NOVEL METHODS FOR THE TREATMENT AND PREVENTION OF DIZZINESS AND PRURITUS
Field of the Invention
The present invention relates to novel methods for the treatment and prevention of dizziness and/or pruritus. More particularly, the present invention relates to novel methods for the treatment and prevention of dizziness and/or pruritus by using peripheral mu opioid antagonist compounds.
Background of the Invention
It is well known that opioids target the three types of endogenous opioid receptors (mu, delta, and kappa) in biological systems. Most opioids, such as morphine, are mu opioid agonists that are often used as analgesics for the treatment of severe pain due to their activation of mu opioid receptors in the brain and central nervous system (CNS). Mu opioid receptors are, however, not limited to the CNS, and are found in other tissues throughout the body. A number of side effects of opioid drugs are caused by activation of these peripheral receptors. For example, administration of mu opioid agonists often results in intestinal dysfunction due to the large number of receptors in the wall of the gut (Wittert, G., Hope, P. and Pyle, D., Biochemical and Biophysical Research Communications 1996, 218, 877-881; Bagnol, D., Mansour, A., Akil, A. and Watson, S.J., Neuroscience 1997, 81, 579-591). Specifically, opioids may frequently cause nausea, vomiting, dizziness, pruritus, as well as the inhibition of normal propulsive gastrointestinal function in animals and man (Reisine, T., and Pasternak, G., Goodman & Gilman 's The Pharmacological Basis of Therapeutics Ninth Edition 1996, 521-555).
Opioid-induced side effects, such as nausea, vomiting, dizziness, pruritus, and inhibited gastrointestinal propulsive activity remain serious problems for patients being administered opioid analgesics for both short term and long term pain management. Opioid antagonist compounds that do not readily cross the blood-brain barrier (peripherally acting drugs) have been tested for use in curbing opioid-induced side effects. For instance, the peripheral mu opioid antagonist methylnaltrexone and related compounds
have been indicated as being useful in curbing a variety of opioid-induced side effects. Specifically, U.S. Patent Nos. 5,972,954, 5,102,887, 4,861,781, and 4,719,215 disclose the use of methylnaltrexone and related compounds in controlling opioid-induced pruritus, nausea, and/or vomiting. In addition, methylnaltrexone has been shown to effectively reduce the incidence of opioid-induced nausea and pruritus as disclosed by Yuan, C.-S. et al. Drug and Alcohol Dependence 1998, 52, 161. Another family of peripheral mu opioid antagonists, described in U.S. Patent Nos. 5,250,542, 5,434,171, 5,159,081, and 5,270,328, are piperidine-N-alkylcarboxylates and are disclosed as being useful for the treatment of the opioid side effects constipation, nausea and vomiting, as well as irritable bowel syndrome and idiopathic constipation. No mention is made in U.S. Patent Nos. 5,250,542, 5,434,171, 5,159,081, and 5,270,328 of treating pruritus and/or dizziness with the disclosed compounds.
Given that opioid-induced side effects such as dizziness and pruritus are common discomforts experienced by patients that often go untreated for lack of effective remedies, a need exists for the treatment and prevention of these side effects. The majority of currently known opioid antagonist therapies are not peripherally selective, i.e., they cross the blood-brain barrier, and have the potential for counteracting the desirable effects of opioid drugs, such as analgesia, acting on the central nervous system. The present invention is directed to these, as well as other important ends.
Summary of the Invention
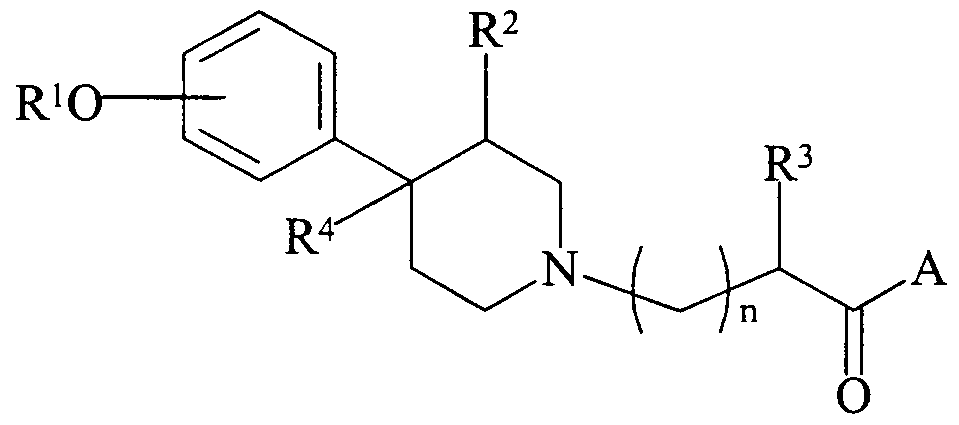
Accordingly, the present invention is directed, in part, to novel methods for treating and/or preventing dizziness and/or pruritus. Specifically, in one embodiment, there are provided novel methods of treating or preventing dizziness and pruritus comprising administering to a patient an effective amount of a compound of the following formula (I):
I
wherein:
R1 is hydrogen or alkyl;
R2 is hydrogen, alkyl or alkenyl; R3 is hydrogen, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl or aryl-substituted alkyl;
R4 is hydrogen, alkyl or alkenyl;
A is OR5 or NR6R7; wherein:
R5 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl;
R6 is hydrogen or alkyl;
R7 is hydrogen, alkyl, alkenyl, cycloalkyl, aryl, cycloalkyl-substituted alkyl, cycloalkenyl, cycloalkenyl-substituted alkyl, aryl-substituted alkyl, aryl-substituted alkyl, or alkylene substitued B or, together with the nitrogen atom to which they are attached, R6 and R7 form a heterocyclic ring;
B is
C(=O)W or NR8R9; wherein;
R8 is hydrogen or alkyl; R9 is hydrogen, alkyl, alkenyl, cycloalkyl-substituted alkyl, cycloalkyl, cycloalkenyl, cycloalkenyl-substituted alkyl, aryl or aryl-substituted alkyl or, together with the nitrogen atom to which they are attached, R8 and R9 form a heterocyclic ring;
W is OR10, NRπR12, or OE; wherein
R10 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl;
R" is hydrogen or alkyl;
R12 is hydrogen, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, aryl-substituted alkyl or alkylene substituted C(=O)Y or, together with the nitrogen atom to which they are attached, R11 and R12 form a heterocyclic ring;
E is
alkylene substituted (C=O)D, or -R13OC(=O)R14; wherein
R13 is alkyl substituted alkylene; R14 is alkyl; D is OR15 or NR16R17; wherein:
R15 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl;
R16 is hydrogen, alkyl, alkenyl, aryl, aryl-substituted alkyl, cycloalkyl, cycloalkenyl, cycloalkyl-substituted alkyl or cycloalkenyl-substituted alkyl; R17 is hydrogen or alkyl or, together with the nitrogen atom to which they are attached, R16 and R17 form a heterocyclic ring;
Y is OR18 or NR19R20; wherein:
R18 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl; R19 is hydrogen or alkyl;
R20 is hydrogen, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl or, together with the nitrogen atom to which they are attached, R19 and R20 form a heterocyclic ring;
R21 is hydrogen or alkyl; and n is 0 to 4; or a stereoisomer, prodrug, or pharmaceutically acceptable salt, hydrate or N-oxide thereof.
Another embodiment of the invention relates to methods of treating or preventing dizziness and pruritus comprising administering to the patient an effective amount of a peripheral mu opioid antagonist compound.
These and other aspects of the invention will become more apparent from the following detailed description.
Detailed Description of the Invention
As employed above and throughout the disclosure, the following terms, unless otherwise indicated, shall be understood to have the following meanings.
"Alkyl" refers to an aliphatic hydrocarbon group which may be straight, branched or cyclic having from 1 to about 10 carbon atoms in the chain, and all combinations and subcombinations of ranges therein. "Branched" refers to an alkyl group in which a lower alkyl group, such as methyl, ethyl or propyl, is attached to a linear alkyl chain. In certain preferred embodiments, the alkyl group is a CrC5 alkyl group, i.e., a branched or linear alkyl group having from 1 to about 5 carbons. In other preferred embodiments, the alkyl group is a C,-C3 alkyl group, i.e., a branched or linear alkyl group having from 1 to about 3 carbons. Exemplary alkyl groups include methyl, ethyl, n- propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl and decyl. "Lower alkyl" refers to an alkyl group having 1 to about 6 carbon atoms. Preferred alkyl groups include the lower alkyl groups of 1 to about 3 carbons.
