WO2002012249A2 - Substituted pyrrole compounds and their use as spla2 inhibitors - Google Patents
Substituted pyrrole compounds and their use as spla2 inhibitors Download PDFInfo
- Publication number
- WO2002012249A2 WO2002012249A2 PCT/US2001/021106 US0121106W WO0212249A2 WO 2002012249 A2 WO2002012249 A2 WO 2002012249A2 US 0121106 W US0121106 W US 0121106W WO 0212249 A2 WO0212249 A2 WO 0212249A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- alkyl
- hydrogen
- formula
- Prior art date
Links
- 0 IC(C=C1)=CC=C*1NC=I Chemical compound IC(C=C1)=CC=C*1NC=I 0.000 description 6
- UOGXZILMRCXMOE-UHFFFAOYSA-N CCOC(c([nH]1)cc2c1[s]cc2OC)=O Chemical compound CCOC(c([nH]1)cc2c1[s]cc2OC)=O UOGXZILMRCXMOE-UHFFFAOYSA-N 0.000 description 1
- OMYRIIOGYVNOAM-UHFFFAOYSA-N CCc([n]1Cc2ccccc2)c(C(C(N)=O)=O)c2c1[s]cc2OC Chemical compound CCc([n]1Cc2ccccc2)c(C(C(N)=O)=O)c2c1[s]cc2OC OMYRIIOGYVNOAM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- This invention relates to novel substituted pyrrole compounds useful for Inflammatory Diseases .
- SPI1A2 human non- pancreatic secretory phospholipase A2
- This invention provides novel substituted pyrrole compounds having potent and selective effectiveness as inhibitors of mammalian S A2.
- This invention is also the use of novel substituted pyrrole compounds useful in the treatment and prevention of Inflammatory Diseases.
- This invention is also the use of novel substituted pyrrole compounds to inhibit mammalian sPLA2 mediated release of fatty acids .
- This invention is also a pharmaceutical composition containing any of the substituted pyrrole compounds of the invention.
- Inflammatory Diseases refers to diseases such as inflammatory bowel disease, sepsis, septic shock, adult respiratory distress syndrome, pancreatitis, trauma- induced shock, bronchial asthma, allergic rhinitis, rheumatoid arthritis, cystic fibrosis, stroke, acute bronchitis, chronic bronchitis, acute bronchiolitis, chronic bronchiolitis, osteoarthritis, gout, spondylarthropathris, ankylosing spondylitis, Reiter's syndrome, psoriatic arthropathy, enterapathric spondylitis, Juvenile arthropathy or juvenile ankylosing spondylitis, Reactive arthropathy, infectious or post- infectious arthritis, gonoccocal arthritis, tuberculous arthritis, viral arthritis, fungal arthritis, syphilitic arthritis, Lyme disease, arthritis associated with "vasculitic syndromes” , polyarteritis nodosa, hyper
- A is 0, NR, SO 2 , SO, or S.
- substituted pyrrole compounds of the invention employ certain defining terms as follows:
- alkyl by itself or as part of another substituent means, unless otherwise defined, a straight or branched chain monovalent hydrocarbon radical such as methyl, ethyl, n-propyl, isopropyl, n-butyl, tertiary butyl, sec-butyl, n-pentyl, and n-hexyl .
- alkenyl employed alone or in combination with other terms means a straight chain or branched monovalent hydrocarbon group having the stated number range of carbon atoms, and typified by groups such as vinyl, propenyl, crotonyl, isopentenyl, and various butenyl isomers .
- hydrocarbyl means an organic group containing only carbon and hydrogen.
- halo means fluoro, chloro, bromo, or iodo.
- heterocyclic radical refers to radicals derived from monocyclic or polycyclic, saturated or unsaturated, substituted or unsubstituted heterocyclic nuclei having 5 to 14 ring atoms and containing from 1 to 3 hetero atoms selected from the group consisting of nitrogen, oxygen or sulfur.
- Typical heterocyclic radicals are pyrrolyl, pyrrolodinyl , piperidinyl, furanyl, thiophenyl, pyrazolyl, imidazolyl, phenylimidazolyl, triazolyl, isoxazolyl, oxazolyl, thiazolyl, thiadiazolyl, indolyl , carbazolyl, norharmanyl, azaindolyl, benzofuranyl , dibenzofuranyl, dibenzothiophenyl , indazolyl, imidazo (1.2-A) pyridinyl, benzotriazolyl, anthranilyl, 1, 2-benzisoxazolyl , benzoxazolyl , benzothiazolyl, purinyl, pyridinyl, dipyridylyl .
- phenylpyridinyl benzylpyridinyl, pyrimidinyl, phenylpyrimidinyl, pyrazinyl, 1, 3 , 5-triazinyl, quinolinyl, phthalazinyl, quinazolinyl,morpholino, thiomorpholino, homopiperazinyl, tetrahydrofuranyl, tetrahydropyranyl, oxacanyl, 1, 3-dioxolanyl , 1, 3-dioxanyl, 1, 4-dioxanyl, tetrahydrothiopheneyl, pentamethylenesulfadyl, 1,3- dithianyl, 1, 4-dithianyl , 1, 4-thioxanyl, azetidinyl, hexamethyleneiminium, heptamethyleneiminium, piperazinyl and quinoxalinyl .
- carbocyclic radical refers to radicals derived from a saturated or unsaturated, substituted or unsubstituted 5 to 14 membered organic nucleus whose ring forming atoms (other than hydrogen) are solely carbon atoms.
- Typical carbocyclic radicals are cycloalkyl, cycloalkenyl, phenyl , spiro [5.5] undecanyl , naphthyl , norbornanyl , bicycloheptadienyl, toluyl, xylenyl, indenyl, stilbenyl, terphenylyl, diphenylet ylenyl, phenyl-cyclohexenyl, acenaphthylenyl , and anthracenyl, biphenyl, dibenzylyl and related dibenzylyl homologues represented by the formula (a) :
- n is a number from 1 to 8.
- non-interfering substituent and “non- interfering group” refer to radicals suitable for substitution at positions 4, 5, 6 and/or 7 of the substituted pyrrole nucleus and on other nucleus positions or substituents (as hereinafter described for Formula I) , and radicals suitable for substitution on the heterocyclic radical and carbocyclic radical as defined above.
- Illustrative non-interfering radicals are Ci-Cs alkyl, C2- Cg alkenyl, C2-C8 alkynyl, C7-C12 aralkyl, C7-C12 alkaryl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, phenyl, toluyl, xylenyl, biphenyl, Ci-Cs alkoxy, C2-C8 alkenyloxy, C2-C8 alkynyloxy, C2-C12 alkoxyalkyl, C2-C12 alkoxyalk loxy, C2- C12 alkylcarbonyl, C2-C12 alkylcarbonylamino, C2-C12 alkoxyamino, C2-C12 alkoxyaminocarbonyl , C1-C12 alkylamino, C1-C12 alkylamino, C1-C12 alkylamino, C1-C12 alkylamino
- organic substituent refers to a monovalent radical consisting of carbon and hydrogen with or without oxygen, nitrogen, sulfur, halogen, or other elements.
- Illustrative organic substituents are C-j_-C8 alkyl, aryl, C7-C14 aralkyl, C7-C1 alkaryl, C3-C8 cycloalkyl, C- ⁇ -Cs alkoxyalkyl and these groups substituted with halogen, -CF3, -OH, ⁇ -C Q alkyl, amino, carbonyl, and -CN.
- substituted group is an organic group substituted with one or more non-interfering substituents.
