WO2000000469A2 - Alkyl ketones as potent anti-cancer agents - Google Patents
Alkyl ketones as potent anti-cancer agents Download PDFInfo
- Publication number
- WO2000000469A2 WO2000000469A2 PCT/US1999/014758 US9914758W WO0000469A2 WO 2000000469 A2 WO2000000469 A2 WO 2000000469A2 US 9914758 W US9914758 W US 9914758W WO 0000469 A2 WO0000469 A2 WO 0000469A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cysteine
- chloromethyl ketone
- ketone
- cys
- dodecyl
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/24—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/25—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/30—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by doubly-bound oxygen atoms
- C07C233/31—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by doubly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C245/00—Compounds containing chains of at least two nitrogen atoms with at least one nitrogen-to-nitrogen multiple bond
- C07C245/12—Diazo compounds, i.e. compounds having the free valencies of >N2 groups attached to the same carbon atom
- C07C245/14—Diazo compounds, i.e. compounds having the free valencies of >N2 groups attached to the same carbon atom having diazo groups bound to acyclic carbon atoms of a carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/18—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by doubly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/22—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/22—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and doubly-bound oxygen atoms bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/46—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having at least one of the nitrogen atoms, not being part of nitro or nitroso groups, further bound to other hetero atoms
- C07C323/48—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having at least one of the nitrogen atoms, not being part of nitro or nitroso groups, further bound to other hetero atoms to nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/51—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/52—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/51—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/57—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being further substituted by nitrogen atoms, not being part of nitro or nitroso groups
- C07C323/58—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being further substituted by nitrogen atoms, not being part of nitro or nitroso groups with amino groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/51—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/57—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being further substituted by nitrogen atoms, not being part of nitro or nitroso groups
- C07C323/58—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being further substituted by nitrogen atoms, not being part of nitro or nitroso groups with amino groups bound to the carbon skeleton
- C07C323/59—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being further substituted by nitrogen atoms, not being part of nitro or nitroso groups with amino groups bound to the carbon skeleton with acylated amino groups bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/51—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/60—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton with the carbon atom of at least one of the carboxyl groups bound to nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/02—Ortho- or ortho- and peri-condensed systems
- C07C2603/04—Ortho- or ortho- and peri-condensed systems containing three rings
- C07C2603/06—Ortho- or ortho- and peri-condensed systems containing three rings containing at least one ring with less than six ring members
- C07C2603/10—Ortho- or ortho- and peri-condensed systems containing three rings containing at least one ring with less than six ring members containing five-membered rings
- C07C2603/12—Ortho- or ortho- and peri-condensed systems containing three rings containing at least one ring with less than six ring members containing five-membered rings only one five-membered ring
- C07C2603/18—Fluorenes; Hydrogenated fluorenes
Definitions
- the present invention relates to alkyl ketone compounds effective for treating tumor cells and particularly effective to induce apoptosis in leukemia cells, breast cancer cells, prostate cancer cells, and brain cancer cells.
- Cancer is a major disease that continues as one of the leading causes of death at any age. In the United States alone, it is anticipated that more than a half a million Americans will die of cancer in 1999. Currently, radiotherapy and chemotherapy are two important methods used in the treatment of cancer.
- Novel alkyl ketone compounds have been found to be potent cytotoxic agents with potent activity against cancer cells.
- certain alkyl ketone compounds were found to exhibit potent cytotoxic activity, particularly against human breast cancer and leukemic cell lines, at micromolar concentrations. These compounds were also effective in inhibiting adhesion and invasion by cancer cells.
- the present invention includes novel compounds and compositions having potent cytotoxic activity.
- the present invention also includes methods for treating tumors by administering to a subject an effective amount of a compound of the invention to inhibit growth and/or induce apoptosis of tumor cells.
- Compositions of the invention contain an effective cytotoxic or inhibitory amount of a compound.
- the compounds of the invention have the following formula I:
