WO1994025465A1 - SUBSTITUTED METHYLENEDIOXY[3',4':6,7]INDOLIZINO-[1,2-b]QUINOLINONES - Google Patents
SUBSTITUTED METHYLENEDIOXY[3',4':6,7]INDOLIZINO-[1,2-b]QUINOLINONES Download PDFInfo
- Publication number
- WO1994025465A1 WO1994025465A1 PCT/US1994/004866 US9404866W WO9425465A1 WO 1994025465 A1 WO1994025465 A1 WO 1994025465A1 US 9404866 W US9404866 W US 9404866W WO 9425465 A1 WO9425465 A1 WO 9425465A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- methyldioxolo
- indolizino
- acetyl
- quinoun
- Prior art date
Links
- XTTRRRKQNATISH-UHFFFAOYSA-N CC(C(C=C1N2Cc3cc(cc4OCOc4c4)c4nc13)=C(C)C2=O)=O Chemical compound CC(C(C=C1N2Cc3cc(cc4OCOc4c4)c4nc13)=C(C)C2=O)=O XTTRRRKQNATISH-UHFFFAOYSA-N 0.000 description 1
- JTNWNBFTNYLYGN-UHFFFAOYSA-N CC1(C(C=C2N3CCC2=O)=C(C)C3=O)OCCO1 Chemical compound CC1(C(C=C2N3CCC2=O)=C(C)C3=O)OCCO1 JTNWNBFTNYLYGN-UHFFFAOYSA-N 0.000 description 1
- PWPXSSVOQCFRAP-UHFFFAOYSA-N Nc1c(C=O)cc2O[IH]Oc2c1 Chemical compound Nc1c(C=O)cc2O[IH]Oc2c1 PWPXSSVOQCFRAP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/22—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains four or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/475—Quinolines; Isoquinolines having an indole ring, e.g. yohimbine, reserpine, strychnine, vinblastine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
Definitions
- This invention relates to antiviral compounds, pharmaceutical compositions thereof, and a method of treating viral infections. More specifically, this invention relates to certain indolizino[ l,2-b]-quinolinyl derivatives which have antiviral activity.
- Camptothecin is an example of one such compound. It is a water-insoluble, cytotoxic alkaloid produced by Camptotheca acuminata trees indigenous to China and Nothapodytes foetida trees indigenous to India. Camptothecin and its close congeners are known to inhibit eukaryotic topoisomerase I. The cytotoxic and antitumor activity of camptothecin and its close congeners is due to this inhibition of eukaryotic topoisomerase I (Cancer Res. 1988, 48, 1722; Molec. Pharmacol.
- camptothecin has been shown to have an effect on viruses by a number of investigators in laboratory settings.
- camptothecin Although camptothecin has demonstrated antiviral activity in in vitro tissue culture systems, camptothecin and its close analogs that have a hydroxylactone moiety cannot be considered as useful in vivo antiviral agents because they inhibit mammalian topoisomerase I, inhibit host cell DNA replication, and are cytotoxic to mammalian cells. Furthermore, camptothecin is not expected to be attractive for drug development as an antiviral agent because of unacceptable dose-limiting toxicity, unpredictable toxicity, poor aqueous solubility, and/or unacceptable shelf life stability.
- the present invention provides a method for treating viral infections, which method comprises administering to an infected host in need thereof an effective amount of a compound of Formula I, or a pharmaceutically acceptable salt thereof, alone or in combination with a carrier, diluent or excipient.
- R O, -OH, and OR 1 ;
- Rl is -COR 4 , or -P(O)(OH)R 5 wherein:
- R3 is -H or lower alkyl
- R 4 is -CR 3 R 6 R 7 ;
- R 5 is OH or CH NH 2 ;
- R6 is H or the side chain of any naturally occuring ⁇ -amino acid
- R7 is -NR9RlO . . _ - v_ whe _ e X is any pharmaceutically acceptable anion
- R8 is lower alkyl
- R9 and R 1 ⁇ are independently selected from the group consisting of -H, -Cl-6 alkyl, and R ⁇ and R 1 ⁇ taken together to form a 5-7 membered saturated heterocyclic ring containing the nitrogen on which R ⁇ and R 1 ⁇ are substituted; and R 11 is -CH 2 R 12 , wherein:
- R 12 is -N(CH 3 ) 2 ⁇ ' ⁇ /
- this invention relates to novel compounds of Formula I, and pharmaceutically acceptable salts thereof.
