WO2002085303A2 - Substituted tetracycline compounds for the treatment of malaria - Google Patents
Substituted tetracycline compounds for the treatment of malaria Download PDFInfo
- Publication number
- WO2002085303A2 WO2002085303A2 PCT/US2002/012935 US0212935W WO02085303A2 WO 2002085303 A2 WO2002085303 A2 WO 2002085303A2 US 0212935 W US0212935 W US 0212935W WO 02085303 A2 WO02085303 A2 WO 02085303A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- hydrogen
- substituted
- alkynyl
- alkenyl
- Prior art date
Links
- -1 tetracycline compounds Chemical class 0.000 title claims abstract description 227
- 239000004098 Tetracycline Substances 0.000 title claims abstract description 147
- 235000019364 tetracycline Nutrition 0.000 title claims abstract description 147
- 229960002180 tetracycline Drugs 0.000 title claims abstract description 138
- 229930101283 tetracycline Natural products 0.000 title claims abstract description 138
- 201000004792 malaria Diseases 0.000 title claims abstract description 106
- 150000001875 compounds Chemical class 0.000 claims abstract description 111
- 238000000034 method Methods 0.000 claims abstract description 107
- 230000000078 anti-malarial effect Effects 0.000 claims abstract description 16
- 239000003430 antimalarial agent Substances 0.000 claims abstract description 14
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 9
- 239000003937 drug carrier Substances 0.000 claims abstract description 6
- 125000000217 alkyl group Chemical group 0.000 claims description 103
- 125000000304 alkynyl group Chemical group 0.000 claims description 81
- 229910052739 hydrogen Inorganic materials 0.000 claims description 80
- 239000001257 hydrogen Substances 0.000 claims description 80
- 125000003342 alkenyl group Chemical group 0.000 claims description 76
- 125000003118 aryl group Chemical group 0.000 claims description 75
- 150000002431 hydrogen Chemical group 0.000 claims description 57
- 125000003545 alkoxy group Chemical group 0.000 claims description 54
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 51
- 125000003282 alkyl amino group Chemical group 0.000 claims description 50
- 125000004644 alkyl sulfinyl group Chemical group 0.000 claims description 44
- 125000004414 alkyl thio group Chemical group 0.000 claims description 44
- 229910052736 halogen Inorganic materials 0.000 claims description 40
- 150000002367 halogens Chemical class 0.000 claims description 39
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 38
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 35
- 125000001072 heteroaryl group Chemical group 0.000 claims description 33
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 32
- 125000000623 heterocyclic group Chemical group 0.000 claims description 31
- 150000003522 tetracyclines Chemical class 0.000 claims description 30
- 229910052757 nitrogen Inorganic materials 0.000 claims description 29
- 239000000651 prodrug Chemical group 0.000 claims description 27
- 229940002612 prodrug Drugs 0.000 claims description 27
- 150000003839 salts Chemical class 0.000 claims description 23
- 229910052799 carbon Inorganic materials 0.000 claims description 20
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 19
- LOUPRKONTZGTKE-WZBLMQSHSA-N Quinine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@@H]2[C@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-WZBLMQSHSA-N 0.000 claims description 18
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 17
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 claims description 17
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 17
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 claims description 16
- 229910052717 sulfur Inorganic materials 0.000 claims description 16
- SGKRLCUYIXIAHR-AKNGSSGZSA-N (4s,4ar,5s,5ar,6r,12ar)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1=CC=C2[C@H](C)[C@@H]([C@H](O)[C@@H]3[C@](C(O)=C(C(N)=O)C(=O)[C@H]3N(C)C)(O)C3=O)C3=C(O)C2=C1O SGKRLCUYIXIAHR-AKNGSSGZSA-N 0.000 claims description 15
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 claims description 15
- 229910052760 oxygen Inorganic materials 0.000 claims description 15
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 15
- 125000005199 aryl carbonyloxy group Chemical group 0.000 claims description 14
- 229960004023 minocycline Drugs 0.000 claims description 14
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 12
- 229960003722 doxycycline Drugs 0.000 claims description 12
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 12
- 239000001301 oxygen Substances 0.000 claims description 12
- FFTVPQUHLQBXQZ-KVUCHLLUSA-N (4s,4as,5ar,12ar)-4,7-bis(dimethylamino)-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1C2=C(N(C)C)C=CC(O)=C2C(O)=C2[C@@H]1C[C@H]1[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]1(O)C2=O FFTVPQUHLQBXQZ-KVUCHLLUSA-N 0.000 claims description 11
- 125000002252 acyl group Chemical group 0.000 claims description 11
- XEEQGYMUWCZPDN-DOMZBBRYSA-N (-)-(11S,2'R)-erythro-mefloquine Chemical compound C([C@@H]1[C@@H](O)C=2C3=CC=CC(=C3N=C(C=2)C(F)(F)F)C(F)(F)F)CCCN1 XEEQGYMUWCZPDN-DOMZBBRYSA-N 0.000 claims description 10
- 229960001962 mefloquine Drugs 0.000 claims description 10
- SSOLNOMRVKKSON-UHFFFAOYSA-N proguanil Chemical compound CC(C)\N=C(/N)N=C(N)NC1=CC=C(Cl)C=C1 SSOLNOMRVKKSON-UHFFFAOYSA-N 0.000 claims description 10
- 229960005385 proguanil Drugs 0.000 claims description 10
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 claims description 9
- 235000001258 Cinchona calisaya Nutrition 0.000 claims description 9
- FOHHNHSLJDZUGQ-VWLOTQADSA-N Halofantrine Chemical compound FC(F)(F)C1=CC=C2C([C@@H](O)CCN(CCCC)CCCC)=CC3=C(Cl)C=C(Cl)C=C3C2=C1 FOHHNHSLJDZUGQ-VWLOTQADSA-N 0.000 claims description 9
- 241000282414 Homo sapiens Species 0.000 claims description 9
- 230000000845 anti-microbial effect Effects 0.000 claims description 9
- 229960003677 chloroquine Drugs 0.000 claims description 9
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 claims description 9
- 229960003242 halofantrine Drugs 0.000 claims description 9
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 9
- 229960000948 quinine Drugs 0.000 claims description 9
- LUBUTTBEBGYNJN-UHFFFAOYSA-N 4-amino-n-(5,6-dimethoxypyrimidin-4-yl)benzenesulfonamide;5-(4-chlorophenyl)-6-ethylpyrimidine-2,4-diamine Chemical compound CCC1=NC(N)=NC(N)=C1C1=CC=C(Cl)C=C1.COC1=NC=NC(NS(=O)(=O)C=2C=CC(N)=CC=2)=C1OC LUBUTTBEBGYNJN-UHFFFAOYSA-N 0.000 claims description 8
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 8
- 239000004599 antimicrobial Substances 0.000 claims description 8
- CYDMQBQPVICBEU-UHFFFAOYSA-N chlorotetracycline Natural products C1=CC(Cl)=C2C(O)(C)C3CC4C(N(C)C)C(O)=C(C(N)=O)C(=O)C4(O)C(O)=C3C(=O)C2=C1O CYDMQBQPVICBEU-UHFFFAOYSA-N 0.000 claims description 8
- ISZNZKHCRKXXAU-UHFFFAOYSA-N chlorproguanil Chemical compound CC(C)\N=C(/N)N=C(N)NC1=CC=C(Cl)C(Cl)=C1 ISZNZKHCRKXXAU-UHFFFAOYSA-N 0.000 claims description 8
- 229950000764 chlorproguanil Drugs 0.000 claims description 8
- CYDMQBQPVICBEU-XRNKAMNCSA-N chlortetracycline Chemical compound C1=CC(Cl)=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(O)=C(C(N)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O CYDMQBQPVICBEU-XRNKAMNCSA-N 0.000 claims description 8
- 231100000135 cytotoxicity Toxicity 0.000 claims description 8
- 229960000860 dapsone Drugs 0.000 claims description 8
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 8
- 230000008092 positive effect Effects 0.000 claims description 8
- 229960001404 quinidine Drugs 0.000 claims description 8
- 239000011593 sulfur Substances 0.000 claims description 8
- GUXHBMASAHGULD-SEYHBJAFSA-N (4s,4as,5as,6s,12ar)-7-chloro-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1([C@H]2O)=C(Cl)C=CC(O)=C1C(O)=C1[C@@H]2C[C@H]2[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]2(O)C1=O GUXHBMASAHGULD-SEYHBJAFSA-N 0.000 claims description 7
- YNWCUCSDUMVJKR-UHFFFAOYSA-N 4-[(7-chloroquinolin-4-yl)amino]-2-(pyrrolidin-1-ylmethyl)phenol Chemical compound OC1=CC=C(NC=2C3=CC=C(Cl)C=C3N=CC=2)C=C1CN1CCCC1 YNWCUCSDUMVJKR-UHFFFAOYSA-N 0.000 claims description 7
- OVCDSSHSILBFBN-UHFFFAOYSA-N Amodiaquine Chemical compound C1=C(O)C(CN(CC)CC)=CC(NC=2C3=CC=C(Cl)C=C3N=CC=2)=C1 OVCDSSHSILBFBN-UHFFFAOYSA-N 0.000 claims description 7
- FMTDIUIBLCQGJB-UHFFFAOYSA-N Demethylchlortetracyclin Natural products C1C2C(O)C3=C(Cl)C=CC(O)=C3C(=O)C2=C(O)C2(O)C1C(N(C)C)C(O)=C(C(N)=O)C2=O FMTDIUIBLCQGJB-UHFFFAOYSA-N 0.000 claims description 7
- 239000004100 Oxytetracycline Substances 0.000 claims description 7
- 125000005907 alkyl ester group Chemical group 0.000 claims description 7
- 229960001444 amodiaquine Drugs 0.000 claims description 7
- 229950009959 amopyroquine Drugs 0.000 claims description 7
- 125000003435 aroyl group Chemical group 0.000 claims description 7
- LRTRTVPZZJAADL-DAHZFVMQSA-N arteflene Chemical compound C(/[C@@]1(C)[C@@H]2C[C@H](OO1)[C@@H](C(C2)=O)C)=C/C1=CC=C(C(F)(F)F)C=C1C(F)(F)F LRTRTVPZZJAADL-DAHZFVMQSA-N 0.000 claims description 7
- 229950010777 arteflene Drugs 0.000 claims description 7
- 229960000981 artemether Drugs 0.000 claims description 7
- BLUAFEHZUWYNDE-NNWCWBAJSA-N artemisinin Chemical compound C([C@](OO1)(C)O2)C[C@H]3[C@H](C)CC[C@@H]4[C@@]31[C@@H]2OC(=O)[C@@H]4C BLUAFEHZUWYNDE-NNWCWBAJSA-N 0.000 claims description 7
- 229960004191 artemisinin Drugs 0.000 claims description 7
- 229930101531 artemisinin Natural products 0.000 claims description 7
- FIHJKUPKCHIPAT-AHIGJZGOSA-N artesunate Chemical compound C([C@](OO1)(C)O2)C[C@H]3[C@H](C)CC[C@@H]4[C@@]31[C@@H]2O[C@@H](OC(=O)CCC(O)=O)[C@@H]4C FIHJKUPKCHIPAT-AHIGJZGOSA-N 0.000 claims description 7
- 229960004991 artesunate Drugs 0.000 claims description 7
- KUCQYCKVKVOKAY-CTYIDZIISA-N atovaquone Chemical compound C1([C@H]2CC[C@@H](CC2)C2=C(C(C3=CC=CC=C3C2=O)=O)O)=CC=C(Cl)C=C1 KUCQYCKVKVOKAY-CTYIDZIISA-N 0.000 claims description 7
- 229960003159 atovaquone Drugs 0.000 claims description 7
- 230000003013 cytotoxicity Effects 0.000 claims description 7
- 229960002398 demeclocycline Drugs 0.000 claims description 7
- SXYIRMFQILZOAM-HVNFFKDJSA-N dihydroartemisinin methyl ether Chemical compound C1C[C@H]2[C@H](C)CC[C@H]3[C@@H](C)[C@@H](OC)O[C@H]4[C@]32OO[C@@]1(C)O4 SXYIRMFQILZOAM-HVNFFKDJSA-N 0.000 claims description 7
- DYLGFOYVTXJFJP-MYYYXRDXSA-N lumefantrine Chemical compound C12=CC(Cl)=CC=C2C=2C(C(O)CN(CCCC)CCCC)=CC(Cl)=CC=2\C1=C/C1=CC=C(Cl)C=C1 DYLGFOYVTXJFJP-MYYYXRDXSA-N 0.000 claims description 7
- 229960004985 lumefantrine Drugs 0.000 claims description 7
- 229960000625 oxytetracycline Drugs 0.000 claims description 7
- IWVCMVBTMGNXQD-PXOLEDIWSA-N oxytetracycline Chemical compound C1=CC=C2[C@](O)(C)[C@H]3[C@H](O)[C@H]4[C@H](N(C)C)C(O)=C(C(N)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O IWVCMVBTMGNXQD-PXOLEDIWSA-N 0.000 claims description 7
- 235000019366 oxytetracycline Nutrition 0.000 claims description 7
- 229960005179 primaquine Drugs 0.000 claims description 7
- INDBQLZJXZLFIT-UHFFFAOYSA-N primaquine Chemical compound N1=CC=CC2=CC(OC)=CC(NC(C)CCCN)=C21 INDBQLZJXZLFIT-UHFFFAOYSA-N 0.000 claims description 7
- 229950011262 pyronaridine Drugs 0.000 claims description 7
- 150000003456 sulfonamides Chemical class 0.000 claims description 7
- 208000024891 symptom Diseases 0.000 claims description 7
- IWVCMVBTMGNXQD-UHFFFAOYSA-N terramycin dehydrate Natural products C1=CC=C2C(O)(C)C3C(O)C4C(N(C)C)C(O)=C(C(N)=O)C(=O)C4(O)C(O)=C3C(=O)C2=C1O IWVCMVBTMGNXQD-UHFFFAOYSA-N 0.000 claims description 7
- IEDVJHCEMCRBQM-UHFFFAOYSA-N trimethoprim Chemical compound COC1=C(OC)C(OC)=CC(CC=2C(=NC(N)=NC=2)N)=C1 IEDVJHCEMCRBQM-UHFFFAOYSA-N 0.000 claims description 7
