USRE45173E1 - Inhibitors of CYP 17 - Google Patents
Inhibitors of CYP 17 Download PDFInfo
- Publication number
- USRE45173E1 USRE45173E1 US13/929,901 US201313929901A USRE45173E US RE45173 E1 USRE45173 E1 US RE45173E1 US 201313929901 A US201313929901 A US 201313929901A US RE45173 E USRE45173 E US RE45173E
- Authority
- US
- United States
- Prior art keywords
- pyridin
- methyl
- imidazolidin
- alkyl
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active, expires
Links
- 239000003112 inhibitor Substances 0.000 title description 24
- 150000001875 compounds Chemical class 0.000 claims abstract description 203
- 150000003839 salts Chemical class 0.000 claims abstract description 48
- -1 -OH Chemical group 0.000 claims description 358
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 128
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 48
- 125000001424 substituent group Chemical group 0.000 claims description 41
- 125000005843 halogen group Chemical group 0.000 claims description 37
- 125000000623 heterocyclic group Chemical group 0.000 claims description 32
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 32
- 125000001072 heteroaryl group Chemical group 0.000 claims description 31
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 29
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 24
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 23
- 125000001153 fluoro group Chemical group F* 0.000 claims description 20
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 18
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 18
- 125000001559 cyclopropyl group Chemical class [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 17
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 17
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 17
- 125000004191 (C1-C6) alkoxy group Chemical class 0.000 claims description 16
- 239000008194 pharmaceutical composition Substances 0.000 claims description 15
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 14
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 13
- 230000001028 anti-proliverative effect Effects 0.000 claims description 11
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 10
- 125000004043 oxo group Chemical class O=* 0.000 claims description 10
- 239000008177 pharmaceutical agent Substances 0.000 claims description 10
- ZVIFCOOHWGNPHJ-UHFFFAOYSA-N 1-(2-chloropyridin-4-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(Cl)N=CC=2)CC1 ZVIFCOOHWGNPHJ-UHFFFAOYSA-N 0.000 claims description 9
- 239000004305 biphenyl Substances 0.000 claims description 9
- 235000010290 biphenyl Nutrition 0.000 claims description 9
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 claims description 8
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 claims description 8
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 claims description 8
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 claims description 8
- 239000002246 antineoplastic agent Substances 0.000 claims description 8
- 125000004500 isothiazol-4-yl group Chemical group S1N=CC(=C1)* 0.000 claims description 8
- 125000002837 carbocyclic group Chemical group 0.000 claims description 7
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 229910052736 halogen Inorganic materials 0.000 claims description 7
- SSCWVAMSDPREDJ-UHFFFAOYSA-N 1-(4-chlorothieno[3,2-c]pyridin-2-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2SC3=CC=NC(Cl)=C3C=2)CC1 SSCWVAMSDPREDJ-UHFFFAOYSA-N 0.000 claims description 6
- IECOQVBOIMTOKS-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-naphthalen-2-ylimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=CC=CC3=CC=2)CC1 IECOQVBOIMTOKS-UHFFFAOYSA-N 0.000 claims description 6
- VPMDSBXKDLGEKK-UHFFFAOYSA-N 1-naphthalen-2-yl-3-pyridin-3-ylimidazolidin-2-one Chemical compound C1CN(C=2C=C3C=CC=CC3=CC=2)C(=O)N1C1=CC=CN=C1 VPMDSBXKDLGEKK-UHFFFAOYSA-N 0.000 claims description 6
- 125000004533 benzofuran-5-yl group Chemical group O1C=CC2=C1C=CC(=C2)* 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims description 6
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 6
- 125000004550 quinolin-6-yl group Chemical group N1=CC=CC2=CC(=CC=C12)* 0.000 claims description 6
- GHGZEBQWFIPOQK-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 GHGZEBQWFIPOQK-UHFFFAOYSA-N 0.000 claims description 5
- VYGWQNWTGXPGLJ-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-(hydroxymethyl)pyridin-3-yl]imidazolidin-2-one Chemical compound OCC1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 VYGWQNWTGXPGLJ-UHFFFAOYSA-N 0.000 claims description 5
- IZKKGIDWKNLUKD-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-4-methyl-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1CN(C=2C=C3SC=NC3=CC=2)C(=O)N1C1=CN=CC=C1C IZKKGIDWKNLUKD-UHFFFAOYSA-N 0.000 claims description 5
- MABTYDJGQDAOJX-UHFFFAOYSA-N 1-(1h-indazol-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3NN=CC3=CC=2)CC1 MABTYDJGQDAOJX-UHFFFAOYSA-N 0.000 claims description 5
- JUARPOUGPBQVFA-UHFFFAOYSA-N 1-(3-amino-1h-indazol-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3NN=C(N)C3=CC=2)CC1 JUARPOUGPBQVFA-UHFFFAOYSA-N 0.000 claims description 5
- TZXFLSJQEVAYJS-UHFFFAOYSA-N 1-(3-methyl-2h-indazol-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C=1C=C2C(C)=NNC2=CC=1N(C1=O)CCN1C1=CN=CC=C1C TZXFLSJQEVAYJS-UHFFFAOYSA-N 0.000 claims description 5
- KGFBSQFLCVYNHF-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(5-methylthiophen-3-yl)imidazolidin-2-one Chemical compound S1C(C)=CC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 KGFBSQFLCVYNHF-UHFFFAOYSA-N 0.000 claims description 5
- KGHCNXUVMVGXGO-UHFFFAOYSA-N 1-[3-(difluoromethyl)-4-fluorophenyl]-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(C(F)=CC=2)C(F)F)CC1 KGHCNXUVMVGXGO-UHFFFAOYSA-N 0.000 claims description 5
- 125000004432 carbon atom Chemical group C* 0.000 claims description 5
- 239000012829 chemotherapy agent Substances 0.000 claims description 5
- ZSXYCWMTEFDCJY-UHFFFAOYSA-N 1-(4-chlorothieno[3,2-c]pyridin-3-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C3=C(Cl)N=CC=C3SC=2)CC1 ZSXYCWMTEFDCJY-UHFFFAOYSA-N 0.000 claims description 4
- KPZYFYMGXLMJIT-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(5-methylthiophen-2-yl)imidazolidin-2-one Chemical compound S1C(C)=CC=C1N1C(=O)N(C=2C(=CC=NC=2)C)CC1 KPZYFYMGXLMJIT-UHFFFAOYSA-N 0.000 claims description 4
- FJVWLBSZAIUEEO-UHFFFAOYSA-N 2-fluoro-5-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzonitrile Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(C(F)=CC=2)C#N)CC1 FJVWLBSZAIUEEO-UHFFFAOYSA-N 0.000 claims description 4
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims description 4
- VUEXWBATPUMKKC-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-(4-methoxypyridin-3-yl)imidazolidin-2-one Chemical compound COC1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 VUEXWBATPUMKKC-UHFFFAOYSA-N 0.000 claims description 3
- KJKGLIHTRDRMEQ-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-(difluoromethyl)pyridin-3-yl]imidazolidin-2-one Chemical compound FC(F)C1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 KJKGLIHTRDRMEQ-UHFFFAOYSA-N 0.000 claims description 3
- OKSLGMYNJOSMDB-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-(trifluoromethyl)pyridin-3-yl]imidazolidin-2-one Chemical compound FC(F)(F)C1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 OKSLGMYNJOSMDB-UHFFFAOYSA-N 0.000 claims description 3
- DSVBQBJKGCAMPS-UHFFFAOYSA-N 1-(1-benzothiophen-2-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2SC3=CC=CC=C3C=2)CC1 DSVBQBJKGCAMPS-UHFFFAOYSA-N 0.000 claims description 3
- GBOORIMELYGUBH-UHFFFAOYSA-N 1-(1-benzothiophen-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=CSC3=CC=2)CC1 GBOORIMELYGUBH-UHFFFAOYSA-N 0.000 claims description 3
- QOIKGKJCMCGGMZ-UHFFFAOYSA-N 1-(1-benzothiophen-5-yl)-3-pyridin-3-ylimidazolidin-2-one Chemical compound C1CN(C=2C=C3C=CSC3=CC=2)C(=O)N1C1=CC=CN=C1 QOIKGKJCMCGGMZ-UHFFFAOYSA-N 0.000 claims description 3
- IFZKKZHWFGSWMX-UHFFFAOYSA-N 1-(1-methylindazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=NN(C)C3=CC=2)CC1 IFZKKZHWFGSWMX-UHFFFAOYSA-N 0.000 claims description 3
- MEDAUGPDIHAEHS-UHFFFAOYSA-N 1-(1h-indazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=NNC3=CC=2)CC1 MEDAUGPDIHAEHS-UHFFFAOYSA-N 0.000 claims description 3
- BNOHQYZPSAGZIK-UHFFFAOYSA-N 1-(2,2-difluoro-1,3-benzodioxol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3OC(F)(F)OC3=CC=2)CC1 BNOHQYZPSAGZIK-UHFFFAOYSA-N 0.000 claims description 3
- WEAKNSAEJJMOFX-UHFFFAOYSA-N 1-(2,3-dihydro-1,4-benzodioxin-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3OCCOC3=CC=2)CC1 WEAKNSAEJJMOFX-UHFFFAOYSA-N 0.000 claims description 3
- JGNMCQJBWINJBW-UHFFFAOYSA-N 1-(2,3-dihydro-1-benzofuran-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3CCOC3=CC=2)CC1 JGNMCQJBWINJBW-UHFFFAOYSA-N 0.000 claims description 3
- JAGPHVFAEPSIMT-UHFFFAOYSA-N 1-(2,3-dihydro-1h-inden-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3CCCC3=CC=2)CC1 JAGPHVFAEPSIMT-UHFFFAOYSA-N 0.000 claims description 3
- AOBJYZSGNJYOIH-UHFFFAOYSA-N 1-(2-methoxypyridin-4-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=NC(OC)=CC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 AOBJYZSGNJYOIH-UHFFFAOYSA-N 0.000 claims description 3
- PCXRWRGMTAGRDC-UHFFFAOYSA-N 1-(2-methylpyridin-4-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=NC(C)=CC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 PCXRWRGMTAGRDC-UHFFFAOYSA-N 0.000 claims description 3
- CZIIROAAZWTZPQ-UHFFFAOYSA-N 1-(3,4-difluorophenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(F)C(F)=CC=2)CC1 CZIIROAAZWTZPQ-UHFFFAOYSA-N 0.000 claims description 3
- GOWSHNVJBJSUFW-UHFFFAOYSA-N 1-(3-amino-1h-indazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C(N)=NNC3=CC=2)CC1 GOWSHNVJBJSUFW-UHFFFAOYSA-N 0.000 claims description 3
- HCDVZLMWWFVJDC-UHFFFAOYSA-N 1-(3-chloro-4-fluorophenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(Cl)C(F)=CC=2)CC1 HCDVZLMWWFVJDC-UHFFFAOYSA-N 0.000 claims description 3
- ZTWAZISELGQBHC-UHFFFAOYSA-N 1-(3-chlorophenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(Cl)C=CC=2)CC1 ZTWAZISELGQBHC-UHFFFAOYSA-N 0.000 claims description 3
- RVXWPFRBHJOZMW-UHFFFAOYSA-N 1-(3-methoxyphenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound COC1=CC=CC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 RVXWPFRBHJOZMW-UHFFFAOYSA-N 0.000 claims description 3
- XOECIXDWPJLUFV-UHFFFAOYSA-N 1-(3-methyl-1,2-benzoxazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=C2C(C)=NOC2=CC=C1N(C1=O)CCN1C1=CN=CC=C1C XOECIXDWPJLUFV-UHFFFAOYSA-N 0.000 claims description 3
- XSPFQCQAZULVBG-UHFFFAOYSA-N 1-(3-methyl-1-benzofuran-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=C2C(C)=COC2=CC=C1N(C1=O)CCN1C1=CN=CC=C1C XSPFQCQAZULVBG-UHFFFAOYSA-N 0.000 claims description 3
- SIBMBTMHXWKETK-UHFFFAOYSA-N 1-(3-methylphenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=CC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 SIBMBTMHXWKETK-UHFFFAOYSA-N 0.000 claims description 3
- DCBOTUHXVJRIDF-UHFFFAOYSA-N 1-(4-chloro-3-fluorophenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(F)C(Cl)=CC=2)CC1 DCBOTUHXVJRIDF-UHFFFAOYSA-N 0.000 claims description 3
- JIEBKLNLPJUTTA-UHFFFAOYSA-N 1-(4-fluoro-3-methylphenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(C)C(F)=CC=2)CC1 JIEBKLNLPJUTTA-UHFFFAOYSA-N 0.000 claims description 3
- GESDEHAECKFTCZ-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-[3-(trifluoromethyl)phenyl]imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(C=CC=2)C(F)(F)F)CC1 GESDEHAECKFTCZ-UHFFFAOYSA-N 0.000 claims description 3
- TUKXNBDKPXOTCQ-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-phenylimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=CC=CC=2)CC1 TUKXNBDKPXOTCQ-UHFFFAOYSA-N 0.000 claims description 3
- JZDPGYYNJHNJMX-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-thieno[3,2-c]pyridin-2-ylimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2SC3=CC=NC=C3C=2)CC1 JZDPGYYNJHNJMX-UHFFFAOYSA-N 0.000 claims description 3
- YYGBZVNZWXECRU-UHFFFAOYSA-N 1-(5-chlorothiophen-2-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2SC(Cl)=CC=2)CC1 YYGBZVNZWXECRU-UHFFFAOYSA-N 0.000 claims description 3
- PGDPVFMKZRXLRQ-UHFFFAOYSA-N 1-[4-fluoro-3-(trifluoromethyl)phenyl]-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(C(F)=CC=2)C(F)(F)F)CC1 PGDPVFMKZRXLRQ-UHFFFAOYSA-N 0.000 claims description 3
- ZBXUQBQZTBEXAP-UHFFFAOYSA-N 1-[5-(difluoromethyl)thiophen-3-yl]-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(SC=2)C(F)F)CC1 ZBXUQBQZTBEXAP-UHFFFAOYSA-N 0.000 claims description 3
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 claims description 3
- LBBSFWPZSANPKV-UHFFFAOYSA-N 2-chloro-4-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzonitrile Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(Cl)C(C#N)=CC=2)CC1 LBBSFWPZSANPKV-UHFFFAOYSA-N 0.000 claims description 3
- PAFBAAWQMHARSA-UHFFFAOYSA-N 3-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzonitrile Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(C=CC=2)C#N)CC1 PAFBAAWQMHARSA-UHFFFAOYSA-N 0.000 claims description 3
- 125000004190 benzothiazol-2-yl group Chemical group [H]C1=C([H])C([H])=C2N=C(*)SC2=C1[H] 0.000 claims description 3
- 125000004540 benzothiazol-5-yl group Chemical group S1C=NC2=C1C=CC(=C2)* 0.000 claims description 3
- 125000000814 indol-3-yl group Chemical group [H]C1=C([H])C([H])=C2N([H])C([H])=C([*])C2=C1[H] 0.000 claims description 3
- 125000004531 indol-5-yl group Chemical group [H]N1C([H])=C([H])C2=C([H])C(*)=C([H])C([H])=C12 0.000 claims description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 3
- 125000004159 quinolin-2-yl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C([H])C(*)=NC2=C1[H] 0.000 claims description 3
- 125000004548 quinolin-3-yl group Chemical group N1=CC(=CC2=CC=CC=C12)* 0.000 claims description 3
- 125000004066 1-hydroxyethyl group Chemical group [H]OC([H])([*])C([H])([H])[H] 0.000 claims description 2
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 2
- 125000004179 3-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(Cl)=C1[H] 0.000 claims description 2
- 125000004207 3-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(OC([H])([H])[H])=C1[H] 0.000 claims description 2
- 125000004199 4-trifluoromethylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C(F)(F)F 0.000 claims description 2
- 125000000040 m-tolyl group Chemical group [H]C1=C([H])C(*)=C([H])C(=C1[H])C([H])([H])[H] 0.000 claims description 2
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 claims description 2
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 claims description 2
- GZDRZDYLAZDMNI-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-(4-ethylpyridin-3-yl)imidazolidin-2-one Chemical compound CCC1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 GZDRZDYLAZDMNI-UHFFFAOYSA-N 0.000 claims 2
- FAJLLVSYQLLMSZ-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-(1-hydroxyethyl)pyridin-3-yl]imidazolidin-2-one Chemical compound CC(O)C1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 FAJLLVSYQLLMSZ-UHFFFAOYSA-N 0.000 claims 2
- LADCXVZNJGIFEL-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-(morpholin-4-ylmethyl)pyridin-3-yl]imidazolidin-2-one Chemical compound O=C1N(C=2C=C3SC=NC3=CC=2)CCN1C1=CN=CC=C1CN1CCOCC1 LADCXVZNJGIFEL-UHFFFAOYSA-N 0.000 claims 2
- RPQVYBRVVKFDJX-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-(pyrrolidin-1-ylmethyl)pyridin-3-yl]imidazolidin-2-one Chemical compound O=C1N(C=2C=C3SC=NC3=CC=2)CCN1C1=CN=CC=C1CN1CCCC1 RPQVYBRVVKFDJX-UHFFFAOYSA-N 0.000 claims 2
- IKRCABNTGWHQFX-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-[(cyclopropylamino)methyl]pyridin-3-yl]imidazolidin-2-one Chemical compound O=C1N(C=2C=C3SC=NC3=CC=2)CCN1C1=CN=CC=C1CNC1CC1 IKRCABNTGWHQFX-UHFFFAOYSA-N 0.000 claims 2
- DPNKARXUFIIVDZ-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-[[(5-methyl-1h-pyrazol-3-yl)amino]methyl]pyridin-3-yl]imidazolidin-2-one Chemical compound N1C(C)=CC(NCC=2C(=CN=CC=2)N2C(N(CC2)C=2C=C3SC=NC3=CC=2)=O)=N1 DPNKARXUFIIVDZ-UHFFFAOYSA-N 0.000 claims 2
- MQBKOTVBYVNCAE-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-4,4-dimethyl-3-pyridin-3-ylimidazolidin-2-one Chemical compound CC1(C)CN(C=2C=C3SC=NC3=CC=2)C(=O)N1C1=CC=CN=C1 MQBKOTVBYVNCAE-UHFFFAOYSA-N 0.000 claims 2
- CINFOQPKFKSAGQ-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-4,5-dimethyl-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound O=C1N(C=2C=C3SC=NC3=CC=2)C(C)C(C)N1C1=CN=CC=C1C CINFOQPKFKSAGQ-UHFFFAOYSA-N 0.000 claims 2
- QRPMKVOSQKQWHH-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(4-methylthieno[3,2-c]pyridin-2-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2SC3=CC=NC(C)=C3C=2)CC1 QRPMKVOSQKQWHH-UHFFFAOYSA-N 0.000 claims 2
- GGNNSFBMCVXUPG-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-[3-(trifluoromethyl)-2h-indazol-6-yl]imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3NN=C(C3=CC=2)C(F)(F)F)CC1 GGNNSFBMCVXUPG-UHFFFAOYSA-N 0.000 claims 2
- RFWDXYNJLCSMGW-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-[5-(trifluoromethyl)thiophen-2-yl]imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2SC(=CC=2)C(F)(F)F)CC1 RFWDXYNJLCSMGW-UHFFFAOYSA-N 0.000 claims 2
- NUDAPMWIKRCEJW-UHFFFAOYSA-N 3-(1,3-benzothiazol-6-yl)-4-methyl-1-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound O=C1N(C=2C=C3SC=NC3=CC=2)C(C)CN1C1=CN=CC=C1C NUDAPMWIKRCEJW-UHFFFAOYSA-N 0.000 claims 2
- CTLOJRTZAGTVJG-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-(4-methylpyridin-3-yl)-4-(trifluoromethyl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1C(F)(F)F CTLOJRTZAGTVJG-UHFFFAOYSA-N 0.000 claims 1
- GBBPGYPMPYVHGF-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-(5-chloro-4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=C(Cl)C=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 GBBPGYPMPYVHGF-UHFFFAOYSA-N 0.000 claims 1
- FJTBZWVYMFORRM-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-(6-fluoro-4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC(F)=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 FJTBZWVYMFORRM-UHFFFAOYSA-N 0.000 claims 1
- PTMRRCYRWCHIPK-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-(2-hydroxypropan-2-yl)pyridin-3-yl]imidazolidin-2-one Chemical compound CC(C)(O)C1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 PTMRRCYRWCHIPK-UHFFFAOYSA-N 0.000 claims 1
- QKMWNBZEGYSPDD-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-(2-morpholin-4-ylethoxy)pyridin-3-yl]imidazolidin-2-one Chemical compound O=C1N(C=2C=C3SC=NC3=CC=2)CCN1C1=CN=CC=C1OCCN1CCOCC1 QKMWNBZEGYSPDD-UHFFFAOYSA-N 0.000 claims 1
- LEFYBCRSYAXHML-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-(dimethoxymethyl)pyridin-3-yl]imidazolidin-2-one Chemical compound COC(OC)C1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 LEFYBCRSYAXHML-UHFFFAOYSA-N 0.000 claims 1
- NBTFCPWXFQDHCV-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-[(1-methylpiperidin-4-yl)methoxy]pyridin-3-yl]imidazolidin-2-one Chemical compound C1CN(C)CCC1COC1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 NBTFCPWXFQDHCV-UHFFFAOYSA-N 0.000 claims 1
- IJKPHOXBXNGLMP-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-[(1-methylpyrrolidin-2-yl)methoxy]pyridin-3-yl]imidazolidin-2-one Chemical compound CN1CCCC1COC1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 IJKPHOXBXNGLMP-UHFFFAOYSA-N 0.000 claims 1
- PQDCNVLLXWMGJD-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-[4-methyl-5-(trifluoromethyl)pyridin-3-yl]imidazolidin-2-one Chemical compound C1=NC=C(C(F)(F)F)C(C)=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 PQDCNVLLXWMGJD-UHFFFAOYSA-N 0.000 claims 1
- KIRQPFUKELDEPO-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-pyridin-3-yl-4-(trifluoromethyl)imidazolidin-2-one Chemical compound FC(F)(F)C1CN(C=2C=C3SC=NC3=CC=2)C(=O)N1C1=CC=CN=C1 KIRQPFUKELDEPO-UHFFFAOYSA-N 0.000 claims 1
- WHUMCYYKWPLBAD-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-4,4-dimethyl-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(C)(C)CN(C=2C=C3SC=NC3=CC=2)C1=O WHUMCYYKWPLBAD-UHFFFAOYSA-N 0.000 claims 1
- PBODJZSDMJUSJM-UHFFFAOYSA-N 1-(2h-benzotriazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3N=NNC3=CC=2)CC1 PBODJZSDMJUSJM-UHFFFAOYSA-N 0.000 claims 1
- GQUYNCHMXBUDQG-UHFFFAOYSA-N 1-(3-chloroimidazo[1,2-a]pyridin-7-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C2=CC3=NC=C(Cl)N3C=C2)CC1 GQUYNCHMXBUDQG-UHFFFAOYSA-N 0.000 claims 1
- YJKLLJFVYCAPKY-UHFFFAOYSA-N 1-(3-cyclopropyl-2h-indazol-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3NN=C(C3=CC=2)C2CC2)CC1 YJKLLJFVYCAPKY-UHFFFAOYSA-N 0.000 claims 1
- QQYCBNXZMHEALT-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(1,2-thiazol-4-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C2=CSN=C2)CC1 QQYCBNXZMHEALT-UHFFFAOYSA-N 0.000 claims 1
- SHMNSOZXGJVYAJ-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-[2-(trifluoromethyl)pyridin-4-yl]imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(N=CC=2)C(F)(F)F)CC1 SHMNSOZXGJVYAJ-UHFFFAOYSA-N 0.000 claims 1
- UAKGVFTWFGKKTA-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-quinolin-7-ylimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3N=CC=CC3=CC=2)CC1 UAKGVFTWFGKKTA-UHFFFAOYSA-N 0.000 claims 1
- IQRRFSPJKPPNJW-UHFFFAOYSA-N 3-(1,3-benzothiazol-6-yl)-1-pyridin-3-yl-4-(trifluoromethyl)imidazolidin-2-one Chemical compound O=C1N(C=2C=C3SC=NC3=CC=2)C(C(F)(F)F)CN1C1=CC=CN=C1 IQRRFSPJKPPNJW-UHFFFAOYSA-N 0.000 claims 1
- SHGNERDGXZEUBB-UHFFFAOYSA-N 3-(1,3-benzothiazol-6-yl)-4,4-dimethyl-1-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)C(C)(C)C1 SHGNERDGXZEUBB-UHFFFAOYSA-N 0.000 claims 1
- KJQBFBVWMPTTNR-UHFFFAOYSA-N 3-[3-(1,3-benzothiazol-6-yl)-2-oxoimidazolidin-1-yl]pyridine-4-carboxamide Chemical compound NC(=O)C1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 KJQBFBVWMPTTNR-UHFFFAOYSA-N 0.000 claims 1
- 102000001854 Steroid 17-alpha-Hydroxylase Human genes 0.000 abstract description 10
- 108010015330 Steroid 17-alpha-Hydroxylase Proteins 0.000 abstract description 10
- 239000002697 lyase inhibitor Substances 0.000 abstract description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 543
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 306
- 238000006243 chemical reaction Methods 0.000 description 207
- 238000002360 preparation method Methods 0.000 description 194
- 238000005160 1H NMR spectroscopy Methods 0.000 description 174
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 168
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 162
- 238000004128 high performance liquid chromatography Methods 0.000 description 147
- 239000000047 product Substances 0.000 description 144
- 238000000746 purification Methods 0.000 description 141
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 140
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 138
- 238000004440 column chromatography Methods 0.000 description 135
- 239000000741 silica gel Substances 0.000 description 135
- 229910002027 silica gel Inorganic materials 0.000 description 135
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 134
- SSJXIUAHEKJCMH-PHDIDXHHSA-N (1r,2r)-cyclohexane-1,2-diamine Chemical compound N[C@@H]1CCCC[C@H]1N SSJXIUAHEKJCMH-PHDIDXHHSA-N 0.000 description 133
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 130
- 239000012043 crude product Substances 0.000 description 127
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 124
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 112
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 110
- HDBCTDNLDAMDMZ-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)NCC1 HDBCTDNLDAMDMZ-UHFFFAOYSA-N 0.000 description 107
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 86
- 229910000160 potassium phosphate Inorganic materials 0.000 description 81
- 235000011009 potassium phosphates Nutrition 0.000 description 81
- 239000011541 reaction mixture Substances 0.000 description 77
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 57
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 57
- 238000004809 thin layer chromatography Methods 0.000 description 57
- 229910000027 potassium carbonate Inorganic materials 0.000 description 55
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 51
- 239000000243 solution Substances 0.000 description 50
- 239000000203 mixture Substances 0.000 description 47
- 230000000694 effects Effects 0.000 description 46
- 230000002829 reductive effect Effects 0.000 description 42
- 235000019439 ethyl acetate Nutrition 0.000 description 39
- 239000000543 intermediate Substances 0.000 description 38
- 229940093499 ethyl acetate Drugs 0.000 description 34
- 238000002953 preparative HPLC Methods 0.000 description 31
- 239000012044 organic layer Substances 0.000 description 30
- 238000011282 treatment Methods 0.000 description 28
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 27
- 238000000034 method Methods 0.000 description 27
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 26
- 239000007832 Na2SO4 Substances 0.000 description 24
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 24
- 229910052938 sodium sulfate Inorganic materials 0.000 description 24
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 24
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 21
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 21
- 206010028980 Neoplasm Diseases 0.000 description 20
- 230000003247 decreasing effect Effects 0.000 description 20
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 19
- 239000005457 ice water Substances 0.000 description 18
- 230000002401 inhibitory effect Effects 0.000 description 18
- 239000002904 solvent Substances 0.000 description 18
- 230000008685 targeting Effects 0.000 description 18
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 17
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 16
- 201000011510 cancer Diseases 0.000 description 15
- GLBVHFJJWWZJHD-UHFFFAOYSA-N 1-pyridin-3-ylimidazolidin-2-one Chemical compound O=C1NCCN1C1=CC=CN=C1 GLBVHFJJWWZJHD-UHFFFAOYSA-N 0.000 description 13
- 108090000623 proteins and genes Proteins 0.000 description 13
- JUJWROOIHBZHMG-UHFFFAOYSA-N pyridine Substances C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 13
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 12
- 102000004169 proteins and genes Human genes 0.000 description 12
- 229910000104 sodium hydride Inorganic materials 0.000 description 12
- 206010060862 Prostate cancer Diseases 0.000 description 11
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 11
- 239000012267 brine Substances 0.000 description 11
- 208000035475 disorder Diseases 0.000 description 11
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 11
- 201000010099 disease Diseases 0.000 description 10
- 239000002244 precipitate Substances 0.000 description 10
- 238000010992 reflux Methods 0.000 description 10
- 238000003786 synthesis reaction Methods 0.000 description 10
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 125000000217 alkyl group Chemical group 0.000 description 9
- 239000012141 concentrate Substances 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- 239000007858 starting material Substances 0.000 description 9
- MOXQDTRQWCBEDW-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)imidazolidin-2-one Chemical compound O=C1NCCN1C1=CC=C(N=CS2)C2=C1 MOXQDTRQWCBEDW-UHFFFAOYSA-N 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 8
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 8
- 239000002253 acid Substances 0.000 description 8
- 239000003098 androgen Substances 0.000 description 8
- 229910052786 argon Inorganic materials 0.000 description 8
- 239000003814 drug Substances 0.000 description 8
- 239000012299 nitrogen atmosphere Substances 0.000 description 8
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 7
- LSVSEOAYZBNKSB-UHFFFAOYSA-N 1-naphthalen-2-ylimidazolidin-2-one Chemical compound O=C1NCCN1C1=CC=C(C=CC=C2)C2=C1 LSVSEOAYZBNKSB-UHFFFAOYSA-N 0.000 description 7
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 7
- 150000002632 lipids Chemical class 0.000 description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 7
- 239000012312 sodium hydride Substances 0.000 description 7
- OUSUJFZWTWVGKW-UHFFFAOYSA-N 2-fluoro-4-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzonitrile Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(F)C(C#N)=CC=2)CC1 OUSUJFZWTWVGKW-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- 208000026310 Breast neoplasm Diseases 0.000 description 6
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 6
- 108091000080 Phosphotransferase Proteins 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 238000005859 coupling reaction Methods 0.000 description 6
- 239000000706 filtrate Substances 0.000 description 6
- 238000009472 formulation Methods 0.000 description 6
- 229940088597 hormone Drugs 0.000 description 6
- 239000005556 hormone Substances 0.000 description 6
- 229910052760 oxygen Inorganic materials 0.000 description 6
- 102000020233 phosphotransferase Human genes 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 239000012258 stirred mixture Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 229960003604 testosterone Drugs 0.000 description 6
- WORJRXHJTUTINR-UHFFFAOYSA-N 1,4-dioxane;hydron;chloride Chemical compound Cl.C1COCCO1 WORJRXHJTUTINR-UHFFFAOYSA-N 0.000 description 5
- LBKOHRGUJNDJCO-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-(4-methylpyridin-3-yl)imidazol-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)C=C1 LBKOHRGUJNDJCO-UHFFFAOYSA-N 0.000 description 5
- KMFDWGDCHCZPIC-UHFFFAOYSA-N 1-(3-methyl-1,2-benzoxazol-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C=1C=C2C(C)=NOC2=CC=1N(C1=O)CCN1C1=CN=CC=C1C KMFDWGDCHCZPIC-UHFFFAOYSA-N 0.000 description 5
- JRSVKXSWTDWIIX-UHFFFAOYSA-N 1-(4-acetyl-3-fluorophenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=C(F)C(C(=O)C)=CC=C1N1C(=O)N(C=2C(=CC=NC=2)C)CC1 JRSVKXSWTDWIIX-UHFFFAOYSA-N 0.000 description 5
- HXXGJLCTRJXISH-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(5-methylthiophen-3-yl)imidazol-2-one Chemical compound S1C(C)=CC(N2C(N(C=3C(=CC=NC=3)C)C=C2)=O)=C1 HXXGJLCTRJXISH-UHFFFAOYSA-N 0.000 description 5
- NGAZLZMOFNRHJR-UHFFFAOYSA-N 1-(5-chloro-1-methylindol-3-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C3=CC(Cl)=CC=C3N(C)C=2)CC1 NGAZLZMOFNRHJR-UHFFFAOYSA-N 0.000 description 5
- AHJNCRDRCJNDHW-UHFFFAOYSA-N 1-[3-(difluoromethyl)-4-fluorophenyl]-3-(4-methylpyridin-3-yl)imidazol-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(C(F)=CC=2)C(F)F)C=C1 AHJNCRDRCJNDHW-UHFFFAOYSA-N 0.000 description 5
- BCMYXYHEMGPZJN-UHFFFAOYSA-N 1-chloro-2-isocyanatoethane Chemical compound ClCCN=C=O BCMYXYHEMGPZJN-UHFFFAOYSA-N 0.000 description 5
- ZMGMDXCADSRNCX-UHFFFAOYSA-N 5,6-dihydroxy-1,3-diazepan-2-one Chemical class OC1CNC(=O)NCC1O ZMGMDXCADSRNCX-UHFFFAOYSA-N 0.000 description 5
- 238000006443 Buchwald-Hartwig cross coupling reaction Methods 0.000 description 5
