WO2003011834A1 - Novel vinyl n-(2-benzoylphenyl)-l-tyrosine derivatives and their use as antidiabetics etc - Google Patents
Novel vinyl n-(2-benzoylphenyl)-l-tyrosine derivatives and their use as antidiabetics etc Download PDFInfo
- Publication number
- WO2003011834A1 WO2003011834A1 PCT/DK2002/000469 DK0200469W WO03011834A1 WO 2003011834 A1 WO2003011834 A1 WO 2003011834A1 DK 0200469 W DK0200469 W DK 0200469W WO 03011834 A1 WO03011834 A1 WO 03011834A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound according
- compound
- ethoxy
- benzoyl
- phenylamino
- Prior art date
Links
- YCWJJCCMTYMSGY-UHFFFAOYSA-N COC(C(Cc(cc1)ccc1OCCN(c1ccccc1CC1)c2c1cccc2)Nc(cccc1)c1C(c1ccccc1)=O)=O Chemical compound COC(C(Cc(cc1)ccc1OCCN(c1ccccc1CC1)c2c1cccc2)Nc(cccc1)c1C(c1ccccc1)=O)=O YCWJJCCMTYMSGY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D223/00—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom
- C07D223/14—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D223/18—Dibenzazepines; Hydrogenated dibenzazepines
- C07D223/22—Dibenz [b, f] azepines; Hydrogenated dibenz [b, f] azepines
- C07D223/24—Dibenz [b, f] azepines; Hydrogenated dibenz [b, f] azepines with hydrocarbon radicals, substituted by nitrogen atoms, attached to the ring nitrogen atom
- C07D223/26—Dibenz [b, f] azepines; Hydrogenated dibenz [b, f] azepines with hydrocarbon radicals, substituted by nitrogen atoms, attached to the ring nitrogen atom having a double bond between positions 10 and 11
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D223/00—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom
- C07D223/14—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D223/18—Dibenzazepines; Hydrogenated dibenzazepines
- C07D223/22—Dibenz [b, f] azepines; Hydrogenated dibenz [b, f] azepines
- C07D223/24—Dibenz [b, f] azepines; Hydrogenated dibenz [b, f] azepines with hydrocarbon radicals, substituted by nitrogen atoms, attached to the ring nitrogen atom
- C07D223/28—Dibenz [b, f] azepines; Hydrogenated dibenz [b, f] azepines with hydrocarbon radicals, substituted by nitrogen atoms, attached to the ring nitrogen atom having a single bond between positions 10 and 11
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/28—1,4-Oxazines; Hydrogenated 1,4-oxazines
- C07D265/34—1,4-Oxazines; Hydrogenated 1,4-oxazines condensed with carbocyclic rings
- C07D265/38—[b, e]-condensed with two six-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D279/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one sulfur atom as the only ring hetero atoms
- C07D279/10—1,4-Thiazines; Hydrogenated 1,4-thiazines
- C07D279/14—1,4-Thiazines; Hydrogenated 1,4-thiazines condensed with carbocyclic rings or ring systems
- C07D279/18—[b, e]-condensed with two six-membered rings
- C07D279/22—[b, e]-condensed with two six-membered rings with carbon atoms directly attached to the ring nitrogen atom
Definitions
- the present invention relates to novel vinyl N-(2-benzoylphenyl)-L tyrosine derivatives , to the use of these compounds as pharmaceutical compositions, to pharmaceutical compositions comprising the compounds and to a method of treatment employing these compounds and compositions. More specifically, the compounds of the invention can be utilised in the treatment and/or prevention of conditions mediated by the Peroxisome Proliferator-Activated Receptors (PPAR). The compounds exert their effects by modulating the PPAR ⁇ response in a partial agonist manner.
- PPAR Peroxisome Proliferator-Activated Receptors
- Coronary artery disease is the major cause of death in Type 2 diabetic and metabolic syndrome patients (i.e. patients that fall within the 'deadly quartet' category of impaired glucose tolerance, insulin resistance, hypertriglyceridaemia and/or obesity).
- hypolipidaemic fibrates and antidiabetic thiazolidinediones separately display moderately effective triglyceride-lowering activities although they are neither potent nor efficacious enough to be a single therapy of choice for the dyslipidaemia often observed in Type 2 diabetic or metabolic syndrome patients.
- the thiazolidinediones also potently lower circulating glucose levels of Type 2 diabetic animal models and humans.
- the fibrate class of compounds are without beneficial effects on glycaemia.
- thiazolidinediones and fibrates exert their action by activating distinct transcription factors of the peroxisome proliferator activated receptor (PPAR) family, resulting in increased and decreased expression of specific enzymes and apolipoproteins respectively, both key-players in regulation of plasma triglyceride content.
- Fibrates on the one hand, are PPAR ⁇ activators, acting primarily in the liver.
- Thiazolidin- ediones are high affinity ligands for PPAR ⁇ acting primarily on adipose tissue.
- Adipose tissue plays a central role in lipid homeostasis and the maintenance of energy balance in vertebrates.
- Adipocytes store energy in the form of triglycerides during periods of nutritional affluence and release it in the form of free fatty acids at times of nutritional deprivation.
- white adipose tissue is the result of a continuous differentiation process throughout life. Much evidence points to the central role of PPAR ⁇ activation in initiating and regulating this cell differentiation.
- Several highly specialised proteins are induced during adipocyte differentiation, most of them being involved in lipid
- PPAR ⁇ is involved in stimulating ⁇ -oxidation of fatty acids.
- a PPAR ⁇ - mediated change in the expression of genes involved in fatty acid metabolism lies at the basis of the phenomenon of peroxisome proliferation, a pleiotropic cellular response, mainly limited to liver and kidney and which can lead to hepatocarcinogenesis in rodents.
- the phenomenon of peroxisome proliferation is not seen in man.
- PPAR ⁇ is also involved in the control of HDL cholesterol levels in rodents and humans. This effect is, at least partially, based on a PPAR ⁇ -mediated transcriptional regulation of the major HDL apolipoproteins, apo A-l and apo A-ll.
- the hypotriglyceridemic action of fibrates and fatty acids also involves PPAR ⁇ and can be summarised as follows: (I) an increased lipolysis and clearance of remnant particles, due to changes in lipoprotein lipase and apo C-lll levels, (II) a stimulation of cellular fatty acid uptake and their subsequent conversion to acyl-CoA derivatives by the induction of fatty acid binding protein and acyl-CoA synthase, (III) an induction of fatty acid ⁇ -oxidation pathways, (IV) a reduction in fatty acid and triglyceride synthesis, and finally (V) a decrease in VLDL production.
- both enhanced catabolism of triglyceride-rich particles as well as reduced secretion of VLDL particles constitutes mechanisms that contribute to the hypolipidemic effect of fibrates.