- (acidic group) means an organic group which when attached to a substituted pyrrole nucleus at positions 4 or 5, through suitable linking atoms (hereinafter defined as the "acid linker"), acts as a proton donor capable of hydrogen bonding.
- acid linker suitable linking atoms
- n 1 to 8
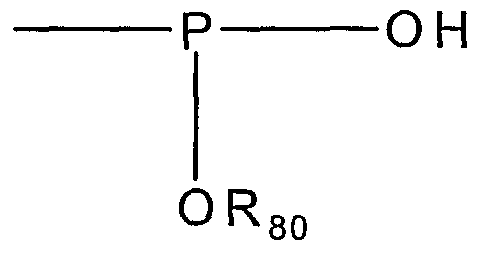
- Rso is a metal or C ⁇ -C8 and Rsi is an organic substituent or -CF3.
- acid linker refers to a divalent linking group symbolized as, -(L a ) ⁇ , which has the function of joining the 4 or 5 position of the substituted pyrrole nucleus to an acidic group in the general relationship:
- acid linker length refers to the number of atoms (excluding hydrogen) in the shortest chain of the linking group - (L a ) - that connects the 4 or 5 position of the substituted pyrrole nucleus with the acidic group.
- the presence of a carbocyclic ring in - (L a ) - counts as the number of atoms approximately equivalent to the calculated diameter of the carbocyclic ring.
- a benzene or cyclohexane ring in the acid linker counts as 2 atoms in calculating the length of - (L a )
- Illustrative acid linker groups are ;
- groups (a) , (b) , and (c) have acid linker lengths of 5, 7, and 2, respectively.
- N-hydroxyfunctional amide group is represented by the formula:
- R 4a is selected from the group consisting of OH, (C ⁇ Cg) alkoxy, and aryloxy; and wherein 4 ⁇ is hydrogen or an organic substituent selected from the group consisting of C]_-Cs alkyl, aryl, C 7 -C 14 aralkyl, C 7 -C 14 alkaryl, C 3 -Cs cycloalkyl, C * -L-C 8 alkoxyalkyl and these groups substitued with halogen, - CF 3 , -OH, C*j_-C 8 alkyl, amino, carbonyl, a d -CN.
- N-hydroxyfunctional amide linker refers to a divalent linking group symbolized as, -(Lh)-, which has the function of joining the 4 position of the substituted pyrrole nucleus to an N-hydroxyfunctional amide group in the general relationship:
- N-hydroxyfunctional amide linker length refer to the number of atoms (excluding hydrogen) in the shortest chain of the linking group - (Lh) - that connects the 4 position of the substituted pyrrole nucleus with the N- hydroxyfunctional amide group.
- the presence of a carbocyclic ring in - (Lh) - counts as the number of atoms approximately equivalent to the calculated diameter of the carbocyclic ring.
- a benzene or cyclohexane ring in the N-hydroxyfunctional amide linker counts as 2 atoms in calculating the length of - (Lh) - ⁇
- Illustrative N- hydroxyfunctional amide linker groups are;
- groups (a) , (b) , and (c) have N- hydroxyfunctional amide linker lengths of 5, 7, and 2, respectively.
- acylamino acid group is represented by the formula:
- R 4c is selected from the group consisting of H, (C]_-Cg) alkyl, (C ⁇ -Cg) alkoxy, heteroaryl and aryl, -CF3; and wherein NR 4 ⁇ is an amino acid residue of either a natural or unnatural amino acid with the nitrogen atom being part of the amino group of the amino acid.
- a typical amino acid is selected from the group comprising isoleucine, valine, phenylalanine, aspartic acid, leucine, glycine, asparagine, cystein, glutamine, glutamic acid, histidine, lysine, methionine, serine, threonine, tryptophan, tyrosine and derivatives thereof.
- amino acid residue refers to the portion of the amino acid group coupled at the nitrogen atom of the amino terminus. It is the amino acid less a hydrogen atom from the amino terminus. It is further illustrated as used herein for the amino acid alanine attached at the nitrogen atom as shown below:
- acylamino acid linker refers to a divalent linking group symbolized as, -(L c )-, which has the function of joining the 4 position of the substituted pyrrole nucleus to an acylamino acid group in the general relationship :
- acylamino acid linker length refer to the number of atoms (excluding hydrogen) in the shortest chain of the linking group - (L c ) - that connects the 4 position, of the substituted pyrrole nucleus with the acidic group.
- the presence of a carbocyclic ring in - (L c ) - counts as the number of atoms approximately equivalent to the calculated diameter of the carbocyclic ring.
- a benzene or cyclohexane ring in the acid linker counts as 2 atoms in calculating the length of -(L c )-.
- Illustrative acylamino acid linker groups are;
- groups (a) , (b) , and (c) have acylamino acid linker lengths of 5, 7, and 2, respectively.
- group radical or
- fragment are synonymous and are intended to indicate functional groups or fragments of molecules attachable to a bond or other fragments of molecules.
- acetamide group represent the acetamide fragment or radical. Structures of groups, radicals or fragments unattached to the substituted pyrrole nucleus have been drawn to show the first line as a connecting bond only.
- amine includes primary, secondary and tertiary amines.
- mammalian include human and domesticated quadrupeds.
- alkylene chain of 1 or 2 carbon atoms refers to the divalent radicals, -CH2-CH2- and -CH2-.
- the present invention provides novel classes of substituted pyrrole compounds useful as SPL 2 inhibitors for the treatment of Inflammatory Diseases.
- Classes of substituted pyrrole compounds of this invention include substituted pyrrole glyoxylamide derivatives, substituted pyrrole-3 -oxime amide derivatives and substituted pyrrole acetamide derivatives.
- the compounds of the invention have the general formula (I) or a pharmaceutically acceptable salt, solvate or prodrug thereof;
- A is 0, S, SO, SO2 , or NR; and wherein R is a non- interfering substitutent ;
- Rl is selected from groups (a) , (b) , and (c) wherein;
- (a) is C7-C20 alkyl, C7-C20 haloalkyl, C7-C20 alkenyl, C7-C20 alkynyl, carbocyclic radical, or heterocyclic radical, or
- (b) is a member of (a) substituted with one or more independently selected non- interfering substituents; or (c) is the group - (L ] _) - n ; where, - ( ⁇ ) - is a divalent linking group of 1 to 8 atoms and where R ⁇ is a group selected from (a) or (b) ;
- R2 is hydrogen, or a group containing 1 to 4 non- hydrogen atoms plus any required hydrogen atoms;
- R3 is -(L3)- Z, where -(L3)- is a divalent linker group selected from a bond or a divalent group selected from: -c-
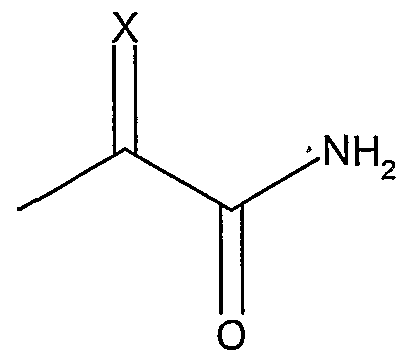
- Z is selected from an amide including acetamide, oxime amide, oxime thioamide, glyoxylamide or a group represented by the formulae,
- R a is independently selected from hydrogen, C ⁇ -C 8 alkyl, aryl, Ci-Cs -alkaryl, C ⁇ -C8 alkoxy, aralkyl and -CN;
- R4 is the group hydrogen, or the group - (L a ) - (acidic group) ; wherein -(L a )-, is an acid linker having an acid linker length of 1 to 8; or the group WR 4e wherein is O, S, or NH, and R 4e is a non- interfering substituent; or the group - (L c ) - (acylamino acid group) ; wherein -(L c )-, is an acylamino acid linker having an acylamino acid linker length of 1 to 8; or the group - (Lh) - (N-hydroxyfunctional amide group) ; wherein -(Lh)-, is an N-hydroxyfunctional amide linker having an N-
- R5 is selected from hydrogen, a non-interfering substituent, or the group, - (L a ) - (acidic group) ; wherein - (L a )-, is an acid linker having an acid linker length of 1 to 8;
- a preferred subclass of compounds of formula (I) are those for which the substitutent A is sulfur or oxygen. Most preferred are compounds of formula (I) wherein the substituent A is sulfur. Another preferred subclass of compounds of formula (I) are those where for R* ] _ the divalent linking group - (L-]_) - is a group represented by any one of the following formulae (la) , (lb) , (lc) , (Id) , (Ie) , or (If) : -c- (la)
- each R ] _ Q is independently hydrogen, C;*-__8 alkyl, C*j__8 haloalkyl or C ⁇ _8 alkoxy.
