WO1997036892A1 - Inhibitors of farnesyl-protein transferase - Google Patents
Inhibitors of farnesyl-protein transferase Download PDFInfo
- Publication number
- WO1997036892A1 WO1997036892A1 PCT/US1997/005060 US9705060W WO9736892A1 WO 1997036892 A1 WO1997036892 A1 WO 1997036892A1 US 9705060 W US9705060 W US 9705060W WO 9736892 A1 WO9736892 A1 WO 9736892A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- aryl
- hydrogen
- independently selected
- substituted
- Prior art date
Links
- 0 C(C1*(Cc2cc3ccccc3cc2)C=*C1)*(CC*1c2ccccc2)C11O*1 Chemical compound C(C1*(Cc2cc3ccccc3cc2)C=*C1)*(CC*1c2ccccc2)C11O*1 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/64—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms, e.g. histidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4178—1,3-Diazoles not condensed 1,3-diazoles and containing further heterocyclic rings, e.g. pilocarpine, nitrofurantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Definitions
- Ras proteins are part of a signalling pathway that links cell surface growth factor receptors to nuclear signals initiating cellular proliferation.
- Biological and biochemical studies of Ras action indicate that Ras functions like a G-regulatory protein.
- Ras In the inactive state, Ras is bound to GDP.
- Ras Upon growth factor receptor activation Ras is induced to exchange GDP for GTP and undergoes a conformational change.
- the GTP-bound form of Ras propagates the growth stimulatory signal until the signal is
- Mutated ras genes (Ha-ras, Ki4a-ras, Ki4b-ras and N-ras) are found in many human cancers, including colorectal carcinoma, exocrine pancreatic carcinoma, and myeloid leukemias. The protein products of these genes are defective in their GTPase activity and constitutively transmit a growth stimulatory signal.
- Ras C-terminus contains a sequence motif termed a "CAAX” or "Cys-Aaa 1 -Aaa 2 -Xaa” box (Cys is cysteine, Aaa is an aliphatic amino acid, the Xaa is any amino acid) (Willumsen et al., Nature 310:583-586 (1984)).
- this motif serves as a signal sequence for the enzymes farnesyl-protein transferase or geranylgeranyl-protein transferase, which catalyze the alkylation of the cysteine residue of the CAAX motif with a C 15 or C 20 isoprenoid, respectively.
- the Ras protein is one of several proteins that are known to undergo post-translational farnesylation.
- farnesylated proteins include the Ras-related GTP-binding proteins such as Rho, fungal mating factors, the nuclear lamins, and the gamma subunit of transducin. James, et al., J. Bi ⁇ l. Chem. 269, 14182 (1994) have identified a peroxisome associated protein Pxf which is also farnesylated. James, et al., have also suggested that there are Ras-related GTP-binding proteins such as Rho, fungal mating factors, the nuclear lamins, and the gamma subunit of transducin. James, et al., J. Bi ⁇ l. Chem. 269, 14182 (1994) have identified a peroxisome associated protein Pxf which is also farnesylated. James, et al., have also suggested that there are
- FPTase farnesyl-protein transferase
- FPP farnesyl diphosphate
- Ras protein substrates
- the peptide derived inhibitors that have been described are generally cysteine containing molecules that are related to the CAAX motif that is the signal for protein prenylation. (Schaber et al., ibid; Reiss et. al., ibid; Reiss et al., PNAS, 88:732-736 (1991)).
- Such inhibitors may inhibit protein prenylation while serving as alternate substrates for the farnesyl-protein transferase enzyme, or may be purely competitive inhibitors (U.S.
- transferase inhibitors are inhibitors of proliferation of vascular smooth muscle cells and are therefore useful in the prevention and thereapy of arteriosclerosis and diabetic disturbance of blood vessels (JP H7- 112930).
- the present invention comprises small molecule imidazolidinone-containing compounds which inhibit the farnesyl- protein transferase.
- the instant compounds lack a thiol moiety and thus offer unique advantages in terms of improved pharmacokinetic behavior in animals, prevention of thiol-dependent chemical reactions, such as rapid autoxidation and disulfide formation with endogenous thiols, and reduced systemic toxicity.
- chemotherapeutic compositions containing these farnesyl transferase inhibitors and methods for their production are further contained in this invention.
- the compounds of this invention are useful in the inhibition of farnesyl-protein transferase and the farnesylation of the oncogene protein Ras.
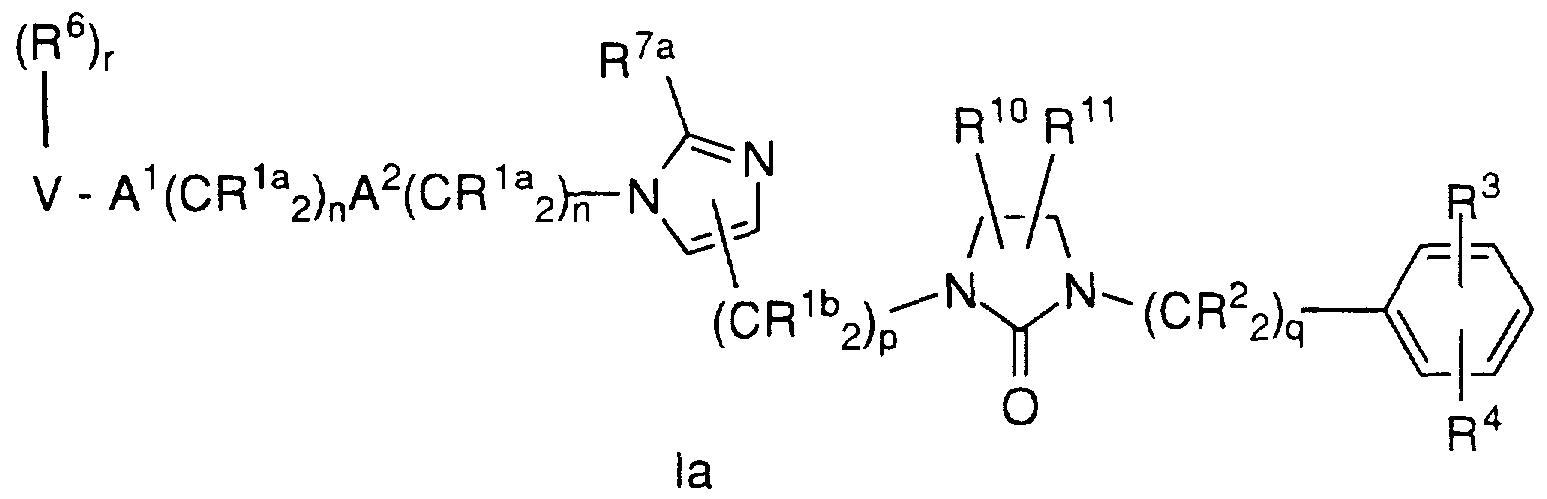
- the inhibitors of farnesyl-protein transferase are illustrated by the formula I: wherein:
- R 1 a , R 1 b and R 2 are independently selected from:
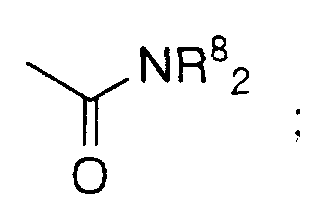
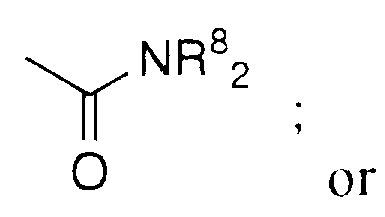
- R 3 and R 4 are independently selected from F, Cl, Br, N(R 8 ) 2 , CF 3 ,
- R 6 is independently selected from:
- heterocycle C 3 -C 10 cycloalkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, perfluoroalkyl, F, Cl, Br, R 8 O-, R 9 S(O) m -.
