US8344136B2 - Process for the preparation of brinzolamide - Google Patents
Process for the preparation of brinzolamide Download PDFInfo
- Publication number
- US8344136B2 US8344136B2 US12/999,054 US99905409A US8344136B2 US 8344136 B2 US8344136 B2 US 8344136B2 US 99905409 A US99905409 A US 99905409A US 8344136 B2 US8344136 B2 US 8344136B2
- Authority
- US
- United States
- Prior art keywords
- formula
- group
- hydrogen
- compound
- chlorine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active, expires
Links
- 0 *N1CC(=O)C2=C(SC(C)=C2)S1(=O)=O Chemical compound *N1CC(=O)C2=C(SC(C)=C2)S1(=O)=O 0.000 description 12
- WFAFJHODEURNEB-UHFFFAOYSA-N CC1=CC(C(=O)CBr)=C(S(N)(=O)=O)S1 Chemical compound CC1=CC(C(=O)CBr)=C(S(N)(=O)=O)S1 WFAFJHODEURNEB-UHFFFAOYSA-N 0.000 description 2
- QCFXXLHCIJNOOO-UHFFFAOYSA-N CC1=CC(C2(CBr)OCCO2)=C(S(N)(=O)=O)S1 Chemical compound CC1=CC(C2(CBr)OCCO2)=C(S(N)(=O)=O)S1 QCFXXLHCIJNOOO-UHFFFAOYSA-N 0.000 description 2
- DVWDNVDSKPUFBA-UHFFFAOYSA-N CC1=CC2=C(S1)S(=O)(=O)NCC21OCCO1 Chemical compound CC1=CC2=C(S1)S(=O)(=O)NCC21OCCO1 DVWDNVDSKPUFBA-UHFFFAOYSA-N 0.000 description 2
- JQTLPQFWGWKTCC-UHFFFAOYSA-N COCCCN1CC(=O)C2=C(SC(C)=C2)S1(=O)=O Chemical compound COCCCN1CC(=O)C2=C(SC(C)=C2)S1(=O)=O JQTLPQFWGWKTCC-UHFFFAOYSA-N 0.000 description 2
- OHVOUOAKGSPWPU-UHFFFAOYSA-N COCCCN1CC2(OCCO2)C2=C(SC(C)=C2)S1(=O)=O Chemical compound COCCCN1CC2(OCCO2)C2=C(SC(C)=C2)S1(=O)=O OHVOUOAKGSPWPU-UHFFFAOYSA-N 0.000 description 2
- JRSYNSSFLFPSPX-UHFFFAOYSA-N CC(=O)C1=C(Cl)SC(Cl)=C1.CC(=O)C1=C(S(N)(=O)=O)SC(Cl)=C1.CC(=O)C1=C(SCC2=CC=CC=C2)SC(Cl)=C1.CC(=O)C1=C(SCl)SC(Cl)=C1.CC(=O)C1=C(SN)SC(Cl)=C1.COCCCN1CC(O)C2=C(SC(Cl)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC(N)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC(N=C(C)C)=C2)S1(=O)=O.COCCCN1CC(OS(=O)(=O)C2=CC=C(C)C=C2)C2=C(SC(N=C(C)C)=C2)S1(=O)=O.NS(=O)(=O)C1=C(C(=O)CBr)C=C(Cl)S1.NS(=O)(=O)C1=C(C(O)CBr)C=C(Cl)S1.O=S1(=O)NCC(O)C2=C1SC(Cl)=C2.O=S=O.O=S=O.O=S=O Chemical compound CC(=O)C1=C(Cl)SC(Cl)=C1.CC(=O)C1=C(S(N)(=O)=O)SC(Cl)=C1.CC(=O)C1=C(SCC2=CC=CC=C2)SC(Cl)=C1.CC(=O)C1=C(SCl)SC(Cl)=C1.CC(=O)C1=C(SN)SC(Cl)=C1.COCCCN1CC(O)C2=C(SC(Cl)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC(N)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC(N=C(C)C)=C2)S1(=O)=O.COCCCN1CC(OS(=O)(=O)C2=CC=C(C)C=C2)C2=C(SC(N=C(C)C)=C2)S1(=O)=O.NS(=O)(=O)C1=C(C(=O)CBr)C=C(Cl)S1.NS(=O)(=O)C1=C(C(O)CBr)C=C(Cl)S1.O=S1(=O)NCC(O)C2=C1SC(Cl)=C2.O=S=O.O=S=O.O=S=O JRSYNSSFLFPSPX-UHFFFAOYSA-N 0.000 description 1
- LXANAHZPFDGBEG-UHFFFAOYSA-N CC(=O)C1=C(S(N)(=O)=O)SC=C1.CC(=O)C1=CSC=C1.CC1(C)COC(C)(C2=C(S(N)(=O)=O)SC=C2)OC1.CC1(C)COC(C)(C2=CSC=C2)OC1.CCOC(C)OC1CN(CCCOC)S(=O)(=O)C2=C1C=C(N)S2.CCOC(C)OC1CN(CCCOC)S(=O)(=O)C2=C1C=CS2.COCCCN1CC(O)C2=C(SC(N)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC=C2)S1(=O)=O.NS(=O)(=O)C1=C(C(=O)CBr)C=CS1.NS(=O)(=O)C1=C(C(O)CBr)C=CS1.O=S1(=O)NCC(O)C2=C1SC=C2.O=S=O.O=S=O Chemical compound CC(=O)C1=C(S(N)(=O)=O)SC=C1.CC(=O)C1=CSC=C1.CC1(C)COC(C)(C2=C(S(N)(=O)=O)SC=C2)OC1.CC1(C)COC(C)(C2=CSC=C2)OC1.CCOC(C)OC1CN(CCCOC)S(=O)(=O)C2=C1C=C(N)S2.CCOC(C)OC1CN(CCCOC)S(=O)(=O)C2=C1C=CS2.COCCCN1CC(O)C2=C(SC(N)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC=C2)S1(=O)=O.NS(=O)(=O)C1=C(C(=O)CBr)C=CS1.NS(=O)(=O)C1=C(C(O)CBr)C=CS1.O=S1(=O)NCC(O)C2=C1SC=C2.O=S=O.O=S=O LXANAHZPFDGBEG-UHFFFAOYSA-N 0.000 description 1
