US8062766B2 - Electroluminescent polymer and organic electroluminescent device using the same - Google Patents
Electroluminescent polymer and organic electroluminescent device using the same Download PDFInfo
- Publication number
- US8062766B2 US8062766B2 US11/600,110 US60011006A US8062766B2 US 8062766 B2 US8062766 B2 US 8062766B2 US 60011006 A US60011006 A US 60011006A US 8062766 B2 US8062766 B2 US 8062766B2
- Authority
- US
- United States
- Prior art keywords
- group
- polymer
- alkyl group
- electroluminescent device
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active, expires
Links
- 0 *[Y]N1C2=CC=C(C)C=C2CC2=C1C=CC(C)=C2.C[Ar]C Chemical compound *[Y]N1C2=CC=C(C)C=C2CC2=C1C=CC(C)=C2.C[Ar]C 0.000 description 15
- CYAGPQZRZOKJBB-UHFFFAOYSA-N CC1=C2N=CC=NC2=C(C2=C3N=CC=NC3=C(C)C=C2)C=C1.CC1=CC=C(C2=CC=C(C3=CC=C(C)S3)S2)S1 Chemical compound CC1=C2N=CC=NC2=C(C2=C3N=CC=NC3=C(C)C=C2)C=C1.CC1=CC=C(C2=CC=C(C3=CC=C(C)S3)S2)S1 CYAGPQZRZOKJBB-UHFFFAOYSA-N 0.000 description 2
- CQGOMURMXYKZMC-UHFFFAOYSA-N CCCCCCCCOC1=C(OCCCCCCCC)C=C2C(=C1)C1=CC(OCCCCCCCC)=C(OCCCCCCCC)C=C1C21C2=CC(C)=CC=C2C2=CC=C(C3=CC=C4C(=C3)OC3=CC(C)=CC=C3N4C3=CC=C(OCC(CC)CCCC)C=C3)C=C21 Chemical compound CCCCCCCCOC1=C(OCCCCCCCC)C=C2C(=C1)C1=CC(OCCCCCCCC)=C(OCCCCCCCC)C=C1C21C2=CC(C)=CC=C2C2=CC=C(C3=CC=C4C(=C3)OC3=CC(C)=CC=C3N4C3=CC=C(OCC(CC)CCCC)C=C3)C=C21 CQGOMURMXYKZMC-UHFFFAOYSA-N 0.000 description 2
- LJNDOHPPVXDTGX-UHFFFAOYSA-N CCCCCCCCOC1=CC2=C(C=C1OCCCCCCCC)C1(C3=CC(C4=CC5=C(C=C4)N(C4=CC=C6C=C(OC)C=CC6=C4)C4=CC=C(C)C=C4CC5)=CC=C3C3=C1C=C(C)C=C3)C1=C2C=C(C)C(C)=C1 Chemical compound CCCCCCCCOC1=CC2=C(C=C1OCCCCCCCC)C1(C3=CC(C4=CC5=C(C=C4)N(C4=CC=C6C=C(OC)C=CC6=C4)C4=CC=C(C)C=C4CC5)=CC=C3C3=C1C=C(C)C=C3)C1=C2C=C(C)C(C)=C1 LJNDOHPPVXDTGX-UHFFFAOYSA-N 0.000 description 2
- SSRQGDWXNLUVRK-UHFFFAOYSA-C C1=CC2=C3C(=C1)O[Al]14(OC5=CC=CC6=C5N1=CC=C6)(OC1=CC=CC5=C1N4=CC=C5)N3=CC=C2.CC1=CC=N2C3=C(C=CC=C13)O[Al]213(OC2=C4C(=CC=C2)C(C)=CC=N41)OC1=C2C(=CC=C1)/C(C)=C\C=N/23.CC1=N2C3=C(C=CC=C3O[AlH]23(OC2=CC=C(C4=CC=CC=C4)C=C2)OC2=CC=CC4=C2N3=C(C)C=C4)C=C1.CC1=N2C3=C(C=CC=C3O[AlH]23(O[SiH2]C2=CC=CC=C2)OC2=CC=CC4=C2N3=C(C)C=C4)C=C1.O=C1C2=C3=C(/C=C\C4=C3=C(C=C2)C2=CC=C3C5=NC6=C(C=CC=C6)N5C(=O)/C5=C/C=C/4C2=C35)C2=NC3=C(C=CC=C3)N12 Chemical compound C1=CC2=C3C(=C1)O[Al]14(OC5=CC=CC6=C5N1=CC=C6)(OC1=CC=CC5=C1N4=CC=C5)N3=CC=C2.CC1=CC=N2C3=C(C=CC=C13)O[Al]213(OC2=C4C(=CC=C2)C(C)=CC=N41)OC1=C2C(=CC=C1)/C(C)=C\C=N/23.CC1=N2C3=C(C=CC=C3O[AlH]23(OC2=CC=C(C4=CC=CC=C4)C=C2)OC2=CC=CC4=C2N3=C(C)C=C4)C=C1.CC1=N2C3=C(C=CC=C3O[AlH]23(O[SiH2]C2=CC=CC=C2)OC2=CC=CC4=C2N3=C(C)C=C4)C=C1.O=C1C2=C3=C(/C=C\C4=C3=C(C=C2)C2=CC=C3C5=NC6=C(C=CC=C6)N5C(=O)/C5=C/C=C/4C2=C35)C2=NC3=C(C=CC=C3)N12 SSRQGDWXNLUVRK-UHFFFAOYSA-C 0.000 description 1
- DNZFTRWLBFTGIY-UHFFFAOYSA-K C1=CC=C(N2C(C3=CC(/C4=N/C5=C(C=CC=C5)N4C4=CC=CC=C4)=CC(/C4=N/C5=C(C=CC=C5)N4C4=CC=CC=C4)=C3)=NC3=C2C=CC=C3)C=C1.CC(C)(C)C1=CC=C(C2=NN=C(C3=CC=C(C4=CC=CC=C4)C=C3)N2C2=CC=CC=C2)C=C1.CC(C)(C)C1=CC=C(C2=NN=C(C3=CC=C(C4=CC=CC=C4)C=C3)O2)C=C1.CC1=N2C3=C(C=CC=C3O[AlH]23(OC2=CC=C(C4=CC=CC=C4)C=C2)OC2=CC=CC4=C2N3=C(C)C=C4)C=C1.CC1=NC2=C(C=CC3=C2N=C(C)C=C3C2=CC=CC=C2)C(C2=CC=CC=C2)=C1 Chemical compound C1=CC=C(N2C(C3=CC(/C4=N/C5=C(C=CC=C5)N4C4=CC=CC=C4)=CC(/C4=N/C5=C(C=CC=C5)N4C4=CC=CC=C4)=C3)=NC3=C2C=CC=C3)C=C1.CC(C)(C)C1=CC=C(C2=NN=C(C3=CC=C(C4=CC=CC=C4)C=C3)N2C2=CC=CC=C2)C=C1.CC(C)(C)C1=CC=C(C2=NN=C(C3=CC=C(C4=CC=CC=C4)C=C3)O2)C=C1.CC1=N2C3=C(C=CC=C3O[AlH]23(OC2=CC=C(C4=CC=CC=C4)C=C2)OC2=CC=CC4=C2N3=C(C)C=C4)C=C1.CC1=NC2=C(C=CC3=C2N=C(C)C=C3C2=CC=CC=C2)C(C2=CC=CC=C2)=C1 DNZFTRWLBFTGIY-UHFFFAOYSA-K 0.000 description 1
- RXPLYMLPNOSCEG-UHFFFAOYSA-N C1=CC=C2OC3=C(C=CC=C3)NC2=C1.CCCCC(CC)COC1=CC=C(Br)C=C1.CCCCC(CC)COC1=CC=C(N2C3=CC=C(Br)C=C3OC3=C2C=CC(Br)=C3)C=C1.CCCCC(CC)COC1=CC=C(N2C3=CC=CC=C3OC3=C2C=CC=C3)C=C1 Chemical compound C1=CC=C2OC3=C(C=CC=C3)NC2=C1.CCCCC(CC)COC1=CC=C(Br)C=C1.CCCCC(CC)COC1=CC=C(N2C3=CC=C(Br)C=C3OC3=C2C=CC(Br)=C3)C=C1.CCCCC(CC)COC1=CC=C(N2C3=CC=CC=C3OC3=C2C=CC=C3)C=C1 RXPLYMLPNOSCEG-UHFFFAOYSA-N 0.000 description 1
- JIIBPSADVANISE-UHFFFAOYSA-D CC1=CC=C2C=CC=C3O[Ga]4(OC(=O)C(C)(C)C)(OC5=C6C(=CC=C5)C=CC(C)=N64)N1=C23.CC1=CC=C2C=CC=C3O[Ga]4(O[Ga]56(OC7=CC=CC8=CC=C(C)N5=C87)OC5=C7C(=CC=C5)C=CC(C)=N76)(OC5=C6C(=CC=C5)C=CC(C)=N64)N1=C23.[H]C([H])([H])C(=O)O[Ga]12(OC3=CC=CC4=CC=C(C)N1=C43)OC1=C3C(=CC=C1)C=CC(C)=N32 Chemical compound CC1=CC=C2C=CC=C3O[Ga]4(OC(=O)C(C)(C)C)(OC5=C6C(=CC=C5)C=CC(C)=N64)N1=C23.CC1=CC=C2C=CC=C3O[Ga]4(O[Ga]56(OC7=CC=CC8=CC=C(C)N5=C87)OC5=C7C(=CC=C5)C=CC(C)=N76)(OC5=C6C(=CC=C5)C=CC(C)=N64)N1=C23.[H]C([H])([H])C(=O)O[Ga]12(OC3=CC=CC4=CC=C(C)N1=C43)OC1=C3C(=CC=C1)C=CC(C)=N32 JIIBPSADVANISE-UHFFFAOYSA-D 0.000 description 1
- MDAKMRBLIZGPRD-UHFFFAOYSA-N CCCCCCCCOC1=CC2=C(C=C1OCCCCCCCC)C1(C3=CC(C4=CC5=C(C=C4)N(C4=CC=C6C=C(OC)C=CC6=C4)C4=CC=C(C)C=C4CC5)=CC=C3C3=C1C=C(C1=CC=C4C(=C1)OC1=C(C=CC(C)=C1)N4C1=CC=C4C=C(OC)C=CC4=C1)C=C3)C1=C2C=C(C)C(C)=C1 Chemical compound CCCCCCCCOC1=CC2=C(C=C1OCCCCCCCC)C1(C3=CC(C4=CC5=C(C=C4)N(C4=CC=C6C=C(OC)C=CC6=C4)C4=CC=C(C)C=C4CC5)=CC=C3C3=C1C=C(C1=CC=C4C(=C1)OC1=C(C=CC(C)=C1)N4C1=CC=C4C=C(OC)C=CC4=C1)C=C3)C1=C2C=C(C)C(C)=C1 MDAKMRBLIZGPRD-UHFFFAOYSA-N 0.000 description 1
- RWUGAABXAQSPML-UHFFFAOYSA-N CCCCCCCCOC1=CC2=C(C=C1OCCCCCCCC)C1(C3=CC(C4=CC5=C(C=C4)N(C4=CC=C6C=C(OC)C=CC6=C4)C4=CC=C(C)C=C4O5)=CC=C3C3=C1C=C(C)C=C3)C1=C2C=C(C)C(C)=C1 Chemical compound CCCCCCCCOC1=CC2=C(C=C1OCCCCCCCC)C1(C3=CC(C4=CC5=C(C=C4)N(C4=CC=C6C=C(OC)C=CC6=C4)C4=CC=C(C)C=C4O5)=CC=C3C3=C1C=C(C)C=C3)C1=C2C=C(C)C(C)=C1 RWUGAABXAQSPML-UHFFFAOYSA-N 0.000 description 1
Images






Classifications
-
- A—HUMAN NECESSITIES
- A47—FURNITURE; DOMESTIC ARTICLES OR APPLIANCES; COFFEE MILLS; SPICE MILLS; SUCTION CLEANERS IN GENERAL
- A47G—HOUSEHOLD OR TABLE EQUIPMENT
