WO2004046117A1 - Pyridazinone derivatives as gsk-3beta inhibitors - Google Patents
Pyridazinone derivatives as gsk-3beta inhibitors Download PDFInfo
- Publication number
- WO2004046117A1 WO2004046117A1 PCT/EP2003/012950 EP0312950W WO2004046117A1 WO 2004046117 A1 WO2004046117 A1 WO 2004046117A1 EP 0312950 W EP0312950 W EP 0312950W WO 2004046117 A1 WO2004046117 A1 WO 2004046117A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- aryl
- oxo
- heteroaryl
- heterocyclyl
- Prior art date
Links
- 0 CN(Cc1ccc(*)cc1Cl)C(C(C(N)=O)=CC(c1ccccc1)=N)=O Chemical compound CN(Cc1ccc(*)cc1Cl)C(C(C(N)=O)=CC(c1ccccc1)=N)=O 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/26—Psychostimulants, e.g. nicotine, cocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/14—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/24—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Definitions
- the present invention relates to compounds according to the general formula (I), with the definitions of the substituents A and Ar given below in the text, as well as their physiologically acceptable salts, methods for producing these compounds and their use as pharmaceuticals.
- These compounds are kinase inhibitors, in particular inhibitors of the kinase GSK- 3 ⁇ (glycogen synthase kinase-3 ⁇ ).
- Pyridazinone derivatives are well known pharmaceuticals, but it has not been reported so far that pyridazinone derivatives can be employed for the inhibition of GSK-3 ⁇ or tau-phosporylation, respectively. Pyridazinone derivatives described in literature differ from those of the present invention due to a different substitution pattern and (partially) different indications.
- WO 03/059891 discloses pyridazinone derivatives that are useful for treating diseases and conditions caused or exacerbated by unregulated p38 MAP Kinase activity and/or TNF activity.
- the compounds described therein can be used, for example, for the treatment of inflammatory conditions, diabetes, Alzheimer's disease or cancer. They differ from the compounds of the present invention in the substitution of the pyridazinone cycle, since the nitrogen at position 2 of the cycle is mostly substituted with alky, aryl or heteroaryl and at position 4 of the cycle there is no amido group defined as substituent (equals substituent A of the compounds of the present invention).
- the object of the present invention is to provide compounds showing this ability.
- A represents A1 or A2
- R is unsubstituted or at least monosubstituted Ci-Cio-alkyl, aryl, aryl-(C ⁇ -C ⁇ 0 - alkyl)-, heteroaryl, heteroaryl-(C ⁇ -C ⁇ o-alkyl)-, heterocyclyl, heterocyclyl-(Cr C 10 -alkyl)-, C 3 -C ⁇ 0 -cycloalkyl, polycycloalkyl, C 2 -C ⁇ 0 -alkenyl or C 2 -C ⁇ 0 - alkinyl,
- substituents are selected from halogen, -CN, C C ⁇ 0 -alkyl, -N0 2 , - OR1 , -C(0)OR1 , -0-C(0)R1 , -NR1 R2, -NHC(0)R1 , -C(0)NR1 R2, -SR1 , - S(0)R1 , -S0 2 R1 , -NHS0 2 R1 , -S0 2 NR1 R2, -C(S)NR1 R2, -NHC(S)R1 ,
- aryl, heterocyclyl and heteroaryl may in turn be at least monosubstituted with Ci-C ⁇ -alkyl, Ci-C ⁇ -alkoxy, halogen, trifluoromethyl, trifluoromethoxy or OH;
- Ar is unsubstituted or at least monosubstituted aryl or heteroaryl
- substituents are selected from halogen, -CN, NO 2 , CrC ⁇ 0 -alkyl, -OR1 , -C(0)OR1 , -0-C(0)R1 , -NR1 R2, -NHC(0)R1 , -C(0)NR1 R2, -NHC(S)R1 , -C(S)NR1 R2, -SR1 , -S(0)R1 , -S0 2 R1 , -NHS0 2 R1 , -S0 2 NR1 R2, -0-S0 2 R1 , -SO 2 -O-RI , aryl, heteroaryl, ary CrC-e-alkyl)-, formyl, trifluoromethyl and trifluoromethoxy,
- aryl and heteroaryl may in turn be at least monosubstituted with C ⁇ -C 6 - alkyl, CrC ⁇ -alkoxy, halogen, trifluoromethyl, trifluoromethoxy or OH;
- R1 and R2 are independently from each other
- heteroaryl is a 5 to 10-membered, aromatic, mono- or bicyclic heterocycle containing one or more heteroatoms selected from N, O und S;
- aryl is phenyl, indanyl, indenyl or naphthyl
- heterocyclyl is a 5 to 10-membered, aliphatic, mono- or bicyclic heterocycle containing one or more heteroatoms selected from N, O and S;
- A is not -C(0)NH(C ⁇ -C ⁇ -alkyl)
- Ar is phenyl which is at least monosubstituted with heterocyclyl or heteroaryl containing nitrogen.
- groups, fragments, residues or substituents such as, for example, aryl, heteroaryl, alkyl etc., may be present several times, they all independently from each other have the meanings indicated and may hence, in each individual case, be identical with or different from each other.
- aryl as well as to any other residue independently from its classification as aryl group, -substituent, -fragment or - residue.
- di(CrCi 0 -alkyl)amino group in which the alkyl substitutents may be identical or different (for instance 2 x ethyl or 1 x propyl and 1 x heptyl).
- a substituent for example aryl
- a substituent may be unsubstituted or at least mono-substituted with a group of further substituents, for example, C ⁇ -C 6 -alkyl, C ⁇ -C 6 -alkoxy, halogen etc.
- a group of further substituents for example, C ⁇ -C 6 -alkyl, C ⁇ -C 6 -alkoxy, halogen etc.
- aryl may be substituted twice with ethyl, aryl may be mono-substituted with methyl or ethoxy, respectively, aryl may be mono-substituted with ethyl or fluoro, respectively, aryl may be substituted twice with methoxy, etc.
- Alkyl, alkenyl and alkynyl residues may be linear or branched. This also applies when they are part of other groups, for example in alkoxy groups (C C ⁇ 0 -alkyl-O-), alkoxycarbonyl groups or amino groups, or when they are substituted.
- alkyl groups are: methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl. This comprises both the n-isomers of these residues and isopropyl, isobutyl, isopentyl, sec-butyl, tert-butyl, neopentyl, 3,3-dimethylbutyl etc.
- alkyl here also includes unsubstituted alkyl residues as well as alkyl residues which are substituted by one or more, for example one, two, three or four, identical or different residues, for example aryl, heteroaryl, alkoxy or halogen.
- the substituents may be present in any desired position of the alkyl group.
- cycloalkyl residues are: cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl. All cycloalkyl groups may be unsubstituted or optionally substituted by one or more further residues, as exemplified above in the case of the alkyl groups.
- alkenyl and alkynyl groups are the vinyl residue, the 1-propenyl residue, the 2-propenyl residue (allyl residue), the 2-butenyl residue, the 2-methyl- 2-propenyl residue, the 3-methyl-2-butenyl residue, the ethynyl residue, the 2- propynyl residue (propargyl residue), the 2-butynyl residue or the 3-butynyl residue.
- alkenyl here also expressly includes cycloalkenyl residues and cycloalkenyl-alkyl-residues (alkyl substituted by cycloalkenyl) containing at least three carbon atoms.
- Examples for cycloalkenyl residues are cyclopentenyl, cyclohexenyl, cycloheptenyl and cyclooctenyl.
- the alkenyl residues may have 1 to 3 conjugated or unconjugated double bonds in a straight or branched chain; the same applies to alkynyl residues in respect of triple bonds.
- the alkenyl and alkinyl residues may be unsubstituted or optionally substituted by one or more further residues, as exemplified above in the case of the alkyl groups.
- polycycloalkyl residues examples are: adamantyl, quinuclidinyl, bornanyl, norbornanyl, bornenyl and norbornenyl. If not stated otherwise, the above-mentioned aryl, heteroaryl and heterocyclic residues may be unsubstituted or may carry one or more, for example one, two, three or four of the substituents indicated in the above definition, which substituents may be in any desired position.
- the substituent may be in the 2-position, the 3-position or the 4- position
- the substituents in disubstituted phenyl residues the substituents may be in 2,3-position, 2,4-position, 2,5-position, 2,6-position, 3,4-position or 3,5-position.
- the substituents In trisubstituted phenyl residues the substituents may be in 2,3,4-position, 2,3,5-position, 2,3,6- position, 2,4,5-position, 2,4,6-position or 3,4,5-position.
- the substituents In fourfold substituted phenyl residues, the substituents may be in the 2,3,4,5-position, the 2,3,4,6- position, or the 2, 3,5,6-position.
- divalent residues may be attached to the adjacent groups by any ring carbon atom.
- a phenylene residue these may be in 1 , 2-position (ortho-phenylene), 1 , 3-position (meta-phenylene) or 1 ,4-position (para-phenylene).
- 5-membered ring aromatics containing one heteroatom such as, for example, thiophene or furan
- the two free bonds may be in 2,3-position, 2,4-position, 2,5-position or 3,4-position.
- a divalent residue derived from pyridine may be a 2,3-, 2,4-, 2,5-, 2,6-, 3,4- or 3,5-pyridinediyl residue.
- the present invention includes all positional isomers, i. e., in the case of a 2,3-pyridinediyl residue, for example, it includes the compound in which the one adjacent group is present in the 2-position and the other adjacent group is present in the 3-position as well as the compound in which the one adjacent group is present in the 3-position and the other adjacent group is present in the 2-position.
- heteroaryl residues, heteroarylene residues, heterocyclyl residues, heterocyclylen residues and rings which are formed by two groups bonded to a nitrogen are preferably derived from completely saturated, partially saturated or completely unsaturated heterocycles (i.e. heterocycloalkanes, heterocycloalkenes, heteroaromatics), which contain one, two, three or four heteroatoms, which may be identical or different; more preferably they are derived from heterocycles which contain one, two, or three, in particular one or two, heteroatoms, which may be identical or different.
- the heterocycles may be monocyclic or polycyclic, for example monocyclic, bicyclic or tricyclic.
- the rings Preferably they are monocyclic or bicyclic.
- the rings preferably are 5- membered rings, 6-membered rings or 7-membered rings.
- aryl and the term “heteroaryl” as used herein comprise bicyclic residues in which both cycles are aromatic as well as bicyclic residues in which only one cycle is aromatic.
- Suitable aliphatic heterocycles include, for example, the saturated heterocycles pyrrolidine, piperidine, piperazine, imidazolidine, pyrazolidine, isothiazolidine, thiazolidine, isoxazolidine, oxazolidine, tetrahydrofuran, dioxolane, 2-oxo-azepane, morpholine and thiomorpholine as well as the partially unsaturated heterocycles isochromamyl, chromamyl, 1 ,2,3,4-tetrahydroisochinolyl and 1 ,2,3,4- tetrahydrochinolyl.
- the degree of saturation of heterocyclic groups is indicated in their individual definitions.
- Substituents which may be derived from these heterocycles may be attached via any suitable carbon atom.
- Residues derived from nitrogen heterocycles may carry a hydrogen atom or a substituent on a ring nitrogen atom, and examples include pyrrole, imidazole, pyrrolidine, morpholine, piperazine residues, etc.
