WO1996017633A1 - Supplemented and unsupplemented tissue sealants, methods of their production and use - Google Patents
Supplemented and unsupplemented tissue sealants, methods of their production and use Download PDFInfo
- Publication number
- WO1996017633A1 WO1996017633A1 PCT/US1995/015876 US9515876W WO9617633A1 WO 1996017633 A1 WO1996017633 A1 WO 1996017633A1 US 9515876 W US9515876 W US 9515876W WO 9617633 A1 WO9617633 A1 WO 9617633A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fibrin
- fibrin sealant
- dressing
- wound
- growth factor
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/36—Blood coagulation or fibrinolysis factors
- A61K38/363—Fibrinogen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L15/00—Chemical aspects of, or use of materials for, bandages, dressings or absorbent pads
- A61L15/16—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons
- A61L15/22—Bandages, dressings or absorbent pads for physiological fluids such as urine or blood, e.g. sanitary towels, tampons containing macromolecular materials
- A61L15/32—Proteins, polypeptides; Degradation products or derivatives thereof, e.g. albumin, collagen, fibrin, gelatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L24/00—Surgical adhesives or cements; Adhesives for colostomy devices
- A61L24/001—Use of materials characterised by their function or physical properties
- A61L24/0015—Medicaments; Biocides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L24/00—Surgical adhesives or cements; Adhesives for colostomy devices
- A61L24/001—Use of materials characterised by their function or physical properties
- A61L24/0036—Porous materials, e.g. foams or sponges
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L24/00—Surgical adhesives or cements; Adhesives for colostomy devices
- A61L24/04—Surgical adhesives or cements; Adhesives for colostomy devices containing macromolecular materials
- A61L24/10—Polypeptides; Proteins
- A61L24/106—Fibrin; Fibrinogen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/40—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices characterised by a specific therapeutic activity or mode of action
- A61L2300/404—Biocides, antimicrobial agents, antiseptic agents
- A61L2300/406—Antibiotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/40—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices characterised by a specific therapeutic activity or mode of action
- A61L2300/412—Tissue-regenerating or healing or proliferative agents
- A61L2300/414—Growth factors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/40—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices characterised by a specific therapeutic activity or mode of action
- A61L2300/416—Anti-neoplastic or anti-proliferative or anti-restenosis or anti-angiogenic agents, e.g. paclitaxel, sirolimus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/40—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices characterised by a specific therapeutic activity or mode of action
- A61L2300/418—Agents promoting blood coagulation, blood-clotting agents, embolising agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/60—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices characterised by a special physical form
- A61L2300/602—Type of release, e.g. controlled, sustained, slow
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L2300/00—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices
- A61L2300/60—Biologically active materials used in bandages, wound dressings, absorbent pads or medical devices characterised by a special physical form
- A61L2300/606—Coatings
- A61L2300/608—Coatings having two or more layers
Definitions
- This invention is directed to unsupplemented and supplemented Tissue Sealants (TS), such as fibrin glue (FG), as well as to methods of their production and use.
- TS Tissue Sealants
- this invention is directed to TSs which do not inhibit full-thickness skin wound healing.
- this invention is directed to TSs which have been supplemented with a growth factor(s) and/or a drug(s), as well as to methods of their production and use.
- the particular growth factor(s) or drug(s) that is selected is a function of its use.
- Wound healing the repair of lesions, begins almost instantly after injury. It requires the successive coordinated function of a variety of cells and the close regulation of degradative and regenerative steps.
- the proliferation, differentiation and migration of cells are important biological processes which underlie wound healing, which also involves fibrin clot formation, resorption of the clot, tissue remodeling, such as fibrosis, endothelialization and epithelialization.
- Wound healing involves the formation of highly vascularized tissue that contains numerous capillaries, many active fibroblasts, and abundant collagen fibrils, but not the formation of specialized skin structures.
- thromboplastin which flows out of injured cells.
- Thromboplastin contacts plasma factor VII to form factor X activator, which then, with factor V and in a complex with phospholipids and calcium, converts prothrombin into thrombin.
- Thrombin catalyzes the release of fibrinopeptides A and B from fibrinogen to produce fibrin monomers, which aggregate to form fibrin filaments.
- Thrombin also activates the transglutaminase, factor XIIIa, which catalyzes the formation of isopeptide bonds to covalently cross-link the fibrin filaments.
- Alpha 2 -antiplasmin is then bound by factor XIII onto the fibrin filaments to thereby protect the filaments from degradation by plasmin (see, for example, Doolittle et al., Ann. Rev. Biochem. 53:195-229
- polypeptide growth factors which exhibit an array of biological activities, are released into the wound where they play a crucial role in healing (see, e.g., Hormonal Proteins and Peptides (Li, C. H., ed.) Volume 7, Academic Press, Inc., New York, N. Y. pp. 231-277 ( 1979) and Brunt et al., Biotechnology 6:25-30 (1988)).
- These activities include recruiting cells, such as leukocytes and fibroblasts, into the injured area, and inducing cell proliferation and differentiation.
- Growth factors that may participate in wound healing include, but are not limited to: platelet-derived growth factors (PDGFs); insulin-binding growth factor- 1 (IGF-1); insulin-binding growth factor-2 (IGF-2); epidermal growth factor (EGF); transforming growth factor- ⁇ (TGF- ⁇ ); transforming growth factor- ⁇ (TGF- ⁇ ); platelet factor 4 (PF-4); and heparin binding growth factors one and two (HBGF-1 and HBGF-2, respectively).
- PDGFs platelet-derived growth factors
- IGF-1 insulin-binding growth factor- 1
- IGF-2 insulin-binding growth factor-2
- EGF epidermal growth factor
- TGF- ⁇ transforming growth factor- ⁇
- TGF- ⁇ transforming growth factor- ⁇
- platelet factor 4 PF-4
- heparin binding growth factors one and two HBGF-1 and HBGF-2, respectively.
- PDGFs are stored in the alpha granules of circulating platelets and are released at wound sites during blood clotting (see, e.g., Lynch et al., J. Clin.
- PDGFs include: PDGF; platelet derived angiogenesis factor (PDAF); TGF- ⁇ ; and PF-4, which is a chemoattractant for neutrophils (Knighton et al., in Growth Factors and Other Aspects of Wound Healing: Biological and Clinical Implications, Alan R. Liss, Inc., New York, New York, pp. 319-329 (1988)).
- PDGF is a mitogen. chemoattractant and a stimulator of protein synthesis in cells of mesenchymal origin, including fibroblasts and smooth muscle cells.
- PDGF is also a nonmitogenic chemoattractant for endothelial cells (see, for example, Adelmann-Grill et al. Eur. J. Cell Biol. 51:322-326 (1990)).
- IGF-1 acts in combination with PDGF to promote mitogenesis and protein synthesis in mesenchymal cells in culture.
- Application of either PDGF or IGF-1 alone to skin wounds does not enhance healing, but application of both factors together appears to promote connective tissue and epithelial tissue growth (Lynch et al., Proc. Natl. Acad. Sci. 76:1279-1283 (1987)) .
- TGF- ⁇ is a chemoattractant for macrophages and monocytes. Depending upon the presence or absence of other growth factors, TGF- ⁇ may stimulate or inhibit the growth of many cell types. For example, when applied in vivo, TGF- ⁇ increases the tensile strength of healing dermal wounds. TGF- ⁇ also inhibits endothelial cell mitosis, and stimulates collagen and glycosaminoglycan synthesis by fibroblasts.
- EGF EGF
- TGF- ⁇ TGF- ⁇
- HBGFs HBGFs
- osteogenin Other growth factors, such as EGF, TGF- ⁇ , the HBGFs and osteogenin are also important in wound healing.
- EGF which is found in gastric secretions and saliva
- TGF- ⁇ which is made by both normal and transformed cells, are structurally related and may recognize the same receptors. These receptors mediate proliferation of epithelial cells. Both factors accelerate reepithelialization of skin wounds.
- Exogenous EGF promotes wound healing by stimulating the proliferation of keratinocytes and dermal fibroblasts (Nanney et al., J. Invest. Dermatol 5 - 3:385-393 (1984) and Coffey et al., Nature 325:817-820 (1987)).
- HBGFs also known as Fibroblast Growth Factors (FGFs), which include acidic HBGF (aHBGF also known as HBFG-1 or FGF-1) and basic HBGF (bHBGF also known as HBGF-2 or FGF-2), are potent mitogens for cells of mesodermal and neuroectodermal lineages, including endothelial cells (see, e.g., Burgess et al., Ann. Rev. Biochem. 58:575-606 (1989)).
- HBGF-1 is chemotactic for endothelial cells and astroglial cells. Both HBGF-1 and HBGF-2 bind to heparin, which protects them from proteolytic degradation.
- the array of biological activities exhibited by the HBGFs suggests that they play an important role in wound healing.
- Basic fibroblast growth factor (FGF-2) is a potent stimulator of angiogenesis and the migration and proliferation of fibroblasts (see, for example, Gospodarowicz et al., Mol Cell Endocinol 46:187-204 (1986) and Gospodarowicz et al., Endo. Rev. 5:95-114 (1985)).
- Acidic fibroblast growth factor (FGF-1) has been shown to be a potent angiogenic factor for endothelial cells (Burgess et al., supra, 1989). However, it has not been established if any FGF growth factor is chemotactic for fibroblasts.
- Growth factors are, therefore, potentially useful for specifically promoting wound healing and tissue repair.
- their use to promote wound healing has yielded inconsistent results (see, e.g., Carter et al., in Growth Factors and
- TGF- ⁇ stimulated the greatest response alone.
- PDGF-bb homodimer and IGF-1 or TGF- ⁇ produced a dramatic increase in connective tissue regeneration and epitheliaiization.
- Tsuboi et al. have reported that the daily application of bFGF to an open wound stimulated wound healing in healing-impaired mice but not in normal mice (J. Exp. Med. 172:245-251 (1990)).
- Surgical adhesives and TSs which contain plasma proteins are used for sealing internal and external wounds, such as in bones and skin, to reduce blood loss and maintain hemostasis.
- Such TSs contain blood clotting factors and other blood proteins.
- FG also called fibrin sealant, is a gel similar to a natural clot which is prepared from plasma. The precise components of each FG are a function of the particular plasma fraction which is used as a starting material. Fractionation of plasma components can be effected by standard protein purification methods, such as ethanol, polyethylene glycol, and ammonium sulfate precipitation, ion exchange, and gel filtration chromatography.
- standard protein purification methods such as ethanol, polyethylene glycol, and ammonium sulfate precipitation, ion exchange, and gel filtration chromatography.
- FG contains a mixture of proteins including traces of albumin, fibronectin and plasminogen. In Canada, Europe and possibly elsewhere, commercially available FG typically also contains aprotinin as a stabilizer.
- FGs generally are prepared from: (1) a fibrinogen concentrate, which contains fibronectin, Factor XIII, and von Willebrand factor; (2) dried human or bovine thrombin; and (3) calcium ions.
- Commercially prepared FGs generally contain bovine components.
- the fibrinogen concentrate can be prepared from plasma by cryoprecipitation followed by fractionation, to yield a composition that forms a sealant or clot upon mixture with thrombin and an activator of thrombin such as calcium ions.
- the fibrinogen and thrombin concentrates are prepared in lyophilized form and are mixed with a solution of calcium chloride immediately prior to use. Upon mixing, the components are applied to a tissue where they coagulate on the tissue surface and form a clot that includes cross-linked fibrin.
- Factor XIII which is present in the fibrinogen concentrate, catalyzes the cross-linking.
- Australian Patent 75097/87 describes a one-component adhesive, which contains an aqueous solution of fibrinogen, factor XIII, a thrombin inhibitor, such as antithrombin III, prothrombin factors, calcium ions, and, if necessary, a plasmin inhibitor.
- Stroetmann U.S. Patent Nos. 4,427,650 and 4,427,651, describes the preparation of an enriched plasma derivative in the form of a powder or sprayable preparation for enhanced wound closure and healing that contains fibrinogen, thrombin and/or prothrombin, and a fibrinolysis inhibitor, and may also contain other ingredients, such as a platelet extract.
- the TSs available in Europe contain proteins of non-human origin such as aprotinin and bovine thrombin. Since these proteins are of non-human origin, people may develop allergic reactions to them.
- heat inactivation is used to inactivate viruses which may be present in the components of the FG. However, this heat inactivation method may produce denatured proteins in the FG which may also be allergenic.
- the ARC/BH FG has advantages over the TSs available in Europe because it does not contain bovine proteins.
- the ARC/BH TS contains human thrombin instead of bovine thrombin and does not contain aprotinin. Since the ARC/BH FG does not contain bovine proteins it should be less allergenic in humans than those TSs available in Europe.
- the ARC/BH FG does not contain bovine proteins it should be less allergenic in humans than those TSs available in Europe.
- ARC/BH FG is virally inactivated by a solvent detergent method which produces fewer denatured proteins and thus is less allergenic than those available in Europe. Therefore, the ARC/BH FG possesses advantages over the TSs which are now commercially available in other countries.
- FG is primarily formulated for clinical topical application and is used to control bleeding, maintain hemostasis and promote wound healing.
- the clinical uses of FG have recently been reviewed (Gibble et al., Transfusion 30:741-747 (1990); Lemer et al., J. Surg. Res. 48: 165-181 (1990)).
- By sealing tissues FG prevents air or fluid leaks, induces hemostasis, and may contribute to wound healing indirectly by reducing or preventing events which may interfere with wound healing such as bleeding, hematomas, infections, etc.
- FG maintains hemostasis and reduces blood loss, it has not yet been shown to possess true wound healing properties. Because FG is suitable for both internal and external injuries, such as bone and skin injuries, and is useful to maintain hemostasis, it is desirable to enhance its wound healing properties.
- FG with a fibrinogen concentration of approximately 39 g/l and a thrombin concentration of 200-600 U/ml has produced clots with significantly increased stress, energy absorption and elasticity values (Byrne et al., Br. J. Surg. 78:841-843 (1991)).
- Demineralized bone matrix is a source of osteoinductive proteins known as bone morphogenetic proteins (BMP), and growth factors which modulate the proliferation of progenitor bone cells (see, e.g. , Hauschka et al.. J.
- BMP-1 BMP-1 through BMP-8.
- BMP-3 and BMP-7 are also known as osteogenin and osteogenic protein-1 (OP-1), respectively.
- DBM materials have little clinical use unless combined with paniculate marrow autografts. There is a limit to the quantity of DBM that can be surgically placed into a recipient's bone to produce a therapeutic effect.
- DBM powder and osteogenin may be washed away by tissue fluids before their osteoinductive potential is expressed.
- seepage of tissue fluids into DBM-packed bone cavities or soft-tissue collapse into the wound bed are two factors that may significantly affect the osteoinductive properties of DBM and osteogenin.
- Soft-tissue collapse into the wound bed may likewise inhibit the proper migration of osteocompetent stem cells into the wound bed.
- Purified BMPs have osteoinductive effects in animals when delivered by a variety of means including FG (Hattori, T., Nippon. Seikeigeka. Gakkai. Zasshi. 64:824-834 (1990); Kawamura et al., Clin. Orthop. Rel Res. 235:302-310 (1988); Schlag et al., Clin. Orthop. Rel. Res. 227:269-285 (1988) and Schwarz etal., Clin. Orthop. Rel. Res. 238:282-287 (1989)) and whole blood clots (Wang et al., J. Cell. Biochem. 15F.Q20 Abstract (1990)).
- FG Heattori, T., Nippon. Seikeigeka. Gakkai. Zasshi. 64:824-834 (1990)
- Kawamura et al. Clin. Orthop. Rel Res. 235:302-310 (1988)
- Schlag et al. Clin. Orthop. Re
- TS also can serve as a "scaffold" which cells can use to move into a wounded area to generate new tissues.
- commercially available preparations of FG and other TSs are too dense to allow cell migration into and through them. This limits their effectiveness in some in vivo uses.
- bone nonunion defects one type of bone wound, called bone nonunion defects, there is a minimal gap above which no new bone formation occurs naturally.
- the treatment for these situations is bone grafting.
- the source of bone autografts is usually limited and the use of allogeneic bones involves a high risk of viral contamination. Because of this situation, the use of demineralized, virally inactivated bone powder is an attractive solution.
- vascular prostheses are frequently made out of polytetrafluoroethylene (PTFE) and are used to replace diseased blood vessels in humans and other animals.
- PTFE polytetrafluoroethylene
- various techniques have been used including seeding of nonautologous endothelial cells onto the prothesis.
- Various substrates which adhere both to the vascular graft and endothelial cells have been investigated as an intermediate substrate to increase endothelial cell seeding. These substrates include preclotted blood (Herring et al., Surgery 84:498-504
- Angiogenesis is the induction of new blood vessels. Certain growth factors such as HBGF-1 and HBGF-2 are angiogenic. However, their in vivo administration attached to: collagen sponges (Thompson et al., Science 241 : 1349-1352 (1988)); beads (Hayek et al., Biochem. Biophys. Res. Commun. 747:876-880
- fibrin gels (0.5-10 mg/ml) implanted subcutaneously in plexiglass chambers induce angiogenesis within 4 days of implantation, compared to empty chambers, or chambers filled with sterile culture medium (Dvorak et al., Lab. Invest. 57:673 (1987)).
- An efficacious, site-directed, drug delivery system is greatly needed in several areas of medicine.
- localized drug delivery is needed in the treatment of local infections, such as in periodontitis, where the systemic administration of antimicrobial agents is ineffective.
- the problem after systemic administration usually lies in the low concentration of the antimicrobial agent which can be achieved at the target site.
- a systemic dose increase may be effective but also may produce toxicity, microbial resistance and drug incompatibility.
- several alternative methods have been devised but none are ideal.
- collagen and/or fibrinogen dispersed in an aqueous medium as an amorphous flowable mass have also been shown to locally deliver drugs (Luck et al., U.S. Reissue Patent 33,375; Luck et al., U.S. Patent 4,978,332).
- the disclosed technology would also be available for the treatment of massed casualties in disaster situation.
- severe natural or man-made disasters occur, local hospitals and clinics may be overwhelmed by the number of individuals requiring trauma care. Combined with the isolating effects of such disasters, the resulting demand for blood and blood products often exceeds the locally available supplies. In many cases, the demand upon the local medical personnel also exceeds the availed number of trained individuals. As a result, less seriously injured persons may be tumed-away or given sub-optimal care.
- the disclosed TS preparations will permit self-treatment in disaster victims, until medical assistance can be provided.
- this invention provides a composition of matter, comprising a TS, wherein the sealant does not inhibit full-thickness skin wound healing.
- this invention provides a composition of matter, comprising: a TS, wherein the total protein concentration of the sealant is less than 30 mg/ml.
- this invention provides a composition of matter comprising a supplemented TS wherein the total protein concentration is less than 30 mg/ml and the supplement is a growth factor(s) and/or a drug(s).
- this invention provides a composition of matter comprising a supplemented TS wherein the total protein concentration is greater than 30 mg/ml and the supplement is a growth factor(s) and/or a drug(s).
- this invention provides a composition of matter that promotes the directed migration of animal cells, comprising: a TS; and an effective concentration of at least one growth factor, wherein the concentration of the growth factor is effective in promoting the directed migration of the animal cells.
- the present invention provides a composition of matter that promotes wound healing, comprising: a TS; and an effective concentration of at least one growth factor, wherein the concentration is effective in promoting wound healing.
- the present invention provides a composition of matter that promotes the endothelialization of a vascular prosthesis, comprising: a TS; and an effective concentration of at least one growth factor, wherein the concentration is effective in promoting the endothelialization of a vascular prosthesis.
- the present invention provides a composition of matter that promotes the proliferation and/or differentiation of animal cells, comprising: a TS; and an effective concentration of at least one growth factor, wherein the concentration is effective in promoting proliferation and/or differentiation of animal cells.
- the present invention provides a composition of matter that promotes the localized delivery of at least one drug, comprising: a TS; and at least one drug.
- the present invention provides a composition of matter that promotes the localized delivery of at least one growth factor, comprising: a TS; and at least one growth factor.
- the present invention provides a process for promoting the healing of wounds, comprising applying to the wound, a composition that contains a TS and an effective concentration of at least one growth factor, wherein the concentration is effective to promote wound healing.
- the present invention provides a process for promoting the endothelialization of a vascular prosthesis, comprising applying to the vascular prosthesis a composition that contains a TS and an effective concentration of at least one growth factor, wherein the concentration is effective to promote the endothelialization of a vascular prothesis.
- the present invention provides a process for promoting the proliferation and/or differentiation of animal cells, comprising placing the cells in sufficient proximity to a TS which contains an effective concentration of at least one growth factor, wherein the concentration is effective in promoting the proliferation and/or differentiation of the cells.
- the present invention provides a process for the localized delivery of at least one drug to a tissue, comprising applying to the tissue a TS which contains at least one drug.
- the present invention provides a process for the localized delivery of at least one growth factor to a tissue, comprising applying to the tissue a TS which contains at least one growth factor.
