WO2004018441A1 - Aryl and heteroaryl morpholine derivatives - Google Patents
Aryl and heteroaryl morpholine derivatives Download PDFInfo
- Publication number
- WO2004018441A1 WO2004018441A1 PCT/US2003/023270 US0323270W WO2004018441A1 WO 2004018441 A1 WO2004018441 A1 WO 2004018441A1 US 0323270 W US0323270 W US 0323270W WO 2004018441 A1 WO2004018441 A1 WO 2004018441A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- alkyl
- disorder
- compound
- morpholin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(*)(C[Al])C1(*)C(*)(*)NC(*)(*)C(*)(*)*1 Chemical compound *C(*)(C[Al])C1(*)C(*)(*)NC(*)(*)C(*)(*)*1 0.000 description 3
- ASEOGQBTBQRHCW-MBMZGMDYSA-N CC(C)Oc1c(CC([C@H]2OCCN(Cc3ccccc3)C2)(c2ccccc2)O)cccc1 Chemical compound CC(C)Oc1c(CC([C@H]2OCCN(Cc3ccccc3)C2)(c2ccccc2)O)cccc1 ASEOGQBTBQRHCW-MBMZGMDYSA-N 0.000 description 1
- JFZHXNMYYRMSLW-PMCHYTPCSA-N COc1c(CC([C@H]2OCCN(Cc3ccccc3)C2)(c2ccccc2)O)cccc1 Chemical compound COc1c(CC([C@H]2OCCN(Cc3ccccc3)C2)(c2ccccc2)O)cccc1 JFZHXNMYYRMSLW-PMCHYTPCSA-N 0.000 description 1
- QPAFLDILQMKTGS-OYKVQYDMSA-N COc1c(CC([C@H]2OCCNC2)(c2ccccc2)O)cccc1 Chemical compound COc1c(CC([C@H]2OCCNC2)(c2ccccc2)O)cccc1 QPAFLDILQMKTGS-OYKVQYDMSA-N 0.000 description 1
- RWMNZSMOBIYZCE-SKCDSABHSA-N OC(Cc(cccc1)c1OC(F)(F)F)([C@H]1OCCN(Cc2ccccc2)C1)c1ccccc1 Chemical compound OC(Cc(cccc1)c1OC(F)(F)F)([C@H]1OCCN(Cc2ccccc2)C1)c1ccccc1 RWMNZSMOBIYZCE-SKCDSABHSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/28—1,4-Oxazines; Hydrogenated 1,4-oxazines
- C07D265/30—1,4-Oxazines; Hydrogenated 1,4-oxazines not condensed with other rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
Definitions
- This invention relates to novel aryl and heteroaryl morpholine compounds, and to their use in selectively inhibiting norepinephrine reuptake.
- Norepinephrine appears to play an important role in the disturbances of vegetative function associated with affective, anxiety and cognitive disorders.
- Atomoxetine hydrochloiide is a selective inhibitor of norepinephrine reuptake, and is marketed for the treatment of attention deficit hyperactivity disorder (ADHD).
- Reboxetine is also a selective norepinephrine reuptake inhibitor, and is marketed for the treatment of depression.
- WO99/15177 discloses the use of Reboxetine to treat ADHD and WOO 1/01973 discloses the use of S,S-Reboxetine to treat ADHD.
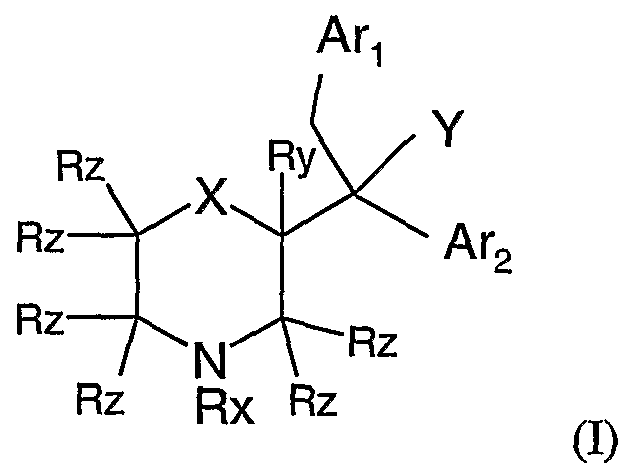
- Rx is H
- Ry is H or C ⁇ -C 4 alkyl; each Rz is independently H or Ci-C alkyl;
- X represents O
- Y represents OH or OR
- R is C ⁇ -C 4 alkyl
- Ax ⁇ is a phenyl ring or a 5- or 6-membered heteroaryl ring each of which may be substituted with 1, 2, 3, 4 or 5 substituents (depending upon the number of available substitution positions) each independently selected from C1-C4 alkyl, O(C ⁇ C4 alkyl), S(C ⁇ -C4 alkyl), halo, hydroxy, pyridyl, thiophenyl and phenyl optionally substituted with 1, 2, 3, 4 or 5 substituents each independently selected from halo,
- a ⁇ 2 is a phenyl ring or a 5- or 6-membered heteroaryl ring each of which may be substituted with 1, 2, 3, 4 or 5 substituents (depending upon the number of available substitution positions) each independently selected from C1-C4 alkyl, O(C ⁇ -C4 alkyl) and halo; wherein each above-mentioned C1-C4 alkyl group is optionally substituted with one or more halo atoms; or a pharmaceutically acceptable salt thereof.
- substituents depending upon the number of available substitution positions
- Arj is phenyl, pyridyl, pyrimidyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, thiophenyl, furanyl, imidazolyl, triazolyl, oxadiazolyl or thiadiazolyl, each of which may be substituted with 1, 2, 3, 4 or 5 substituents (depending upon the number of available substitution positions) each independently selected from C1-C4 alkyl,
- Ar2 is phenyl, pyridyl, pyrimidyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, thiophenyl, furanyl, imidazolyl or triazolyl each of which may be substituted with 1,
- Ry is H or - alkyl; each Rz is independently H or C ⁇ -C 4 alkyl; X represents O; Y represents OH or OR; R is C 1 -C 4 alkyl; and
- Arj and A ⁇ 2 are each independently selected from the group consisting of phenyl, and substituted phenyl; and pharmaceutically acceptable salts thereof.
- the group Ari may be substituted or unsubstituted phenyl.
- Ari may be unsubstituted phenyl or, preferably phenyl substituted with 1, 2, 3, 4 or 5 substituents, preferably with 1 or 2, for example 1, substituent.
- the substituted phenyl group is preferably substituted at the 2- and 5- positions.
- the substituted phenyl group is preferably substituted in the 2- position.
- Suitable substituents include C1-C4 alkyl, O(Cj-C4 alkyl), S(C -C alkyl), halo, and phenyl, optionally substituted with, for example, halo, C1-C4 alkyl, or O(C ⁇ C4 alkyl).
- the group Ar 2 may be substituted or unsubstituted phenyl.
- Ar 2 may be phenyl substituted with 1, 2, 3, 4 or 5 substituents, preferably with 1 substituent.
- Suitable substituents include C1-C4 alkyl, O(C ⁇ -C4 alkyl), and especially, halo.
