US7019142B2 - Process for preparing naphthyridones and intermediates - Google Patents
Process for preparing naphthyridones and intermediates Download PDFInfo
- Publication number
- US7019142B2 US7019142B2 US10/087,756 US8775602A US7019142B2 US 7019142 B2 US7019142 B2 US 7019142B2 US 8775602 A US8775602 A US 8775602A US 7019142 B2 US7019142 B2 US 7019142B2
- Authority
- US
- United States
- Prior art keywords
- compound
- formula
- alkyl
- reaction
- volumes
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related, expires
Links
- 0 [1*]N1CC2([H])C([H])(C)C2([H])C1 Chemical compound [1*]N1CC2([H])C([H])(C)C2([H])C1 0.000 description 5
- SVCPQRXTTGZEAI-UHFFFAOYSA-N CC1=CN(C2=CC=C(F)C=C2F)C2=C(C=C(F)C(Cl)=N2)C1=O Chemical compound CC1=CN(C2=CC=C(F)C=C2F)C2=C(C=C(F)C(Cl)=N2)C1=O SVCPQRXTTGZEAI-UHFFFAOYSA-N 0.000 description 2
- VJVGMYDGDCOOIL-UHFFFAOYSA-N [H]C1(N)C2([H])CN(C3=FC4=C(C=C3F)C(=O)C(C)=CN4C3=CC=C(F)C=C3F)CC12[H] Chemical compound [H]C1(N)C2([H])CN(C3=FC4=C(C=C3F)C(=O)C(C)=CN4C3=CC=C(F)C=C3F)CC12[H] VJVGMYDGDCOOIL-UHFFFAOYSA-N 0.000 description 2
- WVPSKSLAZQPAKQ-UHFFFAOYSA-N [H]C1(N)C2([H])CN(C3=NC4=C(C=C3F)C(=O)C(C(=O)O)=CN4C3=CC=C(F)C=C3F)CC12[H] Chemical compound [H]C1(N)C2([H])CN(C3=NC4=C(C=C3F)C(=O)C(C(=O)O)=CN4C3=CC=C(F)C=C3F)CC12[H] WVPSKSLAZQPAKQ-UHFFFAOYSA-N 0.000 description 1
- DQHQBMIDBBDGAC-UHFFFAOYSA-N [H]C1(N)C2([H])CN(C3=NC4=C(C=C3F)C(=O)C(C)=CN4C3=CC=C(F)C=C3F)CC12[H] Chemical compound [H]C1(N)C2([H])CN(C3=NC4=C(C=C3F)C(=O)C(C)=CN4C3=CC=C(F)C=C3F)CC12[H] DQHQBMIDBBDGAC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/52—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring condensed with a ring other than six-membered
Definitions
- This invention relates to a process for preparing the naphthyridone carboxylic acid, trovafloxacin and derivatives thereof, and intermediates of use therein.
- Trovafloxacin has the formula as disclosed in U.S. Pat. No. 5,164,402.
- the patent also discloses processes for making the compound by using an intermediate of the formula wherein R′ is a nitrogen protecting group, such as tertiary butyloxycarbonyl.
- U.S. Pat. No. 5,475,116 discloses the preparation of other intermediates for use in preparing the naphthyridones of U.S. Pat. No. 5,164,402.
- the present invention relates to a process for preparing a compound of the formula
- R 1 is benzyl, wherein the phenyl of the benzyl may be substituted by one or more of C 1 –C 6 alkyl, C 1 –C 6 alkoxy, halo, nitro, amino or trifluoromethyl, and
- R 2 is C 1 –C 6 alky, trifluoromethyl, or phenyl which may be substituted by one or more of C 1 –C 6 alkyl, C 1 –C 6 alkoxy, halo, nitro, amino or trifluoromethyl, which comprises (a) reducing a compound of the formula wherein R 1 is as defined above, in the presence of iron and a organic solvent under acidic conditions, and (b) acylating the compound of formula III formed: with an acylating agent of the formula R 2 C(O)X wherein R 2 is as defined above, and X is a leaving group.
- the compound of formula III formed in step (a) is not isolated before acylation step (b).
- the invention is further related to a process for preparing a compound of the formula by debenzylating the compound of formula I wherein R 1 and R 2 are as defined above.
- the debenzylation is carried out by reacting a compound of formula I with hydrogen and palladium catalyst in acetic acid and an organic solvent.
- the invention also relates to reacting a compound of the formula IV with a compound of the formula wherein R 3 is C 1 –C 6 alkyl, to form a compound of the formula wherein R 2 is as defined above with reference to formula I.
- the invention relates to hydrolyzing the compound of formula VI with methanesulfonic acid, water and an organic solvent to form the monomethanesulfonic acid salt of the compound of the formula VII, trovafloxacin.
- the invention also relates to hydrolysis of the compound of formula VI with methanesulfonic acid and R 3 OH wherein R 3 is as defined above to form the monomethanesulfonic acid salt of the compound of the formula
- the invention further relates to the intermediates of the formulae wherein R 2 is C 1 –C 6 alkyl, trifluoromethyl, or phenyl which may be substituted by one or more of C 1 –C 6 alkyl, C 1 –C 6 alkoxy, halo, nitro, amino or trifluoromethyl, and
- R 3 is C 1 –C 6 alkyl, and wherein
- R 1 is hydrogen (see formula IV) or benzyl, wherein the phenyl of the benzyl may be substituted by one or more of C 1 –C 6 alkyl, C 1 –C 6 alkoxy, halo, nitro, amino or trifluoromethyl, and
- R 2 is C 1 –C 6 alkyl, trifluoromethyl, or phenyl which may be substituted by one or more of C 1 –C 6 alkyl, C 1 –C 6 alkoxy, halo, nitro, amino or trifluoromethyl.
