GREEN FLUORESCENT LABELED NUCLEOTIDES FOR USE IN PROBES
This invention relates generally to green fluorescent compounds and their use for labeling DNA, and more particularly to green rhodamine dyes and their synthesis and use thereof in labelling DNA. The present invention also relates to the detection and identification of chromosomes or regions of chromosomes by in situ hybridization with DNA probes comprising green fluorescent labels.
Background Of The Invention Labeled DNA probes find particular utility in the identification and study of chromosomes. Alterations in chromosome structure often coincide with, and may be the cause of many inborn genetic disorders and degenerative diseases, including certain cancers. Such alterations may take the form of additional or absent whole chromosomes, or additional or absent portions of chromosomes. Chromosomes may also be rearranged, as by a translocation, so that different chromosomal regions come to be linked to each other. A host of other genetic defects, including inversions, amplifications, and outright deletions, can occur alone or in combination with the above named defects. With the proper labeled DNA probes, detection of chromosomes and any changes therein is possible, and highly desirable.
Chromosome identification by use of DNA probes generally involves hybridization of the probe to a chromosome or region thereof present in a cell sample. One approach to use of DNA probes is the use of "indirect label" probes. An indirect label probe is a nucleic acid probe which has been iabeiled with a moiety which can not be directly identified after hybridization of the probe to the target chromosome. However, the moiety on the probe can be subsequently reacted with a labelled reactive material which can be identified. For example, an indirect label probe is labelled with a hapten, such as biotin. Such a probe, after being hybridized to the target chromosome, can then be reacted with a reactive material, such as an antibody for biotin, having a fluorescent label attached. The subsequent product resulting from the binding of the reactive material to the hapten can then be identified with a fluorescence microscope. This approach is generally referred to as
SUBSTITUTE SHEET
Fluorescence In-Situ Hybridization (hereinafter "FISH"). See, for example, Gray et. al., European Patent Application 0430402.
Another approach to FISH has utilized "direct-label" fluorescent probes, in which a detectable moiety is conjugated directly to the nucleic acid probe. With direct label DNA probes, after hybridization of the probe to the target chromosome, the probe/chromosome hybrid can be directly detected without need for a separate reactive step involving penetration of cellular material by a reactant carrying a detectable label. Direct label DNA probes for chromosome identification are disclosed in U.S. patent application Serial No. 07/762,913, filed September 19, 1991, by M. L. Bittner, L E. Morrison and M. S. Legator, which is herein incorporated by reference.
FISH can potentially lead to high resolution chromosomal analysis. However, most commercially available fluorophore probes require the use of fluorescence microscopes which are extremely sensitive to signal to noise ratios, thereby making routine analysis difficult. Fluorescein is a green fluorescent compound which has been used in FISH. However, fluorescein when used in nucleic acid probes is subject to high photo-oxidation rates and such probes exhibit significant levels of non-specific binding. Further, it is desirable to have different colored fluorescent labelled probes to permit multiple analyses of the same sample. Thus, a better performing green fluorescent label compound for nucleic acid probes is desirable.
In addition, to be suitable for use in iα≤ilii hybridization, a fluorophore label must one, be conjugatable to a nucleic acid without disruption of the nucleic acid strand, and two, not interfere with the hybridization reaction between the nucleic acid and the target chromosome. These requisites are not readily achievable.
Several disclosures of fluorescent labelling compounds and nucleotides containing them exist. However, none of these disclosures are directed to green fluorescent compounds having a rhodamine dye structure. The rhodamine dyes, rhodamine 110 (green), rhodamine B (orange) and rhodamine 6G (red) are well known and commercially available (See Kodak Optical Products Catalog #JJ-169, Eastman Kodak Company, Rochester, NY). European Patent Application 0 272 007, published June 22, 1988, discloses a method of preparing and isolating isomericaily pure 5- and
SUBSTITUTE SHEET
6-succinimidylcarboxylates of certain rhodamine dyes for use in gel elctrophoresis separation of macromolecules, involving DNA sequencing. The 5- and 6-succinimidyl esters disclosed therein are derived from rhodamine dyes having fully substituted amino groups. It does not disclose green fluorescent rhodamine dyes suitable as a green fluorescent label in nucleic acid probes for iα≤jlu identification of chromosomes.
Ward, et al., US Patent No. 4,711,955 discloses attachment of biotin (or other haptens) to nucleotides at the 5-position of pyrimidines, the 8-position of purines, and to the 7-position of 7-deazapurine. The Ward, et al, patent utilizes organo-mercurial methods of biotin attachment to nucleotides. There is, however, no suggestion that label groups other than biotin and haptens could be joined to the modified nucleotide, nor any suggestion of fluorescent direct label DNA probes.
Klevan, et al, US Patent No. 4,828,979, also discloses the modification of mono-nucleotides for subsequent use in labelling reactions. Specifically, dATP and dCTP are derivatized at the 6- and 4-positions, respectively, through a linker arm that varies in length. The modified nucleotides are also joined to biotin, and are then capable of being enzymatically incorporated into poly nucleotides for use as probes. Klevan et al do not disclose use of a fluorescent label in DNA probes.
Musso et al, US Patent No 4,833,251, describes the chemical synthesis and use of indirect label polynucleotide probes labeled with specifically derivatized biotins. The derivatized biotin labels are joined to transaminated cytosine N-4 amino groups on the polynucleotide through a hydrazone linkage. Moreover, Musso et al. is directed to analysis of solid-support bound nucleic acids, and not to use of in-situ hybridization probes.
Wiegant, et al, Nucleic Acids Res. 19, 3237 (1991), discloses the use of fluorescein-dUTP in a nick-translation format to produce fluorescein labeled human nucleic acid probes. The probes are used for in-situ hybridization of human metaphase chromosomes. There is, however no disclosure of other green fluorescent labelled probes.
Urdea and Horn, US Patent No. 4,910,300, teach the synthesis of modified nucleotides for use in oligonucleotide synthesis. There is no
SUBSTITUTE SHEET
disclosure of specific fluorophores or suggestion of synthetic schemes for joining such to nucleic acid probes.
Ruth, US Patent No. 4,948,882, teaches modified nucleotides suitable for oligonucleotide synthesis. Ruth does not specify any particular fluorescent compounds for use in his nucleotides, nor does he disclose fluorescent labelled DNA probes.
Accordingly, a need exists for stable, high emission green fluorescent compounds suitable for use as a nucleotide label, particularly for use in DNA probes for in sjlu hybridization identification of chromosomes or parts thereof. It is the general object of this invention to provide new green emitting rhodamine dye compounds, capable of reacting with nucleotides, and green fluorescent labeled nucleotides for use in probes.
Summary Of The Invention
The present invention describes the synthesis and modification of green emitting rhodamines to yield compounds that are conjugatable to biomolecules for use in fluorescence in-situ hybridization. The present invention provides: (1) green fluorescent label compositions, and labeled probe and nucleotide compositions containing them which are useful for the in ≤ilu detection of a chromosome or a region of a chromosome, (2) methods for the synthesis of such labels, and the nucleotide and probe compositions that incorporate them, and (3) methods for the use of such probe compositions for the in ≤jiu detection of a chromosome or a region of a chromosome.
