WO1999064050A1 - Novel potassium channel drugs and their uses - Google Patents
Novel potassium channel drugs and their uses Download PDFInfo
- Publication number
- WO1999064050A1 WO1999064050A1 PCT/US1999/012777 US9912777W WO9964050A1 WO 1999064050 A1 WO1999064050 A1 WO 1999064050A1 US 9912777 W US9912777 W US 9912777W WO 9964050 A1 WO9964050 A1 WO 9964050A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ligand
- ligands
- linkers
- linker
- compounds
- Prior art date
Links
Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6872—Intracellular protein regulatory factors and their receptors, e.g. including ion channels
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/55—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug, i.e. a dimer, oligomer or polymer of pharmacologically or therapeutically active compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
Definitions
- This invention relates to novel multibinding compounds that bind to potassium ( + ) channels and modulate their activity.
- the compounds of this invention comprise 2-10 K + channel ligands covalently connected by a linker or linkers, wherein the ligands in their monovalent (i.e., unlinked) state bind to one or more types of K + channel.
- the manner of linking the ligands together is such that the multibinding agents thus formed demonstrate an increased biologic and/or therapeutic effect as compared to the same number of unlinked ligands made available for binding to the K + channel.
- the invention also relates to methods of using such compounds and to methods of preparing them.
- the compounds of this invention are particularly useful for treating diseases and conditions of mammals that are mediated by K + channels. Accordingly, this invention also relates to pharmaceutical compositions comprising a pharmaceutically acceptable excipient and an effective amount of a compound of this invention.
- Voltage-regulated potassium channels mediate the flux of K + out of cells in response to changes in membrane potential.
- 28 Voltage-gated K + channels in the open state typically transition to an inactivated state, and must reacquire the ability to respond to an external stimulus during a recovery period.
- An inward rectifying voltage-regulated potassium channel in cardiac muscle is also activated by acetylcholine (i.e., it is gated by more than one type of stimulus).
- 18 A calcium-activated K + channel has been described.
- 16 Potassium channels serve a variety of important cellular functions, including excitability, setting and maintaining the resting potential, repolarizing action potentials, transmembrane transport, volume regulation, signal transduction, and so on. 28 They are implicated in a variety of pathophysiological disorders, including hypertension, cardiac arrhythmogenesis, insulin-dependent diabetes, non-
- Figure 1 illustrates in cross-sectional view the transmembrane domain/subunit organization of various transporter molecules, as it is presently understood by those working in the field of transport physiology. It should be understood that, for purposes of simplification, other subunits that may be involved in or required for transporter activity have been omitted from the diagram.
- voltage-gated ion channels and related proteins are tetrameric structures formed by the noncovalent association of individual subunits (1),(2), or by the interaction of homologous domains of a monomeric protein (3).
- the channels differ as well in the number of transmembrane segments per subunit or per domain.
- Inward-rectifier type K + channels and P 2x purinergic channels have two transmembrane-segments in each subunit,
- Shaker-type K + channels have six transmembrane segments per subunit and Na + and Ca ⁇ channels have six transmembrane segments per domain.
- Neurotransmitter-gated ion channels such as those shown in (4) are organized as pentamers, with each of the subunits having four transmembrane segments/domains.
- the activation gate for potassium channels has not been identified, although a trap door mechanism has been proposed. 81,120
- Potassium channels are structurally similar to, but smaller and simpler than, sodium and calcium ion channels, 98 with the K + channel tetrameric structure being formed by four polypeptides. 3 However, potassium channels represent a diverse class of ion channels. 18 Homotetramers can form, but there is evidence that heterotetramers may be functionally relevant in vivo. 10 The x-ray structure of a bacterial K + channel (which is homologous to mammalian K + channels) has been disclosed. 21 A prokaryotic K + channel was found to have the same structure as a eukaryotic K + channel. 104 The channel has an inverted teepee structure with a large hydrophobic cavity.
- the cavity (10A) is centered in the channel on the cytoplasmic side, and appears to get larger upon channel opening. 21,82 ' 110,114 Voltage-dependent cardiac potassium channel genes have been cloned as cDNAs. 10,113,116 Variability in the potassium channel genes may relate to disease conditions. 14,48,50,70
- N-type and C-type potassium channel inactivation
- Both are partially coupled to activation and are usually voltage insensitive once activation is complete.
- N-type inactivation in Shaker B channels depends on a group of amino acids at the N-terminal that bind to the activated channel and occlude the intracellular mouth of the channel. No sequence similarity has been found among the N-termini of the N-type inactivating channels.
- N-type inactivation is voltage insensitive at positive potentials and competes with drug binding at the intracellular face of the channel.
- C-type inactivation which is less understood, occurs by occlusion of the external mouth of the channel during sustained depolarization.
- C-type inactivation is voltage insensitive at potentials where activation is complete, but recovery from C-type inactivation is voltage sensitive. Both C- and N-type inactivation are coupled or partially coupled to activation, and both require similar degrees of activation to proceed. 40
- potassium channels are recognized as important targets for drug therapy. For example, potassium channels are targeted by certain antidiabetic, antihypertensive and antiarryhthmic drugs.
- Potassium channel antagonists are used for treatment of arrhythmia.
- Antiarrhythmic agents are classified into four classes under the Vaughan Williams classification scheme: Class I (sodium channel blockers); Class II (beta-blockers); Class ⁇ i (potassium channel blockers); and Class IV (calcium channel blockers).
- Class I sodium channel blockers
- Class II beta-blockers
- Class ⁇ i potential channel blockers
- Class IV calcium channel blockers
- an antiarrhythmic agent may have activity in several channels and/or with several receptors.
- 89,92,101 Newer drugs are more selective to specific K + channels, as shown in Table 2. Properties of some known
- K + channel blockers are given in Table 3.
- Table 5 sets forth the principal K + currents and some drugs that block them. 45 The majority of drugs in development are 1 ⁇ blockers. 87,103,112 Some agents appear to be cationic open-channel blockers. 115,118,119
- This invention is directed to novel multibinding compounds that bind to K + channels in mammalian tissues and can be used to treat diseases and conditions mediated by such channels.
- This invention is also directed to general synthetic methods for generating large libraries of diverse multimeric compounds which multimeric compounds are candidates for possessing multibinding properties for potassium channels.
- the diverse multimeric compound libraries provided by this invention are synthesized by combining a linker or linkers with a ligand or ligands to provide for a library of multimeric compounds wherein the linker and ligand each have complementary functional groups permitting covalent linkage.
- the library of linkers is preferably selected to have diverse properties such as valency, linker length, linker geometry and rigidity, hydrophilicity or hydrophobicity, amphiphilicity, acidity, basicity and polarization.
- the library of ligands is preferably selected to have diverse attachment points on the same ligand, different functional groups at the same site of otherwise the same ligand, and the like.
- This invention is also directed to libraries of diverse multimeric compounds which multimeric compounds are candidates for possessing multibinding properties. These libraries are prepared via the methods described above and permit the rapid and efficient evaluation of what molecular constraints impart multibinding properties to a ligand or a class of ligands targeting a potassium channel.
- this invention is directed to a multibinding compound and salts thereof comprising 2 to 10 ligands which may be the same or different and which are covalently attached to a linker or linkers, which may be the same or different, each of said ligands comprising a ligand domain capable of binding to a K + channel.
- the multibinding compounds of this invention are preferably represented by Formula I:
- each L is a ligand that may be the same or different at each occurrence;
- X is a Hhker that may be the same or different at each occurrence;
- p is an integer of from 2 to 10; and
- q is an integer of from 1 to 20; wherein each of said ligands comprises a ligand domain capable of binding to a K + channel.
- q is less than/?.
- the binding of the multibinding compound to a K + channel or channels in a mammal modulates diseases and conditions mediated by the K + channel or channels.
- this invention is directed to a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable excipient and a therapeutically effective amount of one or more multibinding compounds (or pharmaceutically acceptable salts thereof) comprising 2 to 10 ligands which may be the same or different and which are covalently attached to a linker or linkers, which may be the same or different, each of said ligands comprising a ligand domain capable of binding to a K + channel of a cell mediating mammalian diseases or conditions, thereby modulating the diseases or conditions.
- this invention is directed to a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable excipient and a therapeutically effective amount of one or more multibinding compounds represented by
- each L is a ligand that may be the same or different at each occurrence;
- X is a linker that may be the same or different at each occurrence;
- p is an integer of from 2 to 10;
- q is an integer of from 1 to 20; wherein each of said ligands comprises a ligand domain capable of binding to a K + channel of a cell mediating mammalian diseases or conditions, thereby modulating the diseases or conditions.
- q is less than ?.
- this invention is directed to a method for modulating the activity of a K + channel in a biologic tissue, which method comprises contacting a tissue having a K + channel with a multibmding compound (or pharmaceutically acceptable salts thereof) under conditions sufficient to produce a change in the activity of the channel in said tissue, wherein the multibinding compound comprises 2 to 10 ligands which may be the same or different and which are covalently attached to a linker or linkers, which may be the same or different, each of said ligands comprising a ligand domain capable of binding to a K + channel.
- this invention is directed to a method for treating a disease or condition in a mammal resulting from an activity of a K + channel, which method comprises administering to said mammal a therapeutically effective amount of a pharmaceutical composition comprising a pharmaceutically acceptable excipient and one or more multibinding compounds (or pharmaceutically acceptable salts thereof) comprising 2 to 10 ligands which may be the same or different and which are covalently attached to a linker or linkers, which may be the same or different, each of said ligands comprising a ligand domain capable of binding to a K + channel of a cell mediating mammalian diseases or conditions. .
- this invention is directed to a method for treating a disease or condition in a mammal resulting from an activity of a K + channel, which method comprises administering to said mammal a therapeutically effective amount of a pharmaceutical composition comprising a pharmaceutically acceptable excipient and one or more multibinding compounds represented by Formula I:
- each L is a ligand that may be the same or different at each occurrence;
- X is a linker that may be the same or different at each occurrence;/? is an integer of from 2 to 10; and
- q is an integer of from 1 to 20; wherein each of said ligands comprises a ligand domain capable of binding to a K + channel of a cell mediating mammalian diseases or conditions.
- q is less than/?.
- this invention provides processes for preparing the multibinding agents of Formula I. This can be accomplished by combining/? appropriately functionalized ligands with q complementary functionalized linkers under conditions where covalent bond formulation between the ligands and linkers occurs; alternatively, linking portions of/? appropriately functionalized ligands to q complementary functionalized linkers and then completing the synthesis of the ligands in a subsequent step may be performed to prepare these compounds. Another method which may be used involves linking/? appropriately functionalized ligands to portions of the linker(s) and then completing the synthesis of the linker(s) in a subsequent step.
- Coupling one or more of an appropriately functionalized ligand to a complementary functionalized linker, and subsequently coupling one or more additional ligands to said linker or linkers may be done to prepare the claimed compounds. Coupling as above wherein coupling of different appropriately functionalized linkers occurs simulataneously may also be used.
- this invention is directed to a method for identifying multimeric ligand compounds possessing multibinding properties for potassium channels, which method comprises:
- each linker in said library comprises at least two functional groups having complementary reactivity to at least one of the reactive functional groups of the ligand;
- this invention is directed to a method for identifying multimeric ligand compounds possessing multibinding properties for potassium channels, which method comprises:
- each linker comprises at least two functional groups having complementary reactivity to at least one of the reactive functional groups of the ligand;
- the preparation of the multimeric ligand compound library is achieved by either the sequential or concurrent combination of the two or more stoichiometric equivalents of the ligands identified in (a) with the linkers identified in (b). Sequential addition is preferred when a mixture of different ligands is employed to ensure heterodimeric or multimeric compounds are prepared. Concurrent addition of the ligands occurs when at least a portion of the multimer comounds prepared are homomultimeric compounds.
- the assay protocols recited in (d) can be conducted on the multimeric ligand compound library produced in (c) above, or preferably, each member of the library is isolated by preparative liquid chromatography mass spectrometry (LCMS).
- LCMS preparative liquid chromatography mass spectrometry
- this invention is directed to a library of multimeric ligand compounds which may possess multivalent properties for potassium channels, which library is prepared by the method comprising:
- each linker in said library comprises at least two functional groups having complementary reactivity to at least one of the reactive functional groups of the ligand;
- this invention is directed to a library of multimeric ligand compounds which may possess multivalent properties for potassium channels, which library is prepared by the method comprising:
- each linker comprises at least two functional groups having complementary reactivity to at least one of the reactive functional groups of the ligand; and (c) preparing a multimeric ligand compound library by combining at least two stoichiometric equivalents of the library of ligands identified in (a) with the linker or mixture of linkers identified in (b) under conditions wherein the complementary functional groups react to form a covalent linkage between said linker and at least two of said ligands.
- the library of linkers employed in either the methods or the library aspects of this invention is selected from the group comprising flexible linkers, rigid linkers, hydrophobic linkers, hydrophilic linkers, linkers of different geometry, acidic linkers, basic linkers, linkers of different polarization and amphiphilic linkers.
- each of the linkers in the linker library may comprise linkers of different chain length and/or having different complementary reactive groups. Such linker lengths can preferably range from about 2 to lOOA.
- the potassium channel ligand or mixture of ligands is selected to have reactive functionality at different sites on said ligands in order to provide for a range of orientations of said ligand on said multimeric ligand compounds.
- reactive functionality includes, by way of example, carboxylic acids, carboxylic acid halides, carboxyl esters, amines, halides, isocyanates, vinyl unsaturation, ketones, aldehydes, thiols, alcohols, anhydrides, and precursors thereof. It is understood, of course, that the reactive functionality on the ligand is selected to be complementary to at least one of the reactive groups on the linker so that a covalent linkage can be formed between the linker and the ligand.
