USRE49834E1 - Jak1 selective inhibitors and uses thereof - Google Patents
Jak1 selective inhibitors and uses thereof Download PDFInfo
- Publication number
- USRE49834E1 USRE49834E1 US17/526,799 US201717526799A USRE49834E US RE49834 E1 USRE49834 E1 US RE49834E1 US 201717526799 A US201717526799 A US 201717526799A US RE49834 E USRE49834 E US RE49834E
- Authority
- US
- United States
- Prior art keywords
- nrr
- compound
- imidazo
- pyridin
- halo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229940124639 Selective inhibitor Drugs 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims description 258
- 238000000034 method Methods 0.000 claims description 66
- 150000003839 salts Chemical class 0.000 claims description 40
- 125000001424 substituent group Chemical group 0.000 claims description 35
- -1 tetrafluoroborate Chemical compound 0.000 claims description 26
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 22
- 125000000623 heterocyclic group Chemical group 0.000 claims description 22
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 18
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 17
- 230000008569 process Effects 0.000 claims description 16
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical group CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 14
- 239000001257 hydrogen Substances 0.000 claims description 12
- 229910052739 hydrogen Inorganic materials 0.000 claims description 12
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical group [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 11
- JVWXVJOIUKQBOJ-NWDGAFQWSA-N 2-[(2R,5S)-5-(4-methyl-12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,10-pentaen-3-yl)oxan-2-yl]acetonitrile Chemical compound CC1=NC=2C(=C3C(=NC=2)C=CO3)N1[C@H]1CC[C@@H](OC1)CC#N JVWXVJOIUKQBOJ-NWDGAFQWSA-N 0.000 claims description 10
- 239000003054 catalyst Substances 0.000 claims description 10
- 238000005984 hydrogenation reaction Methods 0.000 claims description 10
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 9
- ABAUJVSEICFAOS-NWDGAFQWSA-N 2-[(2R,5S)-5-(4-ethyl-12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,10-pentaen-3-yl)oxan-2-yl]acetonitrile Chemical compound C(C)C1=NC=2C(=C3C(=NC=2)C=CO3)N1[C@H]1CC[C@@H](OC1)CC#N ABAUJVSEICFAOS-NWDGAFQWSA-N 0.000 claims description 8
- WWFMBYPVXNSPOA-JHJVBQTASA-N O1C=CC2=C1C=1N(C(=NC=1C=N2)[C@H](O)C)[C@H]1CC[C@@H](CC1)CC#N Chemical compound O1C=CC2=C1C=1N(C(=NC=1C=N2)[C@H](O)C)[C@H]1CC[C@@H](CC1)CC#N WWFMBYPVXNSPOA-JHJVBQTASA-N 0.000 claims description 7
- UDGHPISWLBXGGY-WDEREUQCSA-N 2-[(2R,5S)-5-[4-(hydroxymethyl)-12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,10-pentaen-3-yl]oxan-2-yl]acetonitrile Chemical compound OCC1=NC=2C(=C3C(=NC=2)C=CO3)N1[C@H]1CC[C@@H](OC1)CC#N UDGHPISWLBXGGY-WDEREUQCSA-N 0.000 claims description 6
- ZFQNHUSKKDBFHY-GRYCIOLGSA-N 2-[(2R,5S)-5-[4-[(1R)-1-hydroxyethyl]-12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,10-pentaen-3-yl]oxan-2-yl]acetonitrile Chemical compound O[C@H](C)C1=NC=2C(=C3C(=NC=2)C=CO3)N1[C@H]1CC[C@@H](OC1)CC#N ZFQNHUSKKDBFHY-GRYCIOLGSA-N 0.000 claims description 6
- UDGHPISWLBXGGY-QWRGUYRKSA-N 2-[(2S,5S)-5-[4-(hydroxymethyl)-12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,10-pentaen-3-yl]oxan-2-yl]acetonitrile Chemical compound OCC1=NC=2C(=C3C(=NC=2)C=CO3)N1[C@H]1CC[C@H](OC1)CC#N UDGHPISWLBXGGY-QWRGUYRKSA-N 0.000 claims description 6
- JKBYTRRGLJOVHV-YHMJZVADSA-N 3-[4-[(1R)-1-hydroxyethyl]-12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,10-pentaen-3-yl]-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide Chemical compound O[C@H](C)C1=NC=2C(=C3C(=NC=2)C=CO3)N1C1CN(CC1)C(=O)NCC(F)(F)F JKBYTRRGLJOVHV-YHMJZVADSA-N 0.000 claims description 6
- UKUHFEJKHOXPQA-SNVBAGLBSA-N 4-[4-[(1R)-1-hydroxyethyl]-12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,4,6,8,10-pentaen-3-yl]-N-(2,2,2-trifluoroethyl)piperidine-1-carboxamide Chemical compound O[C@H](C)C1=NC=2C(=C3C(=NC=2)C=CO3)N1C1CCN(CC1)C(=O)NCC(F)(F)F UKUHFEJKHOXPQA-SNVBAGLBSA-N 0.000 claims description 6
- WKNNRZVOWSSOSY-XYPYZODXSA-N C12=C(C3=C(N=C(N3[C@H]3CC[C@@H](CC3)C#N)CO)C=N2)OC=C1 Chemical compound C12=C(C3=C(N=C(N3[C@H]3CC[C@@H](CC3)C#N)CO)C=N2)OC=C1 WKNNRZVOWSSOSY-XYPYZODXSA-N 0.000 claims description 6
- HIQALKYWCDEOTH-IJLUTSLNSA-N C12=C(N=CC=3N=C(N(C1=3)[C@H]1CC[C@@H](CC1)C#N)[C@@H](C)O)C=CO2 Chemical compound C12=C(N=CC=3N=C(N(C1=3)[C@H]1CC[C@@H](CC1)C#N)[C@@H](C)O)C=CO2 HIQALKYWCDEOTH-IJLUTSLNSA-N 0.000 claims description 6
- 239000003153 chemical reaction reagent Substances 0.000 claims description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 6
- 125000004187 tetrahydropyran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 6
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 5
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 5
- 125000006705 (C5-C7) cycloalkyl group Chemical group 0.000 claims description 4
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 3
- 125000001475 halogen functional group Chemical group 0.000 claims 22
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 92
- 201000010099 disease Diseases 0.000 abstract description 62
- 208000035475 disorder Diseases 0.000 abstract description 30
- 230000000694 effects Effects 0.000 abstract description 22
- 208000027866 inflammatory disease Diseases 0.000 abstract description 17
- 206010028980 Neoplasm Diseases 0.000 abstract description 12
- 201000011510 cancer Diseases 0.000 abstract description 11
- 208000023275 Autoimmune disease Diseases 0.000 abstract description 9
- 230000001613 neoplastic effect Effects 0.000 abstract description 6
- 229940043355 kinase inhibitor Drugs 0.000 abstract description 5
- 239000003757 phosphotransferase inhibitor Substances 0.000 abstract description 5
- HGNXHCJAXKQSOQ-UHFFFAOYSA-N 12-oxa-3,5,8-triazatricyclo[7.3.0.02,6]dodeca-1,3,6,8,10-pentaene Chemical class N1C=NC=2C1=C1C(=NC=2)C=CO1 HGNXHCJAXKQSOQ-UHFFFAOYSA-N 0.000 abstract description 4
- 102000002295 Janus Kinase 1 Human genes 0.000 abstract description 4
- 108010000837 Janus Kinase 1 Proteins 0.000 abstract description 4
- 239000000203 mixture Substances 0.000 description 90
- 239000000243 solution Substances 0.000 description 90
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 69
- 239000003814 drug Substances 0.000 description 52
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 51
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 45
- 229940124597 therapeutic agent Drugs 0.000 description 39
- 238000011282 treatment Methods 0.000 description 35
- 239000000843 powder Substances 0.000 description 29
- 235000002639 sodium chloride Nutrition 0.000 description 29
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 28
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 28
- 229940002612 prodrug Drugs 0.000 description 28
- 239000000651 prodrug Substances 0.000 description 28
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 26
- 239000011541 reaction mixture Substances 0.000 description 25
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 22
- 238000005160 1H NMR spectroscopy Methods 0.000 description 21
- 239000003795 chemical substances by application Substances 0.000 description 20
- 238000010898 silica gel chromatography Methods 0.000 description 19
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- 238000003756 stirring Methods 0.000 description 18
- 239000012299 nitrogen atmosphere Substances 0.000 description 17
- 238000003786 synthesis reaction Methods 0.000 description 17
- 238000006243 chemical reaction Methods 0.000 description 16
- 239000003550 marker Substances 0.000 description 16
- 239000008194 pharmaceutical composition Substances 0.000 description 16
- 208000024891 symptom Diseases 0.000 description 16
- 230000015572 biosynthetic process Effects 0.000 description 15
- 125000005843 halogen group Chemical group 0.000 description 15
- 239000012267 brine Substances 0.000 description 14
- 239000007788 liquid Substances 0.000 description 14
- 239000003921 oil Substances 0.000 description 14
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 14
- 235000019198 oils Nutrition 0.000 description 13
- 239000012074 organic phase Substances 0.000 description 13
- 108091000080 Phosphotransferase Proteins 0.000 description 12
- 239000002552 dosage form Substances 0.000 description 12
- 239000000284 extract Substances 0.000 description 12
- 102000020233 phosphotransferase Human genes 0.000 description 12
- 239000007787 solid Substances 0.000 description 12
- 229940122245 Janus kinase inhibitor Drugs 0.000 description 11
- 239000004012 Tofacitinib Substances 0.000 description 11
- 239000008346 aqueous phase Substances 0.000 description 11
- 239000012453 solvate Substances 0.000 description 11
- 239000003826 tablet Substances 0.000 description 11
- 229960001350 tofacitinib Drugs 0.000 description 11
- UJLAWZDWDVHWOW-YPMHNXCESA-N tofacitinib Chemical compound C[C@@H]1CCN(C(=O)CC#N)C[C@@H]1N(C)C1=NC=NC2=C1C=CN2 UJLAWZDWDVHWOW-YPMHNXCESA-N 0.000 description 11
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 10
- 125000003118 aryl group Chemical group 0.000 description 10
- 125000004432 carbon atom Chemical group C* 0.000 description 10
- 239000000706 filtrate Substances 0.000 description 10
- 238000009472 formulation Methods 0.000 description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- 101150009057 JAK2 gene Proteins 0.000 description 9
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 9
- 210000004369 blood Anatomy 0.000 description 9
- 239000008280 blood Substances 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 9
- 125000004122 cyclic group Chemical group 0.000 description 9
- 125000005842 heteroatom Chemical group 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 8
- 101150069380 JAK3 gene Proteins 0.000 description 8
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 8
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 8
- 102000001253 Protein Kinase Human genes 0.000 description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 8
- 241000219061 Rheum Species 0.000 description 8
- 125000000217 alkyl group Chemical group 0.000 description 8
- 239000002246 antineoplastic agent Substances 0.000 description 8
- 125000001072 heteroaryl group Chemical group 0.000 description 8
- 239000003112 inhibitor Substances 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- 108060006633 protein kinase Proteins 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 7
- MFYSUUPKMDJYPF-UHFFFAOYSA-N 2-[(4-methyl-2-nitrophenyl)diazenyl]-3-oxo-n-phenylbutanamide Chemical compound C=1C=CC=CC=1NC(=O)C(C(=O)C)N=NC1=CC=C(C)C=C1[N+]([O-])=O MFYSUUPKMDJYPF-UHFFFAOYSA-N 0.000 description 7
- 102000042838 JAK family Human genes 0.000 description 7
- 108091082332 JAK family Proteins 0.000 description 7
- 239000004480 active ingredient Substances 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 125000000524 functional group Chemical group 0.000 description 7
- 239000000543 intermediate Substances 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 239000008177 pharmaceutical agent Substances 0.000 description 7
- 102000004169 proteins and genes Human genes 0.000 description 7
- 108090000623 proteins and genes Proteins 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 238000002560 therapeutic procedure Methods 0.000 description 7
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- 241000282412 Homo Species 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 206010003246 arthritis Diseases 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- 229960003957 dexamethasone Drugs 0.000 description 6
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 6
- 229960000485 methotrexate Drugs 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 235000018102 proteins Nutrition 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- 102000004127 Cytokines Human genes 0.000 description 5
- 108090000695 Cytokines Proteins 0.000 description 5
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 5
- 102000004889 Interleukin-6 Human genes 0.000 description 5
- 108090001005 Interleukin-6 Proteins 0.000 description 5
- 102100034195 Thrombopoietin Human genes 0.000 description 5
- 229940034982 antineoplastic agent Drugs 0.000 description 5
- XUZMWHLSFXCVMG-UHFFFAOYSA-N baricitinib Chemical compound C1N(S(=O)(=O)CC)CC1(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 XUZMWHLSFXCVMG-UHFFFAOYSA-N 0.000 description 5
- 229910052799 carbon Inorganic materials 0.000 description 5
- 239000000969 carrier Substances 0.000 description 5
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Natural products OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- 239000005457 ice water Substances 0.000 description 5
- 239000004615 ingredient Substances 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 230000001404 mediated effect Effects 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- SXQFCVDSOLSHOQ-UWTATZPHSA-N (R)-(+)-Lactamide Chemical compound C[C@@H](O)C(N)=O SXQFCVDSOLSHOQ-UWTATZPHSA-N 0.000 description 4
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 4
- 108010036949 Cyclosporine Proteins 0.000 description 4
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- 229940116839 Janus kinase 1 inhibitor Drugs 0.000 description 4
- 208000034578 Multiple myelomas Diseases 0.000 description 4
- 206010035226 Plasma cell myeloma Diseases 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 125000003342 alkenyl group Chemical group 0.000 description 4
- 125000000304 alkynyl group Chemical group 0.000 description 4
- 239000002260 anti-inflammatory agent Substances 0.000 description 4
- 229950000971 baricitinib Drugs 0.000 description 4
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 4
- 239000003246 corticosteroid Substances 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 238000009510 drug design Methods 0.000 description 4
- 238000009509 drug development Methods 0.000 description 4
- 210000003743 erythrocyte Anatomy 0.000 description 4
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 150000002513 isocyanates Chemical class 0.000 description 4
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 description 4
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 208000003476 primary myelofibrosis Diseases 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- 230000004044 response Effects 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 230000002194 synthesizing effect Effects 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 4
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 3
- TZGPACAKMCUCKX-UHFFFAOYSA-N 2-hydroxyacetamide Chemical compound NC(=O)CO TZGPACAKMCUCKX-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N Benzoic acid Natural products OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 3
- 201000004624 Dermatitis Diseases 0.000 description 3
- 208000032027 Essential Thrombocythemia Diseases 0.000 description 3
- 108010008165 Etanercept Proteins 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 206010025323 Lymphomas Diseases 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 208000014767 Myeloproliferative disease Diseases 0.000 description 3
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 3
- 102000038030 PI3Ks Human genes 0.000 description 3
- 108091007960 PI3Ks Proteins 0.000 description 3
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 108010017324 STAT3 Transcription Factor Proteins 0.000 description 3
- 102100024040 Signal transducer and activator of transcription 3 Human genes 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 3
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 3
- 230000005856 abnormality Effects 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 208000007502 anemia Diseases 0.000 description 3
- 229940121363 anti-inflammatory agent Drugs 0.000 description 3
- 239000003435 antirheumatic agent Substances 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 125000003636 chemical group Chemical group 0.000 description 3
- 230000001684 chronic effect Effects 0.000 description 3
- 229960001265 ciclosporin Drugs 0.000 description 3
- 229930182912 cyclosporin Natural products 0.000 description 3
- 108010057085 cytokine receptors Proteins 0.000 description 3
- 102000003675 cytokine receptors Human genes 0.000 description 3
- 230000007812 deficiency Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 239000002988 disease modifying antirheumatic drug Substances 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 229960000403 etanercept Drugs 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical class 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 150000004679 hydroxides Chemical class 0.000 description 3
- 229940075628 hypomethylating agent Drugs 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 229960000598 infliximab Drugs 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 239000002502 liposome Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 3
- 229960001924 melphalan Drugs 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 238000012544 monitoring process Methods 0.000 description 3
- 206010028537 myelofibrosis Diseases 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 235000005985 organic acids Nutrition 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 150000003013 phosphoric acid derivatives Chemical group 0.000 description 3
- 230000026731 phosphorylation Effects 0.000 description 3
- 238000006366 phosphorylation reaction Methods 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 230000002062 proliferating effect Effects 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 210000002345 respiratory system Anatomy 0.000 description 3
- 201000000306 sarcoidosis Diseases 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 238000009097 single-agent therapy Methods 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 229960001967 tacrolimus Drugs 0.000 description 3
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 3
- 238000011200 topical administration Methods 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- 238000000844 transformation Methods 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- HMLGSIZOMSVISS-ONJSNURVSA-N (7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-(2,2-dimethylpropanoyloxymethoxyimino)acetyl]amino]-3-ethenyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound N([C@@H]1C(N2C(=C(C=C)CSC21)C(O)=O)=O)C(=O)\C(=N/OCOC(=O)C(C)(C)C)C1=CSC(N)=N1 HMLGSIZOMSVISS-ONJSNURVSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- KIPSRYDSZQRPEA-UHFFFAOYSA-N 2,2,2-trifluoroethanamine Chemical compound NCC(F)(F)F KIPSRYDSZQRPEA-UHFFFAOYSA-N 0.000 description 2
- CNIIGCLFLJGOGP-UHFFFAOYSA-N 2-(1-naphthalenylmethyl)-4,5-dihydro-1H-imidazole Chemical compound C=1C=CC2=CC=CC=C2C=1CC1=NCCN1 CNIIGCLFLJGOGP-UHFFFAOYSA-N 0.000 description 2
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 2
- FSPQCTGGIANIJZ-UHFFFAOYSA-N 2-[[(3,4-dimethoxyphenyl)-oxomethyl]amino]-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide Chemical compound C1=C(OC)C(OC)=CC=C1C(=O)NC1=C(C(N)=O)C(CCCC2)=C2S1 FSPQCTGGIANIJZ-UHFFFAOYSA-N 0.000 description 2
- YKHQFTANTNMYPP-UHFFFAOYSA-N 2-bromopyridin-3-ol Chemical compound OC1=CC=CN=C1Br YKHQFTANTNMYPP-UHFFFAOYSA-N 0.000 description 2
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- KXMRDHPZQHAXML-UHFFFAOYSA-N 4-[(2-methylpropan-2-yl)oxycarbonylamino]cyclohexane-1-carboxylic acid Chemical compound CC(C)(C)OC(=O)NC1CCC(C(O)=O)CC1 KXMRDHPZQHAXML-UHFFFAOYSA-N 0.000 description 2
- ZQJNPHCQABYENK-UHFFFAOYSA-N 4-methoxycarbonylcyclohexane-1-carboxylic acid Chemical compound COC(=O)C1CCC(C(O)=O)CC1 ZQJNPHCQABYENK-UHFFFAOYSA-N 0.000 description 2
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 2
- 108010003415 Aspartate Aminotransferases Proteins 0.000 description 2
- 102000004625 Aspartate Aminotransferases Human genes 0.000 description 2
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 2
- 208000005024 Castleman disease Diseases 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- 102000004420 Creatine Kinase Human genes 0.000 description 2
- 108010042126 Creatine kinase Proteins 0.000 description 2
- 206010012438 Dermatitis atopic Diseases 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 108010044091 Globulins Proteins 0.000 description 2
- 102000006395 Globulins Human genes 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 201000005569 Gout Diseases 0.000 description 2
- 208000007514 Herpes zoster Diseases 0.000 description 2
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 2
- 206010048643 Hypereosinophilic syndrome Diseases 0.000 description 2
- 208000004575 Infectious Arthritis Diseases 0.000 description 2
- 102000018682 Interleukin Receptor Common gamma Subunit Human genes 0.000 description 2
- 108010066719 Interleukin Receptor Common gamma Subunit Proteins 0.000 description 2
- 102000051628 Interleukin-1 receptor antagonist Human genes 0.000 description 2
- 108700021006 Interleukin-1 receptor antagonist Proteins 0.000 description 2
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 description 2
- 102000015696 Interleukins Human genes 0.000 description 2
- 108010063738 Interleukins Proteins 0.000 description 2
- 108010024121 Janus Kinases Proteins 0.000 description 2
- 102000015617 Janus Kinases Human genes 0.000 description 2
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- 240000007472 Leucaena leucocephala Species 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 2
- 201000002481 Myositis Diseases 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- 208000001388 Opportunistic Infections Diseases 0.000 description 2
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 229920001774 Perfluoroether Polymers 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 2
- 101710151245 Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 2
- 101000744436 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) Trans-acting factor D Proteins 0.000 description 2
- 206010039710 Scleroderma Diseases 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 201000008736 Systemic mastocytosis Diseases 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 239000004098 Tetracycline Substances 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 2
- 102000044159 Ubiquitin Human genes 0.000 description 2
- 108090000848 Ubiquitin Proteins 0.000 description 2
- 206010046851 Uveitis Diseases 0.000 description 2
- 102100025093 Zinc fingers and homeoboxes protein 2 Human genes 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 208000017733 acquired polycythemia vera Diseases 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 230000009056 active transport Effects 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 125000004450 alkenylene group Chemical group 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- 125000004419 alkynylene group Chemical group 0.000 description 2
