WO2004000817A2 - Benzimidazole compounds and their use as estrogen agonists/antagonists - Google Patents
Benzimidazole compounds and their use as estrogen agonists/antagonists Download PDFInfo
- Publication number
- WO2004000817A2 WO2004000817A2 PCT/IB2003/002670 IB0302670W WO2004000817A2 WO 2004000817 A2 WO2004000817 A2 WO 2004000817A2 IB 0302670 W IB0302670 W IB 0302670W WO 2004000817 A2 WO2004000817 A2 WO 2004000817A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- benzoimidazol
- phenol
- methyl
- methoxy
- Prior art date
Links
- 239000000262 estrogen Substances 0.000 title claims abstract description 24
- 229940011871 estrogen Drugs 0.000 title claims abstract description 23
- 239000000556 agonist Substances 0.000 title abstract description 18
- 239000005557 antagonist Substances 0.000 title abstract description 15
- 125000003785 benzimidazolyl group Chemical class N1=C(NC2=C1C=CC=C2)* 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 113
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 33
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 30
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 29
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 24
- 150000003839 salts Chemical class 0.000 claims abstract description 24
- 239000001257 hydrogen Substances 0.000 claims abstract description 19
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 14
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 12
- 150000002367 halogens Chemical class 0.000 claims abstract description 12
- 125000005194 alkoxycarbonyloxy group Chemical group 0.000 claims abstract description 7
- 125000006625 (C3-C8) cycloalkyloxy group Chemical group 0.000 claims abstract description 3
- 125000004104 aryloxy group Chemical group 0.000 claims abstract description 3
- 125000004465 cycloalkenyloxy group Chemical group 0.000 claims abstract description 3
- 239000000203 mixture Substances 0.000 claims description 98
- 238000000034 method Methods 0.000 claims description 83
- -1 -CHO Chemical group 0.000 claims description 62
- 125000001072 heteroaryl group Chemical group 0.000 claims description 26
- PUZPDOWCWNUUKD-UHFFFAOYSA-M sodium fluoride Chemical compound [F-].[Na+] PUZPDOWCWNUUKD-UHFFFAOYSA-M 0.000 claims description 24
- 102000015694 estrogen receptors Human genes 0.000 claims description 22
- 108010038795 estrogen receptors Proteins 0.000 claims description 22
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 19
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 16
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 14
- 241000124008 Mammalia Species 0.000 claims description 14
- 208000001132 Osteoporosis Diseases 0.000 claims description 14
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 claims description 14
- 239000000651 prodrug Substances 0.000 claims description 14
- 229940002612 prodrug Drugs 0.000 claims description 14
- 230000003042 antagnostic effect Effects 0.000 claims description 12
- 239000011775 sodium fluoride Substances 0.000 claims description 12
- 235000013024 sodium fluoride Nutrition 0.000 claims description 12
- 239000000199 parathyroid hormone Substances 0.000 claims description 11
- 125000006554 (C4-C8) cycloalkenyl group Chemical group 0.000 claims description 10
- 210000000988 bone and bone Anatomy 0.000 claims description 10
- 239000003324 growth hormone secretagogue Substances 0.000 claims description 10
- 125000001424 substituent group Chemical group 0.000 claims description 10
- 102000003982 Parathyroid hormone Human genes 0.000 claims description 9
- 108090000445 Parathyroid hormone Proteins 0.000 claims description 9
- 201000010099 disease Diseases 0.000 claims description 9
- 239000000122 growth hormone Substances 0.000 claims description 9
- 125000005842 heteroatom Chemical group 0.000 claims description 9
- 229960001319 parathyroid hormone Drugs 0.000 claims description 9
- 102100033367 Appetite-regulating hormone Human genes 0.000 claims description 8
- 101710111255 Appetite-regulating hormone Proteins 0.000 claims description 8
- 108010051696 Growth Hormone Proteins 0.000 claims description 8
- 208000035475 disorder Diseases 0.000 claims description 8
- 239000008194 pharmaceutical composition Substances 0.000 claims description 8
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 8
- 125000001544 thienyl group Chemical group 0.000 claims description 8
- MCZSFNSZNLRYTB-UHFFFAOYSA-N 4-[2-(2,5-dimethylfuran-3-yl)benzimidazol-1-yl]phenol Chemical compound O1C(C)=CC(C=2N(C3=CC=CC=C3N=2)C=2C=CC(O)=CC=2)=C1C MCZSFNSZNLRYTB-UHFFFAOYSA-N 0.000 claims description 7
- 125000002541 furyl group Chemical group 0.000 claims description 7
- 239000002089 prostaglandin antagonist Substances 0.000 claims description 7
- 239000002522 prostaglandin receptor stimulating agent Substances 0.000 claims description 7
- PXPYVMYIZRSPDS-UHFFFAOYSA-N 4-(2-thiophen-2-ylbenzimidazol-1-yl)phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CS1 PXPYVMYIZRSPDS-UHFFFAOYSA-N 0.000 claims description 6
- HNTFMADEDOCLMD-UHFFFAOYSA-N 4-[1-(4-hydroxyphenyl)benzimidazol-2-yl]-n-(1-phenylethyl)benzamide Chemical compound C=1C=CC=CC=1C(C)NC(=O)C(C=C1)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 HNTFMADEDOCLMD-UHFFFAOYSA-N 0.000 claims description 6
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 6
- KKKUBYHGTXNJEK-UHFFFAOYSA-N 4-[2-(1-ethylpyrrol-2-yl)benzimidazol-1-yl]phenol Chemical compound CCN1C=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 KKKUBYHGTXNJEK-UHFFFAOYSA-N 0.000 claims description 5
- NUXNQMXBOILMFU-UHFFFAOYSA-N 4-[2-(1-methylpyrrol-2-yl)benzimidazol-1-yl]phenol Chemical compound CN1C=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 NUXNQMXBOILMFU-UHFFFAOYSA-N 0.000 claims description 5
- MWLAUYCGCIQJBG-UHFFFAOYSA-N 4-[2-(2-methylfuran-3-yl)benzimidazol-1-yl]phenol Chemical compound O1C=CC(C=2N(C3=CC=CC=C3N=2)C=2C=CC(O)=CC=2)=C1C MWLAUYCGCIQJBG-UHFFFAOYSA-N 0.000 claims description 5
- VHRMCOSEZLZFDO-UHFFFAOYSA-N 4-[2-(3,5-dimethyl-1,2-oxazol-4-yl)benzimidazol-1-yl]phenol Chemical compound CC1=NOC(C)=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 VHRMCOSEZLZFDO-UHFFFAOYSA-N 0.000 claims description 5
- JEKFIHUZBALRRR-UHFFFAOYSA-N 4-[2-(3-bromothiophen-2-yl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=C(Br)C=CS1 JEKFIHUZBALRRR-UHFFFAOYSA-N 0.000 claims description 5
- NQWHPIAGSAZSBZ-UHFFFAOYSA-N 4-[2-(3-chlorothiophen-2-yl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=C(Cl)C=CS1 NQWHPIAGSAZSBZ-UHFFFAOYSA-N 0.000 claims description 5
- NWEHUCOFBRCHAE-UHFFFAOYSA-N 4-[2-(3-ethyl-5-methyl-1,2-oxazol-4-yl)benzimidazol-1-yl]phenol Chemical compound CCC1=NOC(C)=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 NWEHUCOFBRCHAE-UHFFFAOYSA-N 0.000 claims description 5
- GGCVQIHHDQDVBP-UHFFFAOYSA-N 4-[2-(3-methylfuran-2-yl)benzimidazol-1-yl]phenol Chemical compound C1=COC(C=2N(C3=CC=CC=C3N=2)C=2C=CC(O)=CC=2)=C1C GGCVQIHHDQDVBP-UHFFFAOYSA-N 0.000 claims description 5
- MUZDPVNNQNNFOG-UHFFFAOYSA-N 4-[2-(4-methylthiadiazol-5-yl)benzimidazol-1-yl]phenol Chemical compound N1=NSC(C=2N(C3=CC=CC=C3N=2)C=2C=CC(O)=CC=2)=C1C MUZDPVNNQNNFOG-UHFFFAOYSA-N 0.000 claims description 5
- AXXAIOIOCDNIDS-UHFFFAOYSA-N 4-[2-(5-methyl-1,2-oxazol-4-yl)benzimidazol-1-yl]phenol Chemical compound O1N=CC(C=2N(C3=CC=CC=C3N=2)C=2C=CC(O)=CC=2)=C1C AXXAIOIOCDNIDS-UHFFFAOYSA-N 0.000 claims description 5
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 claims description 5
- 125000004644 alkyl sulfinyl group Chemical group 0.000 claims description 5
- 125000004414 alkyl thio group Chemical group 0.000 claims description 5
- 239000003263 anabolic agent Substances 0.000 claims description 5
- 229940124325 anabolic agent Drugs 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- GCWRTGGADFVBTE-UHFFFAOYSA-N 3-methyl-4-[2-(1-methylpyrrol-2-yl)benzimidazol-1-yl]phenol Chemical compound CC1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CN1C GCWRTGGADFVBTE-UHFFFAOYSA-N 0.000 claims description 4
- JWEPQFMXIVSGBU-UHFFFAOYSA-N 4-[1-(4-hydroxyphenyl)benzimidazol-2-yl]-n-(thiophen-2-ylmethyl)benzamide Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(C(=O)NCC=2SC=CC=2)C=C1 JWEPQFMXIVSGBU-UHFFFAOYSA-N 0.000 claims description 4
- CPRVSBVNWMPCSG-UHFFFAOYSA-N 4-[1-(4-hydroxyphenyl)benzimidazol-2-yl]-n-propan-2-ylbenzamide Chemical compound C1=CC(C(=O)NC(C)C)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 CPRVSBVNWMPCSG-UHFFFAOYSA-N 0.000 claims description 4
- GWKUMTMRWOIPDQ-UHFFFAOYSA-N 4-[2-(1-propan-2-ylpyrrol-2-yl)benzimidazol-1-yl]phenol Chemical compound CC(C)N1C=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 GWKUMTMRWOIPDQ-UHFFFAOYSA-N 0.000 claims description 4
- TWJKMVBXWUPJNR-UHFFFAOYSA-N 4-[2-(3,5-dimethyl-1,2-oxazol-4-yl)benzimidazol-1-yl]-3-methylphenol Chemical compound CC1=NOC(C)=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1C TWJKMVBXWUPJNR-UHFFFAOYSA-N 0.000 claims description 4
- QBJRQOOSVWKPKU-UHFFFAOYSA-N 4-[2-(4-methyl-1,2-thiazol-5-yl)benzimidazol-1-yl]phenol Chemical compound C1=NSC(C=2N(C3=CC=CC=C3N=2)C=2C=CC(O)=CC=2)=C1C QBJRQOOSVWKPKU-UHFFFAOYSA-N 0.000 claims description 4
- LPPGDAXBXSIFBT-UHFFFAOYSA-N 4-[2-(furan-2-yl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CO1 LPPGDAXBXSIFBT-UHFFFAOYSA-N 0.000 claims description 4
- 206010028980 Neoplasm Diseases 0.000 claims description 4
- 125000005091 alkenylcarbonylamino group Chemical group 0.000 claims description 4
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 claims description 4
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims description 4
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 4
- 210000003169 central nervous system Anatomy 0.000 claims description 4
- ZRHZEPFBMCFVEY-UHFFFAOYSA-N chembl200021 Chemical compound C1=CC(O)=CC=C1C1=NOC(C(F)(F)F)=C1C1=CC=CC=C1 ZRHZEPFBMCFVEY-UHFFFAOYSA-N 0.000 claims description 4
- 150000002431 hydrogen Chemical group 0.000 claims description 4
- 125000001786 isothiazolyl group Chemical group 0.000 claims description 4
- NPRYQVZOYPELNS-UHFFFAOYSA-N methyl 4-[1-(4-hydroxyphenyl)benzimidazol-2-yl]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 NPRYQVZOYPELNS-UHFFFAOYSA-N 0.000 claims description 4
- HICDQHMHMAYLOS-UHFFFAOYSA-N 4-(2-butan-2-ylbenzimidazol-1-yl)phenol Chemical compound CCC(C)C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 HICDQHMHMAYLOS-UHFFFAOYSA-N 0.000 claims description 3
- VMNOMHRZHCJZJA-UHFFFAOYSA-N 4-[2-(1,2-thiazol-5-yl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=NS1 VMNOMHRZHCJZJA-UHFFFAOYSA-N 0.000 claims description 3
- RXIUHMXQYHLCAV-UHFFFAOYSA-N 4-[2-(2-methylthiophen-3-yl)benzimidazol-1-yl]phenol Chemical compound S1C=CC(C=2N(C3=CC=CC=C3N=2)C=2C=CC(O)=CC=2)=C1C RXIUHMXQYHLCAV-UHFFFAOYSA-N 0.000 claims description 3
- IYAJHEQREHSKPJ-UHFFFAOYSA-N 4-[2-(3-cyclopropyl-1,2-oxazol-4-yl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CON=C1C1CC1 IYAJHEQREHSKPJ-UHFFFAOYSA-N 0.000 claims description 3
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 3
- 208000024172 Cardiovascular disease Diseases 0.000 claims description 3
- 206010057671 Female sexual dysfunction Diseases 0.000 claims description 3
- 125000000304 alkynyl group Chemical group 0.000 claims description 3
- 230000001419 dependent effect Effects 0.000 claims description 3
- 125000000623 heterocyclic group Chemical group 0.000 claims description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 2
- IEUXCIYXPRKDDT-UHFFFAOYSA-N 4-(2-cyclopropylbenzimidazol-1-yl)phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1CC1 IEUXCIYXPRKDDT-UHFFFAOYSA-N 0.000 claims description 2
- REWUJZGEKPQYTC-UHFFFAOYSA-N 4-[2-(1,2-thiazol-4-yl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CSN=C1 REWUJZGEKPQYTC-UHFFFAOYSA-N 0.000 claims description 2
- SUWCEHWCBVMVIJ-UHFFFAOYSA-N 4-[4-phenyl-2-(trifluoromethyl)-1h-imidazol-5-yl]phenol Chemical compound C1=CC(O)=CC=C1C1=C(C=2C=CC=CC=2)N=C(C(F)(F)F)N1 SUWCEHWCBVMVIJ-UHFFFAOYSA-N 0.000 claims description 2
- 208000002874 Acne Vulgaris Diseases 0.000 claims description 2
- 201000004384 Alopecia Diseases 0.000 claims description 2
- 208000024827 Alzheimer disease Diseases 0.000 claims description 2
- 201000001320 Atherosclerosis Diseases 0.000 claims description 2
- 208000023275 Autoimmune disease Diseases 0.000 claims description 2
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 claims description 2
- 206010065687 Bone loss Diseases 0.000 claims description 2
- 125000006374 C2-C10 alkenyl group Chemical group 0.000 claims description 2
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims description 2
- 206010012205 Delayed puberty Diseases 0.000 claims description 2
- 208000016192 Demyelinating disease Diseases 0.000 claims description 2
- 206010013908 Dysfunctional uterine bleeding Diseases 0.000 claims description 2
- 201000009273 Endometriosis Diseases 0.000 claims description 2
- 206010020112 Hirsutism Diseases 0.000 claims description 2
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 2
- 208000013016 Hypoglycemia Diseases 0.000 claims description 2
- 208000007466 Male Infertility Diseases 0.000 claims description 2
- 206010027304 Menopausal symptoms Diseases 0.000 claims description 2
- 206010027514 Metrorrhagia Diseases 0.000 claims description 2
- 206010028116 Mucosal inflammation Diseases 0.000 claims description 2
- 201000010927 Mucositis Diseases 0.000 claims description 2
- 208000008589 Obesity Diseases 0.000 claims description 2
- 208000021384 Obsessive-Compulsive disease Diseases 0.000 claims description 2
- 206010060862 Prostate cancer Diseases 0.000 claims description 2
- 208000004403 Prostatic Hyperplasia Diseases 0.000 claims description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 2
- 208000025844 Prostatic disease Diseases 0.000 claims description 2
- 206010063837 Reperfusion injury Diseases 0.000 claims description 2
- 206010039792 Seborrhoea Diseases 0.000 claims description 2
- 206010040799 Skin atrophy Diseases 0.000 claims description 2
- 208000026928 Turner syndrome Diseases 0.000 claims description 2
- 208000025865 Ulcer Diseases 0.000 claims description 2
- 206010067269 Uterine fibrosis Diseases 0.000 claims description 2
- 206010046798 Uterine leiomyoma Diseases 0.000 claims description 2
- 206010000496 acne Diseases 0.000 claims description 2
- 125000004171 alkoxy aryl group Chemical group 0.000 claims description 2
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 2
- 231100000360 alopecia Toxicity 0.000 claims description 2
- 208000013404 behavioral symptom Diseases 0.000 claims description 2
- 208000035269 cancer or benign tumor Diseases 0.000 claims description 2
- 210000000845 cartilage Anatomy 0.000 claims description 2
- 230000004663 cell proliferation Effects 0.000 claims description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 claims description 2
- 230000007850 degeneration Effects 0.000 claims description 2
- HIVFSHGZYASMMI-UHFFFAOYSA-N ethyl 4-[1-(4-hydroxyphenyl)benzimidazol-2-yl]benzoate Chemical compound C1=CC(C(=O)OCC)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 HIVFSHGZYASMMI-UHFFFAOYSA-N 0.000 claims description 2
- 206010016629 fibroma Diseases 0.000 claims description 2
- 206010049444 fibromatosis Diseases 0.000 claims description 2
- 230000002218 hypoglycaemic effect Effects 0.000 claims description 2
- 208000015181 infectious disease Diseases 0.000 claims description 2
- 208000028867 ischemia Diseases 0.000 claims description 2
- 201000010260 leiomyoma Diseases 0.000 claims description 2
- 206010025135 lupus erythematosus Diseases 0.000 claims description 2
- 230000005906 menstruation Effects 0.000 claims description 2
- 230000002107 myocardial effect Effects 0.000 claims description 2
- 235000020824 obesity Nutrition 0.000 claims description 2
- 230000002611 ovarian Effects 0.000 claims description 2
- 208000006155 precocious puberty Diseases 0.000 claims description 2
- CMGXHSRNTJQAHI-UHFFFAOYSA-N propan-2-yl 4-[1-(4-hydroxyphenyl)benzimidazol-2-yl]benzoate Chemical compound C1=CC(C(=O)OC(C)C)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 CMGXHSRNTJQAHI-UHFFFAOYSA-N 0.000 claims description 2
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 2
- 208000037803 restenosis Diseases 0.000 claims description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 2
- 208000008742 seborrheic dermatitis Diseases 0.000 claims description 2
- 210000000329 smooth muscle myocyte Anatomy 0.000 claims description 2
- 230000009424 thromboembolic effect Effects 0.000 claims description 2
- 206010043778 thyroiditis Diseases 0.000 claims description 2
- 208000010579 uterine corpus leiomyoma Diseases 0.000 claims description 2
- 201000007954 uterine fibroid Diseases 0.000 claims description 2
- 210000001215 vagina Anatomy 0.000 claims description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 claims 2
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 claims 1
- 102000018997 Growth Hormone Human genes 0.000 claims 1
- 125000005708 carbonyloxy group Chemical group [*:2]OC([*:1])=O 0.000 claims 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 abstract description 3
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 abstract description 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 292
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 228
- 239000000243 solution Substances 0.000 description 187
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 137
- 238000006243 chemical reaction Methods 0.000 description 130
- 235000019439 ethyl acetate Nutrition 0.000 description 128
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 116
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 105
- 238000005160 1H NMR spectroscopy Methods 0.000 description 95
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 88
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 86
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 86
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 62
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 61
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 58
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 58
- 239000011541 reaction mixture Substances 0.000 description 52
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 50
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 49
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 47
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 47
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 43
- 239000012267 brine Substances 0.000 description 41
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 41