- the multimeric ligand compound is homomeric (i.e., each of the ligands is the same, although it may be attached at different points) or heterodimeric (i.e., at least one of the ligands is different from the other ligands).
- this invention provides for an iterative process for rationally evaluating what molecular constraints impart multibinding properties to a class of multimeric compounds or ligands targeting a receptor.
- this method aspect is directed to a method for identifying multimeric ligand compounds possessing multibinding properties for potassium channels which method comprises:
- steps (e) and (f) optionally repeating steps (e) and (f) to further elaborate upon said molecular constraints.
- steps (e) and (f) are repeated at least two times, more preferably at from 2-50 times, even more preferably from 3 to 50 times, and still more preferably at least 5-50 times.
- Figure 1 is a highly schematic illustration of the transmembrane organization of various cell membrane transporters.
- Figure 2 illustrates a method for optimizing the linker geometry for presentation of ligands (filled circles) in bivalent compounds:
- Figure 3 shows exemplary linker "core" structures.
- Figure 4 illustrates examples of multi-binding compounds comprising (A) 2 ligands, (B) 3 ligands, (C) 4 ligands, and (D) >4 ligands attached in different formats to a linker.
- Figure 5 illustrates the ligand amiodarone, which may be used in preparing multi- binding compounds. Potentially modifiable positions are indicated by arrows.
- Figure 6 illustrates numerous reactive functional groups and the resulting bonds formed by reaction therebetween.
- FIGS 7 to 21 illustrate convenient methods for preparing the multibinding compounds of this invention.
- the filled circles represent linkers, referred to in the written Examples as "Link”.
- K + channels are considered to be pharmacological receptors: they possess specific binding sites for ligands having agonist and antagonist activities; the binding of ligands to such sites modulates K + flux through the channel; the channel properties (i.e., gating and ion selectivity) are regulatable. Accordingly, diseases or conditions that involve, or are mediated by, K * channels can be treated with pharmacologically active ligands that interact with such channels to initiate, modulate or abrogate transporter activity.
- affinity The interaction of a K + channel and a K + channel-binding ligand may be described in terms of "affinity” and "specificity".
- the "affinity” and “specificity” of any given ligand-K + channel interaction is dependent upon the complementarity of molecular binding surfaces and the energetic costs of complexation (i.e., the net difference in free energy between bound and free states). Affinity may be quantified by the equilibrium constant of complex formation, the ratio of on/off rate constants, and/or by the free energy of complex formation. Specificity relates to the difference in binding affinity of a ligand for different receptors.
- the net free energy of interaction of such ligand with a K + channel is the difference between energetic gains (enthalpy gained through molecular complementarity and entropy gained through the hydrophobic effect) and energetic costs (enthalpy lost through decreased solvation and entropy lost through reduced translational, rotational and conformational degrees of freedom).
- the compounds of this invention comprise 2 to 10 K + charmel-binding ligands covalently linked together and capable of acting as multibinding agents.
- the enhanced activity of these compounds is believed to arise at least in part from their ability to bind in a multivalent manner with multiple ligand binding sites on a K + channel or channels, which gives rise to a more favorable net free energy of binding.
- Multivalent interactions differ from collections of individual monovalent (univalent) interactions by being capable of providing enhanced biologic and/or therapeutic effect. Multivalent binding can amplify binding affinities and differences in binding affinities, resulting in enhanced binding specificity as well as affinity.
- alkyl refers to a monoradical branched or unbranched saturated hydrocarbon chain, preferably having from 1 to 40 carbon atoms, preferably 1-10 carbon atoms, more preferably 1-6 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, secondary butyl, tert-butyl, n-hexyl, n-octyl, n-decyl, n-dodecyl, 2-ethyldodecyl, tetradecyl, and the like, unless otherwise indicated.
- substituted alkyl refers to an alkyl group as defined above having from 1 to 5 substituents selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, -SO-alkyl, -SO-ary
- alkylene refers to a diradical of a branched or unbranched saturated hydrocarbon chain, preferably having from 1 to 40 carbon atoms, preferably 1-10 carbon atoms, more preferably 1-6 carbon atoms. This term is exemplified by groups such as methylene (-CH 2 -), ethylene (-CH 2 CH 2 -), the propylene isomers (e.g., -CH 2 CH 2 CH 2 - and -CH(CH 3 )CH 2 -) and the like.
- substituted alkylene groups include those where 2 substituents on the alkylene group are fused to form one or more cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, heterocyclic or heteroaryl groups fused to the alkylene group; (2) An alkylene group as defined above that is interrupted by 1-20 atoms independently chosen from oxygen, sulfur and NR j -, where R ⁇ is chosen from hydrogen, optionally substituted alkyl, cycloalkyl, alkenyl, cycloalkenyl, alkenyl, cycloalkenyl, alkynyl, aryl, heteroaryl and heterocyclic, or groups selected from carbonyl, carboxyester, carboxyamide and sulfonyl; and (3) An alkylene group as defined above that has both from 1 to 5 substituents as defined above and is also interrupted by 1-20 atoms as defined above.
- substituted alkylenes are chloromethylene (-CH(Cl)-), aminoethylene (-CH(NH 2 )CH 2 -), 2-carboxypropylene isomers (-CH 2 CH(CO 2 H)CH 2 -), ethoxyethyl (-CH 2 CH 2 O-CH 2 CH 2 -), ethylmethylaminoethyl (-CH 2 CH 2 N(CH 3 )CH 2 CH 2 -), l-ethoxy-2-(2-ethoxy-ethoxy)ethane (-CH 2 CH 2 O-CH 2 CH 2 -
- alkaryl or “aralkyl” refers to the groups -alkylene-aryl and -substituted alkylene-aryl in which alkylene and aryl are as defined herein. Such alkaryl groups are exemplified by benzyl, phenethyl and the like.
- alkoxy refers to the groups alkyl-O-, alkenyl-O-, cycloalkyl-O-, cycloalkenyl-O-, and alkynyl-O-, where alkyl, alkenyl, cycloalkyl, cycloalkenyl, and alkynyl are as defined herein.
- Preferred alkoxy groups are alkyl-O- and include, by way of example, methoxy, ethoxy, n-propoxy, iso-propoxy, n-butoxy, tert-butoxy, sec-butoxy, n-pentoxy, n-hexoxy, 1,2-dimethylbutoxy, and the like.
- substituted alkoxy refers to the groups substituted alkyl-O-, substituted alkenyl-O-, substituted cycloalkyl-O-, substituted cycloalkenyl-O-, and substituted alkynyl-O- where substituted alkyl, substituted alkenyl, substituted cycloalkyl, substituted cycloalkenyl and substituted alkynyl are as defined herein.
- alkylalkoxy refers to the groups -alkylene-O-alkyl, alkylene-O-substituted alkyl, substituted alkylene-O-alkyl and substituted alkylene-O-substituted alkyl wherein alkyl, substituted alkyl, alkylene and substituted alkylene are as defined herein.
- Examples of such groups are methylenemethoxy (-CH 2 OCH 3 ), ethylenemethoxy (-CH 2 CH 2 OCH 3 ), n-propylene- iso-propoxy (-CH 2 CH 2 CH 2 OCH(CH 3 ) 2 ), methylene-t-butoxy (-CH 2 -O-C(CH 3 ) 3 ) and the like.
- alkylthioalkoxy refers to the group -alkylene-S-alkyl, alkylene-S- substituted alkyl, substituted alkylene-S-alkyl and substituted alkylene-S-substituted alkyl wherein alkyl, substituted alkyl, alkylene and substituted alkylene are as defined herein.
- Preferred alkylthioalkoxy groups are alkylene-S-alkyl and include, by way of example, methylenethiomethoxy (-CH 2 SCH 3 ), ethylenethiomethoxy n-propylene-iso- thiopropoxy (-CH 2 CE 2 CE 2 SCH(C .
- Alkenyl refers to a monoradical of a branched or unbranched unsaturated hydrocarbon preferably having from 2 to 40 carbon atoms, preferably 2-10 carbon atoms, more preferably 2-6 carbon atoms, and preferably having 1-6 double bonds. This term is further exemplified by such radicals as vinyl, prop-2-enyI, pent-3-enyl, hex-5-enyl, 5-ethyldodec-3,6-dienyl, and the like.
- substituted alkenyl refers to an alkenyl group as defined above having from 1 to 5 substituents selected from the group consisting of alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thiol, thioalkoxy, substituted thioalkoxy, aryl, heteroaryl, heterocyclic, aryloxy, thioaryloxy, heteroaryloxy, thioheteroaryloxy, heterocyclooxy, thioheterocyclooxy, nitro, -SO-alkyl, -SO-substituted alkyl, -SO-aryl, -SO-heteroaryl, -SO ⁇ -alkyl, -SO 2 -substituted
- Alkenylene refers to a diradical of an unsaturated hydrocarbon, preferably having from 2 to 40 carbon atoms, preferably 2-10 carbon atoms, more preferably 2-6 carbon atoms, and preferably having 1-6 double bonds. This term is further exemplified by such radicals as
- substituted alkenylene refers to an alkenylene group as defined above having from 1 to 5 substituents, selected from the group consisting of alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, aminoacyl, aminoacyloxy, oxyacylamino, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, thioaryloxy, heteroaryl, heteroaryloxy, thioheteroaryloxy, heterocyclic, heterocyclooxy, thioheterocyclooxy, nitro, and NRR b , wherein R and R b may be the same or different and are chosen from hydrogen, optionally substituted alkyl, cycloalkyl, alkenyl, cycloalkenyl, alkyny
- substituted alkenylene groups include those where 2 substituents on the alkenylene group are fused to form one or more cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, heterocyclic or heteroaryl groups fused to the alkenylene group.
- Alkynyl refers to a monoradical of an unsaturated hydrocarbon, preferably having from 2 to 40 carbon atoms, preferably 2-10 carbon atoms, more preferably 2-6 carbon atoms, and preferably having 1-6 triple bonds. This term is further exemplified by such radicals as acetylenyl, prop-2-ynyl, pent-3-ynyl, hex-5-ynyl, 5-ethyldodec-3,6-diynyl, and the like.
- substituted alkynyl refers to an alkynyl group as defined above having from 1 to 5 substituents, selected from the group consisting of alkoxy, substituted alkoxy, acyl, acylamino, acyloxy, amino, aminoacyl, aminoacyloxy, oxyacylamino, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, thioaryloxy, heteroaryl, heteroaryloxy, thioheteroaryloxy, heterocyclic, heterocyclooxy, thioheterocycloxy, nitro, -SO-alkyl, -SO-substituted alkyl, -SO-aryl, -SO-heteroaryl, -SO 2 -alkyl, -SO 2 -substit
- Alkynylene refers to a diradical of an unsaturated hydrocarbon radical, preferably having from 2 to 40 carbon atoms, preferably 2-10 carbon atoms, more preferably 2-6 carbon atoms, and preferably having 1-6 triple bonds. This term is further exemplified by such radicals as l,3-prop-2-ynyl, l,5-pent-3-ynyl, l,4-hex-5-ynyl, 5-ethyl-l,12-dodec-3,6-diynyl, and the like.
- acyl refers to the groups -CHO, alkyl-C(O)-, substituted alkyl-C(O)-, cycloalkyl-C(O)-, substituted cycloalkyl-C(O)-, cycloalkenyl-C(O)-, substituted cycloalkenyl- C(O)-, aryl-C(O)-, heteroaryl-C(O)- and heterocyclic-C(O)- where alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, aryl, heteroaryl and heterocyclic are as defined herein.
- acylamino refers to the group -C(O)NRR where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, heterocyclic or where both R groups are joined to form a heterocyclic group (e.g., morpholine) wherein alkyl, substituted alkyl, aryl, heteroaryl and heterocyclic are as defined herein.
- aminoacyl refers to the group -NRC(O)R where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, or heterocyclic wherein alkyl, substituted alkyl, aryl, heteroaryl and heterocyclic are as defined herein.
- aminoacyloxy refers to the group -NRC(O)OR where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, or heterocyclic wherein alkyl, substituted alkyl, aryl, heteroaryl and heterocyclic are as defined herein.
- acyloxy refers to the groups alkyl-C(O)O-, substituted alkyl-C(O)O-, cycloalkyl-C(O)O-, substituted cycloalkyl-C(O)O-, aryl-C(O)O-, heteroaryl-C(O)O-, and heterocyclic-C(O)O- wherein alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, aryl, heteroaryl, and heterocyclic are as defined herein.
- aryl refers to an unsaturated aromatic carbocyclic group of from 6 to 20 carbon atoms having a single ring (e.g., phenyl) or multiple condensed (fused) rings (e.g., naphthyl or anthryl).
- such aryl groups can optionally be substituted with from 1 to 5 substituents selected from the group consisting of acyloxy, hydroxy, thiol, acyl, alkyl, alkoxy, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, substituted alkyl, substituted alkoxy, substituted alkenyl, substituted alkynyl, substituted cycloalkyl, substituted cycloalkenyl, amino, aminoacyl, acylamino, alkaryl, aryl, aryloxy, azido, carboxyl, carboxylalkyl, cyano, halo, nitro, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, aminoacyloxy, oxyacylamino, thioalkoxy, substituted thioaryloxy, thioheteroaryloxy, -
- aryloxy refers to the group aryl-O- wherein the aryl group is as defined above including optionally substituted aryl groups as also defined above.
- arylene refers to a diradical derived from aryl or substituted aryl as defined above, and is exemplified by 1,2-phenylene, 1,3-phenylene, 1,4-phenylene, 1,2-naphthylene and the like.
- amino refers to the group -NH 2
- substituted amino refers to the group -NRR where each R is independently selected from the group consisting of hydrogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl and heterocyclic provided that both R's are not hydrogen.