- 239000004215 Carbon black (E152) Substances 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 108091008606 PDGF receptors Proteins 0.000 description 5
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 5
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 5
- 102000011653 Platelet-Derived Growth Factor Receptors Human genes 0.000 description 5
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 230000008878 coupling Effects 0.000 description 5
- 238000010168 coupling process Methods 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 125000005842 heteroatom Chemical group 0.000 description 5
- 229930195733 hydrocarbon Natural products 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 230000005865 ionizing radiation Effects 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 230000001404 mediated effect Effects 0.000 description 5
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 5
- 238000001959 radiotherapy Methods 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 150000003431 steroids Chemical class 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 229910052717 sulfur Inorganic materials 0.000 description 5
- AYUNIORJHRXIBJ-TXHRRWQRSA-N tanespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCC=C)C(=O)C=C1C2=O AYUNIORJHRXIBJ-TXHRRWQRSA-N 0.000 description 5
- 238000002560 therapeutic procedure Methods 0.000 description 5
- GQBQWKUYLDYPTJ-UHFFFAOYSA-N 1-(1,2-benzoxazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=NOC3=CC=2)CC1 GQBQWKUYLDYPTJ-UHFFFAOYSA-N 0.000 description 4
- AMPKNMFXWAMUCG-UHFFFAOYSA-N 1-(1h-imidazo[4,5-b]pyridin-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CC1=CC=NC=C1N1C(=O)N(C=2C=C3N=CNC3=NC=2)CC1 AMPKNMFXWAMUCG-UHFFFAOYSA-N 0.000 description 4
- MIJIZJMABDAXST-UHFFFAOYSA-N 1-(1h-indol-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3NC=CC3=CC=2)CC1 MIJIZJMABDAXST-UHFFFAOYSA-N 0.000 description 4
- CROYYKZCEYARHH-UHFFFAOYSA-N 1-(2h-benzotriazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one;hydrochloride Chemical compound Cl.CC1=CC=NC=C1N1C(=O)N(C=2C=C3N=NNC3=CC=2)CC1 CROYYKZCEYARHH-UHFFFAOYSA-N 0.000 description 4
- DTVARKQDWVXWDK-UHFFFAOYSA-N 1-(3-acetyl-4-fluorophenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=C(F)C(C(=O)C)=CC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 DTVARKQDWVXWDK-UHFFFAOYSA-N 0.000 description 4
- YDZTUHYCYHHHQT-UHFFFAOYSA-N 1-(3-methylphenyl)-3-[4-(trifluoromethyl)pyridin-3-yl]imidazolidin-2-one Chemical compound CC1=CC=CC(N2C(N(CC2)C=2C(=CC=NC=2)C(F)(F)F)=O)=C1 YDZTUHYCYHHHQT-UHFFFAOYSA-N 0.000 description 4
- BITVQGQQNABWAO-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-naphthalen-2-yl-1,3-diazinan-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=CC=CC3=CC=2)CCC1 BITVQGQQNABWAO-UHFFFAOYSA-N 0.000 description 4
- LAJDGEFZYMPFJK-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-thieno[3,2-c]pyridin-3-ylimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C3=CN=CC=C3SC=2)CC1 LAJDGEFZYMPFJK-UHFFFAOYSA-N 0.000 description 4
- GPMUJAVEZAZKMB-UHFFFAOYSA-N 1-[3-fluoro-4-(methoxyiminomethyl)phenyl]-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=C(F)C(C=NOC)=CC=C1N1C(=O)N(C=2C(=CC=NC=2)C)CC1 GPMUJAVEZAZKMB-UHFFFAOYSA-N 0.000 description 4
- HJWJDMRAWKYGFO-UHFFFAOYSA-N 1-[3-fluoro-4-(n-hydroxy-c-methylcarbonimidoyl)phenyl]-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=C(F)C(C(=NO)C)=CC=C1N1C(=O)N(C=2C(=CC=NC=2)C)CC1 HJWJDMRAWKYGFO-UHFFFAOYSA-N 0.000 description 4
- YJZSUCFGHXQWDM-UHFFFAOYSA-N 1-adamantyl 4-[(2,5-dihydroxyphenyl)methylamino]benzoate Chemical compound OC1=CC=C(O)C(CNC=2C=CC(=CC=2)C(=O)OC23CC4CC(CC(C4)C2)C3)=C1 YJZSUCFGHXQWDM-UHFFFAOYSA-N 0.000 description 4
- ZYYBWMKJCUYWFU-UHFFFAOYSA-N 1-naphthalen-2-yl-3-pyridin-3-yl-1,3-diazinan-2-one Chemical compound C1CCN(C=2C=C3C=CC=CC3=CC=2)C(=O)N1C1=CC=CN=C1 ZYYBWMKJCUYWFU-UHFFFAOYSA-N 0.000 description 4
- HWBMBCXNCGQDJS-UHFFFAOYSA-N 1-pyridin-3-yl-1,3-diazinan-2-one Chemical compound O=C1NCCCN1C1=CC=CN=C1 HWBMBCXNCGQDJS-UHFFFAOYSA-N 0.000 description 4
- APSMUYYLXZULMS-UHFFFAOYSA-N 2-bromonaphthalene Chemical compound C1=CC=CC2=CC(Br)=CC=C21 APSMUYYLXZULMS-UHFFFAOYSA-N 0.000 description 4
- CUYKNJBYIJFRCU-UHFFFAOYSA-N 3-aminopyridine Chemical compound NC1=CC=CN=C1 CUYKNJBYIJFRCU-UHFFFAOYSA-N 0.000 description 4
- IDDJAHXOKSVCFQ-UHFFFAOYSA-N 5-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]-1,2-dihydroindazol-3-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C(=O)NNC3=CC=2)CC1 IDDJAHXOKSVCFQ-UHFFFAOYSA-N 0.000 description 4
- UWZZMRWDOPBSNU-UHFFFAOYSA-N 6-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]-1,2-dihydroindazol-3-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C(C(NN3)=O)=CC=2)CC1 UWZZMRWDOPBSNU-UHFFFAOYSA-N 0.000 description 4
- 206010006187 Breast cancer Diseases 0.000 description 4
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 4
- 108010069236 Goserelin Proteins 0.000 description 4
- 101710113864 Heat shock protein 90 Proteins 0.000 description 4
- 102100034051 Heat shock protein HSP 90-alpha Human genes 0.000 description 4
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 4
- 102000014150 Interferons Human genes 0.000 description 4
- 108010050904 Interferons Proteins 0.000 description 4
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 4
- 101710181812 Methionine aminopeptidase Proteins 0.000 description 4
- 102000029749 Microtubule Human genes 0.000 description 4
- 108091022875 Microtubule Proteins 0.000 description 4
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- 229930012538 Paclitaxel Natural products 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 4
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 150000001721 carbon Chemical group 0.000 description 4
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 229940127089 cytotoxic agent Drugs 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 4
- 229940011871 estrogen Drugs 0.000 description 4
- 239000000262 estrogen Substances 0.000 description 4
- 229960002949 fluorouracil Drugs 0.000 description 4
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 4
- 150000002430 hydrocarbons Chemical class 0.000 description 4
- 210000004688 microtubule Anatomy 0.000 description 4
- BMGQWWVMWDBQGC-IIFHNQTCSA-N midostaurin Chemical compound CN([C@H]1[C@H]([C@]2(C)O[C@@H](N3C4=CC=CC=C4C4=C5C(=O)NCC5=C5C6=CC=CC=C6N2C5=C43)C1)OC)C(=O)C1=CC=CC=C1 BMGQWWVMWDBQGC-IIFHNQTCSA-N 0.000 description 4
- 229950010895 midostaurin Drugs 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- 229960001592 paclitaxel Drugs 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 108010014186 ras Proteins Proteins 0.000 description 4
- 102000016914 ras Proteins Human genes 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 230000003637 steroidlike Effects 0.000 description 4
- 208000011580 syndromic disease Diseases 0.000 description 4
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 4
- 229940086542 triethylamine Drugs 0.000 description 4
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 4
- SOAYXRXPIYDUFV-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-(2,2-dimethoxyethyl)urea Chemical compound COC(OC)CNC(=O)NC1=CC=C2N=CSC2=C1 SOAYXRXPIYDUFV-UHFFFAOYSA-N 0.000 description 3
- GYPRMMAEQJYOPC-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-(2-chloroethyl)urea Chemical compound ClCCNC(=O)NC1=CC=C2N=CSC2=C1 GYPRMMAEQJYOPC-UHFFFAOYSA-N 0.000 description 3
- MNQWCUSURHKAFL-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-(4-chloropyridin-3-yl)imidazolidin-2-one Chemical compound ClC1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 MNQWCUSURHKAFL-UHFFFAOYSA-N 0.000 description 3
- DPCCOLGRQDTVPS-UHFFFAOYSA-N 1-(1-benzothiophen-5-yl)-3-(4-methylpyridin-3-yl)imidazol-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=CSC3=CC=2)C=C1 DPCCOLGRQDTVPS-UHFFFAOYSA-N 0.000 description 3
- BKZMPSSJZDOLEJ-UHFFFAOYSA-N 1-(2-chloro-4-methylquinolin-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C(C)=CC(Cl)=NC3=CC=2)CC1 BKZMPSSJZDOLEJ-UHFFFAOYSA-N 0.000 description 3
- YFJQMNIZVXYKSD-UHFFFAOYSA-N 1-(2-chloroethyl)-3-(3-methylphenyl)urea Chemical compound CC1=CC=CC(NC(=O)NCCCl)=C1 YFJQMNIZVXYKSD-UHFFFAOYSA-N 0.000 description 3
- YYRSVEZMTFUMBT-UHFFFAOYSA-N 1-(2-chloroethyl)-3-(4-methylpyridin-3-yl)urea Chemical compound CC1=CC=NC=C1NC(=O)NCCCl YYRSVEZMTFUMBT-UHFFFAOYSA-N 0.000 description 3
- BMPINTUZIXWLFQ-UHFFFAOYSA-N 1-(2-chloroethyl)-3-naphthalen-2-ylurea Chemical compound C1=CC=CC2=CC(NC(=O)NCCCl)=CC=C21 BMPINTUZIXWLFQ-UHFFFAOYSA-N 0.000 description 3
- GHKZUTAGOUFZGD-UHFFFAOYSA-N 1-(2-chloroethyl)-3-pyridin-3-ylurea Chemical compound ClCCNC(=O)NC1=CC=CN=C1 GHKZUTAGOUFZGD-UHFFFAOYSA-N 0.000 description 3
- KPMDRICDSUOEQZ-UHFFFAOYSA-N 1-(3-chloropropyl)-3-(4-methylpyridin-3-yl)urea Chemical compound CC1=CC=NC=C1NC(=O)NCCCCl KPMDRICDSUOEQZ-UHFFFAOYSA-N 0.000 description 3
- VSKHEVNSMQFOLW-UHFFFAOYSA-N 1-(3-chloropropyl)-3-pyridin-3-ylurea Chemical compound ClCCCNC(=O)NC1=CC=CN=C1 VSKHEVNSMQFOLW-UHFFFAOYSA-N 0.000 description 3
- PGTOGOCRLGQJNM-UHFFFAOYSA-N 1-(3-methylphenyl)imidazolidin-2-one Chemical compound CC1=CC=CC(N2C(NCC2)=O)=C1 PGTOGOCRLGQJNM-UHFFFAOYSA-N 0.000 description 3
- QKJOMYQSLGEGMY-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-1,3-diazinan-2-one Chemical compound CC1=CC=NC=C1N1C(=O)NCCC1 QKJOMYQSLGEGMY-UHFFFAOYSA-N 0.000 description 3
- URLAZVMYVWYOOJ-UHFFFAOYSA-N 1-[4-fluoro-3-(hydroxyiminomethyl)phenyl]-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(C=NO)C(F)=CC=2)CC1 URLAZVMYVWYOOJ-UHFFFAOYSA-N 0.000 description 3
- PDEIOZIVPZNHTN-UHFFFAOYSA-N 1-naphthalen-2-yl-3-[5-(trifluoromethyl)pyridin-3-yl]imidazolidin-2-one Chemical compound FC(F)(F)C1=CN=CC(N2C(N(CC2)C=2C=C3C=CC=CC3=CC=2)=O)=C1 PDEIOZIVPZNHTN-UHFFFAOYSA-N 0.000 description 3
- KAOVUWAWOTVISB-UHFFFAOYSA-N 2-fluoro-5-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzaldehyde Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(C=O)C(F)=CC=2)CC1 KAOVUWAWOTVISB-UHFFFAOYSA-N 0.000 description 3
- CTBHUHPXVVGSNY-UHFFFAOYSA-N 3-(1,3-benzothiazol-6-yl)-1h-imidazol-2-one Chemical compound O=C1NC=CN1C1=CC=C(N=CS2)C2=C1 CTBHUHPXVVGSNY-UHFFFAOYSA-N 0.000 description 3
- WGWQVSSYRQZYBU-UHFFFAOYSA-N 3-bromo-5-chloro-1-methylindole Chemical compound ClC1=CC=C2N(C)C=C(Br)C2=C1 WGWQVSSYRQZYBU-UHFFFAOYSA-N 0.000 description 3
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 3
- OGWKCGZFUXNPDA-CFWMRBGOSA-N 5j49q6b70f Chemical compound C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C=O)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 OGWKCGZFUXNPDA-CFWMRBGOSA-N 0.000 description 3
- 108091006112 ATPases Proteins 0.000 description 3
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 3
- 102000057290 Adenosine Triphosphatases Human genes 0.000 description 3
- OGSPWJRAVKPPFI-UHFFFAOYSA-N Alendronic Acid Chemical compound NCCCC(O)(P(O)(O)=O)P(O)(O)=O OGSPWJRAVKPPFI-UHFFFAOYSA-N 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 3
- 101150051357 CYP17A1 gene Proteins 0.000 description 3
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 3
- 102000001301 EGF receptor Human genes 0.000 description 3
- 108060006698 EGF receptor Proteins 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 3
- NMJREATYWWNIKX-UHFFFAOYSA-N GnRH Chemical class C1CCC(C(=O)NCC(N)=O)N1C(=O)C(CC(C)C)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)CNC(=O)C(NC(=O)C(CO)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C(CC=1NC=NC=1)NC(=O)C1NC(=O)CC1)CC1=CC=C(O)C=C1 NMJREATYWWNIKX-UHFFFAOYSA-N 0.000 description 3
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 3
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 108010014608 Proto-Oncogene Proteins c-kit Proteins 0.000 description 3
- 102000016971 Proto-Oncogene Proteins c-kit Human genes 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 229940127395 Ribonucleotide Reductase Inhibitors Drugs 0.000 description 3
- IIDJRNMFWXDHID-UHFFFAOYSA-N Risedronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CC1=CC=CN=C1 IIDJRNMFWXDHID-UHFFFAOYSA-N 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- 108010017842 Telomerase Proteins 0.000 description 3
- DKJJVAGXPKPDRL-UHFFFAOYSA-N Tiludronic acid Chemical compound OP(O)(=O)C(P(O)(O)=O)SC1=CC=C(Cl)C=C1 DKJJVAGXPKPDRL-UHFFFAOYSA-N 0.000 description 3
- 108010053096 Vascular Endothelial Growth Factor Receptor-1 Proteins 0.000 description 3
- 102000016548 Vascular Endothelial Growth Factor Receptor-1 Human genes 0.000 description 3
- 230000001919 adrenal effect Effects 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- KUFRQPKVAWMTJO-LMZWQJSESA-N alvespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCCN(C)C)C(=O)C=C1C2=O KUFRQPKVAWMTJO-LMZWQJSESA-N 0.000 description 3
- 229960003437 aminoglutethimide Drugs 0.000 description 3
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 3
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 3
- 230000001772 anti-angiogenic effect Effects 0.000 description 3
- 229940046836 anti-estrogen Drugs 0.000 description 3
- 230000001833 anti-estrogenic effect Effects 0.000 description 3
- 239000003886 aromatase inhibitor Substances 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 3
- 150000001540 azides Chemical class 0.000 description 3
- 229960000397 bevacizumab Drugs 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000003246 corticosteroid Substances 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 229960004132 diethyl ether Drugs 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 229960003668 docetaxel Drugs 0.000 description 3
- HESCAJZNRMSMJG-HGYUPSKWSA-N epothilone A Natural products O=C1[C@H](C)[C@H](O)[C@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C HESCAJZNRMSMJG-HGYUPSKWSA-N 0.000 description 3
- GTTBEUCJPZQMDZ-UHFFFAOYSA-N erlotinib hydrochloride Chemical compound [H+].[Cl-].C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 GTTBEUCJPZQMDZ-UHFFFAOYSA-N 0.000 description 3
- 239000000328 estrogen antagonist Substances 0.000 description 3
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 3
- 229960005420 etoposide Drugs 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 229960004421 formestane Drugs 0.000 description 3
- OSVMTWJCGUFAOD-KZQROQTASA-N formestane Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1O OSVMTWJCGUFAOD-KZQROQTASA-N 0.000 description 3
- 229960005277 gemcitabine Drugs 0.000 description 3
- 239000003481 heat shock protein 90 inhibitor Substances 0.000 description 3
- 230000003054 hormonal effect Effects 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 229960000908 idarubicin Drugs 0.000 description 3
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 3
- 229960001101 ifosfamide Drugs 0.000 description 3
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 3
- 229960002411 imatinib Drugs 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 3
- 239000012948 isocyanate Substances 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 229960001428 mercaptopurine Drugs 0.000 description 3
- NWUCJCJMZFLGAF-UHFFFAOYSA-N methyl 2-fluoro-4-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzoate Chemical compound C1=C(F)C(C(=O)OC)=CC=C1N1C(=O)N(C=2C(=CC=NC=2)C)CC1 NWUCJCJMZFLGAF-UHFFFAOYSA-N 0.000 description 3
- BLWFDUMFGUWLIL-UHFFFAOYSA-N methyl 2-fluoro-5-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzoate Chemical compound C1=C(F)C(C(=O)OC)=CC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 BLWFDUMFGUWLIL-UHFFFAOYSA-N 0.000 description 3
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 3
- 229960001156 mitoxantrone Drugs 0.000 description 3
- YKVGKDCHZSPGJK-UHFFFAOYSA-N n-[3-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]phenyl]acetamide Chemical compound CC(=O)NC1=CC=CC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 YKVGKDCHZSPGJK-UHFFFAOYSA-N 0.000 description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 description 3
- 230000002246 oncogenic effect Effects 0.000 description 3
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 3
- 238000002428 photodynamic therapy Methods 0.000 description 3
- 229940002612 prodrug Drugs 0.000 description 3
- 239000000651 prodrug Substances 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000011593 sulfur Substances 0.000 description 3
- 238000001356 surgical procedure Methods 0.000 description 3
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 3
- 229960001278 teniposide Drugs 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 3
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 3
- 229960003048 vinblastine Drugs 0.000 description 3
- XRASPMIURGNCCH-UHFFFAOYSA-N zoledronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CN1C=CN=C1 XRASPMIURGNCCH-UHFFFAOYSA-N 0.000 description 3
- DNXHEGUUPJUMQT-UHFFFAOYSA-N (+)-estrone Natural products OC1=CC=C2C3CCC(C)(C(CC4)=O)C4C3CCC2=C1 DNXHEGUUPJUMQT-UHFFFAOYSA-N 0.000 description 2
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 2
- NBRQRXRBIHVLGI-OWXODZSWSA-N (4as,5ar,12ar)-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1C2=CC=CC(O)=C2C(O)=C(C2=O)[C@@H]1C[C@@H]1[C@@]2(O)C(O)=C(C(=O)N)C(=O)C1 NBRQRXRBIHVLGI-OWXODZSWSA-N 0.000 description 2
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 2
- UMGQVUWXNOJOSJ-KMHUVPDISA-N (e)-2-cyano-3-(3,4-dihydroxyphenyl)-n-[(1r)-1-phenylethyl]prop-2-enamide Chemical compound N([C@H](C)C=1C=CC=CC=1)C(=O)C(\C#N)=C\C1=CC=C(O)C(O)=C1 UMGQVUWXNOJOSJ-KMHUVPDISA-N 0.000 description 2
- FAYAYUOZWYJNBD-UHFFFAOYSA-N 1,3-benzothiazol-6-amine Chemical compound NC1=CC=C2N=CSC2=C1 FAYAYUOZWYJNBD-UHFFFAOYSA-N 0.000 description 2
- GXLGBJPOQCGOSM-UHFFFAOYSA-N 1-(1,3-benzothiazol-2-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2SC3=CC=CC=C3N=2)CC1 GXLGBJPOQCGOSM-UHFFFAOYSA-N 0.000 description 2
- GQQIGOJINAFGNK-UHFFFAOYSA-N 1-(1,3-benzothiazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3N=CSC3=CC=2)CC1 GQQIGOJINAFGNK-UHFFFAOYSA-N 0.000 description 2
- QAUQTQYMZJFHKG-UHFFFAOYSA-N 1-(1,3-benzothiazol-5-yl)-3-pyridin-3-ylimidazolidin-2-one Chemical compound C1CN(C=2C=C3N=CSC3=CC=2)C(=O)N1C1=CC=CN=C1 QAUQTQYMZJFHKG-UHFFFAOYSA-N 0.000 description 2
- SUAPMUGGPOEIDD-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-(6-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=NC(C)=CC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 SUAPMUGGPOEIDD-UHFFFAOYSA-N 0.000 description 2
- AILLYIWQMZKBOI-UHFFFAOYSA-N 1-(1,3-benzothiazol-6-yl)-3-pyridin-3-ylimidazolidin-2-one Chemical compound C1CN(C=2C=C3SC=NC3=CC=2)C(=O)N1C1=CC=CN=C1 AILLYIWQMZKBOI-UHFFFAOYSA-N 0.000 description 2
- YHXUENJXVZJOLK-UHFFFAOYSA-N 1-(1,3-benzoxazol-2-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2OC3=CC=CC=C3N=2)CC1 YHXUENJXVZJOLK-UHFFFAOYSA-N 0.000 description 2
- CQTYBUUVAZPISX-UHFFFAOYSA-N 1-(1-benzothiophen-3-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C3=CC=CC=C3SC=2)CC1 CQTYBUUVAZPISX-UHFFFAOYSA-N 0.000 description 2
- KOMPRMUTJWNJGU-UHFFFAOYSA-N 1-(1-benzothiophen-5-yl)-3-pyridin-3-yl-1,3-diazinan-2-one Chemical compound C1CCN(C=2C=C3C=CSC3=CC=2)C(=O)N1C1=CC=CN=C1 KOMPRMUTJWNJGU-UHFFFAOYSA-N 0.000 description 2
- BLEQHKNJEKXQOO-UHFFFAOYSA-N 1-(1-ethylindol-5-yl)-3-pyridin-3-ylimidazolidin-2-one Chemical compound C=1C=C2N(CC)C=CC2=CC=1N(C1=O)CCN1C1=CC=CN=C1 BLEQHKNJEKXQOO-UHFFFAOYSA-N 0.000 description 2
- AHVQOQXQPBWPAP-UHFFFAOYSA-N 1-(1-methylindol-3-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CC1=CC=NC=C1N1C(=O)N(C=2C3=CC=CC=C3N(C)C=2)CC1 AHVQOQXQPBWPAP-UHFFFAOYSA-N 0.000 description 2
- FYAJFCYSQWLUJV-UHFFFAOYSA-N 1-(1-methylindol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=CN(C)C3=CC=2)CC1 FYAJFCYSQWLUJV-UHFFFAOYSA-N 0.000 description 2
- PRHBACYSLZWPDN-UHFFFAOYSA-N 1-(1h-indol-3-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C3=CC=CC=C3NC=2)CC1 PRHBACYSLZWPDN-UHFFFAOYSA-N 0.000 description 2
- NPUZTWSFCQVBBV-UHFFFAOYSA-N 1-(2,4-difluorophenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C(=CC(F)=CC=2)F)CC1 NPUZTWSFCQVBBV-UHFFFAOYSA-N 0.000 description 2
- WFWLLNUQWVEZOY-UHFFFAOYSA-N 1-(2-chloro-4-methylquinolin-6-yl)-3-pyridin-3-ylimidazolidin-2-one Chemical compound C1=C2C(C)=CC(Cl)=NC2=CC=C1N(C1=O)CCN1C1=CC=CN=C1 WFWLLNUQWVEZOY-UHFFFAOYSA-N 0.000 description 2
- NNFISSVSBCSXJT-UHFFFAOYSA-N 1-(2-chloropyrimidin-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=NC(Cl)=NC=2)CC1 NNFISSVSBCSXJT-UHFFFAOYSA-N 0.000 description 2
- VWNVIVAZXAHJFY-UHFFFAOYSA-N 1-(2-methyl-1,3-benzothiazol-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=C2SC(C)=NC2=CC=C1N(C1=O)CCN1C1=CN=CC=C1C VWNVIVAZXAHJFY-UHFFFAOYSA-N 0.000 description 2
- HDDZDWFXEIUJGZ-UHFFFAOYSA-N 1-(2-methyl-1,3-benzoxazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C=1C=C2OC(C)=NC2=CC=1N(C1=O)CCN1C1=CN=CC=C1C HDDZDWFXEIUJGZ-UHFFFAOYSA-N 0.000 description 2
- LTKWZFZWLLMHMY-UHFFFAOYSA-N 1-(2-methylindazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C2=CC3=CN(C)N=C3C=C2)CC1 LTKWZFZWLLMHMY-UHFFFAOYSA-N 0.000 description 2
- JCUSTPJFYZIYLZ-UHFFFAOYSA-N 1-(3-amino-1-methylindazol-6-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3N(C)N=C(N)C3=CC=2)CC1 JCUSTPJFYZIYLZ-UHFFFAOYSA-N 0.000 description 2
- OSAUEPXGUASKDP-UHFFFAOYSA-N 1-(3-chloro-2-fluorophenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C(=C(Cl)C=CC=2)F)CC1 OSAUEPXGUASKDP-UHFFFAOYSA-N 0.000 description 2
- CSWKWSCUZFAQPF-UHFFFAOYSA-N 1-(3-fluoro-4-methoxyphenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=C(F)C(OC)=CC=C1N1C(=O)N(C=2C(=CC=NC=2)C)CC1 CSWKWSCUZFAQPF-UHFFFAOYSA-N 0.000 description 2
- SRYVZUMYVQXCOO-UHFFFAOYSA-N 1-(3-methyl-2h-indazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=C2C(C)=NNC2=CC=C1N(C1=O)CCN1C1=CN=CC=C1C SRYVZUMYVQXCOO-UHFFFAOYSA-N 0.000 description 2
- FJYFTSQTTQZYAD-UHFFFAOYSA-N 1-(3h-benzimidazol-5-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3N=CNC3=CC=2)CC1 FJYFTSQTTQZYAD-UHFFFAOYSA-N 0.000 description 2
- ASKFCSCYGAFWAB-UHFFFAOYSA-N 1-(4-bromo-2-fluorophenyl)ethanone Chemical compound CC(=O)C1=CC=C(Br)C=C1F ASKFCSCYGAFWAB-UHFFFAOYSA-N 0.000 description 2
- DGVHBANEALMSMO-UHFFFAOYSA-N 1-(4-chloro-2-methylphenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C(=CC(Cl)=CC=2)C)CC1 DGVHBANEALMSMO-UHFFFAOYSA-N 0.000 description 2
- JWKKDTYDCIVBGT-UHFFFAOYSA-N 1-(4-chlorophenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=CC(Cl)=CC=2)CC1 JWKKDTYDCIVBGT-UHFFFAOYSA-N 0.000 description 2
- NINSHVGQARQIAC-UHFFFAOYSA-N 1-(4-fluoro-3-methoxyphenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=C(F)C(OC)=CC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 NINSHVGQARQIAC-UHFFFAOYSA-N 0.000 description 2
- YORXYVZCETZSPD-UHFFFAOYSA-N 1-(4-fluorophenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=CC(F)=CC=2)CC1 YORXYVZCETZSPD-UHFFFAOYSA-N 0.000 description 2
- YIFZVDJTXNRDLV-UHFFFAOYSA-N 1-(4-methoxyphenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=CC(OC)=CC=C1N1C(=O)N(C=2C(=CC=NC=2)C)CC1 YIFZVDJTXNRDLV-UHFFFAOYSA-N 0.000 description 2
- AGOBWSWWFITDDC-UHFFFAOYSA-N 1-(4-methoxythieno[3,2-c]pyridin-2-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C=1C=2C(OC)=NC=CC=2SC=1N(C1=O)CCN1C1=CN=CC=C1C AGOBWSWWFITDDC-UHFFFAOYSA-N 0.000 description 2
- HMHUDXZWHAUFMF-UHFFFAOYSA-N 1-(4-methoxythieno[3,2-c]pyridin-3-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=2C(OC)=NC=CC=2SC=C1N(C1=O)CCN1C1=CN=CC=C1C HMHUDXZWHAUFMF-UHFFFAOYSA-N 0.000 description 2
- QRFUYGTXUWJCSS-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(1,3-thiazol-2-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2SC=CN=2)CC1 QRFUYGTXUWJCSS-UHFFFAOYSA-N 0.000 description 2
- GOTWDPGCPRLWNR-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(1-tritylpyrazol-4-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C2=CN(N=C2)C(C=2C=CC=CC=2)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GOTWDPGCPRLWNR-UHFFFAOYSA-N 0.000 description 2
- CUPDZKNLFJJPRF-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(1h-pyrazol-4-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C2=CNN=C2)CC1 CUPDZKNLFJJPRF-UHFFFAOYSA-N 0.000 description 2
- XOZCBEDYEBXAQI-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(2-methylquinolin-6-yl)imidazolidin-2-one Chemical compound C1=CC2=NC(C)=CC=C2C=C1N(C1=O)CCN1C1=CN=CC=C1C XOZCBEDYEBXAQI-UHFFFAOYSA-N 0.000 description 2
- BJMRHDMVLPJAKV-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(2-pyrrolidin-1-ylpyridin-4-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(N=CC=2)N2CCCC2)CC1 BJMRHDMVLPJAKV-UHFFFAOYSA-N 0.000 description 2
- KVUVETIBKJQSCX-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(3-methylthiophen-2-yl)imidazolidin-2-one Chemical compound C1=CSC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1C KVUVETIBKJQSCX-UHFFFAOYSA-N 0.000 description 2
- SZWJZRHNKZKRAK-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(4-phenylphenoxy)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(OC=2C=CC(=CC=2)C=2C=CC=CC=2)CC1 SZWJZRHNKZKRAK-UHFFFAOYSA-N 0.000 description 2
- CZASMVZUZLJZES-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-[1-(2-trimethylsilylethoxymethyl)benzimidazol-5-yl]imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3N=CN(COCC[Si](C)(C)C)C3=CC=2)CC1 CZASMVZUZLJZES-UHFFFAOYSA-N 0.000 description 2
- IENGHWPBXCGWGW-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-[1-(2-trimethylsilylethoxymethyl)benzotriazol-5-yl]imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3N=NN(COCC[Si](C)(C)C)C3=CC=2)CC1 IENGHWPBXCGWGW-UHFFFAOYSA-N 0.000 description 2
- MLRICNFQACWUMA-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-[1-(2-trimethylsilylethoxymethyl)indazol-5-yl]imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=NN(COCC[Si](C)(C)C)C3=CC=2)CC1 MLRICNFQACWUMA-UHFFFAOYSA-N 0.000 description 2
- HOIOOEJSGWIGRU-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-[1-(2-trimethylsilylethoxymethyl)pyrazolo[3,4-b]pyridin-5-yl]imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=NN(COCC[Si](C)(C)C)C3=NC=2)CC1 HOIOOEJSGWIGRU-UHFFFAOYSA-N 0.000 description 2
- QTKUFJNYXDQFIQ-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-naphthalen-1-yloxyimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(OC=2C3=CC=CC=C3C=CC=2)CC1 QTKUFJNYXDQFIQ-UHFFFAOYSA-N 0.000 description 2
- IJSSVANWRAVUNO-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-quinolin-2-ylimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2N=C3C=CC=CC3=CC=2)CC1 IJSSVANWRAVUNO-UHFFFAOYSA-N 0.000 description 2
- BDIPZADECWOSDW-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-quinolin-3-ylimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=CC=CC3=NC=2)CC1 BDIPZADECWOSDW-UHFFFAOYSA-N 0.000 description 2
- NEFVVCHUAQFFEZ-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-quinolin-6-ylimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=CC=NC3=CC=2)CC1 NEFVVCHUAQFFEZ-UHFFFAOYSA-N 0.000 description 2
- FTYXHLANWJTNKP-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-quinoxalin-6-ylimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3N=CC=NC3=CC=2)CC1 FTYXHLANWJTNKP-UHFFFAOYSA-N 0.000 description 2
- PBFLRCBUTCLQSJ-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-thiophen-2-ylimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2SC=CC=2)CC1 PBFLRCBUTCLQSJ-UHFFFAOYSA-N 0.000 description 2
- DEDJWHSJPXQEPY-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-thiophen-3-ylimidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C2=CSC=C2)CC1 DEDJWHSJPXQEPY-UHFFFAOYSA-N 0.000 description 2
- XNRQIHIOKXQSPG-UHFFFAOYSA-N 1-(5-bromo-2-fluorophenyl)ethanone Chemical compound CC(=O)C1=CC(Br)=CC=C1F XNRQIHIOKXQSPG-UHFFFAOYSA-N 0.000 description 2
- URJYIMAZMXJKSQ-UHFFFAOYSA-N 1-(5-chloro-2-fluorophenyl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C(=CC=C(Cl)C=2)F)CC1 URJYIMAZMXJKSQ-UHFFFAOYSA-N 0.000 description 2
- ZTZDWVMYYZDGPG-UHFFFAOYSA-N 1-(5-fluoro-3-methyl-1-benzothiophen-2-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound S1C2=CC=C(F)C=C2C(C)=C1N(C1=O)CCN1C1=CN=CC=C1C ZTZDWVMYYZDGPG-UHFFFAOYSA-N 0.000 description 2
- IEDBHAALQSYAKF-UHFFFAOYSA-N 1-(5-fluoro-3-methyl-1-benzothiophen-2-yl)-3-pyridin-3-ylimidazolidin-2-one Chemical compound S1C2=CC=C(F)C=C2C(C)=C1N(C1=O)CCN1C1=CC=CN=C1 IEDBHAALQSYAKF-UHFFFAOYSA-N 0.000 description 2
- BYADDAVAVMHSGK-UHFFFAOYSA-N 1-(5-fluoropyridin-3-yl)-3-naphthalen-2-ylimidazolidin-2-one Chemical compound FC1=CN=CC(N2C(N(CC2)C=2C=C3C=CC=CC3=CC=2)=O)=C1 BYADDAVAVMHSGK-UHFFFAOYSA-N 0.000 description 2
- VZJSAXKHNJTZTJ-UHFFFAOYSA-N 1-(5-methoxypyridin-3-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound COC1=CN=CC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 VZJSAXKHNJTZTJ-UHFFFAOYSA-N 0.000 description 2
- WPOYBZXMAUFUCM-UHFFFAOYSA-N 1-(5-methoxypyridin-3-yl)-3-naphthalen-2-ylimidazolidin-2-one Chemical compound COC1=CN=CC(N2C(N(CC2)C=2C=C3C=CC=CC3=CC=2)=O)=C1 WPOYBZXMAUFUCM-UHFFFAOYSA-N 0.000 description 2
- GHMCGSVUGYRXRA-UHFFFAOYSA-N 1-(6-methoxynaphthalen-2-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=CC2=CC(OC)=CC=C2C=C1N(C1=O)CCN1C1=CN=CC=C1C GHMCGSVUGYRXRA-UHFFFAOYSA-N 0.000 description 2
- PMLXLSGKDUNJIP-UHFFFAOYSA-N 1-(6-methoxynaphthalen-2-yl)-3-pyridin-3-yloxyimidazolidin-2-one Chemical compound C1=CC2=CC(OC)=CC=C2C=C1N(C1=O)CCN1OC1=CC=CN=C1 PMLXLSGKDUNJIP-UHFFFAOYSA-N 0.000 description 2
- SBZUUHGKZFVMJI-UHFFFAOYSA-N 1-[1-(4-methylphenyl)sulfonylindol-3-yl]-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N1C2=CC=CC=C2C(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 SBZUUHGKZFVMJI-UHFFFAOYSA-N 0.000 description 2
- HQLNLXHUHVCYQJ-UHFFFAOYSA-N 1-[4-(4-fluorophenyl)phenyl]-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=CC(=CC=2)C=2C=CC(F)=CC=2)CC1 HQLNLXHUHVCYQJ-UHFFFAOYSA-N 0.000 description 2
- FVPIDFPBIHMYTR-UHFFFAOYSA-N 1-[4-fluoro-3-(n-hydroxy-c-methylcarbonimidoyl)phenyl]-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=C(F)C(C(=NO)C)=CC(N2C(N(CC2)C=2C(=CC=NC=2)C)=O)=C1 FVPIDFPBIHMYTR-UHFFFAOYSA-N 0.000 description 2
- RQAVSDINDRNIKL-UHFFFAOYSA-N 1-chloro-3-isocyanatopropane Chemical compound ClCCCN=C=O RQAVSDINDRNIKL-UHFFFAOYSA-N 0.000 description 2
- YFTTXAJBHASNJR-UHFFFAOYSA-N 1-ethyl-6-(2-oxo-3-pyridin-3-ylimidazolidin-1-yl)-3,4-dihydroquinolin-2-one Chemical compound C=1C=C2N(CC)C(=O)CCC2=CC=1N(C1=O)CCN1C1=CC=CN=C1 YFTTXAJBHASNJR-UHFFFAOYSA-N 0.000 description 2
- BNCGWNUFCYLQIB-UHFFFAOYSA-N 1-ethyl-6-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]-3,4-dihydroquinolin-2-one Chemical compound C=1C=C2N(CC)C(=O)CCC2=CC=1N(C1=O)CCN1C1=CN=CC=C1C BNCGWNUFCYLQIB-UHFFFAOYSA-N 0.000 description 2
- UKKJFFSZHPJOBR-UHFFFAOYSA-N 1-imidazo[1,2-a]pyridin-3-yl-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2N3C=CC=CC3=NC=2)CC1 UKKJFFSZHPJOBR-UHFFFAOYSA-N 0.000 description 2
- ZXBYUHDJVRRKCS-UHFFFAOYSA-N 1-imidazo[1,2-a]pyridin-6-yl-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C2=CN3C=CN=C3C=C2)CC1 ZXBYUHDJVRRKCS-UHFFFAOYSA-N 0.000 description 2
- MBHBQHZIZUTEQP-UHFFFAOYSA-N 1-pyridin-3-yl-3-quinolin-6-ylimidazolidin-2-one Chemical compound C1CN(C=2C=C3C=CC=NC3=CC=2)C(=O)N1C1=CC=CN=C1 MBHBQHZIZUTEQP-UHFFFAOYSA-N 0.000 description 2
- QKWWDTYDYOFRJL-UHFFFAOYSA-N 2,2-dimethoxyethanamine Chemical compound COC(CN)OC QKWWDTYDYOFRJL-UHFFFAOYSA-N 0.000 description 2
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 2
