WO2002042273A2 - Acid derivatives useful as serine protease inhibitors - Google Patents
Acid derivatives useful as serine protease inhibitors Download PDFInfo
- Publication number
- WO2002042273A2 WO2002042273A2 PCT/US2001/046884 US0146884W WO0242273A2 WO 2002042273 A2 WO2002042273 A2 WO 2002042273A2 US 0146884 W US0146884 W US 0146884W WO 0242273 A2 WO0242273 A2 WO 0242273A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- substituted
- hydrogen
- alkenyl
- compound
- Prior art date
Links
- 239000003001 serine protease inhibitor Substances 0.000 title abstract description 4
- 239000002253 acid Substances 0.000 title description 32
- 150000001875 compounds Chemical class 0.000 claims abstract description 274
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 59
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 19
- 125000002950 monocyclic group Chemical class 0.000 claims abstract description 10
- 125000004076 pyridyl group Chemical group 0.000 claims abstract description 7
- 125000000217 alkyl group Chemical group 0.000 claims description 129
- -1 phenyloxy Chemical group 0.000 claims description 95
- 239000001257 hydrogen Substances 0.000 claims description 79
- 229910052739 hydrogen Inorganic materials 0.000 claims description 79
- 125000001072 heteroaryl group Chemical group 0.000 claims description 69
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 64
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 60
- 150000002431 hydrogen Chemical class 0.000 claims description 58
- 125000003342 alkenyl group Chemical group 0.000 claims description 50
- 238000000034 method Methods 0.000 claims description 44
- 150000003839 salts Chemical class 0.000 claims description 43
- 125000003118 aryl group Chemical group 0.000 claims description 37
- 125000005017 substituted alkenyl group Chemical group 0.000 claims description 36
- 125000004432 carbon atom Chemical group C* 0.000 claims description 33
- 125000003545 alkoxy group Chemical group 0.000 claims description 32
- 229910052736 halogen Inorganic materials 0.000 claims description 32
- 150000002367 halogens Chemical class 0.000 claims description 32
- 125000004414 alkyl thio group Chemical group 0.000 claims description 27
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 27
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 22
- 229940002612 prodrug Drugs 0.000 claims description 22
- 239000000651 prodrug Substances 0.000 claims description 22
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 21
- 125000001188 haloalkyl group Chemical group 0.000 claims description 21
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 21
- RFEBDZANCVHDLP-UHFFFAOYSA-N 3-[(4-cyanophenyl)methylamino]-6-(trifluoromethyl)quinoxaline-2-carboxylic acid Chemical compound OC(=O)C1=NC2=CC=C(C(F)(F)F)C=C2N=C1NCC1=CC=C(C#N)C=C1 RFEBDZANCVHDLP-UHFFFAOYSA-N 0.000 claims description 20
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 20
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 19
- 125000003282 alkyl amino group Chemical group 0.000 claims description 18
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 18
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 16
- 125000000304 alkynyl group Chemical group 0.000 claims description 15
- 125000000623 heterocyclic group Chemical group 0.000 claims description 14
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 13
- 125000005605 benzo group Chemical group 0.000 claims description 13
- 125000004122 cyclic group Chemical group 0.000 claims description 13
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 13
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 13
- 230000015271 coagulation Effects 0.000 claims description 12
- 238000005345 coagulation Methods 0.000 claims description 12
- 125000005842 heteroatom Chemical group 0.000 claims description 12
- 125000002619 bicyclic group Chemical group 0.000 claims description 11
- 125000004404 heteroalkyl group Chemical group 0.000 claims description 10
- 125000002252 acyl group Chemical group 0.000 claims description 9
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 9
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 9
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 9
- 239000003085 diluting agent Substances 0.000 claims description 8
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 7
- 125000004426 substituted alkynyl group Chemical group 0.000 claims description 7
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 claims description 6
- 239000008194 pharmaceutical composition Substances 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 5
- 230000002757 inflammatory effect Effects 0.000 claims description 5
- 125000000468 ketone group Chemical group 0.000 claims description 5
- 229930194542 Keto Natural products 0.000 claims description 4
- 206010027476 Metastases Diseases 0.000 claims description 4
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 4
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 4
- 235000010290 biphenyl Nutrition 0.000 claims description 3
- 239000004305 biphenyl Substances 0.000 claims description 3
- 239000012530 fluid Substances 0.000 claims description 3
- 125000006555 (C3-C5) cycloalkyl group Chemical group 0.000 claims description 2
- 241000124008 Mammalia Species 0.000 claims description 2
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 claims description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 13
- 208000026278 immune system disease Diseases 0.000 claims 3
- 208000027866 inflammatory disease Diseases 0.000 claims 3
- 230000036770 blood supply Effects 0.000 claims 2
- 229910006074 SO2NH2 Inorganic materials 0.000 claims 1
- 125000002877 alkyl aryl group Chemical group 0.000 claims 1
- 125000004103 aminoalkyl group Chemical group 0.000 claims 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 abstract description 26
- 239000003112 inhibitor Substances 0.000 abstract description 22
- 229910052757 nitrogen Inorganic materials 0.000 abstract description 17
- 125000005647 linker group Chemical group 0.000 abstract description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 abstract description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 243
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 173
- 239000000243 solution Substances 0.000 description 134
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 111
- 235000019439 ethyl acetate Nutrition 0.000 description 86
- 239000000203 mixture Substances 0.000 description 83
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 77
- 238000006243 chemical reaction Methods 0.000 description 74
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 72
- 229910001868 water Inorganic materials 0.000 description 71
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 70
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 56
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 45
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 44
- 239000007787 solid Substances 0.000 description 42
- 239000011541 reaction mixture Substances 0.000 description 41
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 38
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 38
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 37
- 238000005859 coupling reaction Methods 0.000 description 35
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 34
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 33
- 238000010168 coupling process Methods 0.000 description 33
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 28
- 239000012044 organic layer Substances 0.000 description 26
- 239000000047 product Substances 0.000 description 26
- 150000001412 amines Chemical class 0.000 description 25
- 239000000706 filtrate Substances 0.000 description 25
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 24
- 239000012267 brine Substances 0.000 description 24
- 239000000741 silica gel Substances 0.000 description 24
- 229910002027 silica gel Inorganic materials 0.000 description 24
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 24
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 22
- 238000000746 purification Methods 0.000 description 22
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 22
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 21
- 238000001914 filtration Methods 0.000 description 21
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 20
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 20
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 20
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- 238000004128 high performance liquid chromatography Methods 0.000 description 17
- 239000002904 solvent Substances 0.000 description 17
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 16
- 230000008878 coupling Effects 0.000 description 16
- 239000003921 oil Substances 0.000 description 16
- 238000003756 stirring Methods 0.000 description 16
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- 230000000694 effects Effects 0.000 description 15
- 125000001424 substituent group Chemical group 0.000 description 15
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 14
- 239000012043 crude product Substances 0.000 description 14
- QPLDLSVMHZLSFG-UHFFFAOYSA-N Copper oxide Chemical compound [Cu]=O QPLDLSVMHZLSFG-UHFFFAOYSA-N 0.000 description 13
- 102000004190 Enzymes Human genes 0.000 description 13
- 108090000790 Enzymes Proteins 0.000 description 13
- 229940088598 enzyme Drugs 0.000 description 13
- 108060005989 Tryptase Proteins 0.000 description 12
- 102000001400 Tryptase Human genes 0.000 description 12
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 12
- 238000002360 preparation method Methods 0.000 description 12
- 239000000725 suspension Substances 0.000 description 12
- 238000005160 1H NMR spectroscopy Methods 0.000 description 11
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 11
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 11
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 11
- 239000003146 anticoagulant agent Substances 0.000 description 11
- 239000003153 chemical reaction reagent Substances 0.000 description 11
- 238000003818 flash chromatography Methods 0.000 description 11
- 229910052938 sodium sulfate Inorganic materials 0.000 description 11
- 125000005843 halogen group Chemical group 0.000 description 10
- 238000002953 preparative HPLC Methods 0.000 description 10
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 9
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 9
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 9
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 9
- 210000004369 blood Anatomy 0.000 description 9
- 239000008280 blood Substances 0.000 description 9
- 239000003054 catalyst Substances 0.000 description 9
- 201000010099 disease Diseases 0.000 description 9
- 239000000843 powder Substances 0.000 description 9
- 238000010992 reflux Methods 0.000 description 9
- 239000011780 sodium chloride Substances 0.000 description 9
- 239000011550 stock solution Substances 0.000 description 9
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical group CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 8
- 102000012479 Serine Proteases Human genes 0.000 description 8
- 108010022999 Serine Proteases Proteins 0.000 description 8
- 108090000190 Thrombin Proteins 0.000 description 8
- 230000001143 conditioned effect Effects 0.000 description 8
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 8
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- 229960005356 urokinase Drugs 0.000 description 8
- 108010074860 Factor Xa Proteins 0.000 description 7
- 0 N/*=N/c(ccc(N)c1)c1[N+] Chemical compound N/*=N/c(ccc(N)c1)c1[N+] 0.000 description 7
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 7
- 125000004429 atom Chemical group 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 7
- 150000001721 carbon Chemical group 0.000 description 7
- 238000005341 cation exchange Methods 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 238000004440 column chromatography Methods 0.000 description 7
- 238000010511 deprotection reaction Methods 0.000 description 7
- 239000006260 foam Substances 0.000 description 7
- 239000000543 intermediate Substances 0.000 description 7
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 7
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 7
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- 229960004072 thrombin Drugs 0.000 description 7
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 7
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Substances C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 7
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 6
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 6
- BKOOMYPCSUNDGP-UHFFFAOYSA-N 2-methylbut-2-ene Chemical compound CC=C(C)C BKOOMYPCSUNDGP-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- WPYMKLBDIGXBTP-UHFFFAOYSA-N Benzoic acid Natural products OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 6
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 6
- 206010028980 Neoplasm Diseases 0.000 description 6
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 6
- 108010000499 Thromboplastin Proteins 0.000 description 6
- 102000002262 Thromboplastin Human genes 0.000 description 6
- 208000007536 Thrombosis Diseases 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 239000012298 atmosphere Substances 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 201000011510 cancer Diseases 0.000 description 6
- 125000002837 carbocyclic group Chemical group 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 150000002430 hydrocarbons Chemical group 0.000 description 6
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 6
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 6
- 125000004433 nitrogen atom Chemical group N* 0.000 description 6
- 229910052760 oxygen Inorganic materials 0.000 description 6
- 239000000377 silicon dioxide Substances 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- 229910052717 sulfur Inorganic materials 0.000 description 6
- 239000011701 zinc Substances 0.000 description 6
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 5
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 5
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 5
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 5
- 102000015081 Blood Coagulation Factors Human genes 0.000 description 5
- 108010039209 Blood Coagulation Factors Proteins 0.000 description 5
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 5
- 125000002947 alkylene group Chemical group 0.000 description 5
- 229940127219 anticoagulant drug Drugs 0.000 description 5
- 239000003114 blood coagulation factor Substances 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 229960002897 heparin Drugs 0.000 description 5
- 229920000669 heparin Polymers 0.000 description 5
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 150000003254 radicals Chemical class 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 4
- LWRSYTXEQUUTKW-UHFFFAOYSA-N 2,4-dimethoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C(OC)=C1 LWRSYTXEQUUTKW-UHFFFAOYSA-N 0.000 description 4
- ZLGYVWRJIZPQMM-HHHXNRCGSA-N 2-azaniumylethyl [(2r)-2,3-di(dodecanoyloxy)propyl] phosphate Chemical compound CCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OCCN)OC(=O)CCCCCCCCCCC ZLGYVWRJIZPQMM-HHHXNRCGSA-N 0.000 description 4
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 4
- 239000005711 Benzoic acid Substances 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 4
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- 229910019213 POCl3 Inorganic materials 0.000 description 4
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 4
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 206010047249 Venous thrombosis Diseases 0.000 description 4
- 229960001138 acetylsalicylic acid Drugs 0.000 description 4
- 150000001299 aldehydes Chemical class 0.000 description 4
- 125000004450 alkenylene group Chemical group 0.000 description 4
- 125000004419 alkynylene group Chemical group 0.000 description 4
- 235000010233 benzoic acid Nutrition 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- 229940126545 compound 53 Drugs 0.000 description 4
- 229960004643 cupric oxide Drugs 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 4
- 208000014674 injury Diseases 0.000 description 4
- YCJZWBZJSYLMPB-UHFFFAOYSA-N n-(2-chloropyrimidin-4-yl)-2,5-dimethyl-1-phenylimidazole-4-carboxamide Chemical compound CC=1N(C=2C=CC=CC=2)C(C)=NC=1C(=O)NC1=CC=NC(Cl)=N1 YCJZWBZJSYLMPB-UHFFFAOYSA-N 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 239000002464 receptor antagonist Substances 0.000 description 4
- 229940044551 receptor antagonist Drugs 0.000 description 4
- 239000011347 resin Substances 0.000 description 4
- 229920005989 resin Polymers 0.000 description 4
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 4
- 125000004434 sulfur atom Chemical group 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- 125000006168 tricyclic group Chemical group 0.000 description 4
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 4
- QOWBXWFYRXSBAS-UHFFFAOYSA-N (2,4-dimethoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C(OC)=C1 QOWBXWFYRXSBAS-UHFFFAOYSA-N 0.000 description 3
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 3
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 3
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 3
- XDIAMRVROCPPBK-UHFFFAOYSA-N 2,2-dimethylpropan-1-amine Chemical compound CC(C)(C)CN XDIAMRVROCPPBK-UHFFFAOYSA-N 0.000 description 3
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 3
- LFOIDLOIBZFWDO-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(3-phenylmethoxyphenyl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(C=1)=CC=CC=1OCC1=CC=CC=C1 LFOIDLOIBZFWDO-UHFFFAOYSA-N 0.000 description 3
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 3
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 3
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 3
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 3
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 3
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- 239000007832 Na2SO4 Substances 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 108010094028 Prothrombin Proteins 0.000 description 3
- 102100027378 Prothrombin Human genes 0.000 description 3
- 208000006011 Stroke Diseases 0.000 description 3
- 108090000631 Trypsin Proteins 0.000 description 3
- 102000004142 Trypsin Human genes 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 229940121363 anti-inflammatory agent Drugs 0.000 description 3
- 239000002260 anti-inflammatory agent Substances 0.000 description 3
- 229960004676 antithrombotic agent Drugs 0.000 description 3
- LPLAXFBDWKFJGQ-UHFFFAOYSA-N benzyl n-(6-amino-1h-benzimidazol-2-yl)carbamate Chemical compound N1C2=CC(N)=CC=C2N=C1NC(=O)OCC1=CC=CC=C1 LPLAXFBDWKFJGQ-UHFFFAOYSA-N 0.000 description 3
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 3
- 239000001110 calcium chloride Substances 0.000 description 3
- 229910001628 calcium chloride Inorganic materials 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229940125904 compound 1 Drugs 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 229940125833 compound 23 Drugs 0.000 description 3
- 229940125846 compound 25 Drugs 0.000 description 3
- 229940126214 compound 3 Drugs 0.000 description 3
- 229940125878 compound 36 Drugs 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 230000002255 enzymatic effect Effects 0.000 description 3
- 238000013265 extended release Methods 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 238000005194 fractionation Methods 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 150000004820 halides Chemical class 0.000 description 3
- 150000003840 hydrochlorides Chemical class 0.000 description 3
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 3
- 235000019341 magnesium sulphate Nutrition 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 3
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 229940039716 prothrombin Drugs 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- 229960002218 sodium chlorite Drugs 0.000 description 3
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 3
- 229910000162 sodium phosphate Inorganic materials 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 238000001356 surgical procedure Methods 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 3
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 3
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 3
- 239000012588 trypsin Substances 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 2
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 2
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 2
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 2
- VDFVNEFVBPFDSB-UHFFFAOYSA-N 1,3-dioxane Chemical compound C1COCOC1 VDFVNEFVBPFDSB-UHFFFAOYSA-N 0.000 description 2
- RNHDAKUGFHSZEV-UHFFFAOYSA-N 1,4-dioxane;hydrate Chemical compound O.C1COCCO1 RNHDAKUGFHSZEV-UHFFFAOYSA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 2
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 2
- JWYUFVNJZUSCSM-UHFFFAOYSA-N 2-aminobenzimidazole Chemical compound C1=CC=C2NC(N)=NC2=C1 JWYUFVNJZUSCSM-UHFFFAOYSA-N 0.000 description 2
- CSDSSGBPEUDDEE-UHFFFAOYSA-N 2-formylpyridine Chemical class O=CC1=CC=CC=N1 CSDSSGBPEUDDEE-UHFFFAOYSA-N 0.000 description 2
- MHNNAWXXUZQSNM-UHFFFAOYSA-N 2-methylbut-1-ene Chemical compound CCC(C)=C MHNNAWXXUZQSNM-UHFFFAOYSA-N 0.000 description 2
- IAVREABSGIHHMO-UHFFFAOYSA-N 3-hydroxybenzaldehyde Chemical compound OC1=CC=CC(C=O)=C1 IAVREABSGIHHMO-UHFFFAOYSA-N 0.000 description 2
- KYWCWBXGRWWINE-UHFFFAOYSA-N 4-methoxy-N1,N3-bis(3-pyridinylmethyl)benzene-1,3-dicarboxamide Chemical compound COC1=CC=C(C(=O)NCC=2C=NC=CC=2)C=C1C(=O)NCC1=CC=CN=C1 KYWCWBXGRWWINE-UHFFFAOYSA-N 0.000 description 2
- UTCFOFWMEPQCSR-UHFFFAOYSA-N 5-formylsalicylic acid Chemical compound OC(=O)C1=CC(C=O)=CC=C1O UTCFOFWMEPQCSR-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 206010003178 Arterial thrombosis Diseases 0.000 description 2
- 206010003658 Atrial Fibrillation Diseases 0.000 description 2
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 description 2
- 239000004342 Benzoyl peroxide Substances 0.000 description 2
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 208000024172 Cardiovascular disease Diseases 0.000 description 2
- 102000003902 Cathepsin C Human genes 0.000 description 2
- 108090000267 Cathepsin C Proteins 0.000 description 2
- 206010053567 Coagulopathies Diseases 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- 229940127007 Compound 39 Drugs 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 206010051055 Deep vein thrombosis Diseases 0.000 description 2
- 108010049003 Fibrinogen Proteins 0.000 description 2
- 102000008946 Fibrinogen Human genes 0.000 description 2
- 208000032843 Hemorrhage Diseases 0.000 description 2
- 206010062506 Heparin-induced thrombocytopenia Diseases 0.000 description 2
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- 239000005574 MCPA Substances 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 2
- 229910002666 PdCl2 Inorganic materials 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- ATTZFSUZZUNHBP-UHFFFAOYSA-N Piperonyl sulfoxide Chemical group CCCCCCCCS(=O)C(C)CC1=CC=C2OCOC2=C1 ATTZFSUZZUNHBP-UHFFFAOYSA-N 0.000 description 2
- 101800004937 Protein C Proteins 0.000 description 2
- 102000017975 Protein C Human genes 0.000 description 2
- 101800001700 Saposin-D Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 229940122388 Thrombin inhibitor Drugs 0.000 description 2
- 102000003938 Thromboxane Receptors Human genes 0.000 description 2
- 108090000300 Thromboxane Receptors Proteins 0.000 description 2
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 2
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 2
- WHKUVVPPKQRRBV-UHFFFAOYSA-N Trasan Chemical compound CC1=CC(Cl)=CC=C1OCC(O)=O WHKUVVPPKQRRBV-UHFFFAOYSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 2
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 2