- this invention relates to a composition
- a composition comprising a novel compound of Formula I in combination with an acceptable carrier, excipient or diluent, particularly a pharmaceutically acceptable carrier, excipient or diluent
- Aliphatic is intended to include saturated and unsaturated radicals. This includes normal and branched chains, saturated or mono or poly unsaturated chains where both double and triple bonds may be present in any combination.
- the phrases "lower alkyl” and “C1-.5 alkyl” refer to and mean an alkyl group of 1 to 6 carbon atoms in any isomeric form, but particularly the normal or linear form.
- Lower alkoxy means the group lower alkyl-O-.
- Halo means fluoro, chloro, bromo or iodo.
- Acyl means the radical having a terminal carbonyl carbon.
- 5-7 membered saturated heterocyclic ring containing the nitrogen is intended to include saturated rings such as piperidine, pyrrolidine, morpholine, piperazine, and N-alkyl piperazine.
- Salts of any sort may be made from these compounds, provided there is an acidic group present or a sufficiently basic nitrogen.
- Particularly preferred are the pharmaceutically acceptable salts of the instant compounds. These latter salts are those which are acceptable in their application to a pharmaceutical use. By that it is meant that the salt will retain the biological activity of the parent compound and the salt will not have untoward or deleterious effects in its application and use in treating diseases. .
- compositions are prepared in a standard manner.
- the parent compound in a suitable solvent is reacted with an excess of an organic or inorganic acid, in the case of acid addition salts of a base moiety, or an excess of organic or inorganic base in the case where there is an acid group.
- Representative acids are hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, acetic acid, maleic acid, succinic acid or methanesulfonic acid.
- Cationic salts are readily prepared from metal bases such as sodium, potassium, calcium, magnesium, zinc, copper or the like and ammonia.
- Organic bases include the mono or disubstituted amines, ethylenediamine, piperazine, amino acids, caffeine, and the like.
- the ring system of the compounds of the present invention are numbered according to Formula II.
- a chiral center or another form of an isomeric center is created by some combination of substituents, in a compound of the present invention, all forms of such isomer(s) are intended to be covered herein.
- Inventive compounds containing a chiral center may be used as a racemic mixture or the mixture may be separated and an individual enantiomer may be used alone.
- the present invention provides a method for the treatment of viral infections caused by certain DNA viruses comprising administering to an infected animal, preferably a mammal, most preferably a human, in need thereof an effective amount of a compound of Formula I as described hereinabove, or a pharmaceutically acceptable salt thereof, alone or in combination with a carrier, excipient or diluent
- the present invention also provides compounds, and pharmaceutically acceptable salts thereof, which exhibit antiviral activity, said compounds having the structure represented by Formula I hereinabove. More specifically, these compounds and the present method are especially useful in treating the following pathogens in humans:
- Herpes Simplex virus types 1 and 2 HSV-1 and HSV-2;
- CMV Cytomegalovirus
- VZV Varicella Zoster Virus
- a preferred method for treating viral infections according to the present invention uses the following compounds of Formula I:
- Preferred compounds of the present invention include:
- One generic process comprises preparing a 1-keto indolizine adduct and then condensing this fragment with the appropriate substituted aminobenzaldehyde or aminoactophenone.
- Starting materials are commercially available or can be made by published methods.
- the reaction sequences are illustrated by Schemes I-i ⁇ .
