LENTIVIRAL VECTOR COMPOSITIONS AND METHODS OF USE
1.0 BACKGROUND OF THE INVENTION
The present application is a continuation-in-part application of U. S. patent application Serial No. 08/935,312. filed September 22, 1997, the entire contents of which is specifically incorporated herein by reference in its entirety. The United States government has certain rights in the present invention pursuant to grant number HL-59412 from the
National Institutes of Health.
1.1 FIELD OF THE INVENTION
The present invention relates to molecular biology, and in particular, to viral vector compositions useful for the expression of a target gene at high levels in an eukaryotic cell. More particularly, the invention discloses and claims lentivirus packaging and transducing vectors that provide both increased packaging efficiency and increased long-term gene expression. Also provided are methods for using these vector compositions in the recombinant production of selected proteins and in the transfection of particular cell types and cell lines both in vitro and in vivo
1.2 DESCRIPTION OF RELATED ART 1.2.1 VIRAL VECTORS FOR GENE TRANSFER
Viral vectors transduce genes into target cells with high efficiencies owing to specific virus envelope-host cell receptor interaction and viral mechanisms for gene expression. Consequently, viral vectors have been used as vehicles for the transfer of genes into many different cell types including whole embryos, fertilized eggs, isolated tissue samples, and cultured cell lines. Retroviral vectors, capable of integration into the cellular chromosome, have also been used for the identification of developmentally important genes via insertional mutagenesis (Watson et al, 1992). Viral vectors, and retroviral vectors in particular, are also used in therapeutic applications (e.g., gene therapy), in which a gene (or genes) is added to a cell to replace a missing or defective gene or to inactivate a pathogen such as a virus.
1.2.2 LENTIVIRAL VECTORS
An important consideration when using lentiviral vectors for gene transfer into susceptible host cells is that of possible cytopathogenicity upon exposure to some cytotoxic viral proteins. Exposure to HIV-1 proteins may induce cell death or functional unresponsiveness in T cells (Chirmule et al . 1995; Li et al., 1995; Lifson et al., 1986: Macreadie et al, 1996; Nosaka et al, 1 93). It has been observed that direct gene transfer of particular genes into tissue culture cells by the calcium-phosphate DNA co-precipitation method can induce more than 80% cell death. Such cell death was caused mainly by necrosis, with a residual percentage (approximately 2-4%) being caused by apoptosis, or programmed cell death.
Another concern in the development of lentiviral gene transfer systems has been the possibility of generating replication-competent, virulent virus by recombination. Likewise, safety concerns have prompted much effort towards the development of non-viral vector systems, such as liposome-mediated gene transfer, naked DNA injections and gene gun technology. However, all of these non-viral gene transfer methods lack the ability to allow permanent integration of foreign genes into the host cell chromosomes, and are relatively- inefficient.
1.3 DEFICIENCIES IN THE PRIOR ART For long term expression of therapeutic genes in target cells, efficient means of transduction and genome integration are essential. In view of the wide variety of potential genes available for therapy, it is clear that an efficient means of delivering these genes is needed in order to fulfill the promise of gene therapy as a means of treating infectious, as well as non-infectious diseases. While several viral systems including murine retrovirus, adenovirus, parvovirus (adeno-associated virus), vaccinia virus, and herpes virus have been developed in recent years, their use as therapeutic gene transfer vectors has been limited to a few systems and relatively fewer genes (for a review see Nienhuis et al, 1993).
Many factors such as tissue tropism. stability of virus preparations, genome packaging capacity and construct-dependent vector stability all contribute to the difficulties of broad-range use of viral vectors. In addition, in vivo application of viral vectors is often
limited by host immune responses against viral structural proteins and/or transduced gene products.
One of the key issues in human gene therapy is the toxicity and safety to the treatment subjects. Gene therapy applications in humans have met with problems associated with the host immune responses against the gene delivery vehicles or the therapeutic gene products. Viral vectors (e.g., adenovirus) which co-transduce several viral genes together with the therapeutic gene(s) are particularly problematic. For example, readministration is necessary for adenovirus vectors because of the transient nature of viral gene expression. As such, a host immune response to the vector or the therapeutic gene product may be detrimental (Trapnell and Gorziglia, 1994; Tripathy et al., 1996).
Although MLV-vectors have not been reported to induce cytotoxicity and do not elicit strong host immune responses, lentiviral vectors such as HIV-1 which carry several immunostimulatory gene products have the potential to cause cytotoxicity and induce strong immune responses in vivo. The latter are known to induce strong cell-mediated immune responses upon transient exposure (Clerici et al, 1992; Clerici et al, 1994; Pinto et al, 1995; Rowland-Jones et al. 1995). However, this may not be a concern for lentiviral derived transducing vectors, as the latter need not encode any viral genes in the transducing vector.
Given these and other limitations, it is clear that improved vector systems are urgently needed to provide a means of delivering and expressing genes efficiently in mammalian cells. particularly human cells. Such improved vectors would greatly aid the technologies of gene expression and cellular development. Moreover, the creation of such improved vectors is a necessary step toward fully realizing the promise of gene therapy in humans.
2.0 SUMMARY OF THE INVENTION The present invention overcomes these and other inherent limitations in the prior art by providing attenuated lentiviral vector compositions having improved viral packaging and transduction efficiencies that are useful for the delivery of nonlentiviral genes to a target cell. The invention also provides methods for the use of these vectors in delivering one or more transgenes to a target cell, and in particular, to a nondividing cell in a mammal such as a human.
The packaging vectors of the present invention differ from those known previously in that they contain fewer native lentiviral polynucleotide sequences, and hence present a reduced risk of recombination. In particular, the packaging vectors of the present invention are characterized by either (a) the use of a modified but functional major splice donor site, substantialh incapable of serving as a site for homologous recombination, or (b) by the complete omission of the major splice donor site. In an illustrative embodiment, the modified major splice donor site is modified so that it is substantially identical to the major splice donor site of a non-lentiviral retrovirus, such as that of a Rous sarcoma virus (RSV). "Substantially identical" in this context means that minor differences may be tolerated to the extent that they do not significantly impair the functional integrity of the unit.
Other non-essential sequences, such as one or more accessory genes of the source lentivirus may also be deleted in these packaging vectors. Preferably, in the 5' LTR region of the packaging vector, the wild-type promoter and the enhancer element are replaced with a non-homologous promoter (and, optionally, one or more non-homologous enhancer elements).
These modified vectors of the present invention provide significant improvements over those of the prior art by reducing the risk of generating replication-competent virus through recombination with the transducing vector or a defective provirus endogenous to the host or target cell. Preferably, the 5' LTR promoter is a highly inducible promoter, so that the expression of Gag, Pol and Env proteins may be carefully controlled by one of skill in the art employing the vector composition of the present invention. This, together with the inactivation of certain accessory genes, tends to reduce cytotoxicity, and make such vectors more suitable for certain types of gene delivery. Preferably, the Gag and Pol functions are encoded by a first vector and the Env function (preferably, a non HIV-1 -like envelope protein) is encoded by a second vector.
Preferably, Gag expression is enhanced by the operable linking of the gag gene to an enhancer sequence element such as a Kozak sequence.
In a preferred embodiment, the transducing vector is characterized by a functional major splice donor site that differs from that of its source lentivirus. In the latter case, its
major splice donor site need not be identical to that of the packaging vector(s). Such a modification preferably leaves intact a functional packaging signal.
Likewise, the vectors of the invention have a strong nonlentiviral promoter/enhancer in place of the normal 5' LTR, and the gag (except for packaging signals) and pol gene sequences are deleted. Desirably, the env gene sequences are deleted to the extent that this can be done without a substantial loss in yield.
While there may still be regions of sequence identity between the packaging and transducing \ectors that are sufficiently long to present the possibility of homologous recombination, an advantage of the presently disclosed vector system is that it cannot homologously recombine to create a recombinant virus that possesses a functional packaging signal, a functional major splice donor site, and a gag AUG codon. Even if the recombined virus possesses a 5' promoter/enhancer and genes otherwise encoding equivalents of the Gag. Pol and Env proteins, homologous recombination will not occur using the present invention. The first region of significant homology is in the gag gene, after the initiation codon. Hence. if the recombinant virus derives a functional packaging signal and a functional major splice donor site from the transducing vector, it still lacks the gag AUG, since it can crossover to the packaging vector only after the AUG. Contrariwise, if it has the 5' sequence of the packaging vector through the gag AUG, it will lack a functional packaging signal and a functional major splice donor site. A replication-competent virus could still be generated

nonhomologous recombination, or by recombination with a defective endogenous retrovirus.
2.1 LENTIVIRAL VECTOR COMPOSITIONS
The present invention contemplates a gene amplification and transfer system comprising a transducing vector (TV), one or more compatible packaging vectors (HP), and a suitable host cell, the transducing vector and at least one packaging vector being derived from a lentivirus, that allow (1) transfection of the packaging vectors into the host cell to form a packaging cell line that produces essentially (packaging vector RNA)-free viral particles, (2) transfection of the transducing vector into the packaging cell line. (3) the packaging of the transducing vector RNA by the packaging cell line into infectious viral particles, and (4) the administration of the particles to target cells so that such cells are transduced and subsequently express a transgene carried by the transducing vector.
Either the particles are administered directly to the subject, in vivo, or the subject's cells are removed, infected in vitro with the particles, and returned to the body of the subject.
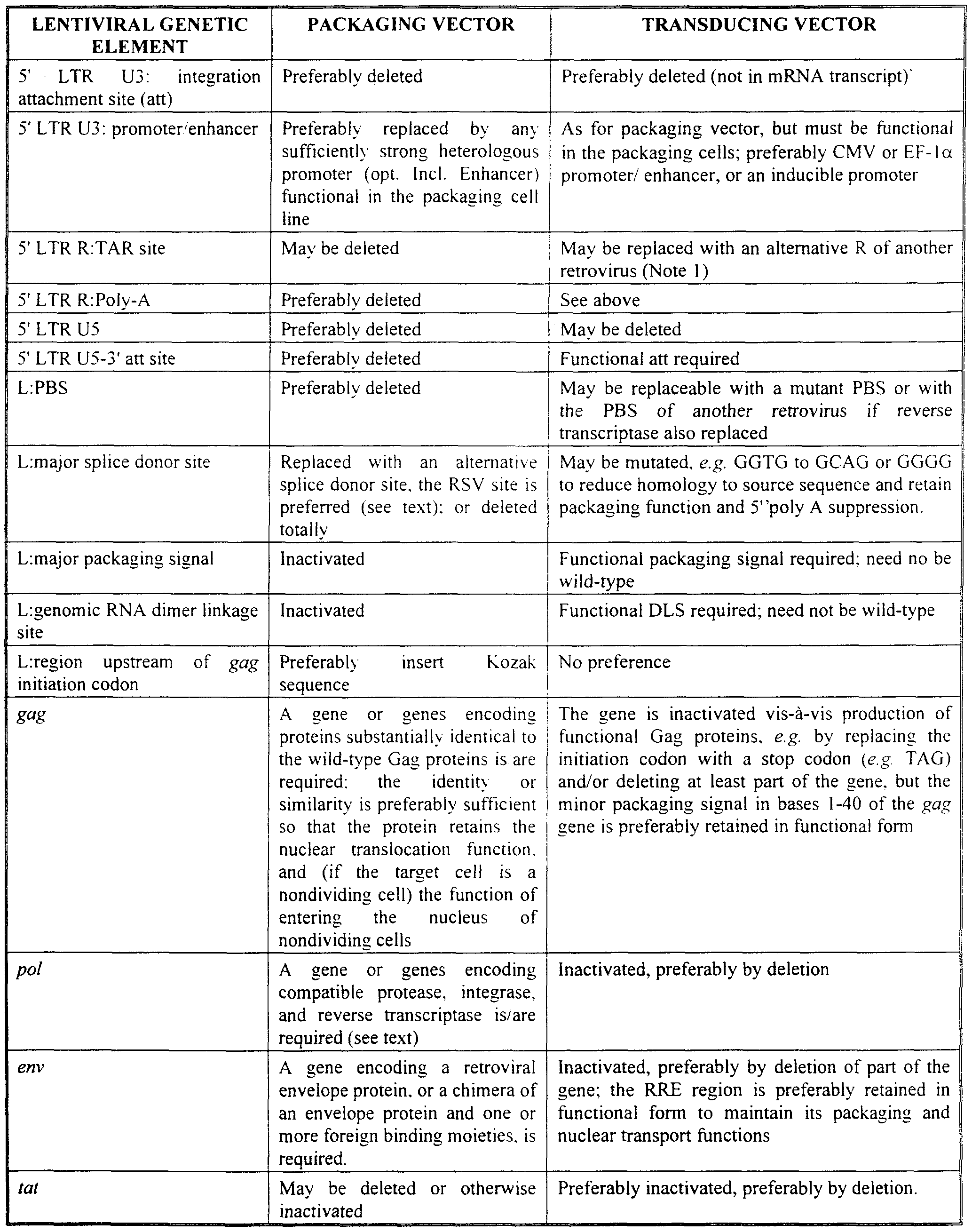
The basic characteristics of the packaging vector and the transducing vector are summarized in the following table:
3.0 BRIEF DESCRIPTION OF THE DRAWINGS
The following drawings form part of the present specification and are included to further demonstrate ceπain aspects of the present invention. The invention may be better understood by reference to the following description taken in conjunction with the accompanying drawings, in which like reference numerals identify like elements, and in which:
FIG. 1A is a simplified schematic illustration showing the HIV-1 genomic structure.
FIG. IB is a simplified schematic illustration of the MV-1 LTR.
FIG. 1C provides simplified schematic illustrations of three I-UV-L LTR deletion constructs.
FIG. ID provides simplified schematic illustrations of three heterologous enhancer/promoter inserts (human CMV IE(a), human CMV IE(b), and Mo-MLV).
FIG. 2 is a graph showing the reverse transcriptase activity of a representative attenuated recombinant HIV-1 tat mutant over time (days post-infection).
FIG. 3A, FIG. 3B, and FIG. 3C show the organization of the HIV-1 genome and a series of HIV-1 mutants containing LTR, tαt, and tte/mutations.
FIG. 4 shows replication efficiencies of several HIV-I recombinants carrying heterologous genes. FIG. 5 shows an HIV-1 transducing vector diagram for the HIV packaging construct l-del.ewv (pHP-ldl).
FIG. 6 shows a Western analysis of HIV-1 proteins in HeLa cells.
FIG. 7 show s seven pHB-1 -derived packaging vector constructs.
FIG. 8 shows six pTV-derived transducing vector constructs. FIG. 9 A shows a pTVψ-derived construct.
FIG. 9B shows a pTVΔ-derived construct.
FIG. 10 shows the Gag processing rates of wild-type HIV-infected MT4 compared with tat-C HIV chronic high producing cells.
FIG. 11 shows a Western analysis of expression of Tat+ and Taf HIV particles and infected cells.
FIG. 12A illustrates the possible cross-over to generate RCV from co-transfection of pHP-dl.28 and pTV-dl.CMVnlacZ.
FIG. 12B similarly illustrates possible crossover with the same packaging vector and a different transducing vector, pTVΔ. FIG. 13A provides a schematic showing a portion of the wild-type HIV-1 sequence, as well as the tatB (wild-type sequence provided in SEQ ID NO:4; the tatB sequence is provided in SEQ ID NO:20).
FIG. 13B provides a schematic showing a portion of the wild-type HIV-1 sequence, as well as the nefA mutations and nefB mutations (wild-type sequence provided in SEQ ID NO:5 and SEQ ID NO:6). The neβ mutations are shown in SEQ ID NO: 18 and SEQ ID NO: 19). The nefA sequence is the same as the wildtype sequence for the sequence shown starting at base 9001 (SEQ ID NO:6 represents the sequences for both wild-type and nefA). For the sequence shown starting at base 8781, the nefA sequence is the same as the nefB sequence shown in SEQ ID NO:5 (i.e. SEQ ID NO:5 represents the sequences for both nefA and neβ in the sequence shown starting at base 8781 ).
FIG. 14 shows a Western analysis of Gag processing in wild-type or tαt HIV-1 infected cell cultures.
FIG. 15 shows a Western analysis indicating the effect of Tat on Gag processing in infected HeLa cells. FIG. 16 shows a Western analysis of the effect of Tat on Gag processing in infected is TE671 cells.
FIG. 17 provides the sequence of a portion of the wild-type HIV-1 sequence, as well as the tatB (wild-type sequence provided in SEQ ID NO:4), and tatA (SEQ ID NO: 16), tatB (SEQ ID NO:20), and tatC (SEQ ID NO: 17). FIG. 18 compares the structures of pHP, wt HIV-1 and pTVΔ.
FIG. 19A, FIG. 19B. and FIG. 19C show the structures of HIV-1 and numerous transducing vector variants, together with the viral titers relative to pTVΔ set at unity. The locations of the SD. the gag AUG codon, and various known or potential packaging signals (stem-loop structures) are indicated. FIG. 20 is a table setting forth the relative titers for the transducing vectors of FIG.
19A, FIG. 19B, and FIG. 19C, but further indicating the number of constructs tested in each sample group, the standard error, and the paired P value.
FIG. 21 is a table comparing wild-type HIV-1, pHP and pTV 5' sequences.
FIG. 22A illustrates the analyses of 5' splice site and SL2 deletion mutants. Schematic illustration of the four stem-loop structure of the HIV packaging signal. SD mutations, and relative vector efficiencies. The relative vector titer of each mutant was determined by normalizing against that of pTV, which was 7.3 ± 0.2 x 10D tu/ml and is arbitrarily set at 1.00.
FIG. 22B shows the Northern analyses of poly (A)+cytoplasmic RNA of SL2 deletion mutants (SD3 and SD4) in comparison to SD1 *. The SD1 mutation effects have been reported previously (Clever et al, 1995). The structure of pTV and its four major RNA species are shown; "F", full-length unspliced, "ss", short intron-spliced, "CMV+s", CMV promoter driven nlacZ transcript plus a spliced RNA population, "Is", long intron-spliced RNA. The hybridization probe used in the Northern analyses is illustrated. FIG. 22C shows the quantitative analyses of viral titer, cytoplasmic full-length viral
RNA, virion RNA, and packaging efficiency of SD3, SD4, and SD1* mutants vs. wt pTV.
For easy comparison, the vector titer, cytoplasmic full-length RNA, packaged virion RNA, and packaging efficiency are all normalized against those of pTV which are set at 1.00.
FIG. 23A shows the analyses of gag AUG and SL4 mutants. Schematic diagram of gag AUG and 5' gag mutants in comparison to a previously reported mutant, gag/env.d\5* (Clever et al, 1995). and relative vector titers.
FIG. 23B shows the Northern analyses of gag AUG and 5' gag mutants. The full- length RNA is denoted by asterisks.
FIG. 23C shows the quantitative comparison of viral titer, cytoplasmic unspliced RNA, packaged virion RNA, and packaging efficiency. FIG. 24A shows the analyses of vector functions of combination mutations in SD,
SA, gag AUG, gag, and env. Shown is a schematic diagram of pTV mutant constructs and their relative titers.
FIG. 24B illustrates the Northern analyses of cytoplasmic RNA of the multiple mutants. FIG. 24C shows the comparison of viral titer, cytoplasmic full-length RNA, packaged virion RNA, and packaging efficiency of the combination pTV mutants.
4.0 DESCRIPTION OF ILLUSTRATIVE EMBODIMENTS
Illustrative embodiments of the invention are described below. In the interest of clarity, not all features of an actual implementation are described in this specification. It will of course be appreciated that in the development of any such actual embodiment, numerous implementation-specific decisions must be made to achieve the developers' specific goals, such as compliance with system-related and business-related constraints, which will vary from one implementation to another. Moreover, it will be appreciated that such a development effort might be complex and time-consuming, but would nevertheless be a routine undertaking for those of ordinary skill in the art having the benefit of this disclosure.
4.1 RETROVIRUSES AND RETROVIRAL VECTORS
The term "retrovirus" is used in reference to RNA viruses that utilize reverse transcriptase during their replication cycle. The retroviral genomic RNA is converted into double-stranded DNA by reverse transcriptase. This double-stranded DNA form of the virus is capable of being integrated into the chromosome of the infected cell; once integrated, it is referred to as a "provirus." The provirus serves as a template for RNA polymerase II and
directs the expression of RNA molecules that encode the structural proteins and enzymes needed to produce new viral particles. At each end of the provirus are structures called "long terminal repeats" or "LTRs." The LTR contains numerous regulatory signals including transcriptional control elements, polyadenylation signals and sequences needed for replication and integration of the viral genome. There are several genera included within the family Retroviridae, including Cisternavirus A, Oncovirus A, Oncovirus B, Oncovirus C, Oncovirus D, Lentivirus, and Spumavirus. Some of the retroviruses are oncogenic (i.e. tumorigenic). while others are not. The oncoviruses induce sarcomas, leukemias, lymphomas, and mammary carcinomas in susceptible species. Retroviruses infect a wide variety of species, and may be transmitted both horizontally and vertically. They are integrated into the host DNA, and are capable of transmitting sequences of host DNA from cell to cell. This has led to the development of retroviruses as vectors for various purposes including gene therapy.
Retroviral vectors derived from the amphotropic Moloney murine leukemia virus (MLV-A), use cell surface phosphate transporter receptors for entry and then permanently integrate into proliferating cell chromosomes. The amphotropic MLV vector system has been well established and is a popular tool for gene delivery (see e.g., Gordon and Anderson, 1994; Miller et al, 1993).
Other retroviruses, including human foam> virus (HFV) and human immnunodeficiency virus (HIV) have gained much recent attention, as their target cells are not limited to dividing cells. Moreover, their restricted host cell tropism can be readily expanded via pseudotyping with vesicular stomatitis virus G (VSV-G) envelope glycoproteins (see e.g.. Bums et al, 1993; Lever, 1996; Russell and Miller, 1996).
While many viral vector systems are available, virtually all of the current human gene therapy that use retroviral vectors are derived from the -amphotropic Moloney murine leukemia virus (M-MuLV), such as pLNL6 (Genbank M63653), see Baker, et al, 1987, for gene transfer (see also Miller and Buttimore, 1986). Among the vectors known in the art, special note may be taken of Chang, U. S. Pat. No. 5,693,508 (1997) that discloses retroviral vectors confining chimeric MoMLV/CMV- IE/HI V-TAR LTRs. The elements essential to the retroviral vector system are viral structural proteins Gag, Pol and Env, the long terminal repeats (LTR), the reverse transcription templates including primer binding site (PBS) and
polypurine tract (PPT), and the packaging signals (psi ψ)The MLV-A vector system is comprised of a packaging cell line expressing Gag, Pol and Env, and a vector construct containing LTRs, PBS. a PPT and the packaging signal sequences. Up to 8 kb of foreign sequences can be inserted into the MLV vector and packaged into virus particles. The commonly used amphotropic MLV packaging cell lines such as PA317, PG-13, ψ-CRIP, GP- AM12 and FLY-AI3 produce 10 07 transducing units per ml after vector DNA transfection (Cosset et al. 1995: Kotani et al, 1994; Lam et al, 1996; Markowitz et al. 1988; Miller and Chen. 1996).
The M-MuLV system has several advantages: 1 ) this specific retrovirus can infect many different cell types; 2) established packaging cell lines are available for the production of recombinant M-MuLV viral particles; and, 3) the transferred genes are permanently integrated into the target cell chromosome. The established M-MuLV vector systems comprise a DNA vector containing a small portion of the retroviral sequence (the viral long terminal repeat or "LTR" and the packaging or "psi" [ψ] signal) and a packaging cell line. The gene to be transferred is inserted into the DNA vector. The viral sequences present on the DNA vector provide the signals necessary for the insertion or packaging of the vector RNA into the \ iral particle and for the expression of the inserted gene. The packaging cell line provides the viral proteins required for particle assembly (Markowitz et al, 1988).
The \ ector DNA is introduced into the packaging cell by any of a variety of techniques (e.g . calcium phosphate coprecipitation, lipofection, electroporation, etc.). The viral proteins produced by the packaging cell mediate the insertion of the vector sequences in the form of RNA into viral particles that are shed into the culture supernatant. The M-MuLV system has been designed to prevent the production of replication-competent virus as a safety measure. The recombinant viral particles produced in these systems can infect and integrate into the target cell but cannot spread to other cells. These safeguards are necessary to prevent the spread of the recombinant virus from the treated patient and to avoid the possibility of helper virus-induced disease (Miller and Buttimore, 19XX; Markowitz et al. 19XX).
After selection, producer cell clones can be established to generate 10 -10 transducing units per ml. Increased transduction efficiencies may be achieved by modification of the transduction protocols through means such as repetitive infection steps,
cocultivation with the producer cell line, centrifugation, and modification of the culture conditions using growth factors and fibronectin, etc. (Kotani et al, 1994; Moritz et al, 1996). Despite these advantages, existing M-MuLV-based retroviral vectors are limited by several intrinsic problems: (a) they do not infect non-dividing cells (Miller et al, 1990);
(b) they produce only low titers of the recombinant virus (Miller and Rosman, 1989; Miller, 1992);
(c) they express foreign proteins at low levels and often get "turned-off ' or inactivated after integration (Miller, 1992); (d) the instability of the enveloped virus particles, as it is both difficult to concentrate in vitro and difficult to manipulate in vivo (Miller, 1992);
(e) the MLV LTR activity is also known to be suppressed in embryonal cells (Challita et al , 1995; Loh et al, 1988); and
(f) long-term expression after viral integration is often restricted by transcription repression, likely due to DNA methylation (Boyes and Bird. 1991 ; Szyf et al,
1990. The low production of recombinant virus produced by the M-MuLV system (e.g.. 10 /ml) compared to the adenoviral system (up to 10 /ml) means that human cells are infected at a very low efficiency. This low efficiency is particularly disadvantageous when the target cell type is represented at very low numbers in the tissue to be infected. Although the hematopoietic stem cell is a preferred target for gene therapy in a large number of disorders, these cells are present at very low frequencies. For example, totipotent embryonic stem cells have been reported to occur at a frequency of 10" to 10" in bone marrow (Glick and Pasternak, 1994). Thus, the low titer produced by existing M-MuLV vector systems is highly inefficient for stem cell infection.
The promoter present in the M-MuLV LTR is quite weak compared with other viral promoters such as the human cytomegalovirus immediate early (CMV-IE), enhancer/promoter. In order to increase expression of the genes carried on the retroviral vector internal promoters possessing stronger activities than the M-MuLV promoter have been utilized. However, the inclusion of an internal promoter to drive the expression of the inserted gene does not always lead to increased levels of expression (Robinson et al. 1995).
Apparently, the activity of the internal promoter is significantly decreased because of interference from the upstream M-MuLV promoter (i.e. transcriptional read-through interference). The dual transcription-unit construct is, however, a common feature in almost all M-MuLV vectors. To create an improved retroviral vector suitable for a wide variety of gene expression studies and gene therapy applications, the clinically-approved gene therapy vector pLNL6 has been modified to allow synthesis of high basal levels of mRNA and increased packaging efficiency. Exemplary vectors and methods have been described in co-pending U.S. Pat. Appl. Serial No. 08/336,132, and Intl. Pat. Appl. Publ. No. PCT/US95/14576 (each specifically incorporated herein by reference in its entirety). However, other limitations remain.
4.2 LENTIVIRUSES AND LENTIVIRAL VECTORS
As used herein, the term "lentivirus" refers to a group (or genus) of retroviruses that give rise to slowly developing diseases. Viruses included within this group include HIV (human immunodeficiency virus; including HIV type 1 , and HIV type 2), the etiologic agent of the human acquired immunodeficiency syndrome (AIDS); visna-maedi, that causes encephalitis (visna) or pneumonia (maedi) in sheep, the caprine arthritis-encephalitis virus, that causes immune deficiency, arthritis, and encephalopathy in goats; equine infectious anemia virus, that causes autoimmune hemolytic anemia, and encephalopathy in horses: feline immunodeficiency virus (FIV). that causes immune deficiency in cats; bovine immune deficiency virus (BIV), that causes lymphadenopathy, lymphocytosis, and possibly central nervous system infection in cattle; and simian immunodeficiency virus (SIV), that cause immune deficiency and encephalopathy in sub-human primates. Diseases caused by these viruses are characterized by a long incubation period and protracted course.
Usually, the viruses latently infect monocytes and macrophages, from which they spread to other cells. HIV, FIV, and SIV also readily infect T lymphocytes (i.e., T-cells).
Lentivirus virions have bar-shaped nucleoids and contain genomes that are larger than other retroviruses. Lentiviruses use tRNAlys as primer for-negative-strand synthesis, rather than the tRNApr0 commonly used by other infectious mammalian retroviruses. The lentiviral
genomes exhibit homology with each other, but not with other retroviruses (see Davis et al. 1990).
An important factor in the disease caused by these viruses is the high mutability of the viral genome that results in the production of mutants capable of evading the host immune response. It is also significant that they are capable of infecting non-dividing cells. Lentiviruses including HIV, SIV, feline immunodeficiency virus (FIV) and equine infectious anemia virus (EIAV) depend on several viral regulatory genes in addition to the simple structural gag-pol-env genes for efficient intracellular replication. Thus, lentiviruses use more complex strategies than classical retroviruses for gene regulation and viral replication, with the packaging signals apparently spreading across the entire viral genome. These additional genes display a web of regulatory functions during the lentiviral life cycle. For example, upon HIV-1 infection, transcription is up-regulated by the expression of Tat through interaction with an RNA target (TAR) in the LTR. Expression of the full-length and spliced mRNAs is then regulated by the function of Rev that interacts with RNA elements present in the gag region and in the env region (RRE)-( Schwartz et al., 1992). Nuclear export of gag-pol and env mRNAs is dependent on the Rev function. In addition to these two essential regulatory genes, a list of accessory genes, including vif vpr, vpx, vpu, and ne are also present in the viral genome. Their effects on efficient virus production and infectivity have been demonstrated, although they are not absolutely required for virus replication (Wong-Staal, 1991 ; Subbramanian and Cohen, 1994; Trono, 1995).
HIV-1 virions contain 60% protein and 2% nucleic acid. The genome consists of two molecules of linear positive-sense single stranded RNA (held together by hydrogen bonds to form a dimer). Even within a single virion, these molecules need not be identical. Hence, genetic variation can occur through recombination between the two viral RNAs of a single virion.
The HIV-1 genome is about 9.7 kb in length. Many HIV-1 pro viral genome sequences have been sequenced in their entirety. The sequence GenBank Ml 9921, LOCUS HIVNL43, Human immunodeficiency virus type 1, NY5/BRU (LAV-1) recombinant clone pNL4-3, 9709 bp ss-RNA, is used as a reference sequence in this discussion. The construction of pNL4-3 has been described in Adachi, et al. (1986). pNL4-3 is a recombinant (infectious) proviral clone that contains DNA from HIV isolates NY5 (5' half and BRU (3'
half). The site of recombination is the EcoRI site at positions 5743-5748. The final sequence is set forth in Dai et al. (1992).
For several reasons, the HIV-1 genome has a high mutation rate. First, there can be recombination between the two RNAs of a single virion. Secondly, a single cell can be infected more than one viral particle simultaneously, and recombination occur between the two viral genomes. Finally, the HIV reverse transcriptase has a high frequency of misincorporation (1 : 1700 to 1 :4000). The replication error rate for HIV is such that each newly synthesized HIV genome carries on average approximately one mutation. For all of these reasons, there is not one HIV-1 sequence, but rather a family of closely related sequences. Different HIV-1 sequences may be identified even in different samples isolated from a single individual. The degree of genetic variation observed is phenomenal—up to 20% within an infected individual. This is essentially due to remorseless cycles of viral replication, most probably due to chronic activation of the immune system. It can be estimated that the number of variants in existence worldwide must be in excess of 10 -1018. and given the nature of RNA viruses even more novel variants should emerge.
HIV-l's are currently divided into two genetic groups based on phylogenetic reconstruction using DNA sequences. The majority of these sequences fall into the M (major) group, while a smaller, but growing, number of sequences are classified as O (outlier). Most HIV-1 strains from around the world can be placed into one of nine nucleotide sequence-defined clades; these clades have been given the letter designations A through I. However, more than a dozen HIV-1 strains isolated from patients have now been shown to ha\ e chimeric genomes in that their gag and env genomic regions cluster with different clades. Interclade recombination is relatively easy to demonstrate because strains from different clades typically differ substantially in their nucleotide sequence identities. For example, the env gene sequences of HIV- I strains of different clades may differ by 20% or more. As might be expected, interclade HIV-1 recombinants have most often been detected in geographic regions where two or more clades are prevalent. At least 17 HIV clades have now been reported in humans: nine HIV-1 clades in the major grouping (A through 1), three HIV-1 group 0 group "outlier" clades, and five HIV-2 clades. Three additional lentiviruses are known in nonhuman primate species (African green monkeys, mandrils, and Syke's monkeys). Thus the potential gene pool for primate lentivirus recombination is on the order
of 20, e.g., 20 gag genes and 20 pol genes. The current HIV-1 clades may have arisen in part through past recombination between some of these genes. Viable recombinants between SIV and HIV ("SHIV' strains) have been genetically engineered.
The principal elements of the HIV-1 genome are set forth below, in the 5' to 3' direction. For further information, see Vaishnav and Wong-Staal, (1991 ). The positions of each element are given according to the Genbank numbering of the complete genome sequence (MI-9921 ) cited above. That means that the numbering begins with the first base of the S' LTR. not with the cap site. The exact positions will vary from strain to strain, and some elements are better defined than others. Note that some genetic elements overlap, and that two (tat and rev) are interrupted. For a compilation of numerous sequences and alignments, at both the nucleic acid and amino acid levels, for many lentiviruses and other retroviruses, see the HIV Sequence Database at http://hiv-web.lanl.gov.
4.3 5' LTR (1-634) Each end of the DNA provirus contains the so-called long terminal repeats (LTRs).
The 5' LTR and 3' LTR regions are essentially identical in the wild-type HIV- I genome. These LTRs are 634-bp non-coding sequences, located at the extreme 5'and 3'ends of the proviral genome, that contain enhancer and promoter regions. The LTRs consist of three distinct coding regions. U3, R, and U5, that can be subdivided into the separate enhancer and promoter regions. The U3 region is 450, the R sequence 100 and the US region some 85 nt long. Transcription initiates at the first base of the R region in the 5'LTR. and polyadenylation occurs immediately after the last R region base in the 3'LTR. The primary transcript is thus about 600 bases shorter than the provirus.
The U3 region includes several features of interest: the integration attachment site (att) at the far 5' end. the promoter TATA box (a segment of DNA, located approximately 19-27 base pairs upstream from the start point of eukaryotic structural genes, to which RNA polymerase binds), promoter (SP 1) regions (promoter binding site for RNA polymerase and reverse transcriptase), the kappa-enhancer (contains two imperfect 1 1 -bp repeats, GGGACTTTCC (SEQ ID NO:XX) and IL-1 and IL2 homologous enhancers.
The R region (454-550) contains the transcription initiation site, the TAR (Tat- activating) region and. the poly A signal (-AATAAAA-); the latter is significant only in the 3' LTR). The primary transcript corresponds to bases 455 to 9626.
The US region contains a polya downstream element and a second integration '15 attachment site at the 3' end. These are sicnificant only in the 3'LTR.
4.4 PRIMER BINDING SITE
Immediately downstream of the 5' LTR is the primer binding site (PBS) (637-651 ) for minus-strand DNA synthesis, called the RNA cap. The PBS is complementary to the 3' end of a Lys transfer RNA (tRNA1> ).
4.5 5' LEADER (L)
The 5' leader (L), the untranslated region between the primer binding site and the initiation codon for gag, has two elements worthy of note. The first is the major 5' splice donor (SD) site (the splice point is at 748) which is used for the processing of full-length genomic RNA to subgenomic mRNA for the synthesis of various viral proteins. The major splice donor site is so called because it acts as the donor site during splicing of the vif. vpr. tat, rev, vpu-env and nef subgenomic RNAs (The Gag-Pol polyprotein is translated from genomic RNA). There are also minor splice donor sites in the vicinity of the first exon of the rev gene.
The other is the major packaging signal (psi) (651-669) which serves as a contact 30 point for the Gag nucleocapsid (Ncp7) protein to bind the RNA and to incorporate it into virus particles. Note that one can define an extended packaging signal extending into the gag gene, to about 820. The 5' leader also contains a sequence that participates in the dimer-linkage structure of 70S RNA. This DLS overlaps with the major packaging signal. A secondary structure model of the leader, and the 5' end of gag, has been prepared (Baudin et al, (1993).
4.6 STRUCTURAL GENES The gag gene encodes a polyprotein (55 kDa) (CDS 790..2292) that is cleaved by the viral protease (see pol) to yield various core and nucelocapsid proteins. The gαg-coding
region extends from the ATG initiation codon at nucteotide 337 to nucleotide 1837 relative to the RNA cap site. The polyprotein is translated from unspliced viral RNA. The precursor Gag protein is cleaved by protease to produce pi 7 (the major matrix MA protein, involved in membrane anchoring, env interaction, and nuclear transport of viral core), p24 (the core capsid CA protein), p7 (the nucleocapsid NC protein, which binds RNA), and p6 (which binds Vpr). A pair of zinc finger motifs in the NC protein binds to the major packaging signal in the viral RNA. The gag gene is believed by some authors to contain one or more minor packaging signals.
The pol gene (CDS est. 2085..5096) encodes a large polyprotein that is a precursor to the virion proteins providing the viral enzyme functions: protease, reverse transcriptase, and integrase. The gag and pol genes overlap

241 nucleotides, and are in different reading frames. A slippage sequence in or upstream of the gag-pol overlap region induces an occasional ribosomal frameshift at a frequencv (about 5%) that ensures that Gag proteins are made in large amounts and Pol proteins in small amounts. Initially, a gag-pol fusion protein (p 190) is created as a result of the ribosomal frameshift, that does not interrupt translation. The viral protease cleaves Gag from Pol, and further digests Gag and plO to separate the various mature proteins. In the case of Pol, the cleavage products are protease (plO), reverse transcriptase (p50), RnaseH (pl5) and integrase (p31). Roughly 50% of the RT remains linked to Rnase H as a single potypeptide (p66). The principal functional form of RT is actually a heterodimer of p66 and p50. All pol gene products are found within the capsid of free HIV- I virions.
Reverse transcriptase is responsible for the synthesis of double-stranded DNA from the viral RNA. Activity of RT is localized to the N-terminus. RT in HIV has an extremely high error rate (about 1/1700 nucleotides). At the 3' end of the pol coding region is the coding region for viral endonuclease/integrase. Integrase functions to integrate the proviral DNA in the host genome.
The env gene (CDS 6221..8785) is located at the 3' end of the genome. It encodes the envelope protein gpl60, some of which is cleaved to yield the envelope proteins gpl20 and gp41. Both function in cell recognition on the outer envelope of a released virus. The C- terminus of gpl20 interacts with the viral receptor CD4 of human T lymphocytes to facilitate the viral entry into the host cell. Only a 12-amino acid sequence in gpl20 is necessary for
binding to CD4; the rest of the protein is mutable. The gpl20 polypeptide contains nine conserved intrachain disulfide bridges and, within this scaffolding, folds into five globular domains (I-V). There are five hypervariable regions (VI -V5) whose sequences vary especially widely among HIV-1 isolates.
4.7 REGULATORY GENES
The tat gene (CDS 5830..6044, 8369..8414) encodes Tat, a trørø-activating protein, the most important activator of of the LTR promoter region. Three functional domains have been identified: an amino terminal amphipathic helix, a cluster of seven cysteine residues, and a stretch of basic amino acids involved in nuclear localization. It is known that conservative mutations of the acidic amino acids of the amphipathic helix are tolerated. Tat mediates the 5' LTR by interacting with its R region, in a segment termed the "TAR" (trans- activating response) element (bases 436-497). The "TAR" element forms a stable stem loop structure that interacts with the Tat protein to prevent premature termination of transcription initiation. Tat is reported in the literature to be absolutely essential for HIV transcription and consequently for viral replication.
The rev gene (CDS 5969..6044, 8369..8643) encodes Rev, another transactivator. Rev is phosphorylated at serine residues, but serine substitution mutants that are not phosphorylated are fully active. The amino terminal 20 amino acids and the carboxy terminal 25 amino acids are known to be dispensable. There are two important domains, a stretch of basic amino acids, which is involved in nuclear localization and in interaction with RRE RNA. and a leucine-rich region, presumed to be involved in transactivation, whose leucines are intolerant of mutation. Rev is a protein whose target is termed RRE (Rev-response element), on the env protein coding region of the mRNA. Interaction of Rev with the RRE region apparently allows for transport of unspliced RNA from the nucleus to the cytoplasm. RRE (7758-7992) is an RNA secondary structure element. Proviruses lacking Rev function remain transcriptionally active but fail to generate new viral particles.
4.8 ACCESSORY GENES The nef gene (CDS 8787..9407) encodes Nef. and overlaps the env gene and the 3'
LTR. Nef may be involved in signal transduction, although this is controversial. There has
also been speculation that Nef down-regulates viral expression. The Nef protein does not appear to be essential to the HIV life cycle in tissue culture.
The vif gene (CDS 5041..5619) encodes Vif, the virion infectivity factor. Vif- deficient mutants are typically much less efficient than wild type HIV at cell-free (as opposed to cell-to-cell) virus transmission. It is not a virion component and the mechanism by which it affects infectivity is unclear.
The vpr gene (CDS 5559..5849) encodes Vpr, a virion protein which accelerates the replication and cytopathic effect of HIV- I in CD4+ T-cells. About I 00 copies of Vpr are associated with each virion. The vpu gene (CDS 6061..6306) encodes Vpu. The vpu gene encodes part of a polycistronic transcript that also includes the env gene. Vpu is a cytoplasmic protein that is thought to facilitate assembly and/or release of viral particles.
4.9 PPT (BASES 9059-9075) Immediately upstream from the 3' LTR is the polypurine tract vital to initiation of positive-strand DNA synthesis.
4.10 3 'LTR (9076..9709)
The 3' LTR is identical to the S' LTR, but is significant mainly by virtue of its poly- A signal (9602..9607), and the "R'"repeat sequence (9S29..9626) allowing RT jumping during DNA synthesis.
