TΓΓLE NOVEL CSAIDS
FIELD OF THE INVENTION
This invention relates to the novel compounds of Formula (I), pharmaceutical compositions and various methods of use of the compounds of Formulas (I).
BACKGROUND OF THE INVENTION
The cyclooxygenase (CO) mediated pathway oxidizes arachidonic acid to produce PGH2 which is in turn metabolized to the prostanoids (PGE2, TxA2, and prostacyclin). These products are produced by various cells including polymorpho-nuclear leukocytes, mast cells and monocytes. The 5-lipoxygenase (5-LO) mediated pathway oxidizes arachidonic acid initially to 5-hydroperoxy-eicosatetraenoic acid (5-HPETE) which is further metabolized to LTA4, the precursor to the peptidoleukotrienes (LTC4, LTD and LTE4) and LTB4. Additionally 5-HPETE is converted to 5-hydroxyeicosatetraenoic acid (5- HETE).
The arachidonic acid oxygenated products, as noted above, have been identified as mediators of various inflammatory conditions. The various inflammatory disease states caused by these mediators and many other conditions, as discussed herein, are all conditions in which a dual inhibitor of both CO and 5-LO would be indicated.
Interleukin-1 (EL-1) and Tumor Necrosis Factor (TNF) are biological substances produced by a variety of cells, such as monocytes or macrophages. IL-1 and TNF affect a wide variety of cells and tissues and these cytokines as well as other leukocyte derived cytokines are important and critical inflammatory mediators of a wide variety of disease states and conditions. The inhibition of these cytokines is of benefit in controlling, reducing and alleviating many of these disease states.
There remains a need for treatment, in this field, for compounds which are cytokine suppresive anti-inflammatory drugs (hereinafter CS AID's), i.e. compounds which are capable of inhibiting cytokines, such as IL-1, IL-6 and TNF; and compounds which are also capable of inhibiting the oxygenation of arachidonic acid by inhibition of enzymes such as lipoxygenase, specifically 5-lipoxygenase (5-LO) and cyclooxygenase (CO) thereby preventing the formation of various leukotrienes and prostaglandins.
SUMMARY OF THE INVENTION
This invention relates to the novel compounds of Formula Q and pharmaceutical compositions comprising a compound of Formula CO and a pharmaceutically acceptable diluent or carrier.
This invention relates to a method of treating an oxygenated polyunsaturated fatty acid mediated disease (hereinafter OPUFA) in an animal in need thereof which comprises administering to such animal, an effective amount of a compound of Formula Q).
This invention also relates to a method of treating a cytokine mediated disease, in an animal in need thereof, which comprises administering to such animal an effective amount of a compound of Formula (II).
This invention specifically relates to a method of inhibiting the production of interleuMn-1 (hereinafter IL-1) in an animal in need thereof which comprises administering to such animal an effective amount of a compound of Formula QI) sufficient to inhibit IL-1. More specifically the inhibition of the production of IL-1 is useful in the treatment, prophylactically or therapeutically, of any disease state in a mammal which is exacerbated or caused by excessive or unregulated IL-1 production.
This invention specifically relates to a method of inhibiting the production of Tumor Necrosis Factor (hereinafter TNF) in an animal in need thereof which comprises administering to such animal, an effective amount of a compound of Formula (II) sufficient to inhibit TNF. More specifically the inhibition of the production of TNF is useful in the treatment, prophylactically or therapeutically, of any disease state in a mammal which is exacerbated or caused by excessive or unregulated TNF production.
DETAILED DESCRIPTION OF THE INVENTION
The compounds of Formula (ϋ are also useful in the treatment of viral infections, where such viruses are sensitive to upregulation by TNF or will elicit TNF production in vivo. The viruses contemplated for treatment herein are those that produce TNF as a result of infection, or those which are sensitive to inhibition, such as by decreased replication, directly or indirectly, by the TNF inhibitors of Formula (II). Such viruses include, but are not limited to; HIV-1, HIV-2 andHIV-3, Cytomegalovirus (CMV), Influenza, adenoviras and the Herpes group of viruses, such as but not limited to, Herpes Zoster and Herpes Simplex.
This invention more specifically relates to a method of treating a mammal, afflicted with a human immunodeficiency virus (HIV), which comprises administering to such mammal an effective TNF inhibiting amount of a compound of Formula 0).
The compounds of Formula QI) may also be used in association with the veterinary treatment of mammals, other than in humans, in need of inhibition of TNF
production. TNF mediated diseases for treatment, therapeutically or prophylactically, in animals include disease states such as those noted above, but in particular viral infections. Examples of such viruses include, but are not limited to, feline immunodeficiency virus (FIV) or other retroviral infection such as equine infectious anaemia virus, caprine arthritis virus, visna virus, maedi virus and other lentiviruses.
A preferred method of this invention is the treatment, therapeutically or prophylactically, of viral infections, in particular where such viruses are sensitive to upregulation by TNF or IL-1 will elicit TNF or IL-1 production in vivo by administering an effective amount of a compound of Formula (II). The compounds of by the structure:
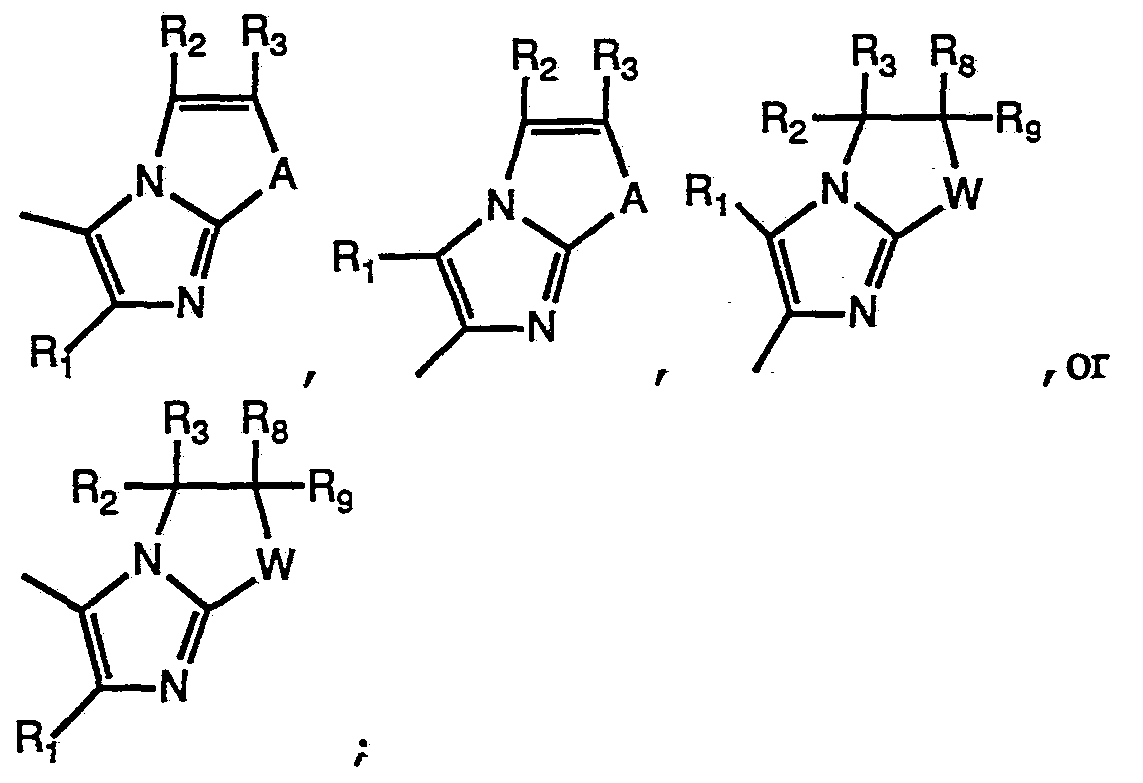
Wi is -(CR4R5)-, or -(CR4R5MCR6R7)- ;
R2, R3, ό, R7, Rδ, and R9 are hydrogen; or one or two of R2, R3, ό, R7» Rδ, and R9 are, independently, hydrogen or Cι_2 alkyl; one of R4 and R5 is OR10 and the other is selected from H, alkyli^, optionally • substituted alkyli-6, aryl, optionally substituted aryl, heteroaryl, or optionally substituted heteroaryl ;
RlO is hydrogen, optionally substituted Ci-6 alkyl, or optionally substituted aryl; provided that when Rio is hydrogen the other of R4 or R5 is other than hydrogen; one of Ri and Ro is 4-pyridyl or C1.4 alkyl-4-pyridyl; and the other of Ri and Rθ is
(a) phenyl ;
(b) mono- or di-substituted phenyl wherein said substituents are independently selected from Cj_4 alkyl, halo, hydroxy , Cj_4 alkoxy, aryloxy, heteroaryloxy, C1.3 alkylthio, C1.3 alkylsulfinyl, C2-.5 1-alkenyl-l- thio, C2-.5 2-alkenyl-l-thio, C2-5 1-alkenyl-l-sulfinyl, C2.5 2-alkenyl-l- sulfinyl, C1-.3 alkylamino, C1.3 dialkylamino, CF3, N-(Cι_3alkanamido), N- (C1.3 alkyl)-N-(Cι_3alkanamido), N-pyrrolidino, N-piperidino, prop-2-ene- 1-oxy, 2,2,2-trihaloethoxy, thiol, acylthio, dithioacyl, thiocarbamyl, dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, phenylthio, phenylsulfinyl, alkoxyalkylthio, alkoxyalkyl-sulfinyl alkylthioalkylthio, acyloxyalkylthio, acyloxyalkylsulfinyl or Z; or
(c) a moiety of the formulae:
wherein Y is selected from
wherein t is 0 or 1; W, \W , R2, R3, R4, R5, Rό, R7, Rs» and R9 are as defined above; A is -CR5=CR7-, -N=C 7-, -S- or -0-;
Ra andRb are independently selected from hydrogen, optionally substituted Cχ-9 alkyl, optionally substituted aryl or optionally substituted heteroaryl;
Zis -S-(CRaRb)rS-Zι; Zi is a functional moiety; or a pharmaceutically acceptable salt thereof.
Preferred mono-substitution of the phenyl ring for compounds of Formula Q) is Ci-4 alkyl, C1-4 alkyl S(0) , m is 0 or 1; C1.4 alkoxy, halo, N-(Cι_3 alkyl) alkanamido, or N-(Ci-3 alkanamido). Preferred di-substitution of the phenyl ring for compounds of Formula Q) is:
(b) disubstituted phenyl wherein said substituents are, independently, Cχ_3 alkylthio, C1-3 alkoxy, halo, Cι_4 alkyl, C]_3 alkylamino, N- (Cι_3alkyl)-N-(Cι_3 alkanamido), Cχ_3 dialkylamino, amino,
N-pyrrolidino or N-piperidino ; or (c) disubstituted phenyl wherein one of said substituents is Cι_3 alkoxy, halo, C1.4 alkyl or CF3, and the other substituent is thiol, alkylthio, al ylsulfinyl, acylthio, dithioacyl, thiocarbamyl, dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio,
alkoxythionothio, arylthio, arylsulfinyl, alkoxyalkylthio, alkoxyalkylsulfinyl, alkylthioalkylthio, acyloxyalkylsulfinyl, acyloxyalkylthio or Z; or
(d) disubstituted phenyl wherein one of said substituents is amino, Ci-3 alkylamino or Cl-3 dialkylamino; and the other substituent is C1.3 alkylthio, Cj_3 alkylsulfinyl, C2-.5 -1-alkenyl-l-thio, C2-.5 1-alkenyl-l- sulfinyl, 03.5 2-alkenyl-l-thio, C3.52-alkenyl-l- sulfinyl, thiol, acylthio, dithioacyl, thiocarbamyl, dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, phenylthio, phenylsulfinyl, alkoxyalkylthio, alkoxyalkylsulfinyl, alkylthioalkylthio, acyloxyalkylthio, acyloxyalkylsulfinyl or Z; or
(e) disubstituted phenyl wherein said substituents are the same and are selected from halo, C1.3 alkoxy, C1.3 alkylamino, Cj_3 dialkylamino, N- pyrrolidino, N-piperidino, 2,2,2- trihaloethoxy, prop-2-ene-l-oxy, hydroxy, Cj_3 alkylthio, C1.3 alkyl-sulfonyl, thiol, acylthio, dithioacyl, thiocarbamyl, dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, phenylthio, phenylsulfinyl, alkoxyalkylthio, alkoxyalkylsulfinyl , alkylthioalkylthio, acyloxyalkylthio, acyloxyalkylsulfinyl or Z;
Preferably, for all the compounds of Formulas (I) when Rj is C1.4 alkyl-4- pyridyl the alkyl substituent is located at the 2-position of the pyridine ring. More preferably the alkyl substituent is methyl.