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 41
- 229910052681 coesite Inorganic materials 0.000 description 37
- 229910052906 cristobalite Inorganic materials 0.000 description 37
- 239000000377 silicon dioxide Substances 0.000 description 37
- 229910052682 stishovite Inorganic materials 0.000 description 37
- 229910052905 tridymite Inorganic materials 0.000 description 37
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 36
- 229940093499 ethyl acetate Drugs 0.000 description 36
- 102000000509 Estrogen Receptor beta Human genes 0.000 description 35
- 108010041356 Estrogen Receptor beta Proteins 0.000 description 34
- 238000003818 flash chromatography Methods 0.000 description 34
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 33
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 31
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 30
- 239000000463 material Substances 0.000 description 30
- 239000007787 solid Substances 0.000 description 30
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 29
- 238000000746 purification Methods 0.000 description 28
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 28
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 27
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 26
- 239000003921 oil Substances 0.000 description 26
- 239000000284 extract Substances 0.000 description 25
- 229910052757 nitrogen Inorganic materials 0.000 description 25
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 24
- 238000010792 warming Methods 0.000 description 23
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 22
- 238000011282 treatment Methods 0.000 description 22
- 238000002360 preparation method Methods 0.000 description 21
- 238000003756 stirring Methods 0.000 description 21
- 239000011324 bead Substances 0.000 description 18
- 239000010410 layer Substances 0.000 description 18
- VUKPHZHQORZZTQ-UHFFFAOYSA-N 2-n-(4-methoxyphenyl)benzene-1,2-diamine;dihydrochloride Chemical compound Cl.Cl.C1=CC(OC)=CC=C1NC1=CC=CC=C1N VUKPHZHQORZZTQ-UHFFFAOYSA-N 0.000 description 17
- 238000005481 NMR spectroscopy Methods 0.000 description 17
- 230000003197 catalytic effect Effects 0.000 description 17
- 230000000694 effects Effects 0.000 description 17
- 239000012467 final product Substances 0.000 description 16
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 16
- 239000000047 product Substances 0.000 description 15
- 108010007005 Estrogen Receptor alpha Proteins 0.000 description 14
- 102000007594 Estrogen Receptor alpha Human genes 0.000 description 14
- HPLDXDRZCMPNFO-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-thiophen-3-ylbenzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CSC=C1 HPLDXDRZCMPNFO-UHFFFAOYSA-N 0.000 description 13
- 229960000583 acetic acid Drugs 0.000 description 13
- 210000004027 cell Anatomy 0.000 description 13
- KNAXKMPOHVBFMJ-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]thiophene-3-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CSC=C1 KNAXKMPOHVBFMJ-UHFFFAOYSA-N 0.000 description 13
- 238000005406 washing Methods 0.000 description 13
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 12
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 12
- 239000012298 atmosphere Substances 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 12
- 239000012948 isocyanate Substances 0.000 description 12
- 150000002513 isocyanates Chemical class 0.000 description 12
- 235000019341 magnesium sulphate Nutrition 0.000 description 12
- 102000005962 receptors Human genes 0.000 description 12
- 108020003175 receptors Proteins 0.000 description 12
- MMJRINZEHFQMLN-UHFFFAOYSA-N 2-n-(4-methoxyphenyl)benzene-1,2-diamine Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1N MMJRINZEHFQMLN-UHFFFAOYSA-N 0.000 description 10
- 235000019441 ethanol Nutrition 0.000 description 10
- 230000011164 ossification Effects 0.000 description 10
- 229920000642 polymer Polymers 0.000 description 10
- 238000004809 thin layer chromatography Methods 0.000 description 10
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 230000027455 binding Effects 0.000 description 9
- 238000012746 preparative thin layer chromatography Methods 0.000 description 9
- 238000010992 reflux Methods 0.000 description 9
- 229910052938 sodium sulfate Inorganic materials 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- 125000004429 atom Chemical group 0.000 description 8
- 125000000753 cycloalkyl group Chemical group 0.000 description 8
- 238000005516 engineering process Methods 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 239000012442 inert solvent Substances 0.000 description 8
- 239000003446 ligand Substances 0.000 description 8
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 8
- 229910000027 potassium carbonate Inorganic materials 0.000 description 8
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 7
- VQQLWBUTTMNMFT-UHFFFAOYSA-N 3-bromothiophene-2-carboxylic acid Chemical compound OC(=O)C=1SC=CC=1Br VQQLWBUTTMNMFT-UHFFFAOYSA-N 0.000 description 7
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 7
- 239000007832 Na2SO4 Substances 0.000 description 7
- 102100038803 Somatotropin Human genes 0.000 description 7
- 230000009471 action Effects 0.000 description 7
- 125000003118 aryl group Chemical group 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 7
- 239000012044 organic layer Substances 0.000 description 7
- 239000011368 organic material Substances 0.000 description 7
- 239000012074 organic phase Substances 0.000 description 7
- 238000004007 reversed phase HPLC Methods 0.000 description 7
- 230000001568 sexual effect Effects 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- XEZNGIUYQVAUSS-UHFFFAOYSA-N 18-crown-6 Chemical compound C1COCCOCCOCCOCCOCCO1 XEZNGIUYQVAUSS-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 6
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N N-phenyl amine Natural products NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 6
- 238000010828 elution Methods 0.000 description 6
- 238000011068 loading method Methods 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- PQLXBRBIKXJXIK-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-(1H-pyrrol-2-yl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CN1 PQLXBRBIKXJXIK-UHFFFAOYSA-N 0.000 description 5
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 5
- FMXSBCWDKMEYSB-UHFFFAOYSA-N 4-[1-(4-methoxyphenyl)benzimidazol-2-yl]benzoic acid Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(C(O)=O)C=C1 FMXSBCWDKMEYSB-UHFFFAOYSA-N 0.000 description 5
- 208000007415 Anhedonia Diseases 0.000 description 5
- 206010006187 Breast cancer Diseases 0.000 description 5
- 208000026310 Breast neoplasm Diseases 0.000 description 5
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- 239000005909 Kieselgur Substances 0.000 description 5
- 206010024419 Libido decreased Diseases 0.000 description 5
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 5
- 208000006262 Psychological Sexual Dysfunctions Diseases 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 5
- 229910052794 bromium Inorganic materials 0.000 description 5
- 150000001735 carboxylic acids Chemical class 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 239000012230 colorless oil Substances 0.000 description 5
- MGHPNCMVUAKAIE-UHFFFAOYSA-N diphenylmethanamine Chemical compound C=1C=CC=CC=1C(N)C1=CC=CC=C1 MGHPNCMVUAKAIE-UHFFFAOYSA-N 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 238000009547 dual-energy X-ray absorptiometry Methods 0.000 description 5
- 239000002834 estrogen receptor modulator Substances 0.000 description 5
- 208000017020 hypoactive sexual desire disease Diseases 0.000 description 5
- 229910052751 metal Inorganic materials 0.000 description 5
- 239000002184 metal Substances 0.000 description 5
- 229910052760 oxygen Inorganic materials 0.000 description 5
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 238000001953 recrystallisation Methods 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 235000013599 spices Nutrition 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 5
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 4
- WGUYBGHDPRGYLS-UHFFFAOYSA-N 2-(4-methoxyphenyl)-1-thiophen-3-ylbenzimidazole Chemical compound C1=CC(OC)=CC=C1C1=NC2=CC=CC=C2N1C1=CSC=C1 WGUYBGHDPRGYLS-UHFFFAOYSA-N 0.000 description 4
- GSAFKDWJWGNOGX-UHFFFAOYSA-N 2-n-(4-methoxy-2-methylphenyl)benzene-1,2-diamine Chemical compound CC1=CC(OC)=CC=C1NC1=CC=CC=C1N GSAFKDWJWGNOGX-UHFFFAOYSA-N 0.000 description 4
- NJEPDWZYBJIKMD-UHFFFAOYSA-N 3-cyclopropyl-1,2-oxazole-4-carboxylic acid Chemical compound OC(=O)C1=CON=C1C1CC1 NJEPDWZYBJIKMD-UHFFFAOYSA-N 0.000 description 4
- NCZPZQIEZCBBHL-UHFFFAOYSA-N 4-[2-(3-methylthiophen-2-yl)benzimidazol-1-yl]phenol Chemical compound C1=CSC(C=2N(C3=CC=CC=C3N=2)C=2C=CC(O)=CC=2)=C1C NCZPZQIEZCBBHL-UHFFFAOYSA-N 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 4
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 4
- 150000008065 acid anhydrides Chemical class 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 150000001556 benzimidazoles Chemical class 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 239000000328 estrogen antagonist Substances 0.000 description 4
- 239000012362 glacial acetic acid Substances 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 150000002500 ions Chemical class 0.000 description 4
- 239000011591 potassium Substances 0.000 description 4
- 229910052700 potassium Inorganic materials 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- WRHZVMBBRYBTKZ-UHFFFAOYSA-N pyrrole-2-carboxylic acid Chemical compound OC(=O)C1=CC=CN1 WRHZVMBBRYBTKZ-UHFFFAOYSA-N 0.000 description 4
- 239000002516 radical scavenger Substances 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 4
- NRQHBNNTBIDSRK-YRNVUSSQSA-N (4e)-4-[(4-methoxyphenyl)methylidene]-2-methyl-1,3-oxazol-5-one Chemical compound C1=CC(OC)=CC=C1\C=C\1C(=O)OC(C)=N/1 NRQHBNNTBIDSRK-YRNVUSSQSA-N 0.000 description 3
- XQDANBKPALOMEF-HYXAFXHYSA-N (nz)-n-(cyclopropylmethylidene)hydroxylamine Chemical compound O\N=C/C1CC1 XQDANBKPALOMEF-HYXAFXHYSA-N 0.000 description 3
- GPQLWAYDIUTFFL-UHFFFAOYSA-N 1,2-bis(4-methoxyphenyl)benzimidazole Chemical compound C1=CC(OC)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(OC)C=C1 GPQLWAYDIUTFFL-UHFFFAOYSA-N 0.000 description 3
- MATKNMWQQPITBJ-UHFFFAOYSA-N 1-(4-methoxy-2-methylphenyl)-2-(1-methylpyrrol-2-yl)benzimidazole Chemical compound CC1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CN1C MATKNMWQQPITBJ-UHFFFAOYSA-N 0.000 description 3
- PXKNPJYKAUUEBQ-UHFFFAOYSA-N 1-(4-methoxy-2-methylphenyl)-2-(2-methylphenyl)benzimidazole Chemical compound CC1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CC=C1C PXKNPJYKAUUEBQ-UHFFFAOYSA-N 0.000 description 3
- BCBUDZZHZSQUPK-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-(2-methylfuran-3-yl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(C)OC=C1 BCBUDZZHZSQUPK-UHFFFAOYSA-N 0.000 description 3
- SFUKZMHWCKKEGE-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-(3-methylthiophen-2-yl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(C)C=CS1 SFUKZMHWCKKEGE-UHFFFAOYSA-N 0.000 description 3
- CSEBUQAWCUJJHA-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-phenylbenzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CC=C1 CSEBUQAWCUJJHA-UHFFFAOYSA-N 0.000 description 3
- DUPKHLXLZQLPGZ-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-thiophen-2-ylbenzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CS1 DUPKHLXLZQLPGZ-UHFFFAOYSA-N 0.000 description 3
- NPKXGPIRFVVWPN-UHFFFAOYSA-N 1-(4-phenylmethoxyphenyl)-2-(1H-pyrrol-2-yl)benzimidazole Chemical compound C=1C=CC=CC=1COC(C=C1)=CC=C1N(C1=CC=CC=C1N=1)C=1C1=CC=CN1 NPKXGPIRFVVWPN-UHFFFAOYSA-N 0.000 description 3
- SKKBPXKSCGYSGD-UHFFFAOYSA-N 2-(2,5-dimethylfuran-3-yl)-1-(4-methoxyphenyl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(C)OC(C)=C1 SKKBPXKSCGYSGD-UHFFFAOYSA-N 0.000 description 3
- SIVDZTOAYPDOTP-UHFFFAOYSA-N 2-(2-chlorophenyl)-1-(4-methoxy-2-methylphenyl)benzimidazole Chemical compound CC1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CC=C1Cl SIVDZTOAYPDOTP-UHFFFAOYSA-N 0.000 description 3
- DAEZSACZCMGYSA-UHFFFAOYSA-N 2-(3-bromothiophen-2-yl)-1-(4-methoxyphenyl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(Br)C=CS1 DAEZSACZCMGYSA-UHFFFAOYSA-N 0.000 description 3
- IPIRBYBEHCAWSX-UHFFFAOYSA-N 2-(3-chlorothiophen-2-yl)-1-(4-methoxyphenyl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(Cl)C=CS1 IPIRBYBEHCAWSX-UHFFFAOYSA-N 0.000 description 3
- JAACRUFVAJSBHL-UHFFFAOYSA-N 2-(4-methoxyphenyl)-1-thiophen-2-ylbenzimidazole Chemical compound C1=CC(OC)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=CS1 JAACRUFVAJSBHL-UHFFFAOYSA-N 0.000 description 3
- HBNSFRKOHHPWGL-UHFFFAOYSA-N 2-chloro-n-[2-(4-methoxy-2-methylanilino)phenyl]benzamide Chemical compound CC1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CC=CC=C1Cl HBNSFRKOHHPWGL-UHFFFAOYSA-N 0.000 description 3
- CFGQZVOVFIZRMN-UHFFFAOYSA-N 2-methylfuran-3-carboxylic acid Chemical compound CC=1OC=CC=1C(O)=O CFGQZVOVFIZRMN-UHFFFAOYSA-N 0.000 description 3
- IJEUISLJVBUNRE-UHFFFAOYSA-N 3,5-dimethyl-1,2-oxazole-4-carboxylic acid Chemical compound CC1=NOC(C)=C1C(O)=O IJEUISLJVBUNRE-UHFFFAOYSA-N 0.000 description 3
- VTISBJQUCOHWDE-UHFFFAOYSA-N 3-(4-methoxyphenyl)-4-phenyl-5-(trifluoromethyl)-1,2-oxazole Chemical compound C1=CC(OC)=CC=C1C1=NOC(C(F)(F)F)=C1C1=CC=CC=C1 VTISBJQUCOHWDE-UHFFFAOYSA-N 0.000 description 3
- JKSQVCXYLAPDTC-UHFFFAOYSA-N 3-bromo-n-[2-(4-methoxyanilino)phenyl]thiophene-2-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(Br)C=CS1 JKSQVCXYLAPDTC-UHFFFAOYSA-N 0.000 description 3
- DJUYSDPPDGMKSC-UHFFFAOYSA-N 3-chloro-n-[2-(4-methoxyanilino)phenyl]thiophene-2-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(Cl)C=CS1 DJUYSDPPDGMKSC-UHFFFAOYSA-N 0.000 description 3
- AVDJIJMAOULDPL-UHFFFAOYSA-N 3-cyclopropyl-4-[1-(4-methoxyphenyl)benzimidazol-2-yl]-1,2-oxazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CON=C1C1CC1 AVDJIJMAOULDPL-UHFFFAOYSA-N 0.000 description 3
- ZZNCBQHXICFHBY-UHFFFAOYSA-N 3-cyclopropyl-n-[2-(4-methoxyanilino)phenyl]-1,2-oxazole-4-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CON=C1C1CC1 ZZNCBQHXICFHBY-UHFFFAOYSA-N 0.000 description 3
- GWXJFENAVVNMTA-UHFFFAOYSA-N 3-ethyl-1,2-oxazole-4-carboxylic acid Chemical compound CCC1=NOC=C1C(O)=O GWXJFENAVVNMTA-UHFFFAOYSA-N 0.000 description 3
- VTFKLWHTNGGHPO-UHFFFAOYSA-N 3-ethyl-4-[1-(4-methoxyphenyl)benzimidazol-2-yl]-5-methyl-1,2-oxazole Chemical compound CCC1=NOC(C)=C1C1=NC2=CC=CC=C2N1C1=CC=C(OC)C=C1 VTFKLWHTNGGHPO-UHFFFAOYSA-N 0.000 description 3
- RSWFHHWVFZWIMV-UHFFFAOYSA-N 3-ethyl-5-methyl-1,2-oxazole-4-carboxylic acid Chemical compound CCC1=NOC(C)=C1C(O)=O RSWFHHWVFZWIMV-UHFFFAOYSA-N 0.000 description 3
- DINVHIBNUCEQHE-UHFFFAOYSA-N 3-ethyl-n-[2-(4-methoxyanilino)phenyl]-1,2-oxazole-4-carboxamide Chemical compound CCC1=NOC=C1C(=O)NC1=CC=CC=C1NC1=CC=C(OC)C=C1 DINVHIBNUCEQHE-UHFFFAOYSA-N 0.000 description 3
- JSGMHIZNTIJCEG-UHFFFAOYSA-N 4-(2-ethylbenzimidazol-1-yl)phenol Chemical compound CCC1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 JSGMHIZNTIJCEG-UHFFFAOYSA-N 0.000 description 3
- IDDOUQZYGAKNIP-UHFFFAOYSA-N 4-(2-thiophen-3-ylbenzimidazol-1-yl)phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CSC=C1 IDDOUQZYGAKNIP-UHFFFAOYSA-N 0.000 description 3
- SULPWCVRDDAISD-UHFFFAOYSA-N 4-[1-(4-methoxy-2-methylphenyl)benzimidazol-2-yl]-3,5-dimethyl-1,2-oxazole Chemical compound CC1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(C)ON=C1C SULPWCVRDDAISD-UHFFFAOYSA-N 0.000 description 3
- LZXWALAZKGWTEL-UHFFFAOYSA-N 4-[1-(4-methoxyphenyl)benzimidazol-2-yl]-3,5-dimethyl-1,2-oxazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(C)ON=C1C LZXWALAZKGWTEL-UHFFFAOYSA-N 0.000 description 3
- CLXXJHBEJXKQNH-UHFFFAOYSA-N 4-[1-(4-methoxyphenyl)benzimidazol-2-yl]-3-methyl-1,2-oxazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CON=C1C CLXXJHBEJXKQNH-UHFFFAOYSA-N 0.000 description 3
- ZLJIBFIUQQLQKB-UHFFFAOYSA-N 4-[1-(4-phenylmethoxyphenyl)benzimidazol-2-yl]-n-propan-2-ylbenzamide Chemical compound C1=CC(C(=O)NC(C)C)=CC=C1C1=NC2=CC=CC=C2N1C(C=C1)=CC=C1OCC1=CC=CC=C1 ZLJIBFIUQQLQKB-UHFFFAOYSA-N 0.000 description 3
- QPGWNXJJOWMXSH-UHFFFAOYSA-N 4-[2-(1H-pyrrol-2-yl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CN1 QPGWNXJJOWMXSH-UHFFFAOYSA-N 0.000 description 3
- NIYAOPBQXQOIGY-UHFFFAOYSA-N 4-[2-(furan-3-yl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=COC=C1 NIYAOPBQXQOIGY-UHFFFAOYSA-N 0.000 description 3
- GUYYAAWZEKHSPP-UHFFFAOYSA-N 4-methoxy-2-methyl-n-(2-nitrophenyl)aniline Chemical compound CC1=CC(OC)=CC=C1NC1=CC=CC=C1[N+]([O-])=O GUYYAAWZEKHSPP-UHFFFAOYSA-N 0.000 description 3
- IYUOBIBLVIIUNB-UHFFFAOYSA-N 4-methoxy-n-[2-(4-methoxyanilino)phenyl]benzamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CC=C(OC)C=C1 IYUOBIBLVIIUNB-UHFFFAOYSA-N 0.000 description 3
- WQCULZRIFWVSGE-UHFFFAOYSA-N 5-[1-(4-methoxyphenyl)benzimidazol-2-yl]thiophene-2-carboxylic acid Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(C(O)=O)S1 WQCULZRIFWVSGE-UHFFFAOYSA-N 0.000 description 3
- HYHZGBOWRWIEGW-UHFFFAOYSA-N 5-methoxycarbonylthiophene-2-carboxylic acid Chemical compound COC(=O)C1=CC=C(C(O)=O)S1 HYHZGBOWRWIEGW-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- 108091026890 Coding region Proteins 0.000 description 3
- 241000557626 Corvus corax Species 0.000 description 3
- 208000004483 Dyspareunia Diseases 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 238000010240 RT-PCR analysis Methods 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- 208000002495 Uterine Neoplasms Diseases 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 150000001350 alkyl halides Chemical class 0.000 description 3
- 150000001448 anilines Chemical class 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 3
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 3
- 229910000024 caesium carbonate Inorganic materials 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 239000003610 charcoal Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 238000010367 cloning Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 3
- CYUGNCLRGFKPAE-UHFFFAOYSA-N dimethyl thiophene-2,5-dicarboxylate Chemical compound COC(=O)C1=CC=C(C(=O)OC)S1 CYUGNCLRGFKPAE-UHFFFAOYSA-N 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 230000001076 estrogenic effect Effects 0.000 description 3
