US20040219565A1 - Oligonucleotides useful for detecting and analyzing nucleic acids of interest - Google Patents
Oligonucleotides useful for detecting and analyzing nucleic acids of interest Download PDFInfo
- Publication number
- US20040219565A1 US20040219565A1 US10/690,487 US69048703A US2004219565A1 US 20040219565 A1 US20040219565 A1 US 20040219565A1 US 69048703 A US69048703 A US 69048703A US 2004219565 A1 US2004219565 A1 US 2004219565A1
- Authority
- US
- United States
- Prior art keywords
- nucleic acid
- nucleic acids
- population
- lna
- exon
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 0 *CC1(C*)OC(*)[C@@H](C)C1C.*CC1(C*)O[C@@H](N2C=C([5*])C(=O)NC2=S)[C@@H](C)C1C.*C[C@]12CO[C@@H](C1C)[C@H](N1C=C([5*])C(=O)NC1=S)O2.I.II.I[IH]I.[5*]C1=CNC(=S)NC1=O.[V]I Chemical compound *CC1(C*)OC(*)[C@@H](C)C1C.*CC1(C*)O[C@@H](N2C=C([5*])C(=O)NC2=S)[C@@H](C)C1C.*C[C@]12CO[C@@H](C1C)[C@H](N1C=C([5*])C(=O)NC1=S)O2.I.II.I[IH]I.[5*]C1=CNC(=S)NC1=O.[V]I 0.000 description 18
- AYHOMQFLDNKCMT-NHWZRCRPSA-N C=NP(OCCC#N)OC1[C@@H]2OC[C@]1(CC)O[C@H]2N1C=NC2=C1N=C(/N=C/N(C)C)NC2.CCC.CC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=C(N)N=C3Cl)O2.CN(C)/C=N/C1=NC2=C(C=N1)N=CN2[C@@H]1O[C@@]2(CO)CO[C@H]1C2O.NC1=NC2=C(C=N1)N=CN2[C@@H]1O[C@@]2(CO)CO[C@H]1C2O Chemical compound C=NP(OCCC#N)OC1[C@@H]2OC[C@]1(CC)O[C@H]2N1C=NC2=C1N=C(/N=C/N(C)C)NC2.CCC.CC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=C(N)N=C3Cl)O2.CN(C)/C=N/C1=NC2=C(C=N1)N=CN2[C@@H]1O[C@@]2(CO)CO[C@H]1C2O.NC1=NC2=C(C=N1)N=CN2[C@@H]1O[C@@]2(CO)CO[C@H]1C2O AYHOMQFLDNKCMT-NHWZRCRPSA-N 0.000 description 2
- JXNHNPYIAGOKDO-UHFFFAOYSA-N C#CC1=CN(C2OC3(CC)COC2C3C)C(=O)NC1=O.CCC12COC(C(N3C=C(I)C(=O)NC3=O)O1)C2C.CCC12COC(C(N3C=C4C=COC4NC3=O)O1)C2O.CCC12COC(C(N3C=C4C=COC4NC3=O)O1)C2OP(OCCC#N)N(C(C)C)C(C)C.CCC12COC(C(N3C=CC(=O)NC3=O)O1)C2C.O=C1NC2OC=CC2=CN1C1OC2(CO)COC1C2O Chemical compound C#CC1=CN(C2OC3(CC)COC2C3C)C(=O)NC1=O.CCC12COC(C(N3C=C(I)C(=O)NC3=O)O1)C2C.CCC12COC(C(N3C=C4C=COC4NC3=O)O1)C2O.CCC12COC(C(N3C=C4C=COC4NC3=O)O1)C2OP(OCCC#N)N(C(C)C)C(C)C.CCC12COC(C(N3C=CC(=O)NC3=O)O1)C2C.O=C1NC2OC=CC2=CN1C1OC2(CO)COC1C2O JXNHNPYIAGOKDO-UHFFFAOYSA-N 0.000 description 1
- YYTZOFULEUZFGR-BOOVNFMYSA-N C.C.C.C.CC(=O)O[C@H]1C(OCC2=CC=CC=C2)[C@](COCC2=CC=CC=C2)(COS(C)(=O)=O)O[C@@H]1OC(C)=O.CC(=O)O[C@H]1C(OCC2=CC=CC=C2)[C@](COCC2=CC=CC=C2)(COS(C)(=O)=O)O[C@H]1N1C=C(C)C(=O)NC1=S.CC1=CC=C(C(=O)N2C(=O)C(C)=CN([C@@H]3O[C@@]4(COCC5=CC=CC=C5)CO[C@H]3C4OCC3=CC=CC=C3)C2=S)C=C1.CC1=CC=C(C(=O)OC2=NC(=S)N([C@@H]3O[C@@]4(COCC5=CC=CC=C5)CO[C@H]3C4OCC3=CC=CC=C3)C=C2C)C=C1.CC1=CN([C@@H]2O[C@@]3(COCC4=CC=CC=C4)CO[C@H]2C3OCC2=CC=CC=C2)C(=S)NC1=O Chemical compound C.C.C.C.CC(=O)O[C@H]1C(OCC2=CC=CC=C2)[C@](COCC2=CC=CC=C2)(COS(C)(=O)=O)O[C@@H]1OC(C)=O.CC(=O)O[C@H]1C(OCC2=CC=CC=C2)[C@](COCC2=CC=CC=C2)(COS(C)(=O)=O)O[C@H]1N1C=C(C)C(=O)NC1=S.CC1=CC=C(C(=O)N2C(=O)C(C)=CN([C@@H]3O[C@@]4(COCC5=CC=CC=C5)CO[C@H]3C4OCC3=CC=CC=C3)C2=S)C=C1.CC1=CC=C(C(=O)OC2=NC(=S)N([C@@H]3O[C@@]4(COCC5=CC=CC=C5)CO[C@H]3C4OCC3=CC=CC=C3)C=C2C)C=C1.CC1=CN([C@@H]2O[C@@]3(COCC4=CC=CC=C4)CO[C@H]2C3OCC2=CC=CC=C2)C(=S)NC1=O YYTZOFULEUZFGR-BOOVNFMYSA-N 0.000 description 1
- SURMSSUCDLDELL-YQYKSQLLSA-N C=NP(OCCC#N)OC1[C@@H]2OC[C@]1(CC)O[C@H]2N1C=NC2=C1N=C(NC(=O)C1=CC=CC=C1)N=C2NC(=O)C1=CC=CC=C1.CC(=O)O[C@H]1C(OCC2=CC=CC=C2)C(COS(C)(=O)=O)(COS(C)(=O)=O)O[C@H]1N1C=NC2=C1N=C(N)N=C2Cl.CCC.CC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=C(N)N=C3Cl)O2.CC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=C(N)N=C3N=[N+]=[N-])O2.NC1=NC2=C(N=CN2[C@@H]2O[C@@]3(CO)CO[C@H]2C3O)C(N)=N1.O=C(NC1=NC2=C(N=CN2[C@@H]2O[C@@]3(CO)CO[C@H]2C3O)C(NC(=O)C2=CC=CC=C2)=N1)C1=CC=CC=C1 Chemical compound C=NP(OCCC#N)OC1[C@@H]2OC[C@]1(CC)O[C@H]2N1C=NC2=C1N=C(NC(=O)C1=CC=CC=C1)N=C2NC(=O)C1=CC=CC=C1.CC(=O)O[C@H]1C(OCC2=CC=CC=C2)C(COS(C)(=O)=O)(COS(C)(=O)=O)O[C@H]1N1C=NC2=C1N=C(N)N=C2Cl.CCC.CC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=C(N)N=C3Cl)O2.CC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=C(N)N=C3N=[N+]=[N-])O2.NC1=NC2=C(N=CN2[C@@H]2O[C@@]3(CO)CO[C@H]2C3O)C(N)=N1.O=C(NC1=NC2=C(N=CN2[C@@H]2O[C@@]3(CO)CO[C@H]2C3O)C(NC(=O)C2=CC=CC=C2)=N1)C1=CC=CC=C1 SURMSSUCDLDELL-YQYKSQLLSA-N 0.000 description 1
- IUSMJJWKRKWFBO-MGNYIMMYSA-N CC(=O)O[C@H]1C(OCC2=CC=CC=C2)C(COS(C)(=O)=O)(COS(C)(=O)=O)O[C@H]1N1C=NC2=C1N=C(N)N=C2Cl.CC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=C(N)N=C3Cl)O2.CC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=C(N)N=C3N=[N+]=[N-])O2.CC[C@]12CO[C@@H](C1OP(N=[Pr])OCCC#N)[C@H](N1C=NC3=C1N=C(NC(=O)C1=CC=CC=C1)N=C3NC(=O)C1=CC=CC=C1)O2.NC1=NC2=C(N=CN2[C@@H]2O[C@@]3(CO)CO[C@H]2C3O)C(N)=N1.O=C(NC1=NC2=C(N=CN2[C@@H]2O[C@@]3(CO)CO[C@H]2C3O)C(NC(=O)C2=CC=CC=C2)=N1)C1=CC=CC=C1.[Pr] Chemical compound CC(=O)O[C@H]1C(OCC2=CC=CC=C2)C(COS(C)(=O)=O)(COS(C)(=O)=O)O[C@H]1N1C=NC2=C1N=C(N)N=C2Cl.CC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=C(N)N=C3Cl)O2.CC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=C(N)N=C3N=[N+]=[N-])O2.CC[C@]12CO[C@@H](C1OP(N=[Pr])OCCC#N)[C@H](N1C=NC3=C1N=C(NC(=O)C1=CC=CC=C1)N=C3NC(=O)C1=CC=CC=C1)O2.NC1=NC2=C(N=CN2[C@@H]2O[C@@]3(CO)CO[C@H]2C3O)C(N)=N1.O=C(NC1=NC2=C(N=CN2[C@@H]2O[C@@]3(CO)CO[C@H]2C3O)C(NC(=O)C2=CC=CC=C2)=N1)C1=CC=CC=C1.[Pr] IUSMJJWKRKWFBO-MGNYIMMYSA-N 0.000 description 1
- KLZGXWAFTKTAQP-FFWOAPSTSA-N CC[C@@]12CO[C@H](C(OC)O1)C2OCC1=CC=C(OC)C=C1.CC[C@]1(COCC2=CC=C(OC)C=C2)O[C@@H]2OC(C)(C)O[C@H]2C1OCC1=CC=C(OC)C=C1.COC1=CC=C(COC2[C@@H]3OC(C)(C)O[C@H]3OC2(CO)CO)C=C1.COC1=CC=C(COC2[C@@H]3OC(C)(C)O[C@H]3OC2(COS(C)(=O)=O)COS(C)(=O)=O)C=C1.COC1=CC=C(COC2[C@H](O)C(OC)OC2(COS(C)(=O)=O)COS(C)(=O)=O)C=C1.COC1=CC=C(COC[C@@]2(COS(C)(=O)=O)OC(OC)[C@@H](O)C2OCC2=CC=C(OC)C=C2)C=C1.COC1=CC=C(COC[C@@]23CO[C@H](C(OC)O2)C3OCC2=CC=C(OC)C=C2)C=C1 Chemical compound CC[C@@]12CO[C@H](C(OC)O1)C2OCC1=CC=C(OC)C=C1.CC[C@]1(COCC2=CC=C(OC)C=C2)O[C@@H]2OC(C)(C)O[C@H]2C1OCC1=CC=C(OC)C=C1.COC1=CC=C(COC2[C@@H]3OC(C)(C)O[C@H]3OC2(CO)CO)C=C1.COC1=CC=C(COC2[C@@H]3OC(C)(C)O[C@H]3OC2(COS(C)(=O)=O)COS(C)(=O)=O)C=C1.COC1=CC=C(COC2[C@H](O)C(OC)OC2(COS(C)(=O)=O)COS(C)(=O)=O)C=C1.COC1=CC=C(COC[C@@]2(COS(C)(=O)=O)OC(OC)[C@@H](O)C2OCC2=CC=C(OC)C=C2)C=C1.COC1=CC=C(COC[C@@]23CO[C@H](C(OC)O2)C3OCC2=CC=C(OC)C=C2)C=C1 KLZGXWAFTKTAQP-FFWOAPSTSA-N 0.000 description 1
- UUFMPTIQSAIWGZ-UHFFFAOYSA-N CN1C=NC2=C1N=C(N)N=C2Cl.CN1C=NC2=C1N=C(N)N=C2N.NC1=NC2=C(N=CN2)C(Cl)=N1 Chemical compound CN1C=NC2=C1N=C(N)N=C2Cl.CN1C=NC2=C1N=C(N)N=C2N.NC1=NC2=C(N=CN2)C(Cl)=N1 UUFMPTIQSAIWGZ-UHFFFAOYSA-N 0.000 description 1
- AGFVIZJFENOJDQ-LZANACOHSA-K COC[C@H]1O[C@@H](C)CC1OP(C)(=O)[O-].COC[C@]12CCO[C@@H](C1OP(C)(=O)[O-])[C@H](C)O2.COC[C@]12CO[C@@H](C1OP(C)(=O)[O-])[C@H](C)O2 Chemical compound COC[C@H]1O[C@@H](C)CC1OP(C)(=O)[O-].COC[C@]12CCO[C@@H](C1OP(C)(=O)[O-])[C@H](C)O2.COC[C@]12CO[C@@H](C1OP(C)(=O)[O-])[C@H](C)O2 AGFVIZJFENOJDQ-LZANACOHSA-K 0.000 description 1
- OYVWBARBSYNNKR-ZIYWRUAHSA-K COC[C@]12CO[C@@H](C1OP(C)(=O)[O-])[C@H](N1C=NC3=C1N=C(N)N=C3)O2.COC[C@]12CO[C@@H](C1OP(C)(=O)[O-])[C@H](N1C=NC3=C1N=C(N)N=C3N)O2.COC[C@]12CO[C@@H](C1OP(C)(=O)[O-])[C@H](N1C=NC3=C1N=CNC3=O)O2 Chemical compound COC[C@]12CO[C@@H](C1OP(C)(=O)[O-])[C@H](N1C=NC3=C1N=C(N)N=C3)O2.COC[C@]12CO[C@@H](C1OP(C)(=O)[O-])[C@H](N1C=NC3=C1N=C(N)N=C3N)O2.COC[C@]12CO[C@@H](C1OP(C)(=O)[O-])[C@H](N1C=NC3=C1N=CNC3=O)O2 OYVWBARBSYNNKR-ZIYWRUAHSA-K 0.000 description 1
- DVNKHDWKDZKTBO-BKHPIQMCSA-N NC1=NC=NC2=C1N=CN2[C@@H]1O[C@@]2(CO)CO[C@H]1C2OCC1=CC=CC=C1.O=C(OC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=CN=C3Cl)O2)C1=CC=CC=C1.O=C1NC=NC2=C1N=CN2[C@@H]1O[C@@]2(COC(=O)C3=CC=CC=C3)CO[C@H]1C2OCC1=CC=CC=C1 Chemical compound NC1=NC=NC2=C1N=CN2[C@@H]1O[C@@]2(CO)CO[C@H]1C2OCC1=CC=CC=C1.O=C(OC[C@]12CO[C@@H](C1OCC1=CC=CC=C1)[C@H](N1C=NC3=C1N=CN=C3Cl)O2)C1=CC=CC=C1.O=C1NC=NC2=C1N=CN2[C@@H]1O[C@@]2(COC(=O)C3=CC=CC=C3)CO[C@H]1C2OCC1=CC=CC=C1 DVNKHDWKDZKTBO-BKHPIQMCSA-N 0.000 description 1
Images
















































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H23/00—Compounds containing boron, silicon, or a metal, e.g. chelates, vitamin B12
Definitions
- the invention relates to nucleic acids and methods for expression profiling of mRNAs, identifying and profiling of particular mRNA splice variants, and detecting mutations, deletions, or duplications of particular exons, e.g., alterations associated with a disease such as cancer, in a nucleic acid sample, e.g., a patient sample.
- the invention furthermore relates to methods for detecting nucleic acids by fluorescence in situ hybridization.
- the field of the invention is oligonucleotides (e.g., oligonucleotide arrays) that are useful for detecting nucleic acids of interest and for detecting differences between nucleic acid samples (e.g, such as samples from a cancer patient and a healthy patient).
- oligonucleotides e.g., oligonucleotide arrays
- nucleic acid samples e.g, such as samples from a cancer patient and a healthy patient.
- DNA chip technology utilizes minituarized arrays of DNA molecules immobilized on solid surfaces for biochemical analyses.
- the power of DNA microarrays as experimental tools relies on the specific molecular recognition via complementary base-pairing, which makes them highly useful for massive parallel analyses.
- microarray technology has thus become the method of choice for many hybridization-based assays, such as expression profiling, SNP detection, DNA re-sequencing, and genotyping on a genomic scale.
- Expression microarrays are capable of profiling gene expression patterns of tens of thousands of genes in a single experiment. Hence, this technology provides a powerful tool for deciphering complex biological systems, and thereby greatly facilitates research in basic biology and living processes, as well as disease diagnostics, theranostics, and drug development.
- the mRNAs are distributed in three frequency classes: (i) superprevalent (10-20% of the total mRNA mass); (ii) intermediate (40-45%); and (iii) low-abundant mRNAs (40-45%). It is therefore of utmost importance that the dynamic range and sensitivity of the expression arrays are optimal, especially when analyzing expression levels of messages or mRNA splice variants belonging to the low-abundant class.
- oligonucleotide microarray manufacturing involves DNA synthesis on solid surfaces using combinatorial chemistry. Most of the current technology is developed by Affymetrix and Rosetta Inpharmatics. Glass is currently preferred as the synthesis support because of its inert chemical properties and low level of intrinsic fluorescence as well as the ability to chemically derivatize the surface. Of the three approaches currently used to manufacture oligonucleotide arrays, the light-directed deprotection method is the most effective one in generating high density microchips. A single round of synthesis involves light-directed deprotection, followed by nucleotide coupling. Photolithographic masking is used to control the regions of the chip designated for illumination.
- Affynietrix uses a combination of photolithography and combinatorial chemistry to manufacture its GeneChip Arrays.
- GeneChip manufacturing begins with a 5-inch square quartz wafer. Initially the quartz is washed to ensure uniform hydroxylation across its surface. The wafer is placed in a bath of silane, which reacts with the hydroxyl groups of the quartz and forms a matrix of covalently linked molecules. The distance between these silane molecules determines the probes' packing density, allowing arrays to hold over 500,000 features within 1.28 square centimeters.
- the principal disadvantage of this method is that a significant amount of chip design work and cost is associated with the mask design.
- oligonucleotide arrays available from Affymetrix are in the range of 5-10 fold more expensive than cDNA microarrays.
- DNA-DNA hybridization using oligonucleotide chips is clearly different from that of cDNA microarrays.
- Hybridizations involving oligos are much more sensitive to the GC content of individual heteroduplexes.
- single base mismatches have a pronounced effect on the hybridization reassociation of short oligos, and point mutations can thus be readily detected using oligo chips.
- cDNA microarrays containing large DNA segments such as cDNAs are generated by physically depositing small amounts of each DNA of interest onto known locations on glass surfaces.
- Two technologies for printing microarrays are (1) mechanical microspotting, and (2) ink-jetting.
- Mechanical microspotting has been extensively used at, e.g., Stanford University, and it utilizes pins or capillaries to deposit small quantities of DNA onto known addresses using motion control systems.
- Recent advances in microspotting technology using modem arraying robots allow for the preparation of 100 microarrays containing over 10,000 features in less than 12 hours.
- a DNA arrayer is relatively easy to set up, and the cost is usually low compared to on-chip oligoarrayers.
- cDNA microarrays are capable of profiling gene expression patterns of tens of thousands of genes in a single experiment.
- the two samples are first labeled using two different fluorescent dyes such as Cy-3 and Cy-5.
- the labeled samples are mixed and hybridized to the clones on the array slide.
- laser excitation of the incorporated, fluorescent target molecules yields an emission with a characteristic spectra, which is measured with a confocal laser scanner.
- the monochrome images from the scanner are imported to the software in which the images are pseudo-colored and merged. Data from a single hybridization is viewed as a normalized ratio in which significant deviations from the ratio are indicative of either increased or decreased expression levels relative to the reference sample. Data from multiple experiments can be examined using any number of data mining tools.
- cDNA microarrays originally developed by Pat Brown and co-workers at the Stanford University, are sensitive, but may not be sufficiently specific with respect to, e.g., discrimination of homologous transcripts in gene families and alternatively spliced isoforms.
- the Affymetrix GeneChip system is specific, but may not be sensitive enough. This lack of sensitivity may explain why Affymetrix uses 1 6 ⁇ 26-mer perfect match capture probes together with 1 6 ⁇ 25-mer mismatch probes per transcript in its expression profiling chips resulting in enormous data sets in genome-wide arrays.
- the functional genomics field is in the process of switching, as they run out of samples, from existing PCR-amplified cDNA fragment libraries for microarraying to custom longmer oligonucleotide arrays comprising transcript-specific oligonucleotide capture probes typically in the range of 30-mers to 80-mers, thus addressing both specificity and sensitivity.
- RNA splicing not only provides functional mRNA, but is also responsible for generating additional diversity.
- RNA-mediated gene regulation is widespread in higher eukaryotes and complex genetic phenomena like RNA interference, co-suppression, transgene silencing, imprinting, methylation, and possibly position-effect variegation and transvection, all involve intersecting pathways based on or connected to RNA signalling (Mattick 2001; EMBO reports 2, 11: 986-991). Recent studies indicate that antisense transcription is a very common phenomenon in the mouse and human genomes (Okazaki et al. 2002; Nature 420: 563-573; Yelin et al. 2003, Nature Biotechnol.). Thus, antisense modulation of gene expression in e.g. human cells may be a common regulatory mechanism.
- the present invention provides novel tools, in which non-naturally occurring nucleic acids, such as LNA oligonucleotides, can be designed to silence or modulate the regulation of a given mRNA by non-coding antisense RNA, by designing a complementary sense LNA oligonucleotide for the regulatory antisense RNA.
