact with
- catalyst support-activator agglomerate comprising a composite of (A) at least one inorganic oxide component selected from SiO 2 , Al 2 O 3 , MgO, AlPO 4 , TiO 2 , ZrO 2 , and
- halogenated organic compounds that can be used in this manner are ethyl, trichloroacetate, chloroform (CHC1 3 ) and n-butylchloride.
- US Patent.5191042 also . refers to the disclosure of Cooper (T. A Cooper, Journ. Am. Chem. Soc, 4158 (1973), which defines in Table 1 an organic halide activity index based on the ability of the halide to oxidize certain vanadium compounds under standard conditions.
- carbon tetrachloride is assigned a reactivity of 1 in tetrahydrofuran at 20 °C.
- other listed halogenated organic compounds have reactivities of from about 0.02 to greater than 200 relative to carbon tetrachloride.
- a halogenated promotor it is prefened to use those having a Cooper Index ranging from about 0.01 up to about 30.
- the use of such promoters, especially in combination with vanadium-based catalysts is generally well known in the art, and for details of use of the such promoters reference may be made to US Patent.5191042 and to other prior art in this field.
- imidazole nucleus shown within the dotted circle is selected from the divalent groups represented by the Formulae la, Ha, Ula, IVa, Va and Via,
- M is a metal from Group 3 to 11 of the Periodic Table or a lanthanide metal
- E l and E 2 are divalent groups independently selected from (i) aliphatic hydrocarbon, (ii) alicyclic hydrocarbon, (iii) aromatic hydrocarbon, (iv) alkyl substituted aromatic hydrocarbon (v) heterocyclic groups and_(vi) heterosubstituted derivatives of said
- D 1 and D 2 can be the same or different donor groups, for example oxygen, sulfur, an amine, an imine or a
- D and D are selected from oxygen, sulfur, an amine of formula
- R and R are hydrogen or (i) aliphatic hydrocarbon, (ii) alicyclic hydrocarbon, (iii) aromatic hydrocarbon, (iv) alkyl substituted aromatic hydrocarbon (v) heterocyclic groups, (vi) heterosubstituted derivatives of said groups (i) to (v), (vii) hydrocarbyl-substituted heteroatom groups and (viii) further imidazole-containing groups.
- -OC 6 H 5 ), tolyloxy (i.e. -OC 6 H 4 (CH 3 )), xylyloxy, mesityloxy, dimethylamino, diethylamino, methylethylamino, thiomethyl, thiophenyl and frimethylsilyl.
- suitable heterosubstituted derivatives of said groups (i) to (v) are 2-chl ⁇ roethyl, 2- bromocyclohexyl, 2-nifrophenyl, 4-ethoxyphenyl, 4-chloro-2-pyridinyl, 4- dimethylaminophenyl and 4-methylaminophenyl.
- hydrocarbyl- substituted heteroatom groups examples include chloro, bromo, fluoro, iodo, nitro, amino, cyano, ether, hydroxyl and silyl, methoxy, ethoxy, phenoxy (i.e. -OC 6 H 5 ), tolyloxy (i.e. -
- DI, El, Z, M, X, L, y and z are as defined above, and wherein the imidazole nucleus within the dotted circle is selected from the divalent groups represented by the Formulae la, Ila, Hla, IVa, Va and Via
- ligands represent some examples of those suitable for making the complexes of Formula C and D in accordance with the present invention.
- ligands can be used to make complexes and catalysts in accordance with the present invention wherein the transition metal is preferably titanium, zirconium, hafnium, vanadium or chromium.
- transition metal complexes that can be employed in the catalyst of the present invention: -
- R1-R 3 alkyl, aryl, etc.
- R1 - R4 alkyl, aryl, etc.
- Z divalent organic or Inorganic radical as -CH2-, -0-,
- Z divalent organic or inorganic radical as -CH2-, -0-, etc.
- X CI, Br, I, NMe2, OR, SR, etc.
- the catalyst of the present invention can, if desired, be utilised on a support material.
- Suitable support materials are, for example, silica, alumina, or zirconia, magnesia, magnesium chloride or a polymer or prepolymer, for example polyethylene, polystyrene, or poly(aminostyrene).
- the catalysts of the present invention can if desired comprise more than one of the defined fransition metal compounds.
- the catalysts of the present invention can also include one or more other catalysts for polymerising 1 -olefins.
- catalysts are other types of transition metal compounds or catalysts, for example, transition metal compounds of the type used in conventional
- the catalysts can be formed in situ in the presence of the support material, or the support material can be pre-impregnated or premixed, simultaneously or sequentially, with one or more of the catalyst components.
- the catalysts of the present invention can if desired be supported on a heterogeneous catalyst, for example, a magnesium halide supported Ziegler Natta catalyst, a Phillips type (chromium oxide) supported catalyst or a supported metallocene catalyst. Formation of the supported catalyst can be achieved for example by treating the fransition metal compounds of the present invention with alumoxane in a suitable inert diluent, for example a volatile hydrocarbon, slurrying a particulate support material with the product and evaporating the volatile diluent.
- the produced supported catalyst is preferably in the form of a free- flowing powder.
- the quantity of support material employed can vary widely, for example from 100,000 to 1 grams per gram of metal present in the transition metal compound.
- the present invention further provides a process for the polymerisation and copolymerisation of 1 -olefins, cycloolefins or dienes, comprising contacting the monomer under polymerisation conditions with the polymerisation catalyst of the present invention.
- Suitable monomers for use in making homopolymers using the polymerisation process of the present invention are, for example, ethylene, propylene, butene, hexene, styrene or conjugated or non-conjugated dienes.
- Prefened monomers are ethylene and propylene.
- Suitable monomers for use in making copolymers using the polymerisation process of the present invention are ethylene, propylene, 1-butene, 1-hexene, 4- methylpentene-1, 1-octene, norbornene, substituted norbornenes, dienes, eg butadiene, ethylidene norbornene, methyl methacrylate, methyl acrylate, butyl acrylate, acrylonitrile, vinyl acetate, vinyl chloride, and styrene.
- a particularly prefened process in accordance with the present invention is the copolymerisation of ethylene and or propylene with comonomers selected from 1- olefms, acrylic acid esters, vinyl esters and vinyl aromatic compounds.