- IOSLINNLJFQMFF-XMMPIXPASA-N [(2R)-1-[[4-[[3-[(4-fluorophenyl)methylsulfanyl]phenoxy]methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound FC1=CC=C(CSC=2C=C(OCC3=CC=C(CN4[C@H](CCC4)CO)C=C3)C=CC=2)C=C1 IOSLINNLJFQMFF-XMMPIXPASA-N 0.000 description 2
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 2
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 150000001447 alkali salts Chemical class 0.000 description 2
- 239000002257 antimetastatic agent Substances 0.000 description 2
- 239000004019 antithrombin Substances 0.000 description 2
- 150000001501 aryl fluorides Chemical class 0.000 description 2
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 description 2
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 2
- 235000019400 benzoyl peroxide Nutrition 0.000 description 2
- 229960003328 benzoyl peroxide Drugs 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-M bisulphate group Chemical group S([O-])(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 2
- 230000000740 bleeding effect Effects 0.000 description 2
- 230000023555 blood coagulation Effects 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 208000026106 cerebrovascular disease Diseases 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 description 2
- 229960003009 clopidogrel Drugs 0.000 description 2
- 229910052681 coesite Inorganic materials 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 229940125961 compound 24 Drugs 0.000 description 2
- 229940127204 compound 29 Drugs 0.000 description 2
- 229940127573 compound 38 Drugs 0.000 description 2
- 229940125936 compound 42 Drugs 0.000 description 2
- 229940125844 compound 46 Drugs 0.000 description 2
- 229940127271 compound 49 Drugs 0.000 description 2
- 229940127113 compound 57 Drugs 0.000 description 2
- 229940125900 compound 59 Drugs 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 208000029078 coronary artery disease Diseases 0.000 description 2
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N coumarin Chemical compound C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 2
- 229910052906 cristobalite Inorganic materials 0.000 description 2
- 239000013058 crude material Substances 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 150000004985 diamines Chemical class 0.000 description 2
- 229960004132 diethyl ether Drugs 0.000 description 2
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 239000002024 ethyl acetate extract Substances 0.000 description 2
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 229940012952 fibrinogen Drugs 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 210000003709 heart valve Anatomy 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- ZTVZLYBCZNMWCF-UHFFFAOYSA-N homocystine Chemical compound [O-]C(=O)C([NH3+])CCSSCCC([NH3+])C([O-])=O ZTVZLYBCZNMWCF-UHFFFAOYSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 2
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 229950004274 ifetroban Drugs 0.000 description 2
- BBPRUNPUJIUXSE-DXKRWKNPSA-N ifetroban Chemical compound CCCCCNC(=O)C1=COC([C@H]2[C@H]([C@@H]3CC[C@H]2O3)CC=2C(=CC=CC=2)CCC(O)=O)=N1 BBPRUNPUJIUXSE-DXKRWKNPSA-N 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 238000002513 implantation Methods 0.000 description 2
- 150000004694 iodide salts Chemical class 0.000 description 2
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical compound NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- 229960005417 ketanserin Drugs 0.000 description 2
- FPCCSQOGAWCVBH-UHFFFAOYSA-N ketanserin Chemical compound C1=CC(F)=CC=C1C(=O)C1CCN(CCN2C(C3=CC=CC=C3NC2=O)=O)CC1 FPCCSQOGAWCVBH-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 229940127215 low-molecular weight heparin Drugs 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- 150000004702 methyl esters Chemical class 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical compound CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 2
- 208000005264 motor neuron disease Diseases 0.000 description 2
- 208000010125 myocardial infarction Diseases 0.000 description 2
- KPSSIOMAKSHJJG-UHFFFAOYSA-N neopentyl alcohol Chemical compound CC(C)(C)CO KPSSIOMAKSHJJG-UHFFFAOYSA-N 0.000 description 2
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 2
- 125000004043 oxo group Chemical group O=* 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 235000021317 phosphate Nutrition 0.000 description 2
- LGRFSURHDFAFJT-UHFFFAOYSA-N phthalic anhydride Chemical compound C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 2
- 229960001006 picotamide Drugs 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 229960000856 protein c Drugs 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- TZSZZENYCISATO-WIOPSUGQSA-N rodatristat Chemical compound CCOC(=O)[C@@H]1CC2(CN1)CCN(CC2)c1cc(O[C@H](c2ccc(Cl)cc2-c2ccccc2)C(F)(F)F)nc(N)n1 TZSZZENYCISATO-WIOPSUGQSA-N 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 229910052682 stishovite Inorganic materials 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 239000003868 thrombin inhibitor Substances 0.000 description 2
- 230000009424 thromboembolic effect Effects 0.000 description 2
- 230000036962 time dependent Effects 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- 230000008733 trauma Effects 0.000 description 2
- 229910052905 tridymite Inorganic materials 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- 230000001960 triggered effect Effects 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 208000019553 vascular disease Diseases 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- XWTYSIMOBUGWOL-UHFFFAOYSA-N (+-)-Terbutaline Chemical compound CC(C)(C)NCC(O)C1=CC(O)=CC(O)=C1 XWTYSIMOBUGWOL-UHFFFAOYSA-N 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical class CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- ZLMVLWAEIINORX-WQDURMRASA-N (2S)-2-amino-5-(diaminomethylideneamino)pentanoic acid 4-nitroaniline 2-(phenylmethoxycarbonylamino)acetic acid (2S)-pyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1.NC1=CC=C([N+]([O-])=O)C=C1.OC(=O)[C@@H](N)CCCNC(N)=N.OC(=O)CNC(=O)OCC1=CC=CC=C1 ZLMVLWAEIINORX-WQDURMRASA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- HYXMHAHVUFTVFZ-UHFFFAOYSA-N (3-formylthiophen-2-yl)boronic acid Chemical compound OB(O)C=1SC=CC=1C=O HYXMHAHVUFTVFZ-UHFFFAOYSA-N 0.000 description 1
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 1
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 1
- SLFNGVGRINFJLK-UHFFFAOYSA-N 1-bromo-2-fluoro-4-methylbenzene Chemical compound CC1=CC=C(Br)C(F)=C1 SLFNGVGRINFJLK-UHFFFAOYSA-N 0.000 description 1
- ROKZAMCDHKVZIQ-UHFFFAOYSA-N 1-bromo-3,3-dimethylbutane Chemical compound CC(C)(C)CCBr ROKZAMCDHKVZIQ-UHFFFAOYSA-N 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical class CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical class CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- ZCBIFHNDZBSCEP-UHFFFAOYSA-N 1H-indol-5-amine Chemical compound NC1=CC=C2NC=CC2=C1 ZCBIFHNDZBSCEP-UHFFFAOYSA-N 0.000 description 1
- NRKYWOKHZRQRJR-UHFFFAOYSA-N 2,2,2-trifluoroacetamide Chemical group NC(=O)C(F)(F)F NRKYWOKHZRQRJR-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- XARVANDLQOZMMJ-CHHVJCJISA-N 2-[(z)-[1-(2-amino-1,3-thiazol-4-yl)-2-oxo-2-(2-oxoethylamino)ethylidene]amino]oxy-2-methylpropanoic acid Chemical compound OC(=O)C(C)(C)O\N=C(/C(=O)NCC=O)C1=CSC(N)=N1 XARVANDLQOZMMJ-CHHVJCJISA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- XTTIQGSLJBWVIV-UHFFFAOYSA-N 2-methyl-4-nitroaniline Chemical compound CC1=CC([N+]([O-])=O)=CC=C1N XTTIQGSLJBWVIV-UHFFFAOYSA-N 0.000 description 1
- HDECRAPHCDXMIJ-UHFFFAOYSA-N 2-methylbenzenesulfonyl chloride Chemical compound CC1=CC=CC=C1S(Cl)(=O)=O HDECRAPHCDXMIJ-UHFFFAOYSA-N 0.000 description 1
- WLAMNBDJUVNPJU-UHFFFAOYSA-N 2-methylbutyric acid Chemical compound CCC(C)C(O)=O WLAMNBDJUVNPJU-UHFFFAOYSA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000006088 2-oxoazepinyl group Chemical group 0.000 description 1
- 125000004638 2-oxopiperazinyl group Chemical group O=C1N(CCNC1)* 0.000 description 1
- 125000004637 2-oxopiperidinyl group Chemical group O=C1N(CCCC1)* 0.000 description 1
- 125000006087 2-oxopyrrolodinyl group Chemical group 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical class BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- GOLORTLGFDVFDW-UHFFFAOYSA-N 3-(1h-benzimidazol-2-yl)-7-(diethylamino)chromen-2-one Chemical compound C1=CC=C2NC(C3=CC4=CC=C(C=C4OC3=O)N(CC)CC)=NC2=C1 GOLORTLGFDVFDW-UHFFFAOYSA-N 0.000 description 1
- UMCMPZBLKLEWAF-BCTGSCMUSA-N 3-[(3-cholamidopropyl)dimethylammonio]propane-1-sulfonate Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCC[N+](C)(C)CCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 UMCMPZBLKLEWAF-BCTGSCMUSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical class OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- GZLGTVRDLCJQTO-UHFFFAOYSA-M 3-methylbutyl(triphenyl)phosphanium;bromide Chemical compound [Br-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(CCC(C)C)C1=CC=CC=C1 GZLGTVRDLCJQTO-UHFFFAOYSA-M 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical class OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 1
- BFWYZZPDZZGSLJ-UHFFFAOYSA-N 4-(aminomethyl)aniline Chemical compound NCC1=CC=C(N)C=C1 BFWYZZPDZZGSLJ-UHFFFAOYSA-N 0.000 description 1
- GVCLNACSYKYUHP-UHFFFAOYSA-N 4-amino-7-(2-hydroxyethoxymethyl)pyrrolo[2,3-d]pyrimidine-5-carbothioamide Chemical compound C1=NC(N)=C2C(C(=S)N)=CN(COCCO)C2=N1 GVCLNACSYKYUHP-UHFFFAOYSA-N 0.000 description 1
- QATKOZUHTGAWMG-UHFFFAOYSA-N 4-fluoro-3-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(F)C(O)=C1 QATKOZUHTGAWMG-UHFFFAOYSA-N 0.000 description 1
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- PHBVTMQLXNCAQO-UHFFFAOYSA-N 5-(aminomethyl)pyridin-2-amine Chemical compound NCC1=CC=C(N)N=C1 PHBVTMQLXNCAQO-UHFFFAOYSA-N 0.000 description 1
- 102000056834 5-HT2 Serotonin Receptors Human genes 0.000 description 1
- 108091005479 5-HT2 receptors Proteins 0.000 description 1
- LSLYOANBFKQKPT-DIFFPNOSSA-N 5-[(1r)-1-hydroxy-2-[[(2r)-1-(4-hydroxyphenyl)propan-2-yl]amino]ethyl]benzene-1,3-diol Chemical compound C([C@@H](C)NC[C@H](O)C=1C=C(O)C=C(O)C=1)C1=CC=C(O)C=C1 LSLYOANBFKQKPT-DIFFPNOSSA-N 0.000 description 1
- MBQFPDJMKXGIFD-UHFFFAOYSA-N 5-nitro-2,1-benzoxazol-3-amine Chemical compound C1=CC([N+]([O-])=O)=CC2=C(N)ON=C21 MBQFPDJMKXGIFD-UHFFFAOYSA-N 0.000 description 1
- UGSBCCAHDVCHGI-UHFFFAOYSA-N 5-nitropyridin-2-amine Chemical compound NC1=CC=C([N+]([O-])=O)C=N1 UGSBCCAHDVCHGI-UHFFFAOYSA-N 0.000 description 1
- RZPOSPORWMDCQT-UHFFFAOYSA-N 6-methoxy-2-tributylstannylpyridine-3-carbaldehyde Chemical compound CCCC[Sn](CCCC)(CCCC)C1=NC(OC)=CC=C1C=O RZPOSPORWMDCQT-UHFFFAOYSA-N 0.000 description 1
- NXSGMAIBPJOSHK-UHFFFAOYSA-N 6-methoxy-3-tributylstannylpyridine-2-carbaldehyde Chemical compound CCCC[Sn](CCCC)(CCCC)C1=CC=C(OC)N=C1C=O NXSGMAIBPJOSHK-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 239000005541 ACE inhibitor Substances 0.000 description 1
- 206010056867 Activated protein C resistance Diseases 0.000 description 1
- 208000004476 Acute Coronary Syndrome Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- ITPDYQOUSLNIHG-UHFFFAOYSA-N Amiodarone hydrochloride Chemical compound [Cl-].CCCCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(I)=C(OCC[NH+](CC)CC)C(I)=C1 ITPDYQOUSLNIHG-UHFFFAOYSA-N 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 206010002388 Angina unstable Diseases 0.000 description 1
- 102100030988 Angiotensin-converting enzyme Human genes 0.000 description 1
- 102000004411 Antithrombin III Human genes 0.000 description 1
- 108090000935 Antithrombin III Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 208000031104 Arterial Occlusive disease Diseases 0.000 description 1
- 206010003162 Arterial injury Diseases 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 239000005528 B01AC05 - Ticlopidine Substances 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 206010005949 Bone cancer Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 1
- NQRYJNQNLNOLGT-UHFFFAOYSA-N C1CCNCC1 Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 1
- XVWHJXJURVOTOS-UHFFFAOYSA-N CC(C)(C)CN(C(C(F)(F)F)=O)C(c(cc1C(O)=O)ccc1C(C=C1)=C(C(OC)=O)NC1=O)=O Chemical compound CC(C)(C)CN(C(C(F)(F)F)=O)C(c(cc1C(O)=O)ccc1C(C=C1)=C(C(OC)=O)NC1=O)=O XVWHJXJURVOTOS-UHFFFAOYSA-N 0.000 description 1
- KIXJALVKFDGNED-UHFFFAOYSA-N CC(C)(C)CNC(c(cc1C(OC)=O)ccc1F)=O Chemical compound CC(C)(C)CNC(c(cc1C(OC)=O)ccc1F)=O KIXJALVKFDGNED-UHFFFAOYSA-N 0.000 description 1
- GSMHJIBIXIWZOH-UHFFFAOYSA-N CC(C)(C)OC(c1cc(C(OCc2ccccc2)=O)ccc1-c1c(C(Nc(cc2)cc3c2nc(N)[n]3C(OC(C)(C)C)=O)=O)nccc1)=O Chemical compound CC(C)(C)OC(c1cc(C(OCc2ccccc2)=O)ccc1-c1c(C(Nc(cc2)cc3c2nc(N)[n]3C(OC(C)(C)C)=O)=O)nccc1)=O GSMHJIBIXIWZOH-UHFFFAOYSA-N 0.000 description 1
- BWAMQMPBVWRBQX-MRXNPFEDSA-N CC(C)(C)[C@@H](CO)NC(c(cc1C(O)=O)ccc1-c(cc1)c(C(NC)=O)nc1OC)=O Chemical compound CC(C)(C)[C@@H](CO)NC(c(cc1C(O)=O)ccc1-c(cc1)c(C(NC)=O)nc1OC)=O BWAMQMPBVWRBQX-MRXNPFEDSA-N 0.000 description 1
- BFWGQUKPFHNZSX-HSZRJFAPSA-N CC(C)(C)[C@@H](CO)NC(c(cc1C(O)=O)ccc1-c(cc1)c(CNc2ccc3nc(N)[nH]c3c2)nc1OC)=O Chemical compound CC(C)(C)[C@@H](CO)NC(c(cc1C(O)=O)ccc1-c(cc1)c(CNc2ccc3nc(N)[nH]c3c2)nc1OC)=O BFWGQUKPFHNZSX-HSZRJFAPSA-N 0.000 description 1
- KQPZRBBUXLOUJN-UHFFFAOYSA-N CC(C)COC(c1cc(C(O)=O)ccc1-c(cc1)c(C(Nc(cc2NC3(C)C(OC(C)(C)C)=O)ccc2N=C3N)=O)nc1OC)=O Chemical compound CC(C)COC(c1cc(C(O)=O)ccc1-c(cc1)c(C(Nc(cc2NC3(C)C(OC(C)(C)C)=O)ccc2N=C3N)=O)nc1OC)=O KQPZRBBUXLOUJN-UHFFFAOYSA-N 0.000 description 1
- ZKYPSULHBXNZLK-UHFFFAOYSA-N CC(C)COC(c1cc(C=O)ccc1O)=O Chemical compound CC(C)COC(c1cc(C=O)ccc1O)=O ZKYPSULHBXNZLK-UHFFFAOYSA-N 0.000 description 1
- UWNODHJECDOWTC-UHFFFAOYSA-N CC(C)c1c(ccnc2N)c2ccc1 Chemical compound CC(C)c1c(ccnc2N)c2ccc1 UWNODHJECDOWTC-UHFFFAOYSA-N 0.000 description 1
- NADPQMQMFJDWFQ-UHFFFAOYSA-N CC1(C)COC(c(cc2C(OC)=O)ccc2O)OC1 Chemical compound CC1(C)COC(c(cc2C(OC)=O)ccc2O)OC1 NADPQMQMFJDWFQ-UHFFFAOYSA-N 0.000 description 1
- ZUEXAVCHHXDXRY-UHFFFAOYSA-N CCOc1ccc(-c(c(C(OCC(C)C)=O)c2)ccc2C(NCC(C)(C)C)=O)c(C(Nc(cc2)cc3c2c(NCc(ccc(OC)c2)c2OC)ncc3)=O)n1 Chemical compound CCOc1ccc(-c(c(C(OCC(C)C)=O)c2)ccc2C(NCC(C)(C)C)=O)c(C(Nc(cc2)cc3c2c(NCc(ccc(OC)c2)c2OC)ncc3)=O)n1 ZUEXAVCHHXDXRY-UHFFFAOYSA-N 0.000 description 1
- MVZHLIJONBYXEA-UHFFFAOYSA-N COC(c1cc(OC)ccc1C(CC1)=C(C=O)N=C1OC)=O Chemical compound COC(c1cc(OC)ccc1C(CC1)=C(C=O)N=C1OC)=O MVZHLIJONBYXEA-UHFFFAOYSA-N 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- 108090000201 Carboxypeptidase B2 Proteins 0.000 description 1
- 102100035023 Carboxypeptidase B2 Human genes 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- HFYFOFMVURXVSD-UHFFFAOYSA-N Cc(cc1)cc2c1[nH]c(-c1ccccn1)n2 Chemical compound Cc(cc1)cc2c1[nH]c(-c1ccccn1)n2 HFYFOFMVURXVSD-UHFFFAOYSA-N 0.000 description 1
- MZZZAWDOYQWKMR-UHFFFAOYSA-N Cc(cc1)cc2c1[nH]c(N)n2 Chemical compound Cc(cc1)cc2c1[nH]c(N)n2 MZZZAWDOYQWKMR-UHFFFAOYSA-N 0.000 description 1
- KBDMNBWTHOKXQA-UHFFFAOYSA-N Cc1c(cccc2N)c2ccn1 Chemical compound Cc1c(cccc2N)c2ccn1 KBDMNBWTHOKXQA-UHFFFAOYSA-N 0.000 description 1
- ZFLFWZRPMDXJCW-UHFFFAOYSA-N Cc1cc(cc(C)cc2)c2[nH]1 Chemical compound Cc1cc(cc(C)cc2)c2[nH]1 ZFLFWZRPMDXJCW-UHFFFAOYSA-N 0.000 description 1
- QEOSOLXDPRSDKG-UHFFFAOYSA-N Cc1cc(cc(C)cc2)c2[n]1C Chemical compound Cc1cc(cc(C)cc2)c2[n]1C QEOSOLXDPRSDKG-UHFFFAOYSA-N 0.000 description 1
- CMBSSVKZOPZBKW-UHFFFAOYSA-N Cc1ccc(N)nc1 Chemical compound Cc1ccc(N)nc1 CMBSSVKZOPZBKW-UHFFFAOYSA-N 0.000 description 1
- RSJLHHCWBUXOKM-UHFFFAOYSA-N Cc1ccc(ccnc2NC)c2c1 Chemical compound Cc1ccc(ccnc2NC)c2c1 RSJLHHCWBUXOKM-UHFFFAOYSA-N 0.000 description 1
- MVHOAOSHABGEFL-UHFFFAOYSA-N Cc1nc(cc(C)cc2)c2[nH]1 Chemical compound Cc1nc(cc(C)cc2)c2[nH]1 MVHOAOSHABGEFL-UHFFFAOYSA-N 0.000 description 1
- 229930186147 Cephalosporin Natural products 0.000 description 1
- 206010008088 Cerebral artery embolism Diseases 0.000 description 1
- 206010008132 Cerebral thrombosis Diseases 0.000 description 1
- 206010008190 Cerebrovascular accident Diseases 0.000 description 1
- 208000032544 Cicatrix Diseases 0.000 description 1
- 102100030563 Coagulation factor XI Human genes 0.000 description 1
- 101710161089 Coagulation factor XI Proteins 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 206010010741 Conjunctivitis Diseases 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- 206010012438 Dermatitis atopic Diseases 0.000 description 1
- 206010012689 Diabetic retinopathy Diseases 0.000 description 1
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 208000005189 Embolism Diseases 0.000 description 1
- 108010014173 Factor X Proteins 0.000 description 1
- 229940123583 Factor Xa inhibitor Drugs 0.000 description 1
- 102000009123 Fibrin Human genes 0.000 description 1
- 108010073385 Fibrin Proteins 0.000 description 1
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 1
- 208000012671 Gastrointestinal haemorrhages Diseases 0.000 description 1
- 108010054964 H-hexahydrotyrosyl-alanyl-arginine-4-nitroanilide Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- 206010058423 Haemangioma-thrombocytopenia syndrome Diseases 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102100025306 Integrin alpha-IIb Human genes 0.000 description 1
- 101710149643 Integrin alpha-IIb Proteins 0.000 description 1
- 206010022562 Intermittent claudication Diseases 0.000 description 1
- 201000001429 Intracranial Thrombosis Diseases 0.000 description 1
- 208000012659 Joint disease Diseases 0.000 description 1
- 208000010299 Kasabach-Merritt syndrome Diseases 0.000 description 1
- 150000000994 L-ascorbates Chemical class 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- 108010028275 Leukocyte Elastase Proteins 0.000 description 1
- 102000016799 Leukocyte elastase Human genes 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- LSDPWZHWYPCBBB-UHFFFAOYSA-N Methanethiol Chemical compound SC LSDPWZHWYPCBBB-UHFFFAOYSA-N 0.000 description 1
- 101100537780 Mus musculus Tpm2 gene Proteins 0.000 description 1
- 208000021642 Muscular disease Diseases 0.000 description 1
- 206010028594 Myocardial fibrosis Diseases 0.000 description 1
- 201000009623 Myopathy Diseases 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 description 1
- YMJLGULXZAWKGM-UHFFFAOYSA-N NC(c1cccc(S)c1)=O Chemical compound NC(c1cccc(S)c1)=O YMJLGULXZAWKGM-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- QKRPTVLPDGXENM-UHFFFAOYSA-N Nc1nc(ccc(S)c2)c2[nH]1 Chemical compound Nc1nc(ccc(S)c2)c2[nH]1 QKRPTVLPDGXENM-UHFFFAOYSA-N 0.000 description 1
- 201000004404 Neurofibroma Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- JCXJVPUVTGWSNB-UHFFFAOYSA-N Nitrogen dioxide Chemical compound O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 239000008118 PEG 6000 Substances 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 208000005764 Peripheral Arterial Disease Diseases 0.000 description 1
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 description 1
- 108010022233 Plasminogen Activator Inhibitor 1 Proteins 0.000 description 1
- 102000001938 Plasminogen Activators Human genes 0.000 description 1
- 108010001014 Plasminogen Activators Proteins 0.000 description 1
- 102100039418 Plasminogen activator inhibitor 1 Human genes 0.000 description 1
- 229920002584 Polyethylene Glycol 6000 Polymers 0.000 description 1
- 229920002594 Polyethylene Glycol 8000 Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 208000032319 Primary lateral sclerosis Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 229940096437 Protein S Drugs 0.000 description 1
- 102000029301 Protein S Human genes 0.000 description 1
- 108010066124 Protein S Proteins 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 208000010378 Pulmonary Embolism Diseases 0.000 description 1
- 206010038563 Reocclusion Diseases 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 108010056373 SK potentiator Proteins 0.000 description 1
- 229910006069 SO3H Inorganic materials 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- GIIZNNXWQWCKIB-UHFFFAOYSA-N Serevent Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1 GIIZNNXWQWCKIB-UHFFFAOYSA-N 0.000 description 1
- 229940122055 Serine protease inhibitor Drugs 0.000 description 1
- 101710102218 Serine protease inhibitor Proteins 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 108010023197 Streptokinase Proteins 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 102100026966 Thrombomodulin Human genes 0.000 description 1
- 108010079274 Thrombomodulin Proteins 0.000 description 1
- 208000032109 Transient ischaemic attack Diseases 0.000 description 1
- GCTFWCDSFPMHHS-UHFFFAOYSA-M Tributyltin chloride Chemical compound CCCC[Sn](Cl)(CCCC)CCCC GCTFWCDSFPMHHS-UHFFFAOYSA-M 0.000 description 1
- 208000007814 Unstable Angina Diseases 0.000 description 1
- 206010046298 Upper motor neurone lesion Diseases 0.000 description 1
- 206010053648 Vascular occlusion Diseases 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 108091005605 Vitamin K-dependent proteins Proteins 0.000 description 1
- YEEZWCHGZNKEEK-UHFFFAOYSA-N Zafirlukast Chemical compound COC1=CC(C(=O)NS(=O)(=O)C=2C(=CC=CC=2)C)=CC=C1CC(C1=C2)=CN(C)C1=CC=C2NC(=O)OC1CCCC1 YEEZWCHGZNKEEK-UHFFFAOYSA-N 0.000 description 1
- JVVXZOOGOGPDRZ-SLFFLAALSA-N [(1R,4aS,10aR)-1,4a-dimethyl-7-propan-2-yl-2,3,4,9,10,10a-hexahydrophenanthren-1-yl]methanamine Chemical compound NC[C@]1(C)CCC[C@]2(C)C3=CC=C(C(C)C)C=C3CC[C@H]21 JVVXZOOGOGPDRZ-SLFFLAALSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- PAAZCQANMCYGAW-UHFFFAOYSA-N acetic acid;2,2,2-trifluoroacetic acid Chemical compound CC(O)=O.OC(=O)C(F)(F)F PAAZCQANMCYGAW-UHFFFAOYSA-N 0.000 description 1
- OOVPFYDKQBSPHP-IUQUCOCYSA-N acetic acid;benzyl n-[(2s)-1-[[2-[[(2s)-5-(diaminomethylideneamino)-1-(4-nitroanilino)-1-oxopentan-2-yl]amino]-2-oxoethyl]amino]-3-methyl-1-oxobutan-2-yl]carbamate Chemical compound CC(O)=O.N([C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCN=C(N)N)C(=O)NC=1C=CC(=CC=1)[N+]([O-])=O)C(=O)OCC1=CC=CC=C1 OOVPFYDKQBSPHP-IUQUCOCYSA-N 0.000 description 1
- RRUDCFGSUDOHDG-UHFFFAOYSA-N acetohydroxamic acid Chemical compound CC(O)=NO RRUDCFGSUDOHDG-UHFFFAOYSA-N 0.000 description 1
- 229960001171 acetohydroxamic acid Drugs 0.000 description 1
- 150000008043 acidic salts Chemical class 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 229940099983 activase Drugs 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical class OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 1
- 239000000808 adrenergic beta-agonist Substances 0.000 description 1
- 238000012382 advanced drug delivery Methods 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 229910001420 alkaline earth metal ion Inorganic materials 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- 102000003801 alpha-2-Antiplasmin Human genes 0.000 description 1
- 108090000183 alpha-2-Antiplasmin Proteins 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229960005260 amiodarone Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 238000002399 angioplasty Methods 0.000 description 1
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000002429 anti-coagulating effect Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000002785 anti-thrombosis Effects 0.000 description 1
- 239000003416 antiarrhythmic agent Substances 0.000 description 1
- 229940065524 anticholinergics inhalants for obstructive airway diseases Drugs 0.000 description 1
- 230000010100 anticoagulation Effects 0.000 description 1
- 229940030600 antihypertensive agent Drugs 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 239000003524 antilipemic agent Substances 0.000 description 1
- 229940127218 antiplatelet drug Drugs 0.000 description 1
- 229960005348 antithrombin iii Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 208000021328 arterial occlusion Diseases 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-L aspartate group Chemical class N[C@@H](CC(=O)[O-])C(=O)[O-] CKLJMWTZIZZHCS-REOHCLBHSA-L 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 201000008937 atopic dermatitis Diseases 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- 229940092705 beclomethasone Drugs 0.000 description 1