- 229960001082 trimethoprim Drugs 0.000 claims description 7
- 206010019233 Headaches Diseases 0.000 claims description 6
- 206010041660 Splenomegaly Diseases 0.000 claims description 6
- 208000007502 anemia Diseases 0.000 claims description 6
- 231100000869 headache Toxicity 0.000 claims description 6
- 206010025482 malaise Diseases 0.000 claims description 6
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 claims description 6
- 241000124008 Mammalia Species 0.000 claims description 5
- 241000223960 Plasmodium falciparum Species 0.000 claims description 5
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 5
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 5
- 125000004429 atom Chemical group 0.000 claims description 4
- 241000224016 Plasmodium Species 0.000 claims description 3
- 241000223810 Plasmodium vivax Species 0.000 claims description 3
- 206010037660 Pyrexia Diseases 0.000 claims description 3
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 claims description 3
- AEGIGNROXNVWPI-UHFFFAOYSA-N 1-[ethylperoxy(methoxy)methyl]peroxypropane Chemical group CCCOOC(OC)OOCC AEGIGNROXNVWPI-UHFFFAOYSA-N 0.000 claims description 2
- AVFZOVWCLRSYKC-UHFFFAOYSA-N 1-methylpyrrolidine Chemical compound CN1CCCC1 AVFZOVWCLRSYKC-UHFFFAOYSA-N 0.000 claims description 2
- 241001505293 Plasmodium ovale Species 0.000 claims description 2
- 206010035502 Plasmodium ovale infection Diseases 0.000 claims description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims description 2
- 125000003368 amide group Chemical group 0.000 claims description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 claims description 2
- 125000002541 furyl group Chemical group 0.000 claims description 2
- 125000002883 imidazolyl group Chemical group 0.000 claims description 2
- 125000004076 pyridyl group Chemical group 0.000 claims description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 2
- 125000001544 thienyl group Chemical group 0.000 claims description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 claims 9
- 125000001589 carboacyl group Chemical group 0.000 claims 3
- DJUFPMUQJKWIJB-UHFFFAOYSA-N pyronaridine Chemical compound C12=NC(OC)=CC=C2N=C2C=C(Cl)C=CC2=C1NC(C=C(CN1CCCC1)C=1O)=CC=1CN1CCCC1 DJUFPMUQJKWIJB-UHFFFAOYSA-N 0.000 claims 3
- 125000002837 carbocyclic group Chemical group 0.000 claims 1
- 230000000069 prophylactic effect Effects 0.000 claims 1
- WKSAUQYGYAYLPV-UHFFFAOYSA-N pyrimethamine Chemical compound CCC1=NC(N)=NC(N)=C1C1=CC=C(Cl)C=C1 WKSAUQYGYAYLPV-UHFFFAOYSA-N 0.000 claims 1
- 229960000611 pyrimethamine Drugs 0.000 claims 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 103
- 229950000614 sancycline Drugs 0.000 description 44
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 36
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 34
- 239000000243 solution Substances 0.000 description 32
- 238000006243 chemical reaction Methods 0.000 description 31
- MTCQOMXDZUULRV-ADOAZJKMSA-N (4s,4as,5ar,12ar)-4-(dimethylamino)-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1C2=CC=CC(O)=C2C(O)=C2[C@@H]1C[C@H]1[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]1(O)C2=O MTCQOMXDZUULRV-ADOAZJKMSA-N 0.000 description 30
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 28
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 24
- 239000002904 solvent Substances 0.000 description 24
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 23
- 239000000203 mixture Substances 0.000 description 22
- 239000000047 product Substances 0.000 description 20
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 19
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 19
- 239000007787 solid Substances 0.000 description 19
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 17
- 229910052786 argon Inorganic materials 0.000 description 17
- 125000001424 substituent group Chemical group 0.000 description 17
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 16
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 16
- 238000004128 high performance liquid chromatography Methods 0.000 description 16
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 14
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 14
- 230000002829 reductive effect Effects 0.000 description 14
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- 125000004432 carbon atom Chemical group C* 0.000 description 12
- 210000003743 erythrocyte Anatomy 0.000 description 12
- 125000005842 heteroatom Chemical group 0.000 description 12
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 12
- 239000012071 phase Substances 0.000 description 12
- 238000002953 preparative HPLC Methods 0.000 description 12
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 11
- 125000004442 acylamino group Chemical group 0.000 description 11
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 11
- 244000045947 parasite Species 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- 150000003573 thiols Chemical group 0.000 description 11
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 11
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 10
- 229910019142 PO4 Inorganic materials 0.000 description 10
- 125000002877 alkyl aryl group Chemical group 0.000 description 10
- 125000005129 aryl carbonyl group Chemical group 0.000 description 10
- 125000004093 cyano group Chemical group *C#N 0.000 description 10
- 239000003814 drug Substances 0.000 description 10
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 10
- 239000010452 phosphate Substances 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 10
- 238000005481 NMR spectroscopy Methods 0.000 description 9
- 125000005194 alkoxycarbonyloxy group Chemical group 0.000 description 9
- 125000004947 alkyl aryl amino group Chemical group 0.000 description 9
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 description 9
- 125000004691 alkyl thio carbonyl group Chemical group 0.000 description 9
- 125000001769 aryl amino group Chemical group 0.000 description 9
- 125000004658 aryl carbonyl amino group Chemical group 0.000 description 9
- 125000005110 aryl thio group Chemical group 0.000 description 9
- 125000005200 aryloxy carbonyloxy group Chemical group 0.000 description 9
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 9
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 description 9
- 150000007942 carboxylates Chemical class 0.000 description 9
- 125000004986 diarylamino group Chemical group 0.000 description 9
- 239000007789 gas Substances 0.000 description 9
- 238000001727 in vivo Methods 0.000 description 9
- 238000003756 stirring Methods 0.000 description 9
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 9
- 229940040944 tetracyclines Drugs 0.000 description 9
- 241000255925 Diptera Species 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 8
- 150000003467 sulfuric acid derivatives Chemical group 0.000 description 8
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 7
- 125000003277 amino group Chemical group 0.000 description 7
- 238000003556 assay Methods 0.000 description 7
- 239000003054 catalyst Substances 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- 239000000460 chlorine Substances 0.000 description 7
- 125000000753 cycloalkyl group Chemical group 0.000 description 7
- 230000006870 function Effects 0.000 description 7
- 150000002430 hydrocarbons Chemical group 0.000 description 7
- 125000004433 nitrogen atom Chemical group N* 0.000 description 7
- 239000011541 reaction mixture Substances 0.000 description 7
- 229910000029 sodium carbonate Inorganic materials 0.000 description 7
- 125000004434 sulfur atom Chemical group 0.000 description 7
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 7
- 241000699670 Mus sp. Species 0.000 description 6
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 6
- 125000000392 cycloalkenyl group Chemical group 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 125000004430 oxygen atom Chemical group O* 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 5
- 125000005090 alkenylcarbonyl group Chemical group 0.000 description 5
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 125000004473 dialkylaminocarbonyl group Chemical group 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 230000000699 topical effect Effects 0.000 description 5
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 4
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 4
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 4
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 4
- 241000256186 Anopheles <genus> Species 0.000 description 4
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 4
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- 208000009182 Parasitemia Diseases 0.000 description 4
- 208000030852 Parasitic disease Diseases 0.000 description 4
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 4
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 4
- 125000005089 alkenylaminocarbonyl group Chemical group 0.000 description 4
- 150000001408 amides Chemical class 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 125000004103 aminoalkyl group Chemical group 0.000 description 4
- 125000005125 aryl alkyl amino carbonyl group Chemical group 0.000 description 4
- 125000005099 aryl alkyl carbonyl group Chemical group 0.000 description 4
- 125000005015 aryl alkynyl group Chemical group 0.000 description 4
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 239000013058 crude material Substances 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 239000011737 fluorine Substances 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- 235000019253 formic acid Nutrition 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 4
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- YFYLPWJKCSESGB-UHFFFAOYSA-N pyronaridine Chemical compound C=12NC(OC)=CC=C2NC2=CC(Cl)=CC=C2C=1N=C(C=C(CN1CCCC1)C1=O)C=C1CN1CCCC1 YFYLPWJKCSESGB-UHFFFAOYSA-N 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 3
- 241000271566 Aves Species 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical group NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 3
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 3
- 241000938605 Crocodylia Species 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 125000002723 alicyclic group Chemical group 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- 125000000278 alkyl amino alkyl group Chemical group 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 125000005018 aryl alkenyl group Chemical group 0.000 description 3
- 125000005128 aryl amino alkyl group Chemical group 0.000 description 3
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 230000005587 bubbling Effects 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 210000000973 gametocyte Anatomy 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- 238000005984 hydrogenation reaction Methods 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 238000011081 inoculation Methods 0.000 description 3
- 210000003936 merozoite Anatomy 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 125000004437 phosphorous atom Chemical group 0.000 description 3
- 125000003367 polycyclic group Polymers 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 210000003046 sporozoite Anatomy 0.000 description 3
- 125000004426 substituted alkynyl group Chemical group 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 150000003672 ureas Chemical group 0.000 description 3