- CEORCTQTTTZSLY-UHFFFAOYSA-N ethyl 3-ethyl-1,2-oxazole-4-carboxylate Chemical compound CCOC(=O)C1=CON=C1CC CEORCTQTTTZSLY-UHFFFAOYSA-N 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 210000002503 granulosa cell Anatomy 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- BHGGUUYCJCSIJD-UHFFFAOYSA-N methyl 4-[1-(4-methoxyphenyl)benzimidazol-2-yl]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(OC)C=C1 BHGGUUYCJCSIJD-UHFFFAOYSA-N 0.000 description 3
- OQLVHSCXQCMLHN-UHFFFAOYSA-N methyl 5-[[2-(4-methoxyanilino)phenyl]carbamoyl]thiophene-2-carboxylate Chemical compound S1C(C(=O)OC)=CC=C1C(=O)NC1=CC=CC=C1NC1=CC=C(OC)C=C1 OQLVHSCXQCMLHN-UHFFFAOYSA-N 0.000 description 3
- JLIRPISIPQMEDU-UHFFFAOYSA-N n-(4-methoxyphenyl)-2-nitroaniline Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1[N+]([O-])=O JLIRPISIPQMEDU-UHFFFAOYSA-N 0.000 description 3
- QLRSMLGUBNUXQI-UHFFFAOYSA-N n-[1-(4-methoxyphenyl)-2-phenylethylidene]hydroxylamine Chemical compound C1=CC(OC)=CC=C1C(=NO)CC1=CC=CC=C1 QLRSMLGUBNUXQI-UHFFFAOYSA-N 0.000 description 3
- LZPGBXUPPODGFW-UHFFFAOYSA-N n-[2-(4-methoxy-2-methylanilino)phenyl]-1-methylpyrrole-2-carboxamide Chemical compound CC1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CC=CN1C LZPGBXUPPODGFW-UHFFFAOYSA-N 0.000 description 3
- MHOIGUVKSLTQPW-UHFFFAOYSA-N n-[2-(4-methoxy-2-methylanilino)phenyl]-2-methylbenzamide Chemical compound CC1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CC=CC=C1C MHOIGUVKSLTQPW-UHFFFAOYSA-N 0.000 description 3
- OKZDKLIKAJFKJT-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-1-methylpyrrole-2-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CC=CN1C OKZDKLIKAJFKJT-UHFFFAOYSA-N 0.000 description 3
- XUZOMSHITOYNQM-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-2-methylthiophene-3-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(C)SC=C1 XUZOMSHITOYNQM-UHFFFAOYSA-N 0.000 description 3
- MZFQTMOBRSQVQW-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-3,5-dimethyl-1,2-oxazole-4-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(C)ON=C1C MZFQTMOBRSQVQW-UHFFFAOYSA-N 0.000 description 3
- SSXYJNSIKNUXJF-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-3-methylthiophene-2-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(C)C=CS1 SSXYJNSIKNUXJF-UHFFFAOYSA-N 0.000 description 3
- XYLCOLFYLQWQRY-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-4-methyl-1,2-thiazole-5-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(C)C=NS1 XYLCOLFYLQWQRY-UHFFFAOYSA-N 0.000 description 3
- HVFRNQLERAMHFN-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-4-methylthiadiazole-5-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(C)N=NS1 HVFRNQLERAMHFN-UHFFFAOYSA-N 0.000 description 3
- VRFJQQLGHBQINQ-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]furan-2-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CC=CO1 VRFJQQLGHBQINQ-UHFFFAOYSA-N 0.000 description 3
- OXGXNQDARRTPKA-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]furan-3-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=COC=C1 OXGXNQDARRTPKA-UHFFFAOYSA-N 0.000 description 3
- OKNMFACCCTWSDB-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]thiophene-2-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CC=CS1 OKNMFACCCTWSDB-UHFFFAOYSA-N 0.000 description 3
- LIROSOXSTPTVIO-UHFFFAOYSA-N n-benzhydryl-4-[1-(4-hydroxyphenyl)benzimidazol-2-yl]benzamide Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(C(=O)NC(C=2C=CC=CC=2)C=2C=CC=CC=2)C=C1 LIROSOXSTPTVIO-UHFFFAOYSA-N 0.000 description 3
- DCOBLQRDHFFDRO-UHFFFAOYSA-N n-benzhydryl-5-[1-(4-hydroxyphenyl)benzimidazol-2-yl]thiophene-2-carboxamide Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(C(=O)NC(C=2C=CC=CC=2)C=2C=CC=CC=2)S1 DCOBLQRDHFFDRO-UHFFFAOYSA-N 0.000 description 3
- XTHJGABSFCQASO-UHFFFAOYSA-N n-benzyl-4-[1-[4-(oxan-2-yloxy)phenyl]benzimidazol-2-yl]benzamide Chemical compound C=1C=C(C=2N(C3=CC=CC=C3N=2)C=2C=CC(OC3OCCCC3)=CC=2)C=CC=1C(=O)NCC1=CC=CC=C1 XTHJGABSFCQASO-UHFFFAOYSA-N 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 150000003180 prostaglandins Chemical class 0.000 description 3
- 238000000159 protein binding assay Methods 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 229940095743 selective estrogen receptor modulator Drugs 0.000 description 3
- 239000000333 selective estrogen receptor modulator Substances 0.000 description 3
- 238000010898 silica gel chromatography Methods 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 235000011152 sodium sulphate Nutrition 0.000 description 3
- 238000012289 standard assay Methods 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 238000005809 transesterification reaction Methods 0.000 description 3
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 3
- 238000001665 trituration Methods 0.000 description 3
- 206010046766 uterine cancer Diseases 0.000 description 3
- 239000003039 volatile agent Substances 0.000 description 3
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 2
- 125000004916 (C1-C6) alkylcarbonyl group Chemical group 0.000 description 2
- IMZYFKBLNPEJBM-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-(1-methylcyclopropyl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1(C)CC1 IMZYFKBLNPEJBM-UHFFFAOYSA-N 0.000 description 2
- KQMKNEUUVYXHQD-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-(1-methylpyrrol-2-yl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CN1C KQMKNEUUVYXHQD-UHFFFAOYSA-N 0.000 description 2
- MPKXKSZJUUJOIG-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-(1-propylpyrrol-2-yl)benzimidazole Chemical compound CCCN1C=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(OC)C=C1 MPKXKSZJUUJOIG-UHFFFAOYSA-N 0.000 description 2
- VIFJWLLIGSXXEW-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-(2-methylthiophen-3-yl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(C)SC=C1 VIFJWLLIGSXXEW-UHFFFAOYSA-N 0.000 description 2
- RUTBPUDUFHNJLO-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-(3-methylfuran-2-yl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(C)C=CO1 RUTBPUDUFHNJLO-UHFFFAOYSA-N 0.000 description 2
- ILAOVOOZLVGAJF-UHFFFAOYSA-N 1-methylpyrrole-2-carboxylic acid Chemical compound CN1C=CC=C1C(O)=O ILAOVOOZLVGAJF-UHFFFAOYSA-N 0.000 description 2
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 2
- CNTHHNPBADVTRY-UHFFFAOYSA-N 2,5-dimethylfuran-3-carboxylic acid Chemical compound CC1=CC(C(O)=O)=C(C)O1 CNTHHNPBADVTRY-UHFFFAOYSA-N 0.000 description 2
- YNSDBFNOAFRKKF-UHFFFAOYSA-N 2-(1-ethylpyrrol-2-yl)-1-(4-methoxyphenyl)benzimidazole Chemical compound CCN1C=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(OC)C=C1 YNSDBFNOAFRKKF-UHFFFAOYSA-N 0.000 description 2
- PYRWEZBEXFMUHH-UHFFFAOYSA-N 2-(4-methoxyphenyl)-1h-benzimidazole Chemical compound C1=CC(OC)=CC=C1C1=NC2=CC=CC=C2N1 PYRWEZBEXFMUHH-UHFFFAOYSA-N 0.000 description 2
- DVGSBIRLXBGROE-UHFFFAOYSA-N 2-(cyclopropylmethyl)-1-(4-methoxyphenyl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1CC1CC1 DVGSBIRLXBGROE-UHFFFAOYSA-N 0.000 description 2
- YWOGIJKIJZJVLQ-UHFFFAOYSA-N 2-(furan-2-yl)-1-(4-methoxyphenyl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CO1 YWOGIJKIJZJVLQ-UHFFFAOYSA-N 0.000 description 2
- NFPWZPXKHGVDQC-UHFFFAOYSA-N 2-(furan-3-yl)-1-(4-methoxyphenyl)benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=COC=C1 NFPWZPXKHGVDQC-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 2
- IKCLCGXPQILATA-UHFFFAOYSA-N 2-chlorobenzoic acid Chemical compound OC(=O)C1=CC=CC=C1Cl IKCLCGXPQILATA-UHFFFAOYSA-N 0.000 description 2
- CLHVBTZQUMNRPL-UHFFFAOYSA-N 2-cyclopropyl-n-[2-(4-methoxyanilino)phenyl]acetamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)CC1CC1 CLHVBTZQUMNRPL-UHFFFAOYSA-N 0.000 description 2
- SMNDYUVBFMFKNZ-UHFFFAOYSA-N 2-furoic acid Chemical compound OC(=O)C1=CC=CO1 SMNDYUVBFMFKNZ-UHFFFAOYSA-N 0.000 description 2
- YJZOPBSZDIBXBG-UHFFFAOYSA-N 2-methylthiophene-3-carboxylic acid Chemical compound CC=1SC=CC=1C(O)=O YJZOPBSZDIBXBG-UHFFFAOYSA-N 0.000 description 2
- CSUMPDPNHRUIIE-UHFFFAOYSA-N 2-n-(4-methoxy-2-methylphenyl)benzene-1,2-diamine;dihydrochloride Chemical compound Cl.Cl.CC1=CC(OC)=CC=C1NC1=CC=CC=C1N CSUMPDPNHRUIIE-UHFFFAOYSA-N 0.000 description 2
- DBWLJKVKKGUSRA-UHFFFAOYSA-N 3-(4-methoxyphenyl)-4-phenyl-5-(trifluoromethyl)-4H-1,2-oxazol-5-ol Chemical compound C1=CC(OC)=CC=C1C1=NOC(O)(C(F)(F)F)C1C1=CC=CC=C1 DBWLJKVKKGUSRA-UHFFFAOYSA-N 0.000 description 2
- SWKIQIDYNFSYLI-UHFFFAOYSA-N 3-ethyl-4-[1-(4-methoxyphenyl)benzimidazol-2-yl]-1,2-oxazole Chemical compound CCC1=NOC=C1C1=NC2=CC=CC=C2N1C1=CC=C(OC)C=C1 SWKIQIDYNFSYLI-UHFFFAOYSA-N 0.000 description 2
- IHCCAYCGZOLTEU-UHFFFAOYSA-N 3-furoic acid Chemical compound OC(=O)C=1C=COC=1 IHCCAYCGZOLTEU-UHFFFAOYSA-N 0.000 description 2
- WGKRMQIQXMJVFZ-UHFFFAOYSA-N 3-iodothiophene Chemical compound IC=1C=CSC=1 WGKRMQIQXMJVFZ-UHFFFAOYSA-N 0.000 description 2
- YBLSBWHFPXDRHC-UHFFFAOYSA-N 3-methyl-1,2-oxazole-4-carboxylic acid Chemical compound CC1=NOC=C1C(O)=O YBLSBWHFPXDRHC-UHFFFAOYSA-N 0.000 description 2
- VRQIXCDZWQMWRJ-UHFFFAOYSA-N 3-methyl-4-[2-(2-methylphenyl)benzimidazol-1-yl]phenol Chemical compound CC1=CC=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1C VRQIXCDZWQMWRJ-UHFFFAOYSA-N 0.000 description 2
- IFLKEBSJTZGCJG-UHFFFAOYSA-N 3-methylthiophene-2-carboxylic acid Chemical compound CC=1C=CSC=1C(O)=O IFLKEBSJTZGCJG-UHFFFAOYSA-N 0.000 description 2
- WEKMIGJSXRCCPG-UHFFFAOYSA-N 4-(1-thiophen-2-ylbenzimidazol-2-yl)phenol Chemical compound C1=CC(O)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=CS1 WEKMIGJSXRCCPG-UHFFFAOYSA-N 0.000 description 2
- LVJZZOCMWDRBGO-UHFFFAOYSA-N 4-(1-thiophen-3-ylbenzimidazol-2-yl)phenol Chemical compound C1=CC(O)=CC=C1C1=NC2=CC=CC=C2N1C1=CSC=C1 LVJZZOCMWDRBGO-UHFFFAOYSA-N 0.000 description 2
- MXMPRHRVFXJSFN-UHFFFAOYSA-N 4-(2-phenylbenzimidazol-1-yl)phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CC=C1 MXMPRHRVFXJSFN-UHFFFAOYSA-N 0.000 description 2
- WXHIHVWVXVVGAX-UHFFFAOYSA-N 4-(2-pyrrolidin-1-ylethoxy)benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1OCCN1CCCC1 WXHIHVWVXVVGAX-UHFFFAOYSA-N 0.000 description 2
- ZRXXSYOHHCRZQW-UHFFFAOYSA-N 4-[1-(3-methoxyphenyl)benzimidazol-2-yl]-n-(thiophen-2-ylmethyl)benzamide Chemical compound COC1=CC=CC(N2C3=CC=CC=C3N=C2C=2C=CC(=CC=2)C(=O)NCC=2SC=CC=2)=C1 ZRXXSYOHHCRZQW-UHFFFAOYSA-N 0.000 description 2
- FJPIZGXJYZIGDZ-UHFFFAOYSA-N 4-[1-(4-hydroxyphenyl)benzimidazol-2-yl]phenol Chemical compound C1=CC(O)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 FJPIZGXJYZIGDZ-UHFFFAOYSA-N 0.000 description 2
- ATTXLOCTCSUPQV-UHFFFAOYSA-N 4-[1-(4-methoxyphenyl)benzimidazol-2-yl]-5-methyl-1,2-oxazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(C)ON=C1 ATTXLOCTCSUPQV-UHFFFAOYSA-N 0.000 description 2
- QBKVTZIBAXQUJR-UHFFFAOYSA-N 4-[1-(4-methoxyphenyl)benzimidazol-2-yl]-n-(1-phenylethyl)benzamide Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(C(=O)NC(C)C=2C=CC=CC=2)C=C1 QBKVTZIBAXQUJR-UHFFFAOYSA-N 0.000 description 2
- CTZPLLXORSXUOF-UHFFFAOYSA-N 4-[1-(4-phenylmethoxyphenyl)benzimidazol-2-yl]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C1=NC2=CC=CC=C2N1C(C=C1)=CC=C1OCC1=CC=CC=C1 CTZPLLXORSXUOF-UHFFFAOYSA-N 0.000 description 2
- VTJAXOWQAKZSMU-UHFFFAOYSA-N 4-[1-[4-(oxan-2-yloxy)phenyl]benzimidazol-2-yl]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C1=NC2=CC=CC=C2N1C(C=C1)=CC=C1OC1OCCCC1 VTJAXOWQAKZSMU-UHFFFAOYSA-N 0.000 description 2
- SKYGCFIPCWFTBH-UHFFFAOYSA-N 4-[2-(1-propylpyrrol-2-yl)benzimidazol-1-yl]phenol Chemical compound CCCN1C=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 SKYGCFIPCWFTBH-UHFFFAOYSA-N 0.000 description 2
- VVQKKJATDFFYEJ-UHFFFAOYSA-N 4-[2-(3-methyl-1,2-oxazol-4-yl)benzimidazol-1-yl]phenol Chemical compound CC1=NOC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 VVQKKJATDFFYEJ-UHFFFAOYSA-N 0.000 description 2
- ZSUQWRBTAXCOMN-UHFFFAOYSA-N 4-[2-(cyclopenten-1-yl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CCCC1 ZSUQWRBTAXCOMN-UHFFFAOYSA-N 0.000 description 2
- SAQXIUNCUUXUBT-UHFFFAOYSA-N 4-[2-(cyclopropylmethyl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1CC1CC1 SAQXIUNCUUXUBT-UHFFFAOYSA-N 0.000 description 2
- JFMUIWOBVJVUAS-UHFFFAOYSA-N 4-methyl-1,2-thiazole-5-carboxylic acid Chemical compound CC=1C=NSC=1C(O)=O JFMUIWOBVJVUAS-UHFFFAOYSA-N 0.000 description 2
- TXOFJBFEUBZSFO-UHFFFAOYSA-N 5-[1-(4-methoxyphenyl)benzimidazol-2-yl]-4-methylthiadiazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(C)N=NS1 TXOFJBFEUBZSFO-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 206010014733 Endometrial cancer Diseases 0.000 description 2
- 206010014759 Endometrial neoplasm Diseases 0.000 description 2
- 101000882584 Homo sapiens Estrogen receptor Proteins 0.000 description 2
- 101001010910 Homo sapiens Estrogen receptor beta Proteins 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 108060001084 Luciferase Proteins 0.000 description 2
- 239000005089 Luciferase Substances 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical class [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 150000001413 amino acids Chemical group 0.000 description 2
- MXMOTZIXVICDSD-UHFFFAOYSA-N anisoyl chloride Chemical compound COC1=CC=C(C(Cl)=O)C=C1 MXMOTZIXVICDSD-UHFFFAOYSA-N 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 238000000065 atmospheric pressure chemical ionisation Methods 0.000 description 2
- UPABQMWFWCMOFV-UHFFFAOYSA-N benethamine Chemical compound C=1C=CC=CC=1CNCCC1=CC=CC=C1 UPABQMWFWCMOFV-UHFFFAOYSA-N 0.000 description 2
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical class C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 2
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Substances N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 238000012875 competitive assay Methods 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 2
- 239000007822 coupling agent Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 238000006210 cyclodehydration reaction Methods 0.000 description 2
- JBDSSBMEKXHSJF-UHFFFAOYSA-N cyclopentanecarboxylic acid Chemical compound OC(=O)C1CCCC1 JBDSSBMEKXHSJF-UHFFFAOYSA-N 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- VFFHLZJSZHWXAJ-VMPITWQZSA-N ethyl (e)-3-pyrrolidin-1-ylprop-2-enoate Chemical compound CCOC(=O)\C=C\N1CCCC1 VFFHLZJSZHWXAJ-VMPITWQZSA-N 0.000 description 2
- ZYLVLVLSCPVZEI-UHFFFAOYSA-N ethyl 3-cyclopropyl-1,2-oxazole-4-carboxylate Chemical compound CCOC(=O)C1=CON=C1C1CC1 ZYLVLVLSCPVZEI-UHFFFAOYSA-N 0.000 description 2
- 239000002024 ethyl acetate extract Substances 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- WFKAJVHLWXSISD-UHFFFAOYSA-N isobutyramide Chemical compound CC(C)C(N)=O WFKAJVHLWXSISD-UHFFFAOYSA-N 0.000 description 2
- 229940047889 isobutyramide Drugs 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- TZWYPBVYNJCMES-UHFFFAOYSA-N methyl 4-[1-[4-(oxan-2-yloxy)phenyl]benzimidazol-2-yl]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1C1=NC2=CC=CC=C2N1C(C=C1)=CC=C1OC1OCCCC1 TZWYPBVYNJCMES-UHFFFAOYSA-N 0.000 description 2
- XWNHJFASCXXDEN-UHFFFAOYSA-N methyl 4-[[2-(4-methoxyanilino)phenyl]carbamoyl]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1C(=O)NC1=CC=CC=C1NC1=CC=C(OC)C=C1 XWNHJFASCXXDEN-UHFFFAOYSA-N 0.000 description 2
- KPPQEZJIMBUYPI-UHFFFAOYSA-N methyl 5-[1-(4-hydroxyphenyl)benzimidazol-2-yl]thiophene-2-carboxylate Chemical compound S1C(C(=O)OC)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 KPPQEZJIMBUYPI-UHFFFAOYSA-N 0.000 description 2
- WEAIPKFSGFOVHW-UHFFFAOYSA-N methyl 5-[1-(4-methoxyphenyl)benzimidazol-2-yl]thiophene-2-carboxylate Chemical compound S1C(C(=O)OC)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(OC)C=C1 WEAIPKFSGFOVHW-UHFFFAOYSA-N 0.000 description 2
- 150000004702 methyl esters Chemical class 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- BDYVCYUXCNZYRW-UHFFFAOYSA-N n-(2-aminophenyl)-4-methoxybenzamide Chemical compound C1=CC(OC)=CC=C1C(=O)NC1=CC=CC=C1N BDYVCYUXCNZYRW-UHFFFAOYSA-N 0.000 description 2
- FNZIOIMZPSWIQA-UHFFFAOYSA-N n-[2-(4-methoxy-2-methylanilino)phenyl]-3,5-dimethyl-1,2-oxazole-4-carboxamide Chemical compound CC1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(C)ON=C1C FNZIOIMZPSWIQA-UHFFFAOYSA-N 0.000 description 2
- JTYDLXKFPDPGBE-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-1-methylcyclopropane-1-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1(C)CC1 JTYDLXKFPDPGBE-UHFFFAOYSA-N 0.000 description 2
- GJOLLBIHQJLUPG-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-1h-pyrrole-2-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CC=CN1 GJOLLBIHQJLUPG-UHFFFAOYSA-N 0.000 description 2
- QHBPSBHHWMQWGZ-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-2,5-dimethylfuran-3-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(C)OC(C)=C1 QHBPSBHHWMQWGZ-UHFFFAOYSA-N 0.000 description 2
- PSSKHTMCAWCVSB-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-3-methyl-1,2-oxazole-4-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CON=C1C PSSKHTMCAWCVSB-UHFFFAOYSA-N 0.000 description 2
- BWZNPJJGYYBMLG-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-3-methylfuran-2-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(C)C=CO1 BWZNPJJGYYBMLG-UHFFFAOYSA-N 0.000 description 2
- JNHRRCLGNHNTJT-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-5-methyl-1,2-oxazole-4-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(C)ON=C1 JNHRRCLGNHNTJT-UHFFFAOYSA-N 0.000 description 2
- NQPFNFPOQAEIBP-UHFFFAOYSA-N n-benzyl-4-[1-(4-hydroxyphenyl)benzimidazol-2-yl]benzamide Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(C(=O)NCC=2C=CC=CC=2)C=C1 NQPFNFPOQAEIBP-UHFFFAOYSA-N 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- 150000002825 nitriles Chemical class 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 230000000849 parathyroid Effects 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 230000035790 physiological processes and functions Effects 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 230000023603 positive regulation of transcription initiation, DNA-dependent Effects 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 238000013268 sustained release Methods 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 150000003512 tertiary amines Chemical class 0.000 description 2
- QERYCTSHXKAMIS-UHFFFAOYSA-N thiophene-2-carboxylic acid Chemical compound OC(=O)C1=CC=CS1 QERYCTSHXKAMIS-UHFFFAOYSA-N 0.000 description 2
- YNVOMSDITJMNET-UHFFFAOYSA-N thiophene-3-carboxylic acid Chemical compound OC(=O)C=1C=CSC=1 YNVOMSDITJMNET-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 210000004291 uterus Anatomy 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- VOAAEKKFGLPLLU-UHFFFAOYSA-N (4-methoxyphenyl)boronic acid Chemical compound COC1=CC=C(B(O)O)C=C1 VOAAEKKFGLPLLU-UHFFFAOYSA-N 0.000 description 1
- WLAMNBDJUVNPJU-BYPYZUCNSA-N (S)-2-methylbutyric acid Chemical compound CC[C@H](C)C(O)=O WLAMNBDJUVNPJU-BYPYZUCNSA-N 0.000 description 1
