WO1994003455A1 - Pyridoindolobenzodiazepines and derivatives as antipsychotics - Google Patents
Pyridoindolobenzodiazepines and derivatives as antipsychotics Download PDFInfo
- Publication number
- WO1994003455A1 WO1994003455A1 PCT/US1993/006823 US9306823W WO9403455A1 WO 1994003455 A1 WO1994003455 A1 WO 1994003455A1 US 9306823 W US9306823 W US 9306823W WO 9403455 A1 WO9403455 A1 WO 9403455A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carbon atoms
- alkyl
- compound
- cycloalkyl
- antipsychotic
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/16—Peri-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
- A61P39/02—Antidotes
Definitions
- This invention relates to pyrido [4',3':2:3]indolo-[1,7-ab][1,5]benzodiazepines, pharmaceutical
- benzodiazepines useful as tranquilizers.
- An objective of the present invention is to provide therapeutically effective antipsychotic drugs with reduced neurologic side effects.
- the present invention provides antipsychotic agents that are structurally dissimilar to traditional neuroleptic antipsychotic drugs. Based on the structural differences between compounds of the present invention and traditional neuroleptic antipsychotic drugs such as haloperidol and chlorpromazine, compounds of the present invention are expected to have a reduced propensity to induce
- R is H, alkyl of 1-10 carbon atoms, cycloalkyl of 3-7 carbon atoms, (CH 2 ) n COR 6 , (CH 2 )nCH(OH)R 8 ; (CH 2 )nCONR 9 R 10 , (CH 2 ) n (cycloalkyl of 3-7 carbon atoms), (CH 2 )n _ adamantyl, (CH 2 ) n N(R 15 ) 2 or (CH 2 )n-W-Ar;
- R 1 , R 2 , R 4 , and R 5 independently are selected from the group H, alkyl of 1-3 carbon atoms, CF 3 , Cl, F, Br, OH, S(O) p R 14 , CN or OCH 3 ;
- R 3 is H, alkyl of 1-3 carbon atoms, cycloalkyl,
- R 6 is H, OH, OR 7 , alkyl of 1-6 carbon atoms,
- R 8 is H, alkyl of 1-3 carbon atoms, cyloalkyl of 3-6 carbon atoms or ;
- R 9 and R 10 independently are selected from the
- Ar is aryl substituted with 0-3 R 12 or heteroaryl substituted with 0-3 R 12 ;
- R 12 is independently selected at each occurrence from the group alkyl of 1-3 carbon atoms, phenyl, halogen, alkoxy, CN, NO 2 , COR 13 , C02R 13 , NR 13 R 14 , and S(O) p R 14 ;
- R 13 and R 14 are each independently selected at each occurrence from the group hydrogen, alkyl of 1-3 carbon atoms and phenyl;
- R 15 is H, alkyl of 1-3 carbon atoms or cycloalkyl of 1-3 carbon atoms;
- W is O, S(O) p , or NH;
- X is O, S, or 2 H;
- n 1-8;
- m 0-3; and
- p is 0-2.
- This invention also provides an intermediate compound, useful for the preparation of compounds of formula (I), having the formula:
- R 1 , R 2 , R 4 , and R 5 independently are selected from the group H, alkyl of 1-3 carbon atoms, CF 3 , Cl, F, Br, OH, S(O) p R 14 , CN, or OCH 3 ;
- R 3 is H, alkyl of 1-3 carbon atoms, cycloalkyl,
- R 6 is H, OH, OR 7 , alkyl of 1-6 carbon atoms,
- R 7 is CH 3 or C 2 H 5 ;
- R 11 is CH 3 O, CH 2 H 5 O, CF 3 , alkyl of 1-9 carbon
- Ar is aryl substituted with 0-3 R 12 or heteroaryl substituted with 0-3 R 12 ;
- R 12 is independently selected at each occurrence from the group alkyl of 1-3 carbon atoms, phenyl, halogen, alkoxy, CN, NO 2 , COR 13 , CO2R 13 , NR 13 R 14 , and S(O) p R 14 ;
- R 13 and R 14 are each independently selected at each occurrence from the group hydrogen, alkyl of 1-3 carbon atoms and phenyl;
- R 15 is H, alkyl of 1-3 carbon atoms or cycloalkyl of 1-3 carbon atoms;
- W is O, S(O) p , or NH;
- X is O, S , or 2 H;
- n 1-8 ;
- m 0-3 ; and
- p 0-2 .