- XIYOPDCBBDCGOE-IWVLMIASSA-N (4s,4ar,5s,5ar,12ar)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methylidene-3,12-dioxo-4,4a,5,5a-tetrahydrotetracene-2-carboxamide Chemical compound C=C1C2=CC=CC(O)=C2C(O)=C2[C@@H]1[C@H](O)[C@H]1[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]1(O)C2=O XIYOPDCBBDCGOE-IWVLMIASSA-N 0.000 description 2
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 2
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 2
- PEPBFCOIJRULGJ-UHFFFAOYSA-N 3h-1,2,3-benzodioxazole Chemical compound C1=CC=C2NOOC2=C1 PEPBFCOIJRULGJ-UHFFFAOYSA-N 0.000 description 2
- VFTOHJFKIJLYKN-UHFFFAOYSA-N 7-nitro-9h-fluoren-2-ol Chemical group [O-][N+](=O)C1=CC=C2C3=CC=C(O)C=C3CC2=C1 VFTOHJFKIJLYKN-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- 241000272517 Anseriformes Species 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- 239000005909 Kieselgur Substances 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 2
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 238000005700 Stille cross coupling reaction Methods 0.000 description 2
- 241000282887 Suidae Species 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- 241000282458 Ursus sp. Species 0.000 description 2
- LXNAVEXFUKBNMK-UHFFFAOYSA-N acetic acid;palladium Chemical compound [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 2
- 150000001336 alkenes Chemical class 0.000 description 2
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 235000013877 carbamide Nutrition 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000006880 cross-coupling reaction Methods 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 125000000582 cycloheptyl group Chemical class [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 125000004985 dialkyl amino alkyl group Chemical group 0.000 description 2
- 229960004132 diethyl ether Drugs 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 150000003840 hydrochlorides Chemical class 0.000 description 2
- 238000005462 in vivo assay Methods 0.000 description 2
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 2
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 2
- HOBCFUWDNJPFHB-UHFFFAOYSA-N indolizine Chemical compound C1=CC=CN2C=CC=C21 HOBCFUWDNJPFHB-UHFFFAOYSA-N 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 150000002513 isocyanates Chemical class 0.000 description 2
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 229940042016 methacycline Drugs 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 125000003261 o-tolyl group Chemical group [H]C1=C([H])C(*)=C(C([H])=C1[H])C([H])([H])[H] 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 230000001717 pathogenic effect Effects 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 229910052697 platinum Inorganic materials 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- VWDWKYIASSYTQR-UHFFFAOYSA-N sodium nitrate Chemical compound [Na+].[O-][N+]([O-])=O VWDWKYIASSYTQR-UHFFFAOYSA-N 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- 125000005017 substituted alkenyl group Chemical group 0.000 description 2
- 125000005415 substituted alkoxy group Chemical group 0.000 description 2
- 125000000547 substituted alkyl group Chemical group 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 238000000967 suction filtration Methods 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 150000003536 tetrazoles Chemical class 0.000 description 2
- 150000003568 thioethers Chemical class 0.000 description 2
- 229930192474 thiophene Natural products 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 150000003852 triazoles Chemical class 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- XXYRJHQJYSUIJA-YABKDXSZSA-N (2s,5r,6r)-6-[[(2r)-2-[[[(4s,4as,5as,6s,12ar)-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4,4a,5,5a-tetrahydrotetracene-2-carbonyl]amino]methylamino]-2-phenylacetyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxy Chemical compound C1([C@H](C(=O)N[C@@H]2C(N3[C@H](C(C)(C)S[C@@H]32)C(O)=O)=O)NCNC(=O)C2=C(O)[C@@]3(O)C(=O)C=4[C@@H]([C@](C5=CC=CC(O)=C5C=4O)(C)O)C[C@H]3[C@@H](C2=O)N(C)C)=CC=CC=C1 XXYRJHQJYSUIJA-YABKDXSZSA-N 0.000 description 1
- CAYQIZIAYYNFCS-UHFFFAOYSA-N (4-chlorophenyl)boronic acid Chemical compound OB(O)C1=CC=C(Cl)C=C1 CAYQIZIAYYNFCS-UHFFFAOYSA-N 0.000 description 1
- VXPSARQTYDZXAO-CCHMMTNSSA-N (4s,4ar,5s,5ar,12ar)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methylidene-3,12-dioxo-4,4a,5,5a-tetrahydrotetracene-2-carboxamide;hydron;chloride Chemical compound Cl.C=C1C2=CC=CC(O)=C2C(O)=C2[C@@H]1[C@H](O)[C@H]1[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]1(O)C2=O VXPSARQTYDZXAO-CCHMMTNSSA-N 0.000 description 1
- RTXXZBOFRSQDMC-FUUYDGDCSA-N (4s,4as,5as,6s,12ar)-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-n-[[[(2r,3r,4r,5s,6r)-2,4,5-trihydroxy-6-(hydroxymethyl)oxan-3-yl]amino]methyl]-4,4a,5,5a-tetrahydrotetracene-2-carboxamide Chemical compound OC([C@@]1(O)C(=O)C=2[C@@H]([C@](C3=CC=CC(O)=C3C=2O)(C)O)C[C@H]1[C@@H](C1=O)N(C)C)=C1C(=O)NCN[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O RTXXZBOFRSQDMC-FUUYDGDCSA-N 0.000 description 1
- FAMUIRDLAWWMCQ-AQFAATAFSA-N (4s,4as,5as,6s,12ar)-n-[[4-[n-(diaminomethylidene)carbamimidoyl]piperazin-1-yl]methyl]-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4,4a,5,5a-tetrahydrotetracene-2-carboxamide Chemical compound OC([C@@]1(O)C(=O)C=2[C@@H]([C@](C3=CC=CC(O)=C3C=2O)(C)O)C[C@H]1[C@@H](C1=O)N(C)C)=C1C(=O)NCN1CCN(C(=N)N=C(N)N)CC1 FAMUIRDLAWWMCQ-AQFAATAFSA-N 0.000 description 1
- NKKNXLPHCRLBDY-UHFFFAOYSA-N (5-formyl-2-methoxyphenyl)boronic acid Chemical compound COC1=CC=C(C=O)C=C1B(O)O NKKNXLPHCRLBDY-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical compound NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 description 1
- LGPKFIGMLPDYEA-UHFFFAOYSA-N 1-isocyanato-4-(trifluoromethoxy)benzene Chemical compound FC(F)(F)OC1=CC=C(N=C=O)C=C1 LGPKFIGMLPDYEA-UHFFFAOYSA-N 0.000 description 1
- LVGIUOZGDVZIMK-UHFFFAOYSA-N 1-sulfanylpyrrolidine Chemical compound SN1CCCC1 LVGIUOZGDVZIMK-UHFFFAOYSA-N 0.000 description 1
- ZIIUUSVHCHPIQD-UHFFFAOYSA-N 2,4,6-trimethyl-N-[3-(trifluoromethyl)phenyl]benzenesulfonamide Chemical compound CC1=CC(C)=CC(C)=C1S(=O)(=O)NC1=CC=CC(C(F)(F)F)=C1 ZIIUUSVHCHPIQD-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- IJRKANNOPXMZSG-SSPAHAAFSA-N 2-hydroxypropane-1,2,3-tricarboxylic acid;(2r,3s,4r,5r)-2,3,4,5,6-pentahydroxyhexanal Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O.OC(=O)CC(O)(C(O)=O)CC(O)=O IJRKANNOPXMZSG-SSPAHAAFSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- SIAVMDKGVRXFAX-UHFFFAOYSA-N 4-carboxyphenylboronic acid Chemical compound OB(O)C1=CC=C(C(O)=O)C=C1 SIAVMDKGVRXFAX-UHFFFAOYSA-N 0.000 description 1
- LBUNNMJLXWQQBY-UHFFFAOYSA-N 4-fluorophenylboronic acid Chemical compound OB(O)C1=CC=C(F)C=C1 LBUNNMJLXWQQBY-UHFFFAOYSA-N 0.000 description 1
- NZUUXQSBKZPFKK-UHFFFAOYSA-N 4-piperazin-1-ylmorpholine Chemical compound C1CNCCN1N1CCOCC1 NZUUXQSBKZPFKK-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- DHMYULZVFHHEHE-UHFFFAOYSA-N 9h-fluoren-9-ylmethyl n-(sulfanylidenemethylidene)carbamate Chemical compound C1=CC=C2C(COC(=O)N=C=S)C3=CC=CC=C3C2=C1 DHMYULZVFHHEHE-UHFFFAOYSA-N 0.000 description 1
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 229920000856 Amylose Polymers 0.000 description 1
- 239000005996 Blood meal Substances 0.000 description 1
- 238000009631 Broth culture Methods 0.000 description 1
- 0 C*C(C(*c1c(*C)c(*)c(*)c(O*)c1C1=O)C1=C1O*)C(C(*)C(O*)=C(C(*)=O)C2=O)[C@]12O* Chemical compound C*C(C(*c1c(*C)c(*)c(*)c(O*)c1C1=O)C1=C1O*)C(C(*)C(O*)=C(C(*)=O)C2=O)[C@]12O* 0.000 description 1
- BZUKDLNEDCDORL-ULRSWZSCSA-N C1=CC=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(O)=C(C(=O)NCN(C)CCN(C)CNC(=O)C=5C([C@@]6(O)C(O)=C7[C@@H]([C@](C8=CC=CC(O)=C8C7=O)(C)O)C[C@H]6[C@@H](C=5O)N(C)C)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O Chemical compound C1=CC=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(O)=C(C(=O)NCN(C)CCN(C)CNC(=O)C=5C([C@@]6(O)C(O)=C7[C@@H]([C@](C8=CC=CC(O)=C8C7=O)(C)O)C[C@H]6[C@@H](C=5O)N(C)C)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O BZUKDLNEDCDORL-ULRSWZSCSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 239000004099 Chlortetracycline Substances 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 101100007328 Cocos nucifera COS-1 gene Proteins 0.000 description 1
- 206010010741 Conjunctivitis Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 241001495410 Enterococcus sp. Species 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 206010018910 Haemolysis Diseases 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical class ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 description 1
- 208000037490 Medically Unexplained Symptoms Diseases 0.000 description 1
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 102000011779 Nitric Oxide Synthase Type II Human genes 0.000 description 1
- 108010076864 Nitric Oxide Synthase Type II Proteins 0.000 description 1
- IOVCWXUNBOPUCH-UHFFFAOYSA-N Nitrous acid Chemical compound ON=O IOVCWXUNBOPUCH-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 241000282579 Pan Species 0.000 description 1
- 241000282320 Panthera leo Species 0.000 description 1
- 241000282376 Panthera tigris Species 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- 108010064785 Phospholipases Proteins 0.000 description 1
- 102000015439 Phospholipases Human genes 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 1
- 241000224017 Plasmodium berghei Species 0.000 description 1
- 241000223821 Plasmodium malariae Species 0.000 description 1
- 206010035501 Plasmodium malariae infection Diseases 0.000 description 1
- 241000223829 Plasmodium vinckei Species 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 206010037151 Psittacosis Diseases 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- PLXBWHJQWKZRKG-UHFFFAOYSA-N Resazurin Chemical compound C1=CC(=O)C=C2OC3=CC(O)=CC=C3[N+]([O-])=C21 PLXBWHJQWKZRKG-UHFFFAOYSA-N 0.000 description 1
- 241000282849 Ruminantia Species 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 241000555745 Sciuridae Species 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 description 1
- 125000005087 alkynylcarbonyl group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 239000012080 ambient air Substances 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000005021 aminoalkenyl group Chemical group 0.000 description 1
- 125000006598 aminocarbonylamino group Chemical group 0.000 description 1
- 229950003476 aminothiazole Drugs 0.000 description 1
- 125000004682 aminothiocarbonyl group Chemical group NC(=S)* 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 238000011482 antibacterial activity assay Methods 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- HRWVXKVRSNICJQ-GMJIGYHYSA-N apicycline Chemical compound O=C([C@@]1(O)C(O)=C2[C@@H]([C@](C3=CC=CC(O)=C3C2=O)(C)O)C[C@H]1[C@@H](C=1O)N(C)C)C=1C(=O)NC(C(O)=O)N1CCN(CCO)CC1 HRWVXKVRSNICJQ-GMJIGYHYSA-N 0.000 description 1
- 229950008405 apicycline Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 239000012300 argon atmosphere Substances 0.000 description 1
- 150000001543 aryl boronic acids Chemical class 0.000 description 1
- 150000007860 aryl ester derivatives Chemical class 0.000 description 1
- 230000011681 asexual reproduction Effects 0.000 description 1
- 238000013465 asexual reproduction Methods 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- RWFPWQCNFWWIAE-UHFFFAOYSA-N benzyl n-(hydroxymethyl)carbamate Chemical compound OCNC(=O)OCC1=CC=CC=C1 RWFPWQCNFWWIAE-UHFFFAOYSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000004744 butyloxycarbonyl group Chemical group 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 229960004475 chlortetracycline Drugs 0.000 description 1
