WO2003037896A1 - N-azabicyclo-substituted hetero-bicyclic carboxamides as nachr agonists - Google Patents
N-azabicyclo-substituted hetero-bicyclic carboxamides as nachr agonists Download PDFInfo
- Publication number
- WO2003037896A1 WO2003037896A1 PCT/US2002/031579 US0231579W WO03037896A1 WO 2003037896 A1 WO2003037896 A1 WO 2003037896A1 US 0231579 W US0231579 W US 0231579W WO 03037896 A1 WO03037896 A1 WO 03037896A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- azabicyclo
- carboxamide
- alkyl
- hept
- substituted
- Prior art date
Links
- 0 CC(N)=C(CCC*=*)N Chemical compound CC(N)=C(CCC*=*)N 0.000 description 3
- CFGKWSDAMXTRHE-ONGXEEELSA-N C[C@@H](c1ccccc1)N(C[C@H](C1)C(O)=O)C1=O Chemical compound C[C@@H](c1ccccc1)N(C[C@H](C1)C(O)=O)C1=O CFGKWSDAMXTRHE-ONGXEEELSA-N 0.000 description 1
- MAKCHOLNXRNXPA-UHFFFAOYSA-N O=C(c1ccc(CCC2)c2c1)NC12NC1N1CC2CC1 Chemical compound O=C(c1ccc(CCC2)c2c1)NC12NC1N1CC2CC1 MAKCHOLNXRNXPA-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
Definitions
- Nicotinic acetylcholine receptors play a large role in central nervous system (CNS) activity. Particularly, they are known to be involved in cognition, learning, mood, emotion, and neuroprotection. There are several types of nicotinic acetylcholine receptors, and each one appears to have a different role in regulating CNS function. Nicotine affects all such receptors, and has a variety of activities.
- the present invention relates to molecules that have a greater effect upon the 7 nAChRs as compared to other closely related members of this large ligand-gated receptor family.
- the invention provides compounds that are active drug
- nAChRs comprise a large family of ligand-gated ion channels that control neuronal
- nAChRs 20 activity and brain function. These receptors have a pentameric structure. In mammals, this gene family is composed of nine alpha and four beta subunits that co- assemble to form multiple subtypes of receptors that have a distinctive pharmacology. Acetylcholine is the endogenous regulator of all of the subtypes, while nicotine non- selectively activates all nAChRs.
- the ⁇ 7 nAChR is one receptor system that has proved to be a difficult target for testing. Native ⁇ 7 nAChR is not routinely able to be stably expressed in most mammalian cell lines (Cooper and Millar, J. Neurochem., 1997, 68(5) :2140-51). Another feature that makes functional assays of oc7 nAChR challenging is that the receptor is rapidly (100 milliseconds) inactivated. This rapid inactivation greatly
- Eisele et al. has indicated that a chimeric receptor formed between the N-terminal ligand binding domain of the ⁇ 7 nAChR (Eisele et al., Nature, 366(6454), p 479-83, 1993), and the pore forming C-terminal domain of the 5-HT 3 receptor expressed well in Xenopus oocytes while retaining nicotinic agonist sensitivity.
- Eisele et al. used the N-terminus of the avian (chick) form of the ⁇ 7 nAChR receptor and the C-terminus of the mouse form of the 5-HT gene.
- the 7 nAChR is a calcium channel while the 5-HT 3 R is a sodium and potassium channel.
- US Patent 6,054,464 discloses azabicyclic esters of carbamic acids useful in therapy, especially in the treatment or prophylaxis of psychotic disorders and intellectual impairment disorders, as well as intermediates and use of intermediates in synthesis.
- US Patent 5,977,144 discloses compositions for benzylidene- and cinnamylidene-anabaseines and methods for using these compositions for treating conditions associated with defects or malfunctioning of nicotinic subtypes brain receptors. These compositions target the ⁇ 7 receptor subtype with little or no activation of the ⁇ 4 ⁇ 2 or other receptor subtypes.
- US Patent 5,599,937 discloses heteroaromatic quinuclidines used for treating diseases related to muscarinic receptor function.
- US Patent 5,561,149 discloses the use of a mono or bicyclic carbocyclic, or heterocyclic carboxylic acid, ester or amide or an imidazolyl carbazol in the manufactxire of a medicament suitable for the treatment of stress-related psychiatric disorders, for increasing vigilance, for the treatment of rhinitis or serotonin-induced disorders and/or coadministration with another active agent to increase the bioavailability thereof, or for nasal administration.
- US Patent 5,543,426 discloses the use of certain 3,7-disubstituted indole compounds for treating depression or cognitive disorders.
- US Patent 5,434,161 discloses imidazopyridines as serotonergic 5-HT 3 antagonists.
- US Patent 5,362,740 discloses dihydrobenzofuran carboxamides useful in treating CNS disorders, but motility disorders, and/or emisis and/or pain in mammals, and/or migraine.
- US Patent 5,352,685 discloses thieno[3,2-b]pyridine derivatives effective for the prevention and therapeutical treatment of the symptoms caused by gastric hypanakinesis, such as heartburn, abdominal distension feeling, anorexia, unpleasant feeling on upper abdomen, abdominalgia, nausea, vomiting, etc. caused by the underlying diseases such as acute and chronic gastritis, stomach and duodenum ulcer, gastroneurosis, gastroptosis, etc.
- US Patent 5,342,845 discloses indole derivatives and drugs. The compound of the invention is disclosed as being effective as a gastrointestinal motor activity regulator, antimigraine, antipsychotic or antianxiety drug and for dementia or orthostatic hypotension.
- US Patent 5,322,951 discloses certain l-(2,3-dihydro-indole)carbonyl intermediates useful for preparing l-(2,3-dihydro)-l-carboxamide final products that possess 5-HT M-receptor antagonist activity.
- US Patent 5,1 5,173 discloses carboxamides useful as antiemetic or antipsychotic agents.
- US Patent 5J 14,947 discloses method for alleviating anxiety using benzobicyclic carboxamides.
- US Patent 5,063,231 discloses method of treatment of visceral pain.
- US Patent 5,039,680 discloses 5-HT 3 antagonists in preventing or reducing dependency on dependency-inducing agents.
- US Patent 5,001,133 discloses substituted benzoic acid heterocyclic amides and esters as being serotonin M antagonists.
- US Patent 4,985,437 discloses the use of certain compounds which act as antagonists of 5-hydroxytryptamine (5-HT) at 5-HT 3 receptors for the treatment of cognitive disorders such as attentional and memory deficits and dementia states.
- 5-hydroxytryptamine (5-HT) 5-hydroxytryptamine
- US Patent 4,973,594 discloses the use of compounds which act as antagonists of 5-hydroxytryptamine (5-HT) at 5-HT 3 receptors for the treatment of depression.
- US Patent 4,937,247 discloses 1-acyl indazoles that are disclosed as having 5- HT antagonist activity.
- US Patent 4,935,511 discloses benzoxazine and benzoxazepin carboxamide 5- HT 3 antagonists properties including CNS, anti-emetic and gastric prokinetic activity and which are void of any significant D receptor binding affinity.
- US Patent 4,933,445 discloses heteroazabenzobicyclic carboxamide 5-HT 3 antagonists properties including CNS, anti-emetic and gastric prokinetic activity.
- US Patent 4,921,982 discloses 5-halo-2,3-dihydro-2,2-dimethylbenzofuran-7- carboxylic acids which are useful as intermediates for 5-HT 3 antagonists.
- US Patent 4,920,227 discloses benzobicyclic carboxamide 5-HT antagonists.
- US Patent 4,920,219 discloses substituted saturated and unsaturated indole quinoline and benzazepine carboxamides and their valuable use as 5-HT 3 antagonists having CNS and gastric prokinetic activity void of any significant D receptor binding properties.
- US Patent 4,920,127 discloses substituted indoles and their use as 5-HT 3 receptor antagonists.
- US Patent 4,910,193 discloses treatment of gastrointestinal disorders.
- US Patent 4,882,327 discloses certain heterocyclic N-substituted carboxamides having 5-HT 3 receptor antagonist activity.
- US Patent 4,863 ,919 discloses a method of enhancing memory or correcting memory deficiency with arylamido (and arylthioamido)-azabicycloalkanes.
- US Patent 4,835,162 discloses agonists and antagonists to nicotine as smoking deterrents.
- US Patent 4,822,795 discloses pharmaceutically useful esters and amides.
- US Patent 4,803 J 99 discloses pharmaceutically useful heterocyclic acid esters and amides or alkylene bridged peperidines as serotonin M antagonists.
- US Patent 4,798,829 discloses l-azabicyclo[3.2.2]nonane derivatives having gastric motility enhancing activity and/or anti-emetic activity and/or 5-HT receptor antagonist activity.
- US Patent 4,797,406 discloses amides and esters containing bridged piperidines and use as serotonin M antagonists.
- US Patent 4,721,720 discloses a method of treating emesis, anxiety and/or irritable bowel syndrome.
- US Patent 4,612,319 discloses bridged quinolizinidinylamides, compositions containing them and methods for their use.
- US Patent 4,605,652 discloses a method of enhancing memory or correcting memory deficiency with arylamido (and arylthioamido)-azabicycloalkanes, and the pharmaceutically acceptable acid addition salts, hydrates and alcoholates thereof.
- WO 01/76576 Al discloses a pharmaceutical composition for treatment of acute, chorine pain and/or neuropathic pain migraines.
- WO 01/60821 Al discloses novel abiarylcarboxamides and their use in therapy, especially in the treatement of prophylaxis of psychotic and intellectual impairment conditions.
- WO 01/36417 Al discloses novel N-azabicyclo-amide derivatives and use in therapy, especially in the treatment of prophylaxis of psychotic disorders and intellectual impairment disorders.
- WO 00/73431 A2 discloses two binding assays to directly measure the affinity and selectivity of compounds at the ⁇ 7 nAChR and the 5-HT 3 R. The combined use of these functional and binding assays may be used to identify compounds that are selective agonists of the ⁇ 7 nAChR.
- WO 97/35860 discloses novel benzimidazol derivatives having an affinity for the serotoninergic 5-HT 3 /5-HT 4 receptors.
- WO 96/33186 discloses substituted dihydrobenzofuran derivatives as 5-HT 4 agonists.
- WO 95/27490 discloses serotonin antagonists (5-HT ) for treating fibromyalgia.
- WO 92/10494 discloses novel compounds having pharmacological activity, to a process for their preparation and their use as pharmaceuticals.
- WO 91/17161 discloses isoquinoline amides and esters as 5-HT 3 receptor antagonists.
- WO 91/09593 discloses 5-HT 3 antagonists for treatment of nausea, bradycardia or hypotension associated myocardial instability.
- WO 90/14347 A as abstracted in chemical abstract 1991 : 143, 158 discloses N- quinuclidinyl-indolecarboxamide derivatives as being antiemetics.
- EP 512 350 A2 discloses 3-(indolyl-2-carboxamido) quinuclidines useful for treating diseases characterized by an excess or enhanced sensitivity to serotonin, e.g., psychosis, nausea, vomiting, dementia or other cognitive diseases, migraine, diabetes.
- the compound may be used to control anxiety, aggression, depression, and pain.
- the compounds are disclosed as serotonin 5-HT antagonists.
- EP 496 064 Al discloses a process for the preparation of substituted benzofuran derivatives.
- the compounds are disclosed as being useful 5-HT receptor antagonists.
- EP 483 836 Al discloses pyrazolo[l,5-a]pyridine-3-carboxylic acid derivatives, their preparation process, and serotonin receptor antagonists containing them as active ingredients.
- EP 403 882 A2 discloses indole derivatives which have pharmacological activities such as 5-HT antagonism and the like.
- EP 279 512 discloses the use of certain 5-HT3 receptro antagonists in the treatement of visceral pain.
- DE 3810552 Al discloses esters and amides of indolyl-, benzo[b]thiophenyl-, benzo[b]furancarboxylic acids or 4-amino-2 methoxy-benzoic acids with N- heterocyclic or N-heterobicyclic alcohols or amines.
- the compounds disclosed have activity against pain especially migraine, as an anti-arrhythmic for gastrointestinal disturbances, stomach disturbances, gastritis ulcer, gall bladder, spastic colon, Crohn's disease, ulcerative colitis, carcinoid syndrome, diarrhea of various types.
- the compounds are also disclosed as speeding stomach emptying, controlling gastro duodenal and gastro esophageal reflux, disturbances of esophageal motility, hiatal hernia, cardiac insufficiency, hypotonic stomach, paralytic ileus, manic depressive psychosis and other psychoses.
- the compounds are also disclosed as useful for stress related diseases, senility, and enhancement of nasal absorption of other agents, e.g., in the treatment of emesis.
- the brain ⁇ 7 nicotinic receptor may be an important therapeutic target for the treatment of Alzheimer's disease using DMXBA which is known as GTS-21.
- the present invention discloses compounds of the Formula I:
- Each R ! is H, alkyl, cycloalkyl, halogenated alkyl, substituted phenyl, or substituted naphthyl; Azabicyclo is
- V VI Ro is H, lower alkyl, substituted lower alkyl, or halogenated lower alkyl
- Each R 2 is independently alkyl, cycloalkyl, substituted phenyl, or substituted naphthyl, or R 2 is absent provided that k , k 5 , or k 6 is 0; k 2 is 0 or 1 ; k 5 and k 6 are independently 0, 1 , or 2; R 2 - is H, alkyl, halogenated alkyl, substituted alkyl, F, Cl, Br, or I;
- W° is a bicyclic moiety and is
- W 1 , W 2 , W 3 , and W 4 are each independently N or C(R 21 ), provided that no more than two of W 1 , W 2 , W 3 , and W 4 are N and further provided when more than two of W 1 , W 2 , W 3 , and W 4 are C(R 2 ⁇ ) that no more than two R 21 are other than H;
- J is N(R 23 ), S, or O;
- Q is N(R 19 ), O, or S;
- Each R 3 is independently H, F, Cl, Br, I, alkyl, halogenated alkyl, substituted alkyl, cycloalkyl, lactam heterocycloalkyl, phenoxy, substituted phenoxy, R 7 , R 9 , -N(R 4 )-aryl, -N(R 4 )-halogenated phenyl, -N(R 4 )-halogenated naphthyl, -O-halogenated phenyl, -O-substituted phenyl, -O-halogenated naphthyl, -O-substituted naphthyl, -O-substituted naphthyl, -S-halogenated phenyl, -S-substituted phenyl, -S-halogenated naphthyl, -S-substituted naphthyl, or alkyl substituted on the ⁇ carbon with R 1 where said
- Each R 4 is H, or alkyl
- Each R 5 is independently H, F, alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, cycloalkyl, -C(O)NH 2 , -CO 2 R ⁇ , or aryl;
- R 6 is H, F, Br, I, Cl, -CN, -CF 3 , -OR 16 , -SR 16 , or -N(R ⁇ 6 ) 2 ;
- Aj is O, S, or NR 19 ,
- A is CR 18 or N
- a 2 and A 3 are independently selected from CR 18 , C(R 18 ) 2 , O, S, N, or NR ⁇ 9 , provided that both A 2 and A 3 are not simultaneously O, simultaneously S, or simultaneously O and S, or
- a and A 3 are independently selected from CR ⁇ 8 , C(R ⁇ 8 ) 2 , O, S, N, or NR 19 , and A is CR ⁇ 8 or N, each 9-membered fused-ring moiety having 0-1 substituent selected from R 2 o and further having 0-3 substituent(s) independently selected from F, Cl, Br, or I, and having a bond directly or indirectly attached to the core molecule where valency allows in either the 6-membered or the 5-membered ring of the fused- ring moiety;
- Each R 8 is independently F, alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, cycloalkyl, -C(O)NH 2 , -CO 2 R,, or aryl;
- Each Rio is independently H, alkyl, cycloalkyl, heterocycloalkyl, alkyl substituted with 1 substituent selected from R 13 , cycloalkyl substituted with 1 substituent selected from R 13 , heterocycloalkyl substituted with 1 substituent selected from R 13 , halogenated alkyl, halogenated cycloalkyl, halogenated heterocycloalkyl, phenyl, or substituted phenyl;
- Each R ⁇ is independently H, alkyl, cycloalkyl, heterocycloalkyl, halogenated alkyl, halogenated cycloalkyl, or halogenated heterocycloalkyl;
- R 13 is -OR formula, -SR admir, -N(R n ) 2 , -C(O)R u , -C(O)NR ⁇ R ⁇ , -CN, -CF 3 , -NR n C(O)Rn, -S(O) 2 N(R u ) 2 , -NR ⁇ S ⁇ R ⁇ , or -NO 2 ;
- R 15 is aryl, R 7 , or R 9 ;
- R 16 is H, alkyl, substituted alkyl, cycloalkyl, halogenated alkyl, substituted phenyl, or substituted naphthyl;
- R 17 is H, alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, -CN, F, Br, Cl, I, -ORi, -C(O)NH 2 , -NHRi, -SR b -CO 2 R b aryl, R 7 , or R 9 , and each of the other two R ⁇ 7 is independently alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, -CN, F, Br, Cl, I, -OR h -C(O)NH 2 , -NHR,, -SR b -CO 2 R l5 aryl, R 7
- fused-ring moiety has 0-3 substituent(s) selected from F, Cl, Br, or I;
- Ri 9 is H, alkyl, halogenated alkyl, substituted alkyl, cycloalkyl, halogenated cycloalkyl, substituted cycloalkyl, phenyl, or phenyl having 1 substituent selected from R 2 o and further having 0-3 substituents independently selected from F, Cl, Br, or
- R 2 o is alkyl, cycloalkyl, heterocycloalkyl, halogenated alkyl, halogenated cycloalkyl, halogenated heterocycloalkyl, -ORn, -SR n , -NR ⁇ R ⁇ , -C(O)R l l5 -C(O)NR u R n , -CN, -NR necessarilyC(O)R ll5 -S(O) 2 NR meaningR n , -NR ⁇ S ⁇ R ⁇ , -NO 2 , alkyl substituted with 1-4 substituent(s) independently selected from F, Cl, Br, I, or R 13 , cycloalkyl substituted with 1-4 substituent(s) independently selected from F, Cl, Br, I, or R 1 , or heterocycloalkyl substituted with 1-4 substituent(s) independently selected from F, Cl, Br, I, or -R 13 ; R 21 is H, F, Cl, Br, I, al
- Each R 22 is independently H, alkyl, substituted alkyl, cycloalkyl, halogenated alkyl, substituted cycloalkyl, heterocycloalkyl, or substituted heterocycloalkyl;
- R 23 is alkyl, halogenated alkyl, substituted alkyl, cycloalkyl, halogenated cycloalkyl, substituted cycloalkyl, heterocycloalkyl, halogenated heterocycloalkyl, substituted heterocycloalkyl, substituted phenyl, naphthyl, substituted naphthyl, R 7 , or
- Embodiments of the invention may include one or more or combination of the following.