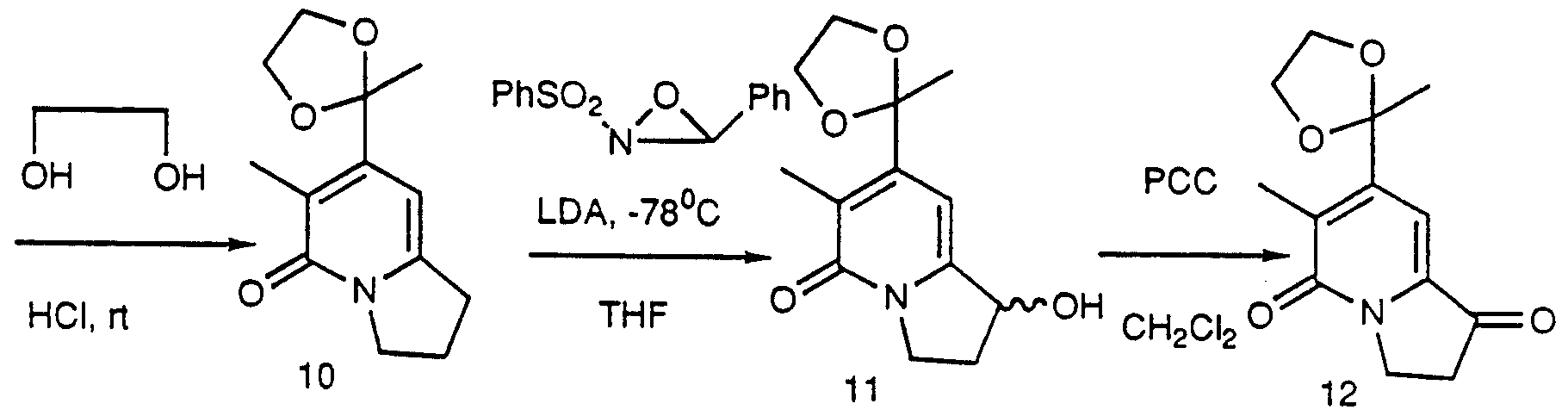
- 2-pyrrolidone 1 can be alkylated with dimethyl sulfate to give ether 2 which can be condensed with acetonedicarboxylate to give indolizine 3.
- Methylation of 3 with methyl iodide at ambient temperature in the inert solvent produced 4 which can be hydrolyzed with an aqueous base to give 5 followed by decarboxylation to produce 6.
- Compound 10 can be oxidized using Davis reagent and base to give alcohol 11 which can be further oxidized with pyridiniumchlorochromate in methylene chloride to give ketone-ketal 12.
- the assay used to test the compounds of the present invention for antiviral activity is well-known. A generalized description of the assay follows.
- Well plates are seeded with the appropriate cells at a concentration of lxl 0 ⁇ cells per well suspended in 0.5 mL of Earle's Minimum Essential Medium (EMEM) containing 10% fetal bovine serum (FBS) and antibiotic and antimycotic solution. After the cells are 80-90% confluent (24 hours), old medium is removed and washed with Hank's buffered saline solution (HBSS). Cells are then infected for 1 hour at 37°C with 100-200 plaque forming units per well of a herpes simplex virus suspended in 250 mL HBSS. Following adsorption, the following are added: A) 250 mlVwell 2 x EMEM containing Human IgG (Sigma Chemical Co., St Louis, Mo.) (ca. 0.1 mg/mL);
- compositions prepared from the compounds of Formula I These compositions have both a human and veterinary utility, and comprise an excipient or carrier which is acceptable for the intended pharmaceutical end use and at least one inventive compound.
- the carrier may be a liquid, or spray, or may be formulated in a solid, non-degradeable or degradeable form for insertion in the rumen.
- Selected excipients and carriers may be employed to prepare compositions acceptable or adaptable for humans use.
- An effective amount of the pharmaceutical compositions of the present invention may be contained in one embodiment, such as in a single pill, capsule, or pre-measured intravenous dose or pre-filled syringe for injection.
- the composition will be prepared in individual dose forms where one unit such as a pill, will contain a sub-optimal dose but the user will be instructed to take two or more unit doses per treatment.
- compositions for later dilution by the end user may also be prepared, for instance for intravenous (IV) formulations and multi-dose injectable formulations.
- Carriers or diluents contemplated for use in these compositions are generally known in the pharmaceutical formulary arts. Reference to useful materials can be found in well known compilations such as Remington's Pharmaceutical Sciences. Mack Publishing Co., Easton, Pa.
- compositions and the pharmaceutical carrier or diluent will, of course, depend upon the intended route of administration, for example whether by intravenous and intramuscular injection, parenterally, topically, orally, or by inhalation.
- parenteral administration the pharmaceutical composition will be in the form of a sterile injectable liquid such as an ampule or an aqueous or nonaqueous liquid suspension.
- the pharmaceutical composition will be in the form of a cream, ointment, liniment, lotion, paste, spray or drops suitable for administration to the skin, eye, ear, nose or genitalia.