- R 1 is H, hydroxyl, (C, - C 30 ) alkyl, (Q - C 30 ) alkenyl, (d - C 30 ) haloalkyl, (d - C 30 ) diazoalkyl, , -CH 2 O-C(O)R 5 , -NR 6 R 7 , or -CH 2 -S- R 9 wherein R 5 is independently aryl, (d - C 30 ) alkyl, (Ci - C 30 ) haloalkyl, (Ci - C 30 ) alkenyl, (Ci - C 30 ) diazoalkyl, (Q - C 24 ) cycloalkyl, or (Ci - C 24 ) cycloalkenyl,
- R 6 is independently H, (d - C 30 ) alkyl, (Ci - C 30 ) haloalkyl, (Ci - C 30 ) diazoalkyl, (Ci - C 30 ) alkenyl, (Ci - C 30 ) haloalkenyl, (Ci - C 2 ) cycloalkyl, or (Ci - C 2 ) cycloalkenyl;
- R 7 is -OR 8 , R 8 is independently (Ci - C 30 ) alkyl, (Ci - C 30 ) haloalkyl, (Ci - C 30 ) diazoalkyl, (Ci - C 30 ) alkenyl, (Ci - C 30 ) haloalkenyl, (Ci - C 2 ) cycloalkyl, or (Ci - C 24 ) cycloalkenyl;
- R 9 is independently (Ci - C 30 ) alkyl, (d - C 30 ) haloalkyl, (Ci - C 30 ) diazoalkyl, (Ci - C 30 ) alkenyl, (Ci - C 30 ) haloalkenyl, (C. - C 24 ) cycloalkyl, (Ci - C 24 ) cycloalkenyl, or -R 10 CO 2 H;
- R 10 is (Ci - C 30 ) alkyl or (Ci - C 30 ) alkenyl, R 2 is Ci or C 2 ; CH 2 or CH 2 CH 2
- R 3 is (C, - C 30 ) alkyl, (Q - C 30 ) haloalkyl, (d - C 30 ) alkenyl, (Ci - C 30 ) haloalkenyl, (Ci - C 2 ) cycloalkyl, (Ci - C 24 ) cycloalkenyl, (Ci - C 24 )aryl, anthroquinonylmethyl, naphthylmethyl, -SR , or -CR ;
- R 11 is independently (Q - C 30 ) alkyl, (d - C 30 ) haloalkyl, (d - C 30 ) alkenyl, or (Ci - C 0 ) haloalkenyl;
- R is aryl substituted methyl;
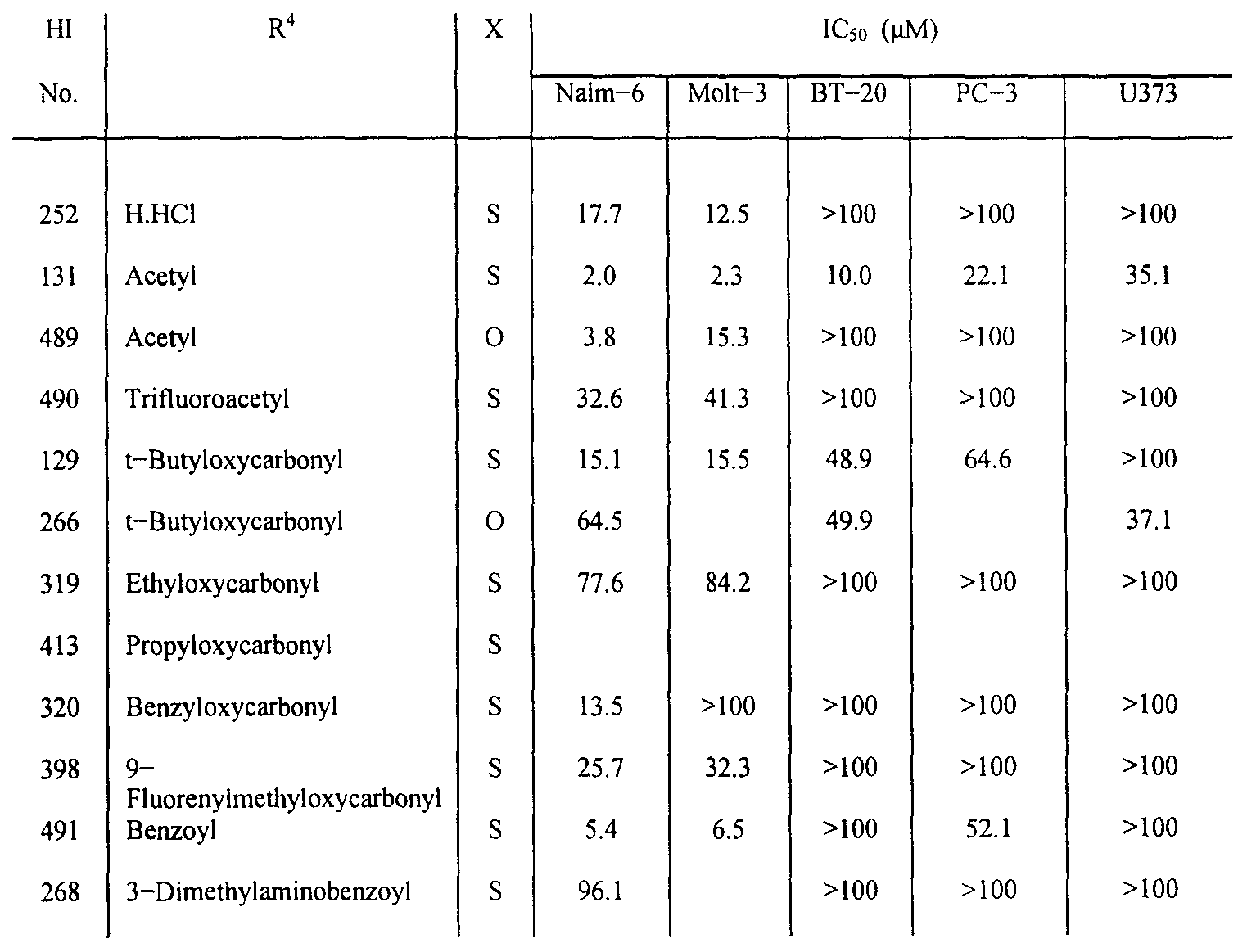
- R 4 is H, -C(O)R 13 , or -C(O)-O-R 14 ;
- R 13 and R 14 are each independently (Ci
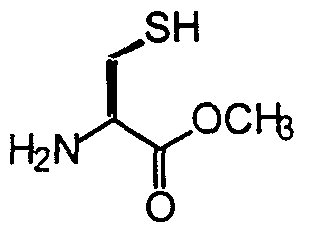
- Preferred compounds of the invention are those where p is the integer 1, R is a haloalkyl, R 2 is Ci, R 3 is a (Ci - C 22 ) alkyl, and R 4 is acetyl. Most preferred is the compound N-Ac-S-dodecyl-Cys chloromethyl ketone (HI— 131).
- Figure 1A is a graph showing survival of primary cancer cells taken from six children with leukemia and treated with different concentrations of compound HI— 131 as a function of drug concentration.
- Figure IB is a graph showing mean survival as a function of drug concentration from the data of Figure 1 A.
- Figures 2A-2F show photographs of apoptosis induced by HI-131 in treated human Leukemia cells.
- FIG. 1A NALM-6 control
- Figure 2B HI-131 treated NALM-6 cells
- FIG. 1 UPN1 control
- Figure 2D HI-131 treated UPN1 cells
- FIG.E UPN2 control
- Figure 2F HI-131 treated UPN2 cells.
- Figure 3 is a bar graph showing induction of apoptosis by HI-131 in treated primary leukemic cells and established NALM-6 and MOLT-3 cell lines.
- Figure 4 is a bar graph showing inhibition of invasive properties of human MDA-MB-231 breast cancer cells by HI-131.
- Figure 5 is a graph showing inhibition of the invasive properties of human U373 (glioblastoma) brain tumor cells by HI-131.
- Figure 6 is a graph showing inhibition of the adhesion of human MDA-
- MB-231 breast tumor cells by HI-131 MB-231 breast tumor cells by HI-131.
- Figure 7 is a graph showing inhibition of the adhesion by human U373 (glioblastoma) cells by HI-131. Detailed Description of the Invention
- the present invention includes novel alkyl ketone compounds having potent activity as cytotoxic agents.
- the compounds of the invention are useful agents for inhibiting growth or inducing apoptosis in tumor cells, for example, leukemia and breast tumor cells.
- alkyl includes both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms. As a preferred embodiment, chains of 1 to 22 carbon atoms are included.
- alkene includes both branched and straight chain aliphatic hydrocarbon groups that have at least one double bond.
- alkoxy includes, saturated and unsaturated, branched and straight chain aliphatic hydrocarbon groups having a specified number of carbon atoms where at least one carbon atom forms a single-bond to an oxygen atom.
- amine includes primary, secondary, and tertiary amines.
- halogen or “halo” substituent includes fluoro, chloro, bromo, and iodo.
- pharmaceutically acceptable salt thereof includes an acid addition salt or a base salt.
- pharmaceutically acceptable carrier includes any material which, when combined with a compound of the invention, allows the compound to retain biological activity, such as the ability to induce apoptosis of leukemia or breast tumor cells, and is non-reactive with the subject's immune system.
- examples include, but are not limited to, any of the standard pharmaceutical carriers such as a phosphate buffered saline solution, water, emulsions such as oil/water emulsions, and various types of wetting agents.
- Compositions comprising such carriers are formulated by well known conventional methods (see, for example, Remington's Pharmaceutical Sciences, Chapter 43, 14th Ed., Mack Publishing Co., Easton, PA).
- Substituted cycloalkyl includes cyclic hydrocarbons having substituents including halo, alkyl, alkenyl, oxyalkyl, oxyalkenyl, haloalkyl, haloalkenyl, and aryl.
- “Substituted cycloalkenyl” includes cyclic hydrocarbons having at least one double bond where substituents include halo, alkyl, alkenyl, oxyalkyl, oxyalkenyl, haloalkyl, haloalkenyl, and aryl.
- Substituted aryl includes aromatic hydrocarbons having substituents including hydroxyl, amino, aminomethyl, halo, alkyl, alkenyl, oxyalkyl, oxyalkenyl, haloalkyl, haloalkenyl, and aryl.
- Treating in the context of this invention means the prevention or reduction in severity of symptoms or effects of a pathological condition, including prolonging life expectancy.
- treatment includes prevention of tumor growth, reduction of tumor size, enhanced tumor cell death, and increased apoptosis.