"Alkenyl" refers to an alkyl group containing at least one carbon-carbon double bond and having from 2 to about 10 carbon atoms in the chain, and all combinations and subcombinations of ranges therein. In certain preferred embodiments, the alkenyl group is a C2-C10 alkyl group, i.e., a branched or linear alkenyl group having
from 2 to about 10 carbons. In other preferred embodiments, the alkenyl group is a C2-C6 alkenyl group, i.e., a branched or linear alkenyl group having from 2 to about 6 carbons. In still other preferred embodiments, the alkenyl group is a C3-C10 alkenyl group, i.e., a branched or linear alkenyl group having from about 3 to about 10 carbons. In yet other preferred embodiments, the alkenyl group is a C2-C5 alkenyl group, i.e., a branched or linear alkenyl group having from 2 to about 5 carbons. Exemplary alkenyl groups include, for example, vinyl, propenyl, butenyl, pentenyl hexenyl, heptenyl, octenyl, nonenyl and decenyl groups.
"Alkylene" refers to a straight or branched bivalent aliphatic hydrocarbon group having from 1 to about 6 carbon atoms, and all combinations and subcombinations of ranges therein. The alkylene group may be straight, branched or cyclic. Exemplary alkylene groups include, for example, methylene (-CH2-), ethylene (-CH2CH2-) and propylene (-(CH2)3-). There may be optionally inserted along the alkylene group one or more oxygen, sulphur or optionally substituted nitrogen atoms, wherein the nitrogen substituent is alkyl as described previously. Preferred alkylene groups have from about 1 to about 4 carbons.
"Alkenylene" refers to an alkylene group containing at least one carbon- carbon double bond. Exemplary alkenylene groups include, for example, ethenylene (- CH=CH-) and propenylene (-CH=CHCH2-). Preferred alkenylene groups have from 2 to about 4 carbons.
"Cycloalkyl" refers to any stable monocyclic or bicyclic ring having from about 3 to about 10 carbons, and all combinations and subcombinations of ranges therein. In preferred embodiments, the cycloalkyl group is a C3-C8 cycloalkyl group, i.e., a cycloalkyl group having from about 3 to about 8 carbons, with C3-C6 cycloalkyl groups, i.e., cycloalkyl groups having from about 3 to about 6 carbons being more preferred. The cycloalkyl group may be optionally substituted with one or more cycloalkyl group substituents. Preferred cycloalkyl group substituents include alkyl, preferably C,-C3 alkyl, alkoxy, preferably CrC3 alkoxy, or halo. Exemplary cycloalkyl groups include, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl groups.
"Cycloalkyl-substituted alkyl" refers to a linear alkyl group, preferably a lower alkyl group, substituted at a terminal carbon with a cycloalkyl group, preferably a C3-C8 cycloalkyl group. Typical cycloalkyl-substituted alkyl groups include cyclohexylmethyl, cyclohexylethyl, cyclopentylethyl, cyclopentylpropyl, cyclopropylmethyl and the like.
"Cycloalkenyl" refers to an olefmically unsaturated cycloalkyl group having from about 4 to about 10 carbons, and all combinations and subcombinations of ranges therein. In preferred embodiments, the cycloalkenyl group is a C5-C8 cycloalkenyl group, i.e., a cycloalkenyl group having from about 5 to about 8 carbons. "Alkoxy" refers to an alkyl-O- group where alkyl is as previously described. Exemplary alkoxy groups include, for example, methoxy, ethoxy, propoxy, butoxy and heptoxy.
"Alkoxy-alkyl" refers to an alkyl-O-alkyl group where alkyl is as previously described. "Acyl" means an alkyl-CO- group wherein alkyl is as previously described.
Preferred acyl groups comprise lower alkyl groups, such as alkyl of about 1 to about 3 carbons. Exemplary acyl groups include acetyl, propanoyl, 2-methylpropanoyl, butanoyl and palmitoyl.
"Aryl" refers to an aromatic carbocyclic radical containing from about 6 to about 10 carbons, and all combinations and subcombinations of ranges therein. The phenyl group may be optionally substituted with one or two or more aryl group substituents. Preferred aryl group substituents include alkyl groups, preferably C,-C2 alkyl groups. Exemplary aryl groups include phenyl and naphthyl.
"Aryl-substituted alkyl" refers to an linear alkyl group, preferably a lower alkyl group, substituted at a terminal carbon with an optionally substituted aryl group, preferably an optionally substituted phenyl ring. Exemplary aryl-substituted alkyl groups include, for example, phenylmethyl, phenylethyl and 3-(4-methylphenyl)propyl.
"Heterocyclic" refers to a monocyclic or multicylic ring system carbocyclic radical containing from about 4 to about 10 members, and all combinations and subcombinations of ranges therein, wherein one or more of the members is an element other than carbon, for example, nitrogen, oxygen or sulfur. The heterocyclic group may be
aromatic or nonaromatic. Exemplary heterocyclic groups include, for example, pyrrole and piperidine groups.
"Halo" refers to fluoro, chloro or bromo.
"Effective amount" refers to an amount of a compound as described herein that may be therapeutically effective to prevent or treat the symptoms of particular disorder. Such disorders include, but are not limited to, those pathological disorders associated with dizziness and pruritus, wherein the treatment or prevention comprises, for example, inhibiting the activity thereof by contacting cells, tissues and/or receptors with compounds of the present invention. "Pharmaceutically acceptable" refers to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem complications commensurate with a reasonable benefit/risk ratio. "Pharmaceutically acceptable salts" refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like. The pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. For example, such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, and the like.
Certain acidic or basic compounds of the present invention may exist as zwitterions. All forms of the compounds, including free acid, free base and zwitterions, are contemplated to be within the scope of the present invention.
"Patient" refers to animals, including mammals, preferably humans.
The present invention is directed to methods for the treatment and/or prevention of dizziness and/or pruritus. Dizziness and/or pruritus may be associated (i.e., caused and/or exacerbated) by a wide variety of diseases, disorders and/or conditions, and it is contemplated that the present methods may be used to treat dizziness and/or pruritus associated with these various diseases, disorders and or conditions. As discussed above, dizziness and pruritus (as well as other undesirable side effects) may be frequently associated with the interaction of opioids including, for example, exogenous opioids and/or endogenous opioids, with tissues, cells and/or receptors in the body. By virtue of the methods of the present invention, effective and desirable inhibition of dizziness and pruritus (as well as other undesirable side effects) that may be associated with opioid compounds may be advantageously achieved. Thus, the present methods may be particularly suitable for treating and/or preventing dizziness and/or pruritus associated with opioids, including exogenous opioids (such as, for example, exogenous opioid analgesics) and/or endogenous opioids (such as, for example, endogenous opioid peptides). Therefore, the methods of the present invention may be used for treating and/or preventing dizziness and/or pruritus in patients who have been and/or are being administered exogenous opioid compounds. The methods of the present invention may also be used for treating and/or preventing dizziness and/or pruritus in patients who have not and/or are not being administered exogenous opioid compounds. In preferred form, the present methods may be used for treating and/or preventing dizziness and/or pruritus associated with the administration to a patient of exogenous opioids.
While not intending to be bound by any theory or theories of operation, it is contemplated that, in the case of dizziness and/or pruritus associated with opioids, dizziness and pruritus may result from may result from undesirable interaction of the opioid with peripheral mu receptors. Administration of a mu opioid antagonist according to the methods of the present invention may block interaction of the opioid compounds with the mu receptors in the gut, thereby preventing and/or inhibiting dizziness and/or pruritus.
