SYNTHESIS AND PURIFICATION OF NATEGLINIDE
FIELD OF THE INVENTION
The present invention relates to a novel process for the synthesis and purification of (-)-N-[trans-4-isopropyl cyclohexane (carbonyl)]-D-phenylalanine, also known as nateglinide. Nateglinide is a pharmaceutical compound having potent hypoglycemic properties.
BACKGROUND OF THE INVENTION The sulfonyl ureas, which exhibit hypoglycemic action particularly tlirough promotion of the secretion of insulin, and a biguanide which exhibits a hypoglycemic action through the metabolism of sugar have hitherto been reported as anti diabetic drugs for oral use. However, they are somewhat unsatisfactory as to their side effects (Diabetes, 19,785, 1970, Ann. Rev. Pharmacol 15,351, 1975). N-Benzoyl phenylalanines and their related compounds possessing hypoglycemic activity have been previously reported (J. Med. Chem. 1988, 31, 2092). Of these compounds (-)-N- [trans-4-isopropyl cyclohexane (carbonyl)j-D-phenylalanine shown in formula I below, and commonly known as nateglinide was found to be more potent than N- benzoyl-D-phenylalanine (J. Med. Chem. 1989, 32, 1436).
In view of the hypoglycemic activity of D-phenylalanine derivatives belonging to the class of compounds known as glinides, numerous reports have been published regarding the process for their preparation and their activity (EP 0196222 (1986); US 4,816,484 (1986, 1989); J.' Med. Chem., (1989) 32, 1436; US RE 34,878; JP
07017899 (95, 17899) 1995). All patents referenced herein are hereby incorporated by reference in their entirety. In case of conflict in teπninology the present disclosure shall control.
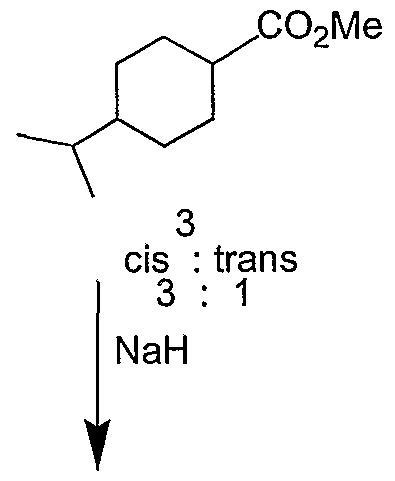
In a process for the synthesis of nateglinide [EP 0196,222 (1986), J. Med. Chem., 1989, 32,1436] cumic acid was converted into nateglinide after undergoing a series of reactions as depicted in Scheme-I.
cis : trans 3 : 1
trans Acid trans : cis 6 : 1 5
N-[(trans-4-isopropyl cyclohexyl)-carbonyl]-D-phenylalanine of the formula I (nateglinide) was prepared in about 9 steps starting from cumic acid of the formula (1). Cumic acid was hydrogenated in acetic acid under 5 kg/cm2 of hydrogen pressure over an Adams catalyst (Pt02) to obtain a mixture of cis and trans (3:1) isopropyl cyclohexyl carboxylic acid of formula (2). The mixture of the acid of formula (2) was esterified by reacting with thionyl chloride and methanol to yield the corresponding ester mixture of formula (3). The isomerization of the ester mixture of formula (3) using sodium hydride gave the corresponding methyl ester of formula (4) in a 6:1 ratio of trans and cis isomer. The basic hydrolysis of the mixture, followed by crystallization provided the pure trans isomer of the acid of formula (5). The acid of formula (5) was coupled with N-hydroxy succinimide (HOSu) of formula (6) and dicyclohexyl carbodiimide (DCC) to yield the compound of formula (7) which was then reacted with the previously prepared D-phenylalanine methyl ester of formula (9) from D-phenylalanine. The synthesized ester of formula (10) was hydrolysed by 2N aqueous sodium hydroxide to the N-substituted phenylalanine derivative I.
The disadvantages of the above described process include:
1) The process is of multi step synthesis, tedious, cumbersome, and difficult to scale up.
2) The catalytic hydrogenation of the cumic acid of formula (1) results in a mixture of predominant cis (3 parts) and minor trans (1 part) of the acid initially and later isomerization is performed.
3) To purify the mixture of the acid of formula (1) high vacuum distillation is performed, which generally leads to decomposition.
4) To generate the acid chloride of the acid of formula (1), thionyl chloride is used, which is corrosive, moisture sensitive, not eco-friendly and difficult to remove by distillation.
5) The methyl ester of formula (3) was purified by high vacuum distillation, an additional and time consuming operation.
6) The isomerization of the mixture of the ester of formula (3) to formula (4) was accomplished with a pyrophoric reagent such as sodium hydride and high vacuum distillation, which is very difficult during a scale up operation.
7) The methyl ester of formula (4) was hydrolysed to the acid of formula (5) by hydrolysis, followed by recrystallization in aqueous methanol which results in loss of the product.