- TWMYPOOISVCBBL-ZFELEUDSSA-N CCN[C@H]1CN(CCCOC)S(=O)(=O)C2=C1C=C(N)S2.COCCCN1CC(=O)C2=C(SC(N)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC(N)=C2)S1(=O)=O.COCCCN1CC(OS(=O)(=O)C2=CC=C(C)C=C2)C2=C(SC(N)=C2)S1(=O)=O.O=S=O.O=S=O.O=S=O.O=S=O Chemical compound CCN[C@H]1CN(CCCOC)S(=O)(=O)C2=C1C=C(N)S2.COCCCN1CC(=O)C2=C(SC(N)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC(N)=C2)S1(=O)=O.COCCCN1CC(OS(=O)(=O)C2=CC=C(C)C=C2)C2=C(SC(N)=C2)S1(=O)=O.O=S=O.O=S=O.O=S=O.O=S=O TWMYPOOISVCBBL-ZFELEUDSSA-N 0.000 description 1
- HWJOKQRHIBNACH-PPHPATTJSA-N CCN[C@H]1CN(CCCOC)S(=O)(=O)C2=C1C=C(N)S2.O=S=O Chemical compound CCN[C@H]1CN(CCCOC)S(=O)(=O)C2=C1C=C(N)S2.O=S=O HWJOKQRHIBNACH-PPHPATTJSA-N 0.000 description 1
- MDFYTZCAFWWLTJ-JTQLQIEISA-N CCN[C@H]1CN(CCCOC)S(=O)(=O)C2=C1C=C(N[SH](=O)=O)S2 Chemical compound CCN[C@H]1CN(CCCOC)S(=O)(=O)C2=C1C=C(N[SH](=O)=O)S2 MDFYTZCAFWWLTJ-JTQLQIEISA-N 0.000 description 1
- ZNVCHIXBODZFAU-UHFFFAOYSA-N COCCCN(CC1(c2c3[s]c(Cl)c2)OCCO1)S3(=O)=O Chemical compound COCCCN(CC1(c2c3[s]c(Cl)c2)OCCO1)S3(=O)=O ZNVCHIXBODZFAU-UHFFFAOYSA-N 0.000 description 1
- ROMUBWYRKIRIQX-UHFFFAOYSA-N COCCCN(CC1(c2c3[s]c(S(N)(=O)=O)c2)OCCO1)S3(=O)=O Chemical compound COCCCN(CC1(c2c3[s]c(S(N)(=O)=O)c2)OCCO1)S3(=O)=O ROMUBWYRKIRIQX-UHFFFAOYSA-N 0.000 description 1
- MWNQVYWQLXVUNH-UHFFFAOYSA-N COCCCN(CC1(c2c3[s]cc2)OCCO1)S3(=O)=O Chemical compound COCCCN(CC1(c2c3[s]cc2)OCCO1)S3(=O)=O MWNQVYWQLXVUNH-UHFFFAOYSA-N 0.000 description 1
- FOMMKUUBLCHWBM-UHFFFAOYSA-N COCCCN1CC(=O)C2=C(SC(Cl)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC(Cl)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC(Cl)=C2)S1(=O)=O.NS(=O)C1=C(C(=O)CBr)C=C(Cl)S1.NS(=O)C1=C(C(O)CBr)C=C(Cl)S1.O=S1(=O)NCC(O)C2=C1SC(Cl)=C2 Chemical compound COCCCN1CC(=O)C2=C(SC(Cl)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC(Cl)=C2)S1(=O)=O.COCCCN1CC(O)C2=C(SC(Cl)=C2)S1(=O)=O.NS(=O)C1=C(C(=O)CBr)C=C(Cl)S1.NS(=O)C1=C(C(O)CBr)C=C(Cl)S1.O=S1(=O)NCC(O)C2=C1SC(Cl)=C2 FOMMKUUBLCHWBM-UHFFFAOYSA-N 0.000 description 1
- OMAKACMCQWBQNK-UHFFFAOYSA-N COCCCN1CC(=O)C2=C(SC(Cl)=C2)S1(=O)=O.COCCCN1CC2(OCCO2)C2=C(SC(Cl)=C2)S1(=O)=O Chemical compound COCCCN1CC(=O)C2=C(SC(Cl)=C2)S1(=O)=O.COCCCN1CC2(OCCO2)C2=C(SC(Cl)=C2)S1(=O)=O OMAKACMCQWBQNK-UHFFFAOYSA-N 0.000 description 1
- ZUMPUHLDUVJPBQ-UHFFFAOYSA-N COCCCN1CC(=O)C2=C(SC(S(N)(=O)=O)=C2)S1(=O)=O.COCCCN1CC2(OCCO2)C2=C(SC(S(N)(=O)=O)=C2)S1(=O)=O Chemical compound COCCCN1CC(=O)C2=C(SC(S(N)(=O)=O)=C2)S1(=O)=O.COCCCN1CC2(OCCO2)C2=C(SC(S(N)(=O)=O)=C2)S1(=O)=O ZUMPUHLDUVJPBQ-UHFFFAOYSA-N 0.000 description 1
- GIBDEIPMAVSTDK-UHFFFAOYSA-N COCCCN1CC(=O)C2=C(SC=C2)S1(=O)=O.COCCCN1CC2(OCCO2)C2=C(SC=C2)S1(=O)=O Chemical compound COCCCN1CC(=O)C2=C(SC=C2)S1(=O)=O.COCCCN1CC2(OCCO2)C2=C(SC=C2)S1(=O)=O GIBDEIPMAVSTDK-UHFFFAOYSA-N 0.000 description 1
- QXIOSUHGJGEXJK-UHFFFAOYSA-N COCCCN1CC2(OCCO2)C2=C(SC(Cl)=C2)S1(=O)=O.COCCCN1CC2(OCCO2)C2=C(SC(S(N)(=O)=O)=C2)S1(=O)=O Chemical compound COCCCN1CC2(OCCO2)C2=C(SC(Cl)=C2)S1(=O)=O.COCCCN1CC2(OCCO2)C2=C(SC(S(N)(=O)=O)=C2)S1(=O)=O QXIOSUHGJGEXJK-UHFFFAOYSA-N 0.000 description 1