- suitable comonomers are 1-butene, 1-hexene, 4-methylpentene-l, methyl methacrylate, methyl acrylate, butyl acrylate, acrylonitrile, vinyl acetate, and styrene.
- Prefened polymerisation processes are the homopolymerisation of ethylene or the homopolymerisation of propylene or copolymerisation of ethylene with one or more of propylene, butene, hexene- 1 and 4-methylpentene-l or copolymerisation of propylene with one or more of ethylene or butene.
- the polymerisation conditions can be, for example, bulk phase, solution phase, slurry phase or gas phase.
- the catalyst can be used to polymerise ethylene under high pressure/high temperature process conditions wherein the polymeric material forms as a melt in supercritical ethylene.
- the polymerisation is conducted under gas phase fluidised or strrred bed conditions.
- Slurry phase polymerisation conditions or gas phase polymerisation conditions are particulariy useful for the production of high-density grades of polyethylene.
- the polymerisation conditions can be batch, continuous or semi- continuous.
- the catalyst is generally fed to the polymerisation zone in the form of a particulate solid.
- This solid can be, for example, an undiluted solid catalyst system formed from the complex A and an activator, or can be the solid complex A alone. In the latter situation, the activator can be fed to the polymerisation zone, for example as a solution, separately from or together with the solid complex.
- the catalyst system or the transition metal complex component of the catalyst system employed in the slurry polymerisation and gas phase polymerisation is supported on a support material.
- the catalyst system is supported on a support material prior to its introduction into the polymerisation zone.
- Suitable support materials are, for example, silica, alumina, zirconia, talc, kieselguhr, magnesia, magnesium chloride and polymers. Impregnation of the support material can be canied out by conventional techniques, for example, by forming a solution or suspension of the catalyst components in a suitable diluent or solvent, and slurrying the support material therewith. The support material thus impregnated with catalyst can then be separated from the diluent for example, by filtration or evaporation techniques.
- the solid particles of catalyst, or supported catalyst are fed to a polymerisation zone either as dry powder or as a slurry in the polymerisation diluent.
- a polymerisation zone is fed to a polymerisation zone as a suspension in the polymerisation diluent.
- the polymerisation zone can be, for example, an autoclave or similar reaction vessel, or a continuous loop reactor, e.g. of the type well know in the manufacture of polyethylene by the Phillips Process.
- the polymerisation process of the present invention is carried out under slurry conditions the polymerisation is preferably carried out at a temperature above 0°C, most preferably above 15°C.
- the polymerisation temperature is preferably maintained below the temperature at which the polymer commences to soften or sinter in the presence of the polymerisation diluent. If the temperature is allowed to go above the latter temperature, fouling of the reactor can occur. Adjustment of the polymerisation within these defined temperature ranges can provide a useful means of controlling the average molecular weight of the producedpolymer.
- a further useful means of controlling the molecular weight is to conduct the polymerisation in the presence of hydrogen gas which acts as chain fransfer agent. Generally, the higher the concentration of hydrogen employed, the lower the average molecular weight of the produced polymer.
- the use of hydrogen gas as a means of controlling the average molecular weight of the polymer or copolymer applies generally to the polymerisation process of the present invention.
- hydrogen can be used to reduce the average molecular weight of polymers or copolymers prepared using gas phase, slurry phase or solution phase polymerisation conditions.
- the quantity of hydrogen gas to be employed to give the desired average molecular weight can be determined by simple "trial and error" polymerisation tests.
- Methods for operating gas phase polymerisation processes are well known in the art. Such methods generally involve agitating (e.g. by stirring, vibrating or fiuidising) a bed of catalyst, or a bed of the target polymer (i.e.
- the catalyst, or one or more of the components employed to form the catalyst can, for example, be introduced into the polymerisation reaction zone in liquid form, for example, as a solution in an inert liquid diluent.
- the transition metal component, or the activator component, or both of these components can be dissolved or slurried in a liquid diluent and fed to the polymerisation zone. Under these circumstances it is prefened the liquid containing the component(s) is sprayed as fine droplets into the polymerisation zone.
- the droplet diameter is preferably within the range 1 to 1000 microns.
- EP-A-0593083 discloses a process for introducing a polymerisation catalyst into a gas phase polymerisation.
- the methods disclosed in EP-A-0593083 can be suitably employed in the polymerisation process of the present invention if desired.
- the present invention also provides a process for the oligomerisation and cooligomerisation of 1 -olefins, comprising contacting the monomeric olefm under oligomerisation conditions with the catalyst of the present invention.
- the oligomerisation and co-oligomerisation reactions of the present invention can be performed under a range of process conditions that are readily apparent to those skilled in the art: as a homogeneous liquid phase reaction in the presence or absence of an inert hydrocarbon diluent such as toluene or heptanes; as a two-phase liquid/liquid reaction; as a slurry process where the catalyst is in a form that displays little or no solubility; as a bulk process in which essentially neat reactant and/or product olefins serve as the dominant medium; as a gas-phase process in which at least a portion of the reactant or product olefin(s) are transported to or from a supported form of the catalyst via the gaseous state.
- Evaporative cooling from one or more monomers or inert volatile liquids is but one method that can be employed to effect the removal of heat from the reaction.
- the (co-)oligomerisation reactions may be performed in the known types of gas-phase reactors, such as circulating bed, vertically or horizontally stirred-bed, fixed-bed, or fluidised-bed reactors, liquid-phase reactors, such as plug-flow, continuously stined tank, or loop reactors, or combinations thereof.
- gas-phase reactors such as circulating bed, vertically or horizontally stirred-bed, fixed-bed, or fluidised-bed reactors
- liquid-phase reactors such as plug-flow, continuously stined tank, or loop reactors, or combinations thereof.
- a wide range of methods for effecting product, reactant, and catalyst separation and/or purification are known to those skilled in the art and may be employed: distillation, filtration, liquid-liquid separation, slurry settling, extraction, etc.
- One or more of these methods may be performed separately from the (co-)oligomerisation reaction or it may be advantageous to integrate at least some with 'a (co-)oligomerisation reaction; a non- limiting example of this would be a process employing catalytic (or reactive) distillation. Also advantageous may be a process which includes more than one reactor, a catalyst kill system between reactors or after the final reactor, or an integrated reactor/separator/purifier. While all catalyst components, reactants, inerts, and products could be employed in the present invention on a once-through basis, it is often economically advantageous to recycle one or more of these materials; in the case of the catalyst system, this might require reconstituting one or more of the catalysts components to achieve the active catalyst system.