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical class CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 2
- ZADAEJSZAKGTSD-UHFFFAOYSA-N 2-bromo-5-fluoro-3-methyl-1-benzothiophene Chemical compound C1=C(F)C=C2C(C)=C(Br)SC2=C1 ZADAEJSZAKGTSD-UHFFFAOYSA-N 0.000 description 2
- AYFJBMBVXWNYLT-UHFFFAOYSA-N 2-bromo-6-methoxynaphthalene Chemical compound C1=C(Br)C=CC2=CC(OC)=CC=C21 AYFJBMBVXWNYLT-UHFFFAOYSA-N 0.000 description 2
- SKCZHWFPCONQQL-UHFFFAOYSA-N 2-fluoro-4-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzaldehyde Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C(F)C(C=O)=CC=2)CC1 SKCZHWFPCONQQL-UHFFFAOYSA-N 0.000 description 2
- NHFDRBXTEDBWCZ-ZROIWOOFSA-N 3-[2,4-dimethyl-5-[(z)-(2-oxo-1h-indol-3-ylidene)methyl]-1h-pyrrol-3-yl]propanoic acid Chemical compound OC(=O)CCC1=C(C)NC(\C=C/2C3=CC=CC=C3NC\2=O)=C1C NHFDRBXTEDBWCZ-ZROIWOOFSA-N 0.000 description 2
- OPNNLECPNPMTMV-UHFFFAOYSA-N 3-bromo-4-(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CC=NC=C1Br OPNNLECPNPMTMV-UHFFFAOYSA-N 0.000 description 2
- FZWUIWQMJFAWJW-UHFFFAOYSA-N 3-bromo-5-methoxypyridine Chemical compound COC1=CN=CC(Br)=C1 FZWUIWQMJFAWJW-UHFFFAOYSA-N 0.000 description 2
- CLPFFLWZZBQMAO-UHFFFAOYSA-N 4-(5,6,7,8-tetrahydroimidazo[1,5-a]pyridin-5-yl)benzonitrile Chemical compound C1=CC(C#N)=CC=C1C1N2C=NC=C2CCC1 CLPFFLWZZBQMAO-UHFFFAOYSA-N 0.000 description 2
- KZCHWOLGQSHZIM-UHFFFAOYSA-N 4-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzoic acid Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=CC(=CC=2)C(O)=O)CC1 KZCHWOLGQSHZIM-UHFFFAOYSA-N 0.000 description 2
- IXZUCLBXKNHBBC-UHFFFAOYSA-N 4-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzonitrile Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=CC(=CC=2)C#N)CC1 IXZUCLBXKNHBBC-UHFFFAOYSA-N 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- BFJIMLOFUSIQBN-UHFFFAOYSA-N 4-methyl-6-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]-1h-quinolin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C(C)=CC(=O)NC3=CC=2)CC1 BFJIMLOFUSIQBN-UHFFFAOYSA-N 0.000 description 2
- IBKMZYWDWWIWEL-UHFFFAOYSA-N 4-methylpyridin-3-amine Chemical compound CC1=CC=NC=C1N IBKMZYWDWWIWEL-UHFFFAOYSA-N 0.000 description 2
- MTOMBUYLSDEPEL-UHFFFAOYSA-N 5-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]-1h-pyridin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=NC(O)=CC=2)CC1 MTOMBUYLSDEPEL-UHFFFAOYSA-N 0.000 description 2
- RDSIMGKJEYNNLF-UHFFFAOYSA-N 5-bromo-1-benzothiophene Chemical compound BrC1=CC=C2SC=CC2=C1 RDSIMGKJEYNNLF-UHFFFAOYSA-N 0.000 description 2
- GLKZKYSZPVHLDK-UHFFFAOYSA-N 5-iodo-1,3-benzothiazole Chemical compound IC1=CC=C2SC=NC2=C1 GLKZKYSZPVHLDK-UHFFFAOYSA-N 0.000 description 2
- XVLLBRYWAUIDSJ-UHFFFAOYSA-N 5-iodo-1-benzothiophene Chemical compound IC1=CC=C2SC=CC2=C1 XVLLBRYWAUIDSJ-UHFFFAOYSA-N 0.000 description 2
- PQUTZPBCQMIHOI-UHFFFAOYSA-N 6-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]-3,4-dihydro-1h-quinolin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3CCC(=O)NC3=CC=2)CC1 PQUTZPBCQMIHOI-UHFFFAOYSA-N 0.000 description 2
- VAVJMZBEDQKNCB-UHFFFAOYSA-N 6-bromo-1-ethyl-3,4-dihydroquinolin-2-one Chemical compound BrC1=CC=C2N(CC)C(=O)CCC2=C1 VAVJMZBEDQKNCB-UHFFFAOYSA-N 0.000 description 2
- NPDAQPXGOAYKAV-UHFFFAOYSA-N 6-bromo-2-chloro-4-methylquinoline Chemical compound C1=C(Br)C=C2C(C)=CC(Cl)=NC2=C1 NPDAQPXGOAYKAV-UHFFFAOYSA-N 0.000 description 2
- IFIHYLCUKYCKRH-UHFFFAOYSA-N 6-bromoquinoline Chemical compound N1=CC=CC2=CC(Br)=CC=C21 IFIHYLCUKYCKRH-UHFFFAOYSA-N 0.000 description 2
- NICZKYFUJVAZLV-UHFFFAOYSA-N 6-iodo-1,3-benzothiazole Chemical compound IC1=CC=C2N=CSC2=C1 NICZKYFUJVAZLV-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- QPMOOTSABMDDMH-UHFFFAOYSA-N 7-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]-3,4-dihydro-1h-quinolin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3NC(=O)CCC3=CC=2)CC1 QPMOOTSABMDDMH-UHFFFAOYSA-N 0.000 description 2
- 102100033793 ALK tyrosine kinase receptor Human genes 0.000 description 2
- 102000005758 Adenosylmethionine decarboxylase Human genes 0.000 description 2
- 108010070753 Adenosylmethionine decarboxylase Proteins 0.000 description 2
- 229940122815 Aromatase inhibitor Drugs 0.000 description 2
- 229940122361 Bisphosphonate Drugs 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 2
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- 229940124766 Cyp17 inhibitor Drugs 0.000 description 2
- 229940123780 DNA topoisomerase I inhibitor Drugs 0.000 description 2
- 229940124087 DNA topoisomerase II inhibitor Drugs 0.000 description 2
- 102000009024 Epidermal Growth Factor Human genes 0.000 description 2
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 2
- QXRSDHAAWVKZLJ-OXZHEXMSSA-N Epothilone B Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C QXRSDHAAWVKZLJ-OXZHEXMSSA-N 0.000 description 2
- 102100038595 Estrogen receptor Human genes 0.000 description 2
- DNXHEGUUPJUMQT-CBZIJGRNSA-N Estrone Chemical compound OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 DNXHEGUUPJUMQT-CBZIJGRNSA-N 0.000 description 2
- 108091008794 FGF receptors Proteins 0.000 description 2
- 102000044168 Fibroblast Growth Factor Receptor Human genes 0.000 description 2
- 102400000932 Gonadoliberin-1 Human genes 0.000 description 2
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 2
- 101500026183 Homo sapiens Gonadoliberin-1 Proteins 0.000 description 2
- 108010031794 IGF Type 1 Receptor Proteins 0.000 description 2
- MPBVHIBUJCELCL-UHFFFAOYSA-N Ibandronate Chemical compound CCCCCN(C)CCC(O)(P(O)(O)=O)P(O)(O)=O MPBVHIBUJCELCL-UHFFFAOYSA-N 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- 102000004317 Lyases Human genes 0.000 description 2
- 108090000856 Lyases Proteins 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- 239000012828 PI3K inhibitor Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 description 2
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 description 2
- 229940079156 Proteasome inhibitor Drugs 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 108090000315 Protein Kinase C Proteins 0.000 description 2
- 102000003923 Protein Kinase C Human genes 0.000 description 2
- 102000009516 Protein Serine-Threonine Kinases Human genes 0.000 description 2
- 108010009341 Protein Serine-Threonine Kinases Proteins 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 2
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 description 2
- 229940123237 Taxane Drugs 0.000 description 2
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 2
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 2
- 239000000317 Topoisomerase II Inhibitor Substances 0.000 description 2
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 2
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 2
- 229940122803 Vinca alkaloid Drugs 0.000 description 2
- AIWRTTMUVOZGPW-HSPKUQOVSA-N abarelix Chemical compound C([C@@H](C(=O)N[C@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)N(C)C(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(O)C=C1 AIWRTTMUVOZGPW-HSPKUQOVSA-N 0.000 description 2
- 108010023617 abarelix Proteins 0.000 description 2
- 229960002184 abarelix Drugs 0.000 description 2
- 229960000853 abiraterone Drugs 0.000 description 2
- GZOSMCIZMLWJML-VJLLXTKPSA-N abiraterone Chemical compound C([C@H]1[C@H]2[C@@H]([C@]3(CC[C@H](O)CC3=CC2)C)CC[C@@]11C)C=C1C1=CC=CN=C1 GZOSMCIZMLWJML-VJLLXTKPSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 235000011054 acetic acid Nutrition 0.000 description 2
- 229940009456 adriamycin Drugs 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 229960004343 alendronic acid Drugs 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 229960002932 anastrozole Drugs 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 230000002280 anti-androgenic effect Effects 0.000 description 2
- 230000000719 anti-leukaemic effect Effects 0.000 description 2
- 239000000051 antiandrogen Substances 0.000 description 2
- 239000002814 antineoplastic antimetabolite Substances 0.000 description 2
- 239000003125 aqueous solvent Substances 0.000 description 2
- 150000001502 aryl halides Chemical class 0.000 description 2
- 229940120638 avastin Drugs 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 150000004663 bisphosphonates Chemical class 0.000 description 2
- QARVLSVVCXYDNA-UHFFFAOYSA-N bromobenzene Chemical compound BrC1=CC=CC=C1 QARVLSVVCXYDNA-UHFFFAOYSA-N 0.000 description 2
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 2
- 229940127093 camptothecin Drugs 0.000 description 2
- 229960004117 capecitabine Drugs 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 230000000973 chemotherapeutic effect Effects 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- ACSIXWWBWUQEHA-UHFFFAOYSA-N clodronic acid Chemical compound OP(O)(=O)C(Cl)(Cl)P(O)(O)=O ACSIXWWBWUQEHA-UHFFFAOYSA-N 0.000 description 2
- 229960002286 clodronic acid Drugs 0.000 description 2
- 229960001334 corticosteroids Drugs 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- 239000003954 decarboxylase inhibitor Substances 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 229960001904 epirubicin Drugs 0.000 description 2
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 2
- QXRSDHAAWVKZLJ-PVYNADRNSA-N epothilone B Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 QXRSDHAAWVKZLJ-PVYNADRNSA-N 0.000 description 2
- 229940082789 erbitux Drugs 0.000 description 2
- 108010038795 estrogen receptors Proteins 0.000 description 2
- 229960003399 estrone Drugs 0.000 description 2
- 229960000255 exemestane Drugs 0.000 description 2
- 229950011548 fadrozole Drugs 0.000 description 2
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 2
- 229960002258 fulvestrant Drugs 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 2
- QTQAWLPCGQOSGP-GBTDJJJQSA-N geldanamycin Chemical class N1C(=O)\C(C)=C/C=C\[C@@H](OC)[C@H](OC(N)=O)\C(C)=C/[C@@H](C)[C@@H](O)[C@H](OC)C[C@@H](C)CC2=C(OC)C(=O)C=C1C2=O QTQAWLPCGQOSGP-GBTDJJJQSA-N 0.000 description 2
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 2
- 229960001442 gonadorelin Drugs 0.000 description 2
- 229960002913 goserelin Drugs 0.000 description 2
- 125000001188 haloalkyl group Chemical group 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 229940022353 herceptin Drugs 0.000 description 2
- 238000001794 hormone therapy Methods 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 229960005236 ibandronic acid Drugs 0.000 description 2
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 2
- 239000000367 immunologic factor Substances 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 229940079322 interferon Drugs 0.000 description 2
- 229940047124 interferons Drugs 0.000 description 2
- 229960004768 irinotecan Drugs 0.000 description 2
- 150000002513 isocyanates Chemical class 0.000 description 2
- 229940043355 kinase inhibitor Drugs 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 229960003881 letrozole Drugs 0.000 description 2
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 2
- KHPKQFYUPIUARC-UHFFFAOYSA-N lumiracoxib Chemical compound OC(=O)CC1=CC(C)=CC=C1NC1=C(F)C=CC=C1Cl KHPKQFYUPIUARC-UHFFFAOYSA-N 0.000 description 2
- 229940124302 mTOR inhibitor Drugs 0.000 description 2
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- NHSHUXFKFGBPJE-UHFFFAOYSA-N methyl 4-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1N1C(=O)N(C=2C(=CC=NC=2)C)CC1 NHSHUXFKFGBPJE-UHFFFAOYSA-N 0.000 description 2
- AFRRJJDSEALKDF-UHFFFAOYSA-N methyl 5-(3-naphthalen-2-yl-2-oxoimidazolidin-1-yl)pyridine-3-carboxylate Chemical compound COC(=O)C1=CN=CC(N2C(N(CC2)C=2C=C3C=CC=CC3=CC=2)=O)=C1 AFRRJJDSEALKDF-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- MVWNGOVAJMONFD-UHFFFAOYSA-N n-[5-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]pyridin-2-yl]acetamide Chemical compound C1=NC(NC(=O)C)=CC=C1N1C(=O)N(C=2C(=CC=NC=2)C)CC1 MVWNGOVAJMONFD-UHFFFAOYSA-N 0.000 description 2
- ZSHAFKCBCJRALK-UHFFFAOYSA-N n-methyl-4-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]benzamide Chemical compound C1=CC(C(=O)NC)=CC=C1N1C(=O)N(C=2C(=CC=NC=2)C)CC1 ZSHAFKCBCJRALK-UHFFFAOYSA-N 0.000 description 2
- XGXNTJHZPBRBHJ-UHFFFAOYSA-N n-phenylpyrimidin-2-amine Chemical class N=1C=CC=NC=1NC1=CC=CC=C1 XGXNTJHZPBRBHJ-UHFFFAOYSA-N 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 125000006574 non-aromatic ring group Chemical group 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 231100000590 oncogenic Toxicity 0.000 description 2
- 229960001756 oxaliplatin Drugs 0.000 description 2
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 229960003978 pamidronic acid Drugs 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 108700017947 pasireotide Proteins 0.000 description 2
- 229960005415 pasireotide Drugs 0.000 description 2
- 239000000816 peptidomimetic Substances 0.000 description 2
- 229940043441 phosphoinositide 3-kinase inhibitor Drugs 0.000 description 2
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 239000002599 prostaglandin synthase inhibitor Substances 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- 239000003207 proteasome inhibitor Substances 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 239000003528 protein farnesyltransferase inhibitor Substances 0.000 description 2
- 150000003222 pyridines Chemical class 0.000 description 2
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 2
- BKXVVCILCIUCLG-UHFFFAOYSA-N raloxifene hydrochloride Chemical compound [H+].[Cl-].C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 BKXVVCILCIUCLG-UHFFFAOYSA-N 0.000 description 2
- 229960002119 raloxifene hydrochloride Drugs 0.000 description 2
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 2
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 229960000759 risedronic acid Drugs 0.000 description 2
- 229960004641 rituximab Drugs 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical class C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 229960001603 tamoxifen Drugs 0.000 description 2
- 239000003277 telomerase inhibitor Substances 0.000 description 2
- 229960004964 temozolomide Drugs 0.000 description 2
- WOYOOJPPGJFXIH-UHFFFAOYSA-N tert-butyl 2-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]-5,7-dihydro-4h-thieno[2,3-c]pyridine-6-carboxylate Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2SC=3CN(CCC=3C=2)C(=O)OC(C)(C)C)CC1 WOYOOJPPGJFXIH-UHFFFAOYSA-N 0.000 description 2
- VUKLKTNZJBIGPR-UHFFFAOYSA-N tert-butyl 5-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]indole-1-carboxylate Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3C=CN(C3=CC=2)C(=O)OC(C)(C)C)CC1 VUKLKTNZJBIGPR-UHFFFAOYSA-N 0.000 description 2
- FPBANVCOFHHNJU-UHFFFAOYSA-N tert-butyl 7-[3-(4-methylpyridin-3-yl)-2-oxoimidazolidin-1-yl]-2-oxo-3,4-dihydroquinoline-1-carboxylate Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=C3N(C(=O)OC(C)(C)C)C(=O)CCC3=CC=2)CC1 FPBANVCOFHHNJU-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 210000001550 testis Anatomy 0.000 description 2
- 229960003433 thalidomide Drugs 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 229960005324 tiludronic acid Drugs 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 2
- 229960000303 topotecan Drugs 0.000 description 2
- 229960000575 trastuzumab Drugs 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- KDQAABAKXDWYSZ-PNYVAJAMSA-N vinblastine sulfate Chemical compound OS(O)(=O)=O.C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 KDQAABAKXDWYSZ-PNYVAJAMSA-N 0.000 description 2
- 229960004982 vinblastine sulfate Drugs 0.000 description 2
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- AQTQHPDCURKLKT-JKDPCDLQSA-N vincristine sulfate Chemical compound OS(O)(=O)=O.C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C=O)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 AQTQHPDCURKLKT-JKDPCDLQSA-N 0.000 description 2
- 229960002110 vincristine sulfate Drugs 0.000 description 2
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 2
- 229960002066 vinorelbine Drugs 0.000 description 2
- 238000010626 work up procedure Methods 0.000 description 2
- 229960004276 zoledronic acid Drugs 0.000 description 2
- AADVCYNFEREWOS-UHFFFAOYSA-N (+)-DDM Natural products C=CC=CC(C)C(OC(N)=O)C(C)C(O)C(C)CC(C)=CC(C)C(O)C(C)C=CC(O)CC1OC(=O)C(C)C(O)C1C AADVCYNFEREWOS-UHFFFAOYSA-N 0.000 description 1
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- DEQANNDTNATYII-OULOTJBUSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-19-[[(2r)-2-amino-3-phenylpropanoyl]amino]-16-benzyl-n-[(2r,3r)-1,3-dihydroxybutan-2-yl]-7-[(1r)-1-hydroxyethyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicosane-4-carboxa Chemical compound C([C@@H](N)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC=CC=2)NC1=O)C(=O)N[C@H](CO)[C@H](O)C)C1=CC=CC=C1 DEQANNDTNATYII-OULOTJBUSA-N 0.000 description 1
- SWDZPNJZKUGIIH-QQTULTPQSA-N (5z)-n-ethyl-5-(4-hydroxy-6-oxo-3-propan-2-ylcyclohexa-2,4-dien-1-ylidene)-4-[4-(morpholin-4-ylmethyl)phenyl]-2h-1,2-oxazole-3-carboxamide Chemical group O1NC(C(=O)NCC)=C(C=2C=CC(CN3CCOCC3)=CC=2)\C1=C1/C=C(C(C)C)C(O)=CC1=O SWDZPNJZKUGIIH-QQTULTPQSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- GSQOBTOAOGXIFL-LFIBNONCSA-N (e)-2-cyano-3-(3,4-dihydroxyphenyl)-n-(3-phenylpropyl)prop-2-enamide Chemical compound C1=C(O)C(O)=CC=C1\C=C(/C#N)C(=O)NCCCC1=CC=CC=C1 GSQOBTOAOGXIFL-LFIBNONCSA-N 0.000 description 1
- GWCNJMUSWLTSCW-SFQUDFHCSA-N (e)-2-cyano-3-(3,4-dihydroxyphenyl)-n-(4-phenylbutyl)prop-2-enamide Chemical compound C1=C(O)C(O)=CC=C1\C=C(/C#N)C(=O)NCCCCC1=CC=CC=C1 GWCNJMUSWLTSCW-SFQUDFHCSA-N 0.000 description 1
- MOWXJLUYGFNTAL-DEOSSOPVSA-N (s)-[2-chloro-4-fluoro-5-(7-morpholin-4-ylquinazolin-4-yl)phenyl]-(6-methoxypyridazin-3-yl)methanol Chemical compound N1=NC(OC)=CC=C1[C@@H](O)C1=CC(C=2C3=CC=C(C=C3N=CN=2)N2CCOCC2)=C(F)C=C1Cl MOWXJLUYGFNTAL-DEOSSOPVSA-N 0.000 description 1
- CYQYBRMAYUBANC-UHFFFAOYSA-N 1,1-dichloroethane;n,n-dimethylformamide Chemical compound CC(Cl)Cl.CN(C)C=O CYQYBRMAYUBANC-UHFFFAOYSA-N 0.000 description 1
- KEIFWROAQVVDBN-UHFFFAOYSA-N 1,2-dihydronaphthalene Chemical class C1=CC=C2C=CCCC2=C1 KEIFWROAQVVDBN-UHFFFAOYSA-N 0.000 description 1
- QZFLXEYQSSOUKY-UHFFFAOYSA-N 1,3-bis(4-methylpyridin-2-yl)imidazolidin-2-one Chemical compound CC1=CC=NC(N2C(N(CC2)C=2N=CC=C(C)C=2)=O)=C1 QZFLXEYQSSOUKY-UHFFFAOYSA-N 0.000 description 1
- YYIWLLRVTGDCMM-UHFFFAOYSA-N 1,3-bis(6-chloropyridin-2-yl)-1,3-diazinan-2-one Chemical compound ClC1=CC=CC(N2C(N(CCC2)C=2N=C(Cl)C=CC=2)=O)=N1 YYIWLLRVTGDCMM-UHFFFAOYSA-N 0.000 description 1
- AICIYIDUYNFPRY-UHFFFAOYSA-N 1,3-dihydro-2H-imidazol-2-one Chemical group O=C1NC=CN1 AICIYIDUYNFPRY-UHFFFAOYSA-N 0.000 description 1
- HUKAUDZEXSHFDY-UHFFFAOYSA-N 1-(1-oxo-2,3-dihydroinden-5-yl)-3-pyridin-3-ylimidazolidin-2-one Chemical compound C=1C=C2C(=O)CCC2=CC=1N(C1=O)CCN1C1=CC=CN=C1 HUKAUDZEXSHFDY-UHFFFAOYSA-N 0.000 description 1
- KLQQLINXJPIDFI-UHFFFAOYSA-N 1-(3,4-dichlorophenyl)-3-[6-(2-piperidin-1-ylethoxy)pyridin-3-yl]imidazolidin-2-one Chemical compound C1=C(Cl)C(Cl)=CC=C1N1C(=O)N(C=2C=NC(OCCN3CCCCC3)=CC=2)CC1 KLQQLINXJPIDFI-UHFFFAOYSA-N 0.000 description 1
- UXSYRPAHXAKPIO-UHFFFAOYSA-N 1-(3,5-dichloropyridin-2-yl)-3-phenyl-1,3-diazinan-2-one Chemical compound ClC1=CC(Cl)=CN=C1N1C(=O)N(C=2C=CC=CC=2)CCC1 UXSYRPAHXAKPIO-UHFFFAOYSA-N 0.000 description 1
- XLDDUPPCVLDNAP-UHFFFAOYSA-N 1-(3-oxo-1,2-dihydroinden-5-yl)-3-pyridin-3-ylimidazolidin-2-one Chemical compound C1=C2C(=O)CCC2=CC=C1N(C1=O)CCN1C1=CC=CN=C1 XLDDUPPCVLDNAP-UHFFFAOYSA-N 0.000 description 1
- NQSXIIMKICOYDQ-UHFFFAOYSA-N 1-(4-methylpyridin-3-yl)-3-(4,5,6,7-tetrahydrothieno[2,3-c]pyridin-2-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2SC=3CNCCC=3C=2)CC1 NQSXIIMKICOYDQ-UHFFFAOYSA-N 0.000 description 1
- NYTAKBBVMIOYLC-UHFFFAOYSA-N 1-(6-chloro-1h-benzimidazol-2-yl)-3-phenylimidazolidin-2-one Chemical compound N=1C2=CC(Cl)=CC=C2NC=1N(C1=O)CCN1C1=CC=CC=C1 NYTAKBBVMIOYLC-UHFFFAOYSA-N 0.000 description 1
- TYIACGUIGLUKHN-UHFFFAOYSA-N 1-(6-fluoropyridin-3-yl)-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound CC1=CC=NC=C1N1C(=O)N(C=2C=NC(F)=CC=2)CC1 TYIACGUIGLUKHN-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- ABDDQTDRAHXHOC-QMMMGPOBSA-N 1-[(7s)-5,7-dihydro-4h-thieno[2,3-c]pyran-7-yl]-n-methylmethanamine Chemical compound CNC[C@@H]1OCCC2=C1SC=C2 ABDDQTDRAHXHOC-QMMMGPOBSA-N 0.000 description 1
- BYMIBQCDHDTAIA-UHFFFAOYSA-N 1-[1-(4-methylphenyl)sulfonylindol-6-yl]-3-(4-methylpyridin-3-yl)imidazolidin-2-one Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N1C2=CC(N3C(N(CC3)C=3C(=CC=NC=3)C)=O)=CC=C2C=C1 BYMIBQCDHDTAIA-UHFFFAOYSA-N 0.000 description 1
- BJHCYTJNPVGSBZ-YXSASFKJSA-N 1-[4-[6-amino-5-[(Z)-methoxyiminomethyl]pyrimidin-4-yl]oxy-2-chlorophenyl]-3-ethylurea Chemical compound CCNC(=O)Nc1ccc(Oc2ncnc(N)c2\C=N/OC)cc1Cl BJHCYTJNPVGSBZ-YXSASFKJSA-N 0.000 description 1
- MGHBDQZXPCTTIH-UHFFFAOYSA-N 1-bromo-2,4-difluorobenzene Chemical compound FC1=CC=C(Br)C(F)=C1 MGHBDQZXPCTTIH-UHFFFAOYSA-N 0.000 description 1
- NNMBNYHMJRJUBC-UHFFFAOYSA-N 1-bromo-3-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=CC(Br)=C1 NNMBNYHMJRJUBC-UHFFFAOYSA-N 0.000 description 1
- DRNWNTAANHEQMK-UHFFFAOYSA-N 1-bromo-3-chloro-2-fluorobenzene Chemical compound FC1=C(Cl)C=CC=C1Br DRNWNTAANHEQMK-UHFFFAOYSA-N 0.000 description 1
- JRGGUPZKKTVKOV-UHFFFAOYSA-N 1-bromo-3-chlorobenzene Chemical compound ClC1=CC=CC(Br)=C1 JRGGUPZKKTVKOV-UHFFFAOYSA-N 0.000 description 1
- PLDWAJLZAAHOGG-UHFFFAOYSA-N 1-bromo-3-methoxybenzene Chemical compound COC1=CC=CC(Br)=C1 PLDWAJLZAAHOGG-UHFFFAOYSA-N 0.000 description 1
- WJIFKOVZNJTSGO-UHFFFAOYSA-N 1-bromo-3-methylbenzene Chemical compound CC1=CC=CC(Br)=C1 WJIFKOVZNJTSGO-UHFFFAOYSA-N 0.000 description 1
- XLQSXGGDTHANLN-UHFFFAOYSA-N 1-bromo-4-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=C(Br)C=C1 XLQSXGGDTHANLN-UHFFFAOYSA-N 0.000 description 1
- RTIPTGMVQIIMKL-UHFFFAOYSA-N 1-bromo-4-chloro-2-methylbenzene Chemical compound CC1=CC(Cl)=CC=C1Br RTIPTGMVQIIMKL-UHFFFAOYSA-N 0.000 description 1
- NHDODQWIKUYWMW-UHFFFAOYSA-N 1-bromo-4-chlorobenzene Chemical compound ClC1=CC=C(Br)C=C1 NHDODQWIKUYWMW-UHFFFAOYSA-N 0.000 description 1
- AITNMTXHTIIIBB-UHFFFAOYSA-N 1-bromo-4-fluorobenzene Chemical compound FC1=CC=C(Br)C=C1 AITNMTXHTIIIBB-UHFFFAOYSA-N 0.000 description 1
- PKJBWOWQJHHAHG-UHFFFAOYSA-N 1-bromo-4-phenylbenzene Chemical group C1=CC(Br)=CC=C1C1=CC=CC=C1 PKJBWOWQJHHAHG-UHFFFAOYSA-N 0.000 description 1
- DLKQHBOKULLWDQ-UHFFFAOYSA-N 1-bromonaphthalene Chemical compound C1=CC=C2C(Br)=CC=CC2=C1 DLKQHBOKULLWDQ-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- HAYAZOLCFXUCGS-UHFFFAOYSA-N 1-fluoro-4-(4-iodophenyl)benzene Chemical group C1=CC(F)=CC=C1C1=CC=C(I)C=C1 HAYAZOLCFXUCGS-UHFFFAOYSA-N 0.000 description 1
- SJJCQDRGABAVBB-UHFFFAOYSA-N 1-hydroxy-2-naphthoic acid Chemical compound C1=CC=CC2=C(O)C(C(=O)O)=CC=C21 SJJCQDRGABAVBB-UHFFFAOYSA-N 0.000 description 1
- 125000006019 1-methyl-1-propenyl group Chemical group 0.000 description 1
- 125000006021 1-methyl-2-propenyl group Chemical group 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- ZESRJSPZRDMNHY-YFWFAHHUSA-N 11-deoxycorticosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 ZESRJSPZRDMNHY-YFWFAHHUSA-N 0.000 description 1
- WHBHBVVOGNECLV-OBQKJFGGSA-N 11-deoxycortisol Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 WHBHBVVOGNECLV-OBQKJFGGSA-N 0.000 description 1
- DBPWSSGDRRHUNT-UHFFFAOYSA-N 17alpha-hydroxy progesterone Natural products C1CC2=CC(=O)CCC2(C)C2C1C1CCC(C(=O)C)(O)C1(C)CC2 DBPWSSGDRRHUNT-UHFFFAOYSA-N 0.000 description 1
- DBPWSSGDRRHUNT-CEGNMAFCSA-N 17α-hydroxyprogesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 DBPWSSGDRRHUNT-CEGNMAFCSA-N 0.000 description 1
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 1
- HYZJCKYKOHLVJF-SFIIULIVSA-N 1H-benzimidazole Chemical class C1=CC=C2N[11CH]=NC2=C1 HYZJCKYKOHLVJF-SFIIULIVSA-N 0.000 description 1
- CKTSBUTUHBMZGZ-SHYZEUOFSA-N 2'‐deoxycytidine Chemical class O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 CKTSBUTUHBMZGZ-SHYZEUOFSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- DCYGAPKNVCQNOE-UHFFFAOYSA-N 2,2,2-triphenylacetic acid Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(C(=O)O)C1=CC=CC=C1 DCYGAPKNVCQNOE-UHFFFAOYSA-N 0.000 description 1
- SHWQRVBJVFBWGP-UHFFFAOYSA-N 2-(1h-imidazol-5-ylmethyl)-9h-carbazole Chemical compound C=1C=C(C2=CC=CC=C2N2)C2=CC=1CC1=CNC=N1 SHWQRVBJVFBWGP-UHFFFAOYSA-N 0.000 description 1
- UGVUZYHUHPXLRY-UHFFFAOYSA-N 2-(2-oxo-3-phenylimidazolidin-1-yl)-3h-benzimidazole-5-carbonitrile Chemical compound O=C1N(C=2NC3=CC=C(C=C3N=2)C#N)CCN1C1=CC=CC=C1 UGVUZYHUHPXLRY-UHFFFAOYSA-N 0.000 description 1
- PXBFMLJZNCDSMP-UHFFFAOYSA-N 2-Aminobenzamide Chemical class NC(=O)C1=CC=CC=C1N PXBFMLJZNCDSMP-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- VTJXFTPMFYAJJU-UHFFFAOYSA-N 2-[(3,4-dihydroxyphenyl)methylidene]propanedinitrile Chemical compound OC1=CC=C(C=C(C#N)C#N)C=C1O VTJXFTPMFYAJJU-UHFFFAOYSA-N 0.000 description 1
- WDFWDWJBSNASHK-UHFFFAOYSA-N 2-[(5-bromobenzimidazol-1-yl)methoxy]ethyl-trimethylsilane Chemical compound BrC1=CC=C2N(COCC[Si](C)(C)C)C=NC2=C1 WDFWDWJBSNASHK-UHFFFAOYSA-N 0.000 description 1
- KOJHRNWNTBDHJI-UHFFFAOYSA-N 2-[(5-bromobenzotriazol-1-yl)methoxy]ethyl-trimethylsilane Chemical compound BrC1=CC=C2N(COCC[Si](C)(C)C)N=NC2=C1 KOJHRNWNTBDHJI-UHFFFAOYSA-N 0.000 description 1
- SDBGBQSQKWGACK-UHFFFAOYSA-N 2-[(5-bromoindazol-1-yl)methoxy]ethyl-trimethylsilane Chemical compound BrC1=CC=C2N(COCC[Si](C)(C)C)N=CC2=C1 SDBGBQSQKWGACK-UHFFFAOYSA-N 0.000 description 1
- XJKQWXNGTAGTSW-UHFFFAOYSA-N 2-[(5-bromopyrazolo[3,4-b]pyridin-1-yl)methoxy]ethyl-trimethylsilane Chemical compound BrC1=CN=C2N(COCC[Si](C)(C)C)N=CC2=C1 XJKQWXNGTAGTSW-UHFFFAOYSA-N 0.000 description 1
- FSPQCTGGIANIJZ-UHFFFAOYSA-N 2-[[(3,4-dimethoxyphenyl)-oxomethyl]amino]-4,5,6,7-tetrahydro-1-benzothiophene-3-carboxamide Chemical compound C1=C(OC)C(OC)=CC=C1C(=O)NC1=C(C(N)=O)C(CCCC2)=C2S1 FSPQCTGGIANIJZ-UHFFFAOYSA-N 0.000 description 1
- 125000004174 2-benzimidazolyl group Chemical group [H]N1C(*)=NC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- BLRCPPIAOOGKDP-UHFFFAOYSA-N 2-benzylidene-3-hydroxybutanedinitrile Chemical class N#CC(O)C(C#N)=CC1=CC=CC=C1 BLRCPPIAOOGKDP-UHFFFAOYSA-N 0.000 description 1
- DRLMMVPCYXFPEP-UHFFFAOYSA-N 2-bromo-1,3-benzothiazole Chemical compound C1=CC=C2SC(Br)=NC2=C1 DRLMMVPCYXFPEP-UHFFFAOYSA-N 0.000 description 1
- RXNZFHIEDZEUQM-UHFFFAOYSA-N 2-bromo-1,3-thiazole Chemical compound BrC1=NC=CS1 RXNZFHIEDZEUQM-UHFFFAOYSA-N 0.000 description 1
- WIFMYMXKTAVDSQ-UHFFFAOYSA-N 2-bromo-1-benzothiophene Chemical compound C1=CC=C2SC(Br)=CC2=C1 WIFMYMXKTAVDSQ-UHFFFAOYSA-N 0.000 description 1
- YYJBWYBULYUKMR-UHFFFAOYSA-N 2-bromo-3-methylthiophene Chemical compound CC=1C=CSC=1Br YYJBWYBULYUKMR-UHFFFAOYSA-N 0.000 description 1
- YFFUYGSLQXVHMB-UHFFFAOYSA-N 2-bromo-4-chloro-1-fluorobenzene Chemical compound FC1=CC=C(Cl)C=C1Br YFFUYGSLQXVHMB-UHFFFAOYSA-N 0.000 description 1
- QPGCPWZFCLBFCG-UHFFFAOYSA-N 2-bromo-4-chlorothieno[3,2-c]pyridine Chemical compound ClC1=NC=CC2=C1C=C(Br)S2 QPGCPWZFCLBFCG-UHFFFAOYSA-N 0.000 description 1
- LMZSCCFROKLQLD-UHFFFAOYSA-N 2-bromo-4-methoxythieno[3,2-c]pyridine Chemical compound COC1=NC=CC2=C1C=C(Br)S2 LMZSCCFROKLQLD-UHFFFAOYSA-N 0.000 description 1
- ZFAJPWYXLYGUJU-UHFFFAOYSA-N 2-bromo-5-chlorothiophene Chemical compound ClC1=CC=C(Br)S1 ZFAJPWYXLYGUJU-UHFFFAOYSA-N 0.000 description 1
- ACDLOOGOFKSUPO-UHFFFAOYSA-N 2-bromo-5-methylthiophene Chemical compound CC1=CC=C(Br)S1 ACDLOOGOFKSUPO-UHFFFAOYSA-N 0.000 description 1
- TUCRZHGAIRVWTI-UHFFFAOYSA-N 2-bromothiophene Chemical compound BrC1=CC=CS1 TUCRZHGAIRVWTI-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- BBVQDWDBTWSGHQ-UHFFFAOYSA-N 2-chloro-1,3-benzoxazole Chemical compound C1=CC=C2OC(Cl)=NC2=C1 BBVQDWDBTWSGHQ-UHFFFAOYSA-N 0.000 description 1
- HLFNGYARDINCQT-UHFFFAOYSA-N 2-chloro-4-iodobenzonitrile Chemical compound ClC1=CC(I)=CC=C1C#N HLFNGYARDINCQT-UHFFFAOYSA-N 0.000 description 1
- KJKIPRQNFDUULB-UHFFFAOYSA-N 2-chloro-4-iodopyridine Chemical compound ClC1=CC(I)=CC=N1 KJKIPRQNFDUULB-UHFFFAOYSA-N 0.000 description 1
- OFUFXTHGZWIDDB-UHFFFAOYSA-N 2-chloroquinoline Chemical compound C1=CC=CC2=NC(Cl)=CC=C21 OFUFXTHGZWIDDB-UHFFFAOYSA-N 0.000 description 1
- CFMZSMGAMPBRBE-UHFFFAOYSA-N 2-hydroxyisoindole-1,3-dione Chemical class C1=CC=C2C(=O)N(O)C(=O)C2=C1 CFMZSMGAMPBRBE-UHFFFAOYSA-N 0.000 description 1
- WBJWXIQDBDZMAW-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carbonyl chloride Chemical compound C1=CC=CC2=C(C(Cl)=O)C(O)=CC=C21 WBJWXIQDBDZMAW-UHFFFAOYSA-N 0.000 description 1
- 125000006020 2-methyl-1-propenyl group Chemical group 0.000 description 1
- GJJVAFUKOBZPCB-UHFFFAOYSA-N 2-methyl-2-(4,8,12-trimethyltrideca-3,7,11-trienyl)-3,4-dihydrochromen-6-ol Chemical compound OC1=CC=C2OC(CCC=C(C)CCC=C(C)CCC=C(C)C)(C)CCC2=C1 GJJVAFUKOBZPCB-UHFFFAOYSA-N 0.000 description 1
- 125000006022 2-methyl-2-propenyl group Chemical group 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- JBIJLHTVPXGSAM-UHFFFAOYSA-N 2-naphthylamine Chemical compound C1=CC=CC2=CC(N)=CC=C21 JBIJLHTVPXGSAM-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000850 2H-chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001698 2H-pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- BYHQTRFJOGIQAO-GOSISDBHSA-N 3-(4-bromophenyl)-8-[(2R)-2-hydroxypropyl]-1-[(3-methoxyphenyl)methyl]-1,3,8-triazaspiro[4.5]decan-2-one Chemical compound C[C@H](CN1CCC2(CC1)CN(C(=O)N2CC3=CC(=CC=C3)OC)C4=CC=C(C=C4)Br)O BYHQTRFJOGIQAO-GOSISDBHSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-N 3-Hydroxy-2-naphthoate Chemical compound C1=CC=C2C=C(O)C(C(=O)O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-N 0.000 description 1
- UZFPOOOQHWICKY-UHFFFAOYSA-N 3-[13-[1-[1-[8,12-bis(2-carboxyethyl)-17-(1-hydroxyethyl)-3,7,13,18-tetramethyl-21,24-dihydroporphyrin-2-yl]ethoxy]ethyl]-18-(2-carboxyethyl)-8-(1-hydroxyethyl)-3,7,12,17-tetramethyl-22,23-dihydroporphyrin-2-yl]propanoic acid Chemical compound N1C(C=C2C(=C(CCC(O)=O)C(C=C3C(=C(C)C(C=C4N5)=N3)CCC(O)=O)=N2)C)=C(C)C(C(C)O)=C1C=C5C(C)=C4C(C)OC(C)C1=C(N2)C=C(N3)C(C)=C(C(O)C)C3=CC(C(C)=C3CCC(O)=O)=NC3=CC(C(CCC(O)=O)=C3C)=NC3=CC2=C1C UZFPOOOQHWICKY-UHFFFAOYSA-N 0.000 description 1
- WMUGCJOURHYBFZ-UHFFFAOYSA-N 3-[3-(1,3-benzothiazol-6-yl)-2-oxoimidazolidin-1-yl]pyridine-4-carbaldehyde Chemical compound O=CC1=CC=NC=C1N1C(=O)N(C=2C=C3SC=NC3=CC=2)CC1 WMUGCJOURHYBFZ-UHFFFAOYSA-N 0.000 description 1
- WNEODWDFDXWOLU-QHCPKHFHSA-N 3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[(2s)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one Chemical compound C([C@@H](N(CC1)C=2C=NC(NC=3C(N(C)C=C(C=3)C=3C(=C(N4C(C5=CC=6CC(C)(C)CC=6N5CC4)=O)N=CC=3)CO)=O)=CC=2)C)N1C1COC1 WNEODWDFDXWOLU-QHCPKHFHSA-N 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- VQWIINXTMHBRDW-UHFFFAOYSA-N 3-bromo-1-(4-methylphenyl)sulfonylindole Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N1C2=CC=CC=C2C(Br)=C1 VQWIINXTMHBRDW-UHFFFAOYSA-N 0.000 description 1
- SRWDQSRTOOMPMO-UHFFFAOYSA-N 3-bromo-1-benzothiophene Chemical compound C1=CC=C2C(Br)=CSC2=C1 SRWDQSRTOOMPMO-UHFFFAOYSA-N 0.000 description 1