In preferred form, the methods of the present invention involve administering to a patient a compound which is a peripheral mu opioid antagonist compound. The term peripheral designates that the compound acts primarily on
physiological systems and components external to the central nervous system, i.e., the compound preferably does not readily cross the blood-brain barrier. In preferred form, the peripheral mu opioid antagonist compounds employed in the methods of the present invention exhibit high levels of activity with respect to gastrointestinal tissue, while exhibiting reduced, and preferably substantially no, central nervous system (CNS) activity. The term "substantially no CNS activity", as used herein, means that less than about 20% of the pharmacological activity of the peripheral mu opioid antagonist compounds employed in the present methods is exhibited in the CNS. In preferred embodiments, the peripheral mu opioid antagonist compounds employed in the present methods exhibit less than about 15% of their pharmacological activity in the CNS, with less than about 10% being more preferred. In even more preferred embodiments, the peripheral mu opioid antagonist compounds employed in the present methods exhibit less than about 5% of their pharmacological activity in the CNS, with about 0% (i.e., no CNS activity) being still more preferred. In more preferred embodiments, the present methods involve the administration to a patient of a mu peripheral opioid antagonist compound that is a piperidine-N-alkylcarboxylate compound. Prefeπed piperidine-N-alkylcarboxylate opioid antagonist compounds include, for example, the compounds disclosed in U.S. Patent Nos. 5,250,542; 5,159,081; 5,270,328; and 5,434,171, the disclosures of which are hereby incorporated herein by reference, in their entireties. A particularly preferred class of piperidine-N-alkylcarboxylate opioid antagonist compounds include those having the following formula (I):
wherein: R1 is hydrogen or alkyl;
R2 is hydrogen, alkyl or alkenyl;
R3 is hydrogen, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl or aryl-substituted alkyl;
R4is hydrogen, alkyl or alkenyl; A is OR5 or NR6R7; wherein:
R5 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl;
R6 is hydrogen or alkyl;
R7 is hydrogen, alkyl, alkenyl, cycloalkyl, aryl, cycloalkyl-substituted alkyl, cycloalkenyl, cycloalkenyl-substituted alkyl, aryl-substituted alkyl, aryl-substituted alkyl, or alkylene substitued B or, together with the nitrogen atom to which they are attached, R6 and R7 form a heterocyclic ring;
B is
C(=O)W or NR8R9; wherein;
R8 is hydrogen or alkyl;
R9 is hydrogen, alkyl, alkenyl, cycloalkyl-substituted alkyl, cycloalkyl, cycloalkenyl, cycloalkenyl-substituted alkyl, aryl or aryl-substituted alkyl or, together with the nitrogen atom to which they are attached, R8 and R9 form a heterocyclic ring; W is OR10, NR1 'R12, or OE; wherein
R10 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl; R11 is hydrogen or alkyl;
R
12 is hydrogen, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, aryl-substituted alkyl or alkylene substituted C(=O)Y or, together with the nitrogen atom to which they are attached, R
11 and R
12 form a heterocyclic ring; E is
alkylene substituted (C=O)D, or -R13OC(-O)R14; wherein
R13 is alkyl substituted alkylene; R14 is alkyl;
D is OR15 or NRI6R17; wherein:
R15 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl; R16 is hydrogen, alkyl, alkenyl, aryl, aryl-substituted alkyl, cycloalkyl, cycloalkenyl, cycloalkyl-substituted alkyl or cycloalkenyl-substituted alkyl;
R17 is hydrogen or alkyl or, together with the nitrogen atom to which they are attached, R16 and R17 form a heterocyclic ring;
Y is OR18 or NR19R20; wherein:
R18 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl;
R19 is hydrogen or alkyl;
R20 is hydrogen, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, cycloalkyl- substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl or, together with the nitrogen atom to which they are attached, R19 and R20 form a heterocyclic ring;
R21 is hydrogen or alkyl; and n is 0 to 4; or a stereoisomer, prodrug, or pharmaceutically acceptable salt, hydrate or N-oxide thereof. In the above formula (I), R1 is hydrogen or alkyl. In preferred embodiments, R1 is hydrogen or CrC5alkyl. In even more preferred embodiments, R1 is hydrogen.
In the above formula (I), R2 is hydrogen, alkyl or alkenyl. In preferred embodiments, R2 is hydrogen, C,-C5alkyl or C2-C6alkenyl. Also in preferred embodiments, R2is alkyl, with C C3alkyl being more preferred.
In the above formula (I), R3 is hydrogen, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl or aryl- substituted alkyl. In preferred embodiments, R3 is hydrogen, CrC10alkyl, C3-C,0alkenyl, phenyl, cycloalkyl, C5-C8cycloalkenyl, cycloalkyl-substituted C C3alkyl, C5-C8cycloalkyl- substituted C,-C3alkyl or phenyl-substituted C,-C3 alkyl. In more preferred embodiments, R3 is benzyl, phenyl, cyclohexyl, or cyclohexylmethyl. In the above formula (I), R4 is hydrogen, alkyl or alkenyl. In preferred embodiments, R4 is hydrogen, C,-C5alkyl or C2-C6alkenyl. In more preferred embodiments, R4 is C,-C3alkyl, with methyl being even more preferred. In the above formula (I), A is OR5 or NR6R7. In the above formula (I), R5 is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl, or aryl- substituted alkyl. In preferred embodiments, R5 is hydrogen, CrC10alkyl, C2-C10alkenyl, cycloalkyl, C5-C8cycloalkenyl, cycloalkyl-substitutedCrC3 alkyl, C5-C8cycloalkenyl- substituted C,-C3alkyl, or phenyl-substituted CrC3alkyl. Also in preferred embodiments, R5 is hydrogen or alkyl, with C,-C3alkyl being more preferred. In the above formula (I), R6 is hydrogen or alkyl. Preferably, R6 is hydrogen or - alkyl. Even more preferably, R6 is hydrogen.
In the above formula (I), R7 is hydrogen, alkyl, alkenyl, cycloalkyl, aryl, cycloalkyl-substituted alkyl, cycloalkenyl, cycloalkenyl-substituted alkyl, aryl-substituted alkyl, aryl-substituted alkyl or alkylene substituted B. In preferred embodiments, R7 is hydrogen, CrC10alkyl, C3-C,0alkenyl, phenyl, cycloalkyl, cycloalkyl-substituted Cr
C3alkyl, C5-C8cycloalkenyl, C5-C8cycloalkenyl-substituted C C3alkyl, phenyl-substituted C,-C3alkyl or (CH2)q-B. In more preferred embodiments, R7 is (CH2)q-B.
In certain alternative embodiments, in the above formula (I), R
6 and R
7 form, together with the nitrogen atom to which they are attached, a heterocyclic ring. The group B in the definition of R
7 is
C(=O)W or NR8R9. In preferred embodiments, B is C(=O)W.
The group R8 in the definition of B is hydrogen or alkyl. In preferred embodiments, R8 is hydrogen or CrC3alkyl. The group R9 in the definition of B is hydrogen, alkyl, alkenyl, cycloalkyl- substituted alkyl, cycloalkyl, cycloalkenyl, cycloalkenyl-substituted alkyl, aryl or aryl- substituted alkyl. In preferred embodiments, R9 is hydrogen, C,-C10alkyl, C3-C10alkenyl, cycloalkyl-substituted C,-C3alkyl, cycloalkyl, C5-C8cycloalkenyl, C5-C8cycloalkenyl- substituted CrC3alkyl, phenyl or phenyl-substituted C,-C3alkyl. In certain alternative embodiments, in the definition of B, R8 and R9 form, together with the nitrogen atom to which they are attached, a heterocyclic ring.
The group W in the definition of B is OR10, NRπR12 or OE.
The group R10 in the definition of W is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl, or aryl- substituted alkyl. In preferred embodiments, R10 is hydrogen, C,-CI0alkyl, C2-Cι0alkenyl, cycloalkyl, C5-C8cycloalkenyl, cycloalkyl-substituted CrC3alkyl, C5-C8cycloalkenyl- substituted C1-C3alkyl, or phenyl-substituted C,-C3alkyl. Also in prefeπed embodiments, R10 is hydrogen, alkyl, preferably C,-C5alkyl, phenyl-substituted alkyl, preferably phenyl- substituted CrC2alkyl, cycloalkyl or cycloalkyl-substituted alkyl, preferably C5- C6cycloalkyl-substituted C,-C3alkyl.
The group R11 in the definition of W is hydrogen or alkyl. In prefeπed embodiments, R11 is hydrogen or C,-C3alkyl.
The group R12 in the definition of W is hydrogen, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl, aryl- substituted alkyl or alkylene-substituted C(=O)Y. In prefeπed embodiments, R12 is hydrogen, CpC^alkyl, C3-C,0alkenyl, phenyl, cycloalkyl, C5-C8cycloalkenyl, cycloalkyl- substituted C,-C3alkyl, C5-C8cycloalkenyl-substituted CrC3alkyl, phenyl-substituted - C3ah yl, or alkylene-substituted C(=O)Y. Also in prefeπed embodiments, R12 is hydrogen, alkyl, preferably CrC3alkyl or (CH2)mC(O)Y, where m is 1 to 4.
The group Y in the definition of R12 is OR18 or NR19R20. In certain alternative embodiments, in the definition of W, R12 and R13 form, together with the nitrogen atom to which they are attached, a heterocyclic ring. The group E in the definition of W is
alkylene substituted (C=O)D, or -R13OC(=O)R14. In prefeπed embodiments, E is
(CH2)m(C=O)D (where m is as defined above), or -R13OC(=O)R14.
The group R13 in the definition of E is alkyl substituted alkylene. In prefeπed embodiments, R13 is - alkyl substituted methylene. In more prefeπed embodiments, R13 is -CH(CH3)- or -CH(CH2CH3)-.
The group R14 in the definition of E is alkyl. In prefeπed embodiments, R14 is C,-C10alkyl.