- the catalyst systems of the present invention can present a variety of advantages over the prior art systems.
- the catalysts are easy to synthesise, have high activity and good catalyst life when employed under conventional industrial . polymerisation conditions.
- the catalysts exhibit single site behaviour which tends to favour the production of narrow molecular weight distribution polymers having uniform properties.
- the vanadium based catalysts of the present invention are capable of making very high molecular weight polymers.
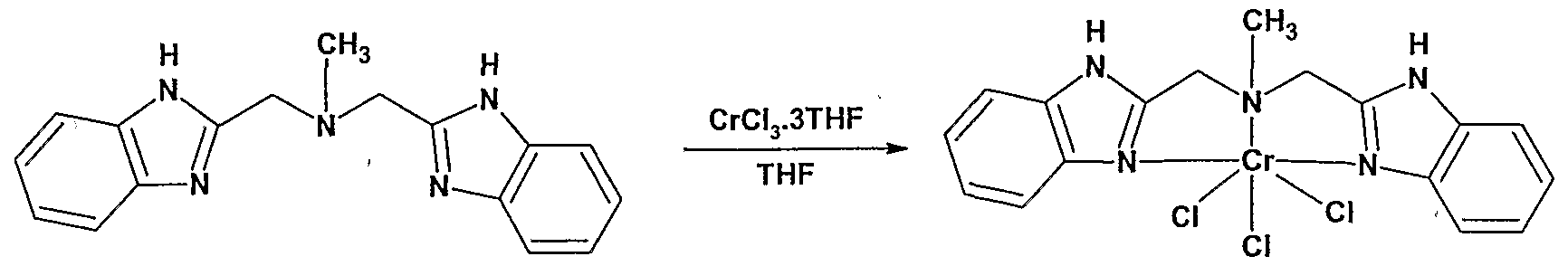
- a further aspect of the present invention provides a novel fransition metal compound having the Formula A
- M is preferably selected from Groups 3 to 7 of the periodic table.
- a prefened novel transition metal compound in accordance with the present has the Formula
- Z is specifically an imidazole-containing group of formula:
- R R 8 , R 9 , R 10 and R ⁇ are independently hydrogen or a monovalent (i) aliphatic hydrocarbon, (ii) alicyclic hydrocarbon, (iii) aromatic hydrocarbon, (iv) alkyl substituted aromatic hydrocarbon (v) heterocyclic groups, (vi) heterosubstituted derivatives of said groups (i) to (v), and (vii) hydrocarbyl-substituted heteroatom groups.
- These defined groups preferably contain 1 to 30, more preferably 2 to 20, most preferably 2 to 12 carbon atoms.
- suitable aliphatic hydrocarbon groups are methyl, ethyl, ethylenyl, butyl, hexyl, isopropyl and tert-butyl.
- heterosubstituted derivatives of said groups (i) to (v) are 2-chloroethyl, 2- bromocyclohexyl, 2-nitro ⁇ henyl, 4-ethoxyphenyl, 4-chloro-2-pyridinyl, 4- dimethyl aminophenyl and 4-methylaminophenyl.
- suitable hydrocarbyl- substituted heteroatom groups are chloro, bromo, fluoro, iodo, nitro, amino, cyano, ether, hydroxyl and silyl, methoxy, ethoxy, phenoxy (i.e. -OC 6 H 5 ), tolyloxy (i.e.
- substituents Ri to ⁇ maybe linked to form cyclic structures.
- Substituents R 2 to R ⁇ may also suitably be inorganic groups such as fluoro, chloro, bromo, iodo, nitro, amino, cyano and hydroxyl.
- Ri is preferably hydrogen, an aliphatic hydrocarbon group, an aromatic hydrocarbon group or is removed to give a formally monoanionic benzimidazole group.
- R 8 to R ⁇ are preferably hydrogen, an aliphatic hydrocarbon group or an aromatic hydrocarbon group.
- M is preferably a metal selected from Groups 3 to 11 of the periodic table, more preferably selected from Sc, Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W, Mn and most preferably V and Cr.
- E is independently selected from divalent (i) aliphatic hydrocarbon, (ii) alicyclic hydrocarbon, (iii) aromatic hydrocarbon, (iv) alkyl substituted aromatic hydrocarbon (v) heterocyclic groups, (vi) heterosubstituted derivatives of said groups (i) to (v), and (vii) hydrocarbyl-substituted heteroatom groups.
- Examples of suitable divalent group R are -CH 2 -, -CH CH 2 -, -CH 2 CH 2 CH 2 -, 1,2-phenylene, tra72S-l,2-cyclopentane, trans-1,2- cyclohexane, 2,3-butane, 1 , 1 '-biphenyl, 1 , 1 '-binaphthyl, and -Si(Me) 2 -.
- E is an aliphatic or aromatic hydrocarbon group. More preferably E is -CH -.
- R 12 or R 13 may be removed, for example by deprotonation when they are hydrogen, to give a formally monoanionic fragments.
- D is an amine of formula -N(R 1 )- as define above.
- R 12 is preferably hydrogen, an aliphatic hydrocarbon, an aromatic hydrocarbon or a further imidazole containing group.
- X is an anionic group and can be, for example, a halide, preferably chloride or bromide; or a hydrocarbyl group, for example, methyl, benzyl or phenyl; a carboxylate, for example, acetate or acetylacetate; an oxide; an amide, for example diethyl amide; an alkoxide, for example, methoxide, ethoxide or phenoxide.
- X can be a non-coordinating or weakly-coordinating anion, for example, tefrafluoroborate, a fluorinated aryl borate or a triflate.
- the anionic groups X may be the same or different and may independently be monoanionic, dianionic or trianionic.
- L is a tortral donor group and can be, for example, a solvate molecule, for example diethyl ether or THF; an amine, for example, diethyl amine, frimethylamine or pyridine; a phosphine, for example trimethyl phosphine or triphenyl phosphine; or an olefm, or a neutral, conjugated or nonconjugated diene, optionally substituted with one or more groups selected from hydrocarbyl or trimethylsilyl groups, said L having up to 40 carbon atoms and forming a .pi.-complex with M.