- this invention provides a process for producing the directed migration of animal cells, comprising: placing in sufficient proximity to the cells, a TS which contains an effective concentration of at least one growth factor, wherein the concentration is effective to produce the desired directed migration of said cells.
- this invention provides a simple to use, fast acting, field-ready fibrin bandage for applying a tissue sealing composition to wounded tissue in a patient, comprising an occlusive backing, affixed to which is a layer of dry materials comprising an effective amount, in combination, of (a) dry, virally-inactivated, purified fibrinogen, (b) dry, virally-inactivated, purified thrombin, and as necessary (c) effective amounts of calcium and/or Factor XIII to produce a tissue-sealing fibrin clot upon hydration.
- this invention provides a method of treating wounded tissue in a patient by applying to said wound a fibrin bandage, comprising: (1) a occlusive backing, affixed to which is a layer of dry materials comprising an effective amount, in combination, of (a) dry, virally-inactivated, purified fibrinogen, (b) dry, virally-inactivated, purified thrombin, and as necessary (c) effective amounts of calcium and/or Factor XIII to produce a tissue-sealing fibrin clot upon hydration.
- this invention provides a simple to use, fast acting, field-ready fibrin dressing for treating wounded tissue in a patient, is formulated as an expandable foam comprising an effective amount, in combination, of (1) virally-inactivated, purified fibrinogen, (2) virally-inactivated, purified thrombin, and as necessary (3) calcium and/or Factor XIII; wherein said composition does not significantly inhibit full-thickness skin wound healing.
- this invention provides a method of treating wounded tissue in a patient by applying to said wound a tissue sealant expandable foam dressing, comprising an effective amount, in combination, of (1) virally-inactivated, purified fibrinogen, (2) virally-inactivated, purified thrombin, and as necessary (3) calcium and/or Factor XIII; wherein said composition does not significantly inhibit full-thickness skin wound healing.
- the TS may be FG.
- FG may be made from the mixing of topical fibrinogen complex (TFC), human thrombin and calcium chloride. Varying the concentration of the TFC has the most significant effect upon the density of the final FG matrix. Varying the concentration of the thrombin has an insignificant effect upon the total protein concentration of the final FG, but has a profound effect upon the time required for the polymerization of the fibrinogen component of the TFC into fibrin. While this effect is well known, it is not generally appreciated that it may be used to maximize the effectiveness of the FG, when it is used alone or supplemented. Because of this effect one can alter the time between the mixing of the FG components and the setting of the FG.
- TFC topical fibrinogen complex
- human thrombin and calcium chloride.
- TFC is a lyophilized mixture of human plasma proteins which have been purified and virally inactivated. When reconstituted TFC contains: Total Protein: 100-130 mg/ml
- Fibrinogen (as clottable protein) 80% of total protein
- Polysorbate-80 0.3% (maximum) pH: 7.1-7.5.
- the reconstituted TFC may also contain trace amounts of fibronectin.
- human thrombin is a lyophilized mixture of human plasma proteins, which have been purified and virally inactivated. When reconstituted it contains:
- Thrombin Potency 300 ⁇ 50 International Units/ml Albumin (Human): 5 mg/ml
- Calcium chloride is added in sufficient concentration to activate the thrombin. As long as there is sufficient calcium, its concentration is not important.
- the composition may contain an inhibiting compound(s) and/or potentiating compound(s), wherein the inhibiting compound(s) inhibit the activities of the sealant that interfere with any of the biological activities of the growth factor, the potentiating compound(s) potentiate, mediate or enhance any of the biological activities of the growth factor, and wherein the concentration of the inhibiting or potentiating compound is effective for achieving the inhibition, potentiation, mediation or enhancement.
- the growth factor-supplemented TSs of this invention are useful for promoting the healing of wounds, especially those that do not readily heal, such as skin ulcers in diabetic individuals, and for delivering growth factors including, but not limited to, FGF-1, FGF-2, FGF-4, PDGFs, EGFs, IGFs, PDGF-bb, BMP-1, BMP-2, OP- 1, TGF- ⁇ , cartilage-inducing factor-A (CIF-A), cartilage-inducing factor-B (CIF-B), osteoid-inducing factor (OIF), angiogenin(s), endothelins, hepatocyte growth factor and keratinocyte growth factor, and providing a medium for prolonged contact between a wound site and the growth factors).
- growth factors including, but not limited to, FGF-1, FGF-2, FGF-4, PDGFs, EGFs, IGFs, PDGF-bb, BMP-1, BMP-2, OP- 1, TGF- ⁇ , carti
- the growth factor-supplemented TS may be used to treat burns and other skin wounds and may comprise a TS and, in addition to the growth factors), an antibiotic(s) and/or an analgesic(s), etc.
- the growth factor-supplemented TS may be used to aid in the engraftment of a natural or artificial graft, such as skin to a skin wound. They may also be used cosmetically, for example in hair transplants, where the TS might contain FGF, EGF, antibiotics and minoxidil, as well as other compounds.
- An additional cosmetic use for the compositions of this invention is to treat wrinkles and scars instead of using silicone or other compounds to do so.
- the TS may contain FGF-1, FGF-4, and/or PDGFs, and fat cells.
- the growth factor-supplemented TSs may be applied to surgical wounds, broken bones or gastric ulcers and other such internal wounds in order to promote healing thereof.
- the TSs of this invention may be used to aid the integration of a graft, whether artificial or natural, into an animal's body as for example when the graft is composed of natural tissue.
- the TSs of this invention can be used to combat some of the major problems associated with certain conditions such as periodontitis. namely persistent infection, bone resorption, loss of ligaments and premature re-epithelialization of the dental pocket.
- this invention provides a mixture of FG, DBM and/or purified BMP's.
- This mixture provides a matrix that allows the cellular components of the body to migrate into it and thus produce osteoinduction where needed.
- the matrix composition in terms of proteins (such as fibrinogen and Factor XIII), enzymes (such as thrombin and plasmin), BMPs, growth factors and DBM and their concentrations are adequately formulated to optimize the longevity of this temporal scaffolding structure and the osteoinduction which needs to occur. All the FG components are biodegradable but during osteogenesis the mixture provides a non-collapsible scaffold that can determine the shape and location of the newly formed bone. Soft tissue collapse into the bony nonunion defect, which is a problem in bone reconstructive surgery, will thus be avoided.
- the use of TS supplemented with growth factors such as CIF-A and CIF-B, infra, which promote cartilage development, will be useful in the reconstruction of lost or damaged cartilage and/or damaged bone.
- an effective concentration of HBGF-1 is added to a FG in order to provide a growth factor-supplemented TS that possesses the ability to promote wound healing.
- an effective amount of a platelet-derived extract is added to a FG.
- an effective concentration of a mixture of at least two growth factors are added to FG and an effective amount of the growth factors- supplemented FG is applied to the wounded tissue.
- drugs, polyclonal and monoclonal antibodies and other compounds may be added to the TS. They accelerate wound healing, combat infection, neoplasia, and/or other disease processes, mediate or enhance the activity of the growth factor in the TS, and/or interfere with TS components which inhibit the activities of the growth factor in the TS.
- antibiotics such as tetracycline and ciprofloxacin
- antiproliferative/cytotoxic drugs such as 5-fluorouracil (5-FU), taxol and/or taxotere
- antivirals such as gangcyclovir, zidovudine, amantidine, vidarabine, ribaravin, trifluridine, acyclovir, dideoxyuridine and antibodies to viral components or gene products
- cytokines such as ⁇ - or ⁇ - or ⁇ -Interferon, ⁇ - or ⁇ -tumor necrosis factor, and interleukins
- colony stimulating factors erythropoietin
- antifungals such as diflucan, ketaconizole and nystatin
- antiparasitic agents such as pentamidine
- anti-inflammatory agents such as ⁇ -1-anti-trypsin and ⁇ -1-antichymotrypsin
- steroids anesthetics
- anesthetics such as pentamidine
- Other compounds which may be added to the TS include, but are not limited to: vitamins and other nutritional supplements; hormones; glycoproteins; fibronectin; peptides and proteins; carbohydrates (both simple and/or complex); proteoglycans; antiangiogenins; antigens; oligonucleotides (sense and/or antisense DNA and/or RNA); BMPs; DBM; antibodies (for example, to infectious agents, tumors, drugs or hormones); and gene therapy reagents. Genetically altered cells and/or other cells may also be included in the TSs of this invention.
- the osteoinductive compounds which can be used in practicing this invention include, but are not limited to: osteogenin (BMP3); BMP-2; OP-1; BMP-2A, -2B, and -7; TGF- ⁇ , HBGF-1 and -2; and FGF-1 and -4.
- BMP3 osteogenin
- BMP-2 OP-1
- BMP-2A, -2B, and -7 TGF- ⁇ , HBGF-1 and -2
- FGF-1 and -4 any which does not destroy the TS can be added to the TSs of this invention.
- poorly water soluble forms of a drug such as the free base of TET, increase the delivery of the drug from the TS more than freely water soluble forms thereof. Therefore, the drug may be bound to an insoluble carrier, such as fibrinogen or activated charcoal, within the TS to prolong the delivery of the drug from the supplemented TS.
- an insoluble carrier such as fibrinogen or activated charcoal
- the supplemented TS can be used in organoids and could contain, for example, growth factors such as FGF- 1 , FGF-2, FGF-4 and OP-1.
- this invention provides a composition that promotes the localized delivery of a poorly water soluble form of an antibiotic(s), such as the free base form of TET, and other drug(s), comprising a TS and an effective concentration of at least one poorly water soluble form of an antibiotic.
- the present invention has several advantages over the previously used TS compositions and methods.
- the first advantage is that the growth factor- and/or drug-supplemented TSs of the present invention have many of the characteristics of an ideal biodegradable carrier, namely: they can be formulated to contain only human proteins thus eliminating or minimizing immunogenicity problems and foreign-body reactions; their administration is versatile; and their removal from the host's tissues is not required because they are degraded by the host's own natural fibrinolytic system.
- a second advantage is that the present invention provides a good way to effectively deliver growth factors and/or drugs for a prolonged period of time to an internal or external wound. It appears that some growth factor receptors must be occupied for at least 12 hours to produce a maximal biological effect. Previously, there was no way to do this. The present invention allows for prolonged contact between the growth factor and its receptors to occur, and thus allows for the production of strong biological effects.
- a third advantage of the present invention is that animal cells can migrate into and through, and grow in the TSs of the present invention. This aids engraftment of the cells to neighboring tissues and prostheses. Based on the composition of the TSs which are available in Europe, it is expected that this is not possible with these formulations. Instead, animal cells must migrate around or digest commercially available TS. Since the importation into the U.S. of commercially available TSs from Europe is illegal (their use in the USA has not been approved by the U.S. FDA).
- a fourth advantage is that because of its initial liquid nature, the TS of the present invention can cover surfaces more thoroughly and completely than many previously available delivery systems. This is especially important for the use of the present invention in coating biomatenals and in the endothelialization of vascular prostheses because the growth factor-supplemented FG will coat the interior, exterior and pores of the vascular prosthesis. As a result of this, plus the ability of endothelial cells to migrate into and through the TS, engraftment of autologous endothelial cells will occur along the whole length of the vascular prosthesis, thereby decreasing its thrombogenicity and antigenicity.
- TSs vascular prostheses
- Previously used TSs for vascular prostheses also primarily were seeded with nonautologous cells which could be rejected by the body and could be easily washed off by the shearing force of blood passing through the prosthesis.
- a fifth advantage is that the supplemented and unsupplemented TS of this invention can be molded and thus can be custom made into almost any desired shape.
- TS such as FG can be supplemented with BMPs and/or
- DBM and can be custom made into the needed shape to most appropriately treat a bone wound. This cannot be done with DBM powder alone because DBM powder will not maintain its shape.
- a sixth advantage is that the AB-supplemented FG of this invention, such as TET-FG, has unexpectedly increased the longevity and stability of the FG compared to that of the unsupplemented FG. This increased stability continues even after appreciable quantities of the AB are no longer remaining in the FG. For example, soaking a newly formed FG clot in a saturated solution of TET produced from free base TET, or in a solution of CIP HCl, produces a FG clot which is stable and preserved even after substantially all the TET or CIP has left the FG clot.
- the AB such as TET or CIP
- the AB inhibits plasminogen which is in the TFC and breaks down the FG.
- the plasminogen is inhibited, its continued inhibition does not appear to depend on appreciable quantities of the TET or CIP remaining in the FG.
- this stabilizing effect one can expect an increased storage shelf life of the TS, and possibly an increased persistence in vivo.
- the seventh advantage of the present invention is a direct result of the prolonged longevity and stability of the TS.
- AB-supplemented FG can be used to produce localized, long term delivery of a drug(s) and/or a growth factor(s). This delivery will continue even after the stabilizing drug, such as TET or CIP, has substantially left the TS.
- the stabilizing drug such as TET or CIP
- drugs such as free base TET, allow for both stabilization of the TS and for prolonged drug delivery.
- Other drugs may do one or the other but not both.
- a compound used for the stabilization of a TS to produce prolonged, localized drug delivery is not previously known in the art.
- An eighth advantage of the present invention is that it allows site-directed angiogenesis to occur in vivo. While others have demonstrated localized non-specific angiogenesis, supra, no one else has used a TS to promote site-directed angiogenesis.
- a ninth advantage of the present invention is that because the components of the TS can be formulated into several forms of simple to use, fast-acting field dressings, it is now possible to control bleeding from hemorrhaging trauma wounds, thereby saving numerous lives that previously would have been lost. Although life-saving methods of treating such wounds are possible by trained medical personal or in fully-equipped clinics and hospitals, the present invention satisfies society's long-felt need for an easy-to-use, first-aid (or even self-applied) treatment that will, in emergency or disaster situations, allow an untrained individual to treat traumatic injuries to control hemorrhage until medical assistance is available.
- Fig. 1 shows Western blots of gels on which samples containing HBGF-1 ⁇ had been incubated with 250 U/ml thrombin in the presence of increasing concentrations of heparin.
- Solutions containing HBGF-1 ⁇ (10 ⁇ g/ml), thrombin (250 ⁇ g/ml), and increasing concentrations of heparin (0, 0.5, 5, 10, 20, and 50 units/ml) were incubated at 37°C for 72 hours. Aliquots were periodically removed from each of the incubating mixtures and were loaded onto 8% SDS polyacrylamide gels that were prepared and run as described by Laemmli (Nature 227:680 (1970)). The gel was then electroblotted onto nitrocellulose and the band corresponding to HBGF-1 ⁇ was identified using an affinity-purified polyclonal rabbit antiserum to HBGF-1 ⁇ .
- each lane contains the following: lane 1 contains SDS-PAGE low molecular weight standards; lane 2 contains biotinylated standards; lane 3 contains 10 ⁇ g/ml HBGF-1 ⁇ ; lane 4 contains 250 U/ml thrombin; and lanes 5-13 contain samples removed from the incubating mixtures at times 0, 1, 2, 4, 6, 8, 24, 48, and 72 hours.
- Fig. 2 shows the incorporation of 3 H-thymidine as a function of relative
- HBGF-1 ⁇ concentration HBGF-1 ⁇ concentration. Samples of the HBGF-1 ⁇ were incubated, as described in Figure 1 and Example 2, in the presence of 250 U/ml thrombin and 5 U/ml heparin for 0, 24 or 72 hours. Dilutions of these samples were then added to NIH 3T3 cells, which were plated as described in Example 3. CPM is plotted v. HBGF-1 concentration.
- Fig. 3. Typical pattern of human umbilical vein endothelial cells after 7 days' growth on FG supplemented with 100 ng/ml of active, wild-type FGF-1. Note the large number of cells and their elongated shape. Compare with the paucity of cells grown on unsupplemented FG ( Figure 5).
- Fig. 4. Typical pattern of human umbilical vein endothelial cells after 7 days' growth on FG supplemented with 10 ng/ml of active, wild-type FGF-1. Note the large number of cells and their elongated shape. Compare with the paucity of cells grown on unsupplemented FG ( Figure 5).
- Fig. 5 Typical pattern of human umbilical vein endothelial cells after 7 days' growth on unsupplemented FG. Note the small number of cells, compared to the number of cells in Figures 3 and 4, which indicates a slower proliferation rate.
- Fig. 6 Typical pattern of human umbilical vein endothelial cells after 7 days' growth on FG supplemented with 100 ng/ml of inactive, mutant FGF-1.
- Fig.7 Typical pattern of human umbilical endothelial cells 24 hours after having been embedded in FG at a concentration of 10 5 cells per ml of FG.
- the protein and thrombin concentrations of the FG were 4 mg/ml and 0.6 NIH units/ml, respectively. Note, their elongated, multipodial morphology and that they formed a cellular network where they came in contact with each other. Compare with the cobblestone shape of similar cells grown in fibronectin (Fig.
- Fig. 8 Typical pattern of human umbilical endothelial cells 48 hours after having been embedded in FG at a concentration of 10 5 cells per ml of FG. The culture conditions were as described in Fig. 7. Note the further accentuated, elongated and multipodial morphology and increasing development of cellular networks. Compare with the cobblestone shaped cells grown in fibronectin (Fig. 10) and note the lack of a cellular network in the latter.
- Fig.9. Typical pattern of human umbilical endothelial cells 24 hours after having been cultured on a surface coated with fibronectin. Note the cobblestone shape of the cells and the lack of cellular networks. Compare to Figure 7.
- Fig. 10 Typical pattern of human umbilical endothelial cells 48 hours after having been cultured in a commonly used film of fibronectin. Note the cobblestone shape of the cells and the lack of cellular networks. Compare to Figure 8.
- FIG. 11 Micrographs of cross sections of PTFE vascular grafts that were explanted from dogs after 7 days (panels A, C, E) or 28 days (panels B, D, F). Prior to implantation, the grafts were either untreated (A and B), coated with FG alone (C and D), or coated with FG supplemented with heparin and HBGF-1 (E and F).
- Untreated controls (A & B) showed minimal mesenchymal tissue ingrowth, with both their interstices filled with, and their luminal surfaces coated with fibrin coagulum.
- the FG-treated grafts showed mesenchymal tissue ingrowth in only the outer half of the grafts' interstices, with the rest being filled with fibrin coagulum. Very few interstitial capillaries were present.
- the grafts treated with FG containing FGF-1 showed more abundant interstitial ingrowth and by 28 days showed numerous capillaries, myofibroblasts and macrophages, with inner capsules consisting of several layers of myofibroblasts beneath confluent endothelial cell layers. Results of similar grafts after 128 days of implantation were similar, with greater numbers of capillaries in the FG + FGF-1 group (data not shown).
- Fig. 12 Scanning electron micrographs of the inner lining of the vascular grafts described in Figure 11 after 28 days of implantation.
- the grafts were either untreated (G), coated with FG alone (H), or coated with FG supplemented with heparin and HBGF-1 (I).
- Untreated control grafts (G) showed sparse areas of endothelial cell coverage amidst areas of thrombus containing red blood cells, platelets, and areas of exposed PTFE graft material (visible in the center and top of the picture).
- Grafts coated with FG alone (H) showed islands of endothelial cells amidst areas of fibrin coagulum.
- grafts treated with FG + HBGF-1 (I) showed confluent endothelial cells oriented along the direction of blood flow.
- Fig. 13 Preparation of disc-shaped implants 1 mm thick and 8 mm in diameter prepared using an aluminum mold.
- Fig. 14 Diagram illustrating intramuscular bioassay for the induction of bone formation in rats by DBM alone, by FG implants or by DBM-FG.
- Fig. 15 Diagram illustrating the induction of bone formation in calvarial implants by DBM-FG.
- Fig. 16 Radio-opacity data at 28 days postoperative from intramuscular implants of DBM-FG, DBM or FG.
- Fig. 17 Radio-opacity data from DBM-FG (30 mg/ml) calvarial implants at 28 days, 3 months and 4 months postoperative.
- Figure 18A is a photograph of a craniotomy site at 28 days post surgery in a treated animal.
- Figure 18B is a photograph of the calvarial wound from an untreated control at 28 days postoperative. Note that only fibrous connective tissue has developed across the craniotomy wound.
- Fig. 19 Photograph from the craniotomy wounds of animals which were treated with DBM particles only.
- Fig. 20 Photograph of new bone formed in the craniotomy site in response to DBM-FG (15 mg/ml).
- Fig. 21 Photograph of new bone formed in the craniotomy site in response to DBM-FG (15 mg/ml). Note that typically more bone marrow formed in craniotomy wounds that had been implanted with DBM-FG disks than with
- the disks contained 100 mg/ml of TET and were placed in closed vessels filled with 2ml of PBS.
- the TET concentration was measured spectrophotometrically in the PBS effluent which had been continuously exchanged at a rate of 3 ml/day.
- the volume of the PBS supernatant had been kept constant at approximately 2 ml.
- the data are the average of two experiments.
- Fig. 24 The release of TET into saliva from 3 ⁇ 6 mm diameter disks containing 50 or 100 TET mg/ml FG at 37°C.