- Those nitrogen heterocyclic residues may also be attached via a ring nitrogen atom, in particular if the respective heterocyclic residue is bonded to a carbon atom.
- Suitable nitrogen heterocycles may also be present as N-oxides or as quarternary salts containing a counterion which is derived from a physiologically acceptable acid.
- Pyridyl residues for example, may be present as pyridine-N-oxides.
- Arylalkyl means an alkyl residue, which in turn is substituted by an aryl residue.
- Heteroarylalkyl means an alkyl residue, which in turn is substituted by a heteroaryl residue.
- Heterocyclylalkyl means an alkyl residue, which in turn is substituted by a heterocyclyl residue.
- alkyl, heteroaryl, heterocyclyl and aryl it is referred to the above-mentioned definitions.
- Halogen is fluorine, chlorine, bromine or iod, preferably fluorine, chlorine or bromine, most preferably fluorine or chlorine.
- the present invention includes all stereoisomeric forms of the compounds of the formula (I). Centers of asymmetry that are present in the compounds of formula (I) all independently of one another have S configuration or R configuration.
- the invention includes all possible enantiomers and diastereomers and mixtures of two or more stereoisomers, for example mixtures of enantiomers and/or diastereomers, in all ratios.
- compounds according to the present invention which may exist as enantiomers may be present in enantiomerically pure form, both as levorotatory and as dextrorotatory antipodes, in the form of racemates and in the form of mixtures of the two enantiomers in all ratios.
- the invention includes both the cis form and the trans form as well as mixtures of these forms in all ratios. All these forms are an object of the present invention.
- the preparation of individual stereoisomers may be carried out, if desired, by separation of a mixture by customary methods, for example by chromatography or crystallization, by the use of stereochemically uniform starting materials for the synthesis or by stereoselective synthesis.
- a derivatization may be carried out before a separation of stereoisomers.
- the separation of a mixture of stereoisomers may be carried out at the stage of the compounds of the formula (I) or at the stage of an intermediate during the synthesis.
- the present invention also includes all tautomeric forms of the compounds of formula (I), in particular keto-enol tautomehsm, i.e. the respective compounds may be present either in their keto form or in their enol form or in mixtures thereof in all ratios.
- the invention also comprises their corresponding physiologically or toxicologically acceptable salts.
- Physiologically acceptable salts are particularly suitable for medical applications, due to their greater solubility in water compared with the starting or base compounds. Said salts must have a physiologically acceptable anion or cation.
- Suitable physiologically acceptable acid addition salts of the compounds of the invention are salts of inorganic acids such as hydrochloric acid, hydrobromic acid, phosphoric acid, metaphosphoric acid, nitric acid, sulfonic acid and sulfuric acid and also of organic acids such as, for example, acetic acid, theophyllinacetic acid, methylene-bis-b-oxynaphthoic acid, benzenesulfonic acid, benzoic acid, citric acid, ethanesulfonic acid, salicylic acid, fumaric acid, gluconic acid, glycolic acid, isethionic acid, lactic acid, lactobionic acid, maleic acid, malic acid, methane- sulfonic acid, succinic acid, p-toluenesulfonic acid, tartaric acid and trifluoroacetic acid.
- Suitable pharmaceutically acceptable basic salts are ammonium salts, alkali metal salts (such as sodium salts and potassium salts
- Salts having a pharmaceutically unacceptable anion are likewise included within the scope of the present invention as useful intermediates for preparing or purifying pharmaceutically acceptable salts and/or for use in nontherapeutic applications, for example in-vitro applications.
- the invention also includes, in addition to the salt forms mentioned, inner salts or betaines (zwitterions).
- the respective salts according to the formula (I) may be obtained by customary methods which are known to the person skilled in the art like, for example by contacting these with an organic or inorganic acid or base in a solvent or dispersant, or by anion exchange or cation exchange with other salts.
- the present invention furthermore includes all solvates of compounds of the formula (I), for example hydrates or adducts with alcohols, active metabolites of the compounds of the formula (I), and also derivatives, which contain physiologically tolerable and cleavable groups, for example esters or amides.
- physiologically functional derivative used herein relates to any physiologically acceptable derivative of an inventive compound of the formula I, for example an ester which on administration to a mammal, for example humans, is capable of forming (directly or indirectly) a compound of the formula I or an active metabolite thereof.
- the physiologically functional derivatives also include prodrugs of the compounds of the invention.
- prodrugs may be metabolized in vivo to a compound of the invention.
- These prodrugs may or may not be active themselves and are also object of the present invention.
- the compounds of the invention may also be present in various polymorphous forms, for example as amorphous and crystalline polymorphous forms. All polymorphous forms of the compounds of the invention are included within the scope of the invention and are another aspect of the invention.
- A is A1 ;
- R is unsubstituted or at least monosubstituted CrCio-alkyl, aryl, aryl-(C ⁇ -C ⁇ o- alkyl)-, heteroaryl, heteroaryl-(Ci-Cio-alkyl)-, heterocyclyl, heterocyclyl- (C-i- Cio-alkyl)-, C 3 -C ⁇ o-cycloalkyl, polycycloalkyl, C 2 -C ⁇ o-alkenyl or C- 2 -C 10 - alkinyl,
- substituents are selected from halogen, -CN, d-Cio-alkyl, -NO 2 , -OR1 , -C(0)OR1 , -0-C(0)R1 , -NR1 R2, -NHC(0)R1 , -C(0)NR1 R2, -SR1 , -S(0)R1 , -SO2RI , -NHSO2RI . -S0 2 NR1 R2, -C(S)NR1 R2, -NHC(S)R1 ,
- heterocyclyl and heteroaryl may in turn be at least monosubstituted with C-i-C ⁇ -alkyl, CrC 6 -alkoxy, halogen, trifluoromethyl, trifluoromethoxy or OH;
- heteroaryl is a 5 to 10-membered, aromatic, mono- or bicyclic heterocycle containing one or more heteroatoms selected from N, O and S;
- aryl is phenyl, indanyl, indenyl or naphthyl
- heterocyclyl is a 5 to 10-membered, aliphatic, mono- Oder bicyclic heterocycle containing one or more heteroatoms selected from N, O and S;
- R is unsubstituted or at least monosubstituted Ci-Cio-alkyl, aryl, aryl-(C ⁇ -C ⁇ o- alkyl)-, heterocyclyl, heterocyclyl-(CrCi 0 -alkyl)-, C 3 -C ⁇ . 0 -cycloalkyl, heteroaryl or heteroaryl-(CrC ⁇ o-alkyl)-,
- substituents are selected from halogen, -CN, C ⁇ -C ⁇ 0 -Alkyl, -NO 2 , -OR1 , -C(0)OR1 , -0-C(0)R1 , -NR1 R2, -NHC(0)R1 , -C(0)NR1 R2, -SR1 , -S(0)R1 , -SO 2 RI , -NHSO 2 RI , -S0 2 NR1 R2, -C(S)NR1 R2, -NHC(S)R1 , -O-SO 2 RI , -SO2-O-RI , oxo, -C(0)R1 , -C(NH)NH 2 , heterocyclyl, C 3 -C 10 - cycloalkyl, aryl-(d ⁇ C 6 -alkyl)-, aryl, heteroaryl, trifluoromethyl, trifluoromethylsulfanyl and trifluoromethoxy,
- heterocyclyl and heteroaryl may in turn be at least monosubstituted with CrC ⁇ -alkyl, d-C- ⁇ -alkoxy, halogen, trifluoromethyl, trifluoroethoxy or OH;
- R1 and R2 are independently from each other
- heteroaryl is a 5 to 10-membered, aromatic, mono- or bicyclic heterocycle containing one or more heteroatoms selected from N, O and S;
- aryl is phenyl, indanyl, indenyl or naphthyl
- heterocyclyl is a 5 to 10-membered, aliphatic, mono- Oder bicyclic heterocycle, containing one or more heteroatoms selected from N, 0 and S;
- Ar is unsubstituted or at least monosubstituted phenyl, pyridinyl, pyrimidinyl, pyrazolyl, thiophenyl, isoxaloyl, benzo[b]thiophenyl, benzodioxolyl or thiazolo[3,2-b][1 ,2,4]-tiazolyl,
- substituents are selected from halogen, -CN, N0 2 , CrCio-alkyl, -OR1 , -C(0)0R1 , -0-C(0)R1 , -NR1 R2, -NHC(0)R1 , -C(0)NR1 R2, -NHC(S)R1 , -C(S)NR1 R2, -SR1 , -S(0)R1 , -S0 2 R1 , -NHS0 2 R1 , -SO 2 NRI R2, -O-SO 2 RI , -SO 2 -O-RI , aryl, heteroaryl, aryl-(d-C 6 -alkyl)-, formyl, trifluoromethyl and trifluoromethoxy,
- aryl and heteroaryl may in turn be at least monosubstituted with d-C 6 - alkyl, C ⁇ -C 6 -alkoxy, halogen, trifluoromethyl, trifluoromethoxy or OH;
- R1 and R2 are independently from each other
- C Cio-alkyl C 3 -C ⁇ 0 -cycloalkyl, aryl, aryl-(d-C 10 -alkyl)-, C 2 -C ⁇ 0 -alkenyl, C 2 -C ⁇ 0 -alkinyl, heterocyclyl, heterocyclyl-(d-do-alkyl)- or heteroaryl, where the substituents are selected from halogen, C ⁇ -C 6 -alkyl, C C 6 -alkoxy, CN, N0 2 , NH 2 , (C C 6 - alkyl) amino-, di(d-C 6 -alkyl)amino-, OH, COOH, -COO-(d-C 6 -alkyl), -CONH 2 , formyl, trifluoromethyl and trifluoromethoxy;
- heteroaryl is a 5 to 10-membered aromatic, mono- or bicyclic heterocycle, containing one or more heteroatoms selected from N, O and S;
- aryl is phenyl, indanyl, indenyl or naphthyl
- heterocyclyl is a 5 to 10-membered aliphatic, mono- oder bicyclic heterocycle, containing one or more heteroatoms selected from N, O and S;
- A is A1 ;
- R is unsubstituted or at least monosubstituted CrCio-alkyl, aryl, aryl-(C ⁇ C ⁇ o- alkyl)-, heterocyclyl, heterocyclyl-(C ⁇ -C ⁇ o-alkyl)-, C 3 -C ⁇ 0 -cycloalkyl, heteroaryl or heteroaryl-(Ci-Cio-alkyl)-,
- substituents are selected from halogen, CrCio-alkyl, -OR1 , -C(0)OR1 , -NR1 R2, -C(0)NR1 R2, -SR1 , -S0 2 R1 , -S0 2 NR1 R2, oxo, -C(0)R1 , -C(NH)NH 2 , heterocyclyl, C 3 -C 10 -cycloalkyl, aryl-(d-C 6 -alkyl)-, aryl, trifluoromethyl and trifluoromethoxy,
- heterocyclyl and heteroaryl may in turn be at least monosubtituted with d-C- 6 -alkyl, d-C- ⁇ -alkoxy, halogen, trifluoromethyl, trifluoromethoxy or OH;
- Ar is unsubstituted or at least monosubstituted phenyl, pyridinyl, pyrimidinyl, pyrazolyl, thiophenyl, isoxazolyl, benzo[b]thiophenyl, benzodioxolyl or thiazolo[3,2-b][1 ,2,4]-tiazolyl,
- substituents are selected from halogen, Ci-Cio-alkyl, -OR1 , -C(0)OR1 , -NR1 R2, -C(0)NR1 R2, aryl, heteroaryl, aryl-(d-C 6 -alkyl)-, trifluoromethyl and Trifluoromethoxy, and aryl and heteroaryl may in turn be at least monosubsituted with C ⁇ -C 6 - alkyl, d-C 6 -alkoxy, halogen, trifluoromethyl or OH;
- R1 und R2 are independently from each other
- d-Cio-alkyl unsubstituted or at least monosubstituted d-Cio-alkyl, C 3 -C ⁇ o ⁇ cycloalkyl, aryl, aryl-(C ⁇ -C ⁇ o-alkyl)-, heterocyclyl, heterocyclyl-(C ⁇ -C ⁇ o-alkyl)- or heteroaryl, where the substituents are selected from halogen, d-C 6 -alkyl, d-Ce-alkoxy, NH 2 , (d-C 6 -alkyl)amino-, di(d-C 6 -alkyl)amino-, OH, trifluoromethyl and trifluoromethoxy;
- heteroaryl is a 5 to 10-membered, aromatic, mono- or bicyclic heterocycle containing one or more heteroatoms selected from N, O und S; heteroaryl is preferably imidazolyl, thiophenyl, furanyl, isoxazolyl, pyridinyl, pyrimidinyl, 1 ,2,3,4- tetrahydrochinolinyl, benzoimidazolyl, indolyl or benzodioxolyl;
- aryl is phenyl, indanyl, indenyl or naphthyl; aryl is preferably phenyl or naphthyl.
