WO1999062897A1 - Hiv integrase inhibitors - Google Patents
Hiv integrase inhibitors Download PDFInfo
- Publication number
- WO1999062897A1 WO1999062897A1 PCT/US1999/012094 US9912094W WO9962897A1 WO 1999062897 A1 WO1999062897 A1 WO 1999062897A1 US 9912094 W US9912094 W US 9912094W WO 9962897 A1 WO9962897 A1 WO 9962897A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- dioxo
- butanoic acid
- substituted
- independently selected
- Prior art date
Links
- ATRJNSFQBYKFSM-UHFFFAOYSA-N Brc(cc[s]1)c1Br Chemical compound Brc(cc[s]1)c1Br ATRJNSFQBYKFSM-UHFFFAOYSA-N 0.000 description 1
- VVVRNQVILCWEHS-UHFFFAOYSA-N CC(c1c(Cc2ccccc2)[nH]cc1)=O Chemical compound CC(c1c(Cc2ccccc2)[nH]cc1)=O VVVRNQVILCWEHS-UHFFFAOYSA-N 0.000 description 1
- JMEUBOXHBMJAFY-UHFFFAOYSA-N CC(c1ccc(N(Cc2ccccc2)S(c2ccccc2)(=O)=O)[s]1)=O Chemical compound CC(c1ccc(N(Cc2ccccc2)S(c2ccccc2)(=O)=O)[s]1)=O JMEUBOXHBMJAFY-UHFFFAOYSA-N 0.000 description 1
- VGUATYBXWKPCMO-UHFFFAOYSA-N CC(c1ccc(NCc2ccccc2)[s]1)=O Chemical compound CC(c1ccc(NCc2ccccc2)[s]1)=O VGUATYBXWKPCMO-UHFFFAOYSA-N 0.000 description 1
- PNRMINDJDJTNJA-UHFFFAOYSA-N CC(c1ccc(Nc2ccccc2)[nH]1)=O Chemical compound CC(c1ccc(Nc2ccccc2)[nH]1)=O PNRMINDJDJTNJA-UHFFFAOYSA-N 0.000 description 1
- RGMVGFUPBCBJSZ-UHFFFAOYSA-N CN(Cc1ccccc1)c1ncc(C(CC(C(O)=O)=O)=O)[s]1 Chemical compound CN(Cc1ccccc1)c1ncc(C(CC(C(O)=O)=O)=O)[s]1 RGMVGFUPBCBJSZ-UHFFFAOYSA-N 0.000 description 1
- VKLXIRLUDYCLDP-UHFFFAOYSA-N Fc1cc(Cc2cc(Br)c[s]2)ccc1 Chemical compound Fc1cc(Cc2cc(Br)c[s]2)ccc1 VKLXIRLUDYCLDP-UHFFFAOYSA-N 0.000 description 1
- AZFCIWCWYTUZRC-UHFFFAOYSA-N NC(N(Cc1ccccc1)Cc1ccccc1)=S Chemical compound NC(N(Cc1ccccc1)Cc1ccccc1)=S AZFCIWCWYTUZRC-UHFFFAOYSA-N 0.000 description 1
- VLDBGWRMAAXGER-UHFFFAOYSA-N OC(C(CC(c1c[s]c(Sc2ccccc2)c1)=O)=O)=O Chemical compound OC(C(CC(c1c[s]c(Sc2ccccc2)c1)=O)=O)=O VLDBGWRMAAXGER-UHFFFAOYSA-N 0.000 description 1
- PWHPVXIPNXXRLL-UHFFFAOYSA-N OC(C(CC(c1ccc(C(c2ccccc2)OCc2ccccc2)[s]1)=O)=O)=O Chemical compound OC(C(CC(c1ccc(C(c2ccccc2)OCc2ccccc2)[s]1)=O)=O)=O PWHPVXIPNXXRLL-UHFFFAOYSA-N 0.000 description 1
- IMIVGSSENVZOCZ-UHFFFAOYSA-N OC(C(CC(c1ccc(C(c2ccccc2)Oc2ccccc2)[s]1)=O)=O)=O Chemical compound OC(C(CC(c1ccc(C(c2ccccc2)Oc2ccccc2)[s]1)=O)=O)=O IMIVGSSENVZOCZ-UHFFFAOYSA-N 0.000 description 1
- BRJJEMWQVHBCBM-UHFFFAOYSA-N OC(C(CC(c1ccc(Cc2cccc(F)c2)[s]1)=O)=O)=O Chemical compound OC(C(CC(c1ccc(Cc2cccc(F)c2)[s]1)=O)=O)=O BRJJEMWQVHBCBM-UHFFFAOYSA-N 0.000 description 1
- CQNPHPWEVXUDFY-UHFFFAOYSA-N OC(C(CC(c1ccc(OCc2ccccc2)[s]1)=O)=O)=O Chemical compound OC(C(CC(c1ccc(OCc2ccccc2)[s]1)=O)=O)=O CQNPHPWEVXUDFY-UHFFFAOYSA-N 0.000 description 1
- UHVRGUQOALWVQN-UHFFFAOYSA-N OC(C(CC(c1cnc(N(Cc2ccccc2)Cc2ccccc2)[s]1)=O)=O)=O Chemical compound OC(C(CC(c1cnc(N(Cc2ccccc2)Cc2ccccc2)[s]1)=O)=O)=O UHVRGUQOALWVQN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/42—Amino or imino radicals substituted by hydrocarbon or substituted hydrocarbon radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/24—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/32—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/34—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/36—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/54—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
- C07D333/60—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/74—Naphthothiophenes
Definitions
- a retrovirus designated human immunodeficiency virus is the etiological agent of the complex disease that includes progressive destruction of the immune system (acquired immune deficiency syndrome; AIDS) and degeneration of the central and peripheral nervous system.
- This virus was previously known as LAV, HTLV-III, or ARV.
- a common feature of retrovirus replication is the insertion by virally-encoded integrase of proviral DNA into the host cell genome, a required step in HIV replication in human T-lymphoid and monocytoid cells.
- Integration is believed to be mediated by integrase in three steps: assembly of a stable nucleoprotein complex with viral DNA sequences; cleavage of two nucleotides from the 3' termini of the linear proviral DNA; covalent joining of the recessed 3' OH termini of the proviral DNA at a staggered cut made at the host target site.
- the fourth step in the process, repair synthesis of the resultant gap may be accomplished by cellular enzymes.
- Nucleotide sequencing of HIV shows the presence of a pol gene in one open reading frame [Ratner, L. et al., Nature, 313, 277(1985)].
- Amino acid sequence homology provides evidence that the pol sequence encodes reverse transcriptase, integrase and an HIV protease [Toh, H. et al., EMBO J. 4, 1267 (1985); Power, M.D. et al., Science, 231, 1567 (1986); Pearl, L.H. et al., Nature, 329, 351 (1987)]. All three enzymes have been shown to be essential for the replication of H ⁇ V.
- antiviral compounds which act as inhibitors of HIV replication are effective agents in the treatment of AIDS and similar diseases, e.g., azidothymidine or AZT.
- Applicants demonstrate that the compounds of this invention are inhibitors of HIV integrase and inhibitors of HIV replication.