Zi is a functional moiety that does not interfere with breakage of the "disulfide bond in-vivo to yield the SH moiety. Preferable Z\ moieties are aryl, optionally substituted aryl, C . alkyl, optionally substituted alkyl , heteroaryl, an optionally substituted heteroaryl, cystiene or glutathione. The optional substituents may be the same as the Ro or Ri phenyl moieties noted above for Formula (I).
Ra and Rb are independently selected from hydrogen, optionally substituted Cι_Q alkyl, optionally substituted aryl, or optionally substituted heteroaryl. The optional substituents for the aryl and heteroaryl ring are the same as the Ro and Ri phenyl moieties noted above for Formula (I), other than Z. Preferably Ra and R are unsubstituted or substituted with C1- alkyl.
Preferably, one of R4 or R5 is hydroxyl, i. e. Rio is hydrogen. When the other of R4 or R5 is aryl, it is preferably an optionally substituted phenyl moiety. Optional substituents groups for R4 or R5 as aryl or heteroaryl are halogen, C1.9 alkyl, halo-substituted C1-9 alkyl, hydroxy-substituted Ci-9alkyl, Ci-6 alkoxy, S(0)nalkylι_6, (CH2)m CO2H, (CH2)mNRnRi2, wherein Rn and R12 are independently selected from hydrogen, aikylι.4, aryl, or Rn and R12 together form a heterocyclic ring of 5 to 7 members, wherein one or two
of the ring members of the heterocyclic ring may additionally be O, N or S and may contain additional unsaturation; n is 0 to 2, and m is 0 to 4. For 5-LO or CO inhibitory activity the 4 or R5 moiety is not substituted with a carboxylic acid moiety.
When R4 or R5 is an optionally substituted alkyl the substituents are selected from halogen, hydroxy, alkoxy, alkylS(0)n, aryl, heteroaryl, CO2H, or NR11R12. For all R4 and R5 substitutions the halogen substituted alkyl moiety may contain more than one halogen selected independently from fluorine, chloride, iodine or bromine; the hydroxy substituted alkyl may also be polyhydroxy substituted.
Preferably when one of R4 or R5 is a substituted phenyl, the substituents are halo, methoxy, carboxylic acid (and salts thereof), or a mono- or di-alkyl substituted methylamine. Preferred heterocyclic rings when Rn and R12 cyclize are a pyrrole, pytrolidine, piperidine, or morpholino ring.
The optional substituents for Rio moieties is the same for the R4 and R5 terms described above. The compounds of Formula (II), a subgenus of the compounds of Formula (I) are also useful in the treatment of an OPUFA mediated diseases and are preferably useful as cytokine inhibitors. The compounds of Formula (II) are represented by the structure:
wherein W2 is -(CR4R5)-, or -(CR4R5MCR6R7)- ;
R2s R3, R , γ, Rδ. and R9 are hydrogen; or one or two of R2 R3, R6» R7. Rg, and R9 are, independently, hydrogen or Cj_2 alkyl; one of R4 and R5 is OR10 and the other is selected from H, alkylι--6, halogen substituted alkyli-^, aryl, optionally substituted aryl ; Rio is hydrogen or Ci-6 alkyl ; provided that when Rio is hydrogen the other of R4 or R5 is other than hydrogen;
R is 4-pyridyl or C1-.4 alkyl-4-pyridyl; RQ is,
(a) phenyl; (b) mono or di-substituted phenyl wherein said substituents are independently selected from C1.4 alkyl, halo, halosubstituted alkyl, C\.A alkoxy, C]_3 alkylthio, C1..3 alkylsulfinyl, C2-5 1-alkenyl-l-thio, C2-.52- alkenyl-1-thio, C2-.5 1-alkenyl-l-sulfinyl, C2-.52-alkenyl-l-sulfinyl, C1.3 alkylamino, Cχ_3 dialkylamino, CF3, N-pyrrolidino, N-piperidino, 2,2,2-
trihaloethoxy, thiol, acylthio, dithioacyl, thiocarbamyl, dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, phenylthio, phenylsulfinyl, alkoxyalkylthio, alkoxyalkylsulfinyl, alkylthioalkylthio, acyloxyalkylthio, acyloxyalkylsulfinyl or Z; provided that is the phenyl is substituted with a C3- alkoxy it is in other than the 4-position;
(c) a moiety of the formulae:
(CR
aR
b)t
J-S'
ζ wherein Y is selected from
wherein t is 0 or 1; W2, Ri, R2, R3, R4, R5, θ. R-7> Rδ> and R9 are as defined above;
A is -CR5=CR7-, -N=CR7-, -S- or -O-;
Ra and R are independently selected from hydrogen, optionally substituted Cι_9 alkyl, optionally substituted aryl or optionally substituted heteroaryl;
Z is -S-(CRaRb)rS-Zι; Zi is a functional moiety; or a pharmaceutically acceptable salt thereof.
Preferable R4 or R5 groups are a substituted aryl or alkyl with halogen, (CH2)m CO2H, or a (CH2)mNRnRi2 moiety, and m is 0 to 4. As in Formula (I) when Ri is a Cι_4 alkyl-4-pyridyl it preferably substituted in the 2-position of the pyridyl ring and the alkyl substituent is preferably methyl. As also the instancein Formula (I) or (II) compounds, the W or W2 term is preferably -(CR4R5)-.
Preferable Ro mono-substitution of a compound of Formula (II) is a C1.3 alkyl, C1.2 alkyl S(0)n, halogen, or CF3 moiety, and n is 0 or 1. If Ro is substituted with a Ci-4 alkoxy moiety it is preferably a methoxy or ethoxy derivative, or if C3-4 alkoxy it is in other than the para position.
Preferable di-substitution of a compound of Formula (II) is:
(a) disubstituted phenyl wherein said substituents are, independently, C .3 alkylthio, Cχ_3 alkoxy, halo, Cj_4 alkyl, Cχ_3 alkylamino, Cχ_3 dialkylamino, amino, N-pyrrolidino or N-piperidino; or
(b) disubstituted phenyl wherein one of said substituents is Cχ_3 alkoxy, halo, C1.4 alkyl or CF3, and the other substituent is thiol, alkylthio, alkylsulfinyl, acylthio, dithioacyl, thiocarbamyl, dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, phenylthio, phenylsulfinyl, alkoxyalkylthio, alkoxyalkylsulfinyl, alkylthioalkylthio, Z, or acyloxyalkylthio; or (c) disubstituted phenyl wherein one of said substituents is amino,
Ci-3 alkylamino or Ci-3 dialkylamino; and the other substituent is Cj_3 alkylthio, C1.3 alkylsulfinyl, C2-.5 -1-alkenyl-l-thio, C2-5 1-alkenyl-l- sulfinyl; C3.52-alkenyl-l-thio, C3.52-alkenyl-l- sulfinyl, thiol, acylthio, dithioacyl, thiocarbamyl, dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, phenylthio, phenylsulfinyl, alkoxyalkylthio, alkoxyalkylsulfinyl, alkylthioalkylthio, acyloxyalkylthio, acyloxyalkylsulfinyl, orZ; or
(d) disubstituted phenyl wherein said substituents are the same and are selected from halo, Cχ_3 alkoxy, Cj_3 alkylamino, Cj_3 dialkylamino, N- pyrrolidino, N-piperidino, 2,2,2-trihaloethoxy, C1.3 alkylthio, thiol, acylthio, dithioacyl, thiocarbamyl , dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, phenylthio, phenylsulfinyl, alkoxyalkylthio, alkoxyalltylsulfinyl , alkylthioalkylthio, acyloxyalkylthio, acyloxyalkylsulfinyl or Z.
It should be noted that the compounds of Formula Q) where R or Rø may be a Cj_3 alkylsulfϊnyl, C2-5 1-alkenyl-l-sulfinyl, C2-.5 -2-alkenyl-l-sulfinyl, alkoxyalkyl¬ sulfinyl, and phenylsulfinyl moiety, may act as prodrugs which are reductively converted in vivo to the corresponding alkylthio or alkenylthio form. It should be noted that the compounds of Formula Q) where Rj or Rø may be a phenyl substituted with an acylthio, dithioacyl, thiocarbamyl, dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, or acyloxyalkylthio may act as prodrugs which are hydrolytically converted in vivo to the corresponding sulfhydryl form. It should be noted that the compounds of Formula (I) where Rj or Rø may be a phenyl substituted with any of the disulfide moieties described herein may act as prodrugs which are oxidatively converted in vivo to the corresponding sulfhydryl farm.
By the term "halo" as used herein is meant all halogens, i.e., chloro, fluoro, bromo and iodo.
By the term "C^alkyl" or "alkyl" groups as used herein is meant to include both straight or branched chain radicals of 1 to 9 carbon atoms, unless the chain length is limited thereto, including, but not limited to methyl, ethyl, n-propyl, isopropyl, n-butyl, sec- butyl, isobutyl, tert-butyl, and the like.
By the term "alkenyl" as used herein is meant to include both straight or branched chain radicals of 1 to 9 carbon atoms, unless the chain length is limited thereto, but not limited to vinyl, 1-propenyl, 2-propenyl, or 3-methyl-2-propenyl. By the term "aryl" as used herein, in any combination, such as "aryloxy", is meant phenyl, or naphthyl.
By the term "heteroaryl" as used herein, in any combination, such as "heteroaryloxy", is meant a 5-10 membered aromatic ring system in which one or more rings contain one or more heteroatoms selected from the group consisting of N, O or S; such as, but not limited, to quinoline, isoquinoline, pyridine, pyrimidine, oxazole, thiazole, thiadiazole, triazole, imidazole.
By the term "sulfinyl" as used herein is meant the oxide of the corresponding sulfide. By the term "thio" as used herein is meant the sulfide. For further clarification, the following table outlines the structural attachment of the atoms of the Rj and Rø substituents of the compounds of Formula (I):
Table 1 Rj or Rø substituents Structural Attachment
Cι_3 alkylsulfinyl [AS(O)-] C2-5 1-alkenyl-l-thio [AA1C=CHS-]
C2-5 1-alkenyl-l-sulfinyl [AA1C=CHS(0)-]
C3.52-alkenyl-l-thio [ACH=CAlCH2S-]
C3.5 2-alkenyl-l-sulfmyl [ACH=CA1CH2S(0)-]
1-acyloxy-l-alkylthio [AC(0)OCH(A1)S-]
NOTE: A and A are hydrogen or alkyl;
Table 2
Additional Rj^ or Rβ Substituents Structural Attachments: acylthio [DC(0)S-] dithioacyl [DC(S)S-] thiocarbamyl [DDlNC(0)S-] dithiocarbamyl [DDlNC(S)S ]
alkylcarbonylalkylthio [DC(0)CH2S-] carbalkoxyalkylthio CBOC(0)CH2S-] alkoxycarbonylthio [BOC(0)S-] alkoxythionothio [BOC(S)S-] alkoxyalkylthio [BOCH2S-] alkoxyalkylsulfinyl [BOCH2S(0)] alkylthioalkylthio [BSCH2S-] disulfide Z [-S(CRaRb)t-S-Zι]
Note: D andD are hydrogen, Ci-9 alkyl, or phenyl; tis 0 or 1
B is Ci-9 alkyl or aryl; Ra, Rb and Zi is aryl, heteroaryl or Ci-9 alkyl (optionally substituted). The hydrogen atoms in the CH2 groups described in Table 2 are, independently, optionally substituted by a Ci-4 alkyl moiety.
By the term "lipoxygenase" as used herein is meant the 5-lipoxygenase, 12- lipoxygenase or 15-lipoxygenase enzymes.