- the TET concentration was measured spectrophotometrically in 0.75 ml of saliva supernatant that had been replaced daily.
- the saliva used in these experiments had been pooled from ten donors, centrifuged, filtered and kept at 4°C.
- TET TET and with 50 and 100 mg/ml TET over a period of 15 days.
- the disks had been kept in 0.75 ml of saliva which had been changed daily.
- the saliva had been pooled from 10 donors. It had then been centrifuged, filtered and stored at
- Fig.26 Antibacterial activity of TET released from TET-supplemented FG.
- Two ml PBS surrounding the 3 ⁇ 6 mm TET-supplemented FG disks was replaced daily.
- 6 mm paper disks impregnated with the collected eluates were incubated for 18 hours at 37°C on agar plates containing E. coli. Then the diameter of the zone of inhibition was measured.
- Fig.27 The release of ciprofloxacin, amoxicillin and metronidazole from FG matrices. Individual 3 x 6 mm diameter disks containing 100 mg/ml of the respective antibiotics were immersed in 2 ml of phosphate-buffered saline at 37°C. The supernatant was replaced daily and the antibiotic concentration was measured spectrophotometrically at 275, 274 and 320 nm, respectively.
- Fig. 28 The release of TET from TET-supplemented FG disks was proportional to the temperature of the PBS bathing the TET-FG disks.
- Fibronectin was added to the lower wells of the modified Boyden's chambers.
- the data are expressed as means +/- S.E. of migrated cells per high power field and demonstrate that, as a function of dose, fibronectin induced the chemotaxis of NIH 3T3 cells toward it.
- Fig.35 Dose-response relationship of the chemotactic response of human dermal fibroblasts (HDFs) to FGF- 1.
- a step gradient of increasing concentrations of FGF-1 was added to the lower wells of the modified Boyden's chambers in the presence of heparin.
- the data are expressed as the means +/- S.E. of migrated cells per high power field and demonstrate that, as a function of dose, FGF-1 induced the chemotaxis of HDFs toward it.
- Fig.36 Dose-response relationship of the chemotactic response of HDFs to FGF-2.
- a step gradient of increasing concentrations of FGF-2 was added to the lower wells of the modified Boyden's chambers.
- the data are expressed as the means +/- S.E. of migrated cells per high power field and demonstrate that, as a function of dose, FGF-2 induced the chemotaxis of HDFs toward it.
- Fig. 37 Dose-response relationship of the chemotactic response of HDFs to FGF-4.
- a step gradient of increasing concentrations of FGF-4 was added to the lower wells of the modified Boyden's chambers.
- the data are expressed as the means +/- S.E. of migrated cells per high power field and demonstrate that, as a function of dose, FGF-4 induced the chemotaxis of HDFs toward it.
- Fig.38 Dose-response relationship of the chemotactic response of HDFs to FGF-4 in solution and in FG.
- FGF-4 was incorporated into FG and placed in the bottom well of chemotaxis chambers. The amount of FGF in the FG was sufficient to result in the indicated concentrations when evenly distributed throughout the FG and medium in the lower chamber.
- Negative controls included medium alone and FG without FGF.
- Medium containing FGF-4 at a concentration of 10 ng/ml in the lower chamber was utilized as a positive control.
- the data are expressed as the means +/- S.E. of migrated cells per high power field and demonstrate that, as a function of dose, FGF-4 released from FG induced the chemotaxis of HDFs toward the FG.
- Fig. 39 Diagram of a self-contained TS Wound Dressing. Description of the Preferred Embodiments
- a wound includes damage to any tissue in a living organism.
- the tissue may be an internal tissue, such as the stomach lining or a bone, or an external tissue, such as the skin.
- a wound may include, but is not limited to, a gastrointestinal tract ulcer, a broken bone, a neoplasia, and cut or abraided skin.
- a wound may be in a soft tissue, such as the spleen, or in a hard tissue, such as bone.
- the wound may have been caused by any agent, including traumatic injury, infection or surgical intervention.
- TS is a substance or composition that, upon application to a wound, seals the wound, thereby reducing blood loss and maintaining hemostasis.
- FG is a composition, prepared from recombinant or plasma proteins, that upon application to a wound forms a clot, thereby sealing the wound, reducing blood loss and maintaining hemostasis.
- FG supra, is a form ofTS.
- supplemented TS includes any TS that, without substantial modification, can serve as a carrier vehicle for the delivery of a growth factor, drug or other compound, or mixtures thereof, and that, by virtue of its adhesive or adsorptive properties, can maintain contact with the site for a time sufficient for the supplemented TS to produce its desired effect, for example to promote wound healing.
- a growth factor-supplemented TS is a TS to which at least one growth factor has been added at a concentration that is effective for its stated purpose.
- the growth factor can. for example, accelerate, promote or improve wound healing, or tissue (re)generation.
- the growth factor-supplemented TSs may also contain additional components, including drugs, antibodies, anticoagulants and other compounds that: 1 ) potentiate, stimulate or mediate the biological activity of the growth factor(s) in the TS; 2) decrease the activities of components of the growth factor-supplemented TS which would inhibit or destroy the biological activities of the growth factors) in the sealant; or 3) allow prolonged delivery of the supplement from the TS; 4) possess other desirable properties.
- a potentiating compound is a compound that mediates or otherwise increases the biological activity of a growth factor in the TS.
- Heparin is an example of a compound that potentiates the biological activity of HBGF-1.
- an inhibiting compound is a compound that inhibits, interferes with, or otherwise destroys a deleterious activity of a component of the
- Inhibiting compounds may exert their effect by protecting the growth factor from degradation.
- An inhibiting compound does not, however, inhibit any activities that are essential for the desired properties, such as, for example, wound healing of the growth factor-supplemented TS.
- An example of an inhibiting compound is heparin.
- a growth factor includes any soluble factor that regulates or mediates cell proliferation, cell differentiation, tissue regeneration, cell attraction, wound repair and/or any developmental or proliferative process.
- the growth factor may be produced by any appropriate means including extraction from natural sources, production through synthetic chemistry, production through the use of recombinant DNA techniques and any other techniques, including virally inactivated, growth factor(s)-rich platelet releasate, which are known to those of skill in the art.
- the term growth factor is meant to include any precursors, mutants, derivatives, or other forms thereof which possess similar biological activity(ies), or a subset thereof, to those of the growth factor from which it is derived or otherwise related.
- HBGF-1 which is also known to those of skill in the art by alternative names, such as endothelial cell growth factor (ECGF) and FGF-1, refers to any biologically active form of HBGF-1, including HBGF-1 ⁇ , which is the precursor of HBGF-l ⁇ and other truncated forms, such as FGF.
- ECGF endothelial cell growth factor
- FGF-1 refers to any biologically active form of HBGF-1, including HBGF-1 ⁇ , which is the precursor of HBGF-l ⁇ and other truncated forms, such as FGF.
- ECGF endothelial cell growth factor
- FGF-1 refers to any biologically active form of HBGF-1, including HBGF-1 ⁇ , which is the precursor of HBGF-l ⁇ and other truncated forms, such as FGF.
- U.S. Patent No. 4,868,113 to Jaye et al., herein incorporated by reference sets forth the amino acid sequences of each form of HBGF
- growth factors may also be known to those of skill in the art by alternative nomenclature. Accordingly, reference herein to a particular growth factor by one name also includes any other names by which the factor is known to those of skill in the art and also includes any biologically active derivatives or precursors, truncated mutant, or otherwise modified forms thereof.
- biological activity refers to one or all of the activities that are associated with a particular growth factor in vivo and/or in vitro.
- a growth factor exhibits several activities, including mitogenic activity (the ability to induce or sustain cellular proliferation) and also non-mitogenic activities, including the ability to induce or sustain differentiation and/or development.
- growth factors are able to recruit or attract particular cells from which the proliferative and developmental processes proceed. For example, under appropriate conditions HBGF-1 can recruit endothelial cells and direct the formation of vessels therefrom. By virtue of this activity, growth factor-supplemented TS may thereby provide a means to enhance blood flow and nutrients to specific sites.
- extended longevity means at least a two fold increase in the visually observable, useful in vitro lifespan of a TS.
- demineralized bone matrix means the organic matrix of bone that remains after bone is decalcified with hydrochloric or another acid.
- BMPs bone morphogenetic proteins
- BMP-2 is also known as BMP-2A.
- BMP-4 is also known as BMP-2B.
- BMP-3 is also known as osteogenin.
- BMP-6 is also known as Vgr-1.
- BMP-7 is also known as OP-1.
- Bone morphogenetic proteins is meant to include, but is not limited to BMP-1 through BMP-8.
- augmentation means using a supplemented or unsupplemented TS to change the internal or external surface contour of a component of an animal's body.
- a damaged bone is a bone which is broken, fractured, missing a portion thereof, or otherwise not healthy, normal bone.
- a deficient bone is a bone which has an inadequate shape or volume to perform its function.
- bone or DBM which is to be used to supplement a TS can be in the form of powder, suspension, strips or blocks or other forms as necessary to perform its desired function.
- organoid means a structure that may be composed of natural, artificial, or a combination of natural and artificial elements, that wholly or in part, replaces the function of a natural organ.
- An example would be an artificial pancreas consisting of a network of capillaries surrounded by cells transfected with an expression vector containing the gene for insulin. Such an organoid would function to release insulin into the bloodstream of a patient with Type I Diabetes.
- the supplement and TS must be selected.
- the supplement and TS may be prepared by methods known to those of skill in the art, may be purchased from a supplier thereof, or may be prepared according to the methods of this application.
- growth factor, drug-or DBM-supplemented FG is prepared.
- the supplement may be added to the fibrinogen, the thrombin, the calcium and/or the water components) before they are mixed to form the TS.
- the supplement(s) can be added to the components as they are being mixed to form the TS.
- the calcium and/or thrombin may be supplied endogenously from body fluids as, for example, those in a wound.
- TS which allows cells to migrate into and/or through it may preferably be used.
- TS such as commercially available FG
- FGs which are well known to those of skill in the art (see, e.g., U.S. Patent Nos.: 4,627,879; 4,377,572; and 4,298,598, all herein incorporated by reference) may be purchased from a supplier or manufacturer thereof, such as IMMUNO AG (Vienna, Austria) and BEHRINGWERKE AG (Germany).
- IMMUNO AG Vehicle
- BEHRINGWERKE AG Germany
- FGs may be supplemented with growth factors, antibiotics and/or other drugs for use in the embodiments of this invention including, but not limited to: in vitro cellular proliferation and/or differentiation; drug delivery; growth factor delivery, etc.
- growth factors antibiotics and/or other drugs
- FG was prepared from ciyoprecipitate from fresh frozen plasma.
- the total protein concentration in the prepared FG is from about 0.01 to 500 mg/ml of FG. In a more preferred embodiment, the total protein concentration in the prepared FG is from about 1 to 120 mg/ml FG. In the most preferred embodiment, the total protein concentration in the prepared FG is from about 4 to 30 mg/ml FG.
- the fibrinogen concentration used to prepare the FG is from about 0.009 to 450 mg/ml of solution. In a more preferred embodiment, the fibrinogen concentration in this preparatory solution is from about 0.9 to 110 mg/ml. In the most preferred embodiment, the fibrinogen concentration in this preparatory solution is from about 3 to 30 mg/ml.
- the thrombin concentration used to prepare the FG is 0.01-350 U/ml. In a more preferred embodiment, the thrombin concentration is 1-175 U/ml. In the most preferred embodiment, the thrombin concentration is 2-4 U/ml.
- the calcium ion concentration be sufficient to allow for activation of the thrombin.
- the USP calcium chloride concentration is 0-100 mM. In a more preferred embodiment, the USP calcium chloride concentration is 1 -40 mM. In the most preferred embodiment, the USP calcium chloride concentration is 2-4 mM.
- the calcium may be supplied by the tissue or body fluids as, for example, in the wound dressing embodiment.
- sterile water for injection should be used.
- concentration(s) of growth factor(s), drugs and other compounds will vary depending on the desired objective, the concentrations must be great enough to allow them to be effective to accomplish their stated purpose.
- the growth factor concentration is from about 1 ng/ml to 1 mg/ml of FG. In a more preferred embodiment, the growth factor concentration is from about 1 ⁇ g/ml to 100 ⁇ g/ml of FG. In the most preferred embodiment, the growth factor concentration is from about 5 ⁇ g/ml to 20 ⁇ g/ml of FG. In a preferred embodiment of this invention the TET or CIP concentration is from 0.01 to 300 mg/ml FG. In a more preferred embodiment of this invention the TET or CIP concentration is 0.01-200 mg/ml.
- the TET or CIP concentration is 1-150 mg/ml.
- the amount of the supplements to be added can be empirically determined by one of skill in the art by testing various concentrations and selecting that which is effective for the intended purpose and the site of application.
- the growth factors may be prepared by any method known to those of skill in the art or may be purchased commercially. Any growth factor may be selected including, but not limited to, for example, growth factors that stimulate the proliferation and/or attraction of certain cell types, such as endothelial cells, fibroblasts, epithelial cells, smooth muscle cells, hepatocytes, and keratinocytes, and/or growth factors which inhibit the growth of the same cell types and smooth muscle cells. Such selection may be dependent upon the particular tissue site for which the growth factor-supplemented TS will be applied and/or the type of effect desired. For example, an EGF-supplemented TS may be preferred for application to wounds in the eye and for treating gastric ulcers while an osteogenin-supplemented TS may be preferred for application to bone fractures and bone breaks in order to promote healing thereof.
- EGF-supplemented TS may be preferred for application to wounds in the eye and for treating gastric ulcers
- an osteogenin-supplemented TS may be preferred for application to bone fractures
- HBGF-1 ⁇ was prepared and added to FG.
- HBGF-1 ⁇ , or HBGF-1 ⁇ , or any other active form of HBGF-1 can be purified from natural sources, from genetically engineered cells that express HBGF-1 or a derivative thereof, or by any method known to those of skill in the art.
- HBGF-1 ⁇ has been prepared using recombinant DNA methodology (Jaye et al., U.S. Patent No. 4,868,113; Jaye et al., J. Biol Chem. 262:16612-16617 (1987)). Briefly, DNA encoding HBGF-1 ⁇ was cloned into a prokaryotic expression vector, a pUC9 derivative, and expressed intracellularly in E.
- HBGF-1 ⁇ was purified from the supernatant using standard methods of protein purification including affinity chromatography on heparin SepharoseTM followed by ion-exchange chromatography on CM-SepharoseTM.
- HBGF-1 In addition to HBGF-1, described above, other growth factors that may be added to the FG include, but are not limited to, HBGF-2, IGF-1, EGF, TGF- ⁇ , TGF- ⁇ , any platelet-derived growth factor or extract, BMPs, and mixtures of any growth factors.
- platelet-derived extracts which serve as rich sources of growth factors, may be added to the TS in addition to or in place of other growth factors, such as HBGF-1.
- a platelet-derived extract prepared by any method known to those of skill in the art, is added to a TS.
- Such an extract has been prepared from plasma derived platelets for use with FG.
- Platelet-Derived Wound Healing Factor may be prepared and added to FG (Knighton et al., Ann. Surg. 204:322-330 (1986)). Briefly, to prepare PDWHF, blood is drawn into anticoagulant solution and platelet-rich plasma is prepared by refrigerated centrifugation. The platelets are isolated and stimulated with thrombin, which releases the contents of the alpha granule contents. The platelets are removed and an effective concentration of the remaining extract is added to a TS. Additional Components of Growth Factor-Supplemented TS.
- the TSs contemplated for use with growth factors contain numerous components, some of which may interfere with the biological activity of the selected growth factor(s).
- thrombin which is an essential component of FG, can act as a proteolytic enzyme and specifically cleave HBGF-1 ⁇ . Therefore, it may be necessary to include additional compounds, such as protease or other inhibitors, that protect the selected growth factor(s) from the action of other components in the TS which interfere with or destroy the biological activity of the growth factor(s).
- Selection of the particular inhibiting compound(s) may be empirically determined by using methods, discussed below, that assess the biological activity of the growth factors) in the TS. Methods to assess biological activity are known to those of skill in the art.
- heparin potentiates the biological activity of HBGF-1 in vivo (see, e.g., Burgess et al., Annu. Rev. Biochem. 58:575-606 (1989)).
- the supplemented TS of the present invention may contain compounds such as drugs, other chemicals, and proteins. These may include, but are not limited to: antibiotics such as TET, ciprofloxacin, amoxicillin, or metronidazole, anticoagulants, such as activated protein C, heparin, prostracyclin (PGI 2 ), prostaglandins, leukotnenes, antithrombin III, ADPase, and plasminogen activator; steroids, such as dexamethasone, inhibitors of prostacyclin, prostaglandins, leukotnenes and/or kinins to inhibit inflammation; cardiovascular drugs, such as calcium channel Mockers; chemoattractants; local anesthetics such as bupivacaine; and antiproliferative/antitumor drugs such as 5-fluorouracil (5-FU), taxol and/or taxotere.
- antibiotics such as TET, ciprofloxacin, amoxicillin,
- supplemental compounds may also include polyclonal, monoclonal or chimeric antibodies, or functional derivatives or fragments thereof. They may be antibodies which, for example, inhibit smooth muscle proliferation, such as antibodies to PDGF, and/or TGF- ⁇ , or the proliferation of other undesirable cell types within and about the area treated with the TS. These antibodies can also be useful in situations where anti-cancer, anti-platelet or anti-inflammatory activity is needed. In general, any antibody whose efficacy would be improved by site-directed delivery may benefit from being used with this TS delivery system.
- composition may be tested by any means known to those of skill in the art (see, e.g., Tsuboi et al., J. Exp. Med. 172:245-251 (1990); Ksander etal., J. Am. Acad. Dermatol 22:781-791 (1990); and Greenhalgh et al., Am. J. Path. 136:1235 (1990)).
- any method including both in vivo and in vitro assays, by which the activity of the selected growth factor(s) in the TS composition can be assessed may be used.
- the activity of HBGF-1 ⁇ has been assessed using two independent in vitro assays. In the first, the proliferation of endothelial cells that had been suspended in a shallow fluid layer covering a plastic surface which had been impregnated with growth factor-supplemented FG was measured. In the second, the incorporation of 3 H-thymidine in cultured fibroblasts in the presence of HBGF-1 was measured.
- FG that had been supplemented with HBGF-1 ⁇ has been tested for its ability to promote healing in vivo using mice as a model system.
- identical punch biopsies were made in the dorsal region of the mice, which were then separated into test, treated control and untreated control groups.
- the wounds in the mice in the test group were treated with the growth factor-supplemented TS.
- the wounds in the mice in the treated control group were treated with unsupplemented TS.
- the wounds in the untreated group were not treated with TS.
- the ability of the growth factor-supplemented TS to induce cell proliferation and to recruit cells may also be assessed by in vitro methods known to those of skill in the art.
- the in vitro assays described above for measuring the biological activity of growth factors and described in detail in the Examples may be used to test the activity of the growth factor in the TS composition.
- the effects of adding inhibiting and/or potentiating compounds can also be assessed.
- HBGF-1 ⁇ in HBGF-1-supplemented FG was specifically cleaved in a stochastic manner, suggesting that a component of the FG preparation, most likely thrombin, was responsible.
- Heparin which is known to bind to HBGF-1 and protect it from certain proteolytic activities, was added to the HBGF-1-supplemented FG.
- the addition of relatively low concentrations of heparin protected HBGF-1 ⁇ from cleavage that would destroy its biological activity in the FG. Therefore, TS compositions that include HBGF-1 may include heparin or some other substance that inhibits the cleavage of HBGF-1 by thrombin or other proteolytic components of the FG.
- heparin has been tested for its ability to inhibit cleavage of HBGF- 1 by thrombin, which is an essential component of FG.
- thrombin which is an essential component of FG.
- mixtures of various concentrations of heparin and HBGF-1-supplemented FG have been prepared, and incubated for various times.
- the biological activity of HBGF-1 in the mixture has been tested and the integrity of the HBGF-1 has been ascertained using western blots of SDS gels. Relatively low concentrations, about a 1:1 molar ratio of heparin:HBGF-1, are sufficient to protect HBGF-1 from degradation in FG.
- a particular compound can be used to potentiate, mediate or enhance the biological activity of a growth factor(s) in TS.
- the growth factor and TS, or the growth factor-supplemented TS Prior to clinical use, is pasteurized or otherwise treated to inactivate any pathogenic contaminants therein, such as viruses.
- Methods for inactivating contaminants are well-known to those of skill in the art and include, but are not limited to, solvent- detergent treatment and heat treatment (see, e.g., Tabor et al., Thrombosis Res. 22:233-238 (1981) and Piszkiewicz et al., Transfusion 28:198-199 (1988)).
- the supplemented TS is applied directly to the wound, other tissue or other desired location.
- it can be applied directly by any means, including spraying on top of the wound.
- It can also be applied internally, such as during a surgical procedure. When it is applied internally, such as to bones, the clot gradually dissolves over time.