- novel alkyl ketone compounds of the invention have the general structure represented by the following formula I:
- X is O or S
- R 1 is H, hydroxyl, (Ci - C 30 ) alkyl, (Ci - C 30 ) alkenyl, (d - C 30 ) haloalkyl, (d - C 30 ) diazoalkyl, , -CH 2 O-C(O)R 5 , -NR 6 R 7 , or -CH 2 -S-
- R y wherein R 5 is independently aryl, (d - C 30 ) alkyl, (d _ C 30 ) haloalkyl, (Ci - C 30 ) alkenyl, (Cj - C 30 ) diazoalkyl, (Ci - C 24 ) cycloalkyl, or (Ci - C 2 ) cycloalkenyl, R 6 is independently H, (Ci - C 30 ) alkyl, (C, - C 30 ) haloalkyl, (Q -
- C 30 diazoalkyl, (C ⁇ - C 30 ) alkenyl, (Ci - C 30 ) haloalkenyl, (Ci - C 4 ) cycloalkyl, or (Ci - C 24 ) cycloalkenyl;
- R 7 is -OR 8 , R 8 is independently (Ci - C 30 ) alkyl, (Ci - C 30 ) haloalkyl, (Ci - C 30 ) diazoalkyl, (Ci - C 30 ) alkenyl, (d - C 30 ) haloalkenyl, (d - C 24 ) cycloalkyl, or (Ci - C2 4 ) cycloalkenyl;
- R 9 is independently (Ci - C 30 ) alkyl, (Ci - C 30 ) haloalkyl, (Ci - C 30 ) diazoalkyl, (Ci - C 30 ) alkenyl, (Ci ⁇ C 30 ) haloalkenyl, (Ci - C 24 ) cycloalkyl, (Ci - C 24 ) cycloalkenyl, or -R 10 CO 2 H;
- R 10 is (Ci - C 30 ) alkyl or (Ci - C 30 ) alkenyl
- R 2 is d or C 2 ; CH 2 or CH 2 CH 2
- R 3 is (d - C 30 ) alkyl, (d - C 30 ) haloalkyl, (d - C 30 ) alkenyl, (d - C 30 ) haloalkenyl, (Ci - C 2 ) cycloalkyl, (Cj - C 24 ) cycloalkenyl, (Ci - C 24 )aryl, anthroquinonylmethyl, naphthylmethyl, -SR , or -CR ;
- R 11 is independently (Ci - C 30 ) alkyl, (Ci - C 30 ) haloalkyl, (Ci - C 0 ) alkenyl, or (d _ C 30 ) haloalkenyl;
- R is aryl substituted methyl;
- R 4 is H, -C(O)R 13 , or -C(O)-O-R 14 ;
- R 13 and R 14 are each independently (Q - C ⁇ 2 ) alkyl, (Ci - Cj ) haloalkyl, (Ci - C ⁇ 2 ) alkenyl, (Ci - C ⁇ 2 ) haloalkenyl, (C 3 - C 12 ) cycloalkyl, or (C 3 - C ⁇ 2 ) cycloalkenyl; or a pharmaceutically acceptable acid addition salt thereof.
- the compounds of formula I are useful for the treatment of cancer, particularly the treatment of leukemia and breast cancer. In the method of the invention, a therapeutic amount of a compound of formula I is administered to a patient for the treatment of cancer.
- a preferred compound of the invention has the structure of formula II:
- a preferred embodiment of the compound of Formula II is that shown as having formula III, where X is S, R 3 is dodecyl, and R 4 is acetyl:
- R 1 is most preferably chloromethyl;
- R 2 is preferably CH 2 ;
- R 3 is preferably a C 12 alkyl;
- R 4 is preferably acetyl; and
- X is preferably S.
- a most preferred compound of formula II is N- Ac-S-dodecy 1-Cys chloromethyl ketone (HI- 131).
- Preferred compounds of the invention having potent anti-cancer affects are the following:
- N-Ac-S-dodecyl-Cys chloromethyl ketone HI-131
- N-Ac-S-pentyl-cysteine chloromethyl ketone HI-224
- N-Ac-S-octyl-cysteine chloromethyl ketone HI-352
- N-Ac-S-tetradecyl-cysteine chloromethyl ketone HI-354
- N-Ac-S-hexadecyl-cysteine chloromethyl ketone (HI-366); N-Ac-S-tr ⁇ ra— tr ans-farnesyl-Cys diazomethyl ketone (HI-367);
- N-Ac-S-benzyloxycarbonyl-cysteine chloromethyl ketone (HI-389); N-Ac-S-2-naphthylmethyl-cysteine chloromethyl ketone (HI-392);
- N-Ac-S-dodecyl-cysteine bromomethyl ketone (HI-488); N-Ac-O-dodecyl-serine chloromethyl ketone (HI-489); N-Trifluoroacetyl-S-dodecyl-cysteine chloromethyl ketone (HI-490); N-Benzoyl-S-dodecyl-cysteine chloromethyl ketone (HI-491).
- Base salts are formed with metals or amines, such as alkali and alkaline earth metals or organic amines.
- metals used as cations are sodium, potassium, magnesium, calcium, and the like.
- heavy metal salts such as for example silver, zinc, cobalt, and cerium.
- suitable amines are N,N'- dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamene, N-methylglucamine, and procaine.
- Pharmaceutically acceptable acid addition salts are formed with organic and inorganic acids.
- suitable acids for salt formation are hydrochloric, suliuric, phosphoric, acetic, citric, oxalic, malonic, salicylic, malic, gluconic, fumaric, succinic, ascorbic, maleic, methanesulfonic, and the like.
- the salts are prepared by contacting the free base form with a sufficient amount of the desired acid to produce either a mono or di, etc. salt in the conventional manner.
- the free base forms may be regenerated by treating the salt form with a base. For example, dilute solutions of aqueous base may be utilized.
- Dilute aqueous sodium hydroxide, potassium carbonate, ammonia, and sodium bicarbonate solutions are suitable for this purpose.
- the free base forms differ from their respective salt forms somewhat in certain physical properties such as solubility in polar solvents, but the salts are otherwise equivalent to their respective free base forms for the purposes of the invention.
- the compounds of the invention are effective cytotoxic agents, for example, against tumor cells such as leukemic and breast cancer cells.
- the cytotoxic effects of alkyl ketone compounds are achieved by contacting cells, such as tumor cells, with micromolar amounts of the inhibitory compound.
- a particularly useful anti-tumor agent is N- Ac-S- dodecy 1-Cys chloromethyl ketone (HI-131) as shown in the Examples below.
- the compounds of the invention can be used in methods of tumor treatment, for example, by administering to a subject a compound of the invention in order to achieve an inhibition of tumor cell growth, a killing of tumor cells, induction of apoptosis, and/or increased patient survival time.
- cytotoxic compounds of the invention are suitable for use in mammals.
- mammals means any class of higher vertebrates that nourish their young with milk secreted by mammary glands, including, for example, humans, rabbits, and monkeys.
- Apoptosis or programmed cellular death, is an active process requiring new protein synthesis. Typically, the process requires ATP, involves new RNA and protein synthesis, and culminates in the activation of endogenous endonucleases that degrade the DNA of the cell, thereby destroying the genetic template required for cellular homeostasis.
- Apoptosis is observed in controlled deletion of cells during metamorphosis, differentiation, and general cell turnover and appears normally to be regulated by receptor-coupled events. For these reasons, apoptosis has been called "programmed cell death" or "cell suicide.” While every cell likely has the genetic program to commit suicide, it is usually suppressed. Under normal circumstances, only those cells no longer required by the organism activate this self-destruction program.
- Apoptotic cell death is characterized by plasma membrane blebbing, cell volume loss, nuclear condensation, and endonucleolytic degradation of DNA at nucleosome intervals. Loss of plasma membrane integrity is a relatively late event in apoptosis, unlike the form of cell death termed necrosis, which can be caused by hypoxia and exposure to certain toxins and which is typically characterized, early-on by increased membrane permeability and cell rupture. As demonstrated in the Examples, the alkyl ketone compounds of the invention are effective agents for inducing apoptosis in tumor cells.
- the compounds of the present invention can be formulated as pharmaceutical compositions and administered to a mammalian host, including a human patient, in a variety of forms adapted to the chosen route of administration.
- the compounds are preferably administered in combination with a pharmaceutically acceptable carrier, and may be combined with or conjugated to specific delivery agents, including targeting antibodies and/or cytokines.
- the compounds can be administered by known techniques, such as orally, parentally (including subcutaneous injection, intravenous, intramuscular, intrasternal or infusion techniques), by inhalation spray, topically, by absorption through a mucous membrane, or rectally, in dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants or vehicles.
- Pharmaceutical compositions of the invention can be in the form of suspensions or tablets suitable for oral administration, nasal sprays, creams, sterile injectable preparations, such as sterile injectable aqueous or oleagenous suspensions or suppositories.
- compositions can be prepared according to techniques well-known in the art of pharmaceutical formulation.
- the compositions can contain microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners or flavoring agents.
- the compositions can contain microcrystalline cellulose, starch, magnesium stearate and lactose or other excipients, binders, extenders, disintegrants, diluents and lubricants known in the art.
- compositions can be prepared according to techniques well-known in the art of pharmaceutical formulation.
- the compositions can be prepared as solutions in saline, using benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons or other solubilizing or dispersing agents known in the art.
- compositions can be formulated according to techniques well-known in the art, using suitable dispersing or wetting and suspending agents, such as sterile oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- suitable dispersing or wetting and suspending agents such as sterile oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- compositions can be prepared by mixing with a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ambient temperatures, but liquefy or dissolve in the rectal cavity to release the drug.
- a suitable non-irritating excipient such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ambient temperatures, but liquefy or dissolve in the rectal cavity to release the drug.
- Preferred administration routes include orally, parenterally, as well as intravenous, intramuscular or subcutaneous routes.
- the compounds of the present invention are administered parenterally, i.e., intravenously or intraperitoneally, by infusion or injection.
- the compounds may be administered directly to a tumor by tumor injection; or by systemic delivery by intravenous injection.
- Solutions or suspensions of the compounds can be prepared in water, isotonic saline (PBS) and optionally mixed with a nontoxic surfactant. Dispersions may also be prepared in glycerol, liquid polyethylene, glycols, DNA, vegetable oils, triacetin and mixtures thereof. Under ordinary conditions of storage and use, these preparations may contain a preservative to prevent the growth of microorganisms.
- the pharmaceutical dosage form suitable for injection or infusion use can include sterile, aqueous solutions or dispersions or sterile powders comprising an active ingredient which are adapted for the extemporaneous preparation of sterile injectable or infusible solutions or dispersions.
- the liquid carrier or vehicle can be a solvent or liquid dispersion medium comprising, for example, water, ethanol, a polyol such as glycerol, propylene glycol, or liquid polyethylene glycols and the like, vegetable oils, nontoxic glyceryl esters, and suitable mixtures thereof.
- the proper fluidity can be maintained, for example, by the formation of liposomes, by the maintenance of the required particle size, in the case of dispersion, or by the use of nontoxic surfactants.
- the prevention of the action of microorganisms can be accomplished by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, thimerosal, and the like.
- isotonic agents for example, sugars, buffers, or sodium chloride.
- Prolonged absorption of the injectable compositions can be brought about by the inclusion in the composition of agents delaying absorption — for example, aluminum monosterate hydrogels and gelatin.
- Sterile injectable solutions are prepared by incorporating the conjugates in the required amount in the appropriate solvent with various other ingredients as enumerated above and, as required, followed by filter sterilization.
- the preferred methods of preparation are vacuum drying and freeze-drying techniques, which yield a powder of the active ingredient plus any additional desired ingredient present in the previously sterile-filtered solutions.
- the compound of the invention can be targeted for specific delivery to the cells to be treated by conjugation of the compounds to a targeting moiety.
- Targeting moiety useful for conjugation to the compounds of the invention include antibodies, cytokines, and receptor ligands expressed on the cells to be treated.
- conjugate means a complex formed with two or more compounds.
- targeting moiety means a compound which serves to deliver the compound of the invention to a specific site for the desired activity.
- Targeting moieties include, for example, molecules which specifically bind molecules present on a cell surface.
- Such targeting moieties useful in the invention include anti-cell surface antigen antibodies.
- Cytokines, including interleukins, factors such as epidermal growth factor (EGF), and the like, are also specific targeting moieties known to bind cells expressing high levels of their receptors.
- Particularly useful targeting moieties for targeting the compounds of the invention to cells for therapeutic activity include those ligands that bind antigens or receptors present on the tumor cells to be treated.
- antigens present on B-lineage cancer cells such as CD 19, can be targeted with anti-CD 19 antibodies such as B43.
- Antibody fragments, including single chain fragments, can also be used.
- IL4 can also be used to target B-cells.
- Cancer cells expressing EGF or IGF receptors can be targeted with the binding ligand.
- Other such ligand-receptor binding pairs are known in the scientific literature for specific cancers. Methods for producing conjugates of the compounds of the invention and the targeting moieties are known.
- the administered dose is that effective to have the desired effect, such as sufficient to reduce or eliminate tumors.
- Appropriate amounts can be determined by those skilled in the art, extrapolating using known methods and relationships, from the in vitro data provided in the Examples.
- the dose of the novel alkyl ketone compounds effective to achieve tumor cell apoptosis, reduction in tumors, and increased survival time is 1-100 mg/kg body weight/dose for a direct targeted administration.
- the effective dose to be administered will vary with conditions specific to each patient. In general, factors such as the disease burden, tumor location (exposed or remote), host age, metabolism, sickness, prior exposure to drugs, and the like contribute to the expected effectiveness of a drug. One skilled in the art will use standard procedures and patient analysis to calculate the appropriate dose, extrapolating from the data provided in the Examples. In general, a dose which delivers about 1-100 mg/kg body weight is expected to be effective, although more or less may be useful.
- compositions of the invention may be administered in combination with other anti-tumor therapies.
- the administered dose of the alkyl ketone compounds may be less than for single drug therapy.
- Tables 3 and 6 and the compounds in Tables 4 and 5 are also described in detail below. All chemicals were purchased from Aldrich Chemical Company (Milwaukee, Wisconsin) and used directly for synthesis without further purification. Anhydrous tetrahydrofuran was dried over sodium and distilled immediately prior to use. Column chromatography was performed using 230-400 mesh silica gel obtained from the Merck Company, with eluant as noted in the experimental procedure.
- the first step of scheme 1 was isoprenylation of the thiol group of N ⁇ Ac- Cys-OH by reaction of the appropriate isoprenyl bromide in a 4 M solution of ammonia in methanol according to the method of Brown and co-workers. This step was carried out in the presence of ethyl acetate when N-Ac-Cys-OH was dodecylated in order to solvate the bromododecane.
- the S- alkylated acid (2a) was activated as the mixed anhydride derivative using isobutylchloroformate and converted to the diazomethyl ketone (3 a) by treatment with diazomethane.
- the diazomethane was converted to the chloromethyl ketone (4a) with HCI in ethyl acetate at 0 °C for 10 minutes.
- the chloromethyl compounds in Tables 1 and 2, 4a-4g, were made from the analogous diazomethyl compounds, 3a-3g, by replacement of the diazomethyl group with a chloromethyl group. The specific synthesis of each two member analogous group will therefore be considered together.
- N-Ac-Cys-OH (1) (0.82 g, 5 mmol) in 4 M ammonia in methanol (35 mL) at 0 °C.
- the reaction was stirred at 0 °C for 3 h then at room temperature for 1 h.
- the solvent was removed under reduced pressure and the residue partitioned between 1- butanol and water.
- the butanol layer was dried (MgSO 4 ) and the solvent removed under reduced pressure.
- the residue was redissolved in methanol and washed with hexane.
- the methanol was then removed under reduced pressure to give N-Ac-S- trans- traws-farnesyl-Cys-OH (2a).
- N-Ac-S-rrar ⁇ -/r ⁇ «s-farnesyl-Cys-OH (2a) (1.84 g, 5 mmol) produced in the previous step was dissolved in dry THF (30 mL) and cooled to - 15 °C. 4-methyl morpholine (0.51 g, 0.55 mL, 5 mmol) and z ' so-butyl chloroformate (0.68 g, 0.65 mL, 5 mmol) were added to the solution. The mixture was stirred at -15 °C for 5 minutes before being filtered by gravity into a solution of diazomethane in ethanolic ether (11 mmol, 30 mL) cooled in an ice bath.
- N-Ac-S- r ⁇ «s-geranyl-Cys-OH (2c) was prepared as described above for N-Ac-S-tttms-frOra-farnesyl-Cys-OH (2a) except that trans-geranyl bromide was used instead of farnesyl bromide.
- N-Ac-S-fr- ⁇ —geranyl-Cys diazomethyl ketone (HI 122) (3c) was prepared as described above for N-AcS-tr ⁇ ns-tr ⁇ ns-famesy 1-Cys diazomethyl ketone (3 a) except that N-Ac-S-/r ⁇ r ⁇ -geranyl-Cys-OH (2c) (2.99 g, 10 mmol) was used instead of N-Ac-S-fr" ⁇ «s-/r ⁇ r ⁇ -farnesyl-Cys-OH (2a).
- N-Ac-S-fr- ⁇ r ⁇ — geranyl-Cys chloromethyl ketone (HI 127) (4c) was prepared as described above for N-Ac-S-tr ⁇ ns- tr ⁇ ns-famesy ⁇ -Cys chloromethyl ketone (4a) except that N-Ac-S-fr- ⁇ r ⁇ -geranyl-Cys diazomethyl ketone (3c) (0.10 g, 0.3 mmol) was used instead of ⁇ N- AcS-tr ⁇ ns- trans- famesy 1-Cys diazomethyl ketone (3a).
- N-Ac-S-(3-methyl-2-butenyl)-Cys-OH (2d) was prepared as described above for N-Ac-S-tr ⁇ s—fr ⁇ TM-farnesyl-Cys-OH (2a) except that 4-bromo-2- methyl-2-butene was used instead of farnesyl bromide.
- N-Ac-S-(3-methyl-2-butenyl)-Cys diazomethyl ketone (3d) was prepared as described above for N-Ac-S-tr ⁇ ns-tr ⁇ ns-famesy ⁇ -Cys diazomethyl ketone (3a) except that N-Ac-S-(3-methyl-2-butenyl)-Cys-OH (2d) (0.69 g, 3 mmol) was used instead of N-AcS-trans- trans- farnesyl-Cys-OH (2a).
- N-Ac-S-(3-methyl-2-butenyl)-Cys chloromethyl ketone (4d) was prepared as described above for ⁇ N-Ac-S-trans-trans-farnesy ⁇ -Cys chloromethyl ketone (4a) except that N-Ac-S-(3-methyl-2-butenyl)-Cys diazomethyl ketone (3d) (0.30 g, 1.2 mmol) was used instead of N-Ac-S-tr ⁇ fls-tr ⁇ ra-farnesyl-Cys diazomethyl ketone (3a).
- N-Ac-Cys-OH (1.62 g, 9.9 mmol) and bromododecane (2.5 g, 10 mmol) were dissolved in a mixture of ethyl acetate (30 mL) and methanol (20 mL).
- the solvent was then removed under reduced pressure and the residue was partitioned between ethyl acetate and 1 M HCI.
- the organic layer was dried over MgSO and the solvent removed under reduced pressure to give N-Ac-S-dodecyl-Cys-OH (2e).
- N-Ac-S-dodecyl-Cys diazomethyl ketone (3e) was prepared as described above for N-AcS-trans- trans-famesy 1-Cys diazomethyl ketone (3a) except that N-Ac-S-dodecyl-Cys-OH (2e) (0.96 g, 2.9 mmol) was used instead ofN-Ac-S- trans- trans-famesyl-Cys-OH (2a).
- the crude product was purified by chromatography on silica gel (1 :3 ethyl acetate/hexane) to give N-Ac-S-dodecyl- Cys diazomethyl ketone (3e).
- N-Ac-S-dodecyl-Cys chloromethyl ketone (4e) was prepared as described above for N-Ac-S-farnesyl-Cys chloromethyl ketone (4a) except that N-Ac-S- dodecyl-Cys diazomethyl ketone (3e) (0.19 g, 0.5 mmol) was used instead of N- Ac-S-tr ⁇ ns- tr ⁇ ns-famesy 1-Cys diazomethyl ketone (3a). N-Ac-S-dodecyl-Cys chloromethyl ketone (4e) was obtained after removal of solvent. 5. N-Boc-S-farnesyl-Cys diazomethyl ketone (HI-82) (3b) and N-Boc-S-farnesyl-Cys chloromethyl ketone (HI-124) (4b)
- S-tr ⁇ r ⁇ - ⁇ r ⁇ r ⁇ -Farnesyl-mercaptoethyl diazomethyl ketone (3f) was prepared as described above for N-AcS-trans-trans-famesy 1-Cys diazomethyl ketone (3a) except that 3-(S-tr ⁇ ns- trans- Farnesyl)-mercaptopropionic acid (2f) was used instead of N-Ac-S-t w- tr ans-famesyl-Cys-OH (2a).
- S-tr ra-?r ⁇ /75—Farnesyl-mercaptomethyl diazomethyl ketone (3g) was prepared as described above for N-Ac-S-tr ⁇ ns- trans- famesy 1-Cys diazomethyl ketone (3a) except that S- rar ⁇ - ⁇ r ⁇ ra-Farnesyl-mercaptoacetic acid (2g) (0.71 g, 2.4 mmol) was used instead of N-Ac-S-fr ⁇ ra , -fr" ⁇ ms , -farnesyl-Cys-OH (2a).
- the first step in scheme 2 was the dodecylation of cysteine methyl ester in a mixture of ethyl acetate and methanol. This was followed by Boc-protection, ester hydrolysis and conversion in turn to the chloromethyl ketone (8).
- S-Dodecyl-Cys chloromethyl ketone hydrochloride (9) was prepared from N-Boc-S-dodecyl-Cys chloromethyl ketone (8), by simply deprotecting the Boc group in a saturated solution of HCI in ethyl acetate.
- Boc-S-dodecyl-Cys-OCH 3 was obtained after removal of solvent.
- N-Boc-S-dodecyl-Cys diazomethyl ketone was prepared as described above for N-AcS-trans- trans-famesy 1-Cys diazomethyl ketone (3 a) except that N-Boc-S-dodecyl-Cys-OH (7) (0.89 g, 2.3 mmol) was used instead of N-Ac-S- trans- trans- farnesyl-Cys-OH (2a).
- the crude product was purified by chromatography on silica gel (1:2 ethyl acetate/hexane) to give N-Boc-S-dodecyl-
- N-Boc-S-dodecyl-Cys chloromethyl ketone (8) was prepared as described above for N-Ac-S-tr ⁇ r ⁇ -fr- ⁇ r ⁇ —farnesyl-Cys chloromethyl ketone (4a) except that N-Boc-S-dodecyl-Cys diazomethyl ketone (0.56 g, 1.4 mmol) was used instead of N-Ac-S-tr ⁇ , -fr * ⁇ — farnesyl-Cys diazomethyl ketone (3a). N-Boc-S-dodecyl- Cys chloromethyl ketone (8) was obtained upon removal of solvent.
- the first step in scheme 3 was the farnesylation of cysteine methyl ester according to the method of Brown et al.
- the farnesylated cysteine methyl ester was then coupled with N-Boc-Gly-OH using EDC/HOBt.
- the ester was hydrolyzed to the acid (11) and the chloromethyl ketone prepared via the diazomethyl ketone (12).
- N-Boc-Gly-S- r ⁇ r ⁇ - ⁇ rara-farnesyl-Cys-OCH (1.25 g, 2.52 mmol) was dissolved in methanol (70 mL) and then a solution of NaOH (3 M, 2.5 mL, 7.56 mmol) at 0 °C was added. The solution was stirred overnight at the same temperature. The solvent was then removed and the residue was dissolved in water (50 mL) that was subsequently acidified to pH 5.
- N-Boc-Gly-S-fr-flra—tr ⁇ ra-farnesy 1-Cys diazomethyl ketone (12) was prepared as described above for N- Ac-S-tr ans- trans- famesy 1-Cys chloromethyl ketone (4a) except that N-Boc-Gly-S- ⁇ s-frara'-farnesyl-Cys-OH (11) (0.39 g, 0.8 mmol) was used instead of N-AcS-tr ⁇ ns-tr ⁇ ns-farnesy ⁇ -Cys diazomethyl ketone (3a).
- N-Boc-Gly-S- r ra- fr ⁇ ra-farnesyl-Cys chloromethyl ketone (13) was prepared as described above for N-Ac-S-tr ⁇ —tr ⁇ «s-farnesy 1-Cys chloromethyl ketone (4a) except that N-Boc-Gly-S-fr ⁇ r ⁇ -tra/75-farnesy 1-Cys diazomethyl ketone (0.21 g, 0.41 mmol) was used instead of ⁇ -Ac-S-tr ⁇ ns- r ⁇ ns-fa esyl-Cys diazomethyl ketone (3 a).
- the straight chain alkyl ketone derivatives (4h-y) were synthesized by a modification of the standard literature procedure. Previously, the standard conditions for making diazomethyl ketones were used, but a closer study of the dodecyl derivative, N-Ac-S-dodecyl-Cys chloromethyl ketone (4e), showed significant formation of the methyl ester as a side-product. Presumably, the mixed anhydride intermediate either could not completely form or was hydrolyzed back to the acid before the diazomethane could react with it. Conducting the mixed anhydride formation at -78 °C increased the stability and longevity of the mixed anhydride and improved yield.
- NMM (0.20 g, 0.22 mL, 2 mmol) and isobutyl chloroformate (0.27 g, 0.26 mL, 2 mmol) was added to the desired N-Ac-S-alkyl-cysteine (2 mmol) in anhydrous THF (20 mL) that had been cooled to -78 °C. The solution was then stirred at -78 °C for 20 min. A solution of diazomethane in ethanolic ether (10 mL) was carefully added and the solution allowed to slowly warm to room temperature. Further portions of diazomethane solution were added until a yellow color persisted.
- the solution was diluted with ether, washed with water and sodium bicarbonate solution and then dried over anhydrous MgSO .
- the solvent was removed under reduced pressure to yield the crude diazomethyl ketone compound.
- the diazomethyl ketone was dissolved in ethyl acetate (20 mL) and cooled in an ice bath.
- a solution of HCI in ethyl acetate (2M, 2 mL) was added and the solution stirred in ice for 5 min until no more diazomethyl ketone could be observed by TLC.
- the solvent was removed under reduced pressure and the residue purified by chromatography on silica gel (ethyl acetate/hexane) to give the pure chloromethyl ketone compounds in Table 3 and Table 6.
- the aldehyde (HI-274) was made from the acid (HI- 208, 2e) via formation of the Weinreb amide (HI-267) by activation of the acid as its' mixed anhydride followed by coupling with N,O-dimethylhydroxylamine.
- the aldehyde was then synthesized by reduction of the Weinreb amide using LiAlFL.
- the thiomethyl ketones (HI-269, 302, 399, 365 & 273) were made by displacement of the halogen of the bromo or chloromethyl ketones with the appropriate thiol in the presence of potassium carbonate in DMF.
- the serine derivative (HI-266) was synthesized by reaction of Boc-Ser-OH with sodium hydride and 1-bromododecane in DMF to give the N-Boc-O- dodecyl-Ser-OH and then with isobutylchloroformate/diazomethane and HCI in ethyl acetate at 0 °C as in the chemistry noted above.
- the acetyl serine derivative (HI-489) was synthesized from HI-266 by removal of the Boc group in saturated HCI in ethyl acetate followed by acetylation using acetic anhydride in dichloromethane in the presence of triethylamine.
- NMR spectra were recorded using a 300 MHz Varian instrument and the chemical shifts reported are in ppm based on tetramethylsilane as the internal standard. Chemical shifts for 13 C NMR are referenced to the chloroform peak at 77.0 ppm. Melting points were done using a Fisher-Jones apparatus and are uncorrected. Fourier Transform Infra-red spectra were recorded on a FT-Nicolet model Protege 460 instrument. GC/MS analysis was done using a Hewlett-Packard GC/MS model 6890 with an HP5973 electron impact mass spectrometer.
- N-Ac-S-trityl-cysteine chloromethyl ketone (HI-350) Pale yellow solid: 1H NMR (CDC1 3 ) ⁇ 1.92 (s, 3H), 2.73 (m, 2H), 3.89 (s, 2H), 4.40 (m, IH), 5.82 (d, IH), 7.21 (m, 15H).
- N-Ac-S-2-naphthylmethyl-cysteine chloromethyl ketone HI-392
- N-Ac-S-dodecyl-cysteine bromomethyl ketone (HI-488) Pale yellow solid: ⁇ NMR (CDC1 3 ) ⁇ 0.84 (t, 3H), 1.22 (m, 18H), 1.53 (m, 2H), 2.02 (s, 3H), 2.50 (t, 2H), 2.92 (m, 2H), 4.12 (s, 2H), 4.89 (m, IH), 6.44 (d, IH).
- N-Ac-S-dodecyl-Cys-N(OCH 3 )-CH 3 (HI-267) Pale yellow oil: ⁇ NMR (CDCI 3 ) ⁇ 0.87 (t, 3H), 1.25 (m, 20H), 2.02 (s, 3H), 2.51 (t, 2H), 2.82 (m, 2H), 3.22 (s, 3H), 3.79 (s, 3H), 5.17 (m, IH), 6.38 (d, IH); MS (El) m/z 314 (M - N(OCH 3 )CH 3 ).
- N-Ac-S-dodecyl-Cys-CH 2 -SPh (HI-269) Pale yellow solid: 1H NMR (CDCI 3 ) ⁇ 0.88 (t, 3H), 1.25 (m, 18H), 1.53 (m, 2H), 1.97 (s, 3H), 2.46 (t, 2H), 2.2 (m, 2H), 5.16 (m, IH), 5.43 (s, 2H), 6.16 (d, IH), 7.33 (s, 5H).
- N-Ac-S-dodecyl-Cys-CH 2 -S-2-naphthyl (HI-302) Pale yellow solid:
- Cytotoxicity of alkyl ketone compounds The cytotoxicity of the alkyl ketone compounds against tumor cells was evaluated in leukemic cells, breast cancer cells, prostate cancer cells, and brain cancer cells.
- Cytotoxicity of various compounds against tumor cells was performed using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) assay (Boehringer Mannheim Corp., Indianapolis, IN). Unless otherwise specified, all cell lines were obtained from the American Type Culture Collection (ATCC). Briefly, exponentially growing cells were seeded into a 96-well plate at a density of 2.5 x 10 4 cells/well and incubated for 36 hours at 37°C prior to drug exposure. On the day of treatment, culture medium was carefully aspirated from the wells and replaced with fresh medium containing the indicated compound at concentrations ranging from 0.1 to 250 ⁇ M. Triplicate wells were used for each treatment. Human leukemic cell lines (NALM-6, MOLT-3) glioblastoma cells
- the absorbence of each well was measured in a microplate reader (Labsystems) at 540 nm and a reference wavelength of 690 nm.
- the OD 5 0 values were compared to those on standard OD 540 - versus - cell number curves generated for each cell line. The percent survival was calculated using the formula:
- a series of compounds was prepared with different aliphatic chain lengths in the R 3 position to determine the effect of chain length on cytotoxicity. As shown in Table 3, compounds with chain lengths of about 5 to 15 were the most effective anti-cancer agents. Preferred lengths were chain lengths of about 11 to 12. Various groups were tested in the R 1 position to further define effective anti- cancer compounds. The results are reported in Table 4. Substitution of a bromomethyl group at the R 1 position produced the most effective compound, consistent with Table I where chloromethyl was the most effective compound. The effect of changing of X and R 4 on cytotoxicity was examined and reported in Table 5.
- Cytotoxicity of HI-131 in Primary Cancer Cells The cytotoxicity of HI-131 against primary cancer cells was evaluated in leukemia cells taken from six children ( Figure 1 A), using the MTT assay described for Example 3. The cells were exposed to HI-131 at concentrations ranging from 0 to 50 ⁇ M. Percent survival was calculated as described for Example 3 and plotted against the HI-131 concentration used in the experiment. A composite concentration survival curve was then prepared from the data ( Figure IB).
- HI-131 Induces Apoptosis of Leukemia Cells
- the ability of HI-131 to induce apoptosis cells was evaluated in human leukemia in NALM-6 cells and primary leukemic cells from 2 patients. Cells were treated with 50 ⁇ M compound HI-131 for 24 hours. After incubatin, the cells were harvested and analyzed for apoptosis by in situ TUNNEL analysis and confocal laser scanning microscopy as described in Sudbeck et al., 1999, Clin. Cancer Res., 5:1589-82. Controls were treated with vehicle alone.
- HI-131 The ability of HI-131 to inhibit invasion by MDA-MB-231 breast cancer cells and U373 glioblastoma cells was evaluated.
- the coated inserts were rehydrated with 0.5 ml serum-free DMEM containing 0.1% bovine serum albumin for 1-2 hours.
- serum-free DMEM containing 0.1% bovine serum albumin
- exponentially growing cells were incubated overnight with HI-131 at various concentrations ranging from 1 ⁇ M to 25 ⁇ M and 2.5 ⁇ M to 25 ⁇ M, respectively.
- the cells were trypsinized, washed twice with serum-free DMEM containing BSA, counted and resuspended at 1 x 10 5 cells/ml.
- a cell suspension containing 5 x 10 4 cells in a serum-free DMEM containing HI-131 or vehicle was added to the Matrigel-coated and rehydrated filter inserts.
- 750 ⁇ l of NIH fibroblast conditioned medium was placed as a chemoattractant in 24-well plates and the inserts were placed in wells and incubated at 37°C for 48 hours. After the incubation period, the filter inserts were removed, the medium was decanted off and the cells on the top side of the filter that did not migrate were scraped off with a cotton-tipped applicator. The invasive cells that migrated to the lower side of the filter were fixed, stained with Hema-3 solutions and counted under microscope.
- IC 50 values were calculated by non-linear regression analysis using Graphpad Prisin Software Version 2.0 (Graphpad Software Inc., San Diego, CA).
- MDA-MB-231 breast cancer cells and U373 glioblastoma cells were highly invasive in Matrigel-coated Boyden chambers.
- HI- 131 inhibited the invasion of both MDA-MB-231 breast cancer cells and U373 glioblastoma cells through the Matrigel matrix in dose-dependent fashion.
- ECM proteins such as laminin, fibronectin, and type IV collagen are thought to play an important role in tumor cell attachment and migration.
- Laminin, fibronectin and collagen have been found in the basal lamina of blood vessels and in the glial limitans externa in the brain that promote the adhesion and invasion of tumor cells in situ (Carbonetto, 1984, Trends Neurosci., 7:382-387; Rutkaet al. J. Neurosurg, 69:155-170; Venstrom, et al, 1993, FASEB J, 7:996-1003).
- the effects of these ECM proteins on integrin- mediated U373 glioblastoma and MDA-MB-231 cell adhesion was examined.
- a human brain tumor cell line derived from an adult patient with glioblastoma, U-373 MG (Cat. #HTB-17) and MDA-MB-231 breast cancer cells (Cat. #HTB-26) were obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained in liquid culture using DMEM supplemented with 10% fetal bovine serum and antibiotics.
- Fibroblast conditioned medium was used as a source of chemoattractant in vitro invasion assays.
- Conditioned medium was prepared as described previously (Albini, et al, 1987, Cancer Res., 47:3239-3245). For the preparation of this conditioned medium NIH/3T3 embryonic fibroblasts (ATCC cat.
- DMSO 0.05%> trypsin (Life Technologies) resuspended in DMEM, incubated at 37°C for 2 hours to allow them to recover from the trypsinization stress and examined for their ability to adhere to plates precoated with ECM proteins.
- adhesion assays cells were centrifuged, washed twice with serum-free DMEM, counted and resuspended in serum-free DMEM to a final concentration of 2.5 x 10 cells/ml. One hundred ⁇ l of the cell suspension containing 2.5x 10 cells were added to each well and cells were allowed to adhere for 1 hour at 37°C in a humidified 5% CO 2 atmosphere. The adherent fraction was quantitated using MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assays.
- MTT 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- the OD 5 0 values were compared to those on standard OD 54 o-versus-cell number curves generated for each cell line.
- the adherent fraction of cells treated with HI-131 was compared to the DMSO-treated control cells and the percent adhesion relative to the control was determined.
Abstract
Description



























Claims




Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002336108A CA2336108A1 (en) | 1998-06-29 | 1999-06-29 | Alkyl ketones as potent anti-cancer agents |
| AU48459/99A AU754086B2 (en) | 1998-06-29 | 1999-06-29 | Alkyl ketones as potent anti-cancer agents |
| JP2000557230A JP2002519341A (en) | 1998-06-29 | 1999-06-29 | Alkyl ketones as potent anticancer agents |
| EP99932068A EP1091934A2 (en) | 1998-06-29 | 1999-06-29 | Alkyl ketones as potent anti-cancer agents |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US9099798P | 1998-06-29 | 1998-06-29 | |
| US60/090,997 | 1998-06-29 | ||
| US9736398P | 1998-08-21 | 1998-08-21 | |
| US60/097,363 | 1998-08-21 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2000000469A2 true WO2000000469A2 (en) | 2000-01-06 |
| WO2000000469A3 WO2000000469A3 (en) | 2000-06-15 |
Family
ID=26782863
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1999/014758 WO2000000469A2 (en) | 1998-06-29 | 1999-06-29 | Alkyl ketones as potent anti-cancer agents |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1091934A2 (en) |
| JP (1) | JP2002519341A (en) |
| AU (1) | AU754086B2 (en) |
| CA (1) | CA2336108A1 (en) |
| WO (1) | WO2000000469A2 (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000000483A2 (en) * | 1998-06-29 | 2000-01-06 | Parker Hugues Institute | Tubulin binding compounds (cobra) |
| US6258841B1 (en) | 1998-06-29 | 2001-07-10 | Parker Hughes Institute | Tubulin binding compounds (COBRA) |
| JP2003527364A (en) * | 2000-03-03 | 2003-09-16 | ティア、メディカ、アクチスカベット | New fatty acid analogs |
| US6734207B2 (en) | 2001-04-20 | 2004-05-11 | Parker Hughes Institute | Cytotoxic compounds |
| US7214831B2 (en) | 2002-05-22 | 2007-05-08 | Errant Gene Therapeutics, Llc | Histone deacetylase inhibitors based on alpha-chalcogenmethylcarbonyl compounds |
| US9744147B2 (en) | 2008-11-11 | 2017-08-29 | Signum Biosciences, Inc. | Isoprenyl compounds and methods thereof |
| AU2014346591B2 (en) * | 2013-11-08 | 2018-09-27 | Promentis Pharmaceuticals, Inc. | Substituted N-acetyl-L-cysteine derivatives and related compounds |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992018465A2 (en) * | 1991-04-15 | 1992-10-29 | The President And Fellows Of Harvard College | Compounds for inhibition of protein methylation |
| WO1995017512A2 (en) * | 1993-12-23 | 1995-06-29 | Cangene Corporation | Proteases from streptomyces and use thereof in protein expression systems |
