WO1997021676A1 - Azetidinone compounds for the treatment of atherosclerosis - Google Patents
Azetidinone compounds for the treatment of atherosclerosis Download PDFInfo
- Publication number
- WO1997021676A1 WO1997021676A1 PCT/EP1996/005588 EP9605588W WO9721676A1 WO 1997021676 A1 WO1997021676 A1 WO 1997021676A1 EP 9605588 W EP9605588 W EP 9605588W WO 9721676 A1 WO9721676 A1 WO 9721676A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oxoazetidin
- alkyl
- compound
- formula
- pyridyl
- Prior art date
Links
- 0 CC(O1)=C(*)OC1=O Chemical compound CC(O1)=C(*)OC1=O 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D205/00—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom
- C07D205/02—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D205/06—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D205/08—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with one oxygen atom directly attached in position 2, e.g. beta-lactams
- C07D205/09—Heterocyclic compounds containing four-membered rings with one nitrogen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with one oxygen atom directly attached in position 2, e.g. beta-lactams with a sulfur atom directly attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invenuon relates to certain novel monocyclic ⁇ -lactam compounds, processes for their preparation, intermediates useful in their preparation, pharmaceuucal compositions containing them and their use in therapy, in parucular in the treatment of atherosclerosis.
- WO 95/00649 (SmithKline Beecham pic) describe the phospholipase A2 enzyme Lipoprotein Associated Phospholipase A2 (Lp-PLA2), the sequence, isolation and purificauon thereof, isolated nucleic acids encoding the enzyme, and recombinant host cells transformed with DNA encoding the enzyme.
- Lp-PLA2 Lipoprotein Associated Phospholipase A2
- Suggested therapeuuc uses for mhibitors of the enzyme mcluded atherosclerosis, diabetes, rheumatoid arthritis, stroke, myocardial infarcuon, reperfusion injury and acute and chronic inflammation.
- a subsequent publicauon from the same group further describes this enzyme (Tew D et al, Arte ⁇ oscler Thromb Vas Biol 1996:16:591-9) wherein it is referred to as LDL-PLA2.
- LDL-PLA2 A later patent applicauon (WO 95/09921, Icos Co orauon) and a related publicauon in Nature (Tjoelker et al, vol 374, 6 April 1995, 549) describe the enzyme PAF-AH which has essentially the same sequence as Lp-PLA2 and suggest that it may have potential as a therapuetic protein for regulating pathological inflammatory events.
- Lp-PLA2 is responsible for the conversion of phosphaudylcholine to lysophosphatidylcholine, during the conversion of low density lipoprotein (LDL) to its oxidised form.
- the enzyme is known to hydrolyse the sn-2 ester of the oxidised phosphaudylcholine to give lysophosphatidylcholine and an oxidauvely modified fatty acid.
- Both products of Lp-PLA2 acuon are biologically acuve with lysophosphatidylcholine, a component of oxidised LDL, known to be a potent chemoattractant for cuculaung monocytes.
- lysophosphaudylchohne is thought play a significant role in atherosclerosis by being responsible for the - accumulauon of cells loaded with cholesterol ester in the arteries. Inhibiuon of the Lp-PLA2 enzyme would therefore be expected to stop the build up of these macrophage enriched lesions (by inhibiuon of the formauon of lysophosphatidylcholine and oxidised free fatty acids) and so be useful in the treatment of atherosclerosis.
- Lp-PLA2 The increased lysophosphatidylcholine content of oxidauvely modified LDL is also thought to be responsible for the endothelial dysfuncuon observed in pauents with atherosclerosis. Inhibitors of Lp-PLA2 could therefore prove beneficial in the treatment of this phenomenon. An Lp-PLA2 inhibitor could also find utility in other disease states that exhibit endothelial dysfuncuon including diabetes, hypertension, angina pecto ⁇ s and after ischaemia and reperfusion. In addition, Lp-PLA2 inhibitors may also have a general applicauon in any disorder that involves acuvated monocytes, macrophages or lymphocytes, as all of these cell types express Lp-PLA 2 . Examples of such disorders include psoriasis.
- Lp-PLA2 inhibitors may also have a general application in any disorder that involves lipid peroxidation in conjunction with Lp-PLA2 activity to produce the two injurious products, lysophosphatidylcholine and oxidatively modified fatty acids.
- Such conditions include the aforementioned conditions atherosclerosis, diabetes, rheumatoid arthritis, stroke, myocardial infarction, reperfusion injury and acute and chronic inflammation. Further such conditions include various neuropsychiauic disorders such as schizophrenia (see Psychopharmacology Bulletin, 31, 159-165, 1995).
- R ⁇ is CR ⁇ -X- i -Y 1 , CR R5- ⁇ 2- ⁇ 2 f or (CH 2 )pX 3 (CH 2 ) q Y 3 ;
- Rl and R 2 which may be the same or different, is each selected from hydrogen, halogen or C(i_8)alkyl; R 4 and R- ⁇ which may be the same or different is each selected from hydrogen and
- C(i_6)alkyl, or R 4 and R 5 may be linked together to form the residue of a C(3_7) cycloalkyl ring;
- X 1 is a linker group and Y 1 is optionally substituted t. ⁇ alkyl C(2-i2)*"""kenyl.
- X 3 is a heteroaryl group and Y 3 is an optionally substituted aryl group, p is an integer from 1 to 6, q is 0 or an an integer from 1 to 6;
- Z is O and R 3 is C(i Vietnamese8)alkyl, arylC( ⁇ _4)alkyl or aryl each of which may be opuonally substituted, or Z is S(O)n in which n is 0, 1 or 2 and R 3 is C ( i _g ) alkyl, C(3_g ) Cycloalkyl, C(3.g)CycloalkylC(i.6)alkyl, aryl, arylC( i _4)alkyl or heteroarylC(i _4)alkyl each of which may be optionally substituted.
- Compounds of formula (I) are inhibitors of Lp-PLA2 and as such are expected to be of use in treating atherosclerosis and the other disease conditions noted elsewhere.
- R* and R 2 include hydrogen, bromo, methyl and ethyl.
- R 1 and R 2 is each hydrogen or one of R 1 and R 2 is hydrogen and the other of R 1 and R 2 is methyl (to give a tra/w-methyl).
- R 1 and R 2 is each hydrogen.
- C(i_g)alkyl for R 3 include methyl, n-butyl, t- butyl and n-hexyl, cyclohexyl and cyclohexylmethyl, suitably methyl, n-butyl, t-butyl or n-hexyl.
- Suitable substituents for the alkyl or cycloalkyl group include halo, hydroxy and carboxy and esters thereof.
- Representative examples of arylC(j,_4)alkyl for R 3 include arylC(i _3)alkyl, preferably arylCH2-
- Representative examples of the aryl group include phenyl and naphthyl, preferably phenyl.
- Suitable examples include benzyl, 2-phenylethyl and 3-phenylpropyl in each of which the phenyl ring may be optionally substituted by up to two substituents.
- Suitable substituents include halo, hydroxy, C(i_6)alkyl, C(i_6)alkoxy, arylC(i_6)alkoxy, carboxy and esters thereof, (C ⁇ alkylthio, (C ⁇ _6)alkylsulphinyl, and (Ci ⁇ alkylsulphonyl.
- aryl for R 3 include phenyl and naphthyl.
- the aryl group is optionally substitued phenyl.
- Suitable substituents for a phenyl or naphthyl ring include halo, hydroxy, C(i_6)alkyl, C(i_6)alkoxy, arylC(i_6)alkoxy, carboxy and esters thereof, (C ⁇ -6)alkylthio, (C i _6)alkylsulphinyl, and (C ⁇ _6)alkylsulphonyl.
- heteroaryl group for inco ⁇ oration into R 3 ⁇ include include pyridyl, pyridyl N-oxide, furanyl, thienyl and thiazolyl.
- the heteroarylalkyi group is heteroarylC(i_3)alkyl, more suitably heteroarylmethyl.
- Preferred values include optionally substitued pyridylmethyl, furanylmethyl, thienylmethyl or thiazolylmethyl.
- Suitable substituents for a heteroaryl ring include halo, hydroxy, C(i_6)alkyl, C(i_6)alkoxy, arylC(i _6)alkoxy, carboxy and esters thereof, (C ⁇ -6)alkylthio, (C ⁇ _6)alkylsulphiny ⁇ and (C ⁇ _6)alkylsulphonyl.
- Z is S(O)n
- n is 1 or 2, more preferably 1.
- Z is SO and R 3 is arylmethyl or heteroarylmethyl, in particular benzyl or furanylmethyl, especially benzyl.
- Suitable esters for inco ⁇ ora ⁇ on into R 3 include pharmaceutically acceptable esters of the formula CO2 . Such esters may be active in their own right and /or be hydrolysable under in vivo conditions in the human body. Suitable pharmaceutically acceptable in vivo hydrolysable ester groups for inco ⁇ oration in R include those which break down readily in the human body to leave the parent acid or its salt.
- suitable values for R include for instance, methyl, ethyl and propyl, (C2"6)alkenyl, for instance allyl. Further examples of suitable values for R include:
- R a is hydrogen, (C ⁇ -6)alkyl, in particular methyl, (C3-7)cycloalkyl, or phenyl;
- R D is (C ⁇ -6)alkyl, (C 1 - 6 )alkoxy(C ⁇ -6)alkyl, phenyl, benzyl, (C3- 7 )cycloalkyl,
- R a and R* 5 together form a 1,2-phenylene group optionally substituted by one or two methoxy groups;
- R c is (C ⁇ -6)alkyl, (C3-7)cycloalkyl, (C ⁇ -6)alkyl(C3- 7 )cycloalkyl;
- R ⁇ * is (C ⁇ -6)alkylene optionally substituted with a methyl or ethyl group
- R e and Rf which may be the same or different is each (C j -gjalkyl or aryl (C 1 -4) alkyl, optionally substituted with e.g. hydroxy;
- RS is (C-[- 6 )alkyl
- R n is hydrogen, (C ⁇ -g)alkyl or phenyl
- Ri is hydrogen or phenyl optionally substituted by up to three groups selected from halogen, (Cj. -6)-alkyl, or (C ⁇ -6)alkoxy;
- Y 4 is oxygen or NH; for instance:
- acyloxyalkyl groups such as acetoxymethyl, isobutyryloxymethyl, pivaloyloxymethyl, benzoyloxymethyl, ⁇ -acetoxyethyl, ⁇ -pivaloyloxyethyl, l-(cyclohexylcarbonyloxy)ethyl, and (l-aminoethyl)carbonyloxymethyl;
- alkoxy/cycloalkoxycarbonyloxyalkyl groups such as ethoxycarbonyloxymethyl, cyclohexyloxycarbonyloxymethyl and ⁇ -ethoxycarbonyloxyethyl
- dialkylaminoalkyl especially di-loweralkylamino alkyl groups such as dimethylaminomethyl, dimethylaminoethyl, diethyiaminomethyl or diethylaminoethyl;
- lactone groups such as phthalidyl and dimethoxyphthalidyl
- R 4 and R- ⁇ when an alkyl group include methyl.
- Representative examples of a (C3_7)cycloalkyl ring include cyclopropyl.
- R 4 and R- ⁇ are both hydrogen or R 4 is hydrogen and R- ⁇ methyl.
- X 4 is CONH.
- x is 0.
- X 1 is CONH.
- Y * * the alkyl chain is unbranched.
- Useful such values of Y** include C(6-io) alkyl. preferably C(g_ ⁇ o) alkyl or C(3_7)cycloalkylC(5_7) alkyl, preferably cyclohexylC(5_7)alkyl.
- Representative examples of ⁇ l include nonyl and cyclohexylhexyl.
- X 2 is:
- X 2 include CO(CH2) y , CONH(CH2) y , COO(CH 2 ) y , CONHCO(CH 2 ) y , CONHO(CH 2 ) y and C ( 1 . 12 )alkylene.
- X 3 is CO or CONR 6 , more preferably CONH.
- y is 1, 2, 5, 6, 7 or 9, preferably 6.
- X 2 is CONH(CH 2 )6-
- heteroaryl rings for inco ⁇ oration into Y 2 include pyridyl and pyridyl N-oxide.
- Suitable substituents for a heteroaryl ring include halo, hydroxy, CQ _g)alkyl and C(i_g)alkoxy.
- Y 2 is 2-pyridyl or 4- pyridyl, preferably in combination with ⁇ l being CONH(CH2)6-
- X 3 include thiazolyl and oxazolyl, in particular
- p is 1.
- Representative examples of aryl rings for inco ⁇ oration into Y 3 include phenyl and naphthyl. Suitable substituents for the aryl ring include halo, hydroxy, C(i_g ) alkyl and C ( i_g ) alkoxy.
- Y 3 is phenyl.
- Representative examples of X 3 - Y 3 include:
- R° is CH 2 CONH ⁇ l in which Y 1 is C(g_ ⁇ o) alkyl or cyclohexylC(5_7)alkyl, in particular nonyl or cyclohexylhexyl.
- C-4 of the ⁇ -lactam ring is a chiral centre which will give rise to the presence of stereoisomers.
- the present invention encompasses all such stereoisomers.
- An additional chiral centre will be introduced in compounds of formula (I) in which z is SO.
- the present invention encompasses all such stereoisomers.
- a further chiral centre will be introduced when R 4 and R are not the same. This will give rise to the existence of extra stereoisomers.
- the present invention encompasses all such stereoisomers.
- the absolute configurations at C-4 and the SO moiety are R and S respectively.
- 'alkyl' and similar terms such as 'alkoxy' includes all straight chain and branched isomers. Representative examples thereof include methyl, ethyl, n-propyl, w ⁇ -propyl, n-butyl, sec-butyl, iso-butyl, t-butyl, n-pentyl and n-hexyl.
- Suitable substituents for an alkyl group include, for example, and unless otherwise defined, halogen, cyano, azido, nitro, carboxy, (C ⁇ _6)alkoxycarbonyl, carbamoyl, mono- or di-(C ⁇ _6)alkylcarbamoyl, sulpho, sulphamoyl, mono- or di-(C ⁇ .
- the term 'aryl' includes, unless otherwise defined, phenyl or naphthyl optionally substituted with up to five, preferably up to three substituents.
- Suitable substituents for an aryl group include, for example, and unless otherwise defined, halogen, cyano, (Ci ⁇ alkyl, (C3-7)cycloalkyI, (C ⁇ -6)alkoxy, halo(C ⁇ -6)alkyl, hydroxy, amino, mono- or di-(C ⁇ -6)alkylamino, acylamino, nitro, carboxy, (C ⁇ -6)alkoxycarbonyl, (Ci -gjalkenyloxycarbonyl, (C ⁇ -6)alkoxycarbonyl(C ⁇ -6)alkyl, carboxy(C ⁇ -6)alkyl, (Ci- ⁇ alkylcarbonyloxy, carboxy(C ⁇ -6)alkyloxy, (Ci- ⁇ t ⁇ lJcoxycarbonyKC i - ⁇
- heterocyclyl' includes aromatic and non-aromatic single or fused rings comprising up to four hetero-atoms in the ring selected from oxygen, nitrogen and sulphur and optionally substituted with up to three substituents.
- the heterocyclic ring comprises from 4 to 7, preferably 5 to 6, ring atoms.
- a fused heterocyclic ring system may include carbocyclic rings and need only include one heterocyclic ring.
- a heteroaryl or a heterocyclyl group may have up to three substituents. Suitable such substituents include those previously mentioned for an aryl group as well as oxo.
- 'halogen' and 'halo' include fluorine, chlorine, bromine and iodine and fluoro, chloro, bromo and iodo, respectively.
- Preferred compounds of formula (I) include:
- N-(Nonyl)-(4-benzylsulphinyl-2-oxoazetidin-l-yl)acetamide (Diastereoisomer 2); and N-(Nonyl)-(4-benzylsulphonyl-2-oxoazetidin- l-yl)acetamide.
- Preferred compounds of formula (I) in which, in R ⁇ , Y 2 is heteroaryl include:
- Preferred compounds of formula (I) in which R° is (CH2)pX 3 (CH2)nY 3 include:
- Impure preparations of the compounds of formula (I) may be used for preparing the more pure forms used in the pharmaceutical compositions.
