WO2003018514A1 - Method of preparing nitroform - Google Patents
Method of preparing nitroform Download PDFInfo
- Publication number
- WO2003018514A1 WO2003018514A1 PCT/SE2002/001551 SE0201551W WO03018514A1 WO 2003018514 A1 WO2003018514 A1 WO 2003018514A1 SE 0201551 W SE0201551 W SE 0201551W WO 03018514 A1 WO03018514 A1 WO 03018514A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- gem
- nitroform
- nitration
- group
- eller
- Prior art date
Links
- 0 *NC(NC(C([N+]([O-])=O)[N+]([O-])=O)=O)=O Chemical compound *NC(NC(C([N+]([O-])=O)[N+]([O-])=O)=O)=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C201/00—Preparation of esters of nitric or nitrous acid or of compounds containing nitro or nitroso groups bound to a carbon skeleton
- C07C201/06—Preparation of nitro compounds
- C07C201/08—Preparation of nitro compounds by substitution of hydrogen atoms by nitro groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C201/00—Preparation of esters of nitric or nitrous acid or of compounds containing nitro or nitroso groups bound to a carbon skeleton
- C07C201/06—Preparation of nitro compounds
- C07C201/12—Preparation of nitro compounds by reactions not involving the formation of nitro groups
Definitions
- the invention relates to a method of preparing trinitromethane or nitroform which is the common name of the compound.
- Nitroform is a valuable compound for producing propellants and explosive components by its high oxygen content and an unstable hydrogen atom, which facilitates its forming of derivatives.
- Nitroform is used, for example, for preparing hydrazinium nitroformat (HNF) which is a salt between hydrazine and nitroform and is used as oxidiser in, inter alia, rocket propellants.
- the oxidiser is chlorine-free, which is increasingly desired in propellant compositions.
- Nitroform is also used as a starting material for producing energetic plasticisers.
- the most well-known method for preparing nitroform is nitration of acetylene with nitric acid, using a mercury catalyst.
- An industrial process for preparing nitroform according to this method is described by A. Wetterholm: Tetrahedron, 1963, Vol. 19, pp 155-163.
- the use of acetylene gas as starting material requires specific measures of precaution in the process, and the use of a mercury catalyst has obvious drawbacks from the environmental point of view.
- An object of the present invention is to provide a simple and safe method for preparing nitroform.
- a starting material dissolved or suspended in sulphuric acid is nitrated.
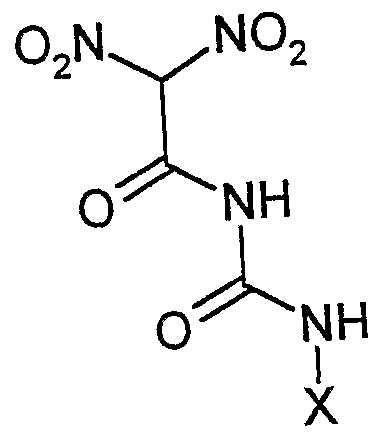
- the starting material is selected from a group consisting of 1) a gem-dinitroacetyl urea with the general formula
- nitrating agent consisting of nitric acid, nitrate salts or nitrogen pentoxide is added to the sulphuric acid solution/suspension. It will thus be easy to control the nitration. Further nitro groups are added to the starting material in nitration, forming a gem-trinitro compound which by hydrolysis splits off nitroform.
- the nitration is carried out at a temperature from -10°C to +80°C, preferably from +10°C to +60°C.
- the sulphuric acid may have a concentration of 70-100%, preferably about 95%.
- the nitric acid which is the preferred nitrating agent, is added as a concentrated acid, especially with a concentration of 85-100%.
- the molar ratio between nitrating agent and substrate may be 2.0-6.0:1 , and is preferably 3.0-4.0:1. Good yields have been obtained with such a low molar ratio as 3.0:1, which in combination with reuse of the sulphuric acid, as will be mentioned below, makes the method economically attractive.
- the nitration can be carried out in an ordinary stain- less steel reactor without any special arrangements.
- the product (the gem-trinitro compound) is hydro- lysed for splitting off nitroform by mixing the reaction mixture with an aqueous medium, for instance pouring it onto crushed ice, diluting it with water or the like.
- the nitroform prepared can be extracted from the reaction mixture using a polar extracting agent which is stable in the environment at issue, for instance methylene chloride or diethyl ether.
- a neutralising agent can then be added to precipitate the corresponding salt of nitroform.
- the sulphuric acid may then be used several times.