- PPAR ⁇ activation was initially reported not to be involved in modulation of glucose or triglyceride levels. (Berger et al.,/ Biol. Chem. , 1999, Vol 274, pp. 6718-6725). Later it has been shown that PPAR ⁇ activation leads to increased levels of HDL cholesterol in dbldb mice (Leibowitz et al. FEBS letters 2000, 473, 333-336). Further, a PPAR ⁇ agonist when dosed to insulin-resistant middle-aged obese rhesus monkeys caused a dramitic dose- dependent rise in serum HDL cholesterol while lowering the levels of small dense LDL, fasting triglycerides and fasting insulin (Oliver et al.
- PPAR ⁇ activation is useful in the treatment and prevention of cardiovascular diseases and conditions including atherosclerosis, hypertriglyceridemia, and mixed dyslipidaemia (PCT publication WO 01/00603 (Chao et al.). A number of compounds have been reported to be useful in the treatment of hyper- glycemia, hyperiipidemia and hypercholesterolemia (U.S. Pat. 5,306,726, PCT Publications nos.
- Glucose lowering as a single approach does not overcome the macrovascular com- plications associated with Type 2 diabetes and metabolic syndrome. Novel treatments of Type 2 diabetes and metabolic syndrome must therefore aim at lowering both the overt hy- pertriglyceridaemia associated with these syndromes as well as alleviation of hyperglycae- mia.
- PPAR ⁇ agonists rosiglitazone and pioglitazone
- PPAR ⁇ agonists are described as full agonists, which lower blood glucose and improve insulin resistance in type 2 diabetic patients, but at the same time it has been reported that they also induce body weight gain, anaemia and oedema limiting the utility of such compounds.
- N-(2-benzoylphenyl)-Z_ tyrosine derivatives have been described in Henkel et al. J. Med Chem. (1998), 41(25), 5020-5036, Collins et al. J. Med Chem. (1998), 41(25), 5037- 5054, Cobb et al. J. Med Chem. (1998), 41(25), 5055-5069, Davis et al. Tetrahedron (1999), 55(39), 11653-11668, WO 9429285, and also in WO 9731907 wherein the derivatives are described as being potent and selective PPAR ⁇ agonists.
- the present invention relates to vinyl N-(2-benzoylphenyl)-L tyrosine derivatives displaying partial PPAR ⁇ agonist activity.
- C 1-6 -alkyl as used herein, alone or in combination, represent a linear or branched, saturated hydrocarbon chain having the indicated number of carbon atoms. Examples of such groups include, but are not limited to methyl, ethyl, n-propyl, isopropyl, butyl, isobutyl, sec-butyl, terf-butyl, pentyl, isopentyl, hexyl, isohexyl and the like.
- C 3 The term "C 3 .
- 6 -cycloalkyl as used herein, alone or in combination, represent a saturated monocyclic hydrocarbon group having the indicated number of carbon atoms.
- examples of such groups include, but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and the like.
- C 2 . 6 -alkenyl represent an olefinically unsaturated branched or straight hydrocarbon group having from 2 to the specified number of carbon atoms and at least one double bond.
- groups include, but are not limited to, vinyl, 1-propenyl, 2-propenyl, allyl, iso-propenyl, 1,3-butadienyl, 1-butenyl, hexenyl, pentenyl and the like.
- C 2 . 6 -alkynyl represent an unsaturated branched or straight hydrocarbon group having from 2 to the specified number of carbon atoms and at least one triple bond.
- examples of such groups include, but are not limited to, 1-propynyl, 2- propynyl, 1-butynyl, 2-butynyl, 1-pentynyl, 2-pentynyI and the like.
- C -6 -alkoxy refers to a straight or branched configuration linked through an ether oxygen having its free valence bond from the ether oxygen.
- linear alkoxy groups are methoxy, ethoxy, propoxy, butoxy, pentoxy, hexoxy and the like.
- branched alkoxy are isopropoxy, sec-butoxy, tert-butoxy, isopentyloxy, isohexyloxy and the like.
- C 3 . 6 -cycloalkoxy as used herein, alone or in combination, represent a saturated monocyclic hydrocarbon group having the indicated number of carbon atoms linked through an ether oxygen having its free valence bond from the ether oxygen.
- Examples of cycloalkoxy groups are cyclopropyloxy, cyclobutyloxy, cyclopentyloxy, cyclohexyloxy and the like.
- C 1-6 -alkylthio refers to a straight or branched monovalent substituent comprising a C 1-6 -alkyl group linked through a divalent sulfur atom having its free valence bond from the sulfur atom and having 1 to 6 carbon atoms e.g. methylthio, ethylthio, propylthio, butylthio, pentylthio and the like.
- C 3-6 -cycloalkylthio represent a saturated monocyclic hydrocarbon group having the indicated number of carbon atoms linked through a divalent sulfur atom having its free valence bond from the sulfur atom.
- Examples of cycloalkoxy groups are cyclopropylthio, cyclobutylthio, cyclopentylthio, cyclohexylthio and the like.
- C 1-6 -alkylamino refers to a straight or branched monovalent substituent comprising a C 1-6 -alkyl group linked through amino having a free valence bond from the nitrogen atom e.g. methylamino, ethylamino, propylamino, butylamino, pentylamino and the like.
- C 3-6 -cycloalkylamino as used herein, alone or in combination, represent a saturated monocyclic hydrocarbon group having the indicated number of carbon atoms linked through amino having a free valence bond from the nitrogen atom e.g. cyclopropylamino, cyclobutylamino, cyclopentylamino, cyclohexylamino and the like.
- aryl refers to an aromatic monocyclic or an aromatic fused bi- or tricyclic hydrocarbon group e.g. phenyl, naphthyl, anthracenyl, phenanthrenyl, azulenyl and the like.
- halogen means fluorine, chlorine, bromine or iodine.
- C ⁇ -dialkylamino refers to an amino group wherein the two hydrogen atoms independently are substituted with a straight or branched, saturated hydrocarbon chain having the indicated number of carbon atoms; such as dimethylamino, N- ethyl-N-methylamino, diethylamino, dipropylamino, N-(n-butyl)-N-methylamino, di(n-pentyl) - amino and the like.
- acyl refers to a monovalent substituent comprising a C 1-6 - alkyl group linked through a carbonyl group; such as e.g. acetyl, propionyl, butyryl, isobutyryl, pivaloyl, valeryl and the like.
- heteroaryl refers to a monovalent substituent comprising a 5-7 membered monocyclic aromatic system or a 8-10 membered bicyclic aromatic system or a 12-16 membered tricyclic containing one or more heteroatoms selected from nitrogen, oxygen and sulfur, e.g.