- heterocyclyl is a 5 to 10-membered, aliphatic, mono- or bicyclic heterocycle containing one or more heteroatoms selected from N, O und S; heterocyclyl is preferably 2-oxo-azepanyl, tetrahydrofuranyl, 1 ,3-dioxolanyl, morpholinyl, piperazinyl or piperidinyl;
- A is A1 ;
- R is unsubstituted or at least monosubstituted d-Cio-alkyl, aryl, aryl-(C ⁇ -C ⁇ o- alkyl)-, heterocyclyl, heterocyclyl-(d-C 10 -alkyl)-, C 3 -C ⁇ o-cycloalkyl, heteroaryl or heteroaryl-(C ⁇ -C ⁇ o-alkyl)-,
- substituents are selected from halogen, C Cio-alkyl, -OR1 , -C(0)OR1 , -NR1 R2, -C(0)NR1 R2, -SR1 , -S0 2 R1 , -S0 2 NR1 R2, oxo,
- -C(0)R1 -C(NH)NH 2 , heterocyclyl, C 3 -C ⁇ 0 -cycloalkyl, aryl-(C C 6 -alkyl)-, aryl, trifluoromethyl and trifluoromethoxy, and aryl, heterocyclyl and heteroaryl may in turn be at least monosubsituted with Ci-Ce-alkyl, C ⁇ -C 6 -alkoxy, halogen, trifluoromethyl, trifluoromethoxy or OH;
- Ar is unsubstituted or at least monosubstituted phenyl, pyridinyl or pyrimidinyl,
- substituents are selected from halogen, Ci-Cio-alkyl, -OR1 , -C(0)OR1 , -NR1 R2, -C(0)NR1 R2, aryl, heteroaryl, aryl-(d-C 6 -alkyl)-, trifluoromethyl and trifluoromethoxy,
- aryl and heteroaryl may in turn be at least monosubsituted with Ci-Ce- alkyl, Ci-C ⁇ -alkoxy, halogen, trifluoromethyl or OH;
- R1 und R2 are independently from each other
- Ci-Cio-alkyl unsubstituted or at least monosubstituted Ci-Cio-alkyl, C 3 -do-cycloalkyl, aryl, aryl-(C ⁇ -C ⁇ o-alkyl)-, heterocyclyl, heterocyclyl-(Ci-Cio-alkyl)- or heteroaryl, where the substituents are selected from halogen, Ci-Ce-alkyl, Ci-Ce-alkoxy, NH 2 , (C ⁇ -C 6 -alkyl)amino-, di(d-C 6 -alkyl)amino-, OH, trifluoromethyl and trifluoromethoxy;
- heteroaryl is a 5 to 10-membered, aromatic, mono- or bicyclic heterocycle containing one or more heteroatoms selected from N, O und S; heteroaryl is preferably imidazolyl, thiophenyl, furanyl, isoxazolyl, pyridinyl, pyrimidinyl, 1 ,2,3,4- tetrahydrochinolinyl, benzoimidazolyl, indolyl or benzodioxolyl;
- aryl is phenyl, indanyl, indenyl or naphthyl; aryl is preferably phenyl or naphthyl.
- heterocyclyl is a 5 to 10-membered, aliphatic, mono- or bicyclic heterocycle containing one or more heteroatoms selected from N, O und S; heterocyclyl is preferably 2-oxo-azepanyl, tetrahydrofuranyl, 1 ,3-dioxolanyl, morpholinyl, piperazinyl or piperidinyl;
- R is unsubstituted or at least monosubstituted aryl-(C ⁇ -C- 6 -alkyl)- or heteroaryl- (Ci-Ce-alkyl)-,
- substituents are selected from halogen, C ⁇ -C 6 -alkyl, -OH, -O-aryl, Ci-Ce-alkoxy, -0-(C ⁇ -C 6 -alkylen)-N(C ⁇ -C 6 -alkyl)2, -C(0)OH, -C(0)0-(C ⁇ -C 6 - alkyl), -NH 2 , -N(C ⁇ -C 6 -alkyl) 2 , -NH(C ⁇ -C 6 -alkyl), -NH(d-C 10 -cycloalkyl), -C(0)NH 2 , -C(0)NH-heteroaryl, -C(0)NH-(Ci-C 6 -alkyl), -S0 2 (d-C e -alkyl),
- aryl, aeterocyclyl and heteroaryl may in turn be at least monosubstituted with d-C 3 -alkyl, C ⁇ -C 3 -alkoxy, fluorine, chlorine, bromine, trifluoromethyl, trifluoromethoxy or OH;
- heteroaryl is imidazolyl, thiophenyl, furanyl, isoxazolyl, pyridinyl, pyrimidinyl, benzoimidazolyl, indolyl or benzodioxolyl;
- aryl is phenyl or naphthyl
- hetrocyclyl is morpholinyl, piperazinyl or piperidinyl;
- A is A1 ;
- Ar is unsubstituted or at least monosubstituted phenyl, pyridin-4-yl or pyrimidin-4-yl,
- substituents are selected from halogen, Ci-Ce-alkyl, -OH, Ci-Ce- alkoxy, -C(0)OH, -C(0)0-(C ⁇ -C 6 -alkyl), -NH 2 , -N(C -C 6 -alkyl) 2 , -NH(d-C 6 - alkyl), -NH(Ci-Cio-cycloalkyl), -NH(heterocyclyl-(d-C 6 -alkyl-)), -NH(aryl-
- C ⁇ -C 6 -alkyl- (C ⁇ -C 6 -alkyl-)), -C(0)NH 2 , -C(0)NH-(d-C 6 -alkyl), aryl, and heteroaryl, and aryl, heterocyclyl and heteroaryl may in turn be at least monosubstituted C ⁇ -C 3 -Alkyl, d-C 3 -alkoxy, fluorine, chlorine, bromine, trifluoromethyl, trifluoromethoxy or OH;
- heteroaryl is pyridinyl or pyrimidinyl
- aryl is phenyl or naphthyl
- hetrocyclyl is morpholinyl, piperazinyl or piperidinyl;
- A is A1 ;
- R is unsubstituted or at least monosubstituted benzyl, phenylethyl-, phenylpropyl-, pyridinylmethyl-, pyridinylethyl- or pyridinylpropyl-,
- substituents are selected from chlorine, bromine, fluorine, trifluoromethyl and carboxy;
- Ar is unsubstituted or at least monosubstituted pyridin-4-yl, pyrimidin-4-yl or phenyl,
- substituents are selected from methylamino-, ethylamino-, propylamino-, butylamino-, hydroxy, methoxy, ethoxy, methyl, ethyl, propyl,
- the convertion with the amine (III) may be carried out using an inert solvent at 0 to 150 °C.
- X is OH
- the compounds of formula (I) may be obtained by acylation of the amine derivatives, either using an acid chloride to be added beforehand or by reaction in the presence of an activating agent.
- the reaction may be carried out by forming an acid chloride according to any methods known to persons skilled in the art or more precisely by the action of oxalyl chloride in toluene, dichloromethane (R.D. MILLER, J. Org. Chem, 56, (4) 1453, (1991)) which, thus formed, will react with the amine (III) in the presence of a base such as pyridine, triethylamine, diisopropylethylamine; the reaction can start at 0°C and when the addition of the acid chloride is complete, the medium is kept stirred at room temperature (G. DAIDONE, Heterocycles, 43, (11), 2385-96, (1996)) or it is heated if necessary.
- a base such as pyridine, triethylamine, diisopropylethylamine
- the reaction may also be carried out in the presence of an activating agent of the carbodiimide type alone (DCC, EDAC) (M. C. DESAI, Tetrahedron Lett., 34, 7685, (1993)) or in the presence of hydroxybenzotriazole and dimethylaminopyridine (J. P. GAMET, Tetrahedron, 40, 1995, (1984), K. BARLOS, J. Org. Chem., 50, 696, (1985)) or according to well known methods of coupling in peptide chemistry (M. BODANSZKY, Principles of Peptide Synthesis; Springer-Verlag, New York, NY, pages 9-58, (1984)) or of forming the amide bond.
- DCC carbodiimide type alone
- EDAC EDAC
- hydroxybenzotriazole and dimethylaminopyridine J. P. GAMET, Tetrahedron, 40, 1995, (1984), K. BARLOS, J. Org. Chem., 50
- the derivatives of formula (II) are obtained by the method described in patent F.R 2481284 and by Y. Shojiro. Chem. Pharm. Bull; 19 (11) p 2354. It is necessary to protect the reactive functional groups.
- the protecting groups are introduced according to any methods known to persons skilled in the art and in particular those described by T.W. GREENE, Protective groups in Organic Synthesis, J. Wiley-lnterscience Publication (1991).
- For the phenols there will be preferably chosen more particularly a benzyl group introduced in the presence of an inorganic base such as sodium carbonate at the reflux temperature of acetone and of acetonitrile (A. R Mac Kenzie, Tetrahedron, 42, 3259, (1986)), which may then be removed by catalytic hydrogenation or more particularly using trifluoroacetic acid under reflux, described in patent W O 9727846.
- the products of general formula (III) may be obtained commercially or by functionalization and protection of the reactive functional groups of commercially available products according to the methods described by Larock, Comprehensive Organic Transformations, VCH, New York, 1999.
- the nitrile functional groups are reduced with hydrogen in the presence of catalysts, BH 3 or more precisely lithium aluminum hydride in solvents such as dioxane or THF (T.M. Koening, Tetrahedron Letters, 35, 1339, (1994)).