- An embodiment of the present invention provides a use of a compound of Formula I for treating a disease or condition, wherein the diseases, disorders, and/or condition is any one or more or combination of the following: cognitive and attention deficit symptoms of Alzheimer's, neurodegeneration associated with diseases such as Alzheimer's disease, pre-senile dementia (mild cognitive impairment), senile dementia, schizophrenia, psychosis, attention deficit disorder, attention deficit hyperactivity disorder, depression, anxiety, general anxiety disorder, post traumatic stress disorder, mood and affective disorders, amyotrophic lateral sclerosis, borderline personality disorder, traumatic brain injury, behavioral and cognitive problems in general and associated with brain tumors, AIDS dementia complex, dementia associated with Down's syndrome, dementia associated with Lewy Bodies, Huntington's disease, Parkinson's disease, tardive dyskinesia, Pick's disease, dysregulation of food intake including bulemia and anorexia nervosa, withdrawal symptoms associated with smoking cessation and dependant drug cessation, Gilles de la Tourette's Syndrome, age-related
- the invention includes treating a mammal suffering from schizophrenia or psychosis by administering compounds of Formula I in conjunction with antipsychotic drugs (also called anti-psychotic agents).
- antipsychotic drugs also called anti-psychotic agents.
- the compounds of the present invention and the antipsychotic drugs can be administered simultaneously or at separate intervals.
- the compounds of the present invention and the antipsychotic drugs can be incorporated into a single pharmaceutical composition.
- two separate compositions i.e., one containing compounds of the present invention and the other containing antipsychotic drugs, can be administered simultaneously.
- the present invention also includes the compounds of the present invention, pharmaceutical compositions containing the active compounds as the free base or as a pharmaceutically acceptable salt and a pharmaceutically acceptable carrier, and methods to treat the identified diseases.
- a further embodiment of the present invention provides a method comprising administering a therapeutically effective amount of a compound of the present invention or a pharmaceutical composition contains said compound to the mammal.
- the compound of Formula I, where Azabicyclo is any one or more of I, II, III, IV, V, or VI.
- the compound of Formula I, where W° is any one or more of (a), (b), or (c).
- the compound of Formula I, where W is C(H), and where V — Z — Y is any one or more of the following: O-C(R 3 V N, O-C(R 5 ) 2 -N(R 4 ), O-C(R 5 ) 2 -S,
- each R 3 is independently any one of the following: H, F, Cl, Br, I, alkyl, halogenated alkyl, substituted alkyl, cycloalkyl, lactam heterocycloalkyl, phenoxy, substituted phenoxy, R 7 , R , -N(R 4 )-aryl, -N(R 4 )-halogenated phenyl, -N(R 4 )-halogenated naphthyl, -O-halogenated phenyl, -O-substituted phenyl, -O-halogenated naphthyl, -O-substituted naphthyl, -O-substituted naphthyl, -S-halogenated phenyl, -S-substituted phenyl, -S-halogenated naphthyl, -S-substituted naphthyl, or alkyl substituted on the ⁇ carbon
- each R 4 is alkyl.
- the compound of Formula I, where one R- t is alkyl and the other is H or alkyl.
- the compound of Formula I, where each R 5 is independently H, F, alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, cycloalkyl, -C(O)NH , -CO Rj, or aryl.
- each R 8 is independently F, alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, cycloalkyl, -C(O)NH 2 , -CO 2 Rj, or aryl.
- R l is H, alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, -CN, F, Br, Cl, I, -CF 3 , -OR u -C(O)NH 2 , -NHRi, -SRi, -CO 2 R ⁇ , aryl, R 7 , or R 9 , and each of the other two R 17 is independently alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, -CN, F, Br, Cl, I, -CF 3 , -ORi, -C(O)NH 2 , -NHR b -
- W° includes any one or more of the following: l,3-oxazolo[4,5-c]pyridin-6-yl, 1,3- oxazolo[5,4-c]pyridin-6-yl, 1 ,3-dioxolo[4,5-c]pyridin-6-yl, l,3-benzoxazol-5-yl, 1,3- benzoxazol-6-yl, l,2-benzisothiazol-5-yl, l,2-benzisothiazol-6-yl, l,3-thiazolo[4,5- c]pyridin-6-yl, l,3-thiazolo[5,4-c]pyridin-6-yl, l,3-benzothiazol-5-yl, 1,3- benzothiazol-6-yl, l,3-benzodioxol-5-yl, lH-benzimidazole-5-y
- W° includes any one or more of the following: l,3-oxazolo[4,5-c]pyridin-6-yl, l,3-oxazolo[5,4-c]pyridin-6-yl, 1,3- dioxolo[4,5-c]pyridin-6-yl, l,3-benzoxazol-5-yl, l,3-benzoxazol-6-yl, 1,2- benzisothiazol-5-yl, l,2-benzisothiazol-6-yl, l,3-thiazolo[4,5-c]pyridin-6-yl, 1,3- thiazolo[5,4-c]pyridin-6-yl, l,3-benzothiazol-5-yl, l,3-benzothiazol-6-yl, 1,3- benzodioxol-5-yl, lH-benzimidazole-5-yl, lH-inda
- W° includes any one or more of the following: l,3-oxazolo[4,5-c]pyridin-6-yl, l,3-oxazolo[5,4-c]pyridin-6-yl, 1,3- dioxolo[4,5-c]pyridin-6-yl, l,3-benzoxazol-5-yl, l,3-benzoxazol-6-yl, 1,2- benzisothiazol-5-yl, l,2-benzisothiazol-6-yl, l,3-thiazolo[4,5-c]pyridin-6-yl, 1,3- thiazolo[5,4-c]pyridin-6-yl, l,3-benzothiazol-5-yl, l,3-benzothiazol-6-yl, 1,3- benzodioxol-5-yl, lH-benzimidazole-5-yl, lH-inda
- Another embodiment includes compounds where W 1 , W 2 , W 3 , and W 4 are each C( ⁇ ) or N. Another embodiment includes compounds where W , W , W , and W 4 are each C(H). Another embodiment includes compounds where J is S.
- the present invention also includes the compounds of the present invention, pharmaceutical compositions containing the active compounds, and methods to treat the identified diseases.
- the present invention also includes a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable excipient.
- the pharmaceutical composition is administered rectally, topically, orally, sublingually, or parenterally for a therapeutically effective interval.
- the pharmaceutical composition is administered to deliver a compound of the present invention in an amount of from about 0.001 to about 100 mg/kg of body weight of said mammal per day.
- the pharmaceutical composition is also administered to deliver a compound of the present invention in an amount of from about 0.1 to about 50 mg/kg of body weight of said mammal per day.
- a pharmaceutical composition comprising a compound of Formula I or a pharmaceutically acceptable salt thereof, an anti-psychotic agent, and a pharmaceutically acceptable excipient.
- the pharmaceutical composition is administered to independently administer said compound and said agent rectally, topically, orally, sublingually, or parenterally for a therapeutically effective interval.
- the pharmaceutical composition is administered to deliver a compound of the present invention in an amount of from about 0.001 to about 100 mg/kg of body weight of said mammal per day.
- the pharmaceutical composition is also administered to deliver a compound of the present invention in an amount of from about 0J to about 50 mg/kg of body weight of said mammal per day.
- the present invention also includes a use of a compound according to Formula I or pharmaceutically acceptable salt thereof for the preparation of a medicament for treating a disease or condition, wherein the mammal would receive symptomatic relief from the administration of a therapeutically effective amount of ⁇ 7 nicotinic acetylcholine receptor agonist.
- the present invention also includes a use of a compound according to Formula I or pharmaceutically acceptable salt thereof for the preparation of a medicament for treating a disease or condition, wherein the mammal would receive symptomatic relief from the administration of a therapeutically effective amount of ⁇ 7 nicotinic acetylcholine receptor agonist, wherein the disease, or condition is any one or more or combination of the following: cognitive and attention deficit symptoms of Alzheimer's, neurodegeneration associated with diseases such as Alzheimer's disease, pre-senile dementia (mild cognitive impairment), senile dementia, schizophrenia, psychosis, attention deficit disorder, attention deficit hyperactivity disorder, depression, anxiety, general anxiety disorder, post traumatic stress disorder, mood and affective disorders, amyotrophic lateral sclerosis, borderline personality disorder, traumatic brain injury, behavioral and cognitive problems in general and associated with brain tumors, AIDS dementia complex, dementia associated with Down's syndrome, dementia associated with Lewy Bodies, Huntington's disease, Parkinson's disease, tardive dyskinesia, Pick's
- the present invention also includes a method for treating a disease or condition in a mammal in need thereof, wherein the mammal would receive symptomatic relief from the administration of an ⁇ 7 nicotinic acetylcholine receptor agonist comprising administering to the mammal a therapeutically effective amount of a compound according to Formula I or pharmaceutically acceptable salt thereof.
- the present invention also includes a method for treating a disease or condition in a mammal in need thereof comprising administering to the mammal a therapeutically effective amount of a compound according to Formula I or pharmaceutically acceptable salt thereof, wherein the disease or condition is any one or more or combination of the following: cognitive and attention deficit symptoms of Alzheimer's, neurodegeneration associated with diseases such as Alzheimer's disease, pre-senile dementia (mild cognitive impairment), senile dementia, schizophrenia, psychosis, attention deficit disorder, attention deficit hyperactivity disorder, depression, anxiety, general anxiety disorder, post traumatic stress disorder, mood and affective disorders, amyotrophic lateral sclerosis, borderline personality disorder, traumatic brain injury, behavioral and cognitive problems in general and associated with brain tumors, AIDS dementia complex, dementia associated with Down's syndrome, dementia associated with Lewy Bodies, Huntington's disease, Parkinson's disease, tardive dyskinesia, Pick's disease, dysregulation of food intake including bulemia and anorexia nervosa, withdrawal symptoms associated with smoking
- the compounds of Formula I (Azabicyclo is I) have optically active centers on the quinuclidine ring.
- the compounds of the present invention include quinuclidines with the 3R configuration and also includes racemic mixtures and compositions of varying degrees of streochemical purities.
- compounds of Formula I include compounds with stereospecificity including:
- the compounds of Formula I (Azabicyclo is II) have optically active center(s) on the [2.2.1] azabicyclic ring at C3 and C4.
- the scope of this invention includes racemic mixtures and the separate stereoisomers of Formula I being endo- S, endo- 4R, exo- S, exo- R:
- endo-AS endo-AR exo- S exo-AR The endo isomer is the isomer where the non-hydrogen substituent at C3 of the [2.2J] azabicyclic compound is projected toward the larger of the two remaining bridges.
- the exo isomer is the isomer where the non-hydrogen substituent at C3 of the [2.2.1] azabicyclic compound is projected toward the smaller of the two remaining bridges.
- the compounds of Formula I (Azabicyclo III) have optically active center(s) on the [2.2J] azabicyclic ring at Cl, C4 and C5.
- the scope of this invention includes racemic mixtures and the separate stereoisomers of Formula I being (1R,4R,5S), ⁇ 1R,4R,5R), (1S,4S,5R), (1S,4S,5S):
- the endo isomer is the isomer where the non-hydrogen substituent at C5 of the [2.2J] azabicyclic compound is projected toward the larger of the two remaining bridges.
- the exo isomer is the isomer where the non-hydrogen substituent at C5 of the [2.2J] azabicyclic compound is projected toward the smaller of the two remaining bridges.
- the compounds of Formula I (Azabicyclo IV) have optically active center(s) on the [2.2J] azabicyclic ring at Cl, C4 and C6.
- the scope of this invention includes racemic mixtures and the separate stereoisomers of Formula I being exo- ⁇ S,4R,6S), exo- ⁇ lR,4S,6R), endo- ⁇ S,4R,6R), and e «_fo-(lR,4S,6S):
- the endo isomer is the isomer where the non-hydrogen substituent at C6 of the [2.2J] azabicyclic compound is projected toward the larger of the two remaining bridges.
- the exo isomer is the isomer where the non-hydrogen substituent at C6 of the [2.2J] azabicyclic compound is projected toward the smaller of the two remaining bridges.
- the compounds of Formula I (Azabicyclo is V) have optically active center(s) on the [3.2J] azabicyclic ring at C3 and C5.
- the scope of this invention includes racemic mixtures and the separate stereoisomers of Formula I being endo- S, 5R, endo-3R, 5S, ex ⁇ -3R, 5R, exo-3S, 5S:
- the compounds of Formula I (Azabicyclo is VI) have optically active centers on the [3.2.2] azabicyclic ring with one center being at C3 when R 2 is absent.
- the scope of this invention includes racemic mixtures and the separate stereoisomers of Formula I being 3(S) and 3(R):
- 3(5) 3(R) The compounds of the present invention having the specified stereochemistry have different levels of activity and that for a given set of values for the variable substitutuents one isomer may be preferred over the other isomers. Although it is desirable that the stereochemical purity be as high as possible, absolute purity is not required.
- This invention involves racemic mixtures and compositions of varying degrees of streochemical purities when the Azabicyclo is substituted with only the amide/thioamide or is substituted with substituents in addition to the amide/thioamide, e.g., k is 1 or 2. This invention involves racemic mixtures and compositions of varying degrees of stereochemical purities.
- racemic mixtures and compositions When racemic mixtures and compositions are referenced, it means racemic mixtures and compositions of varying degree of stereochemical purities. It is preferred to carry out stereoselective syntheses and/or to subject the reaction product to appropriate purification steps so as to produce substantially optically pure materials. Suitable stereoselective synthetic procedures for producing optically pure materials are well known in the art, as are procedures for purifying racemic mixtures into optically pure fractions. Naming a specific isomer includes racemic mixtures thereof within the scope of this invention.
- N-(exo-4(S)-l-azabicyclo[2.2J]hept-3-yl)-l ,3-benzoxazole-5-carboxamide includes N-(ex -4(rac)-l-azabicyclo[2.2J]hept-3-yl)-l,3-benzoxazole-5-carboxamide; N-(3(rac),4(S)-l -azabicyclo[2.2.1 ]hept-3-yl)-l ,3-benzoxazole-5-carboxamide, and N- (l-azabicyclo[2.2J]hept-3-yl)-l,3-benzoxazole-5-carboxamide.
- Stereoselective syntheses and/or subjecting the reaction product to appropriate purification steps produces substantially optically pure materials.
- Suitable stereoselective synthetic procedures for producing optically pure materials are well known in the art, as are procedures for purifying racemic mixtures into optically pure fractions.
- the compound is a racemic mixture, or (ii) the compound has the R stereochemistry at C-3 as discussed herein and stereochemistry is unspecified at C-6.
- R 2 has any definition discussed herein; (iii) R 2 has any definition discussed herein; or
- R 2 - 3 is H; (ii) R 2 - 3 is F, Cl, Br, I, alkyl, halogenated alkyl, substituted alkyl, or substituted phenyl or substituted naphthyl; or
- R . 3 is alkyl, halogenated alkyl, substituted alkyl, or substituted phenyl or substituted naphthyl.
- R 2 - 3 is H
- R - 3 is F, Cl, Br, I, alkyl, halogenated alkyl, substituted alkyl, or substituted phenyl or substituted naphthyl; or (iii) R 2 - 3 is alkyl, halogenated alkyl, substituted alkyl, or substituted phenyl or substituted naphthyl.
- R is absent and where the Azabicyclo has the stereochemistry of 3R, 5R;
- k 5 is 2, where R 2 - a is alkyl, cycloalkyl, substituted phenyl, or substituted naphthyl, and where R 2 - b is alkyl, cycloalkyl, substituted phenyl, or substituted naphthyl; (iv) k 5 is 1, where R 2 is alkyl, cycloalkyl, substituted phenyl, or substituted naphthyl; or
- (v) k 5 is 1 , where R 2 is alkyl, cycloalkyl, substituted phenyl, or substituted naphthyl.
- Another embodiment of compounds of Formula I includes any one or more or combination of the following configurations for compounds:
- k 6 is 2, where each R - a is alkyl, cycloalkyl, substituted phenyl, or substituted naphthyl, and where each R 2 - b is alkyl, cycloalkyl, substituted phenyl, or substituted naphthyl;
- k 6 is 1 , where R 2 is alkyl, cycloalkyl, substituted phenyl, or substituted naphthyl; or
- (iv) k 6 is 1 , where R 2 is alkyl, cycloalkyl, substituted phenyl, or substituted naphthyl.
