WO2002020530A1 - Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors - Google Patents
Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors Download PDFInfo
- Publication number
- WO2002020530A1 WO2002020530A1 PCT/SE2001/001880 SE0101880W WO0220530A1 WO 2002020530 A1 WO2002020530 A1 WO 2002020530A1 SE 0101880 W SE0101880 W SE 0101880W WO 0220530 A1 WO0220530 A1 WO 0220530A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carbamoyl
- thieno
- dichloro
- alkyl
- pyrrole
- Prior art date
Links
- 0 C*C(C)(*C(C)=C)S(C(C)(*)N)(=O)=O Chemical compound C*C(C)(*C(C)=C)S(C(C)(*)N)(=O)=O 0.000 description 4
- ZFBJOTHMHUTHCZ-UHFFFAOYSA-N CC(C)(C)OC(NCC(N(C)Cc1ccccc1)=O)=O Chemical compound CC(C)(C)OC(NCC(N(C)Cc1ccccc1)=O)=O ZFBJOTHMHUTHCZ-UHFFFAOYSA-N 0.000 description 1
- HFUXKDVQXRCOMF-UHFFFAOYSA-N Cc(cc1)ccc1NC(CNC(c1cc([s]c(Cl)c2Cl)c2[nH]1)=O)=O Chemical compound Cc(cc1)ccc1NC(CNC(c1cc([s]c(Cl)c2Cl)c2[nH]1)=O)=O HFUXKDVQXRCOMF-UHFFFAOYSA-N 0.000 description 1
- GBYCGMHLHSSZMY-UHFFFAOYSA-N O=C(c1cc([s]c(Cl)c2Cl)c2[nH]1)NC(Cc1ccccc1)C1C=CC=NC1 Chemical compound O=C(c1cc([s]c(Cl)c2Cl)c2[nH]1)NC(Cc1ccccc1)C1C=CC=NC1 GBYCGMHLHSSZMY-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- the present invention relates to heterocyclic amide derivatives, pharmaceutically acceptable salts and in vivo hydrolysable esters thereof. These heterocyclic amides possess glycogen phosphorylase inhibitory activity and accordingly have value in the treatment of disease states associated with increased glycogen phosphorylase activity and thus are potentially useful in methods for the treatment of a warm-blooded animal such as man.
- the invention also relates to processes for the manufacture of said heterocyclic amide derivatives, to pharmaceutical compositions containing them and to their use in the manufacture of medicaments to inhibit glycogen phosphorylase activity in a warm-blooded animal such as man.
- the liver is the major organ regulating glycaemia in the post-absorptive state. Additionally, although having a smaller role in the contribution to post-prandial blood glucose levels, the response of the liver to exogenous sources of plasma glucose is key to an ability to maintain euglycaemia.
- An increased hepatic glucose output (HGO) is considered to play an important role in maintaining the elevated fasting plasma glucose (FPG) levels seen in type 2 diabetics; particularly those with a FPG >140mg/dl (7.8mM).
- HGO glycogenolysis
- gluconeogenic precursors Hellerstein et al (1997) Am J Physiol, 272: E163).
- Glycogen phosphorylase is a key enzyme in the generation by glycogenolysis of glucose- 1 -phosphate, and hence glucose in liver and also in other tissues such as muscle and neuronal tissue.
- Liver glycogen phosphorylase activity is elevated in diabetic animal models including the db/db mouse and the fa/fa rat (Aiston S et al (2000). Diabetalogia 43, 589-597). Inhibition of hepatic glycogen phosphorylase with chloroindole inhibitors (CP91149 and CP320626) has been shown to reduce both glucagon stimulated glycogenolysis and glucose output in hepatocytes (Hoover et al (1998) J Med Chem 41, 2934-8; Martin et al (1998) PNAS 95, 1776-81, WO 96/39384 and WO 96/39385). Additionally, plasma glucose concentration is reduced, in a dose related manner, in db/db and ob/ob mice following treatment with these compounds.
- Bay K 3401 Studies in conscious dogs with glucagon challenge in the absence and presence of another glycogen phosphorylase inhibitor, Bay K 3401, also show the potential utility of such agents where there is elevated circulating levels of glucagon, as in both Type 1 and Type 2 diabetes. In the presence of Bay R 3401, hepatic glucose output and arterial plasma glucose levels following a glucagon challenge were reduced significantly (Shiota et al, (1997), Am J Physiol, 273: E868).
- ES 2,081,747 discloses that certain amide derivatives of 4H-thieno[3,2-b]pyrroles and 4H-thieno[2,3-b]pyrroles are CCK antagonists and are useful in the treatment of gastric secretion disorders and in the regulation of appetite.
- the compounds disclosed in this document are disclaimed from the compound claims of the present invention.
- US 3,706,810 discloses that certain N-(aminoalkyl) derivatives of thieno[3,2- b]pyrrole-5-carboxamide are useful as analgesic and anti-depressant agents.
- the compounds disclosed in this document are disclaimed from the compound claims of the present invention.
- US 4,751,231 discloses that certain thieno[2,3-b]pyrrole-5-sulfonamides are useful in the treatment of elevated intraocular pressure and glaucoma. Certain amides are disclosed as intermediates. The compounds disclosed in this document are disclaimed from the compound claims of the present invention.
- Q is aryl, substituted aryl, heteroaryl, or substituted heteroaryl; each z and X are independently (C, CH or CH 2 ), N, O or S;
- X 1 is NR a , -CH 2 -, O or S; each — is independently a bond or is absent, provided that both — are not simultaneously bonds;
- R 1 is hydrogen, halogen, -OC C 8 alkyl, -SC C 8 alkyl, -C ⁇ . -C 8 alkyl, -CF 3 , -NH 2 -
- each R a and R b is independently hydrogen or -C ⁇ . -C 8 alkyl; Y is
- R 2 and R 3 are independently hydrogen, halogen, -C ⁇ .-C 8 alkyl, -CN, -C ⁇ C-Si(CH 3 ) 3 , -Od-Csalkyl, -SQ-Qalkyl, -CF 3 , -NH 2 , -NHC r C 8 alkyl,
- A is -NR d R d , -NR a CH 2 CH 2 OR a ,
- the heterocyclic amides of the present invention possess glycogen phosphorylase inhibitory activity and accordingly are expected to be of use in the treatment of type 2 diabetes, insulin resistance, syndrome X, hyperinsulinaemia, hyperglucagonaemia, cardiac ischaemia and obesity, particularly type 2 diabetes.
- the present invention provides a compound of formula (I):
- R and R are independently selected from hydrogen, halo, nitro, cyano, hydroxy, fluoromethyl, difluoromethyl, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, C 2 . 6 alkenyl, C 2-6 alkynyl, C 1-6 alkoxy, C ⁇ -6 alkanoyl, C ⁇ .
- R 6 alkylsulphonyl-N-(C 1 _ 6 alkyl)amino
- R 6 is hydrogen or d- ⁇ alkyl
- R 1 is selected from hydrogen, halo, nitro, cyano, hydroxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, C 2 - 6 alkenyl, C 2-6 alkynyl, C 1-6 alkoxy, C 1-6 alkanoyl, C 1-6 alkanoyloxy, N-(d- 6 alkyl)amino, N,N-(C 1-6 alkyl) 2 amino, C 1-6 alkanoylamino, N-(C 1-6 alkyl)carbamoyl, NN-(C 1 .
- R 1 may be optionally substituted on carbon by one or more groups selected from P and wherein if said heterocyclic group contains an - ⁇ H- moiety that nitrogen may be optionally substituted by a group selected from R;
- R is selected from hydrogen, halo, nitro, cyano, hydroxy, fluoromethyl, difluoromethyl, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, C .
- R a and R b are independently selected from hydrogen or C 1-6 alkyl which is optionally substituted by a group V ;
- F is C 1-6 alkylene optionally substituted by one or more Q or a direct bond
- H is selected from aryl, C 3-8 cycloalkyl and heterocyclic group; wherein H may be optionally substituted on carbon by one or more groups selected from S and wherein if said heterocyclic group contains an -NH- moiety that nitrogen may be optionally substituted by a group selected from T;
- R 3 is hydrogen or C ⁇ -6 alkyl; n is selected from 0-4; wherein the values of R 1 may be the same or different; and wherein the values of R may be the same or different;
- P, S and Q are independently selected from halo, nitro, cyano, hydroxy, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 1-6 alkoxy, d- 6 alkanoyl, d_ 6 alkanoyloxy, N-(C ⁇ _ 6 alkyl)amino, NN-(C 1 _ 6 alkyl) 2 amino, d-ealkanoylamino, N-(d- 6 alkyl)carbamoyl, NN-(C 1-6 alkyl) 2 carbamoyl, N-(C 1 .
- V is selected from halo, nitro, cyano, hydroxy, trifluoromethoxy, trifluoromethyl, amino, carboxy, carbamoyl, mercapto, sulphamoyl, methyl, ethyl, methoxy, ethoxy, acetyl, acetoxy, methylamino, ethylamino, dimethylamino, diethylamino, N-methyl-N-ethylamino, acetylamino, N-methylcarbamoyl, N-ethylcarbamoyl, NN-dimethylcarbamoyl,
- NN-diefhylcarbamoyl N-methyl-N-ethylcarbamoyl, methylthio, ethylthio, methylsulphinyl, ethylsulphinyl, mesyl, ethylsulphonyl, methoxycarbonyl, ethoxycarbonyl, N-methylsulphamoyl, N-ethylsulphamoyl, NN-dimethylsulphamoyl, NN-diethylsulphamoyl, N-methyl-N-ethylsulphamoyl, morpholino, morpholinocarbonyl, N- benzylcarbamoyl, and 4- hydroxypiperidinocarbonyl;
- R, T and U are independently selected from C 1-4 alkyl, C 1-4 alkanoyl, C 1-4 alkylsulphonyl, C 1-4 alkoxycarbonyl, carbamoyl, N-(d- 4 alkyl)carbamoyl, NN-(C 1 .
- R 6 is hydrogen or C 1-6 alkyl
- R 1 is selected from hydrogen, halo, nitro, cyano, hydroxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 1-6 alkoxy, C 1-6 alkanoyl, d- 6 alkanoyloxy, N-(d- 6 alkyl)amino, NN-(C 1-6 alkyl) 2 amino, d- 6 alkanoylamino, N-(C 1-6 alkyl)carbamoyl, NN-(C 1 .
- R 2 is selected from hydrogen, halo, nitro, cyano, hydroxy, fluoromethyl, difluoromethyl, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, C 2 - 6 alkenyl, d ⁇ alkynyl, C 1-6 alkoxy, C 1-6 alkanoyl, C ⁇ -6 alkanoyloxy, N-(C 1-6 alkyl)amino, NN-(C 1 . 6 alkyl) 2 amino, C 1-6 alkanoylamino, NN-(C 1 .
- alkyl) 2 carbamoyl, N-(C 1-6 alkyl)-N-(C 1-6 alkoxy)carbamoyl, C 1-6 alkylS(O) a wherein a is 0 to 2, C 1-6 alkoxycarbonyl, d- 6 alkoxycarbonylamino, N-(C 1-6 alkyl)sulphamoyl, NN-(C 1 .
- E and G are independently selected from a direct bond, -O-, -S-, -SO-, -SO 2 -, -OC(O)-, -C(O)O-, -C(O)-, - ⁇ R ⁇ -NR a C(O)-, -C(O)NR a -, -SO 2 NR a -, -NR a SO 2 -, -NR a C(O)NR
- R a and R b are independently selected from hydrogen or C 1-6 alkyl; F is C 1-6 alkylene optionally substituted by one or more Q or a direct bond; H is selected from aryl, C 3 . 8 cycloalkyl and heterocyclic group; wherein H may be optionally substituted on carbon by one or more groups selected from S and wherein if said heterocyclic group contains an -NH- moiety that nitrogen may be optionally substituted by a group selected from T;
- R 3 is hydrogen or C 1-6 alkyl; n is selected from 0-4; wherein the values of R 1 may be the same or different; and wherein the values of R 3 may be the same or different; P, S and Q are independently selected from halo, nitro, cyano, hydroxy, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 1-6 alkoxy, C 1-6 alkanoyl, C 1-6 alkanoyloxy, N-(C 1-6 alkyl)amino, NN-(C 1 .
- V is selected from halo, nitro, cyano, hydroxy, trifluoromethoxy, trifluoromethyl, amino, carboxy, carbamoyl, mercapto, sulphamoyl, methyl, ethyl, methoxy, ethoxy, acetyl, acetoxy, methylamino, ethylamino, dimethylamino, diethylamino, N-methyl-N-ethylamino, acetylamino, N-methylcarbamoyl, N-ethylcarbamoyl, NN-dimethylcarbamoyl, NN-diethylcarbamoyl, N-methyl-N-ethylcarbamoyl, methylthio, ethylthio, methylsulphinyl, ethylsulphinyl, mesyl, ethylsulphonyl, methoxycarbonyl,
- alkyl includes both straight and branched chain alkyl groups but references to individual alkyl groups such as "propyl" are specific for the straight chain version only.
- d. 6 alkyl includes d_ 4 alkyl, propyl, isopropyl and t-butyl.
- references to individual alkyl groups such as 'propyl' are specific for the straight chained version only and references to individual branched chain alkyl groups such as
- arylC 1-6 alkyl includes arylC 1-4 alkyl, benzyl, 1-phenylethyl and 2-phenylethyl.
- halo refers to fluoro, chloro, bromo and iodo.
- a “heterocyclic group” is a saturated, partially saturated or unsaturated, mono or bicyclic ring containing 4-12 atoms of which at least one atom is chosen from nitrogen, sulphur or oxygen, which may, unless otherwise specified, be carbon or nitrogen linked, wherein a -CH 2 - group can optionally be replaced by a -C(O)-and a ring sulphur atom may be optionally oxidised to form the S-oxide(s).
- heterocyclic group examples and suitable values of the term "heterocyclic group" are morpholino, piperidyl, pyridyl, pyranyl, pyrrolyl, imidazolyl, thiazolyl, indolyl, quinolyl, thienyl, 1,3-benzodioxolyl, 1,3-dioxolanyl, thiadiazolyl, piperazinyl, isothiazolidinyl, 1,3,4-triazolyl, tetrazolyl, pyrrolidinyl, 2-oxazolidinonyl, 5-isoxazolonyl, benz-3-azepinyl, 1,4-benzodioxanyl, thiomorpholino, pyrrolinyl, homopiperazinyl, 3,5-dioxapiperidinyl, 3-pyrazolin-5-onyl, tetrahydropyranyl, benzimidazolyl,
- a "heterocyclic group” is pyridyl, imidazolyl, thiazolyl, quinolyl, thienyl, 1,3-benzodioxolyl, 1,3-dioxolanyl, isothiazolidinyl, 1,3,4-triazolyl, tetrazolyl, 2-oxazolidinonyl, 5-isoxazolonyl, benz-3-azepinyl, hydantoinyl, 1,4-benzodioxanyl, thiomorpholino, 3-pyrazolin-5-onyl, benzimidazolyl, benzthiazolyl, imidazo[l,2-a]pyridyl, pyrimidyl, pyrazinyl, and 2,3-dihydro-l,5-benzothiazepin-4(5H)-one
- Aryl is a partially saturated or unsaturated, mono or bicyclic ring
- aryl is phenyl, naphthyl, 1,2,3,4-tetrahydronaphthyl (tetralinyl) or indanyl. More preferably aryl is phenyl, naphthyl or 1,2,3,4-tetrahydronaphthyl. Most preferably aryl is phenyl, or naphthyl.
- An example of "C 1-6 alkanoyloxy” is acetoxy.
- Examples of "C 1-6 alkoxycarbonyl” include C 1-4 alkoxycarbonyl, methoxycarbonyl, ethoxycarbonyl, n- and t-butoxycarbonyl.
- C 1-6 alkoxycarbonylamino examples include methoxycarbonylamino, ethoxycarbonylamino, n- and t-butoxycarbonylamino.
- Examples of “C 1-6 alkoxy” include methoxy, ethoxy and propoxy.