- carboxy alkyl refers to the group "-C(O)O-alkyl", “-C(O)O-substituted alkyl", “-C(O)O-cycloalkyl", “-C(O)O-substituted cycloalkyl", “-C(O)O-alkenyl”, “-C(O)O- substituted alkenyl”, “-C(O)O-alkynyl” and "-C(O)O-substituted alkynyl” where alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, substituted alkenyl, alkynyl and substituted alkynyl where alkynyl are as defined herein.
- cycloalkyl refers to cyclic alkyl groups of from 3 to 20 carbon atoms having a single cyclic ring or multiple condensed rings.
- Such cycloalkyl groups include, by way of example, single ring structures such as cyclopropyl, cyclobutyl, cyclopentyl, cyclooctyl, and the like, or multiple ring structures such as adamantanyl, and the like.
- substituted cycloalkyl refers to cycloalkyl groups having from 1 to 5 substituents selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, -SO-alkyl, -SO-substituted alkyl
- cycloalkenyl refers to cyclic alkenyl groups of from 4 to 20 carbon atoms having a single cyclic ring or fused rings and at least one point of internal unsaturation.
- suitable cycloalkenyl groups include, for instance, cyclobut-2-enyl, cyclopent-3- enyl, cyclooct-3-enyl and the like.
- substituted cycloalkenyl refers to cycloalkenyl groups having from 1 to 5 substituents selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, aminoacyl, aminoacyloxy, oxyaminoacyl, azido, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, -SO-alkyl,
- halo or halogen refers to fluoro, chloro, bromo and iodo.
- Haloalkyl refers to alkyl as defined above substituted by 1-4 halo groups as defined above, which may be the same or different, such as 3-fluorododecyl, 12,12,12- trifluorododecyl, 2-bromooctyl, -3-bfomo-6-chloroheptyl, and the like.
- heteroaryl refers to an aromatic group of from 1 to 15 carbon atoms and 1 to 4 heteroatoms selected from oxygen, nitrogen and sulfur within at least one ring (if there is more than one ring).
- heteroaryl groups can be optionally substituted with 1 to 5 substituents selected from the group consisting of acyloxy, hydroxy, thiol, acyl, alkyl, alkoxy, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, substituted alkyl, substituted alkoxy, substituted alkenyl, substituted alkynyl, substituted cycloalkyl, substituted cycloalkenyl, amino, aminoacyl, acylamino, alkaryl, aryl, aryloxy, azido, carboxyl, carboxylalkyl, cyano, halo, nitro, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, aminoacyloxy, oxyacylamino, thioalkoxy, substituted thioalkoxy, thioaryloxy, thioheteroaryloxy, -SO
- heteroaryloxy refers to the group heteroaryl-O-.
- heteroarylene refers to the diradical group derived from heteroaryl or substituted heteroaryl as defined above, and is exemplified by the groups 2,6-pyridylene, 2,4- pyridiylene, 1,2-quinolinylene, 1,8-quinolinylene, 1 ,4-benzofuranylene, 2,5-pyridinylene, 1,3- morpholinylene, 2,5-indolenyl, and the like.
- heterocycle or “heterocyclic” refers to a monoradical saturated or unsaturated group having a single ring or multiple condensed rings, from 1 to 40 carbon atoms and from 1 to 10 hetero atoms, preferably 1 to 4 heteroatoms, selected from nitrogen, sulfur, phosphorus, and/or oxygen within the ring.
- heterocyclic groups can be optionally substituted with 1 to 5, and preferably 1 to 3 substituents, selected from the group consisting of alkoxy, substituted alkoxy, cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, acyl, acylamino, acyloxy, amino, aminoacyl, aminoacyloxy, oxyaminoacyl, cyano, halogen, hydroxyl, keto, thioketo, carboxyl, carboxylalkyl, thioaryloxy, thioheteroaryloxy, thioheterocyclooxy, thiol, thioalkoxy, substituted thioalkoxy, aryl, aryloxy, heteroaryl, heteroaryloxy, heterocyclic, heterocyclooxy, hydroxyamino, alkoxyamino, nitro, -SO-alkyl
- heterocyclic groups can have a single ring or multiple condensed rings.
- nitrogen heterocycles and heteroaryls include, but are not limited to, pyrrole, imidazole, pyrazole, pyridine, pyrazine, pyrimidine, pyridazine, indolizine, isoindole, indole, indazole, purine, quinolizine, isoquinoline, quinoline, phthalazine, naphthylpyridine, quinoxaline, quinazoline, cinnoline, pteridine, carbazole, carboline, phenanthridine, acridine, phenanthroline, isothiazole, phenazine, isoxazole, phenoxazine, phenothiazine, imidazolidine, imidazoline, piperidine, piperazine, indoline, morpholino, piperidinyl, tetrahydrofur
- a preferred class of heterocyclics include “crown compounds” which refers to a specific class of heterocyclic compounds having one or more repeating units of the formula
- crown compounds include, by way of example only, [-(CH ⁇ - NH-] 3 , [-((CH 2 ) 2 -O) 4 -((CH 2 ) 2 -NH) 2 ] and the like. Typically such crown compounds can have from 4 to 10 heteroatoms and 8 to 40 carbon atoms.
- heterocyclooxy refers to the group heterocyclic-O-.
- thioheterocyclooxy refers to the group heterocyclic-S-.
- heterocyclene refers to the diradical group derived from a heterocycle as defined herein, and is exemplified by the groups 2,6-morpholino, 2,5-morpholino and the like.
- oxyacylamino refers to the group -OC(O)NRR where each R is independently hydrogen, alkyl, substituted alkyl, aryl, heteroaryl, or heterocyclic wherein alkyl, substituted alkyl, aryl, heteroaryl and heterocyclic are as defined herein.
- thiol refers to the group -SH.
- thioalkoxy refers to the group -S-alkyl.
- substituted thioalkoxy refers to the group -S-substituted alkyl.
- thioaryloxy refers to the group aryl-S- wherein the aryl group is as defined above including optionally substituted aryl groups also defined above.
- heteroaryloxy refers to the group heteroaryl-S- wherein the heteroaryl group is as defined above including optionally substituted aryl groups as also defined above.
- any of the above groups which contain one or more substituents it is understood, of course, that such groups do not contain any substitution or substitution patterns which are sterically impractical and/or synthetically non-feasible.
- the compounds of this invention include all stereochemical isomers arising from the substitution of these compounds.
- Alkyl optionally interrupted by 1-5 atoms chosen from O, S, or N refers to alkyl as defined above in which the carbon chain is interrupted by O, S, or N.
- ethers, sulfides, and a ines for example 1-methoxydecyl, 1-pentyloxynonane, l-(2- isopropoxyethoxy)-4-methylnonane, l-(2-ethoxyethoxy)dodecyl, 2-(t-butoxy)heptyl,
- Heteroarylalkyl refers to heteroaryl as defined above linked to alkyl as defined above, for example pyrid-2-ylmethyl, 8-quinolinylpropyl, and the like.
- Optional or “optionally” means that the subsequently described event or circumstance may or may not occur, and that the description includes instances where said event or circumstance occurs and instances in which it does not.
- optionally substituted alkyl means that alkyl may or may not be substituted by those groups enumerated in the definition of substituted alkyl.
- pharmaceutically acceptable salt refers to salts which retain the biological effectiveness and properties of the multibinding compounds of this invention and which are not biologically or otherwise undesirable.
- the multibinding compounds of this invention are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto.
- Salts derived from inorganic bases include by way of example only, sodium, potassium, lithium, ammonium, calcium and magnesium salts.
- Salts derived from organic bases include, but are not limited to, salts of primary, secondary and tertiary amines, such as alkyl amines, dialkyl amines, trialkyl amines, substituted alkyl amines, di(substituted alkyl) amines, tri(substituted alkyl) amines, alkenyl amines, dialkenyl amines, trialkenyl amines, substituted alkenyl amines, di(substituted alkenyl) amines, tri(substituted alkenyl) amines, cycloalkyl amines, di(cycloalkyl) amines, tri(cycloalkyl) amines, substituted cycloalkyl amines, substituted cycloalkyl amines, substituted
- amines where the two or three substituents, together with the amino nitrogen, form a heterocyclic or heteroaryl group.
- suitable amines include, by way of example only, isopropylamine, trimethyl amine, diethyl amine, tri(wo-propyl) amine, tri(n-propyl) amine, ethanolamine, 2-dimethylaminoethanol, fromemamine, lysine, arginine, histidine, caffeine, procaine, hydrabamine, choline, betaine, ethylenem ' amine, glucosamine, N-alkylglucamines, theobromine, purines, piperazine, piperidine, morpholine, N-ethylpiperidine, and the like.
- carboxylic acid derivatives would be useful in the practice of this invention, for example, carboxylic acid amides, including carboxamides, lower alkyl carboxamides, dialkyl carboxamides,
- Salts derived from inorganic acids include hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
- Salts derived from organic acids include acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, malic acid, malonic acid, succinic acid, maleic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid,
- protecting group refers to any group which when bound to one or more hydroxyl, thiol, amino or carboxyl groups of the compounds prevents reactions from occurring at these groups and which protecting group can be removed by conventional chemical or enzymatic steps to reestablish the hydroxyl, thiol, amino or carboxyl group. See, generally, T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, 2 nd Ed., 1991, John Wiley and Sons, N.Y.
- removable blocking group employed is not critical and preferred removable hydroxyl blocking groups include conventional- substituents such as allyl, benzyl, acetyl, chloroacetyl, thiobenzyl, benzylidine, phenacyl, t-butyl-diphenylsilyl and any other group that can be introduced chemically onto a hydroxyl functionality and later selectively removed either by chemical or enzymatic methods in mild conditions compatible with the nature of the product.
- conventional- substituents such as allyl, benzyl, acetyl, chloroacetyl, thiobenzyl, benzylidine, phenacyl, t-butyl-diphenylsilyl and any other group that can be introduced chemically onto a hydroxyl functionality and later selectively removed either by chemical or enzymatic methods in mild conditions compatible with the nature of the product.
- Preferred removable amino blocking groups include conventional substituents such as t-butyoxycarbonyl (t-BOC), benzyloxycarbonyl (CBZ), fluorenylmethoxycarbonyl (FMOC), allyloxycarbonyl (ALOC) and the like, which can be removed by conventional conditions compatible with the nature of the product.
- t-BOC t-butyoxycarbonyl
- CBZ benzyloxycarbonyl
- FMOC fluorenylmethoxycarbonyl
- ALOC allyloxycarbonyl
- Preferred carboxyl protecting groups include esters such as methyl, ethyl, propyl, t-butyl etc. which can be removed by mild hydrolysis conditions compatible with the nature of the product.
- inert organic solvent or “inert solvent” mean a solvent inert under the conditions of the reaction being described in conjunction therewith [including, for example, benzene, toluene, acetonitrile, tetrahydrofuran (“THF”), dimethylformamide
- DMF chloroform
- CHC1 3 .. chloroform
- methylene chloride or dichloromethane or "CH 2 C1 2 "
- diethyl ether diethyl ether
- ethyl acetate acetone
- methylethyl ketone diethyl ether
- acetone methylethyl ketone
- methanol ethanol
- propanol isopropanol
- tert-butanol dioxane
- pyridine and the like.
- the solvents used in the reactions of the present invention are inert solvents.
- K + channel refers to a structure comprised of integral membrane proteins that functions to allow K + to equilibrate across a membrane according to its electrochemical gradient and at rates that are diffusion limited.
- Ligand denotes a compound that is a binding partner for a K + channel receptor, and is bound thereto, for example, by complementarity.
- the specific region or regions of the ligand molecule that is recognized by the ligand binding site of aKT channel receptor is designated as the "ligand domain".
- a ligand may be either capable of binding to a receptor by itself, or may require the presence of one or more non-ligand components for binding (e.g. ions, a lipid molecule, a solvent molecule, and the like).
- Ligands useful in this invention comprise K + channel modulators such as qui dine, 6,94 glibenclamide, procaine, tetraethyl ammonium, 20 clofilium, 102 melperone, 8 pinacidil, WAY-
- potassium channel ligands that are currently known can be used in the preparation of multibinding compounds of this invention (Table 2)
- portions of the ligand structure that are not essential for molecular recognition and binding activity may be varied substantially, replaced with unrelated structures and, in some cases, omitted entirely without affecting the binding interaction.
- the term "ligand” is not intended to be limited to compounds known to be useful as K + channel receptor-binding compounds (e.g., known drugs), in that ligands that exhibit marginal activity or lack useful activity as monomers can be highly active as multibinding compounds, because of the biological benefit conferred by multivalency.
- the primary requirement for a ligand as defined herein is that it has a ligand domain, as defined above, which is available for binding to a recognition site on a K + channel.
- ligand or "ligands” is intended to include the racemic ligands as well as the individual stereoisomers of the ligands, including pure enantiomers and non-racemic mixtures thereof.
- the scope of the invention as described and claimed encompasses the racemic forms of the ligands as well as the individual enantiomers and non-racemic mixtures thereof.
- ligand binding site denotes a site on a K + channel receptor that recognizes a ligand domain and provides a binding partner for the ligand.
- the ligand binding site may be defined by monomeric or multimeric structures. This interaction may be capable of producing a unique biological effect, for example agonism, antagonism, modulation, or may maintain an ongoing biological event, and the like.
- K + channel ligand binding sites of K + channel receptors that participate in biological multivalent binding interactions are constrained to varying degrees by their intra- and intermolecular associations.
- K + channel ligand binding sites may be covalently joined in a single structure, noncovalently associated in one or more multimeric structures, embedded in a membrane or biopolymer matrix, and so on, and therefore have less translational and rotational freedom than if the same sites were present as monomers in solution.