- QACMXJJLQXUOPQ-UHFFFAOYSA-N 1,2-dichloroethane;3-(ethyliminomethylideneamino)-n,n-dimethylpropan-1-amine Chemical compound ClCCCl.CCN=C=NCCCN(C)C QACMXJJLQXUOPQ-UHFFFAOYSA-N 0.000 description 1
- PCXTYKGTWQCNJI-UHFFFAOYSA-N 1,2-thiazole-4-carboxylic acid Chemical compound OC(=O)C=1C=NSC=1 PCXTYKGTWQCNJI-UHFFFAOYSA-N 0.000 description 1
- GGYXPOOZYZHPLB-UHFFFAOYSA-N 1,2-thiazole-5-carboxylic acid Chemical compound OC(=O)C1=CC=NS1 GGYXPOOZYZHPLB-UHFFFAOYSA-N 0.000 description 1
- IRAQFYMPCQPWHQ-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-[4-(trifluoromethoxy)phenyl]benzimidazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(OC(F)(F)F)C=C1 IRAQFYMPCQPWHQ-UHFFFAOYSA-N 0.000 description 1
- PLALKSRAHVYFOH-UHFFFAOYSA-N 1-(4-methoxyphenyl)-2-phenylethanone Chemical compound C1=CC(OC)=CC=C1C(=O)CC1=CC=CC=C1 PLALKSRAHVYFOH-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- ZUAQMMQOIJTUOQ-UHFFFAOYSA-N 1-[4-[1-(4-hydroxyphenyl)benzimidazol-2-yl]phenyl]ethanone Chemical compound C1=CC(C(=O)C)=CC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 ZUAQMMQOIJTUOQ-UHFFFAOYSA-N 0.000 description 1
- PWKNBLFSJAVFAB-UHFFFAOYSA-N 1-fluoro-2-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1F PWKNBLFSJAVFAB-UHFFFAOYSA-N 0.000 description 1
- DFPYXQYWILNVAU-UHFFFAOYSA-N 1-hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1.C1=CC=C2N(O)N=NC2=C1 DFPYXQYWILNVAU-UHFFFAOYSA-N 0.000 description 1
- DIZKLZKLNKQFGB-UHFFFAOYSA-N 1-methylcyclopropane-1-carboxylic acid Chemical compound OC(=O)C1(C)CC1 DIZKLZKLNKQFGB-UHFFFAOYSA-N 0.000 description 1
- JSZOAYXJRCEYSX-UHFFFAOYSA-N 1-nitropropane Chemical compound CCC[N+]([O-])=O JSZOAYXJRCEYSX-UHFFFAOYSA-N 0.000 description 1
- RQEUFEKYXDPUSK-UHFFFAOYSA-N 1-phenylethylamine Chemical compound CC(N)C1=CC=CC=C1 RQEUFEKYXDPUSK-UHFFFAOYSA-N 0.000 description 1
- APJOSWNBAWMWKX-UHFFFAOYSA-N 1-thiophen-3-ylbenzimidazole Chemical compound S1C=CC(N2C3=CC=CC=C3N=C2)=C1 APJOSWNBAWMWKX-UHFFFAOYSA-N 0.000 description 1
- XQQZRZQVBFHBHL-UHFFFAOYSA-N 12-crown-4 Chemical compound C1COCCOCCOCCO1 XQQZRZQVBFHBHL-UHFFFAOYSA-N 0.000 description 1
- VFTFKUDGYRBSAL-UHFFFAOYSA-N 15-crown-5 Chemical compound C1COCCOCCOCCOCCO1 VFTFKUDGYRBSAL-UHFFFAOYSA-N 0.000 description 1
- KAESVJOAVNADME-UHFFFAOYSA-N 1H-pyrrole Natural products C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 1
- KVVDRQDTODKIJD-UHFFFAOYSA-N 2-cyclopropylacetic acid Chemical compound OC(=O)CC1CC1 KVVDRQDTODKIJD-UHFFFAOYSA-N 0.000 description 1
- ROIMNSWDOJCBFR-UHFFFAOYSA-N 2-iodothiophene Chemical compound IC1=CC=CS1 ROIMNSWDOJCBFR-UHFFFAOYSA-N 0.000 description 1
- DWYHDSLIWMUSOO-UHFFFAOYSA-N 2-phenyl-1h-benzimidazole Chemical compound C1=CC=CC=C1C1=NC2=CC=CC=C2N1 DWYHDSLIWMUSOO-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- BXEAAHIHFFIMIE-UHFFFAOYSA-N 3-chlorothiophene-2-carboxylic acid Chemical compound OC(=O)C=1SC=CC=1Cl BXEAAHIHFFIMIE-UHFFFAOYSA-N 0.000 description 1
- QKCNADNBXWMWDN-UHFFFAOYSA-N 3-ethyl-n-[2-(4-methoxyanilino)phenyl]-5-methyl-1,2-oxazole-4-carboxamide Chemical compound CCC1=NOC(C)=C1C(=O)NC1=CC=CC=C1NC1=CC=C(OC)C=C1 QKCNADNBXWMWDN-UHFFFAOYSA-N 0.000 description 1
- BNYIQEFWGSXIKQ-UHFFFAOYSA-N 3-methylfuran-2-carboxylic acid Chemical compound CC=1C=COC=1C(O)=O BNYIQEFWGSXIKQ-UHFFFAOYSA-N 0.000 description 1
- VQWVFBNWQKPMEI-UHFFFAOYSA-N 4-(2-cyclopentylbenzimidazol-1-yl)phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1CCCC1.C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1CCCC1 VQWVFBNWQKPMEI-UHFFFAOYSA-N 0.000 description 1
- WJCAOXJRPLWDFB-UHFFFAOYSA-N 4-(2-cyclopropylbenzimidazol-1-yl)phenol;4-[2-(4-iodophenyl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1CC1.C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(I)C=C1 WJCAOXJRPLWDFB-UHFFFAOYSA-N 0.000 description 1
- RATSANVPHHXDCT-UHFFFAOYSA-N 4-(trifluoromethoxy)benzoic acid Chemical compound OC(=O)C1=CC=C(OC(F)(F)F)C=C1 RATSANVPHHXDCT-UHFFFAOYSA-N 0.000 description 1
- IKZLOBUOPOAYFI-UHFFFAOYSA-N 4-[1-(4-methoxyphenyl)benzimidazol-2-yl]-1,2-thiazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CSN=C1 IKZLOBUOPOAYFI-UHFFFAOYSA-N 0.000 description 1
- XKEBMBPQSZHGMH-UHFFFAOYSA-N 4-[1-(4-methoxyphenyl)benzimidazol-2-yl]-n-(thiophen-2-ylmethyl)benzamide Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(C(=O)NCC=2SC=CC=2)C=C1 XKEBMBPQSZHGMH-UHFFFAOYSA-N 0.000 description 1
- IBYOEUZAWMRQRH-UHFFFAOYSA-N 4-[2-(2-chlorophenyl)benzimidazol-1-yl]-3-methylphenol Chemical compound CC1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CC=C1Cl IBYOEUZAWMRQRH-UHFFFAOYSA-N 0.000 description 1
- KNZWBWRQMVDWNS-UHFFFAOYSA-N 4-[2-(2-chlorophenyl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=CC=C1Cl KNZWBWRQMVDWNS-UHFFFAOYSA-N 0.000 description 1
- CAXUTBWYPZPROM-UHFFFAOYSA-N 4-[2-(3-ethyl-1,2-oxazol-4-yl)benzimidazol-1-yl]phenol Chemical compound CCC1=NOC=C1C1=NC2=CC=CC=C2N1C1=CC=C(O)C=C1 CAXUTBWYPZPROM-UHFFFAOYSA-N 0.000 description 1
- CLHBKMMAARLOKY-UHFFFAOYSA-N 4-[2-(5-bromothiophen-2-yl)benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(Br)S1 CLHBKMMAARLOKY-UHFFFAOYSA-N 0.000 description 1
- NYLZVZBPQOBWLT-UHFFFAOYSA-N 4-[2-[4-(2-pyrrolidin-1-ylethoxy)phenyl]benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C(C=C1)=CC=C1OCCN1CCCC1 NYLZVZBPQOBWLT-UHFFFAOYSA-N 0.000 description 1
- IVKUSDQYBAMXPS-UHFFFAOYSA-N 4-[2-[4-(trifluoromethoxy)phenyl]benzimidazol-1-yl]phenol Chemical compound C1=CC(O)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(OC(F)(F)F)C=C1 IVKUSDQYBAMXPS-UHFFFAOYSA-N 0.000 description 1
- QBHDSQZASIBAAI-UHFFFAOYSA-N 4-acetylbenzoic acid Chemical compound CC(=O)C1=CC=C(C(O)=O)C=C1 QBHDSQZASIBAAI-UHFFFAOYSA-N 0.000 description 1
- ADCUEPOHPCPMCE-UHFFFAOYSA-N 4-cyanobenzoic acid Chemical compound OC(=O)C1=CC=C(C#N)C=C1 ADCUEPOHPCPMCE-UHFFFAOYSA-N 0.000 description 1
- IRQWEODKXLDORP-UHFFFAOYSA-N 4-ethenylbenzoic acid Chemical compound OC(=O)C1=CC=C(C=C)C=C1 IRQWEODKXLDORP-UHFFFAOYSA-N 0.000 description 1
- GHICCUXQJBDNRN-UHFFFAOYSA-N 4-iodobenzoic acid Chemical compound OC(=O)C1=CC=C(I)C=C1 GHICCUXQJBDNRN-UHFFFAOYSA-N 0.000 description 1
- REIDAMBAPLIATC-UHFFFAOYSA-M 4-methoxycarbonylbenzoate Chemical compound COC(=O)C1=CC=C(C([O-])=O)C=C1 REIDAMBAPLIATC-UHFFFAOYSA-M 0.000 description 1
- NHHQOYLPBUYHQU-UHFFFAOYSA-N 4-methylthiadiazole-5-carboxylic acid Chemical compound CC=1N=NSC=1C(O)=O NHHQOYLPBUYHQU-UHFFFAOYSA-N 0.000 description 1
- MAEBYWXLGMGSHB-UHFFFAOYSA-N 5-[1-(4-methoxyphenyl)benzimidazol-2-yl]-1,2-thiazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=NS1 MAEBYWXLGMGSHB-UHFFFAOYSA-N 0.000 description 1
- AECRWUOQNICJKX-UHFFFAOYSA-N 5-[1-(4-methoxyphenyl)benzimidazol-2-yl]-4-methyl-1,2-thiazole Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=C(C)C=NS1 AECRWUOQNICJKX-UHFFFAOYSA-N 0.000 description 1
- COWZPSUDTMGBAT-UHFFFAOYSA-N 5-bromothiophene-2-carboxylic acid Chemical compound OC(=O)C1=CC=C(Br)S1 COWZPSUDTMGBAT-UHFFFAOYSA-N 0.000 description 1
- VQBXUKGMJCPBMF-UHFFFAOYSA-N 5-methyl-1,2-oxazole-4-carboxylic acid Chemical compound CC=1ON=CC=1C(O)=O VQBXUKGMJCPBMF-UHFFFAOYSA-N 0.000 description 1
- WRDABNWSWOHGMS-UHFFFAOYSA-N AEBSF hydrochloride Chemical compound Cl.NCCC1=CC=C(S(F)(=O)=O)C=C1 WRDABNWSWOHGMS-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 108010039627 Aprotinin Proteins 0.000 description 1
- 206010063659 Aversion Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 208000010228 Erectile Dysfunction Diseases 0.000 description 1
- 101710163275 Estrogen receptor beta-2 Proteins 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 1
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 102000008192 Lactoglobulins Human genes 0.000 description 1
- 108010060630 Lactoglobulins Proteins 0.000 description 1
- 101710128836 Large T antigen Proteins 0.000 description 1
- GDBQQVLCIARPGH-UHFFFAOYSA-N Leupeptin Natural products CC(C)CC(NC(C)=O)C(=O)NC(CC(C)C)C(=O)NC(C=O)CCCN=C(N)N GDBQQVLCIARPGH-UHFFFAOYSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 206010030247 Oestrogen deficiency Diseases 0.000 description 1
- 102000016979 Other receptors Human genes 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 108700020797 Parathyroid Hormone-Related Proteins 0.000 description 1
- 102000043299 Parathyroid hormone-related Human genes 0.000 description 1
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 1
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical class C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 1
- 102000015433 Prostaglandin Receptors Human genes 0.000 description 1
- 108010050183 Prostaglandin Receptors Proteins 0.000 description 1
- 108091027981 Response element Proteins 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 239000005864 Sulphur Substances 0.000 description 1
- STSCVKRWJPWALQ-UHFFFAOYSA-N TRIFLUOROACETIC ACID ETHYL ESTER Chemical compound CCOC(=O)C(F)(F)F STSCVKRWJPWALQ-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 1
- PFCUZDIEKKTHCH-UHFFFAOYSA-N acetonitrile oxide Chemical compound CC#N=O PFCUZDIEKKTHCH-UHFFFAOYSA-N 0.000 description 1
- 239000003377 acid catalyst Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 230000001195 anabolic effect Effects 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 229960004405 aprotinin Drugs 0.000 description 1
- 238000010936 aqueous wash Methods 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- KXDAEFPNCMNJSK-UHFFFAOYSA-N benzene carboxamide Natural products NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 1
- RIIWUGSYXOBDMC-UHFFFAOYSA-N benzene-1,2-diamine;hydron;dichloride Chemical compound Cl.Cl.NC1=CC=CC=C1N RIIWUGSYXOBDMC-UHFFFAOYSA-N 0.000 description 1
- BYBIRLURVZSVME-UHFFFAOYSA-M benzene;copper(1+);trifluoromethanesulfonate Chemical compound [Cu+].C1=CC=CC=C1.[O-]S(=O)(=O)C(F)(F)F BYBIRLURVZSVME-UHFFFAOYSA-M 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 230000037182 bone density Effects 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 230000007177 brain activity Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 230000036996 cardiovascular health Effects 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 230000001149 cognitive effect Effects 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 150000003983 crown ethers Chemical class 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 description 1
- TXWOGHSRPAYOML-UHFFFAOYSA-N cyclobutanecarboxylic acid Chemical compound OC(=O)C1CCC1 TXWOGHSRPAYOML-UHFFFAOYSA-N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 150000001940 cyclopentanes Chemical class 0.000 description 1
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical class O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 1
- PYRZPBDTPRQYKG-UHFFFAOYSA-N cyclopentene-1-carboxylic acid Chemical compound OC(=O)C1=CCCC1 PYRZPBDTPRQYKG-UHFFFAOYSA-N 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- YMGUBTXCNDTFJI-UHFFFAOYSA-N cyclopropanecarboxylic acid Chemical compound OC(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-N 0.000 description 1
- AUQDITHEDVOTCU-UHFFFAOYSA-N cyclopropyl cyanide Chemical compound N#CC1CC1 AUQDITHEDVOTCU-UHFFFAOYSA-N 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical class C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 125000005046 dihydronaphthyl group Chemical group 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 230000009429 distress Effects 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 239000012039 electrophile Substances 0.000 description 1
- JDWLDZUUTIPZHV-UHFFFAOYSA-N ethyl 3-ethyl-5-methyl-1,2-oxazole-4-carboxylate Chemical compound CCOC(=O)C1=C(C)ON=C1CC JDWLDZUUTIPZHV-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000000265 homogenisation Methods 0.000 description 1
- 230000003054 hormonal effect Effects 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical group [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- QYRFJLLXPINATB-UHFFFAOYSA-N hydron;2,4,5,6-tetrafluorobenzene-1,3-diamine;dichloride Chemical compound Cl.Cl.NC1=C(F)C(N)=C(F)C(F)=C1F QYRFJLLXPINATB-UHFFFAOYSA-N 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 201000001881 impotence Diseases 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- GDBQQVLCIARPGH-ULQDDVLXSA-N leupeptin Chemical compound CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C=O)CCCN=C(N)N GDBQQVLCIARPGH-ULQDDVLXSA-N 0.000 description 1
- 108010052968 leupeptin Proteins 0.000 description 1
- 108020001756 ligand binding domains Proteins 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- OTCKOJUMXQWKQG-UHFFFAOYSA-L magnesium bromide Chemical compound [Mg+2].[Br-].[Br-] OTCKOJUMXQWKQG-UHFFFAOYSA-L 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 208000007106 menorrhagia Diseases 0.000 description 1
- CDGNLUSBENXDGG-UHFFFAOYSA-N meta-Cresidine Chemical compound COC1=CC=C(N)C(C)=C1 CDGNLUSBENXDGG-UHFFFAOYSA-N 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- YFKIWUQBRSMPMZ-UHFFFAOYSA-N methane;nickel Chemical compound C.[Ni] YFKIWUQBRSMPMZ-UHFFFAOYSA-N 0.000 description 1
- YJLKRLRSQOCWGR-UHFFFAOYSA-N methyl 4-[1-(4-phenylmethoxyphenyl)benzimidazol-2-yl]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1C1=NC2=CC=CC=C2N1C(C=C1)=CC=C1OCC1=CC=CC=C1 YJLKRLRSQOCWGR-UHFFFAOYSA-N 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 230000008450 motivation Effects 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- QWCLKHXWGCBJPQ-SKCUWOTOSA-N n-[(2r)-1-[(3ar)-3a-[(4-fluorophenyl)methyl]-2-methyl-3-oxo-6,7-dihydro-4h-pyrazolo[4,3-c]pyridin-5-yl]-1-oxo-3-phenylmethoxypropan-2-yl]-2-amino-2-methylpropanamide Chemical compound C([C@@]12CN(CCC1=NN(C2=O)C)C(=O)[C@@H](COCC=1C=CC=CC=1)NC(=O)C(C)(C)N)C1=CC=C(F)C=C1 QWCLKHXWGCBJPQ-SKCUWOTOSA-N 0.000 description 1
- YNTRPKQFSDOKAL-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-1,2-thiazole-4-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=CSN=C1 YNTRPKQFSDOKAL-UHFFFAOYSA-N 0.000 description 1
- YQUHFWXJWXLBFD-UHFFFAOYSA-N n-[2-(4-methoxyanilino)phenyl]-2-methylfuran-3-carboxamide Chemical compound C1=CC(OC)=CC=C1NC1=CC=CC=C1NC(=O)C1=C(C)OC=C1 YQUHFWXJWXLBFD-UHFFFAOYSA-N 0.000 description 1
- HFYZJGHQYGBNGX-UHFFFAOYSA-N n-benzhydryl-4-[1-(4-methoxyphenyl)benzimidazol-2-yl]benzamide Chemical compound C1=CC(OC)=CC=C1N1C2=CC=CC=C2N=C1C1=CC=C(C(=O)NC(C=2C=CC=CC=2)C=2C=CC=CC=2)C=C1 HFYZJGHQYGBNGX-UHFFFAOYSA-N 0.000 description 1
- PVWOIHVRPOBWPI-UHFFFAOYSA-N n-propyl iodide Chemical compound CCCI PVWOIHVRPOBWPI-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Substances [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 1
- 108091008916 nuclear estrogen receptors subtypes Proteins 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- VOXZDWNPVJITMN-WKUFJEKOSA-N oestradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-WKUFJEKOSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- BHAAPTBBJKJZER-UHFFFAOYSA-N p-anisidine Chemical compound COC1=CC=C(N)C=C1 BHAAPTBBJKJZER-UHFFFAOYSA-N 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- DGTNSSLYPYDJGL-UHFFFAOYSA-N phenyl isocyanate Chemical compound O=C=NC1=CC=CC=C1 DGTNSSLYPYDJGL-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 230000001817 pituitary effect Effects 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 208000001685 postmenopausal osteoporosis Diseases 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000004237 preparative chromatography Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 239000000583 progesterone congener Substances 0.000 description 1
- ISYORFGKSZLPNW-UHFFFAOYSA-N propan-2-ylazanium;chloride Chemical compound [Cl-].CC(C)[NH3+] ISYORFGKSZLPNW-UHFFFAOYSA-N 0.000 description 1
- RZWZRACFZGVKFM-UHFFFAOYSA-N propanoyl chloride Chemical compound CCC(Cl)=O RZWZRACFZGVKFM-UHFFFAOYSA-N 0.000 description 1
- FVSKHRXBFJPNKK-UHFFFAOYSA-N propionitrile Chemical compound CCC#N FVSKHRXBFJPNKK-UHFFFAOYSA-N 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003586 protic polar solvent Substances 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- DOYOPBSXEIZLRE-UHFFFAOYSA-N pyrrole-3-carboxylic acid Natural products OC(=O)C=1C=CNC=1 DOYOPBSXEIZLRE-UHFFFAOYSA-N 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000001718 repressive effect Effects 0.000 description 1
- 210000004994 reproductive system Anatomy 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 229920002477 rna polymer Polymers 0.000 description 1
- 239000004576 sand Substances 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 238000006748 scratching Methods 0.000 description 1
- 230000002393 scratching effect Effects 0.000 description 1
- 230000035946 sexual desire Effects 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000003270 steroid hormone Substances 0.000 description 1
- 102000005969 steroid hormone receptors Human genes 0.000 description 1
- 108020003113 steroid hormone receptors Proteins 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000001117 sulphuric acid Substances 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- OGBMKVWORPGQRR-UMXFMPSGSA-N teriparatide Chemical compound C([C@H](NC(=O)[C@H](CCSC)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@@H](N)CO)C(C)C)[C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CNC=N1 OGBMKVWORPGQRR-UMXFMPSGSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- FKKJJPMGAWGYPN-UHFFFAOYSA-N thiophen-2-ylmethanamine Chemical compound NCC1=CC=CS1 FKKJJPMGAWGYPN-UHFFFAOYSA-N 0.000 description 1
- YCGAZNXXGKTASZ-UHFFFAOYSA-N thiophene-2,5-dicarboxylic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)S1 YCGAZNXXGKTASZ-UHFFFAOYSA-N 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 238000003146 transient transfection Methods 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 239000012130 whole-cell lysate Substances 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
- 150000003738 xylenes Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/433—Thidiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/27—Growth hormone [GH], i.e. somatotropin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/08—Antiseborrheics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
- A61P5/30—Oestrogens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
- A61P5/32—Antioestrogens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
- C07D235/08—Radicals containing only hydrogen and carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/18—Benzimidazoles; Hydrogenated benzimidazoles with aryl radicals directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- This invention relates to compounds, in particular benzimidazoles, that are useful as estrogen agonists/antagonists and pharmaceutical uses thereof.