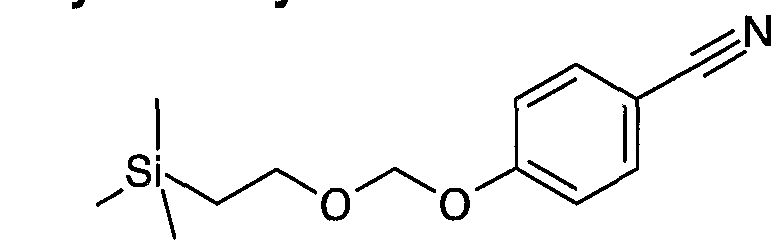
- the phenol functional groups are protected with trimethylsilylethoxymethyl by reacting the starting compound with trimethylsilylethoxymethyl chloride in the presence of sodium hydride in a solvent such as dimethylformamide at room temperature (J. P. WHITTEN, J. Org.
- the derivatives of formula (I) for which the protecting group is an ester can be saponified according to any methods known to persons skilled in the art and in particular by the action of sodium hydroxide on the reflux (L. Anzalone, J. Org. Chem., 50, 2128, (1985).
- the derivatives of formula (I) may be obtained according to route a) by acylating the derivatives of formula (IV) either using an acid chloride, or according to the route b) by acylating the derivatives of formula (IV) or using an anhydride, or according to the route c) by the reaction of an acid in the presence of an activating agent.
- the reaction is carried out in the presence of a base such as pyridine, triethylamine, diisopropylethylamine; the reaction may start at 0°C, and when the addition of the acid chloride is complete, the medium is kept stirred at room temperature (G. DAIDONE, Heterocycles, 43, (11), 2385-96, (1996) or it is heated if necessary.
- a base such as pyridine, triethylamine, diisopropylethylamine
- the reaction is carried out at the reflux temperature of an inert solvent such as xylene or tetrahydrofuran (F. ALBERICIO, Synth. Commun., 31 , (2), 225-32, (2001)) or dichloromethane, (G. PROCTER, Tetrahedron, 51 , (47), 12837-842, (1995)) or in the anhydride itself.
- an inert solvent such as xylene or tetrahydrofuran (F. ALBERICIO, Synth. Commun., 31 , (2), 225-32, (2001)) or dichloromethane, (G. PROCTER, Tetrahedron, 51 , (47), 12837-842, (1995)) or in the anhydride itself.
- the reaction is carried out in the presence of an activating agent of the carbodiimide type alone (DCC, EDAC) (M. C. DESAI, Tetrahedron Lett., 34, 7685, (1993)) or in the presence of hydroxybenzotriazole and dimethylaminopyridine (J. P. GAMET, Tetrahedron, 40, 1995, (1984), K. BARLOS, J. Org. Chem., 50, 696, (1985)) or according to well known methods of coupling in peptide chemistry (M. BODANSZKY, Principles of Peptide Synthesis; Springer- Verlag, New York, NY, pages 9-58, (1984)) or of forming the amide bond.
- DCC carbodiimide type alone
- EDAC EDAC
- hydroxybenzotriazole and dimethylaminopyridine J. P. GAMET, Tetrahedron, 40, 1995, (1984), K. BARLOS, J. Org. Chem., 50, 6
- compounds according to general formula (I) can be prepared by palladium catalyzed coupling according to a reaction of Suzuki (I. Parrot et al., Synthesis; 7; 1999; 1163-1168).
- a compound of formula (IV), where Y1 is halogen, B(OH) or Sn(C C o-alkyl) and Y2 is H or a protecting group, is hereby converted with a compound of formula (V).
- Z may be, for example, B(OH) 2 , B(C ⁇ -C 10 -alkyl) 2 , Sn(C ⁇ -C ⁇ 0 -alkyl) 3 , Zn(C ⁇ -C ⁇ o- alkyl) or halogen.
- Y2 is a protecting group
- said group is removed after the reaction of (IV) and (V) using methods known by a person skilled in the art. All protecting groups known by a person skilled in the art can be used as protecting groups, preferably trimethylsilylethoxymethyl-.
- Pd(triphenylphosphin) 4 Pd-tetrakis-catalyst
- the compounds of formula (I) are isolated and may be purified by known methods, for example by crystallization, chromatography or extraction.
- the use of compounds according to the general formula (I) as pharmaceutical, where the compounds have the above-mentioned preferred, more preferred, even more preferred, even much more preferred, in particular preferred or exceptionally preferred meaning, are also subject of the present invention.
- the compounds of general formula (I) are kinase inhibitors and can therefore be employed for the treatment of diseases, which may result from an abnormal activity of kinases.
- abnormal kinase activity there may be mentioned, for example, that of PI3K, AkT, GSK-3 ⁇ and the like.
- compounds according to the present invention can be used for the inhibition of the kinase GSK-3 ⁇ .
- This effect is particularly relevant for the treatment of metabolic diseases such as type II diabetes or neurodegenerative diseases such as Alzheimer's disease.
- compounds according to the general formula (I) have an inhibitory effect in respect of the phosphorylation of the tau-protein. This effect is particularly relevant for the treatment of neurodegenerative diseases such as Alzheimer's disease.
- diseases which can be treated with the compounds according to the present invention, include: neurodegenerative diseases, strokes, cranial and spinal traumas and peripheral neuropathies, obesity, metabolic diseases, type II diabetes, essential hypertension, atherosclerotic cardiovascular diseases, polycystic ovary syndrome, syndrome X, immunodeficiency or cancer.
- Neurodegenerative diseases are preferably: Alzheimer's disease, Parkinson's disease, frontoparietal dementia, corticobasal degeneration and Pick's disease.
- Compounds according to the present invention are preferably employed for the treatment of metabolic diseases, in particular of type II diabetes.
- the compounds according to the general formula (I) are preferably employed for the treatment of neurodegenerative diseases, in particular of Alzheimer's disease.
- the item treatment also includes prophylaxis, therapy or curing of the above-mentioned diseases.
- compositions for preparing one or more medicaments for prophylaxis and/or treatment of the before-mentioned diseases
- pharmaceutical preparations comprising an effective dose of at least one compound of the formula (I) as well as pharmaceutical preparations comprising an effective dose of at least one compound of the formula (I) for prophylaxis and/or treatment of the before-mentioned diseases
- the amount of a compound according to formula (I) which is required in order to attain the desired biological effect depends on a number of factors, for example the specific compound selected, the intended use, the type of administration and the clinical state of the patient.
- the daily dose is in the range from 0.3 mg to 100 mg (typically from 3 mg to 50 mg) per day per kilogram of body weight, for example 3-10 mg/kg/day.
- An intravenous dose can be, for example, in the range from 0.3 mg to 1.0 mg/kg and can be administered in a suitable manner as an infusion of 10 ng to 100 ng per kilogram per minute.
- Suitable infusion solutions for these purposes may contain, for example, from 0.1 ng to 10 mg, typically from 1 ng to 10 mg per milliliter.
- Individual doses may contain, for example, from 1 mg to 10 g of the active compound.
- ampoules for injections can contain, for example, from 1 mg to 100 mg
- orally administerable individual dose formulations such as, for example, tablets or capsules can contain, for example, from 1.0 to 1000 mg, typically from 10 to 600 mg.
- the abovementioned masses relate to the mass of the free compound on which the salt is based.
- the compound used for the prophylaxis or therapy of the abovementioned conditions may be the compounds according to formula (I) themselves, but they are preferably present in the form of a pharmaceutical composition together with an acceptable carrier.
- the carrier must be naturally acceptable, in the sense that it is compatible with the other ingredients of said composition and is not harmful to the patient's health.
- the carrier may be a solid or a liquid or both and is preferably formulated with the compound as an individual dose, for example as a tablet which may contain from 0.05% to 95% by weight of the active compound.
- Further pharmaceutically active substances may also be present, including further compounds according to formula (I).
- the pharmaceutical compositions of the invention may be prepared according to any of the known pharmaceutical methods which essentially comprise mixing the ingredients with pharmacologically acceptable carriers and/or excipients.
- the pharmaceutical preparations can also contain additives.
- additives can be employed, for example: fillers, binders, lubricants, wetting agents, stabilizers, emulsifiers, dispersants, preservatives, sweeteners, colorants, flavorings, aromatizers, thickeners, diluents, buffer substances, solvents, solubilizers, agents for achieving a depot effect, salts for altering the osmotic pressure, coating agents or antioxidants.
- compositions of the invention may be in form of a pill, tablet, lozenge, coated tablet, granule, capsule, hard or soft gelatin capsule, aqueous solution, alcoholic solution, oily solution, syrup, emulsion suspension pastille suppository, solution for injection or infusion, ointment, tincture, cream, lotion, powder, spray, transdermal therapeutic systems, nasal spray, aerosol mixture, microcapsule, implant, rod or plaster.
- compositions of the invention are those which are suitable for oral, rectal, topical, peroral (e.g. sublingual) and parenteral (e.g. subcutaneous, intramuscular, intradermal or intravenous) administration, although the most suitable manner of administration depends in each individual case on the nature and severity of the condition to be treated and on the nature of the compound according to formula (I) used in each case.
- Sugar-coated formulations and sugar- coated delayed-release formulations are included within the scope of the invention.
- Suitable pharmaceutical compounds for oral administration may be present in separate units as, for example, capsules, cachets, lozenges or tablets, which in each case contain a particular amount of the compound according to formula (I); as powders ( gelatin capsules or cachets) or granules; as solution or suspension in an aqueous or nonaqueous liquid; or as an oil-in-water or water-in-oil emulsion.
- said compositions can be prepared according to any suitable pharmaceutical method which includes a step in which the active compound and the carrier (which may comprise one or more additional components) are contacted.
- compositions are prepared by uniform and homogeneous mixing of the active compound with a liquid and/or finely dispersed solid carrier, after which the product is shaped, if necessary.
- a tablet for example, may be prepared by pressing or shaping a powder or granules of the compound, where appropriate with one or more additional components. Pressed tablets can be prepared by tableting the compound in free-flowing form, for example a powder or granules, mixed, where appropriate, with a binder, lubricant, inert diluent and/or one or more surface active/dispersing agents in a suitable machine.
- Shaped tablets can be prepared by shaping the pulverulent compound, moistened with an inert liquid diluent, in a suitable machine.
- diluents can be used, for example, starch, cellulose, saccharose, lactose or silica.
- the pharmaceutical compositions of the invention may also comprise substances other than diluents, for example one or more lubricants such as magnesium stearate or talc, a coloring, a coating (sugar-coated tablets) or a varnish.
- compositions which are suitable for peroral (sublingual) administration include lozenges which contain a compound according to formula (I) with a flavoring, usually sucrose and gum arabic or tragacanth, and pastilles which comprise the compound in an inert base such as gelatin and glycerol or sucrose and gum arabic.
- Suitable pharmaceutical compositions for parenteral administration preferably comprise sterile aqueous preparations of a compound according to formula (I) which are preferably isotonic with the blood of the intended recipient. These preparations are preferably administered intravenously, although they may also be administered subcutaneously, intramuscularly or intradermally as an injection. Said preparations may preferably be prepared by mixing the compound with water and rendering the obtained solution sterile and isotonic with the blood. Injectable compositions of the invention generally contain from 0.1 to 5% by weight of the active compound.
- sterile compositions for parenteral administration may be preferably solutions which are aqueous or non aqueous, suspensions or emulsions.
