WO2000023114A2 - Polymer conjugates of interferon beta- 1a and their uses - Google Patents
Polymer conjugates of interferon beta- 1a and their uses Download PDFInfo
- Publication number
- WO2000023114A2 WO2000023114A2 PCT/US1999/024201 US9924201W WO0023114A2 WO 2000023114 A2 WO2000023114 A2 WO 2000023114A2 US 9924201 W US9924201 W US 9924201W WO 0023114 A2 WO0023114 A2 WO 0023114A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- beta
- interferon
- polymer
- composition
- activity
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/21—Interferons [IFN]
- A61K38/215—IFN-beta
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/61—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule the organic macromolecular compound being a polysaccharide or a derivative thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S930/00—Peptide or protein sequence
- Y10S930/01—Peptide or protein sequence
- Y10S930/14—Lymphokine; related peptides
- Y10S930/142—Interferon
Definitions
- a major factor limiting the usefulness of these proteinaceous substances for their intended application is that, when given parenterally, they are eliminated from the body within a short time. This can occur as a result of metabolism by proteases or by clearance using normal pathways for protein elimination such as by filtration in the kidneys.
- the oral route of administration of these substances is even more problematic because in addition to proteolysis in the stomach, the high acidity of the stomach destroys them before they reach their intended target tissue.
- the problems associated with these routes of administration of proteins are well known in the pharmaceutical industry, and various strategies are being used in attempts to solve them.
- Another aspect of the invention is a method of inhibiting angiogenesis and neovascularization comprising subject an effective amount of the compositions of the invention.
- the pegylated product of the invention should be particularly effective as an angiogenesis inhibitor.
- the instant invention embraces interferon-beta-la proteins encoded by naturally-occurring DNAs, as well as by non-naturally-occurring DNAs which encode the same protein as encoded by the naturally-occurring DNA. Due to the degeneracy of the nucleotide coding sequences, other polynucleotides may be used to encode interferon-beta- la. These include all, or portions of the above sequences which are altered by the substitution of different codons that encode the same amino acid residue within the sequence, thus producing a silent change. Such altered sequences are regarded as equivalents of these sequences.
- operatively linked a polynucleotide sequence (DNA, RNA) is operatively linked to an expression control sequence when the expression control sequence controls and regulates the transcription and translation of that polynucleotide sequence.
- isolated it is further meant a polynucleotide sequence that is: (i) amplified in vitro by, for example, polymerase chain reaction (PCR); (ii) chemically synthesized; (iii) recombinantly produced by cloning; or (iv) purified, as by cleavage and gel separation.
- PCR polymerase chain reaction
- isolated when applied to polypeptides means a polypeptide or a portion thereof which, by virtue of its origin or manipulation: (i) is present in a host cell as the expression product of a portion of an expression vector; or (ii) is linked to a protein or other chemical moiety other than that to which it is linked in nature; or (iii) does not occur in nature.
- isolated it is further meant a protein that is : (i) chemically synthesized; or (ii) expressed in a host cell and purified away from associated proteins.
- the term generally means a polypeptide that has been separated from other proteins and nucleic acids with which it naturally occurs.
- the polypeptide is also separated from substances such as antibodies or gel matrices (polyacryl amide) which are used to purify it.
- the respective molecules are homologous at that position.
- the percentage homology between two sequences is a function of the number of matching or homologous positions shared by the two sequences divided by the number of positions compared x 100. For instance, if 6 of 10 of the positions in two sequences are matched or are homologous, then the two sequences are 60% homologous.
- angiogenesis and “neovascularization” means, in their broadest sense, the recruitment of new blood vessels. In particular, angiogenesis also refers to the recruitment of new blood vessels at a tumor site.
- a single polymer molecule may be employed for conjugation with an interferon-beta la, although it is also contemplated that more than one polymer molecule can be attached as well.
- Conjugated interferon-beta la compositions of the invention may find utility in both in vivo as well as on-in vivo applications.
- the conjugating polymer may utilize any other groups, moieties, or other conjugated species, as appropriate to the end use application. By way of example, it may be useful in some applications to covalently bond to the polymer a functional moiety imparting UV-degradation resistance, or antioxidation, or other properties or characteristics to the polymer.
- Dlustrative polymers that may usefully be employed to achieve these desirable characteristics are described herein below in exemplary reaction schemes.
- the polymer may be functionalized and then coupled to free amino acid(s) of the peptide(s) to form labile bonds.
- the polymer may be attached anywhere on the interferon-beta la molecule, the most preferred site for polymer coupling is the N-terminus of the interferon- beta-la. Secondary site(s) are at or near the C-terminus and through sugar moieties.
- the invention contemplates as its most preferred embodiments: (i) N-terminally coupled polymer conjugates of interferon-beta-la; (ii) C-terminally coupled polymer conjugates of interferon-beta-la; (iii) sugar-coupled conjugates of polymer conjugates; (iv) as well as N-
- the final amount is a balance between maximizing the extent of the reaction while minimizing non-specific modifications of the product and, at the same time, defining chemistries that will maintain optimum activity, while at the same time optimizing, if possible, the half-life of the protein.
- at least about 50% of the biological activity of the protein is retained, and most preferably 100% is retained.
- the reactions may take place by any suitable method used for reacting biologically active materials with inert polymers, preferably at about pH 5-7 if the reactive groups are on the alpha amino group at the N-terminus.
- the process involves preparing an activated polymer (that may have at least one terminal hydroxyl group) and thereafter reacting the protein with the activated polymer to produce the soluble protein suitable for formulation.
- the above modification reaction can be performed by several methods, which may involve one or more steps.
- the most preferred embodiments of the invention utilize the N- terminal end of interferon-beta- 1 a as the linkage to the polymer.
- Suitable methods are available to selectively obtain an N-terminally modified interferon-beta-la.
- One method is exemplified by a reductive alkylation method which exploits differential reactivity of different types of primary amino groups (the epsilon amino groups on the lysine versus the amino groups on the N-terminal methionine) available for derivatization on interferon- beta-la. Under the appropriate selection conditions, substantially selective derivatization of interferon-beta-la at its N-terminus with a carbonyl group containing polymer can be achieved.
- the glycan on the interferon-beta-la is also in a position that would allow further modification without altering activity.
- Methods for targeting sugars as sites for chemical modification are also well known and therefore it is likely that a polyalkylene glycol polymer can be added directly and specifically to sugars on interferon-beta-la that have been activated through oxidation.
- a polyethyleneglycol-hydrazide can be generated which forms relatively stable hydrazone linkages by condensation with aldehydes and ketones. This property has been used for modification of proteins through oxidized oligosaccharide linkages. See Andresz, H. et al., (1978), Makromol. Chem. 179: 301.
- LFA-3 and CD4 using a procedure such as generating reactive aldehydes on carbohydrate moieties with sodium periodate, forming cystamine conjugates through the aldehydes and inducing cross-linking via the thiol groups on the cystamines. See Pepinsky, B. et al.,
- polyalkylene glycol residues of C1-C4 alkyl polyalkylene glycols preferably polyethylene glycol (PEG), or poly(oxy)alkylene glycol residues of such glycols are advantageously incorporated in the polymer systems of interest.
- the polymer to which the protein is attached can be a homopolymer of polyethylene glycol (PEG) or is a polyoxyethylated polyol, provided in all cases that the polymer is soluble in water at room temperature.
- Non-limiting examples of such polymers include polyalkylene oxide homopolymers such as PEG or polypropylene glycols, polyoxyethylenated glycols, copolymers thereof and block copolymers thereof, provided that the water solubility of the block copolymer is maintained.
- polyoxyethylated polyols include, for example, polyoxyethylated glycerol, polyoxyethylated sorbitol, polyoxyethylated glucose, or the like.
- the glycerol backbone of polyoxyethylated glycerol is the same backbone occurring naturally in, for example, animals and humans in mono-, di-, and triglycerides. Therefore, this branching would not necessarily be seen as a foreign agent in the body.
- dextran As an alternative to polyalkylene oxides, dextran, polyvinyl pyrrolidones, polyacrylamides, polyvinyl alcohols, carbohydrate-based polymers and the like may be used.
- dextran As an alternative to polyalkylene oxides, dextran, polyvinyl pyrrolidones, polyacrylamides, polyvinyl alcohols, carbohydrate-based polymers and the like may be used.
- the polymer need not have any particular molecular weight, but it is preferred that the molecular weight be between about 300 and 100,000, more preferably between 10,000 and 40,000. In particular, sizes of 20,000 or more are best at preventing protein loss due to filtration in the kidneys.
- Polyalkylene glycol derivatization has a number of advantageous properties in the formulation of polymer-interferon-beta la conjugates in the practice of the present invention, as associated with the following properties of polyalkylene glycol derivatives: improvement of aqueous solubility, while at the same time eliciting no antigenic or immunogenic response; high degrees of biocompatibility; absence of in vivo biodegradation of the polyalkylene glycol derivatives; and ease of excretion by living organisms.
- This allows for control in terms of the time course over which the polymer may be cleaved from the interferon-beta 1 a.
- This covalent bond between the interferon-beta-la drug and the polymer may be cleaved by chemical or enzymatic reaction.
- the polymer-interferon-beta- 1 a product retains an acceptable amount of activity.
- the invention contemplates parenteral, nasal, and oral delivery of both the active polymer- interferon-beta- la species and, following hydrolytic cleavage, bioavailability of the interferon-beta-la per se, in in vivo applications.
- reaction schemes described herein are provided for the purposes of illustration only and are not to be limiting with respect to the reactions and structures which may be utilized in the modification of the interferon-beta-la, e.g., to achieve solubility, stabilization, and cell membrane affinity for parenteral and oral administration.
- the reaction of the polymer with the interferon-beta la to obtain the most preferred N-terminal conjugated products is readily carried out using a wide variety of reaction schemes.
- the activity and stability of the interferon-beta-la conjugates can be varied in several ways, by using a polymer of different molecular size. Solubilities of the conjugates can be varied by changing the proportion and size of the polyethylene glycol fragment incorporated in the polymer composition. Utilities
- polyalkylene glycol-derived polymers of value for therapeutic applications of the present invention is their general biocompatibility.
- the polymers have various water solubility properties and are not toxic. They are believed non- immunogenic and non-antigenic and do not interfere with the biological activities of the interferon-beta-la moiety when conjugated under the conditions described herein. They have long circulation in the blood and are easily excreted from living organisms.
- the products of the present invention have been found useful in sustaining the half life of therapeutic interferon-beta la, and may for example be prepared for therapeutic administration by dissolving in water or acceptable liquid medium. Administration is by either the parenteral, aerosol, or oral route. Fine colloidal suspensions may be prepared for parenteral administration to produce a depot effect, or by the oral route while aerosol formulation may be liquid or dry powder in nature. In the dry, lyophilized state or in solution formulations, the interferon-beta- 1 a -polymer conjugates of the present invention should have good storage stability.
- the thermal stability of conjugated interferon-beta-la (Example 3) is advantageous in powder formulation processes that have a dehydration step.
- the present invention contemplates a method of treating an animal subject having or latently susceptible to such condition(s) or disease state(s) and in need of such treatment, comprising administering to such animal an effective amount of a polymer conjugate of the present invention which is therapeutically effective for said condition or disease state.
- Subjects to be treated by the polymer conjugates of the present invention include mammalian subjects and most preferably human subjects.
- animal subjects may be administered polymer conjugates of the invention at any suitable therapeutically effective and safe dosage, as may readily be determined within the skill of the art, and without undue experimentation. Because of the species barriers of Type I interferons, it may be necessary to generate interferon-polymer conjugates as described herein with interferons from the appropriate species.
- the anti-viral activity exhibited by the conjugated proteins may be used in the treatment of viral diseases, such as ECM infection, influenza, and other respiratory tract infections, rabies, and hepatitis. It is also expected that immunomodulatory activities of interferon- beta- la exhibited by the conjugated proteins described herein, may be used in the treatment of autoimmune and inflammatory diseases, such as fibrosis, multiple sclerosis.
- interferons to inhibit formation of new blood vessels (i.e., inhibit angiogenesis and neovascularization) enables conjugates of the invention to be used to treat angiogenic diseases such as diabetic retinopathy, retinopathy of prematurity, macular degeneration, corneal graft rejection, neovascular glaucoma, retrolental fibroplasia, rubeosis and Osier- Webber Syndrome.
- angiogenic diseases such as diabetic retinopathy, retinopathy of prematurity, macular degeneration, corneal graft rejection, neovascular glaucoma, retrolental fibroplasia, rubeosis and Osier- Webber Syndrome.
- interferon has been known for some time and one potential mechanism of interferon action may be to interfere with endothelial cell activity by inhibiting the production or efficacy of angiogenic factors produced by tumor cells.
- Some vascular tumors, such as hemangiomas, are particularly sensitive to treatment with interferon. Treatment with interferon-alpha is the only documented treatment for this disease.
- the polymer-interferon-beta- la conjugates of the invention may be administered per se as well as in the form of pharmaceutically acceptable esters, salts, and other physiologically functional derivatives thereof.