- the TSs may be formulated as a self-contained wound dressing, or fibrin sealant bandage, which contains the necessary thrombin and fibrinogen components of the FG.
- the self-contained dressing or bandage is easy-to-use. requiring no advanced technical knowledge or skill to operate. It can even be self-administered as an emergency first aid measure to preserve life until medical assistance becomes available.
- the self-contained TS wound dressing or fibrin sealant bandage is an advancement over the current technology in that the field-ready preparation can be stored for long periods, and be used to provide rapid TS treatment of a hemorrhaging wound without the time delay associated with solubilization and mixing of the components.
- the self-contained TS wound dressing or fibrin sealant bandage comprises a tissue sealing composition comprising a tissue sealant or fibrin complex of the type previously described.
- the composition may be comprised of purified fibrinogen, thrombin and calcium chloride with sufficient Factor XIII to produce a fibrin clot.
- XIII components are supplied in the form of topical fibrinogen complex (TFC).
- TFC topical fibrinogen complex
- the components are most preferably pathogen-inactivated, purified components derived from human sources.
- the components of the present invention including additives thereto, are treated with a detergent/solvent, and/or otherwise treated, e.g., by pasteurization or ultrafiltration to inactivate any pathogenic contaminants therein, such as viruses.
- Methods for inactivating contaminants are well-known to those of skill in the art and include, but are not limited to, solvent-detergent treatment and heat treatment. Solvent-detergent treatment is particularly advantageous in that the proteinaceous components are not exposed to irreversible heat-denaturation.
- the calcium and/or Factor XIII components may be contained in either the thrombin and/or the fibrinogen component(s), and/or absorbed from the patient's endogenous calcium present in the fluids escaping from the wound.
- Thrombin may also be supplied endogenously.
- Either or both of the thrombin or fibrinogen components can be, but does not have to be, supplemented in each of the following embodiments with one or more growth factors, drugs, inhibiting compounds (to inhibit the activities of the sealant that may interfere with any of the biological activities of the growth factor or drug), and potentiating compounds (to potentiate, mediate or enhance any of the biological activities of the growth factor or drug), compounds which inhibit the breakdown of the fibrin clot, or dyes.
- the growth factor may include, e.g., fibroblast growth factor-1, fibroblast growth factor-2 and fibroblast growth factor-4; platelet-derived growth factor; insulin-binding growth factor-1; insulin-binding growth factor-2; epidermal growth factor; transforming growth factor- ⁇ ; transforming growth factor- ⁇ ; cartilage-inducing factors -A and -B; osteoid-inducing factor; osteogenin and other bone growth factors; collagen growth factor; heparin-binding growth factor-1; heparin-binding growth factor-2; and/or their biologically active derivatives.
- the drug may be an analgesic, antiseptic, antibiotic or other drug(s), such as antiproliferative drugs which can inhibit infection, promote wound healing and/or inhibit scar formation. More than one drug may be added to the composition, to be released simultaneously, or the drug may be released in predetermined time-release manner.
- drugs may include, for example, taxol, tetracycline free base, tetracycline hydrochloride, ciprofloxacin hydrochloride or
- the addition of taxol to the fibrin sealant complex may be particularly advantageous.
- the drug may be a vasoconstrictor, e.g., epinephrine; or the drug may be added to stabilize the tissue sealant or fibrin clot, e.g., aprotinin.
- the supplement(s) is at a concentration in the TS such that it will be effective for its intended purpose, e.g., an antibiotic will inhibit the growth of microbes, an analgesic will relieve pain, etc.
- Dyes, markers or tracers may be added, for example, to indicate the extent to which the fibrin clot may have entered the wound, or to measure the subsequent resorption of the fibrin clot, or the dye may be released from the tissue sealant in a predetermined, time-release manner for diagnostic purposes.
- the dyes, markers or tracers must be physiologically compatible, and may be selected from colored dyes, including water soluble dyes, such as toluidine blue, and radioactive or fluorescent markers or tracers which are known in the art.
- the dyes, markers or tracers may also be compounds which may be chemically coupled to one or more components of the tissue sealant.
- the marker may be selected from among proteinaceous materials which are known in the art, which upon exposure to proteolytic degradation, such as would occur upon exposure to proteases escaping from wounded tissue, change color or develop a color, the intensity of which can be quantified.
- the composition may also be supplemented with effective amounts of demineralized bone matrix and/or bone morphogenic proteins, and/or their biologically compatible derivatives.
- the concentration of the fibrinogen and/or thrombin components of the self-contained TS wound dressing or fibrin sealant bandage may have a significant effect on the density and clotting speed of the final fibrin matrix.
- This principle may be used to satisfy specific uses of the self-contained TS wound dressing or fibrin sealant bandage in specialized situations. For example, the treatment of an arterial wound may require the fibrin clot to set very rapidly and with sufficient integrity to withstand pressurized blood flow. On the other hand, when filling deep crevices in a wound, treatment may require the components to fill the wound completely before the fibrin clot sets.
- the thrombin and fibrinogen components are individually contained in independent quick-evaporating gel layers (e.g., methylcellulose/alcohol/water), wherein the two gel layers are separated from each other by an impermeable membrane, and the pair are covered with an outer, protective, second impermeable membrane.
- the bandage may be coated on the surface that is in contact with the gel in order to insure that the gel pad remains in place during use. (See Figure 39).
- the membrane separating the two gel layers is removed, allowing the two components to mix.
- the outer membrane is then removed and the bandage is applied to the wound site.
- the action of the thrombin and other components of the fibrinogen preparation cause the conversion of the fibrinogen to fibrin, in the manner previously disclosed for other FS applications. This results in a natural inhibition of blood and fluid loss from the wound, and establishes a natural barrier to infection.
- both the thrombin component, and the plastic film separating the thrombin gel and the fibrinogen gel may be omitted.
- the outer impervious plastic film is removed and the bandage applied, as previously described, directly to the wound site.
- the thrombin and calcium naturally present at the wound site then induce the conversion of fibrinogen to fibrin and inhibit blood and fluid loss from the wound as above.
- This alternative embodiment of the gel pack has the advantage of being simpler, cheaper, and easier to produce.
- a patient's wounds have insufficient thrombin to effectively transform the fibrinogen gel into a fibrin tissue sealant.
- the thrombin component must be exogenously supplied, as in the earlier-described gel pack embodiment of the invention.
- a fibrin sealant bandage embodiment is formulated for applying a tissue sealing composition to wounded tissue in a patient, wherein the bandage comprises, in order: (1) an occlusive backing; (2) a physiologically-acceptable adhesive layer on the wound-facing surface of the backing; and (3) a layer of dry materials comprising an effective amount, in combination, of (a) dry, virally-inactivated, purified fibrinogen complex, (b) dry, virally-inactivated, purified thrombin, and as necessary (c) effective amounts of calcium and/or Factor XIII to produce a tissue-sealing fibrin clot upon hydration, wherein the layer of dry materials is affixed to the wound-facing surface of the adhesive layer.
- the occlusive backing and the physiologically-acceptable adhesive layer are one and the same, if the backing layer is sufficiently adhesive to effectively bind the layer of dry materials.
- a removable, waterproof, protective film is placed over the layer of dry materials and the exposed adhesive surface of the bandage for long-term stable storage. In operation the waterproof, protective film is removed prior to the application of the bandage over the wounded tissue.
- the tissue sealant component of the bandage in one embodiment is activated at the time the bandage is applied to the wounded tissue to form a tissue sealing fibrin clot by the patient's endogenous fluids escaping from the hemorrhaging wound.
- the tissue sealant is hydrated and fluid loss from the wound will be significantly diminished within minutes of application of the bandage to the wounded tissue.
- the speed with which the fibrin clot forms and sets may be to some degree dictated by the application, e.g., rapid setting for arterial wounds and hemorrhaging tissue damage, slower setting for treatment of wounds to bony tissue, preferably the fibrin clot will form within twenty minutes after application. More preferably, this effect will be evident within ten minutes after application of the bandage.
- the fibrin clot will form within two to five minutes after application.
- the tissue seal will be substantially formed within 1-2 minutes, more preferably within 1 minute, and most preferably within 30 seconds after application.
- the tissue sealant components are hydrated by a suitable, physiologically-acceptable liquid prior to application of the bandage to the wounded tissue.
- the dry materials may be obtained, for example, by lyophilization or freeze-drying, or suitable, commercially-available materials may be utilized.
- Anhydrous CaCl 2 may also be added to the dry TS components to accelerate the speed of fibrin formation upon hydration of the fibrin sealant bandage.
- the binding of the dry materials to the adhesive or backing layer may be enhanced by adding a binder, preferably a water soluble binder, to the dry components.
- the backing of the fibrin sealant bandage may be of conventional, non-resorbable materials, e.g., a silicone patch or plastic material; or it may be of biocompatible, resorbable materials.
- the backing material may act as more than a delivery device. Its preferred composition is determined by the desired application of the fibrin sealant bandage.
- a non-resorbable backing is appropriate for many external uses, where it provides strength and protection for the fibrin clot.
- the non-resorbable backing is reinforced, e.g., with fibers, to provide extra strength and durability for the protective covering over the fibrin clot.
- the non-resorbable backing may be used to provide strength to the tissue sealing fibrin clot during its formation, e.g., when the hemorrhaging fluids are escaping under pressure, as in an arterial wound. Yet, if such a wound is internal, it is advantageous to remove the backing from the fibrin clot without disturbing the tissue seal. Therefore, a fibrin sealant bandage is provided in which the adhesive layer is of a material having a lower shear strength than that of the fibrin clot, permitting removal of the backing without damage to the fibrin clot or the tissue surrounding the wound.
- a resorbable material is one which is broken down spontaneously or by the body into components which are consumed or eliminated in such a manner as to not significantly interfere with healing and/or tissue regeneration or function, and without causing any other metabolic disturbance. Homeostasis is preserved.
- Materials suitable for preparing the biodegradable backing include proteinaceous substances, e.g., fibrin, collagen, keratin and gelatin, or carbohydrate derived substances, e.g., chitin, chitosan, carboxymethylcellulose or cellulose, and/or their biologically compatible derivatives.
- the adhesive layer if separate from the occlusive backing layer, is selected on the basis of the intended application of the fibrin sealant bandage, and may comprise conventional adhesive materials. Antiseptic may be added to the adhesive layer.
- the adhesive must be sufficient to affix the dry material layer to the occlusive backing, and to maintain an adhesive capability after hydration which is greater than the sheer strength of fibrin.
- the adhesive must be sufficiently sticky to affix the dry material layer to the occlusive backing, but yet have an adhesive capability after hydration which is less than the sheer strength of the fibrin clot.
- the adhesive layer may be of a material which becomes solubilized or less sticky during hydration of the dry materials, permitting removal of the backing from the fibrin clot.
- the dry material layer may be affixed directly to the occlusive bandage.
- the adhesive layer comprises two different adhesives to permit removal after hydration of the occlusive layer without disturbing the tissue sealing fibrin clot.
- the dry, tissue-sealant component materials are affixed to a specific region of the backing, the "inner region,” e.g., the center, with an unencumbered area of adhesive extending beyond the area of dry material, the "outer region.”
- the outer region of adhesive is affixed directly to the skin or tissue surrounding or adjacent to the wound in such a way that the dry material region of the bandage forms a fibrin clot directly over the wound.
- the adhesive layer on the region of backing which is not covered by the dry material layer of the bandage is sufficient to affix the fibrin sealant bandage to the tissue surrounding the wound until its physical removal.
- the adhesive on the outer region must be sufficient to hold the bandage in place, even if fluids are hemorrhaging from the wound under pressure, e.g., an arterial wound.
- the inner region of adhesive is sufficiently sticky to affix the dry material layer to the occlusive backing, but yet have an adhesive capability after hydration which is less than the sheer strength of the fibrin clot.
- the inner region of adhesive is of a material which becomes solubilized or less sticky during hydration of the dry materials, permitting removal of the backing from the fibrin clot.
- the dry material layer may be affixed in the inner region directly to the occlusive bandage, with an adhesive layer added only to the outer layer.
- the backing of the fibrin sealant bandage remains in place affixed to the tissue surrounding the wound until the bandage is physically removed. But upon removal, the backing separates from the tissue sealing fibrin clot without disturbing the tissue seal.
- an independent hydrating layer comprising an effective amount of carbonated water or physiologically-acceptable buffered hydrating agent, such as PBS, or comparable gel, is contained within a rupturable, liquid-impermeable container.
- the rupturable, liquid-impermeable container encapsulating the hydrating layer is affixed directly to the above-described occlusive bandage layer or to the above-described adhesive layer adjacent to the occlusive bandage.
- Affixed to the exposed side (the side which is not attached to the backing or adhesive layer) of the rupturable, liquid-impermeable container encapsulating the hydrating layer is a dry layer of finely-ground, powdered fibrin components, as described above.
- the layer of dry components includes powdered fibrinogen or fibrinogen complex, thrombin, and as necessary sufficient calcium and/or Factor XIII to, upon hydration, form a fibrin clot.
- the dual layers are together covered on all surfaces not in contact with the occlusive backing or adhesive material affixing the layers to the occlusive backing, with an outer, protective, second impermeable membrane.
- the contents are entirely encapsulated within an impermeable container, wherein one side is the occlusive backing material and the other side and all edges are formed by the outer, protective, second impermeable membrane.
- the inner liquid-impermeable container encapsulating the hydrating layer is physically ruptured to release the hydrating material contained therein into the dry fibrin component layer, resulting in a fully-hydrated tissue sealing fibrin clot to inhibit blood and fluid loss from the wound, and to provide a natural barrier to infection.
- the outer, second impermeable membrane retains the released hydrating material in contact with the dry components until a malleable fibrin complex forms, at which time the outer membrane is physically removed and the bandage placed over the wound to form a tissue sealant.
- the outer membrane may be physically removed, and the dual layers forcefully applied to the wound area in a manner which ruptures the inner liquid-impermeable container and releases the hydrating agent into the dry fibrin components so that the tissue sealing fibrin clot is formed directly on the wounded tissue.
- the selected adhesives and backing materials may be determined by the intended application of the bandage.
- the backing may be removable or resorbable, and the adhesive may have the intended purpose upon removal of the bandage of removing the tissue sealant from the wound, or of leaving the tissue sealing fibrin clot undisturbed.
- the adhesive may be a separately bound layer, or the backing may itself act as an adhesive to affix the dry fibrin components.
- the thrombin, calcium and Factor XIII components which are necessary to form the fibrin complex may be affixed as dry material(s) in the dry material layer, or they may be included in liquid or gel form in the hydrating layer.
- fibrin sealant bandage may be divided between the two layers, so long as all of the necessary fibrin-forming components are present, and the dry layer remains non-hydrated until the bandage is used.
- additives such as the previously disclosed growth factors, antibiotics, antiseptics, antiproliferative drugs, etc. may also be included in this embodiment of the fibrin sealant bandage.
- the dry material layer will be hydrated as an expandable, foaming, fibrin tissue sealant.
- the dry material layer may be supplemented with materials which produce gas, and hence foaming, upon contact with the hydrating agent.
- the hydrating layer is in the form of a gel, such as a quick-evaporating gel layers (e.g., methylcellulose/alcohol/water), the rupture of the surrounding impermeable barrier permits the dry material fibrin components to directly contact the hydrating layer as disclosed above to produce the tissue sealing fibrin clot.
- the gel layer in the manner described for a liquid hydrating layer, may comprise any one, or all, of the thrombin, calcium or Factor XIII elements of the fibrin complex, and/or any one of the above-disclosed additives.
- the tissue sealant is delivered as a wound sealing dressing, which need not be affixed to a backing.
- the components are organized essentially as a capsule within a capsule, wherein the term capsule is used to define a broad concept, rather than a material.
- the above- described encapsulated hydrating layer is itself contained within a second encapsulating unit, which contains both the dry fibrin component materials and the encapsulated hydrating layer.
- the inner, liquid-impermeable container encapsulating the hydrating layer is physically ruptured to release the hydrating material contained therein into the dry fibrin component layer, both of which remain completely contained within the outer, second encapsulating unit.
- the integrity of the outer, second encapsulating unit is not broken when the inner container encapsulating the hydrating layer is physically ruptured.
- the mixing of the hydrating layer with the dry fibrin components within the outer encapsulating unit results in a fully-hydrated tissue sealing fibrin clot, which is then released or expelled onto wounded tissue to form a tissue seal.
- the outer encapsulating unit is physically cut or torn, either randomly or at a specific location on the surface, e.g., to form a pour spout to direct the flow of the malleable fibrin mass onto the wound site.
- the hydrating layer is a agent supersaturated with gas
- the mixing of the hydrating agent with the dry fibrin components results in an expandable foaming mixture, which is then applied to the wounded tissue.
- the foaming may, in the alternative, be achieved by hydration of the dry component layer.
- a self-foaming fibrin sealant dressing embodiment for treating wounded tissue in a patient is formulated as an expandable foam comprising a fibrin-forming effective amount, in combination, of (1) virally-inactivated, purified fibrinogen, (2) virally-inactivated, purified thrombin, and as necessary (3) calcium and/or Factor XIII; wherein said composition does not significantly inhibit full-thickness skin wound healing.
- the previously described TS components are stored in a canister or tank with a pressurized propellant, so that the components are delivered to the wound site as an expandable foam, which will within minutes form a fibrin seal.
- Acceptable formulations of the expandable foam embodiment provide the hydrated components of a fibrin clot, which in operation expand up to twenty-fold. The extent of expansion of the tissue sealing fibrin clot, however, is determined by its intended application.
- expandable foam fibrin sealant dressing within the abdomen provides a fibrin tissue sealant to significantly diminish or prevent blood or fluid loss from injured internal tissues organs or blood vessels, while also providing a barrier to infection.
- the expansion of the foam must be controlled to prevent harmful pressure on undamaged tissue, organs or blood vessels. Such a situation may warrant the use of an expandable foam dressing in which the expansion is limited to only 1 - or 2-fold, and not more than 5-10 fold.
- expandable foam fibrin sealant dressing may warrant the use of material which expands at a much greater rate to produce a tight and firm seal over the wounded area.
- Arterial wounds may also respond well to a highly pressurized foam tissue sealant dressing.
- the extent of the expansion of such material may be in the range of above 20-fold, although preferably 10-20 fold, or more preferably 5-10 fold.
- An expansion of less than 5-fold, including 1- to 2-fold may also be applicable to repair of blood vessels or injured bone, for example in small areas, such as the inner ear.
- the set-up time for the formation of the fibrin seal using the expandable foam fibrin dressing is also related to its intended application.
- loss of life may be imminent, such as in a patient who has suffered arterial wounds or damaged heart tissue.
- the fibrin dressing must expand very rapidly and form the fibrin tissue seal as quickly as possible, necessarily before exsanguination.
- the seal will set-up and significantly diminish the patient's fluid loss within 2 minutes or less, more preferably in 1-2 minutes, and most preferably in less than 1 minute.
- not all wounds are immediately life threatening.
- the strength of the tissue sealant repair of bony tissue is more important than a rapid set-up time.
- the composition of the tissue sealing fibrin clot may be modified to permit greater cross-linking or thickening of the fibrin fibrils, or to permit delivery of a more dilute composition which will continue to expand for a longer period of time.
- Such formulations may either permit or require a slightly longer time to set-up the tissue sealing fibrin clot. Although a set-up time of under 1 minute is appropriate for such applications, setup times of 1-2 minutes, or up to 5 minutes would be acceptable. In circumstances recognizable to one of ordinary skill in the art. a long set-up time of 5-10 minutes, or even up to twenty minutes, may be acceptable in non-life threatening situations.
- the delivery devices e.g., canister, tank, etc.
- the canister may comprise either a single or multiple reservoirs. Separate reservoirs, although more expensive, will advantageously permit the hydrated components to remain separated and stable until they are mixed upon application.
- the propellant must be physiologically acceptable, suitable for pharmacological applications, and may include conventionally recognized propellents, for example, CO 2 , N 2 , air or inert gas, such as freon, under pressure.
- dry fibrin components may be supplemented with material(s) which produce gas, and hence foaming, upon contact with the hydrating agent.
- delivery pressure of the expandable foam fibrin dressing from the delivery device when combined with the composition of the fibrin clot itself and its set-up time, determines the extent of expansion of the dressing, the delivery pressure is determined by the nature of the wound being treated. As described above, certain wounds require immediate formation of the tissue sealing fibrin clot to prevent loss of life, while others wounds require slow delivery or time to form extensive cross-links to strengthen the tissue sealing composition. Therefore, delivery pressure may ideally be situation specific.
- Pressure of 1 atmosphere, or less (14.7 lbs/inch 2 ) will provide a low level of expansion and a slower rate of delivery.
- certain life threatening situations may warrant a delivery pressure of 1 -5 atmospheres, or more.
- the delivery pressure chosen corresponds to that of commercially available canister devices.
- the delivery pressure may be important to keep the tissue sealant material from clogging delivery lines or devices.