- R 7 is attached to a substitutable carbon on W and is selected from:
- R 10 and R 11 are independently selected from: H; or
- V is selected from:
- V is not hydrogen if A 1 is S(O) m and V is not hydrogen if A 1 is a bond, n is 0 and A 2 is S(O) m ;
- W is a heterocycle
- Y is aryl or heteroaryl; m is 0, 1 or 2;
- n 0, 1 , 2, 3 or 4;
- p 0, 1 , 2, 3 or 4;
- q 0, 1 , 2, 3 or 4;
- r is 0 to 5, provided that r is 0 when V is hydrogen; and t is 0 or 1; or the pharmaceutically acceptable salts thereof.
- R 1 a and R 2 are independently selected from: hydrogen or C 1 -C 6 alkyl;
- R 1 b is independently selected from:
- heterocycle cycloalkyl, alkenyl, R 8 O-, or -N(R 8 ) 2 ;
- R 3 and R 4 are independently selected from F, Cl, Br, N(R 8 ) 2 , CF 3 ,
- R 6 is independently selected from:
- R 7a is hydrogen or methyl
- R 8 is independently selected from hydrogen, C 1 -C 6 alkyl, benzyl and aryl
- R 9 is independently selected from C 1 -C 6 alkyl and aryl
- R 10 and R 11 are independently selected from: H; or
- V is selected from:
- heterocycle selected from pyrrolidinyl, imidazolyl,
- aryl d) C 1 -C 20 alkyl wherein from 0 to 4 carbon atoms are replaced with a a heteroatom selected from O, S. and N, and
- V is not hydrogen if A 1 is S(O) m and V is not hydrogen if A 1 is a bond, n is 0 and A 2 is S(O) m ; m is 0, 1 or 2;
- n 0, 1, 2, 3 or 4;
- p 0, 1 , 2, 3 or 4;
- q 0, 1 , 2, 3 or 4;
- r is 0 to 5, provided that r is 0 when V is hydrogen; or the pharmaceutically acceptable salts thereof.
- R 1 a and R 2 are independently selected from: hydrogen or C 1 -C 6 alkyl
- R 1 b is independently selected from:
- R 3 and R 4 are independently selected from F, Cl, Br, N(R 8 ) 2 , CF 3 , NO 2 , (R 8 )O-, (R 9 )S(O) m -, (R 8 )C(O)NH-, H 2 N- C(NH)-, (R 8 )C(O)-, (R 8 )OC(O)-, N 3 , CN, (R 9 )OC(O)NR 8 -, C 1 -C 20 alkyl, substituted or
- R 6 is independently selected from:
- perfluoroalkyl F, Cl, R s O-, R 8 C(O)NR 8 -, CN, NO 2 , (R 8 ) 2 N-C(NR 8 )-, R 8 C(O)-, R 8 OC(O)-, -N(R 8 ) 2 , or R 9 OC(O)NR 8 -, and
- R 7 is selected from: hydrogen and C 1 -C 6 alkyl
- R 8 is independently selected from hydrogen, C 1 -C 6 alkyl, benzyl and aryl
- R 9 is independently selected from C 1 -C 6 alkyl and aryl
- R 10 and R 11 are independently selected from:
- V is selected from:
- heterocycle selected from pyrrolidinyl, imidazolyl,
- V is not hydrogen if A 1 is S(O) m and V is not hydrogen if A 1 is a bond, n is 0 and A 2 is S(O) m ;
- W is a heterocycle selected from pyrrolidinyl, pyridinyl, thiazolyl, pyridonyl, 2-oxopiperidinyl, indolyl, quinolinyl, or isoquinolinyl; m is 0, 1 or 2;
- n 0, 1 , 2, 3 or 4;
- p 0, 1 , 2, 3 or 4;
- q 0, 1 , 2, 3 or 4;
- r is 0 to 5, provided that r is 0 when V is hydrogen: and t is 1 ; or the pharmaceutically acceptable salts thereof.
- the inhibitors of farnesyl-protein transferase are illustrated by the formula Ic:
- R 1 b is independently selected from:
- heterocycle cycloalkyl, alkenyl, R 8 O-, or -N(R 8 ) 2 ;
- R 2 are independently selected from: hydrogen or C 1 -C 6 alkyl
- R 3 and R 4 are independently selected from F, Cl, Br, N(R 8 ) 2 , CF 3 ,
- R 6 is independently selected from:
- R 10 and R 11 are : independently selected from: H; or
- n 0, 1 or 2;
- p 0, 1 , 2, 3 or 4;
- q 0, 1 , 2, 3 or 4; or the pharmaceutically acceptable salts thereof.