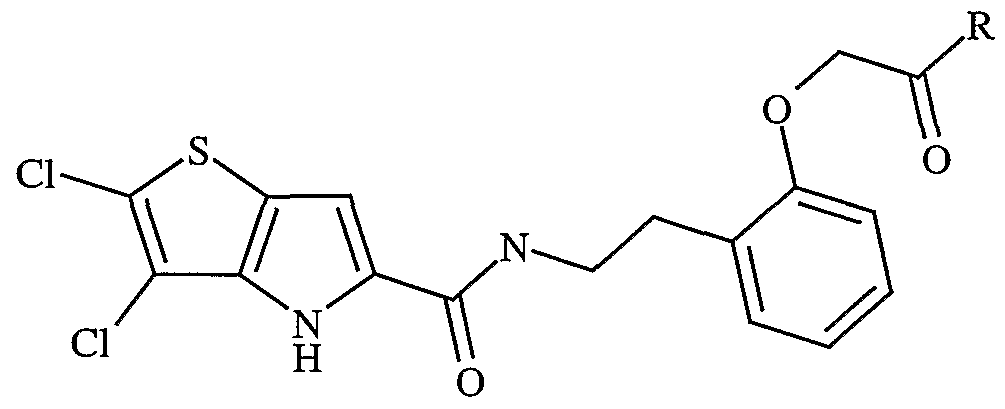
- Examples of “C 1-6 alkanoylamino” include formamido, acetamido and propionylamino. Examples of “C 1 .
- 6 alkylS(O) a wherein a is 0 to 2" include C 1-4 alkylsulphonyl, methylthio, ethylthio, methylsulphinyl, ethylsulphinyl, mesyl and ethylsulphonyl.
- Examples of "C 1-6 alkylsulphonylamino” include methylsulphonylamino, ethylsulphonylamino and propylsulphonylamino.
- C 1-6 alkylsulphonyl-N-(C 1-6 alkyl)amino examples include methylsulphonyl-N-methylamino, ethylsulphonyl-N-methylamino and propylsulphonyl-N-ethylamino.
- C 1-6 alkanoyl examples include C 1-4 alkanoyl, propionyl and acetyl.
- N-(C ⁇ -6 alkyl)amino include methylamino and ethylamino.
- 6 alkyl) 2 amino include di-N-methylamino, di-(N-ethyl)amino and N-ethyl-N-methylamino.
- C 2 _6alkenyl are vinyl, allyl and 1-propenyl.
- C 2 . 6 alkynyl are ethynyl, 1-propynyl and 2-propynyl.
- N-(Ci. 6 alkyl)sulphamoyl are N-(methyl)sulphamoyl and N-(ethyl)sulphamoyl.
- N-(C 1-6 alkyl) 2 sulphamoyl are N,N-(dimethyl)sulphamoyl and N-(methyl)-N-(ethyl)sulphamoyl.
- N-(d.. 6 alkyl)carbamoyl are N-(C 1-4 alkyl)carbamoyl, methylaminocarbonyl and ethylaminocarbonyl.
- Examples of "NN-(d_ 6 a ⁇ kyl) 2 carbamoyl” are NN-(C 1-4 alkyl)carbamoyl, dimethylaminocarbonyl and methylethylaminocarbonyl.
- C 3-8 cycloalkyl ring are cyclopropyl and cyclohexyl.
- (heterocyclic group)d. 6 alkyl) include pyridylmethyl, 3-morpholinopropyl and 2-pyrimid-2-ylethyl.
- Examples of "C 3 _ 8 cycloarkyld- 6 cycloalkyr' include cyclopropylmethyl and 2-cyclohexylpropyl.
- ''N-td-ealky ⁇ sulphamoylamdno'' are N-(methyl)sulphamoylamino and N-(ethyl)sulphamoylamino.
- Examples of "N-(C 1-6 alkyl) 2 sulphamoylamino” are
- C 1-6 alkylsulphonylaminocarbonyl include methylsulphonylaminocarbonyl, ethylsulphonylaminocarbonyl and propylsulphonylaminocarbonyl.
- a suitable pharmaceutically acceptable salt of a compound of the invention is, for example, an acid-addition salt of a compound of the invention which is sufficiently basic, for example, an acid-addition salt with, for example, an inorganic or organic acid, for example hydrochloric, hydrobromic, sulphuric, phosphoric, trifluoroacetic, citric or maleic acid.
- a suitable pharmaceutically acceptable salt of a compound of the invention which is sufficiently acidic is an alkali metal salt, for example a sodium or potassium salt, an alkaline earth metal salt, for example a calcium or magnesium salt, an ammonium salt or a salt with an organic base which affords a physiologically-acceptable cation, for example a salt with methylamine, dimethylamine, trimethylamine, piperidine, morpholine or tris-(2-hydroxyethyl)amine.
- an alkali metal salt for example a sodium or potassium salt
- an alkaline earth metal salt for example a calcium or magnesium salt
- an ammonium salt or a salt with an organic base which affords a physiologically-acceptable cation
- a salt with methylamine, dimethylamine, trimethylamine, piperidine, morpholine or tris-(2-hydroxyethyl)amine for example a salt with methylamine, dimethylamine, trimethylamine, piperidine, morpholine or tris-(2-hydroxye
- An in vivo hydrolysable ester of a compound of the formula (I) containing carboxy or hydroxy group is, for example, a pharmaceutically acceptable ester which is hydrolysed in the human or animal body to produce the parent acid or alcohol.
- Suitable pharmaceutically acceptable esters for carboxy include C 1-6 alkoxymethyl esters for example methoxymethyl, C 1-6 alkanoyloxymethyl esters for example pivaloyloxymethyl, phthalidyl esters,
- C 3-8 cycIoalkoxycarbonyloxyd_ 6 alkyl esters for example 1-cyclohexylcarbonyloxyethyl; l,3-dioxolen-2-onylmethyl esters for example 5-methyl-l,3-dioxolen-2-onylmethyl; and d_ 6 alkoxycarbonyloxyethyl esters for example 1-methoxycarbonyloxyethyl and may be formed at any carboxy group in the compounds of this invention.
- An in vivo hydrolysable ester of a compound of the formula (I) containing a hydroxy group includes inorganic esters such as phosphate esters and ⁇ -acyloxyalkyl ethers and related compounds which as a result of the in vivo hydrolysis of the ester breakdown to give the parent hydroxy group.
- inorganic esters such as phosphate esters and ⁇ -acyloxyalkyl ethers and related compounds which as a result of the in vivo hydrolysis of the ester breakdown to give the parent hydroxy group.
- ⁇ -acyloxyalkyl ethers include acetoxymethoxy and 2,2-dimethylpropionyloxy-methoxy.
- a selection of in vivo hydrolysable ester forming groups for hydroxy include alkanoyl, benzoyl, phenylacetyl and substituted benzoyl and phenylacetyl, alkoxycarbonyl (to give alkyl carbonate esters), dialkylcarbamoyl and N-(dialkylaminoethyl)-N-alkylcarbamoyl (to give carbamates), dialkylaminoacetyl and carboxyacetyl.
- substituents on benzoyl include morpholino and piperazino linked from a ring nitrogen atom via a methylene group to the 3- or 4- position of the benzoyl ring.
- Some compounds of the formula (I) may have chiral centres and/or geometric isomeric centres (E- and Z- isomers), and it is to be understood that the invention encompasses all such optical, diastereoisomers and geometric isomers that possess glycogen phosphorylase inhibitory activity.
- the invention relates to any and all tautomeric forms of the compounds of the formula (I) that possess glycogen phosphorylase inhibitory activity.
- R 1 , R 2 , R 3 , -X-Y-Z-and n are as follows. Such values may be used where appropriate with any of the definitions, claims or embodiments defined hereinbefore or hereinafter.
- R 4 and R 5 are independently selected from hydrogen, halo or d- 6 alkyl.
- R 4 and R 5 are independently selected from hydrogen, chloro, bromo or methyl.
- R 4 and R 5 are independently selected from hydrogen or chloro. More particularly R 4 and R 5 are both chloro.
- R 6 is hydrogen
- R 6 is C 1-6 alkyl.
- R 1 is selected from hydrogen, hydroxy, d_ 6 alkyl, d- 6 alkoxycarbonyl, arylCj-ealkyl and (heterocyclic group)C]. 6 alkyl; wherein R 1 may be optionally substituted on carbon by one or more groups selected from P; and
- P is selected from hydroxy and C 1 . 6 alkylsulphonyl-N-(C 1 . 6 alkyl)amino.
- R 1 is selected from hydrogen, hydroxy, methyl, methoxycarbonyl, benzyl and imidazol-4-ylmethyl; wherein R 1 may be optionally substituted on carbon by one or more groups selected from P; and
- P is selected from hydroxy and mesyl-N-(methyl)amino.
- R 1 is selected from hydrogen, hydroxy, methyl, methoxycarbonyl, mesyl-N-(methyl)aminomethyl, benzyl, hydroxymethyl and imidazol-4-ylmethyl.
- R 1 is selected from hydrogen, mesyl-N-(methyl)aminomethyl or benzyl.
- R 2 is selected from N,N-(C 1 . 4 alkyl) 2 carbamoyl,
- F is C 1-6 alkylene optionally substituted by one or more Q or a direct bond
- H is selected from aryl, C 3-8 cycloalkyl and heterocyclic group; wherein H may be optionally substituted on carbon by one or more groups selected from S and wherein if said heterocyclic group contains an -NH- moiety that nitrogen may be optionally substituted by a group selected from T;
- S is selected from halo, hydroxy, trifluoromethyl, sulphamoyl, ureido, C 1-6 alkyl, C 1-6 alkoxy, NN-(C 1 . 6 alkyl) 2 amino, C 1 . 6 alkanoylamino and aryl; wherein S may be optionally substituted on carbon by one or more groups selected from V;
- N is carbamoyl
- T is independently selected from C 1- alkyl or phenyl. More preferably R 2 is selected from NN-dimethylcarbamoyl,
- E and G are independently selected from a direct bond, -O-, -S-, -C(O)-, - ⁇ R a -, -C(O)NR a -, -NR a SO 2 - and -NR a C(O)O-; wherein R a is hydrogen;
- F is methylene optionally substituted by one or more Q or a direct bond
- H is selected from phenyl, naphthyl, cyclopropyl, thiomorpholino, pyridyl, thiazolyl, isothiazolyl, morpholinyl, 2,3-dihydro-l,5-benzothiazepin-4(5H)-onyl, 5-oxo-3-pyrazolinyl, 2-oxazolidinonyl, 5-hydroxy-l,3,4,5-tetrahydro-benzo[b]azepin-2-onyl, 5-oxo-2-isoxazolinyl, imidazo[l,2-a]pyridinyl, benzothiazolyl, 2,5-dioxoimidazolidinyl, pyrazinyl, pyridazinyl, imidazolyl, benzimidazolyl, tetrazolyl, quinolyl, 1,3-
- T is independently selected from methyl or phenyl.
- R is selected from NN-dimethylcarbamoyl, NN-dimethylsulphamoyl- amino, mesylaminocarbonyl, 2-methoxyphenyl, phenoxy, 2-phenylcyclopropyl, thien-2-yl, 4- fluorophenyl, benzoyl, thiomorpholino, anilinocarbonyl, pyrid-2-ylamino, thiazol-2-yl, benzylsulphonylamino, 2,3-dihydro-l,5-benzothiazepin-4(5H)-one-3-yl, l-phenyl-2,3- dimethyl-5-oxo-3-pyrazolin-4-yl, 3-phenyl-2-oxazolidinon-5-yl, 5-hydroxy-l,3,4,5-tetrahydro- benzo[b]azepin-2-onyl, 3-phenyl-5-oxo-2-isoxazolin-4
- R 2 is selected from NN-dimethylcarbamoyl, NN-dimethylsulphamoylamino, mesylaminocarbonyl, 2-methoxyphenyl, phenoxy, 2- phenylcyclopropyl, thien-2-yl, 4-fluorophenyl, benzoyl, thiomorpholino, anilinocarbonyl, pyrid-2-ylamino, thiazol-2-yl, benzylsulphonylamino, 2,3-dihydro-l,5-benzothiazepin-4(5 ⁇ )- one-3-yl, l-phenyl-2,3-dimethyl-5-oxo-3-pyrazolin-4-yl, 3-phenyl-2-oxazolidinon-5-yl, 5- hydroxy- 1 ,3,4,5-tetrahydro-benzo[b]azepin-2-onyl, 3-phenyl-5-oxo-2-iso
- R 2 is selected from NN-dimethylcarbamoyl, phenoxy, 2- phenylcyclopropyl, thien-2-yl, 4-fluorophenyl, benzoyl, thiomorpholino, anilinocarbonyl, pyrid-2-ylamino or thiazol-2-yl.
- R 3 is hydrogen
- n is selected from 0-3; wherein the values of R 1 may be the same or different; and wherein the values of R 3 may be the same or different.
- n is selected from 0-2; wherein the values of R 1 may be the same or different; and wherein the values of R 3 may be the same or different.
- n is 2; wherein the values of R 1 may be the same or different; and wherein the values of R 3 may be the same or different. In one aspect of the invention preferably n is i.
- n 0.
- preferred compounds of the invention are any one of the Examples or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof.
- R 4 and R 5 are independently selected from hydrogen, halo or C 1-6 alkyl
- R 1 is selected from hydrogen, hydroxy, C 1-6 alkyl, C 1-6 alkoxycarbonyl, aryld- 6 alkyl and (heterocyclic group)C 1-6 alkyl; wherein R 1 may be optionally substituted on carbon by one or more groups selected from P; and
- P is selected from hydroxy and C 1-6 alkylsulphonyl-N-(C 1 . 6 alkyl)amino;
- R 2 is selected from NN-(C 1-4 alkyl) 2 carbamoyl, NN-(C 1 . 6 alkyl) 2 sulphamoylamino, Ci- 6 alkylsulphonylaminocarbonyl and a group -E-F-G-H; wherein E and G are independently selected from a direct bond, -O-, -S-, -C(O)-, -NR a -, -C(O)NR a -, -NR a SO 2 - and -NR a C(O)O-; wherein R a is hydrogen;
- F is C ⁇ -6 alkylene optionally substituted by one or more Q or a direct bond
- H is selected from aryl, C -8 cycloalkyl and heterocyclic group; wherein H may be optionally substituted on carbon by one or more groups selected from S and wherein if said heterocyclic group contains an -NH- moiety that nitrogen may be optionally substituted by a group selected from T;
- S is selected from halo, hydroxy, trifluoromethyl, sulphamoyl, ureido, d-ealkyl, d_ alkoxy, N ) N-(C 1 _ 6 alkyl) 2 amino, C 1-6 alkanoylamino and aryl; wherein S may be optionally substituted on carbon by one or more groups selected from N; Q is hydroxy; V is carbamoyl; and
- X 1 is NR a , -CH 2 -, O or S.
- R 1 is selected from hydrogen, hydroxy, methyl, methoxycarbonyl, mesyl-N-(methyl)aminomethyl and hydroxymethyl ;
- R 2 is selected from NN-dimethylcarbamoyl, NN-dimethylsulphamoylamino, mesylaminocarbonyl, 2-methoxyphenyl, phenoxy, 2-phenylcyclopropyl, thien-2-yl, 4- fluorophenyl, benzoyl, thiomorpholino, anilinocarbonyl, pyrid-2-ylamino, thiazol-2-yl, benzylsulphonylamino, 2,3-dihydro- 1 ,5-benzothiazepin-4(5H)-one-3-yl, 1 -phenyl-2,3- dimethyl-5-oxo-3-pyrazolin-4-yl, 3-phenyl-2-oxazolidinon-5-yl, 5-hydroxy-l ,3, 4,5-tetrahydro- benzo[B]azepin-2-onyl, 3-phenyl-5-oxo-2-isoxa
- R 3 is hydrogen; and n is selected from 0-3; wherein the values of R 1 may be the same or different; or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof.