- the applicants additionally demonstrate that inhibition of integrase in vitro and HIV replication in cells is a direct result of inhibiting the strand transfer reaction catalyzed by the recombinant integrase in vitro and integrase as a component of the preintegration complex in HIV infected cells.
- the particular advantage of the present invention is highly specific inhibition of HIV integrase and HIV replication.
- the compounds of the present invention inhibit integrases of closely related lentiviruses such as HIV 2 and SIV, but not integrases from more distantly related retroviruses, for example RSV. These compounds do not inhibit binding or catalysis of other nucleic acid binding proteins, including enzymatic reactions such as those catalyzed by HIV reverse transcriptase, HIV Rnase H, Influenza transcriptase, Hepatitis C polymerase, Yeast DNA polymerase, DNase I, Eco RI endonuclease, or mammalian polymerase II.
- Zhao et al. (J. Med Chem. vol. 40, pp. 937-941 and 1186- 1194 (1997)) describe hydrazide and arylamide HIV integrase inhibitors.
- Bis-catechols useful for inhibiting HIV integrase are described in LaFemina et al. (Antimicrobial Agents & Chemotherapy, vol. 39, no. 2, pp. 320-324, February 1995).
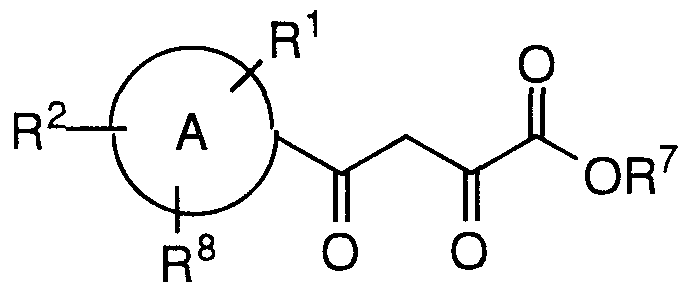
- This invention is concerned with compounds of formula I, combinations thereof, or pharmaceutically acceptable salts thereof, in the inhibition of HIV integrase, the prevention or treatment of infection by HIV and in the treatment of the resulting acquired immune deficiency syndrome (AIDS).
- Compounds of formula I are defined as follows:
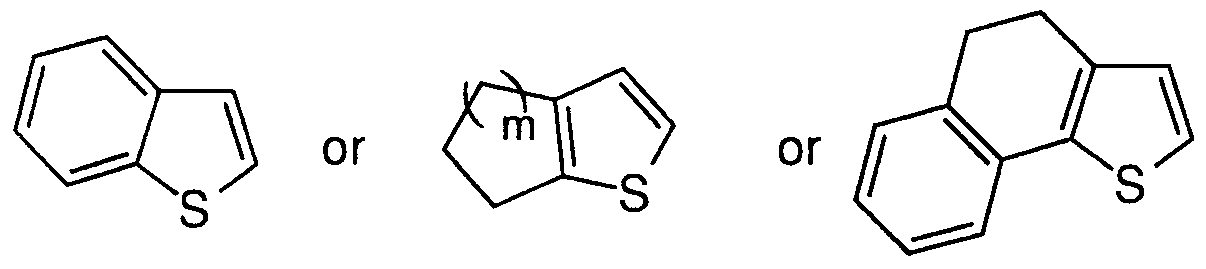
- A is a five-membered heteroaromatic ring containing 1 sulfur atom
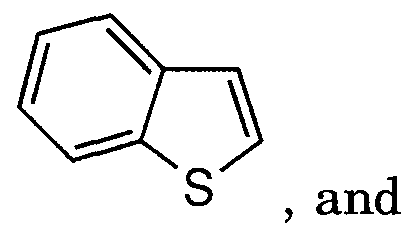
- the heteroaromatic ring may optionally be fused with a phenyl ring or a C4.6 cycloalkyl ring, or with two six membered rings to form:
- R is selected from:
- each R is independently selected from:
- a 5 or 6 membered aromatic or heteroaromatic ring containing 0, 1, 2, 3, or 4 heteroatoms selected from oxygen, nitrogen and sulfur, unsubstituted or substituted on a nitrogen or carbon atom by 1 to 5 substituents selected from: (a) halogen,
- (6) a 5 to 6 membered ring containing 0, 1 or 2 heteroatoms selected from oxygen, nitrogen or sulfur, containing 2 or 3 double bonds, unsubstituted or substituted with 1 or 2 substituents selected from: (a) halogen,