SUBSTITUTE SHEET
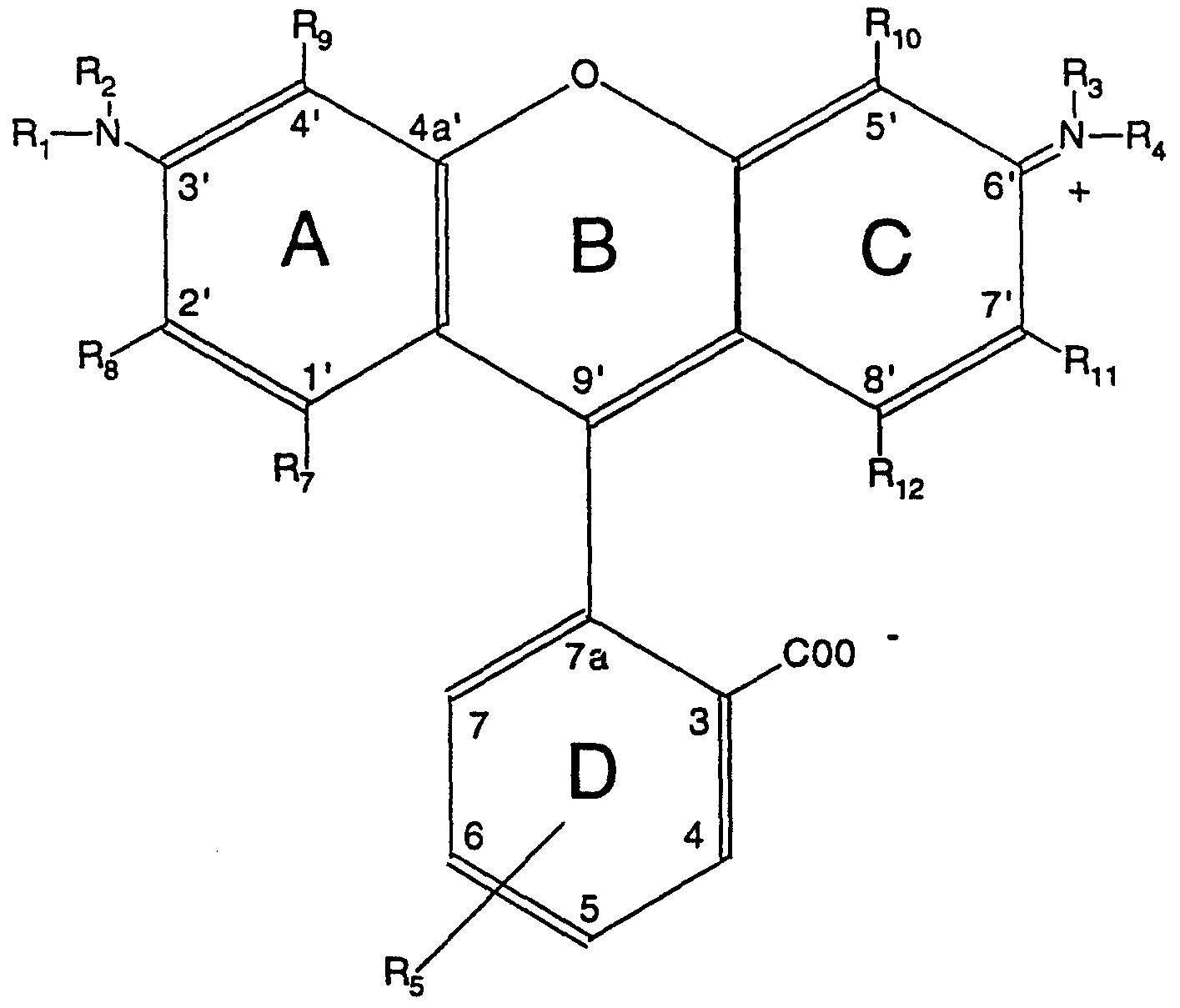
This invention broadly is directed to use as a nucleotide label of a moiety of the following formula:
Re Formula 1 wherein R1 t R2, R3ι and R are hydrogen, R7-R12 are the same or different and are selected from the group consisting of hydrogen, halogen, and alkyl groups, R5 is either a carboxyl group or a lactone linkage to carbon 9', and R6 is a reactive group which can be linked to a nucleotide. For the preferred green fluorescent label moiety of the invention, R7 to R12 are each hydrogen and R6 is a carboxyl group conjugatable to an amine on a nucleotide.
The moiety of the invention can be conjugated to a nucleotide, and after conjugation provides green fluorescent emitting properties to the nucleotide.
The invention also comprises: a iabeiled nucleotide having green fluorescent properties comprising:
(a) a nucleotide triphosphate in either a free nucleotide or a polynucleotide form, and
(b) a green fluorescent moiety of the general formula:
Formula 2 wherein R1 t R2, R3, and R4 are hydrogen, R7, R8, R9, R-|0, R^ and R^ arβ the same or different and are selected from the group consisting of hydrogen, halogen, and alkyl groups of one to eight carbons, and R5 comprises a chemical linkage connected to the nucleotide triphosphate. In this embodiment, the chemical linkage preferably comprises an amide linkage to the nucleotide. In yet another aspect, this invention is directed to a method for preparing a probe for in situ detection of a chromosome or region of a chromosome comprising:
(a) transaminating a number of cytosine bases contained in
DNA sequences having essentially complementary base sequences with respect to the chromosome or chromosome region to be detected to produce DNA sequences having transaminated bases having reactive amine groups; and
SUBSTITUTE SHEET
(b) covalently bonding a fluorescent label comprising a moiety of Formula 1 to at least a portion of the transaminated bases, the number of such bases having labels covalently bonded thereto being sufficient to be detected by optical techniques while essentially retaining the specific binding properties of the probe's DNA sequences with respect to the chromosome or chromosome region to be detected.
In still another aspect, this invention is directed to a method for preparing a probe for iη situ detection of a chromosome or region of a chromosome comprising: (a) producing a labelled nucleotide comprising a moiety of
Formula 2,
(b) purifying the labeled nucleotide by removing impurities resulting from step (a), and
(c) enzymatically incorporating the labeled nucleotide into DNA sequences having essentially complementary base sequences with respect to the chromosome to be detected in sufficient number to allow detection using optical techniques after hybridization of the DNA sequences to a target chromosome.
The fluorescent moieties of the invention provide excellent signal intensity and detectability to labelled nucleotides, such as DNA probes. They are readily detected in FISH procedures with conventional fluorescence microscopes. Furthermore, they can be conjugated to DNA to produce direct label DNA probes, which can be hybridized to chromosomal DNA. Another advantage of the invention is a method of producing the moieties of Formula 1 comprising addition of blocking groups to the unsubstituted amino groups of a rhodamine dye starting material. The blocking groups permit addition of an essential succinimidyl or other reactive group to the starting material in close to quantitive yields. The reactive group is important to achieve conjugation of the label to a nucleic acid. Description Of The Invention
The present invention includes fluorescence emitting, preferably green, rhodamine moieties, their succinimidyl esters and other derivatives capable of conjugation to a nucleotide, and methods of making and using the same. An
SUBSTITUTE SHEET
important feature of the invention is the synthetic method comprising protection of labile amino groups at the 3' and 6' positions of the xanthene-like rings of Formula 1 , which allows the subsequent succinimidylation or addition of another reactive group to take place. The subsequent linking steps which link the green rhodamine structure to a nucleic acid are made nearly quantitative, as a result.
Fluorescent Compound
The green fluorescent compounds of the present invention are defined by Formula 1 above, where the preferred substituent group Re is a carboxyl group, which is suitable for a chemical linkage to a nucleotide triphosphate in either a free or a polynucleotide form.