- the compound of Formula I where the compound is any one or more or combination of the following as the free base, or pharmaceutally acceptable salt thereof as a pure enantiomer or racemic mixture thereof: N-(exo-4(S)-l-azabicyclo[2.2J]hept-3-yl)-l,3-benzoxazole-5-carboxamide; N-(exo-4(S)-l-azabicyclo[2.2J]hept-3-yl)-l,3-benzothiazole-6-carboxamide; N-[(ex ⁇ - S)-l-azabicyclo[2.2J]hept-3-yl]-indane-5-carboxamide; N-(exo-4(S)-l-azabicyclo[2.2J]hept-3-yl)-l,3-benzodioxole-5-carboxamide; N-[(exo- ⁇ S)-l-azabicyclo[2.2J]hept-3-yl]-lH-indazole-5
- the compound of Formula I where the compound is any one or more or combination of the following as the free base, or pharmaceutally acceptable salt thereof as a pure enantiomer or racemic mixture thereof: N-(l -(6-methyl)-azabicyclo[2.2.2]oct-3-yl)-l ,3-benzoxazole-5-carboxamide; N-( 1 -(6-methyl)-azabicyclo[2.2.2]oct-3-yl)-2-methyl- 1 ,3-benzoxazole-5-carboxamide;
- the compound of Formula I where the compound is any one or more or combination of the following as the free base, or pharmaceutally acceptable salt thereof as a pure enantiomer or racemic mixture thereof: N-(2-azabicyclo[2.2J]hept-5-yl)-2-benzoisothiophene-5-benzamide; or N-(2-azabicyclo[2.2J]hept-6-yl))-2-benzoisothiophene-5-benzamide.
- Each Rj is H, alkyl, cycloalkyl, halogenated alkyl, substituted phenyl, or substituted naphthyl; Azabicyclo is
- Ro is H, lower alkyl, substituted lower alkyl, or halogenated lower alkyl;
- Each R 2 is independently alkyl, cycloalkyl, substituted phenyl, or substituted naphthyl, or R 2 is absent provided that k 2 , k 5 , or k 6 is 0; k 2 is 0 or 1 ; k 5 and k 6 are independently 0, 1, or 2;
- R 2 - 3 is H, alkyl, halogenated alkyl, substituted alkyl, F, Cl, Br, or I; W° is a bicyclic moiety and is
- W 1 , W 2 , W 3 , and W 4 are each independently N or C(R 2! ), provided that no more than two of W 1 , W 2 , W 3 , and W 4 are N and further provided when more than two of W 1 , W 2 , W 3 , and W 4 are C(R 21 ) that no more than two R 21 are other than H;
- J is N(R 23 ), S, or O;
- Q is N(R 19 ), O, or S;
- Each R 3 is independently H, F, Cl, Br, I, alkyl, halogenated alkyl, substituted alkyl, cycloalkyl, lactam heterocycloalkyl, phenoxy, substituted phenoxy, R , R 9 , -N(R 4 )-aryl, -N(R 4 )-halogenated phenyl, -N(R 4 )-halogenated naphthyl, -O-halogenated phenyl, -O-substituted phenyl, -O-halogenated naphthyl, -O-substituted naphthyl, -O-substituted naphthyl, -S-halogenated phenyl, -S-substituted phenyl, -S-halogenated naphthyl, -S-substituted naphthyl, or alkyl substituted on the ⁇ carbon with R 15 where said
- Each R 4 is H, or alkyl;
- Each R 5 is independently H, F, alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, cycloalkyl, -C(O)NH 2 , -CO 2 R 1; or aryl;
- Re is H, F, Br, I, Cl, -CN, -CF 3 , -OR 16 , -SR 16 , or -N(R 16 ) 2 ;
- Lower alkyl is both straight- and branched-chain moieties having from 1-4 carbon atoms
- Halogenated lower alkyl is an alkyl moiety having from 1-4 carbon atoms and having 1 to (2n+l) substituent(s) independently selected from F, Cl, Br, or I where n is the maximum number of carbon atoms in the moiety;
- Substituted lower alkyl is an alkyl moiety from 1-4 carbon atoms and having 0-3 substituents independently selected from F, Cl, Br, or I and further having 1 substituent selected from R 7 , R 9 , -OR i0 , -SR 10 , -N(R ⁇ 0 ) 2 , -C(O)R, 0 , -C(O)NR ]0 R ⁇ o, -CN, -NRi 0 C(O)Rio, -S(O) 2 NR 10 R ⁇ o, -NR ⁇ 0 S(O) 2 R, 0 , -NO 2 , phenyl, or substituted phenyl;
- Alkyl is both straight- and branched-chain moieties having from 1-6 carbon atoms
- Halogenated alkyl is an alkyl moiety having from 1-6 carbon atoms and having 1 to (2n+l) substituent(s) independently selected from F, Cl, Br, or I where n is the maximum number of carbon atoms in the moiety;
- Substituted alkyl is an alkyl moiety from 1-6 carbon atoms and having 0-3 substituents independently selected from F, Cl, Br, or I and further having 1 substituent selected from R 7 , R 9 , -OR 10 , -SR 10 , -NR 10 R ⁇ o, -C(O)R 10 , -C(O)NR 10 R ⁇ o, -CN, -NR 10 C(O)R 10 , -S(O) 2 NR 10 R ⁇ o, -NR ⁇ 0 S(O) 2 R 10 , -NO 2 , phenyl, or substituted phenyl;
- Alkenyl is straight- and branched-chain moieties having from 2-6 carbon atoms and having at least one carbon-carbon double bond;
- Halogenated alkenyl is an unsaturated alkenyl moiety having from 2-6 carbon atoms and having 1 to (2n-l) substiruent(s) independently selected from F, Cl, Br, or I where n is the maximum number of carbon atoms in the moiety;
- Substituted alkenyl is an unsaturated alkenyl moiety having from 2-6 carbon atoms and having 0-3 substituents independently selected from F, or Cl, and further having 1 substituent selected from R 7 , R , -OR ⁇ 0 , -SR ⁇ , -N(R ⁇ o) 2 , -C(O)R JO , -C(O)N(Ri 0 ) 2 , -NR 10 C(O)R 10 , -S(O) 2 N(R 10 ) 2 , -NR 10 S(O) 2 R 10 , -CN, phenyl, or substituted phenyl;
- Alkynyl is straight- and branched-chained moieties having from 2-6 carbon atoms and having at least one carbon-carbon triple bond;
- Halogenated alkynyl is an unsaturated alkynyl moiety having from 3-6 carbon atoms and having 1 to (2n-3) substituent(s) independently selected from F, Cl, Br, or I where n is the maximum number of carbon atoms in the moiety;
- Substituted alkynyl is an unsaturated alkynyl moiety having from 3-6 carbon atoms and having 0-3 substituents independently selected from F, or Cl, and further having 1 substituent selected from R 7 , R 9 , -ORio, -SRio, -N(R ⁇ 0 ) 2 , -C(O)R ⁇ 0 , -C(O)N(R 10 ) 2 , -NR,oC(O)R, 0 , -S(O) 2 N(R 10 ) 2 , -NR 10 S(O) 2 R, 0 , -CN, phenyl, or substituted phenyl;
- Cycloalkyl is a cyclic alkyl moiety having from 3-6 carbon atoms
- Lower cycloalkyl is a cyclic alkyl moiety having from 3-4 carbon atoms;
- Halogenated cycloalkyl is a cyclic moiety having from 3-6 carbon atoms and having 1-4 substituents independently selected from F, or Cl;
- Substituted cycloalkyl is a cyclic moiety having from 3-6 carbon atoms and having 0-3 substituents independently selected from F, or Cl, and further having 1 substituent selected from -OR ]0 , -SR 10 , -N(R ⁇ 0 ) 2 , -C(O)R 10 , -CN, -C(O)N(R 10 ) 2 , -NR 10 C(O)R 10 , -S(O) 2 N(R 10 ) 2 , -NR 10 S(O) 2 R 10 , -NO 2 , phenyl, or substituted phenyl;
- Heterocycloalkyl is a cyclic moiety having 4-7 atoms with 1-2 atoms within the ring being -S-, -N(R 19 )-, or -O-;
- Halogenated heterocycloalkyl is a cyclic moiety having from 4-7 atoms with 1-2 atoms within the ring being -S-, -N(R 1 )-, or -O-, and having 1-4 substituents independently selected from F, or Cl;
- Substituted heterocycloalkyl is a cyclic moiety having from 4-7 atoms with 1-2 atoms within the ring being -S-, -N(R 1 )-, or -O- and having 0-3 substituents independently selected from F, or Cl, and further having 1 substituent selected from R 7 , R 9 , -OR 10 , -SRio, -NRioRio, -C(O)R 10 , -C(O)NR 10 R 10 , -CN, -NR 10 C(O)R 10 , -NO 2 , -S(O) NR 1 oR 1 o, -NR 1 oS(O) 2 R 1 o, phenyl, or phenyl having 1 substituent selected from R 2 o and further having 0-3 substituents independently selected from F, Cl, Br, or I;
- Substituted phenoxy is a phenoxy either having 1-3 substituents independently selected from F, Cl, Br, or I, or having 1 substituent selected from R 12 and 0-2 substituents independently selected from F, Cl, Br, or I;
- Aryl is phenyl, substituted phenyl, naphthyl, or substituted naphthyl;
- Substituted phenyl is a phenyl either having 1-4 substituents independently selected from F, Cl, Br, or I, or having 1 substituent selected from R 12 and 0-3 substituents independently selected from F, Cl, Br, or I;
- Substituted naphthyl is a naphthalene moiety either having 1-4 substituents independently selected from F, Cl, Br, or I, or having 1 substituent selected from R ⁇ 2 and 0-3 substituents independently selected from F, Cl, Br, or I, where the substitution can be independently on either only one ring or both rings of said naphthalene moiety;
- R 7 is 5-membered heteroaromatic mono-cyclic moieties containing within the ring 1-3 heteroatoms independently selected from the group consisting of -O-, -N-, -N(R 1 )-, and -S-, and having 0-1 substituent selected from R 2 o and further having 0-3 substituents independently selected from F, Cl, Br, or I, or R 7 is 9-membered fused- ring moieties having a 6-membered ring fused to a 5-membered ring and having the formula
- a t is O, S, or NR 19 ,
- A is CR] 8 or N
- a 2 and A 3 are independently selected from CR 18 , C(R 18 ) 2 , O, S, N, or NR 19 , provided that both A 2 and A 3 are not simultaneously O. simultaneously S, or simultaneously O and S, or
- a 2 and A 3 are independently selected from CRj 8 , C(R 18 ) 2 , O, S, N, or NR 19 , and A is CR 18 or N, each 9-membered fused-ring moiety having 0- 1 substituent selected from R 2 o and further having 0-3 substituent(s) independently selected from F, Cl, Br, or I, and having a bond directly or indirectly attached to the core molecule where valency allows in either the 6-membered or the 5-membered ring of the fused- ring moiety;
- Each R 8 is independently F, alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, cycloalkyl, -C(O)NH 2 , -CO 2 R b or aryl;
- R is 6-membered heteroaromatic mono-cyclic moi
- Each Rio is independently H, alkyl, cycloalkyl, heterocycloalkyl, alkyl substituted with 1 substituent selected from R 13 , cycloalkyl substituted with 1 substituent selected from R 13 , heterocycloalkyl substituted with 1 substituent selected from Rj 3 , halogenated alkyl, halogenated cycloalkyl, halogenated heterocycloalkyl, phenyl, or substituted phenyl;
- Each R ⁇ is independently H, alkyl, cycloalkyl, heterocycloalkyl, halogenated alkyl, halogenated cycloalkyl, or halogenated heterocycloalkyl;
- R ⁇ is -OR ⁇ , -SR ⁇ , alkyl, cycloalkyl, heterocycloalkyl, halogenated alkyl, halogenated cycloalkyl, halogenated heterocycloalkyl, substituted alkyl, substituted cycloalkyl, substituted heterocycloalkyl, -NR ⁇ R ⁇ , -C(O)R ⁇ , -NO 2 , -C(O)NRnRn, -CN, -NR meaningC(O)R n , -S(O) 2 NR ⁇ R ⁇ , or -NRnS ⁇ R ⁇ ;
- R 13 is -OR favor, -SR favor, -N(R n ) 2 , -C(O)RNase, -C(O) R ⁇ Rêt, -CN, -CF 3 ,
- R ⁇ is alkyl, substituted alkyl, halogenated alkyl, -OR ⁇ , -CN, -NO 2 ,
- R 15 is aryl, R 7 , or R 9 ;
- R i6 is H, alkyl, substituted alkyl, cycloalkyl, halogenated alkyl, substituted phenyl, or substituted naphthyl;
- R ⁇ 7 is H, alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, -CN, F, Br, Cl, I, -OR l5 -C(O)NH 2 , -NHR U -SR ⁇ -CO 2 R b aryl, R 7 , or R 9 , and each of the other two R ⁇ is independently alkyl, substituted alkyl, halogenated alkyl, alkenyl, substituted alkenyl, halogenated alkenyl, alkynyl, substituted alkynyl, halogenated alkynyl, -CN, F, Br, Cl, I, -OR,, -C(O)NH 2 , -NHR l 5 -SR,, -CO 2 Rj, aryl, R 7 ,
- R 19 is H, alkyl, halogenated alkyl, substituted alkyl, cycloalkyl, halogenated cycloalkyl, substituted cycloalkyl, phenyl, or phenyl having 1 substituent selected from R 20 and further having 0-3 substituents independently selected from F, Cl, Br, or
- R 2 o is alkyl, cycloalkyl, heterocycloalkyl, halogenated alkyl, halogenated cycloalkyl, halogenated heterocycloalkyl, -OR ⁇ , -SR ⁇ , -NR ⁇ R ⁇ , -C(O)R ⁇ , -C(O)NR n R ⁇ , -CN, -NR ⁇ C(O)RNase, -S(O) 2 NR ⁇ R ⁇ , -NR ⁇ S(O) 2 R ⁇ , -NO 2 , alkyl substituted with 1-4 substituent(s) independently selected from F, Cl, Br, I, or Rj , cycloalkyl substituted with 1-4 substituent(s) independently selected from F, Cl, Br, I, or R ⁇ 3 , or heterocycloalkyl substituted with 1-4 substituent(s) independently selected from F, Cl, Br, I, or -R 13 ;
- R 21 is H, F, Cl, Br, I, alkyl, substituted alkyl, halogenated alkyl, cycloalkyl, -CN, -NR 22 R 22 , -OR 22 , or -SR 22 ;
- Each R 22 is independently H, alkyl, substituted alkyl, cycloalkyl, halogenated alkyl, substituted cycloalkyl, heterocycloalkyl, or substituted heterocycloalkyl;
- R 23 is alkyl, halogenated alkyl, substituted alkyl, cycloalkyl, halogenated cycloalkyl, substituted cycloalkyl, heterocycloalkyl, halogenated heterocycloalkyl, substituted heterocycloalkyl, substituted phenyl, naphthyl, substituted naphthyl, R 7 , or
- R 9 or pharmaceutical composition, pharmaceutically acceptable salt, racemic mixture, or pure enantiomer thereof useful to treat any one of or combination of cognitive and attention deficit symptoms of Alzheimer's, neurodegeneration associated with diseases such as Alzheimer's disease, pre-senile dementia (mild cognitive impairment), senile dementia, schizophrenia, psychosis, attention deficit disorder, attention deficit hyperactivity disorder, mood and affective disorders, amyotrophic lateral sclerosis, borderline personality disorder, traumatic brain injury, behavioral and cognitive problems associated with brain tumors, AIDS dementia complex, dementia associated with Down's syndrome, dementia associated with Lewy Bodies, Huntington's disease, depression, general anxiety disorder, age-related macular degeneration, Parkinson's disease, tardive dyskinesia, Pick's disease, post traumatic stress disorder, dysregulation of food intake including bulemia and anorexia nervosa, withdrawal symptoms associated with smoking cessation and dependant drug cessation, Gilles de la Tourette's Syndrome, glaucoma, neurodegeneration associated with glau
- the invention includes methods of treating a mammal suffering from schizophrenia or psychosis by administering compounds of Formula I in conjunction with antipsychotic drugs.
- the compounds of Formula I and the antipsychotic drugs can be administered simultaneously or at separate intervals.
- the compounds of Formula I and the antipsychotic drugs can be incorporated into a single pharmaceutical composition.
- two separate compositions i.e., one containing compounds of Formula I and the other containing antipsychotic drugs, can be administered simultaneously.
- the present invention also includes the compounds of the present invention, pharmaceutical compositions containing the active compounds, and methods to treat the identified diseases.
- AChR refers to acetylcholine receptor.
- nAChR refers to nicotinic acetylcholine receptor.
- Pre-senile dementia is also known as mild cognitive impairment.
- 5HT R refers to the serotonin- type 3 receptor.
- -btx refers to ⁇ -bungarotoxin.
- FLIPR refers to a device marketed by Molecular Devices, Inc. designed to precisely measure cellular fluorescence in a high throughput whole-cell assay. (Schroeder et. al., J. Biomolecular Screening, 1(2), p 75-80, 1996).
- TLC refers to thin-layer chromatography
- HPLC refers to high pressure liquid chromatography.
- MeOH refers to methanol
- EtOH refers to ethanol.
- IP A refers to isopropyl alcohol.
- THF refers to tetrahydrofuran
- DMSO dimethylsulfoxide
- DMF refers to N,N-dimethylformamide.
- EtOAc refers to ethyl acetate.
- TMS refers to tetramethylsilane.
- TEA refers to triethylamine
- DIEA refers to N,N-diisopropylethylamine.
- MLA refers to methyllycaconitine
- Ether refers to diethyl ether.
- HATU refers to O-(7-azabenzotriazol-l -yl)- ⁇ , ⁇ , ⁇ ', N'-tetramethyluronium hexafluorophosphate.
- CDI refers to carbonyl diimidazole.
- NMO refers to N-methylmorpholine-N-oxide.
- TPAP refers to tetrapropylammonium perruthenate.
- Na 2 SO refers to sodium sulfate.
- K 2 CO 3 refers to potassium carbonate.
- MgSO 4 refers to magnesium sulfate.
- Halogen is F, Cl, Br, or I.