- agonist refers to a ligand that when bound to a K + channel stimulates its activity.
- antagonist refers to a ligand that when bound to a K + channel inhibits its activity.
- Channel block or activation may result from allosteric effects of ligand binding to the channel rather than occupancy of the channel pore. These allosteric effects may produce changes in protein conformation that affect K + binding sites, gating mechanisms and/or the pore region (i.e., ion permeation).
- a potassium channel can exist in several modes: C (closed resting state); C* (activated closed state); O (open state); and I (inactivated state). 44 The probability that a channel will exist in one of these four states changes with voltage.
- a given ligand may have different binding affinities for different states, and be capable of producing agonist or antagonist activity.
- modulatory effect is intended to refer to the ability of a ligand to change the activity of a K + channel through binding to the channel.
- Multibinding agent or “multibinding compound” refers herein to a compound that has from 2 to 10 K + channel ligands as defined herein (which may be the same or different) covalently bound to one or more linkers (which may be the same or different), and is capable of multivalency, as defined below.
- a multibinding compound provides an improved biologic and/or therapeutic effect compared to that of the same number of unlinked ligands available for binding to the ligand binding sites on a K + channel or channels.
- improved "biologic and/or therapeutic effect” include increased ligand-receptor binding interactions (e.g., increased affinity, increased ability to elicit a functional change in the target, improved kinetics), increased selectivity for the target, increased potency, increased efficacy, decreased toxicity, increased therapeutic index, improved duration of action, improved bioavailability, improved pharmacokinetics, improved activity spectrum, and the like.
- the multibinding compounds of this invention will exhibit at least one, and preferably more than one, of the above-mentioned effects.
- Univalency refers to a single binding interaction between one ligand with one ligand binding site as defined herein. It should be noted that a compound having multiple copies of a ligand (or ligands) exhibits univalency when only one ligand of that compound interacts with a ligand binding site. Examples of univalent interactions are depicted below. ⁇ s ⁇ - univalent interaction
- Multivalency refers to the concurrent binding of from 2 to 10 linked ligands, which may be the same or different, and two or more corresponding ligand binding sites, which may be the same or different.
- An example of trivalent binding is depicted below for illustrative purposes.
- library refers to at least 3, preferably from 10 2 to 10 9 and more preferably from 10 2 to 10 4 multimeric compounds. Preferably, these compounds are prepared as a multiplicity of compounds in a single solution or reaction mixture which permits facile synthesis thereof.
- the library of multimeric compounds can be directly assayed for multibinding properties.
- each member of the library of multimeric compounds is first isolated and, optionally, characterized. This member is then assayed for multibinding properties.
- selection refers to a set of multimeric compounds which are prepared either sequentially or concurrently (e.g., combinatorially).
- the collection comprises at least 2 members; preferably from 2 to 10 9 members and still more preferably from 10 to 10 4 members.
- multimeric compound refers to compounds comprising from 2 to 10 ligands covalently connected through at least one linker which compounds may or may not possess multibinding properties (as defined herein).
- pseudohalide refers to functional groups which react in displacement reactions in a manner similar to a halogen.
- Such functional groups include, by way of example, mesyl, tosyl, azido and cyano groups.
- linker refers to a group or groups that covalently links from 2 to 10 ligands (as defined above) in a manner that provides a compound capable of multivalency.
- the linker is a ligand-orienting entity that permits attachment of multiple copies of a ligand (which may be the same or different) thereto.
- linker includes everything that is not considered to be part of the ligand, e.g., ancillary groups such as solubilizing groups, lipophilic groups, groups that alter pharmacodynamics or pharmacokinetics, groups that modify the diffusability of the multibinding compound, spacers that attach the ligand to the linker, groups that aid the ligand-orienting function of the linker, for example, by imparting flexibility or rigidity to the linker as a whole, or to a portion thereof, and so on.
- ancillary groups such as solubilizing groups, lipophilic groups, groups that alter pharmacodynamics or pharmacokinetics, groups that modify the diffusability of the multibinding compound, spacers that attach the ligand to the linker, groups that aid the ligand-orienting function of the linker, for example, by imparting flexibility or rigidity to the linker as a whole, or to a portion thereof, and so on.
- linker does not, however, cover solid inert supports such as beads, glass particles, rods, and the like, but it is to be understood that the multibinding compounds of this invention can be attached to a solid understood that the multibinding compounds of this invention can be attached to a solid support if desired, for example, for use in separation and purification processes and for similar applications.
- linker or linkers that joins the ligands presents them to their array of ligand binding sites. Beyond presenting these ligands for multivalent interactions with ligand binding sites, the linker spatially constrains these interactions to occur within dimensions defined by the linker.
- the linkers used in this invention are selected to allow multivalent binding of ligands to any desired ligand binding sites of a K + channel, whether such sites are located wilhin the cell membrane, interiorly (e.g., within a channel/translocation pore), both interiorly and on the periphery of a channel, at the boundary region between the lipid bilayer and the channel, or at any intermediate position thereof.
- the preferred linker length will vary depending on the distance between adjacent ligand binding sites, and the geometry, flexibility and composition of the linker.
- the length of the linker will preferably be in the range of about 2 A to about lOOA, more preferably from about 2A to about 5 ⁇ A and even more preferably from about 5 to about 20A.
- the ligands are covalently attached to the linker or linkers using conventional chemical techniques.
- the reaction chemistries resulting in such linkage are well known in the art and involve the use of reactive functional groups present on the linker and ligand.
- the reactive functional groups on the linker are selected relative to the functional groups available on the ligand for coupling, or which can be introduced onto the ligand for this purpose. Again, such reactive functional groups are well known in the art.
- reaction between a carboxylic acid of either the linker or the ligand and a primary or secondary amine of the ligand or the linker in the presence of suitable well-known activating agents results in formation of an amide bond covalently linking the ligand to the linker; reaction between an amine group of either the linker or the ligand and a sulfonyl halide of the ligand or the linker results in formation of a sulfonamide bond covalently linking the ligand to the linker; and reaction between an alcohol or phenol group of either the linker or the ligand and an alkyl or aryl halide of the ligand or the linker results in formation of an ether bond covalently linking the Ugand to the linker.
- the linker is attached to the ligand at a position that retains ligand domain-ligand binding site interaction and specifically which permits the ligand domain of the ligand to orient itself to bind to the ligand binding site. Such positions and synthetic protocols for linkage are well known in the art.
- linker embraces everything that is not considered to be part of the ligand.
- the relative orientation in which the ligand domains are displayed depends both on the particular point or points of attachment of the ligands to the linker, and on the framework geometry.
- the determination of where acceptable substitutions can be made on a Hgand is typically based on prior knowledge of structure-activity relationships of the ligand and/or congeners and/or structural information about ligand-receptor complexes (e.g., X-ray crystallography, NMR, and the like).
- Such positions and synthetic protocols for linkage are well known in the art and can be determined by those with ordinary skiU in the art (see, e.g., METHODS OF PREPARATION, Examples 1-29 and Figures 7 to 21.
- linker-ligand conjugate may be tested for retention of activity in a relevant assay system (see Utility and Testing below for representative assays).
- the multibinding compound is a bivalent compound in which two ligands are covalently linked, or a trivalent compound, in which three ligands are covalently linked.
- Linker design is further discussed under METHODS OF PREPARATION.
- “Potency” as used herein refers to the minimum concentration at which a ligand is able to achieve a desirable biological or therapeutic effect.
- the potency of a ligand is typically proportional to its affinity, for its receptor. In some cases, the potency may be non- linearly correlated with its affinity.
- the dose-response curve of each is determined under identical test conditions (e.g., in an in vitro or in vivo assay, in an appropriate animal model (such as a human patient)). The finding that the multibinding agent produces an equivalent biologic or therapeutic effect at a lower concentration than the aggregate unlinked ligand (e.g., on a per weight, per mole or per ligand basis) is indicative of enhanced potency.
- Selectivity is a measure of the binding preferences of a Hgand for different receptors.
- the selectivity of a ligand with respect to its target receptor relative to another receptor is given by the ratio of the respective values of Kj (i.e., the dissociation constants for each ligand-receptor complex) or, in cases where a biological effect is observed below the K d , the ratio of the respective EC 50 s or IC 50 s (i.e., the concentrations that produce 50% of the maximum response for the ligand interacting with the two distinct receptors).
- treatment refers to any treatment of a disease or condition in a mammal, particularly a human, and includes:
- the treatment constitutes prophylactic treatment for the pathologic condition; (ii) inhibiting the disease or condition, i.e., arresting its development; (iii) relieving the disease or condition, i.e., causing regression of the disease or condition; or
- disease or condition which is modulated by treatment with a multibinding K + channel ligand covers all disease states and/or conditions that are generally acknowledged in the art to be usefully treated with a ligand for a K + channel in general, and those disease states and/or conditions that have been found to be usefully treated by a specific multibinding compound of our invention, i.e., the compounds of Formula I.
- disease states include, by way of example only, hypertension, cerebral ischemia, cardiac arrythmias (particularly, arrythmias resulting from potassium-related changes in membrane potential and conduction), cardiac hypertrophy due to systolic or diastolic overload, congestive heart failure, and the like.
- therapeutically effective amount refers to that amount of multibinding compound that is sufficient to effect treatment, as defined above, when administered to a mammal in need of such treatment.
- the therapeutically effective amount will vary depending upon the subject and disease condition being treated, the weight and age of the subject, the severity of the disease condition, the manner of administration and the like, which can readily be determined by one of ordinary skill in the art.
- pharmaceutically acceptable excipient is intended to include vehicles and carriers capable of being coadministered with a multibinding compound to facilitate the performance of its intended function.
- vehicles and carriers capable of being coadministered with a multibinding compound to facilitate the performance of its intended function.
- the use of such media for ' pharmaceutically active substances is well known in the art.
- vehicles and carriers include solutions, solvents, dispersion media, delay agents, emulsions and the like. Any other conventional carrier suitable for use with the multibinding compounds also falls within the scope of the present invention.
- the linker or linkers when covalently attached to multiple copies of the ligands, provides a biocompatible, substantially non-immunogenic multibinding compound.
- the biological activity of the multibinding K + channel compound is highly sensitive to the geometry, composition, size, length, flexibility or rigidity, the presence or absence of anionic or cationic charge, the relative hydrophobicity/hydrophilicity, and similar properties of the linker. Accordingly, the linker is preferably chosen to maximize the biological activity of the compound.
- the Hnker may be biologically "neutral,” i.e., not itself contribute any additional biological activity to the multibinding compound, or it may be chosen to further enhance the biological activity of the compound.
- the linker may be chosen from any organic molecule construct that orients two or more ligands for binding to the receptors to permit multivalency.
- the linker can be considered as a "framework" on which the ligands are arranged in order to bring about the desired ligand-orienting result, and thus produce a multibinding compound.
- different orientations of ligands can be achieved by varying the geometry of the framework (linker) by use of mono- or polycyclic groups, such as aryl and/or heteroaryl groups, or structures incorporating one or more carbon-carbon multiple bonds (alkenyl, alkenylene, alkynyl or alkynylene groups).
- the optimal geometry and composition of frameworks (linkers) used in the multibinding compounds of this invention are based upon the properties of their intended receptors. For example, it is preferred to use rigid cyclic groups (e.g., aryl, heteroaryl), or non-rigid cyclic groups (e.g., cycloalkyl or crown groups) to reduce conformational entropy when such may be necessary to achieve energetically coupled binding.
- rigid cyclic groups e.g., aryl, heteroaryl
- non-rigid cyclic groups e.g., cycloalkyl or crown groups
- hydrophobic hydropbilic characteristics of the linker as well as the presence or absence of charged moieties can readily be controlled by the skilled artisan.
- hydrophobic nature of a linker derived from hexamethylene diamine (H 2 N(CH 2 ) 6 NH 2 ) or related polyamines can be modified to be substantially more hydrophilic by replacing the alkylene group with a poly(oxyalkylene) group such as found in the commercially available
- Different frameworks can be designed to provide preferred orientations of the ligands.
- the identification of an appropriate framework geometry for ligand domain presentation is an important first step in the construction of a multi binding agent with enhanced activity.
- FIG. 2 illustrates a useful strategy for determining an optimal framework display orientation for ligand domains and can be used for preparing the bivalent compounds of this invention.
- Various alternative strategies known to those skilled in the art of molecular design can be substituted for the one described here.
- the ligands are attached to a central core structure such as phenyldiacetylene (Panel A) or cyclohexane dicarboxylic acid (Panel B).
- the ligands are spaced apart from the core by an attaching moiety of variable lengths m and n. If the ligand possesses multiple attachment sites (see discussion below), the orientation of the ligand on the attaching moiety may be varied as well.
- the positions of the display vectors around the central core structures are varied, thereby generating a collection of compounds.
- the process may require the use of multiple copies of the same central core structure or combinations of different types of display cores. It is to be noted that core structures other than those shown here can be used for determining the optimal framework display orientation of the ligands.
- core structures other than those shown here can be used for determining the optimal framework display orientation of the ligands.
- the above-described technique can be extended to trivalent compounds and compounds of higher-order valency.
- linkers that are suitable for use in this invention fall into this category. Others can be readily synthesized by methods known in the art, and as described below. Examples of linkers include aliphatic moieties, aromatic moieties, steroidal moieties, peptides, and the like. Specific examples are peptides or polyamides, hydrocarbons, aromatics, heterocyclics, ethers, lipids, cationic or anionic groups, or a combination thereof.
- linker can be modified by the addition or insertion of anciUary groups into the linker, for example, to change the solubility of the multibinding compound (in water, fats, lipids, biological fluids, etc.), hydrophobicity, hydrophilicity, linker flexibility, antigenicity, stability, and the like.