- C1-C4 alkyl as used herein includes straight and branched chain alkyl groups of 1, 2, 3 or 4 carbon atoms, and may be unsubstituted or substituted. C1-C2 alkyl groups are preferred. Suitable substituents include halo, especially CI and/or F. Thus the term “C1-C4 alkyl” includes haloalkyl. A particularly preferred substituted C1-C4 alkyl group is trifluoromethyl. Similar terms defining different numbers of C atoms (e.g. "C1-C3 alkyl”) take an analogous meaning. When Ry is C1-C4 alkyl it is preferably unsubstituted.
- Rz is C1-C4 alkyl it is preferably unsubstituted.
- R is C1-C4 alkyl it is preferably unsubstituted.
- "5-membered heteroaryl ring” as used herein means a 5-membered aromatic ring including at least one heteroatom independently selected from N, O and S. Preferably there are not more than three heteroatoms in total in the ring. More preferably there are not more than two heteroatoms in total in the ring. More preferably there is not more than one heteroatom in total in the ring.
- the term includes, for example, the groups thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, thiophenyl, furanyl, pyrrolyl, imidazolyl, triazolyl, oxadiazolyl and thiadiazolyl.
- 6-membered heteroaryl ring as used herein means a 6-membered aromatic ring including at least one heteroatom independently selected from N, O and S. Preferably there are not more than three heteroatoms in total in the ring. More preferably there are not more than two heteroatoms in total in the ring. More preferably there is not more than one heteroatom in total in the ring.
- the term includes, for example, the groups pyridyl, pyrimidyl, pyrazinyl, pyridazinyl and triazinyl.
- Halo includes F, CI, Br and I, and is preferably F or CI.
- Pyridyl as used herein includes 2-pyridyl, 3-pyridyl and 4-pyridyl.
- Polyrimidyl as used herein includes 2-pyrimidyl, 4-pyrimidyl and 5- pyrimidyl.
- Pyridazinyl as used herein includes 3-pyridazinyl and 4-pyridazinyl.
- Pyrazinyl as used herein includes 2-pyrazinyl and 3-pyrazinyl.
- Triazinyl as used herein includes 2-(l,3,5-triazinyl), 3-, 5- and 6-(l,2,4- triazinyl) and 4- and 5-(l,2,3-triazinyl).
- Thiazolyl as used herein includes 2-thiazolyl, 4-thiazolyl and 5-thiazolyl.
- Isothiazolyl as used herein includes 3-isothiazolyl, 4-isothiazolyl, and 5- isothiazolyl.
- Oxazolyl as used herein includes 2-oxazolyl, 4-oxazolyl and 5-oxazolyl.
- Isoxazolyl as used herein includes 3-isoxazolyl, 4-isoxazolyl, and 5- isoxazolyl.
- Thiophenyl as used herein includes 2-thiophenyl and 3-thiophenyl.
- Fluanyl as used herein includes 2-furanyl and 3-furanyl.
- Pyrrolyl as used herein includes 2-pyrrolyl and 3-pyrrolyl.
- Imidazolyl as used herein includes 2-imidazolyl and 4-imidazolyl.
- Triazolyl as used herein includes 1-triazolyl, 4-triazolyl and 5-triazolyl.
- Oxadiazolyl as used herein includes 4- and 5-(l,2,3-oxadiazolyl), 3- and 5-(l ,2,4-oxadiazolyl), 3-(l ,2,5-oxadiazolyl), 2-(l ,3,4-oxadiazolyl).
- Thiadiazolyl as used herein includes 4- and 5-(l,2,3-thiadiazolyl), 3- and 5-(l ,2,4-thiadiazolyl), 3-(l ,2,5-thiadiazolyl), 2-(l ,3,4-thiadiazolyl).
- Ry is preferably H or Me. More preferably Ry is H.
- each Rz is preferably H or Me with 0, 1, 2 or 3 of Rz being Me. More preferably only 1 Rz is Me. Most preferably all Rz are H.
- Y is preferably OH or OMe. More preferably, Y is OH.
- R ⁇ and R2 are each independently selected from H, C1-C4 alkyl, O(C ⁇ -C4 alkyl), S(C -C4 alkyl), halo and phenyl;
- R3 is selected from H, C1-C4 alkyl and halo; and pharmaceutically acceptable salts thereof.
- R ⁇ is preferably C1-C3 alkyl (especially trifluoromethyl), O(C ⁇ -C3 alkyl) (especially methoxy or trifluoromethoxy), F or phenyl (Ph).
- R2 is preferably H.
- R2 is also preferably F.
- R3 is preferably H.
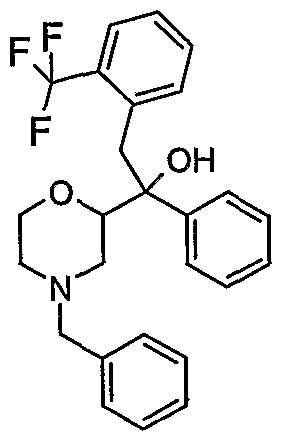
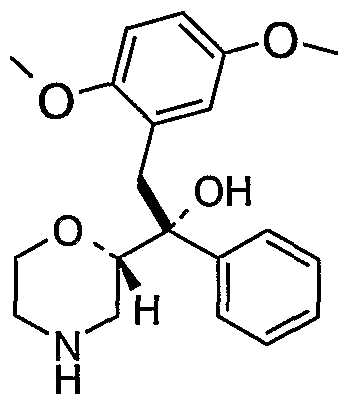
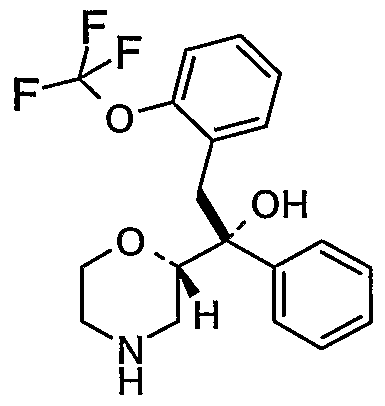
- Especially preferred compounds of the present invention are l-morpholin-2- yl-l-phenyl-2-(2-trifluoromethoxy-phenyl)-ethanol and 2-(5-fluoro-2-methoxy- phenyl)-l-morpholin-2-yl-l-phenyl-ethanol.
- the (S,R) stereoisomer is preferred.
- the preferred salt form is the hydrochloiide salt.
- Compounds of the present invention are selective inhibitors of norepinephrine reuptake.
- Biogenic amine transporters control the amount of biogenic amine neurotransmitters in the synaptic cleft. Inhibition of the respective transporter leads to a rise in the concentration of that neurotransmitter within the synaptic cleft.
- Compounds of Formula I and their pharmaceutically acceptable salts preferably exhibit a Kj value less than 500nM at the norepinephrine transporter as determined using the scintillation proximity assay as described below. More preferred compounds of Formula I and their pharmaceutically acceptable salts exhibit a K ⁇ value less than lOOnM at the norepinephrine transporter.
- More preferred compounds of Formula I and their pharmaceutically acceptable salts exhibit a Kj value less than 50nM at the norepinephrine transporter.