- BPIWEQPTWIHONC-UHFFFAOYSA-N COCCCN1CC2(OCCO2)C2=C(SC(Cl)=C2)S1(=O)=O.NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=C(Cl)S1 Chemical compound COCCCN1CC2(OCCO2)C2=C(SC(Cl)=C2)S1(=O)=O.NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=C(Cl)S1 BPIWEQPTWIHONC-UHFFFAOYSA-N 0.000 description 1
- KETPRZXBLDWTSU-UHFFFAOYSA-N COCCCN1CC2(OCCO2)C2=C(SC(S(N)(=O)=O)=C2)S1(=O)=O.COCCCN1CC2(OCCO2)C2=C(SC=C2)S1(=O)=O Chemical compound COCCCN1CC2(OCCO2)C2=C(SC(S(N)(=O)=O)=C2)S1(=O)=O.COCCCN1CC2(OCCO2)C2=C(SC=C2)S1(=O)=O KETPRZXBLDWTSU-UHFFFAOYSA-N 0.000 description 1
- AYOHKQAUIGLXGP-UHFFFAOYSA-N COCCCN1CC2(OCCO2)C2=C(SC=C2)S1(=O)=O.NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=CS1 Chemical compound COCCCN1CC2(OCCO2)C2=C(SC=C2)S1(=O)=O.NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=CS1 AYOHKQAUIGLXGP-UHFFFAOYSA-N 0.000 description 1
- OFOOUIZKRWATCE-UHFFFAOYSA-N NS(=O)(=O)C1=C(C(=O)CBr)C=C(Cl)S1.NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=C(Cl)S1 Chemical compound NS(=O)(=O)C1=C(C(=O)CBr)C=C(Cl)S1.NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=C(Cl)S1 OFOOUIZKRWATCE-UHFFFAOYSA-N 0.000 description 1
- YBBLULRTZRJIAN-UHFFFAOYSA-N NS(=O)(=O)C1=C(C(=O)CBr)C=CS1.NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=CS1 Chemical compound NS(=O)(=O)C1=C(C(=O)CBr)C=CS1.NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=CS1 YBBLULRTZRJIAN-UHFFFAOYSA-N 0.000 description 1
- NRCOCWSCPXCFEB-UHFFFAOYSA-N NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=C(Cl)S1.O=S1(=O)NCC2(OCCO2)C2=C1SC(Cl)=C2 Chemical compound NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=C(Cl)S1.O=S1(=O)NCC2(OCCO2)C2=C1SC(Cl)=C2 NRCOCWSCPXCFEB-UHFFFAOYSA-N 0.000 description 1
- BDFCRDCREOEJPD-UHFFFAOYSA-N NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=CS1.O=S1(=O)NCC2(OCCO2)C2=C1SC=C2 Chemical compound NS(=O)(=O)C1=C(C2(CBr)OCCO2)C=CS1.O=S1(=O)NCC2(OCCO2)C2=C1SC=C2 BDFCRDCREOEJPD-UHFFFAOYSA-N 0.000 description 1
- NSUGPRFLIOERPF-UHFFFAOYSA-N NS(c([s]1)c(C2(CBr)OCCO2)cc1Cl)(=O)=O Chemical compound NS(c([s]1)c(C2(CBr)OCCO2)cc1Cl)(=O)=O NSUGPRFLIOERPF-UHFFFAOYSA-N 0.000 description 1
- PNPFYHTUGHQXBX-UHFFFAOYSA-N NS(c1c(C2(CBr)OCCO2)cc[s]1)(=O)=O Chemical compound NS(c1c(C2(CBr)OCCO2)cc[s]1)(=O)=O PNPFYHTUGHQXBX-UHFFFAOYSA-N 0.000 description 1
- BEIXXLZNLPKKKG-UHFFFAOYSA-N O=C1CNS(=O)(=O)C2=C1C=C(Cl)S2.O=S1(=O)NCC2(OCCO2)C2=C1SC(Cl)=C2 Chemical compound O=C1CNS(=O)(=O)C2=C1C=C(Cl)S2.O=S1(=O)NCC2(OCCO2)C2=C1SC(Cl)=C2 BEIXXLZNLPKKKG-UHFFFAOYSA-N 0.000 description 1
- NXPYEBXDVYMXCW-UHFFFAOYSA-N O=C1CNS(=O)(=O)C2=C1C=CS2.O=S1(=O)NCC2(OCCO2)C2=C1SC=C2 Chemical compound O=C1CNS(=O)(=O)C2=C1C=CS2.O=S1(=O)NCC2(OCCO2)C2=C1SC=C2 NXPYEBXDVYMXCW-UHFFFAOYSA-N 0.000 description 1
- GABQIEPVQDUMHV-UHFFFAOYSA-N O=S(c([s]1)c2cc1Cl)(NCC21OCCO1)=O Chemical compound O=S(c([s]1)c2cc1Cl)(NCC21OCCO1)=O GABQIEPVQDUMHV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/34—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the present invention relates to a process for the preparation of Brinzolamide, or 2H-thieno[3,2-e]-1,2-thiazin-6-sulfonamide, 4-(ethyl amino)-3,4-dihydro-2-(3-methoxypropyl)-, 1,1-dioxide, (4R)- via intermediates 2,3-dihydro-4H-thieno[3,2-e]-1,2-thiazin-4-ones, 1,1-dioxide.