By. the term "inhibiting the production of IL-1" is meant a) a decrease of excessive in vivo IL-1 levels in a human to normal levels or below normal levels by inhibition of the in vivo release of IL-1 by all cells, including but not limited to monocytes or macrophages; b) a down regulation, at the genomic level, of excessive in vivo IL-1 levels in a human to normal levels or below normal levels; or - - c) a down regulation, by inhibition of the direct synthesis of IL-1 as a postranslational event
By the term "inhibiting the production of TNF" is meant a) a decrease of excessive m vivo TNF levels in a human to normal levels or below normal levels by inhibition of the in vivo release of TNF by all cells, including but not limited to monocytes or macrophages; b) a down regulation, at the genomic level, of excessive in vivo TNF levels in a human to normal levels or below normal levels; or c) a down regulation, by inhibition of the direct synthesis of TNF as a postranslational event.
By the term "TNF mediated disease or disease state" is meant any and all disease states in which TNF plays a role, either by production of TNF itself, or by TNF causing another monokine to be released, such as but not limited to IL-1, or IL-6. A disease
state in which IL-1, for instance is a major component, and whose production or action, is exacerbated or secreted in response to TNF, would therefore be considered a disease stated mediated by TNF.
By the term "cytokine" as used herein is meant any secreted polypeptide that affects the functions of other cells, and is a molecule which modulates interactions between cells in the immune or inflammatory response. A cytokine includes, but is not limited to monokines and lymphokines regardless of which cells produce them. For instance, a monokine is generally referred to as being produced and secreted by a mononuclear cell, such as a macrophage and or monocyte but many other cells produce monokines, such as natural killer cells, fibroblasts, basophils, neutraphils, endothelial cells, brain astrocytes, bone marrow stromal cells, epideral keratinocytes, and β- lymphocytes. Lymphokines are generally referred to as being produced by lymphoctye cells. Examples of cytokines include, but are not limited to, Interleukin-1 (IL-1), Interleukin-6 (IL-6), Tumor Necrosis Factor-alpha (TNFα) and Tumor Necrosis Factor beta (TNFβ). By the term "cytokine interfering or cytokine suppresive amount" is meant an effective amount of a compound of Formula (I) to (HI) which will, when given for the treatment, prophylacticaly or therapeutically, of any disease state which is exacerbated or caused by excessive or unregulated cytokine production, cause a decrease the in vivo levels of the cytokine to normal or below normal levels. The inhibition of a cytokine, contemplated by the present invention, for use in the treatment of a HTV-infected human, must be a cytokine which is implicated in (a) the initiation and/or maintenance of T cell activation and/or activated T cell-mediated HIV gene expression and/or replication, and/or (b) any cytokine-mediated disease associated problem such as cachexia or muscle degeneration. As TNF-β (also known as lymphotoxin) has close structural homology with
TNF-α (also known as cachectin) and since each induces similar biologic responses and binds to the same cellular receptor, both TNF-α and TNF-β are inhibited by the compounds of the present invention and thus are herein referred to collectively as "TNF" unless specifically delineated otherwise. By the term "OPUFA mediated disease or disease state" is meant any disease state which is mediated (or modulated) by oxidation of polyunsaturated fatty acids, specifical the arachidonic acid metabolic pathway. The oxidation of arachidonic acid by such enzymes as the lipoxygenase enzymes or cyclooxgenase enzyme is specifically targeted by the present invention. Such enzymes include, but are not limited to, 5-LO, 12-LO, 15-LO, and CO; which produce the following mediators,including but not limited to, PGE2, LTB4,
LTC4, LTD4, prostaglandins, thromboxane, and prostocyclin.
By the term "OPUFA interfering amount" is meant an effective amount of a compound of Formula which shows a reduction of the in vivo levels of an oxgyenated arachidonic acid metabolite.
The compounds of Formula (I) may be prepared from the known intermediates of Formula (A), as shown below. The compounds of Formula (A) are known compounds and are prepared in Bender etal.. U.S. Patent Application Serial Number 07/255,816, filed October 11, 1988; Bender etal.. U.S. Patent Number 4,175,127, issued November 20, 1979; Bender et l.. U.S. Patent Application Serial Number 07/106,199 filed on July 10, 1987; Bender etal.. U.S. Patent Number 4,803,279, issued February 9, 1989, Bender et al.. U.S. Patent Number 4,719,218, issued January 12, 1988; Bender et al.. U.S. Patent
Number 4,715,310, issued January 14, 1988 the entire disclosures of all of which are hereby incorporated by reference.
Compounds of Formula (A) wherein Rø or Ri is a phenyl substituted with a substituted disulfide moiety are prepared by mild air oxidation of the compounds of Formula (A) wherein the R or Ri is a phenyl substituted with a sulfhydryl group. The non- symmetrical disulfides (Z) wherein Z is -S-S-Zi andZi is aryl, heteroaryl or alkyl, the compounds may be prepared by reaction of the sulfhydryl compound with the appropriate sulfenyl halide in an ethereal solvent to afford compounds of Formula (A) wherein one of Rø or Ri is a phenyl substituted with one or more alkyldithio or aryl-dithio groups. The method of Mukaiyama etal.. Tetrahedron Letters. 56:5907-08 (1968) allows for use of the desired aryl-SH or alkyl-SH reagent treated with diethylazodicarboxylate in 1:1 equivalence at room temperature in a solvent, yielding an adduct which is then treated with 1:1 ratio of the mercaptan of a Formula (A) compound. This process will also yield the disulfide dimer of the compounds of Formula (A). Preferably the disulfide linkage is on the Rø position of the compounds of Formula (A) .
Compounds of Formula (A) wherein R or Ri is phenyl substituted with an alkylthioalkylthio group are prepared by reacting the analogous sulfhydryl compound, prepared as described above, with the appropriate carbonyl component, such as formaldehyde, acetone, or acetaldehyde, using either mineral or Lewis acid catalysis conditions to yield the symmetrical dithioketal. The intermediate hydroxylalkylthio derivative reacts with another sulhydryl containing compound under the acid catalysis conditions to yield what is essentially a "bis" type compound, differing only by the alkyl chain insertion. This process produces the bis disulfide moieties of part (c) Claim 1, for instance, i.e. Formula (A)- S-CRRl-S-Formula (A). The substitution of the alkyl, R or R-, is determined by the reactive carbonyl functional group, wherein R or R- may be Cχ_9 alkyl, aryl or heteroaryl, all optionally substituted.
The nonsymmetrical thioketals can be prepared by the reaction of the metal mercaptan salt, prepared as described above, with a halomethyl thioether to yield compounds
of Formula (A) wherein one of R or Ri is phenyl substituted with one or more alkylthioalkylthio groups. The metal salt reacts with an independent and varying alkyl chain length halomethyl-[CRRl]-thioalkyl[aryl/heteroaryl] compound to yield the "non-bis" type compounds, [Formula (A)-S-CRR1-S-R2], wherein R and R are as defined above for the "bis" compounds, and R-- is a Cj_9 alkyl, aryl or heteroaryl group which may be optionally substituted. A mixture of Rø and Ri linkages is contemplated, as part of the present invention, however, preferably the linkage is on both Rø positions of the compounds of Formula (A).
An alternate method of preparation of the nonsymmetrical disulfide compound, wherein only one component is a compound of Formula (A), and the other half of the disulfide link is an alkyl, aryl or heteroaryl derivative, may be prepared by reaction of a sulfhydryl compound of Formula (A), with the appropriate sulfenyl halide, in an ethereal solvent to afford compounds of Formula (A) wherein one of R or R is phenyl substituted with one or more [alkyl]- dithio groups, i.e. [Formula (A)-S-S-R2], wherein R-R2 are as defined in the above paragraph. The contemplated sulfenyl halide derivatives of alkyl, aryl, or heteroaryl groups may be optionally substituted.
The disulfide compound(s) may also be prepared from the corresponding alkyl sulfoxide compounds, such as methylsulfinyl, propylsulfinyl, iso-propylsulfinyl, wherein the alkyl can be a straight chain or branched derivative having from 1 to 9 carbon atoms, in a solvent, preferably a chlorinated one such as chloroethylene, methylene chloride or
' chloroform, to which is added a carboxcylic acid anhydride, such as trifluroacetic anhydride, or acetic anhydride. The Pummerer rearrangement reaction may require some heating prior to addition of an alkali metal hydroxide, such as sodium hydroxide. If acetic anhydride is used than heating is also likely to be needed during the hydroxide treatment, before addition of iodine solid (I2), which then affords the symmetrical disulfide compound as is noted above.
Mixtures of the sulfoxide compounds may be present in the solution to yield "symmetrical" compounds but with varying substituent groups on the di-heteroaryl-imidazole ring system of the present invention.
The compounds of Formula (A) are represented by the structure:
Wi is -(CR4R5)-, or -(CR4R5MCR6R7)- ;
R2, R3, R4, R5, R6> R7. R8. and R9 are, independently, -H or Cχ_2 alkyl;
one of R and Rø is 4-pyridyl or Cχ.4 alkyl-4-pyridyl; and the other of R and
Rø is
(a) phenyl or monosubstituted phenyl wherein said substituent is Cχ_4 alkyl, halo, hydroxy, Cχ_4 alkoxy, Cχ_3 alkylthio, Cχ_3 alkylsulfinyl, Cχ-3 alkylsulfonyl, C2.5 1-alkenyl-l-thio, C2.52-alkenyl-l-thio, C2.5 1- alkenyl-1-sulfinyl, C2-.52-alkenyl-l-sulfinyl, C2-.5 1-alkenyl-l-sulfonyl, C3. 52-alkenyl-l-sulfonyl, Cχ.3 alkylamino, Cχ.3 dia-lkylamino, CF3, N-(Cχ. 3alkanamido), N-(Cχ_3 alkyI)-N-(Cχ_3alkanamido), N-pyrroIidino, N- piperidino, prop-2-ene-l-oxy, 2,2,2-trihaloethoxy, thiol, acylthio, dithioacyl, thiocarbamyl, dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, phenylthio, phenylsulfinyl, alkoxyalkylthio, alkoxyalkylsulfinyl alkylthioalkylthio, Z, or acyloxyalkylthio;
(b) disubstituted phenyl wherein said substituents are, independently, Cχ.3 alkylthio, Cχ_3 alkoxy, halo, Cχ.4 alkyl, Cχ_3 alkylamino, N- (Cχ_3alkyl)-N-(Cχ_3 alkanamido), Cχ.3 dialkylamino, amino,
N-pyrrolidino or N-piperidino;
(c) disubstituted phenyl wherein one of said substituents is Cχ.3 alkoxy, halo, Cχ_4 alkyl or CF3, and the other substituent is thiol, alkylsulfinyl, acylthio, dithioacyl, thiocarbamyl, dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, phenylthio, phenylsulfinyl, alkoxyalkylthio, alkoxyalkylsulfinyl, alkylthioalkylthio, Z, or acyloxyalkylthio; or
(d) disubstituted phenyl wherein one of said substituents is amino, Cχ-3 alkylamino or Cχ_3 dialkylamino; and the other substituent is Cχ.3 alkylsulfinyl, C2-.5 -1-alkenyl-l-thio, C2-.5 1-alkenyI-l-sulfinyl, C3.52- alkenyl-1-thio, C3.52-alkenyl-l- sulfinyl, thiol, acylthio, dithioacyl, thiocarbamyl, dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, phenylthio, phenylsulfinyl, alkoxyalkylthio, alkoxyalkylsulfinyl, all-ylthioalkylthio, Z, or acyloxyalkylthio; or
(e) disubstituted phenyl wherein said substituents are the same and are selected from halo, Cχ_3 alkoxy, Cχ.3 alkylamino, Cχ_3 dialkylamino, N- pyrrolidino, N-piperidino, 2,2,2-trihaloethoxy, prop-2-ene-l-oxy, hydroxy, Cχ_3 alkylthio, Cχ_3 alkyl-sulfonyl, thiol, acylthio, dithioacyl, thiocarbamyl , dithiocarbamyl, alkylcarbonylalkylthio, carbalkoxyalkylthio, alkoxycarbonylthio, alkoxythionothio, phenylthio, phenylsulfinyl, alkoxyalkylthio, alkoxyalkylsulfinyl , alkylthioalkylthio, or Z,
(f) a moiety of one of the Formulae:
wherein t is 0 or 1; wherein W
a, and Rx - R9 are as defined above; or a pharmaceutically acceptable salt thereof.