- the carbon atom content of various hydrocarbon-containing moieties is indicated by a prefix designating the minimum and maximum number of carbon atoms in the moiety, i.e., the prefix . j indicates a moiety of the integer 'i" to the integer "j" carbon atoms, inclusive.
- Cj- 6 alkyl refers to alkyl of one to six carbon atoms.
- Non-inclusive examples of heteroaryl compounds that fall within the definition of R 7 and R 9 include, but are not limited to, thienyl, benzothienyl, pyridyl, thiazolyl, quinolyl, pyrazinyl, pyrimidyl, imidazolyl, furanyl, benzofuranyl, benzothiazolyl, isothiazolyl, benzisothiazolyl, benzisoxazolyl, benzimidazolyl, indolyl, benzoxazolyl, pyrazolyl, triazolyl, tetrazolyl, isoxazolyl, oxazolyl, pyrrolyl, isoquinolinyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pydridazinyl, triazinyl, isoindolyl, purinyl, oxadiazolyl
- heterocycloalkyl examples include, but are not limited to, tetrahydrofurano, tetrahydropyrano, morpholino, pyrrolidino, piperidino, piperazine, azetidino, azetidinono, oxindolo, dihydroimidazolo, and pyrrolidinono
- Amino protecting group includes, but is not limited to, carbobenzyloxy (CBz), tert butoxy carbonyl (BOC) and the like. Examples of other suitable amino protecting groups are known to person skilled in the art and can be found in "Protective Groups in Organic synthesis," 3rd Edition, authored by Theodora Greene and Peter Wuts.
- ⁇ carbon with R 15 where said ⁇ carbon is determined by counting the longest carbon chain of the alkyl moiety with the C-1 carbon being the carbon attached to the bicyclic moiety W° and the ⁇ carbon being the carbon furthest, e.g., separated by the greatest number of carbon atoms in the chain, from said C-1 carbon:
- Mammal denotes human and other mammals.
- Brine refers to an aqueous saturated sodium chloride solution.
- Equ means molar equivalents.
- IR refers to infrared spectroscopy.
- Lv refers to leaving groups within a molecule, including Cl, OH, or mixed anhydride.
- NMR nuclear (proton) magnetic resonance spectroscopy, chemical shifts are reported in ppm ( ⁇ ) downfield from TMS.
- MS refers to mass spectrometry expressed as m/e or mass/charge unit.
- HRMS refers to high resolution mass spectrometry expressed as m/e or mass/charge unit.
- M+H + refers to the positive ion of a parent plus a hydrogen atom.
- M-H " refers to the negative ion of a parent minus a hydrogen atom.
- M+Na + refers to the positive ion of a parent plus a sodium atom.
- M+K + refers to the positive ion of a parent plus a potassium atom.
- El refers to electron impact.
- ESI refers to elecfrospray ionization.
- Cl refers to chemical ionization.
- FAB refers to fast atom bombardment.
- compositions of the present invention may be in the form of pharmaceutically acceptable salts.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases including inorganic bases and organic bases, and salts prepared from inorganic acids, and organic acids. Salts derived from inorganic bases include aluminum, ammonium, calcium, ferric, ferrous, lithium, magnesium, potassium, sodium, zinc, and the like.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, such as arginine, betaine, caffeine, choline, N, N- dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylamino- ethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, and the like.
- cyclic amines such as arginine, betaine, caffeine, choline, N, N
- Salts derived from inorganic acids include salts of hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, phosphoric acid, phosphorous acid and the like.
- Salts derived from pharmaceutically acceptable organic non-toxic acids include salts of Ci-e alkyl carboxylic acids, di-carboxylic acids, and tri-carboxylic acids such as acetic acid, propionic acid, fumaric acid, succinic acid, tartaric acid, maleic acid, adipic acid, and citric acid, and aryl and alkyl sulfonic acids such as toluene sulfonic acids and the like.
- an effective amount of a compound as provided herein is meant a nontoxic but sufficient amount of the compound(s) to provide the desired effect.
- the exact amount required will vary from subject to subject, depending on the species, age, and general condition of the subject, the severity of the disease that is being treated, the particular compound(s) used, the mode of administration, and the like. Thus, it is not possible to specify an exact “effective amount.” However, an appropriate effective amount may be determined by one of ordinary skill in the art using only routine experimentation.
- the compounds of Formula I have optically active center(s) on the Azabicyclo moiety. Although it is desirable that the stereochemical purity be as high as possible, absolute purity is not required.
- This invention involves racemic mixtures and compositions of varying degrees of stereochemical purities. It is preferred to carry out stereoselective syntheses and/or to subject the reaction product to appropriate purification steps so as to produce substantially optically pure materials. Suitable stereoselective synthetic procedures for producing optically pure materials are well known in the art, as are procedures for purifying racemic mixtures into optically pure fractions.
- the amount of therapeutically effective compound(s) that is adininistered and the dosage regimen for treating a disease condition with the compounds and/or compositions of this invention depends on a variety of factors, including the age, weight, sex and medical condition of the subject, the severity of the disease, the route and frequency of administration, and the particular compound(s) employed, and thus may vary widely.
- the compositions contain well know carriers and excipients in addition to a therapeutically effective amount of compounds of Formula I.
- the pharmaceutical compositions may contain active ingredient in the range of about 0.001 to 100 mg/kg/day for an adult, preferably in the range of about 0J to 50 mg/kg/day for an adult. A total daily dose of about 1 to 1000 mg of active ingredient may be appropriate for an adult.
- compositions for therapeutic use may also comprise one or more non-toxic, pharmaceutically acceptable carrier materials or excipients.
- carrier material or excipient herein means any substance, not itself a therapeutic agent, used as a carrier and/or diluent and/or adjuvant, or vehicle for delivery of a therapeutic agent to a subject or added to a pharmaceutical composition to improve its handling or storage properties or to permit or facilitate formation of a dose unit of the composition into a discrete article such as a capsule or tablet suitable for oral administration.
- Excipients can include, by way of illustration and not limitation, diluents, disintegrants, binding agents, adhesives, wetting agents, polymers, lubricants, glidants, substances added to mask or counteract a disagreeable taste or odor, flavors, dyes, fragrances, and substances added to improve appearance of the composition.
- Acceptable excipients include lactose, sucrose, starch powder, cellulose esters of alkanoic acids, cellulose alkyl esters, talc, stearic acid, magnesium stearate, magnesium oxide, sodium and calcium salts of phosphoric and sulfuric acids, gelatin, acacia gum, sodium alginate, polyvinylpyrrolidone, and/or polyvinyl alcohol, and then tableted or encapsulated for convenient administration.
- Such capsules or tablets may contain a controlled-release formulation as may be provided in a dispersion of active compound in hydroxypropyl- methyl cellulose, or other methods known to those skilled in the art.
- the pharmaceutical composition may be in the form of, for example, a tablet, capsule, suspension or liquid. If desired, other active ingredients may be included in the composition.
- compositions of the present invention may be administered by any suitable route, in the form of a pharmaceutical composition adapted to such a route, and in a dose effective for the treatment intended.
- the compositions may, for example, be administered parenterally, e.g., intravascularly, intraperitoneally, subcutaneously, or intramuscularly.
- parenteral administration e.g., saline solution, dextrose solution, or water may be used as a suitable carrier.
- Formulations for parenteral administration may be in the form of aqueous or non-aqueous isotonic sterile injection solutions or suspensions.
- solutions and suspensions may be prepared from sterile powders or granules having one or more of the carriers or diluents mentioned for use in the formulations for oral administration.
- the compounds may be dissolved in water, polyethylene glycol, propylene glycol, EtOH, corn oil, cottonseed oil, peanut oil, sesame oil, benzyl alcohol, sodium chloride, and/or various buffers.
- Other adjuvants and modes of administration are well and widely known in the pharmaceutical art.
- the serotonin type 3 receptor (5HT R) is a member of a superfamily of ligand- gated ion channels, which includes the muscle and neuronal nAChR, the glycine receptor, and the ⁇ -aminobutyric acid type A receptor.
- the 5HT 3 R exhibits a large degree of sequence homology with ⁇ 7 nAChR but functionally the two ligand-gated ion channels are very different.
- 7 nAChR is rapidly inactivated, is highly permeable to calcium and is activated by acetylcholine and nicotine.
- 5HT 3 R is inactivated slowly, is relatively impermeable to calcium and is activated by serotonin.
- Ondansetron a highly selective 5HT 3 R antagonist
- GTS-21 a highly selective ⁇ 7 nAChR agonist
- ⁇ 7 nAChR is a ligand-gated Ca " channel formed by a homopentamer of ⁇ 7 subunits.
- ⁇ -btx -bungarotoxin
- ⁇ 7 nAChR is expressed at high levels in the hippocampus, ventral tegmental area and ascending cholinergic projections from nucleus basilis to thalamocortical areas.
- ⁇ 7 nAChR agonists increase neurotransmitter release, and increase cognition, arousal, attention, learning and memory.
- Schizophrenia is a complex multifactorial illness caused by genetic and non- genetic risk factors that produce a constellation of positive and negative symptoms.
- the positive symptoms include delusions and hallucinations and the negative symptoms include deficits in affect, attention, cognition and information processing.
- No single biological element has emerged as a dominant pathogenic factor in this disease. Indeed, it is likely that schizophrenia is a syndrome that is produced by the combination of many low penetrance risk factors.
- Clozapine an "atypical" antipsychotic drug, is novel because it is effective in treating both the positive and some of the negative symptoms of this disease. Clozapine 's utility as a drug is greatly limited because continued use leads to an increased risk of agranulocytosis and seizure. No other antipsychotic drug is effective in treating the negative symptoms of schizophrenia. This is significant because the restoration of cognitive functioning is the best predictor of a successful clinical and functional outcome of schizophrenic patients (Green, M.F., Am J Psychiatry, 153:321- 30, 1996).
- One aspect of the cognitive deficit of schizophrenia can be measured by using the auditory event-related potential (P50) test of sensory gating.
- P50 auditory event-related potential
- EEG electroencepholographic
- Normal individuals respond to the first click with greater degree than to the second click.
- schizophrenics and schizotypal patients respond to both clicks nearly the same (Cullum, CM.
- schizophrenics express the same ⁇ 7 nAChR as non-schizophrenics.
- Selective ⁇ 7 nAChR agonists may be found using a functional assay on FLIPR
- FLIPR is designed to read the fluorescent signal from each well of a 96 or 384 well plate as fast as twice a second for up to 30 minutes.
- This assay may be used to accurately measure the functional pharmacology of ⁇ 7 nAChR and 5HT 3 R.
- To conduct such an assay one uses cell lines that expressed functional forms of the ⁇ 7 nAChR using the ⁇ 7/5-HT 3 channel as the drag target and cell lines that expressed functional 5HT 3 R. In both cases, the ligand-gated ion channel was expressed in SH-EP1 cells. Both ion channels can produce robust signal in the FLIPR assay.
- the compounds of the present invention are ⁇ 7 nAChR agonists and maybe used to treat a wide variety of diseases. For example, they may be used in treating schizophrenia, or psychosis.
- Schizophrenia is a disease having multiple aspects.
- drugs are generally aimed at controlling the positive aspects of schizophrenia, such as delusions.
- One drug, Clozapine is aimed at a broader spectrum of symptoms associated with schizophrenia. This drug has many side effects and is thus not suitable for many patients.
- a drug to treat the cognitive and attention deficits associated with schizophrenia.
- schizoaffective disorders or similar symptoms found in the relatives of schizophrenic patients.
- Psychosis is a mental disorder characterized by gross impairment in the patient's perception of reality. The patient may suffer from delusions, and hallucinations, and may be incoherent in speech. His behavior may be agitated and is often incomprehensible to those around him.
- the term psychosis has been applied to many conditions that do not meet the stricter definition given above. For example, mood disorders were named as psychoses.
- the conventional antipsychotic drugs include Chlorpromazine, Fluphenazine, Haloperidol, Loxapine, Mesoridazine, Molindone, Perphenazine, Pimozide, Thioridazine, Thiothixene, and Trifluoperazine. These drugs all have an affinity for the dopamine 2 receptor.
- Atypical antipsychotic drugs generally are able to alleviate positive symptoms of psychosis while also improving negative symptoms of the psychosis to a greater degree than conventional antipsychotics. These drugs may improve neurocognitive deficits. Extrapyramidal (motor) side effects are not as likely to occur with the atypical antipsychotic drugs, and thus, these atypical antipsychotic drugs have a lower risk of producing tardive dyskinesia. Finally these atypical antipsychotic drugs cause little or no elevation of prolactin. Unfortunately, these drugs are not free of side effects.
- the side effects include: agranulocytosis; increased risk of seizures, weight gain, somnolence, dizziness, tachycardia, decreased ejaculatory volume, and mild prolongation of QTc interval.
- the compounds of Formula I and the anti-psychotic drugs can be administered simultaneously or at separate intervals.
- the compounds of Formula I and the anti-psychotic drugs can be inco ⁇ orated into a single pharmaceutical composition, e.g., a pharmaceutical combination therapy composition.
- two separate compositions i.e., one containing compounds of Formula I and the other containing anti-psychotic drugs, can be administered simultaneously.
- anti-psychotic drugs include, but are not limited to, Thorazine, Mellaril, Trilafon, Navane, Stelazine, Permitil, Prolixin, Risperdal, Zyprexa, Seroquel, ZELDOX, Acetophenazine, Carphenazine, Chlorprothixene, Droperidol, Loxapine, Mesoridazine, Molindone, Ondansetron, Pimozide, Prochlorperazine, and Promazine.
- a pharmaceutical combination therapy composition can include therapeutically effective amounts of the compounds of Formula I, noted above, and a therapeutically effective amount of anti-psychotic drugs. These compositions may be formulated with common excipients, diluents or carriers, and compressed into tablets, or formulated elixirs or solutions for convenient oral administration or administered by intramuscular intravenous routes. The compounds can be administered rectally, topically, orally, sublingually, or parenterally and maybe formulated as sustained relief dosage forms and the like.
- compositions containing compounds of Formula I and anti-psychotic drugs are administered on a different schedule.
- One may be administered before the other as long as the time between the two administrations falls within a therapeutically effective interval.
- a therapeutically effective interval is a period of time beginning when one of either (a) the compounds of Formula I, or (b) the anti-psychotic drugs is administered to a human and ending at the limit of the beneficial effect in the treatment of schizophrenia or psychosis of the combination of (a) and (b).
- the methods of administration of the compounds of Formula I and the anti-psychotic drugs may vary. Thus, either agent or both agents may be administered rectally, topically, orally, sublingually, or parenterally.
- the compounds of the present invention are ⁇ 7 nAChR agonists. Therefore, as another aspect of the present invention, the compounds of the present invention may be used to treat a variety of diseases including cognitive and attention deficit symptoms of Alzheimer's, neurodegeneration associated with diseases such as Alzheimer's disease, pre-senile dementia (also known as mild cognitive impairment), and senile dementia.
- diseases including cognitive and attention deficit symptoms of Alzheimer's, neurodegeneration associated with diseases such as Alzheimer's disease, pre-senile dementia (also known as mild cognitive impairment), and senile dementia.
- Alzheimer's disease has many aspects, including cognitive and attention deficits.
- these deficits are treated with cholinesterase inhibitors. These inhibitors slow the break down of acetylcholine, and thereby provide a general nonspecific increase in the activity of the cholinergic nervous system. Since the drugs are nonspecific, they have a wide variety of side effects.
- Neurodegeneration is a common problem associated with diseases such as
- Alzheimer's disease While the current drugs treat some of the symptoms of this disease, they do not control the underlying pathology of the disease. Accordingly, it would be desirable to provide a drug that can slow the progress of Alzheimer's disease.
- Pre-senile dementia mimild cognitive impairment
- Mild cognitive impairment is distinguished from senile dementia in that mild cognitive impairment involves a more persistent and troublesome problem of memory loss for the age of the patient.
- Senile dementia is not a single disease state. However, the conditions classified under this name frequently include cognitive and attention deficits. Generally, these deficits are not treated. Accordingly, there is a need for a drug that provides improvement in the cognitive and attention deficits associated with senile dementia.
- the compounds of the present invention are ⁇ 7 nAChR agonists. Therefore, yet other diseases to be treated with compounds of the present invention include treating the cognitive and attention deficits as well as the neurodegeneration associated with any one or more or combination of the following: attention deficit disorder, attention deficit hyperactivity disorder, depression, anxiety, general anxiety disorder, post traumatic stress disorder, mood and affective disorders, amyotrophic lateral sclerosis, borderline personality disorder, traumatic brain injury, behavioral and cognitive problems associated with brain tumors, AIDS dementia complex, dementia associated with Down's syndrome, dementia associated with Lewy Bodies, Huntington's disease, Parkinson's disease, tardive dyskinesia, Pick's disease, dysregulation of food intake including bulemia and anorexia nervosa, withdrawal symptoms associated with smoking cessation and dependant drug cessation, Gilles de la Tourette's Syndrome, age-related macular degeneration, glaucoma, neurodegeneration associated with glaucoma, or symptoms associated with pain. Attention deficit disorder is generally treated with
- ADHD Attention deficit hyperactivity disorder
- Treatment may include medications such as methylphenidate, dextroamphetamine, or pemoline, which act to decrease impulsivity and hyperactivity and to increase attention. No "cure" for ADHD currently exists. Children with the disorder seldom outgrow it; therefore, there is a need for appropriate medicaments.
- HCA heterocyclic antidepressants
- MAOI's monoamine oxidase inhibitors
- Common side effects from HCA's are sedation and weight gain. In elderly patients with organic brain disease, the side effects from HCA's can also include seizures and behavioral symptoms. The main side effects from using MAOI's occur from dietary and drug interactions. Therefore, agents with fewer side effects would be useful.
- Anxiety disorders (disorders with prominent anxiety or phobic avoidance), represent an area of umet medical needs in the treatment of psychiatric illness. See Diagnostic & Statistical Manual of Mental Disorders, IV (1994), pp 393-394, for various disease forms of anxiety.