8) The coupling of the acid of formula (5) with HOSu of formula (6) was carried out using DCC, which is an acute irritant and a hygroscopic reagent.
Another process for the synthesis of nateglinide is disclosed in US 4,816,484 (1986), US RE 34,878 (1995), and EP 196,222 (1986), and involves transformation of (S)-(- )-perillic acid into nateglinide by the reactions as depicted in Scheme-II. Trans-4- isopropyl cyclohexyl carboxylic acid of formula (5) was prepared by catalytic reduction of S-(-) - perillic acid of formula (11). The hydrogenation is accomplished with an Adams catalyst in an acetic acid medium at 5 kg pressure.
12
Scheme-II
The acid chloride of formula (12) was prepared by treatment of compound of formula (5) with PC15 in Dichloroethane at 40°C. The acid chloride of formula (12) was reacted with D-phenylalanine (8) in acetone using 10% aqueous NaOH to provide nateglinide of formula I. The process appears to be simpler than Scheme-I but it has the following drawbacks:
1) (S)-Perillic acid is an expensive raw material thereby making the process uneconomical.
2) The Adams catalyst Pt02 is not only expensive, but forms a flammable mixture with hydrogen gas thereby making the process not only uneconomical but also dangerous.
3) PC15 used for the acid chloride preparation of formula (12) is extremely corrosive and hygroscopic, and is very difficult to handle during scale up operation.
In yet another process for the synthesis of nateglinide, disclosed in Japan Kokai, Tokkyo Koho JP 07017899 (1995), the trans-isopropyl cyclohexyl carboxylic acid of formula (5) was converted to nateglinide of formula I in a single step using PCI5 as depicted in Scheme-Ill.
Scheme-
The process is straightforward, but it suffers from several drawbacks.
1) The use of dichloroethane (DCE) at the final step of the process is undesirable because it is a class II solvent and is to be avoided in the manufacture of pharmaceutical ingredients.
2) For chlorination, use of PCI5 is not preferred, as explained above.
SUMMARY OF THE INVENTION
In view of the importance gained by nateglinide, there is a need to the development of a simple, safe and economical manufacturing process, which will overcome the
drawbacks encountered in the prior art methods. The present invention fulfills the above described objectives.
We have discovered that nateglinide can be prepared by forming an in-situ mixed anliydride of formula (14) by reaction of the corresponding acid of formula (5) with alkyl chloroformate of the formula (13) wherein R is an alkyl, in a ketonic solvent and reacting said mixed anliydride with a solution of D-phenylalanine and recovering the compound of formula I by acidification. The process is shown in Scheme IV:
Accordingly, the present invention provides a novel process for the preparation of [(trans-4-isopropyl cyclohexyl)-carbonyl]-D-phenylalanine of formula I.
(i) reacting trans-4-isopropyl cyclohexyl carboxylic acid (formula (5))
O II
with an alkyl chloroformate (formula (13))
CIC02R 13 where R represents an alkyl group, in a ketonic solvent in the presence of a base at a temperature in the range of -20° to 30°C to form the mixed anhydride of formula (14); and
(ii) reacting said mixed anhydride with an aqueous alkali salt solution of D- phenylalanine to yield a reaction mixture including [(trans-4-isopropyl cyclohexyl)- carbonyl]-D-phenylalanine.
The compound of formula I may be subsequently recovered and purified by:
(i) diluting the reaction mixture with an aqueous solution of ketone to precipitate the compound of formula I;
(iv) acidifying the resultant reaction mixture to recover the product of formula I, which is a mixture of form B and H polymorphs; and if desired
(v) purifying by conventional methods or without further purification, the resulting compound of formula I.
The resulting compound of formula I, can be converted into the desired polymorph H form of nateglinide by using ethyl methyl ketone and water at 25-35° C temperature.
In a preferred embodiment of the invention, in the compound of formula (13), the alkyl group represents a C1-C10 straight or branched chain alkyl, preferably methyl,
ethyl, propyl, isopropyl, isobutyl, amyl, and isoamyl, or the like. The ketone used as a solvent may be selected from C1-C10 straight and branched chain alkyl ketones, preferably acetone, ethyl methyl ketone, methyl isobutyl ketone, and the like. The reaction may be effected preferably at a temperature in the range of -20 to 30 °C to form the mixed anhydride of formula (14). The mixed anhydride generated in situ is reacted either with an aqueous alkali, preferably sodium, potassium, lithium, and the like, a salt solution of D-phenylalanine such as sodium, potassium, lithium and the like or with organic bases selected from C1-C4 straight or branched chain alkyl preferably from a tertiary amine group, such as triethylamine, tripropylamine and tributylamine, in combination with or without a catalytic amount of N-methyl morpholine, N-ethyl morpholine, N-propyl morpholine and the like. This reaction may be effected in a temperature range of from -40 to 25 °C, and preferably at a temperature in the range of -20 to -15 °C . The aqueous alkali or basic solution may be prepared by reacting D-phenylalanine with the appropriate aqueous alkali or tertiary amine as mentioned above at a temperature between -5 to 0 °C. The addition of the aqueous alkali or basic solution of D-phenylalanine to the mixed anhydride may be effected in one lot at a temperature in the range of - 20 to - 15 °C.