4.11 INFECTIVITY
HIV-1 infects activated and resting lymphocytes, terminally differentiated monocytes and neuronal cells through cellular receptors and co-receptors such as CD4, chemokine receptors and galactosyl ceramide (Harouse et al, 1991; Weiss, 1996). The restricted lentiviral host cell tropism can be expanded by pseudotyping the virus particles with broadly tropic viral envelope proteins from human T cell leukemia virus type I (HTLV-1). amphotropic MLV envelope protein or the vesicular stomatitis virus G glycoprotein (Bums et al, 1993; Landau et al, 1991 ; Page et al, 1990; Spector et al, 1990). Alternatively, a CD4 receptor can be introduced into target cells by adenovirus transduction before HIV vector
transduction in a two-step transduction protocol (Miyake et al, 1996). Naldini, et al. have demonstrated that HIV-I vectors pseudotyped with MLV-A or VSV-G envelope could produce up to 5 X 10" transducing units/ml of vectors capable of infecting nondividing cells such as macrophages and terminally differentiated neurons (Naldini et al, 1996). Infection of nondividing cells by lentiviruses such as HIV-I is mediated by the nuclear localization signal (NLS) in the Gag MA protein (Bukrinsky et al , 1993). Efficient viral entry and integration into non-dividing cells may also require some of the accessory gene products such as Vpr (Fletcher et al, 1996; Heinzinger et al, 1994).
4.12 CYTOTOXICITY
One difficulty related to HIV vector development encountered during the development of the present invention is the cytotoxicity of many HIV gene products to human cells. In particular, it has been difficult to establish continuous cell lines expressing the essential structural proteins Gag. Pol and Env for particle assembly. Cell lines expressing Tat, Rev, Nef have been established. However, expression of Gag, Rev and Vpr has been shown to induce cytopathology. cell death and cell cycle arrest in human cells (See, Emennan, 1996; Miele and Lever. 1995; Nosaka et al, 1993). The development of a tightly inducible system was favored for a lentiviral packaging cell line (Yu et al, 1996). HIV-1 Vpr also induces apoptosis in human cells. The expression of VSV-G protein induces syncytium formation that acain is problematic for establishing a packaging cell line.
4.13 LENTIVIRIS
A "source" or "original" lentivirus is a wild-type lentivirus from which an attenuated and/or replication-defective lentivirus is derived, or which is used as a starting point during construction of the packaging or transducing vector, for the preparation of one or more of the genetic elements of the vector. The genetic element may be employed unchanged, or it may be mutated (but not beyond the point where it lacks a statistically significant sequence similarity to the original element). A vector may have more than one source lentivirus, and the different source lentiviruses may be, e.g., HIV-1 and HIV-2, or HIV and SIV, and so forth.
One may also speak of a "source" or "original" gene, genetic element or protein for a vector gene, genetic element or protein. (The term "genetic element" includes but is not limited to a gene.)
The cognate lentivirus is the wild-type lentivirus with which the vector in question has the greatest percentage sequence identity at the nucleic acid level. Normally, this will be the same as the source lentivirus. However, if a source lentivirus is extensively mutated, it is conceivable that the vector will then more closely resemble some other lentivirus. It is not necessary that the cognate lentivirus be the physical starting point-for the construction; one may choose to synthesize a genetic element, especially a mutant element, directly, rather than to first obtain the original element and then modify it.
One may also speak of a "cognate" protein, gene, or genetic element {e.g., splice donor site or packaging signal). When referring to a cognate protein, percentage sequence identities are of course determined at the amino acid level.
The term "cognate" lentivirus may be difficult to interpret in the extreme case, i.e., if all lentiviral genetic elements have been replaced with surrogate non-lenti viral genetic elements. In this case, the preferred source HIV-1 strain mentioned previously is arbitrarily consider to be the cognate lentivirus.
HIV type 2 (HIV-2) is known to be less pathogenic than HIV-1 in humans, and HIV- 2 infection is associated with natural protection against HIV- I infection. Simian immunodeficiency virus (SIV) also infects human cells; however, it is unclear whether it can cause- AIDS in humans. Thus, both HIV-2 and SIV may be better candidates than HIV-1 for developing lentiviral vectors. It may be advantageous to derive both the packaging and transducing vectors from a lentivirus other than HIV-1 , or to derive one from HIV-1 and the other frovi a lentivirus other than HIV-1. Use of different sources for the two vectors reduces the risk of homologous recombination to generate RCV, and use of a source other than HIV- 1 reduces the health risk if recombination, homologous or otherwise, occurs.
The term "replication" as used herein in reference to a virus or vector, refers not to the normal replication of proviral DNA in a chromosome as a consequence of cell reproduction, or the autnomous replication of a plasmid DNA as a result of the presence of a functional origin of replication, but rather to the completion of a complete viral life cycle wherein infection viral particles containing viral RNA enter a cell, the RNA is reverse transcribed into
DNA, the DNA integrates into the host chromosome as a provirus, the infected cell produces virion proteins and assembles them with full length viral genomic RNA into new, equallv infectious particles.
The term "replication-competent" refers to a. wild-type virus or mutant virus that is capable of replication, such that replication of the virus in an infected cell result in the production of infectious virions which, after infecting another, previously uninfected cell. causes the latter cell to likewise produce such infectious virions. The present invention contemplates the use of replication-defective virus.
A used herein, the term "attenuated virus" refers to any virus (e.g., an attenuated lentivirus that has been modified so that its pathogenicity in the intended subject is substantially reduced. Preferably, the virus is attenuated to the point it is nonpathogenic from a clinical standpoint, i.e., that subjects exposed to the virus do not exhibit a statistically significant increased level of pathology relative to control-subjects.
The present invention contemplates the preparation and use of an attenuated lentivirus. In some embodiments, the attenuated lentivirus is selected from the group consisting of attenuated mutants of human immunodeficiency virus type 1, human immunodeficiency virus type 2, feline immunodeficiency virus, simian
virus, visna-maedi, caprine arthritis-encephatitis virus, equine infectious anemia virus, and bovine immune deficiency virus. Thus, the attenuated virus may be an attenuated HIV-1. attenuated HIV-2, attenuated SIV. or a virus comprised of portions of more than one tentiviral species (e.g.. a hybrid, comprised of portions of HIV-1 and HIV-2, or HIV-1 and SIV, etc.).
A reference virus is a virus whose genome is used in describing the components of a mutant virus. For example, a particular genetic element of the mutant virus may be said to differ from the cognate element of the reference virus by various substitutions, deletions or insertions. It is not necessary that the mutant virus actually be derived from the reference virus.
The preferred reference HIV-1 was mentioned previously. For HIV-2, see LOCUS HIV2ROD, 9671 bp ss-RNA, Human immunodeficiency virus type 2, isolate ROD. completeproviralgenome, ACCESSION Ml 5390 (Clavel et al, 1986).
The preferred reference SIV sequence is LOCUS SIVMM239, 13068 bp ss-RNA, a Simian immunodeficiency virus isolated from a macaque, isolate 239 (Macaca mulatto Mm 239-82); complete proviral genome and flanking sequence, GenBank ACCESSION M33262, (Regier and Desrosiers, 1990). The preferred reference RSV sequence is Genbank locus/accession # AF052428, 9396 bp DNA. the Rous sarcoma virus strain Schmidt-Ruppin B. complete genome.
In the transducing vector, The 5 'LTR and 3 'LTR regions must be sufficiently identical so that ssDNA jumping by the reverse transcriptase will occur. There can be more than one packaging vector, carrying separate structural genes. For example, one vector can encode gag and pol functions, and another vector, env functions.
The packaging vectors and transducing vectors of the present invention are each replication-incompetent viruses. Moreover, the vectors are chosen for incorporation into a given vector system of the present invention are such that it is not possible, without further mutation of the packaging vector(s) or transducing vector, for the cotransfected cells to generate a replication-competent virus by homologous recombination of the packaging vector(s) and transducing vector alone.
4.14 PACKAGING SIGNAL
As used herein, the term "packaging signal" or "packaging sequence" refers to sequences located within the retroviral genome or a vector which are required for, or at least facilitate, insertion of the viral or vector RNA into the viral capsid or particle. The packaging signals in an RNA identify that RNA as one which is to be packaged into a virion. The term "packaging signal" is also used for convenience to refer to a vector DNA sequence that is transcribed into a functional packaging signal. Certain packaging signals may be part of a gene, but are recognized in the form of RNA, rather than as a peptide moiety of the encoded protein.
The major packaging signal is the signal having the predominant effect on whether viral RNA is inserted into the particle. This signal is located in the 5' leader region (spanning the SD site and the gag AUG) of the wild-type lentiviral genome. It is not equivalent to the conventional ψ site of the MLV vectors, in that the latter alone allows efficient MLV vector packaging.
There are also minor packaging signals with a lesser effect on packaging efficiency. Several studies have shown that many sequences in HIV-1, including LTR, TAR, RRE, and in the 5' and 3' gag ORF, the pol ORF, and in the sequences flanking the RRE, contribute to efficient genome packaging, pointing to the complex nature of HIV-1 packaging signals (see e.g., Aidovini and Young. (1990); Kaye et al, (1995); Lever et al. (1989); Richardson et al, (1993).
Earlier studies of the HIV packaging signal demonstrated that a 46 nt (751-796) stem- loop structure derived from the splice donor site to the 5' gag coding region is sufficient to allow packaging of a heterologous Sendai virus RNA but the efficiency was not determined and the location of the insertion was important for the stem-loop conformation (see Hayashi et al. 1992). They further showed that the 46 nt sequence must be inserted in the 5' end of the Sendai RNA to serve as a packaging signal; inserting in the midst of the Sendai RNA destroyed the packaging signal. Secondary structure analysis showed that several stem-loop structural domains can be identified in the 5' untranstated leader region and in the 5' gag coding region (see Baudin et al, (1993).
It was shown that the packaging signals in the 5' end of the HIV genome include TAR and four stem-loops from upstream of the major 5' splice donor site extending into the first 7 arnino acid codons in the gag coding region (see McBride and Panganiban ( 1996); McBride et al, (1997). Parolin et al. demonstrated that up to 653 nt in the gag coding region can enhance RNA packaging efficiency (Parolin et al, 1994). Luban and Goff showed that the first 40 nt of gag coding sequence is strongly influential on the packaging function, and they later reported that the HIV-1 packagng signal requires the very 5' edge of the RNA and sequences downstream of the 170th nt of gag or sequences in pol. Studies indicate that for efficient packaging function, the four stem-loop structure may not be sufficient. Instead, the packaging signal as well as its sequence context consists of the entire packaging signal. This is consistent with the study of Kaye, et al. who have reported that the RRE and env sequences, although not essential to render RNA packaging, may have a positive effect on enhancing the packaging efficiency. Mutation of the gag AUG is detrimental to RNA packaging, thus it is clear that the packaging signal of HIV is not as simple as MLV and RSV.
The key distinction between a packaging vector and a transducing vector is that in the packaging vector, the major packaging signal is inactivated, and, in the transducing vector, the major packaging signal is functional. Ideally, in the packaging vector, all packaging signals would be inactivated, and, in the transducing vector, all packaging signals would be functional. However, countervailing considerations, such as maximizing viral titer, or inhibiting homologous recombination, may render such constructs less desirable.
Using a precise quantitative assay for vector function, it has been found that the 5' major splice donor site, the gag AUG and the extended gag sequences are dispensable for the packaging of a functional HP/TV vector. The highly conserved sequences essential to HIV replication (the SD and gag AUG, and additional coding sequence) have now been deleted from the pTV vector that has greatly improved the safety of the HP/TV vector system and totally eliminated the possibility of generating RCV via homologous recombination at the gag region.
4.15 PACKAGING SYSTEM; PACKAGING VECTORS; PACKAGING CELL LINE
A packaging system is a vector, or a plurality of vectors, that collectively provide in expressible form all of the genetic information required to produce a virion that can encapsulate suitable RNA. transport it from the virion-producing cell, transmit it to a target cell, and, in the target cell, cause the RNA to be reverse transcribed and integrated into the host genome in a such a manner that a transgene incorporated into the aforementioned RNA can be expressed. However, the packaging system must be substantially incapable of packaging itself. Rather, it packages a separate transducing vector that is described below. The general abbreviation for a packaging vector in this specification is HP or pHP.
In the case of an HIV-1 vector, the packaging system will provide functional equivalents of the gag, pol and env genes as discussed below. One may use a single vector that provides all three genes (a "GPE" vector) or a two vector system wherein one vector provides the gag-pol genes (a "GP" vector) and the other vector (an "E" vector) provides the e«v gene. In theory, a three vector system ("G", "P", and "E" vectors) is possible if one is willing to construct distinct gag and pol genes on separate vectors, and operably link them to different regulatable promoters (or one to a regulatable and the other to a constitutive promoter) such that their relative levels of expression can be adjusted appropriately.
The vector or vectors that together compose the packaging system are called the packaging vectors. A packaging cell line is a suitable host cell transfected by a packaging system that, under achievable conditions, produces viral particles. As used herein, the term "packaging cell lines" is typically used in reference to cell lines that express viral structural proteins (e.g.. gag, pol and env), but do not contain a packaging signal. For example, a cell line has been genetically engineered to carry at one chromosomal site within its genome, a 5'- LTR-gag-pol-3'-LTR fragment that lacks a functional psi sequence (designated as Δpsi), and a 5'-LTR- eπv-3'-LTR fragment which is also Δpsi located at another chromosomal site. While both of these segments are transcribed constitutively, because the psi' region is missing and the viral RNA molecules produced are less than full-size, empty viral particles are formed.
If a host cell is transfected by the packaging vector(s) alone, it produces substantially only viral particles without the full-length packaging vector Preferably less than I 0% of the viral particles produced by the packaging cell contain full-length packaging vector-derived RNA. However, since the packaging vector lacks a functional primer binding site, even if these particles infect a new cell, the packaging vector RNA will not be reverse transcribed back into DNA and therefore the new cell will not produce virion. Thus, by itself, the packaging vector is a replication-incompetent virus.
Preferred packaging vectors include vectors selected from the group consisting of pHP-1, pHP-dl.2 and pHP-dl.28, PHP-VSVG, PHP-CMV, pHP-CMVdel.TAR/SD, pHP- CMV-EF I a intron, and pHP-EF.
The pHP construct was made by first replacing the 5'LTR with the CMV-TATA-TAR chimeric promoter, obtained from the BbrPl to Hindlϊl fragment of the chimeric LTR containing CMV IE promoter-TATA box and TAR sequence, that was derived from a recombinant HIV-1 LTR as described previously (Chang and Zhang, (1995) then deleting the rest of the 5' leader sequence extending from the Hindlll site in the end of TAR region to the gag AUG using a synthetic ofigonucleotide containing a splice donor site of Rous sarcoma virus and a conserved Kozak sequence -CCACC- adjacent to the gag AUG.
The Kozak sequence serves to increase the translational efficiency. The gag-pol coding sequence is kept intact. Alternatively, the conserved reverse transcriptase (RT) domain of the pol sequence is replaced with RSV RT domain by PCR amplification and
cloning. The vif, vpr, vpu and env genes were mutated by site-specific mutagenesis to eliminate the AUG initiation codon and some of the coding sequence but not affecting Gag- Pol or Tat/Rev syntheses. The tat coding sequence can also be mutated as described below either by inserting multiple stop codons (e.g. tat-B mutant) or by deleting the initiation AUG codon and part or all of the coding sequence (e g. tat-C mutant) because the pHP can be tot- independent.
A rev independent gag-pol construct can also be made by mutating the nuclear retention signals in the gag-pol coding region as indicated by Schneider, et al. (1997) and in the env coding region including the RRE element. In this rev-independent pHP construct, the rev open reading frame is mutated by removing the intiation codon AUG and deleting the coding sequence. The 3'we PPT-LTR of HIV-1 was entirely deleted from the nef initiation AUG codon which was mutated to contain a new Hindlll site and replaced with a selective marker gene gpt and an SV40 polyadenylation signal.
In some embodiments, the packaging cell and/or cell line contains a transducing vector. The packaging cell line will package the transducing vector into infectious particles. Such a cell line is referred to as a "transgenic virion production cell line".
It is contemplated that packaging may be inducible, as well as non-inducible. In inducible packaging cells and packaging cell lines, lentiviral particles are produced in response to at least one inducer. In non-inducible packaging cell lines and packaging cells, no inducer is required in order for lentiviral particle production to occur.
The packaging vectors necessarily differ from wild-type, replication-competent lentiviral genomes by virtue of the inactivation of at least one packaging signal of the cognate wild-type genome. More than one packaging signal may be inactivated. Preferably, the only lentiviral genes provided by the packaging vector are those encoding structural, or essential regulatory, proteins.
4.16 TRANSDUCING VECTORS
A transducing vector is an expression vector that bears an expressible nonlentiviral gene -of interest and includes at least one functional lentiviral packaging signal, so that, after said transducing vector is transfected into a packaging cell line, the transducing vector is transcribed into RNA. and this RNA is packaged into an infectious viral particle. These
particles, in turn, infect target cells, their RNA is reverse transcribed into DNA, and the DNA is incorporated into the host cell genome as a proviral element, thereby transmitting the gene of interest to the target cells.
As used herein, the term "transduction" refers to the delivery of a gene(s) using a viral or retrovirat vector by means of infection rather than by transfection. In preferred embodiments, retroviral vectors are transduced. For example, an anti-HIV gene carried by a retroviral vector can be transduced into a cell through infection and provirus integration. Thus, a "transduced gene" is a gene that has been introduced into the cell via lentiviral or vector infection and provirus integration. In preferred embodiments, viral vectors (e.g., "transducing vectors") transduce genes into "target,cells" or host cells. It may be convenient to classify transducing vectors as follows: Generation 0 pTV: pTV vectors containing non-replication essential genes or genetic elements, (e.g. vectors previously reported by Naldini, et al. and Shimada, et al. [Naldini. #2755, (1996); Shimada. #470], 1991 ).
Generation 1 pTV: pTV vectors with deletions of all the accessory genes and non-replication essential genetic elements (e.g. vif, vpr, vpu, nef, NF-kB/Spl) Generation 2 pTV: pTV vectors with deletions of replication-essential genetic elements (e.g., gag AUG,
SD site, env sequences, RRE, TAR, such elements are also missing on pHP) Generation 3 pTV: pTV vectors with substitutions of vector-essential genetic elements (complementary substitutions are also present on pHP). In the present invention, various transducing vectors may be used, including pTVψ, pTVψlOO, pTVψl40. pTV. ψ.nlacZ, and pTVψCMV-nlacZ-hyg-di.Smal, pTV , pTV -X, pTV EFnlacZ, PTV EFGFP, PTV CMV-X, pTV CMVnlacZ, pTV SVneo, pTV SVhyg, pTV CMV-GFP, pTV CMV-nlacZ, and pTV CMV-nlacZ-hyg. However, it is not intended that the present invention be limited to these specific transducing vectors. For example, the "pTV -X," indicates that the vector may be comprised of "PTV " in combination with any gene ("X"). Thus, the present invention encompasses transducer
vectors that are suitable for use in the present invention that are linked to any gene of interest (or a "marker gene" or "reporter gene," used to indicate infection or expression of a gene).
One preferred transducing vector pTV is made of a chimeric CMV-TATA-TAR- U5/att-PBS-packaging signal-mutated SD-portion of gαg-portion of em>-mutated nef-PPT- U3/att-R-U5 which exhibits packaging function like the wild type HIV. The U5 sequence was mutated such that all of it was deleted except for the 3'24 nt att site. The 5' chimeric promoter is derived from the NF-kB/Spl deleted CMV-TATA construct of the HIV LTR mutant described previously which directs transcription at the native HIV transcriptional initiation site. The TAR is in the R region that can be mutated at both ends to maintain the repetitive function of the R but significantly different from the wild type HIV R. Alternatively, the R sequence can be replaced. with RSV R so it is completely different from HIV R sequence. Alternatively, the PBS can be modified to become RSV PBS such that the chimeric pHP RT gαg-RSV-RT-/?o/) can initiate minus-strand DNA synthesis using the appropriate tRNA primer. The packaging signal will have conserved stem-loop secondary structure as described by McBride et al. as SLI to SL4 but with mutations in SD (GGTG to GCAG or GGGG) and gag AUG (replaced with ACC or UAG) It was shown that the latter mutations have minimal effect on packaging efficiency. The mutant SD/gagAUG pTV RNA genome is packaged into transducing particles at near 70% wild type efficiency. In preferred embodiments, the vectors of the present invention are capable of "high efficiency transduction." This is intended to encompass transducing vectors capable of transduction at a level of at least 10" /ml, although in particularly preferred embodiments, the vectors are capable of transduction levels of up to 1010/mi. As used herein, the term "low efficiency transduction" refers to transducing vectors capable of transduction at levels less than or equal to 103/ml. As used herein, the term "long-term transduction" refers to vectors that are capable of remaining transduced in host or target cells for time periods that are longer than those observed with other vectors. For example, the present invention provides lentiviral vectors that remain transduced for at least 120 days, more preferably at least one year, most preferably for the life of the subject or the necessary time course of treatment. Long-term gene transduction and high efficiencies of transduction of human cells by the HIV vectors of the present invention were compared with the conventional MLV vector (see Table 5). The
duration of expression is a function of the choice of promoter and the target cell type, more so than the choice of vector.
The term "stable transduction" or "stably transduced" refers to the introduction and integration of foreign DNA into the genome of the transducted cell. The term "stable transductant" refers to a cell that has stably integrated foreign DNA into the genomic DNA.
The term "transient transduction" or "transiently transduced" refers to the introduction of foreign DNA into a cell where the foreign DNA fails to integrate into the genome of the transducted cell. The foreign DNA persists in the nucleus of the transducted cell for several days. During this time the foreign DNA is subject to the regulatory controls that govern the expression of endogenous genes in the chromosomes. The term "transient transductant" refers to cells which have taken up foreign DNA buthave failed to integrate this DNA.
In some preferred embodiments, the target and/or host cells of the present invention are "non-dividing" cells. These cells include cells such as neuronal calls that do not normally divide. However, it is not intended that the present invention be limited to non-dividing cells and may include muscle cells, white blood cells, spleen cells, liver cells, eye cells, epithelial cells, etc.).
In particularly preferred embodiments, the vector and the vector progeny are capable of transducing a plurality of target cells so as to achieve vector titers of at least 103 cfu/ml. The preferred multiplicity of infection (MOI) would be at least one (i.e., one hit on average per cell), more preferably at least two.
4.17 ADAPTATIONS FOR HIV-2 AND SIV DERIVED VECTORS
Based upon the experiments conducted during development of the HIV-1 vector system, HIV-2 and SIV vector systems may be developed (pH2P and PSIVP). To establish a tentiviral vector based on HIV-2 or SIV, the 5'LTR and the untranslated leader sequences of HIV-2ROD and SlVmac239 may be replaced with the recombinant HP-1 enhancer/promoter and a synthetic leader sequence with or without a splice donor site, both obtainable from the pHP vectors. The 3'LTR may be replaced by the SV40 polyadenylation signal. The nef and env genes may both be deleted from the vector. The expression of vpx is preferably included in the-HIV-2/SIV packaging cells because it has been shown that the HIV-2/SIV vpx (or SIVagm vpr) is necessary and sufficient for nuclear import function and does not inhibit cell
cycle progression as does vpr. The VSV-G envelope gene is preferably expressed from a separate expression vector.
Previous studies suggested that SIV or HIV-2 genomes can be assembled into the HIV-1 particles, indicating that the packaging signals of SIV or HIV-2 can be recognized by HIV-1 nucleocapsids. Thus, one may construct a hybrid vector that is essentially an HIV-1 derived vector with SIV or HIV-2 packaging signals (from 3' of the PBS to the extended gag sequences). These HIV-2 and SIV transducing vectors (pTV2 and PTVS) may be tested in co-transfection experiments using pH2P or pSIVP.
Alternatively, one may construct transducing vectors wherein the lentiviral genetic elements are derived sorely from HIV-2 (pTV2) and SIV (pTVS) . However, instead of using modified LTRs, a strong heterologous promoter is preferably used and the transcription initiation site is placed at the beginning of the R-U5 sequence. Sequences in gag-pol and env genes are deleted and the major SD and the gag AUG are mutated. A CMV-driven reporter gene cassette such as the CMV-IE-nlacZ-IRES-hyg from the pTVΔ-nlacZ-hyg vector may be inserted in the nef ORF of the HIV-2 and the SIV vectors. The 3' LTR resembles the native LTR but with a deletion in the U3 except for the 5 'att site.
4.18 MODIFIED MAJOR SPLICE DONOR SITES
A splice donor site is a sequence that directs the splicing of one exon to another exon. Typically, the first exon lies 5' of the second exon. and the splice donor site overlapping and flanking the first exon on its 3' side recognizes a splice acceptor site flanking the second exon on its 5' side. Splice donor sites have a characteristic consensus sequence represented as
(A/C) AG GURAGU (R=purine), (see Jackson, ( 1991 ). The first three bases of the splice donor consensus are the last three bases of the exon. A splice acceptor site is a sequence which acts in conjunction with a splice donor site,
5 so that the intron separating the two sites is removed. The characteristic splice acceptor site is YY-YYYYYYYYNYAG (Y=pyrimidine. N=any base).
In a preferred embodiment, the HIV-1 major SD is replaced with the cognate RSV
SD. The synthetic RSV SD sequence is split into two parts with an Agel site inserted in place of the RSV gag AUG site:
Synthetic RSV SD: (sequence derived from RSV, Gene Bank ACCESSION
#F052428. is underlined)
5'AGCTGGTCGCCCGGTGGATCAAGACCGGTAGCCGTCATAA
AGGTGATTTCGTCGGATC-3' (SEQ ID NO:XX)
(Asel)
The original RSV SD: 5'-
ATTCTGGTCGCCCGGTGGATCAAGCATGGAAGCGTTCATAAAGGT GATTTCGTCCGCGT-3' (SEOIDNO:XX)
The HIV-1 LTR consensus A 5' leader sequences (5'sj is in bold and underlined, the construct was made from HIV-INL4-3, Access # M19921):
5'-GGCTTGCTGAGGTGC-?CACAGCAA.GAGGCGAGAG — GGCGACTGCaIGAGTACGCC-??AAATTTT-3'
The entire 5' leader sequence of HIV-1 consensus A:
GCCTTGAG?TGCTT?AAGTA- TGTGTGCCCGTCTG?TT?T?TGACTCTGGTAACTAGAGATCCCT
CAGACCACT7TAGACTGTGT- AAAAATCTCTAGCAGTGGCGCCCGAACAGG????????????? ???GACTCGAAAGCGAAAG
TTCCAGAGAAG? — TCTCTCGACGCA?-
GGACTCGGCTTGCTGAGGTGC-7CACAGCAAGAGGCGAGAG — CGGCGACTGGTGAGTACGCC-??AAATTTT??-GACTAGCGGAG
GCTAGAAGGAGAGA7A
For reference, the corresponding HIV-2 and SIV sites are as follows:
HIV-2ROD 5' splice junction: (ACCESSION #M 15390)
5^ CAAAAACTGTAGCCGAAAGGGCTTC;CTATCCTACCTTTAGACAGG T AGAAGATTGTGGGAG-3'
SIV 239 (ACCESSION# M33262 M61062-M61093)
ACGGCGTGAGGAGCGGGAGAGGAAGAGGCCTCCGGTTGCA GGTAAGTGCAACACAAAAAAGAAATAGCTGTCTTTTATCCAGGAA GGGGTAATAAGATAGAGTGGGAGATG
The artificially engineered splice donor (SD) site from Rous sarcoma virus (RSV) in the pHP- I construct, a site that is unrelated to HIV sequences, was found to work like the wild- type SD site (i.e., allowing partition of spliced tat and rev, and unspliced gag-pol mRNAs into the cytoplasm). This is a critical factor in some embodiments of the present invention (i.e.. the replacement of the I-IIV SD site with the RSV SD site), as the native leader sequences and the major splice donor site must both be deleted from the HP constructs to decrease the probability of homologous recombination with the transducing vectors (TV).
The splicing junction sequences have been previously studied (Ezzell. 1995; Mount, 1996). In previous studies, it was shown that the first tat coding exon contains positive and negative splicing regulatory elements and the splicing signals can be hundreds of nucleotides away from the splice junction sites (Amendt et al, (1994). Therefore, the success of inserting a functional splice site in the leader region of HP construct using an oligonucleotide sequence containing a small number of nucleotide sequences from RSV 5' splice junction site was surprising.
The splice donor site in the packaging constructs is used solely for the expression of tat and rev genes downstream and serves to stabilize the gag-pol transcript. It is possible that tat and rev functions can be provided in trans and the 5' splice donor site can be totally eliminated. For example, an SV40 promoter with a replication origin can be used in the packaging constructs and the DNA can be transfected into a SV40 large T antigen expressing cell lines such as COS7 cells (African green monkey kidney cells expressing SV40 T Ag).
4.19 MODIFIED PACKAGING SIGNALS
The packaging signal is of course inactivated in the packaging vectors. In the transducing vectors, a functional packaging signal is required, but need not be identical to the source signal.
The packaging signals have a secondary structure; they may be mutated so as to alter the primary sequence while substantially retaining the secondary structure. Applicant has found it possible to mutate the HIV- I major packaging signal by replacing GGTG with GCAG or GGGG. Lentiviral packaging signals may be replaced with nonlentiviral packaging signals, or functional mutants thereof, such as the cognate packaging signal of another virus, such as RSV or MLV. If so. it will generally be necessary to make corresponding mutations in the Gag nucleocapsid protein so that it recognizes the new packaging signal. Thus, one could make a chimera of the Gag nucleocapsid protein and the cognate nucleocapsid protein of the other virus.
Finally, in the case of the packaging vector(s), one may delete the MV-I'maj or packaging signal altogether.
4.20 STRUCTURAL GENES/PROTEINS The terms "Gag protein" and "Gag proteins" refer to any or all proteins, respectively, encoded by the gag gene, including both the ultimate virion proteins and their precursors (i.e., proteins that are processed intracellularly into the ultimate virion proteins.) The terms "Pol protein(s)" and "Env protein(s)" are analogously defined. These terms can be further modified by "-like" or "-equivalent' as elsewhere defined. As noted above, the structural virion genes are the gag, pol and env genes. At least one, and preferably all of these genes is inactivated in the transducing vector. The only part of ga or env necessary to keep is the part that play essential roles in packaging. The first 39 nucleotides of the gag coding sequence excluding the initiation codon and the RPE in the beginning of gp41 coding region of the env sequence are essential to keep. However, site- specific mutagenesis can be performed to further change these sequences to introduce stop
codons in the gag gene and in the env gene and to kill the RRE function of interacting with Rev. This latter changes can further improve the safety of the HP/TV vector system.
However, the packaging vectors must collectively provide genes encoding the functions of the gag, pol and em' genes in order to provide a functional virion. Nonetheless, these genes may differ from the source genes by silent and other functional mutations.
4.21 SILENT MUTATIONS
These may be made almost freely throughout the gene. The only areas where caution is required is where the choice of sequence has regulatory significance, e.g., the slippage region in gag-pol, or the RRE region in env. In some instances, such as in the case of an INS element, it may actually be desirable to inacth ate the regulatory element. In other instances, the regulatory element may be useful, and only silent mutations that leave it functional are desirable.
4.22 FUNCTIONAL MUTATIONS
These are mutations which affect the amino acid sequence of one or more of the encoded polypeptides. but which do not substantially abolish the relevant biological activity of the affected polypeptide(s).
The comments which follow apply not only to mutation of lentiviral proteins, but also to mutation of naturally occurring, nonlentiviral proteins which are acting as the equivalent of a lentiviral protein. For example, instead of using wild-type VSV G protein in place of HIV-1 gp 120, one may use a functional mutant of VSV G protein.
As explained below, while the result of a mutation is not absolutely predictable, some 30 mutations are clearly more likely to be tolerated than others. The accuracy of these predictions is dependent in part on whether a 3-D structure for the protein is known, whether homologous proteins (, functional mutants, naturally occurring or otherwise) have been sequenced, and whether the biologically relevant binding sites of the protein have been identified.
The tremendous natural variation of the HIV-1 genome suggests that it is quite tolerant of multiple mutations in many genes. The following specific guidance is offered:
A general source of 3D structures is the Protein Data Bank, which is searchable on the Internet.
Gag: The overall sequence variability of Gag proteins in HIV-1 isolates is more than 20%. With deliberate mutations, it is likely that a higher degree of sequence variation can be to tolerated. The 3D structure. of the nucteocapsid protein of Gag, complexed to human cyclophilin A. has been determined; Gamble, et al. Cell, 87: 1285-94 (1996); see also PDB structures 1 atv and 1 ncp. Mutational analysis reveals that the zinc finger domains in the NC protein play important roles in RNA encapsulation and HIV infectivity. Mizuno, et al, AIDS Res Hum. Retroviruses, 12:793-800 (1996). . Charged amino acids have also been shown to be involved in RNA packaging and infectivity. Poon, et al, J. Virol, 70:6607-16 (1996). Mutational studies have also been made of the CA proteins, using deletion mutants and chimeras. Carriere, et al, J. Virol, 69:2366-77 (1995). For structure-function relationships in general, see Wills. AIDS, 5:639-54 (1991 ). 3D structures are available for the CA (PDB I afv) and MA (PDB lhiw) proteins. Pol: The overall sequence variability among HIV-1 isolates is more than 20%; in the protease domain, a variability of more than 40% has been observed. The 3D structure of the reverse transcriptase, complexed to an inhibitor, is known, see Kohlstaedt, et al, Science, 256: 1783-90 (1992), and a structural model for the protease has been proposed, see Pearl and Taylor. Nature, 329:351-4 (1987). The polymerase and protease functional domains have been studied by mutagenesis, see Loeb et al. Nature, 329:351-4 (1989); Le Grice, et al, EMBO J.. 10:3905-1 1 (1991). For 3D structures, see also PDB entries lhnv and lrtl (RT), litg (integrase), and lhvk (protease).
Env: For the Env proteins gpl20 and gp41. the overall sequence variability among HIV-1 isolates exceeds 60%. For 3D structures, see PDB entries laik (gp41) and lacy (gpl20 fragment). For discussion of functional domains, see Moulard, et al (1998). Pseudotyping studies have shown that the Env proteins can be entirely replaced by the retroviral but non-lentiviral MoMLV Env proteins, or the unrelated VSV-G protein.
The envelope proteins encoded by the packaging vector may be lentiviral or nonlentiviral proteins. The advantage of a non-lentiviral protein is that it can confer on the produced particles the ability to bind to a cell surface receptor of a class of cells not normally infected by the lentivirus. An example of a non-lentiviral envelope protein of interest is the
vesicular stomatitis virus (VSV) G protein. VSV-G pseudotyped particles are rigid and can be concentrated more than 1000-fold. They also bind to different cells than those bound by HIV- 1 gpl20 typed particles.
Where one of the packaging vectors encodes a non-lentiviral envelope protein, it is referred to as an envelope pseudotyping vector. In preferred embodiments, the pseudotyping vector is selected from the group consisting of pHEF-VSVG, pHEF.A-em', Gibbon ape leukemia virus env, and MLV-Amphotropic env.
Alternative Env proteins: The Env proteins of HIV-1 may be replaced with Env proteins of other lentiviruses, of nonlentiviral retroviruses, of-nonretroviral viruses, or with chimeras of these proteins with other peptides or proteins. Examples are the Env proteins of VSV (G protein), the hemagglutinin protein of influenza virus, the surface antigen (S and preS) of hepatitis B virus, and the Env protein of RSV. These mdofications increase the range of cells which can be transduced with HIV-1 derived vectors.
Chimeric Env Proteins: A chimera may be constructed of an env protein and of a ligand that binds to a specific cell surface receptor, in order to target the vector to cells expressing that receptor. Examples are chimeras including FLA16 (a 6 a.a. peptide which binds integrin receptors), erythropoietin (which binds the erythropoietin receptor), human heregulin (which binds the EGF and related receptors).
Alternatively, the chimera could include an antibody variable light or heavy domain, or both domains joined by suitable peptide linker(a so-called single chain antibody). Such an antibody domain could target any desired cell surface molecule, such as a tumor antigen, the human low-density lipoprotein receptor, or a determinant on human MHC Class I molecules.
Derivatized Env Proteins: Virions may be chemically, enzymatically or physically modified after production in order to alter their cell specificity. Examples of modifications include chemical or enzymatic addition of a ligand which would be recognized by a cell surface receptor (e.g.. addition of lactose so that the virions will transduce human hepatoma cells which express asialoglycoprotein receptors), or incubation of the virus with a biotinylated antibody directed against the vector's Env protein, followed by addition of a streptavidin-linked ligand recognized by the cell-surface receptor. A heterobispecific antibody could be used to link the virion's Env protein to such a ligand.
4.23 REGULATORY GENES
The vector system may provide the regulatory proteins, or surrogates therefor, or wholly omit them. If Tat or Rev equivalents are provided, the corresponding genes may be placed on the transducing vector, or on the same or different helper vector(s). These genes need not be placed on the packaging vectors. Again, silent mutations may be made almost freely.
Functional mutation of Tat and Rev should be feasible. For the 3D structure of Rev, see is PDB entry I rpv: for RR.F-, see I etf and I etg; ffor Tat, see I tiv; for TAR. see lkis.
Tat is a transcriptional factor which acts to bind the polll transcription elongation complex and increases the processivity of transcription.The amino acid sequence of Tat is highly conserved amongst different HIV-1 strains with more than 80% homology. Mutational analysis has shown that the functional domain is in the first coding exon because deletion of the second exon does not affect its transactivation function. The N-terminal domain is highly charged and contains a long stretch of basic amino acids such as arginine which is the characteristic of RNA binding domain. Tat has been shown to bind to the TAR sequence at the loop of a stem-loop structure in the 5' end of the genome. In addition to its transcriptional activation function. Tat has also been shown to enhance reverse transcription, and it has also been shown that Tat can enhance gag protein precursor processing. Therefore, the multiple functions of Tat may indicate that it may be required for high titer vector production. However, Tat may be substituted with different lentiviral transactivators to avoid recombination of HIV sequences.
Rev is also a transcriptional regulator which acts at a post transcriptional step in the nucleus to enhance the export of RRE-containing RNA to the cytoplasm. Its amino acid sequence is highly conserved amongst different HIV-1 strains. Human T cell leukemia virus type I (HTLV-1) encodes a similar protein named Rex. Rex and Rev share low sequence homology (less than 40%) but have similar functions. Mutational analysis have shown that rev function requires both coding exons. Rev binds to RRE in env and interacts with cellular proteins in the nucleus to mediate the nuclear export of the RRE-containing transcripts. The function of Rev is dispensable if RR.E and the inhibitory sequences in the gag-pol and env are mutated.
Although TAR and RRE are known for their functions in mediating Tat and Rev interaction with the viral RNA, these two RNA elements may have other functions unrelated to Tat and Rev interaction which may be important for gene transfer vector function. It is possible that RRE or TAR may contain minor packaging signal to enhance viral RNA encapsulation. The example of RRE mutation on vector function is presented later.
With regard to complete deletion, Tat and Rev have been reported to be absolutely required for ral replication in vitro or in vivo Vaishnav, Y.N., Wong-Staal, F., The Biochemistry of AIDS, Ann. Rev. Biochem., 60:577-630, (1991); Greene, W.C, Regulation of HIV-1 Gene Expression, Annu. Rev. Immunol, 8:453-475, (1990). However, a small element from the Mason-Pfizer monkey virus genome can make human immunodefic ency virus type I expression and replication Rev-independent, Bray, et al. (1994), and this strategy has been used to develop a rev-independent HIV vector system, see Srinivasakumar.et al (1997).
Also, it has been reported that HIV tat mutants with stop codon mutations or deletions in the tat open reading frame can still infect human lymphocytes and macrophages, Chang et al (1995). The requirement for Tat transactivation of HIV- 1 LTR can be diminished if the
LTR enhancer promoter elements are replaced with a chimeric CMV-IE-HIV LTR Robinson. et al (1995).
LTR -and tat mutants of HIV- I have been shonvn to have diminished replication phenotypes (see e.g.. Chang, (1993); Chang and Zhang, (1995); Leonard, et al, (1989).
4.24 ACCESSORY GENES
The accessory proteins of HIV- I may have important functions in viral pathogenesis, see Trono, et al. (1995); but they are dispensable for viral replication in tissue culture. It has been shown that the accessory genes are not essential to the creation of functional packaging and transducing vectors, i.e., they may be completely deleted. Hence, it is unnecessary to consider in detail the guidance offered by the art as to which mutations of the accessory proteins might be functional. Of course, if one chooses to retain an accessory gene, such guidance can be found in the literature on, e.g., sequences of HIV-1 isolates. In general, it is preferable to delete all lentiviral accessory genes when constructing the transducing vector, in order to reduce the risk of homologous recombination to form
RCV. However, certain accessory genes, such as vpr or vpx, may increase transduction efficiency of nondividing cells, in which case there is a countervailing advantage to retaining them in a form in which they encode functional protein. If so, silent mutations, and other functional mutations, may be introduced to reduce the risk of homologous recombination without loss of gene function.
4.25 OTHER GENETIC ELEMENTS
In the packaging vectors (pHP-likes). the 5'LTR can be totally eliminated but a functional promoter will be needed to drive RNA transcription and gag-pol gene expression. Preferably, a strong enhancer/promoter will be used to replace the 5'LTR.
Tat may be needed for high efficiency of Gag-Pol synthesis. In this case, HIV-1 TAR sequence may be retained in the 5' end for enhanced promoter function. In the transducing vectors (pTV-Iikes), the necessary functions for vector production in the 5' LTR is the repetitive sequence R, which serves as annealing sequence for minus-strand DNA jumping to the 3' R, and the attachment site (att) in the 3' end of U5 adjacent to the PBS for provirus integration. The R can be made different from the native HIV R but have the same mutated R in the 3' end. The att site is necessary for integrase recognition and binding and therefore cannot be changed.
Preferably the lentiviral promoter/enhancer elements of the 5 Ltr are replaced with a nonlentiviral-promoter/enhancer in at least one (a) the packaging vectors or (b) the transducing vector. Both the HP 5'LTR and TV S'LTR promoter/enhancers may be replaced with the same or with different promoter/enhancers, e.g., CMV IE in one and EF- I in the other.
In the 5' leader region, no HIV functional elements are necessary for the packaging construct. However, for the transducing vector, several elements are needed, in an order from 5' to 3'including PBS, packaging signal, and dimer linkage sequence (DLS). HIV uses lysine tRNA PBS which may be mutated to a different retroviral PBS such as histidine tRNA or proline tRNA 6f RSV or MLV. However, a coupled change in the RT domain which recognizes the corresponding PBS will also be needed. The packaging signal for MV RNA has been shown to include different areas in the genome. It is possible that site-specific mutations can be made to change the primary sequence but maintain the secondary structure.
The major 5' splice donor site and the gag AUG have been shown by others to be essential for genome packaging. However, it has been demonstrated that both the SD and the gag AUG can be mutated and the modified transducing vector can still be packaged in high efficiency (see examples below). The DLS is not well defined. However, both primary sequence and secondary structure may be necessary for a functional DLS which overlaps the packaging signal between SD and the gag AUG.
In one embodiment, the packaging vector replaces the HIV-1 SD with an RSV SD. The splicing junction sequences have been previously studied, Ezzell, et al, (1995); Mount, (1996). In previous studies, it was shown that the first tat coding exon contains positive and negative splicing regulatory elements and the splicing signals can be hundreds of nucleotides away from the splice junciton sites, Amendt, et al. (1994). Therefore, the success of inserting a functional splice site in the leader region of HP construct using an oliconucteotide sequence containing a small number of nucleotide sequences from RSV 5' splice junction site was a surprise.
4.26 SELECTABLE AND SCREENABLE MARKERS
A vector may contain one or more selectable or screenable markers. Such markers are typically used to determine whether the vector has been successuly introduced into a host or target cell. A selectable marker is a gene whose expression substantially affects whether a cell will survive under particular controllable conditions. A selectable marker may provide for positive selection (cells with the marker are more likely to survive), negative selection (cells with the marker are less likely to survive), or both (the choice of environmental condition dictating whether positive or negative selection occurs).
Selectable markers include those which confer antibiotic resistance (or sensitivity), the ability to utilize a particular nutrient, and resistance (or sensitivity) to high (or low) temperature. Suitable selectable markers include the bacterial neomycin and hygromycin phosphotransferase resistance genes, which confers resistance to G418 and hygromycin, respectively, the bacterial gpt gene, which allows cells tog row in a medium containing mycophenoiic acid, xanthine and aminopterin; the bacterial hisD gene which allows cells to grow in a medium lacking histidine but containing histidinol; the multidrug resistance gene mdr; the hprt and HSV thymidine kinase genes, which allow otherwise hprt- or tk- cells to
grow in a medium containing hypoxanthine, amethopterin and thymidine, and the bacterial genes conferring resistance to puromycin or phleomycin. Positive or negative selection may require the use of a particular strain of host cell for the selection to be effective.
Screenable markers are genes which encode a product whose presence is readily detectable, directly or indirectly, but which do not necessarily affect cell survival. The green fluorescent protein (GFP) is an example. Any cell surface protein not native to the host cell can be used as an immunoscreenable marker. Transformed cells may be segregated out by using a fluorescent antibody to the protein and a cell sorter. Many enzyme-encoding genes are useful as screenable markers, especially those encoding enzymes which can act upon a substrate to provide a colored or luminescent product. The luciferase and beta-galactosidase genes have been especially popular.
A dominant marker encodes an activity which can be detected in any eukaryotic cell line. Examples of dominant selectable markers include the bacterial aminoglycoside 3' phosphotransferase gene (also referred to as the neo gene) which confers resistance to the drug G418 in mammalian cells, the bacterial hygromycin G phosphotransferase (hyg) gene which confers resistance to the antibiotic hygromycin and the bacterial xanthine-guanine phosphoribosyl transferase gene (also referred to as the gpt gene) which confers the ability to grow in the presence of mycophenolic acid. Other selectable markers are not dominant in that their use must be in conjunction with a cell line that lacks the relevant activity. Examples of non-dominant selectable markers include the thymidine kinase (tk) gene which is used in conjunction with tk. cell lines, the CAD gene which is used in conjunction with CAD- deficient cells and the mammalian hypoxanthine-guanine phosphoribosyl transferase (hprt) gene which is used in conjunction with hprt cell lines. A review of the use of markers in mammalian cell lines is provided in Sambrook, (1989).
4.27 REGULATION OF GENE EXPRESSION
The transgene(s) of the transducing vector, and the marker(s) and viral genes (or replacements) of the packaging and transducing vectors, are expressed under the control of regulatory elements. As used herein, the term "regulatory element" refers to a genetic element which controls some aspect of the expression of nucleic acid sequences. For example, a promoter is
a regulatory element which facilitates the initiation of transcription of an operably linked coding region. Other regulatory elements are splicing signals, polyadenylation signals, termination signals, etc. (defined infra). A constitutive promoter is one which is always active at essentially a constant level. Transcriptional control signals in eukaryotes comprise "promoter" and "enhancer" elements. Promoters and enhancers consist of short arrays of DNA sequences that interact specifically with cellular proteins involved in transcription (Maniatis et al. [1987]). Prormoter and enhancer elements have been isolated from a variety of eukaryotic sources including genes in y east, insect and-mammalian cells and viruses (analogous control elements, i.e.. promoters, are also found in prokaryotes). The selection of a particular promoter and enhancer depends on what cell type is to be used to express the protein of interest. Some eukaryotic promoters and enhancers have a broad host range while others are functional in a limited subset of cell types (for review, see, S.D. Voss et al. [1987)). For example, the SV40 early gene enhancer is very active in a wide variety of cell types from many mammalian species and has been widely used for the expression of proteins in mammalian cells (Dijkema et al (1985]). Two other examples of promoter/enhancer elements active in a broad range of mammalian cell types are those from the human elongation factor I a gene (T. Uetsuki et aL, J. Biol. Chem., 264:5791 (1989]; D.W. Kim et al., Gene 91 :217 [1990]; S. Mizushima, and S. Nagata, Nuc. Acids. Res., 18:5322 (1990]) and the long terminal repeats of the Rous sarcoma virus (CM. Gorman et aL. Proe. Natl. Acad. Sci. USA 79:6777 [19821 ) and the human cytomegalovirus (M. Boshart et aL, Cell 41 :521 [19851).
As used herein, the term "promoter/enhancer" denotes a segment of DNA which contains sequences capable of providing both promoter and enhancer functions (i.e., the functions provided by a promoter element and an enhancer element, see above for a discussion of these functions). For example, the long terminal repeats of retroviruses contain both promoter and enhancer functions. The enhancer/promoter may be "endoaenous" or "exogenous" or "hetetologous." An "endogenous" enhancer/promoter is one which is naturally linked with a given gene in the genome. An "exogenous" or "heterolocous" enhancer/promoter is one which is placed in juxtaposition to a gene by means of genetic manipulation (i.e. molecular biological techniques) such that transcription of that gene is
directed by the linked enhancer/promoter. A regulatable promoter is one whose level of activity is subject to regulation by a regulatory molecule. An inducible promoter is one which is normally substantially inactive, but which is activated by the binding of an inducer to an operator site of the promoter. A repressible promoter is one which is normally active, but which is substantially inactivated by the binding of a repressor to an operator site of the promoter. Similar terminology applies to enhancers. The inducer or repressor molecules are typically expressed only in particular tissues, at a particular developmental stage, or under particular environmental conditions (e.g.. damage to the cell, infection, overproduction of a metabolite, absence of a nutrient, etc. ). In the absence of an inducer an inducible promoter may be inactive or may produce a low level of the level of activity in the presence of the inducer will be higher than the basal rate. A tightly inducible promoter is one whose basal level of activity is very low. e.g. , less than 10% of its maximum inducible activity.
Different promoters may have different levels of basal activity in the same or different cell types. When two different promoters are compared in a given cell type in the absence of any inducing factors, if one promoter expresses at a higher level than the other it is said to have a higher basal activity.
The activity of a promoter and/or enhancer is measured by detecting directly or indirectly the level of transcription from the element(s). Direct detection involves quantitating the level of the RNA transcripts produced from that promoter and/or enhancer. Indirect detection involves quantitation of the level of a protein, -often an enzyme, produced from RNA transcribed from the'promoter and/or enhancer. A commonly employed assay for promoter or enhancer activity utilizes the chloramphenicol acetyltransferase (CAT) gene. A promoter and/or enhancer is inserted upstream from the coding region for the CAT gene on a plasmid; the plasmid is introduced into a cell line. The levels of CAT enzyme are measured. The level of enzymatic activity is proportional to the amount of CAT RNA transcribed by the cell line. This CAT assay therefore allows a comparison to be made of the relative strength of different promoters or enhancers in a given cell line. When a romoter is said to express at "hiah" or "low" levels in a cell line this refers to the level of activity relative to another promoter which is used as a reference or standard of promoter activity. Efficient expression of recombinant DNA sequences in eukaryotic cells requires expression of signals directing the efficient termination and polyadenylation of the resulting
transcript. Transcription termination signals are generally found downstream of the polyadenylation signal and are a few hundred nucleotides in length. The term "polyA site" or "polyA sequence" as used herein denotes a DNA sequence that directs both the termination and polyadenylation of the nascent RNA transcript. Efficient polyadenylation of the recombinant transcript is desirable as transcripts lacking a polyA tail are unstable and are rapidly degraded. The polyA signal utilized in an expression vector may be "heterologous" or "endogenous." AnendogenouspolyAsignalisone-thatisfoundnaturallyatthe3'endof the coding region of a given gene in the genome. A heterologous poly A signal is one which is one which is isolated from one gene and placed 3' of another gene. A commonly used heterologous poly A signal is the SV40 poly A signal. The SV40 poly A signal is contained on a 237 bp Bam HIIBcl I restriction fragment and directs both termination and polyadenylation (J.Sambrook et al . supra, at 16.6-16.7).
The cytomegalovirus immediate early promoter-enhancer (CMV-IE) is a strong enhancer/promoter. See Boshart. M., Weber, F.. Jahn. G.. Dorsch-Hasler, K., Fleckenstein. B.. Schaffner, W. A. very strong enhancer is located upstream of an inunediate early gene of human cytomegalovirus. Cell, 41 :521 -530 ( 1985 ). For its incorporation into HIV-1 derived viruses, see Chang, L.J., McNulty, E.. Martin, M. Human immunodeficiency viruses containing heterologous enhancer/promoters are replication competent and exhibit different lymphocyte tropisms. J Virol. 67:743-752 (1993). Another strong promoter-enhancer for eukaryotic gene expression is the elongation factor I alpha promoter enhancer. Kim, D.W., Uetsuki, T., Kaziro, Y., Yamaguchi, N., Sugano, S. Use of the Human Elongation Factor I a Promoter as a Versatile and Efficient Expression System, Gene, 91 :217-223 1996; Mizushima, S., Nagata, S.. PEF-BOS, a Powerful Mammalian Expression Vector. Nucleic Acids Res. , 18:5322 (1990). The internal promoter for a transgene may be the promoter native to that transgene, or a promoter native to the target cell (or viruses infecting the target cell), or another promoter functional in the target cell.
The preferred promoters and enhancers are those exhibiting tissue or cell type sepecificity which can direct the transgene expression in the target cells at the right time(s). For example, a promoter to control human preproinsulin must be operable under control of
carbohydrate in theliver. An example of such a promoter is the rat S-14 liver-specific promoter.
Promoters (and enhancers) may be naturally occurring sequences, or functional mutants thereof including chimeras of natural sequences and mutants thereof. For example, a tissue- specific, development-specific, or otherwise regulatable element of one promoter may be introduced into another promoter.
Chen, et al. Proc. Nat. Acad Sci USA, 93: 10057-62 ( 1996) placed a VSV G gene under the control of a tetracycline-inducible promoter and also-expressed a fusion of the ligand binding domain of the estrogen receptor to a chimeric transcription factor, tTA, obained by fusing the tet repressor (tetr) and the activation domain of HSV virion protein 16.
For the ability to replace the endogenous 5' LTR promoters and enhancers with heterologous ones, such as CMV inu-nediate-earlv enhancer-promoter, see Chang, et al. J. Virol. 67:743-52 (1993).
4.27 TRANSFECTION OF VECTORS
As used herein, the term "vector" is used in reference to nucleic acid molecules that can be used to transfer nucleic acid (e.g., DNA) segment(s) from one cell to another. The term "vehicle" is sometimes used interchangeably with "vector." It is intended that any form of vehicle or vector be encompassed within this definition. For example, vectors include, but are not limited to viral particles, plasmids, transposons, etc.
The term "transfection" as used herein refers to the introduction of foreign DNA into eukaryotic cells. Transfection may be accomplished by a variety of means known to the art including but not limited to calcium phosphate-DNA co-precipitation, DEAE-dextran- mediated transfection, polybrene-mediatea transfection, electroporation, micro injection, liposome fusion, lipofection, protoplast fusion, retroviral infection, and biolistics.
Vectors may contain "viral replicons "or "viral origins of replication." Viral repticons are viral DNA sequences that allow for the extrachromosomat replication of a vector in a host cell expressing the appropriate replication factors. Vectors that contain either the SV40 or polyoma virus origin of replication replicate to high copy number (up to 104 copies/cell) in cells that express the appropriate viral T antigen. Vectors containing the replicons from
bovine papillomavirus or Epstein-Barr virus replicate extrachromosomally at low copy number (-100 copies cell).
4.28 EXPRESSION VECTOR The term "expression vector" as used herein refers to a recombinant DNA molecule containing a desired coding sequence and appropriate nucleic acid sequences necessary for the expression of the operably linked coding sequence in a particular host organism. Nucleic acid sequences necessary for expression in prokaryotes usually include a promoter, an operator (optional), and a ribosome binding site, often along with other sequences. Eukaryotic cells are known to utilize promoters, enhancers, and termination and polyadenylation signals. In some embodiments, "expression vectors" are used in order to permit pseudotyping of the viral envelope proteins.
4.29 HOST CELLS The host cell is a cell into which a vector of interest may be introduced and wherein it may be replicated, and. in the case of an expression vector, in which one or more vector- based genes may be expressed.
It is not necessary that the host cell be infectable by the transducing vector virions of the present inxention. Indeed, it is preferable that they not be so infectable. so the hos cells do not bind the \ irions and thereby reduce the vector production titer. This can be achieved by choosing (or engineering) cells which do not functionally express the receptor to the vector particle envelope protein.
4.30 TARGET CELLS AND ORGANISMS The transducing vector may be administered to a target organism by any route which will permit it to reach the target cells. Such route may be, e.g., intravenous, intramuscular, subcutaneous, or. with an enteric coating, oral. Alternatively, target cells may be removed from the organism, infected, and they (or their progeny) returned to the organism. Or the transducing vector may simply be administered to target cells in culture. The target cells into which the transgene is transferred may be any cell which the transducing vector, after packaging into a virion, is capable of infecting, and in which the
control sequences

ermng expression of the transgene are functional GeneraUy speaking. it will be a eukaryotic cell, preterably a vertebrate cell, more preferably a cell of a mammal or bird. If a mammal, the mammal will preferably belong to one of the orders Aitiodacty a (e g , cows, pigs, goats, sheep). Peπssodact la (e g , horses), Rodenta (e g , rats. mice). Lagomorpha (e g . rabbits). Car vora (e . dogs, cats) or Pπmata (e g . humans, apes. monkeys, lemurs) If a bird, it will preferably be of the orders Anseπforrnes (e g ducks, geese, swans) or Gailifoπnes ( g . quails, grouse, pheasants, turkeys, chickens) Most preferably it will be a human cell
The cells in question may be dividing or non-dividing cells Non-dividing cells of particular interest include neuronal cells and astrocytes Dividing cells of particular interest include hematopoietic stem cells, muscle cells, white blood cells, spleen cells,
cells. epithelial cells and ey e cells
TE67 1, HepG2. HeLa. 293T. and MT4 are of particular mterest for experimental studies. TE671 rhabdomyosarcoma cells can be induced to differentiate into muscle cells by HIV- 1 Vpr HepG2 hepatoma. HeLa cervical carcinoma, 293T human kidney carcnoma and MT4 lymphoma cells are all transformed by HTLV-1 human T cell leukemia virus type 1 MT4 cells are very susceptible to wild-type HIV- I NL4-3 and hence have been used as indicator cell for RCV
4.31 VIRAL ASSA\ S
The successful establishment of the packaging -or-transducing vector in the host (or target) cell may be verified by selecting for the presence of a selectable marker, or screening for the presence of a screenable marker, carried by the vector. The integration of the relevant packaging or transducing vector genes may be determined by collecting genomic DNA. amplifying the gene of interest by PCR, and detecting the amplified sequence with a suitable hybridization probe The production of viral proteins may be detected by an immunoassa , the sample may be a cell lysate or a cell supernatant. An immunoassay by itself cannot determine whether the viral proteins are produced in functional form, although there is greater assurance of this if the antibody used is directed to a confonnational epitope. or is an activity- neutralizing antibody. One may alternatively detect the appropriate messenger RNA by means of a hybridization probe.
The functionality of the produced Gag and Env protein may be determined by examining the cell lysate or supernatant for the presence of viral particies;these may ftu-ther be examined for proper moφhology by means of an electron microscope. It is also possible that antibodies could be used which bind to the formed viral particles, but not to gpl20 or gp41 by itself. The functionality of the Pol reverse transcriptase may be-determined by assaying the viral particles for RT activity. The functionality of the Pol integrase is apparent only in assays which examine whether RNA from viral particles is integrated into the target cell.
Viral particles produced by the packaging cell line may be collected and assayed for total RNA'content. If more specific information is desired as to the nature of any packaged RNA, a suitable hybridization probe may be employed.
In an infectivity assay, the vector is introduced into a first culture of susceptible cells. Then, either a second culture is layered onto the first, so that infectious particles may travel by cell-to-cell contact, or the second culture is exposed to the supernatant of the first culture. The cells of the first and second culture are examined for a least one of the following indicia: RT activity, p24 Gag antigen expression, production of viral particles, and cytotoxic effects. The stringency of the assay is dependent on the susceptible of the cells to infection and to cytotoxicity, and the time allowed for the recombination and spread of the virus in the first and second cultures. Typically, the infectiv ity of the vector or vector system will be compared with that of a wild-type, unattenuated. replication-competent lentivirus.
Animal studies may be used to ascertain the immunogenicity and pathogenicity of the vector system. Some of these assays are described in greater detail below.
4.31.1 MEASUREMENT OF INFECTIVITY OF PACKAGING VECTOR The ability of a packaging vector to generate transmissible virus, as opposed to defective virus, may be measured. One method is described by Mann, et al, Cell, 33: 153-9, (1983). The packaging vector and its wild-type counterpart are independently transfected into suitable host cells, and reverse transcriptase activity in the culture supernatants is assayed over a period of days or weeks. A rapid increase in RT activity over 24-48 hrs is indicative of gene expression after transient transfection. A continued increase is indicative of the efficient spread of virus from the initially transfected cells to the remaining cells on the plate.
A slow or delayed increase could be indicative of either a steady but attenuated spread of virus, or to generation of competent virus by mutation, or by recombination with a cellular sequence capable of providing the missing function. To differentiate these possibilities, one may use various dilutions of culture supernatants from cells previously transfected (days or weeks before) with the vector (or with the control virus), use them to infect fresh cells, and monitor RT activity in the latter. If the latter cells develop high levels of RT activity, it sugoests that nondefective virus was present in the transferred culture supernatant.
4.31.2 MEASUREMENT OF PACKAGING EFFICIENCY
The packaging efficiency of a packaging cell line in the presence or absence of the packageable transducing vector may be measured in a variety of ways. One method is described by Mann, et al, Cell, 33:153-9, (1983). In esssence, total cellular RNA is purified from the culture supernatant of the test and control cell lines, and viral RNA is extracted from purified viral particles released from the test and control cell lines. The two virion preparations are normalized by reference to their reverse transcriptase activity just prior to RNA extraction. The purified RNAs are probed with a virus-specific hybridization probe (e.g., a plasmid containing the entire viral genome) in a slot-blot assay, and the amount of viral RNA in the particles and in the cells is thereby quantified. It is not unusual for ihe packaging efficiency of a packaging cell line to be less than
1% that of a host cell infected by wild-type virus.
4.31.3 MEASUREMENT OF PACKAGING SPECIFICITY
It is also desirable that the packaging cell line be able to efficiently package the highly 20 defective transducing vector into viral particles, and bud the particles into the culture supernatant (in vitro) or extracellular environment (in vivo) without also budding helper virus (the packaging vectors).
One method of measuring this packaging specificity is described by Mann, et al,
Cell, 33:153-9, (1983). In essence, the transducing vector is transfected into the packaging (helper) cell line. After 24 hours, the culture supernatants are used to infect fresh potential host cells (reporter cells). Two days later, selection pressure for the transferred gene is
applied, and 8- 10 days later, the transferred gene-positive colonies or cells are counted. In addition, one determines the reverse transcriptase activity of the supernatant coleicted from the packaging cell lines, and the reverse transcriptase activity of the fresh cells. A transducing vector-specific packaging cell line will pro duce a high transfer gene activity and a low reverse transcriptase activity in the reporter cells. In addition, the reporter cells will not produce reporter gene-positive colony-forrning units (cfus).
4.31.4 ME ASUREMENT OF HELPER ACTIVITY
The ability of a packaging vector to provide all viral functions required in trans may be assayed by cotransfecting host cells with the packaging vector (or control virus) and with a reporter v ector carrying a selectable reporter gene. After 24 hours, culture supernatants of the transfected cells are used to infect a second plate of host cells. Selection pressure for the reporter gene is applied, and reporter-positive colonies are counted. If the helper activity is of wild-type magnitude, the count for the packaging vector should be of the same order of magnitude as that for the control virus, and no reporter activity should be detectable in the second plate when the reporter vector or the control wild-type virus expressing all viral functions is transfected into the host cells of the first plate by itself.
4.31.5 MEASUREMENT OF GENERATION OF REPLICATION-COMPETENT VIRUS Se\ eral sensitive assays are available for the detection of RCV in the present lentiviral vector systems. These include: (1) co-cultivation with a sensitive cell line such as MT4, AA2 or PBLs: (2) the CD4 HeLa MAGI cell assay which relies on Tat transactivation of an integrated LTR-lacZ gene; and (3) a sensitive immunohistochemical staining method for the detection of HIV antigen expression at the individual cell level. As described in the examples below, the latter method was modified and developed for the characterization of "Tat-minus" HIV-1 infection, although all three methods are suitable for the routine titration of infectious HIV-1.
RC-HIV can also be studied in an in vivo model by transduction of humanized SCID/beige mice. In the latter model, a long in vivo incubation time can be performed, mimicking the situation that exists in a human clinical trial. In addition, the possibility of
generating HIV/HERV recombinants may be carefully tested using an artificially constructed HIV/HERV-e«v recombinant.
4.31.6 VIRION STABILITY Since on class of the therapeutic agents of the present invention would be the packaged transducing vectors, the stability of the packaged transducing vectors under adverse conditions, especially those which might be encountered during storage, is of interest. Thermostability may be ascertained by subjected them to elevated (e.g., 37° C) or depressed (e.g., 4° C) temperatures for various periods of time 2, 4, 6 or 8 hrs., or overnight), or to a number (e.g.. 1-6) freeze-thaw cycles, and determining the number of infectious particles remaining as a percentage of the number of such particles prior to treatment. See Bums et al . 1993.
4.31.7 ASSAYS FOR IMMUNOGENICITY A preferred method for deterrninining whether the contemplated vectors, or their gene products, could elicit an immune response in a subject involves evaluating cell-mediated immunity (CMI) using either an immunocompetent mouse model or a a humanized SCID/beige inouse model.
Using a modified hu-PBL-SCID mouse reconstitution protocol, an in vivo model for evaluating CMI against HIV-1 in humans has been developed. SCID/beige mice lacking T, B and natural killer (NK) cell fimctions are severely immunodeficient. This strain of mice can be successfully reconstituted with fresh human peripheral blood lymphocytes (PBLs), and exhibits functional human naive, memory and activated T cell markers for more than 2-3 months (See e.g., copending U.S. Patent Application Serial Nos. 08/848,760, and 08/838,702, both of which are in incorporated by reference). In these experiments, spleen and peripheral blood lymph cytes were harvested 38 days after reconstitution from reconstituted SCID/beige mice, and red blood cells were lysed prior to incubation with anti-mouse 2Kd, anti-human CD45, anti-human CD3, anti-human CD4 and anti-human CD8 labeled antibodies. Reconstituted, human lymphoid cell populations in the spleen and in the peripheral blood of the SCID/beige mice can reach up to 50-80% and 5-12%, respectively.
For the inunune response study, mice repetitively injected with the viral vectors will be analyzed. Their sera will be assayed for Ab response to viral antigens, such as p24 Gag or the pseudotype env (e.g., VSV-G). For cell-mediated immune response study, the mouse splenocytes will be isolated and an in vitro assay for cellular immunity will be performed as described below. T cell response to recall antigen is normally characterized by the production of interferon gamma (IFNγ). This assay requires activation of lymphocytes with the test Ags. such as Gag p24 or Gag-Pol or VSV-G env proteins of the vector.
Upon activation, the Thl lineage of T cells produce interferon gamma (IFN-g) and the measurement of IFN-G production has been shown to be a reliable assay for CMI. Thus, to determine CMI against HIV-1 using the in vivo humanized SCID/beige mouse model, a sensitive ELISPOT assay for the detection of IFN-g producing cells was developed. With the computer assisted imaging system integrated into this protocol, the ELISPOT method was shown to be very convenient and more sensitive than the conventional limiting dilution assay for the determination of the effector T cell precursor frequency. This in vivo model and the ELISPOT assay system were developed for the evaluation of in vivo CMI after lentiviral gene transfer. (See, e.g., PCT/US98/06944).
4.32 PHARMACEUTICAL COMPOSITIONS
In certain embodiments, the present invention concerns formulation of one or more of the lentiviral compositions disclosed herein in pharmaceutically-acceptable solutions for administration to a cell or an animal, either alone, or in combination with one or more other modalities of therapy.
It will also be understood that, if desired, the lentiviral construct, nucleic acid segment. RNA, DNA or PNA compositions as disclosed herein may be administered in combination with other agents as well, such as, e.g., proteins or polypeptides or various pharmaceutically-active agents. In fact, there is virtually no limit to other components that may also be included, given that the additional agents do not cause a significant adverse effect upon contact with the target cells or host tissues. The lentiviral compositions may thus be delivered along with various other agents as required in the particular instance. Such compositions may be purified from host cells or other biological sources, or alternatively may be chemically synthesized as described herein.
Likewise, such compositions may further comprise substituted or derivatized RNA, DNA, or PNA compositions.
Formulation of pharmaceutically-acceptable excipients and carrier solutions is well- known to those of skill in the art. as is the development of suitable dosing and treatment regimens for using the particular compositions described herein in a variety of treatment regimens, including e.g., oral, parenteral, intravenous, intranasal, and intramuscular administration and formulation.
4.32.1 ORAL DELIVERS In certain applications, formulation of the lentiviral vector compositions in a pharmaceutical formulation is contemplated. Such pharmaceutical formulations comprising one or more lentiviral vector constructs as disclosed herein may be delivered via oral administration to an animal. As such, these compositions may be formulated with an inert diluent or with an assimilable edible carrier, or they may be enclosed in hard- or soft-shell gelatin capsule, or they may be compressed into tablets, or they may be incorporated directly with the food of the diet.
The active compounds may even be incorporated with excipients and used in the form of ingestible tablets, buccal tables, troches, capsules, elixirs, suspensions, syrups, wafers, and the like (Mathiowitz et al, 1997; Hwang et al, 1998; U. S. Patent 5,641,515; U. S. Patent 5,580,579 and U. S. Patent 5,792,451, each specifically incorporated herein by reference in its entirety). The tablets, troches, pills, capsules and the like may also contain the following: a binder, as gum tragacanth, acacia, cornstarch, or gelatin; excipients, such as dicalcium phosphate; a disintegrating agent, such as corn starch, potato starch, alginic acid and the like; a lubricant, such as magnesium stearate; and a sweetening agent, such as sucrose, lactose or saccharin may be added or a flavoring agent, such as peppermint, oil of wintergreen, or cherry flavoring. When the dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier. In addition, the lentiviral vector compositions may be incorporated into sustained-release preparations and/or timed- or otherwise controlled-release formulations.
Naturally, the amount of active compound(s) in each therapeutically useful composition may be prepared is such a way that a suitable dosage will be obtained in any given unit dose of the compound. Factors such as solubility, bioavailability, biological half-life, route of administration, product shelf life, as well as other pharmacological considerations will be
contemplated by one skilled in the art of preparing such pharmaceutical formulations, and as such, a variety of dosages and treatment regimens may be desirable.
4.32.2 INJECTABLE DELIVERY In certain circumstances it will be desirable to deliver the pharmaceutical compositions disclosed herein parenterally. intravenously, intramuscularly, or even intraperitoneally as described in U. S. Patent 5,543,158; U. S. Patent 5,641.515 and U. S. Patent 5,399,363 (each specifically incorporated herein by reference in its entirety). Solutions of the active compounds as free base or pharmacologically acceptable salts may be prepared in water suitably mixed with a surfactant, such as hydroxypropylcellulose. Dispersions may also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof and in oils. Under ordinary conditions of storage and use, these preparations contain a preservative to prevent the growth of microorganisms.
The pharmaceutical forms suitable for injectable use include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions (U. S. Patent 5,466,468, specifically incorporated herein by reference in its entirety). In all cases the form must be sterile and must be fluid to the extent that easy syringability exists. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms, such as bacteria and fungi. The carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g., glycerol. propylene glycol, and liquid polyethylene glycol, and the like), suitable mixtures thereof, and/or vegetable oils. Proper fluidity may be maintained, for example, by the use of a coating, such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants. The prevention of the action of microorganisms can be brought about by the addition of one or more various antibacterial and antifungal agents (e.g., parabens, chlorobutanol, phenol, sorbic acid, thimerosal, and the like). In many cases, it will be preferable to include isotonic agents, for example, sugars or sodium chloride. Prolonged absorption of the injectable compositions can be brought about by the use in the compositions of agents delaying absorption, for example, aluminum monostearate and gelatin. For parenteral administration in an aqueous solution, for example, the solution should be suitably buffered if necessary and the liquid diluent first rendered isotonic with sufficient saline
or glucose. These particular aqueous solutions are especially suitable for intravenous, intramuscular, subcutaneous and intraperitoneal administration. In this connection, a sterile aqueous medium that can be employed will be known to those of skill in the art in light of the present disclosure. For example, one dosage may be dissolved in 1 ml of isotonic NaCl solution and either added to 1000 ml of hypodermoclysis fluid or injected at the proposed site of infusion, (see for example, "Remington's Pharmaceutical Sciences" 15th Edition, pages 1035- 1038 and 1570-1580). Some variation in dosage will necessarily occur depending on the condition of the subject being treated. The person responsible for administration will, in any event, determine the appropriate dose for the individual subject. Moreover, for human administration, preparations should meet sterility, pyrogenicity, and the general safety and purity standards as required by FDA Office of Biologies standards.
Sterile injectable solutions are prepared by incorporating the active compounds in the required amount in the appropriate solvent with various of the other ingredients enumerated above, as required, followed by filtered sterilization. Generally, dispersions are prepared by incorporating the various sterilized active ingredients into a sterile vehicle which contains the basic dispersion medium and the required other ingredients from those enumerated above. In the case of sterile powders for the preparation of sterile injectable solutions, the preferred methods of preparation are vacuum-drying and freeze-drying techniques which yield a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof.
The compositions disclosed herein may be formulated in a neutral or salt form. Pharmaceutically-acceptable salts, include the acid addition salts (formed with the free amino groups of the protein) and which are formed with inorganic acids such as, for example, hydrochloric or phosphoric acids, or such organic acids as acetic, oxalic, tartaric, mandelic, and the like. Salts formed with the free carboxyl groups can also be derived from inorganic bases such as, for example, sodium, potassium, ammonium, calcium, or ferric hydroxides, and such organic bases as isopropylamine, trimethylamine, histidine, procaine and the like. Upon formulation, solutions will be administered in a manner compatible with the dosage formulation and in such amount as is therapeutically effective. The formulations are easily administered in a variety of dosage forms such as injectable solutions, drug-release capsules, and the like.
As used herein, "carrier" includes any and all solvents, dispersion media, vehicles, coatings, diluents, antibacterial and antifungal agents, isotonic and absorption delaying agents, buffers, carrier solutions, suspensions, colloids, and the like. The use of such media and agents for pharmaceutical active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, its use in the therapeutic compositions is contemplated. Supplementary active ingredients can also be incorporated into the compositions.
The phrase "pharmaceutically-acceptable" refers to molecular entities and compositions that do not produce an allergic or similar untoward reaction when administered to a human. The preparation of an aqueous composition that contains a protein as an active ingredient is well understood in the art. Typically, such compositions are prepared as injectables, either as liquid solutions or suspensions; solid forms suitable for solution in, or suspension in, liquid prior to injection can also be prepared. The preparation can also be emulsified.
4.32.3 NASAL DELIVERY
In certain embodiments, the pharmaceutical compositions may be delivered by intranasal sprays, inhalation, and/or other aerosol delivery vehicles. Methods for delivering genes, nucleic acids, and peptide compositions directly to the lungs via nasal aerosol sprays has been described e.g., in U. S. Patent 5,756.353 and U. S. Patent 5,804,212 (each specifically incorporated herein by reference in its entirety). Likewise, the delivery of drugs using intranasal microparticle resins (Takenaga et al, 1998) and lysophosphatidyl-glycerol compounds (U. S. Patent 5,725,871, specifically incorporated herein by reference in its entirety) are also well- known in the pharmaceutical arts. Likewise, transmucosal drug delivery in the form of a polytetrafluoroetheylene support matrix is described in U. S. Patent 5,780,045 (specifically incorporated herein by reference in its entirety).
4.32.4 LIPOSOME-, NANOCAPSULE-, AND MICROPARTICLE-MEDIATED DELIVERY
In certain embodiments, the inventors contemplate the use of liposomes, nanocapsules, microparticles, microspheres, lipid particles, vesicles, and the like, for the introduction of the compositions of the present invention into suitable host cells. In particular, the compositions of
the present invention may be formulated for delivery either encapsulated in a lipid particle, a liposome, a vesicle, a nanosphere, or a nanoparticle or the like.
Such formulations may be preferred for the introduction of pharmaceutically-acceptable formulations of the lentiviral constructs disclosed herein. The formation and use of liposomes is generally known to those of skill in the art (see for example, Couvreur et al, 1977; Couvreur. 1988; Lasic, 1998; which describes the use of liposomes and nanocapsules in the targeted antibiotic therapy for intracellular bacterial infections and diseases). Recently, liposomes were developed with improved serum stability and circulation half-times (Gabizon and Papahadjopoulos, 1988; Allen and Choun. 1987; U. S. Patent 5,741,516, specifically incorporated herein by reference in its entirety). Further, various methods of liposome and liposome like preparations as potential daig carriers have been reviewed (Takakura, 1998: Chandran et al, 1997; Margalit, 1995; U. S. Patent 5,567,434; U. S. Patent 5,552,157; U. S. Patent 5,565,213; U. S. Patent 5,738.868 and U. S. Patent 5,795,587, each specifically incorporated herein by reference in its entirety). Liposomes have been used successfully with a number of cell types that are normally resistant to transfection by other procedures including T cell suspensions, primary hepatocyte cultures and PC 12 cells (Renneisen et al. 1990; Muller et al, 1990). In addition, liposomes are free of the DNA length constraints that are typical of viral-based delivery systems. Liposomes have been used effectively to introduce genes, drugs (Heath and Martin, 1986; Heath et al, 1986; Balazsovits et al, 1989; Fresta and Puglisi, 1996), radiotherapeutic agents (Pikul et al, 1987), enzymes (Imaizumi et al, 1990a; Imaizumi et al, 1990b), viruses (Faller and Baltimore. 1984), transcription factors and allosteric effectors (Nicolau and Gersonde, 1979) into a variety of cultured cell lines and animals. In addition, several successful clinical trails examining the effectiveness of liposome-mediated drug delivery have been completed (Lopez-Berestein et al, 1985a; 1985b; Coune, 1988; Sculier et al, 1988). Furthermore, several studies suggest that the use of liposomes is not associated with autoimmune responses, toxicity or gonadal localization after systemic delivery (Mori and Fukatsu, 1992).
Liposomes are formed from phospholipids that are dispersed in an aqueous medium and spontaneously form multilamellar concentric bilayer vesicles (also termed multilamellar vesicles (MLVs). ML Vs generally have diameters of from 25 nm to 4 μm. Sonication of MLVs results
in the formation of small unilamellar vesicles (SUVs) with diameters in the range of 200 to 500 A, containing an aqueous solution in the core.
Liposomes bear resemblance to cellular membranes and are contemplated for use in connection with the present invention as carriers for the peptide compositions. They are widely suitable as both water- and lipid-soluble substances can be entrapped, i.e. in the aqueous spaces and within the bilayer itself, respectively. It is possible that the drug-bearing liposomes may even be employed for site-specific delivery of active agents by selectively modifying the liposomal formulation.
In addition to the teachings of Couvreur et al (1 77; 1988), the following information may be utilized in generating liposomal formulations. Phospholipids can form a variety of structures other than liposomes when dispersed in water, depending on the molar ratio of lipid to water. At low ratios the liposome is the preferred structure. The physical characteristics of liposomes depend on pH, ionic strength and the presence of divalent cations. Liposomes can show low permeability to ionic and polar substances, but at elevated temperatures undergo a phase transition that markedly alters their permeability. The phase transition involves a change from a closely packed, ordered structure, known as the gel state, to a loosely packed, less- ordered structure, known as the fluid state. This occurs at a characteristic phase-transition temperature and results in an increase in permeability to ions, sugars and drugs.
In addition to temperature, exposure to proteins can alter the permeability of liposomes. Certain soluble proteins, such as cytochrome c, bind, deform and penetrate the bilayer, thereby causing changes in permeability. Cholesterol inhibits this penetration of proteins, apparently by- packing the phospholipids more tightly. It is contemplated that the most useful liposome formations for antibiotic and inhibitor delivery will contain cholesterol.
The ability to trap solutes varies between different types of liposomes. For example, MLVs are moderately efficient at trapping solutes, but SUVs are extremely inefficient. SUVs offer the advantage of homogeneity and reproducibility in size distribution, however, and a compromise between size and trapping efficiency is offered by large unilamellar vesicles (LUVs). These are prepared by ether evaporation and are three to four times more efficient at solute entrapment than MLVs. In addition to liposome characteristics, an important determinant in entrapping compounds is the physicochemical properties of the compound itself. Polar compounds are
trapped in the aqueous spaces and nonpolar compounds bind to the lipid bilayer of the vesicle. Polar compounds are released through permeation or when the bilayer is broken, but nonpolar compounds remain affiliated with the bilayer unless it is disrupted by temperature or exposure to lipoprotein. Both types show maximum efflux rates at the phase transition temperature. Liposomes interact with cells via four different mechanisms:
(a) endocytosis by phagocytic cells of the reticuloendothelial system such as macrophages and neutrophils;
(b) adsorption to the cell surface, either by nonspecific weak hydrophobic or electrostatic forces, or by specific interactions with cell-surface components; (c) fusion with the plasma cell membrane by insertion of the lipid bilayer of the liposome into the plasma membrane, with simultaneous release of liposomal contents into the cytoplasm; and
(d) by transfer of liposomal lipids to cellular or subcellular membranes, or vice versa, without any association of the liposome contents. It often is difficult to determine which mechanism is operative and more than one may operate at the same time.
The fate and disposition of intravenously injected liposomes depend on their physical properties, such as size, fluidity, and surface charge. They may persist in tissues for h or days, depending on their composition, and half-lives in the blood range from min to several h. Larger liposomes. such as MLVs and LUVs, are taken up rapidly by phagocytic cells of the reticuloendothelial system, but physiology of the circulatory system restrains the exit of such large species at most sites. They can exit only in places where large openings or pores exist in the capillar endothelium, such as the sinusoids of the liver or spleen. Thus, these organs are the predominate site of uptake. On the other hand, SUVs show a broader tissue distribution but still are sequestered highly in the liver and spleen. In general, this in vivo behavior limits the potential targeting of liposomes to only those organs and tissues accessible to their large size.
These include the blood, liver, spleen, bone marrow, and lymphoid organs.
Targeting is generally not a limitation in terms of the present invention. However. should specific targeting be desired, methods are available for this to be accomplished. Antibodies may be used to bind to the liposome surface and to direct the antibody and its drug contents to specific antigenic receptors located on a particular cell-type surface. Carbohydrate
determinants (glycoprotein or glycolipid cell-surface components that play a role in cell-cell recognition, interaction and adhesion) may also be used as recognition sites as they have potential in directing liposomes to particular cell types. Mostly, it is contemplated that intravenous injection of liposomal preparations would be used, but other routes of administration are also conceivable.
Alternatively , the invention provides for pharmaceutically-acceptable nanocapsule formulations of the compositions of the present invention. Nanocapsules can generally entrap compounds in a stable and reproducible way (Henry-Michelland et al, 1987; Quintanar- Guerrero et al, 1 98: Douglas et al, 1987). To avoid side effects due to intracellular polymeric overloading, such ultrafine particles (sized around 0.1 μm) should be designed using polymers able to be degraded in vivo. Biodegradable polyalkyl-cyanoacrylate nanoparticles that meet these requirements are contemplated for use in the present invention. Such particles may be are easily made, as described (Couvreur et al. 1980; 1988; zur Muhlen et al, 1998; Zambaux et al 1998; Pinto- Alphandry et al, 1995 and U. S. Patent 5,145,684, specifically incorporated herein by reference in its entirety).
4.32.5 ADDITIONAL MODES OF DELIVERY
In addition to the methods of delivery described above, the following techniques are also contemplated as alternative methods of delivering the catalase-expressing or SOD-expressing polynucleotide compositions to a target cell or animal. Sonophoresis (i.e. ultrasound) has been used and described in U. S. Patent 5,656,016 (specifically incorporated herein by reference in its entirety) as a device for enhancing the rate and efficacy of drug permeation into and through the circulatory system. Other drug delivery alternatives contemplated include intraosseous injection (U. S. Patent 5,779,708), microchip devices (U. S. Patent 5,797,898), ophthalmic formulations (Bourlais et al, 1998). transdermal matrices (U. S. Patent 5,770,219 and U. S. Patent 5,783.208) and feedback controlled delivery (U. S. Patent 5,697,899). Each of these references is specifically incorporated herein by reference in its entirety.
4.33 METHODS OF NUCLEIC ACID DELIVERY AND DNA TRANSFECTION In certain embodiments, it is contemplated that one or more lentiviral vector compositions comprising one or more RNA, DNA, PNAs and/or substituted polynucleotide
compositions will be used to transfect an appropriate host cell. Technology for introduction of such polynucleotide compositions into cells is well-known to those of skill in the art.
Several methods for the transfer of expression constructs into cultured mammalian cells also are contemplated by the present invention. These include calcium phosphate precipitation (Graham and Van Der Eb. 1973; Chen and Okayama, 1987; Rippe et al, 1990) DEAE-dextran (Gopal, 1985), electroporation (Wong and Neumann, 1982; Fromm et al, 1985; Tur-Kaspa et al, 1986; Potter et al, 1984; Suzuki et al.. 1998; Vanbever et al, 1998), direct microinjection (Capecchi. 1980; Harland and Weintraub, 1985), DNA-loaded liposomes (Nicolau and Sene. 1982; Fraley et al, 1979; Takura. 1998) and lipofectamine-DNA complexes, cell sonication (Fechheimer et al, 1987), gene bombardment using high velocity microprojectiles (Yang et al. 1990; Klein et al, 1992), and receptor-mediated transfection (Curiel et al, 1991; Wagner et al. 1992; Wu and Wu, 1987; Wu and Wu, 1988). Some of these techniques may be successfully- adapted for in vivo or ex vivo use.
4.34 EXPRESSION IN ANIMAL CELLS
The inventors contemplate that the lentiviral constructs will be preferentially utilized to transformed a suitable host cell. Such cells are preferably animal cells, including mammalian cells such as those obtained from a human or other primate, murine, canine, bovine, equine. epine, or porcine species. The cells may be transformed with one or more lentiviral vectors comprising one or more polynucleotides of interest.
4.35 TRANSGENIC ANIMALS
It is contemplated that in some instances the genome of a transgenic non-human animal of the present invention will have been altered through the stable introduction of one or more of the polynucleotide compositions described herein, either native, synthetically- modified, or mutated. As used herein, the term "transgenic animal" is intended to refer to an animal that has incorporated exogenous DNA sequences into its genome. In designing a heterologous gene for expression in animals, sequences which interfere with the efficacy of gene expression, such as polyadenylation signals, polymerase II termination sequences. hairpins, consensus splice sites and the like, are eliminated. Current advances in transgenic approaches and techniques have permitted the manipulation of a variety of animal genomes
via gene addition, gene deletion, or gene modifications (Franz et al, 1997). For example, mosquitos (Fallon, 1996), trout (Ono et al, 1997), zebrafish (Caldovic and Hackett, 1995), pigs (Van Cott et al, 1997) and cows (Haskell and Bowen, 1995), are just a few of the many animals being studied by transgenics. The creation of transgenic animals that express human proteins such as α-1 -antitrypsin, in sheep (Carver et al, 1993); decay accelerating factor, in pigs (Cozzi et al, 1997); and plasminogen activator, in goats (Ebert et al, 1991 ) have previously been demonstrated. The transgenic synthesis of human hemoglobin (U. S. Patent 5,602,306) and fibrinogen (U. S. Patent 5,639.940) in non-human animals have also been disclosed, each specifically incorporated herein by reference in its entirety. Further, transgenic mice and rat models have recently been described as new directions to study and treat cardiovascular diseases such as hypertension in humans (Franz et al, 1997; Pinto- Siestma and Paul, 1997).
4.36 SELECTION AND CHARACTERIZATION The enzyme luciferase is useful as a screenable marker in the context of the present invention (Kang et al, 1998). In the presence of the substrate luciferin, cells expressing luciferase emit light which can be detected on photographic or x-ray film, in a luminometer (or liquid scintillation counter), by devices that enhance night vision, or by a highly light sensitive video camera, such as a photon counting camera. All of these assays are nondestructive and transformed cells may be cultured further following identification. The photon counting camera is especially valuable as it allows one to identify specific cells or groups of cells which are expressing luciferase and manipulate those in real time. The above techniques also could be utilized if the screenable marker is a protein such as green fluorescent protein (gfp). To confirm the presence of the exogenous DNA or "transgene(s)" in the transformed cells, a variety of assays may be performed. Such assays include, for example, "molecular biological" assays, such as Southern and Northern blotting, RT-PCR™ and PCR™; "biochemical" assays, such as detecting the presence of a protein product, e.g., by immunological means (ELISAs and Western blots) or by enzymatic function assay. While Southern blotting and PCR™ may be used to detect the transgene(s) in question, they do not provide information as to whether the gene is being expressed.
Expression may be evaluated by RT-PCR™ for mRNA and/or specifically identifying the protein products of the introduced genes or evaluating the phenotypic changes brought about by their expression.
Assays for the production and identification of specific proteins may make use of physical-chemical, structural, functional, or other properties of the proteins. Unique physical-chemical or structural properties allow the proteins to be separated and identified by electrophoretic procedures, such as native or denaturing gel electrophoresis or isoelectric focusing, or by chromatographic techniques such as ion exchange or gel exclusion chromatography. The unique structures of individual proteins offer opportunities for use of specific antibodies to detect their presence in formats such as an ELISA assay. Transgenic animals are described that synthesize epitope tagged prion proteins as a method of detecting the expressed protein(s) (U. S. Patent 5,789,655, specifically incorporated herein by- reference in its entirety). Combinations of approaches may be employed with even greater specificity such as western blotting in which antibodies are used to locate individual gene products that have been separated by electrophoretic techniques. Additional techniques may be employed to absolutely confirm the identity of the product of interest such as evaluation by amino acid sequencing following purification. Although these are among the most commonly employed, other procedures may be additionally used.
Assay procedures may also be used to identify the expression of proteins by their functionality . especially the ability of enzymes to catalyze specific chemical reactions involving specific substrates and products. These reactions may be followed by providing and quantifying the loss of substrates or the generation of products of the reactions by physical or chemical procedures. Examples are as varied as the enzyme to be analyzed and may include assays for PAT enzymatic activity by following production of radiolabeled acetylated phosphinothricin from phosphinothricin and l4C-acetyl CoA or for anthranilate synthase activity by following loss of fluorescence of anthranilate, to name two.
Very frequently the expression of a gene product is determined by evaluating the phenotypic results of its expression. These assays also may take many forms including but not limited to analyzing changes in the chemical composition, morphology, or physiological properties of the cells of the animal or human.
4.37 DNA INTEGRATION, RNA EXPRESSION AND INHERITANCE
Genomic DNA may be isolated from animal cell lines or any animal parts to determine the presence of the exogenous gene through the use of techniques well known to those skilled in the art. Note that intact sequences will not always be present, presumably due to rearrangement or deletion of sequences in the cell.
The presence of DNA elements introduced through the methods of this invention may be determined by polymerase chain reaction (PCR™). Using this technique, discreet fragments of DNA are amplified and detected by gel electrophoresis. This type of analysis permits one to determine whether a gene is present in a stable transformant, but does not prove integration of the introduced gene into the host cell genome. Positive proof of DNA integration into the host genome and the independent identities of transformants may be determined using the technique of Southern hybridization. Using this technique, specific DNA sequences that were introduced into the host genome and flanking host DNA sequences can be identified. Hence the Southern hybridization pattern of a given transformant serves as an identifying characteristic of that transformant. In addition it is possible through Southern hybridization to demonstrate the presence of introduced genes in high molecular weight DNA, i.e. confirm that the introduced gene has been integrated into the host cell genome. The technique of Southern hybridization provides information that is obtained using PCR™ e.g., the presence of a gene, but also demonstrates integration into the genome and characterizes each individual transformant. It is contemplated that using the techniques of dot or slot blot hybridization which are modifications of Southern hybridization techniques one could obtain the same information that is derived from PCR™, e.g.. the presence of a gene.
Whereas DNA analysis techniques may be conducted using DNA isolated from any part of an animal. RNA will only be expressed in particular cells or tissue types and hence it will be necessary to prepare RNA for analysis from these tissues. PCR™ techniques may also be used for detection and quantitation of RNA produced from introduced genes. In this application of PCR™ it is first necessary to reverse transcribe RNA into DNA, using enzymes such as reverse transcriptase. and then through the use of conventional PCR™ techniques amplify the DNA. In most instances PCR™ techniques, while useful, will not demonstrate integrity of the RNA product. Further information about the nature of the RNA product may be obtained by Northern blotting. This technique will demonstrate the presence of an RNA species and give information
about the integrity of that RNA. The presence or absence of an RNA species can also be determined using dot or slot blot Northern hybridization. These techniques are modifications of Northern blotting and will only demonstrate the presence or absence of an RNA species.
4.38 SELECTABLE MARKERS
In certain embodiments of the invention, the delivery of a nucleic acid in a cell, and in particular, a lentiviral construct may be identified in vitro or in vivo by including a marker in the expression construct. The marker would result in an identifiable change to the transfected cell permitting ready identification of expression. Usually the inclusion of a drug selection marker aids in cloning and in the selection of transformants, for example, neomycin, puromycin. hygromycin, DHFR, GPT. zeocin and histidinol. Alternatively, enzymes such as herpes simplex virus thymidine kinase (tk) (eukaryotic) or chloramphenicol acetyltransferase (CAT) (prokaryotic) may be employed, as well as markers such as green fluorescent protein, luciferase. and the like. Immunologic markers also can be employed. The selectable marker employed is not believed to be important, as long as it is capable of being expressed simultaneously with the nucleic acid encoding a gene product. Further examples of selectable markers are well known to one of skill in the art.
4.39 MUTAGENESIS OF LENTIVIRAL VECTORS AND POLYNUCLEOTIDE INSERTS In certain embodiments, it is desirable to prepare mutant polypeptides and/or polynucleotides that encode them. In particular embodiments, it may be desirable to alter the lentiviral vector compositions themselves and/or the polynucleotide sequences cloned into those vectors. As such, the inventors comtemplate the use of one or more mutagenesis methods known to those of skill in the art. Indeed, in certain embodiments it may be desirable to introduce one or more mutations into a particular polypeptide sequence, or alternatively, into a DNA sequence encoding the particular polypeptide. The purpose of such mutagenesis may be for producing a mutated peptide with altered biological properties, or for preparing second- or third-generation lentiviral vector compositions having improved trasnfection abilities, or enhanced replication, or infectivity properties. To that end, the present invention encompasses both site-specific mutagenesis methods and random mutagenesis of nucleic acid segments of the present invention. Using
the methods described herein, one may then identify mutants arising from these procedures which have improved activity, increased peptide stability, and or increased viral infectivity, titer, or transfection efficiency.
The means for mutagenizing a DNA segment are well-known to those of skill in the art. Modifications may be made by random, or site-specific mutagenesis procedures. The nucleic acid may be modified by altering its structure through the addition or deletion of one or more nucleotides from the sequence.
Mutagenesis may be performed in accordance with any of the techniques known in the art such as and not limited to synthesizing an oligonucleotide having one or more mutations within the sequence, such as a sequence encoding a particular polypeptide.
In particular, site-specific mutagenesis is a technique useful in the preparation of individual peptides. or biologically functional equivalent proteins or peptides, through specific mutagenesis of the underlying DNA. The technique further provides a ready ability to prepare and test sequence variants, for example, incorporating one or more of the foregoing considerations, by introducing one or more nucleotide sequence changes into the DNA. Site-specific mutagenesis allows the production of mutants through the use of specific oligonucleotide sequences which encode the DNA sequence of the desired mutation, as well as a sufficient number of adjacent nucleotides. to provide a primer sequence of sufficient size and sequence complexity to form a stable duplex on both sides of the deletion junction being traversed. Typically, a primer of about 17 to about 75 nucleotides or more in length is preferred, with about 10 to about 25 or more residues on both sides of the junction of the sequence being altered.
In general, the technique of site-specific mutagenesis is well known in the art, as exemplified by various publications. As will be appreciated, the technique typically employs a phage vector which exists in both a single stranded and double stranded form. Typical vectors useful in site-directed mutagenesis include vectors such as the Ml 3 phage. The phage are readily commercially available and their use is generally well known to those skilled in the art. Double stranded plasmids are also routinely employed in site directed mutagenesis that eliminates the step of transferring the gene of interest from a plasmid to a phage.
Site-directed mutagenesis in accordance herewith is typically performed by first obtaining a single-stranded vector or melting apart of two strands of a double stranded vector which includes within its sequence a DNA sequence which encodes the desired peptide. An oligonucleotide primer bearing the desired mutated sequence is prepared, generally synthetically . This primer is then annealed with the single-stranded vector, and subjected to DNA polymerizing enzymes such as E. coli polymerase I Klenow fragment, in order to complete the synthesis of the mutation-bearing strand. Thus, a heteroduplex is formed wherein one strand encodes the original non-mutated sequence and the second strand bears the desired mutation. This heteroduplex vector is then used to transform or transfect appropriate cells, such as E. coli cells, and clones are selected which include recombinant vectors bearing the mutated sequence arrangement. A genetic selection scheme was devised by Kunkel et al. (1987) to enrich for clones incorporating the mutagenic oligonucleotide. Alternatively, the use of PCR™ with commercially available thermostable enzymes such as Taq polymerase may be used to incorporate a mutagenic oligonucleotide primer into an amplified DNA fragment that can then be cloned into an appropriate cloning or expression vector. The PCR™-mediated mutagenesis procedures of Tomic et al. (1990) and Upender et al. ( 1995) provide two examples of such protocols. A PCR™ employing a thermostable ligase in addition to a thermostable polymerase may also be used to incorporate a phosphorylated mutagenic oligonucleotide into an amplified DNA fragment that may then be cloned into an appropriate cloning or expression vector. The mutagenesis procedure described by Michael (1994) provides an example of one such protocol.
The preparation of sequence variants of the selected peptide-encoding DNA segments using site-directed mutagenesis is provided as a means of producing potentially useful species, but is not meant to be limiting as there are other ways in which sequence variants of peptides and the DNA sequences encoding them may be obtained. For example, recombinant vectors encoding the desired peptide sequence may be treated with mutagenic agents, such as hydroxylamine. to obtain sequence variants.
As used herein, the term "oligonucleotide directed mutagenesis procedure" refers to template-dependent processes and vector-mediated propagation which result in an increase in the concentration of a specific nucleic acid molecule relative to its initial concentration, or in an increase in the concentration of a detectable signal, such as amplification. As used herein,
the term "oligonucleotide directed mutagenesis procedure" is intended to refer to a process that involves the template-dependent extension of a primer molecule. The term template dependent process refers to nucleic acid synthesis of an RNA or a DNA molecule wherein the sequence of the newly synthesized strand of nucleic acid is dictated by the well-known rules of complementary base pairing (see, for example. Watson. 1987). Typically, vector mediated methodologies involve the introduction of the nucleic acid fragment into a DNA or RNA vector, the clonal amplification of the vector, and the recovery of the amplified nucleic acid fragment. Examples of such methodologies are given in U. S. Patent 4,237.224, specifically incorporated herein by reference in its entirety. A number of template dependent processes are available to amplify the target sequences of interest present in a sample. One of the best known amplification methods is the polymerase chain reaction (PCR™) which is described in detail in U. S. Patents 4,683.195. 4.683.202 and 4,800,159 (each of which is specifically incorporated herein by reference in its entirety). Briefly, in PCR™, two primer sequences are prepared which are complementary to regions on opposite complementary strands of the target sequence. An excess of deoxynucleoside triphosphates is added to a reaction mixture along with a DNA polymerase (e.g., Taq polymerase). If the target sequence is present in a sample, the primers will bind to the target and the polymerase will cause the primers to be extended along the target sequence by adding on nucleotides. By raising and lowering the temperature of the reaction mixture, the extended primers will dissociate from the target to form reaction products, excess primers will bind to the target and to the reaction products. The process may then be repeated as necessary. Preferably a reverse transcriptase PCR™ amplification procedure may be performed in order to quantify the amount of mRNA amplified. Polymerase chain reaction methodologies are well known in the art. Another method for amplification is the ligase chain reaction (referred to as LCR), disclosed in Eur. Pat. Appl. Publ. No. 320,308, incorporated herein by reference in its entirety. In LCR, two complementary probe pairs are prepared, and in the presence of the target sequence, each pair will bind to opposite complementary strands of the target such that they abut. In the presence of a ligase, the two probe pairs will link to form a single unit. By temperature cycling, as in PCR™, bound ligated units dissociate from the target and then serve as "target sequences" for ligation of excess probe pairs. U. S. Patent 4,883.750,
specifically incorporated herein by reference in its entirety, describes an alternative method of amplification similar to LCR for binding probe pairs to a target sequence.
Qbeta Replicase™, described in Intl. Pat. Appl. Publ. No. PCT/US87/00880, incorporated herein by reference in its entirety, may also be used as still another amplification method in the present invention. In this method, a replicative sequence of RNA that has a region complementary to that of a target is added to a sample in the presence of an RNA polymerase. The polymerase will copy the replicative sequence that can then be detected.
An isothermal amplification method, in which restriction endonucleases and ligases are used to achieve the amplification of target molecules that contain nucleotide 5'-[α-thio]triphosphates in one strand of a restriction site (Walker et al, 1992. incorporated herein by reference in its entirety), may also be useful in the amplification of nucleic acids in the present invention.
Strand Displacement Amplification (SDA) is another method of carrying out isothermal amplification of nucleic acids that involves multiple rounds of strand displacement and synthesis, i.e., nick translation. A similar method, called Repair Chain Reaction (RCR) is another method of amplification which may be useful in the present invention and is involves annealing several probes throughout a region targeted for amplification, followed by a repair reaction in which only two of the four bases are present. The other two bases can be added as biotinylated derivatives for easy detection. A similar approach is used in SDA.
Sequences can also be detected using a cyclic probe reaction (CPR). In CPR. a probe having 3' and 5' end sequences of non-lentiviral specific DNA and an internal sequence of a lentiviral-specific RNA is hybridized to DNA which is present in a sample. Upon hybridization, the reaction is treated with RNaseH, and the products of the probe identified as distinctive products generating a signal that are released after digestion. The original template is annealed to another cycling probe and the reaction is repeated. Thus, CPR involves amplifying a signal generated by hybridization of a probe to the specific expressed nucleic acid sequence. Still other amplification methods described in Great Britain Pat. Appl. No. 2 202 328, and in Intl. Pat. Appl. Publ. No. PCT/US89/01025, each of which is incorporated herein by
reference in its entirety, may be used in accordance with the present invention. In the former application, "modified" primers are used in a PCR™ like, template and enzyme dependent synthesis. The primers may be modified by labeling them with a capture moiety (e.g.. biotin) and/or a detector moiety (e.g.. enzyme). In the latter application, an excess of labeled probes is added to a sample. In the presence of the target sequence, the probe binds and is cleaved catalytically. After cleavage, the target sequence is released intact to be bound by excess probe. Cleavage of the labeled probe signals the presence of the target sequence.
Other nucleic acid amplification procedures include transcription-based amplification systems (TAS) (Kwoh et al. 1989; Intl. Pat. Appl. Publ. No. WO 88/10315, incorporated herein by reference in its entirety), including nucleic acid sequence based amplification (NASBA) and 3SR. In NASBA, the nucleic acids can be prepared for amplification by standard phenol/chloroform extraction, heat denaturation of a sample, treatment with lysis buffer and minispin columns for isolation of DNA and RNA or guanidinium chloride extraction of RNA. These amplification techniques involve annealing a primer that has one or more polypeptide-specific sequences. Following polymerization, DNA/RNA hybrids are digested with RNase H while double stranded DNA molecules are heat-denatured again. In either case the single stranded DNA is made fully double stranded by addition of second polypeptide-specific primer, followed by polymerization. The double stranded DNA molecules are then multiply transcribed by a polymerase such as T7 or SP6. In an isothermal cyclic reaction, the RNAs are reverse transcribed into double stranded DNA, and transcribed once against with a polymerase such as T7 or SP6. The resulting products, whether truncated or complete, indicate polypeptide-specific sequences.
Eur. Pat. Appl. Publ. No. 329,822, incorporated herein by reference in its entirety, disclose a nucleic acid amplification process involving cyclically synthesizing single-stranded RNA ("ssRNA"), ssDNA, and double-stranded DNA (dsDNA), which may be used in accordance with the present invention. The ssRNA is a first template for a first primer oligonucleotide, which is elongated by reverse transcriptase (RNA-dependent DNA polymerase). The RNA is then removed from resulting DNA:RNA duplex by the action of ribonuclease H (RNase H, an RNase specific for RNA in a duplex with either DNA or RNA). The resultant ssDNA is a second template for a second primer, which also includes the sequences of an RNA polymerase promoter (exemplified by T7 RNA polymerase) 5' to its
homology to its template. This primer is then extended by DNA polymerase (exemplified by the large "Klenow" fragment of E coli DNA polymerase I), resulting as a double-stranded DNA ("dsDNA") molecule, having a sequence identical to that of the original RNA between the primers and having additionally, at one end, a promoter sequence. This promoter sequence can be used by the appropriate RNA polymerase to make many RNA copies of the DNA. These copies can then re-enter the cycle leading to very swift amplification. With proper choice of enzymes, this amplification can be done isothermally without addition of enzymes at each cycle. Because of the cyclical nature of this process, the starting sequence can be chosen to be in the form of either DNA or RNA. Intl. Pat. Appl. Publ. No. WO 89/06700, incorporated herein by reference in its entirety, disclose a nucleic acid sequence amplification scheme based on the hybridization of a promoter primer sequence to a target single-stranded DNA ("ssDNA") followed by transcription of many RNA copies of the sequence. This scheme is not cyclic; i.e. , new templates are not produced from the resultant RNA transcripts. Other amplification methods include "RACE" (Frohman, 1990), and "one-sided PCR™" (Ohara, 1989), which are well- known to those of skill in the art.
4.40 BIOLOGICAL FUNCTIONAL EQUIVALENTS
Modification and changes may be made in the structure of the vector compositions. polynucleotides, polypeptides. and/or peptides of the present invention and still obtain a functional molecule having desirable characteristics. The following is a discussion based upon changing the amino acids of a protein to create an equivalent, or even an improved, second- generation molecule. The amino acid changes may be achieved by changing the codons of the DNA sequence, according to Table 1. For example, certain amino acids may be substituted for other amino acids in a protein structure without appreciable loss of interactive binding capacity with structures such as, for example, antigen-binding regions of antibodies or binding sites on substrate molecules. Since it is the interactive capacity and nature of a protein that defines that protein's biological functional activity, certain amino acid sequence substitutions can be made in a protein sequence, and, of course, its underlying DNA coding sequence, and nevertheless obtain a protein with like properties. It is thus contemplated by the inventors that various changes may be made in the
peptide sequences of the disclosed compositions, or corresponding DNA sequences which encode said peptides without appreciable loss of their biological utility or activity.
In making such changes, the hydropathic index of amino acids may be considered. The importance of the hydropathic amino acid index in conferring interactive biologic function on a protein is generally understood in the art (Kyte and Doolittle, 1982. incorporate herein by reference). It is accepted that the relative hydropathic character of the amino acid contributes to the secondary structure of the resultant protein, which in turn defines the interaction of the protein with other molecules, for example, enzymes, substrates, receptors, DNA, antibodies, antigens, and the like. Each amino acid has been assigned a hydropathic index on the basis of their hydrophobicity and charge characteristics (Kyte and Doolittle, 1982), these are: isoleucine (+4.5); valine (+4.2): leucine (+3.8); phenylalanine (+2.8): cysteine/cystine (+2.5); methionine (+1.9); alanine (+1.8): glycine (-0.4); threonine (-0.7): serine (-0.8); tryptophan (-0.9); tyrosine (-1.3); proline (-1.6): histidine (-3.2); glutamate (-3.5); glutamine (-3.5); aspartate (-3.5); asparagine (-3.5); lysine (-3.9); and arginine (-4.5).
TABLE 1
Amino Acids ( Codons
Alanine Ala A GCA GCC GCG GCU
Cysteine Cys C UGC UGU
Aspartic acid Asp D GAC GAU
Glutamic acid Glu E GAA GAG
Phenylalanine Phe F UUC uuu
Glycine Gly G GGA GGC GGG GGU
Histidine His H CAC CAU
Isoleucine He I AUA AUC AUU
Lysine Lys K AAA AAG
Leucine Leu L UUA UUG CUA cue CUG CUU
Methionine Met M AUG
Asparagine Asn N AAC AAU
Proline Pro P CCA ccc CCG ecu
Glutamine Gin Q CAA CAG
Arginine Arg R AGA AGG CGA CGC CGG CGU
Serine Ser S AGC AGU UCA UCC UCG UCU
Threonine Thr T ACA ACC ACG ACU
Valine Val V GUA GUC GUG GUU
Tryptophan Trp w UGG
Tyrosine Tyr Y UAC UAU
It is known in the art that certain amino acids may be substituted by other amino acids having a similar hydropathic index or score and still result in a protein with similar biological activity, i.e. still obtain a biological functionally equivalent protein. In making such changes, the substitution of amino acids whose hydropathic indices are within ±2 is preferred, those which are within ±1 are particularly preferred, and those within ±0.5 are even more particularly preferred. It is also understood in the art that the substitution of like amino acids can be made effectively on the basis of hydrophilicity. U. S. Patent 4,554,101, incorporated herein by reference, states that the greatest local average hydrophilicity of a protein, as governed
by the hydrophilicity of its adjacent amino acids, correlates with a biological property of the protein.
As detailed in U. S. Patent 4.554,101, the following hydrophilicity values have been assigned to amino acid residues: arginine (+3.0); lysine (+3.0); aspartate (+3.0 ± 1 ); glutamate (+3.0 ± 1 ): serine (+0.3); asparagine (+0.2); glutamine (+0.2); glycine (0); threonine (-0.4); proline (-0.5 + 1 ); alanine (-0.5); histidine (-0.5); cysteine (-1.0); methionine (-1.3): valine (-
1.5); leucine (-1.8); isoleucine (-1.8); tyrosine (-2.3); phenylalanine (-2.5); tryptophan (-3.4).
It is understood that an amino acid can be substituted for another having a similar hydrophilicity value and still obtain a biologically equivalent, and in particular, an immunologically equivalent protein. In such changes, the substitution of amino acids whose hydrophilicity values are within
±2 is preferred, those which are within ±1 are particularly preferred, and those within ±0.5 are even more particularly preferred.
As outlined above, amino acid substitutions are generally therefore based on the relative similarity of the amino acid side-chain substituents, for example, their hydrophobicity, hydrophilicity , charge, size, and the like. Exemplary substitutions which take various of the foregoing characteristics into consideration are well known to those of skill in the art and include: arginine and lysine; glutamate and aspartate; serine and threonine; glutamine and asparagine; and valine, leucine and isoleucine.
4.41 DEFINITIONS AND ABBREVIATIONS
In accordance with the present invention, nucleic acid sequences include and are not limited to DNAs (including genomic and extragenomic DNA); genes; RNAs (including mRNAs, rRNAs and tRNA); nucleosides; and suitable nucleic acid segments either obtained from native sources, chemically synthesized, modified, or otherwise prepared by the hand of man. The following words and phrases have the meanings set forth below.
A, an: In accordance with long standing patent law convention, the words "a" and "an" when used in this application, including the claims, denotes "one or more".
Expression: The combination of intracellular processes, including transcription and translation undergone by a coding DNA molecule such as a structural gene to produce a polypeptide.
Promoter: A recognition site on a DNA sequence or group of DNA sequences that provide an expression control element for a structural gene and to which RNA polymerase specifically binds and initiates RNA synthesis (transcription) of that gene. Structural gene: A gene that is expressed to produce a polypeptide. Transformation: A process of introducing an exogenous DNA sequence (e.g.. a vector, a recombinant DNA molecule) into a cell in which that exogenous DNA is incorporated into a chromosome or is capable of autonomous replication.
Transformed cell: A cell whose DNA has been altered by the introduction of an exogenous DNA molecule into that cell. Transgenic cell: Any cell derived or regenerated from a transformed cell or derived from a transgenic cell.
Vector: A DNA molecule capable of replication in a host cell and/or to which another DNA segment can be operatively linked so as to bring about replication of the attached segment. A plasmid or lentiviral construct of the present invention is an exemplary vector.
The following abbreviations have been utilized throughout: RCR (replication- competent retrovirus); RCV (replication-competent virus); WT (wild-type); PBL (peripheral blood lymphocyte): M (molar); mM (millimolar); μM (micromolar); mol (moles); mmol (millimoles): μmol (micromoles): nmol (nanomoles); g (gravity); gm (grams); mg (milligrams); μg (micrograms); pg (picograms); L (liters); ml (milliliters); μl (microliters); cm (centimeters); mm (millimeters); μm (micrometers); nm (manometers); hr (hour); min (minute); msec (millisecond); °C (degrees Centigrade); AMP (adenosine 5'-monophosphate); cDNA (copy or complimentary DNA); DTT (dithiothreitol); ddH O (double distilled water); dNTP (deoxyribonucleotide triphosphate); rNTP (ribonucleotide triphosphate); ddNTP (dideoxyribonucteotide triphosphate); bp (base pair); kb (kilo base pair); TEM (transmission electron microscope); SEM (scanning electron microscope); TLC (thin layer chromatography); tRNA (transfer RNA); nt (nucleotide); VRC (vanadyl ribonucleoside complex); RNase (ribonuclease); DNase (deoxyribonuclease); poly A (polyriboadenylic acid); PBS (phosphate buffered saline); OD (optical density); HEPES (N-[2- Hydroxyethyl]piperazine-N-[2-ethanesulfonic acid]); HBS (HEPES buffered saline); SDS (sodium dodecyl sulfate); Tris-HCl (tris(Hydroxymethyl]aminomethane-hydrochloride); rpm
(revolutions per minute); ligation buffer (50 mM Tris-HCl, 10 MM MgCl,, 10 mM dithiothreitol, 25 μ/ml bovine serum albumin, and 26 μM NAD+, and pH 7.8); EGTA (ethylene glycol-bis(β-aminoethyl ether) N,N,N',N'-tetraacetic acid); EDTA (ethylenediaminetetracetic acid); ELISA (enzyme linked immunosorbant assay); ELISPOT (enzyme-linked immunosorbent spot assay); LB (Luria-Bertani broth: 10 g tryptone. 5 g yeast extract, and 10 g NaCl per liter, pH adjusted to 7.5 with 1 N NaOH); superbroth (I 2 g tryptone, 24 g yeast extract. 5 g glycerol. 3.8 g KH2PO and 12.5 g, K2HPO per liter); DMEM (Dulbecco's modified Eagle's medium).
5.0 EXAMPLES
The following example is included to demonstrate preferred embodiments of the invention. It should be appreciated by those of skill in the art that the techniques disclosed in the examples which follow represent techniques discovered by the inventor to function well in the practice of the invention, and thus can be considered to constitute preferred modes for its practice. However, those of skill in the art should, in light of the present disclosure, appreciate that many changes can be made in the specific embodiments which are disclosed and still obtain a like or similar result without departing from the spirit and scope of the invention.
5.1 EXAMPLE 1 - PREPARATION AND CHARACTERIZATION OF VIRAL VECTORS 5.1.1 MATERIALS AND METHODS 5.1.1.1 PLASMID DNA CONSTRUCTION
HIV-1 LTR and tat mutations were constructed as described previously (Chang, et al 1993; Chang and Zhang, 1995). Cloned HIV proviruses with heterologous enhancer/promoters were constructed by ligating three fragments from an HIV-1 molecular clone HIVNL4-3 (Adachi et al, 1986), two fragments isolated from the U3-R-CAT plasmids containing inserted heterologous enhancer/promoters and the BamHI plus Pstl digested pT7T318U vector. The proviral segments used in the ligation were as described before (Chang, et al, 1993). The structures of the reconstructed HIV proviral DNAs were'verified by extensive restriction enzyme mapping, and the LTR regions were checked by nucleotide sequencing.
5.1.1.2 RT ASSAY AND P24 ELISA FOR THE DETECTION OF HIV GAG AND POL PRODUCTS
RT assays detect functional reverse transcriptase activity which were performed as described below. The supernatants from transfected cells were spun in a microfuge at 3000 rpm for 5 min before being added to the reaction mixture. Supernatants from virus infections were removed from cultures after the cells had settled. Each reaction mixture contained 10 ml of supernatant and 50 ml of RT cocktail (60 mM Tris-HCl. pH 7.8, 75 mM KC1, 5 mM MgCL, 0.1% Nonidet P-40, 1 mM EDTA. 5 mg/ml poly A and 0.16 mg/ml oligo-dT) and was incubated at 37°C for 1 h. The radioactive products generated in the CAT and RT assays were quantitated by using a Fuji phosphoimager. The results obtained were comparable to those derived by scintillation counting. p24 antigen is derived from p55 gag precursor. The p24 antigen expression was quantified using a commercial ELISA kit from Coulter (Coulter Corp.. Hialeah. FL).
5.1.1.3 RT-PCR AND SEQUENCING OF THE PACKAGED VIRAL GENOMIC RNA Cell-free particles, present in the supernatants of vector producing cells, were harvested (100 μl), centrifuged at top speed for 5 min in a microcentrifuge at room temperature, and filtered through a 0.45 μm-pore-size Eppendorf spin filter. The particles present in the filtrate was dissociated by vortexing in the presence of an equal volume of 8 M LiCl. placed on dry ice for 20 min, transferred to a -20°C freezer for at least 2 h, and centrifuged at top speed in a microcentrifuge at 4°C for 20 min. The RNA pellet was then rinsed with 70% ethanol, dried briefly under vacuum, resuspended in water and reverse transcribed by using an appropriate primer and the RiboClone cDNA Synthesis System (Promega) for the synthesis of the first DNA strand. A control reaction excluding the reverse transcriptase was performed in parallel. The cDNA was amplified by PCR™ using the polymerase and reagents obtained from Perkin Elmer Cetus; 5' and 3' primers (0.1 μmol each) were added to a reaction mixture containing the cDNA (1/20 of the RT product) and amplified for 30 cycles under the following conditions: 94°C for 1 min, 58°C for 1 min and 72°C for 3 min. The product obtained was then subjected to asymmetric PCR™ amplification (i.e., two primers at 10:1 rnolar ratio) to generate single stranded DNA for sequencing as described (Meltzer et al, 1993). Excess primers were removed with a
centricon 100 filtration device (Amicon) after each amplification step. Nucteotide sequencing was performed using Sequenase and protocols supplied by USB.
5.1.1.4 IMMI ΌFLUORESEENT AND IMMUNOHISTOCHEMICAL STAINING For immunofluorescent staining, non-adherent cells were attached to the surface of a microscope cover glass (12 μm circle, Fisher Scientific, Pittsburgh. PA) which had been pretreated with poly-D-lysine (1 mg/ml, Sigma) at room temperature for 10 min. The attached cells were washed with phosphate buffered saline (PBS) three times, fixed in cold acetone and methanol (1 :1) for 5 min, washed three times in PBS, and incubated in blocking (20% FBS. 0.1%Triton X-100 in PBS) solution for 30 min. An HIV patient's serum was used as the primary antibody, which was diluted at 1 :2000 in-blocking solution, and the cells were incubated at room temperature for 1 h or at 4°C overnight with constant shaking. After washing in PBS 4 times for 5 min each, the cells were incubated with normal goat or sheep antisera (1 :200 dilution) at room temperature for 30 min to block non-specific binding. The secondary antibody was FITC-labeled goat anti-human IgG (Fab specific. Sigma Chemical Company, St. Louis, MO). After staining, the cover glass was washed four times in PBS and examined using a fluorescent microscope. For direct immunohistochemical staining, a peroxidase-linked sheep anti-human Ig (Amersham) was used as the secondary antibody. Alternatively , a biotinylated sheep anti-human antibody (Amersham) was used at 1 :2000 dilution and incubated at room temperature for 1 h. The latter step provided a more sensitive method for detection of low level of HIV antigens which was described in detail elsewhere (Chang, and Zhang, 1995).
5.1.1.5 RNA AND PROTEIN ANALYSES Northern analysis was performed as previously described (Robinson, et al, 1995).
For protein analysis, cells were lysed in a buffer containing 50 mM Tris pH 7.4, 300 mM NaCl, 0.5% Triton X-100, 1% (vol./vol.) aprotinin and 1 mM PMSF at 4°C for 10 min and freeze-thawed once. Virus particles were collected by centrifugation in a refrigerated micro centrifuge in a small volume (200 microliters) at 23,000 x g for 1 hr. The supernatant was carefully removed and to the pellet, 20 ml of SDS sample buffer (final 2% SDS, 5% glycerol, 0/001%) BPB. 0.5%) NP-40) was added and the denatured protein was resolved by
polyacrylarnide gel electrophoresis (PAGE) as described previously (Chang et al, 1990). For Western blot analysis, the protein was transferred to a 0.2 micron nitrocellulose filter, stained with Ponceau S to identify the molecular weight marker, and blocked with 10% dried milk in TBS-T (Tris-buffered saline with 0.3%> Tween-20) at room temperature for 30 min to 1 hr. After washed briefly at room temperature, the blot was placed into a "seal-a-meal" bag and incubated with an AIDS patient's serum (diluted at 1 :2,000, or a rabbit polygonal anti- Vpr antibody at 1 : 1.000, or a monoclonal anti-Nef antibody at 1 :1000) in TBS-T containin- 2% dry milk at 4°C overnight. After four washes with TBS-T. the blot was blocked with normal goat sera (the same species as the secondary Ab) at 1 :200 dilution in a shallow tray or in a bag at room temperature for 30 min.
The blot was then transferred to a second bag containing a horseradish peroxidase (HRP) conjugated goat antizhuman (or goat anti-rabbit, or goat anti-mouse) antibody and incubated at room temperature for 1 hr. The blot was washed four times in TBS-T and developed using the chemiluminescence ECL immunodetection reagents from Amersham. The blot was then exposed, to a hyperfilm (Amersham) normally for 1 min and developed.
5.1.1.6 CELLS AND CULTURE CONDITIONS
HeLa cells were propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). H9, CEM, MT4, C8166 and AA2 were obtained from NIH AIDS Research and Reference Reagent Program. Maintenance of the continuous human lymphoid cell lines H9, CEM, MT4, AA2 and the primary human PBLs were as described (Chang, et al, 1993). The Molt3 and THP-1 were obtained from the American Type Culture Collection (Rockville, MD). HeLa clone HL3TI, C8166 and U937 cells were obtained from G. Paviakis, K.-T. Jeang and K. Peden, respectively. HeLa CD4+ clones 1022 and HT-6C (Chesebro and Wehrly, 1988) were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH, from Dr. Bruce Chesebro. The macrophage culture was prepared from HIV-seronegative donors by adherence of PBLs to plastic flasks as described previously with minor modifications (Hassan, et al, 1986). PBLs were prepared using lymphocyte separation medium (Organon Teknika Corp., Durham, NC) by density gradient. The PBLs were resuspended in RPMI 1640 medium supplemented with 20%> heat-inactivated human serum. Approximately 5 x
107 PBLs were attached to a T-75 flask and incubated overnight at 37°C. The next day cells were washed three times with phosphate buffered saline and the attached cells were incubated with 0.02% EDTA in PBS for 5-10 min. The cells were collected with a cell scraper and plated onto a 48-well plate at 5 x 104 cells per well. The viability approached 100%) as determined by trypan blue staining. The initial monocytes were characterized by- Wright's staining and the mature macrophages by both Wright's staining and microscopic examination.
5.1.1.7 PLASMID CONSTRUCTION AND SITE-DIRECTED MUTAGENESIS The tatA, tatB and tatC site-directed mutations were generated by the "Megaprimer" method (Sarkar and Crissman. 1990) using the following mutagenic oligos:
5'-G.ΛATTGGGTGTCGACATAGCGGCCGCTTGTACCAATTGCTATTG-3', (SEQ ID NO:XX)
5'-GGTACAAGCAGTTTAAGGCTAACTTCCTGGATGCTTCC-3' (SEQ ID NO:XX), and
5'-CGACAGAGGAGAGCAAGAAACGGCGCCTCGCGTAGCTAGCGG-3' (SEQ ID NO:XX). respectively.
A fragment containing the tat mutation [EcoRI-Sαel (260 nt)] generated by PCR™ mutagenesis was used to construct the full-length two LTR HIV plasmids. Construction of the tatA and tatC mutations have been described elsewhere (Dimitrov et al, 1993; Amendt et al, 1994). The dl.Spi/CMV tatB macrophage-tropic virus was made by replacing the EcoRI to BamHI fragment in a T-cell tropic construct (pNL4-3, Adachi et al, 1986) with the same fragment from a macrophage-tropic construct (pNLAD8, obtained from Eric Freed). Sequences of the PCR™ fragment and its flanking region in the final constructs were verified by DNA sequencing.
5.1.1.8 TRANSFECTION AND NORTHERN ANALYSES
HeLa cells were transfected using the original Ca (PO4)2-DNA co-precipitation procedure with modifications (Graham and van der Eb, 1973). In brief, HeLa cells were split into 6-well plates 20 h prior to transfection. The plasmid DNA was in 90 ml of ddH20 and mixed with 10 ml of 2.5 M CaCl2 (Mallinckrodt) in a polycarbonate tube. To the DNA
mixture, a 100 ml of BES-buffered solution (50 mM N,N-bis(2-hydroxyethyl]-2- aminoethanesulfonic acid [Calbiochem], 280 mM NaCl, 1.5 mm Na HPO , pH 6.95) was added dropwise. The solution was allowed to sit at room temperature for 45 min to 1 hr before being added to the 2 ml growth culture (pH. 7.1 ). After adding the DNA, the culture was maintained in a 3% CO2 incubator at 37°C overnight. For the CAT assay, HeLa cells were transfected with 3 mg of CAT plasmid in the presence or absence of 0.1 mg of a tat plasmid pSVtat (Peterlin et al, 1986) or pCEP-/α/ (Robinson et al, 1995). For the assay of Tat function using HL3TI cells, transfection was done using 10 mg of DNA of different HIV- 1 constructs. To atenuate virus stocks, HeLa cells were transfected with 10 mg of cloned HIV-1 plasmids and virus was harvested, filtered through a 0.45 μm filter (MILLEX-HV. Millipore Products Division, Bedford. MA) and frozen at -80°C for later use. All transfections were performed in the presence of a control human growth hormone plasmid pXGHS (Nichols Institute Diagnostics). Northern analysis of viral RNA was done as described previously (Chang et al. 1993) and analyzed using a phosphoimager (Fuji, BAS 1000).
5.1.1.9 QUANTITATIVE IMMUNOSTAINING OF HIV-I NFECTED CELLS
Adherent cells were washed with phosphate buffered saline (PBS) three times, fixed in cold acetone and methanol (1 :1) for 2 min. washed three times in PBS, and incubated in blocking solution (20%> FBS, 0.1 %> Triton X-100 in PBS) for 30 min. Non-adherent cells were attached to the surface of a 24-well plate that had been pretreated with poly-D-lysine (1 mg/ml, Sigma) at room temperature for 10 min. An HIV patient serum was used which was diluted at 1 :2000 in a blocking solution containing 20% FBS, 0. 1% Triton X-100 and 2% dry milk in PBS as the first antibody and the incubation was done at room temperature for 1 h or at 4°C overnight with constant shaking. After being washed in PBS for 5 min 4 times, the cells were incubated with a 1 :200 dilution of normal sheep antisera, at room temperature for 30 min to block nonspecific signals. The secondary antibody was a biotinylated sheep anti-human antibody (Amersham) which was used at 1 :2000 dilution and incubated at room temperature for 1 h. The cells were washed four times in PBS-Tween-20 (0.3%) and incubated in the ultra-sensitive ABC staining solution (containing avidin and biotinylated horseradish peroxidase, Pierce Chemical Co.) at room temperature for 30 min. After four
more washes in PBS-Tween-20, the cells were incubated in 3,3'-diaminobenzidine tetrahydrochloride (DAB) solution (Sigma) containing 0.3%> NiCL for 2-3 min. The reaction was stopped by washing cells with tap water for 1 -2 min. Cell staining was scored under an inverted microscope and photographed. To reduce background staining, both the primary and the secondary antisera were preabsorbed with fixed human PBLs. Pretreatment of fixed cells with 0.01% H202 at room temperature for 5 min essentially eliminated all nonspecific background signals. The percentages of positive cells were determined by taking the average of more than three representative counts of 1 ,000 or 10.000 cells.
5.1.1.10 MATERIALS
Unless otherwise indicated, all restriction enzymes were obtained from New England Biolabs and used according to the manufacturers' directions. Unless otherwise indicated, synthetic oligonucleotides were synthesized using an ABI DNA synthesizer, Model No. 391. Non-attenuated HIV strains used included the NL4-3 HIV-1 strain, HIV-1 primary isolates covering the different HIV clades (e.g., 92RWO08. 92HT593, etc.), the ROD strain of HIV-2. and the SlVmac239 strain of SIV, all of which are available from the AIDS Research and Reference Reagent Program.
Regeants for many of the experimental studies were obtained from the following sources: ABI (Applied Biosystems Inc., Foster City. CA); Amersham (Amersham Coφoration. Arlington Heights, IL); ATCC (American Type Culture Collection, Rockville, MD); AIDS Research and Reference Reagent Program (AIDS Research and Reference Reagent Program of the National Institutes of Health, Bethesda, MD); Beckman (Beckman Instruments Inc., Fullerton CA); BM (Boehringer Mannheim Biochemicals, Indianapolis, IN); Bio-101 (Bio-101, Vista, CA); BioRad (BioRad, Richmond, CA); Brinkmann (Brinkmann Instruments Inc. Wesbury, NY); BRL, Gibco BRL and Life Technologies (Bethesda Research Laboratories, Life Technologies, Inc., Gaithersburg, MD); CRI (Collaborative Research Inc. Bedford, MA); Eastman Kodak (Eastman Kodak Co., Rochester, NY); Eppendorf (Eppendorf, Eppendorf North America, Inc., Madison, WI); Falcon (Becton Dickenson Labware, Lincoln Park, NJ); IBI (International Biotechnotogies, Inc., New Haven, CT); ICN. (ICN Biomedicals, Inc., Costa Mesa, CA); Invitrogen (Invitrogen, San Diego, CA); New Brunswick (New Brunswick Scientific Co. Inc., Edison,
NJ); NEB (New England BioLabs Inc., Beverly, MA); NEN (Du Pont NEN Products, Boston, MA); Nichols Institute Diagnostics (Nichols Institute Dia nostics, San Juan Capistrano, CA); Pharmacia (Pharmacia LKB Gaithersburg. MD); Promega (Promega Corporation. Madison. WI); Stratagene (Stratagene Cloning Systems, La Jolla, CA); UVP (UVP, Inc.. San Gabreil, CA); USB (United States Biochemical Corp., Cleveland. OH); Taconic (Taconic. Germantown, NY); Whatman (Whatman Lab. Products Inc. Clifton, NJ).
5.1.2 RESULTS
5.1.2.1 CONSTRUCTION OF ATTENUATED RECOMBINANT HIV-1 CONSTRUCTS As described below, several modified HIV-1 constructs that exhibit reduced cytopathic effects in tissue culture were chosen for use in the 7 development of the present invention.
5.1.2.2 HIV-1 LTR MUTANTS Investigation of virus attenuation was essential to the understanding of viral pathogenesis. the development of preventive vaccines, and development of a safe lentiviral vector system. For production of a safe HIV vector, attenuated mutant molecular constructs of HIV-1 were viewed as better starting materials than wild-type constructs.
One approach to developing these attenuated constructs was establishing mutations in the LTRs of HIV-1. For example, the function of HIV- I LTR enhancer/promoter elements has been studied using recombinant LTRs containing heterologous enhancer/promoters (see FIG. 1). After deleting the regulatory elements including the NF-KB, Sp I binding sites, and/or the TATA box, and inserting a minimal cytomegalovirus enhancer element, delayed replication kinetics has been observed in some CD4+ human lymphoid cell lines (see e.g., Chang et al., 1993). However, these LTR mutations do not severely affect the replication of the full-length HIV- 1 constructs in tissue culture. Although NF-KB and Sp I binding sites in the'HIV-1 LTR are not absolutely required for viral replication and pathogenicity in vivo, a correlation of LTR: mutations with low viral load and prolonged asymptomatic state has been observed for isolates of long term survivors of HIV-1 infection. It was also found that several LTR deletion mutants containing a cytomeaalovir-us enhancer element were capable of attenuating HIV-1 (i.e. the mutants were capable of
infecting human lymphocytes with reduced cytopathic effects when the tat gene also was deleted). Instead of killing the entire culture, infection with these LTR and tat mutants led to rapid cell recovery and establishment of persistent infection. The replication efficiency was not markedly affected by these mutations. By mutating the tat gene, it was also found that the recombinant LTRs (CNtN-IE-HIV-LTR) exhibited increased basal levels of promoter activity which could support virus replication without Tat (Chang and Zhang. 1995: Robinson et al . 1995). These different HIV-1 mutant constructs were useful for the development of lentiviral vectors.
5.1.2.3 REPLICATION-COMPETENT Γ Γ-MINUS MUTANTS
LTR mutants with kB/Spl or Spi deletion and CMV-IE enhancer/promoter insertion have been shown to replicate with delayed kinetics in human lymphocyte culture, including primary PBLs and macrophages (Chang, 1993; Chang and Zhang 1995). As they still exhibit cytopathic effects in culture and thus may be pathogenic in vivo, these constructs are not safe for vaccine use in the present form.
The tat gene was also a target, as it is a gene that is essential for efficient HIV-1 replication. HIV-1 Tat has been implicated in the induction of Kaposi's sarcoma, repression of MHC Class I gene promoter, induction of functional unresponsiveness of T cells, modulation of monocyte function, induction of IL-10 expression, potentiating TNF-induced NF-KB activation and cytotoxicity. and sensitizing T cells to Fas-mediated apoptosis (Chang. 1996; Chirinule et al. 1995; Ensoli et al, 1994; Howeroft et al, 1993; Lafrenie et al. 1996; Westendoφ et al. 1995). To examine whether Tat could be dispensable during HIV-1 replication, a series of tat mutants (two stop-codon mutants, tatA and Tat-B, and a deletion mutant tat-C) were investigated (FIG. 13 A). In FIG. 13 A, the dashes (i.e., — ) indicate bases that are shared with the wild-type sequence, while slashes, (i.e., Ill I) indicate bases that are deleted in the mutant sequence, but are present in the wild-type sequence.
Mutant constructs containing both LTR and tat mutations were established. These LTR/tαt double mutants were generated using the LTR mutant constructs that exhibited enhanced transcriptional activity after inserting heterologous enhancer elements. The recombinant LTR (CMV-IE-HIV-LTR), which has been shown to exhibit increased basal level of promoter activity, can support HIV-1 replication without Tat (Chang, L.J. and
Zhang, C. Virol, 21 1 :157-169 [1995]: Robinson, D., et al, Gene Therap., 2:269-278 [1995]).
During the development. of the present invention, it was determined that the tat-C mutant is more defective than the tat-A and -B mutants, and the dl.Spl/CMV tat-B double mutant is more defective than the dl.Spl/CMV LTR mutant or the dl.Spi/CMV tat-A double mutant reported previously (Chang. L.J. and Zhang. C, Virol, 21 1 :157-169 [19951 ). The dl.Spl/CMV tot-B double mutant infects human lymphoid cell lines with delayed kinetics and exhibited reduced cytopathic effects.
In addition, this double mutant HIV-1 infected primary human PBLs poorly and replicated in primary macrophage culture with reduced kinetics. Based on these results, these already attenuated HIV-1 constructs. dl.Spl/CMV-tdt-B and dl.Spl/CMV tat-C, were chosen for HIV vector development.
5.1.2.4 ATTENUATED LTRJTAT DOUBLE MUTANTS The phenotypes of the LTRJtat mutants were further characterized in human lymphoid cell culture. The tatA or tatB LTR double mutants (Spl deleted and CNFV-IE enhancer inserted) infected human MT4 cells with slightly reduced cytopathic effects. Further, these mutants exhibited delayed replication kinetics when compared with wild-type HIV-1. On the other hand, when cells were infected with the tat-C LTR mutant (Spl /CMV mutant), the cytopathic effect was not so apparent and interestingly, the infected culture recovered rapidly and a persistent infection was established (See, "chr.l and "chr.2," in FIG. 2 and Table 2). In this table, as well as other Figures, descriptions, etc., "chr." indicates chronic infection, while the 1 and 2 indicate that the experiment was repeated twice (i.e., the " 1 " refers to the results of the first experiment, and the "2" refers to the results of the second experiment). In this table, the first column lists the cell line used and the virus used to infect the cells. For example. "MT4/mock" means that MT4 cells were tested without infection with HIV-1 virus (i.e., it was a control). "WT" refers to wild-type virus.
Immunofluorescent staining of cells in the persistent culture using an HIV-1 patient's sera showed that every cell was infected. Continuous output of attenuated infectious virus from these cultures was illustrated by a titration assay on CD4 HeLa cells, and the virus particles were visualized by electron microscopy (TEM and SEM). The persistently infected
culture produced large quantities of fully assembled HIV particles. Virions produced from these high producer cells are tαt-minus and exhibit greatly diminished infectivity. No cytopathic effect has been observed when they were ftulher passed onto human lymphocyte cultures. Interestingly, some cultures recovered from wild-type HIV-1 infection after long term passage also became persistently infected (See. Table 2. AA2/WT [chr.] and Molt3/WT [chr.]). It is possible that the latter persistent cultures were survivors of mutant HIV-1 infection (e.g., pr-minus).
TABLE 2 VIABILITY AND DOUBLING TIME OF TAT HIV-1 INFECTED CULTURES
5.1.2.5 HIV-1 LTR/Γ Γ/NEF TRIPLE MUTANTS
Prolonged asymptomatic survival of macaques infected with a «e/-deleted SIV strain SlVmac239 suggested that the nef gene is a pathogenesis factor (Kestier, H.W. et al, Cell 65:651-662 [1991]). Evidence to strongly support this suggestion came from studies of a
cohort of long term survivors infected with HIV- I through blood transfusion from a single donor in Australia. All the survivors were found to carry HIV-1 strains with multiple deletions in nef and in the U3 region of the 3' LTR (Deacon, N.J., et al, Science 270:988-991 [1995]). The LTR/tαt-minus HIV- 1 constructs were further modified by mutating the nef gene.
To generate nef mutations, site specific mutagenesis was performed in the nef VY to destroy its initiation codon. and a H/«dlll restriction site was generated (-AAGCTT-, nef-A mutant). Also, an additional stop codon was inserted in the nef ORF upstream of the polypurine tract (PPT) in the nef-A mutant, to generate a more defective «e/-minus mutant (nef-B mutant, see below). The nucleotide sequence of pNL4-3 (ΗIV-1 ) from 9001 to 9031 (WT) was 5'- CTCAGGTACCTTTAAGACCAATGACTTACAA-3' (SEQ ID NO:2). while the nef-B mutant sequence generated by site-specific mutagenesis was 5'- CTCAGGTACCTTTAAGACTCTAGATCTAGAA-3' (SEQ ID NO:3). FIG. 13B provides a schematic showing a portion of the wild-type HIV- I sequence, as well as the nef-B mutations (FIG. 13B: wild-type sequence provided in SEQ ID NO:5 and SEQ ID NO:6). The nef-A mutations are also shown in this FIG. 13B. As indicated in this FIG., the nef-A and nef-B mutations contain the same mutations in the sequence shown starting at base 8781 (i.e., SEQ ID NO:5 corresponds the the nef-A sequence and nef-B sequence for this stretch of bases). The nef-A sequence is the same as the wild-type sequence for the sequence shown starting at base 9001 (i.e., SEQ ID NO:6 represents the sequences for both wild-type and nef -A).
Since it is the non-syncytium-inducing, rather than the syncytium-inducing isolates of HIV-1 that are preferentially transmitted during primary infection, the T cell-tropic env gene of the LTR/tat/nef mutant was also substituted with a macrophage-tropic env (HIV ADA). A schematic diagram of these HIV-1 mutants is shown in FIG. 3. These infectious molecular clones are further modified and attenuated by mutating other accessory genes including vpr, vif and vpu. as well as the U3 transcriptional regulatory elements NF-AT, NRT-1, USF and TCF-la. A safe HIV-1 vector construct is developed from these attenuated HIV-1 LTR/tαt/nef mutant constructs with a total deletion of U3 except for the att site.
Additional packaging and transducing vectors derived from mutant HIV-1 LTR, tat and nef constructs established during the development of the present invention were generated and tested for vector function.
Based on the results of experiments with the HIV-1 vectors, HIV-2 and SIV vectors will be constructed using two molecular clones, HIV-2ROD -and SlVmac.
Continued experiments will establish an inducible packaging cell line using the tetracycline (TET-OFF) inducible system.
5.2 EXAMPLE 2 - REPLICATION-COMPETENT HIV-1 VECTORS CARRY ING HETEROLOCOUS FOREIGN GENES
Earlier reports of HIV-1 vector systems demonstrated difficulties in generating high vector titers This w as likely due to multiple modifications in the viral genome during vector construction and the lack of a full understanding of the packaging mechanisms of HIV-1. In addition, vector titers are often construct-dependent. To analyze the ability of HIV-1 vectors carrying heterologous genes to express them at high levels, several "replication-competent" HIV-1 vectors containing different foreign genes which were inserted in the e open reading frame (ORF) in the 3' end of the viral genome were constructed.
The nef gene has been shown to play an important role in viral pathogenesis (Du, Z. et al , Cell 82:665-674 [1995]; Jamieson, B.D., et al, J. Virol, 68:3478-3485 ( 19941 ). Thus, it was considered to be safer to delete the «e allele from the lentiviral vector system to produce useful vectors. Since the nef gene of HIV is dispensable for viral replication in tissue culture, and since the nef ORF does not overlap with other genes, a foreign gene can be inserted into the nef ORF without inactivating the virus.
FIG. 4 shows a comparison of the replication efficiencies of recombinant HIV-1 constructs carrying heterologous foreign genes. In these experiments, TE671 cells were transfected with plasmid DNA; 48 hours later, culture supernatants were used for the in vitro RT (reverse transcriptase) assay. Virus titer (i.e., transduction efficiency) was determined by infecting CD4 HeLa MAGI cells, and blue cell foci were counted under an inverted microscope after X-gal staining. The NIAGI cells carry an integrated LTR-IacZ gene which can be transactivated by transduced HIV-lTat (Kimpton, J. and Emerman, M., J Virol, 66:2232-2239.30 [1992]). The two scales in this FIG. are numerically identical.
In addition, reporter genes including human T cell receptor CD8, T cell costimulator B7-2 (B70). the bacterial hygromycin-B-phosphotransferase (hyg), neomycin- phosphotransferase (neo), xanthine-guanine phosphoribosyltransferase (gpt), puromycin- resistant gene, and histidinol dehydrogenase (hisd) with or without an internal promoter (SV40) were inserted into the nef OPF at the new H dll site or a downstream A7zol site in the nef-A mutant. These hetero logous ΗIV-1 vector constructs were assessed by transfecting human TE671 cells, and quantitatively measuring viral RT expression and transduction efficiencies on a human CD4 cell line. Transduction efficiency was determined by counting the blue nucleated cell foci after X-gal staining. Two independent transfections were done. Representative results are shown in FIG. 4 (the standard deviation is not shown). An insertion of up to 1.5 kb of nucleotide sequences, such as B70 and SV-his, seemed to have no effect on RT production. Furthermore, the infectivity of HIV-SVhis is as high as wild-type HIV- 1.
However, it was surprising to find that the nef-B mutation appeared to have an adverse effect on RT production (See, nefB tested in duplicate, FIG. 4). The cause of this adverse effect is unclear (/' e., it may have been caused by interference with packaging or reverse transcription of the RNA genome), although an understanding of this mechanism is not required in order to use the present invention. Several vectors derived from the nef-B mutant construct showed the same deficiency and thus were reconstructed. A good correlation between RT activity and virus titer was observed in this study, except for pHP-1. which is a packaging vector construct lacking the HIV-1 packaging signals (see below).
These early experiments led to some embodirtdfits of the methods of the present invention for manipulation of the HIV-1 genome for gene expression. For example, it appeared that HIV-1 can sustain extensive changes in the enhancer and promoter region. Indeed, the replacement of the entire U3, except for att, can be tolerated. Partial substitution of the intron region for the regulatory genes (tat and rev) in the env ORF with foreign sequences can affect the splicing efficiency of the singly-spliced messages, although the nearest splice acceptor site is almost I kb away (See e.g., Amendt, B.A., et al, Mol Cell Biol, 14:3960-3970 [1994]). These results suggested that: 1) a modified LTR with reduced homology to wild-type HIV-1 could be used in the vector design; and 2) deletion of the env sequence might interfere with expression of the tαt and rev regulatory genes. In HIV-1 vector
system, the env gene function may be deleted and replaced by the VSV-G envelope gene. As indicated herein, in some cases, it may be desirable to provide additional tat and rev functions for efficient Gag-Pol synthesis. Although an understanding of the mechanism(s) involved is not necessary in order to use the present invention, the study of heterologous replication competant HIV-1 constructs indicated that insertion of foreign sequences in the nef ORF is well tolerated and has minimal effects on viral replication. These advantages led to the development of various embodiments of the lentiviral vector systems of the present invention.
5.3 EXAMPLE 3 -- CONSTRUCTION OF HIV-1 PACKAGING VECTORS
In this Example, HIV-1 packaging and transducing vectors were constructed. Two packaging plasmids. "pHP-1 " and "pHP-VSVG," containing HIV- I env and VSV-G envelope gene respectively, were constructed. FIG. 7 is a structural diaaram of seven different pHP vector constructs, including pHP- 1 and PHP-VSVG. In this Example, attenuated HIV-1 constructs were modified to produce the "pHP-1 " expression vector capable of synthesizing all viral structural proteins, but lacking the packaging signal function. This vector included a strong promoter (in preferred embodiments, it is preferably not a native HIV- 1 LTR), the gag-,pol gene, the RRE element, the tαt and the rev gene. The RRE-Rev interaction is of great importance to the efficient synthesis of the Gag-Pol protein. This dependency may be compensated for if the INS's are deleted and RRE is replaced by a surrogate regulatory element such as the CTE of the Mason- Pfizer monkey virus.
Two approaches to designing the vectors were sidered, namely 1 ) dissecting down the wild-type genome while carefully monitoring vector titers following each modification step, and 2) starting with an over-simplified, inefficient vector construct and building back to restore wild-type function gradually. The goal was to achieve the best efficiency of vector production yet have the vector remain replication-defective to minimize the chance of generating a replication-competent recombinant HIV-1 (RC-HIV). To achieve this, the expression construct pHP-1. which contained a modified 5'HIV-l LTR, a novel major splice donor site derived from RSV, the entire gag-, pol-env, vif, vpr, vpu, tat and rev genes, a
selectable gpt marker gene, and an SV40 polyadenylation signal as shown in FIG. 5 was developed.
The wild type HIV-1 genome contains genetic elements in the 5' to 3' order:
5'LTR(U3RU5)-PBS-Psi-SD-gαg-/? /-v7 -v/7r-t«/-rev-vpu-e«v-nef-PPT-3'LTR
(U3RU5), and the pHP construct contains from 5' to 3': a chimeric CMV-TAR promoter sequence-gαg-/rø/-tαt-rev-PPT-SV40 polya signal.
pHP- I lacks the native HIV- I U3 TATA box. the primer binding site (PBS), polypu ine tract
(PPT). 3' LTR and most of the untranslated 5' leader sequences including the conventional retroviral packaging signal (T) and the major HIV-1 splice donor (SD) site. pHP-1 contains all HIV structural and accessory genes except for the nef gene and thus is capable of expressing the vast majority of the viral proteins, and also contains the bacterial gpt gene. pHP- I provides a provirus capable of mimicking HIV- I infection in terms of the viral proteins expressed yet this virus cannot be packaged into viral particles.
Further mutations introduced by derivatives of the pFIP- 1 provirus, including deletion in the env and in the 5'LTR, vpr, vi, and vpu, greatly reduce the possibility that wild-type HIV will be produced by recombination. Thus. pHP-1 and its derivatives provide excellent HIV packaging vectors. Examples of th pHP-derived packaging vectors include: pHP- dl.Vpr, pHP-Vpr/ala/leu. PHP-dl.eπvΛ pu I, and pHP-dl.emVVpu II. pHP-1 was constructed as follows. First, the Tat-responsive enhancer promoter CMV- TATA-TAR fragment (approximately 400 bp was isolated from dl.kB/Spl-CMV-TATA- TAR HIV (Chang, et al, J. Virol. 61:1 '43 [1993]) by Bbφl-Hindlll digestion, and cloned into EcoRN-BamHl digested pSP72 (Promega) via a linker providing Hindlll and BamHI cohesive sites which contains a modified gag AUG with Kozak translation initiation context and a major splice donor site of Rous sarcoma virus. This linker was formed by annealing the following oligonucleotides:
5 ' -AGCTTGGTCGCCCGGTGGATCAAGACCGOTAGCCGTCATAAAGGTGATTTCGTCG- 3 '
( SEQ I D NO : 9) and
5 ' -GATCCGA33AAATC.-. CTTTATGACGGCTACCGOTCTTGATCCACCGGGCGACCA- 3 ' ( SEQ I D NC : 10 ) .
This first subelone was called pSP-CMV-TAR-SD. Secondly, the gag coding sequence was obtained by PCR from pNL4-3 (a full-length
HIV-1 plasmid) using a 5'primer:
5'-CGGGATCCACCATGGGTGCGAGAGCGTC-3' (SEQ ID NO:l l) and a 3' primer downstream of the Sphl site in the gag gene:
5'-ATCCTATTTGTTCCTGAAGG-3' (SEQ ID NO: 12). The PCR product was digested with BamRl-Sphl (~660 bp) and this fragment was ligated with BamHl-Sphl digested pSP-CMV-TAR-SD to obtain pSP-CMV-TAR-SD-dl.gαg.
Next, the poly-A minus subelone pHP-dl.pA was constructed by ligating the following three fragments: al 112 bp Hpal-Sphl fragment isolated from pSP-CMV-TAR-SD- dl.gαg (contains the promoter-TAR-SD-di.gag), a 7922 bp Sphl-Xhol fragment (dl.gag-pol- env-gpt) of pNLgpt. and a plasmid vector backbone provided by EcoRV -Xhol digested pBS-
KS(-) (Stratagene).
Lastly . pHP- I was made by the following ligation: Notl-Xhol (9059 bp) of pHP- di.pA containing dl.CNfV-TATA-TAR-SD-g<3g-po/-e/?v-gpt, a 422 bp poly-A site from Xhol-Pstl digested pREP9 (Invitrogen), and Notl-Pstl digested pBS-KS(-). The sequence of pHP-1 (12.479 kb) is provided in SEQ ID NO:13; this sequence begins at the promoter of the aλf-BbrΫl site from pNL4-3 (an HIV clone available from the AIDS Research and Reference Reagent Program; the sequence of this recombinant clone is shown in Genbank Accession No. M19921). Additional mutations of pHP-1 to generate pHP-ldl2 and pHPI- dl.28 are described above (See also, FIG. 5). Several additional HP constructs were also made ("pHP-VSVG," "pHP-CMV,"
"pHP-EF," "pHP-CMVdel.TAR/SD,", "pHP-CMV-EFlα-intron", "PHP-dl.Vpr", "pHP- Vpr/ala/leu". "pHP-dl.emWpu I", and "pHP-di.e«v/Vpu II"), each with additional changes (See, FIG. 7). pHP-VSVG was derived from pHP-1, with the HIV-1 env gene being replaced by the VSV-G gene, and with wild-type vpr and tαt, or the vpr and tαt genes mutated by site- specific mutagenesis.
pHP-CMV was derived from pHP-1 with the promoter being replaced by the cytomegalovirus immediate early promoter (CMV-IE) and the tat, rev. env. vpr and vpu deleted. pHP-CMVdel.TAR/SD was derived from pHP-CMV, with the TAR and RSV SD deleted. In other words, this construct lacks any major SD site. pHP-CMV-EF lα-intron was derived from pHP-CMVdel.TAR/SD. with an insertion of the EFlα-intron between the promoter and the Gag AUG. pHP-EF was derived from PHP-CMV by replacing the CMV-IE promoter-enhancer and the synthetic SD site with the human elongation factor Iα (EFlα )'s promoter and enhancer-containing intron (the latter being of course proceeded by a splice donor site). The intron-containing EFlα has been shown to be a stronger promoter than the CW-IE promoter.
The TAR sequence was also deleted. It also contains a poliovirus-derived internal ribosomal entry site (IRES) and the vpr gene. The expression of Vpr may increase the vector transduction efficiency in non-dividing cultures. These constructs were tested for their expression of HIV-1 proteins. Both packaging constructs (i.e. pHP-1 and pHP-VSVG) used a recombinant CMV/HIV-TAR as promoter and a synthetic major splice-donor site. No sequence homology was observed with the HIV-1 genome between TAR (in the 5' end of the RNA) and the gag AUG in these two constructs.
A BamHI site was generated near the gag AUG for the puφose of inserting recombinant HIV-2 and SIV gag-pol sequences in subsequent experiments.
The pHP-VSVG construct with vpr and tat mutations lacks vpr and tat genes, and the VSV-G gene is substituted for the env gene exactly at the env AUG by PCR mutagenesis. These two constructs were the first two packaging plasmids tested.
The construction of these pHP-1 derivatives is described in greater detail below. The three pHP-CMV derivatives were tested, and found to be inefficient in synthesizing HIV proteins, indicating that the pHP-1, pHP-VSVG and pHP-1 dl derivatives are the preferred embodiments of the efficient HIV vector system of the present invention.
5.3.1 PHP-VSVG This clone was made to delete the HIV-1 env gene and replace it with the VSV-G gene, as well as delete the HIV-1 vpr and tat genes. It was constructed by combining the
following four pieces of DNA fragments: 1 ) the recombinant LTR (di.kB/Spl-CMV-TATA- HIV-TAR) gag-pol from Notl to EcoRI fragment of pHP-1 ; 2) a fragment from HIV-1 with deletion in the C-terrninal of Vpr and the Ν-terminal of Tat by PCR using the following two primers: 5'-TAAGAATTCTAGTAGGTTGCTTTCATTGCC-3' (SΕQ ID NO: 14), and
3'-CTTCTCCTTCACTCTCGAGTGATCACTGTCTTCTGCTCTTTC-3' (SΕQ ID
NO: 15), with the second sequence encompassing the env AUG with a new Xhol and a Bcil site 3) the VSV-G gene fragment cut by Sail-Xhal from pBS- VSV-G (obtained from Tom
Hobman of the University of Alberta); and 4) a deleted e«v-gpt-SVpA and plasmid vector backbone from Nhel-Notl digested pHP- 1.
5.3.2 PHP-CMV
This clone was derived from pHP-1 , with the 5'recombinant LTR replaced by a CMV-IΕ enhancer-promoter and the entire env, tat, vpu, rev, vpr, nef deleted, but with the vif gene remaining intact. This clone was constructed by ligation of the following 3 pieces of DNA: 1) the vector pcDNA3.IZeo(+) from Invitrogen cut with Nhel-Xhol; 2) the TARISD- gag-pol from pHP-1 digested with Xbal-EcoRI; and 3) the RRΕ element from pBS-RRΕ digested with EcoRf-Xhol. PBS-RRΕ was constructed by ligating Bg/IIl (nt. 7611) to Hinlll (nt. 8131) of pNL4-3 of HIV-1 with /II-H dIII digested pBS-ΕF. pBS-ΕF was from the PCRed FF1 enhancer promoter cloned into pBS(-).
5.3.3 PHP-CMV-DΕL.TAR SD
This clone is the same as PHP-CMV except that the 5' TAR and splice donor site are deleted. This construction was made by ligating the following two fragments: 1) a 702 bp fragment of Mlul-BamHl digested pcDNA3.I Zeo(+) containing the CMV enhancer; and 2) the vector containing Mlul-BamHl digested pHP-CMV which has deleted TAR and contains the RSV splice donor site.
5.3.4 PHP-CMV-ΕF 1 α-iNTRON - INTRON This clone is similar to pHP-CMV-del.TAR/SD but with an intron from human ΕF-1 a gene inserted between the CMV promoter and the gag AUG. It was made by ligating the
following three DNA fragments: 1) pHP-1 BamHl-EcoRI fragment containing gag-pol and vif; 2) the wI-EcoRl of pcDNAZeonlacZ-RRΕ containing the vector backbone of pcDNA3.IZeo(+), HIV-1 RRΕ and part of the CMV promoter; and 3) the rest of the CMV enhancer promoter was obtained from BamHl-mlul digested pcDNAZeoHGHP2ΕF. a pcDNAZeo3.1 (+) vector containing EFlα intron and the human growth hormone gene. The hGH cloned sequence is available from GenBank (see co-pending U.S. Patent Appl. SN 08/848,760 for additional information regarding this construct).
5.3.5 PHP-1 DL2 AND PHP-1 DL28 To further mutate pHP- 1 for safety reasons (as discussed below), portions of the env gene were deleted by BaTΛ excision. To generate HIV-1 env deletions, pHP-1 was digested at the unique restriction enzyme site Nhel in the env gene, and treated with BaBl exonuclease for 1, 2 or 5 min. The diaested product was self-ligated after T4 DNA polymerase treatment. The self-ligated plasmid DNA was then transformed into competent E. coli DHSα and the plasmid clones containing env deletion were selected. More than six env- deleted pHP-1 clones of different lengths were selected and sequenced, as known in the art. pHP-ldl.2 and pHP-ldl.28 have 2 and 28 nucteotide deletions in the env gene respectively (see FIG. 5). pHP-ldl.28 has been further modified to produce various derivatives. First, the vpr gene was mutated by site-specific mutagenesis so it retains the nuclear localization function but loses its cell cycle arrest function. Specifically, a mutation was made at amino acid 30, changing from Ala to Leu, as described (Mahalingam et al, 1997).
The env/vpu was also mutated by site-specific mutagenesis to delete the env initiation 20 AUG codon and part of the vpu reading frame. These mutations were first made individually and then combined.
Construction of GI (generation one, deletion of vpu, vif, vpr) and G2 (deletion of tαt, rev) pHP vectors based on pHP-dl.28 (nef deleted and env partially deleted):
(a) Construction of env AUG and vpu deletion mutant pHP-dl.erøv/vpu I and pHP- dl. env/vpu
(b) The mutant I has a long deletion from nt 6195 at the vpu amino acid codon 45 to nt. 6330 at the 38th amino acid codon of env gene and a stop codon TAA inserted in the sequence. The primer used was a forward primer:
5'-GTTAATTGATAGACTAGTCTAATATGGGGTACCTG-3' (SEQ ID NO:XX).
The mutant II has a small deletion from nt. 6216 at vpu amino acid codon 52 to nt. 6237 at vpu amino acid codon 59 and env amino acid codon 6 which also has a stop codon mutation TAA at the vpu amino acid codon 50. Note that although these mutations are GI mutations, they are made into the G2 vector pHP-di.28 backbone. Construction of vpr mutants of pHP: tnvo vpr mutants were made, one with frameshift mutation which inactivate the entire vpr function and the other with one amino acid substitution which inactive the cell cycle arrest function of vpr but not the nuclear localization function which can assist transduction of non-dividing cells: pHP-dl.vpr is a frame-shift mutant which was made by EcoRI digestion at nt. 5745, near vpr amino acid codon #62, and resulted in a 4 bp insertion which caused a frameshift. pHP-vpr/ala leu: this is a point mutation which has changed alanine to leucine at a.a. #30. This mutation deleted the cell cycle arrest function but not the nuclear localization function of vpr as reported before (Zhang, #3492, [1997]; Mahalingam, #3791, [1997]). The primer used for mutagenesis is: 5 '-CCTAGGAAAATGTCTAACTAGTTCACTCTTAAGTTCCTC-3' (SEQ ID
NO:XX)
Note that although these mutations are GI mutations, they were made with the G2 vector pHP-dl.28 backbone.
Combination of dl.env/vpu I and di.emVvpull with vpr/ala/leu mutations to generate pHP-vpr/em'/vpul and pHP-vpr/eπv/vpu 11. These two combination mutants were made by ligating the two designated mutant fragments, Spel (in gag) to EcoRI (in vpr) containing the vpr mutation, and EcoRI to Ndel (in env) containing the eπv/vpu mutation, into Ndel and
Spel digested pHP-di.28.
Note that although these mutations are GI mutations, they are made into the G2 vector pHP-dl.28 backbone.
4. Construction of a G2 vector, pHP-dl.Ndel, which has deletion of the following genes, viflvpr/tat/rev/env/vpu : this mutant was made by deleting nt. 5122 to nt. 6399 via Ndel digestion of pHP-dl.28, and resulted in a packaging vector with all the accessory gene functions deleted and the env gene partially deleted. This Ndel-Ndel deletion also included the 3' splice sites SA4. SA5, SA6 and SA7, used for the syntheses of vpr, tat. rev, vpu and env.
5.4 EXAMPLE 4 - CONSTRUCTION OF HIV-1 TRANSDUCING VECTORS (TV)
Two families of transducing vectors were constructed. In the pTVψ vectors, the major packaging signal was modified relative to the source HIV-1 signal. In the pTVΔ vectors, the source major packaging signal was used.
FIG. 8 provides a diagram of six HIV-1 transducing vectors, in which the vector backbone is derived from pNL4-3 and different LTRS. The IRES element shown in FIG. 8 was derived from poliovirus, which could allow bicistronic gene expression. To engineer a packaging signal for the construction of HIV-1 transducing vectors
(TV), an artificial HIV-lψ sequence using four synthetic oligonucleotides was synthesized, which comprised sequences between the PBS and the gag AUG (referred to as "ψlOO" or "PAK140" ) and sequences extending into the gag ORF (referred to as "ψl40" or "PAK140"). These synthetic HIV-lψ sequences contained a mutated SD site (three nucleotides changed in PAKIOO and PAK140, GAGTA ->CATTC) and a mutated gag AUG (Hindlll and BamHI sites inserted upstream of gag AUG in both; PAKIOO stopped just upstream of gag AUG; PAK 140 changed gag AUG to UAG and second codon from GGT to GCC) to avoid possible adverse effects in gene expression. PAKIOO and PAK 140 both started at nt 690 of provirus, i.e.. 5' base of U3=l. The synthetic ψ signals were cloned into the pTVψ vector as shown in FIG. 8, which is comprised of two recombinant LTRs ("dl.kB-CMV/HIV-TAR"), the PBS and 5' leader sequences, an SV40-driven neo resistance gene, and the 3' PPT.
The packaging efficiencies of pTVψlOO and pTVψ 1 40 (FIG. 8. constructs 1 and 2) were tested in a co-transfection experiment. HeLa cells were transfected with pHP-1 and pTVψlOO or pTVψl40 and 48 hours later, the culture supernatants were harvested and used to transduce CD4 HeLa cells (not VSV-0 pseudotyped). G418 resistant colonies were counted 10 days later. As a control, HeLa cells were transfected with wild-type HIV-1 DNA;
48 hours later, the culture supernatant was used to infect CD4 HeLa cells. The titer of the wild-type HIV-1 was determined by a sensitive immunohistochemical staining method using anti-Gag p24 mAb as described by Chang and Zhang (1995). Results of this study showed that both pTV ψ and pTVψl40 were packaged at a very low efficiency (approximately 3 logs of magnitude less than the wild-type HIV-1).
This result indicated that additional HIV- I sequences are needed to improve the packaging function of pTVψl OO and pTVψl40. Therefore, more HIV-1 sequences. including an additional gag sequence and an RRE element, were cloned into pTVψ l40. One such example is shown in FIG. 9A (pTVψ+CMV-nlacZ-hyg). Again, the pTVψ+ was not packaged efficiently, indicating the splice donor site and Gag AUG mutations in pTVψlOO and pTVψl40 are detrimental to HIV packaging. While the tested pTVψs cannot be used as efficient transducing vectors. pTVψs can be efficiently packaged and transduced, as shown below.
Thus, site-specific mutagenesis was performed to change 1-2 nucleotides in the splice donor site, and the Gag AUG in pTVAs using primers:
5'-GCGGCGACTGGGGAGGACGCCAA-3* (SEQ ID NO:7); and 5'-GAAGGAGAGAGTTGGGTGCGAG-3' (SEQ ID NO:8), to generate PTVψSM vectors.
It is desirable to avoid sequence homology betweenn the packaging construct and the transducing vector construct so as to reduce the probability of recombination. However, cotransfection with additional accessory genes such as vpr, nef and vpu may also help to increase the vector titer and the transduction efficiency. Inclusion of accessory genes in the transducing vector does not increase the probability of recombination, provided that such genes are omitted from the packaging vectors. The homology between the preferred pHP and pTV constructs is sufficiently low so that recombination was not detected.
In order to generate a replication-competent HIV-1, the major SD site, the gag AUG and the env sequences must be restored, because they are deleted from the modified pHP and pTV constructs.
In an alternative approach for the construction of an efficient transducing vector the wild- type genome was gradually deleted (pTVΔ). In this embodiment, the two replication- competent HIV-1 vectors, "HIV-1 vectors" and "HIV-1-SVhyg" (see FIG. 4) were used as a
starting point. These two constructs are nef-minus. and exhibited up to 50-70% of the wild- type HIV-1 replication efficiency. A deletion was made starting from the middle of the gag OR.F to the middle of the env ORF. This did not delete the RRE element.
First. pTVΔSVneo was created by digesting PNL-SV with Nhel (with a site located in the middle of the env gene), and Spel (with a site in the middle of the gag gene), and then self- ligated to delete the gag-pol-env. and vif vpti, vpr, tat, and rev genes. pNLSV was created by inserting SVneo in between the nef AUG and the Xhol site in the N-terminus of nef. pTVACMVnlacZ was made by digesting pTVASVneo with Xhol-Kpnl as the vector, which deleted SVneo and part of the nef sequences near the S' end of the PPT of HIV- 1 , the product was then ligated with a Sall-Kpnl fragment containing CW-niacZ sequence from pcDNAzeo-nlacZ. pcDNAzeo-nlacZ was generated by inserting nlacZ of pSP72nlacZ into pcDNA3.1zeo(+).
The two deletion vectors. "pTΔ SVneo" and "pTVΔSVhyg," (see FIG. 8 constructs 3 and 4) were examined for their transduction efficiencies in cotransfection studies. Three additional pTVΔ vectors were also constructed, each containing a different reporter gene: CMV-GFP (green fluorescent protein, pTVΔCMV-GFP), CMV-nlacZ (pTVΔCMV-nlacZ) and CMV-nLacZ-hyg (pTVΔCMV-nlacZ-hyg), as illustrated in FIG. 8 (see FIG. 8 constructs 5 and 6. as well as FIG. 9B).
5.5 EXAMPLE 5 - DETECTION OF SYNTHESIS OF HIV-1 PROTEINS BY PACKAGING CELL LINES 5.5.1 WESTERN BLOT ANALYSIS
In this Example, Western analyses of HIV-1 proteins in HeLa cells transfected with various vector constructs were tested. In this Example, cell lysates were prepared and analyzed by Westem blotting and compared with a wild-type HIV-1 construct (pNL4-3), in order to determine the level of viral proteins synthesized by pHP-1 and PHP-VSVG (with and without Tat), in comparison with wild-type HIV-1. In this Example, the Western blots were performed using serum obtained from an HIV-infected individual, and methods known in the art (See e.g., Ausubel et al. 1992). FIG. 6 shows the results of the Western analyses. In FIG. 6, lane 1 contains cell lysate from MT4 cells infected with HIV-1; lane 2 contains control HeLa cell lysate; lane 3
contains lysate from HeLa cells with pHP-1 ; lane 4 contains wild-type HIV-1 pNL4-3 cell lysates; lanes 5 and 6 contain pHP-VSVG-transfected cell lysates; lane 7 contains pHP- VSVG+Tat cell lysate; and lane 8 contains pHP-VSVG+Vpr cell lysate. As indicated in FIG. 3, the results showed that the level of viral proteins synthesized by pHP- 1 was similar to that of the wild-type pNL4-3 (See, lanes 3 and 4. respectively, in FIG. 6). Similar results were obtained when pHP.1.dl constructs were used.
These results indicated that in the absence of Tat, the recombinant LTR of pHP- VSVG is inactive. Thus, it is likely that the TAR element in the LTR down-regulates transcriptional elongation. These results led to the generation of an inducible packaging cell line using the PHP-VSVG construct as described in Example 4.
5.5.2 REVERSE TRANSCRIPTASE ACTIVITY
Analysis of reverse transcriptase (RT) activity in the transfected culture supernatants indicated that the level of active RT production was reduced 40% for pHP-1 compared with the wild-type construct pNL4-3. RT synthesis of pHP v.v wild type HIV-1NL4-3.
*RT. two independent transfection and assay results are shown; the RT background activity of 50 cpm/ 1 1 was not subtracted.
The expression of Gag-Pol function indicates that tat and rev are functional. Thus, the artificially engineered splice donor (SD) site in the pHP-1 construct, which is unrelated to
HIV sequences, works like the wild-type SD site (i.e. allowing partition of spliced and unspliced mRNAs into the cytoplasm).
The packaging Vector pHP-dl.28 expressed RT at 50-90%) of the wild type level, indicating that the mutations in pHP-dl did not affect the synthesis and function of Gag-Pol. Analyses of RNA expression and packaging function by pHP-CMV and pHP-EF were performed in order to compare these vectors directly with the wild-type HIV-1. These experiments showed that pHP-CMV and pHP-EF do not express Gag-Pol protiens at high efficiencies, indicating that the pHP-1 -derived vectors have important viral sequences that are necessary for efficient vector production. pHP-VSVG did not express HIV-1 proteins unless the Tat transactivating protein is also present (see FIG. 6, lane 6 vs. 1). Thus, although expression of VSV-G and Gag may be cytotoxic, an inducible packaging cell line could be established using pHP-VSVG without a tat plasmid.
5.5.3 PHEF-VSVG Human-elongation factor 1 alpha promoter driven VSV-G envelope and pHEF-A- eπv:EFla promoter driven amphotropic MLV envelope, were also constructed and shown to express high levels of envelope proteins; better than the CMV-IE promoter driven construct.
It should also be noted that overexpression of Gag-Pol may not increase the vector titer because earlier studies have shown that overexpression of Gag-Pol induces protease activation and prevents virus assembly and budding (Karacostas et al, 1993; Park and Morrow, 1991 ). The present example describes vectors that produce measurable amounts of Gag-Pol (e.g.. pHP-1 , pHP-ldel, and pHP-VSVG), as well as vectors that do not express detectable amounts of Gag-Pol (e.g., pHP-CMV and its derivatives). The latter require ftu- ther mutation to be useful as vectors.
5.6 EXAMPLE 6 -- REQUIREMENT FOR HIV-1 TAT FOR EFFICIENT G AL-POL PROTEIN PROCESSING
In this Example, the requirement of Tat for efficient Gag-Pol processing and HIV- vector production in certain packaging systems was demonstrated.
5.6.1 WESTERN ANALYSIS OF TAT' AND TAT' HIV-1 PARTICLES AND INFECTED CELLS
Virus pellets ("P") and cell lysates ("L") were prepared from Tat+ (tat WT) and Taf (tat-B and tat-C) virus-infected cells, and the protein contents were separated by a 10% SDS protein gel. and detected in Western analysis using AIDS patient's serum. The signals were amplified using the Amersham ECL chemiluminescence kit.
In FIG. 1 1. the first three lanes (1-3) indicate the results for mock-infected cells (lane 1), and virus pellets harvested from MT4 cells (lanes 2 and 3 contain viral pellet from cells chronically infected with tatC), and AA2 cells (i.e. CD4+ hybridoma human T and B cells, available from the AIDS Research and Reference Reagent Program) lane 4 contains viral pellet harvested from AA2 cells acutely infected with tatB. Cell lysates and pelleted particles of Tat+ viruses grown in PBL and Molt3 cells, are shown for comparison on lanes 5 and 6 of FIG. 11. In this FIG., protein markers are shown on the left and representative structure proteins of HIV-1 are indicated on the right.
5.6.2 GAG PROCESSING DEFICIENCY OF TAT-MINUS HIV-1 DEMONSTRATED BY METABOLIC LABELING OF CHRONICALLY INFECTED CELLS
WT or t t-minus HIV-1 chronically infected cultures were metabolically labeled with
H- leucine overnight, immunoprecipitated with pooled HIV patient sera, and analyzed by
SDA- PAGE (10%). The relative ratio of Gag p55:p24 is shown at the bottom. Processing of the envelope gplόl to gpl20 was not significantly different between different samples. The
3H- labeled protein bands were quantified using a phosphoimager (BAS I 000). The results are shown in FIG. 14: Lane 1. control MT4; lane 2 & 3, MT4 chronically infected with dl.Spl
CMV tαt-C; lane 4. MT4 acutely infected with WT HIV-1 ; lane 5, C8166 chronically infected with WT HIV-1 ; lane 6, MT4 chronically infected with dl.Spl CMV tat-B; lane 7, AA2 chronically infected with dl.Spl CMV tαt-C.
5.6.3 CELLS CHRONICALLY INFECTED WITH TAT-MINUS HIV-1 ARE DEFICIENT IN GAG PROTEIN PROCESSING DEMONSTRATED BY PULSE-CHASE METABOLIC LABELING The same number of viable cells (3 x 106) was used in each lane of a 10% SDS-
PAGE gel system. Cells were labeled with medium containing bands for HIV-1 Env gpl20,
and Gag p55 and p24. A Fuji phosphoimager was used for quantitation of Gag p55 and p24 of WT- infected MT4 and tαt-C chronic high producer. In FIG. 10, the resultant decrease of p55 and increase of p24 (p55. p24 / pulse-labeled p55) with time (P, 2, 4, 6, 8 hours) were shown 5 and plotted. In FIG. 10, the solid curves demonstrate efficient processing of p55 of HIVNL4-3 with steady increase of p24 and decrease of p55; the dashed curves demonstrate that the amounts of p55 and p24 are not sicnificantly changed with time in the tαt-C high producer cells, indicating a deficiency in Gag processing.
5.6.4 TAT ENHANCES GAG PROCESSING IN HELA CELLS HeLa cells were transfected with plasmid DNA encoding HIV-1 Gag, Rev, Tat,
HTLV Tax Rex, SIV Tat, or HIV Tat exon I as indicated in FIG. 16. The results shown in FIG. 16, clearly demonstrate that Tat enhances p55 to p24 Gag processing. The effect of Tat on Gag processing is TAR-independent as GagTAR-construct which has TAR deletion is also sensitive to this Tat effect. This function of Tat resides in the exon 1 that can be partially restored by SIVTat and HTLV Tax/Rex.
5.6.5 TAT ENHANCES GAG PROCESSING FROM THE PHP- VSV-G
PACKAGING CONSTRUCT
TE671 cells were transfected with plasmids as described above. Cell lysates were harvested 24 hours after DNA removal and analyzed by SDS-PAGE and Western blotting as described using anti-p24 MAb. The result indicated that Gag processing is enhanced by the presence of Tat (See. lane 2 vs. lane 3. and lane 5 vs. lane 6 in FIG. 16).
5.7 EXAMPLE 7 - GENERATION OF AN INDUCIBLE PACKAGING CELL LINE In this Example, an inducible packaging cell line was generated using the pHP-
VSVG, and its derivative construct. First, PHP-VSVG was linearized and transfected into human TE671 cells by electroporation, together with a selective marker. After selection, individual cell clones were tested for Gag-Pol expression by direct extracellular RT assay in the presence or absence of a transfected tat plasmid. The expression of VSV-G protein was detected by immunohistochemical staining.
Briefly, the PHP-VSVG linearized by digestion with Notf, and transfected into the TE671 cells along with pSV2-neo (i.e., with G-418 as the selectable marker). Transfection was accomplished by electroporation, using methods known in the art. Transfected cells were grown in 1 mg/ml of G418 culture in DMEM containing 10% FBS. The induced gag-pol Gag-Pol expression was then determined by direct extracellular RT assays with and without transfected tat plasmid. HIV-1 Gag and RT expression were detected by p24 antigen ELISK or RT (see co-pending U.S. Patent Appl. Ser. No. 08/791,994 and 08/838,702; Chang and Zhang, 1995; Chang et al, 1993).
The expression of Gag-Pol in this inducible cell line still requires Tat function. To make a user-friendly packaging cell line, vpr and tat genes can also be expressed by an inducible promoter. The vpr gene is included because of its function in promoting transduction of nondividing cells. Vpr is a virion-associated protein, and the vpr gene is therefore assigned to the packaging vector so that equivalents of Vpr, like those of Gag, Pol and Env, are produced only in the packaging cell line. A tetracycline-inducible expression vector (a TET-OFF system, suppression of expression in the presence of tetracycline or doxycycline) has been chosen for this puφose. An inducible tat-vpr expression vector has been constructed into the pcDNA3.1/Zeo plasmid with genes arranged in the following order:
-tetOP-tαt-IRES-vpr-IRES-tetR.VP 16-S VpA-(inverted tk-zeo-pA).
Studies of this construct showed co-expression of Tat and Vpr in the absence of tetracycline or doxycycline, indicating that the two internal ribosomal entry sites (IRES) are functional. However, even in the presence of tetracycline or doxycycline, this inducible construct still expresses Tat function, indicating a leaky expression of the tetR.VPlό. As a result, this construct was only used for coexpression of Tat and Vpr in the co-transfection experiments.
A second construct, -tetOP-tot-P2-vpr-SVpA-(inverted tk-zeo-pA), which is up- regulated by a separate tetR.VPlό expression plasmid, has been constructed and used to generate an inducible cell line. tetOP-tαt-P2-vpr-P2-tetR-VP16-SVpA-(inverted tk-zeo-pA) is a clone that expresses HIV-1 Tat and Vpr and the tet tTA operon inducer tetR-VP16 which was made by ligation of the following fragments: tetOP, HIV-1 Tat, internal ribosomal entry site (IRES) P2, HIV- 1 Vpr, IRES P2, tetR-VP16, and the vector pREP9 with EBNA1 gene
sequence deleted. The two tTA plasmids were obtainable from Display Systems Biotechnology, Inc. (now distributed by Clontech). This clone is auto-inducible by the removal of tetracycline or doxycycline (2-10 μg/ml) from the culture media (a Tet-OFF system)(Gossen and Bujard, 1992). As these plasmids use different selective markers (neo, zeo, and hyg) it was possible to co-select them in the same cell. However, a large number of cell clones had to be screened before a stable inducible packaging cell line could be established.
5.8 EXAMPLE 8 -- INTERNAL CMV-IE IN PTVACMVNLACZ PROMOTER EXHIBITS HIGHER PROMOTER ACTIVITY THAN NATIVE CMV-IE
In this Example, the expression of the reporter lacZ gene from the pTV-ΔCMVnlacZ was compared with pcDNAnlacZ (i.e., CMV-IE promoter-driven), 48 hours after transfection of TE671 cells. TE671 cells were transfected with 5 μg of pcDNA3-nlacZ or pTVΔCMVnlacZ, as described above. Following transfection and growth, cells were fixed and stained for β-alactosidase actvity, as described below.
The β-galactosidase activity was detected by the following protocol as published by Kimpton and Emerrnan (Kimpton, J. and Emerman, M., J Virol, 66:2232-2239 [1992]). Briefly, cells were fixed in culture plate at room temperature, with 1% formaldehyde (1.33 ml of 37.6% for final 50 ml) and -0.2% glutaraidehyde (0.4 ml of 25% for final 50 ml) in PBS for 5 minutes. The cells were then washed three times with PBS, and incubated with 500 μl ddH:0 containing 4 mM potassium ferrocyanide (100 μl of 0.4 M for final 10 ml), 4 mM potassium ferricyanide (100 μl of 0.4 M), 2 mM MgCl2 (20 μl 1 M), 0.4 mg/ml X-Gal (200 μl of 20 mg/ml) at 37°C for 50 min to several hours. The blue-staining (i.e. β- galactosidase positive) cells were counted under an inverted microscope. These results indicated greater expression by the pTVΔCMVnlacZ vector, as compared with the pcDNA3- nlacZ. Table 3 shows the results, with more "+" indicating the presence of a relatively greater number of blue-staining cells.
TABLE 3 Β-GALACTOSIDASE ACTIVITY
5.9 EXAMPLE 9 - PRODUCTION EFFICIENCY OF TRANSDUCING VECTORS 5.9.1 PRODI CTION EFFICIENCY OF VSV-G PSEZIDOTYPED VECTORS
In this Example. TE671 cells were transfected with certain packaging and transducing vectors, as identified in the table below.
In this study . VSV-G pseudotyped vectors were produced and the target cells were CD4-minus human cell lines. pHP-VSVG was co-transfected with a PTVΔ plasmid and a tat plasmid (pCEP4tat) into TE671 cells. Culture supernatant was harvested 48 hours later. Tat was included to transactivate both pHP-VSVG and pTVΔ. The production of virus was confirmed by RT assay, and expression of HIV-1 p24 and VSV-G was confirmed by immunohistochemical staining. Virus produced from the transfected cells were harvested without further concentration, and used to infect TE671 cells. After selection with either G418 or hygromycin for 7-10 days, cell colonies were counted under an inverted microscope. The VSV-G pseudoty ped pTVΔSVneo and pTVΔSVhyg both produced transducing titers up 10 /ml without further concentration. This titer was increased to lOVml without concentration, when pHP-dl.2 or pHP-dl.28 were co-transfected with pHEP-VSV-G. This result indicated that PHP-VSVG does not function efficiently. Culture supernatants were harvested 24 hours after removal of transfection solution.
HIV RT activity was detected by an in vitro RT assay and vector titers were determined by transduction and beta-galactosidase assay of TE671 cells 48 hours later.
TE671 cells were also transfected (as described above) with the packaging vector pHP-1 or an ercv-deletion mutant pHP-ldl.2, and compared to the wild-type HIV-1 molecular clone pNL4-3 for their packaging efficiencies. Culture supernatants were collected for RT
assay and for vector titering after 48 hours. The vectors were pseudotyped with the VSV-G envelope and titered on TE671 cells. X-gal stained blue cells were counted after 48 hours.
TABLE 4 PRODUCTION OF HI-TITER HIV-1 DERIVED VECTORS
These results indicated that pHP-1 or pHP-ldl.2 could produce HIV proteins at near the wild-type levels. In addition, both pHP constructs produced higher vector titers than did the wild-type HIVNL4.3 suggesting that the wild-type HIV-1 genome might have interfered with the transducing vector genome for packaging. Also, the presence of additional Tat appears to enhance the vector production. This experiment also showed that the pTVψ vector was poorly packaged and need further modifications.
I l l
TE671 cells transduced with the VSV-G pseudotyped pTVΔCMV-nlacZ vector stained strongly by X-gal and exhibited nuclear β-galactosidase activity. The pTVΔCMV- nlacZ-hyg and PTVΛCMV-GFP did not express.the reporter genes efficiently, whereas
PTVΔCMV-nlacZ did. These transducing vectors were further characterized using dividing and nondividing tissue culture models and a small animal model.
5.10 EXAMPLE 10 -- PRODUCTION EFFICIENCY OF SECOND GENERATION CONSTRUCTS
Comparison of vector titer of different pHP packaging constructs using pTVdeltaCMVnlacZ or pTVdeltaEFnlacZ as transducing reporter gene:
5.10.1 METHODS
TE671 cells were co-transfected with pHP construct (8 μgram per well in a 6-well plate), pTV construct (8 microgram per well), PHEFVSV-G (5 microgram per well as envelope pseudotype) with a tat expression plasmid pCEP-tαt (0.5-1 microgram) and a rev expression plasmid pCMV-rev (0,.5-l microgram). The tat and rev expression plasmids were included because it has been shown that they could enhance the vector titers for most of the pHP constructs and they were necessary for pHP-dl.Ndel which has a tat and rev deletion and for PHP-VSVG which has a tat deletion.
The inventors have shown that the original construct pHP-dl.28 (a GI construct) expressed RT at 50-90%) of the wild type level indicating that the mutations in pHP-dl. did not affect the synthesis and function of Gag-Pol. The relative titers of different pHP mutants are shown below: (all included a co-transfected pTV reporter.transgene) pHP-dl.28 (env, nef deletion, relative titer: 1.00); pNL4-3 (wild type HIV-1 control which in fact produce less vector than pHP-dl.28, relative titer: 0.40); pHP-VSVG (vpr, tat, env and nef deletion. relative titer: 0.014): pHP-dl.e Vvpu I (vpu, env, nef deletion, relative titer: 0.43); pHP- di.emVvpu II (vpu, env, nef deletion, relative titer: 0.38); pHP-dl.vpr (vpr, env, nef deletion, relative titer: 0.85); pHP-vpr/ala/leu (vpr funtional mutation, env, nef deletion, relative titer: 0.85); pHP-vprlenv/vpu I (vpr functional mutation and vpu, env, nef deletion, relative titer: 0.24); pHP- pr/eπv/vpu 11 (vpr functional mutation and vpu, env, nef deletion, relative titer: 0.50); pHP-dl.Ndel (vif, vpr, tat, rev, vpu, env, and nef deletion, relative titer: 0.006).
Thus, as more and more of the essential genes were deleted, such as tat and rev, and sequences such as major splice acceptor sites SA4 at nt.5390, SA5 at nt.5777, SA6 at nt.5960 and SA7 at nt.5976 and the 5' of env coding sequence, the vector efficiency gradually or drastically decreased. Nevertheless, the date showed that the second generation pHP construct such as pHP-vpr/eπv/vpu II can be made with relative titer still at 50% level of the pHP-dl.28 and this is about the same efficiency as using a wild type-HIV-1 as the packaging vector (pNL4-3 titer=40% of pHP-dl.28).
In theory, a G2 pHP construct should contain only gag-pol open reading frames and the RRE regulatory sequences such as the pHP-CMV, pHP-CMV, pHP-CMVdel.TAR/SD. pHP-CMVEFla-intron, or pHP-EF constructs (although the vif gene is still present in all of them). However, these constructs exhibited reduced levels of gag-pol activity as shown by the following summary table.:
5.10.3 METHODS TE671 cells were transfected with 5 microgram of each test HP plasmid and 0.5 microgram of pCEPtat (except for one construct, pHPOCMVEFla-intron, the inventors tested both with and without Tat) and 1 microgram of pCMVrev. The culture supernatant was harvested and p24 level was determined by ELISA as described before.
(The relative level of p24 shown with pHP-dl.28 set at 1.00) pHP-1 (1.00) pHP-dl.28 (1.00) pHP-VSVG (0.008) pHP-dl.vpr (0.34) pHP-dl.e Vvpu I (0.43) pHP-dl.e Vvpu II (1.41) pHP-dl.Ndel (0.007) pHP-CMV (0.05) pHP-CMVdel.TAR/SD (0.03) pHP-CMVEF la-intron (0.21, with Tat) pHP-CMVEF 1 a-intron (0.04, with Tat) pHP-EF (0.27)
It was thus shown that deleting TAR in the 5' LTR as seen in pHP-CMVEF 1 a-intron did not make the pHP construct Tat-independent, suggesting that Tat has alternative effects on gag-pol expression besides promotor transactivation via TAR. In addition, theEFla enhancer promoter and intron construct exhibited the highest level of p24 expression suggesting that the EF-la promoter is a better choice than the CMV promoter in later pHP modifications.
5.11 EXAMPLE 11 -- PRODUCTION OF RC-HIV In order to determine whether an RC-HIV recombinant could be generated, the transfected human TE671 cells (ATCC CRL 8805) were co-cultured with the human lymphoma cell line MT4. MT4 cells are an HTLV- 1 transformed human CD4+ lymphoma cell line, that are very sensitive to HIV-1 infection. These cells are available from the National Institutes of Health AIDS Reagents and Reference Program. Uninfected MT4 cells were added into the co-culture every week during these experiments.
In this Example, it was found that the pHPl packaging construct, but not the env- deleted constructs pHP-ldl.2 (2 nt deletion) and pHP-ldl28 28 nt deletion), produced replication-competent HIV-1 (RCV) after co-transfection with pTV plasmid. Infectious virus was detected from pHP+pTVΔCMVnlacZ MT4 co-culture in 8 days. In addition, no infectious virus was detected from pHP.dl.2 or pHP.dl.28+pTVΔCMVnlacZ MT4 co- culture in 60 days (see Table 4).
TE671 cells were co-transfected with pHP+pTV+pHEF- VSV-G as shown in Table 5, and the culture supernatants were harvested 48 hours after DNA removal for RT assay and vector titer was determined as described before. To detect RCV, the transfected cells were co-cultured with the human MT-4 lymphoblastoid cell line, which is very sensitive to HIV-1 infection, for up to 2 months. The culture supernatants were harvested at different time points after co-culture. To detect replication-competent HIV-1 (RCV), the supernatant from the co- culture was assayed for HIV-1 RT activity and for infectious RCV by passage onto CD4+HeLa cells or uninfected MT4 culture. Infection of CD4+HeLa cells was examined by anti-p24 immunohistochemical staining using pooled AIDS patients' sera, and infection of MT4 cell's by cytopathid effects of RCV and the RT production. A very sensitive assay
which would detect cell-cell transmission of poor replicative virus was also used. After four months of co-culture, the MT4 cells were removed and added to fresh MT4 cells and further cultured for 4 days. The co-cultured MT4 cells were fixed and immunostained with HIV patients' sera. The results showed that both pHP-ldl.2 and pHP-ldl.28 were incapable of producing RC-HIV. In summary, these results indicated that pHP-1 transfected cultures produced replication-competent HIV-1 after 8 days of co-culture. However, no RCV was detected after a 60-day co-culture for either pHP-ldl.2 or pHP-ldl.28 cotransfection. The vector titers produced by pHP-ldl.2 and pHP-ldl.28 were as high as that produced by pHP-1. The 28 nt deletion vector pHP- ldl.28 was shown to be as efficient as pHP-1. and did not produce RCV. based on the sensitive HIV infection assay. Thus, the deletion does not affect vector production efficienc and the ercv-deleted pHP constructs are safe for vector production without generating RCV.
FIG. 12 illustrates the possible cross-over between pHP-dl.28 and pTV- dl.CMVnlacZ. to generate RCV during co-transfection. These results clearly indicate that the recombinants are not infectious, due to the deletion in env and the LTR mutation, and requires two homologous recombination events.
TABLE 5 DETECTION OF REPLICATION-COMPETENT HIV (RCV)
" esu ts o rap ce death and loss o MT4 cells.
'+ to I 1 1 1 ', approximately 10 to 40% of the reporter CD4-HeLa cells were HIV- positive after infection using 1 ml of supernatant.
#The MT4 cells in the TE671/MT4 co-culture were transferred into a fresh MT4 culture on day 46 after co-culture; 12 days later, the MT4 cells were directly immunostained with HIV patients' sera.
'no infectious virus was detected.
5.12 EXAMPLE 12 - IN VITRO TRANSDUCTION OF MITOMYCIN-C-TREATED HUMAN CELLS In this Example, two cell cultures were transduced with HP-TV and observed for its transduction efficiency. TF671 or HeLa cells were treated with the DNA synthesis inhibitor, mitomycin C, at 10 μg/ml for 4 hours, trypsinized and plated into a 6-well culture plate. The cells were transduced with HP-TV HIV vector carrying a nlacZ marker gene in the presence of 4-8 μg/mi polybrene in a total volume of 0.5 ml for 2-3 hours and fed with growth media (DMEM containing 10%> FBS). After 48 hours, the expression of the transduced facz gene was detected by X-gal staining as described above. The results indicated that the HP-TV vector was capable of efficiently transducing mitomycin-C-treated, non-dividing human cells.
The HP/TV lentiviral vectors transduce cells with different efficiencies depending on the cell cycle stage at the time of transduction. To demonstrate this, TE671 was treated with 5 microgram/ml of mitomycin C in DMEM growth media for 2.5 hr and the treated cells were transduced with the pTVdeltaEFnlacZ vector and 48 h later, the transduction efficiency was determined by x-gal staini assay. The result demonstrated that cells were most efficiently transduced at 24-48 hr after mitomycin C treatment, at which time, the cells were arrested at S or G2/M phases. At later stage, when cells entered high chromosomal content (>4N) stage the transduction efficiency became reduced. This result suggests that although HP/TV lentiiral vector transduces post-mitotic cells, the efficiency of gene transduction is still dependent on the cell cycle stage.
5.13 EXAMPLE 13 -- IN VITRO TRANSDUCTION OF PRIMARY NEURONAL CELLS
In this Example, rat neuronal cells were isolated from the brains of Fisher rats according to the method of Ure, et al. The cells were grown in culture medim containing L15CO2 (GIBCO, Grand Island, New York), containing 200 ng/mi 2.5 S nerve growth factor
(NGF), 2.55 rat serum, 1 mg/ml ascorbic acid, and 10 μM cytosine arabinose (Sigma), to inhibit divisions of non-neuronal cells.
In addition to rat neuronal cells, human neurons and astrocytes were obtained from differentiated embryonal neural stem cells provided by Neurospheres, Ltd (Calgary, Alberta. Canada). These cells were infected with the HP-TV vectors carrying the nlacZ reporter gene as described above. Briefly, cells were incubated in culture media containing the HP-TV vector. After two hours of incubation, conditioned media (i.e., supernatant medium harvested from cultured neuronal cells after 24 hours of culture) were added, and the culture continued to incubate for five days. The cells were then fixed with formaldehyde and glutaraidehyde. and incubated with X-gal substrate as described in the β-galactosidase assay described above. The results indicated that the HP -TV vector efficiently transduces primary neuronal cells obtained from rat brains, and human neuronal stem cells (neurons and astrocytes).
5.14 EXAMPLE 14 - IN VIVO TRANSDUCTION OF MUSCLE CELLS In this Example, the HP-TV HIV vector was used- to transduce muscle cell in vivo.
The hind-legs of mice CB-17 SCID/beige mice (Taconic) were intramuscularly injected with 50-100 μl of vectors carrying the nlacZ reporter gene as unconcentrated (10~7ml) or microcentrifuge concentrated (30 x 103/mi) stocks in the presence of 4 μg/ml of polybrene. The mice were sacrificed two days later and the injected tissue was prepared for frozen section and for β-galactosidase analysis. The results showed that HP-TV vector transduced muscle cells efficiently in vivo. In particular, tissues exposed to the concentrated vector stock were transduced at near 100% efficiency at the site of injection. It was also noticed that microcentrifuge concentration increased the infectious virus titer, but not in proportion to the fold of concentration.
5.15 EXAMPLE 15 -- HIV VECTORS ARE MORE EFFICENT THAN AIL V VECTORS
In this Example, HIV vectors were compared with the standard MLV vectors commonly in use. The results obtained in these experiments indicated that HIV vector is more efficient than the MLV vector. In this example, a MLV-derived vector (MFGnlacZ, obtained from Dr. Richard Mulligan) and the HIV-1 derived pHP-ldl.28+pTVΔCMVnlacZ vectors were involved in a long term transduction and gene expression study. Three different
human cells lines (TE671. 293, and HepG2) were used in these experiments. The cells were transduced as described, three times in three days using virus stocks prepared from vector producing cells (transfection of PA317 for MFGnlacZ. approximately 10^ cfu/ml and transfection of 293 for HIV-1 vector, approximate 10^ cfu/ml). The cells were transduced three times and propagated once before staining for beta-galactosidase expression.
Briefly . the transduced cells were grown for 3 days and trypsinized. the number of cells was determined, the cells were then plated into 6-well culture plates and one day later, the cells were stained for beta-galactosidase activity . The number of blue cells were counted and the percentages of blue cell in the wells were determined under an inverted microscope. The results suggest that the HIV-1 derived vectors can transduce all three cell types at 3-10 folds higher efficiencies than the MLV vector. These cells were also passaged for 48 days, and stained for β-galactosidase activity. The results showed that in long term culture, the HP+TV HIV vectors exhibited gene expression stabilit .
Table 6 shows a direct comparison of the transduction efficiences observed at 48 hours and 48 days. As previously mentioned. TE671 are rhabdomyosarcoma cells, 293T are kidney cells, and HepG2 are hepatoma cells. In this table, the numbers indicate the percent of cells transduced after one passage or multiple passages. For the 48 hour samples, the cells were transduced three times and propagated once before staining for β-galactosidase activity as previously described in Example 6.
TABLE 6 COMPARISON OF LONG-TERM TRANSDUCTION EFFICIENCIES
5.16 EXAMPLE 16 - GENE TRANSDUCTION INTO CD34
+ HUMAN HEMATOPOIETIC PRECURSOR CELLS
Gene transfer into the human hematopoietic stem cells (HSCS) has encountered problems of vector transduction efficiency and long term expression stability, (see Barranger, 1996; Brenner. 1996). Amphotropic MLV vectors transduce mouse HSCs quite efficiently but human HSCs poorly due to the low level of cell surface MLV -env receptor expression, (see Orlic et al, 1996; Sabatino, et al., 1997) and possible cis-repressive elements in the MLV LTRs (Challita et al, 1995. In particular, transduction of HSCs in clinical trials has been very difficult (see Dunbar, 1996). Adeno-associated virus vector is capable of transducing hematopoeitic stem cell-derived erythroid cells but only works at extremely high titer, (see Nienhuis et al, 1997). To overcome problems with low amphotropic MLV env receptor on CD34 cells, infectious HIV- I constructs have been pseudotyped with vesicular stomatitis G envelope proteins (VSV-G) and shown to infect CD34 cells quite efficiently, (see Akkina et α/.,1996). However, for obious safety reasons, such replication-competent HIV-1 constructs would never be used in gene therapy application.
The HP/TV vector efficiently transduces actively dividing human cell lines including TE671 (rhabdomyosarcoma), 293T (kidney carcinoma) HepG2 (hepatoma), and HeLa (cervical carcinoma) cells. Non-dividing and terminally differentiated cells such as mitomycin C-treated TE671 or HeLa cells, neruons, monocyte-derived macrophages and muscles can also be efficiently transduced by the HP/TV vectors. In contrast, transduction of metabolically quiescent human peripheral blood lymphocytes or bone marrow blood CD34 stem cells with lentiviral vectors-have not been reported, and, transduction of these cells with viral vectors including AAV, retroviral vectors or lentiviral vectors is extremely inefficient, probably because in the absence of growth factor activation these cells have very low metabolic enzyme and transcriptional activities. Accordingly, viral integration and gene expression do not proceed efficiently.
Nevertheless, transduction of human CD34 derived hematopoietic precursor cells has been demonstrated with the HP/TV vectors carrying either nuclear lacZ or green fluorescent protein (GFP) reporter gene. This has been demonstrated using pTV vector containing human, elongation factor la (EFla) promoter as an internal promoter possibly because EFla
promoter has very high transcriptional activity even in quiescent human hematopoietic precursor cells.
To demonstrate transduction of HSC-derived precursor cells, human PBLs were collected from patients treated with G-CSF (granulocyte-colony stimulating factor) to mobolize bone marrow stem cells and purified through an anti-CD34 antibody affinity column. The collected C34+ cells were washed 2-3 times with RPMI medium containing 10% fetal bovine serum without growth factor supplements, centrifuged at 800 g for 5 min, and resuspended in the same growth medium at 1 x 103 cells/ 100 microliter.
To prepare HP/TV vectors, TE671 cells were transfected with pHP-ldl.28 (8 microgram/well), pTVdi.EFnlacZ or pTVdl.EFGFP (8 microgram/well), pHEF-VSVG (5 microgram/well) and pCEP-tαt (0.2 microgram/well) plasmid DNA in a 6-well culture plate, and 48 hr after DNA was added, culture supernatant was collected and centrifuged at 1000 g for 5 min. The clear supernatant was stored at -80°C for future use. The human CD34 cells were transduced 2-3 times with TV vectors at a multiplicity of infection (MOI) of 10, i.e. approximately 103 cells were transduced with 106 infectious units (IU) of pTV vectors in a final volume of 100 μl in DMEM or RPMI growth medium supplemented with 8 microgram/ml of polybrene for 3 -4 h each time. The 106 IU of pTV vectors were prepared from two ml of vector stocks containing 5 x 105 IU/ml which can be concentrated 30-40 fold in a microfuge spun at 20,800 g at room temperature for 90-120 min. The transduced CD34 cells could be maintained in RPMI supplemented with growth factors for 1-4 days before they were plated into semisolid methylcellulose colony assay media. The plated hematopoietic precursor cells grew and formed colonies in 3-4 weeks-and the expression of transduced nlacZ and GFP genes were assayed by x-Gal calorimetric staining and observed under an 20X inverted fluorescent microscope. For the X-Gal staining, the reaction substrate was prepared in phosphate buffered saline adjusted to pH 8.5 using 150 mM Tris containing 4 mM K-ferrocyanide, 4 mM K- ferricyanide, 2 mM MgCl2. 0.8 mg/ml X-Gal. One ml of the x-Gal substrate was added to each 30 mm dish containing HSC-derived colonies and the dish was incubated at 37°C in a 5% CO incubator for 24-72 hr. The total colonies and the dark blue-stained colonies were counted under an inverted microscope. The GFP expression was observed directly under an inverted fluorescent microscope. The expression efficiency of transduction was determined
to be less than 1% at 3-4 weeks after CD34 cells were plated. However, after 5-6 weeks, the efficiency of expression of the transgene (e.g. GFP gene) increased to more than 20%. To determine the efficiency of transduction of the CD34 cells by the pTV vector, the colonies formed in methylcellulose agar were individually picked up and the genomic DNA extracted and subjected to polymerase chain reaction (PCR) using primers specific to the pTV vector, twenty out of the twenty colonies picked were found to be positive for pTV sequence suggesting that the transduction efficiency had been near 100%>. This study suggests that the CD34 cells can be efficiently transduced by the VSV-G pseudotyped HP/TV vectors. Gene expression is delayed and the level of expression is very low which apparently also depend on the promoter used in the vectors.
5.17 EXAMPLE 17 -- NOVEL PROTOCOL FOR EFFICIENT HSC TRANSDUCTION WITH HP/TV VECTORS
Retroviral vectors transduce HSCs poorly due to reasons including low number of receptors on HSCs, low vector titers. and possible blocks to reverse transcription after entry . (see Sinclair et al, 1997). Protocols to improve transduction efficiency have been developed for retroviral gene transfer into HSCS, for exaxnples, coating culture plates with fibronectin fragment FN30/35, (see Moritz et al. 1996) or adding a pretreatment step using medium containing 5 ng/ml of anti-TGF-beta for 10-20 h, (see Hatzfeld et al, 1996) and applying centrifugal force during infection to increases the reversible binding of virus to the cells, (see Bahnson et al. , 1995). These protocols may or may not improve the poor transduction efficiencies of lentiviral vectors on HSCs as shown. Co-culturing target cells with retroviral producer cells has been shown to improve retroviral transduction efficiency. To improve the efficiency of transducing HSCs with lentiviral vectors, a modified protocol is proposed which combines the growth factor stimulation step with the lentiviral producer cell co-culture step. This protocol will also eliminate the vector concentration step which involves the use of a ultracentrifuge.
The cells used for lentiviral production, TE671, can be modified to express human IL-3, SCF. and flt3 ligand via cDNA co-transfection for the purpose of supporting long term culture and transduction of CD34+/CD38- HSCs. Alternatively, freshly prepared human stromal cells can be modified to become lentiviral vector producer cells by co-transfection
using HP/TV vectors plus pHEF-VSV-G or pHF-V-GALV-e/?v (Gibbon ape leukemia virus) constructs.
Thus. TE671 (or other human cell line) transfectants expressing human IL-3, SCF, and flt3 ligand via transfection, or freshly prepared human stromal cells are co-transfected with HP/TV vector plus pHEF-VSV-G or pHEV-GALV-env (Gibbon ape leukemia virus) constructs and 24-48 hr later, or when the cells become 100% confluent, the transfected cells were treated with mitomycin C (5 microgram/ml) for 2.5 hr, washed and refed with RPMI growth media.
Human IL-3 cDNA was amplified using primers: 5 '-TTTCTAGACCACCATGAGCCGCCTGCCCGTCC-3 ' (SEQ ID NO:XX) and
5 '-AAGGATCCCTAAAAGATCGCGAGGCTC-3' (SEQ ID NO:XX) (Otsuka et
Ω/.. 1988).
Human SCF cDNA was amplified using primers: 5 '-TTTCTAGACCACCATGAAGAAGAC AC AAACTTG-3 ' (SEQ ID NO:XX) and
5 '-AAGGATCCTTACACTTCTTGAAACTC-3 ' (SEQ ID NO:XX) (Martin et al, 1990).
Human flt3 ligand cDNA was amplified using primers: 5 '-TTTCTAGACCACCATGACAGTGCTGGCGCCAG-3 ' (SEQ ID NO:XX) and
5 '-AAGGATCCTCAGTGCTCCACAAGCAGC-3' (SEQ ID NO:XX) (Lyman et al. 1994).
5.18 EXAMPLE 18 -- TRANSDUCING VECTORS Construction of G2 pTV: gag, aug, sd, gag coding sequence, env coding sequence,
RRE and gag/env/RRE deletion mutants: To see if the highly conserved packaging signal, i.e. sequences spanning the gag AUG and the 5' major splice donor, can be changed without affecting packaging function of pTV, the following mutants were constructed and tested for cytoplasmic RNA synthesis (exported from nucleus), and packaging function by virion RNA slot-blot assay, and transduction functions by vector titration.
5.18.1 MUTANT CONSTRUCTION
All mutants were made by the megaprimer site-specific mutagenesis method previously described or by direct DNA molecular cloning.
5.18.1.1 5' SPLICE SITE (SD AT NT. 744) AND GAG AUG (AT NT. 790) MUTATIONS.
The two gag AUG mutants and the two SD mutants were made using primers containing the mutation sequences: pTVdeltaAUGI: 5 '-CTCTCGCACCGGTCTCTCTCCTTC-3' (SEQ ID NO:XX) pTVdeltaAUG2: 5 '-CTCTCGCACCCTACTCTCTCCTTC-3' (SEQ ID NO:XX) pTVdeltaSD 1 : 5 '-GGCGGCGACTGCAGAGTACGCCAA-3' (SEQ ID NO:XX) pTVdeltaSD2: S '-GGCGGCGACTGGGGAGTACGCCAA-S' (SEQ ID NO:XX)
5.18.1.2 GAG CODING SEQUENCE MUTATIONS pTV has a gag-pol-env deletion from nt. 1507-7250. The series of additional gag coding sequence mutants were made by site-specific mutagenesis using primers designed to delete specific lengths of gag coding region as described below:
pTVgag dl. l , deletion of 180 bp, from nt 7430-761 1 using the following primer: 5 "-CTCCAGGTCTGAAGATCTTTGACCCTTCAGTACTC-3, (SEQ ID NO:XX)
pTVgag dl.2, deletion of 361 bp, from nt 7250-761 1 using the following primer: 5 '-CTCCAGGTCTGAAGATCTACTAGTAGTTCCTGCTATG-3' (SEQ ID NO:XX)
pTVgag dl.3 deletion of 591 bp, from nt. 1277-1507 and nt.7250-761 1 using primer:
S '-CTCCAGGTCTGAAGATCTGCCTTCTCTTCTACTACT-S' (SEQ ID NO:XX)
pTVgag dl.4 deletion of 824 bp, from nt. 1044-1507 and nt. 7250-761 1 using primer: 5 '-CTCCAGGTCTGAAGATCTGAGGACTGCTATTGTATT-3' (SEQ ID NO:XX)
pTVgag dl.5 deletion of 1039 bp, from nt. 829-1507 and 7250-761 1 using, primer:
S '-CTCCAGGTCTGAAGATCTCTAATTCTCCCCCGCTT-S' (SEQ ID NO:XX)
5.18.1.3 ENV CODING SEQUENCE AND SPLICE ACCEPTOR 8 (SA8 AT NT. 8369) AND SA9 (AT NT. 8515) MUTATIONS The series of env mutants, some of which contained splice acceptor site 8 & 9 deletion, were made by BaBl deletion at the BamHI site at nt. 8465 and six deletion mutants were isolated and sequenced and their deletions were confirmed as follows: pTVenv di.l . BamHI 2'-12, from nt 8375-8559, between RRE and the CNV promoter but SA8 site (splice acceptor site 8 at nt. 8369) is intact. pTVenv dl.2. BamHI 2'-6: from nt 8355-8586, between R.RE and the CMV promoter. pTVenv dl.3. BamHI 2'-8: from nt 8315-8586, between RRE and the CNV promoter.
15 3. env di. BamHX 2'-12 from nt 8375-8559, between R.RE and the CMV promoter but the
SA8 site (splice acceptor site 8) is intact. pTVenv- dl.4. BamiR 5'-3 from nt. 8160-8604, between RRE and the CMV promoter. pTVenv dl.5. BamHI 51-8 from nt. 8215-8730, between RRE and the CMV promoter. pTVenv dl.6. BamHI 5'-10 from nt. 8214-8785, between RRE and the CMV promoter.
5.18.1.4 RRE AND KRE-/GAG/ENV DELETION MUTATIONS The RRE deletion mutant and the RREIgaglenv deletion mutant were constructed using the following methods and primers:
RRE deletion mutant, deletion pTVdl.RRE: a primer flanking both ends of RRE, with the following seqeunce was used to construct RRE-dl.: 5'- AACCCCAAATCCCCATTCCCACTGCTCTTTTT-3' (SEQ ID NO:XX). The first round PCR generated a 1.3 kbp product which was used as megaprimer to amplify a 2.3 kbp fragment which was digested with Sphl and Notl sites for cloning into pTV vectors. The Sphl-Notl 1350bp was ligated with Sphl and Notl-Xbal 4025 bp and Xbal-Sphl 7332 bp of pTVDnlacZ to generate the RRE deletion mutant.
RRΣ/gaglenv deletion mutant, pTVdl.gαg/e«v/RRE: This deletion starts from gag nt. 829 to env nt. 8785 which was constructed using three fragment ligation approach. The three fragments are: ifosΗII to Bgill 125 bp of pTV gag dl.5
containing 5'leader-gαg-erøv, BgHl to Xbal 4016 bp from pTVDnlacZ, and Xbal to BssHII 6600 bp from pTVDnlacZ as plasmid backbone.
5.18.1.5 COMBINATION OF SDI (GGTG TO GCAG)/ GAG AUG (AUG TO TAG) OR SDI I ENV CODING SEQUENCE /SA DELETION, OR
SDI/RRE/G/1 G7£7V| DELETION MUTATIONS
To make generation 2 pTVs, deletion of more essential sequences such as the SD site coupled with gag AUG, or gag or env coding sequences in the pTV constructs will make the vector system even safer. Surprisingly, in some cases, the combination of mutations did not further decrease vector titer; instead, the combination of mutations increased vector titers
(see below). a) pTVdeltaSDI/AUG2: this mutant was made by site-specific mutagenesis using the existing AUG2 primer: 5'-CTCTCGCACCCTACTCTCTCCTTC-3" (AUG to TAG) (SEQ ID NO:XX) and using the pTVdettaSD I as backbone. b) pTVdeltaSD I I env dl-.6: this mutant was made by restriction enzyme digestion and isolation of DNA fragments containing either the SDI mutation or the env di.6 mutation and ligated with the pTVdeltaCMVnlacZ backbone, c) pTVdeltaSDI/dl.gc/g/emVRRE: this mutant was made by megaprimer mutagenesis as described before using the SDI primer: 5 '- GGCGGCGACTGCAGAGTACGCCAA-3 ' (SEQ ID NO:XX) and a primer residing in the CMV-IE promoter downstream of the dl.gαg/e VRRE region. The amplified fragment containing both SDI mutation and the dl.gtfg/em-'RRE sequence was ligated with two fragments obtained from pTVdeltaCMVnlacZ to generate pTVdeltaSDI 'dl.gαg/ew/RRE. Results and Discussion: The results of analyses of vector RNA, packaging function and vector titer are summarized in the table below:
(full-length/spliced RNAS; Virion RNA levels; relative titers) Control pTVdeltaCMVniUacZ: 1.00)
1. pTVdeltaAUG 1 :(++++/++++; ++; 0.3 5) translation void, steady-state RNA less than wt.
2. pTVdeltaAUG2: 0.72) ++++/++++;++; translation void, steady-state RNA less than wt. 3. PTVdeltaSD I: (++/-++; 0.98) less RNA made and less detected in virions. but wt titer.
4. pTVdeltaSD2: ++; 0.85) less RNA made and less detected in virions, but wt titer.
5. pTVgag dl. 1 : / 1.08) 5
6. pTVgag dl.2: 0.90) 7. 7. pTVgag dl. 3 : 0.8 1 )
8. pTVgag dl. 4: 0.94)
9. pTVgag dl. 5: 1 1 1 l : i 0.65)
10. pTVenv di. 1 : I 1 ; 0.48 ) no spliced RNA.
1 1. pTVenv dl.2: (n.d.; n.d.; 0.65) no spliced RNA. 12. pTVenv dl.3: 0.47) no spliced RNA.
13. pTVenv dl.4: I; 0.60) no spliced RNA.
14. pTVenv dl.5: (n.d.; ! ; 0.64) no spliced RNA.
15. pTVenv dl.6: ( .. /-; i I: 0.44) more fwl-length RNA but less titer than other env dl.. 15 16. pTVdl.RRE: (++@+++++;+; 0. IO) detected 20% virion RNA but less titer.
17. pTVdl.gαg/env/RRE: (++/-; +; 0.02) detected 20% virion RNA but much less titer.
18. PTVdeltaSDIIAUG2: (n.d.; ++; 0.61 )
19. pTVdeltaSDI/e / dl.6: (n.d.;. 1 1 i i !; 1.00). 20. pTVdettaSDI/di.gαg/env/RRE: (n.d.; i i i !; 0.30) detected 80% virion RNA but less titer.
+++-H- represents 100% level with each "+" representing 20%>, representing undetected; n.d., not determined.
The results showed that Northern analyses of cytoplasmic RNA indicated that neither the gag AUG mutants nor the gag deletion mutants have much detrimental effects on MRNA
^ synthesis and the transduction functional analysis showed, as determined by vector titration of vectors on TE671 cells, that one of the two gag AUG mutants (AUG2), and all of the gag coding sequence mutants exhibited have no significant effects on vector titers (less than
350% reduction compared with original pTV vector). However, the gag AUGI mutant pTVdeltaAUGI showed more reduction one of the gag AUG mutants showed more detrimental effects on vector titer compared with "wild type" construct (35% of the wild type vector level was mutated suggesting that the splicing machinery has some effer-ts on the internal enhancer/promoter function, possibly through interfering with transcriptional factor binding to the CMV-IE enhancer/promoter elements.
5.18.1.6 CONSTRUCTION OF 3'U3 DELETION MUTANTS AND ASSAY FOR
VECTOR TITER To generate U3 deletion in the vector system, both the S'U' and the U3 will be deleted except for the att site in the 3' U3 region which is needed for provirus integration. The S' U3 was deleted using the same CMV-TATA-HIV-TAR promoter as illustrated in the construction of pHP-1. The 3'U3 was deleted by megaprimer directed site-specific mutagenesis. 5 different deletion mutants were established as described below:
(a) pTVdl.kB/Spl: this construct was made using a kB/Spl deleted HIV-1 construct as reported by Chang et al. 1993 to replace the 3'LTR of pTVdeltaCMVnlacZ. The kBISpl deleted HIV-1 construct was digested with Kpnl (in the nef region of the genome, nt. 9005) and Ngoml (Νael, nt. 10349) and ligated with Kpnl to Notl and Notl to Ngoml fragments from pTVdeltaCMVnlacZ to generate pTVdl.kBI Spl.
(b) pTV-U3di.l, pTV-U3di.2, pTV-U3di.3, and pTV-U3di.4 were made by megaprimer mutagenesis to generate deletions from nt. 9098-9528 (entire U3 deletion to the beginning of R except for the 5' 24 nt att site), nt.9154-9528 (the 5' sequence of U3 from 9098-9154 was retained), nt. 9098-9512 (the extended 5' TAR sequence in the U3 is retained), and nt. 9154 to 9512 (both 5' and 3' extra sequences in the U3 were retained). These mutants were made using the following primers: primer U3dl. 1 : 5'-GTCTAACCAGAGAGACCCTGGGAGTGAATTAGCCCTTC-
3 ' (SEQ ID ΝO:XX);
primer U3dl.2: 5 '-GTCTAACCAGAGAGACCCCAGGGAAGTAGCCTTGTG-3" (SEQ ID NO:XX); primer U3dl.3: 5 '-CCAGTACAGGCAAAAAGCTGGGAGTGAATTAGCCCTTC- 3 " (SEQ ID KO:XX); primer U3di.4: 5 '-CCAGTACAGGCAAAAAGCCAGGGAAGTAGCCTTGTG-3'
(SEQ ID NO:XX) and using a 5' primer annealed to the EcoRI site of the nlacZ gene: 5' RI primer:
5 '-GTCTAACCAGAGAGACCCTGGGAGTGAATTAGCCCTTC-3' (SΕQ ID NO.XX) and a 3' primer next to the NgoMl site: 3' NgoMl primer: 5'-ATAGAACTCCGTTCTCC-3" (SΕQ ID ΝO:XX)
The PCR amplified ftagment was digested with EcoRI and NgoMl and ligated into EcoRI and Λ'goMI digested pTVdeltaCMVnlacZ to generate the four U3 mutants.
The relative vector titer of these mutants was determined by co-transfection with pHP-dl.28 and pHΕF-VSVG as described above and the transfected culture supernatant was harvested 48 hr later and used to infect TΕ671 and 48 hr after infection, the lacZ gene expression was assayed by x-Gal staining and the blue nucleated cells were counted. The relative vector titer was shown with the pTVdeltaCMVnlacZ set at 1.00.
Table: (relative vector titer)
1. pTVdeltaCMVnlacZ: (1.00 +/- 0.00)
2. pTVdl.kB/Sp I: (1.00 +/- 0.10)
3. pTV-U3dl. I: (0.80 +/-O.24)
4. pTV-U3dl.2: (0.91 +/- 0.24) Is S. pTV-U3dl.3: (1.22 +/- 0.06) 6. pTV-U3dl.4: (0.84 +/- 0.27)
The results showed that the 3'U3, except for the att site, can be deleted from the transducing vector pTV construct without affecting vector titer. The 5' U3 deletion had no effect on vector promoter function as shown in the past. The elimination of U3. sequence from the vector system greatly improved the safety of the HP/TV vector system because U3
is an essential HIV replication element and may play important pathogenesis roles during viral infection.
Therefore, in combination, the inventors have deleted the following HIV-1 essential elements, U3. SD, gag AUG, gag-pol, env, tat, rev and 3' SA sites, and all the accessory genes from the pTV construct.
5.19 EXAMPLE 19 - FUNCTIONAL ANALYSES OF THE PRIMARY HIV PACKAGING SIGN \LS USING THE LENTIVIRAL VECTOR SYSTEM
Lentiv iral vectors infect dividing, non-dividing and terminally differentiated cells and have great potential for gene therapy applications. Recent improvements on the lentiviral transducing \ ectors have included modifications of both LTRs and deletions of most of gag. pol, and env sequences (Gasmi et al, 1999; Kim et al, 1998; Miyoshi et al. 1998;
Mochizuki et al, 1998; Zufferey et al, 1997). The viral sequences in the transducing lentiviral vectors have been minimized down to 40 nt of 5' gag with a SD mutation in the 5' UTR in addition to modifications of both LTRs. Although most HIV sequences have been removed from this simplified transducing vector, the existence of a native 5' UTR and the overlapping 40 nt of gag between the helper vector and the transducing vector still poses safety concerns in therapeutic applications.
As one of the essential elements in HIV replication, the 5' UTR possesses the primer binding site (PBS) and the conventional packaging signal (ψ) spanning from 3' of PBS to the first 40 nt of gag. This ψ region has been demonstrated to form a four stem-looped secondary structure (SL1-SL4) (Baudin et al, 1993; Clever et al, 1995; Harrison and Lever.
1992), and SLI and SL3 have been shown to be more critical than SL2 and SL4 to HIV genome packaging (Clever et al, 1995; Luban and Goff, 1994; McBride and Panganiban. 1996; McBride and Panganiban, 1997). In addition, the inventors have demonstrated that mutations in the SD, which is located in SL2, did not affect genomic RNA packaging (Cui et al, 1999).
5.19.1 THE SL2 HAIRPIN STRUCTURE IS REQUIRED FOR EFFICIENT GENOME PACKAGING Previous studies demonstrated that a point mutation in the HIV SD abrogated most of the RNA splicing without diminishing genome packaging and vector function (Cui et al.
1999). This example examines whether the SD and the adjacent cryptic splice site in SL2 could be deleted without interfering with vector function. By PCR site specific mutagenesis using primers listed in Table 7. a partial and a complete SL2 deletion mutants were constructed and studied (ASD3 and ASD4. FIG. 22A). These pTV transducing vector mutants were assayed for vector efficiency by co-transfection with a helper construct pHP into human TE671 cells as described previously (Chang et al, 1999). The relative vector titer was determined by titration of virus on TE671 cells using β-galactosidase reporter gene assay and normalized against that of the wt vector pTV which is set at 1.00. In contrast to the near wt phenotype of the SD point mutation (SDI). deletions of SD, which also disrupted the SL2 haiφin structure, led to a 60% decrease in vector titer (FIG. 22A). Further analyses of cytoplasmic RNA by Northern blotting revealed that both SD3 and SD4 mutants completely abolished splicing as expected (lanes 4 and 5, FIG. 22B). Similar to the SDI mutant, the cytoplasmic genomic RNA (F) of SD3 and SD4 mutants were also down- regulated by 80% (FIG. 22B and FIG. 22C). This down-regulation of genomic RNA expression was likely due to a decrease in RNA stability and/or pre-termination of RNA transcription, as was demonstrated by others (Ashe et al, 1997; Cui et al, 1999; McBride and Panganiban, 1997). Despite the similar levels of cytoplasmic RNA for all three SD mutants, the vector titers of SD3 and SD4 were about 50%) of the SDI level (FIG. 22 A and FIG. 22C).
TABLE 7
The numbering system used here corresponds to pNL4-3
To further delineate the mutational effects of SDI , SD3 and SD4, virion RNA was examined by slot blot analyses. Virion RNA was pelleted by centrifugation at 14,000 φm for 3 hours. To eliminate plasmid DNA contamination in the virion RNA preparation, the viral pellet was treated with two rounds of DNase I, proteinase K, and phenol/chloroform extraction before used for slot blot hybridization. The amount of packaged virion RNA was analyzed on slot-blots and quantified using a Fuji phosphoimager. Genomic RNA packaging efficiency is defined as the ratio of relative amounts of packaged virion RNA to the corresponding cytoplasmic genomic RNA. As summarized in FIG. 22C, SD3 and SD4 mutants packaged much less virion RNA than did SDL It is apparent that the 50%o decrease in vector titer of SD3 and SD4 compared with SDI was caused by similar decrease in packaging efficiency (FIG. 22C). While this result appears to be different from previous reports that SL2 is not essential for gag binding and packaging (McBride and Panganiban. 1996; McBride and Panganiban. 1997), it is in agreement with a recent study demonstrating that destruction of SL2 stem pairing significantly reduced viral replication as well as packaging (Harrison et al, 1998). Therefore, the conserved splice site sequence of the SD is not critical for virion packaging, but the entire SL2 haiφin is important for maintaining optimal packaging function.
5.19.2 GAG AUG AND THE FIRST 40 NT OF GAG ARE IMPORTANT FOR LENTIVIRAL RNA PACKAGING
Located in SL4, the first 40 nt of gag has been shown to be important for HIV packaging (Luban and Goff, 1997). Using the lentiviral vector system, the contributions of gag AUG, the SL4 stem-loop, and its down-stream G-rich region to lentiviral vector function and packaging were examined. Mutations of gag AUG (PCR™ primers shown in Table 7) resulted in a 30-50%> reduction, whereas deletion of the purine-rich region either by itself or in combination with deletion of the SL4 haiφin structure (Table 7 for PCR™ primers) resulted in an 80%> decline in vector titer (FIG. 23A). About half of the 80%> decrease was attributed to the deletion of the SL4 structure (comparing gaglenv.dll and gαg/env.dl5 in FIG. 23A). Northern analyses revealed that mutation of gag AUG or deletion of the entire gag did not suppress cytoplasmic genomic RNA expression (FIG. 23B). Thus, the marked
decrease in vector titer of the G-rich region deletion (gag.d\6) or along with SL4 deletion (gαg.dl7) was due to a greater than 70% reduction in their packaging efficiencies (FIG. 23C). Mutation of gag AUG, however, caused about 40-50%> decrease in packaging efficiencies (FIG. 23C). The importance of SL4 (the first 40 nt of gag) to genome packaging and the SL4 purine-rich region to viral replication have been reported under different systems (Harrison et al, 1998; Luban and Goff, 1994; McBride and Panganiban, 1997). The contribution of gag AUG to packaging, however, has been unclear. Unlike the observation of Richardson (Richardson et al, 1993) that gag AUG mutations did not affect genome packaging, the gag AUG mutations resulted in about 40-50% reduction in packaging efficiency (FIG. 23C). Interestingly, it has been reported that when nucleotides upstream of gag AUG were mutated to disrupt gag translation, a severe defect in genome packaging occurred, even in the presence of a helper construct for gag production (Luban and Goff, 1994).
5.19.3 COMBINATION OF MUTATIONS IN SD, GAG, ENV, AND RRE STILL PRODUCES A VIABLE TRANSDUCING VECTOR
To further examine whether extensive mutations in other regions of the transducing vector (Cui et al, 1999) could be combined with mutations in the packaging signal region without profound loss of vector function, several combination mutants were constructed and analyzed. The combination of gag AUG mutation (TAG) with any of the splice site mutants (5' splice site SDI or 5' and 3' splice sites
caused further decrease in vector titer (FIG. 24A). Similarly, introducing gag AUG mutation into SDl/gog.dl5/e VRRE, which has most of gag, the entire env and RRE deleted, also reduced vector function (FIG. 24A). Northern analyses indicated that inclusion of the AUG-TAG mutation did not further affect the genomic RNA expression (FIG. 24B). Thus, the further decrease in vector titer after the introduction of AUG mutation is consistent with the observed AUG-TAG mutational effects on packaging function (FIG. 23C and FIG. 24C).
Further deletion of the first 40 nt of gag from SDl/TAG/gαg.dl5/e«v/RRE to generate mutant SDl/gag.dl7/e«v/RRE resulted in a pTV construct with complete deletion of HIV gag and env sequences. Suφrisingly, this vector still exhibited vector function, albeit at a reduced level (up to 105 tu/ml, FIG. 24A). Northern analyses indicated that this construct had more than two-fold increase in cytoplasmic genomic RNA expression compared to the
wt pTV construct (lane 5, FIG. 24B). Thus, the drastic reduction in vector titer was likely- due to the reduced packaging efficiency (FIG. 24C). These results further support that the stem-loop structure of SL4 and its down-stream purine-rich sequences are important for lentiviral genome packaging. In conclusion, it was shown that both SL2 and SL4 in the primary lentiviral packaging signal are important for the vector genome packaging function. The inventors also demonstrated that it is possible to delete all of the HIV gag and env sequences in the lentiviral transducing vector, which further reduced sequence overlap between the transducing pTV and the helper pHP constructs. These modifications should further relieve safety concerns over the lentiviral vector system. Future generations of lentiviral vectors will require functional substitutions of remaining minimal essential FIIV elements in this vector system.
6.0 REFERENCES The following references, to the extent that they provide exemplary procedural or other details supplementary to those set forth herein, are specifically incoφorated herein by- reference.
U.S. Patent 5.272,071.
U.S. Patent 5.654,195. U.S. Patent 5.665,577.
U.S. Patent 5.693,508.
U.S. Patent Appl. Serial No. 08/791,994.
U.S. Patent Appl. Serial No. 08/838,702.
U.S. Patent Appl. Serial No. 08/848,760. Intl. Pat. Appl. Publ. Ser. No. PCT US95/14576.
Intl. Pat. Appl. Publ. Ser. No. PCT US98/06944.
Intl. Pat. Appl. Publ. Ser. No. WO 90/04788.
Intl. Pat. Appl. Publ. Ser. No. WO 91/06667.
Abuchowski. McCoy, Palczuk, Van Es and Davis. J Biol Chem., 252:3582-3586, 1977.
132
Adachi et al. "'Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone," J. Virol,
59, 284-291. 1986. Adams et al. J. Am. Chem. Soc, 105:661, 1983. Agarwal and Riftina, "Synthesis and enzymatic properties of deoxyribooligonucleotides containing methyl and phenylphosphonates linkages," Nucl. Acids Res., 6(9):3009-
3023. 1979. Aidovini and Young. J Virol. 64:1920-1926, 1990. Akasako, et al. Biochemistry, 34:8115-8122, 1995. Akkina et al . "'High-efficiency gene transfer into CD34+ cells with a human immunodeficiency virus type- I -based retroviral vector pseudotyped with vesicular stomatitis virus envelope glycoprotein G," J. Virol, 70:2581 -2585, 1996. Alber, et al. Biochemistry. 26:3751-3758, 1987.
Allen and Choun. "Large unilamellar liposomes with low uptake into the reticuloendothelial system." FEBS Lett., 223:42-46, 1987.
Allen et al. Nature. 327:713, 1987. Altschul et al. '"Gapped BLAST and PSI-BLAST: a new generation of protein database search programs;' Nucl. Acids Res., 25:3389-3402, 1997. Altschul, et al. Nature Genetics, 6:119-129, 1994. Amendt et al. "'Presence of negative and positive cis-acting RNA splicing elements within and flanking the first tat coding exon of human immunodeficiency virus type 1 ," Mol
Cell Biol, 14:3960-3970, 1994. Armitage, Koch, Frydenlund, Orum, Batz, Schuster, "Peptide nucleic acid-anthraquinone conjugates: strand invasion and photoinduced cleavage of duplex DNA," Nucl Acids Res.. 25(22):4674-4678, 1997.
Armitage, Ly. Koch. Frydenlund, Orum, Batz, Schuster, "Peptide nucleic acid-DNA duplexes: long range hole migration from an internally linked anthraquinone," Proc.
Natl Acad. Sci. USA, 94(23):12320-12325, 1997. Ashe, Pearson and Proudfoot, "The HIV-1 5' LTR poly (A) site is inactivated by Ul snRNP interaction with the downstream major splice donor site," Embo. J, 16:5752-63,
1997.
Asseline and Thuong, Tetrahedron Lett. , 30(19):2521-2524, 1989. Atchinson et al. , Science, 194:754-756, 1965. Ayares et α/.. ONAS USA, 83:5199-5203, 1986.
Bahnson et al, "Centrifugal enhancement of retroviral mediated gene transfer." J. Virol Methods, 54: 131 -143. 1995.
Baker, et al . J. Virol, 61 : 1639 1987. Balazsovits et al , "Analysis of the effect of liposome encapsulation on the vesicant properties, acute and cardiac toxicities, and antitumor efficacy of doxorubicin,"
Cancer Chemother. Pharmacol, 23:81 -86, 1989. Barranger, "Hematopoietic stem cell gene transfer," Gene Therapy, 3:379-380, 1996.
Baudin, Marquet, Isel. Darlix. Ehresmann and Ehresmann, "Funcitonal sites in the 5' region of human immunodeficiency virus type 1 RNA form defined structural domains," J.
Mol Biol , 229:382-97. 1993. Beck, Geary. Anderson. Keltner. Shults, Kaufman, Buckley, Corbett, Kupersmith and Miller. N. Engl J. Med , 326:581 -588, 1992.
Behravar, et al, Eur. J. Biochem.. 198:589-592, 1991.
Benvenisty and Reshef. "Direct introduction of genes into rats and expression of the genes."
Proc. Natl. Acad. Sci USA. ; 83(24): 9551-9555, 1986. Berkowitz et al, "51 regions of HIV-1 RNAs are not sufficient for encapsulation: implications for the HIV-1 packaging signal," Virology, 212:718-723, 1995.
Berns et al. In: Virus Persistence. Mehay et al (Ed.), Cambridge Univ. Press, pp. 249-265.
1982. Berns, Pinkerton, Thomas, Hoggan, "Detection of adeno-associated virus (AAV)-specifιc nucleotide sequences in DNA isolated from latently infected Detroit 6 cells," Virol . 68(2):556-560, 1975.
Blackwood and Kadonaga, "Going the distance: a current view of enhancer action," Science.
281(5373):61-63, 1998. Blomer et al. J. Virol, 71 : 6641-6649, 1997. Boffa, "Thrombomodulin in human brain microvasculature," Lupus, 4(2): 165-166, 1995.
Boffa, Berard, Sugi, Mclntyre, "Antiphosphatidylethanolamine antibodies as the only antiphospholipid antibodies detected by ELISA. II. Kininogen reactivity," J.
Rheumatol, 23(8): 1375- 1379, 1996. Boffa, Caφaneto, Allfrey, Proc. Natl Acad. Sci. USA, 92:1901-1905, 1995. Boffa, Morris, Caφaneto, Louissaint, Allfrey, J. Biol Chem., 271 :13228-13233, 1996. Boshart et al, Cell, 41 :521-530, 1985. Bourlais, Acar, Zia, Sado, Needham, Leverge, "Ophthalmic drug delivery systems— recent advances," Prog. Retin Eye Res. , 17(l):33-58, 1998. Bowern, Ramshaw, Clark and Doherty, J. Exp. Med, 260:1532-1543, 1984. Boyes and Bird, Ce/7, 64:1123-1 134, 1991.
Bray et al, "A small element from the Mason-Pfizer monkey virus genome makes human immunodeficiency virus type I expression and replication Rev-independent, " Proc.
Nat. Acad. Sci. USA, 91 :1256-1260, 1994. Brenner "Gene transfer to hematopoietic cells," N. Engl. J. Med., 335:337-339, 1996. Brenner, "Human somatic gene therapy: progress and problems," J. Intern. Med..
237(3):229-239, 1995. Brett and Rumsby, Neurochem. Int., 23:35-44, 1993. Broido et al. Biochem. Biophys. Res. Commun., 119:663, 1984. Bukrinsky et al, Nature, 365:666-669, 1993. Buller, Janik, Sebring, Rose, "Heφes simplex virus types 1 and 2 completely help adenovirus-associated virus replication. J. Virol, 40(l):241-247, 1981. Bums et al, Proc. Natl Acad. Sci. USA, 90:8033-8037, 1993. Caldovic and Hackett Jr., "Development of position-independent expression vectors and their transfer into transgenic fish," Mol. Mar. Biol. Biotechnol, 4(1):51-61, 1995. Capecchi, "High efficiency transformation by direct micro injection of DΝA into cultured mammalian cells," Cell, 22(2):479-488, 1980. Carlsson et al, Nature, 380:207, 1996. Carlsson, Sandier, Jansson, "Influence of the neurotoxin capsaicin on rat pancreatic islets in culture, and on the pancreatic islet blood flow of rats," Eur. J. Pharmacol, 312(1):75- 81, 1996.
Carriere et fl/., J. Virol, 69: 2366-2377, 1995.
Carter et al. In: The Parvoviruses, K.I. Berns (Ed.), Plenum, NY, pp. 67-128, 1983. Caruthers et al. , In: Genetic Engineering, Settlow and Hollander (Eds.), Plenum Press, New
York. 1982. Carver, Dalrymple, Wright, Cottom, Reeves, Gibson. Keenan, Barrass, Scott, Colman, et al . "Transgenic livestock as bioreactors: stable expression of human alpha- 1-antitrypsin by a flock of sheep," Rtotec/mo/ogy N}'. 1 1 (1 1 ): 1263-1270, 1993. Casey. Lo-Hsueh, Lopez, Volgelstein, Stanbridge. "Growth suppression of human breast cancer cells by the introduction of a wild-type p53 gene," Oncogene, 6(10): 1791-7,
1991. Cayouette and Gravel, Invest. Ophthalmol Visual Sci. , 3 ':2022-2028, 1996.
Cech and Brehm, "Replication of the extrachromosomal ribosomal RΝA genes of
Tetrahymena thermophilia," Nucleic Acids Res.. 9(14):3531-43, 1981. Cech, Zaug. Grabowski, "In vitro splicing of the ribosomal RΝA precursor of Tetrahymena: involvement of a guanosine nucleotide in the excision of the intervening sequence," Cell 21(3 Pt 2):487-96, 1981.
Challita et al, 'Multiple modifications in cis elements of the long terminal repeat of retroviral vectors lead to increased expression and decreased DΝA methylation in embryonic carcinoma cells," J. Virol, 69:748-755, 1995. Chamorro, Vila, Ascaso, Elices, Schonewille, Blanc, "Blood pressure and functional recovery in acute ischemic stroke," Stroke, 29(9): 1850-1853, 1998.
Chandran, Roy, Mishra, "Recent trends in drug delivery systems: liposomal drug delivery system— preparation and characterisation," Indian J. Exp. Biol, 35(8):801-809, 1997. Chang and Zhang, "Infection and replication of Tat-minus human immunodeficiency viruses: genetic analyses of TR and tat mutants in primary and long-term human lymphoid cells." Virology, 211 :157-169, 1995.
Chang et al. "Foreign gene delivery and expression in hepatocytes using a hepatitis B virus vector," Hepatology, 14T34A, 1991. Chang et al, "Human immunodeficiency viruses containing heterologous enhancer/promoters are replication competent and exhibit different lymphocyte tropisms," J Virol, 67:743-752, 1993.
Chang, Nunberg, Kaufman, Erlich, Schimke, Cohen, "Phenotypic expression in E colt of a
DNA sequence coding for mouse dihydrofolate reductase," Nature, 275(5681):617-
624. 1978 Chaudhary. Batra, Gallo, Willingham, FitzGerald, Pastan. "A rapid method of cloning functional variable-region antibody genes in Escherichia colt as single-chain lmmunotoxms," Proc Natl Acad Set USA, 87(3) 1066-70, 1990 Chen and Okayama, "High-efficiency transfection of mammalian cells by plasmid DNA,"
Mol C ell Biol . 1.21 A -2152, 1987. Chen et al Proc Natl Acad Set USA, 93:10057-10062, 1996 Chen, McCarty . Bruce. Suzuki and Suzuki, Gene Ther , 5 50-58, 1998
Cheung. Hoggan, Hauswirth, Berns, "Integration of the adeno-associated virus genome into cellular DNA in latently infected human Detroit 6 cells." J Virol , 33(2) 739-748, 1980 Chirmule et al . J Virol , 69:492-498, 1995
Chow. Plumb. Wen, Sohn, Lu, Zhang, Lukacs, Tanswell, Hui, Buchwald, Hu, "Development of an epithelium-specific expression cassette with human DNA regulatory elements for transgene expression in lung airways," Proc Natl Acad Set USA, 94(26): 14695-
14700. 1997 Chπstensen et al J Pept Sci , 1(3): 175-183, 1995.
Chπstensen Johansen, Marker, Thomsen, "Circulating intracellular adhesion molecule-1 (ICAM-1 ) as an early and sensitive marker for virus-induced T cell activation," Chn
Exp Immunol , 102(2):268-273, 1995. Claesseus. et al , Protein Eng , 4:35, 1989 Clapp, D W . "Somatic gene therapy into hematopoietic cells. Current status and future implications," Chn Pennatol , 20(1): 155-168, 1993. Clark, Voulgaropoulou, Fraley, Johnson, "Cell lines for the production of recombinant Adeno- associated virus," Hum Gene Ther., 6:1329-1341. 1995 Claudio. Kress. Factor, Norton and Brosnan, Am J Pathol , 135:1157-1168, 1989. Clavel et al "'Molecular cloning and polymoφhism of the human immunodeficiency virus type 2." Nature, 324:691-695, 1986. Clerici et al J Amer Med Assoc, 271:42-46, 1994. Clerici et al. J Inf Dts., 165:1012-1019, 1992.
Clever et al. J. Virol. 70:5902-5908, 1996.
Clever, Sassetti and Parslow, "'RNA secondary structure and binding sites for gag gene products in the 5' packaging signal of human immunodeficiency virus type 1 ," J. Virol. 69:2101-09. 1995. Coffin, "Retroviridae and their replication," In: Virology, Fields et al. (eds.). New York: Raven Press, pp. 1437-1500, 1990. Constantino and Pelliceian, J Med. Chem., 39:3998-4006, 1996.
Conway, Zolotukhin. Muzyczka, Hayward, Byrne, "Recombinant Adeno-associated virus Type 2 replication and packaging is entirely supported by a Hopes Simplex virus Type 1 amplicon expressing rep and cap," J Virol, 71 :8780-8789, 1997.
Corbeau, et al. Proc. Nat. Acad. Sci. U5A, 93: 14070-14075, 1996. Corey, "Peptide nucleic acids: expanding the scope of nucleic acid recognition," Trends
Biotechnol. 15(6):224-229. 1997. Cosset et al. J. Virol. 69:7430-7436, 1995. Couch et al . "Immunization with types 4 and 7 adenovirus by selective infection of the intestinal tract." Am. Rev. Resp. Dis., 88:394-403, 1963. Coune, "Liposomes as drug delivery system in the treatment of infectious diseases: potential applications and clinical experience," Infection, 16(3): 141-147, 1988. Coupar et al.. "A general method for the construction of recombinant vaccinia virus expressing multiple foreign genes," Gene, 68:1-10, 1988.
Couvreur et al, "Nanocapsules. a New Lysosomotropic Carrier," EEβS Lett., 84:323-326,
1977. Couvreur et al, "Tissue distribution of antitumor drugs associated with polyalkylcyanoacrylate nanoparticles," J. Pharm. Sci., 69(2): 199-202, 1980. Couvreur, "Polyalkyleyanoacrylates as Colloidal Drug Carriers," Crit. Rev. Ther. Drug Carrier Syst., 5: 1-20, 1988. Cozzi, Tucker. Langford, Pino-Chavez, Wright, O'Connell, Young, Lancaster, McLanghlin, Hunt. Bordin. White. "Characterization of pigs transgenic for human decay- accelerating factor," Transplantation, 64(10): 1383-1392, 1997. Crapo, Barry, Foscu and Shelburne, Am. Rev. Respir. Dis., 122:123-143, 1980. Cross, Manning, Stern and Misko, J. Neuroimmnol, 80:121-130, 1997.
Cui, Iwakoma and Chang, "The contributions of viral splice sites and cis-regulatory elements to lentiviral vector function," J Virol , in press, 1999. Cukor et al , In The Paroviruses, K I Berns (Ed ), Plenum, NY, pp. 33-66, 1984. Culver et al Human Gene Ther 2 107, 1991 Curiel, Agarwal, Wagner, Cotten, "Adenovirus enhancement of transfernn-polylysine- mediated gene delivery ," Rroc Natl Acad Set USA, 88(19):8850-8854, 1991 Dai et al , "Mutation of human immunodeficiency virus type I at amino acid 585 on gp41 results in loss of killing by CD8+ A24-restπcted cytotoxic T lymphocytes," J Virol
66, 3151-3154. 1992 Daniel, Lam and Pratt. J Neurol 5c/ . 2 21 1-221, 1981
Davis et al , In: Microbiology. 4th ed , j B Lippincott Co., Philadelphia, PA, pp 1 123-1 151 ,
1990 Day and Tuite, "Post-transcπptional gene regulatory mechanisms in eukaryotes an overview," J Endocnnol , 157(3).361-371, 1998. Deacon et al , Science, 270 988-991, 1995
Deira, Corbacho, Bondia. Lerma, Gascon, Martin, Garcia, Tabernero, "Captopπl hepatotoxicity in a case of renal crisis due to systemic sclerosis," Nephrol Dial
Transplant, 12(8)- 1717-1718. 1997 Delort and Capecchi. "TAXI/UAS a molecular switch to control expression of genes in vivo," Hum Gen Ther , 7.809-820, 1996
Dijkema et α/ , EMBO J , 4 761, 1985 Dimitrov et al. 1993. Doolittle, Science, 214: 149 1981
Dougall and Nick, Endocrinology, 129(5):2376-84, 1991. Douglas, Davis, Ilium, "Nanoparticles in drug delivery," Crit Rev Ther Drug Carrier Syst ,
3(3):233-261, 1987. On et al , Cell, 82:665-674, 1995. Dubensky et al, "Direct transfection of viral and plasmid DNA into the liver or spleen of mice." Proc Nat Acad Sci USA, 81 :7529-7533, 1984. Dueholm et al, J Org Chem , 59:5767-5773, 1994.
Dueholm, Motawia, Pedersen, Nielsen, Lundt. "Synthesis of 3'-alkylthio-2',3'-dideoxy nucleosides with potential anti-HIV activity from 2-deoxy-D-ribose, using a phosphorus pentoxide reagent." Arch. Pharm. (Weinheim), 325(9):597-601, 1992.
Dunbar, "Gene transfer to hematopoietic stem cells: implications for gene therapy of human disease." Annu. Rev. Med., 47: 1 1-20, 1996.
Ebert, Selgrath, DiTullio, Denman, Smith, Memon, Schindler, Monastersky, Vitale, Gordon. "Transgenic production of a variant of human tissue-type plasminogen activator in goat milk: generation of transgenic goats and analysis of expression," Biotechnology NY, 9(9):835-838, 1991. Egholm, Buchardt, Christensen, Behrens, Freier, Driver. Berg, Kim, Norden, Nielsen. "PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen- bonding rules," Nature, 365(6446):566-568. 1993.
Eglitis and Anderson, "Retroviral vectors for introduction of genes into mammalian cells." Biotechniques 6(7):608-614, 1988. Eglitis, Kantoff, Kohn, Karson, Moen, Lothrop, Blaese, Anderson, "Retroviral-mediated gene transfer into hemopoietic cells," Avd. Exp. Med. Biol, 241 :19-27, 1988.
Emennan, Curr. Biol, 6:1096-1103, 1996.
Engelhardt, 1995.
Ensoli et al. Nature, 371 :674-680, 1994. Erzurum, Lemarchand, Rosenfeld, Yoo and Crystal. Nucleic Acids Res., 21 T607-1612, 1993.
Ezzell, "Eukaryotic mRNA processing," The Journal of NIH Research, 7:101-104, 1995.
Faller and Baltimore, "Liposome encapsulation of retrovirus allows efficient super infection of resistant cell lines," J. Virol, 49(l):269-272, 1984.
Fani, Gallo. Fancelli, Mori, Tamburini, Lazcano. "Heterologous gene expression in an escherichia coli population under starvation stress conditions," J. Mol. Evol.
47(3):363-368, 1998.
Fechheimer et al, "Transfection of mammalian cells with plasmid DNA by scrape loading and sonication loading," Proc. Natl. Acad. Sci. USA, 84:8463-8467, 1987.
Ferkol, Lindberg, Chen, Perales, Crawford, Ratnoff, Hanson, "Regulation of the phosphoenolpyruvate carboxykinase/human factor IX gene introduced into the livers of adult rats by receptor-mediated gene transfer," FASEB J, 7(11):1081-1091, 1993.
Ferrari, Samulski, Shenk, Samulski, "Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors," J. Virol, 1996:3227-3234,
1996. Fiers. Contreras, Haegemann, Rogiers, Van de Voorde, Van Heuverswyn, Van Herreweghe. Volckaert, Ysebaert, "Complete nucleotide sequence of SV40 DNA," Nature,
273(5658):1 13-120, 1978. Fletcher et al. EMBO J., 15:6155-6165, 1996. Footer, Egholm, Kron, Coull, Matsudaira, "Biochemical evidence that a D-loop is part of a four-stranded PNA-DNA bundle. Nickel-mediated cleavage of duplex DNA by a Gly-Gly-His bis-PNA," Biochemistry, 35(33):10673-10679, 1996.
Footer. Engholm, Kron, Coull, Matsudaira, Biochemistry, 35: 10673-10679, 1996.
Forster and Symons, "Self-cleavage of plus and minus RNAs of virusoid and a structural model for the active sites," Cell, 49(2):21 1-20, 1987. Fraley et al. "Entrapment of a bacterial plasmid in phospholipid vesicles: Potential for gene transfer." Proc. Nat'l Acad. Sci. USA, 76:3348-3352. 1979.
Franz. Mueller. Haartong, Frey, Katus, "Transgenic animal models: new avenues in cardiovascular physiology," J. Mol. Med., 75(2): 1 15-1 19. 1997. Fresat and Puglisi, "Application of liposomes as potential cutaneous drug delivery systems.
In vitro and in vivo investigation with radioactively labelled vesicles," J. Drug Target. 4(2):95-101, 1996.
Friedmann. "Progress toward human gene therapy," Science, 244: 1275-1281, 1989. Frohman. Downs, Kashio, Brinster, "Tissue distribution and molecular heterogeneity of human growth hormone-releasing factor in the transgenic mouse," Endocrinology.
127(5^:2149-2156, 1990. Frohman, PCR Protocols, a Guide to Methods and Applications XVIII Ed., Academic Press,
1990. Fromm, M.. Taylor, L. P., and Walbot, V., "Expression of genes transferred into monocot and dicot plant cells by electroporation," Proc. Natl. Acad. Sci. USA, 82(17):5824-
5828. 1985.
Fynan, E. F.. Webster, R. G., Fuller, D. H., Haynes, J. R., Santoro, J. C. and Robinson, H. L., "DNA vaccines: protective immunizations by parenteral, mucosal, and gene gun inoculations," Proc. Natl. Acad. Sci. USA 90(24): 1 1478-11482, 1993. Gabizon and Papahadjopoulos, "Liposomes formulations with prolonged circulation time in blood and enhanced uptake by tumors," Proc. Natl Acad. Sci. USA. 85:6949-6953,
1988. Gambacorti-Passerini. Mologni, Bertazzoli, le Coutre, Marchesi, Grignani, Nielsen, "In vitro transcription and translation inhibition by anti-promyelocytic leukemia (PML)/retinoic acid receptor alpha and anti-PML peptide nucleic acid," Blood, 88(4): 141 1-1417, 1996.
Gamble et al. Cell 87: 1285-1294, 1996.
Gasmi, Glynn. Jin. Jolly, Yee and Chen, "Requirements for efficient production and transduction of human immunodeficiency virus type 1 -based vectors." J. Virol, 73: 1828-34, 1999. Gaur, Wiers. Liu, Rothbard and Fathman, Science, 258:1491-1494, 1992.
Gefter et al. "A simple method for polyethylene glycol-promoted hybridization of mouse myeloma cells,"Somatzc Cell Genet., 3(2): 231-236, 1977. Gewirtz, Stem Cells. 3:96, 1993.
Ghosh and Bachhawat. "Targeting of liposomes to hepatocytes," In: Wu G. and C. Wu ed. Liver diseases, targeted diagnosis and therapy using specific receptors and ligands.
New York: Marcel Dekker, pp. 87-104, 1991. Ghosh-Choudhury et al , "Protein IX, a minor component of the human adenovirus capsid, is essential for the packaging of full-length genomes," EMBO J., 6: 1733-1739, 1987. Glick and Pasternak, In: Molecular Biotechnology, American Society for Microbiology, Washington, D.C., p. 412, 1994.
Glinim et al. 'Efficient gene transfer in primitive CD34+/CD381o human bone marrow cells reselected after long-term exposure to GALV-pseudotyped retroviral vector. I- lum," Gene Ther., 8:2079-2086, 1997. Goding, "Monoclonal Antibodies: Principles and Practice," 2nd Edition, Academic Press, Orlando, FL, pp. 60-74, 1986.
Goeddel, Heyneker. Hozumi. Arentzen, Itakura, Yansura, Ross, Miozzari, Crea, Seeburg,
"Direct expression in Escherichia coli of a DNA sequence for human growth hormone," Nature, 281(5732):544-548, 1979. Goeddel, Shepard, Yelverton. Leung, Crea, Sloma, Pestka, "Synthesis of human fibroblast interferon by E. coli." Nucl Acids Res. , 8(18):4057-4074, 1980.
Gold, Schroder, Powell and Kelly. Eur. J. Immunol, 27:2863-2869, 1997. Goldstein and Doi. "Prokaryotic promoters in biotechnology," Biotechnol Annu. Rev.. 1 : 105-
128. 1995. Gomez-Foix et al, "Adenovirus-mediated tranfer of the muscle glycogen phosphorylase gene into hepatocytes confers altered regulation of glycogen metabolism," J. Biol.
Chem., 267(35):25129-25134, 1992. Good and Nielsen, Antisense Nucleic Acid Drug Dev., 7(4):431-437, 1997. Gopal, "Gene transfer method for transient gene expression, stable transfection. and cotransfection of suspension cell cultures," Mol. Cell Biol, 5:1188-1190, 1985. Gordon and Anderson. Curr. Opin. Biotechnol, 5:611-616, 1994. Gorman et al, Proc. Natl. Acad. Sci. USA, 79:6777, 1982. Gossen and Bujard, "Tight control of gene expression in mammalian cells by tetracycline- responsive promoters." Proc. Natl Acad. Sci. USA, 89(12):5547-5551, 1992. Graham and Prevec, "Adenovirus-based expression vectors and recombinant vaccines," Biotechnology. 20:363-390. 1992.
Graham and Prevec. "Manipulation of adenovirus vector," In: Methods in Molecular
Biology: Gene Transfer and Expression Protocol, Ε.J. Murray (ed.), Clifton. NJ:
Humana Press. 7: 109-128. 1991. Graham and van der Εb, "Transformation of rat cells by DNA of human adenovirus 5," Virology, 54(2):536-539, 1973.
Graham et al, "Characteristics of a human cell line transformed by DNA from human adenovirus type 5", J. Gen. Virol, 36:59-72, 1977. Greene, "Regulation of HIV-1 gene expression," Ann. Rev. Immunol, 8:453-475, 1990. Gregoret and Sauer, Proc. Nat. Acad. Sci. USA, 90:4240-4250, 1993.
Gribskov and Burgess, Nucl. Acids Res. 14:6745, 1986, as described by Schwartz and
Dayhoff, eds., In: Atlas of Protein Sequence and Structure, National Biomedical
Research Foundation, pp. 353-358, 1979. Griffith et al. J. Am. Chem. Soc, 1 17:831 -832, 1995. Grossman et al, Nat. Genet., 6:335, 1994.
Guruprasad et al, Protein Eng, 9:849-56. 1996. Guyer et al, J. Biol Chem., 265:17307-17325. 1990. Gwinner et α/., Kidney Int., 48(2):354-62, 1995.
Haaima, Hansen, Christensen, Daho, Nielsen. "Increased DNA binding and sequence discrimination of PNA oligomers containing 2,6-diaminopurine," Nucl. Acids Res..
25(22):4639-4643, 1997. Haaima, Lohse, Buchardt, Nielsen, Angew. Chem.. Int. Ed. Engl, 35:1939-1942, 1996. Handa, Shiroki, Shimojo, "Establishment and characterization of KB cell lines latently infected with adeno-associated viais type 1," Virol. 82(l):84-92, 1977. Hanvey et al. Science, 258:1481-1485, 1992.
Hardy, Kitamura, Harris-Stansil, Dai, Phipps, "Construction of adenovirus vectors through Cre- lox recombination," J. Virol. 71 :1842- 1849. 1997. Harland and Weintraub, "Translation of mammalian mRNA injected into Xenopus oocytes is specifically inhibited by antisense RNA." J. Cell Biol, 101 :1094-1099, 1985. Harlow, E. and Lane, D. "Antibodies: A Laboratory Manual," Cold Spring Harbor
Laboratory, Cold Spring Harbor. NY, 1988. Harouse et al, Science, 25:320-32?, 1991. Harrison and Lever, "The human immunodeficiency virus type 1 packaging signal and major splice donor region have a conserved stable secondary structure," J. Virol, 66:4144- 53, 1992.
Harrison, Miele, Hunter and Lever, "Functional analysis of the core human immunodeficiency virus type 1 packaging signal in a permissive cell line," J. Virol .
72:5886-96, 1998. Haskell and Bowen, "Efficient production of transgenic cattle by retroviral infection of early embryos," Mol. Reprod. Dev, 40(3):386-390, 1995.
Hatzfeld et al. "Increased stable retroviral gene transfer in early hematopoietic progenitors released from quiescence," Human Gene Therapy, 7:207-213, 1996. Hayashi et al. "RNA packaging signal of human immunodeficiency virus type 1," Virology,
188:590-599, 1992. Heath and Martin, "The development and application of protein-liposome conjugation techniques," Chem. Phys. Lipids, 40:347-358. 1986. Heath et al. "Liposome-mediated delivery of pteridine antifolates to cells: in vitro potency of methotrexate and its alpha and gamma substituents," Biochim. Biophys. Ada,
862:72-80, 1986. Heffner and Fracica, in Nitric Oxide and Radicals in the Pulmonary Vasculature, eds. Weir,
Archer and Reeves, pp. 105-133, 1996. Heim and Tsien, "Engineering green fluorescent protein for improved brightness, longer wavelength and fluorescence resonance energy transfer," Curr. Biol, 6:178-182, 1996. Heinzinger et al, Proc. Natl-Acad. Sci. USA. 91 :731 1 -7315, 1994. Henry-Michelland et al, "Attachment of antibiotics to nanoparticles; Preparation, drug- release and antimicrobial activity in vitro." Int. J. Pharm., 35:121-127, 1987. Hersdorffer et al, "Efficient gene transfer in live mice using a unique retroviral packaging line." DNA Cell Biol, 9:713-723, 1990. Herz and Gerard, "Adenovirus-mediated transfer of low density lipoprotein receptor gene acutely accelerates cholesterol clearance in normal mice," Proc. Natl Acad. Sci. USA
90:2812-2816, 1993. Hess, Boiteux. Kruger, "Cooperation of glycolytic enzymes," Adv. Enzyme Regul, 7:149-
167. 1969. Hoffman, Maquire and Bennett, Invest. Ophthalmol Visual Sci, 38:2224-2233, 1997. Hoggan et al, In: Proceeding of the Fourth Lepetit Colloquium, Cacoyac, Mexico, North
Holland, Amsterdam, pp. 243-249, 1972. Hoggan, Fed. Proc., 2A:2A8, 1965.
Honegger. Krenger and Langemann, Neurosci. Lett., 98:327-332, 1989. Hooper, Spitsin, Kean, Champion, Dickson, Chaudhry and Koprowski, Proc. Natl Acad. Sci. USA. 95:675-680, 1998.
Hoover et al (Eds ), "Remington's Pharmaceutical Sciences," 15th Edition, Mack
Publishing Co , Easton, PA, 1975 Horwich et al "Synthesis of hepadenovirus particles that contain replication-defective duck hepatitis B virus genomes in cultured HuH7 cells," J Virol . 64 642-650, 1990 Howeroft et / Science 260 1320-1322, 1993
Hwang, Park Park, "Gastric retentive drug-delivery systems," Crit Rev Thei Drug Carrier
Sy st 1 >(3) 243-284, 1998 Hyrup and \ielsen, "Peptide nucleic acids (PNA) synthesis, properties and potential applications," Bworg Med Chem , 4(1) 5-23, 1996 Hyrup and Nielsen, Bworg Med Chem , 1996 Jackson. \ιιcleιc Acids Res , 19 3715-98, 1991 Jamieson et al J Virol , 68 3478-3485, 1994 Jenkins et al I Biol Chem , 265 19624-19631 , 1990 Johnson et l Crit Rev Biochem & Mol Biol 29(1) 1-68, 1994 Jones and Shenk, "Isolation of deletion and substitution mutants of adenovirus type 5." Cell, 13 181 -188, 1978 Jones and Thirup, Curr Comm Molec Biol , 75-76, 1986
Jooss Yang Fisher, Wilson, "Transduction of dendritic cells by DNA viral vectors directs the immune response to transgene products in muscle fibers," / n ol , 727 4212-4223, 1998 09
Kaneda, Iwai Uchida, "Introduction and expression of the human insulin gene in adult rat liver J Biol Chem , 264(21) 12126-12129, 1989 Karacostas et al Virol , 193 661 -671 , 1993
Karussis. Lehmann, Slavm, Vourka-Karussis, Mizrachi-Koll, Ovadia, Kalland and Abramsky, Proc \atl Acad Sci USA, 90 6400-6404, 1993
Kasid et al Ptoc Natl Acad Set USA, 87 473-477, 1990 Kato et al . "Expression of hepatitis D virus surface antigen in adult rat liver," J Biol Chem ,
266 3361 -3364, 1991 Katz. Taubenberger, Cannella, McFarhn, Raine and McFarland, Ann Neurol , 34 661-669, 1993
Kaye et al, "'Cis-acting sequences involved in human immunodeficiency virus type I RNA packaging," J. Virol. 69:6593-6599, 1995. Kelly and O'Connell. Biochemistry, 32:6828-6835, ????.
Kessler, "Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein," Proc. Natl. Acad. Sci. USA, 93:14082-14087, 1996.
Kestier et al , Cell. 65:651-662. 1991. Kim and Kim. J Neuroscl. 29:100-106, 1991. Kim and Smithies, Nucleic Acids Res., 16:8887-8903, 1988.
Kim et al, "Use of the human elongation factor I a promoter as a versatile and efficient expression system." Gene, 91 :217-223, 1996.
Kim, Mitrophanous. Kingsman and Kingsman, "Minimal requirement for a lentivirus vector based on human immunodeficiency virus type 1," J. Virol , 72:81 1-16. Kimpton and Emerman. J Virol, 66:2232-2239, 1992. Kinnula, Crapo and Raivio, Lab. Invest., 73(1):3-19, 1995. Klein, "Neuron-specific transduction in the rat septohippocampal or nigrostriatal pathway by recombinant adeno-associated virus vectors," Exper. Neurol, 150:183-1 4, 1998. 06 Klein, Kornstein, Sanford. Fromm, "High-velocity microprojectiles for delivering nucleic acids into living cells." Nature, 327:70-73, 1987. Klein, Wolf. Wu, Sanford, "High-velocity microprojectiles for delivering nucleic acids into living cells. 1987," Biotechnology, 24:384-386, 1992.
Kohler and Milstein. Eur. J. Immunol, 6:511-519, 1976. Kohler and Milstein, Nature, 256:495-497, 1975. Kohlstaedt, et al, Science, 256:1783-1790, 1992.
Koppelhus, Zachar, Nielsen. Liu, Eugen-Olsen, Ebbesen, "Efficient in vitro inhibition of HIV-1 gag reverse transcription by peptide nucleic acid (PNA) at minimal ratios of
PNA/RNA," Nucl. Acids Res., 25(11):2167-2173. Kotani et al, Human Gene Ther., 5:19-28, 1994. Kovarik et al, ????.
Kremsky, Wooters, Dougherty, Meyers, Collins, Brown, "Immobilization of DNA via oligonucleotides containing an aldehyde or carboxylic acid group at the 5' terminus,"
Nucl. Acids Res., 15(7):2891-2909, 1987.
Kuby, In: Immunology, 2nd Edition. W.H. Freeman & Company, New York, 1994.
Kunkel et al. Methods Enzymol, 154:367-382, 1987.
Kwoh, Davis, Whitfield, Chappelle, DiMichele, Gingeras, "Transcription-based amplification system and detection of amplified human immunodeficiency virus type 1 with a bead-based sandwich hybridization format," Proc. Natl Acad. Sci USA.
86(41:1173-1177, 1989.
Kyte and Doolittle, "A simple method for displaying the hydropathic character of a protein." J. Mol Biol. 157(1 ): 105-132, 1982.
Lafrenie et al. J. Immunol, 156:1638-1645, 1996. Lam et al, Human Gene Ther.. 7: 1415-1422, 1996.
Landau et al. J. Virol, 65: 162-169, 1991.
Lasic, "Novel applications of liposomes," Trends Biotechnol, 16(7):307-321, 1998.
Laughlin, Tratschin. Coon, Carter, "Cloning of infectious adeno-associated virus genomes in bacterial plasmids," Gene, 23(l ):65-73, 1983. Lawman, et al, J. Biol Chem , 266: 10982-10988, 1991.
Le Gal La Salle et al. , "An adenovirus vector for gene transfer into neurons and glia in the brain." Science, 259:988-990, 1 93. e Grice et al EMBO J., 10:3905-391 1. 1991.
Leonard et al. J. Virol, 6:4919-4924. 1989. Lever et al, J. Virol, 63:4085-4087, 1989.
Lever, Gene Therapy. 3:470-471 , 1996.
Levitt, et al. J. Mol Biol, 226:507, 1992.
U et al, Science, 268:429-431, 1995.
Lifson et al. Science, 232:1123-1 127, 1986. Lim and Sauer, Nαtwre, 339:31-36, 1989.
Urn et al, Biochemistry, 31 :4324-4333, 1992.
Lin et al, Proc. Nat. Acad. Sci. USA, 91 :10265-10269, 1994.
Lipman and Pearson, Science, 277:1435, 1985.
Loeb et al, Nature, 329:351-354, 1989. Loh et al, J. Virol, 62:4086-4095, 1988.
Lopez-Berestein et al, "Liposomal amphotericin B for the treatment of systemic fungal infections in patients with cancer: a preliminary study" J. Infect. Dis., 2151 :704,
1985a. Lopez-Berestein et al, "Protective effect of liposomal-amphotericin B against C. albicans infection in mice," Cancer Drug Delivery. 2: 183, 1985b.
Lossinsky, Badmajew, Robson, Moretz and W sniewski, Acta Neuropathol, 78:359-371. 1989. Lower et al. J. Virol, 69: 141-149, 1995. Lowman and Wells, J. Mot. Biol, 234: 564-578. 1993.
Lu, Xiao, Clapp, Li, Broxmeyer, "High efficiency retroviral mediated gene transducion into single isolated immature and replatable CD34(3+) hematopoietic stem/progenitor cells from human umbilical cord blood." J Exp Med, 178(6):2089-2096, 1993. Luban and Goff, "Mutational analysis of cis-acting packaging signals in human immunodeficiency virus type 1 RNA." J Virol, 68:3784-93, 1994. Lyman et al. "Cloning of the human homologue of the murine flt3 ligang: a growth factor for early hematopoietic progenitor cells." Blood, 83 :2795-2801 , 1994.
Macreadie et al, Mol. Microbiol, 19:1 185-1 192. 1996. Mahalingam et al, "Nuclear import, virion incoφoration, and cell cycle arrest/differentiation are mediated by distinct functional domains of human immunodeficiency virus type I
Vpr," J Virol, 71:6339-47, 1997. Maloy et al.. In: Microbiol Genetics, 2nd Edition. Jones and Barlett Publishers, Boston. MA.
1994. Maniatis et al. "Molecular Cloning: a Laboratory Manual," Cold Spring Harbor Laboratory.
Cold Spring Harbor, NY, 1982. Mann et al. "Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus," Cell, 33:153-159, 1983.
Margalit, "Liposome-mediated drug targeting in topical and regional therapies," Crit. Rev.
Ther. Drug Carrier Syst., 12(2-3):233-261, 1995. Markowitz et al, "A safe packaging line for gene transfer: Separating viral genes on two different plasmids," J. Virol, 62:1 120-1 124, 1988. Markowitz et al, J. Virology, 62: 1120-1124, 1988.
Martin et al.. "'Primary structure and functional expression of rat and human stem cell factor
DNAS." Cell, 63:203-21 1 , 1990. Mathiowitz. Jacob, Jong, Carino, Chickering, Chaturvedi, Santos, Vijayaraghavan, Montgomery, Bassett, Morrell, "Biologically erodable microspheres as potential oral drug delivery systems," Nature. 386(6623):410-414, 1997.
Mathisen, Yu. Johnson, Drazba and Tuohy, J Exp. Med., 186:159-164, 1997. McBride and Panganiban, "Position dependent of functional haiφins important for human immunodeficiency virus type 1 RΝA encapsidation in vivo," J. Virol, 71 :2050-58, 1997. McBride and Panganiban, "The human immunodeficiency virus type I encapsulation site is a multipartite RΝA element composed of functional haiφin structures," J. Virol, 70:2963-2973, 1996. McBride et al, "Efficient encapsulation of humna immunodeficiency virus type I vectors and further. chararterization of cis elements required for encapsulation," J. Virol 71 :4544- 4554. 1997.
McCord, N. Engl J. Med, 312:159-163, 1985. McCord. Proc. Soc. Exp. Biol. Med, 209:112-117. 1995. Michael. Biotechniques, 16:410-412, 1994. Miele and A. M. L. Lever, Gene Ther., 3:357-361 , 1995. Miller and Buttimore, Mol. Cell. Biol, 6:2895, 1986. Miller and Chen, J. Virol, 70:5564-5571 , 1996. Miller and Rosman, BioTechn., 7:980, 1989. Miller et al. Mol. Cell Biol, 10:4239, 1990. Miller et al. Meth. Enzymol, 217:581-599, 1993. Miller. Nature, 357: 455-460, 1992.
Miyake et al. Human Gene Ther., 7:2281-2286, 1996.
Miyoshi, Blomer, Takahashi, Gage and Verma, "Development of a self-inactivating lentivirus vector," J Virol, 72:8150-57, 1998. Mizuno et al. AIDS Res. Hum. Retroviruses, 12: 793-800, 1996. Mizushima and Nagata, "PEF-BOS, a powerful mammalian expression vector, " Nucleic Acids Res., 18:5322, 1990.
Mochizuki. Schwartz. Tanaka, Brady and Reiser, "High-titer human immunodeficiency virus type 1 -based vector systems for gene delivery into nondividing cells," J. Virol ,
72:8873-83. 1998. Morris et al. "A new peptide vector for effcient delivery of oligonucleotides into mammalian cells." Nucleic Acids Res., 25(14):2730-2736, 1997.
Mount, "AT-AC introns: an ATtACk on dogma," Science. 271 : 1690-1692. 1996.
Muller et al . "Efficient transfection and expression of heterologous genes in PC 12 cells,"
Cell. Biol . 9(3):22l -229, 1990. Mulligan, "The Basic Science of Gene Therapy," Science, 260:926-932, 1993. Murata et al. J. Exp. Med, 175:341-351, 1992.
Nadcarni, Pervin. Linhardt, "Directional immobilization of heparin to beaded supports," Anal
Biochem., 222:59-67, 1994. Naidini et al . Proc. Nat. Acad. Sci. USA, 93: 1 1382-1 1388, 1996. Naidini et al. Science. 272:263-267, 1996. Neckers and Whitesell. Amer. J. Physiol, 265:L1, 1993.
Needleman and Wunsch, J. Mol Biol, 48:443, 1970, as revised by Smith and Waterman,
Adv. Appl. Math. 2:482, 1981. Nerurkar. Rose, Stobaugh, Borchardt, "Selective fluorogenic derivatization of a peptide nucleic acid trimer with naphthalene-2,3-dicarboxaldehyde." J. Pharm. Biomed. Anal . 15(7):945-950, 1997.
Neyts, Snoeck, Schols. Balzarini, Esko, Van Schepdael, DeClercq, "Sulfated polymers inhibit the interaction of human cytomegalovirus with cell surface heparan sulfate," Virology,
189:48-58, 1992. Nicolau and Gersonde, "Incoφoration of inositol hexaphosphate into intact red blood cells, I. fusion of effector-containing lipid vesicles with erythrocytes," Naturwissenschaften
(Germany), 66(l l):563-566, 1979. Nicolau and Sene. "Liposome-mediated DNA transfer in eukaryotic cells," Biochem.
Biophys. Ada. 721 : 185-190, 1982. Nicolau et al, "Liposomes as carriers for in vivo gene transfer and expression," Methods Enzymol , 149: 157-176, 1987.
Nielsen, P. E.. Egholm. M.. Berg, R. H., and Buchardt, O, "Peptide nucleic acids (PNAs):
Potential antisense and anti-gene agents," Anti-Cancer Drug Design, 8(l):53-63,
1993. Nielsen, P. E.. Egholm. M.. Berg, R. H., and Buchardt, O, "Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide." Science 254.
1497-1500. 1991. Nienhuis et al. "'Gene transfer into hematopoietic cells," Stem Cells, 1 : 123-134. 1997. Nienhuis et al. In: Hematology. Vol. 16: Viruses and Bone Marrow, N.S. Young, ed., pp.
353-414, 1993. Nilsson and Mosbach. "Immobilized animal cells," Dev. Biol. Stand., 66: 183-93, 1987.
Norton, Piatyszek. Wright. Shay, Corey, "Inhibition of human telomerase activity by peptide nucleic acids." Nat. Biotechnol, 14(5):615-619, 1996. Norton, Waggenspack. Varnum, Corey, Bioorg. Med. Chem., 3:437-445, 1995. Nosaka et o/.. Exp. Cell Res.. 209:89-102, 1993. Ohara, Dort. Gilbert. "One-sided polymerase chain reaction: the amplification of cDNA,"
Proc. Natl Acad. Sci. USA, 86(15):5673-5677, 1989. Okuda, Sakoda, Fujimura and Yanagihara, J Neuroimmunol, 81:201-210, 1998. Ono, Hirose. Miyazaki, Yamamoto, Matsumoto, "Transgenic medaka fish bearing the mouse tyrosinase gene: expression and transmission of the transgene following electroporation of the orange-colored variant," Pigment Cell Res.. 10(3): 168-175.
1997. Orlic et al. , "The level of MRNA encoding the amphotropic retrovirus receptor in mouse and human hematopoietic stem cells is low and correlates with the efficiency of retrovirus transduction." Proc. Natl Acad. Sci. USA, 93:11097-11102, 1996. Orum, Nielsen. Egholm, Berg, Buchardt, Stanley, "Single base pair mutation analysis by
PNA directed PCR™ clamping," Nucl. Acids Res., 21(23):5332-5336, 1993. Orum, Nielsen, Jorgensen, Larrson, Stanley, Koch, "Sequence-specific purification of nucleic acids by PNA-controlled hybrid selection," Biotechniques, 19(3):472-480, 1995. Otsuka et al, "Isolation and characterization of an expressible EDNA encoding human IL-3. Induction of IL-3 MRNA in human T cell clones," J. Immunol, 140:2288-2295,
1988.
Overington. et al. Protein Science, 1 :216-226, 1992.
Padmanabhan. Gudapaty, Liener, Schwartz and Hoidal, Am. Rev. Respir. Dis., 132: 164-167.
1985. Vae et al, J. Virol, 64: 5270-5276, 1990. Paillart et al. J. Virol. 70:8348-8354. 1996.
Paillart et α/.. Proc. Natl Acad Sci. USA, 93:5572-5577, 1996.
Panitch, Hirsch, Schindler and Johnson, Neurology. 37:1097-1102, 1987.
Pardridge, Boado, Kang, "Vector-mediated delivery of a polyamide ("peptide") nucleic acid analogue through the blood-brain barrier in vivo," Proc. Natl Acad. Sci. USA. 92(12):5592-5596, 1995.
Park and Morrow, J. Virol, 65:51 1 1-51 17, 1991.
Parks, Melnick, Rongey, Mayor, "Physical assay and growth cycle studies of a defective adeno-satellite virus," J. Virol, 1 (1 ): 171 -180, 1967. Parolin et al. "Analysis in human immunodeficiency virus type I -vectors of cis-acting sequences that affect gene transfer into human lymphocytes," J. Virol, 68:3888-3895.
1994. Paskind et al. "Dependence of moloney murine leukemia virus production on cell growth."
Virology, 67:242-248, 1975. Paterson and Day, Am. J. Pathol, 52:251-263, 1982. Pearl and Taylor, Nature, 329:351-354, 1987.
Pelletier and Sonenberg, "Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA." Nature, 334:320-325, 1988. Perales et al. Proc. Natl. Acad. Sci. USA, 91 :4086-4090, 1994.
Perry-O'Keefe, Yao, Coull, Fuchs, Egholm, "Peptide nucleic acid pre-gel hybridization: an alternative to southern hybridization," Proc. Natl. Acad. Sci. USA, 93(25): 14670-
14675. 1996. Pikul et al, "In vitro killing of melanoma by liposome-delivered intracellular irradiation.
Arch. Surg, 122(12):1417-1420, 1987. Pinto et al, J. Clin. Invest., 96:867-876, 1995. Pinto- Alphandary, Balland, Couvreur, "A new method to isolate polyalkylcyanoacrylate nanoparticle preparations," J. Drug Target, 3(2):167-169, 1995.
Poon et al, J. Virol, 70: 6607-6616, 1996.
Prokop and Bajpai, "Recombinant DNA Technology I," Conference on Progress in
Recombinant DNA Technology Applications, Potosi, MI, June 3-8, 1990, Ann. N. Y.
Acad. Sci., 646:1-383, 1991. Protein Data Bank or the Cambridge Crystal Structure Data Centre. Sequence databases include the Protein Identification Resource (National Biomedical Research
Foundation), GENBANK (Los Alamos National Laboratory), ENML (European cular Biology and SBASE (International Center for Genetic Engineering and
Biotechnology). NRL-3D (U.S. Naval Research Lab), HSSP (EMBL), 3D-ALI (EMBL), FSSP (EMBL), and the Overington database (J.P. Overington, Pfizer
Central Research). Quintanar-Guerrero, Allemann, Doelker, Fessi, "Preparation and characterization of nanocapsules from preformed polymers by a new process based on emulsification- diffusion technique," Phamr. Res., 15(7): 1056-1062. 1998. Racher et al. Biotechnology Techniques, 9:169-174. 1995.
Ragot et al. "Efficient adenovirus-mediated transfer of a human minidystrophin gene to skeletal muscle of mdx mice," Nature, 361 :647-650, 1993. Raine, In Handbook of Clinical Neurology, eds. Vinken, Bruyn and Klawans (Elsevier, New
York). Vol. 3, 1985. Raine, J. Neuroimmunol , 77:135-152, 1997.
Rao, Invest. Ophthαlmol Visual Sci., 20:159-172. 1981.
Regier and Desrosiers, "The complete nucleotide sequence of a pathogenic molecular clone of simian immunodeficiency virus," AIDS. Hum. Retroviruses, 6, 1221-1231, 1990. Renan, "Cancer genes: current status, future prospects, and applicants in radiotherapy/oncology," Radiother. Oncol, 19:197-218, 1990.
Richardson. Child and Lever, "Packaging of human immunodeficiency virus type 1 RNA requires cis-acting sequences outside the 5' leader region," J. Virol, 67:3997-4005,
1993. Ridgeway. "Mammalian expression vectors," In: Vectors: A survey of molecular cloning vectors and their uses, Rodriguez RL, Denhardt DT, ed., Stoneham: Butterworth, pp.
467-492, 1988.
Rippe et al . "DNA-mediated gene transfer into adult rat hepatocytes in primary culture,"
Mol Cell Biol, 10:689-695, 1990. Rizzo and Lessell. Neurology. 38:185-190, 1988.
Robinson et al , "Retroviral vector with a cmv-IE/HIV-TAR hybrid LTR gives high basal expression levels and is upregulated by HIV- 1 Tat," Gene Therapy, 2:269-278, 1995.
Row land-Jones et al. Nature Med., 1 :59-64, 1995. Rose. Berns. Hoggan, Koczot, "Evidence for a single-stranded adenovirus-associated virus genome: formation of a DNA density hybrid on release of viral DNA," Proc. Natl.
Acad Sci USA, 64(3):863-869, 1969. Rosenberg. Human Gene Therapy, 5: 140, 1994.
Roux et al . "A versatile and potentially general approach to the targeting of specific cell types by retroviruses: Application to the infection of human cells by means of major histocompatibility complex class I and class II antigens by mouse ecotropic murine leukemia virus-derived viruses," Proc. Natl Acad Sci USA, 86:9079-9083, 1989. Rubnitz and Subramani, Mot. Cell. Biol, 4:2253-2258. 1984. Ruf et al . Biochemistry, 33:1565-1572, 1994.
Ruuls, Bauer. Sontrop, Huitinga, Hart and Dijkstra, J. Neuroimmunol, 56:207-217, 1995. Sabatino et al, "Amphotropic or gibbon ape leukemia virus retrovirus binding and transduction correlates with the level of receptor MRNA in human hematopoietic cell lines." Blood Cells Mol. Dis., 23:422-433, 1997.
Sadeck, Fernandes, Silva, Trindade, Chia, Ramos, Leone, "Captopril use in pregnancy and its effects on the fetus and the newborn: case report," (Article in Portugese), Rev. Hosp.
Chn Fac. Med. Sao Paulo, 52(6):328-332, 1997. Salvetti, "Factors influencing recombinant adeno-associated virus production," Hum. Gen. Ther.. 9:695-706, 1998.
Sambrook. Fristch, Maniatis, "Molecular Cloning: A Laboratory Manual," C. Nolan, ed.,
Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989. Sandberg and Terwilliger, Proc. Nat. Acad. Sci. USA, 90:8367-8371, 199?. Sarkar and Crissman, 1990. Sarris, Smith, Shumway, Stinson, Oyer, Robbins, Billingham, Theodore, Moore and Katz, J.
Heart Lung Transplant, 13:940-949, 1994.
Sarver, Cantin. Chang. Zaia. Ladne, Stephens, Rossi, "Ribozymes as potential anti-HIV-1 therapeutic agents," Science, 247(4947): 1222-1225, 1990. Scanlon, Jiao. Funato. Wang, Tone, Rossi, Kashani-Sabet, "Ribozyme-mediated cleavage of c-fos mRNA reduces gene expression of DNA synthesis enzymes and metallothionein." Proc. Natl. Acad. Sci. USA, 88(23): 10591 -5, 1991.
Schneider et al. '"Inactivation of the human immunodeficiency virus type I inhibitory elements allow s Rev-independent expression of Gag and Gag/protease and particle formation." ./ Virol, 71 :4892-4903, 1997. Schraufstatter and Cochrane, in The Lung: Scientific Foundations, 2" Ed., eds. Crystal, West, Weibel and Barnes, pp. 2251-2258, 1997.
Schreiber and Fersht. J. Mot. Biol, 248:478-486, 1995. Schwartz et al. J. Virol. 66: 150-159, 1992. Schwinn, et al. J. Biol Chem., 265:8183-8189, 1990.
Sculier et al. "Pilot study of amphotericin B entrapped in sonicated liposomes in cancer patients with fungal infections," J. Cancer Clin. Oncol, 24(3):527-538, 1988.
Seeger, Batz. Orum. "PNA-mediated purification of PCR™ amplifiable human genomic
DNA from whole blood," Biotechniques, 23(3):512-517, 1997. Segal, In: Biochemical Calculations, 2nd Edition. John Wiley & Sons, New York, 1976. Serrano, Gomez-Lahoz. DePinho, Beach, Bar-Sagi, "Inhibition of ras-induced proliferation and cellular transformation by pl6INK4," Science, 267(5195):249-52. 1995.
Serrano, Harmon. Beach. "A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4," Nαtwre, 366(6456):704-7, 1993. Shaw, Lorens. Dhawan. DalCanto, Tse, Tran, Bonpane, Eswaran, Brocke, Sarvetnik, et al. , Exp.
Med. 185:171 1-1714, 1997. Shen and Huang, Genetics, 112: 441-457, ????.
Shenkin, et al. Proteins: Structure, Function and Genetics, 11 :297-313, 1991.
Shimada et al. J. Clin. Investig, 88: 1043-1047, 1991.
Sinclair et al. "Interaction of vesicular stomatitis virus-G pseuctotyped retrovirus with
CD34(-) and CD34(+)CD38(-) hematopoietic progenitor cells," Gene Therapy. 4:918-927, 1997.
Smokik-Utlaut et al, Mot. Cell. Biol, 3:1204-1211, 1983.
Spector et al. J. Virol. 64:2298-2308, 1990.
Srinivasakumar et al... "The effect of viral regulatory protein expression on gene delivery by- human immunodeficiency virus type I vectors produced in stable packaging cell lines." J. Virol. 71 :5841-5848, 1997. Srivastava, Lusby, Berns, "Nucleotide sequence and organization of the adeno-associated virus
2 genome," ./ Virol, 45(2):555-564, 1983. Stallworth and Waldron, "Cortical blindness as a complication of acute glomerulonephritis."
J.S.C. Med. Assoc, 93(3):99-l 01 , 1997. Stec et «/., J. Am. Chem. Soc, 106:6077-6079, 1984. Stein et α/., Gerce, 72:333-341. 1988.
Stemman, Adv. Immunol, 49:357 -379, 1991.
Stephanz et o/.. Exp. Nephrol 4(3): 151-8, 1996.
Stinchcomb, Struhl, Davis. "Isolation and characterization of a yeast chromosomal replicator," Nature, 282(5734):39, 1979. Stratford-Perricaudet and Perricaudet, "Gene transfer into animals: the promise of adenovirus," p. 51-61, In: Human Gene Transfer, Eds, O. Cohen-Haguenauer and M.
Boiron. Editions John Libbey Eurotext, France, 1991. Stratford-Perricaudet et al, "Evaluation of the transfer and expression in mice of an enzyme-encoding gene using a human adenovirus vector," Hum. Gene Ther.. 1 :241-256, 1990.
Subbramanian and Cohen, J. Virol. 68:6831-6835, 1994.
Sullenger, "Revising messages traveling along the cellular information superhighway,"
Chem. Biol, 2(5):249-253, 1995. Summerford and Samulski, "Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions," J. Virol, 72: 1438-1445, 1998.
Suzuki, Shin. Fjuikura, Matsuzaki, Takata, "Direct gene transfer into rat liver cells by in vivo electroporation." FEBS Lett., 425(3):436-440, 1998. Szyf et al, Mol. Cell. Biol, 10:4396-4400, 1990. Takakura, "Drug delivery systems in gene therapy," Nippon Rinsho, 56(3):691-695, 1998.
Takenaga, Serizawa, Azechi, Ochiai, Kosaka, Igarashi, Mizushima, "Microparticle resins as a potential nasal drug delivery system for insulin," J Controller Release, 52(1-2):81-
87, 1998. Tamayose, Hirai, Shimada, "TA new strategy for large-scale preparation of high-titer recombinant adeno-associated virus by using packaging cell lines and sulfonated cellulose column chromatography." Hum. Gene Ther., 7:507-513, 1996. Tang, White. Gordon, Lumb and Tsan, J. Appl Physiol, 74:1425-1431, 1993. Tannahill et α/., Gastroenterology, 109(3):800-1 1. 1995. Tanswell and Freeman, J. Appl. Physiol. 63:343-352, 1987. Tanswell and Freeman, New Horizons. 3:330-341. 1995.
Temin, "Retrovirus vectors for gene transfer: Efficient integration into and expression of exogenous DNA in vertebrate cell genome," In: Gene Transfer, Kucherlapati (ed.),
Plenum Press, New York, pp. 149-188, 1986. Thiede, Bayerdorffer, Blasczyk, Wittig, Neubauer, "Simple and sensitive detection of mutations in the ras proto-oncogenes using PNA-mediated PCR™ clamping," Nucl
Acids Res., 24(5):983-984, 1996. Thisted, Just. Petersen, Hyldig-Nielsen. Godtfredsen, Cell Vision, 3:358-363, 1996. Thomson et al, Tetrahedron, 51 :6179-6194. 1995. Tomic et al, Nucl. Acids Res., 12:1656, 1990. Top et al, "Immunization with live types 7 and 4 adenovirus vaccines. II. Antibody response and protective effect against acute respiratory disease due to adenovirus type 7,"
J. Infect. Dis., 124:155-160, 1971. Trapnell and Gorziglia, Curr. Opin. Biotechnol. 5:617-625, 1994. Trapp, Peterson, Ransohoff, Rudick, Mork and Bo. N. Engl J. Med, 338:278-285, 1998. Tratschin, Miller, Carter, "Genetic analysis of adeno-associated virus: properties of deletion mutants constructed in vitro and evidence for an adeno-associated virus replication function," J Virol, 51(3):611-619. 1984a. Tratschin, West, Sandbank, Carter, "A human parvovirus, adeno-associated virus, as a eucaryotic vector: transient expression and encapsidation of the procaryotic gene for chloramphenicol acetyltransferase." Mol. Cell. Biol, 4(10):2072-2081, 1984b.
Tripathy et al. Nature Med., 2:545-550. 1996.
Trono, "HIV accessory proteins: leadina roles for the supporting cast," Cell, 2:189-192,
1995. Truong-Le, August, Leong, "Controlled gene delivery by DNA-gelatin nanopspheres," Hum.
Gene Ther., 9(12): 1709-1717, 1998. Tur-Kaspa et al. , "Use of electroporation to introduce biologically active foreign genes into primary rat hepatocytes," Mol. Cell Biol, 6:716-718, 1986. Turrens, Crapo and Freeman, J. Clin. Invest, 73:87-95, 1984. Uetsuki et al. J. Biol. Chem., 264:5791, 1989. Unger, et al. Proteins, 5:335, 1989. Upender et al. Biotechniques, 18:29-31, 1995. Ore et al, Develop. Biol, 154:388-395, 1992.
Vaishnav et al, "The biochemistry of AIDS," Ann. Rev. Biochem., 60:577-630, 1991. Van Cott, Lubon, Russell, Butler, Gwazdauskas, Knight, Drohan, Velander, "Phenotypic and genotypic stability of multiple lines of transgenic pigs expressing recombinant human protein C," Transgenic Res., 6(3):203-212, 1997. van der Veen. Hinton, Incardonna and Hofman, J Neuroimmunol, 77:1-7, 1997.
Vanbever, Fouchard, Jadoul, De Morre, Preat, Marty, "In vivo noninvasive evaluation of hairless rat skin after high-voltage pulse exposure," Skin Parmacol Appl. Skin
Physiol , l l(l):23-34, 1998. Varmus et al, "Retroviruses as mutagens: Insertion and excision of a nontransforming provirus alter the expression of a resident transforming provirus," Cell, 25:23-36,
1981. Vasanthakumar and Ahmed, "Modulation of drug resistance in a daunorubicin resistant subline with oligonucleoside methylphosphonates," Cancer Commun., l(4):225-232, 1989.
Venkatachalarn, et al, J. Biol. Chem., 269:23444-23450, 1994.
Veselkov, Demidov, Frank-Kamenetskii, Nielsen, "PNA as a rare genome-cutter," Nature,
379(6562):214, 1996. Vickers, Griffith, Ramasamy, Risen, Freier, "Inhibition of NF-kappa B specific transcriptional activation by PNA strand invasion," Nucl. Acids Res., 23(15):3003-
3008. 1995.
Wagner, Plank, Zatloukal, Cotten, Birnstiel, "Influenza virus hemagglutinin HA-2 N- terminal fusogenic peptides augment gene transfer by transferrin-polylysine-DNA complexes: toward a synthetic virus-like gene-transfer vehicle," Proc. Natl. Acad.
Sci. USA. 89(17):7934-8, 1992a. Wagner, Zatloukal. Cotten, Kirlappos, Mechtler, Curiel, Birnstiel, "Coupling of adenovirus to transferrin-polylysine/DNA complexes greatly enhances receptor-mediated gene delivery and expression of transfected genes." Proc. Natl Acad. Sci. USA,
89(13 ):6099-6103, 1992. Walker et al . Proc. Natl. Acad. Sci. USA, 89(l):392-396, 1992. Walfher, Dav id-Cu and Lopez, Am. J. Physiol. , 269:L613-L617, 1995. Wang et al, Cytotechnology, 9(l-3):41-9, 1992. Warren and Ward, in 772e Lung: Scientific Foundations, 2" Ed., eds. Crystal, West, Weibel and
Barnes, pp. 2279-2288, 1997. Watson et al. In: Recombinant DNA, 2d Ed., W.H Freeman & Co., NY, pp. 256-263, 1992. Watson, "Fluid and electrolyte disorders in cardiovascular patients," Nurs Clin. North Am.,
22(4):797-803. 1987. Watt et al, Proc. Nat. Acad. Sci. USA, 82:4768-4772, 1985. Webb and Hurskainen, J. Biomol Screen., 1:119-121, 1996.
Webb, Toth. Poehlman, "Influence of physiological factors on the age-related increase in blood pressure in healthy men," Exp. Gerontol, 31(3):341-350, 1996.
Weinberg, Zaaoli, Kadiwar, Yuspa, "p53 gene dosage modifies growth and malignant progression of keratinocytes expressing the v-rasHa oncogene," Cancer Res.,
54(21 V.5584-92, 1994. Weiner, Mackin, Matsui, Orav, Khour, Dawson and Hafler, Science, 259:1321-1324, 1993. Weiss, Science, 272:1885-1886, 1996.
Wells, Biochemistry, 29:8509-8517, 1990. Westendoφ et al, EMBO J., 14:546-554, 1995. Westendoφ et al, Nature, 375:497-500, 1995.
Wettstein, Colombo, Jaenisch, "Non-H2 histocompatibility antigens encoded by Moloney- murine leukemia virus in mov mouse strains are detectable by skin grafting and cytolytic T lymphocytes," J. Immunol, 140(12):4337-4341, 1988.
White, Avraham, Shanley and Groner, J Clin. Invest., 87:2162-2168, 1991. Wilbar and Lipman, Proc. Nat. Acad. Sci. USA, 80:726-730, 1983. Williams, et al... J. Biol Chem., 270:3012-3016, 1995. Wills, AIDS. 5:639-54. 1991. Wispe, Warner, Clark. Dey, Neuman, Glasser, Crapo, Chang and Whitsett. J. Biol Chem.. 267:23937-23941. 1992. Wong and Neumann. "Electric field mediated gene transfer," Biochim. Biophys. Res.
Commun.. 107(2):584-587, 1982. Wong et al. "Appearance of β-lactamase activity in animal cells upon liposome mediated gene transfer." Gene, 10:87-94, 1980.
Wong, T. E.. and Neumann, E., "Electric field mediated gene transfer," Biochim. Biophys.
Res. Commun. 107(2):584-587, 1982. Wong-Staal and Wong-Staal, Microbiol. Rev., 55: 1 93-205, 1991. Wright, Cohn. Fischer and Adler, Environ. Health Perspect., 102:85-90, 1994. Wu and Dean. "Functional significance of loops in the receptor binding domain of Bacillus thuringiensis CrylllA delta-endotoxin," J. Mol. Biol, 255(4):628-640, 1996. Wu and Wu. "Evidence for targeted gene delivery to HepG2 hepatoma cells in vitro."
Biochemistry, 27:887-892, 1988. Wu and Wu. "Receptor-mediated in vitro gene transfections by a soluble DNA carrier system." J. Biol. Chem., 262:4429-4432, 1987.
Wu and Wu. Adv. Drug Delivery Rev., 12:159-167, 1993. Wu, et al, J. Exp. Med, 132:21 1-249, 1970.
Wu. and Dean. "Functional significance of loops in the receptor binding domain of Bacillus thuringiensis CrylllA δ-endotoxin," J. Mol. Biol. 255:628-640, 1996. Xiao and Samulski, "Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector," J Virol, 70:8090-8108, 1996. Xiao, Li, Samulski, "Production of high-titer recombinant adeno-associated virus vectors in the absence of helper Adenovirus," J Virol, 72:2224-2232, 1998. Yang et al. "In vivo and in vitro gene transfer to mammalian somatic cells by particle bombardment," Proc. Natl Acad. Sci. USA, 87:9568-9572, 1990.
Yu et al, J. Virol, 70:4530-4537, 1996.
Yu et al, J. Mol. Biol. 249:388-397, 1995.
Zambaux, Bonneaux. Gref, Maincent, Dellacherie, Alonso, Labrude, Vigneron, "Influence of experimental paparmeters on the characteristics of poly(lactic acid) nanoparticles prepared by a double emulsion method," J. Controlled Release, 50(l-3):31 -40. 1998. Zatloukal, L.. Wagner, E., Cotten, M., Phillips, S., Plank, C, Steinlein, P., Curiel. D. T., and
Birnstiel, M. L., "Transferrinfection: a highly efficient way to express gene constructs in eukaryotic cells," Ann. N Y Acad. Sci., 660: 136-153, 1992. Zelenin et al, "High-velocity mechanical DNA transfer of the chloramphenicol acetyltransferase gene into rodent liver, kidney and mammary gland cells in organ explants and in vivo." FEBS Lett., 280:94-96, 1991.
Zolotukhin, Potter, Hauswirth, Guy, Muzyczka, "A humanized green fluorescent protein cDNA adapted for high level expression in mammalian cells." J. Virol, 70:4646-4654, 1996. Zuckerman, et al, Proc. Nat. Acad. Sci. USA, 89:4505-4509, 1992.
Zufferey, Nagy, Mandel. Naldini and Trono, "Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo," Nat. Biotechnol, 15:871-75, 1997. zur Muhlen, Schwarz, Mehnert, "Solid lipid nanoparticles (SLN) for controlled drug delivery— drug release and release mechanism," Eur. J. Pharm. Biopharm., 45(2): 149-
155, 1998Noss et α/., Trends Biochem. Sci., 11 :287, 1986.
All of the compositions and methods disclosed and claimed herein can be made and executed without undue experimentation in light of the present disclosure. While the compositions and methods of this invention have been described in terms of preferred embodiments, it will be apparent to those of skill in the art that variations may be applied to the compositions and methods and in the steps or in the sequence of steps of the method described herein without departing from the concept, spirit and scope of the invention. More specifically, it will be apparent that certain agents which are both chemically and physiologically related may be substituted for the agents described herein while the same or similar results would be achieved. All such similar substitutes and modifications apparent to those skilled in the art are deemed to be within the spirit, scope and concept of the invention as defined by the appended claims.