WO1995022537A2 - 4-amino derivatives of 5-substituted mycophenolic acid - Google Patents
4-amino derivatives of 5-substituted mycophenolic acid Download PDFInfo
- Publication number
- WO1995022537A2 WO1995022537A2 PCT/US1995/001786 US9501786W WO9522537A2 WO 1995022537 A2 WO1995022537 A2 WO 1995022537A2 US 9501786 W US9501786 W US 9501786W WO 9522537 A2 WO9522537 A2 WO 9522537A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- lower alkyl
- methyl
- hydrogen
- Prior art date
Links
- 0 CCC=C(CC1)*C1C(*)=O Chemical compound CCC=C(CC1)*C1C(*)=O 0.000 description 7
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/87—Benzo [c] furans; Hydrogenated benzo [c] furans
- C07D307/88—Benzo [c] furans; Hydrogenated benzo [c] furans with one oxygen atom directly attached in position 1 or 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Definitions
- the present invention relates to mycophenolic acid derivatives in which the 4-hydroxy group has been replaced by amino substituents.
- the invention includes natural and derivative side chains at the 5 -position.
- the invention is also directed to formulations and methods for treatment.
- MPA Mycophenolic acid
- MPA and certain related compounds having the following structure:
- esters and derivatives of mycophenolic acid are useful in treating auto-immune related disorders, glomerulonephritis and hepatitis, and in preventing allograft rejection.
- anti-inflammatory agents they are useful in treating rheumatoid arthritis.
- anti-tumor agents they are useful in treating solid tumors and malignancies of lymphoreticular origins.
- R 1 is hydrogen or lower alkyl
- R 2 is hydrogen, lower alkyl, -C(O)R 3 , -C(O)NR 4 R 5 , -CO 2 R 6 , or -SO 2 R 3
- R 3 is hydrogen, lower alkyl, halo lower alkyl or optionally
- R 4 is hydrogen, lower alkyl or optionally substituted phenyl
- R 5 is hydrogen, lower alkyl or optionally substituted phenyl
- R 6 is lower alkyl or optionally substituted phenyl
- Z is a side chain selected from Formulae ZA, ZB, ZC, ZD, ZE, ZF, ZG, and ZH:
- Z 1 is H, lower alkyl, halo or CF 3 ;
- Z 2 is H, lower alkyl, lower alkoxy, aryl, or -CH 2 Z 13 , where
- Z 13 is aryl or heteroaryl
- Z 3 is H, lower alkyl, lower alkenyl, lower alkoxy, phenyl, - P(O) (OCH 3 ) 2 , -P(O) (OH) (OCH 3 ), or -S(O) m Z 12 , where
- Z 12 is lower alkyl
- n 0, 1 or 2;
- Z 4 is H, lower alkyl, or phenyl
- n is an integer from 1 to 6
- G 1 is H or lower alkyl
- G 2 is H or lower alkyl
- G 3 is lower alkylene of four to six carbon atoms, or lower alkylene of three to five carbon atoms plus one member that is -O-, -S-, or -N(G 4 )- where G 4 is H or lower alkyl;
- Z 5 is H or lower alkyl
- Z 8 is H or lower alkyl
- D 1 and D 2 together with their adjacent carbon atoms form an optionally substituted, saturated or unsaturated carbocyclic or heterocyclic ring of 3 to 7 atoms;
- G is as defined above;
- Z 5 , Z 8 , and G are as defined above; or
- D 3 is -CH 2 - or -CH 2 CH 2 -;
- G is as defined above;
- Z 6 is H, lower alkyl , lower alkoxy, -COOH, -NH 2 or halo;
- Z 7 is H, lower alkyl , lower alkoxy or halo
- Z 5 and G are as defined above ;
- D 4 is -CH 2 -, -CH 2 CH 2 -, -CH 2 CH 2 CH 2 -, -O-, or -OCH 2 - ;
- the invention relates to a pharmaceutical composition containing a therapeutically effective amount of a compound of Formula I admixed with at least one pharmaceutically acceptable excipient.
- the invention relates to a method of treating immune, inflammatory, tumor, proliferative, viral and psoriatic disorders in a mammal by administering to a mammal in need of such treatment a therapeutically effective amount of a compound of Formula I or a pharmaceutically acceptable salt thereof.
- alkyl refers to a fully saturated monovalent radical of one to twelve carbon atoms containing only carbon and hydrogen, and which may be a cyclic, branched or straight chain radical. This term is further exemplified by radicals such as methyl, ethyl, t-butyl, pentyl, cyclopentyl, cyclohexyl, heptyl, cycloheptyl and
- lower alkyl refers to a monovalent alkyl radical of one to six carbon atoms. This term is further exemplified by such radicals as methyl, ethyl, n-propyl, isopropyl, cyclopropyl, n-butyl, t-butyl, i-butyl (or 2-methylpropyl), isoamyl, pentyl, cyclopentyl, i-pentyl, hexyl and cyclohexyl.
- lower alkenyl refers to an unsaturated monovalent hydrocarbon radical of one to six carbon atoms. This term is further exemplified by such radicals as vinyl, prop-2-enyl, pent-3-enyl, and hex-5-enyl.
- halo refers to fluoro and chloro, unless otherwise specified.
- halo lower alkyl refers to a lower alkyl radical
- halomethyl refers to a methyl radical substituted with one or more chlorine and/or fluorine atoms. This term is further exemplified by such radicals as trichloromethyl, trifluoromethyl, dichloromethyl, fluoromethyl and difluoro-chloromethyl.
- lower alkylene refers to a divalent alkyl radical of one to six carbon atoms. This term is further exemplified by such radicals as methylene, ethylene, n-propylene, i-propylene, n-butylene, t-butylene, i-butylene (or 2-methylpropylene), isoamylene, pentylene, and n-hexylene.
- alkoxy means the group -OR wherein R is lower alkyl.
- lower alkanol means an alcohol of the formula ROH where R is a lower alkyl. This term is further exemplified by such alcohols as methanol, ethanol, n-propanol, i-propanol, n-butanol, t-butanol, i-butanol (or 2-methylpropanol), pentanol, n-hexanol.
- ROH lower alkanol
- This term is further exemplified by such alcohols as methanol, ethanol, n-propanol, i-propanol, n-butanol, t-butanol, i-butanol (or 2-methylpropanol), pentanol, n-hexanol.
- optionally substituted phenyl refers to phenyl and mono-, di-, or tri-substituted phenyl, wherein the optional substituents are lower alkyl, lower alkoxy, hydroxy, trifluoromethyl, or halo.
- This term is further exemplified by such radicals as 2-chlorophenyl, 2-trifluoromethylphenyl, 4-methoxyphenyl, 4-chlorophenyl, 3,4-dimethoxyphenyl, 2-chloro-3,4-dimethoxyphenyl, 4-hydroxyphenyl,
- aryl refers to a monovalent unsaturated aromatic
- carbocyclic radical having a single ring (e.g., phenyl) or two condensed rings (e.g., naphthyl), which can optionally be mono-, di- or
- heteroaryl refers to a monovalent aromatic carbocyclic radical having at least one heteroatom, such as N, O or S, within the ring, such as quinolyl, benzofuranyl, pyridyl, morpholinyl and indolyl, which can optionally be mono-, di- or tri-substituted, independently, with OH, COOH, lower alkyl, lower alkoxy, chloro, fluoro, trifluoromethyl and/or cyano.
- X 1 , X 2 , X 3 , X 4 and X 5 can independently be -CHX a -, -C(O)-, -C(N-X b )-, -C(N-NX d X e ) -, -O-, -S-, -S(O)-, -S(O) 2 - or -NX c -, where
- X a is H, lower alkyl or forms a double bond
- X b is acyl, carbamoyl or ureido
- X c is lower alkyl, C(O)X d , S(O) 2 X d or C(O)NX d X e ; and X d and X e are independently H or lower alkyl;
- a sidechain of Formula ZB in which D 1 and D 2 together represent -CH 2 CH 2 CH 2 CH 2 - , and Z 5 and Z 8 are both hydrogen would be named as a 2-[(2-ethylidene)-2-cyclohex-1-yl]acetic acid derivative.
- a sidechain of Formula ZB in which D 1 and D 2 together represent -CH 2 CH 2 OCH 2 -, and Z 5 and Z 8 are both hydrogen would be named as a 2-[(2-ethylidene)-4-tetrahydropyran-3-yl]acetic acid derivative.
- a “pharmaceutically acceptable salt” may be any salt derived from an inorganic or organic acid or base. Salts may be derived from acids or bases.
- the acid addition salts are derived from inorganic acids, such as hydrochloric acid, hydrobromic acid, sulfuric acid (giving the sulfate and bisulfate salts), nitric acid, phosphoric acid and the like, and organic acids such as acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, malic acid, malonic acid, succinic acid, maleic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, salicylic acid,
- inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid (giving the sulfate and bisulfate salts), nitric acid, phosphoric acid and the like
- organic acids such as acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, malic acid, mal
- the base addition salts are derived from inorganic bases such as sodium hydroxide, potassium hydroxide, lithium hydroxide, ammonia, calcium hydroxide, magnesium hydroxide and the like.
- Cations derived from organic bases include those formed from primary, secondary and tertiary amines, such as isopropylamine, diethylamine, trimethylamine, triethylamine, pyridine, cyclohexylamine, ethylene diamine, monoethanolamine,
- inert organic solvent or “inert solvent” means a solvent inert under the conditions of the reaction being described in conjunction therewith (including, for example, benzene, toluene, acetonitrile, tetrahydrofuran, diethyl ether, chloroform, methylene chloride, pyridine, xylene, dimethylformamide, 1,4-dioxane,
- treatment means any treatment of a disease in a mammal, and includes:
- the term "effective amount” means a dosage sufficient to provide treatment. This will vary depending on the patient and the treatment being effected.
- Steps are isomers that differ only in the way the atoms are arranged in space.
- Enantiomers are a pair of stereoisomers that are non-superimposable mirror images of each other. A 1:1 mixture of a pair of enantiomers is a “racemic” mixture. The term “( ⁇ )” is used to designate a racemic mixture where appropriate.
- Stereoisomers are stereoisomers that have at least two
- stereochemistry at each chiral carbon may be specified by either RS or SR by reference to a single enantiomer of the racemate. In this manner relative stereochemistry is conveyed unambiguously.
- the compounds of the invention may possess one or more asymmetric centers, and can be produced as a racemic mixture or as individual enantiomers or diastereoisomers.
- the number of stereoisomers present in any given compound of Formula I depends upon the number of asymmetric centers present (there are 2 n stereoisomers possible where n is the number of asymmetric centers).
- the individual stereoisomers may be obtained by resolving a racemic or non-racemic mixture of an intermediate at some appropriate stage of the synthesis, or by resolution of the compound of Formula I by conventional means.
- Isolation and purification of the compounds and intermediates described herein can be effected, if desired, by any suitable separation or purification procedure such as, for example, filtration, extraction, crystallization, column chromatography, thin-layer chromatography or thick-layer chromatography, or a combination of these procedures.
- suitable separation and isolation procedures can be found by reference to the examples hereinbelow. However, other equivalent separation or isolation procedures can, of course, also be used.
- the isobenzofuranyl nucleus of the compounds of Formula I is numbered as follows:
- D represents -O-, -S(O) p -, -N(R 9 )-, and the like.
- the compounds of Formula I are prepared from the lower alkyl 4- isocyanato esters of Formula 6, the structure of which is shown below: where Z a is a sidechain of Formula Z as defined in the Summary of the Invention in which G is lower alkoxy.
- the compounds of Formula 6 are then converted to the compounds of Formula I by several different synthetic pathways, depending on the desired substitutions at the 4-position.
- the compounds of Formula 1 may be prepared as described below in Reaction Schemes I to XXII. The preparation of such compounds is also described in more detail in co-pending Application Serial No. 08/???,???, Attorney Docket No. 27960, entitled "5-Substituted Derivatives of
- Step 1 the phenolic hydroxyl group of a mycophenolic acid lower alkyl ester is protected.
- a mycophenolic acid lower alkyl ester of Formula 101 in a solvent (such as ether, ethyl acetate, dimethylamide, or preferably
- dichloromethane is reacted with an equimolar amount of a halogenated protecting group (such as: methoxyethoxymethyl chloride; a sulfonyl chloride, e.g., tosyl chloride, mesyl chloride; or a silyl chloride, e.g., trimethylsilyl chloride, diphenylmethylsilyl chloride, or preferably tert-butyldimethylsilyl chloride) in presence of an equimolar amount of an organic base (such as diisopropylethylamine, triethylamine, or imidazole).
- a halogenated protecting group such as: methoxyethoxymethyl chloride; a sulfonyl chloride, e.g., tosyl chloride, mesyl chloride; or a silyl chloride, e.g., trimethylsilyl chloride, diphenylmethylsilyl chloride
- Step 2 the side chain double bond of a protected mycophenolic acid lower alkyl ester is ozonized to yield an aldehyde.
- a stream of ozonized oxygen is passed through a solution of a protected compound of Formula 102 in a solvent (such as an alcohol, a halocarbon, or preferably a mixture of methanol and dichloromethane).
- a solvent such as an alcohol, a halocarbon, or preferably a mixture of methanol and dichloromethane.
- the reaction takes place at -100 to -40°C (preferably at -80°C), and continues until the presence of excess ozone is detected by the development of a blue color.
- the intermediate hydroperoxide thus formed is reduced without further purification, by the addition of a reducing agent (such as zinc and acetic acid, dimethyl sulfide, or preferably thiourea).
- the reaction takes place at -80°C to 25°C (preferably 0°C) over a period of 12 to 24 hours (preferably 16 hours), to give the corresponding aldehyde of Formula 103.
- Step 3 the aldehyde is converted to a carbinol by addition of an organometallic compound of
- Formula 103a [where M is MgBr or lithium, preferably MgBr (a Grignard reagent); Z 1 is H, lower alkyl or CF 3 , and Z 2 is H or lower alkyl].