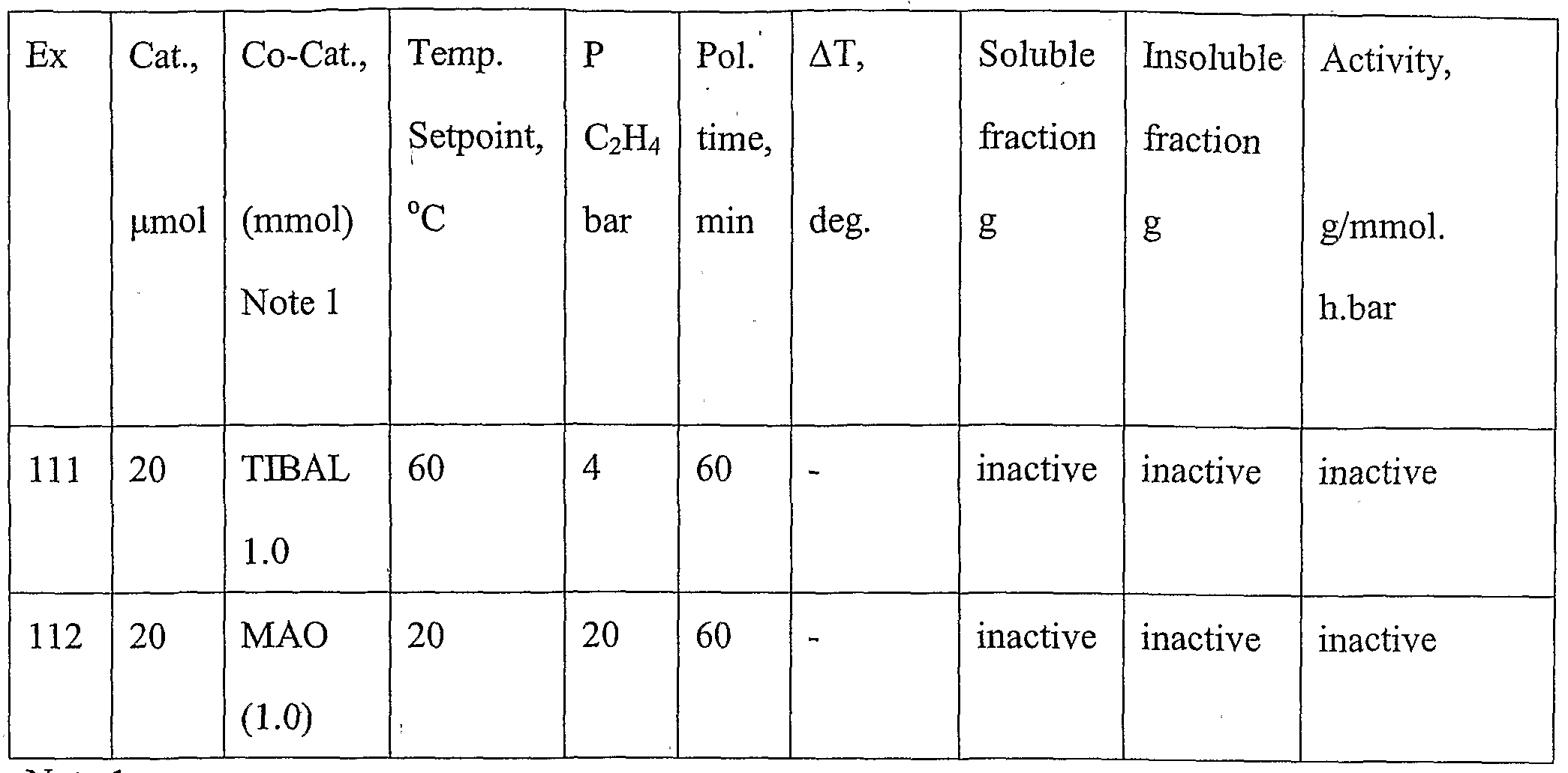
- Temperature set-point is the temperature at the start of the polymerisation reaction.
- the temperature in the reactor is uncontrolled and will change from this point due to the heat formation during the polymerisation reaction.
- ⁇ T is the difference between the temperature in the reactor and the temperature in the cooling bath
- the activity is based on the sum of the soluble and insoluble fractions.
- Example 11 In presence of 10 ml 1-hexene.
- PMAO polymethylalumoxane
- MMAO modified methylalumoxane
- Example 23 In presence of ethylene (1 bar) and propylene (2 bar). GC curves for the soluble fractions in Example 21 are shown in Figure 10; for Example 22 in Figure 11 and for Example 23 in Figure 12.
- Example 24 In presence of ethylene (1 bar) and propylene (2 bar). GC curves for the soluble fractions in Example 21 are shown in Figure 10; for Example 22 in Figure 11 and for Example 23 in Figure 12.
- the ethylene polymerisation reactions were carried out either in a 400 ml "Fischer- • 20 Porter” glass reactor (FPR) equipped with a gas inlet, a catalyst inlet, a mechanical stiner and a digital thermometer or in a IL stainless-steel reactor (SSR) equipped with an integral system for control of reaction temperature, ethylene pressure and ethylene flow.
- FPR "Fischer- • 20 Porter” glass reactor
- SSR IL stainless-steel reactor
- An aliquot of 1-5 ml of the catalyst solution described above was injected in the reactor containing 200 - 300 (FPR) or 400- 800 (SSR) ml solvent (usually toluene, n-
- the reactor content was then poured into a beaker containing 400 ml methanol and a few drops of 2M HCL The polymer was filtered, washed with methanol (if necessary) and dried at 60°C under vacuum.