- 230000004075 alteration Effects 0.000 description 2
- 150000001408 amides Chemical group 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 229960004238 anakinra Drugs 0.000 description 2
- 239000004037 angiogenesis inhibitor Substances 0.000 description 2
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical compound C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 2
- 239000003173 antianemic agent Substances 0.000 description 2
- 229940127079 antineoplastic immunimodulatory agent Drugs 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 201000008937 atopic dermatitis Diseases 0.000 description 2
- 230000001363 autoimmune Effects 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- 230000003115 biocidal effect Effects 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 208000019664 bone resorption disease Diseases 0.000 description 2
- 229960001467 bortezomib Drugs 0.000 description 2
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 2
- 210000000133 brain stem Anatomy 0.000 description 2
- 206010006451 bronchitis Diseases 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000012830 cancer therapeutic Substances 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000011111 cardboard Substances 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 239000008004 cell lysis buffer Substances 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 229960003405 ciprofloxacin Drugs 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 229940099112 cornstarch Drugs 0.000 description 2
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 229960003603 decitabine Drugs 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 239000002158 endotoxin Substances 0.000 description 2
- 229960001433 erlotinib Drugs 0.000 description 2
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 2
- 229940125367 erythropoiesis stimulating agent Drugs 0.000 description 2
- 150000002148 esters Chemical group 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- FEBLZLNTKCEFIT-VSXGLTOVSA-N fluocinolone acetonide Chemical group C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O FEBLZLNTKCEFIT-VSXGLTOVSA-N 0.000 description 2
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 2
- 125000005456 glyceride group Chemical group 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 229960001743 golimumab Drugs 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 150000002431 hydrogen Chemical group 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- XXSMGPRMXLTPCZ-UHFFFAOYSA-N hydroxychloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CCO)CC)=CC=NC2=C1 XXSMGPRMXLTPCZ-UHFFFAOYSA-N 0.000 description 2
- 229960004171 hydroxychloroquine Drugs 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 239000012948 isocyanate Substances 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000011294 monotherapeutic Methods 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 210000000822 natural killer cell Anatomy 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- 201000008482 osteoarthritis Diseases 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 238000001050 pharmacotherapy Methods 0.000 description 2
- 235000021317 phosphate Nutrition 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 208000037244 polycythemia vera Diseases 0.000 description 2
- 229960005205 prednisolone Drugs 0.000 description 2
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 238000002203 pretreatment Methods 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- MUPFEKGTMRGPLJ-ZQSKZDJDSA-N raffinose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)O)O1 MUPFEKGTMRGPLJ-ZQSKZDJDSA-N 0.000 description 2
- 239000011535 reaction buffer Substances 0.000 description 2
- 208000002574 reactive arthritis Diseases 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 125000006413 ring segment Chemical group 0.000 description 2
- 229960004641 rituximab Drugs 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 201000001223 septic arthritis Diseases 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 201000009890 sinusitis Diseases 0.000 description 2
- 208000017520 skin disease Diseases 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- 235000019364 tetracycline Nutrition 0.000 description 2
- 150000003522 tetracyclines Chemical class 0.000 description 2
- 229960003433 thalidomide Drugs 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Chemical compound COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 201000008827 tuberculosis Diseases 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- QCHFTSOMWOSFHM-WPRPVWTQSA-N (+)-Pilocarpine Chemical compound C1OC(=O)[C@@H](CC)[C@H]1CC1=CN=CN1C QCHFTSOMWOSFHM-WPRPVWTQSA-N 0.000 description 1
- HFVMEOPYDLEHBR-UHFFFAOYSA-N (2-fluorophenyl)-phenylmethanol Chemical compound C=1C=CC=C(F)C=1C(O)C1=CC=CC=C1 HFVMEOPYDLEHBR-UHFFFAOYSA-N 0.000 description 1
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 1
- MDKGKXOCJGEUJW-VIFPVBQESA-N (2s)-2-[4-(thiophene-2-carbonyl)phenyl]propanoic acid Chemical compound C1=CC([C@@H](C(O)=O)C)=CC=C1C(=O)C1=CC=CS1 MDKGKXOCJGEUJW-VIFPVBQESA-N 0.000 description 1
- PHOZOHFUXHPOCK-QMMMGPOBSA-N (2s)-2-ethyl-8-methyl-1-thia-4,8-diazaspiro[4.5]decan-3-one Chemical compound N1C(=O)[C@H](CC)SC11CCN(C)CC1 PHOZOHFUXHPOCK-QMMMGPOBSA-N 0.000 description 1
- LJRDOKAZOAKLDU-UDXJMMFXSA-N (2s,3s,4r,5r,6r)-5-amino-2-(aminomethyl)-6-[(2r,3s,4r,5s)-5-[(1r,2r,3s,5r,6s)-3,5-diamino-2-[(2s,3r,4r,5s,6r)-3-amino-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-hydroxycyclohexyl]oxy-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl]oxyoxane-3,4-diol;sulfuric ac Chemical compound OS(O)(=O)=O.N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)O[C@@H]1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)N)O[C@@H]1CO LJRDOKAZOAKLDU-UDXJMMFXSA-N 0.000 description 1
- SGKRLCUYIXIAHR-AKNGSSGZSA-N (4s,4ar,5s,5ar,6r,12ar)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1=CC=C2[C@H](C)[C@@H]([C@H](O)[C@@H]3[C@](C(O)=C(C(N)=O)C(=O)[C@H]3N(C)C)(O)C3=O)C3=C(O)C2=C1O SGKRLCUYIXIAHR-AKNGSSGZSA-N 0.000 description 1
- FFTVPQUHLQBXQZ-KVUCHLLUSA-N (4s,4as,5ar,12ar)-4,7-bis(dimethylamino)-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1C2=C(N(C)C)C=CC(O)=C2C(O)=C2[C@@H]1C[C@H]1[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]1(O)C2=O FFTVPQUHLQBXQZ-KVUCHLLUSA-N 0.000 description 1
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 description 1
- LLNAMUJRIZIXHF-CLFYSBASSA-N (z)-2-methyl-3-phenylprop-2-en-1-ol Chemical compound OCC(/C)=C\C1=CC=CC=C1 LLNAMUJRIZIXHF-CLFYSBASSA-N 0.000 description 1
- HDPNBNXLBDFELL-UHFFFAOYSA-N 1,1,1-trimethoxyethane Chemical compound COC(C)(OC)OC HDPNBNXLBDFELL-UHFFFAOYSA-N 0.000 description 1
- ZGMNAIODRDOMEK-UHFFFAOYSA-N 1,1,1-trimethoxypropane Chemical compound CCC(OC)(OC)OC ZGMNAIODRDOMEK-UHFFFAOYSA-N 0.000 description 1
- FLBAYUMRQUHISI-UHFFFAOYSA-N 1,8-naphthyridine Chemical compound N1=CC=CC2=CC=CN=C21 FLBAYUMRQUHISI-UHFFFAOYSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- FUFLCEKSBBHCMO-UHFFFAOYSA-N 11-dehydrocorticosterone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 FUFLCEKSBBHCMO-UHFFFAOYSA-N 0.000 description 1
- JSFATNQSLKRBCI-VAEKSGALSA-N 15(S)-HETE Chemical compound CCCCC[C@H](O)\C=C\C=C/C\C=C/C\C=C/CCCC(O)=O JSFATNQSLKRBCI-VAEKSGALSA-N 0.000 description 1
- KAESVJOAVNADME-UHFFFAOYSA-N 1H-pyrrole Natural products C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 1
- LATZVDXOTDYECD-UFTFXDLESA-N 2,3-dihydroxybutanedioic acid (3S,4R)-3-ethyl-4-(1,5,7,10-tetrazatricyclo[7.3.0.02,6]dodeca-2(6),3,7,9,11-pentaen-12-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide tetrahydrate Chemical compound O.O.O.O.OC(C(O)C(O)=O)C(O)=O.CC[C@@H]1CN(C[C@@H]1c1cnc2cnc3[nH]ccc3n12)C(=O)NCC(F)(F)F LATZVDXOTDYECD-UFTFXDLESA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- DPZHKLJPVMYFCU-UHFFFAOYSA-N 2-(5-bromopyridin-2-yl)acetonitrile Chemical compound BrC1=CC=C(CC#N)N=C1 DPZHKLJPVMYFCU-UHFFFAOYSA-N 0.000 description 1
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- OZDAOHVKBFBBMZ-UHFFFAOYSA-N 2-aminopentanedioic acid;hydrate Chemical compound O.OC(=O)C(N)CCC(O)=O OZDAOHVKBFBBMZ-UHFFFAOYSA-N 0.000 description 1
- LEACJMVNYZDSKR-UHFFFAOYSA-N 2-octyldodecan-1-ol Chemical compound CCCCCCCCCCC(CO)CCCCCCCC LEACJMVNYZDSKR-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- WHBMMWSBFZVSSR-UHFFFAOYSA-N 3-hydroxybutyric acid Chemical compound CC(O)CC(O)=O WHBMMWSBFZVSSR-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 1
- QISOBCMNUJQOJU-UHFFFAOYSA-N 4-bromo-1h-pyrazole-5-carboxylic acid Chemical compound OC(=O)C=1NN=CC=1Br QISOBCMNUJQOJU-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-N 4-bromobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-N 0.000 description 1
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 1
- 102100030310 5,6-dihydroxyindole-2-carboxylic acid oxidase Human genes 0.000 description 1
- 101710163881 5,6-dihydroxyindole-2-carboxylic acid oxidase Proteins 0.000 description 1
- NMUSYJAQQFHJEW-UHFFFAOYSA-N 5-Azacytidine Natural products O=C1N=C(N)N=CN1C1C(O)C(O)C(CO)O1 NMUSYJAQQFHJEW-UHFFFAOYSA-N 0.000 description 1
- PXRKCOCTEMYUEG-UHFFFAOYSA-N 5-aminoisoindole-1,3-dione Chemical compound NC1=CC=C2C(=O)NC(=O)C2=C1 PXRKCOCTEMYUEG-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- MMRCWWRFYLZGAE-ZBZRSYSASA-N 533u947v6q Chemical compound O([C@]12[C@H](OC(C)=O)[C@]3(CC)C=CCN4CC[C@@]5([C@H]34)[C@H]1N(C)C1=C5C=C(C(=C1)OC)[C@]1(C(=O)OC)C3=C(C4=CC=CC=C4N3)CCN3C[C@H](C1)C[C@@](C3)(O)CC)C(=O)N(CCCl)C2=O MMRCWWRFYLZGAE-ZBZRSYSASA-N 0.000 description 1
- MJZJYWCQPMNPRM-UHFFFAOYSA-N 6,6-dimethyl-1-[3-(2,4,5-trichlorophenoxy)propoxy]-1,6-dihydro-1,3,5-triazine-2,4-diamine Chemical compound CC1(C)N=C(N)N=C(N)N1OCCCOC1=CC(Cl)=C(Cl)C=C1Cl MJZJYWCQPMNPRM-UHFFFAOYSA-N 0.000 description 1
- ULECEJGNDHWSKD-UHFFFAOYSA-N 6-amino-2-[2-[(2-amino-3-phenylpropanoyl)amino]propanoylamino]hexanoic acid Chemical compound NCCCCC(C(O)=O)NC(=O)C(C)NC(=O)C(N)CC1=CC=CC=C1 ULECEJGNDHWSKD-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- GSDSWSVVBLHKDQ-UHFFFAOYSA-N 9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carboxylic acid Chemical compound FC1=CC(C(C(C(O)=O)=C2)=O)=C3N2C(C)COC3=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-UHFFFAOYSA-N 0.000 description 1
- HPLNQCPCUACXLM-PGUFJCEWSA-N ABT-737 Chemical compound C([C@@H](CCN(C)C)NC=1C(=CC(=CC=1)S(=O)(=O)NC(=O)C=1C=CC(=CC=1)N1CCN(CC=2C(=CC=CC=2)C=2C=CC(Cl)=CC=2)CC1)[N+]([O-])=O)SC1=CC=CC=C1 HPLNQCPCUACXLM-PGUFJCEWSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 102100036475 Alanine aminotransferase 1 Human genes 0.000 description 1
- 108010082126 Alanine transaminase Proteins 0.000 description 1
- 206010001935 American trypanosomiasis Diseases 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 1
- 206010053555 Arthritis bacterial Diseases 0.000 description 1
- 206010003267 Arthritis reactive Diseases 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 108010001478 Bacitracin Proteins 0.000 description 1
- 208000009137 Behcet syndrome Diseases 0.000 description 1
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 1
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 101100123850 Caenorhabditis elegans her-1 gene Proteins 0.000 description 1
- 229940122739 Calcineurin inhibitor Drugs 0.000 description 1
- 101710192106 Calcineurin-binding protein cabin-1 Proteins 0.000 description 1
- 102100024123 Calcineurin-binding protein cabin-1 Human genes 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- 206010007134 Candida infections Diseases 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 206010007279 Carcinoid tumour of the gastrointestinal tract Diseases 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- 206010007882 Cellulitis Diseases 0.000 description 1
- 229930186147 Cephalosporin Natural products 0.000 description 1
- 208000024699 Chagas disease Diseases 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- RENMDAKOXSCIGH-UHFFFAOYSA-N Chloroacetonitrile Chemical compound ClCC#N RENMDAKOXSCIGH-UHFFFAOYSA-N 0.000 description 1
- 206010008874 Chronic Fatigue Syndrome Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 208000015943 Coeliac disease Diseases 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 208000023890 Complex Regional Pain Syndromes Diseases 0.000 description 1
- 208000013586 Complex regional pain syndrome type 1 Diseases 0.000 description 1
- 206010010741 Conjunctivitis Diseases 0.000 description 1
- MFYSYFVPBJMHGN-ZPOLXVRWSA-N Cortisone Chemical compound O=C1CC[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 MFYSYFVPBJMHGN-ZPOLXVRWSA-N 0.000 description 1
- MFYSYFVPBJMHGN-UHFFFAOYSA-N Cortisone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)(O)C(=O)CO)C4C3CCC2=C1 MFYSYFVPBJMHGN-UHFFFAOYSA-N 0.000 description 1
- 206010011219 Costochondritis Diseases 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 241001337994 Cryptococcus <scale insect> Species 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- DYDCUQKUCUHJBH-UWTATZPHSA-N D-Cycloserine Chemical compound N[C@@H]1CONC1=O DYDCUQKUCUHJBH-UWTATZPHSA-N 0.000 description 1
- DYDCUQKUCUHJBH-UHFFFAOYSA-N D-Cycloserine Natural products NC1CONC1=O DYDCUQKUCUHJBH-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 229940126190 DNA methyltransferase inhibitor Drugs 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- FMGSKLZLMKYGDP-UHFFFAOYSA-N Dehydroepiandrosterone Natural products C1C(O)CCC2(C)C3CCC(C)(C(CC4)=O)C4C3CC=C21 FMGSKLZLMKYGDP-UHFFFAOYSA-N 0.000 description 1
- 206010048768 Dermatosis Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 1
- 206010012689 Diabetic retinopathy Diseases 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 206010013453 Disseminated tuberculosis Diseases 0.000 description 1
- ZQZFYGIXNQKOAV-OCEACIFDSA-N Droloxifene Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=C(O)C=CC=1)\C1=CC=C(OCCN(C)C)C=C1 ZQZFYGIXNQKOAV-OCEACIFDSA-N 0.000 description 1
- 208000003556 Dry Eye Syndromes Diseases 0.000 description 1
- 206010013774 Dry eye Diseases 0.000 description 1
- 238000008157 ELISA kit Methods 0.000 description 1
- 201000009273 Endometriosis Diseases 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- 208000010201 Exanthema Diseases 0.000 description 1
- 102100034719 Extracellular glycoprotein lacritin Human genes 0.000 description 1
- 208000001640 Fibromyalgia Diseases 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 102100037813 Focal adhesion kinase 1 Human genes 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 206010018001 Gastrointestinal perforation Diseases 0.000 description 1
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 description 1
- ZPACYDRSPFRDHO-ROBAGEODSA-N Gefarnate Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\CCC(=O)OC\C=C(/C)CCC=C(C)C ZPACYDRSPFRDHO-ROBAGEODSA-N 0.000 description 1
- 229920001386 Gefarnate Polymers 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 206010018341 Gliosis Diseases 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 108010069236 Goserelin Proteins 0.000 description 1
- 206010018634 Gouty Arthritis Diseases 0.000 description 1
- 208000009329 Graft vs Host Disease Diseases 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 1
- 229940125497 HER2 kinase inhibitor Drugs 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 208000010496 Heart Arrest Diseases 0.000 description 1
- 206010019280 Heart failures Diseases 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 208000029433 Herpesviridae infectious disease Diseases 0.000 description 1
- 101001090521 Homo sapiens Extracellular glycoprotein lacritin Proteins 0.000 description 1
- 101000746367 Homo sapiens Granulocyte colony-stimulating factor Proteins 0.000 description 1
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 1
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 1
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101000617830 Homo sapiens Sterol O-acyltransferase 1 Proteins 0.000 description 1
- 101000611183 Homo sapiens Tumor necrosis factor Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 241000598436 Human T-cell lymphotropic virus Species 0.000 description 1
- 241000701085 Human alphaherpesvirus 3 Species 0.000 description 1
- 241000701806 Human papillomavirus Species 0.000 description 1
- 241000829111 Human polyomavirus 1 Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- 238000004566 IR spectroscopy Methods 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 1
- 208000010159 IgA glomerulonephritis Diseases 0.000 description 1
- 206010021263 IgA nephropathy Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 102000010781 Interleukin-6 Receptors Human genes 0.000 description 1
- 108010038501 Interleukin-6 Receptors Proteins 0.000 description 1
- OWYWGLHRNBIFJP-UHFFFAOYSA-N Ipazine Chemical compound CCN(CC)C1=NC(Cl)=NC(NC(C)C)=N1 OWYWGLHRNBIFJP-UHFFFAOYSA-N 0.000 description 1
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 1
- 229940121730 Janus kinase 2 inhibitor Drugs 0.000 description 1
- 208000003456 Juvenile Arthritis Diseases 0.000 description 1
- 206010059176 Juvenile idiopathic arthritis Diseases 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- ZCGOMHNNNFPNMX-YHYDXASRSA-N Levocabastinum Chemical compound C1([C@@]2(C(O)=O)CCN(C[C@H]2C)C2CCC(CC2)(C#N)C=2C=CC(F)=CC=2)=CC=CC=C1 ZCGOMHNNNFPNMX-YHYDXASRSA-N 0.000 description 1
- GSDSWSVVBLHKDQ-JTQLQIEISA-N Levofloxacin Chemical compound C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-JTQLQIEISA-N 0.000 description 1
- 229910010084 LiAlH4 Inorganic materials 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- BYBLEWFAAKGYCD-UHFFFAOYSA-N Miconazole Chemical compound ClC1=CC(Cl)=CC=C1COC(C=1C(=CC(Cl)=CC=1)Cl)CN1C=NC=C1 BYBLEWFAAKGYCD-UHFFFAOYSA-N 0.000 description 1
- 201000006836 Miliary Tuberculosis Diseases 0.000 description 1
- 102000004232 Mitogen-Activated Protein Kinase Kinases Human genes 0.000 description 1
- 108090000744 Mitogen-Activated Protein Kinase Kinases Proteins 0.000 description 1
- 102100034256 Mucin-1 Human genes 0.000 description 1
- 108010063954 Mucins Proteins 0.000 description 1
- 108010093825 Mucoproteins Proteins 0.000 description 1
- 102000001621 Mucoproteins Human genes 0.000 description 1
- 229940121743 Muscarinic receptor agonist Drugs 0.000 description 1
- 208000000112 Myalgia Diseases 0.000 description 1
- 206010062207 Mycobacterial infection Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 201000007224 Myeloproliferative neoplasm Diseases 0.000 description 1
- 208000009525 Myocarditis Diseases 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- IJHNSHDBIRRJRN-UHFFFAOYSA-N N,N-dimethyl-3-phenyl-3-(2-pyridinyl)-1-propanamine Chemical compound C=1C=CC=NC=1C(CCN(C)C)C1=CC=CC=C1 IJHNSHDBIRRJRN-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 229910020700 Na3VO4 Inorganic materials 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 206010071579 Neuronal neuropathy Diseases 0.000 description 1
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 102100032028 Non-receptor tyrosine-protein kinase TYK2 Human genes 0.000 description 1
- 206010030154 Oesophageal candidiasis Diseases 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 239000004100 Oxytetracycline Substances 0.000 description 1
- NMLMACJWHPHKGR-NCOIDOBVSA-N P(1),P(4)-bis(uridin-5'-yl) tetraphosphate Chemical compound N1([C@@H]2O[C@@H]([C@H]([C@H]2O)O)COP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@H]([C@@H](O2)N2C(NC(=O)C=C2)=O)O)O)C=CC(=O)NC1=O NMLMACJWHPHKGR-NCOIDOBVSA-N 0.000 description 1
- 108091008606 PDGF receptors Proteins 0.000 description 1
- 206010053869 POEMS syndrome Diseases 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 206010033661 Pancytopenia Diseases 0.000 description 1
- 241000282320 Panthera leo Species 0.000 description 1
- UOZODPSAJZTQNH-UHFFFAOYSA-N Paromomycin II Natural products NC1C(O)C(O)C(CN)OC1OC1C(O)C(OC2C(C(N)CC(N)C2O)OC2C(C(O)C(O)C(CO)O2)N)OC1CO UOZODPSAJZTQNH-UHFFFAOYSA-N 0.000 description 1
- 206010034277 Pemphigoid Diseases 0.000 description 1
- 241000721454 Pemphigus Species 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 102100036056 Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit delta isoform Human genes 0.000 description 1
- 101710204747 Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit delta isoform Proteins 0.000 description 1
- 102000004179 Plasminogen Activator Inhibitor 2 Human genes 0.000 description 1
- 108090000614 Plasminogen Activator Inhibitor 2 Proteins 0.000 description 1
- 102000011653 Platelet-Derived Growth Factor Receptors Human genes 0.000 description 1
- 208000005384 Pneumocystis Pneumonia Diseases 0.000 description 1
- 206010073755 Pneumocystis jirovecii pneumonia Diseases 0.000 description 1
- 206010035664 Pneumonia Diseases 0.000 description 1
- 208000008601 Polycythemia Diseases 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- 108010040201 Polymyxins Proteins 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108091008611 Protein Kinase B Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 201000001263 Psoriatic Arthritis Diseases 0.000 description 1
- 208000036824 Psoriatic arthropathy Diseases 0.000 description 1
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 description 1
- 102000001744 Purinergic P2Y2 Receptors Human genes 0.000 description 1
- 108010029812 Purinergic P2Y2 Receptors Proteins 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 102100033810 RAC-alpha serine/threonine-protein kinase Human genes 0.000 description 1
- 108091030071 RNAI Proteins 0.000 description 1
- ALLWOAVDORUJLA-UHFFFAOYSA-N Rebamipida Chemical compound C=1C(=O)NC2=CC=CC=C2C=1CC(C(=O)O)NC(=O)C1=CC=C(Cl)C=C1 ALLWOAVDORUJLA-UHFFFAOYSA-N 0.000 description 1
- 208000015634 Rectal Neoplasms Diseases 0.000 description 1
- 201000001947 Reflex Sympathetic Dystrophy Diseases 0.000 description 1
- 208000035415 Reinfection Diseases 0.000 description 1
- 208000033464 Reiter syndrome Diseases 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- 208000025747 Rheumatic disease Diseases 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- QCHFTSOMWOSFHM-UHFFFAOYSA-N SJ000285536 Natural products C1OC(=O)C(CC)C1CC1=CN=CN1C QCHFTSOMWOSFHM-UHFFFAOYSA-N 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 206010039705 Scleritis Diseases 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- 102100023085 Serine/threonine-protein kinase mTOR Human genes 0.000 description 1
