WO2004026306A2 - Pyrazole derivatives as transforming growth factor (tgf) inhibitors - Google Patents
Pyrazole derivatives as transforming growth factor (tgf) inhibitors Download PDFInfo
- Publication number
- WO2004026306A2 WO2004026306A2 PCT/IB2003/003933 IB0303933W WO2004026306A2 WO 2004026306 A2 WO2004026306 A2 WO 2004026306A2 IB 0303933 W IB0303933 W IB 0303933W WO 2004026306 A2 WO2004026306 A2 WO 2004026306A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- phenyl
- cycloalkyl
- heteroaryl
- heterocyclic
- Prior art date
Links
- 0 *1N=Nc2ccccc12 Chemical compound *1N=Nc2ccccc12 0.000 description 3
- QRUDEWIWKLJBPS-UHFFFAOYSA-N c1ccc2nn[nH]c2c1 Chemical compound c1ccc2nn[nH]c2c1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 2
- FTNJQNQLEGKTGD-UHFFFAOYSA-N C1Oc(cccc2)c2O1 Chemical compound C1Oc(cccc2)c2O1 FTNJQNQLEGKTGD-UHFFFAOYSA-N 0.000 description 1
- VQORLTKDPVLJIS-UHFFFAOYSA-N CC(C)Cc(cc1)c[n]2c1ncc2 Chemical compound CC(C)Cc(cc1)c[n]2c1ncc2 VQORLTKDPVLJIS-UHFFFAOYSA-N 0.000 description 1
- RVWOTTIWCOILOE-UHFFFAOYSA-N CC(C)Cc1c[n]2nccc2cc1 Chemical compound CC(C)Cc1c[n]2nccc2cc1 RVWOTTIWCOILOE-UHFFFAOYSA-N 0.000 description 1
- AJKVQEKCUACUMD-UHFFFAOYSA-N CC(c1ncccc1)=O Chemical compound CC(c1ncccc1)=O AJKVQEKCUACUMD-UHFFFAOYSA-N 0.000 description 1
- AWWNUOPOBRVLDT-UHFFFAOYSA-N CC1SC1c1ccc2nn[nH]c2c1 Chemical compound CC1SC1c1ccc2nn[nH]c2c1 AWWNUOPOBRVLDT-UHFFFAOYSA-N 0.000 description 1
- CSDSSGBPEUDDEE-UHFFFAOYSA-N O=Cc1ncccc1 Chemical compound O=Cc1ncccc1 CSDSSGBPEUDDEE-UHFFFAOYSA-N 0.000 description 1
- SMWDFEZZVXVKRB-UHFFFAOYSA-N c1cc2cccnc2cc1 Chemical compound c1cc2cccnc2cc1 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 1
- AWBOSXFRPFZLOP-UHFFFAOYSA-N c1cc2n[o]nc2cc1 Chemical compound c1cc2n[o]nc2cc1 AWBOSXFRPFZLOP-UHFFFAOYSA-N 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-N c1ccncc1 Chemical compound c1ccncc1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N c1cncnc1 Chemical compound c1cncnc1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N c1n[nH]c2c1cccc2 Chemical compound c1n[nH]c2c1cccc2 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- IOJUPLGTWVMSFF-UHFFFAOYSA-N c1nc2ccccc2[s]1 Chemical compound c1nc2ccccc2[s]1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention relates to novel pyrazole compounds, including derivatives thereof, to intermediates for their preparation, to pharmaceutical 5 compositions containing them and to their medicinal use.
- the compounds of the , present invention are potent inhibitors of the transforming growth factor ("TGF")- ⁇
- TGF- ⁇ related disease states including, for example, cancer and fibrotic diseases.
- TGF- ⁇ activates both antiproliferative and tumor-promoting signaling 10 cascades.
- Three mammalian TGF- ⁇ isoforms have been identified (TGF- ⁇ l, - ⁇ ll, and - ⁇ lll).
- TGF- ⁇ production promotes tumor progression while its blockade enhances antitumor activity.
- Blockade of TGF- ⁇ enhances antitumor immune responses and inhibits metastasis.
- the present invention provides a novel compound containing a core pyrazole ring substituted with at least one substituted or unsubstituted 2-pyridyl moiety and at least one R 1 moiety as set forth herein, and all pharmaceutically acceptable salts, 20 prodrugs, tautomers, hydrates and solvates thereof.
- the substituted or unsubstituted 2-pyridyl moiety and R 1 moiety can be in an 1 ,2-, 1,3- or 1,4- relationship around the core pyrazole ring; preferably, in an 1,2- or ortho relationship.
- the present invention provides a compound of formula (la): 25
- R 1 , R 3 , R 4 , R 6 and s are each as set forth herein, with the proviso that R contains at least one heteroatom.
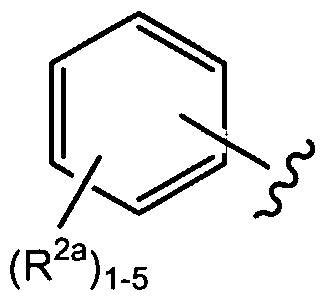
- R 1 is a saturated, unsaturated, or aromatic C 3 -C 20 mono-, bi- or polycyclic ring optionally containing at least one heteroatom selected from the group consisting of N, O and S, wherein R 1 can optionally be further independently substituted with at least one moiety independently selected from the group consisting of: carbonyl, halo, halo(C]-C 6 )alkyl, perhalo(C ⁇ -C 6 )alkyl, perhalo(C ⁇ -C 6 )alkoxy, (C ⁇ -C 6 )alkyl, (C 2 -C 6 )alkenyl, (C 2 -C 6 )alkynyl, hydroxy, oxo, mercapto, (Ci- C 6 )alkylthio, (C ⁇ -C 6 )alkoxy, (C 5 -Ci 0 )aryl or (C 5 -C ⁇ o)heteroaryl, (C 5 -C] 0 )aryl
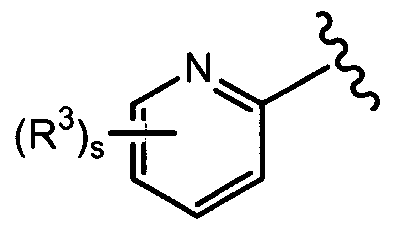
- each R 3 is independently selected from the group consisting of: hydrogen, halo, (C,-C 6 )alkyl, (C 2 -C 6 )alkenyl, (C 2 -C 6 )alkynyl, perhalo(C ⁇ -C 6 )alkyl, phenyl, (C 5 -C ⁇ o)heteroaryl, (C 5 -C,o)heterocyclic, (C 3 -C ⁇ o)cycloalkyl, hydroxy, (C ⁇ -C 6 )alkoxy, perhalo(C ⁇ -C 6 )alkoxy, phenoxy, (C 5 -C ⁇ o)heteroaryl-O-, (C 5 -C ⁇ 0 )heterocyclic-O-, (C 3 -C ⁇ 0 )cycloalkyl-O-, (C,-C 6 )alkyl-S-, (C,-C 6 )alkyl-SO 2 -, (C ⁇ -C
- alkyl, alkenyl, alkynyl, phenyl, heteroaryl, heterocyclic, cycloalkyl, alkoxy, phenoxy, amino of R 3 is optionally substituted by at least one substituent independently selected from (C ⁇ -C 6 )alkyl, (C ⁇ -C 6 )alkoxy, halo(C ⁇ -C 6 )alkyl, halo, H 2 N-, Ph(CH 2 ) ⁇ -6 HN-, and (C,-C 6 )alkylHN-;
- s is an integer from one to five; preferably, one to two; more preferably, one;