- R is H, alkyl of 1-6 carbon atoms, cycloalkyl of 3- 7 carbon atoms, CH 2 -(cycloalkyl of 3-7 carbon atoms), or (CH 2 )nAr;
- R 1 and R 5 are H;
- R 2 is H, CH 3 , Cl, F, or CF 3 ;
- R 3 is H or alkyl of 1-3 carbon atoms
- R 4 is H, CH 3 , or Cl
- Ar is aryl substituted with 0-3 R12;
- R is H, CH 3 , n-hexyl, cyclopropyl,
- R 1 and R 5 are H;
- R 2 is H, CH 3 , Cl, F, or CF 3 ;
- R 3 is H, or alkyl of 1-3 carbon atoms
- R 4 is H, CH 3 , or Cl
- the present invention also provides methods for the
- alkyl is intended to nclude both branched and straight-chain saturated aliphatic
- alkoxy represents an alkyl group of indicated number of carbon atoms attached through an oxygen bridge
- cycloalkyl is intended to include saturated ring groups, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl
- bicyclic ring groups such as
- alkynyl is intended to include hydrocarbon chains of either a straight or branched configuration and one or more triple carbon-carbon bonds which may occur in any stable point along the chain, such as ethynyl, propynyl and the like.
- Cycloalkyl-alkyl is intended to include cycloalkyl attached to alkyl.
- Halo refers to fluoro, chloro, bromo, and iodo;
- Counterion is used to represent a small, negatively charged species such as chloride, bromide, hydroxide, acetate, sulfate, and the like.
- aryl or “aromatic residue” is intended to mean phenyl or naphthyl;
- carbocyclic is intended to mean any stable 5- to 7- membered monocyclic or bicyclic or 7- to 14-membered bicyclic or tricyclic carbon ring, any of which may be saturated, partially unsaturated, or aromatic, for example, indanyl or tetrahydronaphthyl (tetralin).
- aralkyl is intended to mean any aryl group bearing an alkyl group.
- the aralkyl group may be attached at any of its carbon atoms.
- heteroaryl As used herein, the term “heteroaryl” or
- heterocycle is intended to mean a stable 5- to 7- membered monocyclic or bicyclic or 7- to 10-membered bicyclic heterocyclic ring which is either saturated or unsaturated, and which consists of carbon atoms and from 1 to 3 heteroatoms selected from the group consisting of N, O and S and wherein the nitrogen and sulfur
- heteroatoms may optionally be oxidized, and the nitrogen may optionally be quaternized, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom which results in a stable structure.
- the heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable. Examples of such heterocycles include, but are not limited to. pyridyl, pyrimidinyl, furanyl, thienyl, pyrrolyl, pyrazolyl, imidazolyl, tetrazolyl, benzofuranyl,
- heteroarylalkyl is intended to mean heteroaryl bearing an alkyl group.
- heteroarylalkyl may be attached at any of its atoms.
- substituted means that one or more hydrogen on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency is not exceeded, and that the substitution results in a stable compound.
- stable compound or “stable structure” is meant herein a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
- pharmaceutically acceptable salts refer to derivatives of the disclosed compounds that are modified by making acid or base salts.
- compositions of the invention can be prepared by reacting the free acid or base forms of these compounds with a
- the compounds of this invention may be prepared utilizing any of a number of starting materials and methods known to those skilled in the art of organic synthesis.