- the interferon-beta-la preferably is utilized together with one or more pharmaceutically acceptable carrier(s) and optionally any other therapeutic ingredients.
- the carrier(s) must be pharmaceutically acceptable in the sense of being compatible with the other ingredients of the formulation and not unduly deleterious to the recipient thereof.
- the interferon-beta-la is provided in an amount effective to achieve the desired pharmacological effect, as described above, and in a quantity appropriate to achieve the desired daily dose.
- the formulations include those suitable for parenteral as well as non-parenteral administration, and specific administration modalities include oral, rectal, buccal, topical, nasal, ophthalmic, subcutaneous, intramuscular, intravenous, transdermal, intrathecal, intra-articular, intra-arterial, sub-arachnoid, bronchial, lymphatic, vaginal, and intra-uterine administration.
- specific administration modalities include oral, rectal, buccal, topical, nasal, ophthalmic, subcutaneous, intramuscular, intravenous, transdermal, intrathecal, intra-articular, intra-arterial, sub-arachnoid, bronchial, lymphatic, vaginal, and intra-uterine administration.
- Formulations suitable for oral, nasal, and parenteral administration are preferred.
- the formulation advantageously may be administered orally or parenterally.
- the formulation may be advantageously administered orally, rectally, or bronchially.
- the interferon-beta-la When the interferon-beta-la is utilized directly in the form of a powdered solid, the interferon-beta-la may advantageously be administered orally. Alternatively, it may be administered nasally or bronchially, via nebulization of the powder in a carrier gas, to form a gaseous dispersion of the powder which is inspired by the patient from a breathing circuit comprising a suitable nebulizer device.
- the formulations comprising the polymer conjugates of the present invention may conveniently be presented in unit dosage forms and may be prepared by any of the methods well known in the art of pharmacy. Such methods generally include the step of bringing the active ingredient(s) into association with a carrier which constitutes one or more accessory ingredients. Typically, the formulations are prepared by uniformly and intimately bringing the active ingredient(s) into association with a liquid carrier, a finely divided solid carrier, or both, and then, if necessary, shaping the product into dosage forms of the desired formulation.
- Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets, tablets, or lozenges, each containing a predetermined amount of the active ingredient as a powder or granules; or a suspension in an aqueous liquor or a non-aqueous liquid, such as a syrup, an elixir, an emulsion, or a draught.
- a tablet may be made by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine, with the active compound being in a free-flowing form such as a powder or granules which optionally is mixed with a binder, disintegrant, lubricant, inert diluent, surface active agent, or discharging agent.
- Molded tablets comprised of a mixture of the powdered polymer conjugates with a suitable carrier may be made by molding in a suitable machine.
- a syrup may be made by adding the active compound to a concentrated aqueous solution of a sugar, for example sucrose, to which may also be added any accessory ingredient(s).
- a sugar for example sucrose
- Such accessory ingredient(s) may include flavorings, suitable preservative, agents to retard crystallization of the sugar, and agents to increase the solubility of any other ingredient, such as a polyhydroxy alcohol, for example glycerol or sorbitol.
- Formulations suitable for parenteral administration conveniently comprise a sterile aqueous preparation of the active conjugate, which preferably is isotonic with the blood of the recipient (e.g., physiological saline solution).
- Such formulations may include suspending agents and thickening agents or other microparticulate systems which are designed to target the compound to blood components or one or more organs.
- the formulations may be presented in unit-dose or multi-dose form.
- Nasal spray formulations comprise purified aqueous solutions of the active conjugate with preservative agents and isotonic agents. Such formulations are preferably adjusted to a pH and isotonic state compatible with the nasal mucus membranes.
- Formulations for rectal administration may be presented as a suppository with a suitable carrier such as cocoa butter, hydrogenated fats, or hydrogenated fatty carboxylic acid.
- Ophthalmic formulations such as eye drops are prepared by a similar method to the nasal spray, except that the pH and isotonic factors are preferably adjusted to match that of the eye.
- Topical formulations comprise the conjugates of the invention dissolved or suspended in one or more media, such as mineral oil, petroleum, polyhydroxy alcohols, or other bases used for topical pharmaceutical formulations.
- the formulations of this invention may further include one or more accessory ingredient(s) selected from diluents, buffers, flavoring agents, disintegrants, surface active agents, thickeners, lubricants, preservatives
- the present invention contemplates the provision of suitable polymers for in vitro stabilization of interferon-beta la in solution, as a preferred illustrative application of non-therapeutic application.
- the polymers may be employed for example to increase the thermal stability and enzymic degradation resistance of the interferon-beta la.
- EXAMPLE 1 Structure/activity studies of human interferon-beta-la using alanine/serine substitution mutations: Analysis of receptor binding sites and functional domains A. Overview
- JFN-beta- la An extensive mutational analysis of human interferon-beta-la (JFN-beta- la) was undertaken with the aims of mapping residues required for activity and receptor binding.
- the availability of the 3-D crystal structure of human JFN-beta (Karpusas, M. et al. 1997, Proc. Natl. Acad. Sci. 94: 11813-11818) allowed us to identify for alanine (or serine) substitutions the solvent-exposed residues available for receptor interactions, and to retain amino acids involved in intramolecular bonds.
- a panel of 15 alanine substitution mutations were designed that replaced between 2 and 8 residues along distinct regions of each of the helices (A, B, C, D, E) and loops (AB, CD, DE).
- mutant his tagged-JFN-beta expression plasmids were constructed using a wild type JFN-beta gene construct as a template for mutagenesis.
- the mutagenesis strategy involved first introducing unique restriction enzyme cleavage sites throughout the wild type his tagged-JFN beta gene, then replacing distinct DNA sequences between the chosen restriction sites with synthetic ohgonucleotide duplexes, which encoded the alanine (or serine) substitution mutations.
- the mutant JFN genes were subcloned into a plasmid which directed mammalian cell expression in a human 293 kidney cell line.
- JFNAR1/2 complex surface receptor
- IFNAR2/Ig fusion protein comprised of the JFN receptor protein JFNAR2 extracellular domain fused to the hinge, CH2 and CH3 domains of human IgGl.
- JFN-beta alanine (or serine) substituted mutants was to first create a modified JFN-beta gene, which encoded the wild type protein but which carried unique restriction enzyme cleavage sites scattered across the gene. The unique sites were used to exchange wild type sequences for synthetic ohgonucleotide duplexes, which encode the mutated codons.
- JFN-beta cDNA GenBank accession #E00029
- the PCR primers used to subclone the coding sequences of the human IFN-beta gene also allowed us to introduce an enterokinase cleavage site upstream and in frame with the JFN-beta gene (5' PCR primer
- PCR primer 5'-GCCGCTCGAGTTATCAGTTTCGGAGGTAACCTGTAAGTC-3' SEQ ID NO: 4:"BET-022
- flanking restriction enzyme sites BspEI and Xho I
- the mutagenized plasmids were used to transform either the JA221 or XLl-Blue strains of E. coli and recombinant colonies selected for chloramphenicol resistance. Chloramphenicol resistant colonies were further tested for the presence of the desired unique restriction enzyme site by DNA restriction mapping analysis.
- the full set of alanine substitution mutations are depicted in Table 1 (below).
- the names of the mutants specify the structural regions (helices and loops) in which the mutations were introduced.
- the entire panel of alanine (serine) substitutions results in mutation of 65 of the 165 amino acids of human IFN-beta.
- the panel of mutants was created from pCMG275.8 by replacing segments of DNA between the unique restriction sites with synthetic ohgonucleotide duplexes, which carried the genetic coding information depicted in Table 2 (see below).
- gel purified pCMG275.8 vector (cleaved with the appropriate restriction enzyme, as indicated on the list below for each JFN-beta structural region) and ohgonucleotide duplexes (coding strand sequences are shown in Table 2) were ligated together.
- the ligation mixtures were used to transform the JA221 strain of E. coli and recombinant colonies selected for ampicillin resistance. Ampicillin resistant colonies were tested for the presence of the insertion of the mutations by screening for appropriate restriction enzyme sites.
- the line designated IFN- ⁇ shows the wild type human IFN- ⁇ sequence. Alanine or serine substitutions of the IFN- ⁇ residues are shown for each of the mutants and dashes, below relevant regions, indicate wild type sequences. The helices and loop structures are indicated as solid lines below the mutants. The DE loop spans the gap between the D and E helices. Two additional alanine substitution mutants (H93A, H97A and H121A) were generated and analyzed in antiviral assays to assess the effects of mutating these histidines, which chelate zinc in the crustal structure dimer. Both of these mutants retained full wild type activity in antiviral assays, suggesting that zinc- mediated dimer formation is not important for IFN- ⁇ activity. TABLE 2
- DMEM fetal bovine serum
- Dilutions of interferon-beta mutant typically ranged from approximately 1 ⁇ M down to 10 pM. After washing, interferon-beta bound to the plates was detected by adding
- a second receptor binding assay was used to measure the affinity with which the interferon-beta mutants bound to Daudi cells expressing both receptor chains, JFNAR1 and JFNAR2, which together comprise the receptor for interferon-beta.
- This FACS-based assay used a blocking monoclonal antibody directed against the extracellular domain of JFNAR1, EA12 (Biogen, Inc.), to distinguish unoccupied (free) receptor from receptor to which interferon-beta was bound.
- Daudi cells (20 ⁇ l at 2.5 x 10 7 cells/ml) were placed in 96-well V-bottom ELISA plates, and incubated for 1 hour at 4 ° C with various concentrations of interferon-beta mutant (20 ⁇ l in FACS buffer; 5% FBS, 0.1% NaN 3 in PBS). Desirable serial dilutions of interferon-beta mutants ranged from 0.5 ⁇ M down to 0.5 pM. To each well was added 100 ng of biotinylated murine anti-IFNARl monoclonal antibody EA12 (10 ⁇ l), and the plates incubated for an additional 2 minutes at room temperature before being washed twice with FACS buffer (4 °C).
- the cells were then incubated for 30 minutes at 4°C with 50 ⁇ l/well of a 1:200 dilution of R-Phycoerythrin- conjugated streptavidin (Jackson ImmunoResearch), washed twice in FACS buffer, resuspended in 300 ⁇ l FACS buffer containing 0.5% paraformaldehyde, and transferred into 12x75mm polystyrene tubes (Falcon 2052). The samples were then analyzed by flow cytometry on a FACScan (Becton Dickinson).
- FIG. 1 shows the receptor binding affinities for each interferon-beta mutant, determined by this method, expressed as a percentage of the affinity measured for His 6 -wild-type interferon-beta-la in each experiment.
- MTT 2,3-bis[2-Methoxy-4-nitro-5-sulfo-phenyl]-2H-tetrazolium-5-carboxyanilide
- Duplicate experimental points were used for each concentration of interferon-beta mutant tested, and a duplicate set of untreated cells was included in all experiments.
- Cells were incubated for two days at 37 °C in 5% CO 2 incubators, after which 1 ⁇ Ci per well of tritiated thymidine ((methyl- 3 H) thymidine, Amersham TRK758) in 50 ⁇ l medium was added to each well, and incubated for a further 4h.
- Cells were harvested using a LKB plate harvester, and incorporation of tritiated thymidine was measured using a LKB beta plate reader.
- Duplicate experimental values were averaged and the standard deviations determined.
- Histidine tagged-wild-type interferon-beta-la was found to have activities in the antiviral and antiproliferation assays that were each about 3-fold lower than the corresponding activities found for untagged wild-type interferon-beta-la. Because all of the interferon-beta mutants Al-E contain the identical his tag sequence at their N-termini, the effects of the mutations on the properties of the molecule were determined by comparing the activities of these mutants in the antiviral, antiproliferation and binding assays to the activity observed for his tagged-wild-type interferon-beta-la.
- Mutant C 1 possesses antiviral activity that is approximately 6-fold greater than that of wild-type his tagged-interferon-beta-la. This mutant and others of this type are predicted to be useful in reducing the amount of interferon-beta that must be administered to achieve a given level of antiviral effect. Lowering the amount of administered protein is expected to reduce the immunogenicity of the protein and may also reduce side-effects from non-mechanism-based toxicities.
- Mutations in this class are predicted to be advantageous in situations where the therapeutic benefit of interferon-beta administration results from its antiviral effects, and where antiproliferative effects contribute to toxicity or to unwanted side-effects.
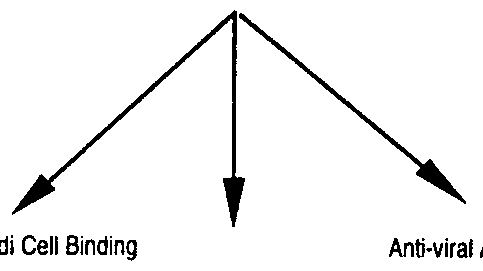
- the relative activities (% wild type) of the alanine substituted mutants in antiviral and antiproliferation assay are compared in Figure 5. Coordinately changed activities (i.e. antiviral and antiproliferation activities that differ by the same factor from the activities of the wild-type his tagged-interferon-beta-la) are seen in most mutants (those lying on the diagonal line).