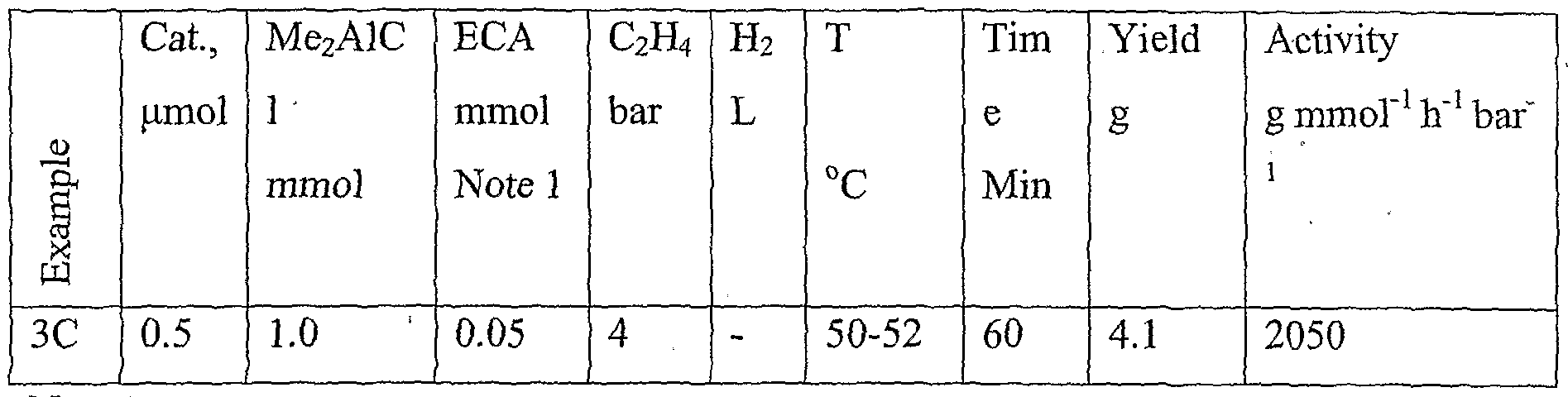
- Me 2 AlCl dimethylaluminium chloride - DMAC
- Runs 53 - 61 the amount of Me2AlCl shown includes the amount of the scavenger (0.5 mmol)
- Example 74 activated catalyst solution aged for 27 hours.
- Me 2 AlCl dimethylaluminium chloride - DMAC; the amount of Me 2 AlCl shown includes the amount of the scavenger (0.5 mmol)
- Figures 16 and 17, respectively, show ethylene uptake as a function of time for Examples 77 and 78.
- Examples 78-81 show ethylene uptake as a function of time for Examples 77 and 78.
- Example 48 - 61 The ethylene copolymerisation tests were canied out using a procedure similar to that described in Example 48 - 61.
- the required amounts of comonomer were preloaded in the polymerisation reactor.
- Table V-2C-FP Polymerisation in toluene. Fisher-Porter glass reactor.
- Example 48 - 61 The ethylene copolymerisation tests were canied out using a procedure similar to that described in Example 48 - 61.
- the required amounts of comonomers were preloaded in the polymerisation reactor.
- Table V-3HC-FP Polymerisation in toluene. Fisher-Porter glass reactor.
- Table V-4HCT-FP Polymerisation in toluene. Fisher-Porter glass reactor.
- Table V-6H-FP Polymerisation in toluene. Fisher-Porter glass reactor.
- the ethylene polymerisation tests were carried out using a procedure similar to that described in Example 48 - 61.
- the solid catalyst was preactivated and fransferred in the reactor ia cannula . .
- Table V-2-SiO2H-FP Polymerisation in toluene. Fisher-Porter glass reactor.
- TIBAL rriisobutyl aluminium
- the ethylene polymerisation test was carried out using a procedure similar to that described in Example 48 - 61. 5 Table V-C1H-FP. Polymerisation in toluene. Fisher-Porter glass reactor.
- Table V-C2H-FP Polymerisation in toluene. Fisher-Porter glass reactor.
- Table V-C1H-FP Polymerisation in toluene. Fisher-Porter glass reactor.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description















































































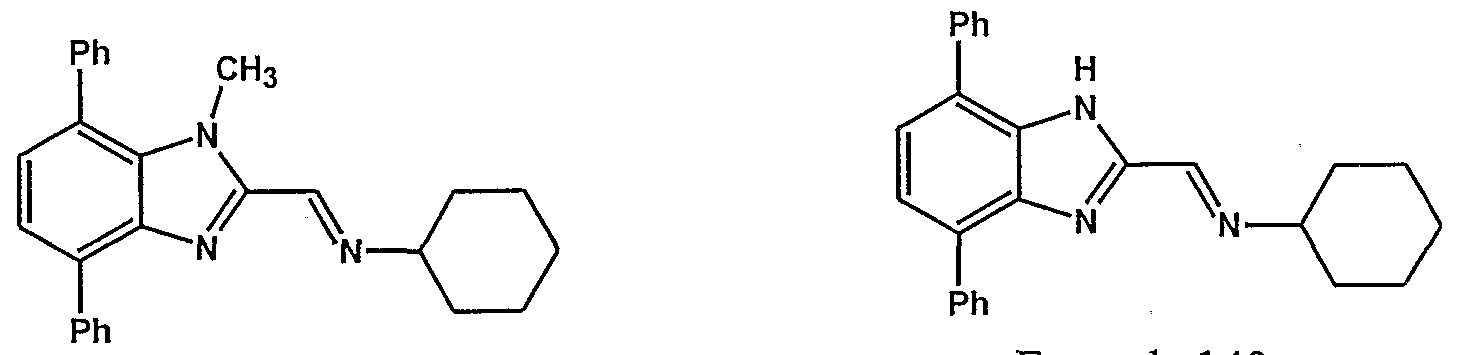













































































































Claims











Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006505990A JP5401011B2 (en) | 2003-03-20 | 2004-03-18 | Polymerization catalyst and oligomerization catalyst |
| BRPI0408549A BRPI0408549B1 (en) | 2003-03-20 | 2004-03-18 | catalysts and polymerization and oligomerization processes |
| EP04721578.5A EP1603957B1 (en) | 2003-03-20 | 2004-03-18 | Polymerisation and oligomerisation catalysts |
| KR1020057017626A KR101273309B1 (en) | 2003-03-20 | 2004-03-18 | Polymerisation and oligomerisation catalysts |
| US10/549,314 US7229943B2 (en) | 2003-03-20 | 2004-03-18 | Polymerisation and oligomerisation catalysts |
| AU2004222079A AU2004222079B2 (en) | 2003-03-20 | 2004-03-18 | Polymerisation and oligomerisation catalysts |
| CA2519854A CA2519854C (en) | 2003-03-20 | 2004-03-18 | Polymerisation and oligomerisation catalysts |
| MXPA05009958A MXPA05009958A (en) | 2003-03-20 | 2004-03-18 | Polymerisation and oligomerisation catalysts. |
| ZA2005/06886A ZA200506886B (en) | 2003-03-20 | 2005-08-26 | Polymerisation and oligomerisation catalysts |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0306430.0A GB0306430D0 (en) | 2003-03-20 | 2003-03-20 | Polymerisation and oligomerisation catalysts |
| GB0306430.0 | 2003-03-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004083263A1 true WO2004083263A1 (en) | 2004-09-30 |
Family
ID=9955183
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2004/001184 WO2004083263A1 (en) | 2003-03-20 | 2004-03-18 | Polymerisation and oligomerisation catalysts |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US7229943B2 (en) |
| EP (1) | EP1603957B1 (en) |
| JP (1) | JP5401011B2 (en) |
| KR (1) | KR101273309B1 (en) |
| CN (1) | CN100491417C (en) |
| AR (1) | AR043667A1 (en) |
| AU (1) | AU2004222079B2 (en) |
| BR (1) | BRPI0408549B1 (en) |
| CA (1) | CA2519854C (en) |
| GB (1) | GB0306430D0 (en) |
| MX (1) | MXPA05009958A (en) |
| RU (1) | RU2343162C2 (en) |
| TW (1) | TWI359697B (en) |
| WO (1) | WO2004083263A1 (en) |
| ZA (1) | ZA200506886B (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005111099A1 (en) * | 2004-05-12 | 2005-11-24 | Ineos Europe Limited | Ethylene copolymers |
| WO2006008438A1 (en) * | 2004-07-15 | 2006-01-26 | Ineos Europe Limited | Polymerisation catalyst |
| WO2006012327A2 (en) * | 2004-06-29 | 2006-02-02 | Exxonmobil Research And Engineering Company | Chromium complexes and their use in olefin polymerization |
| US7011924B2 (en) * | 2003-07-30 | 2006-03-14 | Hynix Semiconductor Inc. | Photoresist polymers and photoresist compositions comprising the same |
| WO2006030193A1 (en) * | 2004-09-14 | 2006-03-23 | Ineos Europe Limited | Polyolefins |