- Particularly preferred as the linking group - (L* j _) - of R ⁇ is an alkylene chain of 1 or 2 carbon atoms, namely, -(CH 2 )- or -(CH 2 -CH 2 )-.
- R ⁇ is a substituted or unsubstituted group selected from the group consisting of 5-C14 cycloalkyl, Cs-C- ⁇ cycloalkenyl, phenyl, naphthyl, norbornanyl, bicycloheptadienyl, toluyl, xylenyl, indenyl , stilbenyl, terphenylyl, diphenylethylenyl, phenyl- cyclohexenyl , acenaphthylenyl, and anthracenyl, biphenyl, bibenzylyl and related bibenzylyl homologues represented by the formula (a) ,- where n is a number from 1 to 8.
- R j _ the combined group - (L-]_) -Rn is selected from the group consisting of
- R 12 is a radical independently selected from halo, Ci-Cs alkyl, C ⁇ -C 8 alkoxy, -S- (C-L-C S alkyl), -O- (C-L-C S alkyl) and C-]_-C 8 haloalkyl where t is a number from 0 to 5 and u is a number from 0 to 4 is the group - (L]_) -R ⁇ ; where, - (L]_) - is a divalent linking group of 1 to 8 atoms and where R]_]_ is a group selected from (a) or (b) .
- R-j_]_ is -(CH 2 )m-R 12 wherein m is an integer from 1 to 6, and R 12 is (d) a group represented by the formula:
- R 13 and R 14 are independently selected from a halogen, Q* . to C 8 alkyl, Ci to C 8 alkyloxy, C-*. to C 8 alkylthio, aryl, heteroaryl, and Ci to C 8 haloalkyl, ⁇ is an oxygen atom or a sulfur atom, L 5 is a bond, -(CH 2 )v-,
- v is an integer from 0 to 2
- ⁇ is -CH 2 - or -(CH 2 ) 2 -
- ⁇ is an oxygen atom or a sulfur atom
- b is an integer from 0 to 3
- d is an integer from 0 to 4
- f, p, and w are independently an integer from 0 to 5
- r is an integer from 0 to 7
- u is an integer from 0 to 4
- Preferred R 2 substituents Preferred R 2 substituents:
- R2 is preferably selected from the group consisting of hydrogen, C2-C4 alkyl, C2-C4 alkenyl, -0- (C1-C3 alkyl), -S-(C ⁇ -C 3 alkyl), -C3-C4 cycloalkyl -CF 3 , halo, -N0 2 , -CN, -SO3.
- Particularly preferred R2 groups are selected from hydrogen, methyl, ethyl, propyl, isopropyl, cyclopropyl, -F, -CF 3 , -Cl, -Br, or -O-CH3.
- R3 substituents A preferred subclass of compounds of formula (I) are those wherein for -(L3)-Z, Z is represented by
- Another preferred subclass of compounds of formula (I) are those wherein Z is an oxime amide group
- R3 is an oxime amide group and R a is hydrogen, methyl or ethyl .
- X is oxygen or sulfur; and R a is selected from hydrogen, Ci-Cs alkyl, aryl, and Ci-Cs alkaryl.
- the linking group -(L 3 )- be a bond.
- R 4 is the group R 4e wherein is oxygen
- R 4e is a non-interfering substituent independently selected from the group consisting of methyl, ethyl, propyl, isopropyl, thiomethyl, C 4 -Cg alkyl, C2 ⁇ Cg alkenyl, C2 ⁇ Cg alkynyl, C 7 - C 12 aralkyl, C 7 -C 12 alkaryl, C3-C8 cycloalkyl, C 3 -C8 cycloalkenyl, phenyl, toluyl, xylenyl, biphenyl, Ci-Cg alkoxy, C 2 ⁇ C alkenyloxy, C2-Cg alkynyloxy, C 2 -C1 2 alkoxyalkyl, C 2 -C 1 2 alkoxyalkyloxy, C 2 -C 12 alkylcarbonyl, 2 _ i
- non-interfering substituents are hydrogen, methyl, ethyl, propyl, and isopropyl.
- N-hydroxyfunctional amide group in the group R 4 is the group:
- R 4a is selected from the group consisting of OH, (C;j_-Cg) alkoxy and aryloxy; and wherein R 4 -* 3 is an organic substituent selected from the group consisting of H, C- ⁇ -Cs alkyl, aryl, C7-C14 aralkyl, C7-C14 alkaryl, C3-C8 cycloalkyl, C -C8 alkoxyalkyl and these groups substituted with halogen, - CF3, -OH, C ⁇ -C8 alkyl, amino, carbonyl, and -CN.
- a more preferred R 4a group is selected from the group consisting of -OH, -OCH3 and -OC 2 H 5 .
- a more preferred R 4b is selected from the group consisting of H, C ⁇ -Cs alkyl, aryl, 0 -014 aralkyl, C -C14 alkaryl, C3-C8 cycloalkyl.
- a most preferred R 4 -- 0 is a group selected from H, CH3 , C2H5 and C3H7.
- R4 is the group - (Lc) - (acylamino acid group)-, wherein - (Lc) - is an acylamino acid linker with an acylamino acid linker length of 2 or 3 , and the "acylamino acid group" is represented by the formula:
- R 4c is selected from the group consisting of H, (C ⁇ -Cg) alkyl, (C ⁇ -Cg) alkoxy, heteroaryl and aryl, -CF3; and wherein NR 4c ⁇ is an amino acid residue of either a natural or unnatural amino acid with the nitrogen atom being part of the amino group of the amino acid; and wherein the amino acid residue is derived from an amino acid selected from the group comprising isoleucine, valine, phenylalanine, aspartic acid, leucine, glycine, asparagine, cystein, glutamine, glutamic acid, histidine, lysine, methionine, serine, threonine, tryptophan, tyrosine and derivatives thereof.