- Mutant Cl shows antiviral activity that is ⁇ 6-fold higher than that of wild- type his tagged-interferon-beta-la, but its activity in the antiproliferation assay is similar to that of wild-type. Mutant Cl thus has antiviral activity that is enhanced by a factor of 5.2 over its antiproliferation activity, relative to wild-type his tagged-interferon-beta-la.
- mutant D displays 65% of wild type activity in the antiviral assay, but only 20% of wild-type activity in the antiproliferation assay, and thus has antiviral activity that is enhanced 3.4- fold over its antiproliferation activity compared to wild type.
- Mutant DEI displays 26% of wild type activity in the antiviral assay but only 8.5% in the antiproliferation assay, and thus has antiviral activity that is enhanced 3.0-fold over its antiproliferation activity compared to wild-type his tagged-interferon-beta-la. When administered at a concentration sufficient to achieve a desired level of antiviral activity, these mutant proteins will show substantially lower levels of antiproliferative activity than the wild-type protein.
- Mutants with antiviral and antiproliferative activities that are low with respect to receptor binding, as compared to wild-type his tagged-interferon-beta-la see Table 4 below.
- Mutant Al displays antiviral and antiproliferative activities that are 2.0-fold and 1.8-fold higher than that observed for wild-type his tagged-interferon-beta-la, but binds to the cognate receptor on Daudi cells with an affinity that is 29-fold higher than wild-type. The binding of this mutant to the JFN-beta receptor is thus enhanced approximately 15-fold compared to the antiviral and antproliferation activities of the protein.
- Elution fractions were analyzed for their absorbance values at 280 nm and the concentration of interferon in the samples estimated from the absorbance using an extinction coefficient of 1.51 for a 1 mg/ml solution.
- 0.5 M sodium phosphate pH 6.0 was added to 50 mM, sodium cyanoborohydride (Aldrich, Milwaukee,
- the PEG-interferon beta-containing elution pool from gel filtration was diluted 1 : 1 with water and loaded at 2 mg interferon beta /ml resin onto an SP- Sepharose ® column.
- the column was washed with 5 mM sodium phosphate pH 5.5, 75 mM NaCl and then the pegylated interferon beta was eluted from the column with 5 mM sodium phosphate pH 5.5, 800 mM NaCl.
- Elution fractions were analyzed for protein content by absorbance at 280 nm.
- the pegylated interferon concentration is reported in interferon equivalents as the PEG moiety did not contribute to absorbance at 280 nm.
- interferon-beta-la samples were tested on human lung carcinoma cells (A549 cells) that had been exposed to encephalomyocarditis (EMC) virus using the procedures involving MTT staining outlined above. Briefly, A549 cells were pretreated for 24 hours with interferon-beta-la or PEG-modified interferon-beta-la (4000, 2000, 1000, 500, 250, 125, 75, 62.5, 31.25, 50, 33.3, 22.2, 14.8, 9.9, 6.6, 4.39 pg/ml) prior to challenge with virus. The assay was performed using duplicate data points for each interferon-beta-la concentration. The standard deviations are shown as error bars in Figure 7.
- Interferon-beta-la was also PEGylated with a 5K PEG-aldehyde moiety that was purchased from Fluka, Inc. (Cat. No. 75936, Ronkonkoma, NY) following the same protocol described for modification with 20K PEG aldehyde except that the reaction contained 2 mg/ml of the 5K PEG. Modification with the 5K PEG was also highly specific for the N-terminus and did not alter the antiviral activity of interferon-beta-la. Like the 20K adduct, the 5K PEG inteferon-beta-la was indistinguishable from the unmodified interferon-beta-la in the antiviral assay.
- the temperature of the cuvette holder was then ramped from 25 ° C to 80 ° C at a rate of 2 ° C/min, and the denaturation of the protein followed by continuous monitoring of absorbance at 280 nm.
- the mid-point of the cooperative unfolding event, Tm was obtained from the melting curves by determining the temperature at which the measured absorbance was mid-way between the values defined by lines extrapolated from the linear regions on each side of the cooperative unfolding transitions.
- EXAMPLE 4 Measurement of interferon-beta-la antiviral activity in the plasma of mice treated with interferon-beta-la and PEGylated interferon-beta-la
- mice (C57B1/6) are injected i.v. through the tail vein with either 50,000 Units of interferon-beta- 1 a or 50,000 Units of PEGylated interferon-beta- 1 a containing the 20K PEG or an equal volume of phosphate buffer given as a control.
- Blood from these mice is obtained via retro-orbital bleeds at different time points after injection (immediately, 0.25, 1, 4, 24 and 48 hours). There are at least 3 mice bled at each time point.
- Whole blood is collected into tubes containing anticoagulant, cells are removed and the resulting plasma frozen until the time of assay. These plasma samples are then tested in anti-viral assays.
- Blood is drawn for pharmacokinetic testing at various time intervals following each injection.
- Blood samples for measurements of the interferon induced biological response marker, serum neopterin, are also drawn following administration of study drug. Evaluations during the study period include clinical observations performed 30 minutes and 1 hour post-dose for signs of toxicitiy. Daily cageside observations will be performed and general appearance, signs of toxicity, discomfort, and changes in behavior will be recorded. Body weights and body temperatures will be recorded at regular intervals through 21 days post-dose. Assay Methods
- CPE cytopathic effect
- the cells are preincubated for 15 to 20 hours with serum samples to allow the induction and synthesis of interferon inducible proteins that then mount an antiviral response. Afterwards EMC virus is added and incubated for a further 30 hours before assessment of cytotoxicity is made using a crystal violet stain.
- An internal interferon beta standard as well as PEG conjugate internal standard is tested concurrently with samples on each assay plate. This standard is calibrated against a natural human fibroblast interferon reference standard (WHO Second International Standard for Interferon, Human Fibroblast, Gb-23-902-53).
- Each assay plate also includes cell growth control wells containing neither interferon beta of any kind nor EMC, and virus control wells contain cells and EMC but no interferon beta. Control plates containing the standard and samples are also prepared to determine the effect, if any, of the samples on cell growth.
- RstripTM software (MicroMath, Inc., Salt Lake City, UT) is used to fit data to pharmacokinetic models. Geometric mean concentrations are plotted by time for each group. Since assay results are expressed in dilutions, geometric means are considered more appropriate than arithmetic means. Serum interferon levels are adjusted for baseline values and non-detectable serum concentrations are set to 5 U/ml, which represents one-half the lower limit of detection.
- concentration data were analyzed by standard model-independent methods (noncompartmental analysis) to obtain pharmacokinetic parameters.
- Area under the curve (AUC) was calculated using the trapezoidal rule.
- Statistical analyses including arithmetic mean and standard deviation, were performed using Microsoft Excel version 5.0 software (Microsoft Corp., Redmond WA). Concentration values reported as below limits of quantitation (BLQ) were not used in the pharmacokinetic analysis. Due to the fact that different computers and computer programs round off or truncate numbers differently, values in some tables (e.g. means, standard deviations, or individual values) may differ slightly from those in other tables, from individually calculated data, or from statistical analysis data. Neither the integrity nor interpretation of the data was affected by these differences.
- pegylated JFN beta-la exhibited higher bioavailability (as measured by the area under the serum concentration-time curve).
- the pegylated JFN beta-la had a higher absolute bioavailability as compared to JFN beta-la when administered by the SC route.
- Table 5 The pharmacokinetic parameters in Table 5.
- Administration of pegylated JFN beta-la by both IV and SC routes results in an increase in the half-life as well as the AUC of JFN beta-la. TABLE 5:
- the mean ( ⁇ std. dev.) peak serum concentrations (Cmax) of JF ⁇ beta-la and pegylated JF ⁇ beta- la were 6400 ( ⁇ 0) and 10800 ( ⁇ 3.5) U/mL, respectively.
- the mean ( ⁇ std. dev.) AUC values were 4453 ( ⁇ 799) and 34373 ( ⁇ 3601) U*hr/mL, respectively.
- the mean ( ⁇ std. dev.) Cmax of IF ⁇ beta-la and pegylated JF ⁇ beta-la were 277 ( ⁇ 75) and 1080 ( ⁇ 381) U/mL, respectively.
- Mean ( ⁇ std. dev.) AUC values were 4753 ( ⁇ 3170) and
- EXAMPLE 8 Anti-Angiogenic Effects of Polymer-Conjugated Interferon Beta-la: Assessment of the ability of PEGylated interferon-beta-la to inhibit endothelial cell proliferation in vitro
- the assay medium is replaced with fresh medium containing 20 ng/ml of human recombinant basic Fibroblast Growth Factor (Becton Dickinson, Cat. # 40060) and various concentrations of conjugated and unconjugated interferon-beta-la proteins or positive control (endostatin can be used as a positive control, as could an antibody to bFGF).
- the final volume is adjusted to 0.5 ml in the 24 well plate or 0.2 ml in the 96 well plate.
- a tumor fragment is implanted subcutaneously in the axillary region of a B6D2F1 mouse.
- the test agent i.e, a PEGylated interferon of the invention
- SC subcutaneously
- IP intraperitoneally
- Source One source, if feasible, for all animals in one experiment.
- Day O Implant tumor. Run bacterial cultures. Test positive control compound in every odd-numbred experiment. Prepare materials. Record deaths daily.
- Day 1 Check cultures. Discard experiment if contaminated. Randomize animals.
- the parameter measured is median survival time Compute mean animal body weights for Day 1 and Day 5, compute Test/Control ratio for all test groups with. The mean animal body weights for staging day and final evaluation day are computed.
- Test/Control ratio is computed for all test groups with > 65 % survivors on Day 5.
- a Test/Control ratio value ⁇ 86% indicates toxicity.
- An excessive body weight change difference (test minus control) may also be used in evaluating toxicity.
- An initial Test/Control ratio greater than or equal to 140% is considered necessary to demonstrate moderate activity.