- heteroaryloxy refers to a heteroaryl as defined herein linked to an oxygen atom having its free valence bond from the oxygen atom e.g. pyrrolyloxy, imidazolyloxy, pyrazolyloxy, triazolyloxy, pyrazinyloxy, pyrimidinyloxy, pyridazinyloxy, isothiazolyloxy, isoxazolyloxy, oxazolyloxy, oxadiazolyloxy, thiadiazolyloxy, quinolinyloxy, isoquinolinyloxy, quinazolinyloxy, quinoxalinyloxy, indoltloxy, benzimidazolyloxy, benzofuranyloxy, pteridinyloxy and purinyloxy carbazolyloxy, phena- zinyloxy, phenanthrolinyloxy, phenothiazinyloxy
- aralkyl refers to a straight or branched saturated carbon chain containing from 1 to 6 carbons substituted with an aromatic carbohydride; such as benzyl, phenethyl, 3-phenylpropyl, 1-naphthylmethyl, 2-(1-naphthyl)ethyl and the like.
- aryloxy refers to phenoxy, 1-naphthyIoxy, 2-naphthyloxy and the like.
- aralkoxy refers to a C 1-6 -alkoxy group substituted with an aromatic carbohydride, such as benzyloxy, phenethoxy, 3-phenylpropoxy, 1-naphthyl- methoxy, 2-(1-naphtyl)ethoxy and the like.
- heteroarylkyl refers to a straight or branched saturated carbon chain containing from 1 to 6 carbons substituted with a heteroaryl group; such as (2- furyl)methyl, (3-furyl)methyl, (2-thienyl)methyl, (3-thienyl)methyl, (2-pyridyl)methyl, 1-methyl- 1-(2-pyrimidyl)ethyl and the like.
- heteroarylkoxy refers to a heteroarylalkyl as defined herein linked to an oxygen atom having its free valence bond from the oxygen atom, e.g.
- arylthio refers to an aryl group linked through a divalent sulfur atom having its free valence bond from the sulfur atom, the aryl group optionally being mono- or polysubstituted with C 1-6 -alkyl, halogen, hydroxy or C 1-6 -alkoxy; e.g. phenylthio, (4-methylphenyl)- thio, (2-chlorophenyl)thio and the like.
- a fused tricyclic ring system refers to a non-aromatic or an aromatic fused tricyclic ringsystem, containing 11-16 ring atoms.
- the ring atoms are independently selected from carbon, nitrogen, oxygen or sulfur.
- a fused tricyclic ring system may be bonded to the rest of the compound through any atom of the ring system capable of such bonding.
- fused tricyclic ring systems are as- indacene, fluorene, phenanthrene, antracene, thianthrene, xanthene, phenoxathiin, carba- zole, carbazoline, phenanthridine, acridine, phenanthroline, phenazine, phenothiazine, phenoxazine, 5H-dibenzo[b,f]azepine, 10,11-dihydro-5 - -dibenzo[b,fjazepine and the like.
- treatment includes treatment, prevention and management of such condition.
- the present invention relates to compounds of the general formula (I):
- A is a fused tricyclic ring system optionally substituted with one or more substituents selected from
- Ri and R 2 are independently hydrogen, halogen, C ⁇ . 6 -alkyl, C 3-6 -cycloalkyl, C 1-6 -alkoxy or C 3- 6 -cycloalkoxy;
- R 3 and R 4 are independently hydrogen or halogen
- R 5 is hydrogen, C 1-6 -alkyl or C 3-6 -cycloalkyl
- a is 1 , 2 or 3;
- the present invention is concerned with compounds of formula (I) wherein A is a fused tricyclic ring system optionally substituted with one or more substituents selected from
- the present invention is concerned with compounds of formula I) wherein A is a fused tricyclic ring system optionally substituted with halogen.
- the present invention is concerned with compounds of for- mula I) wherein A is phenothiazine, phenoxazine, carbazole, carboline, dibenzo[b,f]azepine, 10,11 dihydro-dibenzo[b,f]azepine each of which is optionally substituted with halogen.
- the present invention is concerned with compounds of for- mula I) wherein R ⁇ is H.
- the present invention is concerned with compounds of for- mula I) wherein R 2 is hydrogen.
- the present invention is concerned with compounds of for- mula I) wherein R 3 is hydrogen.
- the present invention is concerned with compounds of for- mula I) wherein R 4 is hydrogen.
- the present invention is concerned with compounds of for- mula I) wherein R 5 is hydrogen or C 1-6 -alkyl.
- the present invention is concerned with compounds of for- mula I) wherein R 5 is hydrogen or methyl.
- the present invention is concerned with compounds of for- mula I) wherein a is 2.
- the present invention is concerned with compounds of for- mula I), which are PPAR ⁇ agonists.
- the present invention is concerned with compounds of for- mula I), which are partial PPAR ⁇ agonists.
- the present invention also encompasses pharmaceutically acceptable salts of the present compounds.
- Such salts include pharmaceutically acceptable acid addition salts, pharmaceutically acceptable base addition salts, pharmaceutically acceptable metal salts, ammonium and alkylated ammonium salts.
- Acid addition salts include salts of inorganic acids as well as organic acids. Representative examples of suitable inorganic acids include hydrochloric, hydrobromic, hydroiodic, phosphoric, sulfuric, nitric acids and the like.
- suitable organic acids include formic, acetic, trichloroacetic, trifluoroacetic, propionic, benzoic, cinnamic, citric, fumaric, glycolic, lactic, maleic, malic, malonic, mandelic, oxalic, picric, pyruvic, salicylic, succinic, methanesulfonic, ethanesulfonic, tartaric, ascorbic, pamoic, bismethylene salicylic, ethanedisulfonic, gluconic, citraconic, aspartic, stearic, palmitic, EDTA, glycolic, p-aminobenzoic, glutamic, benzenesulfonic, p-toluenesulfonic acids, sulphates, nitrates, phosphates, perchlorates, borates, acetates, benzoates, hydroxynaph- thoates, glycero
- compositions include the pharmaceutically acceptable salts listed in J. Pharm. Sci. 1977, 66, 2, which is incorporated herein by reference.
- metal salts include lithium, sodium, potassium, magnesium, zinc, calcium salts and the like.
- amines and organic amines include ammonium, methylamine, di- methylamine, trimethylamine, ethylamine, diethylamine, propylamine, butylamine, tetrame- thylamine, ethanolamine, diethanolamine, triethanolamine, meglumine, ethylenediamine, choline, N,N'-dibenzylethylenediamine, N-benzylphenylethylamine, N-methyl-D-glucamine, guanidine and the like.
- cationic amino acids include lysine, arginine, histidine and the like.
- the pharmaceutically acceptable salts are prepared by reacting the compound of formula I with 1 to 4 equivalents of a base such as sodium hydroxide, sodium methoxide, sodium hydride, potassium t-butoxide, calcium hydroxide, magnesium hydroxide and the like, in solvents like ether, THF, methanol, t-butanol, dioxane, isopropanol, ethanol etc. Mixture of solvents may be used. Organic bases like lysine, arginine, diethanolamine, choline, guandine and their derivatives etc. may also be used.
- a base such as sodium hydroxide, sodium methoxide, sodium hydride, potassium t-butoxide, calcium hydroxide, magnesium hydroxide and the like
- solvents like ether, THF, methanol, t-butanol, dioxane, isopropanol, ethanol etc.