- the purity of intermediate compounds of the present invention is less critical, it will be readily understood that the substantially pure form is preferred as for the compounds of formula (I).
- the compounds of the present invention are obtained in crystalline form.
- solvent of crystallisation may be present in the crystalline product. This invention includes within its scope such solvates.
- some of the compounds of this invention may be crystallised or recrystallised from solvents containing water. In such cases water of hydration may be formed.
- This invention includes within its scope stoichiometric hydrates as well as compounds containing variable amounts of water that may be produced by processes such as lyophilisation.
- different crystallisation conditions may lead to the formation of different polymo ⁇ hic forms of crystalline products.
- This invention includes within its scope all polymo ⁇ hic forms of the compounds of formula (I).
- Compounds of the present invention are inhibitors of the enzyme lipoprotein associated phospholipase A (Lp-PLA2) and as such are expected to be of use in therapy, in particular in the treatment of atherosclerosis.
- Lp-PLA2 lipoprotein associated phospholipase A
- the present invention provides a compound of formula (I) for use in therapy.
- the compounds of formula (I) are inhibitors of lysophosphatidylcholine production by Lp-PLA2 and may therefore also have a general application in any disorder that involves endothelial dysfunction, for example atherosclerosis, diabetes, hypertension, angina pectoris and after ischaemia and reperfusion.
- compounds of formula (I) may have a general application in any disorder that involves lipid peroxidation in conjunction with enzyme activity, for example in addition to conditions such as atherosclerosis and diabetes, other conditions such as rheumatoid arthritis, stroke, inflammatory conditions of the brain such as Alzheimer's Disease, myocardial infarction, reperfusion injury, sepsis, and acute and chronic inflammation. Further such conditions include various neuropsychiauic disorders such as schizophrenia (see Psychopharmacology Bulletin, 31, 159-165, 1995). Further applications include any disorder that involves activated monocytes, macrophages or lymphocytes, as all of these cell types express Lp-PLA j . Examples of such disorders include psoriasis.
- the present invention provides for a method of treating a disease state associated with activity of the enzyme Lp-PLA 2 which method involves treating a patient in need thereof with a therapeutically effective amount of an inhibitor of the enzyme.
- the disease state may be associated with the increased involvement of monocytes, macrophages or lymphocytes; with the formation of lysophosphatidylcholine and oxidised free fatty acids; with lipid peroxidation in conjunction with Lp PLA2 activity; or with endothelial dysfunction.
- Compounds of the present invention may also be of use in treating the above mentioned disease states in combination with anti-hyperlipidaemic or anti- atherosclerotic or anti-diabetic or anti-anginal or anti-inflammatory or anti- hypertension agents.
- examples of the above include cholesterol synthesis inhibitors such as statins, anti-oxidants such as probucol, insulin sensitisers, calcium channel antagonists, and anti-inflammatory drugs such as NSAIDs.
- the compounds of the present invention are usually administered in a standard pharmaceutical composition.
- the present invention therefore provides, in a further aspect, a pharmaceutical composition comprising a compound of formula (I) and a pharmaceutically acceptable carrier.
- Suitable pharmaceutical compositions include those which are adapted for oral or parenteral administration or as a suppository.
- the compounds of formula (I) which are active when given orally can be formulated as liquids, for example syrups, suspensions or emulsions, tablets, capsules and lozenges.
- a liquid formulation will generally consist of a suspension or solution of the compound or pharmaceutically acceptable salt in a suitable liquid car ⁇ ier(s) for example, ethanol, glycerine, non-aqueous solvent, for example polyethylene glycol, oils, or water with a suspending agent, preservative, flavouring or colouring agent.
- a suitable liquid car ⁇ ier(s) for example, ethanol, glycerine, non-aqueous solvent, for example polyethylene glycol, oils, or water with a suspending agent, preservative, flavouring or colouring agent.
- suitable pharmaceutical carrier(s) routinely used for preparing solid formulations. Examples of such carriers include magnesium stearate, starch, lactose, sucrose and cellulose.
- a composition in the form of a capsule can be prepared using routine encapsulation procedures.
- pellets containing the active ingredient can be prepared using standard carriers and then filled into a hard gelatin capsule; alternatively, a dispersion or suspension can be prepared using any suitable pharmaceutical carrier(s), for example aqueous gums, celluloses, silicates or oils and the dispersion or suspension then filled into a soft gelatin capsule.
- Typical parenteral compositions consist of a solution or suspension of the compound of formula (I) in a sterile aqueous carrier or parenterally acceptable oil, for example polyethylene glycol, polyvinyl pyrrolidone, lecithin, arachis oil or sesame oil.
- a sterile aqueous carrier or parenterally acceptable oil for example polyethylene glycol, polyvinyl pyrrolidone, lecithin, arachis oil or sesame oil.
- tiie solution can be lyophilised and then reconstituted with a suitable solvent just prior to administration.
- a typical suppository formulation comprises a compound of formula (I) which is active when administered in this way, with a binding and/or lubricating agent such as polymeric glycols, gelatins or cocoa butter or other low melting vegetable or synthetic waxes or fats.
- a binding and/or lubricating agent such as polymeric glycols, gelatins or cocoa butter or other low melting vegetable or synthetic waxes or fats.
- the composition is in unit dose form such as a tablet or capsule.
- Each dosage unit for oral administration contains preferably from 1 to 500 mg (and for parenteral administration contains preferably from 0.1 to 25 mg) of a compound of the formula (1).
- the daily dosage regimen for an adult patient may be, for example, an oral dose of between 1 mg and 1000 mg, preferably between 1 rag and 500 mg, or an intravenous, subcutaneous, or intramuscular dose of between 0.1 mg and 100 mg, preferably between 0.1 mg and 25 mg, of the compound of the formula (I). the compound being administered 1 to 4 times per day. Suitably the compounds will be administered for a period of continuous therapy, for example for a week or more.
- Compounds of formula (I) may be prepared by adapting processes previously described for ananlogous compounds in International patent applications WO 96/13484, WO 96/19451 and PCT EP96/02765 (SmithKline Beecham pic). Such processes include treating an azeti (II):
- Rl, R 2 , R 3 , and Z are as hereinbefore defined; with an alkylating agent of the formula (HI):
- R ⁇ is as hereinbefore defined; under alkylating conditions.
- Suitable alkylating conditions are well known in the art and include carrying out the reaction in the presence of a suitable base such as sodium hydride or potassium hydroxide optionally with a quaternary ammonium salt such tetrabutyl ammonium bromide, in a suitable alkylating solvent such as tetrahydrofuran (THF), and at a temperature in the range - 10 to 0°C.
- a suitable base such as sodium hydride or potassium hydroxide optionally with a quaternary ammonium salt such tetrabutyl ammonium bromide
- THF tetrahydrofuran
- the preceding alkyiation reaction is conveniently effected on compounds in which n is 0.
- Compounds of formula (I) in which one of R 4 and R ⁇ is alkyl may also be prepared from corresponding compounds of formula (I) where both R 4 and R-> are hydrogen by treatment thereof with an alkylating agent under the conditions described above. Such compounds may be obtained by treating a compound of formula (LT) with an alkylating agent of formula (HI) in which both of R 4 and R 5 is hydrogen, under alkylating conditions as hereinbefore described.
- a second alkyl group for R 4 /R*5 may be introduced by treating a first obtained compound of formula (I) in which one of R 4 and R 5 is hydrogen, with an alkylating agent in the presence of a suitable base such as sodium hydride, potassium hydroxide or lithium hexamethyldisilazide, in a suitable alkylating solvent such as tetrahydrofuran (THF), and at a temperature in the range -80 to 10°C.
- a suitable base such as sodium hydride, potassium hydroxide or lithium hexamethyldisilazide
- THF tetrahydrofuran
- Compounds of formula (II) in which Z is O may be obtained by treating 4- acetoxyazetidinone, 4-benzoyloxyazetidinone or 4-phenylsulfonylazetidinone with a phenol alcohol HOR 3 in the presence of a base such as potassium t-butoxide, in a suitable solvent such as THF at a temperature in the range 0 to 5°C.
- Compounds of formula (IV) in which Z is S may be obtained by treating 4-acetoxyazetidinone with a thiol HSR 3 in the presence of a base such as sodium ethoxide, in a suitable solvent such as ethanol at a temperature in the range 0 to 5°C.
- CONR O may be of the formula (IV):
- a similar process may be used for preparing a compound of formula (I) in which X 2 denotes a group CONR 6 (CH 2 ) y or CONR 6 O(CH 2 ) y , but using an amine NHR 6 (CH 2 ) y Y 1 or a hydroxylamine NH 2 O(CH2) y ⁇ !.
- An acid of formula (IV) in which one of R 4 and R 5 is hydrogen may be obtained by treating a compound of formula (II) with a corresponding 2-bromo (C1.7) alkanoate ester, under alkylating conditions as hereinbefore described; followed by the hydrolysis of the thus formed intermediate ester using standard conditions.
- a second alkyl group may be inu-oduced by alkylating of the first formed monoalkyl ester.
- R 7 is methyl; and x, R 1 , R ⁇ R ⁇ R", R 5 and Z are as hereinbefore defined; using conditions well known in the art for such reactions, for instance heating in toluene in the presence of a catalytic amount of sodium methoxide and an alcohol.
- a compound of formula (VII) in which one of R 4 and R ⁇ is hydrogen may be obtained by treating a compound of formula (II) with a methyl 2-bromoalkanoate, under alkylating conditions as hereinbefore described.
- (CH2) x COO in which x is an integer from 1 to 6 may be prepared by treating a compound of formula (IV) in which R 7 is hydrogen with an alcohol ⁇ ⁇ H or an activated derivative thereof, for instance a tosylate.
- R** is a halogen or other suitable leaving group such as triflate or tosylate and R 1 , R 2 , R 3 , R 4 , R 5 , x and Z are as hereinbefore defined; with an alcohol ⁇ ⁇ H or a suitable salt therof.
- compounds of formula (I) in which Z is S(O)n and n is 0 may be prepared by a process which comprises treating a compound of formula (DC):
- Chirally pure compounds may be prepared by chiral chromatography, from chirally pure intermediates or by chiral synthesis using chiral reagents or catalysis.
- Suitable chiral intermediates may be obtained by resolution or chiral induction or by using chiral reagents, in particular natural chiral molecules, according to methods well known to those skilled in the art.
- a convenient chiral starting material is a penicillin derivative which has the preferred configuration at C-4 of the ⁇ -lactam ring. This is illustrated in the following scheme:
- Sodamide (6.63g) was suspended in Uquid ammonia (100ml) and cooled in a cardice/acetone bath. 4-Picoline (7.3ml) was added, the cooling bath removed and the mixture stirred at reflux for 2hrs. The mixture was cooled again and 5- bromopentylamine hydrobromide (18.53g) added and the mixture allowed to reflux for 5 hrs. The mixture was again cooled, quenched with ammonium chloride (lOg) and the solvent allowed to evaporate overnight. The residue was dissolved in water (100ml), made strongly alkaline with NaOH and extracted with CH,C1 2 (2x100ml).
- N-(6-Cyclohexylhexyl)-4-benzylthio-2-oxoazetidin- 1 -ylacetamide (2.76g) was dissolved in dichloromethane (60ml), cooled to -60°C and a solution of 55-60% m- chloroperbenzoic acid (mCPBA) (1.84g;ca 6.62mM) in dichloromethane (80ml) was added dropwise over 15 mins, then the mixture was stirred at 20°C for 3 hours. The solution was washed with aq. NaHCO/Na.SO 3 , water, dried over MgSO, and evaporated to a colourless solid. This was recrystallised three times from EtOAc (cooling to 20°C only) to give the title compound as a colourless solid m.p. 150- PC, (735mg. 26%).
- mCPBA m- chloroperbenzoic acid
- Example 3 N-(6-Cyclohexylhexyl)-(4-benzyIsulphinyI-2-oxoazetidin-l- yl)acetamide (Diastereoisomer 2)
- the mother liquors from the first two recrystallisations in Example 2 above were combined and evaporated to a solid which was recrystallised from EtOAc, cooling to RT and filtering to remove the first formed solid, then refrigerating to obtain a solid which was recrystallised again from EtOAc to give the title compound as a colourless solid, m.p.
- N-(6-Cyclohexylhexyl)-(4-benzylsulphinyl-2-oxoazetidin- 1 -yl)acetamide (0.95g) was dissolved in dichloromethane (40ml), a solution of 55-60% mCPBA (0.83g) in dichloromethane (40ml) was added and stirred at 20°C for 1.5 hours. The solution was washed with aq NaHCOJNa j SO 3 and brine, dried over MgSO 4 and evaporated to a solid.
- N-(6-(4-pyridyl)hexyl)-4-benzylthio-2-oxoazetidin- l-ylacetamide (3.04g) was dissolved in CH 2 Cl 2 (50ml), cooled to -60°C and a solution of 55-60% m- chloroperbenzoic acid (mCPB A) (2.11 g) in CH 2 C1 2 ( 100ml) was added dropwise over 15 mins. The solution was stirred at 20-25°C for 3 hrs then washed with aq NaHCO j /Na j SO j , brine, dried over MgSO 4 and evaporated to a sticky solid.
- mCPB A m- chloroperbenzoic acid
- Example 12 The mother liquor from the above recrystallisation in Example 12 was evaporated to a solid which was recrystallised twice from ethyl acatate to give the title compound as a colourless solid, m.p. 109-10°C, (1.41g, 46% yield)
- N-(6-(4-Pyridyl)hexyl)-4-benzy lsulphinyl-2-oxoazetidin- 1 -ylacetamide (Dia 1 ) (1.15g) was dissolved in CH 2 Cl 2 (50ml) and a solution of 55-60% CPBA(1.10g;ca 3.5mM) in CH 2 Cl 2 (50ml) added and stirred at 20-25°C for 3hrs and allowed to stand at 20-25°C for 16 h. mCPBA (0.13g) was added and the solution stirred for a further 3 hrs then washed with aq NaHCO/Na-SO,, brine, dried over MgSO 4 and evaporated to an oil.
- Example 22 l-(4-(5-Phenylpentyl)thiazoI-2-ylmethyl)-4-benzylsulphinyl-2- oxoazetidine (Dia l:Dia 2 22:78) 1 -(4-(5-phenylpentyl)thiazol-2-ylmethyl)-4-benzylthio-2-oxoazetidine (1.1 equiv) was dissolved in CH 2 Cl 2 (50ml), cooled to -60°C and a solution of 55-60% m- chloroperbenzoic acid (mCPBA) (2.1 lg) in CH 2 Cl 2 (100ml) was added dropwise over 15 mins.
- mCPBA m- chloroperbenzoic acid
- Enzyme activity was determined by measuring the rate of turnover of the artificial substrate (A) at 37 °C in 50mM HEPES (N-2-hydroxyethylpiperazine-N'-2- ethanesulphonic acid) buffer containing 150mM NaCl, pH 7.4.
- HEPES N-2-hydroxyethylpiperazine-N'-2- ethanesulphonic acid
- Lp-PLA2 was partially purified by density gradient centrifugation of human plasma. Active fractions were pooled and used as the source of Lp-PLA2- The enzyme was pre-incubated at 37 °C with vehicle or test compound for 10 rain in a total volume of 180 ⁇ l. The reaction was then initiated by the addition of 20 ⁇ l lOx substrate (A) to give a final substrate concentration of 20 ⁇ M. The reaction was followed at 405 nm for 20 minutes using a plate reader with automatic mixing. The rate of reaction was measured as the rate of change of absorbance.
- Example 3 The compounds of Example 3, 7, 8, 13, 18 and 22 had IC50 values in the range 5 to
Abstract
Azetidinone compounds of formula (I) in which inter alia: R?0 is CR4R5-X1-Y1¿ or (CH¿2?)pX?2(CH¿2)qY?2; R4 and R5¿ which may be the same or different is each selected from hydrogen and C¿(1-6)?alkyl, or R?4 and R5¿ may be linked together to form the residue of a C¿(3-7)?cycloalkyl ring; X?1¿ is a linker group and Y1 is optionally substituted C¿(1-12)?alkyl C(2-12)alkynyl, C(2-12)alkynyl, C(3-7)-cycloalkylC(1-8) alkyl or an optionally substituted heteroaryl group; and X?2¿ is a heteroaryl group and Y2 is an optionally substituted aryl group, p is an integer from 1 to 6, q is 0 or an integer from 1 to 6; are inhibitors of the phospholipase A2 enzyme Lp PLA2 and are of use in therapy, for instance in treating atherosclerosis.