- the inhibitors of farnesyl-protein transferase are illustrated by the formula Id:
- R 1 b is independently selected from:
- heterocycle cycloalkyl, alkenyl, R 8 O-, or -N(R 8 ) 2 ;
- R 2 are independently selected from: hydrogen or C 1 -C 6 alkyl
- R 3 and R 4 are independently selected from F, Cl, Br, N(R 8 ) 2 , CF 3 ,
- R 8 is independently selected from hydrogen, C 1 -C 6 alkyl, benzyl and aryl; R 9 is independently selected from C 1 -C 6 alkyl and aryl; R 1 b and R 11 are independently selected from: H; or
- n 0, 1 or 2;
- p 0, 1 , 2, 3 or 4;
- q is 0, 1 , 2, 3 or 4; or the pharmaceutically acceptable salts thereof.
- the compounds of the present invention may have asymmetric centers and occur as racemates, racemic mixtures, and as individual diastereomers, with all possible isomers, including optical isomers, being included in the present invention.
- any variable e.g. aryl, heterocycle, R 1 a , R 2 etc.
- its definition on each occurence is independent at every other occurence.
- combinations of substituents/or variables are permissible only if such combinations result in stable compounds.
- alkyl is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms; “alkoxy” represents an alkyl group of indicated number of carbon atoms attached through an oxygen bridge.
- Halogen or “halo” as used herein means fluoro, chloro, bromo and iodo.
- aryl is intended to mean any stable monocyclic or bicyclic carbon ring of up to 7 members in each ring, wherein at least one ring is aromatic.
- aryl elements include phenyl, naphthyl, tetrahydronaphthyl, indanyl, biphenyl, phenanthryl, anthryl or acenaphthyl.
- heterocycle or heterocyclic represents a stable 5- to 7-membered monocyclic or stable 8- to 1 1 - membered bicyclic heterocyclic ring which is either saturated or unsaturated, and which consists of carbon atoms and from one to four heteroatoms selected from the group consisting of N, O, and S, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the heterocyclic ring may be attached at any heteroatom or carbon atom which results in the creation of a stable structure.
- heterocyclic elements include, but are not limited to, azepinyl, benzimidazolyl, benzisoxazolyl, benzofurazanyl, benzopyranyl, benzothiopyranyl, benzofuryl,
- heteroaryl is intended to mean any stable monocyclic or bicyclic carbon ring of up to 7 members in each ring, wherein at least one ring is aromatic and wherein from one to four carbon atoms are replaced by heteroatoms selected from the group consisting of N, O, and S.
- heterocyclic elements include, but are not limited to, benzimidazolyl, benzisoxazolyl, benzofurazanyl, benzopyranyl, benzothiopyranyl, benzofuryl,
- substituted aryl substituted heterocycle
- substituted cycloalkyl are intended to include the cyclic group which is substituted with 1 or 2 substitutents selected from the group which includes but is not limited to F, Cl, Br, CF 3 , NH 2 , N(C 1 -C 6 alkyl) 2 , NO 2 , CN, (C 1 -C 6 alkyl)0-, -OH, (C 1 -C 6
- nitrogen protecting group is as described in U.S. Patent No. 5,424,328 and includes those groups usually employed in the chemistry of peptides, typically a
- triphenylmethyl triphenylmethyl, t-butyloxycarbonyl, acetyl, formyl, di(p- methoxyphenyl)methyl and (p-methoxyphenyl)diphenylmethyl.
- Lines drawn into the ring systems from substituents indicate that the indicated bond may be attached to any of the substitutable ring carbon atoms.
- R 1 a , R 1 b and R 2 are independently selected from: hydrogen, -N(R 8 ) 2 , R 8 C(O)NR 8 - or C 1 -C 6 alkyl unsubstituted or substituted by -N(R 8 ) 2 , R 8 O- or R 8 C(O)NR 8 -.
- R 3 and R 4 are independently selected from: hydrogen, perfluoroalkyl, F, Cl, Br, R 8 O-, R 9 S(O) m -, CN, NO 2 , R 8 2 N-C(NR 8 )-, R 8 C(O)-, R 8 OC(O)-, N 3 , -N(R 8 ) 2 , or R 9 OC(O)NR 8 - and C 1 -C 6 alkyl.
- R 6 is selected from: hydrogen, perfluoroalkyl,
- R 7 is hydrogen
- R 8 is selected from H, C 1 -C 6 alkyl and benzyl.
- R 9 is selected from C 1 -C 6 alkyl.
- R 10 and R 1 1 is selected from H, C 1 -C 6 alkyl and benzyl.
- a 1 and A 2 are independently selected from: a bond, -C(O)NR 8 -, -NR 8 C(O)-, O, -N(R 8 )-, -S(O) 2 N(R 8 )- and- N(R 8 )S(O) 2 -.
- V is selected from hydrogen, heterocycle and aryl. Most preferably, V is phenyl.
- Y is selected from phenyl, furyl, thienyl and pyridyl. Most preferably, Y is phenyl.
- n, p and r are independently 0, 1 , or 2.
- t is 1.
- the substituent (R 6 ) r - V - A 1 (CR 1 a 2 ) n A 2 (CR 1 a 2 ) n - is not H, C 1 -C 6 alkyl or a nitrogen protecting group;
- the pharmaceutically acceptable salts of the compounds of this invention include die conventional non-toxic salts of the compounds of this invention as formed, e.g., from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like: and die salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxy-benzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, trifluoroacetic and the like.
- -N(R 8 ) 2 represents -NHH, -NHCH 3 , -NHC 2 H 5 , etc. It is understood that substituents and substitution patterns on the compounds of the instant invention can be selected by one of ordinary skill in the art to provide compounds that are chemically stable and that can be readily synthesized by techniques known in the art, as well as those methods set forth below, from readily available starting materials.
- the pharmaceutically acceptable salts of the compounds of this invention can be synthesized from the compounds of this invention which contain a basic moiety by conventional chemical methods.
- the salts are prepared either by ion exchange
- Reactions used to generate the compounds of this invention are prepared by employing reactions as shown in Schemes 1 -13, in addition to other standard manipulations such as ester hydrolysis, cleavage of protecting groups, etc., as may be known in the literature or exemplified in the experimental procedures.
- Substituents R and R CH 2 - as shown in the Schemes, represent the substituents R 8 , R 9 and others, depending on the compound of the instant invention that is being synthesized.
- the variable p' represents p-1.