- GAD General anxiety disorder
- Anxiety also includes post-traumatic stress disorder (PTSD), which is a form of anxiety triggered by memories of a traumatic event that directly affected the patient or that the patient may have witnessed.
- PTSD post-traumatic stress disorder
- the disorder commonly affects survivors of traumatic events including sexual assault, physical assault, war, torture, natural disasters, an automobile accident, an airplane crash, a hostage situation, or a death camp.
- the affliction also can affect rescue workers at an airplane crash or a mass shooting, someone who witnessed a tragic accident or someone who has unexpectedly lost a loved one.
- Treatment for PTSD includes cognitive-behavioral therapy, group psychotherapy, and medications such as Clonazepam, Lorazepam and selective serotonin-reuptake inhibitors such as Fluoxetine, Sertraline, Paroxetine, Citalopram and Fluvoxamine. These medications help control anxiety as well as depression.
- Various forms of exposure therapy (such as systemic desensitization and imaginal flooding) have all been used with PTSD patients. Exposure treatment for PTSD involves repeated reliving of the trauma, under controlled conditions, with the aim of facilitating the processing of the trauma. Therefore, there is a need for better pharmaceutical agents to treat post traumatic stress disorder.
- HCA's heterocyclic antidepressant
- MAOI's monoamine oxidase inhibitors
- Benign side effects from the use of lithium include, but are not limited to, weight gain, nausea, diarrhea, polyuria, polydipsia, and tremor. Toxic side effects from lithium can include persistent headache, mental confusion, and may reach seizures and cardiac arrhythmias. Therefore, agents with less side effects or interactions with food or other medications would be useful.
- Borderline personality disorder although not as well known as bipolar disorder, is more common. People having borderline personality disorder suffer from a disorder of emotion regulation. Pharmaceutical agents are used to treat specific symptoms, such as depression or thinking distortions.
- AIDS Acquired immune deficiency syndrome
- HIN infection can cause other problems such as, but not limited to, difficulties in thinking, otherwise known as AIDS dementia complex. Therefore, there is a need to drugs to relieve the confusion and mental decline of persons with AIDS.
- AmyotiOphic lateral sclerosis also known as Lou Gehrig's disease, belongs to a class of disorders known as motor neuron diseases wherein specific nerve cells in the brain and spinal cord gradually degenerate to negatively affect the control of voluntary movement.
- motor neuron diseases wherein specific nerve cells in the brain and spinal cord gradually degenerate to negatively affect the control of voluntary movement.
- Brain tumors are abnormal growths of tissue found inside of the skull. Symptoms of brain tumors include behavioral and cognitive problems. Surgery, radiation, and chemotherapy are used to treat the tumor, but other agents are necessary to address associated symptoms. Therefore, there is a need to address the symptoms of behavioral and cognitive problems.
- Dementia with Lewy Bodies is a neurodegenerative disorder involving abnormal structures known as Lewy bodies found in certain areas of the brain. Symptoms of dementia with Lewy bodies include, but are not limited to, fluctuating cognitive impairment with episodic delirium.
- Parkinson's disease is a neurological disorder characterized by tremor, hypokinesia, and muscular rigidity. Currently, there is no treatment to stop the progression of the disease. Therefore, there is a need of a pharmaceutical agent to address Parkinson's.
- Tardive dyskinesia is associated with the use of conventional antipsychotic drugs. This disease is characterized by involuntary movements most often manifested by puckering of the lips and tongue and/or writhing of the arms or legs. The incidence of tardive dyskinesia is about 5% per year of drug exposure among patients taking conventional antipsychotic drugs. In about 2% of persons with the disease, tardive dyskinesia is severely disfiguring. Currently, there is no generalized treatment for tardive dyskinesia. Furthermore, the removal of the effect-causing drugs is not always an option due to underlying problems. Therefore, there is a need for a pharmaceutical agent to address the symptoms of tardive dyskinesia.
- Pick's disease results from a slowly progressive deterioration of social skills and changes in personality with the resulting symptoms being impairment of intellect, memory, and language. Common symptoms include memory loss, lack of spontaneity, difficulty in thinking or concentrating, and speech disturbances.
- Common symptoms include memory loss, lack of spontaneity, difficulty in thinking or concentrating, and speech disturbances.
- antipsychotic medications may alleviate symptoms in FTD patients who are experiencing delusions or hallucinations. Therefore, there is a need for a pharmaceutical agent to treat the progressive deterioration of social skills and changes in personality and to address the symptoms with fewer side effects.
- Dysregulation of food intake associated with eating disease involve neurophysiological pathways.
- Anorexia nervosa is hard to treat due to patients not entering or remaining in after entering programs.
- Cognitive behavioral therapy has helped patients suffering from bulemia nervosa; however, the response rate is only about 50% and current treatment does not adequately address emotional regulation. Therefore, there is a need for pharmaceutical agents to address neurophysiological problems underlying diseases of dysregulation of food intake.
- Cigarette smoking has been recognized as a major public health problem for a long time. However, in spite of the public awareness of health hazard, the smoking habit remains extraordinarily persistent and difficult to break. There are many treatment methods available, and yet people continue to smoke. Administration of nicotine transdermally, or in a chewing gum base is common treatments. However, nicotine has a large number of actions in the body, and thus can have many side effects. It is clear that there is both a need and a demand of long standing for a convenient and relatively easy method for aiding smokers in reducing or eliminating cigarette consumption. A drug that could selectively stimulate only certain of the nicotinic receptors would be useful in smoke cessation programs.
- Smoke cessation programs may involve oral dosing of the drug of choice.
- the drug may be in the form of tablets. However, it is preferred to administer the daily dose over the waking hours, by administration of a series of incremental doses during the day.
- the preferred method of such administration is a slowly dissolving lozenge, troche, or chewing gum, in which the drug is dispersed.
- Another drug in treating nicotine addiction is Zyban. This is not a nicotine replacement, as are the gum and patch. Rather, this works on other areas of the brain, and its effectiveness is to help control nicotine craving or thoughts about cigarette use in people trying to quit.
- Zyban is not very effective and effective drugs are needed to assist smokers in their desire to stop smoking.
- These drugs may be administered transdermally through the use of skin patches. In certain cases, the drugs may be administered by subcutaneous injection, especially if sustained release formulations are used.
- Drag use and dependence is a complex phenomenon, which cannot be encapsulated within a single definition. Different drugs have different effects, and therefore different types of dependence. Drag dependence has two basic causes, that is, tolerance and physical dependence. Tolerance exists when the user must take progressively larger doses to produce the effect originally achieved with smaller doses. Physical dependence exists when the user has developed a state of physiologic adaptation to a drug, and there is a withdrawal (abstinence) syndrome when the drag is no longer taken. A withdrawal syndrome can occur either when the drug is discontinued or when an antagonist displaces the drug from its binding site on cell receptors, thereby counteracting its effect. Drug dependence does not always require physical dependence.
- Drug dependence often involves psychological dependence, that is, a feeling of pleasure or satisfaction when taking the drug. These feelings lead the user to repeat the drag experience or to avoid the displeasure of being deprived of the drug.
- Drugs that produce strong physical dependence such as nicotine, heroin and alcohol are often abused, and the pattern of dependence is difficult to break. Drugs that produce dependence act on the CNS and generally reduce anxiety and tension; produce elation, euphoria, or other pleasurable mood changes; provide the user feelings of increased mental and physical ability; or alter sensory perception in some pleasurable manner.
- narcotic addiction Among the drags that are commonly abused are ethyl alcohol, opioids, anxiolytics, hypnotics, cannabis (marijuana), cocaine, amphetamines, and hallucinogens.
- the current treatment for drag-addicted people often involves a combination of behavioral therapies and medications. Medications, such as methadone or LAAM (levo-alpha-acetyl-methadol), are effective in suppressing the withdrawal symptoms and drug craving associated with narcotic addiction, thus reducing illicit drug use and improving the chances of the individual remaining in freatment.
- the primary medically assisted withdrawal method for narcotic addiction is to switch the patient to a comparable drag that produces milder withdrawal symptoms, and then gradually taper off the substitute medication.
- the medication used most often is methadone, taken orally once a day. Patients are started on the lowest dose that prevents the more severe signs of withdrawal and then the dose is gradually reduced. Substitutes can be used also for withdrawal from sedatives. Patients can be switched to long-acting sedatives, such as diazepam or phenobarbital, which are then gradually reduced.
- Gilles de la Tourette's Syndrome is an inherited neurological disorder.
- the disorder is characterized by uncontrollable vocal sounds called tics and involuntary movements.
- the symptoms generally manifest in an individual before the person is 18 years of age.
- the movement disorder may begin with simple tics that progress to multiple complex tics, including respiratory and vocal ones.
- Vocal tics may begin as grunting or barking noises and evolve into compulsive utterances.
- Coprolalia involuntary scatologic utterances
- Tics tend to be more complex than myoclonus, but less flowing than choreic movements, from which they must be differentiated. The patient may voluntarily suppress them for seconds or minutes.
- Clonidine may be used for simple and complex tics. Long-term use of Clonidine does not cause tardive dyskinesia; its limiting adverse effect is hypotension. In more severe cases, antipsychotics, such as Haloperidol may be required, but side effects of dysphoria, parkinsonism, akathisia, and tardive dyskinesia may limit use of such antipsychotics. There is a need for safe and effective methods for treating this syndrome.
- Age-related macular degeneration is a common eye disease of the macula which is a tiny area in the retina that helps produce sharp, central vision required for "straight ahead" activities that include reading and driving. Persons with AMD lose their clear, central vision. AMD takes two forms: wet and dry. In dry AMD, there is a slow breakdown of light-sensing cells in the macula. There currently is no cure for dry AMD. In wet AMD, new, fragile blood vessels growing beneath the macula as dry AMD worsens and these vessels often leak blood and fluid to cause rapid damage to the macula quickly leading to the loss of central vision. Laser surgery can treat some cases of wet AMD. Therefore, there is a need of a pharmaceutical agent to address AMD.
- Glaucoma is within a group of diseases occurs from an increase in intraocular pressure causing pathological changes in the optical disk and negatively affects the field of vision.
- Medicaments to treat glaucoma either decrease the amount of fluid entering the eye or increase drainage of fluids from the eye in order to decrease intraocular pressure.
- current drags have drawbacks such as not working over time or causing side effects so the eye-care professional has to either prescribe other drags or modify the prescription of the drug being used. There is a need for safe and effective methods for treating problems manifesting into glaucoma.
- Alpha 7 nicotinic agonists may stimulate the release of inhibitory amino acids such as GABA which will dampen hyperexcitablity.
- Alpha 7 nicotinic agonists are also directly neuroprotective on neuronal cell bodies. Thus alpha 7 nicotinic agonists have the potential to be neuroprotective in glaucoma. Persons afflicted with pain often have what is referred to as the "terrible triad" of suffering from the pain, resulting in sleeplessness and sadness, all of which are hard on the afflicted individual and that individual's family.
- typical and atypical anti-psychotic drugs also called an anti-psychotic agent
- Such combination therapy lowers the effective dose of the anti-psychotic drug and thereby reduces the side effects of the anti-psychotic drugs.
- Some typical anti-psychotic drugs that may be used in the practice of the invention include Haldol.
- Some atypical antipsychotic drugs include Ziprasidone, Olanzapine, Resperidone, and Quetiapine.
- Compounds of Formula I can be prepared as shown in Scheme 1.
- Suitable activating reagents are well known in the art, for examples see Kiso, Y., Yajima, H. "Peptides” pp. 39-91, San Diego, CA, Academic Press, (1995), and include, but are not limited to, agents such as carbodiimides, phosphonium and uronium salts (such as HATU).
- the acid is activated using HATU or is converted to the acyl azide by using DPPA.
- the appropriate amine precursor is added to a solution of the appropriate anhydride or azide to give the desired final compounds.
- the ester (Lv being OMe or OEt) may be reacted directly with the amine precursor in refluxing methanol or ethanol to give the compounds of Formula I.
- the acid is converted into a mixed anhydride by treatment with bis (2-oxo-3-oxazolidinyl) phosphinic chloride in the presence of TEA with CH 2 C1 2 or CHC1 3 as the solvent.
- the resulting anhydride solution is directly reacted with l-azabicyclo[3.2J]octan-3-amine added neat or using DMF or aqueous DMF as solvent.
- the ester (Lv being OMe or OEt) may be reacted directly with the amine in refluxing methanol or ethanol to give the compounds of Formula I.
- 6-substituted-[2.2.2]-3-amines (Azabicyclo I) are known in the art. The preparation of compounds where R is other than H is described in Acta Pol. Pharm. 179-85 (1981). Alternatively, the 6-substituted-[2.2.2]-3-amine can be prepared by reduction of an oxime or an imine of the corresponding 6-substituted-3- quinuclidinone by methods known to one of ordinary skill in the art (see J. Labelled Compds. Radiopharm., 53-60 (1995), J. Med. Chem. 988-995, (1998), Synth. Commun. 1895-191 1 (1992), Synth. Commun. 2009-2015 (1996)).
- the 6-substituted-[2.2.2]-3-amine can be prepared from a 6-substituted-3- hydroxyquinuclidine by Mitsunobu reaction followed by deprotection as described in Synth. Commun. 1895-191 1 (1995).
- the 6-substituted-[2.2.2]-3-amine can be prepared by conversion of a 6-substituted-3-hydroxyquinuclidine into the corresponding mesylate or tosylate, followed by displacement with sodium azide and reduction as described in /. Med. Chem. 587-593 (1975).
- the oximes can be prepared by treatment of the 3-quinuclidinones with hydroxylamine hydrochloride in the presence of base.
- the imines can be prepared by treatment of the 3-quinuclidinones with a primary amine under dehydrating conditions.
- the 3-hydroxyquinuclidines can be prepared by reduction of the 3- quinuclidinones.
- the 6-substituted-3-quinuclidinones can be prepared by known procedures (see /. Gen. Chem. Russia 3791-3795, (1963), J. Chem. Soc. Perkin Trans. 7409-420 (1991), J. Org. Chem. 3982-3996 (2000)).
- Compounds for Azabicyclo II where R 2 is other than H can also be prepared by modification of intermediates described in the synthesis of exo-3 -amino- 1- azabicyclo[2.2J]heptane as the bis(hydro para-toluenesulfonate) salt, described in detail herein.
- Int 6 can be oxidized to the aldehyde and treated with an organometallic reagent to provide Int 20 using procedures described in Tetrahedron (1999), 55, p 13899.
- Int 20 can be converted into the amine using methods described for the synthesis of ex -3-amino-l-azabicyclo[2.2J]heptane as the bis(hydro para- toluenesulfonate) salt. Once the amine is obtained, the desired salt can be made using standard procedures.
- Lv can be -CH 2 Ph, -CH(Me)Ph, -OH, -OMe, or -OCH 2 Ph.
- the respective amine precursors for Azabicyclo III and Azabicyclo IV can be prepared by reduction of an oxime or an imine of the corresponding N-2-azabicyclo[2.2J]- heptanone by methods known to one skilled in the art (see J. Labelled Compds.
- the oximes can be prepared by treatment of the N-2-azabicyclo[2.2J]heptanones with hydroxylamine hydrochloride in the presence of a base.
- the imines can be prepared by treatment of the N-2- azabicyclo[2.2J]-heptanones with a primary amine under dehydrating conditions.
- the N-2-azabicyclo[2.2J]heptanones can be prepared by known procedures (see Jet. Lett. 1419-1422 (1999), J. Med. Chem. 2184-2191 (1992), /. Med. Chem. 706-720 (2000), / Org. Chem., 4602-4616 (1995)).
- exo- and e « o-l-azabicyclo[3.2J]ocran-3-amines are prepared from 1- azabicyclic[3.2J]octan-3-one (Thill, B. P., Aaron, H. S., / Org. Chem., 4376-4380 (1968)) according to the general procedure as discussed in Lewin, A.H., et al., /. Med. Chem., 988-995 (1998).
- thioamides can be prepared from the requisite thioester by direct displacement of the thioester with an amino precursor of the Azabicyclo, for example, see Scheme 2.
- the thioester can be prepared as described in / Organometallic Chem., 95-98 (1987).
- a reagent such and Lawesson's reagent (see Lawesson et. al. in Bull. Soc. Chim.
- Step B Preparation of ethyl E-4-(benzylamino)-2-butenoate (Int 2).
- Ethyl E-4-bromo-2-butenoate (10 mL, 56 mmol, tech grade) is added to a stirred solution of benzylamine (16 mL, 146 mmol) in CH 2 C1 2 (200 mL) at rt.
- the reaction mixture stirs for 15 min, and is diluted with ether (1 L).
- the mixture is washed with saturated aqueous NaHCO 3 solution (3x) and water, dried over Na 2 SO 4 , filtered and concentrated in vacuo.
- the residue is purified by flash chromatography on silica gel.
- Step C Preparation of trar ⁇ -4-nitro- 1 -(phenylmethyl)-3-pyrrolidineacetic acid ethyl ester (Int 3).
- Step D Preparation of tr ⁇ «5 , -4-amino- 1 -(phenylmethyl)-3-pyrrolidineacetic acid ethyl ester (Int 4).
- Step E Preparation of trans-4- ⁇ 1 , 1 -dimethylethoxycarbonylamido)- 1 -
- Di-tert-butyldicarbonate (3.67 g, 16.8 mmol) is added to a stirred solution of Int 4 (2.94 g, 11.2 mmol) in CH 2 C1 2 (30 mL) cooled in an ice bath. The reaction is allowed to warm to rt and stirred overnight. The mixture is concentrated in vacuo. The crude product is purified by flash chromatography on silica gel.