- each R is independently selected from:
- each R is independently selected from: (1) -C ⁇ g alkyl-R 3 , and
- R7 is selected from: (1) -H, and (2) Ci-6 alkyl
- R8 is selected from:
- each n is independently selected from 0, 1 and 2
- each m is independently selected from 0, 1, and 2.
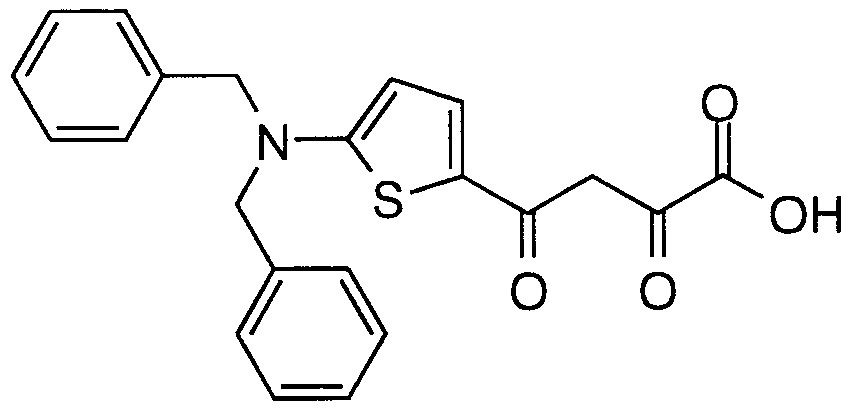
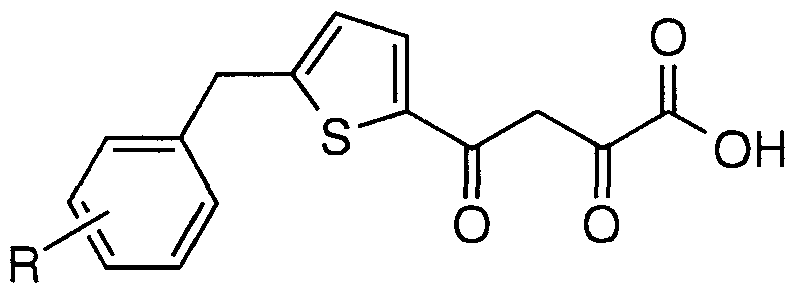
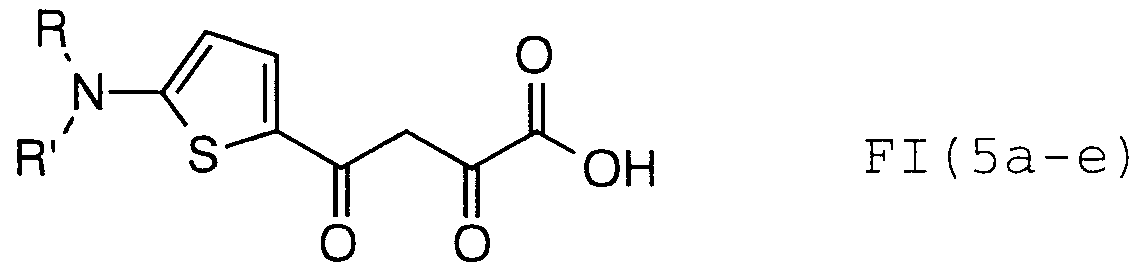
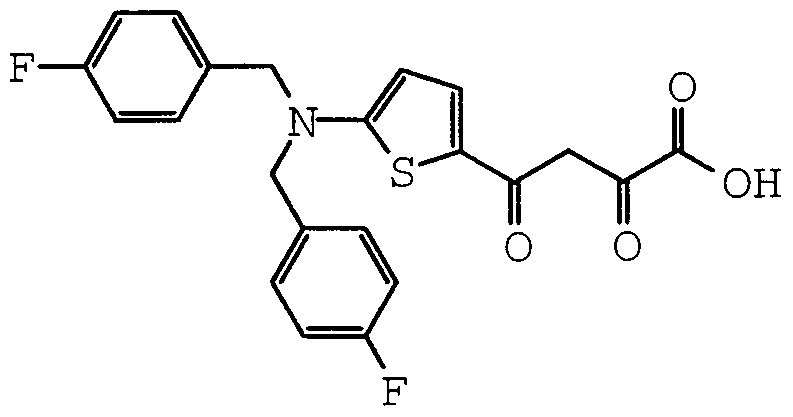
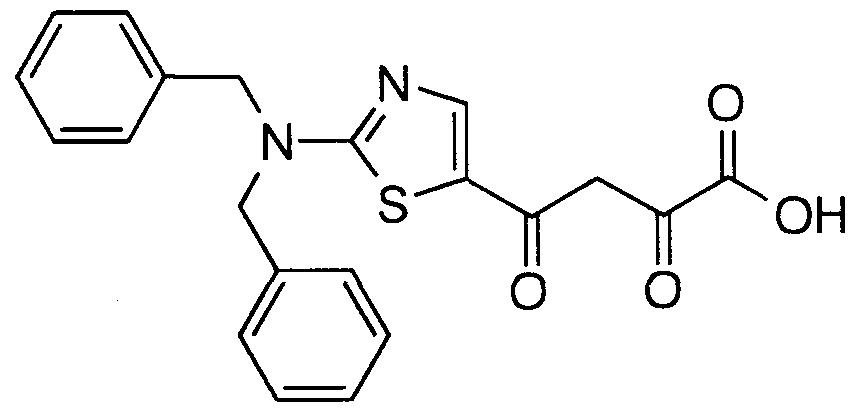
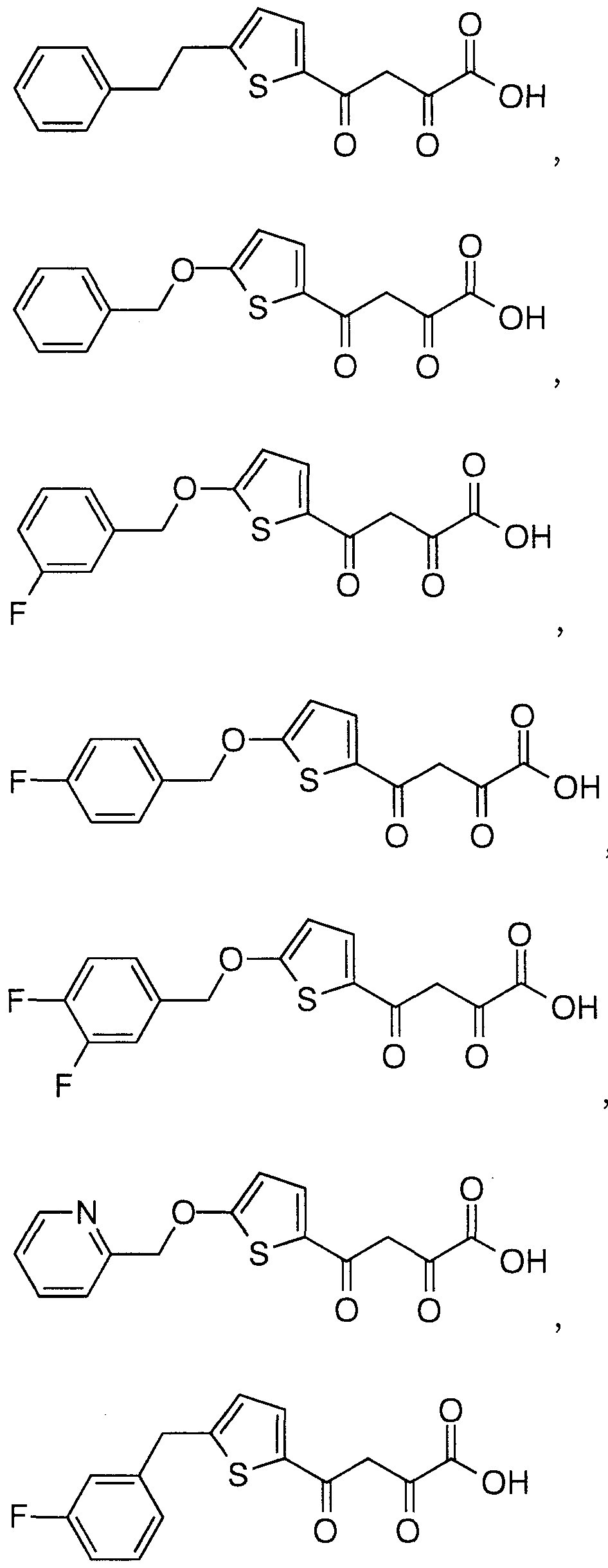
- Particular compounds of structural formula I include:
- structural formula (I) is:
- structural formula (I) is:
- structural formula (I) is:
- structural formula (I) is;
- A is selected from: (1) thienyl
- A is selected from:
- Rl is selected from: (1) -H, (2) -CHg,
- R2 is selected from:
- R2 is selected from:
- R3 is selected from:
- R3 is selected from:
- R4 is selected from: (1) -H,
- R4 is selected from:
- R5 is selected from:
- R5 is selected from: (1) -H,
- R7 is hydrogen
- R ⁇ is selected from: hydrogen, methyl and methoxy.
- compositions useful for inhibiting HIV integrase comprising an effective amount of a compound of this invention, and a pharmaceutically acceptable carrier.
- Pharmaceutical compositions useful for treating infection by HIV, or for treating AIDS or ARC are also encompassed by the present invention, as well as a method of inhibiting HIV integrase, and a method of treating infection by HIV, or of treating AIDS or ARC.
- the present invention is directed to a pharmaceutical composition comprising a therapeutically effective amount of a compound of the present invention in combination with a therapeutically effective amount of an AIDS treatment agent selected from: (1) an AIDS antiviral agent,
- the compounds of the present invention may have asymmetric centers and may occur, except when specifically noted, as mixtures of stereoisomers or as individual diastereomers, or enantiomers, with all isomeric forms being included in the present invention.
- the diketo-acid/ester compounds of the present invention exist as tautomers, and thus by using the phrase "and tautomers thereof in describing compounds of structural formula (I), Applicants also intend the following tautomeric forms of the same compound (la) and (lb):
- any variable e.g., R3, R4, etc.