- A47G9/00—Bed-covers; Counterpanes; Travelling rugs; Sleeping rugs; Sleeping bags; Pillows
- A47G9/10—Pillows
- A47G9/1009—Rigid frame constructions
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/06—Luminescent, e.g. electroluminescent, chemiluminescent materials containing organic luminescent materials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61H—PHYSICAL THERAPY APPARATUS, e.g. DEVICES FOR LOCATING OR STIMULATING REFLEX POINTS IN THE BODY; ARTIFICIAL RESPIRATION; MASSAGE; BATHING DEVICES FOR SPECIAL THERAPEUTIC OR HYGIENIC PURPOSES OR SPECIFIC PARTS OF THE BODY
- A61H39/00—Devices for locating or stimulating specific reflex points of the body for physical therapy, e.g. acupuncture
- A61H39/04—Devices for pressing such points, e.g. Shiatsu or Acupressure
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/02—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/02—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes
- C08G61/10—Macromolecular compounds containing only carbon atoms in the main chain of the macromolecule, e.g. polyxylylenes only aromatic carbon atoms, e.g. polyphenylenes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/12—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/12—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule
- C08G61/122—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G61/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G61/12—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule
- C08G61/122—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides
- C08G61/123—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds
- C08G61/126—Macromolecular compounds containing atoms other than carbon in the main chain of the macromolecule derived from five- or six-membered heterocyclic compounds, other than imides derived from five-membered heterocyclic compounds with a five-membered ring containing one sulfur atom in the ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L65/00—Compositions of macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain; Compositions of derivatives of such polymers
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05B—ELECTRIC HEATING; ELECTRIC LIGHT SOURCES NOT OTHERWISE PROVIDED FOR; CIRCUIT ARRANGEMENTS FOR ELECTRIC LIGHT SOURCES, IN GENERAL
- H05B33/00—Electroluminescent light sources
- H05B33/12—Light sources with substantially two-dimensional radiating surfaces
- H05B33/14—Light sources with substantially two-dimensional radiating surfaces characterised by the chemical or physical composition or the arrangement of the electroluminescent material, or by the simultaneous addition of the electroluminescent material in or onto the light source
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/10—Organic polymers or oligomers
- H10K85/111—Organic polymers or oligomers comprising aromatic, heteroaromatic, or aryl chains, e.g. polyaniline, polyphenylene or polyphenylene vinylene
- H10K85/115—Polyfluorene; Derivatives thereof
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/10—Organic polymers or oligomers
- H10K85/151—Copolymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/02—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/10—Non-macromolecular compounds
- C09K2211/1018—Heterocyclic compounds
- C09K2211/1025—Heterocyclic compounds characterised by ligands
- C09K2211/1029—Heterocyclic compounds characterised by ligands containing one nitrogen atom as the heteroatom
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/14—Macromolecular compounds
- C09K2211/1408—Carbocyclic compounds
- C09K2211/1416—Condensed systems
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/14—Macromolecular compounds
- C09K2211/1408—Carbocyclic compounds
- C09K2211/1425—Non-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/14—Macromolecular compounds
- C09K2211/1408—Carbocyclic compounds
- C09K2211/1433—Carbocyclic compounds bridged by heteroatoms, e.g. N, P, Si or B
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/14—Macromolecular compounds
- C09K2211/1441—Heterocyclic
- C09K2211/1458—Heterocyclic containing sulfur as the only heteroatom
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/14—Macromolecular compounds
- C09K2211/1441—Heterocyclic
- C09K2211/1466—Heterocyclic containing nitrogen as the only heteroatom
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/14—Macromolecular compounds
- C09K2211/1441—Heterocyclic
- C09K2211/1475—Heterocyclic containing nitrogen and oxygen as heteroatoms
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/14—Macromolecular compounds
- C09K2211/1441—Heterocyclic
- C09K2211/1483—Heterocyclic containing nitrogen and sulfur as heteroatoms
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/18—Metal complexes
- C09K2211/186—Metal complexes of the light metals other than alkali metals and alkaline earth metals, i.e. Be, Al or Mg
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K2211/00—Chemical nature of organic luminescent or tenebrescent compounds
- C09K2211/18—Metal complexes
- C09K2211/188—Metal complexes of other metals not provided for in one of the previous groups
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/11—OLEDs or polymer light-emitting diodes [PLED] characterised by the electroluminescent [EL] layers
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/14—Carrier transporting layers
Definitions
- the present invention relates to an electroluminescent polymer and an organic electroluminescent device using the same, and more particularly, to an electroluminescent polymer including a polycyclicamine unit in its polyarylene backbone and an organic electroluminescent device using the same that offers high luminance and efficiency.