- the introduction of one or more poly(ethylene glycol) (PEG) groups onto the linker enhances the hydrophilicity and water solubility of the multibinding compound, increases both molecular weight and molecular size and, depending on the nature of the uhPEGylated linker, may increase the in vivo retention time. Further, PEG may decrease antigenicity and potentially enhances the overall rigidity of the linker.
- PEG poly(ethylene glycol)
- Ancillary groups that enhance the water solubility hydrophiHcity of the linker, and accordingly, the resulting multibinding compounds, are useful in practicing this invention.
- anciUary groups such as, for example, small repeating units of ethylene glycols, alcohols, polyols, (e.g., glycerin, glycerol propoxylate, saccharides, including mono-, oligosaccharides, etc.) carboxylates (e.g., small repeating units of glutamic acid, acrylic acid, etc.), amines (e.g., tefraemylenepentamine), and the like to enhance the water solubiHty and/or hydrophilicity of the multibinding compounds of this invention.
- the ancillary group used to improve water solubility/hydrophiHcity will be a polyether.
- the ancillary group will contain a small number of repeating ethylene oxide (-CH 2 CH 2 O-) units.
- lipophilic ancillary groups within the structure of the linker to enhance the lipophilicity and/or hydrophobicity of the compounds of Formula I is also within the scope of this invention.
- Lipophilic groups useful with the linkers of this invention include, but are not Hmited to, lower alkyl, aromatic groups and polycyclic aromatic groups.
- aromatic groups may be either unsubstituted or substituted with other groups, but are at least substituted with a group which allows their covalent attachment to the linker.
- aromatic groups incorporates both aromatic hydrocarbons and heterocyclic aromatics.
- Other lipophilic groups useful with the linkers of this invention include fatty acid derivatives which may or may not form micelles in aqueous medium and other specific lipophilic groups which modulate interactions between the multibinding compound and biological membranes.
- ancillary groups which result in the compound of Formula I being incorporated into a vesicle, such as a Hposome, or a micelle.
- lipid refers to any fatty acid derivative that is capable of forming a bilayer or miceUe such that a hydrophobic portion of the lipid material orients toward the bilayer while a hydrophilic portion orients toward the aqueous phase.
- HydrophiHc characteristics derive from the presence of phosphato, carboxylic, sulfato, amino, sulfhydryl, nitro and other like groups well known in the art. Hydrophobicity could be conferred by the inclusion of groups that include, but are not limited to, long chain saturated and unsaturated aliphatic hydrocarbon groups of up to 20 carbon atoms and such groups substituted by one or more aryl, heteroaryl, cycloalkyl, and/or heterocyclic group(s).
- Preferred lipids are phosphoglycerides and sphingolipids, representative examples of which include phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, phosphatidylinositol, phosphatidic acid, palmitoyleoyl phosphatidylcholine, lysophosphatidylcholine, lysophosphatidyl-ethanolamine, dipalmitoylphosphatidylchohne, dioleoylphosphatidyl- choline, distearoyl-phosphatidylcholine and dilinoleoylphosphatidylcholine.
- lipid Other compounds lacking phosphorus, such as sphingolipid and glycosphingolipid families, are also within the group designated as lipid. Additionally, the amphipathic lipids described above may be mixed with other lipids including triglycerides and sterols.
- the flexibUity of the linker can be manipulated by the inclusion of anciUary groups which are bulky and/or rigid.
- anciUary groups which are bulky and/or rigid.
- the presence of bulky or rigid groups can hinder free rotation about bonds in the linker, or bonds between the linker and the ancillary group(s), or bonds between the linker and the functional groups.
- Rigid groups can include, for example, those groups whose conformational freedom is restrained by the presence of rings and/or ⁇ -bonds, for example, aryl, heteroaryl and heterocychc groups.
- Other groups which can impart rigidity include polypeptide groups such as oHgo- or polyproline chains.
- Rigidity can also be imparted electrostatically.
- the ancillary groups are either positively or negatively charged, the similarly charged ancUlary groups will force the linker into a configuration affording the maximum distance between each of the like charges.
- the energetic cost of bringing the like-charged groups closer to each other which is inversely related to the square of the distance between the groups, will tend to hold the linker in a configuration that maintains the separation between the like-charged ancUlary groups.
- ancUlary groups bearing opposite charges will tend to be attracted to their oppositely charged counterparts and potentially may enter into both inter- and intramolecular ionic bonds.
- Bulky groups can include, for example, large atoms, ions (e.g., iodine, sulfur, metal ions, etc.) or groups containing large atoms, polycyclic groups, including aromatic groups, non-aromatic groups and structures incorporating one or more carbon-carbon ⁇ -bonds (i.e., alkenes and alkynes). Bulky groups can also include oligomers and polymers which are branched- or straight-chain species. Species that are branched are expected to increase the rigidity of the structure more per unit molecular weight gain than are straight-chain species.
- rigidity is imparted by the presence of alicyclic (e.g., cycloalkyl), aromatic and heterocyclic groups. In other preferred embodiments, this comprises one or more six-membered rings. In still further preferred embodiments, the ring is an aryl group such as, for example, phenyl or naphthyl, or a macrocyclic ring such as, for example, a crown compound.
- the multibinding compounds described herein comprise 2-10 ligands attached covalently to a linker that links the ligands in a manner that allows their multivalent binding to ligand binding sites of K + channels.
- the linker spatially constrains these interactions to occur within dimensions defined by the linker. This and other factors increases the biologic and/or therapeutic effect of the multibinding compound as compared to the same number of ligands used in monobinding form.
- the compounds of this invention are preferably represented by the empirical formula (L) p (X) q where L, X, p and q are as defined above. This is intended to include the several ways in which the ligands can be linked together in order to achieve the objective of multivalency, and a more detailed explanation is provided below.
- the linker may be considered as a framework to which ligands are attached.
- the ligands can be attached at any suitable position on this framework, for example, at the termini of a linear chain or at any intermediate position thereof.
- the simplest and most preferred multibinding compound is a bivalent compound which can be represented as L-X-L, where L is a ligand and is the same or different and X is the linker.
- a trivalent compound could also be represented in a linear fashion, i.e., as a sequence of repeated units L-X-L-X-L, in which L is a ligand and is the same or different at each occurrence, as is X.
- a trivalent compound can also comprise three ligands attached to a central core, and thus be represented as (L) 3 X, where the linker X could include,for example, an aryl or cycloalkyl group.
- Tetravalent compounds can be represented in a linear array:
- multibinding compounds of this invention containing from 5-10 ligands.
- a central Hnker such as an aryl, cycloalkyl or heterocyclyl group, or a crown compound
- the formula (L) p (X) q is also intended to represent a cyclic compound of formula (-L- X-) n ,where n is 2-10.
- m is an integer of from 0 to 20;
- X' at each separate occurrence is -O-, -S-, -S(O)-, - S(O) 2 -, -NR-, -N* R R ' -, -C(O)-, -C(O)O-, -C(O)NH-, -C(S), -C(S)O-, -C(S)NH- or a covalent bond, where R and R ' at each separate occurrence are as defined below for R' and R"; Z is at each separate occurrence selected from alkylene, substituted alkylene, alkylalkoxy, cycloalkylene, substituted cycloalkylene, alkenylene, substituted alkenylene, alkynylene, substituted alkynylene, cycloalkenylene, substituted alkenylene, arylene, substituted arylene, heteroarylene, heterocyclene, substituted heterocyclene,
- n 0, 1 or 2; and R' and R" at each separate occurrence re selected from hydrogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl or heterocychc.
- linker moiety can be optionally substituted at any atom therein by one or more alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl and heterocyclic group.
- the simplest (and preferred) construct is a bivalent compound which can be represented as L-X-L, where L is a K " channel ligand that is the same or different at each occurrence, and X is the linker. Accordingly, examples of the preparation of a bivalent ligand are given below as an illustration of the manner in which multibinding compounds of Formula I are obtained.
- the linker or linkers can be attached to different positions on the ligand molecule to achieve different orientations of the ligand domains and thereby facilitate multivalency.
- the positions that are potentially available for linking a benzofuran such as amiodarone are indicated by arrows in the structure shown in
- Preferred positions of attachment suggested by known SAR are iUustrated in the reaction schemes of Figures 7 to 21.
- Examples of ligands are shown in Table 4.
- Certain K + channel ligands may be chiral and exhibit stereoselectivity.
- the most active enantiomers are preferably used as ligands in the multibinding compounds of this invention.
- the chiral resolution of enantiomers is accomplished by well known procedures that result in the formation of diastereomeric derivatives or salts, followed by conventional separation by chromatographic procedures or by fractional crystallization (see, e.g., Bossert, et al.,Angew. Chem. Int. Ed., 20:762-769 (1981) and U.S. Patent No. 5,571,827 and references cited therein). They may also be obtained by asymmetric synthesis.
- the ligands are covalently attached to the linker using conventional chemical techniques.
- the reaction chemistries resulting in such linkage are well known in the art and involve the coupling of reactive functional groups present on the linker and ligand. In some cases, it may be necessary to protect portions of the ligand that are not involved in linking reactions. Protecting groups for this purpose are well known in the art and are indicated generally in the reaction schemes by the symbols PG and PG'.
- the reactive functional groups on the linker are selected relative to the functional groups on the Hgand that are available for coupling, or can be introduced onto the ligand for this purpose.
- the linker is coupled to ligand precursors, with the completion of ligand synthesis being carried out in a subsequent step.
- functional groups are lacking, they can be created by suitable chemistries that are described in standard organic chemistry texts such as J. March, Advanced Organic Chemistry, 4 th Ed. (Wiley- Interscience, N.Y., 1992). Examples of the chemistry for connecting Hgands by a linker are shown in Figure 6, where R 1 and R 2 represent a Hgand and/or the linking group.
- R 1 and R 2 represent a Hgand and/or the linking group.
- the linker to which the ligands or ligand precursors are attached comprises a "core" molecule having two or more functional groups with reactivity that is complementary to that of the functional groups on the ligand.
- Figure 3 illustrates the diversity of "cores” that are useful for varying the linker size, shape, length, orientation, rigidity, acidity/basicity, hydrophobicity/hydrophilicity, hydrogen bonding characteristics and number of ligands connected. This pictorial representation is intended only to illustrate the invention, and not to limit its scope to the structures shown.
- a solid circle is used to generically represent a core molecule, referred to as "Link" in the
- the solid circle is equivalent to a linker as defined above after reaction.
- the preferred compounds of Formula I are bivalent. Accordingly, and for the purpose of simplicity, most of the figures and reaction schemes below illustrate the synthesis of bivalent K + channel modulators. It should be noted, however, that the same techniques can be used to generate higher order multibinding compounds, i.e., the compounds of the invention where p is 3-10. (See, e.g., Figure 15 and 20.)
- Reactions performed under standard amide coupling conditions are carried out in an inert polar solvent (e.g., DMF, DMA) in the presence of a hindered base (e.g., TEA, DIPEA) and standard amide coupling reagents (e.g., DPP A, PyBOP, HATU, DCC).
- an inert polar solvent e.g., DMF, DMA
- a hindered base e.g., TEA, DIPEA
- standard amide coupling reagents e.g., DPP A, PyBOP, HATU, DCC
- Figures 16 and 17 show ligands coupled to a polypeptide core with a sidechain spacer.
- Solid phase peptide synthesis can be used to produce a wide variety of peptidic core molecules. Techniques well-known to those skilled in the art (including combinatorial methods) are used to vary the distance between ligand attachment sites on the core molecule, the number of attachment sites available for coupling, and the chemical properties of the core molecule.
- Orthogonal protecting groups are used to selectively protect functional groups on the core molecule, thus allowing ancUlary groups to be inserted into the linker of the multibinding compound and/or the preparation of "heterovalomers" (i.e., multibinding compounds with nonidentical ligands).
- AU of the synthetic strategies described above employ a step in which the ligand, attached to spacers or not, is symmetrically linked to functionally equivalent positions on a central core.
- Compounds of Formula I can also be synthesized using an asymmetric linear approach. This strategy is preferred when Unking two or more ligands at different points of connectivity (see, e.g., Figure 18) or when preparing heterovalomers (see, e.g., Figure 19).
- Isolation and purification of the compounds and intermediates described herein can be effected, if desired, by any suitable separation or purification such as, for example, filtration, extraction, crystallization, column chromatography, thin-layer chromatography, thick-layer chromatography, preparative low or high-pressure liquid chromatography or a combination of these procedures. Characterization is preferably by NMR and mass spectroscopy.
- the multibmding compounds of this invention can be used to modulate potassium channels in various tissues including heart, muscle, and neurons. They will typically be used for the treatment of diseases and conditions in mammals that involve or are mediated by K + channels, such as hypertension, cardiac arrythmias, cerebral ischemia, congestive heart failure, and the like.
- the multibinding compounds of this invention are tested in well-known and reliable assays and their activities are compared with those of the corresponding unlinked (i.e., monovalent) ligands.
- the binding affinity is determined by a radioHgand competitive inhibition assay. 23
- the ability of the present compounds to compete with [ 3 H]dofetilide or a similar radioactive ligand in binding to high- and low-affinity binding sites of guinea pig ventricular myocytes is measured in vitro.
- the binding affinity, calculated from competition curves, is compared with that of the monovalent ligand and/or monovalent linker-ligand conjugate.
- Antiarrhythmic effect of compounds of this invention may be determined in vivo in dogs with induced myocardial infarction and reproducibly inducible ventricular tachycardia or ventricular fibrillation. 1,22 Suppression of inducible arrhythmias is measured.
- the antifibrillatory and antiarrhythmic effects of the compounds of this invention may be determined in vivo using a canine model of sudden death. 7 Reduction of the incidence of programmed electrical stimulation (PES) induced ventricular tachycardia and protection against ischemia-induced ventricular fibrillation are measured.