- a reproducible Test/Control ratio value of greater than or equal to 150% is considered significant activity.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Virology (AREA)
- Oncology (AREA)
- Gastroenterology & Hepatology (AREA)
- Communicable Diseases (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Molecular Biology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Neurology (AREA)
- Urology & Nephrology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Ophthalmology & Optometry (AREA)
- Transplantation (AREA)
- Vascular Medicine (AREA)
- Neurosurgery (AREA)
- Pulmonology (AREA)
- Biomedical Technology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
Abstract
Description






Claims
Priority Applications (22)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU14465/00A AU762616B2 (en) | 1998-10-16 | 1999-10-15 | Polymer conjugates of interferon beta-1a and uses |
| SK506-2001A SK287918B6 (en) | 1998-10-16 | 1999-10-15 | Composition containing glycosylated interferon-beta-1a, pharmaceutical composition, use of composition, in-vitro method of prolonging the activity of interferon-beta-1a and use of polymer moiety |
| DE69930015T DE69930015T2 (en) | 1998-10-16 | 1999-10-15 | POLYMER CONJUGATES OF INTERFERON BETA-1A AND ITS USES |
| NZ510689A NZ510689A (en) | 1998-10-16 | 1999-10-15 | Polymer conjugates of interferon beta-1a and uses |
| BRPI9915542A BRPI9915542C1 (en) | 1998-10-16 | 1999-10-15 | composition comprising a glycosylated interferon-beta-1a, pharmaceutical composition comprising the same and an in vitro process of prolonging the activity of interferon-beta-1a in an in vitro system |
| JP2000576887A JP4616478B2 (en) | 1998-10-16 | 1999-10-15 | Interferon-beta-1a polymer conjugates and uses |
| IL14228299A IL142282A0 (en) | 1998-10-16 | 1999-10-15 | Compositions containing polymer conjugates of interferon-beta-1a |
| EEP200100220A EE04967B1 (en) | 1998-10-16 | 1999-10-15 | Glycylated Interferon Beta, its Use and a Pharmaceutical Composition, Method for Prolonging Interferon Beta-1a Activity, and Preparing a Protein of the Invention |
| CA002345138A CA2345138C (en) | 1998-10-16 | 1999-10-15 | Polymer conjugates of interferon beta-1a and their uses |
| SI9930890T SI1121156T1 (en) | 1998-10-16 | 1999-10-15 | Polymer conjugates of interferon beta- 1a and their uses |
| EA200100446A EA004789B9 (en) | 1998-10-16 | 1999-10-15 | Polymer conjugates of interferon beta-1a and their uses |
| EP99970609A EP1121156B1 (en) | 1998-10-16 | 1999-10-15 | Polymer conjugates of interferon beta- 1a and their uses |
| HU0200444A HU229888B1 (en) | 1998-10-16 | 1999-10-15 | Polymer conjugates of interferon betha-1a and uses |
| IL142282A IL142282A (en) | 1998-10-16 | 2001-03-27 | Compositions containing polymer conjugates of interferon-beta-1a |
| IS5913A IS2926B (en) | 1998-10-16 | 2001-03-30 | Polyalkylene glycol derivatives of interferon beta-1A and their use |
| NO20011860A NO20011860L (en) | 1998-10-16 | 2001-04-11 | Polymer conjugates of interferon <beta> -1A and their use |
| US09/832,658 US6962978B2 (en) | 1998-10-16 | 2001-04-11 | Polymer conjugates of interferon beta-1a and uses |
| HK02103771.7A HK1042434B (en) | 1998-10-16 | 2002-05-21 | Polymer conjugates of interferon beta-1a and uses |
| US10/802,540 US7446173B2 (en) | 1998-10-16 | 2004-03-16 | Polymer conjugates of interferon beta-1A and uses |
| CY20061100568T CY1105022T1 (en) | 1998-10-16 | 2006-05-02 | INTERFERON B-1A POLYMER CONJUGATES AND THEIR USE |
| US13/587,242 US9314534B2 (en) | 1998-10-16 | 2012-08-16 | Polymer conjugates of interferon beta-1A and uses |
| US15/131,233 US20160250340A1 (en) | 1998-10-16 | 2016-04-18 | POLYMER CONJUGATES OF INTERFERON BETA-1a AND USES |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10457298P | 1998-10-16 | 1998-10-16 | |
| US60/104,572 | 1998-10-16 | ||
| US12016199P | 1999-02-16 | 1999-02-16 | |
| US60/120,161 | 1999-02-16 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/832,658 Continuation US6962978B2 (en) | 1998-10-16 | 2001-04-11 | Polymer conjugates of interferon beta-1a and uses |
| US13/587,242 Continuation US9314534B2 (en) | 1998-10-16 | 2012-08-16 | Polymer conjugates of interferon beta-1A and uses |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2000023114A2 true WO2000023114A2 (en) | 2000-04-27 |
| WO2000023114A3 WO2000023114A3 (en) | 2000-09-28 |
| WO2000023114A9 WO2000023114A9 (en) | 2001-07-05 |
Family
ID=26801696
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1999/024201 WO2000023114A2 (en) | 1998-10-16 | 1999-10-15 | Polymer conjugates of interferon beta- 1a and their uses |
Country Status (30)
| Country | Link |
|---|---|
| US (5) | US6962978B2 (en) |
| EP (4) | EP2599503B1 (en) |
| JP (5) | JP4616478B2 (en) |
| KR (1) | KR100630849B1 (en) |
| CN (2) | CN101062419B (en) |
| AT (1) | ATE318149T1 (en) |
| AU (1) | AU762616B2 (en) |
| BR (1) | BRPI9915542C1 (en) |
| CA (1) | CA2345138C (en) |
| CY (2) | CY1105022T1 (en) |
| CZ (1) | CZ299164B6 (en) |
| DE (1) | DE69930015T2 (en) |
| DK (2) | DK1121156T3 (en) |
| EA (1) | EA004789B9 (en) |
| EE (2) | EE05201B1 (en) |
| ES (4) | ES2653589T3 (en) |
| HK (2) | HK1042434B (en) |
| HU (1) | HU229888B1 (en) |
| IL (2) | IL142282A0 (en) |
| IS (1) | IS2926B (en) |
| LT (2) | LT2599503T (en) |
| NO (1) | NO20011860L (en) |
| NZ (1) | NZ510689A (en) |
| PL (1) | PL206536B1 (en) |
| PT (2) | PT1656952E (en) |
| SG (2) | SG110047A1 (en) |
| SI (2) | SI1121156T1 (en) |
| SK (1) | SK287918B6 (en) |
| TR (1) | TR200101086T2 (en) |
| WO (1) | WO2000023114A2 (en) |
Cited By (72)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001015736A2 (en) * | 1999-08-27 | 2001-03-08 | Maxygen Aps | Interferon-beta conjugates |
| WO2002036628A2 (en) * | 2000-11-02 | 2002-05-10 | Maxygen Aps | New multimeric interferon beta polypeptides |
| US6531122B1 (en) | 1999-08-27 | 2003-03-11 | Maxygen Aps | Interferon-β variants and conjugates |
| WO2003061577A3 (en) * | 2002-01-18 | 2003-10-02 | Biogen Inc | Polyalkylene glycol with moiety for conjugating biologically active compound |
| WO2004020468A2 (en) * | 2002-08-28 | 2004-03-11 | Maxygen Aps | Interferon beta-like molecules for treatment of cancer |
| WO2004060299A2 (en) | 2002-12-26 | 2004-07-22 | Mountain View Pharmaceuticals, Inc. | Polymer conjugates of interferon-beta with enhanced biological potency |
| WO2004084949A2 (en) * | 2003-03-20 | 2004-10-07 | Xencor | Generating protein pro-drugs using reversible ppg linkages |
| WO2005019260A1 (en) * | 2003-08-25 | 2005-03-03 | Toray Industries, Inc. | INTERFERON-β COMPOSITE |
| EP1575531A2 (en) * | 2002-09-27 | 2005-09-21 | Biogen Idec MA Inc. | THERAPIES FOR CHRONIC INFLAMMATORY DEMYELINATING POLYNEUROPATHY USING INTERFERON-&bgr; |
| EP1608408A1 (en) * | 2003-03-28 | 2005-12-28 | Biopolymed, Inc. | Biologically active material conjugated with biocompatible polymer with 1:1 complex, preparation method thereof and pharmaceutical composition comprising the same |
| EP1628618A2 (en) * | 2002-12-26 | 2006-03-01 | Mountain View Pharmaceuticals, Inc. | Polymer conjugates of cytokines, chemokines, growth factors, polypeptide hormones and antagonists thereof with preserved receptor-binding activity |
| WO2003031464A3 (en) * | 2001-10-10 | 2006-03-02 | Neose Technologies Inc | Remodeling and glycoconjugation of peptides |
| US7144574B2 (en) | 1999-08-27 | 2006-12-05 | Maxygen Aps | Interferon β variants and conjugates |
| WO2008025856A2 (en) | 2006-09-01 | 2008-03-06 | Novo Nordisk Health Care Ag | Modified glycoproteins |
| US7431921B2 (en) | 2000-04-14 | 2008-10-07 | Maxygen Aps | Interferon beta-like molecules |
| WO2009080699A2 (en) * | 2007-12-20 | 2009-07-02 | Merck Serono S.A. | Peg-interferon-beta formulations |
| EP2080771A2 (en) | 2001-02-27 | 2009-07-22 | Maxygen Aps | New interferon beta-like molecules |
| US7645860B2 (en) | 2006-03-31 | 2010-01-12 | Baxter Healthcare S.A. | Factor VIII polymer conjugates |
| WO2010011735A2 (en) | 2008-07-23 | 2010-01-28 | Ambrx, Inc. | Modified bovine g-csf polypeptides and their uses |
| US7691603B2 (en) | 2003-04-09 | 2010-04-06 | Novo Nordisk A/S | Intracellular formation of peptide conjugates |
| US7696163B2 (en) | 2001-10-10 | 2010-04-13 | Novo Nordisk A/S | Erythropoietin: remodeling and glycoconjugation of erythropoietin |
| US7803777B2 (en) | 2003-03-14 | 2010-09-28 | Biogenerix Ag | Branched water-soluble polymers and their conjugates |
| CN101906213A (en) * | 2010-07-23 | 2010-12-08 | 华南理工大学 | Novel protein glycosylation grafting method |
| EP2283896A3 (en) * | 2001-04-09 | 2011-05-25 | Novartis Vaccines and Diagnostics, Inc. | HSA-free formulations of interferon-beta |
| US7982010B2 (en) | 2006-03-31 | 2011-07-19 | Baxter International Inc. | Factor VIII polymer conjugates |
| US7985839B2 (en) | 2006-03-31 | 2011-07-26 | Baxter International Inc. | Factor VIII polymer conjugates |
| US8053561B2 (en) | 2006-03-31 | 2011-11-08 | Baxter International Inc. | Pegylated factor VIII |
| US8063015B2 (en) | 2003-04-09 | 2011-11-22 | Novo Nordisk A/S | Glycopegylation methods and proteins/peptides produced by the methods |
| US8076292B2 (en) | 2001-10-10 | 2011-12-13 | Novo Nordisk A/S | Factor VIII: remodeling and glycoconjugation of factor VIII |
| US8097702B2 (en) | 2004-02-02 | 2012-01-17 | Ambrx, Inc. | Modified human interferon polypeptides with at least one non-naturally encoded amino acid and their uses |
| US8114630B2 (en) | 2007-05-02 | 2012-02-14 | Ambrx, Inc. | Modified interferon beta polypeptides and their uses |
| US8129330B2 (en) | 2002-09-30 | 2012-03-06 | Mountain View Pharmaceuticals, Inc. | Polymer conjugates with decreased antigenicity, methods of preparation and uses thereof |
| US8268967B2 (en) | 2004-09-10 | 2012-09-18 | Novo Nordisk A/S | Glycopegylated interferon α |
| US8361961B2 (en) | 2004-01-08 | 2013-01-29 | Biogenerix Ag | O-linked glycosylation of peptides |
| US8404809B2 (en) | 2005-05-25 | 2013-03-26 | Novo Nordisk A/S | Glycopegylated factor IX |
| EP2621516A2 (en) * | 2010-10-01 | 2013-08-07 | Biogen Idec MA Inc. | Interferon-beta for use as monotherapy or in combination with other cancer therapies |
| WO2013129549A1 (en) | 2012-02-29 | 2013-09-06 | 東レ株式会社 | Body cavity effusion suppressant |
| US8632770B2 (en) | 2003-12-03 | 2014-01-21 | Novo Nordisk A/S | Glycopegylated factor IX |
| US8632771B2 (en) | 2000-06-30 | 2014-01-21 | Regents Of The University Of Minnesota | High molecular weight derivatives of vitamin K-dependent polypeptides |
| US8637640B2 (en) | 2009-07-27 | 2014-01-28 | Baxter International Inc. | Blood coagulation protein conjugates |
| US8637007B2 (en) | 2006-12-15 | 2014-01-28 | Baxter International Inc. | Factor VIIa-polysialic acid conjugate having prolonged in vivo half-life |
| US8642737B2 (en) | 2010-07-26 | 2014-02-04 | Baxter International Inc. | Nucleophilic catalysts for oxime linkage |
| US8716239B2 (en) | 2001-10-10 | 2014-05-06 | Novo Nordisk A/S | Granulocyte colony stimulating factor: remodeling and glycoconjugation G-CSF |
| US8716240B2 (en) | 2001-10-10 | 2014-05-06 | Novo Nordisk A/S | Erythropoietin: remodeling and glycoconjugation of erythropoietin |
| US8791066B2 (en) | 2004-07-13 | 2014-07-29 | Novo Nordisk A/S | Branched PEG remodeling and glycosylation of glucagon-like peptide-1 [GLP-1] |
| US8791070B2 (en) | 2003-04-09 | 2014-07-29 | Novo Nordisk A/S | Glycopegylated factor IX |