The addition of a chilled solution (0°C) of ketone and water mixture (1:1) for dilution of the reaction mixture yields better precipitation of the product during acidification with HC1. The acidification of the reaction mixture is effected to a pH of preferably less than 2, resulting in the isolation of the compound of formula I. The resulting compound of formula I can be converted into the desired polymorph H form of nateglinide by using ethyl methyl ketone and water at 25-35 ° C, as explained in more detail in the Examples below.
The process of the present invention provides a single step synthesis, with overall improvement in the yield and purity, and uses relatively inexpensive reagents which are also relatively easy to handle. The process is also commercially viable and easy to scale up.
The details of the process are provided in the Examples given below which are provided as an illustration of the invention only and therefore should not be construed to limit the scope of the present invention.
Example 1
Preparation of [(Trans-4-isopropyl cycIohexyl)-carbonyl)] -D-phenylalanine of the formula I.
In a 5 liter 4-necked round bottom flask, acetone (800 ml), trans-4-isopropyl cyclohexyl carboxylic acid of formula (5) (obtained from SinoChem Jiang Imp., China or following the procedure of H. Shinkai, M. Nishikawa (J.Med. Chem. 1989, 32:1436) (100 gm) and triethylamine (60.5 gm) were added at 25 to 30°C. The reaction mixture was chilled to -10°C and ethyl chloroformate (obtained from BASF of India) (66.8 gm) was added in one lot at -10°C to the reaction mixture. The temperature was raised to about 5 to 10°C during addition. Chilling was continued and the reaction mixture was maintained at 10 to -20°C for 1 hr 15 min.
In another 5 liter four-necked round bottom flask, water (1000 ml) and sodium hydroxide (24.7 gm) were introduced and cooled to 0°C. To this D-phenylalanine (obtained from Daichi Pure Chemicals, Japan) (101.2 gm) was added in one lot and stirred for 10 minutes, followed by addition of acetone (200 ml) and chilling the solution to -5 to 0°C. D-phenylalanine solution maintained at -5 to 0°C was added to the reaction mixture obtained above in one lot at -20 to -15°C (exothermicity was observed and the temperature rose to about -3 to -2°C). Chilling was continued and the reaction mass was stirred at -2 to -10°C for 30 minutes. The reaction mixture was stirred further for 1 hr at -2 to -10°C preferably at -10°C. The reaction mixture was acidified, with a mixture of cone. HCl (140 ml) and water (140 ml) at -10 to 10°C (pH less than 2). To this mixture 1.5 liters of water was added and stirred at 0 to 10°C for 1 hr. The mixture was filtered with a Buchner funnel, and the solid obtained was slurried in a solution of 300 ml water and again filtered with a Buchner funnel. The solids were washed with water (300 ml x 2) until the pH became neutral. The crude product of formula I was dried at 60 to 70°C until the LOD (loss on drying) reached <2%. The crude product of the formula I appeared as an off- white amorphous solid with a net weight of 160 gm, a melting point of 20°C, a yield of 85%, and a purity of 94 to 95% by HPLC.
Example 2 - Purification of the nateglinide of Example 1
The crude product of formula I (160 gm) obtained in Example 1 was charged into a 5000 ml 4-necked round bottom flask and methanol (800 ml) was added at 25 to 30°C for dissolution. A solution of concentrated hydrochloric acid (5 ml) dissolved in 800 ml water was added to the reaction mixture slowly over 30 minutes at 25 to 30°C. The reaction mixture was maintained for 1 hr at 25 to 35°C and 800 ml of water was added slowly over 30 minutes at 25 to 35°C and the reaction mixture was maintained further at 25 to 35°C for 2 hrs. The precipitated product was filtered and washed with 80 ml solution of MeOH and water (1:1) and then with water until neutral pH was obtained. The product was dried at 60 to 70°C until the LOD reached <2%. The product appeared as white solid, net weight 145 gm, yield 77%, mp 124°C, purity by HPLC 97%. To improve the purity of the product further it was slurried in petroleum ether and methylene chloride mixture as follows.
Example 3 - Purification of the nateglinide of Example 2
Into a 5 liter 4-necked round bottom flask, was introduced 1450 ml of petroleum ether, and 145 gm of the methanol-purified product of Example 2 at 25 to 30°C. To this mixture 145 ml of dichloromethane (DCM) (obtained from Chemplast of India) was added and the reaction mixture was heated to 40 to 45 °C and stirred for 1 hour. The reaction mass was cooled to 35 to 38°C and 110 ml of DCM was added and the reaction mass heated for 30 minutes at 30 to 35°C. The product was filtered and the slurry was washed with 725 ml of petroleum ether. The product was dried at 60 to 70°C until the LOD reached <1%. The dried product appeared as white solid, mp 127 , to 130°C, net weight 108 gm, yield 57%, purity -99% by HPLC. In order to produce the desired, stable, polymorphic form, which is used in formulation, the previously purified B form of the product was converted into H form as follows.