Description
AZETIDINONE COMPOUNDS FOR THE TREATEMENT OF ATHEROSCLEROSIS .
The present invenuon relates to certain novel monocyclic β-lactam compounds, processes for their preparation, intermediates useful in their preparation, pharmaceuucal compositions containing them and their use in therapy, in parucular in the treatment of atherosclerosis.
WO 95/00649 (SmithKline Beecham pic) describe the phospholipase A2 enzyme Lipoprotein Associated Phospholipase A2 (Lp-PLA2), the sequence, isolation and purificauon thereof, isolated nucleic acids encoding the enzyme, and recombinant host cells transformed with DNA encoding the enzyme. Suggested therapeuuc uses for mhibitors of the enzyme mcluded atherosclerosis, diabetes, rheumatoid arthritis, stroke, myocardial infarcuon, reperfusion injury and acute and chronic inflammation. A subsequent publicauon from the same group further describes this enzyme (Tew D et al, Arteπoscler Thromb Vas Biol 1996:16:591-9) wherein it is referred to as LDL-PLA2. A later patent applicauon (WO 95/09921, Icos Co orauon) and a related publicauon in Nature (Tjoelker et al, vol 374, 6 April 1995, 549) describe the enzyme PAF-AH which has essentially the same sequence as Lp-PLA2 and suggest that it may have potential as a therapuetic protein for regulating pathological inflammatory events. It has been shown that Lp-PLA2 is responsible for the conversion of phosphaudylcholine to lysophosphatidylcholine, during the conversion of low density lipoprotein (LDL) to its oxidised form. The enzyme is known to hydrolyse the sn-2 ester of the oxidised phosphaudylcholine to give lysophosphatidylcholine and an oxidauvely modified fatty acid. Both products of Lp-PLA2 acuon are biologically acuve with lysophosphatidylcholine, a component of oxidised LDL, known to be a potent chemoattractant for cuculaung monocytes. As such, lysophosphaudylchohne is thought play a significant role in atherosclerosis by being responsible for the - accumulauon of cells loaded with cholesterol ester in the arteries. Inhibiuon of the Lp-PLA2 enzyme would therefore be expected to stop the build up of these macrophage enriched lesions (by inhibiuon of the formauon of lysophosphatidylcholine and oxidised free fatty acids) and so be useful in the treatment of atherosclerosis.
The increased lysophosphatidylcholine content of oxidauvely modified LDL is also thought to be responsible for the endothelial dysfuncuon observed in pauents with atherosclerosis. Inhibitors of Lp-PLA2 could therefore prove beneficial in the treatment of this phenomenon. An Lp-PLA2 inhibitor could also find utility in other disease states that exhibit endothelial dysfuncuon including diabetes, hypertension, angina pectoπs and after ischaemia and reperfusion.
In addition, Lp-PLA2 inhibitors may also have a general applicauon in any disorder that involves acuvated monocytes, macrophages or lymphocytes, as all of these cell types express Lp-PLA2. Examples of such disorders include psoriasis.
Furthermore, Lp-PLA2 inhibitors may also have a general application in any disorder that involves lipid peroxidation in conjunction with Lp-PLA2 activity to produce the two injurious products, lysophosphatidylcholine and oxidatively modified fatty acids. Such conditions include the aforementioned conditions atherosclerosis, diabetes, rheumatoid arthritis, stroke, myocardial infarction, reperfusion injury and acute and chronic inflammation. Further such conditions include various neuropsychiauic disorders such as schizophrenia (see Psychopharmacology Bulletin, 31, 159-165, 1995).
International patent applications WO 96/13484, WO 96/19451 and PCT EP96 02765 (SmithKline Beecham pic) describe a series of azetidinone derivatives which are inhibitors of Lp PLA2. We have now identified a further series of azetidinone compounds which are distinguished over previous series by tiie substituent at the ring nitrogen and which act as inhibitors of Lp-PLA2-
Accordingly, the present invention provides a compound of formula (I):

Rθ is CR^-X-i-Y1, CR R5-χ2-γ2f or (CH2)pX3(CH2)qY3;
Rl and R2, which may be the same or different, is each selected from hydrogen, halogen or C(i_8)alkyl; R4 and R-^ which may be the same or different is each selected from hydrogen and
C(i_6)alkyl, or R4 and R5 may be linked together to form the residue of a C(3_7) cycloalkyl ring;
X1 is a linker group and Y1 is optionally substituted t.^alkyl C(2-i2)*""kenyl.
C(2-I2)alkynyl, C(3.7)-cycloalkylC(1.8) alkyl; X2 is a linker group and Y2 an optionally substituted heteroaryl group;
X3 is a heteroaryl group and Y3 is an optionally substituted aryl group, p is an integer from 1 to 6, q is 0 or an an integer from 1 to 6;
Z is O and R3 is C(i„8)alkyl, arylC( ι_4)alkyl or aryl each of which may be opuonally substituted, or
Z is S(O)n in which n is 0, 1 or 2 and R3 is C(i _g)alkyl, C(3_g)Cycloalkyl, C(3.g)CycloalkylC(i.6)alkyl, aryl, arylC( i _4)alkyl or heteroarylC(i _4)alkyl each of which may be optionally substituted.
Compounds of formula (I) are inhibitors of Lp-PLA2 and as such are expected to be of use in treating atherosclerosis and the other disease conditions noted elsewhere.
Representative examples of R* and R2 include hydrogen, bromo, methyl and ethyl. Suitably, R1 and R2 is each hydrogen or one of R1 and R2 is hydrogen and the other of R1 and R2 is methyl (to give a tra/w-methyl). Preferably, R1 and R2 is each hydrogen.
Representative examples of C(i_g)alkyl for R3 include methyl, n-butyl, t- butyl and n-hexyl, cyclohexyl and cyclohexylmethyl, suitably methyl, n-butyl, t-butyl or n-hexyl. Suitable substituents for the alkyl or cycloalkyl group include halo, hydroxy and carboxy and esters thereof. Representative examples of arylC(j,_4)alkyl for R3 include arylC(i _3)alkyl, preferably arylCH2- Representative examples of the aryl group include phenyl and naphthyl, preferably phenyl. Suitable examples include benzyl, 2-phenylethyl and 3-phenylpropyl in each of which the phenyl ring may be optionally substituted by up to two substituents. Suitable substituents include halo, hydroxy, C(i_6)alkyl, C(i_6)alkoxy, arylC(i_6)alkoxy, carboxy and esters thereof, (C ^alkylthio, (Cι_6)alkylsulphinyl, and (Ci^alkylsulphonyl.
Representative examples of aryl for R3 include phenyl and naphthyl. Preferably, the aryl group is optionally substitued phenyl. Suitable substituents for a phenyl or naphthyl ring include halo, hydroxy, C(i_6)alkyl, C(i_6)alkoxy, arylC(i_6)alkoxy, carboxy and esters thereof, (C ι-6)alkylthio, (C i _6)alkylsulphinyl, and (Cι_6)alkylsulphonyl.
Representative examples of heteroaryl group for incoφoration into R3~ include include pyridyl, pyridyl N-oxide, furanyl, thienyl and thiazolyl. Suitably, the heteroarylalkyi group is heteroarylC(i_3)alkyl, more suitably heteroarylmethyl. Preferred values include optionally substitued pyridylmethyl, furanylmethyl, thienylmethyl or thiazolylmethyl. Suitable substituents for a heteroaryl ring include halo, hydroxy, C(i_6)alkyl, C(i_6)alkoxy, arylC(i _6)alkoxy, carboxy and esters thereof, (Cι-6)alkylthio, (Cι_6)alkylsulphiny{ and (Cι_6)alkylsulphonyl. Preferably, when Z is S(O)n, n is 1 or 2, more preferably 1. Preferably Z is SO and R3 is arylmethyl or heteroarylmethyl, in particular benzyl or furanylmethyl, especially benzyl.
Suitable esters for incoφoraϋon into R3 include pharmaceutically acceptable esters of the formula CO2 . Such esters may be active in their own right and /or be hydrolysable under in vivo conditions in the human body. Suitable pharmaceutically
acceptable in vivo hydrolysable ester groups for incoφoration in R include those which break down readily in the human body to leave the parent acid or its salt.
Examples of suitable values for R include
 for instance, methyl, ethyl and propyl, (C2"6)alkenyl, for instance allyl. Further examples of suitable values for R include:
for instance, methyl, ethyl and propyl, (C2"6)alkenyl, for instance allyl. Further examples of suitable values for R include:
-CH(Ra)O.CO.Rb;
-CH(Ra)O.CO.ORc;
-CH(Ra)CO.NReRf
-RdNReRf;

CH(Ra)O.CO.C6H4Y4COCH(Ri)NH2 in which:
Ra is hydrogen, (Cι-6)alkyl, in particular methyl, (C3-7)cycloalkyl, or phenyl; RD is (Cι-6)alkyl, (C1-6)alkoxy(Cι-6)alkyl, phenyl, benzyl, (C3-7)cycloalkyl,
(Cι -6)alkyl(C3-7)cycloalkyl, l-amino(Cι -6)alkyl, or l-(Cι-6alkyl)amino(Cι -6)alkyl; or
Ra and R*5 together form a 1,2-phenylene group optionally substituted by one or two methoxy groups; Rc is (Cι -6)alkyl, (C3-7)cycloalkyl, (Cι-6)alkyl(C3-7)cycloalkyl;
Rά* is (C ι -6)alkylene optionally substituted with a methyl or ethyl group;
Re and Rf which may be the same or different is each (Cj-gjalkyl or aryl (C 1 -4) alkyl, optionally substituted with e.g. hydroxy;
RS is (C-[-6)alkyl; Rn is hydrogen, (Cι-g)alkyl or phenyl;
Ri is hydrogen or phenyl optionally substituted by up to three groups selected from halogen, (Cj. -6)-alkyl, or (Cι -6)alkoxy; and
Y4 is oxygen or NH; for instance:
(a) acyloxyalkyl groups such as acetoxymethyl, isobutyryloxymethyl, pivaloyloxymethyl, benzoyloxymethyl, α-acetoxyethyl, α-pivaloyloxyethyl, l-(cyclohexylcarbonyloxy)ethyl, and (l-aminoethyl)carbonyloxymethyl;
(b) alkoxy/cycloalkoxycarbonyloxyalkyl groups, such as ethoxycarbonyloxymethyl, cyclohexyloxycarbonyloxymethyl and α-ethoxycarbonyloxyethyl;
(c) dialkylaminoalkyl, especially di-loweralkylamino alkyl groups such as dimethylaminomethyl, dimethylaminoethyl, diethyiaminomethyl or diethylaminoethyl;
(d) acetamido groups such as N,N-dimethylaminocarbonylmethyl, N,N-(2- hydroxyethyDaminocarbonylmethyl;
(e) lactone groups such as phthalidyl and dimethoxyphthalidyl; and
(f) (5-methyl-2-oxo- 1 ,3-dioxolen-4-y l)methyl.
It will be appreciated by those skilled in the art that the values in the further group of examples include those which have previously been proposed for use as pro- drug esters for various penicillin antibiotics such as ampicillin.
Representative examples of R4 and R-^ when an alkyl group include methyl. Representative examples of a (C3_7)cycloalkyl ring include cyclopropyl. Suitably, R4 and R-^ are both hydrogen or R4 is hydrogen and R-^ methyl.
Suitably, X1 is a direct bond or a group (CH2)XX4 in which X4 is CH2O, CO, COO, CONR6, CONR6CO, or CONHO in which R6 is hydrogen or C(i_6)alkyl, x is 0 (for all except X4 =COO) or an integer from 1 to 6. Suitably X4 is CONH. Suitably x is 0. Preferably, X1 is CONH.
Suitably, in Y**, the alkyl chain is unbranched. Useful such values of Y** include C(6-io) alkyl. preferably C(g_ιo) alkyl or C(3_7)cycloalkylC(5_7) alkyl, preferably cyclohexylC(5_7)alkyl. Representative examples of γl include nonyl and cyclohexylhexyl.
Suitably, X2 is:
(a) a direct bond;
(b) a group X5(CH2)y in which X5 is CO, CONR6, COO, CONR6CO, or CONHO in which R6 is hydrogen or C^.^alkyl and y is 0 or an integer from 1 to 12;
(c) a C(μι 2)alkylene chain optionally interupted by X*^;
(d) a group A-B in which A is a direct bond or χ5 and B is a
 cna**n interupted and or terminated at the end remote from A by one or more groups M selected from O, S(O)n, NR6, alkene or alkyne in which R6 is hydrogen or C(i_6)alkyl and n is 0, 1 or 2.
cna**n interupted and or terminated at the end remote from A by one or more groups M selected from O, S(O)n, NR6, alkene or alkyne in which R6 is hydrogen or C(i_6)alkyl and n is 0, 1 or 2.
Representative examples of X2 include CO(CH2)y, CONH(CH2)y, COO(CH2)y, CONHCO(CH2)y, CONHO(CH2)y and C( 1.12)alkylene. Preferably, X3 is CO or CONR6, more preferably CONH. Preferably, y is 1, 2, 5, 6, 7 or 9, preferably 6. Preferably, X2 is CONH(CH2)6- Representative examples of heteroaryl rings for incoφoration into Y2 include pyridyl and pyridyl N-oxide. Suitable substituents for a heteroaryl ring include halo, hydroxy, CQ _g)alkyl and C(i_g)alkoxy. Suitably, Y2 is 2-pyridyl or 4- pyridyl, preferably in combination with χl being CONH(CH2)6-
Representative examples of X3 include thiazolyl and oxazolyl, in particular

Suitably, p is 1. Representative - values of q - includXe 0 and 5. Representative examples of aryl rings for incoφoration into Y3 include phenyl and naphthyl. Suitable substituents for the aryl ring include halo, hydroxy, C(i_g)alkyl and C(i_g)alkoxy. Suitably, Y3 is phenyl. Representative examples of X3- Y3 include:

It will be readily appreciated by the skilled person that C-4 of the β-lactam ring is a chiral centre which will give rise to the presence of stereoisomers. The present invention encompasses all such stereoisomers. An additional chiral centre will be introduced in compounds of formula (I) in which z is SO. The present invention encompasses all such stereoisomers. A further chiral centre will be introduced when R4 and R are not the same. This will give rise to the existence of extra stereoisomers. The present invention encompasses all such stereoisomers. In preferred compounds of formula (I), the absolute configurations at C-4 and the SO moiety are R and S respectively. In preferred compounds of formula (I) when R4=H, R5=Me, the absolute configuration at the α-carbon (to which R^ is attached) is S.
When used herein, the term 'alkyl' and similar terms such as 'alkoxy' includes all straight chain and branched isomers. Representative examples thereof include methyl, ethyl, n-propyl, wø-propyl, n-butyl, sec-butyl, iso-butyl, t-butyl, n-pentyl and n-hexyl.
Suitable substituents for an alkyl group include, for example, and unless otherwise defined, halogen, cyano, azido, nitro, carboxy, (Cι_6)alkoxycarbonyl, carbamoyl, mono- or di-(Cι _6)alkylcarbamoyl, sulpho, sulphamoyl, mono- or di-(Cτ . 6)alkylsulphamoyl, amino, mono- or di-(Cι_6)alkylamino, acylamino, ureido, (Cι^6)alkoxycarbonylamino, 2,2,2-trichloroethoxycarbonylamino, aryl, heterocyclyl, hydroxy, (Ci.gjalkoxy, acyloxy, oxo, acyl, 2-thienoyl, (Cι_6)alkylthio, (Cι _6)alkylsulphinyl, (Cι_6)alkylsulphonyI, hydroxyimino, (Cι_6)alkoxyimino, hydrazino, hydrazono, benzohydroximoyl, guanidino, amidino and iminoalkylamino. When used herein, the term 'aryl' includes, unless otherwise defined, phenyl or naphthyl optionally substituted with up to five, preferably up to three substituents.