- the present invention provides a compound of formula (I) (as depicted above) wherein:
- R 2 is a group -E-F-G-H; wherein E, F and G are each a direct bond; H is a C 3 _ 12 cycloalkyl which is optionally fused to a benz ring wherein H may be optionally substituted on carbon by one or more groups S which are independently selected from halo, nitro, cyano, hydroxy, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, d ⁇ alkenyl, C 2 - 6 alkynyl, C 1-6 alkoxy, C 1-6 alkanoyl, C 1-6 alkanoyloxy, N-(C 1-6 alkyl)amino, NN-(C 1-6 alkyl) 2 amino, C ⁇ -6 alkanoylamino, N-(C 1-6 alkyl)carbamoyl, NN-(C 1 -
- N is selected from halo, nitro, cyano, hydroxy, trifluoromethoxy, trifluoromethyl, amino, carboxy, carbamoyl, mercapto, sulphamoyl, methyl, ethyl, methoxy, ethoxy, acetyl, acetoxy, methylamino, ethylamino, dimethylamino, diethylamino, N-methyl-N-ethylamino, acetylamino, N-methylcarbamoyl, N-ethylcarbamoyl, NN-dimethylcarbamoyl, NN-diethylcarbamoyl, N-methyl-N-ethylcarbamoyl, methylthio, ethylthio, methylsulphinyl, ethylsulphinyl, mesyl, ethylsulphonyl, methoxycarbonyl,
- R 2 , R 4 , and R 5 are as follows. Such values may be used where appropriate with any of the definitions, claims or embodiments defined hereinbefore or hereinafter.
- R 4 and R 5 are independently selected from hydrogen, halo or d- ⁇ alkyl.
- H is indanyl, 1,2,3,4-tetrahydronaphthyl or cyclopropyl. More preferably H is indanyl or 1,2,3,4-tetrahydronaphthyl. Most preferably H is indanyl.
- S is independently selected from from halo, nitro, cyano, hydroxy, amino, carboxy, carbamoyl, d- 6 alkyl, d_ 6 alkoxy, C 1-6 alkanoyl, d_ 6 alkanoyloxy, N-(d- 6 alkyl)amino, NN-(C 1 . 6 alkyl) 2 amino, d- 6 alkanoylamino, N-(C 1-6 alkyl)carbamoyl, NN-(C 1 . 6 alkyl) 2 carbamoyl, N-(C 1 . 6 alkyl)-N-(C 1-6 alkoxy)carbamoyl, C ⁇ .
- R 2 is a group -E-F-G-H; wherein E, F and G are each a direct bond; and
- H is a cyclic amide of formula
- k is 0, 1, 2 or 3 and 1 is 0, 1, 2 or 3 such that the sum of k and 1 is 2 or 3 and wherein one of the carbon atoms governed by k or 1 may be replaced by sulphur and wherein H is optionally substituted on carbon by one or more groups selected from S and may be independently optionally substituted on nitrogen by a group selected from T;
- S is selected from halo, nitro, cyano, hydroxy, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, d_ 6 alkyl, C 2 - 6 alkenyl, C 2-6 alkynyl, C 1-6 alkoxy, C 1-6 alkanoyl, C 1 . 6 alkanoyloxy, N ⁇ d- ⁇ alk amino, NN-(C 1-6 alkyl) 2 amino, C 1-6 alkanoylamino, N-(C 1-6 alkyl)carbamoyl, NN-(C 1 .
- T and U are independently selected from C 1-4 alkyl, C 1-4 alkanoyl, C ⁇ _ 4 alkylsulphonyl, C 1- alkoxycarbonyl, carbamoyl, N-(C 1 . 4 alkyl)carbamoyl, NN-(C 1 . 4 alkyl)carbamoyl, phenyl, benzyl, benzyloxycarbonyl, benzoyl and phenylsulphonyl wherein T and U may be optionally and independently substituted on carbon by one or more groups selected from V;
- V is selected from halo, nitro, cyano, hydroxy, trifluoromethoxy, trifluoromethyl, amino, carboxy, carbamoyl, mercapto, sulphamoyl, methyl, ethyl, methoxy, ethoxy, acetyl, acetoxy, methylamino, ethylamino, dimethylamino, diethylamino, N-methyl-N-ethylamino, acetylamino, N-methylcarbamoyl, N-ethylcarbamoyl, NN-dimethylcarbamoyl,
- R 4 , R 5 and H are as follows. Such values may be used where appropriate with any of the definitions, claims or embodiments defined hereinbefore or hereinafter.
- R 4 and R 5 are independently selected from hydrogen, halo or C 1-6 alkyl.
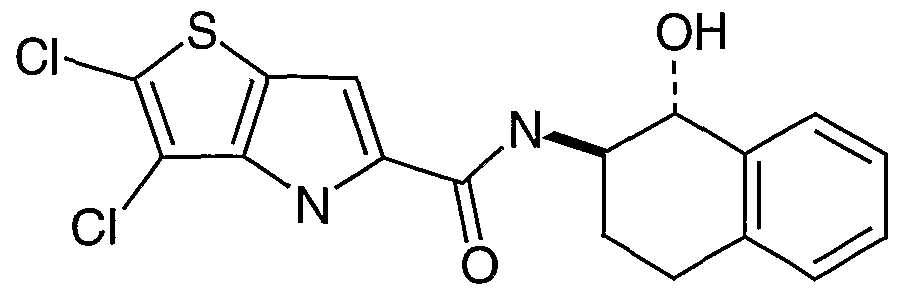
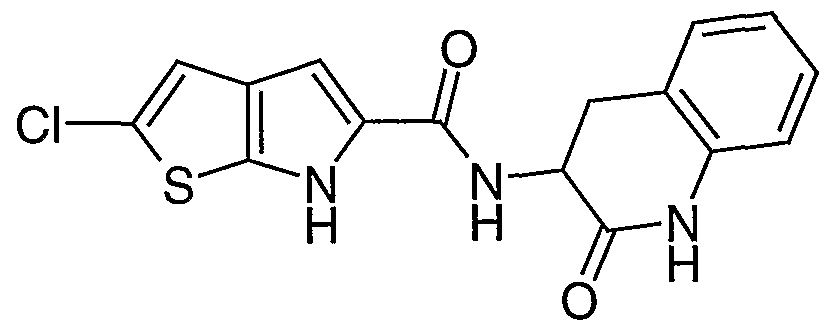
- H is 1,2,3,4-tetrahydroquinolyl, 2-oxo- 1,2,3,4-tetrahydroquinolyl, 4-oxo-2,3,4,5-tetrahydrobenz[l,5]thiazepin-3-yl, 2-oxo-2,3,4,5- tetrahydro-lH-benz[t>]azepinyl, 2,3,4,5-tetrahydro-lH-benz[6]azepinyl or 3-oxo-2,3,4,5- tetrahydro-lH-benz[c]azepinyl each optionally substituted on carbon by one or more groups selected from S wherein S is selected from hydroxy, d_ 6 a ⁇ kyl or d- 6 alkoxy, and each independently optionally substituted on nitrogen by a group selected from T wherein T is selected from d- 4 alkyl or C 1-4 alkanoyl.
- H is 2-oxo-l,2,3,4-tetrahydroquinol-3-yl, l-methyl-2-oxo-l,2,3,4- tetrahydroquinol-3-yl, 4-oxo-2,3,4,5-tetrahydrobenz[l,5]thiazepin-3-yl, 5-hydroxy-2-oxo- 2,3,4,5-tetrahydro-lH-benz[Z>]azepin-4-yl, 2-oxo-2,3,4,5-tetrahydro-lH-benz[ ⁇ ]azepin-3-yl or 3-oxo-2,3 ,4,5-tetrahydro- lH-benz[c]azepin-4-yl.
- the present invention provides a compound of formula (I)
- R is selected from a group -E-F-G-H; wherein E, F and G are each a direct bond;
- H is an unsaturated five membered heterocyclic group containing at least one nitrogen atom and one or two ring atoms selected from oxygen and sulphur and wherein H may be optionally substituted on carbon by one or more groups S which are independently selected from halo, nitro, cyano, hydroxy, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 1-6 alkoxy, C ⁇ -6 alkanoyl, C 1-6 alkanoyloxy, N-(d_ 6 alkyl)amino, NN-(C 1 .
- groups S which are independently selected from halo, nitro, cyano, hydroxy, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ure
- R 3 is hydrogen or C 1-6 alkyl; or a pharmaceutically acceptable salt thereof.
- R 1 , R 3 and H are as follows. Such values may be used where appropriate with any of the definitions, claims or embodiments defined hereinbefore or hereinafter.
- R 1 is selected from hydrogen or benzyl and more preferably benzyl.
- R is hydrogen
- H is 1,3,4-oxadiazolyl, isoxazolyl, oxazolyl or 1,2,4-oxadiazolyl. More preferably H is 5-ethoxycarbonyl-l,3,4-oxadiazol-2-yl, 4- phenylisoxazol-3-yl, 3-phenyl-l,2,4-oxadiazol-5-yl, 4-methoxycarbonyloxazol-5-yl or 3- methylisoxazol-5-yl.
- H may be optionally substituted on carbon by one or more groups S which are independently selected from halo, carboxy, C 1-6 alkyl, d- ⁇ alkoxy, d- ⁇ alkanoyloxy, N-(d. 6 alkyl)amino, NN-(C 1 . 6 alkyl) 2 amino, C 1-6 alkanoylamino, d- 6 alkoxycarbonyl, C 3 _ 8 cycloalkyl and aryl groups.
- S is C 1-6 alkoxy, d- ⁇ aikoxycarbonyl or phenyl.
- R 2 is a group -E-F-G-H; wherein E is a direct bond; F is methylene; wherein G is -C(O) ⁇ R a -, wherein R a is selected from hydrogen or C ⁇ _ 6 alkyl which is optionally substituted by a group V ; H is aryl which may be optionally substituted on carbon by one or more groups selected from S;
- S is selected from halo, nitro, cyano, hydroxy, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, C 2-6 alkenyl, C 2 - 6 alkynyl, C 1-6 alkoxy, C ⁇ -6 alkanoyl, C 1-6 alkanoyloxy, N-(C 1-6 alkyl)amino, NN-(C ⁇ -6 alkyl) 2 amino, d_ 6 alkanoylamino, N ⁇ d-ealky carbamoyl, NN-(C ⁇ .
- V is selected from halo, nitro, cyano, hydroxy, trifluoromethoxy, trifluoromethyl, amino, carboxy, carbamoyl, mercapto, sulphamoyl, methyl, ethyl, methoxy, ethoxy, acetyl, acetoxy, methylamino, ethylamino, dimethylamino, diethylamino, N-methyl-N-ethylamino, acetylamino, N-methylcarbamoyl, N-ethylcarbamoyl, NN-dimethylcarbamoyl,
- NN-diethylcarbamoyl N-methyl-N-ethylcarbamoyl, methylthio, ethylthio, methylsulphinyl, ethylsulphinyl, mesyl, ethylsulphonyl, methoxycarbonyl, ethoxycarbonyl, N-methylsulphamoyl, N-ethylsulphamoyl, NN-dimethylsulphamoyl, NN-diethylsulphamoyl, N-methyl-N-ethylsulphamoyl, morpholino, morpholinocarbonyl, N- benzylcarbamoyl , and 4- hydroxypiperidinocarbonyl; or a pharmaceutically acceptable salt thereof.
- R 1 , R 2 , R 3 , -X-Y-Z-and n are as follows. Such values may be used where appropriate with any of the definitions, claims or embodiments defined hereinbefore or hereinafter.
- H is aryl.
- V is cyano or hydroxy.
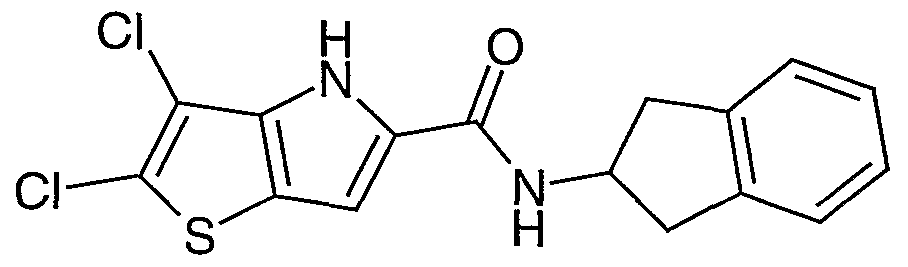
- Specific compounds of the present invention are: 2 ,3 -dichloro-5- [N-(2-phenoxyethyl)carbamoyl] -4H-thieno [3 ,2-b] pyrrole ; 2,3-dichloro-5- ⁇ N-[2-(2-thienyl)ethyl]carbamoyl ⁇ -4H-thieno[3,2-b]pyrrole; 2,3-dichloro-5- ⁇ N-[2-(2-methoxyphenyl)ethyl]carbamoyl ⁇ -4H-thieno[3,2-t3]pyrrole; 2,3-dichloro-5-[N-(2-phenyl-l-cyclopropyl)carbamoyl]-4H-thieno[3,2-t)]pyrrole; 2,3-dichloro-5- ⁇ N-[2-(4-fluorophenyl)ethyl]carbam
- Another aspect of the present invention provides a process for preparing a compound of formula (I) or a pharmaceutically acceptable salt or an in vivo hydrolysable ester thereof which process (wherein R 1 , R 2 , R 3 , -X-Y-Z-and n are, unless otherwise specified, as defined in formula (I)) comprises of: a) reacting an acid of the formula (II):
- Standard peptide coupling reagents known in the art can be employed as suitable coupling reagents, or for example carbonyldiimidazole, l-ethyl-3-(3-dimethylaminopropyl)carbodi-imide hydrochloride and dicyclohexyl-carbodiimide, optionally in the presence of a catalyst such as 1- hydroxybenzotriazole, dimethylaminopyridine or 4-pynOlidinopyridine, optionally in the presence of a base for example triethylamine, di-isopropylethylamine, pyridine, or 2,6-di- ⁇ /£y/-pyridines such as 2,6-lutidine or 2,6-di-tert-butylpyridine.
- Suitable solvents include dimethylacetamide, dichloromethane, benzene, tetrahydrofuran and dimethylformamide.
- the coupling reaction may conveniently be performed at a temperature in the range
- Suitable activated acid derivatives include acid halides, for example acid chlorides, and active esters, for example pentafluorophenyl esters.
- the reaction of these types of compounds with amines is well known in the art, for example they may be reacted in the presence of a base, such as those described above, and in a suitable solvent, such as those described above. The reaction may conveniently be performed at a temperature in the range of -40 to 40°C.
- the acids of formula (II) may be prepared according to Scheme 1:
- aromatic substitution reactions include the introduction of a nitro group using concentrated nitric acid, the introduction of an acyl group using, for example, an acyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; the introduction of an alkyl group using an alkyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; and the introduction of a halogeno group.
- modifications include the reduction of a nitro group to an amino group by for example, catalytic hydrogenation with a nickel catalyst or treatment with iron in the presence of hydrochloric acid with heating; oxidation of alkylthio to alkylsulphinyl or alkylsulphonyl.
- a suitable protecting group for an amino or alkylamino group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an alkoxycarbonyl group, for example a methoxycarbonyl, ethoxycarbonyl or t-butoxycarbonyl group, an arylrnethoxycarbonyl group, for example benzyloxycarbonyl, or an aroyl group, for example benzoyl.
- the deprotection conditions for the above protecting groups necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or alkoxycarbonyl group or an aroyl group may be removed for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an acyl group such as a t-butoxycarbonyl group may be removed, for example, by treatment with a suitable acid as hydrochloric, sulphuric or phosphoric acid or trifluoroacetic acid and an arylmethoxycarbonyl group such as a benzyloxycarbonyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon, or by treatment with a Lewis acid for example boron tris(trifluoroacetate).
- a suitable alternative protecting group for a primary amino group is, for example, a phthaloyl group which may be removed by treatment with an alkylamine, for example dimethylaminopropylamine, or with hydrazine.
- a suitable protecting group for a hydroxy group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an aroyl group, for example benzoyl, or an arylmethyl group, for example benzyl.
- the deprotection conditions for the above protecting groups will necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or an aroyl group may be removed, for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an arylmethyl group such as a benzyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- a suitable protecting group for a carboxy group is, for example, an esterifying group, for example a methyl or an ethyl group which may be removed, for example, by hydrolysis with a base such as sodium hydroxide, or for example a t-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- a base such as sodium hydroxide
- a t-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- the protecting groups may be removed at any convenient stage in the synthesis using conventional techniques well known in the chemical art.
- the compounds defined in the present invention possesses glycogen phosphorylase inhibitory activity. This property may be assessed, for example, using the procedure set out below.