- An organolithium reagent is formed by reaction of a halovinyl
- halovinyl compound of Formula 103a is reacted with magnesium metal in an ethereal solvent (such as ether or preferably tetrahydrofuran).
- an ethereal solvent such as ether or preferably tetrahydrofuran.
- the reaction takes place at 30 to 60°C (preferably 40°C) over a period of 1 to 6 hours (preferably 2 hours).
- the organometallic compound of Formula 103a where M is zinc or cadmium may be prepared by reaction of 103a where M is Li or MgBr with a zinc or cadmium halide, preferably chloride.
- the compound of Formula 103a where M is tin may be prepared by reaction of 103a where M is Li or MgBr with a trialkyl chlorostannane, preferably tributyltin chloride.
- the compound of Formula 103a where M is tin may also be prepared by reaction of 103a where M is trifluoromethanesulfonate by reaction with a compound of formula (R 3 Sn) 2 , where R is alkyl, preferably methyl, in the presence of a palladium catalyst, preferably tetrakis(triphenylphosphine)palladium.
- the compound of Formula 103a where M is trifluoromethanesulfonate may be prepared from a ketone of the formula: by reaction with a strong base (such as sodium hydride or potassium hexamethyldisilazide), followed by reaction of the anion thus produced with trifluoromethanesulfonic anhydride.
- the compound of Formula 103a where M is tin may be prepared by reacting a trialkyl tin hydride (preferably tributyl tin hydride) with an acetylene of the formula
- One molar equivalent of the resultant organometallic reagent is added to a solution of an aldehyde of Formula 103 (in the same solvent system used to make the organometallic reagent).
- the reaction takes place at -80 to 20°C (preferably 0°C) over a period of 5 to 60 minutes (preferably 10 minutes) to give the corresponding carbinol of Formula 104.
- Formula 105 is formed by a Claisen ortho ester rearrangement reaction of a carbinol of Formula 104 and an orthoester of Formula 104a (where Z 3 is H, halo, lower alkyl, lower alkenyl, phenyl, alkoxy or -thio lower alkyl; and Z 4 is H or lower alkyl; or Z 3 and Z 4 taken together with the carbon to which they are attached form cycloalkyl).
- a carbinol of Formula 104 is heated at 50 to 140°C (preferably about 130°C) with about 10 molar equivalents of an orthoester of Formula 104a, in the presence of from 0.05 to 0.25 molar equivalents (preferably 0.10 molar equivalents) of an organic acid catalyst (such as propionic, butyric, or preferably trimethylacetic acid).
- an organic acid catalyst such as propionic, butyric, or preferably trimethylacetic acid.
- One method of preparing individual enantiomers of compounds of Formula 1A is from chiral compounds of Formula 104b, the preparation of which is shown below in Reaction Scheme II.
- Step 1 an aldehyde of Formula 103 is oxidized to the corresponding carboxylic acid of Formula 103f.
- an oxidizing agent for example, chromic acid, silver oxide, bleach, or preferably sodium periodate
- an inert solvent such as toluene, or preferably ethyl acetate
- a catalytic amount for example, about 0.01 molar equivalents
- a catalyst such as ruthenium oxide, or preferably ruthenium trichloride
- Formula 103f is converted to the corresponding acyl halide of Formula 103g.
- a carboxylic acid of Formula 103f is reacted with about one molar equivalent, preferably 1.1 molar equivalents, of an halogenating agent (for example, thionyl chloride, thionyl bromide, or preferably oxalyl chloride), in an inert solvent (such as dichloromethane, or preferably ethyl acetate), in the presence of a catalytic amount (for example, about 0.05 molar equivalents) of dimethylformamide.
- the reaction takes place at 0 to 40°C (preferably 25°C) for 30 minutes to 8 hours (preferably 2 hours), to give the corresponding acyl halide of Formula 103g.
- an acyl halide of Formula 103g is converted to the corresponding keto olefin of Formula 103h by addition of an organometallic compound of Formula 103a.
- An acyl halide of Formula 103g is reacted with about one molar equivalent of a organometallic compound of Formula 103a (where M is cadmium, zinc, tin, or the like, prepared as shown in the preparation of compounds of Formula 104), in an inert solvent (such as dichloromethane, ether, or preferably tetrahydrofuran), optionally in the presence of a catalytic amount (for example, about 0.05 molar equivalents) of a palladium catalyst [preferably tetrakis(triphenylphosphine)palladium].
- the reaction takes place at -10 to 20°C (preferably 0°C) for 30 minutes to 8 hours (preferably 4 hours), to give the corresponding keto olefin of Formula 103h.
- Formula 103h is reduced stereospecifically to the corresponding carbinol of Formula 104b by reduction with borane methyl sulfide in the presence of a catalytic amount of (R)-tetrahydro-1-methyl-3,3-diphenyl-1H,3H-pyrrolo- [1,2-c][1,3,2]oxazaborole.
- a keto olefin of Formula 103h is sterospecifically reduced with about one molar equivalent of borane methyl sulfide in the presence of a catalytic amount (0.05-0.3 molar equivalents) of (R)-tetrahydro-1-methyl-3,3-diphenyl-1H,3H-pyrrolo-[1,2-c][1,3,2]oxazaborole in an inert solvent (preferably a mixture of toluene and dichloromethane).
- the reaction takes place at -30 to 40°C (preferably -20°C) for 1-24 hours (preferably 12 hours), to give the corresponding carbinol of Formula 104b.
- Step 1 an aldehyde of Formula 103 is transformed into an unsaturated aldehyde of Formula 106 by a Wittig reaction with an ylid of Formula 103b (where Z 1 is H or lower alkyl).
- An aldehyde of Formula 103 is reacted with one molar equivalent of an ylid of Formula 103b, in an organic solvent (such as dichloromethane, dimethylformamide or preferably toluene).
- the reaction takes place at 0 to 110°C (preferably 80°C) for 1 to 24 hours (preferably 8 hours) to give the corresponding unsaturated aldehyde of Formula 106.
- an unsaturated aldehyde of Formula 106 is condensed with the anion of an ester of Formula 106a (where Z 3 is H, lower alkyl, lower alkenyl, or phenyl and Z 4 is H, lower alkyl, or phenyl) to give a beta-hydroxy ester of Formula 107.
- An ester of Formula 106a is converted to an alkali metal salt by reacting a solution of the ester in an ethereal solvent (such as ether or preferably tetrahydrofuran) with an equimolar amount of an alkali metal hydride, hexamethyldisilazide or amide (preferably lithium
- ester anion solution 1.0 to 1.5 molar equivalents, preferably 1.0 molar equivalents
- the condensation reaction takes place at a temperature of -100°C to 0°C
- Step 1 the beta-hydroxy group of an ester of Formula 107 is O-alkylated to give the corresponding beta-alkoxy ester (R b ) of Formula 108.
- An ester of Formula 107 is reacted with 1 to 3 (preferably 1.5) molar equivalents of an alkyl halide (preferably an alkyl iodide, such as methyl iodide or n-butyl iodide, preferably methyl iodide) and 1 to 3 (preferably 1.25) molar equivalents of silver oxide, in a polar organic solvent (such as dioxane, dimethylformamide or preferably acetonitrile).
- a polar organic solvent such as dioxane, dimethylformamide or preferably acetonitrile
- Step 1 an alpha-halo alkyl ester of Formula 105 (where Z 1 is H, lower alkyl or CF 3 , Z 2 is H or lower alkyl, Z 3 is H, lower alkyl, lower alkenyl, or phenyl, and Z 4 is halo) is converted to an alpha-hydroxy acid of Formula 1A where Z 4 is hydroxy.
- the reaction takes place by hydrolysis of an alpha-alkanoyloxy ester
- alpha-halo (preferably chloro) ester of Formula 105 is reacted with 1 to 5 (preferably 3) molar equivalents of an alkali metal alkanoate (the metal preferably potassium and the alkanoate preferably acetate) in a polar organic solvent (such as acetonitrile or preferably
- alpha-alkanoyloxy ester is then subjected to basic hydrolysis by reaction with 1 to 5 (preferably 2) molar equivalents of an alkali metal hydroxide (preferably sodium hydroxide) in a mixture of water and an organic solvent (such as methanol, dimethoxyethane or preferably
- Step 1 an unsaturated aldehyde of Formula 106 is reduced and then converted to the corresponding compound of Formula 109 in which R b is a leaving group (a sulfonate or halide, preferably a bromide).
- R b is a leaving group (a sulfonate or halide, preferably a bromide).
- An unsaturated aldehyde of Formula 106 is reacted with from 0.5 to 2 (preferably 1) molar equivalents of a reducing agent (such as sodium cyanoborohydride or preferably sodium borohydride) in an alcoholic solvent (such as ethanol, isopropanol or preferably methanol).
- a reducing agent such as sodium cyanoborohydride or preferably sodium borohydride
- an alcoholic solvent such as ethanol, isopropanol or preferably methanol.
- the allylic alcohol is reacted with from 1 to 1.5 (preferably 1.25) molar equivalents of a sulfonating agent (such as p-toluenesulfonyl chloride) and an organic base, or preferably reacted with a halogenating reagent (such as carbon tetrachloride/triphenylphosphine or preferably N-bromosuccinimide/triphenylphosphine) in an inert organic solvent (such as ether or preferably dichloromethane).
- a sulfonating agent such as p-toluenesulfonyl chloride
- a halogenating reagent such as carbon tetrachloride/triphenylphosphine or preferably N-bromosuccinimide/triphenylphosphine
- an inert organic solvent such as ether or preferably dichloromethane
- an allylic halide or sulfonate of Formula 109 is alkylated with a chiral 4-alkyl N-acyl oxazolidinone of Formula 109a to give the corresponding chiral substituted acyl oxazolidinone of Formula 110.
- hexamethyldisilazide or dialkylamide preferably lithium diisopropylamide
- an inert organic solvent such as ether or preferably tetrahydrofuran.
- the reaction takes place at -100 to -20°C (preferably -80°C) for 5 to 120 minutes (preferably 30 minutes).
- the solution of the salt (1 to 1.5, preferably 1.25 molar equivalents) is then added to a solution of an allylic compound of Formula 109 in the same solvent.
- the alkylation reaction takes place at -100 to 0°C (preferably -80°C) for 30 minutes to 6 hours (preferably 1 hour) to afford the corresponding chiral substituted acyl oxazolidinone of Formula 110.
- a chiral substituted acyl oxazolidinone of Formula 110 is hydrolyzed to the corresponding chiral acid of Formula 1A.
- Use of an acyl oxazolidinone of Formula 109a having a 4-alkyl substituent of the opposite configuration in Reaction Scheme VI, Step 2, followed by hydrolysis as described in Step 3 results in the corresponding chiral acid where Z 3 has the opposite configuration .
- An acyl oxazolidinone of Formula 110 is reacted with from 1.25 to 3.5 (preferably 3.0) molar equivalents of lithium hydroxide, in a mixture of water and a water-miscible organic solvent (such as dioxane or preferably tetrahydrofuran) containing from 6 to 10 (preferably 8) molar equivalents of 30% aqueous hydrogen peroxide.
- the reaction takes place at -20 to 40°C (preferably 20°C) for 1 to 24 hours (preferably 12 hours) to afford the corresponding chiral acid of Formula 1A.
- Step 1 an allylic compound of Formula 109 in which R b is a leaving group (a sulfonate or halide, preferably a bromide) is condensed with an ester of Formula 109b to give the mono- or di-alkyl ester of Formula 112 (where Z 3 is H, lower alkyl, lower alkenyl, or phenyl and Z 4 is H, lower alkyl, or phenyl).
- R b is a leaving group (a sulfonate or halide, preferably a bromide)
- An ester of Formula 109b is converted to an alkali metal salt by reaction with 1.05 to 1.25 (preferably 1.1) molar equivalents of an alkali metal amide (such as sodium hexamethyldisilazide, potassium
- Step 1 a 2-(alkylthio)-4-hexenoic acid ester of Formula 1A (where Z 3 is S-lower alkyl, and Z 4 is H or lower alkyl) is oxidized to give the corresponding 2-(alkylsulfinyl)- or 2-(alkylsulfonyl)-4-hexenoic acid ester of Formula 1A where Z 3 is S(O)lower alkyl or S(O) 2 lower alkyl.
- the reaction can be performed with an acid of Formula 1A where Z 3 is S-lower alkyl, to give the
- An alkylthio-4 -hexenoic acid ester of Formula 1A is reacted with 1.0 to 1.25 (preferably 1.05) molar equivalents of an oxidizing agent (such as oxone ® ) optionally in the presence of an inert support (such as alumina), in a solvent (such as chloroform or preferably dichloromethane).
- an oxidizing agent such as oxone ®
- an inert support such as alumina
- a solvent such as chloroform or preferably dichloromethane
- a 2-(alkylsulfinyl)- or 2-(alkylsulfonyl)-4-hexenoic acid ester of Formula I-ZA-K is hydrolyzed to give the corresponding acid as described with reference to Reaction Scheme X, Step 2.
- Step 1 a protected aldehyde of Formula 103 and a triphenylphosphoranylideneacetate of Formula 103c are combined in a Wittig reaction to give the corresponding alkyl-2-halobutenoate ester of Formula 114.
- An aldehyde of Formula 103 is reacted with 1.0 to 1.5 (preferably
- a 2-halo-4-aryl-2-butenoate ester of Formula 114 (preferably a t-butyl ester) is converted to the corresponding acid (preferably by dissolution in trifluoroacetic acid at room temperature for 1 to 2 hours).
- the acid is isolated and purified by conventional means, then reacted with 0.5 to 3 (preferably 1.6) molar equivalents of a reducing agent (such as sodium cyanoborohydride, sodium borohydride, or preferably borane dimethyl disulfide complex) in an inert solvent (such as methanol, ethanol, isopropanol or preferably THF).
- a reducing agent such as sodium cyanoborohydride, sodium borohydride, or preferably borane dimethyl disulfide complex
- an inert solvent such as methanol, ethanol, isopropanol or preferably THF.