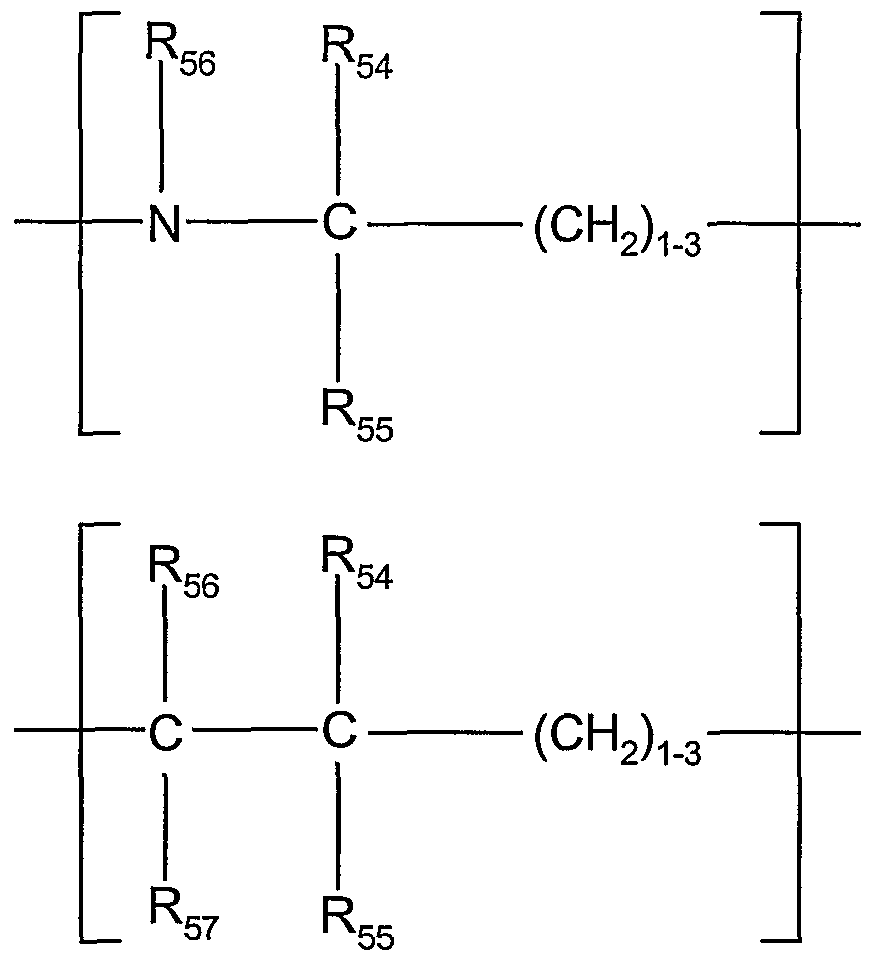
- a preferred acid linker, - (L a ) for R 5 is selected from the group consisting of;
- R 54 , R 55 , Rsg and R5 7 are each independently hydrogen, C ⁇ -C8 alkyl, C ⁇ -C 8 haloalkyl, aryl, C ⁇ -C 8 alkoxy, or halo.
- Preferred (acidic group) for R 5 is selected from the group consisting of -CO 2 H, -SO 3 H and -P(O) (OH) 2 .
- R5 is a non-interfering substituent
- the non-interfering substituent is independently selected from methyl, ethyl, propyl, isopropyl, thiomethyl, -O-methyl, C 4 ⁇ C alkyl, C 2 ⁇ Cg alkenyl, C 2 ⁇ Cg alkynyl, C 7 -C 12 aralkyl, C 7 -C 12 alkaryl, C 3 -C 8 cycloalkyl, C 3 -C 8 cycloalkenyl, phenyl, toluyl, xylenyl, biphenyl, C ⁇ -Cg alkoxy, C 2 ⁇ Cg alkenyloxy, C2-C alkynyloxy, C 2 -C 1 2 alkoxyalkyl, C2-C 12 alkoxyalkyloxy, C 2 - C 12 alkylcarbonyl, C -C 12 alkylcarbon
- non- interfering substituents are methyl, ethyl, propyl, and isopropyl.
- Preferred compounds of the invention are those having the general formula (II) , or a pharmaceutically acceptable salt, solvate or prodrug derivative thereof;
- R22 is selected from hydrogen, methyl, ethyl, propyl, isopropyl, cyclopropyl, -F, -CF3, -Cl, -Br, or -O-CH3.
- R 4 is selected from the group consisting of H,
- R ⁇ 5 is selected from hydrogen, C ⁇ -C8 alkyl, Ci-Cs alkoxy, C ⁇ -C8 alkylthio, C ⁇ -C8 haloalkyl, C ⁇ -C8 hydroxyalkyl , and halo.
- 3 is selected from hydrogen and C ⁇ -C8 alkyl, C ⁇ -C8 alkoxy, -S- (C ⁇ -C8 alkyl), C ⁇ -C8 haloalkyl, C ⁇ -C8, phenyl, halophenyl, hydroxyalkyl, and halo
- t is an integer from 0 to 5.
- substituted pyrrole-3 -oxime amide compounds of the invention represented by compounds of formula (I) or a pharmaceutically acceptable salt, solvate or prodrug thereof;
- A is O, S, or NR, and R is as described previously; R ⁇ ; R , and R5 are as described previously, and
- R3 is represented by the group - (L3) - Z, where - (L3) is a divalent linker group selected from a bond or a divalent group selected from:
- Z is selected from an oxime amide or oxime thioamide group represented by the formulae,
- X is oxygen or sulfur; and R a is selected from hydrogen, C ⁇ -Cs alkyl, aryl, C ⁇ -C8 alkaryl, C ⁇ -Cs alkoxy, aralkyl and -CN;
- R4 is selected from the group consisting of H, (C ⁇ Cg) alkyl, (C ⁇ -Cg) alkoxy, heteroaryl, aryl, - (L c ) - (acylamino acid group) , - (Lh) - (N-hydroxyfunctional amide group) or - (L a ) - (acidic group) .
- substituted pyrrole-3 -amide compounds of the invention represented by compounds of formula (I) or a pharmaceutically acceptable salt, solvate or prodrug thereof;
- A is O, S, or NR, and R is as described previously; R I' R 2' an
- Z is selected from an amide or a thioamide group represented by the formulae,
- X is oxygen or sulfur; and R is selected from hydrogen, C ⁇ -Cs alkyl, aryl, C ⁇ -C8 alkaryl, C ⁇ -Cs alkoxy, aralkyl and -CN; and n is 0, 1, 2 or 3 ;
- R4 is selected from the group consisting of H, (C ⁇ Cg) alkyl, (C ⁇ -Cg) alkoxy, heteroaryl, aryl, - (L c ) - (acylamino acid group) , - (L ) - (N-hydroxyfunctional amide group) or - (L a ) - (acidic group) .
- Preferred specific compounds which are illustrative of the compounds of the invention include compounds represented by the formulae (Cl) , (C2) , (C3) , (C4) , (C5) , (C6) , (C7) , (C8) , (C9) , (CIO), (Cll) , (C12), and (C13) ;
- salts of the above substituted pyrrole compounds represented by formulae (I) and (II) are an additional aspect of the invention.
- various salts may be formed which are more water soluble and physiologically suitable than the parent compound.
- Representative pharmaceutically acceptable salts include but are not limited to, the alkali and alkaline earth salts such as lithium, sodium, potassium, calcium, magnesium, aluminum and the like. Salts are conveniently prepared from the free acid by treating the acid in solution with a base or by exposing the acid to an ion exchange resin.
- salts include the relatively non-toxic, inorganic and organic base addition salts of compounds of the present invention, for example, ammonium, quaternary ammonium, and amine cations, derived from nitrogenous bases of sufficient basicity to form salts with the compounds of this invention (see, for example, S. M. Berge, et al . , "Pharmaceutical Salts," J. Phar. Sci., 66 : 1-19 (1977)).
- the basic group (s) of the compound of the invention may be reacted with suitable organic or inorganic acids to form salts such as acetate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, camsylate, carbonate, chloride, clavulanate, citrate, chloride, edetate, edisylate, estolate, esylate, fluoride, fumarate, gluceptate, gluconate, glutamate, glycolylarsanilate, hexylresorcinate, bromide, chloride, hydroxynaphthoate, iodide, isothionate, lactate, lactobionate, laurate, malate, malseate, mandelate, mesylate, methylbromide, methylnitrate, methylsulfate, mucate, napsylate, nitrate, oleate,
- Certain compounds of the invention may possess one or more chiral centers and may thus exist in optically active forms. Likewise, when the compounds contain an alkenyl or alkenylene group there exists the possibility of cis- and trans- isomeric forms of the compounds.