- R 4 is selected from the group consisting of: hydrogen, halo, halo(C,- C 6 )alkyl, (C,-C 6 )alkyl, (C 2 -C 6 )alkenyl, (C 2 -C 6 )alkynyl, perhalo(C ⁇ -C 6 )alkyl, phenyl, (C 5 -Ci 0 )heteroaryl, (C -C ⁇ 0 )heterocyclic, (C 3 -C ⁇ 0 )cycloalkyl, hydroxy, (C ⁇ -C 6 )alkoxy, perhalo(C ⁇ -C 6 )alkoxy, phenoxy, (C 5 -C i O )heteroaryl-O-, (C 5 -C , O )heterocyclic-O-, (C 3 -C , O )cycloalkyl-O-, (C,-C 6 )alkyl-S-, (C
- alkyl, alkenyl, alkynyl, phenyl, heteroaryl, heterocyclic, cycloalkyl, alkoxy, phenoxy, and amino of R 4 is optionally substituted by at least one substituent independently selected from the group consisting of (C ⁇ -C 6 )alkyl, ( - C 6 )alkoxy, halo(C,-C 6 )alkyl, halo, H 2 N-, Ph(CH 2 ) , -6 -NH-, and (C,-C 6 )alkylNH-;.and
- R 6 is selected from the group consisting of hydrogen, (C]-C )alkyl,
- R 6 is hydrogen or (C,- C 6 )alkyl; more preferably, hydrogen or methyl; where alkyl, alkenyl, alkynyl, phenyl, benzyl, heteroaryl, heterocyclic, cycloalkyl, alkoxy, phenoxy, amino of R 6 is optionally substituted with at least one moiety independently selected from the group consisting of halo, (C ⁇ -C 6 )alkyl, (C 2 -C 6 )alkenyl, (C 2 -C 6 )alkynyl, perhalo(C ⁇ -C 6 )alkyl, (C 3 -C ⁇ o)cycloalkyl, phenyl, benzyl, (C 5 -C ⁇ 0 )heterocyclic, (C 5 -C ⁇ o)he
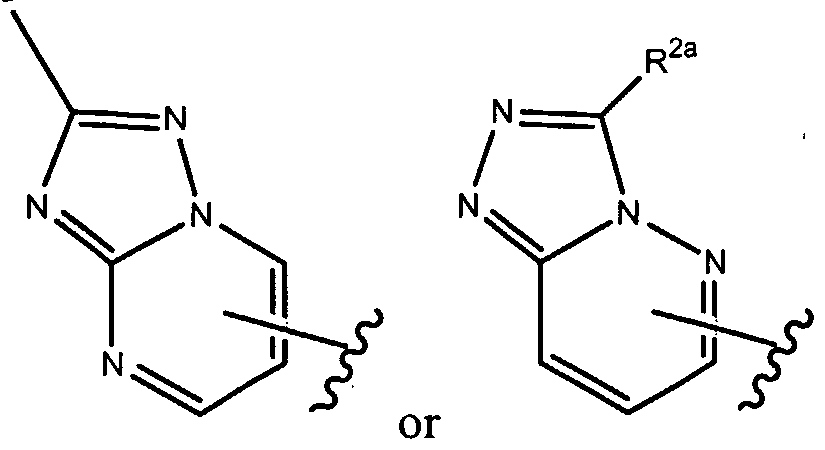
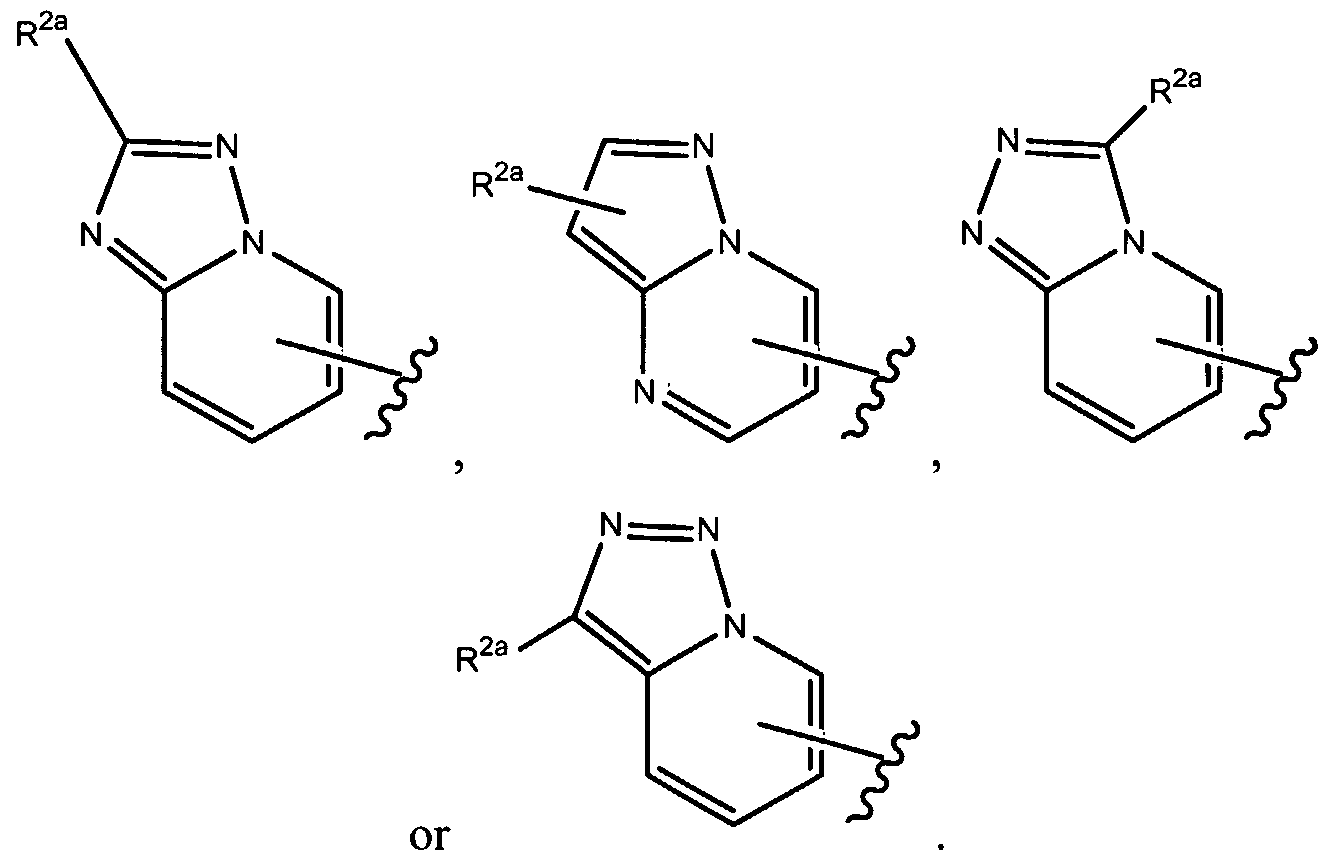
- R 1 of formula (la), as set forth above is
- R of formula (la), as set forth above is
- R 1 of formula (la), as set forth above is
- R of formula (la), as set forth above is
- R , 2a is as set forth herein.
- R , ⁇ of formula (la), as set forth above is
- R 1 of formula (la), as set forth above, is where R 2a is as set forth herein.
- R 1 above can optionally be further substituted by at least one R 2 group, as set forth herein.
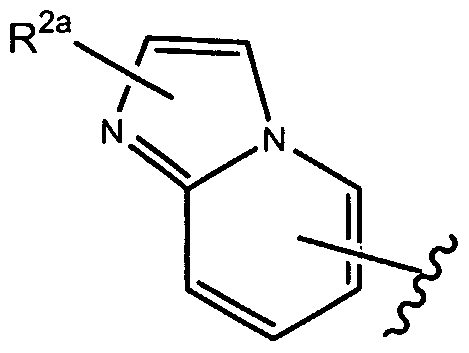
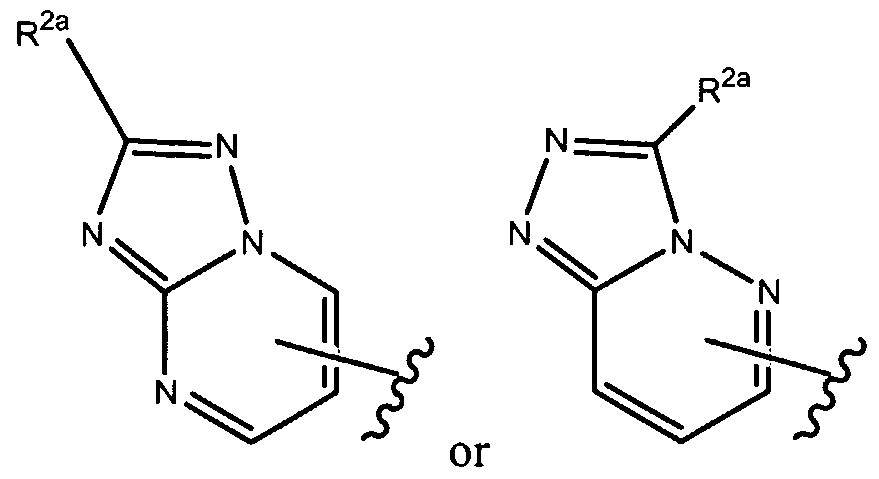
- R 1 of formula (la), as set forth above is
- R 1 of formula (la), as set forth above is
- R of formula (la), as set forth above is
- R 1 of formula (la), as set forth above is
- R of formula (la), as set forth above is
- R , 2a is as set forth herein.
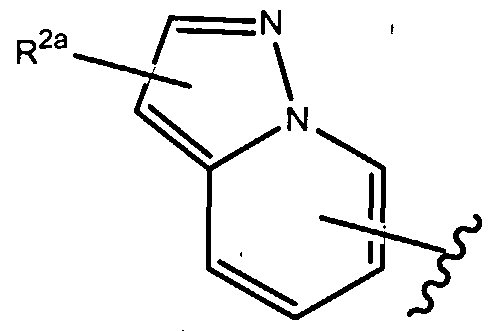
- R 1 of formula (la), as set forth above is
- R 1 of formula (la), as set forth above is
- R of formula (la), as set forth above is
- R 1 of formula (la), as set forth above is
- R 1 of formula (la), as set forth above is
- R of formula (la), as set forth above is
- R 1 of formula (la), as set forth above is where R , 2a i-s as set forth herein.
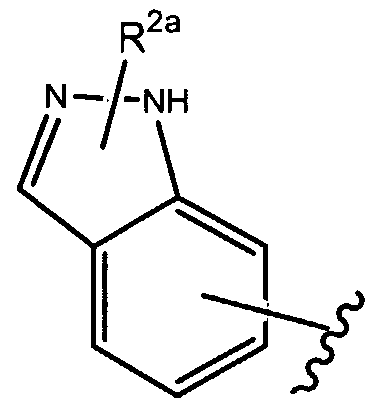
- R' of formula (la), as set forth above is
- R , a is as set forth herein.
- R 1 of formula (la), as set forth above is
- R , 2a j is as set forth herein.
- R 1 of formula (la), as set forth above is
- R , 2a is as set forth herein.
- R of formula (la), as set forth above is
- R 2a is as set forth herein.
- R 1 of formula (la), as set forth above is
- R of formula (la), as set forth above is
- R 2a is as set forth herein.
- R 1 of formula (la), as set forth above is
- R of formula (la), as set forth above is
- R of formula (la), as set forth above is
- R 1 of formula (la), as set forth above, is
- R 1 of formula (la), as set forth above is
- R of formula (la), as set forth above is
- R of formula (la), as set forth above is
- R 1 of formula (la), as set forth above, is selected from the group consisting of:
- R 1 of formula (la), as set forth above, is selected from the group consisting of:
- R 1 of formula (la), as set forth above, is selected from the group consisting of:
- the invention also provides a pharmaceutical composition comprising at , least one compound of the invention and a pharmaceutically acceptable carrier.
- the invention further provides a method of preparation of a compound of the invention.
- the invention still further provides a method of preventing or treating a TGF-related disease state in an animal or human comprising the step of administering a therapeutically effective amount of at least one compound of the invention to the animal or human suffering from the TGF-related disease state.
- the invention still further provides the use of a compound of the invention in the preparation of a medicament for the prevention or treatment of a TGF-related disease state in an animal or human.
- alkyl refers to a linear or branched saturated hydrocarbon (e.g., methyl, ethyl, n-propyl, wopropyl, M-butyl, iso-butyl, secondary-butyl, tertiary- butyl).
- cycloalkyl refers to a mono or bicyclic carbocyclic ring (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, cyclopentenyl, cyclohexenyl, bicyclo[2.2.1]heptanyl, bicyclo[3.2.1]octanyl and bicyclo[5.2.0]nonanyl).
- cycloalkyl refers to a mono or bicyclic carbocyclic ring (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, cyclopentenyl, cyclohexenyl, bicyclo[2.2.1]heptanyl, bicyclo[3.2.1]oc
- halogen refers to includes fluoro, chloro, bromo or iodo or fluoride, chloride, bromide or iodide.
- halo-substituted alkyl or “haloalkyl” refers to an alkyl radical, as set forth above, substituted with one or more halogens, as set forth above, including, but not limited to, chloromethyl, dichloromethyl, fluoromethyl, difluoromethyl, trifluoromethyl, and 2,2,2-trichloroethyl.
- perhaloalkyl refers to an alkyl radical, as set, forth above, where each hyrdrogen of the alkyl group is replaced with a "halogen” or "halo", as set forth above.