- the reaction takes place at 0 to 50°C (preferably 25°C) for 1 to 48 hours (preferably 24 hours) to give
- the allylic alcohol so-produced is reacted with from 1 to 1.5
- a sulfonating agent such as p-toluenesulfonyl chloride
- an organic base or preferably reacted with a halogenating reagent (such as carbon tetrachloride/triphenylphosphine or preferably N-bromosuccinimide/triphenylphosphine) in an inert organic solvent (such as ether or preferably dichloromethane).
- a sulfonating agent such as p-toluenesulfonyl chloride
- a halogenating reagent such as carbon tetrachloride/triphenylphosphine or preferably N-bromosuccinimide/triphenylphosphine
- an inert organic solvent such as ether or preferably dichloromethane
- Step 3 a protected 2-halo-4-aryl-2-butenyl bromide compound of Formula 115 is condensed with a dialkyl malonate of Formula 106b (substituted by Z 4 where Z 4 is hydrogen, lower alkyl, or phenyl), which is hydrolysed and decarboxylated to give the corresponding 4-halo-4-hexenoic acid derivative of Formula 1A where Z 1 is halo.
- a malonic ester of Formula 106b (where Z 4 is H, lower alkyl, or phenyl) is converted to an alkali metal salt by reaction with 1.05 to 1.25 (preferably 1.1) molar equivalents of an alkali metal hydride (preferably sodium hydride) in an organic solvent (such as ether, dioxane or preferably tetrahydrofuran).
- an alkali metal hydride preferably sodium hydride
- organic solvent such as ether, dioxane or preferably tetrahydrofuran
- the dialkyl ester thus produced is then hydrolysed conventionally, using a strong base, preferably aqueous sodium hydroxide, in a protic solvent, preferably ethanol, heating to reflux.
- a strong base preferably aqueous sodium hydroxide
- a protic solvent preferably ethanol
- the dicarboxylic acid thus produced is separated conventionally, and then decarboxylated by heating, preferably in a high-boiling inert solvent, most preferably 1,2- dichlorobenzene, to give the corresponding 4-halo-4-hexenoic acid
- Step 1 a protected phenol of Formula 117 (which can be any of the corresponding protected compounds of Reaction Schemes I to IX, such as Formulae 105, 107, 108, 112, and the like) is deprotected to give the corresponding alkyl ester of Formula 1A as an ester.
- An alkyl ester of Formula 117 (having either an acetal-type or a silyl-type protecting group) is treated with from 0.05 to 0.2 molar equivalents (preferably 0.1 molar equivalents) of an aqueous mineral acid (such as sulfuric, perchloric, or preferably hydrochloric acid), in a water-miscible organic solvent (such as methanol, acetone, or preferably ethanol).
- the reaction takes place at 0 to 50°C (preferably 25°C) over a period of 1 to 6 hours (preferably 2 hours) to give the corresponding free phenol of Formula 1A.
- a compound of Formula 117 is treated with 0.05 to 0.25 molar equivalents (preferably 0.1 molar equivalents) of a Lewis acid (such as zinc chloride or preferably zinc bromide), in a solvent (such as benzene, chloroform, or preferably dichloromethane).
- a Lewis acid such as zinc chloride or preferably zinc bromide
- a solvent such as benzene, chloroform, or preferably dichloromethane
- a compound of Formula 117 is reacted with 1.0 to 1.5 (preferably 1.25) moles of a tetraalkyl ammonium fluoride (preferably tetrabutylammonium fluoride) in an ethereal solvent (such as dioxane or preferably tetrahydrofuran).
- a tetraalkyl ammonium fluoride preferably tetrabutylammonium fluoride
- an ethereal solvent such as dioxane or preferably tetrahydrofuran
- Step 2 a compound of Formula 1A as an ester (prepared as described above) is hydrolyzed to give the corresponding acid of Formula 1A as a carboxylic acid.
- An alkyl ester of Formula 1A is reacted with from 1.5 to 4 molar equivalents (preferably 2 molar equivalents) of an inorganic hydroxide (such as potassium, sodium, or preferably lithium hydroxide) in a mixture of water and an organic solvent (such as tetrahydrofuran, methanol, or preferably dimethoxyethane).
- an inorganic hydroxide such as potassium, sodium, or preferably lithium hydroxide
- an organic solvent such as tetrahydrofuran, methanol, or preferably dimethoxyethane
- the resulting anion is acidified with an aqueous mineral acid (such as hydrochloric acid).
- the acidification takes place at 0 to 40°C (preferably 25°C) over a period of 1 to 10 minutes (preferably 2 minutes) to give the corresponding carboxylic acid of Formula 1A.
- M Li or MgBr.
- the aldehyde of Formula 103 is converted to a carbinol of Formula 202 by addition of an unsaturated cyclic organometallic compound of Formula 201 where M is Li or MgBr, prepared for example as described above with reference to Reaction Scheme I, Step 3.
- One molar equivalent of the organometallic reagent 201 is added to a solution of an aldehyde of Formula 103 (in the same solvent system used to make the organometallic reagent).
- the reaction takes place at -80 to -20°C (preferably -40°C) over a period of 5 to 60 minutes (preferably 15 minutes) to give the corresponding carbinol of Formula 202.
- the racemic compound of Formula 202 may be separated into its two enantiomers by conventional means, for example by conversion into two diastereoisomers that are then separated by crystallization,
- the carbinol is reacted with a chiral isocyanate to give a mixture of diastereoisomeric carbamates, which are separated by chromatography and cleaved to give the pure enantiomers.
- a carbinol of Formula 202 is heated at 30 to 100°C (preferably about 60°C) with 2 to 6 molar equivalents (preferably 4 molar equivalents) of a chiral isocyanate in the presence of 1 to 1.5 molar equivalents (preferably 1.2 molar equivalents) of a strong organic base, for example
- diastereoisomers are then separately cleaved by treatment with 1 to 1.5 molar equivalents (preferably 1.2 molar equivalents) of a trihalosilane, for example trichlorosilane, in the presence of an excess of a tertiary amine, for example triethylamine, in an inert solvent, for example toluene.
- a trihalosilane for example trichlorosilane
- a tertiary amine for example triethylamine
- an inert solvent for example toluene.
- the reaction takes place at a temperature of 90-120°C (preferably 110°C) over a period of 5 minutes to 2 hours (preferably 15 minutes) to give the corresponding enantiomer of the carbinol of Formula 202.
- an alkyl ester of Formula 203 is formed by a Claisen ortho ester reaction of a carbinol of Formula 201 (or an enantiomer thereof) with an appropriately substituted orthoester.
- a carbinol of Formula 202 is heated at 50 to 140°C (preferably 130°C) with a large excess of an orthoester of Formula 104a (see Reaction Scheme I, step 4), in the presence of from 0.05 to 0.25 molar equivalents
- Step 1 an aldehyde of Formula 103 (prepared, for example as described above with reference to Reaction Scheme I, Steps 1 and 2) is transformed into an unsaturated aldehyde of Formula 302 by a Wittig reaction with an ylid of Formula 301 (where Z 5 is H or lower alkyl).
- An aldehyde of Formula 103 is reacted with one molar equivalent of an ylid of Formula 301, in an organic solvent (such as dichloromethane, dimethylformamide or preferably toluene).
- an organic solvent such as dichloromethane, dimethylformamide or preferably toluene.
- the reaction takes place at 0 to 110°C (preferably 80°C) for 1 to 24 hours (preferably 8 hours) to give the corresponding unsaturated aldehyde of Formula 302.
- an unsaturated aldehyde of Formula 302 is converted to the corresponding vinyl carbinol of Formula 303.
- An aldehyde of Formula 302 is reacted with from 1.0 to 1.25
- an organovinyl compound preferably vinylmagnesium bromide
- a solvent such as ether or preferably
- Step 3 a vinyl carbinol of Formula 303 is oxidized to give the corresponding dienone of Formula 304.
- a vinyl carbinol of Formula 303 is reacted with 1.0 to 1.5
- an oxidizing agent such as manganese dioxide, pyridinium chlorochromate or preferably pyridinium dichromate
- a solvent such as pyridine or preferably dichloromethane
- a dienone of Formula 304 is cyclized to give the corresponding cyclopentenone of Formula 305.
- a dienone of Formula 304 reacted with 0.3 to 1.5 (preferably 1.0) molar equivalents of a Lewis acid (such as boron trichloride, tin (IV) chloride or preferably boron trifluoride etherate) in a solvent (such as tetrachloroethane or preferably dichloromethane).
- a Lewis acid such as boron trichloride, tin (IV) chloride or preferably boron trifluoride etherate
- a solvent such as tetrachloroethane or preferably dichloromethane
- a cyclopentenone of Formula 305 is reacted with 1.0 to 1.5
- a reducing agent such as lithium tri-tert-butoxyaluminium hydride or preferably sodium borohydride in the presence of an equimolar amount of cerium trichloride
- cerium trichloride preferably tetrahydrofuran
- Step 6 a cyclopentenol of Formula 306 is transformed to the corresponding vinyl ether of Formula 307.
- a cyclopentenol of Formula 306 is reacted with from 10 to 100
- a vinyl ether of Formula 307 is reacted with from 10 to 100
- An acetaldehyde of Formula 308 is reacted with 1 to 3 (preferably 1.5) molar equivalents of a suitable oxidizing agent (such as silver oxide, Jones reagent or preferably sodium chlorite) in the presence of an oxidizing agent (such as silver oxide, Jones reagent or preferably sodium chlorite) in the presence of an oxidizing agent (such as silver oxide, Jones reagent or preferably sodium chlorite) in the presence of an oxidizing agent (such as silver oxide, Jones reagent or preferably sodium chlorite) in the presence of an oxidizing agent (such as silver oxide, Jones reagent or preferably sodium chlorite) in the presence of an oxidizing agent (such as silver oxide, Jones reagent or preferably sodium chlorite) in the presence of an oxidizing agent (such as silver oxide, Jones reagent or preferably sodium chlorite) in the presence of an oxidizing agent (such as silver oxide, Jones reagent or preferably sodium chlorite) in the presence of an oxidizing agent (
- a phenol such as quinol or preferably resorcinol
- the reaction is conducted in a mixture of water and a water-miscible organic solvent (such as tetrahydrofuran or preferably dioxane) at a pH of from 4 to 6 (preferably 5) at -10 to 25°C (preferably 0°C) for 10 minutes to 2 hours (preferably 30 minutes) to give the corresponding acid of
- R a is a sulphonyloxy protecting group hydrolyzed under basic conditions, using from 1 to 5 (preferably 3) molar equivalents of an alkali metal hydroxide (preferably lithium hydroxide) in a mixture of water and a water-miscible organic solvent (such as dioxane or preferably methanol).
- the reaction takes place at 40 to 100°C (preferably 60°C) for 1 to 48 hours (preferably 12 hours) to afford the corresponding cyclopentene carboxylic acid of Formula 1C.
- an aldehyde of Formula 103 (where R a is a silyl protecting group) is converted to a carbinol by addition of an organometallic compound of Formula 103d (such as a
- M is MgBr or Li
- Z 2 is H or lower alkyl
- TBS is a tert-butyldimethylsilyl protecting group
- the aldehyde of Formula 103 is reacted with from 1.1 to 1.5
- Formula 402 is formed by a Claisen ortho ester rearrangement reaction of a carbinol of Formula 401 and a trialkyl orthoacetate of Formula 104a.
- a carbinol of Formula 401 is heated at 50 to 120°C (preferably about 100°C) with about 10 molar equivalents of an orthoester of Formula 104a, in the presence of from 0.05 to 0.25 (preferably 0.10) molar equivalents of an organic acid catalyst (such as propionic, butyric, or preferably
- an alkyl ester of Formula 402 is reacted with a tetraalkylammonium fluoride and then halogenated to give a protected ester of Formula 403.
- a compound of Formula 402 is reacted with from 2.0 to 3.0 (preferably 2.0) molar equivalents of a tetraalkylammonium (preferably
- tetrabutylammonium fluoride in a solvent (such as dioxane,
- a halogenating agent preferably a brominating agent, such as triphenylphosphine/carbon tetrabromide or preferably
- triphenylphosphine/N-bromosuccinimide in a solvent such as ether or preferably dichloromethane.
- the reaction takes place at from -40 to 0°C (preferably -10°C) for from 1 to 6 hours (preferably 4 hours) to give the corresponding halogenated ester of Formula 403.
- a halogenated ester of Formula 403 is cyclized to give a cycloalkyl ester of Formula 1C.
- a compound of Formula 403 is reacted with from 2.0 to 2.5 (preferably 2.25) molar equivalents of a strong base (such as lithium diisopropylamide, sodium hydride or preferably sodium hexamethyldisilazide) in an ethereal solvent (such as ether, dioxane or preferably tetrahydrofuran)
- a strong base such as lithium diisopropylamide, sodium hydride or preferably sodium hexamethyldisilazide
- an ethereal solvent such as ether, dioxane or preferably tetrahydrofuran
- Step 1 the 2 -methyl group of an alkyl 2-methylbenzoate of Formula 501 (where Z 6 and Z 7 are selected from H, lower alkyl, lower alkoxy, lower alkoxycarbonyl, halo and nitro) is brominated to give the corresponding compound of Formula 502.
- An ester of Formula 501 is reacted with 1.0 to 1.2 (preferably 1.05) molar equivalents of a brominating agent (such as N-bromoacetamide or preferably N-bromosuccinimide), optionally in the presence of an initiator (such as visible light) or from 0.001 to 0.01 (preferably 0.005) molar equivalents of a chemical initiator (such as azobisisobutyronitrile or preferably benzoyl peroxide) in a solvent (such as ethyl formate or preferably carbon tetrachloride).
- a brominating agent such as N-bromoacetamide or preferably N-bromosuccinimide
- an initiator such as visible light
- a chemical initiator such as azobisisobutyronitrile or preferably benzoyl peroxide
- solvent such as ethyl formate or preferably carbon tetrachloride
- the reaction takes place at 40 to 80°C (preferably 75°C) for 30 minutes to to 6 hours (preferably 2 hours) to afford the corresponding alkyl 2-bromomethylbenzoate of Formula 502, which can be purified by conventional means or preferably used directly for the next step.