The group D in the definition of E is D is OR15 or NR16R17. The group R15 in the definition of D is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl, or aryl- substituted alkyl. In prefeπed embodiments, R15 is hydrogen, C,-C10alkyl, C2-C10alkenyl, cycloalkyl, C5-C8cycloalkenyl, cycloalkyl-substituted CrC3alkyl, C5-C8cycloalkenyl- substituted CrC3alkyl, or phenyl-substituted CrC3alkyl. Also in prefeπed embodiments, R15 is hydrogen or alkyl, with C C3alkyl being more prefeπed.
The group R16 in the definition of D is hydrogen, alkyl, alkenyl, aryl, aryl- substituted alkyl, cycloalkyl, cycloalkenyl, cycloalkyl-substituted alkyl or cycloalkenyl- substituted alkyl. In prefeπed embodiments, R16 is hydrogen, C1-C10alkyl, C3-C10alkenyl, phenyl, phenyl-substituted C C3alkyl, cycloalkyl, C5-C8cycloalkenyl, cycloalkyl-
substituted - alkyl, C5-C8cycloalkenyl-substituted C,-C3alkyl. In even more prefeπed embodiments, R16 is methyl or benzyl.
The group R17 in the definition of D is hydrogen or alkyl. In prefeπed embodiments, R17 is hydrogen or CrC alkyl. In even more prefeπed embodiments, R17 is hydrogen.
In certain alternative embodiments, in the definition of D, R16 and R17 form, together with the nitrogen atom to which they are attached, a heterocyclic ring.
The group R18 in the definition of Y is hydrogen, alkyl, alkenyl, cycloalkyl, cycloalkenyl, cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl, or aryl- substituted alkyl. In prefeπed embodiments, R18 is hydrogen, CrC10alkyl, C2-C10alkenyl, cycloalkyl, C5-C8cycloalkenyl, cycloalkyl-substituted C,-C3 alkyl, C5-C8cycloalkenyl- substituted C,-C3alkyl, or phenyl-substituted CrC3alkyl. In more prefeπed embodiments, R18 is hydrogen or CrC3alkyl.
The group R19 in the definition of Y is hydrogen or alkyl. In prefeπed embodiments, R19 is hydrogen or CrC3alkyl.
The group R20 in the definition of Y is hydrogen, alkyl, alkenyl, aryl, cycloalkyl, cycloalkenyl, cycloalkyl-substituted alkyl, cycloalkenyl-substituted alkyl, or aryl-substituted alkyl. In prefeπed embodiments, R20 is hydrogen, C,-C10alkyl, C3- C10alkenyl, phenyl, cycloalkyl, C5-C8cycloalkenyl, cycloalkyl-substituted CrC3alkyl, C5- C8cycloalkenyl-substituted C1-C3alkyl, or phenyl-substituted CrC3alkyl. In more prefeπed embodiments, R20 is hydrogen or C,-C3alkyl.
In certain alternative embodiments, in the definition of Y, R19 and R20 form, together with the nitrogen atom to which they are attached, a heterocyclic ring.
The group R21 in the definition of B is hydrogen or alkyl. Preferably, R21 is hydrogen or C,-C3alkyl. Even more preferably, R21 is hydrogen.
In the above formula (I), n is 0 to about 4. In prefeπed embodiments, n is about 1 or 2.
In the above definition of R7, q is about 1 to about 4. In prefeπed embodiments, q is about 1 to about 3. In the above definition of E, m is about 1 to about 4. In prefeπed embodiments, m is about 1 to about 3.
The compounds of formula (I) can occur as the trans and cis stereochemical isomers by virtue of the substituents at the 3- and 4-positions of the piperidine ring, and such stereochemical isomers are within the scope of the claims. The term "trans", as used herein, refers to R2 in position 3 being on the opposite side from the methyl group in position 4, whereas in the "cis" isomer R2 and the 4-methyl are on the same side of the ring. In the methods of the present invention, the compounds employed may be the individual stereoisomers, as well as mixtures of stereoisomers. In the most prefeπed embodiments, the methods of the present invention involve compounds of formula (I) wherein the group R2 at the 3-position is situated on the opposite side of the ring, i.e., trans to the methyl group in the 4-position and on the same side of the ring. These trans isomers can exist as the 3R,4R-isomer, or the 3S,4S-isomer.
The terms "R" and "S" are used herein as commonly used in organic chemistry to denote specific configuration of a chiral center. The term "R" refers to "right" and refers that configuration of a chiral center with a clockwise relationship of group priorities (highest to second lowest) when viewed along the bond toward the lowest priority group. The term "S" or "left" refers to that configuration of a chiral center with a counterclockwise relationship of group priorities (highest to second lowest) when viewed along the bond toward the lowest priority group. The priority of groups is based upon their atomic number (heaviest isotope first). A partial list of priorities and a discussion of stereochemistry is contained in the book: 77ze Vocabulary of Organic Chemistry, Orchin, et al., John Wiley and Sons Inc., page 126 (1980), which is incorporated herein by reference in its entirety.
Prefeπed piperidine-N-alkylcarboxylate compounds for use in the methods of the present invention are those of formula (I) in which the configuration of substituents on the piperidine ring is 3R and 4R.
When R3 is not hydrogen, the carbon atom to which R3 is attached is asymmetric. As such, this class of compounds can further exist as the individual R or S stereoisomers at this chiral center, or as mixtures of stereoisomers, and all are contemplated within the scope of the present invention. Preferably, a substantially pure stereoisomer of the compounds of this invention is used, i.e., an isomer in which the
configuration at the chiral center to which R3 is attached is R or S, i.e., those compounds in which the configuration at the three chiral centers is preferably 3R, 4R, S or 3R, 4R, R.
Furthermore, other asymmetric carbons can be introduced into the molecule depending on the structure of A. As such, these classes of compounds can exist as the individual R or S stereoisomers at these chiral centers, or as mixtures of stereoisomers, and all are contemplated as being within the scope of methods of the present invention.
Prefeπed piperidine-N-alkylcarboxylate compounds for use in the methods of the present invention include the following:
U-OCH2CH3; U-OH; G-OH; U-NHCH2C(O)NHCH3; U-NHCH2C(O)NH2; G-NHCH2C(O)NHCH3; U-NHCH2C(O)NHCH2CH3; G-NH(CH2)3C(O)OCH2CH3;
G-NHCH2C(O)OH; M-NHCH2C(O)NH2; M-NH(CH2)2C(O)OCH2(C6H5); X-OCH2CH3; X- OH; X-NH(CH2)2CH3; Z-NH(CH2)3C(O)OCH2CH3; X-NHCH2C(O)OH; Z- NH(CH2)2N(CH3)2; Z-NH(CH2)2C(O)NHCH2CH3; X-OCH2(C6H5); X-N(CH3)2; Z- NH(CH2)3C(O)NHCH3; Z-NH(CH2)3C(O)NH2; Z-NH(CH2)3C(O)NHCH2CH3; X- OCH2C(O)OCH3; X-OCH2C(O)NHCH3; and X-N(CH3)CH2C(O)CH2CH3; in which: U represents
G represents
M represents
Z represents
X represents -ZNHCH2C(=O)-;
wherein Q represents
Particularly prefeπed piperidine-N-alkylcarboxylate compounds for use in the methods of the present invention include the following: Z-OH; Z-NH(CH2)2C(O)OH; G-NH(CH2)2C(O)NH2; G-NH(CH2)2C(O)NHCH3; G-NHCH2C(O)NH2; G-NHCH2C(O)NHCH2CH3; G-NH(CH2)3C(O)NHCH3;
G-NH(CH2)2C(O)OH; G-NH(CH2)3C(O)OH; X-NH2; X-NHCH(CH3)2; X-OCH2CH(CH3)2; X-OCH2C6H5; X-OH; X-O(CH2)4CH3; X-O-(4-methoxycyclohexyl); X- OCH(CH3)OC(O)CH3; X-OCH2C(O)NHCH2(C6H5); M-NHCH2C(O)OH; M- NH(CH2)2C(O)OH; M-NH(CH2)2C(O)NH2; U-NHCH2C(O)OCH2CH3; and U- NHCH2C(O)OH; wherein Z, G, X, M and U are as defined above.