- the activity of the compounds is determined by measuring the inhibitory effect of the compounds in the direction of glycogen synthesis, the conversion of glucose- 1 -phosphate into glycogen with the release of inorganic phosphate, as described in EP 0 846464 A2.
- the reactions were in 96well microplate format in a volume of lOO ⁇ l.
- the change in optical density due to inorganic phosphate formation was measured at 620nM in a Labsystems iEMS Reader MF by the general method of (Nordlie R.C and Arion W.J, Methods of Enzymology, 1966, 619-625).
- the reaction is in 50mM HEPES, 2.5mM MgCl 2 , 2.25mM ethylene glycol- bis(b-aminoethyl ether) NNN',N'-tetraacetic acid, lOOmM KC1, 2mM D-(+)-glucose pH7.2, containing 0.5mM dithiothreitol, the assay buffer solution, with 0. lmg type HI glycogen,
- glycogen phosphorylase a from rabbit muscle and 0.5mM glucose- 1 -phosphate.
- GP is pre-incubated in the assay buffer solution with the type HI glycogen at 2.5 mg ml "1 for 30 minutes.
- 40 ⁇ l of the enzyme solution is added to 25 ⁇ l assay buffer solution and the reaction started with the addition of 25 ⁇ l 2mM glucose- 1 -phosphate.
- Compounds to be tested are prepared in lO ⁇ l 10% DMSO in assay buffer solution, with final concentration of 1% DMSO in the assay.
- the non-inhibited activity of GP ⁇ is measured in the presence of lO ⁇ l 10% DMSO in assay buffer solution and maximum inhibition measured in the presence of 30 ⁇ M CP320626 (Hoover et al (1998) J Med Chem 41, 2934-8; Martin et al (1998) P ⁇ AS 95, 1776-81).
- the reaction is stopped after 30min with the addition of 50 ⁇ l acidic ammonium molybdate solution, 12ug ml "1 in 3.48% H 2 SO 4 with 1% sodium lauryl sulphate and lOug ml "1 ascorbic acid. After 30 minutes at room temperature the absorbency at 620nm is measured.
- the activity of the compounds is alternatively determined by measuring the inhibitory effect of the compounds on glycogen degradation, the production of glucose- 1 -phosphate from glycogen is monitored by the multienzyme coupled assay, as described in EP 0 846464 A2, general method of Pesce et al ( Pesce, M A, Bodourian, S H, Harris, R C, and Nicholson, J F (1977) Clinical Chemistry 23, 1171 - 1717).
- the reactions were in 384well microplate format in a volume of 50 ⁇ l.
- the change in fluorescence due to the conversion of the co-factor NAD to NADH is measured at 340nM excitation, 465nm emission in a Tecan Ultra Multifunctional Microplate Reader.
- the reaction is in 50mM HEPES, 3.5mM KH 2 PO 4, 2.5mM MgCl 2 , 2.5mM ethylene glycol-bis(b-aminoethyl ether) NNN'N-tetraacetic acid, lOOmM KC1, 8mM D-(+)-glucose pH7.2, containing 0.5mM dithiothreitol, the assay buffer solution.
- Human recombinant liver glycogen phosphorylase a (hrl GP ⁇ ) 20nM is pre-incubated in assay buffer solution with 6.25mM NAD, 1.25mg type HI glycogen at 1.25 mg ml "1 the reagent buffer, for 30 minutes.
- the coupling enzymes phosphoglucomutase and glucose-6-phosphate dehydrogenase ( Sigma) are prepared in reagent buffer, final concentration 0.25Units per well. 20 ⁇ l of the hrl GPa solution is added to lO ⁇ l compound solution and the reaction started with the addition of 20ul coupling enzyme solution. Compounds to be tested are prepared in lO ⁇ l 5% DMSO in assay buffer solution, with final concentration of 1% DMSO in the assay. The non-inhibited activity of GP ⁇ is measured in the presence of lO ⁇ l 5% DMSO in assay buffer solution and maximum inhibition measured in the presence of 5mgs ml "1 N- ethylmaleimide. After 6 hours at 30°C Relative Fluoresence Units (RFUs) are measured at 340nM excitation, 465nm emission .
- REUs Relative Fluoresence Units
- the assay is performed at a test concentration of inhibitor of lO ⁇ M or lOO ⁇ M. Compounds demonstrating significant inhibition at one or both of these concentrations may be further evaluated using a range of test concentrations of inhibitor to determine an IC 50 , a concentration predicted to inhibit the enzyme reaction by 50%. Activity is calculated as follows :-
- % inhibition (1 - (compound RFUs - fully inhibited RFUs)/ (non-inhibited rate RFUs - fully inhibited RFUs)) * 100.
- Typical IC 50 values for compounds of the invention when tested in the above assay are in the range lOO ⁇ M to InM.
- the inhibitory activity of compounds was further tested in rat primary hepatocytes.
- Rat hepatocytes were isolated by the collagenase perfusion technique, general method of Seglen (P.O. Seglen, Methods Cell Biology (1976) 13 29-83). Cells were cultured on Nunclon six well culture plates in DMEM with high level of glucose containing 10% foetal calf serum, NEAA, Glutamine, penicillin /streptomycin ((100units/100ug)/ml) for 4 to 6 hours. The hepatocytes were then cultured in the DMEM solution without foetal calf serum and with lOnM insulin and lOnM dexamethasone.
- a pharmaceutical composition which comprises a compound of the formula (I), or a pharmaceutically acceptable salt or in vivo hydrolysable ester thereof, as defined hereinbefore in association with a pharmaceutically-acceptable diluent or carrier.
- composition may be in a form suitable for oral administration, for example as a tablet or capsule, for parenteral injection (including intravenous, subcutaneous, intramuscular, intravascular or infusion) as a sterile solution, suspension or emulsion, for topical administration as an ointment or cream or for rectal administration as a suppository.
- parenteral injection including intravenous, subcutaneous, intramuscular, intravascular or infusion
- sterile solution emulsion
- topical administration as an ointment or cream or for rectal administration as a suppository.
- compositions may be prepared in a conventional manner using conventional excipients.
- the compound of formula (I) will normally be administered to a warm-blooded animal at a unit dose within the range 5-5000 mg per square meter body area of the animal, i.e. approximately 0.1-100 mg/kg, and this normally provides a therapeutically-effective dose.
- a unit dose form such as a tablet or capsule will usually contain, for example 1-250 mg of active ingredient.
- a daily dose in the range of 1-50 mg/kg is employed.
- the daily dose will necessarily be varied depending upon the host treated, the particular route of administration, and the severity of the illness being treated. Accordingly the optimum dosage may be determined by the practitioner who is treating any particular patient.
- R 1 may be optionally substituted on carbon by one or more groups selected from P and wherein if said heterocyclic group contains an - ⁇ H- moiety that nitrogen may be optionally substituted by a group selected from R;
- R 2 is selected from hydrogen, halo, nitro, cyano, hydroxy, fluoromethyl, difluoromethyl, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, d.
- E and G are independently selected from a direct bond, -O-, -S-, -SO-, -SO2-, -OC(O)-, -C(O)O-, -C(O)-, - ⁇ R a -, -NR a C(O)-, -C(O)NR a -, -SO 2 NR a -, -NR a SO 2 -, -NR a C(O)NR b -, -OC(O)NR a -, -NR a C(O)O-, -NR a SO 2 NR b -, -SO 2 NR a C(O)- and -C(O)NR a SO 2 -; wherein R a and R b are independently selected from hydrogen or C 1-6 alkyl which is optionally substituted by a group V ;
- F is d- 6 alkylene optionally substituted by one or more Q or a direct bond
- H is selected from aryl, C 3 . 8 cycloalkyl and heterocyclic group; wherein H may be optionally substituted on carbon by one or more groups selected from S and wherein if said heterocyclic group contains an -NH- moiety that nitrogen may be optionally substituted by a group selected from T;
- R 3 is hydrogen or C 1-6 alkyl; n is selected from 0-4; wherein the values of R 1 may be the same or different; and wherein the values of R 3 may be the same or different;
- P, S and Q are independently selected from halo, nitro, cyano, hydroxy, trifluoromethyl, trifluoromethoxy, amino, carboxy, carbamoyl, mercapto, sulphamoyl, ureido, C 1-6 alkyl, C 2 . 6 alkenyl, C 2-6 alkynyl, d_ 6 alkoxy, C 1-6 alkanoyl, C 1-6 alkanoyloxy, N-(C 1-6 alkyl)amino, NN-(d_ 6 alkyl) 2 amino, d.
- V is selected from halo, nitro, cyano, hydroxy, trifluoromethoxy, trifluoromethyl, amino, carboxy, carbamoyl, mercapto, sulphamoyl, methyl, ethyl, methoxy, ethoxy, acetyl, acetoxy, methylamino, ethylamino, dimethylamino, diethylamino, N-methyl-N-ethylamino, acetylamino, N-methylcarbamoyl, N-ethylcarbamoyl, NN-dimethylcarbamo
- X 1 is NR a , -CH 2 -, O or S; or a pharmaceutically acceptable salt or in vivo hydrolysable ester thereof, as defined hereinbefore in the manufacture of a medicament for use in the production of a glycogen phosphorylase inhibitory effect in a warm-blooded animal such as man.
- a compound of the formula (I'), or a pharmaceutically acceptable salt or in vivo hydrolysable ester thereof as defined hereinbefore in the manufacture of a medicament for use in the treatment of type 2 diabetes, insulin resistance, syndrome X, hyperinsulinaemia, hyperglucagonaemia, cardiac ischaemia or obesity in a warm-blooded animal such as man.
- a compound of the formula (I'), or a pharmaceutically acceptable salt or in vivo hydrolysable ester thereof as defined hereinbefore in the manufacture of a medicament for use in the treatment of type 2 diabetes in a warm-blooded animal such as man.
- a method of producing a glycogen phosphorylase inhibitory effect in a warm-blooded animal, such as man, in need of such treatment which comprises administering to said animal an effective amount of a compound of formula (I').
- a method of treating type 2 diabetes, insulin resistance, syndrome X, hyperinsulinaemia, hyperglucagonaemia, cardiac ischaemia or obesity which comprises administering to said animal an effective amount of a compound of formula (I').
- a method of treating type 2 diabetes in a warm-blooded animal, such as man, in need of such treatment which comprises administering to said animal an effective amount of a compound of formula (V).
- the size of the dose required for the therapeutic or prophylactic treatment of a particular cell-proliferation disease will necessarily be varied depending on the host treated, the route of administration and the severity of the illness being treated.
- a unit dose in the range, for example, 1-100 mg/kg, preferably 1-50 mg/kg is envisaged.
- the compounds of formula (I) and their pharmaceutically acceptable salts are also useful as pharmacological tools in the development and standardisation of in vitro and in vivo test systems for the evaluation of the effects of inhibitors of cell cycle activity in laboratory animals such as cats, dogs, rabbits, monkeys, rats and mice, as part of the search for new therapeutic agents.
- the alternative and preferred embodiments of the compounds of the invention described herein also apply. Examples
- temperatures are given in degrees Celsius (°C); operations were carried out at room or ambient temperature, that is, at a temperature in the range of 18-25°C and under an atmosphere of an inert gas such as argon;
- Example #5 2.3-Dichloro-5- ⁇ N-r2-(4-fluorophenyl)ethyllcarbamoyl
- Example #6 2.3-dichloro-5-rN-(N-phenylcarbamoylmethyl)carbamoyll-4H-thienor3.2-
- Example #11 The following compounds were made by the process of Example #11 using 5-carboxy- 3-chloro-4H-thieno[3,2-_7]pyrrole (Method #7) and the appropriate amine:
- Example #12 3-Chloro-5- ⁇ N-r2-(thiomorpholino)ethvncarbamoyll-4H-thieno[3,2-&lpyrrole
- Example #13 3-Chloro-5- ⁇ N-r2-(N-methylmethanesulphonamido)-l-(thiazol-2- vDethyllcarbamoyl I -4H-thieno F3 ,2-fclpyrrole
- Example #14 3-Chloro-5-rN-(benzoylmethyl)carbamoyll-4H-thienor3,2-&lpyrrole
- Example #15 3-Chloro-5- ⁇ N-r2-(2-methoxyphenyl)ethyllcarbamoyli-4H-thienor3.2- bl pyrrole
- Example #16 3-Chloro-5- ⁇ N-r2-(2-thienyl)ethyllcarbamoyl)-4H-thienor3,2-tjlpyrrole
- Example #17 3-Chloro-5-rN-(2-phenyl-l-cvclopropyl)carbamoyll-4H-thienor3.2-t>1pyrrole
- Example #18 3-Chloro-5- ⁇ N-r2-(4-fluorophenyl)ethyllcarbamoyl ⁇ -4H-thienor3.2-61pyrrole
- Example #19 3-Chloro-5-rN-(2-phenoxyethyl)carbamoyll-4H-thienor3,2-b1pyrrole
- Example #20 3-Chloro-5-(N-r2-(l-phenylmethanesulphonamido)ethyllcarbamoyll-4H- thieno ⁇ 3,2-b pyrrole
- Example #21 3-Chloro-5-rN-(4-oxo-2,3,4.5-tetrahydrobenzri,51thiazepin-3-yl)carbamoyll- 4H-thienor3.2
- Example #22 The following compounds were made by the process of Example #22 using 5-carboxy- 2-chloro-4H-thieno[3,2-b]pyrrole (Method #8) and the appropriate amine:
- Example #23 2-Chloro-5-rN-(2-phenyl-l-cvclopropyl)carbamoyll-4H-thienor3.2-/j1pyrrole
- Example #24 2-Chloro-5-rN-(N-phenylcarbamoylmethyl)carbamovn-4H-thienor3 1 2- fclpyrrole
- Example #27 2-Chloro-5-rN-(2-phenoxyethyl)carbamoyll-4H-thienor3.2-61pyrrole
- Example #28 2-Chloro-5- IN- r2-(2-thienyl)ethyllcarbamoyl 1 -4H-thieno ⁇ 3 ,2- ⁇ lpyrrole
- Example #29 2-Chloro-5-(N-r2-(4-fluorophenyl)ethyllcarbamoyll-4H-thienor3.2-t>l ⁇ yrrole
- Example #30 2-Chloro-5- ⁇ N-r2-(N-methylmethanesulphonamido)-l-(thiazol-2- yl)ethyl1carbamoyl)-4H-thienor3,2-/j1pyrrole
- Example #31 2-Chloro-5- ⁇ N-r2-(thiomorpholino)ethyllcarbamoyl
- Example #32 2.3-Dichloro-5-rN-(2,3-dimethyl-5-oxo-l-phenyl-2.5-dihvdro-lH-pyrazol-4- yl)carbamoyl1-4H-thieno[ ⁇ 3,2-t31pyrrole
- Example #33 2,3-Dichloro-5-rN-(4-sulphamoylphenylmethyl)carbamovn-4H-thienor3,2- ⁇ lpyrrole
- Example #37 2,3-Dichloro-5-rN-(5-oxo-3-phenyl-4.5-dihvdroisoxazol-4-yl)carbamoyll-4H- thieno
- Example #38 2.3-Dichloro-5-rN-(5-hvdroxy-2-oxo-2.3.4.5-tetrahvdro- ! ⁇ -benzr61azepin-4- yl)carbamoyl1-4H-thienor3.2-t>1pyrrole
- Example #40 2.3-Dichloro-5-IN-r(4-dimethylaminophenyl)methyllcarbamoyl
- Example #41 5-
- Example #42 2,3-Dichloro-5-
- Example #43 2,3-Dichloro-5-rN-( ⁇ -(K)-hydroxy- -methylphenethyl)carbamoyll-4H- thieno ⁇ 3 ,2-blpyrrole
- Example #45 2,3-Dichloro-5- ⁇ N-r2-(4-hvdroxyphenyl)ethyllcarbamoyl ⁇ 4H-thieno[3,2- t>l pyrrole
- Example #46 2,3-Dichloro-5- ⁇ N-r(benzimidazol-2-yl)methyllcarbamoyl ⁇ -4H-thienor3,2- t>lpyrrole
- Example #50 2,3-Dichloro-5- ⁇ N-[(6-trifluoromethylpyrid-3-yl)methyllcarbamoyl)-4H- thieno ⁇ 32-b ⁇ pyrrole
- Example #51 2.3-Dichloro-5-fN-(2-r(2-pyridazinyl)methyllcarbamoyl ⁇ -4H-thienor3.2- ⁇ lpyrrole
- Example #55 2-Chloro-5-(N- ⁇ 2-r(3-trifluoromethylpyrid-2-yl)aminolethyl ⁇ carbamoyl)-4H- thieno ⁇ ,2-61 pyrrole
- Example #56 2.3-Dichloro-5- ⁇ N-r2-(4-sulphamoylphenyl)ethyllcarbamoyl)-4H-thienor3.2- ⁇ lpyrrole
- Example #57 2.3-Dichloro-5-(N-r2-( ' 2-pyridyl)ethyllcarbamoyll-4H-thienor3.2-t31pyrrole
- Example #58 2.3-Dichloro-5- ⁇ N-(2-f 1 -hvdroxymethyl-2-(4-imidazolyl)ethyllcarbamoyl ⁇ -
- Example #59 2.3-Dichloro-5-(N-(2-r(3-quinolyl)methyllcarbamoyll-4H-thienor3,2- fclpyrrole
- Example #60 5-(N-r3-(4-Acetamidophenoxy)-2-hvdroxypropyl1carbamoyl)-2,3-dichloro-
- Example #64 5- ⁇ N-r2-Benzylthio-l-(hydroxymethyl)ethyllcarbamoyl ⁇ -2.3-dichloro-4H- thienor3.2-/31pyrrole
- Example #65 2.3-Dichloro-5- ⁇ N-r2-(dimethvaminosulphonylamino)ethyllcarbamoyl)-4H- thienof3 ,2-_jlpyrrole
- Example #75 2.3-Dichloro-5-rN-(l-phenylcvclobutyl)methyl)carbamoyll-4H-thienor3,2- b ⁇ pyrrole
- Example #76 2.3-Dichloro-5-rN-( ⁇ -methylphenethyl)carbamoyll-4H-thienor3.2-Z>lpyrrole
- Example #78 5- r N-(N-Benzylcarbamoylmethyl)carbamovH- 2,3-dichloro-4H-thienor3,2- t>lpyrrole
- Example #79 5-rN-(N-Benzyl-N-methylcarbamoylmethyl)carbamoyll-2.3-dichloro-4H- thienor3,2-&lpyrrole
- Example #86 2,3-dichloro-5-(N-[N-methyl-N-(4-methylphenyl)carbamoylmethyll- carbamoyl ⁇ -4H-thienor3 ,2-_>1pyrrole
- Example #87 2.3-dichloro-5-
- Example #91 2.3-dichloro-5-
- Example #92 2,3-dichloro-5- ⁇ N-rN-(2-hvdroxyethyl)carbamoylmethyllcarbamoyl ) -4H- thienor3.2-t>lpyrrole
- Example #93 2,3-dichloro-5- ⁇ N-rN-(3-hvdroxypropyl)carbamoylmethyllcarbamoyll-4H- thienor3,2-Z>lpyrrole
- Example #96 2.3-dichloro-5- ⁇ N-rN-(2.3-dihvdroxypropyl)carbamoylmethyllcarbamoyl)-
- Example #97 The following compounds were made by the process of Example #97 using 5-(N- carboxymethylcarbamoyl)-2,3-dichloro-4H-thieno[3,2-b]pyrrole (Method #12) and the appropriate commercially available amine:
- Example #98 2.3-dichloro-5- ⁇ N-[N-(5-isoquinolyl)carbamoylmethyllcarbamoyl)-4H- thieno ⁇ 32-b ⁇ pyrrole
- Example #101 2.3-dichloro-5- ⁇ N-rN-(2.4-difluorophenyl)-N-methyl-carbamoylmethyll- carbamoyl ⁇ -4H-thieno ⁇ 3 ,2-/31pyrrole
- Example #102 2.3-dichloro-5- ⁇ N-r(L2.3.4-tetrahydro-l-quinolyl)carbonylmethyl1- carbamoyl
- Example #103 2.3-dichloro-5- ⁇ N-rN-(2-cvanoethyl)-N-methylcarbamoylmethyll- carbamoyl ⁇ -4H-thienoF3,2-&1pyrrole
- Example #104 2.3 -dichloro-5 - ⁇ N- rN-(4-hvdroxypiperidino carbamoylmethyll carbamoyl ) -
- Example #106 2,3-dichloro-5-rN-(N-isopropylcarbamoylmethyl)carbamoyn-4H-thienor3,2- /J>lpyrrole
- Example #107 2.3-dichloro-5-rN-(N-isopropyl-N-methylcarbamoylmethyl)carbamoyll-4H- thieno ⁇ 3.2-bl pyrrole
- the two component mixture was separated using bond-elute silica column chromatography (eluent: dichloromethane-dichloromethane/methanol 5% gradient) to afford the less polar product (sulfone) as a white powder (57mg 33%) and the more polar product (sulfoxide) as a white solid (62mg, 37%).