Suitable substituents for an aryl group include, for example, and unless otherwise defined, halogen, cyano, (Ci ^alkyl, (C3-7)cycloalkyI, (Cι -6)alkoxy, halo(Cι-6)alkyl, hydroxy, amino, mono- or di-(Cι-6)alkylamino, acylamino, nitro, carboxy, (Cι-6)alkoxycarbonyl, (Ci -gjalkenyloxycarbonyl, (Cι-6)alkoxycarbonyl(Cι-6)alkyl, carboxy(Cι-6)alkyl, (Ci-^alkylcarbonyloxy, carboxy(Cι-6)alkyloxy, (Ci-βtølJcoxycarbonyKC i -^alkoxy, (Cι-6)alkylthio, (Cι-6)alkylsulphinyl, (C ^alkylsul phony 1, sulphamoyl, mono- and di-(Cj-6)- aikylsulphamoyl, carbamoyl, mono- and di-(Cι ^alkylcarbamoyl, and heterocyclyl. When used herein, the term 'heterocyclyl' includes aromatic and non-aromatic single or fused rings comprising up to four hetero-atoms in the ring selected from oxygen, nitrogen and sulphur and optionally substituted with up to three substituents. Suitably the heterocyclic ring comprises from 4 to 7, preferably 5 to 6, ring atoms. A fused heterocyclic ring system may include carbocyclic rings and need only include one heterocyclic ring. When substituted, a heteroaryl or a heterocyclyl group may have up to three substituents. Suitable such substituents include those previously mentioned for an aryl group as well as oxo.
When used herein, the terms 'halogen' and 'halo' include fluorine, chlorine, bromine and iodine and fluoro, chloro, bromo and iodo, respectively. Preferred compounds of formula (I) include:
N-(6-Cyclohexylhexyl)-(4-benzylsulphinyl-2-oxoazeϋdin-l-yl)acetamide (Diastereoisomer 2);
N-(6-Cyclohexylhexyl)-(4-benzylsulphonyl-2-oxoazetidin-l-yl)acetamide;
N-(Nonyl)-(4-benzylsulphinyl-2-oxoazetidin-l-yl)acetamide (Diastereoisomer 2); and N-(Nonyl)-(4-benzylsulphonyl-2-oxoazetidin- l-yl)acetamide.
Preferred compounds of formula (I) in which, in R^, Y2 is heteroaryl, include:
N-(6-(4-Pyridyl)hexyl)-4-benzylsulphinyl-2-oxoazetidin-l-ylacetamide
(Diastereoisomer 2); and
N-(6-(2-Pyridyl)hexyl)-4-benzylsulphinyl-2-oxoazetidin- 1 -ylacetamide (Diastereoisomer 2).
Preferred compounds of formula (I) in which R° is (CH2)pX3(CH2)nY3 include:
(4-(5-Phenylpentyl)thiazol-2-ylmethyl)-4-benzylsulphinyl-2-oxoazetidine.
Since the compounds of the present invention, in particular compounds of formula (I), are intended for use in pharmaceutical compositions, it will be understood that they are each provided in substantially pure form, for example at least
50% pure, more suitably at least 75% pure and preferably at least 95% pure (% are on a wt wt basis). Impure preparations of the compounds of formula (I) may be used for preparing the more pure forms used in the pharmaceutical compositions. Although
the purity of intermediate compounds of the present invention is less critical, it will be readily understood that the substantially pure form is preferred as for the compounds of formula (I). Preferably, whenever possible, the compounds of the present invention are obtained in crystalline form. When some of the compounds of this invention are allowed to crystallise or are recrystallised from organic solvents, solvent of crystallisation may be present in the crystalline product. This invention includes within its scope such solvates. Similarly, some of the compounds of this invention may be crystallised or recrystallised from solvents containing water. In such cases water of hydration may be formed. This invention includes within its scope stoichiometric hydrates as well as compounds containing variable amounts of water that may be produced by processes such as lyophilisation. In addition, different crystallisation conditions may lead to the formation of different polymoφhic forms of crystalline products. This invention includes within its scope all polymoφhic forms of the compounds of formula (I). Compounds of the present invention are inhibitors of the enzyme lipoprotein associated phospholipase A (Lp-PLA2) and as such are expected to be of use in therapy, in particular in the treatment of atherosclerosis. In a further aspect therefore the present invention provides a compound of formula (I) for use in therapy. The compounds of formula (I) are inhibitors of lysophosphatidylcholine production by Lp-PLA2 and may therefore also have a general application in any disorder that involves endothelial dysfunction, for example atherosclerosis, diabetes, hypertension, angina pectoris and after ischaemia and reperfusion. In addition, compounds of formula (I) may have a general application in any disorder that involves lipid peroxidation in conjunction with enzyme activity, for example in addition to conditions such as atherosclerosis and diabetes, other conditions such as rheumatoid arthritis, stroke, inflammatory conditions of the brain such as Alzheimer's Disease, myocardial infarction, reperfusion injury, sepsis, and acute and chronic inflammation. Further such conditions include various neuropsychiauic disorders such as schizophrenia (see Psychopharmacology Bulletin, 31, 159-165, 1995). Further applications include any disorder that involves activated monocytes, macrophages or lymphocytes, as all of these cell types express Lp-PLAj. Examples of such disorders include psoriasis.
Accordingly, in a further aspect, the present invention provides for a method of treating a disease state associated with activity of the enzyme Lp-PLA2 which method involves treating a patient in need thereof with a therapeutically effective amount of an inhibitor of the enzyme. The disease state may be associated with the increased involvement of monocytes, macrophages or lymphocytes; with the formation of lysophosphatidylcholine and oxidised free fatty acids; with lipid peroxidation in conjunction with Lp PLA2 activity; or with endothelial dysfunction.
Compounds of the present invention may also be of use in treating the above mentioned disease states in combination with anti-hyperlipidaemic or anti- atherosclerotic or anti-diabetic or anti-anginal or anti-inflammatory or anti- hypertension agents. Examples of the above include cholesterol synthesis inhibitors such as statins, anti-oxidants such as probucol, insulin sensitisers, calcium channel antagonists, and anti-inflammatory drugs such as NSAIDs.
In therapeutic use, the compounds of the present invention are usually administered in a standard pharmaceutical composition. The present invention therefore provides, in a further aspect, a pharmaceutical composition comprising a compound of formula (I) and a pharmaceutically acceptable carrier.
Suitable pharmaceutical compositions include those which are adapted for oral or parenteral administration or as a suppository.
The compounds of formula (I) which are active when given orally can be formulated as liquids, for example syrups, suspensions or emulsions, tablets, capsules and lozenges.
A liquid formulation will generally consist of a suspension or solution of the compound or pharmaceutically acceptable salt in a suitable liquid carτier(s) for example, ethanol, glycerine, non-aqueous solvent, for example polyethylene glycol, oils, or water with a suspending agent, preservative, flavouring or colouring agent. A composition in the form of a tablet can be prepared using any suitable pharmaceutical carrier(s) routinely used for preparing solid formulations. Examples of such carriers include magnesium stearate, starch, lactose, sucrose and cellulose.
A composition in the form of a capsule can be prepared using routine encapsulation procedures. For example, pellets containing the active ingredient can be prepared using standard carriers and then filled into a hard gelatin capsule; alternatively, a dispersion or suspension can be prepared using any suitable pharmaceutical carrier(s), for example aqueous gums, celluloses, silicates or oils and the dispersion or suspension then filled into a soft gelatin capsule.
Typical parenteral compositions consist of a solution or suspension of the compound of formula (I) in a sterile aqueous carrier or parenterally acceptable oil, for example polyethylene glycol, polyvinyl pyrrolidone, lecithin, arachis oil or sesame oil. Alternatively, tiie solution can be lyophilised and then reconstituted with a suitable solvent just prior to administration.
A typical suppository formulation comprises a compound of formula (I) which is active when administered in this way, with a binding and/or lubricating agent such as polymeric glycols, gelatins or cocoa butter or other low melting vegetable or synthetic waxes or fats.
Preferably the composition is in unit dose form such as a tablet or capsule.
Each dosage unit for oral administration contains preferably from 1 to 500 mg (and for parenteral administration contains preferably from 0.1 to 25 mg) of a compound of the formula (1).
The daily dosage regimen for an adult patient may be, for example, an oral dose of between 1 mg and 1000 mg, preferably between 1 rag and 500 mg, or an intravenous, subcutaneous, or intramuscular dose of between 0.1 mg and 100 mg, preferably between 0.1 mg and 25 mg, of the compound of the formula (I). the compound being administered 1 to 4 times per day. Suitably the compounds will be administered for a period of continuous therapy, for example for a week or more. Compounds of formula (I) may be prepared by adapting processes previously described for ananlogous compounds in International patent applications WO 96/13484, WO 96/19451 and PCT EP96/02765 (SmithKline Beecham pic). Such processes include treating an azeti (II):

Rl, R2, R3, and Z are as hereinbefore defined; with an alkylating agent of the formula (HI):
R7Rθ
(HI) in which R is a suitable leaving group such as halogen or triflate; and
R^is as hereinbefore defined; under alkylating conditions. Suitable alkylating conditions are well known in the art and include carrying out the reaction in the presence of a suitable base such as sodium hydride or potassium hydroxide optionally with a quaternary ammonium salt such tetrabutyl ammonium bromide, in a suitable alkylating solvent such as tetrahydrofuran (THF), and at a temperature in the range - 10 to 0°C. In compounds of formula (I) in which Z is S(O)n, the preceding alkyiation reaction is conveniently effected on compounds in which n is 0.
Compounds of formula (I) in which n is 1 or 2 can be readily prepared from compounds of formula (I) in which n is 0 by treatment thereof with a suitable oxidising agent such as m-chloroperbenzoic acid. Use of chiral oxidising agents such as (+)- or (-)- 1 , 1 '-bi-2-naphthol / titanium wopropoxide (N Komatsu et al, J Org
Chem, 1993, 58, 7624-7626) can give diastereo isomeric selectivity, if not chirally pure compounds.
Compounds of formula (I) in which one of R4 and R^ is alkyl may also be prepared from corresponding compounds of formula (I) where both R4 and R-> are hydrogen by treatment thereof with an alkylating agent under the conditions described above. Such compounds may be obtained by treating a compound of formula (LT) with an alkylating agent of formula (HI) in which both of R4 and R5 is hydrogen, under alkylating conditions as hereinbefore described.
A second alkyl group for R4/R*5 may be introduced by treating a first obtained compound of formula (I) in which one of R4 and R5 is hydrogen, with an alkylating agent in the presence of a suitable base such as sodium hydride, potassium hydroxide or lithium hexamethyldisilazide, in a suitable alkylating solvent such as tetrahydrofuran (THF), and at a temperature in the range -80 to 10°C.
Compounds of formula (II) in which Z is O may be obtained by treating 4- acetoxyazetidinone, 4-benzoyloxyazetidinone or 4-phenylsulfonylazetidinone with a phenol alcohol HOR3 in the presence of a base such as potassium t-butoxide, in a suitable solvent such as THF at a temperature in the range 0 to 5°C. Compounds of formula (IV) in which Z is S may be obtained by treating 4-acetoxyazetidinone with a thiol HSR3 in the presence of a base such as sodium ethoxide, in a suitable solvent such as ethanol at a temperature in the range 0 to 5°C.
Compounds of formula (IU) may be readily prepared by adapting known synthetic procedures, according to the specific value of X^ . A convenient starting material is an appropriately substituted aryl compound which may then be elaborated to introduce the side chain R7CR4R5 X1 2-. Compounds of formula (I) in which χl denotes a group CONR6 or
CONR O may be of the formula (IV):

(IV) in which: Rl, R2, R3, R4, R5 and Z are as hereinbefore defined; with an amine of the formula (V):
NHR Yl
(V) or a hydroxylamine of the formula (VI):
NH2OY1
(VI) in which R6 and γl are hereinbefore defined, in the presence of an activating agent such as ethyl chloroformate or dicyclohexylcarbodiimide (DCC), in a suitable solvent such as chloroform or dimethyl formamide, at a temperature in the range -10 to 20°C.
A similar process may be used for preparing a compound of formula (I) in which X2 denotes a group CONR6(CH2)y or CONR6O(CH2)y, but using an amine NHR6(CH2)yY1 or a hydroxylamine NH2O(CH2)yγ!. An acid of formula (IV) in which one of R4 and R5 is hydrogen may be obtained by treating a compound of formula (II) with a corresponding 2-bromo (C1.7) alkanoate ester, under alkylating conditions as hereinbefore described; followed by the hydrolysis of the thus formed intermediate ester using standard conditions. A second alkyl group may be inu-oduced by alkylating of the first formed monoalkyl ester.
Compounds of formula (I) in which χ denotes a group (CH2)xCOO in which x is an integer from 1 to 6 may be conveniently prepared by transesterifying a compound of formula (VH):

(vn) in which:
R7 is methyl; and x, R1, R\ R\ R", R5 and Z are as hereinbefore defined; using conditions well known in the art for such reactions, for instance heating in toluene in the presence of a catalytic amount of sodium methoxide and an alcohol. A compound of formula (VII) in which one of R4 and R^ is hydrogen may be obtained by treating a compound of formula (II) with a methyl 2-bromoalkanoate, under alkylating conditions as hereinbefore described. Alternatively, a compound of formula (I) in which χ denotes a group
(CH2)xCOO in which x is an integer from 1 to 6 may be prepared by treating a compound of formula (IV) in which R7 is hydrogen with an alcohol γ θH or an activated derivative thereof, for instance a tosylate.
Compounds of formula (I) in which X 1 is CH2O may be prepared by a suitable ether coupling reaction, for instance treating a compound of formula (VIE):
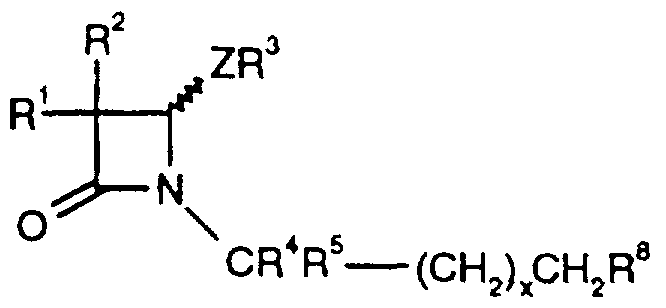
(vm) in which R** is a halogen or other suitable leaving group such as triflate or tosylate and R1, R2, R3, R4, R5, x and Z are as hereinbefore defined; with an alcohol γ θH or a suitable salt therof.
In addition, compounds of formula (I) in which Z is S(O)n and n is 0 may be prepared by a process which comprises treating a compound of formula (DC):

(LX) in which RO, R , R2, χl and γl are as hereinbefore defined; with an alkylating agent of the formula (X):
R8R3
(X) in which R3 and R are as hereinbefore defined; under suitable alkylating conditions, for instance, in a solvent such as acetonitrile, at a temperature in the region 25°C. Compounds of formula (LX) may be obtained from the corresponding 4- acetylthioazetidinone by treatment with silver nitrate and a base in a suitable solvent such as methanol.
Mixtures of diastereoisomeric compounds of formula (I) may be resolved, if so desired, according to procedures well known in the art. For instance sulphoxides (n=l) may be separated by chromatography and/or crystallisation. Chirally pure compounds may be prepared by chiral chromatography, from chirally pure intermediates or by chiral synthesis using chiral reagents or catalysis. Suitable chiral intermediates may be obtained by resolution or chiral induction or by using chiral reagents, in particular natural chiral molecules, according to methods well known to those skilled in the art. For chiral synthesis, a convenient chiral starting material is a penicillin derivative which has the preferred configuration at C-4 of the β-lactam ring. This is illustrated in the following scheme:

The preparation of the starting material (4-methoxybenzyl-6-bromopenicillinate-l- oxide) is described by J. Chem. Soc, Perkin Trans. 1, 1994, 179-188. The present invention will now be illustrated by the following examples. In these, the terms 'diastereoisomer 1' and 'diastereoisomer 2' are used for sulfo xide compounds to refer to the diastereoisomers having R,R/S,S and R,S/S,R configurations, respectively. Such configurations were obtained initially by X-ray analysis of a limited number of compounds and then extrapolated to the remaning compounds on the basis of their H nmr spectra. Unless otherwise specified, all compounds are racemic. Chiral compounds are described as 4R or S, SR or S where the 4 describes the centre at the C4 position in the azetidinone and the S describes the sulfoxide centre.