- Especially preferred compounds of Formula I and their pharmaceutically acceptable salts exhibit a K ⁇ value less than 20nM at the norepinephrine transporter.
- compounds of the present invention selectively inhibit the norepinephrine transporter relative to the serotonin and dopamine transporters by a factor of at least five, more preferably by a factor of at least ten.
- the compounds of the present invention are acid stable.
- they have a reduced interaction (both as substrate and inhibitor) with the liver enzyme Cytochrome P450 (CYP2D6). That is to say, they preferably exhibit less than 75% metabolism via the CYP2D6 pathway according to the CYP2D6 substrate assay described below and they preferably exhibit an IC50 of >6 ⁇ M according to the CYP2D6 inhibitor assay described below. They are indicated for the treatment of disorders associated with norepinephrine dysfunction in mammals, especially humans, including children, adolescents and adults.
- norepinephrine dysfunction refers to a reduction in the amount of norepinephrine neurotransmitter within the synaptic cleft below that which would be considered to be normal or desirable for a species, or an individual within that species.
- disorders associated with norepinephrine dysfunction in mammals refers to disorders which are associated with a reduction in the amount of norepinephrine neurotransmitter within the synaptic cleft below that which would be considered to be normal or desirable for the mammalian species, or an individual within the species, in question.
- the compounds of the present invention are also indicated for the treatment of disorders which are ameliorated by an increase in the amount of norepinephrine neurotransmitter within the synaptic cleft of a mammal above that which would be considered to be normal or desirable for the mammalian species, or an individual within the species, in question.
- treatment refers to both curative and prophylactic treatment of disorders associated with norepinephrine dysfunction.
- Compounds of the present invention may be prepared by conventional organic chemistry techniques from an N-benzyl-ketomorpholine of type 1 by addition of a suitable organometallic derivative (method A), or via the addition of a suitable organometallic reagent to an epoxide of type 2 (method B), as outlined in Scheme 1.
- racemic intermediates of type 1 may be obtained as outlined in Scheme 2 by condensation of an N-benzyl cyanomorpholine 5 (/. Med. Chem. 1993, 36, pp 683 - 689) with a suitable aryl organometallic reagent followed by acid hydrolysis.
- Chiral HPLC separations of the racemic N-benzyl-aryl-ketomorpholine of type 1 gives the required single enantiomer, i.e., the (2S)- N-benzyl-aryl-ketomorpholine of type 6 (Scheme 2).
- the intermediates 3 may be further elaborated using for example organometallic type couplings between an ortho bromide derivative of type 8 and an arylboronic acid as shown in Scheme 4.
- Ari and i ts substituent (Ri) are shown as phenyl and substitution occurs at the 2-position. It will be appreciated that analogous methods could be applied for other possible identities of Ari and Ri and other possible substitution positions. This approach may also be carried out by solid phase synthetic methods as described in more detail in the specific examples below.
- An alternative route for the preparation of the compounds of this invention is method B (see Scheme 1).
- Formation of the intermediate epoxides of type 2 from racemic N-benzyl-ketomorpholines of type 1, may be done using for example trimethyl sulfoxonium iodide and a suitable base, for example sodium hydride.
- Condensation of 2 with a commercially available aryl organometallic, or an aryl organometallic prepared from the corresponding halo aryl derivative gives the intermediates of type 3, as mixtures of diastereoisomers.
- Final deprotections may be done as described above (see scheme 3).
- Final compounds made using method B may be purified using chiral HPLC.
- Y is OR and R is C1-C4 alkyl
- Suitable strong bases will be known to the person skilled in the art and include, for example, sodium hydride.
- suitable alkylating agents will be known to the person skilled in the art and include, for example, C1-C4 alkyl halides such as methyl iodide.
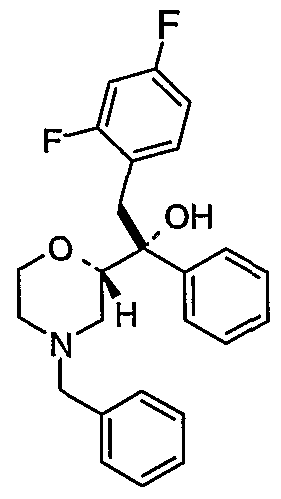
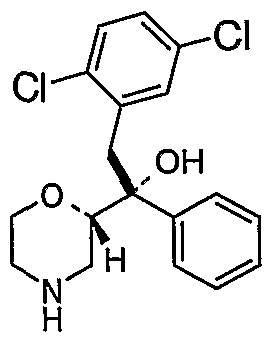
- Preferred intermediates include the compounds
- N-protecting groups will be known to the person skilled in the art as will methods for their removal. Further information on suitable deprotecting groups is contained in the well known text "Protective Groups in Organic Synthesis", Theodora W. Greene and Peter G.M. Wuts, John Wiley & Sons, Inc., New York, 1999, pp.494-653.
- Preferred N-protecting groups include benzyl, allyl, carbamates such as benzyloxycarbonyl (cbz) and t-butyloxycarbonyl (boc) and amides.
- the present invention further provides pharmaceutical compositions comprising a compound of formula (I) or formula (IT) or a pharmaceutically acceptable salt thereof, together with a pharmaceutically acceptable diluent or carrier.
- the present invention provides a compound of formula (I) or formula (JJ) or a pharmaceutically acceptable salt thereof, for use as a pharmaceutical; and a compound of formula (I) or formula (II) or a pharmaceutically acceptable salt thereof, for use as a selective inhibitor of the reuptake of norepinephrine.
- disorders associated with norepinephrine dysfunction in mammals include, for example, nervous system conditions selected from the group consisting of an addictive disorder and withdrawal syndrome, an adjustment disorder (including depressed mood, anxiety, mixed anxiety and depressed mood, disturbance of conduct, and mixed disturbance of conduct and mood), an age-associated learning and mental disorder (including Alzheimer's disease), alcohol addiction, anorexia nervosa, apathy, an attention-deficit disorder (ADD) due to general medical conditions, attention-deficit hyperactivity disorder (ADHD) including the predominantly inattentive type of ADHD and the predominantly hyperactive- impulsive type of ADHD, bipolar disorder, bulimia nervosa, chronic fatigue syndrome, chronic or acute stress, cognitive disorders including mild cognitive impairment (MCI) and cognitive impairment associated with schizophrenia (CIAS), conduct disorder, cyclothymic disorder, dementia of the Alzheimers type (DAT), depression (including a
- the present invention also provides a compound of formula (I) or formula (H) or a pharmaceutically acceptable salt thereof for selectively inhibiting the reuptake of norepinephrine.
- a compound of formula (I) or formula (H) or a pharmaceutically acceptable salt thereof for selectively inhibiting the reuptake of norepinephrine.
- such selective inhibition occurs within mammalian cells (including mammalian cell membrane preparations), especially those found within the central and/or peripheral nervous system.
- Such selective inhibition occurs within the cells of the central nervous system of a mammal, especially a human, in need thereof; and a compound of formula (I) or formula (II) or a pharmaceutically acceptable salt thereof for treating disorders associated with norepinephrine dysfunction in mammals; and the use of a compound of formula (I) or formula (H), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for selectively inhibiting the reuptake of norepinephrine; and the use of a compound of formula (I) or formula (II), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of disorders associated with norepinephrine dysfunction in mammals, including the disorders listed herein.