- the pharmaceutical composition will be in the form of a tablet capsule, powder, pellet, atroche, lozenge, syrup, liquid, or emulsion.
- the pharmaceutical carrier employed may be, for example, either a solid or liquid.
- suitable pharmaceutical carriers or diluents include: for aqueous systems, water; for non-aqueous systems: ethanol, glycerin, propylene glycol, olive oil, com oil, cottonseed oil, peanut oil, sesame oil, liquid paraffins, and mixtures thereof with water, for solid systems: lactose, terra alba, sucrose, talc, gelatin, agar, pectin, acacia, magnesium stearate, stearic acid, kaolin and mannitol; and for aerosol systems: dichlorodifluoromethane, chlorotrifluoroethane and compressed carbon dioxide.
- the instant compositions may include other ingredients such as stabilizers, antioxidants, preservatives, lubricants, suspending agents, viscosity modifiers and the like, provided that the additional ingredients do not have a detrimental effect on the therapeutic action of the instant compositions.
- the carrier or diluent may include time delay material well known to the art, such as glyceryl monostearate or glyceryl distearate alone or with a wax, ethylcellulose, hydroxypropylmethylcellulose, mediylmethacrylate and the like.
- a wide variety of pharmaceutical forms can be employed.
- the preparation can be tableted, placed in a hard gelatin capsule in powder or pellet form or in the form of a troche or lozenge.
- the amount of solid carrier will vary widely but preferably will be from about 25 mg to about 1 gram.
- the preparation will be in the form of a syrup, emulsion, soft gelatin capsule, sterile injectable solution or suspension in an ampule or vial or nonaqueous liquid suspension.
- a pharmaceutically acceptable salt of the compound of Formula I is dissolved in an aqueous solution of an organic or inorganic acid or base.
- the compound of Formula I may be dissolved in a suitable co-solvent or combinations thereof.
- suitable cosolvents include, but are not limited to, alcohol, propylene glycol, polyethylene glycol 300, polysorbate 80, glycerin and the like in concentrations ranging from 0-60% of the total volume. It will be appreciated that the actual preferred dosages of the compounds used in the compositions of this invention will vary according to the particular complex being used, the particular composition formulated, the mode of administration and the particular site, host and disease being treated. It is expected that these compounds will be active in the concentration ranges of two commercial antiviral drugs, Cytovene (ganciclovir) and Zovirax (acyclovir). The latter is manufactured in 200 mg capsules with instructions for treating herpes simplex viral infections by taking one capsule every 4 hours, but not to exceed 5 capsules per day.
- Example 3(A) (0.3 g, 1.2 mmol) was reduced following the procedure of Example 2(E) to give foam 0.24 g (92%).
- a parenteral pharmaceutical composition of this invention suitable for administration by injection 100 mg of a water soluble salt of a compound of Formula I is mixed with 10 ml of 0.9% sterile saline, and the mixture is incorporated into a dosage unit form suitable for administration by injection.