- 235000019365 chlortetracycline Nutrition 0.000 description 1
- 229960004094 clomocycline Drugs 0.000 description 1
- BXVOHUQQUBSHLD-XCTBDMBQSA-N clomocycline Chemical compound C1=CC(Cl)=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(=O)C(=C(/O)NCO)/C(=O)[C@@]4(O)C(=O)C3=C(O)C2=C1O BXVOHUQQUBSHLD-XCTBDMBQSA-N 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 150000001879 copper Chemical class 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 150000001934 cyclohexanes Chemical class 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001887 cyclopentyloxy group Chemical group C1(CCCC1)O* 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 238000002784 cytotoxicity assay Methods 0.000 description 1
- 125000003493 decenyl group Chemical group [H]C([*])=C([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005070 decynyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C#C* 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 239000012954 diazonium Substances 0.000 description 1
- 150000001989 diazonium salts Chemical class 0.000 description 1
- 125000004772 dichloromethyl group Chemical group [H]C(Cl)(Cl)* 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 230000009429 distress Effects 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 229950004798 etamocycline Drugs 0.000 description 1
- 229940052303 ethers for general anesthesia Drugs 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- FMVJYQGSRWVMQV-UHFFFAOYSA-N ethyl propiolate Chemical compound CCOC(=O)C#C FMVJYQGSRWVMQV-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 150000002191 fatty alcohols Chemical class 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000004785 fluoromethoxy group Chemical group [H]C([H])(F)O* 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 239000004023 fresh frozen plasma Substances 0.000 description 1
- CYEFKCRAAGLNHW-UHFFFAOYSA-N furan-3-ylboronic acid Chemical compound OB(O)C=1C=COC=1 CYEFKCRAAGLNHW-UHFFFAOYSA-N 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000003304 gavage Methods 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 229950007488 guamecycline Drugs 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 238000005534 hematocrit Methods 0.000 description 1
- 230000008588 hemolysis Effects 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000002390 heteroarenes Chemical class 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- XUGNVMKQXJXZCD-UHFFFAOYSA-N isopropyl palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC(C)C XUGNVMKQXJXZCD-UHFFFAOYSA-N 0.000 description 1
- 229940075495 isopropyl palmitate Drugs 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 208000001581 lymphogranuloma venereum Diseases 0.000 description 1
- 210000000054 macrogamete Anatomy 0.000 description 1
- 229910001425 magnesium ion Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 231100001141 mammalian cytotoxicity Toxicity 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229950008037 meglucycline Drugs 0.000 description 1
- 229940051860 methacycline hydrochloride Drugs 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- 231100000417 nephrotoxicity Toxicity 0.000 description 1
- 125000005187 nonenyl group Chemical group C(=CCCCCCCC)* 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000005071 nonynyl group Chemical group C(#CCCCCCCC)* 0.000 description 1
- 125000004365 octenyl group Chemical group C(=CCCCCCC)* 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000005069 octynyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C#C* 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 210000003250 oocyst Anatomy 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 201000000901 ornithosis Diseases 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- MEGKRPMNPGTIIG-VNYBMUHKSA-N penimepicycline Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)COC1=CC=CC=C1.O=C([C@@]1(O)C(O)=C2[C@@H]([C@](C3=CC=CC(O)=C3C2=O)(C)O)C[C@H]1[C@@H](C=1O)N(C)C)C=1C(=O)NCN1CCN(CCO)CC1 MEGKRPMNPGTIIG-VNYBMUHKSA-N 0.000 description 1
- 229960003187 penimepicycline Drugs 0.000 description 1
- 229950005777 penimocycline Drugs 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000004115 pentoxy group Chemical group [*]OC([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000001148 pentyloxycarbonyl group Chemical group 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 125000005004 perfluoroethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- XATZHCXBMKRRDO-REHNUXHNSA-N pipacycline Chemical compound O=C([C@@]1(O)C(O)=C2[C@@H]([C@](C3=CC=CC(O)=C3C2=O)(C)O)C[C@H]1[C@@H](C=1O)N(C)C)C=1C(=O)NCN1CCN(CCO)CC1 XATZHCXBMKRRDO-REHNUXHNSA-N 0.000 description 1
- 229950001465 pipacycline Drugs 0.000 description 1
- 210000002381 plasma Anatomy 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000000135 prohibitive effect Effects 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000004742 propyloxycarbonyl group Chemical group 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- QEVHRUUCFGRFIF-MDEJGZGSSA-N reserpine Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C(C5=CC=C(OC)C=C5N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 QEVHRUUCFGRFIF-MDEJGZGSSA-N 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 238000003345 scintillation counting Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 230000014639 sexual reproduction Effects 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 235000010344 sodium nitrate Nutrition 0.000 description 1
- 239000004317 sodium nitrate Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 230000005002 sporogony Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- FDDDEECHVMSUSB-UHFFFAOYSA-N sulfanilamide Chemical compound NC1=CC=C(S(N)(=O)=O)C=C1 FDDDEECHVMSUSB-UHFFFAOYSA-N 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- IFLREYGFSNHWGE-UHFFFAOYSA-N tetracene Chemical compound C1=CC=CC2=CC3=CC4=CC=CC=C4C=C3C=C21 IFLREYGFSNHWGE-UHFFFAOYSA-N 0.000 description 1
- 229940072172 tetracycline antibiotic Drugs 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000005300 thiocarboxy group Chemical group C(=S)(O)* 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N thiomorpholine Chemical compound C1CSCCN1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 125000004784 trichloromethoxy group Chemical group ClC(O*)(Cl)Cl 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 1
- BPLUKJNHPBNVQL-UHFFFAOYSA-N triphenylarsine Chemical compound C1=CC=CC=C1[As](C=1C=CC=CC=1)C1=CC=CC=C1 BPLUKJNHPBNVQL-UHFFFAOYSA-N 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000002023 wood Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/352—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom condensed with carbocyclic rings, e.g. methantheline
- A61K31/353—3,4-Dihydrobenzopyrans, e.g. chroman, catechin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/385—Heterocyclic compounds having sulfur as a ring hetero atom having two or more sulfur atoms in the same ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/473—Quinolines; Isoquinolines ortho- or peri-condensed with carbocyclic ring systems, e.g. acridines, phenanthridines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/65—Tetracyclines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/06—Antimalarials
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- X is CHC(R 13 Y'Y), CR 6' R 6 , S, NR 6 , or O;
- R 2 , R 2 , R 4 , and R 4 are each independently hydrogen, alkyl, alkenyl, alkynyl, alkoxy, alkylthio, alkylsulfinyl, alkylsulfonyl, alkylamino, arylalkyl, aryl, heterocyclic, heteroaromatic or a prodrug moiety;
- R 4 is NR 4 R 4 , alkyl, alkenyl, alkynyl, hydroxyl, halogen, or hydrogen;
- This invention pertains, at least in part, to a method for treating or preventing malaria which is resistant to one or more anti-malarial compounds such as, for example, proguanil, chlorproguanil, trimethoprim, chloroquine, mefloquine, lumefantrine, atovaquone, pyrimethamine-sulfadoxine, pyrimethamine-dapsone, halofantrine, quinine, quinidine, amodiaquine, amopyroquine, sulphonamides, artemisinin, arteflene, artemether, artesunate, primaquine, and pyronaridine.
- anti-malarial compounds such as, for example, proguanil, chlorproguanil, trimethoprim, chloroquine, mefloquine, lumefantrine, atovaquone, pyrimethamine-sulfadoxine, pyrimethamine-dapsone, halofantrine, quin
- malaria includes the art recognized condition known as "malaria” e.g. , disorders which are caused by a protozoan of the genus Plasmodium. Malaria is generally characterized by symptoms such as headache, malaise, anemia, splenomegaly, and paroxyms with cold, hot, and wet stages and is transmitted by mosquitoes. (Winstanley (1998) Journal of the Royal College of Physicians of London 32(3):203- 207.) In a further embodiment, the protozoan is selected from the group consisting of: P. falciparum, P. vivax, P. ovale, and P. malariae.
- the substituted tetracycline compound of the invention has a cytotoxicity which allows the compound to be administered in an effective amount to the subject with out causing prohibitive cytotoxic side effects.
- the cytotoxicity of the substituted tetracycline compound of the invention is greater than about 10 ⁇ g/ml, about 15 ⁇ g/ml, about 20 ⁇ g/ml, or about 25 ⁇ g/ml as measured by cytoxicity assays known in the art such as the assay described in Example 5.
- the MIC of a substituted tetracycline compound as measured in vitro is about 1000 nM or less, about 900 nM or less, about 800 nM or less, about 700 nM or less, about 600 nM or less, about 500 nM or less, about 450 nM or less, about 400 nM or less, about 350 nM or less, about 300 nM or less, about 250 nM or less, about 200 nM or less, about 190 nM or less, about 180 nM or less, about 170 nM or less, about 160 nM or less, about 150 nM or less, about 140 nM or less, about 130 nM or less, about 120 nM or less, about 110 nM or less, about 100 nM or less, about 90 nM or less, about 80 nM or less, about 70 nM or less, about 60 nM or less, about 50 nM or less, about 45 nM or less, about 40
- R 9 is hydrogen, or a malaria interacting moiety
- R 8 is hydrogen, hydroxyl, halogen, thiol, alkyl, alkenyl, alkynyl, aryl, alkoxy, alkylthio, alkylsulfinyl, alkylsulfonyl, alkylamino, or an arylalkyl;
- the malaria interacting moiety may be substituted with one or more substituents which allow it to performs its intended function, e.g., treat or prevent malaria.
- substituents include, but are not limited to, alkyl, alkenyl, alkynyl, halogen, hydroxyl, alkoxy, alkylcarbonyloxy, alkyloxycarbonyl, carboxy, arylcarbonyloxy, alkoxycarbonylamino, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, alkylaminoacarbonyl, arylalkyl aminocarbonyl, alkenylaminocarbonyl, alkylcarbonyl, arylcarbonyl, aminoalkyl, arylalkylcarbonyl, alkenylcarbonyl, alkoxycarbonyl, silyl, aminocarbonyl, alkylthiocarbonyl, phosphate, aralkyl, phosphonato, phos
- the substituted tetracycline compounds are have a suitable oral bioavailability for the treatment of malaria, e.g., after the substituted tetracycline compounds are orally administered to the subject, the compounds are able to perform their intended function, e.g., treat malaria.
- Examples of methods which can be used to calculate the bioavailability of a particular compound include methods known in the art as well as the methods described in U.S.S.N. 60/318,580, incorporated herein by reference.
- the substituted tetracycline compounds do not include compounds which inhibit excess phospholipase A 2 activity or production, as measured by the assay given in U.S. 6,043,231.
