WO2003061670A1 - Compounds useful as a3 adenosine receptor agonists - Google Patents
Compounds useful as a3 adenosine receptor agonists Download PDFInfo
- Publication number
- WO2003061670A1 WO2003061670A1 PCT/GB2003/000304 GB0300304W WO03061670A1 WO 2003061670 A1 WO2003061670 A1 WO 2003061670A1 GB 0300304 W GB0300304 W GB 0300304W WO 03061670 A1 WO03061670 A1 WO 03061670A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- product
- methyl
- ring
- hydrogen
- formula
- Prior art date
Links
- 0 *C(C1O)C=C(CN)C=C1N=*N(*)*=C Chemical compound *C(C1O)C=C(CN)C=C1N=*N(*)*=C 0.000 description 3
- SDECKBOHRPKYMV-UHFFFAOYSA-N CC(c1cccc(CBr)n1)=O Chemical compound CC(c1cccc(CBr)n1)=O SDECKBOHRPKYMV-UHFFFAOYSA-N 0.000 description 1
- XLWPYAZLXACFKX-UHFFFAOYSA-N N#Cc(cc1Br)cc([N+]([O-])=O)c1O Chemical compound N#Cc(cc1Br)cc([N+]([O-])=O)c1O XLWPYAZLXACFKX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- This invention relates to compounds useful as ⁇ 3 adenosine receptor agonists and methods of selectively activating an A3 adenosine receptor in a mammal, particularly a human.
- the present invention also relates to methods of treating various medical disorders with A3 receptor agonists, in particular post- infarct patients, patients with severe angina and related cardiovascular disorders .
- Adenosine an endogenous purine nucleoside, is ubiquitous in mammalian cell types. Adenosine present in the plasma and other extracellular fluids mediates many of its physiological effects via cell surface receptors and is an important regulatory species. Adenosine has the formula:
- Adenosine receptors are generally divided into three major subclasses, Al, A2 and A3, on the basis of the differential affinities of a number of adenosine receptor agonists and antagonists for the receptors, their primary structures and the secondary messenger systems to which they couple.
- WO 95/02604 discloses A3 adenosine receptor agonists and their use as locomotor depressants, hypotensive agents, anxiolytic agents, cerebroprotectants and antiseizure agents.
- US 5573772 and related US 5443836 claim the use of adenosine A3 agonists for applications where ischaemic preconditioning is beneficial, for example cardioprotection.
- WO 98/50047 and WO 99/20284 also relate to ischaemic protection.
- WO 98/50047 claims methods of administering a compound having A3 agonist activity and a compound (whether the same compound or a different one) having Al agonist activity or A2 antagonist activity.
- WO 99/20284 claims a method for preventing or reducing ischaemic heart damage by administration of at least two cardioprotectants, of which one may be an A3 agonist.
- WO 01/19360 claims the use of A3 receptor agonists to achieve the following effect:
- WO 01/083152 relates to the use of adenosine A3 receptor agonists to activate natural killer (NK) cells whilst WO 02/055085 teaches their use to inhibit viral replication.
- WO 02/066020 proposes the use of adenosine A3 receptor agonists to modulate the activity of glycogen synthase kinase 3 ⁇ .
- Adenosine receptor ligands are described in the following documents :
- adenosine receptor agonists which are adenosine analogues characterised by specific variations which make the compounds capable of binding to and acting on one or more adenosine receptors. More particularly, the skilled person knows that there exists a class of adenosine analogue-type A3 receptor agonists .
- Adenosine analogue-type A3 receptor agonists are familiar to the skilled reader and will require no further explanation to the skilled reader. Nonetheless, it may be of assistance to describe that adenosine analogue-type A3 receptor agonists may have an N6 nitrogen which may be identified with the N6 nitrogen of adenosine and is usually substituted by at least one substituent.
- Such agonists include without limitation compounds of the formula:
- D is N or CH; E is O, S or CH2;
- X -- is an N6 substituent; ⁇ 2 (the 4' substituent) is hydroxymethyl , (C1-C3) alkoxymethyl,
- X*- 5 and X ⁇ are each independently hydrogen, alkyl, hydroxyalkyl, alkoxyalkyl, OR a NR a R ⁇ , where R a and R- 3 are independently hydrogen (most preferably ⁇ 3 and X ⁇ are OH) , alkyl, aralkyl, carbamoyl, alkyl carbamoyl, dialkylcarbamoyl, acyl, alkoxycarbonyl, aralkoxycarbonyl, aryloxycarbonyl, or, when ⁇ 3
- R" and R e are independently hydrogen, alkyl, or together with the carbon atom to which they are attached may form a 1,1- cycloalkyl group; alkyl (e.g. trifluoromethyl), (C ] _-C ] _ Q ) alkoxyalkyl, (C-]_-C-]_o) alkoxy, (C ⁇ -C]_o) thioalkoxy, (C-]_-C-]_o) alkylthio, amino, (C- ⁇ -C-LO) alkylamino, -COX 6 R 25 where X 6 is O or NH and R 25 is ⁇ C -C4) alkyl optionally terminally substituted by an aryl or a heteroaryl group [for example phenyl or a 5- or 6-membered heteroaryl group] and additionally or alternatively terminally substituted by hydroxy, (C2-C- ] _o) alkenyl, (C2 ⁇ C-]_ Q ) al
- (C2-C- [ _o) alkenyl or (C2-C- ] _ Q ) alkynyl in either case terminally substituted by an aryl or heteroaryl group [for example phenyl or a 5- or 6-membered heteroaryl group] and, when having a terminal methylic carbon atom, optionally further terminally substituted by hydroxy.
- Alkyl groups comprised in X ⁇ substituents are preferably linear.
- X 2 is mono-N- or di-N,N( (C1-C4) alkylaminocarbonyl, mono-N- or di-, -(C3 ⁇ C5) cycloalkylaminocarbonyl or -(C- ] _-C4) alkyl-N- (C3-C5) cycloalkylaminocarbonyl and especially mono-N- (C1-C ) alkylaminocarbonyl;
- X 3 is OH or NH ;
- X 4 is OH;
- ⁇ 5 is H, halogen, ((C ⁇ -C ⁇ Q) alkyl and especially (C1-C ) alkyl, trifluoromethyl, (C 2 -C ] _o) alkenyl, (C ⁇ -C-LQ) alkynyl, or either of the latter two groups where terminally substituted as described above, ⁇ more preferably being H, chloro, bromo, iodo, (C1-C
- X 3 , X 4 and - ⁇ may be one of the preferred species listed above; most desirably all are preferred.
- the present invention provides an adenosine analogue-type A3 receptor agonist having an N6 nitrogen substituted by a group of the formula -CR 20 R 21 -CYCLE where R 2 *- 1 and R 2 --- are the same or different and H, F or CH3 ;
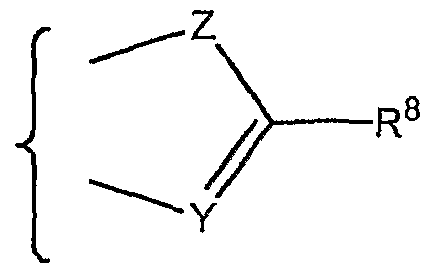
- CYCLE is:
- ring A is a 5- or 6- membered ring characterised by the following features (in which ring positions are numbered relative to the linkage to -CR 20 R 21 -) : i. a carbon atom at the 1-position; ii. carbon atom as CH or a nitrogen atom at position 2; iii. it is 3, 4 fused to ring B; iv. the 5-position ring atom is substituted by a moiety R ⁇ which is H, CH 3 , I, Br, Cl, CF 3 or less preferably OH or NH 2 .
- ring B is a 5 or 6 membered ring characterised by the following features :
- said in-ring heteroatom is joined within the ring secondly to a carbon which is substituted by a moiety R° which is H or another moiety wherein the number of ' atoms which are not hydrogen or halogen is no more than 10;
- the products of the invention include any compound capable of resulting in the delivery of such agonists to adenosine A3 receptors in vivo and include, therefore, salts and prodrugs of such agonists as well as the salts of such prodrugs.
- the term "product of the invention" is to be understood accordingly.
- the invention includes but is not limited to adenosine-5 ' - uronamides which are N6-monosubstituted by -CR R X -CYCLE.
- the uronamides may be ethyl or methyl uronamides .
- the adenosine-5 ' - uronamides may, for example, be 2-substituted by, amongst others, a small substituent such as Cl, Br, I, CH3 or CF3.
- R 1 is C1-C4 alkyl
- R 2 is " selected from hydrogen, halo (e.g. fluoro, chloro, bromo or iodo), CH3 , CF3 , an alkynyl radical of the formula
- R 4 where n is 0 or an integer of from 1 to 4, R 3 is hydrogen or hydroxy, and R 4 is selected from methyl, a substituted or unsubstituted phenyl, a substituted or unsubstituted naphthyl or Het where Het is a 5 or 6 membered heterocyclic aromatic or non- aromatic ring, optionally benzocondensed, containing 1 to 3 heteroatoms selected from oxygen, sulfur and nitrogen linked through a carbon atom or through a nitrogen atom;
- R5 is selected from hydrogen, halo, methyl and CF3; and R ⁇ is selected from hydrogen or amino;
- R 7 is selected from hydrogen, -OR 11 , -C0 2 R 1:L , -COR 11 and -CONR 11 where R 11 is C- ] __4 alkyl; or R ⁇ and R 7 , when taken together with the carbon atoms to which they are attached, form an oxazole ring in which the carbon between the oxygen and the nitrogen of the oxazole may optionally be substituted by an amine group having the formula
- R 9 and R 1 ⁇ 1 which may be the same or different is hydrogen, a C j _-C alkyl radical or a C- ⁇ -C4 alkenyl;
- R 9 and 1 ⁇ are the same.