- L-AIH powder (627 mg, 16.5 mmol) is added in small portions to a stirred solution of Int 5 (3.0 g, 8.3 mmol) in anhydrous THF (125 mL) in a -5°C bath. The mixture is stirred for 20 min in a -5°C bath, then quenched by the sequential addition of water (0.6 mL), 15% (w/v) aqueous NaOH (0.6 mL) and water (1.8 mL). Excess anhydrous K 2 CO 3 is added, and the mixture is stirred for 1 h, then filtered. The filtrate is concentrated in vacuo. The residue is purified by flash chromatography on silica gel.
- Int 6 is a racemic mixture that can be resolved via chromatography using a Diacel chiral pack AD column. From the two enantiomers thus obtained, the
- the methods described herein use the (+)-enantiomer of Int 6 to obtain the optically pure ex -4-S final compounds. However, the methods used are equally applicable to the (-)-enantiomer of Int 6, making non-critical changes to the methods provided herein to obtain the optically pure exo-4-R final compounds.
- Step G Preparation of exo 3-(t ⁇ rt-butoxycarbonylamino)-l- azabicyclo[2.2.1 ]heptane (Int 7).
- TEA 8.0 g, 78.9 mml
- a stirred solution of frit 6 2.5 g, 7.8 mmol
- CH 2 C1 2 50 mL
- CH 3 SO 2 Cl 5.5 g, 47.8 mmol
- the resulting yellow mixture is diluted with saturated aqueous NaHCO 3 solution, extracted with CH 2 C1 2 several times until no product remains in the aqueous layer by TLC.
- the organic layers are combined, washed with brine, dried over Na 2 SO 4 and concentrated in vacuo.
- the residue is dissolved in EtOH (85 mL) and is heated to reflux for 16 h.
- the reaction mixture is allowed to cool to rt, transferred to a Parr bottle and treated with 10% Pd/C catalyst (1.25 g).
- the bottle is placed under an atmosphere of hydrogen (53 psi) for 16 h.
- the mixture is filtered through Celite, and fresh catalyst (10% Pd/C, 1.25 g) is added. Hydrogenolysis continues overnight. The process is repeated three more times until the hydrogenolysis is complete.
- the final mixture is filtered through Celite and concentrated in vacuo.
- the residue is purified by flash chromatography on silica gel.
- Step I Preparation of ethyl 5-hydroxy-6-oxo- 1 ,2,3 ,6-tetrahydropyridine-4- carboxylate (Int 10).
- Absolute EtOH (92.0 mL, 1.58 mol) is added to a mechanically stirred suspension of potassium ethoxide (33.2 g, 395 mmol) in dry toluene (0.470 L).
- 2-pyrrolidinone (33.6 g, 395 mmol) is added, and then a solution of diethyl oxalate (53J mL, 390 mmol) in toluene (98 mL) is added via an addition funnel.
- toluene (118 mL) and EtOH (78 mL) is added sequentially.
- the mixture is heated to reflux for 18 h.
- Step J Preparation of ethyl cw-3-hydroxy-2-oxopiperidine-4-carboxylate (Int 1 1).
- Step K Preparation of cis- 4-(hydroxymethyl)piperidin-3-ol (Int 12).
- Int 11 (3.7 g, 19.9 mmol) as a solid is added in small portions to a stirred solution of L1AIH in THF (80 mL of a 1.0 M solution) in an ice- water bath.
- the mixture is warmed to rt, and then the reaction is heated to reflux for 48 h.
- the mixture is cooled in an ice-water bath before water (3.0 mL, 170 mmol) is added dropwise, followed by the sequential addition of NaOH (3.0 mL of a 15% (w/v) solution) and water (9.0 mL, 500 mmol).
- Excess K 2 CO 3 is added, and the mixture is stirred vigorously for 15 min.
- Step L Preparation of benzyl cw-3-hydroxy-4-(hydroxymethyl)piperidine-l- carboxylate (Int 13).
- N-(benzyloxy carbonyloxy)succinimide (3.04 g, 12.2 mmol) is added to a stirred solution of Int 12 (1.6 g, 12.2 mmol) in saturated aqueous ⁇ aHCO (15 mL) at rt.
- the mixture is stirred at rt for 18 h.
- the organic and aqueous layers are separated.
- Step M Preparation of benzyl c/s-3-hydroxy-4-[(4-methylphenyl)sulfonyl oxymethyl]piperidine- 1 -carboxylate (Int 14).
- E ⁇ r ⁇ -toluenesulfonyl chloride (1.0 g, 5.3 mmol) is added to a stirred solution of Int 13 (3.6 g, 5.3 mmol) in pyridine (10 mL) in a -15°C bath. The mixture is stirred for 4 h, followed by addition of HCl (4.5 mL of a 6.0 M solution). CH 2 C1 2 (5 mL) is added. The organic and aqueous layers are separated. The aqueous layer is extracted with CH 2 C1 2 .
- the pH of the aqueous layer is adjusted to 9 with 50% aqueous NaOH solution.
- the aqueous layer is extracted with CH 2 C1 2 (3X), and the combined organic layers are washed with brine, dried over Na 2 SO 4 , filtered and concentrated in vacuo.
- the crude product is purified by flash chromatography on silica gel. Elution with CHCl 3 -MeOH-NH 4 OH (92:7:1) affords Int 16 as a colorless oil (41 % yield): ⁇ NMR (CDC1 3 ) 64.1, 3.2, 2.8, 2.7-2.5, 2.2, 1.9, 1.5.
- Step P Preparation of e « o-3-amino-l-azabicyclo[2.2J]heptane bis(hydro- /? ⁇ r -toluenesulfonate).
- the resulting oxime (3J mmol) is treated with acetic acid (30 mL) and hydrogenated at 50 psi over PtO 2 (50 mg) for 12 h. The mixture is then filtered and evaporated. The residue is taken up in a minimal amount of water (6 mL) and the pH is adjusted to >12 using solid NaOH. The mixture is then extracted with ethyl acetate (4 X 25 mL), dried over MgSO 4 , filtered, treated with ethereal HCl, and evaporated to give e « ⁇ fo-[3.2J]-Amine.
- This amine can also be prepared according to the following method:
- a suspension (3S)-l-[(S)-l-phenethyl]-5-oxo-3-pyrrolidine-carboxylic acid (82.30 g, 352.8 mmol) in Et 2 O (200 mL) is added in small portions to a slurry of LiAlH 4 (17.41 g, 458.6 mmol) in Et 2 O (700 mL).
- the mixture begins to reflux during the addition.
- the addition funnel containing the suspension is rinsed with Et 2 O (2 x 50 mL), and the mixture is heated in a 50 °C oil bath for an additional 2 h and first allowed to cool to rt and then further cooled using an ice bath.
- the mixture is carefully treated with H 2 O (62 mL).
- Acetyl chloride (270 mL, 3.8 mol) is carefully added to a flask containing chilled (0°C) methanol (1100 mL). After the addition is complete, the acidic solution is stirred for 45 min (0 °C) and then (3R)-l-[(S)-l-phenethyl]-3-
- This foam (10.1 g, 38 mmol) is taken up in MeOH (500 mL), 10% Pd(C) (3.0 g) added and the mixture is hydrogenated (45 psi) overnight. The mixture is filtered and re-subjected to the reduction conditions (9J g, 10% Pd/C, 50 psi). After 5 h, TLC indicates the consumption of the (5i?)-3-oxo-l-[(lS)-l-phenylethyl]-l- azoniabicyclo[3.2J]octane chloride.
- the amine can be coupled to form the appropriate amides or thioamides as a racemic mixture.
- the racemic mixture can then be resolved by chromatography using chiral columns or chiral HPLC, techniques widely known in the art, to provide the requisite resolved enantiomers 3(R) and 3(S) of said amides or thioamides.
- 2-Methyl-l,3-benzoxazole-6-carboxylic acid can be coupled with the exo- or endo-[2.2.l] -Amine using procedures discussed herein to give Example 4(i).
- l-indane-5-yl-ethanone 1.0 g, 6.2 mmol
- the solution is stirred at 55°C for 2 h, followed by cooling to rt.
- Solid sodium bisulfite is added until the solution becomes clear.
- the mixture is diluted with water, followed by aqueous hydrochloric acid (6.0 M). The solid that forms is filtered and washed several times with water.
- Example 6(i) The free base of Example 6(i) is obtained in 99% using the coupling procedure for Example l(i), making non-critical changes.
- Example 6(i) is collected by filtration, washed with acetone, and dried in vacuo overnight to give 113 mg (59%) of Example 6(i) as a white solid: 1H NMR (400 MHz, CD 3 OD) ⁇ 7.72, 7.64, 7.32, 6.71, 4.22-4J9, 3.71- 3.66, 3.43-3.36, 3.25-3J9, 3.03, 2.97, 2.20-2J0, 1.86-1J9.
- 2-Chloro-3-pyridinol (20.0 g, 0J54 mole), ⁇ aHCO 3 (19.5g, 0.232 mole, 1.5 equ), and 150 mL of water are placed in a flask.
- the flask is placed in an oil bath at 90°C, and after 5 min, 37% aqueous formaldehyde (40.5 mL, 0.541 mole, 3.5 equ) is added in six unequal doses in the following order: 12 mL, 3 x 8 L, then 2.2 mL all at 90-min intervals and then the final 2.3 mL after the reaction stirs for 15 h at 90°C.
- the reaction is stirred at 90°C for another 4 h and then cooled by placing the flask in an ice bath.
- the pH of the reaction is then adjusted to 1 using 6N HCl.
- the reaction is stirred for 1.5 h in an ice bath allowing an undesired solid to form.
- the undesired solid is removed by filtration, and the filtrate is extracted seven times with EtOAc.
- the combined organic extracts are concentrated in vacuo, toluene is added to the flask and removed in vacuo to azeotrope water, and then CH 2 Cl 2 is added and removed in vacuo to obtain 2-chloro-6-(hydroxymethyl)-3-pyridinol as a pale yellow solid (81% yield) sufficiently pure for subsequent reaction.
- 4-(Benzylamino)-2-chloro-6-(hydroxymethyl)-3-pyridinol may be produced by amination of 2-chloro-6-(hydroxymethyl)-4-iodo-3-pyridinol (C12) with benzylamine under palladium catalysis.
- Amination of aryl iodides with primary amines such as benzylamine under palladium catalysis is generally described in a review by B.H. Yang and S.L. Buchwald inJ Organomet. Chem., 576, 125-146, 1999 and in greater detail in the references therein.
- 4-(Benzylamino)-2-chloro-6-(hydroxymethyl)-3-pyridinol may be oxidized to 4-(benzylamino)-2-chloro-3-hydroxypyridine-6-carboxaldehyde (C14) under a wide variety of conditions (e.g., TPAP and NMO in CH 2 C1 2 ).
- 4-(Benzylamino)-2-chloro-3- hydroxypyridine-6-carboxaldehyde may be oxidized to produce the corresponding carboxylic acid C15 using an oxidizing reagent such as NaClO 2 and KH 2 PO 4 in DMSO/H 2 O or Ag 2 O, or hydrogen peroxide or ruthenium tetroxide.
- Removal of the benzyl group and the chloro group of Acid C15 may be accomplished by utilizing hydrogen or a hydrogen source (e.g., cyclohexene, cyclohexadiene, ammonium formate, hydrazine, etc.) in the presence of Pd/C or other catalyst, under a variety of conditions and in various solvents, to produce 4-amino-5- hydroxypyridine-2-carboxylic acid (Acid C16).
- hydrogen or a hydrogen source e.g., cyclohexene, cyclohexadiene, ammonium formate, hydrazine, etc.
- Cyclocondensation of Acid C16 with trimethyl orthoformate in the presence of catalytic ⁇ r ⁇ -toluenesulfonic acid may be conducted to produce [l,3]-oxazolo[5,4- c]pyridine-6-carboxylic acid (Acid C17 .
- Acid C21 can be made by the saponification of the methyl ester C20, which can be made pursuant to Wynberg, Hans, et al., Reel. Trav. Chim. Pays-Bas (1968), 87(10), 1006-1010. Acid C2J can then be coupled with the exo- or endo-[2.2 ⁇ ]- Amine using methods discussed herein to provide Example 9 as the free base that can be made into a suitable salt.
- Example 10(i) is collected by filtration, washed with acetone, and dried under high vacuum overnight to give 82 mg (81%) of Example 10(i) as a white solid: ⁇ NMR (400 MHz, CD 3 OD) ⁇ 8.43, 8.35, 8.21, 7.87-7.84, 7.58-7.56, 6.53, 3.81, 3.15, 2.80, 2.70-2.60, 1.75, 1.35.
- Example 1 l(i) is obtained in 81% yield by coupling lH-indazole-6-carboxylic acid with exo- ⁇ 4S)-[2.2.1 ]-3-amine using conditions according to Example 10(i), making non-critical changes.
- the salt is then formed to obtain Example 1 l(i) in 77% yield from the coupling to salt: ⁇ NMR (400 MHz, CD 3 OD) ⁇ 8.15, 8.10, 7.88, 7.63, 6.71 , 4.27-4.25, 3.74-3.69, 3.46-3.36, 3.23-3.21, 3.08, 2.24-2.14, 1.88-1.81.
- Example 12(i) is filtered, washed with acetone and dried in vacuo to afford 209 mg (92%) of Example 12(i) as a white solid: 1H NMR (400 MHz, DMSO- ⁇ ) ⁇ 8.61, 8.18, 7.55, 6.54, 6.25, 3.76, 3.06, 2.89-2.79, 2.63-2.58, 2.46, 1.68, 1.30.
- Example 21 N-(l -(6-methyl)-azabicyclo[2.2.1 ]hept-3-yl)- 1 ,3-benzoxazole-5- carboxamide.
- Example 22 N-(l-(6-methyl)-azabicyclo[2.2J]hept-3-yl)-2-methyl-l,3-benzoxazole- 5-carboxamide.
- Example 27 N-(l-(6-methyl)-azabicyclo[2.2J]hept-3-yl)-l,3-benzodioxole-5- carboxamide.
- Example 28 N-(l-(6-methyl)-azabicyclo[2.2J]hept-3-yl)-l,3-oxazolo[5,4- c]pyridine-6-carboxamide.
- Example 29 N-(l-(6-methyl)-azabicyclo[2.2J]hept-3-yl)-2-benzoisothiophene-5- benzamide.
- Example 30 N-(l-(6-methyl)-azabicyclo[2.2J]hept-3-yl)-lH-indazole-5- carboxamide.
- Example 31 N-(l-(6-methyl)-azabicyclo[2.2J]hept-3-yl)-lH-indazole-6- carboxamide.
- Example 32 N-(l -(6-methyl)-azabicyclo[2.2.1 ]hept-3-yl)- 1 ,3-dioxolo[4,5- c]pyridine-6-carboxamide.
- Example 41 N-(l-(6-methyl)-azabicyclo[2.2.2]oct-3-yl)- 1 ,3-benzoxazole-5- carboxamide.
- Example 42 N-(l -(6-methyl)-azabicyclo[2.2.2]oct-3-yl)-2-methyl- 1 ,3-benzoxazole-
- Example 43 N-(l-(6-methyl)-azabicyclo[2.2.2]oct-3-yl)-l ,3-benzoxazole-6- carboxamide.
- Example 4 N-(l-(6-methyl)-azabicyclo[2.2.2]oct-3-yl)-2-methyl-l ,3-benzoxazole-
- Example 45 N-(l -(6-methyl)-azabicyclo[2.2.2]oct-3-yl)-l ,3-benzothiazole-6- carboxamide.
- Example 46 N-(l-(6-methyl)-azabicyclo[2.2.2]oct-3-yl)indane-5-carboxamide.
- Example 47 N-(l-(6-methyl)-azabicyclo[2.2.2]oct-3-yl)-l,3-benzodioxole-5- carboxamide.
- Example 49 N-(l-(6-methyl)-azabicyclo[2.2.2]oct-3-yl)-2-benzoisothiophene-5- benzamide.
- Example 50 N-(l -(6-methyl)-azabicyclo[2.2.2]oct-3-yl)- lH-indazole-5- carboxamide.
- Example 51 N-(l-(6-methyl)-azabicyclo[2.2.2]oct-3-yl)-lH-indazole-6- carboxamide.
- Example 52 N-(l-(6-methyl)-azabicyclo[2.2.2]oct-3-yl)-l ,3-dioxolo[4,5-c]pyridine-
- Example 61 N-(2-azabicyclo[2.2.1 ]hept-5-yl)- 1 ,3-benzoxazole-5-carboxamide.
- Example 62 N-(2-azabicyclo[2.2.1 ]hept-5-yl)-2-methyl- 1 ,3-benzoxazole-5- carboxamide.
- Example 64 N-(2-azabicyclo[2.2J]hept-5-yl)-2-methyl-l ,3-benzoxazole-6- carboxamide.
- Example 65 N-(2-azabicyclo[2.2J]hept-5-yl)-l,3-benzothiazole-6-carboxamide.
- Example 66 N-(2-azabicyclo[2.2J]hept-5-yl)indane-5-carboxamide.
- Example 68 N-(2-azabicyclo[2.2J]hept-5-yl)-l,3-oxazolo[5,4-c]pyridine-6- carboxamide.
- Example 69 N-(2-azabicyclo[2.2.1 ]hept-5-yl)-2-benzoisothiophene-5-benzamide.
- Example 70 N-(2-azabicyclo[2.2.1]hept-5-yl)-lH-indazole-5-carboxarnide.
- Example 71 N-(2-azabicyclo[2.2.1 ]hept-5-yl)-lH-indazole-6-carboxamide.
- Example 72 N-(2-azabicyclo[2.2.1 ]hept-5-yl)- 1 ,3-dioxolo[4,5-c]pyridine-6- carboxamide.
- Example 81 N-(2-azabicyclo[2.2.1 ]hept-6-yl))- 1 ,3-benzoxazole-5-carboxamide.
- Example 82 N-(2-azabicyclo[2.2J]hept-6-yl))-2 -methyl- l,3-benzoxazole-5- carboxamide.