- a 2-bromomethylbenzoate of Formula 502 is reacted with 1.0 to 1.25 (preferably 1.05) molar equivalents of a triaryl phosphine (preferably triphenyl phosphine) in a solvent (such as dimethylformamide or preferably acetonitrile).
- a solvent such as dimethylformamide or preferably acetonitrile.
- the reaction takes place at 25 to 90°C (preferably 50°C) for 1 to 24 hours (preferably 2 hours) to afford the corresponding phosphonium salt of Formula 503.
- a phosphonium salt of Formula 503 is dissolved or suspended in a solvent (such as dioxane, ether or preferably dimethylformamide) and reacted with 1.0 to 1.25 (preferably 1.05) molar equivalents of a base (such as sodium hydride, triethylamine or preferably 1,5-diazabicyclo[4.3.0]non-5-ene).
- a solvent such as dioxane, ether or preferably dimethylformamide
- a base such as sodium hydride, triethylamine or preferably 1,5-diazabicyclo[4.3.0]non-5-ene.
- the reaction takes place at 0 to 60°C (preferably 25°C) for 1 to 6 hours (preferably 2 hours) to afford the corresponding ylid of Formula 504, which can be isolated by conventional means or its solution can be used directly for the next step.
- an ylid of Formula 504 and a protected aldehyde of Formula 103 are employed in a Wittig reaction to give the corresponding protected substituted benzoic acid alkyl ester of Formula 505 as a mixture of E and Z isomers, from which the desired E isomer of Formula 506 is isolated, as illustrated in Reaction Scheme XV, Step 5.
- a solution of 0.8 to 1.0 (preferably 0.95) molar equivalents of a protected aldehyde of Formula 103 in a solvent (such as ether, dioxane or preferably dimethylformamide) is added to a solution of an ylid of Formula 504 in the same solvent.
- the reaction takes place at 0 to 50°C (preferably 25°C) for 1 to 24 hours (preferably 12 hours) to afford the corresponding protected substituted benzoic acid alkyl ester of Formula 505 as a mixture of E and Z isomers, from which the desired E-isomer of Formula 506 can be isolated by conventional means (such as distillation, chromatography or preferably fractional crystallization).
- Step 1 the alpha carbon of an alkyl 2-alkylbenzoate of Formula 507 (where Z 5 is H or lower alkyl, and Z 6 and Z 7 are selected from H, lower alkyl, lower alkoxy, lower
- a compound of Formula 508 is reacted with from 5 to 20 (preferably 10) molar equivalents of a trialkyl phosphite (preferably triethyl phosphite). The reaction takes place at 100 to 200°C (preferably 150°C) for 1 to 24 hours (preferably 6 hours) to afford the corresponding phosphonate of Formula 509.
- a trialkyl phosphite preferably triethyl phosphite
- a phosphonate of Formula 509 is reacted with 1.0 to 1.5
- a base such as sodium amide, potassium tert-butoxide or preferably sodium hydride
- a solvent such as dioxane, dimethylformamide or preferably dimethoxyethane
- the alkali metal salt is reacted with from 0.9 to 1.1 (preferably 1.0) molar equivalents of a protected aldehyde of Formula 103, dissolved in the same solvent.
- the reaction takes place at 0 to 60°C (preferably 40°C) for 1 to 6 hours (preferably 2 hours) to afford the corresponding protected optionally substituted benzoic acid alkyl ester of Formula 510 as a mixture of E and Z isomers, from which the desired E-isomer of Formula 511 can be isolated by conventional means (such as distillation, chromatography or preferably fractional crystallization).
- Step 1 a protected aldehyde of Formula 103 is converted to a trialkylsilylcarbinol of Formula 513 in a
- a trialkylsilylalkyl-magnesium bromide such as trimethylsilylpropylmagnesium bromide, or preferably trimethylsilylmethylmagnesium bromide
- an ethereal solvent such as ether, dimethoxyethane or preferably tetrahydrofuran
- the reaction takes place at -40 to 40°C (preferably 0°C) for 30 minutes to
- a carbinol of Formula 513 is reacted with from 1.0 to 1.5 (preferably 1.05) molar equivalents of a sulphonyl chloride (such as p-toluenesulphonyl chloride or preferably methanesulphonyl chloride) in the presence of the same molar proportion of a tertiary organic base (such as N-methylpyrrolidine or preferably triethylamine).
- a sulphonyl chloride such as p-toluenesulphonyl chloride or preferably methanesulphonyl chloride
- the reaction takes place at 0 to 40°C (preferably 15°C) for 30 minutes to 4 hours (preferably 2 hours) to afford the corresponding protected alkene of Formula 514 as a mixture of E and Z isomers, from which the desired Z-isomer of Formula 514 can be isolated by conventional means (such as distillation, chromatography or preferably fractional crystallization).
- an alkene of Formula 514 where R" is a silyl protecting group is converted to an alkene of Formula 515 where R a is an acyl group.
- An alkene of Formula 514 is heated at 50-130°C (preferably about 118°C) with a large excess of a mixture (preferably about equimolar) of a carboxylic acid of Formula ROH and an anhydride of Formula (R a ) 2 O (where R a is the desired acyl group), preferably a mixture of acetic acid and acetic anhydride.
- the reaction takes place over a period of 6 to 48 hours
- a protected alkene of Formula 515 is converted to a protected optionally substituted benzoic acid alkyl ester of Formula 511 in a Heck reaction with an alkyl-2-halo-benzoate of Formula 515.
- An alkene of Formula 515 is reacted with 1.0 to 2.0 (preferably 1.25) molar equivalents of an alkyl 2-halobenzoate (such as an alkyl
- the reaction is conducted in the presence of from 0.001 to 0.1 (preferably 0.05) molar equivalents of a palladium catalyst [such as tetrakis (tri-o-tolylphosphine) palladium, or tetrakis (triphenylphosphine)palladium or preferably palladium (II) acetate] optionally in the presence of from 1.0 to 1.25 (preferably 1.05) molar equivalents of a base (such silver carbonate, sodium bicarbonate or preferably triethylamine), in a solvent (such as acetonitrile or preferably dimethylformamide).
- a palladium catalyst such as tetrakis (tri-o-tolylphosphine) palladium, or tetrakis (triphenylphosphine)palladium or preferably palladium (II) acetate
- a base such silver carbonate, sodium bicarbonate or preferably triethylamine
- solvent such as
- the compounds of Formula 1E where Z 6 is nitro are employed as precursors to the corresponding compounds of Formula 1E where Z 6 is amino.
- the nitro compounds are also active as IMPDH inhibitors when tested as described below.
- a nitrobenzoic acid of Formula 1E (where Z 6 is nitro) is reacted with 1.0 to 3.0 (preferably 2.0) molar proportions of a reducing agent (such as sodium hydrosulfite or preferably tin (II) chloride) in hydrochloric acid solution, optionally in the presence of a water-miscible co-solvent (such as methanol or preferably acetic acid).
- a reducing agent such as sodium hydrosulfite or preferably tin (II) chloride
- hydrochloric acid solution optionally in the presence of a water-miscible co-solvent (such as methanol or preferably acetic acid).
- a water-miscible co-solvent such as methanol or preferably acetic acid
- Step 1 a phosphonate of Formula 509 undergoes a base catalyzed condensation (e.g., using 1 molar equivalent of sodium hydride) with tetrahydropyranyloxyacetaldehyde, in a solvent such as dimethylformamide.
- the reaction takes place at 25°C over a period of 1 to 4 hours, to give E/Z mixture from which the desired product of Formula 517 can be isolated by conventional means, such as
- tetrahydropyranyloxy group of a compound of Formula 517 is hydrolyzed in the presence of a catalytic amount of a dilute acid (e.g., HCl) in aqueous tetrahydrofuran.
- a catalytic amount of a dilute acid e.g., HCl
- the reaction takes place at 25°C over a period of 1 to 4 hours, to give the corresponding carbinol of Formula 518.
- Step 3 a carbinol of Formula 518 is converted to the halo (e.g., chloro or bromo) derivative of Formula 519 using 1 molar equivalent of triphenylphosphine and either carbon tetrachloride or carbon tetrabromide, in dichloromethane.
- the reaction takes place at 25°C over a period of 2 hours.
- a halo derivative of Formula 519 undergoes a base-catalyzed ether formation with the indicated phenol, using 5 molar equivalents of potassium carbonate, in
- an ether of Formula 520 is rearranged to give the corresponding ester of Formula IE by a thermal rearrangement catalysed by florisil.
- the rearrangement takes place in toluene at 110°C over a period of one to four days.
- Step 1 an aldehyde of Formula 601, prepared for example as shown in J. Org. Chem.. 1977, p3408, is reduced to a carbinol of Formula 602.
- An aldehyde of Formula 601 is reacted with a reducing agent capable of selectively reducing an aldehyde in the presence of ester groups, preferably from 1 to 2 (preferably 1.5) molar equivalents of sodium borohydride in the presence of from 1 to 2 (preferably 1.5) molar
- a carbinol of Formula 602 is reacted with an equimolar amount of a phenol of Formula 603 in the presence of from 1 to 3 (preferably 2) molar equivalents of a triarylphosphine, preferably triphenylphosphine, plus from 1 to 3 (preferably 1.5) molar equivalents of diethyl azodicarboxylate in an ethereal solvent (preferably tetrahydrofuran).
- the reaction takes place at 0 to 40°C (preferably 25°C) for 1 to 10 hours (preferably 3 hours) to give the corresponding ether of Formula 604.
- An ether of Formula 604 is heated in an inert solvent (preferably toluene) in the presence of about 10 parts by weight of an activated magnesium silicate, preferably Florisil ® .
- the reaction takes place at reflux temperature for 1 to 10 days (preferably 4 days) to give the corresponding diester of Formula 605.
- a diester of Formula 605 is reacted with an excess of an inorganic base, preferably about 50 molar equivalents of lithium hydroxide, in an aqueous solvent (preferably 5:1 methanol:water). The reaction takes place at 0 to 40°C (preferably 25°C) for 1 to 10 days (preferably 2 days) to give the corresponding dicarboxylic acid of Formula 606.
- a dicarboxylic acid of Formula 606 is decarboxylated to give a monocarboxylic acid of Formula 1F.
- a dicarboxylic acid of Formula 606 is heated (optionally in the presence of a high boiling inert solvent, for example tetramethylbenzene, but preferably in the absence of any solvent). The reaction takes place at 160 to 240°C (preferably 195°C) for about 5 minutes to give the
- the phenol of Formula 701 is alkylated with 3-hydroxycyclohexene to give the corresponding ether of Formula 703, by means of the Mitsonobu reaction.
- the Mitsonobu reaction takes place as described with reference to Reaction Scheme XIX, Step 2.
- An alkylated phenol of Formula 704 is reacted with an equimolar amount of t-butyl dimethylsilyl chloride or p-toluenesulfonyl chloride, in the presence of an equimolar amount, respectively, of imidazole or 4-dimethylaminopyridine.
- the reaction takes place in dichloromethane at a temperature of 25°C for 1 to 4 hours to give the corresponding protected phenol of Formula 705.
- a protected phenol of Formula 705 is converted to the corresponding dialdehyde of Formula 706 by ozonolysis.
- the ozonolysis reaction takes place as described with reference to Reaction Scheme I, Step 2.
- an aldehyde of Formula 103 is converted to a carbinol by addition of an organometallic compound of Formula 103e (such as a substituted vinyl organolithium, or preferably a Grignard reagent, where M is MgBr; TBS is a tert-butyldimethylsilyl protecting group; and n is 3-5).
- organometallic compound of Formula 103e such as a substituted vinyl organolithium, or preferably a Grignard reagent, where M is MgBr; TBS is a tert-butyldimethylsilyl protecting group; and n is 3-5).
- a halovinyl (preferably bromovinyl) compound of Formula 103e (where M is halo) is reacted with magnesium metal in an ethereal solvent (such as ether or preferably tetrahydrofuran). The reaction takes place at 30 to
- One molar equivalent of the resultant organometallic reagent is added to a solution of an aldehyde of Formula 103 (in the same solvent system used to make the organometallic reagent).
- the reaction takes place at -80 to 20°C (preferably 0°C) over a period of 5 to 60 minutes (preferably 10 minutes) to give the corresponding silyl-protected carbinol of Formula 801.
- Formula 802 is formed by a Claisen ortho ester rearrangement reaction of a carbinol of Formula 801 and an orthoester compound of Formula 104a (as illustrated in Reaction Scheme I, where Z 3 and Z 4 are H).
- a silyl-protected carbinol of Formula 801 is heated at 50 to 120°C
- a compound of Formula 803 is reacted with from 5 to 30 (preferably
- a carbinol of Formula 803 is converted to a halide (preferably a bromide) of Formula 804, by means of a one-step or a two-step procedure.
- a halide preferably a bromide
- a carbinol of Formula 803 is reacted with from 1.0 to 1.3 (preferably 1.1) molar equivalents of a triaryl (preferably triphenyl) phosphine, and from 1.0 to 1.3 (preferably 1.1) molar
- a halogen source such as N-bromosuccinimide or preferably carbon tetrabromide.
- the reaction is conducted in an inert solvent (such as ether or preferably tetrahydrofuran). The reaction takes place at 0 to
- a carbinol of Formula 803 is converted first into a sulphonate ester (such as a p-toluenesulphonate or preferably a methanesulphonate) by reaction with from 1.0 to 1.5 (preferably 1.3) molar equivalents a sulphonyl halide (preferably methanesulphonyl chloride) in the presence of an equimolar amount of a tertiary organic base (preferably diisopropylethylamine) in a solvent (such as chloroform or preferably dichloromethane).