| WO2006048634A1 (en) * | 2004-11-04 | 2006-05-11 | Ineos Europe Limited | Polymerisation catalysts |
| WO2006096881A1 (en) * | 2005-03-09 | 2006-09-14 | Exxonmobil Chemical Patents Inc. | Methods for oligomerizing olefins |
| US7256296B2 (en) * | 2004-09-22 | 2007-08-14 | Symyx Technologies, Inc. | Heterocycle-amine ligands, compositions, complexes, and catalysts |
| US20100022724A1 (en) * | 2005-08-02 | 2010-01-28 | Grant Berent Jacobsen | Diene Polymerisation |
| US8076524B2 (en) | 2006-02-03 | 2011-12-13 | Exxonmobil Chemical Patents Inc. | Process for generating alpha olefin comonomers |
| EP2567752A1 (en) | 2011-09-08 | 2013-03-13 | IFP Energies Nouvelles | New nickel-based catalytic composition and method for oligomerising olefins using said composition |
| US8648000B2 (en) * | 2006-10-31 | 2014-02-11 | Ineos Commercial Services Uk Limited | Diene polymerisation |
| US11697666B2 (en) | 2021-04-16 | 2023-07-11 | Gilead Sciences, Inc. | Methods of preparing carbanucleosides using amides |
| US11767337B2 (en) | 2020-02-18 | 2023-09-26 | Gilead Sciences, Inc. | Antiviral compounds |
| US12030903B2 (en) | 2020-02-18 | 2024-07-09 | Gilead Sciences, Inc. | Antiviral compounds |
| US12054507B2 (en) | 2020-02-18 | 2024-08-06 | Gilead Sciences, Inc. | Antiviral compounds |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8546128B2 (en) | 2008-10-22 | 2013-10-01 | Life Technologies Corporation | Fluidics system for sequential delivery of reagents |
| US11951474B2 (en) | 2008-10-22 | 2024-04-09 | Life Technologies Corporation | Fluidics systems for sequential delivery of reagents |
| US20120259117A1 (en) * | 2009-06-19 | 2012-10-11 | The Regents Of The University Of California | Organo-metallic frameworks and methods of making same |
| CA2812294A1 (en) | 2010-09-27 | 2012-06-21 | The Regents Of The University Of California | Conductive open frameworks |
| TW201245157A (en) * | 2011-03-15 | 2012-11-16 | Sumitomo Chemical Co | Metal complex and organic electronic device comprising same |
| JP6012055B2 (en) | 2011-09-07 | 2016-10-25 | ダウ コーニング コーポレーションDow Corning Corporation | Titanium-containing complex and condensation reaction catalyst, method for preparing the catalyst, and composition containing the catalyst |
| JP6066355B2 (en) | 2011-09-07 | 2017-01-25 | ダウ コーニング コーポレーションDow Corning Corporation | Zirconium-containing complex and condensation reaction catalyst, method for preparing the catalyst, and composition containing the catalyst |
| WO2013043785A2 (en) | 2011-09-20 | 2013-03-28 | Dow Corning Corporation | Nickel containing hydrosilylation catalysts and compositions containing the catalysts |
| EP2758413B1 (en) | 2011-09-20 | 2018-03-07 | Dow Corning Corporation | Iridium containing hydrosilylation catalysts and compositions containing the catalysts |
| US9480977B2 (en) | 2011-09-20 | 2016-11-01 | Dow Corning Corporation | Ruthenium containing hydrosilylation catalysts and compositions containing the catalysts |
| JP6166266B2 (en) | 2011-10-04 | 2017-07-19 | ダウ コーニング コーポレーションDow Corning Corporation | Iron (III) -containing complex and condensation reaction catalyst, method for preparing the catalyst, and composition containing the catalyst |
| US9139699B2 (en) | 2012-10-04 | 2015-09-22 | Dow Corning Corporation | Metal containing condensation reaction catalysts, methods for preparing the catalysts, and compositions containing the catalysts |
| US9073950B2 (en) | 2011-12-01 | 2015-07-07 | Dow Corning Corporation | Hydrosilylation reaction catalysts and curable compositions and methods for their preparation and use |
| WO2015066693A1 (en) | 2013-11-04 | 2015-05-07 | The Regents Of Thd University Of California | Metal-organic frameworks with a high density of highly charged exposed metal cation sites |
| US10287304B2 (en) | 2014-02-19 | 2019-05-14 | The Regents Of The University Of California | Acid, solvent, and thermal resistant metal-organic frameworks |
| CN107148422A (en) | 2014-03-18 | 2017-09-08 | 加利福尼亚大学董事会 | The mesoscopic material of orderly superlattices comprising micropore metal organic backbone |
| WO2015195179A2 (en) | 2014-03-28 | 2015-12-23 | The Regents Of The University Of California | Metal organic frameworks comprising a plurality of sbus with different metal ions and/or a plurality of organic linking ligands with different functional groups. |
| US9517658B2 (en) | 2014-07-11 | 2016-12-13 | American Axle & Manufacturing, Inc. | Axle assembly with carrier housing having increased strength and reduced mass |
| US10118877B2 (en) | 2014-12-03 | 2018-11-06 | The Regents Of The University Of California | Metal-organic frameworks for aromatic hydrocarbon separations |
| US10058855B2 (en) | 2015-05-14 | 2018-08-28 | The Regents Of The University Of California | Redox-active metal-organic frameworks for the catalytic oxidation of hydrocarbons |