- Organic bases like lysine, arginine, diethanolamine, choline
- acid addition salts wherever applicable are prepared by treatment with acids such as hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid, p-toluenesulphonic acid, methanesulfonic acid, acetic acid, citric acid, maleic acid salicylic acid, hydroxynaphthoic acid, ascorbic acid, palmitic acid, succinic acid, benzoic acid, benzenesulfonic acid, tartaric acid and the like in solvents like ethyl acetate, ether, alcohols, acetone, THF, dioxane etc. Mixture of solvents may also be used.
- acids such as hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid, p-toluenesulphonic acid, methanesulfonic acid, acetic acid, citric acid, maleic acid salicylic acid, hydroxynaphthoic acid, ascorbic
- stereoisomers of the compounds forming part of this invention may be prepared by using reactants in their single enantio eric form in the process wherever possible or by conducting the reaction in the presence of reagents or catalysts in their single enantiomer form or by resolving the mixture of stereoisomers by conventional methods.
- Some of the preferred methods include use of microbial resolution, enzymatic resolution, resolving the diastereomeric salts formed with chiral acids such as mandelic acid, camphorsulfonic acid, tartaric acid, lactic acid, and the like wherever applicable or chiral bases such as brucine, (R)- or (S)-phenylethylamine, cinchona alkaloids and their derivatives and the like.
- the compound of formula I may be converted to a 1:1 mixture of diastereomeric amides by treating with chiral amines, aminoacids, aminoalcohols derived from aminoacids; conventional reaction conditions may be employed to convert acid into an amide; the dia-stereomers may be separated either by fractional crystallization or chromatography and the stereoisomers of compound of formula I may be prepared by hydrolysing the pure diastereomeric amide.
- polymorphs of compound of general formula I forming part of this invention may be prepared by crystallization of compound of formula I under different conditions. For example, using different solvents commonly used or their mixtures for recrystallization; crys- tallizations at different temperatures; various modes of cooling, ranging from very fast to very slow cooling during crystallizations. Polymorphs may also be obtained by heating or melting the compound followed by gradual or fast cooling. The presence of polymorphs may be determined by solid probe nmr spectroscopy, ir spectroscopy, differential scanning calorimetry, powder X-ray diffraction or such other techniques.
- the invention also encompasses prodrugs of the present compounds, which on administration undergo chemical conversion by metabolic processes before becoming active pharmacological substances.
- prodrugs will be functional derivatives of the present compounds, which are readily convertible in vivo into the required compound of the formula (I).
- Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985.
- the invention also encompasses active metabolites of the present compounds.
- the present compounds of formula I can be utilised in the treatment and/or prevention of conditions mediated by nuclear receptors, in particular the Peroxisome Proliferator-Activated Receptors (PPAR).
- nuclear receptors in particular the Peroxisome Proliferator-Activated Receptors (PPAR).
- PPAR Peroxisome Proliferator-Activated Receptors
- the present invention relates to a method of treating and/or preventing Type I or Type II diabetes.
- the present invention relates to the use of one or more compounds of the general formula I or pharmaceutically acceptable salts thereof for the preparation of a medicament for the treatment and/or prevention of Type I or Type II diabetes.
- the present compounds are useful for the treatment and/or prevention of IGT.
- the present compounds are useful for the treatment and/or prevention of Type 2 diabetes.
- the present compounds are useful for the delaying or prevention of the progression from IGT to Type 2 diabetes.
- the present compounds are useful for the delaying or prevention of the progression from non-insulin requiring Type 2 diabetes to insulin requiring Type 2 diabetes.
- the present compounds reduce blood glucose and triglyceride levels and are accordingly useful for the treatment and/or prevention of ailments and disorders such as diabetes and/or obesity.
- the present compounds are useful for the treatment and/or prophylaxis of insulin resistance (Type 2 diabetes), impaired glucose tolerance, dyslipidemia, disorders related to Syndrome X such as hypertension, obesity, insulin resistance, hypergly- caemia, atherosclerosis, hyperiipidemia, coronary artery disease, myocardial ischemia and other cardiovascular disorders.
- the present compounds are effective in decreasing apoptosis in mammalian cells such as beta cells of Islets of Langerhans.
- the present compounds are useful for the treatment of certain renal diseases including glomerulonephritis, glomerulosclerosis, nephrotic syndrome, hypertensive nephrosclerosis.
- the present compounds may also be useful for improving cognitive functions in dementia, treating diabetic complications, psoriasis, polycystic ovarian syndrome (PCOS) and prevention and treatment of bone loss, e.g. osteoporosis.
- PCOS polycystic ovarian syndrome
- the present compounds may also be administered in combination with one or more further pharmacologically active substances e.g., selected from antiobesity agents, antidia- betics, antihypertensive agents, agents for the treatment and/or prevention of complications resulting from or associated with diabetes and agents for the treatment and/or prevention of complications and disorders resulting from or associated with obesity.
- further pharmacologically active substances e.g., selected from antiobesity agents, antidia- betics, antihypertensive agents, agents for the treatment and/or prevention of complications resulting from or associated with diabetes and agents for the treatment and/or prevention of complications and disorders resulting from or associated with obesity.
- the present compounds may be administered in combination with one or more antiobesity agents or appetite regulating agents.
- Such agents may be selected from the group consisting of CART (cocaine am- phetamine regulated transcript) agonists, NPY (neuropeptide Y) antagonists, MC4 (melano- cortin 4) agonists, orexin antagonists, TNF (tumor necrosis factor) agonists, CRF (corticotro- pin releasing factor) agonists, CRF BP (corticotropin releasing factor binding protein) antagonists, urocortin agonists, ⁇ 3 agonists, MSH (melanocyte-stimulating hormone) agonists, MCH (melanocyte-concentrating hormone) antagonists, CCK (cholecystokinin) agonists, se- rotonin re-uptake inhibitors, serotonin and noradrenaline re-uptake inhibitors, mixed serotonin and noradrenergic compounds, 5HT (serotonin) agonists, bombesin agonists, galanin antagonists, growth
- the antiobesity agent is leptin.
- the antiobesity agent is dexamphetamine or amphetamine.
- the antiobesity agent is fenfluramine or dexfenfluramine.
- the antiobesity agent is sibutramine. In a further embodiment the antiobesity agent is orlistat. In another embodiment the antiobesity agent is mazindo! or phentermine.
- Suitable antidiabetics comprise insulin, GLP-1 (glucagon like peptide-1) derivatives such as those disclosed in WO 98/08871 to Novo Nordisk A/S, which is incorporated herein by reference as well as orally active hypoglycaemic agents.