- the present invention provides a method for selectively inhibiting the reuptake of norepinephrine in mammals, comprising administering to a patient in need thereof an effective amount of a compound of formula (I) or formula (TJ) or a pharmaceutically acceptable salt thereof; and a method for treating disorders associated with norepinephrine dysfunction in mammals, comprising administering to a patient in need thereof an effective amount of a compound of formula (I) or formula (H) or a pharmaceutically acceptable salt thereof.
- the present invention includes the pharmaceutically acceptable salts of the compounds of formula (I) and formula (H).
- Suitable salts include acid addition salts, including salts formed with inorganic acids, for example hydrochloric, hydrobromic, nitric, sulphuric or phosphoric acids, or with organic acids, such as organic carboxylic or organic sulphonic acids, for example, acetoxybenzoic, citric, glycolic, o- mandelic-1, mandelic-dl, mandelic d, maleic, mesotartaric monohydrate, hydroxymaleic, fumaric, lactobionic, malic, methanesulphonic, napsylic, naphthalenedisulfonic, naphtoic, oxalic, palmitic, phenylacetic, propionic, pyridyl hydroxy pyruvic, salicylic, stearic, succinic, sulfanilic, tartaric, 2-hydroxyethane sulphonic, tol
- salts are included in the invention. They may serve as intermediates in the purification of compounds or in the preparation of other, for example pharmaceutically acceptable, acid addition salts, or are useful for identification, characterisation or purification. It will be appreciated that compounds of formula (I) and formula (IT) possess one or more asymmetric carbon atoms, and that in the present invention specific individual stereoisomers are preferred. In the present specification, where a structural formula does not specify the stereochemistry at one or more chiral centres, it encompasses all possible stereoisomers and all possible mixtures of stereoisomers (including, but not limited to, racemic mixtures), which may result from stereoisomerism at each of the one or more chiral centers.
- the compounds of the present invention may be used as medicaments in human or veterinary medicine.
- the compounds may be administered by various routes, for example, by oral or rectal routes, topically or parenterally, for example by injection, and are usually employed in the form of a pharmaceutical composition.
- compositions may be prepared by methods well known in the pharmaceutical art and normally comprise at least one active compound in association with a pharmaceutically acceptable diluent, excipient or carrier.
- the active ingredient will usually be mixed with a carrier or diluted by a carrier, and/or enclosed within a carrier which may, for example, be in the form of a capsule, sachet, paper or other container.
- a carrier which may, for example, be in the form of a capsule, sachet, paper or other container.
- the carrier serves as a diluent, it may be solid, semi-solid, or liquid material which acts as a vehicle, excipient or medium for the active ingredient.
- the composition may be in the form of tablets, lozenges, sachets, cachets, elixirs, suspensions, solutions, syrups, aerosols (as a solid or in a liquid medium), ointments containing, for example, up to 10% by weight of the active compound, soft and hard gelatin capsules, suppositories, injection solutions and suspensions and sterile packaged powders.
- suitable carriers are lactose, dextrose, vegetable oils, benzyl alcohols, alkylene glycols, polyethylene glycols, glycerol triacetate, gelatin, carbohydrates such as starch and petroleum jelly, sucrose sorbitol, mannitol, starches, gum acacia,calcium phosphate, alginates, tragacanth, gelatin, syrap, methyl cellulose, methyl- and propyl- hydrobenzoate, talc, magnesium stearate and mineral oil.
- the compounds of formula (I) can also be lyophilized and the lyophilizates obtained used, for example, for the production of injection preparations.
- compositions of the invention may be formulated so as to provide, quick, sustained or delayed release of the active ingredient after administration to the patient by employing procedures well known in the art.
- compositions are preferably formulated in a unit dosage form, each dosage unit containing from about 5 to about 500 mg, more usually about 25 to about 300 mg, of the active ingredient.
- unit dosage form refers to physically discrete units suitable as unitary doses for human subjects and other mammals, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, in association with a suitable pharmaceutical carrier.
- N-benzylethanolamine (172.2 g; 1 equiv. available from Aldrich Chemical Company). 2-Chloroacrylonitrile (100 g; 1 equiv. available from Aldrich Chemical Company) was added dropwise over 2 minutes. The temperature was maintained between 23 °C and 29 °C by means of the ice bath and subsequently a water bath at 15 °C. N-Benzylethanolamine was still detected on TLC after 4.5 h stirring. After one night stirring at room temperature (water bath), no N-benzylethanolamine was detectable by 1H ⁇ MR.
- a 31 double jacket reactor was charged with 4-Benzyl-morpholine-2- carbonitrile (135.05 g; leq) and dry diethyl ether (1.41). Alternatively, toluene may be used in place of diethyl ether.
- phenyl magnesium chloride (2M sol. in tetrahydrofuran, 360 ml, 1.08 equiv., available from Aldrich Chemical Company) was added dropwise over 1 hour. Tm rose to 4°C and came back to 2°C at the end of the addition.
- Tm was progressively raised to 17.5°C over 45 minutes and the mixture stirred at this temperature for another 45 minutes.
- the triphasic suspension was filtered. The organic layer of the mother liquors was eliminated. The filtration cake was then washed with methylene chloride (700 ml). This liquor was charged in the reactor with the acid aqueous layer.
- the two enantiomers may be separated by fractional crystallization from acetonitrile using from 0.55 to 1 equivalent of dibenzoyltartaric acid to generate diastereoisomeric salts of the title compound.
- the crystals may be collected by filtration and neutralized with 30% ⁇ aOH to afford the optically enriched title compound.
- the mixture was post-agitated at about 0°C for 2.5 h, quenched by adding ultra pure water (142.5 L) maintaining 2.1 °C ⁇ Tmass ⁇ 8.7 °C.
- the aqueous layer (176 kg) was separated after 35 minutes of post-stirring allowing the mixture to reach 15 °C and the toluene layer was washed with ultra pure water (142.5 L) and the aqueous layer (162 kg) was separated.
- the organic layer was then concentrated under reduced pressure (150 mbars) maintaining Tmass ⁇ 60 °C in order to distill 162 kg of toluene.
- the toluene layer was cooled to 0°C and a 5 N NaOH aqueous solution (420.1 kg) was slowly added maintaining the temperature at - 2.4 °C ⁇ Tmass ⁇ 11 °C.
- the reaction mixture was post-stirred for lh and the aqueous layer (494.8 kg) was extracted.
- the toluene layer was concentrated under reduced pressure (50 mbars) maintaining Tmass ⁇ 60 °C in order to distill 356.2 kg of toluene and isopropanol (180.4 kg) was added.
- the toluene was stripped off under reduced pressure (100 mbars) maintaining Tmass ⁇ 60 °C in order to distill 186.4 kg of toluene and isopropanol (135 kg) was added again to the mixture.