The preferred synthetic route to produce labelled nucleotides of the invention is:
CDAR (or derivative)
I blocking group addition Blocked rhodamine compound
I esterification with
T N-hydroxy succinimide
Blocked rhodamine succinate i conjugation with T nucleotide containing reactive amino groups Qreen rhodamine |abe||ed nucleotide
The preferred fluorophore starting moiety of the present invention is the green fluorophore dye moiety, 5,(6)-carboxydiaminorhodamine (CDAR). CDAR can be synthesized by any suitable synthetic technique, such as the reaction described in Example 1 below. The residue of CDAR appears in Formula 1, with R7-R12 each being hydrogen. Applicants anticipate that changing any or all of R7-R12 hydrogens on the rings of CDAR would not change the fluorescent color from green. In Formula 1, Rg can be either a carboxyl group or a lactone linked to carbon 9'. Although the lactone form is colorless, the lactone is converted to a carboxyl group during conjugation to the nucleic acid, which results in the green fluorescence properties. A constraint on the use of CDAR or substituted derivatives thereof to derive a nucleotide label is the reactivity of the unsubstituted amino groups,
SUBSTITUTE SHEET
i.e., where R1-R4 are hydrogen, which limit the ability to link such dyes to nucleoside triphosphates, particularly by use of succinimidylation reactions. It is well recognized that typical conditions of succinimidylation chemistry, such as the disuccinimidyl carbonate (DSC) method disclosed in European Patent Application no. 87310256.0, will result in undesired side reactions with the unsubstituted amino groups to yield acylated amino groups. For example, A.K. Ghosh et al., in Tetrahedron Letters 33. (20), 2781 (1992), provide evidence that DSC reacts with unprotected amino groups such as those present on CDAR or its derivatives. Thus, a particularly important aspect of the present invention is the addition of blocking compounds to the amino groups before succinimidylation to produce the desired reactive esters. Thus, the fluorescent moiety of Formula 1 is derived from CDAR or its derivatives by first reacting CDAR or derivative with a blocking group to produce a blocked rhodamine compound. The blocked rhodamine compound can then be reacted with N-hydroxy succinimide to produce a moiety of Formula 1. In fact, selecting a proper blocking group makes it possible to produce a covalent linkage with DNA probe sequences or discrete aminated nucleotides containing reactive amino groups simultaneously with removal of the blocking groups to yield the desired fluorescent labeled compounds. In the present invention, the blocking group is preferably an acid sensitive, but base stable compound; thereby rendering it stable to the base ph conditions necessary for esterification, yet readily removable under the acidic conditions for conjugation to a nucleotide. Any suitable conditions can be used to add the blocking group to the starting material. Preferably, these conditions comprise those used in Example 2 below. Trifiuoroacetyl (TFA) is a preferred blocking group (see Example 2 below). Other blocking groups with known chemical properties similar to TFA, including mono- and multi- chlorinated acetyls such as trichloroacetyl (TCA) and dichloroacetyl (DCA), are also operable. The preferred TFA fluorophore dye intermediate, such as (TFA)CDAR, is competent for subsequent succinimidylation using the methodology of Example 3 below.
The basic fluorescent moiety of Formula 1 employed in the practice of this invention incorporates at least one fluorophore substituent (or group) per molecule and also one reactive substituent (or group) per molecule. In Formula 1, the fluorophore substituent is the four ringed remnant of CDAR.
The reactive substituent is chosen so as to be reactive with and to conjugate
SUBSTITUTE SHEET
to a reactive group incorporated into a linking group in a nucleotide, such as a diethyiamino group.
The reactive substituent of Formula 1 attached to the fluorophore substituent can be chosen to be reactive with either an amino or a carboxyl group present in the linking group of the nucleotide. Preferably, the reactive substituent is carboxyl and the linking group's reactive group is amino to produce an amide linkage. For purposes of reactivity with an amino substituent in a linking group, the carboxyl substituent can be in the acid or salt form, an aldehyde radical or the like. A preferred reactive substituent of Formula 1 is selected from, and exemplified by, the group consisting of isothiocyanates, N-hydroxysuccinimide esters, N_- h yd roxy-7- oxabicyclo[2.2.1 ]hepta-5-ene-2,3-dicarboximide, N-hydroxy-5-norbornene- 2,3-O-dicarboxymide, sulfonyl chlorides, chlorotriazines, hydroxybenzotriazolides, carboxylic acid azides and the like. For purposes of reactivity with a carboxyl substituent in a linking group, the reactive substituent of the fluorescent compound can be a carboxyl- reactive functionality, such as an amino substituent which is in a primary or a secondary form (such as above defined) or the like. A preferred reactive substituent for this conjugation is a primary amino substituent. Linking Compound
The term "linking compound", "linking group" or "chemical linkage" as used herein generally refers to a hydrocarbonaceous moiety suitable for chemical attachment of the fluorescent moiety labels of the invention to a nucleotide, a polynucleotide sequence or a mono-nucleotide. The linking compound therefore must be capable of reacting with the fluorophore compounds of the present invention.
A starting material to produce a linking compound employed in the practice of this invention is a difunctional organic compound, that contains two substituent functional (i.e., reactive) substituents per starting material molecule. At least one of such functional substituents per linking compound molecule is preferably reactive with deoxycytidine nucleotides in a polynucleotide under bisulfite catalyzed aqueous transamination conditions as disclosed in commonly assigned Bittner, et. al. U.S. Patent Application Serial No. 07/762,913. Examples of substituents reactive with a nucleotide
SUBSTITUTE SHEET
include alkyi, amino (primary and secondary), hydrazido, semicarbazido, thiosemicarbazido, and the like. Amino groups are presently most preferred.
The other functional substituent group on the linking compound is reactive with the fluorescent moiety of the invention. This functional group can be immediately reactive, i.e. unblocked, or can be blocked. Examples of suitable unblocked second functional substituent group include amino, carboxyl, phosphate, sulfonate, hydroxyl, hydrazido, semicarbazido, thio¬ semicarbazido and the like. The preferred unblocked second functional substituent include amino (primary or secondary) and carboxyl groups. Examples of suitable blocked second functional substituent group include blocked sulfonate, blocked phosphate and blocked sulfhydryl. Examples of suitable blocking substituents include lower alkyl groups such as methyl, ethyl and propyl.
The two functional substituents present in such a difunctional linking compound can be respective substituents on adjacent carbon atoms relative to each other, or they can be spaced from one another by a plurality of intervening interconnected atoms (preferably carbon atoms). The first and the second functional substituents are connected through a linker or linking moiety. This linking moiety can have any convenient structure but must be non-reactive with other substances that are present in the transamination medium during transamination. The linking moiety is a hydrocarbonaceous divalent group of two to twenty carbons which is acyclic or cyclical and which can optionally incorporate other atoms. It is preferred that only one ether or thio ether group be present. It is presently preferred that the linking compound overall is an organic radical that contains at least two and not more than about a total of about 20 atoms.
Probe Production
The term "probe" or "probe composition" refers to a polynucleotide, or a mixture of polynucieotides, such as a DNA sequence, which has been chemically combined with individual label-containing moieties. Each polynucleotide of a DNA probe is typically single stranded at the time of hybridization to a target chromosome. The probes of the invention comprise a polynucleotide chemically linked to a fluorescent moiety of Formula 1.
SUBSTITUTE SHEET
Any suitable procedure can be employed to prepare the probes of the invention, and suitable procedures are disclosed in Bittner, et. al., U.S. Patent Application 07/762,913. A preferred procedure involves the following steps:
(a) Fragmenting (i.e., disrupting) by any suitable means DNA sequences that are specific to one preselected chromosome or preselected chromosome region into DNA fragments (or segments);
(b) Transaminating deoxycytidine nucleotides existing in the DNA fragments with a linking compound (as above described) to produce DNA fragments containing amino radicals; and
(c) Covalently linking (i.e., chemically bonding) under suitable conditions the amino radicals of the DNA fragments with the fluorescent moieties of Formula 1.