- the oxazole compounds of formula I and the compounds of formula II are most preferred.
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier and an effective amount, e.g. a therapeutically effective amount, including a prophylactically effective amount, of one or more products of the invention.
- the present invention provides a method of selectively activating A3 adenosine receptors in a mammal, which, method comprises acutely or chronically administering to a mammal in need of selective activation of its A3 adenosine receptors a therapeutically effective amount, including a prophylactically effective amount, of one or more products of the invention.
- the present invention provides in one aspect adenosine A3 receptor agonists having a normally mono-substituted N6 nitrogen wherein the substituent is -CR 20 R 21 -CYCLE where R ⁇ and R 21 are the same or different and H, F and CH3; and CYCLE is:
- ring A is a 5- or 6- membered ring characterised by the following features (in which ring positions are numbered relative to the linkage to -CR ( ⁇ R -) : i. a carbon atom at the 1-position; ii . carbon atom in the form of CH- or a nitrogen atom at position 2 ; iii. it is 3 , 4 fused to ring B; iv. the 5-position ring atom is substituted by a moiety R ⁇ * which is H, CH3 , I, Br, Cl, CF3 or less preferably OH or NH . v.
- ring B is a 5 or 6 membered ring characterised by the following features:
- said in-ring heteroatom is joined within the ring secondly to a carbon which is substituted by a moiety R 8 which is H or another moiety wherein the number of atoms which are not hydrogen or halogen is no more than 10;
- the disclosed compounds can exist in different forms, such as salts and esters, for example, and the invention includes all variant forms of the compounds.
- the compounds may be in the form of acid addition salts which, for those compounds for pharmaceutical use, will be pharmaceutically acceptable.
- Exemplary acids include HBr, HCI and HSO2CH3.
- Certain compounds of the invention exist in different tautomeric forms and the invention includes all such tautomers.
- the invention includes prodrugs for the active pharmaceutical species of the invention, for example- in which one or more functional groups are protected or derivatised but can be converted in vivo to the functional group, as in the case of esterified hydroxy groups, for example.
- prodrug represents compounds which are transformed in vivo to the parent compound, for example, by hydrolysis in blood. A thorough discussion is provided in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, Vol. 14 of the A.C.S. Symposium Series, Edward B. Roche, ed. , Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, and Judkins, et al. Synthetic Communications, 26(23), 4351-4367 (1996), each of which is incorporated herein by reference.
- prodrug is to be widely interpreted and includes, inter alia, salts of covalent prodrug molecules .
- such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred. Lists of suitable salts are found in Remington ' s Pharmaceutical Sciences , 17th ed. , Mack Publishing Company, Easton, Pa., US, 1985, p. 1418, the disclosure of which is hereby incorporated by reference.
- the invention thus includes pharmaceutically-acceptable salts of the disclosed compounds and their covalent prodrug molecules wherein the parent compound is modified by making acid or base salts thereof, for example the conventional non-toxic salts or the quaternary ammonium salts which are formed, e.g., from inorganic or organic acids or bases.
- acid addition salts include acetate, adipate, alginate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, citrate, camphorate, camphorsulfonate, cyclopentanepropionate, digluconate, dodecylsulfate, ethanesulfonate, fumarate, glucoheptanoate , glycerophosphate, hemisulfate, heptanoate, hexanoate, hydrochloride, hydrobromide, hydroiodide, 2- hydroxyethanesulfonate, lactate, maleate, methanesulfonate, 2- naphthalenesulfonate, nicotinate, oxalate, pamoate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate
- Base salts include ammonium salts, alkali metal salts such as sodium and potassium salts, alkaline earth metal salts such as calcium and magnesium salts, salts with organic bases such as dicyclohexylamine salts, N-methyl-D-glucamine, and salts with amino acids such as arginine, lysine, and so forth.
- the basic nitrogen-containing groups may be quaternized with such agents as lower alkyl halides, such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl; and diamyl sulfates, long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, aralkyl halides like benzyl and phenethyl bromides and others .
- lower alkyl halides such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides
- dialkyl sulfates like dimethyl, diethyl, dibutyl
- diamyl sulfates long chain halides
- phrases "pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings or animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- Geometric isomers may exist in the products of the present invention.
- the present invention contemplates the various geometric isomers and mixtures thereof resulting from the arrangement of substituents around a carbon—carbon double bond and designates such isomers as of the Z or E configuration, wherein the term “Z” represents substituents on the same side of the carbo —carbon double bond and the term “E” represents substituents on opposite sides of the carbon—carbon double bond.
- the invention therefore includes all variant forms of the defined compounds, for example any substance which, upon administration, is capable of providing directly or indirectly a compound as defined above or providing a species which is capable of existing in equilibrium with such a compound.
- heteromatic ring refers to a ring system which has at least one (e.g. 1, 2 or 3) in-ring heteroatoms and has a conjugated in-ring double bond system.
- heteroatom includes oxygen, sulfur and nitrogen, of which sulfur is less preferred. Examples of such heteroaromatic rings can be seen in CYCLE moieties 1) to 10) below. Such rings are substantially planar.
- alkyl in this specification includes linear and branched alkyl groups, for example methyl, ethyl, n-propyl, isopropyl, tert-butyl, n-pentyl and n-hexyl.
- alkoxy includes groups of which the alkyl part may be linear or branched, for example one of those groups listed in the preceding sentence; alkylene groups may likewise be linear or branched and may, for example, correspond to one of those alkyl groups listed in the preceding sentence.
- the alkyl groups may be (but preferably are not) interrupted by one or more ether linkages .
- halogen herein includes reference to F, Cl, Br and I, of which Cl is often preferred. It will be understood that the invention specifically includes variants of preferred or exemplary compounds in which one or more moieties (e.g. substituents) have been replaced by alternatives described in this application.
- R 0 and R 21 are both the same and/or are H or F (usually both are H) •
- A is a 6-membered ring and B is a 5-membered ring
- R is preferably not H and is more usually -CH3, I or Br or less preferably -CF3 or Cl
- R 8 is H, -R 9 , -OR 9 , -SR 9 , -COR 9 , or more preferably -N0 2 ,
- NR 9 R 10 , -CHR 9 R 10 , -N CR 9 R 10 where R 9 and R 10 are the same, or less preferably different, and are C- ⁇ _-C4 alkyl, C-]_-C4 alkenyl or C-i_-C4 alkoxyalkyl and most preferably ethyl or especially methyl
- CYCLE is a bicyclic ring and more usually a structure of formula (V) where
- G is N, CH, CF, CCH 3 or less preferably CCF 3 ;
- M is preferably but not necessarily H
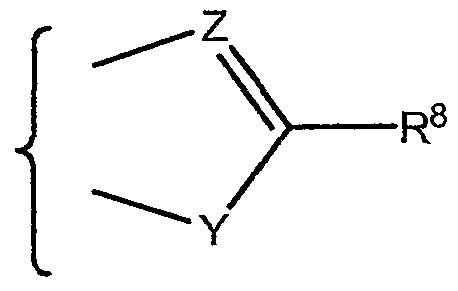
- the 5-membered ring is an oxazole and more preferably Y is 0 and Z is -N-.
- CYCLE moieties may be mentioned:
- R ⁇ a is CH3 or less preferably CF3 __
- any ring A shown above may be fused with any ring B shown above (e.g. ring A of structure 5 may be fused with ring B of structures 2, 6 or 10), e.g. to form a 5/6 (ring A is 5-membered, ring B 6-membered) , 6/6 or 6/5 fused ring.
- Structures 7 and 8 are particularly preferred CYCLE moieties.
- CYCLE is as shown in Formula (I) or Formula (II) (see above, under the heading "Brief Description of the Invention").
- -CR ⁇ R 21 is preferably -CH2-.
- a pre erred class of compounds of the invention have a 4 ' substituent of the formula:
- T is C-j_, C2 C3 or C4 alkyl and T 2 is H.
- a particularly preferred alkyl group is methyl .
- the C2 substituent is hydrogen in some preferred compounds . In other preferred compounds the C2 substituent is halogen, -CH3 or -CF
- the invention provides compounds of formula (III)
- X 1 is -CR 20 R 21 -CYCLE as described above; and D, E, X 2 , X 3 , X 4 and X- ⁇ are as described previously under the heading "Background of the Invention" .
- R and R 2 are as defined previously under the heading "Brief Description of the Invention" and CYCLE is as previously described and is most desirably a bicyclic ring as defined above.
- R is preferably C ] _ - C alkyl (e.g. ethyl or especially methyl), R 2 is preferably hydrogen, halo, methyl or trichloromethyl.
- R , R2 , R 4 and R ⁇ may be as follows:
- R 1 is methyl or ethyl, preferably methyl.