- a last distillation of toluene was performed under reduced pressure (50 mbars) maintaining Tmass ⁇ 60 °C in order to distill 131 kg of toluene and isopropanol (49.4 kg) was finally added to the mixture and the solution was stirred at RT until crystallization (17 minutes).
- Neat (5-Fluoro-2-methoxy-phenyl)-methanol (19.587g, 1 equiv.) was added to neat SOCl 2 (42.2 mL, 4.6 equiv.) at -78°C under a nitrogen atmosphere and the solution was then allowed to warm to room temperature and stirred until evolution of gas had ceased.
- An equivalent volume of anhydrous toluene was added to the flask and the solution heated to 60°C. On cooling the reaction solution was poured onto ice water. The toluene layer was separated and dried (MgSO 4 ) and the solvent removed under reduced pressure.
- Solid magnesium turnings (9.5 g, 28 equiv.) under nitrogen atmosphere at room temperature were stirred vigorously with a magnetic stirring bar overnight. The magnesium was then covered with dry diethyl ether and to the suspension was added 1,2-dibromoethane (50 ⁇ L). A cold bath was then applied followed by dropwise addition of l-chloromethyl-2-methoxy-benzene (18.18 g, 5 equiv. available from Aldrich Chemical Company) in diethyl ether (71 mL) which maintained the temperature at up to 15 °C. The resulting black suspension was stirred at room temperature for 30 minutes and cooled down at -20 °C.
- the active enantiomer was obtained after a further preparative chiral HPLC separation.
- the active enantiomer, a white solid, was next taken up in ethanol and hydrogen chloride was added (large excess of 2M solution in diethyl ether) and the mixture was stirred until it became a clear solution. Then all the volatiles were evaporated in vacuo, to give 447mg of the title compound as white solid.
- the following method may be used. Magnesium turnings (24.2 g, 0.935 mole, 2 eq.) and diethyl ether (300 ml) were loaded in a reactor under N 2 . A solution of 2-trifluoromethoxybenzyl bromide (165 g, 0.647 mole, 1.3 eq.) in diethyl ether (300 ml) was loaded in an addition funnel. Iodine crystals and a small amount of the 2-trifluoromethoxybenzyl bromide solution were added and the reaction mixture was stirred to initiate the reaction. The remainder of the 2- trifluoromethoxybenzyl bromide solution was then added drop- wise maintaining the temperature of the reaction mixture below 35°C.
- the aqueous layer was extracted with diethyl ether (1 L). The organic layers were combined and the filtrates were concentrated under vacuum to about 2 liters. The solution was dried over MgSO 4 , filtered and the filter cake was washed with diethyl ether (200 ml). The filtrate was concentrated under vacuum to orange oil. The residue was twice dissolved in toluene (500 ml) and concentrated to a solid product. The yield of crude title compound was 235 g (103%).
- a stainless steel Buchi hydrogenation reactor was loaded with l-(4-Benzyl-morpholin-2-yl)-l-phenyl-2-(2- trifluoromethoxy-phenyl)-ethanol (230 g, 0.503 mole), methanol (1 L), a suspension of Pd/C (10%, 46 g, 20% loading) in methanol (500 ml), and methanol (500 ml) from equipment rinses.
- a solution of HC1 in ethanol (1.6N, 460 ml, 0.736 mole, 1.5 eq.) was added and the reactor was pressurized with H 2 (3 Bar). The reaction mixture was heated to 40°C and stirred for 3 hours.
- the following method may be used.
- l-(4-Benzyl-morpholin-2-yl)-l-phenyl-2-(2- trifluoromethoxy-phenyl)-ethanol hydrochloiide 150g, 303.7 mmol
- demineralized water 352 mL
- i-PrOH 375 mL
- 5% Pd/C 30 g, 50% water, Johnson & Matthey type 440.
- the heterogeneous reaction mixture was then purged 5 times with 25 psi nitrogen then purged 5 times with 50 psi hydrogen, and the hydrogenation was performed at RT.
- the initial Tmass was 22°C and the maximum
- Tmass during the hydrogenation was 23°C.
- the reactor was stirred vigorously. In- process analysis after 2 hours indicated complete hydrogenolysis.
- the hydrogenation was stopped after 3 hours.
- the nitrogen purged reaction mixture was then filtered at RT through an hyflo filter (56 g), impregnated beforehand with 75 mL of a 50/50 v/v isopropanol/water mixture and washed with 300 mL of a 50/50 v/v isopropanol/water mixture.
- the filtrates were stored overnight at RT.
- the filtrates were concentrated at 40-50°C under reduced pressure (typical 622 g distilled).
- the reaction mixture was cooled to RT and post-agitated.
- the following method may be used. Magnesium turnings (21.6 g, 0.888 mole, 2 eq.) and diethyl ether (300 ml) were loaded in a reactor under N . A solution of 5-fluoro-2-methoxybenzyl chloride (116 g, 0.664 mole, 1.5 eq.) in diethyl ether (200 ml) was loaded in an addition funnel. Iodine crystals and a small amount of the 5-fluoro-2-methoxybenzyl chloride solution were added and the reaction mixture was stirred to initiate the reaction. The remainder of the 5-fluoro-2 methoxybenzyl chloride solution was then added drop- wise maintaining the temperature of the reaction mixture below 28 °C.
- a glass hydrogenation flask was loaded with methanol (1.55 L), Pd/C (10%, 31 g, 20% loading), l-(4- benzyl-morpholin-2-yl)-2-(5-fluoro-2-methoxy-phenyl)-l-phenyl-ethanol (155 g, 0.368 mole) and a solution of HCl in ethanol (2.5N, 233 ml, 0.582 mole, 1.6 eq.).
- the reactor was mounted on a Parr instrument and pressurized with H 2 (49 Psi). The reaction mixture was shaken overnight between 20°C and 15°C. The catalyst was filtered off and washed with methanol (0.5 L).
- the reaction vessel was cooled to room temperature and the reaction mixture taken into methanol (5 ml) and purified by SCX-2 chromatography to obtain the free base as clear oil (50 mg).
- the hydrochloride salt was obtained following general procedures as a white solid (52 mg, 72 % after salt formation.).
- Solid Phase Synthesis of Compounds of the Invention Compounds of the invention wherein Ari is substituted with an aromatic group (i.e. pyridyl, thiophenyl and optionally substituted phenyl) may be prepared by solid phase synthesis using the route shown below (the black dot represents polystyrene resin).
- an aromatic group i.e. pyridyl, thiophenyl and optionally substituted phenyl
- the sequence is preferably performed on a polystyrene resin, without characterisation of the resin-bound intermediates.
- Example 35 l-morpholin-2-yl-l-phenyl-2-(2-pyridin-3-yl-phenyl)-ethanol, RT (6 min gradient) 2.17 min, [M+H] + 361.4
- Example 36 l-morpholin-2-yl-l-phenyl-2-(2-thiophen-3-yl-phenyl)-ethanol, 3.25 min, [M+H] +
- the pharmacological profile of the present compounds may be demonstrated as follows. All of the exemplified compounds above have been found to exhibit a Kj value less than 500nM at the norepinephrine transporter as determined using the scintillation proximity assay described below. Further more, all of the exemplified compounds above have been found to selectively inhibit the norepinephrine transporter relative to the serotonin and dopamine transporters by a factor of at least five using the scintillation proximity assays as described below.