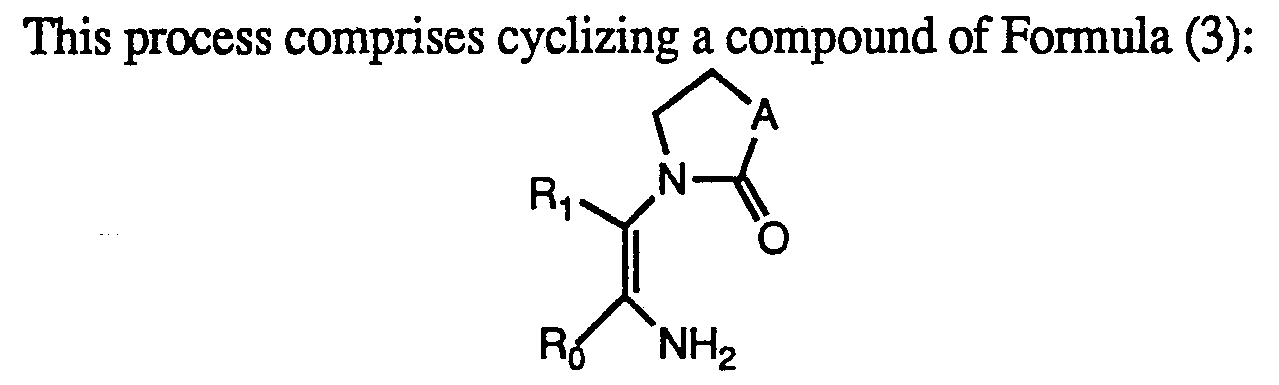
Alternatively the compounds of Formula (A) can be preferably be prepared as outlined in the schematic below. While only a five membered pyrrole is shown the synthesis is also applicable to the six membered nitrogen containing ring. The desired R2-R9 alkyl substituted compounds of Formula (A) are prepared from the correspondingly R2-R9 substituted compounds of Formula (3).
Formula (3) wherein A is (CH2)n and n is 1 or 2; Rx and Rø are as defined for Formula (I) herein. Preferably Rø is a phenyl substituted by a Cχ-4 alkylthio, halogen, Cχ-4 alkyl, or Cx- 4 alkoxy.
The compounds of Formula (3) are prepared by reacting the compounds of Formula (1) and (2):
Formula (1) Formula (2) Formula (3)
Suitable bases include alkyl lithiums such as but not limited to, n-butyl lithium, potassium t-butoxide, lithium diisopropylamide, lithium hexamethylsilylazide, sodium or potassium hydride or potassium hydroxide optionally with a phase transfer catalyst such as tetraethylammonium bromide, or a suitable mixture thereof, e.g. n-butyl-lithium and potassium t-butoxide. Conveniently a compound of Formula (1) is reacted with 1 to 2 mole equivalents, preferably 1.4 to 1.7 mole equivalents of the base before treatment with a compound of Formula (2).
The reaction to form a compound of Formula (3) is in an organic solvent, such as but not limited to, THF, dialkylether, dimethylformamide, toluene, dimethylethylideneurea or tetramethylethylenediamine or a suitable mixture thereof. The reaction should be performed within a temperature range of about -80°C to about 100°C. Preferably the reaction is cooled initially and the temperature is raised to optimize the reaction time of the process.
The compounds of Formula (3) may be isolated on workup and then cyclized to a compound of the Formula (A) with a suitable base as hereinbefore described. An example of such preparation can be found in synthetic Example 3.
Preferably, the compound of the Formula (3) is not isolated, but is formed in situ and cyclized directly to a compound of the Formula (A) under the basic conditions of the reaction mixture. An example of such preparation can be found in synthetic Example 4.
Compounds of Formula (1) are prepared by reacting in the presence of a base a compound of Formula (4), or an acid salt thereof:
RχCH2 Formula (4) wherein Rx is as hereinbefore defined, and L is a suitable leaving group, with a compound of Formula (5)
Formula (5) wherein A is as defined above for Formula (3).
Examples of suitable bases include but are not limited to, potassium carbonate, sodium hydride, sodium hydroxide or lithium diisopropylamide. Suitable leaving groups (L) are well known to those skilled in the art, and include halogens, such as bromine or chloride, oratosylateormesylatemoiety.
The reaction is performed in a solvent, preferably THF, DMF, or mixtures thereof. The reaction may optionally be performed in the presence of water in appropriate cases, where for example when using solid potassium hydroxide together with a phase transfer catalyst as the base. The reaction is conveniently performed at ambient or slightly elevated temperatures. Preferably an aqueous solution of an acid addition salt of a compound
of the Formula (4) is gradually added to a solution of a compound of the Formula (5) and the base.
The compounds of Formula (A) may be themselves used as intermediates to produce other compounds of Formula (A) and such preparations are well described in Bender et al.. U.S. Patent Application Serial Number 07/255,816, filed October 11, 1988; Bender e_t a , U.S. Patent Number 4,175,127, issued November 20, 1979; Bender et al.. U.S. Patent Application Serial Number 07/106,199 filed on July 10, 1987; Bender et al.. U.S. Patent Number 4,803,279, issued February 9, 1989, Bender et al.. U.S. Patent Number 4,719,218, issued January 12, 1988; Bender et al.. U.S. Patent Number 4,715,310, issued January 14, 1988 the entire disclosures of all of which are hereby incorporated by reference.
Compounds of Formula (A) wherein Rø or Ri is a mono- or di-substituted phenyl having a C^alkylsulfinyl, or Cχ.3 alkenylsulfinyl; or wherein R or R- is a di¬ substituted phenyl having at least one

or Cχ_3alkenyl-sulfinyl; or wherein R or Ri is a mono- or di-substituted phenyl having at least one acyloxyalkylsulfinyl, alkoxyalkylsulfinyl or phenyl-sulfinyl substituent are prepared by treatment with appropriate oxidative procedures well known to those skilled in the art and additionally can be found in Bender et al.. U.S. Patent Application Serial Number 07/255,816, filed October 11, 1988; Bender et al.. U.S. Patent Number 4,175,127, issued November 20, 1979; Bender et al.. U.S. Patent Application Serial Number 07/106,199, filed on July 10, 1987; Bender etal.. U.S. Patent Number 4,803,279, issued February 9, 1989, Bender et al.. U.S. Patent Number 4,719,218, issued January 12, 1988; Bender et al.. U.S. Patent Number 4,715,310, issued January 14, 1988; and in Adams et al.. US Patent Application Serial Number 07/537,195, filed June 12, 1990, Attorney's Docket Number SB 14506. Preferably the oxidation is by use of potassium persulfate procedure as described in Adams et al.. USSN
07/537,195, filed June 12, 1990, Attorney's Docket Number SB 14506, whose disclosure is herein incorporated by reference.
The compounds of Formula (A) are used as intermediates to form a 7-hydroxyl or 7 keto moiety by analogous preparation to the methods disclosed in Gallagher et al.,
Tetrahedron Letters. Vol. 30, No. 48, pp. 6599-6602 (1989) the entire disclosure of which is hereby incorporated by reference. The 7-OH and 7-keto comχ>ounds are then used as intermediates to make the final compounds of Formula (I).
The 7-position compounds of Formula (A) containing a 7-hydroxyl or 7-oxo are converted to the protected hydroxyls of Formula (I) or (II) by the schematic illustrated below. The ketal, and 7-position di-subsubstituted compounds are illustrated in the schematic below as well.
X(OH)2, mineral or Lewis acid cat.
SCHEME I Compounds 1 and 2 are prepared, as stated above, by the methods outlined in
Tetrahedron Letters, Gallagher et al.. supra. In scheme I above, the conversion of compound 1 to compound 4 can be accomplished by reaction of compound 1 with an appropriate diol, herein referred to as X(OH)2, using acid catalysts well known to those skilled in the art. Catalysis is preferably conducted with a Lewis acid, for example boron trifluoride etherate, a mineral acid such as HQ, p-toluene suphonic acid, or titanium tetrachlσride; see Greene,T., Protective Groups in Organic Synthesis, Wiley Publishers, p. 116-128 for additional agents. The diol X(OH)2 is a generic formula for X which preferably contains 2 to 3 carbons in a chain which may additionally alkyl substituted, thereby providing for branched diols. Suitable examples would be a 1,2-ethanediol or a l,3-(2-methyl)propandiol moiety. The conversion of compound 1 to 5 can be accomplished using a variety of organometallic reagents which are known to undergo nucleophilic additions to the carbonyl
containing compounds. Examples of such reagents are a suitably substituted organo- magnesium (grignard reagents), -titanium, or -cerium reagents.
The requisite organometallic reagents are either known or are readily available by adaptation of published procedures. The ether compounds 3 and 6 are prepared from compound 2 or 5 respectively using a base catalyzed alkylation (known as the Williamson ether synthesis) when Rio is alkyl or an (hetero)aryl substituted alkyl. Typical alkylation conditions would employ an alkali metal hydride in a dipolar aprotic solvent or an ethereal solvent which is added to an alkyl halide although other properly activated alkylating agents such as mesylates or tosylates may also suffice. The use of an alkali metal alkoxide of a sterically hindered alcohol, for example potassium t-butoxide in t-butanol is also commonly employed. In cases where Rio is aryl the use of a metal catalysis such as cupric oxide, is commonly employed to effect the Ullman reaction for the synthesis of aryl ethers.
More particularly the synthesis of compounds 5 and 6 wherein R5 is a functionalized aryl containing a halo, amino, cyano or carboxy group of Formula (I) and (II) may be prepared from the corresponding benzostabase protected halo aniline (Bonar-Law et al., Tetrahedron Letters. Vol. 31, p 6721 (1990)), see scheme II below. After formation of the organomettalic (illustrated below by lithiation and addition of CeCl3 to form the organocerium) the addition of compound 1 affords the corresponding carbonyl addition product 7. This product may then be converted to the ether moiety, compound 8, using a base initiated alkylation reaction.
Treatment of either compound 7 or 8 with dilute mineral acid or an appropriate source of nucleophilic flouride ion affords the deprotected anilines. These anilines may be diazotized under standard conditions to produce diazonium salts which are then reacted with either halo anions, cyanide or carbon monoxide and the requisite catalyst to afford the halo, cyano or carboxy substitution products respectively. For additional procedural information, see March, J., Advanced Organic Chemistry. 3rd Ed., (1985), pages 646 to 649 (Wiley- Interscience Publishers).
The anilines may also be converted to mono- or di-alkyl amines by reaction with an alkyl halide or by acylation to the amide. Alternatively, when the dialkyl amine is the desired product the preferred route would be to begin with corresponding halo dialkylaniline, forming the organome J'ic reagent directly without the use of the benzostabase protecting group. Similarly when compound 6 is an alkoxy substituted aryl, the alkoxy aryl bromide or iodide may be used to prepare the organometallic reagent which is then added to compound 1. The preparation of all the remaining compounds of Formula (I) and (II) not described herein can be readily achieved as the techniques are well known and can be carried out by one of skill in the art according to the procedures outlined above or in the Examples, infra.
R4 = CO2H, Halo, cyano
SCHEME II
Pharmaceutically acceptable salts and their preparation are well known to those skilled in pharmaceuticals. Pharmaceutically acceptable salts of the compounds of Formula (I) which are useful in the present invention include, but are not limited to hydrochloride, hydrobromide, sulfate or phosphate salts. Preferred pharmaceutically acceptable salts of the compounds of Formula (I) and (II) can be prepared by known techniques such as the method of Bender et al.. U.S. Patent 4,175,127, issued November 20, 1979 the disclosure of which is hereby incorporated by reference.
The compounds of the present invention may contain one or more asymmetric carbon atoms and may exist in racemic and optically active forms. All of these compounds are contemplated to be within the scope of the present invention.
METHOD OF TREATMENT
All of the compounds of Formulas (I) are useful in the methods of the subject invention, i.e. methods of treating an OPUFA disease state, specifically by inhibition of the 5- LO and CO enzymes, and the compounds of Formula (H) are useful for inhibiting cytokines, specifically the production of the IL-1 or TNF in an animal, including humans, in need thereof.
The oxidation of OPUFA's, specifically the arachidonic acid metabolic pathway leading to inflammatory mediators, can be controlled by the 5-LO enzyme, amongst others. That the compounds of Formulas (I) are inhibitors of the 5-lipoxygenase pathway is based on the effects of said compounds on the production of 5-lipoxygenase products in blood gx vivo and on the 5-lipoxygenase in vitro assays, some of which are described hereinafter. The 5-lipoxygenase pathway inhibitory action of the compounds of Formulas (I) may be confirmed by showing that they impair the production of 5-lipoxygenase products such as leukotriene B4 production by RBL-1 cell supernatants.