- alkyl includes saturated monovalent hydrocarbon radicals having straight, branched or cyclic moieties, e.g. methyl, ethyl.
- alkoxy includes O-alkyl groups wherein “alkyl” is defined above.
- the compound of formula III is prepared from the corresponding compound of formula II by reduction in the presence of iron and an organic solvent under acidic conditions.
- the organic solvent is a C 1 –C 6 alcohol, such as ethanol, or an ether such as tetrahydrofuran (THF), and preferably, an alcohol.
- the acidic conditions are obtained by use of a mineral acid, such as hydrochloric acid, or an organic acid, such as acetic acid (AcOH). Acetic acid is preferred since it generally results in increased yields.
- the compound of formula III may then be isolated from the reaction mixture or may be reacted further in situ, without isolation from the reaction mixture. In either case, the further processing is by acylation with an acylating agent of the formula R 2 C(O)X to form the compound of formula I.
- the leaving group X is conveniently a halogen, such as chloro, or the acetoxy group. If the compound of formula III is first isolated, then the acylation may be conducted under conventional acylating conditions, for instance, in the presence of an organic solvent of the type discussed above.
- debenzylation includes removal of R 1 wherein R 1 is benzyl or substituted benzyl.
- the reaction proceeds in accordance with conventional debenzylation of tertiary nitrogen, conveniently by use of hydrogen and palladium catalyst in acetic acid, and in an organic solvent.
- the organic solvent may be a C 1 –C 6 alcoholic solvent, such as ethanol, ethyl acetate, THF or water, or a mixture thereof, such as ethanol and water.
- the compound of formula VI is obtained by coupling the corresponding compound of formula IV with the bicyclic intermediate ester of formula V.
- This coupling reaction may be conducted with or without a solvent.
- the solvent when used, must be inert under the reaction conditions. Suitable solvents are ethyl acetate, acetonitrile, tetrahydrofuran, ethanol, chloroform, dimethylsulfoxide, pyridine, and water, and mixtures thereof.
- the reaction temperature usually ranges from about 20° C. to about 150° C.
- the reaction may advantageously be carried out in the presence of an acid acceptor such as an inorganic or organic base, e.g. an alkali metal or alkaline earth metal carbonate or bicarbonate, or a tertiary amine, e.g. triethylamine, pyridine or picoline.
- an acid acceptor such as an inorganic or organic base, e.g. an alkali metal or alkaline earth metal carbonate or bicarbonate, or a tertiary amine, e.g. triethylamine, pyridine or picoline.
- the mesylate salt of the compound of formula VII, trovafloxacin is formed by hydrolysis of the compound of formula VI with methanesulfonic acid, water and an organic solvent.
- suitable organic solvents include a C 1 –C 6 alcohol, acetone, dimethoxy ethane, glyme, THF, N-methyl-pyrrolidinone, and water, and mixtures thereof.
- the mesylate salt of the compound of formula VIII is obtained by hydrolysis of the compound of formula VI with methanesulfonic acid and a C 1 –C 6 alcohol of the formula R 3 OH, for example ethanol.
- the compound of formula VIII is an intermediate in the preparation of the mesylate salt of a prodrug of trovafloxacin wherein the amino group is substituted by an amino acid or a polypeptide, e.g. dipeptide, as disclosed in U.S. Pat. No. 5,164,402.
- the compound of formula IX in the Reaction Scheme is the intermediate formed in the reaction from compound VI to VII.
- the compound of formula VII and the mesylate salt thereof are useful in the treatment of bacterial infections of broad spectrum, particularly the treatment of gram-positive bacterial strains.
- the active compounds may be administered alone, but will generally be administered in admixture with a pharmaceutical carrier selected with regard to the intended route of administration and standard pharmaceutical practice.
- a pharmaceutical carrier selected with regard to the intended route of administration and standard pharmaceutical practice.
- they can be administered orally or in the form of tablets containing such excipients as starch or lactose, or in capsules either alone or in admixture with excipients, or in the form of elixirs or suspensions containing flavoring or coloring agents.
- they are advantageously contained in an animal feed or drinking water in a concentration of 5–5000 ppm, preferably 25–500 ppm. They can be injected parenterally, for example, intramuscularly, intravenously or subcutaneously.
- a sterile aqueous solution which can contain other solutes, for example, enough salt or flucose to make the solution isotonic.
- compounds can be administered intramuscularly or subcutaneously at dosage levels of about 0.1–50 mg/kg/day, advantageously 0.2–10 mg/kg/day given in a single daily dose or up to 3 divided doses.
- the invention also provides pharmaceutical compositions comprising an antibacterially effective amount of a compound of the formula (I) together with a pharmaceutically acceptable diluent or carrier.
- the compounds of the invention can be administered to humans for the treatment of bacterial diseases by either the oral or parenteral routes, and may be administered orally at dosage levels of about 0.1 to 500 mg/kg/day, advantageously 0.5–50 mg/kg/day given in a single dose or up to 3 divided doses.
- dosage levels are about 0.1–200 mg/kg/day, advantageously 0.5–50 mg/kg/day.
- intramuscular administration may be a single dose or up to 3 divided doses
- intravenous administration can include a continuous drip. Variations will necessarily occur depending on the weight and condition of the subject being treated and the particular route of administration chosen as will be known to those skilled in the art.