- NBMKJKDGKREAPL-DVTGEIKXSA-N beclomethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O NBMKJKDGKREAPL-DVTGEIKXSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- UPABQMWFWCMOFV-UHFFFAOYSA-N benethamine Chemical compound C=1C=CC=CC=1CNCCC1=CC=CC=C1 UPABQMWFWCMOFV-UHFFFAOYSA-N 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- ZPQZRIJTQNWNLG-VXKWHMMOSA-N benzyl n-[2-[(2s)-2-[[(2s)-5-(diaminomethylideneamino)-1-(4-nitroanilino)-1-oxopentan-2-yl]carbamoyl]pyrrolidin-1-yl]-2-oxoethyl]carbamate Chemical compound C([C@H]1C(=O)N[C@@H](CCCN=C(N)N)C(=O)NC=2C=CC(=CC=2)[N+]([O-])=O)CCN1C(=O)CNC(=O)OCC1=CC=CC=C1 ZPQZRIJTQNWNLG-VXKWHMMOSA-N 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 1
- FZGVEKPRDOIXJY-UHFFFAOYSA-N bitolterol Chemical compound C1=CC(C)=CC=C1C(=O)OC1=CC=C(C(O)CNC(C)(C)C)C=C1OC(=O)C1=CC=C(C)C=C1 FZGVEKPRDOIXJY-UHFFFAOYSA-N 0.000 description 1
- 229960004620 bitolterol Drugs 0.000 description 1
- 208000034158 bleeding Diseases 0.000 description 1
- 208000015294 blood coagulation disease Diseases 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 150000001642 boronic acid derivatives Chemical class 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- AOJDZKCUAATBGE-UHFFFAOYSA-N bromomethane Chemical compound Br[CH2] AOJDZKCUAATBGE-UHFFFAOYSA-N 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 229960004436 budesonide Drugs 0.000 description 1
- 150000004648 butanoic acid derivatives Chemical class 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical class C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- 125000003739 carbamimidoyl group Chemical class C(N)(=N)* 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 1
- 229960004195 carvedilol Drugs 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229940124587 cephalosporin Drugs 0.000 description 1
- 150000001780 cephalosporins Chemical class 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 229960005110 cerivastatin Drugs 0.000 description 1
- SEERZIQQUAZTOL-ANMDKAQQSA-N cerivastatin Chemical compound COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 SEERZIQQUAZTOL-ANMDKAQQSA-N 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 239000000812 cholinergic antagonist Substances 0.000 description 1
- 125000004617 chromonyl group Chemical group O1C(=CC(C2=CC=CC=C12)=O)* 0.000 description 1
- 208000023819 chronic asthma Diseases 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 229960004588 cilostazol Drugs 0.000 description 1
- RRGUKTPIGVIEKM-UHFFFAOYSA-N cilostazol Chemical compound C=1C=C2NC(=O)CCC2=CC=1OCCCCC1=NN=NN1C1CCCCC1 RRGUKTPIGVIEKM-UHFFFAOYSA-N 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 230000035602 clotting Effects 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 229940125877 compound 31 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 238000007887 coronary angioplasty Methods 0.000 description 1
- 210000004351 coronary vessel Anatomy 0.000 description 1
- 229960000956 coumarin Drugs 0.000 description 1
- 235000001671 coumarin Nutrition 0.000 description 1
- 125000000332 coumarinyl group Chemical group O1C(=O)C(=CC2=CC=CC=C12)* 0.000 description 1
- 229960000265 cromoglicic acid Drugs 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical compound BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 229960000633 dextran sulfate Drugs 0.000 description 1
- 150000008050 dialkyl sulfates Chemical class 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 102000038379 digestive enzymes Human genes 0.000 description 1
- 108091007734 digestive enzymes Proteins 0.000 description 1
- 125000004611 dihydroisoindolyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 229940113088 dimethylacetamide Drugs 0.000 description 1
- GAFRWLVTHPVQGK-UHFFFAOYSA-N dipentyl sulfate Chemical class CCCCCOS(=O)(=O)OCCCCC GAFRWLVTHPVQGK-UHFFFAOYSA-N 0.000 description 1
- 229960002768 dipyridamole Drugs 0.000 description 1
- IZEKFCXSFNUWAM-UHFFFAOYSA-N dipyridamole Chemical compound C=12N=C(N(CCO)CCO)N=C(N3CCCCC3)C2=NC(N(CCO)CCO)=NC=1N1CCCCC1 IZEKFCXSFNUWAM-UHFFFAOYSA-N 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- VLARUOGDXDTHEH-UHFFFAOYSA-L disodium cromoglycate Chemical compound [Na+].[Na+].O1C(C([O-])=O)=CC(=O)C2=C1C=CC=C2OCC(O)COC1=CC=CC2=C1C(=O)C=C(C([O-])=O)O2 VLARUOGDXDTHEH-UHFFFAOYSA-L 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical class CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- IXTMWRCNAAVVAI-UHFFFAOYSA-N dofetilide Chemical compound C=1C=C(NS(C)(=O)=O)C=CC=1CCN(C)CCOC1=CC=C(NS(C)(=O)=O)C=C1 IXTMWRCNAAVVAI-UHFFFAOYSA-N 0.000 description 1
- 229960002994 dofetilide Drugs 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 238000013171 endarterectomy Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- 229960001123 epoprostenol Drugs 0.000 description 1
- KAQKFAOMNZTLHT-VVUHWYTRSA-N epoprostenol Chemical compound O1C(=CCCCC(O)=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)CCCCC)[C@H](O)C[C@@H]21 KAQKFAOMNZTLHT-VVUHWYTRSA-N 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-N ethanesulfonic acid Chemical class CCS(O)(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-N 0.000 description 1
- MHYCRLGKOZWVEF-UHFFFAOYSA-N ethyl acetate;hydrate Chemical compound O.CCOC(C)=O MHYCRLGKOZWVEF-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 229960001022 fenoterol Drugs 0.000 description 1
- 229950003499 fibrin Drugs 0.000 description 1
- 239000002319 fibrinogen receptor antagonist Substances 0.000 description 1
- 239000003527 fibrinolytic agent Substances 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 230000003176 fibrotic effect Effects 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229960000676 flunisolide Drugs 0.000 description 1
- 229960002714 fluticasone Drugs 0.000 description 1
- MGNNYOODZCAHBA-GQKYHHCASA-N fluticasone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(O)[C@@]2(C)C[C@@H]1O MGNNYOODZCAHBA-GQKYHHCASA-N 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 229960002848 formoterol Drugs 0.000 description 1
- BPZSYCZIITTYBL-UHFFFAOYSA-N formoterol Chemical compound C1=CC(OC)=CC=C1CC(C)NCC(O)C1=CC=C(O)C(NC=O)=C1 BPZSYCZIITTYBL-UHFFFAOYSA-N 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 125000004612 furopyridinyl group Chemical group O1C(=CC2=C1C=CC=N2)* 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 208000030304 gastrointestinal bleeding Diseases 0.000 description 1
- YRSVDSQRGBYVIY-GJZGRUSLSA-N gemopatrilat Chemical compound O=C1N(CC(O)=O)C(C)(C)CCC[C@@H]1NC(=O)[C@@H](S)CC1=CC=CC=C1 YRSVDSQRGBYVIY-GJZGRUSLSA-N 0.000 description 1
- 229950006480 gemopatrilat Drugs 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 150000002315 glycerophosphates Chemical class 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000002008 hemorrhagic effect Effects 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical class CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical class CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 238000011540 hip replacement Methods 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical class I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 1
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 201000004332 intermediate coronary syndrome Diseases 0.000 description 1
- 208000021156 intermittent vascular claudication Diseases 0.000 description 1
- 201000010849 intracranial embolism Diseases 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 229960001361 ipratropium bromide Drugs 0.000 description 1
- KEWHKYJURDBRMN-ZEODDXGYSA-M ipratropium bromide hydrate Chemical compound O.[Br-].O([C@H]1C[C@H]2CC[C@@H](C1)[N@@+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 KEWHKYJURDBRMN-ZEODDXGYSA-M 0.000 description 1
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 1
- 229960002198 irbesartan Drugs 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical class OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 230000000155 isotopic effect Effects 0.000 description 1
- 125000003971 isoxazolinyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 150000003893 lactate salts Chemical class 0.000 description 1
- 108010051044 lanoteplase Proteins 0.000 description 1
- 229950010645 lanoteplase Drugs 0.000 description 1
- 201000010901 lateral sclerosis Diseases 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- CZRQXSDBMCMPNJ-ZUIPZQNBSA-N lisinopril dihydrate Chemical compound O.O.C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 CZRQXSDBMCMPNJ-ZUIPZQNBSA-N 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 229910001416 lithium ion Inorganic materials 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- 239000003055 low molecular weight heparin Substances 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940057948 magnesium stearate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 150000002688 maleic acid derivatives Chemical class 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical class CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- PYRAZCQFEMNXJA-UHFFFAOYSA-N methyl 2-bromo-5-methylbenzoate Chemical compound COC(=O)C1=CC(C)=CC=C1Br PYRAZCQFEMNXJA-UHFFFAOYSA-N 0.000 description 1
- KLDGAVFZSVMKKB-UHFFFAOYSA-N methyl 2-bromo-5-phenylmethoxybenzoate Chemical compound C1=C(Br)C(C(=O)OC)=CC(OCC=2C=CC=CC=2)=C1 KLDGAVFZSVMKKB-UHFFFAOYSA-N 0.000 description 1
- XJKWVPNWHOCFBR-UHFFFAOYSA-N methyl 5-formyl-2-hydroxybenzoate Chemical compound COC(=O)C1=CC(C=O)=CC=C1O XJKWVPNWHOCFBR-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 230000003228 microsomal effect Effects 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- HVOYZOQVDYHUPF-UHFFFAOYSA-N n,n',n'-trimethylethane-1,2-diamine Chemical compound CNCCN(C)C HVOYZOQVDYHUPF-UHFFFAOYSA-N 0.000 description 1
- HXRAMSFGUAOAJR-UHFFFAOYSA-N n,n,n',n'-tetramethyl-1-[(2-methylpropan-2-yl)oxy]methanediamine Chemical compound CN(C)C(N(C)C)OC(C)(C)C HXRAMSFGUAOAJR-UHFFFAOYSA-N 0.000 description 1
- IHFZUIBTENSHSH-UHFFFAOYSA-N n,n-dimethoxy-1-phenylmethanamine Chemical compound CON(OC)CC1=CC=CC=C1 IHFZUIBTENSHSH-UHFFFAOYSA-N 0.000 description 1
- SKEAGJWUKCNICJ-LSDHHAIUSA-N n-[(4-fluoro-3-methoxyphenyl)methyl]-6-[2-[[(2s,5r)-5-(hydroxymethyl)-1,4-dioxan-2-yl]methyl]tetrazol-5-yl]-2-methylpyrimidine-4-carboxamide Chemical compound C1=C(F)C(OC)=CC(CNC(=O)C=2N=C(C)N=C(C=2)C2=NN(C[C@@H]3OC[C@@H](CO)OC3)N=N2)=C1 SKEAGJWUKCNICJ-LSDHHAIUSA-N 0.000 description 1
- LPOIGVZLNWEGJG-UHFFFAOYSA-N n-benzyl-5-(4-methylpiperazin-1-yl)-2-nitroaniline Chemical compound C1CN(C)CCN1C1=CC=C([N+]([O-])=O)C(NCC=2C=CC=CC=2)=C1 LPOIGVZLNWEGJG-UHFFFAOYSA-N 0.000 description 1
- HVAAHUDGWQAAOJ-UHFFFAOYSA-N n-benzylethanamine Chemical group CCNCC1=CC=CC=C1 HVAAHUDGWQAAOJ-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- RIWRFSMVIUAEBX-UHFFFAOYSA-N n-methyl-1-phenylmethanamine Chemical group CNCC1=CC=CC=C1 RIWRFSMVIUAEBX-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- VWPOSFSPZNDTMJ-UCWKZMIHSA-N nadolol Chemical compound C1[C@@H](O)[C@@H](O)CC2=C1C=CC=C2OCC(O)CNC(C)(C)C VWPOSFSPZNDTMJ-UCWKZMIHSA-N 0.000 description 1
- 229960004255 nadolol Drugs 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical class C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229960004398 nedocromil Drugs 0.000 description 1
- RQTOOFIXOKYGAN-UHFFFAOYSA-N nedocromil Chemical compound CCN1C(C(O)=O)=CC(=O)C2=C1C(CCC)=C1OC(C(O)=O)=CC(=O)C1=C2 RQTOOFIXOKYGAN-UHFFFAOYSA-N 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 150000002814 niacins Chemical class 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- LVRLSYPNFFBYCZ-VGWMRTNUSA-N omapatrilat Chemical compound C([C@H](S)C(=O)N[C@H]1CCS[C@H]2CCC[C@H](N2C1=O)C(=O)O)C1=CC=CC=C1 LVRLSYPNFFBYCZ-VGWMRTNUSA-N 0.000 description 1
- 229950000973 omapatrilat Drugs 0.000 description 1
- 229940127234 oral contraceptive Drugs 0.000 description 1
- 239000003539 oral contraceptive agent Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 201000008482 osteoarthritis Diseases 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L persulfate group Chemical group S(=O)(=O)([O-])OOS(=O)(=O)[O-] JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 125000003356 phenylsulfanyl group Chemical group [*]SC1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 125000001557 phthalyl group Chemical group C(=O)(O)C1=C(C(=O)*)C=CC=C1 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-N picric acid Chemical class OC1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-N 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 125000005547 pivalate group Chemical group 0.000 description 1
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 229940127126 plasminogen activator Drugs 0.000 description 1
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 1
- 229920001467 poly(styrenesulfonates) Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920002717 polyvinylpyridine Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- DTGLZDAWLRGWQN-UHFFFAOYSA-N prasugrel Chemical compound C1CC=2SC(OC(=O)C)=CC=2CN1C(C=1C(=CC=CC=1)F)C(=O)C1CC1 DTGLZDAWLRGWQN-UHFFFAOYSA-N 0.000 description 1
- 229960004197 prasugrel Drugs 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 239000003805 procoagulant Substances 0.000 description 1
- 201000008752 progressive muscular atrophy Diseases 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 201000001514 prostate carcinoma Diseases 0.000 description 1
- 229940121649 protein inhibitor Drugs 0.000 description 1
- 239000012268 protein inhibitor Substances 0.000 description 1
- 230000003331 prothrombotic effect Effects 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000006085 pyrrolopyridyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- MIXMJCQRHVAJIO-TZHJZOAOSA-N qk4dys664x Chemical compound O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O MIXMJCQRHVAJIO-TZHJZOAOSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 230000010410 reperfusion Effects 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 229960002917 reteplase Drugs 0.000 description 1
- 108010051412 reteplase Proteins 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- 150000003873 salicylate salts Chemical class 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- 229960004017 salmeterol Drugs 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 108010073863 saruplase Proteins 0.000 description 1
- 231100000241 scar Toxicity 0.000 description 1
- 230000037387 scars Effects 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 208000002320 spinal muscular atrophy Diseases 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- KXCAEQNNTZANTK-UHFFFAOYSA-N stannane Chemical compound [SnH4] KXCAEQNNTZANTK-UHFFFAOYSA-N 0.000 description 1
- 229910000080 stannane Inorganic materials 0.000 description 1
- 238000005797 stannylation reaction Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 229960005202 streptokinase Drugs 0.000 description 1
- 125000005415 substituted alkoxy group Chemical group 0.000 description 1
- 125000005156 substituted alkylene group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 150000003890 succinate salts Chemical class 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960000195 terbutaline Drugs 0.000 description 1
- XIEWEVMWNGAVSF-UHFFFAOYSA-N tert-butyl 2,6-diaminobenzimidazole-1-carboxylate Chemical compound C1=C(N)C=C2N(C(=O)OC(C)(C)C)C(N)=NC2=C1 XIEWEVMWNGAVSF-UHFFFAOYSA-N 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 229960000278 theophylline Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 150000003567 thiocyanates Chemical class 0.000 description 1
- 229960000103 thrombolytic agent Drugs 0.000 description 1
- 230000002537 thrombolytic effect Effects 0.000 description 1
- 230000003558 thrombophilic effect Effects 0.000 description 1
- 230000001732 thrombotic effect Effects 0.000 description 1
- 229960005001 ticlopidine Drugs 0.000 description 1
- PHWBOXQYWZNQIN-UHFFFAOYSA-N ticlopidine Chemical compound ClC1=CC=CC=C1CN1CC(C=CS2)=C2CC1 PHWBOXQYWZNQIN-UHFFFAOYSA-N 0.000 description 1
- FWPIDFUJEMBDLS-UHFFFAOYSA-L tin(II) chloride dihydrate Chemical compound O.O.Cl[Sn]Cl FWPIDFUJEMBDLS-UHFFFAOYSA-L 0.000 description 1
- 229960000187 tissue plasminogen activator Drugs 0.000 description 1
- 230000007838 tissue remodeling Effects 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M toluenesulfonate group Chemical group C=1(C(=CC=CC1)S(=O)(=O)[O-])C LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- 125000005490 tosylate group Chemical group 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 201000010875 transient cerebral ischemia Diseases 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- 229960005294 triamcinolone Drugs 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- QIWRFOJWQSSRJZ-UHFFFAOYSA-N tributyl(ethenyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C=C QIWRFOJWQSSRJZ-UHFFFAOYSA-N 0.000 description 1
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 1
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 1
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical class CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- ACWBQPMHZXGDFX-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=NN1 ACWBQPMHZXGDFX-QFIPXVFZSA-N 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- 230000003966 vascular damage Effects 0.000 description 1
- 208000021331 vascular occlusion disease Diseases 0.000 description 1
- 238000007631 vascular surgery Methods 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 229960005080 warfarin Drugs 0.000 description 1
- PJVWKTKQMONHTI-UHFFFAOYSA-N warfarin Chemical compound OC=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 PJVWKTKQMONHTI-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 229960004764 zafirlukast Drugs 0.000 description 1
- MWLSOWXNZPKENC-SSDOTTSWSA-N zileuton Chemical compound C1=CC=C2SC([C@H](N(O)C(N)=O)C)=CC2=C1 MWLSOWXNZPKENC-SSDOTTSWSA-N 0.000 description 1
- 229960005332 zileuton Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C257/00—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines
- C07C257/10—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines
- C07C257/18—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines having carbon atoms of amidino groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D223/00—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom
- C07D223/02—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D223/06—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom not condensed with other rings with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
Definitions
- the present invention relates to acid derivatives that are inhibitors of serine proteases such as Factor Nlla, Factor LXa, Factor Xa, Factor FXIa, tryptase, and urokinase. These acid derivatives are useful as anticoagulants in treating and preventing cardiovascular diseases, as anti-inflammatory agents, and as metastasis inhibitors in treating cancer.
- thrombin is a proteolytic enzyme that occupies a central position in the coagulation process. Thrombin catalyzes the conversion of fibrinogen to fibrin, is a key effector enzyme for blood clotting, and also is pivotal for other functions, such as activation of helper proteins (including Factors N and N ⁇ i and thrombomodulin), and its own activation.
- helper proteins including Factors N and N ⁇ i and thrombomodulin
- thrombotic diseases Disturbances in the natural balance between pro- and anti-coagulant forces may result in bleeding or thrombotic diseases.
- the series of reactions leading to thrombin production involve a number of coagulation factors present in the blood as precursors (e.g., Factors N ⁇ - XII).
- the coagulation factors are transformed into activated factors (e.g., Factors Vila, LXa, Xa, XIa, etc.).
- Factor Nil forms a complex with a membrane protein called tissue factor, to which Factor NLIa tightly binds.
- tissue factor to which Factor NLIa tightly binds.
- Factor NLIa is present as a complex bound to tissue factor.
- the coagulation factors and tissue factor complexes undergo an ordered chain of reactions that ultimately lead to conversion of Factor X to Factor Xa, and Factor Xa catalyzes the conversion of prothrombin to thrombin.
- An elevated plasma level of coagulation factors is a risk factor for fatal myocardial infarction and associated with coronary artery disease and other abnormalities of the coagulation system, e.g., thrombosis, ischemic vascular disease, intravascular clotting, stroke, embolisms, and so forth.
- antithrombotic agents have been researched and developed for use in treating cardiovascular and other diseases.
- Presently established antithrombotic agents include heparin, coumarin, and aspirin, among others. There are, however, limitations with these agents. For example, both heparin and coumarin have a highly-variable dose-related response, and their anticoagulant effects must be closely monitored to avoid a risk of serious bleeding.
- the present invention provides acid-based compounds useful as inhibitors of Factor Vila, Factor LXa, Factor Xa, Factor FXIa, tryptase, and urokinase. Summary of the Invention
- Acid derivatives are provided that are inhibitors of serine proteases having the Formula I:
- W is selected from C 2 -ioalkyl, C 2 - ⁇ oalkenyl, substituted C 2 . ⁇ oalkyl, substituted C 2 -
- ring B is phenyl or pyridyl
- X 2 is N, CH, or C, provided that X 2 is C when Ri and R 2 join to form a fully unsaturated ring;
- L is ⁇ (CR ⁇ 8 R 19 ) s -Y-(CR 18a R ⁇ 9a )t;
- Z is a 5 to 7-membered monocyclic or 8 to 11-membered bicyclic aryl, heteroaryl, heterocyclo, or cycloalkyl, wherein each Z group is optionally substituted with up to two R 2 o and/or up to one R 21 , except Z is not phenyl substituted with phenyloxy when W is methoxy, s is 0 and Y is -CH 2 -CH -; Ri and R 2 (i) are independently selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, heteroaryl, aryl, heterocyclo, and cycloalkyl; or (ii) are taken together to form an aryl, heteroaryl, cycloalkyl, or heterocyclo, provided that Ri and R 2 do not together form pyrazole when W is methoxy and Z is
- the compounds of this invention are smprisingly selective inhibitors of serine proteases.
- certain selections for the groups "Z-L-" in formula I provide compounds which are particularly selective for inhibition of one or more serine proteases versus other proteases.
- Z-L- is selected from:
- compounds of formula I are particularly selective for inhibition of FVLIa.
- compositions for treating a serine protease disease, an inflammatory or immune condition, or cancer comprising at least one compound of formula I or a pharmaceutically acceptable salt, hydrate or prodrug thereof, and a pharmaceutically acceptable carrier or diluent.
- methods of treating such diseases comprising administering to a mammal in need of such treatment at least one compound of formula I or a pharmaceutically acceptable salt, hydrate or prodrug thereof.
- compositions for use as anticoagulants during the preparation, use, storage, or fractionation of blood and methods of maintaining blood in the fluid phase during its preparation, use, storage, or fractionation.
- alkyl refers to straight or branched chain hydrocarbon groups having 1 to 12 carbon atoms, preferably 1 to 8 carbon atoms. Lower alkyl groups, that is, alkyl groups of 1 to 4 carbon atoms, are most preferred.
- C j.6 alkyl refers to straight and branched chain alkyl groups with one to six carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, t- butyl, n-pentyl, and so forth.
- R' and R" may together form a heterocyclo or heteroaryl ring.
- a substituted alkyl includes an aryl, heterocyclo, cycloalkyl, or heteroaryl substituent
- said ringed systems are as defined below and thus may have zero, one, two, or three substituents, also as defined below.
- alkyl when used in conjunction with another group, e.g., arylalkyl, hydroxyalkyl, etc., the term defines with more specificity a particular substituent that a substituted alkyl will contain.
- arylalkyl refers to a substituted alkyl group having from 1 to 12 carbon atoms and at least one aryl substituent
- lower arylalkyl refers to substituted alkyl groups having 1 to 4 carbon atoms and at least one aryl substituent.
- alkenyl refers to straight or branched chain hydrocarbon groups having 2 to 12 carbon atoms and at least one double bond. Alkenyl groups of 2 to 6 carbon atoms and having one double bond are most preferred.
- alkynyl refers to straight or branched chain hydrocarbon groups having 2 to 12 carbon atoms and at least one triple bond. Alkynyl groups of 2 to 6 carbon atoms and having one triple bond are most preferred.