- 101710173693 Short transient receptor potential channel 1 Proteins 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- 229920002385 Sodium hyaluronate Polymers 0.000 description 1
- HVUMOYIDDBPOLL-XWVZOOPGSA-N Sorbitan monostearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O HVUMOYIDDBPOLL-XWVZOOPGSA-N 0.000 description 1
- 102100021993 Sterol O-acyltransferase 1 Human genes 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 101000697584 Streptomyces lavendulae Streptothricin acetyltransferase Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- 206010051379 Systemic Inflammatory Response Syndrome Diseases 0.000 description 1
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 1
- 108010010057 TYK2 Kinase Proteins 0.000 description 1
- 206010071574 Testicular autoimmunity Diseases 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 102000036693 Thrombopoietin Human genes 0.000 description 1
- 108010041111 Thrombopoietin Proteins 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 208000026317 Tietze syndrome Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108090000340 Transaminases Proteins 0.000 description 1
- 102000003929 Transaminases Human genes 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- LVTKHGUGBGNBPL-UHFFFAOYSA-N Trp-P-1 Chemical compound N1C2=CC=CC=C2C2=C1C(C)=C(N)N=C2C LVTKHGUGBGNBPL-UHFFFAOYSA-N 0.000 description 1
- 241000223109 Trypanosoma cruzi Species 0.000 description 1
- 241000287411 Turdidae Species 0.000 description 1
- 108700036252 Tyrosine Kinase 2 Deficiency Proteins 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 206010046799 Uterine leiomyosarcoma Diseases 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 206010047642 Vitiligo Diseases 0.000 description 1
- ZZXDRXVIRVJQBT-UHFFFAOYSA-M Xylenesulfonate Chemical compound CC1=CC=CC(S([O-])(=O)=O)=C1C ZZXDRXVIRVJQBT-UHFFFAOYSA-M 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- RRDRHWJDBOGQHN-JWCTVYNTSA-N [2-[(2s,5r,8s,11s,14r,17s,22s)-17-[(1r)-1-hydroxyethyl]-22-[[(2s)-2-[[(2s,3r)-3-hydroxy-2-[[(2s)-2-[6-methyloctanoyl(sulfomethyl)amino]-4-(sulfomethylamino)butanoyl]amino]butyl]amino]-4-(sulfomethylamino)butanoyl]amino]-5,8-bis(2-methylpropyl)-3,6,9,12,15 Chemical compound CCC(C)CCCCC(=O)N(CS(O)(=O)=O)[C@@H](CCNCS(O)(=O)=O)C(=O)N[C@H]([C@@H](C)O)CN[C@@H](CCNCS(O)(=O)=O)C(=O)N[C@H]1CCNC(=O)[C@H]([C@@H](C)O)NC(=O)[C@@H](CCNCS(O)(=O)=O)NC(=O)[C@H](CCNCS(O)(=O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CCNCS(O)(=O)=O)NC1=O RRDRHWJDBOGQHN-JWCTVYNTSA-N 0.000 description 1
- 229960003697 abatacept Drugs 0.000 description 1
- 229960000446 abciximab Drugs 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 229940124532 absorption promoter Drugs 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- YTIVTFGABIZHHX-UHFFFAOYSA-L acetylenedicarboxylate(2-) Chemical compound [O-]C(=O)C#CC([O-])=O YTIVTFGABIZHHX-UHFFFAOYSA-L 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 229940119059 actemra Drugs 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 229960002964 adalimumab Drugs 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 201000009961 allergic asthma Diseases 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 208000030961 allergic reaction Diseases 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- 208000004631 alopecia areata Diseases 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 229960004821 amikacin Drugs 0.000 description 1
- LKCWBDHBTVXHDL-RMDFUYIESA-N amikacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1O LKCWBDHBTVXHDL-RMDFUYIESA-N 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229940126575 aminoglycoside Drugs 0.000 description 1
- 229940113720 aminosalicylate Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- 206010002022 amyloidosis Diseases 0.000 description 1
- 230000003444 anaesthetic effect Effects 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 208000022531 anorexia Diseases 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 229960002469 antazoline Drugs 0.000 description 1
- REYFJDPCWQRWAA-UHFFFAOYSA-N antazoline Chemical compound N=1CCNC=1CN(C=1C=CC=CC=1)CC1=CC=CC=C1 REYFJDPCWQRWAA-UHFFFAOYSA-N 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000003092 anti-cytokine Effects 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 230000000845 anti-microbial effect Effects 0.000 description 1
- 230000001062 anti-nausea Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 230000001494 anti-thymocyte effect Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 239000000043 antiallergic agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000005129 aryl carbonyl group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 208000037875 astrocytosis Diseases 0.000 description 1
- 230000007341 astrogliosis Effects 0.000 description 1
- 208000010928 autoimmune thyroid disease Diseases 0.000 description 1
- 230000035578 autophosphorylation Effects 0.000 description 1
- 229940120638 avastin Drugs 0.000 description 1
- 230000003376 axonal effect Effects 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 229960003071 bacitracin Drugs 0.000 description 1
- 229930184125 bacitracin Natural products 0.000 description 1
- CLKOFPXJLQSYAH-ABRJDSQDSA-N bacitracin A Chemical compound C1SC([C@@H](N)[C@@H](C)CC)=N[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]1C(=O)N[C@H](CCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2N=CNC=2)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)NCCCC1 CLKOFPXJLQSYAH-ABRJDSQDSA-N 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 229960003270 belimumab Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000010256 biochemical assay Methods 0.000 description 1
- 239000003124 biologic agent Substances 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 230000036983 biotransformation Effects 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 208000010217 blepharitis Diseases 0.000 description 1
- 238000010322 bone marrow transplantation Methods 0.000 description 1
- BGECDVWSWDRFSP-UHFFFAOYSA-N borazine Chemical compound B1NBNBN1 BGECDVWSWDRFSP-UHFFFAOYSA-N 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 208000000594 bullous pemphigoid Diseases 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical group 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical group 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229940124587 cephalosporin Drugs 0.000 description 1
- 150000001780 cephalosporins Chemical class 0.000 description 1
- 229960003115 certolizumab pegol Drugs 0.000 description 1
- 229940081733 cetearyl alcohol Drugs 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 210000003467 cheek Anatomy 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- JQXXHWHPUNPDRT-BQVAUQFYSA-N chembl1523493 Chemical compound O([C@](C1=O)(C)O\C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)/C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2C=NN1CCN(C)CC1 JQXXHWHPUNPDRT-BQVAUQFYSA-N 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- 229960005091 chloramphenicol Drugs 0.000 description 1
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- 229960003677 chloroquine Drugs 0.000 description 1
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 1
- 230000001713 cholinergic effect Effects 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229940108538 colistimethate Drugs 0.000 description 1
- 108700028201 colistinmethanesulfonic acid Proteins 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 229960004544 cortisone Drugs 0.000 description 1
- QTCANKDTWWSCMR-UHFFFAOYSA-N costic aldehyde Natural products C1CCC(=C)C2CC(C(=C)C=O)CCC21C QTCANKDTWWSCMR-UHFFFAOYSA-N 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 229940109239 creatinine Drugs 0.000 description 1
- 229960000265 cromoglicic acid Drugs 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 description 1
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 1
- 125000006254 cycloalkyl carbonyl group Chemical group 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229960003077 cycloserine Drugs 0.000 description 1
- UWFYSQMTEOIJJG-FDTZYFLXSA-N cyproterone acetate Chemical compound C1=C(Cl)C2=CC(=O)[C@@H]3C[C@@H]3[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 UWFYSQMTEOIJJG-FDTZYFLXSA-N 0.000 description 1
- 229960000978 cyproterone acetate Drugs 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-M decanoate Chemical compound CCCCCCCCCC([O-])=O GHVNFZFCNZKVNT-UHFFFAOYSA-M 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 1
- 206010061428 decreased appetite Diseases 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- FMGSKLZLMKYGDP-USOAJAOKSA-N dehydroepiandrosterone Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC=C21 FMGSKLZLMKYGDP-USOAJAOKSA-N 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- CFCUWKMKBJTWLW-UHFFFAOYSA-N deoliosyl-3C-alpha-L-digitoxosyl-MTM Natural products CC=1C(O)=C2C(O)=C3C(=O)C(OC4OC(C)C(O)C(OC5OC(C)C(O)C(OC6OC(C)C(O)C(C)(O)C6)C5)C4)C(C(OC)C(=O)C(O)C(C)O)CC3=CC2=CC=1OC(OC(C)C1O)CC1OC1CC(O)C(O)C(C)O1 CFCUWKMKBJTWLW-UHFFFAOYSA-N 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 208000033679 diabetic kidney disease Diseases 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- FSBVERYRVPGNGG-UHFFFAOYSA-N dimagnesium dioxido-bis[[oxido(oxo)silyl]oxy]silane hydrate Chemical compound O.[Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O FSBVERYRVPGNGG-UHFFFAOYSA-N 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 229950003529 diquafosol Drugs 0.000 description 1
- 230000009266 disease activity Effects 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- VLARUOGDXDTHEH-UHFFFAOYSA-L disodium cromoglycate Chemical compound [Na+].[Na+].O1C(C([O-])=O)=CC(=O)C2=C1C=CC=C2OCC(O)COC1=CC=CC2=C1C(=O)C=C(C([O-])=O)O2 VLARUOGDXDTHEH-UHFFFAOYSA-L 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000003968 dna methyltransferase inhibitor Substances 0.000 description 1
- 229960003722 doxycycline Drugs 0.000 description 1
- 229950004203 droloxifene Drugs 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- IWCWQNVIUXZOMJ-MISYRCLQSA-N ecabet Chemical compound OC(=O)[C@@](C)([C@@H]1CC2)CCC[C@]1(C)C1=C2C=C(C(C)C)C(S(O)(=O)=O)=C1 IWCWQNVIUXZOMJ-MISYRCLQSA-N 0.000 description 1
- 229950003246 ecabet Drugs 0.000 description 1
- 229960000284 efalizumab Drugs 0.000 description 1
- 229940121647 egfr inhibitor Drugs 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 210000002257 embryonic structure Anatomy 0.000 description 1
- 239000003974 emollient agent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000008387 emulsifying waxe Substances 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940095399 enema Drugs 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 229960002549 enoxacin Drugs 0.000 description 1
- IDYZIJYBMGIQMJ-UHFFFAOYSA-N enoxacin Chemical compound N1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNCC1 IDYZIJYBMGIQMJ-UHFFFAOYSA-N 0.000 description 1
- 239000012055 enteric layer Substances 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 230000001973 epigenetic effect Effects 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- 150000003883 epothilone derivatives Chemical class 0.000 description 1
- 230000010437 erythropoiesis Effects 0.000 description 1
- 201000005655 esophageal candidiasis Diseases 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 1
- LIWAQLJGPBVORC-UHFFFAOYSA-N ethylmethylamine Chemical compound CCNC LIWAQLJGPBVORC-UHFFFAOYSA-N 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 230000003090 exacerbative effect Effects 0.000 description 1
- 201000005884 exanthem Diseases 0.000 description 1
- 229960000255 exemestane Drugs 0.000 description 1
- 239000003172 expectorant agent Substances 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 206010016165 failure to thrive Diseases 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229960004039 finasteride Drugs 0.000 description 1
- DBEPLOCGEIEOCV-WSBQPABSSA-N finasteride Chemical compound N([C@@H]1CC2)C(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)NC(C)(C)C)[C@@]2(C)CC1 DBEPLOCGEIEOCV-WSBQPABSSA-N 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 229960004884 fluconazole Drugs 0.000 description 1
- RFHAOTPXVQNOHP-UHFFFAOYSA-N fluconazole Chemical compound C1=NC=NN1CC(C=1C(=CC(F)=CC=1)F)(O)CN1C=NC=N1 RFHAOTPXVQNOHP-UHFFFAOYSA-N 0.000 description 1
- XRECTZIEBJDKEO-UHFFFAOYSA-N flucytosine Chemical compound NC1=NC(=O)NC=C1F XRECTZIEBJDKEO-UHFFFAOYSA-N 0.000 description 1
- 229960004413 flucytosine Drugs 0.000 description 1
- 229940043075 fluocinolone Drugs 0.000 description 1
- 229960001347 fluocinolone acetonide Drugs 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 229960001048 fluorometholone Drugs 0.000 description 1
- FAOZLTXFLGPHNG-KNAQIMQKSA-N fluorometholone Chemical compound C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@]2(F)[C@@H](O)C[C@]2(C)[C@@](O)(C(C)=O)CC[C@H]21 FAOZLTXFLGPHNG-KNAQIMQKSA-N 0.000 description 1
- 229940124307 fluoroquinolone Drugs 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 229960002390 flurbiprofen Drugs 0.000 description 1
- SYTBZMRGLBWNTM-UHFFFAOYSA-N flurbiprofen Chemical compound FC1=CC(C(C(O)=O)C)=CC=C1C1=CC=CC=C1 SYTBZMRGLBWNTM-UHFFFAOYSA-N 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 description 1
- 229960003779 gefarnate Drugs 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 230000009368 gene silencing by RNA Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 229960003690 goserelin acetate Drugs 0.000 description 1
- 208000024908 graft versus host disease Diseases 0.000 description 1
- 230000036433 growing body Effects 0.000 description 1
- 229960002706 gusperimus Drugs 0.000 description 1
- 231100000640 hair analysis Toxicity 0.000 description 1
- 235000015220 hamburgers Nutrition 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 239000003481 heat shock protein 90 inhibitor Substances 0.000 description 1
- 208000007475 hemolytic anemia Diseases 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 125000006517 heterocyclyl carbonyl group Chemical group 0.000 description 1
- KKLGDUSGQMHBPB-UHFFFAOYSA-N hex-2-ynedioic acid Chemical compound OC(=O)CCC#CC(O)=O KKLGDUSGQMHBPB-UHFFFAOYSA-N 0.000 description 1
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 230000003054 hormonal effect Effects 0.000 description 1
- 238000001794 hormone therapy Methods 0.000 description 1
- 102000057041 human TNF Human genes 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 208000018615 immunodeficiency 35 Diseases 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229940084651 iressa Drugs 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 201000004614 iritis Diseases 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- ISTFUJWTQAMRGA-UHFFFAOYSA-N iso-beta-costal Natural products C1C(C(=C)C=O)CCC2(C)CCCC(C)=C21 ISTFUJWTQAMRGA-UHFFFAOYSA-N 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 201000002215 juvenile rheumatoid arthritis Diseases 0.000 description 1
- 229960000318 kanamycin Drugs 0.000 description 1
- 229930027917 kanamycin Natural products 0.000 description 1
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 1
- 229930182823 kanamycin A Natural products 0.000 description 1
- 229960004125 ketoconazole Drugs 0.000 description 1
- 229960004752 ketorolac Drugs 0.000 description 1
- OZWKMVRBQXNZKK-UHFFFAOYSA-N ketorolac Chemical compound OC(=O)C1CCN2C1=CC=C2C(=O)C1=CC=CC=C1 OZWKMVRBQXNZKK-UHFFFAOYSA-N 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 229960000681 leflunomide Drugs 0.000 description 1
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 1
- 229960004942 lenalidomide Drugs 0.000 description 1
- 229950007278 lenercept Drugs 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- 229960003376 levofloxacin Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 229960002422 lomefloxacin Drugs 0.000 description 1
- ZEKZLJVOYLTDKK-UHFFFAOYSA-N lomefloxacin Chemical compound FC1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNC(C)C1 ZEKZLJVOYLTDKK-UHFFFAOYSA-N 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- DMKSVUSAATWOCU-HROMYWEYSA-N loteprednol etabonate Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)OCCl)(OC(=O)OCC)[C@@]1(C)C[C@@H]2O DMKSVUSAATWOCU-HROMYWEYSA-N 0.000 description 1
- 229960003744 loteprednol etabonate Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 229940124302 mTOR inhibitor Drugs 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 230000036244 malformation Effects 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- 229960004296 megestrol acetate Drugs 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- IZYBEMGNIUSSAX-UHFFFAOYSA-N methyl benzenecarboperoxoate Chemical compound COOC(=O)C1=CC=CC=C1 IZYBEMGNIUSSAX-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 229960000282 metronidazole Drugs 0.000 description 1
- VAOCPAMSLUNLGC-UHFFFAOYSA-N metronidazole Chemical compound CC1=NC=C([N+]([O-])=O)N1CCO VAOCPAMSLUNLGC-UHFFFAOYSA-N 0.000 description 1
- 229960002509 miconazole Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 229940042472 mineral oil Drugs 0.000 description 1
- 229960004023 minocycline Drugs 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 229940066491 mucolytics Drugs 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 229960003816 muromonab-cd3 Drugs 0.000 description 1
- 239000000472 muscarinic agonist Substances 0.000 description 1
- 208000029766 myalgic encephalomeyelitis/chronic fatigue syndrome Diseases 0.000 description 1
- 206010028417 myasthenia gravis Diseases 0.000 description 1
- 208000027531 mycobacterial infectious disease Diseases 0.000 description 1
- DOZYTHNHLLSNIK-JOKMOOFLSA-M mycophenolate sodium Chemical compound [Na+].OC1=C(C\C=C(/C)CCC([O-])=O)C(OC)=C(C)C2=C1C(=O)OC2 DOZYTHNHLLSNIK-JOKMOOFLSA-M 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- 201000005962 mycosis fungoides Diseases 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- PNQCMZHRTCNQMO-UHFFFAOYSA-N n'-iodooxamide Chemical compound NC(=O)C(=O)NI PNQCMZHRTCNQMO-UHFFFAOYSA-N 0.000 description 1
- IDINUJSAMVOPCM-INIZCTEOSA-N n-[(1s)-2-[4-(3-aminopropylamino)butylamino]-1-hydroxy-2-oxoethyl]-7-(diaminomethylideneamino)heptanamide Chemical compound NCCCNCCCCNC(=O)[C@H](O)NC(=O)CCCCCCN=C(N)N IDINUJSAMVOPCM-INIZCTEOSA-N 0.000 description 1
- BLCLNMBMMGCOAS-UHFFFAOYSA-N n-[1-[[1-[[1-[[1-[[1-[[1-[[1-[2-[(carbamoylamino)carbamoyl]pyrrolidin-1-yl]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-[(2-methylpropan-2-yl)oxy]-1-oxopropan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amin Chemical compound C1CCC(C(=O)NNC(N)=O)N1C(=O)C(CCCN=C(N)N)NC(=O)C(CC(C)C)NC(=O)C(COC(C)(C)C)NC(=O)C(NC(=O)C(CO)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C(CC=1NC=NC=1)NC(=O)C1NC(=O)CC1)CC1=CC=C(O)C=C1 BLCLNMBMMGCOAS-UHFFFAOYSA-N 0.000 description 1
- ZBXHNCURDISBRO-UHFFFAOYSA-N n-[[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl]methyl]cyclopropanecarboxamide Chemical compound C1CC1C(=O)NCC(C=1C2=O)=CC=CC=1C(=O)N2C1CCC(=O)NC1=O ZBXHNCURDISBRO-UHFFFAOYSA-N 0.000 description 1
- 229960005016 naphazoline Drugs 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229960005027 natalizumab Drugs 0.000 description 1
- 229960003255 natamycin Drugs 0.000 description 1
- 239000004311 natamycin Substances 0.000 description 1
- 235000010298 natamycin Nutrition 0.000 description 1
- NCXMLFZGDNKEPB-FFPOYIOWSA-N natamycin Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C[C@@H](C)OC(=O)/C=C/[C@H]2O[C@@H]2C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 NCXMLFZGDNKEPB-FFPOYIOWSA-N 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- ZBGPYVZLYBDXKO-HILBYHGXSA-N netilmycin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@]([C@H](NC)[C@@H](O)CO1)(C)O)NCC)[C@H]1OC(CN)=CC[C@H]1N ZBGPYVZLYBDXKO-HILBYHGXSA-N 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 208000004235 neutropenia Diseases 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- XWXYUMMDTVBTOU-UHFFFAOYSA-N nilutamide Chemical compound O=C1C(C)(C)NC(=O)N1C1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 XWXYUMMDTVBTOU-UHFFFAOYSA-N 0.000 description 1
- 229960002653 nilutamide Drugs 0.000 description 1
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 102000037979 non-receptor tyrosine kinases Human genes 0.000 description 1
- 108091008046 non-receptor tyrosine kinases Proteins 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 229960001180 norfloxacin Drugs 0.000 description 1
- OGJPXUAPXNRGGI-UHFFFAOYSA-N norfloxacin Chemical compound C1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNCC1 OGJPXUAPXNRGGI-UHFFFAOYSA-N 0.000 description 1
- 238000002414 normal-phase solid-phase extraction Methods 0.000 description 1
- 102000006255 nuclear receptors Human genes 0.000 description 1
- 108020004017 nuclear receptors Proteins 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 229960001699 ofloxacin Drugs 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 229960000625 oxytetracycline Drugs 0.000 description 1
- IWVCMVBTMGNXQD-PXOLEDIWSA-N oxytetracycline Chemical compound C1=CC=C2[C@](O)(C)[C@H]3[C@H](O)[C@H]4[C@H](N(C)C)C(O)=C(C(N)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O IWVCMVBTMGNXQD-PXOLEDIWSA-N 0.000 description 1
- 235000019366 oxytetracycline Nutrition 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 239000000123 paper Substances 0.000 description 1
- 239000011087 paperboard Substances 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- UOZODPSAJZTQNH-LSWIJEOBSA-N paromomycin Chemical compound N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)O[C@@H]1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)N)O[C@@H]1CO UOZODPSAJZTQNH-LSWIJEOBSA-N 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 235000010603 pastilles Nutrition 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 229960001639 penicillamine Drugs 0.000 description 1
- JLFNLZLINWHATN-UHFFFAOYSA-N pentaethylene glycol Chemical compound OCCOCCOCCOCCOCCO JLFNLZLINWHATN-UHFFFAOYSA-N 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960001190 pheniramine Drugs 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical compound CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 238000003566 phosphorylation assay Methods 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 229960001416 pilocarpine Drugs 0.000 description 1
- 229960002702 piroxicam Drugs 0.000 description 1
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 description 1
- 229940068196 placebo Drugs 0.000 description 1
- 239000000902 placebo Substances 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 201000000317 pneumocystosis Diseases 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 description 1