- solvent or vehicle there may be used water, propylene glycol, polyethylene glycol, vegetable oils, in particular olive oil, organic esters for injection, for example ethyl oleate or other suitable organic solvents.
- These compositions may also contain adjuvants, in particular wetting, isotonizing, emulsifying, dispersing and stabilizing mediums.
- the sterilization may be carried out in several ways, for example by an aseptic filtration, by incorporating sterilizing agents into the composition, by irradiation or by heating. They may also be prepared in the form of sterile solid compositions which may be dissolved at the time of use in sterile water or in any other sterile medium for injection.
- Suitable pharmaceutical compositions for rectal administration are preferably present as individual dose suppositories. These may be prepared by mixing a compound according to formula (I) with one or more conventional solid carriers, for example cocoa butter, and shaping the resulting mixture.
- Suitable pharmaceutical compositions for topical application to the skin are preferably present as ointment, cream, lotion, paste, spray, aerosol or oil.
- Carriers which may be used are petroleum jelly, lanolin, polyethylene glycols, alcohols and combinations of two or more of these substances.
- the active compound is present at a concentration of from 0.1 to 15%, for example from 0.5 to 2%, by weight of the composition.
- Transdermal administration is also possible.
- Suitable pharmaceutical compositions for transdermal administration may be present as individual patches which are suitable for long-term close contact with the epidermis of the patient.
- patches suitably contain the active compound in an optionally buffered aqueous solution, dissolved and/or dispersed in an adhesive or dispersed in a polymer.
- a suitable active compound concentration is from approx. 1 % to 35%, preferably approx. 3% to 15%.
- a particular possibility is the release of the active compound by electro- transport or iontophoresis, as described, for example, in Pharmaceutical Research, 2(6): 318 (1986).
- Tablets containing a dose of 50 mg of active product and having the following composition are prepared according to the usual technique:
- a solution for injection containing 10 mg of active product and having the following composition is prepared:
- Another subject of the present invention is the combination of compounds of the formula (I) with other pharmaceutically active substances not covered by formula
- the compounds of the formula (I) are distinguished by beneficial actions on the metabolism of lipids, and they are particularly suitable for weight reduction and, after weight reduction, for maintaining a reduced weight in mammals and as anorectic agents.
- the compounds are distinguished by their low toxicity and their few side effects.
- the compounds may be employed alone or in combination with other weight- reducing or anorectic active compounds.
- Further anorectic active compounds of this kind are mentioned, for example, in the Rote Liste, Chapter 01 under weight- reducing agents/appetite suppressants, and may also include those active compounds which increase the energy turnover of the organism and thus lead to weight reduction or else those which influence the general metabolism of said organism such that increased calorie intake does not cause an enlargement of the fat depots and a normal calorie intake causes a reduction in the fat depots of said organism.
- the compounds are suitable for the prophylaxis and, in particular, for the treatment of problems of excess weight or obesity.
- the compounds of formula (I) have a beneficial effect on the glucose metabolism, they particularly lower the blood-sugar level and can be used for treatment of type I and type II diabetes.
- the compounds can therefore be used alone or in combination with other blood-sugar lowering active compounds (antidiabetics).
- the compounds of the formula I may be administered in combination with one or more further pharmacologically active substances which may be selected, for example, from the group consisting of antidiabetics, antiadipose agents, blood-pressure-lowering active compounds, lipid reducers and active compounds for the treatment and/or prevention of complications caused by diabetes or associated with diabetes.
- Suitable antidiabetics include insulins, amylin, GLP-1 and GLP-2 derivatives such as, for example, those disclosed by Novo Nordisk A/S in WO 98/08871 and also oral hypoglycemic active compounds.
- Said oral hypoglycemic active compounds preferably include sulfonyl ureas, biguanidines, meglitinides, oxadiazolidinediones, thiazolidinediones, glucosidase inhibitors, glucagon receptor antagonists, GLP-1 agonists, potassium channel openers such as, for example, those disclosed by Novo Nordisk A/S in WO 97/26265 and WO 99/03861 , insulin sensitizers, activators of insulin receptor kinase, inhibitors of liver enzymes involved in the stimulation of gluconeogenesis and/or glycogenolysis, for example glycogen phosphorase inhibitors, modulators of glucose uptake and glucose elimination, lipid metabolism-modifying compounds such as antihyperlipidemic active compounds and antilipidemic active compounds, for example HMGCoA-reductase inhibitors, inhibitors of cholesterol transport/cholesterol uptake, inhibitors of the reabsorption of bile acid or inhibitors of microsomal
- the present compounds are administered in combination with insulin.
- the compounds of the invention are administered in combination with a sulfonylurea such as, for example, tolbutamide, glibenclamide, glimepiride, glipizide, gliquidone, glisoxepide, glibornuride or gliclazide.
- a sulfonylurea such as, for example, tolbutamide, glibenclamide, glimepiride, glipizide, gliquidone, glisoxepide, glibornuride or gliclazide.
- the compounds of the present invention are administered in combination with a biguanidine such as, for example, metformin.
- the compounds of the present invention are administered in combination with a meglitinide such as, for example, repaglinide.
- the compounds of the present invention are administered in combination with a thiazolidinedione such as, for example, troglitazone, ciglitazone, pioglitazone, rosiglitazone or the compounds disclosed by Dr. Reddy's Research Foundation in WO 97/41097, in particular 5-[[4-[(3,4-dihydro-3-methyl-4-oxo-2- quinazolinylmethoxy]phenyl]methyl]-2,4-thiazolidinedione.
- the compounds of the present invention are administered in combination with an -glucosidase inhibitor such as, for example, miglitol or acarbose.
- the compounds of the present invention are administered in combination with an active compound which acts on the ATP-dependent potassium channel of the beta cells, such as, for example, tolbutamide, glibenclamide, glimepiride, glipizide, gliclazide or repaglinide.
- an active compound which acts on the ATP-dependent potassium channel of the beta cells such as, for example, tolbutamide, glibenclamide, glimepiride, glipizide, gliclazide or repaglinide.
- the compounds of the present invention are administered in combination with an antihyperlipidemic active compound or an antilipidemic active compound such as, for example, cholestyramine, colestipol, clofibrate, fenofibrate, gemfibrozil, lovastatin, pravastatin, simvastatin, atorvastatin, cerivastatin, fluvastatin, probucol, ezetimibe or dextrothyroxine.
- an antihyperlipidemic active compound or an antilipidemic active compound such as, for example, cholestyramine, colestipol, clofibrate, fenofibrate, gemfibrozil, lovastatin, pravastatin, simvastatin, atorvastatin, cerivastatin, fluvastatin, probucol, ezetimibe or dextrothyroxine.
- the compounds of the present invention are administered in combination with more than one of the aforementioned compounds, for example in combination with a sulfonylurea and metformin, a sulfonylurea and acarbose, repaglinide and metformin, insulin and a sulfonylurea, insulin and metformin, insulin and troglitazone, insulin and lovastatin, etc.
- the compounds of the invention may be administered in combination with one or more antiadipose agents or appetite-controlling active compounds.
- Such active compounds may be selected from the group consisting of CART agonists, NPY antagonists, MC4 agonists, orexin antagonists, H3 agonists, TNF agonists, CRF agonists, CRF BP antagonists, urocortin agonists, ⁇ 3 agonists, MSH (melanocyte-stimulating hormone) agonists, CCK agonists, serotonin re- uptake inhibitors, mixed serotonin and noradrenalin reuptake inhibitors, 5HT modulators, MAO inhibitors, bombesin agonists, galanin antagonists, growth hormone, growth-hormone-releasing compounds, TRH agonists, uncoupling protein 2 or 3 modulators, leptin agonists, dopamine agonists (bromocriptine, doprexin), lipase/amylase inhibitors, cannabinoid receptor 1 antagonists, modulators of acylation-stimulating protein (ASP), PPAR modulators, RXR modulators
- the antiadipose agent is leptin or modified leptin. In another embodiment, the antiadipose agent is dexamphetamine or amphetamine. In another embodiment, the antiadipose agent is fenfluramine or dexfenfluramine. In yet another embodiment, the antiadipose agent is sibutramine or the mono- and bis-demethylated active metabolite of sibutramine. In another embodiment, the antiadipose agent is orlistate. In another embodiment, the antiadipose agent is mazindol, diethylpropione or phentermine.
- antihypertensive active compounds examples include betablockers such as alprenolol, atenol, timolol, pindolol, propanolol and metoprolol, ACE (angiotensin-converting enzyme) inhibitors such as, for example, benazepril, captopril, enalapril, fosinopril, lisinopril, quinapril and rampril, calcium channel blockers such as nifedipine, felodipine, nicardipine, isradipine, nimodipine, diltiazem and verapamil, and also alphablockers such as doxazosin, urapidil, prazosin and terazosin.
- betablockers such as alprenolol, atenol, timolol, pindolol, propanolol and metoprolol
- ACE angiotens
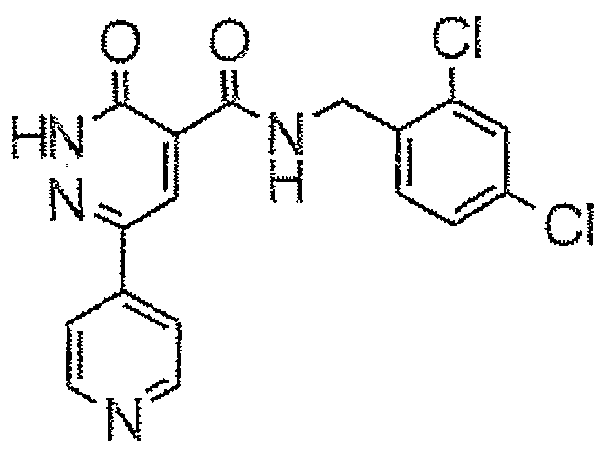
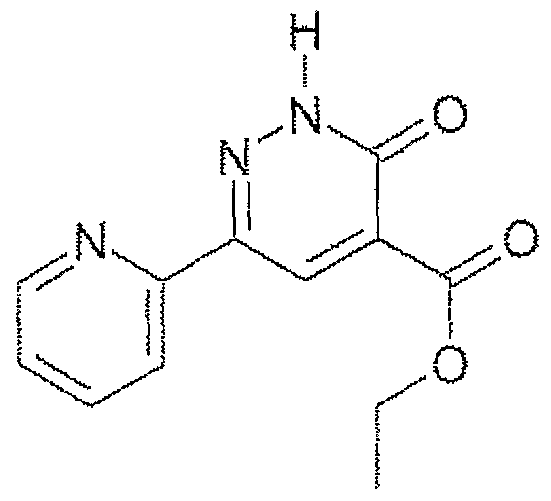
- 0.14 g of 1 -hydroxybenzotriazole, 0.14 cm 3 of 2,4-dichlorobenzylamine and 0.14 cm 3 of triethylamine are added to 0.2 g of 3-oxo-6-phenyl-2,3-dihydro- pyridazine-4-carboxylic acid prepared as described by Y. Shojiro et al., Chem. Pharm. Bull; 19, (11), p. 2354, in 10 cm 3 of dichloromethane.
- 0.2 g of 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride is then added. The mixture is stirred for 48 hours at 19°C. 10 cm 3 of distilled water are added.