- SAYZMFSSIBFGNW-UHFFFAOYSA-N 3-bromo-1-methylindole Chemical compound C1=CC=C2N(C)C=C(Br)C2=C1 SAYZMFSSIBFGNW-UHFFFAOYSA-N 0.000 description 1
- KCMMLUVZVBBEFO-UHFFFAOYSA-N 3-bromo-4-(difluoromethyl)pyridine Chemical compound FC(F)C1=CC=NC=C1Br KCMMLUVZVBBEFO-UHFFFAOYSA-N 0.000 description 1
- QADXKWUCCGPQNR-UHFFFAOYSA-N 3-bromo-4-chloropyridine Chemical compound ClC1=CC=NC=C1Br QADXKWUCCGPQNR-UHFFFAOYSA-N 0.000 description 1
- FXIKIVFBAQCMRY-UHFFFAOYSA-N 3-bromo-4-chlorothieno[3,2-c]pyridine Chemical compound ClC1=NC=CC2=C1C(Br)=CS2 FXIKIVFBAQCMRY-UHFFFAOYSA-N 0.000 description 1
- NMASMRYRBHKNOO-UHFFFAOYSA-N 3-bromo-4-methoxythieno[3,2-c]pyridine Chemical compound COC1=NC=CC2=C1C(Br)=CS2 NMASMRYRBHKNOO-UHFFFAOYSA-N 0.000 description 1
- HEDHNDVPKRVQPN-UHFFFAOYSA-N 3-bromo-5-(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CN=CC(Br)=C1 HEDHNDVPKRVQPN-UHFFFAOYSA-N 0.000 description 1
- DYTMTLPTFUWUSO-UHFFFAOYSA-N 3-bromo-5-chloro-1h-indole Chemical compound ClC1=CC=C2NC=C(Br)C2=C1 DYTMTLPTFUWUSO-UHFFFAOYSA-N 0.000 description 1
- BELDOPUBSLPBCQ-UHFFFAOYSA-N 3-bromo-5-chloropyridine Chemical compound ClC1=CN=CC(Br)=C1 BELDOPUBSLPBCQ-UHFFFAOYSA-N 0.000 description 1
- HNNNBQRRIHKFLI-UHFFFAOYSA-N 3-bromo-5-fluoropyridine Chemical compound FC1=CN=CC(Br)=C1 HNNNBQRRIHKFLI-UHFFFAOYSA-N 0.000 description 1
- STXAVEHFKAXGOX-UHFFFAOYSA-N 3-bromobenzonitrile Chemical compound BrC1=CC=CC(C#N)=C1 STXAVEHFKAXGOX-UHFFFAOYSA-N 0.000 description 1
- NOBDKWLIAQKADB-UHFFFAOYSA-N 3-bromopyridine-4-carbaldehyde Chemical compound BrC1=CN=CC=C1C=O NOBDKWLIAQKADB-UHFFFAOYSA-N 0.000 description 1
- VPDIXJCBBUZNPO-UHFFFAOYSA-N 3-bromopyridine-4-carbonitrile Chemical compound BrC1=CN=CC=C1C#N VPDIXJCBBUZNPO-UHFFFAOYSA-N 0.000 description 1
- ZGIKWINFUGEQEO-UHFFFAOYSA-N 3-bromoquinoline Chemical compound C1=CC=CC2=CC(Br)=CN=C21 ZGIKWINFUGEQEO-UHFFFAOYSA-N 0.000 description 1
- XCMISAPCWHTVNG-UHFFFAOYSA-N 3-bromothiophene Chemical compound BrC=1C=CSC=1 XCMISAPCWHTVNG-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- SMXBHXFTAWZFAU-UHFFFAOYSA-N 3-iodo-4-methylpyridine Chemical compound CC1=CC=NC=C1I SMXBHXFTAWZFAU-UHFFFAOYSA-N 0.000 description 1
- BPQZPYPWXGFNRU-UHFFFAOYSA-N 3-iodoimidazo[1,2-a]pyridine Chemical compound C1=CC=CN2C(I)=CN=C21 BPQZPYPWXGFNRU-UHFFFAOYSA-N 0.000 description 1
- JJYPMNFTHPTTDI-UHFFFAOYSA-N 3-methylaniline Chemical compound CC1=CC=CC(N)=C1 JJYPMNFTHPTTDI-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- QSFREBZMBNRGOK-UHFFFAOYSA-N 4-[(2,5-dihydroxyphenyl)methylamino]benzoic acid methyl ester Chemical compound C1=CC(C(=O)OC)=CC=C1NCC1=CC(O)=CC=C1O QSFREBZMBNRGOK-UHFFFAOYSA-N 0.000 description 1
- QDPVYZNVVQQULH-UHFFFAOYSA-N 4-amino-5-fluoro-3-[6-(4-methylpiperazin-1-yl)-1H-benzimidazol-2-yl]-1H-quinolin-2-one 2-hydroxypropanoic acid hydrate Chemical compound O.CC(O)C(O)=O.C1CN(C)CCN1C1=CC=C(N=C(N2)C=3C(NC4=CC=CC(F)=C4C=3N)=O)C2=C1 QDPVYZNVVQQULH-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- YMQPKONILWWJQG-UHFFFAOYSA-N 4-bromo-1,2-difluorobenzene Chemical compound FC1=CC=C(Br)C=C1F YMQPKONILWWJQG-UHFFFAOYSA-N 0.000 description 1
- AGYWDGVTLKNTBS-UHFFFAOYSA-N 4-bromo-1-chloro-2-fluorobenzene Chemical compound FC1=CC(Br)=CC=C1Cl AGYWDGVTLKNTBS-UHFFFAOYSA-N 0.000 description 1
- AJLIJYGWAXPEOK-UHFFFAOYSA-N 4-bromo-1-fluoro-2-(trifluoromethyl)benzene Chemical compound FC1=CC=C(Br)C=C1C(F)(F)F AJLIJYGWAXPEOK-UHFFFAOYSA-N 0.000 description 1
- SEVMQEIGENUPIE-UHFFFAOYSA-N 4-bromo-1-fluoro-2-methoxybenzene Chemical compound COC1=CC(Br)=CC=C1F SEVMQEIGENUPIE-UHFFFAOYSA-N 0.000 description 1
- VXKYOKPNAXNAFU-UHFFFAOYSA-N 4-bromo-1-fluoro-2-methylbenzene Chemical compound CC1=CC(Br)=CC=C1F VXKYOKPNAXNAFU-UHFFFAOYSA-N 0.000 description 1
- CPENTLJGGGSVAJ-UHFFFAOYSA-N 4-bromo-1-tritylpyrazole Chemical compound C1=C(Br)C=NN1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 CPENTLJGGGSVAJ-UHFFFAOYSA-N 0.000 description 1
- YHQZVEGLXIBPRA-UHFFFAOYSA-N 4-bromo-2-(difluoromethyl)-1-fluorobenzene Chemical compound FC(F)C1=CC(Br)=CC=C1F YHQZVEGLXIBPRA-UHFFFAOYSA-N 0.000 description 1
- FPSAHMIQBUZQQQ-UHFFFAOYSA-N 4-bromo-2-(difluoromethyl)thiophene Chemical compound FC(F)C1=CC(Br)=CS1 FPSAHMIQBUZQQQ-UHFFFAOYSA-N 0.000 description 1
- CJTIWGBQCVYTQE-UHFFFAOYSA-N 4-bromo-2-chloro-1-fluorobenzene Chemical compound FC1=CC=C(Br)C=C1Cl CJTIWGBQCVYTQE-UHFFFAOYSA-N 0.000 description 1
- DWNXGZBXFDNKOR-UHFFFAOYSA-N 4-bromo-2-fluoro-1-methoxybenzene Chemical compound COC1=CC=C(Br)C=C1F DWNXGZBXFDNKOR-UHFFFAOYSA-N 0.000 description 1
- UPCARQPLANFGQJ-UHFFFAOYSA-N 4-bromo-2-fluorobenzaldehyde Chemical compound FC1=CC(Br)=CC=C1C=O UPCARQPLANFGQJ-UHFFFAOYSA-N 0.000 description 1
- HGXWRDPQFZKOLZ-UHFFFAOYSA-N 4-bromo-2-fluorobenzonitrile Chemical compound FC1=CC(Br)=CC=C1C#N HGXWRDPQFZKOLZ-UHFFFAOYSA-N 0.000 description 1
- JFBMFWHEXBLFCR-UHFFFAOYSA-N 4-bromo-2-methylpyridine Chemical compound CC1=CC(Br)=CC=N1 JFBMFWHEXBLFCR-UHFFFAOYSA-N 0.000 description 1
- ABMUSXPGSSMPLK-UHFFFAOYSA-N 4-bromo-2-methylthiophene Chemical compound CC1=CC(Br)=CS1 ABMUSXPGSSMPLK-UHFFFAOYSA-N 0.000 description 1
- QJPJQTDYNZXKQF-UHFFFAOYSA-N 4-bromoanisole Chemical compound COC1=CC=C(Br)C=C1 QJPJQTDYNZXKQF-UHFFFAOYSA-N 0.000 description 1
- HQSCPPCMBMFJJN-UHFFFAOYSA-N 4-bromobenzonitrile Chemical compound BrC1=CC=C(C#N)C=C1 HQSCPPCMBMFJJN-UHFFFAOYSA-N 0.000 description 1
- XRHGYUZYPHTUJZ-UHFFFAOYSA-N 4-chlorobenzoic acid Chemical compound OC(=O)C1=CC=C(Cl)C=C1 XRHGYUZYPHTUJZ-UHFFFAOYSA-N 0.000 description 1
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 description 1
- CFXNVDUEBJYAAZ-UHFFFAOYSA-N 4-iodo-1-propan-2-ylpyrazole Chemical compound CC(C)N1C=C(I)C=N1 CFXNVDUEBJYAAZ-UHFFFAOYSA-N 0.000 description 1
- 125000001826 4H-pyranyl group Chemical group O1C(=CCC=C1)* 0.000 description 1
- 125000004606 5,6,7,8-tetrahydroisoquinolinyl group Chemical group C1(=NC=CC=2CCCCC12)* 0.000 description 1
- 125000004608 5,6,7,8-tetrahydroquinolinyl group Chemical group N1=C(C=CC=2CCCCC12)* 0.000 description 1
- NMUSYJAQQFHJEW-UHFFFAOYSA-N 5-Azacytidine Natural products O=C1N=C(N)N=CN1C1C(O)C(O)C(CO)O1 NMUSYJAQQFHJEW-UHFFFAOYSA-N 0.000 description 1
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- LNBZVSDFWQIXHC-UHFFFAOYSA-N 5-bromo-1-ethylindole Chemical compound BrC1=CC=C2N(CC)C=CC2=C1 LNBZVSDFWQIXHC-UHFFFAOYSA-N 0.000 description 1
- KMJMDIYGNFGEEO-UHFFFAOYSA-N 5-bromo-1-methylbenzimidazole Chemical compound BrC1=CC=C2N(C)C=NC2=C1 KMJMDIYGNFGEEO-UHFFFAOYSA-N 0.000 description 1
- RMCHMXPTUMFFBZ-UHFFFAOYSA-N 5-bromo-1-methylindazole Chemical compound BrC1=CC=C2N(C)N=CC2=C1 RMCHMXPTUMFFBZ-UHFFFAOYSA-N 0.000 description 1
- SBOITLSQLQGSLO-UHFFFAOYSA-N 5-bromo-1-methylindole Chemical compound BrC1=CC=C2N(C)C=CC2=C1 SBOITLSQLQGSLO-UHFFFAOYSA-N 0.000 description 1
- SZRHWHHXVXSGMT-UHFFFAOYSA-N 5-bromo-2,2-difluoro-1,3-benzodioxole Chemical compound C1=C(Br)C=C2OC(F)(F)OC2=C1 SZRHWHHXVXSGMT-UHFFFAOYSA-N 0.000 description 1
- UDWFSJAYXTXMLM-UHFFFAOYSA-N 5-bromo-2,3-dihydro-1-benzofuran Chemical compound BrC1=CC=C2OCCC2=C1 UDWFSJAYXTXMLM-UHFFFAOYSA-N 0.000 description 1
- UMEFRXDFDVRHMJ-UHFFFAOYSA-N 5-bromo-2,3-dihydro-1h-indene Chemical compound BrC1=CC=C2CCCC2=C1 UMEFRXDFDVRHMJ-UHFFFAOYSA-N 0.000 description 1
- KSONICAHAPRCMV-UHFFFAOYSA-N 5-bromo-2,3-dihydroinden-1-one Chemical compound BrC1=CC=C2C(=O)CCC2=C1 KSONICAHAPRCMV-UHFFFAOYSA-N 0.000 description 1
- XPGIBDJXEVAVTO-UHFFFAOYSA-N 5-bromo-2-chloropyrimidine Chemical compound ClC1=NC=C(Br)C=N1 XPGIBDJXEVAVTO-UHFFFAOYSA-N 0.000 description 1
- MMFGGDVQLQQQRX-UHFFFAOYSA-N 5-bromo-2-fluorobenzaldehyde Chemical compound FC1=CC=C(Br)C=C1C=O MMFGGDVQLQQQRX-UHFFFAOYSA-N 0.000 description 1
- GYCNHFWRPJXTSB-UHFFFAOYSA-N 5-bromo-2-fluorobenzonitrile Chemical compound FC1=CC=C(Br)C=C1C#N GYCNHFWRPJXTSB-UHFFFAOYSA-N 0.000 description 1
- MYUQKYGWKHTRPG-UHFFFAOYSA-N 5-bromo-2-fluoropyridine Chemical compound FC1=CC=C(Br)C=N1 MYUQKYGWKHTRPG-UHFFFAOYSA-N 0.000 description 1
- ZBCCSPFGJDVVJB-UHFFFAOYSA-N 5-bromo-2-methyl-1,3-benzoxazole Chemical compound BrC1=CC=C2OC(C)=NC2=C1 ZBCCSPFGJDVVJB-UHFFFAOYSA-N 0.000 description 1
- QFZOHVHNTZTZMJ-UHFFFAOYSA-N 5-bromo-2-methylindazole Chemical compound C1=C(Br)C=CC2=NN(C)C=C21 QFZOHVHNTZTZMJ-UHFFFAOYSA-N 0.000 description 1
- OFKWIQJLYCKDNY-UHFFFAOYSA-N 5-bromo-2-methylpyridine Chemical compound CC1=CC=C(Br)C=N1 OFKWIQJLYCKDNY-UHFFFAOYSA-N 0.000 description 1
- MRQQUDYWELWUQO-UHFFFAOYSA-N 5-bromo-3-methyl-1-benzofuran Chemical compound C1=C(Br)C=C2C(C)=COC2=C1 MRQQUDYWELWUQO-UHFFFAOYSA-N 0.000 description 1
- 125000006163 5-membered heteroaryl group Chemical group 0.000 description 1
- 125000004938 5-pyridyl group Chemical group N1=CC=CC(=C1)* 0.000 description 1
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 1
- RIORRJLLXUYLTE-UHFFFAOYSA-N 6-bromo-1-(4-methylphenyl)sulfonylindole Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N1C2=CC(Br)=CC=C2C=C1 RIORRJLLXUYLTE-UHFFFAOYSA-N 0.000 description 1
- YBCSNSHLPJAYIM-UHFFFAOYSA-N 6-bromo-1-ethyl-4-methylquinolin-2-one Chemical compound C1=C(Br)C=C2C(C)=CC(=O)N(CC)C2=C1 YBCSNSHLPJAYIM-UHFFFAOYSA-N 0.000 description 1
- LFCURAJBHDNUNG-UHFFFAOYSA-N 6-bromo-2,3-dihydro-1,4-benzodioxine Chemical compound O1CCOC2=CC(Br)=CC=C21 LFCURAJBHDNUNG-UHFFFAOYSA-N 0.000 description 1
- TVHRDJPRECFZQO-UHFFFAOYSA-N 6-bromo-2-methylimidazo[1,2-a]pyridine Chemical compound C1=C(Br)C=CC2=NC(C)=CN21 TVHRDJPRECFZQO-UHFFFAOYSA-N 0.000 description 1
- SQRYQSKJZVQJAY-UHFFFAOYSA-N 6-bromo-2-methylquinoline Chemical compound C1=C(Br)C=CC2=NC(C)=CC=C21 SQRYQSKJZVQJAY-UHFFFAOYSA-N 0.000 description 1
- FXPMFQUOGYGTAM-UHFFFAOYSA-N 6-bromoimidazo[1,2-a]pyridine Chemical compound C1=C(Br)C=CC2=NC=CN21 FXPMFQUOGYGTAM-UHFFFAOYSA-N 0.000 description 1
- NOYFLUFQGFNMRB-UHFFFAOYSA-N 6-bromoquinoxaline Chemical compound N1=CC=NC2=CC(Br)=CC=C21 NOYFLUFQGFNMRB-UHFFFAOYSA-N 0.000 description 1
- YVXGKAZJAUWMPM-UHFFFAOYSA-N 6-iodo-2-methyl-1,3-benzothiazole Chemical compound C1=C(I)C=C2SC(C)=NC2=C1 YVXGKAZJAUWMPM-UHFFFAOYSA-N 0.000 description 1
- OASOJRLJBDCVNU-UHFFFAOYSA-N 7-bromoimidazo[1,2-a]pyridine Chemical compound C1=C(Br)C=CN2C=CN=C21 OASOJRLJBDCVNU-UHFFFAOYSA-N 0.000 description 1
- PBCZSGKMGDDXIJ-HQCWYSJUSA-N 7-hydroxystaurosporine Chemical compound N([C@H](O)C1=C2C3=CC=CC=C3N3C2=C24)C(=O)C1=C2C1=CC=CC=C1N4[C@H]1C[C@@H](NC)[C@@H](OC)[C@]3(C)O1 PBCZSGKMGDDXIJ-HQCWYSJUSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- PBCZSGKMGDDXIJ-UHFFFAOYSA-N 7beta-hydroxystaurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3C(O)NC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 PBCZSGKMGDDXIJ-UHFFFAOYSA-N 0.000 description 1
- JJTNLWSCFYERCK-UHFFFAOYSA-N 7h-pyrrolo[2,3-d]pyrimidine Chemical class N1=CN=C2NC=CC2=C1 JJTNLWSCFYERCK-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 101710168331 ALK tyrosine kinase receptor Proteins 0.000 description 1
- 229940097396 Aminopeptidase inhibitor Drugs 0.000 description 1
- YUWPMEXLKGOSBF-GACAOOTBSA-N Anecortave acetate Chemical compound O=C1CC[C@]2(C)C3=CC[C@]4(C)[C@](C(=O)COC(=O)C)(O)CC[C@H]4[C@@H]3CCC2=C1 YUWPMEXLKGOSBF-GACAOOTBSA-N 0.000 description 1
- 102400000068 Angiostatin Human genes 0.000 description 1
- 108010079709 Angiostatins Proteins 0.000 description 1
- 108090000644 Angiozyme Proteins 0.000 description 1
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- QULDDKSCVCJTPV-UHFFFAOYSA-N BIIB021 Chemical compound COC1=C(C)C=NC(CN2C3=NC(N)=NC(Cl)=C3N=C2)=C1C QULDDKSCVCJTPV-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 206010065553 Bone marrow failure Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- 102000009410 Chemokine receptor Human genes 0.000 description 1
- 108050000299 Chemokine receptor Proteins 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- 102100032373 Coiled-coil domain-containing protein 85B Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- OMFXVFTZEKFJBZ-UHFFFAOYSA-N Corticosterone Natural products O=C1CCC2(C)C3C(O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 OMFXVFTZEKFJBZ-UHFFFAOYSA-N 0.000 description 1
- 238000006969 Curtius rearrangement reaction Methods 0.000 description 1
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 1
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 1
- 229940122204 Cyclooxygenase inhibitor Drugs 0.000 description 1
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 1
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- AADVCYNFEREWOS-OBRABYBLSA-N Discodermolide Chemical compound C=C\C=C/[C@H](C)[C@H](OC(N)=O)[C@@H](C)[C@H](O)[C@@H](C)C\C(C)=C/[C@H](C)[C@@H](O)[C@@H](C)\C=C/[C@@H](O)C[C@@H]1OC(=O)[C@H](C)[C@@H](O)[C@H]1C AADVCYNFEREWOS-OBRABYBLSA-N 0.000 description 1
- MWWSFMDVAYGXBV-RUELKSSGSA-N Doxorubicin hydrochloride Chemical compound Cl.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-RUELKSSGSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 102400001047 Endostatin Human genes 0.000 description 1
- 108010079505 Endostatins Proteins 0.000 description 1
- 102400001368 Epidermal growth factor Human genes 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- XOZIUKBZLSUILX-SDMHVBBESA-N Epothilone D Natural products O=C1[C@H](C)[C@@H](O)[C@@H](C)CCC/C(/C)=C/C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C XOZIUKBZLSUILX-SDMHVBBESA-N 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 102000056372 ErbB-3 Receptor Human genes 0.000 description 1
- 102000044591 ErbB-4 Receptor Human genes 0.000 description 1
- DBVJJBKOTRCVKF-UHFFFAOYSA-N Etidronic acid Chemical compound OP(=O)(O)C(O)(C)P(O)(O)=O DBVJJBKOTRCVKF-UHFFFAOYSA-N 0.000 description 1
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 1
- 229940124226 Farnesyltransferase inhibitor Drugs 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 108010082772 GFB 111 Proteins 0.000 description 1
- 101710113436 GTPase KRas Proteins 0.000 description 1
- 102100039788 GTPase NRas Human genes 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- JRZJKWGQFNTSRN-UHFFFAOYSA-N Geldanamycin Natural products C1C(C)CC(OC)C(O)C(C)C=C(C)C(OC(N)=O)C(OC)CCC=C(C)C(=O)NC2=CC(=O)C(OC)=C1C2=O JRZJKWGQFNTSRN-UHFFFAOYSA-N 0.000 description 1
- 229920002971 Heparan sulfate Polymers 0.000 description 1
- 102100024025 Heparanase Human genes 0.000 description 1
- 229940122588 Heparanase inhibitor Drugs 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000600756 Homo sapiens 3-phosphoinositide-dependent protein kinase 1 Proteins 0.000 description 1
- 101000868814 Homo sapiens Coiled-coil domain-containing protein 85B Proteins 0.000 description 1
- 101000744505 Homo sapiens GTPase NRas Proteins 0.000 description 1
- 101000624643 Homo sapiens M-phase inducer phosphatase 3 Proteins 0.000 description 1
- 101000579425 Homo sapiens Proto-oncogene tyrosine-protein kinase receptor Ret Proteins 0.000 description 1
- 101000580039 Homo sapiens Ras-specific guanine nucleotide-releasing factor 1 Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101001117146 Homo sapiens [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 1, mitochondrial Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010062016 Immunosuppression Diseases 0.000 description 1
- 241000764238 Isis Species 0.000 description 1
- 102000010638 Kinesin Human genes 0.000 description 1
- 108010063296 Kinesin Proteins 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 102000008072 Lymphokines Human genes 0.000 description 1
- 108010074338 Lymphokines Proteins 0.000 description 1
- 102100023330 M-phase inducer phosphatase 3 Human genes 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 229940124761 MMP inhibitor Drugs 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 description 1
- 101710087603 Mast/stem cell growth factor receptor Kit Proteins 0.000 description 1
- 102000002274 Matrix Metalloproteinases Human genes 0.000 description 1
- 108010000684 Matrix Metalloproteinases Proteins 0.000 description 1
- GMPKIPWJBDOURN-UHFFFAOYSA-N Methoxyamine Chemical compound CON GMPKIPWJBDOURN-UHFFFAOYSA-N 0.000 description 1
- 238000006845 Michael addition reaction Methods 0.000 description 1
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 1
- QJZRFPJCWMNVAV-HHHXNRCGSA-N N-(3-aminopropyl)-N-[(1R)-1-[7-chloro-4-oxo-3-(phenylmethyl)-2-quinazolinyl]-2-methylpropyl]-4-methylbenzamide Chemical compound NCCCN([C@H](C(C)C)C=1N(C(=O)C2=CC=C(Cl)C=C2N=1)CC=1C=CC=CC=1)C(=O)C1=CC=C(C)C=C1 QJZRFPJCWMNVAV-HHHXNRCGSA-N 0.000 description 1
- AYCPARAPKDAOEN-LJQANCHMSA-N N-[(1S)-2-(dimethylamino)-1-phenylethyl]-6,6-dimethyl-3-[(2-methyl-4-thieno[3,2-d]pyrimidinyl)amino]-1,4-dihydropyrrolo[3,4-c]pyrazole-5-carboxamide Chemical compound C1([C@H](NC(=O)N2C(C=3NN=C(NC=4C=5SC=CC=5N=C(C)N=4)C=3C2)(C)C)CN(C)C)=CC=CC=C1 AYCPARAPKDAOEN-LJQANCHMSA-N 0.000 description 1
- LKJPYSCBVHEWIU-UHFFFAOYSA-N N-[4-cyano-3-(trifluoromethyl)phenyl]-3-[(4-fluorophenyl)sulfonyl]-2-hydroxy-2-methylpropanamide Chemical compound C=1C=C(C#N)C(C(F)(F)F)=CC=1NC(=O)C(O)(C)CS(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-UHFFFAOYSA-N 0.000 description 1
- GPVKLYONJSSZFL-UHFFFAOYSA-N NSC 750259 Natural products CCC(C)C=CC(O)C(O)C(O)C(OC)C(=O)NC1CCCCNC1=O GPVKLYONJSSZFL-UHFFFAOYSA-N 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000006057 Non-nutritive feed additive Substances 0.000 description 1
- MSHZHSPISPJWHW-UHFFFAOYSA-N O-(chloroacetylcarbamoyl)fumagillol Chemical compound O1C(CC=C(C)C)C1(C)C1C(OC)C(OC(=O)NC(=O)CCl)CCC21CO2 MSHZHSPISPJWHW-UHFFFAOYSA-N 0.000 description 1
- 108010016076 Octreotide Proteins 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 229920002556 Polyethylene Glycol 300 Polymers 0.000 description 1
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102100028286 Proto-oncogene tyrosine-protein kinase receptor Ret Human genes 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 108091005682 Receptor kinases Proteins 0.000 description 1
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 description 1
- 101710100963 Receptor tyrosine-protein kinase erbB-4 Proteins 0.000 description 1
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 1
- 101710151245 Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 1
- 206010070308 Refractory cancer Diseases 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- 108091027967 Small hairpin RNA Proteins 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 102000004584 Somatomedin Receptors Human genes 0.000 description 1
- 108010017622 Somatomedin Receptors Proteins 0.000 description 1
- 108050001286 Somatostatin Receptor Proteins 0.000 description 1
- 102000011096 Somatostatin receptor Human genes 0.000 description 1
- 229940121856 Somatostatin receptor antagonist Drugs 0.000 description 1
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 239000005864 Sulphur Substances 0.000 description 1
- JXAGDPXECXQWBC-LJQANCHMSA-N Tanomastat Chemical compound C([C@H](C(=O)O)CC(=O)C=1C=CC(=CC=1)C=1C=CC(Cl)=CC=1)SC1=CC=CC=C1 JXAGDPXECXQWBC-LJQANCHMSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229940123582 Telomerase inhibitor Drugs 0.000 description 1
- 108091033399 Telomestatin Proteins 0.000 description 1
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- 102100037236 Tyrosine-protein kinase receptor UFO Human genes 0.000 description 1
- 108090000848 Ubiquitin Proteins 0.000 description 1
- 102000044159 Ubiquitin Human genes 0.000 description 1
- 208000002495 Uterine Neoplasms Diseases 0.000 description 1
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 1
- 108010053100 Vascular Endothelial Growth Factor Receptor-3 Proteins 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 102100033179 Vascular endothelial growth factor receptor 3 Human genes 0.000 description 1
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 1
- UVIQSJCZCSLXRZ-CKFSHRNPSA-N [(3s,8r,9r,10r,13s,14s)-10,13-dimethyl-17-pyridin-3-yl-2,3,4,7,8,9,11,12,14,15-decahydro-1h-cyclopenta[a]phenanthren-3-yl] acetate Chemical compound C([C@@H]1[C@]2(C)CC[C@H]3[C@@]4(C)CC[C@@H](CC4=CC[C@H]31)OC(=O)C)C=C2C1=CC=CN=C1 UVIQSJCZCSLXRZ-CKFSHRNPSA-N 0.000 description 1
- ODEDPKNSRBCSDO-UHFFFAOYSA-N [2-(hexadecylsulfanylmethyl)-3-methoxypropyl] 2-(trimethylazaniumyl)ethyl phosphate Chemical compound CCCCCCCCCCCCCCCCSCC(COC)COP([O-])(=O)OCC[N+](C)(C)C ODEDPKNSRBCSDO-UHFFFAOYSA-N 0.000 description 1
- MQTBAGAVFDZXKF-UHFFFAOYSA-N [2-fluoro-4-(trifluoromethyl)phenyl]methanamine Chemical compound NCC1=CC=C(C(F)(F)F)C=C1F MQTBAGAVFDZXKF-UHFFFAOYSA-N 0.000 description 1
- 102100024148 [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 1, mitochondrial Human genes 0.000 description 1
- 238000002679 ablation Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 229940037127 actonel Drugs 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 208000020990 adrenal cortex carcinoma Diseases 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 208000007128 adrenocortical carcinoma Diseases 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- PYHXGXCGESYPCW-UHFFFAOYSA-N alpha-phenylbenzeneacetic acid Natural products C=1C=CC=CC=1C(C(=O)O)C1=CC=CC=C1 PYHXGXCGESYPCW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- MDFFNEOEWAXZRQ-UHFFFAOYSA-N aminyl Chemical group [NH2] MDFFNEOEWAXZRQ-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000001548 androgenic effect Effects 0.000 description 1
- 229940030486 androgens Drugs 0.000 description 1
- AEMFNILZOJDQLW-QAGGRKNESA-N androst-4-ene-3,17-dione Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 AEMFNILZOJDQLW-QAGGRKNESA-N 0.000 description 1
- 229960005471 androstenedione Drugs 0.000 description 1
- AEMFNILZOJDQLW-UHFFFAOYSA-N androstenedione Natural products O=C1CCC2(C)C3CCC(C)(C(CC4)=O)C4C3CCC2=C1 AEMFNILZOJDQLW-UHFFFAOYSA-N 0.000 description 1
- 229960001232 anecortave Drugs 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 230000000964 angiostatic effect Effects 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 229940030495 antiandrogen sex hormone and modulator of the genital system Drugs 0.000 description 1
- 229940124623 antihistamine drug Drugs 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000000074 antisense oligonucleotide Substances 0.000 description 1
- 238000012230 antisense oligonucleotides Methods 0.000 description 1
- 150000008209 arabinosides Chemical class 0.000 description 1
- 229940078010 arimidex Drugs 0.000 description 1
- 229940087620 aromasin Drugs 0.000 description 1
- 229940046844 aromatase inhibitors Drugs 0.000 description 1
- 238000006254 arylation reaction Methods 0.000 description 1
- FZCSTZYAHCUGEM-UHFFFAOYSA-N aspergillomarasmine B Natural products OC(=O)CNC(C(O)=O)CNC(C(O)=O)CC(O)=O FZCSTZYAHCUGEM-UHFFFAOYSA-N 0.000 description 1
- 229950004810 atamestane Drugs 0.000 description 1
- PEPMWUSGRKINHX-TXTPUJOMSA-N atamestane Chemical compound C1C[C@@H]2[C@@]3(C)C(C)=CC(=O)C=C3CC[C@H]2[C@@H]2CCC(=O)[C@]21C PEPMWUSGRKINHX-TXTPUJOMSA-N 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 108010023337 axl receptor tyrosine kinase Proteins 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 150000007980 azole derivatives Chemical class 0.000 description 1
- XFILPEOLDIKJHX-QYZOEREBSA-N batimastat Chemical compound C([C@@H](C(=O)NC)NC(=O)[C@H](CC(C)C)[C@H](CSC=1SC=CC=1)C(=O)NO)C1=CC=CC=C1 XFILPEOLDIKJHX-QYZOEREBSA-N 0.000 description 1
- 229950001858 batimastat Drugs 0.000 description 1
- 229930195545 bengamide Natural products 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000004244 benzofuran-2-yl group Chemical group [H]C1=C(*)OC2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 125000004532 benzofuran-3-yl group Chemical group O1C=C(C2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 150000003939 benzylamines Chemical class 0.000 description 1
- 238000012925 biological evaluation Methods 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 230000001851 biosynthetic effect Effects 0.000 description 1
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 230000003182 bronchodilatating effect Effects 0.000 description 1
- MJQUEDHRCUIRLF-TVIXENOKSA-N bryostatin 1 Chemical compound C([C@@H]1CC(/[C@@H]([C@@](C(C)(C)/C=C/2)(O)O1)OC(=O)/C=C/C=C/CCC)=C\C(=O)OC)[C@H]([C@@H](C)O)OC(=O)C[C@H](O)C[C@@H](O1)C[C@H](OC(C)=O)C(C)(C)[C@]1(O)C[C@@H]1C\C(=C\C(=O)OC)C[C@H]\2O1 MJQUEDHRCUIRLF-TVIXENOKSA-N 0.000 description 1
- 229960005539 bryostatin 1 Drugs 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000012512 bulk drug substance Substances 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 229940088954 camptosar Drugs 0.000 description 1
- 230000005773 cancer-related death Effects 0.000 description 1
- 229950002826 canertinib Drugs 0.000 description 1
- OMZCMEYTWSXEPZ-UHFFFAOYSA-N canertinib Chemical compound C1=C(Cl)C(F)=CC=C1NC1=NC=NC2=CC(OCCCN3CCOCC3)=C(NC(=O)C=C)C=C12 OMZCMEYTWSXEPZ-UHFFFAOYSA-N 0.000 description 1
- 125000001589 carboacyl group Chemical group 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 229940047495 celebrex Drugs 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- ZGHQGWOETPXKLY-XVNBXDOJSA-N chembl77030 Chemical compound NC(=S)C(\C#N)=C\C1=CC=C(O)C(O)=C1 ZGHQGWOETPXKLY-XVNBXDOJSA-N 0.000 description 1
- 239000012320 chlorinating reagent Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- AOGYCOYQMAVAFD-UHFFFAOYSA-N chlorocarbonic acid Chemical compound OC(Cl)=O AOGYCOYQMAVAFD-UHFFFAOYSA-N 0.000 description 1
- 125000004773 chlorofluoromethyl group Chemical group [H]C(F)(Cl)* 0.000 description 1
- ZPEIMTDSQAKGNT-UHFFFAOYSA-N chlorpromazine Chemical compound C1=C(Cl)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZPEIMTDSQAKGNT-UHFFFAOYSA-N 0.000 description 1
- 229960001076 chlorpromazine Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- OMFXVFTZEKFJBZ-HJTSIMOOSA-N corticosterone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@H](CC4)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OMFXVFTZEKFJBZ-HJTSIMOOSA-N 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- 239000002875 cyclin dependent kinase inhibitor Substances 0.000 description 1
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- 125000000058 cyclopentadienyl group Chemical group C1(=CC=CC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 1
- 229950006418 dactolisib Drugs 0.000 description 1
- JOGKUKXHTYWRGZ-UHFFFAOYSA-N dactolisib Chemical compound O=C1N(C)C2=CN=C3C=CC(C=4C=C5C=CC=CC5=NC=4)=CC3=C2N1C1=CC=C(C(C)(C)C#N)C=C1 JOGKUKXHTYWRGZ-UHFFFAOYSA-N 0.000 description 1
- ZESRJSPZRDMNHY-UHFFFAOYSA-N de-oxy corticosterone Natural products O=C1CCC2(C)C3CCC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 ZESRJSPZRDMNHY-UHFFFAOYSA-N 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 230000007850 degeneration Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 230000001335 demethylating effect Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 229960003654 desoxycortone Drugs 0.000 description 1
- XOZIUKBZLSUILX-UHFFFAOYSA-N desoxyepothilone B Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC(C)=CCC1C(C)=CC1=CSC(C)=N1 XOZIUKBZLSUILX-UHFFFAOYSA-N 0.000 description 1
- 230000000368 destabilizing effect Effects 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- WMKGGPCROCCUDY-PHEQNACWSA-N dibenzylideneacetone Chemical compound C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 WMKGGPCROCCUDY-PHEQNACWSA-N 0.000 description 1
- 150000001991 dicarboxylic acids Chemical class 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 125000004655 dihydropyridinyl group Chemical group N1(CC=CC=C1)* 0.000 description 1
- OTKJDMGTUTTYMP-UHFFFAOYSA-N dihydrosphingosine Natural products CCCCCCCCCCCCCCCC(O)C(N)CO OTKJDMGTUTTYMP-UHFFFAOYSA-N 0.000 description 1
- LVTYICIALWPMFW-UHFFFAOYSA-N diisopropanolamine Chemical compound CC(O)CNCC(C)O LVTYICIALWPMFW-UHFFFAOYSA-N 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- AAXGWYDSLJUQLN-UHFFFAOYSA-N diphenyl(propyl)phosphane Chemical compound C=1C=CC=CC=1P(CCC)C1=CC=CC=C1 AAXGWYDSLJUQLN-UHFFFAOYSA-N 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- FSIRXIHZBIXHKT-MHTVFEQDSA-N edatrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CC(CC)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FSIRXIHZBIXHKT-MHTVFEQDSA-N 0.000 description 1
- 229950006700 edatrexate Drugs 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 229940120655 eloxatin Drugs 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- ZSWFCLXCOIISFI-UHFFFAOYSA-N endo-cyclopentadiene Chemical group C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 1
- 108060002566 ephrin Proteins 0.000 description 1
- 102000012803 ephrin Human genes 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- XOZIUKBZLSUILX-GIQCAXHBSA-N epothilone D Chemical compound O1C(=O)C[C@H](O)C(C)(C)C(=O)[C@H](C)[C@@H](O)[C@@H](C)CCC\C(C)=C/C[C@H]1C(\C)=C\C1=CSC(C)=N1 XOZIUKBZLSUILX-GIQCAXHBSA-N 0.000 description 1
- 150000003883 epothilone derivatives Chemical class 0.000 description 1
- 229960001433 erlotinib Drugs 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 229960005309 estradiol Drugs 0.000 description 1
- 229930182833 estradiol Natural products 0.000 description 1
- 150000002169 ethanolamines Chemical class 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- LIQODXNTTZAGID-OCBXBXKTSA-N etoposide phosphate Chemical compound COC1=C(OP(O)(O)=O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 LIQODXNTTZAGID-OCBXBXKTSA-N 0.000 description 1
- 229960000752 etoposide phosphate Drugs 0.000 description 1
- 229960004945 etoricoxib Drugs 0.000 description 1
- MNJVRJDLRVPLFE-UHFFFAOYSA-N etoricoxib Chemical compound C1=NC(C)=CC=C1C1=NC=C(Cl)C=C1C1=CC=C(S(C)(=O)=O)C=C1 MNJVRJDLRVPLFE-UHFFFAOYSA-N 0.000 description 1
- 229960005167 everolimus Drugs 0.000 description 1
- 229940085363 evista Drugs 0.000 description 1
- 229940087861 faslodex Drugs 0.000 description 1
- 229940087476 femara Drugs 0.000 description 1
- 229960000556 fingolimod Drugs 0.000 description 1
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- 229960005304 fludarabine phosphate Drugs 0.000 description 1
- 229940043075 fluocinolone Drugs 0.000 description 1
- FEBLZLNTKCEFIT-VSXGLTOVSA-N fluocinolone acetonide Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O FEBLZLNTKCEFIT-VSXGLTOVSA-N 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 229940001490 fosamax Drugs 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229940020967 gemzar Drugs 0.000 description 1
- 235000021474 generally recognized As safe (food) Nutrition 0.000 description 1
- 235000021473 generally recognized as safe (food ingredients) Nutrition 0.000 description 1
- UIVFUQKYVFCEKJ-OPTOVBNMSA-N gimatecan Chemical compound C1=CC=C2C(\C=N\OC(C)(C)C)=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UIVFUQKYVFCEKJ-OPTOVBNMSA-N 0.000 description 1
- 229950009073 gimatecan Drugs 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229940080856 gleevec Drugs 0.000 description 1
- 229940084910 gliadel Drugs 0.000 description 1
- 230000001456 gonadotroph Effects 0.000 description 1
- 229960003690 goserelin acetate Drugs 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 108010037536 heparanase Proteins 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 108091008039 hormone receptors Proteins 0.000 description 1