| US8809501B2 (en) | 2009-07-27 | 2014-08-19 | Baxter International Inc. | Nucleophilic catalysts for oxime linkage |
| US8841439B2 (en) | 2005-11-03 | 2014-09-23 | Novo Nordisk A/S | Nucleotide sugar purification using membranes |
| EP2805965A1 (en) | 2009-12-21 | 2014-11-26 | Ambrx, Inc. | Modified porcine somatotropin polypeptides and their uses |
| EP2805964A1 (en) | 2009-12-21 | 2014-11-26 | Ambrx, Inc. | Modified bovine somatotropin polypeptides and their uses |
| US8911967B2 (en) | 2005-08-19 | 2014-12-16 | Novo Nordisk A/S | One pot desialylation and glycopegylation of therapeutic peptides |
| US8916360B2 (en) | 2003-11-24 | 2014-12-23 | Novo Nordisk A/S | Glycopegylated erythropoietin |
| US8945897B2 (en) | 2010-07-26 | 2015-02-03 | Baxter International Inc. | Materials and methods for conjugating a water soluble fatty acid derivative to a protein |
| US9005625B2 (en) | 2003-07-25 | 2015-04-14 | Novo Nordisk A/S | Antibody toxin conjugates |
| US9029331B2 (en) | 2005-01-10 | 2015-05-12 | Novo Nordisk A/S | Glycopegylated granulocyte colony stimulating factor |
| US9050304B2 (en) | 2007-04-03 | 2015-06-09 | Ratiopharm Gmbh | Methods of treatment using glycopegylated G-CSF |
| US9150848B2 (en) | 2008-02-27 | 2015-10-06 | Novo Nordisk A/S | Conjugated factor VIII molecules |
| US9200049B2 (en) | 2004-10-29 | 2015-12-01 | Novo Nordisk A/S | Remodeling and glycopegylation of fibroblast growth factor (FGF) |
| US9493499B2 (en) | 2007-06-12 | 2016-11-15 | Novo Nordisk A/S | Process for the production of purified cytidinemonophosphate-sialic acid-polyalkylene oxide (CMP-SA-PEG) as modified nucleotide sugars via anion exchange chromatography |
| EP3103880A1 (en) | 2008-02-08 | 2016-12-14 | Ambrx, Inc. | Modified leptin polypeptides and their uses |
| US9707298B2 (en) | 2006-09-15 | 2017-07-18 | Creabilis Therapeutics S.R.L. | Polymer conjugates of Box-A of HMGB1 and Box-A variants of HMGB1 |
| US9795683B2 (en) | 2009-07-27 | 2017-10-24 | Lipoxen Technologies Limited | Glycopolysialylation of non-blood coagulation proteins |
| WO2017194783A1 (en) | 2016-05-13 | 2017-11-16 | Orionis Biosciences Nv | Targeted mutant interferon-beta and uses thereof |
| WO2018141964A1 (en) | 2017-02-06 | 2018-08-09 | Orionis Biosciences Nv | Targeted chimeric proteins and uses thereof |
| US10350301B2 (en) | 2009-07-27 | 2019-07-16 | Baxalta Incorporated | Blood coagulation protein conjugates |
| RU2739261C1 (en) * | 2019-12-31 | 2020-12-22 | Федеральное государственное бюджетное учреждение науки институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова Российской академии наук (ИБХ РАН) | Method for quantitative determination of anti-proliferative activity of human interferon-beta |
| US11273202B2 (en) | 2010-09-23 | 2022-03-15 | Elanco Us Inc. | Formulations for bovine granulocyte colony stimulating factor and variants thereof |
| WO2022200525A1 (en) | 2021-03-26 | 2022-09-29 | Innate Pharma | Multispecific proteins comprising an nkp46-binding site, a cancer antgienge binding site fused to a cytokine for nk cell engaging |
| WO2022258678A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkp30, a cytokine receptor, a tumour antigen and cd16a |
| WO2022258662A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkp46, a cytokine receptor, a tumour antigen and cd16a |
| WO2022258691A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkg2d, a cytokine receptor, a tumour antigen and cd16a |
| US11896643B2 (en) | 2018-02-05 | 2024-02-13 | Orionis Biosciences, Inc. | Fibroblast binding agents and use thereof |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2599503B1 (en) * | 1998-10-16 | 2017-05-17 | Biogen MA Inc. | Polymer conjugates of interferon beta-1A and uses thereof |
| IL142350A0 (en) * | 1998-10-16 | 2002-03-10 | Biogen Inc | Interferon-beta fusion proteins and pharmaceutical compositions containing the same |
| WO2004022593A2 (en) * | 2002-09-09 | 2004-03-18 | Nautilus Biotech | Rational evolution of cytokines for higher stability, the cytokines and encoding nucleic acid molecules |
| US20040062748A1 (en) * | 2002-09-30 | 2004-04-01 | Mountain View Pharmaceuticals, Inc. | Polymer conjugates with decreased antigenicity, methods of preparation and uses thereof |
| ATE384735T1 (en) * | 2003-05-21 | 2008-02-15 | Ares Trading Sa | METHOD FOR CHROMATOGRAPHICAL ANALYSIS OF A PROTEIN SOLUTION |
| JP2007526317A (en) * | 2004-03-01 | 2007-09-13 | エンゾン ファーマスーティカルズ インコーポレイテッド | Interferon-beta polymer conjugate |
| WO2005112911A2 (en) * | 2004-05-21 | 2005-12-01 | The Regents Of The University Of California | Compositions and methods for treating myelin deficiency disorders |
| CA2572751A1 (en) * | 2004-06-30 | 2006-01-12 | Egen Corporation | Pegylated interferon alpha-1b |
| EP2386571B1 (en) | 2005-04-08 | 2016-06-01 | ratiopharm GmbH | Compositions and methods for the preparation of protease resistant human growth hormone glycosylation mutants |
| CN101291684A (en) * | 2005-10-21 | 2008-10-22 | 阿维季尼克斯股份有限公司 | Glycolated and glycosylated poultry derived therapeutic proteins |
| JP2009515933A (en) * | 2005-11-18 | 2009-04-16 | アレス トレーディング ソシエテ アノニム | Interferon in influenza |
| CN101384272B (en) | 2005-12-20 | 2013-05-01 | 杜克大学 | Methods and compositions for delivering active agents with enhanced pharmacological properties |
| CN100475270C (en) * | 2006-01-20 | 2009-04-08 | 清华大学 | Medicine for treating tumor, and application thereof |
| WO2007110231A2 (en) * | 2006-03-28 | 2007-10-04 | Nautilus Biotech, S.A. | MODIFIED INTERFERON-β (IFN-β) POLYPEPTIDES |
| US8637325B2 (en) * | 2009-02-16 | 2014-01-28 | University Of Wyoming | Method and apparatus for pyrolysis-induced cleavage in peptides and proteins |
| EP2049144B8 (en) | 2006-07-21 | 2015-02-18 | ratiopharm GmbH | Glycosylation of peptides via o-linked glycosylation sequences |
| US20100075375A1 (en) | 2006-10-03 | 2010-03-25 | Novo Nordisk A/S | Methods for the purification of polypeptide conjugates |
| AU2007338771A1 (en) * | 2006-12-19 | 2008-07-03 | Merrimack Pharmaceuticals, Inc. | Coadministration of alpha-fetoprotein and an immunomodulatory agent to treat multiple sclerosis |
| KR20100090684A (en) * | 2007-10-09 | 2010-08-16 | 폴리테릭스 리미티드 | Novel conjugated proteins and peptides |
| PL2288715T3 (en) | 2008-04-11 | 2015-03-31 | Merrimack Pharmaceuticals Inc | Human serum albumin linkers and conjugates thereof |
| KR20100137006A (en) | 2008-04-21 | 2010-12-29 | 티슈 리제너레이션 쎄라퓨틱스, 인코포레이티드 | Genetically modified human umbilical cord perivascular cells for prophylaxis against or treatment of biological or chemical agents |
| KR20110112301A (en) | 2008-11-18 | 2011-10-12 | 메리맥 파마슈티컬즈, 인크. | Human serum albumin linkers and conjugates thereof |
| GB0912485D0 (en) * | 2009-07-17 | 2009-08-26 | Polytherics Ltd | Improved conjugation method |
| MX2011011512A (en) * | 2009-04-30 | 2012-02-13 | Novartis Vaccines & Diagnostic | Chimeric factor h binding proteins (fhbp) and methods of use. |
| WO2010142017A1 (en) | 2009-06-09 | 2010-12-16 | Defyrus, Inc . | Administration of interferon for prophylaxis against or treatment of pathogenic infection |
| US20130195799A1 (en) * | 2010-08-19 | 2013-08-01 | Peg Biosciences, Inc. | Synergistic biomolecule-polymer conjugates |
| CA2850469C (en) | 2011-10-01 | 2020-07-07 | Glytech, Inc. | Glycosylated polypeptide and pharmaceutical composition containing same |
| WO2014036520A1 (en) | 2012-08-30 | 2014-03-06 | Merrimack Pharmaceuticals, Inc. | Combination therapies comprising anti-erbb3 agents |
| BR112015024423B1 (en) | 2013-03-29 | 2023-04-25 | Glytech, Inc | GLYCOSYLATED POLYPEPTIDE HAVING INTERFERON ACTIVITY, PHARMACEUTICAL COMPOSITION AND USE OF A GLYCOSYLATED POLYPEPTIDE |
| US11759500B2 (en) | 2014-07-24 | 2023-09-19 | Abion Inc. | PEGylated interferon-beta variant |
| RU2661088C1 (en) * | 2017-05-24 | 2018-07-11 | Федеральное государственное бюджетное учреждение "Государственный научный центр "Институт иммунологии" Федерального медико-биологического агентства России (ФГБУ "ГНЦ Институт иммунологии" ФМБА России) | Method for purification of sustained-release drug based on recombinant interferon beta-1b analogue for the treatment of multiple sclerosis |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1987000056A1 (en) * | 1985-06-26 | 1987-01-15 | Cetus Corporation | Solubilization of proteins for pharmaceutical compositions using polymer conjugation |
| WO1995013090A1 (en) * | 1993-11-10 | 1995-05-18 | Enzon, Inc. | Improved interferon polymer conjugates |
| WO1999055377A2 (en) * | 1998-04-28 | 1999-11-04 | Applied Research Systems Ars Holding N.V. | Polyol-ifn-beta conjugates |
Family Cites Families (91)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4179337A (en) | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| DE2360794C2 (en) * | 1973-12-06 | 1984-12-06 | Hoechst Ag, 6230 Frankfurt | Process for the production of peptides |
| CH596313A5 (en) * | 1975-05-30 | 1978-03-15 | Battelle Memorial Institute | |
| IL47468A (en) | 1975-06-12 | 1979-05-31 | Rehovot Res Prod | Process for the cross-linking of proteins using water soluble cross-linking agents |
| US4002531A (en) | 1976-01-22 | 1977-01-11 | Pierce Chemical Company | Modifying enzymes with polyethylene glycol and product produced thereby |
| US5179017A (en) | 1980-02-25 | 1993-01-12 | The Trustees Of Columbia University In The City Of New York | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4399216A (en) | 1980-02-25 | 1983-08-16 | The Trustees Of Columbia University | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4456748A (en) | 1981-02-23 | 1984-06-26 | Genentech, Inc. | Hybrid human leukocyte interferons |
| EP0070906B1 (en) | 1981-02-04 | 1986-10-15 | Juridical Foundation Japanese Foundation For Cancer Research | Human interferon-beta gene |
| US4414147A (en) | 1981-04-17 | 1983-11-08 | Massachusetts Institute Of Technology | Methods of decreasing the hydrophobicity of fibroblast and other interferons |
| WO1983002461A1 (en) | 1982-01-19 | 1983-07-21 | Cetus Corp | Multiclass hybrid interferons |
| US5149636A (en) | 1982-03-15 | 1992-09-22 | Trustees Of Columbia University In The City Of New York | Method for introducing cloned, amplifiable genes into eucaryotic cells and for producing proteinaceous products |
| US4695543A (en) | 1982-03-23 | 1987-09-22 | Bristol-Myers Company | Alpha Interferon GX-1 |
| US6936694B1 (en) | 1982-05-06 | 2005-08-30 | Intermune, Inc. | Manufacture and expression of large structural genes |
| EP0098110B1 (en) | 1982-06-24 | 1989-10-18 | NIHON CHEMICAL RESEARCH KABUSHIKI KAISHA also known as JAPAN CHEMICAL RESEARCH CO., LTD | Long-acting composition |
| US4470461A (en) | 1982-09-30 | 1984-09-11 | Phillips Petroleum Company | Organic nitro compounds as cosurfactants in enhanced oil recovery processes |
| US4588585A (en) * | 1982-10-19 | 1986-05-13 | Cetus Corporation | Human recombinant cysteine depleted interferon-β muteins |
| GB8334102D0 (en) | 1983-12-21 | 1984-02-01 | Searle & Co | Interferons with cysteine pattern |
| DE3572982D1 (en) | 1984-03-06 | 1989-10-19 | Takeda Chemical Industries Ltd | Chemically modified lymphokine and production thereof |
| GB8412564D0 (en) | 1984-05-17 | 1984-06-20 | Searle & Co | Structure and properties |
| CA1302320C (en) | 1984-06-11 | 1992-06-02 | Hideo Niwa | Expression of human cromosomal interferon-_ in animal cells |