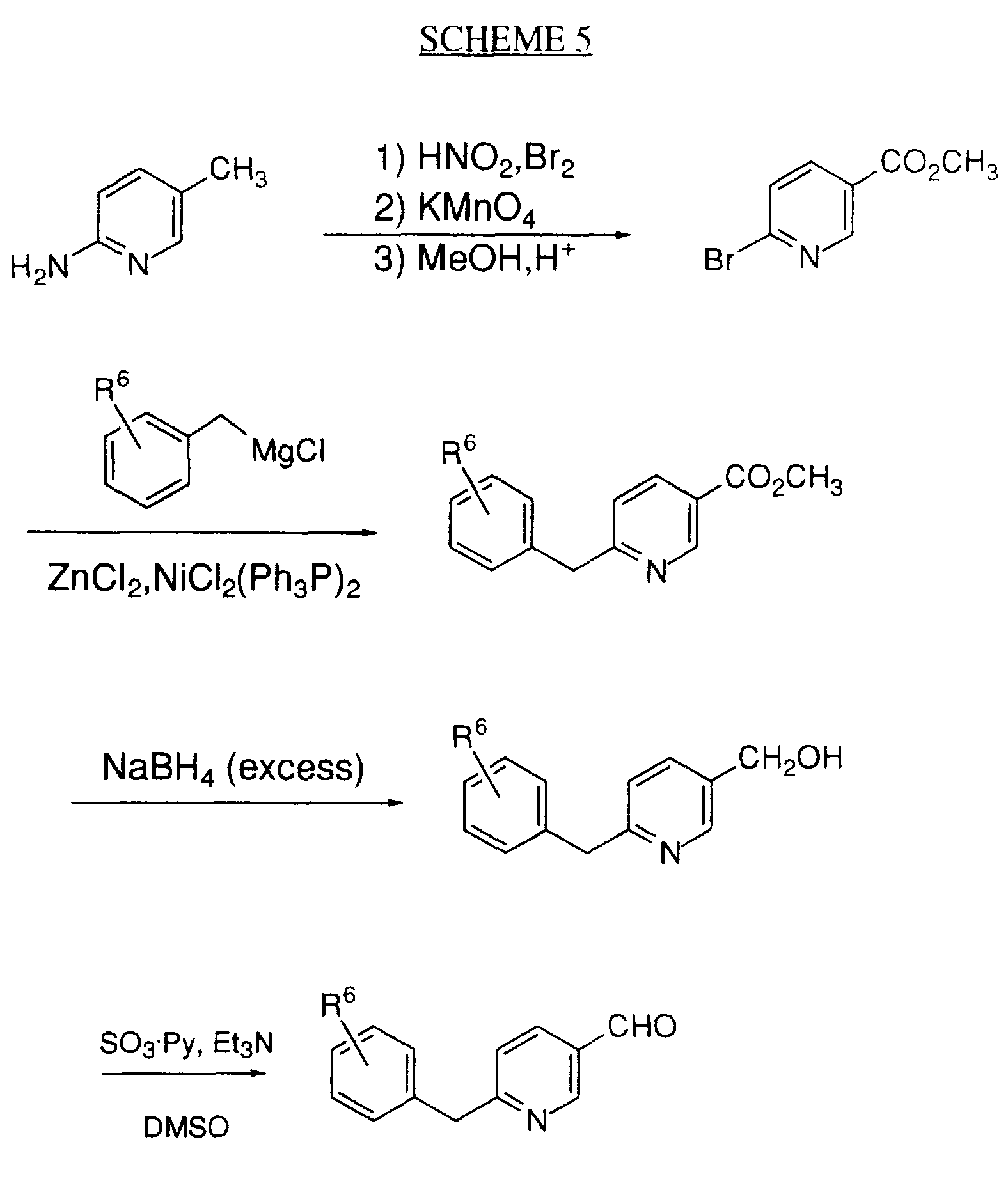
- Schemes 1 -4 illustrates the synthesis of one of the preferred embodiments of the instant invention, wherein the variable W is present as a imidazolyl moiety that is substituted with a suitably substituted benzyl group.
- Substituted protected imidazole alkanols II can be prepared by methods known in the art, such as those described by F. Schneider, Z. Physiol. Chem., 3:206-210 (1961) and C.P. Stewart, Biochem. Journal, 17:130-133(1923).
- imidazolidinone ring can then be formed by reacting intermediate VIII with triphosgene under standard conditions, such as those illustrated, to provide the instant compound IX.
- Scheme 3 illustrates the synthesis of an aldehyde Via which corresponds to aldehyde VI but which has an unsaturated R 10 /R 1 1 moiety. Subsequent reactions provide the instant compound XII.
- the suitably substituted diamine XIII can be reacted with a variety of other aldehydes, such as XIV, as shown in Scheme 9.
- the product XV is first reacted with triphosgene to form the
- the compound XVI is isolated in the salt form, for example, as a trifluoroacetate, hydrochloride or acetate salt, among others.
- Compound XVI can further be selectively protected to obtain XVII which can subsequently be reductively alkylated with a second aldehyde, such as XVIII, to obtain intermediate XIX. Removal of the protecting group, and conversion to cyclized products such as the dihydroimidazole XX can be accomplished by literature procedures, such as those shown.
- the product XXII can be reacted with triphosgene to form the imidazolidinone ring and the protecting groups can be subsequently removed to unmask the hydroxyl group (Schemes 1 1 , 12).
- the alcohol can be oxidized under standard conditions to e.g. an aldehyde, which can then be reacted with a variety of organometallic reagents such as
- the Boc protected amino alcohol XXIII can also be utilized to synthesize 2-aziridinylmethylamides such as XX VII (Scheme 13). Treating XXIII with 1 ,1 '-sulfonyldiimidazole and sodium hydride in a solvent such as dimethylformamide leads to the formation of aziridine XXVII. The aziridine may be reacted with a nucleophile, such as a thiol, in the presence of base to yield, after deprotection, the ring- opened product XXVIII.
- a nucleophile such as a thiol
- the diamine XIII can be reacted with aldehydes derived from amino acids such as O-alkylated tyrosines, according to standard procedures, to obtain compounds such as XXXII, as shown in Scheme 14.
- Intermediate XXXII is first reacted with triphosgene to form the imidazolidinone ring before it is further elaborated.
- R' is an aryl group
- XXXIII can first be hydrogenated to unmask the phenol, and the amine group deprotected with acid to produce XXXIV.
- the amine protecting group in XXXIII can be removed, and O-alkylated phenolic amines such as XXXV produced.