- the residual iron was filtered off and the cake washed with 11.25 L of isopropanol (15 volumes).
- the isopropanol solution was concentrated in vacuo to an oil, 18 L of dichloroethane (24 volumes) was added before bringing the pH to 12 with 8.8 L of 5% sodium hydroxide solution (about 12 volumes).
- the layers were separated and the separated organic layer was dried by magnesium sulfate.
- the resulting dark amber oil was treated with 7.5 L of hexanes (10 volumes) and granulated at 25° C. before collecting the product as a white solid. Drying at 50° C. under vacuum gave 610 g of the title compound (77% yield). Analysis was done by GC/MS, NMR and TLC.
- a Parr Bottle was charged with 150 g of the compound of Example 1, 112 mL of acetic acid (3 equivalents), 1.5 L of methanol (10 volumes) and 15 g of (10% by wt. 50% wet) Pd/C catalyst (0.1 equivalent). The bottle was purged with nitrogen and then brought to 50 psi pressure with hydrogen. The mixture was shaken for 48 hours and recharged with catalyst as necessary during the debenzylation reaction. After TLC indicated that the reaction was complete, the catalyst was filtered off, and the filtrate was concentrated in vacuo to an oil. 3 L of ethyl acetate (20 volumes) was added to the oil, and granulated for an hour. The solid was collected by filtration and dried under vacuum at 50° C. to provide 107 g of the title compound (82% yield) as the acetic acid salt
- a reaction flask was charged with 241.9 g of 7-chloro-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acid ethyl ester, 151.6 g of the acetic acid salt of the compound of Example 2 (1.2 equivalents), 2661 mL of ethyl acetate (11 volumes) and 220 mL of triethylamine (2.5 equivalents). The mixture was heated at refluxing temperature under nitrogen for 6 hours monitored by HPLC or LCMS. After the reaction was completed, the reaction mixture was cooled to ambient temperature. Water (11 volumes) was added and the biphasic mixture was stirred for 17 hours. The white solid was collected by filtration, washed with 2661 mL of water (12 volumes) and oven dried at 50° C. to provide 292 g of the title compound (95% yield).
- the wet cake was mixed with 660 mL of n-butanol (3 volumes), seeded with 0.1 gm of the desired polymorph and heated to 95–100° C. After complete polymorph conversion, in approximately 2 hours, the mixture was cooled to ambient temperature.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
A process for preparing a naphthyridone carboxylic acid and its derivatives makes use of side chain intermediates of formulae I and IV herein.
Description
This is a division of application Ser. No. 09/718,324, filed Nov, 22, 2000 now abandoned, which is a divisional of application Ser. No. 09/236,737, filed Jan. 25, 1999, now U.S. Pat. No. 6,184,380, which claimed the benefit of U.S. Provisional Application No. 60/071,601, filed Jan. 16, 1998, all of which are hereby incorporated herein by reference.
This invention relates to a process for preparing the naphthyridone carboxylic acid, trovafloxacin and derivatives thereof, and intermediates of use therein.
Trovafloxacin has the formula 
as disclosed in U.S. Pat. No. 5,164,402. The patent also discloses processes for making the compound by using an intermediate of the formula 
wherein R′ is a nitrogen protecting group, such as tertiary butyloxycarbonyl.
as disclosed in U.S. Pat. No. 5,164,402. The patent also discloses processes for making the compound by using an intermediate of the formula
wherein R′ is a nitrogen protecting group, such as tertiary butyloxycarbonyl.
U.S. Pat. No. 5,475,116 discloses the preparation of other intermediates for use in preparing the naphthyridones of U.S. Pat. No. 5,164,402.
The present invention relates to a process for preparing a compound of the formula 
wherein R1 is benzyl, wherein the phenyl of the benzyl may be substituted by one or more of C1–C6 alkyl, C1–C6 alkoxy, halo, nitro, amino or trifluoromethyl, and
R2 is C1–C6 alky, trifluoromethyl, or phenyl which may be substituted by one or more of C1–C6 alkyl, C1–C6 alkoxy, halo, nitro, amino or trifluoromethyl, which comprises
(a) reducing a compound of the formula 
wherein R1 is as defined above, in the presence of iron and a organic solvent under acidic conditions, and
(b) acylating the compound of formula III formed: 
with an acylating agent of the formula R2C(O)X wherein R2 is as defined above, and X is a leaving group.
(a) reducing a compound of the formula
wherein R1 is as defined above, in the presence of iron and a organic solvent under acidic conditions, and
(b) acylating the compound of formula III formed:
with an acylating agent of the formula R2C(O)X wherein R2 is as defined above, and X is a leaving group.
In a prefered embodiment of the invention, the compound of formula III formed in step (a) is not isolated before acylation step (b).
The invention is further related to a process for preparing a compound of the formula 
by debenzylating the compound of formula I wherein R1 and R2 are as defined above.
by debenzylating the compound of formula I wherein R1 and R2 are as defined above.
In a preferred embodiment, the debenzylation is carried out by reacting a compound of formula I with hydrogen and palladium catalyst in acetic acid and an organic solvent.