- Further objects of the present invention are the intermediates mentioned above and other intermediates of the synthesis.
- Brinzolamide is a carbonic anhydrase II inhibitor, used to lower intraocular pressure and glaucoma. It is sold by Alcon under the name of Azopt, as 1% ophthalmic suspension.
- EP 527801 claims Brinzolamide and describes a process to prepare it in 14 steps starting from 3-acetylthiophene (scheme 1). It is a synthesis typical of medicinal chemistry not applicable at industrial level, for which no specific preparations are described, because Brinzolamide is not among the preferred compounds of the invention.
- the second reduction (12) occurs in the presence of (+)- ⁇ -chlorodiisopinocamphenylborane, an expensive enantioselective reducing agent, with a stoichiometric excess of 5:1, which requires reaction conditions not easily achievable at industrial scale (3 days of reaction at ⁇ 22° C., difficult work up and chromatography) to isolate the product.
- EP 617038 describes a process for the preparation of Brinzolamide and its analogues starting from 3-acetyl-2,5-dichlorothiophene (scheme 2).
- the sulfonamide is introduced in position 6 through metallation with n-butyl lithium, an expensive raw material, and then with a reaction with sulphurous anhydride and hydroxylamino-O-sulphonic acid.
- the base should be used in substantial excess (2,3 eq.), because the oxydrilic group reacts with the first equivalent. In this case the protection of the oxydrilic group as described in Scheme 1 is not possible without running the risk of racemization of the substrate.
- the present invention describes a process for the preparation of compounds 2,3-dihydro-4H-thieno[3,2-e]-1,2-thiazin-4-ones, 1,1 dioxide and their conversion to Brinzolamide.
- Further objects of the present invention are the above mentioned intermediates, intermediates 3-[2-(halomethyl)-1,3-dioxolan-2-yl]thiophene-2-sulfonamide and 3-[2-(sulphonylmethyl)-1,3-dioxolan-2-yl]thiophene-2-sulfonamide and intermediates 2′,3′-dihydrospiro[1,3-dioxolan-2,4′-thieno[3,2-e][1,2]thiazine], 1′,1′-dioxide.
- R can be hydrogen, alkyl, alkoxy alkyl, haloalkyl, aryl or aryl alkyl, including the steps of
- X and R have the same meaning as above and Z can be a halogen or a sulphonic esther, with an alcohol or a diol to give a ketal of formula 3
- R 1 is an alkyl, linear or branched, or aryl alkyl, and the two groups R 1 can be separated or joined together to form a cycle
- the protection a) of ketone 2 with an appropriate alcohol or diol is preferably performed in a highly boiling solvent such as toluene or xylene, in the presence of an acidic catalyst, preferably chosen among p-toluensulphonic acid, sulphuric acid and boron trifluoride etherate, at a temperature suitable to remove by distillation the water generated during the process.
- a highly boiling solvent such as toluene or xylene
- an acidic catalyst preferably chosen among p-toluensulphonic acid, sulphuric acid and boron trifluoride etherate
- Cyclization b) is performed preferably in the presence of a base to activate the sulphonamidic group and to neutralize the acid generated during the reaction; this base is preferably chosen among potassium carbonate, sodium carbonate, triethylamine, pyridine, potassium hydroxide, sodium hydroxide, sodium hydride, more preferably is potassium carbonate.
- the reaction is performed in a solvent preferably chosen among dimethylsulphoxide, dimethylformamide, N-methylpyrrolidone, or their mixture with THF, toluene and other apolar solvents, more preferably in dimethylsulphoxide, preferably at a temperature between 20° C. and 80° C. In the preferred conditions the reaction takes place in less than one hour.