| WO1998022098A2 (en) * | 1996-11-20 | 1998-05-28 | Qlt Phototherapeutics, Inc. | Cpp32 inhibitors for regulating apoptosis |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5202456A (en) * | 1991-04-15 | 1993-04-13 | The President And Fellows Of Harvard College | Compounds for inhibition of protein methylation |
| JPH05117169A (en) * | 1991-10-29 | 1993-05-14 | Mitsubishi Kasei Corp | Method for producing chloromethyl ketone |
-
1999
- 1999-06-29 WO PCT/US1999/014758 patent/WO2000000469A2/en active IP Right Grant
- 1999-06-29 CA CA002336108A patent/CA2336108A1/en not_active Abandoned
- 1999-06-29 JP JP2000557230A patent/JP2002519341A/en active Pending
- 1999-06-29 EP EP99932068A patent/EP1091934A2/en not_active Withdrawn
- 1999-06-29 AU AU48459/99A patent/AU754086B2/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992018465A2 (en) * | 1991-04-15 | 1992-10-29 | The President And Fellows Of Harvard College | Compounds for inhibition of protein methylation |
| WO1995017512A2 (en) * | 1993-12-23 | 1995-06-29 | Cangene Corporation | Proteases from streptomyces and use thereof in protein expression systems |
| WO1998022098A2 (en) * | 1996-11-20 | 1998-05-28 | Qlt Phototherapeutics, Inc. | Cpp32 inhibitors for regulating apoptosis |
Non-Patent Citations (5)
| Title |
|---|
| CHEN, PING ET AL.: "A Practical Method for the Preparation of alpha'-Chloroketones of N-Carbamate Protected alpha-Aminoacids" TETRAHEDRON LETTERS, vol. 38, no. 18, 1997, pages 3175-3178, XP002120874 * |
| CHEN, YULONG ET AL.: "Solubilization, Partial Purification, and Affinity Labeling of the Membrane-Bound Isoprenylated Protein Endoprotease" BIOCHEMISTRY, vol. 35, no. 10, 1996, pages 3227-3237, XP002120873 * |
| DATABASE CHEMICAL ABSTRACTS [Online] ACS abstract no. 225574, 22 November 1993 (1993-11-22) ANDO, RYOICHI: "Preparation of chloromethyl ketones as inhibitors for serine and thiol protease" XP002120875 & JP 05 117169 A (MITSUBISHI CHEM. IND.) 14 May 1993 (1993-05-14) * |
| DATABASE CHEMICAL ABSTRACTS [Online] ACS Abstract no. 231845, 17 December 1990 (1990-12-17) INAZU, TOSHIYUKI & YAMANOI, TAKASHI: "Synthetic studies on D-erythro-C18-sphingosine" XP002120876 & NOGUSHI KENKYUSHO JIHO, no. 32, 1989, pages 13-26, * |
| JIA, ZONGCHAO ET AL.: "Crystal Structures of Recombinant Rat Cathepsin B and a Cathepsin B-Inhibitor Complex" JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 270, no. 10, 10 March 1995 (1995-03-10), pages 5527-5533, XP002120872 * |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000000483A2 (en) * | 1998-06-29 | 2000-01-06 | Parker Hugues Institute | Tubulin binding compounds (cobra) |
| WO2000000483A3 (en) * | 1998-06-29 | 2000-09-28 | Parker Hugues Inst | Tubulin binding compounds (cobra) |
| US6258841B1 (en) | 1998-06-29 | 2001-07-10 | Parker Hughes Institute | Tubulin binding compounds (COBRA) |
| US6329420B1 (en) | 1998-06-29 | 2001-12-11 | Parker Hughes Institute | Tubulin binding compounds (COBRA) |
| JP2003527364A (en) * | 2000-03-03 | 2003-09-16 | ティア、メディカ、アクチスカベット | New fatty acid analogs |
| US7037937B2 (en) | 2001-04-20 | 2006-05-02 | Parker Hughes Institute | Cytotoxic compounds |
| US6734207B2 (en) | 2001-04-20 | 2004-05-11 | Parker Hughes Institute | Cytotoxic compounds |
| US7214831B2 (en) | 2002-05-22 | 2007-05-08 | Errant Gene Therapeutics, Llc | Histone deacetylase inhibitors based on alpha-chalcogenmethylcarbonyl compounds |
| US9744147B2 (en) | 2008-11-11 | 2017-08-29 | Signum Biosciences, Inc. | Isoprenyl compounds and methods thereof |
| US10314802B2 (en) | 2008-11-11 | 2019-06-11 | Signum Biosciences, Inc. | Isoprenyl compounds and methods thereof |
| AU2014346591B2 (en) * | 2013-11-08 | 2018-09-27 | Promentis Pharmaceuticals, Inc. | Substituted N-acetyl-L-cysteine derivatives and related compounds |
| US10112897B2 (en) | 2013-11-08 | 2018-10-30 | Promentis Pharmaceuticals, Inc. | Substituted N-acetyl-L-cysteine derivatives and related compounds |
| US10358414B2 (en) | 2013-11-08 | 2019-07-23 | Promentis Pharmaceuticals, Inc. | Substituted N-acetyl-L-cysteine derivatives and related compounds |
| AU2018282446B2 (en) * | 2013-11-08 | 2020-07-23 | Promentis Pharmaceuticals, Inc. | Substituted N-acetyl-L-cysteine derivatives and related compounds |
| US10961187B2 (en) | 2013-11-08 | 2021-03-30 | Promentis Pharmaceuticals, Inc. | Substituted N-acetyl-L-cysteine derivatives and related compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2002519341A (en) | 2002-07-02 |
| CA2336108A1 (en) | 2000-01-06 |
| WO2000000469A3 (en) | 2000-06-15 |
| AU4845999A (en) | 2000-01-17 |
| AU754086B2 (en) | 2002-11-07 |
| EP1091934A2 (en) | 2001-04-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20090264487A1 (en) | Hemiasterlin analogs | |
| US6734207B2 (en) | Cytotoxic compounds | |
| KR100667464B1 (en) | Tricyclic derivatives or pharmaceutically acceptable salts thereof, their preparations and pharmaceutical compositions containing them | |
| KR20010021536A (en) | Use of colchinol derivatives as vascular damaging agents | |
| US6482816B1 (en) | Organic arsenic compounds | |
| US6251882B1 (en) | Alkyl ketones as potent anti-cancer agents | |
| WO2007059118A1 (en) | Combretastatin derivatives and related therapeutic methods | |
| AU754086B2 (en) | Alkyl ketones as potent anti-cancer agents | |
| US4904680A (en) | Amino acid derivatives having anti-tumor activity and compositions containing them | |
| KR101068259B1 (en) | Novel betulinic acid derivatives aringcondensed to a heterocyclic group | |
| US20170183297A1 (en) | Omega-3 analogues | |
| JPS6326748B2 (en) | ||
| JP4933683B1 (en) | Novel retinoid derivatives having cytotoxic and / or anti-angiogenic properties | |
| PL184861B1 (en) | Improvement in tolerance of pharmaceutically active beta-amino acids | |
| AU2013350311A1 (en) | Omega-3 analogues | |
| JPH03500880A (en) | Renin inhibitors, methods for their production, methods for their use and compositions containing them | |
| JPH03148246A (en) | Peptidyldifluorodiol renin inhibiting substance | |
| AU636090B2 (en) | Esters of 2-arylmethyl-1-naphthol derivatives as 5-lipoxygenase inhibitors | |
| US6686344B1 (en) | Organic-arsonic compounds | |
| WO1998042700A1 (en) | N-(arginyl)benzenesulphonamide derivatives and use thereof as antithrombotic agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AL AM AT AT AU AZ BA BB BG BR BY CA CH CN CU CZ CZ DE DE DK DK EE EE ES FI FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SK SL TJ TM TR TT UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW SD SL SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AL AM AT AT AU AZ BA BB BG BR BY CA CH CN CU CZ CZ DE DE DK DK EE EE ES FI FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SK SL TJ TM TR TT UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW SD SL SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 48459/99 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2000 557230 Country of ref document: JP Kind code of ref document: A Ref document number: 2336108 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999932068 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999932068 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1999932068 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 48459/99 Country of ref document: AU |