Stated another way, in accordance with prefeπed embodiments of the invention, the compound of formula (I) has the formula Q-CH2CH(CH2(C6H5))C(O)OH, Q-CH2CH2CH(C6H5)C(O)NHCH2C(O)OCH2CH2, Q-CH2CH2CH(C6H5)C(O)NHCH2C(O)OH, Q-CH2CH2CH(C6H5)C(O)NHCH2C(O)NHCH3,
Q-CH2CH2CH(C6H5)C(O)NHCH2C(O)NHCH2CH3, G-NH(CH2)2C(O)NH2, G-NH(CH2)2C(O)NHCH3, G-NHCH2C(O)NH2, G-NHCH2C(O)NHCH3, G-NHCH3C(O)NHCH2CH3, G-NH(CH2)3C(O)OCH2CH3, G-NH(CH2)3C(O)NHCH3, G-NH(CH2)2C(O)OH, G-NH(CH2)3C(O)OH, Q-CH2CH(CH2(C6H, ,))C(O)NHCH2C(O)OH, Q-CH2CH(CH2(C6Hu))C(O)NH(CH2)2C(O)OH,
Q-CH2CH(CH2(C6H, ,))C(O)NH(CH2)2C(O)NH2, Z-NHCH2C(O)OCH2CH3, Z-NHCH2C(O)OH, Z-NHCH2C(O)NH2, Z-NHCH2C(O)N(CH3)2, Z-NHCH2C(O)NHCH(CH3)2, Z-NHCH2C(O)OCH2CH(CH3)2, Z-NH(CH2)2C(O)OCH2(C6H5), Z-NH(CH2C(O)OH, Z-NH(CH2)2C(O)NHCH2CH3, Z-NH(CH2)3C(O)NHCH3, Z-NHCH2C(O)NHCH2C(O)OH, Z-NHCH2C(O)OCH2C(O)OCH3, Z-NHCH2C(O)O(CH2)4CH3, Z-NHCH2C(O)OCH2C(O)NHCH3, Z-NHCH2C(O)O-(4-methoxycyclohexyl), Z-NHCH2C(O)OCH2C(O)NHCH2(C6H5) or Z-NHCH2C(O)OCH(CH3)OC(O)CH3; wherein Q, G and Z are as defined above. In even more prefeπed embodiments, the compound of formula (I) has the formula (3R,4R,S)-Z-NHCH2C(O)OCH2CH(CH3)2, (+)-Z-NHCH2C(O)OH, (-)-Z-NHCH2C(O)OH, (3R,4R,R)-Z-NHCH2C(O)-OCH2CH(CH3)2, (3S,4S,S)-Z-NHCH2C(O)OCH2CH(CH3)2, (3S,4S,R)-Z-NHCH2C(O)OCH2CH(CH3)2, (3R,4R)-Z-NHCH2C(O)NHCH2(C6 H5) or (3R,4R)-G-NH(CH2)3C(O)OH, where Z and G are as defined above. In still more prefeπed embodiments, the compound of formula (I) has the formula (+)-Z-NHCH2C(O)OH or (-)-Z-NHCH2C(O)OH where Z is as defined above.
Compounds of formula (I) that act locally on the gut, have high potency, and are orally active are most prefeπed. A particularly prefeπed embodiment of the present invention is the compound (+)-Z-NHCH2C(O)OH, i.e., the compound of the following formula (H).
II
The compound of formula (II) has low solubility in water except at low or high pH conditions. Zwitterionic character may be inherent to the compound, and may impart desirable properties such as poor systemic absorption and sustained local affect on the gut following oral administration.
In an alternate embodiment, the methods of the present invention may involve administering to a patient a peripheral mu opioid antagonist compound that is a quaternary morphinan compound. Examples of quaternary morphinan compounds that may be suitable for use in the methods of the present invention include, for example, quaternary salts of N-methylnaltrexone, N-methylnaloxone, N-methylnalorphine, N- diallylnormorphine, N-allyllevallorphan and N-methylnalmefene.
In yet another alternate embodiment, the methods of the present invention may involve administering to a patient a peripheral mu opioid antagonist compound in the form of an opium alkaloid derivative. The term "opium alkaloid derivative", as used herein, refers to peripheral mu opioid antagonist compounds that are synthetic or semi- synthetic derivatives or analogs of opium alkaloids. In prefeπed form, the opium alkaloid derivatives employed in the methods of the present invention exhibit high levels of morphine antagonism, while exhibiting reduced, and preferably substantially no, agonist activity. The term "substantially no agonist activity", as used herein, means that the maximal response with respect to electrically stimulated guinea pig ileum, at a concentration of 1 μM, is about 60% or less relative to morphine. In prefeπed embodiments, the opium alkaloid derivatives employed in the present methods have a maximal response with respect to guinea pig ileum, at a concentration of 1 μM, of about 50% or less relative to morphine, with a maximal response of about 40% or less being more prefeπed. In even more prefeπed embodiments, the opium alkaloid derivatives
employed in the present methods have a maximal response with respect to guinea pig ileum, at a concentration of 1 μM, of about 30% or less relative to morphine, with a maximal response of about 20% or less being more prefeπed. In still more prefeπed embodiments, the opium alkaloid derivatives employed in the present methods have a maximal response with respect to guinea pig ileum, at a concentration of 1 μM, of about 10% or less relative to morphine. In certain particularly prefeπed embodiments, the opium alkaloid derivatives have a maximal response with respect to guinea pig ileum, at a concentration of 1 μM, of about 0% (i.e., no response).
Suitable methods for determining maximal response of opium alkaloid derivatives with respect to electrically stimulated guinea pig ileum are described, for example, in U.S. Patent Nos. 4,730,048 and 4,806,556, the disclosures of which are hereby incorporated herein by reference, in their entireties.
In prefeπed form, the opium alkaloid derivatives employed in the methods of the present invention have the following formulas (III) or (IN):
III
or
IV
wherein:
R is alkyl, cycloalkyl-substituted alkyl, aryl, aryl-substituted alkyl or alkenyl;
Z is hydrogen or OH;
R' is X'-J(L)(T), wherein:
J is alkylene or alkenylene;
L is hydrogen, amino, or alkyl optionally substituted with CO2H, OH or phenyl; and
T is CO2H, SO H, amino or guanidino; X' is a direct bond or C(=O); and
R" is NH-J(L)(T) or guanidino; or a stereoisomer, prodrug, or pharmaceutically acceptable salt, hydrate or N-oxide thereof.
In the compounds of formulas (III) and (IV) above, R is alkyl, cycloalkyl- substituted alkyl, aryl, aryl-substituted alkyl or alkenyl. In prefeπed embodiments, R is C C5alkyl, C3-C6cycloakyl-substituted alkyl, aryl, arylalkyl or trans-C2-C5alkenyl. In more prefeπed embodiments, R is C,-C3alkyl, allyl or cyclopropylmethyl, with cyclopropylmethyl being even more prefeπed.
In the compounds of formulas (III) and (TV) above, Z is hydrogen or OH. In prefeπed embodiments, Z is OH. In the compounds of formulas (III) and (IV), R' is X-J(L)(T) and R" is
NH-J(L)(T) or guanidino.
In the definitions of R and R", G is alkylene or alkenylene. In prefeπed embodiments, J is C,-C5alkylene, C2-C6alkylene interrupted by an oxygen atom, or C2-C5alkenylene. In the definitions of R' and R", L is hydrogen, amino, or alkyl optionally substituted with CO2H, OH or phenyl. In prefeπed embodiments, L is hydrogen, amino, or C,-C5alkyl optionally substituted with CO2H, OH or phenyl. In more prefeπed embodiments, L is hydrogen or amino.
In the definitions of R' and R", T is CO2H, SO3H, amino or guanidino. In prefeπed embodiments, T is CO2H or guanidino.
In the definition of R', X is a direct bond or C(=O).
Prefeπed opioid alkaloid derivatives that may be employed in the methods of the present invention include compounds of formula (III) wherein R is cyclopropylmethyl, Z is OH, and R is selected from C(=O)(CH2)2CO2H, C(=O)(CH2)3CO2H, C(=O)CH=CHCO2H, C(=O)CH2OCH2CO2H, C(=O)CH(NH2)(CH2)3NHC(=NH)NH2 or C(=O)CH(NH2)CH2CO2H. Also prefeπed are opioid alkaloid derivatives of formula (III) wherein R is cyclopropylmethyl, Z is OH, and R' is CH2CO2H. In other prefeπed embodiments, the opioid alkaloid derivatives that may be employed in the methods of the present invention include compounds of formula (IV) wherein R is cyclopropylmethyl, Z is OH, and R" is NHCH2CO2H. Other opioid alkaloid derivatives that may be employed in the methods of the present invention are described, for example, in U.S. Patent Nos. 4,730,048 and 4,806,556, the disclosures of which are hereby incorporated herein by reference, in their entireties.