Preparation 1 Methyl (4-benzylthio-2-oxoazetidin-l-yl)acetate a. 4-(Benzyithio)azetidin-2-one
Sodium (8.1g, 0.35mol) was dissolved in ethanol (250ml) and benzyl mercaptan (45.2g, 0.37mol) added dropwise over 20 minutes keeping the temperature between 20°C - 25°C whilst bubbling nitrogen through the mixture. After 15 minutes, the reaction was cooled to 5°C and a solution of 4-acetoxyazetidin-2-one (45.0g, 0.35mol) in ethanol (50ml) was added dropwise over 15 minutes whilst maintaining the temperature at 5°C. The mixture was stirred at room temperature for 60 minutes and evaporated to dryness under reduced pressure. Water (400ral) was added, the mixture extracted with dichloromethane (2x300ml), the extracts dried (MgSO_ι) and evaporated under reduced pressure to an oil. The oil was cooled to -20°C and titurated with ether (400ml) to give a white solid which was isolated by filtration (50.2g, 79%), m.p. 50-51.0°C. 1H NMR 6 (CDC13) 2.86(1H, m, H3a), 3.30 (IH, m, H3b), 3.85 (2H, s, SCH2), 4.68 (IH, m, H4), 7.31 (5H, m, Ph-H). b. Methyl (4-benzylthio-2-oxoazetidin-l-yl)acetate
To a solution of 4-(benzylthio)azetidin-2-one (5.0g, 25mmol), methyl bromoacetate (4.6g, 30mmol) and tetrabutylammonium bromide (0.9g, 0.28mmol ) in dry THF (150ml) was added powdered potassium hydroxide (1.7g, 30mmol). The resulting mixture was stirred for two hours at room temperature before water (50 ml) was added. The solution was extracted with ethyl acetate (3x150ml portions) and the combined extracts dried (MgSO. ) and evaporated. The residue was purified by flash chromatography on silica gel eluted with petroleum ether 60°-80°:ethyl acetate 4: 1 to give methyl (4-benzylthio-2-oxoazetidin-l-yl)acetate as a yellow oil (5g, 70%).
1H NMR δ (CDCI3) 2.96(1H, dd, J=2.5. 16 Hz H3a), 3.24,3.99 (each IH, d, J=18.00 Hz, NCH2). 3*4 <1H- dd- J=5,12.5 Hz H3b). 3*70 (3H, s, OCH3). -77 (2H, s, SCH2). 4.92 (IH, m, H4), 7.28 (5H, m. Ph-H) Preparation 2 (4- Benzyl thio-2-oxoazetidin- 1-yl )acetic acid To a solution of methyl (4-benzylthio-2-oxo-azetidin-l-yl)acetate (2.5g, 9.4mmol) in methanol (80ml) was added, dropwise at 0°C, a solution of 1 N sodium hydroxide (9.9ml, 9.9mmol). The reaction was stirred for 1 hr and evaporated to dryness. Water (50 ml) was added and the solution acidified to pH 3 with dilute hydrochloric acid and extracted with ethyl acetate (3x100ml) . The combined extracts were dried (MgSO4), evaporated and the residue purified by recrystallisation (hexane/ether) to give (4-benzylthio-2- oxo-azetidin-l-yl)acetic acid as a white solid (1.3g, 55%), mp 110-111°C. !H NMR δ (CDCI3) 2.99 (IH, dd, J=6.87,17.5 Hz, H.3a). 3-27, 4.06 (each IH, d, J=18.40 Hz, NCH2), 3.39 (IH, dd, J=5,15.4 Hz, H3b). 3-77 (2H, s, SCH2). 4*9 i (1H- m> H4), 7.27 (5H, m, Ph-H). Preparation s: . (4-BenzyIthio-2-oxoazetidin-l-yl)acetic acid a. 4-(Benzylthio)azetidin-2-one
Sodium (8.1g, 0.35mol) was dissolved in ethanol (250ml) and benzyl mercaptan (45.2g, 0.37mol) added dropwise over 20 minutes keeping the temperature between 20°C - 25°C whilst bubbling nitrogen through the mixture. After 15 minutes, the reaction was cooled to 5°C and a solution of 4-acetoxyazetidin-2-one (45.0g,
0.35mol) in ethanol (50ml) was added dropwise over 15 minutes whilst maintaining
the temperature at 5°C. The mixture was stirred at room temperature for 60 minutes and evaporated to dryness under reduced pressure. Water (400ml) was added, the mixture extracted with dichloromethane (2x300ml), the extracts dried (MgSO4) and evaporated under reduced pressure to an oil. The oil was cooled to -20°C and titurated with ether (400ml) to give a white solid which was isolated by filtration (50.2g, 79%), m.p. 50-51.0°C. b. MetiιyI-(4-benzy.thio-2-oxoazetidin-l-yl) acetate
To a solution of 4-(benzylthio)azeϋdin-2-one (5.0g, 25mmol), methyl bromoacetate (4.6g, 30mmol) and tetrabutylammonium bromide (0.9g, 0.28mmol ) in dry THF (150ml) was added powdered potassium hydroxide (1.7g, 30mmol). The resulting mixture was stirred for two hours at room temperature before water (50 ml) was added. The solution was extracted with ethyl acetate (3x150ml portions) and the combined extracts dried (MgSO4) and evaporated. The residue was purified by flash chromatography on silica gel eluted with petroleum ether 60°-80°:ethyl acetate 4: 1 to give methyl (4-benzylthio-2-oxoazetidin- l-yl)acetate as a yellow oil (5g, 70%).
1H NMR δ (CDC13) 2.96(1H, dd, J=2.5, 16 Hz H3a). 3.24,3.99 (each IH, d, J=18.00 Hz, NCH2). 3*4 ( H- d- J=5.12.5 Hz H3b). 3-70 (3H, s, OCH3). 3*77 (2H, s, SCH2). 4.92 (IH, m, H4), 7.28 (5H, m, Ph-H) c (4-Benzylthio-2-oxoazeridin-l-yl)acetic acid To a solution of methyl (4-benzylthio-2-oxo-azetidin-l-yl)acetate (2.5g, 9.4mmol) in methanol (80ml) was added, dropwise at 0°C, a solution of 1 N sodium hydroxide (9.9ml, 9.9mmoI). The reaction was stirred for 1 hr and evaporated to dryness. Water (50 ml) was added and the solution acidified to pH 3 with dilute hydrochloric acid and extracted with ethyl acetate (3x100ml) . The combined extracts were dried (MgSO4), evaporated and the residue purified by recrystallisation (hexane/ether) to give (4-benzylthio-2-oxo-azetidin- l-yl)acetic acid as a white solid (1.3g, 55%), mp 110-111°C. JH NMR δ (CDC13) 2.99 (IH, dd, J=6.87,17.5 Hz, H3a). *27, 4.06 (each IH, d, J=18.40 Hz, NCH2), 3.39 (IH, dd, J=5,15.4 Hz, H3t>). 3*77 (2H, s, SCH2). 4*91 (IH, m, H4), 7.27 (5H, m, Ph-H). Preparation 4: 6-(4-Pyridyl)hexylamine
Sodamide (6.63g) was suspended in Uquid ammonia (100ml) and cooled in a cardice/acetone bath. 4-Picoline (7.3ml) was added, the cooling bath removed and the mixture stirred at reflux for 2hrs. The mixture was cooled again and 5- bromopentylamine hydrobromide (18.53g) added and the mixture allowed to reflux for 5 hrs. The mixture was again cooled, quenched with ammonium chloride (lOg) and the solvent allowed to evaporate overnight. The residue was dissolved in water (100ml), made strongly alkaline with NaOH and extracted with CH,C12 (2x100ml). The combined organics were partitioned with water at pH7 and the aqueous was washed with CH2C12( 100ml) then basified with NaOH and extracted with CH,C12 (2x 100ml), dried over K O, and evaporated to an orange oil. This was purified by vacuum distillation to give the title compound as a colourless oil (4.94g, 37% yield; b.p. 110-120°C/0.5mm)
'H NMR δ (CDCl,) 1.3-1.7 (8H, m, 4 x CH2), 2.6 (4H, , QLPyr + CH2NH2), 7.10 (2H, m, Pyr-3,5H), 8.48 (IH, m, Pyr-2,6H) Preparation 5: 6-(2-Pyridyl)hexylamine
Treatment of 2-picoline witii sodamine in liquid ammonia followed by 5- bromopentylamine using the procedure for 6-(4-pyridyl)hexylamine gave the title compound in 38% yield, b.p. 250°C at 0.3 mm Hg Preparation 4: 2-Bromomethyl-4-(5-phenylpentyl)thiazole a. 2-Benxoyloxy-4-(5-phenylpentyl)thiazole
A mixture of benzoyloxylthioacetamide (CA8923f) (2.0g, 0.0102moles) and 1-bromo- 6-phenyl-2-hexanone (2.5g, 0.00929moles) in absolute alcohol (2.5ml) was stiired at reflux for 15 minutes. The reaction was cooled and poured into sat.NaHCO3 (aq) and extracted with ethyl acetate (2x50ml). The organic extracts were combined, washed with brine, dried (MgSO4) and evaporated to an orange oil. Purification by column chromatography eluted with 20: 1 to 10:1 P.E.: ethyl acetate gave 2-benxoyloxy-4-(5- phenylpentyl)thiazole as a colourless oil (2.39g, 71%). b. 2-Hydroxymethyl-4-(5-phenylpentyl)thiazole 2-Benxoyloxy-4-(5-phenylpentyI)thiazole (2.29g, 0.00627moles) was treated with 5% ethanolic potassium hydroxide (100ml) and the mixture was heated on a steam bath for 10 minutes. The mixture was reduced in volume to 25ml under reduced pressure, water (100ml) was added and the mixture was reduced to 50ml under reduced pressure. The residue was acidified to pH7 with dilute HCl and extracted with diethyl ether (x2). The organic extracts were combined and washed with sat.NaHCO , brine, dried (MgSO4) and evaporated to a yellow oil. Purification by column chromatography eluted with 1:1 P.E.: ethyl acetate gave 2-hydroxymethyl-4-(5-phenylpentyl)thiazole as a yellow oil (1.55g, 94%). c 2-Bromomethyl-4-(5-phenylpentyl)thiazole A mixture of 2-hydroxymethyl-4-(5-phenylpentyl)thiazole (1.47g, 0.00562moles) and triphenylphosphine (1.59g, 0.00606moles) in dry CH2C12 (25ml) was cooled to 0°C and treated with solid N-bromosuccinimide (1.08g, 0.00606moles) in portions over 10 minutes maintaining the temperature at 0°C. The cooling bath was removed and the reaction was stirred for lh. Purification by column chromatography eluted with 10:1 to 5:1 P.E.: ethyl acetate gave the product as a colourless oil. The oil was dissolved in diethyl ether and washed with saLNaHCO3 (aq), brine, dried (MgSO4) and evaporated to give 2-bromomethyl-4-(5-phenylpentyl)thiazole as colourless oil (1.52g, 83%).
Example 1 N-(6-CydohexyIhexyl)-(4-beιuyIthio-2-oxoazetidin-l-yI)acetamide
A mixture of (4-benzylthio-2-oxoazetidine-lyl)acetic acid (2.06g), 6- (cyclohexyl)hexylamine (1.50g), dicyclohexylcarbodiimide (1.69g) and 1- hydroxybenzotriazole (1.1 lg) in dry dimethylformamide (DMF) (20ml) was stirred at 20°C for 3 hours. Ethyl acetate (50ml) was added, the mixture filtered and the filtrate evaporated to an oil. This oil was taken up in ethyl acetate, washed with aq. NaHCO3 solution and brine, dried over MgSO4 and evaporated to an oil which was purified by chromatography on silica gel (40-60 Petroleum ether/ethyl acetate) to give the title compound as a colourless oil (2.89g, 85% yield).
'H NMR δ (CDC13) 0.7-1.8 (19H, m, 4 x CH2 + Cyclohexyl CH, CH2). 2.95 (IH, dd, J=2, 15 Hz, H3). -25 (2H, m, NHCH2). 3-38 (IH, dd, J=5, 15 Hz, H3), -55, 3.73 (each IH, d, J=17 Hz, NCH2), 3*^2 ( H, s, SCH ), 4.82 (IH, m, H4), 6.06 (IH, br s, NH), 7.2-7.4 (5H, m, Ph-H)
Example 2 N-(6-Cydohexylhexyl)-4-berifcZyIsuIphinyl-2-oxoazetidin-l-ylacetaιnide (Diastereoisomer 1)
N-(6-Cyclohexylhexyl)-4-benzylthio-2-oxoazetidin- 1 -ylacetamide (2.76g) was dissolved in dichloromethane (60ml), cooled to -60°C and a solution of 55-60% m- chloroperbenzoic acid (mCPBA) (1.84g;ca 6.62mM) in dichloromethane (80ml) was added dropwise over 15 mins, then the mixture was stirred at 20°C for 3 hours. The solution was washed with aq. NaHCO/Na.SO3, water, dried over MgSO, and evaporated to a colourless solid. This was recrystallised three times from EtOAc (cooling to 20°C only) to give the title compound as a colourless solid m.p. 150- PC, (735mg. 26%).
Η NMR δ (CDC13) 0.8-1.8 (19H, m, 4 x CH2 + Cyclohexyl CH, CH2), 2.95 (IH, dd, J=5, 15 Hz, H3). *22 (2H. m, NHCH2). 3-46 (IH, dd, J=2, 15 Hz, H3). 3.72, 4.10 (each IH, d, J=17 Hz, NCH2). 3-90, 4.06 (each IH, d, J=13 Hz, SOCH2). 4.53 (IH, m, H4). 6.62 (lH. br s, NH), 7.2-7.5 (5H, m, Ph-H) v ^, 1791 cm'1
Found: C, 66.4; H, 8.1; N, 6.6%; C2 H36N2O3S requires: C, 66.6; H, 8.4; N, 6.5% Example 3 N-(6-Cyclohexylhexyl)-(4-benzyIsulphinyI-2-oxoazetidin-l- yl)acetamide (Diastereoisomer 2) The mother liquors from the first two recrystallisations in Example 2 above were combined and evaporated to a solid which was recrystallised from EtOAc, cooling to RT and filtering to remove the first formed solid, then refrigerating to obtain a solid which was recrystallised again from EtOAc to give the title compound as a colourless solid, m.p. 135-7°C, (1.32 g, 46%) 'H NMR δ (CDCI3) 0.8-1.8 (19H, m, 4 x CH2 + Cyclohexyl CH, CH2), 2.88 (IH, dd, J=2, 15 Hz, H3), 3.2 (3H, m, H3 + NHCH2). 3.91, 4.20(each IH, d, J=17 Hz, NCH2). 4.00, 4.24 (each IH, d, J=13 Hz, SOCH2). 4.62 (IH, m, H4), 7.2-7.5 (6H, m, Ph-H +
NH); v ^ 1793 cm-1
Found: C, 66.2; H, 8.2; N, 6.5%; C24H36N2O3S requires: C, 66.6; H, 8.4; N, 6.5% Example 4 N-(6-Cyclohexylhexyl)-(4-benzylsulphonyl-2-oxoazetidin-l- y acetamide
N-(6-Cyclohexylhexyl)-(4-benzylsulphinyl-2-oxoazetidin- 1 -yl)acetamide (0.95g) was dissolved in dichloromethane (40ml), a solution of 55-60% mCPBA (0.83g) in dichloromethane (40ml) was added and stirred at 20°C for 1.5 hours. The solution was washed with aq NaHCOJNajSO3 and brine, dried over MgSO4 and evaporated to a solid. This was purified by chromatography on silica gel (40-60 Pet ether/EtOAc) then recrystallisation from EtOAc to give the title compound as a colourless solid, m.p. 129-30°C, (630 mg, 64%) 'H NMR δ (CDCI3) 0.8-1.8 (19H, m, 4 x CH2 + cyclohexyl CH, CH,), 3.01 (IH, dd, j=2, 15 Hz, H3), 3.12 (IH, dd, J=5, 15 Hz, H3), 3.25 (2H, m, NHCH2), 3-8 . 3*97 (each IH, d, j=17 Hz, NCH2), 4- 5 (2H, s, SO2CH2), 4-86 (IH, m, H4), 5.96 (IH, br s, NH), 6.9, 7.4 (5H, m, Ph-H ); v ^ 1796 cm" 1
Found: C, 64.2; H, 7.9; N, 6.4%; C24H36N2O4S requires: C, 64.3; H, 8.1; N, 6.2% Example 5 N-(Nonyl)-(4-benzyIthio-2-oxoazetidin-l-yl)acetamide
Treatment of (4-benzylthio-2-oxoazetidine-lyl)acetic acid with nonylamine under the conditions described for Example 1 gave the title compound as a colourless soild, m.p. 64-65°C, 88% yield
1H NMR δ (CDC13) 0.88 (3H, t, J=6.8Hz, CH3), 1.28 (12H, m, 6xCH2). 1-50 (2H, m, NCH2CH2). 2*93- 2-97 (IH, dd, J=2.4, 15.4Hz, H3). 3.24 (2H, m, NHCH2). 3-36, 3.40 (IH, dd, J=5.2, 15.4Hz, H3), 3.57, 3.72 (each IH, d, J=16.7Hz, NCH2), 3 *82 (2H, s, SCH2). 4*81 ( H, m, H4), 6.0 (IH, m, NH), 7.30-7.33 (5H, m, Ph-H); v ^ 1773 cm" 1
Found: C, 67.0; H, 8.4; N, 7.6%; C2JH32N2O2S requires: C, 67.0; H, 8.6; N, 7.4%
Treatment of N-(nonyl)-(4-benzylthio-2-oxoazetidin-l-yl)acetamide with mCPBA as described for Examples 2 and 3 gave the following two compounds (Examples 6 and 7) after separation by crystallisation.