- Organic electroluminescent devices can be classified according to the molecular weight of their materials and manufacturing processes.
- Organic electroluminescent devices can be manufactured from low molecular weight compounds and large molecular weight compounds.
- Low molecular weight compounds can be layered by vacuum deposition and can be easily purified to a high degree.
- color pixels can be easily obtained in a low molecular weight device.
- advantages of low molecular weight organic electroluminescent devices they still require further improvements for practical application, for example, in quantum efficiency and color purity, and there is a need to prevent crystallization of thin layers.
- Various studies on such electroluminescent displays using low molecular weight compounds have been actively undertaken, especially in Japan and the U.S.A.
- ⁇ -conjugated polymers have an alternating structure of single bonds ( ⁇ -bonds) and double bonds ( ⁇ -bonds), where ⁇ -electrons are evenly distributed to move freely in the polymer chain. Accordingly, ⁇ -conjugated polymers have semiconductor properties and can emit light of a visible range corresponding to the HOMO-LUMO energy bandgap, via proper molecular designing, when applied to an emissive layer of an electroluminescent device.
- Such a polymer can be easily formed as a thin layer in the manufacture of electroluminescent devices by spin coating or printing, at low costs and has a high glass transition temperature that allows the thin layer to have good mechanical properties.
- Such polymer-based electroluminescent devices are expected to be more commercially competitive than low molecular weight electroluminescent devices in the near future.
- the present invention provides an electroluminescent polymer with improved emission properties and stability that includes a polycyclicamine unit offering high charge mobility and that can emit blue light in its polyarylene backbone.
- the present invention also provides an organic electroluminescent device with an organic layer formed from the electroluminescent polymer.
- an electroluminescent polymer including: 1 through 99 mole % of a repeating unit represented by Formula (1) below; and 99 through 1 mole % of a repeating unit represented by Formula (2) below:
- R 1 and R 2 are each independently a hydrogen atom, a C 1-12 linear alkyl group, a C 1-12 branched alkyl group or a C 1-12 alkoxy group; Y is phenylene, naphthalene or anthracene, but Y is not phenylene when X is O, S or CH 2 ; and R is a hydrogen atom, a C 1-12 linear alkyl group, a C 1-12 branched alkyl group, a C 1-12 alkoxy group or a C 5-30 cycloalkyl group, or a C 6-14 aromatic group which is unsubstituted or substituted with at least one of a C 1-12 alkyl group, a C 1-12 alkoxy group, and —N(R′)(R′′) where R′ and R′′ are independently a hydrogen atom or a C 1-12 alkyl group, the polymer having a degree of polymerization in the range of 10 to 2,000.
- an organic electroluminescent device including an organic layer between a pair of electrodes, the organic layer containing the above-mentioned electroluminescent polymer.
- an organic electroluminescent device including an organic layer between a first electrode and a second electrode, the organic layer including an emissive layer and a hole transport layer between the emissive layer and the second electrode, at least one of the emissive layer and the hole transport layer containing the above-mentioned electroluminescent polymer.
- FIGS. 1A through 1F are cross-sectional views of structures of organic electroluminescent devices according to embodiments of the present invention.
- FIG. 2A is a reaction scheme illustrating the synthesis of poly(2′,3′,6′,7′ tetraoctyloxyspirofluorene-co-iminodibenzyl) of formula (4) according to an embodiment of the present invention
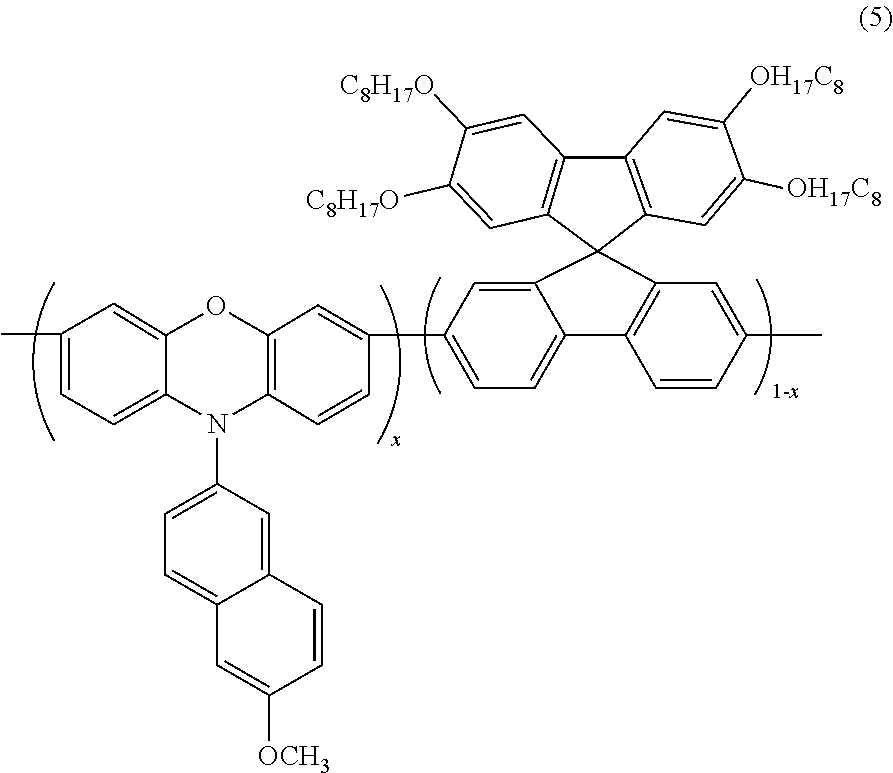
- FIG. 2B is a reaction scheme illustrating the synthesis of poly(2′,3′,6′,7′ tetraoctyloxyspirofluorene-co-phenoxazine) of formula (5) according to an embodiment of the present invention.