- alkylene refers to bivalent straight or branched chain hydrocarbon groups having 1 to 12 carbon atoms, preferably 1 to 8 carbon atoms, e.g., ⁇ -CH 2 - ⁇ n , wherein n is 1 to 12, preferably 1-8. Lower alkylene groups, that is, alkylene groups of 1 to 4 carbon atoms, are most preferred.
- alkenylene and alkynylene refer to bivalent radicals of alkenyl and alknyl groups, respectively, as defined above. When reference is made to a substituted alkylene, alkenylene, or alkynylene group, these groups are substituted with one to three substitutents as defined above for alkyl groups.
- a ringed substituent of an alkyl, alkenyl, alkynyl, alkylene, alkenylene, or alkynylene may be joined at a terminal atom or an available intermediate (branch or chain) atom and thus may comprise, for example, the groups
- alkoxy refers to an alkyl group as defined above having one, two or three oxygen atoms (-O-) in the alkyl chain.
- alkoxy includes the groups -O-C ⁇ _ 12 alkyl, -Ci- ⁇ alkylene-O- -ealkyl, -C ⁇ - 4 alkylene-O-Ci- 4 alkylene-O-Ci- 4 alkyl, O-C ⁇ alkylene-O-C ⁇ alkylene-O-C j ⁇ alkyl, and so forth.
- alkylthio refers to an alkyl group as defined above bonded through one or more sulfur (-S-) atoms.
- alkylthio includes the groups -S-Cj. jj alkyl, -S 1.6 alkylene-S-C 1.6 alkyl, etc.
- alkylamino refers to an alkyl group as defined above bonded through one or more nitrogen (-NR-) groups.
- alkylamino refers to straight and branched chain groups and thus, for example, includes the groups -NH(C j _ 12 alk l) and -N(C w alkyl) z
- monovalent C j.2 alkylamino includes the groups -
- a lower alkylamino comprises an alkylamino having from one to four carbon atoms.
- the carbon atoms of said groups are substituted with one to three substituents as defined above for alkyl groups.
- the carbon and/or nitrogen atoms of these groups are substituted with one to three substitutents appropriately selected from the group of substituents recited above for alkyl groups.
- the alkoxy, alkylthio, or alkylamino groups may be monovalent or bivalent.
- a monovalent alkoxy includes groups such as - O-C j. ⁇ alkyl and -C 1 _ 6 alkylene-O-C 1 _ 6 alkyl
- a bivalent alkoxy includes groups such as -O-C 1 12 alkylene- and -C ⁇ g alkylene-O-C ⁇ g alkylene-, etc.
- heteroalkyl is used herein to refer saturated and unsaturated straight or branched chain hydrocarbon groups having 2 to 12 carbon atoms, preferably 2 to 8 carbon atoms, wherein one, two or three carbon atoms in the straight chain are replaced by a heteroatom (O, S or N).
- heteroalkyl includes alkoxy, alkylthio, and alkylamino groups, as defined above, as well as alkyl groups having a combination of heteroatoms selected from O, S, or N.
- heteroalkyl herein may be monovalent or bivalent, and for example, may comprise the groups -O- (CH 2 ) 2 -5NH-(CH 2 )2- or -O-(CH 2 )2- 5 NH-CH 3 , etc.
- a "substituted heteroalkyl” has one to three substituents appropriately selected from those recited above for alkyl groups.
- acyl refers to a carbonyl group ( p ) linked to an organic radical including an alkyl, alkenyl, alkynyl, substituted alkyl, substituted alkenyl, or substituted alkynyl group, as defined above.
- alkoxycarbonyl refers to a carboxy or ester group ( — c ° ) linked to an organic radical including an alkyl, alkenyl, alkynyl, substituted alkyl, substituted alkenyl, or substituted alkynyl group, as defined above.
- halo or halogen refers to chloro, bromo, fiuoro and iodo.
- haloalkyl means an alkyl having one or more halo substituents, e.g., including trifluoromethyl.
- haloalkoxy means an alkoxy group having one or more halo substituents.
- haloalkoxy includes -OCF 3 .
- sulfonyl refers to a sulphoxide group (i.e., -S(O) 1 . 2 -) linked to an organic radical including an alkyl, alkenyl, alkynyl, substituted alkyl, substituted alkenyl, or substituted alkynyl group, as defined above.
- the organic radical to which the sulphoxide group is attached may be monovalent (e.g., -SO 2 -alkyl), or bivalent
- sulfonamide refers to the group -S(O) 2 NR'R", wherein R' and R" may be hydrogen or alkyl, alkenyl, alkynyl, substituted alkyl, substituted alkenyl, or substituted alkynyl, as defined above. R' and R" may be monovalent or bivalent (e.g., -SO 2 -NH-alkylene, etc.)
- aryl refers to phenyl, biphenyl, 1-naphthyl and 2-naphthyl, with phenyl being preferred.
- said ring may in turn be substituted with one to three of halogen, haloalkyl, haloalkoxy, cyano, nitro, hydroxy, alkoxy, alkylthio, amino, alkylamino, phenyl, benzyl, phenyloxy, and benzyloxy.
- cycloalkyl refers to fully saturated and partially unsaturated hydrocarbon rings of 3 to 9, preferably 3 to 7 carbon atoms.
- a cycloalkyl When a cycloalkyl is substituted with a further ring, said ring may in turn be substituted with one to three of halogen, haloalkyl, haloalkoxy, cyano, nitro, hydroxy, alkoxy, alkylthio, amino, alkylamino, phenyl, benzyl, phenyloxy, and benzyloxy.
- heterocyclo refers to substituted and unsubstituted non-aromatic 3 to 7 membered monocyclic groups, 7 to 11 membered bicyclic groups, and ' 10 to 15 membered tricyclic groups which have at least one heteroatom (O, S or N) in at least one of the rings.
- Each ring of the heterocyclo group containing a heteroatom can contain one or two oxygen or sulfur atoms and/or from one to four nitrogen atoms, provided that the total number of heteroatoms in each ring is four or less, and further provided that the ring contains at least one carbon atom.
- the fused rings completing the bicyclic and tricyclic groups may contain only carbon atoms and may be saturated, partially saturated, or unsaturated.
- the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen atoms may optionally be quaternized.
- the heterocyclo group may be attached at any available nitrogen or carbon atom.
- a heterocyclo When a heterocyclo is substituted with a further ring, said ring may in turn be substituted with one to three of halogen, haloalkyl, haloalkoxy, cyano, nitro, hydroxy, alkoxy, alkylthio, amino, alkylamino, phenyl, benzyl, phenyloxy, and benzyloxy.
- Exemplary monocyclic groups include azetidinyl, pyrrolidinyl, oxetanyl, imidazolinyl, oxazolidinyl, isoxazolinyl, thiazolidinyl, isothiazolidinyl, tetrahydrofuranyl, piperidinyl, piperazinyl, 2-oxopiperazinyl, 2-oxopiperidinyl, 2- oxopyrrolodinyl, 2-oxoazepinyl, azepinyl, 4-piperidonyl, tetrahydropyranyl, mo ⁇ holinyl, thiamoipholinyl, thiamo ⁇ holinyl sulfoxide, thiamo ⁇ holinyl sulfone, 1,3-dioxolane and tetrahydro-lJ-dioxothienyl and the like.
- Exemplary bicyclic heterocyclo groups include quinuclidinyl.
- heteroaryl refers to substituted and unsubstituted aromatic 5 or 6 membered monocyclic groups, 9 or 10 membered bicyclic groups, and 11 to 14 membered tricyclic groups which have at least one heteroatom (O, S or N) in at least one of the rings.
- Each ring of the heteroaryl group containing a heteroatom can contain one or two oxygen or sulfur atoms and/or from one to four nitrogen atoms provided that the total number of heteroatoms in each ring is four or less and each ring has at least one carbon atom.
- the fused rings completing the bicyclic and tricyclic groups may contain only carbon atoms and may be saturated, partially saturated, or unsaturated.
- the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen atoms may optionally be quaternized.
- Heteroaryl groups which are bicyclic or tricyclic must include at least one fully aromatic ring but the other fused ring or rings may be aromatic or non-aromatic.
- the heteroaryl group may be attached at any available nitrogen or carbon atom of any ring.
- a heteroaryl When a heteroaryl is substituted with a further ring, said ring may in turn be substituted with one to three of halogen, haloalkyl, haloalkoxy, cyano, nitro, hydroxy, alkoxy, alkylthio, amino, alkylamino, phenyl, benzyl, phenyloxy, and benzyloxy.
- Exemplary monocyclic heteroaryl groups include pyrrolyl, pyrazolyl, pyrazolinyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, isothiazolyl, furanyl, thienyl, oxadiazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl and the like.
- Exemplary bicyclic heteroaryl groups include indolyl, benzothiazolyl, benzodioxolyl, benzoxaxolyl, benzothienyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuranyl, chromonyl, coumarinyl, benzopyranyl, cinnolinyl, quinoxalinyl, indazolyl, pyrrolopyridyl, furopyridinyl, dihydroisoindolyl, tetrahydroquinolinyl and the like.
- Exemplary tricyclic heteroaryl groups include carbazolyl, benzidolyl, phenanthrollinyl, acridinyl, phenanthridinyl, xanthenyl and the like.
- the term "carbocyclic” refers to optionally substituted aromatic or non- aromatic 3 to 7 membered monocyclic and 7 to 11 membered bicyclic groups, in which all atoms of the ring or rings are carbon atoms.
- metal ion refers to alkali metal ions such as sodium, potassium or lithium and alkaline earth metal ions such as magnesium and calcium, as well as zinc and aluminum.
- H rl2 bond appearing as a dash as in ⁇ "C — C j t should be understood that such bonds may be selected from single or double bonds, as appropriate given the selections for adjacent atoms and bonds.
- bonds linking Ri to X 2 and X 2 to C 6 are single bonds; and when X 2 is C, one of the bonds linking X 2 to an adjacent atom is a double bond, i.e., either a bond to R t or to C 6 is a double bond.
- a linker such as a methylene group
- linker group "L” is inserted into the formula I in the same direction set forth in the text.
- L is recited as -CH 2 -Y-
- the Y group is attached to the C 6 carbon atom i.e., to which X is
- salts form salts which are also within the scope of this invention. Unless otherwise indicated, reference to an inventive compound is understood to include reference to salts thereof.
- the term “salt(s)” denotes acidic and/or basic salts formed with inorganic and/or organic acids and bases.
- the term “salt(s) may include zwitterions (inner salts), e.g., when a compound of formula I contains both a basic moiety, such as an amine or a pyridine or imidazole ring, and an acidic moiety, such as a carboxylic acid.
- Salts of the compounds of the formula I may be formed, for example, by reacting a compound of the formula I with an amount of acid or base, such as an equivalent amount, in a medium such as one in which the salt precipitates or in an aqueous medium followed by lyophilization.
- Exemplary acid addition salts include acetates (such as those formed with acetic acid or trihaloacetic acid, for example, trifluoroacetic acid), adipates, alginates, ascorbates, aspartates, benzoates, benzenesulfonates, bisulfates, borates, butyrates, citrates, camphorates, camphorsulfonates, cyclopentanepropionates, digluconates, dodecylsulfates, ethanesulfonates, fumarates, glucoheptanoates, glycerophosphates, hemisulfates, heptanoates, hexanoates, hydrochlorides (formed with hydrochloric acid), hydrobromides (formed with hydrogen bromide), hydroiodides, 2- hydroxyethanesulfonates, lactates, maleates (formed with maleic acid), methanesulfonates (formed with methanesul
- Exemplary basic salts include ammonium salts, alkali metal salts such as sodium, lithium, and potassium salts; alkaline earth metal salts such as calcium and magnesium salts; barium, zinc, and aluminum salts; salts with organic bases (for example, organic amines) such as trialkylamines such as triethylamine, procaine, dibenzylamine, N-benzyl- ⁇ -phenethylamine, 1-ephenamine, N,N'-dibenzylethylene- diamine, dehydroabietylamine, N-ethylpiperidine, benzylamine, dicyclohexylamine or similar pharmaceutically acceptable amines and salts with amino acids such as arginine, lysine and the like.
- organic bases for example, organic amines
- trialkylamines such as triethylamine, procaine, dibenzylamine, N-benzyl- ⁇ -phenethylamine, 1-ephenamine, N,N'-d
- Basic nitrogen-containing groups may be quaternized with agents such as lower alkyl halides (e.g., methyl, ethyl, propyl, and butyl chlorides, bromides and iodides), dialkyl sulfates (e.g., dimethyl, diethyl, dibutyl, and diamyl sulfates), long chain halides (e.g., decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides), aralkyl halides (e.g., benzyl and phenethyl bromides), and others.
- Preferred salts include monohydrochloride, hydrogensulfate, methanesulfonate, phosphate or nitrate.
- Prodrags and solvates of the inventive compounds are also contemplated.
- the term "prodrug” denotes a compound which, upon administration to a subject, undergoes chemical conversion by metabolic or chemical processes to yield a compound of the formula I, and/or a salt and/or solvate thereof.
- Various forms of prodrags are well known in the art. For examples of such prodrug derivatives, see: a) Design of Prodrugs. edited by H. Bundgaard, (Elsevier, 1985) and Methods in Enzymology, Nol.42, p. 309-396, edited by K. Widder, et al.
- prodrags comprise compounds wherein the upper ring substituent -CO R 3 is a group that will hydrolyze in the body to compounds where said substituent is -CO 2 H.
- prodrugs are preferably administered orally since hydrolysis in many instances occurs principally under the influence of the digestive enzymes. Parenteral administration may be used where the ester per se is active, or in those instances where hydrolysis occurs in the blood.
- physiologically hydrolyzable esters of compounds of formula I include C h alky-benzyl, 4-methoxybenzyl, indanyl, phthalyl, methoxymethyl, C ⁇ g alkanoyloxy-C ⁇ g alkyl, e.g. acetoxymethyl, pivaloyloxymethyl or propionyloxymethyl, C ⁇ g alkoxycarbonyloxy-C ⁇ g alkyl, e.g.
- esters used, for example, in the penicillin and cephalosporin arts. Such esters may be prepared by conventional techniques known in the art.
- inventive compounds may exist in their tautomeric form, in which hydrogen atoms are transposed to other parts of the molecules and the chemical bonds between the atoms of the molecules are consequently rearranged. It should be understood that the all tautomeric forms, insofar as they may exist, are included within the invention. Additionally, inventive compounds may have trans and cis isomers and may contain one or more chiral centers, therefore existing in enantiomeric and diastereomeric forms. The invention includes all such isomers, as well as mixtures of cis and trans isomers, mixtures of diastereomers and racemic mixtures of enantiomers (optical isomers).
- any one of the isomers or a mixture of more than one isomer is intended.
- the processes for preparation can use racemates, enantiomers or diastereomers as starting " materials.
- enantiomeric or diastereomeric products are prepared, they can be separated by conventional methods for example, chromatographic or fractional crystallization.
- the compounds of the instant invention may, for example, be in the free or hydrate form, and may be obtained by methods exemplified by the following descriptions.
- X 2 is N, CH, or C, provided that X 2 is C when Ri and R 2 join to form a fully unsaturated ring;
- L is -(CH 2 ) S -Y-;
- Ri and R 2 are independently selected from hydrogen, lower alkyl, aryl and arylalkyl; or (ii) are taken together to form an aryl, heteroaryl, cycloalkyl, or heterocyclo; wherein when Ri and R 2 individually or together form a heteroaryl, aryl, heterocyclo or cycloalkyl, said cyclic group is optionally substituted with up to two R 26 ;
- R- t is hydrogen or lower alkyl
- R 5 is hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, heterocyclo or heteroaryl;
- R 6 is selected from C ⁇ - 6 alkyl, more preferably C 2 - 6 alkyl, phenyl, and benzyl;
- R 3 o at each occurrence is selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, cycloalkyl, and phenyl;
- R 3 ⁇ and R 32 at each occurrence are independently selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, and cycloalkyl; m and n are independently 0, 1 or 2; and s is 0, 1 or 2.
- More preferred compounds are those compounds having the formulae:
- X 3 is CH or N
- R 5 is Ci- 6 alkyl, -CH(CH 2 OH)C(CH 3 ) 3 , or C ⁇ - 2 alkyl substituted with C 5 - 6 cycloalkylene; either (a) s is 0 and Z is selected from
- R26 s C 2 - 6 straight or branched alkenyl, -OR 0 or -NR 3 ⁇ R 32 , and R 27 is hydrogen, or R 26 and R 27 together form a fused benzo ring;
- R 3 o is C1- 5 straight or branched chain alkyl, C 2 - 6 straight or branched alkenyl, C 3 -
- R 3 ⁇ and R 2 are selected from hydrogen and lower alkyl.
- Z is selected from:
- R 5 is hydrogen or hydroxymethyl; and R 26 is C ⁇ - 3 alkoxy or NH(Ci- 4 alkyl).
- the compounds of the invention may be prepared by the exemplary processes described in the following Schemes A through D. Methods for making intermediates including appropriately-protected amine-coupling components are shown in Schemes E through G and I through X, and Scheme H shows a method for making an unprotected amine-coupling component. These amines may be coupled to substrates to make compounds of formula I and deprotected, when necessary or desired, as shown in Schemes A-D and the Examples. Exemplary reagents and procedures for these reactions appear hereinafter and in the working Examples. Starting materials are commercially available or can be readily prepared by one of ordinary skill in the art as shown herein or as described in the literature. For all of the schemes, the groups Ri-R 2 , W, X, Z, r, s etc., are as described herein for a compound of formula I, unless otherwise indicated.
- Compounds of formula la can be made by reacting acid 1 with an amine having the desired group Z, i.e., Z-NHR 13 .
- the 2-position acid group is suitably protected (P'), and the reaction is carried out in the presence of coupling reagent(s) such as DCC/HOBT/DMAP, EDC/DMAP, or DIC/HOAT to afford the corresponding amide compound.
- the group P' optionally may be deprotected to afford the compound of formula la wherein R 3 is hydrogen, or the group P' may be retained wherein P' comprises the desired group R 3 .
- the group P' may be deprotected to afford the group CO 2 H, with the group CO 2 H then converted to another desired R 3 group.
- the compound having the acid group CO 2 H may be reacted with a halide having the desired R 3 group, i.e., X-R 3 where X is Cl, Br, or I, in the presence of base, or the acid compound may be coupled with an alcohol such as R 3 OH in a coupling reagent. It may be necessary or desired to protect additional functional groups besides the 2-position acid before performing the coupling reaction, as one skilled in the field will appreciate. Those additional protecting groups can be removed after the coupling using appropriate deprotecting conditions. Preparation of acids 1 , wherein R ⁇ and R 2 together form an unsaturated carbocyclic or heterocyclic ring, is described in WO 99/041231, inco ⁇ orated herein by reference, and described in the Examples that appear hereinafter.
- the aldehyde 2, wherein the 2-position acid group is suitably protected (P') can be coupled with an amine Z-NHR 13 in the presence of a reducing reagent such as sodium triacetoxyborohydride, to afford the corresponding amine compound having the group CO 2 P'.
- a reducing reagent such as sodium triacetoxyborohydride
- the compound of formula lb is provided, having the desired group R 3 .
- Preparation of aldehydes 2, wherein Ri and R 2 together form an unsaturated carbocyclic or heterocyclic ring is described in WO 99/041231, inco ⁇ orated herein, and further shown in the Examples hereinafter.
- Aryl fluoride 3a is reacted with amine 4 in DMSO in the presence of a base such as DLEA to afford intermediate 5.
- a base such as DLEA
- triflate 3b is reacted with amine 4 in the presence of a suitable palladium reagent to afford intermediate 5.
- Selective deprotection of the P" group of compound 5 affords acid 6.
- Acid 6 is reacted with an appropriate amine in the presence of suitable coupling reagents.
- Compounds having the desired group R 3 are obtained as described in Scheme A, i.e., by optional deprotection, further reaction or coupling, to afford compounds of formula Lla, above.
- Aryl fluoride 3a is reacted with amine 7 in DMSO in the presence of a base such as DLEA to afford compound 8, where Ri is defined as above except where Ri and R form a ring, the ring is a heterocyclo.
- a base such as DLEA
- Selective deprotection of the P" group affords acid 9.
- Reaction of acid 9 with an amine Z-NHR ⁇ 3 in the presence of coupling reagent(s) such as DCC/HOBT/DMAP, EDC/DMAP, or DIC/HOAT affords the corresponding amide compound.
- coupling reagent(s) such as DCC/HOBT/DMAP, EDC/DMAP, or DIC/HOAT affords the corresponding amide compound.
- Compounds having the desired group R 3 are obtained as described in Scheme A, i.e., by optional deprotection, further reaction or coupling, to afford compounds of formula Lib, above.
- Compound 10 was prepared according to J. Med. Chem., Vol. 42 (1999), at pp. 3510-3519, from 2-methyl-4-nitroaniline. A mixture of compound 10 and 1-(1,1- dimethylethoxy)-N,N, N', N'-tetramethyl-methanediamine in dry DMF (10 mL) was stirred at 70°C for 2h under N 2 . After cooling to rt, the reaction mixture was treated with hexane, and the solid was collected by filtration and washed with hexane to give compound JJ as black crystals. Compound ⁇ was converted to compound 13 in two alternate ways. In one approach, compound ⁇ was converted to 13 .
- compound JJ was converted to 13 by first mixing compound JJ and 2,4-dimethoxylbenzylamine in DMF and stirring the mixture at 140°C for 3h. The solvent was removed by vacuum distillation and residue treated with EtOAc. The orange solid was collected by filtration and washed with hexane to give compound 12. To a solution of compound Y ⁇ in anisole was added TFA. The reaction mixture was stirred at 90°C for lh and the solvent removed under reduced pressure. The residue was treated with sat'd NaHCO 3 (30 mL) and the product collected by filtration and washed with water to afford compound 13.
- Compound 20 was synthesized from compound 19 following the procedure described in Osborn, et al., J.Chem. Soc. (1956), at 4191, and compounds 21_a and 21b were prepared according to Poradowska et al., Synthesis, (1975), at p. 732.
- Compound 2 a and phthalic acid anhydride 22 were powdered and mixed well. Heating the mixture for 2 h at 130 to 150°C and finally 2 min to 220°C finished the reaction. The cooled solid material of crude compound 23 was powdered and washed with ether/DCM (10:1) and dried yielding compound 23 as a beige powder.
- Nitro and carboxylic acid starting materials (e.g. 48) were dissolved in DCM and N.N-DMF (10:1). lJ'-carbonyldiimidazole (1.2 equiv) was added, and the reaction stirred at rt for 5 h. Ammonium hydroxide (2 equiv) was then added. After stirring overnight at rt, the reaction was concentrated, washed with base and extracted with EtOAc to yield compound 49. Compound 49 was then hydrogenated at 40 psi on the PARR shaker in the presence of Pd/C catalyst. Filtration and concentration yielded the appropriate Z-amine coupling s same or similar
- the resulting compound 54 was dissolved in EtOAc and Pd/C catalyst was added. The mixture was placed on the PARR shaker at 40 psi for 2h. Filtering off the catalyst yielded the desired compound 55 in 80% yield.
- the inventive compounds are inhibitors of the activated coagulation serine proteases known as Factor Vila, Factor LXa, Factor Xa, Factor Xla, and thrombin and also inhibit other serine proteases, such as trypsin, tryptase, and urokinase.
- the compounds are useful for treating or preventing those processes, which involve the production or action of Factor NLIa, Factor LXa, Factor Xa, Factor Xla, thrombin, trypsin, and/or tryptase.
- they are useful as metastasis inhibitors in treating cancer.
- the term “treating” or “treatment” encompasses prevention, partial alleviation, or cure of the disease or disorder.
- inventive compounds are useful in treating consequences of atherosclerotic plaque rupture including cardiovascular diseases associated with the activation of the coagulation cascade in thrombotic or thrombophilic states.
- cardiovascular diseases associated with the activation of the coagulation cascade in thrombotic or thrombophilic states.