- R 2 is hydrogen, halogen, notably chloro, CH3 or
- R 2 may be an alkynyl radical of the formula
- R 3 is hydrogen.
- R 4 is selected from methyl, unsubstituted phenyl or a heterocyclic moiety selected from pyridyl, thienyl, furyl, imidazolyl, thiazolyl, pyrazoyl and triazoyl.
- Thienyl is the preferred heterocyclic radical .
- alkynyl radicals are - ⁇ -R 4 in which R 4 is unsubstituted phenyl or thienyl; -s- (CH2) n -CHR 3 R 4 where n is 2 , R 3 is hydrogen and R 4 is methyl or unsubstituted phenyl; and - ⁇ - (CH2) n -CHR R 4 where n is O, R 3 is hydroxy and R 4 is phenyl .
- R 4 is a substituted phenyl or a substituted naphthyl, this may be substituted with from 1 to 3 substituents selected from halo (fluoro, chloro, bromo and iodo) , C]_-Cg alkyl,
- C ⁇ -Cg haloalkyl C- ⁇ -Cg alkoxy, C ⁇ -C haloalkoxy, C2 ⁇ Cg alkoxycarbonyl, C2 ⁇ C alkoxyalkyl, C- ⁇ -Cg alkylthio, thio, CHO, cyanomethyl, nitro, cyano, hydroxy, carboxy, C2 ⁇ C acyl, amino, c l -c 3 monoalkylamino, C2 ⁇ C dialkylamino, methylenedioxy; aminocarbonyl .
- the preferred halo is chloro.
- the preferred C]_-Cg alkyl are methyl or ethyl.
- the preferred C ⁇ -Cg haloalkyl is trifluoromethyl.
- the preferred C ] _-Cg alkoxy are methoxy or ethoxy.
- the preferred C-j_-Cg haloalkoxy are trifluoromethoxy or difluoromethoxy.
- Tne preferrd C2-Cg alkoxycarbonyl are methoxycarbonyl or ethoxycarbonyl.
- the preferred C ⁇ -C alkoxyalkyl are methoxymethyl, methoxyethyl or ethoxymethyl.
- the preferred C- ⁇ -Cg alkylthio is methylthio.
- the preferred C2 ⁇ C acyl is acetyl.
- the preferred C -C3 monoalkylamino are methylamino, ethylamino, isopropylamino.
- the preferred C2-C5 dialkylamino are dimethylamino , diethylamino, methylethylamino, methylisopropylamino, diisopropylamin
- R ⁇ is selected from bromo, iodo and methyl.
- ⁇ iodo or methyl in first sub-class, R5 is iodo and in a second sub-class R- ⁇ is methyl.
- R- ⁇ is bromo .
- R ⁇ and R 7 are not joined together to form an oxazole ring.
- R 5 and R 7 may each be hydrogen.
- R ⁇ is hydrogen and R 7 is as defined above, preferably -COR 11 where R 11 is C-j__4 alkyl, for example methyl.
- R , R 2 , and R ⁇ are as defined above, R ⁇ is hydrogen or amino, and R 7 is selected from hydrogen, -OR 11 , -CO2R 11 , -COR 11 and -CONR 11 where R 11 is C- ] __ alkyl.
- R 1 is methyl or ethyl, more preferably methyl.
- R 2 is hydrogen, halogen notably chloro, CH3 or
- R 2 is the formula
- R 3 is hydrogen.
- R 4 is selected from methyl, unsubstituted phenyl or a heterocyclic moiety selected from pyridyl, thienyl, furyl, imidazolyl, thiazolyl, pyrazoyl and triazoyl.
- Thienyl is the preferred heterocyclic radical.
- R ⁇ is selected from bromo, iodo and methyl.
- R- ⁇ is iodo or methyl; in first sub-class, R ⁇ is iodo and in a second sub-class R ⁇ is methyl.
- R- ⁇ is bromo.
- R ⁇ and B may each be hydrogen.
- ⁇ may be hydrogen and R 7 be as defined above, preferably -COR 11 where R 11 is C ] __4 alkyl, for example methyl .
- One preferred group of compounds within this aspect of the invention are those in which:
- R 1 is methyl or ethyl, preferably methyl
- R 2 is H or halogen (e.g. chloro), CH3, CF3 or, less
- 'R 4 preferably, an alkynyl radical of the formula where n, R 3 are as defined above and R 4 is selected from methyl, unsubstituted phenyl or a heterocyclic moiety selected from pyridyl, thienyl, furyl, imidazolyl, thiazolyl, pyrazoyl and triazoyl .
- R-> is iodo or methyl or is bromo; R ⁇ and R 7 are hydrogen.
- R 2 may be - ⁇ -R 4 in which R 4 is unsubstituted phenyl or thienyl; - ⁇ - (CH2) n -CHR 3 R 4 where n is 2, R 3 is hydrogen and R 4 is methyl or unsubstituted phenyl; and - ⁇ - (CH2) n -CHR R 4 where n is
- R 3 is hydroxy and R 4 is phenyl.
- Compounds of this aspect of the invention include:
- R 1 , R , and R ⁇ are as defined above under the heading "Brief Description of the Invention"
- one of Y and Z is oxygen and the other of Y and Z is nitrogen
- R° is as defined above under the heading "Brief Description of the Invention” as defined above and where
- R 1 is methyl or ethyl, preferably methyl.
- R 2 is hydrogen, or halogen, notably chloro, CH3
- R 2 may be an alkynyl radical of the formula
- R 3 is hydrogen.
- R 4 is selected from methyl, unsubstituted phenyl or a heterocyclic moiety selected from pyridyl, thienyl, furyl, imidazolyl, thiazolyl, pyrazoyl and triazoyl.
- Thienyl is the preferred heterocyclic radical.
- R ⁇ is selected from bromo, iodo and methyl
- R ⁇ is iodo or methyl; in first sub-class, R5 is iodo and in a second sub-class R ⁇ is methyl.
- R5 is bromo .
- Y is oxygen and Z is nitrogen and in another, Y is nitrogen and Z is oxygen. It is presently preferred that Y is 0 and Z is N.
- R 1 is methyl or ethyl, preferably methyl
- R 2 is H, halogen (e.g. chloro) CH3 or CF3, or less preferably is an alkynyl radical of the formula
- R 4 is selected from methyl, unsubstituted phenyl or a heterocyclic moiety selected from pyridyl, thienyl, furyl, imidazolyl, thiazolyl, pyrazoyl and triazoyl (e.g.
- R5 is iodo, chloro, bromo or methyl;
- R ⁇ is iodo or methyl. In another class of compounds R- ⁇ is bromo. It is preferred that R 2 is H or halo .
- R 1 is C ⁇ -C4 alkyl
- R 2 is hydrogen, halo (e.g. chloro, bromo or iodo) CH3 or CF3, or less preferably is an alkynyl radical of the formula
- n is 0 or an integer of from 1 to 4
- R 3 is hydrogen or hydroxy
- R 4 is selected from methyl, a substituted or unsubstituted phenyl, a substituted or unsubstituted naphthyl or Het where Het is a 5 or 6 membered heterocyclic aromatic or non- aromatic ring, optionally benzocondensed, containing 1 to 3 heteroatoms selected from oxygen, sulfur and nitrogen linked through a carbon atom or through a nitrogen atom;
- R ⁇ is selected from hydrogen, halo, methyl or less preferably CF3 ;
- R8 is as defined above under the heading "Brief
- R 9 and R 1 ⁇ which may be the same or different, are selected from hydrogen, a C -C4 alkyl radical or a C -C4 alkenyl radical; one of Y and Z is nitrogen and the other of Y and Z is oxygen; and
- R 4 is a substituted phenyl or a substituted naphthyl, this may be with from 1 to 3 substituents selected from halo (fluoro, chloro, bromo and iodo) , C; ] _-Cg alkyl, C-j_-Cg haloalkyl, C- ] _-Cg alkoxy, C -Cg haloalkoxy, C2 ⁇ C alkoxycarbonyl,
- the preferred halo is chloro.
- the preferred C- ⁇ -Cg alkyl are methyl or ethyl.
- the preferred C-]_-Cg haloalkyl is trifluoromethyl.
- the preferred C- ] _-Cg alkoxy are methoxy or ethoxy.
- the preferred C ] _-Cg haloalkoxy are trifluoromethoxy or difluoromethoxy.
- Tne preferred C2 ⁇ C alkoxycarbonyl are methoxycarbonyl or ethoxycarbonyl.
- the preferred C2 ⁇ Cg alkoxyalkyl are methoxymethyl, methoxyethyl or ethoxymethyl.
- the preferred C--_-Cg alkylthio is methylthio.
- the preferred C2 ⁇ C acyl is acetyl .
- the preferred C1-C3 monoalkylamino are methylamino, ethylamino, isopropylamino.
- the preferred C2 ⁇ C dialkylamino are dimethylamine diethylamino, methylethylamino, methylisopropylamino, diisopropylamino .
- R is methyl or ethyl, preferably methyl.
- R 2 is hydrogen, halogen, notably chloro, CH3 or CF3.
- R 2 is an alkynyl radical of the formula
- R 3 is hydrogen.
- R 4 is selected from methyl, unsubstituted phenyl or a heterocyclic moiety selected from pyridyl, thienyl, furyl, imidazolyl, thiazolyl, pyrazoyl and triazoyl.