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 description 1
- 229920002503 polyoxyethylene-polyoxypropylene Polymers 0.000 description 1
- 229940113124 polysorbate 60 Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229960000688 pomalidomide Drugs 0.000 description 1
- UVSMNLNDYGZFPF-UHFFFAOYSA-N pomalidomide Chemical compound O=C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O UVSMNLNDYGZFPF-UHFFFAOYSA-N 0.000 description 1
- 208000017805 post-transplant lymphoproliferative disease Diseases 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 229940124606 potential therapeutic agent Drugs 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000000634 powder X-ray diffraction Methods 0.000 description 1
- 229960002847 prasterone Drugs 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 239000006041 probiotic Substances 0.000 description 1
- 230000000529 probiotic effect Effects 0.000 description 1
- 235000018291 probiotics Nutrition 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- KCXFHTAICRTXLI-UHFFFAOYSA-N propane-1-sulfonic acid Chemical compound CCCS(O)(=O)=O KCXFHTAICRTXLI-UHFFFAOYSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- UORVCLMRJXCDCP-UHFFFAOYSA-M propynoate Chemical compound [O-]C(=O)C#C UORVCLMRJXCDCP-UHFFFAOYSA-M 0.000 description 1
- 229950008679 protamine sulfate Drugs 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 238000000163 radioactive labelling Methods 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 206010037844 rash Diseases 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000003753 real-time PCR Methods 0.000 description 1
- 229950004535 rebamipide Drugs 0.000 description 1
- 229940044601 receptor agonist Drugs 0.000 description 1
- 239000000018 receptor agonist Substances 0.000 description 1
- 229940075993 receptor modulator Drugs 0.000 description 1
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 1
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 1
- 206010038038 rectal cancer Diseases 0.000 description 1
- 229940100618 rectal suppository Drugs 0.000 description 1
- 239000006215 rectal suppository Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 201000001275 rectum cancer Diseases 0.000 description 1
- 210000003289 regulatory T cell Anatomy 0.000 description 1
- 229930002330 retinoic acid Natural products 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229940120975 revlimid Drugs 0.000 description 1
- 230000000552 rheumatic effect Effects 0.000 description 1
- 206010039083 rhinitis Diseases 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- XMSXOLDPMGMWTH-UHFFFAOYSA-N rivoglitazone Chemical compound CN1C2=CC(OC)=CC=C2N=C1COC(C=C1)=CC=C1CC1SC(=O)NC1=O XMSXOLDPMGMWTH-UHFFFAOYSA-N 0.000 description 1
- 229950010764 rivoglitazone Drugs 0.000 description 1
- 239000011435 rock Substances 0.000 description 1
- 229960000215 ruxolitinib Drugs 0.000 description 1
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 description 1
- 231100000279 safety data Toxicity 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229940116351 sebacate Drugs 0.000 description 1
- CXMXRPHRNRROMY-UHFFFAOYSA-L sebacate(2-) Chemical compound [O-]C(=O)CCCCCCCCC([O-])=O CXMXRPHRNRROMY-UHFFFAOYSA-L 0.000 description 1
- 230000000580 secretagogue effect Effects 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 208000002491 severe combined immunodeficiency Diseases 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 201000002314 small intestine cancer Diseases 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 229940010747 sodium hyaluronate Drugs 0.000 description 1
- RSIJVJUOQBWMIM-UHFFFAOYSA-L sodium sulfate decahydrate Chemical compound O.O.O.O.O.O.O.O.O.O.[Na+].[Na+].[O-]S([O-])(=O)=O RSIJVJUOQBWMIM-UHFFFAOYSA-L 0.000 description 1
- YWIVKILSMZOHHF-QJZPQSOGSA-N sodium;(2s,3s,4s,5r,6r)-6-[(2s,3r,4r,5s,6r)-3-acetamido-2-[(2s,3s,4r,5r,6r)-6-[(2r,3r,4r,5s,6r)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2- Chemical compound [Na+].CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 YWIVKILSMZOHHF-QJZPQSOGSA-N 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000001587 sorbitan monostearate Substances 0.000 description 1
- 235000011076 sorbitan monostearate Nutrition 0.000 description 1
- 229940035048 sorbitan monostearate Drugs 0.000 description 1
- 238000011476 stem cell transplantation Methods 0.000 description 1
- 230000003637 steroidlike Effects 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-L suberate(2-) Chemical compound [O-]C(=O)CCCCCCC([O-])=O TYFQFVWCELRYAO-UHFFFAOYSA-L 0.000 description 1
- 238000000859 sublimation Methods 0.000 description 1
- 230000008022 sublimation Effects 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 229960001940 sulfasalazine Drugs 0.000 description 1
- NCEXYHBECQHGNR-QZQOTICOSA-N sulfasalazine Chemical compound C1=C(O)C(C(=O)O)=CC(\N=N\C=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-QZQOTICOSA-N 0.000 description 1
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 229960004492 suprofen Drugs 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 238000013269 sustained drug release Methods 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000002889 sympathetic effect Effects 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- KKEYFWRCBNTPAC-UHFFFAOYSA-L terephthalate(2-) Chemical compound [O-]C(=O)C1=CC=C(C([O-])=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-L 0.000 description 1
- IWVCMVBTMGNXQD-UHFFFAOYSA-N terramycin dehydrate Natural products C1=CC=C2C(O)(C)C3C(O)C4C(N(C)C)C(O)=C(C(N)=O)C(=O)C4(O)C(O)=C3C(=O)C2=C1O IWVCMVBTMGNXQD-UHFFFAOYSA-N 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 229940040944 tetracyclines Drugs 0.000 description 1
- MHXBHWLGRWOABW-UHFFFAOYSA-N tetradecyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCC MHXBHWLGRWOABW-UHFFFAOYSA-N 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 229960000707 tobramycin Drugs 0.000 description 1
- NLVFBUXFDBBNBW-PBSUHMDJSA-N tobramycin Chemical compound N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NLVFBUXFDBBNBW-PBSUHMDJSA-N 0.000 description 1
- 229960003989 tocilizumab Drugs 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- 102000035160 transmembrane proteins Human genes 0.000 description 1
- 108091005703 transmembrane proteins Proteins 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- 229960005294 triamcinolone Drugs 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 1
- CWMFRHBXRUITQE-UHFFFAOYSA-N trimethylsilylacetylene Chemical group C[Si](C)(C)C#C CWMFRHBXRUITQE-UHFFFAOYSA-N 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- IHIXIJGXTJIKRB-UHFFFAOYSA-N trisodium vanadate Chemical compound [Na+].[Na+].[Na+].[O-][V]([O-])([O-])=O IHIXIJGXTJIKRB-UHFFFAOYSA-N 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 229960000497 trovafloxacin Drugs 0.000 description 1
- WVPSKSLAZQPAKQ-CDMJZVDBSA-N trovafloxacin Chemical compound C([C@H]1[C@@H]([C@H]1C1)N)N1C(C(=CC=1C(=O)C(C(O)=O)=C2)F)=NC=1N2C1=CC=C(F)C=C1F WVPSKSLAZQPAKQ-CDMJZVDBSA-N 0.000 description 1
- 229940046728 tumor necrosis factor alpha inhibitor Drugs 0.000 description 1
- 239000002452 tumor necrosis factor alpha inhibitor Substances 0.000 description 1
- 208000019206 urinary tract infection Diseases 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 229960003824 ustekinumab Drugs 0.000 description 1
- 229960004914 vedolizumab Drugs 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- KDQAABAKXDWYSZ-PNYVAJAMSA-N vinblastine sulfate Chemical compound OS(O)(=O)=O.C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 KDQAABAKXDWYSZ-PNYVAJAMSA-N 0.000 description 1
- 229960004982 vinblastine sulfate Drugs 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- IQDSXWRQCKDBMW-NSFJATOBSA-N vintriptol Chemical compound C([C@@H](C[C@@](O)(CC)C1)C[C@@]2(C3=C(OC)C=C4N(C)[C@H]5[C@@]([C@@H]([C@]6(CC)C=CCN7CC[C@]5([C@H]67)C4=C3)O)(O)C(=O)N[C@@H](CC=3C4=CC=CC=C4NC=3)C(=O)OCC)C(=O)OC)N1CCC1=C2NC2=CC=CC=C12 IQDSXWRQCKDBMW-NSFJATOBSA-N 0.000 description 1
- 229950003415 vintriptol Drugs 0.000 description 1
- 229950005839 vinzolidine Drugs 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 229960005289 voclosporin Drugs 0.000 description 1
- BICRTLVBTLFLRD-PTWUADNWSA-N voclosporin Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C=C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O BICRTLVBTLFLRD-PTWUADNWSA-N 0.000 description 1
- 108010057559 voclosporin Proteins 0.000 description 1
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 1
- 229960000237 vorinostat Drugs 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
- 229940071104 xylenesulfonate Drugs 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- YKPUWZUDDOIDPM-VURMDHGXSA-N zucapsaicin Chemical compound COC1=CC(CNC(=O)CCCC\C=C/C(C)C)=CC=C1O YKPUWZUDDOIDPM-VURMDHGXSA-N 0.000 description 1
- 229960002860 zucapsaicin Drugs 0.000 description 1
- 150000003952 β-lactams Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/14—Ortho-condensed systems
- C07D491/147—Ortho-condensed systems the condensed system containing one ring with oxygen as ring hetero atom and two rings with nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4155—1,2-Diazoles non condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
Definitions
- This invention relates to novel 1H-furo[3,2-b]imidazo[4,5-d]pyridine derivatives, their pharmaceutically acceptable salts, solvates, hydrates and polymorphs thereof as selective Jak1 kinase inhibitors.
- the invention also provides compositions comprising a compound of this invention and the use of such compositions in methods of treating diseases and conditions associated with Jak1 and are useful in treating disorders related to Jak1 activities such as an autoimmune disease or disorder, an inflammatory disease or disorder, and a cancer or neoplastic disease or disorder.
- the protein kinases represent a large family of proteins that play a central role in the regulation of a wide variety of cellular processes and maintenance of cellular function.
- a partial, non-limiting, list of these kinases include: non-receptor tyrosine kinases such as the Janus kinase family (Jak1, Jak2, Jak3 and Tyk2); receptor tyrosine kinases such as platelet-derived growth factor receptor kinase (PDGFR); and serine/threonine kinases such as b-RAF.
- Aberrant kinase activity has been observed in many disease states including benign and malignant proliferative disorders as well as diseases resulting from inappropriate activation of the immune and nervous systems.
- the compounds of this invention selectively inhibit the activity of one or more protein kinases over other related kinases, and are thus expected to be useful in the treatment of diseases mediated by the selectively inhibited kinase(s) while avoiding the undesirable side effects associated with the inhibition of the related kinase(s).
- the Janus kinase family comprises 4 known family members: Jak 1, 2, 3, and tyrosine kinase 2 (Tyk2). These cytoplasmic tyrosine kinases are associated with membrane cytokine receptors such as common gamma-chain receptors and the glycoprotein 130 (gp 130) trans-membrane proteins (Murray, J. Immunol. 178(5):2623-2629, 2007). Almost 40 cytokine receptors signal through combinations of these 4 Jak family members and their 7 downstream substrates: the signal transduction activators of transcription (STAT) family members (Ghoreschi et al., Immunol Rev. 228(1):273-287, 2009).
- STAT signal transduction activators of transcription
- Jak activation via trans- and auto-phosphorylation.
- the Jak family kinases in turn phosphorylate cytokine receptor residues, creating binding sites for sarcoma homology 2 (SH2) containing proteins, such as the STAT factors and other regulators, which are subsequently activated by Jak phosphorylation.
- SH2 sarcoma homology 2
- STAT factors such as the STAT factors and other regulators
- STATs sarcoma homology 2
- STATs enter the nucleus initiating expression of survival factors, cytokines, chemokines, and molecules that facilitate leukocyte cellular trafficking (Schindler et al., J. Biol. Chem. 282(28):20059-20063, 2007).
- Jak activation also results in cell proliferation via phosphoinositide 3-kinase (PI3K) and protein kinase B-mediated pathways.
- PI3K phosphoinositide 3-kinase
- Jak3 and Jak1 are components of the common gamma-chain cytokine receptor complexes, and blockade of either inhibits signaling by inflammatory cytokines: interleukin (IL)-2, 4, 7, 9, 15, and 21 (Ghoreschi et al., Immunol. Rev. 228(1):273-287, 2009).
- IL interleukin
- other pathologically relevant cytokines such as IL-6
- Jak1 blockade inhibits signaling of many pro-inflammatory cytokines (Guschin et al., EMBO J. 14(7):1421-1429, 1995).
- Jak1 and Jak2 Humans deficient in Jak1 and Jak2 have not been described. Mice lacking Jak1 die perinatally (Schindler et al., J. Biol Chem. 282(28):20059-20063, 2007). Jak2 deficiency in mice is also lethal, with Jak2 ⁇ / ⁇ embryos dying between Day 12 and Day 13 after conception because of deficits in erythropoiesis (Neubauer et al., Cell 93(3):397-409, 1998). Jak3 deficiency has been described in humans and presents as severe combined immunodeficiency in the first few months of life, with symptoms such as failure to thrive, severe and recurrent infections, thrush, and diarrhea.
- Tyk2-deficiency additionally has been described in humans, manifesting with impaired antimicrobial responses, elevated serum IgE, and atopic dermatitis (Minegishi et al., Immunity 25(5):745-755, 2006).
- Jak1 and Jak2 Given the high degree of structural similarity between Jak1 and Jak2 (Williams et al., J. Mal. Biol. 387(1):219-232, 2009), the literature suggests that the majority of Jak1 inhibitors also inhibit Jak2 (lncyte Corp. press release, 10 Nov. 2010; Changelian et al., Science 302(5646):875-878, 2003).
- Anti-cytokine therapies have become standard in the treatment of RA.
- Jak1 inhibition is an effective therapy for the treatment of signs and symptoms of RA.
- Multiple clinical trials administering Pfizer's Jak 1/3 inhibitor tofacitinib (Kremer et al., Arthritis Rheum. 60(7):1895-1905, 2009; Riese et al. Best Pract. Res. Clin. Rheumatol. 24(4):513-526, 2010), Incyte/Lilly's Jak1/2 inhibitor INCB-28050/LY3009104 (lncyte Corp. press release, 10 Nov. 2010), or Galapagos' Jak1 inhibitor GLP0634 (Galapagos Nev. press release, 22 Nov. 2011) have demonstrated statistically significant efficacy in this disease.
- Tofacitinib an inhibitor of Jak1, and Jak3, has been approved in the United States and additional countries around the world for the indication of adult patients with moderately to severely active RA who have had an inadequate response or intolerance to methotrexate (MTX), used as monotherapy or in combination with MTX or other non-biologic DMARDs.
- MTX methotrexate
- Safety data from Phase 2 and Phase 3 studies in patients (Fleischmann, Curr. Opin. Rheumatol. 24(3):335-341, 2012; Kremer et al., Arthritis Rheum. 64(4):970-981, 2012; Fleischmann et al., Arthritis Rheum.
- Epstein-Barr virus-associated post-transplant lymphoproliferative disorder has been observed at an increased rate in renal transplant patients treated with tofacitinib and concomitant immunosuppressive medications. Gastrointestinal perforations in patients receiving tofacitinib also were reported. In addition, laboratory abnormalities have been described, including dose-related decreases in absolute neutrophil counts as well as hemoglobin. Furthermore, small increases in liver transaminases (alanine aminotransferase [ALT], aspartate aminotransferase [AST]) and serum creatinine, and elevated LDL, HDL, and total cholesterol levels have been reported.
- ALT lanine aminotransferase
- AST aspartate aminotransferase
- Jak blockade may be effective in managing disease and achieving remission
- the first generation Jak inhibitors such as tofacitinib and baricitinib
- Jak inhibitors have failed to reach their full potential, at least partly due to their tolerability and safety issues that limit dose.
- the first generation Jak inhibitors tofacitinib and baricitinib have been characterized as Jak1/Jak3 and Jak1/Jak2 inhibitors, respectively (Fridman et al., J. Immunol., 184:5298-5307, 2010; Meyer et al., J. lnflamm. (Lond.) 7:41, 2010; and Taylor et al., Rheumatology 52:i44-i55, 2013).
- these first generation Jak inhibitors have failed to reach their full potential due to tolerability issues that limited dose (Fleischmann et al., Curr. Opin. Rheumatol.
- JAKs are known to play roles in the regulation of over forty pathways (Murray, J. Immunol. 178:2623-2629, 2007).
- these inhibitors may not be optimally selective for kinases within the JAK family. For instance, incidence of severe anemia was reported to be a dose limiting factor during Tofacitinib Phase II development in RA (Pfizer, Investigators Brochure.
- Jak1 selective inhibitors are on-going (Zak et al. J. Med. Chem. 2013, 56, 4764-4785; Menet et al. Future Med. Chem. 2015, 7, 203-235; WO2013/007768).
- Prominent Jak1 selective compounds in development are GLP0634, ABT-494 (WO2015/061665), and the compound in a recent patent publication from Incyte (WO2015/168246), but no Jak1 selective inhibitor has been approved yet.
- novel 1H-furo[3,2-b]imidazo[4,5-d]pyridine derivatives are described as Jak1 selective inhibitors. These compounds, and compositions comprising a compound of this invention are useful in treating disorders related to Jak1 activities such as an autoimmune disease or disorder, or an inflammatory disease or disorder, and a cancer or neoplastic disease or disorder.
- This invention discloses novel 1H-furo[3,2-b]imidazo[4,5-d]pyridine derivatives their pharmaceutically acceptable salts, solvates, hydrates and polymorphs thereof as selective Jak1 kinase inhibitors.
- the invention also provides compositions comprising a compound of this invention and the use of such compositions in methods of treating diseases and conditions associated with Jak1.
- R 1 is H, halo, or C 1-3 alkyl optionally substituted with 1, 2, or 3 substituents independently selected from the group consisting of halo, OH, CN, OR, NHR, NRR′, N(R)C( ⁇ O)R′, N(R)C( ⁇ O)(O)R′, OC( ⁇ O)NRR′, C( ⁇ O)R, C( ⁇ O)NRR′, N(R)S(O) 2 R′, S(O) 2 R, and S(O) 2 NRR 1 ;
- R 2 is H, halo, or C 1-3 alkyl
- Cy is C 3-7 cycloalkyl, 3-7 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl, each optionally substituted with 1, 2, or 3 substituents independently selected from the group consisting of R 3 , oxo, halo, OH, CN, OR, NHR, NRR′, N(R)C( ⁇ O)R′, N(R)C( ⁇ O)(O)R′, OC( ⁇ O)NRR′, C( ⁇ O)R, C( ⁇ O)NRR′, N(R)S(O) 2 R′, S(O) 2 R, and S(O) 2 NRR′, wherein R 3 is C 1-3 alkyl optionally substituted with 1, 2, or 3 substituents independently selected from the group consisting of halo, OH, CN, OR, NHR, NRR′, N(R)C( ⁇ O)R′, N(R)C( ⁇ O)(O)R′, OC( ⁇ O)NRR′
- R, R′ each is independently H, or C 1-3 alkyl optionally substituted with 1, 2, or 3 substituents independently selected from the group consisting of halo, OH, and CN.
- the compounds of this invention, and compositions comprising them, are useful for treating or lessening the severity of Jak1 modulated diseases, disorders, or symptoms thereof.
- the invention in another aspect, relates to a method of treating a disease or disease symptom in a subject in need thereof including administering to the subject an effective amount of a compound of formula I herein, or pharmaceutically acceptable salt, solvate or hydrate thereof (or composition thereof).
- the disease or disease symptom can be any of those modulated by Jak1.
- the disease or disease symptom can be, for example, an autoimmune disease or disorder such as rheumatoid arthritis or an inflammatory disease or disorder, and cancer or neoplastic proliferative disease or disorder (e.g., including those delineated herein).
- the invention relates to a compound of formula A1-14:
- the invention relates to a process of preparing a compound of formula
- R 2 is H, and R 1 and Cy are as defined above.
- the (C 1-6 alkyl) 3 oxonium tetrafluoroborate can be triethyloxonium tetrafluoroborate.
- the invention relates to a process of preparing compound of formula V comprising reducing a compound of formula VII:
- the hydrogenation catalyst can be palladium on carbon.
- the invention relates to a process preparing compound of formula VII comprising contacting a compound of formula A1-14:
- the base can be N,N-Diisopropylethylamine.
- ameliorate and “treat” are used interchangeably and both mean decrease, suppress, attenuate, diminish, arrest, or stabilize the development or progression of a disease (e.g., a disease or disorder delineated herein).
- disease is meant any condition or disorder that damages or interferes with the normal function of a cell, tissue, or organ.
- marker is meant any alteration that is associated with a disease or disorder.
- compound as used herein, is also intended to include pharmaceutically acceptable salts, prodrugs, and prodrug salts of a compound of formulae herein.
- the term also includes any solvates, hydrates, and polymorphs of any of the foregoing.
- the specific recitation of “prodrug,” “prodrug salt,” “solvate,” “hydrate,” or “polymorph” in certain aspects of the invention described in this application shall not be interpreted as an intended omission of these forms in other aspects of the invention where the term “compound” is used without recitation of these other forms.
- a salt of a compound of this invention is formed between an acid and a basic group of the compound, such as an amino functional group, or a base and an acidic group of the compound, such as a carboxyl functional group.
- the compound is a pharmaceutically acceptable acid addition salt.
- prodrug means a derivative of a compound that can hydrolyze, oxidize, or otherwise react under biological conditions (in vitro or in vivo) to provide a compound of this invention.
- Prodrugs may only become active upon such reaction under biological conditions, or they may have activity in their unreacted forms.
- Examples of prodrugs contemplated in this invention include, but are not limited to, analogs or derivatives of compounds of any one of the formulae disclosed herein that comprise biohydrolyzable moieties such as amides, esters, carbamates, carbonates, and phosphate analogues.
- Prodrugs can typically be prepared using well-known methods, such as those described by Burger's Medicinal Chemistry and Drug Discovery (1995) 172-178, 949-982 (Manfred E. Wolff ed., 5th ed); see also Goodman and Gilman's, The Pharmacological basis of Therapeutics, 8th ed., McGraw-Hill, Int. Ed. 1992, “Biotransformation of Drugs”.