- alkenyl refers to a linear or branched hydrocarbon chain radical containing at least two carbon atoms and at least one double bond. Examples include, but are not limited to, ethenyl, 1-propenyl, 2-propenyl (allyl), iso- propenyl, 2 -methyl- 1-propenyl, 1-butenyl, and 2-butenyl.
- alkynyl refers to a linear or branched hydrocarbon chain radical having at least one triple bond including, but not limited to, ethynyl, propynyl, and butynyl.
- aryl refers to an aromatic radical such as, for example, phenyl, naphthyl, tetrahydronaphthyl, and indanyl.
- heteroaryl refers to an aromatic group containing at least one heteroatom selected from O, S and N.
- heteroaryl groups include, but are not limited to, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, thienyl, furyl, imidazolyl, pyrrolyl, oxazolyl (e.g., 1,3-oxazolyl, 1 ,2-oxazolyl), thiazolyl (e.g., 1,2-thiazolyl, 1,3 -thiazolyl), pyrazolyl, tetrazolyl, triazolyl (e.g., 1,2,3- triazolyl, 1 ,2,4-triazolyl), oxadiazolyl (e.g., 1,2,3-oxadiazolyl), thiadiazolyl (e.g., 1,3,4-thiadiazolyl), quinolyl, is
- heterocyclic groups include, but are not limited to, azetidinyl, tetrahydrofuranyl, imidazolidinyl, pyrrolidinyl, piperidinyl, piperazinyl, oxazolidinyl, thiazolidinyl, pyrazolidinyl, thiomorpholinyl, tetrahydrothiazinyl, tetrahydro-thiadiazinyl, morpholinyl, oxetanyl, tetrahydrodiazinyl, oxazinyl, oxcithiazinyl, indolinyl, isoindolinyl, quincuclidinyl, chromanyl, isochromanyl, benzocazinyl, and the like.
- Examples of monocyclic saturated or unsaturated ring systems are tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, imidazolidin-1-yl, imidazolidin-2-yl, imidazolidin-4-yl, pyrrolidin-1-yl, pyrrolidin-2-yl, pyrrolidin-3- yl, piperidin-1-yl, piperidin-2-yl, piperidin-3-yl, piperazin-1-yl, piperazin-2-yl, piperazin-3-yl, l,3-oxazolidin-3-yl, isothiazolidine, l,3-thiazolidin-3-yl, l,2-pyrazolidin-2-yl, 1,3-pyrazolidin-l-yl, thiomo holin-yl, 1 ,2-tetrahydrothiazin-2- yl, l,3-tetrahydrothiazin
- the term "pharmaceutically acceptable acid addition salt” refers to non-toxic acid addition salts, i.e., salts derived from pharmacologically acceptable anions, such as the hydrochloride, hydrobromide, hydroiodide, nitrate, sulfate, bisulfate, phosphate, acid phosphate, acetate, lactate, citrate, acid citrate, tartrate, bitartrate, succinate, maleate, fumarate, gluconate, saccharate, benzoate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate and pamoate [i.e. , 1 ,1 '-methylene-bis-(2-hydroxy-3-naphthoate)]salts.
- pharmacologically acceptable anions such as the hydrochloride, hydrobromide, hydroiodide, nitrate, sulfate
- the term "pharmaceutically acceptable base addition salt” refers to non-toxic base addition salts, i.e., salts derived from such pharmacologically acceptable cations such as alkali metal cations (e.g., potassium and sodium) and alkaline earth metal cations (e.g., calcium and magnesium), ammonium or water- soluble amine addition salts such as N-methylglucamine-(meglumine), and the lower alkanolammonium and other base salts of pharmaceutically acceptable organic amines.
- alkali metal cations e.g., potassium and sodium
- alkaline earth metal cations e.g., calcium and magnesium
- ammonium or water- soluble amine addition salts such as N-methylglucamine-(meglumine)
- the lower alkanolammonium and other base salts of pharmaceutically acceptable organic amines such as N-methylglucamine-(meglumine
- suitable substituent refers to a chemically and pharmaceutically acceptable functional group, i.e., a moiety that does not negate the inhibitory and/or therapeutic activity of the inventive compounds.
- suitable substituents may be routinely selected by those skilled in the art.
- substituents include, but are not limited to, carbonyl,
- TGF-related disease state refers to any disease , state mediated by the production of TGF- ⁇ .
- Ph refers to phenyl
- a saturated, unsaturated, or aromatic C 3 -C 20 mono-, bi- or polycyclic ring optionally containing at least one heteroatom refers to, but is not limited to,
- R 2a is independently se ected from the group consisting o 3 f: (C ⁇ -C 6 )alkyl, (C 2 -C 6 )alkenyl, (C 2 -C 6 )alkynyl, (C 3 -C ⁇ 0 )cycloalkyl, (C 5 -C 10 )aryl, (C,-C 6 )alkylaryl, amino, carbonyl, carboxyl, (C 2 -C 6 )acid, (C]-C 6 )ester, (C 5 -C ⁇ 0 )heteroaryl,
- reaction schemes illustrate the preparation of the compounds of the present invention.
- a compound of the invention may be prepared by methods analogous to those described in U.S. Application Nos. 10/094,717, 10/094,760, and 10/115,952 and WO 02/40476.
- R 1 , R 3 , R 4 , R 6 , R 2a and s in the reaction schemes and the discussion that follow are defined above.
- Scheme 1 refers to the preparation of compounds of the formula la.
- a compound of the formula III was prepared from aldehydes of the formula II by first treatment with an aromatic amine, such as aniline, in a polar solvent. Suitable solvents include ethyl acetate, isopropyl acetate, or tetrahydrofuran, preferably isopropyl acetate.
- the resulting reaction mixture is heated to a temperature from about 50°C to about 100°C, preferably about 60°C, and then slowly treated with phosphorous acid diphenyl ester. The temperature of the reaction mixture was maintained for a period from about 30 minutes to about 3 hours, preferably about 1 hour and then cooled to ambient temperature overnight.
- a compound of formula II was prepared according to Preparation E, set forth below.
- a compound of the formula V was prepared from a compound of the formula
- aforesaid reaction was run at a temperature from about 0°C to about 100°C, preferably about 22°C (ambient temperature), for a period from about 30 minutes to about 5 hours, preferably about 2 hours.
- acid such as hydrochloric acid
- a compound of formula la was prepared from a compound of formula V by treatment with an excess of dimethylformamide dimethylacetal, DMF-DMA, and heating neat at a temperature from about 60°C to about 100°C, preferably about 80°C, for a period of about 30 minutes to about 4 hours, preferably about 2 hours.
- an appropriate hydrazine such as H 2 NNHR 6
- Suitable solvents for the aforesaid reaction include methanol and ethanol.
- the aforesaid reaction was conducted at temperature of about 0°C to about 50°C, preferably about 22°C (ambient temperature), for about 1 to about 4 hours, preferably about 2 hours.
- a compound of formula Va can be prepared from a compound of formula V by reaction with a base such as lithium bis(trimethylsilyl)amide in a solvent such as tetrahydrofuran at a temperature of -78°C followed by addition of a simple ester of activated carboxylic acid (R 4 CO 2 H) such as an acid chloride or acyl imidazole.
- a compound of formula la is then formed by addition of an appropriate hydrazine, such as H 2 NNHR 6 .
- a compound of formula la can be prepared by treatment of V with a base such as sodium hydride in a solvent such as pyridine followed by addition of a thioisocyanate, SCNR 4 .
- a compound of formula la is then formed by addition of an appropriate hydrazine, such as H 2 NNHR 6 .
- a compound of formula la can be prepared according to the procedures set forth in WO 00/31063 and WO 02/72576. "'''
- Scheme 2 refers to the preparation of compounds of the formula V, which are intermediates in the preparation of compounds of formula la in Scheme 1.
- a compound of the formula V was prepared from a compound of the formula VI by treatment with a base, such as butyl lithium, at 'a temperature of about
- reaction was run at a temperature from about -78°C to about 0°C, preferably about -20°C, for a period from about 1 hour to about 10 hours, preferably about 3 hours.
- the compound of formula V can be prepared according to the methods of Davies, I. W.; Marcoux, J.F.; Corley, E. G.; Journet, M.; Cai, D.-W.; Palucki, M.; Wu, J.; Larsen, R. D.; Rossen, K.; Pye, P. J.; Di Michele, L.; Dormer, P.; Reider, P. J.; J. Ore. Chem.. Vol. 65, pp. 8415-8420 (2000).
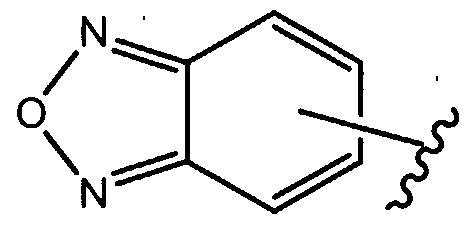
- Scheme 3 refers to the preparation of compounds of the formula la where R 1 is
- Scheme 4 refers to the preparation of compounds of the formula V, which are intermediates in the preparation of compounds of formula la in Scheme 1.
- a compound of the formula V was prepared from compound, of the formula X, which is either commercially available or prepared according to Preparation B, as set forth below, by reaction with a compound of the formula R'-Cl, in the presence of a catalyst such as palladium II acetate, a base (e.g. , potassium tert-butoxide, and AMPHOS® (i.e., 2-dicyclohexylphosphino-2'-(N,N- dimethylamino)biphenyl, commercially available from Strem Chemicals, ⁇
- a catalyst such as palladium II acetate
- a base e.g. , potassium tert-butoxide, and AMPHOS® (i.e., 2-dicyclohexylphosphino-2'-(N,N- dimethylamino)biphenyl
- Preparation A refers to the preparation of compounds of the formula IV, which are intermediates useful in the preparation of compounds of the formula la.
- R is a simple alkyl group such as methyl or ethyl.
- compounds of the formula XII were prepared from a compound of the formula XI, wherein X is a chloride or bromide, by an alkoxycarbonylation reaction.
- Suitable conditions include metal-halogen exchange with butyl lithium in a solvent such as tetrahydrofuran at a temperature of about 0°C, for a period of time of about 30 minutes, followed by the addition of ethylchloroformate at a temperature of about 0°C, followed by a period of time of about 2.4 hours at about 50°C.
- a solvent such as tetrahydrofuran
- ethylchloroformate at a temperature of about 0°C
- 2.4 hours at about 50°C.
- the compound of the formula IV was prepared from a compound of the formula XII by a two-step process.
- Suitable reducing agents include lithium borohydride, sodium borohydride, lithium aluminum hydride, and borane in tetrahydrofuran.
- Suitable solvents for the aforesaid reaction include methanol, ethanol, tetrahydrofuran, diethyl ether, and dioxane.