- the organic phase is washed again with 3 times 10 cm 3 of aqueous normal hydrochloric acid solution and then with 10 cm 3 of saturated aqueous sodium chloride solution.
- the organic phase is then dried over magnesium sulfate.
- the mixture is filtered through a sinter funnel and then evaporated under reduced pressure (2 kPa; 45°C).
- the residue is taken up with 10 cm 3 of diisopropyl ether.
- the insoluble material is filtered off through a sinter funnel and then rinsed again with 10 cm 3 of diisopropyl ether.
- reaction mixture is then poured onto a solution of 10 cm 3 of dichloromethane containing 0.19 cm 3 of triethylamine and 0.21 cm 3 of 2,4-dichlorobenzylamine.
- the reaction medium is stirred for 12 hours at 19°C and then filtered through a sinter funnel, rinsed with 10 cm 3 of dichloromethane, 10 cm 3 of distilled water and with 10 cm 3 of aqueous normal hydrochloric acid solution.
- the organic phase is dried over magnesium sulfate, filtered through a sinter funnel and then evaporated under reduced pressure (2 kPa; 45°C).
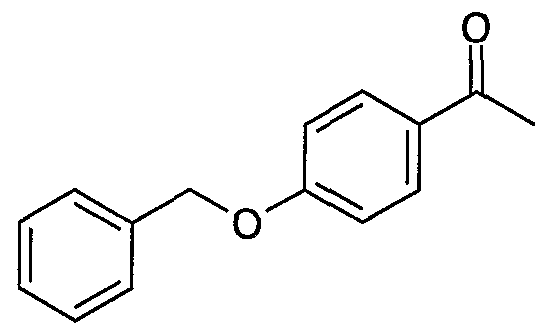
- the solid residue is triturated in 20 cm 3 of pentane, suction-filtered through a sinter funnel and oven- dried under reduced pressure (10 kPa; 20°C). 23.1 g of 4-benzyloxyacetophenone are obtained in the form of a white solid melting at 99°C.
- the product obtained is triturated in 150 cm 3 of ethanol, filtered through a sinter funnel, washed with twice 50 cm 3 of ethanol and 50 cm 3 of isopropyl ether to give, after drying under reduced pressure (2 kPa; 55°C), 5.6 g of diethyl hydroxy[2-(4-benzyloxyphenyl)-2-oxoethyl]malonate melting at 80°C.
- N-(2,4-dichlorobenzyl)-3-oxo-6-[4-(hydroxy)phenyl]-2,3-dihydropyri- dazine-4-carboxamide is obtained in the form of a yellow solid melting at a temperature above 260°C.
- Oxalyl chloride is added to a solution a 8.73 g of 6-chloro-3-oxo-2,3- dihydro-pyridazine-4-carboxylic acid and 1 ml DMF in 250 ml THF at 5- 10°C and the mixture is stirred at room temperature for 2 h. Afterwards, it is evaporated to dryness, the residue dissolved in 450 ml THF and 13.8 g potassium carbonate and a solution of 7.2 g 4-chloro-benzyl amide in THF are added. The solvent is distilled off after 2 h of stirring at room temperature, the residue suspended in 100 ml water and a pH of 6.4 is adjusted. The obtained precipitate is sucked off, suspended again in 50 ml
- the cortex sections having a thickness of 300 ⁇ m are prepared from 8-10- week old male OFA rats (Iffa-Credo), sacrificed by decapitation. They are incubated in 5 ml of DMEM medium containing pyruvate and glucose 4.5 g/l at 37°C for 40 min. The sections are then washed twice with the medium, distributed into microtubes (50 ⁇ l in 500 ⁇ l of medium with or without test compounds), and incubated at 37°C, with stirring. Two hours later, the experiment is stopped by centrifugation.
- the sections are washed, sonicated and centrifuged at 18300 g, for 15 min at 4°C.
- concentration of proteins in supernatant is determined by a commercial assay (BCA Protein Assay, Pierce) based on the Lowry method.
- the samples denatured beforehand for 10 min at 70°C, are separated on a 4-12% Bis-Tris vertical gel in the presence of MOPS-SDS buffer and electrotransferred onto nitrocellulose membrane.
- Immunolabeling is performed with their monoclonal antibody AD2 which specifically recognizes the phosphorylated epitopes Ser396/404 of the Tau protein.
- the immunoactive proteins are visualized by adding a second antibody directed against the mouse ⁇ y a s and coupled to peroxidase and a chemiluminescent substrate.
- the autoradiograms obtained are finally quantified using the 'GeneTools' software from Syngene (GeneGnome, Ozyme) in order to determine an IC 50 .
- the compounds of formula (I) have a very advantageous activity and in particular some compounds have an IC 50 of less than 100 ⁇ M.
- GSK- ⁇ activity is measured using human recombinant GSK-3B and a primed (pre-phosphorylated) substrate peptide (derived from glycogen synthase and containing the phosphorylation sites 3a, b, and c) on basis of the AlphaScreen technology in 384-well plate format (small volume plate, white, GREINER).
- a primed (pre-phosphorylated) substrate peptide derived from glycogen synthase and containing the phosphorylation sites 3a, b, and c
- 384-well plate format small volume plate, white, GREINER
- phospho-glycogen synthase peptide 34 nM in kinase buffer (20 mM Hepes, pH 7,4, 10 mM MgCI, 200 mM EDTA, 1 mM DTT, 0,1 mg/ml BSA, 10 ⁇ M ATP) are incubated at room temperature for 60 min.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Immunology (AREA)
- Psychiatry (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Psychology (AREA)
- Child & Adolescent Psychology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Description
























































































Claims




Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MXPA05005270A MXPA05005270A (en) | 2002-11-19 | 2003-11-19 | Pyridazinone derivatives as gsk-3beta inhibitors. |
| AU2003283414A AU2003283414A1 (en) | 2002-11-19 | 2003-11-19 | Pyridazinone derivatives as gsk-3beta inhibitors |
| DE60315354T DE60315354T2 (en) | 2002-11-19 | 2003-11-19 | PYRIDAZINONE DERIVATIVES AS GSK-3BETA HEMMER |
| EP03775372A EP1581505B1 (en) | 2002-11-19 | 2003-11-19 | Pyridazinone derivatives as gsk-3beta inhibitors |
| CA002506022A CA2506022A1 (en) | 2002-11-19 | 2003-11-19 | Pyridazinone derivatives as gsk-3beta inhibitors |
| BR0316720-8A BR0316720A (en) | 2002-11-19 | 2003-11-19 | Pyridazinone Derivatives as gsk-3beta Inhibitors |
| JP2004552660A JP2006509748A (en) | 2002-11-19 | 2003-11-19 | Pyridazinone derivatives as GSK-3 beta inhibitors |
| NO20052887A NO20052887L (en) | 2002-11-19 | 2005-06-14 | Pyridazinone derivatives such as GSK-3Beta inhibitors. |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0214443A FR2847253B1 (en) | 2002-11-19 | 2002-11-19 | NOVEL DERIVATIVES OF PYRIDAZINONES AS MEDICAMENTS AND PHARMACEUTICAL COMPOSITIONS COMPRISING THEM |
| FR0214443 | 2002-11-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004046117A1 true WO2004046117A1 (en) | 2004-06-03 |
Family
ID=32187711
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2003/012949 WO2004046130A1 (en) | 2002-11-19 | 2003-11-19 | Pyridazinone derivatives as cdk2-inhibitors |
| PCT/EP2003/012950 WO2004046117A1 (en) | 2002-11-19 | 2003-11-19 | Pyridazinone derivatives as gsk-3beta inhibitors |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2003/012949 WO2004046130A1 (en) | 2002-11-19 | 2003-11-19 | Pyridazinone derivatives as cdk2-inhibitors |
Country Status (19)
| Country | Link |
|---|---|
| EP (2) | EP1581505B1 (en) |
| JP (1) | JP2006509748A (en) |
| KR (1) | KR20050083918A (en) |
| CN (1) | CN1741999A (en) |
| AR (2) | AR042202A1 (en) |
| AT (2) | ATE369356T1 (en) |
| AU (2) | AU2003283414A1 (en) |
| BR (1) | BR0316720A (en) |
| CA (2) | CA2518917A1 (en) |
| CO (1) | CO5700722A2 (en) |
| DE (2) | DE60315516T2 (en) |
| FR (1) | FR2847253B1 (en) |
| MA (1) | MA27814A1 (en) |
| MX (1) | MXPA05005270A (en) |
| NO (1) | NO20052887L (en) |
| PL (1) | PL376129A1 (en) |
| RU (1) | RU2005119151A (en) |
| TW (2) | TW200418478A (en) |
| WO (2) | WO2004046130A1 (en) |
Cited By (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005085230A1 (en) * | 2004-03-02 | 2005-09-15 | Sanofi-Aventis Deutschland Gmbh | 4-benzimidazol-2-yl-pyridazine-3-one-derivatives, production and use thereof in medicaments |
| WO2005111018A1 (en) | 2004-05-18 | 2005-11-24 | Sanofi-Aventis Deutschland Gmbh | Pyridazinone derivatives, methods for producing them and their use as pharmaceuticals |
| WO2007127475A2 (en) * | 2006-04-28 | 2007-11-08 | Northwestern University | Pyridazines for demyelinating diseases and neuropathic pain |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| US7470689B2 (en) | 2004-03-02 | 2008-12-30 | Aventis Pharma S.A. | 4-benzimidazol-2-ylpyridazin-3-one derivatives |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2010099217A1 (en) | 2009-02-25 | 2010-09-02 | Braincells, Inc. | Modulation of neurogenesis using d-cycloserine combinations |
| WO2010101849A1 (en) * | 2009-03-02 | 2010-09-10 | Irm Llc | N- (hetero)aryl, 2- (hetero)aryl-substituted acetamides for use as wnt signaling modulators |
| EP2258357A2 (en) | 2005-08-26 | 2010-12-08 | Braincells, Inc. | Neurogenesis with acetylcholinesterase inhibitor |
| EP2275096A2 (en) | 2005-08-26 | 2011-01-19 | Braincells, Inc. | Neurogenesis via modulation of the muscarinic receptors |
| US7888357B2 (en) | 2001-08-31 | 2011-02-15 | Northwestern University | Anti-inflammatory and protein kinase inhibitor compositions and related methods for downregulation of detrimental cellular responses and inhibition of cell death |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| EP2314289A1 (en) | 2005-10-31 | 2011-04-27 | Braincells, Inc. | Gaba receptor mediated modulation of neurogenesis |
| WO2011063115A1 (en) | 2009-11-19 | 2011-05-26 | Braincells Inc. | Combination of nootropic agent with one or more neurogenic or neurogenic sensitizing agents for stimulating or increasing neurogenesis |
| WO2011091033A1 (en) | 2010-01-20 | 2011-07-28 | Braincells, Inc. | Modulation of neurogenesis by ppar agents |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| EP2377530A2 (en) | 2005-10-21 | 2011-10-19 | Braincells, Inc. | Modulation of neurogenesis by PDE inhibition |
| EP2377531A2 (en) | 2006-05-09 | 2011-10-19 | Braincells, Inc. | Neurogenesis by modulating angiotensin |