- Example #113 The following compounds were made by the process of Example #113 using 5- carboxy-2,3-dichloro-4H-thieno[3,2-b]pyrrole (Method #9) and the appropriate amine:
- Example #115 2.3-Dichloro-5-rN-(4-phenylisoxazol-3-ylmethyl)carbamoyll-4H-thienor3.2- tjlpyrrole
- Example #116 2.3-Dichloro-5-('N-(2-r2-(2-morpholinoethoxy)phenyllethyllcarbamoyl)-4H- thieno ⁇ 3 ,2-/31 pyrrole
- Example #117 2,3-Dichloro-5-(N- ⁇ 2-r2-(methoxycarbonylmethoxy)phenyllethyl ⁇ - carbamoyl)-4H-thienoF3,2-/j1pyrrole
- Example #123 2.3-Dichloro-5-(N-(2-r2-(N-methylcarbamoylmethoxy)phenyllethyll- carbamoyl)-4H-thienor3 1 2-/jlpyrrole
- Example #125 2.3-Dichloro-5-(N- ⁇ 2-r2-(morpholinocarbonylmethoxy)phenyllethyl
- Example #126 5-(N- ⁇ 2-r2-(N-Benzylcarbamoylmethoxy)phenyllethyl ⁇ carbamoyl)-2.3- dichloro-4H-thieno[3,2-&lpyrrole
- Example #127 2,3-Dichloro-5-(N- ⁇ 2-r2-(4-hvdroxypiperidinocarbonylmethoxy)phenvn- ethyl)carbamoyl)-4H-thienor3,2-/J>lpyrrole
- the reaction mixture was diluted with ethyl acetate (75ml) washed with dilute citric acid, water and brine, dried over magnesium sulphate and concentrated.
- the crude material was purified by bond elute silica column chromatography (eluent - DCM/Ethyl acetate gradient 0-50%) to give the title compound (147mg, 34%).
- the reaction mixture was diluted with ethyl acetate (50ml), water (20ml) was added and the p ⁇ adjusted to 7 with dilute hydrochloric acid.
- the organic fraction was separated washed with water and brine, dried over magnesium sulphate and concentrated.
- the crude material was purified by bond elute silica column chromatography (eluent - Ethyl acetate ) to give the title compound as a solid (128mg, 67%).
- the reaction mixture was diluted with ethyl acetate (100ml), water (20ml) was added and the p ⁇ adjusted to 7 with dilute hydrochloric acid.
- the organic fraction was separated washed with water and brine, dried over magnesium sulphate and concentrated.
- the crude material was purified by bond elute silica column chromatography (eluent - Ethyl acetate ) to give the title compound as a solid (72mg, 34%)
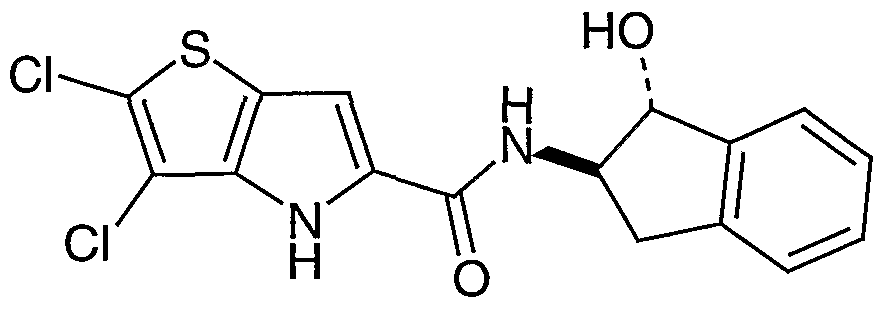
- 2,3-Dichloro-5- [N-( 1 -hydroxyindan-2-yl)carbamoyl] -4H-thieno[3 ,2-t)]pyrrole (Example #133) was subject to preparative ⁇ PLC under the following conditions to give 2,3- dichloro-5-[N-((-/S,2S)-l-hydroxyindan-2-yl)carbamoyl]-4H-thieno[3,2-t3]pyrrole as a white solid (9 mg) and 2,3-dichloro-5-[N-((H-,2i-)-l-hydroxyindan-2-yl)carbamoyl]-4H- thieno[3,2-t)]pyrrole as a white solid (12 mg).
- Example #141 2,3-Dichloro-5-[N-(3-methylisoxazol-5-yl)methyllcarbamoyll-4H- thienor3.2-/jlpyrrole
- Example #142 2,3-Dichloro-5-rN-(4-hvdroxy-l,l-dioxotetrahvdrothiophen-3-yl)carbamoyll-
- Example #143 2.3-dichloro-5-(N- ⁇ N-methyl-N-r(l-oxo-1.2.3.4-tetrahvdronaphth-2- yl)methyllcarbamoylmethyllcarbamoyl)-4H-thieno[3,2-t)lpyrrole
- Triethylamine (lOlmg, l.Ommol) was added to a suspension 5-[N-(l-aminoindan-2- yl)carbamoyl]-2,3-dichloro-4H-thieno[3,2-j]pyrrole trifluoroacetic acid salt (Example #150, 240mg, 0.5mmol) in dichloromethane (4ml), followed by acetyl chloride (47mg, 0.6mmol) dissolved in dichloromethane (1ml) and the reaction stirred at room temperature for 6 hours during which a white solid precipitated.
- the starting materials for the Examples above are either commercially available or are readily prepared by standard methods from known materials. For example the following reactions are illustrations but not limitations of the preparation of some of the starting materials used in the above reactions.
- Methanolic sodium methoxide solution (28%) (5 ml, 25.9 mmol) was diluted with MeO ⁇ (5 ml) and was cooled to -25°C under nitrogen.
- Aldehyde Aldehyde ref. Gronowitz et al. Tetrahedron Vol.32 1976 p.1403
- N-(tert-Butoxycarbonyl)glycine (875mg, 5mmol) was dissolved in DMF (7ml) containing DIPEA (3.5ml, 20mmol) and benzylamine (536mg, 5mmol). The mixture was allowed to stand for one minute before addition of 0-(7-Azabenzotriazol-l-yl)-NNN',N'- tetramethyluronium hexafluorophosphate (HATU) (2.09g, 5.5mmol). The solution was allowed to stand for approximately 18 hours before being partitioned between ethyl acetate (50ml) and water (50ml).
- HATU 0-(7-Azabenzotriazol-l-yl)-NNN',N'- tetramethyluronium hexafluorophosphate
- Aqueous ammonium chloride (10%, 200 ml) was added and the solution was extracted using diethyl ether.
- the aqueous layer was acidified and re-extracted using diethyl ether.
- the combined organic layers were dried, filtered and concentrated under reduced pressure.
- the residue (5.30 g, 22.6 mmol) was dissolved in ethanol (100 ml) containing pyridine (18 ml) and hydroxylamine hydrochloride (1.88 g, 27.1 mmol).
- the mixture was heated under reflux in an inert atmosphere for 2.5 hours before being cooled and concentrated under reduced pressure.
- the residue was suspended in water and cooled to 0°C.
- the mixture was acidified to pH 4 using aqueous hydrochloric acid (2.0 M).
- the resulting solid (9.2 g, 42.8 mmol) was dissolved in dry DMF (30 ml) and the solution was heated on a steam bath for 2 hours. Ammonium chloride (2.22 g, 41.5 mmol) was added to the hot solution along with sodium azide (2.68 g, 56.3 mmol). The mixture was cooled and left to stir for over 48 hours. The solution was filtered and basified to p ⁇ 8 using aqueous potassium bicarbonate. The aqueous layer was washed using EtOAc before being acidified using dilute aqueous hydrochloric acid. A white solid was filtered off.
- N-Benzyloxycarbonylglycine (2.09 g) was dissolved in toluene (40 ml) and DMF (5 drops).
- Oxalyl chloride (1.3 ml) was added and the mixture was stirred at ambient temperature for 2 hours.
- the solution was diluted with diethyl ether (25 ml) and was added dropwise to a solution of l-amino-3-phenoxy-2-propanol (1.67 g) in diethyl ether (25 ml).
- Sodium hydroxide (0.4 g) was dissolved in water (1.5 ml) and was added to the mixture. The solution was stirred for greater than 48 hours before being filtered.
- N-t-Butoxycarboylglycine (1.92 g, 1.1 mmol) was dissolved in dry EtOAc (20 ml) and the solution was cooled to -25°C.
- N-Methyl morpholine (1.1 g, 1.1 mmol) was added and the mixture was stirred for 2 minutes before the addition of ethyl chloroformate (1.08 g, 1.0 mmol).
- a suspension of 5-amino-3- methylisothiazole hydrochloride (1.5 g, 1.0 mmol) was added in triethylamine (1.01 g, 1.0 mmol) and dry EtOAc (10 ml). The mixture was stirred at ambient temperature for 17 hours before being filtered.
- Ethanolamine (6.0 ml) was suspended in dry xylene (100 ml) and the mixture was stirred at ambient temperature with sodium hydride (3 g). After 20 minutes the solution was cooled to 0°C. 3,6-Dichloropyridazine (15.0 g) was added. The solution was warmed to ambient temperature and was stirred for 15 hours before being extracted with chloroform. The chloroform was concentrated and the residue was taken up in EtOAc before being filtered. Ethanolic HCl was added and a white solid was filtered off. This compound (550 mg) was dissolved in MeOH (130 ml) and palladium on charcoal (5%, 200 mg) was added. The suspension was stirred under an atmosphere of hydrogen for 6 hours before being filtered. The filtrate was concentrated and the residue was triturated using diethyl ether. The title compound was isolated as a white solid (340 mg).
- Ethylene diamine (30 ml) was dissolved in DCM (130 ml) with di-t-Zmtyl dicarbonate (13.5 g). The mixture was stirred at ambient temperature for 30 minutes before the solvent was decanted off. The residue was washed with water and dried. The product (6.1 g) was dissolved in ethanol (15 ml). Chloroacetic acid (5.77 g) solution in aqueous sodium hydroxide (1 M, 61 ml) was added dropwise, followed by more aqueous sodium hydroxide (1 M, 31 ml). The mixture was stirred for 17 hours before being washed with diethyl ether.
- Benzyl chloroformate (5 g) was added to the aqueous phase and the mixture was stirred at ambient temperature for 4 hours. The solution was washed with diethyl ether and the aqueous layer was acidified using aqueous citric acid (30%). The solution was extracted using EtOAc. The combined organic layers were washed with brine, then water before being dried, filtered and concentrated. The residue was triturated using diethyl ether/petrol mixture and the required intermediate was isolated as a white solid. Following repeated procedure on a larger scale, this solid (28 g) was dissolved in ethanol (250 ml) containing palladium on charcoal (10%, 4.6 g). The suspension was stirred under an atmosphere of hydrogen until the reaction was complete.
- the mixture was filtered and the filtrate was concentrated.
- the residue (3 g, was treated with potassium cyanate (1.3 g) in water (30 ml) and the mixture was heated under reflux for 2 hours. An excess of concentrated hydrochloric acid was added and the mixture was heated for a short time before being concentrated.
- the residue was taken up in water (50 ml) and was loaded onto basic Amberlite® IRA resin. The resin was washed with water until the eluent was found to be neutral. Dilute aqueous hydrochloric acid was used to elute the crude product from the resin. The acidic fractions were concentrated and the residue was triturated with ethanol. The title compound was isolated as orange crystals (950 mg).