- diseases include arterial thrombosis, coronary artery disease, acute coronary syndromes, myocardial infarction, unstable angina, ischemia resulting from vascular occlusion cerebral infarction, stroke and related cerebral vascular diseases (including cerebrovascular accident and transient ischemic attack).
- the compounds are useful in treating or preventing formation of atherosclerotic plaques, transplant atherosclerosis, peripheral arterial disease and intermittent claudication.
- the compounds can be used to prevent restenosis following arterial injury induced endogenously (by rapture of an atherosclerotic plaque), or exogenously (by invasive cardiological procedures such as vessel wall injury resulting from angioplasty).
- inventive compounds are useful in preventing venous thrombosis, coagulation syndromes, deep vein thrombosis (DNT), disseminated intravascular coagulopathy, Kasabach-Merritt syndrome, pulmonary embolism, cerebral thrombosis, atrial fibrillation, and cerebral embolism.
- DNT deep vein thrombosis
- pulmonary embolism cerebral thrombosis
- atrial fibrillation and cerebral embolism.
- the compounds are useful in treating peripheral arterial occlusion, thromboembolic complications of surgery (such as hip replacement, endarterectomy, introduction of artificial heart valves, vascular grafts, and mechanical organs), implantation or transplantation of organ, tissue or cells, and thromboembolic complications of medications (such as oral contraceptives, hormone replacement, and heparin, e.g., for treating heparin-induced thrombocytopenia).
- the inventive compounds are useful in preventing thrombosis associated with artificial heart valves, stents, and ventricular enlargement including dilated cardiac myopathy and heart failure.
- the compounds are also useful in treating thrombosis due to confinement (i.e. immobilization, hospitalization, bed rest etc.).
- These compounds are also useful in preventing thrombosis and complications in patients genetically predisposed to arterial thrombosis or venous thrombosis (including activated protein C resistance, FNieiden, Prothrombin 20210, elevated coagulation factors FNFI, FNHI, FLX, FX, FXI, prothrombin, TAFI and fibrinogen), elevated levels of homocystine, and deficient levels of antithrombin, protein C, and protein S.
- the inventive compounds may be used for treating heparin- intolerant patients, including those with congenital and acquired antithrombin in deficiencies, heparin-induced thrombocytopenia, and those with high levels of polymo ⁇ honuclear granulocyte elastase.
- the present compounds may also be used to inhibit blood coagulation in connection with the preparation, storage, fractionation, or use of whole blood.
- the compounds may be used to maintain whole and fractionated blood in the fluid phase such as required for analytical and biological testing, e.g., for ex vivo platelet and other cell function studies, bioanalytical procedures, and quantitation of blood-containing components.
- the compounds may be used as anticoagulants in extraco ⁇ eal blood circuits, such as those necessary in dialysis and surgery (such as coronary artery bypass surgery); for maintaining blood vessel patency in patients undergoing transluminal coronary angioplasty, vascular surgery including bypass grafting, arterial reconstruction, atherectomy, vascular graft and stent patency, tumor cell metastasis, and organ, tissue, or cell implantation and transplantation.
- extraco ⁇ eal blood circuits such as those necessary in dialysis and surgery (such as coronary artery bypass surgery); for maintaining blood vessel patency in patients undergoing transluminal coronary angioplasty, vascular surgery including bypass grafting, arterial reconstruction, atherectomy, vascular graft and stent patency, tumor cell metastasis, and organ, tissue, or cell implantation and transplantation.
- the inventive compounds are useful as anti-inflammatory agents, in treating chronic asthma, allergic rhinitis, inflammatory bowel disease, psoriasis, conjunctivitis, atopic dermatitis, pancreatis, rheumatoid arthritis, osteoarthritis, septic shock, and chronic inflammatory joint diseases, diseases of joint cartilage destruction, and/or vascular damage due to bacterial and/or viral infections.
- the inventive compounds may be useful for treating diabetic retinopathy or motor neuron diseases such as amyotrophic lateral sclerosis, progressive muscular atrophy, and primary lateral sclerosis.
- the inventive compounds may be useful for tissue remodeling diseases and for treating plaque instability and sequelli.
- these compounds may be useful for treating fibrotic diseases and conditions, for example, fibrosis, scleroderma, pulmonary fibrosis, liver cirrhosis, myocardial fibrosis, neurofibromas, and hypertrophic scars.
- the compounds of the present invention are useful in treating cancer and preventing the prothrombotic complications of cancer.
- the compounds are useful in treating tumor growth, as an adjunct to chemotherapy, and for treating diseases involving metastases including, but not limited to cancer, more particularly, cancer of the lung, prostate, colon, breast, ovaries, and bone. These compounds may also be useful in preventing angiogenesis.
- inventive compounds may also be used in combination with other antithrombotic or anticoagulant drugs such as thrombin inhibitors, platelet aggregation inhibitors such as aspirin, clopidogrel, ticlopidine or CS-747, warfarin, low molecular weight heparins (such as LONENOX), GPIIb/GPLIIa blockers, PAI-1 inhibitors such as XR-330 and T-686, inhibitors of ⁇ -2-antiplasmin such as anti- ⁇ -2- antiplasmin antibody and thromboxane receptor antagonists (such as ifetroban), prostacyclin mimetics, phosphodiesterase (PDE) inhibitors, such as dipyridamole or cilostazol, PDE inhibitors in combination with thromboxane receptor antagonists/thiOmboxane A synthetase inhibitors (such as picotamide), serotonin-2- receptor antagonists (such as ketanserin), fibrinogen receptor antagonists, hypolipidemic agents,
- microsomal triglyceride transport protein inhibitors such as disclosed in U.S. Patent Nos. 5,739,135, 5,712,279 and 5,760,246), antihypertensive agents such as angiotensin-converting enzyme inhibitors (e.g., captopril, lisinopril or fosinopril); angiotensin-JJ receptor antagonists (e.g., irbesartan, losartan or valsartan); and/or ACE/NEP inhibitors (e.g., omapatrilat and gemopatrilat); ⁇ -blockers (such as propranolol, nadolol and carvedilol), PDE inhibitors in combination with aspirin, ifetroban, picotamide, ketanserin, or clopidogrel and the like.
- microsomal triglyceride transport protein inhibitors such as disclosed in U.S. Patent Nos. 5,739,135, 5,712,2
- inventive compounds are also useful in combination with anti-arrhythmic agents such as for atrial fibrillation, for example, amiodarone or dofetilide.
- the inventive compounds may be used in combination with prothrombolytic agents, such as tissue plasminogen activator (natural or recombinant), streptokinase, reteplase, activase, lanoteplase, urokinase, prourokinase, anisolated streptokinase plasminogen activator complex (ASP AC), animal salivary gland plasminogen activators, and the like.
- tissue plasminogen activator natural or recombinant
- streptokinase reteplase
- activase lanoteplase
- urokinase prourokinase
- anisolated streptokinase plasminogen activator complex ASP AC
- animal salivary gland plasminogen activators and the like.
- inventive compounds may also be used in combination with ⁇ -adrenergic agonists such as albuterol, terbutaline, formoterol, salmeterol, bitolterol, pilbuterol, or fenoterol; anticholinergics such as ipratropium bromide; anti-inflammatory cortiocosteroids such as beclomethasone, triamcinolone, budesonide, fluticasone, flunisolide or dexamethasone; and anti-inflammatory agents such as cromolyn, nedocromil, theophylline, zileuton, zafirlukast, monteleukast and pranleukast.
- ⁇ -adrenergic agonists such as albuterol, terbutaline, formoterol, salmeterol, bitolterol, pilbuterol, or fenoterol
- anticholinergics such as ipratropium bromide
- inventive compounds may also be useful in combination with other anticancer strategies and chemotherapies such as taxol and/or cisplatin.
- the compounds may act synergistically with one or more of the above agents.
- the inventive compounds may act synergistically with the above agents to prevent reocclusion following a successful thrombolytic therapy and/or reduce the time to reperfusion.
- reduced doses of thrombolytic agent(s) may be used, therefore minimizing potential hemorrhagic side effects.
- the compounds of formula I may be administered by any means suitable for the condition to be treated, which may depend on the need for site-specific treatment or quantity of drug to be delivered.
- Systematic treatment is typically preferred for cancerous conditions, although other modes of delivery are contemplated.
- the compounds may be delivered orally, such as in the form of tablets, capsules, granules, powders, or liquid formulations including syrups; sublingually; bucally; transdermally; parenterally, such as by subcutaneous, intravenous, intramuscular or intrasternal injection or infusion (e.g., as sterile injectable aqueous or non-aqueous solutions or suspensions); nasally such as by inhalation spray; rectally such as in the form of suppositories, or in the form of liposome particles.
- Dosage unit formulations containing non-toxic, pharmaceutically acceptable vehicles or diluents may be administered.
- the compounds may be administered in a form suitable for immediate release or extended release. Immediate release or extended release may be achieved with suitable pharmaceutical compositions or, particularly in the case of extended release, with devices such as subcutaneous implants or osmotic pumps.
- compositions for oral administration include suspensions which may contain, for example, microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners or flavoring agents such as those known in the art; and immediate release tablets which may contain, for example, microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and/or lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants such as those known in the art.
- the inventive compounds may be orally delivered by sublingual and/or buccal administration, e.g., with molded, compressed, or freeze-dried tablets.
- compositions may include fast-dissolving diluents such as mannitol, lactose, sucrose, and/or cyclodextrins.
- fast-dissolving diluents such as mannitol, lactose, sucrose, and/or cyclodextrins.
- high molecular weight excipients such as celluloses (ANICEL®) or polyethylene glycols (PEG); an excipient to aid mucosal adhesion such as hydroxypropyl cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), sodium carboxymethyl cellulose (SCMC), and/or maleic anhydride copolymer (e.g., GA ⁇ TREZ®); and agents to control release such as polyacrylic copolymer (e.g., CARBOPOL 934®).
- Lubricants, glidants, flavors, coloring agents and stabilizers may also be added for ease of fabrication and use.
- compositions for nasal aerosol or inhalation administration include solutions which may contain, for example, benzyl alcohol or other suitable preservatives, abso ⁇ tion promoters to enhance abso ⁇ tion and/or bioavailability, and/or other solubilizing or dispersing agents such as those known in the art.
- compositions for parenteral administration include injectable solutions or suspensions which may contain, for example, suitable non-toxic, parenterally acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- suitable non-toxic, parenterally acceptable diluents or solvents such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- compositions for rectal administration include suppositories which may contain, for example, suitable non-irritating excipients, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures but liquefy and/or dissolve in the rectal cavity to release the drug.
- suitable non-irritating excipients such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures but liquefy and/or dissolve in the rectal cavity to release the drug.
- the effective amount of a compound of the present invention may be determined by one of ordinary skill in the art.
- the specific dose level and frequency of dosage for any particular subject may vary and will depend upon a variety of factors, including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the species, age, body weight, general health, sex and diet of the subject, the mode and time of administration, rate of excretion, drag combination, and severity of the particular condition.
- An exemplary effective amount of compounds of formula I may be within the dosage range of about 0.1 to about 100 mg/kg, preferably about 0.2 to about 50 mg/kg and more preferably about 0.5 to about 25 mg/kg (or from about 1 to about 2500 mg, preferably from about 5 to about 2000 mg) on a regimen in single or 2 to 4 divided daily doses.
- Compound was prepared as a 5 mM stock in DMSO, diluted further in DMSO and added directly to the assays.
- the DMSO concentration for all these studies was less than 1% and compared to DMSO vehicle controls.
- Human Factor NLIa Human Factor NLIa was obtained from Enzyme Research Labs (Cat.# HFNJJA 1640). Human recombinant tissue factor (L ⁇ ONL ⁇ from Dade Behring Cat.# B4212-100; "20 ml vial”) was diluted with 8 ml of H 2 O per vial and diluted further 1:30 into the 302 ⁇ l final assay volume.
- Tissue factor activated FNLIa enzymatic activity was measured in a buffer containing 150 mM ⁇ aCl, 5mM CaCl , 1 mM CHAPS and 1 mg/ml PEG 6000 (pH 7.4) with 1 nM FNLIa and 100 ⁇ M D-he-Pro- Arg-AFC (Enzyme Systems Products, Km > 200 ⁇ M) 0.66% DMSO.
- the assay (302 ⁇ l total volume) was incubated at RT for 2 hr prior to reading fluorome-ric signal (Ex 405 / Em 535) using a Victor 2 (Wallac) fluorescent plate reader.
- Tryptase inhibition activity was measured using either isolated human skin tryptase or recombinant human tryptase prepared from the human recombinant beta- protryptase expressed by baculovirus in insect cells.
- the expressed beta-protryptase was purified using sequential immobilized heparin affinity resin followed by an immunoaffinity column using an anti-tryptase monoclonal antibody.
- the protryptase was activated by auto-catalytic removal of the N-terminal in the presence of dextran sulfate followed by dipeptidyl peptidase I (DPPI) removal of the two N-terminal amino acids to give the mature active enzyme (Sakai et al, J. Clin.
- DPPI dipeptidyl peptidase I
- the substrate (10 ⁇ l) was then added to each well to give a final concentration of 100 ⁇ M benzyloxycarbonyl-glycine-proline- arginine-p-nitroaniline (CBz-Gly-Pro-Arg-pNA).
- the microplate was then shaken on a platform vortex mixer at a setting of 800 (Sarstedt TPM-2). After a total of three minutes incubation, 10 ⁇ l of the working stock solution of tryptase was added to each well. The microplate was vortexed again for one minute and then incubated without shaking at RT for an additional 2 minutes.
- the microplate was read on a microplate reader (Molecular Devices UN max) in the kinetic mode (405 nm wavelength) over twenty minutes at RT.
- IC 50 the fraction of control activity
- FCA fraction of control activity
- the inventive compounds demonstrated activity as inhibitors of Factors NLIa, LXa, Xa, Xla, LXa, tryptase and/or urokinase.
- Ph phenyl
- Boc tert-butoxycarbonyl
- CBZ carbobenzyloxy or carbobenzoxy or benzyloxycarbonyl
- NMM N-methyl mo ⁇ holine
- NaHCO 3 sodium bicarbonate
- NaBH(OAc) 3 sodium triacetoxyborohydride
- EDC palladium on carbon
- EDC or EDC.HCl
- EDCI or EDCI.HCl
- EDAC 3-ethyl-3'- (dimethylamino)propyl- carbodiimide hydrochloride (or l-(3-dimethylaminopropyl)- 3-ethylcarbodiimide hydrochloride)
- HOBT or HOBT.H 2 O 1-hydroxybenzotriazole hydrate
- HOAT l-Hydroxy-7-azabenzotriazole
- DIAD diisopropyl azodicarboxylate
- DLEA diisopropylethylamine
- TBS t-butyldimethylsilyl
- Tf trifluoromethanesulfonyl
- Step 3B was repeated using 26 mg (0.032 mmol) of compound 3A to make another 13 mg of crude compound 3B, and the combined crade product (19 mg and 13 mg) was chromatographed over silica gel using 10% and then 15% MeOH in methylene chloride to give 22 mg of pure compound 3B as a glassy solid.
- Step C A mixture of compound 3B (19 mg, 0.031 mmol), 2.5 mL of MeOH, and 33 ⁇ L of 1.0 N HC1 was hydrogenated for 20 min. in the presence of 6 mg of 10% Pd/C. After filtration and concentration of the filtrate, the residue was lyophilized - , resort _-
- the compound of Example 8 was prepared in the same manner described above for Example 7, and the compounds of Examples 9- 11 were prepared in the manner described above for Example 3, using an appropriate amine-coupling component.
- Compounds of Examples 12-27 were synthesized via automation using a TEC AN liquid handler for reagents and starting material addition and Procedures A and B below.
- the desired amine-coupling components were prepared as set forth in the previous schemes, as known in the field, and/or as set forth in the literature, i.e., see, e.g., Yatsunami et al, S. Eur. Pat. Appl. No.
- reaction mixtures were purified via solid phase extraction using a SCX cation exchange column (CUBCXHL5R3, 500MG/3ML/50PKG) as follows: 1). Columns were conditioned with 1.5 mL of 1:1 CH 3 CN-iPr 2 OH solution; 2). Reaction mixture (1 mL) was loaded on to SCX columns; and 3). Columns were eluted with 1.5 mL of 1: 1 CH 3 CN-iPr OH solution into 9 mm x 80 mm microtubes. The CH 3 CN-iPr 2 OH solutions were concentrated using a speed vac for 12h.
- SCX cation exchange column CUBCXHL5R3, 500MG/3ML/50PKG
- reaction mixtures were purified via solid phase extraction using a SCX cation exchange column (CUBCXHL5R3, 500MG/3ML/50PKG) as follows: 1). Columns were conditioned with 1.5 mL of 1:1 CH 3 CN-iPr 2 OH solution; 2). Reaction mixture (1 mL) was loaded on to SCX columns; 3). Columns were rinsed with 1.5 mL of a 1:1 CH 3 CN-iPr 2 OH solution; and
- Example 54 The resulting product was purified by prep HPLC.
- the tert-butyl protecting group was removed by treating the compound with TFA in DCM.
- the trifluoro acetamide protecting group was removed by dissolving the compound in DMSO and adding 10 eq. of solid NaOH. The mixture was stirred for 12 h at rt and purified by prep HPLC to give Example 54.
- Step C The process of Step C, above, was followed, adding 3.65 mL of 2-methyl-2- butene to a solution of compound 70E (2.71 g, 6.06 mmol) in 33 mL of tBuOH:CH 3 CN:H 2 O (6: 1:2), and using 2.36 g (26.04 mmol) of NaClO 2 ,L00 g (7.27 mmol) of NaH 2 PO 4 H O, and stirring the reaction for 30 min before adding cold H 2 O (90 mL). The aq. solution was extracted with EtOAc (150 mL), and the organic extract washed with H 2 O (2x) and sat. NaCl (2x), dried (MgSO ), filtered, and cone, to give 2.85 g (100%) of compound 70F.
- Step I Procedures for Examples 70-160 having Formula Ik, above (Table 8)
- a TEC AN liquid handler was used to add 35 ⁇ L of DMF to each of a number of reaction tubes (12 mm x 65 mm) in a mini -reactor. Then, to each reaction tube was added 144 ⁇ L (0.036 mmol) of a 2.5 M solution of an amine in DMF, the amine having the desired groups R 4 and R 5 . Insoluble amines were added by hand. 5J ⁇ L (0.037 mmol) of TEA was added by hand to any tubes which contained an amine salt. The TECAN was then used to add 150 ⁇ L (18 mg, 0.030 mmol) of a stock solution of scaffold Compound 70H in DMF. This addition was followed by 150 ⁇ L of a stock solution containing 5.2 ⁇ L (0.033 mmol) of DIC and 4.5 mg (0.033 mmol) of HO At in DMF. The tubes were sealed and shaken for 24 h.
- reaction mixtures were purified by solid phase extraction using a SCX cation exchange column (CUBCXHL5R3, 500 MG/3 ML/ 50 PKG) as follows:
- Example 172 was purified with HPLC using YMC S5 ODS 20X100 column to give 53 mg of Example 172 as a white lyophilate.
- MS, m z (M+l) + 506.
- 1H-NMR 500 MHz), ⁇ 9.86 (s, IH, NH), 8.13 (s, IH, ArH), 8.01 (S, IH, ArH), 7.72 (dd, IH, ArH), 6.67 (d, IH, ArH), 7.53 (d, IH, ArH), 7.45 (d, IH, ArH), 7.35 (dd, IH, ArH), 7.28 (d, IH, ArH), 6.96 (d, IH, ArH), 5.36 (s, IH, OCHO), 4.04 (s, 3H, OCH 3 ), 3.79 (d, 2H, OCH 2 ), 3.68 (d, 2H, OCH 2 ), 1.28 (s, 3H, CH 3 ), 0.80 (s, 3H, CH 3 ).
- the aldehyde from Step D was oxidized to a carboxylic acid by dissolving 115 mg (0.27 mmol) of the aldehyde, 97 mg (1.07 mmol) of sodium chlorite, and 0J3 mL of (1.21 mmol) 2-methyl-2-butene in 15 mL t-BuOH/CH 3 CN/H 2 O (6: 1:2) at 0°C. After a solution formed, 56 mg (0.40 mmol) of NaH 2 PO4*H 2 O was added. The reaction mixture was stirred for 2 h at rt and diluted with water. The organic layer was extracted with EtOAc, dried and concentrated to give a quantitative yield of the desired acid.
- Example 173 The acid was then coupled to the 1 -amino isoquinoline amine coupling component and deprotected as described in Example 171, step F, to give Example 173.
- 1H NMR (500 MHz) confirmed the cis product: ⁇ 8.25 (s, IH, ArH), 8.24 (d, IH, ArH), 7.90 (s, IH, ArH), 7.85 (d, IH, ArH), 7.53 (d, IH, ArH), 7.38 (d, IH, ArH), 7.37 (d, IH, ArH), 7J4 (d, IH, ArH), 6.99 (d, IH, ArH), 6.97 (d, IH, ' ArH), 6.45 (d, IH, CH), 5.74 (m, IH, CH), 4.05 (s, 3H, OCH 3 ), 2.20 (m, 2H, CH 2 ), 1.66 (m, IH, CH), 0.88 (d, 6H, 2CH 3 ).
- Example 174C was converted to the corresponding 2-carboxylic acid using standard conditions. The acid was then coupled to the aminobenzimidazole amine- coupling component as in Example 70, step 70b, and deprotected using the DCM LLFA, (1 : 1) and saponification to give Example 174.
- Example 175 100 mg (0.33 mmol) of the aldehyde from Step A was oxidized to a carboxylic acid using conditions as described in Example 173, Step F. The acid was coupled to the amino benzimidazole amine-coupling component and deprotected following step D of Example 174 to give Example 175.
- Compound 176B was hydrogenated at 40 psi in the presence of Pd/C catalyst overnight to remove the benzyl group to provide compound 177 A.
- Step B Example 177
- Step F Example 179
- Trifluoroacetic anhydride (1.4 mL, 10 mmol) was added to compound 191D (443 mg, 1 mmol) in 3 mL of DMF and the solution was stirred for 19 h at rt. The solvent was removed in vacuo and the residue taken up in EtOAc. The EtOAc was washed with water (2x), cautiously with dilute NaHCO 3 (2x), and water (2x), dried (sodium sulfate), and concentrated to give 525 mg of crade compound 191E as a yellow oil.
- Compound 194 A was pre pared from (3 -hydroxybenzaldehyde) as described: G. M. Kesera, et al., Tetrahedron, Nol. 48 (1992), at pp. 913-922.
- Example 195 Treatment of Example 195 (10 mg, 0.016 mmol) in 1 mL of MeOH and 0.4 mL of ether with excess etheral diazomethane for 1 h provided 9.3 mg of Example 196 as a glassy residue.
- Step E Example 201
- Compound 207A was prepared using the general procedure for the stannylation of pyridine carboxaldehydes described in J. Heterocyclic Chem , Vol. 31 , (1994), at p. 1161.
- N,N,N'-trimethylethylenediamine (6.12 ml, 48 mmol) in THF (150 ml) at -78°C was added 2.0M nBuLi (22 ml, 44 mmol). Approximately 15 min. later, 2-pyridinecarboxaldehyde (4.3 g, 40 mmol) was added and the mixture was stirred at -78 °C for 15 min.
- Step G Example 207
- Compound 207F was taken up in 20%TFA CH 2 C1 2 and stirred at rt for 3 h.
- N,N-Dimethylamine (36 ⁇ L, 0.072 mmol) was added to a solution of compound 191H (15 mg, 0.018 mmol) in DMSO (225 ⁇ L). After 24 h of shaking the reaction mixture was placed on top of a 300 mg SCX cation exchange column (CUBCX1HL3R3) which had been conditioned with MeOH (1.5 mL). The column was washed with MeOH (1.5 mL). The product was eluted with 2.0 M NHJMeOH (1.5 mL). The eluant was cone, to give 9.6 mg of (compound 229 A).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Physical Education & Sports Medicine (AREA)
- Cardiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Dermatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Neurology (AREA)
- Gastroenterology & Hepatology (AREA)
- Otolaryngology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Biomedical Technology (AREA)
- Hospice & Palliative Care (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Ophthalmology & Optometry (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description