Example 4 - Conversion of nateglinide to H form
In a 5000 ml four necked round bottom flask, was introduced the 108 grams of petroleum ether-DCM purified product of Example 3 and Ethyl methyl ketone (540 ml) were added at 25 to 35°C. Suspended extraneous fibrous particles were filtered
through a candy (candle-shaped) filter and a 2 micron polypropylene micro filter (Millipore). To the clear solution water (2160 ml) was added at 25 to 30°C under stirring and the reaction mass stirred for 24 hours at 25 to 30°C. The reaction mixture was then cooled to 15°C and the precipitate of the product filtered. The product was dried at 60 to 70°C until the LOD reached <1%. The dried product appeared as white crystalline solid, weighed 84 gm, with a yield of 44%, and mp 137 to 138°C, purity >99% by HPLC.
Example 5 - Analysis of nateglinide of Example 4
The product from Example 4 was subjected to analysis. The specific rotation [G;]20 D =
-9.2 (c=l in methanol).
The IR (KBr) spectrum shows the absorption at 3300-3400 cm"1 (O-H, N-H absorption), 3100 cm"1 & (C-H-str), 2900 cm"1 (C-H-str), 1710 cm"1 (COOH),1650 cm"' (CONH)
The 1H NMR (CDC13) TMS as internal standard shows δ 7.1-7.4 (m, 5H), 6.1 (d, 2H),
4.8 (q, 2H), 3.2-3.4 (m, 2H), 2.0-2.1 (m, 1H), 1.6-1.9 (m, 4H), 1.2-1.5 (m, 3H), 0.9-
1.3 (m, 2H), 0.8 (d, 6H).
The 13C MR (CDC13) shows the following signals at δ 178, 174, 138, 130, 128, 126, 54, 46, 44, 38, 34, 30, 20 ppm.
The CIMS shows m/z 317 (M+).
The elemental analysis theoretical %C 71.98; %H 8.50; %N 4.41; Observed values
%C 71.82; %H 8.45; %N 4.35.
Example 6 - Preparation of [(Trans-4-isopropyl cyclohexyl)-carbonyl)]-D- phenylalanine of formula I
In this example, the nateglinide of Example 1 was prepared in a scaled up process. In a 1000 liter (glass-lined) reactor was charged 120 liters of acetone, 15 kg of trans-4- isopropyl cyclohexyl carboxylic acid of formula (5), 9.4 kg of triethylamine, at 25 to 30°C. The reaction mixture was chilled to -10°C and 10.05 kg of ethyl chloroformate was added in one lot at -10°C to the reaction mixture. The temperature increased to
about 5 to 10°C during addition. The reaction mixture was maintained at 10 to -20°C for 1 hr 15 min.
In a separate 1000 liter stainless steel reactor was introduced 150 liters of water and 3.7 kg of sodium hydroxide. The mixture was cooled to 0°C. 15.3 kg of D- phenylalanine was added in one lot and the mixture stirred for 10 minutes, followed by the addition of 30 liters of acetone. The solution was chilled to -5 to 0°C. D-phenylalanine solution maintained at -5 to 0°C was added to the reaction mixture obtained above in one lot at -20 to -15°C (exothermicity was observed and the temperature rose to about -3 to -2°C). The reaction mass was chilled and stirred at -2 to -10°C for 30 minutes. The reaction mixture was diluted with a chilled (0°C) solution of 30 liters of acetone and 30 liters of water. The reaction mass was stirred further for 1 hr at -2 to -10°C, preferably towards -10°C.
The reaction mixture was then acidified with a solution of cone. HCl (16 kg) diluted with water (20 liters) at -10 to 10°C (pH less than 2). To this mixture was added 250 liters of water and the mixture stirred at 0 to 10°C for 1 hr. The solids obtained were filtered by centrifuge and then slurried in a solution of 25 liters of acetone and 25 liters of water and filtered again. The product was then washed with 10 liters of 1:1 mixtures of acetone and water and then with water (60 liters x 3) until the pH became neutral. The crude product of the formula I obtained was dried at 60 to 70°C until the LOD reached <2%. The crude product of the formula I appeared as an off-white amorphous solid, net weight 22 kg, mp 120°C, yield 78%, purity 94 to 95% by HPLC.
Example 7 - Purification of the nateglinide of Example 6
The crude product of Example 6 (22 kg) was charged into 500 liter GL reactor and 110 liters of methanol were added at 25 to 30°C for dissolution. A solution of cone, hydrochloric acid (650 ml) dissolved in 110 liters of water was added to the reaction mixture slowly over 30 minutes at 25 to 30°C. The reaction mixture was maintained for 1 hr at 25 to 35°C and 110 liters of water were then added slowly over 30 minutes at 25 to 35°C, and the reaction mixture maintained further at 25 to 35°C for 2 hrs. The precipitated product obtained was filtered and washed with 10 liters of a 1:1 mixture of methanol and water and then with water, until a neutral pH was obtained. The product obtained was dried at 60 to 70°C till until the LOD reached <2%. The
product appeared as a white solid, net weight 20 kg, yield 71%, mp 124°C, purity by HPLC 97%.