- the R- and S- isomers and mixtures thereof, including racemic mixtures as well as mixtures of cis- and trans- isomers, are contemplated by this invention. Additional asymmetric carbon atoms can be present in a substituent group such as an alkyl group. All such isomers as well as the mixtures thereof are intended to be included in the invention.
- a particular stereoisomer is desired, it can be prepared by methods well known in the art by using stereospecific reactions with starting materials which contain the asymmetric centers and are already resolved or, alternatively by methods which lead to mixtures of the stereoisomers and subsequent resolution by known methods.
- a racemic mixture may be reacted with a single enantiomer of some other compound. This changes the racemic form into a mixture of diastereomers and diastereomers, because they have different melting points, different boiling points, and different solubilities can be separated by conventional means, such as crystallization.
- Prodrugs are derivatives of the compounds of the invention which have chemically or metabolically cleavable groups and become by solvolysis or under physiological conditions the compounds of the invention which are pharmaceutically active in vivo.
- Derivatives of the compounds of this invention have activity in both their acid and base derivative forms, but the acid derivative form often offers advantages of solubility, tissue compatibility, or delayed release in a mammalian organism (see, Bundgard, H., Design of Prodrugs, pp. 7-9, 21-24, Elsevier, Amsterdam 1985) .
- Prodrugs include acid derivatives well known to practitioners of the art, such as, for example, esters prepared by reaction of the parent acidic compound with a suitable alcohol, or amides prepared by reaction of the parent acid compound with a suitable amine.
- Simple aliphatic or aromatic esters derived from acidic groups pendent on the compounds of this invention are preferred prodrugs.
- double ester type prodrugs such as (acyloxy) alkyl esters or ( (alkoxycarbonyl) oxy) alkyl esters.
- Particularly preferred esters as prodrugs are methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, tert-butyl, morpholinoethyl, and N, N-diethylglycolamido .
- N, N-diethylglycolamido ester prodrugs may be prepared by reaction of the sodium salt of a compound of Formula (I) (in a medium such as dimethylformamide) with 2-chloro-N,N- diethylacetamide (available from Aldrich Chemical Co., Milwaukee, Wisconsin USA; Item No. 25,099-6) .
- Morpholinylethyl ester prodrugs may be prepared by reaction of the sodium salt of a compound of Formula (I) (in a medium such as dimethylformamide) 4- (2- chloroethyl) morpholine hydrochloride (available from Aldrich Chemical Co., Milwaukee, Wisconsin USA, Item No. C4, 220-3) .
- the substituted pyrrole-3 -glyoxylamide derivative compounds of the invention are generally prepared by appending the glyoxylamide group to the 3 position of the substituted pyrrole nucleus (prepared as described infra) , generating intermediates which are themselves compounds of the invention. Alternatively, this is followed by coupling the "acidic group” or the “acylamino acid group” or the "N-hydroxyfunctional amide group” at the 4 or 5 position depending on the starting material , to the intermediate above to form other compounds of the invention.
- Preparation of the 3- glyoxylamide intermediates 7 or 7a is as shown in scheme 1: Scheme 1
- a compound of formula 1 (scheme 1) wherein A is O, NR, SO, SO 2 , or S is reacted with ethyl azidoacetate .
- the compound 1 is 4-methoxythiophen-3-carbaldehyde (CAS #82069-74-7) and can be synthesized in two steps from the corresponding 4-methoxythiophen-3 -carboxylic acid methylester (purchased from Maybridge Chemical Company) using lithiumaluminum hydride reduction to the alcohol, followed by oxidation with for example, TPAP (tetra-n-propylammonium perruthenate (VI) ) (CAS# 114615-86-9, Aldrich Chemical Co., Milwaukee, U.S.A.) in combination with 4-methylmorpohline- N-oxide in methylene chloride.
- TPAP tetra-n-propylammonium perruthenate
- the use of molecular sieves in the oxidation of alcohol to aldehyde as above is optional but preferred.
- the resulting starting material compound (1) is isolated by methods known to one of skill in the art, including but not limited to chromatography, crystallization, or distillation.
- Other compounds of formula 1 wherein A is NR, SO, or SO 2 may be purchased where available or made by methods known to one of skill in the art.
- the reaction to form compound (2) is catalyzed by a base, preferably a strong base such as sodium ethoxide purchased or generated as needed by dissolving sodium metal in absolute ethanol.
- the reagents are added at about -20 °C and allowed to warm up to about 30-50 °c over 30 to 60 minutes.
- the reaction mixture is worked-up by dilution with water and filtration, or by aqueous extraction as necessary to afford the azidoacrylate compound of formula (2) .
- the compound of formula (2) is ring closed to afford the compound of formula (3) under heating, and preferably in the presence of a catalyst.
- the preferred reaction conditions include heating compound (2) in refluxing toluene or other suitable solvent in the presence of a catalytic amount of a catalyst, e.g. rhodium acetate dimer.
- the product substituted pyrrole compound (3) is isolated by concentration followed by recrystallization. Other methods of isolation are within the purview of this invention and are known to one of skill in the art.
- the compound of formula (3) is transformed to the compound of formula (4) or analog thereof, by reacting compound (3) with an organic halide in the presence of a base such as for example sodium hydride and in the presence of a solvent such for example dimethylformamide .
- a base such as for example sodium hydride
- a solvent such for example dimethylformamide .
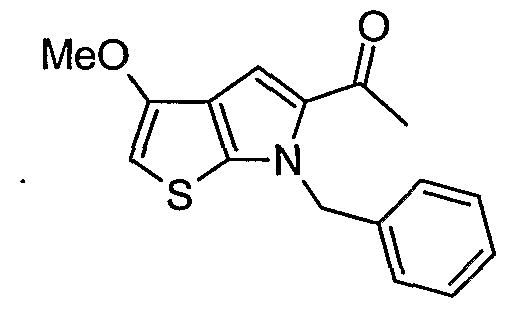
- treatment of compound (3) with sodium hydride in DMF followed by addition of benzylbromide results in compound (4) upon work-up.
- the compound of formula (4) is converted to the ketone, for example, the methylketone .
- Preparation of compound (5) is accomplished for example, by reaction of compound (4) with ⁇ -chloro- ⁇ - methylene [bis (cyclopentadienyl) -titanium] dimethylaluminum (Tebbe reagent) .
- the formation of the methylketone derivative using the Tebbe reagent involves addition of about 1.2 molar equivalent of Tebbe reagent to a cold (about 0 °C) solution of compound (4) in tetrahydrofuran followed by warming to about room temperature.
- a cold (about 0 °C) solution of compound (4) in tetrahydrofuran followed by warming to about room temperature.
- the reaction mixture is quenched by addition of saturated aqueous potassium carbonate. This likely results in vigorous evolution of gas..
- the product (5) is isolated upon aqueous. extractive work up procedures and standard purification methods, i.e. chromatography and/or crystallization.
- the compound of formula (5) may be converted to the compound of formula (6) or (6a) .
- the compound of formula (6) may be obtained for example, by use of borane-tetrahydrofuran complex (CAS# 14044-65-6, Aldrich Chemical Co.) or other method suitable for the reduction of compound (5) in THF or other suitable solvent or solvent mixture.
- the compound of formula (6a) may be obtained by the use of mild and/or selective reducing agents and/or reaction conditions which do not affect the methoxy substituent at position 4 or 5 depending on the starting material employed.