- PES programmed electrical stimulation
- the antiarrhythmic effect of the compounds of this invention may be determined in vivo using the mouse chloroform model. 8 The percentage of animals showing normal sinus rhythm is measured.
- the antiarrhythmic effect of the compounds of this invention may be determined in vivo using the rat coronary ligation model. 8 Ventricular extrasy stolens occurring during the 30 minutes following the procedure are counted.
- the antiarrhythmic effect of the compounds of this invention may be determined in vitro or in vivo using rat coronary artery ligation/reperfusion models. 8 In the in vitro model, excised rat hearts are retrogradely perfused with a solution of the compound to be tested, then the coronary artery is ligated, followed by reperfusion. In the in vivo evaluation, the compound is ad ⁇ iistered i.p., then the coronary artery is ligated, followed by reperfusion. In both models the incidence and time to onset during reperfusion of ventricular extrasy stolen, tachyarrhythmia and fibriUation are measured. The antiarrhythmic effect of the compounds of this invention may be determined in vivo using an anesthetized rat model of ventricular arrhythmias. 9 The time to onset of ventricular extrasystoles is measured.
- the antiarrhythmic effect of the compounds of this invention may be determined in vivo using a canine myocardial infarction model where compound is administered 24 hours after ligation of the left anterior descending coronary artery. 11 Right ventricular effective refractory period, monophasic action potential duration and reduction of PES induced ventricular tachycardia and ventricular fibrillation are measured.
- the ability of the compounds of this invention to prolong the action potential (achieve a slower onset of active state block) and recover faster from block may be determined in vitro using rabbit ventricular myocytes. 13 ' 30 Development of block during a long depolarizing clamp and recovery from block are measured.
- the ability of the compounds of this invention to suppress repolarization arrhythmias may be determined in vitro using canine epicardium midmyocardium and endocardium and canine cardiac Purki ⁇ je fibers and in vivo using anesthetized rabbits. 26,58,62
- the ability of the compounds of this invention to suppress arrhythmias may be determined in vivo using the feline coronary occlusion and left stellate ganglion stimulation model, the conscious canine model of transient ischemia during exercise in the presence of a healed MI and the conscious canine model of complete occlusion after recent MI. 52,59
- the ability of the compounds of this invention to prolong action potential duration may be determined in vivo and in vitro using guinea pig hearts, 57 and in vitro using calf cardiac Purkinje fibers.
- the ability of the compounds of this invention to prevent atrial fibrillation (AF) may be determined using a canine model of sustained vagotonic AF. 68 Prevention of AF induction is measured. Reverse use dependence may also be determined.
- the effect of compounds of this invention on tachycardia may be determined in vitro using rabbit right atrial preparations. 2 Micro-electrode techniques are used to measure the ability to prolong the refractory period and thus prevent initiation of tachycardia.
- the effect of the compounds of this invention on tachyarrhythmias may be determined in vitro using guinea pig right ventricular papillary muscle. 27 The action potential duration at different extracellular potassium concentrations is measured.
- the effect of compounds of this invention on various specific potassium current may be determined in vitro using guinea pig ventricular myocytes and sinoatrial node cells, human atrial myocytes, canine ventricular muscle and Purkinje fibers, guinea pig papillary muscle, single voltage clamped guinea pig ventricular myocytes and human ventricular endomyocardium. 6,12,17,25,29,32,33,37,51,66,69,71
- the ability of compounds of this invention to inhibit potassium currents in a non- cardiac preparation may be determined using rat taste receptor cells. 60 Selectivity and/or Specificity:
- the ability of compounds of this invention to modulate the KATP channel may be determined using a 86 Rb efflux assay. 15,64 Thus, this is a potency assay.
- the selectivity and/or specificity of the compounds of this invention may be determined using CHO cell lines expressing specific recombinant potassium channel subtypes. 34 .
- the selectivity of compounds of this invention for various potassium channel currents may be determined in vitro using cloned K channels expressed in cells or ventricular myocytes. 12,34
- Antivasoconstrictor activity is determined as described in Brittain, et al., Physiologist, 28:325 (1985) as the concentration of a compound required to produce 50% vasorelaxation in KCl-contracted rabbit thoracic aorta strips in the presence of calcium.
- concentration of a compound required to inhibit coronary vasoconstriction induced by a thromboxane mimetic U-46619, i.e., 9, 11 -methanoepoxy-PGHj
- guinea pig Langendorff heart preparation is measured as described in Eltze, et al., Chirality, 2:233-240 (1990).
- Antihypertensive activity is determined in male spontaneously hypertensive rats by measurement of mean arterial blood pressure (Rovnyak, et al., J. Med. Chem., 35:3254-3263 (1992)). Tissue selectivity
- Selectivity for vascular smooth muscle as compared with cardiac muscle can be assessed by comparing the concentration of a multibinding compound that produces a 50% increase in coronary blood flow in an isolated guinea-pig heart with that required to inhibit myocardial contractility. See, e.g., Osterrieder, W. and Hoick, M., J. Cardiovasc Pharm., 13:754-9 (1989); and Cremers, et al., J. Cardiovasc. Pharm., 29:692-696 (1997).
- factors such as the proper juxtaposition of the individual Hgands of a multibinding compound with respect to the relevant array of binding sites on a target or targets is important in optimizing the interaction of the multibinding compound with its target(s) and to maximize the biological advantage through multivalency.
- One approach is to identify a library of candidate multibinding compounds with properties spanning the multibinding parameters that are relevant for a particular target. These parameters include: (1) the identity of ligand(s), (2) the orientation of ligands, (3) the valency of the construct, (4) linker length, (5) linker geometry, (6) linker physical properties, and (7) linker chemical functional groups.
- a single ligand or set of Ugands is (are) selected for incorporation into the libraries of candidate multibinding compounds which library is directed against a particular biological target or targets.
- the only requirement for the ligands chosen is that they are capable of interacting with the selected target(s).
- ligands may be known drugs, modified forms of known drugs, substructures of known drugs or substrates of modified forms of known drugs (which are competent to interact with the target), or other compounds.
- Ligands are preferably chosen based on known favorable properties that may be projected to be carried over to or amplified in multibinding forms. Favorable properties include demonstrated safety and efficacy in human patients, appropriate PK/ADME profiles, synthetic accessibUity, and desirable physical properties such as solubUity, logP, etc.
- ligands which display an unfavorable property from among the previous list may obtain a more favorable property through the process of multibinding compound formation; i.e., ligands should not necessarily be excluded on such a basis.
- a ligand that is not sufficiently potent at a particular target so as to be efficacious in a human patient may become highly potent and efficacious when presented in multibinding form.
- a ligand that is potent and efficacious but not of utility because of a non-mechanism-related toxic side effect may have increased therapeutic index (increased potency relative to toxicity) as a multibinding compound.
- Compounds that exhibit short in vivo half-lives may have extended half-lives as multibinding compounds.
- Physical properties of ligands that limit their usefulness e.g. poor bioavailabUity due to low solubility, hydrophobicity, hydrophUicity
- each Ugand at which to attach the ligand to the linker.
- the selected points on the Hgand/linker for attachment are functionalized to contain complementary reactive functional groups. This permits probing the effects of presenting the ligands to their receptor (s) in multiple relative orientations, an important multibinding design parameter.
- the only requirement for choosing attachment points is that attaching to at least one of these points does not abrogate activity of the ligand.
- Such points for attachment can be identified by structural information when avaUable. For example, inspection of a co-crystal structure of a protease inhibitor bound to its target allows one to identify one or more sites where linker attachment wUl not preclude the enzyme: inhibitor interaction.
- positions of attachment that do abrogate the activity of the monomeric ligand may also be advantageously included in candidate multibinding compounds in the library provided that such compounds bear at least one ligand attached in a manner which does not abrogate intrinsic activity. This selection derives from, for example, heterobivalent interactions within the context of a single target molecule.
- a receptor antagonist ligand bound to its target receptor and then consider modifying this ligand by attaching to it a second copy of the same ligand with a Hnker which allows the second ligand to interact with the same receptor molecule at sites proximal to the antagonist binding site, which include elements of the receptor that are not part of the formal antagonist binding site and/or elements of the matrix surrounding the receptor such as the membrane.
- the most favorable orientation for interaction of the second ligand molecule with the receptor/matrix may be achieved by attaching it to the linker at a position which abrogates activity of the Hgand at the formal antagonist binding site.
- Another way to consider this is that the SAR of individual ligands within the context of a multibinding structure is often different from the SAR of those same ligands in momomeric form.
- bivalent interaction focused on bivalent interactions of dimeric compounds bearing two copies of the same ligand joined to a single linker through different attachment points, one of which may abrogate the binding/activity of the monomeric ligand. It should also be understood that bivalent advantage may also be attained with heterodimeric constructs bearing two different ligands that bind to common or different targets.
- a 5HT 4 receptor antagonist and a bladder-selective muscarinic M 3 antagonist may be joined to a Hnker through attachment points which do not abrogate the binding affinity of the monomeric ligands for their respective receptor sites.
- the dimeric compound may achieve enhanced affinity for both receptors due to favorable interactions between the 5HT 4 ligand and elements of the M 3 receptor proximal to the formal M 3 antagonist binding site and between the M 3 ligand and elements of the 5HT 4 receptor proximal to the formal 5HT 4 antagonist binding site.
- the dimeric compound may be more potent and selective antagonist of overactive bladder and a superior therapy for urinary urge incontinence.
- linkages that are possible at those points.
- the most preferred types of chemical linkages are those that are compatible with the overall structure of the ligand (or protected forms of the ligand) readUy and generally formed, stable and intrinsically inocuous under typical chemical and physiological conditions, and compatible with a large number of available linkers. Amide bonds, ethers, amines, carbamates, ureas, and sulfonamides are but a few examples of preferred linkages.
- Linkers spanning relevant multibinding parameters through selection of valency, linker length, linker geometry, rigidity, physical properties, and chemical functional groups
- Linker length Linkers are chosen in a range of lengths to aUow the spanning of a range of inter-ligand distances that encompass the distance preferable for a given divalent interaction.
- the preferred distance can be estimated rather precisely from high-resolution structural information of targets, typically enzymes and soluble receptor targets.
- high-resolution structural information is not avaUable (such as 7TM G-protein coupled receptors)
- preferred linker distances are 2-20 A, with more preferred linker distances of 3-12 A.
- preferred linker distances are 20-100 A, with more preferred distances of 30-70 A.
- Linker geometry and rigidity The combination of Hgand attachment site, linker length, linker geometry, and linker rigidity determine the possible ways in which the ligands of candidate multibinding compounds may be displayed in three dimensions and thereby presented to their binding sites.
- Linker geometry and rigidity are nominaUy determined by chemical composition and bonding pattern, which may be controlled and are systematically varied as another sparining function in a multibinding array. For example, linker geometry is varied by attaching two ligands to the ortho, meta, and para positions of a benzene ring, or in cis- or tr ⁇ /w-arrangements at the 1,1- vs. 1,2- vs. 1,3- vs.
- Linker rigidity is varied by controlling the number and relative energies of different conformational states possible for the linker.
- a divalent compound bearing two Ugands joined by 1 ,8-octyl Hnker has many more degrees of freedom, and is therefore less rigid than a compound in which the two ligands are attached to the 4,4' positions of a biphenyl Hnker.
- Linker physical properties The physical properties of linkers are nominally determined by the chemical constitution and bonding patterns of the linker, and linker physical properties impact the overall physical properties of the candidate multibinding compounds in which they are included.
- a range of linker compositions is typically selected to provide a range of physical properties (hydrophobicity, hydrophUicity, amphiphUicity, polarization, acidity, and basicity) in the candidate multibinding compounds.
- the particular choice of linker physical properties is made within the context of the physical properties of the ligands they join and preferably the goal is to generate molecules with favorable PK/ADME properties.
- linkers can be selected to avoid those that are too hydrophUic or too hydrophobic to be readUy absorbed and/or distributed in vivo.
- Linker chemical functional groups are selected to be compatible with the chemistry chosen to connect linkers to the ligands and to impart the range of physical properties sufficient to span initial examination of this parameter. Combinatorial synthesis
- n being determined by the sum of the number of different attachment points for each ligand chosen
- m linkers by the process outlined above
- a library of (n ⁇ )m candidate divalent multibinding compounds is prepared which spans the relevant multibinding design parameters for a particular target. For example, an array generated from two ligands, one which has two attachment points (Al, A2) and one which has three attachment points (Bl, B2, B3) joined in all possible combinations provide for at least 15 possible combinations of multibinding compounds:
- combinatorial libraries can employ solid phase chemistries well known in the art wherein the ligand and/or linker is attached to a solid support.
- the combinatorial Ubary is prepared in the solution phase.
- candidate multibinding compounds are optionally purified before assaying for activity by, for example, chromatographic methods (e.g., HPLC).
- Various methods are used to characterize the properties and activities of the candidate multibinding compounds in the library to determine which compounds possess multibinding properties. Physical constants such as solubUity under various solvent conditions and logD/clogD values can be determined. A combination of NMR spectroscopy and computational methods is used to determine low-energy conformations of the candidate multibinding compounds in fluid media. The abUity of the members of the library to bind to the desired target and other targets is determined by various standard methods, which include radioligand displacement assays for receptor and ion channel targets, and kinetic inhibition analysis for many enzyme targets. In vitro efficacy, such as for receptor agonists and antagonists, ion channel blockers, and antimicrobial activity, can also be determined. Pharmacological data, including oral absorption, everted gut penetration, other pharmacokinetic parameters and efficacy data can be determined in appropriate models. In this way, key structure-activity relationships are obtained for multibinding design parameters which are then used to direct future work.