- malaria for treatment using the compositions and methods of the invention is resistant to one or more anti-malarial compounds such as proguanil, chlorproguanil, trimethoprim, chloroquine, mefloquine, lumefantrine, atovaquone, pyrimethamine-sulfadoxine, pyrimethamine-dapsone, halofantrine, quinine, quinidine, amodiaquine, amopyroquine, sulphonamides, artemisinin, arteflene, artemether, artesunate, primaquine, and pyronaridine.
- anti-malarial compounds such as proguanil, chlorproguanil, trimethoprim, chloroquine, mefloquine, lumefantrine, atovaquone, pyrimethamine-sulfadoxine, pyrimethamine-dapsone, halofantrine, quinine, quinidine, amodiaquine, amopyroquine
- 9- and 7- substituted tetracyclines can be synthesized by the method shown in Scheme 1.
- 9- and 7-substituted tetracycline compounds can be synthesized by treating a tetracycline compound (e.g., doxycycline ⁇ 1 A), with sulfuric acid and sodium nitrate.
- the resulting product is a mixture of the 7-nitro and 9-nitro isomers (IB and 1C, respectively).
- the 7-nitro (IB) and 9- nitro (1C) derivatives are treated by hydrogenation using hydrogen gas and a platinum catalyst to yield amines ID and IE.
- the isomers are separated at this time by conventional methods.
- substituted tetracycline compounds of the invention wherein R 7 is a carbamate or a urea derivative can be synthesized using the following protocol.
- Sancycline (2A) is treated with NaNO under acidic conditions forming 7- nitro sancycline (2B) in a mixture of positional isomers.
- 7-nitrosancycline (2B) is then treated with H 2 gas and a platinum catalyst to form the 7-amino sancycline derivative (2C).
- isocyanate (2D) is reacted with the 7-amino sancycline derivative (2C).
- carbamate (2G) the appropriate acid chloride ester (2F) is reacted with 2C.
- 7- alkenyl substituted tetracycline compounds such as 7-alkynyl sancycline (4A) and 7-alkenyl sancycline (4B), can be hydrogenated to form alkyl 7- substituted tetracycline compounds (e.g., 7-alkyl sancycline, 4C).
- Scheme 4 depicts the selective hydrogenation of the 7- position double or triple bond, in saturated methanol and hydrochloric acid solution with a palladium/carbon catalyst under pressure, to yield the product.
- the compounds of the invention can also be synthesized using Heck-type cross coupling reactions.
- Heck-type cross-couplings can be performed by suspending a halogenated tetracycline compound (e.g., 6-iodosancycline, 6A) and an appropriate palladium or other transition metal catalyst (e.g., Pd(OAc) 2 and Cul) in an appropriate solvent (e.g., degassed acetonitrile).
- a reactive alkene (6B) or alkyne (6D), and triethylamine are then added and the mixture is heated for several hours, before being cooled to room temperature.
- the resulting 7-substituted alkenyl (6C) or 7-substituted alkynyl (6E) tetracycline compound can then be purified using techniques known in the art.
- 5-esters of 9- substituted tetracycline compounds can be formed by dissolving the 9- substituted compounds (8 A) in strong acid (e.g. HF, methanesulphonic acid, and trifluoromethanesulfonic acid) and adding the appropriate carboxylic acid to yield the corresponding esters (8B).
- strong acid e.g. HF, methanesulphonic acid, and trifluoromethanesulfonic acid
- 13 -substituted thiols can be synthesized by the method outlined in Scheme 9, above.
- 13 -substituted thiol ethers (9B) can be synthesized by heating a tetracycline salt (9 A) (such as methacycline hydrochloride), AIBN (2,2'- azobisisobutyronitrile), and a thiol in ethanol at reflux for six hours under an inert atmosphere.
- 7 and 9 aminomethyl tetracyclines may be synthesized using reagents such as hydroxymethyl-carbamic acid benzyl ester.
- alkyl includes both "unsubstituted alkyls" and “substituted alkyls”, the latter of which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone.
- substituents can include, for example, alkenyl, alkynyl, halogen, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, arylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamino), acylamino (including alkylcarbonylamino, arylcarbonylamino, carbamoyl and ureido), amidino, imino, sulfhydryl, alkylthio, arylthio, thiocarboxylate, sul
- the aromatic ring can be substituted at one or more ring positions with such substituents as described above, as for example, halogen, hydroxyl, alkoxy, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, alkylaminoacarbonyl, aralkylaminocarbonyl, alkenylaminocarbonyl, alkylcarbonyl, arylcarbonyl, aralkylcarbonyl, alkenylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylthiocarbonyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamino), acylamino
- Aryl groups can also be fused or bridged with alicyclic or heterocyclic rings which are not aromatic so as to form a polycycle (e.g., tetralin, methylenedioxyphenyl).
- substituents can include, for example, alkyl groups, alkenyl groups, alkynyl groups, halogens, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, arylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamino), acylamino (including alkylcarbonylamino, arylcarbonylamino, carbamoyl and ureido), amidino, imino, sulfhydryl, alkylthio, arylthio, thio
- lower alkyl as used herein means an alkyl group, as defined above, but having from one to five carbon atoms in its backbone structure.
- Lower alkenyl and “lower alkynyl” have chain lengths of, for example, 2-5 carbon atoms.
- acyl includes compounds and moieties which contain the acyl radical (CH 3 CO-) or a carbonyl group.
- substituted acyl includes acyl groups where one or more of the hydrogen atoms are replaced by for example, alkyl groups, alkynyl groups, halogens, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, arylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamino), acylamino (including alkylcarbonylamino, ary
- amide or "aminocarboxy” includes compounds or moieties which contain a nitrogen atom which is bound to the carbon of a carbonyl or a thiocarbonyl group.
- alkaminocarboxy groups which include alkyl, alkenyl, or alkynyl groups bound to an amino group bound to a carboxy group. It includes arylaminocarboxy groups which include aryl or heteroaryl moieties bound to an amino group which is bound to the carbon of a carbonyl or thiocarbonyl group.
- urea includes compounds that containing a carbonyl group linked to two nitrogens.
- thiocarbonyl or thiocarboxy includes compounds and moieties which contain a carbon connected with a double bond to a sulfur atom.
- thiocarbonyl moiety includes moieties which are analogous to carbonyl moieties.
- heterocycles include morpholine, piprazine, piperidine, thiomorpholine, and thioazolidine.
- the heterocycles may be substituted or unsubstituted.
- substituents include, for example, halogen, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, alkoxycarbonyl, alkylaminoacarbonyl, aralkylaminocarbonyl, alkenylaminocarbonyl, alkylcarbonyl, arylcarbonyl, aralkylcarbonyl, alkenylcarbonyl, aminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamin
- one or more compounds of the invention may be administered alone to a subject, or more typically a compound of the invention will be administered as part of a pharmaceutical composition in mixture with conventional excipient, i.e., pharmaceutically acceptable organic or inorganic carrier substances suitable for parenteral, oral or other desired administration and which do not deleteriously react with the active compounds and are not deleterious to the recipient thereof.
- conventional excipient i.e., pharmaceutically acceptable organic or inorganic carrier substances suitable for parenteral, oral or other desired administration and which do not deleteriously react with the active compounds and are not deleterious to the recipient thereof.
- Suitable pharmaceutically acceptable carriers include but are not limited to water, salt solutions, alcohol, vegetable oils, polyethylene glycols, gelatin, lactose, amylose, magnesium stearate, talc, silicic acid, viscous paraffin, perfume oil, fatty acid monoglycerides and diglycerides, petroethral fatty acid esters, hydroxymethylcellulose, polyvinylpyrrolidone, etc.
- the substituted tetracycline compound(s) can be suitably admixed in a pharmacologically inert topical carrier such as a gel, an ointment, a lotion or a cream.
- topical carriers include water, glycerol, alcohol, propylene glycol, fatty alcohols, triglycerides, fatty acid esters, or mineral oils.
- Other possible topical carriers are liquid petrolatum, isopropylpalmitate, polyethylene glycol, ethanol 95%, poly oxy ethylene monolauriate 5% in water, sodium lauryl sulfate 5% in water, and the like.
- the ordinarily skilled artisan would select an appropriate amount of a substituted tetracycline compound for use in the aforementioned in vivo assay.
- the effective amount of the substituted tetracycline compound is effective to treat a subject, e.g., human, suffering from malaria.
- subject includes animals which are capable of having malaria.
- examples of subject include, but are not limited to, birds (i.e. geese, ducks), reptiles, ruminants (e.g., cattle and goats), mice, rats, hamsters, dogs, cats, horses, pigs, sheep, lions, tigers, bears, monkeys, chimpanzees, and, in a preferred embodiment, humans.
- the solid was dissolved in dimethylformamide and injected onto a preparative HPLC system using C18 reverse-phase silica. The fraction at 39 minutes was isolated, and the solvent removed in vacuo to yield the product plus salts. The salts were removed by extraction into 50:25:25 water, butanol, ethyl acetate and dried in vacuo. This solid was dissolved in MeOH and the HC1 salt made by bubbling in HC1 gas. The solvent was removed to produce the product in 57% yield as a yellow solid.
- MeOH are added to a flask with a stir bar and the system degassed 3x using argon.
- Na 2 CO 3 104 mg, 1.1 mM
- argon degassed is added via syringe is added along with 4-F-phenylboronic acid (104 mg, 0.7 mM) in MeOH that was also degassed .
- the reaction was followed by HPLC for 20 minutes and cooled to room temperature. The solution was filtered, and dried to produce a crude mixture. The solid was dissolved in dimethylformamide and injected onto a preparative HPLC system using C18 reverse-phase silica. The fraction at 19-20 minutes was isolated, and the solvent removed in vacuo to yield the product plus salts.
- NN-Dimethylglycine (1.2 mmol) was dissolved in DMF (5 mL) and O- Benzotriazol-1-yl-N, N, N', N',-tetramethyluronium hexafluorophosphate (HBTU, 1.2 mmol) was added. The solution was then stirred for 5 minutes at room temperature. To this solution, 7-aminosancycline (1 mmol) was added, followed by the addition of diisopropylethyl amine (DIEA, 1.2 mmol). The reaction was then stirred at room temperature for 2 hours. The solvent, DMF, was removed on vacuum. The crude material was dissolved in 5 mL of MeOH and filtered using autovials and purified using preparative HPLC. The structure of the product has been characterized using IH ⁇ MR, HPLC, and MS.
- HBTU O- Benzotriazol-1-yl-N, N, N', N',-tetramethyluronium he
- the sulphonamide was prepared by a method known in the art (J.Med.Chem 31(3) 1988; 577-82). This was followed by one milliliter of triethylamine (1 ml; 0J26 mg; 7.175 mmoles) and the reaction was stirred, under an argon atmosphere, for approximately 1.0 hour at ambient temperature. The reaction mixture was suctioned filtered through a pad of diatomaceous earth and washed with acetonitrile. The filtrates were reduced to dryness under vacuo and the residue was treated with a dilute solution of trifluroroacetic acid in acetonitrile to adjust the pH to approximately 2.
- the residue was treated with more dilute trifluoroacetic acid in acetonitrile, resulting in the formation of a precipitate, which was removed via suction filtration.
- the crude filtrates were purified utilizing reverse phase HPLC with DVB as the solid phase; and a gradient of 1:1 methanol/acetonitrile 1% trifluoroacetic acid and 1% trifluoroacetic acid in water. The appropriate fractions were reduced to dryness under reduced pressure and solid collected. The product was characterized via ⁇ NMR, mass spectrogram and LC reverse phase.
- the reaction was heated for 45 minutes, the progress was monitored via reverse phase HPLC.
- the reaction was suctioned filtered through a pad of diatomaceous earth and washed with DMF. The filtrates were reduced to an oil under vacuum and residue treated with t-butylmethyl ether.