The pathophysiological role of arachidonic acid metabolites has been the focus of recent intensive studies. In addition to the well-described phlogistic activity (i.e. general inflammatory activity) of prostaglandins, the more recent description of similar activity for eicosanoids has broadened the interest in these products as mediators of inflammation. These mediators produce inflammatory conditions such as rheumatoid arthritis, osteoarthritis, bronchial inflammation, inflammatory bowel disease, ulcerative colitis, asthma, cardiovascular disorders, glaucoma, emphysema, acute respiratory distress syndrome, lupus, gout, psoriasis, dermatitis, pyresis, pain and other allergic oriented disorders such as allergic rhinitis, allergic conjunctivitis, food allergies, and uticaria. Additional conditions such as blood platelet aggregation, and notably conditions resulting from thrombosis, including total or partial thrombosis, coronary thrombosis, phlebitis and phlebothrombosis are also implicated in the arachidonic acid pathway. Other disease states for which a 5-LO inhibitor would be useful is in the treatment
of myocardial infarctions, rejection of organ transplants, tissue trauma, multiple sclerosis, atherosclerosis, vasculitis, glomerulo-nephritis, and immune complex disease, as well as use in the optical areas, particularly for general inflammation of the cαrneal anterior and posterior segments due to disease or surgery, such as post surgical inflammation or uveitis. The compounds of Formula Q) are also useful for treating disease states mediated by the cyclooxygenase pathway metabolism of arachidonic acid in an animal, including humans, in need thereof. That the compounds of Formula (I) are inhibitors of cyclooxygenase products is based upon assays which effect the production of the PGE2 products, and assays with human monocytes, the assays of which are described herein. The disease states associated with the CO metabolic pathway are typically those considered for the non-steroidal antϋnflammatory drugs (nsaids), whose primary mode of action is by CO inhibition. The primary diseases of interest, but not limited thereto, are the various arthritic conditions, pyresis and pain.
Interleukin-1 QL-l) has been demonstrated to mediate a variety of biological activities thought to be important in immunoregulation and other physiological conditions such as inflammation [See, e.g., Dinarello et al., Rev. Infect. Disease. 6, 51 (1984)]. The myriad of known biological activities of IL-1 include the activation of T helper cells, induction of fever, stimulation of prostaglandin or collagenase production, neutrophil chemotaxis, induction of acute phase proteins and the suppression of plasma iron levels. The compounds of Formulas QI) are useful as inhibitors of cytokines, specifically IL-1. The inhibitory activity of a compound of Formula (Q) on the production of the IL-1 in vitro, on the human monocyte, may be determined as described in an assay herein.
There are many disease states in which excessive or unregulated IL-1 production is implicated in exacerbating and/or causing the disease. These include riieumatoid arthritis, osteoarthritis, endotoxemia and/or toxic shock syndrome, other acute or chronic inflammatory disease states such as the inflammatory reaction induced by endotoxin or inflammatory bowel disease; tuberculosis, atherosclerosis, muscle degeneration, cachexia, psoriatic arthritis, Reite s syndrome, rheumatoid arthritis, gout, traumatic arthritis, rubella arthritis, and acute synovitis. Recent evidence also links IL-1 activity to diabetes and pancreatic β cells.
Dinarello, J. Clinical Immunology.5 (5), 287-297 (1985), reviews the biological activities which have been attributed to IL-1. It should be noted that some of these effects have been described by others as indirect effects of IL-1. The discovery of a compound which specifically inhibits TNF production will not only contribute to the understanding of how this molecule is synthesized, processed and secreted, but will also provide a therapeutic approach for diseases in which excessive or unregulated TNF production is implicated.
Excessive or unregulated TNF production is implicated in mediating or exacerbating a number of diseases including rheumatoid arthritis, rheumatoid spondylitis, osteoarthritis, gouty arthritis and other arthritic conditions; sepsis, septic shock, endotoxic shock, gram negative sepsis, toxic shock syndrome, adult respiratory distress syndrome, cerebral malaria, chronic pulmonary inflammatory disease, silicosis, pulmonary sarcoidosis, bone resorption diseases, reperfusion injury, graft vs. host reaction, allograft rejections, fever and myalgias due to infection, such as influenza, cachexia secondary to infection or malignancy, cachexia, secondary to acquired immune deficiency syndrome (AIDS), AIDS, ARC (AIDS related complex), keloid formation, scar tissue formation, Crohn's disease, ulcerative colitis, or pyresis.
AIDS results from the infection of T lymphocytes with Human Immunodeficiency Virus (HIV). At least three types or strains of HIV have been identified, i.e., HIV-1, HIV-2 and HIV-3. As a consequence of HIV infection, T-cell mediated immunity is impaired and infected individuals manifest severe opportunistic infections and/or unusual neoplasms. HIV entry into the T lymphocyte requires T lymphocyte activation. Other viruses, such as HtV-l, HIV-2 infect T lymphocytes after T Cell activation and such virus protein expression and/or replication is mediated or maintained by such T cell activation. Once an activated T lymphocyte is infected with HIV, the T lymphocyte must continue to be maintained in an activated state to permit HIV gene expression and/or HIV replication. Monokines, specifically TNF, are implicated in activated T-cell mediated HTV protein expression and/or virus replication by playing a role in maintaining T lymphocyte activation. Therefore, interference with monokine activity such as by inhibition of monokine production, notably TNF, in an HTV-infected individual aids in limiting the maintenance of T cell activation, thereby reducing the progression of HIV infectivity to previously uninfected cells which results in a slowing or elimination of the progression of immune dysfunction caused by
HIV infection. Monocytes, macrophages, and related cells, such as kupffer and glial cells, have also been implicated in maintenance of the HIV infection. These cells, like T-cells, are targets for viral replication and the level of viral replication is dependent upon the activation state of the cells. [See Rosenberg et al.. The Immunopathogenesis of HTV Infection, Advances in Immunology, Vol. 57, (1989)]. Monokines, such as TNF, have been shown to activate HIV replication in monocytes and/or macrophages [See Poli, et al.. Proc. Natl. Acad. Sci., 87:782-784 (1990)], therefore, inhibition of monokine production or activity aids in limiting HIV progression as stated above for T-cells. Additional studies have identified TNF- α as a common factor in the activation of HTV in vitro and has provided a clear mechanism of action via the nuclear factor KB, a nuclear regulatory protein found in the cytoplasm of cells
(Osborn, et al., PNAS (86) 2336-2340). This evidence suggests that a reduction of TNF synthesis may have an antiviral effect in HIV infections, by reducing the transcription and thus virus production.
TNF has also been implicated in various roles with other viral infections, such as the cytomegalia virus (CMV), influenza virus, adenovirus, and the herpes family of viruses, such as Herpes Zoster and Herpes Simplex I and II, for similar reasons as those noted above. TNF also alters the properties of endothelial cells and has various pro- coagulant activities, such as producing an increase in tissue factor pro-coagulant activity and suppression of the anticoagulant protein C pathway as well as down-regulating the expression of thrombomodulin. TNF also has pro-inflammatory activities which together with its early production (during the initial stage of an inflammatory event) make it a likely mediator of tissue injury in several important disorders including but not limited to, myocardial infarction, stroke and circulatory shock. Of specific importance may be TNF-induced expression of adhesion molecules, such as intercellular adhesion molecule (ICAM) or endothelial leukocyte adhesion molecule (ELAM) on endothelial cells.
TNF is also believed to be an important mediator of many other inflammatory states or diseases. Therefore, inhibitors of TNF production would have utility in any inflammatory state or disease in which abnormal levels of TNF are produced. Abnormal levels of TNF constitute levels of 1) free (not cell bound) TNF, greater than or equal to 1 picogram per ml; 2) any cell associated TNF; or 3) the presence of TNF mRNA above basal levels in cells or tissues in which TNF is produced. In addition, the present invention attributes many biological disease states noted herein to IL-1 activity. These disease states are also considered appropriate disease states of TNF activity and hence compounds of Formulas (H) are also useful in their treatment as well, and should not be considered solely a -limitation to IL-1 activity alone.
It has also been discovered that the compounds of Formulas (II) are useful for treating disease states mediated by the cytokine TNF in an animal, including mammals, in need thereof. The inhibitory effect of a compound of Formulas (H) on the production of the TNF in_- vitro, on the human monocyte, may be determined by the assay which is described herein.
PHARMACEUTICAL COMPOSITIONS
This invention further relates to the use of a compound of Formula T) or a pharmaceutically acceptable salt thereof in the manufacture of a medicament for the treatment of prophylactically or therapeutically, any disease state in an animal, including humans, which is caused by or exacerbated by OPUFA metabolizing enzymes, such as 5-LO or CO. This invention further relates to the use of a compound of Formula (II), or pharmaceutically acceptable salts thereof in the manufacture of a medicament for the treatment of prophylactically or therapeutically, any disease state in an animal, including humans, which is exacerbated or caused by excessive or unregulated IL-1, or TNF production.
This invention also relates to a pharmaceutical composition comprising an effective, non-toxic amount of a compound of Formulas (I) or (II) and a pharmaceutically acceptable carrier or diluent The compounds of Formula (I) and (II) are administered in conventional dosage forms prepared by combining a compound of Formula (I) and (II) with standard pharmaceutical carriers according to conventional procedures. The compounds of Formula (I) and (II) may also be administered in conventional dosages in combination with a known, second therapeutically active compound. These procedures may involve mixing, granulating and compressing or dissolving the ingredients as appropriate to the desired preparation. The pharmaceutical carrier employed may be, for example, either a solid or liquid. Exemplary of solid carriers are lactose, terra alba, sucrose, talc, gelatin, agar, pectin, acacia, magnesium stearate, stearic acid and the like. Exemplary of liquid carriers are syrup, peanut oil, olive oil, water and the like. Similarly, the carrier or diluent may include time delay material well known to the art, such as glyceryl mono-stearate or glyceryl distearate alone or with a wax.
A wide variety of pharmaceutical forms can be employed. Thus, if a solid carrier is used, the preparation can be tableted, placed in a hard gelatin capsule in powder or pellet form or in the form of a troche or lozenge. The amount of solid carrier will vary widely but preferably will be from about 25 mg. to about 1 g. When a liquid carrier is used, the preparation will be in the form of a syrup, emulsion, soft gelatin capsiile, sterile injectable liquid such as an ampule or nonaqueous liquid suspension.
To obtain a stable water soluble dose form of an insoluble Formula (I) or (II) compound, a pharmaceutically acceptable salt of the Formula (I) or (II) compound is dissolved in an aqueous solution of an organic or inorganic acid, such as a 0.3 M solution of succinic acid or citric acid.
All applicable dosage ranges, formulations, applications, i.e. topical, oral, parenteral, etc. apply equally to the compounds of Formulas (I) and (II).
The compounds of Formula (I) may be administered topically. Thus, the compounds of Formula (I) may be administered topically in the treatment or prophylaxis of inflammation in an animal, including man and other mammals, and may be used in the relief or prophylaxis of 5-lipoxygenase pathway mediated diseases such as rheumatoid arthritis, rheumatoid spondylitis, osteoarthritis, gouty arthritis and other arthritic conditions, inflamed joints, eczema, psoriasis or other inflammatory skin conditions such as sunburn; inflammatory eye conditions including conjunctivitis; pyresis, pain and other conditions associated with inflammation. For those disease states indicated above which are also mediated by a cytokine the compounds of Formula (II) may be administered topically. The amount of a compound of Formula (I) or (II), for all methods of use disclosed herein, required for therapeutic effect on topical administration will, of course, vary
with the coπφound chosen, the nature and severity of the inflammatory condition, whether eicosanoid or cytokine mediated, and the animal undergoing treatment, and is ultimately at the discretion of the physician. A suitable, topical, anti-inflammatory dose of an active ingredient, ϊ.e., a compound of Formula Q) or QI) is 0.1 mg to 150 mg, administered one to four, preferably two or three times daily.
By topical administration is meant non-systemic administration and includes the application of a compound of Formula Q) or QJ) externally to the epidermis, to the buccal cavity and instillation of such a compound into the ear, eye and nose, and where the compound does not significantly enter the blood stream. By systemic administration is meant oral, intravenous, ήitraperitoneal and intramuscular administration.