The invention also relates to reacting a compound of the formula IV with a compound of the formula 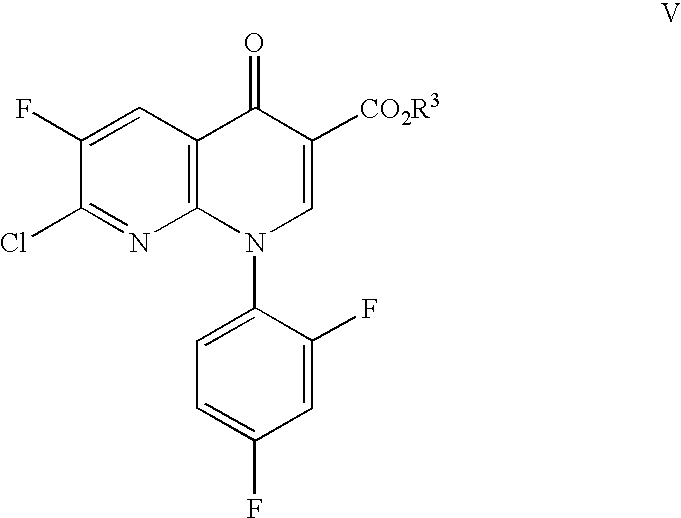
wherein R3 is C1–C6 alkyl, to form a compound of the formula 
wherein R2 is as defined above with reference to formula I.
wherein R3 is C1–C6 alkyl, to form a compound of the formula
wherein R2 is as defined above with reference to formula I.
The invention relates to hydrolyzing the compound of formula VI with methanesulfonic acid, water and an organic solvent to form the monomethanesulfonic acid salt of the compound of the formula VII, trovafloxacin.
The invention also relates to hydrolysis of the compound of formula VI with methanesulfonic acid and R3OH wherein R3 is as defined above to form the monomethanesulfonic acid salt of the compound of the formula 
The invention further relates to the intermediates of the formulae 
wherein R2 is C1–C6 alkyl, trifluoromethyl, or phenyl which may be substituted by one or more of C1–C6 alkyl, C1–C6 alkoxy, halo, nitro, amino or trifluoromethyl, and
wherein R2 is C1–C6 alkyl, trifluoromethyl, or phenyl which may be substituted by one or more of C1–C6 alkyl, C1–C6 alkoxy, halo, nitro, amino or trifluoromethyl, and
R3 is C1–C6 alkyl,
and 
wherein
and
wherein
R1 is hydrogen (see formula IV) or benzyl, wherein the phenyl of the benzyl may be substituted by one or more of C1–C6 alkyl, C1–C6 alkoxy, halo, nitro, amino or trifluoromethyl, and
R2 is C1–C6 alkyl, trifluoromethyl, or phenyl which may be substituted by one or more of C1–C6 alkyl, C1–C6 alkoxy, halo, nitro, amino or trifluoromethyl.
The term “alkyl”, as used herein, includes saturated monovalent hydrocarbon radicals having straight, branched or cyclic moieties, e.g. methyl, ethyl.
The term “alkoxy”, as used herein, includes O-alkyl groups wherein “alkyl” is defined above.
The processes of the invention are depicted in the following reaction scheme. Unless indicated otherwise, R1, R2, R3 and X are as defined above. 
The compound of formula III is prepared from the corresponding compound of formula II by reduction in the presence of iron and an organic solvent under acidic conditions. The organic solvent is a C1–C6 alcohol, such as ethanol, or an ether such as tetrahydrofuran (THF), and preferably, an alcohol. The acidic conditions are obtained by use of a mineral acid, such as hydrochloric acid, or an organic acid, such as acetic acid (AcOH). Acetic acid is preferred since it generally results in increased yields.
The compound of formula III may then be isolated from the reaction mixture or may be reacted further in situ, without isolation from the reaction mixture. In either case, the further processing is by acylation with an acylating agent of the formula R2C(O)X to form the compound of formula I. The leaving group X is conveniently a halogen, such as chloro, or the acetoxy group. If the compound of formula III is first isolated, then the acylation may be conducted under conventional acylating conditions, for instance, in the presence of an organic solvent of the type discussed above.
The compound of formula I is subjected to debenzylation to form the compound of formula IV. It is understood that in the context of the invention, debenzylation includes removal of R1 wherein R1 is benzyl or substituted benzyl. The reaction proceeds in accordance with conventional debenzylation of tertiary nitrogen, conveniently by use of hydrogen and palladium catalyst in acetic acid, and in an organic solvent. The organic solvent may be a C1–C6 alcoholic solvent, such as ethanol, ethyl acetate, THF or water, or a mixture thereof, such as ethanol and water.
The compound of formula VI is obtained by coupling the corresponding compound of formula IV with the bicyclic intermediate ester of formula V. This coupling reaction may be conducted with or without a solvent. The solvent, when used, must be inert under the reaction conditions. Suitable solvents are ethyl acetate, acetonitrile, tetrahydrofuran, ethanol, chloroform, dimethylsulfoxide, pyridine, and water, and mixtures thereof.
The reaction temperature usually ranges from about 20° C. to about 150° C.
The reaction may advantageously be carried out in the presence of an acid acceptor such as an inorganic or organic base, e.g. an alkali metal or alkaline earth metal carbonate or bicarbonate, or a tertiary amine, e.g. triethylamine, pyridine or picoline.
The mesylate salt of the compound of formula VII, trovafloxacin, is formed by hydrolysis of the compound of formula VI with methanesulfonic acid, water and an organic solvent. Examples of suitable organic solvents include a C1–C6 alcohol, acetone, dimethoxy ethane, glyme, THF, N-methyl-pyrrolidinone, and water, and mixtures thereof.