Example 6 N-(Nonyl)-(4-benzyIsulphinyl-2-oxoazetidin-l-yl)acetamide (Diastereoisomer 1)
Colourless solid, m.p. 162-164°C, 22% yield
1H NMR δ (CDCI3) 0.88 (3H, t, J=6.8Hz, CH3), 1.28 (12H, m, 6xCH2). 1*50 (2H, m,
NCH2CH2)> *93, 2.97 (IH, dd, J=4.8, 14.8Hz, H3). 3.22 (2H, m, NHCH2). 3*44- 3.48 (IH, dd, J=2.0, 14.8Hz, H3). 3-73, 4.10 (each IH, d, J=17.6Hz, NCH2). 3-90. 4.05 (each IH, d, J=12.8Hz, SOCH2), 4.53 (IH, m, H4), 6.6 (IH, m, NH), 7.26-7.40 (5H, m, Ph-H); v ^ 1791 cm"1
Found: C, 64.0; H, 7.8; N, 7.4%; C21H32N2O3S requires: C, 64.3; H. 8.2; N, 7.1% Example 7 N-(NonyI)-(4-benzylsulphinyl-2-oxoazetidin-l-yl)acetamide (Diastereoisomer 2)
Colourless solid, m.p. 107-108°C, 34% yield
1H NMR δ (CDCI3) 0.88 (3H, t, J=6.8Hz, CH3), 1.28 (12H, m, 6xCH2), 1-52 (2H, m, NCH2CH2). 2-85, 2.89 (IH, dd, J=2.4, 15.3Hz, H3), 3-15, 3.19 (IH, dd, J=5.4, 15.3Hz, H3), -26 (2H, m, NHCH2). 3.91, 4.23 (each IH, d, J=17.1Hz, NCH2). 4*<X 4.20 (each IH, d, J=13Hz, SOCH2), 4*62 (IH, m, H4), 7.2 (IH, m, NH), 7.26-7.40 (5H. ra. Ph-H); v^ 1794 cm" 1
Found: C. 63.9; H, 8.0; N, 7.2%; C21H32N2O3S requires: C, 64.3; H, 8.2; N, 7.1% Example 8 N-(Nonyl)-(4-benzylsulphonyl-2-oxoazetidin-l-yl)acetamide Treatment of N-(nonyl)-(4-benzylsulphinyl-2-oxoazetidin-l-yl)acetamide with mCPBA as described for Example 4 gave the title compound as a colourless solid, m.p. 125-126°C, 76% yield
1H NMR δ (CDCI3) 0.88 (3H, t, J=6.8Hz, CH3). 1.28 (12H, m, 6XCH2). 1-50 (2H, m, NCH2CH2), 2.97, 3.03 (IH, dd, J=2.4, i5.4Hz. H3), 3*09. - 5 (IH, dd, J=5.1, 15.4Hz, H3), -24 (2H, m, NHCH2), 3-83, 3.98 (each IH, d, J=16.9Hz, NCH2)- 4*32. 4.39 (each IH, d, J=14.3Hz, SO2CH2), 4.85 (IH, m, H*), 5.95 (IH, m, NH), 7.41 (5H, m, Ph-H) v,^ 1796 cm'1
Found: C, 61.4; H, 7.6; N, 7.0%; C21H32N2O4S requires: C, 61.7; H, 7.9; N, 6.9% Example 11 N-(6-( PyridyI)hexyl)^-benzyIthio-2-oxoazetidin-l-ylacetamide A mixture of 4-benzylthio-2-oxoazetidine- lylacetic acid (2.26g), 6-(4- pyridyDhexylamine (1.60g), dicyclohexylcarbodiimide (1.86g) and 1-
hydroxybenzotriazole (1.22g) in dry dimethylformamide (20ml) was stirred at 20- 25°C for 3 hrs. Ethyl acetate (50ml) was added, the mixture was filtered and the filtrate evaporated to an oil. This oil was taken up in EtOAc, washed with NaHCO3, brine, dried over MgSO4 and evaporated to an orange oil which was purified by chromatography on silica gel (EtOAc/EtOH) to give the title compound as a colourless oil (3.22g. 87% yield)
Η NMR δ (CDC13) 1.2-1.7 (8H, m, 4 x CHJ, 2.59 (2H, t, J=8 Hz, CH2Ph), 2.95 (IH, dd, J=2, 15 Hz. H3), 3-24 (2H, m, NHCH2), -37 (IH, dd, J=5, 15 Hz, H,), 3.56, 3.72 (each IH, d, =17 Hz, NCH2), 3-81 (2H, s, SCH2), 4.82 (IH, m, HJ, 6.18 (IH, br s, NH), 7.09 (2H, m, Pyr-3,5H), 7.30 (5H, m, Ph-H), 8.47 (IH, m, Pyr-2,6H) Example 12 N-(6-(4-PyridyI)hexyl)-4-benzylsulphinyl-2-oxoazetidin-l- ylacetamide (Dia 1)
N-(6-(4-pyridyl)hexyl)-4-benzylthio-2-oxoazetidin- l-ylacetamide (3.04g)was dissolved in CH2Cl2(50ml), cooled to -60°C and a solution of 55-60% m- chloroperbenzoic acid (mCPB A) (2.11 g) in CH2C12 ( 100ml) was added dropwise over 15 mins. The solution was stirred at 20-25°C for 3 hrs then washed with aq NaHCOj/NajSOj, brine, dried over MgSO4 and evaporated to a sticky solid. This was recrystallised twice from ethyl acetate to give the title compound as a colourless solid m.p. 167-8°C, (0.90g; 30%) Η NMR δ (CDC13) 1.2-1.7 (8H, m, 4 x CH2), 2.59 (2H, t, J=8 Hz, CH2Ph), 2.97 (IH, dd, J=5, 15 Hz, H,), 3.23 (2H, m, NHCH2). 3.47 (IH, dd, J=2, 15 Hz, H3), 3.68, 4.15 (each IH, d, J=17 Hz, NCH2), 3.89, 4.07 (each IH, d, J=13 Hz, SOCH2), 4.50 (IH, m, HJ, 6.75 (IH, br s, NH), 7.10 (2H, m, Pyr-3,5H), 7.25, 7.40 (5H, 2 x m, Ph-H), 8.48 (2H, br s, Pyr-2,6H); v ^ 1792 cm" 1 Found: C, 64.4; H, 6.7; N, 9.8%; C^H^N^S requires: C, 64.6; H. 6.8; N, 9.8% Example 13 N-(6-(4-Pyridyl)hexyl)-4-benzylsulphinyI-2-oxoazetidin-l- ylacetamide (95% Dia 2)
The mother liquor from the above recrystallisation in Example 12 was evaporated to a solid which was recrystallised twice from ethyl acatate to give the title compound as a colourless solid, m.p. 109-10°C, (1.41g, 46% yield)
Η NMR δ (CDC13) 1.3-1.8 (8H, m. 4 x CH2), 2.59 (2H, t, J=8 Hz, CH2Ph). 2.92 (IH, dd, J=2, 15 Hz, H,), 3-25 (3H, m. NHCH2 +H,), 3-87, 4.27 (each IH, d, J=17 Hz, NCH2). 3-98, 4.19 (each IH. d. J=13 Hz, SOCH2), 4.61 (IH, m, HJ, 7.10 (2H, m, Pyr-3,5H), 7.25,7.38 (6H, 2 x m, Ph-H + NH), 8.47 (2H, m, Pyr-2,6H); v <-<, 1793 cm'1
Found: C, 64.3; H, 6.6; N, 9.8%; C^N^S requires: C, 64.6; H, 6.8; N, 9.8%
Example 14 N-(6-(4-Pyridyl)hexyl)-4-benzylsulphonyI-2-oxoazetidin-l- ylacetamide
N-(6-(4-Pyridyl)hexyl)-4-benzy lsulphinyl-2-oxoazetidin- 1 -ylacetamide (Dia 1 ) (1.15g) was dissolved in CH2Cl2(50ml) and a solution of 55-60% CPBA(1.10g;ca 3.5mM) in CH2Cl2(50ml) added and stirred at 20-25°C for 3hrs and allowed to stand at 20-25°C for 16 h. mCPBA (0.13g) was added and the solution stirred for a further 3 hrs then washed with aq NaHCO/Na-SO,, brine, dried over MgSO4 and evaporated to an oil. This oil was chromatographed on silica gel (EtOAc/EtOH) to give the title compound as a solid which was recrystallised from EtOAc/Et.,0 to give a colourless solid m.p. 106-8°C, (0.38g, 31%)
'H NMR δ (CDC13) 1.3-1.8 (8H, m, 4 x CH2). 2.60 (2H, t, J=8 Hz, CH2Pyr), 3.00 (IH, dd, J=2, 15 Hz. H,), 3.11 (IH, dd, J=5, 15 Hz. H,), 3.25 (2H, m, NHCH2), 3.86, 3.94 (each IH, d, J=17 Hz, NCH2), 4.34 (2H, s, SO2CH2), 4.83 (IH, m, HJ, 6.10 (IH, br s, NH), 7.10 (2H, m, Pyr-3,5H), 7.41 (5H, m, Ph-H), 8.47 (2H, m, Pyr-6H); v ^ 1797 cm"1
Found: C, 62.0; H, 6.5; N, 9.5%; C^H^O.S requires: C, 62.3; H, 6.6; N, 9.5% Example 15 N-(6-(4-Pyridyl)hexyl)-4-benzylsulphonyI-2-oxoazetidin-l- yiacetamide N-(pyridyl)oxide N-(6-(4-Pyridyl)hexyl)-4-benzylsulphonyl-2-oxoazetidin- 1 -ylacetamide N- (pyridyl)oxide was also produced in the oxidation of N-(6-(4-Pyridyl)hexyl)-4- benzylsulphinyl-2-oxoazetidin-l-ylacetamide with mCPBA (details in Example 14 above) and was obtained after chromatography and recrystallisation from ethyl acetate as a colourless solid (0.44g, 35%), m.p. 132-4°C. 'H NMR δ (CDCl.) 1.3-1.8 (8H, m, 4 x CHJ, 2.60 (2H, t, J=8 Hz, CH2Pyr), 3.00 (IH, dd, J=2, 15 Hz. H,), 3.12 (IH, dd, J=5, 15 Hz, H3), 3.25 (2H, m, NHCH2), 3.91 (2H, s, NCH2), 4.36 (2H, s, SO2CH2), 4.83 (IH, m, HJ. 6.22 (IH, br s, NH), 7.09 (2H, m, Pyr-3,5H). 7.41 (5H, m, Ph-H), 8.12 (2H, m, Pyr-2,6H); v c=0 1783 cm"1 Found; C, 59.6; H, 6.2; N, 9.1%; CMH29N3O5S requires: C, 60.1; H, 6.4; N, 9.1% Example 16 N-(6-(2-Pyridyl)hexyl -berifcZylt ^-2-oxoazetidin-l-ylacetarnide Treatment of of 4-benzylthio-2-oxoazetidine- lylacetic acid with 6-(2- pyridyl)hexylamine, dicyclohexylcarbodiimide and 1 -hydroxybenzotriazole in dry dimethylformamide as described for Example 11 gave the title compound as a colourless oil in 90% yield. Η NMR δ (CDC13) 1.3-1.8 (8H, m, 4 x CHJ, 2.77 (2H, t, J=8 Hz, CH2Pyr), 2.94 (IH, dd, J=2, 15 Hz, HJ, 3.23 (2H, m, NHCH2), 3.36 (IH, dd, J=5, 15 Hz, HJ, 3.54, 3.75 (each IH, d, J=17 Hz, NCH2), 3.81 (2H, s, SCH2), 4.83 (IH, m, HJ, 6.29 (IH, br s, NH), 7.15 (2H, m, Pyr-3,5H), 7.30 (5H, m, Ph-H), 7.59 (IH, m, Pyr-4H), 8.50 (IH, m, Pyr-6H) Examples 17 and 18 were prepared by the methods described for Examples 12 and 13 and Examples 19 and 20 were prepared by the methods described for Examples 14 and 15.