- FIG. 2C is a reaction scheme illustrating the synthesis of poly(2′,3′,6′,7′ tetraoctyloxyspirofluorene-co-iminodibenzyl-phenoxazine) of formula (6) according to an embodiment of the present invention
- An embodiment of the present invention provides an electroluminescent polymer, having a degree of polymerization in the range of 10 to 2,000, including: 1 through 99 mole % of a repeating unit represented by Formula (1) below; and 99 through 1 mole % of a repeating unit represented by Formula (2) below:
- Ar is a C 6-26 aromatic group or a C 4-20 heteroaromatic group including at least one heteroatom in the aromatic ring, where the aromatic group or the heteroaromatic group is unsubstituted or substituted with at least one of a C 1-12 alkyl group, a C 1-12 alkoxy group, and —N(R′)(R′′) where R′ and R′′ are each independently a hydrogen atom or a C 1-12 alkyl group;
- R 1 and R 2 are hydrogen atoms, C 1-12 linear alkyl groups, C 1-12 branched alkyl groups or C 1-12 alkoxy groups
- Y is phenylene, naphthalene or anthracene, but Y is not phenylene when X is O, S or CH 2
- R is a hydrogen atom, a C 1-12 linear alkyl group, a C 1-12 branched alkyl group, a C 1-12 alkoxy group or a C 5-30 cycloalkyl group, or a C 6-14 aromatic group which is unsubstituted or substituted with at least one of a C 1-12 alkyl group, a C 1-12 alkoxy group, and —N(R′)(R′′) where R′ and R′′ are independently a hydrogen atom or a C 1-12 alkyl group.
- the electroluminescent polymer may further include 1 through 99 mole % of a repeating unit represented by Formula (3) below:
- polycyclicamine monomers which offer high charge mobility and are able to emit blue light are incorporated into the polyarylene backbone by copolymerization with arylene monomers to improve the blue electroluminescent property of a final polymer product.
- the Ar unit may have a structure selected from the group consisting of Formulas (1a) through (1aa) below, and preferably Formula 1o, 1q or 1r:
- R 3 , R 4 , R 5 , and R 6 are each independently hydrogen, a C 1-12 alkyl group, a C 1-12 alkoxy group, a C 6-20 alkyl group or —N(R′)(R′′) where R′ and R′′ are each independently a hydrogen atom or a C 1-12 alkyl group.
- a fluorene structure exhibits more fluorescence than other aromatic structures and greater chemical flexibility due to soluble moiety at the 9-9′ position that is highly likely to accept various substituents, including an alkyl group. This is the reason why the fluorene structure is preferable for the arylene unit.
- FIGS. 2A through 2C illustrates reaction schemes illustrating the synthesis of electroluminescent polymers from arylene (Ar) units, 2′,3′,6′,7′-tetraoctyloxyspirofluorene according to embodiments of the present invention.
- an electroluminescent polymer according to an embodiment of the present invention has a weight average molecular weight of 10,000-2,000,000.
- the weight average molecular weight of the electroluminescent polymer affects thin film formation and the lifetime of an electroluminescent device manufactured from the polymer. If the electroluminescent polymer has a weight average molecular weight less than 10,000, crystallization is likely to occur in the manufacture and the driving of a device. On the other hand, it is impractical to obtain an electroluminescent polymer having a weight average molecular weight greater than 2,000,000 via a Pd(0) or Ni(0)-mediated aryl coupling reaction.
- a narrower molecular weight distribution (MWD) of an electroluminescent polymer is known to be advantageous in various aspects, especially for the longer lifetime of a device.
- the electroluminescent polymer according to an embodiment of the present invention has a MWD of 1.5 to 5.
- an electroluminescent polymer according to an embodiment of the present invention is represented by Formulas (4) through (6) below:
- x is a real number ranging from 0.01 to 0.99, preferably, from 0.01 to 0.20.
- x is a real number ranging from 0.01 to 0.99, preferably, from 0.01 to 0.20.
- x is a real number ranging from 0.01 to 0.99, preferably, from 0.01 to 0.20.
- Examples of an unsubstituted alkyl group used as a substituent in the current embodiment of the present invention may include methyl, ethyl, propyl, isobutyl, sec-butyl, tert-butyl, pentyl, iso-amyl, hexyl, etc.
- At least one hydrogen atom from the alkyl group may be substituted with a halogen atom, a hydroxyl group, a nitro group, a cyano group, a substituted or unsubstituted amino group (—N(R′)(R′′) where R′ and R′′ are each independently a hydrogen atom or a C 1-12 alkyl group), an amidino group, a hydrazine group, a hydrazon group, a carboxyl group, a sulfonic acid group, a phosphoric acid group, a C 1-20 alkyl group, a C 1-20 alkyl halide group, a C 1-20 alkenyl group, a C 1-20 alkynyl group, a C 1-20 heteroalkyl group, a C 5-30 aryl group, a C 5-30 arylalkyl group, a C 5-30 heteroaryl group, or a C 5-30 heteroarylalkyl group.
- An aryl group used as a substituent in the compound of the current embodiment of the present invention, has a carbocycle aromatic structure containing at least one aromatic ring, wherein the aromatic rings may adhere together using a pendant or be fused.
- the aryl group for example, may be an aromatic group, such as phenyl, naphthyl, tetrahydronaphthyl, etc.
- at least one hydrogen atom of the aryl group may be substituted with a substituent.
- a heteroaryl group used as a substituent in the compound of the current embodiment of the present invention, includes 1, 2, or 3 hetero atoms selected from the group consisting of N, O, P, and S. Also, the heteroaryl group has an aromatic ring structure having 5-30 ring atoms, wherein the rest of the ring atoms are carbon atoms. The ring atoms may adhere together using a pendant or be fused. As in the case of the C 1-30 alkyl group, at least one hydrogen atom of the heteroaryl group may be substituted with a substituent.
- An alkoxy group, used as a substituent in the compound of the current embodiment of the present invention is a radical-O-alkyl group, wherein the alkyl group is the same as described above.
- Examples of the alkoxy group include methoxy, ethoxy, propoxy, isobutyloxy, sec-butyloxy, pentyloxy, iso-amyloxy, hexyloxy, etc., and as in the case of the alkyl group, at least one hydrogen atom of the alkoxy group may be substituted with a substituent.
- An arylalkyl group used as a substituent in the compound of the current embodiment of the present invention, is formed by substituting one or more of a plurality of hydrogen atoms of the above described aryl group with lower alkyl, such as methyl, ethyl, propyl, etc.
- alkyl such as methyl, ethyl, propyl, etc.
- the arylalkyl group include benzyl, phenylethyl, etc.
- at least one hydrogen atom of the arylalkyl group may be substituted with a substituent.
- a heteroarylalkyl group used in the compound of the current embodiment of the present invention, is formed by substituting one or more a plurality of hydrogen atoms of the heteroaryl group with lower alkyl, wherein the heteroaryl group of the heteroarylalkyl group is the same as described above. As in the case of the alkyl group, at least one hydrogen atom of the heteroarylalkyl group may be substituted with a substituent.