- Thienyl is the preferred heterocyclic radical.
- R ⁇ is selected from bromo, iodo and methyl.
- ⁇ is iodo or methyl; in first subclass, R5 is iodo and in a second sub-class R ⁇ is methyl.
- R ⁇ is bromo.
- Y is oxygen and Z is nitrogen and in another aspect, Y is nitrogen and Z is oxygen. It is presently preferred that Y is 0 and Z is N.
- the invention concerns compounds in which: R 1 is methyl or ethyl, preferably methyl;
- R 2 is H, halo (e.g. Cl, Br or I), CH3 or CF3 , or less • preferably is an alkynyl radical of the formula
- n, R 3 are as defined above and R 4 is selected from methyl, unsubstituted phenyl or a heterocyclic moiety selected from- pyridyl, thienyl, furyl, imidazolyl, thiazolyl, pyrazoyl and triazoyl (e.g.
- R 2 may be - ⁇ -R 4 in which R 4 is unsubstituted phenyl or thienyl; - ⁇ - (CH2) n -CHR 3 R 4 where n is 2 , R 3 is hydrogen and R 4 is methyl or unsubstituted phenyl; and - ⁇ - (CH2) n -CHR R 4 where n is O, R 3 is hydroxy and R 4 is phenyl) .
- R5 is iodo, chloro, bromo or methyl; and
- Y is 0 and Z is N. It is also preferred that R 2 is hydrogen or halo .
- R ⁇ is iodo or methyl; in a first sub-class R-> is iodo and in a second sub-class R ⁇ is methyl. In another class of compounds R ⁇ is bromo.
- Another aspect of this invention resides in methods of treating a mammal having a disease or condition mediated by an A3 adenosine receptor by administering a therapeutically effective amount of a product of the invention to the mammal .
- the present invention provides a method of selectively activating A3 adenosine receptors in a mammal, which method comprises acutely or chronically administering to a mammal in need of selective activation of its A3 adenosine receptors a therapeutically effective amount, including a prophylactically effective amount, of a compound which binds with the A3 receptor so as to stimulate an A3 receptor-dependent response .
- A3 adenosine receptor agonists can be used in the treatment of any disease, state or condition involving the release of cyclic adenosine monophosphate or the release of inositol-1, 4, 5-triphosphate (IP3), diacylglycerol (DAG), and free radicals and subsequent arachidonic acid cascades.
- IP3 inositol-1, 4, 5-triphosphate
- DAG diacylglycerol
- free radicals and subsequent arachidonic acid cascades can be treated in accordance with the present inventive method, wherein one of the above-described compounds is acutely administered, e.g., within about a few minutes to about an hour of the onset or realization of symptoms.
- the method also has utility in the treatment of chronic disease states and conditions, in particular those conditions and disease states wherein prophylactic or therapeutic administration of one of the above-described compounds will prevent the further onset of symptoms or will reduce recovery time.
- diseases states and conditions that may be treated in accordance with the present inventive method include inflammatory disorders, such as vascular inflammation and arthritis, allergies,- asthma, wound healing, stroke, cardiac infarct, cardiac failure, acute spinal cord injury, acute head injury or trauma, seizure, neonatal hypoxia (cerebral palsy; prophylactic treatment involves chronic exposure through placental circulation) , hypoxia and chronic hypoxia due to arteriovenous malformations and occlusive cerebral artery disease, severe neurological disorders related to excitotoxicity, Parkinson's disease, Huntington's chorea, and other diseases of the central nervous system (CNS) , cardiac disease, kidney disease, and contraception.
- Particular disease states which may be treated with the compounds of the invention are cardiac infarct and hypoxia.
- the above compounds may be used to treat malignant hypotension.
- the administration of IB- MECA results in a significant increase (e.g., about 10-30%) in basal or systemic blood pressure (e.g., from about 70 mm Hg to about 90 mm Hg) .
- the above compounds may also be used to treat and/or protect against a variety of disorders, including, for example, seizures, transient ischemic shock, strokes, focal ischemia originating from thrombus or cerebral hemorrhage, global ischemia originating from cardiac arrest, trauma, neonatal palsy, hypovolemic shock, and hyperglycemia and associated neuropathies .
- the present inventive method includes the administration to an animal, such as a mammal, particularly a human, in need of the desired A3 receptor-dependent response of an effective amount, e.g., a therapeutically effective amount, of one or more of the aforementioned present inventive compounds or pharmaceutically acceptable salts or derivatives thereof, alone or in combination with one or more other pharmaceutically active compounds .
- an effective amount e.g., a therapeutically effective amount
- the compounds of the invention will normally be administered orally, intravenously, subcutaneously, buccally, rectally, dermally, nasally, tracheally, bronchially, by any other parenteral route, as an oral or nasal " spray or via inhalation,
- the compounds may be administered in the form of pharmaceutical preparations comprising prodrug or active compound either as a free compound or, for example, a pharmaceutically acceptable non-toxic organic or inorganic acid or base addition salt, in a pharmaceutically acceptable dosage form.
- the compositions may be administered at varying doses .
- the most preferred routes of administration are injection and infusion, especially intravenous administration.
- the compounds of the invention may be combined and/or co- administered with any antithrombotic agent, such as the antiplatelet agents acetylsalicylic acid, ticlopidine, clopidogrel, thromboxane receptor and/or synthetase inhibitors, fibrinogen receptor antagonists, prostacyclin mimetics and phosphodiesterase inhibitors and ADP-receptor (P2 T) antagonists .
- any antithrombotic agent such as the antiplatelet agents acetylsalicylic acid, ticlopidine, clopidogrel, thromboxane receptor and/or synthetase inhibitors, fibrinogen receptor antagonists, prostacyclin mimetics and phosphodiesterase inhibitors and ADP-receptor (P2 T) antagonists .
- antithrombotic agent such as the antiplatelet agents acetylsalicylic acid, ticlopidine, clopidogrel, thromboxane receptor and/or synthet
- the compounds of the invention may be combined and/or co- administered with thrombolytics such as tissue plas inogen activator (natural, recombinant or modified) , streptokinase, urokinase, prourokinase, anisoylated plasminogen-streptokinase activator complex (APSAC) , animal salivary gland plasminogen activators, and the like, in the treatment of thrombotic diseases, in particular myocardial infarction.
- tissue plas inogen activator naturally, recombinant or modified
- streptokinase urokinase
- prourokinase prourokinase
- anisoylated plasminogen-streptokinase activator complex APSAC
- animal salivary gland plasminogen activators and the like
- the pharmaceutical compounds of the invention may be administered orally or parenterally ( “parenterally” as used herein, refers to modes of administration which include intravenous, intramuscular, intraperitoneal, intrasternal, subcutaneous and intraarticular injection and infusion of which intavenous is most preferred.) to a host to obtain a desired effect, for example protection against ischaemia or a cardioprotectant effect.
- parenterally refers to modes of administration which include intravenous, intramuscular, intraperitoneal, intrasternal, subcutaneous and intraarticular injection and infusion of which intavenous is most preferred.
- parenterally refers to modes of administration which include intravenous, intramuscular, intraperitoneal, intrasternal, subcutaneous and intraarticular injection and infusion of which intavenous is most preferred.
- parenterally refers to modes of administration which include intravenous, intramuscular, intraperitoneal, intrasternal, subcutaneous and intraarticular injection and infusion of which intavenous is most preferred.
- Actual dosage levels of active ingredients in the pharmaceutical compositions of this invention may be varied so as to obtain an amount of the active compound (s) that is effective to achieve the desired therapeutic response for a particular patient, compositions, and mode of administration.
- the selected dosage level will depend upon the activity of the particular compound, the route of administration, the severity of the condition being treated and the condition and prior medical history of the patient being treated. However, it is within the skill of the art to start doses of the compound at levels lower than required for to achieve the desired therapeutic effect and to gradually increase the dosage until the desired effect is achieved.
- Another aspect of this invention is directed to methods of reducing tissue damage (e.g., substantially preventing tissue damage, inducing tissue protection) resulting from ischemia or hypoxia comprising administering to a mammal in need of such treatment a therapeutically effective amount of a product of the invention.
- tissue damage e.g., substantially preventing tissue damage, inducing tissue protection
- Preferred ischemic/hypoxic tissues taken individually or as a group are cardiac, brain, liver, kidney, lung, gut, skeletal muscle, spleen, pancreas, nerve, spinal cord, retina tissue, the vasculature, or intestinal tissue, an especially preferred ischemic/hypoxic tissue is cardiac tissue.
- the products of the invention are administered to prevent perioperative myocardial ischemic injury.
- the products of this invention are administered prophylactically.
- the ischemic/hypoxic damage may occur during organ transplantation.
- the compounds of this invention are administered prior to, during or shortly after, cardiac surgery or non-cardiac surgery (e.g., a three to four day infusion).
- a product of the invention is administered locally.