- PCR polymerase chain reaction
- Human dopamine transporter GenBank M95167. Reference: Nandenbergh DJ, Persico AM and Uhl GR. A human dopamine transporter cDNA predicts reduced glycosylation, displays a novel repetitive element and provides racially- dimorphic Taql RFLPs. Molecular Brain Research (1992) volume 15, pages 161- 166. Human norepinephrine transporter: GenBank M65105. Reference: Pacholczyk T, Blakely, RD and Amara SG. Expression cloning of a cocaine- and antidepressant-sensitive human noradrenaline transporter. Nature (1991) volume 350, pages 350-354.
- Human serotonin transporter GenBank L05568. Reference: Ramamoorthy S, Bauman AL, Moore KR, Han H, Yang-Feng T, Chang AS, Ganapathy V and Blakely RD. Antidepressant- and cocaine-sensitivehuman serotonin transporter: Molecular cloning, expression, and chromosomal localization. Proceedings of the National Academy of Sciences of the USA (1993) volume 90, pages 2542-2546.
- PCR products were cloned into a mammalian expression vector (eg pcDNA3.1 (Invitrogen)) using standard ligation techniques.
- the constructs were then used to stably transfect HEK293 cells using a commercially available lipofection reagent (LipofectamineTM - Invitrogen) following the manufacture' s protocol.
- Scintillation proximity assays for determining the affinity of test ligands at the norepinephrine transporter.
- the compounds of the present invention are norepinephrine reuptake inhibitors, and possess excellent activity in, for example, a scintillation proximity assay (e.g. J. Gobel, D.L. Saussy and A. Goetz, J. Pharmacol. Toxicolo. (1999), 42, 237-244).
- a scintillation proximity assay e.g. J. Gobel, D.L. Saussy and A. Goetz, J. Pharmacol. Toxicolo. (1999), 42, 237-244
- Cell pastes from large scale production of HEK-293 cells expressing cloned human norepinephrine transporters were homogenized in 4 volumes 50mM Tris- HCl containing 300mM NaCl and 5mM KC1, pH 7.4.
- the homogenate was centrifuged twice (40,000g, lOmin, 4°C) with pellet re-suspension in 4 volumes of Tris-HCl buffer containing the above reagents after the first spin and 8 volumes after the second spin.
- the suspended homogenate was centrifuged (lOOg, lOrnin, 4°C) and the supernatant kept and re-centrifuged (40,000g, 20min, 4°C).
- the pellet was resuspended in Tris-HCl buffer containing the above reagents along with 10%w/v sucrose and O.lmM phenylmethylsulfonyl fluoride (PMSF).
- the membrane preparation was stored in aliquots (1ml) at -80°C until required.
- the protein concentration of the membrane preparation was determined using a bicinchoninic acid (BCA) protein assay reagent kit (available from Pierce).
- BCA bicinchoninic acid
- Each well of a 96 well microtitre plate was set up to contain the following: 50 ⁇ l 2nM [N-methyl- 3 H]-Nisoxetine hydrochloride (70-87Ci/mmol, from NEN
- Test compound 25 ⁇ l Test compound, assay buffer (total binding) or lO ⁇ M Desipramine HCl
- microtitre plates were incubated at room temperature for 10 hours prior to reading in a Trilux scintillation counter.
- the results were analysed using an automatic spline fitting programme (Multicalc, Packard, Milton Keynes, UK) to provide Ki values for each of the test compounds.
- test compound to compete with [ 3 H]-citalopram for its binding sites on cloned human serotonin transporter containing membranes has been used as a measure of test compound ability to block serotonin uptake via its specific transporter (Ramamoorthy, S., Giovanetti, E., Qian, Y., Blakely, R., (1998)
- Membrane preparation is essentially similar to that for the norepinephrine transporter containing membranes as described above.
- the membrane preparation was stored in aliquots (1ml) at -70°C until required.
- the protein concentration of the membrane preparation was determined using a BCA protein assay reagent kit.
- Each well of a 96 well microtitre plate was set up to contain the following: 50 ⁇ l 2nM [ 3 H]-Citalopram (60-86Ci/mmol, Amersham Biosciences)
- KC1 25 ⁇ l Diluted compound, assay buffer (total binding) or lOO ⁇ M Fluoxetine (nonspecific binding) 50 ⁇ l WGA PVT SPA Beads (40mg/ml)
- microtitre plates were incubated at room temperature for 10 hours prior to reading in a Trilux scintillation counter.
- the results were analysed using an automatic spline fitting programme (Multicalc, Packard, Milton Keynes, UK) to provide Ki (nM) values for each of the test compounds.
- test compound The ability of a test compound to compete with [ 3 H]-WTN35,428 for its binding sites on human cell membranes containing cloned human dopamine transporter has been used as a measure of the ability of such test compounds to block dopamine uptake via its specific transporter (Ramamoorthy et al 1998 supra).
- Each well of a 96well microtitre plate was set up to contain the following: 50 ⁇ l 4nM [ 3 H]-WIN35,428 (84-87Ci/mmol, from NEN Life Science Products)
- the acid stability of a compound according to the present invention was determined as a solution in buffer at 6 different pH values (HCl 0.1N, pH 2, pH 4, pH 6, pH 7, and pH 8) at 40°C over a time course of 72 hours. Samples were taken at the beginning of the study and after 3, 6 and 24 hours and analysed by capillary electrophoresis. The original sample used in this study contained 0.8% of the undesired epimer as internal standard. The samples taken at the different time points during the study did not show any significant change in the percentage of the undesired epimer. This assay confirms that compounds of the present invention are chemically and configurationally stable under acidic conditions.
- Cytochrome P450 2D6 (CYP2D6) is a mammalian enzyme which is commonly associated with the metabolism of around 30% of pharmaceutical compounds. Moreover, this enzyme exhibits genetic polymorphism, resulting in the presence of both normal and poor metabolizers in the population.
- a low involvement of CYP2D6 in the metabolism of compounds i.e. the compound being a poor substrate of CYP2D6 is desirable in order to reduce any variability from subject to subject in the pharmacokinetics of the compound.
- compounds with a low inhihibitor potential for CYP2D6 are desirable in order to avoid drug-drug interactions with co-administered drugs that are substrates of CYP2D6.
- Compounds may be tested both as substrates and as inhibitors of this enzyme by means of the following assays.
- This assay determines the extent of the CYP2D6 enzyme involvement in the total oxidative metabolism of a compound in microsomes.
- Preferred compounds of the present invention exhibit less than 75% total metabolism via the CYP2D6 pathway.
- HLM human liver microsomes
- the extent of oxidative metabolism in human liver microsomes (HLM) is determined after a 30 minute incubation in the absence and presence of Quinidine, a specific chemical inhibitor of CYP2D6. The difference in the extent of metabolism in absence and presence of the inhibitor indicates the involvement of CYP2D6 in the metabolism of the compound.