- This has a high potential in target identification, target validation and therapeutic use of LNA oligonucleotides as modulating and silencing sense nucleic acid agents. Misplaced control of alternative splicing can cause disease
- transcripts The detection of the detailed structure of all transcripts is an important goal for molecular characterization of a cell or tissue. Without the ability to detect and quantify the splice variants present in one tissue, the transcript content or the protein content cannot be described accurately. Molecular medical research shows that many cancers result in altered levels of splice variants, so an accurate method to detect and quantify these transcripts is required. Mutations that produce an aberrant splice form can also be the primary cause of such severe diseases such as spinal muscular dystrophy and cystic fibrosis.
- Desirable methods can distinguish between mRNA splice variants and quantitate the amount of each variant in a sample.
- Other desirable methods can detect differences in expressions patterns between patient nucleic acid samples and nucleic acid standards.
- the present invention demonstrates the usefulness of LNA-modified oligonucleotides in the construction of highly specific and sensitive microarrays for expression profiling (e.g., mRNA splice variant detection) and comparative genomic hybridization.
- the invention provides novel technology platforms based on nucleic acids with LNA or other high affinity nucleotides for sensitive and specific assessment of alternative splicing using microarray technology.
- LNA microarrays are able to discriminate between highly homologous as well as differentially spliced transcripts.
- the invention furthermore provides methods for highly sensitive and specific nucleic acid detection by fluorescence in situ hybridization using LNA-modified oligonucleotides.
- the present methods greatly facilitate the analysis of gene expression patterns from a particular species, tissue, cell type.
- the analysis of the human spliceome provides important information for pharmacogenetics.
- the present methods are highly valuable in medical research and diagnostics as well as in drug development and toxicological studies.
- the invention features populations of high affinity nucleic acids that have duplex stabilizing properties and thus are useful for a variety of nucleic acid detection, amplification, and hybridization methods (e.g., expression or mRNA splice variant profiling).
- Some of these oligonucleotides contain novel nucleotides created by combining specialized synthetic nucleobases with an LNA backbone, thus creating high affinity oligonucleotides with specialized properties such as reduced sequence discrimination for the complementary strand or reduced ability to form intramolecular double stranded structures.
- the invention also provides improved methods for identifying nucleic acids in a sample and for classifying a nucleic acid sample by comparing its pattern of hybridization to an array to the corresponding pattern of hybridization of one or more standards to the array (e.g., comparative genomic hybridization).
- Other desirable modified bases have decreased ability to self-anneal or to form duplexes with oligonucleotides containing one or more modified bases.
- the invention also provides arrays of nucleic acids containing these modified bases that have a decreased variance in melting temperature and/or an increased capture efficiency compared to naturally-occurring nucleic acids. These arrays as well as the oligonucleotides in solution can be used in a variety of applications for the detection, characterization, identification, and/or amplification of one or more target nucleic acids. These oligonucleotides and oligonucleotides of the invention in general can also be used for solution assays, such as homogeneous assays.
- the invention features a non-naturally-occurring nucleic acid with a melting temperature that is at least 3, 5, 8, 10, 12, 15, 20, 25, 30, 35, or 40° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides.
- the nucleic acid hybridizes to a first region within a first exon of a target nucleic acid and to a second region within a second exon of the target nucleic acid that is adjacent to the first exon.
- the invention provides a non-naturally-occurring nucleic acid with a melting temperature that is at least 3, 5, 8, 10, 12, 15, 20, 25, 30, 35, or 4 0 ° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides.
- the nucleic acid hybridizes to a first region within an exon of a target nucleic acid and to a second region within an intron of the target nucleic acid that is adjacent to the exon.
- the invention features a non-naturally-occurring nucleic acid with a melting temperature that is at least 3, 5, 8, 10, 12, 15, 20, 25, 30, 35, or 40° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides.
- the nucleic acid hybridizes to a first region within a first intron of a target nucleic acid and to a second region within a second intron of the target nucleic acid that is adjacent to the first intron.
- the invention provides a nucleic acid that is a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10, 25, 50, 100, 150, 200, 500, 800, 1000, or 1200% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of the nucleic acid.
- the nucleic acid hybridizes to a first region within a first exon of a target nucleic acid and to a second region within a second exon of the target nucleic acid that is adjacent to the first exon.
- the invention features a nucleic acid that is a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10, 25, 50, 100, 150, 200, 500, 800, 1000, or 1200% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of the nucleic acid.
- the nucleic acid hybridizes to a first region within an exon of a target nucleic acid and to a second region within an intron of the target nucleic acid that is adjacent to the exon.
- the invention provides a nucleic acid that is a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10, 25, 50, 100, 150, 200, 500, 800, 1000, or 1200% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of the nucleic acid.
- the nucleic acid hybridizes to a first region within a first intron of a target nucleic acid and to a second region within a second intron of the target nucleic acid that is adjacent to the first intron.
- the nucleic acids of the invention featuring a non-naturally occurring nucleic acid exhibit increased duplex stability due to slower rates of dissociation of the nucleic acid complexes (the off-rate) (Christensen et al. 2001, Biochem. J. 354: 481-484).
- the structure of desirable adenosine, thymine, guanine and cytosine analogs are those disclosed in PCT Publication No. WO 97/12896, Formula 5, 6, 7, 8, 9, 10, 11, 12 and 13. These modified bases may be incorporated as part of an LNA, DNA, or RNA unit and used any of the oligomers of the invention.
- the invention features a nucleic acid that is an LNA (i.e., a nucleic acids with one or more LNA units) and that hybridizes to a first region within a first exon of a target nucleic acid and to a second region within a second exon of the target nucleic acid that is adjacent to the first exon.
- LNA i.e., a nucleic acids with one or more LNA units
- the invention features a nucleic acid that is an LNA and that hybridizes to a first region within an exon of a target nucleic acid and to a second region within an intron of the target nucleic acid that is adjacent to the exon.
- the invention provides nucleic acid that is an LNA and that hybridizes to a first region within a first intron of a target nucleic acid and to a second region within a second intron of the target nucleic acid that is adjacent to the first intron.
- the length of the segment of the nucleic acid hybridizing to the first region and the length of the segment of the nucleic acid hybridizing to the second region are between 3 and 50 nucleotides, 10 and 40 nucleotides, or 20 and 30 nucleotides, inclusive.
- the length of the segment of the nucleic acid hybridizing to the first region and the length of the segment of the nucleic acid hybridizing to the second region may be the same length or different lengths.
- the nucleic acid containing LNA units are symmetrically spaced on both sides of a junction between either two exons, an exon and an intron, or two introns, or alternatively, the nucleic acid containing LNA units are spaced on both sides of a junction based on equalized duplex melting temperatures of the segments.
- the nucleic acid has one or more LNA units within 5, 4, 3, 2, or 1 nucleotides of a junction between either two exons, an exon and an intron, or two introns.
- the invention features a population of nucleic acids that includes one or more nucleic acids of any one of the above aspects.
- the invention features a non-naturally-occurring nucleic acid with a melting temperature that is at least 3, 5, 8, 10, 12, 15, 20, 25, 30, 35, or 40° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides.
- the nucleic acid hybridizes to only one exon or to only one intron of a target nucleic acid.
- the invention features a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10, 25, 50, 100, 150, 200, 500, 800, 1000, or 1200% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of the nucleic acid.
- the nucleic acid hybridizes to only one exon or to only one intron of a target nucleic acid.
- the invention features a nucleic acid that is an LNA and that hybridizes to only one exon or to only one intron of a target nucleic acid.
- nucleic acid does not hybridize to both an exon and an intron.
- the invention features a population of nucleic acids that includes one or more nucleic acids of any one of the above aspects.
- the invention features a pharmaceutical composition that includes one or more of the nucleic acids of the invention and a pharmaceutically acceptable carrier, such as one of the carriers described herein.
- the invention features a population of two or more nucleic acids of the invention.
- the populations of nucleic acids of the invention may contain any number of unique molecules.
- the population may contain as few as 10, 10 2, 10 4 , or 10 5 unique molecules or as many as 107, 108, 109 or more unique molecules.
- at least 1, 5, 10, 50, 100 or more of the polynucleotide sequences are a non-naturally-occurring sequence.
- at least 20, 40, or 60% of the unique polynucleotide sequences are non-naturally-occurring sequences.
- the nucleic acids are all the same length; however, some of the molecules may differ in length.
- the length of one or more nucleic acids is between 15 and 150 nucleotides, 5 and 100 nucleotides, 20 and 80 nucleotides, or 30 and 60 nucleotides in length, inclusive.
- the nucleic acid is 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 30, 40, 50 nucleotides or at least 60, 70, 80, 90, 100, 120, or 130 nucleotides in length.
- the nucleic acid is between 8 and 40 nucleotides, such as between 9 and 30, or 12 and 25, or 15 and 20 nucleotides.
- At least 5, 10, 15, 20, 30, 40, 50, 60, or 70% of the nucleotides in the nucleic acid are LNA units.
- every second nucleotide, every third, every fourth nucleotide, every fifth nucleotide, or every sixth nucleotide in the nucleic acid is an LNA unit.
- every second and every third nucleotide, (ii) every second and every fourth nucleotide, (iii) every second and every fifth nucleotide, (iv) every second and every sixth nucleotide, (v) every third and every fourth nucleotide, (vi) every third and every fifth nucleotide, (vii) every third and every sixth nucleotide, (viii) every fourth and every fifth nucleotide, (ix) every fourth and every sixth nucleotide, and/or (x) every fifth and every sixth nucleotide in the nucleic acid is an LNA unit.
- every second, every third, and every fourth nucleotide in the nucleic acid is an LNA unit.
- the nucleic acids of the invention have one or more of the following substitution patterns which is repeated throughout the nucleic acids: XxXx, xXxX, XxxXxx, xXxxXx, xxXxxX, XxxxXxxx, xXxxxXxx, xxXxxxXx, or xxxXxxxX in which “X” denotes an LNA unit and “x” denotes a DNA or RNA unit.
- the nucleotides that are not LNA units are naturally-occuring DNA or RNA nucleotides.
- the nucleic acid comprises two or more contiguous LNA units. Desirably, the nucleic acid comprises at least 2, 3, 4, 5, 6, 7, or 8 contiguous LNA units. In desirable embodiments, the number of contiguous LNA units is between 5 and 20% or 10 and 15% of the total length of the nucleic acid. In a particular embodiment, 5 contiguous nucleotides of a 50-mer merged probe are LNA units. In one embodiment, the nucleic acid does not have greatly extended stretches of modified DNA or RNA residues, e.g. greater than about 4, 5, 6, 7, or 8 consecutive modified DNA or RNA residues. According to this embodiment, one or more non-modified DNA or RNA units are present after a consecutive stretch of about 3, 4, 5, 6, 7, or 8 modified nucleic acids.
- nucleic acids have an LNA substitution pattern that results in the formation of negligible secondary structure by the nucleic acids with itself.
- the nucleic acids do not form hairpins or do not form other secondary structures that would otherwise inhibit or prevent their binding to a target nucleic acid.
- opposing nucleotides in a palindrome pair or opposing nucleotides in inverted repeats or in reverse complements are not both LNA units.
- the nucleic acids in the first population form less than 3, 2, or 1 intramolecular base-pairs or base-pairs between two identical molecules.
- the nucleic acid does not have LNA-5-nitroindole: LNA-5-nitroindole intramolecular base-pairs.
- At least one LNA unit (e.g., at least 2, 3, 4, 5, 6, 7, 8, 9, or 10 LNA units) in the nucleic acid hybridizes to a first region within a first exon of a target nucleic acid and at least one LNA unit (e.g., at least 2, 3, 4, 5, 6, 7, 8, 9, or 10 LNA units) in the nucleic acid hybridizes to a second region within a second exon of the target nucleic acid that is adjacent to the first exon.
- the number of LNA units that bind to each region can be the same or different.
- the 5′ terminal nucleotide of the nucleic acid is or is not an LNA unit.
- the 3′ terminal nucleotide of the nucleic acid is not an LNA unit (e.g., the nucleic acid may contain a 3′ terminal naturally-occurring nucleotide).
- the nucleic acid can distinguish between different nucleic acids (e.g., mRNA splice variants) that cannot be distinguished using a naturally-occurring control nucleic acid (e.g., a control nucleic acid that consists of only 2′-deoxynucleotides such as a control nucleic acid of the same length as the nucleic acid of the invention).
- a naturally-occurring control nucleic acid e.g., a control nucleic acid that consists of only 2′-deoxynucleotides such as a control nucleic acid of the same length as the nucleic acid of the invention.
- the hybridization intensity of the nucleic acid for an exon of interest is at least 2, 3, 4, 5, 6, or 10 fold greater than the hybridization intensity of the nucleic acid for another exon in the same target nucleic acid (e.g., mRNA) or in another nucleic acid.
- the hybridization intensity of the nucleic acid for target nucleic acid is at least 2, 3, 4, 5, 6, or 10 fold greater than the hybridization intensity for a non-target nucleic acid with less than 99, 95, 90, 80, 70, or 60% sequence identity to the target nucleic acid.
- nucleic acids of the population or all of the nucleic acids of a subpopulation of the population are the same length.
- the population includes one or more nucleic acids of a different length.
- longer nucleic acids contain one or more nucleotides with universal bases.
- nucleotides with universal bases can be used to increase the thermal stability of nucleic acids that would otherwise have a thermal stability lower than some or all of the nucleic acids in the population.
- one or more nucleic acids have a universal base located at the 5′ or 3′ terminus of the nucleic acid.
- one or more (e.g., 2, 3, 4, 5, 6, or more) universal bases are located at the 5′ and 3′ termini of the nucleic acid. Desirably, all of the nucleic acids in the population have the same number of universal bases. Desirable universal bases include inosine, pyrene, 3-nitropyrrole, and 5-nitroindole.
- the nucleic acid has at least one LNA A or LNA T. In some embodiments, each nucleic acid has at least one LNA A or LNA T. Desirably, all of the adenine and thymine-containing nucleotides in the LNA are LNA A and LNA T, respectively. In some embodiments, a nucleic acid with a increased capture efficiency or melting temperature compared to a control nucleic acid has at least one LNA T or LNA C. In some embodiments, all of the thymine and cytosine-containing nucleotides in the LNA are LNA T and LNA C, respectively.
- a nucleic acid with an increased specificity or decreased self-complementarity compared to a control nucleic acid has at least one LNA A or LNA C. In some embodiments, all of the adenine and cytosine-containing nucleotides in the LNA are LNA A and LNA C, respectively.
- At least 10, 20, 25, 30, 40, 50, 60, 70, 80, 90, or 100% of the nucleic acids in the population have one ore more LNA units.
- the LNA has at least one 2,6-diaminopurine, 2-aminopurine, 2-thio-thymine, 2-thio-uracil, inosine, or hypoxanthine base.
- the LNA has a nucleotide with a 2′0, 4° C.-methylene linkage between the 2′ and 4′ position of a sugar moiety.
- one or more nucleic acids in the first population are LNA/DNA, LNA/RNA, or LNA/DNA/RNA chimeras.
- the variance in the melting temperature of the population is at least 10, 20, 30, 40, 50, 60, or 70% less than the variance in the melting temperature of the corresponding control population of nucleic acids of the same length with 2′-deoxynucleotides (e.g., DNA nucleotides) instead of LNA units or other modified or non-naturally-occurring units.
- the standard deviation in melting temperature is less than 10, 9.5, 9, 8.5, 8, 7.5, 7, 6.5, or 6.
- the range in melting temperatures for nucleic acids in the population is less than 70, 60, 50, 40, 30, or 20° C.
- the variance in the melting temperature of the population is less than 59, 50, 40, 30, 25, 20, 15, 10, or 5° C.
- the nucleic acids are covalently bonded to a solid support.
- the nucleic acids are in a predefined arrangement.
- the first population has at least 10; 100; 1,000; 5,000; 10,000; 100,000; or 1,000,000 different nucleic acids.
- the nucleic acids in the population together hybridize to at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, or 100% of the exons of a target nucleic acid.
- the population includes nucleic acids that together hybridize to at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, or 100% of the nucleic acids expressed by a particular cell or tissue.
- the population includes nucleic acids that together hybridize to at least one exon from at least 1,5, 10, 20, 25, 30, 40, 50, 60, 70, 80, 90, or 100% of the nucleic acid sequences expressed by a particular cell or tissue at a given point in time (e.g., an expression array with sequences corresponding to the sequences of mRNA molecules expressed by a particular cell type or a cell under a particular set of conditions).
- the plurality of nucleic acids are used as PCR primers or FISH probes.
- Desirable modified bases of the present invention when incorporated into the central position of a 9-mer oligonucleotide (all other eight residues or units being natural DNA or RNA units with natural bases) exhibit a T m difference equal to or less than about 15, 12, 10, 9, 8, 7, 6, 5, 4, 3 or 2° C. upon hybridizing to the four complementary oligonucleotide variants that are identical except for the unit corresponding to the LNA unit, where each variant has one of the natural bases uracil, cytosine, thymine, adenine or guanine. That is, the highest and the lowest T m (referred to herein as the T m differential) obtained with such four complementary sequences is 15,12, 10,9,8,7,6,5,4, 3 or 2° C. or less.
- Modified nucleic acid oligomers of the invention desirably contain at least one LNA unit, such as an LNA unit with a modified nucleobase.
- Modified nucleobases or nucleosidic bases desirably base-pair with adenine, guanine, cytosine, uracil, or thymine.
- Exemplary oligomers contain 2 to 100, 5 to 100, 4 to 50, 5 to 50, 5 to 30, or 8 to 15 nucleic acid units.
- one or more LNA units with natural nucleobases are incorporated into the oligonucleotide at a distance from the LNA unit having a modified base of 1 to 6 (e.g., 1 to 4) bases.
- At least two LNA units with natural nucleobases are flanking an LNA unit having a modified base.
- at least two LNA units independently are positioned at a distance from the LNA unit having the modified base of 1 to 6 (e.g., 1 to 4 bases).
- Desirable modified nucleobases or nucleosidic bases for use in nucleic acid compositions of the invention include optionally substituted carbon alicyclic or carbocyclic aryl groups (i.e., only carbon ring members), particularly multi-ring carbocyclic aryl groups such as groups having 2, 3, 4, 5, 6, 7, or 8 linked, particularly fused carbocyclic aryl moieties.
- Optionally substituted pyrene is also desirable.
- Such nucleobases or nucleosidic bases can provide significant performance results, as demonstrated in the examples which follow.
- Heteroalicyclic and heteroaromatic nucleobases or nucleosidic bases also are suitable.
- the carbocyclic moiety is linked to the 1′-position of the LNA unit through a linker (e.g., a branched or straight alkylene or alkenylene).
- Desirable LNA units have a carbon or hetero alicyclic ring with four to six ring members, e.g. a furanose ring, or other alicyclic ring structures such as a cyclopentyl, cycloheptyl, tetrahydropyranyl, oxepanyl, tetrahydrothiophenyl, pyrrolidinyl, thianyl, thiepanyl, piperidinyl, and the like.
- at least one ring atom of the carbon or hetero alicyclic group is taken to form a further cyclic linkage to thereby provide a multi-cyclic group.
- the cyclic linkage may include one or more, typically two atoms, of the carbon or hetero alicyclic group.
- the cyclic linkage also may include one or more atoms that are substituents, but not ring members, of the carbon or hetero alicyclic group.
- Other desirable LNA units are compounds having a substituent on the 2′-position of the central sugar moiety (e.g., ribose or xylose), or derivatives thereof, which favors the C3′-endo conformation, commonly referred to as the North (or simply N for short) conformation.
- LNA units include ENA (2′-O,4′-C-ethylene-bridged nucleic acids such as those disclosed in WO 00/47599) units as well as non-bridged riboses such as 2′-F or 2′—O—methyl.
- an exemplary control nucleic acid has ⁇ -D-2-deoxyribose instead of one or more bicyclic or sugar groups of a LNA unit or other modified or non-naturally-occurring units in a nucleic acid of the first population.
- the nucleic acid or population of the invention and the control nucleic acid or population only have naturally-occurring nucleobases.
- the capture efficiency of the corresponding control nucleic acid is calculated as the average capture efficiency for all of the nucleic acids that have either A, T, C, G or mC (methyl Cytosin) in each position corresponding to a non-naturally-occurring nucleobase in the nucleic acid in the first population.
- the invention features a complex of one or more target nucleic acids and nucleic acid of the invention (e.g., nucleic acid probes) in which one or more target nucleic acids are hybridized to a plurality of nucleic acids of the invention. Desirably, at least 2, 3, 4, 5, 6, 7, 10, 15, 20, 30, or 40 different target nucleic acids are hybridized.
- the target nucleic acids are cDNA molecules reverse transcribed from a patient sample or cRNA molecules amplified from a patient sample using a T7 RNA polymerase-based linear amplification system or the like. The target nucleic acids are labeled prior to hybridization to the nucleic acids of invention.
- the invention features a method for detecting the presence of one or more target nucleic acids in a sample.
- This method involves incubating a nucleic acid sample with one or more nucleic acids of the invention under conditions that allow at least one target nucleic acid to hybridize to at least one of the nucleic acids of the invention. Desirably, hybridization is detected for at least 2, 3, 4, 5, 6, 8, 10, or 12 target nucleic acids.
- the method further includes contacting the target nucleic acid with a second nucleic acid or a population of second nucleic acids that binds to a different region of the target molecule than the first nucleic acid.
- the method further involves identifying one or more hybridized target nucleic acids and/or determining the amount of one or more hybridized target nucleic acids.
- the method further includes determining the presence or absence of an mRNA splice variant of interest in the sample and/or determining the presence or absence of a mutation, deletion, and/or duplication of an exon of interest.
- the mutation, deletion, and/or duplication is indicative of a disease, disorder, or condition, such as cancer.
- the method is repeated under one or more different incubation conditions.
- the method is repeated at 1, 3, 5, 8, 10, 15, 20, 30, 40 or more different temperatures, cation concentrations (e.g., concentrations of monovalent cations such as Na+ and K + or divalent cations such as Mg 2+ and Ca 2+ ), denaturants (e.g., hydrogen bond donors or acceptors that interfere with the hydrogen bonds keeping the base-pairs together such as formamide or urea).