- the instant compounds are useful as pharmaceutical agents for mammals, especially for humans. These compounds may be administered to patients for use in the treatment of cancer. Examples of the type of cancer which may be treated with the compounds of this invention include, but are not limited to, colorectal carcinoma, exocrine pancreatic carcinoma, myeloid leukemias and neurological tumors.
- Such tumors may arise by mutations in the ras genes themselves, mutations in the proteins that can regulate Ras formation (i.e., neurofibromen (NF-1 ), neu, scr, abl , lck, fyn) or by other mechanisms.
- NF-1 neurofibromen
- NF-1 neurofibromen
- neu e.g., neu, scr, abl , lck, fyn
- the compounds of the instant invention inhibit farnesyl- protein transferase and the farnesylation of the oncogene protein Ras.
- the instant compounds may also inhibit tumor angiogenisis, thereby affecting the growth of tumors (J. Rak et al. Cancer Research, 55:4575- 4580 (1995)).
- tumor angiogenisis Such anti-angiogenisis properties of the instant
- the compounds of this invention are also useful for inhibiting other proliferative diseases, both benign and malignant, wherein Ras proteins are aberrantly activated as a result of oncogenic mutation in other genes (i.e., the Ras gene itself is not activated by mutation to an oncogenic form) with said inhibition being accomplished by the administration of an effective amount of the compounds of the invention to a mammal in need of such treatment.
- a component of NF- 1 is a benign proliferative disorder.
- the instant compounds may also be useful in the treatment of certain viral infections, in particular in the treatment of hepatitis delta and related viruses (J.S. Glenn et al. Science, 256: 1331 - 1333 (1992).
- the compounds of the instant invention are also useful in the prevention of restenosis after percutaneous transluminal coronary angioplasty by inhibiting neointimal formation (C. Indolfi et al. Nature medicine, 1 :541 -545(1995).
- the instant compounds may also be useful in the treatment and prevention of polycystic kidney disease (D.L. Schaffner et al. American Journal of Pathology, 142: 1051 -1060 (1993) and B. Cowley, Jr. et al.FASEB Journal, 2:A3160 (1988)).
- the compounds of this invention may be administered to mammals, preferably humans, either alone or, preferably, in
- the compounds can be administered orally or parenterally, including the intravenous intramuscular, intraperitoneal, subcutaneous, rectal and topical routes of administration.
- the selected compound may be administered, for example, in the form of tablets or capsules, or as an aqueous solution or suspension.
- carriers which are commonly used include lactose and corn starch, and lubricating agents, such as magnesium stearate, are commonly added.
- useful diluents include lactose and dried com starch.
- the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening and/or flavoring agents may be added.
- sterile solutions of the active ingredient are usually prepared, and the pH of the solutions should be suitably adjusted and buffered.
- the total concentration of solutes should be controlled in order to render the preparation isotonic.
- the present invention also encompasses a pharmaceutical composition useful in the treatment of cancer, comprising the
- compositions of this invention include aqueous solutions comprising compounds of this invention and pharmacologically acceptable carriers, e.g., saline, at a pH level, e.g., 7.4.
- pharmacologically acceptable carriers e.g., saline
- the solutions may be introduced into a patient's intramuscular blood-stream by local bolus injection.
- the daily dosage will normally be determined by the prescribing physician with the dosage generally varying according to the age, weight, and response of the individual patient, as well as the severity of the patient's symptoms.
- a suitable amount of compound is administered to a mammal undergoing treatment for cancer.
- Administration occurs in an amount between about 0.1 mg/kg of body weight to about 60 mg/kg of body weight per day, preferably of between 0.5 mg/kg of body weight to about 40 mg/kg of body weight per day.
- the compounds of the instant invention are also useful as a component in an assay to rapidly determine the presence and quantity of farnesyl-protein transferase (FPTase) in a composition.
- FPTase farnesyl-protein transferase
- composition to be tested may be divided and the two
- mixtures which comprise a known substrate of FPTase (for example a tetrapeptide having a cysteine at the amine terminus) and farnesyl pyrophosphate and, in one of the mixtures, a compound of the instant invention.
- FPTase for example a tetrapeptide having a cysteine at the amine terminus
- farnesyl pyrophosphate for example a tetrapeptide having a cysteine at the amine terminus
- the chemical content of the assay mixtures may be determined by well known
- inhibitors of FPTase absence or quantitative reduction of the amount of substrate in the assay mixture without the compound of the instant invention relative to the presence of the unchanged substrate in the assay containing the instant compound is indicative of the presence of FPTase in the composition to be tested.
- potent inhibitor compounds of the instant invention may be used in an active site titration assay to determine the quantity of enzyme in the sample.
- a series of samples composed of aliquots of a tissue extract containing an unknown amount of farnesyl-protein transferase, an excess amount of a known substrate of FPTase (for example a tetrapeptide having a cysteine at the amine terminus) and farnesyl pyrophosphate are incubated for an appropriate period of time in the presence of varying concentrations of a compound of the instant invention.
- the concentration of a sufficiently potent inhibitor i.e., one that has a Ki substantially smaller than the
- concentration of enzyme in the assay vessel required to inhibit the enzymatic activity of the sample by 50% is approximately equal to half of the concentration of the enzyme in that particular sample.
- Step B Preparation of 1 -triphenylmethyl-4-(acetoxymethyl)- imidazole
- Step A Alcohol from Step A was suspended in 500 mL of pyridine. Acetic anhydride (74 mL) was added dropwise, and the reaction was stirred for 48 hours during which it became homogeneous. The solution was poured into 2 L of EtOAc, washed with water (3 x 1 L), 5% aq. HCl soln. (2 x 1 L), sat. aq. NaHCO 3 , and brine, then dried (Na 2 SO 4 ), filtered, and concentrated in vacuo to provide the crude product. The acetate was isolated as a white powder (85.8 g) which was sufficiently pure for use in the next step. Step C: Preparation of 1-(4-cyanobenzyl)-5-(acetoxymethyl)- imidazole hydrobromide
- Step D Preparation of 1-(4-cyanobenzyl)-5-(hydroxymethyl)- imidazole .