For use as a starting material in the preparation of the inventive probes, the starting DNA sequence(s) can be obtained by various techniques, for example, from (a) DNA that is separated by flow sorting a plurality of a single preselected chromosome of a multi-chromosomal genome; (b) a chromosome library of a preselected chromosome, and (c) an inter species hybrid which incorporates DNA of a preselected chromosome. A preferred starting chromosomal DNA is a chromosome library which has been prepared by standard methods and is available from traditional sources known to those in the art, such as the American Type Culture Collection (ATCC) or other repositories of human or other cloned genetic material. ATCC deposits are available from the American Type Culture Collection, 12301 Parklawn Drive, Rockville, Maryland.
Regional chromosomal DNA is used in the practice of this invention, and can be derived either directly or indirectly from one preselected region of a chromosome of a multi-chromosomal genome. Such starting regional chromosomal DNA is typically in the form of at least one DNA sequence. It is presently preferred that each such sequence or sequences incorporate a plurality of at least one DNA repeated segment and preferably a plurality (i.e., at least two) of structurally differing DNA repeated segments. Preferably, such regional DNA sequence is unique relative to other regions of the total genome of the organism under study. The starting regional DNA incorporates a
SUBSTITUTE SHEET
multiplicity of DNA segments that occur individually at various locations in and throughout the individual preselected region of one chromosome and that are representative of DNA occurring in the preselected region. A presently preferred genome is the human genome The DNA segments are derived from a particular preselected starting chromosomal DNA or starting regional chromosomal DNA by fragmenting. After fragmenting, the DNA segments preferably have an average size that is within a range of about 20 to about 600 bp with a preferred average size range of about 150 to about 600 bp, a more preferred average size range being about 200 to about 400 bp, and a presently most preferred average segment size being about 300 bp. Each of these DNA segments, or fragments, is believed to be complementary to one or more DNA sequences which occur in the particular preselected chromosome or preselected chromosome region. In the present invention it is preferred to form fragmented DNA fragments by sonication of a specific chromosomal DNA. Sonication conditions utilize an aqueous dispersion of starting specific chromosomal DNA that is in the range of about 0.05 to about 4 mg per ml. The ultrasonic frequency applied is about 20,000 cycles per second and is applied for about 1 to about 10 minutes with the tube containing the sample preferably immersed in a cooled bath (dry ice and ethanol) to reduce heating of the sample. In the case of a Branson Sonifier Model 450 (Danbury, CT) with the icrotip located about 2 to about 5 mm from the bottom of the tube in an aqueous solution, a suitable output power is in the range of about 25 to about 30 watts. Preferably, such ultrasonic energy is applied using a cycle of 80% on, 20% off, for a total time of about 5 minutes.
If chromosomally or regionally specific DNA is obtained, for example, from a commercial source in an already suitably fragmented state, then a separate fragmenting step is not needed before a subsequent transaminating processing step is undertaken.
The probes of the invention are useful for the identification of chromosomes, of aneuploidy or abnormal chromosomes and of dicentric chromosomes, for sex determinations of organisms, for controls used with other nucleic acid probes and for other suitable applications.
SUBSTITUTE SHEET
Transamination of Chromosome Probes
The term "chromosome paint" or "painting probe", refers to a probe or probe composition, such as the polynucleotide embodiments of this invention, which is adapted to hybridize under hybridizing conditions with a target which comprises one predetermined chromosome of a multi-chromosomal genome. Typically, one painting probe of this invention can be combined with a second so as to make possible the simultaneous staining and detection of two predetermined chromosomes. In contrast, a "chromosome enumeration probe" refers to a DNA probe which is adapted to hybridize with a target region of a chromosome, such as a centromere region.
To permit labeling of DNA probes with the fluorescent moieties of the invention, preferably a fraction of the total deoxycytidine bases in the chromosomal DNA are transaminated with an amino group of a difunctional linking compound (as above described) in the carbon atom 4 position of the amino group of cytosines (i.e., deoxycytidine nucleotides). About 0.2 to about 8 mole percent of all nucleotides contained in such a mixture of starting DNA sequences or DNA fragments are transaminated. The most effective percentage of amination in any given instance is typically influenced by the particular fluorescent label moiety used. The transamination is conveniently accomplished under aqueous liquid phase conditions in the presence of a bisulfite catalyst with denatured DNA sequences or segments. The linking compound is dissolved in the aqueous transaminating medium. The technique and advantages of employing a chaotrope in the transamination are taught in Bittner et al., copending U.S. Serial No. 07/762,913.
Enzymatic (Direct. Label Probe Compositions
Synthesis of fluorescent labeled depxynucleoside triphosphates (fiuoro- dNTPs)
The fluorescent compounds of the present invention can be attached to dNTPs through linkers such as those used, as described above, in direct fluorescent probe labelling methodology. Preferably, the compounds have a reactive carboxy group that is chemically reacted to the reactive group moiety of the linker, which is previously attached to the nucieoside triphosphate at a
SUBSTITUTE SHEET
suitably accessible site. The fluorescent moieties of the invention can also be linked to mono- and di-phosphates.
Suitable examples of nucleotides possessing attached linkers having a reactive substituent moiety are the aminated deoxyribonucieotides 5-(3- aminoallyl)-2'deoxyuridine-5'-triphosphate (AAdUTP, available from Sigma, cat. no. A5910, N4-(3-aminopropyl)-2'-deoxycytidine-5'-triphosphate (APdCTP, see Cruickshank, U.S. Pat. No. 5,091,519), and N6-(6-aminohexyl)- 2'-deoxyadenosine-5'-triphosphate (AHdATP, available from Life Technologies, Inc., Cat. N0.9514SA). Synthesis of the fluoro-dNTPs can be carried out as described in
Example 9 below. The method preferably comprises reacting a reactive substituent of the fluorescent compound with the aminated deoxyribonucleotide in a borate buffer solution. The borate buffer also acts to remove the TFA blocking groups, yielding the "conjugated" fluoro-dNTP. Fluorescently labeled dNTP's prepared in this way are competent for enzymatic incorporation into poiynucleotides by a variety of methods well known to those skilled in the art. For example, we have carried out the enzymatic incorporation of the fluoro-dNTP's by nick translation using a modification of a standard nick translation protocol, available from Life Technologies (See Gibco/BRL-Life Technologies, Inc. Cat. No. 8160SB).
In situ Hybridization and staining
The chromosome painting probe, the chromosome enumeration probes and the enzymatically labeled probe compositions of this invention are well suited for use in hybridization procedures as stains for and to identify respective preselected chromosomes or chromosome regions. The process involves the three sequential steps of (a) contacting a specimen believed to contain such a chromosome or chromosome region (including fragments thereof) under hybridizing conditions with a probe of the invention which will hybridize with the target DNA of a chromosome or chromosome region to produce hybrids between the target DNA and the probe DNA segments present in the probe composition, (b) separating from the resulting specimen residual portions of the probe composition, and (c) examining the resulting specimen.
SUBSTITUTE SHEET
An in situ hybridization procedure can involve a particular specimen which contains all or only a fraction of the preselected chromosome or chromosome region. Currently, we prefer to use the probes of this invention in in situ hybridization procedures of the type that are commonly and conveniently carried out on specimens which have preliminarily been prepared and mounted on a slide, such as a slide comprised of glass or the like.