- the wounds may be not only life-threatening but extensive, involving large, jagged openings in tissue or bone with significant internal damage, often with accompanying serious burns.
- Such wounds may present numerous severed arteries and blood vessels in addition to extensive areas of wounded tissue.
- Example 1 The following examples are included for illustrative purposes only and are not intended to limit the scope of the invention.
- Example 1
- HBGF-1 ⁇ and other bacterial proteins was loaded onto a 2.6 cm diameter by 10 cm high column of Heparin-SepharoseTM (Pharmacia Fine Chemicals, Upsala, Sweden). The column was washed with 5 column volumes of 0.15 M NaCl in 20 mM phosphate buffer, pH 7.3, and then was eluted with a 0.15 M NaCl in 20 mM phosphate buffer to 2.0 M NaCl gradient.
- the eluate was monitored by UV absorption at 280 nm. Three peaks of UV absorbing material eluted and were analyzed by SDS polyacrylamide gel electrophoresis. Peak number three electrophoresed as a single band at about 17,400 daltons and contained substantially pure HBGF-1 ⁇ .
- peak number three which contained the growth factor activity, was dialyzed overnight against 20 mM histidine, 0.15 M NaCl, pH 7.5.
- Two mg of protein was loaded onto a 1 ml CM-SepharoseTM (Pharmacia, Upsala. Sweden) ion exchange column. The column was washed with 10 bed volumes (0.5 ml/min) of 20 mM histidine, 0.15 M NaCl, pH 7.5 and eluted with a gradient of
- HBGF-1 ⁇ was identified by SDS polyacrylamide gel electrophoresis. This purified HBGF-1 was used to supplement FG in subsequent examples.
- HBGF-1 Stability of HBGF-1 It was necessary to add an ingredient to the FG that would inhibit or prevent the digestion of HBGF-1 ⁇ by thrombin (Lobb, Biochem. 27:2572-2578 (1988)), which is a component of FG. Heparin, which adsorbs to HBGF-1. was selected and tested to determine whether it could protect HBGF-1 from digestion by thrombin and any other proteolytic components of the FG. The stability of HBGF-1 in the presence of increasing concentrations of heparin was assessed.
- Example 3 After the incubation was complete, the samples were thawed and separated on 15% SDS polyacrylamide gels under reducing conditions according to the method of Laemmli (Nature 227:680 (1970)). The gel was then electroblotted onto nitrocellulose and the band corresponding to HBGF-1 was identified using an affinity-purified polyclonal rabbit antiserum to HBGF-1. The Western blots are shown in Fig. 1 on which the HBGF-1 ⁇ band at 17,400 mw can be seen. The results indicated that in the presence of concentrations of heparin as low as 5 U/ml, HBGF-1 ⁇ was protected from digestion by thrombin. In addition, as described in Example 3, its biological activity was not altered. Example 3
- HBGF-1 The biological activity of HBGF-1 in the incubation mixture that contained 5 U/ml of heparin, and was described in Example 2, was measured using an 3 H-thymidine incorporation assay with NIH 3T3 cells.
- NIH 3T3 cells were introduced into 96 well plates and were incubated at 37°C under starvation conditions in Dulbecco's Modified Medium (DMEM; GIBCO, Grand Island, New York) with 0.5% fetal bovine serum (BCS; GIBCO, Grand Island, New York) until the cells reached 30 to 50% confluence. Two days later, varying dilutions of HBGF-1 from the samples prepared in Example 2 were added to each well without changing the medium. Diluent (incubation buffer) was added in place of growth factor for the negative controls and DMEM with 10% BCS, which contains growth factors needed for growth, was added in place of the HBGF-1 sample for the positive controls.
- DMEM Dulbecco's Modified Medium
- BCS fetal bovine serum
- HBGF-1 biological activity in HBGF-1 in the presence of thrombin and heparin was also measured by observing endothelial cell proliferation.
- the surfaces of petri dishes were impregnated with the HBGF-1 supplemented FG.
- a shallow layer of endothelial cells was added and the number of cells was measured. Over time the number of cells increased. In addition, the cells appeared to be organizing into vessels.
- HBGF-1 retains its biological activities in FG that includes heparin, which protects HBGF-1 from the degradative activity of thrombin and may also potentiate the biological activity of the HBGF-1 in the growth factor-supplemented FG.
- HBGF-1 Diffusion from a FG Clot A FG clot was formed in a 5 ml plastic test tube by mixing 0.3 ml of the fibrinogen complex containing 10 U/ml heparin and thrombin and 40 mM CaCl 2 .
- Four test tubes were set up as follows:
- Each clot was covered with 0.2 M histidine buffer, pH 7.3. Thirty ⁇ l samples of the overlying buffer were removed from each tube every two hours and were run on a western blot.
- FGF-1 acidic fibroblast growth factor
- the cells seeded onto the FG layer were maintained for 7 days in DMEM containing 10% fetal bovine serum (FBS).
- FBS fetal bovine serum
- human umbilical endothelial cells 10 5 or more cells per ml, were embedded in FG, the protein concentration of which was 4 mg/ml.
- the concentration of thrombin in the FG was adjusted to 0.6 NIH U/ml.
- the culture medium used in all of the experiments was Ml 99 (Sigma Chemical Co., St. Louis, MO) supplemented with 10% fetal bovine serum, 10 ⁇ g/ml streptomycin, 100 U/ml penicillin, 1 ng/ml FGF-1 and 10 U/ml heparin.
- Control cells As a control, an identical cell suspension was cultured on a surface coated with fibronectin at 10 ⁇ g/cm 2 . Control cells acquired a cobblestone shape and maintained this morphology for at least 5 days. Figures 9 and 10 show this situation at 24 and 48 hours, respectively.
- PMEXNEO-3T3-2.2 cells are fibroblast cells that contain a modified genome with the potential to express genetically engineered proteins (Forough et al., J. Biol Chem. 268:2960-2968 (1993)). To determine the behavior of these cells in FG, 10 5 cells per well were cultured under three conditions: (1) embedded in FG; (2) on the surface of FG; and (3) in the absence of FG (controls). The experiments were carried out in duplicate in 24-well plates in
- DMEM media (Sigma Chemical Co., St. Louis, MO) supplemented with 10% FBS.
- the FG protein concentration was 4 mg/ml.
- the medium was supplemented with 1.5% FBS was used as negative controls.
- the first study examined the in vivo washout characteristics of HBGF-1-supplemented FG suspension applied to expanded PTFE grafts implanted into rabbit aortas.
- Similar grafts were implanted into the aortaileac position in dogs.
- HBGF-1 an angiogenic factor, was used in studies.
- Other growth factors such as a FGF, FGF-4 and/or OP-1 can also be used as a supplement (s) for the vascular grafts.
- the modified FG was sterilely prepared by adding approximately 1 ng/cm 2 area of the inner and outer graft surfaces of human recombinant I25 I-HBGF-1, 20 ⁇ g/cm 2 porcine intestinal mucosal heparin, and 2.86 mg/cm 2 fibrinogen to 2.86 ⁇ 10 -2 U/cm 2 reconstituted, commercially available, human thrombin (1000 U/ml) to induce polymerization.
- the 125 I-HBGF-1 was specifically prepared as follows. Fibrinogen was reconstituted by adding 500 mg of fibrinogen into 25 ml of PBS to produce a fibrinogen concentration of 20 mg/ml of PBS. Three ml of this solution which contained 60 mg fibrinogen were placed into 12 Eppendorf plastic tubes and maintained at -70°C. Each of these aliquots was used individually.
- the thrombin was reconstituted by diluting a commercially available preparation thereof (Armour Pharmaceutical Co., Kankakee, IL) at a concentration of 1000 U/ml by a factor of 1 :10 in sterile solution to produce a concentration of 100 U/ml. This thrombin solution was again diluted 1 :10 to produce a solution of 10 U/ml.
- the bovine heparin (Upjohn, Kalamazoo, MI) was reconstituted by diluting the preparation at a concentration of 1000 U/ml by a factor of 1 :1000 using normal saline.
- One end of the expanded PTFE graft was placed over a plastic 3-way stopcock nozzle and was secured there with a 2-0 silk tie.
- the PTFE was then encircled with a 3 ⁇ 3 cm square of ParafilmTM which was then crimped there with a straight hemostat to establish a watertight seal.
- a second 2-0 silk tie was positioned over the parafilm adjacent to the stopcock to form another seal.
- a straight hemostat was then used to clamp the distal 2 mm of the PTFE/parafilm to seal this end.
- Equal volumes of fibrinogen and thrombin solution prepared as described above were mixed and allowed to react for approximately 30 seconds which is when polymerization occurs. The thrombin-polymerized fibrin is then opaque.
- the fibrin/thrombin mixture was aspirated into a one cc syringe.
- NOTE The volume of this graft was 0.42 ml.
- the syringe was attached to the stopcock and the mixture was injected by hand over a period of 5 seconds until the liquid was seen to "sweat" through the PTFE interstices and filled the space between the PTFE and the ParafilmTM.
- the 3-way stopcock was closed to the PTFE graft for 3 minutes and a scalpel blade was used to cut the ligature at the end of the PTFE over the stopcock.
- the PTFE graft/parafilm was removed from the stopcock and a hemostat was used to remove the PTFE from the parafilm envelope.
- a number 3 embolectomy catheter was passed through the graft five times until the graft lumen was completely clear.
- the growth factor-supplemented FG-treated PTFE graft was allowed to dry overnight for about 12 hours under a laminar flow hood. The treated graft was then ready for implantation.
- this HBGF-supplemented FG was pressure perfused into a 34 mm (24 mm + 5 mm at each end) ⁇ 4 mm (internal diameter) thin-walled, expanded PTFE graft thereby coating the graft's luminal surface and extending through the nodes to the graft's outer surfaces.
- the lumen of the graft was cleared as stated above.
- These grafts were then interposed into the infrarenal abdominal aortas of 24, 3-5 Kg New Zealand white rabbits. In the first study, the animals were sacrificed and specimens were explanted at 0 time (to correct for losses due to surgical manipulation) and after 5, 30, and 60 min, and 1, 7, 14, and 30 days.
- Residual radioactivity was determined by gamma counting. Remaining 125 I-HBGF-1, corrected for spontaneous decay, is expressed as a percentage of the zero time value.
- the second study evaluated the effects of the applied HBGF-1-supplemented FG suspension on: the rate of endothelialization of widely expanded 60 ⁇ m internodal distance expanded PTFE grafts implanted into canine aorta-iliac positions; the proliferative activity of these endothelial cells as a function of time; and the relative contributions of the HBGF-1 and the FG in stimulating the observed endothelial cell proliferation.
- Three groups of 50 ⁇ 4 mm non-reinforced expanded PTFE grafts were implanted in the aortailiac position of 12 dogs.
- Plasma reduced platelets were prepared and pelleted. The supernatant plasma was removed. The pelleted platelets were washed, suspended in buffer containing 50 mM histidine and 0.15 M sodium chloride at pH 6.5. and treated with bovine thrombin. After treatment, the supernatant was collected by centrifugation and aliquots were frozen at -80°C. The extract was thawed and mixed with FG or other TSs. The platelet extract obtained in this manner was biologically active since it increased the incorporation of radioactive labeled thymidine into the DNA of proliferating NIH3T3 fibroblasts compared to the controls.
- Example 10 To evaluate the effect of platelet extract on wound healing, experiments identical to those carried out below in Example 10 with HBGF-1 ⁇ were carried out with platelet extract in diabetic mice. From the results of these experiments is clear that, given the low concentration of growth factors in the platelet extract, a dose larger than 100 ⁇ g of platelet extract protein per wound needs to be used to promote wound healing.
- mice Female C57BL/K s J-db/db mice were obtained from Jackson Laboratories (Bar Harbor, ME) and were 8 to 12 weeks old at the start of the experiment. They were housed in separate cages after surgery in an animal care facility.
- mice are used as a model of impaired wound healing in diabetic humans because the metabolic abnormalities seen in these mice are similar to those found in human diabetics.
- the healing impairment characterized by markedly delayed cellular infiltration, granulation tissue formation, and time required for wound closure suggest that healing in this mouse model may be relevant to the healing impairment seen in human diabetes.
- the concentrated topical fibrinogen complex (TFC) used in this study was produced from fresh frozen pooled human plasma.
- the TFC product (American Red Cross—Baxter Hyland Division, Los Angeles, CA) was supplied in lyophilized form. After reconstitution with 3.3 ml of sterile water, the protein characteristics of the TFC solution used in this study were: total protein, 120 mg/ml; fibrinogen, 90 mg/ml; fibronectin, 13.5 mg/ml; Factor XIII, 17 U/ml; and plasminogen, 2.2 ⁇ g/ml.
- Topical bovine thrombin (5000-unit vial, Armour Pharmaceutical Co.,
- Kankakee, IL was reconstituted with 5 ml sterile water and was serially diluted in 80 mM calcium chloride solution (American Reagent Laboratories, Shirley, NY) to a concentration of 15 U/ml.
- TFC was mixed with 0.015 ml of thrombin.
- the FG that was produced had a protein concentration of approximately 60 mg/ml.
- mice were anesthetized with a mixture consisting of 7 ml ketamine hydrochloride (100 mg/ml; Ketaset, Aveco Co., Inc., Fort Dodge, IA), 3 ml xylazine (20 mg/ml; Rompun, Mobey Corp., Shawnee, KA), and 20 ml physiological saline, at a dose of 0.1 ml per 100 g body wt, administered intramuscularly.
- the dorsal hair was clipped, and the skin was washed with povidone-iodine solution and wiped with 70% alcohol solution.
- Two full-thickness, round surgical wounds (6 mm diameter) were made on the lower back of the mouse, one on each side, equidistant from the midline.
- the medial edges of the two wounds were separated by a margin of at least 1.5 cm of unwounded skin.
- the dressing was a transparent semipermeable adhesive polyurethane dressing (OpsiteTM, Smith and Nephew, Massillon, OH). Tincture of Benzoin compound (Paddock Laboratories, Minneapolis, MN) was applied at the periphery of the wound area prior to application of the dressing. There was a margin of at least 0.5 cm of skin surrounding the wound edge over which no tincture of benzoin was applied to avoid the possible inflammatory effects of benzoin on the raw wound. No further treatments were applied to the wound for the duration of the experiment.
- mice were divided into 4 treatment groups, with each mouse serving as its own control:
- Group I The wound on one side of the animal was treated with FG (60 mg/ml) while the contralateral wound received no treatment. Both wounds were covered with OpsiteTM.
- Group II Diluted FG (1.0 mg/ml) was topically applied to the wound on one side while the contralateral wound received no treatment. Both wounds were covered with OpsiteTM.
- Group III FG (60 mg/ml) was topically applied over both wounds. The wound on one side was left uncovered while the contralateral wound was covered with OpsiteTM.
- Group IV No topical treatment was applied over the wounds. The wound on one side of the animal was left uncovered while the wound on the contralateral side was covered with OpsiteTM.
- the animals were euthanized on Day 9 of the experiment.
- the wounds were excised down to the muscle layer, including a margin of 0.5 mm of unwounded skin, and were placed in buffered 10% formalin solution.
- the specimens were submitted to a histology laboratory for processing. Specimens were embedded in paraffin, and the midportion of the wound was cut in 5- ⁇ m sections.
- the slides were stained with hematoxylin and eosin, or with Masson's trichrome for histologic analysis. Each slide was given a histological score ranging from 1 to 15, with 1 corresponding to no healing and 15 corresponding to a scar with organized collagen fibers (Table 1).
- the scoring scale was based on scales used by previous investigators. The criteria used previously were modified and were further defined to more precisely reflect the extent of: reepithelialization, degree of cellular invasion, granulation tissue formation, collagen deposition, vascularity, and wound contraction.
- the histologic score was assigned
- the paired t test was used for comparison of paired means in the different treatment groups.
- the analyses were performed using the RS/1 Release 3.0 statistical software package (BBN Software Products Corporation).
- mice (1) when applied over open wounds, FG at a concentration formulated for hemostasis (60 mg/ml) resulted in lower histological scores at Day 9 which indicated slower rates of wound healing compared to that of untreated wounds; (2) dilution of the FG protein concentration to 1 mg/ml resulted in a higher histological score at Day 9 which indicated a faster rate of wound healing; and (3) application of a semipermeable dressing (OpsiteTM) per se significantly retarded wound closure in this animal model by itself.
- FG a concentration formulated for hemostasis
- the total protein concentration of FG is an important variable when comparing the results of studies using FG. Beneficial effects of fibrin in promoting wound healing and tissue repair have been reported, but lower concentrations of fibrinogen have been used in the present studies than is commonly found in commercial preparations.
- FG at a concentration of 60 mg/ml delayed wound closure (Group I).
- HBGF- IB growth factor-supplemented FG The effect of HBGF- IB growth factor-supplemented FG on the rate of wound repair in diabetic mice was assessed.
- the methods used in this experiment were similar to those just described above.
- Two 6 mm full-thickness skin biopsies on the dorsal part of each of 6 test mice were filled with FG to which 5 ⁇ g of HBGF-1 ⁇ had been added.
- Identical biopsies in six mice were left untreated, and in six control mice were filled with unsupplemented FG. After 9 days, all of the mice were sacrificed and histological preparations of 5 micron thick slices from each of the wounds and surrounding skin were prepared and stained with hematoxylin and eosin.
- TFC Concentrated human TFC
- human thrombin Boxter Hyland Division, Glendale, CA
- solvent detergent method New York Blood Center
- Human thrombin 1000 U vial was reconstituted with 3.3 ml sterile water, and was serially diluted in 40 mM calcium chloride solution (American
- Topical bovine thrombin (5000 U vial, Armour Pharmaceutical Co., Kankakee, IL) was reconstituted with 5 ml sterile water, and was serially diluted in 40 mM calcium chloride solution to a concentration of 15 U/ml. Bovine thrombin was used for preparing implants for intramuscular bioassay.
- the fibrinogen should be present at a concentration of 1 to 120 mg/ml FG, more preferably from 3 to 60 mg/ml FG, most preferably from 10 to 30 mg/ml FG.
- DBM should be present at an approximate concentration of about 1 to 1000 mg/ml FG, more preferably from 50 to 500 mg/ml FG, most preferably from 300-500 mg/ml FG.
- the particle size of demineralized bone powder should be from 0.01 to 1000 microns, preferably from 20-500 microns and most preferably from 70-250 microns.
- the osteoinductive growth factor(s) or BMPs should be present at a concentration(s) of about 1 to 100 ⁇ g/ml wherein the concentration(s) is effective to accomplish its desired purpose.
- Growth factors which may be used as osteoinductive substances in this embodiment include, but are not limited to: osteogenin (BMP3); BMP-2; OP-1; HBGF-1; HBGF-2; BMP 2A, 2B and 7; FGF-1; FGF-4; and TGF- ⁇ .
- drugs such as antibiotics, can be used to supplement the TS for use in bone repair.
- Rat DBM was prepared as follows. The epiphyses of the long bones of rats were removed leaving only the diaphyses behind. The diaphyses were split, if necessary, and the bone marrow was then thoroughly flushed with deionized water (Milli-Q Water Purification SystemTM, Millipore Corporation, Bedford, MA). The diaphyses were then washed at room temperature. At 4 °C, 1000 mis of deionized water was added to 100 g of bone. The mixture was stirred for 30 minutes and the water was decanted. This step was repeated for two hours.
- deionized water Milli-Q Water Purification SystemTM, Millipore Corporation, Bedford, MA
- the bone was then milled to make bone powder.
- the powder was sieved and 74 to 420 micron size particles were collected.
- Ten gram aliquots of the bone powder were placed in 250 ml centrifuge bottles. Eighty mis of 0.5 N HCl was added to each bottle slowly in order to avoid frothing. The contents of each bottle were then stirred gently. After 15 minutes, an additional 100 mis more of 0.5 N HCl was added to each bottle over the course of 10 minutes. The bottles were then stirred gently for an additional
- the pellets were then washed with 180 mis of deionized water by stirring to produce an even suspension.
- the suspension was then centrifuged for an additional 15 minutes. The supernatant was then decanted as before. The washing was repeated until the pH of the supernatant equaled the pH of the deionized water.
- pellets were then frozen at -180°C in a freezer. They were then lyophilized using standard procedures.
- Disk-shaped implants 1 mm thick and 8 mm in diameter were produced using a 4-piece aluminum mold (Figure 13). Twenty-five mg of rat DBM powder was added into the mold chamber. Thirty ⁇ l of TFC was then pipetted onto the DBM and mixed until the DBM had absorbed all of the solution. The concentrations of TFC which were used were 10, 20, 40, 80, or 120 mg/ml. Thirty ⁇ l of thrombin solution (15 U/ml in 40 mM calcium chloride solution) was then added to the DBM-TFC complex, was mixed, and was compressed into a disk-shape using a piston-shaped lid. It was determined that 25 mg of DBM powder had a volume of 20 ⁇ l. After DBM had been added to the FG, the final protein concentrations were as follows:
- DBM-TFC complex Fifty mg was poured into an aluminum mold, to which 60 ⁇ l of TFC was then added to the DBM and mixed until fully absorbed. Sixty ⁇ l of thrombin was then added to the DBM-TFC complex, mixed and compressed into a disk-shape with a diameter of 1 cm and a thickness of 2 mm using a piston-shaped lid. The disk was then cut manually into the desired shape (triangle, square or donut).