The mesylate salt of the compound of formula VIII is obtained by hydrolysis of the compound of formula VI with methanesulfonic acid and a C1–C6 alcohol of the formula R3OH, for example ethanol. The compound of formula VIII is an intermediate in the preparation of the mesylate salt of a prodrug of trovafloxacin wherein the amino group is substituted by an amino acid or a polypeptide, e.g. dipeptide, as disclosed in U.S. Pat. No. 5,164,402.
The compound of formula IX in the Reaction Scheme is the intermediate formed in the reaction from compound VI to VII.
The compound of formula VII and the mesylate salt thereof (the active compounds) are useful in the treatment of bacterial infections of broad spectrum, particularly the treatment of gram-positive bacterial strains.
The active compounds may be administered alone, but will generally be administered in admixture with a pharmaceutical carrier selected with regard to the intended route of administration and standard pharmaceutical practice. For example, they can be administered orally or in the form of tablets containing such excipients as starch or lactose, or in capsules either alone or in admixture with excipients, or in the form of elixirs or suspensions containing flavoring or coloring agents. In the case of animals, they are advantageously contained in an animal feed or drinking water in a concentration of 5–5000 ppm, preferably 25–500 ppm. They can be injected parenterally, for example, intramuscularly, intravenously or subcutaneously. For parenteral administration, they are best used in the form of a sterile aqueous solution which can contain other solutes, for example, enough salt or flucose to make the solution isotonic. In the case of animals, compounds can be administered intramuscularly or subcutaneously at dosage levels of about 0.1–50 mg/kg/day, advantageously 0.2–10 mg/kg/day given in a single daily dose or up to 3 divided doses.
The invention also provides pharmaceutical compositions comprising an antibacterially effective amount of a compound of the formula (I) together with a pharmaceutically acceptable diluent or carrier.
The compounds of the invention can be administered to humans for the treatment of bacterial diseases by either the oral or parenteral routes, and may be administered orally at dosage levels of about 0.1 to 500 mg/kg/day, advantageously 0.5–50 mg/kg/day given in a single dose or up to 3 divided doses. For intramuscular or intravenous administration, dosage levels are about 0.1–200 mg/kg/day, advantageously 0.5–50 mg/kg/day. While intramuscular administration may be a single dose or up to 3 divided doses, intravenous administration can include a continuous drip. Variations will necessarily occur depending on the weight and condition of the subject being treated and the particular route of administration chosen as will be known to those skilled in the art.
The following Examples illustrate the invention. The abbreviations used mean the following: GC=gas chromatography; MS=mass spectometry; TLC=thin layer chromatography, HPLC=high performance liquid chromatography; LCMS=liquid chromatography mass spectometry; and NMR=nuclear magnetic resonance.
A 3-necked round bottom flask, equiped with a thermometer, a overhead stirrer and a condenser with nitrogen purge, was charged with 768 g of nitrocyclopropane, 5.75 L of isopropanol (7.5 volumes), 1.79 L of acetic acid (9.1 equivalents) and 1153 g of iron powder (6 equivalents). The reaction mixture was heated at 50° C. until the reaction was completed by GC/MS analysis (about 6 hours). 448 mL of acetic anhydride (1.4 equivalents) was added and stirred at 50° C. for 15 minutes before cooling. The reaction mixture was diluted with 8 L isopropanol (10.5 volumes) and stirred for 30 minutes. The residual iron was filtered off and the cake washed with 11.25 L of isopropanol (15 volumes). The isopropanol solution was concentrated in vacuo to an oil, 18 L of dichloroethane (24 volumes) was added before bringing the pH to 12 with 8.8 L of 5% sodium hydroxide solution (about 12 volumes). The layers were separated and the separated organic layer was dried by magnesium sulfate. The resulting dark amber oil was treated with 7.5 L of hexanes (10 volumes) and granulated at 25° C. before collecting the product as a white solid. Drying at 50° C. under vacuum gave 610 g of the title compound (77% yield). Analysis was done by GC/MS, NMR and TLC.
A Parr Bottle was charged with 150 g of the compound of Example 1, 112 mL of acetic acid (3 equivalents), 1.5 L of methanol (10 volumes) and 15 g of (10% by wt. 50% wet) Pd/C catalyst (0.1 equivalent). The bottle was purged with nitrogen and then brought to 50 psi pressure with hydrogen. The mixture was shaken for 48 hours and recharged with catalyst as necessary during the debenzylation reaction. After TLC indicated that the reaction was complete, the catalyst was filtered off, and the filtrate was concentrated in vacuo to an oil. 3 L of ethyl acetate (20 volumes) was added to the oil, and granulated for an hour. The solid was collected by filtration and dried under vacuum at 50° C. to provide 107 g of the title compound (82% yield) as the acetic acid salt
A reaction flask was charged with 241.9 g of 7-chloro-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acid ethyl ester, 151.6 g of the acetic acid salt of the compound of Example 2 (1.2 equivalents), 2661 mL of ethyl acetate (11 volumes) and 220 mL of triethylamine (2.5 equivalents). The mixture was heated at refluxing temperature under nitrogen for 6 hours monitored by HPLC or LCMS. After the reaction was completed, the reaction mixture was cooled to ambient temperature. Water (11 volumes) was added and the biphasic mixture was stirred for 17 hours. The white solid was collected by filtration, washed with 2661 mL of water (12 volumes) and oven dried at 50° C. to provide 292 g of the title compound (95% yield).