- a sulphonate ester such as a p-toluenesulphonate or preferably a methanesulphonate
- a sulphonyl halide preferably methanesulphonyl chloride
- a tertiary organic base preferably diisopropylethylamine
- solvent such as chloroform or preferably dichloromethane
- the so-obtained sulphonate ester is then reacted with from 5 to 20 (preferably 20) molar equivalents of an alkali metal halide (preferably lithium bromide) in a solvent (t-such as 2-butanone or preferably acetone).
- a solvent such as 2-butanone or preferably acetone.
- the reaction takes place at 0 to 56°C (preferably at reflux) for 30 to 180 minutes (preferably 90 minutes) to afford the corresponding halide of Formula 804.
- a halogenated carbinol/alkyl ester of Formula 804 is deprotected at the phenolic group to give the corresponding halogenated carbinol/alkyl ester of Formula 805.
- the deprotection reaction takes place as described above with reference to Reaction Scheme X, Step 1.
- Step 6 a halogenated carbinol/alkyl ester of Formula 805 is subjected to a base-induced cyclization reaction to afford the product of Formula 1H.
- a compound of Formula 805 is reacted with from 2.0 to 2.5 (preferably 2.3) molar equivalents of a strong base (such as lithium diisopropylamide, sodium hydride or preferably sodium hexamethyldisilazide) in a solvent (such as dioxane or preferably tetrahydrofuran).
- a strong base such as lithium diisopropylamide, sodium hydride or preferably sodium hexamethyldisilazide
- a solvent such as dioxane or preferably tetrahydrofuran
- the cycloalkyl ester of Formula I-ZH-A1 may then be hydrolyzed to give the corresponding acid of Formula 1H.
- the hydrolysis takes place as described above with reference to Reaction Scheme X, Step 2.
- Formula 302 (where Z 5 is methyl) undergoes an aldol reaction with the bromo-alkyl oxazolidinone of Formula 806 (where q is 1 or 2), which can be prepared by analogy with the reactions described in J. Amer. Chem. Soc , 103, 2127 , 1981, to give the acyloxazolidinone of Formula 807.
- An oxazolidinone of Formula 806 is reacted with an equimolar amount of a base (such as lithium diisopropylamide or preferably di-n-butylboryl trifluoromethane sulphonate/triethylamine), and then with an aldehyde of Formula 302.
- a base such as lithium diisopropylamide or preferably di-n-butylboryl trifluoromethane sulphonate/triethylamine
- an acyloxazolidinone of Formula 807 is hydrolyzed to the carboxylic acid of Formula 808.
- An acyloxazolidinone of Formula 807 is reacted with 1-5 (preferably 3) molar equivalents of lithium hydroxide in 3 :1 tetrahydrofuran containing 5-20 (preferably 12) molar equivalents of hydrogen peroxide.
- the reaction takes place at -10 to 25°C (preferably 0°) for 5 to 60 minutes (preferably 30 minutes) to give the corresponding carboxylic acid of Formula 808.
- a phenol of Formula 809 is esterified to give the corresponding ester of Formula 810.
- a phenol of Formula 809 is treated with methanol in the presence of 0.05 to 0.2 (preferably 0.1) molar equivalents of an acid catalyst
- reaction takes place at 0 to
- a methyl ester of Formula 810 undergoes an intramolecular cyclization reaction to give the corresponding cyclized ester of Formula 1H.
- a methyl ester of Formula 810 is treated with 1.9 to 2.5 (preferably 2.0) molar equivalents of a strong base (such as lithium diisopropylamide or preferably sodium hydride) in tetrahydrofuran (or preferably
- a strong base such as lithium diisopropylamide or preferably sodium hydride
- reaction takes place at -10 to 25°C (preferably 0°) for 1-12 hours (preferably 2 hours) to give the corresponding cyclized ester of Formula IH, which may be hydrolyzed to give the corresponding acid of Formula IH, using the method described with respect to Reaction Scheme X, Step 2.
- esters of Formula 1 can be prepared as described in U.S. Patents Nos. 4,727,069 and 4,753,935, incorporated herein by reference, by deprotection of a precursor (e.g., as described with reference to Reaction Scheme X, Steps 1) or as described below by attachment of a leaving group and its replacement by the desired ester.
- the compound of Formula 1 may be prepared from the corresponding carboxylic acid by reaction with a large excess of an alcohol of the formula GH, where G is lower alkoxy, preferably methanol, with a catalytic amount of an acid catalyst, (such as methanesulfonic acid, sulfuric acid, hydrochloric acid and p-toluenesulfonic acid), preferably p-toluenesulfonic acid).
- an acid catalyst such as methanesulfonic acid, sulfuric acid, hydrochloric acid and p-toluenesulfonic acid
- the reaction is carried out in the temperature range from about 0°C to 40°C, preferably at about 20°C, for about 1 to 7 days, preferably about 24 hours.
- the lower alkyl ester of Formula 1 is isolated and purified by conventional means.
- a compound of Formula l is reacted in an organic solvent, preferably dichloromethane, with about 1 to 3 molar equivalents, preferably about 2 molar equivalents, of a base, preferably pyridine, and with a slight excess, preferably about 1.1 molar equivalents, of a sulfonic anhydride, (such as a halo lower alkyl sulfonic anhydride, halomethyl sulfonic anhydride, and halosulfonic anhydride, or preferably trifluoromethane- sulfonic anhydride or fluorosulfonic anhydride) or a sulfonyl halide, (such as trifluoromethylsulfonyl bromide, preferably trifluoromethylsulfonyl chloride).
- a sulfonic anhydride such as a halo lower alkyl sulfonic anhydride, halomethyl sulfonic anhydride, and halos
- the reaction is carried out in an inert solvent, preferably dichloromethane, in the temperature range from about -20°C to 20°C, preferably at about 0°C, for about 15 to 45 minutes, preferably about 30 minutes.
- an inert solvent preferably dichloromethane
- the trifluoromethylsulfonyl reaction product, a compound of Formula 2 is isolated and purified by conventional means.
- a compound of Formula 2 is reacted with about 1 to 3 molar
- Step 3 the cyano compound of Formula 3 is hydrolyzed to the carboxy compound of Formula 4.
- a compound of Formula 3 is hydrolyzed by reacting it with about 1 to 10 molar equivalents, preferably about 4 molar equivalents, of an inorganic base (e.g., sodium hydroxide, lithium hydroxide, or potassium hydroxide, preferably sodium hydroxide,) in a large excess of organic solvent, preferably in about 3:2 water-.methanol solution.
- the reaction is carried out in the temperature range from about 40°C to 130°C, preferably at about the reflux temperature of the 3:2 water/methanol solvent, for about 1 to 3 hours, preferably about 2 hours.
- the reaction solution is distilled, and an additional of about 1 to 1.6 molar equivalents, preferably about 1.3 molar equivalents, of an inorganic base (e.g., sodium hydroxide, lithium hydroxide, or potassium hydroxide, preferably sodium hydroxide,) is added and the reaction is continued in the temperature range from about 40°C to 130°C, preferably at about the reflux temperature of the remaining solution, for about 1 to 3 days, preferably about 2 days.
- an inorganic base e.g., sodium hydroxide, lithium hydroxide, or potassium hydroxide, preferably sodium hydroxide,
- the reaction product, a compound of Formula 4 is isolated and purified by conventional means.
- the compound of Formula 4 may be prepared by reacting a corresponding compound of Formula 2 with a catalytic amount of 1,1'-bis (diphenylphosphine)ferrocene palladium dichloride in a large excess of an alkanol (preferably methanol) in an organic solvent (preferably
- reaction product which is a diester of a compound of Formula 4 is then hydrolyzed by reacting it with about 1 to 10 molar equivalents, preferably about 4 molar equivalents, of an inorganic base, preferably aqueous lithium hydroxide, in a large excess of an organic solvent, preferably 4:1 methanol/water solution.
- the solution is heated to a temperature range from about 30°C to 80°C, preferably from about 50°C to 60°C, for about 1 to 10 hours, preferably for about 2 to 6 hours.
- the reaction product, a compound of Formula 4 is isolated and purified by conventional means.
- a carboxy derivative of Formula 4 is converted to the ester of Formula 5.
- the compound of Formula 4 is reacted in a large excess of a compound of the formula GH, where G is lower alkoxy, preferably methanol, with catalytic amounts of an acid catalyst, preferably p-toluenesulfonic acid.
- the reaction is carried out in the temperature range from about 0°C to 40°C, preferably at about 20°C, for about 4 hours to 3 days, preferably about 24 hours.
- the reaction product, a 4 -carboxy derivative of Formula 5 is isolated and purified by conventional means.
- a compound of Formula 5 is reacted with about 1 to 3 molar
- an organic base preferably triethylamine
- an organic solvent preferably dimethylformamide
- chlorophosphate in the temperature range from about -20°C to 20°C, preferably at about 0°C, allowing it to warm to the temperature range from about 0°C to 40°C, preferably at about 20°C, allowing the reaction to proceed for about 0.5 to 2 hours, preferably about 1 hour.
- the reaction mixture is recooled to the temperature range from about -20°C to 20°C, preferably at about 0°C, and a large excess of sodium azide is added and the reaction proceeds for about 10 to 30 hours, preferably about 18 hours.
- the isocyanato reaction product, a compound of Formula 6, is isolated and purified by conventional means.
- a compound of Formula 5 is reacted with about 1 to 3 molar equivalents, preferably about 2 molar equivalents, of an organic base, preferably triethylamine, in a large excess of an organic solvent, preferably dimethylformamide, and with a slight excess, preferably 1.2 molar equivalents, of a diphenyl or dialkyl phosphoroazide, preferably diphenyl phosphoroazide, in the temperature range from about 0°C to 40°C, preferably at about 20°C, for about 12 to 36 hours, preferably for about 24 hours.
- the isocyanato reaction product, a compound of Formula 6, is isolated and purified by conventional means.
- Z a represents a sidechain of Formula Z as defined in the Summary of the
- Z a and Z b are as defined above.
- a compound of Formula 6 is hydrolyzed with about 1 to 20 molar equivalents, preferably 10 molar equivalents, of an inorganic base, preferably lithium hydroxide monohydrate, in an inert organic solvent, preferably 3:10 water: 1,4-dioxane.
- the reaction is carried out in the temperature range from about 0°C to 40°C, preferably at about 20°C, for about 1 to 3 hours, preferably about 2 hours.
- the reaction product, a 4-amino compound of Formula IA where Z is Z b is isolated and purified by conventional means, preferably column chromatography.
- a compound of Formula IA is esterified with a lower alkanol of formula GH, where G is lower alkoxy, as described in the preparation of a compound of Formula I as an ester.
- a compound of Formula 6 is reacted with a large excess of an amine compound of the formula HNR 4 R 5 , where R 4 and R 5 are as defined in the Summary of Invention, for example, methylamine, dimethylamine, methylphenylamine, ammonia, and the like, in an inert organic solvent, preferably
- reaction is carried out in the temperature range from about 0°C to 40°C, preferably at about 20°C, for about 30 minutes to 2 hours, preferably about 1 hour.
- the reaction product, a 4-(optionally substituted ureido) ester of Formula 1B is isolated and purified by conventional means.
- An ester of Formula IB is hydrolyzed by reacting with about 1 to 10 molar equivalents, preferably about 4 molar equivalents of an inorganic base, preferably aqueous lithium hydroxide, in a large excess of an organic solvent, preferably 4:1 methanol/water.
- the solution is heated to a temperature range from about 30°C to 80°C, preferably from about 50°C to 60°C, for about 1 to 10 hours, preferably for about 2 to 6 hours.
- the reaction product, a 4-(optionally substituted ureido) acid compound of Formula IB is isolated and purified by conventional means.
- a compound of Formula IA is reacted in a large excess of an inert organic solvent, preferably dichloromethane, with about 1 to 6 molar equivalents, preferably about 2.5 molar equivalents, of an anhydride compound of the formula (R 3 C(O)) 2 O or of a acyl chloride of the formula R 3 C(O)Cl, where R 3 is as defined in the Summary of the Invention.
- the reaction is carried out in the temperature range from about 0°C to 40°C, preferably at about 20°C, for about 30 minutes to 2 hours, preferably about 1 hour.
- the reaction product, a carbamate ester of Formula IC is isolated and purified by conventional means, preferably by recrystallization.
- a compound of Formula IC as an ester is hydrolyzed as described in the preparation of a compound of Formula 1B to give the corresponding compound of Formula IC as a carboxylic acid.
- a compound of Formula IC is reacted with about 1 to 10 molar equivalents, preferably about 4.5 molar equivalents, of a weak base, preferably potassium carbonate, and with about 1 to 10 molar equivalents, preferably about 4 molar equivalents, of a lower alkyl bromide or iodide, preferably an iodide, in an inert organic solvent, preferably
- the reaction is carried out in the temperature range from about 0°C to 40°C, preferably at about 20°C, for about 12 to 48 hours, preferably about 24 hours.
- the organic layer is purified to give a carbamate ester of Formula ID where R 1 is lower alkyl and G is lower alkoxy.
- a carbamate ester of Formula ID where R 1 is lower alkyl is hydrolyzed as described in the preparation of a compound of Formula IB to give the corresponding carboxylic acid of Formula ID where R 1 is lower alkyl.
- An amido ester of Formula ID is hydrolyzed by reacting with about 1 to 10 molar equivalents, preferably about 4 molar equivalents of an inorganic base (for example sodium hydroxide, preferably lithium
- the reaction takes place with (a) a reaction time of 20 to 120 hours, preferably 50 to 100 hours and most preferably 100 hours and (b) an initial pot temperature range of 114 to 120°C increasing to a final pot temperature range of 118 to 130°C, preferably an initial pot temperature range of 115 to 118°C increasing to a final pot temperature range of 118 to 125°C, each depending on solute concentration and atmospheric pressure, and most preferably an initial pot temperature of 116°C increasing to a final pot temperature of 121°C with a ratio of the acid compound of Formula I (where G is hydroxy) to toluene of lgm:2ml at one atmosphere of pressure.
- Some of the compounds of Formula I may be converted to corresponding base addition salts by virtue of the presence of a carboxylic acid group.