| JP2019503407A (en) | 2015-11-27 | 2019-02-07 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | Covalent organic structure having woven structure |
| KR102545533B1 (en) * | 2016-05-27 | 2023-06-21 | 에스케이이노베이션 주식회사 | Oligomerization Catalyst And Process For Preparing Ethylene Oligomer Using Thereof |
| WO2018208375A1 (en) * | 2017-05-09 | 2018-11-15 | Exxonmobil Chemical Patents Inc. | Linear alpha olefin process including solvent purification using dividing wall distillation column |
| SG11202102679QA (en) | 2018-09-18 | 2021-04-29 | Nikang Therapeutics Inc | Fused tricyclic ring derivatives as src homology-2 phosphatase inhibitors |
| CN112090447B (en) * | 2019-06-17 | 2023-05-26 | 中国石油天然气股份有限公司 | Alpha-diamido palladium catalyst and preparation method and application thereof |
| CN116970136B (en) * | 2023-09-20 | 2024-02-09 | 山西大学 | Benzimidazolyl covalent organic framework, preparation method and application thereof, proton conductor, and preparation method and application thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000219704A (en) * | 1999-01-29 | 2000-08-08 | Tosoh Corp | Catalyst for olefin polymerization and preparation of polyolefin by using the same |
| WO2000069923A1 (en) * | 1999-05-14 | 2000-11-23 | The Dow Chemical Company | Transition metal complexes and olefin polymerization process |
| US6281303B1 (en) * | 1999-07-27 | 2001-08-28 | Eastman Chemical Company | Olefin oligomerization and polymerization catalysts |
| WO2001074831A1 (en) * | 2000-03-31 | 2001-10-11 | Polimeri Europa S.P.A. | Complex polymerization catalysts for the homopolymerization of ethylene and for the copolymerization of ethylene |
-
2003
- 2003-03-20 GB GBGB0306430.0A patent/GB0306430D0/en not_active Ceased
-
2004
- 2004-03-18 KR KR1020057017626A patent/KR101273309B1/en active IP Right Grant
- 2004-03-18 CA CA2519854A patent/CA2519854C/en not_active Expired - Lifetime
- 2004-03-18 JP JP2006505990A patent/JP5401011B2/en not_active Expired - Fee Related
- 2004-03-18 BR BRPI0408549A patent/BRPI0408549B1/en active IP Right Grant
- 2004-03-18 RU RU2005132270/04A patent/RU2343162C2/en not_active IP Right Cessation
- 2004-03-18 US US10/549,314 patent/US7229943B2/en not_active Expired - Lifetime
- 2004-03-18 WO PCT/GB2004/001184 patent/WO2004083263A1/en active Application Filing
- 2004-03-18 AU AU2004222079A patent/AU2004222079B2/en not_active Ceased
- 2004-03-18 CN CNB2004800075676A patent/CN100491417C/en not_active Expired - Lifetime
- 2004-03-18 EP EP04721578.5A patent/EP1603957B1/en not_active Expired - Lifetime
- 2004-03-18 MX MXPA05009958A patent/MXPA05009958A/en unknown
- 2004-03-19 AR ARP040100930A patent/AR043667A1/en unknown
- 2004-03-19 TW TW093107473A patent/TWI359697B/en not_active IP Right Cessation
-
2005
- 2005-08-26 ZA ZA2005/06886A patent/ZA200506886B/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000219704A (en) * | 1999-01-29 | 2000-08-08 | Tosoh Corp | Catalyst for olefin polymerization and preparation of polyolefin by using the same |
| WO2000069923A1 (en) * | 1999-05-14 | 2000-11-23 | The Dow Chemical Company | Transition metal complexes and olefin polymerization process |
| US6281303B1 (en) * | 1999-07-27 | 2001-08-28 | Eastman Chemical Company | Olefin oligomerization and polymerization catalysts |
| WO2001074831A1 (en) * | 2000-03-31 | 2001-10-11 | Polimeri Europa S.P.A. | Complex polymerization catalysts for the homopolymerization of ethylene and for the copolymerization of ethylene |
Non-Patent Citations (1)
| Title |
|---|
| PATENT ABSTRACTS OF JAPAN vol. 2000, no. 11 3 January 2001 (2001-01-03) * |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7011924B2 (en) * | 2003-07-30 | 2006-03-14 | Hynix Semiconductor Inc. | Photoresist polymers and photoresist compositions comprising the same |
| WO2005111099A1 (en) * | 2004-05-12 | 2005-11-24 | Ineos Europe Limited | Ethylene copolymers |
| US7297805B2 (en) | 2004-06-29 | 2007-11-20 | Exxonmobil Research And Engineering Company | Chromium complexes and their use in olefin polymerization |
| WO2006012327A3 (en) * | 2004-06-29 | 2006-04-13 | Exxonmobil Res & Eng Co | Chromium complexes and their use in olefin polymerization |
| WO2006012327A2 (en) * | 2004-06-29 | 2006-02-02 | Exxonmobil Research And Engineering Company | Chromium complexes and their use in olefin polymerization |
| US7498390B2 (en) * | 2004-06-29 | 2009-03-03 | Exxonmobil Research And Engineering Company | Chromium complexes and their use in olefin polymerization |
| US7842638B2 (en) | 2004-07-15 | 2010-11-30 | Ineos Europe Limited | Polymerisation catalyst |
| WO2006008438A1 (en) * | 2004-07-15 | 2006-01-26 | Ineos Europe Limited | Polymerisation catalyst |
| WO2006030193A1 (en) * | 2004-09-14 | 2006-03-23 | Ineos Europe Limited | Polyolefins |
| US7989565B2 (en) | 2004-09-14 | 2011-08-02 | Ineos Europe Limited | Polyolefins |