| US5231176A (en) | 1984-08-27 | 1993-07-27 | Genentech, Inc. | Distinct family DNA encoding of human leukocyte interferons |
| FI90990C (en) | 1984-12-18 | 1994-04-25 | Boehringer Ingelheim Int | Recombinant DNA molecule, transformed host organism, and method for producing interferon |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US4885166A (en) | 1985-06-11 | 1989-12-05 | Ciba-Geigy Corporation | Hybrid interferons |
| US4766106A (en) | 1985-06-26 | 1988-08-23 | Cetus Corporation | Solubilization of proteins for pharmaceutical compositions using polymer conjugation |
| US4917888A (en) | 1985-06-26 | 1990-04-17 | Cetus Corporation | Solubilization of immunotoxins for pharmaceutical compositions using polymer conjugation |
| US4935233A (en) | 1985-12-02 | 1990-06-19 | G. D. Searle And Company | Covalently linked polypeptide cell modulators |
| JP2514950B2 (en) | 1986-03-10 | 1996-07-10 | エフ・ホフマン―ラ ロシユ アーゲー | Chemically modified protein, its production method and intermediate |
| JPS63152393A (en) | 1986-07-03 | 1988-06-24 | Takeda Chem Ind Ltd | Glycosyl derivative |
| US5183746A (en) | 1986-10-27 | 1993-02-02 | Schering Aktiengesellschaft | Formulation processes for pharmaceutical compositions of recombinant β- |
| US4894226A (en) | 1986-11-14 | 1990-01-16 | Cetus Corporation | Solubilization of proteins for pharmaceutical compositions using polyproline conjugation |
| US5004605A (en) | 1987-12-10 | 1991-04-02 | Cetus Corporation | Low pH pharmaceutical compositions of recombinant β-interferon |
| US4904584A (en) | 1987-12-23 | 1990-02-27 | Genetics Institute, Inc. | Site-specific homogeneous modification of polypeptides |
| US5135919A (en) | 1988-01-19 | 1992-08-04 | Children's Medical Center Corporation | Method and a pharmaceutical composition for the inhibition of angiogenesis |
| US4847325A (en) | 1988-01-20 | 1989-07-11 | Cetus Corporation | Conjugation of polymer to colony stimulating factor-1 |
| US4847625A (en) * | 1988-02-16 | 1989-07-11 | Ford Aerospace Corporation | Wideband, aperture-coupled microstrip antenna |
| CA1340810C (en) | 1988-03-31 | 1999-11-02 | Motoo Yamasaki | Polypeptide derivatives of human granulocyte colony stimulating factor |
| GB8824591D0 (en) | 1988-10-20 | 1988-11-23 | Royal Free Hosp School Med | Fractionation process |
| WO1990005534A1 (en) | 1988-11-23 | 1990-05-31 | Genentech, Inc. | Polypeptide derivatives |
| ATE135370T1 (en) | 1988-12-22 | 1996-03-15 | Kirin Amgen Inc | CHEMICALLY MODIFIED GRANULOCYTE COLONY EXCITING FACTOR |
| US5116964A (en) * | 1989-02-23 | 1992-05-26 | Genentech, Inc. | Hybrid immunoglobulins |
| US5166322A (en) * | 1989-04-21 | 1992-11-24 | Genetics Institute | Cysteine added variants of interleukin-3 and chemical modifications thereof |
| US5286637A (en) | 1989-08-07 | 1994-02-15 | Debiopharm, S.A. | Biologically active drug polymer derivatives and method for preparing same |
| JPH04218000A (en) | 1990-02-13 | 1992-08-07 | Kirin Amgen Inc | Modified polypeptide |
| US5252714A (en) | 1990-11-28 | 1993-10-12 | The University Of Alabama In Huntsville | Preparation and use of polyethylene glycol propionaldehyde |
| GEP20033082B (en) | 1991-03-15 | 2003-10-27 | Amgen Inc | Pegylation of Polypeptides |
| US5595732A (en) | 1991-03-25 | 1997-01-21 | Hoffmann-La Roche Inc. | Polyethylene-protein conjugates |
| AU2147192A (en) | 1991-06-28 | 1993-01-25 | Genentech Inc. | Method of stimulating immune response using growth hormone |
| US5281698A (en) | 1991-07-23 | 1994-01-25 | Cetus Oncology Corporation | Preparation of an activated polymer ester for protein conjugation |
| AU3423293A (en) | 1991-12-19 | 1993-07-19 | Baylor College Of Medicine | Pva or peg conjugates of peptides for epitope-specific immunosuppression |
| ZA933926B (en) | 1992-06-17 | 1994-01-03 | Amgen Inc | Polyoxymethylene-oxyethylene copolymers in conjuction with blomolecules |
| US5382657A (en) | 1992-08-26 | 1995-01-17 | Hoffmann-La Roche Inc. | Peg-interferon conjugates |
| WO1994005332A2 (en) | 1992-09-01 | 1994-03-17 | Berlex Laboratories, Inc. | Glycolation of glycosylated macromolecules |
| US5306344A (en) * | 1992-12-02 | 1994-04-26 | Edward C. Levy Company | Mixture of portland cement concrete materials for freeze/thaw resistance |
| US5298410A (en) | 1993-02-25 | 1994-03-29 | Sterling Winthrop Inc. | Lyophilized formulation of polyethylene oxide modified proteins with increased shelf-life |
| US5681811A (en) | 1993-05-10 | 1997-10-28 | Protein Delivery, Inc. | Conjugation-stabilized therapeutic agent compositions, delivery and diagnostic formulations comprising same, and method of making and using the same |
| US5359030A (en) | 1993-05-10 | 1994-10-25 | Protein Delivery, Inc. | Conjugation-stabilized polypeptide compositions, therapeutic delivery and diagnostic formulations comprising same, and method of making and using the same |
| US5874075A (en) | 1993-10-06 | 1999-02-23 | Amgen Inc. | Stable protein: phospholipid compositions and methods |
| US5641656A (en) | 1993-10-22 | 1997-06-24 | University Of Connecticut | Nucleic acids encoding avian interferon (IFN) proteins and recombinant methods using them |
| US5643575A (en) | 1993-10-27 | 1997-07-01 | Enzon, Inc. | Non-antigenic branched polymer conjugates |
| US5605976A (en) | 1995-05-15 | 1997-02-25 | Enzon, Inc. | Method of preparing polyalkylene oxide carboxylic acids |
| US5951974A (en) | 1993-11-10 | 1999-09-14 | Enzon, Inc. | Interferon polymer conjugates |
| EP1090645B1 (en) | 1994-02-08 | 2005-12-07 | Amgen Inc., | Oral delivery of chemically modified proteins |
| US5545723A (en) | 1994-03-15 | 1996-08-13 | Biogen Inc. | Muteins of IFN-β |
| US5639725A (en) | 1994-04-26 | 1997-06-17 | Children's Hospital Medical Center Corp. | Angiostatin protein |
| GB9415379D0 (en) | 1994-07-29 | 1994-09-21 | Smithkline Beecham Plc | Novel compounds |
| US5824784A (en) * | 1994-10-12 | 1998-10-20 | Amgen Inc. | N-terminally chemically modified protein compositions and methods |
| US5738846A (en) | 1994-11-10 | 1998-04-14 | Enzon, Inc. | Interferon polymer conjugates and process for preparing the same |
| WO1996017929A1 (en) | 1994-12-07 | 1996-06-13 | Novo Nordisk A/S | Polypeptide with reduced allergenicity |
| US5672662A (en) | 1995-07-07 | 1997-09-30 | Shearwater Polymers, Inc. | Poly(ethylene glycol) and related polymers monosubstituted with propionic or butanoic acids and functional derivatives thereof for biotechnical applications |
| US5702717A (en) | 1995-10-25 | 1997-12-30 | Macromed, Inc. | Thermosensitive biodegradable polymers based on poly(ether-ester)block copolymers |
| US5908621A (en) | 1995-11-02 | 1999-06-01 | Schering Corporation | Polyethylene glycol modified interferon therapy |
| US5723125A (en) | 1995-12-28 | 1998-03-03 | Tanox Biosystems, Inc. | Hybrid with interferon-alpha and an immunoglobulin Fc linked through a non-immunogenic peptide |
| US5747639A (en) | 1996-03-06 | 1998-05-05 | Amgen Boulder Inc. | Use of hydrophobic interaction chromatography to purify polyethylene glycols |
| JP2001508783A (en) | 1997-01-29 | 2001-07-03 | ポリマスク・ファーマシューティカルズ・パブリック・リミテッド・カンパニー | PEGylation method |
| US5990237A (en) | 1997-05-21 | 1999-11-23 | Shearwater Polymers, Inc. | Poly(ethylene glycol) aldehyde hydrates and related polymers and applications in modifying amines |
| ES2297889T3 (en) | 1997-07-14 | 2008-05-01 | Bolder Biotechnology, Inc. | DERIVATIVES OF HORMONE OF GROWTH AND RELATED PROTEINS. |
| US6180095B1 (en) | 1997-12-17 | 2001-01-30 | Enzon, Inc. | Polymeric prodrugs of amino- and hydroxyl-containing bioactive agents |
| US5981709A (en) | 1997-12-19 | 1999-11-09 | Enzon, Inc. | α-interferon-polymer-conjugates having enhanced biological activity and methods of preparing the same |
| US5985263A (en) | 1997-12-19 | 1999-11-16 | Enzon, Inc. | Substantially pure histidine-linked protein polymer conjugates |
| US5965119A (en) | 1997-12-30 | 1999-10-12 | Enzon, Inc. | Trialkyl-lock-facilitated polymeric prodrugs of amino-containing bioactive agents |
| DK1061954T3 (en) | 1998-03-12 | 2004-10-18 | Nektar Therapeutics Al Corp | Polyethylene glycol derivatives with proximal reactive groups |
| US6703381B1 (en) | 1998-08-14 | 2004-03-09 | Nobex Corporation | Methods for delivery therapeutic compounds across the blood-brain barrier |
| IL142350A0 (en) * | 1998-10-16 | 2002-03-10 | Biogen Inc | Interferon-beta fusion proteins and pharmaceutical compositions containing the same |
| EP2599503B1 (en) * | 1998-10-16 | 2017-05-17 | Biogen MA Inc. | Polymer conjugates of interferon beta-1A and uses thereof |
| US7144574B2 (en) | 1999-08-27 | 2006-12-05 | Maxygen Aps | Interferon β variants and conjugates |
| BR0013638A (en) * | 1999-08-27 | 2002-05-14 | Maxygen Aps | New interferon beta-like molecules |
| US6531122B1 (en) | 1999-08-27 | 2003-03-11 | Maxygen Aps | Interferon-β variants and conjugates |
| CA2394980C (en) | 1999-12-22 | 2008-05-13 | Shearwater Corporation | Sterically hindered derivatives of water soluble polymers |
| US9506008B2 (en) | 2013-12-23 | 2016-11-29 | Exxonmobil Research And Engineering Company | Method for improving engine fuel efficiency |
-
1999
- 1999-10-15 EP EP12198907.3A patent/EP2599503B1/en not_active Expired - Lifetime
- 1999-10-15 CA CA002345138A patent/CA2345138C/en not_active Expired - Lifetime
- 1999-10-15 CZ CZ20011329A patent/CZ299164B6/en not_active IP Right Cessation
- 1999-10-15 EE EEP200700058A patent/EE05201B1/en unknown
- 1999-10-15 JP JP2000576887A patent/JP4616478B2/en not_active Expired - Lifetime
- 1999-10-15 AT AT99970609T patent/ATE318149T1/en active
- 1999-10-15 PT PT60034154T patent/PT1656952E/en unknown
- 1999-10-15 HU HU0200444A patent/HU229888B1/en unknown
- 1999-10-15 EA EA200100446A patent/EA004789B9/en not_active IP Right Cessation
- 1999-10-15 ES ES10182477.9T patent/ES2653589T3/en not_active Expired - Lifetime
- 1999-10-15 PL PL347968A patent/PL206536B1/en not_active IP Right Cessation
- 1999-10-15 DK DK99970609T patent/DK1121156T3/en active
- 1999-10-15 BR BRPI9915542A patent/BRPI9915542C1/en not_active IP Right Cessation
- 1999-10-15 IL IL14228299A patent/IL142282A0/en active IP Right Grant
- 1999-10-15 DK DK06003415.4T patent/DK1656952T3/en active
- 1999-10-15 AU AU14465/00A patent/AU762616B2/en not_active Expired
- 1999-10-15 NZ NZ510689A patent/NZ510689A/en not_active IP Right Cessation
- 1999-10-15 TR TR2001/01086T patent/TR200101086T2/en unknown
- 1999-10-15 SG SG200302608A patent/SG110047A1/en unknown
- 1999-10-15 EP EP99970609A patent/EP1121156B1/en not_active Expired - Lifetime
- 1999-10-15 WO PCT/US1999/024201 patent/WO2000023114A2/en active IP Right Grant
- 1999-10-15 ES ES06003415.4T patent/ES2447772T3/en not_active Expired - Lifetime
- 1999-10-15 KR KR1020017004800A patent/KR100630849B1/en active Protection Beyond IP Right Term
- 1999-10-15 SK SK506-2001A patent/SK287918B6/en not_active IP Right Cessation
- 1999-10-15 EP EP10182477.9A patent/EP2260872B1/en not_active Expired - Lifetime
- 1999-10-15 SI SI9930890T patent/SI1121156T1/en unknown
- 1999-10-15 ES ES99970609T patent/ES2257884T3/en not_active Expired - Lifetime
- 1999-10-15 EE EEP200100220A patent/EE04967B1/en unknown
- 1999-10-15 ES ES12198907.3T patent/ES2630278T3/en not_active Expired - Lifetime
- 1999-10-15 SG SG2008070138A patent/SG2008070138A/en unknown
- 1999-10-15 DE DE69930015T patent/DE69930015T2/en not_active Expired - Lifetime
- 1999-10-15 CN CN200710112119.8A patent/CN101062419B/en not_active Expired - Lifetime
- 1999-10-15 EP EP06003415.4A patent/EP1656952B1/en not_active Expired - Lifetime
- 1999-10-15 CN CNB998122025A patent/CN1329082C/en not_active Expired - Lifetime
- 1999-10-15 PT PT99970609T patent/PT1121156E/en unknown
- 1999-10-15 LT LTEP12198907.3T patent/LT2599503T/en unknown
- 1999-10-15 SI SI9931074T patent/SI1656952T1/en unknown
-
2001
- 2001-03-27 IL IL142282A patent/IL142282A/en active Protection Beyond IP Right Term
- 2001-03-30 IS IS5913A patent/IS2926B/en unknown
- 2001-04-11 US US09/832,658 patent/US6962978B2/en not_active Expired - Lifetime
- 2001-04-11 NO NO20011860A patent/NO20011860L/en not_active Application Discontinuation
-
2002
- 2002-05-21 HK HK02103771.7A patent/HK1042434B/en not_active IP Right Cessation
-
2004
- 2004-03-16 US US10/802,540 patent/US7446173B2/en not_active Expired - Lifetime
-
2006
- 2006-05-02 CY CY20061100568T patent/CY1105022T1/en unknown
- 2006-10-12 JP JP2006279334A patent/JP2007063283A/en active Pending
- 2006-10-16 HK HK06111314.0A patent/HK1089393A1/en not_active IP Right Cessation
-
2008
- 2008-03-20 US US12/077,605 patent/US20090104150A1/en not_active Abandoned
-
2010
- 2010-09-14 JP JP2010206172A patent/JP5396358B2/en not_active Expired - Lifetime
-
2011
- 2011-02-10 JP JP2011027941A patent/JP2011093937A/en not_active Withdrawn
-
2012
- 2012-08-16 US US13/587,242 patent/US9314534B2/en not_active Expired - Fee Related
-
2014
- 2014-03-14 CY CY20141100207T patent/CY1115125T1/en unknown
- 2014-03-26 JP JP2014064230A patent/JP5863865B2/en not_active Expired - Lifetime
-
2016
- 2016-04-18 US US15/131,233 patent/US20160250340A1/en not_active Abandoned
-
2017
- 2017-10-05 LT LTPA2017030C patent/LTC2599503I2/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1987000056A1 (en) * | 1985-06-26 | 1987-01-15 | Cetus Corporation | Solubilization of proteins for pharmaceutical compositions using polymer conjugation |
| WO1995013090A1 (en) * | 1993-11-10 | 1995-05-18 | Enzon, Inc. | Improved interferon polymer conjugates |
| WO1999055377A2 (en) * | 1998-04-28 | 1999-11-04 | Applied Research Systems Ars Holding N.V. | Polyol-ifn-beta conjugates |
Cited By (134)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7238344B2 (en) | 1999-08-27 | 2007-07-03 | Maxygen, Inc. | Interferon-β variants and conjugates |
| US7338788B2 (en) | 1999-08-27 | 2008-03-04 | Maxygen Aps | Interferon-β variants and conjugates |
| US7144574B2 (en) | 1999-08-27 | 2006-12-05 | Maxygen Aps | Interferon β variants and conjugates |
| US6531122B1 (en) | 1999-08-27 | 2003-03-11 | Maxygen Aps | Interferon-β variants and conjugates |
| WO2001015736A2 (en) * | 1999-08-27 | 2001-03-08 | Maxygen Aps | Interferon-beta conjugates |
| WO2001015736A3 (en) * | 1999-08-27 | 2003-05-22 | Maxygen Aps | Interferon-beta conjugates |
| US7431921B2 (en) | 2000-04-14 | 2008-10-07 | Maxygen Aps | Interferon beta-like molecules |
| US8632771B2 (en) | 2000-06-30 | 2014-01-21 | Regents Of The University Of Minnesota | High molecular weight derivatives of vitamin K-dependent polypeptides |
| US9050372B2 (en) | 2000-06-30 | 2015-06-09 | Regents Of The University Of Minnesota | High molecular weight derivatives of vitamin K-dependent polypeptides |
| WO2002036628A2 (en) * | 2000-11-02 | 2002-05-10 | Maxygen Aps | New multimeric interferon beta polypeptides |
| WO2002036628A3 (en) * | 2000-11-02 | 2003-01-03 | Maxygen Aps | New multimeric interferon beta polypeptides |
| EP2080771A2 (en) | 2001-02-27 | 2009-07-22 | Maxygen Aps | New interferon beta-like molecules |
| JP2015044882A (en) * | 2001-04-09 | 2015-03-12 | ノバルティス バクシンズ アンド ダイアグノスティックス,インコーポレーテッド | HSA-FREE INTERFERON-β FORMULATIONS |
| EP2283897A3 (en) * | 2001-04-09 | 2011-06-15 | Novartis Vaccines and Diagnostics, Inc. | HSA-free formulations of interferon-beta |
| EP2283896A3 (en) * | 2001-04-09 | 2011-05-25 | Novartis Vaccines and Diagnostics, Inc. | HSA-free formulations of interferon-beta |
| CZ305994B6 (en) * | 2001-04-09 | 2016-06-08 | Novartis Vaccines & Diagnostics, Inc. | HSA-free compositions containing beta-interferon |
| US7696163B2 (en) | 2001-10-10 | 2010-04-13 | Novo Nordisk A/S | Erythropoietin: remodeling and glycoconjugation of erythropoietin |
| WO2003031464A3 (en) * | 2001-10-10 | 2006-03-02 | Neose Technologies Inc | Remodeling and glycoconjugation of peptides |
| AU2002360264B2 (en) * | 2001-10-10 | 2009-03-19 | Novo Nordisk A/S | Remodeling and glycoconjugation of peptides |
| US8076292B2 (en) | 2001-10-10 | 2011-12-13 | Novo Nordisk A/S | Factor VIII: remodeling and glycoconjugation of factor VIII |
| AU2009202405B2 (en) * | 2001-10-10 | 2011-09-22 | Novo Nordisk A/S | Remodelling and glycoconjugation of peptides |
| US7416858B2 (en) | 2001-10-10 | 2008-08-26 | Neose Technologies, Inc. | Pharmaceutical compositions of glycoconjugates |
| US8716240B2 (en) | 2001-10-10 | 2014-05-06 | Novo Nordisk A/S | Erythropoietin: remodeling and glycoconjugation of erythropoietin |
| US8716239B2 (en) | 2001-10-10 | 2014-05-06 | Novo Nordisk A/S | Granulocyte colony stimulating factor: remodeling and glycoconjugation G-CSF |
| NO339857B1 (en) * | 2002-01-18 | 2017-02-06 | Biogen Ma Inc | Conjugate containing polyalkylene glycol, pharmaceutical composition containing same and use of same for the treatment of disease |
| EP3669887A1 (en) * | 2002-01-18 | 2020-06-24 | Biogen MA Inc. | Polyalkylene polymer compounds and uses thereof |
| WO2003061577A3 (en) * | 2002-01-18 | 2003-10-02 | Biogen Inc | Polyalkylene glycol with moiety for conjugating biologically active compound |
| JP2016014022A (en) * | 2002-01-18 | 2016-01-28 | バイオゲン エムエー インコーポレイテッド | Polyalkylene glycol with moiety for conjugating biologically active compound |
| EP3025726A1 (en) * | 2002-01-18 | 2016-06-01 | Biogen MA Inc. | Polyalkylene polymer compounds and uses thereof |
| US8017733B2 (en) | 2002-01-18 | 2011-09-13 | Biogen Idec Ma Inc. | Polyalkylene polymer compounds and uses thereof |
| EA011829B1 (en) * | 2002-01-18 | 2009-06-30 | Байоджен Айдек Ма Инк. | Polyalkylene glycol having a moiety for conjugation of a biologically active compound |
| US8524660B2 (en) | 2002-01-18 | 2013-09-03 | Biogen Idec Ma Inc. | Polyalkylene polymer compounds and uses thereof |
| NO344612B1 (en) * | 2002-01-18 | 2020-02-10 | Biogen Ma Inc | Pharmaceutical composition comprising interferon beta (IFN β) and conjugate comprising reaction products of an activated polyalkylene glycol polymer and a process for their preparation. |
| JP2013136756A (en) * | 2002-01-18 | 2013-07-11 | Biogen Idec Ma Inc | Polyalkylene glycol with moiety for conjugating biologically active compound |
| JP2012255156A (en) * | 2002-01-18 | 2012-12-27 | Biogen Idec Ma Inc | Polyalkylene glycol with moiety for conjugating biologically active compound |
| EA009783B1 (en) * | 2002-01-18 | 2008-04-28 | Байоджен Айдек Ма Инк. | Polyalkylene glycolhaving a moiety for conjugation of a biologically active compound |
| WO2004020468A3 (en) * | 2002-08-28 | 2004-06-10 | Maxygen Aps | Interferon beta-like molecules for treatment of cancer |
| WO2004020468A2 (en) * | 2002-08-28 | 2004-03-11 | Maxygen Aps | Interferon beta-like molecules for treatment of cancer |
| EP1575531A2 (en) * | 2002-09-27 | 2005-09-21 | Biogen Idec MA Inc. | THERAPIES FOR CHRONIC INFLAMMATORY DEMYELINATING POLYNEUROPATHY USING INTERFERON-&bgr; |
| EP1575531A4 (en) * | 2002-09-27 | 2008-01-23 | Biogen Idec Inc | Therapies for chronic inflammatory demyelinating polyneuropathy using interferon-beta |
| US8129330B2 (en) | 2002-09-30 | 2012-03-06 | Mountain View Pharmaceuticals, Inc. | Polymer conjugates with decreased antigenicity, methods of preparation and uses thereof |
| US9125880B2 (en) | 2002-12-26 | 2015-09-08 | Mountain View Pharmaceuticals, Inc. | Polymer conjugates of interferon-beta with enhanced biological potency |
| EP1628618A4 (en) * | 2002-12-26 | 2009-09-09 | Mountain View Pharmaceuticals | Polymer conjugates of cytokines, chemokines, growth factors, polypeptide hormones and antagonists thereof with preserved receptor-binding activity |
| JP2011051991A (en) * | 2002-12-26 | 2011-03-17 | Mountain View Pharmaceuticals Inc | Polymer combo of antagonist in which cytokine, chemokine, growth factor, polypeptide hormone and receptor bonding activity are preserved |
| EP1667708A2 (en) * | 2002-12-26 | 2006-06-14 | Mountain View Pharmaceuticals, Inc. | Polymer conjugates of interferon- beta with enhanced biological potency |
| AU2003303636B2 (en) * | 2002-12-26 | 2010-08-05 | Mountain View Pharmaceuticals, Inc. | Polymer conjugates of cytokines, chemokines, growth factors, polypeptide hormones and antagonists thereof with preserved receptor-binding activity |
| EP1667708A4 (en) * | 2002-12-26 | 2009-01-14 | Mountain View Pharmaceuticals | Polymer conjugates of interferon- beta with enhanced biological potency |
| WO2004060299A2 (en) | 2002-12-26 | 2004-07-22 | Mountain View Pharmaceuticals, Inc. | Polymer conjugates of interferon-beta with enhanced biological potency |
| EP1628618A2 (en) * | 2002-12-26 | 2006-03-01 | Mountain View Pharmaceuticals, Inc. | Polymer conjugates of cytokines, chemokines, growth factors, polypeptide hormones and antagonists thereof with preserved receptor-binding activity |
| NO339917B1 (en) * | 2002-12-26 | 2017-02-13 | Mountain View Pharmaceuticals Inc | Method of increasing in vitro biological potency for non-glycosylated interferon beta with enhanced biological potency, conjugate, pharmaceutical composition and kit |
| JP2006519170A (en) * | 2002-12-26 | 2006-08-24 | マウンテン ビュー ファーマシューティカルズ,インコーポレイテッド | Polymer conjugates of cytokines, chemokines, growth factors, polypeptide hormones, and their antagonists with conserved receptor binding activity |
| TWI406672B (en) * | 2002-12-26 | 2013-09-01 | Mountain View Pharmaceuticals | Polymer conjugates of interferon-beta with enhanced biological potency |
| US8247381B2 (en) | 2003-03-14 | 2012-08-21 | Biogenerix Ag | Branched water-soluble polymers and their conjugates |
| US7803777B2 (en) | 2003-03-14 | 2010-09-28 | Biogenerix Ag | Branched water-soluble polymers and their conjugates |
| WO2004084949A3 (en) * | 2003-03-20 | 2004-12-29 | Xencor Inc | Generating protein pro-drugs using reversible ppg linkages |
| WO2004084949A2 (en) * | 2003-03-20 | 2004-10-07 | Xencor | Generating protein pro-drugs using reversible ppg linkages |
| AU2004224466B2 (en) * | 2003-03-28 | 2008-01-03 | Biopolymed Inc. | Biologically active material conjugated with biocompatible polymer with 1:1 complex, preparation method thereof and pharmaceutical composition comprising the same |
| EP1608408A1 (en) * | 2003-03-28 | 2005-12-28 | Biopolymed, Inc. | Biologically active material conjugated with biocompatible polymer with 1:1 complex, preparation method thereof and pharmaceutical composition comprising the same |
| EP1608408A4 (en) * | 2003-03-28 | 2008-02-27 | Biopolymed Inc | Biologically active material conjugated with biocompatible polymer with 1:1 complex, preparation method thereof and pharmaceutical composition comprising the same |
| US8791070B2 (en) | 2003-04-09 | 2014-07-29 | Novo Nordisk A/S | Glycopegylated factor IX |
| US8063015B2 (en) | 2003-04-09 | 2011-11-22 | Novo Nordisk A/S | Glycopegylation methods and proteins/peptides produced by the methods |
| US8853161B2 (en) | 2003-04-09 | 2014-10-07 | Novo Nordisk A/S | Glycopegylation methods and proteins/peptides produced by the methods |