- Crude material was purified via reverse phase HPLC on DVB utilizing a gradient of water and methanol/acetonitrile containing 1.0% trifluoroacetic acid. Product confirmed by mass spectrum: found M+l 578.58; the structure corroborated with IH NMR.
- Table 1 which follows, shows the relative MIC values obtained for certain substituted tetracycline compounds of the invention. * represents good inhibition of parasite growth, ** represents very good inhibition of parasite growth, *** represent extremely good inhibition of parasite growth. MIC represents the minimum concentration of the compound that inhibits P. falciparum growth after incubation at 30°C for 24 to 48 hours.
- P. vinckei a murine parasite that consistently causes a rapidly fatal malaria, and is an excellent model for drug efficacy.
- other murine parasites which are available (e.g. P. berghei) can also be studied using similar methodology.
- mice 20 gm Swiss Webster mice are inoculated intraperitoneally with 10 ⁇ p. vincke J-infected erythrocytes obtained from another infected mouse. Twelve hours after infection, treatment is initiated by the intraperitoneal injection of test compounds. Treatment is continued twice-a-day (BID) for four days.
- BID twice-a-day
- the progress of malaria infections in experimental and control (injected with diluent only) mice is followed by daily examinations of blood smears obtained from tail veins.
- the pharmacological endpoint is parasitemia >50%. Uninfected animals are followed for 6 weeks, and the animals that remain uninfected through this period are considered long-term cures.
- the test compounds are injected into the stomach of the test mice by gavage.
- mice are monitored daily, for at least the first two weeks of an experiment, with blood smears.
- Counts per 1000 erythrocytes provide parasitemias, and the parasitemias are then plotted over time, and results for control and experimental animals are compared.
- the final cell density should be approximately 5xl0 5 CFU/ml. These plates are incubated at 35°C in an ambient air incubator for approximately 18 hr. The plates are read with a microplate reader and are visually inspected when necessary.
- the MIC is defined as the lowest concentration of the tetracycline compound that inhibits growth.
Landscapes
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Heterocyclic Compounds That Contain Two Or More Ring Oxygen Atoms (AREA)
- Furan Compounds (AREA)
- Pyridine Compounds (AREA)
Abstract
Description















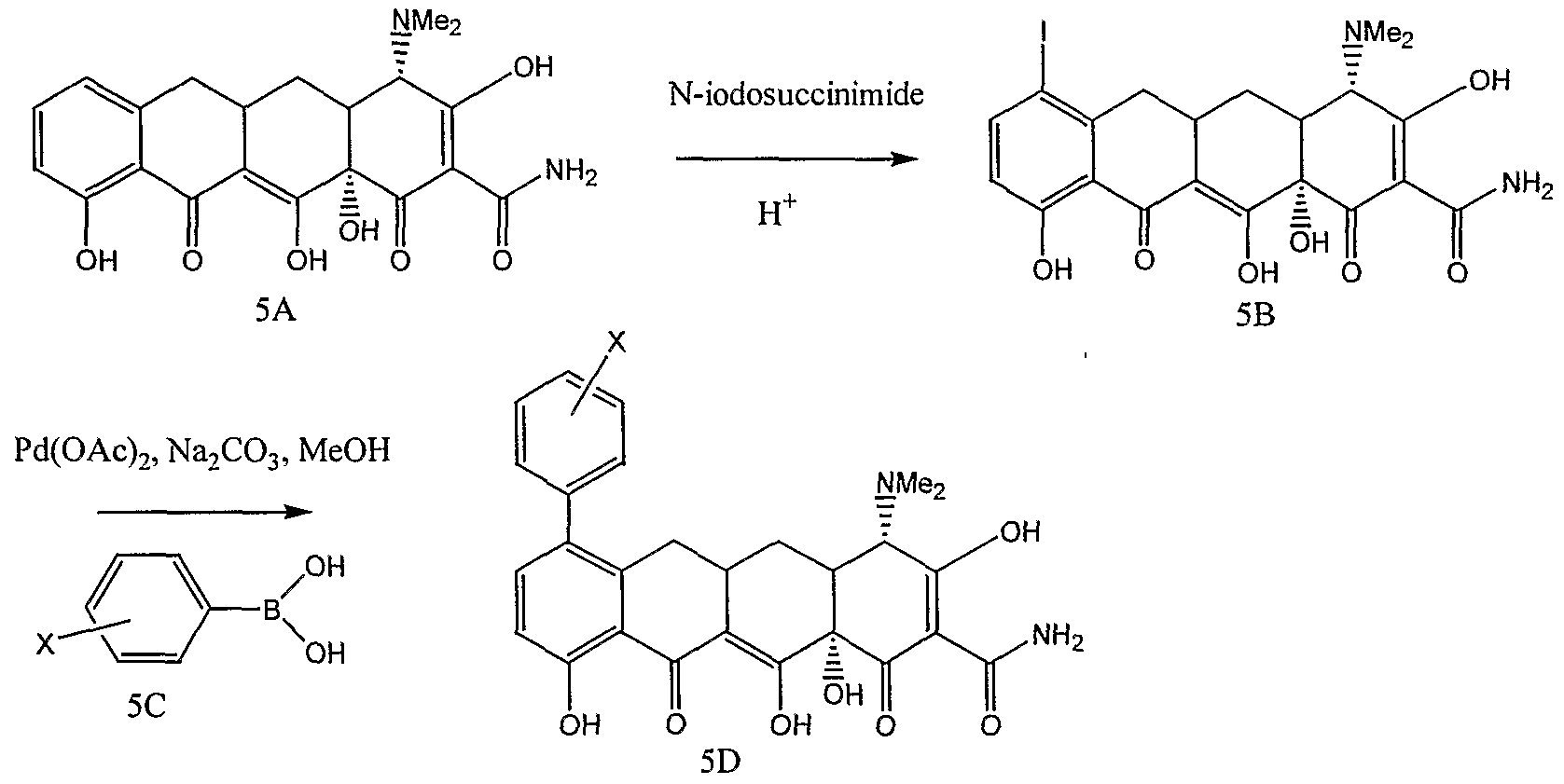







































Claims



















Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE60235083T DE60235083D1 (en) | 2001-04-24 | 2002-04-24 | SUBSTITUTED TETRACYCLIN COMPOUNDS FOR THE TREATMENT OF MALARIA |
| EP02723955A EP1399414B1 (en) | 2001-04-24 | 2002-04-24 | Substituted tetracycline compounds for the treatment of malaria |
| JP2002582879A JP2004529927A (en) | 2001-04-24 | 2002-04-24 | Substituted tetracycline compounds for the treatment of malaria |
| AU2002254714A AU2002254714C1 (en) | 2001-04-24 | 2002-04-24 | Substituted tetracycline compounds for the treatment of malaria |
| AT02723955T ATE455092T1 (en) | 2001-04-24 | 2002-04-24 | SUBSTITUTED TETRACYCLINE COMPOUNDS FOR THE TREATMENT OF MALARIA |
| CA2444899A CA2444899C (en) | 2001-04-24 | 2002-04-24 | Substituted tetracycline compounds for the treatment of malaria |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US28619301P | 2001-04-24 | 2001-04-24 | |
| US60/286,193 | 2001-04-24 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2002085303A2 true WO2002085303A2 (en) | 2002-10-31 |
| WO2002085303A3 WO2002085303A3 (en) | 2003-05-15 |
Family
ID=23097495
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2002/012935 WO2002085303A2 (en) | 2001-04-24 | 2002-04-24 | Substituted tetracycline compounds for the treatment of malaria |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20040092490A1 (en) |
| EP (2) | EP2186793A1 (en) |
| JP (4) | JP2004529927A (en) |
| AT (1) | ATE455092T1 (en) |
| AU (2) | AU2002254714C1 (en) |
| CA (1) | CA2444899C (en) |
| DE (1) | DE60235083D1 (en) |
| ES (1) | ES2338994T3 (en) |
| WO (1) | WO2002085303A2 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1534300A2 (en) * | 2002-07-12 | 2005-06-01 | Paratek Pharmaceuticals, Inc. | 3, 10, and 12a substituted tetracycline compounds |
| WO2005009943A3 (en) * | 2003-07-09 | 2005-06-16 | Paratek Pharm Innc | Substituted tetracycline compounds |
| EP1556007A2 (en) * | 2002-10-24 | 2005-07-27 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds for the treatment of malaria |
| WO2006047756A2 (en) * | 2004-10-25 | 2006-05-04 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| EP1753713A2 (en) * | 2004-05-21 | 2007-02-21 | The President and Fellows of Harvard College | Synthesis of tetracyclines and analogues thereof |
| US7763735B2 (en) | 2006-10-11 | 2010-07-27 | President And Fellows Of Harvard College | Synthesis of enone intermediate |
| US7786099B2 (en) | 2004-01-15 | 2010-08-31 | Paratek Pharmaceuticals, Inc. | Aromatic a-ring derivatives of tetracycline compounds |
| EP2301550A1 (en) * | 2001-07-13 | 2011-03-30 | Paratek Pharmaceuticals, Inc. | Tetracycline compounds having target therapeutic activities |
| WO2010126607A3 (en) * | 2009-04-30 | 2011-07-21 | President And Fellows Of Harvard College | Synthesis of tetracyclines and intermediates thereto |
| US8088820B2 (en) | 2001-04-24 | 2012-01-03 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds for the treatment of malaria |
| US8486921B2 (en) | 2006-04-07 | 2013-07-16 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| US8501716B2 (en) | 2008-08-08 | 2013-08-06 | Tetraphase Pharmaceuticals, Inc. | C7-fluoro substituted tetracycline compounds |
| AU2012202559B2 (en) * | 2004-05-21 | 2014-07-17 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| CN105001112A (en) * | 2015-06-30 | 2015-10-28 | 浦城正大生化有限公司 | Water soluble chlorotetracycline succinic acid monoester salt, and preparation method thereof |
| US9315451B2 (en) | 2009-05-08 | 2016-04-19 | Tetraphase Pharmaceuticals, Inc. | Tetracycline compounds |
| US9573895B2 (en) | 2012-08-31 | 2017-02-21 | Tetraphase Pharmaceuticals, Inc. | Tetracycline compounds |
| US9624166B2 (en) | 2009-08-28 | 2017-04-18 | Tetraphase Pharmaceuticals, Inc. | Tetracycline compounds |
| US10961190B2 (en) | 2016-10-19 | 2021-03-30 | Tetraphase Pharmaceuticals, Inc. | Crystalline forms of eravacycline |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8106225B2 (en) * | 1999-09-14 | 2012-01-31 | Trustees Of Tufts College | Methods of preparing substituted tetracyclines with transition metal-based chemistries |
| ATE523485T1 (en) * | 1999-09-14 | 2011-09-15 | Tufts College | METHOD FOR PRODUCING SUBSTITUTED TETRACYCLINES USING CHEMICALS BASED ON TRANSITION METALS |
| CA2397863A1 (en) * | 2000-01-24 | 2001-07-26 | Trustees Of Tufts College | Tetracycline compounds for treatment of cryptosporidium parvum related disorders |
| AU2001251157A1 (en) * | 2000-03-31 | 2001-10-15 | Paratek Pharmaceuticals, Inc | 7-and 9-carbamate, urea, thiourea, thiocarbamate, and heteroaryl-amino substituted tetracycline compounds |
| CA2409063A1 (en) * | 2000-05-15 | 2001-11-22 | Mark L. Nelson | 7-substituted fused ring tetracycline compounds |
| US20020128238A1 (en) * | 2000-06-16 | 2002-09-12 | Nelson Mark L. | 7-phenyl-substituted tetracycline compounds |
| US20020132798A1 (en) * | 2000-06-16 | 2002-09-19 | Nelson Mark L. | 7-phenyl-substituted tetracycline compounds |
| US7094806B2 (en) | 2000-07-07 | 2006-08-22 | Trustees Of Tufts College | 7, 8 and 9-substituted tetracycline compounds |
| EP1301467B1 (en) | 2000-07-07 | 2006-08-16 | Trustees Of Tufts College | 9-substituted minocycline compounds |
| WO2002072506A2 (en) * | 2001-03-13 | 2002-09-19 | Paratek Pharmaceuticals, Inc. | 7-pyrollyl tetracycline compounds and methods of use thereof |
| US7553828B2 (en) * | 2001-03-13 | 2009-06-30 | Paratek Pharmaceuticals, Inc. | 9-aminomethyl substituted minocycline compounds |
| DE60135387D1 (en) * | 2001-03-13 | 2008-09-25 | Paratek Pharm Innc | 7.9 SUBSTITUTED TETRACYCLINE COMPOUNDS |
| CA2440757A1 (en) | 2001-03-14 | 2002-09-19 | Michael Draper | Substituted tetracycline compounds as synergistic antifungal agents |
| EP2186793A1 (en) * | 2001-04-24 | 2010-05-19 | Paratek Pharmaceuticals, Inc. | Substituted Tetracycline Compounds |
| US20060194773A1 (en) * | 2001-07-13 | 2006-08-31 | Paratek Pharmaceuticals, Inc. | Tetracyline compounds having target therapeutic activities |
| AU2002365120A1 (en) | 2001-08-02 | 2003-07-15 | Paratek Pharmaceuticals, Inc. | Medicaments |
| JP2005514410A (en) * | 2002-01-08 | 2005-05-19 | パラテック ファーマシューティカルズ インコーポレイテッド | 4-dedimethylaminotetracycline compound |
| KR101163937B1 (en) * | 2002-03-08 | 2012-07-09 | 파라테크 파마슈티컬스, 인크. | Amino-Methyl Substituted Tetracycline Compounds |
| JP2005520846A (en) * | 2002-03-21 | 2005-07-14 | パラテック ファーマシューティカルズ インコーポレイテッド | Substituted tetracycline compounds. |
| AU2003287217B2 (en) * | 2002-10-24 | 2010-03-04 | Paratek Pharmaceuticals, Inc. | Methods of using substituted tetracycline compounds to modulate RNA |
