WO1991000257A1 - Novel potent inducers of terminal differentiation and methods of use thereof - Google Patents
Novel potent inducers of terminal differentiation and methods of use thereof Download PDFInfo
- Publication number
- WO1991000257A1 WO1991000257A1 PCT/US1990/002690 US9002690W WO9100257A1 WO 1991000257 A1 WO1991000257 A1 WO 1991000257A1 US 9002690 W US9002690 W US 9002690W WO 9100257 A1 WO9100257 A1 WO 9100257A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- same
- different
- group
- independently
- Prior art date
Links
- 0 C*C(*(C(C(C)C)(N)I)O)C(C)=O Chemical compound C*C(*(C(C(C)C)(N)I)O)C(C)=O 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C243/00—Compounds containing chains of nitrogen atoms singly-bound to each other, e.g. hydrazines, triazanes
- C07C243/24—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids
- C07C243/26—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids with acylating carboxyl groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C243/28—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids with acylating carboxyl groups bound to hydrogen atoms or to acyclic carbon atoms to hydrogen atoms or to carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/02—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
- C07C233/04—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with carbon atoms of carboxamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C233/05—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with carbon atoms of carboxamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/02—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
- C07C233/04—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with carbon atoms of carboxamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C233/07—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with carbon atoms of carboxamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/34—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by amino groups
- C07C233/35—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by amino groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
- C07C233/36—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by amino groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom having the carbon atom of the carboxamide group bound to a hydrogen atom or to a carbon atom of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C259/00—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups
- C07C259/04—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids
- C07C259/06—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids having carbon atoms of hydroxamic groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C259/00—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups
- C07C259/04—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids
- C07C259/10—Compounds containing carboxyl groups, an oxygen atom of a carboxyl group being replaced by a nitrogen atom, this nitrogen atom being further bound to an oxygen atom and not being part of nitro or nitroso groups without replacement of the other oxygen atom of the carboxyl group, e.g. hydroxamic acids having carbon atoms of hydroxamic groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D231/28—Two oxygen or sulfur atoms
- C07D231/30—Two oxygen or sulfur atoms attached in positions 3 and 5
- C07D231/32—Oxygen atoms
- C07D231/36—Oxygen atoms with hydrocarbon radicals, substituted by hetero atoms, attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/60—Three or more oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D243/00—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms
- C07D243/06—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4
- C07D243/08—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4 not condensed with other rings
Definitions
- Cancer is a disorder in which a population of cells has become, in varying degrees, unresponsive to the control mechanisms which normally govern proliferation and differentiation.
- chemotherapeutic treatment of cancer a) blocking hormone-dependent tumor cell proliferation by interference with the production or peripheral action of sex hormones; and b) killing cancer cells directly by exposing them to cytotoxic substances, which injure both neoplastic and normal cell populations.
- cancer therapy is also being attempted by the induction of terminal differentiation of the neoplastic cells (1).
- differentiation has been reported by exposure of cells to a variety of stimuli, including: cyclic AMP and retinoic acid (2,3), aclarubicin and other anthracyclines (4).
- neoplastic transformation does not necessarily destroy the potential of cancer cells to differentiate (1,5,6).
- tumor cells which do not respond to the normal regulators of proliferation and appear to be blocked in the expression of their differentiation program, and yet can be induced to differentiate and cease replicating.
- agents including some relatively simple polar compounds (5,7-9), derivatives of vitamin D and retinoic acid (10-12), steroid hormones (13), growth factors (6,14), proteases (15,16), tumor promoters (17,18), and inhibitors of DNA or RNA synthesis (4,19-24), can induce various transformed cell lines and primary human tumor explants to express more differentiated characteristics.
- HMBA human erythroleukemia cells
- HMBA Upon addition of HMBA to MELC (745A-DS19) in culture, there is a latent period of 10 to 12 hours before commitment to terminal differentiation is detected. Commitment is defined as the capacity of cells to express terminal differentiation despite removal of inducer (25). Upon continued exposure to HMBA there is progressive recruitment of cells to differentiate. Recently, the present inventors reported that MELC cell lines made resistant to relatively low levels of vincristine become markedly more sensitive to the inducing action of HMBA and can be induced to differentiate with little or no latent period (26).
- HMBA is capable of inducing phenotypic changes consistent with differentiation in a broad variety of cells lines (5).
- the characteristics of the drug induced effect have been most extensively studied in the murine erythroleukemia cell system (MELC) (5,25,27,28).
- MELC induction of differentiation is both time and concentration dependent.
- the minimum concentration required to demonstrate an effect in vitro in most strains is 2 to 3 mM; the minimum duration of continuous exposure generally required to induce differentiation in a substantial portion (>20%) of the population without continuing drug exposure is about 36 hours.
- HMBA protein kinase C is involved in the pathway of inducer-mediated differentiation.
- the in vitro studies provided a basis for evaluating the potential of HMBA as a cytodifferentiation agent in the treatment of human cancers (30).
- phase I clinical trials with HMBA have been completed (31-36). Recently, the first evidence was reported that this compound can induce a therapeutic response in patients with cancer (35,36).
- phase I clinical trials demonstrate that the potential efficacy of HMBA is limited, in part, by dose-related toxicity which prevents achieving optimal blood levels and by the need for intravenous administration of large quantities of the agent, over prolonged periods.
- the present invention provides new chemical inducers which are 2-20 times more active than HMBA. It has unexpectedly been found that compounds having two or more nonpolar components connected by a polar group and having groups on the termini of the compound are effective inducers of terminal differentiation. For instance, bis-hexamethylene triacetamide, which comprises three acetamide groups connected by two six-carbon chains, has been found to be a potent inducer of terminal differentiation in MELC.
- This new class of compounds of the present invention may be useful for selectively inducing terminal differentiation of neoplastic cells and therefore aid in treatment of tumors in patients.
- the invention provides a class of compounds having the structure:
- each of A, A,, A 2 , A 3 , and A 4 represent a polar group which comprises a nitrogen, sulfur or oxygen atom wherein each of A, A 1 , A 2 , A 3 , and A 4 may independently be the same as, or different from, the others of A, A 1 , A 2 , A 3 , and A 4 ; wherein each of R and R 1 is a hydrogen atom; a lower alkyl, alkenyl, or alkynyl group; or a group having the structure:
- each of R 2 and R 3 being a hydrogen atom or a lower alkyl, alkenyl, or alkynyl group; and wherein each of R, R 1 , R 2 and R 3 may independently be the same as, or different from the other of R, R 1' R 2 and R 3 ; wherein each of [R-A] and [A 4 -R 1 ] have a dipole moment greater than about 2.7 Debye units; wherein each of B, B 1 , B 2 , and B 3 represents a nonpolar group which comprises at least 4 atoms in a chain, the termini of which chains are attached to A and A 1 , A 1 and A 2 , A 2 and A 3 , and A 3 and A 4 , respectively; wherein each such atom is oxygen, nitrogen, carbon, or sulfur and wherein each of B, B 1 , B 2 , and B 3 may independently be the same as, or different from the others of B, B 1' B 2 and B 3 ; and wherein each of a and b
- R1 and R2 may be the same or different and each is a lower alkyl group.
- R is H or a lower alkyl group.
- This invention further provides a compound having the structure:
- n is an integer which is greater than 1 and R 1 and R 2 may be the same or different and each is H or a lower alkyl group.
- X1 and X2 may independently be the same or different and each is N(CH 3 ) 2' NH-phenyl, or HNCH 3 .
- This invention also provides a compound having the structure:
- R1 and R2 may be the same or different and each is H or a lower alkyl group and n is an integer from 1 to 10.
- This invention further provides a compound having the structure:
- R1, R2, R3, R4, R5, R6 may independently be the same or different from each other and is H or a lower alkyl group; wherein X is methyl or phenyl; and n is an integer from 1 to about 15.
- R1 and R2 may be the same or different and is H or CH3.
- the invention also provides a compound having the structure:
- R 1 and R 2 may be the same or different and is H or
- R 1 and R 2 are the same or different and is H or lower alkyl group.
- R1 and R1 may independently be the same or different and each may be H or a lower alkyl group; wherein R3 and R4 may independently be the same or different and each may be CH3 or OH; and wherein n is 5 or 6.
- R 1 and R 2 is the same or different and is hydrogen, lower alkyl, alkenyl, alkynyl, an amide or hydroxyamide.
- This invention further provides a compounds having the following structures:
- the invention also concerns a method of selectively inducing terminal differentiation of neoplastic cells and thereby inhibiting proliferation of such cells which comprises contacting the cells under suitable condition with an amount of the compound effective to selectively induce terminal differentiation.
- the invention provides a method of treating a patient having a tumor characterized by proliferation of neoplastic cells which comprises administering to the patient an amount of the compound effective to selectively induce terminal differentiation of such neoplastic cells, thereby inhibiting their proliferation, and suppressing oncogenicity.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier and the compound in an amount less than an amount which cause toxicity in the patient.
- HMBA hexamethylene bisacetamide
- SBDA suberic acid bis-N-methyl diacetamide
- Figure 3 Optimal concentration of IC-135 for inducement of terminal differentiation.
- Figure 4 Comparison of HMBA and IC-135 on DS19 cells.
- the invention provides a class of compounds having the structure:
- each A, A 1 , A 2 , A 3 , and A 4 represent a polar group which comprises a nitrogen, sulfur or oxygen atom and wherein each of A, A 1 , A 2 , A 3 , and A 4 may independently be the same as, or different from, the others of A, A 1 , A 2 , A 3 , and A 4 ; wherein each of R and R 1 is a hydrogen atom; a lower alkyl, alkenyl, or alkynyl group; or a group having the structure:
- each of R 2 and R 3 being a hydrogen atom or a lower alkyl, alkenyl, or alkynyl group; and wherein each of R, R 1 , R 2 and R 3 may independently be the same as, or different from, the others of R, R 1 , R 2 and R 3 ; wherein each of [R-A] and [A 4 -R 1 ] have a dipolar moment greater than about 2.7 Debye units; wherein each of B, B 1 , B 2 , and B 3 represents a nonpolar group which comprises at least 4 atoms in a chain, the termini of which chains are attached to A and A 1 , A 1 , and A 2 , A 2 and A 3 , and A 3 and A 4 , respectively; wherein each such atom is oxygen, nitrogen, carbon, or sulfur and wherein each of B, B 1 , B 2 , and B 3 may independently be the same as, or different from the others of B, B 1 , B 2 , and B 3 ; and wherein
- the compounds of the present invention are made up of two components.
- One component comprises a polar group, i.e. functional groups with significant dipole moments, such as amides, sulfoxides, amine oxides and related functional groups.
- the terminal portions of the compound each have dipole moments greater than about 2.7 debye units.
- the polar groups within the compound represented by -A 1 , -A 2 - and -A 3 -, have significant dipolar moments but not necessarily in excess of 2.7 debye units.
- the polar groups are carbonyl radicals or bivalent radicals of an amide, a sulfoxide or a amine oxide.
- Each polar group need not necessarily be the same as the other polar groups.
- the poplar groups within the compound are the same as each other and the terminal polar groups are the same.
- all the polar groups are amide groups attached to the compound at the nitrogen atom or at the carbon atom of the carbonyl radical.
- the amide group may comprise one or more hydrocarbon substituents, such as a lower alkyl or alkenyl groups, including branched or unbranched groups.
- the term "lower alkyl or alkenyl group” is intended to include saturated and unsaturated hydrocarbon groups with 1 to about 5 carbon atoms.
- R 4 is a hydrogen atom or a lower alkyl or alkenyl group
- R 2 and R 3 each is hydrogen atom, a methyl group or a ethyl group.
- the compound also requires at least two nonpolar sections, designated B and B 1 , which are attached to and connect polar groups. Additional nonpolar sections may also be present, e.g. B 2 when a is 1 and B 3 when b is 1.
- the nonpolar sections may comprise linear saturated hydrocarbon chains, linear unsaturated hydrocarbon chains containing one or more double or triple bonds, or saturated or unsaturated hydrocarbon chains containing one or more lower alkyl or alkenyl groups or small carbocyclic rings as substituents.
- the nonpolar groups are hydrocarbon chains comprising 4 to 7 methylene groups, especially preferred are hydrocarbon chains containing 6 carbon atoms.
- R is hydrogen or a methyl group and x is 5 or 6.
- Other compounds of this invention include a compound having the structure:
- R1 and R2 may be the same or different and each is a lower alkyl group. Also provided by this invention is a compound having the structure:
- R is H or a lower alkyl group.
- This invention further provides a compound having the structure:
- n is an integer which is greater than 1 and R 1 and R 2 may be the same or different and each is H or a lower alkyl group.
- X1 and X2 may independently be the same or different and each is N(CH 3 ) 2 , NH-phenyl, or HNCH 3 .
- This invention also provides a compound having the structure: or a compound having the structure:
- R1 and R2 may be the same or different and each is H or a lower alkyl group; and n is an integer from 1 to 10.
- This invention further provides a compound having the structure: wherein R1, R2, R3, R4, R5, R6 may independently be the same or different from each other and is H or a lower alkyl group; wherein X is methyl or phenyl; and n is an integer from 1 to about 15.
- R1 and R2 may be the same or different and is H or CH3.
- the invention also provides a compound having the structure:
- R 1 and R 2 may be the same or different and is H or
- R 1 and R 2 are the same or different and is H or lower alkyl group.
- R1 and R1 may independently be the same or different and each may be H or a lower alkyl group; wherein R3 and R4 may independently be the same or different and each may be CH3 or OH; and wherein n is 5 or 6.
- R 1 and R 2 is the same or different and is hydrogen, lower alkyl, alkenyl, alkynyl, an amide or hydroxyamide.
- This invention further provides a compounds having the following structures:
- the invention also concerns a method of selectively inducing terminal differentiation of neoplastic cells and thereby inhibiting proliferation of such cells which comprises contacting the cells under suitable conditions with an amount of the compounds shown above effective to selectively induce terminal differentiation in the cells.
- a method of selectively inducing terminal differentiation of neoplastic cells and thereby inhibiting proliferation of such cells comprises contacting the cells under suitable conditions with an amount of a compound effective to selectively induce terminal differentiation, the compound having the structure:
- X is phenyl or methyl and n is an integer from 1 to 15.
- the contacting must be performed continuously for a prolonged period of time, i.e. for at least 48 hours, preferably for about 4-5 days or longer.
- the method may be practiced in vivo or in vitro. If the method is practiced in vitro, contacting may be effected by incubating the cells with the compound.
- the concentration of the compound in contact with the cells should be from about 1 ⁇ M to abount 25 mM, preferably from about 4 ⁇ M to about 5 mM.
- the concentration depends upon the individual compound.
- compound 12 of Table 1 should have a concentration from about 0.1 to about 2 mM, preferably from about 0.5 to about 0.7 mM.
- the optimal concentration is as low as 5 ⁇ M to about 5 mM.
- Another factor determining the preferable range is the state of the tumor cells.
- the range of effective concentration of compound 12 from Table 1 is from about 0.01 to about 0.3 mM with a preferable range of about 0.05 to about 0.1 mM.
- the invention also concerns a method of treating a patient having a tumor characterized by proliferation of neoplastic cells which comprises administering to the patient an amount of the compounds shown above, or with a compound having the structure:
- X is phenyl or methyl and n is an integer from 1 to 15, effective to selectively induce terminal differentiation of such neoplastic cells thereby inhibiting their proliferation, and suppressing oncogenicity.
- the method of the present invention is intended for the treatment of human patients with tumors. However, it is also likely that the method would be effective in the treatment of tumors in other animals.
- the term tumor is intended to include any cancer caused by the proliferation of neoplastic cells, such as lung cancer, acute lymphoid myeloma, bladder melanoma, renal carcinoma, breast carcinoma, or colorectal carcinoma.
- the administration of the compound to the patient may be effected orally or parenterally. To date, administration intravenously has proven to be effective.
- the administration of the compound must be performed continuously for a prolonged period of time, such as for at least 3 days preferably more than 5 days.
- the administration is effected continuously for at least 10 days and is repeated at intervals wherein at each interval the administration is continuously effected for at least 10 days.
- the administration may be effected at intervals as short as 5-10 days, up to about 25-35 days and continuously for at least 10 days during each such interval.
- the optimal interval period will vary depending on the type of patient and tumor. For example, in the incidence of acute leukemia, the so called myelodysplastic syndrome, continuous infusion would seem to be indicated so long as the patient tolerated the drug without toxicity and there was a positive response.
- the amount of the compound administered to the patient is less than an amount which would cause toxicity in the patient.
- the compounds have the structures:
- the amount of the compound which is administered to the patient is less than the amount which causes a concentration of the compound in the patient's plasma to equal or exceed the toxic level of the compound.
- the concentration of the compound in the patient's plasma is maintained at about 1.0 mM. It has been found with HMBA, that administration of the compound in an amount from about 5 gm/m 2 /day to about 30 gm/m 2 /day, particularly about 20 gm/m 2 /day, is effective without producing toxicity in the patient.
- the optimal amount of the compounds is substantially lower than 30 gm/m 2 /day, and may even be lower than 1 gm/m 2 /day.
- the optimal amount of the compound which should be administered to the patient in the practice of the present invention will depend on the particular compound used and the type of cancer being treated.
- homologs are molecules having substantial structural similarities to the above-described compounds and analogs are molecules having substantial biological similarities regardless of structural similarities.
- the invention also concerns a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier, such as sterile pyrogen-free water, and any of the compounds listed above in an amount less than an amount which if administered intravenously or orally to a patient would cause the patient's blood plasma concentration of the compound to exceed toxic levels.