In a reaction flask, 220 g of the compound of Example 3, 1.76 L of n-butanol (8 volumes), 1.54 L of water (7 volumes) and 141 mL of 70% methanesulfonic acid (3.0 equivalents) were mixed. The mixture was heated at reflux for 21 hours, and the reaction was monitored by HPLC or LCMS. After complete reaction, the mixture was cooled to 50° C. and filtered to make it speck-free. The filtrated was cooled to 0–5° C. and granulated for 2 hours. The solid was collected by filtration, washed with 220 mL of water (1 volume) and 660 mL n-butanol (3 volumes). The wet cake was mixed with 660 mL of n-butanol (3 volumes), seeded with 0.1 gm of the desired polymorph and heated to 95–100° C. After complete polymorph conversion, in approximately 2 hours, the mixture was cooled to ambient temperature. The solid was filtered, washed with 100 mL of n-butanol (0.5 volumes) and dried in a nitrogen atmosphere to provide 200 g of (1α, 5α, 6α)-7-(6-amino-3-azabicyclo[3.1.0]hex-3-yl)-1-(2,4-difluorophenyl)-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acid, monomethanesulfonate (87% yield).
0.8 mL of methanesulfonic acid (2.7 equivalents) was added dropwise to a solution of 2.2 g of the compound of Example 3 in 10 mL of ethanol (4.5 volumes). The resulting reaction mixture was heated at refluxing temperature for 40 hours, monitored by GCMS. After the reaction was completed, it was diluted with ethyl acetate (20 mL) and washed with (3×10 ml) 1M sodium hydroxide solution. The organic layer was separated, dried over anhydrous magnesium sulphate and filtered. The filtrate was concentrated in vacuo to provide 1.37 g of (1α, 5α, 6α)-7-(6-amino-3-azabicyclo[3.1.0]hex-3-yl)-1-(2,4-difluorophenyl)-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acid ethyl ester, monomethanesulfonate (96% yield).
Claims (4)
1. A process for the preparation of a compound of the formula 
wherein R2 is C1–C6 alkyl, trifluoromethyl, or phenyl which may be substituted by one or more of C1–C6 alkyl, C1–C6 alkoxy, halo, nitro, amino or trifluoromethyl, and
R3 is C1–C6 alkyl,
which comprises reacting a compound of the formula 
with a compound of the formula 
2. A process comprising hydrolysis of the compound of formula VI with methanesulfonic acid, water and an organic solvent to form a monomethanesulfonic acid salt of a compound of the formula 
3. A process comprising hydrolysis of the compound of formula VI with methanesulfonic acid and R3OH wherein R3 is C1–C6 alkyl to form a monomethanesulfonic acid salt of a compound of the formula 
4. A compound of the formula 
wherein
R2 is C1–C6 trifluoromethyl, or phenyl which may be substituted by one or more of C1–C6 alkyl, C1–C6 alkoxy, halo, nitro, amino or trifluoromethyl, and
R3 is C1–C6 alkyl.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/087,756 US7019142B2 (en) | 1998-01-16 | 2002-03-04 | Process for preparing naphthyridones and intermediates |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US7160198P | 1998-01-16 | 1998-01-16 | |
| US09/236,737 US6184380B1 (en) | 1999-01-25 | 1999-01-25 | Process for preparing naphthyridones and intermediates |
| US71832400A | 2000-11-22 | 2000-11-22 | |
| US10/087,756 US7019142B2 (en) | 1998-01-16 | 2002-03-04 | Process for preparing naphthyridones and intermediates |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US71832400A Division | 1998-01-16 | 2000-11-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| US20020095043A1 US20020095043A1 (en) | 2002-07-18 |
| US7019142B2 true US7019142B2 (en) | 2006-03-28 |
Family
ID=27371924
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/087,756 Expired - Fee Related US7019142B2 (en) | 1998-01-16 | 2002-03-04 | Process for preparing naphthyridones and intermediates |
Country Status (1)
| Country | Link |
|---|---|
| US (1) | US7019142B2 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11274082B2 (en) | 2019-05-31 | 2022-03-15 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| US11458149B1 (en) | 2019-05-31 | 2022-10-04 | Ikena Oncology, Inc. | TEAD inhibitors and uses thereof |
Citations (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0230274A2 (en) | 1986-01-21 | 1987-07-29 | Bayer Ag | Derivatives of 7-(azabicycloalkyl)-quinoloncarboxylic acid and -naphthyridoncarboxylic acid |
| EP0413455A2 (en) | 1989-08-16 | 1991-02-20 | Pfizer Inc. | Azabicyclo quinolone carboxylic acids |
| US5102667A (en) | 1989-11-23 | 1992-04-07 | Rhone-Poulenc Sante | Isoindolone derivatives, their preparation and the pharmaceutical compositions containing them |
| US5164402A (en) * | 1989-08-16 | 1992-11-17 | Pfizer Inc | Azabicyclo quinolone and naphthyridinone carboxylic acids |
| WO1993018001A1 (en) | 1992-03-02 | 1993-09-16 | Pfizer Inc. | Preparation of intermediates in the synthesis of quinoline antibiotics |
| US5391763A (en) | 1990-07-11 | 1995-02-21 | Pfizer Inc. | 3-aza-bicyclo[3.1.0]hexanes which are intermediates for anti-bacterial azabicyclo quinolone carboxylic acids |
| US5475116A (en) | 1994-04-29 | 1995-12-12 | Pfizer Inc. | Aza bicyclo[3,1,0]hexane intermediates useful in the synthesis of quinolones |
| WO1997000268A1 (en) * | 1995-06-15 | 1997-01-03 | Pfizer Inc. | Process for preparing derivatives of azabicyclo naphthyridine carboxylic acid comprising a dipeptide |
| WO1997007800A1 (en) | 1995-08-29 | 1997-03-06 | Pfizer Inc. | Zwitterionic forms of trovafloxacin |
| US5623078A (en) | 1995-06-23 | 1997-04-22 | Chisso Corporation | Process for producing an intermediate of a new quinolone compound |
| WO1997019921A1 (en) | 1995-11-30 | 1997-06-05 | Pfizer Limited | Process for preparing dioxoazabicyclohexanes |
| EP0818445A1 (en) | 1996-07-09 | 1998-01-14 | Pfizer Inc. | Preparation of intermediates useful in the synthesis of quinoline antibiotics |
| US5728711A (en) | 1994-04-07 | 1998-03-17 | Pfizer Inc. | Treatment of H. pylori infections |
| US5763454A (en) | 1995-06-06 | 1998-06-09 | Pfizer, Inc. | Crystal form of anhydrous 7-( 1α,5α,6α!-6-amino-3-azabicyclo 3.1.0!hex-3-yl)-6-fluoro-1-(2,4-difluorophenyl)-1,4-dihydro-4-oxo-1,8 naphthyridine-3-carboxylic acid, methanessulfonic acid salt |
| US5847158A (en) | 1996-07-09 | 1998-12-08 | Pfizer Inc. | Process for preparing 2,4-dioxo-3-azabicyclo 3.1.0!Hexanes |
| WO1999006368A1 (en) | 1997-08-02 | 1999-02-11 | Bayer Aktiengesellschaft | Special 3-azabicyclo[3.1.0]hexanes, method for producing and modifying the same, and their use |
| EP0930297A1 (en) * | 1998-01-16 | 1999-07-21 | Pfizer Products Inc. | A process for preparing naphthyridones and intermediates |
| US5929240A (en) | 1994-12-12 | 1999-07-27 | Pfizer Inc. | Process and intermediates for preparing naphthyridonecarboxylic acid salts |
| US6066647A (en) * | 1996-07-29 | 2000-05-23 | Pfizer Inc. | Zwitterionic forms of trovafloxacin |
| US6184380B1 (en) * | 1999-01-25 | 2001-02-06 | Pfizer Inc. | Process for preparing naphthyridones and intermediates |
| US6194424B1 (en) * | 1996-04-17 | 2001-02-27 | Bayer Aktiengesellschaft | Arylacetamides and their use as medicaments |
-
2002
- 2002-03-04 US US10/087,756 patent/US7019142B2/en not_active Expired - Fee Related
Patent Citations (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0230274A2 (en) | 1986-01-21 | 1987-07-29 | Bayer Ag | Derivatives of 7-(azabicycloalkyl)-quinoloncarboxylic acid and -naphthyridoncarboxylic acid |
| SU1538897A3 (en) | 1986-01-21 | 1990-01-23 | Байер Аг (Фирма) | Method of producing derivatives of quinoline- or naphthyrydine carbolic acid or their acid-additive salts, or their hydrates |
| US5229396A (en) | 1989-08-16 | 1993-07-20 | Pfizer Inc. | Anti-bacterial azabicyclo quinolone carboxylic acids |
| WO1991002526A1 (en) | 1989-08-16 | 1991-03-07 | Pfizer Inc. | Azabicyclo quinolone carboxylic acids |
| US5164402A (en) * | 1989-08-16 | 1992-11-17 | Pfizer Inc | Azabicyclo quinolone and naphthyridinone carboxylic acids |
| EP0413455A2 (en) | 1989-08-16 | 1991-02-20 | Pfizer Inc. | Azabicyclo quinolone carboxylic acids |
| US5102667A (en) | 1989-11-23 | 1992-04-07 | Rhone-Poulenc Sante | Isoindolone derivatives, their preparation and the pharmaceutical compositions containing them |
| US5391763A (en) | 1990-07-11 | 1995-02-21 | Pfizer Inc. | 3-aza-bicyclo[3.1.0]hexanes which are intermediates for anti-bacterial azabicyclo quinolone carboxylic acids |
| WO1993018001A1 (en) | 1992-03-02 | 1993-09-16 | Pfizer Inc. | Preparation of intermediates in the synthesis of quinoline antibiotics |
| US5298629A (en) | 1992-03-02 | 1994-03-29 | Pfizer Inc. | Intermediates in the synthesis of quinoline antibiotics |
| HUT70497A (en) | 1992-03-02 | 1995-10-30 | Pfizer | Preparation of intermediates in the synthesis of quinoline antibiotics |
| US5728711A (en) | 1994-04-07 | 1998-03-17 | Pfizer Inc. | Treatment of H. pylori infections |
| US5475116A (en) | 1994-04-29 | 1995-12-12 | Pfizer Inc. | Aza bicyclo[3,1,0]hexane intermediates useful in the synthesis of quinolones |
| US5929240A (en) | 1994-12-12 | 1999-07-27 | Pfizer Inc. | Process and intermediates for preparing naphthyridonecarboxylic acid salts |
| US5763454A (en) | 1995-06-06 | 1998-06-09 | Pfizer, Inc. | Crystal form of anhydrous 7-( 1α,5α,6α!-6-amino-3-azabicyclo 3.1.0!hex-3-yl)-6-fluoro-1-(2,4-difluorophenyl)-1,4-dihydro-4-oxo-1,8 naphthyridine-3-carboxylic acid, methanessulfonic acid salt |
| WO1997000268A1 (en) * | 1995-06-15 | 1997-01-03 | Pfizer Inc. | Process for preparing derivatives of azabicyclo naphthyridine carboxylic acid comprising a dipeptide |