- biohydrolyzable moiety means a functional group (e.g., amide, ester, carbamate, carbonate, or phosphate analogue, that either: 1) does not destroy the biological activity of the compound and confers upon that compound advantageous properties in vivo, such as uptake, duration of action, or onset of action; or 2) is itself biologically inactive but is converted in vivo to a biologically active compound.
- a functional group e.g., amide, ester, carbamate, carbonate, or phosphate analogue
- a prodrug salt is a compound formed between an acid and a basic group of the prodrug, such as an amino functional group, or a base and an acidic group of the prodrug, such as a carboxyl functional group.
- the prodrug salt is a pharmaceutically acceptable salt.
- Particularly favored prodrugs and prodrug salts are those that increase the bioavailability of the compounds of this invention when such compounds are administered to a mammal (e.g., by allowing an orally administered compound to be more readily absorbed into the blood) or which enhance delivery of the parent compound to a biological compartment (e.g., the brain or central nervous system) relative to the parent species.
- Preferred prodrugs include derivatives where a group that enhances aqueous solubility or active transport through the gut membrane is appended to the structure of formulae described herein. See, e.g., Alexander, J. et al. Journal of Medicinal Chemistry 1988, 31, 318-322; Bundgaard, H.
- pharmaceutically acceptable refers to a component that is, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and other mammals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salt means any non-toxic salt that, upon administration to a recipient, is capable of providing, either directly or indirectly, a compound or a prodrug of a compound of this invention.
- Acids commonly employed to form pharmaceutically acceptable salts include inorganic acids such as hydrogen bisulfide, hydrochloric, hydrobromic, hydroiodic, sulfuric and phosphoric acid, as well as organic acids such as para-toluenesulfonic, salicylic, tartaric, bitartaric, ascorbic, maleic, besylic, fumaric, gluconic, glucuronic, formic, glutamic, methanesulfonic, ethanesulfonic, benzenesulfonic, lactic, oxalic, para-bromophenylsulfonic, carbonic, succinic, citric, benzoic and acetic acid, and related inorganic and organic acids.
- inorganic acids such as hydrogen bisulfide, hydrochloric, hydrobromic, hydroiodic, sulfuric and phosphoric acid
- organic acids such as para-toluenesulfonic, salicylic, tartaric, bitartaric, as
- Such pharmaceutically acceptable salts thus include sulfate, pyrosulfate, bisulfate, sulfite, bisulfite, phosphate, monohydrogenphosphate, dihydrogenphosphate, metaphosphate, pyrophosphate, chloride, bromide, iodide, acetate, propionate, decanoate, caprylate, acrylate, formate, isobutyrate, caprate, heptanoate, propiolate, oxalate, malonate, succinate, suberate, sebacate, fumarate, maleate, butyne-1,4-dioate, hexyne-1,6-dioate, benzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, phthalate, terephthalate, sulfonate, xylenesulfonate, phenylacetate, phenylpropionate,
- Suitable bases for forming pharmaceutically acceptable salts with acidic functional groups of prodrugs of this invention include, but are not limited to, hydroxides of alkali metals such as sodium, potassium, and lithium; hydroxides of alkaline earth metal such as calcium and magnesium; hydroxides of other metals, such as aluminum and zinc; ammonia, and organic amines, such as unsubstituted or hydroxy-substituted mono-, di-, or trialkylamines; dicyclohexylamine; tributyl amine; pyridine; N-methyl,N-ethylamine; diethylamine; triethylamine; mono-, bis-, or tris-(2-hydroxy-lower alkyl amines), such as mono-, bis-, or tris-(2-hydroxyethyl)amine, 2-hydroxy-tert-butylamine, or tris-(hydroxymethyl)methylamine, N, N,-di-lower alkyl-N-(hydroxy lower alkyl)
- hydrate means a compound which further includes a stoichiometric or non-stoichiometric amount of water bound by non-covalent intermolecular forces.
- solvate means a compound which further includes a stoichiometric or non-stoichiometric amount of solvent such as water, acetone, ethanol, methanol, dichloromethane, 2-propanol, or the like, bound by non-covalent intermolecular forces.
- polymorph means solid crystalline forms of a compound or complex thereof which may be characterized by physical means such as, for instance, X-ray powder diffraction patterns or infrared spectroscopy. Different polymorphs of the same compound can exhibit different physical, chemical and/or spectroscopic properties. Different physical properties include, but are not limited to stability (e.g., to heat, light or moisture), compressibility and density (important in formulation and product manufacturing), hygroscopicity, solubility, and dissolution rates (which can affect bioavailability).
- Differences in stability can result from changes in chemical reactivity (e.g., differential oxidation, such that a dosage form discolors more rapidly when comprised of one polymorph than when comprised of another polymorph) or mechanical characteristics (e.g., tablets crumble on storage as a kinetically favored polymorph converts to thermodynamically more stable polymorph) or both (e.g., tablets of one polymorph are more susceptible to breakdown at high humidity).
- chemical reactivity e.g., differential oxidation, such that a dosage form discolors more rapidly when comprised of one polymorph than when comprised of another polymorph
- mechanical characteristics e.g., tablets crumble on storage as a kinetically favored polymorph converts to thermodynamically more stable polymorph
- both e.g., tablets of one polymorph are more susceptible to breakdown at high humidity.
- Different physical properties of polymorphs can affect their processing. For example, one polymorph might be more likely to form solvates or might be more difficult to filter or wash free of impurities than another
- substantially free of other stereoisomers means less than 25% of other stereoisomers, preferably less than 10% of other stereoisomers, more preferably less than 5% of other stereoisomers and most preferably less than 2% of other stereoisomers, or less than “X”% of other stereoisomers (wherein X is a number between 0 and 100, inclusive) are present.
- Methods of obtaining or synthesizing diastereomers are well known in the art and may be applied as practicable to final compounds or to starting material or intermediates. Other embodiments are those wherein the compound is an isolated compound.
- at least X % enantiomerically enriched means that at least X % of the compound is a single enantiomeric form, wherein X is a number between 0 and 100, inclusive.
- stable compounds refers to compounds which possess stability sufficient to allow manufacture and which maintain the integrity of the compound for a sufficient period of time to be useful for the purposes detailed herein (e.g., formulation into therapeutic products, intermediates for use in production of therapeutic compounds, isolatable or storable intermediate compounds, treating a disease or condition responsive to therapeutic agents).
- Stepoisomer refers to both enantiomers and diastereomers.
- halo or halogen refers to any radical of fluorine, chlorine, bromine or iodine.
- alk or “alkyl” refer to straight or branched chain hydrocarbon groups having 1 to 12 carbon atoms, preferably 1 to 8 carbon atoms.
- the expression “lower alkyl” refers to alkyl groups of 1 to 4 carbon atoms (inclusive).
- arylalkyl refers to a moiety in which an alkyl hydrogen atom is replaced by an aryl group.
- alkenyl refers to straight or branched chain hydrocarbon groups of 2 to 10, preferably 2 to 4, carbon atoms having at least one double bond. Where an alkenyl group is bonded to a nitrogen atom, it is preferred that such group not be bonded directly through a carbon bearing a double bond.
- alkoxy refers to an —O-alkyl radical.
- alkylenedioxo refers to a divalent species of the structure —O—R—O—, in which R represents an alkylene.
- alkynyl refers to straight or branched chain hydrocarbon groups of 2 to 10, preferably 2 to 4, carbon atoms having at least one triple bond. Where an alkynyl group is bonded to a nitrogen atom, it is preferred that such group not be bonded directly through a carbon bearing a triple bond.
- alkylene refers to a divalent straight chain bridge of 1 to 5 carbon atoms connected by single bonds (e.g., —(CH 2 ) x —, wherein x is 1 to 5), which may be substituted with 1 to 3 lower alkyl groups.
- alkenylene refers to a straight chain bridge of 2 to 5 carbon atoms having one or two double bonds that is connected by single bonds and may be substituted with 1 to 3 lower alkyl groups.
- exemplary alkenylene groups are —CH ⁇ CH—CH ⁇ CH—, —CH 2 —CH ⁇ CH—, —CH 2 —CH ⁇ CH—CH 2 —, —C(CH 3 ) 2 CH ⁇ CH— and —CH(C 2 H 5 )—CH ⁇ CH—.
- alkynylene refers to a straight chain bridge of 2 to 5 carbon atoms that has a triple bond therein, is connected by single bonds, and may be substituted with 1 to 3 lower alkyl groups.
- exemplary alkynylene groups are —C ⁇ C—, —CH 2 —C ⁇ C—, —CH(CH 3 )C ⁇ C— and —C ⁇ C—CH(C 2 H 5 )CH 2 —.
- cycloalkyl and “cycloalkenyl” as employed herein includes saturated and partially unsaturated cyclic, respectively, hydrocarbon groups having 3 to 12 carbons, preferably 3 to 8 carbons, and more preferably 3 to 6 carbons.
- aryl refers to aromatic cyclic groups (for example 6 membered monocyclic, 10 membered bicyclic or 14 membered tricyclic ring systems) which contain 6 to 14 carbon atoms.
- exemplary aryl groups include phenyl, naphthyl, biphenyl and anthracene.
- Heteroaryl refers to a monocyclic or fused ring (i.e., rings which share an adjacent pair of atoms) group of 5 to 12 ring atoms containing one, two, three or four ring heteroatoms selected from N, O, or S, the remaining ring atoms being C, and, in addition, having a completely conjugated pi-electron system, wherein 0, 1, 2, 3, or 4 atoms of each ring may be substituted by a substituent.
- heteroaryl groups examples, without limitation, of heteroaryl groups are pyrrole, furan, thiophene, imidazole, oxazole, thiazole, pyrazole, pyridine, pyrimidine, quinoline, quinazoline, isoquinoline, purine and carbazole.
- heterocycle refers to fully saturated or partially unsaturated cyclic groups, for example, 3 to 7 membered monocyclic, 7 to 12 membered bicyclic, or 10 to 15 membered tricyclic ring systems, which have at least one heteroatom in at least one ring, wherein 0, 1, 2 or 3 atoms of each ring may be substituted by a substituent.
- Each ring of the heterocyclic group containing a heteroatom may have 1, 2, 3 or 4 heteroatoms selected from nitrogen atoms, oxygen atoms and/or sulfur atoms, where the nitrogen and sulfur heteroatoms may optionally be oxidized and the nitrogen heteroatoms may optionally be quaternized.
- the heterocyclic group may be attached at any heteroatom or carbon atom of the ring or ring system.
- heterocyclyl refers to fully saturated or partially unsaturated cyclic groups, for example, 3 to 7 membered monocyclic, 7 to 12 membered bicyclic, or 10 to 15 membered tricyclic ring systems, which have at least one heteroatom in at least one ring, wherein 0, 1, 2 or 3 atoms of each ring may be substituted by a substituent.
- Each ring of the heterocyclyl group containing a heteroatom may have 1, 2, 3 or 4 heteroatoms selected from nitrogen atoms, oxygen atoms and/or sulfur atoms, where the nitrogen and sulfur heteroatoms may optionally be oxidized and the nitrogen heteroatoms may optionally be quaternized.
- the heterocyclyl group may be attached at any heteroatom or carbon atom of the ring or ring system.
- substituted refers to a group “substituted” on any functional group delineated herein, e.g., alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkenyl, aryl, heterocyclyl, or heteroaryl group at any atom of that group.
- Suitable substituents include, without limitation halogen, CN, NO 2 , OR 15 , SR 15 , S(O) 2 OR 15 , NR 15 R 16 , C 1 -C 2 perfluoroalkyl, C 1 -C 2 perfluoroalkoxy, 1,2-methylenedioxy, C(O)OR 15 , C(O)NR 15 R 16 , OC(O)NR 15 R 16 , NR 15 C(O)NR 15 R 16 , C(NR 16 )NR 15 R 16 , NR 15 C(NR 16 )NR 15 R 16 , S(O) 2 NR 15 R 16 , R 17 , C(O)R 17 , NR 15 C(O)R 17 , S(O)R 17 , S(O) 2 R 17 , R 16 , oxo, C(O)R 16 , C(O)(CH 2 )nOH, (CH 2 )nOR 15 , (CH 2 )nC(O)NR 15 R 16
- Each R 15 is independently hydrogen, C 1 -C 4 alkyl or C 3 -C 6 cycloalkyl.
- Each R 16 is independently hydrogen, alkenyl, alkynyl, C 3 -C 6 cycloalkyl, aryl, heterocyclyl, heteroaryl, C 1 -C 4 alkyl or C 1 -C 4 alkyl substituted with C 3 -C 6 cycloalkyl, aryl, heterocyclyl or heteroaryl.
- Each R 17 is independently C 3 -C 6 cycloalkyl, aryl, heterocyclyl, heteroaryl, C 1 -C 4 alkyl or C 1 -C 4 alkyl substituted with C 3 -C 6 cycloalkyl, aryl, heterocyclyl or heteroaryl.
- Each C 3 -C 6 cycloalkyl, aryl, heterocyclyl, heteroaryl and C 1 -C 4 alkyl in each R 15 , R 16 and R 17 can optionally be substituted with halogen, CN, C 1 -C 4 alkyl, OH, C 1 -C 4 alkoxy, NH 2 , C 1 -C 4 alkylamino, C 1 -C 4 dialkylamino, C 1 -C 2 perfluoroalkyl, C 1 -C 2 perfluoroalkoxy, or 1,2-methylenedioxy.
- oxo refers to an oxygen atom, which forms a carbonyl when attached to carbon, an N-oxide when attached to nitrogen, and a sulfoxide or sulfone when attached to sulfur.
- acyl refers to an alkylcarbonyl, cycloalkylcarbonyl, arylcarbonyl, heterocyclylcarbonyl, or heteroarylcarbonyl substituent, any of which may be further substituted by substituents.
- the compounds of this invention may contain one or more asymmetric centers and thus occur as racemates and racemic mixtures, single enantiomers, individual diastereomers and diastereomeric mixtures, as well as cis and trans geometric isomers. All such isomeric forms of these compounds are expressly included in the present invention.
- the compounds of this invention may also be represented in multiple tautomeric forms, in such instances, the invention expressly includes all tautomeric forms of the compounds described herein. All such isomeric forms of such compounds are expressly included in the present invention. All crystal forms of the compounds described herein are expressly included in the present invention.
- the present invention provides a compound of Formula I:
- R 1 is H, halo, or C 1-3 alkyl optionally substituted with 1, 2, or 3 substituents independently selected from the group consisting of halo, OH, CN, OR, NHR, NRR′, N(R)C( ⁇ O)R′, N(R)C( ⁇ O)(O)R′, OC( ⁇ O)NRR′, C( ⁇ O)R, C( ⁇ O)NRR′, N(R)S(O) 2 R′, S(O) 2 R, and S(O) 2 NRR′;
- R 2 is H, halo, or C 1-3 alkyl
- Cy is C 3-7 cycloalkyl, 3-7 membered heterocyclyl, phenyl, or 5-6 membered heteroaryl, each optionally substituted with 1, 2, or 3 substituents independently selected from the group consisting of R 3 , oxo, halo, OH, CN, OR, NHR, NRR′, N(R)C( ⁇ O)R′, N(R)C( ⁇ O)(O)R′, OC( ⁇ O)NRR′, C( ⁇ O)R, C( ⁇ O)NRR′, N(R)S(O) 2 R′, S(O) 2 R, and S(O) 2 NRR′, wherein R 3 is C 1-3 alkyl optionally substituted with 1, 2, or 3 substituents independently selected from the group consisting of halo, OH, CN, OR, NHR, NRR′, N(R)C( ⁇ O)R′, N(R)C( ⁇ O)(O)R′, OC( ⁇ O)NRR′
- R, R′ each is independently H, or C 1-3 alkyl optionally substituted with 1, 2, or 3 substituents independently selected from the group consisting of halo, OH, and CN.
- Cy can be C 5-7 cycloalkyl, or 5-7 membered heterocyclyl, each optionally substituted with 1, 2, or 3 substituents independently selected from the group consisting of R 3 , oxo, halo, OH, CN, OR, NHR, NRR′, N(R)C( ⁇ O)R′, N(R)C( ⁇ O)(O)R′, OC( ⁇ O)NRR′, C( ⁇ O)R, C( ⁇ O)NRR′, N(R)S(O) 2 R′, S(O) 2 R, and S(O) 2 NRR′, wherein R 3 is C 1-3 alkyl optionally substituted with 1, 2, or 3 substituents independently selected from the group consisting of halo, OH, CN, OR, NHR, NRR′, N(R)C( ⁇ O)R′, N(R)C( ⁇ O)(O)R′, OC( ⁇ O)NRR′, C( ⁇ O)R, C( ⁇ O)
- R 2 can be hydrogen
- Representative compounds of the invention are depicted in Table 1.
- the stereochemistry at the chiral carbon atoms is independently either RS, R, or S, unless specified.
- the stereochemistry shows only one of the trans or cis isomers, and the structures of their respective isomers are not shown.
- the structures depicted herein, including the Table 1 structures may contain certain —NH—, —NH 2 (amino) and —OH (hydroxyl) groups where the corresponding hydrogen atom(s) do not explicitly appear; however they are to be read as —NH—, —NH 2 or —OH as the case may be.
- a stick bond is drawn and is meant to depict a methyl group.
- the methods delineated herein contemplate converting compounds of one formula to compounds of another formula.
- the process of converting refers to one or more chemical transformations, which can be performed in situ, or with isolation of intermediate compounds.
- the transformations can include reacting the starting compounds or intermediates with additional reagents using techniques and protocols known in the art, including those in the references cited herein.
- Intermediates can be used with or without purification (e.g., filtration, distillation, sublimation, crystallization, trituration, solid phase extraction, and chromatography).
- compositions comprising an effective amount of a compound of any of the formulae herein, or a pharmaceutically acceptable salt, solvate, hydrate, polymorph or prodrug, if applicable, of said compound; and an acceptable carrier.
- a composition of this invention is formulated for pharmaceutical use (“a pharmaceutical composition”), wherein the carrier is a pharmaceutically acceptable carrier.
- the carrier(s) must be “acceptable” in the sense of being compatible with the other ingredients of the formulation and, in the case of a pharmaceutically acceptable carrier, not deleterious to the recipient thereof in amounts typically used in medicaments.
- Pharmaceutically acceptable carriers, adjuvants and vehicles that may be used in the pharmaceutical compositions of this invention include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, polyethylene glycol and wool fat.
- ion exchangers alumina, aluminum stearate, lecithin
- serum proteins such as human serum albumin
- buffer substances such as phosphat
- compositions of the invention include those suitable for oral, rectal, nasal, topical (including buccal and sublingual), vaginal or parenteral (including subcutaneous, intramuscular, intravenous and intradermal) administration.
- the compound of the formulae herein is administered transdermally (e.g., using a transdermal patch).
- Other formulations may conveniently be presented in unit dosage form, e.g., tablets and sustained release capsules, and in liposomes, and may be prepared by any methods well known in the art of pharmacy. See, for example, Remington's Pharmaceutical Sciences, Mack Publishing Company, Philadelphia, Pa. (17th ed. 1985).
- Such preparative methods include the step of bringing into association with the molecule to be administered ingredients such as the carrier that constitutes one or more accessory ingredients.
- ingredients such as the carrier that constitutes one or more accessory ingredients.
- the compositions are prepared by uniformly and intimately bringing into association the active ingredients with liquid carriers, liposomes or finely divided solid carriers or both, and then if necessary shaping the product.
- compositions of the present invention suitable for oral administration may be presented as discrete units such as capsules, sachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution or a suspension in an aqueous liquid or a non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion, or packed in liposomes and as a bolus, etc.
- Soft gelatin capsules can be useful for containing such suspensions, which may beneficially increase the rate of compound absorption.
- a tablet may be made by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent, preservative, surface-active or dispersing agent.
- Molded tablets may be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- the tablets optionally may be coated or scored and may be formulated so as to provide slow or controlled release of the active ingredient therein.
- compositions of pharmaceutically active ingredients are known in the art and described in several issued US patents, some of which include, but are not limited to, U.S. Pat. Nos. 4,369,172; and 4,842,866, and references cited therein.
- Coatings can be used for delivery of compounds to the intestine (see, e.g., U.S. Pat. Nos. 6,638,534, 5,217,720, and 6,569,457, 6,461,631, 6,528,080, 6,800,663, and references cited therein).
- a useful formulation for the compounds of this invention is the form of enteric pellets of which the enteric layer comprises hydroxypropylmethyl cellulose acetate succinate.
- carriers that are commonly used include lactose and corn starch.
- Lubricating agents such as magnesium stearate, are also typically added.
- useful diluents include lactose and dried cornstarch.
- aqueous suspensions are administered orally, the active ingredient is combined with emulsifying and suspending agents. If desired, certain sweetening and/or flavoring and/or coloring agents may be added.
- compositions suitable for topical administration include lozenges comprising the ingredients in a flavored basis, usually sucrose and acacia or tragacanth; and pastilles comprising the active ingredient in an inert basis such as gelatin and glycerin, or sucrose and acacia.
- compositions suitable for parenteral administration include aqueous and non-aqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- the formulations may be presented in unit-dose or multi-dose containers, for example, sealed ampules and vials, and may be stored in a freeze dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets.
- Such injection solutions may be in the form, for example, of a sterile injectable aqueous or oleaginous suspension.
- This suspension may be formulated according to techniques known in the art using suitable dispersing or wetting agents (such as, for example, Tween 80) and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example, as a solution in 1,3-butanediol.
- the acceptable vehicles and solvents that may be employed are mannitol, water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- Fatty acids, such as oleic acid and its glyceride derivatives are useful in the preparation of injectables, as are natural pharmaceutically-acceptable oils, such as olive oil or castor oil, especially in their polyoxyethylated versions.
- These oil solutions or suspensions may also contain a long-chain alcohol diluent or dispersant.
- compositions of this invention may be administered in the form of suppositories for rectal administration.
- These compositions can be prepared by mixing a compound of this invention with a suitable non-irritating excipient which is solid at room temperature but liquid at the rectal temperature and therefore will melt in the rectum to release the active components.
- suitable non-irritating excipient include, but are not limited to, cocoa butter, beeswax and polyethylene glycols.
- compositions of this invention may be administered by nasal aerosol or inhalation.
- Such compositions are prepared according to techniques well-known in the art of pharmaceutical formulation and may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other solubilizing or dispersing agents known in the art.