- a pharmaceutically acceptable carrier such as sterile pyrogen-free water
- MELC 745A-DS19 cells and the variants of MELC derived from this cell line namely, the vincristine-resistant MELC V3.17 and VCR.C(2) 15 cell lines (26), and the dimethylsulfoxide-resistant cell line, DR10 (37), were maintained in alpha minimal essential medium containing 10% fetal calf serum (16).
- Cell cultures for all experiments were initiated with cells in logarithmic growth phase (day 2 cultured cells) at a density of 10 5 cells/ml. Inducer compounds were added in the final concentrations indicated below, dissolved in culture medium without fetal calf serum unless otherwise indicated. Cell density and benzidine reactively were determined as described (16).
- HMBA compound 1 of Table 1, (9) was obtained from Aldrich Chemical Co.
- Compound 5 (Table 1) was synthesized from meso-2,3-dimethylsuccinic acid in six steps. Reduction of the dimethyl ester with LiAlH 4 afforded the meso-2,3-dimethylbutanediol (92% yield). This was converted to the bis-tosylate, and then to the bis-phtalimide. Deprotection and acetylation as before gave compound 5 as an oil (61% yield from the diol).
- Compound 10 (Table 1) was prepared by making a solution of 19.8gm commercial bis-hexamethylenetriamine in 500 ml of 1,4-dioxane at room temperature under argon. Then 44.8 ml. of triethylamine was added, and 20.3 ml. of acetyl chloride was slowly added with stirring. After two hours of stirring at room temperature the triethylamine hydrochloride was removed by filtration and the filtrate was concentrated in vacuo. The product triacetyl compound was isolated as a clear viscous oil at about 90% yield by chromatography on basic alumina using isopropanol/ethyl acetate/dichloromethane in the ratio 2/3/5. On thin layer plates of basic alumina with this solvent mixture the product had an R f of ca. 0.6.
- Compound 12 (Table 1) was prepared by simple dialkylation of dimethyl malonate with the know N,N'-dimethyl-6-bromohexanecarboxamide (39).
- n is an integer from 1 to 15 are made as follows.
- the suspension immediately became a solution.
- the solution was heated at 110oC for an extra 10 minutes, cooled down, filtered, and acidified.
- the clear water solution was extracted with chloroform (10 ⁇ 50 ml). The combined chloroform extracts were dried over anhydrous magnesium sulfate, and evaporated.
- R1, R2, R3, R4, R5, R6 may independently be the same or different from each other and is H or a lower alkyl group; wherein X is methyl or phenyl; and n is an integer from 1 to about 15, were made as follows.
- R1 and R6 is each H and R2, R3, R4, and R5 is each CH3, to a chloroform (200 ml) solution of suberoyl chloride (8 g; 38 mmol) was added at room temperature to 1,1-dimethylhydrazine (11.5 ml; 9.1 g; 152 mmol). The mixture was stirred at room temperature for about three hours. The solvent was evaporated, and the residue was dissolved in methanol (400 ml).
- heterocyclic compounds may be alkylated after they are formed from malonic chloride and N,N 1 -dialkylahydrazine.
- R 1 and R 2 may be the same or different and is H or CH 3 may be made as follows.
- the heterocyclic part of the compound may be made from malonic chloride and N,N 1 - dealkylethylenediamine.
- the heterocyclic compound may then be alkylated with the corresponding alkyl bromide under known conditions.
- the acid may be synthesized from the corresponding acid via acidic chloride with hydroxylamine hydrochloride in a solution of tetrahydrofuran and water.
- the acid may be made from 1,4- benzenediacrylic acid by an addition of bromine, and following elimination with base.
- the reaction suspension was stirred at room temperature for about one hour and evaporated to a volume of 20 ml.
- the suspension was cooled and filtered.
- the crude solid product was slurried in methanol (200 ml) and the suspension was cooled and filtered.
- the filtrate was dried over anhydrous magnesium sulfate, and evaporated to a solid residue.
- the solid was slurried in ether (250 ml) and recovered by filtration.
- the yield of trans-2-butene-1,4-di-(N-hydroxylcarboxamide) was 3.8 g (63%).
- the suspension was stirred at room temperature for about one hour and filtered.
- the solid was slurried in methanol (2 liters) and the suspension then was boiled and filtered.
- the methanol solution was concentrated to a volume of about 20 ml.
- the solid product was separated by filtration and washed with ether (5 ⁇ 20 ml).
- the yield of trans,trans-1,3-butadiene-1,4-(N-hydroxylcarboxamide) was 3.1 g (52%).
- the suspension was stirred at room temperature for about two hours and filtered.
- the crude product was slurried in water (50 ml) and the resulting suspension was boiled (about 10 minutes).
- the solid was separated by filtration, and slurried in acetone (200 ml) and boiled.
- the hot suspension was filtered, and the solid was washed with acetone (5 ⁇ 50 ml), and then with ether (5 ⁇ 20 ml).
- the yield of diphenyl-4,4'-di-(N-hydroxylcarbox-amide) was 4 .9 g (88%) .
- the acid was suspended in benzene (500 ml), and oxalyl chloride (11.2 ml; 16.3 g; 0.129 mol) and a few drops of DMF were added. The mixture was stirred at room temperature for about 3 hours. The solvent was evaporated and the oily residue was dissolved in chloroform (100 ml). The chloroform solution of the acid chloride was slowly added to a stirred solution of O-behzylhydroxylamine (16.3 g; 0.132 mol) in chloroform (300 ml). A white precipitate was formed immediately. The suspension was filtered, and concentrated hydrochloric acid was added (50 ml) to the chloroform filtrate and again filtered.
- the chloroform layer was washed with concentrated hydrochloric acid (3 ⁇ 100 ml), water (3 ⁇ 100 ml), and dried over anhydrous magnesium sulfate. The chloroform was evaporated yielding pure N,N'-dibenzyloxy-6, 6-di(ethoxycarbonyl)-1,11-undecanedicarboxamide (17.1 g; 89%).
- the benzyloxyamide (6 g; 0.01 mol) was dissolved in methanol (50 ml), and 5 % Pd-C (200 mg) was added. The methanol suspension was hydrogenated at room temperature overnight. The catalyst was separated by filtration and methanol was evaporated yielding pure undecane-6,6-di(ethylcarboxylate)-1,11-di(N-hydroxylcarboxamide) (3.9 g; 93%). The overall yield was 55.4%.
- R1 and R1 may independently be the same or different and is H or a lower alkyl group
- R3 and R4 may independently be the same or different and each is CH3 or OH
- n is 5 or 6
- the dibenzyl ester (18 g; 0.032 mol) was dissolved in methanol (50 ml) and catalyst (200mg of 5 % Pd-C) was added. The suspension was hydrogenated overnight, and the catalyst was removed by filtration. The methanol was evaporated yielding pure acid (11.5 g; 94%).
- the acid 11 g; 0.0286 mol was suspended in benzene (500 ml), and oxalyl chloride (10ml; 14.5 g; 0.14 mol) and a few drops of DMF were added. The mixture was stirred at room temperature for about 3 hours and evaporated to an oily residue.
- N-benzyloxyamide (6 g; 0.01 mol) was dissolved in methanol (509 ml) and catalyst (200 mg of 5% Pd-C) was added. The suspension was hydrogenated at room temperature overnight. The catalyst was removed by filtration and the methanol was evaporated yielding pure hydroxamic acid (3.95 g; 95%). The overall yield of 1,3-dimethyl-5,5-di(5-pentyl-n-hydroxylcaboxamide)barbituric acid was 23.4%.
- R1 and R2 independently be the same or different and each may is H or a lower alkyl group and n is an integer from 1 to 10 is synthesized as follows.
- the mixture was stirred at 80oC for about 30 minutes.
- Iodomethane (23.3 ml; 53.2 g; 380 mmol) was added to this cool solution at a temperature of about 5oC and the reaction was continued at 60oC overnight.
- the solvent was evaporated, and the residue was dissolved in water (50 ml).
- the product was more than 95% pure by NMR(DMSO).
- HO-N(CH3)-CO-(CH2)6-CO-n(CH3)-OH was made by slowly adding a solution of suberoyl chloride (5 g; 23.7 mmol) in tetrahydrofuran (20 ml) to a stirred water solution of potassium hydroxide (5.3 g; 94.8 mmol) and N-methylhydroxylamine hydrochloride (4 g; 47.4 mmol) at about 5oC.
- the reaction mixture was stirred at room temperature for about one hour.
- the organic layer was separated and the water layer was extracted with chloroform (5 ⁇ 50 ml).
- the combined chloroform layers were dried over anhydrous magnesium sulfate and evaporated.
- the product was purified by crystallization from acetone.
- the yield of hexane-1,6-di(N-hydroxyl-N-methylcarboxamide was 2.7 g (46%).
- Each compound in Table 1 was assayed 3 or more times to determine effectiveness as an inducer of MELC (745A-DS19) cell line.
- the cell density of MELC in culture for 5 d. without inducer was 2.0 to 2.6 ⁇ 10 6 cells/ml.
- the cell density of MELC in culture for 5 d. with inducer was 1.2 to 2.0 ⁇ 10 6 cells/ml.
- Compounds 11 and 12 were dissolved in absolute ethyl alcohol.
- the final concentration of ethyl alcohol in the cultured medium ranged between 0.1 and 3%. This concentration of ethyl alcohol had no effect on cell growth of MELC in culture without inducer. All other compounds were dissolved in culture medium without fetal calf serum. The indicated optimal concentration represents the final concentration in the culture medium.
- HMBA hexamethylene bisacetamide
- Table 1 compound 1
- N-methyl acetamide Table 1, compound 2
- HMBA induces erythroid differentiation in MELC at an optimal concentration of 5 mM (8).
- the present inventors previously showed that the optimum number of methylene groups in the apolar chain is six (27).
- HMBA and SBDA have their polar groups separated by identical methylene bridges, and they have a similar ratio of polar to apolar hydrophobic moieties. Many structurally related compounds have been examined, but only a few showed comparable or greater activity (Table 1, compounds 6 through 12). It is clear that the structures of active compounds may differ sufficiently from HMBA to make it likely that they will also display different pharmacokinetics.
- One of the more active of these hybrid compounds is a dimmer of HMBA.
- Bishexeunethylene triacetamide (BHTA; compound 10) is about 2 fold more active as inducer, on a molar basis, than HMBA ( Figure 1C).
- the most active of the hybrid compounds assayed in this study is one in which two pentamethylene carboxyamides are linked by the dimethyl ester of malonic acid (compound 12).
- This compound with 4 exposed polar groups balanced by two apolar pentamethylene domains, is roughly 10 fold more active than HMBA.
- 0.6 mM compound 12 is about as effective as 5.0 mM HMBA, inducing over 90% of cells to differentiate after 5 d. in culture (Fig. 2).
- Polymethylenediamine derivatives carrying propionyl groups instead of the acetyl groups of HMBA are also active, but methoxycarbonyl groups are less effective and bulky pivaloyl groups lead to loss of activity.
- HMBA can have a double bond (cis or trans) or a triple bond in the center and retain its activity (27).
- Replacement of the polymethylene chain with a cyclohexane ring leads to inactivity (27), although compound 9, with a longer chain interrupted by an amide group, is active.
- Diamides of dicarboxylic acids such as suberic acid, are active with either one or two methyl groups on each nitrogen (compounds 7 , 8, and 9), but not with a methyl and a methoxyl substituent, and they are also active with one (but not two) ethyl groups on each nitrogen.
- Suberic diamides of pyrollidine, of morpholine, or of piperazine are inactive. This shows that there is a limit, to the amount of carbon tolerated on the ends of the polar groups.
- the compounds assayed for inducing activity with vincristine- resistant MELC include compounds 1, 3, 4, 8, 10, 11 and 12 (Table 1).
- vincristine-resistant MELC were induced at lower concentrations than were required for induction of vincristine-resistant MELC were induced more rapidly than the vincristine-sensitive cells.
- 0.1 mM compound 12 was the optimal concentration for inducing vincristine-resistant MELC (VCR.C(2) 15).
- VCR.C(2) 15 the optimal concentration for inducing vincristine-resistant MELC
- Table 2 shows the cell densities, B+%, and percent of cells committed for cell lines DS19 and V3.17 grown in 1 mM to 5 mM of HMBA and IC-135.
- Figures 3, 4, 5A and B, and 6A and B are graphical representations of the data presented in Table 2.
- Table 3 shows cell counts for days 1, 2 and 5 and percentage of cells committed and benzidine reactive (B+) at day 5 for cell liens DS-19, V3.17 and DR-10 grown in 5 mM of HMBA and 0.5 to 3 mM of IC-135.
- IC-135 is more reactive in the tested cell lines at lower concentrations than HMBA.
- Table 4 shows the results of several structurally different compounds when assayed 3 or more times to determine effectiveness as an inducer of MELC (745A-DS19) cell line.
- the cell density of MELC in culture for 5 d. without inducer was 2.0 to 2.6 ⁇ 10 6 cells/ml. Discussion
- agents which can induce transformed cells to differentiate and suppress their oncogenicity has important implications for the treatment of cancer. While a number of agents have been identified that can induce tumor cells in vitro to express features of their differentiated phenotype and to decrease their proliferative capacity (4,10-24), these agents have generally proved to be relatively ineffective or two toxic when evaluated in clinical trials (40).
- the hybrid polar/apolar compound, HMBA has been one of the best characterized with respect to its in vitro inducing activity in MELC and in a number of other transformed cell lines, as well as for certain human tumor explants (30). It can trigger a differentiation program in transformed cells which is similar to that of their normal lineage (5).
- the observed positive therapeutic responses to HMBA albeit largely transient, occurred despite relatively low serum concentrations of HMBA ( ⁇ 1 mM), compared to the optimum demonstrated in vitro (4 to 5 mM) (35,36).
- the present invention provides a new group of hybrid polar/apolar compounds which are as active or even more active, on a molar basis, than HMBA.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The invention provides compounds, several of which belong to a class having two or more nonpolar components connected by a polar group and having polar groups on the termini of the compound. The invention also concerns a method of selectively inducing termini differentiation of neoplastic cells and thereby inhibiting proliferation of such cells which comprises contacting the cells under suitable condition with an amount of the compound effect to selectively induce terminal differentiation. Moreover, the invention provides a method of treating a patient having a tumor characterized by proliferation of neoplastic cells which comprises administering to the patient an amount of the compound effective to selectively induce terminal differentiation of such neoplastic cells, thereby inhibiting their proliferation and suppressing oncogenicity. Lastly, the present invention provides a pharmaceutical composition comprising a pharmaceutically acceptable carrier and the compound in an amount effective less than an amount which would cause toxicity in the patient.
Description
NOVEL POTENT INDUCERS OF
TERMINAL DIFFERENTIATION AND METHODS OF USE THEREOF
This is a continuation-in-part application of U.S. Serial No. 374,343, filed June 30, 1989, which in turn is a continuation-in-part of application U.S. Serial No.270,963, filed November 14, 1988, the contents of which are hereby incorporated by reference into the present application.
This application also claims the benefit of PCT International Application No. PCT/US89/05203, filed November 14, 1989, under 35 U.S.C. § 119.
Background of the Invention
Throughout this application various publications are referenced by arabic numerals within parentheses. Full citations for these publications may be found at the end of the specification immediately preceding the claims. The disclosures of these publications in their entireties are hereby incorporated by reference into this application in order to more fully describe the state of the art to which this invention pertains.
Cancer is a disorder in which a population of cells has become, in varying degrees, unresponsive to the control mechanisms which normally govern proliferation and differentiation. For many years there have been two principal strategies for chemotherapeutic treatment of cancer: a) blocking hormone-dependent tumor cell proliferation by interference with the production or peripheral action of sex hormones; and b) killing cancer cells directly by exposing them to cytotoxic substances,
which injure both neoplastic and normal cell populations.
Relatively recently, cancer therapy is also being attempted by the induction of terminal differentiation of the neoplastic cells (1). In cell culture models differentiation has been reported by exposure of cells to a variety of stimuli, including: cyclic AMP and retinoic acid (2,3), aclarubicin and other anthracyclines (4).
There is abundant evidence that neoplastic transformation does not necessarily destroy the potential of cancer cells to differentiate (1,5,6). There are many examples of tumor cells which do not respond to the normal regulators of proliferation and appear to be blocked in the expression of their differentiation program, and yet can be induced to differentiate and cease replicating. A variety of agents, including some relatively simple polar compounds (5,7-9), derivatives of vitamin D and retinoic acid (10-12), steroid hormones (13), growth factors (6,14), proteases (15,16), tumor promoters (17,18), and inhibitors of DNA or RNA synthesis (4,19-24), can induce various transformed cell lines and primary human tumor explants to express more differentiated characteristics.
Early studies by the present inventors identified a series of polar compounds that were effective inducers of differentiation in a number of transformed cell lines (8,9). Of these, the most effective inducer, until recently, was the hybrid polar/apolar compound N,N'-hexamethylene bisacetamide (HMBA) (9). The use of polar/apolar compounds to induce murine erythroleukemia cells (MELC) to undergo erythroid differentiation with suppression of oncogenicity has proved a useful model to study inducer-mediated
differentiation of transformed cells (5,7-9). HMBA-induced MELC terminal erythroid differentiation is a multistep process. Upon addition of HMBA to MELC (745A-DS19) in culture, there is a latent period of 10 to 12 hours before commitment to terminal differentiation is detected. Commitment is defined as the capacity of cells to express terminal differentiation despite removal of inducer (25). Upon continued exposure to HMBA there is progressive recruitment of cells to differentiate. Recently, the present inventors reported that MELC cell lines made resistant to relatively low levels of vincristine become markedly more sensitive to the inducing action of HMBA and can be induced to differentiate with little or no latent period (26).
HMBA is capable of inducing phenotypic changes consistent with differentiation in a broad variety of cells lines (5). The characteristics of the drug induced effect have been most extensively studied in the murine erythroleukemia cell system (MELC) (5,25,27,28). MELC induction of differentiation is both time and concentration dependent. The minimum concentration required to demonstrate an effect in vitro in most strains is 2 to 3 mM; the minimum duration of continuous exposure generally required to induce differentiation in a substantial portion (>20%) of the population without continuing drug exposure is about 36 hours.
The primary target of action of HMBA is not known. There is evidence that protein kinase C is involved in the pathway of inducer-mediated differentiation (29). The in vitro studies provided a basis for evaluating the potential of HMBA as a cytodifferentiation agent in the treatment of human cancers
(30). Several phase I clinical trials with HMBA have been completed (31-36). Recently, the first evidence was reported that this compound can induce a therapeutic response in patients with cancer (35,36). These phase I clinical trials demonstrate that the potential efficacy of HMBA is limited, in part, by dose-related toxicity which prevents achieving optimal blood levels and by the need for intravenous administration of large quantities of the agent, over prolonged periods.
The present invention provides new chemical inducers which are 2-20 times more active than HMBA. It has unexpectedly been found that compounds having two or more nonpolar components connected by a polar group and having groups on the termini of the compound are effective inducers of terminal differentiation. For instance, bis-hexamethylene triacetamide, which comprises three acetamide groups connected by two six-carbon chains, has been found to be a potent inducer of terminal differentiation in MELC.
This new class of compounds of the present invention may be useful for selectively inducing terminal differentiation of neoplastic cells and therefore aid in treatment of tumors in patients.
Summary of the Invention
The invention provides a class of compounds having the structure:
[R-A]-B-A1-B1-[A2-B2-]a[A3-B3-]b[A4-R1] wherein each of A, A,, A2, A3, and A4 represent a polar group which comprises a nitrogen, sulfur or oxygen atom wherein each of A, A1, A2, A3, and A4 may independently be the same as, or different from, the others of A, A1, A2, A3, and A4; wherein each of R and R1 is a hydrogen atom; a lower alkyl, alkenyl, or alkynyl group; or a group having the structure:

wherein each of R2 and R3 being a hydrogen atom or a lower alkyl, alkenyl, or alkynyl group; and wherein each of R, R1, R2 and R3 may independently be the same as, or different from the other of R, R1' R2 and R3; wherein each of [R-A] and [A4-R1] have a dipole moment greater than about 2.7 Debye units; wherein each of B, B1, B2, and B3 represents a nonpolar group which comprises at least 4 atoms in a chain, the termini of which chains are attached to A and A1, A1 and A2, A2 and A3, and A3 and A4, respectively; wherein each such atom is oxygen, nitrogen, carbon, or sulfur and wherein each of B, B1, B2, and B3 may independently be the same as, or different
from the others of B, B1' B2 and B3; and wherein each of a and b is independently 0 or 1.
Other compounds of this invention include a compound having the structure:

or a compound having the structure:

Also provided by this invention is a compound having the structure:

This invention further provides a compound having the structure:

wherein n is an integer which is greater than 1 and R1 and R2 may be the same or different and each is H or a lower alkyl group.
A compound having the structure:

This invention also provides a compound having the structure:

or a compound having the structure:

wherein R1 and R2 may be the same or different and each is H or a lower alkyl group and n is an integer from 1 to 10.
This invention further provides a compound having the structure:

wherein R1, R2, R3, R4, R5, R6 may independently be the same or different from each other and is H or a lower alkyl group; wherein X is methyl or phenyl; and n is an integer from 1 to about 15.
A compound having the structure:

is also provided by this invention wherein R1 and R2 may be the same or different and is H or CH3.
The invention also provides a compound having the structure:

wherein R1 and R2 may be the same or different and is H or
CH 3.
A compound having the structure:

is provided by this invention, wherein X has the structure:

or
-CH=CH-CH=CH-;

wherein R1 and R2 are the same or different and is H or lower alkyl group.
Further provided by this invention is a compound having the structure:

wherein R1 and R1 may independently be the same or different and each may be H or a lower alkyl group; wherein R3 and R4 may independently be the same or different and each may be CH3 or OH; and wherein n is 5 or 6.
A compound having the structure:

This invention further provides a compounds having the following structures:




The invention also concerns a method of selectively inducing terminal differentiation of neoplastic cells and thereby inhibiting proliferation of such cells which comprises contacting the cells under suitable condition with an amount of the compound effective to selectively induce terminal differentiation.
Moreover, the invention provides a method of treating a patient having a tumor characterized by proliferation of neoplastic cells which comprises administering to the patient an amount of the compound effective to selectively induce terminal differentiation of such neoplastic cells, thereby inhibiting their proliferation, and suppressing oncogenicity.
Lastly, the present invention provides a pharmaceutical composition comprising a pharmaceutically acceptable carrier and the compound in an amount less than an amount which cause toxicity in the patient.
Brief Description of the Figures
Figure 1: Comparison of the hybrid polar/apolar compounds,
(A) hexamethylene bisacetamide (HMBA) ; (B) suberic acid bis-N-methyl diacetamide (SBDA) ; and
(C) bis-hexamethylene triacetamide (BHTA) as inducers of differentiation of vincristinesensitive (745A-DS19) (●) and vincristineresistant (VCR.C(2) 15) (▲) MELC for HMBA and
SBDA and (V3.17) for BHTA. Each compound was added to the cells in culture at the final concentration indicated. Benzidine reactive cells (left panel of each section of this figure) were determined after 4 d. of culture and commitment (right panel of each section of this figure) after 2 d. of culture. Figure 2: Concentration dependent curves of inducer activity of compound 12. with vincristine- sensitive (745A-DS19) (●) and vincristine- resistant (VCR.C(2) 15) (▲) MELC. The compound was added to the cultures at the final concentrations indicated. Benzidine reactive cells were determined after 5 d. in culture and commitment after 2 d. of culture.
Figure 3: Optimal concentration of IC-135 for inducement of terminal differentiation.
Figure 4: Comparison of HMBA and IC-135 on DS19 cells.
Figure 5A
and B: Comparison of % cell committed for HMBA and IC- 135 on V3.17 and DS19 cell lines.
Figure
6A and B: Comparison of B+% for HMBA and IC-135 on V3.17 and DS19 cell lines.
Detailed Description of the Invention
The invention provides a class of compounds having the structure:
[R-A]-B-A1-B1-[A2-B2-]a[A3-B3-]b[A4-R1]
wherein each A, A1, A2, A3, and A4 represent a polar group which comprises a nitrogen, sulfur or oxygen atom and wherein each of A, A1, A2, A3, and A4 may independently be the same as, or different from, the others of A, A1, A2, A3, and A4; wherein each of R and R1 is a hydrogen atom; a lower alkyl, alkenyl, or alkynyl group; or a group having the structure:

The compounds of the present invention are made up of two components. One component comprises a polar group, i.e. functional groups with significant dipole moments, such as amides, sulfoxides, amine oxides and related functional groups.
The terminal portions of the compound, represented by R-A and A4-R1, each have dipole moments greater than about 2.7 debye units. The polar groups within the compound, represented by -A1, -A2- and -A3-, have significant dipolar moments but not necessarily in excess of 2.7 debye units. In the preferred embodiments, the polar groups are carbonyl radicals or bivalent radicals of an amide, a sulfoxide or a amine oxide. Each polar group need not necessarily be the same as the other polar groups. In the most preferred embodiments, the poplar groups within the compound are the same as each other and the terminal polar groups are the same. Preferably, all the polar groups are amide groups attached to the compound at the nitrogen atom or at the carbon atom of the carbonyl radical. The amide group may comprise one or more hydrocarbon substituents, such as a lower alkyl or alkenyl groups, including branched or unbranched groups. The term "lower alkyl or alkenyl group" is intended to include saturated and unsaturated hydrocarbon groups with 1 to about 5 carbon atoms.
The embodiments where a and b are 0 and A is a carbonyl radical or a group having the structure:

wherein R4 is a hydrogen atom or a lower alkyl or alkenyl group, have proven to be most useful embodiments to date.
Particularly preferred are compounds where a and b are O, A is a carbonyl radical and R has the structure:

The compound also requires at least two nonpolar sections, designated B and B1, which are attached to and connect polar groups. Additional nonpolar sections may also be present, e.g. B2 when a is 1 and B3 when b is 1. The nonpolar sections may comprise linear saturated hydrocarbon chains, linear unsaturated hydrocarbon chains containing one or more double or triple bonds, or saturated or unsaturated hydrocarbon chains containing one or more lower alkyl or alkenyl groups or small carbocyclic rings as substituents. In one of the preferred embodiments, the nonpolar groups are hydrocarbon chains comprising 4 to 7 methylene groups, especially preferred are hydrocarbon chains containing 6
carbon atoms.
Some of the preferred compounds for the practice of the present invention are those having the structures:




o

or


or a compound having the structure:

wherein R1 and R2 may be the same or different and each is a lower alkyl group.
Also provided by this invention is a compound having the structure:

This invention further provides a compound having the structure:

wherein n is an integer which is greater than 1 and R1 and R2 may be the same or different and each is H or a lower alkyl group.
A compound having the structure:

This invention also provides a compound having the structure:
 or a compound having the structure:
or a compound having the structure:

wherein R1 and R2 may be the same or different and each is H or a lower alkyl group; and n is an integer from 1 to 10.
This invention further provides a compound having the structure:
 wherein R1, R2, R3, R4, R5, R6 may independently be the same or different from each other and is H or a lower alkyl group; wherein X is methyl or phenyl; and n is an integer from 1 to about 15.
wherein R1, R2, R3, R4, R5, R6 may independently be the same or different from each other and is H or a lower alkyl group; wherein X is methyl or phenyl; and n is an integer from 1 to about 15.
A compound having the structure:

is also provided by this invention wherein R1 and R2 may be the same or different and is H or CH3.
The invention also provides a compound having the structure:

CH 3.
A compound having the structure:

is provided by this invention, wherein X has the structure:
or


Further provided by this invention is a compound having the structure:

wherein R1 and R1 may independently be the same or different and each may be H or a lower alkyl group; wherein R3 and R4 may independently be the same or different and each may be CH3 or OH; and wherein n is 5 or 6.
A compound having the structure:

This invention further provides a compounds having the following structures:
 and
and
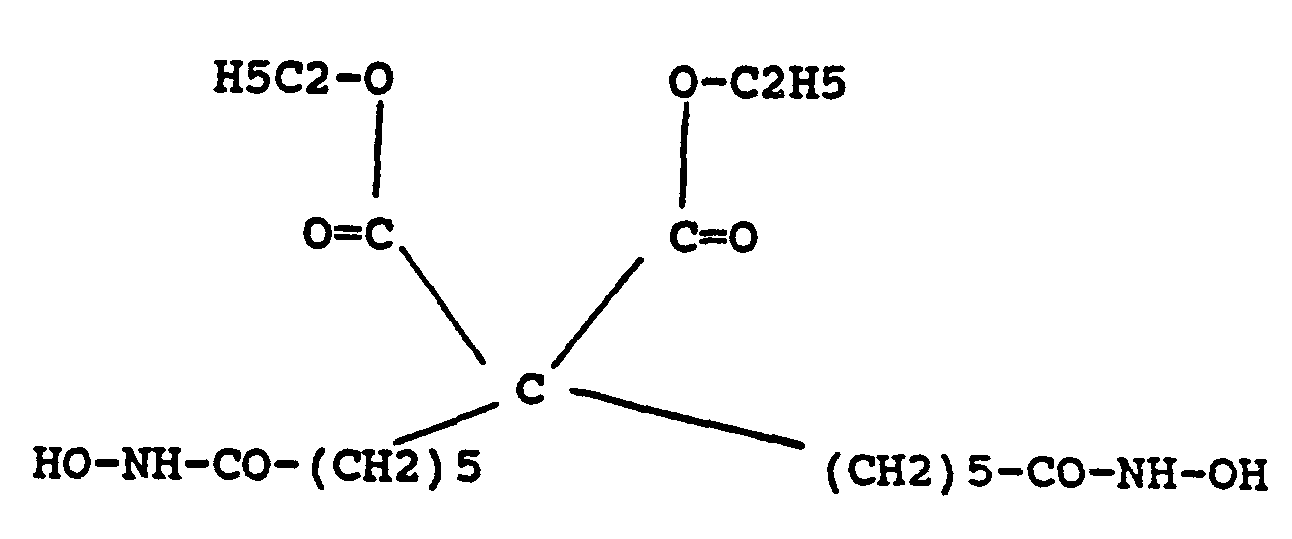


The invention also concerns a method of selectively inducing terminal differentiation of neoplastic cells and thereby inhibiting proliferation of such cells which comprises contacting the cells under suitable conditions with an amount of the compounds shown above effective to selectively induce terminal differentiation in the cells.
A method of selectively inducing terminal differentiation of neoplastic cells and thereby inhibiting proliferation of such cells is further provided by this invention which comprises contacting the cells under suitable conditions with an amount of a compound effective to selectively induce terminal differentiation, the compound having the structure:

wherein X is phenyl or methyl and n is an integer from 1 to 15.
The contacting must be performed continuously for a prolonged period of time, i.e. for at least 48 hours, preferably for about 4-5 days or longer.
The method may be practiced in vivo or in vitro. If the method is practiced in vitro, contacting may be effected by incubating the cells with the compound. The concentration of the compound in contact with the cells should be from about 1 μM to abount 25 mM, preferably from about 4 μM to about 5 mM.
However, for the compound having the structure:

The concentration depends upon the individual compound. For example, compound 12 of Table 1 should have a concentration from about 0.1 to about 2 mM, preferably from about 0.5 to about 0.7 mM. However, for the compounds listed in Table 4, the optimal concentration is as low as 5μM to about 5 mM. Another factor determining the preferable range is the state of the tumor cells. Thus, in cells which have low levels of vincristine resistance, the range of effective concentration of compound 12 from Table 1 is from about 0.01 to about 0.3 mM with a preferable range of about 0.05 to about 0.1 mM.
The invention also concerns a method of treating a patient having a tumor characterized by proliferation of neoplastic cells which comprises administering to the patient an amount
of the compounds shown above, or with a compound having the structure:

The method of the present invention is intended for the treatment of human patients with tumors. However, it is also likely that the method would be effective in the treatment of tumors in other animals. The term tumor is intended to include any cancer caused by the proliferation of neoplastic cells, such as lung cancer, acute lymphoid myeloma, bladder melanoma, renal carcinoma, breast carcinoma, or colorectal carcinoma. The administration of the compound to the patient may be effected orally or parenterally. To date, administration intravenously has proven to be effective. The administration of the compound must be performed continuously for a prolonged period of time, such as for at least 3 days preferably more than 5 days. In the most preferred embodiments, the administration is effected continuously for at least 10 days and is repeated at intervals wherein at each interval the administration is continuously effected for at least 10 days. For example, the administration may be effected at intervals as short as 5-10 days, up to about 25-35 days and
continuously for at least 10 days during each such interval. The optimal interval period will vary depending on the type of patient and tumor. For example, in the incidence of acute leukemia, the so called myelodysplastic syndrome, continuous infusion would seem to be indicated so long as the patient tolerated the drug without toxicity and there was a positive response. The amount of the compound administered to the patient is less than an amount which would cause toxicity in the patient. In the certain embodiments, wherein the compounds have the structures:


or

This invention, in addition to the above listed compounds, is intended to encompass the use of homologs and analogs of such compounds. In this context, homologs are molecules having substantial structural similarities to the above-described compounds and analogs are molecules having substantial biological similarities regardless of structural similarities.
The invention also concerns a pharmaceutical composition comprising a pharmaceutically acceptable carrier, such as sterile pyrogen-free water, and any of the compounds listed above in an amount less than an amount which if administered intravenously or orally to a patient would cause the patient's blood plasma concentration of the compound to exceed toxic levels.
The invention is illustrated in the Experimental Detail section which follows. This section is set forth to aid in an understanding of the invention but is not intended to, and should not be construed to, limit in any way the invention as set forth in the claims which follow thereafter.
Experimental Details
Methods
Cells and Materials
MELC 745A-DS19 cells and the variants of MELC derived from this cell line, namely, the vincristine-resistant MELC V3.17 and VCR.C(2) 15 cell lines (26), and the dimethylsulfoxide-resistant cell line, DR10 (37), were maintained in alpha minimal essential medium containing 10% fetal calf serum (16). Cell cultures for all experiments were initiated with cells in logarithmic growth phase (day 2 cultured cells) at a density of 105 cells/ml. Inducer compounds were added in the final concentrations indicated below, dissolved in culture medium without fetal calf serum unless otherwise indicated. Cell density and benzidine reactively were determined as described (16).
Commitment to terminal differentiation, characterized by limited cell division (colony size <32 cells) and accumulation of hemoglobin (benzidine reactive colonies) was assayed by a colony cloning assay using 2% methylcellulose as described (25).


 Chemistry
Chemistry
HMBA, compound 1 of Table 1, (9) was obtained from Aldrich Chemical Co.
The preparation and characterization of compounds 1, 2, 6, 7 , 8 and 9 of Table 1 (9,27) was previously described. All new amides were purified by chromatography on alumina with 5% methanol in methylene chloride, and were judged pure by thin layer chromatography (single spot) and 1H NMR spectroscopy. Final products were characterized by 1H NMR, infrared, and CI mass spectroscopy, while intermediates were characterized by 1H NMR spectra. The data were consistent with the assigned structures (expected infrared and NMR signals, M + 1 mass spectra).
For the synthesis of compound 3 (Table 1), the known 3-methyl-1,5-dibromopentane (38) was converted to the bis-phthalimide derivative, and this was cleaved with hydrazine to afford 3-methyl-1,5-diaminopentane, isolated as the dihydrochloride (m.p. 123-126º). This was converted to compound 3 with acetic anhydride and triethylamine in dioxane.
Compound 4, (Table 1) (m.p. 67-68º) was obtained in quantitative yield by similar acetylation of commerically available 2-methyl-1,5-diaminopentane.
Compound 5 (Table 1) was synthesized from meso-2,3-dimethylsuccinic acid in six steps. Reduction of the dimethyl ester with LiAlH4 afforded the meso-2,3-dimethylbutanediol (92% yield). This was converted to the bis-tosylate, and then to the bis-phtalimide. Deprotection
and acetylation as before gave compound 5 as an oil (61% yield from the diol).
Compound 10 (Table 1) was prepared by making a solution of 19.8gm commercial bis-hexamethylenetriamine in 500 ml of 1,4-dioxane at room temperature under argon. Then 44.8 ml. of triethylamine was added, and 20.3 ml. of acetyl chloride was slowly added with stirring. After two hours of stirring at room temperature the triethylamine hydrochloride was removed by filtration and the filtrate was concentrated in vacuo. The product triacetyl compound was isolated as a clear viscous oil at about 90% yield by chromatography on basic alumina using isopropanol/ethyl acetate/dichloromethane in the ratio 2/3/5. On thin layer plates of basic alumina with this solvent mixture the product had an Rf of ca. 0.6.
The mass spectrum (chemical ionization, NH3 carrier) showed peaks at 342 (100%, M + 1), 227 (10%) and 115 (22%). The infrared spectrum (thin film on NaCl) had bands at 3288, 2931, 2858, 1627, 1560, 1437, 1373, and 1292 cm-1. In the proton NMR (CDCl3) the acetyl groups appeared at 6.10 as a broad signal, while the methylene protons appeared as multiples with the expected intensities in the regions of 3.12 to 3.30 and 1.21 to 1.54.

bis-Hexamethylenetriacetamide (IC-135) For preparation of the triamide compound 11 (Table 1),
dimethyl malonate was dialkylated with ethyl 6-bromohexanoate under standard conditions. The resulting tetraester was then hydrolyzed and thermally monodecarboxylated to 1,7,13-tridecanetricarboxylic acid. Treatment with thionyl chloride followed by diethylamine afforded compound H (Table 1).
Compound 12 (Table 1) was prepared by simple dialkylation of dimethyl malonate with the know N,N'-dimethyl-6-bromohexanecarboxamide (39).
The compound having the structure:

was made as follows. 1,3-dimethylbarbituric acid (5g; 0.032 mol) in DMF (100 ml) was slowly added to a suspension of sodium hydride (1.5g; 0.064 mol) in DMF (300 ml). The suspension was stirred at 80ºC for five hours. N,N-Dimethyl 6-bromohexanoylamide (14.3 g; 0.064 mol) in DMF (100 ml) was added to the cool suspension with stirring. The resulting suspension was stirred at 120ºC overnight. The DMF was evaporated and the residue was partitioned between chloroform and water (100-100 ml). The chloroform layer was separated, and the water layer was extracted with chloroform (5 × 50 ml). The combined chloroform layers were dried over anhydrous magnesium sulfate, and evaporated. The oily residue was purified by column chromatography on silica gel in ethyl acetate-tetrahydrofuran (2:1). The yield of 1,3-
dimethyl-5,5-di(N,N-dimethyl-5-pentylcaboxamide)barbituric acid was 4.1 g (30%).
% NMR (CDCl3, 200 MHz) δ3.33(s, bar, N-CH3,6H), 2.98 (s, N-CH3, 6H), 2.93 (s, N-CH3, 6H), 2.24 (t, J=7 Hz, CH2CON, 4H), 1.96 (m, 4H), 1.58 (m, 4H), 1.28 (m, 4H), 1.08 (m, 4H).
The compound having the structure:
was made as follows. Diethyl malonate (50.6 ml; 53.3 g; 0.33 mol) in DMF (150 ml) was added slowly to a cool suspension of sodium hydride (16 g; 0.67 mol) in DMF (1 liter). The suspension was carefully heated with stirring at 80º CC (about one hour). The clear mixture was cooled, and N,N-dimethyl 6-bromohexanoylamide (146.5 g; 0.66 mol) in DMF (200 ml) was slowly added at room temperature. The suspension was stirred at 110ºC for two hours, and the DMF was evaporated. The semisolid residue was partitioned between chloroform and water (about 200-200 ml). The organic layer was separated, and the water layer was extracted with chloroform (5 × 100 ml). The combined chloroform layers were dried over anhydrous magnesium sulfated and evaporated. The oily residue was purified by column chromatography on silica gel in ethyl acetate-tetrahydrofuran (4:1). The yield of diethyl undecane-6,6-di(ethylcarboxylate)-1,11-di(N,N-dimethlycarboxamide) was
106 g (76%) .
1H-NMR(CDCl3 , 200 MHz) δ4 . 11 (q, J=7.0 Hz , 4H) , 2.96 (t,J=7.5Hz , 1H) , 2.89 (s, 6H) , 2.25 (t, J=7.2Hz , 4H) , 1.88 (m,
4H) , 1.59 (m, 4H) , 1.16-1.31 (m, 14H) .
IR (NaCl, CH2Cl2) 2937, 2866, 1728, 1642, 1498, 1462, 1399, 1369, 1265, 1153, 1097, 1029 cm-1.
MS (FAB, glycerol) m/e=443 (40%, M+1+), 398 (25%), 295 (80%), 142 (45%).
The compound having the structure:

was made as follows. 1,3,5,-trimethylbarbituric acid (1.7g; 10 mmol) in DMF (20ml) was added at room temperature to a suspension of sodium hydride (0.24 g; 10 mmol) in DMF (100 ml). The suspension was stirred at 90°C for one hour. N,N-Dimethyl 6-bromohexanoylamide (2.22 g; 10 mmol) in DMF (20 ml) was added to a cool mixture (about 5ºC). The suspension was stirred at 110ºC overnight. DMF was evaporated, and the residue was partitioned between chloroform and water (50-50 ml). The chloroform layer was separated, and the water layer was extracted with chloroform. The combined chloroform layers were dried over anhydrous magnesium sulfate and evaporated. The oily residue was purified by
column chromatography on silica gel in ethyl acetate-tetrahydrofuran. The yield of 1,3-dimethyl-5-methyl-5-(5-pentyl-N,N-dimethylcarboxamide)barbituric acid was 1.5 g(48%).
1H NMR (CDCl3, 200MHz) 53.23(s, bar.H, N-CH3,6H), 2.99 (s, N-CH3, 3H), 2.94 (s, N-CH3, 3H), 2.26 (t, J=7.2 Hz, CH2CON, 2H), 1.98 (m, 2H), 1.62 (m, 2H), 1.52 (m, C-Me, 3H), 1.32 (m, 2H), 1.11 (m, 2H).
The compounds having the structure:

The compounds having the structure:

1H NMR (CDCl3, 200 MHz) 57.38 (q, J=8 Hz), 3.03 (s, N- CH3,6H), 2.94 (s,N-CH3,6H), 2.8 (d, J=8 Hz, N-CH3,6H), 2.32
(t, J=7.2 Hz, CH2CON, 4H), 2.82 (m, 4H), 2.62 (m, 4H), 1.25 (m, 8H).
When X1 and X2 are the same and each is N(CH3)2, the hexamethylamide (2 g; 4.9 mol) in DMF (10 ml) was added at room temperature to a suspension of sodium hydride (0.24 g;
0.01 mol) in DMF (20 ml). The mixture was stirred at 80ºC for one hour. Methyl iodide (3.5 ml; 8 g; 56 mmol) was added with stirring to the cool reaction mixture. The reaction was continued at 60ºC overnight. The solvent was evaporated and the solid residue was dissolved in water (50 ml) and the water solution was extracted with 8:2 chloroform-methanol (5 × 50 ml). The combined organic layers were dissolved in acetone (200 ml) and filtered through a short column of silica gel. Acetone was evaporated yielding pure undecane-1,6,6,11-tetra-(N,N-dimethylcarboxamide) (1.36 g; 63%).
% NMR (CDCl3) 52.99 (s, N-CH3, 6H), 2.94 (s, N-CH3, 6H), 2.92 (s, N-CH3, 6H), 2.88 (s, N-CH3, 6H), 2.28 (t, J=7.2 Hz, CH2CON, 4H), 1.86 (m, 4H), 1.62 (m, 4H), 1.32 (m, 8H).
The compounds having the structure:

wherein n is an integer from 1 to 15 are made as follows. A dibarbituric acid (3.66 g; 0.01 mol for n = 2) was suspended in 9:1 water-dioxane (60 ml) and heated up to 100ºC. To this suspension was added at 110ºC a solution of potassium hydroxide (5.6 g; 0.1 mol/20 ml water for n=2). The suspension immediately became a solution. The solution was heated at 110ºC for an extra 10 minutes, cooled down, filtered, and acidified. The clear water solution was extracted with chloroform (10 × 50 ml). The combined
chloroform extracts were dried over anhydrous magnesium sulfate, and evaporated. The yields of the alkane-1,1,n,n,-tetra-(N-methylcarboxamide) were for: n=2, 85%; n=3, 79%; n=4, 83%; n=5, 81%; n=6, 74%; n=7, 83%.
1H NMR (CDCl3, 200 MHz) for compounds having n=2, 57.92 (q, J=4.6 Hz, NH-Me, 4H), 2.85 (d, J=4.6 Hz, N-CH3, 12H), 1.85 (s, CH2, 4H), 1.43 (s, C-CH3, 6H).
The compounds having the structure:

wherein R1, R2, R3, R4, R5, R6 may independently be the same or different from each other and is H or a lower alkyl group; wherein X is methyl or phenyl; and n is an integer from 1 to about 15, were made as follows. When R1 and R6 is each H and R2, R3, R4, and R5 is each CH3, to a chloroform (200 ml) solution of suberoyl chloride (8 g; 38 mmol) was added at room temperature to 1,1-dimethylhydrazine (11.5 ml; 9.1 g; 152 mmol). The mixture was stirred at room temperature for about three hours. The solvent was evaporated, and the residue was dissolved in methanol (400 ml). The methanol was evaporated and the solid residue was dissolved in water (100 ml). The water solution was extracted with hexanes (5 × 50 ml), and with chloroform (5 × 50 ml). The combined extracts were dried over anhydrous magnesium sulfate and evaporated. The solid residue was crystallized from acetone. The yield of hexane-1,6-di(N2,N2-dimethyl-carboxhydrazide) was 6.5 g (66%). When R1, R2, R3, R4, R5, and R6 is each CH3, to a suspension
of sodium hydride ( 1.4 g; 57.9 mmol) in DMF (60 ml) at room temperature was added tetramethylhydrazide (5 g; 19.3 mmol) followed by methyl iodide (31.5 ml; 71.8 g; 579 mmol). The reaction mixture was stirred at 60*C for five hours. The DMF was evaporated, and the solid residue was dissolved in water (50 ml) and extracted with chloroform (5 × 50 ml). The chloroform extracts were dried over anhydrous magnesium sulfate and evaporated. The product was purified by crystallization from hexanes. The yield of hexane-1,6-di(N1,N2,2-trimethylcarboxhydrazide) was 3.7 g (67 %).
The compounds having the structure:

The compounds having the structure:

wherein R1 and R2 may be the same or different and is H or CH3 may be made as follows. The heterocyclic part of the compound may be made from malonic chloride and N,N1- dealkylethylenediamine. The heterocyclic compound may then be alkylated with the corresponding alkyl bromide under known conditions.
The compounds having the structure:
 and
and

may be synthesized from the corresponding acid via acidic chloride with hydroxylamine hydrochloride in a solution of tetrahydrofuran and water. The acid may be made from 1,4- benzenediacrylic acid by an addition of bromine, and following elimination with base.
The compound having the structure:

1H NMR (DMSO-d6, 200 MHz), 11.05 (broad s, OH, 2H), 9.12 (broad s, NH, 2H), 7.77 (d, J=8.2 Hz, arom. H, 2H), 7.62 (d, J=8.2 Hz, arom. H, 2H), 7.47 (d, J=15.8 Hz, CH, 2H), 6.53 (d, J=15.8 Hz, CH, 2H).
The compound having the structure:

was made as follows. A suspension of trans-B-hydromuconic acid (5 g; 0.0347 mol), oxalyl chloride (12 ml; 17.6 g; 0.139 mol) in benzene (250 ml) and a few drops of DMF was stirred at room temperature until the reaction suspension became clear (about one hour). The solvent was evaporated, and the oily residue was dissolved in tetrahydrofuran (50 ml). The tetrahydrofuran solution was slowly added into cool (about 5ºC) aqueous solution of hydroxylamine hydrochloride (4.8 g; 0.0694 mol) and potassium hydroxide (7.8 g; 0.1388 mol). The reaction suspension was stirred at room temperature for about one hour and evaporated to a volume of 20 ml. The suspension was cooled and filtered. The crude solid product was slurried in methanol (200 ml) and the suspension was cooled and filtered. The filtrate was dried over anhydrous magnesium sulfate, and evaporated to a solid residue. The solid was slurried in ether (250 ml) and recovered by filtration. The yield of trans-2-butene-1,4-di-(N-hydroxylcarboxamide) was 3.8 g (63%).
1H NMR (DMSO-d6, 200 MHz) , δ 10.85 (s , OH, 2H) , 9.08 (s , NH, 2H) , 7. 11 (d of d, J1=11.2 Hz , J2=2 .8 Hz , CH, 2H) , 6. 17 (d
of d, J1=11.4 Hz, J2=2.8 Hz).
The compound having the structure:

was made as follows. A solution of trans, trans-muconic acid (5 g; 0.035 mol), oxalyl chloride (12.3 ml, 17.9 g; 0.141 mol) and a few drops N,N-dimethyl formamide in benzene (250 ml) was stirred at room temperature for three hours. The solvent was removed under reduced pressure. The oily residue was dissolved in tetrahydrofuran (50 ml) and the tetrahydrofuran solution was added dropwise to the stirred solution of hydroxylamine hydrochloride (4.9 g; 0.074 mol) and potassium hydroxide (7.9 g; 0.141 mol) at about 5ºC A white precipitate was formed immediately. The suspension was stirred at room temperature for about one hour and filtered. The solid was slurried in methanol (2 liters) and the suspension then was boiled and filtered. The methanol solution was concentrated to a volume of about 20 ml. The solid product was separated by filtration and washed with ether (5 × 20 ml). The yield of trans,trans-1,3-butadiene-1,4-(N-hydroxylcarboxamide) was 3.1 g (52%).
1H NMR (DMSO-d6, 200 MHz) , 10.46 (s , 2H, OH) , 8.75 (s , 2H, NH) , 5.53 (t, J=3 .4 Hz , CH, 2H) , 2 .70 (d, J=3 .4 Hz , 4H, CH2) .
The compound having the structure:

was made as follows. A suspension of 1,4-phenylenediacrylic acid (10 g; 0.184 mol) and thionyl chloride (13.4 ml; 21.9 g; 0.184 mol) in benzene (500 ml) was refluxed for five hours. The clear solution was cooled, and the solvent was evaporated. The oily residue was dissolved in tetrahydrofuran (100 ml) and slowly added into cooled (about 5ºC) aqueous (100 ml) solution of hydroxylamine hydrochloride (6.4 g; 0.092 mol) and potassium hydroxide (10.3 g; 0.184 mol). A white precipitate was formed immediately. The suspension was stirred at room temperature (two hours), and filtered. The crude product was slurried in boiling water (100 ml), and the suspension was filtered. The solid was slurried in acetone (200 ml) and boiled. The hot suspension was filtered, and the solid was washed with acetone (5 × 20 ml), and then with ether (5 × 20 ml). The
yield of benzene-1 , 4-di [trans-ethene-2- (N-hydroxulcarboxamide) ] was 6.2 g (54%) .
1H NMR (DMSO-d6, 200 Hz) , δ 10.82 (s, OH, 2H) , 9.17 (s , NH, 2H) , 7.58 (s, C6H4, 4H) , 7.45 (d, J=15. 8 Hz , CH, 4H) , 6.51 (d, J=15.8 Hz) .
The compound having the structure:

was made as follows. A suspension of 4,4-diphenyldicarboxylic acid (5 g; 20.6 mmol) and thionyl chloride (7.3 ml; 12 g; 0.1 mmol) in benzene (250 ml) was refluxed overnight. The clear solution was cooled, and the solvent was evaporated. The solid residue was dissolved in tetrahydrofuran (100 ml) and was added slowly into a cool (about 5ºC) aqueous (100 ml) solution of hydroxylamine hydrochloride (2.9 g; 41.7 mmol) and potassium hydroxide (4.65 g; 83 mmol). A white precipitate was formed immediately. The suspension was stirred at room temperature for about two hours and filtered. The crude product was slurried in water (50 ml) and the resulting suspension was boiled (about 10 minutes). The solid was separated by filtration, and slurried in acetone (200 ml) and boiled. The hot suspension was filtered, and the solid was washed with acetone (5 × 50 ml), and then with ether (5 × 20 ml). The yield of diphenyl-4,4'-di-(N-hydroxylcarbox-amide) was
4 .9 g (88%) .
1H NMR (DMSO-d6, 200 MHz), δ 11.33 (s, OH, 2H) , 9.12 (broad s, NH, 2H), 7.87 (d, J=8.2 Hz, arom. H, 2H), 7.80 (d, J=8.4 Hz, arom. H, 2H).
The compound having the structure:

(5 × 50 ml). The combined chloroform layers were dried over anhydrous magnesium sulfate, and evaporated. The oily residue was purified by column chromatography on silica gel in hexanes-ethyl acetate. The yield of undecane-6,6-di(ethylcarboxylate)-1,11-di(benzylσarboxylate) was 41 g (72
%) . Dibenzyl 6,6-di(ethoxycarbonyl)-1,11-undecanedicarboxylate (20 g; 35.2 mmol) was dissolved in methanol (150 ml) and 5 % Pd-C (200 mg) was added. The reaction
suspension was hydrogenated at room temperature overnight. The catalyst was removed by filtration, and the methanol was evaporated yielding pure 6,6-(ethylcarboxylate)-1,11-undecanedicarboxylic acid (12.5 g; 93%).
The acid was suspended in benzene (500 ml), and oxalyl chloride (11.2 ml; 16.3 g; 0.129 mol) and a few drops of DMF were added. The mixture was stirred at room temperature for about 3 hours. The solvent was evaporated and the oily residue was dissolved in chloroform (100 ml). The chloroform solution of the acid chloride was slowly added to a stirred solution of O-behzylhydroxylamine (16.3 g; 0.132 mol) in chloroform (300 ml). A white precipitate was formed immediately. The suspension was filtered, and concentrated hydrochloric acid was added (50 ml) to the chloroform filtrate and again filtered. The chloroform layer was washed with concentrated hydrochloric acid (3 × 100 ml), water (3 × 100 ml), and dried over anhydrous magnesium sulfate. The chloroform was evaporated yielding pure N,N'-dibenzyloxy-6, 6-di(ethoxycarbonyl)-1,11-undecanedicarboxamide (17.1 g; 89%).
The benzyloxyamide (6 g; 0.01 mol) was dissolved in methanol (50 ml), and 5 % Pd-C (200 mg) was added. The methanol suspension was hydrogenated at room temperature overnight. The catalyst was separated by filtration and methanol was evaporated yielding pure undecane-6,6-di(ethylcarboxylate)-1,11-di(N-hydroxylcarboxamide) (3.9 g; 93%). The overall yield was 55.4%.
1H NMR (DMSO-d6, 200 MHz), 10.30 (broad s, OH, 2H), 8.75 (broad s, NH, 2H), 4.10 (q, J=7 Hz, COOCH2CH3, 4H), 1.90 (t, J=7.2 HZ, CH2CONHOH, 4H), 1.71 (m, 4H), 1.45 (m, 4H), 1.18
(m, 8H), 1.14 (t, J=7 Hz, COOCH2CH3, 6H)
The compounds having the structure:

wherein R1 and R1 may independently be the same or different and is H or a lower alkyl group, wherein R3 and R4 may independently be the same or different and each is CH3 or OH, and wherein n is 5 or 6 were made as follows. 1,3-dimethylbarbituric acid (15.6 g; 0.1 mol) was slowly added at room temperature to a suspension of sodium hydride (4.8 g; 0.2 mol) in N,N-dimethylformamide (300 ml). The reaction was heated with stirring at 100ºC for about three hours. A DMF (200 ml) solution of benzyl 6-bromohexanoate (57 g; 0.2 mol) was slowly added to the cool suspension with vigorous stirring. The suspension was heated at 110ºC overnight, and the solvent was evaporated. The residue was partitioned between chloroform and water (300-300 ml). The chloroform layer was separated and the water layer was extracted with chloroform (5 × 50 ml). The combined chloroform layers were dried over anhydrous magnesium sulfate, and evaporated. The oily residue was purified by column chromatography on silica gel in ethyl acetate-tetrahydrofuran (9:1). The yield of
dibenzyl ester was 18 g (32%).
The dibenzyl ester (18 g; 0.032 mol) was dissolved in methanol (50 ml) and catalyst (200mg of 5 % Pd-C) was added. The suspension was hydrogenated overnight, and the catalyst was removed by filtration. The methanol was evaporated yielding pure acid (11.5 g; 94%). The acid (11 g; 0.0286 mol) was suspended in benzene (500 ml), and oxalyl chloride (10ml; 14.5 g; 0.14 mol) and a few drops of DMF were added. The mixture was stirred at room temperature for about 3 hours and evaporated to an oily residue. The oily residue was dissolved in chloroform (100ml) and poured into a chloroform (100 ml) solution of O-benzyl-hydroxylamine (14.4 g; 0.117 mol). A white precipitate was formed immediately. After two hours stirring at room temperature the suspension was filtered. Concentrated hydrochloric acid (50 ml) was added to the filtrate and again filtered. The chloroform layer was separated and washed with concentrated hydrochloric acid (3 × 100 ml), water (3 × 100 ml) and dried over anhydrous magnesium sulfate. The chloroform was evaporated yielding pure N-benzyloxyamide (14 g; 82%).
The N-benzyloxyamide (6 g; 0.01 mol) was dissolved in methanol (509 ml) and catalyst (200 mg of 5% Pd-C) was added. The suspension was hydrogenated at room temperature overnight. The catalyst was removed by filtration and the methanol was evaporated yielding pure hydroxamic acid (3.95 g; 95%). The overall yield of 1,3-dimethyl-5,5-di(5-pentyl-n-hydroxylcaboxamide)barbituric acid was 23.4%.
1H NMR (DMSO-d6, 200 MHz), δ 10.30 (s, OH, 2H) , 8.60 (broad s, NH, 2H), 3.18 (s, N-CH3, 6H), 1.85 (t, J=7.2 Hz,
CH2CONHOH, 4H), 1.80 (t, J=7 Hz, CH2-bar., 4H), 1.39 (m, 4H), 1.10 (m, 8H). The compound having the structure:

wherein R1 and R2 independently be the same or different and each may is H or a lower alkyl group and n is an integer from 1 to 10 is synthesized as follows. The tetramethylamide (2 g; 6.4 mmol; for n=2) in DMF (10 ml) was slowly added to a suspension of sodium hydride (0.9; 38 mmol) in DMF (50 ml). The mixture was stirred at 80ºC for about 30 minutes. Iodomethane (23.3 ml; 53.2 g; 380 mmol) was added to this cool solution at a temperature of about 5ºC and the reaction was continued at 60ºC overnight. The solvent was evaporated, and the residue was dissolved in water (50 ml). The water solution was extracted with 20% methanol in chloroform (10 × 30 ml). The combined organic extracts were dried over anhydrous magnesium sulfate and evaporated. The product was purified by short column chromatography in acetone. The yields of the alkane-1,1,n,n-tetra-(N,N-dimethylcarboxamide) were for: n=2, 1.3 g (55%); n=3 (52%); n=4, 51%; n=5 (53%); n=6 (53%).
1H NMR (CDCl3, 200 MHz) for compound having n=2, δ 2.94 (s, N-Me, 12 H), 2.87 (s, N-Me, 12H), 1.82 (s, CH2, 4H), 1.32 (s, C-Me, 6H).
The compound having the structure:

was synthesized as follows. N-methy1anyline (7.3 ml; 7.219 g; 67.3 mmol) was slowly added to a solution of pimeloyl chloride (5 ml; 6.025 g; 30.6 mmol) in chloroform (500 ml). The suspension was stirred at room temperature for about three hours, and the solid was separated by filtration. The chloroform filtrate was washed with concentrated hydrochloric acid (3 × 100 ml), water (3 × 100 ml) and dried over anhydrous magnesium sulfate. The chloroform was evaporated, the solid residue was slurried in hexanes and filtered. The yield of pentane-1,5-di(N-methyl-N-phenylcarboxamide) was 6.3 g (90%).
1H NMR (DMSO-d6, 200 MHz), δ 7.30 (m, Ph, 10 H), 3.12 (s, N-Me, 6H), 1.93 (t, J=7 Hz, CH2CON, 4H), 3.31 (m, 4H), 1.01 (m, 2H).
The compound having the structure:

1H NMR (CDCl3, 200 MHz), δ 7.59 (d, J=7.4 Hz, arom. 2H), 7.28 (t, J=7.4 HZ, arom. 2H), 7.26 (s, NH, 1H), 7.10 (t, J=7.4 Hz,arom. 1H), 4.26 (q, J=7 Hz, COOCH2CH3, 2H), 2.99 (s, N-Me, 6H), 2.95 (s, N-Me, 6H), 2.24 (t, J=5.2 Hz, CH2CON, 4H) 1.85 (m, 4H), 1.60 (m, 4H), 1.65 (m, 8H).
The compounds having the structure:


was synthesized as follows. A solution of tetraphthaloyl chloride (5 g; 24 mmol) in tetrahydrofuran (50 ml) was slowly added (about 30 minutes) to a stirred solution of potassium hydroxide (5.52 g; 98.6 mmol) and hydroxylamine hydrochloride (3.4 g; 48.9 mmol) in water (100 ml) and the reaction was maintained at a temperature below 5ºC The reaction suspension was stirred an additional hour at room temperature. The suspension was evaporated, and the solid was slurried in boiling methanol (about 1 liter). The hot methanol suspension was filtered, the filtrate was dried over anhydrous magnesium sulfate and the methanol was evaporated. The solid residue was slurried in ethyl acetate (30 ml). The solid was separated by filtration and washed with ether (5 × 10 ml) and hexanes (5 × 10 ml). The yield of benzene-1,4-di(N-hydroxylcarboxamide) was 3.2 g; (66%).
The compound having the structure:
HO-N(CH3)-CO-(CH2)6-CO-n(CH3)-OH was made by slowly adding a solution of suberoyl chloride (5 g; 23.7 mmol) in tetrahydrofuran (20 ml) to a stirred water solution of potassium hydroxide (5.3 g; 94.8 mmol) and N-methylhydroxylamine hydrochloride (4 g; 47.4 mmol) at about 5ºC. The reaction mixture was stirred at room temperature
for about one hour. The organic layer was separated and the water layer was extracted with chloroform (5 × 50 ml). The combined chloroform layers were dried over anhydrous magnesium sulfate and evaporated. The product was purified by crystallization from acetone. The yield of hexane-1,6-di(N-hydroxyl-N-methylcarboxamide was 2.7 g (46%).
Each compound in Table 1 was assayed 3 or more times to determine effectiveness as an inducer of MELC (745A-DS19) cell line. The cell density of MELC in culture for 5 d. without inducer was 2.0 to 2.6 × 106 cells/ml. The cell density of MELC in culture for 5 d. with inducer was 1.2 to 2.0 × 106 cells/ml. Compounds 11 and 12 were dissolved in absolute ethyl alcohol. The final concentration of ethyl alcohol in the cultured medium ranged between 0.1 and 3%. This concentration of ethyl alcohol had no effect on cell growth of MELC in culture without inducer. All other compounds were dissolved in culture medium without fetal calf serum. The indicated optimal concentration represents the final concentration in the culture medium.
Results
New Hybrid Polar/Aoolar Compounds Active as
Cytodifferentiation Agents
In earlier studies (8), the present inventors reported that fairly high concentrations of certain polar organic solvents induce MELC to undergo erythroid differentiation. The question arose as to how the effectiveness of polar compounds might be increased so that similar concentrations of these solvent-like molecules could induce
differentiation. Although the mechanisms were not clear, and are still incompletely understood, the following hypothesis was considered: perhaps, at the target site of action, more than one solvent molecule must bind or interact. if this were so, the well-known chelate effect could provide more effective compounds. Instead of binding two or more independent solvent molecules, the cellular target might interact more efficiently with a single molecule carrying two or more solvent-like groups. Provided these groups were held in the right relationship one to the other, binding of one would carry the other into the target region providing a high, effective concentration. Such a chelate effect is well precedented; it accounts for the strong binding of metal ions by chelating ligands, and is the basis for much of the catalytic activity of enzyme.
This concept led the present inventors to new effective cytodifferentiating agents. The best studied to date is hexamethylene bisacetamide (HMBA, Table 1, compound 1), consisting of two acetamide molecules linked at nitrogen by a six carbon polyethylene chain (5,9). N-methyl acetamide (Table 1, compound 2) , another of the polar organic solvents, was shown to be effective, but only at high concentration (50 mM) , whereas HMBA induces erythroid differentiation in MELC at an optimal concentration of 5 mM (8). The present inventors previously showed that the optimum number of methylene groups in the apolar chain is six (27). In the present invention, it was found that a four or five carbon chain is comparably effective if extra carbons are provided as branching methyl groups (Table 1, compounds 3, 4 and 5). This suggests that the important factor is not simply the length of the chain but also the number of hydrophobic hydrocarbon units in it.
Acetamide can also be dimerized by linking the methyl groups of the molecule. Suberic acid bis-N-methylamide (SBDA; Table 1, compound 7), can be thought of as N-methylacetamide linked at the acetyl group by four methylene units. SBDA is comparable in activity as an inducer to HMBA (Figure 1A and B). Since the structures of SBDA and HMBA are different, it is likely that the metabolic fates of these two compounds are completely different, and their similarity in effectiveness means that the compounds themselves are the principal active agents, rather than their catabolic products.
HMBA and SBDA have their polar groups separated by identical methylene bridges, and they have a similar ratio of polar to apolar hydrophobic moieties. Many structurally related compounds have been examined, but only a few showed comparable or greater activity (Table 1, compounds 6 through 12). It is clear that the structures of active compounds may differ sufficiently from HMBA to make it likely that they will also display different pharmacokinetics. One of the more active of these hybrid compounds is a dimmer of HMBA. Bishexeunethylene triacetamide (BHTA; compound 10) is about 2 fold more active as inducer, on a molar basis, than HMBA (Figure 1C). The most active of the hybrid compounds assayed in this study is one in which two pentamethylene carboxyamides are linked by the dimethyl ester of malonic acid (compound 12). This compound, with 4 exposed polar groups balanced by two apolar pentamethylene domains, is roughly 10 fold more active than HMBA. For example, 0.6 mM compound 12 is about as effective as 5.0 mM HMBA, inducing over 90% of cells to differentiate after 5 d. in culture (Fig. 2).
Polymethylenediamine derivatives carrying propionyl groups instead of the acetyl groups of HMBA are also active, but methoxycarbonyl groups are less effective and bulky pivaloyl groups lead to loss of activity. The present inventors previously showed that HMBA can have a double bond (cis or trans) or a triple bond in the center and retain its activity (27). Replacement of the polymethylene chain with a cyclohexane ring leads to inactivity (27), although compound 9, with a longer chain interrupted by an amide group, is active.
Diamides of dicarboxylic acids, such as suberic acid, are active with either one or two methyl groups on each nitrogen (compounds 7 , 8, and 9), but not with a methyl and a methoxyl substituent, and they are also active with one (but not two) ethyl groups on each nitrogen. Suberic diamides of pyrollidine, of morpholine, or of piperazine are inactive. This shows that there is a limit, to the amount of carbon tolerated on the ends of the polar groups.
These findings indicate that for optimal activity, two, or even better, three or four uncharged polar groups of limited bulk must be connected by chains of about 5 to 6 carbons.
Increased Sensitivity of Vincristine-Resistant MELC to Polar/Applar Compounds
As previously reported, MELC resistant to relatively low concentrations of vincristine show a marked increase in sensitivity to HMBA (Fig. 1A) (26). The dose-response of
MELC to several of the new polar compounds identified as being as or more active than HMBA was examined. The compounds assayed for inducing activity with vincristine-
resistant MELC include compounds 1, 3, 4, 8, 10, 11 and 12 (Table 1). In each instance, vincristine-resistant MELC were induced at lower concentrations than were required for induction of vincristine-resistant MELC were induced more rapidly than the vincristine-sensitive cells. For the most active of the compounds, 0.1 mM compound 12 was the optimal concentration for inducing vincristine-resistant MELC (VCR.C(2) 15). For example, 0.1 mM compound 12 induced only 6% commitment of over 95% of V.C.R. (2) 15 cells after 2 d. and the accumulation of over 80% benzidine-sensitive (DS-19) MELC, 0.1 mM compound 12 induced only 6% commitment by d. 2 and 4% benzidine reactive cells by d. 5. At concentrations of compound 12 as low as 0.05, 35% of VCR.C(2) 15 became benzidine reactive by d. 2, compared to only 2% of DS-19.
Effect of Polar/Apolar Compounds on MELC Resistant to Inducer Mediated Terminal Cell Division
Several of the new polar/apolar compounds were evaluated as inducers of MELC cell line DR10, which is resistant to induction by dimethylsulfoxide and can be induced by HMBA to accumulate hemoglobin but not commit to terminal cell division (37). Compounds 7, 8 and 10, assayed as inducers of DR10, caused accumulation of hemaglobin, but not commitment to terminal cell division. Thus, these new hybrid polar/apolar compounds are similar to HMBA in their effect on DR10 cells.
Cell cultures were grown in the presence of different concentrations of Hexamethylenebisacetamide (HMBA)

and compound 10, bis-Hexamethylenetriacetamide (IC-135).
At 1, 2 and 5 days, the cell densities and the percentage of cells that were benzidine reactive (B+) were measured. Table 2 shows the cell densities, B+%, and percent of cells committed for cell lines DS19 and V3.17 grown in 1 mM to 5 mM of HMBA and IC-135. Figures 3, 4, 5A and B, and 6A and B are graphical representations of the data presented in Table 2.
Table 3 shows cell counts for days 1, 2 and 5 and percentage of cells committed and benzidine reactive (B+) at day 5 for cell liens DS-19, V3.17 and DR-10 grown in 5 mM of HMBA and 0.5 to 3 mM of IC-135.
As can be seen in Table 2 and 3 and Figures 3-6, IC-135 is more reactive in the tested cell lines at lower concentrations than HMBA.
Table 4 shows the results of several structurally different compounds when assayed 3 or more times to determine effectiveness as an inducer of MELC (745A-DS19) cell line. The cell density of MELC in culture for 5 d. without inducer was 2.0 to 2.6 × 106 cells/ml.
Discussion
The development of agents which can induce transformed cells to differentiate and suppress their oncogenicity has important implications for the treatment of cancer. While a number of agents have been identified that can induce tumor cells in vitro to express features of their differentiated phenotype and to decrease their proliferative capacity (4,10-24), these agents have generally proved to be relatively ineffective or two toxic when evaluated in clinical trials (40).
Among cytodifferentiation agents, the hybrid polar/apolar compound, HMBA, has been one of the best characterized with respect to its in vitro inducing activity in MELC and in a number of other transformed cell lines, as well as for certain human tumor explants (30). It can trigger a differentiation program in transformed cells which is similar to that of their normal lineage (5).
The recent finding that vincristine-resistant MELC are more sensitive to HMBA and to other active hybrid polar/apolar compounds provides an approach toward identifying the mechanisms of HMBA action (26). The lack of a latent period for inducer-mediated differentiation of vincristine-resistant cells suggests that an early effect of inducers may involve alternations (e.g. new proteins, or modification of existing proteins such as phosphorylation) which are constitutively expressed in the vincristine-resistant cell lines, and may, therefore, be identified and characterized.
The observed positive therapeutic responses to HMBA, albeit largely transient, occurred despite relatively low serum
concentrations of HMBA (<1 mM), compared to the optimum demonstrated in vitro (4 to 5 mM) (35,36). The present invention provides a new group of hybrid polar/apolar compounds which are as active or even more active, on a molar basis, than HMBA.