- the compound of formula (6 ⁇ or (6a) may be reacted with oxalyl chloride followed by reaction with ammonia (THF solution saturated with ammonia) to afford the compound of formula (7) or the corresponding analog compound- of formula (7a) .
- the compound of formula (7a) may be demethylated for example with boron tribromide in methylene chloride or other suitable solvent to afford the compound of formula (8a) .
- the compound of formula (8a) may be o-acylated, o-alkylated, o-arylated or otherwise converted to a compound represented within the scope of R 4 .
- the compound of formula (9a) may be converted to the free acid (10a) by hydrolysis.
- the free acid (10a) may in turn be converted to the acid salt or to derivatives such as the ester or amide by procedures known to one of skill in the art or found in general reference texts (J. March Advanced Organic Chemistry, Wiley Interscience publishers, New York, N.Y, 1985, and R. C. Larock Co prehensive Organic Transformations, VCH Publishers, New York, N.Y,
- the N- hydroxyfunctional amide group may be introduced via the acid (10a) or acid salt thereof, by reaction with for example hydroxylamine hydrochloride or substituted hydroxylamine hydrochloride to afford the N- hydroxyfunctional amide compound of formula (10b) .
- Substituted pyrrole-3 -acetamide derivative SPL 2 inhibitors may be lithiated at the (3) position with an organolithium reagent e.g. n- butyllithium, followed by quenching the lithiated intermediate with ethylene oxide for example, to afford upon hydrolysis, the terminal alcohol derivative (lOd) at position (3) (scheme 2) :
- organolithium reagent e.g. n- butyllithium
- the resulting alcohol intermediate (lOd) itself a compound of the invention may be converted by oxidation to the acid and further converted to the ester (11) .
- Conversion of the alcohol intermediate (lOd) to compound ester via an intermediate acid may be accomplished by oxidation of the alcohol with sodium hypochlorite in buffered t-butanol for example, followed by esterification of the incipient acid to the ester (11) . Methods for these conversions are known to one of skill in the art and may be found in general reference texts disclosed herein.
- the ester (11) may be converted to the acetamide derivative (12) or other substituted acetamide compound.
- N-substituted methylchloroaluminum amides result in the corresponding substituted acetamides (see Weinreb supra) .
- the 3 -substituted amide or acetamide substituted pyrrole compounds described above may be converted to the corresponding 4-substituted acylamino acid compounds or the 4-substituted N-hydroxyfunctional amide compounds as described previously for the glyoxylamide compounds .
- the compound of formula (7) is heated with hydroxylamine hydrochloride (when R is H) in a THF/methanol mixture for about 1 to 8 hours or until the reaction is deemed complete,
- the reaction product compound (7c) a compound of the invention, is isolated by chromatography or other known laboratory procedure.
- Substituted oximes such as when R is methyl, ethyl, phenyl or other non-interfering substituent may be prepared by reaction of the corresponding substituted hydroxylamine hydrochloride or free base with the glyoxylamide (e.g. compound 7) as described supra .
- ester i.e. methylester of the acid compound (10a) , or the acid salts thereof, may be converted to the corresponding oxime or substituted oxime functionality at position (3) by the method described above.
- the ester functionality at the (4) or (5) position on the substituted pyrrole nucleus, as in for example, compound (13) may be converted to the acid by hydrolysis using lithium hydroxide or other known ester hydrolysis methods to afford a compound of formula (14) . See, for example, J. March Advanced Organic Chemistry, Wiley Interscience publishers, New York, N.Y, 1985, and R. C. Larock Comprehensive Organic Transformations, VCH Publishers, New York, N.Y, 1989.
- the oxime compounds prepared as described above may be converted to the N-hydroxyfunctional amide at the 4 or 5 position, via the free acid, the ester or the acid salt functionalities at the 4 or 5 position.
- scheme 3 shows the conversion of the free acid compound (14) to the N-hydroxyfunctional amide compound (15) .
- the compound (14) and analogs thereof may be converted to the acylamino acid compound (16) and corresponding homologs thereof by procedures described supra .
- substituted pyrrole compounds described herein are believed to achieve their beneficial therapeutic action principally by direct inhibition of mammalian (including human) SPLA2 , and not by acting as antagonists for arachidonic acid, nor other active agents below arachidonic acid in the arachidonic acid cascade, such as 5-lipoxygenases, cyclooxygenases, and etc.
- the method of the invention for inhibiting SPLA 2 mediated release of fatty acids comprises contacting mammalian SPLA 2 with a therapeutically effective amount of substituted pyrrole compounds corresponding to formulae (I) or (II) as described herein including salt or a prodrug derivative thereof.
- Another aspect of this invention is a method for treating Inflammatory Diseases such as inflammatory bowel disease, septic shock, adult respiratory distress syndrome, pancreatitis, trauma, bronchial asthma, allergic rhinitis, rheumatoid arthritis, osteoarthritis, and related diseases which comprises administering to a mammal (including a human) a therapeutically effective dose of the substituted pyrrole compound of the invention (see, formulae I and II) .
- Inflammatory Diseases such as inflammatory bowel disease, septic shock, adult respiratory distress syndrome, pancreatitis, trauma, bronchial asthma, allergic rhinitis, rheumatoid arthritis, osteoarthritis, and related diseases
- the compounds of this invention are useful for inhibiting SPLA 2 mediated release of fatty acids such as arachidonic acid.
- inhibiting is meant the prevention or therapeutically significant reduction in release of SPLA2 initiated fatty acids by the compounds of the invention.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- the specific dose of a compound administered according to this invention to obtain therapeutic or prophylactic effects will, of course, be determined by the particular circumstances surrounding the case, including, for example, the compound administered, the route of administration and the condition being treated.
- Typical daily doses will contain a non-toxic dosage level of from about 0.01 mg/kg to about 50 mg/kg of body weight of an active compound of this invention.
- compounds of the invention (per Formula I or II) or pharmaceutical formulations containing these compounds are in unit dosage form for administration to a mammal .
- the unit dosage form can be a capsule or tablet itself, or the appropriate number of any of these.
- the quantity of Active ingredient in a unit dose of composition may be varied or adjusted from about 0.1 to about 1000 milligrams or more according to the particular treatment involved. It may be appreciated that it may be necessary to make routine variations to the dosage depending on the age and condition of the patient. The dosage will also depend on the route of administration.
- the compound can be administered by a variety of routes including oral, aerosol, rectal, transdermal, subcutaneous, intravenous, intramuscular, and intranasal .
- Pharmaceutical formulations of the invention are prepared by combining (e.g., mixing) a therapeutically effective amount of the substituted pyrrole compound of the invention together with a pharmaceutically acceptable carrier or diluent therefor.
- the present pharmaceutical formulations are prepared by known procedures using well known and readily available ingredients.
- the Active ingredient will usually be admixed with a carrier, or diluted by a carrier, or enclosed within a carrier which may be in the form of a capsule, sachet, paper or other container .
- a carrier which may be in the form of a capsule, sachet, paper or other container .
- the carrier serves as a diluent, it may be a solid, semi-solid or liquid material which acts as a vehicle, or can be in the form of tablets, pills, powders, lozenges, elixirs, suspensions, emulsions, solutions, syrups, aerosols (as a solid or in a liquid medium) , or ointment, containing, for example, up to 10% by weight of the active compound.
- the compounds of the present invention are preferably formulated prior to administration.
- the carrier may be a solid, liquid, or mixture of a solid and a liquid.
- the compounds of the invention may be dissolved in at a concentration of 2 mg/ml in a 4% dextrose/0.5% Na citrate aqueous solution.