- cation concentrations e.g., concentrations of monovalent cations such as Na+ and K + or divalent cations such as Mg 2+ and Ca 2+
- denaturants e.g., hydrogen bond donors or acceptors that interfere with the hydrogen bonds keeping the base-pairs together such as formamide or urea.
- the method also includes identifying the target nucleic acid hybridized to the nucleic acids of the invention and/or determining the amount of the target nucleic acid hybridized to the nucleic acids of the invention.
- the target nucleic acids are labeled with a fluorescent group.
- the labeling is repeated using different fluorescent groups (e.g., labelling for so-called dye-swap labeling experiments).
- the determination of the amount of bound target nucleic acid involves one or more of the following: (i) adjusting for the varying intensity of the excitation light source used for detection of the hybridization, (ii) adjusting for photobleaching of the fluorescent group, and/or (iii) comparing the fluorescent intensity of the target nucleic acid(s) hybridized to the nucleic acids of the invention of nucleic acids to the fluorescent intensity of adifferent sample of nucleic acids hybridized to the nucleic acids of the invention (e.g., a different sample hybridized to the same population of nucleic acids of the invention on the same or a different solid support such as the same chip or a different chip).
- this comparison in fluorescent intensity involves adjusting for a difference in the amount of the nucleic acids of the invention used for hybridization to each sample and/or adjusting for a difference in the buffer (e.g., a difference in Mg 2+ concentration) used for hybridization to each sample or scaling for different labeling efficiencies with different fluorochromes.
- a difference in the buffer e.g., a difference in Mg 2+ concentration
- the target nucleic acids are cDNA molecules reverse transcribed from a patient sample or cRNA molecules amplified using a T7 RNA polymerase-based linear amplification system or the like from a patient sample.
- the sample has nucleic acids that are amplified using one or more primers specific for an exon of a target nucleic acid, and the method involves determining the presence or absence of an mRNA splice variant with the exon in the sample.
- one or more of the primers are specific for an exon or exon-exon junction of a pathogen of interest, and the method involves determining the presence or absence of a nucleic acid with the exon in the sample.
- the nucleic acids of the invention are covalently bonded to a solid support by reaction of a nucleoside phosphoramidite with an activated solid support, and subsequent reaction of a nucleoside phosphoramide with an activated nucleotide or nucleic acid bound to the solid support.
- the solid support or the growing nucleic acid bound to the solid support is activated by illumination, a photogenerated acid, or electric current.
- the invention features a method for amplifying a target nucleic acid molecule.
- the method involves (a) incubating a first nucleic acid of the invention with a target nucleic acid under conditions that allow the first nucleic acid to bind the target nucleic acid; and (b) extending the first nucleic acid with the target nucleic acid as a template.
- the method further involves contacting the target nucleic acid with a second nucleic acid (e.g., a second nucleic acid of the invention) that binds to a different region of the target nucleic acid than the first nucleic acid.
- the sequence of the target nucleic acid is known or unknown.
- the invention features a method of detecting a nucleic acid of a pathogen (e.g., a nucleic acid in a sample such as a blood or urine sample from a mammal).
- a pathogen e.g., a nucleic acid in a sample such as a blood or urine sample from a mammal.
- This method involves contacting a nucleic acid probe of the invention (e.g., a probe specific for an exon or a mRNA from a particular pathogen or family of pathogens) with a nucleic acid sample under conditions that allow the probe to hybridize to at least one nucleic acid in the sample.
- the probe is desirably at least 60, 70, 80, 90, 95, or 100% complementary to a nucleic acid of a pathogen (e.g., a bacteria, virus, or yeast such as any of the pathogens described herein). Hybridization between the probe and a nucleic acid in the sample is detected, indicating that the sample contains the corresponding nucleic acid from a pathogen.
- the method is used to determine what strain of a pathogen has infected a mammal (e.g., a human) by determining whether a particular nucleic acid is present in the sample.
- the probe has a universal base in a position corresponding to a nucleotide that varies among-different strains of a pathogen, and thus the probe detects the presence of a nucleic acid from any of a multiple of pathogenic strains.
- the invention features a method for classifying a test nucleic acid sample including target nucleic acids. This method involves (a) incubating a test nucleic acid sample with a one or more nucleic acids of the invention under conditions that allow at least one of the nucleic acids in the test sample to hybridize to at least one nucleic acid of the invention, (b) detecting a hybridization pattern of the test nucleic acid sample, and (c) comparing the hybridization pattern to a hybridization pattern of a first nucleic acid standard, whereby the comparison indicates whether or not the test sample has the same classification as the first standard.
- the method also includes comparing a hybridization pattern of the test nucleic acid sample to a hybridization pattern of a second standard.
- a hybridization pattern of the test nucleic acid sample is compared to at least 3, 4, 5, 8, 10, 15, 20, 30, 40, or more standards.
- the method also includes identifying the hybridized target nucleic acid and/or determining the amount of hybridized target nucleic acid.
- the target nucleic acids are labeled with a fluorescent group.
- the first nucleic acid standard is labeled with a different fluorescent group. The fluorescence of the target nucleic acids and the first nucleic acid standard can be detected simultaneously or sequentially.
- the method further includes determining the presence or absence of an mRNA splice variant of interest in the sample and/or determining the presence or absence of a mutation, deletion, and/or duplication of an exon of interest.
- the mutation, deletion, and/or duplication is indicative of a disease, disorder, or condition, such as cancer.
- the determination of the amount of bound target nucleic acid involves one or more of the following: (i) adjusting for the varying intensity of the excitation light source used for detection of the hybridization, (ii) adjusting for photobleaching of the fluorescent group, and/or (iii) comparing the fluorescent intensity of the target nucleic acid(s) hybridized to the nucleic acids of the invention to the fluorescent intensity of a different sample of nucleic acids hybridized to the nucleic acids of the invention (e.g., a different sample hybridized to same set of nucleic acids of the invention on the same or a different solid support such as the same chip or a different chip).
- this comparison in fluorescent intensity involves adjusting for a difference in the amount of the plurality used for hybridization to each sample and/or adjusting for a difference in the buffer (e.g., a difference in Mg 2+ concentration) used for hybridization to each sample.
- a difference in the buffer e.g., a difference in Mg 2+ concentration
- the nucleic acids in the population together hybridize to at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, or 100% of the exons of a target nucleic acid.
- the population includes nucleic acids that together hybridize to at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, or 100% of the nucleic acids expressed by a particular cell or tissue.
- the population includes nucleic acids that together hybridize to at least one exon from at least 1, 5, 10, 20, 25, 30, 40, 50, 60, 70, 80, 90, or 100% of the nucleic acid sequences expressed by a particular cell or tissue at a given point in time (e.g., an expression array with sequences corresponding to the sequences of mRNA molecules expressed by a particular cell type or a cell under a particular set of conditions).
- the method further includes using a nucleic acid or a region of a nucleic acid that is present in a first test sample but not present in a first standard or not present in a second test sample as a probe or primer for the detection, amplification, or characterization of the nucleic acid.
- the method is repeated under one or more different incubation or hybridization conditions.
- the method is repeated at 1, 3, 5, 8, 10, 15, 20, 30, 40 or more different temperatures, cation concentrations (e.g., concentration of monovalent cations such as Na+ and K + or divalent cations such as Mg 2+ and Ca 2 ),′ denaturants (e.g., hydrogen bond donors or acceptors that interfere with the hydrogen bonds keeping the base-pairs together such as formamide or urea).
- the sample has nucleic acids that are amplified using one or more primers specific for an exon of a target nucleic acid, and the method involves determining the presence or absence of an mRNA splice variant with the exon in the sample.
- one or more of the primers are specific for an exon or exon-exon junction of a pathogen of interest, and the method involves determining the presence or absence of a nucleic acid with the exon in the sample.
- the comparison of the hybridization pattern of a patient nucleic acid sample to that of one or more standards is used to determine whether or not a patient has a particular disease, disorder, condition, or infection or an increased risk for a particular disease, disorder, condition, or infection.
- the comparison is used to determine what pathogen has infected a patient and to select a therapeutic for the treatment of the patient.
- the comparison is used to select a therapeutic for the treatment or prevention of a disease or disorder in the patient.
- the comparison is used to include or exclude the patient from a group in a clinical trial.
- the comparison is used to compare the expression of nucleic acids (e.g., mRNA splice forms associated with toxicity) in the presence and absence of a candidate compound (e.g., a lead compound for drug development).
- a candidate compound e.g., a lead compound for drug development
- the comparison is used to determine differences in expression of nucleic acids (e.g., mRNA splice variants) under particular conditions (e.g., under different environmental stress conditions) or at different developmental time points.
- the expression of one or more members from a particular enzyme class e.g., protein kinase splice variants is measured.
- the nucleic acids of the invention are covalently bonded to a solid support by reaction of a nucleoside phosphoramidite with an activated solid support, and subsequent reaction of a nucleoside phosphoramide with an activated nucleotide or nucleic acid bound to the solid support.
- the solid support or the growing nucleic acid bound to the solid support is activated by illumination, a photogenerated acid, or electric current.
- nucleic acids of the invention offers a means to “fine tune” the chemical, physical, biological, pharmacokinetic, and pharmacological properties of the nucleic acids thereby facilitating improvement in their safety and efficacy profiles when used as a therapeutic drug.
- the invention features the use of one ore more nucleic acids of the invention for the detection, amplification, or classification of a nucleic acid of interest or a population of nucleic acids of interest.
- the invention features the use of one or more nucleic acids of the invention for alternative mRNA splice variant detection, expression profiling, comparative genomic hybridization, or real-time PCR.
- the nucleic acids are used to determine the amount of one or more target nucleic acids (e.g., mRNA splice variants) in a sample.
- fluorescently labeled RT-PCR products from the amplification of a test nucleic acid sample are hybridized to a population of nucleic acids of the invention.
- the amount of one or more RT-PCR products is measured to determine the amount of the corresponding nucleic acid in the initial sample.
- the invention features the use of a nucleic of the invention as a PCR primer or FISH probe.
- the invention features a method of selecting a nucleic acid for a population of nucleic acids. This method involves (a) determining the melting temperature of a nucleic acid of the invention, determining the ability of the nucleic acid to self-anneal, determining the ability of the nucleic acid to hybridize to one or more exons or introns of a target nucleic acid, and/or determining the ability of the nucleic acid to hybridize to a non-target nucleic acid, and (b) selecting the nucleic acid for inclusion or exclusion from the population based on the determination in step (a).
- step (a) is performed for at least 2, 3, 4, 5, 6, 10, 20, 50, 100, 200, 500, 1,000, 5,000 or more nucleic acids, and a subset of the nucleic acids are selected for inclusion in the population based on the determination in step (a).
- the nucleic acids with the highest melting temperatures and/or ability to hybridize to one or more exons or introns of a target nucleic acid are selected.
- the nucleic acids with the lowest ability to self-anneal and/or hybridize to a non-target nucleic acid are selected.
- the invention also features a variety of databases. These databases are useful for storing the information obtained in any of the methods of the invention. These databases may also be used in the diagnosis of disease or an increased risk for a disease or in the selection of a desirable therapeutic for a particular patient or class of patients.
- the invention provides an electronic database including at least 1, 10, 10 2 , 10 3 , 5 ⁇ 10 3 , 10 4 , 10 5 , 10 6 , 10 7 , 10 8 , or 10 9 records of a nucleic acid of interest or a population of nucleic acids of interest (e.g., one or more nucleic acids in a standard or in a test nucleic acid sample) correlated to records of its hybridization pattern to a plurality of nucleic acids of the invention under one or more incubation conditions (e.g., one or more temperatures, denaturant concentrations, or salt concentrations).
- a nucleic acid of interest e.g., one or more nucleic acids in a standard or in a test nucleic acid sample
- incubation conditions e.g., one or more temperatures, denaturant concentrations, or salt concentrations.
- the invention features computer including the database of the above aspect and a user interface (i) capable of displaying a hybridization pattern for a nucleic acid of interest or a population of nucleic acids of interest whose record is stored in the computer or (ii) capable of displaying a nucleic acid of interest (e.g., displaying the polynucleotide sequence or another identifying characteristic of the nucleic acid of interest) or a population of nucleic acids of interest that produces a hybridization pattern whose record is stored in the computer.
- a user interface capable of displaying a hybridization pattern for a nucleic acid of interest or a population of nucleic acids of interest whose record is stored in the computer or a user interface (i) capable of displaying a hybridization pattern for a nucleic acid of interest or a population of nucleic acids of interest whose record is stored in the computer or (ii) capable of displaying a nucleic acid of interest (e.g., displaying the polynucleotide sequence or
- oligonucleotides which are complementary to a specific target messenger RNA (mRNA) sequence, such as a specific mRNA splice variant.
- mRNA target messenger RNA
- oligonucleotides with a modified backbone such as LNA or phosphorothioate
- the invention features the use of a nucleic acid of the invention for the manufacture of a pharmaceutical composition for treatment of a disease curable by an antisense or RNAi technology.
- the invention provides a method for inhibiting the expression of a target nucleic acid in a cell.
- the method involves introducing into the cell a nucleic acid of the invention in an amount sufficient to specifically attenuate expression of the target nucleic acid.
- the introduced nucleic acid has a nucleotide sequence that is essentially complementary to a region of desirably at least 20 nucleotides of the target nucleic acid.
- the cell is in a mammal.
- the invention provides a method for preventing, stabilizing, or treating a disease, disorder, or condition associated with a target nucleic acid in a mammal.
- This method involves introducing into the mammal a nucleic acid of the invention in an amount sufficient to specifically attenuate expression of the target nucleic acid, wherein the introduced nucleic acid has a nucleotide sequence that is essentially complementary to a region of desirably at least 20 nucleotides of the target nucleic acid.
- the invention provides a method for preventing, stabilizing, or treating a pathogenic infection in a mammal by introducing into the mammal a nucleic acid of the invention in an amount sufficient to specifically attenuate expression of a target nucleic acid of a pathogen.
- the introduced nucleic acid has a nucleotide sequence that is essentially complementary to a region of desirably at least 20 nucleotides of the target nucleic acid.
- the mammal is a human.
- the introduced nucleic acid is single stranded or double stranded.
- nucleic acids may be administered to the mammal in a single dose or multiple doses.
- the doses may be separated from one another by, for example, one week, one month, one year, or ten years. It is to be understood that, for any particular subject, specific dosage regimes should be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the compositions.
- Exemplary mammals that can be treated using the methods of the invention include humans, primates such as monkeys, animals of veterinary interest (e.g., cows, sheep, goats, buffalos, and horses), and domestic pets (e.g., dogs and cats).
- Exemplary cells in which one or more target genes can be silenced using the methods of the invention include invertebrate, plant, bacteria, yeast, and vertebrate (e.g., mammalian or human) cells.
- Optimum dosages for gene silencing applications may vary depending on the relative potency of individual oligonucleotides, and can generally be estimated based on EC 50 values found to be effective in in vitro and in vivo animal models. In general, dosage is from 0.001 ⁇ g to 100 g per kg of body weight (e.g., 0.001 ⁇ g/kg to 1 g/kg), and may be given once or more daily, weekly, monthly or yearly, or even once every 2 to 20 years (U.S.P.N. 6,440,739). Persons of ordinary skill in the art can easily estimate repetition rates for dosing based on measured residence times and concentrations of the drug in bodily fluids or tissues.
- oligonucleotide is administered in maintenance doses, ranging from 0.001 ug to 100 g per kg of body weight (e.g., 0.001 1 g/kg to 1 g/kg), once or more daily, to once every 20 years.
- conventional treatments may be used in combination with the nucleic acids of the present invention.
- Suitable carriers include, but are not limited to, saline, buffered saline, dextrose, water, glycerol, ethanol, and combinations thereof.
- the composition can be adapted for the mode of administration and can be in the form of, for example, a pill, tablet, capsule, spray, powder, or liquid.
- the pharmaceutical composition contains one or more pharmaceutically acceptable additives suitable for the selected route and mode of administration.
- compositions may be administered by, without limitation, any parenteral route including intravenous, intra-arterial, intramuscular, subcutaneous, intradermal, intraperitoneal, intrathecal, as well as topically, orally, and by mucosal routes of delivery such as intranasal, inhalation, rectal, vaginal, buccal, and sublingual.
- the pharmaceutical compositions of the invention are prepared for administration to vertebrate (e.g., mammalian) subjects in the form of liquids, including sterile, non-pyrogenic liquids for injection, emulsions, powders, aerosols, tablets, capsules, enteric coated tablets, or suppositories.
- the invention features a method of synthesizing a nucleic acid.
- This method involves synthesizing a 2-thio-uridine nucleoside or nucleotide of formula IV using a compound of formula VIII, IX, X, XI, or XII as shown below.
- the nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.
- nucleobase thiolation is performed on the O 2 position of compound XI to form compound IV.
- sulphurization on both O 2 and 04 in compound VIII generates a 2,4-dithio-uridine nucleoside or nucleotide of formula X which is converted into compound IV.
- a cyclic ether of formula XI is transferred into compound IV or a 2—O—alkyl-uridine nucleoside or nucleotide of formula XII through reaction with the 5′ position.
- a 2—O—alkyl-uridine nucleoside or nucleotide of formula XII is generated by direct alkylation of a uridine nucleoside or nucleotide of formula VIII.
- R 4 and R 2 are each independently alkyl (e.g., methyl or ethyl), acyl (e.g., acetyl or benzoyl), or any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl).
- R 5 is any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, trityl(triphenylmethyl), acetyl, benzoyl, or benzyl.
- R 5 is hydrogen, alkyl (e.g., methyl or ethyl), 1-propynyl, thiazol-2-yl, pyridine-2-yl, thien-2-yl, imidazol-2-yl, (4/5-methyl)-thiazol-2-yl, 3-(iodoacetamido)propyl, 4-[N,N-bis(3-aminopropyl)amino]butyl), or halo (e.g., chloro, bromo, iodo, fluoro).
- alkyl e.g., methyl or ethyl
- 1-propynyl thiazol-2-yl
- pyridine-2-yl thien-2-yl
- imidazol-2-yl (4/5-methyl)-thiazol-2-yl
- 3-(iodoacetamido)propyl 4-[N,N-bis(3-aminopropyl)
- the group —OR 3′ in the formulas IV, VIII, IX, X, XI, and XII is any of the groups listed for R 3 or R 3′ in formula IIa or formula Ib or listed for R 3 or R 3* in formula Ia, Scheme A, or Scheme B, or the group —OR 3 or R 3′ in the formulas IV, VIII, IX, X, XI, and XII is selected from the group consisting of H, —OH, P(Q(CH 2 ) 2 CN)N(iPr) 2 , P(O(CH 2 ) 2 CN)N(iPr) 2 , phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted
- the group —OR 5 ′ in the formulas IV, and VIII, IX, X, and XII is any of the groups listed for R 5 or R 5′ in formula Ia or formula Ib or listed for R 5 or R 5* in formula Ia, Scheme A, or Scheme B, or the group —OR 5 ′ or R 5 ′ 0 in the formulas IV, and VIII, IX, X, and XII is selected from the group consisting of H, —OH, P(O(CH 2 ) 2 CN)N(iPr) 2 , P(O(CH 2 ) 2 CN)N(iPr) 2 , phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted
- acetyl or benzoyl aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trity
- the invention features a method of synthesizing a nucleic acid.
- This method involves synthesizing a 2-thiopyrimidine nucleoside or nucleotide of formula IV using a compound of formula III or compounds of the formula I, II, and III as shown below.
- the nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.
- lewis acid-catalyzed condensation of a substituted sugar of formula I and a substituted 2-thio-uracil of formula II results in a substituted 2-thio-uridine nucleoside or nucleotide of the formula III.
- a compound of formula III is converted into a LNA 2-thiouridine nucleoside or nucleotide of formula IV.
- R 4 ′ and R 5′ are, e.g., methanesulfonyloxy, p-toluenesulfonyloxy, or any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, trityl(triphenylmethyl), acetyl, benzoyl, or benzyl, R′′ is, e.g., acetyl, benzoyl, alkoxy (e.g., methoxy).
- R 2 is, e.g.,acetyl or benzoyl
- R 3 is any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, trityl(triphenylmethyl), acetyl, or benzoyl.
- R 5 is hydrogen, alkyl (e.g.
- methyl or ethyl 1-propynyl, thiazol-2-yl, pyridine-2-yl, thien-2-yl, imidazol-2-yl, (4/5-methyl)-thiazol-2-yl, 3-(iodoacetamido)propyl, 4-[N,N-bis(3-aminopropyl)amino]butyl), or halo (e.g., chloro, bromo, iodo, fluoro).
- halo e.g., chloro, bromo, iodo, fluoro
- the group —OR 3′ in the formulas I, III, and IV is any of the groups listed for R 3 or R 3′ in formula Ia or formula Ib or listed for R 3 or R 3* in formula Ia, Scheme A, or Scheme B, or the group —OR 3′ or R 3′ in the formulas I, III, and IV is selected from the group consisting of H, —OH, P(O(CH 2 ) 2 CN)N(iPr) 2 , phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., me
- acetyl or benzoyl aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trity
- R 5′ in the formulas I, III, and IV is any of the groups listed for R 5 or R 5′ in formula Ia or formula Ib or listed for R 5 or R 5* in formula Ia, Scheme A, or Scheme B, or R 5′ in the formulas I, III, and IV is selected from the group consisting of H, —OH, P(O(CH 2 ) 2 CN)N(iPr) 2 , phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g.,
- acetyl or benzoyl aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trity
- the invention features a method of synthesizing a nucleic acid.
- This method involves synthesizing a 2-thiopyrimidine nucleoside or nucleotide of formula IV using a compound of formula VII, compounds of the formula V, VI, and VII, or compounds of the formula I, V, VI, and VII as shown below.
- the nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.
- a 2-thio-uridine nucleoside or nucleotide of the formula IV is synthesized through ring-synthesis of the nucleobase by reaction of an amino sugar of the formula V and a substituted isothiocyanate of the formula VI.
- R 4 ′ and R 5′ are each idenpendently, e.g., methanesulfonyloxy, p-toluenesulfonyloxy, or any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, trityl(triphenylmethyl), acetyl, benzoyl, or benzyl.