- the conversion is accomplished by treatment with a stoichiometric amount of an appropriate base, such as potassium carbonate, sodium bicarbonate, ammonia, ethylenediamine, monoethanolamine, diethanolamine, triethanolamine and the like.
- an appropriate base such as potassium carbonate, sodium bicarbonate, ammonia, ethylenediamine, monoethanolamine, diethanolamine, triethanolamine and the like.
- the free acid is dissolved in a polar organic solvent such as ethanol, methanol or ethyl acetate, and the base added in water, ethanol, methanol or isopropanol.
- the temperature is maintained at about 0°C to 50°C.
- the resulting salt precipitates spontaneously or may be brought out of solution with a less polar solvent.
- the base addition salts of the compounds of Formula I may be decomposed to the corresponding free acids by treating with at least a stoichiometric amount of a suitable acid, such as hydrochloric acid or sulfuric acid, typically in the presence of aqueous solvent, and at a temperature of between about 0°C and 50°C.
- a suitable acid such as hydrochloric acid or sulfuric acid
- the free acid form is isolated by conventional means, such as extraction with an organic solvent.
- some of the compounds of Formula I may be converted to the acid addition salts by the substitution of an organic or inorganic acid for the base in the above procedure.
- the acid salts can be decomposed to the corresponding free bases by similar treatment with an appropriate base.
- R 1 is hydrogen or lower alkyl
- R 2 is hydrogen, lower alkyl, -C(O)R 3 , -C(O)NR 4 R 5 , -CO 2 R 6 , or -SO 2 R 3
- R 3 is hydrogen, lower alkyl, halo lower alkyl or optionally substituted phenyl
- R 4 is hydrogen, lower alkyl or optionally substituted phenyl
- R 5 is hydrogen, lower alkyl or optionally substituted phenyl
- R 6 is lower alkyl or optionally substituted phenyl
- Z is a side chain selected from Formulae ZA, ZB, ZC, ZD, ZE, ZF, ZG, and ZH:
- Z 1 is H, lower alkyl, halo or CF 3 ;
- Z 2 is H, lower alkyl, lower alkoxy, aryl, or -CH 2 Z 13 , where
- Z 13 is aryl or heteroaryl
- Z 3 is H, lower alkyl, lower alkenyl, lower alkoxy, phenyl,
- Z 12 is lower alkyl
- n 0, 1 or 2;
- Z 4 is H, lower alkyl, or phenyl,
- n is an integer from 1 to 6
- G 1 is H or lower alkyl
- G 2 is H or lower alkyl
- G 3 is lower alkylene of four to six carbon atoms, or lower alkylene of three to five carbon atoms plus one member that is -O-, -S-, or -N(G 4 )- where G 4 is H or lower alkyl;
- Z 5 is H or lower alkyl
- Z 8 is H or lower alkyl
- D 1 and D 2 together with their adjacent carbon atoms form an optionally substituted, saturated or unsaturated carbocyclic or heterocyclic ring of 3 to 7 atoms;
- G is as defined above;
- Z 5 , Z 8 , and G are as defined above; or
- D 3 is -CH 2 - or -CHjCHz-; and G is as defined above; or
- Z 6 is H, lower alkyl, lower alkoxy, -COOH, -NH 2 or halo;
- Z 7 is H, lower alkyl, lower alkoxy or halo
- D 4 is -CH 2 -, -CH 2 CH 2 -, -CH 2 CH 2 CH 2 - , -O-, or -OCH 2 -;
- R 2 is -C(O)R 3 wherein R 3 is lower alkyl, halo lower alkyl or optionally substituted phenyl; or
- R 2 is -C(O)R 3 , where R 3 is lower alkyl, halo lower alkyl or optionally substituted phenyl, and G is lower alkoxy, with a compound of the formula R 1 X, where R 1 is lower alkyl and X is iodine or bromine, to form a compound of Formula I wherein R 1 is lower alkyl; or
- one preferred category includes the compounds where Z is a sidechain of Formula ZA.
- a preferred group includes the compounds where Z 1 is hydrogen, especially where R 1 is hydrogen and R 2 is hydrogen or -C(O)R 3 .
- a preferred subgroup includes the compounds where R 2 , Z 2 and Z 3 are all hydrogen, especially where Z 4 is methyl.
- Another preferred subgroup includes the compounds where R 2 , Z 3 and Z 3 are all hydrogen, especially where Z 2 is methyl.
- Another preferred category includes the compounds where Z is a sidechain of Formula ZB, especially where R 1 is hydrogen and R 2 is hydrogen or -C(O)R 3 .
- a preferred group includes the compounds where D 1 and D 2 together with the their adjacent carbon atoms form a saturated carbocyclic ring of 5 or 6 carbon atoms.
- a preferred subgroup includes the compounds where D 1 and D 2 together represent -CH 2 CH 2 CH 2 -, especially where Z 5 and Z 8 are both hydrogen.
- a preferred class within this subgroup includes the compounds in which R 1 and R 2 are both hydrogen.
- Another preferred subgroup includes the compounds where D 1 and D 2 together represent -CH 2 -CH 2 CH 2 CH 2 -, especially where Z 5 and Z 8 are both hydrogen.
- a preferred class within this subgroup includes the compounds in which R 1 and R 2 are both hydrogen.
- Yet another preferred subgroup includes the compounds where D 1 and D 2 together represent -CH 2 CH 2 OCH 2 -, especially where Z 5 and Z 8 are both
- a preferred class within this subgroup includes the compounds in which R 1 and R 2 are both hydrogen.
- the compounds are useful as immunosuppressive agents, anti-inflammatory agents, anti-tumor agents, anti-proliferative agents, anti-viral agents and anti-psoriatic agents in mammals, whether domestic (cattle, pigs, sheep, goats, horses), pets (cats, dogs), or preferably humans.
- the compounds are inhibitors of inosine monophosphate dehydrogenase (IMPDH) and thus inhibit de novo purine synthesis; they have anti-proliferative effects (e.g., against smooth muscle cells and both B and T lymphocytes) and inhibit antibody formation and the glycosylation of cell adhesion molecules in lymphocytes and endothelial cells.
- IMPDH inosine monophosphate dehydrogenase
- the compounds are useful in treating auto-immune related disorders, for example: Type I Diabetes Mellitus;
- Inflammatory Bowel Disease e.g., Crohn's Disease and ⁇ lcerative Colitis
- Systemic Lupus Erythematosus Chronic Active Hepatitis; Multiple Sclerosis
- Grave 's Disease Hashimoto's Thyroiditis
- Behcet's Syndrome Myasthenia Gravis
- Sjogren's Syndrome Pernicious Anemia
- the compounds are also useful as therapeutic immunosuppressive agents in the treatment of Asthma, Immunohemolytic Anemia, Glomerulonephritis, and Hepatitis.
- Preventative uses of the compounds as immunosuppressive agents include the treatment of allograft rejection, for example, in cardiac, lung, pancreatic, renal, liver, skin and corneal allografts, and prevention of Graft vs. Host Disease.
- the compounds are useful for inhibiting proliferative responses to vascular injury, for example, stenosis following an insult to a blood vessel wall in post-angioplasty restenosis, and post-cardiac by-pass surgery restenosis.
- the compounds are useful as anti-inflammatory agents, for example, in treating Rheumatoid Arthritis, Juvenile Rheumatoid Arthritis and Uveitis.
- the compounds are useful in treating solid tumors and malignancies of lymphoreticular origin.
- the compounds' utility for treatment of solid tumors includes: cancers of the head and neck, including squamous cell carcinoma; lung cancer, including small cell and non-small cell lung carcinoma; mediastinal tumors;
- esophageal cancer including squamous cell carcinoma and adenocarcinoma; pancreatic cancer; cancer of the hepatobiliary system, including
- hepatocellular carcinoma including cholangiocarcinoma, gall bladder carcinoma and biliary tract carcinoma; small intestinal carcinoma, including
- adenocarcinoma adenocarcinoma, sarcoma, lymphoma and carcinoids
- colorectal cancer including colon carcinoma and rectal carcinoma
- metastatic carcinoma adenocarcinoma, sarcoma, lymphoma and carcinoids
- cancers of the genitourinary system including ovarian cancer, uterine sarcoma, and renal cell, ureteral, bladder, prostate, urethral, penile, testicular, vulvar, vaginal, cervical, endometrial, and fallopian tube carcinoma; breast cancer; endocrine system cancer; soft tissue sarcomas; malignant mesotheliomas; skin cancer, including squamous cell carcinoma, basal cell carcinoma and melanoma; cancer of the central nervous system; malignant bone tumors; and plasma cell neoplasms.
- lymphoreticular origin the compounds are useful in treating, for example: Lymphomas and Leukemias, including B, T and promonocyte cell line
- Chronic Lymphocytic Leukemia Acute Lymphocytic Leukemia and Hairy Cell Leukemia.
- the compounds are useful in treating, for example: retroviruses, including Human T-leukemia Viruses, Types I and II (HTLV-1 and HTLV-2) , Human Immuno Deficiency Viruses, Types I and II
- Herpes Viruses including EBV infected B-lymphocytes, CMV infection, Herpes Virus Type 6, Herpes Simplex, Types 1 and 2, (HSV-1, HSV-2) and Herpes Zoster.
- the compounds are useful in treating, for example, psoriasis and psoriatic arthritis.
- IMPDH Inosine 5'-Monophosphate Dehydrogenase
- Initial animal screening tests to determine anti-inflammatory activity potential include the adjuvant arthritis assay, e.g., according to the method of Pearson, Proc Soc Exp. Biol . Med. , 91:95-101 (1956). Also, in vitro tests, for example those using synovial explants from patients with rheumatoid arthritis, Dayer, et al., J. Exp. Med. , 145:1399-1404 (1977), are useful in determining whether compounds exhibit anti- inflammatory activity.
- Autoimmune activity is determined, for example, utilizing
- host disease are conducted, e.g., as described by Storb, Deeg, Whitehead, et al., "Methotrexate and cyclosporin compared with cyclosporin alone for prophylaxis of acute graft versus host disease after marrow transplantation for leukemia.” New England J. Med. , 314:729-735 (1986).
- Immunosuppressive activity is determined by both in vivo and in vitro procedures.
- In vivo activity is determined, e.g., utilizing a modification of the Jerne hemolytic plaque assay, [Jerne, et al., "The agar plaque technique for recognizing antibody producing cells,” Cell -bound Antibodies, Amos, B. and Kaprowski, H. editors (Wistar Institute Press, Philadelphia) 1963, p. 109].
- In vitro activity is determined, e.g., by an adaptation of the procedure described by Greaves, et al., "Activation of human T and B lymphocytes by polyclonal mitogens," Nature, 248:698-701 (1974).
- Anti-viral activity is determined, for example, by the procedure described by Smee, et al. ["Anti-Herpesvirus Activity of the Acyclic
- Anti-viral activity can likewise be determined by measurement of reverse transcriptase activity, for example, according to the method described by Chen et al., Biochem. Pharm. , 36:4361 (1987).
- a large scale clinical trial can be conducted, e.g., as described by Volberding, P.A., et al. "Zidovudine in asymptomatic human immunodeficiency virus infection: a controlled trial in persons with fewer than 500 CD4 positive cells per cubic millimeter, " New England J. Med., 322(14):941-949 (1990).
- a smaller scale (Phase I) clinical trial can be conducted, e.g., as described by Browne, et al., "2 ',3'-Didehydro-3'-deoxythymidine (d4T) in Patients with AIDS or AIDS-Related Complex:
- Tests for systemic activity in psoriasis can be carried out, for example, as described by Spatz, et al., "Mycophenolic acid in psoriasis," Bri tish Journal of Dermatology, 98:429 (1978).
- Tests for anti-tumor activity can be performed, for example, as described by Carter, et al. ["Mycophenolic acid: an anticancer compound with unusual properties,” Nature, 223:848 (1969)].
- In vitro activity for treating stenosis is demonstrated, for example, by inhibiting the proliferation of smooth muscle cells, as established by the following human arterial smooth muscle cell proliferation assay.
- Human smooth muscle cells are grown in culture.
- a test group is treated with the test compound added at selected concentrations in fresh media. Both groups receive 2 ⁇ Ci tritiated thymidine ( 3 HTdR), a radioisotope label.
- 3 HTdR 2 ⁇ Ci tritiated thymidine
- the cells are harvested and the amount of label incorporated into DNA is counted by scintillation; this is compared for the test and control groups, the amount being proportional to cell proliferation.
- Inhibition of smooth muscle proliferation is established when the test group has a lower radioisotope count than the control group.
- the concentrations of test compound required to inhibit proliferation by 50% (the IC 50 ), and to inhibit proliferation by more than 95% are determined.
- In vivo activity for treating stenosis is demonstrated, for example, in rat and pig models for arterial stenosis.
- a test group is treated with the test compound, starting 6 days before and continuing for 14 days after injury to the left carotid artery; the test group is compared to a control group receiving vehicle without the test compound.
- Injury is achieved by a gentle perfusion of air through a 10 mm long section of the left artery. The right artery is left intact.
- Arterial cross-sections (10 ⁇ m) are taken from both the left and right arteries of each subject, and the area of the vessel wall (endothelium, intima, media) is measured, The amount of vascular proliferation is calculated by subtracting the mean area of the intact, right carotid artery from the mean area of the injured, left carotid artery. Reduction in vascular proliferation is established when the test group shows less proliferation than the control group.
- the compounds of Formula I are administered at a therapeutically effective dosage, e.g., a dosage sufficient to provide treatment for the disease states previously described.
- Administration of the compounds of the invention or the pharmaceutically acceptable salts thereof can be via any of the accepted modes of administration for agents that serve similar utilities.
- the compounds can be used both prophylactically (e.g., to prevent allograft rejection) and therapeutically.
- a daily dose is from about 0.01 to 100.0 mg/kg of body weight, preferably about 0.1 to 64.3 mg/kg of body weight, and most preferably about 0.3 to 43.0 mg/kg of body weight.