- the hydrolysis c) of the ketal to carbonyl is preferably performed with an acidic catalyst, preferably with hydrochloric acid, sulphuric acid, hydrobromic acid, more preferably with aqueous hydrochloric acid, in the presence of water and an organic solvent preferably chosen among toluene, acetone, ethanol, methanol, THF, more preferably toluene, preferably at a temperature between 0° C. and 80° C. In the preferred conditions the reaction takes place in 8 hours.
- an acidic catalyst preferably with hydrochloric acid, sulphuric acid, hydrobromic acid, more preferably with aqueous hydrochloric acid
- an organic solvent preferably chosen among toluene, acetone, ethanol, methanol, THF, more preferably toluene, preferably at a temperature between 0° C. and 80° C. In the preferred conditions the reaction takes place in 8 hours.
- the invention includes an optional step of transformation of the group X in a group X with a different meaning and/or the transformation of the group R in a group R with a different meaning after one of the steps a), b) or c), or on the open ketale 3, on the cyclised ketal 4 or on the ketone 1.
- X is hydrogen or chlorine
- this can be efficiently transformed in sulphonamide or one of its synthetic precursors according to what is described in Organic Process Research & Development 1999, 3, 114-120 or J. Org. Chem. 1997, 62, 9372-9375, or J. Org. Chem. 1993, 58, 1672-1679, or J. Org. Chem. 1991, 56, 763-769.
- the process object of this invention is advantageous because it allows to insert a sulphonamidic functional group using a stoichiometric quantity of butyl lithium, without having this reacting with the carbonylic group, protected under a ketal form.
- R When in the compounds of formula 3 or 4 R is hydrogen, this can be transformed in alkyl, alkoxy alkyl, haloalkyl, aryl or aryl alkyl after a reaction with an alkyl halide, a sulphate or a sulphonate, preferably after a reaction with 1,3-dichloropropane, 1,3-dibromopropane, 1,3-diiodopropane, 1-chloro-3-methoxypropane or 1-bromo-3-methoxypropane, more preferably with 1-chloro-3-methoxypropane or 1-bromo-3-methoxypropane.
- This procedure is advantageous because it allows to insert an alkyl, alkoxy alkyl, aryl or aryl alkyl group, preferably 3-chloropropyl, 3-bromopropyl, 3-iodopropyl o 3-methoxypropyl, more preferably 3-methoxypropyl, in the same conditions of the cyclization reaction of step b).
- R is 3-chloropropyl, 3-bromopropyl, 3-iodopropyl
- this can be transformed in 3-methoxypropyl after a reaction with sodium or potassium methylate, or with methanol in the presence of a strong base.
- a particularly preferred embodiment of the invention is a process for the preparation of 2-(3-methoxypropyl)-4-oxo-3,4-dihydro-2H-thieno[3,2-e][1,2]thiazine, 1,1-dioxide of formula 5
- X is hydrogen, chlorine, or sulphonamide, comprising the steps of
- X is hydrogen, halogen, thiol, thioether, or amide or halogenide of sulphonic acid
- R is hydrogen, alkyl, alkoxy alkyl, haloalkyl, aryl or aryl alkyl
- Z is a halogen or sulphonic esther
- R 1 is an alkyl, linear o branched, or aryl alkyl, and the two groups R 1 can be separated or joined together to form a cycle; of the compounds of formula 4
- the compound of formula 2 is suspended in toluene (10-20 volumes) and ethylene glycol (5-10 equivalents); p-toluensulphonic acid is added in catalytical quantity. The mixture is heated until reflux with azeotropic removal of water for 5-12 hours, until complete conversion (HPLC). At the end of the reaction the solution is cooled down at room temperature. Triethylamine and water are added (at least double compared with glycol) and the phases are separated. The organic phase is washed with water and concentrated under vacuum to give the desired product 7 as a light-colored solid with HPLC purity above 95% and with a yield of 80-95%.
- the desired compound is prepared according to general procedure 1 starting from 3-(bromoacetyl)thiophene-2-sulphonammide with a yield of 87%.
- the desired compound is prepared according to general procedure 1 starting from 3-(bromoacetyl)-5-chlorothiophene-2-sulphonamide with a yield of 93%.
- the product 8 is isolated through distillation of the solvent under vacuum, abtaining a solid with a HPLC purity of 80-95% and a yield of 90-97%.
- the desired compound is prepared according to general procedure 2 starting from 3-[2-(bromomethyl)-1,3-dioxolan-2-yl]thiophene-2-sulphonamide of example 1 with a yield of 90%.
- the desired compound is prepared according to general procedure 2 starting from 3-[2-(bromomethyl)-1,3-dioxolan-2-yl]-5-chlorothiophen-2-sulphonammide of example 2 with a yield of 97%.
- the compound of structure 3 is dissolved in DMSO (5-10 volumes); potassium carbonate is added (1.2-2.5 equivalents) and the mixture is heated for one hour at 50-60° C. When the conversion is complete, 1-chloro-3-methoxypropane (1.5-3 equivalents) is added to the suspension and the mixture is heated again at 60° C. for 2-8 hours.
- the mixture is recovered with water and toluene.
- the phases are separated and the organic phase is washed with water.
- the product 9 is isolated through distillation of the solvent under vacuum, obtaining a solid with a HPLC assay of 85-95% and a yield of 90-99%.
- the desired compound is prepared according to the general procedure 3 starting from 3-[2-(bromomethyl)-1,3-dioxolan-2-yl]thiophene-2-sulphonamide of example 1 with a yield of 90%.