To accomplish identification of a preselected chromosome or chromosome region in such a slide mounted specimen using a probe of this invention, the following illustrative procedure can be carried out. Preferably, such slide mounted specimen is preliminarily processed to dehydrate at least partially and also denature at least partially the DNA that is presumed to be present therein. Conventional denaturing and dehydrating procedures and materials can be employed, followed by a sequential hybridization step carried out under hybridizing conditions. First, the slide mounted specimen is contacted with the probe composition of this invention. Next, the combination of the specimen and the treating probe composition that is in contact therewith are incubated at about 30 to 45°C for an appropriate time. Next, the resulting hybrid-containing specimen is subjected to a liquid washing procedure to remove the unreacted, residual treating probe composition. See Bhatt et al. in Nucleic Acids Research 16: 3951-3961 (1988). If desired, a counterstain can then be incorporated into the mounting medium.
The slides can be viewed immediately after processing under a fluorescence microscope using conventional filters, or they can be stored at room temperature for several days or the like before examination. It is an advantage of the invention that the preferred probes having a CDAR based green fluorescent label can be identified using filter sets widely available for detection of fluorescein labels.
Those skilled in the art will appreciate that in in Situ, hybridization of a slide mounted specimen, the sequence of (a) contacting and (b) separating (as above indicated) can be advantageously carried out more than once before the step (c) (examining) as above indicated is carried out. In each such repeat of steps (a) and (b) (each of which is conveniently carried out as above described herein), a different probe composition is employed, with a probe composition of this invention being employed on one repeat, and with another (different) probe composition being employed in each of the other repeats,
SUBSTITUTE SHEET
each such other probe composition being targeted to a different predetermined fractional region of said genome.
Those skilled in the art will readily appreciate that the green fluorescent nucleotides of the invention will produce emitted light of a color which can contrast with color of light emitted by a different fluorophore label of a different probe composition. Therefore, the invention permits use of at least two such color contrasting fluorophore labeled probes, sequentially or simultaneously to distinguish karyotypes, genomes, or specific chromosomes or regions thereof. It will also be appreciated that such different fluorescent compounds with different emitted colors can also be combined in new individual probe compositions. Such new probe compositions can be made by combining fluorescentiy labeled probe compositions which comprise different emission colors but which can hybridize to the same target. For example, the green emitting nucleotides of the present invention, and the orange-red compounds of the prior art can be combined in specific ratios that result in probe compositions of many different colors. Different combinations of such fluorescent labeled probe compositions can be used to identify individual chromosomes when the probe compositions comprise whole chromosome paints or chromosome enumerator probes. A set of such combined labeled probe compositions can be used simultaneously to identify most if not all the chromosomes of a particular karyotype.
Probe Mixtures
It is a feature and advantage of the probe compositions of this invention that they can be combined with other probe compositions, such as those labeled with N, N, N', N' tetramethyl - 5, (6) - carboxy - 3', 6' - diamino rhodamine (N1 hydroxysuccinimide ester) (CTMR, with orange fluorescence) and the like, without adversely affecting the chemical structure or the functional capacity thereof. For example, orange (CTMR) and green (CDAR) fluorescent probes targeted to 2 different chromosomes or chromosome regions can be used to treat the same sample concomitantly. Thus, even though mixed probe compositions incorporate complex DNA segment mixtures under hybridizing conditions, these individual segments only hybridize with complementary target DNA so that the desired specific chromosomal identification is achieved. The other probe compositions used in conjunction with the probes of the invention should preferably be suitable
SUBSTITUTE SHEET
for usage in in situ hybridization under comparable hybridization conditions (relative to a probe composition of this invention).
The following examples should be considered illustrative and not as limiting the invention.
Example 1. Synthesis 5J6.-Carboxydiamiπorhodamine (CDAR)
A 250 ml two-necked flask was charged with 3-aminophenol (25.0 g, 0. 224 mol), trimellitic anhydride (20.0g, 0.104 mol) and concentrated sulfuric acid (100 mis). The side arm was fitted with a teflon temperature probe (inserted directly into the reaction mixture) connected to a Model 210 temperature controller (J-Kem Electronics Inc.) and the mixture magnetically stirred under a stream of nitrogen while being heated to 180 °C (+ or - 2 °C).
The mixture took approximately 45 mins to reach 180 °C, after which the temperature was maintained at 180 "C for a further 4hr. The resulting dark- red solution was cooled to room temperature, then added in portions to magnetically stirred ice-cold water (400 ml). The reaction flask was rinsed with water (2 X 50 ml). After stirring for some time the resulting precipitate was collected by vacuum filtration (sinter glass funnel), washed with limited quantities of ice-cold water and air dried to give approximately 50 g of a brick- red solid. Thin Layer Chromatography [TLC-acetonotrile-acetic acid-water (8:1:1, v/v/v)] indicated that this solid was impure, and consisted of one major and several minor green fluorescing components. Further purification was achieved by dissolving a portion of the brick- red solid (~ 10 g) in concentrated ammonia - water (1 : 4, v/v, 150 mis), cooling on ice and precipitating the dye by addition of concentrated sulfuric acid to pH 2. The precipitate was then collected by vacuum filtration, washed with cold
SUBSTITUTE SHEET
water and air dried to yield approximately 7 grams of material. Final purification was achieved by crystallization from boiling 0.5 M aqueous HCI (30 ml per gram of dye) with charcoal treatment (0.5 g per 10 g dye) and hot filtration through a fluted filter paper (- 50 % recovery of deep red crystals). TLC analysis with System E revealed one major component (the carboxydiaminorhodamine, Rf = 0.40) and one minor component (Rf = 0.71) both with green fluorescence.
Additional checks of composition were performed using TLC and spectral analysis. Using chloroform-methanol-water, (12:7:1 , v/v/v) ,Rf was determined to be 0.27 for CDAR, and 0.32 for the minor component
UV absorption maximum was at 498 nm (in 0.1 M Na2HPO4/NaH2PO , pH 7.4), and fluorescence emission maximum was at 523 nm, with excitation at 450 nm. In methanol, absorption maximum was 503 nm and emission maximum was at 528 nm. Example 2; Synthesis of Bis(N-Trifluoroacetyl)-5,(6)-carboχy-3'.6'- diaminorhodamine [Bis(TFA)CDAR]
Trifluoroacetic anhydride (TFAA) (12.0 g, 57.2 mmol) was added in portions to a mixture of acetonitrile - pyridinβ (2 : 1 v/v; 120 mis) in a round bottom flask. The mixture was thoroughly cooled to -10°C (ethanol/liquid nitrogen). Purified CDAR (2.0 g, 5.4 mmol) was then added in 0.4 g portions over a period of 1.5 hrs.
After each addition of dye, the fluorescence of the solution gradually fades to give a wine-red solution. The solution was then allowed to warm to 10°C for 0.5 hr, then recooled to 0°C and the reaction quenched with 5 ml of water. The solution was evaporated using first an aspirator, then a vacuum pump. The residue was then coevaporated twice with 40 ml of toluene. The residue
SUBSTITUTE SHEET
was dissolved in 200 ml ethyl acetate and extracted twice with 100 ml 1 M HCI. The aqueous extracts were back-extracted with 100 mi ethyl acetate, then the organic phase extracts were combined and dried over sodium sulfate. Care is required during the extractions because the free carboxylic acid will extract into an aqueous phase under some conditions and significant losses can occur. The residue was again evaporated to dryness, followed by coevaporation with toluene to remove any remaining pyridine. This process may leave a product ranging from a yellow powder to a dark oily residue. The dark oily residue was converted to a powder by dissolving the residue in a minimum of ethanol, diluting with toluene, then evaporating. This results in a fine yellow solid on the walls of the flask. The solid can be removed with a spatula. The desired product appears as a major, mobile TLC component (Rf = 0.34 using chloroform - methanol (4:1, v/v)) that yellows after standing under the UV lamp. A significant quantity of material remains at baseline. Example 3. Synthesis of bis.N-Trifluoroacetvn-5.-6)-carboxy-3'.6'- diaminorhodamine-.N'-hydroxysuccinimide. ester fbisfTFA.CDAR-NHS]
Bis(TFA)CDAR as 1.76 grams of pale-yellow powder from step 2 above, was placed in a rotary evaporator flask and dissolved in 30 mis of anhydrous N-N dimethylformamide (DMF) to which 0.536g N-hydroxysuccinimide (NHS) was added. The mixture was cooled to 0°C and 0.588 gram N.N'- diisopropylcarbodiimide (DIPC) in 3ml DMF was added to the magnetically stirred solution. DIPC (in DIPC/NHS/DMF/ 0° to 25°C) produces good purity product in good yield . After 1 hour the ice-bath was removed and the mixture stirred at room temperature for 6 hr. A further 0.25 g of DIPC and 0.2 g of NHS were added and the mixture was stirred at room temperature overnight.