- R′′ is, e.g., acetyl or benzoyl or alkoxy (e.g., methoxy), and Ris, e.g., acetyl or benzoyl
- R 3′ is any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, trityl(triphenylmethyl), acetyl, or benzoyl.
- R 5 are R 6 each idenpendently, e.g., hydrogen or alkyl (e.g. methyl or ethyl).
- R 6 can also be, e.g., an appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl).
- R 5 is hydrogen or methyl
- R 6 is methyl or ethyl.
- the group —OR 3 in the formulas I, V, VII, and IV is any of the groups listed for R 3 or R 3 ′ in formula Ia or formula Ib or listed for R 3 or R 3* in formula Ia, Scheme A, or Scheme B, or the group —ORor R 3 in the formulas I, V, VII, and IV is selected from the group consisting of H, —OH, P(O(CH 2 ) 2 CN)N(iPr) 2 , phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g.,
- acetyl or benzoyl aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trity
- R 5′ in the formulas I, V, VII, and IV is any of the groups listed for R 5 or R 5 in formula Ia or formula Ib or listed for R 5 or R 5* in formula Ia, Scheme A, or Scheme B, or R 5 in the formulas I, V, VII, and IV is selected from the group consisting of H, —OH, P(O(CH 2 ) 2 CN)N(iPr) 2 , phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g.,
- acetyl or benzoyl aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trity
- the invention features a method of synthesizing a nucleic acid.
- This method involves synthesizing a 2-thiopyrimidine nucleoside as shown below.
- the method further comprises reacting one or both compounds of the formula 4 with a phosphodiamidite (e.g., 2-cyanoethyl tetraisopropylphosphorodiamidite) to produce the corresponding nucleoside phosphoramidite.
- a phosphodiamidite e.g., 2-cyanoethyl tetraisopropylphosphorodiamidite
- the nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.
- a glycosyl-donor is coupled to a nucleobase as shown in pathway A.
- ring synthesis of the nucleobase is performed as show in pathway B.
- LNA-T diol is modified as shown in pathway C.
- R is hydrogen, methyl, 1-propynyl, thiazol-2-yl, pyridine-2-yl, thien-2-yl, imidazol-2-yl, (4/5-methyl)-thiazol-2-yl, 3-(iodoacetamido)propyl, 4-[N,N-bis(3-aminopropyl)amino]butyl, or halo (e.g., chloro, bromo, iodo, fluoro).
- halo e.g., chloro, bromo, iodo, fluoro
- R 1 , R 2 , and R 3 are each any appropriate protecting group such as acetyl, benzyl, silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl).
- the invention features a method of synthesizing a nucleic acid.
- This method involves synthesizing a 2-thiopyrimidine nucleoside or nucleotide of formula 4 using a compound of formula 3, compounds of the formula 2 and 3, or compounds of the formula 1, 2, 3, and 4 as shown below.
- the nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.
- This method can also be performed using any other appropriate protecting groups instead of Bn (benzyl), Ac (acetyl), or Ms (methansulfonyl).
- the method further comprises reacting one or both compounds of the formula 4 with a phosphodiamidite (e.g., 2-cyanoethyl tetraisopropylphosphorodiamidite) to produce the corresponding nucleoside phosphoramidite.
- a phosphodiamidite e.g., 2-cyanoethyl tetraisopropylphosphorodiamidite
- the invention features a method of synthesizing a nucleic acid.
- This method involves synthesizing a nucleoside or nucleotide of formula 10 or 11 using a compound of any one of the formula 6-9, compounds of the formula 5 and any one of the formulas 6-9, or compounds of the formula 4, 5, and any one of the formulas 6-9 as shown below.
- the nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.
- This method can also be performed using any other appropriate protecting groups instead of DMT, Bn, Ac, or Ms.
- a compound of formula 4 is used as a glycosyl donor in a coupling reaction with silylated hypoxantine to form a compound of the formula 5.
- a compound of the formula 5 is used in a ring closing reaction to form a compound of the formula 6.
- deprotection of the 5′-hydroxy group of compound 6 is performed by displacing the 5′—O—mesyl group with sodium benzoate to produce a compound of the formula 7 that is converted into a compound of the formula 8 after saponification of the 5′-benzoate.
- compound 8 is converted to a DMT-protected compound 9 prior to debenzylation of the 3′—O—hydroxy group.
- a phosphoramidite of the formula 11 is generated by phosphitylation of a nucleoside of the formula 10.
- the R 1 is H or P(O(CH 2 ) 2 CN)N(iPr) 2 .
- the group R 1 or —OR 1 is any of the groups listed for R 3 or R 3 in formula Ia or formula Ib or listed for R 3 or R 3* in formula Ia, Scheme A, or Scheme B, or the group —OR 1 or R 1 is selected from the group consisting of-OH, P(O(CH 2 ) 2 CN)N(iPr) 2 , phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), optionally substituted aryl,
- acetyl or benzoyl aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trity
- the invention features a method of synthesizing a nucleic acid.
- This method involves synthesizing a nucleoside or nucleotide of formula 20 or 21 as shown below, in which compound 4 is the same sugar shown in the above aspect.
- the nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.
- This method can also be performed using any other appropriate protecting groups instead of DMT, Bn, Bz (benzoyl), Ac, or Ms. Additionally, the method can be performed with any other halogen (e.g., fluoro or bromo) instead of chloro.
- a solution of compound 14 in aqueous 1,4-dioxane is treated with sodium hydroxide to give a bicyclic compound 15.
- sodium benzoate is used for displacement of 5′-mesylate of compound 15 to give compound 16.
- compound 17 is formed by reaction of compound 16 with sodium azide.
- compound 18 is produced by saponification of the 5′-benzoate of compound 17.
- hydrogenation of compound 18 produces compound 19.
- the peracelation method is used to benzolylate the 2- and 6-amino groups of compound 19, yielding 20, which is desirably converted into the phosphoramidite compound 21.
- the invention features a derivative of a compound of the formula 20 or 21 as described in the above aspect in which 3′-OH or —OP(O(CH 2 ) 2 CN)N(iPr) 2 group is replaced by any other group is selected from the group consisting of phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g.
- acetyl or benzoyl aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trity
- the invention features a method of synthesizing a nucleic acid.
- This method involves synthesizing a nucleoside or nucleotide of formula 20 or 21 as shown below.
- the nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.
- This method can also be performed using any other appropriate protecting groups instead of DMT.
- compound 17 is formed by reaction of compound 7 withl,3-dichloro-1,1,3,3-tetraisopropyldisiloxane.
- compound 18 is formed by reaction of compound 17 with phenoxyacetic anhydride.
- compound 19 is generated by reaction of compound 18 with acid.
- compound 20 is produced by reacting compound 19 with DMT-Cl.
- compound 20 is reacted with 2-cyanoethyl tetraisopropylphosphorodiamidite to give the phosphoramidite 21.
- acetyl or benzoyl aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trity
- the invention features a method of synthesizing a nucleic acid.
- This method involves synthesizing a nucleoside or nucleotide of formula 24 or 25 as shown below.
- the nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.
- This method can also be performed using any other appropriate protecting groups instead of Bz, Bn, and DMT. Additionally, the method can be performed with any other halogen (e.g., fluoro or bromo) instead of chloro.
- the compound 16 is formed from compounds 4, 14, and 15 as illustrated in an aspect above.
- the 5′—O—benzoyl group of compound 16 is hydrolyzed by aqueous sodium hydroxyde to give compound 22.
- Compound 23 is desirably produced by incubation of compound 22 in the presence of paladium hydroxide and ammonium formate.
- the 2-amine of compound 23 is selectively protected with an amidine group after treatment with N,N-dimethylformamide dimethyl acetal to yield compound 24.
- the diol 24 is 5′—O—DMT protected and 3′—O—phosphitylated produce the phosphoramidite LNA-2AP compound 25.
- compound 25 has one of the following groups instead of the P(O(CH 2 ) 2 CN)N(iPr) 2 group: any of the groups listed for R 3 or R 3′ in formula Ia or formula Ib or listed for R 3 or R 3* in formula Ia, Scheme A, or Scheme B, or a group selected from the group consisting of-OH, phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g.
- acetyl or benzoyl aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trity
- the invention features a nucleic acid of the invention that includes a compound of the formula 6 pCor the product of a compound of the formula 6 pC treated with ammonia as described herein.
- the invention features a method of synthesizing a nucleic acid that involves performing one or more of the steps described herein for the synthesis of a compound of the formula 6 pCor the product of a compound of theformula 6 pC treated with ammonia.
- the invention features a method of synthesizing a nucleic acid.
- This method involves one or more of any of the nucleosides or nucleotides of the invention with (i) any other nucleoside or nucleotide of the invention, (ii) any other nucleoside or nucleotide of formula Ia, formula Ib, formula Ia, Scheme A, or Scheme B, and/or (iii) any naturally-occurring nucleoside or nucleotide.
- the method involves reacting one or more nucleoside phosphoramidites of any of the above aspects with a nucleotide or nucleic acid.
- the invention provides a method for the synthesis of a population of nucleic acids (e.g., a population of nucleic acids of the invention) on a solid support.
- This method involves the reaction of a plurality of nucleoside phosphoramidites with an activated solid support (e.g., a solid support with an activated linker) and the subsequent reaction of a plurality of nucleoside phosphoramidites with activated nucleotides or nucleic acids bound to the solid support.
- an activated solid support e.g., a solid support with an activated linker
- the solid support or the growing nucleic acid bound to the solid support is activated by illumination, a photogenerated acid, or electric current.
- one or more spots or regions e.g., a region with an area of less than 1 cm 2, 0.1 cm2, 0.01 cm2, 1 mm2, or 0.1 mm that desirably contains one particular nucleic acid monomer or oligomer
- the solid support are irradiated to produce a photogenerated acid that removes the 5′-OH protecting group of one or more nucleic acid monomers or oligomers to which a nucleotide is subsequently added.
- an electric current is applied to one or more spots or regions (e.g., a region with an area of less than 1 cm 2,0.1 cm2,0.01 cm2, 1 mm2, or 0. mm2 that desirably contains one particular nucleic acid monomer or oligomer) on the solid support to remove an electrochemically sensitive protecting group of one or more nucleic acid monomers or oligomers to which a nucleotide is subsequently added.
- spots or regions e.g., a region with an area of less than 1 cm 2,0.1 cm2,0.01 cm2, 1 mm2, or 0. mm2 that desirably contains one particular nucleic acid monomer or oligomer
- one or more spots or regions (e.g., a region with an area of less than 1 cm2, 0.1 cm2, 0.01 cm2, 1 mm2, or 0.1 mm2 that desirably contains one particular nucleic acid monomer or oligomer) on the solid support are irradiated to remove a photosensitive protecting group of one or more nucleic acid monomers or oligomers to which a nucleotide is subsequently added.
- the solid support e.g., chip, coverslip, microscope glass slide, quartz, or silicon
- the solid support is less than 1, 0.5, 0.1. or 0.05 mm thick.
- the invention features a method of reacting a population of nucleic acids of the invention with one or more nucleic acids.
- This method involves incubating an immobilized population of nucleic acids of the invention with a solution that includes one or more probes (e.g., at least 2, 3, 4, 5, 10, 15, 20, 30, 40, 50, 60, 80, 100, or 150 different nucleic acids) and one or more target nucleic acids (e.g., at least 2, 3, 4, 5, 10, 15, 20, 30, 40, 50, 60, 80, 100, or 150 different target nucleic acids).
- probes e.g., at least 2, 3, 4, 5, 10, 15, 20, 30, 40, 50, 60, 80, 100, or 150 different nucleic acids
- target nucleic acids e.g., at least 2, 3, 4, 5, 10, 15, 20, 30, 40, 50, 60, 80, 100, or 150 different target nucleic acids.
- the incubation is performed in the presence of a ligase under conditions that allow the ligase to covalently react one or more immobilized nucleic acids with one or more nucleic acid probes in solution that hybridize to the same target nucleic acid. Desirably, at least 2, 5, 10, 15, 20, 30, 40, 50, 80, or 100 pairs of immobilized nucleic acids and nucleic acid probes are ligated. In various embodiments, the incubation occurs between 15 and 45° C., such as between 20 and 40° C. or between 25 and 35° C.
- a nucleic acid probe or primer specifically hybridizes to a target nucleic acid but does not substantially hybridize to non-target molecules, which include other nucleic acids in a cell or biological sample having a sequence that is less than 99, 95, 90, 80, or 70% identical or complementary to that of the target nucleic acid.
- the amount of the these non-target molecules hybridized to, or associated with, the nucleic acid probe or primer, as measured using standard assays is 2-fold, desirably 5-fold, more desirably 10-fold, and most desirably 50-fold lower than the amount of the target nucleic acid hybridized to, or associated with, the nucleic acid probe or primer.
- the amount of a target nucleic acid hybridized to, or associated with, the nucleic acid probe or primer, as measured using standard assays is 2-fold, desirably 5-fold, more desirably 10-fold, and most desirably 50-fold greater than the amount of a control nucleic acid hybridized to, or associated with, the nucleic acid probe or primer.
- the nucleic acid probe or primer is substantially complementary (e.g., at least 80, 90, 95, 98, or 100% complementary) to a target nucleic acid or a group of target nucleic acids from a cell.
- the probe or primer is homologous to multiple RNA or DNA molecules, such as RNA or DNA molecules from the same gene family.
- the probe or primer is homologous to a large number of RNA or DNA molecules.
- the probe or primer binds to nucleic acids which have polynucleotide sequences that differ in sequence at a position that corresponds to the position of a universal base in the probe or primer.
- control nucleic acids include nucleic acids with a random sequence or nucleic acids known to have little, if any, affinity for the nucleic acid probe or primer.
- the target nucleic acid is an RNA, DNA, or cDNA molecule.
- the association constant (Ka) of the nucleic acid toward a complementary target molecule is higher than the association constant of the complementary strands of the double stranded target molecule.
- the melting temperature of a duplex between the nucleic acid and a complementary target molecule is higher than the melting temperature of the complementary strands of the double stranded target molecule.
- the LNA-pyrene is in a position corresponding to the position of a non-base (e.g., a unit without a base) in another nucleic acid, such as a target nucleic acid.
- a non-base e.g., a unit without a base
- incorporation of pyrene in a DNA strand that is hybridized against the four natural bases decreases the T m by ⁇ 4.5° C. to ⁇ 6.8° C.; however, incorporation of pyrene in a DNA strand in a position opposite a non-base only decreases the T m by ⁇ 2.3° C. to ⁇ 4.6° C., most likely due to the better accommodation of the pyrene in the B-type duplex (Matray and Kool, J. Am.
- incorporation on LNA-pyrene into a nucleic acid in a position opposite a non-base (e.g., a unit without a base or a unit with a small group such as a noncyclic group instead of a base) in a target nucleic acid may also minimize any potential decrease in T m due to the pyrene substitution
- the number of molecules in the plurality of nucleic acids of the invention is at least 2, 4, 5, 6, 7, 8, or 10-fold greater than the number of molecules in the test nucleic acid sample.
- a LNA is a triplex-forming oligonucleotide.
- the target nucleic acids e.g., cDNA molecules reverse transcribed from a patient sample or cRNA molecules amplified from a patient sample using a T7 RNA polymerase-based amplification system or the like
- an enzyme such as a uracil-DNA glycosylase (e.g., E. coli uracil-DNA glycosylase) or using chemical hydrolysis such as alkaline hydrolysis.
- the average size of the fragmented nucleic acids is between 300 and 50 nucleic acids, such as approximately 300, 200, 100, or 50 nucleotides.
- the present invention has a variety of advantages related to nucleic acid analysis methods.
- the ability to equalize melting temperatures of a series of nucleic acids is generally applicable and desirable in all situations where more than one sequence is used simultaneously (e.g. DNA arrays with more than one capture probe, PCR and especially multiplex PCR, homogeneous assays such as Taqman and Molecular beacon).
- Sample preparation of specific sequences e.g., DNA or RNA extraction using capture probes on filters or magnetic beads
- melting temperature equalization of specific probe sequences is useful.
- the invention provides high affinity nucleotides (e.g., LNA and other high affinity nucleotides with a modified base and/or backbone) that can be used, e.g., arraysof the invention.
- the nucleic acids of the invention containing LNA units exhibited a suprising ability to discriminate between different mRNA splice variants compared to naturally-occurring nucleic acids.
- universal bases can be added as part of flanking regions in capture probes (e.g., probes of an array) to stabilize hybridization with high affinity nucleotides in the capture probes.
- Replacement of one or more DNA-t nucleotides with LNA-T and/or replacement of one or more DNA-a nucleotides with LNA-A reduces the variability of melting temperatures for capture probes of similar length but different GC and AT content by desirably at least 10, 20, 30, 40 or 50%. Additionally, replacement of one or more DNA-t nucleotides with LNA-T and/or replacement of one or more DNA-c with LNA-C increases the stability of a large number of capture probes, while desirably avoiding self-complementary sequences with LNA:LNA base-pairs within a capture probe that would otherwise reduce or eliminate the binding of target molecules to the probe. Although a general T and C substitution may not reduce the variability of melting temperatures of the probes, this substitution increases the melting temperature and binding efficiency of many capture probes that contain these two nucleotides.
- the invention also provides a general substitution algorithm for enhancement of the hybridization signal of a test nucleic acid sample by inclusion of high affinity monomers (e.g., LNA and other high affinity nucleotides with a modified base and/or backbone) in the array.
- high affinity monomers e.g., LNA and other high affinity nucleotides with a modified base and/or backbone
- This method increases the stability and binding affinity of capture probes while avoiding substitutions in positions that may form self-complementary base-pairs which may otherwise inhibit binding to a target molecule.
- the substitution algorithm is broadly useful for specialized arrays, as well as for PCR primers and FISH probes.
- LNA Locked Nucleoside Analogues
- nucleoside analogues e.g., bicyclic nucleoside analogues, e.g., as disclosed in WO 9914226
- LNA nucleoside and LNA nucleotide e.g., LNA nucleoside and LNA nucleotide.
- LNA includes the compounds as described in the present specificatiion including the compounds described in Example 17.
- the term “monomeric LNA” may, e.g., refer to the monomers LNA A, LNA T, LNA C, or any other LNA monomers.
- LNA unit is meant an individual LNA monomer (e.g., an LNA nucleoside or LNA nucleotide).or an oligomer (e.g., an oligonucleotide or nucleic acid) that includes at least one LNA monomer.
- LNA units as disclosed in WO 99/14226 are in general particularly desirable modified nucleic acids for incorporation into an oligonucleotide of the invention.
- the nucleic acids may be modified at either the 3′ and/or 5′ end by any type of modification known in the art. For example, either or both ends may be capped with a protecting group, attached to a flexible linking group, attached to a reactive group to aid in attachment to the substrate surface, etc.
- Desirable LNA units and their method of synthesis also are disclosed in WO 0056746, WO 0056748, WO 0066604, Morita et al., Bioorg. Med. Chem. Lett. 12(1):73-76, 2002; Hakansson et al., Bioorg. Med. Chem. Lett. 11 (7):935-938, 2001; Koshkin et al., J. Org. Chem. 66(25):8504-8512, 2001; Kvaerno et al., J. Org. Chem. 66(16):5498-5503, 2001; Hakansson et al., J. Org. Chem.
- LNA modified oligonucleotide a oligonucleotide comprising at least one LNA monomeric unit of the general scheme A, described infra, having the below described illustrative examples of modifications:
- X is selected from —O—, —S—, —N(R N )—, —C(R 6 R 6* )—, —O—C(R 7 R 7* )—, —C(R 6 R 6* )—O—, —S—C(R 7 R 7* )—, —C(R 1 R 6* )—S—, —N(R N* )-C(R 7 R 7* )—, —C(R 6 R 6 *)—N(R N* )—, and —C(R 6 R 6* )—C(R 7 R 7* ).
- B is selected from a modified base as discussed above e.g. an optionally substituted carbocyclic aryl such as optionally substituted pyrene or optionally substituted pyrenylmethylglycerol, or an optionally substituted heteroalicylic or optionally substituted heteroaromatic such as optionally substituted pyridyloxazole, optionally substituted pyrrole, optionally substituted diazole or optionally substituted triazole moieties; hydrogen, hydroxy, optionally substituted C 1-4 -alkoxy, optionally substituted C 1-4 -alkyl, optionally substituted C 1-4 -acyloxy, nucleobases, DNA intercalators, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands.
- an optionally substituted carbocyclic aryl such as optionally substituted pyrene or optionally substituted pyrenylmethylglycerol, or an optionally substituted heteroalicylic or optionally substituted heteroar
- P designates the radical position for an internucleoside linkage to a succeeding monomer, or a 5′-terminal group, such internucleoside linkage or 5′-terminal group optionally including the substituent R 5 .
- One of the substituents R 2 , R 2* , R 3 , and R 3* is a group P* which designates an internucleoside linkage to a preceding monomer, or a 2′/3′-terminal group.
- Each of the substituents R 1* , R 2 , R 2* , R 3 , R 4* , R 5 , R 5* , R 6* and R 6 , R 7 , and R 7* which are present and not involved in P, P* or the biradical(s), is independently selected from hydrogen, optionally substituted C 1-12 -alkyl, optionally substituted C 2-12 -alkenyl, optionally substituted C 2-12 -alkynyl, hydroxy, C 1-12 -alkoxy, C 2-12 -alkenyloxy, carboxy, C 1-12 -alkoxycarbonyl, C 1-12 -alkylcarbonyl, formyl, aryl, aryloxy-carbonyl, aryloxy, arylcarbonyl, heteroaryl, heteroaryloxy-carbonyl, heteroaryloxy, heteroarylcarbonyl, amino, mono- and di(C 1-6 -alkyl)amino, carbamoyl,
- Exemplary 5′, 3′, and/or 2′ terminal groups include —H, —OH, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g.
- acetyl or benzoyl aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trity
- references herein to a nucleic acid unit, nucleic acid residue, LNA unit, or similar term are inclusive of both individual nucleoside units and nucleotide units and nucleoside units and nucleotide units within an oligonucleotide.