- the orally active hypoglycaemic agents preferably comprise sulphonylureas, bigua- nides, meglitinides, glucosidase inhibitors, glucagon antagonists such as those disclosed in WO 99/01423 to Novo Nordisk A/S and Agouron Pharmaceuticals, Inc., GLP-1 agonists, potassium channel openers such as those disclosed in WO 97/26265 and WO 99/03861 to Novo Nordisk A S which are incorporated herein by reference, DPP-IV (dipeptidyl peptidase- IV) inhibitors, inhibitors of hepatic enzymes involved in stimulation of gluconeogenesis and/or glycogenolysis, glucose uptake modulators, compounds modifying the lipid metabolism such as antihyperlipidemic agents and antilipidemic agents as HMG CoA inhibitors (statins), compounds lowering food intake, RXR agonists and agents acting on the ATP-dependent potassium channel of the ⁇ -cells.
- the present compounds are administered in combination with a sulphonylurea eg. tolbutamide, glibenclamide, glipizide or glicazide.
- a sulphonylurea eg. tolbutamide, glibenclamide, glipizide or glicazide.
- the present compounds are administered in combination with a biguanide eg. metformin.
- the present compounds are administered in combination with a meglitinide eg. repaglinide or senaglinide.
- the present compounds are administered in combination with an ⁇ -glucosidase inhibitor eg. miglitol or acarbose.
- an agent acting on the ATP-dependent potassium channel of the ⁇ -cells eg. tolbutamide, glibenclamide, glipizide, glicazide or repaglinide.
- the present compounds may be administered in combination with nateglinide.
- the present compounds are administered in combination with an antihyperlipidemic agent or antilipidemic agent eg. cholestyramine, colestipol, clofi- brate, gemfibrozil, lovastatin, pravastatin, simvastatin, probucol or dextrothyroxine.
- an antihyperlipidemic agent or antilipidemic agent eg. cholestyramine, colestipol, clofi- brate, gemfibrozil, lovastatin, pravastatin, simvastatin, probucol or dextrothyroxine.
- the present compounds are administered in combination with more than one of the above-mentioned compounds eg. in combination with a sulphony- lurea and metformin, a sulphonylurea and acarbose, repaglinide and metformin, insulin and a sulphonylurea, insulin and metformin, insulin, insulin and lovastatin, etc.
- the present compounds may be administered in combination with one or more antihypertensive agents.
- antihypertensive agents are ⁇ -blockers such as alprenolol, atenolol, timolol, pindolol, propranolol and metoprolol, ACE (angiotensin converting enzyme) inhibitors such as benazepril, captopril, enalapril, fosinopril, lisinopril, quinapril and ramipril, calcium channel blockers such as nifedipine, felodipine, nicardipine, isradipine, nimodipine, diltiazem and verapamil, and ⁇ -blockers such as doxazosin, urapidil, prazosin and terazosin.
- ⁇ -blockers such as alprenolol, atenolol, timolol, pin
- compositions comprising, as an active ingredient, at least one compound of the formula I or any optical or geometric isomer or tautomeric form thereof including mixtures of these or a pharmaceutically acceptable salt thereof together with one or more pharmaceutically acceptable carriers or diluents.
- the invention relates to the use of compounds of the general formula I or their tautomeric forms, their stereoisomers, their polymorphs, their pharmaceutically acceptable salts or pharmaceutically acceptable solvates thereof for the preparation of a pharmaceutical composition for the treatment and/or prevention of conditions mediated by nuclear receptors, in particular the Peroxisome Proliferator-Activated Receptors (PPAR) such as the conditions mentioned above.
- PPAR Peroxisome Proliferator-Activated Receptors
- the present invention also relates to a process for the preparation of the above said novel compounds, their derivatives, their analogs, their tautomeric forms, their stereoisomers, their polymorphs, their pharmaceutically acceptable salts or pharmaceutically acceptable solvates.
- compositions according to the invention may be formulated with pharmaceutically acceptable carriers or diluents as well as any other known adjuvants and excipients in accordance with conventional techniques such as those disclosed in Remington: The Science and Practice of Pharmacy, 19 th Edition, Gennaro, Ed., Mack Publishing Co., Easton, PA, 1995.
- the compositions may appear in conventional forms, for example capsules, tablets, aerosols, solutions, suspensions or topical applications.
- compositions include a compound of formula I or a pharmaceutically acceptable acid addition salt thereof, associated with a pharmaceutically acceptable excipient which may be a carrier or a diluent or be diluted by a carrier, or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container.
- a pharmaceutically acceptable excipient which may be a carrier or a diluent or be diluted by a carrier, or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container.
- the active compound will usually be mixed with a carrier, or diluted by a carrier, or enclosed within a carrier which may be in the form of a ampoule, capsule, sachet, paper, or other container.
- the carrier When the carrier serves as a diluent, it may be solid, semi-solid, or liquid material which acts as a vehicle, excipient, or medium for the active compound.
- the active compound can be adsorbed on a granular solid container for example in a sachet.
- suitable carriers are water, salt solutions, alcohols, polyethylene glycols, polyhydroxyethoxylated castor oil, peanut oil, olive oil, gelatine, lactose, terra alba, sucrose, cyclodextrin, amylose, magnesium stearate, talc, gelatin, agar, pectin, acacia, stearic acid or lower alkyl ethers of cellulose, silicic acid, fatty acids, fatty acid amines, fatty acid monoglycerides and diglycerides, pentaerythritol fatty acid esters, polyoxyethylene, hydroxymethylcellulose and polyvinylpyrrolidone.
- the carrier or diluent may include any sustained release material known in the art, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax.
- the formulations may also include wetting agents, emulsifying and suspending agents, preserving agents, sweetening agents or flavouring agents.
- the formulations of the invention may be formulated so as to provide quick, sustained, or delayed release of the active ingredient after administration to the patient by employing procedures well known in the art.
- compositions can be sterilized and mixed, if desired, with auxiliary agents, emulsifiers, salt for influencing osmotic pressure, buffers and/or colouring substances and the like, which do not deleteriously react with the active compounds.
- the route of administration may be any route, which effectively transports the active compound to the appropriate or desired site of action, such as oral, nasal, pulmonary, trans- dermal or parenteral e.g. rectal, depot, subcutaneous, intravenous, intraurethral, intramuscular, intranasal, ophthalmic solution or an ointment, the oral route being preferred.
- the preparation may be tabletted, placed in a hard gelatin capsule in powder or pellet form or it can be in the form of a troche or lozenge. If a liquid carrier is used, the preparation may be in the form of a syrup, emulsion, soft gelatin capsule or sterile injectable liquid such as an aqueous or non-aqueous liquid suspension or solution.
- the preparation may contain a compound of formula I dissolved or suspended in a liquid carrier, in particular an aqueous carrier, for aerosol application.
- a liquid carrier in particular an aqueous carrier
- the carrier may contain additives such as solubilizing agents, e.g. propylene glycol, surfactants, absorption enhancers such as lecithin (phosphatidylcholine) or cyclodextrin, or preservatives such as parabenes.