Example 8 - Purification of the nateglinide of Example 7
To improve the purity of the product of Example 7 further it was slurried in petroleum ether and methylene chloride mixture as follows.
A 500 liter glass lined reactor was charged with 200 liters of petroleum ether and 20 kg of the product from above (methanol purified) at 25 to 30°C. To mixture was added 20 liters of dichloromethane. The reaction mixture was heated to 40 to 45°C and stirred for 1 hour. The reaction mass was cooled to 35 to 38°C and 15 liters of DCM added followed by stirring for 30 minutes at 30 to 35°C. The resultant product was filtered and washed with 100 liters of petroleum ether, and the product dried at 60 to 70°C until the LOD reached <1%. The dried product appeared as white solid, mp 127 to 130°C, net weight 16 kg, yield 57%, purity -99% by HPLC.
Example 9 - Conversion of the nateglinide of Example 8 to H form polymorph
To produce the desired stable polymorphic form which is used in formulation, the product of Example 8 was converted into H form as follows.
Into a 500 liter glass lined reactor was added 16 kg of the product of Example 8 and 80 liters of ethyl methyl ketone at 25 to 35°C. The mixture was filtered to remove suspended extraneous particles through a candy filter and a 2 micron polypropylene micro filter (Millipore). To the resultant clear solution, 320 liters of water were added at 25 to 30°C under stirring and the reaction mass stirred for 24 hours at 25 to 30°C. The mixture was cooled to 15°C and the resultant the precipitate product filtered. The product was dried at 60 to 70°C until the LOD reached <1%. The dried product appeared as a white crystalline solid, weighing 12.5 kg, with a yield of 44%, mp 137 to 138°C, and purity >99% by HPLC.
The specific rotation [c]20 D= -9.2 (c=l in methanol).
The IR (KBr) spectrum shows the absorption at 3300-3400 cm"1 (O-H, N-H absorption), 3100 cm"1 & (C-H-str), 2900 (C-H-str) cm"1 , 1710 cm"1 (COOH),1650 cm"1 (CONH)
The 1H NMR (CDC1 ) TMS as internal standard shows δ 7.1-7.4 (m, 5H), 6.1 (d, 2H), 4.8 (q, 2H), 3.2-3.4 (m, 2H), 2.0-2.1 (m, 1H), 1.6-1.9 (m, 4H), 1.2-1.5 (m, 3H), 0.9- 1.3 (m, 2H), 0.8 (d, 6H).
The 13C NMR (CDC13) shows the following signals at δ 178, 174, 138, 130, 128, 126, 54, 46, 44, 38, 34, 30, 20 ppm. The CIMS shows m/z 317 (M+).
The elemental analysis theoretical %C 71.98; %H 8.50; %N 4.41; Observed values %C 71.82; %H 8.45; %N 4.35.
Example 10
Examples 1-5 were repeated, except that the amount of reactants and solvents were reduced and potassium hydroxide was substituted for sodium hydroxide. Flask 1 - contents amount acetone 200 ml trans-4-isopropyl cyclohexyl carboxylic acid 25 gm triethylamine 15.1 gm ethyl chloroformate 16.6 gm
Flask 2 - contents amount water 250 ml potassium hydroxide 8.6 gm
D-phenylalanine 25.4 gm acetone 50 ml
The dried product appeared as a white crystalline solid, weighed 20 gm, yield 44%, mp 137 to 138°C, purity >99% by HPLC.
The specific rotation [α]20 D= -9.2 (c=l in methanol).
The IR (KBr) spectrum shows the absorption at 3300-3400 cm"1 (O-H, N-H absorption), 3100 cm"1 & (C-H-str), 2900 (C-H-str) cm"1 , 1710 cm"1 (COOH),1650 cm"1 (CONH) The 1H NMR (CDC13) TMS as internal standard shows δ 7.1-7.4 (m, 5H), 6.1 (d, 2H),
4.8 (q, 2H), 3.2-3.4 (m, 2H), 2.0-2.1 (m, 1H), 1.6-1.9 (m, 4H), 1.2-1.5 (m, 3H), 0.9-
1.3 (m, 2H), 0.8 (d, 6H).
The 13C NMR (CDCI3) shows the following signals at δ 178, 174, 138, 130, 128, 126, 54, 46, 44, 38, 34, 30, 20 ppm. The CIMS shows m/z 317 (M+).
The elemental analysis theoretical %C 71.98; %H 8.50; %N 4.41; Observed values %C 71.82; %H 8.45; %N 4.35.