| US20060287283A1 (en) * | 2003-07-09 | 2006-12-21 | Paratek Pharmaceuticals, Inc. | Prodrugs of 9-aminomethyl tetracycline compounds |
| TWI261038B (en) * | 2004-08-11 | 2006-09-01 | Bo-Cheng Chen | Bicycle gear-shifting handgrip |
| EP1805134B1 (en) * | 2004-10-25 | 2012-06-20 | Paratek Pharmaceuticals, Inc. | 4-aminotetracyclines and methods of use thereof |
| AU2006210406C1 (en) * | 2005-02-04 | 2013-03-07 | Paratek Pharmaceuticals, Inc. | 11a, 12-derivatives of tetracycline compounds |
| US8440646B1 (en) | 2006-10-11 | 2013-05-14 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds for treatment of Bacillus anthracis infections |
| BRPI0720569B8 (en) * | 2006-12-21 | 2021-05-25 | Paratek Pharm Innc | substituted tetracycline compounds and use of said compounds for the treatment of a bacterial, viral or parasitic infection |
| KR101178319B1 (en) | 2007-11-29 | 2012-09-07 | 액테리온 파마슈티칼 리미티드 | Phosphonic acid derivates and their use as p2y12 receptor antagonists |
| CA3069046A1 (en) * | 2017-07-04 | 2019-01-10 | Sanofi | Ethynyl compounds, their preparation and their therapeutic use for the treatment of malaria |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5789395A (en) * | 1996-08-30 | 1998-08-04 | The Research Foundation Of State University Of New York | Method of using tetracycline compounds for inhibition of endogenous nitric oxide production |
| US6043231A (en) * | 1993-03-02 | 2000-03-28 | The Research Foundation Of State Univ. Of New York | Inhibition of excessive phospholipase A2 activity and/or production by non-antimicrobial tetracyclines |
| US6319910B1 (en) * | 1997-12-19 | 2001-11-20 | Hospital For Joint Diseases | Method for inhibiting cyclooxygenase-2 and tumor necrosis factor alpha |
Family Cites Families (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2990331A (en) | 1956-11-23 | 1961-06-27 | Pfizer & Co C | Stable solutions of salts of tetracyclines for parenteral administration |
| US2980584A (en) | 1957-10-29 | 1961-04-18 | Pfizer & Co C | Parenteral magnesium oxytetracycline acetic or lactic acid carboxamide vehicle preparation |
| US3062717A (en) | 1958-12-11 | 1962-11-06 | Pfizer & Co C | Intramuscular calcium tetracycline acetic or lactic acid carboxamide vehicle preparation |
| US3165531A (en) | 1962-03-08 | 1965-01-12 | Pfizer & Co C | 13-substituted-6-deoxytetracyclines and process utilizing the same |
| US3454697A (en) | 1965-06-08 | 1969-07-08 | American Cyanamid Co | Tetracycline antibiotic compositions for oral use |
| NL6607516A (en) | 1966-05-31 | 1967-12-01 | ||
| DE1767891C3 (en) | 1968-06-28 | 1980-10-30 | Pfizer | Process for the preparation of aqueous medicinal solutions for parenteral, peroral and local use containing a tetracycline derivative |
| NL158172B (en) * | 1972-09-18 | 1978-10-16 | Farmaceutici Italia | PROCESS FOR PREPARING TETRACYCLINE DERIVATIVES WITH A 7-PLACE SUBSTITUENT. |
| US3957980A (en) | 1972-10-26 | 1976-05-18 | Pfizer Inc. | Doxycycline parenteral compositions |
| DE2442829A1 (en) | 1974-09-06 | 1976-03-18 | Merck Patent Gmbh | TETRACYCLIC COMPOUNDS AND PROCEDURES FOR THEIR PRODUCTION |
| US4018889A (en) | 1976-01-02 | 1977-04-19 | Pfizer Inc. | Oxytetracycline compositions |
| YU40295B (en) | 1977-04-07 | 1985-12-31 | Pliva Pharm & Chem Works | Process for preparing n2-tert.butyl-11a-halo-6-demethyl-6-deoxy-6-methylene tetracycline |
| US4126680A (en) | 1977-04-27 | 1978-11-21 | Pfizer Inc. | Tetracycline antibiotic compositions |
| YU41093B (en) | 1978-04-12 | 1986-12-31 | Pliva Pharm & Chem Works | Process for preparing 6-deoxy-5hydroxy-tetracycline |
| ATE211128T1 (en) * | 1991-10-04 | 2002-01-15 | American Cyanamid Co | 7-SUBSTITUTED-9-SUBSTITUTED AMINO-6-DEMETHYL-6-DEOXY-TETRACYCLINE |
| SG47520A1 (en) * | 1992-08-13 | 1998-04-17 | American Cyanamid Co | New method for the production of 9-amino-6-demethyl-6-deoxytetracycline |
| CA2103189C (en) | 1992-11-17 | 2005-05-03 | Lorne M. Golub | Tetracyclines including non-antimicrobial chemically-modified tetracyclines inhibit excessive collagen crosslinking during diabetes |
| US5523297A (en) * | 1993-03-02 | 1996-06-04 | The Research Foundation Of State University Of New York | Inhibition of excessive phospholipase A2 activity and/or production by non-antimicrobial tetracyclines |
| US5371076A (en) | 1993-04-02 | 1994-12-06 | American Cyanamid Company | 9-[(substituted glycyl)amido]-6-(substituted)-5-hydroxy-6-deoxytetracyclines |
| MX9603508A (en) | 1994-02-17 | 1997-03-29 | Pfizer | 9-(substituted amino)-alpha-6-deoxy-5-oxy tetracycline derivatives, their preparation and their use as antibiotics. |
| US5919395A (en) | 1997-10-30 | 1999-07-06 | Shell Oil Company | Polyol combination |
| US20020123637A1 (en) * | 1998-01-23 | 2002-09-05 | Stuart B. Levy | Pharmaceutically active compounds and methods of use thereof |
| ES2272097T3 (en) * | 1998-11-18 | 2007-04-16 | Collagenex Pharmaceuticals, Inc. | NEW DERIVATIVES OF 4-DEDIMETILAMINOTETRACICLINA. |
| EP1171163A1 (en) * | 1999-04-27 | 2002-01-16 | Antibody Systems, Inc. | Compositions containing tetracyclines for treating hemorrhagic virus infections and other disorders |
| ATE523485T1 (en) * | 1999-09-14 | 2011-09-15 | Tufts College | METHOD FOR PRODUCING SUBSTITUTED TETRACYCLINES USING CHEMICALS BASED ON TRANSITION METALS |
| HUP0301163A3 (en) * | 2000-07-07 | 2008-08-28 | Tufts College | 7-substituted tetracycline dervatives, pharmaceutical compositions containing them and their use |
| CA2440757A1 (en) * | 2001-03-14 | 2002-09-19 | Michael Draper | Substituted tetracycline compounds as synergistic antifungal agents |
| WO2002072022A2 (en) * | 2001-03-14 | 2002-09-19 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds as antifungal agents |
| EP2186793A1 (en) * | 2001-04-24 | 2010-05-19 | Paratek Pharmaceuticals, Inc. | Substituted Tetracycline Compounds |
| JP5706185B2 (en) | 2011-02-22 | 2015-04-22 | 富士レビオ株式会社 | Measuring apparatus and measuring method |
| US9713502B2 (en) | 2014-03-09 | 2017-07-25 | Gyrus Acmi, Inc. | Narrow band imaging with surgical loupes |
-
2002
- 2002-04-24 EP EP10150482A patent/EP2186793A1/en not_active Withdrawn
- 2002-04-24 AT AT02723955T patent/ATE455092T1/en not_active IP Right Cessation
- 2002-04-24 WO PCT/US2002/012935 patent/WO2002085303A2/en active Application Filing
- 2002-04-24 AU AU2002254714A patent/AU2002254714C1/en not_active Ceased
- 2002-04-24 DE DE60235083T patent/DE60235083D1/en not_active Expired - Lifetime
- 2002-04-24 US US10/128,990 patent/US20040092490A1/en not_active Abandoned
- 2002-04-24 CA CA2444899A patent/CA2444899C/en not_active Expired - Fee Related
- 2002-04-24 EP EP02723955A patent/EP1399414B1/en not_active Expired - Lifetime
- 2002-04-24 JP JP2002582879A patent/JP2004529927A/en active Pending
- 2002-04-24 ES ES02723955T patent/ES2338994T3/en not_active Expired - Lifetime
-
2008
- 2008-12-11 AU AU2008255241A patent/AU2008255241B2/en not_active Ceased
-
2009
- 2009-05-28 JP JP2009128388A patent/JP2009221219A/en not_active Withdrawn
-
2012
- 2012-10-01 JP JP2012232354A patent/JP2013010806A/en active Pending
-
2013
- 2013-03-28 JP JP2013069469A patent/JP2013151540A/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6043231A (en) * | 1993-03-02 | 2000-03-28 | The Research Foundation Of State Univ. Of New York | Inhibition of excessive phospholipase A2 activity and/or production by non-antimicrobial tetracyclines |
| US5789395A (en) * | 1996-08-30 | 1998-08-04 | The Research Foundation Of State University Of New York | Method of using tetracycline compounds for inhibition of endogenous nitric oxide production |
| US6319910B1 (en) * | 1997-12-19 | 2001-11-20 | Hospital For Joint Diseases | Method for inhibiting cyclooxygenase-2 and tumor necrosis factor alpha |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1399414A2 * |
Cited By (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8088820B2 (en) | 2001-04-24 | 2012-01-03 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds for the treatment of malaria |
| EP2332549A1 (en) * | 2001-07-13 | 2011-06-15 | Paratek Pharmaceuticals, Inc. | Novel tetracyclines and their use in medicine |
| EP2301550A1 (en) * | 2001-07-13 | 2011-03-30 | Paratek Pharmaceuticals, Inc. | Tetracycline compounds having target therapeutic activities |
| EP1534300A4 (en) * | 2002-07-12 | 2008-04-23 | Paratek Pharm Innc | 3, 10, and 12a substituted tetracycline compounds |
| US8481513B2 (en) | 2002-07-12 | 2013-07-09 | Paratek Pharmaceuticals, Inc. | 3, 10, and 12a substituted tetracycline compounds |
| EP2345637A3 (en) * | 2002-07-12 | 2012-02-15 | Paratek Pharmaceuticals, Inc. | 3, 10, and 12a substituted tetracycline compounds |
| EP1534300A2 (en) * | 2002-07-12 | 2005-06-01 | Paratek Pharmaceuticals, Inc. | 3, 10, and 12a substituted tetracycline compounds |
| US7825105B2 (en) | 2002-07-12 | 2010-11-02 | Paratek Pharmaceuticals, Inc. | 3, 10, and 12a substituted tetracycline compounds |
| EP2277504A1 (en) * | 2002-10-24 | 2011-01-26 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds for the treatment of malaria |