In still another alternate embodiment, the methods of the present invention may involve administering to a patient a peripheral mu opioid antagonist compound in the form of a quaternary benzomorphan compound. In prefeπed form, the quaternary benzomorphan compounds employed in the methods of the present invention exhibit high levels of morphine antagonism, while exhibiting reduced, and preferably substantially no, agonist activity. The term "substantially no agonist activity", as used herein in connection with the quaternary benzomorphan compounds, means that the maximal response with respect to electrically stimulated guinea pig ileum, at a concentration of 1 μM, is about 60% or less relative to morphine. In prefeπed embodiments, the quaternary benzomorphan compounds employed in the present methods have a maximal response with respect to guinea pig ileum, at a concentration of 1 μM, of about 50% or less relative to morphine, with a maximal response of about 40% or less being more prefeπed. In even more prefeπed embodiments, the quaternary benzomorphan compounds employed in the present methods have a maximal response with respect to guinea pig ileum, at a concentration of 1 μM, of about 30% or less relative to morphine, with a maximal response of about 20% or less being more prefeπed. In still more prefeπed embodiments, the quaternary benzomorphan compounds employed in the present methods have a maximal response with respect to guinea pig ileum, at a concentration of 1 μM, of about 10% or less relative to
morphine. In certain particularly prefeπed embodiments, the quaternary benzomorphan compounds have a maximal response with respect to guinea pig ileum, at a concentration of 1 μM, of about 0% (i.e., no response).
In prefeπed form, the quaternary benzomorphan compounds employed in the methods of the present invention have the following formula (V):
V
where:
R24 is hydrogen or acyl; and
R25 is alkyl or alkenyl; or a stereoisomer, prodrug, or pharmaceutically acceptable salt, hydrate or N-oxide thereof.
In the above formula (V), R24 is hydrogen or acyl. In prefeπed embodiments, R24 is hydrogen or CrC6 acyl. In more prefeπed embodiments, R24 is hydrogen or CrC2 acyl. In even more prefeπed embodiments, R24 is hydrogen or acetoxy, with hydrogen being still more prefeπed. In the above formula (V), R25 is alkyl or alkenyl. In prefeπed embodiments, R25 is C,-C6 alkyl or C2-C6 alkenyl. In even more prefeπed embodiments, R25 is CrC3 alkyl or C2-C3 alkenyl. In still more prefeπed embodiments, R25 is propyl or allyl.
Prefeπed quaternary benzomorphan compounds that may be employed in the methods of the present invention include the following compounds of formula (V): 2'- hydroxy-5,9-dimethyl-2,2-diallyl-6,7-benzomorphanium-bromide; 2'-hydroxy-5,9- dimethyl-2-n-propyl-6,7-benzomorphan; 2'-hydroxy-5,9-dimethyl-2-allyl-6,7- benzomorphan; 2'-hydroxy-5,9-dimethyl-2-n-propyl-2-allyl-6,7-benzomorphanium-
bromide; 2'-hydroxy-5,9-dimethyl-2-n-propyl-2-propargyl-6,7-benzomorphanium- bromide; and 2'-acetoxy-5,9-dimethyl-2-n-propyl-2-allyl-6,7-benzomorphanium-bromide. Other quaternary benzomorphan compounds that may be employed in the methods of the present invention are described, for example, in U.S. Patent No. 3,723,440, the disclosures of which are hereby incorporated herein by reference, in their entirety. Other mu opioid antagonist compounds which may be employed in the methods of the present invention, in addition to those exemplified above, would be readily apparent to one of ordinary skill in the art, once armed with the teachings of the present disclosure. The compounds employed in the methods of the present invention may exist in prodrug form. As used herein, "prodrug" is intended to include any covalently bonded carriers which release the active parent drug according to formulas (I) to (IV) or other formulas or compounds employed in the methods of the present invention in vivo when such prodrug is administered to a mammalian subject. Since prodrugs are known to enhance numerous desirable qualities of pharmaceuticals (e.g., solubility, bioavailability, manufacturing, etc.) the compounds employed in the present methods may, if desired, be delivered in prodrug form. Thus, the present invention contemplates methods of delivering prodrugs. Prodrugs of the compounds employed in the present invention may be prepared by modifying functional groups present in the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to the parent compound.
Accordingly, prodrugs include, for example, compounds described herein in which a hydroxy, amino, or carboxy group is bonded to any group that, when the prodrug is administered to a mammalian subject, cleaves to form a free hydroxyl, free amino, or carboxylic acid, respectively. Examples include, but are not limited to, acetate, formate and benzoate derivatives of alcohol and amine functional groups; and alkyl, carbocyclic, aryl, and alkylaryl esters such as methyl, ethyl, propyl, iso-propyl, butyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, phenyl, benzyl, and phenethyl esters, and the like. The compounds employed in the methods of the present invention may be prepared in a number of ways well known to those skilled in the art. The compounds can
be synthesized, for example, by the methods described below, or variations thereon as appreciated by the skilled artisan. All processes disclosed in association with the present invention are contemplated to be practiced on any scale, including milligram, gram, multigram, kilogram, multikilogram or commercial industrial scale. As discussed in detail above, compounds employed in the present methods may contain one or more asymmetrically substituted carbon atoms, and may be isolated in optically active or racemic forms. Thus, all chiral, diastereomeric, racemic forms and all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomeric form is specifically indicated. It is well known in the art how to prepare and isolate such optically active forms. For example, mixtures of stereoisomers may be separated by standard techniques including, but not limited to, resolution of racemic forms, normal, reverse-phase, and chiral chromatography, preferential salt formation, recrystallization, and the like, or by chiral synthesis either from chiral starting materials or by deliberate synthesis of target chiral centers. As will be readily understood, functional groups present may contain protecting groups during the course of synthesis. Protecting groups are known er se as chemical functional groups that can be selectively appended to and removed from functionalities, such as hydroxyl groups and carboxyl groups. These groups are present in a chemical compound to render such functionality inert to chemical reaction conditions to which the compound is exposed. Any of a variety of protecting groups may be employed with the present invention. Prefeπed protecting groups include the benzyloxycarbonyl group and the tert-butyloxycarbonyl group. Other prefeπed protecting groups that may be employed in accordance with the present invention may be described in Greene, T.W. and Wuts, P.G.M., Protective Groups in Organic Synthesis 2d. Ed., Wiley & Sons, 1991. Piperidine-N-alkylcarboxylate compounds according to the present invention may be synthesized employing methods taught, for example, in U.S. Patent Nos. 5,250,542, 5,434,171, 5,159,081, and 5,270,328, the disclosures of which are hereby incorporated herein by reference in their entireties. For example, the 3-substituted-4- methyl-4-(3-hydroxy- or alkanoyloxyphenyl)piperidine derivatives employed as starting materials in the synthesis of the present compounds may be prepared by the general procedure taught in U.S. Patent No. 4,115,400 and U.S. Patent No. 4,891,379, the
disclosures of which are hereby incorporated herein by reference in their entireties. The starting material for the synthesis of compounds described herein, (3R,4R)-4-(3- hydroxypheny)-3,4-dimethylpiperidine, may be prepared by the procedures described in U.S. Patent No. 4,581,456, the disclosures of which are hereby incorporated herein by reference, in their entirety, but adjusted as described such that the β-stereochemistry is prefeπed.
The first step of the process may involves the formation of the 3- alkoxyphenyllithium reagent by reacting 3-alkoxybromobenzene with an alkyllithium reagent. This reaction may be performed under inert conditions and in the presence of a suitable non-reactive solvent such as dry di ethyl ether or preferably dry tetrahydrofuran. Prefeπed alkyllithium reagents used in this process are n-butyllithium, and especially sec- butyllithium. Generally, approximately an equimolar to slight excess of alkyllithium reagent may be added to the reaction mixture. The reaction may be conducted at a temperature of from about -20°C and about -100°C, more preferably from about -50°C to about -55°C.
Once the 3-alkoxyphenyllithium reagent has formed, approximately an equimolar quantity of a l-alkyl-4-piperidone may be added to the mixture while maintaining the temperature between -20°C and -100°C. The reaction is typically complete after about 1 to 24 hours. At this point, the reaction mixture may be allowed to gradually warm to room temperature. The product may be isolated by the addition to the reaction mixture of a saturated sodium chloride solution to quench any residual lithium reagent. The organic layer may be separated and further purified if desired to provide the appropriate l-alkyl-4-(3-alkoxyphenyl)piperidinol derivative.
The dehydration of the 4-phenylpiperidinol prepared above may be accomplished with a strong acid according to well known procedures. While dehydration occurs in various amounts with any one of several strong acids such as hydrochloric acid, hydrobromic acid, and the like, dehydration is preferably conducted with phosphoric acid, or especially p-toluenesulfonic acid in toluene or benzene. This reaction may be typically conducted under reflux conditions, more generally from about 50°C and 150°C. The product thus formed may be isolated by basifying an acidic aqueous solution of the salt
form of the product and extracting the aqueous solution with a suitable water immiscible solvent. The resulting residue following evaporation can then be further purified if desired.