- An aryloxy group used in the compound of the current embodiment of the present invention is a radical-O-aryl, wherein the aryl is the same as described above.
- Examples of the aryloxy group include phenoxy, naphthoxy, anthracenyloxy, phenanthrenyloxy, fluorenyloxy, indenyloxy, etc.
- at least one hydrogen atom of the aryloxy group may be substituted with a substituent.
- a heteroaryloxy group used in the compound of the current embodiment of the present invention is a radical-O-heteroaryl, wherein the heteroaryl is the same as described above.
- heteroaryloxy group examples include a benzyloxy group, a phenylethyloxy group, etc. and as in the case of the C 1-30 alkyl group, at least one hydrogen atom of the heteroaryloxy group may be substituted with a substituent.
- a cycloalkyl group used in the compound of the current embodiment of the present invention has a monovalent monocyclic structure having 5-30 carbon atoms. As in the case of the C 1-30 alkyl group, at least one hydrogen atom of the cycloalkyl group may be substituted with a substituent.
- a heterocycloalkyl group used in the compound of the current embodiment of the present invention, includes 1, 2, or 3 hetero atoms selected from the group consisting of N, O, P, and S. Also, the heterocycloalkyl group has a monovalent monocyclic structure having 5-30 ring atoms, wherein the rest of the ring atoms are carbon. As in the case of the alkyl group, at least one hydrogen atom of the cycloalkyl group may be substituted with a substituent.
- An amino group used in the compound of the current embodiment of the present invention is —N(R′)(R′′), where R′ and R′′ are independently a hydrogen atom or a C 1-12 alkyl group.
- FIGS. 1A through 1F are schematic views of structures of organic electroluminescent devices according to embodiments of the present invention.
- an emissive layer 12 containing the electroluminescent polymer is formed on a first electrode 10 and a second electrode 14 is formed on the emissive layer 12 .
- an emissive layer 12 containing the electroluminescent polymer is formed on a first electrode 10 , a hole blocking layer 13 is formed on the emissive layer 12 , and a second electrode 14 is formed on the hole blocking layer 13 .
- an organic electroluminescent device is manufactured in the same manner as in FIG. 1B , except that a hole injection layer 11 (or also called a buffer layer) is formed between the first electrode 10 and the emissive layer 12 .
- a hole injection layer 11 or also called a buffer layer
- an organic electroluminescent device has the same structure as in FIG. 1C , except that an electron transport layer 15 is formed on the emissive layer 12 instead of the hole blocking layer 13 .
- an organic electroluminescent device has the same structure as in FIG. 1C , except that an electron transport layer 15 is formed on the hole-blocking layer 13 .
- an organic electroluminescent device has the same structure as in FIG. 1E , except that a hole transport layer 16 is additionally formed between the hole injection layer 11 and the emissive layer 12 .
- the hole transport layer 16 prevents impurities from penetrating from the hole injection layer 11 into the emissive layer 12 .
- the organic electroluminescent devices having the structures of FIGS. 1A through 1F according to embodiments of the present invention can be manufactured using conventional methods, and is not limited thereto.
- a patterned first electrode 10 is formed on a substrate (not shown).
- the substrate may be a substrate used in a conventional organic electroluminescent device, preferably a glass substrate or a transparent plastic substrate having transparent and waterproof properties, a smooth surface, and which is easy to handle.
- the thickness of the substrate may be in the range of 0.3 to 1.1 mm.
- the anode is formed of a conductive metal or an oxide thereof for easy hole injection.
- a conductive metal or an oxide thereof for easy hole injection.
- indium tin oxide, indium zinc oxide, nickel, platinum, aurum, iridium, etc. may be used.
- the substrate, on which the first electrode 10 is formed, is washed and treated with UV/ozone.
- an organic solvent such as isopropanol, acetone, etc. is used for the washing.
- the hole injection layer 11 is selectively formed on the first electrode 10 .
- the hole injection layer 11 may be formed of a conventionally-used material, such as poly(3,4-ethylenedioxythiophene)/polystyrene parasulfonate, starburst based material, copper phthalocyanine, polythiophene, polyaniline, polyacetylene, polypyrrole, polyphenylene vinylene, or derivatives thereof.
- the upper portion of the first electrode 10 is spin coated using such a material and dried to form the hole injection layer 11 .
- the thickness of the hole injection layer 11 is in the range of 300 to 2000 ⁇ and preferably in the range of 500 to 1100 ⁇ .
- the drying temperature of the first electrode 10 when forming the hole injection layer 11 may be in the range of 100 to 250° C.
- An emissive layer 12 is formed by coating the upper portion of the hole injection layer 11 with a composition for forming the emissive layer using a spin coating method, etc. and drying the coated hole injection layer 11 .
- the composition for forming an emissive layer may be formed of a polymer in 0.5-20% of weight and a solvent in 99.5-80% of weight, both containing the repeating units of Formulas (1) and (2).
- Any solvent that can dissolve an emissive polymer may be used, and examples of this solvent include toluene, chlorobenzene, xylene, etc.
- the composition for forming an emissive layer may further include a dopant.
- the amount of the dopant may differ based on a material used to form an emissive layer, but it may be in the range of 30 to 80 parts by weight based on 100 parts by weight of the material (weight of the host and the dopant.) If the amount of the dopant is outside the above range, emissive properties of the electroluminescent device deteriorate.
- the dopant may be formed of arylamine, a peryl based compound, a pyrrole based compound, a hydrazone based compound, a carbazole based compound, a stilbene based compound, a starburst based compound, an oxadiazole based compound, etc.
- the thickness of the emissive layer 12 may be controlled by regulating the density of the compound for forming an emissive layer and the rotating speed while spin coating.
- the thickness of the emissive layer 12 may be in the range of 100 to 1000 ⁇ , and preferably in the range of 500 to 1000 ⁇ . When the thickness of the emissive layer 12 is less than 100 ⁇ , emissive efficiency deteriorates. When the thickness of the emissive layer 12 is greater than 1000 ⁇ , driving voltage increases.
- a hole transport layer 16 may be selectively formed between the hole injection layer 11 and the emissive layer 12 . Any material having hole transport properties may be used to form the hole transport layer 16 . An example of such a material is polytriphenylamine, etc.
- the thickness of the hole transport layer 16 may be in the range of 100 to 1,000 ⁇ .
- a hole blocking layer 13 and/or an electron transport layer 15 are formed on the upper portion of the emissive layer 12 using deposition or spin coating.
- the hole blocking layer 13 prevents excitons formed from the emissive material from moving to the electron transport layer 15 or holes from moving to the electron transport layer 15 .
- the hole blocking layer 13 may be formed of LiF, BaF 2 or MgF 2 , a phenanthroline based compound (e.g.: BCP available from UDC), an imidazole based compound, a triazole based compound, an oxadiazole based compound (e.g.: PBD), aluminum complex (available from UDC), or BAlq having the below formulas, etc.
- a phenanthroline based compound e.g.: BCP available from UDC
- an imidazole based compound e.g.: imidazole based compound
- a triazole based compound e.g.: PBD
- an oxadiazole based compound e.g.: PBD
- aluminum complex available from UDC
- BAlq having the below formulas, etc.