- the dosage range would be about 0.7 mg to 7 g per day, preferably about 7.0 mg to 4.5 g per day, and most preferably about 21 mg to 3.0 g per day.
- the amount of active compound administered will, of course, be dependent on the subject and disease state being treated, the severity of the affliction, the manner and schedule of administration (e.g., oral administration one day prior to cancer
- any pharmaceutically acceptable mode of administration can be used.
- the compounds of Formula I can be administered either alone or in combination with other pharmaceutically acceptable excipients, including solid, semi-solid, liquid or aerosol dosage forms, such as, for example, tablets, capsules, powders, liquids, injectables, suspensions, suppositories, aerosols or the like.
- the compounds of Formula I can also be administered in sustained or controlled release dosage forms, including depot injections, osmotic pumps, pills, transdermal (including electrotransport) patches, and the like, for the prolonged administration of the compound at a predetermined rate, preferably in unit dosage forms suitable for single administration of precise dosages.
- compositions will typically include a conventional pharmaceutical carrier or excipient and a compound of Formula I or a pharmaceutically acceptable salt thereof.
- these compositions may include other medicinal agents, pharmaceutical agents, carriers, adjuvants, etc., such as multidrug resistance modifying agents, steroids, immunosuppressants such as
- cyclosporine A azathioprene, rapamycin, FK-506, brequinar, leflunomide and vincrystine.
- the pharmaceutically acceptable composition will contain about 0.1% to 90%, preferably about 0.5% to 50%, by weight of a compound or salt of Formula I, the remainder being suitable pharmaceutical excipients, carriers, etc.
- One preferred manner of administration for the conditions detailed above is oral, using a convenient daily dosage regimen which can be adjusted according to the degree of affliction.
- oral using a convenient daily dosage regimen which can be adjusted according to the degree of affliction.
- a pharmaceutically acceptable composition is formed by the incorporation of any of the normally employed excipients, such as, for example, mannitol, lactose, starch, povidone, magnesium stearate, sodium saccharine, talcum, cellulose, croscarmellose sodium, glucose, gelatin, sucrose, magnesium carbonate, and the like.
- excipients such as, for example, mannitol, lactose, starch, povidone, magnesium stearate, sodium saccharine, talcum, cellulose, croscarmellose sodium, glucose, gelatin, sucrose, magnesium carbonate, and the like.
- Such compositions take the form of solutions, suspensions, tablets, dispersible tablets, pills, capsules, powders, sustained release formulations and the like.
- compositions will take the form of a pill or tablet and thus the composition will contain, along with the active ingredient, a diluent such as lactose, sucrose, dicalcium phosphate, or the like; a lubricant such as magnesium stearate or the like; a disintegrant such as croscarmellose sodium or the like; and a binder such as starch, gum acacia, polyvinylpyrrolidine, gelatin, cellulose and derivatives thereof, and the like.
- a diluent such as lactose, sucrose, dicalcium phosphate, or the like
- a lubricant such as magnesium stearate or the like
- a disintegrant such as croscarmellose sodium or the like
- binder such as starch, gum acacia, polyvinylpyrrolidine, gelatin, cellulose and derivatives thereof, and the like.
- Liquid pharmaceutically administrable compositions can, for example, be prepared by dissolving, dispersing, etc. an active compound as defined above and optional pharmaceutical adjuvants in a carrier, such as, for example, water, saline, aqueous dextrose, glycerol, glycols, ethanol, and the like, to thereby form a solution or suspension.
- a carrier such as, for example, water, saline, aqueous dextrose, glycerol, glycols, ethanol, and the like
- the pharmaceutical composition to be administered may also contain minor amounts of nontoxic auxiliary substances such as wetting agents, suspending agents, emulsifying agents, or solubilizing agents, pH buffering agents and the like, for example, sodium acetate, sodium citrate, cyclodextrine derivatives, polyoxyethylene, sorbitan monolaurate or stearate, etc.
- composition or formulation to be administered will, in any event, contain a quantity of the active compound in an amount effective to alleviate the symptoms of the subject being treated.
- Dosage forms or compositions containing active ingredient in the range of 0.005% to 95% with the balance made up from pharmaceutically acceptable carrier may be prepared.
- a pharmaceutically acceptable composition is formed by the incorporation of any of the normally employed excipients, such as, for example pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, talcum, povidone, cellulose derivatives, croscarmellose sodium, glucose, sucrose, magnesium carbonate, sodium saccharin, talcum and the like.
- excipients such as, for example pharmaceutical grades of mannitol, lactose, starch, magnesium stearate, talcum, povidone, cellulose derivatives, croscarmellose sodium, glucose, sucrose, magnesium carbonate, sodium saccharin, talcum and the like.
- Such compositions take the form of solutions, suspensions, tablets, capsules, powders, sustained release formulations and the like.
- Such compositions may contain 0.01%-95% active ingredient, preferably 0.1-50%.
- triglycerides is preferably encapsulated in a gelatin capsule.
- ester solutions and the preparation and encapsulation thereof, are disclosed in U.S. Patents Nos. 4,328,245; 4,409,239; and 4,410,545.
- the solution e.g. in a polyethylene glycol
- a pharmaceutically acceptable liquid carrier e.g. water
- liquid or semi-solid oral formulations may be prepared by dissolving or dispersing the active compound or salt in vegetable oils, glycols, triglycerides, propylene glycol esters (e.g. propylene carbonate) and the like, and encapsulating these solutions or suspensions in hard or soft gelatin capsule shells.
- Parenteral administration is generally characterized by injection, either subcutaneously, intramuscularly or intravenously.
- injectables can be prepared in conventional forms, either as liquid solutions or
- compositions to be administered may also contain minor amounts of non-toxic auxiliary substances such as wetting or emulsifying agents, pH buffering agents, solubility enhancers, and the like, such as for example, sodium acetate, polyoxyethylene, sorbitan monolaurate, triethanolamine oleate, cyclodextrins, etc.
- non-toxic auxiliary substances such as wetting or emulsifying agents, pH buffering agents, solubility enhancers, and the like, such as for example, sodium acetate, polyoxyethylene, sorbitan monolaurate, triethanolamine oleate, cyclodextrins, etc.
- a more recently devised approach for parenteral administration employs the implantation of a slow-release or sustained-release system, such that a constant level of dosage is maintained. See, e.g., U.S. Patent No. 3,710,795.
- composition will comprise 0.2-2% of the active agent in solution.
- Formulations of the active compound or a salt may also be
- the particles of the formulation have diameters of less than 50 microns, preferably less than 10 microns.
- R 1 is Methyl.
- R 3 is -CF 3
- Z is ZA, in which Z 1 is Methyl.
- Z 2 , Z 3 , and Z 4 are Hydrogen, and G is Methoxy
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Immunology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Transplantation (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description






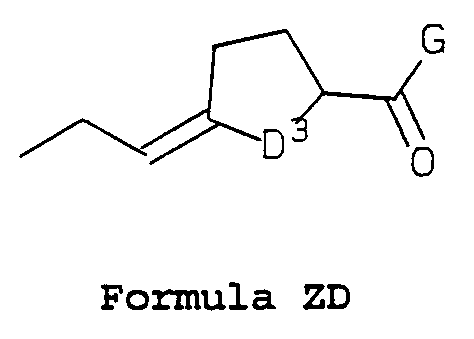


























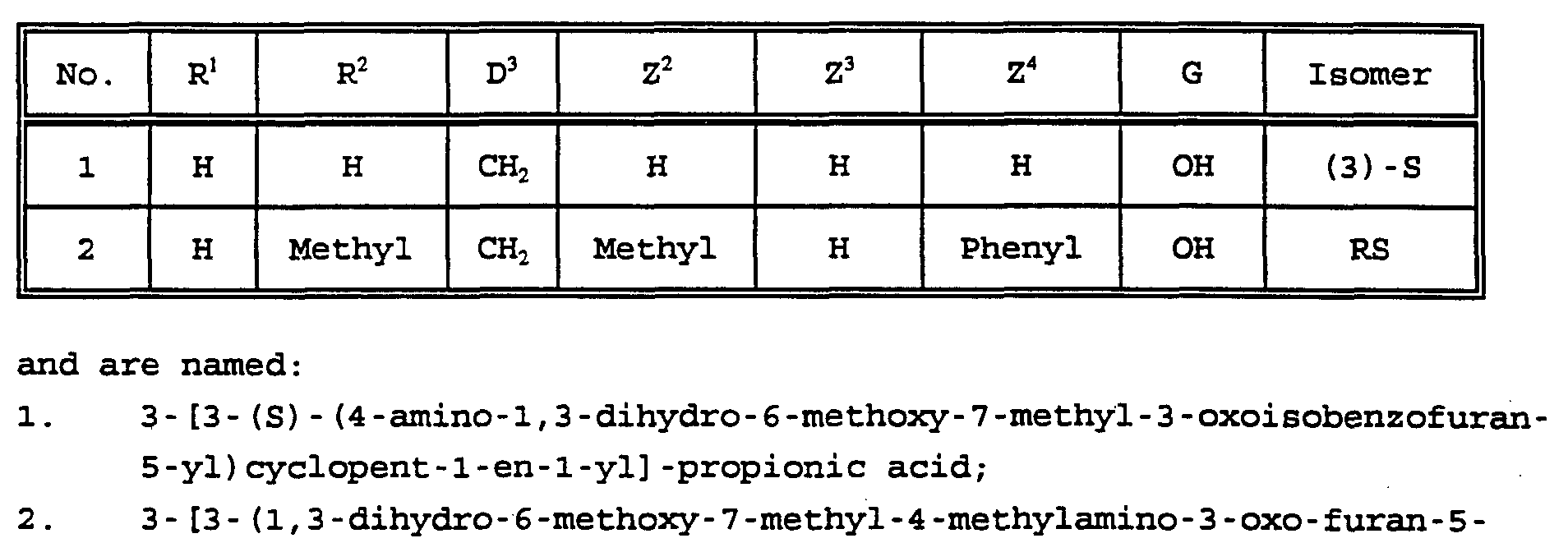

































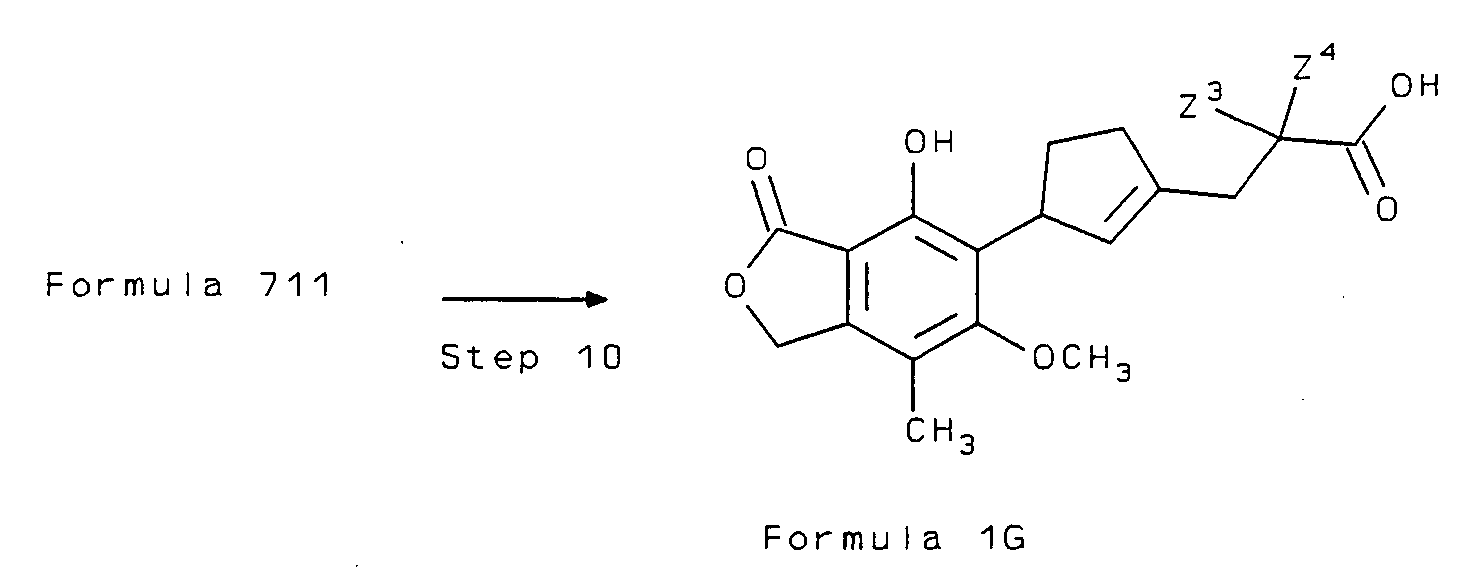



















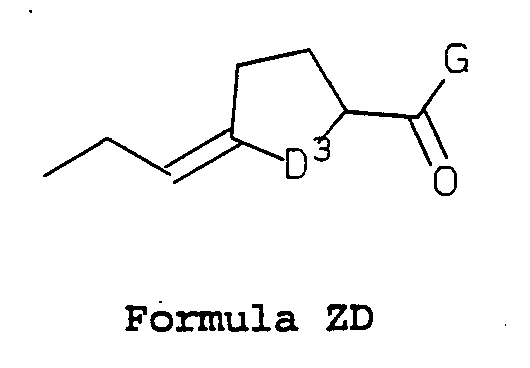






































Claims











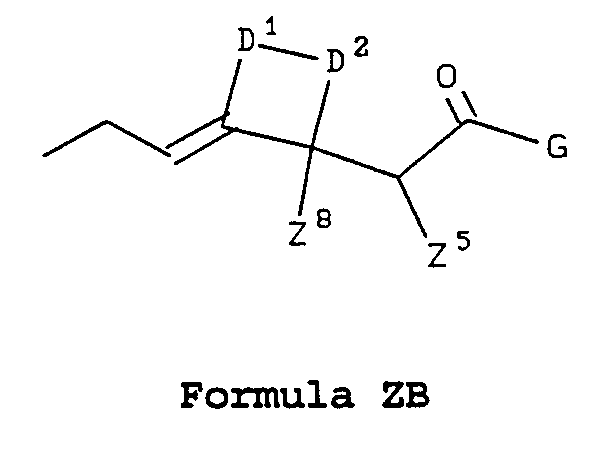






Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX9603484A MX9603484A (en) | 1994-02-18 | 1995-02-16 | 4-amino derivatives of 5-substituted mycophenolic acid. |
| BR9506838A BR9506838A (en) | 1994-02-18 | 1995-02-16 | Derivatives of mycophenolic acid substituted in position 5 |
| EP95910983A EP0745072B1 (en) | 1994-02-18 | 1995-02-16 | 4-amino derivatives of 5-substituted mycophenolic acid |
| DE69502380T DE69502380T2 (en) | 1994-02-18 | 1995-02-16 | 4-AMINO DERIVATIVES OF 5-SUBSTITUTED MYCOPHENOLIC ACIDS |
| AU18753/95A AU1875395A (en) | 1994-02-18 | 1995-02-16 | 4-amino derivatives of 5-substituted mycophenolic acid |
| JP7521867A JPH09509173A (en) | 1994-02-18 | 1995-02-16 | 4-Amino derivative of 5-substituted mycophenolic acid |
| DK95910983T DK0745072T3 (en) | 1994-02-18 | 1995-02-16 | 4-amino derivatives of 5-substituted mycophenolic acid |
| SI9530105T SI0745072T1 (en) | 1994-02-18 | 1995-02-16 | 4-amino derivatives of 5-substituted mycophenolic acid |
| FI963220A FI963220A (en) | 1994-02-18 | 1996-08-16 | 4-Amino derivatives of 5-substituted mycophenolic acid |
| HK98111768A HK1010784A1 (en) | 1994-02-18 | 1998-11-05 | 4-amino derivatives of 5-substituted mycophenolic acid |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/198,732 US5512568A (en) | 1994-02-18 | 1994-02-18 | Method of using 4-amino derivatives of 5-substituted mycophenolic acid |
| US08/198,732 | 1994-02-18 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO1995022537A2 true WO1995022537A2 (en) | 1995-08-24 |