- 3-Hydroxyphenylacetic acid (30.4 ml) was dissolved in MeOH (160 ml). Concentrated sulphuric acid (1.6 ml) was added and the mixture was heated under reflux for 6 hours. The mixture was concentrated to low bulk before the addition of toluene (120 ml). The mixture was washed with water, saturated aqueous sodium bicarbonate and brine. The combined organic layers were reduced in volume by one half before being stirred with ammonia solution (180 ml) for 16 hours. The mixture was concentrated and filtered. The solid was washed with water and was dried.
- the mixture was filtered and the filtrate was concentrated.
- the residual oil was purified by flash column chromatography, using a gradient of MeOH in EtOAc as eluent, to afford both cis- and trans- isomers of the required intermediate.
- the trans- isomer (1.6 g) was dissolved in dry diethyl ether (180 ml). The solution was heated under reflux using Soxhlet equipment with lithium aluminium hydride (2.0 g) for 16 hours. The solution was cooled and water (2 ml) was added with sodium hydroxide solution (3 M, 2 ml), followed by more water (6 ml). The metal residues were removed by filtration and the filtrate was extracted using DCM. The combined organic layers were dried, filtered and concentrated. The residue was triturated using ethanol to afford the title compound as a white solid.
- N-(Benzyloxycarbonyl)-2-(2-hydroxyphenyl)ethylamine (542mg, 2mmol) dissolved in DMF 5ml was treated with potassium carbonate (325 mesh, 350mg, 2.5mmol) and 2- chloroethyl p-toluenesulphonate (484ul,2.6mmol) and the mixture was stirred at 60C overnight.
- Isoamyl nitrite (15 ml, 108 mmol) was added to a solution of indan-l,2-dione (12 g, 90 mmol) in methanol (380 ml) at 45°C followed by concentrated HCl (12 ml) dropwise over 5 minutes. The reaction mixture was stirred for 3 hours at room temperature. Excess isoamyl nitrite (1 ml) and concentrated HCl (1 ml) was added and the suspension stirred for a further 15 minutes. On cooling to room temperature a white precipitate formed.
- Tetralone (1 eq, 100 mmol, 15 g) and methylamine hydrochloride (2.5 eq, 250 mmol, 15 g) and paraformaldehyde (1 eq, 100 mmol, 3 g) in ethanol (100 ml) were heated under reflux for 16 hours.
- the reaction mixture was cooled and filtered to afford 2- (methylaminomethyl)-3,4,dihydro-l-(2H)-naphthalenone as a solid (14 g).
- the filtrate was concentrated and the residue was taken up in ethyl acetate before being washed with aqueous sodium bicarbonate, dried, filtered and concentrated.
- the residue was left to stand in a solution of hydrogen bromide in acetic acid for 30 minutes.
- the mixture was triturated with diethyl ether and the solvent was decanted off.
- the residue was treated with ammonia solution and this was extracted with chloroform.
- the chloroform was concentrated and the residue was recrystallised from ethanol to afford the title compound.
- (+/-) tr ⁇ 7M-2-bromo ⁇ l-hydroxyindan (21.0g, O.lmol) and potassium phthalimide (42.0g, 0.22 mol ) in dry DMF (120ml) was heated at 100°C for 5 hours.
- the reaction mixture was cooled and evaporated to an oil which was triturated with ethyl acetate and filtered.
- the filtrates were evaporated and purified by chromatography on silica with 2: 1 iso- hexane:ethyl acetate as eluent to give (+l-)-trans -l-hydroxy-2-phthalimido indan as a pale yellow amorphous powder (17.7g, 63%).
- the crade product was purified by chromatography on silica with 4: 1 w ⁇ -hexane: ethyl acetate as eluent to give the title compound as a white solid (16.1g, 96%); NMR 1.42(9H, s), 2.78(1H, dd), 3.0(1H, dd), 4.3-4.42(lH, m), 4.78-4.9(lH, m), 4.9-5.0(lH, m), 6.3(1H, d), 7.0-7.25(4H, m).
- the above formulations may be obtained by conventional procedures well known in the pharmaceutical art.
- the tablets (a)-(c) may be enteric coated by conventional means, for example to provide a coating of cellulose acetate phthalate.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Child & Adolescent Psychology (AREA)
- Emergency Medicine (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Endocrinology (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Indole Compounds (AREA)
- Pyrrole Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Enzymes And Modification Thereof (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description






















































































































Claims



Priority Applications (24)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EEP200300083A EE200300083A (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrole amides as glycogen phosphorylase inhibitors |
| CA002417594A CA2417594A1 (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| US10/344,506 US20030232875A1 (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| DK01961577T DK1317459T3 (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolylamides as glucogen phosphorylase inhibitors |
| JP2002525151A JP2004508376A (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolylamides as glycogen phosphorylase inhibitors |
| AU2001282833A AU2001282833B2 (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| SI200130112T SI1317459T1 (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| BR0113606-2A BR0113606A (en) | 2000-09-06 | 2001-08-31 | Compound, pharmaceutical composition, and use of a compound or pharmaceutically acceptable salt or in vivo hydrolysable ester thereof |
| HU0400784A HUP0400784A3 (en) | 2000-09-06 | 2001-08-31 | Bicyclic heterocondensated pyrrolcarboxamide derivatives, their use and pharmaceutical compositions containing them |
| AU8283301A AU8283301A (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| NZ524011A NZ524011A (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| PL01361024A PL361024A1 (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| KR1020037003265A KR100802369B1 (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| MXPA03001512A MXPA03001512A (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors . |
| SK259-2003A SK2592003A3 (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| DE60102710T DE60102710T2 (en) | 2000-09-06 | 2001-08-31 | BICYCLIC PYRROLYL AMIDE AS GLUCOSE PHOSPHORYLASE HEMMER |
| IL15429101A IL154291A0 (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| UA2003021026A UA73781C2 (en) | 2000-09-06 | 2001-08-31 | Dicyclic pyrolylamides as inhibitors of glucogenphosphorylase |
| EP01961577A EP1317459B1 (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| AT01961577T ATE263772T1 (en) | 2000-09-06 | 2001-08-31 | BIZYCLIC PYRROLYL AMIDES AS GLUCOGEN PHOSPHORYLASE INHIBITORS |
| IS6727A IS2110B (en) | 2000-09-06 | 2003-02-24 | Bi-ring pyrrolylamide as glucogenophosphorylase inhibitors |
| NO20031024A NO20031024D0 (en) | 2000-09-06 | 2003-03-05 | Bicyclic pyrrolyl amides as glucogenic phosphorylase inhibitors |
| BG107624A BG107624A (en) | 2000-09-06 | 2003-03-10 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
| HK03107519A HK1055299A1 (en) | 2000-09-06 | 2003-10-16 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0021831.3 | 2000-09-06 | ||
| GBGB0021831.3A GB0021831D0 (en) | 2000-09-06 | 2000-09-06 | Chemical compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2002020530A1 true WO2002020530A1 (en) | 2002-03-14 |
Family
ID=9898927
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/SE2001/001880 WO2002020530A1 (en) | 2000-09-06 | 2001-08-31 | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors |
Country Status (31)
| Country | Link |
|---|---|
| US (1) | US20030232875A1 (en) |
| EP (1) | EP1317459B1 (en) |
| JP (1) | JP2004508376A (en) |
| KR (1) | KR100802369B1 (en) |
| CN (2) | CN1264846C (en) |
| AT (1) | ATE263772T1 (en) |
| AU (2) | AU8283301A (en) |
| BG (1) | BG107624A (en) |
| BR (1) | BR0113606A (en) |
| CA (1) | CA2417594A1 (en) |
| CZ (1) | CZ2003616A3 (en) |
| DE (1) | DE60102710T2 (en) |
| DK (1) | DK1317459T3 (en) |
| EE (1) | EE200300083A (en) |
| ES (1) | ES2217183T3 (en) |
| GB (1) | GB0021831D0 (en) |
| HK (1) | HK1055299A1 (en) |
| HU (1) | HUP0400784A3 (en) |
| IL (1) | IL154291A0 (en) |
| IS (1) | IS2110B (en) |
| MX (1) | MXPA03001512A (en) |
| NO (1) | NO20031024D0 (en) |
| NZ (1) | NZ524011A (en) |
| PL (1) | PL361024A1 (en) |
| PT (1) | PT1317459E (en) |
| RU (1) | RU2003104013A (en) |
| SK (1) | SK2592003A3 (en) |
| TR (1) | TR200401659T4 (en) |
| UA (1) | UA73781C2 (en) |
| WO (1) | WO2002020530A1 (en) |
| ZA (1) | ZA200301013B (en) |
Cited By (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1125580A2 (en) * | 2000-01-24 | 2001-08-22 | Pfizer Products Inc. | Methods of treating diabetic cardiomyopathy using glycogen phosphorylase inhibitors |
| WO2002064565A1 (en) * | 2001-02-13 | 2002-08-22 | Aventis Pharma Deutschland Gmbh | Acylated 1,2,3,4-tetrahydronaphthyl amines and their use as pharmaceutical |
| WO2003074485A2 (en) * | 2002-03-06 | 2003-09-12 | Astrazeneca Ab | Indole-amide derivatives and their use as glycogen phosphorylase inhibitors |
| WO2003074513A2 (en) * | 2002-03-06 | 2003-09-12 | Astrazeneca Ab | Indole amide derivatives and their use as glycogen phosphorylase inhibitors |
| WO2003074532A1 (en) * | 2002-03-06 | 2003-09-12 | Astrazeneca Ab | Heterocyclic amide derivatives as inhibitors of glycogen phosphorylase |
| WO2003074531A1 (en) * | 2002-03-06 | 2003-09-12 | Astrazeneca Ab | Heterocyclic amide derivatives having glycogen phosphorylase inhibitory activity |
| EP1388535A1 (en) * | 2002-08-07 | 2004-02-11 | Aventis Pharma Deutschland GmbH | Acylated arylcycloalkylamines and their use as pharmaceuticals |
| WO2004041780A2 (en) * | 2002-11-07 | 2004-05-21 | Pfizer Products Inc. | N-(indole-2-carbonyl) amides as anti-diabetic agents |
| WO2004092158A1 (en) * | 2003-04-17 | 2004-10-28 | Pfizer Products Inc. | Carboxamide derivatives as anti-diabetic agents |
| WO2004113345A1 (en) * | 2003-06-20 | 2004-12-29 | Japan Tobacco Inc. | Fused pyrrole compound and medicinal use thereof |
| WO2005013981A1 (en) * | 2003-08-07 | 2005-02-17 | Astrazeneca Ab | Heterocyclic amide derivatives which possess glycogen phosphorylase inhibitory activity |
| WO2005013975A1 (en) * | 2003-08-07 | 2005-02-17 | Astrazeneca Ab | Indolamide derivatives which possess glycogen phosphorylase inhibitory activity |
| WO2004104001A3 (en) * | 2003-05-21 | 2005-03-03 | Osi Pharm Inc | Pyrrolopyridine-2-carboxylic acid amide inhibitors of glycogen phoshorylase |
| WO2005018637A1 (en) * | 2003-08-22 | 2005-03-03 | Astrazeneca Ab | Heterocyclic amide derivatives which possess glycogen phosphorylase inhibitory activity |
| WO2005019172A1 (en) * | 2003-08-22 | 2005-03-03 | Astrazeneca Ab | Indol-2-amides as glycogen phosphorylase inhibitors |
| WO2005020985A1 (en) * | 2003-08-29 | 2005-03-10 | Astrazeneca Ab | Indolamide derivatives which possess glycogen phosphorylase inhibitory activity |
| WO2005020986A1 (en) * | 2003-08-29 | 2005-03-10 | Astrazeneca Ab | Heterocyclic amide derivatives which posses glycogen phosphorylase inhibitory activity |
| WO2005020987A1 (en) * | 2003-08-30 | 2005-03-10 | Astrazeneca Ab | Heterocyclic amide derivatives which posses glycogen phosphorylase inhibitory activity |
| JP2006505541A (en) * | 2002-10-03 | 2006-02-16 | アストラゼネカ アクチボラグ | Methods and intermediates for producing thienopyrrole derivatives |
| WO2006053274A2 (en) * | 2004-11-15 | 2006-05-18 | Bristol-Myers Squibb Company | 2-amino-1-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| WO2006055463A2 (en) * | 2004-11-15 | 2006-05-26 | Bristol-Myers Squibb Company | 2-amino-3-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| WO2006055462A1 (en) * | 2004-11-15 | 2006-05-26 | Bristol-Myers Squibb Company | 2-amino-4-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| US7057046B2 (en) * | 2002-05-20 | 2006-06-06 | Bristol-Myers Squibb Company | Lactam glycogen phosphorylase inhibitors and method of use |
| WO2006059165A1 (en) * | 2004-12-02 | 2006-06-08 | Prosidion Limited | Pyrrolopyridine-2-carboxylic acid amide derivative useful as inhibitor of glycogen phosphorylase |
| WO2006082401A1 (en) * | 2005-02-05 | 2006-08-10 | Astrazeneca Ab | Chemical compounds |
| US7138415B2 (en) | 2002-03-06 | 2006-11-21 | Astrazeneca Ab | Indolamid derivatives which possess glycogenphosphorylase inhibitory activity |
| US7166636B2 (en) | 2002-03-06 | 2007-01-23 | Astrazeneca Ab | Indole-amid derivatives which possess glycogen phosphorylase inhibitory activity |
| US7179839B2 (en) | 2001-02-13 | 2007-02-20 | Sanofi-Aventis Deutschland Gmbh | Acylated indanyl amines and their use as pharmaceuticals |
| US7186735B2 (en) | 2002-08-07 | 2007-03-06 | Sanofi-Aventis Deutschland Gmbh | Acylated arylcycloalkylamines and their use as pharmaceuticals |
| US7223786B2 (en) | 2004-11-15 | 2007-05-29 | Bristol-Myers Squibb Company | 2-aminonaphthalene derivatives and related glycogen phosphorylase inhibitors |
| EP1841426A1 (en) * | 2005-01-19 | 2007-10-10 | Merck & Co., Inc. | Tertiary carbinamines having substituted heterocycles, which are active as inhibitors of beta-secretase, for the treatment of alzheimer's disease |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| US7307174B2 (en) | 2002-10-03 | 2007-12-11 | Astrazeneca Ab | Process and intermediates for the preparation of thienopyrrole derivatives |
| US7405210B2 (en) | 2003-05-21 | 2008-07-29 | Osi Pharmaceuticals, Inc. | Pyrrolopyridine-2-carboxylic acid amide inhibitors of glycogen phosphorylase |
| WO2010057141A2 (en) * | 2008-11-17 | 2010-05-20 | The Regents Of The University Of Michigan | Alphavirus inhibitors and uses thereof |
| US7884124B2 (en) | 2006-06-30 | 2011-02-08 | Sepracor Inc. | Fluoro-substituted inhibitors of D-amino acid oxidase |
| US7893098B2 (en) | 2003-12-29 | 2011-02-22 | Sepracor Inc. | Pyrrole and pyrazole DAAO inhibitors |
| US7902252B2 (en) | 2007-01-18 | 2011-03-08 | Sepracor, Inc. | Inhibitors of D-amino acid oxidase |