- Step E Preparation of 1 -(4-cyanobenzyl)-5-imidazole- carboxaldehyde
- Step G Preparation of ( ⁇ )-ethyl 2-[(phenylmethyl)imino]-4- hexynoate
- Step H Preparation of ( ⁇ )-ethyl 2-[(tert-butoxycarbonyl)amino]-4- hexynoate
- Step K Preparation of ( ⁇ )-2-(te rt-butoxycarbonylamino)-4-hexynal
- a suspension of lithium aluminum hydride (1.56 g, 41.1 mmol) in ether (150 mL) was stirred at room temperature for 30 minutes.
- the solution was cooled to -55 °C under nitrogen, and a solution of the product from Step J (11.10 g, 41.1 mmol) in ether (150 mL) was added over 15 min, maintaining the temperature below -50 °C.
- the reaction was warmed to 5 °CC then recooled to -40 °C.
- a solution of potassium hydrogen sulfate (21.8 g) in 25 mL water was slowly added, maintaining the temperature below -35 °C.
- Step M Preparation of ( ⁇ )-2-[(1 -(4-cyanobenzyl)-5- imidazolylmethyl)amino]-1-[(3-chlorophenyl)amino]-4- hexyne
- Step N Preparation of ( ⁇ )-4-(2-butynyl)-1-(3-chlorophenyl)-3-[ 1 - (4-cyanobenzyl)-5-imidazolylmethyl]-2-imidazolidinone hydrochloride
- Step A N-Methoxy-N-methyl 2(S)-(tert-butoxycarbonylamino)- hexanamide
- Step D Preparation of (S)-2-[(1-(4-cyanobenzyl)-5- imidazolylmethyl)amino]-1-[(2,3-dimethylphenyl)amino]-4- hexane
- Step E Preparation of (S)-4- n-butyl-3-[1 -(4-cyanobenzyl)-5- imidazolylmethyl]-1-(2,3-dimethylphenyl)-2- imidazolidinone hydrochloride
- the product was taken up in methanol and injected onto a preparative HPLC column and purified with a mixed gradient of 25%- 55% acetonitrile/0.1 % TFA; 75%-45% 0.1 % aqueous TFA over 50 minutes. After concentration in vacuo, the resultant product was partitioned between dichloromethane and aq. NaHCO 3 soln., and the aqueous phase was extracted with CH 2 CI 2 . The organic solution was washed with brine, dried (Na 2 SO 4 ), filtered, and concentrated to dryness to provide the product free base, which was taken up in CH 2 CI 2 and treated with excess 1 M HCl/ether solution. After concentrated in vacuo, the titled product (33.7 mg) was isolated as a white powder. FAB mass spectrum m/e 442 (M+1).
- Step B Preparation of N-[ 1-(4-cyanobenzyl)-5-imidazolylmethyl]- N'-(3-ch lorophenyl)ethylene diamine
- the amine hydrochloride from Step A (978 mg) was partitioned between dilute aqueous NaHCO 3 solution and methylene chloride. The aqueous layer was washed with three portions of CH 2 CI 2 , and the combined organics were dried (Na 2 SO 4 ), filtered, and
- Step C Preparation of 1 -(3-chlorophenyl)-3-[ 1 -(4-cyanobenzyl)-5- imidazolylmethyl]-2-imidazolidinone hydrochloride
- Step A Preparation of (S)-2-[(4-chloro-1 -(4-cyanobenzyl)-5- imidazolylmemyl)amino]-1 -[(2-chloro-5,6- dimethylphenyl)aminol-4-hexane hydrochloride
- Step B Preparation of 4-(S)-4-n-butyl-3-[4-chloro-1 -(4- cyanobenzyl)-5-imidazolylmethyl]-1 -(2-chloro-5,6- dimethylphenyl)-2-imidazolidinone hydrochloride
- the product was purified by silica gel chromatography (2.5-5% MeOH/CH 2 Cl 2 ), then taken up in CH 2 CI 2 and treated with excess 1 M HCl/ether solution. After concentrated in vacuo, the titled product ( 18 mg) was isolated as a white powder.
- Step A Preparation of (S)-2-(tert-butoxycarbonylamino)-N- methoxy-N-methyl-4-(methylthio)butanamide
- Step B Preparation of (S)-2-(tert-butoxycarbonylamino)-N- methoxy-N-methyl-4-(methanesulfonyl)butanamide
- Step D Preparation of (S)-2-(tert-butoxycarbonylamino)-N-(3- chlorobenzyl)-4-(methanesulfonyl)butanamine
- 3-chlorobenzylamine 0.628 mL, 5.14 mmol
- crushed molecular sieves 1.5 g
- sodium triacetoxyborohydride 2.73 g, 12.9 mmol
- the reaction was stirred ovemight, allowing it to warm to room temperature.
- Step E Preparation of (S)-2-(tert-butoxycarbonylamino)-N- (benzyloxycarbonyl)-N-(3-chlorobenzyl)-4- (methanesulfonyl)butanamine
- Step F Preparation of (S)-2-[(1 -(4-cyanobenzyl)-5- imidazolylmethyl)ammo]-N-(berizyloxycarbonyl)-N-(3- chlorobenzyl)-4-(methanesulfonyl)butanamine
- Step H Preparation of (S)-1 -(3-chlorobenzyl)-3-[ 1 -(4- cyanobenzyl)-5-imidazolylmethyl]-4-[(2- methanesulfonyl)ethyl]-2-imidazolidinone hydrochloride
- triethylamine 0.15 mL, 1.0 mmol
- triphosgene 31 mg, 0.10 mmol
- Bovine FPTase was assayed in a volume of 100 ⁇ l containing 100 mM N-(2- hydroxy ethyl) piperazine-N'-(2-ethane sulfonic acid) (HEPES), pH 7.4, 5 mM MgCl 2 , 5 mM dithiothreitol (DTT), 100 mM [ 3 H]-farnesyl diphosphate ([ 3 H]-FPP; 740 CBq/mmol, New England Nuclear), 650 nM Ras-CVLS and 10 ⁇ g/ml FPTase at 31 °C for 60 min. Reactions were initiated with FPTase and stopped with 1 ml of 1.0 M HCL in ethanol.
- Precipitates were collected onto filter-mats using a TomTec Mach II cell harvestor, washed with 100% ethanol, dried and counted in an LKB ⁇ - plate counter.
- the assay was linear with respect to both substrates, FPTase levels and time; less than 10% of the [ 3 H]-FPP was utilized during the reaction period.