- the starting material according to a) above can be produced by nitration of barbituric acid to gem-5,5-dinitrobarbituric acid which is then hydrolysed with water to form gem-dinitroacetyl urea or is treated with a ring-opening nucleophile, for instance ammonia, methanol, ethanol, isopropanol or an amine, to form substituted gem-dinitroacetyl urea.
- a ring-opening nucleophile for instance ammonia, methanol, ethanol, isopropanol or an amine
- the starting material according to b) above can correspondingly be produced by nitration of 2-amino-4,6-dihydroxypyrirnidine followed by hydrolysis with water, as described in Latypov et al: A new convenient route to gem-dinitroalifatic compounds, Conference Proceedings, 31st International Conference of ICT: 2000.
- the starting material according to c) is a commercially available product.
- Potassium dinitroacetyl urea (6.8 g) was dissolved in sulphuric acid (20 ml, 95% concentration). Concentrated nitric acid (1.4 ml) was added by drops. The temperature was increased to about 40°C for one hour. The reaction mixture was poured onto 60 g crushed ice and then extracted with 2x50 ml diethyl ether. The ether phase was dried with sodium sulphate. Potassium hydroxide dissolved in ethanol was added. A yellow precipitate of potassium nitroform (4.6 g; 68% yield) was obtained.
- 4,6-dihydroxypyrimidine (4 g) was dissolved in sulphuric acid (20 ml, 95% concentration). Fuming nitric acid was added (6 ml) during cooling with ice and was then kept at room temperature for 12 hours. The reaction mixture was poured onto crushed ice and extracted with methylene chloride. Yield of nitroform 60%, measured by UV spectroscopy in methylene chloride.
- Dinitroacetyl urea (3 g) was dissolved in sulphuric acid (20 ml, 95% concentration). Concentrated nitric acid (0.57 ml) was added. The reaction mixture was stirred for one hour at 20°C. Then it was poured onto 77 g crushed ice, heated to 50°C for 10 minutes and extracted with 2 x 125 ml methylene chloride. Yield of nitroform 62%.
- Example 4 Dinitroacetyl urea (3 g) was dissolved in sulphuric acid (20 ml, 95% concentration). Concentrated nitric acid (0.57 ml) was added. The reaction mixture was stirred for one hour at 20°C with a cover (covering layer) consisting of 30 ml methylene chloride. The methylene chloride was separated from the reaction mixture and mixed with 30 ml n-heptane. The mixture was evaporated to 20 ml and then trinitroacetyl urea precipitated. The trinitroactyl urea could now easily be hydrolysed with potassium hydroxide for splitting off potassium nitroform.
Abstract
Description

Claims



Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SE0102908-1 | 2001-08-31 | ||
| SE0102908A SE519778C2 (en) | 2001-08-31 | 2001-08-31 | Methods of preparing nitroform |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2003018514A1 true WO2003018514A1 (en) | 2003-03-06 |
| WO2003018514A9 WO2003018514A9 (en) | 2004-04-22 |
Family
ID=20285198
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/SE2002/001551 WO2003018514A1 (en) | 2001-08-31 | 2002-08-30 | Method of preparing nitroform |
Country Status (2)
| Country | Link |
|---|---|
| SE (1) | SE519778C2 (en) |
| WO (1) | WO2003018514A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105111087A (en) * | 2015-08-19 | 2015-12-02 | 南京理工大学 | Trinitroethanol preparation method with improved process safety |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3007960A (en) * | 1958-11-10 | 1961-11-07 | Purdue Research Foundation | N-trinitroalkyl-n-nitroaminoalkyl acids and derivatives thereof |
| US3125606A (en) * | 1964-03-17 | Process for the manufacture of | ||
| US3491160A (en) * | 1968-02-05 | 1970-01-20 | Escambia Chem Corp | Process for producing nitroform |
| US4122124A (en) * | 1977-12-05 | 1978-10-24 | Rockwell International Corporation | Production of trinitromethane |
| WO1997038967A1 (en) * | 1996-04-16 | 1997-10-23 | Arco Chemical Technology, L.P. | Reducing tetranitromethane in compositions containing nitroaromatic compounds |
-
2001
- 2001-08-31 SE SE0102908A patent/SE519778C2/en not_active IP Right Cessation
-
2002
- 2002-08-30 WO PCT/SE2002/001551 patent/WO2003018514A1/en not_active Application Discontinuation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3125606A (en) * | 1964-03-17 | Process for the manufacture of | ||