- microsomal incubation mixture (total volume 0.1 mL) contained the NCE (4 ⁇ M), ⁇ -NADPH (1 mM), microsomal proteins (0.5 mg/mL), and Quinidine
- the extent of CYP2D6 involvement was calculated comparing the extent of metabolism in absence and in presence of quinidine in the incubation.
- timeO and time30 correspond to the 0 and 30 minutes incubation time.
- the % of CYP2D6 involvement was calculated as follows : ( extent of metabolism without inhibitor) - (% extent of metabolism with inhibitor) % extent of metabolism without inhibitor
- the CYP2D6 inhibitor assay evaluates the potential for a compound to inhibit CYP2D6. This is performed by the measurement of the inhibition of the bufuralol l'-hydroxylase activity by the compound compared to a control.
- the 1'- hydroxylation of bufuralol is a metabolic reaction specific to CYP2D6.
- Preferred compounds of the present invention exhibit an IC5 0 higher than 6 ⁇ M for CYP2D6 activity, the ICso being the concentration of the compound that gives 50 % of inhibition of the CYP2D6 activity.
- Human liver microsomes (mixture of 20 different donors, mixed gender) were acquired from Human Biologies (Scottsdale, AZ).
- ⁇ -NADPH was purchased from Sigma (St Louis, MO).
- Bufuralol was purchased from Ultrafine (Manchester, UK). All the other reagents and solvents were of analytical grade.
- Microsomal incubation mixture (total volume 0.1 mL) contained bufuralol 10 ⁇ M, ⁇ -NADPH (2 mM), microsomal proteins (0.5 mg/mL), and the new chemical entity (NCE) (0, 5, and 25 ⁇ M) in 100 mM sodium phosphate buffer pH 7.4.
- the mixture was incubated in a shaking waterbath at 37 °C for 5 minutes.
- the reaction was terminated by the addition of methanol (75 ⁇ L).
- the samples were vortexed and the denaturated proteins were removed by centrifugation. The supernatant was analyzed by liquid chromatography connected to a fluorescence detector.
- the formation of the 1 ' -hydroxybufuralol was monitored in control samples (0 ⁇ M NCE) and in the samples incubated in presence of the NCE.
- the stock solution of NCE was prepared in a mixture of Acetonitrile/Water to reach a final concentration of acetonitrile in the incubation below 1.0%.
- Solvent A and Solvent B consisted of a mixture of 0.02 M potassium dihydrogenophosphate buffer pH3/ methanol in the proportion 90/10 (v/v) for solvent A and 10/90 (v/v) for solvent B. The run time was 7.5 minutes. Formation of 1' -hydroxybufuralol was monitored by fluorimetric detection with extinction at ⁇ 252 nm and emission at ⁇ 302 nm.
- the IC50 of the NCE for CYP2D6 was calculated by the measurement of the percent of inhibition of the formation of the 1' -hydroxybufuralol in presence of the NCE compared to control samples (no NCE) at a known concentration of the NCE.
- the IC50 is calculated from the percent inhibition of the formation of the 1 '- hydroxybufuralol as follows (assuming competitive inhibition):
- NCE Concentration x( l00- Percent of inhibition) Percent of inhibition The IC50 estimation is assumed valid if inhibition is between 20% and 80% (Moody GC, Griffin SJ, Mather AN, McGinnity DF, Riley RJ. 1999. Fully automated analysis of activities catalyzed by the major human liver cytochrome P450 (CYP) enzymes: assessment of human CYP inhibition potential. Xenobiotica, 29(1): 53-75).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Neurology (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description


































































Claims






Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03748975A EP1534694A1 (en) | 2002-08-23 | 2003-08-18 | Aryl and heteroaryl morpholine derivatives |
| AU2003268024A AU2003268024A1 (en) | 2002-08-23 | 2003-08-18 | Aryl and heteroaryl morpholine derivatives |
| US10/524,921 US7354920B2 (en) | 2002-08-23 | 2003-08-18 | Aryl and heteroaryl morpholine derivatives |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0219687.1A GB0219687D0 (en) | 2002-08-23 | 2002-08-23 | Benzyl morpholine derivatives |
| GB0219687.1 | 2002-08-23 | ||
| US60/415,305 | 2002-09-30 | ||
| US41530302P | 2002-10-01 | 2002-10-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004018441A1 true WO2004018441A1 (en) | 2004-03-04 |
Family
ID=9942871
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2003/023270 Ceased WO2004018441A1 (en) | 2002-08-23 | 2003-08-18 | Aryl and heteroaryl morpholine derivatives |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US7354920B2 (en) |
| EP (1) | EP1534694A1 (en) |
| AR (1) | AR041026A1 (en) |
| AU (1) | AU2003268024A1 (en) |
| GB (1) | GB0219687D0 (en) |
| PE (1) | PE20040975A1 (en) |
| TW (1) | TW200413339A (en) |
| WO (1) | WO2004018441A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005047272A1 (en) | 2003-11-10 | 2005-05-26 | Eli Lilly And Company | Morpholine derivatives as norepinephrine reuptake inhibitors |
| WO2005066144A1 (en) * | 2003-12-23 | 2005-07-21 | Eli Lilly And Company | Morpholine derivatives as norepinephrine reuptake inhibitors |
| WO2005060949A3 (en) * | 2003-12-12 | 2005-09-09 | Lilly Co Eli | Selective norepinephrine reuptake inhibitors for the treatment of hot flashes, impulse control disorders and personality change due to a general medical condition |
| WO2007005935A3 (en) * | 2005-07-06 | 2009-04-16 | Molecular Neuroimaging Llc | Norepinephrine transporter radiotracers and methods of syntheses thereof |
| US7659394B2 (en) | 2004-04-30 | 2010-02-09 | Pfizer Inc | Substituted morpholine compounds for the treatment of central nervous system disorders |
| WO2017172368A1 (en) | 2016-03-31 | 2017-10-05 | Oncternal Therapeutics, Inc. | Indoline analogs and uses thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0721777A2 (en) * | 1995-01-11 | 1996-07-17 | Eli Lilly And Company | Use of tomoxetine for the treatment of attention deficit/hyperactivity disorder |
| EP0756869A2 (en) * | 1995-07-24 | 1997-02-05 | Eli Lilly And Company | Use of duloxetine or of N-alkyl-3-phenyl-(2-alkylthiophenoxy)-propylamines for treating attention-deficit/hyperactivity disorder |
| WO1999064009A1 (en) * | 1998-06-11 | 1999-12-16 | Merck Sharp & Dohme Limited | Use of an nk-1 receptor antagonist for treating cognitive disorders |
| US6274579B1 (en) * | 1998-01-21 | 2001-08-14 | Glaxo Wellcome Inc. | Pharmaceutically active morpholinol |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE759013R (en) | 1969-11-17 | 1971-05-17 | Ici Ltd | DERIVATIVES OF |