- its definition on each occurrence is independent of its definition at every other occurrence.
- combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- the compounds of the present inventions are useful in the inhibition of HrV integrase, the prevention or treatment of infection by human immunodeficiency virus (HIV) and the treatment of consequent pathological conditions such as AIDS.
- Treating AIDS or preventing or treating infection by HFv 7 is defined as including, but not limited to, treating a wide range of states of HIV infection: AIDS, ARC (AIDS related complex), both symptomatic and asymptomatic, and actual or potential exposure to HIV.
- the compounds of this invention are useful in treating infection by HIV after suspected past exposure to HIV by e.g., blood transfusion, exchange of body fluids, bites, accidental needle stick, or exposure to patient blood during surgery.
- the compounds of this invention are useful in the preparation and execution of screening assays for antiviral compounds.
- the compounds of this invention are useful for isolating enzyme mutants, which are excellent screening tools for more powerful antiviral compounds.
- the compounds of this invention are useful in establishing or determining the binding site of other antivirals to HIV integrase, e.g., by competitive inhibition.
- the compounds of this invention are commercial products to be sold for these purposes.
- the present invention also provides for the use of a compound of structural formula (I) to make a pharmaceutical composition useful for inhibiting HIV integrase and in the treatment of AIDS or ARC.
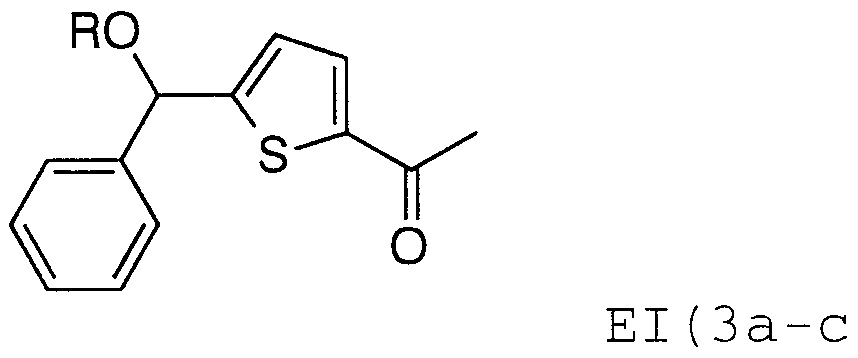
- AI ( 3 ) i. NaOH, MeOH-H 2 0-THF
- the compounds of the present invention may be administered in the form of pharmaceutically acceptable salts.
- pharmaceutically acceptable salt is intended to include all acceptable salts such as acetate, lactobionate, benzenesulfonate, laurate, benzoate, malate, bicarbonate, maleate, bisulfate, mandelate, bitartrate, mesylate, borate, methylbromide, bromide, methylnitrate, calcium edetate, methylsulfate, camsylate, mucate, carbonate, napsylate, chloride, nitrate, clavulanate, N-methylglucamine, citrate, ammonium salt, dihydrochloride, oleate, edetate, oxalate, edisylate, pamoate (embonate), estolate, palmitate, esylate, pantothenate, fumarate, phosphate/diphosphate, gluceptate, polygalacturonate,
- pharmaceutically acceptable salts of the compounds of this invention include those formed from cations such as sodium, potassium, aluminum, calcium, lithium, magnesium, zinc, and from bases such as ammonia, ethyl enediamine, N-methyl- glutamine, lysine, arginine, ornithine, choline, N,N'-dibenzylethylene- diamine, chloroprocaine, diethanolamine, procaine, N-benzylphenethyl- amine, diethylamine, piperazine, tris(hydroxymethyl)aminomethane, and tetramethylammonium hydroxide.
- bases such as ammonia, ethyl enediamine, N-methyl- glutamine, lysine, arginine, ornithine, choline, N,N'-dibenzylethylene- diamine, chloroprocaine, diethanolamine, procaine, N-benzylphenethyl- amine, diethylamine, pipe
- esters can be employed, e.g. acetate, maleate, pivaloyloxymethyl, and the like, and those esters known in the art for modifying solubility or hydrolysis characteristics for use as sustained release or prodrug formulations.
- the compounds of the present invention may be administered orally, parenterally (including subcutaneous injections, intravenous, intramuscular, intrasternal injection or infusion techniques), by inhalation spray, or rectally, in dosage unit formulations containing conventional non-toxic pharmaceutically- acceptable carriers, adjuvants and vehicles.
- administering a should be understood to mean providing a compound of the invention or a prodrug of a compound of the invention to the individual in need of treatment.
- a method of treating and a pharmaceutical composition for treating HIV infection and AIDS involves administering to a patient in need of such treatment a pharmaceutical composition comprising a pharmaceutical carrier and a therapeutically-effective amount of a compound of the present invention.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- compositions may be in the form of orally-aclministrable suspensions or tablets, nasal sprays, sterile injectible preparations, for example, as sterile injectible aqueous or oleagenous suspensions or suppositories.
- these compositions When administered orally as a suspension, these compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may contain microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners/flavoring agents known in the art.
- these compositions may contain microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants known in the art.
- compositions When administered by nasal aerosol or inhalation, these compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art.
- the injectible solutions or suspensions may be formulated according to known art, using suitable non-toxic, parenterally- acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution, or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- suitable non-toxic, parenterally- acceptable diluents or solvents such as mannitol, 1,3-butanediol, water, Ringer's solution or isotonic sodium chloride solution, or suitable dispersing or wetting and suspending agents, such as sterile, bland, fixed oils, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- compositions When rectally administered in the form of suppositories, these compositions may be prepared by mixing the drug with a suitable non-irritating excipient, such as cocoa butter, synthetic glyceride esters of polyethylene glycols, which are solid at ordinary temperatures, but liquefy and/or dissolve in the rectal cavity to release the drug.
- a suitable non-irritating excipient such as cocoa butter, synthetic glyceride esters of polyethylene glycols, which are solid at ordinary temperatures, but liquefy and/or dissolve in the rectal cavity to release the drug.
- the compounds of this invention can be administered orally to humans in a dosage range of 1 to 1000 mg/kg body weight in divided doses.
- One preferred dosage range is 0.1 to 200 mg/kg body weight orally in divided doses.
- Another preferred dosage range is 0.5 to 100 mg/kg body weight orally in divided doses.
- the compositions are preferably provided in the form of tablets containing 1.0 to 1000 milligrams of the active ingredient, particularly 1.0, 5.0, 10.0, 15.0. 20.0, 25.0, 50.0, 75.0, 100.0, 150.0, 200.0, 250.0, 300.0, 400.0, 500.0, 600.0, 750.0, 800.0, 900.0, and 1000.0 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the present invention is also directed to combinations of the HIV integrase inhibitor compounds with one or more agents useful in the treatment of AIDS.
- the compounds of this invention may be effectively administered, whether at periods of pre-exposure and or post-exposure, in combination with effective amounts of the AIDS antivirals, imunomodulators, antiinfectives, or vaccines, such as those in the following table.