- injectable solutions or suspensions preferably aqueous solutions with the active compound dissolved in polyhydroxylated castor oil.
- Tablets, dragees, or capsules having talc and/or a carbohydrate carrier or binder or the like are particularly suitable for oral application.
- Preferable carriers for tablets, dragees, or capsules include lactose, corn starch, and/or potato starch.
- a syrup or elixir can be used in cases where a sweetened vehicle can be employed.
- a typical tablet which may be prepared by conventional tabletting techniques may contain:
- the pharmaceutical composition of the invention may comprise the compound of formula (I) in combination with further pharmacologically active substances such as those described in the foregoing.
- the compounds of the invention may be administered to a mammal, especially a human in need of such treatment, prevention, elimination, alleviation or amelioration of diseases related to the regulation of blood sugar.
- mammals include also animals, both domestic animals, e.g. household pets, and non-domestic animals such as wildlife.
- a typical oral dosage is in the range of from about 0.001 to about 100 mg/kg body weight per day, preferably from about 0.01 to about 50 mg/kg body weight per day, and more preferred from about 0.05 to about 10 mg/kg body weight per day administered in one or more dosages such as 1 to 3 dosages.
- the exact dosage will depend upon the frequency and mode of administration, the sex, age, weight and general condition of the subject treated, the nature and severity of the condition treated and any concomitant diseases to be treated and other fac- tors evident to those skilled in the art.
- a typical unit dosage form for oral administration one or more times per day such as 1 to 3 times per day may contain of from 0.05 to about 1000 mg, preferably from about 0.1 to about 500 mg, and more preferred from about 0.5 mg to about 200 mg.
- the compounds used as starting materials are either known compounds or compounds, which can readily be prepared by methods known per se.
- the structures of the compounds are confirmed by either elemental analysis (MA) nuclear magnetic resonance (NMR), mass spectrometry (MS) or optical rotation.
- NMR shifts ( ⁇ ) are given in parts per million (ppm) and only selected peaks are given, mp is melting point and is given in °C.
- Column chromatography was carried out using the technique described by W.C. Still et al, J. Org. Chem. 1978, 43, 2923-2925 on Merck silica gel 60 (Art 9385). The optical rotation was measured on a Advanced Laser Polarimeter.
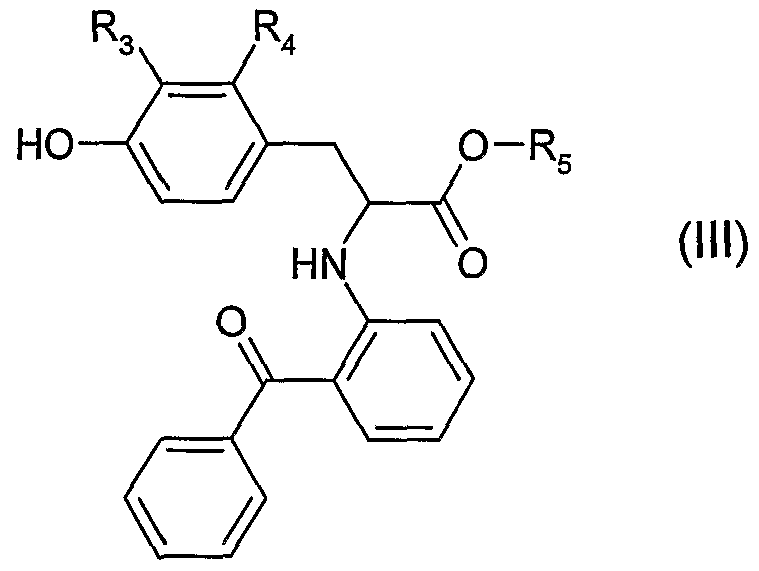
- R 3 , R , and R 5 are defined as above, except that R 5 is not hydrogen under Mitsunobu conditions, using a reagent such as triphenylphosphine/diethylazodicarboxylate and the like to obtain a compound of formula I, wherein A, R ⁇ R 2 , R 3, R 4 , R 5 and a are defined as above, except that R 5 is not hydrogen.
- Step B Reacting a compound of formula IV wherein L is a leaving group such as p- toluenesulfonate, methanesulfonate, halogen, triflate and the like and wherein A, R-i, R 2 and a are defined as above with a compound of formula III wherein R 3 , R , R 5 and a are defined as above, except that R 4 is not hydrogen to give a compound of formula I wherein A, R ⁇ R 2 , R 3, R 4 , R 5 and a are defined as above, except that R is not hydrogen.
- L is a leaving group such as p- toluenesulfonate, methanesulfonate, halogen, triflate and the like and wherein A, R-i, R 2 and a are defined as above with a compound of formula III wherein R 3 , R , R 5 and a are defined as above, except that R 4 is not hydrogen to give a compound of formula I wherein A, R ⁇
- reaction mixture was added more triphenylphosphine (53 mg, 0.20 mmol) and diethylazodicarboxylate (35 mg, 0.20 mmol), and the mixture was stirred for another 84 h.
- the reaction mixture was diluted with water and the product extracted with dichloromethane (x3). The organic layers were combined, washed with water, dried (MgSO 4 ) and evaporated.
- the crude product was then purified by column chromatography on silica (eluent: dichloromethane).
- reaction mixture was diluted with water and the product extracted with ethyl acetate (x3). The organic layers were combined, washed with water, dried (MgSO 4 ) and evaporated. The crude product was then purified by column chromatography on silica (eluent:toluene/ethyl acetate (8:1)) to give 500 mg (83%) of the title compound.
- the reaction mixture was added more triphenylphosphine (53 mg, 0.20 mmol) and diethylazodicarboxylate (35 mg, 0.20 mmol), and the mixture was stirred for another 80 h.
- the reaction mixture was diluted with water (10 mL) and the product extracted with dichloromethane (25 mL x 2). The organic layers were combined, washed with water, dried (MgSO 4 ) and evaporated.
- the crude product was then purified by column chromatography on silica (eluent: dichloromethane) to give 105 mg, (73%) of the title compound.
- the reaction mixture was added more triphenylphosphine (47 mg, 0.18 mmol) and diethyla- zodicarboxylate (31 mg, 0.18 mmol), and the mixture was stirred for another 3 h.
- the reaction mixture was diluted with water (10 mL) and the product extracted with dichloromethane (25 mL x 2). The organic layers were combined, washed with water, dried (MgSO 4 ) and evaporated.
- the crude product was then purified by column chromatography on silica (eluent: dichloromethane) to give 82 mg, (57%) of the title compound.
- the PPAR transient transactivation assays are based on transient transfection into human HEK293 cells of two plasmids encoding a chimeric test protein and a reporter protein respectively.
- the chimeric test protein is a fusion of the DNA binding domain (DBD) from the yeast GAL4 transcription factor to the ligand binding domain (LBD) of the human PPAR proteins.
- the GAL4 DBD will direct the chimeric protein to bind only to Gal4 enhancers (of which none existed in HEK293 cells).