- Example 83 N-(2 ⁇ azabicyclo[2.2.1 ]hept-6-yl))- 1 ,3-benzoxazole-6-carboxamide.
- Example 84 N-(2-azabicyclo[2.2J]hept-6-yl))-2-methyl-l,3-benzoxazole-6- carboxamide.
- Example 85 N-(2-azabicyclo[2.2.1 ]hept-6-yl))- 1 ,3-benzothiazole-6-carboxamide.
- Example 86 N-(2-azabicyclo[2.2J]hept-6-yl))indane-5-carboxamide.
- Example 87 N-(2-azabicyclo[2.2.1 ]hept-6-yl))- 1 ,3-benzodioxole-5-carboxamide.
- Example 88 N-(2-azabicyclo[2.2J]hept-6-yl))-l ,3-oxazolo[5,4-c]pyridine-6- carboxamide.
- Example 90 N-(2-azabicyclo[2.2J]hept-6-yl))-lH-indazole-5-carboxamide.
- Example 91 N-(2-azabicyclo[2.2J]hept-6-yl))-lH-indazole-6-carboxamide.
- Example 92 N-(2-azabicyclo[2.2.1 ]hept-6-yl))-l ,3-dioxolo[4,5-c]pyridine-6- carboxamide.
- Example 101 N-[(3R,5i?)- 1 -azabicyclo[3.2J]oct-3-yl]-l ,3-benzoxazole-5- carboxamide.
- Example 102 N-[(3R,5R)-l-azabicyclo[3.2J]oct-3-yl]-2-methyl-l,3-benzoxazole-5- carboxamide.
- Example 103 N-[(3R,5R)-l-azabicyclo[3.2J]oct-3-yl]-l,3-benzoxazole-6- carboxamide.
- Example 104 N-[(3R,5R)-l-azabicyclo[3.2J]oct-3-yl]-2-methyl-l,3-benzoxazole-6- carboxamide.
- Example 106 N-[(3R,5R)-1 -azabicyclo[3.2.1 ]oct-3-yl]indane-5-carboxamide.
- Example 107 N-[(3R,5R)-l-azabicyclo[3.2J]oct-3-yl]-l,3-benzodioxole-5- carboxamide.
- Example 108 N-[(3R,5R)-l-azabicyclo[3.2J]oct-3-yl]-l,3-oxazolo[5,4-c]pyridine-6- carboxamide.
- Example 109 N- [(3R,5R)- 1 -azabicyclo [3.2 J ] oct-3 -yl] -2-benzoisothiophene-5 - benzamide.
- Example 110 N-[(3R,5R)-l-azabicyclo[3.2J]oct-3-yl]-lH-indazole-5-carboxamide.
- Example 111 N-[(3R,5R)-l-azabicyclo[3.2J]oct-3-yl]-lH-indazole-6-carboxamide.
- Example 112 N-[(3R,5R)-l-azabicyclo[3.2J]oct-3-yl]-l,3-dioxolo[4,5-c]pyridine-6- carboxamide.
- Example 121 N-((3R)-l-azabicyclo[3.2.2]nonan-3-yl)-l ,3-benzoxazole-5- carboxamide.
- Example 122 N-((3R)-l-azabicyclo[3.2.2]nonan-3-yl)-l,3-benzothiazole-6- carboxamide.
- Example 124 N-((3R)-l-azabicyclo[3.2.2]nonan-3-yl)-l,3-benzodioxole-5- carboxamide.
- Example 125 N-[(3R)-l-azabicyclo[3.2.2]nonan-3-yl]-lH-indazole-6-carboxamide.
- Example 126 N-[(3R)-l-azabicyclo[3.2.2]nonan-3-yl]-l ,3-dioxolo[4,5-c]pyridine-6- carboxamide.
- the benzothiazole and benzimidazole intermediates can be prepared using the methods as shown in Scheme 4 or Scheme 5, respectively.
- the benzoxazole intermediates can be prepared using methods described in Campaigne, E.; Van Nerth, J. ⁇ ., J. Org. Chem., 1958, 23, 1344-1346, whereby the requisite o-aminophenol is treated with diethyloxalate.
- An alternate preparation of these compounds utilizes an approach described in Pol. J. Pharm., 1984, 683-688, wherein the o-aminophenol is treated with glycolic acid.
- the resultant alcohol is then oxidized with KMnO to afford the desired benzoxazole-2-carboxylic acid derivative. Similar approaches can be followed to afford the desired benzothiazole and benzimidazole derivatives.
- the coupling of the Azabicyclo moiety occurs using either the carboxylic acids, such as C1012 or C1013. or the ethyl esters, such as C1001. ClOl l. or C1017. as shown in Scheme 6.
- 2-Carboethoxy derivatives can be directly coupled to the amino-azabicyclo moiety upon heating in the ester in ethanol at reflux.
- An alternate route entails subjecting the ester to hydrolysis providing a carboxylic acid.
- the carboxylic acid can then be coupled to the amino-azabicyclo moiety using a variety of amide bond coupling reagents.
- ⁇ 7-5HT ⁇ receptor The cDNA encoding the N-terminal 201 amino acids from the human 7 nAChR that contain the ligand binding domain of the ion channel is fused to the cDNA encoding the pore forming region of the mouse 5HT 3 receptor as described by Eisele JL, et al., Chimaeric nicotinic-serotonergic receptor combines distinct ligand binding and channel specificities, Nature (1993), Dec. 2;366(6454):479-83, and modified by Groppi, et al., WO 00/73431.
- the chimeric ⁇ 7-5HT 3 ion channel is inserted into pGS175 and pGS179 which contain the resistance genes for G-418 and hygromycin B, respectively. Both plasmids were simultaneously transfected into SH- EP1 cells and cell lines were selected that were resistant to both G-418 and hyrgromycin B. Cell lines expressing the chimeric ion channel were identified by their ability to bind fluorescent ⁇ -bungarotoxin on their cell surface. The cells with the highest amount of fluorescent ⁇ -bungarotoxin binding were isolated using a Fluorescent Activated Cell Sorter (FACS).
- FACS Fluorescent Activated Cell Sorter
- Cell lines that stably expressed the chimeric ⁇ 7-5HT 3 were identified by measuring fluorescent ⁇ -bungarotoxin binding after growing the cells in minimal essential medium containing nonessential amino acids supplemented with 10% fetal bovine serum, L-glutamine, 100 units/ml penicillin/streptomycin, 250 ng/mg fungizone, 400 ⁇ g/ml hygromycin B, and 400 ⁇ g/ml G-418 at 37° C with 6% CO 2 in a standard mammalian cell incubator for at least 4 weeks in continuous culture.
- the cells were incubated with the dye for 60 min at 37° C and then washed with a modified version of Earle's balanced salt solution (MMEBSS) as described in WO 00/73431.
- MMEBSS Earle's balanced salt solution
- the ion conditions of the MMEBSS is adjusted to maximize the flux of calcium ion through the chimeric ⁇ 7-5HT 3 ion channel as described in WO 00/73431.
- the activity of compounds on the chimeric ⁇ 7-5HT 3 ion channel is analyzed on FLIPR.
- the instrument is set up with an excitation wavelength of 488 nanometers using 500 milliwatts of power. Fluorescent emission is measured above 525 nanometers with an appropriate F-stop to maintain a maximal signal to noise ratio.
- Agonist activity of each compound is measured by directly adding the compound to cells expressing the chimeric ⁇ 7-5HT 3 ion channel and measuring the resulting increase in intracellular calcium that is caused by the agonist-induced activation of the chimeric ion channel.
- the assay is quantitative such that concentration-dependent increase in intracelluar calcium is measured as concentration-dependent change in Calcium Green fluorescence.
- the effective concentration needed for a compound to cause a 50% maximal increase in intracellular calcium is te ⁇ ned the EC 5 o.
- the following examples of the present invention have EC 50 values from about 180 nM to about 5700 nM: Example 1, Example 5, Example 6, Example 7, Example 10, Example 11, Example 12, and Example 105.
- Another way for measuring ⁇ 7 nAChR agonist activity is to determine binding constants of a potential agonist in a competition binding assay.
- ⁇ 7 nAChR agonists there is good correlation between functional EC 5 o values using the chimeric ⁇ 7-5HT 3 ion channel as a drug target and binding affinity of compounds to the endogenous ⁇ 7 nAChR.
- Binding Assay For saturation studies, 0.4 mL homogenate are added to test tubes containing buffer and various concentrations of radioligand, and are incubated in a final volume of 0.5 mL for 1 hour at 25 °C. Nonspecific binding is determined in tissues incubated in parallel in the presence of 0.05 mis MLA for a final concentration of 1 ⁇ M, added before the radioligand. In competition studies, drugs are added in increasing concentrations to the test tubes before addition of 0.05 mis [ ⁇ H]-MLA for a final concentration 3.0 to 4.0 nM. The incubations are terminated by rapid vacuum filtration through Whatman GF/B glass filter paper mounted on a 48 well Brandel cell harvester.
- Filters are pre-soaked in 50 mM Tris HCl pH 7.0 - 0.05 % polyethylenimine. The filters are rapidly washed two times with 5 mL aliquots of cold 0.9%) saline and then counted for radioactivity by liquid scintillation spectrometry. Data Analysis.
- Ki inhibition constant
Abstract
Description














































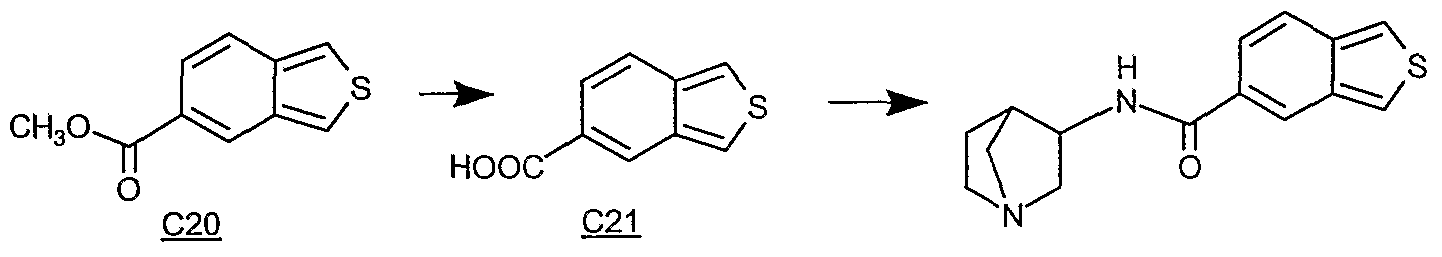











Claims






Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MXPA04003986A MXPA04003986A (en) | 2001-10-26 | 2002-10-17 | N-azabicyclo-substituted hetero-bicyclic carboxamides as nachr agonists. |
| BR0213760-7A BR0213760A (en) | 2001-10-26 | 2002-10-17 | Compound, pharmaceutical composition, use of compound and method for treating disease or condition |
| JP2003540177A JP2005511574A (en) | 2001-10-26 | 2002-10-17 | N-azabicyclo-substituted heterobicyclic carboxamides as NACHR agonists |
| CA002464194A CA2464194A1 (en) | 2001-10-26 | 2002-10-17 | N-azabicyclo-substituted hetero-bicyclic carboxamides as nachr agonists |
| EP02784010A EP1438308A1 (en) | 2001-10-26 | 2002-10-17 | N-azabicyclo-substituted hetero-bicyclic carboxamides as nachr agonists |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US34443601P | 2001-10-26 | 2001-10-26 | |
| US60/344,436 | 2001-10-26 | ||
| US34267401P | 2001-12-21 | 2001-12-21 | |
| US60/342,674 | 2001-12-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003037896A1 true WO2003037896A1 (en) | 2003-05-08 |
Family
ID=26993131
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2002/031579 WO2003037896A1 (en) | 2001-10-26 | 2002-10-17 | N-azabicyclo-substituted hetero-bicyclic carboxamides as nachr agonists |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP1438308A1 (en) |
| JP (1) | JP2005511574A (en) |
| BR (1) | BR0213760A (en) |
| CA (1) | CA2464194A1 (en) |
| MX (1) | MXPA04003986A (en) |
| WO (1) | WO2003037896A1 (en) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004039366A1 (en) * | 2002-11-01 | 2004-05-13 | Pharmacia & Upjohn Company Llc | Nicotinic acetylcholine agonists in the treatment of glaucoma and retinal neuropathy |
| WO2004099206A1 (en) * | 2003-05-12 | 2004-11-18 | Novartis Ag | Isoquinoline-3-carboxylic acid amides and pharmaceutical uses thereof |
| WO2006001894A1 (en) * | 2004-04-22 | 2006-01-05 | Memory Pharmaceutical Corporation | Indoles, 1h-indazoles, 1,2-benzisoxazoles, 1,2-benzoisothiazoles, and preparation and uses thereof |
| WO2006089100A1 (en) | 2005-02-17 | 2006-08-24 | Amr Technology, Inc. | Benzoxazole carboxamides for treating cinv and ibs-d |
| US7304087B2 (en) | 2002-10-24 | 2007-12-04 | Glaxo Group Limited | 1-acyl-pyrrolidine derivatives for the treatment of viral infections |
| WO2008019363A2 (en) * | 2006-08-07 | 2008-02-14 | Albany Molecular Research, Inc. | 2-alkylbenzoxazole carboxamides as 5ht3 modulators |
| US7396833B2 (en) | 2003-12-22 | 2008-07-08 | Memory Pharmaceuticals Corporation | Indoles, 1H-indazoles, 1,2-benzisoxazoles, and 1,2-benzisothiazoles, and preparation and uses thereof |
| US7429664B2 (en) | 2002-09-25 | 2008-09-30 | Memory Pharmaceuticals Corporation | Indazoles, benzothiazoles, and benzoisothiazoles, and preparation and uses thereof |
| US7579362B2 (en) | 2002-09-04 | 2009-08-25 | Novartis Ag | Aza-bicycloalkyl ethers and their use as alpha7-nAChR agonists |
| US7625924B2 (en) | 2004-12-22 | 2009-12-01 | Memory Pharmaceuticals Corporation | Nicotinic alpha-7 receptor ligands and preparation and uses thereof |
| US7632831B2 (en) | 2004-05-07 | 2009-12-15 | Memory Pharmaceuticals Corporation | 1H-indazoles, benzothiazoles, 1,2-benzoisoxazoles, 1,2-benzoisothiazoles, and chromones and preparation and uses thereof |
| US7732477B2 (en) | 2001-12-27 | 2010-06-08 | Bayer Schering Pharma Aktiengesellschaft | 2-heteroarylcarboxylic acid amides |
| US7750022B2 (en) | 2003-08-13 | 2010-07-06 | Neurosearch A/S | Quinuclidine derivatives and their pharmaceutical use |
| US7863271B2 (en) | 2006-08-07 | 2011-01-04 | Albany Molecular Research, Inc. | 2-aminobenzoxazole carboxamides as 5HT3 modulators |
| US8048885B2 (en) | 2005-12-16 | 2011-11-01 | Novartis Ag | Organic compounds |
| US8173667B2 (en) | 2005-10-21 | 2012-05-08 | Novartis Ag | 1-aza-bicycloalkyl derivatives |
| AU2007271504B2 (en) * | 2006-07-01 | 2012-05-24 | Merck Patent Gmbh | Indazole derivatives for treating HSP90-induced diseases |
| US8263619B2 (en) | 2004-03-25 | 2012-09-11 | Memory Pharmaceuticals Corporation | Indazoles, benzothiazoles, benzoisothiazoles, benzisoxazoles, and preparation and uses thereof |
| US8609662B2 (en) | 2004-07-14 | 2013-12-17 | Novartis Ag | 3-(heteroaryl-oxy)-2-alkyl-1-aza-bicycloalkyl derivatives as alpha. 7-nachr ligands for the treatment of CNS diseases |
| US8759346B2 (en) | 2005-12-16 | 2014-06-24 | Novartis Ag | Organic compounds |
| US8933090B2 (en) | 2004-06-18 | 2015-01-13 | Novartis Ag | 1-aza-bicyclo[3.3.1]nonanes |
| US9108961B2 (en) | 2010-05-17 | 2015-08-18 | Forum Pharmaceuticals, Inc. | Crystalline form of (R)-7-chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide hydrochloride |
| US9585877B2 (en) | 2012-05-08 | 2017-03-07 | Forum Pharmaceuticals, Inc. | Methods of maintaining, treating or improving cognitive function |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT2540297E (en) | 2008-11-19 | 2015-08-26 | Forum Pharmaceuticals Inc | Treatment of cognitive disorders with (r)-7-chloro-n-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide and pharmaceutically acceptable salts thereof |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4605652A (en) * | 1985-02-04 | 1986-08-12 | A. H. Robins Company, Inc. | Method of enhancing memory or correcting memory deficiency with arylamido (and arylthioamido)-azabicycloalkanes |
| DE3724059A1 (en) * | 1986-07-30 | 1988-02-18 | Sandoz Ag | USE OF mono- and bicyclic carbocyclic and heterocyclic CARBONSAEUREESTERN and amides OF NITROGEN CONTAINING ALCOHOLS AND AMINES AND THEIR SAEUREADDITIONSSALZEN and quaternary ammonium salts, AND OF IMIDAZOLYL carbazoles for treating psychiatric disorders such as SOCIAL withdrawal symptoms, bipolar disorders, PSYCHOSIS AND OTHER RELATED TO STRESS STANDING DISEASES, DISEASES OF WAKE STATE LIKE GERIATRIC DISEASES, REMOTE FOR TREATING RHINITIS, LUNG EMBOLICS AND IMPROVING THE NASAL RESORPTION OF OTHER ACTIVE SUBSTANCES .......... |
| EP0323077A1 (en) * | 1987-12-24 | 1989-07-05 | JOHN WYETH & BROTHER LIMITED | Heterocyclic compounds |
| EP0327335A1 (en) * | 1988-02-01 | 1989-08-09 | Synthelabo | Arylamido(and arylthioamido)-azabicycloalkanes for enhancing memory or correcting memory deficiency |