- Purified compounds were dissolved in 100% dimethyl sulfoxide (DMSO) and were diluted 20-fold into the assay. Percentage inhibition is measured by the amount of
- Human FPTase was prepared as described by Omer el al., Biochemistry 32:5167-5176 (1993). Human FPTase activity was assayed as described above with the exception that 0.1 % (w/v)
- polyethylene glycol 20,000, 10 ⁇ M ZnCl 2 and 100 nM Ras-CVIM were added to the reaction mixture. Reactions were performed for 30 min., stopped with 100 ⁇ l of 30% (v/v) trichloroacetic acid (TCA) in ethanol and processed as described above for the bovine enzyme.
- TCA trichloroacetic acid
- the cell line used in this assay is a v-ras line derived from either Ratl or NIH3T3 cells, which expressed viral Ha-ras p21.
- the assay is performed essentially as described in DeClue, J.E. el al., Cancer Research 51 :712-717, (1991). Cells in 10 cm dishes at 50-75%
- the cells are labelled in 3 ml methionine-free DMEM supple- meted with 10% regular DMEM, 2% fetal bovine serum and 400 mCi[ 35 S]methionine (1000 Ci/mmol).
- the cells are lysed in 1 ml lysis buffer (1 % NP40/20 mM HEPES, pH 7.5/5 mM MgCl 2 /1mM DTT/10 mg/ml aprotinen/2 mg/ml leupeptin/2 mg/ml antipain/0.5 mM PMSF) and the lysates cleared by centrifugation at 100,000 x g for 45 min. Aliquots of lysates containing equal numbers of acid-precipitable counts are bought to 1 ml with IP buffer (lysis buffer lacking DTT) and immunoprecipitated with the ras-specific monoclonal antibody Y 13-259 (Furth, M.E. el al., J. Virol.
- Rat 1 cells transformed with either v-ras, v-raf, or v-mos are seeded at a density of 1 x 10 4 cells per plate (35 mm in diameter) in a 0.3% top agarose layer in medium A (Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum) over a bottom agarose layer (0.6%). Both layers contain 0.1 % methanol or an appropriate concentration of the instant compound (dissolved in methanol at 1000 times the final concentration used in the assay).
- the cells are fed twice weekly with 0.5 ml of medium A containing 0.1 % methanol or the concentration of the instant compound.
- Photomicrographs are taken 16 days after the cultures are seeded and comparisons are made.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Urology & Nephrology (AREA)
- Virology (AREA)
- Ophthalmology & Optometry (AREA)
- Gastroenterology & Hepatology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description















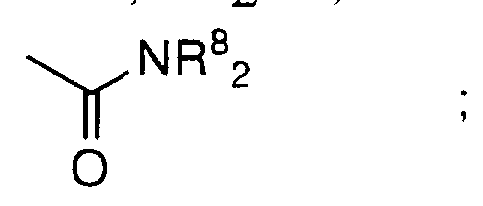

















Claims


















Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU25932/97A AU716153B2 (en) | 1996-04-03 | 1997-03-27 | Inhibitors of farnesyl-protein transferase |
| EP97917669A EP0891353A4 (en) | 1996-04-03 | 1997-03-27 | Inhibitors of farnesyl-protein transferase |
| JP09535441A JP2000507581A (en) | 1996-04-03 | 1997-03-27 | Farnesyl-protein transferase inhibitor |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US1477796P | 1996-04-03 | 1996-04-03 | |
| US60/014,777 | 1996-04-03 | ||
| GBGB9616736.6A GB9616736D0 (en) | 1996-07-09 | 1996-07-09 | Inhibitors of farnesyl-protein transferase |
| GB9616736.6 | 1996-08-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997036892A1 true WO1997036892A1 (en) | 1997-10-09 |
Family
ID=26309841
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1997/005060 WO1997036892A1 (en) | 1996-04-03 | 1997-03-27 | Inhibitors of farnesyl-protein transferase |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP0891353A4 (en) |
| JP (1) | JP2000507581A (en) |
| AU (1) | AU716153B2 (en) |
| CA (1) | CA2250192A1 (en) |
| WO (1) | WO1997036892A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0880320A1 (en) * | 1996-01-30 | 1998-12-02 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| WO1999038862A1 (en) * | 1998-02-02 | 1999-08-05 | Lg Chemical Ltd. | Farnesyl transferase inhibitors having a piperidine structure and process for preparation thereof |
| FR2780892A1 (en) * | 1998-07-08 | 2000-01-14 | Sod Conseils Rech Applic | USE OF PRENYLTRANSFERASE INHIBITORS FOR THE PREPARATION OF A MEDICINAL PRODUCT FOR TREATING CONDITIONS RESULTING FROM MEMBRANE FIXATION OF HETEROTRIMERIC PROTEIN |
| US7259174B2 (en) | 2004-05-25 | 2007-08-21 | National Health Research Institutes | Imidazolidinone compounds |
| WO2009011910A2 (en) * | 2007-07-18 | 2009-01-22 | Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Serices | Imidazolidinone compounds, methods to inhibit deubiquitination and methods of treatment |
| US7501445B2 (en) | 2001-08-21 | 2009-03-10 | National Health Research Institutes | Imidazolidinone compounds |
| US7504517B2 (en) | 2006-06-20 | 2009-03-17 | Wyeth | Kv1.5 potassium channel inhibitors |
| US8518968B2 (en) | 2009-12-04 | 2013-08-27 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Hydrazone and diacyl hydrazine compounds and methods of use |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6297239B1 (en) * | 1997-10-08 | 2001-10-02 | Merck & Co., Inc. | Inhibitors of prenyl-protein transferase |
| JP2014040374A (en) * | 2010-12-15 | 2014-03-06 | Taisho Pharmaceutical Co Ltd | Substance for inhibition of glycine transporter |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3017356A (en) * | 1958-03-03 | 1962-01-16 | Petrolite Corp | Process of inhibiting corrosion |
| US4661602A (en) * | 1985-03-29 | 1987-04-28 | G. D. Searle & Co. | Substituted alkyl imidazole derivatives |
| US4810800A (en) * | 1986-05-06 | 1989-03-07 | Merrell Dow Pharmaceuticals Inc. | Novel imidazole dopamine beta hydroxylase inhibitors |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995009001A1 (en) * | 1993-09-30 | 1995-04-06 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| US5439918A (en) * | 1994-03-14 | 1995-08-08 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| EP0783517A2 (en) * | 1994-09-29 | 1997-07-16 | Merck & Co. Inc. | Thiol-free inhibitors of farnesyl-protein transferase |