- United States Patent No. 4,438,120 which is herein incorporated by reference, discloses starting materials and methods useful in the preparation of the compounds of this invention.
- Scheme 1 shows a method for the preparation of compounds of formula (I) wherein X is oxygen. Starting materials for the synthesis of compounds of this invention according to Scheme 1 are
- the free base of (4) can be obtained by treating the hydrochloride salt of (4) with a base, such as sodium hydroxide.
- a base such as sodium hydroxide.
- Compounds of formula (4) are compounds of formula (I) wherein X is oxygen. These may be further purified, if desired, utilizing standard techniques such as recrystallization and chromatography.
- a compound of formula (3) may be obtained as a by-product of the reaction of Scheme 1.
- Compounds of formula (3) are disclosed in U.S. Patent No. 4,438,120.
- the proportion of (3) to (4) produced in this reaction may vary according to the substituents R 1 ,R 2 ,R 4 and R 5 .
- the separation of (3) and (4) from the mixture can be effected by exploiting the difference in their
- solubilities in standard solvents or by high performance liquid chromatography are solubilities in standard solvents or by high performance liquid chromatography.
- R of formula (1) cannot be (CH 2 ) n COR 6 .
- this group can be introduced at a later stage, into compounds of formula (4) where R is H.
- an R group such as (CH 2 ) n COR 6 can be introduced onto the amine nitrogen atom of a compound of formula (4) wherein R is H by alkylation, or by Michael reaction.
- a compound of formula (4) (R is H) can be alkylated with an alkyl halide, RX, wherein R is alkyl, such as 1-bromodecane, or (CH 2 ) n COR 6 in the presence of a base such as potassium carbonate in a solvent such as
- a compound of formula (4) (R is H) can be condensed with an ⁇ , ⁇ -unsaturated carbonyl compound such as methyl vinyl ketone in a solvent such as
- a compound of formula (4) wherein R is not H can be prepared by treating a compound of formula (4) wherein R is H with an acyl halide such as benzoyl chloride in the presence of a base such as
- a compound of formula (4) wherein R is not H and R 3 is H can be alkylated at the 9-position with an alkyl halide, R 3 X, such as methyl iodide in the presence of a base such as sodium hydride in a solvent such as
- compounds of formula (5) can be prepared by heating a compound of formula (4) with borane in tetrahydrofuran at a temperature between 20 and 80 °C, isolating the borane-amine complex formed and heating it with 1-octene in a solvent such as xylene at a temperature between 120-150 °C, followed by standard work-up, isolation and purification procedures.
- Compounds of formula (6) wherein R is alkyl, aralkyl, cycloalkyl or cycloalkylalkyl can be prepared from the corresponding compound of formula (4) by heating said corresponding compound with phosphorus pentasulfide in the presence of a solvent such as pyridine or toluene at a temperature between 90 and 100°C.
- HRMS resolution mass spectra
- Reagents were purchased from commercial sources and, where necessary, purified prior to use according to the general procedures outlined by D. D. Perrin and W.
- THF tetrahydrofuran
- dichloromethane and the combined dichloromethane extracts were washed with water, dried over MgSO 4 and stripped of the solvent under reduced pressure to yield a viscous liquid which was dissolved in a minimum quantity of tetrahydrofuran and the solution added to 0.5N solution of hydrogen chloride in anhydrous ether (1000 ml).
- the salt that separated was filtered off, washed with ether, suspended in 2-propanol (500 ml) and refluxed with stirring overnight.
- the hot mixture was filtered and the collected solid was washed with 2-propanol, then with ether and boiled again with 2-propanol (200 ml).
- the hot mixture was filtered and the collected solid was washed with ether and then suspended in 500 ml of IN NaOH solution and stirred at room temperature for 30 minutes and the mixture then
- the solid can be suspended in an excess of ammonium hydroxide (28-30%) and the mixture heated with stirring on a steam bath for 30 minutes and filtered. The collected solid was washed with water, pressed dry and boiled with 2-propanol, and the hot mixture filtered.