- Another aspect of this invention is directed to methods of reducing myocardial tissue damage (e.g., substantially preventing tissue damage, inducing tissue protection) during surgery (e.g., coronary artery bypass grafting (CABG) surgeries, vascular surgeries, percutaneous transluminal coronary angioplasty (PTCA) or any percutaneous transluminal coronary intervention (PTCI) , organ transplantation, or other non-cardiac surgeries) comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- CABG coronary artery bypass grafting
- PTCA percutaneous transluminal coronary angioplasty
- PTCI percutaneous transluminal coronary intervention
- Another aspect of this invention is directed to methods of reducing myocardial tissue damage (e.g., substantially preventing tissue damage, inducing tissue protection) in patients presenting with ongoing cardiac syndromes (acute coronary syndromes, e.g., myocardial infarction or unstable angina) or cerebral ischemic events (e.g., stroke) comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- myocardial tissue damage e.g., substantially preventing tissue damage, inducing tissue protection
- ongoing cardiac syndromes e.g., myocardial infarction or unstable angina
- cerebral ischemic events e.g., stroke
- Another aspect of this invention is directed to chronic methods of reducing myocardial tissue damage (e.g., substantially preventing tissue damage, inducing tissue protection) in a patient with diagnosed coronary heart disease (e.g., previous myocardial infarction or unstable angina) or patients who are at high risk for myocardial infarction (e.g. age > 65 and two or more risk factors for coronary heart disease) comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- myocardial tissue damage e.g., substantially preventing tissue damage, inducing tissue protection
- a patient with diagnosed coronary heart disease e.g., previous myocardial infarction or unstable angina
- patients who are at high risk for myocardial infarction e.g. age > 65 and two or more risk factors for coronary heart disease
- Another aspect of this invention is directed to methods of preventing ischemic/hypoxic damage comprising the chronic oral administration to a mammal in need of such treatment of a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating cardiovascular diseases comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating arteriosclerosis comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating arrhythmia comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating angina pectoris comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating cardiac hypertrophy comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating renal diseases comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating diabetic complications comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating restenosis comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating organ hypertrophies or hyperplasias comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating septic shock and other inflammatory diseases (septicemia, endotoxcemia) comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- septic shock and other inflammatory diseases septicemia, endotoxcemia
- Another aspect of this invention is directed to methods for treating cerebro ischemic disorders comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating myocardial stunning comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating myocardial dysfunction comprising administering to a mammal a therapeutically effective amount of a product of the invention.
- Another aspect of this invention is directed to methods for treating cerebrovascular diseases comprising administering to a mammal a therapeutically effective amount of a product of the invention. Further applications of the products of the invention are described in the prior art documents mentioned under the heading "Background of the Invention" .
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier and an effective amount, e.g., a therapeutically effective amount, including a prophylactically effective amount, of one or more of the aforesaid compounds.
- the products of the invention may be formulated into pharmaceutical compositions as described in WO 95/02604, the contents of which are incorporated herein by reference.
- a pharmaceutical composition including a compound of the invention, in admixture with a pharmaceutically- acceptable adjuvant, diluent or carrier.
- Proposed compositions are intavenous formulations . These formulations typically contain a compound of the invention or a salt thereof.
- compositions of this invention for parenteral injection suitably comprise pharmaceutically acceptable sterile aqueous or nonaqueous solutions, dispersions, suspensions or emulsions as well as sterile powders for reconstitution into sterile injectable solutions or dispersions just prior to use.
- suitable aqueous and nonaqueous carriers, diluents, solvents or vehicles include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol and the like) , and suitable mixtures thereof, vegetable oils (such as olive oil) and injectable organic esters such as ethyl oleate.
- Proper fluidity can be maintained, for example, by the use of coating materials such as lecithin, by the maintenance of the required particle size in the case of dispersions and by the use of surfactants.
- These compositions may also contain adjuvants such as preservative, wetting agents, emulsifying agents and dispersing agents .
- Prevention of the action of microorganisms may be ensured by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutanol or phenol sorbic acid. It may also be desirable to include isotonic agents such as sugars or sodium chloride, for example.
- Prolonged absorption of the injectable pharmaceutical form may be brought about by the inclusion of agents (for example aluminum monostearate and gelatin) which delay absorption.
- the absorption of the drug in order to prolong the effect of the drug, it is desirable to slow the absorption of the drug from subcutaneous or intramuscular injection. This may be accomplished by the use of a liquid suspension of crystalline or amorphous material with poor water solubility. The rate of absorption of the drug then depends upon its rate of dissolution which, in turn, may depend upon crystal size and crystalline form. Alternatively, delayed absorption of a parenterally administered drug form is accomplished by dissolving or suspending the drug in an oil vehicle.
- Injectable depot forms are suitably made by forming microencapsule matrices of the drug in biodegradable polymers, for example polylactide-polyglycolide. Depending upon the ratio of drug to polymer and the nature of the particular polymer employed, the rate of drug release can be controlled. Examples of other biodegradable polymers include poly(orthoesters) and poly (anhydrides) . Depot injectable formulations may also prepared by entrapping the drug in liposomes or microemulsions which are compatible with body tissues.
- the injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable media just prior to use.
- Solid dosage forms for oral administration include capsules, tablets, pills, powders and granules.
- the active compound is typically mixed with at least one inert, pharmaceutically acceptable excipient or carrier such as sodium citrate or dicalcium phosphate and/or one or more: a) fillers or extenders such as starches, lactose, sucrose, glucose, mannitol and silicic acid; b) binders such as carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidone, sucrose and acacia; c) humectants such as glycerol; d) disintegrating agents such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates and sodium carbonate; e) solution retarding agents such as paraffin; f) absorption accelerators such as quaternary ammonium compounds; g) wetting agents such as cetyl alcohol and glycerol monostearate;
- the dosage form may also comprise buffering agents.
- Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycol, for example.
- oral formulations contain a dissolution aid.
- the dissolution aid is not limited as to its identity so long as it is pharmaceutically acceptable.
- examples include nonionic surface active agents , such as sucrose fatty acid esters , glycerol fatty acid esters, sorbitan fatty acid esters (e.g., sorbitan trioleate) , polyethylene glycol, polyoxyethylene hydrogenated castor oil, polyoxyethylene sorbitan fatty acid esters, polyoxyethylene alkyl ethers, methoxypolyoxyethylene alkyl ethers, polyoxyethylene alkylphenyl ethers, polyethylene glycol fatty acid esters, polyoxyethylene alkylamines, polyoxyethylene alkyl thioethers, polyoxyethylene polyoxypropylene copolymers, polyoxyethylene glycerol fatty acid esters, pentaerythritol fatty acid esters, propylene glycol monofatty acid esters , polyoxyethylene propylene glycol monofatty acid esters,
- the solid dosage forms of tablets, dragees, capsules, pills, and granules can be prepared with coatings and shells such as enteric coatings and other coatings well known in the pharmaceutical formulating art . They may optionally contain opacifying agents and may also be of a composition such that they release the active ingredient (s) only, or preferentially, in a certain part of the intestinal tract, and/or in delayed fashion. Examples of embedding compositions which can be used include polymeric substances and waxes.
- the products of the invention may also be in micro- encapsulated form, if appropriate, with one or more of the above-mentioned excipients .
- the active compound may be in finely divided form, for example it may be micronised.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art such as water or other solvents, solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, dimethyl formamide, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor, and sesame oils) , glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan and mixtures thereof.
- inert diluents commonly used in the art such as water or other solvents, solubilizing agents and emulsifiers such as ethyl alcohol
- the oral compositions may also include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring and perfuming agents.
- Suspensions in addition to the active compounds, may contain suspending agents such as ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar, and tragacanth and mixtures thereof.
- compositions for rectal or vaginal administration are preferably suppositories which can be prepared by mixing the compounds of this invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at room temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at room temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- Compounds of the present invention can also be administered in the form of liposomes.
- liposomes are generally derived from phospholipids or other lipid substances. Liposomes are formed by mono- or multi- lamellar hydrated liquid crystals which are dispersed in an aqueous medium.
- any non-toxic, physiologically acceptable and metabolisable lipid capable of forming liposomes can be used.
- the present compositions in liposome form can contain, in addition to a compound of the present invention, stabilisers, preservatives, excipients and the like.
- the preferred lipids are the phospholipids and the phosphatidyl cholines (lecithins) , both natural and synthetic. Methods to form liposomes are known in the art, for example, Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, N.Y. (1976) , p 33 et seq.
- Dosage forms for topical administration of a compound of this invention include powders, sprays, ointments and inhalants.
- the active compound is mixed under sterile conditions with a pharmaceutically acceptable carrier and any needed preservatives, buffers or propellants which may be required.
- Ophthalmic formulations, eye ointments, powders and solutions are also contemplated as being within the scope of this invention.
- the compounds of the invention are orally active, have rapid onset of activity and low toxicity.
- the compounds of the invention have the advantage that they may be more efficacious, be less toxic, be longer acting, have a broader range of activity, be more potent, produce fewer side effects, be more easily absorbed than, or that they may have other useful pharmacological properties over, compounds known in the prior art .
- Suitable doses and dosage regimens can be determined by conventional range-finding techniques known to those of ordinary skill in the art. Generally, treatment is initiated with smaller dosages, which are less than the optimum dose of the compound. Thereafter, the dosage is increased by small increments until the optimum effect under the circumstances is reached. For convenience, the total daily dosage may be divided and administered in portions during the day if desired.
- the compounds of the invention may be administered by any suitable means. A preferred method of administration is by IV injection.
- the present invention provides for a wide range of selective A3 receptor-dependent responses .