- Topical administration of the pharmaceutical compositions of this invention is especially useful when the desired treatment involves areas or organs readily accessible by topical application.
- the pharmaceutical composition should be formulated with a suitable ointment containing the active components suspended or dissolved in a carrier.
- Carriers for topical administration of the compounds of this invention include, but are not limited to, mineral oil, liquid petroleum, white petroleum, propylene glycol, polyoxyethylene polyoxypropylene compound, emulsifying wax and water.
- the pharmaceutical composition can be formulated with a suitable lotion or cream containing the active compound suspended or dissolved in a carrier.
- Suitable carriers include, but are not limited to, mineral oil, sorbitan monostearate, polysorbate 60, cetyl esters wax, cetearyl alcohol, 2-octyldodecanol, benzyl alcohol and water.
- the pharmaceutical compositions of this invention may also be topically applied to the lower intestinal tract by rectal suppository formulation or in a suitable enema formulation. Topically-transdermal patches and iontophoretic administration are also included in this invention.
- Particularly favored derivatives and prodrugs are those that increase the bioavailability of the compounds of this invention when such compounds are administered to a mammal (e.g., by allowing an orally administered compound to be more readily absorbed into the blood) or which enhance delivery of the parent compound to a biological compartment (e.g., the brain or central nervous system) relative to the parent species.
- Preferred prodrugs include derivatives where a group that enhances aqueous solubility or active transport through the gut membrane is appended to the structure of formulae described herein. See, e.g., Alexander, J. et al. Journal of Medicinal Chemistry 1988, 31, 318-322; Bundgaard, H.
- Application of the subject therapeutics may be local, so as to be administered at the site of interest.
- Various techniques can be used for providing the subject compositions at the site of interest, such as injection, use of catheters, trocars, projectiles, pluronic gel, stents, sustained drug release polymers or other device which provides for internal access.
- the invention provides a method of impregnating an implantable drug release device comprising the step of contacting said drug release device with a compound or composition of this invention.
- Implantable drug release devices include, but are not limited to, biodegradable polymer capsules or bullets, non-degradable, diffusible polymer capsules and biodegradable polymer wafers.
- the invention provides an implantable medical device coated with a compound or a composition comprising a compound of this invention, such that said compound is therapeutically active.
- a composition of the present invention further comprises a second therapeutic agent.
- the second therapeutic agent includes any compound or therapeutic agent known to have or that demonstrates advantageous properties when administered alone or with a compound of any of the formulae herein.
- Drugs that could be usefully combined with these compounds include other kinase inhibitors and/or other therapeutic agents for the treatment of the diseases and disorders discussed above.
- the second therapeutic agent is an agent useful in the treatment or prevention of a disease or condition selected from cancer and neoplastic diseases or disorders, or autoimmune and inflammatory diseases or disorders.
- the invention provides separate dosage forms of a compound of this invention and a second therapeutic agent that are associated with one another.
- association with one another means that the separate dosage forms are packaged together or otherwise attached to one another such that it is readily apparent that the separate dosage forms are intended to be sold and administered together (within less than 24 hours of one another, consecutively or simultaneously).
- the compound of the present invention is present in an effective amount.
- effective amount refers to an amount which, when administered in a proper dosing regimen, is sufficient to reduce or ameliorate the severity, duration or progression of the disorder being treated, prevent the advancement of the disorder being treated, cause the regression of the disorder being treated, or enhance or improve the prophylactic or therapeutic effect(s) of another therapy.
- An effective amount of a compound of this invention can range from about 0.001 mg/kg to about 500 mg/kg, more preferably 0.01 mg/kg to about 50 mg/kg, more preferably 0.1 mg/kg to about 2.5 mg/kg.
- Effective doses will also vary, as recognized by those skilled in the art, depending on the diseases treated, the severity of the disease, the route of administration, the sex, age and general health condition of the patient, excipient usage, the possibility of co-usage with other therapeutic treatments such as use of other agents and the judgment of the treating physician.
- an effective amount of the second therapeutic agent is between about 20% and 100% of the dosage normally utilized in a monotherapy regime using just that agent.
- an effective amount is between about 70% and 100% of the normal monotherapeutic dose.
- the normal monotherapeutic dosages of these second therapeutic agents are well known in the art. See, e.g., Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), each of which references are entirely incorporated herein by reference.
- the invention provides a method of treating a subject suffering from or susceptible to a disease or disorder or symptom thereof (e.g., those delineated herein) comprising the step of administering to said subject an effective amount of a compound or a composition of this invention.
- a disease or disorder or symptom thereof e.g., those delineated herein
- Such diseases are well known in the art and are also disclosed herein.
- the method of treating involves treatment of a disorder that is mediated by the Jak1 protein kinase.
- the method of treating involves treatment of a disorder that is mediated primarily by the Jak1 protein kinase, but also to some extent by the Jak2 protein kinase.
- the invention provides a method of treating a disease in a subject comprising administering to the subject a compound of any of the formulae herein.
- invention provides a method of treating a disease in a subject comprising administering to the subject a composition comprising a compound of any of the formulae herein.
- invention provides a method of treating a disease in a subject comprising administering to the subject a composition comprising a compound of any of the formulae herein.
- the disease is mediated by the Jak1 kinase.
- the condition may be an inflammatory disease/disorder, an autoimmune disease/disorder, such as, but not limited to rheumatoid arthritis (RA), juvenile idiopathic arthritis, osteoarthritis, multiple sclerosis, allergic asthma, chronic obstructive pulmonary disease, bronchitis, experimental allergic encephalomyelitis, Crohn's disease, vasculitis, cardiomyopathy, ankylosing spondylitis (AS), glomerulonephritis, insulin-dependent diabetes, psoriatic arthritis, psoriasis, plaque psoriasis, ulcerative colitis, systemic lupus erythematosus (SLE), diabetic nephropathy, peripheral neuropathy, uveitis, fibrosing alveolitis, type I diabetes, juvenile diabetes, Castleman disease, neutropenia, endometriosis, autoimmune thyroid disease, rheumato
- JAK-associated diseases include inflammation and inflammatory diseases or disorders
- inflammatory diseases of the eye e.g., sarcoidosis, inflammatory diseases of the eye (e.g., ulceris, uveitis, scleritis, conjunctivitis, blepharitis, or related disease)
- inflammatory diseases of the respiratory tract e.g., the upper respiratory tract including the nose and sinuses such as rhinitis or sinusitis or the lowe respiratory tract including bronchitis, chronic obstructive pulmonary disease, and the like
- inflammatory myopathy such as myocarditis and other inflammatory diseases.
- the disease is, cancer, a proliferative or other neoplastic disease, such as, but not limited to, breast cancer, Castleman's disease, colon and colorectal cancers, gastric cancer, gastrointestinal carcinoid tumor, gastrointestinal stromal tumor, glioblastoma, head and neck cancer, Kaposi's sarcoma, liver cancer, lung cancer, melanoma, pancreatic cancer, prostate cancer, renal cancer, rectal cancer, small intestine cancer, thyroid cancer, uterine leiomyosarcoma, lymphomas and leukemias such as acute lymphoblastic leukemia, acute myelogenous leukemia, multiple myeloma, cutaneous T cell lymphoma, cutaneous B cell lymphoma, myelodysplastic syndrome (MDS), myeloproliferative disorders (MPDs) such as polycythemia vera (PV), essential thrombocythemia (ET), myelofibrosis with myeloid
- the myeloproliferative disorder is post-essential thrombocythemia melofibrosis (Post-ET MF) or post-polycythemia versa myelofibrosis (Post-PV MF),
- JAK-associated diseases include ischemia reperfusion injuries or a disease or condition related to an inflammatory ischemic event such as stroke or cardiac arrest, endotoxin-driven disease state (e.g., complications after bypass surgery of chronic endotoxin states contributing to chronic cardiac failure), anorexia, sclerodermitis, fibrosis, conditions associated with hypoxia or astrogliosis such as diabetic retinopathy, cancer, or neurodegeneration, and other inflammatory disease such as systemic inflammatory response syndrome and septic shock.
- an inflammatory ischemic event such as stroke or cardiac arrest
- endotoxin-driven disease state e.g., complications after bypass surgery of chronic endotoxin states contributing to chronic cardiac failure
- anorexia exia
- sclerodermitis e.g., complications after bypass surgery of chronic endotoxin states contributing to chronic cardiac failure
- fibrosis fibrosis
- conditions associated with hypoxia or astrogliosis such as diabetic
- JAK-associated disease include gout and increased prostate size due to, e.g., benign prostate hypertrophy or benign prostatic hyperplasia, as well as bone resorption diseases such as osteoporosis or osteoarthritis, bone resorption diseases associated with: hormonal imbalance and/or hormonal therapy, autoimmune disease (e.g., osseous sarcoidosis).
- benign prostate hypertrophy or benign prostatic hyperplasia as well as bone resorption diseases such as osteoporosis or osteoarthritis, bone resorption diseases associated with: hormonal imbalance and/or hormonal therapy, autoimmune disease (e.g., osseous sarcoidosis).
- JAK-associated diseases or conditions include ameliorating the dermatological side effects of other pharmaceuticals by administration of the compound of the invention.
- numerous pharmaceutical agents result in unwanted allergic reaction which can manifest as acneiform rash or related dermatitis.
- Example pharmaceutical agents that have such undesirable side effects include anti-cancer drugs such as gefitinib, cetuximab, erlotinib, and the like.
- the compounds of the invention may be administered systemically or topically (e.g., localized to the vicinity of the dermatitis) in combination with pharmaceutical agent having the undesirable dermatological side effect.
- compositions of the invention include topical formulations containing the compound of the invention and a further pharmaceutical agent which can cause dermatitis, skin disorders, or related side effects.
- the method of this invention is used to treat a subject suffering from or susceptible to a disease or condition.
- diseases, disorders or symptoms thereof include, for example, those modulated by the Jak1 protein kinase.
- the disease or disease symptom can be, for example, rheumatoid arthritis, cancer or proliferation disease or disorder.
- Methods delineated herein include those wherein the subject is identified as in need of a particular stated treatment. Identifying a subject in need of such treatment can be in the judgment of a subject or a health care professional and can be subjective (e.g. opinion) or objective (e.g. measurable by a test or diagnostic method).
- the compounds of the formulae herein can be used to treat subjects having a disease or disorder who have been treated with and developed resistance to other therapeutic agents.
- the methods herein include those where a subject resistant to treatment with methotrexate or anti-TNF-alpha therapy.
- the invention provides a method of modulating the activity of the Jak1 protein kinase in a cell comprising contacting a cell with one or more compounds of any of the formulae herein.
- the above method of treatment comprises the further step of co-administering to said patient one or more second therapeutic agents.
- second therapeutic agent may be made from any therapeutic agent known to be useful for indications herein.
- additional therapeutic may include chemotherapeutics, anti-inflammatory agents, steroids, immunosuppressants, as well as PI3Kdelta, mTOR, BCR-ABl, FLT-3, RAF and FAK kinase inhibitors, and the like.
- Additional therapeutic agents include but are not limited to agents for treatment of diseases, disorders or symptoms thereof including for example, (1) agents that modulate human immune system or are anti-inflammatory agents selected from the group consisting of, but not limited to, aspirin, acetaminophen, aminosalicylate, antithymoyte globulin, ciprofloxacin, corticosteroid, cyclosporine, deoxyspergualin, daclizuma, metronidazole, probiotic, tacrolimus, ibuprofen, naproxen, piroxicam, prednisolone, dexamethasone, anti-inflammatory steroid, methotrexate, chloroquine, azathioprine, hydroxychloroquine, mycophnolate, muromonab-CD3, penicillamine, sulfasalazine, leflunomide, tacrolimus, tocilzumab, anakinra, abatacept, certolizumab
- a nucleic acid therapy such as antisense or RNAi; nuclear receptor ligands (e.g., agonists and/or antagonists. All-trans retinoic acid or boxarotene); epigenetic targeting agents such as histone deacetylase inhibitors (e.g., vorinostat), hypomethylating agents (e.g., decitabine), regulators of protein stability such as HSP90 inhibitors, ubiquitin and/or ubiquitin like conjugating or deconjugating molecules.
- histone deacetylase inhibitors e.g., vorinostat
- hypomethylating agents e.g., decitabine
- regulators of protein stability such as HSP90 inhibitors, ubiquitin and/or ubiquitin like conjugating or deconjugating molecules.
- the additional pharmaceutical agent is selected from IMiDs, an anti-IL-6 agent, an anti-TNF-alpha agent, a hypomethylating agent, and a biologic response modifier (RBM).
- RBM is generally a substance made from living organisms to treat disease. Examples of RBMs include IL-2, GM-CSF, CSF, monoclonal antibodies such as abciximab, etanercept, infliximab, rituximab, trastuzumab, and high dose ascorbate.
- the hypomethylating agent is a DNA methyltransferase inhibitor such as 5 azacytidine and decitabine.
- IMiDs include thalidomide, lenalidomide, pomalidomide, CC-11006, and CC-10015.
- the additional pharmaceutical agents include anti-thymocyte globulin, recombinant human granulocyte colony-stimulating factor (G-CSF), granulocyte-moncyte CSF (GM-CSF), a erythropoiesis-stimulating agent (ESA), and cyclosporine.
- G-CSF recombinant human granulocyte colony-stimulating factor
- GM-CSF granulocyte-moncyte CSF
- ESA erythropoiesis-stimulating agent
- the additional therapeutic agent is an additional JAK inhibitor.
- the additional JAK inhibitor is tofacitinib, ruxolitinib or baricitinib.
- one or more JAK inhibitors of the invention can be used in combination with one or more other cancer therapeutic agents in the treatment of cancer, such as multiple myeloma, and may improve the treatment benefit as compared to the benefit shown by the other cancer therapeutic agents, without exacerbating of their toxic effects.
- additional pharmaceutical agents used in the treatment of multiple myeloma can include, without limition, melphalan, melphalan plus prednisone (MP), doxorubicin, dexamethasone, and bortezomib.
- Additional agents used in the treatment of multiple myeoloma include BRC-ABL, FLT-3, RAF, MEK, PI3K, mTOR inhibitors. Additive or synergistic effects are desirable outcomes of combining a JAK inhibitor of the current invention with an additional agent.
- agents such as dexamethasone or other agents may be reversible upon treatment with a JAK inhibitor of the present invention.
- the agents can be combined with the present compounds in a single or continuous dosage form, or the agents can be administered simultaneously or sequentially as separate dosage forms.
- a corticosteroid such as dexamethasone is administered to a patient in combination with at least one JAK inhibitor of the invention where the dexamethasone is administered intermittently as opposed to continuously.
- combinations of one or more JAK inhibitors of the invention with other therapeutic agents can be administered to a patient prior to, during, and/or after a bone marrow transplantation or stem cell transplantation.
- the additional therapeutic agent is fluocinolone acetonide or remexolone.
- the additional therapeutic is a corticosteroid such as triamcinolone, dexamethasone, fluocinolone, cortisone, prednisolone, or flumetholone.
- the additional therapeutic agent includes Dehydrex, Civamide, sodium hyaluronate, cyclosporine, ARG101, AGR1012, ecabet sodium, gefarnate, 15-(s)-hydroxyeicosatetraenoic acid, cevilemine doxycycline, minocycline, iDestrin, cyclosporine A, oxytetracycline, voclosporin, ARG103, RX-10045, DYN15, rivoglitazone, TB4, OPH-01, PCS101, REV1-31, Lacritin, rebamipide, OT-551, PAI-2, pilocarpine, tacrolimus, pimercrolimus, loteprednol etabonate, rituximan, diquafosol tetrasodium, KLS-0611, dehydroepiandrosterone, anakinra, efalizuma, my
- the additional therapeutic agent is an anti-angiogenic agent, cholinergic agent, TRP-1 receptor modulator, a calcium channel blocker, a mucin secretagogue, MUC1 stimulant, a calcineurin inhibitor, a P2Y2 receptor agonist, a muscarinic receptor agonist, and a tetracycline derivative.
- the additional therapeutic agents include demulcent eye drops, which include, but not limited to, compositions containing polyvinylalchol, hyroxypropyl methylcellulose, glycerin, polyethylene glycol (e.g., PEG400), or carboxymethyl cellose,
- the additional therapeutic agent is a mucolytic drug, such as N-acetyl-systeine, which can interact with the mucoproteins and decrease the viscositiy of the tear film.
- the additional therapeutic agent includes an antibiotic, antiviral, antifungal, anesthetic, anti-inflammatory agents including steroidal and non-steroidal anti-inflammatories, and anti-allergic agents.
- suitable medicaments include aminoglycosides such as amikacin, gentamycin, tobramycin, streptomycin, netilmycin, and kanamycin; fluoroquinolones such as ciprofloxacin, norfloxacin, ofloxacin, trovafloxacin, lomefloxacin, levofloxacin, and enoxacin; naphthyridine; sulfonamides; polymyxin; chloramphenicol; neomycin; paramomycin; colistimethate; bacitracin, vanocomycin; tetracyclines; rifampin and its derivatives; cycloserine; beta-lactams; cephalosporins; emphotericins; flucon
- co-administered means that the second therapeutic agent may be administered together with a compound of this invention as part of a single dosage form (such as a composition of this invention comprising a compound of the invention and an second therapeutic agent as described above) or as separate, multiple dosage forms.
- the additional agent may be administered prior to, consecutively with, or following the administration of a compound of this invention.
- both the compounds of this invention and the second therapeutic agent(s) are administered by conventional methods.
- composition of this invention comprising both a compound of the invention and a second therapeutic agent to a subject does not preclude the separate administration of that same therapeutic agent, any other second therapeutic agent or any compound of this invention to said subject at another time during a course of treatment.
- Effective amounts of these second therapeutic agents are well known to those skilled in the art and guidance for dosing may be found in patents and published patent applications referenced herein, as well as in Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), and other medical texts. However, it is well within the skilled artisan's purview to determine the second therapeutic agent's optimal effective-amount range.
- the effective amount of the compound of this invention is less than its effective amount would be where the second therapeutic agent is not administered. In another embodiment, the effective amount of the second therapeutic agent is less than its effective amount would be where the compound of this invention is not administered. In this way, undesired side effects associated with high doses of either agent may be minimized. Other potential advantages (including without limitation improved dosing regimens and/or reduced drug cost) will be apparent to those of skill in the art.
- the invention provides the use of a compound of any of the formulae herein alone or together with one or more of the above-described second therapeutic agents in the manufacture of a medicament, either as a single composition or as separate dosage forms, for treatment or prevention in a subject of a disease, disorder or symptom set forth above.
- Another aspect of the invention is a compound of the formulae herein for use in the treatment or prevention in a subject of a disease, disorder or symptom thereof delineated herein.
- the methods herein include those further comprising monitoring subject response to the treatment administrations.
- monitoring may include periodic sampling of subject tissue, fluids, specimens, cells, proteins, chemical markers, genetic materials, etc. as markers or indicators of the treatment regimen.
- the subject is prescreened or identified as in need of such treatment by assessment for a relevant marker or indicator of suitability for such treatment.
- the invention provides a method of monitoring treatment progress.
- the method includes the step of determining a level of diagnostic marker (Marker) (e.g., any target or cell type delineated herein modulated by a compound herein) or diagnostic measurement (e.g., screen, assay) in a subject suffering from or susceptible to a disorder or symptoms thereof delineated herein, in which the subject has been administered a therapeutic amount of a compound herein sufficient to treat the disease or symptoms thereof.
- the level of Marker determined in the method can be compared to known levels of Marker in either healthy normal controls or in other afflicted patients to establish the subject's disease status.
- a second level of Marker in the subject is determined at a time point later than the determination of the first level, and the two levels are compared to monitor the course of disease or the efficacy of the therapy.
- a pre-treatment level of Marker in the subject is determined prior to beginning treatment according to this invention; this pre-treatment level of Marker can then be compared to the level of Marker in the subject after the treatment commences, to determine the efficacy of the treatment.
- a level of Marker or Marker activity in a subject is determined at least once. Comparison of Marker levels, e.g., to another measurement of Marker level obtained previously or subsequently from the same patient, another patient, or a normal subject, may be useful in determining whether therapy according to the invention is having the desired effect, and thereby permitting adjustment of dosage levels as appropriate. Determination of Marker levels may be performed using any suitable sampling/expression assay method known in the art or described herein. Preferably, a tissue or fluid sample is first removed from a subject. Examples of suitable samples include blood, urine, tissue, mouth or cheek cells, and hair samples containing roots. Other suitable samples would be known to the person skilled in the art.
- Determination of protein levels and/or mRNA levels (e.g., Marker levels) in the sample can be performed using any suitable technique known in the art, including, but not limited to, enzyme immunoassay, ELISA, radiolabelling/assay techniques, blotting/chemiluminescence methods, real-time PCR, and the like.
- kits for use to treat diseases, disorders, or symptoms thereof, including those delineated herein comprise: a) a pharmaceutical composition comprising a compound of any of the formula herein or a pharmaceutically acceptable salt thereof; or a prodrug, or a pharmaceutically acceptable salt of a prodrug thereof; or a hydrate, solvate, or polymorph thereof, wherein said pharmaceutical composition is in a container; and b) instructions describing a method of using the pharmaceutical composition to treat the disease, disorder, or symptoms thereof, including those delineated herein.
- the container may be any vessel or other sealed or sealable apparatus that can hold said pharmaceutical composition.
- Examples include bottles, divided or multi-chambered holders bottles, wherein each division or chamber comprises a single dose of said composition, a divided foil packet wherein each division comprises a single dose of said composition, or a dispenser that dispenses single doses of said composition.
- the container can be in any conventional shape or form as known in the art which is made of a pharmaceutically acceptable material, for example a paper or cardboard box, a glass or plastic bottle or jar, a re-sealable bag (for example, to hold a “refill” of tablets for placement into a different container), or a blister pack with individual doses for pressing out of the pack according to a therapeutic schedule.
- the container employed can depend on the exact dosage form involved, for example a conventional cardboard box would not generally be used to hold a liquid suspension. It is feasible that more than one container can be used together in a single package to market a single dosage form.
- tablets may be contained in a bottle, which is in turn contained within a box.
- the container is a blister pack.
- the kit may additionally comprising information and/or instructions for the physician, pharmacist or subject.
- Such memory aids include numbers printed on each chamber or division containing a dosage that corresponds with the days of the regimen which the tablets or capsules so specified should be ingested, or days of the week printed on each chamber or division, or a card which contains the same type of information.