While it is possible for an active ingredient to be administered alone as the raw chemical, it is preferable to present it as a pharmaceutical formulation. The active ingredient may comprise, for topical administration, from 0.001% to 10% w/w, e.g. from 1% to 2% by weight of the formulation although it may comprise as much as 10% w/w but preferably not in excess of 5% w/w and more preferably from 0.1% to 1% w/w of the formulation.
The topical formulations of the present invention comprise an active ingredient together with one or more acceptable carrier(s) therefor and optionally any other therapeutic ingrεdient(s). The caπier(s) must be 'acceptable' in the sense of being compatible with the other ingredients of the formulation and not deleterious to the recipient thereof. Formulations suitable for topical administration include liquid or semi-liquid preparations suitable for penetration through the skin to the site of inflammation such as liniments, lotions, creams, ointments or pastes, and drops suitable for administration to the eye, ear or nose.
Drops according to the present invention may comprise sterile aqueous or oily solutions or suspensions and may be prepared by dissolving the active ingredient in a suitable aqueous solution of a bactericidal and/or fungicidal agent and/or any other suitable preservative, and preferably including a surface active agent. The resulting solution may then be clarified by filtration, transferred to a suitable container which is then sealed and sterilized by autoclaving or maintaining at 98-10OC. for half an hour. Alternatively, the solution may be sterilized by filtration and transferred to the container by an aseptic technique. Examples of bactericidal and fungicidal agents suitable for inclusion in the drops are phenylmercuric nitrate or acetate (0.002%), benzalkonium chloride (0.01%) and chlorhexidine acetate (0.01%). Suitable solvents for the preparation of an oily solution include glycerol, diluted alcohol and propylene glyco Lotions according to the present invention include those suitable for application to the skin or eye. An eye lotion may comprise a sterile aqueous solution optionally containing a bactericide and may be prepared by methods similar to those for the preparation of drops. Lotions or liniments for application to the skin may also include an agent to hasten drying and
to cool the skin, such as an alcohol or acetone, and/or a moisturizer such as glycerol or an oil such as castor oil or arachis oil.
Creams, ointments or pastes according to the present invention are semi-solid formulations of the active ingredient for external application. They may be made by mixing the active ingredient in finely-divided or powdered form, alone or in solution or suspension in an aqueous or non-aqueous fluid, with the aid of suitable machinery, with a greasy or non- greasy basis. The basis may comprise hydrocarbons such as hard, soft or liquid paraffin, glycerol, beeswax, a metallic soap; a mucilage; an oil of natural origin such as almond, corn, arachis, castor or olive oil; wool fat or its derivatives, or a fatty acid such as steric or oleic acid together with an alcohol such as propylene glycol or macrogels. The formulation may incorporate any suitable surface active agent such as an anionic, cationic or non-ionic surfactant such as sorbitan esters or polyoxyethylene derivatives thereof. Suspending agents such as natural gums, cellulose derivatives or inorganic materials such as silicaceous silicas, and other ingredients such as lanolin, may also be included. The methods of the subject invention may be carried out by delivering the monokine activity interfering agent parenterally. The term 'parenteral' as used herein includes intravenous, intramuscular, subcutaneous intranasal, intrarectal, intravaginal or intraperitoneal administration. The subcutaneous and intramuscular forms of parenteral administration are generally preferred. Appropriate dosage forms for such administration may be prepared by conventional techniques.
For all methods of use disclosed herein, for the compounds of Formulas (I) and (II), the daily oral dosage regimen will preferably be from about .1 to about 80 mg/kilogram of total body weight, preferably from about .5 to 30 mg kg, more preferably from about lmg to 15mg. The daily parenteral dosage regimen will preferably be from about .1 to about 80 mg per kilogram (kg) of total body weight, preferably from about .5 to about 30 mg kg, and more preferably from about lmg to 15mg/kg.
The compounds of Formula (I) and (II) may also be administered by inhalation. By "inhalation" is meant intranasal and oral inhalation administration. Appropriate dosage forms for such administration, such as an aerosol formulation or a metered dose inhaler, may be prepared by conventional techniques. The preferred daily dosage amount of a compound of Formula (I) administered by inhalation for all methods disclosed herein, is from about .01 mg kg to about 1 mg kg per day.
It will be recognized by one of skill in the art that the form and character of the pharmaceutically acceptable carrier or diluent is dictated by the amount of active ingredient with which it is to be combined, the route of administration and other well-known variables. It will also be recognized by one of skill in the art that the optimal quantity and spacing of individual dosages of a compound of Formula (I) or (H), or a pharmaceutically acceptable salt thereof will be determined by the nature and extent of the condition being
treated, the form, route and site of administration, and the particular patient being treated, and that such optimums can be determined by conventional techniques.
It will also be appreciated by one of skill in the art that the optimal course of treatment, i.e., the number of doses of a compound of Formula Q and (II) or the pharmaceutically acceptable salts thereof given per day for a defined number of days, can be ascertained by those skilled in the art using conventional course of treatment determination tests.
EXAMPLES Without further elaboration, it is believed that one skilled in the art can, using the preceding description, utilize the present invention to its fullest extent The following Examples are, therefore, to be construed as merely illustrative and not a limitation of the scope of the present invention in any way.
EXAMPLE A Inhibitory Effect of compounds of Formula (I) on in vitro IL-1 Production by Human Monocytes The effects of compounds of Formula (I) on the in vitro production of IL-1 by human monocytes are examined using the following protocol..
Bacterial lipopolysaccharide (LPS) is used to induce IL-1 production by human peripheral blood monocytes. IL-1 activity is measured by its ability to stimulate a Interleukin
2 O-L-2) producing cell line (EL-4) to secrete IL-2, in concert with A23187 ionophore, according to the method of Simon et al., J. Immunol. Methods. 84. 85, (1985). Human peripheral blood monocytes is isolated and purified from either fresh blood preparations from volunteer donors, or from blood bank buffy coats, according to the procedure of Colotta et a , J. Immunol.. 132.936 (1984). 1 X 106 of such monocytes were plated in 24-well plates at a concentration of 1-2 million/ml per well. The cells were allowed to adhere for 2 hours, after which time non-adherent cells is removed by gentle washing. Test compounds were then added to the cells for 1 hour (hr) before the addition of lipopolysaccharide (50 ng ml), and the cultures are incubated at 37°C for an additional 24 hours. At the end of the incubation period, culture supernatants were removed and clarified of cells and all debris. Culture supernatants were immediately assayed for IL-1 biological activity in the manner described above, as well as forprostaglandin and/or leukotriene concentrations by radioimmunoassay. The compound of Example 7 demonstrated an IC50 of 0.29μM.
UTILΠΎ EXAMPLE B
In the tests used to determine activity as 5-lipoxygenase pathway inhibitors, male Balb/c mice (20-28 g), are used. AU mice were obtained from Charles River Breeding Laboratories, Kingston, N.Y. Within a single experiment, mice were age matched.
Reagents were employed as follows:
Compounds of Formula (I) are used as the free base. The compounds were dissolved in acid saline. Compounds are administered by lavage at the indicated dose in a final volume of 10 ml/kg. For in vitro experiments, compounds are dissolved at appropriate concentrations in ethanol (final concentration 1.0%) and then diluted to final concentrations using the buffers indicated in the text
Arachidonic Acid-Induced Mouse Ear Inflammation Arachidonic acid in acetone (2 mg/20 ml) is applied to the inner surface of the left ear. The thickness of both ears is then measured with a dial micrometer one hour after treatment, and the data is expressed as the change in thickness (10"3 cm) between treated and untreated ears.
Test compounds are given orally in acid/saline at the times indicated prior to the topical application of arachidonic acid.
Assay of 5-Lipoχygenase Activities The 5-lipoxygenase (5-LO) is isolated from extracts of RBL-1 cells. These cells are obtained from the American Type Culture Collection (#CRL 1378) and are grown at 37° with 5% CO2 in spinner culture using Eagles essential medium (MEM) supplemented medium with 10% heat inactivated fetal calf serum. The cells were collected from culture by centrifugation at 2,000xg for 20 minutes and then washed twice with 50mM sodium phosphate (pH 7.0) which contains ImM EDTA and 0.1% gelatin. After this wash, the cells were resuspended in fresh phosphate buffer to achieve a concentration of 5X10^ cells/ml. This suspension is disrupted by nitrogen cavitation using the Parr bomb at 750psi for 10 minutes. The broken cells are then centrifuged at 10,000xg for 20 minutes. The supernatant was collected and centrifuged at 100,000 xg for 60 minutes. This supernatant was collected and stored at -70°C until assayed.
The inhibition of 5-lipoxygenase activity is measured by one of two assays, the radiotracer extent assay either measured after 90 seconds at 20°C or measured according to the method of G. K. Hogaboom et al.. Molecular Pharmacol. 30. 510-519 (1986) or the continuous O2 consumption assay. The results from either assay are comparable if not identical. Compounds were dissolved in ethanol with the final concentration of ethanol being 1% in the assay.
The radiotracer extent assay examines the 5-lipoxygenase products [transLTB4 (DI-HETE), 5HETE and 5HPETE] produced after a 90 second incubation at 20°C. Aliquots
(40mL) of the supernatant are preincubated with the inhibitor or vehicle for 10 minutes in 25mM BisTris buffer (pH 7.0) which also contains ImM EDTA, ImM ATP, 50mM NaCl, 5% ethylene gylcol and 100 mg/ml of sonicated phosphatidylcholine (total volume 0.238 ml).
The 5-lipoxygenase reaction is initiated by the addition of CaCl2 (2mM) and 1-C14- arachidonic acid (25mM; 100,000dpm))(final volume 0.25ml). After 90 seconds, the reaction is terminated by the addition of two volumes (0.5ml) of ice chilled acetone. The sample is allowed to deproteinize on ice for 10 minutes prior to centrifuging at 1,000 xg for 10 minutes. The deproteinized supernatants are dried under argon and then redissolved in 200 mL of ethanol. These samples are then analyzed by reverse phase HPLC as described by G.K. Hogaboom et al.. Molecular Pharmacol. 30: 510-519 (1986), herein incorporated by reference. The compound-mediated inhibition of 5-lipoxygenase activity is described as the concentration of compound causing a 50% inhibition of product synthesis. The second assay for assessing inhibition of the 5-lipoxygenase activity is a continuous assay which monitors the consumption of O2 as the reaction progresses. The 5- lipoxygenase enzyme (200mL) is preincubated with the inhibitor or its vehicle in 25mM BisTris buffer (pH 7.0) which contains ImM EDTA, ImM ATP, 5mM NaCl and 5% ethylene glycol for 2 minutes at 20°C (total volume 2.99 ml). Arachidonic acid (lOmM) and CaCl2 (2mM) are added to start the reaction, and the decrease in O2 concentration follows with time using a Clark-type electrode and the Yellow Spring O2 monitor (type 53)(Yellow Springs,
OH). The optimum velocity is calculated from progress curves. The compound mediated inhibition of 5-Iipoxygenase activity is described as the concentration of compound causing a 50% inhibition of optimum velocity for the vehicle-treated sample. The compound of Example 7 demonstrated a 6% inhibition at 20μM/ml.
LTC-4 /PGE2 Production from Human Monocytes in vitro a) Cell Preparation: Human monocytes are prepared from leukosource packs supplied by the American Red Cross (Philadelphia,Pa). The leukosource packs are fractionated by a two-step procedure described by F. Colatta et al., J. Immunol. 132.936 (1984), herein incorporated by reference, that uses sequential sedimentation on Ficoll followed by sedimentation on Percoll. The monocyte fraction which results from this technique is composed of greater than 85% monocytes (with the remainder being neutrophils and lymphocytes). The monocytes (1.5 X 10^) are placed into polypropylene tubes and used as a suspended culture. The assay buffer consisted of RPMI 1640 buffer, [Moore, G. E. et al., JAMA. 199.519 (1967) herein incorporated by reference] 1% human AB serum, 2mM glutamine, 100 U/ml Penicillin/Streptomycin, 25 mM HEPES [4-(2-hydroxyethyl)-l- piperarine-ethanesulfonic acid], and ImM CaCl2- b) LTC4/PGE2 Production: Monocytes (0.9ml/tube) were dispensed into 12 X 75 mm polypropylene tubes (as a suspended culture). Compounds (lOOul of a 10X stock of the compound of interest) dissolved in the assay media are added per tube (performed in duplicate). The cells are incubated for about 45 minutes at about 7°C with constant agitation in a humidified incubator. A23187 calcium ionophore (2uM final concentration) used to
stimulate the cells, is added and the monocytes are incubated an additional 15 minutes. Supernatants are then collected from each tube, clarified by centrifugation, divided into two aliquots and stored at -70°C until assayed. c) Radio-immunoassay: Supernatants are assayed for LTC4 production and PGE2 by radioimmunassay; which is performed using a New England Nuclear Leukotriene [3-HJ-LTC4 and [125rj_PGE2 RIA Kit according to the manufacturer's (New England Nuclear, Boston Massachusetts) instructions. The compound-mediated inhibition of LTC4 is described as the concentration of compound causing a 50% inhibition of LTC4 production.