- benzodiazepin-8(9H)-one (5.02 g) in benzene (300 ml) under nitrogen. After the addition was complete the mixture was refluxed for 2 hours, cooled to 15-20 °C stirred and treated with 20% aqueous sodium hydroxide (150 ml) added dropwise initially and rapidly after the excess of the reducing agent had been destroyed. The mixture was transferred to a separatory funnel and shaken vigorously after the addition of water (150 ml). The organic layer was removed and the aqueous layer extracted once with benzene and the combined organic extracts were washed with water, dried over MgSO 4 and stripped of the solvent under reduced pressure.
- compositions possess psychotropic properties, particularly antipsychotic activity of good duration, while lacking the typical movement disorder side-effects of standard antipsychotic agents.
- these compounds may be useful in the treatment of
- PCP phencyclidine
- a Coulbourn Instruments large modular test cage 25 ⁇ 30 ⁇ 30 cm
- a pole suspended from the center of the ceiling and a stainless steel grid floor.
- a 0.75 mA pulsed current (250 msec on, 750 msec off) is delivered to the grid floor by a Coulbourn Instruments programmable shocker.
- Test Procedure Each trial lasts 25 seconds. Ten seconds after the rat is placed in the testing
- footshock is delivered for 15 seconds.
- the rat is immediately removed from the testing apparatus. If the rat climbs the pole within the first 10 seconds, footshock is avoided and an avoidance response is
- Drug testing is initiated after rats are well trained to consistently avoid footshock. Rats are tested for conditioned avoidance responding at various time intervals (30-360 minutes) after oral administration of test compound.
- inhibition of conditioned avoidance responding is expressed as a percentage of the corresponding Drug Vehicle (control) value.
- the percent antagonism is used to calculate ED 50 values when appropriate.
- a Peak ED 50 value ⁇ 20 mg/kg corresponds to +++; a value between 20-50 mg/kg corresponds to ++; a value between 50-100 corresponds to +; a value >100 corresponds to -.
- catalepsy at various time intervals (30-360 minutes) after oral administration of test compound.
- each rat is placed with its front paws over a 10 cm high horizontal bar.
- the intensity of catalepsy is measured by the length of time it takes the animal to move both forelegs to the table. A time of 20 seconds is considered maximal catalepsy.
- ED 50 mg/kg PO
- Dosage forms (compositions) suitable for administration ranges from 1 mg to 2000 mg.
- the active ingredient can be administered orally in solid dosage forms, such as capsules, tablets, and powders, or in liquid dosage forms, such as elixirs, syrups, and suspensions; it can also be administered parenterally in sterile liquid dosage forms.
- Gelatin capsules contain the active ingredient and powdered carriers, such as lactose, sucrose, mannitol, starch, cellulose derivatives, magnesium stearate, stearic acid, and the like. Similar diluents can be used to make compressed tablets. Both tablets and capsules can be manufactured as sustained release products to provide for continuous release of medication over a period of hours. Compressed tablets can be sugar coated or film coated to mask any unpleasant taste and protect the tablet from the atmosphere, or enteric-coated for selective disintegration in the
- Liquid dosage forms for oral administration can contain coloring and flavoring to increase patient acceptance.
- water a suitable oil, saline, aqueous dextrose (glucose), and related sugar solutions and glycols such as propylene glycol or polyethylene glycols are suitable carriers for parenteral solutions.
- saline aqueous dextrose (glucose)
- glycols such as propylene glycol or polyethylene glycols are suitable carriers for parenteral solutions.
- Solutions for parenteral administration preferably contain a water soluble salt of the active ingredient, suitable stabilizing agents, and if necessary, buffer substances.
- suitable stabilizing agents such as sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite
- bisulfite, sodium sulfite, or ascorbic acid are suitable stabilizing agents.
- citric acid and its salts and sodium EDTA are also used.
- parenteral solutions can contain
- preservatives such as benzalkonium chloride, methyl- or propyl-paraben, and chlorobutanol.
- Suitable pharmaceutical carriers are described in
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Toxicology (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description











Claims

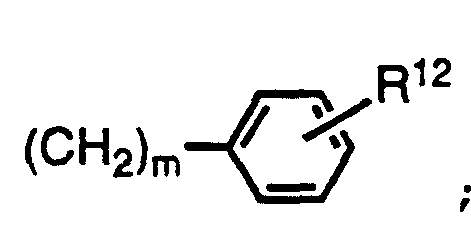


Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR9306896A BR9306896A (en) | 1992-07-29 | 1993-07-23 | Pyridoindole benzodiazepine compounds pharmaceutical compositions method of treating physiological or drug-induced psychosis or dyskinesia and intermediate compounds |
| AU46853/93A AU4685393A (en) | 1992-07-29 | 1993-07-23 | Pyridoindolobenzodiazepines and derivatives as antipsychotics |
| JP6505349A JPH08500579A (en) | 1992-07-29 | 1993-07-23 | Pyridoindolobenzodiazepines and derivatives as antipsychotics |
| KR1019950700217A KR950702562A (en) | 1992-07-29 | 1993-07-23 | Pyridoindolobenzodiazepine and its derivatives as antipsychotics (PYRIDOINDOLOBEN ZODIAZEPINES AND DERIVATIVES AS ANTIPSYCHOTICS) |
| SK403-95A SK40395A3 (en) | 1992-07-29 | 1993-07-23 | Pyridoindolobenzodiazepines and derivatives as antipsychotics |
| EP93917295A EP0652878A1 (en) | 1992-07-29 | 1993-07-23 | Pyridoindolobenzodiazepines and derivatives as antipsychotics |
| FI950303A FI950303A0 (en) | 1992-07-29 | 1995-01-24 | Pyridoindolobenzodiazepines and derivatives as antipsychotics |
| NO950327A NO950327L (en) | 1992-07-29 | 1995-01-27 | Pyridoindolobenzodiazepines and derivatives as antipsychotic agents |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US07/921,051 | 1992-07-29 | ||
| US07/921,051 US5321023A (en) | 1992-07-29 | 1992-07-29 | Pyridoindolobenzodiazepines and derivatives as antipsychotics |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994003455A1 true WO1994003455A1 (en) | 1994-02-17 |
Family
ID=25444846
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1993/006823 WO1994003455A1 (en) | 1992-07-29 | 1993-07-23 | Pyridoindolobenzodiazepines and derivatives as antipsychotics |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US5321023A (en) |
| EP (1) | EP0652878A1 (en) |
| JP (1) | JPH08500579A (en) |
| KR (1) | KR950702562A (en) |
| CN (1) | CN1100424A (en) |
| AU (1) | AU4685393A (en) |
| BR (1) | BR9306896A (en) |
| CA (1) | CA2141332A1 (en) |
| CZ (1) | CZ19895A3 (en) |
| FI (1) | FI950303A0 (en) |
| HR (1) | HRP931082A2 (en) |
| HU (1) | HUT69393A (en) |
| IL (1) | IL106352A0 (en) |
| MX (1) | MX9304552A (en) |
| NO (1) | NO950327L (en) |
| NZ (1) | NZ254650A (en) |
| PL (1) | PL307231A1 (en) |
| RU (1) | RU95107706A (en) |
| SK (1) | SK40395A3 (en) |
| TW (1) | TW260670B (en) |
| WO (1) | WO1994003455A1 (en) |
| ZA (1) | ZA935486B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6219605B1 (en) | 1995-12-12 | 2001-04-17 | Trw Airbag Systems Gmbh & Co. Kg | Air bag system with variable activation time point |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8367655B2 (en) * | 2009-07-03 | 2013-02-05 | Daya Drug Discoveries, Inc. | Pyridoindolobenzox—and thiazepine derivatives and uses thereof |
| WO2014137849A1 (en) * | 2013-03-04 | 2014-09-12 | Daya Drug Discoveries, Inc | Pentacyclic pyridoindolo[b,e]azepine derivatives and uses thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4438120A (en) * | 1982-11-12 | 1984-03-20 | E. I. Du Pont De Nemours & Company | Pyridoindolobenzodiazepine tranquilizers |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3373168A (en) * | 1964-06-02 | 1968-03-12 | Hoffmann La Roche | Certain benzazepinoxyridoindole compounds and their preparation |
| US3457271A (en) * | 1964-06-02 | 1969-07-22 | Hoffmann La Roche | 11-lower alkyl-hexahydro-1-benzazepino (3,2,1-h,i)pyrido(4,3-b)indole |
| US3764684A (en) * | 1971-08-11 | 1973-10-09 | M Finizio | Novel indolobenzazepine derivatives, useful as tranquilizers |
| US3790675A (en) * | 1972-04-19 | 1974-02-05 | Endo Lab | Certain indolobenzazepine derivatives as analgesics |
| US3983123A (en) * | 1973-01-22 | 1976-09-28 | Endo Laboratories, Inc. | Trans-octahydropyridoindolobenzazepines |
| US3890327A (en) * | 1973-05-11 | 1975-06-17 | Du Pont | Trans-1,2,3,4,4a,8,9,14a-octahydropyrido(r',3':2,3)indolo(1,7-ab)+8 1)benzazepine |
| US3932650A (en) * | 1973-01-22 | 1976-01-13 | E. I. Du Pont De Nemours & Company | Trans-octahydropyridoindolobenzazepines as central nervous system depressants |
| CA1026331A (en) * | 1973-12-06 | 1978-02-14 | Joel G. Berger | Substituted indolobenzazepines |
-
1992
- 1992-07-29 US US07/921,051 patent/US5321023A/en not_active Expired - Lifetime
-
1993
- 1993-07-15 IL IL106352A patent/IL106352A0/en unknown
- 1993-07-23 AU AU46853/93A patent/AU4685393A/en not_active Abandoned
- 1993-07-23 EP EP93917295A patent/EP0652878A1/en not_active Ceased
- 1993-07-23 JP JP6505349A patent/JPH08500579A/en active Pending
- 1993-07-23 CZ CZ95198A patent/CZ19895A3/en unknown
- 1993-07-23 CA CA002141332A patent/CA2141332A1/en not_active Abandoned
- 1993-07-23 WO PCT/US1993/006823 patent/WO1994003455A1/en not_active Application Discontinuation
- 1993-07-23 HU HU9500193A patent/HUT69393A/en unknown
- 1993-07-23 PL PL93307231A patent/PL307231A1/en unknown
- 1993-07-23 RU RU95107706/14A patent/RU95107706A/en unknown
- 1993-07-23 KR KR1019950700217A patent/KR950702562A/en not_active Application Discontinuation
- 1993-07-23 NZ NZ254650A patent/NZ254650A/en unknown
- 1993-07-23 BR BR9306896A patent/BR9306896A/en not_active Application Discontinuation
- 1993-07-23 SK SK403-95A patent/SK40395A3/en unknown
- 1993-07-27 HR HR07/921,051A patent/HRP931082A2/en not_active Application Discontinuation
- 1993-07-28 MX MX9304552A patent/MX9304552A/en unknown
- 1993-07-29 TW TW082106067A patent/TW260670B/zh active
- 1993-07-29 ZA ZA935486A patent/ZA935486B/en unknown
- 1993-07-29 CN CN93109416A patent/CN1100424A/en active Pending
-
1995
- 1995-01-24 FI FI950303A patent/FI950303A0/en not_active Application Discontinuation
- 1995-01-27 NO NO950327A patent/NO950327L/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4438120A (en) * | 1982-11-12 | 1984-03-20 | E. I. Du Pont De Nemours & Company | Pyridoindolobenzodiazepine tranquilizers |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6219605B1 (en) | 1995-12-12 | 2001-04-17 | Trw Airbag Systems Gmbh & Co. Kg | Air bag system with variable activation time point |