| EP2298772A1 (en) * | 2006-10-18 | 2011-03-23 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds |
| US8053603B2 (en) | 2006-01-06 | 2011-11-08 | Sunovion Pharmaceuticals Inc. | Tetralone-based monoamine reuptake inhibitors |
| US8097760B2 (en) | 2006-03-31 | 2012-01-17 | Sunovion Pharmacuticals Inc. | Preparation of chiral amides and amines |
| WO2012027331A1 (en) | 2010-08-27 | 2012-03-01 | Ironwood Pharmaceuticals, Inc. | Compositions and methods for treating or preventing metabolic syndrome and related diseases and disorders |
| WO2013138352A1 (en) | 2012-03-15 | 2013-09-19 | Synergy Pharmaceuticals Inc. | Formulations of guanylate cyclase c agonists and methods of use |
| US8669291B2 (en) | 2007-05-31 | 2014-03-11 | Sunovion Pharmaceuticals Inc. | Phenyl substituted cycloalkylamines as monoamine reuptake inhibitors |
| US8877975B2 (en) | 2006-01-06 | 2014-11-04 | Sunovion Pharmaceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| WO2014197720A2 (en) | 2013-06-05 | 2014-12-11 | Synergy Pharmaceuticals, Inc. | Ultra-pure agonists of guanylate cyclase c, method of making and using same |
| EP2923706A1 (en) | 2009-12-03 | 2015-09-30 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase useful for the treatment of hypercholesterolemia |
| WO2016030534A1 (en) | 2014-08-29 | 2016-03-03 | Tes Pharma S.R.L. | INHIBITORS OF α-AMINO-β-CARBOXYMUCONIC ACID SEMIALDEHYDE DECARBOXYLASE |
| WO2017004500A1 (en) * | 2015-07-02 | 2017-01-05 | Genentech, Inc. | Bicyclic lactams and methods of use thereof |
| WO2018069532A1 (en) | 2016-10-14 | 2018-04-19 | Tes Pharma S.R.L. | Inhibitors of alpha-amino-beta-carboxymuconic acid semialdehyde decarboxylase |
| WO2020104456A1 (en) | 2018-11-20 | 2020-05-28 | Tes Pharma S.R.L | INHIBITORS OF α-AMINO-β-CARBOXYMUCONIC ACID SEMIALDEHYDE DECARBOXYLASE |
| US10947226B2 (en) | 2016-10-17 | 2021-03-16 | Genentech, Inc. | Bicyclic pyridone lactams and methods of use thereof |
| US11072607B2 (en) | 2016-12-16 | 2021-07-27 | Genentech, Inc. | Inhibitors of RIP1 kinase and methods of use thereof |
| US11634436B2 (en) | 2018-04-20 | 2023-04-25 | Genentech, Inc. | Pyridine lactam compounds and methods of use thereof |
| EP4066896A4 (en) * | 2019-11-27 | 2024-03-27 | Riken | G9a inhibitor |
| RU2827714C1 (en) * | 2015-07-02 | 2024-10-01 | Ф. Хоффманн-Ля Рош Аг | Bicyclic lactams and methods of use thereof |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10215907A1 (en) * | 2002-04-11 | 2003-11-06 | Aventis Pharma Gmbh | Acyl-4-carboxyphenyl-urea derivatives, processes for their preparation and their use |
| DE10215908B4 (en) * | 2002-04-11 | 2005-08-18 | Aventis Pharma Deutschland Gmbh | Acyl-3-carboxyphenyl-urea derivatives and their use as medicaments |
| DE10225635C1 (en) * | 2002-06-07 | 2003-12-24 | Aventis Pharma Gmbh | N-benzoylureido-cinnamic acid derivatives, process for their preparation and their use |
| DE50311954D1 (en) * | 2002-06-17 | 2009-11-12 | Saltigo Gmbh | Process for the preparation of mono-N-sulfonylated diamines |
| EP1523471B1 (en) * | 2002-07-11 | 2009-09-16 | Sanofi-Aventis Deutschland GmbH | Urea-substituted and urethane-substituted acylureas, methods for the production thereof and their use as medicaments |
| US7453002B2 (en) | 2004-06-15 | 2008-11-18 | Bristol-Myers Squibb Company | Five-membered heterocycles useful as serine protease inhibitors |
| US20090298745A1 (en) * | 2004-12-02 | 2009-12-03 | Gerard Hugh Thomas | Treatment of Diabetes with Glycogen Phosphorylase Inhibitors |
| WO2006082400A1 (en) * | 2005-02-05 | 2006-08-10 | Astrazeneca Ab | Indan-amide derivatives with glycogen phosphorylase inhibitory activity |
| JP6974331B2 (en) * | 2016-02-05 | 2021-12-01 | デナリ セラピューティクス インコーポレイテッドDenali Therapeutics Inc. | Compounds, compositions and methods |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2081747A1 (en) * | 1993-09-07 | 1996-03-01 | Esteve Labor Dr | Amide derivatives of thienopyrroles, their preparation and application as drugs |
| WO1996039384A1 (en) * | 1995-06-06 | 1996-12-12 | Pfizer, Inc. | Substituted n-(indole-2-carbonyl)-glycinamides and derivatives as glycogen phosphorylase inhibitors |
| EP0846464A2 (en) * | 1996-12-05 | 1998-06-10 | Pfizer Inc. | Use of glycogen phosphorylase inhibitor for reducing non-cardiac tissue damage resulting from ischemia |
| WO1999046268A1 (en) * | 1998-03-12 | 1999-09-16 | Novo Nordisk A/S | MODULATORS OF PROTEIN TYROSINE PHOSPHATASES (PTPases) |
| US5998463A (en) * | 1998-02-27 | 1999-12-07 | Pfizer Inc | Glycogen phosphorylase inhibitors |
| EP1088824A2 (en) * | 1999-09-30 | 2001-04-04 | Pfizer Products Inc. | Bicyclic pyrrolyl amides as glycogen phosphorylase inhibitors |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3706810A (en) * | 1970-09-15 | 1972-12-19 | American Cyanamid Co | N-morpholinoalkyl-thieno(3,2-b)pyrrole-5-carboxamides |
| US4692522A (en) * | 1985-04-01 | 1987-09-08 | Merck & Co., Inc. | Benzofused lactams useful as cholecystokinin antagonists |
| US4599198A (en) * | 1985-08-02 | 1986-07-08 | Pfizer Inc. | Intermediates in polypeptide synthesis |
| US4668769A (en) * | 1985-08-02 | 1987-05-26 | Hoover Dennis J | Oxa- and azahomocyclostatine polypeptides |
| US4720503A (en) * | 1985-08-02 | 1988-01-19 | Merck & Co., Inc. | N-substituted fused-heterocyclic carboxamide derivatives as dual cyclooxygenase and lipoxygenase inhibitors |
| FR2601368B1 (en) * | 1986-07-08 | 1989-04-07 | Synthelabo | NITROFURAN DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATION. |
| DE3629545A1 (en) * | 1986-08-30 | 1988-03-10 | Bayer Ag | DIHYDROPYRIDINE COMPOUNDS, METHOD FOR THEIR PRODUCTION AND THEIR USE |
| US4751231A (en) * | 1987-09-16 | 1988-06-14 | Merck & Co., Inc. | Substituted thieno[2,3-b]pyrrole-5-sulfonamides as antiglaucoma agents |
| US5863903A (en) * | 1994-03-09 | 1999-01-26 | Novo Nordisk A/S | Use of hydroxy alkyl piperidine and pyrrolidine compounds to treat diabetes |
| FR2723739B1 (en) * | 1994-08-19 | 1997-02-14 | Sanofi Sa | GLYCINAMIDE DERIVATIVES, METHODS FOR THEIR PREPARATION AND MEDICINAL PRODUCTS CONTAINING THEM. |
| CO5271699A1 (en) * | 2000-01-24 | 2003-04-30 | Pfizer Prod Inc | PROCEDURE FOR THE TREATMENT OF CARDIOMIOPATIA USING INHIBITORS OF THE GLUCOGENO FOSFORILASA |
| US6555569B2 (en) * | 2000-03-07 | 2003-04-29 | Pfizer Inc. | Use of heteroaryl substituted N-(indole-2-carbonyl-) amides for treatment of infection |
| US7057046B2 (en) * | 2002-05-20 | 2006-06-06 | Bristol-Myers Squibb Company | Lactam glycogen phosphorylase inhibitors and method of use |
-
2000
- 2000-09-06 GB GBGB0021831.3A patent/GB0021831D0/en not_active Ceased
-
2001
- 2001-08-31 JP JP2002525151A patent/JP2004508376A/en not_active Withdrawn
- 2001-08-31 WO PCT/SE2001/001880 patent/WO2002020530A1/en active IP Right Grant
- 2001-08-31 BR BR0113606-2A patent/BR0113606A/en not_active IP Right Cessation
- 2001-08-31 EP EP01961577A patent/EP1317459B1/en not_active Expired - Lifetime
- 2001-08-31 DE DE60102710T patent/DE60102710T2/en not_active Expired - Fee Related
- 2001-08-31 KR KR1020037003265A patent/KR100802369B1/en not_active IP Right Cessation
- 2001-08-31 CN CNB018183220A patent/CN1264846C/en not_active Expired - Fee Related
- 2001-08-31 AU AU8283301A patent/AU8283301A/en active Pending
- 2001-08-31 IL IL15429101A patent/IL154291A0/en unknown
- 2001-08-31 NZ NZ524011A patent/NZ524011A/en unknown
- 2001-08-31 MX MXPA03001512A patent/MXPA03001512A/en active IP Right Grant
- 2001-08-31 HU HU0400784A patent/HUP0400784A3/en unknown
- 2001-08-31 PT PT01961577T patent/PT1317459E/en unknown
- 2001-08-31 PL PL01361024A patent/PL361024A1/en unknown
- 2001-08-31 US US10/344,506 patent/US20030232875A1/en not_active Abandoned
- 2001-08-31 UA UA2003021026A patent/UA73781C2/en unknown
- 2001-08-31 CN CNA2006100818966A patent/CN1896078A/en active Pending
- 2001-08-31 CA CA002417594A patent/CA2417594A1/en not_active Abandoned
- 2001-08-31 ES ES01961577T patent/ES2217183T3/en not_active Expired - Lifetime
- 2001-08-31 RU RU2003104013/04A patent/RU2003104013A/en not_active Application Discontinuation
- 2001-08-31 EE EEP200300083A patent/EE200300083A/en unknown
- 2001-08-31 SK SK259-2003A patent/SK2592003A3/en unknown
- 2001-08-31 CZ CZ2003616A patent/CZ2003616A3/en unknown
- 2001-08-31 DK DK01961577T patent/DK1317459T3/en active
- 2001-08-31 AT AT01961577T patent/ATE263772T1/en not_active IP Right Cessation
- 2001-08-31 TR TR2004/01659T patent/TR200401659T4/en unknown
- 2001-08-31 AU AU2001282833A patent/AU2001282833B2/en not_active Ceased
-
2003
- 2003-02-05 ZA ZA200301013A patent/ZA200301013B/en unknown
- 2003-02-24 IS IS6727A patent/IS2110B/en unknown
- 2003-03-05 NO NO20031024A patent/NO20031024D0/en not_active Application Discontinuation
- 2003-03-10 BG BG107624A patent/BG107624A/en unknown
- 2003-10-16 HK HK03107519A patent/HK1055299A1/en not_active IP Right Cessation
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2081747A1 (en) * | 1993-09-07 | 1996-03-01 | Esteve Labor Dr | Amide derivatives of thienopyrroles, their preparation and application as drugs |
| WO1996039384A1 (en) * | 1995-06-06 | 1996-12-12 | Pfizer, Inc. | Substituted n-(indole-2-carbonyl)-glycinamides and derivatives as glycogen phosphorylase inhibitors |
| EP0846464A2 (en) * | 1996-12-05 | 1998-06-10 | Pfizer Inc. | Use of glycogen phosphorylase inhibitor for reducing non-cardiac tissue damage resulting from ischemia |
| US5998463A (en) * | 1998-02-27 | 1999-12-07 | Pfizer Inc | Glycogen phosphorylase inhibitors |
| WO1999046268A1 (en) * | 1998-03-12 | 1999-09-16 | Novo Nordisk A/S | MODULATORS OF PROTEIN TYROSINE PHOSPHATASES (PTPases) |
| EP1088824A2 (en) * | 1999-09-30 | 2001-04-04 | Pfizer Products Inc. | Bicyclic pyrrolyl amides as glycogen phosphorylase inhibitors |
Cited By (107)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1125580A3 (en) * | 2000-01-24 | 2002-11-27 | Pfizer Products Inc. | Methods of treating diabetic cardiomyopathy using glycogen phosphorylase inhibitors |
| EP1125580A2 (en) * | 2000-01-24 | 2001-08-22 | Pfizer Products Inc. | Methods of treating diabetic cardiomyopathy using glycogen phosphorylase inhibitors |
| US6867184B2 (en) | 2000-01-24 | 2005-03-15 | Pfizer, Inc. | Methods of treating diabetic cardiomyopathy using glycogen phosphorylase inhibitors |
| US8163751B2 (en) | 2001-02-13 | 2012-04-24 | Sanofi-Aventis Deutschland Gmbh | Acylated indanyl amines and their use as pharmaceuticals |
| WO2002064565A1 (en) * | 2001-02-13 | 2002-08-22 | Aventis Pharma Deutschland Gmbh | Acylated 1,2,3,4-tetrahydronaphthyl amines and their use as pharmaceutical |
| US6949556B2 (en) | 2001-02-13 | 2005-09-27 | Aventis Pharma Deutschland Gmbh | Acylated 1,2,3,4-tetrahydronaphthyl amines and their use as pharmaceutical agents |
| US7179839B2 (en) | 2001-02-13 | 2007-02-20 | Sanofi-Aventis Deutschland Gmbh | Acylated indanyl amines and their use as pharmaceuticals |
| AU2002250920B2 (en) * | 2001-02-13 | 2007-06-28 | Sanofi-Aventis Deutschland Gmbh | Acylated 1,2,3,4-tetrahydronaphthyl amines and their use as pharmaceutical |
| US7713963B2 (en) | 2001-02-13 | 2010-05-11 | Sanofi-Aventis Deutschland Gmbh | Acylated indanyl amines and their use as pharmaceuticals |
| US7169927B2 (en) | 2002-03-06 | 2007-01-30 | Astrazeneca Ab | Indole-amide derivatives and their use as glycogen phosphorylase inhibitors |
| WO2003074531A1 (en) * | 2002-03-06 | 2003-09-12 | Astrazeneca Ab | Heterocyclic amide derivatives having glycogen phosphorylase inhibitory activity |
| US7115648B2 (en) | 2002-03-06 | 2006-10-03 | Astrazeneca Ab | Indole-amide derivatives and their use as glycogen phosphorylase inhibitors |
| WO2003074513A3 (en) * | 2002-03-06 | 2003-12-31 | Astrazeneca Ab | Indole amide derivatives and their use as glycogen phosphorylase inhibitors |
| US7122567B2 (en) | 2002-03-06 | 2006-10-17 | Astrazeneca Ab | Heterocyclic amide derivatives having glycogen phosphorylase inhibitory activity |
| US7129249B2 (en) | 2002-03-06 | 2006-10-31 | Astrazeneca Ab | Heterocyclic amide derivatives as inhibitors of glycogen phoshorylase |
| US7332515B2 (en) | 2002-03-06 | 2008-02-19 | Astrazeneca Ab | Indole-amid derivatives which possess glycogen phosphorylase inhibitory activity |
| WO2003074485A2 (en) * | 2002-03-06 | 2003-09-12 | Astrazeneca Ab | Indole-amide derivatives and their use as glycogen phosphorylase inhibitors |
| WO2003074485A3 (en) * | 2002-03-06 | 2003-12-04 | Astrazeneca Ab | Indole-amide derivatives and their use as glycogen phosphorylase inhibitors |
| US7138415B2 (en) | 2002-03-06 | 2006-11-21 | Astrazeneca Ab | Indolamid derivatives which possess glycogenphosphorylase inhibitory activity |
| WO2003074513A2 (en) * | 2002-03-06 | 2003-09-12 | Astrazeneca Ab | Indole amide derivatives and their use as glycogen phosphorylase inhibitors |
| US7166636B2 (en) | 2002-03-06 | 2007-01-23 | Astrazeneca Ab | Indole-amid derivatives which possess glycogen phosphorylase inhibitory activity |
| WO2003074532A1 (en) * | 2002-03-06 | 2003-09-12 | Astrazeneca Ab | Heterocyclic amide derivatives as inhibitors of glycogen phosphorylase |
| US7057046B2 (en) * | 2002-05-20 | 2006-06-06 | Bristol-Myers Squibb Company | Lactam glycogen phosphorylase inhibitors and method of use |
| WO2004014842A1 (en) * | 2002-08-07 | 2004-02-19 | Aventis Pharma Deutschland Gmbh | Acylated arylcycloalkylamines and their use as pharmaceuticals |
| US7186735B2 (en) | 2002-08-07 | 2007-03-06 | Sanofi-Aventis Deutschland Gmbh | Acylated arylcycloalkylamines and their use as pharmaceuticals |
| CN1304365C (en) * | 2002-08-07 | 2007-03-14 | 塞诺菲-安万特德国有限公司 | Acylated arylcycloalkylamines and their use as pharmaceuticals |
| EP1388535A1 (en) * | 2002-08-07 | 2004-02-11 | Aventis Pharma Deutschland GmbH | Acylated arylcycloalkylamines and their use as pharmaceuticals |
| US7307174B2 (en) | 2002-10-03 | 2007-12-11 | Astrazeneca Ab | Process and intermediates for the preparation of thienopyrrole derivatives |