UTILΠΎ EXAMPLE C
Inhibitory Effect of compounds of Formula (I) on vitro TNF production by Human Monocytes
Section I: Assay set-up The effects of compounds of Formula (II) on the in vitro production of TNF by human monocytes are examined using the following protocol.
Human χ>eripheral blood monocytes are isolated and purified from either blood bank buffy coats or plateletpheresis residues, according to the procedure of Colotta, R. et al., £. Immunol.. 132(2):936 (1984). The monocytes are plated at a density of 1 X 106 cells/ml medium/well in 24-well multi-dishes. The cells are allowed to adhere for 1 hour after which time the supernatant is aspirated and 1 ml fresh medium (RPMI-1640 (Whitaker Biomedical Products, Whitaker, CA) containing 1% fetal calf serum and penicillin and streptomycin at 10 units/ml is added. The cells are incubated for 45 minutes in the presence or absence of test compounds at lnM-lOuM dose ranges (compounds were solubilized in Dimethyl- sulfoxide/Ethanol such that the final solvent concentration in the culture medium is 0.5%
Dimethyl sulfoxide 0.5% Ethanol). Bacterial lipopolysaccharide (E. coli 055:B5 [LPS] from Sigma Chemicals Co.) is then added at 100 ng ml in 10 ml Phosphate Buffered Saline (PBS) and cultures incubated for 16-18 hours at 37°C in a 5% CO2 incubator. At the end of the incubation period, culture supernatants are removed from the cells, centrifuged at 3000 revolutions per minute (rpm) to remove cell debris and .05 ml of the supernatant is assayed for TNF activity using the radioimmunoassay described below.
Section II: Radioimmunoassay procedure for TNF activity
The assay buffer consists of 0.01M NaPO 0.15M NaCl, 0.025M EDTA and 0.1% sodium azide at pH 7.4. Human recombinant TNF (rhTNF) obtained using the procedure of Chen et al., Nature. 330:581-583 (1987) is iodinated by a modified Chloramine-T method described in Section in below. To samples (50 μl culture supernatants) or rhTNF standards, a 1 9000 dilution of polyclonal rabbit anti-rhTNF (Genzyme, Boston, MA) and 8000 cpm of
125ι_χNFis added in a final volume of 400 μl buffer and incubated overnight (18 hours) at 4°C. Normal rabbit serum and goat anti-rabbit IgG (Calbiochem) are titered against each other for maximum precipitation of the anti-rhTNF. The appropriate dilutions of carrier normal rabbit serum (1/200), goat anti-rabbit IgG (1/4) and 25 Units heparin (Calbiochem) are allowed to precipitate and 200 μl of this complex was added per assay tube and incubated overnight at 4°C. Tubes are centrifuged for 30 minutes at 2000 rpm, supernatants are carefully aspirated, and radioactivity associated with the pellets is measured in a Beckman Gamma 5500 counter. The logit-log linear transformation curve is used for the calculations. The concentrations of TNF in the samples are read from a standard curve of rhTNF that is linear in the 157 to 20,000 pg ml range.
Section HI: Radioiodination of rhTNF
Iodination of rhTNF is performed using a modified chloramine-T method of Frolik et al., J. Biol. Chem.. 259:10995-11000 (1984). Briefly, 5 mg of rhTNF in 5 ml of 20MM Tris ph 7.5, is diluted with 15 ml of 0.5M KPO4 and 10 ml of carrier free
To initiate the reaction, a 5ml aliquot of a lOOmg/ml (aqueous) chloramine-T solution is added. After 2 minutes at room temperature, an additional 5 ml aliquot is added followed 1.5 minutes later by a final 5 ml addition of chloramine-T. The reaction is stopped 1 minute later by sequential addition of 20 ml of 50mM Sodium Metabisulfite, 100 ml of 120mM Potassium Iodide and 200 ml of 1.2 mg ml Urea. The contents are mixed and the reaction mixture is passed over a pre-packed Sephadex G-25 column (PD 10 Pharmacia), equilibrated and eluted with Phosphate Buffered Saline pH 7.4 containing 0.25% gelatin. The peak radioactivity containing fractions are pooled and stored at -20°C. Specific activity of
125I-TNF is 80-100 mCi mg protein. Biological activity of iodinated TNF is measured by the L929 cytotoxicity assay of Neale, M.L. et al., Eur. J. Can.
Clin. Oncol..25(1):133-137 (1989) and has been found to be 80% that of unlabeled TNF.
Section IV: Measurement of TNF- ELIS A:
Levels of TNF are also measured using a modification of the basic sandwich ELIS A assay method described in Winston et al., Current Protocols in Molecular Biology.
Page 11.2.1, Ausubel et al., Ed. (1987) John Wiley and Sons, New York, USA The ELIS A employed a murine monoclonal anti-human TNF antibody, described below, as the capture antibody and a polyclonal rabbit anti-human TNF , described below, as the second antibody. For detection, aperoxidase-conjugated goat anti-rabbit antibody (Boehringer Mannheim, Indianopolis, Indiana, USA, Catalog # 605222) is added followed by a substrate for peroxidase (lmg/ml orthophenylenediamine with 0.1% urea peroxide). TNF levels in samples are calculated from a standard curve generated with recombinant human TNF
produced in E. Coli (obtained from SmithKline Beecham Pharmaceuticals, King of Prussia, PA, USA).
Section V: Production of anti-human TNF antibodies: Monoclonal antibodies to human TNF are prepared from spleens of B ALB/c mice immunized with recombinant human TNF using a modification of the method of Kohler and Millstein. Nature 256:495 (1975), the entire disclosure of which is hereby incorporated by reference. Polyclonal rabbit anti-human TNF antibodies are prepared by repeated immunization of New Zealand White (NZW) rabbits with recombinant human TNF emulsified in complete Freund's adjuvant (DIFCO, IL., USA).
S YNTHEΗC EXAMPLES EXAMPLE 1
5.6-Dihvdro-2-(4-methylthiophenyl)-3-(4-pyridinyl)-7H-pyrrolo[1.2-a1imidazol-7-ol (Intermediate 7-ol compound)
a.) 5.6-Dihvdro-2-f 4-methylthiophenylV 3-(4-pyridinyl 7H-pyrrolor 1.2-a] - imidazol-7-ol.
To a solution of 5,6^dihydro-2-(4-fluorophenyl)-3-(4-pyridinyl)-7H-pyrrolo[l,2- a]imidazol-7-ol (0.85 grams (hereinafter g), 2.9 millimoles(hereinafter mmol)) in DMF (10 milliLiters (hereinafter mL)) was added sodium thiomethoxide (0.30 g, 4.4 mmol). The resulting mixture was heated at 120°C for 48 h, then allowed to cool. The mixture was concentrated under reduced pressure, and the residue was partitioned between Η2O and CH2CI2. The organic extract was washed with saturated aqueous NaCl and dried (MgS04). The solvent was removed in vαcuo, and the residue was recrystallized twice from MeOH to provide a light tan solid (0.19 g, 20%). m.p. 229 - 230°C iH NMR (CDCI3) : δ 8.59 (d, 2H); 7.40 (d, 2H); 7.19 (2 overlapping d, 4H); 6.18 (br d, 1H); 5.27 (m, 1H); 4.27 (m, 1H); 3.94 (m, 1H); 2.90 (m, 1H); 2.63 (m, 1H); 2.50 (s, 3H). CIMS (NH3); m/e (rel. int.) : 324 [(M+H)+, 100], 308 (11).
Anal. Calc. for C18HX7N3OS : C 66.85, H 5.30, N 12.99, S 9.91; found : C 66.78, H 5.55, N 12.95, S 9.58.
EXAMPLE 2 5.6-Dihvdro-2-(4-fluorophenylV3-('4-DyridinylV7H-pyπOlori.2-a1imidazol-7-ol
(Intermediate 7-ol compound)
a l-f5.6-Dihvdro-2-f4-fluorophenvD-3-(4-pyridinvn-7H-pyrrolon.2-a1-
imidazol-7-yl 1 - l-(4-nitrophenyl methanol.
To a solution of 5,6-dihydro-2-(4-fluorophenyl)-3-(4-pyridinyl)-7H-pyrrolo[l,2- a]imidazole (15.0 g, 0.054 moles (hereinafter mol)) in CH2CI2 (50 mL) at 0°C was added methoxyethoxymethyl chloride (30 mL, 0.26 mol). The resulting mixture was allowed to warm to room temperature and stirred for 1 hour (hereinafter h). Ether was added, and the mixture was decanted (3x). The residue was dissolved in EtOH (400 mL), and to this solution were added triethylamine (40 mL, 0.29 mol) and 4-nitrobenzaldehyde (15.0 g, 0.10 mol). The resulting mixture was heated at reflux for 48 h, then allowed to cool and concentrated under reduced pressure. The residue was partitioned between H2O and CH2CI2. The organic extract was washed with saturated aqueous NaCl and dried (MgS04). The solvent was removed in vacuo, and the residue was triturated with EtOAc. The orange solid which formed was collected by filtration to afford the title compound (8.0 g, 34%) which was used without further purification.
b) 5.6-Dihydro-2-r4-fluorophenylV3-r4-pyridinyl)-7H-pyrτolori.2-alimidazol-7-one.
To a solution of Jones reagent (25 mL) in acetone (250 mL) was added l-{5,6-dihydro-2-(4- fluorophenyl)-3-(4-pyriά nyl)-7H-pyπolo[l,2-a]imidazol-7-yl}-l-(4-nitrophenyl)methanol (5.0 g, 12 mmol). The resulting mixture was stirred at room temperature for 30 min, then the pΗ was adjusted to 7 - 8 with 2.5 N NaOΗ. The solid material was removed from the acetone solution by decantation and partitioned between 2.5 N NaOΗ and 1 : 2 CΗ2CI2/
Et2θ. This mixture was filtered, and the layers were separated. The organic extract was combined with the acetone solution and evaporated under reduced pressure. The residue was partitioned between 2.5 N NaOH and CH2CI2, and the organic extract was washed with saturated aqueous NaCl and dried (MgS04). The solvent was removed in vacuo, and the residue was triturated with Et2θ to provide the title compound as an orange solid (1.5 g, 43%), which was used without further purification.
c) 5.6-Dihydro-2-(4-fluorophenyl)-3-(4-pyridinyl -7H-pyrrolo-ri.2-a]imidazol-7-ol. To a solution of 5,6^dihydro-2-(4-fluorophenyl)-3-(4-pyridinyl)-7-H-pyrrolo[l,2- a]imidazol-7-one (crude product prepared above) in MeOH (15 mL) was added sodium borohydride (1.5 g, 40 mmol), and the resulting mixture was stirred at room temperature for 15 min. The mixture was concentrated under reduced pressure, and the residue was partitioned between H2O and CH2CI2. The organic extract was washed with saturated aqueous NaCl and dried (MgSθ4). The solvent was removed in vacuo, and the residue was triturated sparingly with EtOAc and copiously with Et2θ. The solid which formed (0.90 g, 26%) was recrystallized from MeOH to afford the title compound as a white solid. H NMR (DMSO-d6) : 8.58 (d, 2H); 7.45 (dd, 2H); 7.36 (d, 2H); 7.16 (apparent t, 2H); 5.76 (d, 1H); 4.99 (m, 1H); 4.16 (m, 1H); 3.95 (m, 1H); 2.82 (m, 1H); 2.30 (m, 1H).
CIMS (NH3); m/e (rel. int.) : 296 [(M+H)+, 100].