| US4920219A (en) * | 1988-11-29 | 1990-04-24 | Rorer Pharmaceutical Corp. | Substituted saturated and unsaturated indole quinoline and benzazepine carboxamides and their use as pharmacological agents |
| US4920227A (en) * | 1988-11-29 | 1990-04-24 | Rorer Pharmaceutical Corp. | Benzobicyclic carboxamide 5-HT3 antagonists |
| JPH04247081A (en) * | 1991-02-01 | 1992-09-03 | Takeda Chem Ind Ltd | 5-membered heterocyclic acid amide |
| US5686461A (en) * | 1993-03-05 | 1997-11-11 | Glaxo Wellcome S.P.A. | Indole derivatives |
| WO2001036417A1 (en) * | 1999-11-18 | 2001-05-25 | Astrazeneca Ab | New use and novel n-azabicyclo-amide derivatives |
| WO2001060821A1 (en) * | 2000-02-18 | 2001-08-23 | Astrazeneca Ab | Novel biarylcarboxamides |
-
2002
- 2002-10-17 BR BR0213760-7A patent/BR0213760A/en not_active IP Right Cessation
- 2002-10-17 EP EP02784010A patent/EP1438308A1/en not_active Withdrawn
- 2002-10-17 MX MXPA04003986A patent/MXPA04003986A/en unknown
- 2002-10-17 JP JP2003540177A patent/JP2005511574A/en not_active Withdrawn
- 2002-10-17 CA CA002464194A patent/CA2464194A1/en not_active Abandoned
- 2002-10-17 WO PCT/US2002/031579 patent/WO2003037896A1/en not_active Application Discontinuation
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4605652A (en) * | 1985-02-04 | 1986-08-12 | A. H. Robins Company, Inc. | Method of enhancing memory or correcting memory deficiency with arylamido (and arylthioamido)-azabicycloalkanes |
| DE3724059A1 (en) * | 1986-07-30 | 1988-02-18 | Sandoz Ag | USE OF mono- and bicyclic carbocyclic and heterocyclic CARBONSAEUREESTERN and amides OF NITROGEN CONTAINING ALCOHOLS AND AMINES AND THEIR SAEUREADDITIONSSALZEN and quaternary ammonium salts, AND OF IMIDAZOLYL carbazoles for treating psychiatric disorders such as SOCIAL withdrawal symptoms, bipolar disorders, PSYCHOSIS AND OTHER RELATED TO STRESS STANDING DISEASES, DISEASES OF WAKE STATE LIKE GERIATRIC DISEASES, REMOTE FOR TREATING RHINITIS, LUNG EMBOLICS AND IMPROVING THE NASAL RESORPTION OF OTHER ACTIVE SUBSTANCES .......... |
| EP0323077A1 (en) * | 1987-12-24 | 1989-07-05 | JOHN WYETH & BROTHER LIMITED | Heterocyclic compounds |
| EP0327335A1 (en) * | 1988-02-01 | 1989-08-09 | Synthelabo | Arylamido(and arylthioamido)-azabicycloalkanes for enhancing memory or correcting memory deficiency |
| US4920219A (en) * | 1988-11-29 | 1990-04-24 | Rorer Pharmaceutical Corp. | Substituted saturated and unsaturated indole quinoline and benzazepine carboxamides and their use as pharmacological agents |
| US4920227A (en) * | 1988-11-29 | 1990-04-24 | Rorer Pharmaceutical Corp. | Benzobicyclic carboxamide 5-HT3 antagonists |
| JPH04247081A (en) * | 1991-02-01 | 1992-09-03 | Takeda Chem Ind Ltd | 5-membered heterocyclic acid amide |
| US5686461A (en) * | 1993-03-05 | 1997-11-11 | Glaxo Wellcome S.P.A. | Indole derivatives |
| WO2001036417A1 (en) * | 1999-11-18 | 2001-05-25 | Astrazeneca Ab | New use and novel n-azabicyclo-amide derivatives |
| WO2001060821A1 (en) * | 2000-02-18 | 2001-08-23 | Astrazeneca Ab | Novel biarylcarboxamides |
Non-Patent Citations (2)
| Title |
|---|
| A. ORJALES ET AL.: "Benzimidazole-2-carboxylic acid amides and esters: a new structural class of 5-HT3 ligands", EUR. J. MED. CHEM., vol. 34, no. 5, 1999, pages 415 - 422, XP002225452 * |
| PATENT ABSTRACTS OF JAPAN vol. 017, no. 025 18 January 1993 (1993-01-18) * |
Cited By (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7732477B2 (en) | 2001-12-27 | 2010-06-08 | Bayer Schering Pharma Aktiengesellschaft | 2-heteroarylcarboxylic acid amides |
| US8884017B2 (en) | 2001-12-27 | 2014-11-11 | Bayer Intellectual Property Gmbh | 2-heteroarylcarboxylic acid amides |
| US8076355B2 (en) | 2001-12-27 | 2011-12-13 | Bayer Schering Pharma Aktiengesellschaft | 2-heteroarylcarboxylic acid amides |
| US9012451B2 (en) | 2002-09-04 | 2015-04-21 | Novartis Ag | Aza-bicycloalkyl ethers and their use as ALPHA7-nachr agonists |
| US7579362B2 (en) | 2002-09-04 | 2009-08-25 | Novartis Ag | Aza-bicycloalkyl ethers and their use as alpha7-nAChR agonists |
| US9567343B2 (en) | 2002-09-04 | 2017-02-14 | Novartis Ag | Aza-bicyloalkyl ethers and their use as alpha7-nachr agonists |
| US9849117B2 (en) | 2002-09-04 | 2017-12-26 | Novartis Ag | Aza-bicycloalkyl ethers and their use as alpha7-nachr agonists |
| US8236803B2 (en) | 2002-09-04 | 2012-08-07 | Novartis Ag | Aza-bicycloalkyl ethers and their use as alpha7-nAChR agonists |
| US7943773B2 (en) | 2002-09-25 | 2011-05-17 | Memory Pharmaceuticals Corporation | Indazoles, benzothiazoles, and benzoisothiazoles, and preparation and uses thereof |
| US8252811B2 (en) | 2002-09-25 | 2012-08-28 | Memory Pharmaceuticals Corporation | Indazoles, benzothiazoles, and benzoisothiazoles, and preparation and uses thereof |
| US8134003B2 (en) | 2002-09-25 | 2012-03-13 | Memory Pharmaceuticals Corporation | Indazoles, benzothiazoles, and benzoisothiazoles, and preparation and uses thereof |
| US7429664B2 (en) | 2002-09-25 | 2008-09-30 | Memory Pharmaceuticals Corporation | Indazoles, benzothiazoles, and benzoisothiazoles, and preparation and uses thereof |
| US7304087B2 (en) | 2002-10-24 | 2007-12-04 | Glaxo Group Limited | 1-acyl-pyrrolidine derivatives for the treatment of viral infections |
| WO2004039366A1 (en) * | 2002-11-01 | 2004-05-13 | Pharmacia & Upjohn Company Llc | Nicotinic acetylcholine agonists in the treatment of glaucoma and retinal neuropathy |
| CN100341874C (en) * | 2003-05-12 | 2007-10-10 | 诺瓦提斯公司 | Isoquinoline-3-carboxylic acid amides and pharmaceutical uses thereof |
| AU2004235971C1 (en) * | 2003-05-12 | 2008-12-04 | Novartis Ag | Isoquinoline-3-carboxylic acid amides and pharmaceutical uses thereof |
| AU2004235971B2 (en) * | 2003-05-12 | 2008-04-03 | Novartis Ag | Isoquinoline-3-carboxylic acid amides and pharmaceutical uses thereof |
| JP2006525976A (en) * | 2003-05-12 | 2006-11-16 | ノバルティス アクチエンゲゼルシャフト | Isoquinoline-3-carboxylic acid amides and their use as pharmaceuticals |
| WO2004099206A1 (en) * | 2003-05-12 | 2004-11-18 | Novartis Ag | Isoquinoline-3-carboxylic acid amides and pharmaceutical uses thereof |
| US7683075B2 (en) | 2003-05-12 | 2010-03-23 | Novartis Ag | Isoquinoline-3-carboxylic acid amides and pharmaceutical uses thereof |
| US7750022B2 (en) | 2003-08-13 | 2010-07-06 | Neurosearch A/S | Quinuclidine derivatives and their pharmaceutical use |
| US7964600B2 (en) | 2003-12-22 | 2011-06-21 | Memory Pharmaceuticals Corporation | Indoles, 1H-indazoles, 1,2-benzisoxazoles, and 1,2-benzisothiazoles, and preparation and uses thereof |
| US7396833B2 (en) | 2003-12-22 | 2008-07-08 | Memory Pharmaceuticals Corporation | Indoles, 1H-indazoles, 1,2-benzisoxazoles, and 1,2-benzisothiazoles, and preparation and uses thereof |
| US7790722B2 (en) | 2003-12-22 | 2010-09-07 | Memory Pharmaceuticals Corporation | Indoles, 1H-indazoles, 1,2-benzisoxazoles, and 1,2-benzisothiazoles, and preparation and uses thereof |
| US8158629B2 (en) | 2003-12-22 | 2012-04-17 | Memory Pharmaceuticals Corporation | Indoles, 1H-indazoles, 1,2-benzisoxazoles, and 1,2-benzisothiazoles, and preparation and uses thereof |
| US8691841B2 (en) | 2004-03-25 | 2014-04-08 | Memory Pharmaceuticals Corporation | Indazoles, benzothiazoles, benzoisothiazoles, benzisoxazoles, and preparation and use thereof |
| US8486937B2 (en) | 2004-03-25 | 2013-07-16 | Memory Pharmaceuticals Corporation | Indazoles, benzothiazoles, benzoisothiazoles, benzisoxazoles, and preparation and use thereof |
| US8263619B2 (en) | 2004-03-25 | 2012-09-11 | Memory Pharmaceuticals Corporation | Indazoles, benzothiazoles, benzoisothiazoles, benzisoxazoles, and preparation and uses thereof |
| US7488737B2 (en) | 2004-04-22 | 2009-02-10 | Memory Pharmaceutical Corporation | Indoles, 1H-indazoles, 1,2-benzisoxazoles, 1,2-benzoisothiazoles, and preparation and uses thereof |
| US7902217B2 (en) | 2004-04-22 | 2011-03-08 | Memory Pharmaceuticals Corporation | Indoles, 1H-indazoles, 1,2-benzisoxazoles, 1,2-benzoisothiazoles, and preparation and uses thereof |
| JP2011241227A (en) * | 2004-04-22 | 2011-12-01 | Memory Pharmaceuticals Corp | Indole, 1h-indazole, 1,2-benzisoxazole, 1,2-benzoisothiazole, and preparation and uses thereof |
| JP2007534692A (en) * | 2004-04-22 | 2007-11-29 | メモリー・ファーマシューティカルズ・コーポレイション | Indole, 1H-indazole, 1,2-benzisoxazole, 1,2-benzisothiazole, and their preparation and use |
| WO2006001894A1 (en) * | 2004-04-22 | 2006-01-05 | Memory Pharmaceutical Corporation | Indoles, 1h-indazoles, 1,2-benzisoxazoles, 1,2-benzoisothiazoles, and preparation and uses thereof |
| US7632831B2 (en) | 2004-05-07 | 2009-12-15 | Memory Pharmaceuticals Corporation | 1H-indazoles, benzothiazoles, 1,2-benzoisoxazoles, 1,2-benzoisothiazoles, and chromones and preparation and uses thereof |
| US9475811B2 (en) | 2004-06-18 | 2016-10-25 | Novartis Ag | 1-aza-bicyclo[3.3.1]nonanes |
| US8933090B2 (en) | 2004-06-18 | 2015-01-13 | Novartis Ag | 1-aza-bicyclo[3.3.1]nonanes |
| US9657010B2 (en) | 2004-07-14 | 2017-05-23 | Novartis Ag | Substituted quinuclidines as alpha 7-nicotinic acetylcholine receptor activity modulators |
| US8609662B2 (en) | 2004-07-14 | 2013-12-17 | Novartis Ag | 3-(heteroaryl-oxy)-2-alkyl-1-aza-bicycloalkyl derivatives as alpha. 7-nachr ligands for the treatment of CNS diseases |
| US7625924B2 (en) | 2004-12-22 | 2009-12-01 | Memory Pharmaceuticals Corporation | Nicotinic alpha-7 receptor ligands and preparation and uses thereof |
| WO2006089100A1 (en) | 2005-02-17 | 2006-08-24 | Amr Technology, Inc. | Benzoxazole carboxamides for treating cinv and ibs-d |
| US7781430B2 (en) | 2005-02-17 | 2010-08-24 | Albany Molecular Research, Inc. | Benzoxazole carboxamides for treating CINV and IBS-D |
| US7307094B2 (en) | 2005-02-17 | 2007-12-11 | Amr Technology, Inc. | Benzoxazole carboxamides for treating CINV and IBS-D |
| US8173667B2 (en) | 2005-10-21 | 2012-05-08 | Novartis Ag | 1-aza-bicycloalkyl derivatives |
| US9206181B2 (en) | 2005-12-16 | 2015-12-08 | Novartis Ag | 1-aza-bicyclo[3.3.1] non-4-yl)-[5-(1H-indol-5-yl)-heteroaryl]-amines as cholinergic ligands of the n-AChR for the treatment of psychotic and neurodegenerative disorders |
| US8048885B2 (en) | 2005-12-16 | 2011-11-01 | Novartis Ag | Organic compounds |
| US8759346B2 (en) | 2005-12-16 | 2014-06-24 | Novartis Ag | Organic compounds |
| US8637517B2 (en) | 2005-12-16 | 2014-01-28 | Novartis Ag | Organic compounds |
| US8722903B2 (en) | 2006-07-01 | 2014-05-13 | Merck Patent Gmbh | Indazole derivatives for the treatment of HSP9O-induced diseases |
| AU2007271504B2 (en) * | 2006-07-01 | 2012-05-24 | Merck Patent Gmbh | Indazole derivatives for treating HSP90-induced diseases |
| US7863271B2 (en) | 2006-08-07 | 2011-01-04 | Albany Molecular Research, Inc. | 2-aminobenzoxazole carboxamides as 5HT3 modulators |
| WO2008019363A2 (en) * | 2006-08-07 | 2008-02-14 | Albany Molecular Research, Inc. | 2-alkylbenzoxazole carboxamides as 5ht3 modulators |
| WO2008019363A3 (en) * | 2006-08-07 | 2008-04-03 | Amr Technology Inc | 2-alkylbenzoxazole carboxamides as 5ht3 modulators |
| US8278301B2 (en) | 2006-08-07 | 2012-10-02 | Albany Molecular Research, Inc. | 2-alkylbenzoxazole carboxamides as 5HT3 modulators |
| US7553846B2 (en) | 2006-08-07 | 2009-06-30 | Albany Molecular Research, Inc. | 2-alkylbenzoxazole carboxamides as 5-HT3 modulators |
| US9108961B2 (en) | 2010-05-17 | 2015-08-18 | Forum Pharmaceuticals, Inc. | Crystalline form of (R)-7-chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide hydrochloride |
| US9273044B2 (en) | 2010-05-17 | 2016-03-01 | Forum Pharmaceuticals, Inc. | Crystalline form of (R)-7-chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide hydrochloride monohydrate |
| US9550767B2 (en) | 2010-05-17 | 2017-01-24 | Forum Pharmaceuticals, Inc. | Crystalline form of (R)-7-chloro-N-(quinuclidin-3-yl)benzo[b]thiophene-2-carboxamide hydrochloride monohydrate |
| US9585877B2 (en) | 2012-05-08 | 2017-03-07 | Forum Pharmaceuticals, Inc. | Methods of maintaining, treating or improving cognitive function |
Also Published As
| Publication number | Publication date |
|---|---|
| MXPA04003986A (en) | 2004-07-23 |
| CA2464194A1 (en) | 2003-05-08 |
| JP2005511574A (en) | 2005-04-28 |
| BR0213760A (en) | 2004-10-19 |
| EP1438308A1 (en) | 2004-07-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6858613B2 (en) | Fused bicyclic-N-bridged-heteroaromatic carboxamides for the treatment of disease | |
| US6911543B2 (en) | Azabicyclic-substituted fused-heteroaryl compounds for the treatment of disease | |
| US7001900B2 (en) | Azabicyclic compounds for the treatment of disease | |
| US6894042B2 (en) | Azabicyclic compounds for the treatment of disease | |
| US6951868B2 (en) | Azabicyclic-phenyl-fused-heterocyclic compounds for treatment of disease | |
| WO2003037896A1 (en) | N-azabicyclo-substituted hetero-bicyclic carboxamides as nachr agonists | |
| US6849620B2 (en) | N-(azabicyclo moieties)-substituted hetero-bicyclic aromatic compounds for the treatment of disease | |
| US20060116395A1 (en) | 1H-pyrazole and 1h-pyrole-azabicyclic compounds for the treatment of disease | |
| JP2005511574A6 (en) | N-azabicyclo-substituted heterobicyclic carboxamides as NACHR agonists | |
| US20040147522A1 (en) | Compounds having both alpha7 nicotinic agonist activity and 5HT3 antagonist activity for the treatment of CNS diseases | |
| US20030236279A1 (en) | Substituted-aryl compounds for treatment of disease | |
| AU2002339957A1 (en) | Azabicyclic-substituted fused-heteroaryl compounds for the treatment of disease |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LU MC NL PT SE SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2464194 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2004/003986 Country of ref document: MX Ref document number: 2003540177 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002784010 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002784010 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2002784010 Country of ref document: EP |