- 229940088013 hycamtin Drugs 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229960001330 hydroxycarbamide Drugs 0.000 description 1
- 229950006905 ilmofosine Drugs 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000004245 indazol-3-yl group Chemical group [H]N1N=C(*)C2=C([H])C([H])=C([H])C([H])=C12 0.000 description 1
- 125000004537 indazol-5-yl group Chemical group N1N=CC2=CC(=CC=C12)* 0.000 description 1
- 125000002249 indol-2-yl group Chemical group [H]C1=C([H])C([H])=C2N([H])C([*])=C([H])C2=C1[H] 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 229940084651 iressa Drugs 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004254 isoquinolin-1-yl group Chemical group [H]C1=C([H])C2=C([H])C([H])=C([H])C([H])=C2C(*)=N1 0.000 description 1
- 125000004551 isoquinolin-3-yl group Chemical group C1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 125000004552 isoquinolin-4-yl group Chemical group C1=NC=C(C2=CC=CC=C12)* 0.000 description 1
- 150000002537 isoquinolines Chemical class 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 150000003951 lactams Chemical group 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- YROQEQPFUCPDCP-UHFFFAOYSA-N losoxantrone Chemical compound OCCNCCN1N=C2C3=CC=CC(O)=C3C(=O)C3=C2C1=CC=C3NCCNCCO YROQEQPFUCPDCP-UHFFFAOYSA-N 0.000 description 1
- 229950008745 losoxantrone Drugs 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229950005069 luminespib Drugs 0.000 description 1
- 229960000994 lumiracoxib Drugs 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- OCSMOTCMPXTDND-OUAUKWLOSA-N marimastat Chemical compound CNC(=O)[C@H](C(C)(C)C)NC(=O)[C@H](CC(C)C)[C@H](O)C(=O)NO OCSMOTCMPXTDND-OUAUKWLOSA-N 0.000 description 1
- 229940121386 matrix metalloproteinase inhibitor Drugs 0.000 description 1
- 239000003771 matrix metalloproteinase inhibitor Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- DLILIUSWDLJMCE-UHFFFAOYSA-N methyl 4-bromo-2-fluorobenzoate Chemical compound COC(=O)C1=CC=C(Br)C=C1F DLILIUSWDLJMCE-UHFFFAOYSA-N 0.000 description 1
- CZNGTXVOZOWWKM-UHFFFAOYSA-N methyl 4-bromobenzoate Chemical compound COC(=O)C1=CC=C(Br)C=C1 CZNGTXVOZOWWKM-UHFFFAOYSA-N 0.000 description 1
- DXOPSTSVRPCTOS-UHFFFAOYSA-N methyl 5-bromo-2-fluorobenzoate Chemical compound COC(=O)C1=CC(Br)=CC=C1F DXOPSTSVRPCTOS-UHFFFAOYSA-N 0.000 description 1
- AAJZXPWBILCHAW-UHFFFAOYSA-N methyl 5-bromopyridine-3-carboxylate Chemical compound COC(=O)C1=CN=CC(Br)=C1 AAJZXPWBILCHAW-UHFFFAOYSA-N 0.000 description 1
- NQMRYBIKMRVZLB-UHFFFAOYSA-N methylamine hydrochloride Chemical compound [Cl-].[NH3+]C NQMRYBIKMRVZLB-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000000394 mitotic effect Effects 0.000 description 1
- ZAHQPTJLOCWVPG-UHFFFAOYSA-N mitoxantrone dihydrochloride Chemical compound Cl.Cl.O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO ZAHQPTJLOCWVPG-UHFFFAOYSA-N 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical compound CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 1
- VYGYNVZNSSTDLJ-HKCOAVLJSA-N monorden Natural products CC1CC2OC2C=C/C=C/C(=O)CC3C(C(=CC(=C3Cl)O)O)C(=O)O1 VYGYNVZNSSTDLJ-HKCOAVLJSA-N 0.000 description 1
- 230000036457 multidrug resistance Effects 0.000 description 1
- XXHOHJTVFUJJMT-UHFFFAOYSA-N n-(3-bromophenyl)acetamide Chemical compound CC(=O)NC1=CC=CC(Br)=C1 XXHOHJTVFUJJMT-UHFFFAOYSA-N 0.000 description 1
- LFMGMKPLBDZJLP-UHFFFAOYSA-N n-(4-bromopyridin-2-yl)acetamide Chemical compound CC(=O)NC1=CC(Br)=CC=N1 LFMGMKPLBDZJLP-UHFFFAOYSA-N 0.000 description 1
- MJFCOXATGBYERZ-UHFFFAOYSA-N n-(5-bromopyridin-2-yl)acetamide Chemical compound CC(=O)NC1=CC=C(Br)C=N1 MJFCOXATGBYERZ-UHFFFAOYSA-N 0.000 description 1
- BLCLNMBMMGCOAS-UHFFFAOYSA-N n-[1-[[1-[[1-[[1-[[1-[[1-[[1-[2-[(carbamoylamino)carbamoyl]pyrrolidin-1-yl]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-[(2-methylpropan-2-yl)oxy]-1-oxopropan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amin Chemical compound C1CCC(C(=O)NNC(N)=O)N1C(=O)C(CCCN=C(N)N)NC(=O)C(CC(C)C)NC(=O)C(COC(C)(C)C)NC(=O)C(NC(=O)C(CO)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C(CC=1NC=NC=1)NC(=O)C1NC(=O)CC1)CC1=CC=C(O)C=C1 BLCLNMBMMGCOAS-UHFFFAOYSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 150000002790 naphthalenes Chemical class 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 229950010159 nemorubicin Drugs 0.000 description 1
- CTMCWCONSULRHO-UHQPFXKFSA-N nemorubicin Chemical compound C1CO[C@H](OC)CN1[C@@H]1[C@H](O)[C@H](C)O[C@@H](O[C@@H]2C3=C(O)C=4C(=O)C5=C(OC)C=CC=C5C(=O)C=4C(O)=C3C[C@](O)(C2)C(=O)CO)C1 CTMCWCONSULRHO-UHQPFXKFSA-N 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 1
- OSTGTTZJOCZWJG-UHFFFAOYSA-N nitrosourea Chemical compound NC(=O)N=NO OSTGTTZJOCZWJG-UHFFFAOYSA-N 0.000 description 1
- 229940085033 nolvadex Drugs 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 125000003518 norbornenyl group Chemical group C12(C=CC(CC1)C2)* 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 229960002700 octreotide Drugs 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- QNDVLZJODHBUFM-WFXQOWMNSA-N okadaic acid Chemical compound C([C@H](O1)[C@H](C)/C=C/[C@H]2CC[C@@]3(CC[C@H]4O[C@@H](C([C@@H](O)[C@@H]4O3)=C)[C@@H](O)C[C@H](C)[C@@H]3[C@@H](CC[C@@]4(OCCCC4)O3)C)O2)C(C)=C[C@]21O[C@H](C[C@@](C)(O)C(O)=O)CC[C@H]2O QNDVLZJODHBUFM-WFXQOWMNSA-N 0.000 description 1
- VEFJHAYOIAAXEU-UHFFFAOYSA-N okadaic acid Natural products CC(CC(O)C1OC2CCC3(CCC(O3)C=CC(C)C4CC(=CC5(OC(CC(C)(O)C(=O)O)CCC5O)O4)C)OC2C(O)C1C)C6OC7(CCCCO7)CCC6C VEFJHAYOIAAXEU-UHFFFAOYSA-N 0.000 description 1
- 238000011474 orchiectomy Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 229940127084 other anti-cancer agent Drugs 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 238000002638 palliative care Methods 0.000 description 1
- VMZMNAABQBOLAK-DBILLSOUSA-N pasireotide Chemical compound C([C@H]1C(=O)N2C[C@@H](C[C@H]2C(=O)N[C@H](C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@H](C(N[C@@H](CC=2C=CC(OCC=3C=CC=CC=3)=CC=2)C(=O)N1)=O)CCCCN)C=1C=CC=CC=1)OC(=O)NCCN)C1=CC=CC=C1 VMZMNAABQBOLAK-DBILLSOUSA-N 0.000 description 1
- NEEFMPSSNFRRNC-HQUONIRXSA-N pasireotide aspartate Chemical compound OC(=O)[C@@H](N)CC(O)=O.OC(=O)[C@@H](N)CC(O)=O.C([C@H]1C(=O)N2C[C@@H](C[C@H]2C(=O)N[C@H](C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@H](C(N[C@@H](CC=2C=CC(OCC=3C=CC=CC=3)=CC=2)C(=O)N1)=O)CCCCN)C=1C=CC=CC=1)OC(=O)NCCN)C1=CC=CC=C1 NEEFMPSSNFRRNC-HQUONIRXSA-N 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- WVUNYSQLFKLYNI-AATRIKPKSA-N pelitinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC1=CC=C(F)C(Cl)=C1 WVUNYSQLFKLYNI-AATRIKPKSA-N 0.000 description 1
- WBXPDJSOTKVWSJ-ZDUSSCGKSA-N pemetrexed Chemical compound C=1NC=2NC(N)=NC(=O)C=2C=1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 WBXPDJSOTKVWSJ-ZDUSSCGKSA-N 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- JLFNLZLINWHATN-UHFFFAOYSA-N pentaethylene glycol Chemical compound OCCOCCOCCOCCOCCO JLFNLZLINWHATN-UHFFFAOYSA-N 0.000 description 1
- XDRYMKDFEDOLFX-UHFFFAOYSA-N pentamidine Chemical compound C1=CC(C(=N)N)=CC=C1OCCCCCOC1=CC=C(C(N)=N)C=C1 XDRYMKDFEDOLFX-UHFFFAOYSA-N 0.000 description 1
- 229960004448 pentamidine Drugs 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- SZFPYBIJACMNJV-UHFFFAOYSA-N perifosine Chemical compound CCCCCCCCCCCCCCCCCCOP([O-])(=O)OC1CC[N+](C)(C)CC1 SZFPYBIJACMNJV-UHFFFAOYSA-N 0.000 description 1
- 229950010632 perifosine Drugs 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 1
- FAIAAWCVCHQXDN-UHFFFAOYSA-N phosphorus trichloride Chemical compound ClP(Cl)Cl FAIAAWCVCHQXDN-UHFFFAOYSA-N 0.000 description 1
- 230000002165 photosensitisation Effects 0.000 description 1
- 239000003504 photosensitizing agent Substances 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 229960004293 porfimer sodium Drugs 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 229950003608 prinomastat Drugs 0.000 description 1
- YKPYIPVDTNNYCN-INIZCTEOSA-N prinomastat Chemical compound ONC(=O)[C@H]1C(C)(C)SCCN1S(=O)(=O)C(C=C1)=CC=C1OC1=CC=NC=C1 YKPYIPVDTNNYCN-INIZCTEOSA-N 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 238000011471 prostatectomy Methods 0.000 description 1
- 229940121649 protein inhibitor Drugs 0.000 description 1
- 239000012268 protein inhibitor Substances 0.000 description 1
- 150000003834 purine nucleoside derivatives Chemical class 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 239000002718 pyrimidine nucleoside Substances 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 150000003248 quinolines Chemical class 0.000 description 1
- AECPBJMOGBFQDN-YMYQVXQQSA-N radicicol Chemical compound C1CCCC(=O)C[C@H]2[C@H](Cl)C(=O)CC(=O)[C@H]2C(=O)O[C@H](C)C[C@H]2O[C@@H]21 AECPBJMOGBFQDN-YMYQVXQQSA-N 0.000 description 1
- 229930192524 radicicol Natural products 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- 229940099538 rapamune Drugs 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 208000016691 refractory malignant neoplasm Diseases 0.000 description 1
- 239000003488 releasing hormone Substances 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- OAKGNIRUXAZDQF-TXHRRWQRSA-N retaspimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(O)C1=CC(O)=C2NCC=C OAKGNIRUXAZDQF-TXHRRWQRSA-N 0.000 description 1
- 229930002330 retinoic acid Natural products 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 238000011808 rodent model Methods 0.000 description 1
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 description 1
- QXKJWHWUDVQATH-UHFFFAOYSA-N rogletimide Chemical compound C=1C=NC=CC=1C1(CC)CCC(=O)NC1=O QXKJWHWUDVQATH-UHFFFAOYSA-N 0.000 description 1
- VHXNKPBCCMUMSW-FQEVSTJZSA-N rubitecan Chemical compound C1=CC([N+]([O-])=O)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VHXNKPBCCMUMSW-FQEVSTJZSA-N 0.000 description 1
- 229950009213 rubitecan Drugs 0.000 description 1
- 229950008902 safingol Drugs 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical class O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229950003647 semaxanib Drugs 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- 229940112726 skelid Drugs 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 239000004055 small Interfering RNA Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- OTKJDMGTUTTYMP-ZWKOTPCHSA-N sphinganine Chemical compound CCCCCCCCCCCCCCC[C@@H](O)[C@@H](N)CO OTKJDMGTUTTYMP-ZWKOTPCHSA-N 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- UXXQOJXBIDBUAC-UHFFFAOYSA-N tandutinib Chemical compound COC1=CC2=C(N3CCN(CC3)C(=O)NC=3C=CC(OC(C)C)=CC=3)N=CN=C2C=C1OCCCN1CCCCC1 UXXQOJXBIDBUAC-UHFFFAOYSA-N 0.000 description 1
- 229950007866 tanespimycin Drugs 0.000 description 1
- 229940120982 tarceva Drugs 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- YVSQVYZBDXIXCC-INIZCTEOSA-N telomestatin Chemical compound N=1C2=COC=1C(N=1)=COC=1C(N=1)=COC=1C(N=1)=COC=1C(N=1)=COC=1C(=C(O1)C)N=C1C(=C(O1)C)N=C1[C@@]1([H])N=C2SC1 YVSQVYZBDXIXCC-INIZCTEOSA-N 0.000 description 1
- 229960000235 temsirolimus Drugs 0.000 description 1
- HKIKRUZACUCNBF-UHFFFAOYSA-N tert-butyl 2-bromo-5,7-dihydro-4h-thieno[2,3-c]pyridine-6-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCC2=C1SC(Br)=C2 HKIKRUZACUCNBF-UHFFFAOYSA-N 0.000 description 1
- UOCVSZYBRMGQOL-UHFFFAOYSA-N tert-butyl 5-bromo-2,3-dihydroindole-1-carboxylate Chemical compound BrC1=CC=C2N(C(=O)OC(C)(C)C)CCC2=C1 UOCVSZYBRMGQOL-UHFFFAOYSA-N 0.000 description 1
- XOIQAACOGWWWGH-UHFFFAOYSA-N tert-butyl 6-bromo-2-oxo-3,4-dihydroquinoline-1-carboxylate Chemical compound BrC1=CC=C2N(C(=O)OC(C)(C)C)C(=O)CCC2=C1 XOIQAACOGWWWGH-UHFFFAOYSA-N 0.000 description 1
- LIUWGGLMAGXFGU-UHFFFAOYSA-N tert-butyl 7-bromo-2-oxo-3,4-dihydroquinoline-1-carboxylate Chemical compound C1=C(Br)C=C2N(C(=O)OC(C)(C)C)C(=O)CCC2=C1 LIUWGGLMAGXFGU-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000005958 tetrahydrothienyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 235000010384 tocopherol Nutrition 0.000 description 1
- 229960001295 tocopherol Drugs 0.000 description 1
- 229930003799 tocopherol Natural products 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- 229930003802 tocotrienol Natural products 0.000 description 1
- 239000011731 tocotrienol Substances 0.000 description 1
- 235000019148 tocotrienols Nutrition 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 229960001612 trastuzumab emtansine Drugs 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 229960005294 triamcinolone Drugs 0.000 description 1
- GFNANZIMVAIWHM-OBYCQNJPSA-N triamcinolone Chemical compound O=C1C=C[C@]2(C)[C@@]3(F)[C@@H](O)C[C@](C)([C@@]([C@H](O)C4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 GFNANZIMVAIWHM-OBYCQNJPSA-N 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- KVJXBPDAXMEYOA-CXANFOAXSA-N trilostane Chemical compound OC1=C(C#N)C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@@]32O[C@@H]31 KVJXBPDAXMEYOA-CXANFOAXSA-N 0.000 description 1
- 229960001670 trilostane Drugs 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- TUCIOBMMDDOEMM-RIYZIHGNSA-N tyrphostin B42 Chemical compound C1=C(O)C(O)=CC=C1\C=C(/C#N)C(=O)NCC1=CC=CC=C1 TUCIOBMMDDOEMM-RIYZIHGNSA-N 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 229960002004 valdecoxib Drugs 0.000 description 1
- LNPDTQAFDNKSHK-UHFFFAOYSA-N valdecoxib Chemical compound CC=1ON=C(C=2C=CC=CC=2)C=1C1=CC=C(S(N)(=O)=O)C=C1 LNPDTQAFDNKSHK-UHFFFAOYSA-N 0.000 description 1
- 229960000241 vandetanib Drugs 0.000 description 1
- YCOYDOIWSSHVCK-UHFFFAOYSA-N vatalanib Chemical compound C1=CC(Cl)=CC=C1NC(C1=CC=CC=C11)=NN=C1CC1=CC=NC=C1 YCOYDOIWSSHVCK-UHFFFAOYSA-N 0.000 description 1
- 229940099039 velcade Drugs 0.000 description 1
- YTZALCGQUPRCGW-ZSFNYQMMSA-N verteporfin Chemical compound N1C(C=C2C(=C(CCC(O)=O)C(C=C3C(CCC(=O)OC)=C(C)C(N3)=C3)=N2)C)=C(C=C)C(C)=C1C=C1C2=CC=C(C(=O)OC)[C@@H](C(=O)OC)[C@@]2(C)C3=N1 YTZALCGQUPRCGW-ZSFNYQMMSA-N 0.000 description 1
- JXLYSJRDGCGARV-CFWMRBGOSA-N vinblastine Chemical compound C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-CFWMRBGOSA-N 0.000 description 1
- NMDYYWFGPIMTKO-HBVLKOHWSA-N vinflunine Chemical compound C([C@@](C1=C(C2=CC=CC=C2N1)C1)(C2=C(OC)C=C3N(C)[C@@H]4[C@@]5(C3=C2)CCN2CC=C[C@]([C@@H]52)([C@H]([C@]4(O)C(=O)OC)OC(C)=O)CC)C(=O)OC)[C@H]2C[C@@H](C(C)(F)F)CN1C2 NMDYYWFGPIMTKO-HBVLKOHWSA-N 0.000 description 1
- 229960000922 vinflunine Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 229940061392 visudyne Drugs 0.000 description 1
- 229960001771 vorozole Drugs 0.000 description 1
- XLMPPFTZALNBFS-INIZCTEOSA-N vorozole Chemical compound C1([C@@H](C2=CC=C3N=NN(C3=C2)C)N2N=CN=C2)=CC=C(Cl)C=C1 XLMPPFTZALNBFS-INIZCTEOSA-N 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
- 229940033942 zoladex Drugs 0.000 description 1
- 229940002005 zometa Drugs 0.000 description 1
- CGTADGCBEXYWNE-JUKNQOCSSA-N zotarolimus Chemical compound N1([C@H]2CC[C@@H](C[C@@H](C)[C@H]3OC(=O)[C@@H]4CCCCN4C(=O)C(=O)[C@@]4(O)[C@H](C)CC[C@H](O4)C[C@@H](/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C3)OC)C[C@H]2OC)C=NN=N1 CGTADGCBEXYWNE-JUKNQOCSSA-N 0.000 description 1
- 229950009819 zotarolimus Drugs 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Definitions
- the present invention relates to cyclic urea derivatives and their use for the treatment of various disease conditions mediated by the regulation of 17 ⁇ -hydroxylase/C 17,20 -lyase.
- Cancer is characterized by an increase in the number of abnormal cells derived from a given normal tissue, invasion of adjacent tissues by these abnormal cells, or lymphatic or blood-borne spread of malignant cells to regional lymph nodes and to distant sites (i.e., metastasis).
- Prostate cancer is currently the most common non-skin cancer and the second leading cause of cancer-related death in men after lung cancer.
- the primary course of treatment for patients diagnosed with organ-confined prostate cancer is usually prostatectomy or radiotherapy.
- These treatments for prostate and breast cancer are highly invasive and characterized by undesirable and serious side effects.
- a large percent of individuals who receive localized treatments such as surgery or radiotherapy may suffer from recurring cancer and widespread metastases.
- surgery and radiation therapies there are several drawbacks to chemotherapy, including the fact that almost all chemotherapeutic agents are toxic, and chemotherapy causes significant, and often dangerous, side effects, such as severe nausea, bone marrow depression, and immunosuppression. Additionally, many tumor cells are resistant or become resistant to chemotherapeutic agents through multi-drug resistance.
- Treatments such as hormone therapy are another option for individuals diagnosed with hormone-dependent, hormone-responsive, or hormone-sensitive cancers, such as prostate or breast cancer.
- hormone-dependent, hormone-responsive, or hormone-sensitive cancers such as prostate or breast cancer.
- hormone-sensitive cancers such as prostate or breast cancer.
- some individuals who have been administered current hormone therapy treatments may not show a significant response to such treatments and some may suffer from relapsing of cancer.
- chemo-refractory and hormone-refractory cancer patients are left with very few treatment options and there remains an unmet need for more effective was to treat cancer such as, but not limited to, prostate cancer and breast cancer.
- the biosynthetic cascade also leads to the formation of gluco- and mineralcorticoids.
- Steroid-type compounds and non-steroid-type compounds are already known as steroid C 17,20 -lyase inhibitors.
- the steroid-type compounds are disclosed in, for example, WO 92/15404, WO 93/20097, EP-A 288053, EP-A 413270 and the like.
- non-steroid-type compounds for example, in WO94/27989, WO96/14090 and WO97/00257 azole derivatives are described in WO95/09157 1H-benzimidazole derivatives are described in U.S. Pat. No. 5,491,161, dihydronaphthalene derivatives are described in WO99/18075, and naphthalene derivatives are shown in WO99/54309.
- Non-steroidal small molecule inhibitors have been described for example in BMC 2004,12, (4313), YM116, 2-(1H-imidazol-4-ylmethyl)-9H-carbazole, and their effects in decreasing adrenal androgen synthesis by inhibiting C17-20 lyase activity in NCI-H295 human adrenocortical carcinoma cells has been described by Ideyama Y, Kudoh M, Tanimoto K, Susaki Y, Nanya T, Nakahara T, Ishikawa H, Fujikura T, Akaza H, Shikama H in “Jpn. J. Pharmacol., 1999, 79:No. 2(213-20)”.
- Novel non-steroidal inhibitor of cytochrome P450 (17 alpha-hydroxylase/C17-20 lyase), YM116, and its role in decreased prostatic weights by reducing the serum concentrations of testosterone and adrenal androgens in rats has been reported by Ideyama Y, Kudoh M, Tanimoto K, Susaki Y, Nanya T, Nakahara T, Ishikawa H, Yoden T, Okada M, Fujikura T, Shikama H Proc. Am. Assoc. Cancer Res., 1998, 39:89 Meet. (384)
- the compounds described herein have been shown to be inhibitors of 17 ⁇ -hydroxylase/C 17,20 -lyase.
- One embodiment of the present invention provides compounds of Formula (I) or (II)
- n 1, 2, or 3;
- phenyl optionally substituted with 1 to 3 substituents selected from halo, -CN, -OH, (C 1 -C 6 )alkyl, halo-substituted (C 1 -C 6 )alkyl, (C 1 -C 6 )alkoxy, -NH 2 , -NH(C 1 -C 4 )alkyl, -N((C 1 -C 4 )alkyl) 2 , -NHC(O)-(C 1 -C 4 )alkyl, -C(O)NH 2 , -C(O)-NH(C 1 -C 4 )alkyl, -C(O)—N((C 1 -C 4 )alkyl) 2 , or a 5- to 6-membered heterocycle,
- R 54 is (C 1 -C 4 )alkyl, halo-substituted (C 1 -C 4 )alkyl, or -CH 2 OH, or two R 54 taken together with the carbon atom(s) to which they are attached form a 3- to 6-membered fully or partially saturated carbocyclic ring or two R 54 attached to adjacent carbons taken together with the carbons to which they are attached form a fused phenyl;
- p 0, 1, 2, or 3;
- q 0, 1 or 2;
- A is a 5- to 10-membered heteroaryl containing one or more nitrogen atom(s), where said heteroaryl is optionally substituted with 1 to 3 substituents each independently selected from halo, -OH, -CN, (C 1 -C 4 )alkyl, halo-substituted (C 1 -C 4 )alkyl, hydroxy-substituted (C 1 -C 4 )alkyl, -(CH 2 ) r O (C 1 -C 4 )alkyl, -(CH 2 ) r CH(O(C 1 -C 4 )alkyl) 2 , -NH(C 1 -C 4 )alkyl, -(CH 2 ) s NH(C 1 -C 4 )alkyl, -(CH 2 ) r N((C 1 -C 4 )alkyl) 2 , -(CH 2 ) r NH(C 3 -C 6 )cycloal
- r 0, 1 or 2;
- s is 1 or 2; or a pharmaceutically acceptable salt thereof
- A is a 5-membered heteroaryl containing 1 to 2 nitrogen atom(s), where said heteroaryl is optionally substituted with (C 1 -C 4 )alkyl or halo-substituted (C 1 -C 4 )alkyl; or a pharmaceutically acceptable salt thereof.
- A is a 6-membered heteroaryl containing 1 to 2 nitrogen atom(s), where said heteroaryl is optionally substituted with (C 1 -C 4 )alkyl or halo-substituted (C 1 -C 4 )alkyl; or a pharmaceutically acceptable salt thereof.
- A is a pyridine, where said pyridine is optionally substituted with 1 to 3 substituents each independently selected from halo, -CN, (C 1 -C 4 )alkyl, halo-substituted (C 1 -C 4 )alkyl, hydroxy-substituted (C 1 -C 4 )alkyl, -(CH 2 ) r O(C 1 -C 4 )alkyl, -(CH 2 ) r CH(O(C 1 -C 4 )alkyl) 2 , -(CH 2 ) r NH(C 1 -C 4 )alkyl, -(CH 2 ) r N((C 1 -C 4 )alkyl) 2 , -(CH 2 ) r NH(C 3 -C 6 )cycloalkyl), -NH 2 , -NHC(O)-(C 1 -C 4 )alkyl, -
- A is an optionally substituted pyridin-3-yl.
- the compound of Formula (I) or (II) is a compound of Formula (Ia) or (Ib)
- R 52 is CF 3 or a (C 1 -C 4 )alkyl substituted with a 5- to 6-membered partially or fully saturated heterocycle (preferably, the heterocycle is attached to the alkyl group via a nitrogen ring atom, e.g. pyrrolidin-1-ylmethyl).
- n is 1, and p is 0.
- Another embodiment of the present invention provides compounds of Formula (Ia) or (Ib).
- n 1, 2, or 3;
- phenyl optionally substituted with 1 to 3 substituents selected from halo, -CN, -OH, (C 1 -C 6 )alkyl, halo-substituted (C 1 -C 6 )alkyl, (C 1 -C 6 )alkoxy, -NH 2 , -NH(C 1 -C 4 )alkyl, -N((C 1 -C 4 )alkyl) 2 , -NHC(O)-(C 1 -C 4 )alkyl, -C(O)NH 7 , -C(O)-NH(C 1 -C 4 )alkyl, -C(O)-N((C 1 -C 4 )alkyl) 2 , or a 5- to 6-membered heterocycle,
- R 54 is (C 1 -C 4 )alkyl (e.g., -CH 3 ), halo-substituted (C 1 -C 4 )alkyl (e.g., -CF 3 ), or -CH 2 OH, or two R 54 taken together with the carbon atom(s) to which they are attached form a 3- to 6-membered fully or partially saturated carbocyclic ring (e.g., for a compound of Formula (Ia) when q is 2, the two R 54 on adjacent carbons may form a fused cycloalkenyl ring; and for compounds of Formula (Ib), when p is 2 or 3, two R 54 on adjacent carbons may form a fused fully or partially saturated cycloalkyl ring or two R 54 on the same carbon atom may form a spiral ring);
- p 0, 1, 2, or 3;
- q 0, 1 or 2;
- R 50 , R 51 and R 52 are each independently H, halo, -OH, -CN, (C 1 -C 4 )alkyl, halo-substituted (C 1 -C 4 )alkyl, hydroxy-substituted (C 1 -C 4 )alkyl, -(CH 2 ), -O(C 1 -C 4 )alkyl, —(CH 2 ), -CH(O(C 1 -C 4 )alkyl) 2 , -NH 2 , -NH(C 1 -C 4 )alkyl, -N((C 1 -C 4 )alkyl) 2 , -NHC(O)-(C 1 -C 4 )alkyl, -C(O)NH 2 , -C(O)-NH(C 1 -C 4 )alkyl, -C(O)-NH(C 1 -C 4 )alkyl, -C(O)
- r 0, 1 or 2;
- R 50 , R 51 , and R 52 are H and R 53 is phenyl, R 53 is not unsubstituted or substituted with halogen or CF 3 , or a pharmaceutically acceptable salt thereof.
- a compound of Formula (Ia) is provided having the definitions above.
- compounds of Formula (Ib) is provided where n is 1.
- R 50 is H or methyl (more preferably, R 50 is H);
- R 51 is H, halo, methyl, trifluoromethyl, methoxy, or -C(O)OCH 3 ;
- R 52 is halo, -CN, methyl, ethyl, methoxy, hydroxymethyl, 1-hydroxyethyl, 2-hydroxypropan-2-yl, difluoromethyl, trifluoromethyl, dimethoxymethyl, -NH 2 , or -NHC(O)CH 3 .
- R 53 is
- a phenyl optionally substituted with 1 of 2 substituents each independently selected form fluoro, chloro, cyano, methyl, difluoromethyl, trifluoromethyl, methoxy, or -C(O)NHCH 3 ;
- a fused phenyl selected from naphthalen-2-yl, naphthalen-1-yl, 1H-indol-5-yl, 1H-indol-6-yl, benzothiazol-5-yl, benzothiazol-6-yl, 1,2,3,4-tetrahydro-quinolin-6-yl, benzo[b]thiophen-5-yl, quinolin-6-yl, quinolin-7-yl indan-5-yl, 1,2-dihydroquinolin-6-yl, 1H-indazol-5-yl, 1H-indazol-6-yl, benzofuran-5-yl, 2,3-dihydrobenzo[1,4]dioxin-6-yl, 2,3-dihydro-benzofuran-5-yl, benzo[1,3]dioxol-5-yl, 1,2,3,4-tetrahydro-quinolin-7-yl,
- a 5- to 6-membered heteroaryl selected from thiophen-2-yl, thiophen-3-yl, pyridin-3-yl, pyridin-4-yl, pyrimidin-5-yl, 1H-pyrazol-4-yl, thiazol-2-yl, or isothiazol-4-yl, where said 5- to 6-membered heteroaryl is optionally substituted with 1 to 3 substituents each independently selected from fluoro, chloro, methyl, ethyl, isopropyl, hydroxy, difluoromethyl, trifluoromethyl, methoxy, -NH 2 , -NHC(O)CH 3 , -C(O)NHCH 3 , or pyrrolidin-1-yl; or
- a fused heteroaryl selected from benzo[b]thiophen-2-yl, benzo[b]thiophen-3-yl, quinolin-2-yl, quinolin-3-yl, benzooxazol-2-yl, benzothiazol-2-yl, 4,5,6,7-tetrahydro-thieno[2,3-c]pyridin-2-yl, imidazo[1,2-a]pyridin-3-yl, imidazo[1,2-a]pyridin-6-yl, imidazo[1,2-a]pyridin-7-yl, 3H-imidazo[4,5-b]pyridin-6-yl, thieno[3,2-c]pyridin-2-yl, thieno[3,2-c]pyridin-3-yl, or 1H-indol-3-yl, where said fused heteroaryl is optionally substituted with 1 to 4 substituents each independently selected from fluoro, chloro, cyano, methyl,
- R 53 is
- a phenyl optionally substituted with 1 to 2 substituents each independently selected from fluoro, chloro, methyl, methoxy, trifluoromethyl, difluoromethyl, or cyano;
- a fused phenyl selected from naphthalen-2-yl, quinolin-6-yl, 3,4-dihydro-2-oxo-quinolin-6-yl, benzo[b]thiophen-5-yl, benzo[d]isoxazol-5-yl, 1H-indazol-6-yl, 1H-indazol-5-yl, benzothiazol-6-yl, 1,2-dihydro-3-oxo-indazol-6-yl, indan-5-yl, 1H-benzotriazol-5-yl, benzofuran-5-yl, 2,3-dihydro-benzo[1,4]dioxin-6-yl, 2,3-dihydro-benzofuran-5-yl, or benzo[1,3]dioxol-5-yl where said fused phenyl is optionally substituted with 1 to 2 substituents each independently selected from chloro, fluoro,
- a 5- to 6-membered heteroaryl selected from isothiazol-4-yl, thiophen-2-yl, thiophen-3-yl, or pyridin-4-yl, where said isothiazol-4-yl, said thiophen-2-yl, said thiophen-3-yl, and said pyridin-4-yl are optionally substituted with fluoro, chloro, methyl, trifluoromethyl, difluoromethyl, or methoxy; or
- a fused heteroaryl selected from thieno[3,2-c]pyridin-2-yl, thieno[3,2-c]pyridin-3-yl, thieno[3,2-c]pyridin-2-yl, imidazo[1,2-a]pyridin-7-yl, or benzo [b]thiophen-2-yl, where said fused heteroaryl is optionally substituted with 1 to 2 substituents each independently selected from fluoro, chloro, methyl, difluoromethyl, trifluoromethyl, cyclopropyl, or amino; or a pharmaceutically acceptable salt thereof.
- R 53 is phenyl, 4-chloro-3-fluoro-phenyl, m-tolyl, 3-methoxy-phenyl, 3-chloro-4-fluoro-phenyl, 4-fluoro-3-methyl-phenyl, 3-trifluoromethyl-phenyl, 3-chloro-phenyl, 4-fluoro-3-trifluoromethyl-phenyl, 3-difluoromethyl-4-fluoro-phenyl, 3-cyano-4-fluorophenyl, 3-cyanophenyl, 3-chloro-4-cyanophenyl, 3,4-difluoro-phenyl, 4-trifluoromethyl-phenyl; or a pharmaceutically acceptable salt thereof.
- R 53 is naphthalen-2-yl, benzo[b]thiophen-5-yl, 3-methyl-benzo[d]isoxazol-5-yl, 1H-indazol-5-yl, 1-methyl-1H-indazol-5-yl, 3-amino-1H-indazol-5-yl, 1H-indazol-6-yl, 3-amino-1H-indazol-6-yl, 3-methyl-1H-indazol-6-yl, 3-trifluoromethyl-1H-indazol-6-yl, benzothiazol-6-yl, 1,2-dihydro-3-oxo-indazol-6-yl, indan-5-yl, 1H-benzotriazol-5-yl, 3-methyl-benzofuran-5-yl, 2,3-dihydro-benzo[1,4]dioxin-6-yl, 2,3-dihydro-benzofuran-5-yl, or
- R 53 is benzothiazol-6-yl, 3-methyl-benzofuran-5-yl, 1H-indazol-6-yl, 3-methyl-1H-indazol-6-yl, or 3-trifluoromethyl-1H-indazol-6-yl; or a pharmaceutically acceptable salt thereof.
- R 53 is 5-methyl-thiophen-2-yl, 5-chloro-thiophen-2-yl, 5-trifluoromethyl-thiophen-2-yl, 5-difluoromethyl-thiophen-3-yl, 5-methyl-thiophen-3-yl, 2-methyl-pyridin-4-yl, 2-trifluoromethyl-pyridin-4-yl, 2-chloro-pyridin-4-yl, or 2-methoxy-pyridin-4-yl; or a pharmaceutically acceptable salt thereof.
- R 53 is 4-chloro-thieno[3,2-c]pyridin-2-yl, 4-chloro-thieno[3,2-c]pyridin-3-yl, thieno[3,2-c]pyridin-2-yl, 3-chloro-imidazo[1,2-a]pyridin-7-yl, benzo[b]thiophen-2-yl, or 4-methylthieno[3,2-c]pyridin-2-yl; or a pharmaceutically acceptable salt thereof.
- R 54 is -CH 3 or CF 3 .
- Particular compounds include: 1-Benzothiazol-6-yl-3-(4-methyl-pyridin-3-yl)-imidazolidin-2-one; 1-(2-Chloro-pyridin-4-yl)-3-(4-methyl-pyridin-3-yl)-imidazolidin-2-one; 1-(4-Chloro-thieno[3,2-c]pyridin-2-yl)-3-(4-methyl-pyridin-3-yl)-imidazolidin-2-one; 1-(1H-Indazol-6-yl)-3-(4-methyl-pyridin-3-yl)-imidazolidin-2-one; 1-(3-Difluoromethyl-4-fluoro-phenyl)-3-(4-methyl-pyridin-3-yl)-1,3-dihydro-imidazol-2-one; 1-(4-Methyl-pyridin-3-yl)-3-(5-methyl-thiophen-3-yl)-1,3
- Other compounds include those described in the Example section below, in particular, those compounds having an IC 50 less than 1 ⁇ M (or 1,000 nM), preferably, less than 500 nM, more preferably, less than 100 nM.
- a pharmaceutical composition which comprises a compound of Formula (I), (II), (Ia) or (Ib) or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable carrier or excipient.
- the pharmaceutical composition optionally comprises at least one additional pharmaceutical agent (suitable pharmaceutical agents are described herein below).
- a method of treating a disease, disorder, or syndrome mediated by Cyp17 inhibition comprises administering a compound according to Formula (I), (II), (Ia) or (Ib), or a pharmaceutical composition comprising the compound of Formula (I), (II), (Ia) or (Ib) and pharmaceutically acceptable excipients, to a subject in need thereof.
- Another aspect of the present invention includes a compound according to Formula (I), (II), (Ia) or (Ib) for use in therapy (e.g., the use of a compound of Formula (Ia) or (Ib) for the treatment of a disease, disorder, or syndrome mediated by Cyp17 inhibition).
- Yet another aspect of the present invention includes a method for treating a disease, disorder or syndrome mediated by Cyp17 inhibition comprising the step of administering
- composition comprising at least one additional pharmaceutical agent and a pharmaceutically acceptable carrier or excipient
- said at least one additional pharmaceutical agent is all anticancer agent, chemotherapy agent, or antiproliferative compound.
- the first and second compositions may be administered either simultaneously or sequentially in any order.
- the disease, disorder, or syndrome is selected from the group consisting of cancer (in particular, prostate cancer) and inflammation.
- alkyl refers to a hydrocarbon radical of the general formula C n H 2n+1 .
- the alkane radical may be straight or branched.
- (C 1 -C 6 )alkyl refers to a monovalent, straight, or branched aliphatic group containing 1 to 6 carbon atoms (e.g., methyl, ethyl, n-propyl, i-propyl, n-butyl, s-butyl, t-butyl, n-pentyl, 1-methylbutyl, 2-methylbutyl, 3-methylbutyl, neopentyl, 3,3-dimethylpropyl, hexyl, 2-methylpentyl, and the like).
- alkyl portion i.e., alkyl moiety
- acyl e.g., alkanoyl
- alkylamino dialkylamino
- alkylthio group alkyl portion of an alkoxy, acyl (e.g., alkanoyl), alkylamino, dialkylamino, and alkylthio group
- Halo-substituted alkyl refers to an alkyl group, as defined above, substituted with at least one halogen atom.