Example 11
Example 10 was repeated, except that the reactants and solvents and amounts were adjusted as follows: Flask 1 - contents amount acetone 200 ml trans-4-isopropyl cyclohexyl carboxylic acid 25 gm triethylamine 15.1 gm ethyl chloroformate 16.6 gm
Flask 2 - contents amount water 250 ml lithium hydroxide 6.0 gm
D-phenylalanine 25.4 gm acetone 50 ml
The dried product appeared as white crystalline solid, weighed 20 gm, yield 44%, mp 137 to 138°C, purity >99% by HPLC.
The specific rotation [α]20 D= -9.2 (c=l in methanol).
The IR (KBr) spectrum shows the absorption at 3300-3400 cm"1 (O-H, N-H absorption), 3100 cm"1 & (C-H-str), 2900 (C-H-str) cm"1 , 1710 cm"1 (COOH),1650 cm"1 (CONH) The 1H NMR (CDC13) TMS as internal standard shows δ 7.1 -7.4 (m, 5H), 6.1 (d, 2H),
4.8 (q, 2H), 3.2-3.4 (m, 2H), 2.0-2.1 (m, 1H), 1.6-1.9 (m, 4H), 1.2-1.5 (m, 3H), 0.9-
1.3 (m, 2H), 0.8 (d, 6H).
The 13C NMR (CDC13) shows the following signals at δ 178, 174, 138, 130, 128, 126,
54, 46, 44, 38, 34, 30, 20 ppm. The CIMS shows m/z 317 (M+-).
The elemental analysis theoretical %C 71.98; %H 8.50; %N 4.41; Observed values
%C 71.82; %H 8.45; %N 4.35.
Example 12
Example 11 was repeated, except that the reactants and solvents and amounts were adjusted as follows:
Flask 1 - contents amount acetone 200 ml trans-4-isopropyl cyclohexyl carboxylic acid 25 gm triethylamine 15.6 gm methyl chloroformate 14.58 gm
Flask 2 - contents amount water 250 ml sodium hydroxide 6.17 gm
D-phenylalanine 25.4 gm acetone 50 ml
The dried product appeared as a white crystalline solid, weighed 20 gm, yield 44%, mpl37 to 138°C, purity >99% by HPLC. The specific rotation [(X]20 Ό ~ -9.2 (c=l in methanol).
The IR (KBr) spectrum shows the absorption at 3300-3400 cm"1 (O-H, N-H absorption), 3100 cm"1 & (C-H-str), 2900 (C-H-str) cm"1 , 1710 cm"1 (COOH),1650 cm"1 (CONH)
The 1H NMR (CDC13) TMS as internal standard shows δ 7.1-7.4 (m, 5H), 6.1 (d, 2H), 4.8 (q, 2H), 3.2-3.4 (m, 2H), 2.0-2.1 (m, IH), 1.6-1.9 (m, 4H), 1.2-1.5 (m, 3H), 0.9-
1.3 (m, 2H), 0.8 (d, 6H).
The 13C NMR (CDC13) shows the following signals at δ 178, 174, 138, 1 0, 128, 126,
54, 46, 44, 38, 34, 30, 20 ppm.
The CIMS shows m/z 317 (M+). The elemental analysis theoretical %C 71.98; %H 8.50; %N 4.41; Observed values
%C 71.82; %H 8.45; %N 4.35.
Example 13 Example 12 was repeated, except that the reactants and solvents and amounts were adjusted as follows: Flask 1 - contents amount acetone 160 ml
trans-4-isopropyl cyclohexyl carboxylic acid 20 gm tripropyl amine 17.6 gm ethyl chloroformate 13.28 gm Flask 2 - contents amount water 200 ml sodium hydroxide 6.88 gm
D-phenylalanine 20.32 gm acetone 40 ml
The dried product appeared as a white crystalline solid, weighed 16 gm, yield 44%, mpl37 to 138°C, purity >99% by HPLC.
The specific rotation [ce]2°D= -9.2 (c=l in methanol). The IR (KBr) spectrum shows the absorption at 3300-3400 cm"1 (O-H, N-H absorption), 3100 cm"1 & (C-H-str), 2900 (C-H-str) cm"1, 1710 cm"1 (COOH),1650 cm"1 (CONH)
The !H NMR (CDC13) TMS as internal standard shows δ 7.1-7.4 (m, 5H), 6.1 (d, 2H),
4.8 (q, 2H), 3.2-3.4 (m, 2H), 2.0-2.1 (m, IH), 1.6-1.9 (m, 4H), 1.2-1.5 (m, 3H), 0.9- 1.3 (m, 2H), 0.8 (d, 6H).
The 13C NMR (CDCI3) shows the following signals at δ 178, 174, 138, 130, 128, 126,
54, 46, 44, 38, 34, 30, 20 ppm.
The CIMS shows m/z 317 (M+).
The elemental analysis: theoretical %C 71.98; %H 8.50; %N 4.41; Observed values %C 71.82; %H 8.45; %N 4.35.