- EXAMPLE 7 Oral Composition To prepare an oral pharmaceutical composition of this invention, 100 mg of a compound of Formula I is mixed with 750 mg of lactose, and the mixture is incorporated into an oral dosage unit form, such as a hard gelatin capsule, which is suitable for oral administration.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Virology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description









Claims




Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002161980A CA2161980A1 (en) | 1993-05-03 | 1994-05-03 | Substituted methylenedioxy¬3',4':6,7|indolizino-¬1,2-b|quinolinones |
| AU66704/94A AU684777B2 (en) | 1993-05-03 | 1994-05-03 | Substituted methylenedioxy(3',4':6,7)indolizino-(1,2-b)quinolinones |
| NZ266094A NZ266094A (en) | 1993-05-03 | 1994-05-03 | Quinolinone derivatives; compounds, pharmaceutical formulations |
| JP6524634A JPH08509742A (en) | 1993-05-03 | 1994-05-03 | Substituted methylenedioxy [3 ', 4': 6,7] indolizino [1,2-b] quinolinone |
| EP94915450A EP0699201A1 (en) | 1993-05-03 | 1994-05-03 | SUBSTITUTED METHYLENEDIOXY 3',4':6,7]INDOLIZINO- 1,2-$i(b)]QUINOLINONES |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US5713393A | 1993-05-03 | 1993-05-03 | |
| US08/057,133 | 1993-05-03 | ||
| US21365794A | 1994-03-15 | 1994-03-15 | |
| US08/213,657 | 1994-03-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994025465A1 true WO1994025465A1 (en) | 1994-11-10 |
Family
ID=26736111
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1994/004866 WO1994025465A1 (en) | 1993-05-03 | 1994-05-03 | SUBSTITUTED METHYLENEDIOXY[3',4':6,7]INDOLIZINO-[1,2-b]QUINOLINONES |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP0699201A1 (en) |
| JP (1) | JPH08509742A (en) |
| AU (1) | AU684777B2 (en) |
| CA (1) | CA2161980A1 (en) |
| MX (1) | MX9403275A (en) |
| NZ (1) | NZ266094A (en) |
| WO (1) | WO1994025465A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5883255A (en) * | 1990-10-31 | 1999-03-16 | Smithkline Beecham Corporation | Substituted indolizino 1,2-b!quinolinones |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5191297A (en) * | 1975-02-07 | 1976-08-10 | 77 arukokishikaruboniru 88 mechiruindorijino * 1*22b * kinorin 9 * 11h * onnoseiho | |
| US4914205A (en) * | 1987-06-25 | 1990-04-03 | Kabushiki Kaisha Yakult Honsha | Camptothecin derivatives |
| US4981968A (en) * | 1987-03-31 | 1991-01-01 | Research Triangle Institute | Synthesis of camptothecin and analogs thereof |
| WO1992007856A1 (en) * | 1990-10-31 | 1992-05-14 | Smithkline Beecham Corporation | SUBSTITUTED INDOLIZINO[1,2-b]QUINOLINONES |
| US5225404A (en) * | 1989-11-06 | 1993-07-06 | New York University | Methods of treating colon tumors with tumor-inhibiting camptothecin compounds |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69209969T2 (en) * | 1991-10-29 | 1996-09-12 | Glaxo Wellcome Inc | Water soluble camptothecin derivatives |
| JPH05191297A (en) * | 1992-01-10 | 1993-07-30 | Fujitsu Ltd | Serial/parallel conversion circuit |
-
1994
- 1994-05-03 JP JP6524634A patent/JPH08509742A/en active Pending
- 1994-05-03 WO PCT/US1994/004866 patent/WO1994025465A1/en not_active Application Discontinuation
- 1994-05-03 NZ NZ266094A patent/NZ266094A/en unknown
- 1994-05-03 MX MX9403275A patent/MX9403275A/en unknown
- 1994-05-03 AU AU66704/94A patent/AU684777B2/en not_active Ceased
- 1994-05-03 EP EP94915450A patent/EP0699201A1/en not_active Withdrawn
- 1994-05-03 CA CA002161980A patent/CA2161980A1/en not_active Abandoned
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5191297A (en) * | 1975-02-07 | 1976-08-10 | 77 arukokishikaruboniru 88 mechiruindorijino * 1*22b * kinorin 9 * 11h * onnoseiho | |
| US4981968A (en) * | 1987-03-31 | 1991-01-01 | Research Triangle Institute | Synthesis of camptothecin and analogs thereof |
| US4914205A (en) * | 1987-06-25 | 1990-04-03 | Kabushiki Kaisha Yakult Honsha | Camptothecin derivatives |
| US5225404A (en) * | 1989-11-06 | 1993-07-06 | New York University | Methods of treating colon tumors with tumor-inhibiting camptothecin compounds |