- halogen atom is fluoro
- common haloalkyl groups include fluoromethyl, difluoromethyl, trifluoromethyl, 2,2,2-trifluoroethyl, 2,2,2,1,1-pentafluoroethyl, and the like.
- Mixed halogen substitution are also included (e.g., chlorofluoromethyl).
- alkenyl refers to a monovalent group derived from a hydrocarbon having at least one carbon-carbon double bond.
- C 2 -C 6 -alkenyl refers to a monovalent group derived from a hydrocarbon having two to six carbon atoms and comprising at least one carbon-carbon double bond.
- the alkenyl group can be unbranched or branched. Representative examples of alkenyl include vinyl, 1-propenyl, 2-propenyl, 1-methyl-1-propenyl, 1-methyl-2-propenyl, 2-methyl-1-propenyl, 2-methyl-2-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, and so on.
- alkynyl refers to a monovalent group derived from a hydrocarbon having at least one carbon-carbon triple bond.
- C 2 -C 6 -alkynyl refers to a monovalent group derived from a hydrocarbon having two to six carbon atoms and comprising at least one carbon-carbon triple bond.
- the alkynyl group can be unbranched or branched. Representative examples include ethynyl, propynyl, butyn-1-yl, butyn-2-yl, and so on.
- hydroxy-substituted alkyl refers to an alkyl group, as defined above, substituted with one or more hydroxyl (-OH) groups (e.g., -CH 2 OH, -CH(OH) 2 , -CH(OH)-CH(OH, -CH(OH)-CH 3 , and so on).
- -OH hydroxyl
- the alkyl group is substituted with 1 to 2 hydroxyl groups, more preferably one hydroxyl group.
- Halogen or “halo” may be fluorine, chlorine, bromine or iodine (preferred halogens as substituents are fluorine and chlorine).
- oxo or -C(O)- refers to a carbonyl group.
- a ketone, aldehyde, or part of an acid, ester, amide, lactone, or lactam group for example, a ketone, aldehyde, or part of an acid, ester, amide, lactone, or lactam group.
- partially or fully saturated carbocyclic ring refers to nonaromatic rings that are either partially or fully hydrogenated and may exist as a single ring, bicyclic ring or a spiral ring. Unless specified otherwise, the carbocyclic ring is generally a 3- to 8-membered ring.
- partially or fully saturated carbocyclic rings include groups such as cyclopropyl, cyclopropenyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclopentadienyl, cyclohexyl, cyclohexenyl, cyclohexadienyl, norbornyl (bicyclo[2.2.1]heptyl), norbornenyl, bicyclo[2.2.2]octyl, and the like.
- partially saturated or fully saturated heterocyclic ring refers to nonaromatic rings that are either partially or fully hydrogenated and may exist as a single ring, bicyclic ring or a spiral ring.
- the heterocyclic ring is generally a 3- to 6-membered ring containing 1 to 3 heteroatoms (preferably 1 or 2 heteroatoms) each independently selected from sulfur, oxygen and/or nitrogen.
- Partially saturated or fully saturated heterocyclic rings include groups such as epoxy, aziridinyl, tetrahydrofuranyl, dihydrofuranyl, dihydropyridinyl, pyrolidinyl, N-methylpyrrolidinyl, imidazolidinyl, imidazolinyl, piperidinyl, piperazinyl, pyrazolidinyl, 2H-pyranyl, 4H-pyranyl, 2H-chromenyl, oxazinyl, morpholino, thiomorpholino, tetrahydrothienyl, tetrahydrothienyl 1,1-dioxide, and the like.
- the heterocyclic ring may be attached via any ring member.
- fused phenyl refers to a phenyl group fused to another ring, such as another phenyl (i.e., naphthalene (e.g., naphthalen-2-yl, naphthalen-1-yl), a partially or fully saturated cycloalkyl (e.g., indan-5-yl, 2,3-dihydro-1H-indenyl, or tetrahydronaphthalenyl, etc.), a heteroaryl (e.g., 1H-indol-5-yl, 1H-indol-6-yl, benzothiazol-5-yl, benzothiazol-6-yl, benzo[b]thiophen-5-yl, quinolin-6-yl, quinolin-7-yl, isoquinolin-5-yl, isoquinolin-6-yl isoquinolin-7-yl, isoquinolin-8-yl, indazol
- heteroaryl or “heteroaromatic ring” refers to aromatic moieties containing at least one heteratom (e.g., oxygen, sulfur, nitrogen or combinations thereof) within a 5- to 6-membered aromatic ring system (e.g., pyrrolyl, pyridyl, pyrazolyl, thienyl, furanyl, oxazolyl, imidazolyl, tetrazolyl, triazinyl, pyrimidyl, pyrazinyl, thiazolyl, isothiazolyl, etc.).
- a typical single heteroaryl ring is generally a 5- to 6-membered ring containing one to three heteroatoms each independently selected from oxygen, sulfur and nitrogen.
- fused heteroaryl refers to a heteroaryl group fused to another ring, such as another heteroaryl (e.g. purinyl, thieno[3,2-c]pyridinyl (e.g., thieno[3,2-c]pyridin-2-yl and thieno[3,2-c]pyridin-3-yl), imidazo[1,2-a]pyridinyl (e.g., imidazo[1,2-a]pyridin-3-yl, imidazo[1,2-a]pyridin-6-yl, imidazo[1,2-a]pyridin-7-yl and 3H-imidazo[4,5-b]pyridin-6-yl), or benzo[b]thiophenyl, etc.), phenyl (e.g., benzo[b]thiophen-2-yl, benzo[b]thiophen-3-yl, quinolin-2-yl, quinolin-3-yl, benzooxazol-2
- the fused heteroaryl can be substituted on any of the atoms within the fused system.
- an imidazo[1,2-a]pyridinyl group may be substituted on the imidazole or pyridine portion of the fused system.
- terapéuticaally effective amount means an amount of a compound of the present invention that (i) treats or prevents the particular disease, condition, or disorder, (ii) attenuates, ameliorates, or eliminates one or more symptoms of the particular disease, condition, or disorder, or (iii) prevents or delays the onset of one or more symptoms of the particular disease, condition, or disorder described herein.
- animal refers to humans (male or female), companion animals (e.g., dogs, cats and horses), zoo animals, marine animals, birds and other similar animal species.
- phrases “pharmaceutically acceptable” indicates that the substance or composition must be compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the mammal being treated therewith.
- treating embrace both preventative, i.e., prophylactic, and palliative treatment.
- compound of the present invention refers to compounds of Formula (I), (II), (Ia) and (Ib), prodrugs thereof, pharmaceutically acceptable salts of the compounds, and/or prodrugs, and hydrates or solvates of the compounds, salts, and/or prodrugs, as well as, all stereoisomers (including diastereoisomers and enantiomers), tautomers and isotopically labeled compounds.
- the present invention provides compounds and pharmaceutical formulations thereof that are useful in the treatment of diseases, conditions and/or disorders modulated by the inhibition of 17 ⁇ -hydroxylase/C 17,20 -lyase.
- Compounds of the present invention may be synthesized by synthetic routes that include processes analogous to those well-known in the chemical arts, particularly in light of the description contained herein.
- the starting materials are generally available from commercial sources such as Aldrich Chemicals (Milwaukee, Wis.) or are readily prepared using methods well known to those skilled in the art (e.g., prepared by methods generally described in Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, New York (1967-1999 ed.), or Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, including supplements (also available via the Beilstein online database)).
- reaction schemes depicted below provide potential routes for synthesizing the compounds of the present invention as well as key intermediates.
- Examples section below For a more detailed description of the individual reaction steps, see the Examples section below.
- Those skilled in the art will appreciate that other synthetic routes may be used to synthesize the inventive compounds.
- specific starting materials and reagents are depicted in the schemes and discussed below, other starting materials and reagents can be easily substituted to provide a variety of derivatives and/or reaction conditions.
- many of the compounds prepared by the methods described below can be further modified in light of this disclosure using conventional chemistry well known to those skilled in the art.
- R is represented by -A or the following group
- Step-1 & 2
- Steps 1 and 2 may be synthesized using methods analogous to those described by Kak-Shan Shia, et al., in J. Med. Chem., 2002, 45, 1644-1655 using the desired starting materials which are available commercially or synthesized using known procedures described in the art.
- 2-chloroalkyl isocyanates can be prepared using the methods described by C K Johnson in J Org Chem (1967), 32(5), 1508-10. The reaction times in certain cases were prolonged to increase the % yield as compared to reported yields in the above mentioned J Med Chem reference.
- Step-2 obtained as described above may be converted into the desired products by reacting with the appropriate alkyl or aryl halides preferably chloro/bromo alkyl or an derivatives using conditions well know to those of skill in the art, e.g., the Buchwald-Hartwig C-N coupling conditions or NaH/DMF, and the like.
- Preferred conditions are those known as the ‘Buchwald-Hartwig” reaction, e.g., in the presence of (a) a catalyst, such as copper iodide, (b) a base, such as potassium phosphate or cesium carbonate; and (c) a ligand, such as trans-1,2-diamino cyclohexane, in the presence of suitable solvents (e.g., 1,4-dioxane) at temperatures ranging from about room temperature to the refluxing temperature of the solvent.
- suitable solvents e.g., 1,4-dioxane
- R 53 and R may be introduced in the reverse.
- R 53 -NH 2 is used as the starting material.
- the R group is then introduced in step 3 by using R-X instead of R 53 -X. See, e.g., Example 79 in the Example section below for a more detailed description.
- Scheme 2 describes how one could make the starting material (SM-1) above where R 54 is other than hydrogen.
- the desired chloro carboxylic acid is first converted to its corresponding acid chloride derivative using procedures well-known to those of skill in the art.
- the carboxylic acid derivative may be treated with thionyl chloride in the presence of dimethylformamide (DMF) and a solvent (e.g., dichloromethane (DCM)).
- DMF dimethylformamide
- DCM dichloromethane
- Other chlorinating agents may be used, e.g., phosphorous trichloride or phosphorous pentachloride.
- the acid chloride can then be converted to its corresponding azide by treatment with sodium azide.
- the azide is then converted to the desired isocyanate (SM-1) by the Curtius rearrangement, e.g., heating the azide at elevated temperatures.
- SM-1 isocyanate
- Scheme 3 describes a synthesis for compounds of the present invention having an 1H-imidazol-2(3H)-one core (compounds of Formula (I) or (Ia).
- R is represented by -A or the following moiety
- aromatic or heteroaromatic amines in particular, 6-benzothiazolyl amine, 5-benzo[b]thiophenyl amine, 2-difluoromethyl-1-fluoro-phen-4-yl amine, 2-methyl-thiophene-4-yl amine, and the like
- reagents such as triphosgene, triethylamine (TEA) and suitable solvents (e.g., THF) to provide the corresponding 1-substituted 3-(2,2-dimethoxy-ethyl)-urea intermediate compounds.
- Step 2 1-substituted-1H-imidazol-2(3H)-one intermediates can be prepared by methods analagous to those known in art, such as the procedures described by T. Hafner, et al., in Synthesis (2007) 9, 1403-1411.
- the products of Step 2 obtained as described above may be converted into the desired products by reacting with the desired aryl halides preferably iodo or bromo aryl derivatives using conditions such as the Buchwald-Hartwig C-N coupling conditions as described in Scheme 1 above.
- Scheme 4 provides an alternative synthesis for preparing compounds of Formula (II) or (Ib).
- the desired R 53 group may be attached to the desired amino carboxylate compound via Buchwald-Hartwig C-N coupling conditions or NaH/DMF, and the like.
- the cyclic urea is then formed using methods analogous to those described by Kak-Shan Shia, et al., in J. Med. Chem., 2002, 45, 1644-1655.
- the pyridine derivative may then be coupled to the imidazoline via a Buchwald-Hartwig C-N coupling reaction described previously.
- the unsymmetrical disubstituted-1H-imidazolin-2(31-1)-ones can be prepared by other methods discussed by T. Hafner, et al., in Synthesis (2007) 9, 1403-1411 (e.g., Brazier, S. A, et al., J Chem. Soc. (1912), 101, 2352 and Schonherr, H. J., et al, Chem Ber (1970), 103, 1037).
- Scheme 5 provides another alternative synthesis for preparing compounds of Formula (II) or (Ib).
- R is represented by -A or the following group
- Intermediate (I-5a) may be formed via a Michael addition of the desired amine (R 53 -NH 2 ) to the desired acrylic acid using procedures well-known to those of skill in the art.
- a suitable solvent e.g., toluene
- the amino acid intermediate (I-5a) may then be cyclized to form the cyclic urea intermediate (I-5b).
- the cyclic urea intermediate (I-5b) may be formed by treating the amino acid intermediate (I-5a) with an activating agent (e.g., diphenyl phosphoryl azide (DPPA)) in the presence of an amine (e.g., triethylamine) and appropropriate solvent (e.g., toluene) at elevated temperatures.
- an activating agent e.g., diphenyl phosphoryl azide (DPPA)
- amine e.g., triethylamine
- solvent e.g., toluene
- Example section below provides a more detailed description of the synthetic schemes as well as other alternative processes for making compounds of the present invention which could be easily modified (e.g., substituting different starting materials) by those of skill in the art.
- the compounds and intermediates described herein may be isolated and used as the compound per se or its salt.
- Many of the compounds represented by Formula (I), (II), (Ia), and (Ib) are capable of forming acid addition salts, particularly pharmaceutically acceptable acid addition salts.
- Pharmaceutically acceptable acid addition salts of the compound of Formula (I), (II), (Ia) and (Ib) include those of inorganic acids, for example, hydrohalic acids such as hydrochloric acid, hydrobromic acid or hydroiodic acid, nitric acid, sulfuric acid, phosphoric acid; and organic acids, for example aliphatic monocarboxylic acids such as formic acid, acetic acid, propionic acid and butyric acid, aliphatic hydroxy acids such as lactic acid, citric acid, tartaric acid or malic acid, dicarboxylic acids such as maleic acid or succinic acid, aromatic carboxylic acids such as benzoic acid, p-chlorobenzoic acid, diphenylacetic acid or triphenylacetic acid, aromatic hydroxy acids such as o-hydroxybenzoic acid, p-hydroxybenzoic acid, 1-hydroxynaphthalene-2-carboxylic acid or 3-hydroxynaphthalene-2-carboxylic acid, and sulfonic
- compositions of the present invention which contain acidic, e.g. carboxyl, groups, are also capable of forming salts with bases, in particular pharmaceutically acceptable bases such as those well known in the art; suitable such salts include metal salts, particularly alkali metal or alkaline earth metal salts such as sodium, potassium, magnesium or calcium salts, or salts with ammonia or pharmaceutically acceptable organic amines or heterocyclic bases such as ethanolamines, benzylamines or pyridine. These salts may be prepared from compounds of Formula (I), (II), (Ia) and (Ib) by known salt-forming procedures.
- the compounds exist in individual optically active isomeric forms or as mixtures thereof, e.g. as racemic or diastereomeric mixtures.
- the present invention embraces both individual optically active R and S isomers as well as mixtures, e.g. racemic or diastereomeric mixtures, thereof.
- the present invention includes all pharmaceutically acceptable isotopically-labeled compounds of the present invention wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes suitable for inclusion in the compounds of the invention comprises isotopes of hydrogen, such as 2 H and 3 H, carbon, such as 11 C, 13 C and 14 C, chlorine, such as 36 Cl fluorine, such as 18 F, iodine, such as 123 I and 125 I, nitrogen, such as 13 N and 15 N, oxygen, such as 15 O, 17 O and 18 O, phosphorus, such as 32 P, and sulphur, such as 35 S.
- hydrogen such as 2 H and 3 H
- carbon such as 11 C, 13 C and 14 C
- chlorine such as 36 Cl fluorine, such as 18 F
- iodine such as 123 I and 125 I
- nitrogen such as 13 N and 15 N
- oxygen such as 15 O, 17 O and 18 O
- phosphorus such as 32 P
- sulphur such as 35 S.
- substitution with heavier isotopes such as deuterium, i.e. 2 H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances.
- Isotopically-labeled compounds of the present invention can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations Sections using an appropriate isotopically-labeled reagent in place of the non-labeled reagent previously employed.
- solvates are considered pharmaceutical compositions, e.g., a compound of Formula (I), (II), (Ia) or (Ib) (or pharmaceutically acceptable salt thereof) in combination with all excipient, wherein the excipient is a solvent.
- Compounds of the present invention are useful for treating diseases, conditions and disorders mediated by the regulation of 17 ⁇ -hydroxylase/C 17,20 -lyase (e.g., cancer (in particular, prostate cancer) or inflammation); consequently, the compounds of the present invention (including the compositions and processes used therein) may be used in the manufacture of a medicament for the therapeutic applications described herein.
- another embodiment of the present invention is a pharmaceutical composition comprising a therapeutically effective amount of a compound of the present invention and a pharmaceutically acceptable excipient, diluent or carrier.
- a typical formulation is prepared by mixing a compound of the present invention and a carrier, diluent or excipient.
- Suitable carriers, diluents and excipients are well known to those skilled in the art and include materials such as carbohydrates, waxes, water soluble and/or swellable polymers, hydrophilic or hydrophobic materials, gelatin, oils, solvents, water, and the like.
- the particular carrier, diluent or excipient used will depend upon the means and purpose for which the compound of the present invention is being applied. Solvents are generally selected based on solvents recognized by persons skilled in the art as safe (GRAS) to be administered to a mammal.
- GRAS solvents recognized by persons skilled in the art as safe
- safe solvents are non-toxic aqueous solvents such as water and other non-toxic solvents that are soluble or miscible in water.
- Suitable aqueous solvents include water, ethanol, propylene glycol, polyethylene glycols (e.g., PEG400, PEG300), etc. and mixtures thereof.
- the formulations may also include one or more buffers, stabilizing agents, surfactants, wetting agents, lubricating agents, emulsifiers, suspending agents, preservatives, antioxidants, opaquing agents, glidants, processing aids, colorants, sweeteners, perfuming agents, flavoring agents and other known additives to provide an elegant presentation of the drug (i.e., a compound of the present invention or pharmaceutical composition thereof) or aid in the manufacturing of the pharmaceutical product (i.e., medicament).
- buffers stabilizing agents, surfactants, wetting agents, lubricating agents, emulsifiers, suspending agents, preservatives, antioxidants, opaquing agents, glidants, processing aids, colorants, sweeteners, perfuming agents, flavoring agents and other known additives to provide an elegant presentation of the drug (i.e., a compound of the present invention or pharmaceutical composition thereof) or aid in the manufacturing of the pharmaceutical product (i.e., medicament).
- the formulations may be prepared using conventional dissolution and mixing procedures.
- the bulk drug substance i.e., compound of the present invention or stabilized form of the compound (e.g., complex with a cyclodextrin derivative or other known complexation agent)
- a suitable solvent in the presence of one or more of the excipients.
- the compound of the present invention is typically formulated into pharmaceutical dosage forms to provide an easily controllable dosage of the drug and to give the patient an elegant and easily handleable product.
- the pharmaceutical composition (or formulation) for application may be packaged in a variety of ways depending upon the method used for administering the drug.
- an article for distribution includes a container having deposited therein the pharmaceutical formulation in an appropriate form.
- Suitable containers are well-known to those skilled in the art and include materials such as bottles (plastic and glass), sachets, ampoules, plastic bags, metal cylinders, and the like.
- the container may also include a tamper-proof assemblage to prevent indiscreet access to the contents of the package.
- the container has deposited thereon a label that describes the contents of the container. The label may also include appropriate warnings.
- a Cyp17 inhibitor of the present invention may be usefully combined with at least one additional pharmacologically active compound, particularly in the treatment of cancer.
- a compound of the present invention may be administered simultaneously, sequentially or separately in combination with one or more agents selected from chemotherapy agents, e.g. mitotic inhibitors such as a taxane (e.g., paclitaxel or docetaxel), a vinca alkaloid (e.g., vincristine, vinblastine, vinorelbine or vinflunine) or other anticancer agents, e.g. cisplatin, 5-fluorouracil or 5-fluoro-2-4(1H,3H)-pyrimidinedione (5FU), flutamide or gemcitabine.
- chemotherapy agents e.g. mitotic inhibitors such as a taxane (e.g., paclitaxel or docetaxel), a vinca alkaloid (e.g., vincristine, vinblastine, vinorelbine or vinflun
- a compound of the present invention may also be used in combination with other antiproliferative compounds.
- antiproliferative compounds include, but are not limited to aromatase inhibitors; antiestrogens; topoisomerase I inhibitors; topoisomerase II inhibitors; microtubule active compounds; alkylating compounds; compounds which induce cell differentiation processes; cyclooxygenase inhibitors; MMP inhibitors; mTOR inhibitors; antineoplastic antimetabolites; platin compounds; compounds targeting/decreasing a protein or lipid kinase activity and further anti-angiogenic compounds; compounds which target, decrease or inhibit the activity of a protein or lipid phosphatase; gonadorelin agonists; anti-androgens; methionine aminopeptidase inhibitors; bisphosphonates; biological response modifiers; antiproliferative antibodies; heparanase inhibitors; inhibitors of Ras oncogenic isoforms; telomerase inhibitors; proteasome inhibitors; compounds
- tumor treatment approaches including surgery, ionizing radiation, photodynamic therapy, implants, e.g. with corticosteroids, hormones, or they may be used as radiosensitizers.
- implants e.g. with corticosteroids, hormones, or they may be used as radiosensitizers.
- anti-inflammatory and/or antiproliferative treatment combination with anti-inflammatory drugs is included. Combination is also possible with antihistamine drug substances, bronchodilatatory drugs, NSAID or antagonists of chemokine receptors.
- aromatase inhibitor as used herein relates to a compound which inhibits the estrogen production, i.e. the conversion of the substrates androstenedione and testosterone to estrone and estradiol, respectively.
- the term includes, but is not limited to steroids, especially atame-stane, exemestane and formestane and, in paricular, non-steroids, especially aminoglutethimide, roglethimide, pyridoglutethimide, trilostane, testolactone, ketokonazole, vorozole, fadrozole, anastrozole and letrozole.
- Exemestane can be administered, e.g., in the form as it is marketed, e.g.
- AROMASIN Formestane can be administered, e.g., in the form as it is marketed, e.g. under the trademark LENTARON. Fadrozole can be administered, e.g., in the form as it is marketed, e.g. un-der the trademarkAFEMA. Anastrozole can be administered, e.g., in the form as it is marketed, e.g. under the trademark ARIMIDEX. Letrozole can be administered, e.g., in the form as it is marketed, e.g. under the trademark FEMARA or FEMAR. Amino glutethimide can be administered, e.g., in the form as it is marketed, e.g. under the trademark, ORIMETEN.
- a combination of the invention comprising a chemo-therapeutic agent which is an aromatase inhibitor is particularly useful for the treatment of hormone receptor positive tumors, e.g., breast tumors.
- anti-estrogen as used herein relates to a compound which antagonizes the effect of estrogens at the estrogen receptor level.
- the term includes, but is not limited to tamoxifen, fulvestrant, raloxifene and raloxifene hydrochloride.
- Tamoxifen can be administered, e.g., in the form as it is marketed, e.g. under the trademark NOLVADEX.
- Raloxifene hydrochloride can be administered, e.g., in the form as it is marketed, e.g. under the trademark EVISTA.
- Fulvestrant can be formulated as disclosed in U.S. Pat. No.
- 4,659,516 or it can be administered, e.g., in the form as it is marketed, e.g. under the trademark FASLODEX.
- a combination of the invention comprising a chemotherapeutic agent which is an anti-estrogen is particularly useful for the treatment of estrogen receptor positive tumors, e.g. breast tumors.
- anti-androgen as used herein relates to any substance which is capable of inhibiting the biological effects of androgenic hormones and includes, but is not limited to, bicalutamide (CASODEX), which can be formulated, e.g. as disclosed in U.S. Pat. No. 4,636,505.
- CASODEX bicalutamide
- gonadorelin agonist as used herein includes, but is not limited to abarelix, goserelin and goserelin acetate. Goserelin is disclosed in U.S. Pat. No. 4,100,274 and can be administered. e.g., in the form as it is marketed, e.g. under the trademark ZOLADEX. Abarelix can be formulated, e.g. as disclosed in U.S. Pat. No. 5,843,901.
- topoisomerase I inhibitor includes, but is not limited to topotecan, gimatecan, irinotecan, camptothecin and its analogues, 9-nitrocamptothecin and the macromolecular camptothecin conjugate PNU-166148 (compound Al in WO99/17804).
- Irinotecan can be administered, e.g. in the form as it is marketed, e.g. under the trademark CAMPTOSAR.
- Topotecan can be administered, e.g., in the form as it is marketed, e.g. under the trademark HYCAMTIN.
- topoisomerase II inhibitor includes, but is not limited to the anthracyclines such as doxorubicin (including liposomal formulation, e.g. CAELYX), daunorubicin, epirubicin, idarubicin and nemorubicin, the anthraquinones mitoxantrone and losoxantrone, and the podophillotoxines etoposide and teniposide.
- Etoposide can be administered, e.g. in the form as it is marketed, e.g. under the trademark ETOPOPHOS.
- Teniposide can be administered, e.g. in the form as it is marketed, e.g.
- Doxorubicin can be administered, e.g. in the form as it is marketed, e.g. under the trademark ADRIBLASTIN or ADRIAMYCIN.
- Epirubicin can be administered, e.g. in the form as it is marketed, e.g. under the trademark FARMORUBICIN.
- Idarubicin can be administered, e.g. in the form as it is marketed, e.g. under the trademark ZAVEDOS.
- Mitoxantrone can be administered, e.g. in the form as it is marketed, e.g. under the trademark NOVANTRON.
- microtubule active compound relates to microtubule stabilizing, microtubule destabilizing compounds and microtublin polymerization inhibitors including, but not limited to taxanes, e.g. paclitaxel and docetaxel, vinca alkaloids, e.g., vinblastine, especially vinblastine sulfate, vincristine especially vincristine sulfate, and vinorelbine, discodermolides, cochicine and epothilones and derivatives thereof, e.g. epothilone B or D or derivatives thereof.
- Paclitaxel may be administered e.g. in the form as it is marketed, e.g. TAXOL.
- Docetaxel can be administered, e.g., in the form as it is marketed, e.g. under the trademark TAXOTERE.
- Vinblastine sulfate can be administered, e.g., in the form as it is marketed, e.g. under the trademark VINBLASTIN R.P.
- Vincristine sulfate can be administered, e.g., in the form as it is marketed, e.g. under the trademark FARMISTIN.
- Discodermolide can be obtained, e.g., as disclosed in U.S. Pat. No. 5,010,099.
- Epothilone derivatives which are disclosed in WO 98/10121, U.S. Pat. No. 6,194,181, WO 98/25929, WO 98/08849, WO 99/43653, WO 98/22461 and WO 00/31247.
- Epothilone A and/or B are also included.
- alkylating compound includes, but is not limited to, cyclophosphamide, ifosfamide, melphalan or nitrosourea (BCNU or Gliadel).
- Cyclophosphamide can be administered, e.g., in the form as it is marketed, e.g. under the trademark CYCLOSTIN.
- Ifosfamide can be administered, e.g., in the form as it is marketed, e.g., under the trademark HOLOXAN.
- antimetabolite includes, but is not limited to, 5-Fluorouracil or 5-FU, capecitabine, gemcitabine, DNA demethylating compounds, such as 5-azacy-tidine and decitabine, methotrexate and edatrexate, and folic acid antagonists such as pemetrexed.
- Capecitabine can be administe-red, e.g., in the form as it is marketed, e.g. under the trademark XELODA.
- Gemcitabine can be administered, e.g., in the form as it is marketed, e.g. under the trademark GEMZAR.
- platinum compound as used herein includes, but is not limited to, carboplatin, cis-platin, cisplatinum and oxaliplatin.
- Carboplatin can be administered, e.g., in the form as it is marketed, e.g. under the trademark CARBOPLAT.
- Oxaliplatin can be administered, e.g., in the form as it is marketed, e.g. under the trademark ELOXATIN.
- compounds targeting/decreasing a protein or lipid kinase activity includes, but is not limited to, protein tyrosine kinase and/or serine and/or threonine kinase inhibitors or lipid kinase inhibitors, e.g., a) compounds targeting, decreasing or inhibiting the activity of the platelet-derived growth factor-receptors (PDGFR), such as compounds which target, decrease or inhibit the activity of PDGFR, especially compounds which inhibit the PDGF receptor, e.g.
- PDGFR platelet-derived growth factor-receptors
- a N-phenyl-2-pyrimidine-amine derivative e.g. imatinib, SU101, SU6668 and GFB-111
- imatinib h
- imatinib or nilotinib AN 107 ); PD180970; AG957; NSC 680410: PD173955 from ParkeDavis; or dasatinib (BMS-354825); i) compounds targeting, decreasing or inhibiting the activity of members of the protein kinase C (PKC) and Raf family of serine/threonine kinases, members of the MEK, SRC, JAK, FAK, PDK1, PKB/Akt, and Ras/MAPK family members, and/or members of the cyclin-dependent kinase family (CDK) and are especially those staurosporine derivatives disclosed in U.S. Pat. No. 5,093,330, e.g.
- PKC protein kinase C
- Raf family of serine/threonine kinases members of the MEK, SRC, JAK, FAK, PDK1, PKB/Akt, and Ras/MAPK family members
- CDK cycl
- examples of further compounds include e.g. UCN-01, safingol, BAY 43-9006, Bryostatin 1, Perifosine; Ilmofosine; RO 318220 and RO 320432; GO 6976; Isis 3521; LY333531/LY379196; isochinoline compounds such as those disclosed in WO 00/09495; FTIs; BEZ235 (a PI3K inhibitor) or AT7519 (CDK inhibitor); j) compounds targeting, decreasing or inhibiting the activity of protein-tyrosine kinase inhibitors, such as compounds which target, decrease or inhibit the activity of protein-tyrosine kinase inhibitors include imatinib mesylate (GLEEVEC) or tyrphostin.
- GLEEVEC imatinib mesylate
- a tyrphostin is preferably a low molecular weight (mw ⁇ 1500) compound, or a pharmaceutically acceptable salt thereof, especially a compound selected from the benzylidenemalonitrile class or the S-arylbenzenemalonirile or bisubstrate quinoline class of compounds, more especially any compound selected from the group consisting of Tyrphostin A23/RG-50810; AG 99; Tyrphostin AG 213: Tyrphostin AG 1748; Tyrphostin AG 490; Tyrphostin B44; Tyrphostin B44 (+) enantiomer; Tyrphostin AG 555; AG 494; Tyrphostin AG 556, AG957 and adaphostin (4- ⁇ [(2,5-dihydroxyphenyl)methyl]amino ⁇ -benzoic acid adamantyl ester; NSC 680410, adaphostin); k) compounds targeting, decreasing or inhibiting the activity of the epidermal growth factor family of receptor
- EGF receptor ErbB2, ErbB3 and ErbB4 or bind to EGF or EGF related ligands, and are in particular those compounds, proteins or monoclonal antibodies generically and specifically disclosed in WO 97/02266, e.g. the compound of ex. 39, or in EP 0 564 409, WO 99/03854, EP 0520722, EP 0 566 226, EP 0 787 722, EP 0 837 063, U.S. Pat. No. 5,747,498, WO 98/10767, WO 97/30034, WO 97/49688, WO 97/38983 and, especially, WO 96/30347 (e.g.
- WO 96/33980 e.g. compound ZD 1839
- WO 95/03283 e.g. compound ZM105180
- trastuzumab Herceptin
- cetuximab Erbitux
- Iressa Tarceva
- OSI-774 CI-1033
- EKB-569 E1.1, E2.4, E2.5, E6.2, E6.4, E2.11, E6.3 or E7.6.3, and 7H-pyrrolo-[2,3-d]pyrimidine derivatives which are disclosed in WO 03/013541
- 1) compounds targeting, decreasing or inhibiting the activity of the c-Met receptor such as compounds which target, decrease or inhibit the activity of c-Met, especially compounds which inhibit the kinase activity of c-Met receptor, or antibodies that target the extra-cellular domain of c-Met or bind to HGF.
- anti-angiogenic compounds include compounds having another mechanism for their activity, e.g. unrelated to protein or lipid kinase inhibition e.g. thalidomide (THALOMID) and TNP-470.
- TAALOMID thalidomide
- TNP-470 TNP-470.
- Compounds which target, decrease or inhibit the activity of a protein or lipid phosphatase are e.g., inhibitors of phosphatase 1, phosphatase 2A, or CDC25, e.g. okadaic acid or a derivative thereof.
- Compounds which induce cell differentiation processes are e.g. retinoic acid, or tocopherol or tocotrienol.
- cyclooxygenase inhibitor as used herein includes, but is not limited to, e.g. Cox-2 inhibitors, 5-alkyl substituted 2-arylaminophenylacetic acid and derivatives, such as celecoxib (CELEBREX), rofecoxib (VIOXX), etoricoxib, valdecoxib or a 5-alkyl-2-arylaminophenylacetic acid, e.g. 5-methyl-2-(2′-chloro-6′-fluoroanilino)phenyl acetic acid, lumiracoxib.
- Cox-2 inhibitors 5-alkyl substituted 2-arylaminophenylacetic acid and derivatives, such as celecoxib (CELEBREX), rofecoxib (VIOXX), etoricoxib, valdecoxib or a 5-alkyl-2-arylaminophenylacetic acid, e.g. 5-methyl-2-(2′-chloro-6′-fluor
- bisphosphonates as used herein includes, but is not limited to, etridonic, clodronic, tiludronic, pamidronic, alendronic, ibandronic, risedronic and zoledronic acid.
- Etridonic acid can be administered, e.g., in the form as it is marketed, e.g. under the trademark DIDRONEL.
- Clodronic acid can be administered, e.g., in the form as it is marketed, e.g. under the trademark BONEFOS.
- titaniumudronic acid can be administered, e.g., in the form as it is marketed, e.g. under the trademark SKELID.
- “Pamidronic acid” can be administered, e.g. in the form as it is marketed, e.g. under the trademark AREDIA.
- “Alendronic acid” can be administered, e.g., in the form as it is marketed, e.g. under the trademark FOSAMAX.
- “Ibandronic acid” can be administered, e.g., in the form as it is marketed, e.g. under the trademark BONDRANAT.
- “Risedronic acid” can be administered, e.g., in the form as it is marketed, e.g. under the trademark ACTONEL.
- “Zoledronic acid” can be administered, e.g. in the form as it is marketed, e.g. under the trademark ZOMETA.
- mTOR inhibitors relates to compounds which inhibit the mammalian target of rapamycin (mTOR) and which possess antiproliferative activity such as sirolimus (Rapamune), everolimus (Certican ⁇ ), CCI-779 and ABT578.
- heparanase inhibitor refers to compounds which target, decrease or inhibit heparin sulfate degradation.
- the term includes, but is not limited to, PI-88.
- biological response modifier refers to a lymphokine or interferons, e.g. interferon.
- inhibitor of Ras oncogenic isoforms e.g. H-Ras, K-Ras, or N-Ras, as used herein refers to compounds which target, decrease or inhibit the oncogenic activity of Ras e.g. a “farnesyl transferase inhibitor” e.g. L-744832, DK8G557 or R115777 (Zarnestra).
- telomerase inhibitor refers to compounds which target, decrease or inhibit the activity of telomerase.
- Compounds which target, decrease or inhibit the activity of telomerase are especially compounds which inhibit the telomerase receptor, e.g. telomestatin.
- methionine aminopeptidase inhibitor refers to compounds which target, decrease or inhibit the activity of methionine aminopeptidase.
- Compounds which target, decrease or inhibit the activity of methionine aminopeptidase are e.g. bengamide or a derivative thereof.
- proteasome inhibitor refers to compounds which target, decrease or inhibit the activity of the proteasome.
- Compounds which target, decrease or inhibit the activity of the proteasome include e.g. Bortezomid (Velcade) and MLN 341.
- matrix metalloproteinase inhibitor or (“MMP” inhibitor) as used herein includes, but is not limited to, collagen peptidomimetic and nonpeptidomimetic inhibitors, tetracycline derivatives, e.g.
- compounds used in the treatment of hematologic malignancies includes, but is not limited to, FMS-like tyrosine kinase inhibitors e.g.
- FMS-like tyrosine kinase receptors FMS-like tyrosine kinase receptors
- ara-c 1-b-D-arabinofuransylcytosine
- ALK inhibitors e.g. compounds which target, decrease or inhibit anaplastic lymphoma kinase.
- Compounds which target, decrease or inhibit the activity of FMS-like tyrosine kinase receptors (Flt-3R) are especially compounds, proteins or antibodies which inhibit members of the Flt-3R receptor kinase family, e.g. PKC412, TKI258, midostaurin, a staurosporine derivative, SU11248 and MLN518.
- HSP90 inhibitors includes, but is not limited to, compounds targeting, decreasing or inhibiting the intrinsic ATPase activity of HSP90; degrading, targeting, decreasing or inhibiting the HSP90 client proteins via the ubiquitin proteosome pathway.
- Compounds targeting, decreasing or inhibiting the intrinsic ATPase activity of HSP90 are especially compounds, proteins or antibodies which inhibit the ATPase activity of HSP90 e.g., 17-allylamino,17-demethoxygeldanamycin (17AAG), a geldanamycin derivative; other geldanamycin related compounds; radicicol and HDAC inhibitors.
- An example HSP90 inhibitor is AUY922.
- antiproliferative antibodies includes, but is not limited to, trastuzumab (Herceptin), Trastuzumab-DM1,erbitux, bevacizumab (Avastin), rituximab (Rituxan), PRO64553 (anti-CD40) and 2C4 Antibody.
- antibodies is meant e.g. intact monoclonal antibodies, polyclonal antibodies, multispe-cific antibodies formed from at least 2 intact antibodies, and antibodies fragments so long as they exhibit the desired biological activity.
- compounds of formula (I) can be used in combination with standard leukemia therapies, especially in combination with therapies used for the treatment of AML.
- compounds of formula (I) can be administered in combination with, e.g., farnesyl transferase inhibitors and/or other drugs useful for the treatment of AML, such as Daunorubicin, Adriamycin, Ara-C, VP-16, Teniposide, Mitoxantrone, Idarubicin, Carboplatinum and PKC412.
- antigenemic compounds includes, for example, Ara-C, a pyrimidine analog, which is the 2-alpha-hydroxy ribose (arabinoside) derivative of deoxycytidine. Also included is the purine analog of hypoxanthine, 6-mercaptopurine (6-MP) and fludarabine phosphate.
- Somatostatin receptor antagonists refers to compounds which target, treat or inhibit the somatostatin receptor such as octreotide, and SOM230 (pasireotide).
- Tumor cell damaging approaches refer to approaches such as ionizing radiation.
- ionizing radiation means ionizing radiation that occurs as either electromagnetic rays (such as X-rays and gamma rays) or particles (such as alpha and beta particles). Ionizing radiation is provided in, but not limited to, radiation therapy and is known in the art. See Hellman, Principles of Radiation Therapy, Cancer, in Principles and Practice of Oncology, Devita et al., Eds., 4 th Edition, Vol. 1, pp. 248-275 (1993).