- the reporter plas- mid contained a Gal4 enhancer driving the expression of the firefly luciferase protein.
- HEK293 cells expressed the GAL4-DBD-PPAR-LBD fusion protein.
- the fusion protein will in turn bind to the Gal4 enhancer controlling the luciferase expression, and do nothing in the absence of ligand.
- luciferase pro- tein Upon addition to the cells of a PPAR ligand luciferase pro- tein will be produced in amounts corresponding to the activation of the PPAR protein.
- the amount of luciferase protein is measured by light emission after addition of the appropriate substrate.
- HEK293 cells were grown in DMEM + 10% FCS. Cells were seeded in 96-well plates the day before transfection to give a confluency of 50-80 % at transfection. A total of 0,8 ⁇ g DNA containing 0,64 ⁇ g pM1 ⁇ / ⁇ LBD, 0,1 ⁇ g pCMV ⁇ Gal, 0,08 ⁇ g pGL2(Gal4) 5 and 0,02 ⁇ g pADVANTAGE was transfected per well using FuGene transfection reagent according to the manufacturers instructions (Roche). Cells were allowed to express protein for 48 h followed by addition of compound.
- Plasmids Human PPAR ⁇ , ⁇ and ⁇ was obtained by PCR amplification using cDNA synthesized by reverse transcription of mRNA from human liver, adipose tissue and plancenta re- spectively. Amplified cDNAs were cloned into pCR2.1 and sequenced.
- the ligand binding domain (LBD) of each PPAR isoform was generated by PCR (PPAR ⁇ : aa 167 - C-terminus; PPAR ⁇ : aa 165 - C-terminus; PPAR ⁇ : aa 128 - C-terminus) and fused to the DNA binding domain (DBD) of the yeast transcription factor GAL4 by subcloning fragments in frame into the vector pM1 (Sadowski et al. (1992), Gene 118, 137) generating the plasmids pMl ⁇ LBD, pMl ⁇ LBD and pM1 ⁇ . Ensuing fusions were verified by sequencing.
- the reporter was constructed by inserting an oligonucleotide encoding five repeats of the GAL4 recognition sequence (5 x CGGAGTACTGTCCTCCG(AG)) (Webster et al. (1988), Nucleic Acids Res. 16, 8192) into the vector pGL2 promotor (Promega) generating the plasmid pGL2(GAL4) 5 .
- pCMV ⁇ Gal was purchased from Clontech and pADVANTAGE was purchased from Promega.
- Luciferase assay Medium including test compound was aspirated and 100 ⁇ l PBS incl. 1mM Mg++ and Ca++ was added to each well. The luciferase assay was performed using the LucLite kit according to the manufacturers instructions (Packard Instruments). Light emission was quantified by counting on a Packard LumiCounter. To measure ⁇ - galactosidase activity 25 ⁇ l supernatant from each transfection lysate was transferred to a new microplate. ⁇ -galactosidase assays were performed in the microwell plates using a kit from Promega and read in a Labsystems Ascent Multiscan reader. The ⁇ -galactosidase data were used to normalize (transfection efficiency, cell growth etc.) the luciferase data.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Heart & Thoracic Surgery (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
- Nitrogen- Or Sulfur-Containing Heterocyclic Ring Compounds With Rings Of Six Or More Members (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Indole Compounds (AREA)
Abstract
Description















Claims

Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003517026A JP2005503377A (en) | 2001-07-30 | 2002-07-05 | Novel vinyl N- (2-benzoylphenyl) -L-tyrosine derivatives and their use in antidiabetic agents, etc. |
| EP02745184A EP1414806A1 (en) | 2001-07-30 | 2002-07-05 | Novel vinyl n-(2-benzoylphenyl)-l-tyrosine derivatives and their use as antidiabetics etc |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DKPA200101156 | 2001-07-30 | ||
| DKPA200101156 | 2001-07-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003011834A1 true WO2003011834A1 (en) | 2003-02-13 |
Family
ID=8160645
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/DK2002/000469 WO2003011834A1 (en) | 2001-07-30 | 2002-07-05 | Novel vinyl n-(2-benzoylphenyl)-l-tyrosine derivatives and their use as antidiabetics etc |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP1414806A1 (en) |
| JP (1) | JP2005503377A (en) |
| WO (1) | WO2003011834A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004048333A1 (en) * | 2002-11-26 | 2004-06-10 | Shenzhen Chipscreen Biosciences Ltd. | Substituted arylalcanoic acid derivatives as ppar pan agonists with potent antihyperglycemic and antihyperlipidemic activity |
| WO2005056536A1 (en) * | 2003-12-10 | 2005-06-23 | Ranbaxy Laboratories Limited | Antidiabetic agents which exhibit activity against ppar |
| WO2006010775A1 (en) * | 2004-07-30 | 2006-02-02 | Laboratorios Salvat, S.A. | Tyrosine derivatives as ppar-gamma-modulators |
| WO2006067086A1 (en) | 2004-12-20 | 2006-06-29 | Adamed Sp. Z O.O. | New 3-phenylpropionic acid derivatives for the treatment of diabetes |
| WO2006077206A1 (en) | 2005-01-19 | 2006-07-27 | Adamed Sp. Z O.O. | New 3-phenylpropionic acid derivatives and their use as ppar- gamma rezeptor liganden |
| US7220766B2 (en) | 2005-01-20 | 2007-05-22 | Adamed, Sp. Z.O.O. | 3-phenylpropionic acid derivatives |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997031907A1 (en) * | 1996-02-28 | 1997-09-04 | Glaxo Group Limited | Substituted 4-hydroxy-phenylalcanoic acid derivatives with agonist activity to ppar-gamma |
| WO2000063153A1 (en) * | 1999-04-20 | 2000-10-26 | Novo Nordisk A/S | New compounds, their preparation and use |
| EP1167357A1 (en) * | 1999-04-06 | 2002-01-02 | Sankyo Company, Limited | Alpha-substituted carboxylic acid derivatives |
-
2002
- 2002-07-05 EP EP02745184A patent/EP1414806A1/en not_active Withdrawn
- 2002-07-05 WO PCT/DK2002/000469 patent/WO2003011834A1/en not_active Application Discontinuation
- 2002-07-05 JP JP2003517026A patent/JP2005503377A/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997031907A1 (en) * | 1996-02-28 | 1997-09-04 | Glaxo Group Limited | Substituted 4-hydroxy-phenylalcanoic acid derivatives with agonist activity to ppar-gamma |