| GB1412546A (en) | 1973-08-08 | 1975-11-05 | Ici Ltd | Morpholine derivatives |
| GB2167407B (en) | 1984-11-22 | 1988-05-11 | Erba Farmitalia | Enantiomers of phenoxy derivatives of benzyl morpholine and salts thereof |
| AU9214498A (en) | 1997-09-23 | 1999-04-12 | Eli Lilly And Company | Treatment of attention-deficit/hyperactivity disorder |
| US6503905B1 (en) | 1998-12-29 | 2003-01-07 | Pfizer Inc | 3,3-biarylpiperidine and 2,2-biarylmorpholine derivatives |
| GEP20043296B (en) | 1999-02-23 | 2004-01-12 | Pfizer Prod Inc | Monoamine Reuptake Inhibitors for Treatment of CNS Disorders |
| DE60022692T2 (en) | 1999-07-01 | 2006-06-22 | Pharmacia & Upjohn Co. Llc, Kalamazoo | (S, S) reboxetine for the treatment of migraine headache |
| WO2004017977A2 (en) * | 2002-08-23 | 2004-03-04 | Eli Lilly And Company | 2- (phenoxymethyl)- and 2- (phenylthiomethyl)-morpholine derivatives for use as selective norepinephrine reuptake inhibitors |
| AU2003261245A1 (en) * | 2002-08-23 | 2004-03-11 | Eli Lilly And Company | Benzyl morpholine derivatives |
| GB0319793D0 (en) * | 2003-08-22 | 2003-09-24 | Lilly Co Eli | Pyridinylmorpholine derivatives |
| DE602004019698D1 (en) * | 2003-12-23 | 2009-04-09 | Lilly Co Eli | MORPHOLINE DERIVATIVES AS INHIBITORS OF THE RECOVERY OF NOREPINEPHRIN |
-
2002
- 2002-08-23 GB GBGB0219687.1A patent/GB0219687D0/en not_active Ceased
-
2003
- 2003-08-08 TW TW092121850A patent/TW200413339A/en unknown
- 2003-08-18 AU AU2003268024A patent/AU2003268024A1/en not_active Abandoned
- 2003-08-18 US US10/524,921 patent/US7354920B2/en not_active Expired - Lifetime
- 2003-08-18 EP EP03748975A patent/EP1534694A1/en not_active Withdrawn
- 2003-08-18 WO PCT/US2003/023270 patent/WO2004018441A1/en not_active Ceased
- 2003-08-20 AR ARP030103012A patent/AR041026A1/en unknown
- 2003-08-21 PE PE2003000852A patent/PE20040975A1/en not_active Application Discontinuation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0721777A2 (en) * | 1995-01-11 | 1996-07-17 | Eli Lilly And Company | Use of tomoxetine for the treatment of attention deficit/hyperactivity disorder |
| EP0756869A2 (en) * | 1995-07-24 | 1997-02-05 | Eli Lilly And Company | Use of duloxetine or of N-alkyl-3-phenyl-(2-alkylthiophenoxy)-propylamines for treating attention-deficit/hyperactivity disorder |
| US6274579B1 (en) * | 1998-01-21 | 2001-08-14 | Glaxo Wellcome Inc. | Pharmaceutically active morpholinol |
| WO1999064009A1 (en) * | 1998-06-11 | 1999-12-16 | Merck Sharp & Dohme Limited | Use of an nk-1 receptor antagonist for treating cognitive disorders |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2004289616B2 (en) * | 2003-11-10 | 2010-08-05 | Eli Lilly And Company | Morpholine derivatives as norepinephrine reuptake inhibitors |
| KR100783855B1 (en) * | 2003-11-10 | 2007-12-10 | 일라이 릴리 앤드 캄파니 | Morpholine derivatives as norepinephrine reuptake inhibitors |
| EA009960B1 (en) * | 2003-11-10 | 2008-04-28 | Эли Лилли Энд Компани | Morpholine derivatives as norepinephrine reuptake inhibitors |
| US7423037B2 (en) | 2003-11-10 | 2008-09-09 | Eli Lilly And Company | Morpholine derivatives as norepinephrine reuptake inhibitors |
| WO2005047272A1 (en) | 2003-11-10 | 2005-05-26 | Eli Lilly And Company | Morpholine derivatives as norepinephrine reuptake inhibitors |
| EP2223916A1 (en) | 2003-11-10 | 2010-09-01 | Eli Lilly and Company | Morpholine derivatives as norepinephrine reuptake inhibitors |
| WO2005060949A3 (en) * | 2003-12-12 | 2005-09-09 | Lilly Co Eli | Selective norepinephrine reuptake inhibitors for the treatment of hot flashes, impulse control disorders and personality change due to a general medical condition |
| WO2005066144A1 (en) * | 2003-12-23 | 2005-07-21 | Eli Lilly And Company | Morpholine derivatives as norepinephrine reuptake inhibitors |
| US7595315B2 (en) | 2003-12-23 | 2009-09-29 | Eli Lilly And Company | Morpholine derivatives as norepinephrine reuptake inhibitors |
| US7659394B2 (en) | 2004-04-30 | 2010-02-09 | Pfizer Inc | Substituted morpholine compounds for the treatment of central nervous system disorders |
| WO2007005935A3 (en) * | 2005-07-06 | 2009-04-16 | Molecular Neuroimaging Llc | Norepinephrine transporter radiotracers and methods of syntheses thereof |
| WO2017172368A1 (en) | 2016-03-31 | 2017-10-05 | Oncternal Therapeutics, Inc. | Indoline analogs and uses thereof |
| EP3795563A1 (en) | 2016-03-31 | 2021-03-24 | Oncternal Therapeutics, Inc. | Indoline analogs and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US7354920B2 (en) | 2008-04-08 |
| GB0219687D0 (en) | 2002-10-02 |
| AU2003268024A1 (en) | 2004-03-11 |
| PE20040975A1 (en) | 2004-12-29 |
| AR041026A1 (en) | 2005-04-27 |
| EP1534694A1 (en) | 2005-06-01 |
| US20060003998A1 (en) | 2006-01-05 |
| TW200413339A (en) | 2004-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1534291B1 (en) | 2-(phenylthiomethyl)- morpholine derivatives for use as selective norepinephrine reuptake inhibitors | |
| WO2005105763A1 (en) | Substituted morpholine compounds for the treatment of central nervous system disorders | |
| EP1567501B1 (en) | Propanamine derivatives as serotonin and norepinephrine reuptake inhibitors | |
| WO2004018441A1 (en) | Aryl and heteroaryl morpholine derivatives | |
| EP1716126B1 (en) | Morpholine derivatives as norepinephrine reuptake inhibitors | |
| US7488728B2 (en) | Pyridinylmorpholine derivatives | |
| EP1682523B1 (en) | Morpholine derivatives as norepinephrine reuptake inhibitors | |
| US7294623B2 (en) | Benzyl morpholine derivatives | |
| EP1622874B1 (en) | 3,4-dihydro-1h-quinolin-2-one derivatives as norepinephrine reuptake inhibitors | |
| HK1095135B (en) | Morpholine derivatives as norepinephrine reuptake inhibitors | |
| MXPA06005226A (en) | Morpholine derivatives as norepinephrine reuptake inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2006003998 Country of ref document: US Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10524921 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003748975 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003748975 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10524921 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: JP |
|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) |