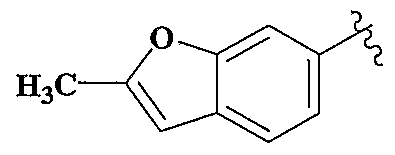




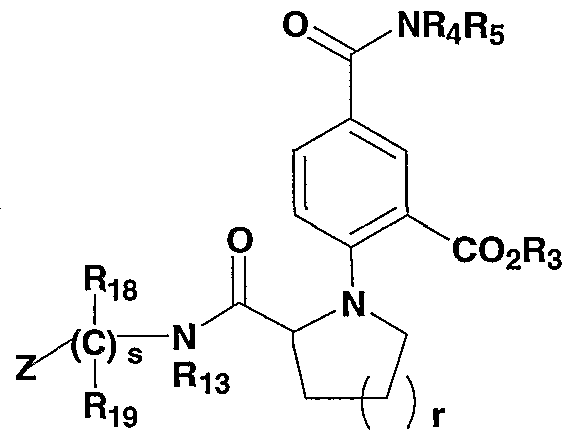





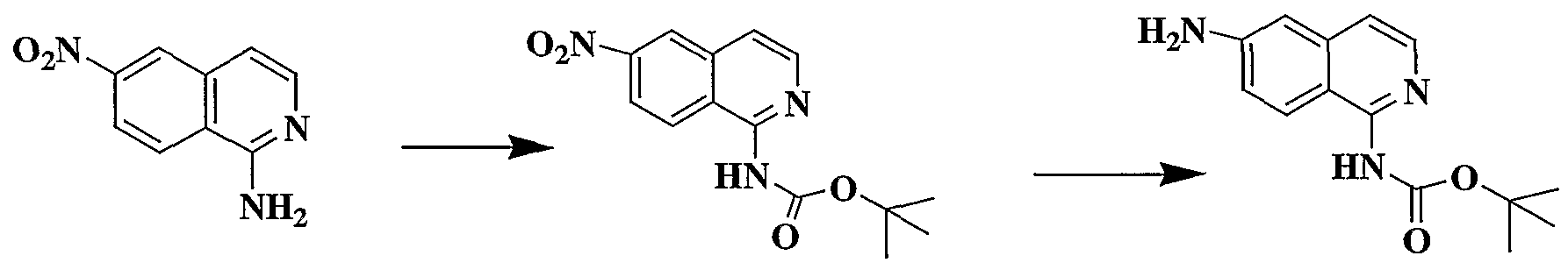








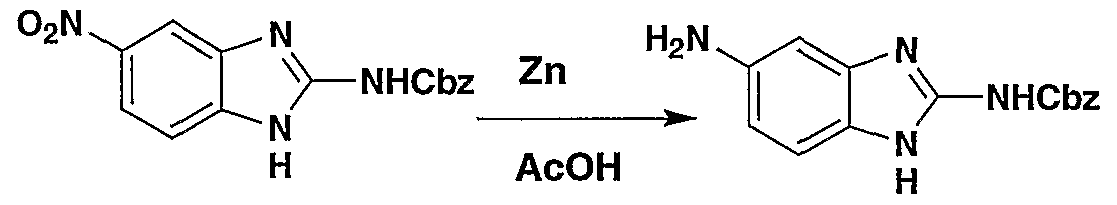



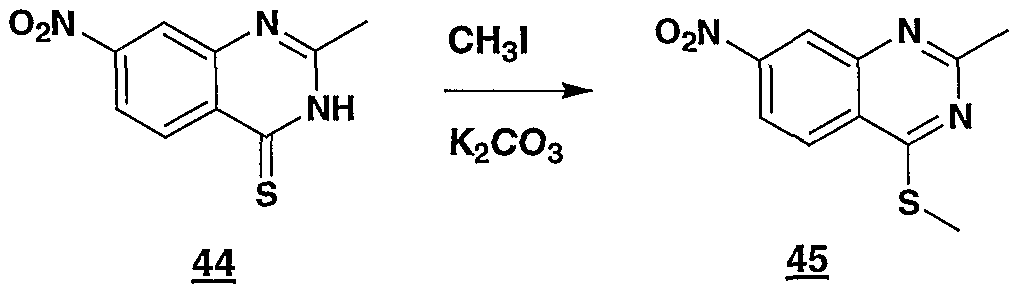







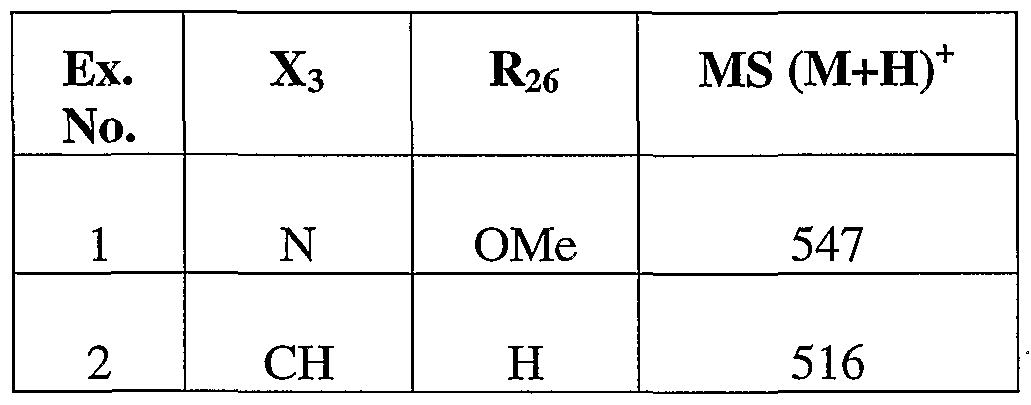






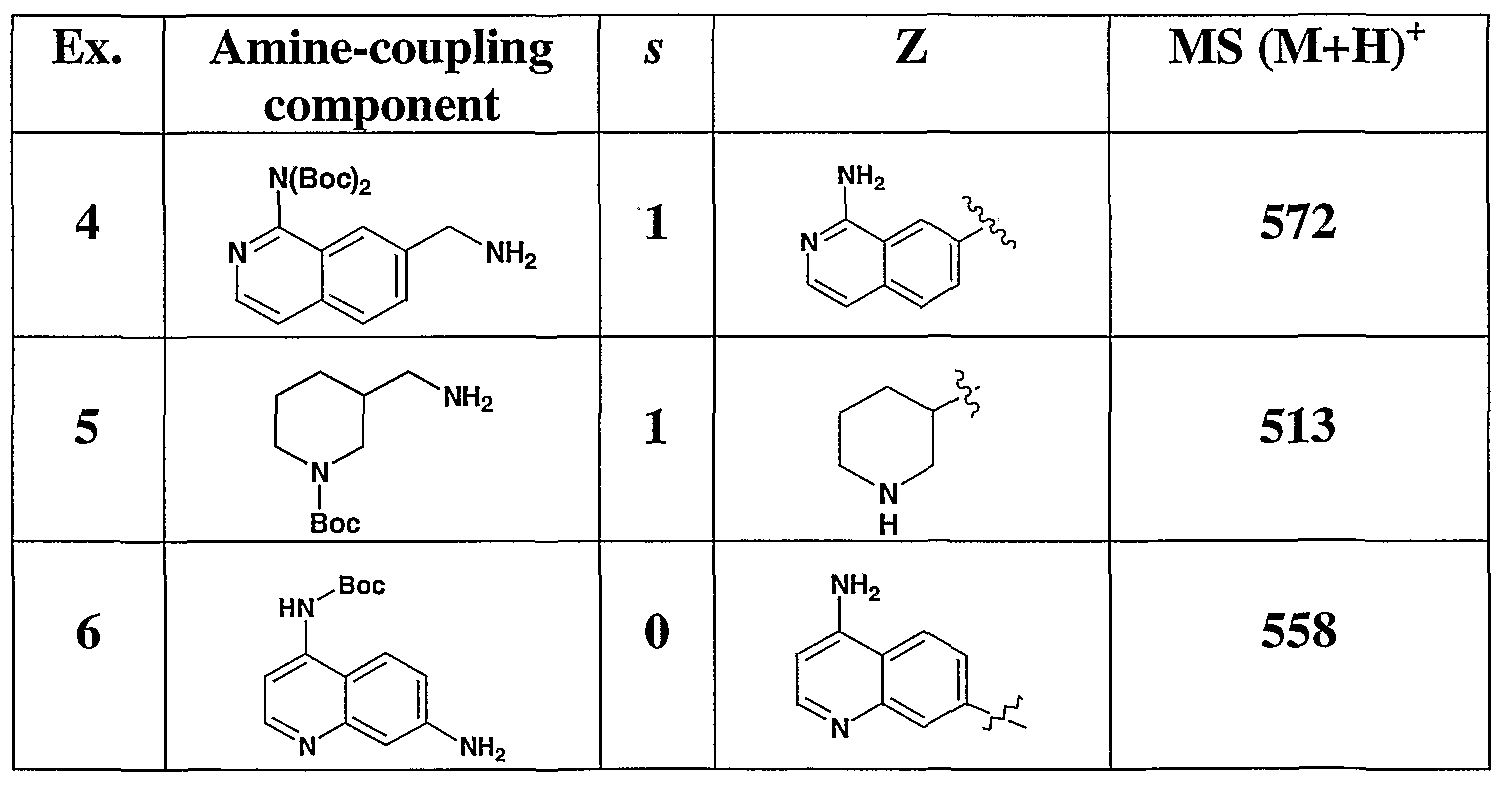


























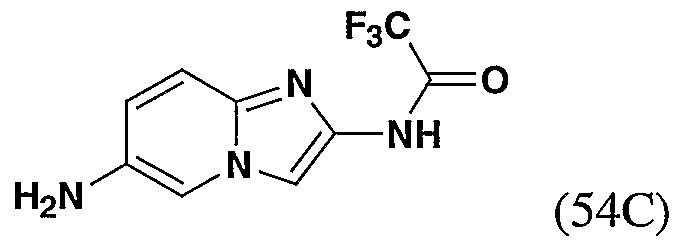














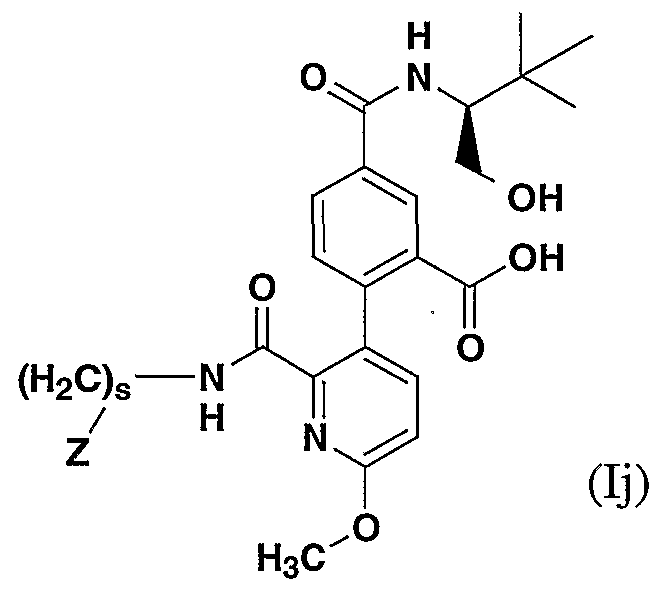










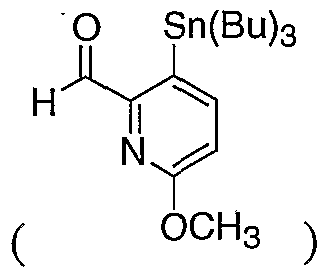














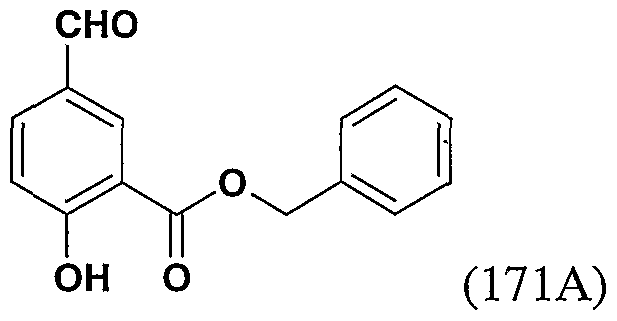



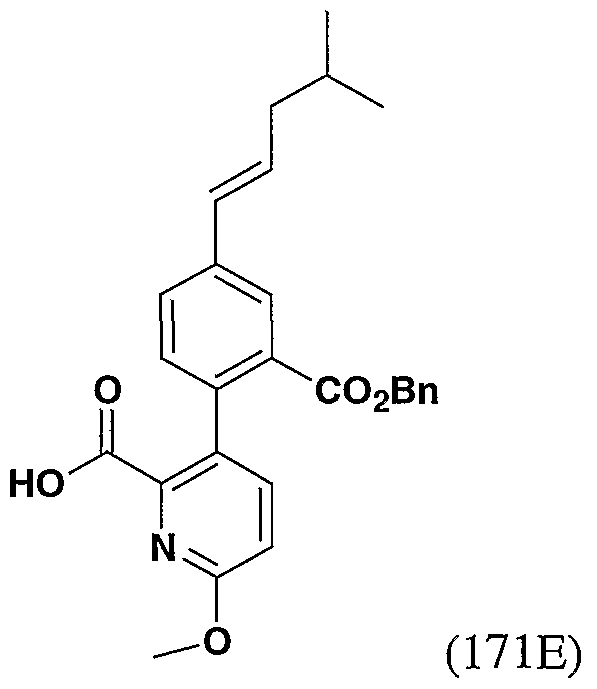

























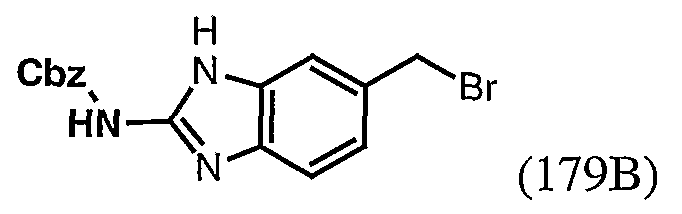









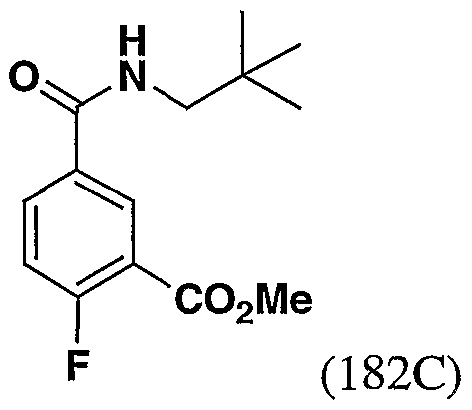



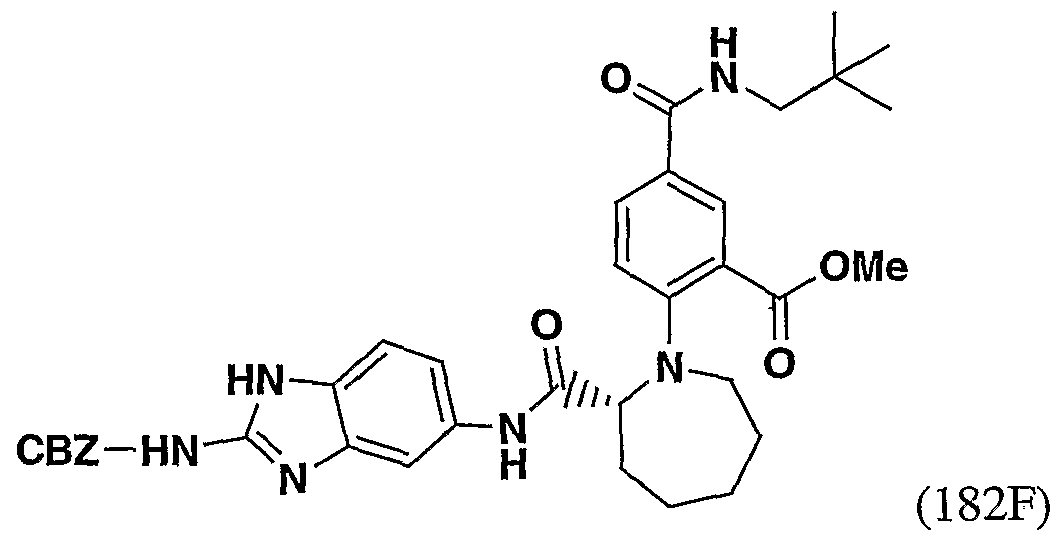



























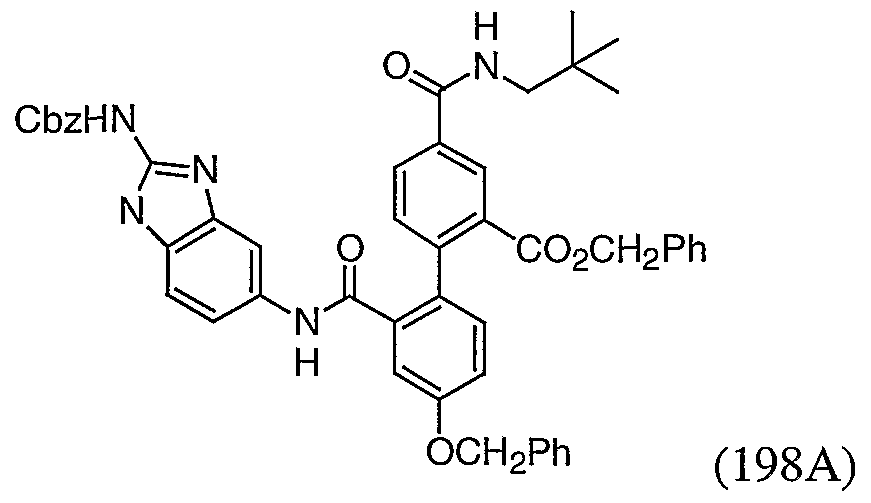

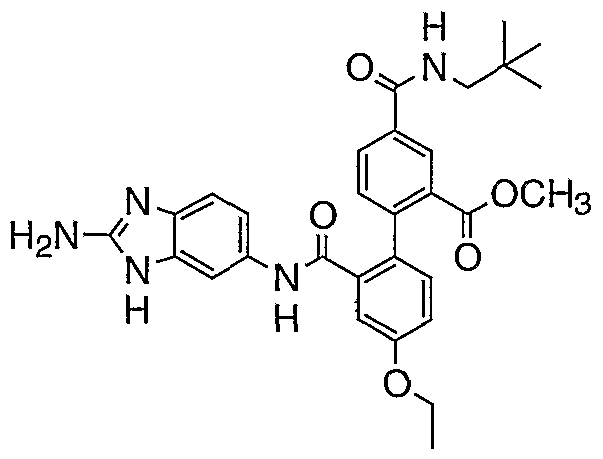


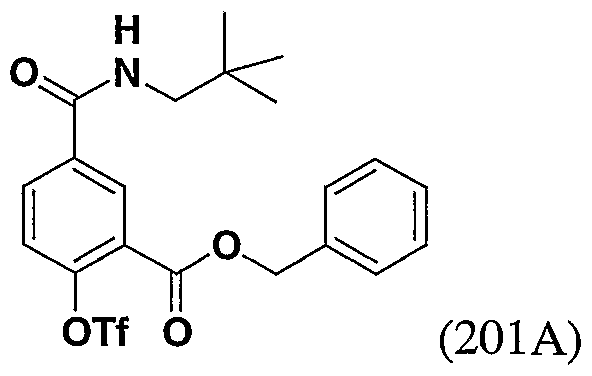













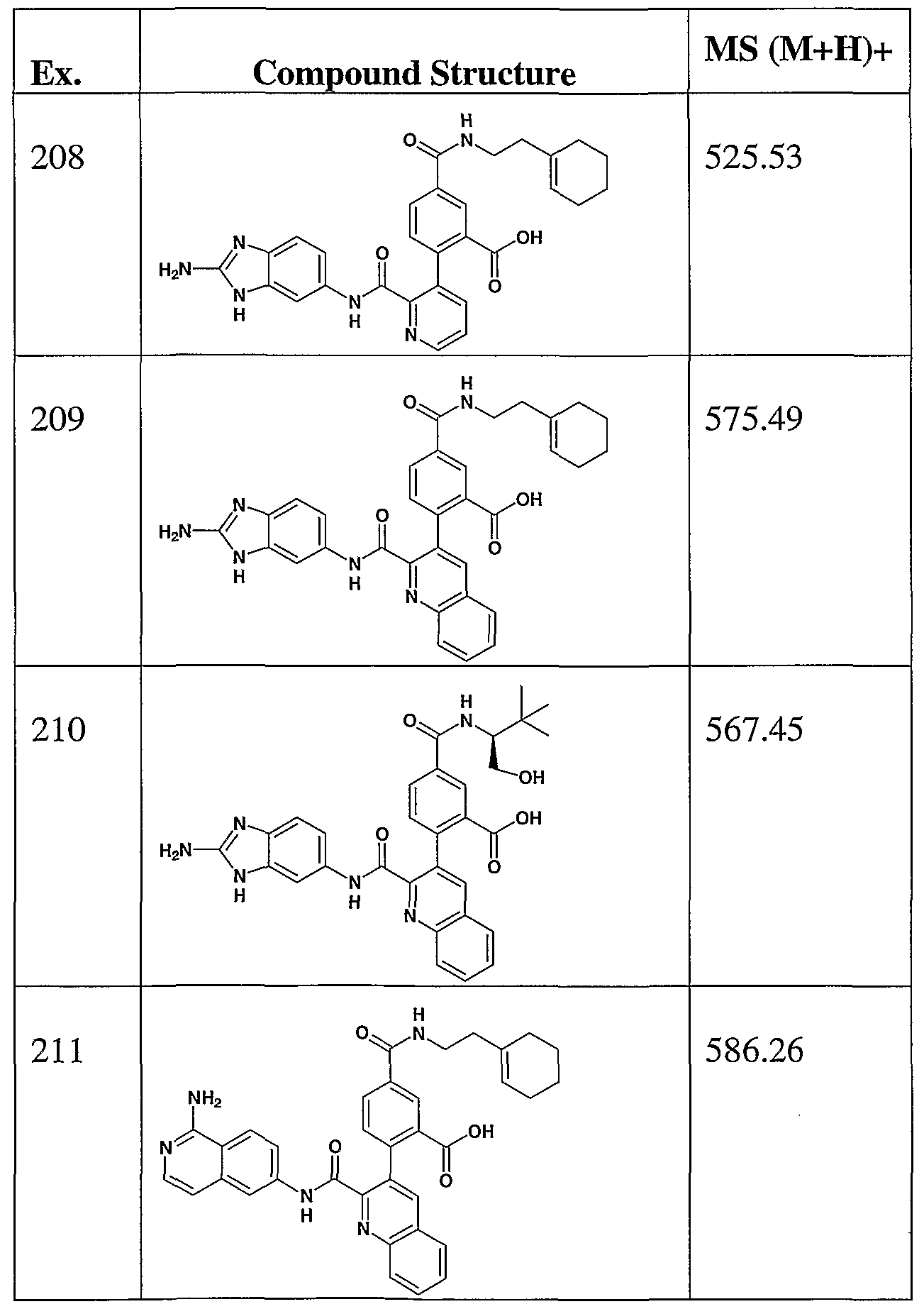
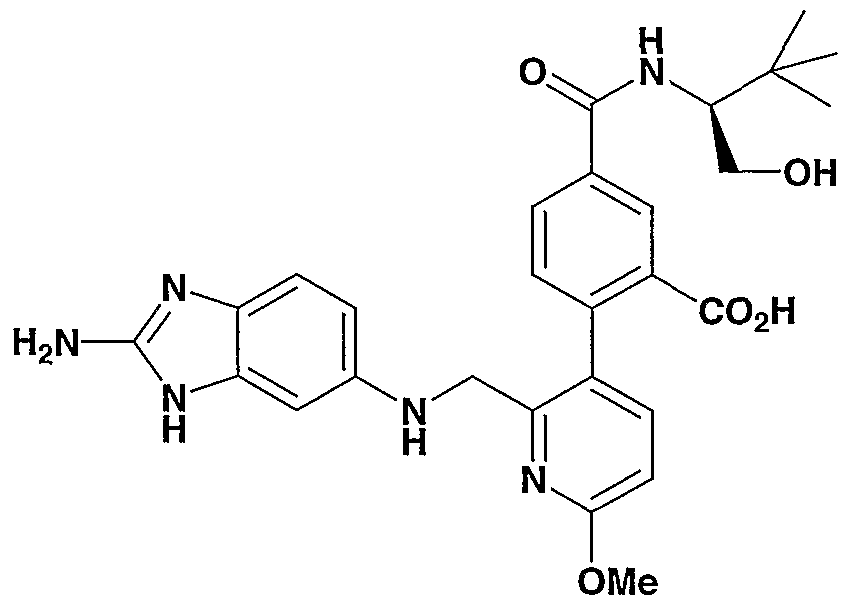