- implants were placed in a sterile nylon bag having a mesh size of 70 microns and measuring 1 cm ⁇ 1 cm.
- the animals were anesthetized with a mixture consisting of 10 ml ketamine hydrochloride (Vetalar, 100 mg/ml, Parke-Davis, Morris Plains, NJ), 5 ml xylazine (Rompun, 20 mg/ml, Mobay Corporation, Shawnee, KN), and 1 ml physiologic saline (0.9% NaCl), at a dose of 0.1 ml per 100 gm body weight, administered intramuscularly.
- the operative site of the animal was prepped with 70% alcohol solution, followed by povidone-iodine solution. The surgical procedure was then performed using aseptic technique.
- Intramuscular Bioassay A midline ventral incision was made and a space was created between the pectoralis muscles with blunt dissection. A nylon envelope containing the designated experimental material was inserted into the intramuscular space and secured with a 3-0 Dexon suture (Figure 14). The same procedure was then repeated at the contralateral side. The skin was then closed with staples. The implants were harvested after four weeks, were x-rayed and were prepared for histology.
- each rat was identified by ear punches and returned to its cage where they were ambulatory within 2-3 hours.
- the rats were euthanized in a carbon dioxide chamber.
- a skin incision was made around the experimental recipient bed (i.e., pectoralis major or calvaria) and the soft tissues were reflected from the recipient beds.
- the craniotomies with 3-4 mm contiguous bone were recovered from the fronto-occipito-parietal complex.
- sharp and blunt dissection was used to recover the implanted nylon envelopes.
- the implants were radiographed using X-OMATLTM high contrast Kodak x-ray film (Eastman Kodak Company, Rochester, NY) in a Minishot Benchtop
- Radio-opacity measurements of some DBM disks were higher than DBM-FG disks but the other measurements were well within the range of measurements for DBM-FG disks. Thirty out of 32 FG disks which were not supplemented with DBM (93.75%) did not develop radio-opacity.
- DBM-FG disks in the form of squares, triangles or donuts were also markedly radio-opaque as compared to FG disks which were not supplemented with DBM.
- the original shapes of the implants were generally retained.
- the intramuscular bioassay was positive for DBM and DBM-FG implants, as evidenced by formation of ossicles with a central cavity filled with marrow and resorption of previously implanted DBM particles.
- Non-treated 8 mm craniotomy wounds showed only fibrous connective tissue developing across the craniotomy wound (Figure 18). Histology of DBM implants showed DBM particles to be scattered all over the field. Some DBM particles migrated over and under the edges of host bone ( Figure 19). Most DBM particles were, however, within the confines of the craniotomy wound and were surrounded by loose connective tissue that was well vascularized. Active resorption of DBM by osteoclasts was noted. A lot of DBM particles were also noted to be populated by live cells. New osteoid and bone laid down by osteoblasts were quite evident.
- FG The natural biocompatibility and biodegradability of FG are characteristics that make it an ideal delivery vehicle for DBM and BMPs. FG facilitated the shaping of DBM into the desired form to fill bony defects, maintained DBM within the defect, and may have been synergistic with DBM. Furthermore, soft tissue prolapse did not occur and bony contour was maintained. DBM-supplemented FG possessed an appropriate microarchitecture, biodegradation profile and release kinetics to support osteoblast recruitment and osteoregeneration.
- the DBM-FG matrix Since the shape of the DBM-FG matrix determined the morphology of the newly formed bone, when possible, the DBM-FG matrix should be made of a predetermined shape. However, the DBM-FG matrix in liquid form can be delivered or injected into an irregularly shaped defect where it will polymerize and encourage bone formation in the DBM-FG-filled area.
- TFC human topical fibrinogen concentrate
- Freeze-dried thrombin concentrate supplied by The American Red Cross/Baxter-Hyland, Inc., Glendale, CA, was reconstituted with 3.5 ml of a 40 mM solution of calcium chloride prepared in water for injection. The resulting solution contained approximately 250 U/ml.
- TET-FG was formulated by mixing the desired weight of TET with 1 ml of reconstituted TFC solution and with 1 ml of reconstituted thrombin solution in the presence of injection quality calcium chloride (purchased from American
- the TET was in the free base form and was purchased from Sigma Chemical Company (St. Louis, MO).
- the TET-FG was formed by mixing TFC and thrombin through a DuofloTM dispenser (Hamaedics, CA) onto a Millipore membrane in a 12 mm diameter Millipore culture plate (Millipore Corporation, Bedford, MA). The mixture was allowed to set for one hour at 22°C. Six mm diameter disks containing the TET-FG and the Millipore membrane were cut from the latter using a 6 mm punch biopsy. The TET-FG-containing disks were used for the TET release studies.
- TET phosphate buffered saline
- saliva was measured using 24-well cell culture plates (Coming Glass Works, Corning, NY) under two different sets of conditions. In one condition, the static mode, 2 ml of PBS or 0.75 ml of saliva was replaced daily in the 24-well cell culture plates. In the other condition, the continuous exchange mode,
- TET release from the TET-FG was measured with PBS having been exchanged at a rate of approximately 3 ml per day.
- the samples were stored at -20°C until analyzed.
- the saliva had been collected from 10 different people, had been pooled, and clarified by centrifugation at 5000g. It was then filtered through a 0.45 ⁇ m pore sized membrane and was stored at 4°C for daily use.
- the eluted TET was thawed and was analyzed spectrophotometrically at 320 nm and/or biologically by the inhibition of E. coli growth on agar plates.
- Ciprofloxacin HCl CIP
- Amoxicillin AMO
- Metronidazole MET
- FG containing CIP HCl, AMO or MET were prepared as before for TET.
- the AB-FG disks were placed in individual wells in a 24-well cell culture plate and were covered with 2 ml of PBS that was collected, replaced daily and stored at -20°C as before, until analyzed.
- the concentrations of CIP, AMO and MET in the eluates were measured spectrophotometrically at 275, 274 and 320 nm, respectively, and were compared to standard curves containing 0 to 50 ug/ml of the corresponding AB.
- the maintenance of the structural integrity of the FG and the TET-FG disks was estimated by visual observation and physical inspection by "poking" the disks with a fine spatula.
- the porous membrane which had been cut out while making the disks remained attached to the TET-FG and was used to help position the disks during the evaluation of their structural integrity. Pictures of top and lateral views of the disks were also taken and were used in the evaluation.
- the structural integrity of FG and TET-FG were measured under both sterile and non-sterile conditions.
- the PBS and saliva were stored frozen until analyzed.
- the same procedure was used except that the entire process was run under sterile conditions.
- the sterility of the system was tested by incubating 0.2 ml of sample and 2 ml of broth at 37°C and the turbidity of the broth was monitored for 48 hours. Lack of turbidity indicated sterility of the system.
- the stability of the CIP-, AMO-, and MET-FG were studied as above but under non-sterile conditions only.
- the TET- and CIP-induced FG stabilization can be exploited for controlling the total release time not only for these ABs, but also for other drugs or "supplements" added to FG whose release rate and/or total release duration depends on the integrity of the FG matrix.
- CIP- induced FG stabilization can be exploited for controlling the total release time of TET, CIP and other drugs or supplements which have been added to the TET-FG or CIP-FG matrices.
- FG was supplemented with 50 mg/ml of TET free base and was shaped as 6 ⁇ 2.5 mm disks for this study.
- the protein concentration of FG was adjusted to 60 mg/ml.
- the disks were placed in 2 ml of PBS, pH 7.3 and were allowed to stand at 4, 23 and 37°C. To wash the disks, the PBS was replaced even
- FG was adjusted to 60, 30 and 15 mg/ml. Each disk was placed in 3 ml of distilled water. The water was replaced with the same volume of water every 10 minutes for a total of one hour. The TET concentration in the collected samples was determined spectrophotometrically against a standard curve as before.
- the TET release rate was inversely proportional to the FG protein concentration.
- TET-FG and CIP-FG protected mice from death caused by S. aureaus 202A for at least 48 hours after the administration of the AB-supplemented FG.
- the fibrinogen was solubilized with sterile water or, for one group with water saturated with 5-FU at a concentration of 17 mg/ml.
- Thrombin solutions were made with sterile water, and then were diluted in 40 mM CaCl 2 to a concentration of 15 U/ml, or Thrombin was dissolved in 40 mM CaCl 2 saturated with 5-FU in a concentration of 17 mg/ml.
- Control FG clots did not contain 5-FU and were produced by mixing 200 ⁇ l of TFC solution (at 60 mg/ml) with 200 ⁇ l of Thrombin solution (at 15 U/ml) and allowing 20 minutes to polymerize. These clots were made in 12 by 75 mm test tubes and then were placed in 10 mis of 0.05 M Histidine, 0.15 M NaCl, pH 7.3 (Buffer).
- FG clots containing saturated levels of liquid 5-FU were produced by mixing 200 ⁇ l of TFC (60 mg/ml + 17 mg/ml 5-FU) with 200 ⁇ l Thrombin solution (15 U/ml + 17 mg/ml 5-FU) and allowing 20 minutes for the clots to fully polymerize.
- the addition of saturated levels of 5-FU in both the TFC and Thrombin solutions somewhat altered clot formation producing a clot that was translucent, as compared to the control FG clots which were quite opaque.
- the clots that were formed were physically the same as those made with FG alone except in color. Clots were formed in 12 by 75 mm test tubes and then placed in
- a second group of FG clots were made that contained an amount of solid anhydrous 5-FU equal to the amount included in clots formed with saturated solutions of 5-FU. These clots were formed by the addition of 7 mg of solid anhydrous 5-FU to 200 ⁇ l of TFC (60 mg/ml) and 200 ⁇ l of Thrombin (15 U/ml).
- the final group contained 50 mg of solid anhydrous 5-FU per clot. Due to the increased mass of 5-FU (50 mg instead of 7 mg) the previously used method did not work. Instead of producing a homogenous clot, a clot was formed with the majority of the 5-FU having settled to the bottom of the test tube. To avoid this problem the bottom of the test tube was first coated with 100 ⁇ l of TFC
- the final total protein concentration of the FG in all groups was 30 mg/ml. Each group contained 10 replicates. Each duplicate was incubated at 37°C in 10 mis of buffer. Buffer was exchanged for 10 mis of fresh solution at 5, 10, 22, 33, 52, 75 and 114 hours. Aliquots of the eluate buffer were then examined in a spectrophotometer at a wavelength setting of 260 nm. Previous experiments had demonstrated that 5-FU absorbed strongly at this wavelength, while eluates from control FG clots did not.
- 5-FU-FG mixture can also be formulated into an injectable form (data not shown). It would further be expected that the use of an analog or other form of 5-FU that was less soluble in the surrounding aqueous medium than the anhydrous form, and/or had a slower dissolution rate, would result in a further increase in delivery times.
- the result of this process is a sustainable delivery of the antiproliferative/cytotoxic drug 5-FU from fibrin clots for at least 10 times longer than is possible using the drug in the aqueous form.
- This technology i.e., the use of a solid form of the drug, preferably one with a low solubility and/or dissolution rate
- DMEM Dulbecco's Modified Eagle's Medium
- Antibiotic-Antimycotic solution was purchased from GIBCO (Grand Island, N.Y.).
- FGF-1 and -4 FGF-4 were a kind gift of Reginald Kidd, Plasma
- FGF-2 also known as basic FGF or bFGF
- bFGF basic FGF
- Millicell-PCF (12.0 ⁇ m) inserts were purchased from Millipore, Inc. (Bedford, MA). Heparin was obtained from the UpJohn Company (Kalamazoo, MI).
- NIH/3T3 fibroblasts at passage 126 were purchased from the American
- Type Culture Collection Rockville, MD. Cultures from passages 129-133 were used in the chemotaxis assays. Cultures were propagated in DMEM supplemented with 10% Calf serum and approximately 1 % antibiotic antimycotic solution. Human dermal fibroblasts (HDFs) were purchased from Clonetics, Inc. (San Diego, CA) at passage 2. Cultures from passages 3-5 were used in the chemotaxis assays. Cultures were cultivated in DMEM supplemented with 20% FBS (Hyclone Laboratories, Inc., Logan, UT) and approximately 1% antibiotic antimycotic solution (Gibco, Grand Island, N.Y.).
- FBS Hyclone Laboratories, Inc., Logan, UT
- the procedure used to determine cellular chemotaxis was a combination of two known methodologies.
- a modification of Boyden's chamber was used as follows: Millicell-PCF (Millipore, Inc., Bedford, MA) (12.0 ⁇ m) 12.0 mm diameter inserts were placed in individual wells of 24 well plates to create the upper and lower chemotaxis chambers. Chemotaxis results were arrived at by performing checkerboard analysis for every combination of cells and growth factors. Concentrations ranging from .1, 1, 10, 100 ng/ml with/without added heparin (10 U/ml) were used for FGF-1 , FGF-2 (no heparin) and FGF-4 with all the cell types mentioned in the materials section.
- Chemotaxis chambers and cells were utilized as described above. Fifty ⁇ l of 8 mg/ml Topical Fibrinogen Complex (TFC, American Red Cross, Rockville, MD) was added to the bottom of 24 well plates. Forty ⁇ l of test growth factor +/- heparin at a final concentration of 10 U/ml (FGF-1, FGF-4 with heparin, FGF-2 alone) was added to the TFC and thoroughly mixed. Ten ⁇ l of bovine Thrombin (Armour Pharmaceutical Company, Kankakee, IL) was added and mixed thoroughly. The components were allowed to gel at room temperature for approximately 30 minutes. Total volume in the lower
- the assay was performed at 37°C in a 5%CO 2 humidified chamber for approximately 24 hours. At the end of 24 hours, the filters were removed, fixed and stained and the number of cells on the underside of the filter was enumerated as described above.
- NIH 3T3 fibroblasts The ability of NIH 3T3 fibroblasts to migrate towards various well known chemotactic agents was determined to ensure that the cells used in this assay retained this capacity.
- Fibronectin was the most effective chemotactic agent tested for both NIH 3T3 and HDFs with maximal responses occurring at 20 ⁇ g/ml ( Figure 31 , Table 7). Thereafter, fibronectin at 20 ⁇ g/ml was used as the positive control for migration.
- the FGFs produced a profound chemotactic response in HDFs.
- a very good distinction was obtained between the negative control and the concentration of FGF which elicited a maximal migratory response: 18, 12 and 10 fold in response to FGF-1, -2 and -4, respectively.
- the stimulation of chemotaxis by growth factors was not as high for NIH 3T3 cells as it was for HDFs, possibly due to the high passage number of the available stock cultures of the NIH 3T3 cells as compared to the HDFs.
- FGF-1, FGF-2 and FGF-4 were found to be potent stimulators of fibroblast chemotaxis. Directed migration of fibroblasts by one or a combination of the above growth factors could result in fibroblast presence in the site of injury, thereby leading to fibroplasia and the laying down of collagen and an extracellular matrix. Thus, aside from it's well recognized angiogenic properties, FGF's may have a role in wound healing, acting either alone or in a combination with PDGF, IGF-I, TGF- ⁇ and/or other factors.
- the present invention of incorporating FGFs into FG allows for the prolonged exposure of cells to the FGFs and can be applied to a wound.
- the resulting fibrin coating mimics the natural response to tissue injury, while delivering the growth factor directly to the wound site.
- FG which contained FGF-1 was used to line artificial vascular grafts (Example 8, herein). When these grafts were placed into the vessels of rabbits, the FGF-1 was released for a period of up to 28 days.
- the effect of the incorporation of FGF-1 into the graft walls was the total endothelialization of the artificial grafts within the same period (Greisler et al.
- the TS contains and delivers angiogenic substances, such as Fibroblast Growth Factor- 1 (FGF-1), in an amount such that its concentration which is released from the supplemented TS is effective to induce angiogenesis.
- FGF-1 Fibroblast Growth Factor- 1
- This embodiment is used in a controlled manner to revascularize body areas which have been deprived of an adequate blood supply such as cardiac, brain and muscle tissue, and the retina.
- This embodiment is used to restore or improve circulation to implanted organs or re-attached limbs.
- This embodiment can be used to generate a vascular network or "vascular bed" for: the generation of artificial organs or organoids, the delivery and/or localization of and/or nourishment of cells used in gene therapy, or as a target of gene therapy, for the nourishment and/or localization of cells for tissue augmentation.
- This embodiment also precludes the necessity of implantation of a device or substance which may induce a foreign body or other excessive inflammatory reaction which could compromise the blood vessel formation or the function of the underlying organ(s).
- the invention consists of a formulation of fibrinogen, (suitable for the formation of fibrin) with or without fibronectin and/or collagen, into which is placed an appropriate concentration of an angiogenic substance, such as FGF-1.
- the fibrinogen may also contain stabilizers to protect against the proteolytic activity of Thrombin.
- heparin sulfate (1-1000 U/ml) may be used as the stabilizer in the range of concentration of from 1 ng/ml to 1 mg/ml.
- the angiogenic substance is contained, in an appropriate concentration, in the thrombin, calcium, or water components.
- the concentration of the FGF-1 in the TS should be from 0.1 ng/ml to 1 mg/ml, more preferably from 1 ng/ml to 100 ⁇ g/ml, most preferably from 100 ng/ml to 10 ⁇ g/ml.
- the FGF-1, or other angiogenic substance will induce blood vessel formation within the body of the deposited FG.
- the FG will be naturally biodegraded leaving the intact blood vessel(s).
- the TS contains and delivers a cartilage promoting factor(s), such as cartilage-inducing factors-A and/or -B (CIF-A and CIF-B, respectively, which are also known as TGF-B, and TGF-B, respectively) and/or another, factor(s) such as Osteoid-Inducing Factor (OIF) in an amount such that the concentration of the inducing factor(s) which is released from the supplemented TS is effective to induce cartilage formation.
- cartilage promoting factor(s) such as cartilage-inducing factors-A and/or -B (CIF-A and CIF-B, respectively, which are also known as TGF-B, and TGF-B, respectively
- another factor(s) such as Osteoid-Inducing Factor (OIF)
- the concentration of the inducing factors should be 0.1 ng/ml to 1 mg/ml, more preferably from 1 ng/ml to 500 ng/ml, most preferably from 100 to 250 ng/ml.
- This embodiment may also contain drugs, such as antibiotics, and other growth factors, such as EGF, PDGF, and bFGF in the TS.
- the cartilage inducing substance is contained in an appropriate concentration in the fibrinogen or thrombin or calcium or water component(s) which are used to prepare the TS.
- the supplemented TS can either be pre-shaped to the desired final cartilage form prior to implantation or it can be implanted into the body of the recipient in the liquid form as the TS is mixed and polymerizes. The resulting form may then be sculpted as desired to produce the required shape of cartilage needed.
- the Cartilage Inducing TS (CI-TS) mixture can also be used to precoat a conventional implant, with the result being a conventional implant with a coating of living cartilage.
- the CI-TS is then implanted into the body of the recipient. This implantation can be heterotopic or orthotopic. After an appropriate interval, the CI-TS is be replaced by living cartilage with the form of the original CI-TS implant.
- Such implants can be used to replace damaged or lost cartilage, or to improve the tissue integration and/or function of an artificial implant. Examples of such uses include the replacement or reconstruction of nasal or ear tissue, the generation of a functional joint surface on a bone implant grown in vivo, or the generation of a similar surface on an artificial implant.
- the repair of cartilage damaged by disease, such as rheumatoid arthritis, can also be accomplished using the CI-TS to produce a new and smooth cartilage surface to the arthus.
- Implants intended for space filling applications in Plastic/Reconstructive surgery can also be either formed from CI-TS, or coated with
- this invention is an advancement because it permits the generation of cartilaginous tissue which is required to fully mimic the body's natural make-up. This results in improved joint repair, artificial joints and other implants, both for orthopedic and other applications.
- this embodiment can be used: to produce improved orthopedic implants or improved plastic/reconstructive implants: for joint repair for traumatic, congenital or pathologically damaged or dysfunctional cartilage; to produce coatings of pacemaker implants and wires to increase their tissue integration and to reduce foreign body reactions. Similar coatings could also be applied to any implantable device for the similar purposes.
- This embodiment is a self-contained TS wound dressing, or bandage, which contains both the thrombin and fibrinogen components of the FG.
- the calcium is contained in either the thrombin and/or the fibrinogen component(s).
- Either or both of the thrombin or fibrinogen compone/ts can be, but does not have to be, supplemented with a growth factors), such as a FGF or bFGF, or a drug(s) such as, an analgesic, antibiotic or other drug(s), which ca/ inhibit infection, promote wound healing and/or inhibit scar formation.