- the present invention also relates to benzimidazoles that are selective for the ER ⁇ receptor and pharmaceutical uses thereof.
- the estrogen receptor plays a central role in regulating a diverse array of normal physiological processes involved in the development and function of the reproductive system, as well as many other aspects of health, such as bone density, cardiovascular health, etc.
- estradifen display mixed agonist/antagonist action; that is they are either estrogen agonists, estrogen antagonists or a partial estrogen antagonist when binding to the estrogen receptors of different tissues.
- Estrogen is the agent of choice in preventing osteoporosis or post menopausal bone loss in women, it is the only treatment that unequivocally reduces fractures.
- estrogen stimulates the uterus and is associated with an increased risk of endometrial cancer. Although the risk of endometrial cancer is thought to be reduced by concurrent use of a progestin, there remains concern about possible increased risk of breast cancer with the use of an estrogen. It would be desirable to be able to produce ligands which are recognizable by and able to bind to the estrogen receptor. Further, it would be desirable to produce ligands having estrogen-like function, but which are devoid of unwanted side-effects of estrogenic compounds.
- osteoporosis is greatly ameliorated by the use of fully active estrogens; however, due to the recognized risk of uterine cancer in patients treated chronically with active estrogens, it is not clinically advisable to treat osteoporosis with fully active estrogens for prolonged periods.
- estrogen binds to a single estrogen receptor (ER) in cells, causing conformationai changes that result in release from ' ' heat shock proteins and binding of the receptor as a dimer to the so-called estrogen response element in the promoter region of a variety of genes.
- ER ⁇ a second estrogen receptor
- ER ⁇ a second estrogen receptor
- ER ⁇ has been identified and cloned (Katzenellenbogen and Korach Endocrinology 138, 861-2 (1997).
- ER ⁇ and the classical ER, renamed ER ⁇ , have significantly different amino acid sequences in the ligand binding domain and carboxy-terminal transactivation domains ( -56% amino acid identity) and only 20% homology in their amino-terminal transactivation domain. This suggests that some ligands may have higher affinity to one receptor over the other. Further, ligand-dependent conformationai changes of the two receptors, and interaction with co-factors, will result in very different biological actions of a single ligand.
- a ligand that acts as an agonist on ER ⁇ may very well serve as an antagonist on ER ⁇ .
- An example of such behavior has been described by Paech et al. (Science 277, 1508-1510, 1997).
- organs in which there is a high proportion of ER ⁇ receptors include the uterus and the hypothalmus.
- ER ⁇ is highly expressed in large amounts in ovaries and bone.
- ER-selective modulators would possess significant clinical utility. Further, ER-selective modulators that have the capacity to selectively bind or activate the ER subtypes, ER ⁇ and ER ⁇ , would be useful in elucidating the biology of the two receptors and will assist in the development of estrogen pharmaceuticals with improved tissue selectivity.
- the invention relates to a compound of formula (I):
- R 1 and R 2 are each independently selected from the group consisting of (C C 6 )alkyl; phenyl; (C 2 -C 6 )heteroaryl; (C 3 -C 8 )cycloalkyl; and (C 4 -C 8 )cycloalkenyl ;
- R 12 and R 13 are each independently selected from the group consisting of hydrogen; (d - C 7 )alkyl; (C 3 -C 8 )cycloalkyl; (C 4 -C 8 )cycloalkenyl;(C 6 -C 10 ) aryl; (C 2 - C 10 )alkenyl, (C 2 -C 10 )alkynyl; (C 2 -C 4 )heteroaryl; (C C 6 )alkylaryl; (C C 6 ) alkyl(C 2 - C 6 )heteroaryl; (C 2 -C 6 )alkoxyaryl ; (C 2 -C 6 ) alkoxy(C 2 -C 6 )heteroaryl; or R 12 and R 13 taken together form a three to eight membered heterocyclic ring having 1 to 3 heteroatoms; n is from 0 to 5; and x is 1 or 2;
- R 1 and R 2 are each independently a group of the formula:
- R 7 , R 8 , R 10 and R 1 are each independently hydrogen; hydroxy; (d- C 6 ) alkyl; (C C 6 )alkoxy; or halogen; R 9 is hydroxy; (d-C 6 ) alkoxy; (C C 6 )alkoxycarbonyloxy; (C C 6 )alkylcarbonyloxy; (C 3 -C 8 )cycloalkoxy; (C 4 -C 8 )cycloalkenyloxy; or (C 6 -C 12 ) aryloxy; and
- R 3 , R 4 , R 5 and R 6 are each independently hydrogen, hydroxy; (d-C 6 )alkyl;
- alkyl refers to straight or branched, monovalent, saturated aliphatic chains having the number of carbon atoms designated and includes, but is not limited to methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl, pentyl, isopentyl, and hexyl.
- alkenyl refers to straight or branched chain hydrocarbon groups of 2 to 10 carbon atoms having at least one double bond.
- alkynyl refers to straight of branched chain hydrocarbon groups of 2 to 10 carbon atoms having at least one triple bond.
- aryl refers to monocylic and polycyclic aromatic groups, or fused ring systems having at least one aromatic ring, having from 5 to 14 backbone atoms.
- aryl groups include, without limitation, phenyl, naphthyl, dihydronaphthyl, > tetrahydronapthyl, and the like.
- Cycloalkyl means a cyclic hydrocarbon. Examples of cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl. Preferred cycloalkyl groups are (C 3 -C 8 )cycloalkyl.
- cycloalkyl group it is also possible for the cycloalkyl group to have one or more double bonds, but is not aromatic.
- Cycloalkyl having at least one double bond are herein referred to as "cycloalkenyF'groups.
- Examples of cycloalkyl groups having at least one double bond include cyclopentenyl, cyclohexenyl, cyclohexadienyl, cyclobutadienyl, and the like.
- Heteroaryl means an aryl ring containing one or more heteroatoms. If the heteroaryl group contains more than one heteroatom, the heteroatom may be the same or different.
- heteroaryl groups include pyridyl, pyrimidinyl, , imidazolyl, thienyl, furyl, pyrazinyl, pyrrolyl, pyranyl, isobenzofuranyl, chromenyl, xanthenyl, indolyl, isoindolyl, indolizinyl, triazolyl, pyridazinyl, indazolyl, purinyl, quinolizinyl, isoquinolyl, quinolyl, phthalazinyl, naphthyridinyl, quinoxalinyl, isothiazolyl, benzo[b]thienyl, isooxazolyl, isothiazolyl and thiodiazolyl.
- heteroatoms include pyr
- substituted means that a hydrogen atom on a molecule has been replaced with a different atom or molecule.
- the atom or molecule replacing the atom is denoted as a "substituent.”
- substituted specifically envisions and allows for substitutions that are common in the art. However, it is generally understood by those skilled in the art that the substituents should be selected so as to not adversely affect the pharmacological characteristics or adversely interfere with the use of the medicament.
- Suitable substituents include halogen; (C C 6 )alkyl; (C 3 -C 8 )cycloalkyl; (C 4 -C 8 )cycloalkenyl; (d-C 6 )alkoxy; hydroxy; R 12 CO 2 , R 12 R 13 NCO, R 12 R 13 N; (C C 6 ) alkylcarbonyl, CHO, cyano, thio; (C C 6 )alkylthio; (C ⁇ -C 6 )alkylsulfonyl; (C
- heterocycloalkyl cycloalkylalkyl
- heteroarylalkyl the term defines with more specificity at least one of the groups that a substituted alkyl will contain. In other words, in these instances the specifically named groups are bonded directly through a substituted or unsubstituted alkyl chain, as defined.
- An "estrogen agonist/antagonist” is a compound that acts as an agonist at some receptors and an antagonist at other receptor. Estrogen agonists/antagonists are also known as selective estrogen receptor modulators (SERMs).
- prodrug refers to compounds that are drug precursors which, following administration, release the drug in vivo via some chemical or physiological process (e.g. a prodrug on being brought to the physiological pH or through enzyme action is converted to the desired drug form).
- Estrogen Receptor refers to ER ⁇ and/or the ER ⁇ .
- Estrogen Receptor Modulators are compounds that bind to the ER ⁇ and/or the ER ⁇ receptors and function as estrogen agonists/estrogen antagonists.
- An "ER ⁇ selective estrogen receptor modulator” is a compound that selectively binds to the ER ⁇ receptor.
- selective it is meant that the compound exhibits at least 5 times the binding affinity for the ER ⁇ than the ER ⁇ receptor as indicated by IC 50 in a competitive binding assay.
- more selective it is meant that the compound exhibits at least 50 times the binding affinity for the ER ⁇ than the ER ⁇ receptor as indicated by IC 50 in a competitive binding assay.
- terapéuticaally antagonizing or agonizing as used in the present specification, it is meant that the compound is selective or more selective for the ER ⁇ receptor and exhibits agonist and/or antagonist activity.
- therapeutically effective amount means an amount of a compound of the present invention that (i) treats or prevents the particular disease, condition, or disorder, (ii) attenuates, ameliorates, or eliminates one or more symptoms of the particular disease, condition, or disorder, or (iii) prevents or delays the onset of one or more symptoms of the particular disease, condition, or disorder described herein.
- phrases "pharmaceutically acceptable” indicates that the substance or composition must be compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the mammal being treated therewith.
- pharmaceutically-acceptable salt refers to nontoxic anionic salts containing anions such as (but not limited to) chloride, bromide, iodide, sulfate, bisulfate, phosphate, acetate, maleate, fumarate, oxalate, lactate, tartrate, citrate, gluconate, methanesulfonate and 4-toluene-sulfonate. Where more than one basic moiety exists the expression includes multiple salts (e.g., di-salt).
- the expression also refers to nontoxic cationic salts such as (but not limited to) sodium, potassium, calcium, magnesium, ammonium or protonated benzathine (N,N'- dibenzylethylenediamine), choline, ethanolamine, diethanolamine, ethylenediamine, meglamine (N-methyl-glucamine), benethamine (N-benzylphenethylamine), piperazine or tromethamine (2-amino-2-hydroxymethyl-1 ,3-propanediol).
- nontoxic cationic salts such as (but not limited to) sodium, potassium, calcium, magnesium, ammonium or protonated benzathine (N,N'- dibenzylethylenediamine), choline, ethanolamine, diethanolamine, ethylenediamine, meglamine (N-methyl-glucamine), benethamine (N-benzylphenethylamine), piperazine or tromethamine (2-amino-2-hydroxymethyl-1 ,
- Hypoactive sexual desire disorder is a disorder in which sexual fantasies and desire for sexual activity are persistently or recurrently diminished or absent, causing marked distress or interpersonal difficulties.
- Hypoactive sexual desire disorder may be lifelong or acquired, generalized (global) or situational (partner-specific).
- Sexual desire is a complex psychosomatic process based on brain activity (the "generator” or “motor” running in a rheostatic cyclic fashion), a poorly defined hormonal milieu, and cognitive scripting that includes sexual aspiration and motivation. Desynchronization of these components results in hypoactive sexual desire disorder.
- Sexual anhedonia (decreased or absent pleasure in sexual activity) is not an official diagnosis. It is almost always classified under hypoactive sexual desire disorder, because loss of pleasure almost always results in loss of desire (although loss of desire may occur first). The cause is likely to be depression or.drugs if anhedonia is acquired and global (with all partners in all situations); interpersonal factors if anhedonia is confined to one partner or one situation; or repressive Confors (eg. guilt, shame) due to family dysfunction or childhood trauma if anhedonia is lifelong. Sexual aversion is the probable diagnosis in lifelong cases. Dyspareunia is painful coitus or attempted coitus. Dyspareunia is usually introital but may also occur before, during, or after intercourse. Causes include menopausal involution with dryness and thinning of the mucosa. Pain during or after coitus is the chief complaint.
- the invention relates to a compound of formula (I), wherein R 1 is phenyl or (C 2 -C 6 ) heteroaryl.
- the (C 2 -C 6 ) heteroaryl is thienyl; furyl; pyrrolyl; isoxazolyl; isothiazolyl or thiodiazolyl.
- the invention relates to a compound according to formula (I), wherein R 1 is phenyl optionally substituted by R 12 CO 2 or R 12 R 13 NC(O).
- the invention relates to a compound according to formula(l), wherein R 2 is a group of formula (II)
- the invention relates to a compound according to formula (I) wherein R 2 is a group of formula (ll),wherein R 7 , R 8 , R 10 and R 11 are hydrogen and R 9 is hydroxy or (d-C 6 )alkoxy.
- the invention relates to a compound according to formula (I), wherein R 3 , R 4 , R 5 and R 6 are hydrogen.
- the invention relates to a compound according to formula (I) wherein R 1 is phenyl or (C 2 -C 6 )heteroaryl; R 2 is a group of formula (II); and R 3 , R 4 , R 5 and R 6 are hydrogen.
- the (C 2 -C 6 )heteroaryl is thienyl; furyl; pyrrolyl; isoxazolyl; isothiazoyl or thiodiazolyl, R 7 , R 8 , R 10 and R 11 are hydrogen; and R 9 is hydroxy or (C 1 -C 6 )alkoxy.
- the invention relates to a compound of Formula (I), wherein R is phenyl optionally substituted by R 2 CO 2 or R 12 R 13 NC(O); R 2 is a group of formula (II); and R 3 , R 4 , R 5 and R 6 are hydrogen.
- the invention relates to a compound of formula (I) or the pharmaceutically accepted salts thereof, wherein the compound of formula (I) is selected from the group consisting of:
- the invention in a second aspect, relates to a pharmaceutical composition for antagonizing or agonizing an estrogen receptor in a mammal comprising an estrogen receptor antagonizing or agonizing effective amount of a compound of formula (I) according to claim 1 , or a pharmaceutically accepted salt thereof, and a pharmaceutically acceptable carrier.
- the invention in a third aspect, relates to a pharmaceutical composition for selectively antagonizing or agonizing an ER ⁇ estrogen receptor in a mammal comprising an ER ⁇ estrogen receptor antagonizing or agonizing effective amount of a compound of formula (I) or the pharmaceutically acceptable salts thereof and a pharmaceutically acceptable carrier.
- the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising an agent selected from the group consisting of an anabolic agent; a growth hormone; a growth hormone secretagogue; a prostaglandin agonist/antagonist; a parathyroid hormone; sodium fluoride; and a mixture thereof; the pharmaceutical composition further comprising a compound of formula (I)
- the invention in a fifth aspect, relates to a method of treating a condition which presents with low bone mass in a mammal comprising administering to the mammal a compound of formula (I) , a prodrug thereof or a pharmaceutically acceptable salt, or a stereoisomeric mixture of said compound, salt or prodrug.
- the condition is osteoporosis.
- the invention in a sixth aspect, relates to a kit comprising: a) an amount of a compound of Formula (I) as defined in claim 1 ; b) an amount of a second compound comprising an anabolic agent; a growth hormone; a growth hormone secretagogue; a prostaglandin agonist/antagonist; a parathyroid hormone; sodium fluoride; or a mixture thereof; and c) a container.
- the invention in a seventh aspect, relates to a method of treating a disease mediated by the estrogen receptor in a mammal , comprising administering to the mammal a therapeutically effective amount of a compound of formula (l) according to claim 1 in a pharmaceutically effective carrier.
- the disease is selected from the group consisting of perimenopausal or postmenopausal syndrome, osteoporosis, atrophy of skin or vagina, elevated serum cholesterol levels, cardiovascular disease, Alzheimer's disease, estrogen dependent cancers, including breast or uterine cancer, a prostatic disease, benign prostatic hyperplasia, prostate cancer, obesity, endometriosis, bone loss, uterine fibrosis, aortal smooth muscle cell proliferation, acne, hirsutism, dysfunctional uterine bleeding, dysmenorrehea, male infertility, male erectile dysfunction (MED), psychological and behavioral symptoms during menstruation, ulcerative mucositis, uterine fibroid disease, restenosis, atherosclerosis, musculoaponeurotic fibromatosis, alopecia, autoimmune disease, cartilage degeneration, delayed puberty, demyelinating disease, dysmyelinating disease, hypoglycemia, lupus erythemato
- a prostatic disease
- the present invention relates to compounds that have activity as estrogen receptor modulators, as well as pharmaceutical compositions containing one or more of such compounds and methods of use related to the same.
- the compounds of this invention have utility in the treatment of a wide range of estrogen-related conditions.
- the compounds of this invention may be administered as a therapeutic and/or prophylactic agent.
- Certain compounds within the class of estrogen receptor modulators as described herein were found to be selective for the ER ⁇ receptor, and certain compounds within the class of ER ⁇ selective compounds were found to be more selective for the ER ⁇ receptor.
- the compounds of the invention have an IC 50 with respect to ER ⁇ and/or ER ⁇ of no more than 500 nanomolar.
- some compounds of formula (I) were selective for the ER ⁇ receptor.
- an assay may be performed as described in the present specification in the section entitled "assay for estrogen receptor binding activity".
- Preferred compounds compounds of formula (I) that are selective for the ER ⁇ receptor are compounds of formula (I) in which R-i represents a five membered heteroaryl ring having up to 3 heteroatoms independently selected from N, O and S.
- Suitable five membered heteroaryl rings include, but are not limited to, thienyl ; furyl; pyrrolyl; isoxazolyl; and thiodazolyl groups; preferably thienyl, furyl, pyrrolyl and isoxazolyl groups .
- the five membered heteroaryl ring selected as R may optionally be substituted.
- the substituted R ⁇ groups include, but are not limited to, 1- methyl-1 H-pyrrol-2-yl; 3,5-dimethyl-isoxazol-4-yl; 3-bromo-thiopen-2-yl; and 1-ethyl- 1 H-pyrrol-2-yl.
- R ⁇ represents a five membered heteroaryl ring having up to 3 heteroatoms independently selected from N, O and S; and R 9 represents OH.
- Compounds of formula (I) that are selective for the ER ⁇ receptor include, but are not limited to: 4-(2-thiophen-3-yl-benzoimidazol-1-yl-phenol;
- the compounds of this invention may be administered to mammals (including humans) orally or parenterally in the conventional form of preparations, such as capsules, microcapsules, tablets, granules, powder, troches, pills, suppositories, injections, suspensions and syrups.
- Suitable formulations may be prepared by methods commonly employed using conventional organic or inorganic additives, such as excipients, binders, disintegrators, lubricants, flavoring agents, stabilizers, , dispersing agents, diluents, preservatives, and a base wax.
- the amount of the active ingredient in the preparation may be at a level that will exhibit the desired therapeutic effect.
- the active ingredient may be usually administered once to four times a day with a unit dosage of 0.1 mg to 50 mg in human patients, but the above dosage may be properly varied depending on the age, body weight and medical condition of the patient and the type of administration.
- the following compounds were found to have activity as estrogen agonists/antagonists: 4-(5-phenyl-2- trifluoromethyl-3H-imidazol-4-yl)-phenol; 4-[5-[(4-hydroxy-phenyl)2-trifluoromethyl-3- H-imidazol-4-yl]-phenoI; 4[5-[(4-methoxy-phenyI)-2-trifluoromethyl-1 H-imidazol-4-yl]- phenol; and 4-(4-phenyl-5-trifluoromethyl-isoxazol-3-yl)-phenol.
- the 4-(5-phenyl-2- trifluoromethyI-3H-imidazol-4-yl)-phenol; 4-[5[(4-hydroxy-phenyl)2-trifluoromethyl-3-H- imidazol-4-yl]-phenol; 4[5-[(4-methoxy-phenyl)-2-trifluoromethyl-1 H-imidazol-4-yl]- phenol compounds may be prepared by the methods described in "Preparation and Anti-inflammatory Activity of Some Nonacidic Trisubstituted Imidazoles", Journal of Medicinal Chemistry. 1974, Vol. 17, No.11.
- the 4-(4-phenyl-5-trifluoromethyl- isoxazol-3-yl)-phenol compound may be prepared by the methods described in Example 62.
- the compounds of the present invention may also be used in combination with other agents to provide sustained therapeutic and prophylactic effects.
- the compounds of the present invention may be used with other agents including, but not limited to, an anabolic agent; a growth hormone; a growth hormone secretagogue; a prostaglandin agonist/antagonist; a parathyroid hormone; sodium fluoride; or a mixture thereof. Any prostaglandin agonist/antagonist may be used in combination with the compounds of this invention.
- prostaglandin agonist/antagonist refers to compounds which bind to prostaglandin receptors (e.g., An S. et al., Cloning and Expression of the EP 2 Subtype of Human Receptors for Prostaglandin E 2 ,
- U.S. patent 4,621 ,100 the disclosure of which is incorporated herein by reference, discloses substituted cyclopentanes useful for bone formation activity
- U.S. patent 5,216,183 the disclosure of which is incorporated herein by reference, discloses cyclopentanones useful for bone formation activity.
- sodium fluoride may be used in combination with the compounds of this invention.
- the term "sodium fluoride” refers to sodium fluoride in all its forms (e.g., slow release sodium fluoride, sustained release sodium fluoride). Sustained release sodium fluoride is disclosed in U.S. patent 4,904,478, the disclosure of which is incorporated herein by reference.
- the activity of sodium fluoride is readily determined by those skilled in the art of biological protocols (e.g., see Eriksen E.F. et al., Bone Histomorphometrv. Raven Press, New York, 1994, pages 1-74; Grier S.J. et.
- parathyroid hormone refers to parathyroid hormone, fragments or metabolites thereof and structural analogs thereof which can stimulate bone formation and increase bone mass.
- parathyroid hormone related peptides and active fragments and analogs of parathyroid related peptides See PCT publication no. WO 94/01460.
- Such bone anabolic functional activity is readily determined by those skilled in the art of standard assays (e.g., see Eriksen E.F. et al., Bone Histomorphometry, Raven Press, New York, 1994, pages 1- 74; Grier S.J. et.
- growth hormone secretagogue refers to a compound which stimulates the release of growth hormone or mimics the action of growth hormone (e.g., increases bone formation leading to increased bone mass). Such actions are readily determined by those skilled in the art of standard assays well known to those of skill in the art. A variety of these compounds are disclosed in the following published PCT patent applications: WO 95/14666; WO 95/13069; WO 94/19367; WO 94/13696; and WO 95/34311.
- growth hormones or growth hormone secretagogues will be known to those skilled in the art.
- a preferred growth hormone secretagogue is N-[1 (R)-[1 ,2- Dihydro-1-methanesulfonylspiro[3H-indole-3,4'-piperidin]-1'-yI)carbonyl]-2- (phenylmethyloxy)ethyl]-2-amino-2-methylpropanamide:MK-677.
- growth hormone secretagogues include 2-amino-N-(2-(3a-(R)-benzyl-2-methyl-3-oxo-2 ) 3,3a,4,6,7-hexahydro- pyrazolo-[4,3-c]pyridin-5-yl)-1 -(R)-benzyloxymethyl-2-oxo-ethyl)-isobutyramide or its L-tartaric acid salt; 2-amino-N-(1-(R)-benzyloxymethyl-2-(3a-(R)-(4-fluoro-benzyl)-2-methyl-3- oxo-2,3,3a,4,6,7-hexahydro-pyrazolo[4,3-c]pyridin-5-yl)-2-oxo-ethyl)isobutyramide;
- this invention relates to a kit comprising: a. an amount of a compound of formula (I) a prodrug thereof or a pharmaceutically acceptable salt of said compound or said prodrug, or a steroisomer or diastereomeric mixture of a compound of formula (I), prodrug or salt and a pharmaceutically acceptable carrier or diluent in a first unit dosage form; b. an amount of a second compound an anabolic agent; a growth hormone; a growth hormone secretagogue; a prostaglandin agonist/antagonist; a parathyroid hormone; sodium fluoride; or a mixture thereof; and c. a container.
- Suitable second compounds for use in the kit as defined above are described in the specification above.
- prodrugs and pharmaceutically acceptable salts may be formed from the compounds used as the second compounds in the combinations and kits of the invention. All of such prodrugs and pharmaceutically acceptable salts so formed are within the scope of this invention.
- Preparations 1 , 2 and 3 describe the preparation of materials that are used to prepare compounds according to the present invention.
- carboxylic acids which are not commercially available may be synthesized.
- the variables X,Y, and Z are each independently nitrogen, oxygen or sulfur.
- the carboxylic acid compound of Formula (XV) is treated with an excess amount of a strong base, such as lithium diisopropylamide (LDA) or butyl lithium, in an inert solvent, such as tetrahydrofuran (THF), dimethyl ether (DME), dioxane, or a mixture thereof at a temperature of from about -78°C to about 100° C, preferably about room temperature, for a period between about 1 hour to about 24 hours, preferably about 12 hours and is then alkylated with an alkyl halide, at a temperature of from about - 78°C to about 100 °C, preferably about room temperature, for a time period of between about 1 hour to about 24 hours, preferably about 12 hours, to give carboxylic acid of Formula (X)
- a strong base such as lithium diisopropylamide
- the 3,5 disubstituted carboxy isoxazoles may be prepared by treatment of a vinylogous carbamate with a nitrile oxide compound such as acetonitrile oxide, propionitrile or cyclopropane carbonitrile to give esters (XVIII).
- a nitrile oxide compound such as acetonitrile oxide, propionitrile or cyclopropane carbonitrile
- esters (XVIII) esters
- Nitrile oxides may be synthesized by methods known to those of skill in the art and as described in Journal of the American Chemical Society 1960, 82, 5339-42; and Journal of Medicinal Chemistry. 1976, 19, 562-565, which are incorporated by reference in their entirety.
- the ester may be converted to the corresponding carboxylic acid compound of Formula (XIX) by treatment with lithium hydroxide
- LiOH lithium hydroxide
- NaOH sodium hydroxide
- KOH potassium hydroxide
- reaction 1 the nitrobenzene compound of Formula (I) wherein X is a halogen (including chlorine, fluorine, or bromine, preferably fluorine), is reacted with an amine compound having the Formula NH 2 R 2 to produce a nitroaniline compound of Formula (II).
- R 2 is a phenyl substituent
- the amine could be chosen to be an aniline compound.
- the reaction is conducted in the presence of a base, such as potassium carbonate, potassium tert-butoxide, powdered sodium hydroxide, or powdered potassium' hydroxide to give the corresponding nitroaniline compound of Formula (II).
- the reaction may be conducted at a temperature between about room temperature to about 200°C, preferably at' about 160°C, for a time period from between about 2 to 24 hours, preferably about 12 hours.
- the reaction may be conducted neat or in a solvent. Suitable solvents include dimethyl sulphoxide (DMSO), dimethylformamide (DMF) or a mixture thereof.
- reaction 2 of preparation 3 the nitroaniline compound of Formula (II) is reduced to the amine functionality upon treatment with hydrogen gas in the presence of a metal such as palladium, platinum or nickel to give aniline compounds of Formula (III).
- a metal such as palladium, platinum or nickel
- Either pure metal or metal on carbon, such as palladium on carbon, nickel on carbon, or platinum on carbon may be used.
- the reaction is conducted at a temperature between about 0°C to about 100°C, preferably room temperature, for a time period between about 1 to about 24 hours, preferably about 12 hours.
- the aniline compound of Formula (III), prepared according to the method of Preparation 3, is coupled with carboxylic acid compounds having an appropriate R 1 substituent in the presence of an appropriate coupling agent, such as 1-propanephosphonic acid cyclic anhydride (PPAA), 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), or a mixture thereof, and a catalytic amount of an additive such as 1-hydroxybenzotriazole (HOBt) or 4- dimethylaminopyridine (DMAP) at a temperature between about 0°C to about 60 °C, preferably about room temperature, for a time period between about 1 to about 36 hours, preferably about 12 hours.
- an appropriate coupling agent such as 1-propanephosphonic acid cyclic anhydride (PPAA), 1-(3- dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), or a mixture thereof
- PPAA 1-propanephosphonic
- Carboxylic acids may be obtained commercially or may be prepared analogously according to the methods described in Preparation 1.
- the aniline compounds of Formula (III) may be treated with an acid chloride compound or an acid anhydride compound of the corresponding carboxylic compounds having the appropriate R 1 substituent, the reaction with the acid chloride or acid anhydride being conducted in the presence of a tertiary amine base such as triethylamine or 4-dimethylaminopyridine (DMAP) to give amide compounds of Formula (IV).
- Suitable acid chloride and acid anhydrides are commercially available or can be prepared from corresponding carboxylic acids by procedures analogous to those described in reference to Preparation 1. Any unreacted aniline compounds of Formula (III) from the aforementioned reactions may be optionally removed by treatment with a scavenger reagent, such as polymer supported isocyanate.
- the amide compound of Formula (IV) is cyclodehydrated upon treatment with an acid, such as acetic acid or hydrochloric acid, at temperature from about room temperature to about 100°C, preferably at about 75°C, to give the benzimidazole compounds (V).
- an acid such as acetic acid or hydrochloric acid
- R 1 and/or R 2 have a hydroxyl substituent; or if R 3 , R 4 , R 5 or R 6 are hydroxyl, it is preferable to protect the hydroxyl substituents through the use of protecting groups for all hydroxyls. Protection may be effected by treatment of the compound containing the hydroxyl substituent with a strong base such as sodium hydride (NaH), sodium hexamethyldisilazide (NaHMDS) or potassium hexamethyldisilazide (KHMDS) and reaction with an electrophile such as an alkyl halide, such as methyl iodide or benzyl bromide.
- a strong base such as sodium hydride (NaH), sodium hexamethyldisilazide (NaHMDS) or potassium hexamethyldisilazide (KHMDS)
- an electrophile such as an alkyl halide, such as methyl iodide or benzyl bromid
- the reaction may take place in an inert solvent, such as diethyl ether, dimethylformamide (DMF), tetrahydrofuran (THF), toluene or a mixture thereof, at a temperature of from about 0°C to about 100 °C, preferably at room temperature.
- an inert solvent such as diethyl ether, dimethylformamide (DMF), tetrahydrofuran (THF), toluene or a mixture thereof
- a metal catalyst such as platinum, nickel or palladium, preferably palladium
- an inert solvent such as tetrahydrofuran (THF), ethanol (EtOH) or methanol (MeOH), preferably EtOH at a temperature of room temperature to 100 °C, preferably at room temperature.
- Methyl ether protecting groups can be removed by treatment with boron tribromide in an inert solvent such as methylene chloride or 1 ,2 dichloroethane, preferably methylene chloride at a temperature of between about -78° C to reflux, preferably at about 0 °C.
- Preferred protecting groups are methyl and benzyl ethers. Tetrahydropyranyl (THP) protecting groups may also be used.
- the THP protecting group may be introduced using dihydropyran with a suitable acid catalyst, such as sulphuric acid, para-toluene sulfonic acid (TsOH) or pyridinium para-toluene sulphonate (PPTS), in an inert solvent, such as methylene chloride, THF or 1 ,2 dichlorethane, preferably methylene chloride.
- a suitable acid catalyst such as sulphuric acid, para-toluene sulfonic acid (TsOH) or pyridinium para-toluene sulphonate (PPTS)
- an inert solvent such as methylene chloride, THF or 1 ,2 dichlorethane, preferably methylene chloride.
- the reaction can be run at a temperature of from about 0 to about 85 °C, preferably at room temperature.
- the THP group may be removed by treatment with and acid such as acetic acid, trifluoroacetic acid (TFA), hydrochloric acid (HCI), para- toluene sulfonic acid (TsOH), PPTS or magnesium bromide (MgBr 2 ) in the presence of a protic solvent such as trifluouroacetic acid, water, methanol or ethanol.
- acid such as acetic acid, trifluoroacetic acid (TFA), hydrochloric acid (HCI), para- toluene sulfonic acid (TsOH), PPTS or magnesium bromide (MgBr 2 )
- a protic solvent such as trifluouroacetic acid, water, methanol or ethanol.
- Triethylsilane may optionally be added to the reaction.
- the pyrrolyl compound of Formula (VII) is prepared from the pyrrole compound of Formula (VI) by treatment with a strong base, such as potassium tert-butoxide or potassium hexamethyldisilazide (KHDMS), in the presence of a suitable crown ether, such as 18-crown-6 for potassium bases, 15- crown-5 for sodium bases, and 12-crown-4 for lithium bases, at a temperature of from about -78°C to room temperature, preferably about 0°C for a time period of from about 30 minutes to about 24 hours, preferably about 1 hour.
- a strong base such as potassium tert-butoxide or potassium hexamethyldisilazide (KHDMS)
- KHDMS potassium tert-butoxide or potassium hexamethyldisilazide
- alkyl halide having the desired alkyl substituent such as methyl iodide in an inert solvent such as THF, DMF, dioxane, dimethoxy ethane (DME) or a mixture thereof, at a temperature of from about -78°C to about 100 °C, preferably at about room temperature, for a time period from about 1 hour to about 72 hours, preferably about 24 hours.
- pyrrolyl compounds may be analogously prepared by the methods described in schemes 1 and 2, above.
- reaction 1 the aniline compound of Formula (VIII) may be treated with an acid chloride or acid anhydride in the presence of a tertiary amine base such as triethyiamine or dimethylamino pyridine (DMAP) with subsequent cyclodehyd ration to give a benzimidazole (IX).
- a tertiary amine base such as triethyiamine or dimethylamino pyridine (DMAP) with subsequent cyclodehyd ration to give a benzimidazole (IX).
- the reaction is conducted in an inert solvent such as methylene chloride, THF, DMF or a mixture thereof, preferably methylene chloride.
- the aniline compound may be treated with a carboxylic acid and an appropriate coupling agent, as described in reference to reaction scheme 1 , with subsequent cyclodehydration.
- Carboxylic acids which are not available commercially may be prepared according to preparation 1. Suitable acid chloride and acid anhydrides
- the benzimidazole compound may be arylated by treatment with an aromatic or heteroaromatic halide in the presence of a suitable metal catalyst such as tris(dibenzylideneacetone)dipalladium(0) with the appropriate additives, such as 1 ,10 phenanthroline, copper(l)trifluoromethane sulfonate benzene, cesium carbonate, or a mixture thereof to produce a compound of Formula (X).
- a suitable metal catalyst such as tris(dibenzylideneacetone)dipalladium(0)
- additives such as 1 ,10 phenanthroline, copper(l)trifluoromethane sulfonate benzene, cesium carbonate, or a mixture thereof to produce a compound of Formula (X).
- arylated it is meant that the R 2 substituent in the compound of Formula (X) is an aryl or heteroaryl groups, such as phenyl or thienyl.
- the reaction may be conducted in a suitable solvent, such as xylene, DMF, or a mixture thereof, at a temperature of from about 0 to about 165°C, preferably about 135°C for a time period of between about 1 to about 72 hours, preferably about 48 hours.
- the benzimidazole compound of Formula (IX) may be arylated by treatment with a boronic acid in the presence of a suitable catalyst, such as copper(ll) acetate to give an arylated compound of Formula (X).
- a suitable catalyst such as copper(ll) acetate
- the reaction may be conducted in the presence of a base, such as pyridine, Et 3 N or 1 ,4 diazobicylo[2.2.2]octane, in an inert solvent, such as toluene, methylene chloride or a mixture thereof, at a temperature of from about 0°C to about 100°C, preferably about room temperature.
- the reaction may be conducted for a time period of from about 1 hour to about 72 hours, preferably 36 hours.
- the benzimidazole ester of Formula (XII) may be prepared from a benzimidazole alkyl ester compound of Formula (XI) by transesterification and subsequent deprotection.
- methyl esters are the benzimidazole alkyl ester compounds of Formula (XI).
- Transesterification may be accomplished by treatment with boron tribromide (BBr 3 ) in a solvent, such as methylene chloride, chloroform, 1 ,2 dichloroethane or a mixture thereof, followed by the addition of the alcohol having the desired alkyl substituent, for the transesterification.
- a solvent such as methylene chloride, chloroform, 1 ,2 dichloroethane or a mixture thereof
- the benzimidazole alkyl ester compound of Formula (XI) may be hydrolyzed to a carboxylic acid compound of Formula (XIII) and coupled with the appropriate alcohol under conditions as described in reference to the coupling reaction as described in reference to scheme 1 , reaction 1 , except using the appropriate alcohol in place of the aniline.
- the benzimidazole amide compound of Formula (XIV) may be prepared from the carboxylic acid compound of Formula (XIII) by coupling with an appropriate amine compound under conditions as described in reference to the coupling reaction as described in reference to reaction scheme 1.
- the subject invention also includes isotopically-labelled compounds, which are identical to those recited in Formulas (I) but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine and chlorine, such as 2 H, 3 H, 13 C, 1 C, 15 N, 8 0, 1 ; 7 0, 31 P, 32 P, 35 S, 18 F, and 36 CI, respectively.
- Compounds of the present invention which contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of this invention.
- Certain isotopically-labelled compounds of the present invention for example those into which radioactive isotopes such as 3 H and 14 C are incorporated, are useful in drug and/or substrate tissue distribution assays. Tritiated, i.e., 3 H, and carbon-14, i.e., 14 C, isotopes are particularly preferred for their ease of preparation and detectability.
- Isotopically labelled compounds of Formula (I) of this invention and prodrugs thereof can generally be prepared by carrying out the procedures disclosed in the Schemes and/or in the Examples below, by substituting a readily available isotopically labelled reagent for a non-isotopically labelled reagent.
- compounds of the present invention may act as antagonists or agonists. The antagonist/agonist activity of the compounds may be determined by any method known in the art.
- estrogenic activity in human breast cancer MCF7 cells and primary rat granulosa cells may be assessed by transient transfection of an estrogen responsive ERE3-TK-lux luciferase reporter vector essentially as has been described previously in other cell backgrounds, as in Petersen DN, Tkalcevic GT, Koza-Taylor PH, Turi TG & Brown TA (1998) Identification of estrogen receptor ⁇ 2. a functional variant of estrogen receptor expressed in normal rat tissue. Endocrinology 139: 1082-1092, incorporated herein by reference in its entirety.
- the MCF7 cell activity was considered to be mediated through ER ⁇ and the granulosa activity was considered to be mediated through
- ER ⁇ .MCF7 cells may be obtained from ATCC (Manassas, VA) and transfected with Lipofectamine Plus (Gibco/BRL, Rockville, MD) as described by the manufacturers. Luciferase may be measured 24 hours after compound addition.
- Primary rat granulosa cells may be isolated and transfected with ERE3-TK-lux as described in O'Brien ML, Park K, In Y, & Park-Sarge O-K (1999) Characterization of estrogen receptor- ⁇ (ER ⁇ ) messenger ribonucleic acid and protein expression in rat granulosa cells. Endocrinology 140: 4530-4541, incorporated herein by reference in its entirety.
- the invention has been described in detail with particular reference to specific embodiments thereof, but it will be understood that various modifications can be effected within the scope of the invention.
- cDNA cloning of human ER ⁇ and ER ⁇ The coding region of human ER ⁇ was cloned by reverse transcriptase polymerase chain reaction (RT-PCR) from human breast cancer cell mRNA using EXPAND High Fidelity PCR System according to manufacturer's instructions (Boehringer-Mannheim, Indianapolis, IN). The coding region of human ER ⁇ was cloned by RT-PCR from human testes and pituitary mRNA using EXPAND High Fidelity PCR System according to manufacturer's instructions (Boehringer-Mannheim, Indianapolis, IN). PCR products were cloned into pCR2.1 TA Cloning Kit (Invitrogen, Carlsbad, CA) and sequenced. Each receptor coding region was subcloned into the mammalian expression vector pcDNA3 ((Invitrogen, Carlsbad, CA).
- RT-PCR reverse transcriptase polymerase chain reaction
- 293T cells Mammalian cell expression. Receptor proteins were overexpressed in 293T cells. These cells, derived from HEK293 cells (ATCC, Manassas, VA), have been engineered- to stably- express large T antigen and can therefore replicate plasmids containing a SV40 origin of replication to high copy numbers. 293T cells were transfected with either hER ⁇ -pcDNA3 or hER ⁇ -pcDNA3 using lipofectamine as described by the manufacturer (Gibco/BRL, Bethesda, MD). Cells were harvested in phosphate buffered saline (PBS) with 0.5 mM EDTA at 48 h post-transfection.
- PBS phosphate buffered saline
- Step A (4-Methoxy-phenyl)-(2-nitro-phenyl)-amine 1-Fluoro-2-nitrobenzene (37.6g, 28.0 ml, 0.266 mmol), p-anisidine (32.8g, 0.266) and K 2 CO 3 (55.0 g, 0.399 mol) were combined in flask and heated at 160°C overnight under an atmosphere of N 2 . The mixture was cooled to ca. 90°C and water (200ml) was added slowly to the reaction. The mixture was partitioned with EtOAc (1 L). The remaining solid was stirred for 45 minutes in EtOAc (200 ml), MeOH (200 ml) and i water (200 ml) until complete dissolution occurred.
- 3,5-Dimethyl-isoxazole-4-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide was prepared in a procedure analogous to that described in example 1 step C except that 3,5-dimethylisoxazol-4-carboxylic acid (0.083 g, 0.59 mmol) was used instead of 3-thiophenecarboxylic acid MS (M+1) 338.
- lsothiazole-4-carboxylic acid can be prepared according to the procedure of H.P. Benschop, A.M. Oosten, D.H.J.M. Platenburg and C. Van Hooidonk J. Med. Chem. 1970, 13(6), 1208.
- 2-lsothiazol-4-yl-1-(4-methoxy-ph ⁇ enyl)-1 H-benzoimidazole was prepared in a procedure analogous to that described in example 1 step D except isothiazole-4- carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide was used instead of thiophene-3-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide. MS (M+1) 308.
- 4-(2-lsothiazol-4-yl-benzoimidazol-1-yl)-phenol (0.015g, 0.051 mmol) was prepared in a procedure analogous to that described in example 1 step E except that 2-isothiazol- 4-yl-1-(4-methoxy-phenyl)-1 H-benzoimidazole was used instead of 1-(4-methoxy- phenyl)-2-thiophen-3-yl-1 H-benzoimidazole.
- 4-Methyl-isothiazole-5-carboxylic acid can be prepared according to the procedure of M.P.L Caton, D.H. Jones, R.SIack and K.R.H. Wooldridge J. Chem. Soc. 1964, 446.
- 3-Chloro-thiophene-2-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide was prepared in a procedure analogous to that as described in example 5 step A except that 3-chloro-thiophene-2-carboxylic acid (0.094g, 0.052 mmol) was used 5 instead of 3-bromothiophene-2-carboxylic acid.
- Step B 1 -(4-Methoxy-phenyl)-2-(1 H-pyrrol-2-yl)-1 H-benzoimidazole
- Furan-3-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide was prepared in a.procedure-analogous-to that as described in example 5 step A. except that furan-3- carboxylic acid (0.076, 0.069 mmol) was used , instead of 3-bromothiophene-2- carboxylic acid. MS (M+1) 309.
- Step B 2-Furan-3 : yl-1-(4-methoxy-phenyl)-1 H-benzoimidazole
- 2-Furan-3-yl-1-(4-methoxy-phenyl)-1 H-benzoimidazole was prepared in a procedure analogous to that as described in example 1 step D except that furan-3-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide was used instead of thiophene-3- carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide. MS (M+1) 291.
- 3-Metf ⁇ yl-furan-2-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide was prepared in a procedure analogous that as described in example 5 step A except that 3-methyl-furan-2-carboxylic (0.086g, 0.69 mmol) acid was used instead of 3- bromothiophene-2-carboxyli ⁇ acid.
- Furan-2-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide was prepared in a procedure analogous to that as described in example 5 step A except that furan-2- carboxylic acid (0.078 g, 0.685 mmol) was used instead of 3-bromothiophene-2- carboxylic acid.
- 2-Furan-2-yl-1-(4-methoxy-phenyl)-1 H-benzoimidazole was prepared in a procedure analogous to that described in example 1 step D except that furan-2-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide was used instead of thiophene-3- carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide.
- StepB 3-Ethyl-isoxazole-4-carboxylic acid
- MeOH/THF MeOH/THF
- 5N NaOH 5N NaOH
- the reaction mixture was stirred at room temperature overnight.
- Step E 4-[2-(3-Ethyl-isoxazol-4-yl)-benzoimidazol-1-yl]-phenol
- Step E 4-[2-(3-Cyclopropyl-isoxazol-4-yl)-benzoimidazoi-1-yl]-phenol
- Step A 1 -(4-Methoxy-phenyl)-2-(1 -propyl-1 H-pyrrol-2-yl)-1 H-benzoimidazole
- the reaction was stirred 0 at room temperature for 60 hours, quenched with sat. NH CI (1 ml), and diluted with CH 2 CI 2 (20ml). The mixture was washed with sat. NH Cl (1x20 ml) and the aqueous washing was extracted with CH 2 CI 2 (1x20 ml).
- Step B (
- Step C 0 1 -Methyl-1 H-pyrrole-2-carboxylic acid [2-(4-methoxy-2-methyl-phenylamino)-phenyl]- amide
- Isocyanate scavenger beads (Argonaut Technologies) were added to the reaction mixture to remove excess N-(4-methoxy-2-methyl- 0 phenyl)-benzene-1 ,2-diamine and the suspension was stirred for several hours. The beads were removed by filtration, and the filtrate was concentrated by vacuum to give 1 -methyl-1 H-pyrrole-2-carboxylic acid [2-(4-methoxy-2-methyl-phenylamino)-phenyl]- amide which was taken on directly into the next. MS (M+1) + 336. Step D
- reaction material was diluted with CH 2 CI 2 , washed with saturated sodium bicarbonate and extracted into CH 2 CI 2 .
- the combined organic material was dried (MgSO 4 ), filtered, and concentrated, giving 3, 5-dimethyl- isoxazole-4-carboxylic acid [2-(4-methoxy-2-methyl-phenylamino)-phenyl]-amide.
- 1-(4-Methoxy-phenyl)-2-(3-methyl-thiophen-2-yl)-1 H-benzoimidazole was prepared in a procedure analogous to that as described in example 1step D except that 3-methyl- thiophene-2-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide was used instead of thiophene-3-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide.
- Step C 4-[2-(3-Methyl-thiophen-2-yl)-benzoimidazol-1 -yl]-phenol
- lsothiazole-5-carboxylic acid can be prepared according to the procedure of M.P.L. Caton, D.H. Jones, R.SIack and K.R.H. Wooldridge J. Chem. Soc. 1964, 446.
- Step A lsothiazole-5-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide lsothiazole-5-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide was prepared in a procedure analogous to that as described in example 5 step A except that isothiazole-5-carboxylic acid (0.067g, 0.521 mmol) was used instead of 3- bromothiophene-2-carboxylic acid.
- 2-lsothiazol-5-yl-1-(4-methoxy-phenyl)-1 H-benzoimidazole was prepared in a procedure analogous to that as described in example 1 step D except thatisothiazole-5-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]-amide was used instead of thiophene-3-carboxylic acid [2-(4-methoxy-phenylamino)-phenyl]- amide.
- 4-(2-lsothiazol-5-yl-benzoimidazol-1-yl)-phenol was prepared in a procedure analogous to that as described in example 1 step E except that 2-isothiazol-5-yl-1- (4-methoxy-phenyl)-1 H-benzoimidazole was used instead of 1-(4-methoxy-phenyl)- 2-thiophen-3-yl-1 H-benzoimidazole.
- Step l To a solution of acid (1.5 eq.), Et 3 N (5.0 eq.), DMAP and N-(4-methoxy-phenyl)- benzene-1 ,2-diamine di-hydrochloride (1.0 eq) in CH 2 CI 2 (1.5ml) was added PPAA (2.0eq.). The reaction was stirred overnight at room temperature. Unreacted N-(4- methoxy-phenyl)-benzene-1 ,2-diamine was scavenged with polymer supported isocyanate (Argonaut Technologies). The polymer beads are removed via filtration and the volatiles are removed under a stream of N 2 . Step 2
- Examples 30-45 were prepared according to the above procedure using the appropriate carboxylic acid as defined in each example.
- 4-(2-pyrrolidin-1-yl-ethoxy)-benzoic acid can be prepared according to the reference Jones, Charles D.; Jevnikar, Mary G.; Pike, Andrew J.; Peters, Mary K.; Black, Larry J.; et al. J.Med.Chem.1984, 27 (8); 1057-1066.
- 4- ⁇ 2-[4-(2-Pyrrolidin-1 -yl-ethoxy)-phenyl]-benzoimidazol-1 -yl ⁇ -phenol was prepared in according to the general procedure using 4-(2-pyrrolidin-1-yl-ethoxy)-benzoic acid as the carboxylic acid and the final product was purified by recrystallisation from hot MeOH. MS (M+1) 400.
- 4-[2-(2-Chloro-phenyl)-benzoimidazol-1-yl]-phenol was prepared according to the general procedure using o-chlorobenzoic acid except that no purification of the final product was needed and polymer supported isocyanate beads were added during step 1.
- Example 36 4-(2-Cyclopentyl-benzoimidazol-1-yl)-phenol 4-(2-Cyclopentyl-benzoimidazol-1-yl)-phenol was prepared according to the general procedure using cyclopentanecarboxylic acid except no purification of the final product was required and all solutions were dried using Alltech spice filters.
- Example 39 4-(2-Trifluoromethyl-benzoimidazol-1-yl)-phenol 4-(2-Trifluoromethyl-benzoimidazol-1-yl)-phenol was prepared according to the general procedure using trifluoroacetic acid except that the final product was purified by reverse phase HPLC and MgSO 4 was used to dry all solutions. MS (MH) + 279; 1 H NMR (acetone) ⁇ H 9.03 (s, 1 H), 7.86-7.89 (m, 1 H), 7.42-7.46 (m, 4H), 7.20-7.23 (m, 1H), 7.10-7.13 (d, 2H).
- 4-[1-(4-Hyroxy-phenyl)-1H-benzoimidazol-2-yl]-benzonitrile was prepared according to the general procedure using 4-cyano benzoic acid, except that the product of step 2 was purified by preparative TLC, all solutions were dried using Na 2 SO 4 and the final product crystallized upon standing requiring no further purification.
- N-(4-methoxy-2-methyl-phenyl)-benzene-1 ,2-diamine (0.200 g, 0.877 mmol)
- otoluic acid (0.178 g, 1.3155 mmol)
- triethyiamine 0.13 mL, 4.385 mmol
- 4-dimethylami ⁇ opyridine in methylene chloride (2 mL)
- 1-propanephosphonic acid cyclic anhydride (1.05 mL, 1.754 mmol) 5 as a 50% solution in ethyl acetate.
- the white solid, the diacyl compound, was filtered and washed with ethyl acetate and acetone. The filtrate and decanted oil were combined. Additional white solid precipitated and was filtered.
- the concentrated filtrate was dissolved in a mixture of methylene chloride and ethyl acetate (30 mL, 1:1 v/v) and methanol (2 mL). The solution was cooled over night in a refrigerator and feathery white crystals (desired product) formed and were filtered.
- the filtrate was concentrated and the residue was dissolved in methylene chloride (10 mL) and ethyl acetate (5 mL). Methylene chloride was slowly removed on the rotary evaporator until cloudy.
- TLC indicated mostly starting material and 450 mg of cesium carbonate from a new source was added. The material was stirred while heating at 65°C for 4 h. No change was seen by TLC and heating continued over weekend. The solvent was stripped and purification of the crude residue by silica gel flash chromatography, eluting with diethyl ether, gave 14 mg of an impure yellow oil. Further purification be preparatory TLC (1.0 mm), eluting with diethyl ether, gave 7 mg of pure 2-(4-Methoxy-phenyl)-1-thiophen-3-yl-1 H-benzoimidazole.
- N- [2-(4-methoxy-phenylamino)-phenyl]-terephthalamic acid methyl ester (28 g, 0.074 mol) was heated in glacial acetic acid (225ml) at 80°C under an atmosphere of nitrogen over night. Once the reaction material cooled to room temperature, heptane was added (400ml) and some of the acetic acid/heptane mixture was evaporated. Additional heptane (2x200ml) was added and the remaining acetic acid was evaporated. The resulting light brown solid was triturated with Et 2 O and the product was filtered as a white solid (14.4 g, 40.2 mmol) of pure title compound.
- the reaction was neutralized to pH7 with saturated sodium bicarbonate and diluted with ethyl acetate (25ml), forming a white solid in the process. After filtering the solid, the organic phase of the biphasic filtrate was washed with saturated sodium bicarbonate and the brine. The organic extract was then dried (MgSO ) and concentrated by vacuum. The crude residue was triturated with diethyl ether to give the title compound (0.376g, 1.09 mmol).
- Example 54 4-ri-(4-Hvdroxy-phenyl)-1H-benzoimidazol-2-vn-benzoic acid isopropyl ester Following the method outlined in Example 53, except replacing ethanol with isopropyl alcohol yielded title compound. MS (MH) + 373; 1 H NMR (CDCI 3 ) ⁇ H 7.94-7.96 (d, 2H), 7.85-7.887 (d, 1H), 7.63-7.65 (d, 2H), 7.20-7.34 (m, 3H), 7.13-7.15 (d, 2H), 6.92-6.94 (d, 2H), 5.18-5.23 (m, 1 H), 1.32-1.34 (d, 6H).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Endocrinology (AREA)
- Diabetes (AREA)
- Immunology (AREA)
- Hematology (AREA)
- Neurosurgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Obesity (AREA)
- Dermatology (AREA)
- Zoology (AREA)
- Cardiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Gastroenterology & Hepatology (AREA)
- Urology & Nephrology (AREA)
- Heart & Thoracic Surgery (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Emergency Medicine (AREA)
- Reproductive Health (AREA)
- Vascular Medicine (AREA)
- Hospice & Palliative Care (AREA)
Abstract
Description












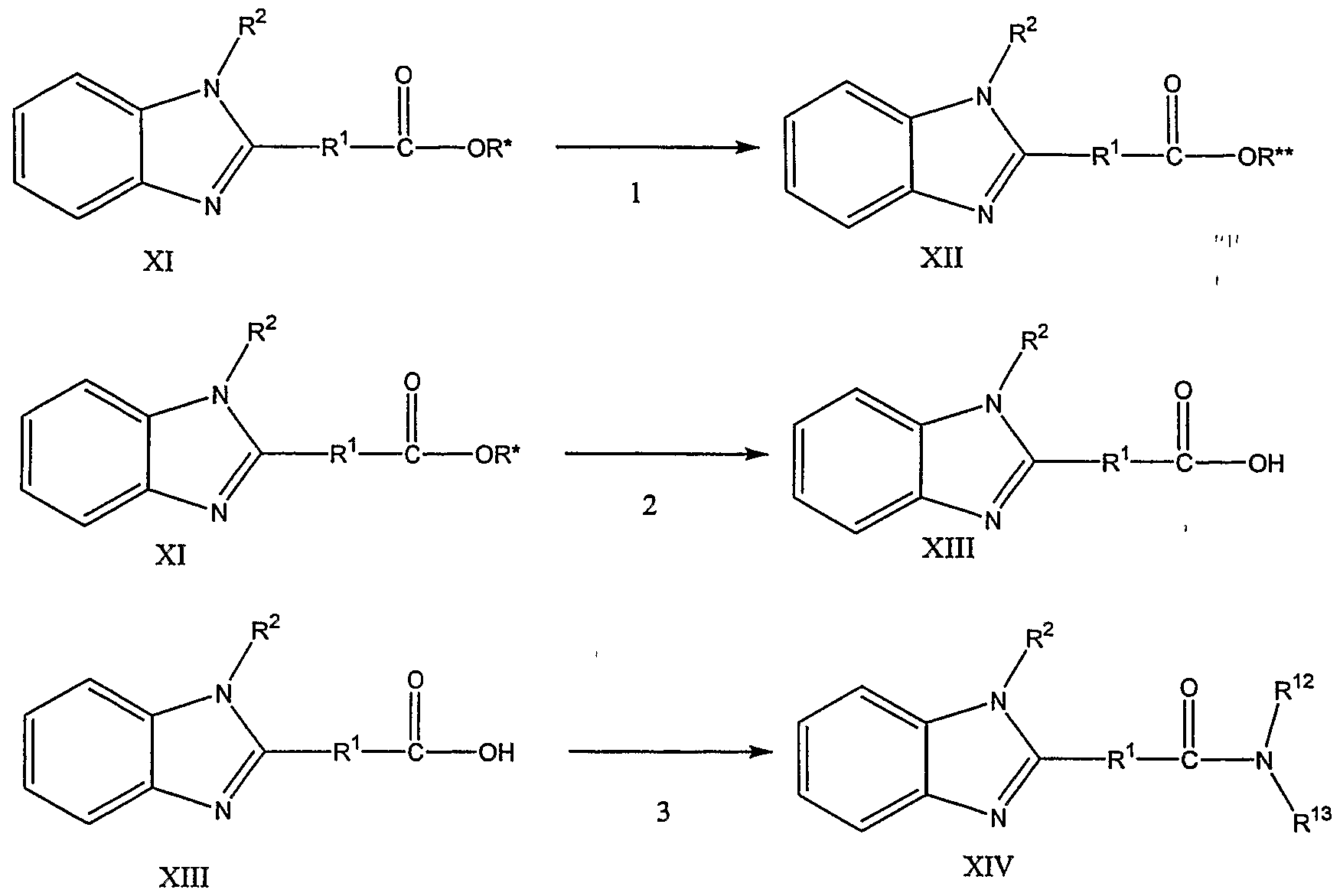
Claims


Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03732945A EP1517897A2 (en) | 2002-06-24 | 2003-06-12 | Benzimidazole compounds and their use as estrogen agonists/antagonists |
| AU2003238597A AU2003238597A1 (en) | 2002-06-24 | 2003-06-12 | Benzimidazole compounds and their use as estrogen agonists/antagonists |
| MXPA04011123A MXPA04011123A (en) | 2002-06-24 | 2003-06-12 | Benzimidazole compounds and their use as estrogen agonists/antagonists. |
| CA002487266A CA2487266A1 (en) | 2002-06-24 | 2003-06-12 | Benzimidazole compounds and their use as estrogen agonists/antagonists |
| BR0312069-4A BR0312069A (en) | 2002-06-24 | 2003-06-12 | Benzimidazole compounds and their use as estrogen agonists / antagonists |
| JP2004515164A JP2005534675A (en) | 2002-06-24 | 2003-06-12 | Benzimidazole compounds and their use as estrogen agonists / antagonists |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US39133702P | 2002-06-24 | 2002-06-24 | |
| US60/391,337 | 2002-06-24 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004000817A2 true WO2004000817A2 (en) | 2003-12-31 |
| WO2004000817A3 WO2004000817A3 (en) | 2004-05-21 |
Family
ID=30000694
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2003/002670 WO2004000817A2 (en) | 2002-06-24 | 2003-06-12 | Benzimidazole compounds and their use as estrogen agonists/antagonists |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20040002524A1 (en) |
| EP (1) | EP1517897A2 (en) |
| JP (1) | JP2005534675A (en) |
| AU (1) | AU2003238597A1 (en) |
| BR (1) | BR0312069A (en) |
| CA (1) | CA2487266A1 (en) |
| MX (1) | MXPA04011123A (en) |
| WO (1) | WO2004000817A2 (en) |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004031159A1 (en) * | 2002-09-25 | 2004-04-15 | Wyeth | Substituted 4-(indazol-3-yl)phenols as estrogen receptor (er) ligands and their use in the treatment of inflammatory diseases |
| JP2006505522A (en) * | 2002-08-08 | 2006-02-16 | スミスクライン ビーチャム コーポレーション | Thiophene compound |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| US7354927B2 (en) | 2004-09-07 | 2008-04-08 | Wyeth | 6H-[1]benzopyrano[4,3-b]quinolines and their use as estrogenic agents |
| JP2008545663A (en) * | 2005-05-27 | 2008-12-18 | クイーンズ ユニバーシティ アット キングストン | Treatment of protein folding disorders |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2009026658A1 (en) * | 2007-08-29 | 2009-03-05 | The University Of Sydney | Ppar agonists |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010051085A1 (en) * | 2008-10-30 | 2010-05-06 | Oncotherapy Science, Inc. | 7-hydroxy-benzoimidazole-4-yl-methanone derivatives and pbk inhibitors containing the same |
| CN1919839B (en) * | 2006-09-01 | 2010-05-12 | 西北师范大学 | Preparation technique of 2-chloromethylbenzimidazole |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| CN102149688A (en) * | 2008-09-11 | 2011-08-10 | 霍夫曼-拉罗奇有限公司 | New benzimidazole derivatives |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| CN102781930A (en) * | 2009-12-18 | 2012-11-14 | 田边三菱制药株式会社 | Novel anti-platelet agent |
| CN103119042A (en) * | 2010-08-25 | 2013-05-22 | 株式会社新药 | Novel heterocyclic compound, and composition for treating inflammatory diseases using same |
| CN108884053A (en) * | 2015-12-15 | 2018-11-23 | 斯坦福大学托管董事会 | The method of prevention and/or treatment related with aging cognitive disorder and neuroinflamation |
| CN109053584A (en) * | 2018-09-12 | 2018-12-21 | 南京大学 | The preparation and its application of one kind 1,2- diaryl benzimidazole derivative |
| US11465998B2 (en) | 2019-04-25 | 2022-10-11 | Regents Of The University Of Minnesota | Therapeutic compounds and methods of use thereof |
| US11891382B2 (en) | 2017-04-26 | 2024-02-06 | Basilea Pharmaceutica International AG | Processes for the preparation of furazanobenzimidazoles and crystalline forms thereof |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7318925B2 (en) | 2003-08-08 | 2008-01-15 | Amgen Fremont, Inc. | Methods of use for antibodies against parathyroid hormone |
| AU2003282780A1 (en) | 2003-08-08 | 2005-03-07 | Abgenix, Inc. | Antibodies directed to parathyroid hormone (pth) and uses thereof |
| CA2569738A1 (en) * | 2004-06-07 | 2005-12-22 | Duramed Pharmaceuticals, Inc. | Dispenser for progestin used for acute and maintenance treatment of dub |
| JP2015044778A (en) * | 2013-08-29 | 2015-03-12 | 花王株式会社 | Hair growth inhibitor |
| CN105669614A (en) * | 2016-01-12 | 2016-06-15 | 四川大学 | Diarylamine-containing furoylamide compound and its use in pesticide |
| CN111712495A (en) * | 2017-12-22 | 2020-09-25 | 拜耳公司 | Hydroxy isoxazolines and derivatives thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1174493A (en) * | 1967-05-10 | 1969-12-17 | Manufactures J R Bottu | New Benzimidazoles and process for their preparation |
| WO2002040467A1 (en) * | 2000-11-17 | 2002-05-23 | Smithkline Beecham Corporation | Compounds |
| WO2002046168A1 (en) * | 2000-12-07 | 2002-06-13 | Astrazeneca Ab | Therapeutic benzimidazole compounds |
| WO2003050095A1 (en) * | 2001-12-05 | 2003-06-19 | Wyeth | Substituted benzoxazoles and analogues as estrogenic agents |
| WO2003066629A2 (en) * | 2002-02-06 | 2003-08-14 | Vertex Pharmaceuticals Incorporated | Heteroaryl compounds useful as inhibitors of gsk-3 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3932389A (en) * | 1974-12-11 | 1976-01-13 | Pfizer Inc. | 2-Descarboxy-2-(tetrazol-5-yl)-11-desoxy-15-substituted-.omega.-pentanorprostaglandins |
| US3982016A (en) * | 1975-08-06 | 1976-09-21 | Pfizer Inc. | Bone deposition by 16-aryl-13,14-dihydro-PGE2 p-biphenyl esters |
| US4018892A (en) * | 1975-08-06 | 1977-04-19 | Pfizer Inc. | Bone deposition by 16-aryl-13,14-dihydro-PGE2 p-biphenyl esters |
| US4132847A (en) * | 1977-07-22 | 1979-01-02 | Pfizer Inc. | 4-Pyrone prostaglandin antagonists |
| US4219483A (en) * | 1978-09-11 | 1980-08-26 | Pfizer Inc. | 4-Pyrone prostaglandin antagonists |
| US4621100A (en) * | 1981-09-25 | 1986-11-04 | The Upjohn Company | Treatment of osteoporosis with prostaglandins |
| GB8612796D0 (en) * | 1986-05-27 | 1986-07-02 | Fbc Ltd | Fungicides |
| US5216183A (en) * | 1988-04-19 | 1993-06-01 | Teijin Limited | Cyclopentanone/cyclopentenone derivative |
| JPH0772181B2 (en) * | 1989-06-26 | 1995-08-02 | 株式会社大塚製薬工場 | Benzimidazole derivative |
| US5552426A (en) * | 1994-04-29 | 1996-09-03 | Eli Lilly And Company | Methods for treating a physiological disorder associated with β-amyloid peptide |
| US7115645B2 (en) * | 2000-01-14 | 2006-10-03 | Schering Aktiengesellschaft | 1,2 diarylbenzimidazoles and their pharmaceutical use |
-
2003
- 2003-06-11 US US10/460,565 patent/US20040002524A1/en not_active Abandoned
- 2003-06-12 EP EP03732945A patent/EP1517897A2/en not_active Withdrawn
- 2003-06-12 MX MXPA04011123A patent/MXPA04011123A/en not_active Application Discontinuation
- 2003-06-12 CA CA002487266A patent/CA2487266A1/en not_active Abandoned
- 2003-06-12 AU AU2003238597A patent/AU2003238597A1/en not_active Abandoned
- 2003-06-12 WO PCT/IB2003/002670 patent/WO2004000817A2/en not_active Application Discontinuation
- 2003-06-12 JP JP2004515164A patent/JP2005534675A/en active Pending
- 2003-06-12 BR BR0312069-4A patent/BR0312069A/en not_active IP Right Cessation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1174493A (en) * | 1967-05-10 | 1969-12-17 | Manufactures J R Bottu | New Benzimidazoles and process for their preparation |
| WO2002040467A1 (en) * | 2000-11-17 | 2002-05-23 | Smithkline Beecham Corporation | Compounds |
| WO2002046168A1 (en) * | 2000-12-07 | 2002-06-13 | Astrazeneca Ab | Therapeutic benzimidazole compounds |
| WO2003050095A1 (en) * | 2001-12-05 | 2003-06-19 | Wyeth | Substituted benzoxazoles and analogues as estrogenic agents |
| WO2003066629A2 (en) * | 2002-02-06 | 2003-08-14 | Vertex Pharmaceuticals Incorporated | Heteroaryl compounds useful as inhibitors of gsk-3 |
Non-Patent Citations (9)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 113, no. 23, 3 December 1990 (1990-12-03), Columbus, Ohio, US; abstract no.: 211905p, KAMISSAROV V N: "Synthesis of 1-(3,5-di-tert-butyl-4-hydroxyphenyl)- 2-substituted benzimidazoles" page 759 XP002254128 & KHIM. GETEROTSIKL. SOEDIN., no. 4, 1990, pages 483-485, * |
| CHEMICAL ABSTRACTS, vol. 58, no. 1, 7 January 1963 (1963-01-07), Columbus, Ohio, US; abstract no.: 515g, SETO S ET AL: "Reaction of N,N'-(3,5-cyclohexadiene-1,2 diylidene)dibenzamide" XP002254127 & YAKUGAKU ZASSHI, vol. 82, 1962, pages 590-594, * |
| DATABASE CAPLUS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; XP002254129 accession no. STN Database accession no. 1989:478007 & JP 64 000074 A (OTSUKA PHARMACEUTICAL FACTORY, INC.) 5 January 1989 (1989-01-05) * |
| DATABASE CAPLUS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; XP002254130 accession no. STN Database accession no. 1991:408806 & JP 03 031264 A (OTSUKA PHARMACEUTICAL FACTORY, INC.) 12 February 1991 (1991-02-12) * |
| DATABASE CHEMCATS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; XP002254131 accession no. STN & "ChemBridge Product List (Catalog)" 17 January 2002 (2002-01-17), CHEMBRIDGE CORPORATION , SAN DIEGO, CA, USA * |
| ERSAN S ET AL: "Studies on analgesic and anti-inflammatory activities of 1-dialkylaminomethyl-2-(p-substituted phenyl)-5-substituted benzimidazole derivatives" ARZNEIMITTEL-FORSCHUNG, vol. 47(II), no. 7, July 1997 (1997-07), pages 834-836, XP002254125 * |
| GRELLA G ET AL: "Sintesi ed actività coleretica di acidi 3-(2-aril-5R-benzimidazol-1-il)butanoici" IL FARMACO, EDIZIONE SCIENTIFICA, vol. 42, no. 7, July 1987 (1987-07), pages 475-490, XP002254126 * |
| UZUNOGLU S ET AL: "Synthesis and activities of 5-substituted-2-(p-substituted phenyl)-1-dialkylaminomethyl benzimidazole derivatives" IL FARMACO, vol. 52, no. 10, October 1997 (1997-10), pages 619-623, XP002254124 * |
| YUN Y K ET AL: "Polymer-assisted parallel solution phase synthesis of substituted benzimidazoles" SYNLETT, no. 5, May 2002 (2002-05), pages 739-742, XP002254123 * |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006505522A (en) * | 2002-08-08 | 2006-02-16 | スミスクライン ビーチャム コーポレーション | Thiophene compound |
| US7241791B2 (en) | 2002-09-25 | 2007-07-10 | Wyeth | Substituted 4-(indazol-3-yl)phenols |
| WO2004031159A1 (en) * | 2002-09-25 | 2004-04-15 | Wyeth | Substituted 4-(indazol-3-yl)phenols as estrogen receptor (er) ligands and their use in the treatment of inflammatory diseases |
| US7354927B2 (en) | 2004-09-07 | 2008-04-08 | Wyeth | 6H-[1]benzopyrano[4,3-b]quinolines and their use as estrogenic agents |
| JP2008545663A (en) * | 2005-05-27 | 2008-12-18 | クイーンズ ユニバーシティ アット キングストン | Treatment of protein folding disorders |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| CN1919839B (en) * | 2006-09-01 | 2010-05-12 | 西北师范大学 | Preparation technique of 2-chloromethylbenzimidazole |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2009026658A1 (en) * | 2007-08-29 | 2009-03-05 | The University Of Sydney | Ppar agonists |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| CN102149688A (en) * | 2008-09-11 | 2011-08-10 | 霍夫曼-拉罗奇有限公司 | New benzimidazole derivatives |
| WO2010051085A1 (en) * | 2008-10-30 | 2010-05-06 | Oncotherapy Science, Inc. | 7-hydroxy-benzoimidazole-4-yl-methanone derivatives and pbk inhibitors containing the same |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| US9533983B2 (en) | 2009-12-18 | 2017-01-03 | Mitsubishi Tanabe Pharma Corporation | Antiplatelet agent |
| CN102781930A (en) * | 2009-12-18 | 2012-11-14 | 田边三菱制药株式会社 | Novel anti-platelet agent |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| EP2610255A4 (en) * | 2010-08-25 | 2014-02-12 | Neopharm Co Ltd | Novel heterocyclic compound, and composition for treating inflammatory diseases using same |
| CN103119042A (en) * | 2010-08-25 | 2013-05-22 | 株式会社新药 | Novel heterocyclic compound, and composition for treating inflammatory diseases using same |
| US8809541B2 (en) | 2010-08-25 | 2014-08-19 | Neopharm Co., Ltd. | Heterocyclic compound, and composition for treating inflammatory diseases using same |
| EP2610255A2 (en) * | 2010-08-25 | 2013-07-03 | Neopharm Co., Ltd. | Novel heterocyclic compound, and composition for treating inflammatory diseases using same |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| CN108884053A (en) * | 2015-12-15 | 2018-11-23 | 斯坦福大学托管董事会 | The method of prevention and/or treatment related with aging cognitive disorder and neuroinflamation |
| CN108884053B (en) * | 2015-12-15 | 2022-01-04 | 斯坦福大学托管董事会 | Method for preventing and/or treating age-related cognitive disorders and neuroinflammation |
| US11891382B2 (en) | 2017-04-26 | 2024-02-06 | Basilea Pharmaceutica International AG | Processes for the preparation of furazanobenzimidazoles and crystalline forms thereof |
| CN109053584A (en) * | 2018-09-12 | 2018-12-21 | 南京大学 | The preparation and its application of one kind 1,2- diaryl benzimidazole derivative |
| CN109053584B (en) * | 2018-09-12 | 2022-03-18 | 南京大学 | Preparation and application of 1, 2-diaryl benzimidazole derivatives |
| US11465998B2 (en) | 2019-04-25 | 2022-10-11 | Regents Of The University Of Minnesota | Therapeutic compounds and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| MXPA04011123A (en) | 2005-02-14 |
| CA2487266A1 (en) | 2003-12-31 |
| BR0312069A (en) | 2005-03-29 |
| WO2004000817A3 (en) | 2004-05-21 |
| AU2003238597A8 (en) | 2004-01-06 |
| JP2005534675A (en) | 2005-11-17 |
| AU2003238597A1 (en) | 2004-01-06 |
| EP1517897A2 (en) | 2005-03-30 |
| US20040002524A1 (en) | 2004-01-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1517897A2 (en) | Benzimidazole compounds and their use as estrogen agonists/antagonists | |
| US20030220377A1 (en) | Indole compounds and their use as estrogen agonists/antagonists | |
| JP5376956B2 (en) | Treatment of Duchenne muscular dystrophy | |
| TWI333950B (en) | New benzimidazole derivatives | |
| JP5554988B2 (en) | Inhibitors of histone deacetylase | |
| JP4970538B2 (en) | Benzimidazole derivatives, processes for their preparation, their use as FXR agonists and formulations containing them | |
| US20030087902A1 (en) | Substituted indoles, pharmaceutical compounds containing such indoles and their use as PPAR-gamma binding agents | |
| JP6552061B2 (en) | Negative allosteric modulators (NAMS) of metabotropic glutamate receptors and their use | |
| CA2769474C (en) | Acyl guanidine derivatives modulating the hedgehog protein signaling pathway | |
| JP2001501957A (en) | Aminothiophene carboxamide | |
| KR100682144B1 (en) | New retinoids for the treatment of emphysema | |
| OA12631A (en) | Anthranilic acid amides with a heteroarylsulfonyl side chain, process for their preparation, their use as medicaments or diagnostic agents and pharmaceutical preparations comprising said compounds. | |
| JPWO2006077821A1 (en) | Aromatic sulfone compounds as aldosterone receptor modulators | |
| EP0498723A1 (en) | Nitrogen containing bicycle compounds, method for their preparation, their use as pharmaceutical and compositions containing them | |
| JP2000256323A (en) | 2-oxoquinoline compound and its medicinal use | |
| KR20050013255A (en) | Tricyclic steroid hormone nuclear receptor modulators | |
| CN110678450B (en) | Isoxazole derivatives as nuclear receptor agonists and uses thereof | |
| EP2238110A1 (en) | 5.6-bisaryl-2-pyridine-carboxamide derivatives, preparation thereof and therapeutic application thereof as antagonists for urotensine ii receptors | |
| WO2022218440A1 (en) | Fxr regulator, preparation method therefor, pharmaceutical composition thereof and use thereof | |
| CA2722258C (en) | N-acylthiourea and n-acylurea inhibitors of the hedgehog protein signalling pathway | |
| JP2003523961A (en) | 1,2-Diarylbenzimidazoles for the treatment of diseases associated with microglial activation | |
| KR20040023591A (en) | Alkoxycarbonylamino benzoic acid or alkoxycarbonylamino tetrazolyl phenyl derivatives as ip antagonists | |
| CN108602775B (en) | Mast cell modulators and uses thereof | |
| JP2002522423A (en) | Imidazole and related compounds as α1A agonists | |
| JP3142581B2 (en) | New pyrrole derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2004/011123 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2487266 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003732945 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004515164 Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003732945 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2003732945 Country of ref document: EP |