| US8063047B2 (en) | 2004-11-02 | 2011-11-22 | Centre National De La Recherche Scientifique (Cnrs) | Pyridazine compounds and methods |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012003189A1 (en) * | 2010-06-29 | 2012-01-05 | Irm Llc | Compositions and methods for modulating the wnt signaling pathway |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| US8158627B2 (en) | 2006-04-28 | 2012-04-17 | Northwestern University | Compositions and treatments using pyridazine compounds and cholinesterase inhibitors |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| US8367672B2 (en) | 2004-11-02 | 2013-02-05 | Universite De Strasbourg | Pyridazine compounds, compositions and methods |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| AU2012203023C1 (en) * | 2009-03-02 | 2013-11-07 | Novartis Ag | N-(hetero)aryl, 2- (hetero)aryl-substituted acetamides for use as Wnt signaling modulators |
| US9408845B2 (en) | 2006-04-28 | 2016-08-09 | Northwestern University | Formulations containing pyridazine compounds |
| US10941160B2 (en) | 2013-10-18 | 2021-03-09 | Celgene Quanticel Research, Inc. | Bromodomain inhibitors |
| US11040035B2 (en) | 2016-05-25 | 2021-06-22 | Bayer Pharma Aktiengesellschaft | 3-oxo-2,6-diphenyl-2,3-dihydropyridazine-4-carboxamides |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1598348A1 (en) | 2004-05-18 | 2005-11-23 | Aventis Pharma Deutschland GmbH | Novel pyridazinone derivatives as inhibitors of CDK2 |
| AU2005329423A1 (en) | 2004-09-20 | 2006-09-28 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-CoA desaturase inhibitors |
| BRPI0515478A (en) | 2004-09-20 | 2008-07-22 | Xenon Pharmaceuticals Inc | heterocyclic derivatives and their use as mediators of stearoyl coa desaturase |
| US7777036B2 (en) | 2004-09-20 | 2010-08-17 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as therapeutic agents |
| MX2007003318A (en) | 2004-09-20 | 2007-05-18 | Xenon Pharmaceuticals Inc | Heterocyclic derivatives for the treatment of diseases mediated by stearoyl-coa desaturase enzymes. |
| AR051202A1 (en) | 2004-09-20 | 2006-12-27 | Xenon Pharmaceuticals Inc | HETEROCICLIC DERIVATIVES AND THEIR USE AS INHIBITORS OF ESTEAROIL-COA DESATURASA |
| TW200626154A (en) | 2004-09-20 | 2006-08-01 | Xenon Pharmaceuticals Inc | Heterocyclic derivatives and their use as therapeutic agents |
| EP2316458A1 (en) | 2004-09-20 | 2011-05-04 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives for inhibiting human stearoyl-coa-desaturase |
| CN101208089A (en) | 2005-06-03 | 2008-06-25 | 泽农医药公司 | Aminothiazole derivatives as human stearoyl-CoA desaturase inhibitors |
| US9212146B2 (en) | 2008-03-18 | 2015-12-15 | Shanghai Institute Of Materia Medica, Chinese Academy Of Sciences | Substituted pyridazinones for the treatment of tumors |
| CN101538245B (en) | 2008-03-18 | 2011-02-16 | 中国科学院上海药物研究所 | One-class pyridazinone compounds and preparation method and application thereof |
| GB201201100D0 (en) * | 2012-01-20 | 2012-03-07 | Medical Res Council | Polypeptides and methods |
| CN106458910B (en) * | 2014-02-19 | 2020-11-06 | 亚倍生技制药公司 | Mitochondrial aldehyde dehydrogenase 2(ALDH2) binding polycyclic amides and their use for treating cancer |
| JOP20190193A1 (en) * | 2017-02-09 | 2019-08-08 | Bayer Pharma AG | 2-heteroaryl-3-oxo-2,3-dihydropyridazine-4-carboxamides for the treatment of cancer |
| EP3713922A1 (en) * | 2017-11-21 | 2020-09-30 | Bayer Aktiengesellschaft | Sulphur substituted 3-oxo-2,3-dihydropyridazine-4-carboxamides |
| CN111533697B (en) * | 2020-05-07 | 2023-03-31 | 成都大学 | 4-aminopyridazinone compound and preparation method thereof |
| CN111393374A (en) * | 2020-05-08 | 2020-07-10 | 张建蒙 | Oxo-dihydropyridazine derivative and application thereof in antitumor drugs |
| CN111533735A (en) * | 2020-05-08 | 2020-08-14 | 张建蒙 | Oxo-dihydropyridazine thiazole derivative and application thereof in antitumor drugs |
| WO2022113003A1 (en) | 2020-11-27 | 2022-06-02 | Rhizen Pharmaceuticals Ag | Cdk inhibitors |
| WO2022149057A1 (en) | 2021-01-05 | 2022-07-14 | Rhizen Pharmaceuticals Ag | Cdk inhibitors |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5418233A (en) * | 1991-10-18 | 1995-05-23 | Karl Thomae Gmbh | Heterobiarly derivatives and pharmaceutical compositions thereof |
| JPH09216883A (en) * | 1996-02-09 | 1997-08-19 | Fujisawa Pharmaceut Co Ltd | Pyrazolopyridine compound and medicine containing the same compound |
| EP1061077A1 (en) * | 1998-03-02 | 2000-12-20 | Kowa Co., Ltd. | Novel pyridazine derivatives and drugs containing the same as the active ingredient |
| WO2002022587A1 (en) * | 2000-09-18 | 2002-03-21 | Eisai Co., Ltd. | Pyridazinones and triazinones and medicinal use thereof |
| WO2002050065A2 (en) * | 2000-12-21 | 2002-06-27 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE4127404A1 (en) * | 1991-08-19 | 1993-02-25 | Thomae Gmbh Dr K | CYCLIC IMINODERIVATES, MEDICAMENTS CONTAINING SUCH COMPOUNDS AND METHOD FOR THE PRODUCTION THEREOF |
| AU2010399A (en) * | 1997-12-24 | 1999-07-19 | Affymetrix, Inc. | Methods of using chemical libraries to search for new kinase inhibitors |
| US6440959B1 (en) * | 1999-04-21 | 2002-08-27 | Hoffman-La Roche Inc. | Pyrazolobenzodiazepines |
-
2002
- 2002-11-19 FR FR0214443A patent/FR2847253B1/en not_active Expired - Fee Related
-
2003
- 2003-11-19 EP EP03775372A patent/EP1581505B1/en not_active Expired - Lifetime
- 2003-11-19 EP EP03811383A patent/EP1611121B1/en not_active Expired - Lifetime
- 2003-11-19 CN CNA2003801050578A patent/CN1741999A/en active Pending
- 2003-11-19 JP JP2004552660A patent/JP2006509748A/en active Pending
- 2003-11-19 DE DE60315516T patent/DE60315516T2/en not_active Expired - Lifetime
- 2003-11-19 RU RU2005119151/04A patent/RU2005119151A/en not_active Application Discontinuation
- 2003-11-19 DE DE60315354T patent/DE60315354T2/en not_active Expired - Lifetime
- 2003-11-19 AT AT03811383T patent/ATE369356T1/en not_active IP Right Cessation
- 2003-11-19 AR ARP030104278A patent/AR042202A1/en unknown
- 2003-11-19 AT AT03775372T patent/ATE368651T1/en not_active IP Right Cessation
- 2003-11-19 KR KR1020057009083A patent/KR20050083918A/en not_active Application Discontinuation
- 2003-11-19 AR ARP030104280A patent/AR042089A1/en unknown
- 2003-11-19 AU AU2003283414A patent/AU2003283414A1/en not_active Abandoned
- 2003-11-19 TW TW092132354A patent/TW200418478A/en unknown
- 2003-11-19 MX MXPA05005270A patent/MXPA05005270A/en unknown
- 2003-11-19 AU AU2003296586A patent/AU2003296586A1/en not_active Abandoned
- 2003-11-19 WO PCT/EP2003/012949 patent/WO2004046130A1/en active IP Right Grant
- 2003-11-19 CA CA002518917A patent/CA2518917A1/en not_active Abandoned
- 2003-11-19 CA CA002506022A patent/CA2506022A1/en not_active Abandoned
- 2003-11-19 BR BR0316720-8A patent/BR0316720A/en not_active Application Discontinuation
- 2003-11-19 TW TW092132459A patent/TW200503727A/en unknown
- 2003-11-19 PL PL03376129A patent/PL376129A1/en unknown
- 2003-11-19 WO PCT/EP2003/012950 patent/WO2004046117A1/en active IP Right Grant
-
2005
- 2005-05-17 MA MA28287A patent/MA27814A1/en unknown
- 2005-05-18 CO CO05048069A patent/CO5700722A2/en not_active Application Discontinuation
- 2005-06-14 NO NO20052887A patent/NO20052887L/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5418233A (en) * | 1991-10-18 | 1995-05-23 | Karl Thomae Gmbh | Heterobiarly derivatives and pharmaceutical compositions thereof |
| JPH09216883A (en) * | 1996-02-09 | 1997-08-19 | Fujisawa Pharmaceut Co Ltd | Pyrazolopyridine compound and medicine containing the same compound |
| EP1061077A1 (en) * | 1998-03-02 | 2000-12-20 | Kowa Co., Ltd. | Novel pyridazine derivatives and drugs containing the same as the active ingredient |
| WO2002022587A1 (en) * | 2000-09-18 | 2002-03-21 | Eisai Co., Ltd. | Pyridazinones and triazinones and medicinal use thereof |
| WO2002050065A2 (en) * | 2000-12-21 | 2002-06-27 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
Non-Patent Citations (1)
| Title |
|---|
| PATENT ABSTRACTS OF JAPAN vol. 1997, no. 12 25 December 1997 (1997-12-25) * |
Cited By (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7888357B2 (en) | 2001-08-31 | 2011-02-15 | Northwestern University | Anti-inflammatory and protein kinase inhibitor compositions and related methods for downregulation of detrimental cellular responses and inhibition of cell death |
| US8088774B2 (en) | 2001-08-31 | 2012-01-03 | Northwestern University | Anti-inflammatory and protein kinase inhibitor compositions and related methods for downregulation of detrimental cellular responses and inhibition of cell death |
| WO2005085230A1 (en) * | 2004-03-02 | 2005-09-15 | Sanofi-Aventis Deutschland Gmbh | 4-benzimidazol-2-yl-pyridazine-3-one-derivatives, production and use thereof in medicaments |
| US7754713B2 (en) | 2004-03-02 | 2010-07-13 | Aventis Pharma S.A. | 4-benzimidazol-2-ylpyridazin-3-one derivatives |
| US7470689B2 (en) | 2004-03-02 | 2008-12-30 | Aventis Pharma S.A. | 4-benzimidazol-2-ylpyridazin-3-one derivatives |
| US7727989B2 (en) | 2004-03-02 | 2010-06-01 | Sanofi-Aventis Deutschland Gmbh | 4-benzimidazol-2-yl-pyridazine-3-one-derivatives, production and use thereof in medicaments |
| US7709466B2 (en) | 2004-05-18 | 2010-05-04 | Sanofi-Aventis Deutschland Gmbh | Pyridazinone derivatives, methods for their production and their use as pharmaceuticals |
| WO2005111018A1 (en) | 2004-05-18 | 2005-11-24 | Sanofi-Aventis Deutschland Gmbh | Pyridazinone derivatives, methods for producing them and their use as pharmaceuticals |
| US8367672B2 (en) | 2004-11-02 | 2013-02-05 | Universite De Strasbourg | Pyridazine compounds, compositions and methods |