- the electron transport layer 15 may be formed of an oxazole based compound, an isooxazole based compound, a triazole based compound, an isothiazole based compound, an oxadiazole based compound, a thiadiazole based compound, a perylene based compound, aluminum complex (e.g.: Alq3 (tris(8-quinolinolato)-aluminum), BAlq, SAlq, Almq3, gallium complex (e.g: Gaq′2OPiv, Gaq′2OAc, 2(Gaq′2)), etc.
- the thickness of the hole blocking layer 13 may be in the range of 100 to 1,000 ⁇ , and the thickness of the electron transport layer 15 may be in the range of 100 to 1,000 ⁇ . If the thicknesses of the hole blocking layer 13 and the electron transport layer 15 are outside the above ranges, the properties of the hole blocking layer 13 and the electron transport layer 15 respectively deteriorate.
- a second electrode 14 is formed on the above product, and the final product is soldered to complete an organic electroluminescent device.
- the material used to form the second electrode 14 is not specifically limited, and the second electrode 14 may be formed by depositing a metal having a small work function, such as Li, Ca, Ca/Al, LiF/Ca, BaF 2 /Ca, LiF/Al, Al, Mg, or Mg ally.
- the thickness of the second electrode 14 may be in the range of 50 to 3,000 ⁇ .
- the polymer of Formula (1) is used as a material to form an emissive layer while manufacturing the organic electroluminescent device, but due to its chemical properties, it may also be used as a material to form a hole transport layer. Also, the polymer may be used as an intermediate in bio-field.
- the method of preparing an organic electroluminescent device according to the current embodiment of the present invention does not require a special apparatus, and the organic electroluminescent device may be manufactured using a conventional method of manufacturing an organic electroluminescent device using an emissive polymer.
- a Schlenk flask was fully evacuated and refluxed with nitrogen gas to completely remove moisture. 330 mg (1.2 mmol) of Ni(COD) 2 and 187 mg (1.2 mmol) of bipyridal were injected into the Schlenk flask in a glove box. Next, the flask was fully evacuated again and refluxed with nitrogen gas. 5 ml of anhydrous dimethyl formamide (DMF), 0.13 ml (1.2 mmol) of 1,5-cyclooctadiene (COD), and 5 ml of anhydrous toluene were added to the flask under nitrogen atmosphere. The reaction mixture was stirred at 80° C.
- DMF dimethyl formamide
- COD 1,5-cyclooctadiene
- a Schlenk flask was fully evacuated and refluxed with nitrogen gas to completely remove moisture. 330 mg (1.2 mmol) of Ni(COD) 2 and 187 mg (1.2 mmol) of bipyridal were injected into the Schlenk flask in a glove box. Next, the flask was fully evacuated again and refluxed with nitrogen gas. 5 ml of anhydrous dimethyl formamide (DMF), 0.13 ml (1.2 mmol) of 1,5-cyclooctadiene (COD), and 5 ml of anhydrous toluene were added to the flask under nitrogen atmosphere. The reaction mixture was stirred at 80° C.
- DMF dimethyl formamide
- COD 1,5-cyclooctadiene
- a Schlenk flask was fully evacuated and refluxed with nitrogen gas to completely remove moisture. 330 mg (1.2 mmol) of Ni(COD) 2 and 187 mg (1.2 mmol) of bipyridal were injected into the Schlenk flask in a glove box. Next, the flask was fully evacuated again and refluxed with nitrogen gas. 5 ml of anhydrous dimethyl formamide (DMF), 0.13 ml (1.2 mmol) of 1,5-cyclooctadiene (COD), and 5 ml of anhydrous toluene were added to the flask under nitrogen atmosphere. The reaction mixture was stirred at 80° C.
- DMF dimethyl formamide
- COD 1,5-cyclooctadiene
- a Schlenk flask was fully evacuated and refluxed with nitrogen gas to completely remove moisture. 330 mg (1.2 mmol) of Ni(COD) 2 and 187 mg (1.2 mmol) of bipyridal were injected into the Schlenk flask in a glove box. Next, the flask was fully evacuated again and refluxed with nitrogen gas. 5 ml of anhydrous dimethyl formamide (DMF), 0.13 ml (1.2 mmol) of 1,5-cyclooctadiene (COD), and 5 ml of anhydrous toluene were added to the flask under nitrogen atmosphere. The reaction mixture was stirred at 80° C.
- DMF dimethyl formamide
- COD 1,5-cyclooctadiene
- n 0.9.
- Reaction processes of poly(2′,3′,6′,7′-tetraoctyloxyspirofluorene-co-phenoxazine), as the electroluminescent polymer of Formula (7) are as follows.
- a Schlenk flask was fully evacuated and refluxed with nitrogen gas to completely remove moisture. 880 mg (3.2 mmol) of Ni(COD) 2 and 500 mg (3.2 mmol) of bipyridal were injected into the Schlenk flask in a glove box. Next, the flask was fully evacuated again and refluxed with nitrogen gas. 10 ml of anhydrous dimethyl formamide (DMF), 346 mg (3.2 mmol) of 1,5-cyclooctadiene (COD), and 10 ml of anhydrous toluene were added to the flask under nitrogen atmosphere. The reaction mixture was stirred at 80° C.
- DMF dimethyl formamide
- COD 1,5-cyclooctadiene
- ITO indium-tin oxide
- ITO transparent electrode
- Batron P 4083 available from Bayer Co.
- NTSID9 prepared in Preparation Example 1 was dissolved in 5 g of toluene to obtain an electroluminescent polymer solution.
- the electroluminescent polymer solution was spin coated on the buffer layer, backed, and placed in a vacuum oven to fully remove the solvent and form an emissive layer. Prior to the spin coating, the electroluminescent polymer solution was filtered through a 0.2 mm filter. The concentration of electroluminescent polymer solution and the spinning rate were controlled to form an emissive layer having a thickness of about 80 nm.
- the resulting electroluminescent device had a single stack structure in which ITO, PEDOT, the electroluminescent polymer, Ca, and Al were sequentially stacked upon one another.
- the emissive area was 4 mm 2 .
- An electroluminescent device was manufactured in the same manner as in Example 4, except that 0.05 g of NTS9 of Preparation Example 2 was used instead of 0.05 g of NTSID9 of Preparation Example 1, to prepare an electroluminescent polymer solution.
- An electroluminescent device was manufactured in the same manner as in Example 4, except that 0.05 g of NTS29 of Preparation Example 3 was used instead of 0.05 g of NTSID9 of Preparation Example 1, to prepare an electroluminescent polymer solution.
- An electroluminescent device was manufactured in the same manner as in Example 4, except that 0.05 g of poly(2′,3′,6′,7′-tetraoctyloxyspirofluorene) of Comparative Example 1 was used, instead of 0.05 g of NTSID9 of Preparation Example 1, to prepare the electroluminescent polymer solution.
- An electroluminescent device was manufactured in the same manner as in Example 4, except that 0.05 g of poly(2′,3′,6′,7′-tetraoctyloxyspirofluorene-co-phenoxazine) in a mole ratio of 90:10 was used, instead of 0.05 g of NTSID9 of Preparation Example 1, to prepare the electroluminescent polymer solution.
- the electroluminescent properties were measured using the electroluminescent devices of Examples 4-6 and Comparative Examples 3-4. For this measurement, a forward bias voltage was applied as a direct current (DC) driving voltage. The results are shown in Table 1 below.