- the supplement(s) is at a concentration in the TS such that it will be effective for its intended purpose, e.g., an antibiotic will inhibit the growth of microbes, an analgesic will relieve pain.
- the thrombin and fibrinogen are separated from each other by an impermeable membrane, and the pair are covered with another such membrane.
- the thrombin and fibrinogen are contained in a quick evaporating gel (e.g., methylcellulose/alcohol/water).
- the bandage may be coated on the surface that is in contact with the gel in order to insure that the gel pad remains in place during use. (See Figure 39).
- the membrane separating the two components is removed, allowing the two components to mix.
- the outer membrane is then removed and the bandage is applied to the wound site.
- the action of the thrombin and other components of the fibrinogen preparation cause the conversion of the fibrinogen to fibrin, just as they do with any application of FS. This results in a natural inhibition of blood and fluid loss from the wound, and the establishment of a natural barrier to infection.
- the thrombin component and the plastic film separating the Thrombin gel and the Fibrinogen gel may be omitted.
- the calcium that was previously in the Thrombin gel may or may not be included in the Fibrinogen gel as desired.
- the outer impervious plastic film is removed and the bandage applied, as previously described, directly to the wound site.
- the Thrombin and calcium naturally present at the wound site then induce the conversion of fibrinogen to fibrin and inhibit blood and fluid loss from the wound as above.
- This embodiment has the advantage of being simpler, cheaper, and easier to produce. However, there may be circumstances in which a patient's wounds have insufficient thrombin. In those cases, the previous embodiment of the invention should be used.
- This embodiment is an advancement over the current technology as it permits the rapid application of TS to a wound without the time delay associated with solubilization and mixing of the components. It also requires no technical knowledge or skill to operate. These characteristics make it ideal for use in field applications, such as in trauma packs for soldiers, rescue workers, ambulance/paramedic teams, firemen, in first aid kits for the general public, and by emergency room personnel in hospitals. A small version may also be useful for use by the general public.
- This embodiment uses supplemented TS as a coating for the surfaces of orthopedic devices and other biomatenals which are to be implanted into an animal's body.
- these devices are urinary catheters, intravascular catheters, sutures, vascular prostheses, intraocular lenses, contact lenses, heart valves, shoulder/elbow/hip/knee replacement devices, total artificial hearts, etc.
- these biomaterials may become sites for bacterial adhesion and colonization, which eventually may lead to clinical infection that will endanger the life of the animal. To minimize this problem. the biomaterial is coated with a supplemented TS.
- the TS can be supplemented with: a growth factoids); a drug(s), such as an antibiotic; BMP; and/or cultured cells, etc.
- antibiotics that may be incorporated into the TS include, but are not limited to: the penicillins: cephalosporins; tetracyclines; chloramphenicols; metronidazoles; and aminoglycosides.
- growth factors which may be incorporated into the TS include but are not limited to FGF, PDGF, TGF- ⁇ .
- BMPs which may be incorporated into the TS include, but are not limited to, BMP 1 through 8.
- DBM can also be added to the TS.
- cultured cells which may be incorporated into the TS include, but are not limited to, endothelial cells, osteoblasts, fibroblasts, etc.
- the supplement(s) may be contained in either the thrombin, fibrinogen. calcium or water components).
- concentration of the supplement in the TS is adequate such that it will be effective for its intended purpose, e.g., an antibiotic will inhibit the growth of microbes on the biomaterial, a growth factor will induce the growth of the desired cell type(s) in the TS and/or on the surface of the biomaterial.
- This invention is an improvement for existing biomaterial products, which include titanium and titanium alloy devices (such as fixation plates, shoulder/elbow/hip/knee replacement devices, osseointegrated dental implants, etc), solid silicone products
- Silastic nasal implants such as Silastic nasal implants, liquid and/or gel silicone products (such as breast implants and testicular implants), and natural or synthetic polymers used as conventional materials in healing a wound site, which may have various forms, such as monofilaments, fibrous assemblies (such as cotton, paper, nonwoven fabrics), films, sponges, bags, etc.
- FG is produced from 3 components: fibrinogen (for example as TFC); and thrombin, both of which may be in the lyophilized form; as well as calcium.
- fibrinogen for example as TFC
- thrombin both of which may be in the lyophilized form; as well as calcium.
- the lyophilized fibrinogen is reconstituted with sterile water, while the thrombin component is reconstituted with calcium chloride solution.
- a supplement may be added to any of the three components prior to mixing. Appropriate volumes of the fibrinogen and thrombin containing calcium are mixed to produce the FG.
- the FG is then applied to the biomaterial's surface as a coating thereof as, for example, by spraying, painting, etc. Alternatively, the implant is dipped in the FG while it is still liquid.
- a supplement may also be added to the FG before or after it has been coated on a biomaterial surface.
- a FG-coated implant is soaked in an antibiotic solution for a specified period of time so that the antibiotic will diffuse into the TS.
- Another example is coating a device with TS after which cultured cells are seeded onto the fibrin coating. Coating the surface of biomaterials, which will be implanted into an animal, with supplemented TS will serve several purposes, including: the inhibition of bacterial adhesion to the biomaterial; the inhibition of growth of bacteria adhered to the biomaterial: local immune stimulation and/or normalization; the promotion of would healing; and the promotion of engraftment of the biomaterial to the surrounding tissue.
- the TSs may be formulated as a self-contained wound dressing, or fibrin sealant bandage, which contains the necessary thrombin and fibrinogen components of the FG.
- the self-contained dressing or bandage is easy-to-use, requiring no advanced technical knowledge or skill to operate.
- the present inventors have prepared a fibrin sealant bandage for applying a tissue sealing composition to wounded tissue in a patient, wherein the bandage comprises, in order: (1) an occlusive backing; (2) a pharmacologically-acceptable adhesive layer on the wound-facing surface of the backing; and (3) a layer of dry materials comprising an effective amount, in combination, of (a) dry, virally-inactivated, purified tissue fibrinogen complex, (b) dry, virally-inactivated, purified thrombin, affixed to the wound-facing surface of the adhesive layer or backing, and (c) calcium chloride.
- a removable, waterproof, soft plastic, protective film was placed over the layer of dry materials and the exposed adhesive surface of the bandage for stable storage purposes. In operation the waterproof, protective film is removed prior to the application of the bandage over the wounded tissue. The bandage was applied with pressure until the TS has formed over the target area.
- the fibrin sealant bandage was tested using a conventional, adhesive silicone patch measuring 6 cm X 5 cm, having a total area of 30 cm 2 .
- the dry components were placed over the adhesive patch to a depth of V. cm, so that the total volume of fibrin formed by the TS upon hydration equaled 15 cc (30 cm 2 ⁇ 1 ⁇ 2 cm).
- the materials used were: 360 mg of topical fibrinogen complex (TFC), described previously; approximately 340 U thrombin, also described previously: and 88 mg CaCl 2 (40 mM).
- TFC topical fibrinogen complex
- the binding capacity of the bandage for the dry material layer was, in part, dependent upon applying the dry materials as a uniformly-ground, fine powder.
- the calcium chloride was ground to a fine powder and mixed with the finely ground lyophilized TFC and thrombin, and applied as a powder to the adhesive side of the silicone patch and allowed to adhere to form the fibrin sealant patch.
- the dry materials were mixed and ground together.
- the fibrin sealant patch was applied to a damp cellulose sponge, representative of a tissue wound, so that the fibrin sealant component was adjacent to the surface of the sponge.
- the sponge had been previously dampened with room-temperature distilled H 2 O.
- Fibrin formation began to develop within 30 seconds of application. Within three minutes of application, a fibrin gel had formed affixing the tissue sealing fibrin clot to the sponge. This first patch hydrated by the endogenously available liquid was labeled FSB#1.
- Patch FSB#3 was prepared the same as FSB#1, but absent the thrombin component.
- Patch FSB#4 was prepared the same as FSB#1, but absent the TFC component.
- Patch FSB#5 was prepared the same as FSB#1, but absent the calcium chloride component. The results of each test were evaluated over time. As shownbelow in Table 10, a clotted gel formed when the fibrin components were hydrated with PBS, but remained in solution when either the fibrinogen or thrombin components were deleted from fibrin sealant bandage composition. Similarly, although a weak, watery gel was formed after 30 minutes when the calcium component was deleted from the fibrin sealant bandage and from the hydrating fluid, the composition was unable to develop into a tissue sealing fibrin clot.
- the fibrin sealant bandage formulated on silicone patches as described above, were also found to effectively form fibrin seals when tested on gelatin surfaces and in vivo on rat tissue. Based on the successful formation of the fibrin seal to a variety of materials and textures, including basic in vivo testing on an uninjured rat, animal studies will be conducted as described in the previous Examples evaluating the TS composition to optimize the hemostatic utility of the fibrin sealant bandage, and to establish delivery kinetics of supplementary components to be added, e.g. , growth hormones, drugs. antibiotics, antiseptics, etc.
- the present inventors have prepared a self-foaming fibrin sealant dressing for applying a tissue sealing composition to wounded tissue in a patient, wherein the dressing is applied as an expandable foam comprising an effective amount, in combination, of (1) virally-inactivated, purified fibrinogen complex, (2) virally-inactivated, purified thrombin, (3) calcium, and (4) a physiologically acceptable hydration agent; wherein said composition does not significantly inhibit full-thickness skin wound healing.
- the previously described TS components will be stored in a canister or tank with a pressurized propellant, so that the components are delivered to the wound site as an expandable foam, which will within minute(s) form a fibrin seal.
- a bench model test system is prepared from standard Amicon pressure chambers to determine optimal particle size. Particle size has proven to be important. Preliminary experiments have revealed that a reduction in particle size of the TFC, fibrin and calcium components results in a significant reduction in the time required to hydrate the reagents.
- Testing is also relevant to determining the feasibility of combining all of the reagents within a single reservoir, or whether it is more advantageous to maintain each component in a separate reservoir until application. Although probably more expensive, the latter canister prototype (having multiple separate reservoirs) may prove advantageous, in terms of stability and long-term storage.
- the test system consists of one or two pressure vessels driven by a pressurized reservoir containing the pharmaceutically acceptable hydrating agent (e.g., water or PBS), and pressurized compressed gas cylinders.
- the reagents are placed into the appropriate chamber(s) and the reservoir charged with hydrating agent saturated with the propellant at the desired pressure. Mixing of water and the reagents in their reservoirs is accomplished by opening connecting valves.
- the output is directed into either a single line, or in the case in which the components remain separated, into the joining piece of a Hemedics Fibrin Sealant Dispenser.
- the TFC was rehydrated with 3 cc dH 2 O, and warmed to 37oC to the concentrations shown in Table 11.
- the thrombin was rehydrated with 0.5 cc CaCl 2 solution (100 mM) to the concentrations shown in Table 11.
- the hydrated components were mixed and carbonated water (10 cc) was added to produce the volumes shown in Table 11.
- the resulting foaming mixture was placed in a vacuum jar to increase the foaming. Vacuum pressure was applied until the foam dried.
- the result was a permanent, integrated, foamy mass of fibrin, which expanded approximately 5-fold, and which was both self-adherent and adherent to adjacent textured surfaces.
- the foam was also quantitatively measured in calibrated plastic beakers. After two minutes, the volume of the foam was measured and the mass was gently probed to determine that it had set. The quantitative measurements of the expansion of the self- foaming fibrin sealant is indicated in Table 11. Once set, the expandable foam was no longer adhesive to new surfaces.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Surgery (AREA)
- Materials Engineering (AREA)
- Hematology (AREA)
- Dispersion Chemistry (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Materials For Medical Uses (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description

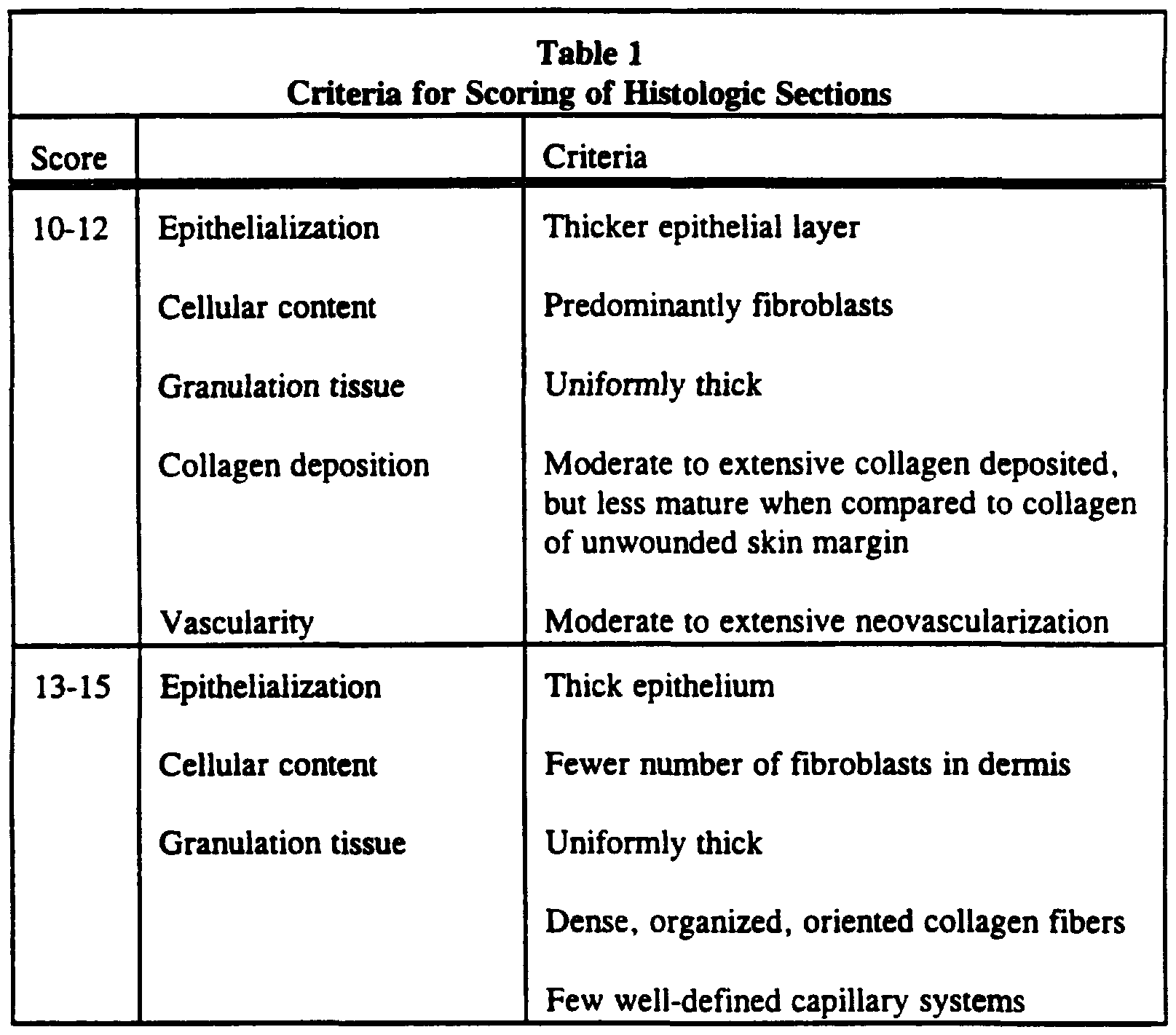






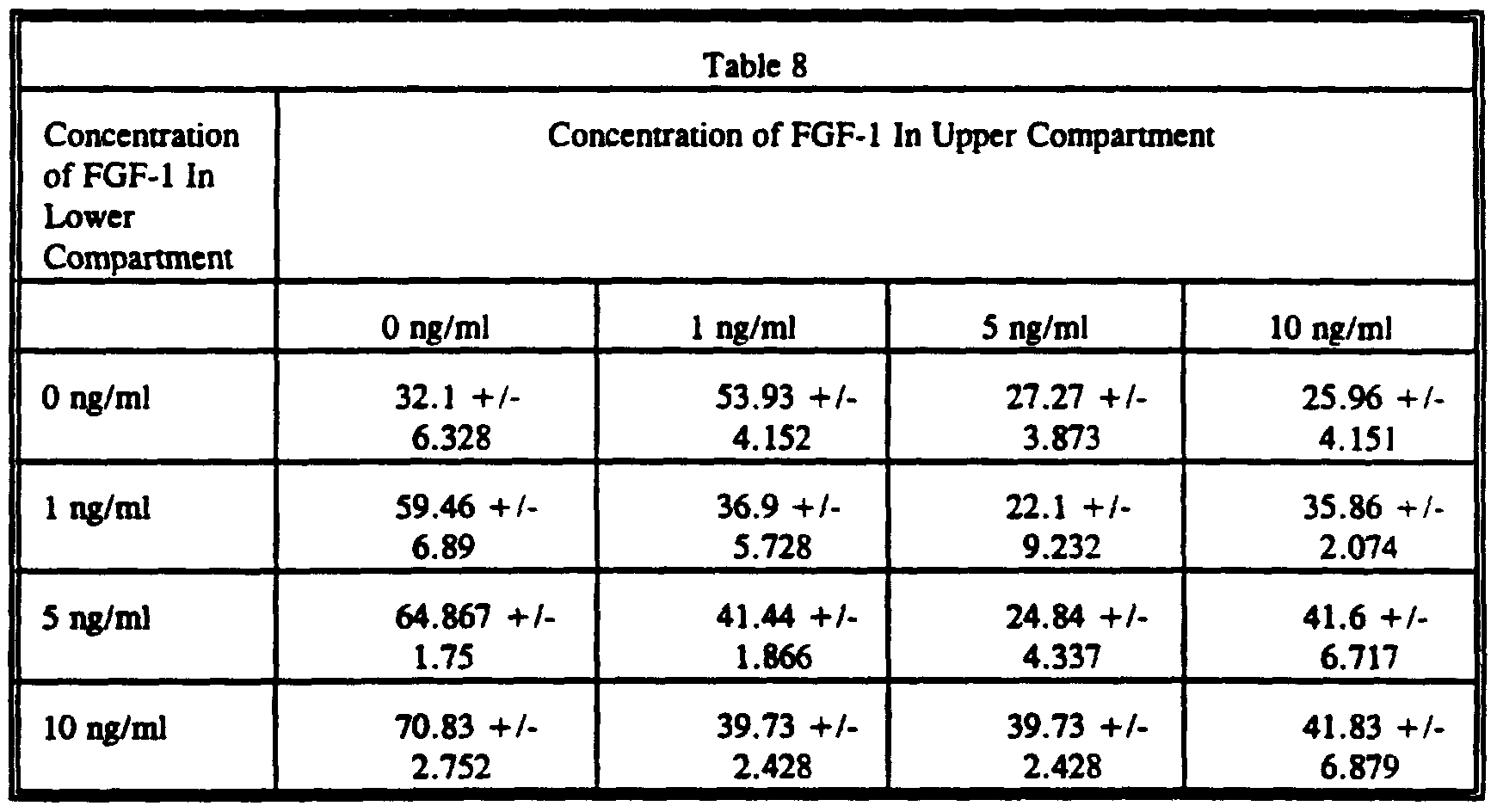


Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU45100/96A AU717906B2 (en) | 1994-12-07 | 1995-12-06 | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| CA002207289A CA2207289C (en) | 1994-12-07 | 1995-12-06 | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| JP51775396A JP4223548B2 (en) | 1994-12-07 | 1995-12-06 | Supplemental or non-complementary tissue sealants, their preparation and use |
| EP95943692A EP0796115A1 (en) | 1994-12-07 | 1995-12-06 | Supplemented and unsupplemented tissue sealants, methods of their production and use |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US35100694A | 1994-12-07 | 1994-12-07 | |
| US08/351,006 | 1994-12-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1996017633A1 true WO1996017633A1 (en) | 1996-06-13 |
| WO1996017633A9 WO1996017633A9 (en) | 1996-09-26 |
Family
ID=23379191
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1995/015876 WO1996017633A1 (en) | 1994-12-07 | 1995-12-06 | Supplemented and unsupplemented tissue sealants, methods of their production and use |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP0796115A1 (en) |
| JP (1) | JP4223548B2 (en) |
| AU (1) | AU717906B2 (en) |
| CA (1) | CA2207289C (en) |
| WO (1) | WO1996017633A1 (en) |
Cited By (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0869804A1 (en) * | 1995-06-07 | 1998-10-14 | American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| WO1999015209A2 (en) * | 1997-09-19 | 1999-04-01 | Baxter Aktiengesellschaft | Fibrin sponge |
| WO1999056798A1 (en) * | 1998-05-07 | 1999-11-11 | Genzyme Corporation | Compositions comprising hemostatic compounds and bioabsorbable polymers |
| US6096309A (en) * | 1997-06-18 | 2000-08-01 | Cohesion Technologies, Inc. | Compositions containing thrombin and microfibrillar nanometer collagen, and methods for preparation and use thereof |
| EP1032367A2 (en) * | 1997-11-17 | 2000-09-06 | Haemacure Corporation | Fibrin sealants or adhesives comprising a hyaluronic acid derivative material |
| WO2000053592A1 (en) * | 1999-03-05 | 2000-09-14 | Asahi Kasei Kabushiki Kaisha | Osteogenesis promoters |
| WO2001062278A1 (en) * | 2000-02-22 | 2001-08-30 | Hogy Medical Co. Ltd. | Hemostatic soluble cellulose fibers containing coagulating protein for treating wound and process for producing the same |
| WO2001097872A1 (en) * | 2000-06-22 | 2001-12-27 | Austin Sam L | Bioadhesive compositions and methods of preparation and use |
| US6921532B1 (en) | 2000-06-22 | 2005-07-26 | Spinal Restoration, Inc. | Biological Bioadhesive composition and methods of preparation and use |
| USRE39192E1 (en) | 1990-11-27 | 2006-07-18 | American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| USRE39298E1 (en) | 1990-11-27 | 2006-09-19 | The American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| USRE39321E1 (en) | 1990-11-27 | 2006-10-03 | The American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| US7189410B1 (en) | 1990-11-27 | 2007-03-13 | The American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| US7241730B2 (en) | 1998-08-27 | 2007-07-10 | Universitat Zurich | Enzyme-mediated modification of fibrin for tissue engineering: fibrin formulations with peptides |
| US7247609B2 (en) | 2001-12-18 | 2007-07-24 | Universitat Zurich | Growth factor modified protein matrices for tissue engineering |