Example 14
Example 13 was repeated, except that the reactants and solvents and amounts were adjusted as follows: Flask 1 - contents amount acetone 200 ml trans-4-isopropyl cyclohexyl carboxylic acid 25 gm tributyl amine 28.5 gm ethyl chloroformate 16.6 gm
Flask 2 - contents amount water 250 ml sodium hydroxide 6.17 gm
D-phenylalanine 25.4 gm acetone 50 ml
The dried product appeared as a white crystalline solid, weighed 20 gm, yield 44%, mp 1 7 to 138°C, purity >99% by HPLC.
The specific rotation [O;]20 D= -9.2 (c=l in methanol).
The IR (KBr) spectrum shows the absorption at 3300-3400 cm"1 (O-H, N-H absorption), 3100 cm"1 & (C-H-str), 2900 (C-H-str) cm"1 , 1710 cm"1 (COOH),1650 cm"1 (CONH) The ]HNMR (CDC13) TMS as internal standard shows δ 7.1-7.4 (m, 5H), 6.1 (d, 2H),
4.8 (q, 2H), 3.2-3.4 (m, 2H), 2.0-2.1 (m, IH), 1.6-1.9 (m, 4H), 1.2-1.5 (m, 3H), 0.9-
1.3 (m, 2H), 0.8 (d, 6H).
The 13C NMR (CDC13) shows the following signals at δ 178, 174, 138, 130, 128, 126,
54, 46, 44, 38, 34, 30, 20 ppm. The CIMS shows m/z 317 (M+') .
The elemental analysis theoretical %C 71.98; %H 8.50; %N 4.41; Observed values
%C 71.82; %H 8.45; %N 4.35.
Example 15 Example 14 was repeated, except that the reactants and solvents and amounts were adjusted as follows:
Flask 1 - contents amount
Ethyl methyl ketone 200 ml trans-4-isopropyl cyclohexyl carboxylic acid 25 gm triethylamine 15.6 gm ethyl chloroformate 16.6 gm
Flask 2 - contents amount water 250 ml sodium hydroxide 6.17 gm
D-phenylalanine 25.4 gm
Ethyl methyl ketone 50 ml
The dried product appeared as a white crystalline solid, weighed 18.5 gm, yield 40%, mp 137 to 138°C, purity >99% by HPLC. The specific rotation [c]20 D= -9.2 (c=l in methanol).
The IR (KBr) spectrum shows the absorption at 3300-3400 cm"1 (O-H, N-H absorption), 3100 cm"1 & (C-H-str), 2900 (C-H-str) cm"1 , 1710 cm"1 (COOH),1650 cm"1 (CONH)
The 1H NMR (CDC13) TMS as internal standard shows δ 7.1-7.4 (m, 5H), 6.1 (d, 2H), 4.8 (q, 2H), 3.2-3.4 (m, 2H), 2.0-2.1 (m, IH), 1.6-1.9 (m, 4H), 1.2-1.5 (m, 3H), 0.9- 1.3 (m, 2H), 0.8 (d, 6H).
The 13C NMR (CDC13) shows the following signals at δ 178, 174, 138, 130, 128, 126, 54, 46, 44, 38, 34, 30, 20 ppm. The CIMS shows m/z 317 (M+). The elemental analysis theoretical %C 71.98; %H 8.50; %N 4.41; Observed values %C 71.82; %H 8.45; %N 4.35.
Example 16 Example 15 was repeated, except that the reactants and solvents and amounts were adjusted as follows:
Flask 1 - contents amount methyl ethyl ketone 120 ml trans-4-isopropyl cyclohexyl carboxylic acid 25 gm tributyl amine 17.2 gm methyl chloroformate 8.75 gm
Flask 2 - contents amount water 150 ml potassium hydroxide 5.19 gm
D-phenylalanine 15.28 gm methyl ethyl ketone 30 ml
The dried product appeared as a white crystalline solid, weighed 12 gm, yield 44%, mpl37 to 138°C, purity >99% by HPLC. The specific rotation [o;]20 D= -9.2 (c=l in methanol).
The IR (KBr) spectrum shows the absorption at 3300-3400 cm"1 (O-H, N-H absorption), 3100 cm"1 & (C-H-str), 2900 (C-H-str) cm"1 , 1710 cm"1 (COOH),1650 cm"1 (CONH) .
The 'HNMR (CDCI3) TMS as internal standard shows δ 7.1-7.4 (m, 5H), 6.1 (d, 2H), 4.8 (q, 2H), 3.2-3.4 (m, 2H), 2.0-2.1 (m, IH), 1.6-1.9 (m, 4H), 1.2-1.5 (m, 3H), 0.9- 1.3 (m, 2H), 0.8 (d, 6H).
The 13C NMR (CDC13) shows the following signals at δ 178, 174, 138, 130, 128, 126, 54, 46, 44, 38, 34, 30, 20 ppm. The CIMS shows m/z 317 (M+).
The elemental analysis theoretical %C 71.98; %H 8.50; %N 4.41; Observed values %C 71.82; %H 8.45; %N 4.35.