Also Published As
| Publication number | Publication date |
|---|---|
| MX9304552A (en) | 1994-05-31 |
| HU9500193D0 (en) | 1995-03-28 |
| FI950303A (en) | 1995-01-24 |
| FI950303A0 (en) | 1995-01-24 |
| HRP931082A2 (en) | 1997-06-30 |
| CZ19895A3 (en) | 1995-11-15 |
| RU95107706A (en) | 1997-03-20 |
| SK40395A3 (en) | 1995-12-06 |
| KR950702562A (en) | 1995-07-29 |
| EP0652878A1 (en) | 1995-05-17 |
| CA2141332A1 (en) | 1994-02-17 |
| CN1100424A (en) | 1995-03-22 |
| HUT69393A (en) | 1995-09-28 |
| PL307231A1 (en) | 1995-05-15 |
| NZ254650A (en) | 1996-03-26 |
| ZA935486B (en) | 1995-01-30 |
| BR9306896A (en) | 1998-12-08 |
| US5321023A (en) | 1994-06-14 |
| JPH08500579A (en) | 1996-01-23 |
| NO950327D0 (en) | 1995-01-27 |
| IL106352A0 (en) | 1993-11-15 |
| AU4685393A (en) | 1994-03-03 |
| TW260670B (en) | 1995-10-21 |
| NO950327L (en) | 1995-01-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2002246726B2 (en) | Substituted pyrroloquinolines and pyridoquinolines as serotonin agonists and antagonists | |
| US7081455B2 (en) | Substituted heterocycle fused gamma-carbolines | |
| US7071186B2 (en) | Substituted heterocycle fused gamma-carbolines | |
| US6849640B2 (en) | Therapeutic 1H-pyrido [4,3-b] indoles | |
| US5563152A (en) | Pyrrolo-pyridine derivatives | |
| US20050239768A1 (en) | Substituted hexahydro-pyridoindole derivatives as serotonin receptor agonists and antagonists | |
| AU2002246726A1 (en) | Substituted pyrroloquinolines and pyridoquinolines as serotonin agonists and antagonists | |
| KR20030070073A (en) | Substituted Pyridoindoles As Serotonin Agonists and Antagonists | |
| KR20050043935A (en) | Hydroxy alkyl substituted 1,3,8-triazaspiro[4.5]decan-4-one derivatives useful for the treatment of orl-1 receptor mediated disorders | |
| CZ111497A3 (en) | Diazepinoindoles as phosphodiesterase iv inhibitors | |
| US5321023A (en) | Pyridoindolobenzodiazepines and derivatives as antipsychotics | |
| AU3018000A (en) | Process for preparing 8-cyclopentyl-6-ethyl-3-(substituted) -5,8- dihydro-4H-1,2,3A,7,8-pentazza-as indacenes and intermediates useful therein | |
| AU2002246725A1 (en) | Aryl and aminoaryl substituted serotonin receptor agonist and antagonist ligands |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BB BG BR BY CA CZ FI HU JP KP KR KZ LK MG MN MW NO NZ PL RO RU SD SK UA VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 950303 Country of ref document: FI |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1995-198 Country of ref document: CZ Ref document number: 1993917295 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2141332 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 254650 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 40395 Country of ref document: SK |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993917295 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1995-198 Country of ref document: CZ |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1993917295 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1993917295 Country of ref document: EP |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV1995-198 Country of ref document: CZ |