- the members of the library which exhibit multibinding properties can be readUy deteirnined by conventional methods. First those members which exhibit multibinding properties are identified by conventional methods as described above including conventional assays (both in vitro and in vivo).
- each member of the library can be encrypted or tagged with appropriate information allowing determination of the structure of relevant members at a later time.
- each member of the library can be encrypted or tagged with appropriate information allowing determination of the structure of relevant members at a later time. See, for example, Dower, et al., International Patent Application PubHcation No. WO 93/06121; Brenner, et al., Proc. Natl. Acad. Sci., USA, 89:5181 (1992); GaUop, et al., U.S. Patent No. 5,846,839; each of which are incorporated herein by reference in its entirety.
- the structure of relevant multivalent compounds can also be determined from soluble and untagged libaries of candidate multivalent compounds by methods known in the art such as those described by Hindsgaul, et al., Canadian Patent Application No. 2,240,325 which was published on July 11, 1998. Such methods couple frontal affinity chromatography with mass spectroscopy to determine both the structure and relative binding affinities of candidate multibinding compounds to receptors.
- an optional component of the process is to ascertain one or more promising multibinding "lead” compounds as defined by particular relative ligand orientations, linker lengths, linker geometries, etc. Additional libraries can then be generated around these leads to provide for further information regarding strucmre to activity relationships. These arrays typically bear more focused variations in linker structure in an effort to further optimize target affinity and/or activity at the target (antagonism, partial agonism, etc.), and/or alter physical properties. By iterative redesign/analysis using the novel principles of multibinding design along with classical medicinal chemistry, biochemistry, and pharmacology approaches, one is able to prepare and identify optimal multibinding compounds that exhibit biological advantage towards their targets and as therapeutic agents.
- suitable divalent linkers include, by way of example only, those derived from dicarboxylic acids, disulfonylhalides, dialdehydes, diketones, dihalides, diisocyanates,diamines, diols, mixtures of carboxylic acids, sulfonylhalides, aldehydes, ketones, halides, isocyanates, amines and diols.
- carboxylic acid, sulfonylhalide, aldehyde, ketone, halide, isocyanate, amine and diol functional group is reacted with a complementary functionality on the ligand to form a covalent linkage.
- complementary functionality is well known in the art as illustrated in the following table: COMPLEMENTARY BINDING CHEMISTRIES
- Second Reactive Group Linkage hydroxyl isocyanate urethane amine epoxide ⁇ -hydroxyami ⁇ e sulfonyl halide amine sulfonamide carboxyl acid amine amide hydroxyl alkyl/aryl halide ether aldehyde amine/NaCNBH 4 amine ketone amine/NaCNBH 4 amine amine isocyanate carbamate
- Exemplary linkers include the following linkers identified as X-l through X-418 as set forth below:
- L-1 ligands are benzofuran compounds (e.g., 7A-1, 7B-1 or 7C-1 of Examples 1-3). Phenylmethane sulfonamide structures are designated L-2 ligands (e.g., 8A-1, 8B-1, 8C-1, 9A-1, 9B-1, lOA-1, lOB-1, or 11-1 of Examples 4-11).
- L-3 ligands are azimUide compounds (e.g., 12-1, 12-3 of Examples 12-13).
- L-4 ligands are tedisam compounds (e.g., 13-1 of Example 14).
- Combinations of ligands (L) and linkers (X) per this invention include, by way example only, homo- and hetero-dimers wherein a first ligand is selected from L-1 through L-4 above and the second ligand and linker is selected from the following:
- compositions which contain, as the active ingredient, one or more of the compounds of Formula I above or a pharmaceutically acceptable salt thereof and one or more pharmaceutically acceptable excipients, carriers, diluents, permeation enhancers, solubilizers and adjuvants.
- the compounds may be administered alone or in combination with other therapeutic agents (e.g., other antihypertensive drugs, diuretics and the like).
- Such compositions are prepared in a manner well known in the pharmaceutical art (see, e.g., Remington 's Pharm. Sci., Mack Publishing Co., Philadelphia, PA, 17 th Ed. (1985) and "Modern Pharm. ", Marcel Dekker, Inc., 3 rd Ed. (G.S. Banker & C.T. Rhodes, Eds.).
- the compounds of Formula I may be administered by any of the accepted modes of administration of agents having similar utilities, for example, by oral, parenteral, rectal, buccal, intranasal or transdermal routes. The most suitable route will depend on the nature and severity of the condition being treated. Oral adrninistration is a preferred route for the compounds of this invention.
- the active ingredient is usuaUy diluted by an excipient or enclosed within such a carrier which can be in the form of a capsule, sachet, paper or other container.
- the excipient serves as a diluent, it can be a solid, semi-solid, or liquid material, which acts as a vehicle, carrier or medium for the active ingredient.
- compositions can be in the form of tablets, pills, powders, lozenges, sachets, cachets, elixirs, suspensions, emulsions, solutions, syrups, aerosols (as a solid or in a liquid medium), ointments containing, for example, up to 10% by weight of the active compound, soft and hard gelatin capsules, suppositories, sterile injectable solutions, and sterile packaged powders.
- Pharmaceutically acceptable salts of the active agents may be prepared using standard procedures known to those skilled in the art of synthetic organic chemistry and described, e.g., by J. March, Advanced Organic Chem. Reactions, Mechanisms and Structure, 4 th Ed. (N.Y.: Wiley-Interscience, 1992).
- excipients include lactose, dextrose, sucrose, sorbitol, mannitol, starches, gum acacia, calcium phosphate, alginates, tragacanth, gelatin, calcium silicate, microcrystalline cellulose, polyvinylpyrrolidone, cellulose, sterile water, syrup, and methyl cellulose.
- the formulations can additionally include: lubricating agents such as talc, magnesium stearate, and mineral oil; wetting agents; emulsifying and suspending agents; preserving agents such as methyl- and propylhydroxy-benzoates; sweetening agents; and flavoring agents.
- compositions of the invention can be formulated so as to provide quick, sustained or delayed release of the active ingredient after administration to the patient by employing procedures known in the art.
- Controlled release drug delivery systems for oral administration include osmotic pump systems and dissolutional systems containing polymer-coated reservoirs or drug-polymer matrix formulations. Examples of controlled release systems are given in U.S. Patent Nos. 3,845,770; 4,326,525; 4,902514; and 5,616,345.
- Another preferred formulation for use in the methods of the present invention employs transdermal delivery devices ("patches"). Such transdermal patches may be used to provide continuous or discontinuous infusion of the compounds of the present invention in controlled amounts.
- the construction and use of transdermal patches for the delivery of pharmaceutical agents is well known in the art. See, e.g., U.S. Patent Nos. 5,023,252; 4,992,445 and 5,001,139. Such patches may be constructed for continuous, pulsatile, or on demand delivery of pharmaceutical agents.
- compositions are preferably formulated in a unit dosage form.
- unit dosage forms refers to physically discrete units suitable as unitary dosages for human subjects and other mammals, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, in association with a suitable pharmaceutical excipient (e.g., a tablet, capsule, ampoule).
- the active compound is effective over a wide dosage range and is generally administered in a pharmaceutically effective amount.
- each dosage unit contains from 1-250 mg of a compound of Formula I, and for parenteral ad ⁇ iistration, preferably from 0.1 to 60 mg of a compound of Formula I or a pharmaceutically acceptable salt thereof.
- the amount of the compound actually administered will be determined by a physician, in the light of the relevant circumstances, including the condition to be treated, the chosen route of administration, the actual compound administered and its relative activity, the age, weight, and response of the individual patient, the severity of the patient's symptoms, and the like.
- the principal active ingredient is mixed with a pharmaceutical excipient to form a solid preformulation composition containing a homogeneous mixture of a compound of the present invention.
- a pharmaceutical excipient for preparing solid compositions such as tablets, the principal active ingredient is mixed with a pharmaceutical excipient to form a solid preformulation composition containing a homogeneous mixture of a compound of the present invention.
- these preformulation compositions as homogeneous, it is meant that the active ingredient is dispersed evenly throughout the composition so that the composition may be readily subdivided into equally effective unit dosage forms such as tablets, pills and capsules.
- the tablets or pills of the present invention may be coated or otherwise compounded to provide a dosage form affording the advantage of prolonged action.
- the tablet or pill can comprise an inner dosage and an outer dosage component, the latter being in the form of an envelope over the former.
- the two components can be separated by an enteric layer which serves to resist disintegration in the stomach and permit the inner component to pass intact into the duodenum or to be delayed in release.
- enteric layers or coatings such materials including a number of polymeric acids and mixtures of polymeric acids with such materials as shellac, cetyl alcohol, and cellulose acetate.
- liquid forms in which the novel compositions of the present invention may be incorporated for administration orally or by injection include aqueous solutions, suitably flavored syrups, aqueous or oil suspensions, and flavored emulsions with edible oils such as corn oil, cottonseed oil, sesame oil, coconut oil, or peanut oil, as well as elixirs and similar pharmaceutical vehicles.
- compositions for inhalation or insufflation include solutions and suspensions in pharmaceutically acceptable, aqueous or organic solvents, or mixtures thereof, and powders.
- the liquid or solid compositions may contain suitable pharmaceutically acceptable excipients as described supra.
- the compositions are aclrninistered by the oral or nasal respiratory route for local or systemic effect.
- Compositions in preferably pharmaceutically acceptable solvents may be nebulized by use of inert gases. Nebulized solutions may be inhaled directly from the nebulizing device or the nebulizing device may be attached to a face mask tent, or intermittent positive pressure breathing machine. Solution, suspension, or powder compositions may be administered, preferably orally or nasally, from devices which deliver the formulation in an appropriate manner.
- the above ingredients are mixed and filled into hard gelatin capsules in 340 mg quantities.
- the components are blended and compressed to form tablets, each weighing 240 mg.
- a dry powder inhaler formulation is prepared containing the following components: Ingredient Wei ht %
- the active ingredient is mixed with the lactose and the mixture is added to a dry powder inhaling appliance.
- Formulation Example 4 Tablets, each containing 30 mg of active ingredient, are prepared as follows:
- the active ingredient, starch, and magnesium stearate are blended, passed through a o. 20 mesh U.S. sieve, and filled into hard gelatin capsules in 150 mg quantities.
- the active ingredient is passed through a No. 60 mesh U.S. sieve and suspended in the saturated fatty acid glycerides previously melted using the minimum heat necessary. The mixture is then poured into a suppository mold of nominal 2.0 g capacity and allowed to cool.
- Suspensions each containing 50 mg of medicament per 5.0 mL dose are made as follows: Ingredient Amount
- the active ingredient, sucrose and xanthan gum are blended, passed through a No. 10 mesh U.S. sieve, and then mixed with a previously made solution of the microcrystalline cellulose and sodium carboxymethyl cellulose in water.
- the sodium benzoate, flavor, and color are diluted with some of the water and added with stirring. Sufficient water is then added to produce the required volume.
- a subcutaneous formulation may be prepared as follows:
- Direct techniques usuaUy involve placement of a drug delivery catheter into the host's ventricular system to bypass the blood-brain barrier.
- a drug delivery catheter into the host's ventricular system to bypass the blood-brain barrier.
- One such hnplantable delivery system used for the transport of biological factors to specific anatomical regions of the body is described in U.S. Patent 5,011,472 which is herein incorporated by reference.
- Indirect techniques usually involve formulating the compositions to provide for drug latentiation by the conversion of hydrophUic drugs into Hpid-soluble drugs.
- Latentiation is generally achieved through blocking of the hydroxy, carbonyl, sulfate, and primary amine groups present on the drug to render the drug more lipid soluble and amenable to transportation across the blood-brain barrier.
- the delivery of hydrophilic drugs may be enhanced by intra-arterial infusion of hypertonic solutions which can transiently open the blood-brain barrier.
- Example 10B Preparation of 1,8-di- [N-ethyl N'-[2-[4-[methylsulfonylamino]benzoylaminoethyl]- amino] 3,5-dioxaoctane, 10B-2, in which Link is (CH 2 ) 2 (O(CH 2 ) 2 ) 2 . A.
- N-Methyl 2-[4-[2-butylbe ⁇ zofuran-3-ylcarbonyl]-2,6-diiodophenoxy]ethylamine (15- 4), prepared according to procedures described in Eur. J. Med. Chem., 1974, 19-25, (3 mmol) is dissolved in MeCN (30 riiL), and l,3,5-tri(bromomethyl)benzene (1 mmol) and K 2 CO 3
- N-Methyl 2-[4-[2-butylbenzofuran-3-ylcarbonyl]-2,6- diiodophenoxyjethylamine (15-4), prepared according to procedures described in Eur. J. Med. Chem., 1974, 19-25, (5 mmol) is dissolved in EtOH (25 mL) and ethyl bromoacetate (5 mmol) and dusopropylethylamine (10 mmol) are added. The progress of the reaction is followed by tic. When it is complete, the reaction is added to water and extracted with EtOAc. The extract is washed with dilute HCI, the dried and the solvent is evaporated under reduced pressure.
- N-Methyl N-(4-aminophenylethyl) 2-[4-(methylsulfonylamino)phenoxy]- ethylamine,18-l the preparation of which is described in Examples 4A and 4B above, (2 mmol) is dissolved in dry CH 2 C1 2 (25 mL); diisopropylethylamine (10 mmol) and 3- bromopropanesulfonyl chloride (2 mmol) are added. The progress of the reaction is followed by tic. When it is complete, the reaction is added to water and extracted with EtOAc. The extract is washed and dried and the solvent is evaporated under reduced pressure.
- Example 24 A Using the procedure of Example 24 A, except that 4-bromobutanesulfonyl chloride is employed instead of 6-bromohexanesulfonyl chloride, there is prepared the compound 19-9, in which Link is (CH 2 ) 3 .