| JP2006505541A (en) * | 2002-10-03 | 2006-02-16 | アストラゼネカ アクチボラグ | Methods and intermediates for producing thienopyrrole derivatives |
| CN100378106C (en) * | 2002-10-03 | 2008-04-02 | 阿斯利康(瑞典)有限公司 | Process and intermediates for the preparation of the thienopyrrole derivatives |
| US7411074B2 (en) | 2002-10-03 | 2008-08-12 | Astrazeneca Ab | Process and intermediates for the preparation of the thienopyrrole derivatives |
| US6992101B2 (en) | 2002-11-07 | 2006-01-31 | Pfizer, Inc. | N-(indole-2-carbonyl) and H-thieno[2,3-b]pyrrole-5-carboxamide anti-diabetic agents |
| WO2004041780A2 (en) * | 2002-11-07 | 2004-05-21 | Pfizer Products Inc. | N-(indole-2-carbonyl) amides as anti-diabetic agents |
| WO2004041780A3 (en) * | 2002-11-07 | 2005-05-19 | Pfizer Prod Inc | N-(indole-2-carbonyl) amides as anti-diabetic agents |
| US6992092B2 (en) | 2003-04-17 | 2006-01-31 | Pfizer Inc. | Anti-diabetic agents |
| WO2004092158A1 (en) * | 2003-04-17 | 2004-10-28 | Pfizer Products Inc. | Carboxamide derivatives as anti-diabetic agents |
| US7405210B2 (en) | 2003-05-21 | 2008-07-29 | Osi Pharmaceuticals, Inc. | Pyrrolopyridine-2-carboxylic acid amide inhibitors of glycogen phosphorylase |
| US8158622B2 (en) | 2003-05-21 | 2012-04-17 | Prosidion Limited | Pyrrolopyridine-2-carboxylic acid amide inhibitors of glycogen phosphorylase |
| EA009215B1 (en) * | 2003-05-21 | 2007-12-28 | Прозидион Лимитед | Pyrrolopyridine-2-carboxylic acid amide inhibitors of glycogen phosphorylase |
| WO2004104001A3 (en) * | 2003-05-21 | 2005-03-03 | Osi Pharm Inc | Pyrrolopyridine-2-carboxylic acid amide inhibitors of glycogen phoshorylase |
| WO2004113345A1 (en) * | 2003-06-20 | 2004-12-29 | Japan Tobacco Inc. | Fused pyrrole compound and medicinal use thereof |
| WO2005013981A1 (en) * | 2003-08-07 | 2005-02-17 | Astrazeneca Ab | Heterocyclic amide derivatives which possess glycogen phosphorylase inhibitory activity |
| WO2005013975A1 (en) * | 2003-08-07 | 2005-02-17 | Astrazeneca Ab | Indolamide derivatives which possess glycogen phosphorylase inhibitory activity |
| WO2005018637A1 (en) * | 2003-08-22 | 2005-03-03 | Astrazeneca Ab | Heterocyclic amide derivatives which possess glycogen phosphorylase inhibitory activity |
| WO2005019172A1 (en) * | 2003-08-22 | 2005-03-03 | Astrazeneca Ab | Indol-2-amides as glycogen phosphorylase inhibitors |
| WO2005020986A1 (en) * | 2003-08-29 | 2005-03-10 | Astrazeneca Ab | Heterocyclic amide derivatives which posses glycogen phosphorylase inhibitory activity |
| WO2005020985A1 (en) * | 2003-08-29 | 2005-03-10 | Astrazeneca Ab | Indolamide derivatives which possess glycogen phosphorylase inhibitory activity |
| WO2005020987A1 (en) * | 2003-08-30 | 2005-03-10 | Astrazeneca Ab | Heterocyclic amide derivatives which posses glycogen phosphorylase inhibitory activity |
| US7893098B2 (en) | 2003-12-29 | 2011-02-22 | Sepracor Inc. | Pyrrole and pyrazole DAAO inhibitors |
| WO2006055462A1 (en) * | 2004-11-15 | 2006-05-26 | Bristol-Myers Squibb Company | 2-amino-4-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| WO2006053274A3 (en) * | 2004-11-15 | 2006-07-06 | Bristol Myers Squibb Co | 2-amino-1-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| US7223786B2 (en) | 2004-11-15 | 2007-05-29 | Bristol-Myers Squibb Company | 2-aminonaphthalene derivatives and related glycogen phosphorylase inhibitors |
| US7214704B2 (en) | 2004-11-15 | 2007-05-08 | Bristol-Myers Squibb Company | 2-Amino-1-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| US7226942B2 (en) | 2004-11-15 | 2007-06-05 | Bristol-Myers Squibb Company | 2-amino-4-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| WO2006055463A3 (en) * | 2004-11-15 | 2006-12-28 | Bristol Myers Squibb Co | 2-amino-3-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| US7365061B2 (en) | 2004-11-15 | 2008-04-29 | Bristol-Myers Squibb Company | 2-Amino-3-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| WO2006053274A2 (en) * | 2004-11-15 | 2006-05-18 | Bristol-Myers Squibb Company | 2-amino-1-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| WO2006055463A2 (en) * | 2004-11-15 | 2006-05-26 | Bristol-Myers Squibb Company | 2-amino-3-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| WO2006059165A1 (en) * | 2004-12-02 | 2006-06-08 | Prosidion Limited | Pyrrolopyridine-2-carboxylic acid amide derivative useful as inhibitor of glycogen phosphorylase |
| US8664397B2 (en) | 2004-12-02 | 2014-03-04 | Prosidion Limited | Pyrrolopyridine-2-carboxylic acid amide derivative useful as inhibitor of glycogen phosphorylase |
| EP1841426A4 (en) * | 2005-01-19 | 2010-04-07 | Merck Sharp & Dohme | Tertiary carbinamines having substituted heterocycles, which are active as inhibitors of beta-secretase, for the treatment of alzheimer's disease |
| US8338614B2 (en) | 2005-01-19 | 2012-12-25 | Merck, Sharp & Dohme Corp. | Tertiary carbinamines having substituted heterocycles which are active as β-secretase inhibitors for the treatment of alzheimer's disease |
| EP1841426A1 (en) * | 2005-01-19 | 2007-10-10 | Merck & Co., Inc. | Tertiary carbinamines having substituted heterocycles, which are active as inhibitors of beta-secretase, for the treatment of alzheimer's disease |
| WO2006082401A1 (en) * | 2005-02-05 | 2006-08-10 | Astrazeneca Ab | Chemical compounds |
| US9868718B2 (en) | 2006-01-06 | 2018-01-16 | Sunovion Pharmaceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| US8877975B2 (en) | 2006-01-06 | 2014-11-04 | Sunovion Pharmaceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| US10562878B2 (en) | 2006-01-06 | 2020-02-18 | Sunovion Pharamceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| US8053603B2 (en) | 2006-01-06 | 2011-11-08 | Sunovion Pharmaceuticals Inc. | Tetralone-based monoamine reuptake inhibitors |
| US8097760B2 (en) | 2006-03-31 | 2012-01-17 | Sunovion Pharmacuticals Inc. | Preparation of chiral amides and amines |
| EP2351568A2 (en) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Uses of dpp-iv inhibitors |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| US7884124B2 (en) | 2006-06-30 | 2011-02-08 | Sepracor Inc. | Fluoro-substituted inhibitors of D-amino acid oxidase |
| EP2298772A1 (en) * | 2006-10-18 | 2011-03-23 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds |
| US8492405B2 (en) | 2006-10-18 | 2013-07-23 | Takeda Pharmaceutical Company Limited | Glucokinase-activating fused heterocyclic compounds and methods of treating diabetes and obesity |
| US7902252B2 (en) | 2007-01-18 | 2011-03-08 | Sepracor, Inc. | Inhibitors of D-amino acid oxidase |
| US9586888B2 (en) | 2007-05-31 | 2017-03-07 | Sunovion Pharmaceuticals Inc. | Phenyl substituted cycloalkylamines as monoamine reuptake inhibitors |
| US8669291B2 (en) | 2007-05-31 | 2014-03-11 | Sunovion Pharmaceuticals Inc. | Phenyl substituted cycloalkylamines as monoamine reuptake inhibitors |
| WO2010057141A3 (en) * | 2008-11-17 | 2010-09-16 | The Regents Of The University Of Michigan | Alphavirus inhibitors and uses thereof |
| WO2010057141A2 (en) * | 2008-11-17 | 2010-05-20 | The Regents Of The University Of Michigan | Alphavirus inhibitors and uses thereof |
| EP2923706A1 (en) | 2009-12-03 | 2015-09-30 | Synergy Pharmaceuticals Inc. | Agonists of guanylate cyclase useful for the treatment of hypercholesterolemia |
| WO2012027331A1 (en) | 2010-08-27 | 2012-03-01 | Ironwood Pharmaceuticals, Inc. | Compositions and methods for treating or preventing metabolic syndrome and related diseases and disorders |
| EP3708179A1 (en) | 2012-03-15 | 2020-09-16 | Bausch Health Ireland Limited | Formulations of guanylate cyclase c agonists and methods of use |
| EP4309673A2 (en) | 2012-03-15 | 2024-01-24 | Bausch Health Ireland Limited | Formulations of guanylate cyclase c agonists and methods of use |
| WO2013138352A1 (en) | 2012-03-15 | 2013-09-19 | Synergy Pharmaceuticals Inc. | Formulations of guanylate cyclase c agonists and methods of use |
| EP4424697A2 (en) | 2013-06-05 | 2024-09-04 | Bausch Health Ireland Limited | Ultra-pure agonists of guanylate cyclase c, method of making and using same |
| WO2014197720A2 (en) | 2013-06-05 | 2014-12-11 | Synergy Pharmaceuticals, Inc. | Ultra-pure agonists of guanylate cyclase c, method of making and using same |
| WO2016030534A1 (en) | 2014-08-29 | 2016-03-03 | Tes Pharma S.R.L. | INHIBITORS OF α-AMINO-β-CARBOXYMUCONIC ACID SEMIALDEHYDE DECARBOXYLASE |
| US9708272B2 (en) | 2014-08-29 | 2017-07-18 | Tes Pharma S.R.L. | Inhibitors of α-amino-β-carboxymuconic acid semialdehyde decarboxylase |
| US11254644B2 (en) | 2014-08-29 | 2022-02-22 | Tes Pharma S.R.L. | Inhibitors of alpha-amino-beta-carboxymuconic acid semialdehyde decarboxylase |
| US10513499B2 (en) | 2014-08-29 | 2019-12-24 | Tes Pharma S.R.L. | Inhibitors of alpha-amino-beta-carboxymuconic acid semialdehyde decarboxylase |
| AU2016287581B2 (en) * | 2015-07-02 | 2019-06-06 | F. Hoffmann-La Roche Ag | Bicyclic lactams and methods of use thereof |
| KR20180023988A (en) * | 2015-07-02 | 2018-03-07 | 에프. 호프만-라 로슈 아게 | Bicyclic lactams and their use |
| RU2827714C1 (en) * | 2015-07-02 | 2024-10-01 | Ф. Хоффманн-Ля Рош Аг | Bicyclic lactams and methods of use thereof |
| RU2716136C2 (en) * | 2015-07-02 | 2020-03-06 | Ф. Хоффманн-Ля Рош Аг | Bicyclic lactams and methods for use thereof |
| EP3760625A1 (en) * | 2015-07-02 | 2021-01-06 | F. Hoffmann-La Roche AG | Bicyclic lactams as receptor-interacting protein-1 (rip1) kinase inhibitors for treating e.g. inflammatory diseases |
| AU2019203444B2 (en) * | 2015-07-02 | 2021-01-21 | F. Hoffmann-La Roche Ag | Bicyclic lactams and methods of use thereof |
| WO2017004500A1 (en) * | 2015-07-02 | 2017-01-05 | Genentech, Inc. | Bicyclic lactams and methods of use thereof |
| US10988459B2 (en) | 2015-07-02 | 2021-04-27 | Genentech, Inc. | Bicyclic lactams and methods of use thereof |
| KR102104911B1 (en) * | 2015-07-02 | 2020-04-28 | 에프. 호프만-라 로슈 아게 | Bicyclic lactam and its use method |
| TWI788830B (en) * | 2015-07-02 | 2023-01-01 | 瑞士商赫孚孟拉羅股份公司 | Bicyclic lactams and methods of use thereof |
| TWI763630B (en) * | 2015-07-02 | 2022-05-11 | 瑞士商赫孚孟拉羅股份公司 | Bicyclic lactams and methods of use thereof |
| WO2018069532A1 (en) | 2016-10-14 | 2018-04-19 | Tes Pharma S.R.L. | Inhibitors of alpha-amino-beta-carboxymuconic acid semialdehyde decarboxylase |
| US10947226B2 (en) | 2016-10-17 | 2021-03-16 | Genentech, Inc. | Bicyclic pyridone lactams and methods of use thereof |
| US11072607B2 (en) | 2016-12-16 | 2021-07-27 | Genentech, Inc. | Inhibitors of RIP1 kinase and methods of use thereof |
| US11634436B2 (en) | 2018-04-20 | 2023-04-25 | Genentech, Inc. | Pyridine lactam compounds and methods of use thereof |
| WO2020104456A1 (en) | 2018-11-20 | 2020-05-28 | Tes Pharma S.R.L | INHIBITORS OF α-AMINO-β-CARBOXYMUCONIC ACID SEMIALDEHYDE DECARBOXYLASE |
| EP4066896A4 (en) * | 2019-11-27 | 2024-03-27 | Riken | G9a inhibitor |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1317459B1 (en) | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors | |
| AU2001282833A1 (en) | Bicyclic pyrrolyl amides as glucogen phosphorylase inhibitors | |
| US7129249B2 (en) | Heterocyclic amide derivatives as inhibitors of glycogen phoshorylase | |
| AU2008273017C1 (en) | Heterocyclic compounds useful as Raf kinase inhibitors | |
| JP5639197B2 (en) | Heterocycloalkyl-containing thienopyrimidines for pharmaceutical compositions | |
| EP1658069B1 (en) | Heterocyclic amide derivatives which possess glycogen phosphorylase inhibitory activity | |
| KR20030045187A (en) | Tetrahydrobenzazepine Derivatives Useful as Modulators of Dopamine D3 Receptors(Antipsychotic Agents) | |
| EP1658067B1 (en) | Indolamide derivatives which possess glycogen phosphorylase inhibitory activity | |
| JP5814948B2 (en) | Substituted fused imidazole derivatives, pharmaceutical compositions thereof, and methods of use | |
| WO2003074531A1 (en) | Heterocyclic amide derivatives having glycogen phosphorylase inhibitory activity | |
| WO2003074513A2 (en) | Indole amide derivatives and their use as glycogen phosphorylase inhibitors | |
| WO2013044360A1 (en) | Inhibitors of protein tyrosine kinase activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PH PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 154291 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003/01013 Country of ref document: ZA Ref document number: 200301013 Country of ref document: ZA Ref document number: 524011 Country of ref document: NZ Ref document number: 2417594 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 191/MUMNP/2003 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10344506 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/A/2003/001512 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001282833 Country of ref document: AU Ref document number: 2001961577 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2592003 Country of ref document: SK Ref document number: PV2003-616 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020037003265 Country of ref document: KR Ref document number: 03018834 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002525151 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 10762401 Country of ref document: BG Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020037003265 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1-2003-500097 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 2003104013 Country of ref document: RU Kind code of ref document: A Ref country code: RU Ref document number: RU A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 018183220 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2003-616 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001961577 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2001961577 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 524011 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 524011 Country of ref document: NZ |