- EDG binders refers a class of immunosuppressants that modulates lymphocyte recirculation, such as FTY720.
- ribonucleotide reductase inhibitors refers to pyrimidine or purine nucleoside analogs including, but not limited to, fludarabine and/or cytosine arabinoside (ara-C), 6-thioguanine, 5-fluorouracil, cladribine, 6-mercaptopurine (especially in combination with ara-C against ALL) and/or pentostatin.
- Ribonucleotide reductase inhibitors are especially hydroxyurea or 2-hydroxy-1H-isoindole-1,3-dione derivatives, such as PL-1, PL-2, PL-3, PL-4, PL-5, PL-6, PL-7 or PL-8 mentioned in Nandy et al., Acta Oncologica, Vol. 33, No. 8, pp. 953-961 (1994).
- S-adenosylmethionine decarboxylase inhibitors includes, but is not limited to the compounds disclosed in U.S. Pat. No. 5,461,076.
- VEGF vascular endothelial growth factor
- WO 98/35958 e.g. 1-(4-chloroanilino)-4-(4-pyridylmethyl)phthalazine or a pharmaceutically acceptable salt thereof, e.g. the succinate, or in WO 00/09495, WO 00/27820, WO 00/59509, WO 98/11223, WO 00/27819 and EP 0 769 947; those as described by Prewett et al, Cancer Res, Vol. 59, pp. 5209-5218 (1999); Yuan et al., Proc Natl Acad Sci USA, Vol. 93, pp.
- Photodynamic therapy refers to therapy which uses certain chemicals known as photosensitizing compounds to treat or prevent cancers.
- Examples of photodynamic therapy includes treatment with compounds, such as e.g. VISUDYNE and porfimer sodium.
- Angiostatic steroids refers to compounds which block or inhibit angiogenesis, such as, e.g., anecortave, triamcinolone, hydrocortisone, 11-epihydrocotisol, cortexolone, 17-hydroxyprogesterone, corticosterone, desoxycorticosterone, testosterone, estrone and dexamethasone.
- angiogenesis such as, e.g., anecortave, triamcinolone, hydrocortisone, 11-epihydrocotisol, cortexolone, 17-hydroxyprogesterone, corticosterone, desoxycorticosterone, testosterone, estrone and dexamethasone.
- Implants containing corticosteroids refers to compounds, such as e.g. fluocinolone, dexamethasone.
- “Other chemotherapeutic compounds” include, but are not limited to, plant alkaloids, hormonal compounds and antagonists; biological response modifiers, preferably lymphokines or interferons: antisense oligonucleotides or oligonucleotide derivatives; shRNA or siRNA; or miscellaneous compounds or compounds with other or unknown mechanism of action.
- reaction was monitored by TLC (10% MeOH in chloroform).
- the reaction mixture was filtered through a celite bed and the bed was washed with chloroform.
- the organic layer was concentrated and purification by column chromatography (using silica gel of mesh size of 60-120, 20% ethyl acetate in hexane as eluent) afforded 48 mg (28% yield) of 1-naphthalen-2-yl-3-pyridin-3-yl-imidazolidin-2-one.
- 1-pyridin-3-yl-tetrahydro-pyrimidin-2-one (I-10b: 118 mg, 0.724 mmol) was reacted with 2-bromo-naphthalene (150 mg, 0.724 mmol), 1,4-dioxane (5 mL), copper iodide (13.79 mg, 0.0724 mmol), trans-1,2-diamino cyclohexane (24.865 mg, 0.2172 mmol) and potassium carbonate (200 mg, 1.448 mmol) to afford the crude product. Purification by column chromatography on silica gel (1% MeOH in chloroform) afforded 75 mg of the product (35.71% yield).
- 1-pyridin-3-yl-tetrahydro-pyrimidin-2-one (I-10b: 67.8 mg, 0.3831 mmol) was reacted with 5-iodo-benzo[b]thiophene (100 mg, 0.3831 mmol), 1,4-dioxane (5 mL), copper iodide (7.29 mg, 0.03831 mmol), trans-1,2-diamino cyclohexane (13.15 mg, 0.1149 mmol) and potassium carbonate (106.1 mg, 0.7662 mmol) to afford 15 mg of the product (17.5% yield).
- Step 3 Preparation of 1-Naphthalen-2-yl-3-(5-trifluoromethyl-pyridin-3- ⁇ -imidazolidin-2-one (79A)
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract

where R53, R54, p, q, and n are as defined herein. The compounds of the present invention have been found to be useful as 17α-hydroxylase/C17,20-lyase inhibitors.
Description

wherein

wherein n, R50, R51, R52, R53, R54 and q are as defined above; or a pharmaceutically acceptable salt thereof. In a particular embodiment of the compound of Formula (Ib), R52 is CF3 or a (C1-C4)alkyl substituted with a 5- to 6-membered partially or fully saturated heterocycle (preferably, the heterocycle is attached to the alkyl group via a nitrogen ring atom, e.g. pyrrolidin-1-ylmethyl). In another particular embodiment of the compound of Formula (Ib), n is 1, and p is 0.

wherein:

In Scheme I above, R is represented by -A or the following group

Step-1 & 2:


In Scheme 3 above, R is represented by -A or the following moiety

Step 1:



| DIPA: | Diisopropylamine | ||
| DPPA: | Diphenylphosphoryl Azide | ||
| DCM: | Dichloromethane | ||
| DCE: | Dichloroethane | ||
| DMF: | N,N-Dimethylformamide | ||
| DMSO: | Dimethylsulfoxide | ||
| TEA: | Triethylamine | ||
| THF: | Tetrahydrofuran | ||
| NaBH(OAc)3: | Sodium triacetoxy borohydride | ||
| PTSA: | Para toluene sulphonic acid | ||
| TES: | Triethyl silane | ||
| LiHMDS: | Lithium bis(trimethylsilyl)amide | ||
| TEA: | Triethyl amine | ||
| Pd2(dba)3: | Tris(dibenzylideneacetone)dipalladium(0) | ||
| TLC: | Thin Layer Chromatography | ||
| NMR: | Nuclear Magnetic Resonance | ||
| LCMS: | Liquid chromatography Mass spectrometry | ||
| HPLC: | High Performance Liquid Chromatography | ||



















































































































































































































































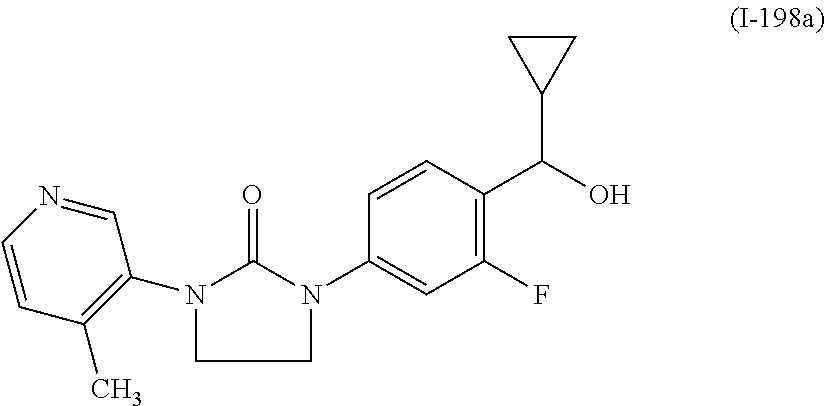















| CYP | Cytochrome P450 | ||
| CPM | Counts per minute | ||
| Cyt b5 | Cytochrome b5 | ||
| DMSO | Dimethyl sulfoxide | ||
| DHEA | Dehydroepiandrosterone | ||
| NADPH | Nicotinamide adenine dinucleotide phosphate | ||
- Sample: Enzyme reaction in presence of inhibitor.
- Positive control: Enzyme reaction without inhibitor but containing DMSO at 1% final concentration.
- Blank-Contains all reagents except enzyme.
% Inhibition=100%−% Lyase activity
| TABLE 1 | |||
| rCYP17 | |||
| Lyase IC 50 | Lyase | ||
| Example | nM | % Inh @ | |
| No. | Final compounds | Human/Rat | 100 nM |
| 1A | 1-Naphthalen-2-yl-3-pyridin-3- | 34 nM/35 nM | 84% |
| yl-imidazolidin-2-one | |||
| 2A | 1-(1-Ethyl-1H-indol-5-yl)-3- | —/— | 23% |
| pyridin-3-yl-imidazolidin-2-one | |||
| 3A | 1-(6-Methoxy-naphthalen-2-yl)- | —/— | 31% |
| 3-pyridin-3-yl-imidazolidin-2- | |||
| one | |||
| 4A | 1-Benzothiazol-6-yl-3-pyridin-3- | 77 nM/— | 59% |
| yl-imidazolidin-2-one | |||
| 5A | 1-Ethyl-6-(2-oxo-3-pyridin-3-yl- | —/— | 51% |
| imidazolidin-1-yl)-3,4-dihydro- | |||
| 1H-quinolin-2-one | |||
| 6A | 1-(5-Fluoro-3-methyl- | —/157 nM | 51% |
| benzo[b]thiophen-2-yl)-3- | |||
| pyridin-3-yl-imidazolidin-2-one | |||
| 7A | 1-Benzo[b]thiophen-5-yl-3- | —/16 nM | 87% |
| pyridin-3-yl-imidazolidin-2-one | |||
| 8A | 1-Pyridin-3-yl-3-quinolin-6-yl- | —/— | 48% |
| imidazolidin-2-one | |||
| 9A | 1-Benzothiazol-5-yl-3-pyridin-3- | —/— | 18% |
| yl-imidazolidin-2-one | |||
| 10A | 1-Naphthalen-2-yl-3-pyridin-3- | —/— | 11% |
| yl-tetrahydro-pyrimidin-2-one | |||
| 11A | 1-(2-Chloro-4-methyl-quinolin- | —/97 nM | 59% |
| 6-yl)-3-pyridin-3-yl- | |||
| imidazolidin-2-one | |||
| 12A | 1-(1-Hydroxyimino-indan-5-yl)- | —/— | 17% |
| 3-pyridin-3-yl-imidazolidin-2- | |||
| one | |||
| 13A | 1-Benzo[b]thiophen-5-yl-3- | —/— | 9% |
| pyridin-3-yl-tetrahydro- | |||
| pyrimidin-2-one | |||
| 14A | 1-(4-Methyl-pyridin-3-yl)-3- | 17 nM/2 nM | 102% |
| naphthalen-2-yl-imidazolidin-2- | |||
| one | |||
| 15A | 1-(4-Methyl-pyridin-3-yl)-3-(5- | 10 nM/— | — |
| methyl-thiophen-2-yl)- | |||
| imidazolidin-2-one | |||
| 16A | 1-Benzothiazol-6-yl-3-(4- | 6 nM/5 nM | 94% |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 17A | 1-Ethyl-6-[3-(4-methyl-pyridin- | —/21 nM | 83% |
| 3-yl)-2-oxo-imidazolidin-1-yl]- | |||
| 3,4-dihydro-1H-quinolin-2-one | |||
| 18A | 1-(4-Fluoro-phenyl)-3-(4- | 76 nM/12 nM | 88% |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 19A | 1-Biphenyl-4-yl-3-(4-methyl- | —/11 nM | 91% |
| pyridin-3-yl)-imidazolidin-2-one | |||
| 20A | 1-Ethyl-4-methyl-6-[3-(4- | —/56 nM | 69% |
| methyl-pyridin-3-yl)-2-oxo- | |||
| imidazolidin-1-yl]-1H-quinolin- | |||
| 2-one | |||
| 21A | 1-(2-Chloro-4-methyl-quinolin- | —/5 nM | 98% |
| 6-yl)-3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 22A | 4-Methyl-6-[3-(4-methyl- | —/24 nM | 85% |
| pyridin-3-yl)-2-oxo- | |||
| imidazolidin-1-yl]-1H-quinolin- | |||
| 2-one | |||
| 23A | 1-(4-Methyl-pyridin-3-yl)-3- | —/20 nM | 85% |
| quinolin-2-yl-imidazolidin-2-one | |||
| 24A | 1-(4-Methyl-pyridin-3-yl)-3- | 72 nM/— | 98% |
| quinolin-3-yl-imidazolidin-2-one | |||
| 25A | 1-(6-Methoxy-naphthalen-2-yl)- | —/— | 89% |
| 3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 26A | 1-Benzo[b]thiophen-2-yl-3-(4- | 15 nM/— | 100% |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 27A | 1-Benzo[b]thiophen-5-yl-3-(4- | 24 nM/— | 101% |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 28A | 1-(1H-Indazol-5-yl)-3-(4- | 4 nM/52 nM | 61% |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 29A | 1-(3-Methyl-benzofuran-5-yl)-3- | 3 nM/— | 98% |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 30A | 1-(6-Fluoro-pyridin-3-yl)-3-(4- | —/— | 76% |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 31A | 1-(4-Methyl-pyridin-3-yl)-3- | 108 nM/— | 74% |
| thiophen-3-yl-imidazolidin-2- | |||
| one | |||
| 32A | 1-(5-Methoxy-pyridin-3-yl)-3- | —/— | 73% |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 33A | 1-(5-Fluoro-3-methyl- | —/— | — |
| benzo[b]thiophen-2-yl)-3-(4- | |||
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 34A | 1-(2-Chloro-pyrimidin-5-yl)-3- | —/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 35A | 1-(4-Methyl-pyridin-3-yl)-3- | —/— | — |
| (1H-pyrazol-4-yl)-imidazolidin- | |||
| 2-one | |||
| 36A | 1-(4-Methyl-pyridin-3-yl)-3- | —/— | — |
| thiazol-2-yl-imidazolidin-2-one | |||
| 37A | 1-(4-Methoxy-phenyl)-3-(4- | —/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 38A | 1-(4-Chloro-phenyl)-3-(4- | 70 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 39A | 4-[3-(4-Methyl-pyridin-3-yl)-2- | —/— | — |
| oxo-imidazolidin-1-yl]- | |||
| benzonitrile | |||
| 40A | 1-Benzooxazol-2-yl-3-(4- | —/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 41A | 1-(5-Chloro-thiophen-2-yl)-3-(4- | 10 nM/3 nM | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 42A | 1-(4-Methyl-pyridin-3-yl)-3- | —/— | — |
| thiophen-2-yl-imidazolidin-2- | |||
| one | |||
| 43A | 1-(6-Hydroxy-pyridin-3-yl)-3-(4- | —/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 44A | N-Methyl-4-[3-(4-methyl- | —/— | — |
| pyridin-3-yl)-2-oxo- | |||
| imidazolidin-1-yl]-benzamide | |||
| 45A | 1-(3,4-Difluoro-phenyl)-3-(4- | 27 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 46A | 1-(3-Chloro-4-fluoro-phenyl)-3- | 10 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 47A | 1-Benzothiazol-2-yl-3-(4- | —/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 48A | 1-(1H-Indol-5-yl)-3-(4-methyl- | —/— | — |
| pyridin-3-yl)-imidazolidin-2-one | |||
| 49A | 1-(2,3-Dihydro- | 30 nM/— | — |
| benzo[1,4]dioxin-6-yl)-3-(4- | |||
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 50A | 1-(4-Methyl-pyridin-3-yl)-3-(4- | 51 nM/— | — |
| trifluoromethyl-phenyl)- | |||
| imidazolidin-2-one | |||
| 51A | 1-(2,3-Dihydro-benzofuran-5- | 14 nM/— | — |
| yl)-3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 52A | 1-(4-Methyl-pyridin-3-yl)-3- | 96 nM/5 nM | 97% |
| quinolin-6-yl-imidazolidin-2-one | |||
| 53A | 1-(3-Fluoro-4-methoxy-phenyl)- | 136 nM/— | — |
| 3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 54A | 1-(4-Chloro-3-fluoro-phenyl)-3- | 15 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 55A | 1-(4-Methyl-pyridin-3-yl)-3-m- | 9 nM/7 nM | — |
| tolyl-imidazolidin-2-one | |||
| 56A | 1-(3-Methoxy-phenyl)-3-(4- | 25 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 57A | 1-(4-Methyl-pyridin-3-yl)-3- | —/— | — |
| (4,5,6,7-tetrahydro-thieno[2,3- | |||
| c]pyridin-2-yl)-imidazolidin-2- | |||
| one | |||
| 58A | 1-(2-Chloro-pyridin-4-yl)-3-(4- | 27 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 59A | 1-(4-Fluoro-3-methyl-phenyl)-3- | 17 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 60A | 1-(5-Chloro-2-fluoro-phenyl)-3- | —/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 61A | 1-(2,4-Difluoro-phenyl)-3-(4- | —/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 62A | 1-Benzothiazol-5-yl-3-(4- | 426 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 63A | 6-[3-(4-Methyl-pyridin-3-yl)-2- | —/— | — |
| oxo-imidazolidin-1-yl]-3,4- | |||
| dihydro-1H-quinolin-2-one | |||
| 64A | 1-(2,2-Difluoro- | 35 nM/— | — |
| benzo[1,3]dioxol-5-yl)-3-(4- | |||
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 65A | 7-[3-(4-Methyl-pyridin-3-yl)-2- | —/— | — |
| oxo-imidazolidin-1-yl]-3,4- | |||
| dihydro-1H-quinolin-2-one | |||
| 66A | N-{3-[3-(4-Methyl-pyridin-3- | —/— | — |
| yl)-2-oxo-imidazolidin-1-yl]- | |||
| phenyl}-acetamide | |||
| 67A | 1-(4-Methyl-pyridin-3-yl)-3-(3- | —/— | — |
| methyl-thiophen-2-yl)- | |||
| imidazolidin-2-one | |||
| 68A | 1-(4-Methyl-pyridin-3-yl)-3-(2- | —/— | — |
| methyl-quinolin-6-yl)- | |||
| imidazolidin-2-one | |||
| 69A | 1-(4-Methyl-pyridin-3-yl)-3- | 34 nM/— | — |
| phenyl-imidazolidin-2-one | |||
| 70A | 1-(4-Methyl-pyridin-3-yl)-3-(3- | 6.5 nM/2 nM | — |
| trifluoromethyl-phenyl)- | |||
| imidazolidin-2-one | |||
| 71A | 1-(1-Isopropyl-1H-pyrazol-4-yl)- | —/— | — |
| 3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 72A | 1-(2-Methoxy-pyridin-4-yl)-3- | 9.4 nM/10 nM | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 73A | 1-Imidazo[1,2-a]pyridin-7-yl-3- | 109 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 74A | 1-(4-Fluoro-3-methoxy-phenyl)- | 76 nM/— | — |
| 3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 75A | N-{5-[3-(4-Methyl-pyridin-3- | >10 μM | — |
| yl)-2-oxo-imidazolidin-1-yl]- | |||
| pyridin-2-yl}-acetamide | |||
| 76A | 1-(2-Amino-pyridin-4-yl)-3-(4- | 192 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 77A | 1-(4-Methyl-pyridin-3-yl)-3- | 195 nM/— | — |
| quinoxalin-6-yl-imidazolidin-2- | |||
| one | |||
| 78A | 1-(5-Difluoromethyl-thiophen-3- | 9.3 nM/— | — |
| yl)-3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one, trifluoro- | |||
| acetic acid | |||
| 79A | 1-Naphthalen-2-yl-3-(5- | —/— | 3.6% |
| trifluoromethyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 80A | 1-(5-Chloro-pyridin-3-yl)-3- | —/— | 57% |
| naphthalen-2-yl-imidazolidin-2- | |||
| one | |||
| 81A | 1-(5-Fluoro-pyridin-3-yl)-3- | —/— | 82% |
| naphthalen-2-yl-imidazolidin-2- | |||
| one | |||
| 82A | 1-(5-Methoxy-pyridin-3-yl)-3- | —/— | — |
| naphthalen-2-yl-imidazolidin-2- | |||
| one | |||
| 83A | 5-(3-Naphthalen-2-yl-2-oxo- | —/— | — |
| imidazolidin-1-yl)-nicotinic acid | |||
| methyl ester | |||
| 84A | 1-Benzothiazol-6-yl-3-(4-chloro- | 85 nM/— | — |
| pyridin-3-yl)-imidazolidin-2-one | |||
| 85A | 1-(4-Methyl-pyridin-3-yl)-3-(2- | 1,263 nM/— | — |
| pyrrolidin-1-yl-pyridin-4-yl)- | |||
| imidazolidin-2-one | |||
| 86A | 1-(3-Chloro-2-fluoro-phenyl)-3- | 1,200 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 87A | 1-(4′-Fluoro-biphenyl-4-yl)-3-(4- | 255 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 88A | 1-(3-Chloro-phenyl)-3-(4- | 12 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 89A | 1-(4-Chloro-2-methyl-phenyl)-3- | 326 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 90A | 1-(4-Fluoro-3-trifluoromethyl- | 5 nM/— | — |
| phenyl)-3-(4-methyl-pyridin-3- | |||
| yl)-imidazolidin-2-one | |||
| 91A | 1-(3-Difluoromethyl-4-fluoro- | 4.3 nM/— | — |
| phenyl)-3-(4-methyl-pyridin-3- | |||
| yl)-imidazolidin-2-one | |||
| 91B | 1-(3-Difluoromethyl-4-fluoro- | 22 nM/— | — |
| phenyl)-3-(4-methyl-pyridin-3- | |||
| yl)-1,3-dihydro-imidazol-2-one | |||
| 92A | 1-(4-Methyl-pyridin-3-yl)-3-(5- | 21 nM/— | — |
| methyl-thiophen-3-yl)-1,3- | |||
| dihydro-imidazol-2-one | |||
| 92B | 1-(4-Methyl-pyridin-3-yl)-3-(5- | 12 nM/— | — |
| methyl-thiophen-3-yl)- | |||
| imidazolidin-2-one | |||
| 93A | 1-(2-Methyl-benzooxazol-5-yl)- | >10 μM/— | — |
| 3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 94A | 1-Imidazo[1,2-a]pyridin-6-yl-3- | 110 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 95A | 1-(3-Methyl-1H-indazol-5-yl)-3- | 119 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 96A | N-{4-[3-(4-Methyl-pyridin-3- | >10 μM/— | — |
| yl)-2-oxo-imidazolidin-1-yl]- | |||
| pyridin-2-yl}-acetamide | |||
| 97A | 1-(4-Methoxy-thieno[3,2- | 356 nM/— | — |
| c]pyridin-2-yl)-3-(4-methyl- | |||
| pyridin-3-yl)-imidazolidin-2-one | |||
| 98A | 1-(4-Chloro-thieno[3,2- | 18 nM/— | — |
| c]pyridin-2-yl)-3-(4-methyl- | |||
| pyridin-3-yl)-imidazolidin-2-one | |||
| 99A | 1-(4-Chloro-thieno[3,2- | 22 nM/— | — |
| c]pyridin-3-yl)-3-(4-methyl- | |||
| pyridin-3-yl)-imidazolidin-2-one | |||
| 100A | 1-(4-Methyl-pyridin-3-yl)-3-(2- | 46 nM/— | — |
| methyl-pyridin-4-yl)- | |||
| imidazolidin-2-one | |||
| 101A | 1-(3-Methyl-benzo[d]isoxazol-5- | 46 nM/— | — |
| yl)-3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 102A | 1-(3-Methyl-1H-indazol-6-yl)-3- | 7.5 nM/5 nM | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one, | |||
| trifluoroacetic acid | |||
| 103A | 2-Fluoro-5-[3-(4-methyl-pyridin- | 19 nM/— | — |
| 3-yl)-2-oxo-imidazolidin-1-yl]- | |||
| benzonitrile | |||
| 104A | 1-(2-Methyl-imidazo[1,2- | 1,991 nM/— | — |
| a]pyridin-6-yl)-3-(4-methyl- | |||
| pyridin-3-yl)-imidazolidin-2-one | |||
| 105A | 1-(2-Methyl-benzothiazol-6-yl)- | 1,469 nM/— | — |
| 3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 106A | 3-[3-(4-Methyl-pyridin-3-yl)-2- | 35 nM/— | — |
| oxo-imidazolidin-1-yl]- | |||
| benzonitrile, trifluoroacetic acid | |||
| 107A | 1-(1H-Indol-3-yl)-3-(4-methyl- | 334 nM/— | — |
| pyridin-3-yl)-imidazolidin-2-one | |||
| 108A | 1-(1H-Benzoimidazol-5-yl)-3-(4- | 363 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 109A | 1-Benzo[b]thiophen-3-yl-3-(4- | 158 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 110A | 1-(4-Methoxy-thieno[3,2- | >10 μM/— | — |
| c]pyridin-3-yl)-3-(4-methyl- | |||
| pyridin-3-yl)-imidazolidin-2-one | |||
| 111A | 1-(3-Methyl-benzo[d]isoxazol-6- | 106 nM/— | — |
| yl)-3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 112A | 2-Chloro-4-[3-(4-methyl- | 28 nM/— | — |
| pyridin-3-yl)-2-oxo- | |||
| imidazolidin-1-yl]-benzonitrile | |||
| 113A | 1-Benzo[d]isoxazol-5-yl-3-(4- | 230 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 114A | 1-(1-Methyl-1H-indazol-5-yl)-3- | 24 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 115A | 1-(1-Methyl-1H-indol-3-yl)-3- | 173 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one, | |||
| trifluoroacetic acid | |||
| 116A | 1-(1-Methyl-1H-benzoimidazol- | >10 μM/— | — |
| 5-yl)-3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one, | |||
| trifluoroacetic acid | |||
| 117A | 5-[3-(4-Methyl-pyridin-3-yl)-2- | 94 nM/— | — |
| oxo-imidazolidin-1-yl]-1,2- | |||
| dihydro-indazol-3-one | |||
| 118A | 1-(3-Amino-1H-indazol-5-yl)-3- | 17 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 119A | 1-Imidazo[1,2-a]pyridin-3-yl-3- | 3,198 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 120A | 1-(4-Methyl-pyridin-3-yl)-3- | 4 μM/— | — |
| thieno[3,2-c]pyridin-2-yl- | |||
| imidazolidin-2-one | |||
| 121A | 1-(1H-Indazol-6-yl)-3-(4- | 2.5 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 122A | 1-(3H-Imidazo[4,5-b]pyridin-6- | 383 nM/— | — |
| yl)-3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one, | |||
| trifluoroacetic acid | |||
| 123A | 1-(3-Amino-1H-indazol-6-yl)-3- | 10 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 124A | 1-Benzothiazol-6-yl-3-(4- | 23 nM/12 nM | — |
| methoxy-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 125A | 1-Benzothiazol-6-yl-3-(4- | 17 nM/— | — |
| difluoromethyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 126A | 1-Benzothiazol-6-yl-3-(4- | 25 nM/— | — |
| hydroxymethyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 127A | 1-Benzothiazol-6-yl-3-(6- | >10 μM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 128A | 1-Benzothiazol-6-yl-3-(4- | 44 nM/— | — |
| trifluoromethyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 129A | 3-(3-Benzothiazol-6-yl-2-oxo- | 202 nM/— | — |
| imidazolidin-1-yl)- | |||
| isonicotinonitrile | |||
| 130A | 1-(4-Methyl-pyridin-3-yl)-3- | >10 μM/— | — |
| naphthalen-2-yl-imidazolidin-2- | |||
| one | |||
| 131A | 1-m-Tolyl-3-(4-trifluoromethyl- | 103 nM/— | — |
| pyridin-3-yl)-imidazolidin-2-one | |||
| 132A | 1-(2-Methyl-2H-indazol-5-yl)-3- | >10 μM | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 133A | 1-(4-Methyl-pyridin-3-yl)-3- | 1,970 nM/— | — |
| naphthalen-1-yl-imidazolidin-2- | |||
| one | |||
| 134A | 1-(1-Methyl-1H-indol-5-yl)-3- | 950 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 135A | 6-[3-(4-Methyl-pyridin-3-yl)-2- | 15.9 nM/— | — |
| oxo-imidazolidin-1-yl]-1,2- | |||
| dihydro-indazol-3-one | |||
| 136A | 1-(4-Methyl-pyridin-3-yl)-3- | 500 nM/— | — |
| thieno[3,2-c]pyridin-3-yl- | |||
| imidazolidin-2-one | |||
| 137A | 1-(5-Chloro-1-methyl-1H-indol- | 1,554 nM/— | — |
| 3-yl)-3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 138A | 1-Indan-5-yl-3-(4-methyl- | 13 nM/— | — |
| pyridin-3-yl)-imidazolidin-2-one | |||
| 139A | 1-Benzo[b]thiophen-5-yl-3-(4- | 83 nM/— | — |
| methyl-pyridin-3-yl)-1,3- | |||
| dihydro-imidazol-2-one | |||
| 140A | 2-Fluoro-4-[3-(4-methyl-pyridin- | 97 nM/— | — |
| 3-yl)-2-oxo-imidazolidin-1-yl]- | |||
| benzonitrile | |||
| 141A | 1-(1H-Benzotriazol-5-yl)-3-(4- | 13 nM/— | — |
| methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one, | |||
| hydrochloride | |||
| 142A | 1-Benzothiazol-6-yl-3-(4- | 21 nM/— | — |
| methyl-pyridin-3-yl)-1,3- | |||
| dihydro-imidazol-2-one | |||
| 143A | 1-(3-Amino-1-methyl-1H- | 116 nM/— | — |
| indazol-6-yl)-3-(4-methyl- | |||
| pyridin-3-yl)-imidazolidin-2-one | |||
| 144A | 1-(1H-Indol-6-yl)-3-(4-methyl- | 174 nM/— | — |
| pyridin-3-yl)-imidazolidin-2-one | |||
| 145A | 1-(3-Chloro-imidazo[1,2- | 31 nM/— | — |
| a]pyridin-7-yl)-3-(4-methyl- | |||
| pyridin-3-yl)-imidazolidin-2-one | |||
| 146A | 1-Methyl-3-[3-(4-methyl- | >10 μM | — |
| pyridin-3-yl)-2-oxo- | |||
| imidazolidin-1-yl]-1H-indole-4- | |||
| carbonitrile | |||
| 147A | 1-Hydroxymethyl-3,3-dimethyl- | 346 nM/— | — |
| 5-[3-(4-methyl-pyridin-3-yl)-2- | |||
| oxo-imidazolidin-1-yl]-1,3- | |||
| dihydro-indol-2-one | |||
| 148A | 1-(4-Methyl-pyridin-3-yl)-3-(2- | 25 nM/— | — |
| trifluoromethyl-pyridin-4-yl)- | |||
| imidazolidin-2-one | |||
| 149A | 1-Benzothiazol-6-yl-3-(4- | 37 nM/— | — |
| dimethoxymethyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 150A | N-[3-(3-Benzothiazol-6-yl-2- | 496 nM/— | — |
| oxo-imidazolidin-1-yl)-pyridin- | |||
| 4-yl]-acetamide | |||
| 151A | 1-Benzothiazol-6-yl-3-(5-chloro- | 13.3 nM/— | — |
| 4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 152A | 1-(4-Amino-pyridin-3-yl)-3- | 7 nM/— | — |
| benzothiazol-6-yl-imidazolidin- | |||
| 2-one hydrochloride | |||
| 153A | 1-(benzo[d]thiazol-6-yl)-3-(4- | 280 nM | — |
| methyl-5- | |||
| (trifluoromethyl)pyridin-3- | |||
| yl)imidazolidin-2-one, | |||
| trifluoroacetic acid salt | |||
| 154A | 1-(isothiazol-4-yl)-3-(4- | 89 nM | — |
| methylpyridin-3-yl)imidazolidin- | |||
| 2-one | |||
| 155A | 1-(4-methylpyridin-3-yl)-3-(5- | 2.7 nM | — |
| (trifluoromethyl)thiophen-2- | |||
| yl)imidazolidin-2-one | |||
| 156A | 1-(benzo[d]thiazol-6-yl)-3-(4-(2- | >10 μM | — |
| hydroxypropan-2-yl)pyridin-3- | |||
| yl)imidazolidin-2-one | |||
| 157A | 1-(4-methylpyridin-3-yl)-3-(4- | 21 nM | — |
| methylthieno[3,2-c]pyridin-2- | |||
| yl)imidazolidin-2-one | |||
| 158A | 1-(benzo[d]thiazol-6-yl)-3-(4-(1- | 38 nM | — |
| hydroxyethyl)pyridin-3- | |||
| yl)imidazolidin-2-one | |||
| 159A | 1-(benzo[d]thiazol-6-yl)-3-(4- | 3.4 nM | — |
| ethylpyridin-3-yl)imidazolidin-2- | |||
| one | |||
| 160A | 1-(4-methylpyridin-3-yl)-3-(3- | 34 nM | — |
| (trifluoromethyl)-1H-indazol-6- | |||
| yl)imidazolidin-2-one | |||
| 161A | 1-(3-cyclopropyl-1H-indazol-6- | 82 nM | — |
| yl)-3-(4-methylpyridin-3- | |||
| yl)imidazolidin-2-one | |||
| 162A | 1-(4-methylpyridin-3-yl)-3- | 317 nM | — |
| (quinolin-7-yl)imidazolidin-2- | |||
| one | |||
| 163A | 3-Benzothiazol-6-yl-4,4- | >10 μM | — |
| dimethyl-1-(4-methyl-pyridin-3- | |||
| yl)-imidazolidin-2-one | |||
| 164A | 1-Benzothiazol-6-yl-4,4- | 70 nM/— | — |
| dimethyl-3-pyridin-3-yl- | |||
| imidazolidin-2-one | |||
| 165A | 1-Benzothiazol-6-yl-4,4- | 1,153 nM/— | — |
| dimethyl-3-(4-methyl-pyridin-3- | |||
| yl)-imidazolidin-2-one | |||
| 166A | 3-Benzothiazol-6-yl-4-methyl-1- | 32 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 167A | 1-Benzothiazol-6-yl-4-methyl-3- | 38 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 168A | 1-Benzothiazol-6-yl-4-ethyl-3- | 384 nM/— | — |
| (4-methyl-pyridin-3-yl)-1,3- | |||
| dihydro-imidazol-2-one | |||
| 169A | 1-Benzothiazol-6-yl-4-ethyl-3- | —/— | — |
| pyridin-3-yl-1,3-dihydro- | |||
| imidazol-2-one | |||
| 170A | 1-Benzothiazol-6-yl-3-pyridin-3- | —/— | — |
| yl-4-trifluoromethyl- | |||
| imidazolidin-2-one | |||
| 171A | 1-Benzothiazol-6-yl-3-(4- | —/— | — |
| methyl-pyridin-3-yl)-1,3- | |||
| dihydro-benzoimidazol-2-one | |||
| 172A | 1-Benzothiazol-6-yl-4-methyl-3- | 326 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 173A | 3-Benzothiazol-6-yl-1-pyridin-3- | —/— | — |
| yl-4-trifluoromethyl- | |||
| imidazolidin-2-one | |||
| 174A | 1-Benzothiazol-6-yl-3-(4- | —/— | — |
| methyl-pyridin-3-yl)-4- | |||
| trifluoromethyl-imidazolidin-2- | |||
| one | |||
| 175A | 1-Benzothiazol-6-yl-4,5- | 85 nM/— | — |
| dimethyl-3-(4-methyl-pyridin-3- | |||
| yl)-imidazolidin-2-one | |||
| 176A | 1-Benzothiazol-6-yl-3-(4- | 39 nM/— | — |
| pyrrolidin-1-ylmethyl-pyridin-3- | |||
| yl)-imidazolidin-2-one | |||
| 177A | 1-Benzothiazol-6-yl-3-(4- | 32 nM/— | — |
| morpholin-4-ylmethyl-pyridin-3- | |||
| yl)-imidazolidin-2-one | |||
| 178A | 1-Benzothiazol-6-yl-3-(4- | 19 nM/— | — |
| cyclopropylaminomethyl- | |||
| pyridin-3-yl)-imidazolidin-2-one | |||
| 179A | 1-Benzothiazol-6-yl-3-(6-fluoro- | —/— | — |
| 4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 180A | 1-Benzothiazol-6-yl-3-[4-(2- | —/— | — |
| morpholin-4-yl-ethoxy)-pyridin- | |||
| 3-yl]-imidazolidin-2-one | |||
| 181A | 1-Benzothiazol-6-yl-3-[4-(1- | 3064 nM/— | — |
| methyl-pyrrolidin-2-ylmethoxy)- | |||
| pyridin-3-yl]-imidazolidin-2-one | |||
| 182A | 1-Benzothiazol-6-yl-3-{4-[(5- | 27 nM/— | — |
| methyl-1H-pyrazol-3-ylamino)- | |||
| methyl]-pyridin-3-yl}- | |||
| imidazolidin-2-one | |||
| 183A | 1-Benzothiazol-6-yl-3-(3H- | >10 μM/— | — |
| imidazo[4,5-b]pyridin-6-yl)- | |||
| imidazolidin-2-one | |||
| 184A | 1-Benzothiazol-6-yl-3-[4-(1- | >10 μM/— | — |
| methyl-piperidin-4-ylmethoxy)- | |||
| pyridin-3-yl]-imidazolidin-2-one | |||
| 185A | 3-(3-Benzothiazol-6-yl-2-oxo- | >10 μM/— | — |
| imidazolidin-1-yl)- | |||
| isonicotinamide | |||
| 186A | 1-Benzothiazol-6-yl-3- | >10 μM/— | — |
| imidazo[1,2-a]pyrazin-5-yl- | |||
| imidazolidin-2-one | |||
| 187A | 1-Benzothiazol-6-yl-3-(1H- | >10 μM/— | — |
| pyrrolo[2,3-b]pyridin-5-yl)- | |||
| imidazolidin-2-one | |||
| 188A | 3-Benzothiazol-6-yl-3′-methyl- | 195 nM/— | — |
| 4,5-dihydro-3H,3′H- | |||
| [1,4′]biimidazolyl-2-one | |||
| 189A | 1-Benzothiazol-6-yl-3-(4- | 280 nM/— | — |
| methyl-5-trifluoromethyl- | |||
| pyridin-3-yl)-imidazolidin-2-one | |||
| trifluoro-acetic acid | |||
| 190A | 1-Isothiazol-4-yl-3-(4-methyl- | 89 nM/— | — |
| pyridin-3-yl)-imidazolidin-2-one | |||
| 191A | 1-Benzothiazol-6-yl-3-pyridin-2- | >10 μM/— | — |
| yl-imidazolidin-2-one | |||
| 192A | 1-(4-Methyl-pyridin-3-yl)-3-(5- | 2.7 nM/— | — |
| trifluoromethyl-thiophen-2-yl)- | |||
| imidazolidin-2-one | |||
| 193A | 1-Benzothiazol-6-yl-3-[4-(1- | >10 μM/— | — |
| hydroxy-1-methyl-ethyl)- | |||
| pyridin-3-yl]-imidazolidin-2-one | |||
| 194A | 1-(4-Methyl-pyridin-3-yl)-3-(4- | 21 nM/— | — |
| methyl-thieno[3,2-c]pyridin-2- | |||
| yl)-imidazolidin-2-one | |||
| 195A | 1-Benzothiazol-6-yl-3-[4-(1- | 38 nM/— | 8% |
| hydroxy-ethyl)-pyridin-3-yl]- | |||
| imidazolidin-2-one | |||
| 196A | 1-Benzothiazol-6-yl-3-(4-ethyl- | 3.4 nM/— | — |
| pyridin-3-yl)-imidazolidin-2-one | |||
| 197A | 1-(4-Methyl-pyridin-3-yl)-3-(3- | 34 nM/— | — |
| trifluoromethyl-1H-indazol-6- | |||
| yl)-imidazolidin-2-one | |||
| 198A | 1-(3-Cyclopropyl-1H-indazol-6- | 82 nM/— | — |
| yl)-3-(4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
| 199A | 1-(4-Methyl-pyridin-3-yl)-3- | 317 nM/— | — |
| quinolin-7-yl-imidazolidin-2-one | |||
| 200A | 3-Benzothiazol-6-yl-4,4- | >10 μM/— | — |
| dimethyl-1-(4-methyl-pyridin-3- | |||
| yl)-imidazolidin-2-one | |||
| 201A | 1-Benzothiazol-6-yl-4,4- | 70 nM/— | — |
| dimethyl-3-pyridin-3-yl- | |||
| imidazolidin-2-one | |||
| 202A | 1-Benzothiazol-6-yl-4,4- | 1153 nM/— | — |
| dimethyl-3-(4-methyl-pyridin-3- | |||
| yl)-imidazolidin-2-one | |||
| 203A | 3-Benzothiazol-6-yl-4-methyl-1- | 32 nM/— | — |
| (4-methyl-pyridin-3-yl)- | |||