- the desired compound is prepared according to general procedure 3 starting from 3-[2-(bromomethyl)-1,3-dioxolan-2-yl]-5-chlorothiofen-2-sulphonamide of example 2 with a yield of 99%.
- the aqueous phase is added to an aqueous solution of sodium acetate (6-8 equivalents) and hydroxylamino-O-sulphonic acid (2-4 equivalents) and stirred at room temperature for 2-8 hours.
- ethyl acetate is added and the phases are separated.
- the organic phase is washed with a solution of bicarbonate and water.
- the desired compound is prepared according to general procedure 4 starting from 2′-(3-methoxypropyl)-2′,3′-dihydrospiro[1,3-dioxolan-2,4′-thieno[3,2-e][1,2]thiazin], 1′,1′-dioxide of example 5 with a yield of 76%.
- the desired compound is prepared according to general procedure 4 starting from 6′-chloro-2′-(3-methoxypropyl)-2′,3′-dihydrospiro[1,3-dioxolan-2,4′-thieno[3,2-e][1,2]thiazin], 1′,1′-dioxide of example 6 with a yield of 89%.
- the compound of formula 5 is dissolved in toluene (10-20 volumes) and an aqueous solution of hydrochloric acid 2-12 N is added. The mixture is stirred at a temperature which can vary between 20° C. and 80° C. for a time between 2 and 16 ore, until complete hydrolysis. The phases are separated and the product 1 is isolated through distillation of the organic solvent under vacuum, obtaining a solid with a HPLC assay of 85-95% and a yield of 65-99%.
- the desired compound is prepared according to the general procedure 5 starting from 2′,3′-dihydrospiro[1,3-dioxolan-2,4′-thieno[3,2-e][1,2]thiazin], 1′,1′-dioxide of example 3 with a yield of 66%.
- the desired compound is prepared according to general procedure 5 starting from 6′-chloro-2′,3′-dihydrospiro[1,3-dioxolan-2,4′-thieno[3,2-e][1,2]thiazin], 1′,1′-dioxide of example 4 with a yield of 95%.
- the desired compound is prepared according to the general procedure 5 starting from 2′-(3-methoxypropyl)-2′,3′-dihydrospiro[1,3-dioxolan-2,4′-thieno[3,2-e][1,2]thiazin], 1′,1′-dioxide of example 5 with a yield of 97%.
- the desired compound is prepared according to the general procedure 5 starting from 6′-chloro-2′-(3-methoxypropyl)-2′,3′-dihydrospiro[1,3-dioxolan-2,4′-thieno[3,2-e][1,2]thiazin], 1′,1′-dioxide of example 6 with a yield of 99%.
- the desired compound is prepared according to the general procedure 5 starting from 2′-(3-methoxypropyl)-2′,3′-dihydrospiro[1,3-dioxolan-2,4′-thieno[3,2-e][1,2]thiazin]-6′-sulphonamide, 1′,1′-dioxide of examples 7 or 8 with a quantitative yield.
Abstract
Description






where X can be hydrogen, halogen, thiol, thioether, or amide or halogenide of sulphonic acid,
R can be hydrogen, alkyl, alkoxy alkyl, haloalkyl, aryl or aryl alkyl,
including the steps of
- a) reaction of a compound of formula 2

wherein X and R have the same meaning as above and Z can be a halogen or a sulphonic esther,
with an alcohol or a diol to give a ketal of formula 3

wherein X, R and Z have the same meaning as above and R1 is an alkyl, linear or branched, or aryl alkyl, and the two groups R1 can be separated or joined together to form a cycle,
- b) cyclization of the compound of formula 3 to form a compound of formula 4

wherein X, R and R1 have the same meaning as above,
- c) hydrolysis of a compound of formula 4.
X is preferably hydrogen, chlorine, bromine, sulfonamide;
R is preferably hydrogen, 3-chloropropyl, 3-bromopropyl, 3-iodopropile o 3-methoxypropyl, preferably 3-methoxypropyl;
X is preferably chlorine or bromine, more preferably bromine;
R1 is preferably methyl, ethyl, isopropyl, t-butyl, benzyl, or a chain formed by ethylene, propylene, 2,2-dimethylpropylene, more preferably ethylene.

wherein X is hydrogen, chlorine, or sulphonamide, comprising the steps of
- a) Reaction of the compound of formula 6

- wherein X is hydrogen or chlorine, with ethylene glycol in the presence of an acid, to obtain the compound 7

- wherein X is hydrogen or chlorine,
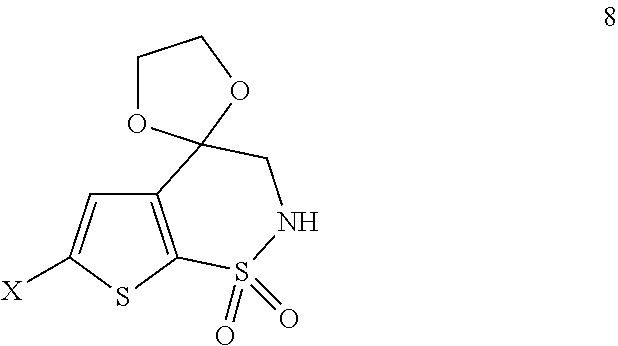
- b1) Cyclization of the compound of formula 7 in the presence of a base to obtain the compound of formula 8

- wherein X is hydrogen or chlorine;
- b2) Reaction of the compound of formula 8 with 1-chloro-3-methoxypropane or 1-bromo-3-methoxypropane to obtain the compound of formula 9

- wherein X is hydrogen or chlorine;
- b3) Optional conversion of the group X in a sulphonamide;
- c) Hydrolysis of the compound of formula 9, wherein X is sulphonamide, hydrogen or chlorine.