1. Sporn, M.B., Roberts, A.B., Driscoll, J.S. (1985) in Cancer: Principles and Practice of Oncology, eds. Hellman, S., Rosenberg, S.A. & DeVita, V.T., Jr., Ed. 2, (J.B. Lippincott, Philadelphia), P. 49.
2. Breitman, T.R., Selonick, S.E., and Collins, S.J.
Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA., 77:2936-2940, 1980.
3. Olsson, I.L., and Breitman, T.R. Induction of differentiation of the human histiocytic lymphoma cell line U.937 by retinoic acid and cyclic adenosine 3':5'- monophosphate-inducing agents. Cancer Res., 42:3924- 3927, 1982.
4. Schwartx, E.L. & Sartorelli, A.C. (1982) Cancer Res.,
42, 2651-2655.
5. Marks, P.A., Sheffery, M. & Rifkind, R.A. (1987) Cancer Res. 47. 659.
6. Sachs, L. (1978) Nature (Lond.) 274, 535.
7. Friend, C., Scher, W., Holland, J.W. & Sato, T. (1971) Proc. Natl. Acad. Sci. (USA), 68, 378-382.
8. Tanaka, M., Levy, J., Terada, M., Breslow, R., Rifkind, R.A. & Marks, P.A. (1975) Proc. Natl. Acad. Sci. (USA), 72, 1003-1006.
9. Reuben, R.C., Wife, R.L., Breslow, R., Rifkind, R.A. & Marks, P.A. (1976) Proc. Natl. Acad. Sci. (USA), 73,
862-866.
10. Abe, E., Miyaura, C., Sakagami, H., Takeda, M., Konno, K., Yamazaki, T., Yoshika, S. & Suda, T. (1981) Proc. Natl. Acad. Sci. (USA), 78, 4990-4994.
11. Schwartz, E.L., Snoddy, J.R., Kreutter, D., Rasmussen, H. & Sartorelli, A.C. (1983) Proc. Am. Assoc. Cancer Res.. 24. 18.
12. Tanenaga, K., Hozumi, M. & Sakagami, Y. (1980) Cancer Res.. 12, 914-919.
13. Lotem, J. & Sachs, L. (1975) Int. J. Cancer. 15. 731- 740.
14. Metcalf, D. (1985) Scienct, 229. 16-22.
15. Scher, W., Scher, B.M. & Waxman, S. (1983) EXP.
Hematpl., 11, 490-498.
16. Scher, W., Scher, B.M. & Waxman, S. (1982) Biochem. & Biophys. Res. Comm.. 109, 348-354.
17. Huberman, E. & Callaham, M.F. (1979) Proc. Natl. Acad.
Sci. (USA), 26, 1293-1297.
18. Lottem, J. & Sachs, L. (1979) Proc. Natl. Acad. Sci.
(USA), 26, 5158-5162.
19. Terada, M., Epner, E., Nudel, U., Salmon, J., Fibach,
E., Rifkind, R.A. & Marks, P.A. (1978) Proc. Natl. Acad. Sci. (USA), 75, 2795-2799. 20. Morin, M.J. & Sartorelli, A.C. (1984) Cancer Res.. 44, 2807-2812.
21. Schwartz, E.L., Brown, B.J., Nierenberg, M., Marsh, J.C. & Sartorelli, A.C. (1983) Cancer Res., 43. 2725- 2730.
22. Sugano, H., Furusawa, M., Kawaguchi, T. & Ikawa, Y.
(1973) Bibl. Hematol., 39, 943-954.
23. Ebert, P.S., Wars, I. & Buell, D.N. (1976) Cancer Res., 22, 1809-1813.
24. Hayashi, M., Okabe, J. & Hozumi, M. (1979) Gann, 70.
235-238.
25. Fibach, E., Reuben, R.C., Rifkind, R.A. & Marks, P.A.
(1977) Cancer Res., 37, 440-444.
26. Melloni, E., Pontremoli, S., Damiani, G., Viotti, P., Weich, N., Rifkind, R.A. & Marks, P.A. (1988) Proc. Natl. Acad. Sci. (USA), 85, 3835-3839.
27. Reuben, R., Khanna, P.L., Gazitt, Y., Breslow, R., Rifkind, R.A. & Marks, P.A. (1978) J. Biol. Chem.. 253. 4214-4218.
28. Marks, P.A. and Rifkind, R.A. Hexamethylene Biscetamide Induced Differentiation of Transformed Cells: Molecular and Cellular Effects and Therapeutic
Application. International Journal of Cell Cloning, 6:230-240, 1988.
29. Melloni, E., Pontremoli, S., Michetti, M., Sacco, O., Cakiroglu, A.G., Jackson, J.F., Rifkind, R.A. & Marks, P.A. Proc. Natl. Acad. Sciences (USA) 84, 5282-5286, 1987.
30. Marks, P.A. & Rifkind, R.A. (1984) Cancer. 21, 2766- 2769.
31. Egorin, M.J., Sigman, L.M. VanEcho, D.A., Forrest, A., Whitacre, M.Y. & Aisner, J. (1987) Cancer Res., 47. 617-623.
32. Rowinsky, E.W., Ettinger, D.S., Grochow, L.B., Brundrett, R.B., Cates, A.E. & Donehower, R.C. (1986) J. Clin. Oncol., 4, 1835-1844.
33. Rowinsky, E.L. Ettinger, D.S., McGuire, W.P., Noe, D.A., Grochow, L.B. & Donehower, R.C. (1987) Cancer Res., 47, 5788-5795.
34. Callery, P.S., Egorin, M.J., Geelhaar, L.A. & Nayer, M.S.B. (1986) Cancer Res.. 46, 4900-4903.
35. Young, C.W. Fanucchi, M.P., Walsh, T.B., Blatzer, L., Yaldaie, S., Stevens, Y.W., Gordon, C., Tong, W., Rifkind, R.A. & Marks, P.A. (1988) Cancer Res.. 48, 7304-7309.
36. Andreeff, M., Young, C., Clarkson, B., Fetten, J., Rifkind, R.A. & Marks, P.A. (1988) Blood. 72, 186a.
37. Ohta, Y., Tanaka, M., Terada, M., Miller, O.J., Bank, A., Marks, P.A. & Rifkind, R.A. (1976) Proc. Natl. Acad. Sci. (USA) 73, 1232-1236.
38. Cartledge, F.K. & Nguyen, T.B. (1986) J. Org. Chem.,
51, 2206-2210.
39. Hoffmann-LaRoche and Co., A.G. British Patent 1, 138- 529 (1969).
40. Dmitrovsky, E., Markman, M. & Marks, P.A. (1989) in Cancer Chemotherapy and Biological Therapy. Annual 11, eds., D.L. Longo, H.M. Pinedo, B.A. Chabner, eds., Excepta Medica, Publisher, in press.
Claims
1. A compound having the structure:
[R-A]-B-A1-B1-[A2-B2-]a[A3-B3-]b[A4-R1] wherein each A, A1, A2, A3, and A4 represent a polar group which comprises a nitrogen, sulfur or oxygen atom and wherein each of A, A1, A2, A3, and A4 may independently be the same as, or different from, the others of A, A1, A2, A3, and A4; wherein each of R and R1 is a hydrogen atom; a lower alkyl, alkenyl, or alkynyl group; or a group having the structure:

and wherein each of a and b is independently 0 or 1.
2. A compound of claim 1 having the structure:

3. A compound of claim 1, wherein each of A, A1, A2, A3 and A4 is a carbonyl radical or a bivalent radical of an amide, a sulfoxide, or an amine oxide.
4. A compound of claim 1, wherein each of A, A1, A2, A3 and A4 is a carbonyl radical or a bivalent radical of an amide.
5. A compound of claim 4, wherein all of A, A1, A2, A3, and A4 are the same.
6. A compound of claim 5, wherein all of A, A1, A2, A3 and
A4 have the structure:

7. A compound of claim 4, wherein all of A, A1, A2, A3 and A4 are a carbonyl radicals.
8. A compound of claim 1, wherein R and R1 are the same and are hydrogen atoms, methyl groups, ethyl groups or groups having the structure:

each of R2 and R3 each is hydrogen atom, a methyl group an ethyl group and being the same or different.
9. A compound of claim 8, wherein all of A, A1, A2, A3, and A4 are carbonyl radicals and R and R1 are:

10. A compound of claim 1, wherein each of B, B1, B2, and B3 further comprises a nonpolar substituent attached to an atom of a chain.
11. A compound of claim 1, wherein each of B, B1, B2, and B3 comprises 4 to 7 atoms connected in a chain.
12. A compound of claim 1, wherein each of B, B1, B2, and B3 may comprise a hydrocarbon chain.
13. A compound of claim 1 having the structure:

wherein each of R, R1, and R4 is a hydrogen atom or a methyl, ethyl, or propyl group, R and R1 are the same or different; and wherein y is 4, 5, or 7.
14. A compound of claim 13, wherein R and R1 are hydrogen atoms, each R4 is an ethyl group, and y is 4, 5 or 6.
15. A compound of claim 13 having the structure:

wherein y is 4, 5 or 6.
16. A compound of claim 15 having the structure:

17. A compound of claim 1, having the structure:

wherein each of R2 and R3 is a hydrogen atom or a methyl, ethyl of propyl group and is the same or different, and wherein each of y and y1 is independently 4, 5, 6 or 7.
18. A compound of claim 17, wherein R2 is a hydrogen atom, R3 is an ethyl group and each of y and yl is independently 4, 5 or 6.
19. A compound of claim 17, wherein A1 is
or



20. A compound of claim 19 having the structure:

21. A compound of claim 18 having the structure:

wherein each of y and y1 is independently 5 or 6.
22. A compound of claim 21, wherein A1 is: or



23 . A compound having the structure:

24 . A compound having the structure:

25. A compound having the structure:

26. A compound having the structure:

wherein n is an integer which is greater than 1 and R1 and R2 may be the same or different and each is H or a lower alkyl group.
27. A compound having the structure:

28. A compound having the structure: 
29. A compound having the structure:  wherein R1 and R2 may be the same or different and each is H or a lower alkyl group; and n is an integer from 1 to 10.
wherein R1 and R2 may be the same or different and each is H or a lower alkyl group; and n is an integer from 1 to 10.
30. A compound having the structure:  wherein R1, R2, R3, R4, R5, R6 may independently be the same or different from each other and is H or a lower alkyl group; wherein X is methyl or phenyl; and n is an integer from 1 to about 15.
wherein R1, R2, R3, R4, R5, R6 may independently be the same or different from each other and is H or a lower alkyl group; wherein X is methyl or phenyl; and n is an integer from 1 to about 15.
31. A compound having the structure:

wherein R1 and R2 may be the same or different and is H or CH3.
32. A compound having the structure:

wherein R1 and R2 may be the same or different and is
H or CH 3.
33. A compound having the structure:  wherein X has the structure:
wherein X has the structure: 


-CH=CH-CH=CH-; wherein R1 and R2 are the same or different and is H or lower alkyl group.
34. A compound having the structure:

wherein R1 and R1 may independently be the same or different and each may be H or a lower alkyl group; wherein R3 and R4 may independently be the same or different and each may be CH3 or OH; and wherein n is 5 or 6.
35. A compound having the structure:

wherein R1 and R2 is the same or different and is hydrogen, lower alkyl, alkenyl, alkynyl, an amide or hydroxyamide.
36. A compound having the structure:

37. A compound having the structure:

38. A compound having the structure: 
39. A compound having the structure:

40. A method of selectively inducing terminal differentiation of neoplastic cells and thereby inhibiting proliferation of such cells which comprises contacting the cells under suitable conditions with an amount of a compound effective to selectively induce terminal differentiation, the compound having the structure:
[R-A]-B-A1-B1-[A2-B2-]a[A3-B3 ]b[A4-R1] wherein each A, A1, A2, A3, and A4 represent a polar group which comprises a nitrogen, sulfur or oxygen atom and wherein each of A, A1, A2, A3, and A4 may independently be the same as, or different from, the others of A, A1, A2, A3, and A4; wherein each of R and R1 is a hydrogen atom; a lower alkyl, alkenyl, or alkynyl group; or a group having the structure:

wherein each of R2 and R3 being a hydrogen atom or a lower alkyl, alkenyl, or alkynyl group; and wherein each of R, R1, R2 and R3 may independently be the same as, or different from, the others of R, R1, R2 and R3; wherein each of [R-A] and [A4-R1] have a dipolar moment greater than about 2.7 Debye units; wherein each of B, B1, B2, and B3 represents a nonpolar group which comprises at least 4 atoms in a chain, the termini of which chains are attached to A and A1, A1 and A2, A2 and A3, and A3 and A4, respectively; wherein each such atom is oxygen, nitrogen, carbon, or sulfur and wherein each of B, B1, B2, and B3 may independently be the same or different from each of B, B1, B2, and B3 ; and wherein each of a and b is independently 0 or 1.
41. A method of claim 40, wherein the compound has the structure:

42. A method of claim 40, wherein the contacting is effected continuously for at least 48 hours.
43. A method of claim 40, wherein the compound has the structure:

44. A method of claim 43, wherein the compound has the structure:

45. A method of claim 43, wherein the amount of the compound is from about 0.5mM to about 25mM.
46. A method of claim 40, wherein the compound has the structure:

or 

and wherein each of y and y1 is independently 5, 6 or 7.
47. A method of claim 46 , wherein the compound has the structure:

49. A method of selectively inducing terminal differentiation of neoplastic cells and thereby inhibiting proliferation of such cells which comprises contacting the cells under suitable conditions with an amount of a compound of claims 23-39, effective to selectively induce terminal differentiation.
50. A method of selectively inducing terminal differentiation of neoplastic cells and thereby inhibiting proliferation of such cells which comprises contacting the cells under suitable conditions with an amount of a compound effective to selectively induce terminal differentiation, the compound having the structure:

wherein X is phenyl or methyl and n is an integer from 1 to 15.
51. A method of claim 49 or 50 wherein the effective amount of the compound is from about 2μM to about 6000 μM.
52. A method of claim 51, wherein the contacting is effected continuously for at least 48 hours.
53. A method of treating a patient having a tumor characterized by proliferation of neoplastic cells which comprises administering to the patient an amount of a compound effective to selectively induce terminal differentiation of such neoplastic cells and thereby inhibit their proliferation, the compound having the structure:
[R-A]-B-A1-B1-[A2-B2-]a[A3-B3-]b[A4-R1] wherein each of A, A1, A2, A3, and A4 represent a polar group which comprises a nitrogen, sulfur or oxygen atom and wherein each of A, A1, A2, A3, and A4 may independently be the same as, or different from, the others of A, A1, A2, A3, and A4; wherein each of R and R1 is a hydrogen atom; a lower alkyl, alkenyl, or alkenyl group; or a group having the structure:

wherein each of R2 and R3 being a hydrogen atom or a lower alkyl, alkenyl, or alkynyl group; and wherein each of R, R1, R2 and R3; wherein each of [R-A] and [A4-R1] have a dipolar moment greater than about 2.7 Debye units; wherein each of B, B1, B2, and B3 represents a nonpolar group of which comprises least 4 atoms in a chain, the termini of which chains are attached to A and A1, A1 and A2 A2 and A3, and A3 and A4, respectively; wherein each such atom is oxygen, nitrogen, carbon, or sulfur and wherein each of B, B1, B2, and B3 may independently be the same as, or different from the others of B, B1, B2 and B3; and wherein each of a and b is independently 0 or 1.
54. A method of claim 53, wherein the compound has the structure:

55. A method of claim 53, wherein the compound has the structure:

56. A method of claim 55, wherein the compound has the structure:

57. A method of claim 51, wherein the compound has the structure:


and wherein each of y and y1 is independently 5, 6 or 7.
58. A method of claim 57, wherein the compound has the structure:

59. A method of claim 57, wherein the compound has a structure:

60. A method of treating patient having a tumor characterized by proliferation of neoplastic cells which comprises administering to the patient an amount of a compound effective to selectively induce terminal differentiation, which neoplastic cells and thereby inhibit their proliferation, the compound having the structure:

wherein X is phenyl or methyl and n is an integer from 1 to 15.
61. A method of treating patient having a tumor characterized by proliferation of neoplastic cells which comprises administering to the patient an amount of a compound of claims 23-39, in an amount effective to selectively induce terminal differentiation of neoplastic cells and thereby inhibit their proliferation.
62. A method of claim 60 or 61, wherein the amount of the compound is less than an amount which causes toxicity in the patient.
63. A method of claim 62, wherein the tumor is lung cancer, acute lymphoid myeloma, bladder melanoma, renal carcinoma, breast carcinoma or colorectal carcinoma.
64. A method of claim 60 or 61, wherein the administering is effected intravenously.
65. A method of claim 60 or 61, wherein the administering is effected orally.
66. A pharmaceutical composition comprising a pharmaceutically acceptable carrier and the compound of claim 1 or 23-39, in an amount effective to selectively induce terminal differentiation of suitable neoplastic cells and less than an amount which causes toxicity in a patient.
67. A pharmaceutical composition of claim 66, wherein the amount of the compound is from about 0.1 gm/m2/day to about 30 gm/m2/day.
68. A pharmaceutical composition of claim 66, wherein the compound has the structure:

69. A pharmaceutical composition of claim 66, wherein the amount of the compound is not more than about 30 gm/m2/day.
70. A pharmaceutical composition of claim 66, wherein the compound has the structure:

71. A pharmaceutical composition of claim 70, wherein the amount of the compound is not more than about 30 gm/m2/day.
72. A pharmaceutical composition of claim 66 , wherein the compound has the structure:

73. A pharmaceutical composition of claim 72 , wherein the amount of the compound is not more than about 30 gm/m2/day.
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES91901442T ES2128314T3 (en) | 1989-11-14 | 1990-11-14 | NEW POWERFUL TERMINAL DIFFERENTIATION INDUCTORS AND METHOD OF USE THEREOF. |
| EP91901442A EP0594577B1 (en) | 1989-11-14 | 1990-11-14 | Novel potent inducers of terminal differentiation and method of use thereof |
| PCT/US1990/006649 WO1991008191A1 (en) | 1989-11-14 | 1990-11-14 | Novel potent inducers of terminal differentiation and method of use thereof |
| AT91901442T ATE175185T1 (en) | 1989-11-14 | 1990-11-14 | NEW POTENT INDUCTORS OF TERMINAL DIFFERENTIATION AND METHOD OF USE THEREOF |
| JP03501924A JP3083842B2 (en) | 1989-11-14 | 1990-11-14 | Novel and potent terminal differentiation inducer and method of using the same |
| CA002068224A CA2068224C (en) | 1989-11-14 | 1990-11-14 | Novel potent inducers of terminal differentiation and method of use thereof |
| AU69726/91A AU643248B2 (en) | 1989-11-14 | 1990-11-14 | Novel potent inducers of terminal differentiation and method of use thereof |
| DE69032871T DE69032871T2 (en) | 1989-11-14 | 1990-11-14 | NEW POTENTIAL INDUCTORS OF TERMINAL DIFFERENTIATION AND METHOD FOR THEIR USE |
| US08/424,556 US5608108A (en) | 1988-11-14 | 1995-04-17 | Potent inducers of terminal differentiation and method of use thereof |
| US08/476,624 US5840960A (en) | 1988-11-14 | 1995-06-07 | Potent inducers of terminal differentiation and method of use thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US374,343 | 1989-06-30 | ||
| US07/374,343 US5055608A (en) | 1988-11-14 | 1989-06-30 | Novel potent inducers of thermal differentiation and method of use thereof |
| PCT/US1989/005203 WO1990009092A2 (en) | 1988-11-14 | 1989-11-14 | Novel potent inducers of terminal differentiation and method of use thereof |
| USPCT/US89/05203 | 1989-11-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1991000257A1 true WO1991000257A1 (en) | 1991-01-10 |
Family
ID=26780126
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1990/002690 WO1991000257A1 (en) | 1988-11-14 | 1990-05-14 | Novel potent inducers of terminal differentiation and methods of use thereof |
Country Status (2)
| Country | Link |
|---|---|
| AU (1) | AU5927190A (en) |
| WO (1) | WO1991000257A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0594577A1 (en) * | 1989-11-14 | 1994-05-04 | Sloan-Kettering Institute For Cancer Research | Novel potent inducers of terminal differentiation and method of use thereof |
| EP0642509A4 (en) * | 1991-10-04 | 1995-01-31 | Sloan Kettering Inst Cancer | Novel potent inducers of terminal differentiation and methods of use thereof. |
| USRE38506E1 (en) | 1991-10-04 | 2004-04-20 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and methods of use thereof |
| WO2011019393A2 (en) * | 2009-08-11 | 2011-02-17 | President And Fellows Of Harvard College | Class- and isoform-specific hdac inhibitors and uses thereof |
| US8178579B2 (en) | 2001-05-09 | 2012-05-15 | President And Fellows Of Harvard College | Dioxanes and uses thereof |
| US8304451B2 (en) | 2006-05-03 | 2012-11-06 | President And Fellows Of Harvard College | Histone deacetylase and tubulin deacetylase inhibitors |
| US8329946B2 (en) | 1996-03-26 | 2012-12-11 | President And Fellows Of Harvard College | Histone deacetylases, and uses related thereto |
| US8435780B2 (en) | 2000-03-03 | 2013-05-07 | President And Fellows Of Harvard College | Class II human histone deacetylases, and uses related thereto |
| US8440716B2 (en) | 2008-07-23 | 2013-05-14 | President And Fellows Of Harvard College | Deacetylase inhibitors and uses thereof |
| US8754237B2 (en) | 2006-02-14 | 2014-06-17 | President And Fellows Of Harvard College | Bifunctional histone deacetylase inhibitors |
| US8999289B2 (en) | 2005-03-22 | 2015-04-07 | President And Fellows Of Harvard College | Treatment of protein degradation disorders |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2279973A (en) * | 1940-05-08 | 1942-04-14 | Du Pont | Stabilization of organic substances |
| US2346665A (en) * | 1940-05-08 | 1944-04-18 | Du Pont | Acid |
| US3450673A (en) * | 1965-09-07 | 1969-06-17 | Ashland Oil Inc | Polyurethane compositions from diaminimides |
| US4442305A (en) * | 1981-08-24 | 1984-04-10 | The United States Of America As Represented By The United States Department Of Energy | Polycatecholamide chelating agents |
| US4611053A (en) * | 1985-02-15 | 1986-09-09 | Sasa Michiyuki Mitch | Polyhydroxamide polymer |
| US4801748A (en) * | 1985-03-08 | 1989-01-31 | Osaka University | Catalytic process for preparing amide from nitrile, amine and water |
| US4863967A (en) * | 1986-06-16 | 1989-09-05 | Research Corporation | N,N-diaminophthalamides |
-
1990
- 1990-05-14 WO PCT/US1990/002690 patent/WO1991000257A1/en unknown
- 1990-05-14 AU AU59271/90A patent/AU5927190A/en not_active Abandoned
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2279973A (en) * | 1940-05-08 | 1942-04-14 | Du Pont | Stabilization of organic substances |
| US2346665A (en) * | 1940-05-08 | 1944-04-18 | Du Pont | Acid |
| US3450673A (en) * | 1965-09-07 | 1969-06-17 | Ashland Oil Inc | Polyurethane compositions from diaminimides |
| US4442305A (en) * | 1981-08-24 | 1984-04-10 | The United States Of America As Represented By The United States Department Of Energy | Polycatecholamide chelating agents |
| US4611053A (en) * | 1985-02-15 | 1986-09-09 | Sasa Michiyuki Mitch | Polyhydroxamide polymer |
| US4801748A (en) * | 1985-03-08 | 1989-01-31 | Osaka University | Catalytic process for preparing amide from nitrile, amine and water |
| US4863967A (en) * | 1986-06-16 | 1989-09-05 | Research Corporation | N,N-diaminophthalamides |
Non-Patent Citations (10)
| Title |
|---|
| ARZNEIM-FORSCH, Vol. 35, No. 7, "Quantitative Analysis of Biological Activities of Diactinomyein Analogs and methotrexate Derivatives with Van Der Waals Volume", PRABHAKER et al., 1985, pages 1030-3. * |
| CHEM. ABSTRACTS, Vol. 105, No. 9, 78501 "A Facile Synthesis of Aliphatic Dihydroxamic Acids of General Formula RONR-CO-(CH) -CO-NR'OR", BROWN et al; & SYNTH. COMUN., Vol. 15, No. 13, 1159-64, 1985. * |
| CHEMICAL ABSTRACTS, Vol. 101, No. 7, 54665t, "Synthesis of some Dihydroxamic Acid Siderophores", DAS et al; & J. CHEM. ENG. DATA, 29(3), 345-48, 1984. * |
| CHEMICAL ABSTRACTS, Vol. 98, No. 23, "Antibypanosomal and Antimycotic Effect of Various Hydroxamic Acids", TABERNO et al., see Abstract No. 191329v; & ACTA. CIENT, VENEZ, 32(5), 411-16. * |
| INORGANIC CHEMISTRY, Vol. 25, No. 21, "Design of Metal Chelates with Biological Activity", BROWN et al., 1986, pp. 3792-3796. * |
| INT. J. CANCER, Vol. 23, No. 1, "Induction of Erythroid Differentiation in Murine Erythroleukemia Cells by Nitrogen Substituted Polymethylene Diamides", HOZUMI et al., pp. 119-22. * |
| J. BIOL. CHEM., Vol. 253, No. 12, "Inducers of Erytholeukemic Differentiation", REUBEN et al., June 1978, pp. 4214-18. * |
| J. ORG. CHEM., Vol. 46, No. 25, "Lipophilic Enterobactin Analogs", WEITL et al., 1981, pp. 5234-7. * |
| ORGANIC CHEMISTRY:, MORRISON AND BOYD, 3rd Ed., ALLYN AND BARON INC. p. 755. * |
| PROC. NATL. ACAD. SCI. USA, Vol. 72, No. 3, "Induction of Erythroid Differentiation...", etc. TANAKA et al., March 1975, pp. 1003-6. * |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0594577B1 (en) * | 1989-11-14 | 1998-12-30 | Sloan-Kettering Institute For Cancer Research | Novel potent inducers of terminal differentiation and method of use thereof |
| EP0594577A1 (en) * | 1989-11-14 | 1994-05-04 | Sloan-Kettering Institute For Cancer Research | Novel potent inducers of terminal differentiation and method of use thereof |
| EP0642509A4 (en) * | 1991-10-04 | 1995-01-31 | Sloan Kettering Inst Cancer | Novel potent inducers of terminal differentiation and methods of use thereof. |
| EP0642509A1 (en) * | 1991-10-04 | 1995-03-15 | Sloan-Kettering Institute For Cancer Research | Novel potent inducers of terminal differentiation and methods of use thereof |
| USRE38506E1 (en) | 1991-10-04 | 2004-04-20 | Sloan-Kettering Institute For Cancer Research | Potent inducers of terminal differentiation and methods of use thereof |
| US8329945B2 (en) | 1996-03-26 | 2012-12-11 | President And Fellows Of Harvard College | Histone deacetylases, and uses related thereto |
| US8426592B2 (en) | 1996-03-26 | 2013-04-23 | President And Fellows Of Harvard College | Histone deacetylases, and uses related thereto |
| US8399233B2 (en) | 1996-03-26 | 2013-03-19 | President And Fellows Of Harvard College | Histone deacetylases, and uses related thereto |
| US8362084B2 (en) | 1996-03-26 | 2013-01-29 | President And Fellows Of Harvard College | Histone deacetylases, and uses related thereto |
| US8329946B2 (en) | 1996-03-26 | 2012-12-11 | President And Fellows Of Harvard College | Histone deacetylases, and uses related thereto |
| US8895284B2 (en) | 2000-03-03 | 2014-11-25 | President And Fellows Of Harvard College | Class II human histone deacetylases, and uses related thereto |
| US8435780B2 (en) | 2000-03-03 | 2013-05-07 | President And Fellows Of Harvard College | Class II human histone deacetylases, and uses related thereto |
| US8178579B2 (en) | 2001-05-09 | 2012-05-15 | President And Fellows Of Harvard College | Dioxanes and uses thereof |
| US8999289B2 (en) | 2005-03-22 | 2015-04-07 | President And Fellows Of Harvard College | Treatment of protein degradation disorders |
| US10172905B1 (en) | 2005-03-22 | 2019-01-08 | President And Fellows Of Harvard College | Treatment of protein degradation disorders |
| US9572854B2 (en) | 2005-03-22 | 2017-02-21 | President And Fellows Of Harvard College | Treatment of protein degradation disorders |
| US8754237B2 (en) | 2006-02-14 | 2014-06-17 | President And Fellows Of Harvard College | Bifunctional histone deacetylase inhibitors |
| US8304451B2 (en) | 2006-05-03 | 2012-11-06 | President And Fellows Of Harvard College | Histone deacetylase and tubulin deacetylase inhibitors |
| US9434686B2 (en) | 2008-07-23 | 2016-09-06 | President And Fellows Of Harvard College | Deacetylase inhibitors and uses thereof |
| US8440716B2 (en) | 2008-07-23 | 2013-05-14 | President And Fellows Of Harvard College | Deacetylase inhibitors and uses thereof |
| WO2011019393A2 (en) * | 2009-08-11 | 2011-02-17 | President And Fellows Of Harvard College | Class- and isoform-specific hdac inhibitors and uses thereof |
| US8716344B2 (en) | 2009-08-11 | 2014-05-06 | President And Fellows Of Harvard College | Class- and isoform-specific HDAC inhibitors and uses thereof |
| US9540317B2 (en) | 2009-08-11 | 2017-01-10 | President And Fellows Of Harvard College | Class- and isoform-specific HDAC inhibitors and uses thereof |
| US10059657B2 (en) | 2009-08-11 | 2018-08-28 | President And Fellows Of Harvard College | Class-and isoform-specific HDAC inhibitors and uses thereof |
| WO2011019393A3 (en) * | 2009-08-11 | 2011-06-16 | President And Fellows Of Harvard College | Class- and isoform-specific hdac inhibitors and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| AU5927190A (en) | 1991-01-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5175191A (en) | Potent inducers of terminal differentiation and methods of use thereof | |
| US5608108A (en) | Potent inducers of terminal differentiation and method of use thereof | |
| US5055608A (en) | Novel potent inducers of thermal differentiation and method of use thereof | |
| US6087367A (en) | Potent inducers of terminal differentiation and methods of use thereof | |
| US5700811A (en) | Potent inducers of terminal differentiation and method of use thereof | |
| Marks et al. | Polar/apolar chemical inducers of differentiation of transformed cells: strategies to improve therapeutic potential. | |
| USRE38506E1 (en) | Potent inducers of terminal differentiation and methods of use thereof | |
| FI88289B (en) | ANALOGIFICATION OF THE PHARMACEUTICAL FRAMEWORK OF THE THERAPEUTIC ANALYSIS | |
| AU643248B2 (en) | Novel potent inducers of terminal differentiation and method of use thereof | |
| JPH07503718A (en) | Novel amidine derivatives, their preparation and use as LTB↓4 antagonists | |
| BRPI0611966A2 (en) | pharmaceutically acceptable compound or solvate, hydrate or salt thereof, pharmaceutical composition, process, and methods of inhibiting sphingosine kinase in a patient in need of such inhibition, to treat a disorder and disease | |
| WO1991000257A1 (en) | Novel potent inducers of terminal differentiation and methods of use thereof | |
| JPS5832847A (en) | (3-aminopropoxy)bibenzyl compound | |
| JPH04139127A (en) | Medicine for preventing or treating cardiopathy | |
| US4897423A (en) | Dinitrobenzenesulfonamides | |
| WO1990009092A2 (en) | Novel potent inducers of terminal differentiation and method of use thereof | |
| AU708115B2 (en) | Novel potent inducers of terminal differentiation and methods of use thereof | |
| US4853415A (en) | 2-(substituted sulfamyl) derivatives of 4-nitrobenzamide as radiation sensitizers | |
| CA2574103A1 (en) | Novel potent inducers of terminal differentiation and methods of use thereof | |
| CA2818871A1 (en) | Integrin-linked kinase inhibitors | |
| JPS5998075A (en) | Novel nitrosamine compound and pharmaceutical containing said compound as active component |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU DK FI HU JP KR NO |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB IT LU NL SE |