- Abacavir (1592U89) Glaxo Wellcome HIV infection, AIDS, ARC (RT inhibitor)
- Cidofovir Gilead Science CMV retinitis, herpes, papilloma virus
- Ribavirin (Costa Mesa, CA) positive, LAS, ARC
- Isethionate (IM & IV) (Rosemont, IL)
- Preferred combinations are simultaneous or alternating treatments of with a compound of the present invention and an inhibitor of H ⁇ protease and/or a non-nucleoside inhibitor of HIV reverse transcriptase.
- An optional fourth component in the combination is a nucleoside inhibitor of HP/ reverse transcriptase, such as AZT, 3TC, ddC or ddl.
- H ⁇ protease is indinavir, which is the sulfate salt of N-(2(R)-hydroxy- l(S)-indanyl)-2(R)-phenylmethyl-4-(S)- hydroxy-5-(l-(4-(3-pyridyl-methyl)-2(S)-N'-(t-butylcarboxamido)- piperazinyl))-pentaneamide ethanolate, and is synthesized according to U.S. 5,413,999.
- Indinavir is generally administered at a dosage of 800 mg three times a day.
- Other preferred protease inhibitors are nelfinavir and ritonavir.
- HP7 reverse transcriptase Another preferred inhibitor of HP/ protease is saquinavir which is administered in a dosage of 600 or 1200 mg tid.
- Preferred non- nucleoside inhibitors of HP7 reverse transcriptase include efavirenz.
- the preparation of ddC, ddl and AZT are also described in EPO 0,484,071. These combinations may have unexpected effects on limiting the spread and degree of infection of HP7.
- Preferred combinations include those with the following (1) indinavir with efavirenz, and, optionally, AZT and/or 3TC and/or ddl and/or ddC; (2) indinavir, and any of AZT and/or ddl and/or ddC and/or 3TC, in particular, indinavir and AZT and 3TC; (3) stavudine and 3TC and/or zidovudine; (4) zidovudine and lamivudine and 141W94 and 1592U89; (5) zidovudine and lamivudine.
- the compound of the present invention and other active agents may be administered separately or in conjunction.
- the administration of one element may be prior to, concurrent to, or subsequent to the administration of other agent(s).
- Indinavir is an inhibitor of HP7 protease and is the sulfate salt of N-(2(R)-hydroxy-l(S)-indanyl)-2(R)-phenylmethyl-4-(S)-hydroxy-5- (l-(4-(3-pyridyl-methyl)-2(S)-N'-(t-butylcarboxamido)-piperazinyl))- pentaneamide ethanolate, and is synthesized according to U.S. 5,413,999.
- Indinavir is generally administered at a dosage of 800 mg three times a day.
- aq is aqueous; Ac represents acetyl; ACN is acetonitrile; Bn represents benzyl; DMF is dimethyl formamide; DMSO is dimethyl sulfoxide; Et represents ethyl; IPA is isopropyl alcohol; Me represents methyl; NaHMDS represents sodium hexamethyl disilamide; rt, RT both represent room temperature; sat represents saturated; THF is tetrahydrofuran; TLC is thin layer (Si ⁇ 2) chromatography.
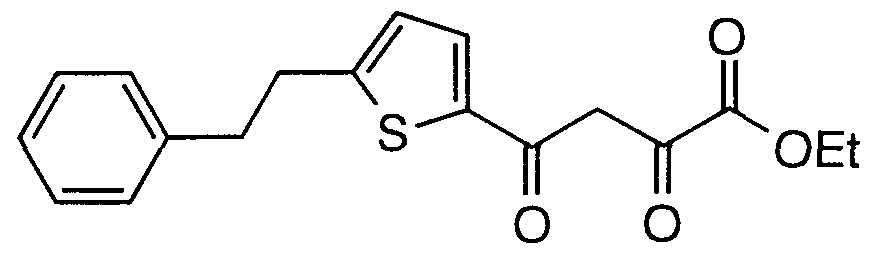
- Step A Preparation of ethyl 2,4-dioxo-4-(5-phenethynylthiophen-2- yl)butanoate AI(2)
- Step B Preparation of ethyl 2,4-dioxo-4-(5-phenethylthiophen-2- yDbutanoate AI(3)
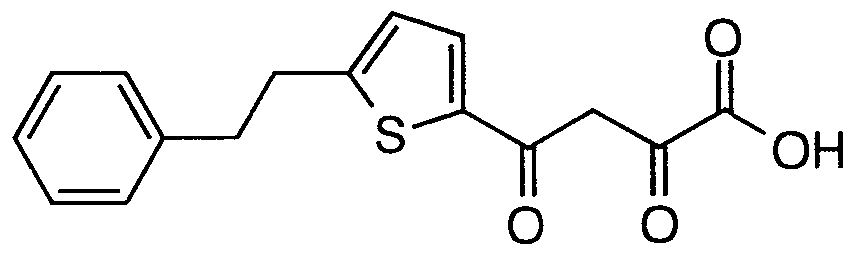
- Step C Preparation of 2,4-dioxo-4-(5-phenethylthiophen-2- yl)butanoic acid AI(4)
- a suspension of sodium hydride (538 mg, 22.4 mmol) in anhydrous DMSO (30 mL) was stirred at 60 °C under an atmosphere of argon for 1 hr.
- the resultant mixture was cooled to rt, benzyl alcohol (2.32 mL, 22.40 mmol) and 2-acetyl-5-chlorothiophene (3.01 g, 18.74 mmol) was added.
- the mixture was heated under an atmosphere of argon at 85 °C overnight.
- the product mixture was concentrated under vacuum, and the residue partitioned between ethyl acetate and dilute aqueous HCl.
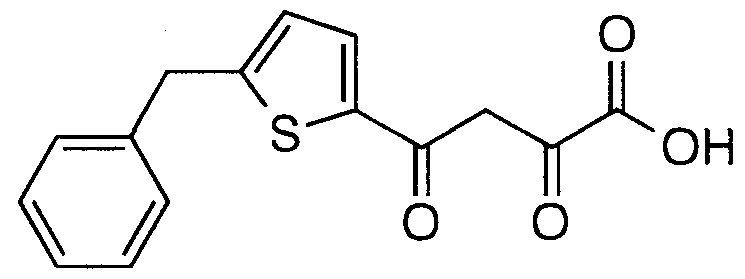
- Step B Preparation of 2,4-dioxo-4-(5-benzyloxythiophen-2- yDbutanoic acid AII(4a)
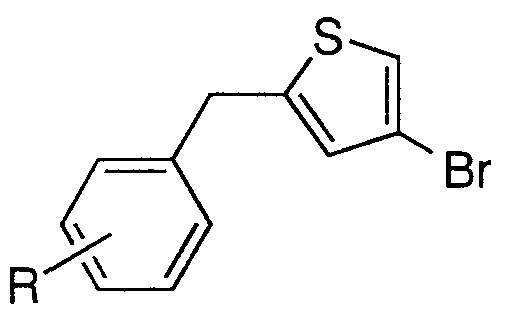
- Step A Preparation of (5-bromothiophen-2-yl)-(3- fluorophenyDmethanol BI(2a)
- the resultant solution was diluted with dichloromethane, and neutralized with dilute HCl.
- the organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum to provide the title compound as brown oil.
- the oil was used in the following step without further purification.