- Exemplary dosages range from about 0.1 to about 100 mg/kg body weight of the animal being treated/day.
- Therapeutically effective dosages range from about 0.01 to about 10 mg/kg body weight/day.
- This invention is also directed to pharmaceutical compositions which comprise a therapeutically effective amount of a product of the invention and a pharmaceutically acceptable carrier, vehicle or diluent.
- This invention is also directed to pharmaceutical compositions for the reduction of tissue damage resulting from ischemia or hypoxia which comprise a therapeutically effective amount of a product of the invention.
- kits for use in treating a mammal having or at risk of having a disease or condition resulting from, for example, ischemia or hypoxia which may be ameliorated by an A3 agonist comprises a) a suitable dosage form, such as, for example, an injectable parenteral solution particularly adapted for intravenous or intramuscular injection, comprising a compound of Formula I; and b) instructions describing a method of using the dosage form to reduce tissue damage resulting from ischemia or hypoxia.
- Yet another aspect of this invention is combinations of a product of the invention and one or more other compounds as described below.
- This invention is also directed to a pharmaceutical combination composition comprising: a therapeutically effective amount of a composition comprising a first product, said first product being a product of the invention; a second compound, said second compound being a cardiovascular agent; and, optionally, a pharmaceutical carrier, vehicle or diluent.
- tissue damage e.g., substantially preventing tissue damage, inducing tissue protection
- administering to a mammal a first product, said first compound being a product of the invention; and a second product, said second product being a cardiovascular agent wherein the amounts of the first and second compounds result in a therapeutic effect.
- kits comprising: a. a product of the invention and a pharmaceutically- acceptable carrier, vehicle or diluent in a first unit dosage form; b. a cardiovascular agent and a pharmaceutically acceptable carrier, vehicle or diluent in a second unit dosage form; and c. means for containing said first and second dosage forms wherein the amounts of the first and second compounds result in a therapeutic effect.
- the invention therefore includes methods of treatment in which a product of the invention and one or more other therapeutic agents are administered to a mammal. Also included are products including both a product of the invention and one or more other therapeutic agents .
- Said other therapuetic agent (s) e.g., agents having a cardiovascular effect
- metformin or other 5 adenosine A3 receptor agonists.
- cardiovascular agents include angiotensin II (All) receptor antagonists, C5a inhibitors, soluble complement receptor type 1 (sCRl) or analogues, partial fatty acid oxidation (PFOX) inhibitors (specifically, ranolazine) , acetyl CoA carboxylase activators, alonyl CoA decarboxylase inhibitors, 5' AMP- activated protein kinase (AMPK) inhibitors, adenosine nucleoside inhibitors, anti-apoptotic agents (e.g., caspase inhibitors), monophosphoryl lipid A or analogues, nitric oxide synthase activators/inhibitors, protein kinase C activators (specifically, protein kinase E) , protein kinase delta inhibitor, poly (ADP ribose) synthetase (PARS, PARR) inhibitors, metformin
- a patient is administered, in effective amounts, a product of the invention and a thrombolytic. Sometimes, but not always, one of these two active agents is administered more or less immediately after the other.
- This invention is also directed to a pharmaceutical combination composition comprising: a therapeutically effective amount of a composition comprising a first product, said first product being a product of the invention; a second product, said second product being a glycogen phosphorylase inhibitor; and, optionally, a pharmaceutical carrier, vehicle or diluent.
- tissue damage e.g., substantially preventing tissue damage, inducing tissue protection
- a mammal a. a first compound, said first compound being a product of the invention said second compound being a glycogen phosphorylase inhibitor wherein the amounts of the first and second compounds result in a therapeutic effect.
- kits comprising: a. a product of the invention and a pharmaceutically acceptable carrier, vehicle or diluent in a first unit dosage form; b. a glycogen phosphorylase inhibitor and a pharmaceutically acceptable carrier, vehicle or diluent in a second unit dosage form; and b. means for containing said first and second dosage forms wherein the amounts of the first and second compounds result in a therapeutic effect.
- This invention is also directed to a pharmaceutical combination composition
- a pharmaceutical combination composition comprising: a therapeutically effective amount of a composition comprising a product of the invention; an aldose reductase inhibitor; and, optionally, a pharmaceutical carrier, vehicle or diluent.
- Another aspect of this invention are methods of reducing tissue damage (e.g., substantially preventing tissue damage, inducing tissue protection) resulting from or which could result from ischemia or hypoxia comprising administering to a mammal a. a product of the invention and c. an aldose reductase inhibitor wherein the amounts of said product and said inhibitor result in a therapeutic effect.
- kits comprising: a.
- a preferred aldose reductase inhibitor is zopolrestat : , 3 , 4-dihydro-4-oxo-3- [ [5-trifluoromethyl) -2-benzothiazolyl] methyl] -1-phthalazineacetic acid.
- ischemic or hypoxic tissues taken individually or as a group are wherein the ischemic/hypoxic tissue is cardiac, brain, liver, kidney, lung, gut, skeletal muscle, spleen, pancreas, nerve, spinal cord, retina tissue, the vasculature, or intestinal tissue.
- An especially preferred ischemic or hypoxic tissue is cardiac tissue.
- the combinations are administered to prevent perioperative myocardial ischemic injury.
- the combinations of this invention are administered prophylactically.
- the ischemic/hypoxic damage may occur during organ transplantation.
- the combinations of this invention are administered prior to, during and/or shortly after, cardiac surgery or non-cardiac surgery. In one aspect of this invention the combinations are administered locally.
- myocardial tissue damage is reduced during or after surgery.
- myocardial tissue damage is reduced in patients presenting with ongoing cardiac or cerebral ischemic events.
- myocardial tissue damage is reduced by chronic administration of the above combinations in a patient with diagnosed coronary heart disease.
- the term "reduction” is intended ' to include partial prevention or prevention which, although greater than that which would result from taking no compound or from taking a placebo, is less than 100% in addition to substantially total prevention.
- the term "damage resulting from ischemia or hypoxia "as employed herein refers to conditions directly associated with reduced blood flow or oxygen delivery to tissue, for example due to a clot or obstruction of blood vessels which supply blood to the subject tissue and which result, inter alia, in lowered oxygen transport to such tissue, impaired tissue performance, tissue dysfunction and/or necrosis and/or apoptosis.
- the oxygen carrying capacity of the blood or organ perfusion medium may be reduced, e.g., in a hypoxic environment, such that oxygen supply to the tissue is lowered, and impaired tissue performance, tissue dysfunction, and/or tissue necrosis and/or apoptosis ensues.
- treating includes preventative (e.g., prophylactic) and palliative treatment .
- Compounds of the invention may be synthesized by any suitable means.
- synthesis of adenosine analogues is well known in the art and is described in the documents listed above under the heading "Background of the Invention".
- guidance may be found in the "Compound Synthesis” sections of WO 95/02604 as well as of corresponding US 5773423 and US 5688774, which sections are included herein by reference.
- the reader is also referred to reaction schemes A to I and examples of WO 92/05177 and corresponding US 5561134 and US 5736554, all of which disclosures are incorporated herein by reference.
- a first method comprises reacting a compound of the formula L-CR 0R 1 -CYCLE, where L is a leaving group, with a compound H2 -AR , where the nitrogen of H2 - is the N6 nitrogen of an adenosine A3 receptor agonist and ARA represents the remainder of the adenosine A3 receptor agonist.
- a second method comprises reacting a compound of the formula H 2 N -CR 20 R 21 -CYCLE with a compound of the formula
- C6-L-ARA where ARA again represents the residue of an adenosine A3 receptor agonist, excluding the N6 nitrogen, and C6-L represents a leaving" group substituted on the C6 carbon of ARA.
- reactive functional groups of ARA e.g. hydroxy or amino groups constituting X 3 and X 4 of formula III
- X 3 and X 4 of formula III may be protected.
- Suitable leaving groups include chloro and bromo. Chloro is often convenient- for the second method, as in the case of an
- ARA residue as illustrated by formula IV in which R 2 is H and R 1 is Me.
- This method uses as a starting material the known 5 ' -N- alkylcarboxamidoadenosines such as 5 ' -N- methylcarboxamidoadenosine, which may be protected as necessary prior to reaction.
- the 2-picolyl reactant may be made using the reaction scheme of Figure 1, which may be generalised where necessary.
- the 2-alkenyl substituted compounds of the invention may be synthesised using the synthetic reaction scheme illustrated in Figure 2, which may be generalized where necessary.
- the 6-chloro-2-iodopurine-9-riboside (Compound A) is already known, J. Med. Chem., 2000, vol43 , page 4137.
- step 1 this is protected in a manner known per se to yield compound B which is then oxidised to give acid C ⁇ Acid C is reacted with the R 1 amine (R- ⁇ - ⁇ ) to give uronamide D.
- the 2 iodo uronamide is then reacted with triethylamine, bis (triphenylphosphine)palladium dichloride and Cui in catalytic amount using as solvent a mixture of acetonitrile/DMF 2:1 and to this mixture is added the terminal alkyne and the reaction takes place under N2 atmosphere at roo . temperature (ref J. Med. Chem. 1995, vol 38, 1462-1472) to result in compound E.
- the coupling of the 2-picolylamine with the 6-chloro derivative is then accomplished as described above (compound F) , and deprotection with HCI IN at 70 °C yields the final compound, G.