- the compounds delineated herein can be assessed for their biological activity using protocols known in the art, including for example, those delineated herein. Certain of the compounds herein demonstrate unexpectedly superior attributes (e.g., inhibition of P450, metabolic stability, pharmacokinetic properties, etc.) making them superior candidates as potential therapeutic agents.
- Step 1 A solution of trans-4-(Boc-amino)cyclohexane carboxylic acid (A1-1) (62 g, 0.256 mol, 1.0 eq) in THF (1500 mL) was treated with NMM (64.6 g, 0.64 mol, 2.5 eq) in nitrogen atmosphere. The mixture was cooled to ⁇ 78° C., and isobutyl chloroformate (33.6 g, 0.33 mol, 1.3 eq) was added dropwise. After stirring at ⁇ 78° C. for 1 hr, NH 3 (gas) was bubbled through the mixture for about 20 mins. After that the reaction temperature rose to ⁇ 30° C., then stirring at ⁇ 30° C. for 1 hr. The resulting slurry was filtered, washed by water (3*200 mL), and oven dried to give compound A1-2 as white powder (58 g, yield 93.5%). MS-ESI:[M+1] + : 243.1
- Step 2 A solution of compound A1-2 (74 g, 0.306 mol, 1.0 eq) in DCM (1000 mL) was treated with triethylamine (77.2 g, 0.64 mol, 2.5 eq). The mixture was cooled to 0° C. in ice-bath, and TFAA (80.9 g, 0.383 mol, 1.25 eq) was added dropwise. The ice bath was removed after addition and the reaction temperature rose to 20° C., then stirring at 20° C. for 2 hrs, water (300 mL) was added, and then the aqueous phase was extracted twice with DCM.
- Step 3 To a solution of compound A1-3 (10 g, 44.6 mmol, 1.0 eq) in DCM (50 mL), was added TFA (20 g). The reaction mixture was stirred for 2 hrs at room temperature until TLC showed the reaction was complete, then concentrated under vacuum. Ice-water (30 mL) was added and the solution was treated with aqueous sodium hydroxide solution (4 mol/L) to pH 10. Then the aqueous phase was extracted six times with DCM/methanol (10/1). The combined extracts was dried over anhydrous sodium sulfate, concentrated to give compound A1-4 as an off-white solid (5.1 g, yield 91.9%). MS-ESI:[M+1] + : 125.1.
- Step 4 In nitrogen atmosphere, to a solution of 2-Bromo-3-hydroxypyridine (A1-5) (225 g, 1.293 mol, 1.0 eq), trimethylsilylacetylene (153.3 g, 1.592 mol, 1.23 eq) in 1,4-dioxane (2500 mL) was added CuI (25 g) and Pd(PPh3) 2 Cl 2 (45 g). The reaction mixture was stirred for 30 mins at 25° C., then cooled to 10° C. and triethylamine (363 g, 3.594 mol, 2.78 eq) was added dropwise. After stirring for 4 hrs at 60° C., the solution was cooled and concentrated under vacuum.
- Step 5 To a solution of compound A1-6 (105 g, 0.55 mol, 1.0 eq) in DCM (1000 mL) was added m-chloroperoxybenzoic acid (85%, 230 g, 1.13 mol, 12.06 eq) in portions below 25° C. After stirring overnight at room temperature, saturated sodium bicarbonate solution was added to pH 7-8 in ice-bath. The resulting mixture was filtered, and the filtrate was separate, and was extracted twice with DCM. The combined organic layers was washed with saturated sodium bicarbonate solution and brine, dried over anhydrous sodium sulfate, concentrated to give compound A1-7 as a brown liquid (115 g, yield 100%).
- Step 6 A solution of compound A1-7 (115 g, 0.55 mol, 1.0 eq) in toluene (400 mL) was added to phosphorus oxychloride (400 mL) in ice-bath below 30° C. The reaction mixture was stirred for 2 hrs at 90° C., cooled to room temperature, and concentrated. The residue was slowly added saturated sodium bicarbonate solution to pH 7-8 below 20° C., and the mixture was extracted twice with MTBE. The combined organic layers was washed with brine, dried over anhydrous sodium sulfate, concentrated and purified by silica gel column chromatography to give compound A1-8 as a yellow liquid (73 g, yield 58.7%).
- Step 7 To a solution of compound A1-8 (73 g, 0.323 mol, 1.0 eq) in THF (400 mL) was added aqueous sodium hydroxide solution (300 mL, 4 mol/L). After stirring for 1 hr at 50° C., the reaction mixture was cooled to room temperature and 1000 mL water was added. The mixture was extracted twice with MTBE. The combined organic layers was washed with brine, dried over anhydrous sodium sulfate, concentrated and recrystallized from ethyl acetate and petroleum ether to give compound A1-9 as an off-white powder (30 g, yield 60.5%).
- Step 8 Compound A1-9 (9.21 g, 60 mmol, 1.0 eq) was dissolved in methanol (150 mL), then water (150 mL) and sodium hydroxide (24 g, 10 eq) were added. After stirring for 1 hr at 50° C., the reaction mixture was cooled to 20° C. and concentrated. The residue was extracted three times with DCM, then the combined organic layers was dried over anhydrous sodium sulfate and concentrated to give compound A1-10 as a yellow liquid (5.5 g, yield 61.5%). MS-ESI:[M+1] + : 150
- Step 9 Compound A1-10 (5.5 g, 37 mmol, 1.0 eq) was added to 40% HBr aq (150 mL). The reaction mixture was heated to reflux for 18 hrs, cooled and concentrated. The residue was treated with saturated sodium bicarbonate solution (100 mL) to pH 7-8. After stirring for 20 min, the precipitate was filtered, washed with water, and oven dried to give compound A1-11 as an off-white powder (3.1 g, yield 62%). MS-ESI:[M+1] + : 136
- Step 10 In nitrogen atmosphere, a mixture of compound A1-11 (1.68 g, 12.4 mmol, 1.0 eq) in 120 mL DCM was cooled to ⁇ 5° C., and tetrabutyl-ammonium nitrate (5.17 g, 17 mmol, 1.36 eq) in DCM (30 mL) was added drop wise below 0° C., then TFAA (5.17 g, 20 mmol, 1.6 eq) was added all at once. After addition, the reaction mixture was stirred at ⁇ 5° C. for 1 hr and then warmed up to 25° C. and stirred for 15 hrs.
- Step 11 A mixture of compound A1-12 (1.37 g, 7.61 mmol, 1.0 eq) and propionic acid (50 mL) was heated to 110° C., then fuming nitric acid (0.65 mL) was added dropwise at 110° C. to 120° C. After stirring for 30 mins at 125° C. and cooled to room temperature, ether (100 mL) was added, and the solid was filtered, washed with ether and dried under vacuum to give compound A1-13 as yellow a powder (1.2 g, yield 87.6%). MS-ESI:[M+1] + : 181.
- Step 12 To a solution of compound A1-13 (1.2 g, 6.67 mmol, 1.0 eq) in 1,2-dichloroethane (50 mL), was added phosphorus oxychloride (15 mL) below 20° C., then stirred for 2 hrs at 95° C. in nitrogen atmosphere, cooled to 25° C. and concentrated. The residue was slowly added saturated sodium bicarbonate solution to pH 7-8 below 20° C., and the mixture was extracted twice with MTBE. The combined organic layers was washed with brine, dried over anhydrous sodium sulfate, concentrated to give compound A1-14 as a light-yellow powder (0.8 g, yield 60.4%),
- Step 13 To a solution of A1-14 (280 mg, 1.41 mmol, 1.0 eq) in n-butanol (20 mL) was added compound A1-4 (290 mg, 2.34 mmol, 1.66 eq) and DIPEA (403 mg, 3.12 mmol, 2.21 eq). The reaction mixture was stirred for 1 hr at 135° C., concentrated and purified by silica gel column chromatography to give A1-15 as a yellow powder (320 mg, yield 79.4%). MS-ESI:[M+1] + : 287.1.
- Step 14 To a solution of A1-15 (320 mg, 1.12 mmol, 1.0 eq) in methanol (15 mL), was added 10% Pd/C (0.3 g, 50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrates was concentrated under vacuum, and A1-16 was obtained as a yellow oil (286 mg, yield 100%). MS-ESI: [M+1] + : 257.1
- Example 2 was made using the same procedure as Example 1 except that (R)-(+)-Lactamide is replaced by 2-hydroxyacetamide in step 15): 40 mg of title compound as a light-yellow powder, MS-ESI: [M+1] + : 297.4
- Step 1 In nitrogen atmosphere, to a solution of trans-1,4-cyclohexane-dicarboxylic acid monomethyl ester (A3-1) (100 g, 0.538 mol, 1.0 eq) and triethylamine (57.4 g, 0.568 mol, 1.055 eq) in t-butyl alcohol (1000 mL) was added dropwise diphenylphosphoryl azide (155 g, 0.563 mol, 1.047 eq) at room temperature. The mixture was refluxed over 16 hrs. Upon completion by TLC, the mixture was then cooled and concentrated. Water (1000 mL) was added, and the mixture was extracted three times with MTBE.
- A3-1,4-cyclohexane-dicarboxylic acid monomethyl ester (A3-1) (100 g, 0.538 mol, 1.0 eq) and triethylamine (57.4 g, 0.568 mol, 1.055 eq) in
- Step 2 A suspension of LiAlH 4 (9.0 g, 0.236 mol, 1.12 eq) in THF (500 mL) was cooled to 0° C. in ice-bath, and then added a solution of compound A3-2 (54.3 g, 0.211 mol, 1.0 eq) in THF (200 mL) while keeping the temperature below 10° C. The reaction mixture was stirred overnight at room temperature, and then quenched with sodium sulfate decahydrate (27 g) at 15° C. to 25° C., filtered and the filtrate was concentrated to give compound A3-3 as a white powder (43 g, yield 89%).
- Step 3 A mixture of compound A3-3 (11.5 g, 0.05 mol, 1.0 eq) and triethylamine (7.6 g, 0.075 mol, 1.5 eq) in DCM (200 mL), was added methylsufonyl chloride (6.9 g, 0.06 mol, 1.2 eq) dropwise below 10° C. After stirring for 2 hrs at room temperature, water (300 mL) was added. The mixture was extracted twice with ethyl acetate. The combined extracts was washed with brine, dried over anhydrous sodium sulfate, concentrated to give compound A3-4 as yellow liquid (16.0 g, yield 100%). MS-ESI:[M+1] + : 307.1
- Step 4 To a solution of compound A3-4 (16.0 g, 0.05 mol, 1.0 eq) in DMSO (150 mL) was added sodium cyanide (7.0 g, 0.143 mol, 2.86 eq) in portions below 20° C. After stirring for 5 hrs at 85° C., the mixture was cooled to room temperature, ice-water (500 mL) was added. The mixture was extracted twice with MTBE. The combined extracts was washed three times with brine, dried over anhydrous sodium sulfate, concentrated and purified by silica gel column chromatography to give compound A3-5 as white powder (9.3 g, yield 78%). MS-ESI:[M+1] + : 238.1.
- Step 5 To a solution of compound A3-5 (1.1 g, 4.6 mmol, 1.0 eq) in DCM (10 mL) was added TFA (6 g). The reaction mixture was stirred for 2 hrs at room temperature, then concentrated under vacuum. Ice-water (15 mL) was added and the solution was treated with aqueous sodium hydroxide solution (4 mol/L) to pH 10. Then the aqueous phase was extracted five times with DCM/methanol (10/1). The combined extracts was dried over anhydrous sodium sulfate, concentrated to give compound A3-6 as a yellow oil (0.55 g, yield 87.7%). MS-ESI:[M+1] + : 138.1.
- Step 6 to step 8 are the same as step 13 to step 15 in Example 1 except that the amine A1-4 is replaced by A3-6 to make the title compound: 70 mg of light-yellow powder (Yield: 0.565%). MS-ESI: [M+1] + : 325.5
- Step 1 In a round bottom flask, triethylamine (188 g, 1.86 mol, 1.0 eq) was added dropwise to a stirred solution of di-tert-butyl dicarbonate (162 g, 0.744 mol, 1.2 eq) and compound A4-1 (100 g, 0.62 mol, 1.0 eq) in water (500 mL) and 1,4-dioxane (500 mL). After stirring for 18 hrs at room temperature, the solution was extracted with MTBE (500 mL*2) and the aqueous phase was cooled on ice and carefully acidified to pH 3 by slow addition of 10% citric acid solution.
- MTBE 500 mL*2
- Step 2 A solution of compound A4-2 (40 g, 0.153 mmol, 1.0 eq) in THF (600 mL) was treated with 4-methylmorpholine (17 g, 0.168, 1.1 eq) at room temperature. The resulting mixture was cooled to 0° C. before being treated with isobutyl chloroformate (22.7 g, 0.166 mmol, 1.08 eq) dropwise. The resulting reaction mixture was stirred at 0° C. for an addition 20 mins before being filtered and washed with THF. Then the clear filtrate solution was cooed to 0° C., and treated with a solution of NaBH 4 (11.2 g, 0.295 mol, 1.93 eq) in water (100 mL).
- Step 3 A solution of compound of A4-3 (25 g, 0.1 mol, 1.0 eq) in toluene (300 mL) and acetic acid (150 mL) was heated to reflux for 5 hrs and then cooled, concentrated under vacuum. The residual was added saturated sodium bicarbonate solution to pH 7-8 in ice-bath. Then the mixture was extracted three times with ethyl acetate, and the combined extracts was washed with brine, dried over anhydrous sodium sulfate, concentrated and recrystallized by ethyl acetate and PE to give compound A4-4 as a white powder (8.0 g, yield 37.2%).
- Step 4 A solution of tributyl phosphine (72.9 g, 0.36 mol, 1.0 eq) in nitromethane (500 mL), was added dropwise chloroacetonitrile (27.2 g, 0.36 mol, 1.0 eq) in nitrogen atmosphere. The resulting reaction mixture was stirred for 16 hrs at room temperature, then concentrated. The residual oil solidified when a small amount of ethyl acetate was added. The solid was recrystallized by ethyl acetate and DCM to afford compound A4-5 as a white powder (95 g, yield 95%).
- Step 5 To a solution of dry compound A4-5 (8.3 g, 30 mmol, 3.0 eq) in N,N-dimethylacetamide (30 mL) in nitrogen atmosphere, was added solid Potassium tert-butoxide (3.1 g, 28 mmol, 2.8 eq) in portions at 0° C. The resulting mixture was gradually warmed to 30° C. and stirred for 2 hrs. The resulting ylide solution was then treated with compound A4-4 (2.15 g, 10 mmol, 1.0 eq), and stirred overnight at 70° C. After cooled to room temperature, the resulting slurry was poured into the mixture of ice-water (100 mL) and saturated sodium bicarbonate solution (100 mL).
- Step 6 To a solution of compound A4-6 (7.5 g, 10 mmol, 1.0 eq) in methanol (200 mL), was added 10% Pd/C (0.5 g,50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrates was concentrated under vacuum, and purified by silica gel column chromatography to give compound A4-7 as off-white powder (1.6 g, yield 66.7%). MS-ESI:[M+1] + : 241.1
- Step 7 To a solution of compound A4-7 (1.6 g, 6.67 mmol, 1.0 eq) in DCM (20 mL), was added TFA (10 g, 88.5 mmol, 13.2 eq). The reaction mixture was stirred for 2 hrs at room temperature until TLC showed the reaction was complete, then concentrated under vacuum. Water (20 mL) was added and the solution was treated with aqueous sodium hydroxide solution (4 mol/L) to pH 10. Then the aqueous phase was extracted six times with DCM/methanol (10/1). The combined extracts was dried over anhydrous sodium sulfate, concentrated to give compound A4-8 as light-brown oil (950 mg, yield 100%). MS-ESI:[M+1] + : 141.1
- Step 8 To a solution of compound A1-14 (prepared as step 4 to 12 in example 1) (600 mg, 3.0 mmol, 1.0 eq) in n-butanol (15 mL), was added compound A4-8 (950 mg, 6.7 mmol, 2.26 eq) and DIPEA (1.36 g, 10.5 mmol, 3.5 eq). The reaction mixture was stirred for 1 hr at 135° C., concentrated and purified by silica gel column chromatography to give compound A4-9 (2R,5S) as light-yellow powder (254 mg, yield 28.0%).MS-ESI: [M+1] + : 303.1.
- Step 9 To a solution of compound A4-9 (254 g, 0.84 mmol, 1.0 eq) in methanol (20 mL), was added 10% Pd/C (0.15 g,50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrates was concentrated under vacuum, and compound A4-10 was obtained as yellow oil (230 mg, yield 100%). MS-ESI:[M+1] + : 273.1
- Step 10 A solution of D-Lactamide (388 mg, 4.2 mmol, 5.0 eq) and Et3O—BF 4 (1.3 g, 6.72 mmol, 8.0 eq) in THF (10 mL) was stirred for 30 mins at room temperature in nitrogen atmosphere. Then the above solution was added to the mixture of compound A4-10 (230 mg, 0.84 mmol, 1.0 eq) in ethanol (10 mL). After stirring for 3 hrs at 85° C. until HPLC showed the reaction was complete, the mixture was concentrated, added water and extracted four times with ethyl acetate. The organic phases was discarded and the aqueous phase was treated with saturated sodium bicarbonate solution to pH 8, extracted twice with ethyl acetate.
- Step 1 To a solution of compound A1-14 (prepared as step 4 to 12 in example 1) (820 mg, 4.13 mmol, 1.0 eq) in n-butanol (15 mL), was added compound A5-3 (1.0 g, 5.37 mmol, 1.3 eq) and DIPEA (1.6 g, 12.4 mmol, 3.0 eq). The reaction mixture was stirred for 1 hr at 135° C., concentrated and purified by silica gel column chromatography to give compound A5-4 as yellow powder (1.32 g, yield 91.8%). MS-ESI:[M+1] + : 349.1
- Step 2 To a solution of compound A5-4 (1.32 g, 3.8 mmol, 1.0 eq) in DCM (15 mL), was added a solution of HCl in ethanol (30% w/w) (15 mL). The reaction mixture was stirred for 2 hrs at room temperature until TLC showed the reaction was complete, then concentrated under vacuum. Ice-water (20 mL) was added and the solution was treated with aqueous sodium hydroxide solution (4 mol/L) to pH 10. Then the aqueous phase was extracted three times with DCM. The combined extracts was dried over anhydrous sodium sulfate, concentrated to give compound A5-5 as yellow powder (950 mg, yield 100%). MS-ESI:[M+1] + : 249.1
- Step 3 A mixture of 2,2,2-trifluoroethylamine (A5-1) (1.21 g, 12.2 mmol, 1.0 eq) and pyridine (2.4 g, 30.5 mmol, 2.5 eq) in DCM (50 mL) was cooled to 0° C., and treated with triphosgene (1.34 g, 4.52 mmol, 0.37 eq) in DCM (50 mL) dropwise below 5° C. After addition, the reaction mixture was stirred at 35° C. for 1 hr and then 25° C. for 2 hrs. The isocyanate (A5-2) solution was used for next step without purification.
- Step 4 A mixture of compound A5-5 (0.95 g, 3.8 mmol, 1.0 eq) and pyridine (0.45 g, 5.7 mmol, 1.5 eq) in DCM (60 mL) was cooled to 10° C., and treated with the isocyanate (A5-2) solution (12.2 mmol, 3.2 eq) dropwise. The reaction mixture was heated to reflux for 3 h, and then cooled. Saturated sodium bicarbonate solution (200 mL) was added, the mixture was extracted twice with DCM. The combined extracts was washed brine, dried over anhydrous sodium sulfate, concentrated and purified by silica gel column chromatography to give compound A5-6 as yellow powder (850 mg, yield 60%). MS-ESI:[M+1] + : 374.3
- Step 5 To a solution of compound A5-6 (850 mg, 2.28 mmol, 1.0 eq) in methanol (80 mL), was added 10% Pd/C (0.45 g, 50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrate was concentrated under vacuum, and compound A5-7 was obtained as brown oil (800 mg, yield 100%). MS-ESI:[M+1] + : 344.3
- Step 6 A solution of D-Lactamide (1.27 g,13.68 mmol,6.0 eq) and Et3O—BF4 (3.53 g, 18.24 mmol, 8.0 eq) in THF (20 mL) was stirred 30 min at room temperature in nitrogen atmosphere. Then the above solution was added to the mixture of compound A5-7 (800 mg, 2.28 mmol, 1.0 eq) in ethanol (20 mL). After stirring for 5 hrs at 85° C. until HPLC showed the reaction was complete, the mixture was concentrated, added HCl (1 mol/L, 30 mL) and extracted four times with ethyl acetate.
- Step 1 In nitrogen atmosphere, to a solution of compound A1-14 (500 mg, 2.0 mmol, 1.0 eq) in butyl alcohol (8 mL), were added compound A4-8 (350 mg, 2.5 mmol, 1.0 eq) and DIPEA (403 mg, 8.25 mmol, 3.3 eq). The reaction mixture was stirred 2 hrs at 135° C., then concentrated and purified by silica gel column chromatography to give compound A4-9 as yellow solid (194 mg, yield 25.6%). MS-ESI:[M+1] + : 302.3
- Step 2 To a solution of compound A4-9 (97 mg, 1.0 mmol) in methanol (15 mL), was added 10% Pd/C (50 mg, 50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrate was concentrated to give compound A4-10 as yellow oil (535 mg, yield: 100%). MS-ESI: [M+1] + : 272.5
- Step 3 A solution of glycolamide (126 mg, 1.6 mmol, 5.0 eq) and Et3O—BF4 (310 mg, 1.6 mmol, 5.0 eq) in THF (10 mL) was stirred 30 mins at room temperature in nitrogen atmosphere. Then the above solution was added to the mixture of compound A4-10 (88 mg, 0.32 mmol, 1.0 eq) in ethanol (10 mL). After stirring 12 hrs at 85° C., the mixture was concentrated, added water and extracted three times with ethyl acetate. The organic phases were discarded and the aqueous phase was treated with saturated sodium bicarbonate solution (100 mL) to pH: 8, then the mixture was extracted twice with ethyl acetate. The second organic phase was dried over anhydrous sodium sulfate, concentrated and purified by silica gel column chromatography to give the title compound as an off-white powder (70 mg, yield: 70%). MS-ESI: [M+1]+: 313.5
- Step 1 In nitrogen atmosphere, to a solution of compound A1-14 (500 mg, 2.0 mmol, 1.0 eq) in butyl alcohol (8 mL), were added compound A4-8 (350 mg, 2.5 mmol, 1.0 eq) and DIPEA (403 mg, 8.25 mmol, 3.3 eq). The reaction mixture was stirred 2 hrs at 135° C., then concentrated and purified by silica gel column chromatography to give compound A8-1 as yellow solid (67 mg, yield 8.84%).