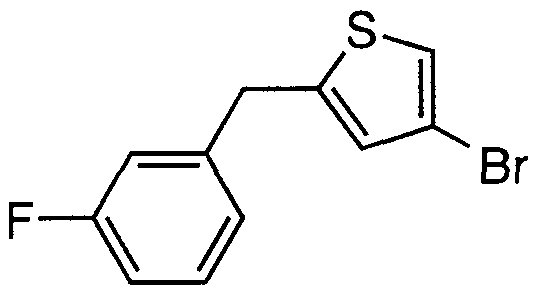
- Step B Preparation of 2-bromo-5-(3-fluorobenzyl)thiophene BI(3a)
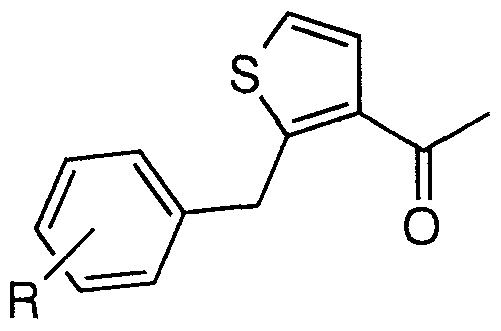
- Step C Preparation of 2-acetyl-5-(3-fluorobenzyl)thiophene BI(4a)
- the resultant solution was diluted with ether, and neutralized with dilute HCl.
- the organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum.
- the residue was subjected to column chromatography on silica gel eluting with 20% ethyl acetate in hexane. Collection and concentration of appropriate fractions provide the title compound as clear pale yellow oil.
- Step D Preparation of ethyl 2,4-dioxo-4-[5-(3-fluorobenzyl)thiophen-
- the resultant solution was diluted with ethyl acetate, and neutralized with dilute HCl.
- the organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum. The residue was triturated with hexane. The precipitate was filtered to provide the title compound as yellow solid.
- Step E Preparation of 2,4-dioxo-4-[5-(3-fluorobenzyl)thiophen-2- yl]butanoic acid BI(6a)
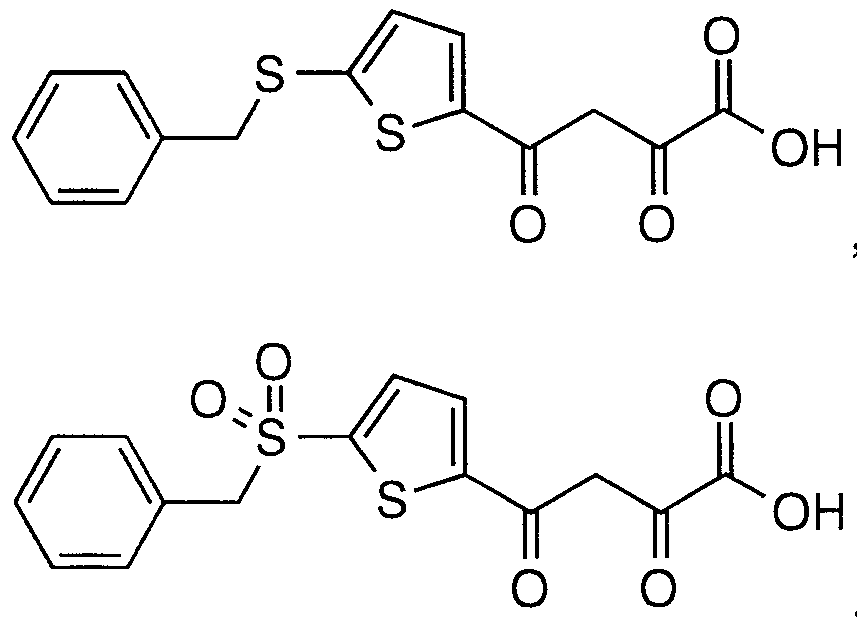
- Step A Preparation of 2-acetyl-5-phenylsufanylthiophene BII(2)
- Step A Preparation of (4-bromothiophen-2-yl)-(3- fluorophenyDmethanol CI(2a)
- Step C Preparation of 2,4-dioxo-4-[5-(3-fluorobenzyl)thiophen-3- yl]butanoic acid CI(6a)
- the resultant solution was diluted with ether, and washed successively with aq. NaOH, and brine.
- the organic extract was dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum.
- the residue was subjected to column chromatography on silica gel eluting with hexane. Collection and concentration of appropriate fractions provided the title compound.
- Step B Preparation of 4-acetyl-2-phenylsulfanylthiophene CII(2)
- the resultant solution was diluted with ether, and neutralized with dilute HCl.
- the organic extract was washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated under vacuum.
- the residue was subjected to column chromatography on silica gel eluting with 20% ethyl acetate in hexane. Collection and concentration of appropriate fractions provide the title compound as clear pale yellow oil.
Abstract
Description































































































































































Claims
























Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000552109A JP2002517390A (en) | 1998-06-03 | 1999-06-01 | HIV integrase inhibitor |
| AU42255/99A AU757409B2 (en) | 1998-06-03 | 1999-06-01 | Hiv integrase inhibitors |
| CA002333771A CA2333771A1 (en) | 1998-06-03 | 1999-06-01 | Hiv integrase inhibitors |
| EP99926095A EP1086091A4 (en) | 1998-06-03 | 1999-06-01 | Hiv integrase inhibitors |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US8784698P | 1998-06-03 | 1998-06-03 | |
| US60/087,846 | 1998-06-03 | ||
| GB9814925.5 | 1998-07-09 | ||
| GBGB9814925.5A GB9814925D0 (en) | 1998-07-09 | 1998-07-09 | HIV integrase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999062897A1 true WO1999062897A1 (en) | 1999-12-09 |
Family
ID=26314001
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1999/012094 WO1999062897A1 (en) | 1998-06-03 | 1999-06-01 | Hiv integrase inhibitors |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1086091A4 (en) |
| JP (1) | JP2002517390A (en) |
| AU (1) | AU757409B2 (en) |
| CA (1) | CA2333771A1 (en) |
| WO (1) | WO1999062897A1 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6333323B1 (en) | 1998-03-26 | 2001-12-25 | Shionogi & Co., Ltd. | Indole derivatives with antiviral activity |
| US6492423B1 (en) | 1998-07-27 | 2002-12-10 | Istituto Di Ricerche Di Biologia Molecolare Pangeletti Spa | Diketoacid-derivatives as inhibitors of polymerases |
| WO2003035076A1 (en) * | 2001-10-26 | 2003-05-01 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Dihydroxypyrimidine carboxamide inhibitors of hiv integrase |
| US6620841B1 (en) | 1998-12-25 | 2003-09-16 | Shionogi & Co., Ltd. | Aromatic heterocycle compounds having HIV integrase inhibiting activities |
| WO2005087759A1 (en) * | 2004-03-10 | 2005-09-22 | The Government Of The United States Of America, As Represented By The Secretary Of The Department Of Health And Human Services | Qiunolin-4-ones as inhibitors of retroviral integrase for the treatment of hiv, aids and aids related complex (arc) |
| US7169780B2 (en) | 2001-10-26 | 2007-01-30 | Istitute Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | N-substituted hydroxypyrimidinone carboxamide inhibitors of HIV integrase |
| US7323460B2 (en) | 2002-03-15 | 2008-01-29 | Merck & Co., Inc. | N-(substituted benzyl)-8-hydroxy-1,6-naphthyridine-7-carboxamides useful as HIV integrase inhibitors |
| EP2033952A1 (en) | 2001-03-01 | 2009-03-11 | Shionogi&Co., Ltd. | Nitrogen-containing Heteroaryl compounds having HIV Integrase Inhibitory Activity |
| EP2045242A1 (en) | 2002-08-13 | 2009-04-08 | Shionogi&Co., Ltd. | Heterocyclic compounds having inhibitory activity against HIV integrase |
| WO2010047774A2 (en) * | 2008-10-20 | 2010-04-29 | The Texas A & M University System | Inhibitors of mycobacterium tuberculosis malate synthase, methods of marking and uses thereof |
| US7888375B2 (en) | 2006-07-19 | 2011-02-15 | The University Of Georgia Research Foundation, Inc | Pyridinone diketo acids: inhibitors of HIV replication |
| US8703801B2 (en) | 2009-12-07 | 2014-04-22 | University Of Georgia Research Foundation, Inc. | Pyridinone hydroxycyclopentyl carboxamides: HIV integrase inhibitors with therapeutic applications |