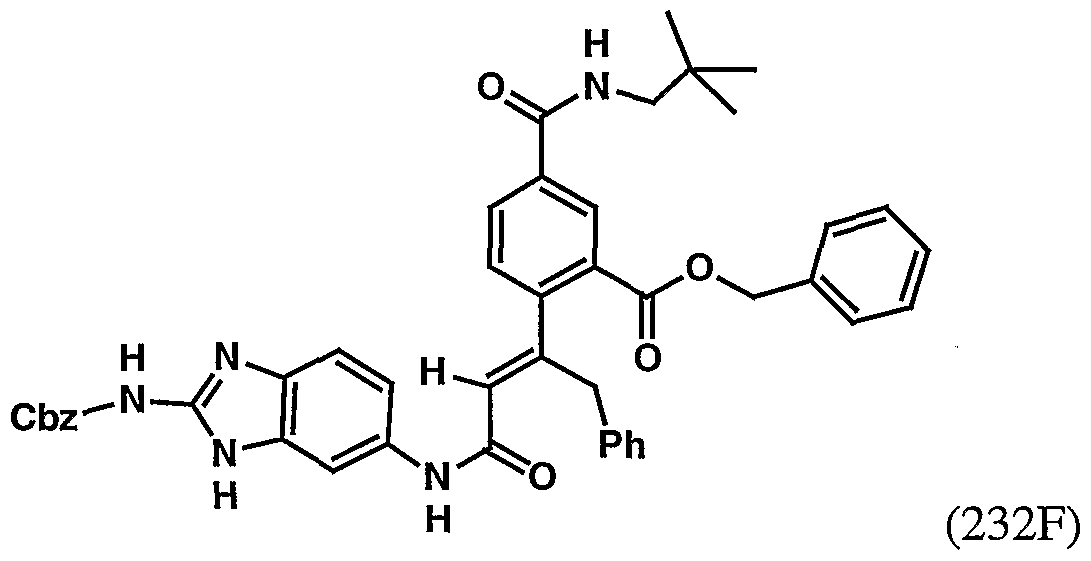

Claims






































Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2002227269A AU2002227269A1 (en) | 2000-11-07 | 2001-11-07 | Acid derivatives useful as serine protease inhibitors |
| JP2002544409A JP2004514669A (en) | 2000-11-07 | 2001-11-07 | Acid derivatives useful as serine protease inhibitors |
| HU0400651A HUP0400651A2 (en) | 2000-11-07 | 2001-11-07 | Acid derivatives useful as serine protease inhibitors and pharmaceutical compositions containing them |
| CA002428191A CA2428191A1 (en) | 2000-11-07 | 2001-11-07 | Acid derivatives useful as serine protease inhibitors |
| EP01996145A EP1332131A2 (en) | 2000-11-07 | 2001-11-07 | Acid derivatives useful as serine protease inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US24639200P | 2000-11-07 | 2000-11-07 | |
| US60/246,392 | 2000-11-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2002042273A2 true WO2002042273A2 (en) | 2002-05-30 |
| WO2002042273A3 WO2002042273A3 (en) | 2002-08-29 |
Family
ID=22930465
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2001/046884 WO2002042273A2 (en) | 2000-11-07 | 2001-11-07 | Acid derivatives useful as serine protease inhibitors |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US6642252B2 (en) |
| EP (1) | EP1332131A2 (en) |
| JP (1) | JP2004514669A (en) |
| AU (1) | AU2002227269A1 (en) |
| CA (1) | CA2428191A1 (en) |
| HU (1) | HUP0400651A2 (en) |
| WO (1) | WO2002042273A2 (en) |
Cited By (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1394147A1 (en) * | 2001-06-04 | 2004-03-03 | Eisai Co., Ltd. | Carboxylic acid derivative and medicine comprising salt or ester of the same |
| US6730684B1 (en) | 1999-10-08 | 2004-05-04 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| US6762201B1 (en) | 1999-10-08 | 2004-07-13 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| US6765005B2 (en) | 1999-04-19 | 2004-07-20 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| WO2004096784A1 (en) * | 2003-04-28 | 2004-11-11 | Astrazeneca Ab | New heterocyclic amides exhibiting an inhibitory activity at the vanilloid receptor 1 (vr1). |
| WO2004101535A1 (en) * | 2003-05-16 | 2004-11-25 | Novartis Ag | Pharmaceutical composition comprising valsartan |
| US6846819B1 (en) | 1999-10-08 | 2005-01-25 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| WO2005016928A1 (en) * | 2003-08-15 | 2005-02-24 | Banyu Pharmaceutical Co., Ltd. | Imidazopyridine derivatives |
| US6906192B2 (en) | 2000-11-07 | 2005-06-14 | Bristol Myers Squibb Company | Processes for the preparation of acid derivatives useful as serine protease inhibitors |
| US6964970B2 (en) | 1999-06-01 | 2005-11-15 | Affinium Pharmaceuticals, Inc. | Antibacterial compounds |
| EP1594845A2 (en) * | 2003-02-11 | 2005-11-16 | Bristol-Myers Squibb Company | Benzene acetamide compounds useful as serine protease inhibitors |
| WO2005108399A1 (en) * | 2004-05-10 | 2005-11-17 | Banyu Pharmaceutical Co., Ltd. | Imidazopyridine compound |
| WO2005123680A1 (en) | 2004-06-15 | 2005-12-29 | Bristol-Myers Squibb Company | Six-membered heterocycles useful as serine protease inhibitors |
| US7049310B2 (en) | 2001-04-06 | 2006-05-23 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| WO2006066173A2 (en) * | 2004-12-17 | 2006-06-22 | Eli Lilly And Company | Novel mch receptor antagonists |
| US7071216B2 (en) | 2002-03-29 | 2006-07-04 | Chiron Corporation | Substituted benz-azoles and methods of their use as inhibitors of Raf kinase |
| US7138412B2 (en) | 2003-03-11 | 2006-11-21 | Bristol-Myers Squibb Company | Tetrahydroquinoline derivatives useful as serine protease inhibitors |
| WO2007070818A1 (en) * | 2005-12-14 | 2007-06-21 | Bristol-Myers Squibb Company | Six-membered heterocycles useful as serine protease inhibitors |
| WO2008031508A1 (en) | 2006-09-13 | 2008-03-20 | Sanofi-Aventis | TARTRATE DERIVATIVES FOR USE AS COAGULATION FACTOR IXa INHIBITORS |
| US7423150B2 (en) | 2003-10-16 | 2008-09-09 | Novartis Ag | Substituted benzazoles and methods of their use as inhibitors of Raf kinase |
| US7511035B2 (en) | 2005-01-25 | 2009-03-31 | Glaxo Group Limited | Antibacterial agents |
| US7544835B2 (en) | 2001-04-20 | 2009-06-09 | Eisai R&D Management Co., Ltd. | Carboxylic acid derivative and salt thereof |
| US7652041B2 (en) | 2005-01-14 | 2010-01-26 | Millennium Pharmaceuticals, Inc. | Cinnamide and hydrocinnamide derivatives with kinase inhibitory activity |
| US7781595B2 (en) | 2003-09-22 | 2010-08-24 | S*Bio Pte Ltd. | Benzimidazole derivatives: preparation and pharmaceutical applications |
| US7893096B2 (en) | 2003-03-28 | 2011-02-22 | Novartis Vaccines And Diagnostics, Inc. | Use of small molecule compounds for immunopotentiation |
| US8097758B2 (en) | 2006-09-13 | 2012-01-17 | Sanofi-Aventis | Isoserine derivatives for use as coagulation factor IXa inhibitors |
| US8299108B2 (en) | 2002-03-29 | 2012-10-30 | Novartis Ag | Substituted benzazoles and methods of their use as inhibitors of raf kinase |
| US8604055B2 (en) | 2004-12-31 | 2013-12-10 | Dr. Reddy's Laboratories Ltd. | Substituted benzylamino quinolines as cholesterol ester-transfer protein inhibitors |
| US8895545B2 (en) | 2006-07-20 | 2014-11-25 | Debiopharm International Sa | Acrylamide derivatives as Fab I inhibitors |
| US8901105B2 (en) | 2012-06-19 | 2014-12-02 | Debiopharm International Sa | Prodrug derivatives of (E)-N-methyl-N-((3-M ethylbenzofuran-2-yl)methyl)-3-(7-oxo-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)acrylamide |
| WO2015063093A1 (en) * | 2013-10-30 | 2015-05-07 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| US9040558B2 (en) | 2004-12-31 | 2015-05-26 | Dr. Reddy's Laboratories Ltd. | Substituted benzylamino quinolines as cholesterol ester-transfer protein inhibitors |
| WO2015123091A1 (en) | 2014-02-11 | 2015-08-20 | Merck Sharp & Dohme Corp. | Factor xia inhibitors |
| WO2015123093A1 (en) | 2014-02-11 | 2015-08-20 | Merck Sharp & Dohme Corp. | Factor xia inhibitors |
| WO2015164308A1 (en) | 2014-04-22 | 2015-10-29 | Merck Sharp & Dohme Corp. | FACTOR XIa INHIBITORS |
| US9242969B2 (en) | 2013-03-14 | 2016-01-26 | Novartis Ag | Biaryl amide compounds as kinase inhibitors |
| WO2016015593A1 (en) | 2014-07-28 | 2016-02-04 | Merck Sharp & Dohme Corp. | FACTOR XIa INHIBITORS |
| WO2016046158A1 (en) * | 2014-09-24 | 2016-03-31 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| WO2016046156A1 (en) * | 2014-09-24 | 2016-03-31 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| WO2016029216A3 (en) * | 2014-08-22 | 2016-05-12 | Biocryst Pharmaceuticals, Inc. | Method for producing amidine derivatives |
| EP2914580A4 (en) * | 2012-11-05 | 2016-06-01 | Lg Life Sciences Ltd | Thioaryl derivatives as gpr120 agonists |
| US9573969B2 (en) | 2014-09-12 | 2017-02-21 | Novartis Ag | Compounds and compositions as kinase inhibitors |
| WO2017074832A1 (en) | 2015-10-29 | 2017-05-04 | Merck Sharp & Dohme Corp. | FACTOR XIa INHIBITORS |
| WO2017074833A1 (en) | 2015-10-29 | 2017-05-04 | Merck Sharp & Dohme Corp. | Macrocyclic spirocarbamate derivatives as factor xia inhibitors, pharmaceutically acceptable compositions and their use |
| US9862737B2 (en) | 2007-02-16 | 2018-01-09 | Debiopharm International Sa | Salts, prodrugs and polymorphs of fab I inhibitors |
| WO2018039094A1 (en) | 2016-08-22 | 2018-03-01 | Merck Sharp & Dohme Corp. | Pyridine-1-oxide derivatives and their use as factor xia inhibitors |
| US10112915B2 (en) | 2015-02-02 | 2018-10-30 | Forma Therapeutics, Inc. | 3-aryl bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10183934B2 (en) | 2015-02-02 | 2019-01-22 | Forma Therapeutics, Inc. | Bicyclic [4,6,0] hydroxamic acids as HDAC inhibitors |
| CN109879808A (en) * | 2019-03-05 | 2019-06-14 | 北京工业大学 | One kind chalcones derivative of base containing five-membered azole heterocycle and preparation method and medical usage |
| US10421742B2 (en) | 2015-07-09 | 2019-09-24 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| US10555935B2 (en) | 2016-06-17 | 2020-02-11 | Forma Therapeutics, Inc. | 2-spiro-5- and 6-hydroxamic acid indanes as HDAC inhibitors |
| US10626092B2 (en) | 2016-05-02 | 2020-04-21 | Mei Pharma, Inc. | Polymorphic forms of 3-[(2-butyl-1-(2-diethylamino-ethyl)-1H-benzoimidazol-5-yl]-N-hydroxy-acrylamide and uses thereof |
| US10647661B2 (en) | 2017-07-11 | 2020-05-12 | Vertex Pharmaceuticals Incorporated | Carboxamides as modulators of sodium channels |
| US10751351B2 (en) | 2016-02-26 | 2020-08-25 | Debiopharm International S.A. | Medicament for treatment of diabetic foot infections |
| US12011449B2 (en) | 2016-09-19 | 2024-06-18 | Novartis Ag | Therapeutic combinations comprising a c-RAF inhibitor |
| US12036227B2 (en) | 2017-05-02 | 2024-07-16 | Novartis Ag | Combination therapy |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1350511B1 (en) * | 2000-12-25 | 2008-09-10 | Daiichi Sankyo Company, Limited | Medicinal compositions containing aspirin |
| KR20100028674A (en) * | 2001-06-29 | 2010-03-12 | 가부시키가이샤 시세이도 | Composite powders and skin preparations for external use containing the same |
| EP1594505A4 (en) * | 2003-02-11 | 2008-08-13 | Bristol Myers Squibb Co | Phenylglycine derivatives useful as serine protease inhibitors |
| AU2004249671A1 (en) * | 2003-05-20 | 2004-12-29 | Genentech, Inc. | Benzofuran inhibitors of factor VIIa |
| JP2007505160A (en) * | 2003-05-20 | 2007-03-08 | ジェネンテック・インコーポレーテッド | Acylsulfamide inhibitors of factor VIIa |
| DE10323898A1 (en) * | 2003-05-26 | 2004-12-23 | Wilex Ag | Hydroxyamidine and hydroxyguanidine compounds as urokinase inhibitors |
| ATE405553T1 (en) * | 2004-12-08 | 2008-09-15 | Bristol Myers Squibb Co | HETEROCYCLIC COMPOUNDS AS INHIBITORS OF FACTOR VIIA |
| PE20061117A1 (en) * | 2005-01-10 | 2006-10-13 | Bristol Myers Squibb Co | DERIVATIVES OF PHENYLGLYCINAMIDE AS INHIBITORS OF FACTOR VIIa |
| EP2987796B1 (en) | 2005-02-16 | 2018-08-08 | Anacor Pharmaceuticals, Inc. | Halogen-substituted boronophthalides for the treatment of infections |
| US7767657B2 (en) * | 2005-02-16 | 2010-08-03 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules |
| ATE455103T1 (en) * | 2005-06-24 | 2010-01-15 | Bristol Myers Squibb Co | PHENYLGLYCINAMIDE AND PYRIDYLGLYCINAMIDE DERIVATIVES USED AS ANTICOAGULATION AGENTS |
| PE20071132A1 (en) * | 2005-12-23 | 2007-12-14 | Bristol Myers Squibb Co | MACROCYCLIC COMPOUNDS AS INHIBITORS OF FACTOR VIIA |
| EP2061756B1 (en) | 2006-06-08 | 2013-09-25 | Bristol-Myers Squibb Company | 2-aminocarbonylphenylamino-2-phenilacetamides as factor viia inhibitors useful as anticoagulants |
| PE20081775A1 (en) * | 2006-12-20 | 2008-12-18 | Bristol Myers Squibb Co | MACROCYCLIC COMPOUNDS AS INHIBITORS OF FACTOR VIIA |
| EP2099783A1 (en) * | 2006-12-20 | 2009-09-16 | Brystol-Myers Squibb Company | Bicyclic lactam factor viia inhibitors useful as anticoagulants |
| JP5964965B2 (en) | 2011-08-18 | 2016-08-03 | ドクター レディズ ラボラトリーズ リミテッド | Substituted heterocyclic amine compounds as cholesteryl ester transfer protein (CETP) inhibitors |
| MX352074B (en) | 2011-09-27 | 2017-11-08 | Dr Reddys Laboratories Ltd | 5 - benzylaminomethyl - 6 - aminopyrazolo [3, 4 -b] pyridine derivatives as cholesteryl ester -transfer protein (cetp) inhibitors useful for the treatment of atherosclerosis. |
| WO2018002673A1 (en) | 2016-07-01 | 2018-01-04 | N4 Pharma Uk Limited | Novel formulations of angiotensin ii receptor antagonists |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3995045A (en) * | 1975-09-25 | 1976-11-30 | E. R. Squibb & Sons, Inc. | 2'-[(3,6-Dihydro-phenyl-1(2H)pyridinyl)alkylaminocarbonyl][1,1'-biphenyl]-2-carboxylic acids |
| GB2056968A (en) * | 1979-08-21 | 1981-03-25 | Fujisawa Pharmaceutical Co | Piperazine derivative, processes for the preparation thereof and pharmaceutical composition comprising the same |
| WO1998057937A2 (en) * | 1997-06-19 | 1998-12-23 | The Du Pont Merck Pharmaceutical Company | Inhibitors of factor xa with a neutral p1 specificity group |
| WO1999041231A1 (en) * | 1998-02-17 | 1999-08-19 | Ono Pharmaceutical Co., Ltd. | Amidino derivatives and drugs containing the same as the active ingredient |
| WO2001070678A2 (en) * | 2000-03-24 | 2001-09-27 | Merck Patent Gmbh | Substituted biphenyl derivatives |
| WO2002034711A1 (en) * | 2000-10-20 | 2002-05-02 | Biocryst Pharmaceuticals, Inc. | Biaryl compounds as serine protease inhibitors |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5137818B2 (en) | 1972-09-01 | 1976-10-18 | ||
| US4786755A (en) | 1985-06-03 | 1988-11-22 | Warner-Lambert Company | Diphenic acid monoamides |
| JPH0623151B2 (en) | 1989-06-02 | 1994-03-30 | 田辺製薬株式会社 | Biphenyl derivative, production method thereof and synthetic intermediate thereof |
| US5612341A (en) | 1995-06-07 | 1997-03-18 | Biotech Research Laboratories | Brominated hexahydroxybiphenyl derivatives |
| AU712082B2 (en) | 1996-02-28 | 1999-10-28 | Merck & Co., Inc. | Fibrinogen receptor antagonists |
| IL123986A (en) | 1997-04-24 | 2011-10-31 | Organon Nv | Serine protease inhibiting antithrombotic agents and pharmaceutical compositions comprising them |
| US5783705A (en) | 1997-04-28 | 1998-07-21 | Texas Biotechnology Corporation | Process of preparing alkali metal salys of hydrophobic sulfonamides |
| US6358959B1 (en) | 1999-01-26 | 2002-03-19 | Merck & Co., Inc. | Polyazanaphthalenone derivatives useful as alpha 1a adrenoceptor antagonists |
| ATE259363T1 (en) | 1999-09-25 | 2004-02-15 | Smithkline Beecham Plc | PIPERAZINE DERIVATIVES AS 5-HT1B ANTAGONISTS |
| EP1242366A1 (en) | 1999-12-15 | 2002-09-25 | Axys Pharmaceuticals, Inc. | Salicylamides as serine protease and factor xa inhibitors |
| NZ523553A (en) | 2000-07-12 | 2004-04-30 | Akzo Nobel Nv | Thrombin inhibitors comprising an aminoisoquinoline group |
| DE10046272A1 (en) | 2000-09-19 | 2002-03-28 | Merck Patent Gmbh | New biphenyl-substituted amino-(iso)quinoline derivatives, are factor Xa and factor VIIa inhibitors useful e.g. for treating thrombosis, myocardial infarction, inflammation, angina pectoris or restenosis |
-
2001
- 2001-11-07 EP EP01996145A patent/EP1332131A2/en not_active Withdrawn
- 2001-11-07 CA CA002428191A patent/CA2428191A1/en not_active Abandoned
- 2001-11-07 US US10/052,927 patent/US6642252B2/en not_active Expired - Lifetime
- 2001-11-07 HU HU0400651A patent/HUP0400651A2/en unknown
- 2001-11-07 AU AU2002227269A patent/AU2002227269A1/en not_active Abandoned
- 2001-11-07 WO PCT/US2001/046884 patent/WO2002042273A2/en not_active Application Discontinuation
- 2001-11-07 JP JP2002544409A patent/JP2004514669A/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3995045A (en) * | 1975-09-25 | 1976-11-30 | E. R. Squibb & Sons, Inc. | 2'-[(3,6-Dihydro-phenyl-1(2H)pyridinyl)alkylaminocarbonyl][1,1'-biphenyl]-2-carboxylic acids |
| GB2056968A (en) * | 1979-08-21 | 1981-03-25 | Fujisawa Pharmaceutical Co | Piperazine derivative, processes for the preparation thereof and pharmaceutical composition comprising the same |
| WO1998057937A2 (en) * | 1997-06-19 | 1998-12-23 | The Du Pont Merck Pharmaceutical Company | Inhibitors of factor xa with a neutral p1 specificity group |
| WO1999041231A1 (en) * | 1998-02-17 | 1999-08-19 | Ono Pharmaceutical Co., Ltd. | Amidino derivatives and drugs containing the same as the active ingredient |
| WO2001070678A2 (en) * | 2000-03-24 | 2001-09-27 | Merck Patent Gmbh | Substituted biphenyl derivatives |
| WO2002034711A1 (en) * | 2000-10-20 | 2002-05-02 | Biocryst Pharmaceuticals, Inc. | Biaryl compounds as serine protease inhibitors |
Non-Patent Citations (2)
| Title |
|---|
| G.KESERU ET AL: "Synthesis of Plagiochins C and D" TETRAHEDRON, vol. 48, no. 5, 1992, pages 913-922, XP001080011 * |
| KOHRT J T ET AL: "An efficient synthesis of 2-(3-(4-amidinophenylcarbamoyl)naphthalen- 2 -yl)-5-((2,2-methylpropyl)carbamo yl)benzoic acid: a factor VIIa inhibitor discovered by the Ono Pharmaceutical Company" TETRAHEDRON LETTERS, ELSEVIER SCIENCE PUBLISHERS, AMSTERDAM, NL, vol. 41, no. 32, 5 August 2000 (2000-08-05), pages 6041-6044, XP004243509 ISSN: 0040-4039 * |
Cited By (132)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6765005B2 (en) | 1999-04-19 | 2004-07-20 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| US6964970B2 (en) | 1999-06-01 | 2005-11-15 | Affinium Pharmaceuticals, Inc. | Antibacterial compounds |
| US6762201B1 (en) | 1999-10-08 | 2004-07-13 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| US6730684B1 (en) | 1999-10-08 | 2004-05-04 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| US6846819B1 (en) | 1999-10-08 | 2005-01-25 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| US7557125B2 (en) | 1999-10-08 | 2009-07-07 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| US7524843B2 (en) | 1999-10-08 | 2009-04-28 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| US6906192B2 (en) | 2000-11-07 | 2005-06-14 | Bristol Myers Squibb Company | Processes for the preparation of acid derivatives useful as serine protease inhibitors |
| US7049310B2 (en) | 2001-04-06 | 2006-05-23 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| US7250424B2 (en) | 2001-04-06 | 2007-07-31 | Affinium Pharmaceuticals, Inc. | Fab I inhibitors |
| US7544835B2 (en) | 2001-04-20 | 2009-06-09 | Eisai R&D Management Co., Ltd. | Carboxylic acid derivative and salt thereof |
| EP1394147A1 (en) * | 2001-06-04 | 2004-03-03 | Eisai Co., Ltd. | Carboxylic acid derivative and medicine comprising salt or ester of the same |
| US7687664B2 (en) | 2001-06-04 | 2010-03-30 | Eisai R&D Management Co., Ltd. | Carboxylic acid derivative, a salt thereof or an ester of them, and medicament comprising it |
| EP1394147A4 (en) * | 2001-06-04 | 2007-10-24 | Eisai R&D Man Co Ltd | Carboxylic acid derivative and medicine comprising salt or ester of the same |
| US7071216B2 (en) | 2002-03-29 | 2006-07-04 | Chiron Corporation | Substituted benz-azoles and methods of their use as inhibitors of Raf kinase |
| US8614330B2 (en) | 2002-03-29 | 2013-12-24 | Novartis Vaccines And Diagnostics, Inc. | Substituted benz-azoles and methods of their use as inhibitors of RAF kinase |
| US7728010B2 (en) | 2002-03-29 | 2010-06-01 | Novartis Vaccines And Diagnostics, Inc. | Substituted benz-azoles and methods of their use as inhibitors of Raf kinase |
| US8299108B2 (en) | 2002-03-29 | 2012-10-30 | Novartis Ag | Substituted benzazoles and methods of their use as inhibitors of raf kinase |
| EP1594845A4 (en) * | 2003-02-11 | 2008-05-21 | Bristol Myers Squibb Co | Benzene acetamide compounds useful as serine protease inhibitors |
| JP2006517589A (en) * | 2003-02-11 | 2006-07-27 | ブリストル−マイヤーズ スクイブ カンパニー | Benzeneacetamide compounds useful as serine protease inhibitors |
| EP1594845A2 (en) * | 2003-02-11 | 2005-11-16 | Bristol-Myers Squibb Company | Benzene acetamide compounds useful as serine protease inhibitors |
| US7138412B2 (en) | 2003-03-11 | 2006-11-21 | Bristol-Myers Squibb Company | Tetrahydroquinoline derivatives useful as serine protease inhibitors |
| US7709646B2 (en) | 2003-03-11 | 2010-05-04 | Bristol-Myers Squibb Company | Tetrahydroquinoline derivatives useful as serine protease inhibitors |
| US7893096B2 (en) | 2003-03-28 | 2011-02-22 | Novartis Vaccines And Diagnostics, Inc. | Use of small molecule compounds for immunopotentiation |
| WO2004096784A1 (en) * | 2003-04-28 | 2004-11-11 | Astrazeneca Ab | New heterocyclic amides exhibiting an inhibitory activity at the vanilloid receptor 1 (vr1). |
| WO2004101535A1 (en) * | 2003-05-16 | 2004-11-25 | Novartis Ag | Pharmaceutical composition comprising valsartan |
| EP1657242A1 (en) * | 2003-08-15 | 2006-05-17 | Banyu Pharmaceutical Co., Ltd. | Imidazopyridine derivatives |
| EP1657242A4 (en) * | 2003-08-15 | 2008-10-29 | Banyu Pharma Co Ltd | Imidazopyridine derivatives |
| US7504412B2 (en) | 2003-08-15 | 2009-03-17 | Banyu Pharmaceuticals, Co., Ltd. | Imidazopyridine derivatives |
| WO2005016928A1 (en) * | 2003-08-15 | 2005-02-24 | Banyu Pharmaceutical Co., Ltd. | Imidazopyridine derivatives |
| US8551988B2 (en) | 2003-09-22 | 2013-10-08 | Mei Pharma, Inc. | Benzimidazole derivatives: preparation and pharmaceutical applications |
| US9717713B2 (en) | 2003-09-22 | 2017-08-01 | Mei Pharma, Inc. | Benzimidazole derivatives: preparation and pharmaceutical applications |
| US7781595B2 (en) | 2003-09-22 | 2010-08-24 | S*Bio Pte Ltd. | Benzimidazole derivatives: preparation and pharmaceutical applications |
| US10736881B2 (en) | 2003-09-22 | 2020-08-11 | Mei Pharma, Inc. | Benzimidazole derivatives: preparation and pharmaceutical applications |
| US9024029B2 (en) | 2003-09-22 | 2015-05-05 | Mei Pharma, Inc. | Benzimidazole derivatives: preparation and pharmaceutical applications |
| US10201527B2 (en) | 2003-09-22 | 2019-02-12 | Mei Pharma, Inc. | Benzimidazole derivatives: preparation and pharmaceutical applications |
| US9402829B2 (en) | 2003-09-22 | 2016-08-02 | Mei Pharma, Inc. | Benzimidazole derivatives: preparation and pharmaceutical applications |
| US7423150B2 (en) | 2003-10-16 | 2008-09-09 | Novartis Ag | Substituted benzazoles and methods of their use as inhibitors of Raf kinase |
| US8415382B2 (en) | 2003-10-16 | 2013-04-09 | Novartis Vaccines And Diagnostics, Inc. | Substituted benzazoles and methods of their use as inhibitors of RAF Kinase |
| AU2005240942B2 (en) * | 2004-05-10 | 2010-08-05 | Banyu Pharmaceutical Co., Ltd. | Imidazopyridine compound |
| US7566781B2 (en) | 2004-05-10 | 2009-07-28 | Banyu Pharmaceutical Co., Ltd. | Imidazopyridine compound |
| WO2005108399A1 (en) * | 2004-05-10 | 2005-11-17 | Banyu Pharmaceutical Co., Ltd. | Imidazopyridine compound |
| WO2005123680A1 (en) | 2004-06-15 | 2005-12-29 | Bristol-Myers Squibb Company | Six-membered heterocycles useful as serine protease inhibitors |
| JP4834663B2 (en) * | 2004-06-15 | 2011-12-14 | ブリストル−マイヤーズ スクイブ カンパニー | 6-membered heterocycles useful as serine protease inhibitors |
| US7838543B2 (en) | 2004-12-17 | 2010-11-23 | Eli Lilly And Company | MCH receptor antagonists |
| WO2006066173A3 (en) * | 2004-12-17 | 2006-07-27 | Lilly Co Eli | Novel mch receptor antagonists |
| WO2006066173A2 (en) * | 2004-12-17 | 2006-06-22 | Eli Lilly And Company | Novel mch receptor antagonists |
| US8604055B2 (en) | 2004-12-31 | 2013-12-10 | Dr. Reddy's Laboratories Ltd. | Substituted benzylamino quinolines as cholesterol ester-transfer protein inhibitors |
| US9040558B2 (en) | 2004-12-31 | 2015-05-26 | Dr. Reddy's Laboratories Ltd. | Substituted benzylamino quinolines as cholesterol ester-transfer protein inhibitors |
| US7652041B2 (en) | 2005-01-14 | 2010-01-26 | Millennium Pharmaceuticals, Inc. | Cinnamide and hydrocinnamide derivatives with kinase inhibitory activity |
| US7759340B2 (en) | 2005-01-25 | 2010-07-20 | Glaxo Group Limited | Antibacterial agents |
| US7511035B2 (en) | 2005-01-25 | 2009-03-31 | Glaxo Group Limited | Antibacterial agents |
| WO2007070818A1 (en) * | 2005-12-14 | 2007-06-21 | Bristol-Myers Squibb Company | Six-membered heterocycles useful as serine protease inhibitors |
| US8163749B2 (en) | 2005-12-14 | 2012-04-24 | Bristol-Myers Squibb Company | Six-membered heterocycles useful as serine protease inhibitors |
| US8895545B2 (en) | 2006-07-20 | 2014-11-25 | Debiopharm International Sa | Acrylamide derivatives as Fab I inhibitors |
| US8263621B2 (en) | 2006-09-13 | 2012-09-11 | Sanofi | Tartrate derivatives for use as coagulation factor IXa inhibitors |
| US8097758B2 (en) | 2006-09-13 | 2012-01-17 | Sanofi-Aventis | Isoserine derivatives for use as coagulation factor IXa inhibitors |
| WO2008031508A1 (en) | 2006-09-13 | 2008-03-20 | Sanofi-Aventis | TARTRATE DERIVATIVES FOR USE AS COAGULATION FACTOR IXa INHIBITORS |
| US9862737B2 (en) | 2007-02-16 | 2018-01-09 | Debiopharm International Sa | Salts, prodrugs and polymorphs of fab I inhibitors |
| US8901105B2 (en) | 2012-06-19 | 2014-12-02 | Debiopharm International Sa | Prodrug derivatives of (E)-N-methyl-N-((3-M ethylbenzofuran-2-yl)methyl)-3-(7-oxo-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)acrylamide |
| US10035813B2 (en) | 2012-06-19 | 2018-07-31 | Debiopharm International Sa | Prodrug derivatives of (E)-N-methyl-N-((3-methylbenzofuran-2-yl)methyl)-3-(7-oxo-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)acrylamide |
| US9447044B2 (en) | 2012-11-05 | 2016-09-20 | Lg Life Sciences Ltd. | Thioaryl derivatives as GPR120 agonists |
| TWI610917B (en) * | 2012-11-05 | 2018-01-11 | 南韓商Lg化學股份有限公司 | Thioaryl derivatives as gpr120 agonists |
| EP2914580A4 (en) * | 2012-11-05 | 2016-06-01 | Lg Life Sciences Ltd | Thioaryl derivatives as gpr120 agonists |
| US10709712B2 (en) | 2013-03-14 | 2020-07-14 | Novartis Ag | Biaryl amide compounds as kinase inhibitors |
| US9242969B2 (en) | 2013-03-14 | 2016-01-26 | Novartis Ag | Biaryl amide compounds as kinase inhibitors |
| US9694016B2 (en) | 2013-03-14 | 2017-07-04 | Novartis Ag | Biaryl amide compounds as kinase inhibitors |
| US10245267B2 (en) | 2013-03-14 | 2019-04-02 | Novartis Ag | Biaryl amide compounds as kinase inhibitors |
| CN105849109A (en) * | 2013-10-30 | 2016-08-10 | 拜耳制药股份公司 | Substituted oxopyridine derivatives |
| WO2015063093A1 (en) * | 2013-10-30 | 2015-05-07 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| US9765070B2 (en) | 2013-10-30 | 2017-09-19 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| WO2015123093A1 (en) | 2014-02-11 | 2015-08-20 | Merck Sharp & Dohme Corp. | Factor xia inhibitors |
| WO2015123091A1 (en) | 2014-02-11 | 2015-08-20 | Merck Sharp & Dohme Corp. | Factor xia inhibitors |
| WO2015164308A1 (en) | 2014-04-22 | 2015-10-29 | Merck Sharp & Dohme Corp. | FACTOR XIa INHIBITORS |
| WO2016015593A1 (en) | 2014-07-28 | 2016-02-04 | Merck Sharp & Dohme Corp. | FACTOR XIa INHIBITORS |
| WO2016029216A3 (en) * | 2014-08-22 | 2016-05-12 | Biocryst Pharmaceuticals, Inc. | Method for producing amidine derivatives |
| US9573969B2 (en) | 2014-09-12 | 2017-02-21 | Novartis Ag | Compounds and compositions as kinase inhibitors |
| US9809610B2 (en) | 2014-09-12 | 2017-11-07 | Novartis Ag | Compounds and compositions as kinase inhibitors |
| US10071995B2 (en) | 2014-09-24 | 2018-09-11 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| WO2016046156A1 (en) * | 2014-09-24 | 2016-03-31 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| US10077265B2 (en) | 2014-09-24 | 2018-09-18 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| WO2016046158A1 (en) * | 2014-09-24 | 2016-03-31 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| US10494354B2 (en) | 2015-02-02 | 2019-12-03 | Forma Therapeutics, Inc. | 3-aryl-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10239845B2 (en) | 2015-02-02 | 2019-03-26 | Forma Therapeutics, Inc. | 3-aryl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10494351B2 (en) | 2015-02-02 | 2019-12-03 | Forma Therapeutics, Inc. | 3-aryl-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10214501B2 (en) | 2015-02-02 | 2019-02-26 | Forma Therapeutics, Inc. | 3-alkyl bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10494352B2 (en) | 2015-02-02 | 2019-12-03 | Forma Therapeutics, Inc. | 3-aryl-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US11891365B2 (en) | 2015-02-02 | 2024-02-06 | Valo Health, Inc. | 3-alkyl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US11702412B2 (en) | 2015-02-02 | 2023-07-18 | Valo Health, Inc. | Bicyclic [4,6,0] hydroxamic acids as HDAC inhibitors |
| US10377726B2 (en) | 2015-02-02 | 2019-08-13 | Forma Therapeutics, Inc. | 3-aryl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10407418B2 (en) | 2015-02-02 | 2019-09-10 | Forma Therapeutics, Inc. | Bicyclic [4,6,0] hydroxamic acids as HDAC inhibitors |
| US10414738B2 (en) | 2015-02-02 | 2019-09-17 | Forma Therapeutics, Inc. | 3-alkyl bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10421732B2 (en) | 2015-02-02 | 2019-09-24 | Forma Therapeutics, Inc. | 3-alkyl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US11279681B2 (en) | 2015-02-02 | 2022-03-22 | Valo Health, Inc. | 3-alkyl bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10421731B2 (en) | 2015-02-02 | 2019-09-24 | Forma Therapeutics, Inc. | 3-alkyl bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10428031B2 (en) | 2015-02-02 | 2019-10-01 | Forma Therapeutics, Inc. | 3-alkyl bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10442776B2 (en) | 2015-02-02 | 2019-10-15 | Forma Therapeutics, Inc. | 3-alkyl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10501424B2 (en) | 2015-02-02 | 2019-12-10 | Forma Therapeutics, Inc. | 3-aryl-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10450284B2 (en) | 2015-02-02 | 2019-10-22 | Forma Therapeutics, Inc. | 3-aryl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10457652B2 (en) | 2015-02-02 | 2019-10-29 | Forma Therapeutics, Inc. | 3-alkyl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10464909B2 (en) | 2015-02-02 | 2019-11-05 | Forma Therapeutics, Inc. | 3-alkyl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10464910B2 (en) | 2015-02-02 | 2019-11-05 | Forma Therapeutics, Inc. | 3-alkyl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10472337B2 (en) | 2015-02-02 | 2019-11-12 | Forma Therapeutics, Inc. | 3-aryl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10479772B2 (en) | 2015-02-02 | 2019-11-19 | Forma Therapeutics, Inc. | 3-aryl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10494353B2 (en) | 2015-02-02 | 2019-12-03 | Forma Therapeutics, Inc. | 3-aryl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10183934B2 (en) | 2015-02-02 | 2019-01-22 | Forma Therapeutics, Inc. | Bicyclic [4,6,0] hydroxamic acids as HDAC inhibitors |
| US10214500B2 (en) | 2015-02-02 | 2019-02-26 | Forma Therapeutics, Inc. | 3-alkyl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US11274085B2 (en) | 2015-02-02 | 2022-03-15 | Valo Health, Inc. | 3-aryl-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10450283B2 (en) | 2015-02-02 | 2019-10-22 | Forma Therapeutics, Inc. | 3-alkyl bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10513501B2 (en) | 2015-02-02 | 2019-12-24 | Forma Therapeutics, Inc. | 3-alkyl bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US11274084B2 (en) | 2015-02-02 | 2022-03-15 | Valo Health, Inc. | 3-aryl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10988450B2 (en) | 2015-02-02 | 2021-04-27 | Valo Early Discovery, Inc. | 3-alkyl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10870645B2 (en) | 2015-02-02 | 2020-12-22 | Valo Early Discovery, Inc. | Bicyclic [4,6,0] hydroxamic acids as HDAC inhibitors |
| US10829461B2 (en) | 2015-02-02 | 2020-11-10 | Valo Early Discovery, Inc. | 3-aryl-4-amido-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10112915B2 (en) | 2015-02-02 | 2018-10-30 | Forma Therapeutics, Inc. | 3-aryl bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10829462B2 (en) | 2015-02-02 | 2020-11-10 | Valo Early Discovery, Inc. | 3-alkyl bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US10822316B2 (en) | 2015-02-02 | 2020-11-03 | Valo Early Discovery, Inc. | 3-aryl-bicyclic [4,5,0] hydroxamic acids as HDAC inhibitors |
| US11180471B2 (en) | 2015-07-09 | 2021-11-23 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| US10421742B2 (en) | 2015-07-09 | 2019-09-24 | Bayer Pharma Aktiengesellschaft | Substituted oxopyridine derivatives |
| WO2017074832A1 (en) | 2015-10-29 | 2017-05-04 | Merck Sharp & Dohme Corp. | FACTOR XIa INHIBITORS |
| WO2017074833A1 (en) | 2015-10-29 | 2017-05-04 | Merck Sharp & Dohme Corp. | Macrocyclic spirocarbamate derivatives as factor xia inhibitors, pharmaceutically acceptable compositions and their use |
| US10751351B2 (en) | 2016-02-26 | 2020-08-25 | Debiopharm International S.A. | Medicament for treatment of diabetic foot infections |
| US10626092B2 (en) | 2016-05-02 | 2020-04-21 | Mei Pharma, Inc. | Polymorphic forms of 3-[(2-butyl-1-(2-diethylamino-ethyl)-1H-benzoimidazol-5-yl]-N-hydroxy-acrylamide and uses thereof |
| US11730721B2 (en) | 2016-06-17 | 2023-08-22 | Valo Health, Inc. | 2-spiro-5- and 6-hydroxamic acid indanes as HDAC inhibitors |
| US10555935B2 (en) | 2016-06-17 | 2020-02-11 | Forma Therapeutics, Inc. | 2-spiro-5- and 6-hydroxamic acid indanes as HDAC inhibitors |
| US10874649B2 (en) | 2016-06-17 | 2020-12-29 | Valo Early Discovery, Inc. | 2-spiro-5- and 6-hydroxamic acid indanes as HDAC inhibitors |
| WO2018039094A1 (en) | 2016-08-22 | 2018-03-01 | Merck Sharp & Dohme Corp. | Pyridine-1-oxide derivatives and their use as factor xia inhibitors |
| US12011449B2 (en) | 2016-09-19 | 2024-06-18 | Novartis Ag | Therapeutic combinations comprising a c-RAF inhibitor |
| US12036227B2 (en) | 2017-05-02 | 2024-07-16 | Novartis Ag | Combination therapy |
| US11603351B2 (en) | 2017-07-11 | 2023-03-14 | Vertex Pharmaceuticals Incorporated | Carboxamides as modulators of sodium channels |
| US10647661B2 (en) | 2017-07-11 | 2020-05-12 | Vertex Pharmaceuticals Incorporated | Carboxamides as modulators of sodium channels |
| CN109879808A (en) * | 2019-03-05 | 2019-06-14 | 北京工业大学 | One kind chalcones derivative of base containing five-membered azole heterocycle and preparation method and medical usage |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2428191A1 (en) | 2002-05-30 |
| HUP0400651A2 (en) | 2004-06-28 |
| EP1332131A2 (en) | 2003-08-06 |
| AU2002227269A1 (en) | 2002-06-03 |
| WO2002042273A3 (en) | 2002-08-29 |
| US20030166685A1 (en) | 2003-09-04 |
| JP2004514669A (en) | 2004-05-20 |
| US6642252B2 (en) | 2003-11-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6642252B2 (en) | Acid derivatives useful as serine protease inhibitors | |
| US6713467B2 (en) | Acid derivatives useful as serine protease inhibitors | |
| TWI586651B (en) | Substituted tetrahydroisoquinoline compounds as factor xia inhibitors | |
| JP2739776B2 (en) | Antithrombotic amidinophenylalanine and amidinopyridylalanine derivatives | |
| JP2846963B2 (en) | Antithrombotic amidinotetrahydropyridylalanine derivative | |
| EA001280B1 (en) | Substituted n-[(aminoiminomethyl or aminomethyl)phenyl]propyl amides | |
| JP2017525744A (en) | Diamide macrocycle which is an FXIA inhibitor | |
| US20060173057A1 (en) | Methods of treating factor VIIa-associated conditions with compounds having an amine nucleus | |
| JP2009511516A (en) | Imidazole derivatives as inhibitors of TAFIa | |
| CZ20031554A3 (en) | Guanidine and amidine derivatives functioning as XA factor inhibitors | |
| CZ301855B6 (en) | N-guanidinoalkylamides, process of their preparation, use and pharmaceutical mixtures in which they are comprised | |
| US7144895B2 (en) | Benzene acetamide compounds useful as serine protease inhibitors | |
| US9657006B2 (en) | Macrocyclic factor VIIa inhibitors | |
| WO2005030706A1 (en) | Amide type carboxamide derivative | |
| JP2001514257A (en) | Protease inhibitor | |
| US6846838B2 (en) | Ureido-substituted aniline compounds useful as serine protease inhibitors | |
| CN110759901A (en) | Tetrahydroisoquinoline derivatives, and preparation method and application thereof | |
| EA008688B1 (en) | New 4-oxo-4,6,7,8,-tetrahydropyrrolo[1,2-a]pyrazine-6-carboxamide compounds, a process for their preparation and pharmaceutical compositions containing them | |
| CZ2003452A3 (en) | Data processing system, with an Internet connection facility has a structured program loaded in memory for use in assigning predetermined data from a multiplicity of data to hierarchical memory addresses | |
| JPH0672867A (en) | Antilipemic agent |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PH PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002227269 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002544409 Country of ref document: JP Ref document number: 2428191 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001996145 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001996145 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2001996145 Country of ref document: EP |