- Example 2 Example 3
- Example 2 Maximum 460 475 470 437, 464 479 emission wavelength (nm) Maximum 2.0 3.8 3.1 1.8 4.8 efficiency (cd/A) Driving 3.4 2.8 3.0 4.0 3.2 voltage (V)
- the electroluminescent devices manufactured in Examples 4 through 6 and Comparative Examples 3 and 4 exhibit typical rectifying diode characteristics.
- the electroluminescent devices of Examples 1 through 3 including the polymers according to embodiments of the present invention show stable voltage-current density properties that stay at initial levels even after operations are repeatedly performed.
- an iminodibenzyl unit contains an ethylene group, and thus has a twisted structure in its polymer backbone. Hence, shift to short wavelength is easier. Also, a polymer having long lifetime was obtained due to low interface effect and high film packing density using bulkily fused to nitrogen aromatic structure in a penoxazine unit.
- the electroluminescent devices of Examples 1 through 3 containing polymers have remarkably lower y value compared to the electroluminescent device of Comparative Example 2, hence the electroluminescent devices of Examples 1 through 3 have wide blue area.
- an electroluminescent polymer according to the present invention shows strong and stable emissive properties.
- the electroluminescent polymer according to the present invention is used for organic layers, in particular, an emissive layer, of an electroluminescent device, color purity, luminance and efficiency of the organic electroluminescent device are improved.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Rehabilitation Therapy (AREA)
- General Health & Medical Sciences (AREA)
- Pain & Pain Management (AREA)
- Physical Education & Sports Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Otolaryngology (AREA)
- Pulmonology (AREA)
- Electroluminescent Light Sources (AREA)
- Polyoxymethylene Polymers And Polymers With Carbon-To-Carbon Bonds (AREA)
Abstract

-
- where Ar, X, Y, and R are the same as described in the detailed description of the invention and the claims.
Description

-
- where Ar is a C6-26 aromatic group or a C4-20 heteroaromatic group including at least one heteroatom in the aromatic ring, where the aromatic group or the heteroaromatic group is unsubstituted or substituted with at least one of a C1-12 alkyl group, a C1-12 alkoxy group, and —N(R′)(R″) where R′ and R″ are each independently a hydrogen atom or a C1-12alkyl group;

where R1 and R2 are each independently a hydrogen atom, a C1-12 linear alkyl group, a C1-12 branched alkyl group or a C1-12alkoxy group; Y is phenylene, naphthalene or anthracene, but Y is not phenylene when X is O, S or CH2; and R is a hydrogen atom, a C1-12 linear alkyl group, a C1-12 branched alkyl group, a C1-12 alkoxy group or a C5-30 cycloalkyl group, or a C6-14 aromatic group which is unsubstituted or substituted with at least one of a C1-12 alkyl group, a C1-12 alkoxy group, and —N(R′)(R″) where R′ and R″ are independently a hydrogen atom or a C1-12alkyl group, the polymer having a degree of polymerization in the range of 10 to 2,000.

where Ar is a C6-26 aromatic group or a C4-20 heteroaromatic group including at least one heteroatom in the aromatic ring, where the aromatic group or the heteroaromatic group is unsubstituted or substituted with at least one of a C1-12 alkyl group, a C1-12 alkoxy group, and —N(R′)(R″) where R′ and R″ are each independently a hydrogen atom or a C1-12 alkyl group;

where R1 and R2 are hydrogen atoms, C1-12 linear alkyl groups, C1-12 branched alkyl groups or C1-12 alkoxy groups;
Y is phenylene, naphthalene or anthracene, but Y is not phenylene when X is O, S or CH2; and
R is a hydrogen atom, a C1-12 linear alkyl group, a C1-12 branched alkyl group, a C1-12 alkoxy group or a C5-30 cycloalkyl group, or a C6-14 aromatic group which is unsubstituted or substituted with at least one of a C1-12 alkyl group, a C1-12 alkoxy group, and —N(R′)(R″) where R′ and R″ are independently a hydrogen atom or a C1-12 alkyl group.

where X′ is O or S; and Y and R are as described above.




where R3, R4, R5, and R6 are each independently hydrogen, a C1-12 alkyl group, a C1-12 alkoxy group, a C6-20 alkyl group or —N(R′)(R″) where R′ and R″ are each independently a hydrogen atom or a C1-12 alkyl group.








| TABLE 1 | ||
| Example | ||
| Comparative | Comparative | |||||
| Example 4 | Example 5 | Example 6 | Example 3 | Example 4 | ||
| Emissive | NTSID9 of | NTS9 of | NTS29 of | Compound of | Compound of |
| polymer | Preparation | Preparation | Preparation | Comparative | Comparative |
| Example 1 | Example 2 | Example 3 | Example 1 | Example 2 | |
| Maximum | 460 | 475 | 470 | 437, 464 | 479 |
| emission | |||||
| wavelength | |||||
| (nm) | |||||
| Maximum | 2.0 | 3.8 | 3.1 | 1.8 | 4.8 |
| efficiency | |||||
| (cd/A) | |||||
| Driving | 3.4 | 2.8 | 3.0 | 4.0 | 3.2 |
| voltage | |||||
| (V) | |||||
| Half-lifetime | 17,000 | 46,000 | 37,400 | 720 | 31,000 |
| (hr) | |||||
| (@100 cd/m2) | |||||
| Color | (0.16, 0.10) | (0.15, 0.30) | (0.15, 0.26) | (0.15, 0.20) | (0.15, 0.32) |
| coordinates | |||||
| (x, y) | |||||
Claims (8)








Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020060008238A KR101328972B1 (en) | 2006-01-26 | 2006-01-26 | Electroluminescent Polymer And Organic-electroluminescent Device Manufactured By Using The Same |
| KR10-2006-0008238 | 2006-01-26 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20070173633A1 US20070173633A1 (en) | 2007-07-26 |
| US8062766B2 true US8062766B2 (en) | 2011-11-22 |
Family
ID=38286380
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/600,110 Active 2029-10-27 US8062766B2 (en) | 2006-01-26 | 2006-11-16 | Electroluminescent polymer and organic electroluminescent device using the same |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US8062766B2 (en) |
| JP (1) | JP5390072B2 (en) |
| KR (1) | KR101328972B1 (en) |
| CN (1) | CN101007941B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20110272678A1 (en) * | 2008-11-28 | 2011-11-10 | Sumitomo Chemical Company, Limited | Organic electroluminescent device and method for manufacturing the same |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101223720B1 (en) * | 2006-06-05 | 2013-01-17 | 삼성디스플레이 주식회사 | Polymer compound and organic light-emitting device employing the same |
| JP5256445B2 (en) * | 2006-11-02 | 2013-08-07 | 名古屋市 | Dibenzazepine copolymer |
| DE102008049037A1 (en) * | 2008-09-25 | 2010-04-22 | Merck Patent Gmbh | New polymers with low polydispersity |
| IT1393059B1 (en) * | 2008-10-22 | 2012-04-11 | Eni Spa | LOW-GAP PI-CONJUGATED COPOLYMERS CONTAINING BENZOTRIAZOLIC UNITS |
| KR101107489B1 (en) * | 2009-08-03 | 2012-01-19 | 부경대학교 산학협력단 | A phenothiazine-based polymer and organic light-emitting device using the polymer |
| KR101137386B1 (en) | 2009-10-09 | 2012-04-20 | 삼성모바일디스플레이주식회사 | Polymer and organic light emitting diode comprising the same |
| KR101174880B1 (en) * | 2010-04-02 | 2012-08-17 | 삼성디스플레이 주식회사 | Polymer and Organic Light Emitting Device containing the polymer |