- a “modified base” or other similar term refers to a composition (e.g., a non-naturally occuring nucleobase or nucleosidic base) which can pair with a natural base (e.g., adenine, guanine, cytosine, uracil, and/or thymine) and/or can pair with a non-naturally occurring nucleobase or nucleosidic base.
- the modified base provides a T m differential of 15, 12, 10, 8, 6, 4, or 2° C. or less as described herein.
- Exemplary modified bases are described in EP 1 072 679 and WO 97/12896.
- nucleobase is meant the naturally occurring nucleobases adenine (A), guanine (G), cytosine (C), thymine (T) and uracil (U) as well as non-naturally occurring nucleobases such as xanthine, diaminopurine, 8-oxo-N 6 -methyladenine, 7-deazaxanthine, 7-deazaguanine, N 4 ,N 4 -ethanocytosin, N′,N′-ethano-2,6-diaminopurine, 5-methylcytosine (mC), 5-(C 3 —C 6 )-alkynyl-cytosine, 5-fluorouracil, 5-bromouracil, pseudoisocytosine, 2-hydroxy-5-methyl-4-triazolopyridin, isocytosine, isoguanine, inosine and the “non-naturally occurring” nucleobases described in Benner
- nucleobase thus includes not only the known purine and pyrimidine heterocycles, but also heterocyclic analogues and tautomers thereof. Further naturally and non-naturally occurring nucleobases include those disclosed in U.S. Pat. No. 3,687,808 (Merigan, et al.), in Chapter 15 by Sanghvi, in Antisense Research and Application, Ed. S. T. Crooke and B.
- nucleosidic base or “base unit” is further intended to include compounds such as heterocyclic compounds that can serve like nucleobases including certain “universal bases” that are not nucleosidic b.ases in the most classical sense but serve as nucleosidic bases.
- universal bases are 3-nitropyrrole, optionally substituted indoles (e.g., 5-nitroindole), and optionally substituted hypoxanthine.
- Other desirable universal bases include, pyrrole, diazole or triazole derivatives, including those universal bases known in the art.
- a “substituted” group such as a nucleobase or nucleosidic base and the like may be substituted by other than hydrogen at one or more available positions, typically 1 to 3 or 4 positions, by one or more suitable groups such as those disclosed herein.
- suitable groups that may be present on a “substituted” group include e.g.
- alkanoyl such as a C 1-6 alkanoyl group such as acyl and the like; carboxamido; alkyl groups including those groups having 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5, or 6 carbon atoms; alkenyl and alkynyl groups including groups having one or more unsaturated linkages and from 2 to 12 carbon, or 2, 3, 4, 5 or 6 carbon atoms; alkoxy groups including those having one or more oxygen linkages and from 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5 or 6 carbon atoms; aryloxy such as phenoxy; alkylthio groups including those moieties having one or more thioether linkages and from 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5 or 6 carbon atoms; alkylsulfinyl groups including those moieties having one or more sulfinyl groups including those moieties having one or more sulfinyl groups including those moieties having one or more sulfin
- oxy-LNA monomer or unit any nucleoside or nucleotide which contains an oxygen atom in a 2′-4′ linkage.
- a “non-oxy-LNA” monomer or unit is broadly defined as any nucleoside or nucleotide which does not contain an oxygen atom in a 2′-4′-linkage.
- non-oxy-LNA monomers include 2′-deoxynucleotides (DNA) or nucleotides (RNA) or any analogues of these monomers which are not oxy-LNA, such as for example the thio-LNA and amino-LNA described herein with respect to formula Ia and in Singh et al. J. Org. Chem. 1998, 6, 6078-9, and the derivatives described in Susan M. Freier and Karl-Heinz Altmann, Nucleic Acids Research, 1997, vol 25, pp 4429-4443.
- universal base is meant a naturally-occurring or desirably a non-naturally occurring compound or moiety that can pair with a natural base (e.g., adenine, guanine, cytosine, uracil, and/or thymine), and that has a T m differential of 15, 12, 10, 8, 6, 4, or 2° C. or less as described herein.
- a natural base e.g., adenine, guanine, cytosine, uracil, and/or thymine
- oligonucleotide oligomer
- oligo is meant a successive chain of monomers (e.g., glycosides of heterocyclic bases) connected via internucleoside linkages.
- the linkage between two successive monomers in the oligo consist of 2 to 4, desirably 3, groups/atoms selected from —CH 2 —, —O—, —S—, —NR H _, >C ⁇ O, >C ⁇ NR H , >C ⁇ S, —Si(R′′) 2 —, —SO—, —S(O) 2 —, —P(O) 2 —, —PO(BH 3 )—, —P(O,S)—, —P(S) 2 —, —PO(R′′)—, —PO(OCH 3 )—, and —PO(NHR H )—, where RH is selected from hydrogen and C 1-4 -alkyl, and R′′ is selected from C 1-6 -
- ucceeding monomer is meant the neighboring monomer in the 5′-terminal direction
- preceding monomer is meant the neighboring monomer in the 3′-terminal direction
- LNA spiked oligo an oligonucleotide, such as a DNA oligonucleotide, wherein at least one unit (and preferably not all units) has been substituted by the corresponding LNA nucleoside monomer.
- T m is used in reference to the “melting temperature.”
- the melting temperature is the temperature at which 50% of a population of double-stranded nucleic acid molecules becomes dissociated into single strands.
- the equation for calculating the T m of nucleic acids is well-known in the art.
- a nucleic acid compound that has a T m differential of a specified amount means the nucleic acid exhibits that specified T m differential when incorporated into a specified 9-mer oligonucleotide with respect to the four complementary variants, as defined immediately below.
- a T m differential provided by a particular modified base is calculated by the following protocol (steps a) through d)):
- a T m differential for a particular modified base is determined by subtracting the highest T m value determined in steps a) through d) immediately above from the lowest T m value determined by steps a) through d) immediately above.
- T m is meant the variance in the values of the melting temperatures for a population of nucleic acids.
- the T m for each nucleic acid is determined by experimentally measuring or computationally predicting the temperature at which 50% of a population double-stranded molecules with the sequence of the nucleic acid becomes dissociated into single strands.
- the T m is the temperature at which 50% of a population of 100% complementary double-stranded molecules with the sequence of the nucleic acid becomes dissociated into single strands.
- the T m of this “modified” nucleic acid is approximated by determining the T m for each possible double stranded molecule in which one strand is the modified nucleic acid and the other strand has either A, T, C, or G in each position corresponding to a nucleobase other than A, T, C, G, or U in the modified nucleic acid.
- the modified nucleic acid has the sequence XMX in which X is 0, 1, or more A, T, C, G or U bases and M is any other nucleobase or nucleosidic base
- the T m is calculated for each possible double stranded molecule in which one strand is XMX and the other strand is X′YX′ in which X′ is the base complementary to the corresponding X base and Y is either A, T, C, or G.
- the average is then calculated for the T m values for each possible double stranded molecule (i.e., four possible duplexes per modified nucleobase or nucleoside base in the modified nucleic acid) and used as the approximate T m value for the modified nucleic acid.
- capture efficiency is meant the amount of target nucleic acid(s) bound to a particular nucleic acid or a population of nucleic acids. Standard methods can be used to calculate the capture efficiency by measuring the amount of bound target nucleic acid(s) and/or measuring the amount of unbound target nucleic acid(s).
- the capture efficiency of a nucleic acid or nucleic acid population of the invention is typically compared to the capture efficiency of a control nucleic acid or nucleic acid population under the same incubation conditions (e.g., using same buffer and temperature).
- a control nucleic acid may have ⁇ -D-2-deoxyribose instead of one or more bicyclic or sugar groups of a LNA unit or other modified or non-naturally-occurring units in a nucleic acid of the invention.
- the nucleic acid of the invention and the control nucleic acid only have naturally-occurring nucleobases.
- the capture efficiency of the corresponding control nucleic acid is calculated as the average capture efficiency for all of the nucleic acids that have either A, T, C, or G in each position corresponding to a non-naturally-occurring nucleobase in the nucleic acid of the invention.
- Monomers are referred to as being “complementary” if they contain nucleobases that can form hydrogen bonds according to Watson-Crick base-pairing rules (e.g., G with C, A with T, or A with U) or other hydrogen bonding motifs such as for example diaminopurine with T, inosine with C, and pseudoisocytosine with G.
- Watson-Crick base-pairing rules e.g., G with C, A with T, or A with U
- other hydrogen bonding motifs such as for example diaminopurine with T, inosine with C, and pseudoisocytosine with G.
- substantially complementarity is meant having a sequence that is at least 60, 70, 80, 90, 95, or 100% complementary to that of another sequence. Sequence complementarity is typically measured using sequence analysis software with the default parameters specified therein (e.g., Sequence Analysis Software Package of the Genetics Computer Group, University of Wisconsin Biotechnology Center, 1710 University Avenue, Madison, Wis. 53705). This software program matches similar sequences by assigning degrees of homology to various substitutions, deletions, and other modifications.
- the term “homology” refers to a degree of complementarity. There can be partial homology or complete homology (i.e., identity). A partially complementary sequence that at least partially inhibits a completely complementary sequence from hybridizing to a target nucleic acid is referred to using the functional term “substantially homologous.”
- substantially homologous refers to a probe that can hybridize to a strand of the double-stranded nucleic acid sequence under conditions of low stringency, e.g. using a hybridization buffer comprising 20% formamide in 0.8M saline/0.08M sodium citrate (SSC) buffer at a temperature of 37° C. and remaining bound when subject to washing once with that SSC buffer at 37° C.
- SSC sodium citrate
- substantially homologous refers to a probe that can hybridize to (i.e., is the complement of) the single-stranded nucleic acid template sequence under conditions of low stringency, e.g. using a hybridization buffer comprising 20% formamide in 0.8M saline/0.08M sodium citrate (SSC) buffer at a temperature of 37° C. and remaining bound when subject to washing once with that SSC buffer at 37° C.
- SSC sodium citrate
- an internal probe is meant a nucleic acid (e.g., a probe or primer) that hybridizes to either only one exon or only one intron of a nucleic acid (e.g., mRNA).
- the internal probe may hybridize to the 5′ end of the exon or intron, the 3′ end of the exon or intron, or between the 5′ end and the 3′ end of the exon or intron.
- the internal probe is at least 90, 95, 96, 97, 98, 99, or 100% identical to the corresponding region of a target nucleic acid.
- merged probe is meant a nucleic acid (e.g., a probe or primer) that hybridizes to more than one exon and/or intron of a nucleic acid (e.g., mRNA). Desirably, the merged probe hybridizes to two consecutive exons (e.g., exons in a mature mRNA transcript that may or may not be consecutive in the corresponding DNA molecule). In another desirable embodiment, the merged probe hybridizes to an exon and the consecutive intron. In desirable embodiments, the merged probe hybridizes to the same number of nucleotides in each exon or to the same number of nucleotides in the exon and intron.
- a nucleic acid e.g., a probe or primer
- the length of the region of the merged probe that hybridizes to one exon differs by less than 60, 40, 20, 10, or 5% from the length of the region of the merged probe that hybridizes to the other exon or to the intron.
- the merged probe is at least 90, 95, 96, 97, 98, 99, or 100% identical to the corresponding region of a target nucleic acid.
- poly-T 20 tail is meant a DNA polymer consisting of 20 DNA-t units added by polymerase chain reaction as a tail to a nucleic acid sequence, which is subsequently cloned in a plasmid vector allowing in vitro synthesis of poly(A)20 polyadenylated RNA.
- RNA or DNA unit e.g., a naturally-occurring RNA or DNA unit.
- corresponding unmodified reference nucleobase is meant a nucleobase that is not part of an LNA unit and is in the same orientation as the nucleobase in an LNA unit.
- mutation is meant an alteration in a naturally-occurring or reference nucleic acid sequence, such as an insertion, deletion, frameshift mutation, silent mutation, nonsense mutation, or missense mutation.
- the amino acid sequence encoded by the nucleic acid sequence has at least one amino acid alteration from a naturally-occurring sequence.
- selecting substantially partitioning a molecule from other molecules in a population.
- the partitioning provides at least a 2-fold, desirably, a 30-fold, more desirably, a 100-fold, and most desirably, a 1,000-fold enrichment of a desired molecule relative to undesired molecules in a population following the selection step.
- the selection step may be repeated a number of times, and different types of selection steps may be combined in a given approach.
- the population desirably contains at least 109 molecules, more desirably at least 101, 1013, or 1014 molecules and, most desirably, at least 1015 molecules.
- a “population” is meant more than one nucleic acid.
- a “population” according to the invention desirably means more than 101, 102, 103, or 104 different molecules.
- photochemically active groups compounds which are able to undergo chemical reactions upon irradiation with light.
- functional groups are quinones, especially 6-methyl-1,4-naphtoquinone, anthraquinone, naphtoquinone, and 1,4-dimethyl-anthraquinone, diazirines, aromatic azides, benzophenones, psoralens, diazo compounds, and diazirino compounds.
- thermochemically reactive group is meant a functional group which is able to undergo thermochemically-induced covalent bond formation with other groups.
- functional parts of thermochemically reactive groups are carboxylic acids, carboxylic acid esters such as activated esters, carboxylic acid halides such as acid fluorides, acid chlorides, acid bromide, acid iodides, carboxylic acid azides, carboxylic acid hydrazides, sulfonic acids, sulfonic acid esters, sulfonic acid halides, semicarbazides, thiosemicarbazides, aldehydes, ketones, primary alcohols, secondary alcohols, tertiary alcohols, phenols, alkyl halides, thiols, disulphides, primary amines, secondary amines, tertiary amines, hydrazines, epoxides, maleimides, and boronic acid derivatives.
- chelating group is meant a molecule that contains more than one binding site and frequently binds to another molecule, atom, or ion through more than one binding site at the same time.
- functional parts of chelating groups are iminodiacetic acid, nitrilotriacetic acid, ethylenediamine tetraacetic acid (EDTA), and aminophosphonic acid.
- reporter group is meant a group which is detectable either by itself or as a part of an detection series.
- functional parts of reporter groups are biotin, digoxigenin, fluorescent groups (e.g., groups which are able to absorb electromagnetic radiation, e.g.
- Ligand is meant a compound which binds.
- Ligands can comprise functional groups such as aromatic groups (such as benzene, pyridine, naphthalene, anthracene, and phenanthrene), heteroaromatic groups (such as thiophene, furan, tetrahydrofuran, pyridine, dioxane, and pyrimidine), carboxylic acids, carboxylic acid esters, carboxylic acid halides, carboxylic acid azides, carboxylic acid hydrazides, sulfonic acids, sulfohic acid esters, sulfonic acid halides, semicarbazides, thiosemicarbazides, aldehydes, ketones, primary alcohols, secondary alcohols, tertiary alcohols, phenols, alkyl halides, thiols, disulphides, primary amines, secondary amines, tertiary amines, hydrazines
- DNA intercalators photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands correspond to the “active/functional” part of the groups in question.
- DNA intercalators, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands are typically represented in the form M-K- where M is the “active/functional” part of the group in question and where K is a spacer through which the “active/functional” part is attached to the 5- or 6-membered ring.
- the group B in the case where B is selected from DNA intercalators, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands, has the form M-K-, where M is the “active/functional” part of the DNA intercalator, photochemically active group, thermochemically active group, chelating group, reporter group, and ligand, respectively, and where K is an optional spacer comprising 1-50 atoms, desirably 1-30 atoms, in particular 1-15 atoms, between the 5- or 6-membered ring and the “active/functional” part.
- spacer is meant a thermochemically and photochemically non-active distance-making group and is used to join two or more different moieties of the types defined above. Spacers are selected on the basis of a variety of characteristics including their hydrophobicity, hydrophilicity, molecular flexibility and length (e.g., Hermanson et. al., “Immobilized Affinity Ligand Techniques,” Academic Press, San Diego, Calif. (1992). Generally, the length of the spacers is less than or about 400 A, in some applications desirably less than 100 A.
- the spacer thus, comprises a chain of carbon atoms optionally interrupted or terminated with one or more heteroatoms, such as oxygen atoms, nitrogen atoms, and/or sulphur atoms.
- the spacer K may comprise one or more amide, ester, amino, ether, and/or thioether functionalities, and optionally aromatic or mono/polyunsaturated hydrocarbons, polyoxyethylene such as polyethylene glycol, oligo/polyamides such as poly- ⁇ -alanine, polyglycine, polylysine, peptides, oligosaccharides, or oligo/polyphosphates.
- the spacer may consist of combined units thereof.
- the length of the spacer may vary, taking into consideration the desired or necessary positioning and spatial orientation of the “active/functional” part of the group in question in relation to the 5- or 6-membered ring.
- the spacer includes a chemically cleavable group. Examples of such chemically cleavable groups include disulphide groups cleavable under reductive conditions and peptide fragments cleavable by peptidases.
- target nucleic acid or “nucleic acid target” is meant a particular nucleic acid sequence of interest.
- the “target” can exist in the presence of other nucleic acid molecules or within a larger nucleic acid molecule.
- solid support any rigid or semi-rigid material to which a nucleic acid binds or is directly or indirectly attached.
- the support can be any porous or non-porous water insoluble material, including without limitation, membranes, filters, chips, slides, wafers, fibers, magnetic or nonmagnetic beads, gels, tubing, strips, plates, rods, polymers, particles, microparticles, capillaries, and the like.
- the support can have a variety of surface forms, such as wells, trenches, pins, channels and pores.
- an “array” is meant a fixed pattern of at least two different immobilized nucleic acids on a solid support.
- the array includes at least 102, more desirably, at least 103, and, most desirably, at least 1 different nucleic acids.
- antisense nucleic acid is meant a nucleic acid, regardless of length, that is complementary to a coding strand or mRNA of interest.
- the antisense molecule inhibits the expression of only one nucleic acid, and in other embodiments, the antisense molecule inhibits the expression of more than one nucleic acid.
- the antisense nucleic acid decreases the expression or biological activity of a nucleic and or encoded protein by at least 20, 40, 50, 60, 70, 80, 90, 95, or 100%.
- An antisense molecule can be introduced, e.g., to an individual cell or to whole animals, for example, it may be introduced systemically via the bloodstream.
- a region of the antisense nucleic acid or the entire antisense nucleic acid is at least 70, 80, 90, 95, 98, or 100% complementary to a coding sequence, regulatory region (5′ or 3′ untranslated region), or an mRNA of interest.
- the region of complementarity includes at least 5, 10, 20, 30, 50, 75, 100, 200, 500, 1000, 2000 or 5000 nucleotides or includes all of the nucleotides in the antisense nucleic acid.
- the antisense molecule is less than 200, 150, 100, 75, 50, or 25 nucleotides in length. In other embodiments, the antisense molecule is less than 50,000; 10,000; 5,000; or 2,000 nucleotides in length. In certain embodiments, the antisense molecule is at least 200, 300, 500, 1000, or 5000 nucleotides in length.
- the number of nucleotides in the antisense molecule is contained in one of the following ranges: 5-15 nucleotides, 16-20 nucleotides, 21-25 nucleotides, 26-35 nucleotides, 36-45 nucleotides, 46-60 nucleotides, 61-80 nucleotides, 81-100 nucleotides, 101-150 nucleotides, or 151-200 nucleotides, inclusive.
- the antisense molecule may contain a sequence that is less than a full-length sequence or may contain a full-length sequence.
- double stranded nucleic acid is meant a nucleic acid containing a region of two or more nucleotides that are in a double stranded conformation.
- the double stranded nucleic acids consist entirely of LNA units or a mixture of LNA units, ribonucleotides, and/or deoxynucleotides.
- the double stranded nucleic acid may be a single molecule with a region of self-complementarity such that nucleotides in one segment of the molecule base-pair with nucleotides in another segment of the molecule.
- the double stranded nucleic acid may include two different strands that have a region of complementarity to each other.
- the regions of complementarity are at least 70, 80, 90, 95, 98, or 100% identical.
- the region of the double stranded nucleic acid that is present in a double stranded conformation includes at least 5, 10, 20, 30, 50, 75, 100, 200, 500, 1000, 2000 or 5000 nucleotides or includes all of the nucleotides in the double stranded nucleic acid.
- Desirable double stranded nucleic acid molecules have a strand or region that is at least 70, 80, 90, 95, 98, or 100% identical to a coding region or a regulatory sequence (e.g., a transcription factor binding site, a promoter, or a 5′ or 3′ untranslated region) of a nucleic acid of interest.
- the double stranded nucleic acid is less than 200, 150, 100, 75, 50, or 25 nucleotides in length. In other embodiments, the double stranded nucleic acid is less than 50,000; 10,000; 5,000; or 2,000 nucleotides in length.
- the double stranded nucleic acid is at least 200, 300, 500, 1000, or 5000 nucleotides in length.
- the number of nucleotides in the double stranded nucleic acid is contained in one of the following ranges: 5-15 nucleotides, 16-20 nucleotides, 21-25 nucleotides, 26-35 nucleotides, 36-45 nucleotides, 46-60 nucleotides, 61-80 nucleotides, 81-100 nucleotides, 101-150 nucleotides, or 151-200 nucleotides, inclusive.
- the double stranded nucleic acid may contain a sequence that is less than a full-length sequence or may contain a full-length sequence.
- the double stranded nucleic acid inhibits the expression of only one nucleic acid, and in other embodiments, the double stranded nucleic acid molecule inhibits the expression of more than one nucleic acid.
- the nucleic acid decreases the expression or biological activity of a nucleic acid of interest or a protein encoded by a nucleic acid of interest by at least 20, 40, 50, 60, 70, 80, 90, 95, or 100%.
- a double stranded nucleic acid can be introduced, e.g., to an individual cell or to whole animals, for example, it may be introduced systemically via the bloodstream.
- the double stranded nucleic acid or antisense molecule includes one or more LNA nucleotides, one or more universal bases, and/or one or more modified nucleotides in which the 2′ position in the sugar (e.g., ribose or xylose) contains a halogen (such as fluorine group) or contains an alkoxy group (such as a methoxy group) which increases the half-life of the double stranded nucleic acid or antisense molecule in vitro or in vivo compared to the corresponding double stranded nucleic acid or antisense molecule in which the corresponding 2′ position contains a hydrogen or an hydroxyl group.
- a halogen such as fluorine group
- alkoxy group such as a methoxy group
- the double stranded nucleic acid or antisense molecule includes one or more linkages between adjacent nucleotides other than a naturally-occurring phosphodiester linkage.
- linkages include phosphoramide, phosphorothioate, and phosphorodithioate linkages.
- the double stranded or antisense molecule is purified.
- a factor is substantially pure when it is at least 50%, by weight, free from proteins, antibodies, and naturally-occurring organic molecules with which it is naturally associated. Desirably, the factor is at least 75%, more desirably, at least 90%, and most desirably, at least 99%, by weight, pure.
- a substantially pure factor may be obtained by chemical synthesis, separation of the factor from natural sources, or production of the factor in a recombinant host cell that does not naturally produce the factor. Nucleic acids and proteins may be purified by one skilled in the art using standard techniques such as those described by Ausubel et al.
- the factor is desirably at least 2, 5, or 10 times as pure as the starting material, as measured using polyacrylamide gel electrophoresis, column chromatography, optical density, HPLC analysis, or western analysis (Ausubel et al., supra). Desirable methods of purification include immunoprecipitation, column chromatography such as immunoaffinity chromatography, magnetic bead immunoaffinity purification, and panning with a plate-bound antibody.
- treating, stabilizing, or preventing a disease, disorder, or condition is meant preventing or delaying an initial or subsequent occurrence of a disease, disorder, or condition; increasing the disease-free survival time between the disappearance of a condition and its reoccurrence; stabilizing or reducing an adverse symptom associated with a condition; or inhibiting or stabilizing the progression of a condition.
- at least 20, 40, 60, 80, 90, or 95% of the treated subjects have a complete remission in which all evidence of the disease disappears.
- the length of time a patient survives after being diagnosed with a condition and treated with a nucleic acid of the invention is at least 20, 40, 60, 80, 100, 200, or even 500% greater than (i) the average amount of time an untreated patient survives or (ii) the average amount of time a patient treated with another therapy survives.
- treating, stabilizing, or preventing cancer is meant causing a reduction in the size of a tumor, slowing or preventing an increase in the size of a tumor, increasing the disease-free survival time between the disappearance of a tumor and its reappearance, preventing an initial or subsequent occurrence of a tumor, or reducing an adverse symptom associated with a tumor.
- the number of cancerous cells surviving the treatment is at least 20, 40, 60, 80, or 100% lower than the initial number of cancerous cells, as measured using any standard assay.
- the decrease in the number of cancerous cells induced by administration of a nucleic acid of the invention is at least 2, 5, 10, 20, or 50-fold greater than the decrease in the number of non-cancerous cells.
- the number of cancerous cells present after administration of a nucleic acid of the invention is at least 2, 5, 10, 20, or 50-fold lower than the number of cancerous cells present prior to the administration of the compound or after administration of a buffer control.
- the methods of the present invention result in a decrease of 20, 40, 60, 80, or 100% in the size of a tumor as determined using standard methods. Desirably, at least 20, 40, 60, 80, 90, or 95% of the treated subjects have a complete remission in which all evidence of the cancer disappears. Desirably, the cancer does not reappear or reappears after at least 5, 10, 15, or 20 years.
- Exemplary cancers that can be treated, stabilized, or prevented using the above methods include prostate cancers, breast cancers, ovarian cancers, pancreatic cancers, gastric cancers, bladder cancers, salivary gland carcinomas, gastrointestinal cancers, lung cancers, colon cancers, melanomas, brain tumors, leukemias, lymphomas, and carcinomas. Benign tumors may also be treated or prevented using the methods and nucleic acids of the present invention.
- infection is meant the invasion of a host animal by a pathogen (e.g., a bacteria, yeast, or virus).
- the infection may include the excessive growth of a pathogen that is normally present in or on the body of an animal or growth of a pathogen that is not normally present in or on the animal.
- an infection can be any situation in which the presence of a pathogen population(s) is damaging to a host.
- an animal is “suffering” from an infection when an excessive amount of a pathogen population is present in or on the animal's body, or when the presence of a pathogen population(s) is damaging the cells or other tissue of the animal.
- the number of a particular genus or species of pathogen is at least 2, 4, 6, or 8 times the number normally found in the animal.
- a bacterial infection may be due to gram positive and/or gram negative bacteria.
- the bacterial infection is due to one or more of the following bacteria: Chlamydophila pneumoniae, C. psittaci, C. abortus, Chlamydia trachomatis, Simkania negevensis, Parachlamydia acanthamoebae, Pseudomonas aeruginosa, P. alcaligenes, P. chlororaphis, P. fluorescens, P. luteola, P. mendocina, P. monteilii, P. oryzihabitans, P. pertocinogena, P.
- ducreyi Pasteurella multocida, P. haemolytica, Branhamella catarrhalis, Helicobacter pylori, Campylobacter fetus, C. jejuni, C. coli, Borrelia burgdorferi, V. cholerae, V parahaemolyticus, Legionella pneumophila, Listeria monocytogenes, Neisseria gonorrhea, N. meningitidis, Kingella dentrificans, K. kingae, K. oralis, Moraxella catarrhalis, M atlantae, M lacunata, M. nonliquefaciens, M.
- osloensis M phenylpyruvica, Gardnerella vaginalis, Bacteroidesfragilis, Bacteroides distasonis , Bacteroides 3452A homology group, Bactero ides vulgatus, B. ovalus, B. thetaiotaomicron, B. uniformis, B. eggerthii, B. splanchnicus, Clostridium difficile, Mycobacterium tuberculosis, M. avium, M intracellulare, M leprae, C. diphtheriae, C. ulcerans, C. accolens, C. afermentans, C. amycolatum, C. argentorense, C. auris, C.
- agalactiae S. pyogenes, Enterococcus avium, E. casseliflavus, E. cecorum, E. dispar, E. durans, E. faecalis, E. faecium, E. flavescens, E. gallinarum, E. hirae, E. malodoratus, E. mundtii, E. pseudoavium, E. raffinosus, E. solitarius, Staphylococcus aureus, S. epidermidis, S. saprophyticus, S. intermedius, S. hyicus, S. haemolyticus, S. hominis , and/or S. saccharolyticus .
- a nucleic acid is administered in an amount sufficient to prevent, stabilize, or inhibit the growth of a pathogenic bacteria or to kill the bacteria.
- the viral infection relevant to the methods of the invention is an infection by one or more of the following viruses: West Nile virus (e.g., Samuel, “Host genetic variability and West Nile virus susceptibility,” Proc. Natl. Acad. Sci. USA Aug.
- West Nile virus e.g., Samuel, “Host genetic variability and West Nile virus susceptibility,” Proc. Natl. Acad. Sci. USA Aug.
- mammal in need of treatment is meant a mammal in which a disease, disorder, or condition is treated, stabilized, or prevented by the administration of a nucleic acid of the invention.
- FIG. 1 shows the structures of selected nucleotide monomers: DNA (T), LNA (TL), pyrene DNA (Py), 2′-OMe-RNA [2′-OMe(T)], abasic LNA (AbL), phenyl LNA (17a), and pyrenyl LNA (17d).
- FIG. 2 illustrates the chemical structures of Selective Binding Complementary (SBC) nucleotides.
- FIG. 3 shows the base-pairing between modified bases and naturally-occurring nucleotides. These modified bases may be incorporated as part of an LNA, DNA, or RNA unit and used any of the oligomers of the invention.
- FIGS. 4A and 4B show the sensitivity of 50-mer LNA capture probes compared to 50-mer DNA capture probes.
- SWI 5-specific 50-mer DNA oligonucleotides (green bars) and 50-mer capture probes with an LNA nucleotide incorporated at every third nucleotide position (red bars) were printed at the oligo concentration indicated below.
- the slides were hybridized at 65° C. in 3 ⁇ SSC (FIG. 4A) and at 70° C. in 3 ⁇ SSC (FIG. 4B).
- FIGS. 5A and 5B show the specificity of 40-mer LNA capture probes (red bars) compared to DNA capture probes (green bars).
- the hybridizations were carried out at 65° C. in 3 ⁇ SSC. Bars 1 and 7 represent perfectly matched duplexes, bars 2 and 8, 3, and 9, 4 and 10, 5 and 11, and 6 and 12 represent duplexes with 1, 2, 3, 4, 5 mismatches, respectively.
- the in vitro RNA used was SW15 in FIG. 5A and TH114 in FIG. 5B.
- FIG. 6 shows the detection principle for alternative exon skipping in the C. elegans let-2 gene using LNA oligonucleotide capture probes and comparative expression profiling.
- FIG. 7 shows the detection of alternative splicing of C. elegans Let-2 exon 9 and 10 using LNA-modified capture probes.
- FIGS. 8A and 8B show the comparison of DNA and LNA-modified oligonucleotide capture probes in the specific capture of the C. elegans T01 D3.3 mRNA, exon 4.
- FIG. 9 illustrates the LNA exon-exon junction (merged) probe concept.
- FIGS. 10A and 10B show the capture probe specificity for the C. elegans T10D3.3 mRNA, exon 4 (FIG. 10A) and exon 5 (FIG. 10B) as validated by short complementary target oligonucleotides.
- FIG. 11 shows the construction of the recombinant splice variants in the in vitro transcription vector.
- the small bars show the location of the hybridization for the oligonucleotide capture probes used in this example. The sequences of the capture probes are described herein.
- FIGS. 12A LNA probes
- 12B DNA control probes
- FIG. 13 shows the sensitivity of 50-mer LNA capture probes compared to 50-mer DNA capture probes.
- the slides were hybridized at 65° C. in 3 ⁇ SSC.
- FIG. 14 is a bar graph of the signal intensities of a patient DNA sample hybridized to an array of the invention.
- the names of the probes in FIGS. 14 and 17 match, although the numbers used in FIG. 14 are abbreviated, e.g., probe No. 10580 Menkes.14 50NH2C 6-2 .LNA in FIG. 17 corresponds to the second probe counted from the left “14.2” LNA in the lower graph of FIG. 14.
- FIG. 15 is a graph comparing the spot intensity for probes of the invention with different LNA substitution patterns.
- FIG. 16 is a bar graph of the spot intensity for LNA probes for different exons.
- FIG. 17 is a table of comparative genome hybridization (CGH) capture probe sequences.
- FIG. 18 is a flow chart of the steps of oligo design software of the invention.
- the OligoDesign software features LNA modified oligonucleotide secondary structure prediction, LNA spiked oligonucleotide melting temperature prediction, genome wide cross hybridization prediction, secondary structure prediction of the target, and recognition and filtering of the target in the genome. These features are determined for each possible probe of the query gene and presented to an artificial neural network. The probes are then ranked according to the neural network prediction and the top scoring probes are returned.
- FIGS. 19A-19F are a schematic illustration of the OligoDesign software of the invention.
- FIG. 20 illustrates photo-activated immobilization of nucleic acids of the invention, which enables polarized coupling of anthraquinone (AQ)-linked LNA oligonucleotides onto the polymer surface. No pretreatment of the slide is needed. A covalent bond is formed between the oligonucleotide and the polymer using a UV source, e.g. Stratalinker.
- a UV source e.g. Stratalinker.
- FIG. 21 illustrates an injection-molded polymer slide. Finger indents ease slide handling.
- the slide has a well-defined printing and hybridization window, frosted surface for identification and orientation, and space for barcodes.
- FIG. 22 illustrates spot quality on different slides that can be used to immobilize nucleic acids of the invention.
- the hydrophobic slide surface ensures that extremely homogenous spots are generated when hydrophilic spotting solution is applied to the surface.
- a high spot quality is obtained on the ImmobilizerTM polymer slide compared to a glass slide when using a spot-to-spot distance of 150 ⁇ M.
- the high-quality arrays simplify downstream image analyses.
- FIG. 23 is a schematic illustration of a method of the invention.
- FIG. 24 is a table of exemplary target nucleic acids (Holstege et al. (1998)(Cell 95, 717-728, and Causton et al. (2001) Mol. Biol. Cell 12, 323-337).
- FIG. 25 is a graph of Cy5 intensity.
- Yeast actin 1-specific 50-mer capture probes were synthesized as DNA and DNA/LNA mixmer oligonucleotides.
- LNA-substituted mixmer capture probes contain an LNA at every 4 th , 5 th , and 6 th nucleotide position (LNA — 4, LNA — 5, LNA — 6).
- On-chip melting profiles demonstrate a 8-10° C. increase in T m obtained with LNA capture probes.
- FIG. 26A illustrates the heat-shock response in yeast.
- the array was hybridized with Cy3-labeled standard and Cy5-labelled heat-shock yeast cDNA.
- FIG. 26B also illustrates the heat-shock response in yeast.
- the microarray data were normalized using yeast actin 1.
- the ssa4 gene encoding heat shock protein HSP70 is up-regulated over 2-fold. Expression of the gual gene is down-regulated.
- FIG. 27A compares expression of wild-type and ssa4 mutant yeast. The array was hybridized with Cy3-labeled wild-type and Cy5-labelled ssa4 mutant yeast cDNA.
- FIG. 27B also compares wild-type and ssa4 yeast. The hybridization data were normalized using yeast actin 1. ssa4 is detected in the wild-type yeast strain, but not in the ssa4 knock-out strain.
- FIG. 28 illustrates mRNA splicing
- FIG. 29 is a picture showing gel electrophoresis of fragmented cDNA from the yeast wild-type strain.
- the molecular marker (lane 1 and 9) is from Life technologies, USA.
- Lanes 2-8 represents the UDG-fragmented cDNA 1-7 according to the different dUTP/dTTP ratios in Table 18.
- FIG. 30 is a graph of the log ratios of the normalized fluorescence intensities from the wild-type yeast strain (signal) and those from the ⁇ ssa4 yeast strain (noise) as a function of capture probe position in the 3′ region of the SSA4 mRNA.
- FIG. 31 is a schematic illustration of mRNA splicing.
- FIG. 32 is a schematic illustration of alternative mRNA splicing.
- FIG. 33 is a schematic illustration of probes of the invention.
- FIG. 34 is a schematic illustration of probes of the invention.
- FIG. 35 illustrates an exemplary computer for use in the methods of the invention.
- FIGS. 36 a - 36 d show the sensitivity and specificity of LNA oligonucleotide capture probes (black solid bars) compared to DNA capture probes (white, open bars) on expression microarrays. Fluorescence intensity is shown in arbitrary units (relative measurements). The arrays comprising 50-mer and 40-mer perfect match and 1-5 mismatch capture probes were hybridized at 65° C. in 3 ⁇ SSC with Cy3-labelled cDNA from 10 ⁇ g C. elegans total RNA spiked with yeast a) SWI5 RNA and c) THI4 RNA.
- 36 b and 36 d demonstrate the improved mismatch discrimination with the 50-mer LNA probes by increasing the hybridization temperature from 65° C. to 70° C. hybridized with Cy3-labelled cDNA from 10 ⁇ g C. elegans total RNA spiked with yeast b) SWI5 RNA and d) THI4 RNA.
- FIG. 37 shows the expected (black, solid bars) and observed (white, open bars) fold-of-change in the expression levels of the Cy3-ULS-labelled yeast HSP78 spike RNA as measured by on-chip capture using three different 25-mer oligonucleotide capture probes (DNA control, LNA-T substituted, LNA — 3 substituted in which every third nucleotide was substituted with an LNA monomer).
- DNA control DNA control, LNA-T substituted, LNA — 3 substituted in which every third nucleotide was substituted with an LNA monomer.
- one ng of HSP78 in vitro spike RNA or 200 pg HSP78 in vitro spike RNA was used, respectively.
- the fold change of the HSP78 RNA in the two hybridizations in the comparison is 5-fold.
- FIG. 38 shows the measured intensity levels by on-chip capture using three different 25-mer oligonucleotide capture probe designs (DNA control, LNA_T substituted and LNA C and T substituted probes).
- DNA control LNA_T substituted and LNA C and T substituted probes.
- One (1) ng biotin-labeled HSP78 target was used in the hybridization experiments, followed by staining with Streptavidin Phycoerythrin.
- the LNA_T and LNA_TC substituted 25-mer capture probes show a significantly enhanced on-chip capture of the HSP78 RNA target, compared to the DNA 25-mer control probes under four different hybridization stringency conditions in dicated on the graph.
- FIG. 39 shows the detection of alternatively spliced mRNAs using LNA-substituted 50-mer oligonucleotide capture probes. Parts per million (ppm) calculations indicate spike transcripts per total transcripts in the hybridisation mix. Calculations are based on an average C. elegans RNA being 1000 nucleotides as in Hill et al. (2000) Science 290:809-812.
- the 50-mer LNA-DNA mixmer capture probes substituted with an LNA nucleotide at every third nucleotide position, are able to provide highly accurate measurements for fold-changes in the expression of three homologous, alternatively spliced mRNA variants in the concentration range of 1000 ppm to 10 ppm.
- the quantification of the splice isoforms was carried out using a set of both internal, exon-specific probes and merged, splice junction specific probes, printed onto microarrays and hybridized with complex cDNA target pools spiked with different cloned artificial splice isoforms in which the middle exon was either alternatively skipped or excluded completely resulting in the three different splice isoforms; 01-INS3-03, 01-INS4-03 and 01-03.
- FIG. 40 shows the detection of alternatively spliced mRNAs using LNA-substituted 40-mer oligonucleotide capture probes. Parts per million (ppm) calculations indicate spike transcripts per total transcripts in the hybridisation mix. Calculations are based on an average C. elegans RNA being 1000 nucleotides as in Hill et al. (2000) Science 290:809-812.
- the 40-mer LNA-DNA mixmer capture probes substituted with an LNA nucleotide at every third nucleotide position, are able to provide highly accurate measurements for fold-changes in the expression of three homologous, alternatively spliced mRNA variants in the concentration range of 1000 ppm to 10 ppm.
- the quantification of the splice isoforms was carried out using a set of both internal, exon-specific probes and merged, splice junction specific probes, printed onto microarrays and hybridized with complex cDNA target pools spiked with different cloned artificial splice isoforms in which the middle exon was either alternatively skipped or excluded completely resulting in the three different splice isoforms; 01-INS3-03, 01-INS4-03 and 01-03.
- FIGS. 41A-41E show the comparison of different LNA/DNA mixmer oligonucleotide probes in the detection of human satellite-2 repeats by fluorescence in situ hybridization. Experiment conditions: 6.4 pmoles of Cy3 labeled probe was hybridized for 30 minutes at 37° C., after simultaneous denaturation of the target and the probe at 75° C. for 5 minutes.
- B. LNA-3 giving bright signals on chromosomes 1, 16 and 9,
- C Dispersed LNA giving signals on chromosomes 1 and 16 only,
- D. LNA Block giving smaller signals on chromosome 1
- E. DNA control oligonucleotide FISH probe giving no signals on any of the chromosomes.
- FIG. 42 Illustrates the hybridisation of the Cy3-labelled human telomere repeat specific, LNA-2 substituted oligonucleotide probe on human metaphase chromosomes resulted in prominent signals on the telomeres.
- Alternative splicing is the process by which different mature messenger RNAs are produced from the same pre-mRNA. Because the mRNA composition of a given cell determines the proteins present in a cell, this process is an important aspect of a cells gene expression profile.
- Current investigations of transcriptomes i.e., the total complexity of RNA transcripts produced by an organism indicate that at least 50-60% of the genes of complex eukaryotes produce more than one splice variant.
- the present invention provides a novel method for detecting and quantifying the levels of splice variants in complex mRNA pools using LNA discriminating probes and high-throughput LNA oligonucleotide microarray technology.
- the detection concept which uses internal LNA exon probes and/or splice-variant specific exon-exon junction or exon-intron or intron-exon (so-called merged) probes is depicted in FIG. 9.
- exon-specific (or intron-specific) LNA oligonucleotide probes are designed and used to detect the relative levels of a given exon (or intron) in complex mRNA pools using oligonucleotide microarray technology or similar techniques.
- Exon-exon LNA junction probes are designed for multiple or all possible exon-exon combinations or exon-intron combinations.
- the LNA discriminating probes are highly specific and superior compared to DNA oligonucleotides due to the higher ⁇ T m of LNA probes.
- each sub-element i.e., exon structure or exon-intron structure
- a given alternatively spliced mRNA isoform thus giving the exact composition of the mRNA.
- the ratios of each splice variant can be quantified using the combined readouts from both internal and merged LNA probes and control probes.
- the invention is applicable both in single fluor (single channel) or comparative two-fluor (two channel) microarray hybridizations.
- RNA molecules may be constructed in an in vitro transcription vector for the production of clean IVT RNA.
- Both internal and junction-specific LNA oligonucleotide capture probes are designed, synthesized, and spotted onto, e.g., Exiqon's polymer microarray platform.
- the resulting splice-specific microarray is used to validate the LNA discriminating probe concept by spiking the in vitro RNAs individually as well as in different ratios into a complex RNA background for fluorochrome-labelling and array hybridization.
- the internal and merged probes of the invention can also be used in any standard method for the analysis of mRNA splice variants (see, for example, Yeakley et al., Nature Biotechnology 20:353-358, 2002; Clark et al., Science 296:907-910, 2002; Mutch et al., Genome Biology 2(12):preprint00009.1-0009.31, 2001).
- the internal and/or merged probes of the invention can also be used for gene expression profiling of alternative splice variants, oligonucleotide expression microarrays, real-time PCR, and profiling of alternatively spliced mRNAs using microtiterplate assays or fiber-optic arrays.
- RNA splicing is now widely recognized as a means to generate protein diversity.
- Alternative splicing is a key mechanism for regulating gene expression, and any mutation or defect in its regulation can impact considerably cell functions. Therefore, it is likely to be an important source of novel gene and protein targets implicated in human pathology. Industry has long recognized the need for innovative discovery technologies that focuses on the origin of disease for the development of novel diagnostics and therapeutics.
- Comparative Genomic Hybridization is a powerful technology for detection of unbalanced chromosome rearrangements and holds much promise for screening and identification of interstitial submicroscopic rearrangements that otherwise cannot be detected using classical cytogenetic or FISH technologies.
- the adaptation of CGH onto an oligo microarray platform allows detection of small single exon deletion/duplications on a genome wide scale.
- microarrays that can detect, e.g., single exon aberrations. This detection can be achieved by employing LNA mixmer oligos as capture probes for individual exons in selected genes.
- a model system for these methods is the Menkes loci.
- Menkes disease is a lethal-X linked recessive disorder associated with copper metabolism disturbance leading to death in early childhood.
- the Menkes locus has been mapped to Xq13.
- the gene spans about 150 kb genomic region, contains 23 exons, and encodes a 8.5 kb gene transcript.
- the gene for Menkes disease (now designated as A TP7A) encodes a 1500 amino acid membrane-bound Cu-binding P-type ATPase (ATP7A).
- ATP7A P-type ATPase
- the 8.5 kb transcript is expressed in all tissues from normal individuals (though only trace amounts are present in liver), but is diminished or absent in Menkes disease patients.
- Several different kinds of mutations, like chromosome aberrations, point mutations and partial gene deletions affecting A TP7A have been identified in MD patients.
- 50-mer capture probes with LNA spiked in every second, third, and fourth position have been designed for every exon (23 exons) representing ATP7A, using the OligoDesign software tool, described herein.
- the C6-amino-linked capture probes were spotted onto Immobilizer slides and hybridized with patient samples with Cy3 fluorescent dye and a known reference genomic sample with Cy5. After mixing equal amounts of the labelled DNA, the probe is hybridized it to array.
- the ratio of Cy5 signal to Cy3 for each clone indicates differences in chromosome/DNA material. For example, the Cy5 signal is higher than Cy3 if the patient genome has a deletion, and is lower if there is duplication. In regions that are unchanged, the Cy5:Cy3 ratio is 1:1.
- LNA oligonucleotide-based CGH makes it possible to assess a large number of chromosomal aberrations that are being screen for in the cytogenetic clinic.
- standard FISH analysis typically only detects large chromosomal rearrangements.
- an array that contains a series of overlapping probes is used to detect a chromosomal deletion in a nucleic acid sample, such as a patient sample.
- Clinical diagnosis is a key element in healthcare management and point-of-care.
- a large number of analyses in the hospitals are based on the use of robust, cost efficient, sensitive and highly specific diagnostic tests.
- diagnosis of various diseases is performed with a high selectivity and reliability, resulting in confirmation of medical diagnosis, choice of therapy and follow-up treatment as well as prevention.
- clinical diagnosis also contributes to the control of healthcare costs.
- the field of clinical diagnostics involves analyzing biological fluid samples (blood, urine, etc.) or biopsies collected from patients in order to establish the diagnosis of diseases, whether of infectious, metabolic, endocrine or cancerous origin.
- Medical analysis of infectious diseases involves testing and identifying the micro-organisms causing the infection e.g.
- testing for and identifying a micro-organism in blood and determining its susceptibility to antibiotics or detecting an antigen-antibody reaction produced as a response to an attack by a micro-organism in the human body e.g. testing for antibodies for the diagnosis of hepatitis.
- the accurate diagnosis of metabolic and endocrine diseases and cancers, resulting in a disease phenotype with a bodily imbalance involves the measurement of diagnostic substances or elements present in the biological fluids or biopsies. These substances are examined and results are interpreted with reference to known normal values.
- RNA levels in clinical diagnosis have been used successfully in accurate quantification of RNA levels in clinical diagnosis as well as in microbiological control.
- the applications are wide-ranging and include methods for quantification of the regulation and expression of drug resistance markers in tumour cells, monitoring of the responses to chemotherapy, measuring the biodistribution and transcription of gene-encoded therapeutics, molecular assessment of the tumor stage in a given cancer, detecting circulating tumor cells in cancer patients and detection of bacterial and viral pathogens.
- RT-PCR reverse transcription polymerase chain reaction
- RNA levels are real-time nucleic acid sequence based amplification (NASBA) combined with molecular beacon detection molecules.
- NASBA real-time nucleic acid sequence based amplification
- NASBA is a singe-step isothermal RNA-specific amplification method that amplifies mRNA in a double stranded DNA environment, and this method has recently proven useful in the detection of various mRNAs and in the detection of both viral and bacterial RNA in clinical samples.
- van't Veer et al. (Nature 2002: 415, 31) describe the successful use of microarrays in obtaining digital mRNA signatures from breast tumors and the use of these signatures in the precise prediction of the clinical outcome of breast cancer in patients.
- Locked nucleic acid (LNA) oligonucleotides constitute a novel class of bicyclic RNA analogs having an exceptionally high affinity and specificity toward their complementary DNA and RNA target molecules. Besides increased thermal stability, LNA-containing oligonucleotides show significantly increased mismatch discrimination, and allow full control of the melting temperature across microarray hybridizations.
- the LNA chemistry is completely compatible with conventional DNA phosphoramidite chemistry and thus LNA substituted oligonucleotides can be designed to optimize performance.
- LNA oligonucleotides would be well-suited for large-scale clinical studies providing highly accurate genotyping by direct competitive hybridization of two allele-specific LNA probes to e.g. microarrays of immobilized patient amplicons.
- the use of LNA substituted oligonucleotides would increase both sensitivity and specificity in detection and quantification of mRNA levels in clinical samples, either by quantitative RT-PCR, quantitative NASBA or oligonucleotide microarrays, compared with DNA probes.
- Application of LNA oligonucleotides into diagnostic kits would thus significantly enhance their performance.
- LNA substituted oligonucleotides would increase the sensititity and specificity in the detection of alternatively spliced mRNA isoforms and non-coding RNAs either by homogeneous assays (Taqman assay, Lightcycler assay, NASBA) or by oligonucleotide microarrays in a massive parallel analysis setup.
- T m melting temperatures
- Similar melting temperatures also allow the same hybridization conditions to be used for multiple experiments, which is particularly useful for assays involving hybridization to nucleic acids of varying “AT” content.
- current methods often require less stringent conditions for hybridization of nucleic acids with high “AT” content compared to nucleic acids with low “AT” content. Due to this variation in hybridization stringency, current methods may require significant trial and error to optimize the hybridization conditions for each experiment.
- LNA nucleotide analogs increase their binding affinity for DNA and RNA.
- the stability of duplexes can generally be ranked as follows: DNA:DNA ⁇ DNA:RNA ⁇ RNA:RNA ⁇ LNA:DNA ⁇ LNA:RNA ⁇ LNA:LNA.
- the DNA:DNA duplex is thus the least stable and the LNA:LNA duplex the most stable.
- the affinity of the LNA nucleotides A and T corresponds approximately to the affinity of DNA G and C to their complementary bases.
- the capture efficiency of one or more nucleic acids can be increased by including any of the high affinity nucleotides (e.g., LNA units) described herein within the nucleic acids.
- the examples herein also provide algorithms for optimizing the substitution patterns of the nucleic acids to minimize self-complementarity that may otherwise inhibit the binding of the nucleic acids to target molecules.
- LNA A and LNA T substitutions are made to equalize the melting temperatures of the nucleic acids.
- LNA A and LNA C substitutions are made to minimize self-complementarity and to increase specificity.
- LNA C and LNA T substitutions also minimize self-complementarity.
- oligonucleotides containing LNA C and LNA T are desirable because these modified nucleotides are easy to synthesis and are especially useful for applications such as antisense technology in which minimizing cost is especially desirable.
- Capture probes for the Saccharomyces cerevisiae genes SWI5 (YDR146C) and THI4 (YGR144W) were designed as 50-mer standard DNA and different LNA/DNA “mixmer” oligonucleotides (i.e., oligonucleotides containing both LNA and DNA nucleotides) respectively, for comparison (Table 2).
- LNA/DNA “mixmer” oligonucleotides i.e., oligonucleotides containing both LNA and DNA nucleotides
- 40-mer oligonucleotides were designed as truncated versions of the 50-mer capture probes (Table 2).
- LNA oligoarrays The specificity of the LNA oligoarrays was addressed by introducing 1-5 consecutive mismatches positioned in the middle of 40-mer LNA/DNA mixmer capture probes with LNA in every fourth position.
- in vitro synthesized yeast RNA for either SWI5 or THI4 was spiked into Caenorhabditis elegans total RNA for cDNA target synthesis.
- Amplification of the yeast genes was performed using standard PCR with yeast genomic DNA as the template.
- a forward primer containing a restriction enzyme site and a reverse primer containing a universal linker sequence were used.
- the reverse primer was exchanged with a nested primer containing a poly-T20 tail and a restriction enzyme site.
- the DNA fragments were ligated into the pTRIamp 18 vector (Ambion, USA) using the Quick Ligation Kit (New England Biolabs, USA) according to the supplier's instructions and transformed into E. coli DH-5 ⁇ by standard methods.
- the PCR clones were sequenced using M13 forward and M13 reverse primers on an ABI 377 (Applied Biosystems, USA). Synthesis of in vitro RNA was carried out using the MEGAscriptTM T7 Kit (Ambion, USA) according to the manufacturer's instructions.
- microarrays were printed on Immobilizerm MicroArray Slides (Exiqon, Denmark) using the Biochip One Arrayer from Packard Biochip technologies (Packard, USA). The arrays were printed with a spot volume of 2 ⁇ 300 pl of a 10 ⁇ M capture probe solution. Four replicas of the capture probes were printed on each slide.
- RNA Ten ng of S. cerevisiae in vitro synthesized RNA (either SWI5 or THI4) was combined with 10 ⁇ g of C. elegans total RNA and 5 ⁇ g oligo dT primer (T20VN) in an RNase free, pre-siliconized 1.5 mL tube, and the final volume was adjusted with DEPC-water to 8 ⁇ L. The reaction mixture was heated at +70° C.
- First strand cDNA synthesis was carried out by adding 1 ⁇ L of SuperscriptTM II (Invitrogen, 200 U/mL), mixing, and incubating the reaction mixture for one hour at 42° C. An additional 1 ⁇ L of SuperscriptTM II was added, and the cDNA synthesis reaction mixture was incubated for an additional one hour at 42° C.; the reaction was stopped by heating at 70° C. for 5 minutes, and quenching on ice for 2 minutes. The RNA was hydrolyzed by adding 3 ⁇ L of 0.5 M NaOH, and incubating at 70° C. for 15 minutes.
- SuperscriptTM II Invitrogen, 200 U/mL
- the samples were neutralized by adding 3 ⁇ L of 0.5 M HCl, and purified by adding 450 ⁇ L 1 ⁇ TE buffer, pH 7.5 to the neutralized sample and transferring the samples onto a Microcon-30 concentrator.
- the samples were centrifuged at 14000 ⁇ g in a microcentrifuge for 8 minutes, the flow-through was discarded and the washing step was repeated twice by refilling the filter with 450 ⁇ l 1 ⁇ TE buffer and by spinning for ⁇ 12 minutes. Centrifugation was continued until the volume was reduced to 5 ⁇ L, and finally the labelled cDNA probe was eluted by inverting the Microcon-30 tube and spinning at 1000 ⁇ g for 3 minutes.
- the arrays were hybridized overnight using the following protocol.
- the Cy3TM or Cy5TM-labelled cDNA samples were combined in one tube followed by addition of 3 ⁇ L 21 ⁇ SSC (3 ⁇ SSC final), 0.5 ⁇ L 1 M HEPES, pH 7.0 (25 mM final), 25 ⁇ g yeast tRNA (1.25 ⁇ g/tL final), 10 ⁇ g PolyA blocker (0.5 ⁇ g/mL final), 0.6 ⁇ L 10% SDS (0.3% final), and DEPC-treated water to 20 ⁇ L final volume.
- the labelled cDNA target sample was filtered in a Millipore 0.22 micron spin column according to the manufacturer's instructions (Millipore, USA), and the probe was denatured by incubating the reaction at 100° C. for 2 minutes.
- the sample was cooled at 20-25° C. for 5 minutes by spinning at maximum speed in a microcentrifuge.
- a LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on ImmobilizerTM MicroArray Slide, and the hybridization mixture was applied to the array from the side. An aliquot of 30 ⁇ L of 3 ⁇ SSC was added to both ends of the hybridization chamber, and the ImmobilizerTM MicroArray Slide was placed in the hybridization chamber.
- the chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol.
- the Lifterslip coverslip was washed off in 2 ⁇ SSC, pH 7.0 containing 0.1% SDS at room temperature for one minute, followed by washing of the microarrays subsequently in 1.0 ⁇ SSC, pH 7.0 at room temperature for one minute, and then in 0.2 ⁇ SSC, pH 7.0 at room temperature for one minute. Finally, the slides were washed for 5 seconds in 0.05 ⁇ SSC, pH 7.0. The slides were then dried by centrifugation in a swinging bucket rotor at approximately 600 rpm for 5 minutes.
- the 40-mer perfect match LNA capture probes showed a 5-fold to 14-fold increase in the intensity levels compared to DNA oligonucleotides under standard hybridization conditions. Capture probes of other lengths and/or with other LNA substitution patterns can be used similarly.
- TABLE 2 DNA and LNA-modified SWI5 (YDR146C) and THI4 (YGR144W) oligonucleotide capture probes. LNA modifications are depicted by uppercase letters in the sequence, mt denotes the number of mismatches (bolded) in the center of the oligonucleotide with respect to its target cDNA (mRNA), and “mC” denotes LNA methyl cytosine.
- ESTs (expressed sequence tags) desirably indicate different isoforms. If ESTs were only different from the gene annotation(s), this could simply mean that the prediction is wrong, and nothing more. Desirably, there are different EST splice indications in different developmental stages.
- Two gene prediction algorithms e.g., GeneFinder and Genie
- Exons of interest e.g., exons being skipped and their flanking exons
- C. elegans gene TOID3.3 desirably exceed 70 base-pairs.
- Other genes of interest may be selected using one or more of the above criteria or using other criteria, such as the medical relevance of the gene.
- oligonucleotide For each region, an oligonucleotide with a palindromic value below 100 (default value in PICK70, value based on Smith-Waterman algorithm) and with a melting temperature closest to 80° C. was picked. The location of the oligonucleotide within the region was then compared to the list made using the above BLAST search. If the oligonucleotide did not coincide with a BLAST hit exceeding around 25 (consecutive) base-pairs, this oligonucleotide sequence was chosen as a 50-mer capture probe. Otherwise, a new oligo sequence was picked from the PICK70 output.
- oligonucleotide sequences were “BLASTed” against the complete C. elegans genome. The matches were run against a list made from the GenBank reports of the complete genome, indexing whether positions in the genome were genic or intergenic.
- every fourth DNA nucleotide was substituted with an LNA nucleotide, as shown in Table 3.
- the oligonucleotides were synthesized with an anthraquinone (AQ) moiety at the 5′-end of each oligonucleotide (e.g., as described in allowed U.S. Ser. No. 09/611,833), followed by a hexaethyleneglycol tetramer linker region and the LNA/DNA mixmer capture oligonucleotide sequence.
- AQ anthraquinone
- C. elegans wild-type strain (Bristol-N2) was maintained on nematode growth medium (NG) plates seeded with Escherichia coli strain OP50 at 20° C., and the eggs and L1 larvae were prepared as described in Hope, I. A. (ed.) “ C. elegans —A Practical Approach “, Oxford University Press 1999. The samples were immediately flash frozen in liquid N 2 and stored at ⁇ 80° C. until RNA isolation.
- NG nematode growth medium
- a 100 1 aliquot of packed C. elegans worms from a L1 larvae population was homogenized using the FastPrep Bio101 from Kem-En-Tec for 1 minute at speed 6 followed by isolation of total RNA from the extracts using the FastPrep Bio101 kit (Kem-En-Tec) according to the manufacturer's instructions.
- a 50 ⁇ l aliquot of packed C. elegans eggs was homogenized in lysis buffer (RNeasy total RNA purification kit, QIAGEN) containing quartz sand for 3 minutes using a Pellet Pestle Motor followed by isolation of total RNA according to the manufacturer's manual.
- RNA from worms (L1 larvae) as well as eggs was ethanol precipitated for 24 hours at ⁇ 20° C. by addition of 2.5 volumes of 96% EtOH and 0.1 volume of 3M Na-acetate, pH 5.2 (Ambion, USA), followed by centrifugation of the total RNA sample for 30 minutes at 13200 rpm.
- the total RNA pellet was air-dried and redissolved in 6 ⁇ l (worms) or 2.5 ⁇ l (eggs) of diethylpyrocarbonate (DEPC)-treated water (Ambion, USA) and stored at ⁇ 80° C.
- RNA 1.5 ⁇ g from eggs or 1 ⁇ g total RNA from worms (L1 larvae) were mixed with 5 ⁇ g oligo(dT)1 2-18 primer (Amersham Pharmacia Biotech, USA) and 0.5 ⁇ g of random hexamers, pd(N) 6 (Amersham Pharmacia Biotech, USA) and DEPC-treated water to a final volume of 7 ⁇ l. The mixture was heated at 70° C.
- the primers were pre-annealed at 37° C. for 5 minutes, followed by addition of 400 units of Superscipt II reverse transcriptase (Invitrogen, USA).
- First strand cDNA synthesis was carried out at 37° C. for 30 minutes, followed by 2 hours at 42° C., and the reaction was stopped by incubation at 70° C. for 5 minutes, followed by incubation on ice for 5 minutes.
- Unincorporated dNTPs were removed by gel filtration using MicroSpin S-400 HR columns as described below.
- the column was pre-spun for 1 minute at 735 ⁇ g in a 1.5 ml tube, and the column was placed in a new 1.5 ml tube.
- the cDNA sample was slowly applied to the top center of the resin and spun at 735 ⁇ g for 2 minutes.
- the eluate was collected.
- the volume of the eluate was adjusted to 50 ⁇ l with TE-buffer pH 7.0 before being used as the template for linear PCR.
- PCR reactions were carried out using the following program: 95° C. for 5 minutes followed by 30 cycles of PCR using the following cycling program (denaturation at 95° C. for 45 seconds, annealing at 60° C. for 30 seconds, and extension at 72° C. for 1 minute) followed by a final extension step at 72° C. for 10 minutes and incubation on ice for 5 minutes.
- PCR reactions were carried out using the following program: 95° C. for 5 minutes followed by 30 cycles of PCR using the following cycling program (deriaturation at 95° C. for 45 seconds annealing at 60° C. for 30 seconds extension at 72° C. for 1 minute) followed by a final extension step at 72° C. for 10 minutes and incubation on ice for 5 minutes.
- the capture probes were first diluted to a 10 ⁇ M final concentration in 100 mM Na-phosphate buffer pH 7.0 and spotted on Euray COP microarray slides using the Biochip Arrayer One (Packard Biochip Technologies) with a spot volume of 300 ⁇ l and 300 ⁇ m between the spots.
- the capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker for 90 seconds at full power (Stratagene, USA).
- Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides for 1 ⁇ 2 hour in 30 % acetone before rising in milli-Q H 2 O. After washing, the slides were centrifuged at 800 rpm for 2 minutes and stored in a slide box until microarray hybridization.
- the slides were hybridized with 2.5 ⁇ l of the Cy3-labelled and 2.5 ⁇ l of the Cy5-labelled target preparation from eggs and worms, respectively, as described above (see “Reverse transcription (RT)-PCR′′ section) in 25 ⁇ l of hybridization solution, containing 25 mM HEPES, pH 7.0, 3 ⁇ SSC, 0.3% SDS, and 25 ⁇ g of yeast tRNA.
- the target probe was filtered in a Millipore 0.22 micron spin column (Ultrafree-MC, Millipore, USA), denatured by incubation at 100° C. for 5 minutes, cooled at room temperature for 5 minutes, and then carefully applied onto the prepared microarray. One-third of a cover slip was laid over the microarray, and the hybridization was performed for 16-18 hours at 65° C. in a hybridization chamber (DieTech, model Joe deRisi, USA).
- the slides were washed sequentially by plunging gently in 2 ⁇ SSC/0.1% SDS at room temperature until the cover slip falls off into the washing solution, then in 1 ⁇ SSC pH 7.0 (150 mM NaCl, 15 mM Sodium Citrate) at room temperature for 1 minute, then in 0.2 ⁇ SSC, pH 7.0 (30 mM NaCl, 3 mM Sodium Citrate) at room temperature for 1 minute, and finally in 0.05 ⁇ SSC (7.5 mM NaCl, 0.75 mM Sodium Citrate) for 5 seconds, followed by drying of the slides by spinning at 500 rpm for 5 minutes. The slides were stored in a slide box in the dark until scanning.
- the C. elegans let-2 gene microarray was scanned in an ArrayWoRx Scanner (Applied Precision, USA) using an exposure time of 5 seconds, resolution of 5.0, and high (high level) sensitivity.
- the hybridization data were analyzed using the ArrayVision image analysis software package 5.1 (IMAGING Research Inc., USA).
- the detection principle for alternative exon skipping the C. elegans let-2 gene is shown in FIG. 6.
- FIG. 7 analysis of the comparative hybridization data from the C. elegans Let-2 exon 8-1 1 array demonstrates detection of alternative exon skipping of the let-2 exon 9 (eggs) and exon 10 (L1 larvae) using LNA-modified 50-mer capture probes. Capture probes of other lengths and/or with other LNA substitution patterns can be used similarly.
- Capture Probe Design The design method of exon-specific capture probes for the C. elegans gene T01D3.3 exon 4 has been described in example 2.
- Poly(A) + RNA was isolated from the worm samples using the Pick-Pen (Bio-Nobile, Finland) Starter kit combined with the KingFisher mRNA purification kit (Thermol.absystems, Finland) according to the manufacturer's instructions. The yield was 1-2 ⁇ g poly(A) + RNA from approximately 50 mg of C. elegans worms.
- First strand cDNA synthesis was carried out by adding 1 ⁇ L of SuperscriptTM II (Invitrogen, 200 U/mL), mixing, and incubating the reaction mixture for one hour at 42° C. An additional 1 ⁇ L of SuperscriptTM II was added and the cDNA synthesis reaction mixture was incubated for an additional one hour at 42° C.; the reaction was stopped by heating at 70° C. for 5 minutes, and quenching on ice for 2 minutes. The RNA was hydrolyzed by adding 3 ⁇ L of 0.5 M NaOH and incubating at 70° C. for 15 minutes.
- SuperscriptTM II Invitrogen, 200 U/mL
- the samples were neutralized by adding 3 ⁇ L of 0.5 M HCl and purified by adding 450 ⁇ L 1 ⁇ TE buffer, pH 7.5 to the neutralized sample and transferring the samples onto a Microcon-30 concentrator.