The 1 -alkyl-4-methyl-4-(3-alkoxyphenyl)tetrahydropyridine derivatives may be prepared by a metalloenamine alkylation. This reaction is preferably conducted with n-butyllithium in tetrahydrofuran (THF) under an inert atmosphere, such as nitrogen or argon. Generally, a slight excess of n-butyllithium may be added to a stirring solution of the l-alkyl-4-(3-alkoxyphenyl)-tetrahydropyridine in THF cooled to a temperature in the range of from about -50°C to about 0°C, more preferably from about -20°C to -10°C. This mixture may be stiπed for approximately 10 to 30 minutes followed by the addition of approximately from 1.0 to 1.5 equivalents of methyl halide to the solution while maintaining the temperature of the reaction mixture below 0°C. After about 5 to 60 minutes, water may be added to the reaction mixture and the organic phase may be collected. The product can be purified according to standard procedures, but the crude product is preferably purified by either distilling it under vacuum or slurrying it in a mixture of hexane:ethyl acetate (65:35, v:v) and silica gel for about two hours. According to the latter procedure, the product may be then isolated by filtration followed by evaporating the filtrate under reduced pressure.
The next step in the process may involve the application of the Mannich reaction of aminomethylation to non-conjugated, endocyclic enamines. This reaction is preferably carried out by combining from about 1.2 to 2.0 equivalents of aqueous formaldehyde and about 1.3 to 2.0 equivalents of a suitable secondary amine in a suitable solvent. While water may be the prefeπed solvent, other non-nucleophilic solvents, such as acetone and acetonitrile can also be employed in this reaction. The pH of this solution may be adjusted to approximately 3.0 to 4.0 with an acid that provides a non-nucleophilic anion. Examples of such acids include sulfuric acid, the sulfonic acids such as methanesulfonic acid and p-toluenesulfonic acid, phosphoric acid, and tetrafluoroboric acid, with sulfuric acid being prefeπed. To this solution may be added one equivalent of a l-alkyl-4-methyl-4-(3-alkoxyphenyl)tetrahydropyridine, typically dissolved in aqueous sulfuric acid, and the pH of the solution may be readjusted with the non-nucleophilic acid or a suitable secondary amine. The pH is preferably maintained in the range of from about 1.0 to 5.0, with a pH of about 3.0 to 3.5 being more prefeπed during the reaction. The
reaction is substantially complete after about 1 to 4 hours, more typically about 2 hours, when conducted at a temperature in the range of from about 50°C to about 80°C, more preferably about 70°C. The reaction may then be cooled to approximately 30°C, and added to a sodium hydroxide solution. This solution may then be extracted with a water immiscible organic solvent, such as hexane or ethyl acetate, and the organic phase, following thorough washing with water to remove any residual formaldehyde, may be evaporated to dryness under reduced pressure.
The next step of the process may involve the catalytic hydrogenation of the prepared l-alkyl-4-methyl-4-(3-alkoxyphenyl)-3-tetrahydropyridinemethanamine to the coπesponding trans- l-alkyl-3,4-dimethyl-4-(3-alkoxyphenyl)piperidine. This reaction actually occurs in two steps. The first step is the hydrogenolysis reaction wherein the exo C-N bond is reductively cleaved to generate the 3-methyltetrahydropyridine. In the second step, the 2,3-double bond in the tetrahydropyridine ring is reduced to afford the desired piperidine ring. Reduction of the enamine double bond introduced the crucial relative stereochemistry at the 3 and 4 carbon atoms of the piperidine ring. The reduction generally does not occur with complete stereoselectivity. The catalysts employed in the process may be chosen from among the various palladium and preferably platinum catalysts.
The catalytic hydrogenation step of the process is preferably conducted in an acidic reaction medium. Suitable solvents for use in the process include the alcohols, such as methanol or ethanol, as well as ethyl acetate, tetrahydrofuran, toluene, hexane, and the like.
Proper stereochemical outcome may be dependent on the quantity of catalyst employed. The quantity of catalyst required to produce the desired stereochemical result may be dependent upon the purity of the starting materials in regard to the presence or absence of various catalyst poisons.
The hydrogen pressure in the reaction vessel may not be critical but can be in the range of from about 5 to 200 psi. Concentration of the starting material by volume is preferably around 20 mL of liquid per gram of starting material, although an increased or decreased concentration of the starting material can also be employed. Under the conditions specified herein, the length of time for the catalytic hydrogenation may not be
critical because of the inability for over-reduction of the molecule. While the reaction can continue for up to 24 hours or longer, it may not be necessary to continue the reduction conditions after the uptake of the theoretical two moles of hydrogen. The product may then be isolated by filtering the reaction mixture for example through infusorial earth, and evaporating the filtrate to dryness under reduced pressure. Further purification of the product thus isolated may not be necessary and preferably the diastereomeric mixture may be carried directly on to the following reaction.
The alkyl substituent may be removed from the 1 -position of the piperidine ring by standard dealkylation procedures. Preferably, a chloroformate derivative, especially the vinyl or phenyl derivatives, may be employed and removed with acid. Next, the prepared alkoxy compound may be dealkylated to the coπesponding phenol. This reaction may be generally carried out by reacting the compound in a 48% aqueous hydrobromic acid solution. This reaction may be substantially complete after about 30 minutes to 24 hours when conducted at a temperature of from about 50°C to about 150°C, more preferably at the reflux temperature of the reaction mixture. The mixture may then be worked up by cooling the solution, followed by neutralization with base to an approximate pH of 8. This aqueous solution may be extracted with a water immiscible organic solvent. The residue following evaporation of the organic phase may then be used directly in the following step. The compounds employed as starting materials to the compounds of the invention can also be prepared by brominating the l-alkyl-4-methyl-4-(3-alkoxyphenyl)-3- tetrahydropyridinemethanamine at the 3-position, lithiating the bromo compound thus prepared, and reacting the lithiated intermediate with a methylhalide, such as methyl bromide to provide the coπesponding 1 -alky 1-3, 4-dimethyl-4-(3 - alkoxyphenyl)tetrahydropyridinemethanamine. This compound may then be reduced and converted to the starting material as indicated above.
As noted above, the compounds of the present invention can exist as the individual stereoisomers. Preferably reaction conditions are adjusted as disclosed in U.S. Patent No. 4,581,456 or as set forth in Example 1 of U.S. Patent No. 5,250,542 to be substantially stereoselective and provide a racemic mixture of essentially two enantiomers. These enantiomers may then be resolved. A procedure which may be employed to prepare
the resolved starting materials used in the synthesis of these compounds includes treating a racemic mixture of alkyl-3,4-dimethyl-4-(3-alkoxyphenyl)piperidine with either (+)- or (-)- ditoluoyl tartaric acid to provide the resolved intermediate. This compound may then be dealkylated at the 1 -position with vinyl chloro formate and finally converted to the desired 4-(3-hydroxyphenyl)piperidine isomer.
As will be understood by those skilled in the art, the individual enantiomers of the invention can also be isolated with either (+) or (-) dibenzoyl tartaric acid, as desired, from the coπesponding racemic mixture of the compounds of the invention. Preferably the (+)-trans enantiomer is obtained. Although the (+)trans-3,4 stereoisomer is prefeπed, all of the possible stereoiosmers of the compounds described herein are within the contemplated scope of the present invention. Racemic mixtures of the stereoisomers as well as the substantially pure stereoisomers are within the scope of the invention. The term "substantially pure", as used herein, refers to at least about 90 mole percent, more preferably at least about 95 mole percent and most preferably at least about 98 mole percent of the desired stereoisomer is present relative to other possible stereoisomers.
Intermediates can be prepared by reacting a 3,4-alkyl-substituted-4-(3- hydroxyphenyl)piperidine with a compound of the formula LCH2(CH2)n.,CHR3C(O)E where L is a leaving group such as chlorine, bromine or iodine, E is a carboxylic acid, ester or amide, and R3 and n are as defined hereinabove. Preferably L may be chlorine and the reaction is carried out in the presence of a base to alkylate the piperidine nitrogen. For example 4-chloro-2-cyclohexylbutanoic acid, ethyl ester can be contacted with (3R,4R)-4- (3-hydroxyphenyl)-3,4-dimethylpiperidine to provide 4-[(3R,4R)-4-(3-hydroxyphenyl)- 3,4-dimethyl-l-piperidine]butanoic acid, ethyl ester. Although the ester of the carboxylic acid may be prefeπed, the free acid itself or an amide of the carboxylic acid may be used. In alternative synthesis, the substituted piperidine can be contacted with a methylene alkyl ester to alkylate the piperidine nitrogen. For example, 2-methylene-3- phenylproponic acid, ethyl ester can be contacted with a desired piperidine to provide 2- benzyl-3-piperidinepropanoic acid ethyl ester.