| US7429241B2 (en) | 2005-09-29 | 2008-09-30 | Codman & Shurtleff, Inc. | Dural graft and method of preparing the same |
| US7597687B2 (en) | 2004-10-29 | 2009-10-06 | Spinal Restoration, Inc. | Injection of fibrin sealant including an anesthetic in spinal applications |
| US7601685B2 (en) | 1998-08-27 | 2009-10-13 | Eidgenossische Technische Hochschule Zurich | Growth factor modified protein matrices for tissue engineering |
| WO2010019997A1 (en) | 2008-08-18 | 2010-02-25 | Angioblast Systems, Inc. | Monoclonal antibody stro-4 |
| WO2011083154A1 (en) | 2010-01-08 | 2011-07-14 | Profibrix Bv | Dry powder fibrin sealant |
| EP2399991A1 (en) | 2005-04-12 | 2011-12-28 | Angioblast Systems Incorporated | Isolation of adult multipotential cells by tissue non-specific alkaline phosphatase |
| US8124075B2 (en) | 2004-07-16 | 2012-02-28 | Spinal Restoration, Inc. | Enhanced biological autologous tissue adhesive composition and methods of preparation and use |
| US8206448B2 (en) | 2004-10-29 | 2012-06-26 | Spinal Restoration, Inc. | Injection of fibrin sealant using reconstituted components in spinal applications |
| US8226942B2 (en) | 2007-12-28 | 2012-07-24 | Kuros Biosurgery Ag | PDGF fusion proteins incorporated into fibrin foams |
| US8318674B2 (en) | 2005-01-06 | 2012-11-27 | Kuros Biosurgery Ag | Local treatment of bone defects |
| US8403923B2 (en) | 2004-10-29 | 2013-03-26 | Spinal Restoration, Inc. | Injection of fibrin sealant in the absence of corticosteroids in spinal applications |
| US8419722B2 (en) | 2004-10-29 | 2013-04-16 | Spinal Restoration, Inc. | Apparatus and method for injection of fibrin sealant in spinal applications |
| US8575101B2 (en) | 2005-01-06 | 2013-11-05 | Kuros Biosurgery Ag | Supplemented matrices for the repair of bone fractures |
| US8961947B2 (en) | 2007-04-13 | 2015-02-24 | Kuros Biosurgery Ag | Polymeric tissue sealant |
| US20160287659A1 (en) * | 2006-11-27 | 2016-10-06 | Haemostatix Limited | Biogel |
| US9827205B2 (en) | 2008-12-12 | 2017-11-28 | Mallinckrodt Pharma Ip Trading D.A.C. | Dry powder fibrin sealant |
| US9833538B2 (en) | 2015-08-07 | 2017-12-05 | Xcede Technologies, Inc. | Adhesive compositions and related methods |
| US9956311B2 (en) | 2012-02-03 | 2018-05-01 | Xcede Technologies, Inc. | Tissue patch |
| KR101878769B1 (en) | 2017-04-03 | 2018-08-16 | 조석형 | Low molecular CM-β-1,3-Glucan calcium salt hemostatic agent powder |
| EP3424514A1 (en) | 2007-08-06 | 2019-01-09 | Mesoblast, Inc. | Method for generating, repairing and/or maintaining connective tissue in vivo |
| DE102017126149A1 (en) * | 2017-11-08 | 2019-05-09 | Johannes Gutenberg-Universität Mainz | Active substance-containing layer composite and method for its production |
| US10588998B2 (en) | 2015-08-07 | 2020-03-17 | Xcede Technologies, Inc. | Adhesive compositions and related methods |
| US10589001B2 (en) | 2011-03-16 | 2020-03-17 | Kuros Biosurgery Ag | Pharmaceutical formulation for use in spinal fusion |
| US10660945B2 (en) | 2015-08-07 | 2020-05-26 | Victor Matthew Phillips | Flowable hemostatic gel composition and its methods of use |
| WO2020061067A3 (en) * | 2018-09-17 | 2020-07-23 | Board Of Regents, The University Of Texas System Texas System | Compositions and methods for treating bone injury |
| US10751444B2 (en) | 2015-08-07 | 2020-08-25 | Victor Matthew Phillips | Flowable hemostatic gel composition and its methods of use |
| WO2021078986A1 (en) * | 2019-10-23 | 2021-04-29 | Fibriant B.V. | Fibrinogen as adjuvant for antimicrobial agents and therapy |
| US10994047B2 (en) | 2014-01-08 | 2021-05-04 | Haemostatix Limited | Peptide dendrimers comprising fibrinogen-binding peptides |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK2052746T3 (en) * | 2004-10-20 | 2014-12-08 | Ethicon Inc | Absorbable hemostatic patch |
| US9358318B2 (en) | 2004-10-20 | 2016-06-07 | Ethicon, Inc. | Method of making a reinforced absorbable multilayered hemostatic wound dressing |
| KR100701552B1 (en) * | 2006-06-23 | 2007-03-30 | 한국과학기술연구원 | Method for manufacturing biodegradable polyester polymer material in the form of filament and sheet using compressed gas |
| EP2111239B1 (en) | 2006-12-15 | 2013-03-06 | Lifebond Ltd. | Gelatin-transglutaminase hemostatic dressings and sealants |
| CN102124058B (en) | 2008-06-18 | 2014-05-28 | 生命连结有限公司 | Improved cross-linked compositions |
| EP2515957B1 (en) | 2009-12-22 | 2015-07-29 | Lifebond Ltd | Modification of enzymatic crosslinkers for controlling properties of crosslinked matrices |
| CN103118713B (en) | 2010-08-05 | 2016-06-01 | 生命连结有限公司 | Dry composition wound dressing and binding agent |
| US11998654B2 (en) | 2018-07-12 | 2024-06-04 | Bard Shannon Limited | Securing implants and medical devices |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0081990A1 (en) * | 1981-12-11 | 1983-06-22 | Johnson & Johnson Products Inc. | Film dressing with fabric backing |
| GB2185747A (en) * | 1985-11-12 | 1987-07-29 | Dow Corning Sa | Methods of making dressings |
| WO1992009301A1 (en) * | 1990-11-27 | 1992-06-11 | The American National Red Cross | Tissue sealant and growth factor containing compositions that promote accelerated wound healing |
| WO1992013565A1 (en) * | 1991-01-31 | 1992-08-20 | Shaw, Robert, Francis | Growth factor containing matrix for the treatment of cartilage lesions |
| EP0562864A1 (en) * | 1992-03-25 | 1993-09-29 | JOHNSON & JOHNSON MEDICAL, INC. | Heteromorphic sponges containing active agents |
| WO1994020133A1 (en) * | 1993-03-12 | 1994-09-15 | The American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
-
1995
- 1995-12-06 CA CA002207289A patent/CA2207289C/en not_active Expired - Lifetime
- 1995-12-06 JP JP51775396A patent/JP4223548B2/en not_active Expired - Lifetime
- 1995-12-06 AU AU45100/96A patent/AU717906B2/en not_active Withdrawn - After Issue
- 1995-12-06 EP EP95943692A patent/EP0796115A1/en not_active Ceased
- 1995-12-06 WO PCT/US1995/015876 patent/WO1996017633A1/en active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0081990A1 (en) * | 1981-12-11 | 1983-06-22 | Johnson & Johnson Products Inc. | Film dressing with fabric backing |
| GB2185747A (en) * | 1985-11-12 | 1987-07-29 | Dow Corning Sa | Methods of making dressings |
| WO1992009301A1 (en) * | 1990-11-27 | 1992-06-11 | The American National Red Cross | Tissue sealant and growth factor containing compositions that promote accelerated wound healing |
| WO1992013565A1 (en) * | 1991-01-31 | 1992-08-20 | Shaw, Robert, Francis | Growth factor containing matrix for the treatment of cartilage lesions |
| EP0562864A1 (en) * | 1992-03-25 | 1993-09-29 | JOHNSON & JOHNSON MEDICAL, INC. | Heteromorphic sponges containing active agents |
| WO1994020133A1 (en) * | 1993-03-12 | 1994-09-15 | The American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
Cited By (60)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7189410B1 (en) | 1990-11-27 | 2007-03-13 | The American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| USRE39321E1 (en) | 1990-11-27 | 2006-10-03 | The American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| USRE39298E1 (en) | 1990-11-27 | 2006-09-19 | The American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| USRE39192E1 (en) | 1990-11-27 | 2006-07-18 | American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| EP0869804A4 (en) * | 1995-06-07 | 2001-08-16 | American Nat Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| EP0869804A1 (en) * | 1995-06-07 | 1998-10-14 | American National Red Cross | Supplemented and unsupplemented tissue sealants, methods of their production and use |
| US6280727B1 (en) | 1997-06-18 | 2001-08-28 | Cohesion Technologies, Inc. | Compositions containing thrombin and microfibrillar collagen and methods for preparation and use thereof |
| US6096309A (en) * | 1997-06-18 | 2000-08-01 | Cohesion Technologies, Inc. | Compositions containing thrombin and microfibrillar nanometer collagen, and methods for preparation and use thereof |
| WO1999015209A3 (en) * | 1997-09-19 | 1999-06-17 | Immuno Ag | Fibrin sponge |
| WO1999015209A2 (en) * | 1997-09-19 | 1999-04-01 | Baxter Aktiengesellschaft | Fibrin sponge |
| US6548729B1 (en) | 1997-09-19 | 2003-04-15 | Baxter Aktiengesellschaft | Fibrin sponge |
| EP1032367A4 (en) * | 1997-11-17 | 2005-02-02 | Haemacure Corp | Fibrin sealants or adhesives comprising a hyaluronic acid derivative material |
| EP1032367A2 (en) * | 1997-11-17 | 2000-09-06 | Haemacure Corporation | Fibrin sealants or adhesives comprising a hyaluronic acid derivative material |
| WO1999056798A1 (en) * | 1998-05-07 | 1999-11-11 | Genzyme Corporation | Compositions comprising hemostatic compounds and bioabsorbable polymers |
| US7601685B2 (en) | 1998-08-27 | 2009-10-13 | Eidgenossische Technische Hochschule Zurich | Growth factor modified protein matrices for tissue engineering |
| US7241730B2 (en) | 1998-08-27 | 2007-07-10 | Universitat Zurich | Enzyme-mediated modification of fibrin for tissue engineering: fibrin formulations with peptides |
| WO2000053592A1 (en) * | 1999-03-05 | 2000-09-14 | Asahi Kasei Kabushiki Kaisha | Osteogenesis promoters |
| WO2001062278A1 (en) * | 2000-02-22 | 2001-08-30 | Hogy Medical Co. Ltd. | Hemostatic soluble cellulose fibers containing coagulating protein for treating wound and process for producing the same |
| US7351422B2 (en) | 2000-02-22 | 2008-04-01 | Hogy Medical Co., Ltd. | Hemostatic soluble cellulose fibers containing coagulating protein for treating wound and process for producing the same |
| US7235255B2 (en) | 2000-06-22 | 2007-06-26 | Spinal Restoration, Inc. | Biological bioadhesive compositions and methods of preparation and use |
| AU2001273623B2 (en) * | 2000-06-22 | 2006-06-08 | Spinal Restoration, Inc. | Bioadhesive compositions and methods of preparation and use |
| US6921532B1 (en) | 2000-06-22 | 2005-07-26 | Spinal Restoration, Inc. | Biological Bioadhesive composition and methods of preparation and use |
| US6468527B2 (en) | 2000-06-22 | 2002-10-22 | Sam L. Austin | Biological bioadhesive composition and methods of preparation and use |
| US7229633B2 (en) | 2000-06-22 | 2007-06-12 | Spinal Restoration, Inc. | Biological bioadhesive compositions and methods of preparation and use |
| WO2001097872A1 (en) * | 2000-06-22 | 2001-12-27 | Austin Sam L | Bioadhesive compositions and methods of preparation and use |
| US8034618B2 (en) | 2001-12-18 | 2011-10-11 | Eldgenossische Technische Hochschule Zurich | PTH containing cell growth matrix |
| US7247609B2 (en) | 2001-12-18 | 2007-07-24 | Universitat Zurich | Growth factor modified protein matrices for tissue engineering |
| US8124075B2 (en) | 2004-07-16 | 2012-02-28 | Spinal Restoration, Inc. | Enhanced biological autologous tissue adhesive composition and methods of preparation and use |
| US7597687B2 (en) | 2004-10-29 | 2009-10-06 | Spinal Restoration, Inc. | Injection of fibrin sealant including an anesthetic in spinal applications |
| US8419722B2 (en) | 2004-10-29 | 2013-04-16 | Spinal Restoration, Inc. | Apparatus and method for injection of fibrin sealant in spinal applications |
| US8986304B2 (en) | 2004-10-29 | 2015-03-24 | Bsnc Holding, Llc | Injection of fibrin sealant using reconstituted components in spinal applications |
| US8206448B2 (en) | 2004-10-29 | 2012-06-26 | Spinal Restoration, Inc. | Injection of fibrin sealant using reconstituted components in spinal applications |
| US8403923B2 (en) | 2004-10-29 | 2013-03-26 | Spinal Restoration, Inc. | Injection of fibrin sealant in the absence of corticosteroids in spinal applications |
| US8575101B2 (en) | 2005-01-06 | 2013-11-05 | Kuros Biosurgery Ag | Supplemented matrices for the repair of bone fractures |
| US8318674B2 (en) | 2005-01-06 | 2012-11-27 | Kuros Biosurgery Ag | Local treatment of bone defects |
| EP2399991A1 (en) | 2005-04-12 | 2011-12-28 | Angioblast Systems Incorporated | Isolation of adult multipotential cells by tissue non-specific alkaline phosphatase |
| EP3327116A1 (en) | 2005-04-12 | 2018-05-30 | Mesoblast, Inc. | Isolation of adult multipotential cells by tissue non-specific alkaline phosphatase |
| US8367405B2 (en) | 2005-04-12 | 2013-02-05 | Mesoblast, Inc. | Isolation of adult multipotential cells by tissue non-specific alkaline phosphatase |
| US7429241B2 (en) | 2005-09-29 | 2008-09-30 | Codman & Shurtleff, Inc. | Dural graft and method of preparing the same |
| US20160287659A1 (en) * | 2006-11-27 | 2016-10-06 | Haemostatix Limited | Biogel |
| US9180222B2 (en) | 2007-04-13 | 2015-11-10 | Kuros Biosurgery Ag | Polymeric tissue sealant |
| US8961947B2 (en) | 2007-04-13 | 2015-02-24 | Kuros Biosurgery Ag | Polymeric tissue sealant |
| EP3424514A1 (en) | 2007-08-06 | 2019-01-09 | Mesoblast, Inc. | Method for generating, repairing and/or maintaining connective tissue in vivo |
| US8226942B2 (en) | 2007-12-28 | 2012-07-24 | Kuros Biosurgery Ag | PDGF fusion proteins incorporated into fibrin foams |
| WO2010019997A1 (en) | 2008-08-18 | 2010-02-25 | Angioblast Systems, Inc. | Monoclonal antibody stro-4 |
| EP3121597A1 (en) | 2008-08-18 | 2017-01-25 | Mesoblast, Inc. | Monoclonal antibody stro-4 |
| US9827205B2 (en) | 2008-12-12 | 2017-11-28 | Mallinckrodt Pharma Ip Trading D.A.C. | Dry powder fibrin sealant |
| WO2011083154A1 (en) | 2010-01-08 | 2011-07-14 | Profibrix Bv | Dry powder fibrin sealant |
| US10589001B2 (en) | 2011-03-16 | 2020-03-17 | Kuros Biosurgery Ag | Pharmaceutical formulation for use in spinal fusion |
| US9956311B2 (en) | 2012-02-03 | 2018-05-01 | Xcede Technologies, Inc. | Tissue patch |
| US10994047B2 (en) | 2014-01-08 | 2021-05-04 | Haemostatix Limited | Peptide dendrimers comprising fibrinogen-binding peptides |
| US10588998B2 (en) | 2015-08-07 | 2020-03-17 | Xcede Technologies, Inc. | Adhesive compositions and related methods |
| US9833538B2 (en) | 2015-08-07 | 2017-12-05 | Xcede Technologies, Inc. | Adhesive compositions and related methods |
| US10660945B2 (en) | 2015-08-07 | 2020-05-26 | Victor Matthew Phillips | Flowable hemostatic gel composition and its methods of use |
| US10722611B2 (en) | 2015-08-07 | 2020-07-28 | Xcede Technologies, Inc. | Adhesive compositions and related methods |
| US10751444B2 (en) | 2015-08-07 | 2020-08-25 | Victor Matthew Phillips | Flowable hemostatic gel composition and its methods of use |
| KR101878769B1 (en) | 2017-04-03 | 2018-08-16 | 조석형 | Low molecular CM-β-1,3-Glucan calcium salt hemostatic agent powder |
| DE102017126149A1 (en) * | 2017-11-08 | 2019-05-09 | Johannes Gutenberg-Universität Mainz | Active substance-containing layer composite and method for its production |
| WO2020061067A3 (en) * | 2018-09-17 | 2020-07-23 | Board Of Regents, The University Of Texas System Texas System | Compositions and methods for treating bone injury |
| WO2021078986A1 (en) * | 2019-10-23 | 2021-04-29 | Fibriant B.V. | Fibrinogen as adjuvant for antimicrobial agents and therapy |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0796115A1 (en) | 1997-09-24 |
| JPH10510183A (en) | 1998-10-06 |
| CA2207289A1 (en) | 1996-06-13 |
| AU717906B2 (en) | 2000-04-06 |
| AU4510096A (en) | 1996-06-26 |
| CA2207289C (en) | 2005-04-12 |
| JP4223548B2 (en) | 2009-02-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2207289C (en) | Supplemented and unsupplemented tissue sealants, methods of their production and use | |
| US6117425A (en) | Supplemented and unsupplemented tissue sealants, method of their production and use | |
| US6559119B1 (en) | Method of preparing a tissue sealant-treated biomedical material | |
| US7196054B1 (en) | Methods for treating wound tissue and forming a supplemented fibrin matrix | |
| US6197325B1 (en) | Supplemented and unsupplemented tissue sealants, methods of their production and use | |
| US6054122A (en) | Supplemented and unsupplemented tissue sealants, methods of their production and use | |
| AU696691B2 (en) | Supplemented and unsupplemented tissue sealants, methods of their production and use | |
| WO1996017633A9 (en) | Supplemented and unsupplemented tissue sealants, methods of their production and use | |
| WO1996040174A1 (en) | Supplemented and unsupplemented tissue sealants, methods of their production and use | |
| US6124273A (en) | Chitin hydrogels, methods of their production and use | |
| CA2097063C (en) | Tissue sealant and growth factor containing compositions that promote accelerated wound healing | |
| AU733471B2 (en) | Supplemented and unsupplemented tissue sealants, methods of their production and use | |
| AU778583B2 (en) | Supplemented and unsupplemented tissue sealants, methods of their production and use | |
| AU5053200A (en) | Supplemented and unsupplemented tissue sealants, methods of their production and use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA JP |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| COP | Corrected version of pamphlet |
Free format text: PAGES 1/31-31/31,DRAWINGS,REPLACED BY NEW PAGES BEARING THE SAME NUMBER;DUE TO LATE TRANSMITTAL BY THE RECEIVING OFFICE |
|
| ENP | Entry into the national phase |
Ref document number: 2207289 Country of ref document: CA Kind code of ref document: A Ref document number: 2207289 Country of ref document: CA |
|
| REEP | Request for entry into the european phase |
Ref document number: 1995943692 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1995943692 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1995943692 Country of ref document: EP |