- Solid form formulations include powders, tablets and capsules.
- a solid carrier can be one or more substances which may also act as flavoring agents, lubricants, solubilisers, suspending agents, binders, tablet disintegrating agents and encapsulating material .
- Tablets for oral administration may contain suitable excipients such as calcium carbonate, sodium carbonate, lactose, calcium phosphate, together with disintegrating agents, such as maize, starch, or alginic acid, and/or binding agents, for example, gelatin or acacia, and lubricating agents such as magnesium stearate, stearic acid, or talc.
- suitable excipients such as calcium carbonate, sodium carbonate, lactose, calcium phosphate
- disintegrating agents such as maize, starch, or alginic acid
- binding agents for example, gelatin or acacia
- lubricating agents such as magnesium stearate, stearic acid, or talc.
- the carrier is a finely divided solid which is in admixture with the finely divided Active ingredient.
- the Active ingredient is mixed with a carrier having the necessary binding properties in suitable proportions and compacted in the shape and size desired.
- the powders and tablets preferably contain from about 1 to about 99 weight percent of the Active ingredient which is the novel compound of this invention.
- Suitable solid carriers are magnesium carbonate, magnesium stearate, talc, sugar lactose, pectin, dextrin, starch, gelatin, tragacanth, methyl cellulose, sodium carboxymethyl cellulose, low melting waxes, and cocoa butter.
- Sterile liquid form formulations include suspensions, emulsions, syrups and elixirs.
- the Active ingredient can be dissolved or suspended in a pharmaceutically acceptable carrier, such as sterile water, sterile organic solvent or a mixture of both.
- a pharmaceutically acceptable carrier such as sterile water, sterile organic solvent or a mixture of both.
- the Active ingredient can often be dissolved in a suitable organic solvent, for instance aqueous propylene glycol.
- Other compositions can be made by dispersing the finely divided Active ingredient in aqueous starch or sodium carboxymethyl cellulose solution or in a suitable oil .
- Active ingredient refers to a compound according to Formula (I) or (II) or a pharmaceutically acceptable salt, solvate, or prodrug thereof.
- Hard gelatin capsules are prepared using the following ingredients:
- a tablet is prepared using the ingredients below:
- the components are blended and compressed to form tablets each weighing 665 mg
- Formulation 3 An aerosol solution is prepared containing the following components :
- the active compound is mixed with ethanol and the mixture added to a portion of the propellant 22, cooled to -30°C and transferred to a filling device. The required amount is then fed to a stainless steel container and diluted with the remainder of the propellant. The valve units are then fitted to the container.
- Tablets each containing 60 mg of Active ingredient, are made as follows:
- the Active ingredient, starch and cellulose are passed through a No. 45 mesh U.S. sieve and mixed thoroughly.
- the aqueous solution containing polyvinylpyrrolidone is mixed with the resultant powder, and the mixture then is passed through a No. 14 mesh U.S. sieve.
- the granules so produced are dried at 50 °C and passed through a No. 18 mesh U.S. sieve.
- the sodium carboxymethyl starch, magnesium stearate and talc, previously passed through a No . 60 mesh U.S. sieve, are then added to the granules which, after mixing, are compressed on a tablet machine to yield tablets each weighing 150 mg.
- Capsules each containing 80 mg of Active ingredient, are made as follows:
- Suppositories each containing 225 mg of Active ingredient, are made as follows:
- the Active ingredient is passed through a No. 60 mesh U.S. sieve and suspended in the saturated fatty acid glycerides previously melted using the minimum heat necessary. The mixture is then poured into a suppository mold of nominal 2 g capacity and allowed to cool .
- Suspensions each containing 50 mg of Active ingredient per 5 ml dose, are made as follows:
- the Active ingredient is passed through a No. 45 mesh U.S. sieve and mixed with the sodium carboxymethyl cellulose and syrup to form a smooth paste.
- the benzoic acid solution, flavor and color are diluted with a portion of the water and added, with stirring. Sufficient water is then added to produce the required volume .
- An intravenous formulation may be prepared as follows : Active ingredient 100 mg
- the solution of the above ingredients generally is administered intravenously to a subject at a rate of 1 ml per minute.
- Bovine Serum Albumin (fatty acid free) (1 g/L) (Sigma A-7030, product of Sigma Chemical Co., St. Louis MO, USA) TRIS HCl (3.94 g/L) pH 7.5 (adjust with NaOH) ENZYME BUFFER -
- a measured volume of racemic dipheptanoyl thio PC supplied in chloroform at a concentration of 100 mg/ml is taken to dryness and redissolved in 10 millimolar
- reaction Buffer is added to the solution, then DTNB to give the Reaction Mixture.
- the reaction mixture thus obtained contains ImM diheptanoly thio-PC substrate, 0.29 mm Triton X- 100TM detergent, and 0.12 mm DTMB in a buffered aqueous solution at pH 7.5.
- test compound or solvent blank
- SPLA2 10 microliters
- % inhibition measured at 405 nanometers generated by enzyme reactions containing inhibitors relative to the uninhibited control reactions was determined. Each sample was titrated in triplicate and result values were averaged for plotting and calculation of IC50 values. IC50 values were determined by plotting log concentration versus inhibition values in the range from 10-90% inhibition.
Abstract
Description































































Claims























Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP01958850A EP1307461A2 (en) | 2000-08-04 | 2001-07-20 | Substituted pyrrole compounds and their use as spla2 inhibitors |
| JP2002518224A JP2004505982A (en) | 2000-08-04 | 2001-07-20 | Novel sPLA2 inhibitors |
| US10/332,480 US6730694B1 (en) | 2001-07-20 | 2001-07-20 | sPLA2 inhibitors |
| AU2001280461A AU2001280461A1 (en) | 2000-08-04 | 2001-07-20 | Substituted pyrrole compounds and their use as spla2 inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US22339800P | 2000-08-04 | 2000-08-04 | |
| US60/223,398 | 2000-08-04 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2002012249A2 true WO2002012249A2 (en) | 2002-02-14 |
| WO2002012249A3 WO2002012249A3 (en) | 2002-07-18 |
Family
ID=22836318
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2001/021106 WO2002012249A2 (en) | 2000-08-04 | 2001-07-20 | Substituted pyrrole compounds and their use as spla2 inhibitors |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP1307461A2 (en) |
| JP (1) | JP2004505982A (en) |
| AU (1) | AU2001280461A1 (en) |
