WO1993010076A1 - Synthesis and optical resolution of the taxol side chain and related compounds - Google Patents
Synthesis and optical resolution of the taxol side chain and related compounds Download PDFInfo
- Publication number
- WO1993010076A1 WO1993010076A1 PCT/US1992/009911 US9209911W WO9310076A1 WO 1993010076 A1 WO1993010076 A1 WO 1993010076A1 US 9209911 W US9209911 W US 9209911W WO 9310076 A1 WO9310076 A1 WO 9310076A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- racemic
- side chain
- groups
- formula
- group
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims description 41
- HYJVYOWKYPNSTK-UONOGXRCSA-N (2r,3s)-3-benzamido-2-hydroxy-3-phenylpropanoic acid Chemical compound N([C@H]([C@@H](O)C(O)=O)C=1C=CC=CC=1)C(=O)C1=CC=CC=C1 HYJVYOWKYPNSTK-UONOGXRCSA-N 0.000 title claims description 36
- 230000015572 biosynthetic process Effects 0.000 title claims description 24
- 238000003786 synthesis reaction Methods 0.000 title claims description 19
- 230000003287 optical effect Effects 0.000 title description 8
- 238000000034 method Methods 0.000 claims abstract description 164
- 229940123237 Taxane Drugs 0.000 claims abstract description 121
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical group C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 claims abstract description 99
- 239000000203 mixture Substances 0.000 claims abstract description 79
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 claims abstract description 54
- 238000002425 crystallisation Methods 0.000 claims abstract description 53
- 229930012538 Paclitaxel Natural products 0.000 claims abstract description 31
- 229960001592 paclitaxel Drugs 0.000 claims abstract description 31
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 claims abstract description 31
- 230000001747 exhibiting effect Effects 0.000 claims abstract description 20
- 230000008878 coupling Effects 0.000 claims abstract description 14
- 238000010168 coupling process Methods 0.000 claims abstract description 14
- 238000005859 coupling reaction Methods 0.000 claims abstract description 14
- 238000004519 manufacturing process Methods 0.000 claims abstract description 6
- 230000002194 synthesizing effect Effects 0.000 claims abstract description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 73
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 63
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 57
- -1 carboxylic acid epoxide Chemical class 0.000 claims description 56
- 125000000623 heterocyclic group Chemical group 0.000 claims description 51
- 239000002904 solvent Substances 0.000 claims description 47
- 230000008025 crystallization Effects 0.000 claims description 46
- 238000006243 chemical reaction Methods 0.000 claims description 43
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 claims description 36
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 36
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 35
- 125000000217 alkyl group Chemical group 0.000 claims description 34
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 34
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 33
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 31
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 30
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 27
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 27
- 150000003944 halohydrins Chemical class 0.000 claims description 27
- 239000001257 hydrogen Substances 0.000 claims description 27
- 229910052739 hydrogen Inorganic materials 0.000 claims description 27
- 239000002243 precursor Substances 0.000 claims description 27
- UGUUDTWORXNLAK-UHFFFAOYSA-N azidoalcohol Chemical group ON=[N+]=[N-] UGUUDTWORXNLAK-UHFFFAOYSA-N 0.000 claims description 26
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 25
- 150000002924 oxiranes Chemical class 0.000 claims description 25
- 239000012039 electrophile Substances 0.000 claims description 23
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 22
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 claims description 22
- 229960001701 chloroform Drugs 0.000 claims description 20
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 19
- 230000032050 esterification Effects 0.000 claims description 19
- 238000005886 esterification reaction Methods 0.000 claims description 19
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 claims description 18
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 18
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 18
- 238000003476 Darzens condensation reaction Methods 0.000 claims description 17
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 17
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 claims description 16
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 16
- 125000004423 acyloxy group Chemical group 0.000 claims description 16
- 125000003342 alkenyl group Chemical group 0.000 claims description 16
- 125000000304 alkynyl group Chemical group 0.000 claims description 16
- 125000000392 cycloalkenyl group Chemical group 0.000 claims description 16
- 238000005984 hydrogenation reaction Methods 0.000 claims description 16
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims description 15
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 15
- 239000011877 solvent mixture Substances 0.000 claims description 15
- OVMSOCFBDVBLFW-VHLOTGQHSA-N 5beta,20-epoxy-1,7beta,13alpha-trihydroxy-9-oxotax-11-ene-2alpha,4alpha,10beta-triyl 4,10-diacetate 2-benzoate Chemical compound O([C@@H]1[C@@]2(C[C@H](O)C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)O)C(=O)C1=CC=CC=C1 OVMSOCFBDVBLFW-VHLOTGQHSA-N 0.000 claims description 14
- 150000001336 alkenes Chemical class 0.000 claims description 14
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 claims description 14
- 230000001590 oxidative effect Effects 0.000 claims description 14
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 14
- 239000003960 organic solvent Substances 0.000 claims description 13
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 13
- 230000008707 rearrangement Effects 0.000 claims description 13
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 claims description 12
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 claims description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 12
- 150000002431 hydrogen Chemical group 0.000 claims description 12
- 239000012038 nucleophile Substances 0.000 claims description 12
- 239000003795 chemical substances by application Substances 0.000 claims description 11
- 239000012454 non-polar solvent Substances 0.000 claims description 11
- 239000007800 oxidant agent Substances 0.000 claims description 11
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 11
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 claims description 11
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 11
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 claims description 10
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 claims description 10
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 10
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims description 9
- 125000004103 aminoalkyl group Chemical group 0.000 claims description 9
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 9
- 239000012374 esterification agent Substances 0.000 claims description 9
- 125000002541 furyl group Chemical group 0.000 claims description 9
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 claims description 9
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 claims description 9
- DYMRYCZRMAHYKE-UHFFFAOYSA-N n-diazonitramide Chemical compound [O-][N+](=O)N=[N+]=[N-] DYMRYCZRMAHYKE-UHFFFAOYSA-N 0.000 claims description 9
- 125000001544 thienyl group Chemical group 0.000 claims description 9
- 125000004001 thioalkyl group Chemical group 0.000 claims description 9
- 150000003573 thiols Chemical class 0.000 claims description 9
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 8
- KFSLWBXXFJQRDL-UHFFFAOYSA-N Peracetic acid Chemical compound CC(=O)OO KFSLWBXXFJQRDL-UHFFFAOYSA-N 0.000 claims description 8
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 claims description 8
- 229910052736 halogen Inorganic materials 0.000 claims description 8
- 150000002367 halogens Chemical group 0.000 claims description 8
- 239000012433 hydrogen halide Substances 0.000 claims description 8
- 229910000039 hydrogen halide Inorganic materials 0.000 claims description 8
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 8
- 150000004965 peroxy acids Chemical class 0.000 claims description 8
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 8
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 claims description 7
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 claims description 7
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 claims description 7
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 claims description 7
- 229930014667 baccatin III Natural products 0.000 claims description 7
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 claims description 7
- 229910000041 hydrogen chloride Inorganic materials 0.000 claims description 7
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 7
- WRBRIRNGWAUZRF-UHFFFAOYSA-N 2-methyl-2-[(2-methylpropan-2-yl)oxy]propane;sodium Chemical compound [Na].CC(C)(C)OC(C)(C)C WRBRIRNGWAUZRF-UHFFFAOYSA-N 0.000 claims description 6
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 claims description 6
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 claims description 6
- 239000002253 acid Substances 0.000 claims description 6
- 239000000010 aprotic solvent Substances 0.000 claims description 6
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 6
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 6
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 claims description 6
- LULAYUGMBFYYEX-UHFFFAOYSA-N metachloroperbenzoic acid Natural products OC(=O)C1=CC=CC(Cl)=C1 LULAYUGMBFYYEX-UHFFFAOYSA-N 0.000 claims description 6
- 229910000105 potassium hydride Inorganic materials 0.000 claims description 6
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 claims description 6
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 claims description 6
- 229910000104 sodium hydride Inorganic materials 0.000 claims description 6
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims description 5
- 239000003444 phase transfer catalyst Substances 0.000 claims description 5
- 239000012312 sodium hydride Substances 0.000 claims description 5
- 239000008096 xylene Substances 0.000 claims description 5
- XYPISWUKQGWYGX-UHFFFAOYSA-N 2,2,2-trifluoroethaneperoxoic acid Chemical compound OOC(=O)C(F)(F)F XYPISWUKQGWYGX-UHFFFAOYSA-N 0.000 claims description 4
- LJGHYPLBDBRCRZ-UHFFFAOYSA-N 3-(3-aminophenyl)sulfonylaniline Chemical compound NC1=CC=CC(S(=O)(=O)C=2C=C(N)C=CC=2)=C1 LJGHYPLBDBRCRZ-UHFFFAOYSA-N 0.000 claims description 4
- YNJSNEKCXVFDKW-UHFFFAOYSA-N 3-(5-amino-1h-indol-3-yl)-2-azaniumylpropanoate Chemical compound C1=C(N)C=C2C(CC(N)C(O)=O)=CNC2=C1 YNJSNEKCXVFDKW-UHFFFAOYSA-N 0.000 claims description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 4
- SQQRAHOSGYMFCS-UHFFFAOYSA-J [Na+].Cl[Ru](Cl)Cl.[O-]I(=O)(=O)=O Chemical group [Na+].Cl[Ru](Cl)Cl.[O-]I(=O)(=O)=O SQQRAHOSGYMFCS-UHFFFAOYSA-J 0.000 claims description 4
- 229910052744 lithium Inorganic materials 0.000 claims description 4
- 229940073584 methylene chloride Drugs 0.000 claims description 4
- SEDZOYHHAIAQIW-UHFFFAOYSA-N trimethylsilyl azide Chemical compound C[Si](C)(C)N=[N+]=[N-] SEDZOYHHAIAQIW-UHFFFAOYSA-N 0.000 claims description 4
- RMHGCKSCDKUSLM-UHFFFAOYSA-N (5z)-5-diazo-2,3,4,4a,6,7,8,9-octahydrobenzo[7]annulene Chemical compound [N-]=[N+]=C1CCCCC2=CCCCC12 RMHGCKSCDKUSLM-UHFFFAOYSA-N 0.000 claims description 3
- SGUVLZREKBPKCE-UHFFFAOYSA-N 1,5-diazabicyclo[4.3.0]-non-5-ene Chemical compound C1CCN=C2CCCN21 SGUVLZREKBPKCE-UHFFFAOYSA-N 0.000 claims description 3
- ZVHVKUZIHOULCY-UHFFFAOYSA-N C(C)(CC)OC(C)CC.[Na] Chemical compound C(C)(CC)OC(C)CC.[Na] ZVHVKUZIHOULCY-UHFFFAOYSA-N 0.000 claims description 3
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 claims description 3
- QIQRVIPQIJRSAD-UHFFFAOYSA-N [Li].CC(C)(C)OC(C)(C)C Chemical compound [Li].CC(C)(C)OC(C)(C)C QIQRVIPQIJRSAD-UHFFFAOYSA-N 0.000 claims description 3
- 229940022682 acetone Drugs 0.000 claims description 3
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 claims description 3
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 claims description 3
- 229910052808 lithium carbonate Inorganic materials 0.000 claims description 3
- 229910000103 lithium hydride Inorganic materials 0.000 claims description 3
- JILPJDVXYVTZDQ-UHFFFAOYSA-N lithium methoxide Chemical compound [Li+].[O-]C JILPJDVXYVTZDQ-UHFFFAOYSA-N 0.000 claims description 3
- AZVCGYPLLBEUNV-UHFFFAOYSA-N lithium;ethanolate Chemical compound [Li+].CC[O-] AZVCGYPLLBEUNV-UHFFFAOYSA-N 0.000 claims description 3
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 claims description 3
- 239000001095 magnesium carbonate Substances 0.000 claims description 3
- 229910000021 magnesium carbonate Inorganic materials 0.000 claims description 3
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 claims description 3
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 claims description 3
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 claims description 2
- 125000003545 alkoxy group Chemical group 0.000 claims description 2
- 150000001805 chlorine compounds Chemical group 0.000 claims description 2
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 claims description 2
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 claims description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 claims 4
- 150000004702 methyl esters Chemical class 0.000 claims 4
- MYHNUQVKTSTVDI-UHFFFAOYSA-N 2-methyl-2-[(2-methylpropan-2-yl)oxy]propane;potassium Chemical compound [K].CC(C)(C)OC(C)(C)C MYHNUQVKTSTVDI-UHFFFAOYSA-N 0.000 claims 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical group CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims 2
- GAXLZEYAJZDKNS-UHFFFAOYSA-N C(C)(CC)OC(C)CC.[K] Chemical compound C(C)(CC)OC(C)CC.[K] GAXLZEYAJZDKNS-UHFFFAOYSA-N 0.000 claims 2
- 229910000042 hydrogen bromide Inorganic materials 0.000 claims 2
- ANYSGBYRTLOUPO-UHFFFAOYSA-N lithium tetramethylpiperidide Chemical compound [Li]N1C(C)(C)CCCC1(C)C ANYSGBYRTLOUPO-UHFFFAOYSA-N 0.000 claims 2
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 claims 2
- BDAWXSQJJCIFIK-UHFFFAOYSA-N potassium methoxide Chemical compound [K+].[O-]C BDAWXSQJJCIFIK-UHFFFAOYSA-N 0.000 claims 2
- FTTATHOUSOIFOQ-UHFFFAOYSA-N 1,2,3,4,6,7,8,8a-octahydropyrrolo[1,2-a]pyrazine Chemical compound C1NCCN2CCCC21 FTTATHOUSOIFOQ-UHFFFAOYSA-N 0.000 claims 1
- HOJZAHQWDXAPDJ-UHFFFAOYSA-N 3-anilino-2-hydroxypropanoic acid Chemical compound OC(=O)C(O)CNC1=CC=CC=C1 HOJZAHQWDXAPDJ-UHFFFAOYSA-N 0.000 claims 1
- HYJVYOWKYPNSTK-UHFFFAOYSA-N 3-benzamido-2-hydroxy-3-phenylpropanoic acid Chemical compound C=1C=CC=CC=1C(C(O)C(O)=O)NC(=O)C1=CC=CC=C1 HYJVYOWKYPNSTK-UHFFFAOYSA-N 0.000 claims 1
- 125000002456 taxol group Chemical group 0.000 claims 1
- 239000000243 solution Substances 0.000 description 70
- 239000013078 crystal Substances 0.000 description 46
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 45
- 239000000047 product Substances 0.000 description 22
- UYJLJICUXJPKTB-UHFFFAOYSA-N methyl 3-benzamido-2-hydroxy-3-phenylpropanoate Chemical compound C=1C=CC=CC=1C(C(O)C(=O)OC)NC(=O)C1=CC=CC=C1 UYJLJICUXJPKTB-UHFFFAOYSA-N 0.000 description 18
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 16
- 238000002390 rotary evaporation Methods 0.000 description 15
- 239000000126 substance Substances 0.000 description 14
- 239000002585 base Substances 0.000 description 13
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- HAFFKTJSQPQAPC-DTWKUNHWSA-N methyl (2r,3s)-3-phenyloxirane-2-carboxylate Chemical compound COC(=O)[C@@H]1O[C@H]1C1=CC=CC=C1 HAFFKTJSQPQAPC-DTWKUNHWSA-N 0.000 description 11
- 125000004429 atom Chemical group 0.000 description 10
- 238000001914 filtration Methods 0.000 description 9
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 8
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 8
- VLKYMHSOQDMQNF-UHFFFAOYSA-N methyl 3-azido-2-hydroxy-3-phenylpropanoate Chemical group COC(=O)C(O)C(N=[N+]=[N-])C1=CC=CC=C1 VLKYMHSOQDMQNF-UHFFFAOYSA-N 0.000 description 8
- 230000037361 pathway Effects 0.000 description 8
- 239000003208 petroleum Substances 0.000 description 8
- 230000002950 deficient Effects 0.000 description 7
- 239000002274 desiccant Substances 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 239000003921 oil Substances 0.000 description 7
- 239000012044 organic layer Substances 0.000 description 7
- 238000010898 silica gel chromatography Methods 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 230000000259 anti-tumor effect Effects 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 238000002844 melting Methods 0.000 description 6
- 230000008018 melting Effects 0.000 description 6
- 238000007142 ring opening reaction Methods 0.000 description 6
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 6
- 235000017557 sodium bicarbonate Nutrition 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- 238000010189 synthetic method Methods 0.000 description 6
- DBXFAPJCZABTDR-KUEXGRMWSA-N Cephalomannine Natural products O=C(O[C@@H]1C(C)=C2[C@@H](OC(=O)C)C(=O)[C@]3(C)[C@@H](O)C[C@@H]4[C@](OC(=O)C)([C@H]3[C@H](OC(=O)c3ccccc3)[C@@](O)(C2(C)C)C1)CO4)[C@@H](O)[C@H](NC(=O)/C(=C\C)/C)c1ccccc1 DBXFAPJCZABTDR-KUEXGRMWSA-N 0.000 description 5
- PVALSANGMFRTQM-DTWKUNHWSA-N [(2s,3r)-3-phenyloxiran-2-yl]methanol Chemical group OC[C@@H]1O[C@@H]1C1=CC=CC=C1 PVALSANGMFRTQM-DTWKUNHWSA-N 0.000 description 5
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzenecarboxaldehyde Natural products O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 5
- DBXFAPJCZABTDR-WBYYIXQISA-N cephalomannine Chemical compound O([C@@H]1[C@]2(O)C[C@@H](C(=C([C@@H](OC(C)=O)C(=O)[C@]3(C)[C@@H](O)C[C@H]4OC[C@]4([C@H]31)OC(C)=O)C2(C)C)C)OC(=O)[C@H](O)[C@@H](NC(=O)C(/C)=C/C)C=1C=CC=CC=1)C(=O)C1=CC=CC=C1 DBXFAPJCZABTDR-WBYYIXQISA-N 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- 239000012452 mother liquor Substances 0.000 description 5
- 229910052938 sodium sulfate Inorganic materials 0.000 description 5
- 235000011152 sodium sulphate Nutrition 0.000 description 5
- 125000001424 substituent group Chemical group 0.000 description 5
- IKGZHVRGUUZPII-CXRLMVSZSA-N (2R,3S)-3-[benzoyloxy(1-ethoxyethyl)amino]-2-hydroxy-3-phenylpropanoic acid Chemical compound C(C1=CC=CC=C1)(=O)ON([C@H]([C@@H](O)C(=O)O)C1=CC=CC=C1)C(C)OCC IKGZHVRGUUZPII-CXRLMVSZSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- 239000005711 Benzoic acid Substances 0.000 description 4
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- PVALSANGMFRTQM-IUCAKERBSA-N [(2s,3s)-3-phenyloxiran-2-yl]methanol Chemical group OC[C@@H]1O[C@H]1C1=CC=CC=C1 PVALSANGMFRTQM-IUCAKERBSA-N 0.000 description 4
- 229910052783 alkali metal Inorganic materials 0.000 description 4
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 235000010233 benzoic acid Nutrition 0.000 description 4
- 238000001816 cooling Methods 0.000 description 4
- 238000006735 epoxidation reaction Methods 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 230000008020 evaporation Effects 0.000 description 4
- 239000000284 extract Substances 0.000 description 4
- JUINSXZKUKVTMD-UHFFFAOYSA-N hydrogen azide Chemical compound N=[N+]=[N-] JUINSXZKUKVTMD-UHFFFAOYSA-N 0.000 description 4
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- 230000003647 oxidation Effects 0.000 description 4
- 238000007254 oxidation reaction Methods 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- 238000012552 review Methods 0.000 description 4
- 238000006798 ring closing metathesis reaction Methods 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- 229940001593 sodium carbonate Drugs 0.000 description 4
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- 238000004809 thin layer chromatography Methods 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 4
- OOCCDEMITAIZTP-QPJJXVBHSA-N (E)-cinnamyl alcohol Chemical compound OC\C=C\C1=CC=CC=C1 OOCCDEMITAIZTP-QPJJXVBHSA-N 0.000 description 3
- YWLXLRUDGLRYDR-SKXCCXORSA-N 10-dab iii Chemical compound O([C@H]1C2[C@@](C([C@H](O)C3=C(C)[C@@H](O)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 YWLXLRUDGLRYDR-SKXCCXORSA-N 0.000 description 3
- FBXGQDUVJBKEAJ-UHFFFAOYSA-N 4h-oxazin-3-one Chemical compound O=C1CC=CON1 FBXGQDUVJBKEAJ-UHFFFAOYSA-N 0.000 description 3
- 241000196324 Embryophyta Species 0.000 description 3
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 3
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 3
- 230000003213 activating effect Effects 0.000 description 3
- 239000003513 alkali Substances 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 238000011914 asymmetric synthesis Methods 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 239000012069 chiral reagent Substances 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 150000004141 diterpene derivatives Chemical class 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- QABLOFMHHSOFRJ-UHFFFAOYSA-N methyl 2-chloroacetate Chemical group COC(=O)CCl QABLOFMHHSOFRJ-UHFFFAOYSA-N 0.000 description 3
- 229930014626 natural product Natural products 0.000 description 3
- 239000012299 nitrogen atmosphere Substances 0.000 description 3
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 3
- 235000011181 potassium carbonates Nutrition 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- YBCAZPLXEGKKFM-UHFFFAOYSA-K ruthenium(iii) chloride Chemical compound [Cl-].[Cl-].[Cl-].[Ru+3] YBCAZPLXEGKKFM-UHFFFAOYSA-K 0.000 description 3
- 238000007127 saponification reaction Methods 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 3
- 229940063683 taxotere Drugs 0.000 description 3
- 150000003952 β-lactams Chemical class 0.000 description 3
- UUFQTNFCRMXOAE-UHFFFAOYSA-N 1-methylmethylene Chemical compound C[CH] UUFQTNFCRMXOAE-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- JIQILHWIXUIMIO-UHFFFAOYSA-N OC.COC=O Chemical compound OC.COC=O JIQILHWIXUIMIO-UHFFFAOYSA-N 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 240000002834 Paulownia tomentosa Species 0.000 description 2
- 235000010678 Paulownia tomentosa Nutrition 0.000 description 2
- 241000202349 Taxus brevifolia Species 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 229910000102 alkali metal hydride Inorganic materials 0.000 description 2
- 150000008046 alkali metal hydrides Chemical class 0.000 description 2
- 150000004703 alkoxides Chemical class 0.000 description 2
- OOCCDEMITAIZTP-UHFFFAOYSA-N allylic benzylic alcohol Natural products OCC=CC1=CC=CC=C1 OOCCDEMITAIZTP-UHFFFAOYSA-N 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 150000001540 azides Chemical class 0.000 description 2
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 2
- WPUJEWVVTKLMQI-UHFFFAOYSA-N benzene;ethoxyethane Chemical compound CCOCC.C1=CC=CC=C1 WPUJEWVVTKLMQI-UHFFFAOYSA-N 0.000 description 2
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 210000004027 cell Anatomy 0.000 description 2
- XENVCRGQTABGKY-ZHACJKMWSA-N chlorohydrin Chemical compound CC#CC#CC#CC#C\C=C\C(Cl)CO XENVCRGQTABGKY-ZHACJKMWSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 239000010779 crude oil Substances 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000007865 diluting Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- GCSAXWHQFYOIFE-UHFFFAOYSA-N dipyridin-2-yl carbonate Chemical compound C=1C=CC=NC=1OC(=O)OC1=CC=CC=N1 GCSAXWHQFYOIFE-UHFFFAOYSA-N 0.000 description 2
- 229930004069 diterpene Natural products 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- FJKIXWOMBXYWOQ-UHFFFAOYSA-N ethenoxyethane Chemical compound CCOC=C FJKIXWOMBXYWOQ-UHFFFAOYSA-N 0.000 description 2
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 2
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 239000003517 fume Substances 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 230000001965 increasing effect Effects 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 2
- UYJLJICUXJPKTB-GJZGRUSLSA-N methyl (2s,3s)-3-benzamido-2-hydroxy-3-phenylpropanoate Chemical compound N([C@H]([C@H](O)C(=O)OC)C=1C=CC=CC=1)C(=O)C1=CC=CC=C1 UYJLJICUXJPKTB-GJZGRUSLSA-N 0.000 description 2
- 230000000877 morphologic effect Effects 0.000 description 2
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 2
- YCOZIPAWZNQLMR-UHFFFAOYSA-N pentadecane Chemical compound CCCCCCCCCCCCCCC YCOZIPAWZNQLMR-UHFFFAOYSA-N 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 239000012258 stirred mixture Substances 0.000 description 2
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 238000010792 warming Methods 0.000 description 2
- 239000011592 zinc chloride Substances 0.000 description 2
- 235000005074 zinc chloride Nutrition 0.000 description 2
- OOCCDEMITAIZTP-DAXSKMNVSA-N (Z)-cinnamyl alcohol Chemical compound OC\C=C/C1=CC=CC=C1 OOCCDEMITAIZTP-DAXSKMNVSA-N 0.000 description 1
- MQLACMBJVPINKE-UHFFFAOYSA-N 10-[(3-hydroxy-4-methoxyphenyl)methylidene]anthracen-9-one Chemical compound C1=C(O)C(OC)=CC=C1C=C1C2=CC=CC=C2C(=O)C2=CC=CC=C21 MQLACMBJVPINKE-UHFFFAOYSA-N 0.000 description 1
- TYLVGQKNNUHXIP-MHHARFCSSA-N 10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)C=4C=CC=CC=4)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 TYLVGQKNNUHXIP-MHHARFCSSA-N 0.000 description 1
- OKDGRDCXVWSXDC-UHFFFAOYSA-N 2-chloropyridine Chemical class ClC1=CC=CC=N1 OKDGRDCXVWSXDC-UHFFFAOYSA-N 0.000 description 1
- 229930190007 Baccatin Natural products 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 241000218631 Coniferophyta Species 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical group [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 235000015241 bacon Nutrition 0.000 description 1
- HUMNYLRZRPPJDN-KWCOIAHCSA-N benzaldehyde Chemical group O=[11CH]C1=CC=CC=C1 HUMNYLRZRPPJDN-KWCOIAHCSA-N 0.000 description 1
- 238000006480 benzoylation reaction Methods 0.000 description 1
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 1
- 229910010277 boron hydride Inorganic materials 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000011210 chromatographic step Methods 0.000 description 1
- 238000011097 chromatography purification Methods 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 239000007859 condensation product Substances 0.000 description 1
- 150000004292 cyclic ethers Chemical class 0.000 description 1
- UVJHQYIOXKWHFD-UHFFFAOYSA-N cyclohexa-1,4-diene Chemical compound C1C=CCC=C1 UVJHQYIOXKWHFD-UHFFFAOYSA-N 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 125000004188 dichlorophenyl group Chemical group 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- SRCZQMGIVIYBBJ-UHFFFAOYSA-N ethoxyethane;ethyl acetate Chemical compound CCOCC.CCOC(C)=O SRCZQMGIVIYBBJ-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- CELWCAITJAEQNL-UHFFFAOYSA-N oxan-2-ol Chemical compound OC1CCCCO1 CELWCAITJAEQNL-UHFFFAOYSA-N 0.000 description 1
- 238000013031 physical testing Methods 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 238000011085 pressure filtration Methods 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000006462 rearrangement reaction Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000013341 scale-up Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 150000004579 taxol derivatives Chemical class 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012795 verification Methods 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/66—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety
- C07C69/73—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids
- C07C69/732—Esters of carboxylic acids having esterified carboxylic groups bound to acyclic carbon atoms and having any of the groups OH, O—metal, —CHO, keto, ether, acyloxy, groups, groups, or in the acid moiety of unsaturated acids of unsaturated hydroxy carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/16—Preparation of optical isomers
- C07C231/20—Preparation of optical isomers by separation of optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/14—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms condensed with carbocyclic rings or ring systems
Definitions
- This invention relates to the racemic synthesis of taxane side chains and derivatives thereof, such as th taxol side chain. This invention also relates to the resolution of racemic mixtures of taxane side chains to obtain enantiomers in substantially optically pure form.
- this invention relates to the semisynthesis of taxanes such as taxol by coupling the resolved substantially optically pure taxane side chain to a taxane ring nucleus.
- Taxanes are alkaloids possessing a taxane nucleus.
- the taxane nucleus comprises the three ring structure shown below which is also identified as 4,8,12,15,15-pentamethyl-tricyclo [9.3.1.0 3*8 ] pentadecane:
- Taxane nucleus molecules such as baccatin-Ill and 10-desacetylbaccatin-III are inactive compounds as antitumor agents. However, attachment of the C-13 side chain to the molecule confers antitumor activity to the product. For instance, the core diterpene nucleus of taxol is baccatin-III. Thus, baccatin-III and 10- desacetylbaccatin-III are used to prepare taxol or similarly active compounds semi-synthetically by attachment of the C-13 taxol side chain.
- taxol Among the taxane molecules that have been studied most with respect to their antitumor activity are taxol, taxotere, 10-desacetyltaxol, cephalomannine and 10- desacetylcephalomannine.
- taxol taxotere
- 10-desacetyltaxol cephalomannine
- 10- desacetylcephalomannine The structures of these taxanes are shown below:
- R 8 H
- Taxanes such as taxol are believed to exert their antitumor activity by inducing tubulin polymerization and forming extremely stable and nonfunctional microtubules, which has an antiproliferative effect on taxane sensitive cells.
- Rowinsky et al. Journal of the National Cancer Institute. Vol. 82, No. 15, pp. 1247-59 (1990) ; Suffnes ⁇ , Gann Monographs Cancer Research, Vol. 36, pp. 21-24 (1989).
- the taxane known as taxol was first reported to be isolated from the stem bark of the western yew Taxus brevifolia. a slow growing conifer. Its structure was elucidated by ani et al., Journal of the American Chemical Society . Vol. 93, pp. 2325-27 (1971) .
- Taxanes such as taxol
- Taxanes are presently obtained in extremely low yield from the bark of T. brevifolia (0.004-0.016%). Because the level of occurrence is so low, large numbers of trees must be harvested to provide sufficient material for even a single course of therapy. Consequently, the availability of trees is insufficient to meet the demand for taxol and related taxanes.
- taxol and related taxanes are through semisynthesis.
- semisynthetic methods rely upon a source of the taxane ring nucleus, such as provided by 10-desacetylbaccatin III and baccatin-III, which are readily isolated from plant matter.
- the taxane ring nucleus is then coupled to an optically pure side chain that is chemically synthesized and that confers the desired pharmacological activity to the product. Denis et al., J. Or ⁇ . Chem.. Vol 55, pp. 1957-59 (1990) .
- a chiral molecule is any molecule that is not superimposable on its mirror image.
- the elements that characterize chiral molecules are chiral centers, chiral axes, chiral planes or a combination of these elements.
- the most commonly occurring cases in organic chemistry are those molecules that contain chiral centers or chiral atoms such as a carbon atom. Such molecules may have greater than one chiral atom.
- a chiral carbon atom can exist as two unique spacial dispositions of the four different substituents or groups that are chemically bonded to that atom.
- the two distinct mirror image arrangements of a chiral molecule are known in the art as enantiomers. These enantiomers are nonsuperimposable mirror images of each other. Thus, the enantiomers are said to exist in right- and left-handed forms.
- a mixture of equal amounts of each enantio er is called a racemic mixture or racemate. Any synthetic method that results in a racemic mixture is a racemic synthesis. Conversely, any synthetic method that results in the preponderance of one enantiomer over the other is known as an enantioselective synthesis.
- One method to obtain an enantioselective synthesis is to use asymmetric synthesis techniques.
- a two-step analysis is needed to designate a chiral atom as either R or S.
- the substituents around the chiral atom are prioritized in decreasing order according to their atomic number.
- the rules of priority with respect to substituent groups with multiple atoms or with double or triple bonds are described in the Cahn- Ingold-Prelog method. See Cahn, An Introduction to the Sequence Rule. J. Chem. Ed. , Vol. 41, p. 116 (1964).
- S econd the three-dimensional structure of the molecule must be visualized so that the group of lowest priority i as far as possible from the sight of the viewer.
- the R configuration exists when the sequence of the other group in decreasing order of priority is viewed in the clockwis (right-handed) direction.
- the S configuration exists whe the sequence obtained is viewed as being in the counterclockwise (left-handed) direction.
- one characteristic of compounds tha contain one or more chiral atoms is that they can be optically active.
- An optically active molecule is one that rotates plane-polarized light in a characteristic manner when such light is passed through a solution of th optically active molecule. If a substance rotates plane- polarized light to the right, it is designated the dextrorotary or "d" form. Such designation is indicated by a "+ ⁇ sign in the front of the degrees of rotation. I a substance rotates plane-polarized light to the left, it is the levorotary or "1" form and it is indicated with a • » -" sign before the degrees of rotation.
- One enantiomer will rotate plane-polarized light in a positive direction, while the other enantiomer will rotate plane-polarized light in the negative direction.
- These compounds may simply be referred to as ⁇ • +" or "-”.
- a racemic mixture of the + and - enantiomers may be referred to as n ⁇ " .
- the direction of rotation of plane polarized light by a particular enantiomer is independent of that enantiomer's R/S designation.
- optical purity of a molecule is generally expressed in terms of percent enantiomeric excess.
- Enantiomeric excess is a term that describes the preponderance of one enantiomer of a molecule over the other enantiomer. For example, an enantiomeric excess of 0% applies to a racemic mixture, and an enantiomeric excess of 100% applies to an optically pure compound.
- diastereomeric excess is a term used to describe the preponderance of one diastereomer over the ⁇ other in a mixture of diastereomeric forms of a chiral molecule.
- taxol and other taxanes depend upon a source of optically pure side chain.
- the naturally occurring and thus desired form of the taxol side chain is the "-" enantiomer, which is also described as the (2R, 3S)-isomer.
- Antitumor activity of taxanes requires such a side chain attached to position 13 of the taxane nucleus.
- the structure of the taxol side chain i.e. the (2R,3S)-N-benzoyl-3-phenylisoserine group, is shown below:
- These methods are deficient in that the chiral reagents employed are extremely expensive, and the amount of optically active side chain produced is extremely small.
- asymmetric synthetic methods require extremely specific starting materials in order to be effective.
- an additional deficiency of these methods is that only a limited number of starting materials and their derivatives may be successfully employed to produce the taxol side chain, thereby limiting the number of different compounds that may be prepared for clinical evaluation.
- Holton, U.S. Patent 5,015,744 refers to a method of producing a taxol side chain precursor in the form of an optically pure oxazinone.
- the oxazinone is then contacted with a taxane nucleus in the form of an alcohol to provide a taxol intermediate, which upon mild hydrolysis, is reported to produce taxol.
- the optically pure oxazinone precursor is derived from an optically pure acyclic taxol side chain, which in turn is derived from an optically pure ⁇ -lactam that has been resolved by the crystallization of diastereomeric ⁇ -lactam Mosher's esters.
- This method is deficient in that the indirect resolution of the optically pure ⁇ -lactam enantiomer requires the use of expensive chiral reagents as well as two additional reaction steps to obtain the appropriate enantiomer.
- Direct crystallization techniques if they are to be successful, depend upon the formation of a special crystalline form of the racemic compound that is called a conglomerate.
- a conglomerate is a mixture of two crystalline enantiomers that are separable by physical means.
- the melting point of the resolved enantiomers must be at least 20°C higher than the melting point of the racemic mixture.
- a conglomerate forms only if there is a difference in the solubility of the racemate relative to that of the enantiomers.
- a compound when it forms a conglomerate, aggregates into two distinct crystalline forms. Each crystal of a conglomerate contains only one of the two enantiomers in substantially pure form.
- the enantiomers of a conglomerate-forming compound and its derivatives may be resolved into their substantially optically pure forms by direct crystallization methods.
- novel methods for the synthesis of the racemic taxane side chain and derivatives thereof This invention also provides for an economical method for resolving the substantially optically pure enantiomers of the taxane side chain and its derivatives.
- This invention also provides for the semisynthetic production of taxanes such as taxol.
- the invention provides a method for the production of a substantially optically pure taxane side chain having the following formula:
- R 1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, quinoline, C1-C8 hydroxyalky1, C1-C6 aminoalkyl, and 2-, 3-, or 4-pyridino, or
- U and V are independently selected from the group consisting of hydrogen, halogen, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or aryl-N-amido, C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino, C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl- ⁇ or arylurea, trichloromethyl, and trifluoromethyl;
- R 2 is selected from the group consisting of C1-
- the method comprises the steps of preparing a racemic mixture of enantiomers of the taxane side chain capable of exhibiting conglomerate behavior an resolving the racemic taxane side chain into its substantially optically pure enantiomers.
- Taxanes such a taxol may be produced in accordance with this invention b synthesis of the taxane side chain capable of exhibiting conglomerate behavior, resolution of the side chain into its optically pure enantiomers and coupling the substantially optically pure (2R,3S)-taxane side chain to the taxane ring nucleus.
- This invention also provides a method of producing as a novel intermediate, a halohydrin composition of methyl threo-3-chloro-2-hydroxy-3- phenylpropionate having the formula:
- This invention provides a method wherein the racemic taxane side chain is synthesized in a form capable of exhibiting conglomerate behavior upon crystallization. That property of the synthesized racemic taxane side chain allows for the substantially optically pure enantiomers of the conglomerate forming side chain to be resolved by physical means such as manual sorting, localized crystallization, differentiated crystallization, and entrainment procedures.
- this invention provides a method of semisynthesizing taxanes such as taxol.
- the substantially optically pure (2R,3S)-taxane side chain is prepared for coupling and then coupled to a taxane ring nucleus.
- the present invention contemplates multiple methods of synthesizing the racemic taxane side chain 1.
- the structure of the preferred taxol side chain methyl ester, N-Benzoyl-3-phenylisoserine methyl ester, also referred to in the art as beta- or ⁇ -amido ester, is depicted below:
- the starting materials are an electrophile and a haloester.
- the electrophile has the following formula:
- R 1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, quinoline, C1-C8 hydroxyalkyl, C1-C6 aminoalkyl, and 2-, 3-, or 4-pyridino.
- the R 1 group of the electrophile 2 has the following formula:
- U and V are independently selected from the group consisting of hydrogen, halogen, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or aryl-N-amido , C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino, C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl- or arylurea, trichloromethyl, and trifluoromethyl.
- U and V are selected from the group consistin of hydrogen, halogen, azide, amino, trichloromethyl, and trifluoromethyl.
- U and V are both hydrogen, i.e., R 1 of the electrophile 2 is a phenyl group and the electrophile is benzaldehyde.
- the haloester preferably has the formula:
- X 1 is selected from the group consisting of chloride, bromide, or iodide and R 2 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, and C7-C12 alkylphenyl.
- R 2 is a methyl group and the haloester is methyl chloroacetate.
- the initial step of the synthesis to make a taxane side chain precursor is a Darzens condensation-like reaction between the electrophile and the haloester to make a first heterocyclic epoxide ring in which the groups R 1 and C0 2 R 2 are trans with the following formula:
- the heterocycle 5 in which the groups R 1 and CO. R 2 are trans is the trans-epoxide, methyl trans-3- phenyloxiranecarboxylate having the following formula:
- the base employed is of sufficient strength to generate an anionic form of the haloester known as an enolate, while preventing saponification of the ester moiety.
- Suitable bases include the alkali carbonate bases, such as sodium carbonate, potassium carbonate, or cesium carbonate; amin bases, such as triethylamine, diisopropylethylamine, 1,5-diazabicyclo [4.3.0]non-5-ene and diazobicyclo[5.4.0]undec-7-ene, l,4-diazabicyclo[2.2.0]octane, and alkali metal amide bases such as lithium diisopropylamide, lithium hexamethyldisilamide, lithium tetra ethylpiperidide, and alkali metal hydrides such as sodium or potassium hydride.
- the base is an alkali metal alkoxide selected from the group consisting of sodium methoxide, sodium ethoxide, sodium sec-butyloxide, and sodium tert-butyloxide.
- the sodium counterion may be replaced with a potassium counterion, although some changes in basicity and reaction selectivity may result.
- the solvent used in the reaction combining the electrophile and the haloester is preferably one normally associated with Darzens condensation reactions performed under basic conditions.
- Preferred solvents are alcohols, such as propanol, isopropanol, or butanol; ethers, such as diethyl ether; cyclic ethers, such as tetrahydrofuran; dipolar aprotic solvents, such as dimethylformamide, dimethylsulfoxide, N-methylpyrrolidinone or hexamethylphosphoramide; and mixtures thereof. More preferred solvents are methanol, ethanol, and mixtures thereof.
- the stereochemistry of the product formed by the Darzens condensation reaction between the electrophile and haloester is highly dependent upon the particular bases and solvents used to carry out the reaction. It is preferred that a base and solvent be chosen which would lead to a Darzens Condensation product 5 whose stereochemistry is such that the groups R 1 and C0 2 R 2 are trans. It has been found that a trans epoxide may be obtained by using an alkali metal alkoxide in the corresponding alcoholic solvent.
- reaction temperature of between about -30°C and about +40°C is preferred. More preferably, the reaction is performed at a temperature between about -20°C and about +20°C. Most preferably, the reaction of the electrophile and the haloester is performed between about -10°C and about +10°C.
- a suitable base/solvent solution is sodium metal in methanol chilled to about 0°C in an ice- salt bath.
- a mixture of the electrophile and haloester such as benzaldehyde and methyl chloroacetate, at a rate to maintain the reaction temperature at or about 0°C.
- the ratio of electrophile to haloester is from 1:1 to 1:3. More preferably, 1:1.5.
- the Darzens condensation reaction is sufficiently complete after stirring for about 15 hours at ambient temperature.
- the reaction product may be purified by conventional means, such as diluting the mixture with water, then extracting with diethyl ether, followed by drying over anhydrous magnesium sulfate, removing the solvent and distilling the residue.
- the racemic epoxide 5 is cleaved with a gaseous hydrogen halide of the formula HX 2 wherein X 2 is either chloride or bromide; and thereby caused to undergo a syn-ring opening to form a halohydrin having the following formula:
- halohydrin 6 is the previously
- nonpolar solvents include benzene, toluene, xylene, pentane, hexane, heptane, methylene chloride, chloroform, ethyl ether and tetrahydrofuran. More preferred nonpolar solvents include benzene, toluene, xylene, pentane, hexane, and heptane. Most preferred nonpolar solvents include benzene, toluene, and xylene.
- syn-ring opening may be accomplished by bubbling hydrogen chloride gas through a solution of 5a in dry benzene. After removing the excess hydrogen chloride by stirring under partial vacuum and removing the solvent, the residue can be trituated with petroleum ether-benzene to yield the chlorohydrin.
- the halohydrin 6 is treated with base to form a second racemic heterocyclic epoxide ring in which the groups R 1 and CO,R 2 are cis with the following formula:
- R 1 and CO,R 2 are cis is the cis-epoxide methyl cis-3- phenyloxiranecarboxylate (i.e. the stereoisomer of 5a) , which has the following formula:
- the intramolecular S N 2 ring closure of the halohydrin 6 is performed with a base to generate a heterocycle 7 in which the groups R 1 and C0 2 R 2 are cis.
- the base is selected from the group consisting of alkali or alkaline earth metal carbonates, such as sodium carbonate, lithium carbonate, potassium carbonate, cesium carbonate and magnesium carbonate; alkali metal alkoxides, such as sodium methoxide, sodium ethoxide, sodium tert-butyloxide, lithium methoxide, lithium ethoxide, and lithium tert-butyloxide; and alkali metal hydrides, such as sodium hydride, potassium hydride, and lithium hydride.
- these bases may be used with or without suitable phase transfer catalysts.
- intramolecular S N 2 ring closure can be effected in the presence of a phase transfer catalyst such as Aliquot 336, which is manufactured by the Henkel Corporation.
- a phase transfer catalyst such as Aliquot 336, which is manufactured by the Henkel Corporation.
- phase transfer catalysts see Tung and Speziale, Chemistry And Industry, p. 1985 (1963) , which is incorporated herein by reference. Without wishing to be bound by any theory, it is believed that the closure of such a halohydrin may be caused to proceed via two distinct mechanistic pathways, an ionic and a nonionic pathway.
- the ionic pathway arises from partial or complete dissociation of the halogen from the remainder of the halohydrin, followed by capture of the carbonium ion so formed by the alkoxide.
- the second pathway is nonionic and involves the direct displacement of the halogen with the alkoxide. It is preferred in this method of the invention that the closure proceed via the nonionic pathway. More preferably, the closure of the halohydrin proceeds via a nonionic s N 2-like pathway.
- the character of the reaction pathway may be influenced by the nature of the solvent used in the reaction. A solvent mixture that is sufficiently polar to permit heterocycle formation via a nonionic mechanism, yet avoid the ionic pathway is preferred.
- the solvent mixture comprises organic solvents and water.
- Suitable organic solvents to mix with water may be selected from the group consisting of dimethylformamide, dimethylsulfoxide, methanol, ethanol, isopropanol, and acetone.
- the ratio of organic solvent to water is preferably 60-90:40-10. A more preferred ratio is about 70:30. The most preferred ratio is about 60:40.
- S N 2 ring closure of 6 can be effected by adding sodium carbonate to a suspension of 6 in water followed by addition of acetone, which causes the halohydrin 6 to solubilize.
- the mixture is stirred at between 10°C and 60°C, preferably at about 50°C for about 2 hours before removing the acetone in vacuo and extracting the residue with diethyl ether.
- the ethereal extracts are in turn washed with water, dried over a desiccant such as anhydrous magnesium sulfate, and concentrated.
- the resulting crude oil is distilled to « * obtain the product 7.
- a racemic heterocycle 7 with cis stereochemistry about the groups R 1 and C0 2 R 2 is selectively cleaved into the hydroxy azide side chain precursor by conventional means, such as selective cleavage with sodium azide in aqueous methanol- methyl formate or with Lewis acid mediated processes such as trimethylsilyl azide with a catalytic amount of zinc chloride.
- Lewis acid mediated processes such as trimethylsilyl azide with a catalytic amount of zinc chloride.
- the hydroxy azide is methyl 3-Azido- 2-hydroxy-3-phenylpropionate and has the following formula:
- steps 8a These steps generally comprise treating the «*• heterocycle 7 wherein the groups R 1 and C0 2 R 2 are cis with a nucleophile, such as sodium azide in aqueous methanol- methyl formate or azidotrimethylsilane with a catalytic amount of zinc chloride, to form the hydroxy azide side chain precursor 8.
- a nucleophile such as sodium azide in aqueous methanol- methyl formate or azidotrimethylsilane with a catalytic amount of zinc chloride.
- nucleophiles include alkali or alkaline earth metal azides.
- the organic solvent from the solution containing the hydroxy azide 8 is first evaporated and the product is extracted into ether.
- the ether layer is dried over sodium sulfate and subsequently evaporated to give a product that is sufficiently pure (by NMR analysis) for the next step of the reaction sequence.
- the trans epoxide 5 undergoes a syn-ring opening with HN 3 preferably in a nonpolar aprotic solvent such as benzene, toluene, hexane, tetrahydrofuran , diethyl ether, methylene chloride or chloroform to directly form the hydroxy azide 8.
- a sample of trans epoxide 5a, methyl trans-3-phenyloxiranecarboxylate was treated with excess hydrogen azide dissolved in benzene containing a few drops of boron trifluoride etherate for 7 days at room temperature.
- the excess hydrogen azide was then quenched by addition of solid anhydrous sodium bicarbonate, the ⁇ > resulting mixture filtered, and the solvent removed by evaporation.
- the hydroxy azide product 8a was purified b silica gel chromatography with 10% ethyl acetate in hexane.
- the hydroxy azide also called an azido alcohol
- the hydroxy azide is caused to undergo an esterification/hydrogenation/rearrangement to form the racemic taxane side chain.
- the hydroxy1 group of the azido alcohol 8 is protected by conversion to the corresponding azido ester having the following formula:
- COR 4 group is the hydroxyl protecting group wherein R 4 is selected from the group consisting of R 1 (as described above) or OR 5 , wherein R 5 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl.
- the esterification is a benzoylation, i.e., R 4 is a phenyl group and the resulting azido ester is ( ⁇ )- methyl threo-3-azido-2-benzoyl-3-phenylpropionate.
- the hydroxyl protecting group chosen must be capable of subsequently undergoing transfer from the O
- such reagents include but are not limited to acyl transfer catalysts, such as pyridine or dimethylaminopyridine; and dehydrating agents, such as dicyclohexylcarbodiimide, sulfonyl chloride, carbonyldiimidazole, oxalyl chloride, triphenylphosphine/BrCl 3 C, and 2-chloropyridinium salts.
- acyl transfer catalysts such as pyridine or dimethylaminopyridine
- dehydrating agents such as dicyclohexylcarbodiimide, sulfonyl chloride, carbonyldiimidazole, oxalyl chloride, triphenylphosphine/BrCl 3 C, and 2-chloropyridinium salts.
- the azido group is hydrogenated by conversion from its nascent form (i.e., N 3 ) into its desired form, such as an amine group (i.e., NH 2 ) , by reduction.
- the reduction is performed either by using hydrogen gas and a hydrogenation catalyst, hydrogen generating source, such as 1,4-cyclohexadiene or ammonium formate and a hydrogenation catalyst, or by using a hydride source such as the boron hydride reagents.
- the rearrangement proceeds as the amine group formed in the hydrogenation- reduction attacks the hydroxyl protecting group, i.e., the COR 4 group.
- This attack transfers the COR 4 protecting group from the hydroxyl group site of the azido ester to the amine group site (i.e., an 0 >N transfer), and results in the formation of the desired taxane side chain having the following formula:
- Preferred taxane side chains synthesized in this manner contemplated by this invention can be selected from the group consisting of compounds having the formula (1) , wherein R 1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, quinoline, C1-C8 hydroxyalkyl, C1-C6 aminoalkyl, and 2-, 3-, or 4-pyridino; R 2 is selected from the group consisting of C1-C8 linear or branched alkyl,
- R 4 is selected from the group consisting of R 1 or OR 5 wherein R 5 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl,
- R 1 of the taxane side chain is • selected from the group having the formula
- U and V are independently selected from the group consisting of hydrogen, halogen, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or aryl-N-amido, C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino , C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl- or arylurea, trichloromethyl, and trifluoromethyl;
- R 2 is selected from the group consisting of C1-C
- R 1 of the taxane side chain has the formula:
- U and V are selected from the group consisting of hydrogen, halogen, azide, amino, trichloromethyl, and trifluoromethyl;
- R 2 is a methyl group; and
- R 4 is a phenyl grou .
- R 1 and R 4 are phenyl groups and
- R 2 is a methyl group. This is the taxol side chain (la) .
- Another method of preparing the taxane side chain according to the present invention starts with the epoxidation of an appropriately substituted racemic hydroxylated olefin in which the groups R 1 and CH 2 0H are trans.
- the olefin has the formula: -OH
- the olefin in which the groups R 1 and CH 2 OH are trans is the trans-cinnamyl alcohol, 3-phenyl-2- propen-1-ol, with the following formula:
- racemic hydroxy epoxide is ( ⁇ )-trans- 3-phenyloxiranemethanol with the following formula:
- racemic hydroxy epoxide 11 in which the groups R 1 and CH 2 OH are trans is then oxidized and esterified.
- the racemic hydroxy epoxide 11 is oxidized with a mild oxidant, such as ruthenium trichloride-sodium periodate, to form a racemic carboxylic acid epoxide in which the groups R 1 and C0 2 H are trans ° having the following formula:
- racemic carboxylic acid epoxide 12 is in turn converted with an esterification agent such as ethereal diazomethane into the racemic heterocycle in which the groups R 1 and C0 2 Me are trans with the following formul :
- R 1 is a phenyl group. This is equivalent to 5a, which is produced in the Darzens condensation of the preferred embodiment described above.
- the racemic heterocycle 12a in which the groups R 1 and C0 2 Me are trans formed in this embodiment is equivalent to the heterocyclic epoxide ring 5 produced in the Darzens condensation of the preferred embodiment described above, where R 2 is a methyl group.
- the taxane side chain 1 may then be formed according to the methods described above for converting the epoxide 5 to the hydroxy azide side chain precursor and then to the taxane side chain.
- Another method of producing the taxane side chain according to the present invention starts with the epoxidation of an appropriately substituted hydroxylated olefin in which the groups R 1 and CH 2 OH are cis.
- the cis-olefin has the formula:
- R 1 is as described above. More preferably, R 1 is a phenyl group, i.e., the olefin is the cis-cinnamyl alcohol, cis-3-phenyl-2-propen-l-ol.
- the racemic olefin 13 is treated with an epoxidizing agent such as meta-chloroperbenzoic acid or another peracid such as perbenzoic acid, peracetic acid, performic acid, peroxytrifluoroacetic acid, or peroxyphthalic acid to provide a racemic hydroxy epoxide in which the groups R 1 and CH 2 OH are cis having the following formula:
- an epoxidizing agent such as meta-chloroperbenzoic acid or another peracid such as perbenzoic acid, peracetic acid, performic acid, peroxytrifluoroacetic acid, or peroxyphthalic acid
- R 1 is as described above. More preferably, R 1 is phenyl group, i.e., the racemic hydroxy epoxide is ( ⁇ )- cis-3-phenyloxiranemethanol.
- the olefin 13 in which the groups R 1 and CH 2 OH are cis is provided in a 0.5M to 2.5M organic
- solvent solution such as chloroform, methylene chloride, trichloromethane containing sodium or potassium bicarbonate to maintain an alkaline reaction.
- solvent solution such as chloroform, methylene chloride, trichloromethane containing sodium or potassium bicarbonate to maintain an alkaline reaction.
- the olefin/organic solvent solution is about 1.5M. The resulting mixture is stirred while slowly
- the organic layer is preferably sequentially washed with about a 20% sodium bisulfite solution to consume the unreacted peracid, a saturated sodium bicarbonate solution to remove carboxylic
- racemic hydroxy epoxide is oxidized with a mild oxidant, such as ruthenium trichloride-sodium periodate, to form a racemic carboxylic acid epoxide in which the groups R 1 and C0 2 H are cis having the following formula:
- racemic carboxylic acid epoxide 15 is in turn converted with an esterification agent such as ethereal diazomethane into a racemic heterocycle in which the groups R 1 and C0 2 Me are cis with the following formula:
- the taxane side chain 1 may the be formed according to the methods described above for converting the epoxide 7 to the hydroxy azide side chain precursor 8 and then to the taxane side chain 1.
- racemic taxol side chain and its derivatives can be made to crystallize into conglomerates. It is contemplated by this invention that a racemic taxane side chain or taxane side chain precursor/intermediate whose attachment at the C-13 point of the taxane ring nucleus confers anti-tumor activity in the resulting taxane, will likewise be capable of exhibiting conglomerate behavior. Thus, it is also contemplated by this invention that the substantially optically pure enantiomers of these side chains can be resolved by direct crystallization methods.
- Another method of determining whether a racemic compound will exhibit conglomerate behavior involves physical testing of the melting points of the racemic and the pure enantiomers. As discussed above, generally if the melting point of the resolved enantiomer is at least 20°C higher than the melting point of the racemic mixture, the racemic compound will crystallize in the form of a conglomerate.
- Another method of determining conglomerate behavior involves conducting a visual morphological appearance analysis of the crystals from a racemate.
- each crystal of a conglomerate is either completely + or completely -.
- the + and crystals are themselves non-superimposable mirror images of each other. These enantiomerically pure crystalline forms can thus be distinguished visually with the aid of even a low power microscope.
- the practitioner can view a group of crystals of the racemic compound and determine whether they exist in right and left-handed forms. If so, the compound has crystallized into a conglomerate.
- the resolution of the substantially optically pure enantiomers of the conglomerate forming racemate is achieved by direct crystallization methods such as manual sorting of the conglomerate crystals, by localized and differentiated crystallization techniques, and by an entrainment procedure as depicted in Figure 1.
- the entrainment procedure is preferred in that it allows for the resolution of the optically pure side chain in industrial scale quantities.
- substantially optically pure means any molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 50%. More preferably, “substantially optically pure” means any molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 75%. Most preferably, “substantially optically pure” means any molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 90%.
- Resolution by manual sorting involves separating the + and - forms of the conglomerate on the basis of the morphological appearance of crystals formed during a simple crystallization, while the mother liquor remains racemic.
- the crystals of the + form and the - form may be sufficiently different in crystalline appearance that they may be separated by manual means.
- the crystalline forms are in fact mirror images of each other and can thus be distinguished by the right-handed and left-handed nature of the crystalline forms.
- Resolution by localized crystallization involves the simultaneous exposure of a solution that is supersaturated with respect to a racemic compound dissolved in the solvent to seed crystals of both + and - enantiomers that are placed in geographically separated locations in the crystallization vessel, and which serves as seed crystals for the further crystallization of the like enantiomers from the racemate.
- a solution that is supersaturated with respect to a racemic compound dissolved in the solvent to seed crystals of both + and - enantiomers that are placed in geographically separated locations in the crystallization vessel, and which serves as seed crystals for the further crystallization of the like enantiomers from the racemate.
- Such crystals may be grown by adding to a solution that is supersaturated with respect to a racemic compound, a large seed crystal of one enantiomer, such as the + enantiomer. As crystallization proceeds, the + enantiomer aggregates and crystallizes directly upon the seed crystal to give rise to even larger crystals of that enantiomer, while the - enantiomer forms smaller crystals distributed about the crystallization flask. Following removal of the solvent, the compound may be resolved by simply sifting the formed crystals. For a detailed description of differentiated crystallization, see Collet et al., Chemical Reviews. Vol. 60, pp. 215-230 (1980).
- Seed crystals of that enantiomer in excess are then added to the solution, also called the "mother liquor.”
- the steps of dissolving the racemic mixture in a warm solvent along with a slight excess of one enantiomer, cooling the solvent and adding seed crystals of the enantiomer in excess has the effect of creating a solution that is supersaturated with respect to the enantiomer whose seed crystals are added.
- the enantiomer in excess should be present in suf icient quantities to achieve a 1%-15% excess in solution of tha enantiomer by weight. More preferably, 3%-8% by weight.
- the enantiomer in exces that supersaturates the solution, for example the + enantiomer, begins to crystallize upon its seed crystals.
- the crystallization is allowed to proceed for an appropriate duration; for example, 1 to 3 hours was sufficient to perform the entrainment protocol as depicte in Figure 1.
- the crystallization should proceed for a duration of time such that 5% to 10% of the total mass of taxol side chain present in solution crystallizes upon the seed crystal as a single enantiomer
- the practitioner may then remove the crystals by any convenient means, and determine the mass of the + enantiomer removed. Now, however, the solution mother liquor is saturated with respect to the - enantiomer. Th practitioner therefore adds an amount of the racemic compound equal to the amount of the + enantiomer removed above, then warms the solution to completely dissolve the freshly added racemic compound in the solution.
- the solution vessel is warmed to about 35°C- 50°C. After sufficient warming to dissolve solids, the solution is cooled to a crystallization temperature. Preferably, the solution is cooled to a crystallization temperature of 10°C-30°C. Most preferably, 15°C-25°C.
- a resolution by entrainment is therefore an iterative procedure that may be performed on any scale and with any number of iterations as desired.
- a resolution by entrainment is useful on either a laborator or industrial scale.
- the taxol side chain or its derivatives are preferably dissolved in a solvent, although supercooled melts may also be applicable. More preferably, the side chain or its derivatives are dissolved in a warmed solvent in order to create a supersaturated solution. Any solvent in which the side chain dissolves and in which each enantiomer has a unique solubility relative to that of the racemate may be used in the method of the present invention.
- Preferred solvents may be selected from the group consisting of dimethylsulfoxide, dimethylformamide, dimethylacetamide, N-methylpyrolidinone, chloroform, trichloromethane, methylene chloride, acetone, methanol, ethanol, isopropanol and water and mixtures thereof.
- a seed crystal is added to the cooled supersaturated solution.
- the seed crystal may be added in any convenient manner. For example, the seed crystal may be simply dropped into a solution of material to be resolved, or it may be adhered to a retractable object, such as a glass rod or chemically inert line or string, to which other material subsequently crystallizes. Generally, the highest optical purity will be obtained with the slowest crystallization rates.
- the crystallization rate is often highly temperature dependent, with the crystallization rate increasing as temperature falls since the solubility of the compound generally decreases as temperature decreases. Further, the level of saturation is highly dependent upon the solvent chosen and the compound in question. To obtain an enantiomeric purity of greater than 90%, the solvent, temperature and crystallization rate must be adjusted accordingly.
- the crystallized material may be removed by any convenient method. Commonly used methods of removing » solid material from liquid include decanting or centrifugation. Preferred methods include gravity, vacuu or pressure filtration. Alternatively, an object containing adhered crystals may be simply removed from th crystallization liquor.
- the resolution of the taxane side chain through entrainment according to the process of the present invention is capable of providing large quantities of substantially optically pure enantiomers. Such quantities may be obtained by using the substantially optically pure enantiomers obtained as seed crystals to purify additional racemic solutions of the side chain. These resolutions can be repeated until the desired quantities of substantially pure enantiomers are obtained.
- This invention also contemplates the synthesis and resolution of the substantially optically pure taxane side chains such as the taxol side chain and derivatives thereof suitable for coupling to a taxane ring nucleus such as l ⁇ -desacetylbaccatin III and baccatin III, which are depicted below:
- R 8 H 10-Desacetyl Baccatin
- R 8 Ac Baccatin III
- Additional taxane ring nucleus formulas include:
- a and B are independently hydrogen or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy or A and B together form an oxo
- L and D are independently hydrogen or hydroxy or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy
- E and F are independently hydrogen or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy or; E and F together from an oxo
- G is hydrogen or hydroxy or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy or G and M together form an ox or methylene or G and M together form an oxirane or M and F together form an oxetane
- J is hydrogen, hydroxy, or lower alkanoyloxy, aleknoyloxy, alkynoyloxy
- the above taxane ring nuclei may be obtained from plant matter and then prepared by conventional means such as described in Denis et al. , Journal American Chemical Society. Vol. 110, pp. 5917-19 (1988).
- Denis et al., U.S. Patent 4,924,011 refer to the preparation of the taxane ring nucleus 7- triethylsilylbaccatin-III.
- the optically pure side chain must be prepared for coupling to the taxane ring nucleus to produce the taxane.
- R 6 is a hydroxyl protecting group selected from the group consisting of ethoxyethyl, methoxymethyl, ethoxymethyl, 2- trimethylsilylethoxymethyl, 2,2,2-trichloroethoxymethyl, methylthiomethy1, 2-methoxyethoxymethyl, tetrahydropyranol, trimethylsilyl, isopropyldimethysilyl, tert-butyldimethylsilyl, and triphenylmethyldimethylsilyl; R 1 , R 2 , R 4 are as described above.
- hydroxyl protecting groups are selected from the group consisting of ethoxyethyl, methoxymethyl, ethoxymethyl, 2- trimethylsilylethoxymethyl and 2,2,2- trichloroethoxy ethyl. These hydroxyl protecting groups and others are described in Denis et al., J. Org. Chem.. 51, p. 46-50 (1986); Denis et al., J. Am. Chem. Soc.. Vol. 110, pp 5917-19 (1988) and Swindell et al., J. Med. Chem.. Vol. 34, pp. 1176-84 (1991).
- This protected taxane side chain free carboxyli acid is then coupled to the taxane ring nucleus to provide the desired taxane.
- This coupling is achieved through the use of an activating reagent.
- Preferred activating reagents include dicyclohexylcarbodiimide,
- R 7 is selected from the group consisting of dichlorophenyl, 1,3,5-trichlorophenyl, 2-nitrophenyl, •a_ . .
- Example 8 ( ⁇ )-Trans-3-phenyloxiranemethanol (lla) by epoxidation
- the mixture was magnetically stirred while adding a chloroform (100 mL) solution of 85% meta-chloroperbenzoic acid (9.07 g, 45.0 mmol) dropwise over the period of one hour.
- the mixture was then heated to reflux for 3.5 hours and kept overnight at ambient temperature.
- the contents of the flask were cooled in an ice bath and the precipitated benzoic acid removed by filtration.
- the organic layer was washed sequentially with 20% sodium bisulfite solution (35 mL) to consume excess peracid, saturated sodium bicarbonate solution (2 x 50 mL) to remove excess benzoic acid, and finally with saturated sodium chloride solution (50 mL) to remove excess water from the organic phase.
- the organic layer was then dried over magnesium sulfate, filtered to remove the desiccant and the solution concentrated by rotary evaporation to an oil.
- the solution at ice4 temperature was adjusted to pH 1-2 with 10% hydrochloric acid solution and the resulting cold aqueous solution extracted with precooled diethyl ether (7 x 30 -fflL) • Tne black or organic extract was dried over sodium sulfate and quickly filtered through a short funnel packed with flash silica, followed by washes with cold diethyl ether (2 20 mL) .
- the almost clear ether layer at ice temperature was immediately treated with excess diazomethane, after which the reaction was allowed to gradually warm to ambient temperature and sit in a fume hood overnight. Anhydrous magnesium sulfate was slowly added to the resulting solution, the mixture filtered, and the solvent removed by rotary evaporation.
- Crystals of N-Benzoyl-3-phenylisoserine methyl ester la were grown by dissolving the racemate (0.5 g) in dimethylsulfoxide (2.0 mL) and allowing the solvent to slowly concentrate by evaporation at ambient temperature. The crystals were sorted by viewing under a low power stereoscopic microscope.
- Racemic taxol side chain methyl ester la (10 g) was dissolved in warm ethanol (150 mL) and the solution cooled to ambient temperature. Seed crystals of the
- (2R,3S)-taxol side chain methyl ester (3 g) of size greater than 30 mesh were added and stirring of the mixture was carried out for 0.5.-1.0 hour.
- the crystals that deposited were filtered, washed with cold ethanol (2 x 5 mL) and dried. The solid was then sifted through a 30 mesh sieve whereupon 5.6 g of crystalline (2R,3S)-taxol side chain methyl ester of optical purity of greater than
- Example 19 Taxol (2R,3S)-N-Benzoyl-O-(1-ethoxyethyl)-3- phenylisoserine 20 (42.8 mg, 0.12 mmol) in anhydrous toluene (1 mL) was introduced under an nitrogen atmosphere into a 5 mL round-bottomed flask equipped with a magnetic stirrer. Di-2-pyridyl carbonate (25.9 mg, 0.12 mmol) was then added and the mixture left to react for 4 to 5 minutes. 4-Dimethylaminopyridine (4.9 mg, 0.04 mmol) and 7-triethylsilylbaccatin III (14 mg, 0.02 mmol) were then added in a single portion.
- the colorless and homogeneous solution was left for 3 to 4 minutes, and then heated for 10 hours at 72°-74°C. After cooling, the reaction mixture was diluted with ethyl acetate (10 mL) and the organic solution as washed successively with saturated aqueous sodium bicarbonate solution (3 x 5 mL) , water (2 x 5 mL) and finally with saturated sodium chloride solution (5 mL) . The organic phase was dried over anhydrous sodium sulfate, the desiccant removed by filtration, and the ° solvent removed by rotary evaporation.
- the residue obtained was purified by analytical thin-layer chromatography on silica, eluting with an diethyl ether/methylene chloride (5:95 by volume) mixture, 4 runs being performed.
- the product (8.4 mg, 40% yield) was obtained in the form of a mixture of two epimers in the ratio 60:40, melting at 169°-173°C, after recrystallization from a methylene chloride/pentane mixture.
- the above product (72 mg, 0.009 mmol) was introduced at 0°C under an nitrogen atmosphere into a 10 mL round-bottomed flask equipped with a magnetic ⁇ tirrer.
- Ethanolic hydrochloric acid solution (3.6 mL) , coole beforehand to 0 °C, was added the mixture stirred at 0 °C for 30 hours.
- the reaction mixture was then diluted by adding ethyl acetate (20 mL) and water (10 mL) at 0°C. Following separation of the two phases, the organic layer was washed with water (5 x 5 mL) and with saturated sodium chloride solution (2 x 5 mL) and then dried over anhydrous sodium sulfate.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Analytical Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Epoxy Compounds (AREA)
Abstract
This invention relates to a method for the production of a substantially optically pure taxane side chain comprising the steps of synthesizing a racemic mixture of enantiomers of the taxane side chain that is capable of exhibiting conglomerate behavior and resolving the substantially optically pure enantiomers by direct crystallization methods. This invention also relates to the semisynthesis of taxanes such as taxol through coupling the substantially optically pure taxane side chain to a taxane ring nucleus.
Description
SYNTHESIS AND OPTICAL RESOLUTION OF THE TAXOL SIDE CHAIN AND RELATED COMPOUNDS
•v*.
TECHNICAL FIELD OF THE INVENTION
***
This invention relates to the racemic synthesis of taxane side chains and derivatives thereof, such as th taxol side chain. This invention also relates to the resolution of racemic mixtures of taxane side chains to obtain enantiomers in substantially optically pure form.
10 In addition, this invention relates to the semisynthesis of taxanes such as taxol by coupling the resolved substantially optically pure taxane side chain to a taxane ring nucleus.
BACKGROUND OF THE INVENTION
15 Taxanes are alkaloids possessing a taxane nucleus. The taxane nucleus comprises the three ring structure shown below which is also identified as 4,8,12,15,15-pentamethyl-tricyclo [9.3.1.03*8] pentadecane:

Several members of the taxane series of molecules, of which taxol, taxotere, cephalomannine,
30
,|N desacetylcephalomannine are members, possess antitumor activity. Some of the taxanes, and in particular taxol, have found use in the treatment of ovarian cancer and leukemia. MacGuire et al., Annals of Internal Medicine, Vol. Ill, p. 273 (1989) .
35
Taxane nucleus molecules such as baccatin-Ill
and 10-desacetylbaccatin-III are inactive compounds as antitumor agents. However, attachment of the C-13 side chain to the molecule confers antitumor activity to the product. For instance, the core diterpene nucleus of taxol is baccatin-III. Thus, baccatin-III and 10- desacetylbaccatin-III are used to prepare taxol or similarly active compounds semi-synthetically by attachment of the C-13 taxol side chain.
Among the taxane molecules that have been studied most with respect to their antitumor activity are taxol, taxotere, 10-desacetyltaxol, cephalomannine and 10- desacetylcephalomannine. The structures of these taxanes are shown below:
TAXANE MOLECULES

R8 = Ac, R" = C6HS Taxol R8 = H, R9 = (CH3) 3CO Taxotere R8 = H, R9 = C6HS 10-
Desacetyl
Taxol
R8 = AC, R >°* - = CH3CH = C (CH3) Cephalomann ine
R8 = H, R9 = CH3CH = C (CH3) 10-
Desacetyl cephalomann ine
AC = CCH, Acetate
Taxanes such as taxol are believed to exert their antitumor activity by inducing tubulin polymerization and forming extremely stable and nonfunctional microtubules, which has an antiproliferative effect on taxane sensitive cells. Rowinsky et al., Journal of the National Cancer Institute. Vol. 82, No. 15, pp. 1247-59 (1990) ; Suffnesε, Gann Monographs Cancer Research, Vol. 36, pp. 21-24 (1989). The taxane known as taxol was first reported to be isolated from the stem bark
of the western yew Taxus brevifolia. a slow growing conifer. Its structure was elucidated by ani et al., Journal of the American Chemical Society . Vol. 93, pp. 2325-27 (1971) .
Taxanes, such as taxol, are presently obtained in extremely low yield from the bark of T. brevifolia (0.004-0.016%). Because the level of occurrence is so low, large numbers of trees must be harvested to provide sufficient material for even a single course of therapy. Consequently, the availability of trees is insufficient to meet the demand for taxol and related taxanes.
Furthermore, wild populations of trees grow under highly variable conditions, which result in inconsistent levels of taxanes produced in the bark. T. brevifoliaf therefore, represents a nonrenewable and inconsistent source of taxanes. Unfortunately, this difficulty in obtaining adequate supplies of taxol has significantly limited the extent of clinical investigations despite its reported activity against melanoma and ovarian cancer. There have been several attempts to isolate taxanes from plant matter. For example, Wani et al. ,
Journal of the American Chemical Society. Vol 93, 2325-27 (1971) , National Cancer Institute Natural Products Branch paper NSC #125973 dated July 15, 1983, Witherup et al., Journal of Natural Products, Vol. 53, No. 5, pp. 1249-53 (1990), and Witherup et al. , Journal of Liquid
Chromatoσraphv. Vol. 12, No. 11, 2117-32 (1989), refer to the purification of taxol. In addition, Senilh et al. , Journal of Natural Products . Vol. 46, No. 1, pp. 131-37 (1984) , refers to the purification of taxol and cephalomannine. These attempts are deficient in that the taxanes are isolated in relatively low yield, approximately 50% based upon the theoretical yield of taxanes.
There have been attempts to chemically synthesize taxanes, whose characteristic diterpene ring
system nucleus is a difficult synthetic target. Syntheti efforts to obtain taxol have been hampered by the high degree of structural complexity and the profuse stereochemical attributes of the molecule. Holton et al., Journal of the American Chemical Society. Vol. 110, pp. 6558-60 (1988) and Swindell et al., Journal of Organic Chemistry. Vol. 55, pp. 3-5 (1990) , teach various attempted syntheses of the taxanes. These syntheses are deficient because of their complexity. Further, as discussed above, the mere presence of the diterpene nucleus in these synthetic products is inadequate to exhibit "taxol-like" activity. Thus, these final products lack sufficient pharmacological activity to serve as an effective antitumor agent.
Another approach to obtain taxol and related taxanes is through semisynthesis. Generally, semisynthetic methods rely upon a source of the taxane ring nucleus, such as provided by 10-desacetylbaccatin III and baccatin-III, which are readily isolated from plant matter. The taxane ring nucleus is then coupled to an optically pure side chain that is chemically synthesized and that confers the desired pharmacological activity to the product. Denis et al., J. Orσ. Chem.. Vol 55, pp. 1957-59 (1990) .
In the synthesis and semisynthesis of the taxane compounds (and organic chemicals in general) , a number of stereochemical terms apply. A chiral molecule is any molecule that is not superimposable on its mirror image. The elements that characterize chiral molecules are chiral centers, chiral axes, chiral planes or a combination of these elements. The most commonly occurring cases in organic chemistry are those molecules that contain chiral centers or chiral atoms such as a carbon atom. Such molecules may have greater than one chiral atom.
A chiral carbon atom can exist as two unique spacial dispositions of the four different substituents or
groups that are chemically bonded to that atom. The two distinct mirror image arrangements of a chiral molecule are known in the art as enantiomers. These enantiomers are nonsuperimposable mirror images of each other. Thus, the enantiomers are said to exist in right- and left-handed forms. A mixture of equal amounts of each enantio er is called a racemic mixture or racemate. Any synthetic method that results in a racemic mixture is a racemic synthesis. Conversely, any synthetic method that results in the preponderance of one enantiomer over the other is known as an enantioselective synthesis. One method to obtain an enantioselective synthesis is to use asymmetric synthesis techniques.
Stereoisomers that are not enantiomers of one another are called diastereomers of one another. Mixtures of diastereomers are known as diastereomeric mixtures.
A number of nomenclature systems have been developed to describe the arrangement of groups about a chiral atom. A commonly used protocol for the specification of configuration is the Cahn-Ingold-Prelog method. See Morrison and Boyd, Organic Chemistry. 3rd edition, published by Allyn and Bacon, Inc. , Boston. According to this method, the prefixes R (right-handed) and S (left-handed) in front of the substituents or compound name are used to designate the absolute configuration of the substituents about the chiral atom(s) .
A two-step analysis is needed to designate a chiral atom as either R or S. First, the substituents around the chiral atom are prioritized in decreasing order according to their atomic number. The rules of priority with respect to substituent groups with multiple atoms or with double or triple bonds are described in the Cahn- Ingold-Prelog method. See Cahn, An Introduction to the Sequence Rule. J. Chem. Ed. , Vol. 41, p. 116 (1964). Second, the three-dimensional structure of the molecule
must be visualized so that the group of lowest priority i as far as possible from the sight of the viewer. The R configuration exists when the sequence of the other group in decreasing order of priority is viewed in the clockwis (right-handed) direction. The S configuration exists whe the sequence obtained is viewed as being in the counterclockwise (left-handed) direction.
In general, one characteristic of compounds tha contain one or more chiral atoms is that they can be optically active. An optically active molecule is one that rotates plane-polarized light in a characteristic manner when such light is passed through a solution of th optically active molecule. If a substance rotates plane- polarized light to the right, it is designated the dextrorotary or "d" form. Such designation is indicated by a "+■■ sign in the front of the degrees of rotation. I a substance rotates plane-polarized light to the left, it is the levorotary or "1" form and it is indicated with a •»-" sign before the degrees of rotation. One enantiomer will rotate plane-polarized light in a positive direction, while the other enantiomer will rotate plane-polarized light in the negative direction. These compounds may simply be referred to as ■•+" or "-". A racemic mixture of the + and - enantiomers may be referred to as n±" . The direction of rotation of plane polarized light by a particular enantiomer is independent of that enantiomer's R/S designation.
The optical purity of a molecule is generally expressed in terms of percent enantiomeric excess. "Enantiomeric excess" is a term that describes the preponderance of one enantiomer of a molecule over the other enantiomer. For example, an enantiomeric excess of 0% applies to a racemic mixture, and an enantiomeric excess of 100% applies to an optically pure compound. Similarly, "diastereomeric excess" is a term used to describe the preponderance of one diastereomer over the
β other in a mixture of diastereomeric forms of a chiral molecule.
The semisynthetic preparation of taxol and other taxanes depends upon a source of optically pure side chain. The naturally occurring and thus desired form of the taxol side chain is the "-" enantiomer, which is also described as the (2R, 3S)-isomer. Antitumor activity of taxanes requires such a side chain attached to position 13 of the taxane nucleus. Generally, the structure of the taxol side chain, i.e. the (2R,3S)-N-benzoyl-3-phenylisoserine group, is shown below:

(la) For example, Senilh et al., C. R. Seances Acad. Sci Ser. 2., Vol. 299, pp. 1039-43 (1984), F. Gueritte-Voegelin, Tetrahedron. Vol. 42, pp. 4451-60 (1986), Colin et al., European patent Application 0 253 278, Colin et al., European patent Application 0 253 739, Denis et al. , Journal of Organic Chemistry. Vol. 55, pp. 1957-59 (1990) , Denis et al.. Journal of Organic Chemistry. Vol. 51, pp. 46-50 (1988), and Ojima et al.. Journal of Organic Chemistry. Vol. 56, pp. 1681-83 (1991), teach the semisynthesis of taxol from the coupling of the taxol side chain to naturally procured lQ-desacetyl baccatin-III. Each of the methods described are deficient because the optically pure side chain is synthesized using costly and complex asymmetric synthesis techniques.
The attempts to synthesize the (2R,3S)-N- benzoyl-3-phenylisoserine side chain of taxol using
asymmetric synthesis techniques generally rely upon the use of chiral reagents that impart asymmetry to the final product. See, e.g., Denis et al., J. Org. Chem.. Vol. 51, pp. 46-50 (1986); Denis et al., J. Org. Chem.. Vol. 55, pp. 1957-59 (1990); Ojima et al., J. Org. Chem.. Vol. 56, pp. 1681-83 (1991); Holton, U.S. Patent 5,015,744, issued May 14, 1991; and Denis et al., U.S. Patent 4,924,011, issued May 8, 1990. These methods are deficient in that the chiral reagents employed are extremely expensive, and the amount of optically active side chain produced is extremely small. Also, asymmetric synthetic methods require extremely specific starting materials in order to be effective. Thus, an additional deficiency of these methods is that only a limited number of starting materials and their derivatives may be successfully employed to produce the taxol side chain, thereby limiting the number of different compounds that may be prepared for clinical evaluation.
There have been attempts to prepare structural analogs of the taxol side chain. See, e.g., Swindell et al.. Journal of Medicinal Chemistry. Vol. 34, pp. 1176-84 (1991) . These attempts are also deficient in that they rely on the use of expensive chiral starting materials.
Generally, there are numerous ways to separate the enantiomers of a particular racemic compound. In the majority of cases, separation of a racemic mixture is carried out by diastereomeric compound crystallization or by chromatography techniques such as those described by Daicel Chemical Industries, Ltd., in Application Guide for Chiral Column Selection (1989) , or by kinetic resolution techniques. These methods are expensive, laborious and yield universally low quantities of optically pure material.
For example, Holton, U.S. Patent 5,015,744, refers to a method of producing a taxol side chain precursor in the form of an optically pure oxazinone. The
oxazinone is then contacted with a taxane nucleus in the form of an alcohol to provide a taxol intermediate, which upon mild hydrolysis, is reported to produce taxol. The optically pure oxazinone precursor is derived from an optically pure acyclic taxol side chain, which in turn is derived from an optically pure β-lactam that has been resolved by the crystallization of diastereomeric β-lactam Mosher's esters. This method is deficient in that the indirect resolution of the optically pure β-lactam enantiomer requires the use of expensive chiral reagents as well as two additional reaction steps to obtain the appropriate enantiomer.
There have also been attempts to resolve racemic mixtures of the taxol side chain and its analogs into enantiomers by kinetic resolution. Kinetic resolution methods rely upon the selective reactivity of one enantiomer of a racemic mixture with another chiral substance such as a chiral chemical reagent or an enzyme. See, e.g., Honig et al. , Tetrahedron, Vol. 46, no. 11, pp. 3841-50 (1990) . These methods are deficient in that the yield of the optically active taxol side chain is too low to be preparatively useful.
There have been attempts to resolve racemic mixtures of compounds unrelated to the taxol side chain by direct crystallization methods. These methods allow for industrial scale resolutions of such compounds. For example, these methods have historically been applied to the resolution of amino acids produced industrially. Collet et al.. Chemical Reviews. Vol. 80, No. 3 (1980). Industrially useful direct crystallization techniques include manual sorting of the crystalline enantiomers and localized crystallization, differentiated crystallization and entrainment procedures.
Direct crystallization techniques, if they are to be successful, depend upon the formation of a special crystalline form of the racemic compound that is called a
conglomerate. A conglomerate is a mixture of two crystalline enantiomers that are separable by physical means. Generally, for a racemic compound to exhibit conglomerate behavior, the melting point of the resolved enantiomers must be at least 20°C higher than the melting point of the racemic mixture. Brittain, Pharmaceutical
Research, Vol. 7, pp. 683-90 (1990). Thus, a conglomerate forms only if there is a difference in the solubility of the racemate relative to that of the enantiomers. A compound, when it forms a conglomerate, aggregates into two distinct crystalline forms. Each crystal of a conglomerate contains only one of the two enantiomers in substantially pure form. As such, the enantiomers of a conglomerate-forming compound and its derivatives may be resolved into their substantially optically pure forms by direct crystallization methods.
Although conglomerate formation is thus a much desired property for resolving a racemate on an industrial scale, it is estimated that only about 5-10% of all racemic compounds form a conglomerate. Brittain, supra. Hence, it is unknown and would be highly unexpected that a synthetically prepared racemic taxane side chain would exhibit conglomerate behavior prior to the present invention. Furthermore, the conditions for resolving the substantially optically pure enantiomers of a conglomerate forming compound such as the selection of solvents, temperatures and rates of crystallization are stringent.
SUMMARY OF THE INVENTION In accordance with the present invention, there are provided novel methods for the synthesis of the racemic taxane side chain and derivatives thereof. This invention also provides for an economical method for resolving the substantially optically pure enantiomers of the taxane side chain and its derivatives. This invention also provides for the semisynthetic production of taxanes such as taxol.
The invention provides a method for the production of a substantially optically pure taxane side chain having the following formula:

(1) wherein R1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, quinoline, C1-C8 hydroxyalky1, C1-C6 aminoalkyl, and 2-, 3-, or 4-pyridino, or

(3) wherein U and V are independently selected from the group consisting of hydrogen, halogen, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or aryl-N-amido, C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino, C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl-
ϋ or arylurea, trichloromethyl, and trifluoromethyl; R2 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, and C7-C12 alkylphenyl; R4 is selected from the group consisting of R1 or OR5, wherein R5 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl.
The method, in general, comprises the steps of preparing a racemic mixture of enantiomers of the taxane side chain capable of exhibiting conglomerate behavior an resolving the racemic taxane side chain into its substantially optically pure enantiomers. Taxanes such a taxol may be produced in accordance with this invention b synthesis of the taxane side chain capable of exhibiting conglomerate behavior, resolution of the side chain into its optically pure enantiomers and coupling the substantially optically pure (2R,3S)-taxane side chain to the taxane ring nucleus.
This invention also provides a method of producing as a novel intermediate, a halohydrin composition of methyl threo-3-chloro-2-hydroxy-3- phenylpropionate having the formula:

(6a) This invention provides a method wherein the racemic taxane side chain is synthesized in a form capable of exhibiting conglomerate behavior upon crystallization. That property of the synthesized racemic taxane side chain
allows for the substantially optically pure enantiomers of the conglomerate forming side chain to be resolved by physical means such as manual sorting, localized crystallization, differentiated crystallization, and entrainment procedures. In addition, this invention provides a method of semisynthesizing taxanes such as taxol. The substantially optically pure (2R,3S)-taxane side chain is prepared for coupling and then coupled to a taxane ring nucleus.
Accordingly, it is thus an object of the present invention to provide a method for the efficient racemic synthesis of the taxane side chain capable of exhibiting conglomerate behavior and derivatives thereof.
It is a further object of the present invention to provide a method for the synthesis of the racemic taxane side chain capable of exhibiting conglomerate behavior and derivatives thereof that allows flexibility in the selection of starting materials.
It is also an object of the present invention to provide an economical source of the enantiomerically pure (2R,3S)-taxane side chain and derivatives thereof.
It is thus an additional object of the present invention to provide an economically viable method for the resolution of racemic taxane side chain and derivatives thereof. It is a further object of this invention to provide an economically viable method for the semisynthesis of taxanes such as taxol.
It is an additional object of this invention to provide a novel intermediate that may be used to synthesize the taxane side chain and taxanes such as taxol.
Other objects and features of this invention will become apparent to those skilled in the art from the following description.
<-* BRIEF DESCRIPTION OF THE DRAWINGS
Certain objects and advantages of this inventio will be apparent upon consideration of the accompanying drawing which generally depicts the preferred entrainment protocol used to resolve the taxol side chain la. DETAILED DESCRIPTION OF THE INVENTION
SYNTHESES OF THE RACEMIC TAXOL SIDE CHAIN The present invention contemplates multiple methods of synthesizing the racemic taxane side chain 1. The structure of the preferred taxol side chain methyl ester, N-Benzoyl-3-phenylisoserine methyl ester, also referred to in the art as beta- or β-amido ester, is depicted below:
PhCO

(la) The names assigned to the chemical compounds recited herein are based on the IUPAC rules of chemical nomenclature.
In the preferred method of the present invention, the starting materials are an electrophile and a haloester. Preferably, the electrophile has the following formula:

(2) wherein R1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, quinoline, C1-C8 hydroxyalkyl, C1-C6 aminoalkyl, and 2-, 3-, or 4-pyridino.
More preferably, the R1 group of the electrophile 2 has the following formula:

(3) wherein U and V are independently selected from the group consisting of hydrogen, halogen, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or aryl-N-amido , C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino, C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl- or arylurea, trichloromethyl, and trifluoromethyl.
Preferably, U and V are selected from the group consistin of hydrogen, halogen, azide, amino, trichloromethyl, and trifluoromethyl. Most preferably, U and V are both hydrogen, i.e., R1 of the electrophile 2 is a phenyl group and the electrophile is benzaldehyde.
The haloester preferably has the formula:

(4) wherein X1 is selected from the group consisting of chloride, bromide, or iodide and R2 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, and C7-C12 alkylphenyl. Most preferably, X1 is chloride and R2 is a methyl group and the haloester is methyl chloroacetate.
The initial step of the synthesis to make a taxane side chain precursor is a Darzens condensation-like reaction between the electrophile and the haloester to make a first heterocyclic epoxide ring in which the groups R1 and C02R2 are trans with the following formula:

(5) wherein R1 and R2 are as described above. Preferably, the heterocycle 5 in which the groups R1 and CO.R2 are trans is the trans-epoxide, methyl trans-3- phenyloxiranecarboxylate having the following formula:
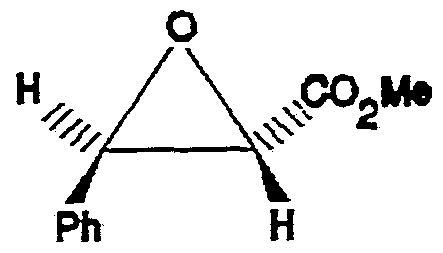
(5a) It has been found that a particularly advantageous method of synthesizing the heterocycle 5 involves reacting an electrophile, such as an aliphatic or aromatic aldehyde- or ketone-like molecule, with a haloester, as similarly shown in Rietveld et al. , Archives of Toxicology. Vol. 61, No. 5, pp. 362-72 (1988), which is incorporated herein by reference. Other suitable methods are described in Organic Synthesis, by Fuhrop and Penzlin, published by Verlag Chemie, and in Organic Synthetic Methods, published by John Wiley, New York, both of which are incorporated herein by reference.
Reactions of this sort are commonly performed under basic conditions. Preferably, the base employed is
of sufficient strength to generate an anionic form of the haloester known as an enolate, while preventing saponification of the ester moiety. Suitable bases include the alkali carbonate bases, such as sodium carbonate, potassium carbonate, or cesium carbonate; amin bases, such as triethylamine, diisopropylethylamine, 1,5-diazabicyclo [4.3.0]non-5-ene and diazobicyclo[5.4.0]undec-7-ene, l,4-diazabicyclo[2.2.0]octane, and alkali metal amide bases such as lithium diisopropylamide, lithium hexamethyldisilamide, lithium tetra ethylpiperidide, and alkali metal hydrides such as sodium or potassium hydride. Preferably, the base is an alkali metal alkoxide selected from the group consisting of sodium methoxide, sodium ethoxide, sodium sec-butyloxide, and sodium tert-butyloxide. Alternatively, the sodium counterion may be replaced with a potassium counterion, although some changes in basicity and reaction selectivity may result. The solvent used in the reaction combining the electrophile and the haloester is preferably one normally associated with Darzens condensation reactions performed under basic conditions. Preferred solvents are alcohols, such as propanol, isopropanol, or butanol; ethers, such as diethyl ether; cyclic ethers, such as tetrahydrofuran; dipolar aprotic solvents, such as dimethylformamide, dimethylsulfoxide, N-methylpyrrolidinone or hexamethylphosphoramide; and mixtures thereof. More preferred solvents are methanol, ethanol, and mixtures thereof.
In this method of the invention, the stereochemistry of the product formed by the Darzens condensation reaction between the electrophile and haloester is highly dependent upon the particular bases and solvents used to carry out the reaction. It is preferred that a base and solvent be chosen which would lead to a Darzens Condensation product 5 whose
stereochemistry is such that the groups R1 and C02R2 are trans. It has been found that a trans epoxide may be obtained by using an alkali metal alkoxide in the corresponding alcoholic solvent.
Those of skill in the art will understand that this reaction and all other reactions described herein ma proceed best at an optimum temperature. Generally, chemical reactions proceed faster at relatively higher temperatures, and slower at relatively lower temperatures. However, a loss of reaction selectivity is often associated with increased reaction rates occurring at higher temperatures. Thus, a balance must be found between the speed of reaction and the yield of the desired product. In the case of the reaction of the electrophile and the haloester, it has been found that a reaction temperature of between about -30°C and about +40°C is preferred. More preferably, the reaction is performed at a temperature between about -20°C and about +20°C. Most preferably, the reaction of the electrophile and the haloester is performed between about -10°C and about +10°C.
For example, a suitable base/solvent solution is sodium metal in methanol chilled to about 0°C in an ice- salt bath. To this solution, can be added a mixture of the electrophile and haloester, such as benzaldehyde and methyl chloroacetate, at a rate to maintain the reaction temperature at or about 0°C. Preferably, the ratio of electrophile to haloester is from 1:1 to 1:3. More preferably, 1:1.5. The Darzens condensation reaction is sufficiently complete after stirring for about 15 hours at ambient temperature. The reaction product may be purified by conventional means, such as diluting the mixture with water, then extracting with diethyl ether, followed by drying over anhydrous magnesium sulfate, removing the solvent and distilling the residue. In the next step of this synthetic route, the
racemic epoxide 5 is cleaved with a gaseous hydrogen halide of the formula HX2 wherein X2 is either chloride or bromide; and thereby caused to undergo a syn-ring opening to form a halohydrin having the following formula:

(6) wherein X2, R1 and R2 are as described above. More preferably, the halohydrin 6 is the previously
15 unknown threo-chlorohydrin, methyl-threo-chloro-2-hydroxy- 3-phenylpropionate with the following formula:

25
(6a) Solvents and reaction conditions that may be used to accomplish the syn-ring opening and cleave the racemic heterocycle 5 are those that provide for the
30 survival of the ester moiety and allow for the nucleophillic attack of the halide component of the hydrogen halide to provide the correct stereochemistry and regiochemistry. Nonpolar solvents must be used to accomplish these goals. For a discussion of suitable
35. types of solvents, see Tung & Speziale, J. Orσ. Chem.. pp.
2009-12 (1963) , which is incorporated herein by reference. Preferred nonpolar solvents include benzene, toluene, xylene, pentane, hexane, heptane, methylene chloride, chloroform, ethyl ether and tetrahydrofuran. More preferred nonpolar solvents include benzene, toluene, xylene, pentane, hexane, and heptane. Most preferred nonpolar solvents include benzene, toluene, and xylene.
For example, syn-ring opening may be accomplished by bubbling hydrogen chloride gas through a solution of 5a in dry benzene. After removing the excess hydrogen chloride by stirring under partial vacuum and removing the solvent, the residue can be trituated with petroleum ether-benzene to yield the chlorohydrin.
In the next step, the halohydrin 6 is treated with base to form a second racemic heterocyclic epoxide ring in which the groups R1 and CO,R2 are cis with the following formula:

(7) wherein R1 and R2 are as described above.
Preferably, the heterocycle in which the groups
R1 and CO,R2 are cis is the cis-epoxide methyl cis-3- phenyloxiranecarboxylate (i.e. the stereoisomer of 5a) , which has the following formula:

(7a) The steps of 1) opening the first heterocycle 5 to form a halohydrin 6 and 2) closing the halohydrin 6 to form a second heterocyclic ring 7 are performed in a manner to ensure the correct regiochemistry and stereochemistry of functional groups in the final side chain. For example, the group R1 and the C02R2 group must be cis to one another in the second heterocycle 7 if the heterocyclic ring, upon opening, is to yield the correct stereochemistry of the final side chain.
As stated above, the intramolecular SN2 ring closure of the halohydrin 6 is performed with a base to generate a heterocycle 7 in which the groups R1 and C02R2 are cis. Preferably, the base is selected from the group consisting of alkali or alkaline earth metal carbonates, such as sodium carbonate, lithium carbonate, potassium carbonate, cesium carbonate and magnesium carbonate; alkali metal alkoxides, such as sodium methoxide, sodium ethoxide, sodium tert-butyloxide, lithium methoxide, lithium ethoxide, and lithium tert-butyloxide; and alkali metal hydrides, such as sodium hydride, potassium hydride, and lithium hydride. Additionally, these bases may be used with or without suitable phase transfer catalysts. For instance, intramolecular SN2 ring closure can be effected in the presence of a phase transfer catalyst such as Aliquot 336, which is manufactured by the Henkel Corporation. For a discussion of phase transfer
catalysts, see Tung and Speziale, Chemistry And Industry, p. 1985 (1963) , which is incorporated herein by reference. Without wishing to be bound by any theory, it is believed that the closure of such a halohydrin may be caused to proceed via two distinct mechanistic pathways, an ionic and a nonionic pathway. The ionic pathway arises from partial or complete dissociation of the halogen from the remainder of the halohydrin, followed by capture of the carbonium ion so formed by the alkoxide. The second pathway is nonionic and involves the direct displacement of the halogen with the alkoxide. It is preferred in this method of the invention that the closure proceed via the nonionic pathway. More preferably, the closure of the halohydrin proceeds via a nonionic sN2-like pathway. In general, the character of the reaction pathway may be influenced by the nature of the solvent used in the reaction. A solvent mixture that is sufficiently polar to permit heterocycle formation via a nonionic mechanism, yet avoid the ionic pathway is preferred. The solvent mixture comprises organic solvents and water. Suitable organic solvents to mix with water may be selected from the group consisting of dimethylformamide, dimethylsulfoxide, methanol, ethanol, isopropanol, and acetone. The ratio of organic solvent to water is preferably 60-90:40-10. A more preferred ratio is about 70:30. The most preferred ratio is about 60:40.
For example, SN2 ring closure of 6 can be effected by adding sodium carbonate to a suspension of 6 in water followed by addition of acetone, which causes the halohydrin 6 to solubilize. The mixture is stirred at between 10°C and 60°C, preferably at about 50°C for about 2 hours before removing the acetone in vacuo and extracting the residue with diethyl ether. The ethereal extracts are in turn washed with water, dried over a desiccant such as anhydrous magnesium sulfate, and concentrated. The resulting crude oil is distilled to
«* obtain the product 7.
Once a racemic heterocycle 7 with cis stereochemistry about the groups R1 and C02R2 has been prepared, it is selectively cleaved into the hydroxy azide side chain precursor by conventional means, such as selective cleavage with sodium azide in aqueous methanol- methyl formate or with Lewis acid mediated processes such as trimethylsilyl azide with a catalytic amount of zinc chloride. See Denis et al., J. Org. Chem.. 55, 1957-59 (1990) , Denis et al., J. Org. Chem.. 51, 46-50 (1986), which are incorporated herein by reference. The hydroxy azide side chain precursor has the following formula:

(8) wherein R1 and R2 are as described above.
Preferably, the hydroxy azide is methyl 3-Azido- 2-hydroxy-3-phenylpropionate and has the following formula:

( 8a ) These steps generally comprise treating the
«*• heterocycle 7 wherein the groups R1 and C02R2 are cis with a nucleophile, such as sodium azide in aqueous methanol- methyl formate or azidotrimethylsilane with a catalytic amount of zinc chloride, to form the hydroxy azide side chain precursor 8. Other nucleophiles include alkali or alkaline earth metal azides.
For example, in Denis et al., J. Org. Chem.. 55, p. 1959-64 (1990) , a sample of the epoxide 7a in methanol- water (8:1) was treated with methyl formate and sodium azide and then stirred under argon at 50°C for 46 hours. The crude product was extracted with ether, washed with water, dried over a desiccant, and the solvent was removed by evaporation. The product was then purified by silica gel chromatography with 10% ethyl acetate in hexane to give the hydroxy azide 8a. An improved procedure eliminates the chromatographic purification and the expenses associated with this process. In this improved procedure, the organic solvent from the solution containing the hydroxy azide 8 is first evaporated and the product is extracted into ether. The ether layer is dried over sodium sulfate and subsequently evaporated to give a product that is sufficiently pure (by NMR analysis) for the next step of the reaction sequence.
In an alternative embodiment of this invention, the trans epoxide 5 undergoes a syn-ring opening with HN3 preferably in a nonpolar aprotic solvent such as benzene, toluene, hexane, tetrahydrofuran , diethyl ether, methylene chloride or chloroform to directly form the hydroxy azide 8. For example, a sample of trans epoxide 5a, methyl trans-3-phenyloxiranecarboxylate, was treated with excess hydrogen azide dissolved in benzene containing a few drops of boron trifluoride etherate for 7 days at room temperature. The excess hydrogen azide was then quenched by addition of solid anhydrous sodium bicarbonate, the
■> resulting mixture filtered, and the solvent removed by evaporation. The hydroxy azide product 8a was purified b silica gel chromatography with 10% ethyl acetate in hexane.
Once the heterocycle has been opened with the nucleophile to form the azide side chain precursor 8, the hydroxy azide, also called an azido alcohol, is caused to undergo an esterification/hydrogenation/rearrangement to form the racemic taxane side chain.
Generally, in the esterification step, the hydroxy1 group of the azido alcohol 8 is protected by conversion to the corresponding azido ester having the following formula:

(9) wherein the COR4 group is the hydroxyl protecting group wherein R4 is selected from the group consisting of R1 (as described above) or OR5, wherein R5 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl. Preferably, the esterification is a benzoylation, i.e., R4 is a phenyl group and the resulting azido ester is (±)- methyl threo-3-azido-2-benzoyl-3-phenylpropionate.
The hydroxyl protecting group chosen must be capable of subsequently undergoing transfer from the O
(hydroxyl group) ro the N (azido group - in either its nascent or subsequently converted form) , which is next
phase of the esterification/hydrogenation/rearrangement. See Protective Groups in Organic Synthesis, by Theodora W Greene, published by John Wiley, New York, 1981, incorporated herein by reference, for a description of additional suitable protecting groups. The esterification may be facilitated by the us of chemical reagents to increase the rate of azido ester formation. Generally, such reagents include but are not limited to acyl transfer catalysts, such as pyridine or dimethylaminopyridine; and dehydrating agents, such as dicyclohexylcarbodiimide, sulfonyl chloride, carbonyldiimidazole, oxalyl chloride, triphenylphosphine/BrCl3C, and 2-chloropyridinium salts. Once the hydroxyl group has been protected by esterification, the hydrogenation/rearrangement is performed. The azido group is hydrogenated by conversion from its nascent form (i.e., N3) into its desired form, such as an amine group (i.e., NH2) , by reduction. Generally, the reduction is performed either by using hydrogen gas and a hydrogenation catalyst, hydrogen generating source, such as 1,4-cyclohexadiene or ammonium formate and a hydrogenation catalyst, or by using a hydride source such as the boron hydride reagents.
As the reaction progresses, the rearrangement proceeds as the amine group formed in the hydrogenation- reduction attacks the hydroxyl protecting group, i.e., the COR4 group. This attack transfers the COR4 protecting group from the hydroxyl group site of the azido ester to the amine group site (i.e., an 0 >N transfer), and results in the formation of the desired taxane side chain having the following formula:

(1) Preferred taxane side chains synthesized in this manner contemplated by this invention can be selected from the group consisting of compounds having the formula (1) , wherein R1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, quinoline, C1-C8 hydroxyalkyl, C1-C6 aminoalkyl, and 2-, 3-, or 4-pyridino; R2 is selected from the group consisting of C1-C8 linear or branched alkyl,
C3-C8 cycloalkyl, and C7-C12 alkylphenyl; and R4 is selected from the group consisting of R1 or OR5 wherein R5 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl,
C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl.
More preferably, R1 of the taxane side chain is • selected from the group having the formula

(3) wherein U and V are independently selected from the group consisting of hydrogen, halogen, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or aryl-N-amido, C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino , C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl- or arylurea, trichloromethyl, and trifluoromethyl; R2 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, and C7-C12 alkylphenyl; and R4 is selected from the same group as R1.
Even more preferably R1 of the taxane side chain has the formula:

wherein U and V are selected from the group consisting of hydrogen, halogen, azide, amino, trichloromethyl, and trifluoromethyl; R2 is a methyl group; and R4 is a phenyl grou .
Most preferably R1 and R4 are phenyl groups and
R2 is a methyl group. This is the taxol side chain (la) .
For example, Denis et al., J. Org. Chem.. 55, 1959-64 (1990) , which is incorporated by reference, describe a one-pot benzoylation-hydrogenation in ethyl acetate of the hydroxy azide 8a, to form the taxol side chain methyl ester la. A mixture of hydroxy azide, benzoyl chloride, triethylamine and 4-
(dimethylamino)pyridine in ethyl acetate was stirred under argon at 20°C for 4 hours, whereupon methanol was added. After being stirred for an additional 3 hours, the reaction mixture was treated with 10% palladium on carbon and then placed under a hydrogen atmosphere. The resulting mixture was stirred for 68 hours and then processed with dichloromethane in the usual manner to afford the crude product, which was purified by silica gel chromatography with 5% ether in dichloromethane to give the taxol side chain methyl ester la.
It has been observed that the chromatography step can usually be avoided by carefully monitoring the hydrogenation/rearrangement reaction by TLC to insure it goes to completion. This method has been modified for "scale-up" to include first dilution with ethyl acetate, then washes first with dilute aqueous acid (i.e. 5% HC1) and second with saturated sodium bicarbonate solution. Subsequent drying of the ethyl acetate layer over a desiccant (sodium or magnesium sulfate) and evaporation of the solvent gives a product that can be purified by crystallization (from alcohol or ethyl acetate/hexane mixtures) . Another method of preparing the taxane side chain according to the present invention starts with the epoxidation of an appropriately substituted racemic hydroxylated olefin in which the groups R1 and CH20H are trans. Preferably, the olefin has the formula:
-OH

(10) wherein R1 is as described above.
More preferably, the olefin in which the groups R1 and CH2OH are trans is the trans-cinnamyl alcohol, 3-phenyl-2- propen-1-ol, with the following formula:

(10a) The racemic olefin in which the groups R1 and CH2OH are trans 10 is treated with an epoxidizing agent such as meta-chloroperbenzoic acid or another peracid, such as perbenzoic acid, peracetic acid, performic acid, peroxytrifluoroacetic acid, or peroxyphthalic acid to provide a racemic hydroxy epoxide in which the groups R1 and CH,OH are trans with the following formula:

(11) wherein R1 is as described above.
More preferably, the racemic hydroxy epoxide is (±)-trans- 3-phenyloxiranemethanol with the following formula:

(lla) The racemic hydroxy epoxide 11 in which the groups R1 and CH2OH are trans is then oxidized and esterified. First, the racemic hydroxy epoxide 11 is oxidized with a mild oxidant, such as ruthenium trichloride-sodium periodate, to form a racemic carboxylic acid epoxide in which the groups R1 and C02H are trans
° having the following formula:

(12 wherein R1 is as described above. Other mild oxidants are described in H.O. House, Modern Synthetic Reactions, pp. 292-96 (2d Ed. , W.A. Benjamin, Menlo Park, CA) .
The racemic carboxylic acid epoxide 12 is in turn converted with an esterification agent such as ethereal diazomethane into the racemic heterocycle in which the groups R1 and C02Me are trans with the following formul :

(12a) Most preferred in the racemic heterocycle in which the groups R1 and CO,Me are trans, R1 is a phenyl group. This is equivalent to 5a, which is produced in the Darzens condensation of the preferred embodiment described above.
These ruthenium trichloride oxidation and diazomethane esterification steps are described by Denis et al. , J. Org. Chem.. 51, p. 46-50 (1986), which is
incorporated herein by reference.
The racemic heterocycle 12a in which the groups R1 and C02Me are trans formed in this embodiment is equivalent to the heterocyclic epoxide ring 5 produced in the Darzens condensation of the preferred embodiment described above, where R2 is a methyl group. Thus, the taxane side chain 1 may then be formed according to the methods described above for converting the epoxide 5 to the hydroxy azide side chain precursor and then to the taxane side chain. Another method of producing the taxane side chain according to the present invention starts with the epoxidation of an appropriately substituted hydroxylated olefin in which the groups R1 and CH2OH are cis.
Preferably, the cis-olefin has the formula:
R1χ -OH
H" H
(13) wherein R1 is as described above. More preferably, R1 is a phenyl group, i.e., the olefin is the cis-cinnamyl alcohol, cis-3-phenyl-2-propen-l-ol.
The racemic olefin 13 is treated with an epoxidizing agent such as meta-chloroperbenzoic acid or another peracid such as perbenzoic acid, peracetic acid, performic acid, peroxytrifluoroacetic acid, or peroxyphthalic acid to provide a racemic hydroxy epoxide in which the groups R1 and CH2OH are cis having the
following formula:

(14
10 wherein R1 is as described above. More preferably, R1 is phenyl group, i.e., the racemic hydroxy epoxide is (±)- cis-3-phenyloxiranemethanol.
Preferably, the olefin 13 in which the groups R1 and CH2OH are cis is provided in a 0.5M to 2.5M organic
15 solvent solution such as chloroform, methylene chloride, trichloromethane containing sodium or potassium bicarbonate to maintain an alkaline reaction. Most preferably, the olefin/organic solvent solution is about 1.5M. The resulting mixture is stirred while slowly
20 adding, for example, a chloroform solution of meta-chloroperbenzoic acid or another peracid. The mixture is heated to reflux for 3-6 hours and then left undisturbed for 8-24 hours to allow sufficient precipitation of benzoic acid. The mixture is then cooled
25 in an ice bath and the benzoic acid and unreacted peracid precipitate removed by filtration. The organic layer is preferably sequentially washed with about a 20% sodium bisulfite solution to consume the unreacted peracid, a saturated sodium bicarbonate solution to remove carboxylic
30 acid and finally with a saturated sodium chloride solution to remove water. The organic layer is further dried over desiccant such as anhydrous magnesium sulfate and concentrated by rotary evaporation to an oil. The oil is ,j. subjected to flash silica gel chromatography using ethyl acetate in petroleum ether as eluant to obtain 14.
The racemic hydroxy epoxide 14 in which the groups R1 and CH2OH are cis is then oxidized and esterified. First, the racemic hydroxy epoxide is oxidized with a mild oxidant, such as ruthenium trichloride-sodium periodate, to form a racemic carboxylic acid epoxide in which the groups R1 and C02H are cis having the following formula:

(15) wherein R1 is as described above. Other mild oxidants are described in H.O. House, Modern Synthetic Reactions, pp. 292-96 (2d Ed. , W.A. Benjamin, Menlo Park, CA) .
The racemic carboxylic acid epoxide 15 is in turn converted with an esterification agent such as ethereal diazomethane into a racemic heterocycle in which the groups R1 and C02Me are cis with the following formula:

(16) wherein R1 is as described above.
Oxidation and esterification steps such as these are described by Denis et al., J. Org. Chem.. 51, p. 46-50
(1986) , which is incorporated herein by reference. The racemic heterocycle 16 formed in this embodiment is equivalent to that which is produced in SN2 ring closure of the halohydrin 6, which is 7, wherein R2 is a methyl group. Thus, the taxane side chain 1 may the be formed according to the methods described above for converting the epoxide 7 to the hydroxy azide side chain precursor 8 and then to the taxane side chain 1.
RESOLUTION OF THE OPTICALLY PURE TAXOL SIDE CHAIN Surprisingly, it has now advantageously been found that the racemic taxol side chain and its derivatives can be made to crystallize into conglomerates. It is contemplated by this invention that a racemic taxane side chain or taxane side chain precursor/intermediate whose attachment at the C-13 point of the taxane ring nucleus confers anti-tumor activity in the resulting taxane, will likewise be capable of exhibiting conglomerate behavior. Thus, it is also contemplated by this invention that the substantially optically pure enantiomers of these side chains can be resolved by direct crystallization methods.
There are several ways to determine whether a racemic compound will exhibit conglomerate behavior. In the course of the present invention, X-ray crystallography was utilized to analyze five separate crystals from a racemic mixture of enantiomers of the taxol side chain la. Successful mathematical solution of the X-ray data was only possible, for the case of a single enantiomer in the asymmetric unit of the acentric space group _?21. In the case of the taxol side chain la, the individual crystals analyzed were thus each comprised of only a single enantiomer, i.e., the racemic taxol side chain had crystallized in the form of a conglomerate. Thus, it was discovered that the enantiomers of the racemic taxol side chain are resolvable by industrial scale direct crystallization methods, which heretofore have never been
used in the production of taxanes.
Another method of determining whether a racemic compound will exhibit conglomerate behavior involves physical testing of the melting points of the racemic and the pure enantiomers. As discussed above, generally if the melting point of the resolved enantiomer is at least 20°C higher than the melting point of the racemic mixture, the racemic compound will crystallize in the form of a conglomerate.
Another method of determining conglomerate behavior involves conducting a visual morphological appearance analysis of the crystals from a racemate. Generally, each crystal of a conglomerate is either completely + or completely -. Further, the + and crystals are themselves non-superimposable mirror images of each other. These enantiomerically pure crystalline forms can thus be distinguished visually with the aid of even a low power microscope. To determine whether a given racemic compound forms a conglomerate, the practitioner can view a group of crystals of the racemic compound and determine whether they exist in right and left-handed forms. If so, the compound has crystallized into a conglomerate.
The resolution of the substantially optically pure enantiomers of the conglomerate forming racemate is achieved by direct crystallization methods such as manual sorting of the conglomerate crystals, by localized and differentiated crystallization techniques, and by an entrainment procedure as depicted in Figure 1. The entrainment procedure is preferred in that it allows for the resolution of the optically pure side chain in industrial scale quantities.
Direct crystallization methods for the resolution of the enantiomers of the conglomerate provide a high degree of optical purity of the final product. As used herein, "substantially optically pure" means any
molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 50%. More preferably, "substantially optically pure" means any molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 75%. Most preferably, "substantially optically pure" means any molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 90%.
Resolution by manual sorting involves separating the + and - forms of the conglomerate on the basis of the morphological appearance of crystals formed during a simple crystallization, while the mother liquor remains racemic. For example, the crystals of the + form and the - form may be sufficiently different in crystalline appearance that they may be separated by manual means. The crystalline forms are in fact mirror images of each other and can thus be distinguished by the right-handed and left-handed nature of the crystalline forms.
Resolution by localized crystallization involves the simultaneous exposure of a solution that is supersaturated with respect to a racemic compound dissolved in the solvent to seed crystals of both + and - enantiomers that are placed in geographically separated locations in the crystallization vessel, and which serves as seed crystals for the further crystallization of the like enantiomers from the racemate. For a description of localized crystallization and its applications to both laboratory and industrial work, see Collet et al. , Chemical Reviews. Vol. 60, pp. 215-30 (1980) , which is incorporated herein by reference. The use of differentiated crystallization depends upon the formation of crystals of a different size for each enantiomer, while the mother liquor remains racemic. Such crystals may be grown by adding to a solution that is supersaturated with respect to a racemic compound, a large seed crystal of one enantiomer, such as
the + enantiomer. As crystallization proceeds, the + enantiomer aggregates and crystallizes directly upon the seed crystal to give rise to even larger crystals of that enantiomer, while the - enantiomer forms smaller crystals distributed about the crystallization flask. Following removal of the solvent, the compound may be resolved by simply sifting the formed crystals. For a detailed description of differentiated crystallization, see Collet et al., Chemical Reviews. Vol. 60, pp. 215-230 (1980).
Industrial scale separation of enantiomers from a conglomerate forming racemate is most readily practiced using a procedure known as resolution by entrainment. Th separation of enantiomers is based on the property of conglomerate forming compounds that the solubility of a particular enantiomer will be less than that of the corresponding racemic compound. Crystallization of one enantiomer, such as the + enantiomer, results in the mother liquor being enriched in the opposite - enantiomer. For a detailed description of resolution by entrainment, see Collet et al.. Chemical Reviews. Vol. 60, pp. 215-30 (1980).
The following general entrainment protocol is followed for the resolution of a racemic mixture (± mixture) into its substantially optically pure + and - enantiomers. First, an amount of the ± mixture to be resolved is completely dissolved in a warmed solvent along with a small amount of one enantiomer, which is now in excess. The solution is then cooled to a crystallization temperature. Seed crystals of that enantiomer in excess are then added to the solution, also called the "mother liquor." The steps of dissolving the racemic mixture in a warm solvent along with a slight excess of one enantiomer, cooling the solvent and adding seed crystals of the enantiomer in excess, has the effect of creating a solution that is supersaturated with respect to the enantiomer whose seed crystals are added. Preferably, the
enantiomer in excess should be present in suf icient quantities to achieve a 1%-15% excess in solution of tha enantiomer by weight. More preferably, 3%-8% by weight.
As the solution cools, the enantiomer in exces that supersaturates the solution, for example the + enantiomer, begins to crystallize upon its seed crystals. The crystallization is allowed to proceed for an appropriate duration; for example, 1 to 3 hours was sufficient to perform the entrainment protocol as depicte in Figure 1. Generally, the crystallization should proceed for a duration of time such that 5% to 10% of the total mass of taxol side chain present in solution crystallizes upon the seed crystal as a single enantiomer The practitioner may then remove the crystals by any convenient means, and determine the mass of the + enantiomer removed. Now, however, the solution mother liquor is saturated with respect to the - enantiomer. Th practitioner therefore adds an amount of the racemic compound equal to the amount of the + enantiomer removed above, then warms the solution to completely dissolve the freshly added racemic compound in the solution.
Preferably, the solution vessel is warmed to about 35°C- 50°C. After sufficient warming to dissolve solids, the solution is cooled to a crystallization temperature. Preferably, the solution is cooled to a crystallization temperature of 10°C-30°C. Most preferably, 15°C-25°C.
Upon cooling, seed crystals of the - enantiomer are added to the solution, which now supersaturated with respect to the - enantiomer, begins to precipitate crystals of -. The process may now be repeated as described above, with the addition of racemic material followed by seed crystals of the + enantiomer. See Figure 1 for a more detailed example of this procedure.
A resolution by entrainment is therefore an iterative procedure that may be performed on any scale and with any number of iterations as desired. Thus, a
resolution by entrainment is useful on either a laborator or industrial scale.
In the entrainment method of the present invention, the taxol side chain or its derivatives are preferably dissolved in a solvent, although supercooled melts may also be applicable. More preferably, the side chain or its derivatives are dissolved in a warmed solvent in order to create a supersaturated solution. Any solvent in which the side chain dissolves and in which each enantiomer has a unique solubility relative to that of the racemate may be used in the method of the present invention. Preferred solvents may be selected from the group consisting of dimethylsulfoxide, dimethylformamide, dimethylacetamide, N-methylpyrolidinone, chloroform, trichloromethane, methylene chloride, acetone, methanol, ethanol, isopropanol and water and mixtures thereof. As stated above, a seed crystal is added to the cooled supersaturated solution. The seed crystal may be added in any convenient manner. For example, the seed crystal may be simply dropped into a solution of material to be resolved, or it may be adhered to a retractable object, such as a glass rod or chemically inert line or string, to which other material subsequently crystallizes. Generally, the highest optical purity will be obtained with the slowest crystallization rates. The crystallization rate is often highly temperature dependent, with the crystallization rate increasing as temperature falls since the solubility of the compound generally decreases as temperature decreases. Further, the level of saturation is highly dependent upon the solvent chosen and the compound in question. To obtain an enantiomeric purity of greater than 90%, the solvent, temperature and crystallization rate must be adjusted accordingly.
The crystallized material may be removed by any convenient method. Commonly used methods of removing
» solid material from liquid include decanting or centrifugation. Preferred methods include gravity, vacuu or pressure filtration. Alternatively, an object containing adhered crystals may be simply removed from th crystallization liquor. The resolution of the taxane side chain through entrainment according to the process of the present invention is capable of providing large quantities of substantially optically pure enantiomers. Such quantities may be obtained by using the substantially optically pure enantiomers obtained as seed crystals to purify additional racemic solutions of the side chain. These resolutions can be repeated until the desired quantities of substantially pure enantiomers are obtained.
For instance, approximately 4.0 g of - or (2R,3S)-taxol side chain methyl ester and approximately 3.2 g of + or (2S,3R)-taxol side chain methyl ester were obtained after only nine such cycles while starting with approximately 10 g of the racemate conglomerate and approximately 0.4 g of the (2R,3S)-enantiomer. See Figure 1- Furthermore, the isolated products were greater that 95% optically pure; the (2R,3S)-isomer specific rotation [α- 2^ - 47.2° (MeOH) , the (2S,3R)-isomer specific rotation α]2^ + 47.7° (MeOH) .
PREPARATION OF THE TAXANE SIDE CHAIN FOR COUPLING TO THE TAXANE RING NUCLEUS
This invention also contemplates the synthesis and resolution of the substantially optically pure taxane side chains such as the taxol side chain and derivatives thereof suitable for coupling to a taxane ring nucleus such as lθ-desacetylbaccatin III and baccatin III, which
are depicted below:

OCOC8H5
(17)
R8 = H 10-Desacetyl Baccatin
III
R8 = Ac Baccatin III
Ac = CCH, Acetate
Additional taxane ring nucleus formulas include:

(18) wherein A and B are independently hydrogen or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy or A and B together form an oxo; L and D are independently
hydrogen or hydroxy or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy; E and F are independently hydrogen or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy or; E and F together from an oxo; G is hydrogen or hydroxy or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy or G and M together form an ox or methylene or G and M together form an oxirane or M and F together form an oxetane; J is hydrogen, hydroxy, or lower alkanoyloxy, aleknoyloxy, alkynoyloxy, or aryloylox or I is hydrogen, hydroxy, or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy; or I and J taken together form an oxo; and K is hydrogen, hydroxy or lower alkoxy, alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy; and P and Q are independently hydrogen or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy or P and Q together form an oxo.
The above taxane ring nuclei may be obtained from plant matter and then prepared by conventional means such as described in Denis et al. , Journal American Chemical Society. Vol. 110, pp. 5917-19 (1988). For example, Denis et al., U.S. Patent 4,924,011, refer to the preparation of the taxane ring nucleus 7- triethylsilylbaccatin-III.
After the racemic taxane side chain has been successfully resolved into its optically pure form, the optically pure side chain must be prepared for coupling to the taxane ring nucleus to produce the taxane.
The resolved (2R,3S)-taxane side chain 1 is first converted to its hydroxyl protected form of the
following formula :

(19) by attachment of a hydroxyl protecting group R6, wherein R6 is a hydroxyl protecting group selected from the group consisting of ethoxyethyl, methoxymethyl, ethoxymethyl, 2- trimethylsilylethoxymethyl, 2,2,2-trichloroethoxymethyl, methylthiomethy1, 2-methoxyethoxymethyl, tetrahydropyranol, trimethylsilyl, isopropyldimethysilyl, tert-butyldimethylsilyl, and triphenylmethyldimethylsilyl; R1, R2, R4 are as described above. Most preferred hydroxyl protecting groups are selected from the group consisting of ethoxyethyl, methoxymethyl, ethoxymethyl, 2- trimethylsilylethoxymethyl and 2,2,2- trichloroethoxy ethyl. These hydroxyl protecting groups and others are described in Denis et al., J. Org. Chem.. 51, p. 46-50 (1986); Denis et al., J. Am. Chem. Soc.. Vol. 110, pp 5917-19 (1988) and Swindell et al., J. Med. Chem.. Vol. 34, pp. 1176-84 (1991).
Thereafter, saponification of the R2 group back to hydrogen is performed to form the protected taxane side
° chain free carboxylic acid of the following formula:

(20)
10 This protected taxane side chain free carboxyli acid is then coupled to the taxane ring nucleus to provide the desired taxane. This coupling is achieved through the use of an activating reagent. Preferred activating reagents include dicyclohexylcarbodiimide,
15 carbonyldiimidizole, di-2-pyridyl carbonate, oxalyl chloride, sulfonyl chloride or any of the activating or dehydrating agents described in Compendium Of Organic Synthetic Methods. Vols. 1-6, §107 (Wiley & Sons, NY).
Alternatively, the coupling of the taxane side
20 chain free carboxylic acid to the taxane nucleus may be achieved through formation of an activated ester of the following formula:
25

30
(21) wherein R7 is selected from the group consisting of dichlorophenyl, 1,3,5-trichlorophenyl, 2-nitrophenyl, •a_ . .
JJ 4-nιtrophenyl and 2 ,4-dmιtrophenyl. Methodologies for
° the formation of such activated esters are described in Compendium Of Organic Synthetic Methods. Vols. 1-6, §107 (Wiley & Sons, NY); Holton, U.S. Patent 5,015,744, issue May 14, 1991 and Denis et al., U.S. Patent 4,924,011, issued May 8, 1990, which are incorporated herein by reference.
The examples that follow, given without implied limitation, show how the invention can be put into practice.
Example 1 (±)-Methyl trans-3-phenyloxiranecarboxylate
(5a) by Darzens Condensation
To a solution of sodium metal (76.0 g, 3.3 g atom) in methanol (1000 ml) , which had been chilled to 0°C in an ice-salt bath, was added a mixture of benzaldehyde 2 (233.5 g, 2.2 mol) and methyl chloroacetate 4 (358.7 g, 3.3 mol) over a period of 3-4 hours at a rate so as to maintain the temperature of the reaction at about 0°C. The mixture was then stirred overnight at ambient temperature. After concentration by rotary evaporation to about 500 ml of volume, the mixture was poured into water (1500 ml) . The product was extracted with diethyl ether (4 x 300 ml) and dried over anhydrous magnesium sulfate. The solvent was then removed on a rotary evaporator and the residue distilled (108-110°C, 0.6 mm Hg) to yield (±)- methyl trans-3-phenyloxiranecarboxylate 5a (288.3 g, 73%).
Example 2
(±)-Methyl-threo-3-chloro-2-hydroxy-3-phenylpropionate
(6a) by Syn-Ring Opening
Hydrogen chloride gas was bubbled through a solution of (±)-methyl trans-3-phenyloxiranecarboxylate 5a (89.1 g, 0.50 mol) in dry benzene (1000 ml) at ambient temperature until the starting material was consumed as detected by thin layer chromatography. The reaction was generally judged to be complete in 3-4 hours. The excess hydrogen chloride was then removed by stirring the mixture
under a partial vacuum. The colorless residue following removal of the solvent by rotary evaporation was triturated with petroleum ether-benzene to form the substantially pure chlorohydrin (±)-6a (69.8 g, 65%).
Example 3 (±)-Methyl cis-3-phenyloxiranecarboxylate (7a) by SN2 Ring
Closure
Sodium carbonate (3.5 g, 7% solution) was added to a suspension of the threo-chlorohydrin 6a (2.15 g, 0.01 mmol) in water (46.5 ml) followed by addition of acetone (50 ml) . The mixture was stirred at 50°C for 2 hours before removing the acetone in vacuo and extracting the residue with diethyl ether (2 x 100 ml) . The ethereal extracts were in turn washed with water (3 x 50 ml) , dried over anhydrous magnesium sulfate, and evaporated to an oil on a rotary evaporator. Distillation (110-112°C, 0.6 mm Hg) of the resulting crude oil afforded the substantially pure compound (±)-7a (0.90 g, 50%).
Example 4
(±)-methyl-3-azido-2-hydroxy-3-phenylpropionate (8a) by selective cleavage of (7a)
To a solution of (±)-methyl cis-3- phenyloxiranecarboxylate 7a (27.0 g, 0.15 mol) in methanol (712 mL) was added methyl formate (126 mL) followed by addition of sodium azide (49.2 g, 0.76 mol) in water (88 mL) . The whole mixture was stirred under nitrogen at 50°C for 46 hours before concentrating the reaction by rotary evaporation. The product was extracted into diethyl ether (4 x 50 mL) , and the combined ethereal extracts dried over sodium sulfate and rotary evaporated to give 31.2 g (93%) of (±)-methyl threo-3-azido-2-hydroxy-3-phenylpropionate that was sufficiently pure (>95% by NMR analysis) for the next step.
Example 5
(±)-methyl-3-azido-2-hydroxy-3-phenylpropionate (8a) by
Syn-Ring opening of (5a)
(±)-Methyl trans-3-phenyloxiranecarboxylate 5a (17.82 g, 0.1 mol) was added to a solution of benzene (200 ml) containing hydrogen azide (0.5 mol) . A few drops (5- 10 drops) of boron trifluoride etherate were added and the reaction allowed to stand at room temperature for 7 days. Anhydrous sodium carbonate was then added to consume the excess hydrogen azide, followed by filtration of the resulting mixture. The solvent was then removed by rotary evaporation and the residue chromatographed on silica gel while eluting with 10% ethyl acetate in hexane to give recovered (±)-methyl trans-3-phenyloxirane carboxylate (5.8 g, 33%) and a mixture of erythro and threo azide alcohols. The latter two compounds were separated by passage through a second column of silica gel to give (±)- methyl-3-azido-2-hydroxy-3-phenylpropionate 8a (8.5 g, 38%) .
Example 6 (±)-Methyl threo-3-azido-2-benzoyl-3-phenylpropionate (9)
Benzoyl chloride (2.55 mL, 22.0 mmol) and 4- dimethylaminopyridine (25 mg) were added to a solution of (±)-methyl threo-3-azido-2-hydroxy-3-phenylpropionate 8a (4.42 g, 20.0 mmol) and triethyla ine (2.42 g, 24.0 mmol) in methylene chloride (50 mL) . After stirring for l hour at room temperature, the reaction was quenched by addition of water (5 mL) , the methylene chloride layer was separated, and the aqueous phase extracted with methylene chloride (3 x 10 mL) . The combined organic phases were washed with saturated sodium bicarbonate solution (3 x 25 mL) and dried over sodium sulfate. The raw product (6.6 g) obtained by rotary evaporation of the solvent was crystallized from petroleum ether-ethyl acetate to give pure (±)-methyl threo-3-azido-2-hydroxy-3-phenylpropionate 9 (6.40 g, 98%) .
Example 7
(±)-N-Benzoyl-3-phenylisoserine methyl ester: (±)-taxol side chain methyl ester (la)
A mixture of (±)-methyl threo-3-azido-2-hydroxy 3-phenylpropionate 9 (3.00 g, 9.22 mmol) and 10% palladiu on carbon (600 mg) in methanol (185 mL) was vigorously stirred under 1 atmosphere of hydrogen. After 15 hours, the hydrogen was replaced with a nitrogen atmosphere, the mixture was filtered, and the filtrate was allowed to stand at room temperature for 72 hours. The methanol was then removed by rotary evaporation to leave a crude solid that was purified by precipitation from methylene chloride with cyclohexane (2.46 g, 89%) . Crystallization of this material from chloroform yielded 1.65 g (60%) of (±)-N- Benzoyl-3-phenylisoserine methyl ester (racemic taxol side chain methyl ester) la.
Example 8 (±)-Trans-3-phenyloxiranemethanol (lla) by epoxidation A 500 ml, three-necked flask fitted with a reflux condenser and a dropping funnel was charged with a chloroform (25 mL) , trans-cinnamyl alcohol 10a (5.00 g, 37.0 mmol) and sodium bicarbonate (4.20 g, 50.0 mmol) . The mixture was magnetically stirred while adding a chloroform (100 mL) solution of 85% meta-chloroperbenzoic acid (9.07 g, 45.0 mmol) dropwise over the period of one hour. The mixture was then heated to reflux for 3.5 hours and kept overnight at ambient temperature. The contents of the flask were cooled in an ice bath and the precipitated benzoic acid removed by filtration. The organic layer was washed sequentially with 20% sodium bisulfite solution (35 mL) to consume excess peracid, saturated sodium bicarbonate solution (2 x 50 mL) to remove excess benzoic acid, and finally with saturated sodium chloride solution (50 mL) to remove excess water from the organic phase. The organic layer was then dried over magnesium sulfate, filtered to remove the desiccant
and the solution concentrated by rotary evaporation to an oil. Chromatography of the raw product on silica gel while eluting with 35% ethyl acetate in petroleum ether provided fractions of pure product, which upon rotary evaporation gave (±)-trans-3-phenyloxiranemethanol 11a (4.46 g, 82%) .
Example 9
(±)-Methyl trans-3-phenyloxiranecarboxylate (12a) by oxidation/esterification
Sodium bicarbonate (8.40 g, 100.0 mmol), sodium periodate (12.81 g, 59.9 mmol) and ruthenium trichloride (0.12 g. 0.58 mmol) were added to a vigorously stirred mixture of (±)-trans-3-phenyloxiranemethanol 11a (3.00 g, 19.9 mmol) in a solvent system comprised of carbon tetrachloride:acetonitrile:water (1:1:1.5, 140 mL) . The reaction was stirred at ambient temperature for 44 hours prior to dilution with water (100 mL) . The solution at ice4 temperature was adjusted to pH 1-2 with 10% hydrochloric acid solution and the resulting cold aqueous solution extracted with precooled diethyl ether (7 x 30 -fflL) • Tne black or organic extract was dried over sodium sulfate and quickly filtered through a short funnel packed with flash silica, followed by washes with cold diethyl ether (2 20 mL) . The almost clear ether layer at ice temperature was immediately treated with excess diazomethane, after which the reaction was allowed to gradually warm to ambient temperature and sit in a fume hood overnight. Anhydrous magnesium sulfate was slowly added to the resulting solution, the mixture filtered, and the solvent removed by rotary evaporation. The raw product was purified by flash silica gel chromatography while eluting with 35% ethyl acetate in petroleum either to give (±)-methyl trans-3-phenyloxiranecarboxylate 12a (3.31 g, 93%) upon rotary evaporation of the appropriate column fractions. Example 10
° (±)-cis-3-phenyloxiranemethanol (14) by epoxidation
A 250 mL three-necked flask fitted with a reflu condenser, stir bar and a dropping funnel was charged wit a chloroform solution (25 mL) of cis-3-phenyl-2-propene-l-ol 13 (5.00 g, 37.0 mmol.) and sodium bicarbonate (4.20 g, 50.0 mmol). The mixture was stirred while adding a chloroform solution (100 mL) of 85 -chloroperbenzoic acid (9.10 g, 45.0 mmol) dropwise over a period of 1 hour. The mixture was heated to reflux for
3.5 hours and then allowed to sit overnight. The content of the flask were cooled in an ice bath and the precipitate removed by filtration. The organic layer was sequentially washed with a 20% sodium bisulfite solution
(35 ml) , a saturated sodium bicarbonate solution (2 x 50 ml) and finally a saturated sodium chloride solution (50 ml) . The organic layer was dried over anhydrous magnesium sulfate and concentrated to an oil by rotary evaporation.
The oil was subjected to flash silica gel chromatography using 35% ethyl acetate in petroleum ether as eluant. The major fraction was identified as the compound (±)-14 (4.60 g, 82%).
Example 11
(±)- Methyl cis-3-phenyloxiranecarboxylate (16) by oxidation/esterification
A 2 L, one-necked flask fitted with a stir bar was charged with (±)-cis-3-phenyloxiranemethanol 14 (3.71 g, 20.9 mmol), and a mixture of carbon tetrachloride:acetonitrile:water (1:1:1.5, 150 mL) . Sodium bicarbonate (8.82 g, 105 mmol), sodium periodate (13.46 g, 63.0 mmol) and ruthenium trichloride (0.13 g, 0.61 mmol) were added to the vigorously stirred mixture. The reaction was stirred at ambient temperature for 44 hours before diluting with water (100 mL) . Following cooling of the reaction to 0°C, the pH was adjusted to about 1-2 with 10% hydrochloric acid solution.. The cold aqueous solution was extracted with precooled diethyl
ϋ ether (7 x 30 mL) . The black organic solution was dried over anhydrous sodium sulfate and quickly filtered throug a short funnel packed with flash silica and washed with cold diethyl ether (2 x 20 mL) . The almost clear ether layer at ice temperature was immediately treated with excess diazomethane. The solution was gradually warmed t room temperature and placed and left in a fume hood overnight. Anhydrous magnesium sulfate was slowly added to the solution, the mixture filtered and the solvent evaporated. Flash silica gel chromatography using 35% ethyl acetate in petroleum ether as eluant delivered the title compound (±)-15 as a yellow oil (3.32 g, 88%).
Example 12
Verification of Conglomerate Formation by X-Ray
Crysta11ography
Conglomerate formation by the synthesized racemic taxol side chain la was demonstrated through X-ray crystal structure analysis wherein successful solution of the structure was possible only in the acentric space group P2ι.
Data were collected on five crystals all of which were found to belong to the acentric monoclinic space group P2» with unit cell constants a = 5.414(4), b = 7.813(1), c = 17.802(7)A, β = 90.87(4)°. After initial solution of the structure with one quadrant of data out to θ = 25°, an investigation of absolute configuration was carried out by collecting a full sphere of data out to θ = 10°. Refinement of both enantiomers utilizing the full sphere data for each data set revealed that enantiomerically pure crystals of each enantiomer were present in the conglomerate. This and other procedures for the determination of absolute configuration are described in Stout & Jensen, X-ray Structure Determination. A Practical Guide, pp. 410-412 (Wiley & sons, New York, 1989) .
Example 13
Resolution of (±)-N-Benzoyl-3-phenylisoserine methyl este
(la) by Entrainment
Racemic taxol side chain methyl ester la
(approx. 10 g) and the (2R, 3S)-enantiomer (approx. 0.4 g) were dissolved in a minimal volume of dimethylsulfoxide
(7.5-10.0 ml) with warming. The solution was then cooled to about 15-20°C and seed crystals (approx. 10 mg) of the
(2R, 3S)-enantiomer were added. The stirred solution was allowed to crystallize for 1-3 hours and the (2R, 3S)-enantiomer (about 0.8 g) was recovered by filtration.
Additional racemic side chain (0.8 g) was then dissolved in the filtrate, the solution cooled to 15-20°C, and seed crystals (approx. 10 mg) of the (2S, 3R)-taxol side chain methyl ester were added. The stirred solution was again allowed to crystallize for 1-3 hours before recovering the
(2S, 3R)-enantiomer (about 0.8 g) by filtration. This cycle of adding more racemic side chain equal to the amount of substantially pure product isolated and subsequent collection of additional quantities of the (2R, 3S)- and (2S, 3R)-isomers was repeated several times.
After nine such cycles, about 4.0 g of the (2R, 3S)-isomer and 3.2 g of the (2S, 3R)-enantiomer were obtained, each in greater than 95% optical purity.
Example 14 Resolution of (±)-N-Benzoyl-3-phenylisoserine methyl ester
(la) by Manual Sorting of Conglomerate
Crystals of N-Benzoyl-3-phenylisoserine methyl ester la (taxol side chain methyl ester) were grown by dissolving the racemate (0.5 g) in dimethylsulfoxide (2.0 mL) and allowing the solvent to slowly concentrate by evaporation at ambient temperature. The crystals were sorted by viewing under a low power stereoscopic microscope.
Example 15
Resolution of (±)-N-Benzoyl-3-phenylisoserine methyl ester (la) by Localized Crystallization
Crystals of (±)-N-Benzoyl-3-phenylisoserine methyl ester la (1.0 g) were dissolved in ethanol (20 mL) in a 100 mL beaker. Seed crystals of the (2R,3S)-isomer
(-15 mg) were placed on one side of the beaker and seed crystals of the (2S,3R)-isomer (-15 mg) on the opposite side. Crystallization was allowed to proceed until approximately 0.3-0.4 g total of taxol side chain methyl ester had crystallized. The individual crystals within each locale of the beaker were then collected with a flat spatula and dried. Optical purity of each enantiomer was determined to be greater than 80%.
Example 16
Resolution of (±)-N-Benzoyl-3-phenylisoserine methyl ester (la) by Differentiated Crystallization
Racemic taxol side chain methyl ester la (10 g) was dissolved in warm ethanol (150 mL) and the solution cooled to ambient temperature. Seed crystals of the
(2R,3S)-taxol side chain methyl ester (3 g) of size greater than 30 mesh were added and stirring of the mixture was carried out for 0.5.-1.0 hour. The crystals that deposited were filtered, washed with cold ethanol (2 x 5 mL) and dried. The solid was then sifted through a 30 mesh sieve whereupon 5.6 g of crystalline (2R,3S)-taxol side chain methyl ester of optical purity of greater than
85% was obtained. Example 17
(2R,3S)-N-Benzoyl-0-(1-ethoxyethyl)-3-phenylisoserine methyl ester (19) ; Hydroxyl Group Protection
(2R,3S)-N-benzoyl-3-phenylisoserine methyl ester ("-" la) (299 mg, 1.0 mmol), dry methylene chloride (10 mL) , pyridiniun p-toluenesulfonate (25.1 mg, 0.1 mmol) and ethyl vinyl ether (721 mg, 10.0 mmol) were introduced successively into a 25 mL round-bottomed flask equipped with a magnetic stirrer. Pyridine (1 drop) was added when the reaction (ambient temperature) was judged complete by TLC analysis and the reaction mixture was then diluted by
adding methylene chloride (25 mL) . The organic phase was washed with water (2 x 15 mL) , and then with saturated sodium chloride solution (15 mL) and dried over anhydrous sodium sulfate. After filtration and removal of the solvents by rotary evaporation, N-benzoyl-0-(l- ethoxyethyl)-3-phenylisoserine methyl ester 19 (370 mg, 99%) was obtained in the form of an equimolar mixture of two epi ers.
Example 18
(2R,3S)-N-Benzoyl-O-(1-ethoxyethyl)-3-phenylisoserine (20) ; Saponification of Ester
Hydrolysis of (2R,3S)-N-benzoyl-3-phenylisoserine methyl ester 19 (127 mg, 0.37 mmol) was effected by treatment with potassium carbonate (127 mg, 0.92 mmol) in methanol (4 mL) and water (2 mL) for 15 hours at ambient temperature to give 122 mg (100%) of the corresponding free carboxylic acid 20.
Example 19 Taxol (2R,3S)-N-Benzoyl-O-(1-ethoxyethyl)-3- phenylisoserine 20 (42.8 mg, 0.12 mmol) in anhydrous toluene (1 mL) was introduced under an nitrogen atmosphere into a 5 mL round-bottomed flask equipped with a magnetic stirrer. Di-2-pyridyl carbonate (25.9 mg, 0.12 mmol) was then added and the mixture left to react for 4 to 5 minutes. 4-Dimethylaminopyridine (4.9 mg, 0.04 mmol) and 7-triethylsilylbaccatin III (14 mg, 0.02 mmol) were then added in a single portion. The colorless and homogeneous solution was left for 3 to 4 minutes, and then heated for 10 hours at 72°-74°C. After cooling, the reaction mixture was diluted with ethyl acetate (10 mL) and the organic solution as washed successively with saturated aqueous sodium bicarbonate solution (3 x 5 mL) , water (2 x 5 mL) and finally with saturated sodium chloride solution (5 mL) . The organic phase was dried over anhydrous sodium sulfate, the desiccant removed by filtration, and the
° solvent removed by rotary evaporation. The residue obtained was purified by analytical thin-layer chromatography on silica, eluting with an diethyl ether/methylene chloride (5:95 by volume) mixture, 4 runs being performed. The product (8.4 mg, 40% yield) was obtained in the form of a mixture of two epimers in the ratio 60:40, melting at 169°-173°C, after recrystallization from a methylene chloride/pentane mixture. The above product (72 mg, 0.009 mmol) was introduced at 0°C under an nitrogen atmosphere into a 10 mL round-bottomed flask equipped with a magnetic εtirrer. 0.5% Ethanolic hydrochloric acid solution (3.6 mL) , coole beforehand to 0 °C, was added the mixture stirred at 0 °C for 30 hours. The reaction mixture was then diluted by adding ethyl acetate (20 mL) and water (10 mL) at 0°C. Following separation of the two phases, the organic layer was washed with water (5 x 5 mL) and with saturated sodium chloride solution (2 x 5 mL) and then dried over anhydrous sodium sulfate. After filtration of the desiccant, the solvents were removed by rotary evaporation, and the residue obtained (72 mg) was purified by preparative thin layer chromatography on silica, eluting with a methylene chloride/methanol (90:10 by volume) mixture. 54 mg (0.063 mmol, 91% yield) of taxol was thereby obtained.
While we have hereinbefore described a number of embodiments of this invention, it is apparent that the basic instructions can be altered to provide other embodiments which utilize the methods and compositions of this invention. Therefore, it will be appreciated that the scope of this invention is defined by the claims appended hereto rather than by the specific embodiments which have been presented hereinbefore by way of example.
Claims
1. A method for the production of taxanes comprising the steps of: preparing a racemic mixture of enantiomers of the taxane side chain capable of exhibiting conglomerate behavior having the formula:

(1) wherein R1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, guinoline, C1-C8 hydroxyalkyl, C1-C6 aminoalkyl, and 2-, 3-, or 4-pyridino, or

(3) wherein U and V are independently selected from the group consisting of hydrogen, chloride, bromide, iodide, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or •*• aryl-N-amido, C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino, C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl- or arylurea, trichloromethyl, and trifluoromethyl; R2 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, and C7-C12 alkylphenyl; R4 is selected from the group consisting of R1 or OR5, wherein R5 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl; resolving the racemic taxane side chain into its substantially optically pure enantiomers; preparing the substantially optically pure (2R,3S)-taxane side chain for coupling to a taxane ring nucleus of the formula:

(18) wherein A and B are independently hydrogen or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy or A and B together form an oxo; L and D are independently hydrogen or hydroxy or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy; E and F are independently hydrogen or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy or; E and F together from an oxo; G is hydrogen or hydroxy or lower alkanoyloxy, alkenoyloxy, » alkynoyloxy, or aryloyloxy or G and M together form an oxo or methylene or G and M together form an oxirane or M and F together form an oxetane; J is hydrogen, hydroxy, or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy or I is hydrogen, hydroxy, or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy; or I and J taken together form an oxo; and K is hydrogen, hydroxy or lower alkoxy, alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy; and P and Q are independently hydrogen or lower alkanoyloxy, alkenoyloxy, alkynoyloxy, or aryloyloxy or P and Q together form an oxo; and coupling the substantially optically pure taxane side chain to the taxane ring nucleus.
2. The method according to claim 1 wherein the racemic mixture of enantiomers of the taxane side chain (1) capable of exhibiting conglomerate behavior is synthesized by: providing a racemic heterocycle having the following formula:

(5) in which the R1 and C02R2 groups are trans; opening the ring (5) with a hydrogen halide having the formula HX2, wherein X2 is either chloride or bromide, in a nonpolar solvent to form a halohydrin having the formula:

(6) forming a ring structure by closing the halohydrin with a base in a solvent mixture to form a racemic heterocycle in which the R1 and C02R2 groups are cis having the formula:

(7) opening the ring of the heterocycle (7) in which the R1 and C02R2 groups are cis with a nucleophile to form a racemic hydroxy azide side chain precursor having the
<- formula:

(8) performing an esterification/hydrogenation/rearrangement of the racemic hydroxy azide side chain precursor (8) to provide the racemic taxane side chain (1) .
3. The method according to claim 1 wherein the racemic mixture of enantiomers of the taxane side chain (1) capable of exhibiting conglomerate behavior is synthesized by providing a racemic heterocycle having the following formula:

(5) in which the R1 and C02R2 groups are trans; opening the ring (5) with HN3 in an nonpolar aprotic solvent to form a hydroxy azide side chain precursor ° having the formula:

(8 performing an esterification/hydrogenation/rearrangement of the hydroxy azide side chain precursor (8) to provide the racemic taxane side chain (1) .
4. The method according to either one of claims 2 or 3 wherein the racemic heterocycle (5) in which the R1 and C02R2 groups are trans is formed by a Darzens Condensation under basic conditions in a solvent between an electrophile having the formula:

(2) wherein R1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, quinoline, C1-C8 hydroxyalkyl, C1-C6 aminoalkyl, and 2-, 3-, or 4-pyridino, or

(3) wherein U and V are independently selected from the group consisting of hydrogen, chloride, bromide, iodide, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or aryl-N-amido, C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino, C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl- or arylurea, trichloromethyl, and trifluoromethyl; and a haloester having the formula:

(4) wherein X1 is selected from the group consisting of chloride, bromide, or iodide and R2 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, and C7-C12 alkylphenyl.
5. The method according to either one of claims 2 or 3 wherein the racemic heterocycle (5) in which the groups R1 and C02R2 are trans, where R2 is a methyl group, is synthesized by: epoxidizing a racemic hydroxylated olefin in which the groups R1 and CH2OH are trans having the formula:
-OH
,r *H
(10) with an epoxidizing agent to form a racemic epoxide in which the groups R1 and CH2OH are trans having the formula:

(11) oxidizing the racemic epoxide (11) in which the groups R1 and CH2OH are trans with an oxidant to form a racemic carboxylic acid epoxide in which the groups R1 and C02H are ° trans having the formula:

(12) converting the racemic carboxylic epoxide (12) in which the groups R1 and C02H are trans with an esterification agent into said racemic heterocycle (5) in which the groups R1 and C02Me are trans.
6. The method of claim 1 wherein the racemic mixture of enantiomers of the taxane side chain (1) capable of exhibiting conglomerate behavior, wherein R2 is a methyl group, is synthesized by: epoxidizing a hydroxylated olefin in which the groups R1 and CH2OH are cis having the formula:
-OH
H^
(13) with an epoxidizing agent to form a racemic epoxide in o which the groups R1 and CH2OH are cis having the formula:

(14) oxidizing the racemic epoxide (14) in which the groups R1 and CH2OH are cis with an oxidant to form a racemic carboxylic acid epoxide in which the groups R1 and C02H are cis having the formula:

(15) converting the racemic carboxylic epoxide (15) in which the groups R1 and C0,H are cis with an esterification agent into a racemic heterocycle in which the groups R1 and Cθ2Me
SUBSTITUTESHEET » are cis having the formula:

(16)
10 opening the ring of the racemic heterocycle (16) in which the groups Rl and CO-jMe are cis with a nucleophile to form a racemic hydroxy azide side chain precursor with the following formula wherein R2 is a methyl group:
15

(8) performing an esterification/hydrogenation/rearrangement " of the hydroxy azide side chain precursor (8) to provide the racemic taxane side chain methyl ester (1) .
7. A method of synthesizing a racemic mixture of enantiomers of the taxane side chain (1) capable of 0
5 exhibiting conglomerate behavior having the formula:

(1) wherein R1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, quinoline, C1-C8 hydroxyalkyl, C1-C6 aminoalkyl, and 2-, 3-, or 4-pyridino, or

(3) wherein U and V are independently selected from the group consisting of hydrogen, chloride, bromide, iodide, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or aryl-N-amido, C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino, C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl- or arylurea, trichloromethyl, and trifluoromethyl; R2 is
UBSTITUTESHEET selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, and C7-C12 alkylphenyl; R4 is selected from the group consisting of R1 or OR5, wherein R5 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl; comprising the steps of: performing a Darzens Condensation under basic conditions in a solvent between an electrophile having the formula:

(2) wherein R1 is as described above; and a haloester having the formula:

(4) wherein X1 is selected from the group consisting of chloride, bromide or iodide and R2 is as described above; to form a racemic heterocycle in which the groups R1 and
SUBSTITUTE SHEE C02R2 are trans having the formula:

(5) opening the ring of a racemic heterocycle in which the R1 and C02R2 groups are trans with a hydrogen halide having the formula HX2, wherein X2 is either chloride or bromide, in a nonpolar solvent to form a halohydrin having the formul :

(6) forming a ring structure by closing the halohydrin with a base in a solvent mixture to form a racemic heterocycle in
SUBSTITUTESHEET β which the R1 and C02R2 groups are cis having the formula:

(7) opening the ring of the heterocycle (7) in which the R1 and C02R2 groups are cis with a nucleophile to form a racemic azide side chain precursor having the formula:

(8) performing an esterification/hydrogenation/rearrangement of the racemic azide side chain precursor (8) to provide the racemic taxane side chain (1) .
8. A method of synthesizing a racemic mixture of enantiomers of the taxane side chain (1) capable of
SUBSTITUTESHEET exhibiting conglomerate behavior having the formula:

(1) wherein R1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, quinoline, C1-C8 hydroxyalkyl, C1-C6 aminoalkyl, and 2-, 3-, or 4-pyridino, or

(3) wherein U and V are independently selected from the group consisting of hydrogen, chloride, bromide, iodide, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or aryl-N-amido, C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino, C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl- or arylurea, trichloromethyl, and trifluoromethyl; R2 is a
SUBSTITUTESHEET methyl group; R4 is selected from the group consisting of R1 or OR5, wherein R5 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl; comprising the steps of: epoxidizing a racemic hydroxylated olefin in which the groups R1 and CH2OH are trans having the formula:
-OH
(10) with an epoxidizing agent to form a racemic epoxide in which the groups R1 and CH2OH are trans having the formula:

(11) oxidizing the racemic epoxide (11) in which the groups R1 and CH2OH are trans with an oxidant to a racemic carboxylic acid epoxide in which the groups R1 and C02H are
SUBSTITUTESHEET «* trans having the formula:

(12) converting the racemic carboxylic epoxide (12) in which the groups R1 and C02H are trans with an esterification agent into a racemic heterocycle in which the groups R1 and C02Me are trans having the following formula:

(12a) opening the ring of a racemic heterocycle (12a) in which the R1 and C02Me groups are trans with a hydrogen halide having the formula HX2, wherein X2 is either chloride or bromide, in a nonpolar solvent to form a halohydrin having the formula:

(6) wherein R2 is a methyl group; forming a ring structure by closing the halohydrin (6) with a base in a solvent mixture to form a racemic heterocycle in which the R1 and C02R2 groups are cis having the formula:

(7) wherein R2 is a methyl group; opening the ring of the heterocycle (7) in which the R1 and COjMe groups are cis with a nucleophile to form a racemic hydroxy azide side chain precursor having the formula:

(8) wherein R2 is a methyl group; performing an esterification/hydrogenation/rearrangement of the racemic azide side chain precursor (8) to provide the racemic taxane side chain methyl ester.
9. A halohydrin composition of methyl threo-3- Chloro-2-hydroxy-3-phenylpropionate having the formula:

(6a)
10. A method for the synthesis of the composition according to claim 9, comprising the steps of: performing a Darzens Condensation under basic conditions in a solvent between an electrophile having the formula:

(2) wherein R1 is a phenyl group; and a haloester having the formula:

(4) wherein X1 is a chloride and R2 is a methyl group; to form a racemic heterocycle in which the phenyl and
CO- te groups are trans having the formula:

(5a) opening the ring of a racemic heterocycle (5a) in which
SUBSTITUTESHEET the phenyl and C02Me groups are trans with hydrogen chloride in a nonpolar solvent to form the halohydrin compound (6a) of claim 9.
11. method of preparing taxol having the formula:

wherein R8 and Ac are acetate groups and R9 is a phenyl group; comprising the steps of: providing a halohydrin methyl threo-3-chloro-2-hydroxy-3- phenylpropionate having the formula:

( 6a) forming a ring structure by closing the halohydrin (6a) with a base in a solvent mixture to form a racemic heterocycle in which the phenyl and C02Me groups are cis
SUBSTITUTESHEET ° having the formula:

(7a) opening the ring of the heterocycle (7a) in which the phenyl and C02Me groups are cis with a nucleophile to form a racemic azide side chain precursor having the formula:

SUBSTITUTESHEET behavior having the formula :

OH
(la) resolving the racemic taxol side chain methyl ester into its substantially optically pure enantiomer (2R,3S)-N- benzoyl-3-phenylisoserine; preparing the substantially optically pure (2R,3S)-N- benzoyl-3-phenylisoserine for coupling to a taxane ring nucleus baccatin III, having the formula:

OCOCβH6
(17) wherein R8 and Ac are acetate groups; and coupling the prepared taxol side chain to the taxol ring nucleus.
12. The method according to claim l wherein the taxane side chain capable of exhibiting conglomerate
SUBSTITUTE SHEET behavior is the taxol side chain (2R,3S)-N-benzoyl-3- phenylisoserine having the following formula:

(la) 13. The method according to either of claims 5, 6, or 8 wherein the epoxidizing agent is selected from the group consisting of meta-chloroperbenzoic acid or another peracid, such as perbenzoic acid, peracetic acid, performic acid, peroxytrifluoroacetic acid, or peroxyphthalic acid.
14. The method according to either of claims 5, 6, or 8 wherein the oxidant is ruthenium trichloride-sodium periodate.
15. The method according to either of claims 5, 6, or 8 wherein the esterification agent is ethereal diazomethane.
16. The method according to either of claims 3, 8, 9 or 11 wherein the hydrogen halide is hydrogen chloride or hydrogen bromide.
17. The method according to either of claims 2, 7, 8 or 10 wherein the nonpolar solvent is selected from the group consisting of: benzene, toluene, xylene, pentane, hexane, heptane, methylene chloride, chloroform, ethyl ether and tetrahydrofuran.
SUBSTITUTESHEET u 18. The method according to either of claims 2, 7, 8 or 10 wherein the base is selected from the group consisting of: sodium carbonate, lithium carbonate, potassium carbonate, cesium carbonate, magnesium carbonate, sodium methoxide, sodium ethoxide, sodium tert-butyloxide, lithium methoxide, lithium ethoxide, lithium tert-butyloxide, sodium hydride, potassium hydride, and lithium hydride.
19. The method according to either of claims 2, 7, 8 or 11 wherein the base is used with phase transfer catalysts.
20. The method according to either of claims 2, 7, 8 or 11 wherein the solvent mixture is comprised of an organic solvent selected from the group consisting of dimethylformamide, dimethylsulfoxide, methanol, ethanol, isopropanol, and acetone; and water.
21. The method according to claim 20 wherein the ratio of organic solvent to water in the solvent mixture is from about 60:90 to about 40:10.
22. The method according to claim 20 wherein the ratio of organic solvent to water in the solvent mixture is about 70:30.
23. The method according to claim 20 wherein the ratio of organic solvent to water in the solvent mixture is about 60:40.
24. The method according to either claims 2, 6, 7, 8 or 11 wherein the nucleophile is selected from the group consisting of sodium azide or azidotrimethylsilane.
25. The method according to claim 2 wherein the
SUBSTITUTE SHEET ° nonpolar aprotic solvent is selected from the group consisting of benzene, toluene, hexane, tetrahydrofuran, diethyl ether, methylene chloride and chloroform.
26. The method according to either of claims 4, 7, or 10 wherein the base used is selected from the group consisting of sodium carbonate, potassium carbonate, cesium carbonate, triethylamine, diisopropylethyla ine, 1,5-diazabicyclo[4.3.0]non-5-ene, diazobicyclo[5.4.0]undec-7-ene, 1,4-diazabicyclo[2.2.0]octane, lithium diisopropylamide, lithium hexamethyldisilamide, lithium tetramethylpiperidide, sodium hydride, potassium hydride, sodium methoxide, sodium ethoxide, sodium sec-butyloxide, sodium tert-butyloxide, potassium methoxide, potassium ethoxide, potassium sec-butyloxide, potassium tert-butyloxid .
27. The method according to either of claims 4, 7, or 10 wherein the solvent is selected from the group consisting of: propanol, isopropanol, butanol, diethyl ether, tetrahydrofuran, dimethylformamide, dimethylsulfoxide, N-methylpyrrolidinone, hexamethylphosphoramide, methanol, ethanol, and mixtures thereof.
28. The method according to either of claims 4, 7, or 10 wherein the Darzens Condensation reaction between the electrophile (2) and the haloester (4) is performed at a reaction temperature of between about -30°C and about +40°C.
29. The method according to either of claims 4, 7, or 10 wherein the Darzens Condensation reaction between the electrophile (2) and the haloester (4) is performed at a reaction temperature of between about -20°c and about
SUBSTITUTESHEET ■> +20°C.
30. The method according to either of claims 4, 7, or 10 wherein the Darzens Condensation reaction between the electrophile (2) and the haloester (4) is performed a a reaction temperature of between about -10°C and about +10°C.
31. The method according to either of claims l or 11 wherein substantially optically pure means any molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 50%.
32. The method according to either of claims 1 or 11 wherein substantially optically pure means any molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 75%.
33. The method according to either of claims 1 or 11 wherein substantially optically pure means any molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 90%.
34. The method according to either of claims l or 11 wherein the racemic taxane side chain capable of forming a conglomerate is resolved by direct crystallization means.
35. The method according to claim 34 wherein the conglomerate of substantially optically pure enantiomers is sorted by manual means.
36. The method according to claim 34 wherein the substantially optically pure enantiomers are resolved by localized crystallization techniques.
37.. The method according to claim 34 wherein the
SUBSTITUTESHEET * substantially optically pure enantiomers are resolved by differentiated crystallization techniques.
38. The method according to claim 34 wherein the substantially optically pure enantiomers are resolved by entrainment procedures.
39. The method according to claim 38 wherein in each iteration of the entrainment procedure, the enantiomer in excess is present in sufficient quantities to achieve a 1% to 15% excess of that enantiomer by weight in solution.
40. The method according to claim 38 wherein in each iteration of the entrainment procedure, the enantiomer in excess is present in sufficient quantities to achieve a 3% to 8% excess of that enantiomer by weight in solution.
41. The method according to claim 38 wherein in each iteration of the entrainment procedure, the solution is cooled to a crystallization temperature of about 10°C to about 30°C.
42. The method according to claim 38 wherein in each iteration of the entrainment procedure, the solution is cooled to a crystallization temperature of about 15°C to about 25°C.
43. The method according to claim 38 wherein the entrainment procedure is performed utilizing a solvent selected from the group consisting of dimethylsulfoxide, dimethylformamide, dimethylacetamide, N- methylpyrolidinone, chloroform, trichloromethane, methylene chloride, acetone, methanol, ethanol, isopropanol and water and mixtures thereof.
44.. A method for the production of a substantially
SUBSTITUTESHEET ■> optically pure taxane side chain having the formula:

(1) wherein R1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, quinoline, C1-C8 hydroxyalkyl, C1-C6 aminoalkyl, and 2-, 3-, or 4-pyridino, or

(3) wherein U and V are independently selected from the group consisting of hydrogen, chloride, bromide, iodide, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or aryl-N-amido, C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino, C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl- or arylurea, trichloromethyl, and trifluoromethyl; R2 is
SUBSTITUTESHEET ° selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, and C7-C12 alkylphenyl; K* is selected from the group consisting of R1 or OR5, wherein R5 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl; comprising the steps of: preparing a racemic mixture of enantiomers of the taxane side chain capable of exhibiting conglomerate behavior; and resolving the racemic taxane side chain into its substantially optically pure enantiomers.
45. The method according to claim 44 wherein the racemic mixture of enantiomers of the taxane side chain (1) capable of exhibiting conglomerate behavior is synthesized by: providing a racemic heterocycle having the following formula:

(5) in which the R1 and C02R2 groups are trans; opening the ring (5) with a hydrogen halide having the formula HX2, wherein X2 is either chloride or bromide, in a
SUBSTITUTE SHEET nonpolar solvent to form a halohydrin having the formula:
OH
R1 H c COgR2
(6) forming a ring structure by closing the halohydrin (6) with a base in a solvent mixture to form a racemic heterocycle in which the R1 and C02R2 groups are cis having the formula:

(7) opening the ring of the heterocycle (7) in which the R1 and C02R2 groups are cis with a nucleophile to form a
SUBSTITUTESHEET ° racemic azide side chain precursor having the formula:

(8) performing an esterification/hydrogenation/rearrangement of the racemic azide side chain precursor (8) to provide the racemic taxane side chain (1) .
46. The method according to claim 44 wherein the racemic mixture of enantiomers of the taxane side chain capable of exhibiting conglomerate behavior is synthesized by: providing a racemic heterocycle having the following formula:

(5) in which the R1 and C02R2 groups are trans; opening the ring (5) with HN3 in an nonpolar aprotic solvent to form a hydroxy azide side chain precursor
SUBSTITUTESHEET β having the formula:

(8) performing an esterification/hydrogenation/rearrangement of the hydroxy azide (8) to provide the racemic taxane side chain (1) .
47. The method according to either one of claims 45 or 46 wherein the racemic heterocycle (5) in which the R1 and C02R2 groups are trans is formed by a Darzens Condensation under basic conditions in a solvent between an electrophile having the formula:

(2) wherein R1 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, C3-C8 cycloalkenyl, C1-C8 linear or branched alkenyl, C1-C8 linear or branched alkynyl, C5-C20 aryl, indole, thiophenyl, furanyl, quinoline, C1-C8 hydroxyalkyl, C1-C6
SUBSTITUTESHEET aminoalkyl, and 2-, 3-, or 4-pyridino, or

(3) wherein U and V are independently selected from the group consisting of hydrogen, chloride, bromide, iodide, hydroxyl, thiol, nitro, azide, amino, C2-C8 alkyl- or aryl-N-amido, C2-C8 alkyl- or arylcarboxylate, C1-C8 carboalkoxy, C1-C8 carboaryloxy, C2-C8 alkyl- or aryl-s-thiocarboxylate, C1-C4 alkoxy, C1-C8 monoalkylamino, C1-C8 dialkylamino, C1-C8 linear or branched alkyl, C1-C8 thioalkyl, or C1-C8 alkyl- or arylcarbonate, C1-C8 alkyl- or arylcarbamate, C1-C8 alkyl- or arylurea, trichloromethyl, and trifluoromethyl; and a haloester having the formula:

(4) wherein X1 is a halogen such as chloride, bromide, or iodide and R2 is selected from the group consisting of C1-C8 linear or branched alkyl, C3-C8 cycloalkyl, and C7-C12 alkylphenyl.
SUBSTITUTE SHEET
48. The method according to either one of claims 45 or 46 wherein the racemic heterocycle (5) in which the groups R1 and C02R2 are trans, wherein R2 is a methyl group, is synthesized by: epoxidizing a racemic hydroxylated olefin in which the groups R1 and CH2OH are trans having the formula:
H, -OH
R'
(10) with an epoxidizing agent to form a racemic epoxide in which the groups R~ and CH2OH are trans having the formula:

(ID oxidizing the racemic epoxide (11) in which the groups R1 and CH2OH are trans with an oxidant to form a racemic carboxylic acid epoxide in which the groups R1 and C02H are
SUBSTITUTESHEET β trans having the formula:

(12) converting the racemic carboxylic epoxide (12) in which the groups R1 and C02H are trans with an esterification agent into said racemic heterocycle (5) in which the groups R1 and CO-jMe are trans.
49. The method of claim 44 wherein the racemic mixture of enantiomers of the taxane side chain (l) capable of exhibiting conglomerate behavior, wherein R2 is a methyl group, is synthesized by: epoxidizing a hydroxylated olefin in which the groups R1 and CH2OH are cis having the formula:

(13) with an epoxidizing agent to form a racemic epoxide in
SUBSTITUTESHEET which the groups R1 and CH2OH are cis having the formula:

(14) oxidizing the racemic epoxide (14) in which the groups R1 and CH2OH are cis with an oxidant to form a racemic carboxylic acid epoxide in which the groups R1 and C02H are cis having the formula:

(15) converting the racemic carboxylic epoxide (15) in which the groups R1 and C02H are cis with an esterification agent into a racemic heterocycle in which the groups R1 and C02Me
SUBSTITUTESHEET 0 are cis having the formula:

(16)
10 opening the ring of the racemic heterocycle (16) in which the groups R1 and COjMe are cis with a nucleophile to form a racemic hydroxy azide side chain precursor with the following formula wherein R2 is a methyl group:
15

(8) performing an esterification/hydrogenation/rearrangement
9•. . . .
'-' of the hydroxy azide side chain precursor (8) to provide the racemic taxane side chain methyl ester (1) .
50. The method according to claim 44 wherein the taxane side chain capable of exhibiting conglomerate 0 behavior is the taxol side chain (2R,3S)-N-benzoyl-3-
5
SUBSTITUTESHEET ° phenylisoserine having the following formula:

(la)
51. The method according to either of claims 48 or 49 wherein the epoxidizing agent is selected from the group consisting of meta-chloroperbenzoic acid or another peracid, such as perbenzoic acid, peracetic acid, performic acid, peroxytrifluoroacetic acid, or peroxyphthalic acid.
52. The method according to either of claims 48 or 49 wherein the oxidant is ruthenium trichloride-sodium periodate.
53. The method according to either of claims 48 or 49 wherein the esterification agent is ethereal diazomethane.
54. The method according to claim 45 wherein the hydrogen halide is hydrogen chloride or hydrogen bromide.
55. The method according to claims 45 wherein the nonpolar solvent is selected from the group consisting of: benzene, toluene, xylene, pentane, hexane, heptane, methylene chloride, chloroform, ethyl ether and tetrahydrofuran.
56. The method according to claim 45 wherein the base is selected from the group consisting of:
SUBSTITUTESHEET ° sodium carbonate, lithium carbonate, potassium carbonate, cesium carbonate, magnesium carbonate, sodium methoxide, sodium ethoxide, sodium tert-butyloxide, lithium methoxide, lithium ethoxide, lithium tert-butyloxide, sodium hydride, potassium hydride, and lithium hydride.
57. The method according to claim 45 wherein the base is used with phase transfer catalysts.
58. The method according to claim 45 wherein the solvent mixture is comprised of an organic solvent selected from the group consisting of dimethylformamide, dimethylsulfoxide, methanol, ethanol, isopropanol, and acetone; and water.
59. The method according to claim 58 wherein the ratio of organic solvent to water in the solvent mixture is from about 60:90 to about 40:10.
60. The method according to claim 58 wherein the ratio of organic solvent to water in the solvent mixture is about 70:30.
61. The method according to claim 58 wherein the ratio of organic solvent to water in the solvent mixture is about 60:40.
62. The method according to either claims 45 or 49 wherein the nucleophile is selected from the group consisting of sodium azide or azidotrimethylsilane.
63. The method according to claim 46 wherein the nonpolar aprotic solvent is selected from the group consisting of benzene, toluene, hexane, tetrahydrofuran, diethyl ether, methylene chloride and chloroform.
SUBSTITUTESHEET o 64. The method according to claim 47 wherein the base used is selected from the group consisting of sodium carbonate, potassium carbonate, cesium carbonate, triethylamine, diisopropylethylamine, 1,5-diazabicyclo [4.3.0]non-5-ene, diazobicyclo[5.4.0]undec-7-ene, l,4-diazabicyclo[2.2.0]octane, lithium diisopropylamide, lithium hexamethyldisilamide, lithium tetramethylpiperidide, sodium hydride, potassium hydride, sodium methoxide, sodium ethoxide, sodium sec-butyloxide, sodium tert-butyloxide, potassium methoxide, potassium ethoxide, potassium sec-butyloxide, potassium tert- butyloxide.
65. The method according to claim 47 wherein the solvent is selected from the group consisting of: propanol, isopropanol, butanol, diethyl ether, tetrahydr-:.furan, dimethylformamide, dimethylsulfoxide, N-methylp/rrolidinone, hexamethylphosphoramide, methanol, ethanol, and mixtures thereof.
66. The method according to claim 47 wherein the
Darzens Condensation reaction between the electrophile (2) and the haloester (4) is performed at a reaction temperature of between about -30°C and about +40°C.
67. The method according to claim 47 wherein the
Darzens Condensation reaction between the electrophile (2) and the haloester (4) is performed at a reaction temperature of between about -20°C and about +20°C.
68. The method according to claim 47 wherein the Darzens Condensation reaction between the electrophile (2) and the haloester (4) is performed at a reaction temperature of between about -10°C and about +10°C.
69. The method according to claim 44 wherein
SUBSTITUTESHEET substantially optically pure means any molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 50%.
70. The method according to claim 44 wherein substantially optically pure means any molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 75%.
71. The method according to claim 44 wherein substantially optically pure means any molecular system which possesses an enantiomeric or diastereomeric excess of greater than about 90%.
72. The method according to claim 44 wherein the racemic taxane side chain capable of forming a conglomerate is resolved by direct crystallization means.
73. The method according to claim 72 wherein the conglomerate of substantially optically pure enantiomers is sorted by manual means.
74. The method according to claim 72 wherein the substantially optically pure enantiomers are resolved by localized crystallization techniques.
75. The method according to claim 72 wherein the substantially optically pure enantiomers are resolved by differentiated crystallization techniques.
76. The method according to claim 72 wherein the substantially optically pure enantiomers are resolved by entrainment procedures.
77. The method according to claim 76 wherein in each iteration of the entrainment procedure, the enantiomer in excess is present in sufficient quantities to achieve a 1% ° to 15% excess of that enantiomer by weight in solution.
78. The method according to claim 76 wherein in each iteration of the entrainment procedure, the enantiomer in excess is present in sufficient quantities to achieve a 3% to 8% excess of that enantiomer by weight in solution.
79. The method according to claim 76 wherein in each iteration of the entrainment procedure, the solution is cooled to a crystallization temperature of about 10°C to about 30°C.
80. The method according to claim 76 wherein in each iteration of the entrainment procedure, the solution is cooled to a crystallization temperature of about 15°C to about 25°C.
81. The method according to claim 76 wherein the entrainment procedure is performed utilizing a solvent selected from the group consisting of dimethylsulfoxide, dimethylformamide, dimethylacetamide, N- methylpyrolidinone, chloroform, trichloromethane, methylene chloride, acetone, methanol, ethanol, isopropanol and water and mixtures thereof.
SUBSTITUTE SHEET
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US79713691A | 1991-11-22 | 1991-11-22 | |
| US797,136 | 1991-11-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1993010076A1 true WO1993010076A1 (en) | 1993-05-27 |
Family
ID=25170010
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1992/009911 WO1993010076A1 (en) | 1991-11-22 | 1992-11-19 | Synthesis and optical resolution of the taxol side chain and related compounds |
Country Status (2)
| Country | Link |
|---|---|
| AU (1) | AU3140093A (en) |
| WO (1) | WO1993010076A1 (en) |
Cited By (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2707165A1 (en) * | 1993-07-06 | 1995-01-13 | Rhone Poulenc Rorer Sa | Application of taxoids to the treatment of parasitic diseases. |
| US5580899A (en) * | 1995-01-09 | 1996-12-03 | The Liposome Company, Inc. | Hydrophobic taxane derivatives |
| US5589502A (en) * | 1994-11-17 | 1996-12-31 | Tanabe Seiyaku Co., Ltd. | Baccatin derivatives and processes for preparing the same |
| FR2740451A1 (en) * | 1995-10-27 | 1997-04-30 | Seripharm | NEW INTERMEDIARIES FOR THE HEMISYNTHESIS OF TAXANES, THEIR METHODS OF PREPARATION AND THEIR USE IN THE GENERAL SYNTHESIS OF TAXANES |
| EP0797988A2 (en) | 1993-07-19 | 1997-10-01 | Angiotech Pharmaceuticals, Inc. | Anti-angiogenic compositions and methods of use |
| US5677470A (en) * | 1994-06-28 | 1997-10-14 | Tanabe Seiyaku Co., Ltd. | Baccatin derivatives and processes for preparing the same |
| US6051600A (en) * | 1995-09-12 | 2000-04-18 | Mayhew; Eric | Liposomal hydrolysis-promoting hydrophobic taxane derivatives |
| RU2164501C1 (en) * | 2000-07-03 | 2001-03-27 | Шугина Галина Александровна | Method of cleaning polluted subsurface water |
| US6392063B1 (en) | 1995-09-12 | 2002-05-21 | The Liposome Company, Inc. | Hydrolysis-promoting hydrophobic taxane derivatives |
| US6759397B2 (en) | 2000-05-01 | 2004-07-06 | The University Of British Columbia | Ginsenoside chemotherapy |
| US7407973B2 (en) | 2003-10-24 | 2008-08-05 | Intermune, Inc. | Use of pirfenidone in therapeutic regimens |
| EP1982720A1 (en) | 2002-05-20 | 2008-10-22 | The Regents of the University of California | Ribaviren for use to treat cancer |
| EP2014307A2 (en) | 2001-03-13 | 2009-01-14 | Angiotech International Ag | Micellar drug delivery vehicles and uses thereof |
| EP2093235A1 (en) | 2006-02-08 | 2009-08-26 | Alios Biopharma Inc. | Hyperglycosylated variants of interferon alfacon-1 |
| EP2108390A2 (en) | 2008-03-31 | 2009-10-14 | Cordis Corporation | Device for local and/or regional delivery employing liquid formulations of therapeutic agents |
| EP2123312A2 (en) | 2008-05-19 | 2009-11-25 | Cordis Corporation | Extraction of solvents from drug containing polymer reservoirs |
| WO2009143454A2 (en) | 2008-05-22 | 2009-11-26 | Kereos, Inc. | Combination antitumor therapy |
| WO2010005850A1 (en) | 2008-07-08 | 2010-01-14 | The J. David Gladstone Institutes | Methods and compositions for modulating angiogenesis |
| EP2181704A2 (en) | 2002-12-30 | 2010-05-05 | Angiotech International Ag | Drug delivery from rapid gelling polymer composition |
| US7846940B2 (en) | 2004-03-31 | 2010-12-07 | Cordis Corporation | Solution formulations of sirolimus and its analogs for CAD treatment |
| US7875677B2 (en) | 2001-04-20 | 2011-01-25 | The University Of British Columbia | Micellar drug delivery systems for hydrophobic drugs |
| US7875284B2 (en) | 2006-03-10 | 2011-01-25 | Cook Incorporated | Methods of manufacturing and modifying taxane coatings for implantable medical devices |
| US7919108B2 (en) | 2006-03-10 | 2011-04-05 | Cook Incorporated | Taxane coatings for implantable medical devices |
| US7989490B2 (en) | 2004-06-02 | 2011-08-02 | Cordis Corporation | Injectable formulations of taxanes for cad treatment |
| EP2380606A1 (en) | 2006-06-30 | 2011-10-26 | Cook Incorporated | Methods of manufacturing and modifying taxane coatings for implantable medical devices |
| EP2390262A1 (en) | 2003-05-16 | 2011-11-30 | Intermune, Inc. | Synthetic chemokine receptor ligands and methods of use thereof |
| US8313521B2 (en) | 1995-06-07 | 2012-11-20 | Cook Medical Technologies Llc | Method of delivering an implantable medical device with a bioabsorbable coating |
| WO2012162007A1 (en) | 2011-05-25 | 2012-11-29 | Cordis Corporation | Expandable devices coated with a rapamycin composition |
| WO2012162010A1 (en) | 2011-05-25 | 2012-11-29 | Cordis Corporation | Expandable devices coated with a paclitaxel composition |
| US8409601B2 (en) | 2008-03-31 | 2013-04-02 | Cordis Corporation | Rapamycin coated expandable devices |
| US8420110B2 (en) | 2008-03-31 | 2013-04-16 | Cordis Corporation | Drug coated expandable devices |
| WO2013151774A1 (en) | 2012-04-04 | 2013-10-10 | Halozyme, Inc. | Combination therapy with an anti - hyaluronan agent and a tumor - targeted taxane |
| WO2013188763A1 (en) | 2012-06-15 | 2013-12-19 | The Brigham And Women's Hospital, Inc. | Compositions for treating cancer and methods for making the same |
| WO2014004760A1 (en) | 2012-06-28 | 2014-01-03 | Covidien Lp | Post-processing of a medical device to control morphology and mechanical properties |
| US8642063B2 (en) | 2008-08-22 | 2014-02-04 | Cook Medical Technologies Llc | Implantable medical device coatings with biodegradable elastomer and releasable taxane agent |
| US8691780B2 (en) | 2005-02-17 | 2014-04-08 | The Board Of Trustees Of The Leland Stanford Junior University | Txr1 and enhanced taxane sensitivity based on the modulation of a pathway mediated thereby |
| US8710095B2 (en) * | 2002-04-30 | 2014-04-29 | Bionumerik Pharmaceuticals, Inc. | Drugs for prophylaxis or mitigation of taxane-induced neurotoxicity |
| WO2014160185A2 (en) | 2013-03-14 | 2014-10-02 | The Board Of Trustees Of The Leland Stanford Junior University | Mitochondrial aldehyde dehydrogenase-2 modulators and methods of use thereof |
| WO2015081282A1 (en) | 2013-11-27 | 2015-06-04 | Redwood Bioscience, Inc. | Hydrazinyl-pyrrolo compounds and methods for producing a conjugate |
| WO2015127137A1 (en) | 2014-02-19 | 2015-08-27 | Aldea Pharmaceuticals, Inc. | Mitochondrial aldehyde dehydrogenase 2 (aldh2) binding polycyclic amides and their use for the treatment of cancer |
| EP3056196A1 (en) | 2006-03-22 | 2016-08-17 | SynCore Biotechnology CO., LTD | Treatment of triple receptor negative breast cancer |
| US9579390B2 (en) | 2012-11-12 | 2017-02-28 | Redwood Bioscience, Inc. | Compounds and methods for producing a conjugate |
| WO2017053920A1 (en) | 2015-09-25 | 2017-03-30 | Zy Therapeutics Inc. | Drug formulation based on particulates comprising polysaccharide-vitamin conjugate |
| WO2017083637A1 (en) | 2015-11-12 | 2017-05-18 | The Board Of Trustees Of The Leland Stanford Junior University | Cell-penetrating, guanidinium-rich oligophosphotriesters for drug and probe delivery |
| WO2017115301A1 (en) | 2015-12-30 | 2017-07-06 | Syncore Biotechnology Co., Ltd. | Treatment of breast cancer using a combination of a cationic liposomal formulation of taxane, a non-liposomal formulation of taxane and a further active agent |
| WO2018017190A2 (en) | 2016-06-01 | 2018-01-25 | Baxalta Incorporated | Formulations of polyalkylene oxide-asparaginase and methods of making and using the same |
| EP3300745A1 (en) | 2013-02-15 | 2018-04-04 | The Regents of the University of California | Chimeric antigen receptor and methods of use thereof |
| WO2018127082A1 (en) | 2017-01-05 | 2018-07-12 | Syncore Biotechnology Co., Ltd. | Treatment of pancreatic cancer |
| WO2018136570A1 (en) | 2017-01-18 | 2018-07-26 | F1 Oncology, Inc. | Chimeric antigen receptors against axl or ror2 and methods of use thereof |
| US10087139B2 (en) | 2015-03-03 | 2018-10-02 | Toray Fine Chemicals Co., Ltd. | 3-Phenylisoserine derivative production method |
| EP3388082A1 (en) | 2017-04-13 | 2018-10-17 | Galera Labs, LLC | Combination cancer immunotherapy with pentaaza macrocyclic ring complex |
| WO2018218004A1 (en) | 2017-05-24 | 2018-11-29 | The Board Of Regents Of The University Of Texas System | Linkers for antibody drug conjugates |
| WO2019152661A1 (en) | 2018-01-31 | 2019-08-08 | Galera Labs, Llc | Combination cancer therapy with pentaaza macrocyclic ring complex and platinum-based anticancer agent |
| EP3539563A1 (en) | 2012-07-19 | 2019-09-18 | Redwood Bioscience, Inc. | Antibody specific for cd22 and methods of use thereof |
| US10426753B2 (en) | 2014-04-03 | 2019-10-01 | Invictus Oncology Pvt. Ltd. | Supramolecular combinatorial therapeutics |
| WO2019195586A1 (en) | 2018-04-06 | 2019-10-10 | The Regents Of The University Of California | Methods of treating egfrviii expressing glioblastomas |
| WO2019195596A1 (en) | 2018-04-06 | 2019-10-10 | The Regents Of The University Of California | Methods of treating glioblastomas |
| WO2019222435A1 (en) | 2018-05-16 | 2019-11-21 | Halozyme, Inc. | Methods of selecting subjects for combination cancer therapy with a polymer-conjugated soluble ph20 |
| WO2020148612A1 (en) | 2019-01-14 | 2020-07-23 | Ignite Immunotherapy, Inc. | Recombinant vaccinia virus and methods of use thereof |
| WO2020165730A1 (en) | 2019-02-14 | 2020-08-20 | Ignite Immunotherapy, Inc. | Recombinant vaccinia virus and methods of use thereof |
| WO2020206033A1 (en) | 2019-04-02 | 2020-10-08 | Kenjockety Biotechnology, Inc. | Efflux pump-cancer antigen multi-specific antibodies and compositions, reagents, kits and methods related thereto |
| WO2021116943A1 (en) | 2019-12-12 | 2021-06-17 | Ignite Immunotherapy, Inc. | Variant oncolytic vaccinia virus and methods of use thereof |
| WO2021155028A1 (en) | 2020-01-29 | 2021-08-05 | Kenjockety Biotechnology, Inc. | Anti-mdr1 antibodies and uses thereof |
| WO2021247426A1 (en) | 2020-06-04 | 2021-12-09 | Kenjockety Biotechnology, Inc. | Anti-abcg2 antibodies and uses thereof |
| WO2021247423A1 (en) | 2020-06-04 | 2021-12-09 | Kenjockety Biotechnology, Inc. | Abcg2 efflux pump-cancer antigen multi-specific antibodies and compositions, reagents, kits and methods related thereto |
| US11208632B2 (en) | 2016-04-26 | 2021-12-28 | R.P. Scherer Technologies, Llc | Antibody conjugates and methods of making and using the same |
| WO2022013696A2 (en) | 2020-07-14 | 2022-01-20 | Pfizer Inc. | Recombinant vaccinia virus |
| WO2022051390A1 (en) | 2020-09-02 | 2022-03-10 | Kenjockety Biotechnology, Inc. | Anti-abcc1 antibodies and uses thereof |
| WO2022103603A1 (en) | 2020-11-13 | 2022-05-19 | Kenjockety Biotechnology, Inc. | Anti-mrp4 (encoded by abcc4 gene) antibodies and uses thereof |
| US11660266B2 (en) | 2018-04-11 | 2023-05-30 | Ohio State Innovation Foundation | Methods and compositions for sustained release microparticles for ocular drug delivery |
| WO2023114658A1 (en) | 2021-12-13 | 2023-06-22 | Kenjockety Biotechnology, Inc. | Anti-abcb1 antibodies |
| WO2023159220A1 (en) | 2022-02-18 | 2023-08-24 | Kenjockety Biotechnology, Inc. | Anti-cd47 antibodies |
| US11897928B2 (en) | 2018-07-18 | 2024-02-13 | Manzanita Pharmaceuticals, Inc. | Conjugates for delivering an anti-cancer agent to nerve cells, methods of use and methods of making thereof |
| US12187810B2 (en) | 2020-11-20 | 2025-01-07 | R.P. Scherer Technologies, Llc | Glycoside dual-cleavage linkers for antibody-drug conjugates |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991013066A2 (en) * | 1990-02-21 | 1991-09-05 | Rhone-Poulenc Rorer S.A. | METHOD FOR PREPARING CIS-β-PHENYLGLYCIDIC-(2R,3R) ACID |
-
1992
- 1992-11-19 WO PCT/US1992/009911 patent/WO1993010076A1/en active Application Filing
- 1992-11-19 AU AU31400/93A patent/AU3140093A/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991013066A2 (en) * | 1990-02-21 | 1991-09-05 | Rhone-Poulenc Rorer S.A. | METHOD FOR PREPARING CIS-β-PHENYLGLYCIDIC-(2R,3R) ACID |
Non-Patent Citations (3)
| Title |
|---|
| JOURNAL OF ORGANIC CHEMISTRY vol. 28, no. 7, June 1963, EASTON US pages 2009 - 2012 C. C. TUNG, A. J. SPEZIALE 'Epoxide Studies. The Ring Opening of cis- and trans-N,N-Diethylphenylglycidamide' cited in the application * |
| JOURNAL OF ORGANIC CHEMISTRY vol. 51, no. 1, 10 January 1986, EASTON US pages 46 - 50 A. E. GREENE 'An Efficient, Enantioselective Synthesis of the Taxol Side Chain' cited in the application * |
| JOURNAL OF ORGANIC CHEMISTRY vol. 55, no. 6, 16 March 1990, EASTON US pages 1957 - 1959 A. E. GREENE 'An Improved Synthesis of the Taxol Side Chain and of RP 56976' cited in the application * |
Cited By (99)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2707165A1 (en) * | 1993-07-06 | 1995-01-13 | Rhone Poulenc Rorer Sa | Application of taxoids to the treatment of parasitic diseases. |
| EP2226085A1 (en) | 1993-07-19 | 2010-09-08 | Angiotech Pharmaceuticals, Inc. | Anti-angiogenic compositions and methods of use |
| US7820193B2 (en) | 1993-07-19 | 2010-10-26 | Angiotech Pharmaceuticals, Inc. | Anti-angiogenic compositions and methods of use |
| EP0797988A2 (en) | 1993-07-19 | 1997-10-01 | Angiotech Pharmaceuticals, Inc. | Anti-angiogenic compositions and methods of use |
| US5677470A (en) * | 1994-06-28 | 1997-10-14 | Tanabe Seiyaku Co., Ltd. | Baccatin derivatives and processes for preparing the same |
| US5589502A (en) * | 1994-11-17 | 1996-12-31 | Tanabe Seiyaku Co., Ltd. | Baccatin derivatives and processes for preparing the same |
| US5608073A (en) * | 1994-11-17 | 1997-03-04 | Tanabe Seiyaku Co., Ltd. | Baccatin derivatives and processes for preparing the same |
| US6291690B1 (en) | 1995-01-09 | 2001-09-18 | The Liposome Company, Inc. | Hydrophobic taxane derivatives |
| US5939567A (en) * | 1995-01-09 | 1999-08-17 | The Liposome Company, Inc. | Synthesis of hydrophobic taxane derivatives |
| US5580899A (en) * | 1995-01-09 | 1996-12-03 | The Liposome Company, Inc. | Hydrophobic taxane derivatives |
| US8313521B2 (en) | 1995-06-07 | 2012-11-20 | Cook Medical Technologies Llc | Method of delivering an implantable medical device with a bioabsorbable coating |
| US6482850B2 (en) | 1995-09-12 | 2002-11-19 | The Liposome Company | Hydrolysis-promoting hydrophobic taxane derivatives |
| US6051600A (en) * | 1995-09-12 | 2000-04-18 | Mayhew; Eric | Liposomal hydrolysis-promoting hydrophobic taxane derivatives |
| US6392063B1 (en) | 1995-09-12 | 2002-05-21 | The Liposome Company, Inc. | Hydrolysis-promoting hydrophobic taxane derivatives |
| US6265587B1 (en) | 1995-10-27 | 2001-07-24 | Societe D'etude Et De Recherche En Ingenierie Pharmaceutique Seripharm | Intermediary compounds for the hemisynthesis of taxanes and preparation processes therefor |
| US6825365B2 (en) | 1995-10-27 | 2004-11-30 | Novasep | Intermediates for the hemisynthesis of taxanes and preparation processes therefor |
| WO1997015562A1 (en) * | 1995-10-27 | 1997-05-01 | Societe D'etude Et De Recherche En Ingenierie Pharmaceutique Seripharm | Intermediary compounds for the hemisynthesis of taxanes and preparation processes therefor |
| FR2740451A1 (en) * | 1995-10-27 | 1997-04-30 | Seripharm | NEW INTERMEDIARIES FOR THE HEMISYNTHESIS OF TAXANES, THEIR METHODS OF PREPARATION AND THEIR USE IN THE GENERAL SYNTHESIS OF TAXANES |
| US6759397B2 (en) | 2000-05-01 | 2004-07-06 | The University Of British Columbia | Ginsenoside chemotherapy |
| RU2164501C1 (en) * | 2000-07-03 | 2001-03-27 | Шугина Галина Александровна | Method of cleaning polluted subsurface water |
| EP2014307A2 (en) | 2001-03-13 | 2009-01-14 | Angiotech International Ag | Micellar drug delivery vehicles and uses thereof |
| US7875677B2 (en) | 2001-04-20 | 2011-01-25 | The University Of British Columbia | Micellar drug delivery systems for hydrophobic drugs |
| US8710095B2 (en) * | 2002-04-30 | 2014-04-29 | Bionumerik Pharmaceuticals, Inc. | Drugs for prophylaxis or mitigation of taxane-induced neurotoxicity |
| EP1982720A1 (en) | 2002-05-20 | 2008-10-22 | The Regents of the University of California | Ribaviren for use to treat cancer |
| EP2181704A2 (en) | 2002-12-30 | 2010-05-05 | Angiotech International Ag | Drug delivery from rapid gelling polymer composition |
| EP2390262A1 (en) | 2003-05-16 | 2011-11-30 | Intermune, Inc. | Synthetic chemokine receptor ligands and methods of use thereof |
| US7407973B2 (en) | 2003-10-24 | 2008-08-05 | Intermune, Inc. | Use of pirfenidone in therapeutic regimens |
| US7846940B2 (en) | 2004-03-31 | 2010-12-07 | Cordis Corporation | Solution formulations of sirolimus and its analogs for CAD treatment |
| US8557272B2 (en) | 2004-03-31 | 2013-10-15 | Cordis Corporation | Device for local and/or regional delivery employing liquid formulations of therapeutic agents |
| US8003122B2 (en) | 2004-03-31 | 2011-08-23 | Cordis Corporation | Device for local and/or regional delivery employing liquid formulations of therapeutic agents |
| US7932265B2 (en) | 2004-03-31 | 2011-04-26 | Cordis Corporation | Solution formulations of sirolimus and its analogs for CAD treatment |
| US7989490B2 (en) | 2004-06-02 | 2011-08-02 | Cordis Corporation | Injectable formulations of taxanes for cad treatment |
| US9402804B2 (en) | 2004-06-02 | 2016-08-02 | CARDINAL HEALTH SWITZERLAND 515 GmbH | Injectable formulations of taxanes for cad treatment |
| US8691780B2 (en) | 2005-02-17 | 2014-04-08 | The Board Of Trustees Of The Leland Stanford Junior University | Txr1 and enhanced taxane sensitivity based on the modulation of a pathway mediated thereby |
| EP2093235A1 (en) | 2006-02-08 | 2009-08-26 | Alios Biopharma Inc. | Hyperglycosylated variants of interferon alfacon-1 |
| US7919108B2 (en) | 2006-03-10 | 2011-04-05 | Cook Incorporated | Taxane coatings for implantable medical devices |
| US7875284B2 (en) | 2006-03-10 | 2011-01-25 | Cook Incorporated | Methods of manufacturing and modifying taxane coatings for implantable medical devices |
| US8147540B2 (en) | 2006-03-10 | 2012-04-03 | Cook Medical Technologies Llc | Taxane coatings for implantable medical devices |
| EP3056196A1 (en) | 2006-03-22 | 2016-08-17 | SynCore Biotechnology CO., LTD | Treatment of triple receptor negative breast cancer |
| EP2380606A1 (en) | 2006-06-30 | 2011-10-26 | Cook Incorporated | Methods of manufacturing and modifying taxane coatings for implantable medical devices |
| EP2108390A2 (en) | 2008-03-31 | 2009-10-14 | Cordis Corporation | Device for local and/or regional delivery employing liquid formulations of therapeutic agents |
| US8409601B2 (en) | 2008-03-31 | 2013-04-02 | Cordis Corporation | Rapamycin coated expandable devices |
| US8420110B2 (en) | 2008-03-31 | 2013-04-16 | Cordis Corporation | Drug coated expandable devices |
| US8871240B2 (en) | 2008-03-31 | 2014-10-28 | Cordis Corporation | Rapamycin coated expandable devices |
| US8273404B2 (en) | 2008-05-19 | 2012-09-25 | Cordis Corporation | Extraction of solvents from drug containing polymer reservoirs |
| EP2123312A2 (en) | 2008-05-19 | 2009-11-25 | Cordis Corporation | Extraction of solvents from drug containing polymer reservoirs |
| EP2859916A2 (en) | 2008-05-22 | 2015-04-15 | Galera Therapeutics, LLC | Combination of an antimitotic agent and a selective superoxide dismutase mimetic in antitumor therapy |
| WO2009143454A2 (en) | 2008-05-22 | 2009-11-26 | Kereos, Inc. | Combination antitumor therapy |
| WO2010005850A1 (en) | 2008-07-08 | 2010-01-14 | The J. David Gladstone Institutes | Methods and compositions for modulating angiogenesis |
| US8642063B2 (en) | 2008-08-22 | 2014-02-04 | Cook Medical Technologies Llc | Implantable medical device coatings with biodegradable elastomer and releasable taxane agent |
| WO2012162010A1 (en) | 2011-05-25 | 2012-11-29 | Cordis Corporation | Expandable devices coated with a paclitaxel composition |
| WO2012162007A1 (en) | 2011-05-25 | 2012-11-29 | Cordis Corporation | Expandable devices coated with a rapamycin composition |
| US10137104B2 (en) | 2012-04-04 | 2018-11-27 | Halozyme, Inc. | Combination therapy with an anti-hyaluronan agent and therapeutic agent |
| WO2013151774A1 (en) | 2012-04-04 | 2013-10-10 | Halozyme, Inc. | Combination therapy with an anti - hyaluronan agent and a tumor - targeted taxane |
| US9913822B2 (en) | 2012-04-04 | 2018-03-13 | Halozyme, Inc. | Combination therapy with an anti-hyaluronan agent and therapeutic agent |
| WO2013188763A1 (en) | 2012-06-15 | 2013-12-19 | The Brigham And Women's Hospital, Inc. | Compositions for treating cancer and methods for making the same |
| WO2014004760A1 (en) | 2012-06-28 | 2014-01-03 | Covidien Lp | Post-processing of a medical device to control morphology and mechanical properties |
| US9956385B2 (en) | 2012-06-28 | 2018-05-01 | The Spectranetics Corporation | Post-processing of a medical device to control morphology and mechanical properties |
| EP3539563A1 (en) | 2012-07-19 | 2019-09-18 | Redwood Bioscience, Inc. | Antibody specific for cd22 and methods of use thereof |
| US9579390B2 (en) | 2012-11-12 | 2017-02-28 | Redwood Bioscience, Inc. | Compounds and methods for producing a conjugate |
| EP3613439A1 (en) | 2013-02-15 | 2020-02-26 | The Regents Of The University Of California | Chimeric antigen receptor and methods of use thereof |
| EP4303232A2 (en) | 2013-02-15 | 2024-01-10 | The Regents of The University of California | Chimeric antigen receptor and methods of use thereof |
| EP3881868A1 (en) | 2013-02-15 | 2021-09-22 | The Regents Of The University Of California | Chimeric antigen receptor and methods of use thereof |
| EP3300745A1 (en) | 2013-02-15 | 2018-04-04 | The Regents of the University of California | Chimeric antigen receptor and methods of use thereof |
| WO2014160185A2 (en) | 2013-03-14 | 2014-10-02 | The Board Of Trustees Of The Leland Stanford Junior University | Mitochondrial aldehyde dehydrogenase-2 modulators and methods of use thereof |
| WO2015081282A1 (en) | 2013-11-27 | 2015-06-04 | Redwood Bioscience, Inc. | Hydrazinyl-pyrrolo compounds and methods for producing a conjugate |
| WO2015127137A1 (en) | 2014-02-19 | 2015-08-27 | Aldea Pharmaceuticals, Inc. | Mitochondrial aldehyde dehydrogenase 2 (aldh2) binding polycyclic amides and their use for the treatment of cancer |
| US10426753B2 (en) | 2014-04-03 | 2019-10-01 | Invictus Oncology Pvt. Ltd. | Supramolecular combinatorial therapeutics |
| US10087139B2 (en) | 2015-03-03 | 2018-10-02 | Toray Fine Chemicals Co., Ltd. | 3-Phenylisoserine derivative production method |
| WO2017053920A1 (en) | 2015-09-25 | 2017-03-30 | Zy Therapeutics Inc. | Drug formulation based on particulates comprising polysaccharide-vitamin conjugate |
| WO2017083637A1 (en) | 2015-11-12 | 2017-05-18 | The Board Of Trustees Of The Leland Stanford Junior University | Cell-penetrating, guanidinium-rich oligophosphotriesters for drug and probe delivery |
| WO2017115301A1 (en) | 2015-12-30 | 2017-07-06 | Syncore Biotechnology Co., Ltd. | Treatment of breast cancer using a combination of a cationic liposomal formulation of taxane, a non-liposomal formulation of taxane and a further active agent |
| US11208632B2 (en) | 2016-04-26 | 2021-12-28 | R.P. Scherer Technologies, Llc | Antibody conjugates and methods of making and using the same |
| US11788066B2 (en) | 2016-04-26 | 2023-10-17 | R.P. Scherer Technologies, Llc | Antibody conjugates and methods of making and using the same |
| WO2018017190A2 (en) | 2016-06-01 | 2018-01-25 | Baxalta Incorporated | Formulations of polyalkylene oxide-asparaginase and methods of making and using the same |
| EP4019007A1 (en) | 2016-06-01 | 2022-06-29 | Servier IP UK Limited | Formulations of polyalkylene oxide-asparaginase and methods of making and using the same |
| WO2018127082A1 (en) | 2017-01-05 | 2018-07-12 | Syncore Biotechnology Co., Ltd. | Treatment of pancreatic cancer |
| WO2018136570A1 (en) | 2017-01-18 | 2018-07-26 | F1 Oncology, Inc. | Chimeric antigen receptors against axl or ror2 and methods of use thereof |
| EP3388082A1 (en) | 2017-04-13 | 2018-10-17 | Galera Labs, LLC | Combination cancer immunotherapy with pentaaza macrocyclic ring complex |
| WO2018218004A1 (en) | 2017-05-24 | 2018-11-29 | The Board Of Regents Of The University Of Texas System | Linkers for antibody drug conjugates |
| WO2019152661A1 (en) | 2018-01-31 | 2019-08-08 | Galera Labs, Llc | Combination cancer therapy with pentaaza macrocyclic ring complex and platinum-based anticancer agent |
| WO2019195586A1 (en) | 2018-04-06 | 2019-10-10 | The Regents Of The University Of California | Methods of treating egfrviii expressing glioblastomas |
| WO2019195596A1 (en) | 2018-04-06 | 2019-10-10 | The Regents Of The University Of California | Methods of treating glioblastomas |
| US11660266B2 (en) | 2018-04-11 | 2023-05-30 | Ohio State Innovation Foundation | Methods and compositions for sustained release microparticles for ocular drug delivery |
| WO2019222435A1 (en) | 2018-05-16 | 2019-11-21 | Halozyme, Inc. | Methods of selecting subjects for combination cancer therapy with a polymer-conjugated soluble ph20 |
| US11897928B2 (en) | 2018-07-18 | 2024-02-13 | Manzanita Pharmaceuticals, Inc. | Conjugates for delivering an anti-cancer agent to nerve cells, methods of use and methods of making thereof |
| WO2020148612A1 (en) | 2019-01-14 | 2020-07-23 | Ignite Immunotherapy, Inc. | Recombinant vaccinia virus and methods of use thereof |
| WO2020165730A1 (en) | 2019-02-14 | 2020-08-20 | Ignite Immunotherapy, Inc. | Recombinant vaccinia virus and methods of use thereof |
| WO2020206033A1 (en) | 2019-04-02 | 2020-10-08 | Kenjockety Biotechnology, Inc. | Efflux pump-cancer antigen multi-specific antibodies and compositions, reagents, kits and methods related thereto |
| WO2021116943A1 (en) | 2019-12-12 | 2021-06-17 | Ignite Immunotherapy, Inc. | Variant oncolytic vaccinia virus and methods of use thereof |
| WO2021155028A1 (en) | 2020-01-29 | 2021-08-05 | Kenjockety Biotechnology, Inc. | Anti-mdr1 antibodies and uses thereof |
| WO2021247423A1 (en) | 2020-06-04 | 2021-12-09 | Kenjockety Biotechnology, Inc. | Abcg2 efflux pump-cancer antigen multi-specific antibodies and compositions, reagents, kits and methods related thereto |
| WO2021247426A1 (en) | 2020-06-04 | 2021-12-09 | Kenjockety Biotechnology, Inc. | Anti-abcg2 antibodies and uses thereof |
| WO2022013696A2 (en) | 2020-07-14 | 2022-01-20 | Pfizer Inc. | Recombinant vaccinia virus |
| WO2022051390A1 (en) | 2020-09-02 | 2022-03-10 | Kenjockety Biotechnology, Inc. | Anti-abcc1 antibodies and uses thereof |
| WO2022103603A1 (en) | 2020-11-13 | 2022-05-19 | Kenjockety Biotechnology, Inc. | Anti-mrp4 (encoded by abcc4 gene) antibodies and uses thereof |
| US12187810B2 (en) | 2020-11-20 | 2025-01-07 | R.P. Scherer Technologies, Llc | Glycoside dual-cleavage linkers for antibody-drug conjugates |
| WO2023114658A1 (en) | 2021-12-13 | 2023-06-22 | Kenjockety Biotechnology, Inc. | Anti-abcb1 antibodies |
| WO2023159220A1 (en) | 2022-02-18 | 2023-08-24 | Kenjockety Biotechnology, Inc. | Anti-cd47 antibodies |
Also Published As
| Publication number | Publication date |
|---|---|
| AU3140093A (en) | 1993-06-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1993010076A1 (en) | Synthesis and optical resolution of the taxol side chain and related compounds | |
| US5770745A (en) | Synthesis of taxol, taxol analogs and their intermediates with variable A-ring side chain structures and compositions thereof | |
| JP3394284B2 (en) | Semi-synthesis of taxane derivatives using metal alkoxides and oxazinones | |
| JP2500198B2 (en) | New β-lactam | |
| US20010037020A1 (en) | Method for preparation of taxol | |
| US6350886B1 (en) | β-lactams, methods for the preparation of taxanes, and sidechain-bearing taxanes | |
| JPH03232869A (en) | Preparation of taxole using oxazinone | |
| HU225507B1 (en) | Intermediary compounds for hemisynthesis of taxanes and preparation of them | |
| EP1786798B1 (en) | One pot synthesis of taxane derivatives and their conversion to paclitaxel and docetaxel | |
| JP3445973B2 (en) | Intermediates and methods useful for semisynthesis of paclitaxel and its analogs | |
| US7358378B2 (en) | Processes for the preparation of paclitaxel | |
| US6048990A (en) | Method for selective acylation of C-2'-O-protected-10-hydroxy-taxol at the C-10 position | |
| US6448417B1 (en) | Methods and useful intermediates for paclitaxel synthesis from C-7, C-10 di-cbz 10-deacetylbaccatin III | |
| US6399783B1 (en) | Methods for the esterification of alcohols and compounds useful therefor as potential anticancer agents | |
| KR101009467B1 (en) | Taxane derivatives useful for the synthesis of docetaxel and preparation methods thereof | |
| CN101675028A (en) | Preparation (2R, 3S)-method of 3-phenylisoserine methyl esters acetate | |
| US20090292131A1 (en) | Processes for preparation of taxanes and intermediates thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AT AU BB BG BR CA CH CS DE DK ES FI GB HU JP KP KR LK LU MG MN MW NL NO PL RO RU SD SE |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL SE BF BJ CF CG CI CM GA GN ML MR SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| 122 | Ep: pct application non-entry in european phase | ||
| NENP | Non-entry into the national phase |
Ref country code: CA |