- Step 2 To a solution of compound A8-1 (67 mg, 1.0 mmol) in methanol (10 mL), was added 10% Pd/C (30 mg, 50% wet). Hydrogenation was carried out under atmospheric pressure at room temperature until hydrogen uptake ceased. The catalyst was filtered and washed by methanol. The filtrate was concentrated to give compound A8-2 as yellow oil (60 mg, yield: 100%).
- Step 3 A solution of glycolamide (105 mg, 1.33 mmol, 6.0 eq) and Et 3 O—BF 4 (258 mg, 1.33 mmol, 6.0 eq) in THF (10 mL) was stirred 30 mins at room temperature in nitrogen atmosphere. Then the above solution was added to the mixture of compound A8-2 (60 mg, 0.221 mmol, 1.0 eq) in ethanol (10 mL). After stirring 12 hrs at 85° C., the mixture was concentrated, added water and extracted three times with ethyl acetate. The organic phases was discarded and the aqueous phase was treated with saturated sodium bicarbonate solution (100 mL) to pH: 8, then the mixture was extracted twice with ethyl acetate. The second organic phases was dried over anhydrous sodium sulfate, concentrated and purified by silica gel column chromatography to give the title compound as an off-white powder (21 mg, yield: 30.5%).
- Base Reaction buffer 20 mM Hepes (pH 7.5), 10 mM MgCl 2 , 1 mM EGTA, 0.02% Brij35, 0.02 mg/ml BSA, 0.1 mM Na 3 VO 4 , 2 mM DTT, 1% DMSO.
- Required cofactors are added individually to each kinase reaction
- Example B2 Human Whole Blood p-STAT3 Assay
- IL-6 R&D systems; Cat #206-IL
- TPO Thrombopoietin
- Examples 3, 4 and 7 showed selective inhibition of IL-6 induced STAT3 phosphorylation, but not TPO induced STAT3 phosphorylation as shown below based on the range of IC50: +: >100 ⁇ M; ++: 20-100 ⁇ M; +++: 5-20 ⁇ M; ++++: ⁇ 5 ⁇ M.
- Example 11 also showed some activity in the TPO induced STAT3 phosphorylation assay.
Abstract
Description

or a pharmaceutically acceptable salt thereof; or a prodrug, or a pharmaceutically acceptable salt of a prodrug thereof; or a hydrate, solvate, or polymorph thereof; wherein:
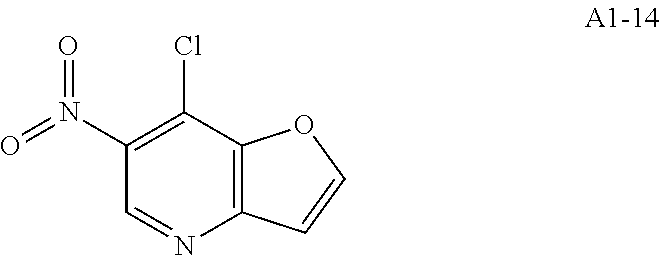
useful for the process of making compounds of formula I.

comprising contacting a compound of formula V:

and a compound of formula VI:

in the presence of a (C1-6 alkyl)3oxonium tetrafluoroborate at sufficient temperature, and for sufficient time to produce a compound of formula I:

wherein R2 is H, and R1 and Cy are as defined above. The (C1-6 alkyl)3oxonium tetrafluoroborate can be triethyloxonium tetrafluoroborate.

in the presence of a hydrogenation catalyst and hydrogen gas at sufficient temperature, sufficient pressure and for sufficient time to produce a compound of formula V wherein Cy is as defined above. The hydrogenation catalyst can be palladium on carbon.

and a compound of formula VIII:
Cy-NH2 VIII
in the presence of a base at sufficient temperature, and for sufficient time to produce a compound of formula VII wherein Cy is as defined above. The base can be N,N-Diisopropylethylamine.

or a pharmaceutically acceptable salt thereof; or a prodrug, or a pharmaceutically acceptable salt of a prodrug thereof; or a hydrate, solvate, or polymorph thereof; wherein:
| TABLE 1 | |||
 |
1 | ||
 |
2 | ||
 |
3 | ||
 |
4 | ||
 |
5 | ||
 |
6 | ||
 |
7 | ||
 |
8 | ||
 |
9 | ||
 |
10 | ||
 |
11 | ||
- trans-4-[2-[(R)-1-Hydroxyethyl]-1H-furo[3,2-b] imidazo[4,5-d]pyridin-1-yl]cyclohexanecarbonitrile (1);
- trans-4-[2-(Hydroxymethyl)furo[3,2-b]imidazo[4,5-d]pyridin-1-yl]cyclohexanecarbonitrile (2);
- 2-[trans-4-[2-[(R)-1-Hydroxyethyl]furo[3,2-b]imidazo[4,5-d]pyridin-1-yl]cyclohexyl]acetonitrile (3);
- 2-[(2R,5S)-5-[2-[(R)-1-Hydroxyethyl]furo[3,2-b]imidazo[4,5-d]pyridin-1-yl]tetrahydropyran-2-yl]acetonitrile (4);
- 3-[2-[(R)-1-Hydroxyethyl]-1H-furo[3,2-b]imidazo[4,5-d]pyridin-1-yl]-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide (5);
- (R)-4-[2-(1-Hydroxyethyl)-1H-furo[3,2-b]imidazo[4,5-d]pyridin-1-yl]-N-(2,2,2-trifluoroethyl)piperidine-1-carboxamide (6);
- 2-[(2R,5 S)-5-[2-(Hydroxymethyl)furo[3,2-b]imidazo[4,5-d]pyridin-1-yl]tetrahydropyran-2-yl]acetonitrile (7);
- 2-[(2S,5 S)-5-[2-(Hydroxymethyl)furo[3,2-b]imidazo[4,5-d]pyridin-1-yl]tetrahydropyran-2-yl]acetonitrile (8),
- 2-[(2R,5S)-5-[2-Ethylfuro[3,2-b]imidazo[4,5-d] pyridin-1-yl] tetrahydropyran-2-yl]acetonitrile (9),
- 2-[(2R,5S)-5-[2-Furo[3,2-b]imidazo[4,5-d] pyridin-1-yl] tetrahydropyran-2-yl]acetonitrile (10),
- 2-[(2R,5S)-5-[2-Methylfuro[3,2-b]imidazo[4,5-d] pyridin-1-yl] tetrahydropyran-2-yl]acetonitrile (11).

















| Example | Jak1 | Jak2 |
| 1 | ++ | NT |
| 2 | ++ | NT |
| 3 | ++++ | ++ |
| 4 | ++++ | ++ |
| 5 | + | + |
| 6 | + | + |
| 7 | ++++ | ++ |
| 8 | ++ | + |
| 9 | ++++ | ++ |
| 11 | ++++ | +++ |
| Example | IL-6 | TPO |
| 3 | ++++ | + |
| 4 | ++++ | + |
| 7 | ++++ | + |
| 11 | ++++ | ++ |
Claims (22)







Cy-NH2 VIII




Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/526,799 USRE49834E1 (en) | 2016-10-03 | 2017-09-30 | Jak1 selective inhibitors and uses thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662403660P | 2016-10-03 | 2016-10-03 | |
| US17/526,799 USRE49834E1 (en) | 2016-10-03 | 2017-09-30 | Jak1 selective inhibitors and uses thereof |
| US16/333,994 US10738060B2 (en) | 2016-10-03 | 2017-09-30 | JAK1 selective inhibitors and uses thereof |
| PCT/US2017/054668 WO2018067422A1 (en) | 2016-10-03 | 2017-09-30 | Novel jak1 selective inhibitors and uses thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| USRE49834E1 true USRE49834E1 (en) | 2024-02-13 |
Family
ID=61831161
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/333,994 Ceased US10738060B2 (en) | 2016-10-03 | 2017-09-30 | JAK1 selective inhibitors and uses thereof |
| US17/526,799 Active USRE49834E1 (en) | 2016-10-03 | 2017-09-30 | Jak1 selective inhibitors and uses thereof |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/333,994 Ceased US10738060B2 (en) | 2016-10-03 | 2017-09-30 | JAK1 selective inhibitors and uses thereof |
Country Status (21)
| Country | Link |
|---|---|
| US (2) | US10738060B2 (en) |
| EP (1) | EP3509591B1 (en) |
| JP (1) | JP7089141B2 (en) |
| KR (1) | KR102399848B1 (en) |
| CN (2) | CN108366994B (en) |
| AU (3) | AU2017339417C1 (en) |
| BR (1) | BR112019005969A2 (en) |
| CA (1) | CA3039178A1 (en) |
| DK (1) | DK3509591T3 (en) |
| EA (1) | EA201990523A1 (en) |
| ES (1) | ES2901216T3 (en) |
| HK (1) | HK1253040A1 (en) |
| HR (1) | HRP20211965T1 (en) |
| HU (1) | HUE058120T2 (en) |
| IL (3) | IL265358B (en) |
| MX (1) | MX2019003649A (en) |
| NZ (1) | NZ751284A (en) |
| PL (1) | PL3509591T3 (en) |
| PT (1) | PT3509591T (en) |
| RS (1) | RS62695B1 (en) |
| WO (1) | WO2018067422A1 (en) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7839138B2 (en) * | 2007-01-29 | 2010-11-23 | Electro Scientific Industries, Inc. | Adjustable force electrical contactor |
| HRP20211965T1 (en) | 2016-10-03 | 2022-03-18 | Highlightll Pharmaceutical (Hainan) Co., Ltd. | Novel jak1 selective inhibitors and uses thereof |
| PT3568396T (en) * | 2017-01-11 | 2021-02-09 | Leo Pharma As | Novel amino-imidazopyridine derivatives as janus kinase inhibitors and pharmaceutical use thereof |
| EP3873433A1 (en) * | 2018-10-31 | 2021-09-08 | Incyte Corporation | Combination therapy for treatment of hematological diseases |
| KR20210137087A (en) * | 2019-03-05 | 2021-11-17 | 인사이트 코포레이션 | JAK1 pathway inhibitors for the treatment of chronic lung allograft dysfunction |
| WO2020191041A2 (en) * | 2019-03-19 | 2020-09-24 | Incyte Corporation | Biomarkers for vitiligo |
| EP3981771A4 (en) * | 2019-06-06 | 2023-03-15 | Hangzhou Highlightll Pharmaceutical Co., Ltd. | Synthesis method of furoimidazopyridine compound, crystal form of furoimidazopyridine compound, and crystal form of salt thereof |
| CA3142629A1 (en) * | 2019-06-06 | 2020-12-10 | Highlightll Pharmaceutical (Hainan) Co., Ltd | Method for synthesizing furoimidazopyridine compound, polymorphic substance and polymorphic substance of salt |
| US20230192617A1 (en) | 2020-05-19 | 2023-06-22 | Bayer Cropscience Aktiengesellschaft | Azabicyclic(thio)amides as fungicidal compounds |
| JP2023529475A (en) | 2020-06-10 | 2023-07-10 | バイエル、アクチエンゲゼルシャフト | Azabicyclyl-substituted heterocycles as novel fungicides |
| WO2022260945A1 (en) * | 2021-06-07 | 2022-12-15 | The Regents Of The University Of California | Compositions and methods for treating celiac disease |
| WO2023035913A1 (en) * | 2021-09-13 | 2023-03-16 | Hangzhou Highlightll Pharmaceutical Co., Ltd | Methods of treating cns disorders |
| CN114213424B (en) * | 2021-12-30 | 2023-05-26 | 杭州高光制药有限公司 | Synthesis method of furan [3,2-b ] pyridine derivative |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1104764A1 (en) | 1998-08-12 | 2001-06-06 | Hokuriku Seiyaku Co., Ltd. | 1h-imidazopyridine derivatives |
| US20070185152A1 (en) | 2004-03-02 | 2007-08-09 | Smithkline Beecham Corporation | Inhibitors of akt activity |
| US20120122846A1 (en) | 2010-10-08 | 2012-05-17 | Abbott Laboratories | FURO[3,2-d]PYRIMIDINE COMPOUNDS |
| WO2013024895A1 (en) | 2011-08-12 | 2013-02-21 | Nissan Chemical Industries, Ltd. | Tricyclic heterocyclic compounds and jak inhibitors |
| WO2014071031A1 (en) | 2012-11-01 | 2014-05-08 | Incyte Corporation | Tricyclic fused thiophene derivatives as jak inhibitors |
| WO2015168246A1 (en) | 2014-04-30 | 2015-11-05 | Incyte Corporation | Processes of preparing a jak1 inhibitor and new forms thereto |
| US20150342952A1 (en) | 2014-05-30 | 2015-12-03 | Incyte Corporation | TREATMENT OF CHRONIC NEUTROPHILIC LEUKEMIA (CNL) AND ATYPICAL CHRONIC MYELOID LEUKEMIA (aCML) BY INHIBITORS OF JAK1 |
| US10738060B2 (en) | 2016-10-03 | 2020-08-11 | TLL Pharmaceutical, LLC | JAK1 selective inhibitors and uses thereof |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4369172A (en) | 1981-12-18 | 1983-01-18 | Forest Laboratories Inc. | Prolonged release therapeutic compositions based on hydroxypropylmethylcellulose |
| DE3583799D1 (en) | 1985-01-11 | 1991-09-19 | Abbott Lab Ltd | SOLID PREPARATION WITH SLOW RELEASE. |
| JP2773959B2 (en) | 1990-07-10 | 1998-07-09 | 信越化学工業株式会社 | Colon release solid preparation |
| UA73092C2 (en) | 1998-07-17 | 2005-06-15 | Брістол-Майерс Сквібб Компані | Tablets with enteric coating and method for their manufacture |
| AU4800999A (en) | 1998-07-28 | 2000-02-21 | Tanabe Seiyaku Co., Ltd. | Preparation capable of releasing drug at target site in intestine |
| US6461631B1 (en) | 1999-11-16 | 2002-10-08 | Atrix Laboratories, Inc. | Biodegradable polymer composition |
| US6800663B2 (en) | 2002-10-18 | 2004-10-05 | Alkermes Controlled Therapeutics Inc. Ii, | Crosslinked hydrogel copolymers |
| CN101282974A (en) * | 2005-08-04 | 2008-10-08 | 西特里斯药业公司 | Benzimidazole derivatives as SIRTUIN modulators |
| KR20120081164A (en) * | 2009-09-29 | 2012-07-18 | 엑스커버리 홀딩 컴퍼니 엘엘씨 | Pi3k(delta) selective inhibitors |
| WO2012022045A1 (en) * | 2010-08-20 | 2012-02-23 | Hutchison Medipharma Limited | Pyrrolopyrimidine compounds and uses thereof |
| WO2013007768A1 (en) | 2011-07-13 | 2013-01-17 | F. Hoffmann-La Roche Ag | Tricyclic heterocyclic compounds, compositions and methods of use thereof as jak inhibitors |
| WO2015061665A1 (en) | 2013-10-24 | 2015-04-30 | Abbvie Inc. | Jak1 selective inhibitor and uses thereof |
-
2017
- 2017-09-30 HR HRP20211965TT patent/HRP20211965T1/en unknown
- 2017-09-30 CN CN201780004442.5A patent/CN108366994B/en active Active
- 2017-09-30 CN CN202110514751.5A patent/CN113214278B/en active Active
- 2017-09-30 JP JP2019517991A patent/JP7089141B2/en active Active
- 2017-09-30 RS RS20211521A patent/RS62695B1/en unknown
- 2017-09-30 PL PL17858944T patent/PL3509591T3/en unknown
- 2017-09-30 US US16/333,994 patent/US10738060B2/en not_active Ceased
- 2017-09-30 DK DK17858944.6T patent/DK3509591T3/en active
- 2017-09-30 PT PT178589446T patent/PT3509591T/en unknown
- 2017-09-30 EA EA201990523A patent/EA201990523A1/en unknown
- 2017-09-30 EP EP17858944.6A patent/EP3509591B1/en active Active
- 2017-09-30 MX MX2019003649A patent/MX2019003649A/en unknown
- 2017-09-30 NZ NZ751284A patent/NZ751284A/en unknown
- 2017-09-30 US US17/526,799 patent/USRE49834E1/en active Active
- 2017-09-30 AU AU2017339417A patent/AU2017339417C1/en active Active
- 2017-09-30 KR KR1020197009016A patent/KR102399848B1/en active IP Right Grant
- 2017-09-30 HU HUE17858944A patent/HUE058120T2/en unknown
- 2017-09-30 CA CA3039178A patent/CA3039178A1/en active Pending
- 2017-09-30 WO PCT/US2017/054668 patent/WO2018067422A1/en unknown
- 2017-09-30 BR BR112019005969A patent/BR112019005969A2/en unknown
- 2017-09-30 ES ES17858944T patent/ES2901216T3/en active Active
-
2018
- 2018-09-27 HK HK18112420.5A patent/HK1253040A1/en unknown
-
2019
- 2019-03-13 IL IL265358A patent/IL265358B/en unknown
-
2022
- 2022-02-17 AU AU2022201058A patent/AU2022201058B2/en active Active
- 2022-02-17 AU AU2022201061A patent/AU2022201061B2/en active Active
- 2022-03-10 IL IL291265A patent/IL291265B2/en unknown
- 2022-03-10 IL IL291267A patent/IL291267B2/en unknown
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1104764A1 (en) | 1998-08-12 | 2001-06-06 | Hokuriku Seiyaku Co., Ltd. | 1h-imidazopyridine derivatives |
| US20070185152A1 (en) | 2004-03-02 | 2007-08-09 | Smithkline Beecham Corporation | Inhibitors of akt activity |
| US20120122846A1 (en) | 2010-10-08 | 2012-05-17 | Abbott Laboratories | FURO[3,2-d]PYRIMIDINE COMPOUNDS |
| WO2013024895A1 (en) | 2011-08-12 | 2013-02-21 | Nissan Chemical Industries, Ltd. | Tricyclic heterocyclic compounds and jak inhibitors |
| WO2014071031A1 (en) | 2012-11-01 | 2014-05-08 | Incyte Corporation | Tricyclic fused thiophene derivatives as jak inhibitors |
| WO2015168246A1 (en) | 2014-04-30 | 2015-11-05 | Incyte Corporation | Processes of preparing a jak1 inhibitor and new forms thereto |
| US20150344497A1 (en) | 2014-04-30 | 2015-12-03 | Incyte Corporation | Processes of preparing a jak1 inhibitor and new forms thereto |
| US20150342952A1 (en) | 2014-05-30 | 2015-12-03 | Incyte Corporation | TREATMENT OF CHRONIC NEUTROPHILIC LEUKEMIA (CNL) AND ATYPICAL CHRONIC MYELOID LEUKEMIA (aCML) BY INHIBITORS OF JAK1 |
| US10738060B2 (en) | 2016-10-03 | 2020-08-11 | TLL Pharmaceutical, LLC | JAK1 selective inhibitors and uses thereof |
Non-Patent Citations (4)
| Title |
|---|
| [No Author Listed] Bioisosteres. In: Medicinal Chemistry, Second Edition. 2003. You et al., Eds. Section 3, Chapter 24:583-5. |
| Extended European Search Report for EP App. No. 17858944.6 dated Feb. 25, 2020. 9 pages. |
| International Search Report and Written Opinion for PCT/US2017/054668 dated Dec. 15, 2017. 8 pages. |
| Silverman, The Organic Chemistry of Drug Design and Drug Action, Second Edition. 2004:29-34. |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| USRE49834E1 (en) | Jak1 selective inhibitors and uses thereof | |
| EP3555097B1 (en) | Imidazo[4,5-d]pyrrolo[2,3-b]pyridine compounds as inhibitors of janus kinases | |
| JP2020097595A (en) | Novel salt for treatment of inflammatory disorders and pharmaceutical composition thereof | |
| KR102336291B1 (en) | 3-(2-aminopyrimidin-4-yl)-5-(3-hydroxypropynyl)-1h-pyrrolo[2,3-c]pyridine derivatives as nik inhibitors for the treatment of cancer | |
| TWI406864B (en) | Chemical compounds | |
| US8912178B2 (en) | mTOR selective kinase inhibitors | |
| EP3652169A1 (en) | Pyrazolo and triazolo bicyclic compounds as jak kinase inhibitors | |
| EP3571198B1 (en) | Bicyclic amines as jak kinase inhibitors | |
| CN112851682B (en) | Substituted pyridine amide compound and application thereof | |
| US20210101881A1 (en) | Pyrimidine compound, preparation method thereof and medical use thereof | |
| KR20220161396A (en) | Preparation, Compositions, and Crystal Forms of Substituted Pyridinone-Pyridinyl Compounds | |
| WO2019070167A1 (en) | Epidermal growth factor receptor inhibitors | |
| WO2018130563A1 (en) | Novel amino-imidazopyridine derivatives as janus kinase inhibitors and pharmaceutical use thereof | |
| US10766883B2 (en) | Aminopyrazoles as janus kinase inhibitors | |
| EA040948B1 (en) | SELECTIVE JAK1 INHIBITORS AND THEIR USE |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| FEPP | Fee payment procedure |
Free format text: ENTITY STATUS SET TO UNDISCOUNTED (ORIGINAL EVENT CODE: BIG.); ENTITY STATUS OF PATENT OWNER: SMALL ENTITY |
|
| FEPP | Fee payment procedure |
Free format text: ENTITY STATUS SET TO SMALL (ORIGINAL EVENT CODE: SMAL); ENTITY STATUS OF PATENT OWNER: SMALL ENTITY |
|
| AS | Assignment |
Owner name: HANGZHOU HIGHLIGHTII PHARMACEUTICAL CO., LTD, CHINA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:HIGHLIGHTII PHARMACEUTICAL (HAINAN) CO., LTD;REEL/FRAME:060592/0056 Effective date: 20220520 Owner name: HIGHLIGHTII PHARMACEUTICAL (HAINAN) CO., LTD, CHINA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:TLL PHARMACEUTICAL, LLC;REEL/FRAME:060829/0642 Effective date: 20210113 |