| WO (1) | WO2002012249A2 (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7884124B2 (en) | 2006-06-30 | 2011-02-08 | Sepracor Inc. | Fluoro-substituted inhibitors of D-amino acid oxidase |
| US7893098B2 (en) | 2003-12-29 | 2011-02-22 | Sepracor Inc. | Pyrrole and pyrazole DAAO inhibitors |
| US7902252B2 (en) | 2007-01-18 | 2011-03-08 | Sepracor, Inc. | Inhibitors of D-amino acid oxidase |
| US8053603B2 (en) | 2006-01-06 | 2011-11-08 | Sunovion Pharmaceuticals Inc. | Tetralone-based monoamine reuptake inhibitors |
| US8097760B2 (en) | 2006-03-31 | 2012-01-17 | Sunovion Pharmacuticals Inc. | Preparation of chiral amides and amines |
| US8669291B2 (en) | 2007-05-31 | 2014-03-11 | Sunovion Pharmaceuticals Inc. | Phenyl substituted cycloalkylamines as monoamine reuptake inhibitors |
| US8877975B2 (en) | 2006-01-06 | 2014-11-04 | Sunovion Pharmaceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0114014A2 (en) * | 1982-12-16 | 1984-07-25 | Adir | Thieno(2,3-b) pyrrole derivatives, process for their preparation and pharmaceutical compositions containing them |
| FR2564467A1 (en) * | 1984-05-21 | 1985-11-22 | Adir | Novel derivatives of thieno[2,3-b]pyrrole, process for their preparation and the pharmaceutical compositions which contain them |
| FR2565981A1 (en) * | 1984-06-15 | 1985-12-20 | Adir | Novel thieno[2,3-b]pyrrole derivatives, process for their preparation and the pharmaceutical compositions which contain them |
| EP0675110A1 (en) * | 1994-04-01 | 1995-10-04 | Eli Lilly And Company | 1H-Indole-3-glyoxylamide sPLA2 inhibitors |
| WO1998025609A1 (en) * | 1996-12-10 | 1998-06-18 | Eli Lilly And Company | PYRROLES AS sPLA2 INHIBITORS |
| WO1999009978A1 (en) * | 1997-08-28 | 1999-03-04 | Eli Lilly And Company | Method for treatment of non-rheumatoid athritis |
| WO1999016453A1 (en) * | 1997-09-26 | 1999-04-08 | Eli Lilly And Company | Method for the treatment of cystic fibrosis |
| WO1999040914A1 (en) * | 1998-02-17 | 1999-08-19 | Astrazeneca Uk Limited | Bicyclic pyrrole derivatives as mcp-1 inhibitors |
| WO1999051605A1 (en) * | 1998-03-31 | 1999-10-14 | Shionogi & Co., Ltd. | PYRROLO[1,2-a]PYRAZINE sPLA2 INHIBITOR |
| WO1999059999A1 (en) * | 1998-05-21 | 1999-11-25 | Shionogi & Co., Ltd. | PYRROLO[1,2-b]PYRIDAZINE DERIVATIVES HAVING sPLA2 INHIBITORY EFFECT |
| WO2000000201A1 (en) * | 1998-06-30 | 2000-01-06 | Eli Lilly And Company | BICYCLIC sPLA2 INHIBITORS |
-
2001
- 2001-07-20 EP EP01958850A patent/EP1307461A2/en not_active Withdrawn
- 2001-07-20 AU AU2001280461A patent/AU2001280461A1/en not_active Abandoned
- 2001-07-20 WO PCT/US2001/021106 patent/WO2002012249A2/en active Application Filing
- 2001-07-20 JP JP2002518224A patent/JP2004505982A/en not_active Withdrawn
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0114014A2 (en) * | 1982-12-16 | 1984-07-25 | Adir | Thieno(2,3-b) pyrrole derivatives, process for their preparation and pharmaceutical compositions containing them |
| FR2564467A1 (en) * | 1984-05-21 | 1985-11-22 | Adir | Novel derivatives of thieno[2,3-b]pyrrole, process for their preparation and the pharmaceutical compositions which contain them |
| FR2565981A1 (en) * | 1984-06-15 | 1985-12-20 | Adir | Novel thieno[2,3-b]pyrrole derivatives, process for their preparation and the pharmaceutical compositions which contain them |
| EP0675110A1 (en) * | 1994-04-01 | 1995-10-04 | Eli Lilly And Company | 1H-Indole-3-glyoxylamide sPLA2 inhibitors |
| WO1998025609A1 (en) * | 1996-12-10 | 1998-06-18 | Eli Lilly And Company | PYRROLES AS sPLA2 INHIBITORS |
| WO1999009978A1 (en) * | 1997-08-28 | 1999-03-04 | Eli Lilly And Company | Method for treatment of non-rheumatoid athritis |
| WO1999016453A1 (en) * | 1997-09-26 | 1999-04-08 | Eli Lilly And Company | Method for the treatment of cystic fibrosis |
| WO1999040914A1 (en) * | 1998-02-17 | 1999-08-19 | Astrazeneca Uk Limited | Bicyclic pyrrole derivatives as mcp-1 inhibitors |
| WO1999051605A1 (en) * | 1998-03-31 | 1999-10-14 | Shionogi & Co., Ltd. | PYRROLO[1,2-a]PYRAZINE sPLA2 INHIBITOR |
| WO1999059999A1 (en) * | 1998-05-21 | 1999-11-25 | Shionogi & Co., Ltd. | PYRROLO[1,2-b]PYRIDAZINE DERIVATIVES HAVING sPLA2 INHIBITORY EFFECT |
| WO2000000201A1 (en) * | 1998-06-30 | 2000-01-06 | Eli Lilly And Company | BICYCLIC sPLA2 INHIBITORS |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7893098B2 (en) | 2003-12-29 | 2011-02-22 | Sepracor Inc. | Pyrrole and pyrazole DAAO inhibitors |
| US8053603B2 (en) | 2006-01-06 | 2011-11-08 | Sunovion Pharmaceuticals Inc. | Tetralone-based monoamine reuptake inhibitors |
| US8877975B2 (en) | 2006-01-06 | 2014-11-04 | Sunovion Pharmaceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| US9868718B2 (en) | 2006-01-06 | 2018-01-16 | Sunovion Pharmaceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| US10562878B2 (en) | 2006-01-06 | 2020-02-18 | Sunovion Pharamceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| US8097760B2 (en) | 2006-03-31 | 2012-01-17 | Sunovion Pharmacuticals Inc. | Preparation of chiral amides and amines |
| US7884124B2 (en) | 2006-06-30 | 2011-02-08 | Sepracor Inc. | Fluoro-substituted inhibitors of D-amino acid oxidase |
| US7902252B2 (en) | 2007-01-18 | 2011-03-08 | Sepracor, Inc. | Inhibitors of D-amino acid oxidase |
| US8669291B2 (en) | 2007-05-31 | 2014-03-11 | Sunovion Pharmaceuticals Inc. | Phenyl substituted cycloalkylamines as monoamine reuptake inhibitors |
| US9586888B2 (en) | 2007-05-31 | 2017-03-07 | Sunovion Pharmaceuticals Inc. | Phenyl substituted cycloalkylamines as monoamine reuptake inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2001280461A1 (en) | 2002-02-18 |
| JP2004505982A (en) | 2004-02-26 |
| WO2002012249A3 (en) | 2002-07-18 |
| EP1307461A2 (en) | 2003-05-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6974831B2 (en) | sPLA2 inhibitors | |
| US6608099B1 (en) | Indole sPLA2 inhibitors | |
| US6451839B1 (en) | Indole sPLA2 inhibitors | |
| US6730694B1 (en) | sPLA2 inhibitors | |
| US6831095B1 (en) | Hydroxyfunctional amide 1h-indole derivatives active as sPLA2 inhibitors | |
| EP1423366A1 (en) | Cyclopenta b ! indole derivatives as spla2 inhibitors | |
| US6391908B1 (en) | Oxime amide indole type sPLA2 inhibitors | |
| WO2002012249A2 (en) | Substituted pyrrole compounds and their use as spla2 inhibitors | |
| US20040063941A1 (en) | Novel spla2 inhibitors | |
| EP1220839B1 (en) | Hydroxyfunctional amide 1h-indole derivatives active as spla2 inhibitors | |
| US6930123B2 (en) | sPLA2 inhibitors | |
| EP1358156B1 (en) | Benz(g) indoles and their use as spla2 inhibitors | |
| US20040092543A1 (en) | Novel spla2 inhibitors | |
| EP1349836B1 (en) | Tetracyclic derivatives as spla2 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10332480 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001958850 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001958850 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |