Process For Preparing A Chiral Nucleoside Analogue
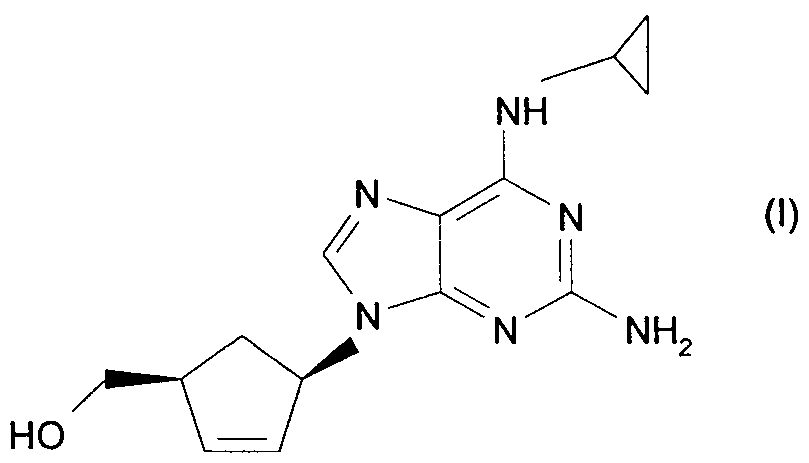
The present invention relates to a new process for the preparation of the chiral nucleoside analogue (1S, 4R)-4-[2-amino-6-(cyclopropylamino)-9H purin-9-yl]-2- cyclopentene-1 -methanol (compound of Formula (I)).
The compound of formula (I) is described as having potent activity against human immunodeficiency virus (HIV) and hepatitis B virus (HBV) in EPO34450.
Results presented at the 34th Interscience Conference on Antimicrobial Agents and Chemotherapy (October 4-7, 1994) demonstrate that the compound of formula I has significant activity against HIV comparable to, and if not better than, some current anti HIV drugs, such as zidovudine and didanosine.
Currently the compound of Formula (I) is undergoing clinical investigation to determine its safety and efficacy in humans. Therefore, there exists at the present time a need to supply large quantities of this compound for use in clinical trials.
Current routes of synthesising the compound of formula (I) involve multiple steps and are relatively expensive. It will be noted that the compound has two centres of asymmetry and it is essential that any route produces the compound of formula (I) substantially free of the corresponding enantiomer, preferably the compound of formula (I) is greater than 95% w/w free of the corresponding enantiomer.
Processes proposed for the preparation of the compound of formula (I) generally start from a pyrimidine compound, coupling with a 4-amino-2-cyclopentene-1- methanol analogue, cyciisation to form the imidazole ring and then introduction of
the cyclopropylamine group into the 6 position of the purine, such routes include those suggested in EPO434450 and WO9521161. Essentially both routes disclosed in the two prior patent applications involve the following steps:-
(i) coupling (1S, 4R)-4-amino-2-cyclopentene-1 -methanol to N-(4,6-dichloro-5- formamido-2-pyrimidinyl) acetamide or a similar analogue thereof, for example N- (2-amino-4,6-dichloro-5-pyrimidinyl) formamide;
(ii) ring closure of the resultant compound to form the intermediate (1 S, 4R)-4- (2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1 -methanol;
(iii) substituting the halo group by a cyclopropylamino group on the 6 position of the purine ring.
The above routes are multi-step processes. By reducing the number of processing steps significant cost savings can be achieved due to the length of time to manufacture the compound being shortened and the waste streams minimised.
An alternative process suggested in the prior art involves the direct coupling of carbocyclic ribose analogues to the N atom on the 9 position of 2-amino-6-chloro purine. For example WO91/15490 discloses a single step process for the formation of the (1S, 4R)- 4-(2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1- methanol intermediate by reacting (1S, 4R)-4-hydroxy-2-cyclopentene-1 -methanol, in which the allylic hydroxyl group has been activated as an ester or carbonate and the other hydroxyl group has a blocking group attached (for example 1 ,4- bis- methylcarbonate) with 2-amino-6-chloropurine.
However we have found that when synthesising (1S, 4R)-4-(2-amino-6-chloro-9H- purin-9-yl)-2-cyclopentene-1- methanol by this route a significant amount of an N- 7 isomer is formed (i.e. coupling has occurred to the nitrogen at the 7- position of the purine ring) compared to the N-9 isomer desired. Further steps are therefore required to convert the N-7 product to the N-9 product, or alternatively removing the N-7 product, adding significantly to the cost.
We have found that by using a transition metal catalysed process for the direct coupling of a compound of formula (II) or (III),
wherein X is H or a blocking group and Y is H or an activating group, with 2-amino- 6-(cyclopropylamino)purine, the compound of formula (I) is produced in excellent yield and with excellent N9-selectivity.
This process offers significant cost savings compared to previous processes described and represents the first aspect of the present invention. The major commercial advantages are the length of time to manufacture the compound of formula (I) is significantly reduced using the process of the invention compared to earlier processes described, and wastage is significantly reduced.
Accordingly presented as the first feature of the present invention is a process for the preparation of a compound of formula (I) comprising reacting a compound of formula (II) or (III),
wherein X is H or a blocking group and Y is H or an activating group, with 2- amino-6-(cyclopropylamino) purine in the presence of a catalyst.
It will be understood that the process described above could also be used where the compound of formula (II) or (III) used is a mixture of enantiomers, particularly a racemic mixture, and that the compound formed would be a mixture of enantiomers of the compound of formula (I). The mixture of enantiomers of the compound of formula (I) may then be resolved to the compound of formula (I) by chiral chromatographic techniques or by reaction with stereoselective enzymes, for
example esterifying the compound of formula (I) and resolving by processing with an esterase, as described in EP 034450.
Therefore accordingly presented as a second feature of the invention is a process for the preparation of a mixture of enantiomers of a compound of formula (I) comprising reacting a mixture of enantiomer of a compound of formula (II) or (III),
wherein X is H or a blocking group and Y is H or an activating group, with 2-amino- 6-(cyciopropylamino) purine. An optional further step is resolving the product of the invention to a compound of formula (I) substantially free of the corresponding enantiomer.
2-Amino-6-cyclopropylamino-9H-purine can be prepared by treating 2-amino-6- chloropurine with cyclopropylamine, or in accordance with the methods described in US5420115.
(1S, 4R)- 4-Hydroxy-2-cyclopentene-1 -methanol and the activated and/or blocked version of (1S, 4R)-4-hydroxy-2-cyclopentene-1-methanol may be prepared in accordance with reactions described in WO91/15490 or Hodgson et al. in J.Chem. Soc. Perkin Trans. 1 1994; 3373-3378..
(1 R,2R)-2-Hydroxy-3-cyclopentene-1 -methanol may be prepared in accordance with the procedures described in WO92/18444.
The preferred catalyst is a transition metal catalyst, preferably a palladium compound but may also be a derivative of other transition metals (such as nickel, molybdenum, tungsten), preferably in the presence of a ligand such as a phosphine. Preferred transition metal catalyst are tetrakis (triphenylphosphine) palladium, or tris(dibenzylideneacetone) dipalladium, in the presence of triphenylphosphine
Suitable activated derivatives of the allylic alcohol group of the compound of formula (II) or (III) include esters (such as acetate); carbonates (such as methylcarbonate); carbamates, preferably RR 'NOC wherein R and R1 independently selected from C1-6alkyl, aryl or heteroaryl; or phosphates, preferably (RO)2OP wherein each R is independently selected from C1-6alkyl, aryl and heteroaryl. Alternatively the allylic alcohol may be activated in situ by the addition of a metal salt, for example stannous chloride (see, for example, Y. Masuyama et al, Chem.Lett. 1995 p. 1121 and references contained therein).
A base such as sodium hydride or cesium carbonate may optionally be added, particularly where the activated derivative is a simple ester such as acetate.
For the compound of formula (III) a preferred activitating blocking groups are cylic carbonates (such as used for the compound of formula (IV).
Suitable blocking groups may be any such groups recognised in the art of organic chemistry as suitable for protecting primary hydroxymethyl groups, suitable blocking groups include those reported by T W Green in Protecting Groups in Organic Synthesis, Chapter 7, page 10. J. Wiley and sons, New York, 1981 , and include esters, ethers and carbonates (such as methyl carbonate).
Suitable solvents include, for example, dimethylsulphoxide, N,N- dimethylformamide, N,N-dimethylacetamide or tetrahydrofuran preferably at temperatures between 0° and 150°C.
Glossary
The following abbreviations are used:
DMF = N, N-dimethyl formamide DMSO = dimethyl sulfoxide
Example 1 (1 S. 4R)-4-[2-Amino-6-(cvclopropylamino)-9H purin-9-vπ-2-cvclopentene-1 - methanol
Triphenylphosphine (14mg) was added, under nitrogen, to a mixture of (1S.4R)- 4-hydroxy-2-cyclopentene -1 -methanol bis(methylcarbonate) (91 mg), 2-amino-6- (cyclopropylamino) purine (90mg), tris(dibenzylideneacetone)dipalladium (12mg) and dry DMF (2ml) and the resulting solution stirred at room temperature for 40 min.
The DMF was removed at 60° in vacuo and the residue partitioned between ethyl acetate (25ml.) and 20% sodium chloride solution (10ml.). The ethyl acetate solution was washed with 20% sodium chloride (2x12ml.) and with saturated sodium chloride solution, then dried (MgSO4) and the solvent removed in vacuo.