| EP3042894A1 (en) | 2001-08-10 | 2016-07-13 | Shionogi & Co., Ltd. | Antiviral agent |
| US10414747B2 (en) | 2016-10-04 | 2019-09-17 | Merck Sharp & Dohme Corp. | Benzo[b]thiophene compounds as sting agonists |
| US10793557B2 (en) | 2018-04-03 | 2020-10-06 | Merck Sharp & Dohme Corp. | Sting agonist compounds |
| US11285131B2 (en) | 2017-08-04 | 2022-03-29 | Merck Sharp & Dohme Corp. | Benzo[b]thiophene STING agonists for cancer treatment |
| US11312772B2 (en) | 2017-08-04 | 2022-04-26 | Merck Sharp & Dohme Corp. | Combinations of PD-1 antagonists and benzo [b] thiophene STING agonists for cancer treatment |
| US11702430B2 (en) | 2018-04-03 | 2023-07-18 | Merck Sharp & Dohme Llc | Aza-benzothiophene compounds as STING agonists |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4336397A (en) * | 1980-12-29 | 1982-06-22 | Merck & Co., Inc. | 2,4-Dioxo-4-substituted-1-butanoic acid derivatives useful in treating urinary tract calcium oxalate lithiasis |
| US4386092A (en) * | 1980-01-16 | 1983-05-31 | Yoshitomi Pharmaceutical Industries, Ltd. | Heterocyclic-substituted oxoalkanoic acid derivatives |
| US5470862A (en) * | 1995-02-03 | 1995-11-28 | Ohmeda Pharmaceutical Products Division Inc. | Substituted pyrazolyl compounds and methods employing such compounds |
| US5618830A (en) * | 1994-10-17 | 1997-04-08 | Merck & Co., Inc. | Dioxobutanoic acid derivatives as inhibitors of influenza endonuclease |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL137974A0 (en) * | 1998-03-26 | 2001-10-31 | Shionogi & Co | Indole derivatives having antiviral activity |
-
1999
- 1999-06-01 CA CA002333771A patent/CA2333771A1/en not_active Abandoned
- 1999-06-01 EP EP99926095A patent/EP1086091A4/en not_active Withdrawn
- 1999-06-01 JP JP2000552109A patent/JP2002517390A/en not_active Withdrawn
- 1999-06-01 WO PCT/US1999/012094 patent/WO1999062897A1/en not_active Application Discontinuation
- 1999-06-01 AU AU42255/99A patent/AU757409B2/en not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4386092A (en) * | 1980-01-16 | 1983-05-31 | Yoshitomi Pharmaceutical Industries, Ltd. | Heterocyclic-substituted oxoalkanoic acid derivatives |
| US4336397A (en) * | 1980-12-29 | 1982-06-22 | Merck & Co., Inc. | 2,4-Dioxo-4-substituted-1-butanoic acid derivatives useful in treating urinary tract calcium oxalate lithiasis |
| US5618830A (en) * | 1994-10-17 | 1997-04-08 | Merck & Co., Inc. | Dioxobutanoic acid derivatives as inhibitors of influenza endonuclease |
| US5470862A (en) * | 1995-02-03 | 1995-11-28 | Ohmeda Pharmaceutical Products Division Inc. | Substituted pyrazolyl compounds and methods employing such compounds |
Non-Patent Citations (4)
| Title |
|---|
| DATABASE HCAPLUS 1 January 1900 (1900-01-01), LIN BOR SHENG, SCHEBLEIN J W, BAGLEY J R: "Substituted Pyrazolyl Compounds and Methods Employing These Compounds", XP001052762, Database accession no. 1996:13275 * |
| DATABASE HCAPLUS 1 January 1900 (1900-01-01), SALEH R M: "Use of Ethyl 2-Thenoylpyruvate in the Synthesis of Heterocycles and their Derivatives", XP001052763, Database accession no. 1991:228839 * |
| DATABASE HCAPLUS 1 January 1900 (1900-01-01), YANBORISOV T N, ET AL: "Synthesis and Pharmacological Activity of Heteroylpyruvic Acids and Their Derivatives", XP001055000, Database accession no. 1998:808874 * |
| See also references of EP1086091A4 * |
Cited By (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6506787B2 (en) | 1998-03-26 | 2003-01-14 | Shionogi & Co., Ltd. | Indole derivatives having an antiviral activity |
| US6333323B1 (en) | 1998-03-26 | 2001-12-25 | Shionogi & Co., Ltd. | Indole derivatives with antiviral activity |
| US6716605B2 (en) | 1998-03-26 | 2004-04-06 | Shionogi & Co., Ltd. | Indole derivatives having an antiviral activity |
| US6492423B1 (en) | 1998-07-27 | 2002-12-10 | Istituto Di Ricerche Di Biologia Molecolare Pangeletti Spa | Diketoacid-derivatives as inhibitors of polymerases |
| US7098201B2 (en) | 1998-12-25 | 2006-08-29 | Shionogi & Co., Ltd. | Heteroaromatic derivatives having an inhibitory activity against HIV integrase |
| US6620841B1 (en) | 1998-12-25 | 2003-09-16 | Shionogi & Co., Ltd. | Aromatic heterocycle compounds having HIV integrase inhibiting activities |
| US6645956B1 (en) | 1998-12-25 | 2003-11-11 | Shionogi & Co., Ltd. | Heteroaromatic derivatives having an inhibitory activity against HIV integrase |
| EP2033952A1 (en) | 2001-03-01 | 2009-03-11 | Shionogi&Co., Ltd. | Nitrogen-containing Heteroaryl compounds having HIV Integrase Inhibitory Activity |
| EP3042894A1 (en) | 2001-08-10 | 2016-07-13 | Shionogi & Co., Ltd. | Antiviral agent |
| US9572813B2 (en) | 2001-08-10 | 2017-02-21 | Shionogi & Co., Ltd. | Antiviral agent |
| US7169780B2 (en) | 2001-10-26 | 2007-01-30 | Istitute Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | N-substituted hydroxypyrimidinone carboxamide inhibitors of HIV integrase |
| US7217713B2 (en) | 2001-10-26 | 2007-05-15 | Istituto Di Richerche Di Biologia Molecolare P. Angeletti S.P.A. | N-substituted hydroxypyrimidinone carboxamide inhibitors of HIV integrase |
| US7232819B2 (en) | 2001-10-26 | 2007-06-19 | Istituto Di Ricerche Di Biologia P. Angeletti S.P.A. | Dihydroxypyrimidine carboxamide inhibitors of HIV integrase |
| US7435734B2 (en) | 2001-10-26 | 2008-10-14 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | N-substituted hydroxypyrimidinone carboxamide inhibitors of HIV integrase |
| US7459452B2 (en) | 2001-10-26 | 2008-12-02 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Dihydroxypyrimidine carboxamide inhibitors of HIV integrase |