Example 17
Example 16 was repeated, except that the reactants and solvents and amounts were adjusted as follows:
Flask 1 - contents amount acetone 500 ml trans-4-isopropyl cyclohexyl carboxylic acid 50 gm triethylamine 31.5 gm ethyl chloroformate 33.5 gm
Into a second 2 liter four-necked round bottom flask were introduced 400 ml water and 200 ml acetone at room temperature. To this 51 gm D-phenylalanine was added in one lot and stirred for 10 minutes, followed by addition of 50 gm triethylamine and stirring the solution at 25-30°C . The solution was chilled to -5 to 0°C and a solution of N-methyl morpholine (0.1 gm) dissolved in mixture of 9ml water and 1 ml acetone was added to the reaction mixture at 0-5°C and maintained for 10 minutes under stirring.
D-phenylalanine solution maintained at -5 to 0°C was added to the reaction mixture obtained above in one lot at -20 to -15°C (exothermicity was observed and the temperature rose to about -3 to -2°C). Chilling was continued and the reaction mass was stirred at -2 to -10°C for 30 minutes. The reaction mixture was stirred further for 1 hr at -2 to -10°C preferably at -10°C.
The reaction mixture was diluted with water (600ml) and acidified, with a mixture of cone. HCl (70 ml) and water (400 ml) slowly in about 45-60 minutes, at -10 to 10°C (pH less than 2). The reaction mixture was maintained at 0 to 10°C for 1 hr under stirring. The product was filtered and the solid obtained was slurried in a solution of 1000 ml water, stirred for 30 minutes and filtered. The washing was repeated with
water (100 ml x 2) until the pH became neutral and the product was suck dried for 1 hour and the wet product of the formula I was taken to next step of converting into H form, without drying. The crude wet product of the formula I appeared as an off- white amorphous solid, purity >99% by HPLC. To produce the desired, stable, polymorphic form, which is used in formulation, the wet crude was converted into H form as follows.
In a 2000 ml four necked round bottom flask, 170 gm of the crude wet product produced above was charged and 250 ml ethyl methyl ketone were added at 25 to 35°C. The suspended extraneous particles were filtered through candy and micro filters. To the clear solution 1000 ml water was added at 25 to 30°C under stirring and the reaction mass was stirred for 24 hours at 25 to 30°C. The reaction mixture was cooled to 15°C and the precipitate of the product filtered. The product was dried at 60 to 70°C until LOD reached <1%. The dried product appeared as a white crystalline solid, weighed 64 gm, yield 68%, and mp 137 to 138°C, purity >99% by HPLC. The specific rotation [o;] D = -9.2 (c=l in methanol).
The IR (KBr) spectrum shows the absorption at 3300-3400 cm"1 (O-H, N-H absorption), 3100 cm"1 & (C-H-str), 2900 cm"1 (C-H-str), 1710 cm"1 (COOH),1650 cm"1 (CONH) The !H NMR (CDC13) TMS as internal standard shows δ 7.1-7.4 (m, 5H), 6.1 (d, 2H), 4.8 (q, 2H), 3.2-3.4 (m, 2H), 2.0-2.1 (m, IH), 1.6-1.9 (m, 4H), 1.2-1.5 (m, 3H), 0.9- 1.3 (m, 2H), 0.8 (d, 6H).
The 13C NMR (CDC13) shows the following signals at δ 178, 174, 138, 130, 128, 126, 54, 46, 44, 38, 34, 30, 20 ppm. The CIMS shows m/z 317 (M+). The elemental analysis theoretical %C 71.98; %H 8.50; %N 4.41; Observed values %C 71.82; %H 8.45; %N 4.35.
Example 18
Example 17 was repeated except the step of adding N-methyl morpholine was omitted.
The dried product appeared as a white crystalline solid, weighed 64 gm, yield 68%, and mp 137 to 138°C, purity >99% by HPLC.
The specific rotation [α] π20 D= -9.2 (c=l in methanol). The IR (KBr) spectrum shows the absorption at 3300-3400 cm"1 (O-H, N-H absorption), 3100 cm"1 & (C-H-str), 2900 cm"1 (C-H-str), 1710 cm"1 (COOH),1650 cm"1 (CONH) The 1H NMR (CDC13) TMS as internal standard shows δ 7.1-7.4 (m, 5H), 6.1 (d, 2H), 4.8 (q, 2H), 3.2-3.4 (m, 2H), 2.0-2.1 (m, IH), 1.6-1.9 ( , 4H), 1.2-1.5 (m, 3H), 0.9- 1.3 (m, 2H), 0.8 (d, 6H).
The 13C NMR (CDC13) shows the following signals at δ 178, 174, 138, 130, 128, 126, 54, 46, 44, 38, 34, 30, 20 ppm. The CIMS shows m/z 317 (M+).
The elemental analysis theoretical %C 71.98; %H 8.50; %N 4.41; Observed values %C 71.82; %H 8.45; %N 4.35.