| EP1167357A1 (en) * | 1999-04-06 | 2002-01-02 | Sankyo Company, Limited | Alpha-substituted carboxylic acid derivatives |
| WO2000063153A1 (en) * | 1999-04-20 | 2000-10-26 | Novo Nordisk A/S | New compounds, their preparation and use |
Non-Patent Citations (5)
| Title |
|---|
| BROWN K K ET AL: "A NOVEL N-ARYL TYROSINE ACTIVATOR OF PEROXISOME PROLIFERATOR-ACTIVATED RECEPTOR-GAMMA REVERSES THE DIABETIC PHENOTYPE OF THE ZUCKER DIABETIC FATTY RAT", DIABETES, NEW YORK, NY, US, vol. 48, July 1999 (1999-07-01), pages 1415 - 1424, XP000985787, ISSN: 0012-1797 * |
| COBB J E ET AL: "N-(2-Benzoylphenyl)-L-tyrosine PPAR gamma agonists. 3. Structure-activity relationship and optimization of the N-aryl substituent", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 41, no. 25, 3 December 1998 (1998-12-03), pages 5055 - 5069, XP002156427, ISSN: 0022-2623 * |
| COLLINS J L ET AL: "n-(2-BENZOYLPHENYL)-L-TYROSINE PPAR-GAMMA AGONISTS. 2. STRUCTURE-ACTIVITY RELATIONSHIP AND OPTIMIZATION OF THE PHENYL ALKYL ETHER MOIETY", JOURNAL OF MEDICINAL AND PHARMACEUTICAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. EASTON, US, vol. 41, no. 25, 1998, pages 5037 - 5054, XP002211273 * |
| HENKE B R ET AL: "N-(2-BENZOYLPHENYL)-L-TYROSINE PPARGAMMA AGONISTS. 1. DISCOVERY OF A NOVEL SERIES OF POTENT ANTIHYPERGLYCEMIC AND ANTIHYPERLIPIDEMIC AGENTS", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 41, no. 25, 1998, pages 5020 - 5036, XP000864731, ISSN: 0022-2623 * |
| SORBERA L A ET AL: "FARGLITAZAR Antidiabetic PPARgamma agonist. GI-262570", DRUGS OF THE FUTURE, BARCELONA, ES, vol. 26, no. 4, 2001, pages 354 - 363, XP008005620, ISSN: 0377-8282 * |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004048333A1 (en) * | 2002-11-26 | 2004-06-10 | Shenzhen Chipscreen Biosciences Ltd. | Substituted arylalcanoic acid derivatives as ppar pan agonists with potent antihyperglycemic and antihyperlipidemic activity |
| US7268157B2 (en) | 2002-11-26 | 2007-09-11 | Shenzhen Chipscreen Biosciences, Ltd. | Substituted arylalcanoic acid derivatives as PPAR pan agonists with potent antihyperglycemic and antihyperlipidemic activity |
| WO2005056536A1 (en) * | 2003-12-10 | 2005-06-23 | Ranbaxy Laboratories Limited | Antidiabetic agents which exhibit activity against ppar |
| WO2006010775A1 (en) * | 2004-07-30 | 2006-02-02 | Laboratorios Salvat, S.A. | Tyrosine derivatives as ppar-gamma-modulators |
| US7423172B2 (en) | 2004-07-30 | 2008-09-09 | Laboratorios Salvat, S.A. | Tyrosine derivatives as PPAR-γ-modulators |
| EA013227B1 (en) * | 2004-07-30 | 2010-04-30 | Лабораториос Салват, С.А. | Tyrosine derivatives as ppar-gamma-modulators |
| WO2006067086A1 (en) | 2004-12-20 | 2006-06-29 | Adamed Sp. Z O.O. | New 3-phenylpropionic acid derivatives for the treatment of diabetes |
| US7919515B2 (en) | 2004-12-20 | 2011-04-05 | Adamed Sp. Z O.O. | 3-phenylpropionic acid derivatives |
| WO2006077206A1 (en) | 2005-01-19 | 2006-07-27 | Adamed Sp. Z O.O. | New 3-phenylpropionic acid derivatives and their use as ppar- gamma rezeptor liganden |
| US7309791B2 (en) | 2005-01-19 | 2007-12-18 | Adamed Sp. Z.O.O | 3-phenylpropionic acid derivatives |
| US7629370B2 (en) | 2005-01-19 | 2009-12-08 | Adamed Sp. Z O.O. | 3-phenylpropionic acid derivatives |
| US7220766B2 (en) | 2005-01-20 | 2007-05-22 | Adamed, Sp. Z.O.O. | 3-phenylpropionic acid derivatives |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2005503377A (en) | 2005-02-03 |
| EP1414806A1 (en) | 2004-05-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6569901B2 (en) | Alkynyl-substituted propionic acid derivatives, their preparation and use | |
| EP1438283A1 (en) | Dicarboxylic acid derivatives, their preparation and therapeutic use | |
| AU3958000A (en) | New compounds, their preparation and use | |
| EP1276710A1 (en) | New compounds, their preparation and use | |
| EP1414778A1 (en) | Novel vinyl carboxylic acid derivatives and their use as antidiabetics etc. | |
| US7091245B2 (en) | Compounds, their preparation and use | |
| EP1745014B1 (en) | Novel compounds, their preparation and use | |
| US7968723B2 (en) | Compounds, their preparation and use | |
| EP1578716A1 (en) | Dicarboxylic acid derivatives as ppar-agonists | |
| EP1254102A1 (en) | Alkynylsubstituted propionic acid derivatives and their use against diabetes and obesity | |
| US6869967B2 (en) | Peroxisome proliferator-activated receptor (PPAR) active vinyl carboxylic acid derivatives | |
| WO2003011814A1 (en) | Novel vinyl n-(2-benzoylphenyl)-l-tyrosine derivatives and their use as antidiabetics etc | |
| US7067530B2 (en) | Compounds, their preparation and use | |
| US6972294B1 (en) | Compounds, their preparation and use | |
| EP1414806A1 (en) | Novel vinyl n-(2-benzoylphenyl)-l-tyrosine derivatives and their use as antidiabetics etc | |
| US7220877B2 (en) | Compounds, their preparation and use | |
| US6509374B2 (en) | Compounds, their preparation and use | |
| US7816385B2 (en) | Dimeric dicarboxylic acid derivatives, their preparation and use | |
| US20030055076A1 (en) | Novel compounds, their preparation and use | |
| AU2002316815A1 (en) | Novel vinyl carboxylic acid derivatives and their use as antidiabetics etc. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG UZ VN YU ZA ZM ZW Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BY BZ CA CH CN CO CR CU CZ DE DM DZ EC EE ES FI GB GD GE GH HR HU ID IL IN IS JP KE KG KP KR LC LK LR LS LT LU LV MA MD MG MN MW MX MZ NO NZ OM PH PL PT RU SD SE SG SI SK SL TJ TM TN TR TZ UA UG UZ VN YU ZA ZM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LU MC NL PT SE SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ UG ZM ZW AM AZ BY KG KZ RU TJ TM AT BE BG CH CY CZ DK EE ES FI FR GB GR IE IT LU MC PT SE SK TR BF BJ CF CG CI GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002745184 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003517026 Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002745184 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2002745184 Country of ref document: EP |