The residue was dissolved in methanol (10ml.), potassium carbonate (17mg) added and the mixture stirred under nitrogen for 15h.
The solvent was removed in vacuo and the residue chromatographed on silica gel
(Merck 9385), eluting with dichloromethane-methanol [(95:5) increasing to (90:10)] to give the title compound (53mg) as a cream foam.
δ(DMSO-d6): 7.60 (s.1 H); 7.27 (s,1 H); 6.10 (dt,1 H); 5.86 (dt, 1 H); 5.81 (s,2H); 5.39 (m,1H); 4.75 (t,1H); 3.44 (t,2H); 3.03 (m, 1H): 2.86 (m,1H);2.60 (m,1H); 1.58 (dt, 1 H); 0.65 (m, 2H); 0.57 (m,2H).
TLC SiO2/CHCI3-MeOH (4:1 ) Rf 0.38; det. UN., KMnO4
Example 2
(1 S. 4R)-4-[2-Amino-6-(cyclopropylamino )-9H-purin-9-vπ-2-cvclopentene-1 ■ methanol
A stirred mixture of 2-amino-6-(cyclopropylamino)purine (100mg) and cesium carbonate (175mg) in dry DMSO (5ml) was heated at 60° under nitrogen for 2h. The mixture was cooled to room temperature then tetrakis(triphenylphosphine) palladium (85mg) and a solution of (1S,4R)-4-hydroxy-2-cyclopentene-1 -methanol diacetate (79mg) in DMSO (1 ml) were added. The mixture was then heated at 65° for 2.25h.
Methanol (10ml) and potassium carbonate (210mg) were added and the mixture stirred at 40° for 45min, after which the solid was filtered off on Celite and the filtrate evaporated to low volume at 90° in vacuo. The residual gum was triturated with dichloromethane (2x10ml) and the resulting brown solid residue chromatographed in silica gel (Merck 9385), eluting with dichloromethane- methanol (9:1 ) to give the title compound (26mg) as a yellow foam, identical by NMR to the product of Example 1. Tic SiO2/CHCI3-MeOH (4:1 ) Rf 0.38; det UN., KMnO4
Example 3
(1 S. 4R)-4-r2-Amino-6-(cvclopropylaminoV9H-purin-9-yl1-2-cvclopentene-1 ■ methanol
A stirred mixture of 2-amino-6-(cyclopropylamino)purine (1.00g) and cesium carbonate (1.75 g) in dry DMSO (50 ml_) was heated at 60°c under nitrogen for 2 h. The mixture was cooled to room temperature then tetrakis(triphenylphosphine) palladium (0.85 g) and a solution of (1R,2R)-2-hydroxy-3-cyclopentene-1- methanol diacetate (0.79 g) in DMSO (10 ml_) was added. The mixture was heated at 65°C for 2.5 h then allowed to cool to room temperature overnight. Methanol (100 ml_) and potassium carbonate (2.10 g) were added and the mixture was stirred at 40°C for 1 h after which the mixture was filtered through Celite. The filtrate was evaporated to low volume at 90°C in vacuo to give a brown oil. Dichloromethane (2x100 ml) was added to the oil and evaporated. The resulting crude oil was chromatographed on silica gel (elution with dichloromethane- methanol (9:1 )) to give the title compound as a yellow foam (0.12 g), identical by ΝMR to the product of Example 1. TLC: (silica gel, methylene chloride/methanol 9/1 (v/v); Rf = 0.45. Rf of reference product, 0.45).
Example 4
2-Amino-6-cvclopropylamino-9H-purine
A suspension of 2-amino-6-chloropurine (5.0g) in methanol (15ml) was treated with cyclopropylamine (12.5ml). The suspension was heated at reflux for 28h. The resultant solution was allowed to cool to 40°C and was evaporated in vacuo to give a yellow-orange foam. The foam was dissolved in isopropanol (200ml) and methanol (50ml) and stirred with potassium carbonate (5.0g). The suspension was cooled to 0-5°C and stirred for 30 minutes. The suspension was filtered, the filtrate was evaporated and the residue was dried in a vacuum oven at 45°C for 64h to give an orange foam (7.11g). A portion of this foam (1.0g) was recrystallised from acetonitrile (10ml) and isopropanol (1.0ml). The solid was isolated by filtration and dried in a vacuum oven at 45°C to give a pale orange solid (0.622g).
δ (DMSO-d6): 7.61 (s, 1 H); 7.15 (s, 1 H); 5.6 (s,2H); 3.01 (m, 1 H); 0.52-0.70 (m, 4H) M.Pt. 150-152°C.