| US5623078A (en) | 1995-06-23 | 1997-04-22 | Chisso Corporation | Process for producing an intermediate of a new quinolone compound |
| WO1997007800A1 (en) | 1995-08-29 | 1997-03-06 | Pfizer Inc. | Zwitterionic forms of trovafloxacin |
| WO1997019921A1 (en) | 1995-11-30 | 1997-06-05 | Pfizer Limited | Process for preparing dioxoazabicyclohexanes |
| US6194424B1 (en) * | 1996-04-17 | 2001-02-27 | Bayer Aktiengesellschaft | Arylacetamides and their use as medicaments |
| EP0818445A1 (en) | 1996-07-09 | 1998-01-14 | Pfizer Inc. | Preparation of intermediates useful in the synthesis of quinoline antibiotics |
| US5847158A (en) | 1996-07-09 | 1998-12-08 | Pfizer Inc. | Process for preparing 2,4-dioxo-3-azabicyclo 3.1.0!Hexanes |
| US6066647A (en) * | 1996-07-29 | 2000-05-23 | Pfizer Inc. | Zwitterionic forms of trovafloxacin |
| WO1999006368A1 (en) | 1997-08-02 | 1999-02-11 | Bayer Aktiengesellschaft | Special 3-azabicyclo[3.1.0]hexanes, method for producing and modifying the same, and their use |
| EP0930297A1 (en) * | 1998-01-16 | 1999-07-21 | Pfizer Products Inc. | A process for preparing naphthyridones and intermediates |
| US6184380B1 (en) * | 1999-01-25 | 2001-02-06 | Pfizer Inc. | Process for preparing naphthyridones and intermediates |
Non-Patent Citations (4)
| Title |
|---|
| Braish et al., "Construction of the (1 alpha, 5 alpha, 6 alpha)-6-amino-3-azabicyclo[3.1.0]hexane Ring System," Synlett, 11, pp. 1100-1102 (1991). |
| Bundgaard, et al., Design of prodrugs, Elsevier, pp. 27-30. * |
| Kiso, et. al. , "A Flouride Ion Deprotection strategy in Peptide synthesis, combination with selective deprotection using the Dilute Methanesulfonic acid of alpha-amino protecting groups", Col. 36 (1988), Chem. Pharm. Communications, pp. 5024-5027. * |
| US 1998-7160 P, Patent Application, "Process for preparing napthyridones and intermediates", Chiu et. al., pp. 1-7. * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11274082B2 (en) | 2019-05-31 | 2022-03-15 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| US11458149B1 (en) | 2019-05-31 | 2022-10-04 | Ikena Oncology, Inc. | TEAD inhibitors and uses thereof |
| US11760728B2 (en) | 2019-05-31 | 2023-09-19 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| US11925651B2 (en) | 2019-05-31 | 2024-03-12 | Ikena Oncology, Inc. | TEAD inhibitors and uses thereof |
| US12577208B2 (en) | 2019-05-31 | 2026-03-17 | EHE Foundation | TEAD inhibitors and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US20020095043A1 (en) | 2002-07-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6184380B1 (en) | Process for preparing naphthyridones and intermediates | |
| US7166721B2 (en) | Preparation of 1H-imidazo[4,5-C] quinolin 4-amines via novel 1H-imidazo[4,5-c] quinolin 4-cyano and 1H-imidazo[4,5-c] quinolin 4-carboxamide intermediates | |
| US20070060755A1 (en) | Preparation of 1H-imidazo [4,5-C] quinolin-4-amines via 1H-imidazo [4,5-C] quinolin-4-phthalimide intermediates | |
| EP0132845A2 (en) | Novel 1,8-Naphthyridine derivatives, and process for preparation thereof | |
| EP0297858A2 (en) | Bridged-diazabicycloalkyl quinolone carboxylic acids and esters | |
| EP0695755A1 (en) | Pyrrolocarbazoles | |
| US7019142B2 (en) | Process for preparing naphthyridones and intermediates | |
| AU694149B2 (en) | Process and intermediates for preparing naphthyridonecarboxylic acid salts | |
| EP0930297B1 (en) | A process for preparing naphthyridones and intermediates | |
| EP0812838A1 (en) | Pyridonecarboxylic acid derivative substituted by bicyclic amino group, ester thereof, salt thereof, and bicyclic amine as intermediate therefor | |
| US6114531A (en) | Process for preparing quinolone and naphthyridone carboxylic acids | |
| US11414386B2 (en) | Process for the preparation of ivacaftor and its intermediates | |
| US6080756A (en) | Polymorphs of the prodrug 6-N-(L-ALA-L-ALA)-trovafloxacin | |
| US5929240A (en) | Process and intermediates for preparing naphthyridonecarboxylic acid salts | |
| US6359137B1 (en) | Process for preparing trovafloxacin acid salts | |
| US7834027B2 (en) | Gemifloxacin process and polymorphs | |
| CZ11999A3 (en) | Process for preparing naphthyridones and intermediates for such preparation process | |
| HK1019879A (en) | A process for preparing naphthyridones and intermediates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| REMI | Maintenance fee reminder mailed | ||
| LAPS | Lapse for failure to pay maintenance fees | ||
| STCH | Information on status: patent discontinuation |
Free format text: PATENT EXPIRED DUE TO NONPAYMENT OF MAINTENANCE FEES UNDER 37 CFR 1.362 |
|
| FP | Lapsed due to failure to pay maintenance fee |
Effective date: 20100328 |