| US7256296B2 (en) * | 2004-09-22 | 2007-08-14 | Symyx Technologies, Inc. | Heterocycle-amine ligands, compositions, complexes, and catalysts |
| WO2006048634A1 (en) * | 2004-11-04 | 2006-05-11 | Ineos Europe Limited | Polymerisation catalysts |
| WO2006099053A1 (en) * | 2005-03-09 | 2006-09-21 | Exxonmobil Chemical Patents Inc. | Methods for oligomerizing olefins |
| WO2006096881A1 (en) * | 2005-03-09 | 2006-09-14 | Exxonmobil Chemical Patents Inc. | Methods for oligomerizing olefins |
| US20100022724A1 (en) * | 2005-08-02 | 2010-01-28 | Grant Berent Jacobsen | Diene Polymerisation |
| US8124698B2 (en) * | 2005-08-02 | 2012-02-28 | Ineos Europe Limited | Diene polymerisation |
| US8076524B2 (en) | 2006-02-03 | 2011-12-13 | Exxonmobil Chemical Patents Inc. | Process for generating alpha olefin comonomers |
| US8648000B2 (en) * | 2006-10-31 | 2014-02-11 | Ineos Commercial Services Uk Limited | Diene polymerisation |
| EP2567752A1 (en) | 2011-09-08 | 2013-03-13 | IFP Energies Nouvelles | New nickel-based catalytic composition and method for oligomerising olefins using said composition |
| US11767337B2 (en) | 2020-02-18 | 2023-09-26 | Gilead Sciences, Inc. | Antiviral compounds |
| US12030903B2 (en) | 2020-02-18 | 2024-07-09 | Gilead Sciences, Inc. | Antiviral compounds |
| US12054507B2 (en) | 2020-02-18 | 2024-08-06 | Gilead Sciences, Inc. | Antiviral compounds |
| US11697666B2 (en) | 2021-04-16 | 2023-07-11 | Gilead Sciences, Inc. | Methods of preparing carbanucleosides using amides |
Also Published As
| Publication number | Publication date |
|---|---|
| ZA200506886B (en) | 2006-05-31 |
| AU2004222079B2 (en) | 2008-01-31 |
| KR101273309B1 (en) | 2013-06-11 |
| KR20050118196A (en) | 2005-12-15 |
| CA2519854A1 (en) | 2004-09-30 |
| US7229943B2 (en) | 2007-06-12 |
| BRPI0408549B1 (en) | 2020-01-21 |
| BRPI0408549A (en) | 2006-03-07 |
| AR043667A1 (en) | 2005-08-03 |
| CA2519854C (en) | 2011-11-22 |
| GB0306430D0 (en) | 2003-04-23 |
| EP1603957A1 (en) | 2005-12-14 |
| RU2343162C2 (en) | 2009-01-10 |
| US20060094588A1 (en) | 2006-05-04 |
| TW200427511A (en) | 2004-12-16 |
| JP2006520834A (en) | 2006-09-14 |
| CN1761688A (en) | 2006-04-19 |
| AU2004222079A1 (en) | 2004-09-30 |
| RU2005132270A (en) | 2006-08-10 |
| CN100491417C (en) | 2009-05-27 |
| MXPA05009958A (en) | 2006-03-09 |
| TWI359697B (en) | 2012-03-11 |
| EP1603957B1 (en) | 2015-10-28 |
| JP5401011B2 (en) | 2014-01-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1603957B1 (en) | Polymerisation and oligomerisation catalysts | |
| US7842638B2 (en) | Polymerisation catalyst | |
| EP1776396B1 (en) | Polymerisation and oligomerisation catalysts | |
| EP1789459B1 (en) | Polyolefins | |
| WO2005111099A1 (en) | Ethylene copolymers | |
| US7358209B2 (en) | Polymerisation catalysts | |
| CA2412990C (en) | Olefin trimerisation using a catalyst comprising a source of chromium, molybdenum or tungsten and a ligand containing at least one phosphorous, arsenic or antimony atom bound to at least one (hetero)hydrocarbyl group | |
| WO1999019335A1 (en) | Novel polymerisation catalysts | |
| CA2617436A1 (en) | Diene polymerisation catalysts comprising two or more transition metals | |
| WO2006048634A1 (en) | Polymerisation catalysts | |
| JP5550808B2 (en) | Diene polymerization | |
| US20040087436A1 (en) | Novel polymerisation catalysts | |
| WO2006030192A1 (en) | Polyolefins | |
| EP1767549A1 (en) | Polymerisation catalysts | |
| WO2006030199A1 (en) | Polymerisation catalysts |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004721578 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004222079 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005/06886 Country of ref document: ZA Ref document number: 3808/DELNP/2005 Country of ref document: IN Ref document number: 200506886 Country of ref document: ZA |
|
| ENP | Entry into the national phase |
Ref document number: 2004222079 Country of ref document: AU Date of ref document: 20040318 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004222079 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2006094588 Country of ref document: US Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10549314 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2519854 Country of ref document: CA Ref document number: PA/a/2005/009958 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006505990 Country of ref document: JP Ref document number: 1020057017626 Country of ref document: KR Ref document number: 20048075676 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005132270 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004721578 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057017626 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: PI0408549 Country of ref document: BR |
|
| WWP | Wipo information: published in national office |
Ref document number: 10549314 Country of ref document: US |