| WO1995022537A3 WO1995022537A3 (en) | 1995-10-26 |
Family
ID=22734572
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1995/001786 WO1995022537A2 (en) | 1994-02-18 | 1995-02-16 | 4-amino derivatives of 5-substituted mycophenolic acid |
Country Status (22)
| Country | Link |
|---|---|
| US (2) | US5512568A (en) |
| EP (1) | EP0745072B1 (en) |
| JP (1) | JPH09509173A (en) |
| CN (1) | CN1141039A (en) |
| AT (1) | ATE165826T1 (en) |
| AU (1) | AU1875395A (en) |
| BR (1) | BR9506838A (en) |
| CA (1) | CA2183531A1 (en) |
| CO (1) | CO4340732A1 (en) |
| DE (1) | DE69502380T2 (en) |
| DK (1) | DK0745072T3 (en) |
| ES (1) | ES2116078T3 (en) |
| FI (1) | FI963220A (en) |
| HK (1) | HK1010784A1 (en) |
| IL (1) | IL112666A (en) |
| LV (1) | LV12149B (en) |
| MX (1) | MX9603484A (en) |
| PH (1) | PH31426A (en) |
| SI (1) | SI0745072T1 (en) |
| TW (1) | TW438788B (en) |
| WO (1) | WO1995022537A2 (en) |
| ZA (1) | ZA951293B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2583678A2 (en) | 2004-06-24 | 2013-04-24 | Novartis Vaccines and Diagnostics, Inc. | Small molecule immunopotentiators and assays for their detection |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6514979B1 (en) | 1999-03-03 | 2003-02-04 | University Of Maryland Biotechnology Institute | Synergistic combinations of guanosine analog reverse transcriptase inhibitors and inosine monophosphate dehydrogenese inhibitors and uses therefor |
| US6811998B2 (en) * | 1999-06-25 | 2004-11-02 | Roche Diagnostics Operations, Inc. | Conjugates of uncompetitive inhibitors of inosine monophosphate dehydrogenase |
| US20050187170A1 (en) * | 2003-06-16 | 2005-08-25 | Biocryst Pharmaceuticals, Inc. | Enhancing the efficiency of RNA polymerase inhibitors by using inosine monophosphate dehydrogenase inhibitors |
| US12011429B2 (en) | 2016-06-02 | 2024-06-18 | Steven Baranowitz | Prevention and treatment of viral infections |
| US10603299B2 (en) | 2016-06-02 | 2020-03-31 | Steven Baranowitz | Prevention and treatment of viral infections |
| CA3026241A1 (en) | 2016-06-02 | 2017-12-07 | Steven Baranowitz | Prevention and treatment of viral infections |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4686234A (en) * | 1985-11-27 | 1987-08-11 | Syntex (U.S.A) Inc. | Mycophenolic acid derivatives in the treatment of inflammatory diseases, in particular rheumatoid arthritis |
| US4725622A (en) * | 1986-01-23 | 1988-02-16 | Syntex (U.S.A.) Inc. | Mycophenolic acid derivatives in the treatment of rheumatoid arthritis |
| US5380879A (en) * | 1994-02-18 | 1995-01-10 | Syntex (U.S.A.) Inc. | Derivatives of mycophenolic acid |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3825571A (en) * | 1971-07-31 | 1974-07-23 | Chugai Pharmaceutical Co Ltd | Mycophenolic acid derivatives |
| JPS5542995B2 (en) * | 1972-02-24 | 1980-11-04 | ||
| JPS5529994B2 (en) * | 1972-03-02 | 1980-08-07 | ||
| US4959387A (en) * | 1986-01-23 | 1990-09-25 | Syntex (U.S.A.) Inc. | Mycophenolic acid derivatives in the treatment of rheumatoid arthritis |
| US4753935A (en) * | 1987-01-30 | 1988-06-28 | Syntex (U.S.A.) Inc. | Morpholinoethylesters of mycophenolic acid and pharmaceutical compositions |
| US4727069A (en) * | 1987-01-30 | 1988-02-23 | Syntex (U.S.A.) Inc. | Heterocyclic aminoalkyl esters of mycophenolic acid, derivatives thereof and pharmaceutical compositions |
| US4748173A (en) * | 1987-01-30 | 1988-05-31 | Syntex (U.S.A.) Inc. | Heterocyclic aminoalkyl esters of mycophenolic acid and derivatives thereof and pharmaceutical compositions |
| US4861776A (en) * | 1987-01-30 | 1989-08-29 | Syntex (U.S.A) Inc. | Heterocyclic aminoalkyl esters of mycophenolic acid and derivatives thereof, compositions and use |
| JPH0637486B2 (en) * | 1988-05-18 | 1994-05-18 | 三共株式会社 | Novel antibiotic phthalexin and method for producing the same |
| US5247083A (en) * | 1992-07-10 | 1993-09-21 | Syntex (U.S.A.) Inc. | Direct esterification of mycophenolic acid |
-
1994
- 1994-02-18 US US08/198,732 patent/US5512568A/en not_active Expired - Fee Related
-
1995
- 1995-02-16 IL IL11266695A patent/IL112666A/en not_active IP Right Cessation
- 1995-02-16 BR BR9506838A patent/BR9506838A/en not_active Application Discontinuation
- 1995-02-16 ES ES95910983T patent/ES2116078T3/en not_active Expired - Lifetime
- 1995-02-16 AT AT95910983T patent/ATE165826T1/en not_active IP Right Cessation
- 1995-02-16 DK DK95910983T patent/DK0745072T3/en active
- 1995-02-16 EP EP95910983A patent/EP0745072B1/en not_active Expired - Lifetime
- 1995-02-16 CN CN95191688A patent/CN1141039A/en active Pending
- 1995-02-16 DE DE69502380T patent/DE69502380T2/en not_active Expired - Fee Related
- 1995-02-16 AU AU18753/95A patent/AU1875395A/en not_active Abandoned
- 1995-02-16 WO PCT/US1995/001786 patent/WO1995022537A2/en active IP Right Grant
- 1995-02-16 CO CO95005927A patent/CO4340732A1/en unknown
- 1995-02-16 SI SI9530105T patent/SI0745072T1/en unknown
- 1995-02-16 CA CA002183531A patent/CA2183531A1/en not_active Abandoned
- 1995-02-16 ZA ZA951293A patent/ZA951293B/en unknown
- 1995-02-16 PH PH49981A patent/PH31426A/en unknown
- 1995-02-16 JP JP7521867A patent/JPH09509173A/en not_active Ceased
- 1995-02-16 MX MX9603484A patent/MX9603484A/en not_active IP Right Cessation
- 1995-02-16 TW TW084101405A patent/TW438788B/en not_active IP Right Cessation
- 1995-05-26 US US08/452,245 patent/US5538969A/en not_active Expired - Fee Related
-
1996
- 1996-08-16 FI FI963220A patent/FI963220A/en unknown
-
1998
- 1998-07-27 LV LVP-98-157A patent/LV12149B/en unknown
- 1998-11-05 HK HK98111768A patent/HK1010784A1/en not_active IP Right Cessation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4686234A (en) * | 1985-11-27 | 1987-08-11 | Syntex (U.S.A) Inc. | Mycophenolic acid derivatives in the treatment of inflammatory diseases, in particular rheumatoid arthritis |
| US4725622A (en) * | 1986-01-23 | 1988-02-16 | Syntex (U.S.A.) Inc. | Mycophenolic acid derivatives in the treatment of rheumatoid arthritis |
| US5380879A (en) * | 1994-02-18 | 1995-01-10 | Syntex (U.S.A.) Inc. | Derivatives of mycophenolic acid |
Non-Patent Citations (1)
| Title |
|---|
| JOURNAL OF MEDICINAL CHEMISTRY, vol. 33, 1990 pages 833-838, P. H. NELSON; E. EUGUI, C. C. WANG, A. C. ALLISON 'Synthesys and Immunosuppressive Activity of Some Side-Chain Variants of Mycophenolic Acid' cited in the application * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2583678A2 (en) | 2004-06-24 | 2013-04-24 | Novartis Vaccines and Diagnostics, Inc. | Small molecule immunopotentiators and assays for their detection |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0745072B1 (en) | 1998-05-06 |
| LV12149B (en) | 1998-12-20 |
| US5512568A (en) | 1996-04-30 |
| EP0745072A1 (en) | 1996-12-04 |
| CA2183531A1 (en) | 1995-08-24 |
| CO4340732A1 (en) | 1996-07-30 |
| DE69502380D1 (en) | 1998-06-10 |
| FI963220A0 (en) | 1996-08-16 |
| AU1875395A (en) | 1995-09-04 |
| ATE165826T1 (en) | 1998-05-15 |
| LV12149A (en) | 1998-10-20 |
| DE69502380T2 (en) | 1998-10-08 |
| ZA951293B (en) | 1996-08-16 |
| CN1141039A (en) | 1997-01-22 |
| SI0745072T1 (en) | 1998-10-31 |
| US5538969A (en) | 1996-07-23 |
| FI963220A (en) | 1996-10-16 |
| MX9603484A (en) | 1998-01-31 |
| DK0745072T3 (en) | 1999-01-18 |
| ES2116078T3 (en) | 1998-07-01 |
| IL112666A0 (en) | 1995-05-26 |
| BR9506838A (en) | 1997-09-30 |
| IL112666A (en) | 2000-01-31 |
| TW438788B (en) | 2001-06-07 |
| HK1010784A1 (en) | 1999-06-25 |
| JPH09509173A (en) | 1997-09-16 |
| PH31426A (en) | 1998-10-29 |
| WO1995022537A3 (en) | 1995-10-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5536747A (en) | 6-substituted mycophenolic acid and derivatives | |
| US5493030A (en) | 5-substituted derivatives of mycophenolic acid | |
| US5380879A (en) | Derivatives of mycophenolic acid | |
| CA2352786C (en) | Benzofuran derivatives, process for the preparation of the same and uses thereof | |
| US20040167171A1 (en) | Benzo-fused 5-membered hetrocycle compounds, process for preparation of the same, and use thereof | |
| EP0745072B1 (en) | 4-amino derivatives of 5-substituted mycophenolic acid | |
| US5554612A (en) | 4-amino 6-substitutes mycophenolic acid and derivatives | |
| JPH09124631A (en) | Benzofuran derivative and medicine composition containing it | |
| JP4364467B2 (en) | Benzofuran derivatives, their production and use | |
| EP0420571A2 (en) | 6-and/or 7-substituted -1,2,3,4,4A,9B-hexahydro-8-hydroxydibenzofuran-3-ols as inhibitors of leukotriene biosynthesis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/1996/003484 Country of ref document: MX Ref document number: 95191688.2 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AM AT AU BB BG BR BY CA CH CN CZ DE DK EE ES FI GB GE HU JP KE KG KP KR KZ LK LR LT LU LV MD MG MN MW MX NL NO NZ PL PT RO RU SD SE SI SK TJ TT UA UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): KE MW SD SZ UG AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AM AT AU BB BG BR BY CA CH CN CZ DE DK EE ES FI GB GE HU JP KE KG KP KR KZ LK LR LT LU LV MD MG MN MW MX NL NO NZ PL PT RO RU SD SE SI SK TJ TT UA UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): KE MW SD SZ UG AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1995910983 Country of ref document: EP Ref document number: 2183531 Country of ref document: CA Ref document number: 963220 Country of ref document: FI |
|
| WWP | Wipo information: published in national office |
Ref document number: 1995910983 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1995910983 Country of ref document: EP |