- the alkyne used in the phenyl alkyne; other alkynes can be used in analogous ,manner.
- the alkenyl compounds may be prepared analogously.
- Each P is a protecting group, which may be taken together to represent a bridging protecting group such as an isopropylidene radical.
- the protecting group may be removed by conventional means, for example by treatment with an acid.
- L is a leaving group which may for example be selected from chloro, bromo or iodo or tosylates. Preferably the leaving group is chloro .
- This method thus uses as a starting material the known 2 ' , 3 ' -0-isopropylidene-6-chloropurine-5 ' -alkyluronamide and equivalents.
- This reaction step is also shown more specifically in the reaction scheme of Figure 4.
- the benzoxazole reactant may be made using the reaction scheme of Figure 3 , which may be generalised where necessary.
- the 2-alkynyl substituted compounds of the invention may be synthesised using the synthetic reaction scheme . illustrated in Figure 5 which may be generalized where necessary.
- 2 ' ,3 ' -O-Isopropylideneguanosine-5 ' -carboxylic acid A (J " . Org. Chem. 1999, 64, 293-295) is reacted with triethylamine, isopropenylchloroformate and methylamine at 0 S C yielding compound B.
- This is reacted with phosphoryl chloride to obtain compound C.
- C is treated with isoamyl nitrite, Cui, CH2I2 a d I2 to give compound D.
- the 2 iodo uronamide is then reacted with triethylamine, bis (triphenylphosphine) palladium dichloride and Cui in catalytic amount using as solvent a mixture of acetonitrile/DMF 2:1 and to this mixture is added the terminal alkyne and the reaction takes place under 2 atmosphere at room temperature (ref J. Med. Chem. 1995, vol 38, 1462-1472) to result in compound E.
- the coupling of the benzoxazole reactant with the 6-chloro derivative is then accomplished as described above.
- the alkyne used in the phenyl alkyne; other alkynes can be used in analogous manner.
- the alkenyl compounds may be prepared analogously.
- Aqueous H 2 0 2 (30%, 2.6 mL) was added to 2,4-lutidine (5 mL, 43.2 mmol) in acetic ' acid (15 mL) , and the mixture was stirred for 3 hours at 90 -C. The mixture was cooled, and a . second portion of aqueous H 2 0 2 (30%, 1.1 mL) was added, after which the mixture was stirred for another 20 hours at 90 a C. The solvent was evaporated (toluene was used to remove remaining traces of acetic acid by means of azeotropic destination) . The pH was adjusted to 10 with NaOH 10 M, CH 3 CN was added (10 mL) and precipitated materials were filtered off.
- N-Methyl-1' -deoxy-1' - [6-chloro-2- (2-phenyl-l-ethynyl) -9H- purin-9-yl] -2 ' , 3 ' -O-isopropylidene- ⁇ -D-ribofuranuronamide (210 mg, 0.47 mmol), (4-iodo-2-pyridyl)methylamine (219 mg, 0.93 mmol), and triethylamine (0.2 mL, 1.41 mmol) were dissolved in absolute ethanol (3.0 L) . The solution was stirred at 65 a C for 16 h in a sealed vessel.
- Examples 1 to 8 were evaluated for their pharmacological effect. These compounds were firstly evaluated for binding to human A 3 receptors expressed in Chinese Hamster Ovary cells (CHO cells). [ 125 I] -AB-MECA (0.3nM) binding to membrane preparations was examined using 60 min incubation at room temperature. The displacement of binding by the adenosine analogues was determined, non-specific binding being measured from the displacement by IB-MECA (10" 7 M) .
- Ai receptor functional activity was determined from the negative inotropic response of guinea-pig isolated paced left atria set up in tissue baths containing Krebs-bicarbonate solution gassed with 5% C0 2 in oxygen at 37°C. Dose-related inhibition of atrial developed tension (negative inotropy) was observed commencing at 300nM. The IC 50 value (concentration for 50% inhibition of contractions) was 7500nM.
- Compound 2's A 2 receptor functional activity was determined from the relaxation response of guinea-pig isolated tracheal spirals set up in tissue baths containing Krebs-bicarbonate solution gassed with 5% C0 2 in oxygen at 37°C. The tissue was pre-contracted with carbachol (lOOnM) and when the tension had reached a plateau, increasing concentrations of compound 9 were introduced cumulatively. There were dose-related relaxations indicative of A 2 receptor activity commencing at l ⁇ M. The IC 50 (concentration for 50% inhibition of the carbachol-induced contraction) was 2O,00OnM.
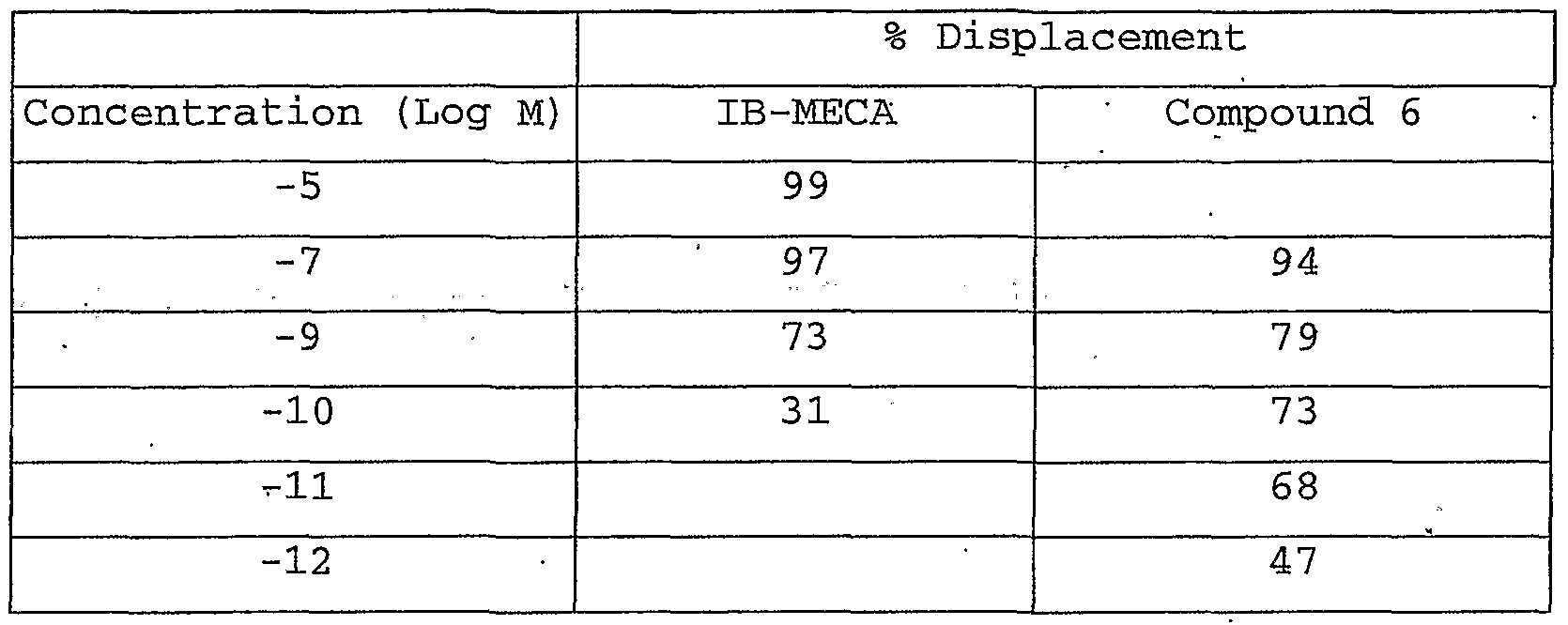
- Table 2 shows displacement of [ 125 I] -AB-MECA (0.3nM) from human A 3 receptors transfected into Chinese Hamster Ovary (CHO) cells by IB-MECA and compound 6.
- Table 3 shows the activity of compound 6 at Ai receptors of guinea-pig atria compared with the standard non-selective agonist, NECA (N-ethylcarboxamidoadenosine) .
- Table 4 shows the activity of compound 6 at A 2 receptors of guinea-pig trachea compared with the standard non-selective agonist, NECA (N-ethylcarboxamidoadenosine) .
- the activity is expressed as the relaxation of the trachea precontracted with carbachol (lOOnM) obtained in cumulative concentration-response curves.
- carbachol carbachol
- a maximum effective concentration of NECA was added and the responses to compound 6 were expressed as a percentage of this maximum response.
- the IC25 concentration concentration for 25% of the maximum response to NECA was then calculated.
- a full concentration-response curve for NECA was obtained and the IC25 value for NECA calculated. This data is represented graphically in Figure 7.
- Compound 6 has picomolar potency for binding to the human A 3 receptor. Based on functional tests in atrial and tracheal tissues, the selectivity over Ai and A 2 receptors is 6.25x10 s and 5.6xl0 6 , respectively.
- Guinea-pig isolated left atria were set up in tissue baths - containing Krebs-bicarbonate solution gassed with 5% C0 2 in " oxygen at 37°C and electrically paced at 2Hz with pulses of threshold voltage +50% and- 5ms pulse width. Developed tension was recorded. After equilibrium, they were exposed to 30 min of simulated ischaemia by gassing with 5% C0 2 in nitrogen and removing the glucose substrate which was replaced with choline chloride (7mM) to maintain isotonicity. Pacing was continued throughout. After 30 min, the tissues were reoxygenated and glucose was returned.