| imidazolidin-2-one | |||
Claims (21)




Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/929,901 USRE45173E1 (en) | 2009-06-26 | 2013-06-28 | Inhibitors of CYP 17 |
| US14/257,433 US20140228394A1 (en) | 2009-06-26 | 2014-04-21 | Inhibitors of cyp 17 |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN01500/CHE/2009 | 2009-06-26 | ||
| IN1500CH2009 | 2009-06-26 | ||
| IN2181/DEL/2009 | 2009-10-21 | ||
| IN2181DE2009 | 2009-10-21 | ||
| US12/824,845 US8263635B2 (en) | 2009-06-26 | 2010-06-28 | Inhibitors of CYP 17 |
| US13/929,901 USRE45173E1 (en) | 2009-06-26 | 2013-06-28 | Inhibitors of CYP 17 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/824,845 Reissue US8263635B2 (en) | 2009-06-26 | 2010-06-28 | Inhibitors of CYP 17 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/257,433 Continuation US20140228394A1 (en) | 2009-06-26 | 2014-04-21 | Inhibitors of cyp 17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| USRE45173E1 true USRE45173E1 (en) | 2014-09-30 |
Family
ID=42357251
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/824,845 Ceased US8263635B2 (en) | 2009-06-26 | 2010-06-28 | Inhibitors of CYP 17 |
| US13/929,901 Active 2031-01-01 USRE45173E1 (en) | 2009-06-26 | 2013-06-28 | Inhibitors of CYP 17 |
| US14/257,433 Abandoned US20140228394A1 (en) | 2009-06-26 | 2014-04-21 | Inhibitors of cyp 17 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/824,845 Ceased US8263635B2 (en) | 2009-06-26 | 2010-06-28 | Inhibitors of CYP 17 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/257,433 Abandoned US20140228394A1 (en) | 2009-06-26 | 2014-04-21 | Inhibitors of cyp 17 |
Country Status (44)
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9339501B2 (en) | 2011-04-28 | 2016-05-17 | Novartis Ag | 17a-hydroxylase/C17,20-lyase inhibitors |
| WO2021026454A1 (en) | 2019-08-08 | 2021-02-11 | Laekna Limited | Method of treating cancer |
Families Citing this family (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT2445502T (en) | 2009-06-25 | 2017-09-22 | Alkermes Pharma Ireland Ltd | Heterocyclic compounds for the treatment of neurological and psychological disorders |
| JP5998137B2 (en) | 2010-08-04 | 2016-09-28 | ペルフィキュア ファーマシューティカルズ,インコーポレイテッド | Combination therapy to treat prostate cancer |
| EP2627648A1 (en) * | 2010-09-16 | 2013-08-21 | Novartis AG | 17aHYDROXYLASE/C17,20-LYASE INHIBITORS |
| SG191724A1 (en) | 2011-01-11 | 2013-08-30 | Glaxosmithkline Llc | Combination |
| BR112014004268A8 (en) * | 2011-08-26 | 2023-01-17 | Meiji Seika Pharma Co Ltd | Method for producing pest control agent |
| US8969586B2 (en) | 2011-09-27 | 2015-03-03 | Bristol-Myers Squibb Company | Substituted bicyclic heteroaryl compounds |
| CN102702223A (en) * | 2012-05-28 | 2012-10-03 | 盛世泰科生物医药技术(苏州)有限公司 | Method for preparing 2-bromo-4-chlorothieno[3, 2-C]pyridine |
| JP2015518888A (en) | 2012-06-06 | 2015-07-06 | ノバルティス アーゲー | Combination of a 17-alpha-hydroxylase (C17,20-lyase) inhibitor and a specific PI-3K inhibitor for the treatment of tumor diseases |
| WO2014158875A1 (en) | 2013-03-14 | 2014-10-02 | Pellficure Pharmaceuticals Inc. | Novel therapy for prostate carcinoma |
| TW201534586A (en) * | 2013-06-11 | 2015-09-16 | Orion Corp | Novel CYP17 inhibitors/antiandrogens |
| JP2016526538A (en) | 2013-06-20 | 2016-09-05 | バイエル・クロップサイエンス・アクチェンゲゼルシャフト | Aryl sulfide and aryl sulfoxide derivatives as acaricides and insecticides |
| RU2016116915A (en) * | 2013-10-01 | 2017-11-13 | Новартис Аг | COMBINATION |
| JOP20200094A1 (en) | 2014-01-24 | 2017-06-16 | Dana Farber Cancer Inst Inc | Antibody molecules to pd-1 and uses thereof |
| JOP20200096A1 (en) | 2014-01-31 | 2017-06-16 | Children’S Medical Center Corp | Antibody molecules to tim-3 and uses thereof |
| SI3116909T1 (en) | 2014-03-14 | 2020-03-31 | Novartis Ag | Antibody molecules to lag-3 and uses thereof |
| US10093620B2 (en) | 2014-09-12 | 2018-10-09 | Pellficure Pharmaceuticals, Inc. | Compositions and methods for treatment of prostate carcinoma |
| US9783527B2 (en) * | 2014-09-16 | 2017-10-10 | Abbvie Inc. | Indazole ureas and method of use |
| MA41044A (en) | 2014-10-08 | 2017-08-15 | Novartis Ag | COMPOSITIONS AND METHODS OF USE FOR INCREASED IMMUNE RESPONSE AND CANCER TREATMENT |
| PE20171067A1 (en) | 2014-10-14 | 2017-07-24 | Novartis Ag | ANTIBODY MOLECULES BINDING AND USES OF PD-L1 |
| WO2016100882A1 (en) | 2014-12-19 | 2016-06-23 | Novartis Ag | Combination therapies |
| WO2016145102A1 (en) | 2015-03-10 | 2016-09-15 | Aduro Biotech, Inc. | Compositions and methods for activating "stimulator of interferon gene" -dependent signalling |
| BR112018000925A2 (en) | 2015-07-17 | 2018-09-11 | Toyama Chemical Co., Ltd | nitrogen-containing heterocyclic compound |
| EP3317301B1 (en) | 2015-07-29 | 2021-04-07 | Novartis AG | Combination therapies comprising antibody molecules to lag-3 |
| EP3878465A1 (en) | 2015-07-29 | 2021-09-15 | Novartis AG | Combination therapies comprising antibody molecules to tim-3 |
| JP6878405B2 (en) | 2015-07-29 | 2021-05-26 | ノバルティス アーゲー | Combination therapy with antibody molecule against PD-1 |
| CR20180234A (en) | 2015-11-03 | 2018-09-11 | Janssen Biotech Inc | Antibodies specifically binding pd-1 and their uses |
| JP2019503349A (en) | 2015-12-17 | 2019-02-07 | ノバルティス アーゲー | Antibody molecules against PD-1 and uses thereof |
| WO2017211818A1 (en) * | 2016-06-06 | 2017-12-14 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Proteasome inhibitors |
| JP2019525896A (en) | 2016-06-06 | 2019-09-12 | マックス−プランク−ゲゼルシャフト・ツア・フェルデルング・デア・ヴィッセンシャフテン・エー・ファオ | Proteasome inhibitor |
| US11098077B2 (en) | 2016-07-05 | 2021-08-24 | Chinook Therapeutics, Inc. | Locked nucleic acid cyclic dinucleotide compounds and uses thereof |
| EP3528799A1 (en) | 2016-10-24 | 2019-08-28 | Pellficure Pharmaceuticals, Inc. | Pharmaceutical compositions of 5-hydroxy-2-methylnaphthalene-1, 4-dione |
| AU2017385292B2 (en) | 2016-12-27 | 2020-05-14 | Fujifilm Corporation | Antitumor agent and bromodomain inhibitor |
| CN108264509B (en) * | 2016-12-30 | 2021-05-04 | 复旦大学 | Substituted benzothieno [2,3-c ] tetrahydropyridine derivatives, preparation method and application thereof |
| UY37695A (en) | 2017-04-28 | 2018-11-30 | Novartis Ag | BIS 2’-5’-RR- (3’F-A) (3’F-A) CYCLE DINUCLEOTIDE COMPOUND AND USES OF THE SAME |
| WO2018237173A1 (en) | 2017-06-22 | 2018-12-27 | Novartis Ag | Antibody molecules to cd73 and uses thereof |
| CN111278816B (en) | 2017-09-04 | 2024-03-15 | C4医药公司 | Dihydro quinolinones |
| AU2019230014A1 (en) | 2018-03-05 | 2020-09-17 | Alkermes Pharma Ireland Limited | Aripiprazole dosing strategy |
| US11542275B2 (en) * | 2018-03-22 | 2023-01-03 | Aurigene Discovery Technologies Limited | Substituted imidazolidin-2-one derivatives as PRMT5 inhibitors |
| AR126019A1 (en) | 2018-05-30 | 2023-09-06 | Novartis Ag | ANTIBODIES AGAINST ENTPD2, COMBINATION THERAPIES AND METHODS OF USE OF ANTIBODIES AND COMBINATION THERAPIES |
| JP2022548881A (en) | 2019-09-18 | 2022-11-22 | ノバルティス アーゲー | ENTPD2 Antibodies, Combination Therapy and Methods of Using Antibodies and Combination Therapy |
| TW202321210A (en) * | 2021-07-22 | 2023-06-01 | 瑞士商諾華公司 | Compounds and compositions for the treatment of coronaviral related diseases |
| CN115304591B (en) * | 2022-07-08 | 2024-10-01 | 广州中医药大学(广州中医药研究院) | Bridged ring steroid synthetase inhibitor and preparation method and application thereof |
Citations (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3832352A (en) | 1971-05-03 | 1974-08-27 | Ciba Geigy Corp | 2-(thi)oxo-3-imidazolyl(2)-tetrahydroimidazoles |
| US5145845A (en) | 1991-05-14 | 1992-09-08 | Warner-Lambert Co. | Substituted 2-carboxylindoles having pharmaceutical activity |
| US5519036A (en) | 1993-02-22 | 1996-05-21 | Karl Thomae Gmbh | Cyclic derivatives pharmaceutical compositions containing these compounds and processes for preparing them |
| JPH08176111A (en) | 1994-12-20 | 1996-07-09 | Kyorin Pharmaceut Co Ltd | N,n'-di-substituted ethylenediamine derivative and production of n,n'-di-substituted imidazolidinone derivative using the same |
| WO1997008150A1 (en) | 1995-08-22 | 1997-03-06 | The Du Pont Merck Pharmaceutical Company | Substituted cyclic ureas and derivatives thereof useful as retroviral protease inhibitors |
| US5789425A (en) | 1993-09-17 | 1998-08-04 | Kyorin Pharmaceutical Co., Ltd. | Imidazolidinone derivatives, their acid adducts and therapeutic drugs for senile dementia |
| WO2001054694A1 (en) | 2000-01-28 | 2001-08-02 | Bristol-Myers Squibb Company | Tetrahydropyrimidone inhibitors of fatty acid binding protein and method |
| WO2002020493A2 (en) | 2000-09-06 | 2002-03-14 | Chugai Seiyaku Kabushiki Kaisha | A traceless solid-phase synthesis of 2-imidazolones |
| WO2003057220A1 (en) | 2002-01-08 | 2003-07-17 | Glaxo Group Limited | Cyclic urea derivatives with 5-ht2c receptor activity |
| WO2004009558A2 (en) | 2002-07-24 | 2004-01-29 | Ptc Therapeutics, Inc. | Ureido substituted benzoic acid compounds, their use for nonsense suppression and the treatment of diseases caused by such mutations |
| WO2004026859A1 (en) | 2002-09-18 | 2004-04-01 | Pfizer Products Inc. | Novel imidazole compounds as transforming growth factor (tgf) inhibitors |
| WO2006078698A1 (en) | 2005-01-19 | 2006-07-27 | Cengent Therapeutics, Inc. | 2-imidazolone and 2-imidazolidinone heterocyclic inhibitors of tyrosine phosphatases |
| WO2007109330A2 (en) | 2006-03-21 | 2007-09-27 | Epix Delaware, Inc. | S1p receptor modulating compounds |
| WO2008094556A2 (en) | 2007-01-30 | 2008-08-07 | Biogen Idec Ma Inc. | Imidazolone compounds as tgf-beta family type i receptors, alk5 and/or alk4 antagonists |
| WO2009078992A1 (en) | 2007-12-17 | 2009-06-25 | Amgen Inc. | Linear tricyclic compounds as p38 kinase inhibitors |
| WO2009097567A1 (en) | 2008-01-30 | 2009-08-06 | Cephalon, Inc. | Substituted spirocyclic piperidine derivatives as histamine-3 (h3) receptor ligands |
| US20090264650A1 (en) | 2005-03-31 | 2009-10-22 | Nobuo Cho | Prophylactic/Therapeutic Agent for Diabetes |
| WO2009143039A2 (en) | 2008-05-19 | 2009-11-26 | Schering Corporation | Heterocyclic compounds as factor ixa inhibitors |
| WO2009158473A1 (en) | 2008-06-25 | 2009-12-30 | Envivo Pharmaceuticals, Inc. | 5- and 6-membered heterocyclic compounds |
| WO2009156484A2 (en) | 2008-06-27 | 2009-12-30 | Novartis Ag | Organic compounds |
| WO2010045303A2 (en) | 2008-10-16 | 2010-04-22 | Schering Corporation | Pyrrolidine, piperidine and piperazine derivatives and methods of use thereof |
| US20100222588A1 (en) | 2009-02-27 | 2010-09-02 | Thesis Chemistry, Llc | Method for Manufacture of 2-Oxoimidazolidines |
| WO2011017534A2 (en) | 2009-08-07 | 2011-02-10 | Tokai Pharmaceuticals, Inc. | Treatment of prostate cancer |
| US20110039893A1 (en) | 2006-10-11 | 2011-02-17 | Takeda Pharmaceutical Company Limited | Gsk-3beta inhibitor |
| WO2011059969A2 (en) | 2009-11-13 | 2011-05-19 | Tokai Pharmaceuticals, Inc. | Mammalian metabolites of steroids |
| WO2011088160A2 (en) | 2010-01-15 | 2011-07-21 | Biomarin Pharmaceutical Inc. | Novel cyp17 inhibitors |
| WO2011098776A1 (en) | 2010-02-15 | 2011-08-18 | Cambridge Enterprise Limited | 5-ht receptor modulators |
Family Cites Families (65)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CH590267A5 (en) * | 1972-10-04 | 1977-07-29 | Ciba Geigy Ag | |
| DE2701794A1 (en) * | 1976-01-21 | 1977-07-28 | Ciba Geigy Ag | OXIGENATED N-ARYL DIAZACYCLES |
| GB1524747A (en) | 1976-05-11 | 1978-09-13 | Ici Ltd | Polypeptide |
| ATE28864T1 (en) | 1982-07-23 | 1987-08-15 | Ici Plc | AMIDE DERIVATIVES. |
| GB8327256D0 (en) | 1983-10-12 | 1983-11-16 | Ici Plc | Steroid derivatives |
| AU606677B2 (en) | 1987-04-22 | 1991-02-14 | Merrell Dow Pharmaceuticals Inc. | 17 beta -(cyclopropylimino) androst-5-EN-3 beta- Ol and related compounds useful as C17-20 lyase inhibitors |
| US5093330A (en) | 1987-06-15 | 1992-03-03 | Ciba-Geigy Corporation | Staurosporine derivatives substituted at methylamino nitrogen |
| US5010099A (en) | 1989-08-11 | 1991-04-23 | Harbor Branch Oceanographic Institution, Inc. | Discodermolide compounds, compositions containing same and method of preparation and use |
| US4966898A (en) | 1989-08-15 | 1990-10-30 | Merrell Dow Pharmaceuticals Inc. | 4-substituted 17β-(cyclopropylamino)androst-5-en-3β-ol and related compounds useful as C17-20 lyase inhibitors |
| US5395855A (en) | 1990-05-07 | 1995-03-07 | Ciba-Geigy Corporation | Hydrazones |
| JPH06507355A (en) | 1991-03-01 | 1994-08-25 | エレクトロスタティック テクノロジー インコーポレーテッド | Powder coating method for manufacturing thin laminates for circuit boards, etc. |
| NZ243082A (en) | 1991-06-28 | 1995-02-24 | Ici Plc | 4-anilino-quinazoline derivatives; pharmaceutical compositions, preparatory processes, and use thereof |
| GB9300059D0 (en) | 1992-01-20 | 1993-03-03 | Zeneca Ltd | Quinazoline derivatives |
| US5457102A (en) | 1994-07-07 | 1995-10-10 | Janssen Pharmaceutica, N.V. | Pyrroloimidazolyl and imidazopyridinyl substituted 1H-benzimidazole derivatives |
| JP2742331B2 (en) | 1992-03-31 | 1998-04-22 | ブリテイツシユ・テクノロジー・グループ・リミテツド | 17-substituted steroids useful for cancer treatment |
| TW225528B (en) | 1992-04-03 | 1994-06-21 | Ciba Geigy Ag | |
| DK0666868T4 (en) | 1992-10-28 | 2006-09-18 | Genentech Inc | Use of anti-VEGF antibodies to treat cancer |
| US5270329A (en) * | 1992-12-10 | 1993-12-14 | Eli Lilly And Company | Antitumor compositions and methods of treatment |
| GB9310635D0 (en) | 1993-05-21 | 1993-07-07 | Glaxo Group Ltd | Chemical compounds |
| GB9314893D0 (en) | 1993-07-19 | 1993-09-01 | Zeneca Ltd | Quinazoline derivatives |
| KR960704852A (en) | 1993-09-30 | 1996-10-09 | 오노다 마사요시 | Azole derivatives and pharmaceutical compositions thereof |
| GB2282377B (en) | 1993-09-30 | 1997-09-03 | British Tech Group | Synthesis of 17-(3-pyridyl) steroids |
| WO1996014090A1 (en) | 1994-11-07 | 1996-05-17 | Janssen Pharmaceutica N.V. | Compositions comprising carbazoles and cyclodextrins |
| EP1110953B1 (en) | 1995-03-30 | 2009-10-28 | Pfizer Products Inc. | Quinazoline derivatives |
| GB9508538D0 (en) | 1995-04-27 | 1995-06-14 | Zeneca Ltd | Quinazoline derivatives |
| US5747498A (en) | 1996-05-28 | 1998-05-05 | Pfizer Inc. | Alkynyl and azido-substituted 4-anilinoquinazolines |
| US5843901A (en) | 1995-06-07 | 1998-12-01 | Advanced Research & Technology Institute | LHRH antagonist peptides |
| US5880141A (en) | 1995-06-07 | 1999-03-09 | Sugen, Inc. | Benzylidene-Z-indoline compounds for the treatment of disease |
| AU6015796A (en) | 1995-06-14 | 1997-01-15 | Yamanouchi Pharmaceutical Co., Ltd. | Fused imidazole derivatives and medicinal composition thereof |
| DK0836605T3 (en) | 1995-07-06 | 2002-05-13 | Novartis Ag | Pyrrolopyrimidines and Methods for their Preparation |
| US5760041A (en) | 1996-02-05 | 1998-06-02 | American Cyanamid Company | 4-aminoquinazoline EGFR Inhibitors |
| GB9603095D0 (en) | 1996-02-14 | 1996-04-10 | Zeneca Ltd | Quinazoline derivatives |
| NZ332119A (en) | 1996-04-12 | 2001-08-31 | Warner Lambert Co | Quinazoline compounds which are irreversible inhibitors of tyrosine kinases |
| WO1997044339A1 (en) * | 1996-05-20 | 1997-11-27 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| DE69734513T2 (en) | 1996-06-24 | 2006-07-27 | Pfizer Inc. | PHENYLAMINO-SUBSTITUTED TRICYCL DERIVATIVES FOR THE TREATMENT OF HYPERPROLIFERATIVE DISEASES |
| JP2001500851A (en) | 1996-08-30 | 2001-01-23 | ノバルティス アクチエンゲゼルシャフト | Process for producing epothilone and intermediate products obtained during the process |
| CA2264908C (en) | 1996-09-06 | 2006-04-25 | Obducat Ab | Method for anisotropic etching of structures in conducting materials |
| DE19638745C2 (en) | 1996-09-11 | 2001-05-10 | Schering Ag | Monoclonal antibodies against the extracellular domain of the human VEGF receptor protein (KDR) |
| AU4342997A (en) | 1996-09-13 | 1998-04-02 | Sugen, Inc. | Use of quinazoline derivatives for the manufacture of a medicament in the reatment of hyperproliferative skin disorders |
| EP0837063A1 (en) | 1996-10-17 | 1998-04-22 | Pfizer Inc. | 4-Aminoquinazoline derivatives |
| HU229833B1 (en) | 1996-11-18 | 2014-09-29 | Biotechnolog Forschung Gmbh | Epothilone d production process, and its use as cytostatic as well as phytosanitary agents |
| US6441186B1 (en) | 1996-12-13 | 2002-08-27 | The Scripps Research Institute | Epothilone analogs |
| CO4950519A1 (en) | 1997-02-13 | 2000-09-01 | Novartis Ag | PHTHALAZINES, PHARMACEUTICAL PREPARATIONS THAT UNDERSTAND THEM AND THE PROCESS FOR THEIR PREPARATION |
| CO4940418A1 (en) | 1997-07-18 | 2000-07-24 | Novartis Ag | MODIFICATION OF A CRYSTAL OF A DERIVATIVE OF N-PHENYL-2-PIRIMIDINAMINE, PROCESSES FOR ITS MANUFACTURE AND USE |
| NZ501822A (en) | 1997-10-02 | 2001-12-21 | Yukijirushi Nyugyo Kabushiki K | Therapeutic agents for male and female sex hormone dependent diseases |
| GB9721069D0 (en) | 1997-10-03 | 1997-12-03 | Pharmacia & Upjohn Spa | Polymeric derivatives of camptothecin |
| US6194181B1 (en) | 1998-02-19 | 2001-02-27 | Novartis Ag | Fermentative preparation process for and crystal forms of cytostatics |
| NZ506742A (en) | 1998-02-25 | 2003-09-26 | Sloan Kettering Inst Cancer | Synthesis of desoxyepothilones and hydroxy acid derivative intermediates |
| DE69924717T2 (en) | 1998-04-23 | 2006-03-09 | Takeda Pharmaceutical Co. Ltd. | Naphthalenic derivatives, their preparation and their use |
| ATE459616T1 (en) | 1998-08-11 | 2010-03-15 | Novartis Ag | ISOCHINOLINE DERIVATIVES WITH ANGIOGENESIS-INHIBITING EFFECT |
| GB9824579D0 (en) | 1998-11-10 | 1999-01-06 | Novartis Ag | Organic compounds |
| UA71587C2 (en) | 1998-11-10 | 2004-12-15 | Шерінг Акцієнгезелльшафт | Anthranilic acid amides and use thereof as medicaments |
| NZ511722A (en) | 1998-11-20 | 2004-05-28 | Kosan Biosciences Inc | Recombinant methods and materials for producing epothilone and epothilone derivatives |
| IL143596A0 (en) | 1998-12-22 | 2002-04-21 | Genentech Inc | Vascular endothelial cell growth factor antagonists and uses thereof |
| BR0009507A (en) | 1999-03-30 | 2002-01-15 | Novartis Ag | Phthalazine derivatives for the treatment of inflammatory diseases |
| JP2002302488A (en) * | 2000-03-30 | 2002-10-18 | Takeda Chem Ind Ltd | Substituted 1,3-thiazole compound, its production method and use thereof |
| NO312876B1 (en) * | 2000-07-21 | 2002-07-15 | Balans Man As | Adjustable chair |
| AR035885A1 (en) | 2001-05-14 | 2004-07-21 | Novartis Ag | DERIVATIVES OF 4-AMINO-5-FENIL-7-CYCLLOBUTILPIRROLO (2,3-D) PYRIMIDINE, A PROCESS FOR ITS PREPARATION, A PHARMACEUTICAL COMPOSITION AND THE USE OF SUCH DERIVATIVES FOR THE PREPARATION OF A PHARMACEUTICAL COMPOSITION |
| GB0119249D0 (en) | 2001-08-07 | 2001-10-03 | Novartis Ag | Organic compounds |
| US20040198773A1 (en) | 2001-09-26 | 2004-10-07 | Barry Hart | Substituted 3-pyridyl oxazoles as c17,20 lyase inhibitors |
| GB0418900D0 (en) | 2004-08-24 | 2004-09-29 | Btg Int Ltd | Novel salt forms |
| DK2206719T3 (en) | 2005-03-02 | 2015-01-26 | Univ Maryland | A pharmaceutical composition comprising 3-BETA-HYDROXY-17- (1-H-benzimidazol-1-yl) androsta-5, 16-DIEN |
| CA2838089A1 (en) | 2006-08-25 | 2008-02-28 | Cougar Biotechnology, Inc. | Methods for treating cancer comprising the administration of a vitamin d compound and an additional therapeutic agent, and compositions containing the same |
| WO2008154382A1 (en) | 2007-06-06 | 2008-12-18 | University Of Maryland, Baltimore | Hdac inhibitors and hormone targeted drugs for the treatment of cancer |
| CN101084881B (en) | 2007-06-23 | 2012-08-29 | 淮北辉克药业有限公司 | Targeted quick-releasing effervescence preparation and preparation method thereof |
-
2010
- 2010-06-24 GE GEAP201012550A patent/GEP20156250B/en unknown
- 2010-06-24 ES ES10730132.7T patent/ES2475945T3/en active Active
- 2010-06-24 MA MA34559A patent/MA33451B1/en unknown
- 2010-06-24 PL PL10730132T patent/PL2445903T3/en unknown
- 2010-06-24 BR BRPI1015568A patent/BRPI1015568B8/en active IP Right Grant
- 2010-06-24 JO JO2010210A patent/JO2892B1/en active
- 2010-06-24 AU AU2010264698A patent/AU2010264698C1/en active Active
- 2010-06-24 DK DK10730132.7T patent/DK2445903T3/en active
- 2010-06-24 CN CN201080028351.3A patent/CN102803250B/en active Active
- 2010-06-24 JP JP2012516752A patent/JP5456891B2/en active Active
- 2010-06-24 SI SI201030633T patent/SI2445903T1/en unknown
- 2010-06-24 RS RS20140323A patent/RS53384B/en unknown
- 2010-06-24 SG SG2011084407A patent/SG176105A1/en unknown
- 2010-06-24 CA CA2765983A patent/CA2765983C/en active Active
- 2010-06-24 MX MX2011013771A patent/MX2011013771A/en active IP Right Grant
- 2010-06-24 EA EA201200049A patent/EA021011B1/en not_active IP Right Cessation
- 2010-06-24 KR KR1020127001913A patent/KR101360725B1/en active IP Right Grant
- 2010-06-24 MY MYPI2011005620A patent/MY155570A/en unknown
- 2010-06-24 EP EP10730132.7A patent/EP2445903B1/en active Active
- 2010-06-24 AR ARP100102237A patent/AR078228A1/en unknown
- 2010-06-24 PE PE2011002130A patent/PE20121048A1/en not_active Application Discontinuation
- 2010-06-24 PT PT107301327T patent/PT2445903E/en unknown
- 2010-06-24 ME MEP-2014-65A patent/ME01886B/en unknown
- 2010-06-24 NZ NZ596488A patent/NZ596488A/en not_active IP Right Cessation
- 2010-06-24 WO PCT/EP2010/059029 patent/WO2010149755A1/en active Application Filing
- 2010-06-24 UY UY0001032730A patent/UY32730A/en not_active Application Discontinuation
- 2010-06-24 UA UAA201115301A patent/UA105794C2/en unknown
- 2010-06-25 TW TW099120966A patent/TWI492942B/en not_active IP Right Cessation
- 2010-06-28 US US12/824,845 patent/US8263635B2/en not_active Ceased
-
2011
- 2011-11-15 ZA ZA2011/08373A patent/ZA201108373B/en unknown
- 2011-11-29 TN TNP2011000611A patent/TN2011000611A1/en unknown
- 2011-12-01 IL IL216741A patent/IL216741A/en not_active IP Right Cessation
- 2011-12-19 CU CU2011000235A patent/CU24039B1/en active IP Right Grant
- 2011-12-20 CR CR20110684A patent/CR20110684A/en unknown
- 2011-12-21 DO DO2011000399A patent/DOP2011000399A/en unknown
- 2011-12-21 NI NI201100223A patent/NI201100223A/en unknown
- 2011-12-22 CO CO11177195A patent/CO6480927A2/en active IP Right Grant
- 2011-12-22 CL CL2011003266A patent/CL2011003266A1/en unknown
- 2011-12-22 HN HN2011003371A patent/HN2011003371A/en unknown
-
2012
- 2012-01-26 EC EC2012011625A patent/ECSP12011625A/en unknown
- 2012-06-12 HK HK12105692.6A patent/HK1164876A1/en not_active IP Right Cessation
-
2013
- 2013-06-28 US US13/929,901 patent/USRE45173E1/en active Active
-
2014
- 2014-04-21 US US14/257,433 patent/US20140228394A1/en not_active Abandoned
- 2014-06-24 HR HRP20140593AT patent/HRP20140593T1/en unknown
- 2014-06-26 CY CY20141100465T patent/CY1115426T1/en unknown
- 2014-07-10 SM SM201400091T patent/SMT201400091B/en unknown
Patent Citations (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3832352A (en) | 1971-05-03 | 1974-08-27 | Ciba Geigy Corp | 2-(thi)oxo-3-imidazolyl(2)-tetrahydroimidazoles |
| US5145845A (en) | 1991-05-14 | 1992-09-08 | Warner-Lambert Co. | Substituted 2-carboxylindoles having pharmaceutical activity |
| US5519036A (en) | 1993-02-22 | 1996-05-21 | Karl Thomae Gmbh | Cyclic derivatives pharmaceutical compositions containing these compounds and processes for preparing them |
| EP0719773B1 (en) | 1993-09-17 | 2001-11-21 | Kyorin Pharmaceutical Co., Ltd. | Imidazolidinone derivative, acid-addition salt thereof, and remedy for senile dementia |
| US5789425A (en) | 1993-09-17 | 1998-08-04 | Kyorin Pharmaceutical Co., Ltd. | Imidazolidinone derivatives, their acid adducts and therapeutic drugs for senile dementia |
| JPH08176111A (en) | 1994-12-20 | 1996-07-09 | Kyorin Pharmaceut Co Ltd | N,n'-di-substituted ethylenediamine derivative and production of n,n'-di-substituted imidazolidinone derivative using the same |
| WO1997008150A1 (en) | 1995-08-22 | 1997-03-06 | The Du Pont Merck Pharmaceutical Company | Substituted cyclic ureas and derivatives thereof useful as retroviral protease inhibitors |
| WO2001054694A1 (en) | 2000-01-28 | 2001-08-02 | Bristol-Myers Squibb Company | Tetrahydropyrimidone inhibitors of fatty acid binding protein and method |
| WO2002020493A2 (en) | 2000-09-06 | 2002-03-14 | Chugai Seiyaku Kabushiki Kaisha | A traceless solid-phase synthesis of 2-imidazolones |
| WO2003057220A1 (en) | 2002-01-08 | 2003-07-17 | Glaxo Group Limited | Cyclic urea derivatives with 5-ht2c receptor activity |
| US20050154028A1 (en) | 2002-01-08 | 2005-07-14 | Bromidge Steven M. | Cyclic urea derivatives with 5-ht2c receptor activity |
| WO2004009558A2 (en) | 2002-07-24 | 2004-01-29 | Ptc Therapeutics, Inc. | Ureido substituted benzoic acid compounds, their use for nonsense suppression and the treatment of diseases caused by such mutations |
| US20060167065A1 (en) | 2002-07-24 | 2006-07-27 | Wilde Richard G | Ureido substituted benzoic acid compounds and their use for nonsense suppression and the treatment of disease |
| US7405233B2 (en) | 2002-07-24 | 2008-07-29 | Ptc Therapeutics, Inc. | Ureido substituted benzoic acid compounds and their use for nonsense suppression and the treatment of disease |
| WO2004026859A1 (en) | 2002-09-18 | 2004-04-01 | Pfizer Products Inc. | Novel imidazole compounds as transforming growth factor (tgf) inhibitors |
| WO2006078698A1 (en) | 2005-01-19 | 2006-07-27 | Cengent Therapeutics, Inc. | 2-imidazolone and 2-imidazolidinone heterocyclic inhibitors of tyrosine phosphatases |
| US20090264650A1 (en) | 2005-03-31 | 2009-10-22 | Nobuo Cho | Prophylactic/Therapeutic Agent for Diabetes |
| WO2007109330A2 (en) | 2006-03-21 | 2007-09-27 | Epix Delaware, Inc. | S1p receptor modulating compounds |
| US20110039893A1 (en) | 2006-10-11 | 2011-02-17 | Takeda Pharmaceutical Company Limited | Gsk-3beta inhibitor |
| WO2008094556A2 (en) | 2007-01-30 | 2008-08-07 | Biogen Idec Ma Inc. | Imidazolone compounds as tgf-beta family type i receptors, alk5 and/or alk4 antagonists |
| WO2009078992A1 (en) | 2007-12-17 | 2009-06-25 | Amgen Inc. | Linear tricyclic compounds as p38 kinase inhibitors |
| WO2009097567A1 (en) | 2008-01-30 | 2009-08-06 | Cephalon, Inc. | Substituted spirocyclic piperidine derivatives as histamine-3 (h3) receptor ligands |
| WO2009143039A2 (en) | 2008-05-19 | 2009-11-26 | Schering Corporation | Heterocyclic compounds as factor ixa inhibitors |
| WO2009158473A1 (en) | 2008-06-25 | 2009-12-30 | Envivo Pharmaceuticals, Inc. | 5- and 6-membered heterocyclic compounds |
| WO2009156484A2 (en) | 2008-06-27 | 2009-12-30 | Novartis Ag | Organic compounds |
| WO2010045303A2 (en) | 2008-10-16 | 2010-04-22 | Schering Corporation | Pyrrolidine, piperidine and piperazine derivatives and methods of use thereof |
| US20100222588A1 (en) | 2009-02-27 | 2010-09-02 | Thesis Chemistry, Llc | Method for Manufacture of 2-Oxoimidazolidines |
| WO2011017534A2 (en) | 2009-08-07 | 2011-02-10 | Tokai Pharmaceuticals, Inc. | Treatment of prostate cancer |
| WO2011059969A2 (en) | 2009-11-13 | 2011-05-19 | Tokai Pharmaceuticals, Inc. | Mammalian metabolites of steroids |
| WO2011088160A2 (en) | 2010-01-15 | 2011-07-21 | Biomarin Pharmaceutical Inc. | Novel cyp17 inhibitors |
| WO2011098776A1 (en) | 2010-02-15 | 2011-08-18 | Cambridge Enterprise Limited | 5-ht receptor modulators |
Non-Patent Citations (17)
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9339501B2 (en) | 2011-04-28 | 2016-05-17 | Novartis Ag | 17a-hydroxylase/C17,20-lyase inhibitors |
| WO2021026454A1 (en) | 2019-08-08 | 2021-02-11 | Laekna Limited | Method of treating cancer |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| USRE45173E1 (en) | Inhibitors of CYP 17 | |
| US8946260B2 (en) | 17α-hydroxylase/C17,20-lyase inhibitors | |
| US8507676B2 (en) | Heterocyclic oxime compounds | |
| US8859535B2 (en) | Hydroxy substituted isoquinolinone derivatives | |
| JP5330498B2 (en) | Hydroxamate-based inhibitors of deacetylase B | |
| US9339501B2 (en) | 17a-hydroxylase/C17,20-lyase inhibitors | |
| US20130324526A1 (en) | [1,2,4] triazolo [4,3-b] pyridazine compounds as inhibitors of the c-met tyrosine kinase | |
| BOCK et al. | Patent 2765983 Summary |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: NOVARTIS AG, SWITZERLAND Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:BOCK, MARK GARY;GAUL, CHRISTOPH;REEL/FRAME:033339/0234 Effective date: 20100325 Owner name: NOVARTIS AG, SWITZERLAND Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:AURIGENE DISCOVERY TECHNOLOGIES LIMITED;REEL/FRAME:033339/0377 Effective date: 20100407 Owner name: AURIGENE DISCOVERY TECHNOLOGIES LIMITED, INDIA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:GUMMADI, VENKATESHWAR RAO;SENGUPTA, SAUMITRA;REEL/FRAME:033339/0341 Effective date: 20100407 |
|
| FPAY | Fee payment |
Year of fee payment: 4 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 8TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1552); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 8 |
|
| FEPP | Fee payment procedure |
Free format text: 11.5 YR SURCHARGE- LATE PMT W/IN 6 MO, LARGE ENTITY (ORIGINAL EVENT CODE: M1556); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 12TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1553); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 12 |