| US9527819B2 (en) | 2004-11-02 | 2016-12-27 | Northwestern University | Pyridazine compounds, compositions and methods |
| US9663493B2 (en) | 2004-11-02 | 2017-05-30 | Northwestern University | Pyridazine compounds, compositions and methods |
| US8063047B2 (en) | 2004-11-02 | 2011-11-22 | Centre National De La Recherche Scientifique (Cnrs) | Pyridazine compounds and methods |
| US8933076B2 (en) | 2004-11-02 | 2015-01-13 | Centre National De La Recherche Scientifique (Cnrs) | Pyridazine compounds, compositions and methods |
| EP2258357A2 (en) | 2005-08-26 | 2010-12-08 | Braincells, Inc. | Neurogenesis with acetylcholinesterase inhibitor |
| EP2258358A2 (en) | 2005-08-26 | 2010-12-08 | Braincells, Inc. | Neurogenesis with acetylcholinesterase inhibitor |
| EP2258359A2 (en) | 2005-08-26 | 2010-12-08 | Braincells, Inc. | Neurogenesis by muscarinic receptor modulation with sabcomelin |
| EP2275096A2 (en) | 2005-08-26 | 2011-01-19 | Braincells, Inc. | Neurogenesis via modulation of the muscarinic receptors |
| EP2275095A2 (en) | 2005-08-26 | 2011-01-19 | Braincells, Inc. | Neurogenesis by muscarinic receptor modulation |
| EP2377530A2 (en) | 2005-10-21 | 2011-10-19 | Braincells, Inc. | Modulation of neurogenesis by PDE inhibition |
| EP2314289A1 (en) | 2005-10-31 | 2011-04-27 | Braincells, Inc. | Gaba receptor mediated modulation of neurogenesis |
| US9408845B2 (en) | 2006-04-28 | 2016-08-09 | Northwestern University | Formulations containing pyridazine compounds |
| US8158627B2 (en) | 2006-04-28 | 2012-04-17 | Northwestern University | Compositions and treatments using pyridazine compounds and cholinesterase inhibitors |
| WO2007127475A3 (en) * | 2006-04-28 | 2008-11-06 | Univ Northwestern | Pyridazines for demyelinating diseases and neuropathic pain |
| WO2007127475A2 (en) * | 2006-04-28 | 2007-11-08 | Northwestern University | Pyridazines for demyelinating diseases and neuropathic pain |
| EP2377531A2 (en) | 2006-05-09 | 2011-10-19 | Braincells, Inc. | Neurogenesis by modulating angiotensin |
| EP2382975A2 (en) | 2006-05-09 | 2011-11-02 | Braincells, Inc. | Neurogenesis by modulating angiotensin |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2010099217A1 (en) | 2009-02-25 | 2010-09-02 | Braincells, Inc. | Modulation of neurogenesis using d-cycloserine combinations |
| US8546396B2 (en) | 2009-03-02 | 2013-10-01 | Irm Llc | N-(hetero)aryl, 2-(hetero)aryl—substituted acetamides for use as Wnt signaling modulators |
| EP2623493A1 (en) * | 2009-03-02 | 2013-08-07 | Irm Llc | N-(hetero)aryl,2-(hetero)aryl-substituted acetamides for use as WNT signaling modulators |
| US10251893B2 (en) | 2009-03-02 | 2019-04-09 | Novartis Ag | N-(hetero)aryl, 2-(hetero)aryl-substituted acetamides for use as Wnt signaling modulators |
| AU2010221493C1 (en) * | 2009-03-02 | 2013-11-07 | Novartis Ag | N- (hetero)aryl, 2- (hetero)aryl-substituted acetamides for use as Wnt signaling modulators |
| AU2012203023C1 (en) * | 2009-03-02 | 2013-11-07 | Novartis Ag | N-(hetero)aryl, 2- (hetero)aryl-substituted acetamides for use as Wnt signaling modulators |
| CN102369187A (en) * | 2009-03-02 | 2012-03-07 | Irm责任有限公司 | N- (hetero)aryl, 2- (hetero)aryl-substituted acetamides for use as wnt signaling modulators |
| EA021225B1 (en) * | 2009-03-02 | 2015-05-29 | Новартис Аг | N-(HETERO)ARYL,2-(HETERO)ARYL-SUBSTITUTED ACETAMIDES FOR USE AS Wnt SIGNALING MODULATORS |
| AU2010221493B2 (en) * | 2009-03-02 | 2012-06-07 | Novartis Ag | N- (hetero)aryl, 2- (hetero)aryl-substituted acetamides for use as Wnt signaling modulators |
| JP2012519210A (en) * | 2009-03-02 | 2012-08-23 | アイアールエム・リミテッド・ライアビリティ・カンパニー | N- (hetero) aryl, 2- (hetero) aryl substituted acetamides for use as WNT signaling regulators |
| KR101359873B1 (en) | 2009-03-02 | 2014-02-06 | 아이알엠 엘엘씨 | N-(hetero)aryl,2-(hetero)aryl-substituted acetamides for use as wnt signaling modulators |
| JP2014129381A (en) * | 2009-03-02 | 2014-07-10 | Irm Llc | N-(hetero)aryl, 2-(hetero)aryl substituted acetamides for using as wnt signaling regulator |
| US9238646B2 (en) | 2009-03-02 | 2016-01-19 | Novartis Ag | N-(hetero)aryl, 2-(hetero)aryl-substituted acetamides for use as WNT signaling modulators |
| WO2010101849A1 (en) * | 2009-03-02 | 2010-09-10 | Irm Llc | N- (hetero)aryl, 2- (hetero)aryl-substituted acetamides for use as wnt signaling modulators |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011063115A1 (en) | 2009-11-19 | 2011-05-26 | Braincells Inc. | Combination of nootropic agent with one or more neurogenic or neurogenic sensitizing agents for stimulating or increasing neurogenesis |
| WO2011091033A1 (en) | 2010-01-20 | 2011-07-28 | Braincells, Inc. | Modulation of neurogenesis by ppar agents |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| US9181235B2 (en) | 2010-06-29 | 2015-11-10 | Novartis Ag | Substituted pyridines for modulating the WNT signaling pathway |
| AU2011271479B2 (en) * | 2010-06-29 | 2014-08-14 | Irm Llc | Compositions and methods for modulating the Wnt signaling pathway |
| WO2012003189A1 (en) * | 2010-06-29 | 2012-01-05 | Irm Llc | Compositions and methods for modulating the wnt signaling pathway |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US10941160B2 (en) | 2013-10-18 | 2021-03-09 | Celgene Quanticel Research, Inc. | Bromodomain inhibitors |
| US11884680B2 (en) | 2013-10-18 | 2024-01-30 | Celgene Quanticel Research, Inc. | Bromodomain inhibitors |
| US11040035B2 (en) | 2016-05-25 | 2021-06-22 | Bayer Pharma Aktiengesellschaft | 3-oxo-2,6-diphenyl-2,3-dihydropyridazine-4-carboxamides |
Also Published As
| Publication number | Publication date |
|---|---|
| TW200418478A (en) | 2004-10-01 |
| ATE369356T1 (en) | 2007-08-15 |
| PL376129A1 (en) | 2005-12-12 |
| MA27814A1 (en) | 2006-04-03 |
| EP1611121A1 (en) | 2006-01-04 |
| CA2506022A1 (en) | 2004-06-03 |
| CA2518917A1 (en) | 2004-06-03 |
| EP1581505B1 (en) | 2007-08-01 |
| ATE368651T1 (en) | 2007-08-15 |
| JP2006509748A (en) | 2006-03-23 |
| KR20050083918A (en) | 2005-08-26 |
| NO20052887D0 (en) | 2005-06-14 |
| BR0316720A (en) | 2005-10-18 |
| FR2847253B1 (en) | 2007-05-18 |
| WO2004046130A1 (en) | 2004-06-03 |
| EP1581505A1 (en) | 2005-10-05 |
| FR2847253A1 (en) | 2004-05-21 |
| AU2003283414A1 (en) | 2004-06-15 |
| AR042089A1 (en) | 2005-06-08 |
| DE60315516T2 (en) | 2008-04-17 |
| CO5700722A2 (en) | 2006-11-30 |
| TW200503727A (en) | 2005-02-01 |
| DE60315354D1 (en) | 2007-09-13 |
| AR042202A1 (en) | 2005-06-15 |
| AU2003296586A1 (en) | 2004-06-15 |
| NO20052887L (en) | 2005-07-29 |
| CN1741999A (en) | 2006-03-01 |
| EP1611121B1 (en) | 2007-08-08 |
| RU2005119151A (en) | 2006-01-20 |
| DE60315354T2 (en) | 2008-05-08 |
| DE60315516D1 (en) | 2007-09-20 |
| MXPA05005270A (en) | 2005-07-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2004046117A1 (en) | Pyridazinone derivatives as gsk-3beta inhibitors | |
| US7727989B2 (en) | 4-benzimidazol-2-yl-pyridazine-3-one-derivatives, production and use thereof in medicaments | |
| US7709466B2 (en) | Pyridazinone derivatives, methods for their production and their use as pharmaceuticals | |
| JP4792455B2 (en) | Novel 4-benzimidazol-2-ylpyridazin-3-one derivatives | |
| KR100814599B1 (en) | Imidazole compounds for the treatment of neurodegenerative disorders | |
| US20040142932A1 (en) | Substituted pyridazinones | |
| US20050020594A1 (en) | Substituted pyridazinones | |
| CZ392597A3 (en) | Imidazole compounds, process of their preparation pharmaceutical compositions containing thereof and treatment method | |
| JP6446433B2 (en) | Pyrimidine substituted pyrrolidine derivatives, pharmaceutical compositions and uses thereof | |
| US20120289518A1 (en) | Derivatives of 2-oxoalkyl-1-piperazin-2-one, preparation method thereof and therapeutic use of same | |
| US7309701B2 (en) | Pyridazinone derivatives as pharmaceuticals and pharmaceutical compositions containing them | |
| EP1667679B1 (en) | Aminobenzimidazoles as selective melanin concentrating hormone receptor antagonists for the treatment of obesity and related disorders | |
| US7462613B2 (en) | Pyridazinone derivatives as pharmaceuticals and pharmaceutical compositions containing them | |
| JP2009529575A (en) | Pyridine-containing large heterocyclic compounds as kinase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 539916 Country of ref document: NZ Ref document number: 168517 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200503825 Country of ref document: ZA Ref document number: 2506022 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 929/CHENP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2005/005270 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004552660 Country of ref document: JP Ref document number: P20050444A Country of ref document: HR Ref document number: 05048069 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003283414 Country of ref document: AU Ref document number: 1020057009083 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2005/0419 Country of ref document: YU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20038A50578 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003775372 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2005119151 Country of ref document: RU Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 376129 Country of ref document: PL |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057009083 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1-2005-500867 Country of ref document: PH |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003775372 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0316720 Country of ref document: BR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2003775372 Country of ref document: EP |