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Urology & Nephrology (AREA)
- Cardiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Cell Biology (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hematology (AREA)
- Immunology (AREA)
- Analytical Chemistry (AREA)
- Food Science & Technology (AREA)
- Physics & Mathematics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Microbiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Investigating Or Analysing Biological Materials (AREA)
Abstract
Description









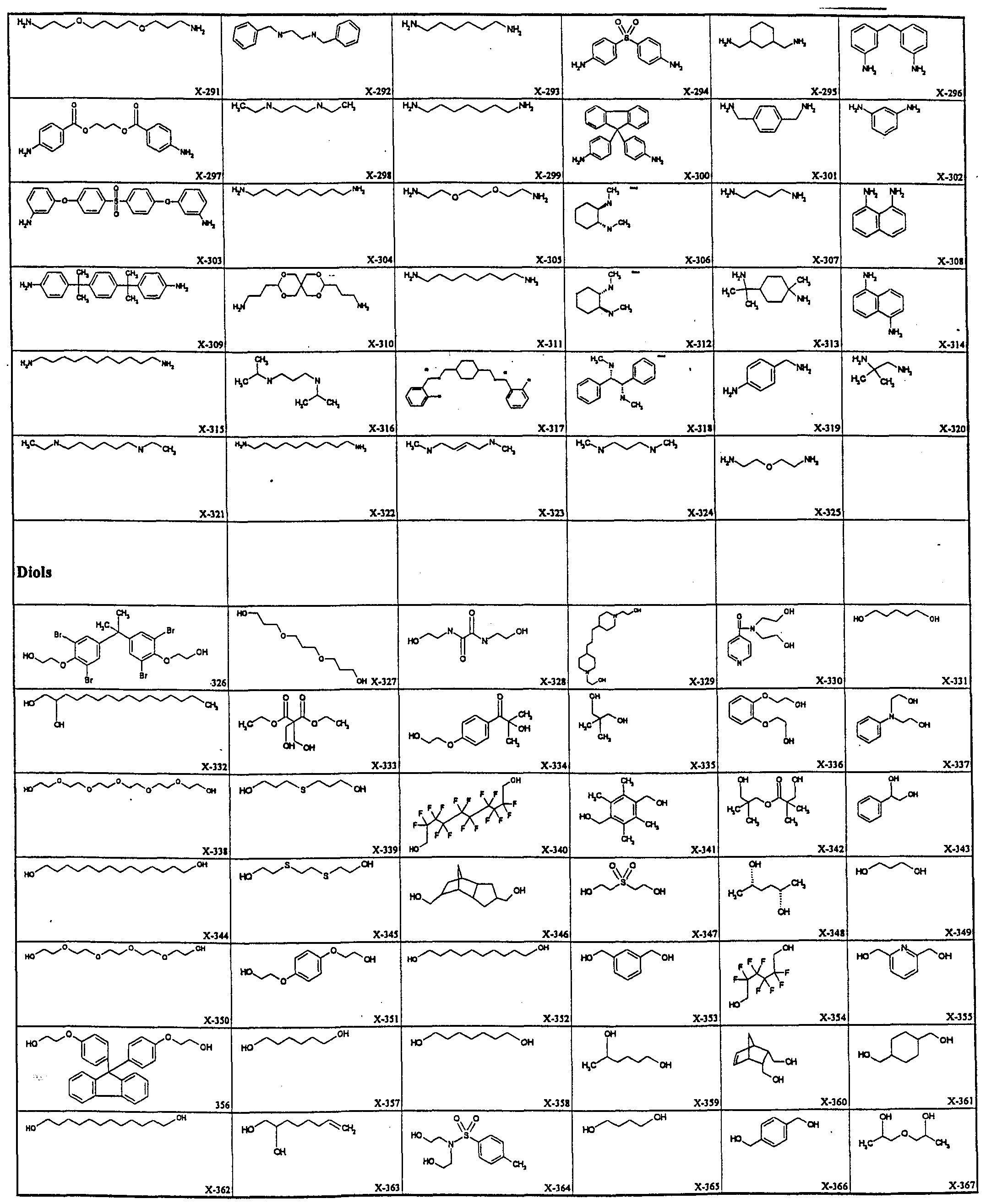








Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000553118A JP2002517466A (en) | 1998-06-08 | 1999-06-07 | Novel potassium channel drugs and their uses |
| CA002318745A CA2318745A1 (en) | 1998-06-08 | 1999-06-07 | Novel potassium channel drugs and their uses |
| EP99927330A EP1086063A4 (en) | 1998-06-08 | 1999-06-07 | Novel potassium channel drugs and their uses |
| AU44264/99A AU4426499A (en) | 1998-06-08 | 1999-06-07 | Novel potassium channel drugs and their uses |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US8846598P | 1998-06-08 | 1998-06-08 | |
| US60/088,465 | 1998-06-08 | ||
| US9306898P | 1998-07-16 | 1998-07-16 | |
| US60/093,068 | 1998-07-16 | ||
| US11386498P | 1998-12-24 | 1998-12-24 | |
| US60/113,864 | 1998-12-24 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO1999064050A1 true WO1999064050A1 (en) | 1999-12-16 |
| WO1999064050A8 WO1999064050A8 (en) | 2000-02-24 |
| WO1999064050A9 WO1999064050A9 (en) | 2000-04-20 |
Family
ID=27375970
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1999/012777 WO1999064050A1 (en) | 1998-06-08 | 1999-06-07 | Novel potassium channel drugs and their uses |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP1086063A4 (en) |
| JP (1) | JP2002517466A (en) |
| AR (1) | AR015310A1 (en) |
| AU (1) | AU4426499A (en) |
| CA (1) | CA2318745A1 (en) |
| SG (1) | SG80037A1 (en) |
| WO (1) | WO1999064050A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1147768A1 (en) * | 2000-04-18 | 2001-10-24 | LTS Lohmann Therapie-Systeme AG | Transdermal therapeutic system for administering dofetilide |
| WO2002057792A2 (en) * | 2000-12-29 | 2002-07-25 | Neogenesis Pharmaceuticals, Inc. | Affinity selection-based screening of hydrophobic proteins |
| JP2004509915A (en) * | 2000-09-29 | 2004-04-02 | ソルベイ・フアーマシユーチカルズ・ベー・ブイ | Sustained-release pharmaceutical composition independent of ionic strength |
| WO2008112287A1 (en) * | 2007-03-12 | 2008-09-18 | Nektar Therapeutics | Oligomer-beta blocker conjugates |
| US8410167B2 (en) | 2008-04-17 | 2013-04-02 | Sanofi | Use of dronedarone for the preparation of a medicament for use in the prevention of cardiovascular hospitalization or of mortality |
| US8602215B2 (en) | 2010-06-30 | 2013-12-10 | Sanofi | Methods for reducing the risk of an adverse dronedarone/beta-blockers interaction in a patient suffering from atrial fibrillation |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992005802A1 (en) * | 1990-09-28 | 1992-04-16 | Neorx Corporation | Polymeric carriers for release of covalently linked agents |
| US5545568A (en) * | 1992-09-14 | 1996-08-13 | The Regents Of The University Of California | Solid phase and combinatorial synthesis of compounds on a solid support |
-
1999
- 1999-06-07 WO PCT/US1999/012777 patent/WO1999064050A1/en not_active Application Discontinuation
- 1999-06-07 AU AU44264/99A patent/AU4426499A/en not_active Abandoned
- 1999-06-07 EP EP99927330A patent/EP1086063A4/en not_active Withdrawn
- 1999-06-07 JP JP2000553118A patent/JP2002517466A/en not_active Withdrawn
- 1999-06-07 CA CA002318745A patent/CA2318745A1/en not_active Abandoned
- 1999-06-08 SG SG9902714A patent/SG80037A1/en unknown
- 1999-06-08 AR ARP990102703A patent/AR015310A1/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992005802A1 (en) * | 1990-09-28 | 1992-04-16 | Neorx Corporation | Polymeric carriers for release of covalently linked agents |
| US5545568A (en) * | 1992-09-14 | 1996-08-13 | The Regents Of The University Of California | Solid phase and combinatorial synthesis of compounds on a solid support |
Non-Patent Citations (4)
| Title |
|---|
| JUNG G. (ED.): "COMBINATORIAL PEPTIDE AND NONPEPTIDE LIBRARIES.", 1 January 1996, WEINHEIM, VCH., DE, ISBN: 978-3-527-29380-3, article BUNIN B A, PLUNKETT M J, ELLMAN J A: "15 SYNTHESIS AND EVALUATION OF THREE 1,4-BENZODIAZEPINE LIBRARIES", pages: 405 - 424, XP002923778, 022202 * |
| S.B.SHUKER ET AL.: "Discovering High-Affinity Ligands for Proteins: SAR by NMR", SCIENCE, AMERICAN ASSOCIATION FOR THE ADVANCEMENT OF SCIENCE, US, no. 274, 1 January 1996 (1996-01-01), US, pages 1531 - 1534, XP002074440, ISSN: 0036-8075, DOI: 10.1126/science.274.5292.1531 * |
| SALATA J J, ET AL.: "A NOVEL BENZODIAZEPINE THAT ACTIVATES CARDIAC SLOW DELAYED RECTIFIER K+ CURRENTS", MOLECULAR PHARMACOLOGY, AMERICAN SOCIETY FOR PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, US, vol. 53, 1 January 1998 (1998-01-01), US, pages 220 - 230, XP002923779, ISSN: 0026-895X * |
| See also references of EP1086063A4 * |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1147768A1 (en) * | 2000-04-18 | 2001-10-24 | LTS Lohmann Therapie-Systeme AG | Transdermal therapeutic system for administering dofetilide |
| JP2004509915A (en) * | 2000-09-29 | 2004-04-02 | ソルベイ・フアーマシユーチカルズ・ベー・ブイ | Sustained-release pharmaceutical composition independent of ionic strength |
| WO2002057792A2 (en) * | 2000-12-29 | 2002-07-25 | Neogenesis Pharmaceuticals, Inc. | Affinity selection-based screening of hydrophobic proteins |
| WO2002057792A3 (en) * | 2000-12-29 | 2003-11-06 | Neogenesis Pharmaceuticals Inc | Affinity selection-based screening of hydrophobic proteins |
| WO2008112287A1 (en) * | 2007-03-12 | 2008-09-18 | Nektar Therapeutics | Oligomer-beta blocker conjugates |
| US9782488B2 (en) | 2007-03-12 | 2017-10-10 | Nektar Therapeutics | Oligomer-beta blocker conjugates |
| US10251959B2 (en) | 2007-03-12 | 2019-04-09 | Nektar Therapeutics | Oligomer-beta blocker conjugates |
| US10881738B2 (en) | 2007-03-12 | 2021-01-05 | Nektar Therapeutics | Oligomer-beta blocker conjugates |
| US8410167B2 (en) | 2008-04-17 | 2013-04-02 | Sanofi | Use of dronedarone for the preparation of a medicament for use in the prevention of cardiovascular hospitalization or of mortality |
| US9107900B2 (en) | 2008-04-17 | 2015-08-18 | Sanofi | Use of dronedarone for the preparation of a medicament for use in the prevention of cardiovascular hospitalization or of morality |
| US8602215B2 (en) | 2010-06-30 | 2013-12-10 | Sanofi | Methods for reducing the risk of an adverse dronedarone/beta-blockers interaction in a patient suffering from atrial fibrillation |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1086063A1 (en) | 2001-03-28 |
| CA2318745A1 (en) | 1999-12-16 |
| AR015310A1 (en) | 2001-04-18 |
| WO1999064050A8 (en) | 2000-02-24 |
| JP2002517466A (en) | 2002-06-18 |
| EP1086063A4 (en) | 2001-03-28 |
| SG80037A1 (en) | 2001-04-17 |
| WO1999064050A9 (en) | 2000-04-20 |
| AU4426499A (en) | 1999-12-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6355805B1 (en) | β3-adrenergic receptor agonists | |
| US20030044845A1 (en) | Novel therapeutic agents for membrane transporters | |
| WO1999063984A9 (en) | Novel sodium channel drugs and uses | |
| US6420354B1 (en) | Sodium channel drugs and uses | |
| WO1999064033A9 (en) | Phosphodiesterase-v modulator drugs and their uses | |
| US20030073127A1 (en) | Novel calcium channel drugs and uses | |
| US6479498B1 (en) | Sodium channel drugs and uses | |
| EP1086063A1 (en) | Novel potassium channel drugs and their uses | |
| ZA200004564B (en) | Novel potassium channel drugs and their uses. | |
| EP1085863A1 (en) | Cross-reference to related applications |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US US US UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999927330 Country of ref document: EP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: C1 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US US US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: C1 Designated state(s): GH GM KE LS MW SD SL SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| CFP | Corrected version of a pamphlet front page | ||
| CR1 | Correction of entry in section i |
Free format text: PAT. BUL. 50/99 UNDER (81) ADD "AE, ZA" |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: C2 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US US US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: C2 Designated state(s): GH GM KE LS MW SD SL SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| COP | Corrected version of pamphlet |
Free format text: PAGES 1/24-24/24, DRAWINGS, REPLACED BY NEW PAGES 1/23-23/23; DUE TO LATE TRANSMITTAL BY THE RECEIVING OFFICE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 505889 Country of ref document: NZ |
|
| ENP | Entry into the national phase |
Ref document number: 2318745 Country of ref document: CA Ref country code: CA Ref document number: 2318745 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 44264/99 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2000/04564 Country of ref document: ZA Ref document number: 200004564 Country of ref document: ZA |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 553118 Kind code of ref document: A Format of ref document f/p: F |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999927330 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1999927330 Country of ref document: EP |