Step b3) of conversion of the group X in sulphonamide can be performed on the compounds of formula 8 where X is hydrogen or chlorine and is preferably performed with a reaction with butyl lithium in tetrahydrofuran at −40° C., followed by bubbling of sulphurous anhydride in the solution until an acid pH is achieved, removal of the solvent and addition of hydroxylamino-O-sulphonic acid in water in the presence of sodium acetate.
- i. Reduction of the carbonyl in position 4 to an alcohol;
- ii. Stereoselective reduction of the carbonyl in position 4 to an alcohol;
- iii. Transformation of the alcohol in position 4 obtained at i and ii in halogenide or sulphonic esther;
- iv. Substitution of the halogenide or sulphonic esther obtained at iii with an amine, preferably ethylamine;
- v. Reaction of the ketone with an amine, preferably ethylamine, to form the ketimine in position 4;
- vi. Reduction of the ketimine in position 4 obtained at v to amine;
- vii. Stereoselective reduction of the ketimine in position 4;
- viii. Transformation of the group X in a group X with a different meaning;
- ix. Transformation of the group R in a group R with a different meaning.
- x. Resolution of the racemate.

wherein X is hydrogen, halogen, thiol, thioether, or amide or halogenide of sulphonic acid,
R is hydrogen, alkyl, alkoxy alkyl, haloalkyl, aryl or aryl alkyl;
Z is a halogen or sulphonic esther;
R1 is an alkyl, linear o branched, or aryl alkyl, and the two groups R1 can be separated or joined together to form a cycle;
of the compounds of formula 4

wherein X, R and R1 have the above mentioned meaning;
and of the compounds of formula 1

wherein X and R have the above mentioned meaning, provided that in the compounds of formula 1 when X is hydrogen or chlorine, R is not hydrogen or 3-methoxypropyl and when X is sulphonamide, R is not 3-methoxypropyl. These compounds are useful intermediates in the present process for the preparation of Brinzolamide.
-
- 1) The protection a) of the carbonyl activates the reaction of intramolecular cyclization b);
- 2) The protection a) of the carbonyl allows to perform the transformation of groups X and R (for example changing X=Cl or H in X=SO2NH2) without involving the first functional group;
- 3) The process uses cheap raw materials, commercially available or easily synthesized,
- 4) All the reactions have high yields and can be easily transferred at industrial scale.













Claims (12)









Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IT001084A ITMI20081084A1 (en) | 2008-06-16 | 2008-06-16 | PROCESS FOR THE PREPARATION OF BRINZOLAMIDE |
| ITMI2008A001084 | 2008-06-16 | ||
| ITMI2008A1084 | 2008-06-16 | ||
| PCT/IB2009/052538 WO2010004457A2 (en) | 2008-06-16 | 2009-06-15 | Process for the preparation of brinzolamide |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20110118461A1 US20110118461A1 (en) | 2011-05-19 |
| US8344136B2 true US8344136B2 (en) | 2013-01-01 |
Family
ID=40301798
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/999,054 Active 2029-09-27 US8344136B2 (en) | 2008-06-16 | 2009-06-15 | Process for the preparation of brinzolamide |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8344136B2 (en) |
| EP (1) | EP2303856B1 (en) |
| CN (1) | CN102056914B (en) |
| IT (1) | ITMI20081084A1 (en) |
| WO (1) | WO2010004457A2 (en) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20130129211A (en) * | 2010-12-03 | 2013-11-27 | 다우 아그로사이언시즈 엘엘씨 | Processes for the preparation of enamines |
| CN103087081A (en) * | 2011-11-03 | 2013-05-08 | 山东威智医药工业有限公司 | Method for preparation of brinzolamide and intermediates thereof |
| CN102936236A (en) * | 2012-11-13 | 2013-02-20 | 江苏吉贝尔药业有限公司 | Method for preparing brinzolamide (3- acetyl-5-chloro-2-(benzylthiol) thiophene) |
| KR101564401B1 (en) | 2014-04-10 | 2015-11-02 | 한국화학연구원 | Process for the preparation of brinzolamide |
| CN113354665B (en) * | 2021-03-05 | 2022-04-22 | 株洲壹诺生物技术有限公司 | Method for synthesizing key intermediate of brinzolamide |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992016525A1 (en) | 1991-03-14 | 1992-10-01 | Korea Research Institute Of Chemical Technology | Thiophenesulfonylurea derivatives |
| US5240923A (en) | 1990-04-09 | 1993-08-31 | Alcon Laboratories, Inc. | Sulfonamides useful as carbonic anhydrase inhibitors |
| US5470973A (en) | 1994-10-03 | 1995-11-28 | Alcon Laboratories, Inc. | Synthesis of sulfonamide intermediates |
| US5585377A (en) | 1990-04-09 | 1996-12-17 | Alcon Laboratories, Inc. | Sulfonamides useful as carbonic anhydrase inhibitors |
-
2008
- 2008-06-16 IT IT001084A patent/ITMI20081084A1/en unknown
-
2009
- 2009-06-15 WO PCT/IB2009/052538 patent/WO2010004457A2/en active Application Filing