TLC (chloroform - methanol (4:1, v/v)) then revealed that the reaction had gone
SUBSTITUTE SHEET
to approximately 90 % completion. After recooling to 0°C, the reaction was quenched with 1.5 ml glacial acetic acid, and the DMF was removed in vacuo. The residue was dissolved in 250 ml ethyl acetate and extracted twice with 250 mis of 2M NH4CI solution, then twice with 250 ml 5% w/v sodium hydrogen carbonate solution. Caution is required because this solution tends to form emulsions and the trifluoroacetylamino functions are very susceptible to aqueous base hydrolysis. Further extractions with 2M NH4CI (2 X 250 ml) were then carried out. All the aqueous extracts were kept and separately back- extracted with ethyl acetate The organic phases were then combined, dried with Na2SO4 and evaporated to yield an orange solid. The sodium hydrogen bicarbonate extraction selectively extracts most of the highly yellow fluorescent side-product that migrates slightly below the desired CDAR-NHS on TLC, without significant removal of CDAR - caution is still needed in these extractions to minimize base hydrolysis of the TFA groups. At this point the product still shows some baseline impurities on TLC (chloroform - methanol at either 9:1, v/v or 4:1, v/v). The impure solid product was dissolved in ethyl acetate with a minimum of ethanol added to effect complete dissolution. Final purification was achieved by flash chromatography on a 12 X 3.0 cm column of packed silica in chloroform - acetic acid (99:1, v/v) After application of the dissolved product the column was eluted with chloroform - acetic acid (99 : 1 , v/v). The desired product should elute at the solvent front. Problems with elution profile can usually be overcome by using chloroform plus 2% v/v methanol to elute the product, which is a pale-yellow oil. The product was precipitated from ethyl acetate - petroleum ether (4 ml + 80 ml), and collected by filtration on a sintered glass funnel as a pale-yellow powder. Additional product was obtained by washing the sintered glass funnel and flask with ethyl acetate. The powder contained one major component and two extremely minor components, as judged by TLC (Chloroform - methanol (9:1, v/v)). In addition, the powder yielded a satisfactory 1 H NMR spectrum that was indicative of an approximately 1:1 mixture of regioisomers. 5 mgs of the major product dissolved completely in 500 μl dimethyl sulfoxide (DMSO) to give a straw-colored solution and was successfully used in a variety of labelling applications.
Example 4. Regeneration of CDAR from bis.N-Trifluoroacetyl.-5..6.-carboxv- 3'.6'-diaminorhodamine-.N'-hydroxysuccinimide, ester fbis(TFA.CDAR-NHS1
A 5 mg portion of bis(TFA)CDAR-NHS prepared in example 3, was dissolved in 100 μl methanol. Upon solution, 100 μl aqueous 0.1 M K2CO3
SUBSTITUTE SHEET
was added. The solution immediately begins to turn yellow/green indicating partial hydrolysis of the trifiuoroacetyl protecting groups. The simultaneous deprotection/ regeneration reaction proceeded for several hours. After 5-6 hr the reaction appeared to be complete by as judged by TLC (chloroform - methanol - water (12 : 7 : 1 , v/v/v), or acetonitrile - acetic acid - water ( 8 : 1 : 1, v/v/v)). A single major component could be detected along with traces of a higher Rf green fluorescing material.
Example 5. Human Chromosome - Specific DNA Probes
Human chromosome-specific DNA were obtained as recombinant phage libraries from Lawrence Livermore National Laboratories (LLNL) constructed as described in Van Dilla, M.A. et al. (Biotechnology 4: 537-552, 1986). These libraries were amplified by growth on an £ coli host strain. The amplified phage were purified, their DNA was extracted, and this DNA was digested with the restriction enzyme Hind III. Insert DNA was purified away from the lambda vector DNA and cloned into the Hind III site of the plasmid vector pBS (Stratagene, La Jolla, CA). The resulting plasmids were transformed into an E. coli strain, DH5α (Bethesda Research Libraries, Gaithersburg, Maryland).
After propagation of the E. coli strain in conventional fermenters, plasmid DNA was extracted from the bacterial cell pellets (see Bittner et al. copending U.S. Serial No. 07/762,912 filed on Sep 20, 1991). The cells were thoroughly resuspended in 3 times the cell pellet mass (M) (in milliliters) of a solution containing 50nτM glucose (filter sterilized), 10 mM NaEDTA (pH 7.5-
8.0), and 25mM Tris-HCI (pH 8.0). The cells were lysed with vigorous swirling after the addition of 6xM (in milliliters) in a solution containing 0.2 M NaOH, and 1% (w/v) sodium dodecylsulfate (SDS). When the solution cleared,
4.5xM (in milliliters) of a solution containing 55.5 ml of glacial acetic acid and
147.5 grams of potassium acetate in a final volume of 500 ml was mixed thoroughly resulting in the production of a flocculent precipitate. The supernatant was separated from the flocculent precipitate and this supernatant centrifuged for 15 minutes at 7000 x g to remove residual precipitate.
Nucleic acid was precipitated from the supernatant with one volume of ethanol followed by centrifugation for 10 minutes at 7000 x g, and the nucleic acid pellets were resuspended in a total of 0.54xM (in milliliters). The nucleic
SUBSTITUTE SHEET
acid was then extracted with 1/2 volume of neutralized phenol and 1/2 volume of chloroform and precipitated with two volumes of ethanol. The nucleic acid was resuspended in 0.3xM (in milliliters) of a solution of 50 mM Tris HCI (pH 7.0) and 100 mM sodium acetate. 0.77xM (in microliters) of 10 mg/ml RNase (heat treated) was then added and allowed to digest for 30 minutes at room temperature or overnight at 4°C. 0.615xM (in microliters) of a solution of Proteinase K (20mg/ml) was then added and incubated at 55°C for three hours. DNA was extracted with 1/2 volume of neutralized phenol and 1/2 volume of chloroform and precipitated with two volumes of ethanol. DNA was resuspended in 0.415xM (in milliliters) of water, and 0.05xM milliliters of 5 M NaCI and 0.155xM milliliters of 50% (w/v) polyethyleneglycol (PEG) (molecular weight 6000-8000) were added, incubated on ice water for one hour and precipitated by centrifugation for 15 minutes at 7,000 x g. The DNA was resuspended in 0.04xM milliliters of water and 1/10 volume of 3M sodium acetate and extracted with 1/2 volume of neutralized phenol and 1/2 volume of chloroform and precipitated with two volumes of ethanol. The purified DNA was resuspended in 0.0476xM milliliters of deionized H20. The
DNA concentration was determined by fluorometry.
Finally, the purified DNA was disrupted into small fragments of approximately 300 base pairs by sonication using a Branson Sonifier 450
(Danbury, Connecticut). This size of fragments has been empirically determined to be the optimum for DNA probes used for in situ hybridization.
Four milligrams of the purified plasmid DNA prepared above was resuspended in 2 mis of water and immersed in a dry ice/ethanol bath to prevent boiling during sonication. The microtip of the sonication device was immersed in this solution until the tip was 2-5mm from the bottom of the tube.
Sonication was carried out at an output power of 25-30 watts, discontinuously, with an 80% duty cycle (on 80% of time, off 20% of time), for a period of 5 minutes. Following sonication, the DNA was precipitated by the addition of 0.2 ml of 3 M sodium acetate (pH 5.5) and 4 ml of ethanol. The precipitate was recovered by centrifugation for 5 minutes at 8,000 x g and vacuum dried.
Example 6. Bisulfite Catalyzed Transamination of DNA
DNA obtained by the method of Example 1 was transaminated by the addition of ethylenediamine to the C4 carbon atom of the base cytosine. This reaction is catalyzed by sodium bisulfite. To prepare the bisulfite buffer, 1.7 ml
SUBSTITUTE SHEET
of fuming HCI was slowly. added to 1 ml deionized H20 on ice. 1 ml fresh ethylenediamine was then slowly added on ice. After dissolution of the ethylenediamine, the solution was warmed to room temperature and 0.475 g sodium metabisulfite was added. Fuming HCI was then slowly added to the bisulfite mixture until the pH reached 7.0. Deionized water was added to a final volume of 5.0 ml. To transaminate DNA, 1 milligram of sonicated DNA was resuspended in 0.3 ml H20. The DNA was denatured by boiling at
100°C .for 5 minutes then quickly chilled in an ice water bath. The transamination reaction was initiated by the addition of 0.3 ml of this DNA solution to 2.7 ml of bisulfite buffer, and the reaction was incubated at 37°C for
2 days. The DNA solution was desalted by routine dialysis against 5-10 millimolar sodium borate (pH 8.0). After dialysis, 0.3 ml of 3 M sodium acetate (pH 5.5) was added to the dialysate. The aminated DNA was precipitated with 2.5 volumes of ethanol and recovered after centrifugation at 8,000 x g for 10 minutes. The pellets were vacuum dried and rehydrated at a concentration of
3 mg/ml DNA. This solution was stored at -80°C until use.
Example 7. Labelling of Chromosome 1 Probe with bis (TFA. CDAR-NHS
50 μg of unlabeled whole chromosome paint (fragmented polynucleotide directed to human chromosome 1) were evaporated to dryness in a 2.5 ml screw-cap Eppendorf tube. The dried pellet was redissolved in 50 μl deionized water and denatured for 5 min at 95 C followed by quick chill on ice. To the tube on ice was added 100 mM sodium borate buffer (pH 9.0, 350 μl, prepared from boric acid) and the mixture was vortexed briefly and warmed to room temperature. 100 μl of a stock solution of bis (TFA) CDAR-NHS in DMSO (stock solution was 20 mM, 6.63 mgs dye per 500 ul DMSO) was then added and the mixture vortexed briefly, covered with silver foil and placed on the rotary mixer for 18 hr. The DNA was precipitated by addition of 50 μl of 3M sodium acetate, followed by 1.250 ml absolute ethanol and chilling to - 60°C for 2 hr. After centrifugation, the supernatant was removed, the pellet washed with 70 % aqueous ethanol, and dried in the vacuum concentrator. The deep-red pellet, which was sometimes sluggish to dissolve, was periodically vortexed in 100 μl of deionized water. Final purification was achieved using a Biospin 30 spun column (BioRad, 7326006). A 5μl aliquot of the eluant diluted to 1005 μl with 20 mM sodium hydroxide showed absorbance at 260 nm. and 508 nm. to give an A260A508 ratio of 6.54 (after correction).
Therefore, 2.8% of the bases were labeled with CDAR. The absorbance at
SUBSTITUTE SHEET
260 nm was corrected for dye absorbance as follows; A26o(corrected) = A26O - (A508 X 0-271 ). Fluorescence in 0.1 M phosphate buffer was absorption maximum at 509 nm and emission max at 533 nm.
Example 8. Labelling of Chromosome Enumerator (CEP-8) Probe with bis (TFA. CDAR-NHS
50 μg of a nucleic acid hybridization probe directed to a specified region of human chromosome 8 (CEP-8 DNA) were evaporated to dryness in a 2.5 ml screw-cap Eppendorf tube. Preparation of CEP-8 DNA is described in Bittner et al. copending U.S. Serial No. 07/762,912 filed on Sep 20, 1991. The pellet was redissolved in deionized water (50 μl) and denatured for 5 min at 95°C followed by quick chill on ice. To the tube on ice was added 100 mM sodium borate buffer (pH 8.5, 350 μl, prepared from boric acid) and the mixture was vortexed briefly and warmed to room temperature. 100 μl of a stock solution of CDAR-NHS in DMSO (1. 5 - 2.0 mgs dye/ 100 μl DMSO, 23 - 30 mM) was then added and the mixture vortexed briefly, covered with silver foil and placed on the rotary mixer for 16 hr. Just after addition of the dye the solution was almost colorless (pale-yellow) since the yellow/green fluorescence of the dye only slowly develops with mixing overnight. To ensure complete deprotection of the dye, 1M sodium carbonate solution (one- tenth volume, 50 μl) was added and the mixing continued for 6 - 24 hr. The mixture was split into two lots and the DNA was precipitated by addition of sodium acetate (3M, 28 μl) followed by absolute ethanol (688 μl) to each and chilling to - 60 C for 2 hr. After centrifugation, the supernatant was removed, the pellet washed with 70 % aqueous ethanol, and dried in the vacuum concentrator. The deep-red pellets, which were sometimes sluggish to dissolve, were periodically vortexed in 50 μl of deionized water each. Final purification was achieved using a spun column (BioRad, Biospin 30). The eluants were combined and a 5μl aliquot of the solution diluted to 1005 μl with 20 mM sodium hydroxide showed a A260A508 ratio of 6.54 after correction, or that 2.8% of the bases were labeled with CDAR.
Example 9 Preparation of N4-(3-Aminopropyl)-2'-deoxycytidine-5'- triphosphate-carboxy diaminorhodamine conjugate. 6.6 mgs of bis (TFA) (CDAR-NHS), (prepared as described in example
4) was dissolved in 500 μl DMSO and added to a solution containing N4-(3- aminoρropyl)-2'-deoxycytidine-5'-triphosphate (APdCTP, see
SUBSTITUTE SHEET
Cruickshank, U.S. Pat. No. 5,091,519) dissolved in 1.5 ml. 0.1 M sodium borate buffer (pH 9). The 2.5 ml Eppendorf tube containing the pale-yellow mixture was wrapped in silver foil, and allowed to react on a rotary mixer at room temperature overnight.
Analysis of a 25 μl aliquot of the reaction mixture by high pressure liquid c romatography (HPLC) then indicated that a complete conversion of the starting material had been achieved to give two new compounds with retention times (Rt = 18.03 min.) of 37.0 min. and 38.3 min., (isomeric mixture) in a ratio of 1 to 1. To ensure complete removal of the trifiuoroacetyl protecting groups, 200 μl of 1 M sodium carbonate solution was added and the mixture rotary mixed for a further 6 hr (since no change in retention time was observed by HPLC, this treatment was probably not necessary). The products were purified by chromatography on column of DEAE
Sephadex A-25 (1.5cm X 27 cm) first equilibrated with 0.05 M triethyla monium bicarbonate (TEAB) buffer. Solvent was supplied from a two-chamber gradient mixer (Pharmacia GM-1) to a peristaltic pump (Pharmacia P-1, set to pump 100-120 ml per hr, 10X, 1.5 setting, 3.1 mm tubing) to a 3-way valve (Pharmacia, LV-3) then to the column (Econo column, BioRad). From the column the eluent travelled to a Holochrome UV/VIS
SUBSTITUTE SHEET
detector (1 AUFS, 552 nm) which was connected to a Kipp & Zonen chart recorder (chart speed 2 mm/min, 20 mV setting). Finally, eluant fractions (7ml, 220 drop) were collected on a Gilson microfractionator.
The sample was diluted to 2 mi with 0.05 M TEAB and applied via the 3-way valve to the top of the column. The column was then washed with 0.05 M TEAB (250 ml) before a gradient was started. The front chamber of the mixer was filled with 0.05 M TEAB and the rear with 0.25 M TEAB (250 ml). Next, the front chamber was filled with 0.25 M TEAB and the rear chamber with 0.5 M TEAB. Finally a gradient of 0.5 M to 0.75 M buffer is run until the product is eluted. Various dye containing components and side-products are eluted prior to elution of the product.
Frequently the sample application process took place in the afternoon and the first stage of the gradient was carried out overnight in which the eluant chambers were filled and the pump turned down to 1X, 5 setting. This flow rate would usually fill roughly 65 fraction tubes with 7 ml of liquid. The next stage of the gradient was then started the following morning.
The fractions containing pure (HPLC) product (in fact a mixture of the two isomers, the 5- and 6- carboxydiaminorhodamine product peaks) were pooled and evaporated to dryness on a rotary evaporator (bath temp. > 30 C) under oil vacuum pump pressure. The residue was co-evaporated twice with absolute ethanol, dissolved in several mis of deionized water and quantitated by absorbance at 503 nm. Assuming an extinction coefficient of 60,000 at 503 nm, the yield was 0.516 μmol (26 %). Finally the samples were evaporated to dryness on a Savant vacuum concentrator and stored at -20 C. Example 10. Enzvmatic Incorporation of Novel Fluorescent labeled
Nucleotides (fluoro-dNTP's) into DNA Probes
The enzymatic incorporation of the fluoro-dNTP's by nick translation was carried out using a modification of a standard nick translation protocol (Life Technologies, Bethesda MD) as follows. To a 1.5 ml Eppendorf tube cooled on ice was added (sequentially), 68 μl deionized water, 2.5 μl 0.2 mM spectrum orange α'UTP, 0.2 mM each deoxycytosine triphosphate (dCTP), deoxy adenosine triphosphate (dATP), deoxyguanosine triphosphate (dGTP) (10 μl), 0.2 mM deoxythymidine triphosphate (dTTP) (7.5 μl), 1 μg/μl Alu1 restricted CEP template (2.0 μl), E. Coli DNA polymerase I solution (10 μl).
SUBSTITUTE SHEET
The mixture was vortexed briefly and spun for 10 sec on the microcentrifuge to mix. The tube was then incubated for 2 hr at 16 C followed by quenching with 0.3 M ethylenediaminetetraacetic acid (EDTA) (10 μl), and then vortexiπg and centrifuging. Each reaction mixture was then purified on a Biospin gel filtration column (Biorad cat. # 732-6006) following instructions supplied by the manufacturer to give a colorless or near colorless eluant (120 μl). The DNA was then precipitated by addition of 4M sodium acetate (12 μl) and ethanol (280 μl) and stored at -20 C for at least 2 hr. The supernatant was removed by pipette and the invisible pellet dried on a Savant "SpeedVac" refrigerated vacuum concentrator. The DNA pellet was redissolved in deionized water (20 μl) by carefully rolling the solvent over the sides of the tube with a Pasteur pipette. Assuming a 50% recovery the solution contains 50 ng of labeled probe per μl.
Example 11. Detection of the Centromere Region of Chromosome #8 bv In Situ Hybridization using Novel Labeled DNA Probes
The probe composition of the preceding Example 8 was used to identify (detect) the centromere region of human chromosome #8 as follows:
The target DNA consisted of cultured normal white blood cells that were treated to arrest the cells in metaphase. These cells were dropped onto a microscope slide from a distance of about 2 to 3 feet to break open the nuclei and expose the chromosomes. Unbroken cells and interphase nuclei were also present on the slide surface. Before hybridizing, the slide was placed in a denaturing solution consisting of 70% formamide/0.3 M NaCI/30mM sodium citrate, pH 7.0, for 2 minutes at 70°C. The slide was then dehydrated by passage through 70%, 85%, and 100% ethanol baths (2 minutes each). The slide was then warmed to approximately 40°C.
The hybridization mix that was placed on each slide was always 55% formamide/10% dextran sulfate/0.15 M NaCI/15 mM sodium citrate, pH 7.0. The concentrations of probe added to the basic hybridization mix varied to determine the optimal concentration needed to obtain acceptable signal intensity and specificity. The reaction mixture also contained 4.5 μg of sonicated human placental DNA added as carrier and blocking DNA. In addition to the unlabeled human placental DNA, approximately 96 ng of fluorescein labeled, sonicated human placental DNA was added as a genomic counterstain. The preparation of such genomic counterstain is
SUBSTITUTE SHEET
taught in copending Morrison et al. U.S. Serial No. 07/762,928 filed on September 19, 1991. Ten μl of the completed hybridization mixture was denatured by heating at 70°C for from 5 to 15 minutes and then incubated at 37°C for 5 minutes. The mix was applied directly to the slide, covered with a glass coverslip whose edges were sealed with rubber cement, and allowed to hybridize overnight at 42°C in a humidified chamber.
On the following day the glass cover slip was displaced so that the unbound probe could be removed from the slide by a series of washes: three times for 15 minutes each at 45°C in 0.3 MNaCI/30mM sodium citrate 50% formamide v/v, pH 7.0, followed by a single wash for 15 minutes at 45°C in 0.3 M NaCI/30mM sodium citrate (2XSCC), pH 7.0, followed by another wash for 15 minutes at 45°C in "PN buffer", which is 0.1 M sodium phosphate/0.1% NP40 detergent (Calbiochem catalog # 492015). Finally, the slide was washed twice in PN buffer for 2 minutes at room temperature, then air dried. Ten microliters of antifade solution was placed over the target cells and a coverslip was placed over that. The slides were viewed with a fluorescence microscope. Bright green fluorescence was observed at the target area of the chromosome. Fluorescence from non-specific binding was not observed. Comparative Example 1
This comparative example was an attempt to produce CDAR-NHS directly by reaction of CDAR with Disuccinimidyl carbonate. To a partial solution of 34.0 mg of CDAR, from Example 1, and 64.0 mg of DSC in 0.25 ml of 1M solution of DMF was added 122.17 mg of DMAP solution. The deep red solution immediately turned light orange, then upon stirring at room temperature, darkened somewhat but still remained somewhat pale. After 1.5 hours, the reaction mixture was examined by thin layer chromatography (CH3CN: acetic acid: H20; 8:1:1) which revealed no starting material and a complex mixture of products, including a major component which appeared orange on THC and other colored components. The direct reaction was unsuccessful in producing CDAR-NHS. While preferred embodiments of the invention have been described, the invention is capable of various modifications. Therefore, the invention should not be considered limited to the precise details set forth herein, but should be construed as covering all embodiments and modifications within the scope of the following claims.
SUBSTITUTE SHEET