| US3007960A (en) * | 1958-11-10 | 1961-11-07 | Purdue Research Foundation | N-trinitroalkyl-n-nitroaminoalkyl acids and derivatives thereof |
| US3491160A (en) * | 1968-02-05 | 1970-01-20 | Escambia Chem Corp | Process for producing nitroform |
| US4122124A (en) * | 1977-12-05 | 1978-10-24 | Rockwell International Corporation | Production of trinitromethane |
| WO1997038967A1 (en) * | 1996-04-16 | 1997-10-23 | Arco Chemical Technology, L.P. | Reducing tetranitromethane in compositions containing nitroaromatic compounds |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105111087A (en) * | 2015-08-19 | 2015-12-02 | 南京理工大学 | Trinitroethanol preparation method with improved process safety |
Also Published As
| Publication number | Publication date |
|---|---|
| SE519778C2 (en) | 2003-04-08 |
| WO2003018514A9 (en) | 2004-04-22 |
| SE0102908D0 (en) | 2001-08-31 |
| SE0102908L (en) | 2003-03-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9296664B2 (en) | Energetic active composition comprising a dihydroxylammonium salt or diammonium salt of a bistetrazolediol | |
| US9598380B2 (en) | Facile method for preparation of 5-nitrotetrazolates using a batch system | |
| CN106188009A (en) | 3,4 dinitro 1 (1H tetrazolium 5 base) 1H pyrazoles 5 amine are containing energy ion salt preparation method and performance | |
| US7981393B2 (en) | Method of producing salts of dinitramidic acid | |
| CN112194625B (en) | 1, 4-dinitroamino-3, 5-dinitropyrazole oxalyldihydrazine nitrate, preparation method and application thereof | |
| JPH04504844A (en) | Method for producing dinitrotoluene using an inorganic salt as a phase separating agent | |
| WO2003018514A1 (en) | Method of preparing nitroform | |
| Coburn et al. | An improved synthesis of 3, 6‐diamino‐1, 2, 4, 5‐tetrazine. I | |
| GB2355714A (en) | Ammonium 3,5-diaminopicrate | |
| US3125606A (en) | Process for the manufacture of | |
| RU2147577C1 (en) | Method of preparing n-(2-nitroxyethyl)nicotineamide | |
| US5659080A (en) | Synthetic method for forming ammonium dinitrammide (ADN) | |
| US20110160450A1 (en) | Explosive | |
| Langlet et al. | Formation of Nitroform in the Nitration of Gem‐Dinitro Compounds | |
| ZA200302347B (en) | Method for producing DNDA. | |
| KR102619242B1 (en) | Method for preparing salt derivatives of 3-dinitromethyl-5-nitramino-1,2,4-triazolate(ndnt), salt derivatives of 3-dinitromethyl-5-nitramino-1,2,4-triazolate thereby | |
| CN115340501B (en) | Energetic ionic salt based on bitriazole compounds and synthesis method thereof | |
| US3006957A (en) | Process for preparation of bis(trinitroethyl)amine | |
| KR102507950B1 (en) | Method for preparing salt derivatives of 4-nitramino-3-(5-dinitromethyl-1,2,4-oxadiazolyl)-furazanate(ndnf), salt derivatives of 4-nitramino-3-(5-dinitromethyl-1,2,4-oxadiazolyl)-furazanate thereby | |
| US4233250A (en) | Process for synthesizing the alkali metal salts of dinetromethane | |
| US7563889B1 (en) | 3,3,7,7,-tetrakis(difluoramino)octahydro-1,5-diazocinium salts and method for making the same | |
| US7632943B1 (en) | Method for making 3,3,7,7-tetrakis(difluoramino)octahydro-1,5-dinitro-1,5-diazocine (HNFX) | |
| CN113402522B (en) | Fluorine-containing [1,2,4] triazole [4,3-b ] [1,2,4,5] tetrazine ammonium nitrate and preparation method thereof | |
| CN113105406B (en) | Energy-containing molecules of wheel-shaped triazine nitrate and synthetic method thereof | |
| US8444783B1 (en) | 3,3,7,7-tetrakis(difluoramino)octahydro-1,5-diazocinium salts |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BY BZ CA CH CN CO CR CU CZ DE DM DZ EC EE ES FI GB GD GE GH HR HU ID IL IN IS JP KE KG KP KR LC LK LR LS LT LU LV MA MD MG MN MW MX MZ NO NZ OM PH PL PT RU SD SE SG SI SK SL TJ TM TN TR TZ UA UG US UZ VC VN YU ZA ZM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LU MC NL PT SE SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ UG ZM ZW AM AZ BY KG KZ RU TJ TM AT BE BG CH CY CZ DK EE ES FI FR GB GR IE IT LU MC PT SE SK TR BF BJ CF CG CI GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| COP | Corrected version of pamphlet |
Free format text: PAGE 6, CLAIMS, DELETED |
|
| 122 | Ep: pct application non-entry in european phase | ||
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: JP |