- 2009-06-15 CN CN200980121617.6A patent/CN102056914B/en not_active Expired - Fee Related
- 2009-06-15 EP EP09786425.0A patent/EP2303856B1/en not_active Not-in-force
- 2009-06-15 US US12/999,054 patent/US8344136B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5240923A (en) | 1990-04-09 | 1993-08-31 | Alcon Laboratories, Inc. | Sulfonamides useful as carbonic anhydrase inhibitors |
| US5585377A (en) | 1990-04-09 | 1996-12-17 | Alcon Laboratories, Inc. | Sulfonamides useful as carbonic anhydrase inhibitors |
| WO1992016525A1 (en) | 1991-03-14 | 1992-10-01 | Korea Research Institute Of Chemical Technology | Thiophenesulfonylurea derivatives |
| US5470973A (en) | 1994-10-03 | 1995-11-28 | Alcon Laboratories, Inc. | Synthesis of sulfonamide intermediates |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102056914B (en) | 2015-06-17 |
| EP2303856B1 (en) | 2016-12-28 |
| ITMI20081084A1 (en) | 2009-12-17 |
| EP2303856A2 (en) | 2011-04-06 |
| WO2010004457A3 (en) | 2010-07-29 |
| WO2010004457A2 (en) | 2010-01-14 |
| CN102056914A (en) | 2011-05-11 |
| US20110118461A1 (en) | 2011-05-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4903819B2 (en) | Hydroxyacetic acid ester derivatives, their production and use as synthetic intermediates | |
| US8344136B2 (en) | Process for the preparation of brinzolamide | |
| US8158808B2 (en) | Synthesis and preparations of duloxetine salts | |
| US8952173B2 (en) | Method for the resolution of 2-amino-6-propylamino-4,5,6,7-tetrahydrobenzothiazol and intermediate compounds | |
| US20100009977A1 (en) | Process for the preparation of (R)-(+)-4-(Ethylamino)-3,4-dihydro-2-(3-methoxypropyl)-2H-thieno[3,2-e]-1,2-thiazine-6-sulfonamide-1,1-dioxide. | |
| KR101564401B1 (en) | Process for the preparation of brinzolamide | |
| US20070238883A1 (en) | Process for the preparation of(s)-(+)-N,N-dimethyl-3-(1-Naphthalenyloxy)-3-(2-Thienyl)propanamine, A duloxetine intermediate | |
| US9221771B2 (en) | Method for the preparation of substituted oxazolidinones | |
| US20050209262A1 (en) | Process for producing optically active sulfoxide | |
| US6495691B1 (en) | Process for the preparation of tetrahydrothieno[3,2-c]pyridine derivatives | |
| SU922109A1 (en) | Process for producing r,s-2,3,5,6-tetrahydro-6-phenylimidazo(2,1,b)-thiazole | |
| EP0439404A2 (en) | Preparation of 2-(2'-thienyl) alkylamines and derivatives thereof and synthesis of 4,5,6,7- thieno [3,2-c] pyridine derivatives therefrom | |
| WO2009074883A2 (en) | Improved process for preparing duloxetine | |
| EP0342118A1 (en) | 2-(2-nitrovinyl)thiophene reduction and synthesis of thieno (3,2-c)pyridine derivatives | |
| JP5198877B2 (en) | Method for producing benzazepinones | |
| WO2010103550A2 (en) | Process for the preparation of intermediates | |
| US7799952B2 (en) | Stereoselective method for the production of (R)-Dimepranol | |
| EP0846111B1 (en) | Syntheses of optically pure compounds useful as garft inhibitors and their intermediates | |
| EP0497695A1 (en) | N-2-chlorobenzyl-2-oxo and N-2-chlorobenzyl-2,2-dioxo-1,2,3-oxathiazolidine derivatives, their preparation and synthesis of thieno[3,2-c]pyridine derivatives therefrom | |
| US20060074238A1 (en) | Asymmetric process for the preparation of diarylethylpiperazines derivatives and novel asymmetric diarylmethylamines as intermediates | |
| US20050222433A1 (en) | Process for preparing 3-hydroxythiolane | |
| JP2004224714A (en) | Method for producing isoxazolidine-3-thione derivative | |
| CA2080863A1 (en) | Use of substituted tetrahydrothiophenes, some of which are known, as medicaments, new active substances and processes for their preparation | |
| US20040002600A1 (en) | Process for the conversion of penam ring system to cepham ring system | |
| JPH08217779A (en) | Production of hexahydrothieno(3,4-d)imidazole-2,4-diones |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: PHF S.A., SWITZERLAND Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:FALCHI, ALESSANDRO;DE LUCCHI, OTTORINO;CASTELLIN, ANDREA;SIGNING DATES FROM 20101126 TO 20101213;REEL/FRAME:025612/0402 |
|
| STCF | Information on status: patent grant |
Free format text: PATENTED CASE |
|
| FEPP | Fee payment procedure |
Free format text: PAT HOLDER NO LONGER CLAIMS SMALL ENTITY STATUS, ENTITY STATUS SET TO UNDISCOUNTED (ORIGINAL EVENT CODE: STOL); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| AS | Assignment |
Owner name: F.I.S. - FABBRICA ITALIANA SINTETICI S.P.A., ITALY Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:PHF SA;REEL/FRAME:037959/0335 Effective date: 20160219 |
|
| FPAY | Fee payment |
Year of fee payment: 4 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 8TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1552); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 8 |