| WO2003035076A1 (en) * | 2001-10-26 | 2003-05-01 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Dihydroxypyrimidine carboxamide inhibitors of hiv integrase |
| US7820660B2 (en) | 2001-10-26 | 2010-10-26 | Instituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | N-substituted hydroxypyrimidinone carboxamide inhibitors of HIV integrase |
| US7323460B2 (en) | 2002-03-15 | 2008-01-29 | Merck & Co., Inc. | N-(substituted benzyl)-8-hydroxy-1,6-naphthyridine-7-carboxamides useful as HIV integrase inhibitors |
| EP2045242A1 (en) | 2002-08-13 | 2009-04-08 | Shionogi&Co., Ltd. | Heterocyclic compounds having inhibitory activity against HIV integrase |
| WO2005087759A1 (en) * | 2004-03-10 | 2005-09-22 | The Government Of The United States Of America, As Represented By The Secretary Of The Department Of Health And Human Services | Qiunolin-4-ones as inhibitors of retroviral integrase for the treatment of hiv, aids and aids related complex (arc) |
| US7776883B2 (en) | 2004-03-10 | 2010-08-17 | The United States Of America As Represented By The Department Of Health And Human Services | Quinolin-4-ones as inhibitors of retroviral integrase for the treatment of HIV, AIDS and AIDS related complex (ARC) |
| US7888375B2 (en) | 2006-07-19 | 2011-02-15 | The University Of Georgia Research Foundation, Inc | Pyridinone diketo acids: inhibitors of HIV replication |
| US8664255B2 (en) | 2008-10-20 | 2014-03-04 | The Texas A&M University System | Inhibitors of mycobacterium tuberculosis malate synthase, methods of making and uses thereof |
| WO2010047774A3 (en) * | 2008-10-20 | 2010-08-19 | The Texas A & M University System | Inhibitors of mycobacterium tuberculosis malate synthase, methods of marking and uses thereof |
| WO2010047774A2 (en) * | 2008-10-20 | 2010-04-29 | The Texas A & M University System | Inhibitors of mycobacterium tuberculosis malate synthase, methods of marking and uses thereof |
| US8703801B2 (en) | 2009-12-07 | 2014-04-22 | University Of Georgia Research Foundation, Inc. | Pyridinone hydroxycyclopentyl carboxamides: HIV integrase inhibitors with therapeutic applications |
| US10414747B2 (en) | 2016-10-04 | 2019-09-17 | Merck Sharp & Dohme Corp. | Benzo[b]thiophene compounds as sting agonists |
| US10703738B2 (en) | 2016-10-04 | 2020-07-07 | Merck Sharp & Dohme Corp. | Benzo[b]thiophene compounds as STING agonists |
| US10730849B2 (en) | 2016-10-04 | 2020-08-04 | Merck Sharp & Dohme Corp. | Benzo[b]thiophene compounds as STING agonists |
| US11285131B2 (en) | 2017-08-04 | 2022-03-29 | Merck Sharp & Dohme Corp. | Benzo[b]thiophene STING agonists for cancer treatment |
| US11312772B2 (en) | 2017-08-04 | 2022-04-26 | Merck Sharp & Dohme Corp. | Combinations of PD-1 antagonists and benzo [b] thiophene STING agonists for cancer treatment |
| US10793557B2 (en) | 2018-04-03 | 2020-10-06 | Merck Sharp & Dohme Corp. | Sting agonist compounds |
| US11702430B2 (en) | 2018-04-03 | 2023-07-18 | Merck Sharp & Dohme Llc | Aza-benzothiophene compounds as STING agonists |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2333771A1 (en) | 1999-12-09 |
| EP1086091A1 (en) | 2001-03-28 |
| AU757409B2 (en) | 2003-02-20 |
| AU4225599A (en) | 1999-12-20 |
| JP2002517390A (en) | 2002-06-18 |
| EP1086091A4 (en) | 2001-10-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6262055B1 (en) | HIV integrase inhibitors | |
| WO1999062897A1 (en) | Hiv integrase inhibitors | |
| US6306891B1 (en) | HIV integrase inhibitors | |
| EP1326611B1 (en) | Aza- and polyaza-naphthalenyl-carboxamides useful as hiv integrase inhibitors | |
| US6841558B2 (en) | Aza-and polyaza-naphthalenyl carboxamides useful as HIV intergrase inhibitors | |
| AU756826C (en) | HIV integrase inhibitors | |
| CA2425440C (en) | Aza- and polyaza-naphthalenyl carboxamides useful as hiv integrase inhibitors | |
| US7087610B2 (en) | Benzothiazole antiviral agents | |
| US6380249B1 (en) | HIV integrase inhibitors | |
| EP1943221B1 (en) | Piperazine amidines as antiviral agents | |
| US7569573B2 (en) | Diketo acids with nucleobase scaffolds: anti-HIV replication inhibitors targeted at HIV integrase | |
| US20070287712A1 (en) | Piperazine Enamines as Antiviral Agents | |
| EP1082121A1 (en) | Hiv integrase inhibitors | |
| US8450361B2 (en) | Substituted indole and azaindole oxoacetyl piperazinamide derivatives | |
| US7501419B2 (en) | 4-Squarylpiperazine derivatives as antiviral agents | |
| AU2004200446A1 (en) | Substituted 6-Benzyl-4-Oxopyrimidines, process for their preparation and pharmaceutical compositions containing them | |
| US20060223834A1 (en) | Diketo acids on nucleobase scaffolds as inhibitors of Flaviviridae | |
| US6890942B2 (en) | Acyl sulfonamides as inhibitors of HIV integrase | |
| AU6109500A (en) | Alpha-hydroxy-gamma-(((carbocyclic-or heterocyclic-substituted)amino)carbonyl)alkanamide derivatives and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AU AZ BA BB BG BR BY CA CN CU CZ EE GD GE HR HU ID IL IN IS JP KG KR KZ LC LK LR LT LV MD MG MK MN MX NO NZ PL RO RU SG SI SK SL TJ TM TR TT UA US UZ VN YU ZA |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 42255/99 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 552109 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999926095 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2333771 Country of ref document: CA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999926095 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 42255/99 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1999926095 Country of ref document: EP |