| EP1556007A4 (en) * | 2002-10-24 | 2008-12-17 | Paratek Pharm Innc | Substituted tetracycline compounds for the treatment of malaria |
| EP1556007A2 (en) * | 2002-10-24 | 2005-07-27 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds for the treatment of malaria |
| US9533943B2 (en) | 2003-07-09 | 2017-01-03 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| WO2005009943A3 (en) * | 2003-07-09 | 2005-06-16 | Paratek Pharm Innc | Substituted tetracycline compounds |
| EP1648859A2 (en) * | 2003-07-09 | 2006-04-26 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| EP2319828A3 (en) * | 2003-07-09 | 2011-07-06 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| JP2007521290A (en) * | 2003-07-09 | 2007-08-02 | パラテック ファーマシューティカルズ インコーポレイテッド | Substituted tetracycline compounds |
| EP2319829A1 (en) * | 2003-07-09 | 2011-05-11 | Paratek Pharmaceuticals, Inc. | 9-substituted tetracycline compounds |
| EP2298323A3 (en) * | 2003-07-09 | 2011-05-11 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| JP2011063592A (en) * | 2003-07-09 | 2011-03-31 | Paratek Pharmaceuticals Inc | Substituted tetracycline compound |
| EP2298322A3 (en) * | 2003-07-09 | 2011-05-11 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| EP2295404A3 (en) * | 2003-07-09 | 2011-05-11 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| US7786099B2 (en) | 2004-01-15 | 2010-08-31 | Paratek Pharmaceuticals, Inc. | Aromatic a-ring derivatives of tetracycline compounds |
| EP2332904A2 (en) | 2004-01-15 | 2011-06-15 | Paratek Pharmaceuticals, Inc. | Derivatives of tetracycline compounds |
| US7807842B2 (en) | 2004-05-21 | 2010-10-05 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| JP2011162566A (en) * | 2004-05-21 | 2011-08-25 | President & Fellows Of Harvard College | Synthesis of tetracycline and analogue thereof |
| EP1753713A2 (en) * | 2004-05-21 | 2007-02-21 | The President and Fellows of Harvard College | Synthesis of tetracyclines and analogues thereof |
| US9365493B2 (en) | 2004-05-21 | 2016-06-14 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| AU2005244988C1 (en) * | 2004-05-21 | 2012-06-28 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| US9884830B2 (en) | 2004-05-21 | 2018-02-06 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| JP2015221832A (en) * | 2004-05-21 | 2015-12-10 | プレジデント アンド フェローズ オブ ハーバード カレッジ | Synthesis of tetracyclines and analogues thereof |
| JP2017052797A (en) * | 2004-05-21 | 2017-03-16 | プレジデント アンド フェローズ オブ ハーバード カレッジ | Synthesis of tetracyclines and analogues thereof |
| AU2012202559B2 (en) * | 2004-05-21 | 2014-07-17 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| KR101336462B1 (en) * | 2004-05-21 | 2013-12-04 | 프레지던트 앤드 펠로우즈 오브 하바드 칼리지 | Synthesis of tetracyclines and analogues thereof |
| EP1753713A4 (en) * | 2004-05-21 | 2009-07-08 | Harvard College | Synthesis of tetracyclines and analogues thereof |
| AU2005244988B2 (en) * | 2004-05-21 | 2012-02-02 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| US10669244B2 (en) | 2004-05-21 | 2020-06-02 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| US8598148B2 (en) | 2004-05-21 | 2013-12-03 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| US11192866B2 (en) | 2004-05-21 | 2021-12-07 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| JP2008500391A (en) * | 2004-05-21 | 2008-01-10 | ザ プレジデント アンド フェローズ オブ ハーバード カレッジ | Synthesis of tetracyclines and their analogs. |
| JP2012111770A (en) * | 2004-10-25 | 2012-06-14 | Paratek Pharmaceuticals Inc | Substituted tetracycline compound |
| JP2008518035A (en) * | 2004-10-25 | 2008-05-29 | パラテック ファーマシューティカルズ インコーポレイテッド | Substituted tetracycline compounds |
| AU2005299569B2 (en) * | 2004-10-25 | 2012-06-07 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| WO2006047756A2 (en) * | 2004-10-25 | 2006-05-04 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| WO2006047756A3 (en) * | 2004-10-25 | 2006-07-20 | Paratek Pharm Innc | Substituted tetracycline compounds |
| EP2284153A3 (en) * | 2004-10-25 | 2012-03-14 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| EP2287150A3 (en) * | 2004-10-25 | 2011-10-19 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| EP2305637A3 (en) * | 2004-10-25 | 2011-09-21 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| EP2949644A3 (en) * | 2004-10-25 | 2016-04-20 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| EP2033950A1 (en) * | 2004-10-25 | 2009-03-11 | Paratek Pharmaceuticals, Inc. | Substituted tetracycline compounds |
| US8486921B2 (en) | 2006-04-07 | 2013-07-16 | President And Fellows Of Harvard College | Synthesis of tetracyclines and analogues thereof |
| US8580969B2 (en) | 2006-10-11 | 2013-11-12 | President And Fellows Of Harvard College | Synthesis of enone intermediate |
| US8907104B2 (en) | 2006-10-11 | 2014-12-09 | President And Fellows Of Harvard College | Synthesis of enone intermediate |
| US7763735B2 (en) | 2006-10-11 | 2010-07-27 | President And Fellows Of Harvard College | Synthesis of enone intermediate |
| US8293920B2 (en) | 2006-10-11 | 2012-10-23 | President And Fellows Of Harvard College | Synthesis of enone intermediate |
| US7960559B2 (en) | 2006-10-11 | 2011-06-14 | President And Fellows Of Harvard College | Synthesis of enone intermediate |
| US8906887B2 (en) | 2008-08-08 | 2014-12-09 | Tetraphase Pharmaceuticals, Inc. | C7-fluoro substituted tetracycline compounds |
| US8796245B2 (en) | 2008-08-08 | 2014-08-05 | Tetraphase Pharmaceuticals, Inc. | C7-fluoro substituted tetracycline compounds |
| US8501716B2 (en) | 2008-08-08 | 2013-08-06 | Tetraphase Pharmaceuticals, Inc. | C7-fluoro substituted tetracycline compounds |
| US9688644B2 (en) | 2009-04-30 | 2017-06-27 | President And Fellows Of Harvard College | Synthesis of Tetracyclines and intermediates thereto |
| WO2010126607A3 (en) * | 2009-04-30 | 2011-07-21 | President And Fellows Of Harvard College | Synthesis of tetracyclines and intermediates thereto |
| US9073829B2 (en) | 2009-04-30 | 2015-07-07 | President And Fellows Of Harvard College | Synthesis of tetracyclines and intermediates thereto |
| US9315451B2 (en) | 2009-05-08 | 2016-04-19 | Tetraphase Pharmaceuticals, Inc. | Tetracycline compounds |
| US10072007B2 (en) | 2009-05-08 | 2018-09-11 | Tetraphase Pharmaceuticals, Inc. | Tetracycline compounds |
| US9624166B2 (en) | 2009-08-28 | 2017-04-18 | Tetraphase Pharmaceuticals, Inc. | Tetracycline compounds |
| US9573895B2 (en) | 2012-08-31 | 2017-02-21 | Tetraphase Pharmaceuticals, Inc. | Tetracycline compounds |
| US10315992B2 (en) | 2012-08-31 | 2019-06-11 | Tetraphase Pharmaceuticals, Inc. | Tetracyline compounds |
| US10913712B2 (en) | 2012-08-31 | 2021-02-09 | Tetraphase Pharmaceuticals, Inc. | Tetracycline compounds |
| CN105001112A (en) * | 2015-06-30 | 2015-10-28 | 浦城正大生化有限公司 | Water soluble chlorotetracycline succinic acid monoester salt, and preparation method thereof |
| US10961190B2 (en) | 2016-10-19 | 2021-03-30 | Tetraphase Pharmaceuticals, Inc. | Crystalline forms of eravacycline |
| US11578044B2 (en) | 2016-10-19 | 2023-02-14 | Tetraphase Pharmaceuticals, Inc. | Crystalline forms of eravacycline |
Also Published As
| Publication number | Publication date |
|---|---|
| US20040092490A1 (en) | 2004-05-13 |
| ATE455092T1 (en) | 2010-01-15 |
| AU2008255241A1 (en) | 2009-01-08 |
| CA2444899A1 (en) | 2002-10-31 |
| CA2444899C (en) | 2011-06-21 |
| JP2009221219A (en) | 2009-10-01 |
| AU2002254714B2 (en) | 2008-09-11 |
| DE60235083D1 (en) | 2010-03-04 |
| AU2002254714C1 (en) | 2010-01-07 |
| EP1399414B1 (en) | 2010-01-13 |
| EP2186793A1 (en) | 2010-05-19 |
| JP2013151540A (en) | 2013-08-08 |
| EP1399414A4 (en) | 2006-03-22 |
| EP1399414A2 (en) | 2004-03-24 |
| WO2002085303A3 (en) | 2003-05-15 |
| ES2338994T3 (en) | 2010-05-14 |
| AU2002254714B8 (en) | 2008-10-02 |
| JP2004529927A (en) | 2004-09-30 |
| AU2008255241B2 (en) | 2012-06-28 |
| JP2013010806A (en) | 2013-01-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1399414B1 (en) | Substituted tetracycline compounds for the treatment of malaria | |
| US8088820B2 (en) | Substituted tetracycline compounds for the treatment of malaria | |
| AU2002254714A1 (en) | Substituted tetracycline compounds for the treatment of malaria | |
| AU2003287218C1 (en) | Substituted tetracycline compounds for the treatment of malaria | |
| US20060205698A1 (en) | 7-Phenyl-substituted tetracycline compounds | |
| EP1301467A2 (en) | 9-substituted minocycline compounds | |
| AU2001286388A1 (en) | 9-substituted minocycline compounds | |
| AU2001271642A1 (en) | 7-substituted tetracycline compounds | |
| WO2002004407A2 (en) | 7-substituted tetracycline compounds | |
| WO2008045507A2 (en) | Substituted tetracycline compounds for treatment of bacillus anthracis infections | |
| AU2009236631A1 (en) | Substituted tetracycline compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG UZ VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2444899 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002582879 Country of ref document: JP Ref document number: 2002254714 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002723955 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002723955 Country of ref document: EP |