Example 17 N-(6-(2-Pyridyl)hexyI)-4-benzylsulphinyl-2-oxoazetidin-l- ylacetamide (Dia 1)
Colourless solid, m.p. 126-30°C, 30% yield
Η NMR δ (CDCl,) 1.3-1.9 (8H, m, 4 x CH2), 2.77 (2H, t, J=8 Hz, CH2Pyr), 2.95 (IH, dd, J=5, 15 Hz, H,), 3.22 (2H, m. NHCH2), 3.45 (IH, dd, J=2, 15 Hz. HJ, 3.75, 4.09 (each IH, d, J=17 Hz, NCH2), 3.90, 4.06 (each IH, d, J=13 Hz, SOCH2), 4.54 (IH, m, HJ, 6.75 (IH, br s, NH), 7.10 (2H, m, Pyr-3,5H), 7.30 (5H, m, Ph-H), 7.59 (IH, m, Pyr-4H), 8.50 (IH, m, Pyr-6H); v c=0 1791 cm" 1 Found: C, 64.5; H, 6.6; N, 9.8%; C^H^OjS requires: C, 64.6; H, 6.8; N, 9.8% Example 18 N-(6-(2-Pyridyl)hexyI)-4-benzyIsulphinyl-2-oxoa2etidin-l- yiacetamide (92% Dia 2) Colourless solid, m.p. 93-6°C, 20% yield
Η NMR δ (CDCl,) 1.3-1.9 (8H, m, 4 x CHJ, 2.78 (2H, t, J=8 Hz, CH2Pyr), 2.86 (IH, dd, J=2, 15 Hz, HJ, 3.25 (3H, m, NHCH2 + H,). 3.92, 4.23 (each IH, d, J=17 Hz, NCH,), 4-0, 4.20 (each IH, d, J=13 Hz, SOCHJ, 4.63 (IH, m, HJ, 7.10 (2H, m, Pyr-3,5H). 7.30 (6H, m, Ph-H + NH), 7.58 (IH, m, Pyr-4H), 8.50 (IH, m. Pyr-6H); v M 1793 cm'1
Found: C, 64.5; H, 6.7; N, 9.8%; C.JHj.N.OjS requires: C, 64.6; H, 6.8; N. 9.8%
Example 19 N-(6-(2-Pyridyl)hexyl)-4-benzylsulphonyl-2-oxoazetidin-l- ylacetamide
Colourless solid, m.p. 116-8°C. 9% yield Η NMR δ (CDCl,) 1.3-1.8 (8H, m.4 x CH2), 2.78 (2H. t. J=8 Hz. CH2Pyr), 2.99 (IH, dd, J=2, 15 Hz, H,), 3.11 (IH, dd, J=5, 15 Hz. H,), 3.24 (2H, m, NHCH2), 3.82, 4.01 (each IH, d, J=17 Hz, NCH2). 4.36 (2H, s, SO2CH2), 4.87 (IH, m, HJ, 6.26 (IH, br s, NH), 7.12 (2H, m, Pyr-3,5H , 7.41 (5H, m. Ph-H), 7.60 (IH, m, Pyr-4H), 8.50 (IH, m, Pyr-6H); v c=0 1792 cm" 1 Found: C, 62.2; H, 6.5; N, 9.4%; CBUJi3OtS requires: C, 62.3; H, 6.6; N, 9.5% Example 20 N-(6-(2.PyridyI)hexyl)-4-benzylsulphonyl-2-oxoazetidin-l- ylacetamide N-(pyridyl)oxide Colourless solid, m.p. 153-4°C, 61% yield 'H NMR δ (CDCl,) 1.3-1.8 (8H, m, 4 x CH2), 2.96 (3H, m, CH2Pyr + H3), 3.13 (IH, dd, J=5, 15 Hz, H,). 3.25 (2H, m, NHCH2), 3.85, 4.09 (each IH, d, J=17 Hz, NCH2), 4.38 (2H, s, SO2CH2), -92 (IH, m, HJ, 6.61 (IH, br s, NH), 7.2 (3H, m, Pyr- 3.4.5H), 7.41 (5H, m, Ph-H), 8.26 (IH, m, Pyr-6H); v ^ 1793 cm"1 Found: C, 60.0; H, 6.3; N, 9.1%; C-jH^N.OjS requires: C, 60.1; H, 6.4; N, 9.1% Example 21 l-(4-(5-Phenylpentyl)thiazol-2-ylmethyl)-4-benzylthio-2- oxoazetidine
To a mixture of 2-bromomethyl-4-(5-phenylpentyl)thiazole (0.5 g), 4-benzylthio- azetidin-2-one (0.3 g) and tetra-n-butylammonium bromide (0.05 g) in dry tetrahydrofuran (THF), cooled to 10°C, powdered potassium hydroxide (0.096 g) was added. The mixture was warmed to 20°C and stirred for 4 hours. Ethyl acetate and brine were added and the organic solution separated, washed and evapotated to give a brown oil. Chromatography on silica gel using petroleum ether/ethyl acetate gave the title compound as a pale yellow oil (0.48 g, 68% yield). *H nmr δ (CDC13) 1.40 (2H, m, CHJ, 1.63-1.75 (4H, m, 2 C U), 2.59 (2H, t, J=7.6 Hz, PhCH,). 2.74 (2H, t, J=7.6Hz, thiazoIvlCH-.). 2.93, 2.97 (IH, dd, J=2.4, 15.2 Hz, Hj), 3.33, 3.37 (IH, dd, J=5.2, 15.2 Hz, HJ, 3.77 (2H, s, SCH--). 4.33, 4.76 (each IH, d, J=16.4 Hz, NCϋϊ), 4.78 (IH, , H4), 6.83 (IH, s, thiazole-H), 7.14-7.29 (10H, m, 2xPh-H); v ^ 1772 cm 1
Example 22 l-(4-(5-Phenylpentyl)thiazoI-2-ylmethyl)-4-benzylsulphinyl-2- oxoazetidine (Dia l:Dia 2 22:78) 1 -(4-(5-phenylpentyl)thiazol-2-ylmethyl)-4-benzylthio-2-oxoazetidine (1.1 equiv) was dissolved in CH2Cl2(50ml), cooled to -60°C and a solution of 55-60% m- chloroperbenzoic acid (mCPBA) (2.1 lg) in CH2Cl2(100ml) was added dropwise over 15 mins. The solution was stirred at 20-25°C for 3 hrs then washed with aq NaHCO Na2SO3, brine, dried over MgSO4 and solvent removed under reduced pressure to give the title compound as a mixture of diastereoisomers in the form of a colourless oil in 87% yield, v c=0 1785 cm'1
Found: C, 66.2; H, 6.2; N, 5.9%; C2SH2ltN2θ2S requires: C, 66.3; H, 6.2; N, 6.2% Example 23 l-(4-(5-Phenylpentyl)thiazol-2-ylmethyl)-4-benzylsulphonyI-2- oxoazetidine
Treatment of l-(4-(5-phenylpentyl)thiazol-2-ylmethyl)-4-benzylsulfinyl-2- oxoazetidine with mCPBA (1.1 equiv) under the conditions described for Example 2 gave the titie compound in the form of a colourless oil in 91% yield. 1H nmr δ (CDCl,) 1.40 (2H, m, £H2), 1.61-1.73 (4H, m, 2x£H2), 2.60 (2H, t, J=7.8 Hz, PhC±y, 2.74 (2H, t, J=7.6Hz, thiazolyl£H2), 3.15 (2H, d, J=4Hz, 2xH3), 4.35 (2H, m, SO^H,), 4-50, 4.97 (each IH, d, J=16.4 Hz, NCϋ2), 4.84 (IH, m, HJ, 6.86 (IH, s, thiazole-H), 7.14-7.39 (10H, m, 2xPh-H); v ^ 1792 cm" 1
Found: C, 64.1; H, 6.2; N, 6.0%; C^H^N.O.S requires: C, 64.1; H, 6.0; N, 6.0% Example 24 4-Methylthio-l-((5-phenyloxazol-2-yI)methyl)azetidin-2-one Treatment of 4-methylthioazetidin-2-one with 2-bromomethyl-5-phenyloxazole in the presence of lithium hexamethyldisilazide in THF at -70°C gave the title compound as a yellow gum.
Found: C, 61.3; H, 5.2; N, 10.2%; CI4HuN2O2S requires: C, 61.3; H, 5.1; N, 10.2% Example 25 4-Methylsulfinyl-l-((5-phenyloxazol-2-yl)methyl)azetidin-2-one Treatment of 4-methylthio-l-((5-phenyloxazol-2-yl)methyl)azetidin-2-one with mCPBA (1 eq) in dichloromethane at -78°C following the procedure of Example 2 gave the titie compound as a mixture of diastereoisomers as a yellow gum. m z 290 (M*), 227, 158, 103, 77.
Found: C, 56.3; H, 4.9; N, 9.5%; C,4H14N2O,S (+ 0.4 H2O + 0.02 CH2C12) requires: C, 56.2; H, 5.0; N, 9.4%; (solvents identified by nmr) Example 26 4-Methylsulfinyl-l-((5-phenyloxazoI-2-yl)methyI)azetidin-2-one Treatment of 4-methylthio-l-((5-phenyloxazol-2-yl)methyl)azetidin-2-one with mCPBA (2 eq) in dichloromethane at 20°C gave the title as a cream solid Found: C, 54.8; H, 4.7; N, 9.2%; C1 H14N2O S requires: C, 54.9; H, 4.6; N, 9.1%;
Biological Data
Screen for Lp-PLA inhibition.
Enzyme activity was determined by measuring the rate of turnover of the artificial substrate (A) at 37 °C in 50mM HEPES (N-2-hydroxyethylpiperazine-N'-2- ethanesulphonic acid) buffer containing 150mM NaCl, pH 7.4.

(A)
Assays were performed in 96 well titre plates.
Lp-PLA2 was partially purified by density gradient centrifugation of human plasma. Active fractions were pooled and used as the source of Lp-PLA2- The enzyme was pre-incubated at 37 °C with vehicle or test compound for 10 rain in a total volume of 180 μl. The reaction was then initiated by the addition of 20 μl lOx substrate (A) to give a final substrate concentration of 20 μM. The reaction was followed at 405 nm for 20 minutes using a plate reader with automatic mixing. The rate of reaction was measured as the rate of change of absorbance.
Results:
The compounds of Example 3, 7, 8, 13, 18 and 22 had IC50 values in the range 5 to
200nM.
Claims
1. A compound of formula (I):

R is CR^ -Y1, CR4R5-X2-Y2, or (CH2)pX3(CH2)qY3;
Rl and R2, which may be the same or different, is each selected from hydrogen, halogen or C(i_g)alkyl; R4 and R-5 which may be the same or different is each selected from hydrogen and
C(i_6)alkyl, or R4 and R5 may De »nked together to form the residue of a C(3_7) cycloalkyl ring;
X1 is a linker group and Y1 is optionally substituted C(\.\ 2)alkyl C(2-i2)aUceny1 >
C(2-i2)alkynyl, C(3.7)-cycloalkylC(1.8) alkyl; X2 is a linker group and Y2 an optionally substituted heteroaryl group;
X3 is a heteroaryl group and Y3 is an optionally substituted aryl group, p is an integer from 1 to 6, q is 0 or an an integer from 1 to 6;
Z is O and R3 is C(i_8)alkyl, arylC(1_4)alkyl or aryl each of which may be optionally substituted, or Z is S(O)n in which n is 0, 1 or 2 and R3 is C(i_g)alkyl, C(3_8)Cycloalkyl,
C(3_8)cycloalkylC(i_6)alkyl, aryl, arylC(i _4)alkyl or heteroarylC^.^alkyl each of which may be optionally substituted.
2. A compound of formula (I) as claimed in claim 1 in which R and R2 is each hydrogen.
3. A compound of formula (I) as claimed in claim 1 or 2 in which Z is SO and R3 is arylmethyl or heteroarylmethyl
4. A compound as claimed in claim 3 in which R3 is benzyl or furanylmethyl.
5. A compound as claimed in claim 4 in which R3 includes a carboxy or ester substituent.
6. A compound as claimed in any one of claims 1 to 5 in which R4 and R-5 are both hydrogen or R4 is hydrogen and R^ methyl.
7. A compound as claimed in any one of claims 1 to 6 in which: χl is a direct bond or a group (CH2)XX4 in which X4 is CH2O, CO, COO, CONR6, CONR^O, or CONHO in which R6 is hydrogen or C(i.6)alkyl, x is 0 (for all except X4 =COO) or an integer from 1 to 6; or (ii) X2 is: (a) a direct bond;
(b) a group χ5(CH2)y in which X5 is CO, CONR6, COO, CONR6CO, or CONHO in which R6 is hydrogen or C(i_ *,alkyl and y is 0 or an integer from 1 to 12;
(c) a C(i-i2)alkylene chain optionally interupted by X5;
(d) a group A-B in which A is a direct bond or X*5 and B is a C(i_i2)alkylene chain interupted and/or terminated at the end remote from A by one or more groups M selected from O, S(O)n, NR6, alkene or alkyne in which R6 is hydrogen or C(i_6)al yl and n is 0, 1 or 2.
8. A compound as claimed in claim 7 in which X1 is CONH or X2 is CONH(CH2)6-
9. A compound as claimed in any one of claims 1 to 8 in which Y1 is 0(5.10) alkyl, C(3_7)cycloalkylC(5_7) alkyl or Y2 is pyridyl or pyridyl N-oxide.
10. A compound as claimed in any one of claims 1 to 9 in which X3 is:

11. A compound of formula (I) selected from: N-(6-Cyclohexylhexyl)-(4-benzylthio-2-oxoazetidin- 1 -yl)acetamide;
N-(6-Cyclohexylhexy l)-4-benzylsulphinyl-2-oxoazetidin- 1 -ylacetamide
(Diastereoisomer 1);
N-(6-Cyclohexylhexyl)-(4-benzylsulphinyl-2-oxoazetidin-l-yl)acetamide
(Diastereoisomer 2); N-(6-Cyclohexylhexyl)-(4-benzylsulphonyl-2-oxoazetidin- 1 -yl)acetamide;
N-(T^onyl)-(4-benzylthio-2-oxoazetidin-l-yl)acetamide;
N-(Nonyl)-(4-benzylsulphinyl-2-oxoazetidin-l-yl)acetamide (Diastereoisomer 1);
N-(Nonyl)-(4-benzylsulphinyl-2-oxoazetidin- 1 -yl)acetamide (Diastereoisomer 2);
N-(Nonyl)-(4-benzylsulphonyl-2-oxoazetidin- 1 -yl)acetamide; N-(6-(4-Pyridyl)hexyl)-4-benzylthio-2-oxoazetidin-l -ylacetamide;
N-(6-(4-Pyridyl)hexyl)-4-benzylsulphinyl-2-oxoazetidin-l -ylacetamide (Dia 1);
N-(6-(4-Pyridyl)hexyl)-4-benzylsulphinyl-2-oxoazetidin- 1-ylacetamide (Dia 2);
N-(6-(4-Pyridyl)hexyl)-4-benzylsulphonyl-2-oxoazetidin- 1 -ylacetamide;
N-(6-(4-Pyridyl)hexyl)-4-benzylsulphonyl-2-oxoazetidin- 1 -ylacetamide N- (pyridyl)oxide;
N-(6-(2-Pyridyl)hexyl)-4-benzylthio-2-oxoazetidin-l-ylacetamide;
N-(6-(2-Pyridyl)hexyl)-4-benzylsulphinyl-2-oxoazetidin-l-ylacetamide (Dia 1);
N-(6-(2-Pyridyl)hexyl)-4-benzylsulphinyl-2-oxoazetidin- 1 -ylacetamide (Dia 2);
N-(6-(2-Pyridyl)hexyl)-4-benzylsulphonyl-2-oxoazetidin-l-ylacetamide; N-(6-(2-PyridyI)hexyl)-4-benzylsulphonyl-2-oxoazetidin- 1 -ylacetamide N- (pyridyl)oxide; l-(4-(5-Phenylpentyl)ti iazol-2-ylmethyl)-4-benzylthio-2-oxoazeϋdine; l-(4-(5-Phenylpentyl)thiazol-2-ylmethyl)-4-benzylsulphinyl-2-oxoazeticline ; l-(4-(5-Phenylpentyl)thiazol-2-ylmethyl)-4-benzylsulphonyl-2-oxoazetidine; 4-Methylthio-l-((5-phenyloxazol-2-yl)methyl)azetidin-2-one;
4-Methylsulfinyl-l-((5-phenyloxazol-2-yl)methyl)azetidin-2-one; and 4-Methylsulfιnyl-l-((5-phenyloxazol-2-yl)methyl)azeϋdin-2-one;
12. A pharmaceutical composition comprising a compound of formula (I) and a pharmaceutically acceptable carrier.
13. A compound of formula (I) for use in therapy.
14. The use of a compound of formula (I) as defined in claim 1 in the manufacture of a medicament for treating atherosclerosis.
15. The use of a compound of formula (I) as defined in claim 1 in the manufacture of a medicament for treating diabetes, hypertension, angina pectoris, after ischaemia, reperfusion, rheumatoid arthritis, stroke, myocardial infarction, reperfusion injury, sepsis, and acute and chronic inflammation, inflammatory conditions of the brain such as Alzheimer's Disease, neuropsychiauic disorders such as schizophrenia, and psoriasis.
16. A method of treating a disease state associated with activity of the enzyme Lp-PLA2 which method involves treating a patient in need thereof with a therapeutically effective amount of an inhibitor of the enzyme.
17. A method as claimed in claim 15 in which the disease state is associated with: (a) the increased involvement of monocytes, macrophages or lymphocytes; (b) the formation of lysophosphatidylcholine and oxidised free fatty acids;
(c) lipid peroxidation in conjunction with Lp PLA2 activity; or
(d) endothelial dysfunction.
18. A process for preparing a compound of formula (I) as defined in claim 1 which comprises treating an azetidone of formula (II): 
(II) in which:
Rl, R2, R3, and Z are as hereinbefore defined; with an alkylating agent of the formula (IH):
R7Rθ
(Lϋ) in which R7 is a suitable leaving group such as halogen or triflate; and Rθ-s as hereinbefore defined; under alkylating conditions.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP96942375A EP0865429A1 (en) | 1995-12-08 | 1996-12-04 | Azetidinone compounds for the treatment of atherosclerosis |
| JP9521747A JP2000505063A (en) | 1995-12-08 | 1996-12-04 | Azetidinone compounds for the treatment of atherosclerosis |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB9525148.4A GB9525148D0 (en) | 1995-12-08 | 1995-12-08 | Novel compounds |
| GB9525148.4 | 1995-12-08 | ||
| GB9525150.0 | 1995-12-08 | ||
| GB9525149.2 | 1995-12-08 | ||
| GBGB9525150.0A GB9525150D0 (en) | 1995-12-08 | 1995-12-08 | Novel compounds |
| GBGB9525149.2A GB9525149D0 (en) | 1995-12-08 | 1995-12-08 | Novel compounds |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997021676A1 true WO1997021676A1 (en) | 1997-06-19 |
Family
ID=27268020
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1996/005588 WO1997021676A1 (en) | 1995-12-08 | 1996-12-04 | Azetidinone compounds for the treatment of atherosclerosis |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP0865429A1 (en) |
| JP (1) | JP2000505063A (en) |
| WO (1) | WO1997021676A1 (en) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997041098A1 (en) * | 1996-04-26 | 1997-11-06 | Smithkline Beecham Plc | Azetidinone derivatives for the treatment of atherosclerosis |
| US6414179B1 (en) | 2000-02-18 | 2002-07-02 | Bristol-Myers Squibb Company | Alpha-and beta-substituted trifluoromethyl ketones as phospholipase inhibitors |
| WO2002096415A2 (en) * | 2001-05-25 | 2002-12-05 | Schering Corporation | Use of azetidinone substituted derivatives in the treatment of alzheimer's disease |
| US6492550B2 (en) | 2000-02-18 | 2002-12-10 | Bristol-Myers Squibb Company | Alpha-substituted thio, -oxo trifluoromethylketones as phospholipase inhibitors |
| DE10134478A1 (en) * | 2001-07-16 | 2003-02-06 | Morphochem Ag | New N-(thizolylmethyl)-azetidinone derivatives, useful as medicaments, especially antibiotics, or plant protectants |
| US6627757B2 (en) | 2001-03-28 | 2003-09-30 | Schering Corporation | Enantioselective synthesis of azetidinone intermediate compounds |
| US6924391B2 (en) | 2000-05-11 | 2005-08-02 | Bristol-Myers Squibb Company | Alpha-amino,-thio,-oxo substituted ketones as phospholipase inhibitors |
| EP1686119A1 (en) | 2000-02-16 | 2006-08-02 | Smithkline Beecham Plc | Pyrimidine-4-one derivatives as ldl-pla2 inhibitors |
| WO2008140449A1 (en) | 2007-05-11 | 2008-11-20 | Thomas Jefferson University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| WO2012076435A1 (en) | 2010-12-06 | 2012-06-14 | Glaxo Group Limited | Pyrimidinone compounds for use in the treatment of diseases or conditions mediated by lp - pla2 |
| WO2012080497A2 (en) | 2010-12-17 | 2012-06-21 | Glaxo Group Limited | Methods of treatment and prevention of eye diseases |
| WO2013014185A1 (en) | 2011-07-27 | 2013-01-31 | Glaxo Group Limited | Bicyclic pyrimidone compounds |
| WO2013013503A1 (en) | 2011-07-27 | 2013-01-31 | Glaxo Group Limited | 2,3-dihydroimidazo[1,2-c] pyrimidin-5(1h)-one compounds use as lp-pla2 inhibitors |
| WO2014114248A1 (en) | 2013-01-25 | 2014-07-31 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| WO2014114694A1 (en) | 2013-01-25 | 2014-07-31 | Glaxosmithkline Intellectual Property Development Limited | 2,3-dihydroimidazol[1,2-c]pyrimidin-5(1h)-one based lipoprotein-associated phospholipase a2 (lp-pla2) inhibitors |
| WO2014114249A1 (en) | 2013-01-25 | 2014-07-31 | Glaxosmithkline Intellectual Property Development Limited | Bicyclic pyrimidone compounds as inhibitors of lp-pla2 |
| US8962633B2 (en) | 2007-05-11 | 2015-02-24 | Thomas Jefferson University | Methods of treatment and prevention of metabolic bone diseases and disorders |
| US9029383B2 (en) | 2007-05-11 | 2015-05-12 | The Trustees Of The University Of Pennsylvania | Methods of treatment of skin ulcers |
| WO2016012917A1 (en) | 2014-07-22 | 2016-01-28 | Glaxosmithkline Intellectual Property Development Limited | 1,2,3,5-tetrahydroimidazo[1,2-c]pyrimidine derivatives useful in the treatment of diseases and disorders mediated by lp-pla2 |
| WO2016012916A1 (en) | 2014-07-22 | 2016-01-28 | Glaxosmithkline Intellectual Property Development Limited | 1,2,3,5-tetrahydroimidazo[1,2-c]pyrimidine derivatives useful in the treatment of diseases and disorders mediated by lp-pla2 |
| AU2014200542B2 (en) * | 2007-05-11 | 2016-08-25 | Rowan University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| WO2021089032A1 (en) | 2019-11-09 | 2021-05-14 | 上海赛默罗生物科技有限公司 | Tricyclic dihydroimidazopyrimidone derivative, preparation method therefor, pharmaceutical composition and use thereof |
| WO2022233302A1 (en) | 2021-05-07 | 2022-11-10 | 上海赛默罗生物科技有限公司 | Pyrimidinone derivative and preparation method therefor, pharmaceutical composition, and use |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10348022A1 (en) * | 2003-10-15 | 2005-05-25 | Imtm Gmbh | New dipeptidyl peptidase IV inhibitors for the functional influence of different cells and for the treatment of immunological, inflammatory, neuronal and other diseases |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0199630A1 (en) * | 1985-04-10 | 1986-10-29 | Merck & Co. Inc. | Substituted azetidinones, pharmaceutical compositions containing them, and their use for the manufacture of anti-inflammatory and antidegenerative medicaments |
| GB2266527A (en) * | 1992-03-17 | 1993-11-03 | Merck & Co Inc | Substituted azetidinones useful in the treatment of leukemia |
| WO1995002579A1 (en) * | 1993-07-12 | 1995-01-26 | Zeneca Limited | Substituted azetidinone amide derivatives as anti-inflammatory and antidegenerative agents |
| WO1996013484A1 (en) * | 1994-10-29 | 1996-05-09 | Smithkline Beecham Plc | Azetidinone derivatives for the treatment of atherosclerosis |
| WO1996019451A1 (en) * | 1994-12-22 | 1996-06-27 | Smithkline Beecham Plc | Substituted azetidin-2-ones for treatment of atherosclerosis |
-
1996
- 1996-12-04 EP EP96942375A patent/EP0865429A1/en not_active Withdrawn
- 1996-12-04 JP JP9521747A patent/JP2000505063A/en active Pending
- 1996-12-04 WO PCT/EP1996/005588 patent/WO1997021676A1/en not_active Application Discontinuation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0199630A1 (en) * | 1985-04-10 | 1986-10-29 | Merck & Co. Inc. | Substituted azetidinones, pharmaceutical compositions containing them, and their use for the manufacture of anti-inflammatory and antidegenerative medicaments |
| GB2266527A (en) * | 1992-03-17 | 1993-11-03 | Merck & Co Inc | Substituted azetidinones useful in the treatment of leukemia |
| WO1995002579A1 (en) * | 1993-07-12 | 1995-01-26 | Zeneca Limited | Substituted azetidinone amide derivatives as anti-inflammatory and antidegenerative agents |
| WO1996013484A1 (en) * | 1994-10-29 | 1996-05-09 | Smithkline Beecham Plc | Azetidinone derivatives for the treatment of atherosclerosis |
| WO1996019451A1 (en) * | 1994-12-22 | 1996-06-27 | Smithkline Beecham Plc | Substituted azetidin-2-ones for treatment of atherosclerosis |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997041098A1 (en) * | 1996-04-26 | 1997-11-06 | Smithkline Beecham Plc | Azetidinone derivatives for the treatment of atherosclerosis |
| US9266841B2 (en) | 2000-02-16 | 2016-02-23 | Glaxo Group Limited | Compounds |
| BG66014B1 (en) * | 2000-02-16 | 2010-10-29 | Smithkline Beecham P.L.C. | Pyrimidine-4-one derivatives as ldl-pla2 inhibitors |
| US8871775B2 (en) | 2000-02-16 | 2014-10-28 | Glaxo Group Limited | Compounds |
| EP1686119A1 (en) | 2000-02-16 | 2006-08-02 | Smithkline Beecham Plc | Pyrimidine-4-one derivatives as ldl-pla2 inhibitors |
| US6414179B1 (en) | 2000-02-18 | 2002-07-02 | Bristol-Myers Squibb Company | Alpha-and beta-substituted trifluoromethyl ketones as phospholipase inhibitors |
| US6492550B2 (en) | 2000-02-18 | 2002-12-10 | Bristol-Myers Squibb Company | Alpha-substituted thio, -oxo trifluoromethylketones as phospholipase inhibitors |
| US6924391B2 (en) | 2000-05-11 | 2005-08-02 | Bristol-Myers Squibb Company | Alpha-amino,-thio,-oxo substituted ketones as phospholipase inhibitors |
| US6627757B2 (en) | 2001-03-28 | 2003-09-30 | Schering Corporation | Enantioselective synthesis of azetidinone intermediate compounds |
| WO2002096415A3 (en) * | 2001-05-25 | 2003-01-16 | Schering Corp | Use of azetidinone substituted derivatives in the treatment of alzheimer's disease |
| WO2002096415A2 (en) * | 2001-05-25 | 2002-12-05 | Schering Corporation | Use of azetidinone substituted derivatives in the treatment of alzheimer's disease |
| DE10134478B4 (en) * | 2001-07-16 | 2007-10-31 | Priaton Gmbh | Process for the preparation of thiazole-substituted β-lactams |
| US6900312B2 (en) * | 2001-07-16 | 2005-05-31 | Priaton Gmbh | Thiazole-substituted β-lactams |
| DE10134478A1 (en) * | 2001-07-16 | 2003-02-06 | Morphochem Ag | New N-(thizolylmethyl)-azetidinone derivatives, useful as medicaments, especially antibiotics, or plant protectants |
| AU2014200542B2 (en) * | 2007-05-11 | 2016-08-25 | Rowan University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| EP2152865A4 (en) * | 2007-05-11 | 2010-11-03 | Univ Jefferson | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| EP2152865A1 (en) * | 2007-05-11 | 2010-02-17 | Thomas Jefferson University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| WO2008140449A1 (en) | 2007-05-11 | 2008-11-20 | Thomas Jefferson University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| AU2007353454B2 (en) * | 2007-05-11 | 2013-10-31 | Rowan University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| AU2007353454C1 (en) * | 2007-05-11 | 2014-03-27 | Rowan University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| AU2007353454C8 (en) * | 2007-05-11 | 2014-04-24 | Rowan University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| EP2977452A2 (en) | 2007-05-11 | 2016-01-27 | Thomas Jefferson University | Methods of treatment and prevention of neurodegenerative diseases and disorders |
| US9029383B2 (en) | 2007-05-11 | 2015-05-12 | The Trustees Of The University Of Pennsylvania | Methods of treatment of skin ulcers |
| US8962633B2 (en) | 2007-05-11 | 2015-02-24 | Thomas Jefferson University | Methods of treatment and prevention of metabolic bone diseases and disorders |
| WO2012076435A1 (en) | 2010-12-06 | 2012-06-14 | Glaxo Group Limited | Pyrimidinone compounds for use in the treatment of diseases or conditions mediated by lp - pla2 |
| WO2012080497A2 (en) | 2010-12-17 | 2012-06-21 | Glaxo Group Limited | Methods of treatment and prevention of eye diseases |
| WO2013013503A1 (en) | 2011-07-27 | 2013-01-31 | Glaxo Group Limited | 2,3-dihydroimidazo[1,2-c] pyrimidin-5(1h)-one compounds use as lp-pla2 inhibitors |
| WO2013014185A1 (en) | 2011-07-27 | 2013-01-31 | Glaxo Group Limited | Bicyclic pyrimidone compounds |
| WO2014114249A1 (en) | 2013-01-25 | 2014-07-31 | Glaxosmithkline Intellectual Property Development Limited | Bicyclic pyrimidone compounds as inhibitors of lp-pla2 |
| WO2014114694A1 (en) | 2013-01-25 | 2014-07-31 | Glaxosmithkline Intellectual Property Development Limited | 2,3-dihydroimidazol[1,2-c]pyrimidin-5(1h)-one based lipoprotein-associated phospholipase a2 (lp-pla2) inhibitors |
| WO2014114248A1 (en) | 2013-01-25 | 2014-07-31 | Glaxosmithkline Intellectual Property Development Limited | Compounds |
| WO2016012917A1 (en) | 2014-07-22 | 2016-01-28 | Glaxosmithkline Intellectual Property Development Limited | 1,2,3,5-tetrahydroimidazo[1,2-c]pyrimidine derivatives useful in the treatment of diseases and disorders mediated by lp-pla2 |
| WO2016012916A1 (en) | 2014-07-22 | 2016-01-28 | Glaxosmithkline Intellectual Property Development Limited | 1,2,3,5-tetrahydroimidazo[1,2-c]pyrimidine derivatives useful in the treatment of diseases and disorders mediated by lp-pla2 |
| WO2021089032A1 (en) | 2019-11-09 | 2021-05-14 | 上海赛默罗生物科技有限公司 | Tricyclic dihydroimidazopyrimidone derivative, preparation method therefor, pharmaceutical composition and use thereof |
| WO2022233302A1 (en) | 2021-05-07 | 2022-11-10 | 上海赛默罗生物科技有限公司 | Pyrimidinone derivative and preparation method therefor, pharmaceutical composition, and use |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0865429A1 (en) | 1998-09-23 |
| JP2000505063A (en) | 2000-04-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1997021676A1 (en) | Azetidinone compounds for the treatment of atherosclerosis | |
| CN104603118B (en) | It is used as the thiazole and related derivatives through formamide or sulfonamide substitutions of orphan nuclear receptor ROR γ instrumentalities | |
| US6875867B2 (en) | Process for preparing chiral diol sulfones and dihydroxy acid HMG CoA reductase inhibitors | |
| US11180488B2 (en) | Isoxazole o-linked carbamoyl cyclohexyl acids as LPA antagonists | |
| US6071899A (en) | Azetidinone derivatives for the treatment of atherosclerosis | |
| JP2002543190A (en) | Pyrimidinone compounds | |
| KR102247765B1 (en) | Bace1 inhibitors | |
| JP2004511473A (en) | Pyrimidinone derivatives and their use in the treatment of atherosclerosis | |
| CN102627609A (en) | Heterocyclic aspartyl protease inhibitors | |
| JPH11500415A (en) | Substituted azetidin-2-ones for treating atherosclerosis | |
| JPH05132458A (en) | Novel substituted azetidinones as antiphlo- gistic and antidenaturant | |
| TWI690515B (en) | Dihydropyrimidin-2-one compound and pharmaceutical use thereof | |
| WO1997021675A1 (en) | Monocyclic beta-lactame derivatives for treatment of atherosclerosis | |
| JP2018538295A (en) | New processes and intermediates | |
| EP1858898A1 (en) | Hydrazinocarbonyl-thieno[2,3-c]pyrazoles, preparation method, compositions containing same and use | |
| JPH0196181A (en) | 2-oxo-1-(((substituted sulfonyl)amino)carbonyl) azetidines | |
| EP0900199A1 (en) | Azetidinone derivatives for the treatment of atherosclerosis | |
| EP0915843A1 (en) | Azetidinone derivatives for the treatment of atheroscleroses | |
| JP2003524628A (en) | Pyrimidinone derivatives for the treatment of atherosclerosis | |
| WO2005009392A2 (en) | Dihydropyrimidone inhibitors of calcium channel function | |
| JP2023523770A (en) | Imidazopyrimidines as modulators of IL-17 | |
| WO2003015786A1 (en) | 2, 5-substituted 1-(aminocarbonylalkyl) -pyrimidin-4-one derivatives with lp-pla2 inhinitory activity for the treatment of atherosclerosis | |
| Cremonesi et al. | Asymmetric synthesis of 1, 3-thiazolidine-derived spiro-β-lactams via a Staudinger reaction between chiral ketenes and imines | |
| JP2023513121A (en) | ADAMTS inhibitors, their manufacture and pharmaceutical use | |
| CZ20004587A3 (en) | Tetrahydroquinoline derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): JP US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1996942375 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996942375 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1996942375 Country of ref document: EP |