| IL117580A0 (en) * | 1995-03-29 | 1996-07-23 | Merck & Co Inc | Inhibitors of farnesyl-protein transferase and pharmaceutical compositions containing them |
-
1997
- 1997-03-27 EP EP97917669A patent/EP0891353A4/en not_active Withdrawn
- 1997-03-27 WO PCT/US1997/005060 patent/WO1997036892A1/en not_active Application Discontinuation
- 1997-03-27 AU AU25932/97A patent/AU716153B2/en not_active Ceased
- 1997-03-27 JP JP09535441A patent/JP2000507581A/en active Pending
- 1997-03-27 CA CA002250192A patent/CA2250192A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3017356A (en) * | 1958-03-03 | 1962-01-16 | Petrolite Corp | Process of inhibiting corrosion |
| US4661602A (en) * | 1985-03-29 | 1987-04-28 | G. D. Searle & Co. | Substituted alkyl imidazole derivatives |
| US4810800A (en) * | 1986-05-06 | 1989-03-07 | Merrell Dow Pharmaceuticals Inc. | Novel imidazole dopamine beta hydroxylase inhibitors |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0891353A4 * |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0880320A1 (en) * | 1996-01-30 | 1998-12-02 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| EP0880320A4 (en) * | 1996-01-30 | 1999-06-16 | Merck & Co Inc | Inhibitors of farnesyl-protein transferase |
| WO1999038862A1 (en) * | 1998-02-02 | 1999-08-05 | Lg Chemical Ltd. | Farnesyl transferase inhibitors having a piperidine structure and process for preparation thereof |
| FR2780892A1 (en) * | 1998-07-08 | 2000-01-14 | Sod Conseils Rech Applic | USE OF PRENYLTRANSFERASE INHIBITORS FOR THE PREPARATION OF A MEDICINAL PRODUCT FOR TREATING CONDITIONS RESULTING FROM MEMBRANE FIXATION OF HETEROTRIMERIC PROTEIN |
| US7501445B2 (en) | 2001-08-21 | 2009-03-10 | National Health Research Institutes | Imidazolidinone compounds |
| US7259174B2 (en) | 2004-05-25 | 2007-08-21 | National Health Research Institutes | Imidazolidinone compounds |
| US7504517B2 (en) | 2006-06-20 | 2009-03-17 | Wyeth | Kv1.5 potassium channel inhibitors |
| US7803827B2 (en) | 2006-06-20 | 2010-09-28 | Wyeth Llc | Kv1.5 potassium channel inhibitors |
| WO2009011910A2 (en) * | 2007-07-18 | 2009-01-22 | Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Serices | Imidazolidinone compounds, methods to inhibit deubiquitination and methods of treatment |
| WO2009011910A3 (en) * | 2007-07-18 | 2009-04-30 | Us Gov Health & Human Serv | Imidazolidinone compounds, methods to inhibit deubiquitination and methods of treatment |
| US8637560B2 (en) | 2007-07-18 | 2014-01-28 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Imidazolidinone compounds, methods to inhibit deubiquitination and methods of treatment |
| US8518968B2 (en) | 2009-12-04 | 2013-08-27 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Hydrazone and diacyl hydrazine compounds and methods of use |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0891353A4 (en) | 2001-08-08 |
| JP2000507581A (en) | 2000-06-20 |
| CA2250192A1 (en) | 1997-10-09 |
| AU716153B2 (en) | 2000-02-17 |
| EP0891353A1 (en) | 1999-01-20 |
| AU2593297A (en) | 1997-10-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6066738A (en) | Inhibitors of farnesyl-protein transferase | |
| US5914341A (en) | Inhibitors of farnesyl-protein transferase | |
| US5968965A (en) | Inhibitors of farnesyl-protein transferase | |
| US5859012A (en) | Inhibitors of farnesyl-protein transferase | |
| US5925651A (en) | Inhibitors of farnesyl-protein transferase | |
| US5780492A (en) | Inhibitors of farnesyl-protein transferase | |
| US5885995A (en) | Inhibitors of farnesyl-protein transferase | |
| AU707139B2 (en) | Inhibitors of farnesyl-protein transferase | |
| WO1997036900A1 (en) | Inhibitors of farnesyl-protein transferase | |
| AU715667B2 (en) | Inhibitors of farnesyl-protein transferase | |
| WO1997036605A1 (en) | Inhibitors of farnesyl-protein transferase | |
| WO1997027752A1 (en) | Inhibitors of farnesyl-protein transferase | |
| WO1997036886A1 (en) | Inhibitors of farnesyl-protein transferase | |
| AU707416B2 (en) | Inhibitors of farnesyl-protein transferase | |
| AU2660797A (en) | Inhibitors of farnesyl-protein transferase | |
| WO1997036593A1 (en) | Inhibitors of farnesyl-protein transferase | |
| AU716153B2 (en) | Inhibitors of farnesyl-protein transferase | |
| AU703988B2 (en) | Inhibitors of farnesyl-protein transferase | |
| US6028201A (en) | Inhibitors of farnesyl-protein transferase | |
| AU2555797A (en) | Inhibitors of farnesyl-protein transferase | |
| US5981562A (en) | Inhibitors of farnesyl-protein transferase | |
| WO1997036591A1 (en) | Inhibitors of farnesyl-protein transferase | |
| WO1996031525A2 (en) | Inhibitors of farnesyl-protein transferase | |
| AU5428596A (en) | Inhibitors of farnesyl-protein transferase | |
| EP0837857A2 (en) | Inhibitors of farnesyl-protein transferase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AU AZ BA BB BG BR BY CA CN CU CZ EE GE HU IL IS JP KG KR KZ LC LK LR LT LV MD MG MK MN MX NO NZ PL RO RU SG SI SK TJ TM TR TT UA US UZ VN YU AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1997917669 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2250192 Country of ref document: CA Ref country code: CA Ref document number: 2250192 Kind code of ref document: A Format of ref document f/p: F |
|
| WWP | Wipo information: published in national office |
Ref document number: 1997917669 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1997917669 Country of ref document: EP |