- the aforesaid reaction was run at a temperature from about 0°C to about 100°C, preferably about 65°C, for a period from about 10 minutes to about 1 hour, preferably about 30 minutes.
- the resulting primary alcohol was then oxidized to the corresponding aldehyde of the formula IV by treatment with an oxidizing agent, such as N-methyl mo ⁇ holine N-oxide/TPAP, Dess-Martin reagent, PCC or oxalyl chloride-DMSO, preferably oxalyl chloride- DMSO.
- an oxidizing agent such as N-methyl mo ⁇ holine N-oxide/TPAP, Dess-Martin reagent, PCC or oxalyl chloride-DMSO, preferably oxalyl chloride- DMSO.
- Suitable solvents for the aforesaid reaction include chloroform, tetrahydrofuran, or dichloromethane.
- the aforesaid reaction was conducted at a temperature from about -78°C to about 22°C for a time from about 15 minutes to about 3 hours, preferably about 1 hour.
- Preparation B refers to the preparation of compounds of the formula X, which are intermediates useful in the preparation of compounds of the formula la.
- a compound of formula XIII was prepared from a compound of the formula II by reaction with methyl magnesium bromide in a polar solvent such as a mixture of tetrahydrofuran and toluene. The aforesaid reaction was run at a temperature from about -78°C to about 0°C, preferably about -60°C, for a period from about 10 minutes to about 1 hour, preferably about 40 minutes, followed by a period of about 90 minutes at a temperature of about -10°C.
- the compound of formula II was prepared according to Preparation E, set forth below.
- the compound of formula X was prepared from a compound of the formula XIII by treatment with an oxidizing agent, such as N-methyl mo ⁇ holine N- oxide/TPAP, Dess-Martin reagent, PCC or oxalyl chloride-DMSO, preferably oxalyl chloride-DMSO.
- an oxidizing agent such as N-methyl mo ⁇ holine N- oxide/TPAP, Dess-Martin reagent, PCC or oxalyl chloride-DMSO, preferably oxalyl chloride-DMSO.
- Suitable solvents for the aforesaid reaction include chloroform, tetrahydrofuran, or dichloromethane.
- the aforesaid reaction was conducted at a temperature from about -78°C to about 22°C for a time from about 15 minutes to , about 3 hours, preferably about 1 hour.
- Preparation C refers to the preparation of compounds of the formula VII, which are intermediates useful in the preparation of compounds of the formula la.
- R is a simple alkyl group such as methyl or ethyl.
- compounds of the formula XV were prepared from a compound of the formula XIV, which may be prepared according to a procedure described in Preparation A or are commercially available, by treatment with a base such as lithium hydroxide, in a polar protic solvent. Suitable solvents for the aforesaid reaction included methanol, ethanol, and water. The aforesaid reaction was conducted at a temperature from about 0°C to about 30°C preferably about 22°C (room temperature) for a time from about 15 minutes to about 3 hours, preferably about 1 hour.
- the compound of the formula VII was prepared from a compound of the formula XV by reaction with a suitable activating agent and a compound of the formula and a base.
- Suitable activating agents included thionyl chloride, carbonyldiimidazole, EDCI and DCC, preferably oxalyl chloride.
- Suitable bases included triethylamine, Hunig's base, or DBU, preferably triethyl amine.
- Suitable solvents for the aforesaid reaction include methylene chloride, N,N'- dimethylformamide, tetrahydrofuran, and a mixture thereof, preferably methylene chloride.
- the aforesaid reaction was conducted at a temperature from about 0°C to about 30°C, preferably about 22°C (room temperature) for a time from about 6 hours to about 48 hours, preferably about 12 hours.
- Preparation D refers to the preparation of compounds of the formula VIII, which is an intermediate useful in the preparation of compounds of formula (la), where R 1 is
- R is (C ⁇ -C 6 )alkyl.
- the compound of formula VIII was prepared from a compound of formula XVI by treatment with an alkyl halide, such as methyl iodide, in the presence of a base such as sodium hydride, in a polar aprotic solvent such as N,N' -dimethylformamide.
- alkyl halide such as methyl iodide
- a base such as sodium hydride
- a polar aprotic solvent such as N,N' -dimethylformamide
- Preparation E refers to the preparation of compounds of the formula II, which are intermediates useful in the preparation of compounds of formula (la).
- R is a simple alkyl group such as methyl or ethyl.
- compounds of the formula XVIII were prepared from heteroarylhalides of the formula XVII, wherein X is a chloride or bromide, according to the procedure described for the preparation of compound XII from compound XI in Preparation A.
- the compound of the formula II was prepared from a compound of the formula XVIII according to the two-step process described for the preparation of compound IV from compound XII in Preparation A. '
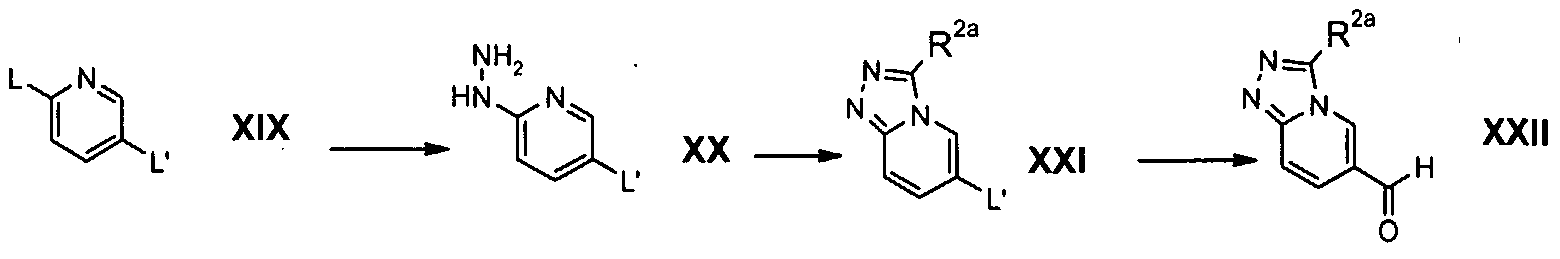
- Preparation F refers to the preparation of compounds of the formula XXII which are intermediates in the preparation of compounds of the formula (la).
- compounds of formula XXII were prepared from compounds of formula XXI by a formylation reaction. Suitable conditions for formylation include metal halogen exchange with isopropylmagnesium chloride in a solvent such as tetrahydrofuran at a temperature of about 0°C, for a period of time of about 30 minutes, followed by the addition of N,N-dimethylformamide at a temperature of about 0°C, followed by a period of time of about 2.5 hours at a temperature of about 50°C.
- a compound of the invention which is basic in nature is capable of forming a wide variety of different salts with various inorganic and organic acids. Although such salts must be pharmaceutically acceptable for administration to animals and humans, it is often desirable in practice to initially isolate a compound of the invention from the reaction mixture as a pharmaceutically unacceptable salt and then simply convert the latter back to the free base compound by treatment with an alkaline reagent, and subsequently convert the free base to a pharmaceutically acceptable acid addition salt.
- the acid addition salts of the base compounds of this invention are readily prepared by treating the base compound with a substantially equivalent amount of the chosen mineral or organic acid in an aqueous solvent medium or in a suitable organic solvent such as, for example, methanol or ethanol. Upon careful evaporation of the solvent, the desired solid salt is obtained.
- the acids which can be used to prepare the pharmaceutically acceptable acid addition salts of the base compounds of this invention are those which form non- toxic acid addition salts, i.e., salts containing pharmacologically acceptable anions, such as chloride, bromide, iodide, nitrate, sulfate or bisulfate, phosphate or acid phosphate, acetate, lactate, citrate or acid citrate, tartrate or bitartrate, succinate, maleate, fumarate, gluconate, saccharate, benzoate, methanesulfonate and pamoate [i.e., l,l'-methylene-bis-(2-hydroxy-3-naphthoate)] salts.
- a compound of the invention which is also acidic in nature e.g., contains a
- COOH or tetrazole moiety is capable of forming base salts with various pharmacologically acceptable cations.
- salts must be pharmaceutically acceptable for administration to animals and humans, it is often desirable in practice to initially isolate a compound of the invention from the reaction mixture as a pharmaceutically unacceptable salt and then simply convert the latter back to the free acid compound by treatment with an acidic reagent, and subsequently convert the free acid to a pharmaceutically acceptable base addition salt.
- pharmaceutically acceptable base addition salts include the alkali metal or alkaline-earth metal salts and particularly, the sodium and potassium salts. These salts can be prepared by conventional techniques.
- the chemical bases which can be used as reagents to prepare the pharmaceutically acceptable base addition salts of this invention are those which form non-toxic base salts with the herein described acidic compounds of the invention.
- These non-toxic base salts include salts derived from such pharmacologically acceptable cations as sodium, potassium, calcium and magnesium, etc.
- These salts can easily be prepared by treating the corresponding acidic compounds with an aqueous solution containing the desired pharmacologically acceptable cations, and then evaporating the resulting solution to dryness, preferably under reduced pressure.
- they may also be prepared by mixing lower alkanolic solutions of the acidic compounds and the desired alkali metal alkoxide together, and then evaporating the resulting solution to dryness in the same manner as before.
- stoichiometric quantities of reagents are preferably employed in order to ensure completeness of reaction and maximum product yields.
- Isotopically-labeled compounds are also encompassed by the present invention.
- an "isotopically-labeled compound” refers to a compound of the invention including pharmaceutical salts, prodrugs thereof, each as described herein, in which one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be inco ⁇ orated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine and chlorine, such as 2 H, 3 H, 13 C, 14 C, l 5 N, 18 0, 17 0, 31 P, 32 P, 35 S, 18 F, and 36 C1, respectively.
- the compounds may be useful in drug and/or substrate tissue distribution assays.
- Tritiated ( 3 H) and carbon-14 ( 14 C) labeled compounds are particularly preferred for their ease of preparation and detectability. Further, substitution with heavier isotopes such as deuterium ( 2 H) can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements and, hence, may be preferred in some circumstances.
- Isotopically labeled compounds of the invention can be prepared by any means known in the art. Stereoisomers (e.g., cis and trans isomers) and all optical isomers of a compound of the invention (e.g., R and S enantiomers), as well as racemic, diastereomeric and other mixtures of such isomers are contemplated by the present invention.
- Stereoisomers e.g., cis and trans isomers
- optical isomers of a compound of the invention e.g., R and S enantiomers
- racemic, diastereomeric and other mixtures of such isomers are contemplated by the present invention.
- the compounds, salts, prodrugs, tautomers, hydrates, and solvates of the present invention can exist in several tautomeric forms, including the enol and imine form, and the keto and enamine form and geometric isomers and mixtures thereof.
- Tautomers exist as mixtures of a tautomeric set in solution. In solid form, usually one tautomer predominates. Even though one tautomer may be described, the present invention includes all tautomers of the present compounds.
- the present invention also includes atropisomers of the present invention.
- Atropisomers refer to compounds of the invention that can be separated into rotationally restricted isomers.
- a compound of the invention as described above, can be used in the manufacture of a medicament for the prophylactic or therapeutic treatment of a
- TGF-related disease state in an animal or human in an animal or human.
- a compound of the invention is a potent inhibitor of transforming growth factor (“TGF”)- ⁇ signaling pathway and are therefore of use in therapy.
- TGF transforming growth factor
- the present invention provides a method of preventing or treating a TGF-related disease in an animal or human comprising the step of administering a therapeutically effective amount of at least one compound of the invention to the animal or human suffering from the TGF-related disease state.
- the term "therapeutically effective amount” refers to an amount of a compound of the invention required to inhibit the TGF- ⁇ signaling pathway. As would be understood by one of skill in the art, a “therapeutically effective amount” will vary from patient to patient and will be determined on a case by case basis. Factors to consider include, but are not limited to, the patient being treated, weight, health, compound administered, etc.
- disease states that can be treated by inhibition of the TGF- ⁇ signaling pathway.
- diseases states include, but are not limited to, all types of cancer (e.g., breast, lung, colon, prostate, ovarian, pancreatic, melanoma, all hematological malignancies, etc.) as well as all types of fibrotic diseases (e.g., glomerulonephritis, diabetic nephropathy, hepatic fibrosis, pulmonary fibrosis, arterial hype ⁇ lasia and restenosis, scleroderma, and dermal scarring).
- the present invention also provides a pharmaceutical composition comprising at least one compound of the invention and at least one pharmaceutically acceptable; carrier.
- the pharmaceutically acceptable carrier may be any such carrier known in the art including those described in, for example, Remington's Pharmaceutical Sciences, Mack Publishing Co., (A. R. Gennaro edit. 1985).
- a pharmaceutical composition of the invention may be prepared by conventional means known in the art including, for example, mixing at least one compound of the invention with a pharmaceutically acceptable carrier.
- a pharmaceutical composition of the invention may be used in the prevention or treatment of a TGF-related disease state, as described above, in an animal or human.
- a compound of the invention may be formulated as a pharmaceutical composition for oral, buccal, intranasal, parenteral (e.g., intravenous, intramuscular or subcutaneous), topical, or rectal administration or in a form suitable for administration by inhalation or insufflation.
- the pharmaceutical composition may take the form of, for example, a tablet or capsule prepared by conventional means with a pharmaceutically acceptable excipient such as a binding agent (e.g., pregelatinized maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose); filler (e.g., lactose, microcrystalline cellulose or calcium phosphate); lubricant (e.g., magnesium stearate, talc or silica); disintegrant (e.g., potato starch or sodium starch glycolate); or wetting agent (e.g., sodium lauryl sulphate).
- a binding agent e.g., pregelatinized maize starch, polyvinylpyrrolidone or hydroxypropyl methylcellulose
- filler e.g., lactose, microcrystalline cellulose or calcium phosphate
- lubricant e.g., magnesium stearate, talc or silica
- disintegrant e.g.,
- Liquid preparations for oral administration may take the form of a, for example, solution, syrup or suspension, or they may be presented as a dry product for constitution with water or other suitable vehicle before use.
- Such liquid preparations may be prepared by conventional means with a pharmaceutically acceptable additive such as a suspending agent (e.g., sorbitol syrup, methyl cellulose or hydrogenated edible fats); emulsifying agent (e.g., lecithin or acacia); non-aqueous vehicle (e.g., almond oil, oily esters or ethyl alcohol); and preservative (e.g., methyl or propyl p-hydroxybenzoates or sorbic acid).
- a suspending agent e.g., sorbitol syrup, methyl cellulose or hydrogenated edible fats
- emulsifying agent e.g., lecithin or acacia
- non-aqueous vehicle e.g., almond oil, oily esters or ethyl
- the composition may take the form of tablets or lozenges formulated in conventional manner.
- a compound of the present invention may also be formulated for sustained delivery according to methods well known to those of ordinary skill in the art. Examples of such formulations can be found in United States Patents 3,538,214, 4,060,598, 4,173,626, 3,119,742, and 3,492,397, which are herein inco ⁇ orated by reference in their entirety.
- a compound of the invention may be formulated for parenteral administration by injection, including using conventional catheterization techniques or infusion.
- Formulations for injection may be presented in unit dosage form, e.g., in ampules or in multi-dose containers, with an added preservative.
- the compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain a formulating agent such as a suspending, stabilizing and/or dispersing agent.
- the active ingredient may be in powder form for reconstitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
- a compound of the invention may also be formulated in rectal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter or other glycerides.
- a compound of the invention may be conveniently delivered in the form of a solution or suspension from a pump spray container that is squeezed or pumped by the patient or as an aerosol spray presentation from a pressurized container or a nebulizer, with the use of a suitable propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- a suitable propellant e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- the dosage unit may be determined by providing a valve to deliver a metered amount.
- the pressurized container or nebulizer may contain a solution or suspension of the compound of the invention.
- Capsules and cartridges for use in an inhaler or insufflator may be formulated containing a powder mix of a compound of the invention and a suitable powder base such as lactose or starch.
- a proposed dose of a compound of the invention for oral, parenteral or , buccal administration to the average adult human for the treatment of a TGF-related disease state is about 0.1 mg to about 2000 mg, preferably, about 0.1 mg to about 200 mg of the active ingredient per unit dose which could be administered, for example, 1 to 4 times per day.
- Aerosol formulations for treatment of the conditions referred to above in the average adult human are preferably arranged so that each metered dose or "puff of aerosol contains about 20 ⁇ g to about 10,000 ⁇ g, preferably, about 20 ⁇ g to about lOOO ⁇ g of a compound of the invention.
- the overall daily dose with an aerosol will be within the range from about lOO ⁇ g to about 100 mg, preferably, about lOO ⁇ g to about 10 mg.
- Administration may be several times daily, for example 2, 3, 4 or 8 times, giving for example, 1 , 2 or 3 doses each time.
- Aerosol combination formulations for treatment of the conditions referred to above in the average adult human are preferably arranged so that each metered dose or "puff of aerosol contains from about 0.01 mg to about 1000 mg, preferably, about 0.01 mg to about 100 mg of a compound of this invention, more preferably from about 1 mg to about 10 mg of such compound. Administration may be several times daily, for example 2, 3, 4 or 8 times, giving for example, 1, 2 or 3 doses each time.
- Aerosol formulations for treatment of the conditions referred to above in the average adult human are preferably arranged so that each metered dose or "puff of aerosol contains from about 0.01 mg to about 20,000 mg, preferably, about 0.01 mg to about 2000 mg of a compound of the invention, more preferably from about 1 mg to about 200 mg. Administration may be several times daily, for example 2, 3, 4 or 8 times, giving for example, 1 , 2 or 3 doses each time.
- a compound of the invention may be formulated as an ointment or cream.
- prodrugs refers to a ' pharmacologically inactive derivative of a parent drug molecule that requires biotransformation, either spontaneous or enzymatic, within the organism to release the active drug.
- Prodrugs are variations or derivatives of the compounds of this invention which have groups cleavable under metabolic conditions. Prodrugs become the compounds of the invention which are pharmaceutically active in vivo, when they undergo solvolysis under physiological conditions or undergo enzymatic degradation.
- Prodrug compounds of this invention may be called single, double, triple etc., depending on the number of biotransformation steps required to release the active drug within the organism, and indicating the number of functionalities present in a precursor-type form.
- Prodrug forms often offer advantages of solubility, tissue compatibility, or delayed release in the mammalian organism (see, Bundgard, Design of Prodrugs, pp. 7-9, 21-24, Elsevier, Amsterdam 1985 and Silverman, The Organic Chemistry of Drug Design and Drug Action, pp. 352-401, Academic Press, San Diego, Calif, 1992).
- Prodrugs commonly known in the art include acid derivatives well known to practitioners of the art, such as, for example, esters prepared by reaction of the parent acids with a suitable alcohol, or amides prepared by reaction of the parent acid compound with an amine, or basic groups reacted to form an acylated base derivative.
- the prodrug derivatives of this invention may be combined with other features herein taught to enhance bioavailability. For example, a compound of the invention having free amino, amido, hydroxy or carboxylic groups can be converted into prodrugs.
- Prodrugs include compounds wherein an amino acid residue, or a polypeptide chain of two or more (e.g., two, three or four) amino acid residues which are covalently joined through peptide bonds to free amino, hydroxy or carboxylic acid groups of compounds of the invention.
- the amino acid residues include the 20 naturally occurring amino acids commonly designated by three letter symbols and also include, 4-hydroxyproline, hydroxylysine, demosine, isodemosine, 3-methylhistidine, norvalin, beta-alanine, gamma-aminobutyric acid, citrulline homocysteine, homoserine, ornithine and methionine sulfone.
- Prodrugs also include compounds wherein carbonates, carbamates, amides and alkyl esters which are covalently bonded to the above substituents of a compound of the invention through the carbonyl carbon prodrug sidechain.
- a compound of the invention, as described herein, whether alone or as part of a , pharmaceutical composition may be combined with another compound(s) of the invention and/or with another therapeutic agent(s).
- Suitable therapeutic agent(s) include, but are not limited to, standard non-steroidal anti-inflammatory agents (hereinafter NSAID's) (e.g, piroxicam, diclofenac), propionic acids (e.g., naproxen, flubiprofen, fenoprofen, ketoprofen and ibuprofen), fenamates (e.g.
- NSAID's e.g, piroxicam, diclofenac
- propionic acids e.g., naproxen, flubiprofen, fenoprofen, ketoprofen and ibuprofen
- fenamates e.g.
- mefenamic acid indomethacin, sulindac, apazone
- pyrazolones e.g., phenylbutazone
- salicylates e.g., aspirin
- COX-2 inhibitors e.g., celecoxib, valdecoxib, rofecoxib and etoricoxib
- analgesics and intraarticular therapies e.g., corticosteroids
- hyaluronic acids e.g., hyalgan and synvisc
- anticancer agents e.g., endostatin and angiostatin
- cytotoxic drugs e.g., adriamycin, daunomycin, cis-platinum, etoposide, taxol, taxotere
- alkaloids e.g., vincristine
- antimetabolites e.g., methotrexate
- cardiovascular agents e.g., calcium channel block
- a compound of the invention exhibits an in vitro IC50 value of less than about 10 ⁇ M.
- Example 7 exhibits a T ⁇ RI IC50 value of about 51 nM.
- the compounds of the present invention also possess differential activity (i.e. are selective for) for T ⁇ RI over T ⁇ RII and T ⁇ RIII. Selectivity is measured in standard assays as a IC 50 ratio of inhibition in each assay.
- TGF- ⁇ Type II Receptor (T ⁇ RII) Kinase Assay Protocol Phosphorylation of myelin basic protein (MBP) by the T ⁇ RII kinase was measured as follows: 80 microliters of MBP (Upstate Biotechnology #13-104) diluted in kinase reaction buffer (KRB) containing 50 mM MOPS, 5 mM MgC , pH 7.2 to yield a final concentration of 3 micromolar MBP was added to each well of a Millipore 96-well multiscreen-DP 0.65 micron filtration plate (#MADPNOB50). 20 microliters of inhibitor diluted in KRB was added to appropriate wells to yield the desired final concentration (10 - 0.03 micromolar).
- KRB kinase reaction buffer
- MADPNOB50 Millipore 96-well multiscreen-DP 0.65 micron filtration plate
- ALK-5 (T ⁇ Rf) Kinase Assay Protocol The kinase assays were performed with 65 nM GST-ALK5 and 84 nM GST-Smad3 in 50 mM HEPES, 5 mM MgCl 2 ,1 mM CaCl 2 , 1 mM dithiothreitol, and 3 _M ATP. Reactions were incubated with 0.5 Ci of [ 33 P]_ATPfor 3 h at 30°C. Phosphorylated protein was captured on P-81 paper (Whatman, Maidstone,' England), washed with 0.5% phosphoric acid, and counted by liquid scintillation.
- Smad3 or Smadl protein was also coated onto FlashPlate Sterile Basic Microplates (PerkinElmer Life Sciences, Boston, MA). Kinase assays were then performed in Flash-Plates with same assay conditions using either the kinase domain of ALK5 with Smad3 as substrate or the kinase domain of ALK6 (BMP receptor) with Smadl as substrate. Plates were washed three times with phosphate buffer and counted by TopCount (Packard Bio-science, Meriden, CT). (Laping, N.J. et al. Molecular Pharmacology 62:58-64 (2002)). The following Examples illustrate the preparation of the compounds of the present invention. Melting points are uncorrected.
- NMR data are reported in parts per million (d) and are referenced to the deuterium lock signal from the sample solvent (deuteriochloroform unless otherwise specified).
- Mass Spectral data were obtained using a Micromass ZMD APCI Mass Spectrometer equipped with a Gilson gradient high performance liquid chromatograph. The following solvents and gradients were used for the analysis. Solvent A; 98% water/2% acetonirile/0.01% formic acid and solvent B; acetonitrile containing 0.005% formic acid. Typically, a gradient was run over a period of about 4 minutes starting at 95% solvent A and ending with 100% solvent B.
- the mass spectrum of the major eluting component was then obtained in positive or negative ion mode scanning a molecular weight range from 165 AMU to 1100 AMU. Specific rotations were measured at room temperature using the sodium D line (589 nm). Commercial reagents were utilized without further purification.
- THF refers to tetrahydrofuran.
- DMF refers to N,N-dimethylformamide.
- Chromatography refers to column chromatography performed using 32-63 mm silica gel and executed under nitrogen pressure (flash chromatography) conditions. Room or ambient temperature refers to 20-25 °C. All non-aqueous reactions were run under a nitrogen atmosphere for convenience and to maximize yields. Concentration at reduced pressure means that a rotary evaporator was used.
- protecting groups may be required during preparation. After the target molecule is made, the protecting group can be removed by methods well known to those of ordinary skill in the art, such as described in Greene and Wuts, "Protective Groups in Organic Synthesis” (2 nd Ed, John Wiley & Sons 1991).
- Step A Preparation of [(6-Methyl-pyridyl-2-yl)-phenylamino-methyll-phosphonic acid diphenyl ester
- Step B Preparation of l-(6-Methyl-pyridyl-2-ylV2-quinolin-4-yl-ethanone
- a 2L round-bottom flask was charged with [(6-Methyl-pyridyl-2-yl)- phenylamino-methyl]-phosphonic acid diphenyl ester (24.5 g, 57 mmol), quinoline- 4-carbaldehyde (11.65 g, 74.1 mmol), 90 mL of tetrahydrofuran, and 180 mL of isopropylalcohol.
- Step C Preparation of 4- 3-(6-Methyl-pyridyl-2-ylVlH-pyrazol-4-yl-quinoline
- Example 5 2-(4-Benzo[l,3]dioxol-5-yl-l-methyl-lH-pyrazol-3-yl)-6-methyl-pyridine
- HPLC t R 4.45 min
- LC-MS 294 (M+l).
- Example 6 4-[ 1 -Methyl-3-(6-methyl-pyridin-2-yl)- 1 H-pyrazol-4-yl]-quinoline
- HPLC t R 4.17 min
- LC-MS 301 (M+l).
- Example 8
- Example 9 l-Methyl-6-[l-methyl-3-(6-methyl-pyridin-2-yl)-lH-pyrazol-4-yl]-lH-benzotriazole
- HPLC t R 3.79 min
- LC-MS 305 (M+l).
- Example 10 4-(l -Methyl-3-pyridin-2-yl-lH-pyrazol-4-yl)-quinoline M il
- Example 12 6-[3-(6-Methyl-pyridin-2-yl)-lH-pyrazol-4-yl]-2-trifluoromethyl-[l,2,4]triazolo[l,5- a]pyridine
- HPLC t R 4.77 min
- LC-MS 345 (M+l).
- Example 14 3-Methyl-6-[3-(6-methyl-pyridin-2-yl)-lH-pyrazol-4-yl]-[l,2,4]triazolo[4,3- a]pyridine
- HPLC t R 2.77 min
- LC-MS 291 (M+l).
- Example 15 2-Methyl-6-[3-(6-methyl-pyridin-2-yl)-l H-pyrazol-4-yl]-[ 1 ,2,4]triazolo[ 1 ,5- a]pyridine
- HPLC t R 3.38 min
- LC-MS 291 (M+l).
- Step B Preparation of 4-Methoxy-6-[3-(6-methyl-p idin-2-ylVlH-pyrazol-4-v ⁇ ⁇
- 2-(4-Methoxy-quinolin-6-yl)-l-(6-methyl- pyridin-2-yl)-ethanone 70 mg, 0.17 mmol
- N,N-dimethylformamide dimethyl acetal 1.25 mmol
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Diabetes (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Dermatology (AREA)
- Urology & Nephrology (AREA)
- Endocrinology (AREA)
- Vascular Medicine (AREA)
- Gastroenterology & Hepatology (AREA)
- Pulmonology (AREA)
- Emergency Medicine (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description













































Claims






Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT03797443T ATE450258T1 (en) | 2002-09-18 | 2003-09-08 | NEW PYRAZOLE COMPOUNDS AS TRANSFORMING GROWTH FACTOR (TGF) INHIBITORS |
| DE60330362T DE60330362D1 (en) | 2002-09-18 | 2003-09-08 | NEW PYRAZOL COMPOUNDS AS AN INHIBITOR OF THE TRANSFORMING GROWTH FACTOR (TGF) |
| BR0314302-3A BR0314302A (en) | 2002-09-18 | 2003-09-08 | Pyrazole Compounds as Transforming Growth Factor (TGF) Inhibitors |
| CA2496295A CA2496295C (en) | 2002-09-18 | 2003-09-08 | Novels pyrazole compounds as transforming growth factor (tgf) inhibitors |
| MXPA05002376A MXPA05002376A (en) | 2002-09-18 | 2003-09-08 | Pyrazole derivatives as transforming growth factor (tgf) inhibitors. |
| EP03797443A EP1542684B1 (en) | 2002-09-18 | 2003-09-08 | Novel pyrazole compounds as transforming growth factor (tgf) inhibitors |
| EA200500377A EA200500377A1 (en) | 2002-09-18 | 2003-09-08 | PYRAZOL DERIVATIVES AS INHIBITORS OF THE TRANSFORMING GROWTH FACTOR (TGF) |
| AP2005003263A AP2005003263A0 (en) | 2002-09-18 | 2003-09-08 | Pyrazole derivatives as transforming growth factor(TGF) inhibitors. |
| AU2003259475A AU2003259475A1 (en) | 2002-09-18 | 2003-09-08 | Pyrazole derivatives as transforming growth factor (TGF) inhibitors |
| JP2004568904A JP4519657B2 (en) | 2002-09-18 | 2003-09-08 | Novel pyrazole compounds as transforming growth factor (TGF) inhibitors |
| NO20050838A NO20050838L (en) | 2002-09-18 | 2005-02-16 | Novel pyrazole compounds as transforming growth factor (TGF) inhibitors |
| IS7698A IS7698A (en) | 2002-09-18 | 2005-02-17 | Pyrazole compounds that inhibit growth factor (TGF) metabolism |
| HR20050247A HRP20050247A2 (en) | 2002-09-18 | 2005-03-16 | Pyrazole derivatives as transforming growth factor (tgf) inhibitors |
| TNP2005000082A TNSN05082A1 (en) | 2002-09-18 | 2005-03-18 | PYRAZOLE DERIVATIVES AS INHIBITORS OF TRANSFORMING GROWTH FACTOR (TGF) |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US41214602P | 2002-09-18 | 2002-09-18 | |
| US60/412,146 | 2002-09-18 | ||
| US48454303P | 2003-07-02 | 2003-07-02 | |
| US60/484,543 | 2003-07-02 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004026306A2 true WO2004026306A2 (en) | 2004-04-01 |
| WO2004026306A3 WO2004026306A3 (en) | 2004-07-01 |
Family
ID=32033585
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2003/003933 WO2004026306A2 (en) | 2002-09-18 | 2003-09-08 | Pyrazole derivatives as transforming growth factor (tgf) inhibitors |
Country Status (27)
| Country | Link |
|---|---|
| US (4) | US6958354B2 (en) |
| EP (2) | EP2165708A3 (en) |
| JP (1) | JP4519657B2 (en) |
| KR (1) | KR20050044807A (en) |
| CN (1) | CN1681501A (en) |
| AP (1) | AP2005003263A0 (en) |
| AR (1) | AR041272A1 (en) |
| AT (1) | ATE450258T1 (en) |
| AU (1) | AU2003259475A1 (en) |
| BR (1) | BR0314302A (en) |
| CA (1) | CA2496295C (en) |
| CO (1) | CO5550456A2 (en) |
| DE (1) | DE60330362D1 (en) |
| EA (1) | EA200500377A1 (en) |
| EC (1) | ECSP055679A (en) |
| ES (1) | ES2335099T3 (en) |
| HR (1) | HRP20050247A2 (en) |
| IS (1) | IS7698A (en) |
| MA (1) | MA27440A1 (en) |
| MX (1) | MXPA05002376A (en) |
| NO (1) | NO20050838L (en) |
| OA (1) | OA12929A (en) |
| PA (1) | PA8583201A1 (en) |
| PE (1) | PE20040988A1 (en) |
| PL (1) | PL375979A1 (en) |
| UY (1) | UY27985A1 (en) |
| WO (1) | WO2004026306A2 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004111036A1 (en) * | 2003-06-16 | 2004-12-23 | Smithkline Beecham Corporation | 4- (heterocyclyl- fused phenyl)- 3- (phenyl or pyrid -2- yl) pyrazoles as inhibitors of the alk-5- receptor |
| EP1596656A2 (en) * | 2003-02-12 | 2005-11-23 | Biogen Idec MA Inc. | Pyrazoles and methods of making and using the same |
| WO2006026305A1 (en) * | 2004-08-31 | 2006-03-09 | Biogen Idec Ma Inc | Pyrimidinylpyrazoles as tgf-beta inhibitors |
| WO2006028029A1 (en) * | 2004-09-07 | 2006-03-16 | Sankyo Company, Limited | Substituted biphenyl derivative |
| WO2006044509A2 (en) * | 2004-10-15 | 2006-04-27 | Biogen Idec Ma Inc. | Methods of treating vascular injuries |
| WO2009016460A2 (en) * | 2007-08-01 | 2009-02-05 | Pfizer Inc. | Pyrazole compounds and their use as raf inhibitors |
| US7781428B2 (en) | 2006-10-31 | 2010-08-24 | Pfizer Inc. | Pyrazoline compounds |
| WO2016210292A1 (en) | 2015-06-25 | 2016-12-29 | Children's Medical Center Corporation | Methods and compositions relating to hematopoietic stem cell expansion, enrichment, and maintenance |
| WO2017161001A1 (en) | 2016-03-15 | 2017-09-21 | Children's Medical Center Corporation | Methods and compositions relating to hematopoietic stem cell expansion |
| WO2020003219A1 (en) * | 2018-06-29 | 2020-01-02 | Oat & Iil India Laboratories Private Limited | Substituted pyrazole derivatives as insecticides and fungicides |
| US10548883B2 (en) | 2016-06-13 | 2020-02-04 | Genfleet Therapeutics (Shanghai) Inc. | Benzotriazole-derived α and β-unsaturated amide compound used as TGF-β RI inhibitor |
| EP3539957A4 (en) * | 2016-11-14 | 2020-05-13 | Jiangsu Hengrui Medicine Co., Ltd. | 3,4-bipyridyl pyrazole derivative, and preparation method therefor and medical application thereof |
| US10822337B2 (en) | 2015-04-01 | 2020-11-03 | Rigel Pharmaceuticals, Inc. | TGF-β inhibitorC |
| US10952996B2 (en) | 2018-12-11 | 2021-03-23 | Theravance Biopharma R&D Ip, Llc | ALK5 inhibitors |
| US11590116B2 (en) | 2019-11-22 | 2023-02-28 | Theravance Biopharma R&D Ip, Llc | Substituted pyridines and methods of use |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2499429C (en) | 2002-09-18 | 2010-09-21 | Pfizer Products Inc. | Novel oxazole and thiazole compounds as transforming growth factor (tgf) inhibitors |
| DE60328028D1 (en) * | 2002-09-18 | 2009-07-30 | Pfizer Prod Inc | IMIDAZOL COMPOUNDS AS INHIBITORS OF THE TRANSFORMING GROWTH FACTOR (TWF) |
| WO2004026306A2 (en) * | 2002-09-18 | 2004-04-01 | Pfizer Products Inc. | Pyrazole derivatives as transforming growth factor (tgf) inhibitors |
| PA8595001A1 (en) * | 2003-03-04 | 2004-09-28 | Pfizer Prod Inc | NEW CONDENSED HETEROAROMATIC COMPOUNDS THAT ARE INHIBITORS OF THE TRANSFORMING GROWTH FACTOR (TGF) |
| TW200639163A (en) * | 2005-02-04 | 2006-11-16 | Genentech Inc | RAF inhibitor compounds and methods |
| CN101330914A (en) * | 2005-12-16 | 2008-12-24 | 爱尔康公司 | Control of intraocular pressure using ALK5 modulation agents |
| CN106132950B (en) | 2014-01-01 | 2018-11-09 | 麦迪威森技术有限责任公司 | Aminopyridines and application method |
| CA3039220A1 (en) * | 2016-10-26 | 2018-05-03 | Icahn School Of Medicine At Mount Sinai | Method for increasing cell proliferation in pancreatic beta cells, treatment method, and composition |
| EP3717475B1 (en) | 2017-11-20 | 2023-06-07 | Icahn School of Medicine at Mount Sinai | Kinase inhibitor compounds and compositions and methods of use |
| ES2960412T3 (en) * | 2017-12-13 | 2024-03-04 | Genfleet Therapeutics Shanghai Inc | Crystalline form and salt form of the TGF-?RI inhibitor and method of preparation thereof |
| JP2021510153A (en) | 2018-01-05 | 2021-04-15 | アイカーン スクール オブ メディシン アット マウント サイナイ | Methods, Therapeutic Methods, and Compositions to Increase Pancreatic Beta Cell Proliferation |
| US11866427B2 (en) | 2018-03-20 | 2024-01-09 | Icahn School Of Medicine At Mount Sinai | Kinase inhibitor compounds and compositions and methods of use |
| EP3886854A4 (en) | 2018-11-30 | 2022-07-06 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
| CN110467601B (en) * | 2019-08-29 | 2021-07-02 | 杭州市西溪医院 | Pyrazole bipyridyl ketone compound, intermediate, preparation method and application thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0388691A1 (en) * | 1989-03-22 | 1990-09-26 | Sterling Drug Inc. | Azole-1-alkanamides as antiarrhytmic agents and preparation thereof |
| WO2002062794A2 (en) * | 2001-02-02 | 2002-08-15 | Glaxo Group Limited | Compounds |
| WO2002062787A1 (en) * | 2001-02-02 | 2002-08-15 | Glaxo Group Limited | Pyrazoles as tgf inhibitors |
| WO2002066462A1 (en) * | 2001-02-02 | 2002-08-29 | Glaxo Group Limited | Pyrazole derivatives against tgf overexpression |
| WO2002088107A1 (en) * | 2001-04-26 | 2002-11-07 | Eisai Co., Ltd. | Nitrogenous fused-ring compound having pyrazolyl group as substituent and medicinal composition thereof |
Family Cites Families (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE640616A (en) | 1962-12-19 | |||
| US3492397A (en) | 1967-04-07 | 1970-01-27 | Warner Lambert Pharmaceutical | Sustained release dosage in the pellet form and process thereof |
| US4060598A (en) | 1967-06-28 | 1977-11-29 | Boehringer Mannheim G.M.B.H. | Tablets coated with aqueous resin dispersions |
| US3538214A (en) | 1969-04-22 | 1970-11-03 | Merck & Co Inc | Controlled release medicinal tablets |
| US4173626A (en) | 1978-12-11 | 1979-11-06 | Merck & Co., Inc. | Sustained release indomethacin |
| JP3734180B2 (en) * | 1994-12-28 | 2006-01-11 | エーザイ株式会社 | New pyrazole derivatives |
| US6514977B1 (en) | 1997-05-22 | 2003-02-04 | G.D. Searle & Company | Substituted pyrazoles as p38 kinase inhibitors |
| AU7726898A (en) | 1997-05-22 | 1998-12-11 | G.D. Searle & Co. | Pyrazole derivatives as p38 kinase inhibitors |
| EP0983260A2 (en) | 1997-05-22 | 2000-03-08 | G.D. Searle & Co. | 3(5)-HETEROARYL SUBSTITUTED PYRAZOLES AS p38 KINASE INHIBITORS |
| US6465493B1 (en) | 1999-04-09 | 2002-10-15 | Smithkline Beecham Corporation | Triarylimidazoles |
| AR029803A1 (en) | 2000-02-21 | 2003-07-16 | Smithkline Beecham Plc | IMIDAZOLS REPLACED WITH PIRIDILE AND PHARMACEUTICAL COMPOSITIONS THAT UNDERSTAND THEM |
| GB0007405D0 (en) | 2000-03-27 | 2000-05-17 | Smithkline Beecham Corp | Compounds |
| PE20020506A1 (en) | 2000-08-22 | 2002-07-09 | Glaxo Group Ltd | PIRAZOLE DERIVATIVES FUSED AS PROTEIN KINASE INHIBITORS |
| GB0027987D0 (en) | 2000-11-16 | 2001-01-03 | Smithkline Beecham Plc | Compounds |
| JP2004517068A (en) | 2000-11-16 | 2004-06-10 | スミスクライン・ビーチャム・コーポレイション | Compound |
| GB0100762D0 (en) | 2001-01-11 | 2001-02-21 | Smithkline Beecham Plc | Novel use |
| EP1370557B1 (en) | 2001-03-09 | 2005-11-16 | Pfizer Products Inc. | Benzimidazole anti-inflammatory compounds |
| AR039241A1 (en) | 2002-04-04 | 2005-02-16 | Biogen Inc | HETEROARILOS TRISUSTITUIDOS AND METHODS FOR ITS PRODUCTION AND USE OF THE SAME |
| US20050245520A1 (en) | 2002-07-31 | 2005-11-03 | Nerina Dodic | 2-Phenylpyridin-4-yl derivatives as alk5 inhibitors |
| DE60328028D1 (en) | 2002-09-18 | 2009-07-30 | Pfizer Prod Inc | IMIDAZOL COMPOUNDS AS INHIBITORS OF THE TRANSFORMING GROWTH FACTOR (TWF) |
| EP1542995A1 (en) | 2002-09-18 | 2005-06-22 | Pfizer Products Inc. | Novel isothiazole and isoxazole compounds as transforming growth factor (tgf) inhibitors |
| BR0314577A (en) | 2002-09-18 | 2005-08-09 | Pfizer Prod Inc | Triazole derivatives as transforming growth factor (tgf) inhibitors |
| CA2499429C (en) | 2002-09-18 | 2010-09-21 | Pfizer Products Inc. | Novel oxazole and thiazole compounds as transforming growth factor (tgf) inhibitors |
| WO2004026306A2 (en) | 2002-09-18 | 2004-04-01 | Pfizer Products Inc. | Pyrazole derivatives as transforming growth factor (tgf) inhibitors |
| PA8595001A1 (en) | 2003-03-04 | 2004-09-28 | Pfizer Prod Inc | NEW CONDENSED HETEROAROMATIC COMPOUNDS THAT ARE INHIBITORS OF THE TRANSFORMING GROWTH FACTOR (TGF) |
-
2003
- 2003-09-08 WO PCT/IB2003/003933 patent/WO2004026306A2/en active Application Filing
- 2003-09-08 AP AP2005003263A patent/AP2005003263A0/en unknown
- 2003-09-08 OA OA1200500078A patent/OA12929A/en unknown
- 2003-09-08 CA CA2496295A patent/CA2496295C/en not_active Expired - Fee Related
- 2003-09-08 ES ES03797443T patent/ES2335099T3/en not_active Expired - Lifetime
- 2003-09-08 KR KR1020057004598A patent/KR20050044807A/en not_active Application Discontinuation
- 2003-09-08 AU AU2003259475A patent/AU2003259475A1/en not_active Abandoned
- 2003-09-08 DE DE60330362T patent/DE60330362D1/en not_active Expired - Lifetime
- 2003-09-08 JP JP2004568904A patent/JP4519657B2/en not_active Expired - Fee Related
- 2003-09-08 CN CNA038222655A patent/CN1681501A/en active Pending
- 2003-09-08 BR BR0314302-3A patent/BR0314302A/en not_active IP Right Cessation
- 2003-09-08 PL PL03375979A patent/PL375979A1/en not_active Application Discontinuation
- 2003-09-08 EP EP09177298A patent/EP2165708A3/en not_active Withdrawn
- 2003-09-08 MX MXPA05002376A patent/MXPA05002376A/en active IP Right Grant
- 2003-09-08 EP EP03797443A patent/EP1542684B1/en not_active Expired - Lifetime
- 2003-09-08 EA EA200500377A patent/EA200500377A1/en unknown
- 2003-09-08 AT AT03797443T patent/ATE450258T1/en not_active IP Right Cessation
- 2003-09-15 PE PE2003000940A patent/PE20040988A1/en not_active Application Discontinuation
- 2003-09-16 UY UY27985A patent/UY27985A1/en not_active Application Discontinuation
- 2003-09-16 AR ARP030103356A patent/AR041272A1/en unknown
- 2003-09-17 PA PA20038583201A patent/PA8583201A1/en unknown
- 2003-09-17 US US10/667,189 patent/US6958354B2/en not_active Expired - Fee Related
-
2005
- 2005-02-16 NO NO20050838A patent/NO20050838L/en unknown
- 2005-02-17 IS IS7698A patent/IS7698A/en unknown
- 2005-03-16 HR HR20050247A patent/HRP20050247A2/en not_active Application Discontinuation
- 2005-03-16 CO CO05024529A patent/CO5550456A2/en not_active Application Discontinuation
- 2005-03-16 EC EC2005005679A patent/ECSP055679A/en unknown
- 2005-03-18 MA MA28154A patent/MA27440A1/en unknown
- 2005-09-23 US US11/234,564 patent/US7151110B2/en not_active Expired - Fee Related
-
2006
- 2006-11-08 US US11/557,638 patent/US7638537B2/en not_active Expired - Fee Related
-
2009
- 2009-11-12 US US12/617,027 patent/US20100056571A1/en not_active Abandoned
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0388691A1 (en) * | 1989-03-22 | 1990-09-26 | Sterling Drug Inc. | Azole-1-alkanamides as antiarrhytmic agents and preparation thereof |
| WO2002062794A2 (en) * | 2001-02-02 | 2002-08-15 | Glaxo Group Limited | Compounds |
| WO2002062787A1 (en) * | 2001-02-02 | 2002-08-15 | Glaxo Group Limited | Pyrazoles as tgf inhibitors |
| WO2002066462A1 (en) * | 2001-02-02 | 2002-08-29 | Glaxo Group Limited | Pyrazole derivatives against tgf overexpression |
| WO2002088107A1 (en) * | 2001-04-26 | 2002-11-07 | Eisai Co., Ltd. | Nitrogenous fused-ring compound having pyrazolyl group as substituent and medicinal composition thereof |
Non-Patent Citations (5)
| Title |
|---|
| DATABASE CAPLUS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; retrieved from STN, accession no. 1993:472619 Database accession no. 119:72619 XP002262349 * |
| DATABASE CAPLUS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; retrieved from STN, accession no. 1996:607249 Database accession no. 125:247806 XP002262351 * |
| DATABASE CAPLUS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; retrieved from STN, accession no. 2000:881141 Database accession no. 134:29414 XP002262350 * |
| DATABASE CROSSFIRE BEILSTEIN [Online] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; Database accession no. 1181440 XP002262348 * |
| DATABASE CROSSFIRE BEILSTEIN [Online] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; Database accession no. 1220267 XP002262347 * |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1596656A2 (en) * | 2003-02-12 | 2005-11-23 | Biogen Idec MA Inc. | Pyrazoles and methods of making and using the same |
| JP2006517592A (en) * | 2003-02-12 | 2006-07-27 | バイオジェン・アイデック・エムエイ・インコーポレイテッド | Pyrazole and methods of making and using them |
| EP1596656A4 (en) * | 2003-02-12 | 2006-10-18 | Biogen Idec Inc | Pyrazoles and methods of making and using the same |
| WO2004111036A1 (en) * | 2003-06-16 | 2004-12-23 | Smithkline Beecham Corporation | 4- (heterocyclyl- fused phenyl)- 3- (phenyl or pyrid -2- yl) pyrazoles as inhibitors of the alk-5- receptor |
| WO2006026305A1 (en) * | 2004-08-31 | 2006-03-09 | Biogen Idec Ma Inc | Pyrimidinylpyrazoles as tgf-beta inhibitors |
| WO2006028029A1 (en) * | 2004-09-07 | 2006-03-16 | Sankyo Company, Limited | Substituted biphenyl derivative |
| WO2006044509A2 (en) * | 2004-10-15 | 2006-04-27 | Biogen Idec Ma Inc. | Methods of treating vascular injuries |
| WO2006044509A3 (en) * | 2004-10-15 | 2006-08-17 | Biogen Idec Inc | Methods of treating vascular injuries |
| US7781428B2 (en) | 2006-10-31 | 2010-08-24 | Pfizer Inc. | Pyrazoline compounds |
| CN101815712A (en) * | 2007-08-01 | 2010-08-25 | 辉瑞有限公司 | Pyrazole compound and as the purposes of RAF inhibitor |
| WO2009016460A3 (en) * | 2007-08-01 | 2009-03-26 | Pfizer | Pyrazole compounds and their use as raf inhibitors |
| WO2009016460A2 (en) * | 2007-08-01 | 2009-02-05 | Pfizer Inc. | Pyrazole compounds and their use as raf inhibitors |
| US7772246B2 (en) | 2007-08-01 | 2010-08-10 | Pfizer Inc. | Pyrazole compounds as RAF inhibitors |
| US10822337B2 (en) | 2015-04-01 | 2020-11-03 | Rigel Pharmaceuticals, Inc. | TGF-β inhibitorC |
| US11466018B2 (en) | 2015-04-01 | 2022-10-11 | Rigel Pharmaceuticals, Inc. | TGF-β inhibitors |
| WO2016210292A1 (en) | 2015-06-25 | 2016-12-29 | Children's Medical Center Corporation | Methods and compositions relating to hematopoietic stem cell expansion, enrichment, and maintenance |
| WO2017161001A1 (en) | 2016-03-15 | 2017-09-21 | Children's Medical Center Corporation | Methods and compositions relating to hematopoietic stem cell expansion |
| EP4049665A1 (en) | 2016-03-15 | 2022-08-31 | Children's Medical Center Corporation | Methods and compositions relating to hematopoietic stem cell expansion |
| US10548883B2 (en) | 2016-06-13 | 2020-02-04 | Genfleet Therapeutics (Shanghai) Inc. | Benzotriazole-derived α and β-unsaturated amide compound used as TGF-β RI inhibitor |
| US10899741B2 (en) | 2016-11-14 | 2021-01-26 | Jiangsu Hengrui Medicine Co., Ltd. | 3,4-bipyridyl pyrazole derivative, and preparation method therefor and medical application thereof |
| EP3539957A4 (en) * | 2016-11-14 | 2020-05-13 | Jiangsu Hengrui Medicine Co., Ltd. | 3,4-bipyridyl pyrazole derivative, and preparation method therefor and medical application thereof |
| WO2020003219A1 (en) * | 2018-06-29 | 2020-01-02 | Oat & Iil India Laboratories Private Limited | Substituted pyrazole derivatives as insecticides and fungicides |
| US10952996B2 (en) | 2018-12-11 | 2021-03-23 | Theravance Biopharma R&D Ip, Llc | ALK5 inhibitors |
| US11730720B2 (en) | 2018-12-11 | 2023-08-22 | Theravance Biopharma R&D Ip, Llc | ALK5 inhibitors |
| US11590116B2 (en) | 2019-11-22 | 2023-02-28 | Theravance Biopharma R&D Ip, Llc | Substituted pyridines and methods of use |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7638537B2 (en) | Pyrazole compounds as transforming growth factor (TGF) inhibitors | |
| EP1542990B1 (en) | Novel imidazole compounds as transforming growth factor (tgf) inhibitors | |
| US7053095B2 (en) | Triazole compounds as transforming growth factor (TGF) inhibitors | |
| US7030125B2 (en) | Isothiazole and isoxazole compounds as transforming growth factor (TGF) inhibitors | |
| US7417041B2 (en) | Imidazopyrimidines as transforming growth factor (TGF) inhibitors | |
| EP1542994A1 (en) | Novel triazole and oxazole compounds as transforming growth factor (tgf) inhibitor | |
| ZA200502277B (en) | Novel isothiazole and isoxazole compounds as transforming growth factor (TGF) inhibitors. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003259475 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200501335 Country of ref document: ZA |
|
| ENP | Entry into the national phase |
Ref document number: 2496295 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 651/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 167096 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2005/002376 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003797443 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2005/0220 Country of ref document: YU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 538831 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20050247A Country of ref document: HR Ref document number: 375979 Country of ref document: PL Ref document number: 05024529 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AP/P/2005/003263 Country of ref document: AP Ref document number: 1020057004598 Country of ref document: KR Ref document number: 2004568904 Country of ref document: JP Ref document number: 200500377 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 8693 Country of ref document: GE Ref document number: 20050094 Country of ref document: UZ Ref document number: 20038222655 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057004598 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003797443 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1-2005-500482 Country of ref document: PH |