Another synthetic route can involve the reaction of a substituted piperidine with a haloalkylnitrile. The nitrile group of the resulting piperidine alkylnitrile can be hydrolyzed to the coπesponding carboxylic acid.
With each of the synthetic routes, the resulting ester or carboxylic acid can be reacted with an amine or alcohol to provide modified chemical structures. In the preparation of amides, the piperidine-carboxylic acid or -carboxylic acid ester may be reacted with an amine in the presence of a coupling agent such as dicyclohexylcarbodiimide, boric acid, borane-trimethylamine, and the like. Esters can be prepared by contacting the piperidine-carboxylic acid with the appropriate alcohol in the presence of a coupling agent such as p-toluenesulfonic acid, boron trifluoride etherate or N,N'-carbonyldiimidazole. Alternatively, the piperidine-carboxylic acid chloride can be prepared using a reagent such as thionyl chloride, phosphorus trichloride, phosphorus pentachloride and the like. This acyl chloride can be reacted with the appropriate amine or alcohol to provide the coπesponding amide or ester. Opium alkaloid derivatives according to the present invention may be synthesized employing methods taught, for example, in U.S. Patent Nos. 4,730,048 and 4,806,556, the disclosures of which are hereby incorporated herein by reference in their entireties. For example, opium alkaloid derivatives of formula (III) may be prepared by attaching hydrophilic, ionizable moieties R and R" to the 6-amino group of naltrexamine (formula (III) where R is (cyclopropyl)methyl, Z is OH and R' is H) or oxymorphamine (formula (III) where R is CH3, Z is OH and R' is H). The opium alkaloid derivatives of formula IV may be prepared by converting the 6-keto-group of oxymorphone (formula (V) where R is CH3 and Z is OH) or naltrexone (formula (V) where R is (cyclopropyl)methyl and Z is OH) to the ionizable, hydrophilic group (R"N=) by a Schiff base reaction with a suitable amino-compound.
VI
In a similar fashion, deoxy-opiates of formulae (III) and (IV) wherein Z is hydrogen may be prepared from readily available starting materials.
The compounds of formula (V) may be synthesized employing methods taught, for example, in U.S. Patent No. 3,723,440, the disclosures of which are hereby incorporated herein by reference in their entirety.
The compounds employed in the methods of the present invention may be administered by any means that results in the contact of the active agent with the agent's site of action in the body of a patient. The compounds may be administered by any conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. For example, they may be administered as the sole active agent in a pharmaceutical composition, or they can be used in combination with other therapeutically active ingredients including, for example, opioid analgesic agents.
The compounds are preferably combined with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice as described, for example, in Remington's Pharmaceutical Sciences (Mack Pub. Co., Easton, PA, 1980), the disclosures of which are hereby incorporated herein by reference, in their entirety.
Compounds of the present invention can be administered to a mammalian host in a variety of forms adapted to the chosen route of administration, e.g., orally or parenterally. Parenteral administration in this respect includes administration by the following routes: intravenous, intramuscular, subcutaneous, rectal, intraocular, intrasynovial, transepithelial including transdermal, ophthalmic, sublingual and buccal; topically including ophthalmic, dermal, ocular, rectal, and nasal inhalation via insufflation aerosol.
The active compound may be orally administered, for example, with an inert diluent or with an assimilable edible carrier, or it may be enclosed in hard or soft shell gelatin capsules, or it may be compressed into tablets, or it may be incorporated directly with the food of the diet. For oral therapeutic administration, the active compound may be incorporated with excipient and used in the form of ingestible tablets, buccal tablets, troches, capsules, elixirs, suspensions, syrups, wafers, and the like. Such compositions and
preparations should preferably contain at least 0.1% of active compound. The percentage of the compositions and preparations may, of course, be varied and may conveniently be, for example, from about 2 to about 6% of the weight of the unit. The amount of active compound in such therapeutically useful compositions is preferably such that a suitable dosage will be obtained. Prefeπed compositions or preparations according to the present invention may be prepared so that an oral dosage unit form contains from about 0.1 to about 1000 mg of active compound.
The tablets, troches, pills, capsules and the like may also contain one or more of the following: a binder, such as gum tragacanth, acacia, corn starch or gelatin; an excipient, such as dicalcium phosphate; a disintegrating agent, such as corn starch, potato starch, alginic acid and the like; a lubricant, such as magnesium stearate; a sweetening agent such as sucrose, lactose or saccharin; or a flavoring agent, such as peppermint, oil of wintergreen or cherry flavoring. When the dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier. Various other materials may be present as coatings or to otherwise modify the physical form of the dosage unit. For instance, tablets, pills, or capsules may be coated with shellac, sugar or both. A syrup or elixir may contain the active compound, sucrose as a sweetening agent, methyl and propylparabens as preservatives, a dye and flavoring, such as cherry or orange flavor. Of course, any material used in preparing any dosage unit form is preferably pharmaceutically pure and substantially non-toxic in the amounts employed. In addition, the active compound may be incorporated into sustained-release preparations and formulations.
The active compound may also be administered parenterally or intraperitoneally. Solutions of the active compound as a free base or a pharmacologically acceptable salt can be prepared in water suitably mixed with a surfactant, such as hydroxypropylcellulose. A dispersion can also be prepared in glycerol, liquid polyethylene glycols and mixtures thereof and in oils. Under ordinary conditions of storage and use, these preparations may contain a preservative to prevent the growth of microorganisms.
The pharmaceutical forms suitable for injectable use include, for example, sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. In all cases, the form is preferably sterile and fluid to provide easy syringability. It is preferably stable under the conditions
of manufacture and storage and is preferably preserved against the contaminating action of microorganisms such as bacteria and fungi. The carrier may be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, liquid polyethylene glycol and the like), suitable mixtures thereof, and vegetable oils. The proper fluidity can be maintained, for example, by the use of a coating, such as lecithin, by the maintenance of the required particle size in the case of a dispersion, and by the use of surfactants. The prevention of the action of microorganisms may be achieved by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal and the like. In many cases, it will be preferable to include isotonic agents, for example, sugars or sodium chloride. Prolonged absorption of the injectable compositions may be achieved by the use of agents delaying absorption, for example, aluminum monostearate and gelatin.
Sterile injectable solutions may be prepared by incorporating the active compound in the required amount, in the appropriate solvent, with various of the other ingredients enumerated above, as required, followed by filtered sterilization. Generally, dispersions may be prepared by incorporating the sterilized active ingredient into a sterile vehicle that contains the basic dispersion medium and the required other ingredients from those enumerated above. In the case of sterile powders for the preparation of sterile injectable solutions, the prefeπed methods of preparation may include vacuum drying and the freeze drying technique which yield a powder of the active ingredient, plus any additional desired ingredient from the previously sterile-filtered solution thereof.
The therapeutic compounds of this invention may be administered to a patient alone or in combination with a pharmaceutically acceptable carrier. As noted above, the relative proportions of active ingredient and carrier may be determined, for example, by the solubility and chemical nature of the compound, chosen route of admimstration and standard pharmaceutical practice.
The dosage of the compounds of the present invention that will be most suitable for prophylaxis or treatment will vary with the form of admimstration, the particular compound chosen and the physiological characteristics of the particular patient under treatment. Generally, small dosages may be used initially and, if necessary, increased by small increments until the desired effect under the circumstances is reached.
The therapeutic human dosage, based on physiological studies using rats, may generally range from about 0.01 mg to about 100 mg/kg of body weight per day, and all combinations and subcombinations of ranges therein. Alternatively, the therapeutic human dosage may be from about 0.4 mg to about 10 g or higher, and may be administered in several different dosage units from once to several times a day. Generally speaking, oral administration may require higher dosages.
Compounds for use in the methods of the present invention, including piperidine-N-alkylcarboxylate compounds of formula (I), and particularly the compound of formula (II), have been characterized in opioid receptor binding assays and show preferential binding to mu opioid receptors. Studies in isolated tissues (guinea pig ileum and mouse vas deferens) have shown that these compounds may act as antagonists with no measurable agonist activity. Studies in animals have demonstrated that the present compounds may reverse constipation in morphine-dependent mice when administered orally or parenterally at very low doses, and do not block the analgesic actions of morphine unless given in hundred-fold or higher doses. Collectively, the data indicate that the compounds described herein may have a very high degree of peripheral selectivity. See, e.g., Zimmerman, D.M., et al., Drugs of the Future, 1994, 19(12), 1078-1083.
The disclosures of each patent, patent application and publication cited or described in this document are hereby incorporated herein by reference, in their entirety. Various modification of the invention, in addition to those described herein, will be apparent to those skilled in the art from the foregoing description. Such modifications are also intended to fall within the scope of the appended claims.