- Table 5 shows a comparison of the effects of IB-MECA and compound 6 on atrial- contractile function after 30 min simulated ischaemia. This data is also represented graphically in Figure 8.
- Guinea pig hearts were perfused by the Langendorff method.
- the cut aortic stump was perfused reterogradely with Krebs r solution gassed with 5% C0 2 in oxygen at 37°C at a constant flow rate of 7ml/min to perfuse the coronary circulation.
- the heart was jacketed at 37°C and the spontaneous force of contraction was measured by attaching a clip to the apex of the heart which was connected. to a tension transducer. Coronary perfusion pressure was also monitored.
- the pK a values for compounds 6 and 7 are given in Table 6 below.
- the partition coefficient profile for compound 6 is given in Table 7 below.
- the partition measurements were based on- a long chain ester (propylene glycol dipelargonate-PGDP) /water model.
- Table 6 shows pK a values of compounds 6 and 7 calculated using methanol and dimethylformamide co-solvents respectively. All experiments carried out in ionic strength water (0.15 KC1) .
- Table 7 shows Log P value of compound 6 (partition solvent: propylene glycol dipelargonate (PGDP) .
Abstract
Description





































































Claims































Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003561614A JP2006502088A (en) | 2002-01-25 | 2003-01-27 | Compounds useful as A3 adenosine receptor agonists |
| EP20030700933 EP1469864A1 (en) | 2002-01-25 | 2003-01-27 | Compounds useful as a-3 adenosine receptor agonists |
| CA002474337A CA2474337A1 (en) | 2002-01-25 | 2003-01-27 | Compounds useful as a3 adenosine receptor agonists |
| US10/899,625 US7414036B2 (en) | 2002-01-25 | 2004-07-26 | Compounds useful as A3 adenosine receptor agonists |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0201849.7 | 2002-01-25 | ||
| GB0201849A GB0201849D0 (en) | 2002-01-25 | 2002-01-25 | A�- adenosine receptor agonists |
| GB0201919A GB0201919D0 (en) | 2002-01-28 | 2002-01-28 | A�-Adenosine receptor agonists |
| GB0201919.8 | 2002-01-28 | ||
| GB0212438.6 | 2002-05-29 | ||
| GB0212438A GB0212438D0 (en) | 2002-05-29 | 2002-05-29 | Compounds |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/899,625 Continuation-In-Part US7414036B2 (en) | 2002-01-25 | 2004-07-26 | Compounds useful as A3 adenosine receptor agonists |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003061670A1 true WO2003061670A1 (en) | 2003-07-31 |
Family
ID=27617147
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2003/000304 WO2003061670A1 (en) | 2002-01-25 | 2003-01-27 | Compounds useful as a3 adenosine receptor agonists |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP1469864A1 (en) |
| JP (1) | JP2006502088A (en) |
| CA (1) | CA2474337A1 (en) |
| WO (1) | WO2003061670A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005012323A2 (en) * | 2003-07-31 | 2005-02-10 | Trigen Limited | Compounds useful as a3 adenosine receptor agonists |
| US7737126B2 (en) | 2004-05-24 | 2010-06-15 | Glaxo Group Limited | Purine derivative |
| WO2011002917A1 (en) | 2009-06-30 | 2011-01-06 | Pgxhealth, Llc | Alkoxy-carbonyl-amino-alkynyl-adenosine compounds and derivatives thereof as a2a r agonists |
| US7985740B2 (en) | 2005-07-19 | 2011-07-26 | Glaxo Group Limited | Purine derivatives as agonists of the adenosine A2A receptor |
| WO2020247546A1 (en) | 2019-06-03 | 2020-12-10 | Biointervene, Inc. | Adenosine analogs for the treatment of disease |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE504592T1 (en) | 2006-03-27 | 2011-04-15 | Otsuka Chemical Co Ltd | TREHALOSE COMPOUND AND DRUGS CONTAINING THE COMPOUND |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0704215A2 (en) * | 1994-06-02 | 1996-04-03 | Takeda Chemical Industries, Ltd. | Inhibitor of vascular permeability enhancer |
| US5688774A (en) * | 1993-07-13 | 1997-11-18 | The United States Of America As Represented By The Department Of Health And Human Services | A3 adenosine receptor agonists |
| EP1241176A1 (en) * | 2001-03-16 | 2002-09-18 | Pfizer Products Inc. | Purine derivatives for the treatment of ischemia |
-
2003
- 2003-01-27 EP EP20030700933 patent/EP1469864A1/en not_active Withdrawn
- 2003-01-27 CA CA002474337A patent/CA2474337A1/en not_active Abandoned
- 2003-01-27 JP JP2003561614A patent/JP2006502088A/en active Pending
- 2003-01-27 WO PCT/GB2003/000304 patent/WO2003061670A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5688774A (en) * | 1993-07-13 | 1997-11-18 | The United States Of America As Represented By The Department Of Health And Human Services | A3 adenosine receptor agonists |
| EP0704215A2 (en) * | 1994-06-02 | 1996-04-03 | Takeda Chemical Industries, Ltd. | Inhibitor of vascular permeability enhancer |
| EP1241176A1 (en) * | 2001-03-16 | 2002-09-18 | Pfizer Products Inc. | Purine derivatives for the treatment of ischemia |
Non-Patent Citations (1)
| Title |
|---|
| LI, HESHUI ET AL: "Synthesis of 6-(5-hydroxy-2-pyridylamino)-9-ribofuranosylpurine and its derivatives", ZHONGGUO YAOWU HUAXUE ZAZHI (1998), 8(2), 116-121, 1998, XP009010054 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005012323A2 (en) * | 2003-07-31 | 2005-02-10 | Trigen Limited | Compounds useful as a3 adenosine receptor agonists |
| WO2005012323A3 (en) * | 2003-07-31 | 2005-05-26 | Trigen Ltd | Compounds useful as a3 adenosine receptor agonists |
| US7737126B2 (en) | 2004-05-24 | 2010-06-15 | Glaxo Group Limited | Purine derivative |
| US7985740B2 (en) | 2005-07-19 | 2011-07-26 | Glaxo Group Limited | Purine derivatives as agonists of the adenosine A2A receptor |
| WO2011002917A1 (en) | 2009-06-30 | 2011-01-06 | Pgxhealth, Llc | Alkoxy-carbonyl-amino-alkynyl-adenosine compounds and derivatives thereof as a2a r agonists |
| WO2020247546A1 (en) | 2019-06-03 | 2020-12-10 | Biointervene, Inc. | Adenosine analogs for the treatment of disease |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2006502088A (en) | 2006-01-19 |
| EP1469864A1 (en) | 2004-10-27 |
| CA2474337A1 (en) | 2003-07-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7414036B2 (en) | Compounds useful as A3 adenosine receptor agonists | |
| WO2005012323A2 (en) | Compounds useful as a3 adenosine receptor agonists | |
| CN109803972B (en) | 1H-imidazole [4,5-H ] quinazoline compound as protein kinase inhibitor | |
| JP6802251B2 (en) | Cyanopyrrolidine as a DUB inhibitor for the treatment of cancer | |
| CA2976741C (en) | 1-cyano-pyrrolidine compounds as usp30 inhibitors | |
| AU2013305390C1 (en) | Dihydropyrimidine compounds and their application in pharmaceuticals | |
| EP3131897B1 (en) | Factor ixa inhibitors | |
| JP2021522275A (en) | 2-Amino-pyridine or 2-amino-pyrimidine derivative as a cyclin-dependent kinase inhibitor | |
| JP2019534261A (en) | Spiro ring compounds | |
| US10493060B2 (en) | Spirocyclic compounds | |
| JP7139568B2 (en) | Dihydropyrimidine compound and its preparation method and use | |
| CA3120514A1 (en) | Cyclic ureas | |
| KR19990023007A (en) | Aromatic compounds and pharmaceutical compositions containing them | |
| WO2022087011A1 (en) | Bicyclic compounds | |
| CA3134635A1 (en) | Pyrrole compounds | |
| JP2022531899A (en) | Modified cyclic dinucleoside compound as a STING modulator | |
| WO2003061670A1 (en) | Compounds useful as a3 adenosine receptor agonists | |
| KR20230144550A (en) | Inhibitors of cGAS activity as therapeutic agents | |
| AU2003202076A1 (en) | Compounds useful as A3 adenosine receptor agonists | |
| JP2023532027A (en) | Novel compound and pharmaceutical composition for prevention or treatment of resistant cancer containing the same | |
| TW202322808A (en) | Compounds, compositions and methods of treating disorders | |
| US20230374008A1 (en) | Bicyclic compounds | |
| TW202409010A (en) | Inhibitors of human respiratory syncytial virus and metapneumovirus |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003202076 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003561614 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1024/KOLNP/2004 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2474337 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10899625 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003700933 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003700933 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 165930 Country of ref document: IL |