| KR101202351B1 (en) | 2010-07-16 | 2012-11-16 | 삼성디스플레이 주식회사 | Dendrimer and organic light emitting device using the same |
| KR101830784B1 (en) * | 2011-09-09 | 2018-02-22 | 삼성전자주식회사 | Polymer and organic light emitting diode comprising the same |
| CN104024372B (en) | 2011-12-23 | 2015-08-12 | 第一毛织株式会社 | For organic optoelectronic device compound, comprise the organic illuminating element of this compound and comprise the display unit of this organic illuminating element |
| KR101788090B1 (en) * | 2014-11-28 | 2017-11-15 | 삼성에스디아이 주식회사 | Polymer, organic layer composition, organic layer, and method of forming patterns |
| CN107922402B (en) * | 2015-08-14 | 2021-12-31 | 默克专利有限公司 | Phenoxazine derivatives for organic electroluminescent devices |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62225518A (en) * | 1986-03-28 | 1987-10-03 | Mitsubishi Chem Ind Ltd | Organic semiconductor |
| US6169163B1 (en) | 1995-07-28 | 2001-01-02 | The Dow Chemical Company | Fluorene-containing polymers and compounds useful in the preparation thereof |
| JP2003165829A (en) * | 2001-11-30 | 2003-06-10 | Toppan Printing Co Ltd | Polymer and organic thin film element using the same |
| US20040072989A1 (en) * | 2002-06-21 | 2004-04-15 | Samsung Sdi Co., Ltd. | Blue electroluminescent polymer and organic-electroluminescent device using the same |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100552683B1 (en) * | 2002-06-21 | 2006-02-20 | 삼성에스디아이 주식회사 | Blue electroluminescent polymer and organic-electroluminescent device manufactured by using the same |
| US6916902B2 (en) * | 2002-12-19 | 2005-07-12 | Dow Global Technologies Inc. | Tricyclic arylamine containing polymers and electronic devices therefrom |
| KR20070073454A (en) * | 2006-01-05 | 2007-07-10 | 삼성에스디아이 주식회사 | Organic electroluminescence device |
-
2006
- 2006-01-26 KR KR1020060008238A patent/KR101328972B1/en active IP Right Grant
- 2006-08-30 CN CN2006101263391A patent/CN101007941B/en active Active
- 2006-11-16 US US11/600,110 patent/US8062766B2/en active Active
-
2007
- 2007-01-24 JP JP2007014343A patent/JP5390072B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62225518A (en) * | 1986-03-28 | 1987-10-03 | Mitsubishi Chem Ind Ltd | Organic semiconductor |
| US6169163B1 (en) | 1995-07-28 | 2001-01-02 | The Dow Chemical Company | Fluorene-containing polymers and compounds useful in the preparation thereof |
| JP2003165829A (en) * | 2001-11-30 | 2003-06-10 | Toppan Printing Co Ltd | Polymer and organic thin film element using the same |
| US20040072989A1 (en) * | 2002-06-21 | 2004-04-15 | Samsung Sdi Co., Ltd. | Blue electroluminescent polymer and organic-electroluminescent device using the same |
Non-Patent Citations (1)
| Title |
|---|
| Stéphan et al., "Blue light electroluminescent devices based on a copolymer derived from fluorene and carbazole", Synthetic Metals 106 (1999) pp. 115-119. |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20110272678A1 (en) * | 2008-11-28 | 2011-11-10 | Sumitomo Chemical Company, Limited | Organic electroluminescent device and method for manufacturing the same |
| US8698133B2 (en) * | 2008-11-28 | 2014-04-15 | Sumitomo Chemical Company, Limited | Organic electroluminescent device comprising a polymer obtained by polymerizing a polymerizable compound capable of exhibiting charge transportability and method for manufacturing the same |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5390072B2 (en) | 2014-01-15 |
| US20070173633A1 (en) | 2007-07-26 |
| KR101328972B1 (en) | 2013-11-13 |
| CN101007941B (en) | 2012-08-29 |
| KR20070078200A (en) | 2007-07-31 |
| CN101007941A (en) | 2007-08-01 |
| JP2007204748A (en) | 2007-08-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8062766B2 (en) | Electroluminescent polymer and organic electroluminescent device using the same | |
| US6984461B2 (en) | Blue electroluminescent polymer and organic-electroluminescent device using the same | |
| US7172823B2 (en) | Blue electroluminescent polymer and organo-electroluminescent device employing the same | |
| US7648777B2 (en) | Blue electroluminescent compound and organic electroluminescent device using the same | |
| US7521525B2 (en) | Blue luminescent polymer and organoelectroluminescent device using the same | |
| KR101146976B1 (en) | Blue electroluminescent polymer and organoelectroluminescent device employing the same | |
| US7524567B2 (en) | Spirofluorene-based polymer and organic electroluminescent device using the same | |
| JPH10324870A (en) | High-molecular phosphor and organic electroluminescent element | |
| KR101243918B1 (en) | Blue electroluminescent polymer, a method for preparing the blue electroluminescent polymer and an organoelectroluminescent device employing the same | |
| US7872411B2 (en) | Organic electroluminescence device using a copolymer and a phosphorescent compound | |
| US8257839B2 (en) | Polyvinyl pyrrole host material, luminescent layer comprising the same, and organic electroluminescent device comprising the luminescent layer | |
| KR100552683B1 (en) | Blue electroluminescent polymer and organic-electroluminescent device manufactured by using the same | |
| US7569287B2 (en) | Blue electroluminescent polymer and organic electroluminescent device using the same | |
| US6495644B1 (en) | Electroluminiscent conjugated polymers modified with high electronegative heterocyclic moieties and their applications in polymeric light emitting diodes | |
| KR101193178B1 (en) | An organic light emitting device and a method for preparing the same | |
| KR20060091520A (en) | Dihydrophenazine compound and organo-electroluminescent device employing the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: SAMSUNG SDI CO., LTD., KOREA, REPUBLIC OF Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:SON, JHUN-MO;KIM, YU-JIN;PARK, SANG-HOON;AND OTHERS;REEL/FRAME:018613/0528 Effective date: 20061113 |
|
| AS | Assignment |
Owner name: SAMSUNG MOBILE DISPLAY CO., LTD., KOREA, REPUBLIC Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:SAMSUNG SDI CO., LTD.;REEL/FRAME:022034/0001 Effective date: 20081210 Owner name: SAMSUNG MOBILE DISPLAY CO., LTD.,KOREA, REPUBLIC O Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:SAMSUNG SDI CO., LTD.;REEL/FRAME:022034/0001 Effective date: 20081210 |
|
| STCF | Information on status: patent grant |
Free format text: PATENTED CASE |
|
| FEPP | Fee payment procedure |
Free format text: PAYOR NUMBER ASSIGNED (ORIGINAL EVENT CODE: ASPN); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY |
|
| AS | Assignment |
Owner name: SAMSUNG DISPLAY CO., LTD., KOREA, REPUBLIC OF Free format text: MERGER;ASSIGNOR:SAMSUNG MOBILE DISPLAY CO., LTD.;REEL/FRAME:029241/0599 Effective date: 20120702 |
|
| FPAY | Fee payment |
Year of fee payment: 4 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 8TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1552); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 8 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 12TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1553); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 12 |