| WO1992007856A1 (en) * | 1990-10-31 | 1992-05-14 | Smithkline Beecham Corporation | SUBSTITUTED INDOLIZINO[1,2-b]QUINOLINONES |
Non-Patent Citations (3)
| Title |
|---|
| CHEMICAL ABSTRACTS, Vol. 77, issued 1972, HORWITZ et al., "Camptothecin, Mechanism of Inhibition of Adenovirus Formation", see page 7079, col. 1, Abstract No. 70781u, Virology, Vol. 48, No. 3, pages 690-8 (1972). * |
| CHEMICAL ABSTRACTS, Vol. 84, issued 1976, ATHERTON et al., "Interferon Induction by Viruses and Polynucleotides", see page 428, Abstract No. 103727f, J. Gen. Virol. 1975, Pt 3. 297-3054. * |
| See also references of EP0699201A4 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5883255A (en) * | 1990-10-31 | 1999-03-16 | Smithkline Beecham Corporation | Substituted indolizino 1,2-b!quinolinones |
Also Published As
| Publication number | Publication date |
|---|---|
| MX9403275A (en) | 1995-01-31 |
| NZ266094A (en) | 1997-10-24 |
| JPH08509742A (en) | 1996-10-15 |
| AU684777B2 (en) | 1998-01-08 |
| CA2161980A1 (en) | 1994-11-10 |
| EP0699201A1 (en) | 1996-03-06 |
| AU6670494A (en) | 1994-11-21 |
| EP0699201A4 (en) | 1996-01-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3447292B2 (en) | Imidazopyridines and their use for the treatment of gastrointestinal diseases | |
| CA3040919A1 (en) | Substituted pyridinone-containing tricyclic compounds, and methods using same | |
| ZA200305275B (en) | Aryl ether substituted imidazoquinolines. | |
| KR20060127909A (en) | Polycyclic agents for the treatment of respiratory syncytial virus infections | |
| EP1727817B1 (en) | Azabicyclooctan-3-one derivatives and use thereof | |
| JPH0633268B2 (en) | Water-soluble camptothecin analogue | |
| EP2057149A2 (en) | Pseudobase benzo [c]phenanthridines with improved efficacy, stability, and safety | |
| WO2011134283A1 (en) | Matrinic acid/ matrine derivatives and preparation methods and uses thereof | |
| FI67686B (en) | PROCEDURE FOR THE FRAMEWORK OF THERAPEUTIC ANIMAL PRODUCTS | |
| US20070293527A9 (en) | Polycyclic compounds as potent alpha2-adrenoceptor antagonists | |
| EP0555347A1 (en) | SUBSTITUTED INDOLIZINO 1,2-b]QUINOLINONES | |
| JP2003528876A (en) | Alkylated imidazopyridine derivatives | |
| JPH09508635A (en) | Aza cyclic derivative | |
| WO1993016698A1 (en) | SUBSTITUTED FURO[3',4':6,7]INDOLIZINO[1,2-b]QUINOLINONES | |
| EP0828743A1 (en) | Water soluble camptothecin analogs | |
| JP5922157B2 (en) | (-)-Huperzine A process and related compositions and methods of treatment | |
| US5883255A (en) | Substituted indolizino 1,2-b!quinolinones | |
| TW202102502A (en) | Fused polycyclic pyridone compounds as influenza virus replication inhibitors | |
| WO1994025465A1 (en) | SUBSTITUTED METHYLENEDIOXY[3',4':6,7]INDOLIZINO-[1,2-b]QUINOLINONES | |
| FI59097B (en) | PROCEDURE FOR THE FRAMSTATING OF THERAPEUTIC ANVAENDBARA PYRIMIDO (4,5-B) QUINOLIN-4 (3H) -ON-2-CARBOXYL SYRADERIVAT | |
| ES2636469T3 (en) | New derivatives of pyrido [3,4-c] [1,9] phenanthroline and 11,12-dihydropyrid [3,4-c] [1,9] phenanthroline and their use, particularly for the treatment of cancer | |
| CN113603689B (en) | Polycyclic pyridone compounds, pharmaceutical compositions and uses thereof | |
| RU2745985C1 (en) | Anticoronavirus therapeutic agent - substituted 7-hydroxy-3,4,12,12a-tetrahydro-1h-[1,4]oxazino[3,4-c]pyrido[2,1-f][1,2,4]triazine-6,8-dione for the prevention and treatment of covid-19 | |
| WO1993020818A1 (en) | SUBSTITUTED INDOLIZINO[1,2-b]QUINOLINONES | |
| EP0637960A1 (en) | SUBSTITUTED INDOLIZINO 1,2-b]QUINOLINONES |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 94192467.X Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BB BG BR BY CA CN CZ FI HU JP KP KR KZ LK MG MN MW NO NZ PL RO RU SD SI SK UA US US VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 266094 Country of ref document: NZ Ref document number: 2161980 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1994915450 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994915450 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1994915450 Country of ref document: EP |