| US7691603B2 (en) | 2003-04-09 | 2010-04-06 | Novo Nordisk A/S | Intracellular formation of peptide conjugates |
| US9005625B2 (en) | 2003-07-25 | 2015-04-14 | Novo Nordisk A/S | Antibody toxin conjugates |
| WO2005019260A1 (en) * | 2003-08-25 | 2005-03-03 | Toray Industries, Inc. | INTERFERON-β COMPOSITE |
| US7691975B2 (en) | 2003-08-25 | 2010-04-06 | Toray Industries, Inc. | Interferon-β complex |
| US8916360B2 (en) | 2003-11-24 | 2014-12-23 | Novo Nordisk A/S | Glycopegylated erythropoietin |
| US8632770B2 (en) | 2003-12-03 | 2014-01-21 | Novo Nordisk A/S | Glycopegylated factor IX |
| US8361961B2 (en) | 2004-01-08 | 2013-01-29 | Biogenerix Ag | O-linked glycosylation of peptides |
| US8232371B2 (en) | 2004-02-02 | 2012-07-31 | Ambrx, Inc. | Modified human interferon polypeptides and their uses |
| US8097702B2 (en) | 2004-02-02 | 2012-01-17 | Ambrx, Inc. | Modified human interferon polypeptides with at least one non-naturally encoded amino acid and their uses |
| US8791066B2 (en) | 2004-07-13 | 2014-07-29 | Novo Nordisk A/S | Branched PEG remodeling and glycosylation of glucagon-like peptide-1 [GLP-1] |
| US8268967B2 (en) | 2004-09-10 | 2012-09-18 | Novo Nordisk A/S | Glycopegylated interferon α |
| US9200049B2 (en) | 2004-10-29 | 2015-12-01 | Novo Nordisk A/S | Remodeling and glycopegylation of fibroblast growth factor (FGF) |
| US10874714B2 (en) | 2004-10-29 | 2020-12-29 | 89Bio Ltd. | Method of treating fibroblast growth factor 21 (FGF-21) deficiency |
| US9029331B2 (en) | 2005-01-10 | 2015-05-12 | Novo Nordisk A/S | Glycopegylated granulocyte colony stimulating factor |
| US8404809B2 (en) | 2005-05-25 | 2013-03-26 | Novo Nordisk A/S | Glycopegylated factor IX |
| US8911967B2 (en) | 2005-08-19 | 2014-12-16 | Novo Nordisk A/S | One pot desialylation and glycopegylation of therapeutic peptides |
| US8841439B2 (en) | 2005-11-03 | 2014-09-23 | Novo Nordisk A/S | Nucleotide sugar purification using membranes |
| US8067543B2 (en) | 2006-03-31 | 2011-11-29 | Baxter International Inc. | Factor VIII polymer conjugates |
| US8053561B2 (en) | 2006-03-31 | 2011-11-08 | Baxter International Inc. | Pegylated factor VIII |
| US8071724B2 (en) | 2006-03-31 | 2011-12-06 | Baxter International Inc. | Factor VIII polymer conjugates |
| US8071727B2 (en) | 2006-03-31 | 2011-12-06 | Baxter International Inc. | Factor VIII polymer conjugates |
| US7985839B2 (en) | 2006-03-31 | 2011-07-26 | Baxter International Inc. | Factor VIII polymer conjugates |
| US7985838B2 (en) | 2006-03-31 | 2011-07-26 | Baxter International Inc. | Factor VIII polymer conjugates |
| US8071725B2 (en) | 2006-03-31 | 2011-12-06 | Baxter International Inc. | Factor VIII polymer conjugates |
| US7645860B2 (en) | 2006-03-31 | 2010-01-12 | Baxter Healthcare S.A. | Factor VIII polymer conjugates |
| US7982010B2 (en) | 2006-03-31 | 2011-07-19 | Baxter International Inc. | Factor VIII polymer conjugates |
| US8071726B2 (en) | 2006-03-31 | 2011-12-06 | Baxter International Inc. | Factor VIII polymer conjugates |
| US8003760B2 (en) | 2006-03-31 | 2011-08-23 | Baxter International Inc. | Factor VIII polymer conjugates |
| US11020458B2 (en) | 2006-03-31 | 2021-06-01 | Takeda Pharmaceutical Company Limited | Factor VIII polymer conjugates |
| US8071728B2 (en) | 2006-03-31 | 2011-12-06 | Baxter International Inc. | Factor VIII polymer conjugates |
| WO2008025856A2 (en) | 2006-09-01 | 2008-03-06 | Novo Nordisk Health Care Ag | Modified glycoproteins |
| US9707298B2 (en) | 2006-09-15 | 2017-07-18 | Creabilis Therapeutics S.R.L. | Polymer conjugates of Box-A of HMGB1 and Box-A variants of HMGB1 |
| US8637007B2 (en) | 2006-12-15 | 2014-01-28 | Baxter International Inc. | Factor VIIa-polysialic acid conjugate having prolonged in vivo half-life |
| US9050304B2 (en) | 2007-04-03 | 2015-06-09 | Ratiopharm Gmbh | Methods of treatment using glycopegylated G-CSF |
| US8114630B2 (en) | 2007-05-02 | 2012-02-14 | Ambrx, Inc. | Modified interferon beta polypeptides and their uses |
| US9493499B2 (en) | 2007-06-12 | 2016-11-15 | Novo Nordisk A/S | Process for the production of purified cytidinemonophosphate-sialic acid-polyalkylene oxide (CMP-SA-PEG) as modified nucleotide sugars via anion exchange chromatography |
| WO2009080699A2 (en) * | 2007-12-20 | 2009-07-02 | Merck Serono S.A. | Peg-interferon-beta formulations |
| US9138403B2 (en) | 2007-12-20 | 2015-09-22 | Merck Serono Sa | PEG-interferon-beta formulations |
| WO2009080699A3 (en) * | 2007-12-20 | 2009-11-26 | Merck Serono S.A. | Peg-interferon-beta formulations |
| EP3103880A1 (en) | 2008-02-08 | 2016-12-14 | Ambrx, Inc. | Modified leptin polypeptides and their uses |
| US9150848B2 (en) | 2008-02-27 | 2015-10-06 | Novo Nordisk A/S | Conjugated factor VIII molecules |
| WO2010011735A2 (en) | 2008-07-23 | 2010-01-28 | Ambrx, Inc. | Modified bovine g-csf polypeptides and their uses |
| EP3225248A1 (en) | 2008-07-23 | 2017-10-04 | Ambrx, Inc. | Modified bovine g-csf polypeptides and their uses |
| US10138283B2 (en) | 2008-07-23 | 2018-11-27 | Ambrx, Inc. | Modified bovine G-CSF polypeptides and their uses |
| US10350301B2 (en) | 2009-07-27 | 2019-07-16 | Baxalta Incorporated | Blood coagulation protein conjugates |
| US8637640B2 (en) | 2009-07-27 | 2014-01-28 | Baxter International Inc. | Blood coagulation protein conjugates |
| US11564992B2 (en) | 2009-07-27 | 2023-01-31 | Takeda Pharmaceutical Company Limited | Nucleophilic catalysts for oxime linkage |
| US10414793B2 (en) | 2009-07-27 | 2019-09-17 | Baxalta Incorporated | Nucleophilic catalysts for oxime linkage |
| US8809501B2 (en) | 2009-07-27 | 2014-08-19 | Baxter International Inc. | Nucleophilic catalysts for oxime linkage |
| US10576160B2 (en) | 2009-07-27 | 2020-03-03 | Baxalta Incorporated | Nucleophilic catalysts for oxime linkage |
| US9731024B2 (en) | 2009-07-27 | 2017-08-15 | Baxalta Incorporated | Nucleophilic catalysts for oxime linkage |
| US11040109B2 (en) | 2009-07-27 | 2021-06-22 | Takeda Pharmaceutical Company Limited | Blood coagulation protein conjugates |
| US9795683B2 (en) | 2009-07-27 | 2017-10-24 | Lipoxen Technologies Limited | Glycopolysialylation of non-blood coagulation proteins |
| US9492555B2 (en) | 2009-07-27 | 2016-11-15 | Baxalta Incorporated | Nucleophilic catalysts for oxime linkage |
| EP2805965A1 (en) | 2009-12-21 | 2014-11-26 | Ambrx, Inc. | Modified porcine somatotropin polypeptides and their uses |
| EP2805964A1 (en) | 2009-12-21 | 2014-11-26 | Ambrx, Inc. | Modified bovine somatotropin polypeptides and their uses |
| CN101906213A (en) * | 2010-07-23 | 2010-12-08 | 华南理工大学 | Novel protein glycosylation grafting method |
| CN101906213B (en) * | 2010-07-23 | 2012-12-05 | 华南理工大学 | Novel protein glycosylation grafting method |
| US8642737B2 (en) | 2010-07-26 | 2014-02-04 | Baxter International Inc. | Nucleophilic catalysts for oxime linkage |
| US8945897B2 (en) | 2010-07-26 | 2015-02-03 | Baxter International Inc. | Materials and methods for conjugating a water soluble fatty acid derivative to a protein |
| US11273202B2 (en) | 2010-09-23 | 2022-03-15 | Elanco Us Inc. | Formulations for bovine granulocyte colony stimulating factor and variants thereof |
| EP2621516A4 (en) * | 2010-10-01 | 2014-04-09 | Biogen Idec Inc | Interferon-beta for use as monotherapy or in combination with other cancer therapies |
| EP2621516A2 (en) * | 2010-10-01 | 2013-08-07 | Biogen Idec MA Inc. | Interferon-beta for use as monotherapy or in combination with other cancer therapies |
| WO2013129549A1 (en) | 2012-02-29 | 2013-09-06 | 東レ株式会社 | Body cavity effusion suppressant |
| WO2017194783A1 (en) | 2016-05-13 | 2017-11-16 | Orionis Biosciences Nv | Targeted mutant interferon-beta and uses thereof |
| WO2018141964A1 (en) | 2017-02-06 | 2018-08-09 | Orionis Biosciences Nv | Targeted chimeric proteins and uses thereof |
| US11896643B2 (en) | 2018-02-05 | 2024-02-13 | Orionis Biosciences, Inc. | Fibroblast binding agents and use thereof |
| RU2739261C1 (en) * | 2019-12-31 | 2020-12-22 | Федеральное государственное бюджетное учреждение науки институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова Российской академии наук (ИБХ РАН) | Method for quantitative determination of anti-proliferative activity of human interferon-beta |
| WO2022200525A1 (en) | 2021-03-26 | 2022-09-29 | Innate Pharma | Multispecific proteins comprising an nkp46-binding site, a cancer antgienge binding site fused to a cytokine for nk cell engaging |
| WO2022258678A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkp30, a cytokine receptor, a tumour antigen and cd16a |
| WO2022258662A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkp46, a cytokine receptor, a tumour antigen and cd16a |
| WO2022258691A1 (en) | 2021-06-09 | 2022-12-15 | Innate Pharma | Multispecific proteins binding to nkg2d, a cytokine receptor, a tumour antigen and cd16a |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9314534B2 (en) | Polymer conjugates of interferon beta-1A and uses | |
| EP1121382B1 (en) | Interferon-beta fusion proteins and uses | |
| JP2002527491A5 (en) | ||
| EP1739090A2 (en) | Interferon-beta fusion proteins and uses | |
| MXPA01003791A (en) | Polymer conjugates of interferon beta-1a and uses |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 99812202.5 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2000 14465 Country of ref document: AU Kind code of ref document: A |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM EE ES FI GB GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM EE ES FI GB GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14465/00 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2345138 Country of ref document: CA Ref document number: 2345138 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 510689 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 142282 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09832658 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2000 576887 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 5062001 Country of ref document: SK Ref document number: PV2001-1329 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001/01086 Country of ref document: TR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020017004800 Country of ref document: KR Ref document number: PA/a/2001/003791 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200100446 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999970609 Country of ref document: EP |
|
| AK | Designated states |
Kind code of ref document: C2 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CR CU CZ DE DK DM EE ES FI GB GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: C2 Designated state(s): GH GM KE LS MW SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| COP | Corrected version of pamphlet |
Free format text: PAGES 59-63, CLAIMS, REPLACED BY NEW PAGES 59-63; PAGES 1/8-8/8, DRAWINGS, REPLACED BY NEW PAGES 1/10-10/10; DUE TO LATE TRANSMITTAL BY THE RECEIVING OFFICE |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999970609 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2001-1329 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020017004800 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 14465/00 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1999970609 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020017004800 Country of ref document: KR |