EXAMPLE 3
6.7-Dihvdro-2-(4-methylthiophenyl -3-(4-pyridinyl)-5H-pyrroloπ.2-a]imidazole. (Intermediate compound of Formula (A) a) To a vigorously stirred suspension of potassium hydroxide (341.0g,6.09 mol) and tetraethylammonium bromide (51.2g, 0.24mol) in tetrahydrofuran (TΗF) 2.01) was added 2- pyrrolidinone (97.2 ml, 1.28 mol) at 20°C. A thick white slurry formed and the temperature rose to 27°C within 30 minutes. The reaction mixture was stirred mechanically for a total of 100 minutes between 20-
30°C before 4-picolyl chloride hydrochloride (200.0g, 1.22mol) in demineralized water (120ml) was added over 25 minutes. The temperature rose to 40°C and was not allowed to rise above this. The reaction mixture was stirred for 120 minutes after this addition and was then filtered through Celite. The reaction flask and filtered solids were washed with TΗF (400ml) and the washings combined with the filtrate. Any aqueous material carried over during the filtration was separated before the organic solution was concentrated to a volume of 800ml by atmospheric distillation of the TΗF. The solution was cooled to 20°C at which point 60-80 petrol (500ml) was added. The solution was stirred for 10 minutes when a further 500ml quantity of 60-80 petrol was added. This mixture was stirred for a further 10 minutes when a final 600ml quantity of 60-80 petrol was added. The mixture was cooled to 5°C for 16 hours before the product was isolated by filtration, washed with 60-80 petrol (400ml), and dried at 40°C, 100 mmΗg for 24 hours. Hence l-(4-picolyl)-2-pyrrolidinone 186.0g (86%) was obtained as a pale brown granular crystalline solid; m.p. 82-84°C; HPLC assay 96.1%; M+, 176.0947. CχøH 2N2θ requires 176.0950; m/z 176, (M+), 147 (M+ - C2H5), 119 (147-CO) and 903 (119 - HCN); v maximum (KBr) 2950, 1690 (C=0), 1600,
1450, 1420, 1300 and 1280 cm"1; 6H(270 MHz, CDCI3) 1.85 (2H, m, -CH2CH2CH2-), 2.20 (2H, t, -CH2C(0), 3.10 (2H, t, -CH2CH2NRRI), 4.25 (2H, s, PyCH2-), 6.95 (2H, m, Ar(3,5)) and 8.30 (2H, m, Ar(2,6). b) To a solution of l-(4-picolyl)-2-pyrrolidinone (20.0g, 0.1 Mmol) in dry THF (260ml) was added n-butyllithium (50.0ml of a 2.5 M solution in hexane 0.125mol) at 0 to -
10°C. The addition required 10 minutes. Potassium tertbutoxide (12.7g, 0.114mol) in THF (65ml) was then added at 0 to 10°C over 5 minutes and the resultant golden yellow suspension stirred for 10 minutes. At this point 4-methylthiobenzonitrile (18.6g, 0.125mol) in THF (31ml) was added over 5 minutes at 0° to -10°C. When the addition was complete the reaction mixture was allowed to warm to ambient temperature over 30 minutes. After this period the reaction mixture was heated under reflux for 120 minutes and the cooled to 30°C before demineralised water (80ml) was added. The resultant mobile solution was stirred for 30 minutes and the aqueous layer then allowed 30 minutes to separate before it was removed.
The solvent was exchanged with ethyl acetate via a put and take distillation where 140ml solvent was removed and the replaced with 140ml ethyl acetate. This process was continued until the base temperature reached 77°C. A further 45ml ethyl acetate was added and the solution cooled to 50°C before 60-80 petrol (87ml) was added. The product crystallized on cooling to room temperature and after stirring for 3 hours the suspension was cooled to 0-5°C and stirred for a further 2 hours. The product was then isolated by filtration, washed with 60-80 petrol (40ml) and then dried at 40°C, lOOmmHg for 24 hours. Hence 6,7-dihydro-2-(4-methylthiophenyl)-3-(4-pyridinyl)-5H-pyrrolo [1,2-a] imidazole was obtained as a pale yellow crystalline solid; 17.6g, 50%; m.p. 172°C; HPLC assay 95.6%; δH (270MHz, CDC13) 2.50 (3H, S, -SMe). 2.70 (2H, m, -CH2CH2, CH2-) 3.00 (2H, t, - CH2CH2CH2NRRI), 4.05 (2H, t, -CH2CH2CH2NRR1), 7.20 (2H, m, MeS Ar), 7.30 (2H, m, 3,5-Py), 7.50 (2H, m, Me S Ar) and 8.60 (2H, m, 2,6-Py).
EXAMPLE 4 6.7-Dihvdro-2-(4-methylthiophenyl -3-(4-pyridinyl')-5H-pyrrolo I"1.2-a1 imidazole (Intermediate compound of Formula (A) a) To a solution of l-(4-ρicolyl)-2-pyrrolidinone (2.01g, 11.4mmol) in THF (285ml) was added n-butyllithium (8.70ml of a 2.5M solution in hexane, 21.7mmol) at -70°C. The resultant yellow suspension was stirred for 90 minutes between -30 to -70°C before 4- methylthiobenzonitrile (2.72g, 18.3mmol) in THF (40ml) was added at -65°C. The reaction mixture was stirred with warming to room temperature over 15 minutes and was then stirred for a further 21 hours. After this time ammonia (720μl of a 35% w/w aqueous solution) was added which caused the reaction mixture to change from blood red to yellow in color. This solution was stirred for 30 minutes before the solvent was removed in vacuo and the residue chromatographed on silica gel using ethyl acetate: triethylamine - 96:4 as elutant Hence Z-l- ammo-l-(4-memyltMophenyl)-2-(4-pyridyl)-2-(l-(2-pyrrolidinoyl))ethene (1.2g, 32%) was obtained as a free flowing yellow powder, m.p.220-222°C (from ethyl acetate) M+ 325.1271. C18H19N3OS requires 325.1249. v maximum (nujol mull) 3500-3300 (N-H), 1669 (C-O), 1632 (C-C) and 1566 cm"1; δH (270 MHz, d6-DMSO) 2.10 (2H, m, -CH2CH2CH2-), 2.40
(2H, t, -CH2CH2CH2C(0)-), 2.50 (3H, s, -SMe), 3.50 (2H, t, -CH2CH2CH2C(0)-), 5.70 (2H, s, -NH2), 6.50 (2H, m, 3,5-Py), 7.25 (4H, m, MeS Ar) and 8.05 (2H, m, 2,6-Py); m/z 325(M+), 308 (M-NH3), 268 (M-C3H5O) and 150 (CgHgNS). b) To a suspension of Z-l-amino-l-(4-methylthiophenyl)-2-(4-pyπdyl)-2-{l-(2- ρyrrolidinoyl)}ethene (114mg, 0.351mmol) in THF (8.8ml) was added n-butyllithium (249μl of a 2.5M solution in hexane, 0.491mmol) at -40°C. The resultant dark red solution was allowed to warm to room temperature over 30 minutes and was then stirred at this temperature
for 19 hours. After this time the color changed to light yellow. At this point the reaction mixture was assayed by HPLC and found to contain the title compound 88mg, 82%.
EXAMPLE 5 2-(fluorophenyl)-6.7-dihvdro-3-(4-pyridinyl)-5H-pyrrolori.2-a1imidazole (Intermediate compound of Formula (A)
To a solution of l-(4-picolyl)-2-pyrrolidinone (56mg, 0.318mmol) in dry THF (8ml) was added n-butyllithium (472μl of a 1.0M solution in hexane, 0.477mmol) at -80°C. The resultant cloudy bright yellow solution was stirred between -50 to -80°C for 50 minutes before p-fluorobenzonitrile (61mg, 0.8097mmol) was added in THF 93ml) at -80°C. The reaction mixture was then allowed to warm to room temperature when it became dark red. It was stirred for 18 hours before the solvent was removed in vacuo and the residue chromatographed on silica gel using ethyl acetate:methanol - 4:1 as elutant Hence the title compound was obtained (7mg, 7%).
EXAMPLE 6 2-(4-Bromophenyl)-6.7-dihvdro-3-(4-pyridinyl)-5H-pyriOlori.2-a1imidazole (Intermediate compound of Formula (A)
To a solution of l-(4-picolyl)-2-pyrrolidinone (2.66g, 15.1mmol) in dry THF (76ml) was added n-butyllithium (7.26ml of a 2.5M solution in hexane, 18.1mmol) at -40°C. A solution of potassium tert butoxide (1.69g, 15.1mmol) in THF (8.5ml) was then added and the resultant golden yellow suspension stirred at -40°C for 10 minutes. At this point a solution of 4-bromobenzonitrile (5.50g, 30.2mmol) in THF (50ml) was added at -40°C and the reaction mixture then allowed to warm to room temperature. After stirring for 18 hours the reaction mixture was concentrated to dryness and the residue chromatographed on silica gel using ethyl acetate:methanol - 5:1 as elutant Hence the title compound was obtained as a yellow crystalline solid (0.71g, 14%); M+ 339.0371. CX7H14N3 79Brrequires 339.0371 M+ 341.0387. CX7H14N3 81βr requires 341.0351. δH (270MHz, CDCI3) 2.65 (2H, m, - CH2,CH2CH2-), 3.00 (2H, t, -CH^C^C^NRR"), 4.00 (2H, t, -CH2CH2CH2NRR*), 7.25 (2H, m, 3,5-Py), 7.40 (4H, m, Br-Ar) and 8.60 (2H, m, 2,6-Py); m/z 339 (M+), 341 (M+) 259 (M-HBr), 310 (M-C2H5) and 312 (M-C2H5).
EXAMPLE 7
5.6-Dihvdro-2-(4-fluorophenyl -7-methyl-3-(4-pyridinyl)-7H-pyπOlori.2-a1imidazol-7-ol. (A compound of Formula I and II)
5.6-Dihvdro-2-f4-fluorophenv -3-(4-pyridyl -7-methyl-7H-pyrrolori.2- 1imidazol-7-ol. To a stirring solution of 5,6-dihydro-2-(4-fluorophenyl)-3-(4-pyridyi)-7H-pyrrolo[l,2-fl]- imidazol-7-one (100 mg, 0.34 mmol) in toluene (10 mL) under an argon atmosphere was added trimethylaluminum (1.7 mL of 2 M solution in toluene, 3.41 mmol). The resulting mixture was heated at reflux for 4 h, then cooled to 0°C. To the cooled mixture was slowly added MeOΗ, followed by CΗ2CI2. The mixture was washed successively with H2O, saturated aqueous NaCl and dried (MgS04). The solvent was removed in vacuo, and the residue was purified by flash chromatography, eluting with 2% MeOH/ CH2CI2 to afford a light tan solid (47 mg, 45%) which was recrystallized from MeOH and dried in vacuo. m.p. 246 - 248°C
1H_NMR (CDCI3) : 8.60 (m, 2H); 7.48 (apparent dd, 2H); 7.20 (m, 2H); 7.02 (apparent t, 2H); 4.19 (m, 1H); 3.95 (m, 1H); 2.81 - 2.58 (m, 2H); 1.80 (s, 3H).
CIMS: m/e (rel. int.) : 310 (M+H)+.
Anal. Calc. for CιgHi6FN30-l/2 H20 : C 67.91, H 5.38, N 13.19; found : C 67.95, H 5.09, N 13.05.
EXAMPLE 8 5.6-Dihvdro-2-r4-fluorophenyD-7-methoxy-7-methyl-3-(4-pyridinyl)-7H-pyrrolo- π.2-a1imidazol-7-ol. (A compound of Formula I and H) To a stirring solution of the 5,6-Dihydro-2-(4-fluorophenyl)-7-methyl-3-(4- pyridinyl)-7H-pyrrolo[l,2-a]imidazol-7-ol (1.0 g, 3.4mmol) prepared in Example 7, in DMF (2ml) is added NaH (16.8mg, 0.35mmol of a 50% suspension in oil). After the gas evolution ceases Mel (0.022ml, 0.32mmol) is added and stirring is continued for 1 h at which point the reaction mixture is evaporated to dryness under reduced pressure and chromatographed on silica gel to yield the desired methyl ether derivative.
The above description fully discloses the invention including preferred embodiments thereof. Modifications and improvements of the embodiments specifically disclosed herein are within the scope of the following claims. Without further elaboration, it is believed that one skilled in the art can, using the preceding description, utilize the present invention to its fullest extent Therefore the Examples herein are to be construed as merely illustrative and not a limitation of the scope of the present invention in any way. The embodiments of the invention in which an exclusive property or privilege is claimed are defined as follows.