- the samples were centrifuged at 14000 ⁇ g in a microcentrifuge for ⁇ 8 minutes, the flow-through was discarded, and the washing step was repeated twice by refilling the filter with 450 ⁇ l 1 ⁇ TE buffer and by spinning for ⁇ 12 minutes. Centrifugation was continued until the volume was reduced to 5 ⁇ L, and finally the labelled cDNA probe was eluted by inverting the Microcon-30 tube and spinning at 1000 ⁇ g for 3 minutes.
- the C. elegans gene T01D3.3/exon 4 capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by either a hexaethyleneglycol-2 or a hexaethyleneglycol-4 (HEG2/HEG4) linker (Table 4).
- the capture probes were first diluted to a 10 ⁇ M final concentration in 100 mM Na-phosphate buffer pH 7.0, followed by a two-fold dilution series (10 ⁇ M, 5 ⁇ M, 2.5 uM, 1.25 ⁇ M, 0.625 ⁇ M, 0.31 ⁇ M, and 0.155 ⁇ M) and spotted on Exiqon's polycarbonate microarray slides using the Biochip Arrayer One (Packard Biochip Technologies, USA) with a spot volume of 3 ⁇ 300 pl and 400 ⁇ m between the spots.
- the capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker for 90 seconds at full power (Stratagene, USA).
- Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides for 24 hours in milli-Q H 2 O. After washing, the slides were dried in an oven at 37° C. for 30 minutes and stored in a slide box until microarray hybridization.
- the arrays were hybridized overnight using the following protocol.
- the Cy3TM-labelled cDNA sample was combined with 3 1L 21 ⁇ SSC (3 ⁇ SSC final), 0.5 ⁇ L 1 M HEPES, pH 7.0 (25 mM final), 25 ⁇ g yeast tRNA (1.25 ⁇ g/mL final), 10 ⁇ g PolyA blocker (0.5 ⁇ g/ ⁇ L final), 0.6 ⁇ L 10% SDS (0.3% final), and DEPC-treated water to 20 ⁇ L final volume.
- the labelled cDNA target sample was filtered in a Millipore 0.22 micron spin column according to the manufacturer's instructions (Millipore, USA), and the probe was denatured by incubating the reaction at 100° C. for 2 minutes.
- the sample was cooled at 20-25° C. for 5 minutes by spinning at maxium speed in a microcentrifuge, and then carefully applied on top of the microarray.
- a cover slip was laid over the microarray and the hybridization was performed for 16 hours at 63° C. in a hybridization chamber (Coming, USA) submerged in a water bath, with an aliquot of 30 ⁇ L of 3 ⁇ SSC added to both ends of the hybridization chamber to prevent evaporation. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol.
- the coverslip was washed off in 2 ⁇ SSC, pH 7.0 containing 0.1% SDS at room temperature for one minute, followed by washing of the microarrays subsequently in 1.0 x SSC, pH 7.0 at room temperature for one minute, and then in 0.2 ⁇ SSC, pH 7.0 at room temperature for one minute. Finally, the slides were washed for 5 seconds in 0.05 ⁇ SSC, pH 7.0. The slides were then dried by centrifugation in a swinging bucket rotor at approximately 600 rpm for 5 minutes.
- the C. elegans gene TOl D3.3 exon 4 array was scanned in an ArrayWoRx Scanner (Applied Precision, USA) using an exposure time of 5 seconds, resolution of 5.0, and high (high level) sensitivity.
- the hybridization data were analyzed using the ArrayVision image analysis software package 5.1 (IMAGING Research Inc., USA). As shown in FIGS. 8A and 8B, analysis of the hybridization data from the C. elegans gene 26/T01D3.3 exon 4 array demonstrates that the use of LNA-modified capture probes for the C.
- Capture probe design Exon-specific capture probes for the C. elegans gene T01D3.3 exons 4 and 5 were designed as described in Example 2.
- the C. elegans gene T01D3.3/exon 4-5 capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by either a hexaethyleneglycol-2 or a hexaethyleneglycol-4 (HEG2/HEG4) linker (Table 5).
- the capture probes were first diluted to a 10 ⁇ M final concentration in 100 mM Na-phosphate buffer pH 7.0, followed by a two-fold dilution series (10 ⁇ M, 5 ⁇ M, 2.5 ⁇ M, 1.25 ⁇ M, 0.625 1M, 0.31, ⁇ M, and 0.155 ⁇ M) and spotted on Euray polycarbonate microarray slides using the Biochip Arrayer One (Packard Biochip Technologies) with a spot volume of 3 ⁇ 300 pl and 400 ⁇ m between the spots.
- the capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker for 90 seconds at full power (Stratagene, USA).
- Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides for 24 hours in milli-Q H 2 O. After washing, the slides were dried in an oven at 37° C. for 30 minutes, and stored in a slide box until microarray hybridization.
- the slides were hybridized with a high (saturated) concentration of 1 ⁇ M of each gene T10D3.3, exon 4 or 5 target oligo (Table 5) in 50 ⁇ l of hybridization solution, containing 25 mM HEPES, pH 7.0, 3 ⁇ SSC, 0.22% SDS, and 0.8 ⁇ g/ ⁇ l of poly(A) blocker.
- the target probes were filtered in a Millipore 0.45 micron spin column (Ultrafree-MC, Millipore, USA), denatured by incubation at 100° C. for 2 minutes, cooled at room temperature for 5 minutes, and then carefully applied onto the prepared microarray. One-half of a cover slip was laid over the microarray, and the hybridization was performed for 16-18 hours at 63° C. in a hybridization chamber (Coming, USA).
- the slides were washed sequentially by plunging gently in 1 ⁇ SSCT (150 mM NaCl, 15 mM Sodium Citrate+Tween 20) at room temperature for one minute, then in 0.2 ⁇ SSCT (30 mM NaCl, 3 mM Sodium Citrate+Tween 20) at room temperature for one minute, and finally in Milli Q water, followed by drying of the slides in an oven at 37° C. for 30 minutes.
- the slides were Cy5 labelled using a Cy5-straptavidin target.
- the C. elegans gene T01D3.3 exon 4-5 microarray was scanned in an ArrayWoRx Scanner (Applied Precision, USA) using an exposure time of 5 seconds, resolution of 5.0, and high (high level) sensitivity.
- the hybridization data were analyzed using the ArrayVision image analysis software package 5.1 (IMAGING Research Inc., USA).
- FIGS. 10A and 10B analysis of the hybridization data from the C. elegans gene 26/TOI D3.3 array demonstrates that both the DNA as well as the LNA capture probes for the C. elegans TOID3.3 exons 4 (FIG. 10A) and exon 5 (FIG.
- LNA-modified oligonucleotide capture probes every third DNA nucleotide was substituted with an LNA nucleotide.
- the probes designed to capture the junction of the recombinant splice variants were designed with LNA modifications in a block of five consecutive LNAs nucleotides, two on the 5′ side of the splice junction and three on the 3′ side of the splice junction. All capture probes are shown in Table 6. TABLE 6 Internal, exon-specific and merged, exon—exon junction specific oligonucleotide capture probes.
- the splice variant capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by a hexaethyleneglycol-2 (HEG2) linker.
- the capture probes were first diluted to a 20 ⁇ M final concentration in 100 mM Na-phosphate buffer pH 7.0, and spotted on the Immobilizer polymer microarray slides (Exiqon, Denmark) using the Biochip Arrayer One (Packard Biochip Technologies, USA) with a spot volume of 2 ⁇ 300 pl and 300 ⁇ m between the spots.
- the capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker with 2300 ⁇ joules (Stratagene, USA).
- Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides two times 15 minutes in 1 ⁇ SSC. After washing, the slides were dried by centrifugation at 1000 ⁇ g for 2 minutes, and stored in a slide box until microarray hybridization.
- Genomic DNA was prepared from a wild-type standard laboratory strain of Saccharomyces cerevisiae using the Nucleon MiY DNA extraction kit (Amersham Biosciences, USA) according to the supplier's instructions. Amplification of the partial yeast gene was performed using standard PCR with yeast genomic DNA as the template. In the first step of amplification, a forward primer containing a restriction enzyme site and a reverse primer containing a universal linker sequence were used. In this step, 20 base-pairs were added to the 3′-end of the amplicon, next to the stop codon. In the second step of amplification, the reverse primer was exchanged with a nested primer containing a poly-T 20 tail and a restriction enzyme site.
- the SWI5 amplicon contains 730 bp of the SWI5 ORF plus a 20 bp universal linker sequence and a poly-A 20 tail.
- the PCR primers used were YDR146C-For-EcoRI (acgtgaattcaaatacagacaatgaaggagatga) (SEQ ID NO:), YDR146C-Rev-Uni (gatccccgggaattgccatgttacctttgattagtttttcattggc (SEQ ID NO:)), and Uni-polyT-BamHI (acgtggatccttttttttttttttttttttttgatccccgggaattgccatg (SEQ ID NO:)).
- the PCR amplicon was cleaved with the restriction enzymes EcoRI and BamHI.
- the DNA fragment was ligated into the pTRIampl 8 vector (Ambion, USA) using the Quick Ligation Kit (New England Biolabs, USA) according to the supplier's instructions and transformed into E. coli DH-5 ⁇ (by standard methods.
- the Arabidopsis thaliana Rubisco small subunit ssu2b gene fragment (gil 7064721) was amplified from genomic DNA using primers named DJ305 (5′-ACTATGATGGACGATACTGGAC-3′ (SEQ ID NO:)) and DJ306 (5′-ATTGGATCGATCCGATGATCCTAATGAAGGC-3′ (SEQ ID NO:)), containing ClaI restriction site linkers.
- the purified PCR fragment was digested with ClaI and then cloned into the swi5 (gI:7839148) vector at the unique ClaI site (atcgat) giving each insert a flanking sequence from the original yeast SWI5 insert (named exon01 and exon 03, FIG. 11).
- the product was inserted in the reverse orientation, so that the insert sequence is as follows: AtcgatCCGATGATCCTAATGAAGGCGCCCGGGTACTCCTTCTTGCATTCTTCAACTTCC (SEQ ID NO:) TTCAACACTTGAGCGGAGTCGGTGCATCCGAACAATGGAAGCTTCCACATTGTCCAG TATCGTCCATCATAGTatcgat.
- the Arabidopsis thaliana Lea gene (gil 526423) was amplified from genomic DNA with primers named DJ307 (5′-GGAATTATCGATGTGTGATAGGATCAGTGTTCAG-3′ (SEQ ID NO:)), and DJ308 (5′-AATTGGATCGATATTAGCAGTCTCCTTCGCC-3′ (SEQ ID NO:)), including the ClaI linker sites as above.
- the PCR fragment was digested with ClaI cloned into the yeast SWI5 IVT construct as above at the unique ClaI site.
- the fragment was inserted in the forward orientation, resulting in the following insert sequence: atcgatGTGTGATAGGTTCAGTGTTCAGGGCTGTCCAAGGAACGTATGAGCATGCGAGA (SEQ ID NO:) GACGCTGTAGTTGGAAAAACCCACGAAGCGGCTGAGTCTACCAAAGAAGGAGCTCA GATAGCTTCAGAGAAAGCGGTTGGAGCAAAGGACGCAACCGTCGAGAAAGCTAAG GAAACCGCTGATTATACTGCGGAGAAGGTGGGTGAGTATAAAGACTATACGGTTGA TAAAGCTAAAGAGGCTAAGGACACAACTGCAGAGAAGGCGAAGGAGACTGCTAATa tcgat.
- RNA from the splice variants were made using the MEGAscriptTM high yield transcription kit according to the manufacturer's instructions (Ambion, USA). The yield of IVT RNA was quantified at a Nanodrop spectrophotometer (Nanodrop Technologies, USA, FIG. 11). Isolation of total RNA from C. elegans C. elegans wild-type strain (Bristol-N2) was maintained on nematode growth medium (NG) plates seeded with Escherichia coli strain OP50 at 20° C., and the mixed stages of the nematode were prepared as described by Hope (ed.) (“ C. elegans —A Practical Approach”, Oxford University Press 1999). The samples were immediately flash frozen in liquid N 2 and stored at ⁇ 80° C. until RNA isolation.
- NG nematode growth medium
- a 100 ⁇ l aliquot of packed C. elegans worms from a mixed stage population was homogenized using the FastPrep Bio101 from Kem-En-Tec for one minute, speed 6 followed by isolation of total RNA from the extracts using the FastPrep Biol ⁇ kit (Kem-En-Tec) according to the manufacturer's instructions.
- RNA pellet was air-dried and redissolved in 10 ⁇ l of diethylpyrocarbonate (DEPC)-treated water (Ambion, USA) and stored at ⁇ 80° C.
- DEPC diethylpyrocarbonate
- Unincorporated dNTPs were removed by gel filtration using MicroSpin S-400 HR columns as described below.
- the column was pre-spun for one minute at 1500 ⁇ g in a 1.5 ml tube, and the column was placed in a new 1.5 ml tube.
- the cDNA sample was slowly applied to the top center of the resin and spun 1500-x g for 2 minutes. The eluate was collected.
- RNA was degraded by adding 3 ⁇ l of 0.5 M NaOH.
- the solution was mixed well and incubated at 70° C. for 15 minutes.
- the solution was neutralized by adding 3 ⁇ l of 0.5 M HCl and mixed well.
- the labelled cDNA target sample was filtered in Millipore 0.22 ⁇ l filter spin column (Ultrafree-MC, Millipore, USA) according to the manufacturer's instructions, followed by incubation of the reaction mixture at 100° C. for 2-5 minutes.
- the cDNA probes were cooled at room temperature for 2-5 minutes by spinning at maxium speed in a microcentrifuge.
- a LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on Immobilizerm MicroArray Slide, and the hybridization mixture was applied to the array from the side.
- An aliquot of 30 ⁇ L of 3 ⁇ SSC was added to both ends of the hybridization chamber, and the ImmobilizerTM MicroArray Slide was placed in the hybridization chamber (DieTech, USA). The chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol.
- the slides were washed sequentially by plunging gently in 2 ⁇ SSC/0.1% SDS at room temperature until the cover slip falls of into the washing solution, then in 1 ⁇ SSC pH 7.0 (150 mM NaCl, 15 mM Sodium Citrate) at room temperature for one minute, then in 0.2 ⁇ SSC, pH 7.0 (30 mM NaCl, 3 mM Sodium Citrate) at room temperature for one minute, and finally in 0.05 ⁇ SSC (7.5 mM NaCl, 0.75 mM Sodium Citrate) for 5 seconds, followed by drying of the slides by spinning at 1000 ⁇ g for 2 minutes.
- the slides were stored in a slide box in the dark until scanning.
- the splice variant microarray was scanned in a ScanArray 4000XL confocal laser scanner (Packard Instruments, USA). The hybridization data were analyzed using the GenePix Pro 4.01 microarray analysis software (Axon, USA).
- microarrays were printed on ImmobilizerTM MicroArray Slides (Exiqon, Denmark) using the Biochip One Arrayer from Packard Biochip technologies (Packard, USA). The arrays were printed with a spot volume of 2 ⁇ 300 pl of a 10 ⁇ M (final concentration) capture probe dilution. Four replicas of the capture probes were printed on each slide
- the arrays were hybridized overnight using the following protocol.
- the desired amount of biotin-labelled oligonucleotide was combined in one tube followed by addition of 3 ⁇ L 21 ⁇ SSC (3 ⁇ SSC final), 0.5 1L 1 M HEPES, pH 7.0 (25 mM final), 25 ⁇ g yeast tRNA (1.25 ⁇ g/ ⁇ L final), 0.6 ⁇ L 10% SDS (0.3% final), and DEPC-treated water to 20 ⁇ L final volume.
- the biotin-labelled target sample was filtered in a Millipore 0.22 micron spin column according to the manufacturer's instructions (Millipore, USA), and the probe was denatured by incubating the reaction at 100° C. for 2 minutes.
- the sample was cooled at 20-25° C. for 5 minutes by spinning at maxium speed in a microcentrifuge.
- a LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on ImmobilizerTM MicroArray Slide, and the hybridization mixture was applied to the array from the side.
- An aliquot of 30 ⁇ L of 3 ⁇ SSC was added to both ends of the hybridization chamber, and the ImmobilizerTM MicroArray Slide was placed in the hybridization chamber.
- the chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol.
- the Lifterslip coverslip was washed off in 2 ⁇ SSC, pH 7.0 containing 0.1% SDS at room temperature for 1 minute, followed by washing of the microarrays subsequently in 10.1 ⁇ SSC, pH 7.0 at room temperature for 1 minute, and then in 0.2 ⁇ SSC, pH 7.0 at room temperature for 1 minute. Finally, the slides were washed for 5 seconds in 0.05 ⁇ SSC, pH 7.0. The slides were then dried by centrifugation in a swinging bucket rotor at approximately 200 G for 2 minutes.
- LNA-modified Oligonucleotides in Microarrays Provides Significantly Improved Sensitivity in Comparative Genome Hybridization (CGH).
- This example demonstrates the advantages of using LNA oligonucleotide microarrays in Comparative Genome Hybridization (CGH) experiments.
- Capture probes for all 23 exons of the Menkes gene (A TP7A) were designed as 50-mer standard DNA and different LNA/DNA mixmer oligonucleotides, respectively, for comparison (FIG. 17).
- the C6-amino-linked capture probes were applied to Immobilizer slides and hybridized with patient DNA samples labelled with a Cy3 fluorescent dye.
- Fluorescent Cy3 labelled patient genomic DNA was hybridized to microarrays spotted with the CGH capture probes listed in FIG. 17. Compared to DNA capture probes, capture probes with LNA in every second position (LNA-2) had a significantly better capture rate of non-amplified labelled genomic patient DNA as shown in FIGS. 14-16. Capture probes of other lengths and/or with other LNA substitution patterns can be used similarly.
- This example demonstrates the use of the C. elegans LNA tox oligoarray in gene expression profiling experiments in the nematode Caenorhabditis elegans .
- the C. elegans tox oligoarray monitors the expression of a selection of 1 10 genes relevant for general stress response and for the metabolism of toxic compounds. Two different capture probes for each of these target genes were designed and included in the LNA tox array.
- the C. elegans LNA tox oligoarray contained capture probes providing control for cDNA synthesis efficiency and the developmental stage of the nematode. Capture probes for constitutively expressed genes for data set normalization were also included on the C. elegans LNA tox oligoarray.
- microarrays were printed on ImmobilizerM MicroArray Slides (Exiqon, Denmark) using the Biochip One Arrayer from Packard Biochip technologies (Packard, USA). The arrays were printed with a spot volume of 2 ⁇ 300 pl of a 10 1M capture probe solution. Four replicas of the capture probes were printed on each slide.
- First strand cDNA synthesis was carried out by adding ⁇ L of SuperscriptTM II (Invitrogen, 200 U/mL), mixing, and incubating the reaction mixture for 1 hour at 42° C. An additional 1 ⁇ L of SuperscriptTM II was added, and the cDNA synthesis reaction mixture was incubated for an additional 1 hour at 42° C.; the reaction was stopped by heating at 70° C. for 5 minutes, and quenching on ice for 2 minutes. The RNA was hydrolyzed by adding 3 ⁇ L of 0.5 M NaOH, and incubating at 70° C. for 15 minutes.
- SuperscriptTM II Invitrogen, 200 U/mL
- the samples were neutralized by adding 3 ⁇ L of 0.5 M HCl, and purified by adding 450 ⁇ L 1 ⁇ TE buffer, pH 7.5 to the neutralized sample and transferring the samples onto a Microcon-30 concentrator.
- the samples were centrifuged at 14000 ⁇ g in a microcentrifuge for 8 minutes, the flow-through was discarded, and the washing step was repeated twice by refilling the filter with 450 ⁇ l 1 ⁇ TE buffer and by spinning for ⁇ 12 minutes. Centrifugation was continued until the volume was reduced to 5 ⁇ L, and finally the labelled cDNA probe was eluted by inverting the Microcon-30 tube and spinning at 1 000xg for 3 minutes.
- the arrays were hybridized overnight using the following protocol.
- the Cy3TM and Cy5TM-labelled cDNA samples were combined in one tube followed by addition of 3 ⁇ L 21 ⁇ SSC (3 ⁇ SSC final), 0.5 ⁇ L 1 M HEPES, pH 7.0 (25 mM final), 25 ⁇ g yeast tRNA (1.25 ⁇ g/ ⁇ L final), 0.6 ⁇ L 10% SDS (0.3% final), and DEPC-treated water to 20 ⁇ L final volume.
- the labelled cDNA target sample was filtered in a Millipore 0.22 micron spin column according to the manufacturer's instructions (Millipore, USA), and the probe was denatured by incubating the reaction at 100° C. for 2 minutes. The sample was cooled at 20-25° C.
- the Lifterslip coverslip was washed off in 2 ⁇ S SC, pH 7.0 containing 0.1% SDS at room temperature for 1 minute, followed by washing of the microarrays subsequently in 10.1 ⁇ SSC, pH 7.0 at room temperature for 1 minute, and then in 0.2 ⁇ SSC, pH 7.0 at room temperature for 1 minute. Finally, the slides were washed for 5 seconds in 0.05 ⁇ SSC, pH 7.0. The slides were then dried by centrifugation in a swinging bucket rotor at approximately 200 G for 2 minutes. The slide was then ready for scanning.
- the splice variant capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by a hexaethyleneglycol-2 (HEG2) linker.
- the capture probes were first diluted to a 20 uM final concentration in 100 mM Na-phosphate buffer pH 7.0, and spotted on the Immobilizer polymer microarray slides (Exiqon, Denmark) using the Biochip Arrayer One (Packard Biochip Technologies, USA) with a spot volume of 2 ⁇ 300 pl and 300 ⁇ m between the spots.
- the capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker with 2300 ⁇ joules (Stratagene, USA).
- Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides two times 15 minutes in 1 ⁇ SSC. After washing, the slides were dried by centrifugation at 1000 ⁇ g for 2 minutes, and stored in a slide box until microarray hybridization.
- Genomic DNA was prepared from a wild type standard laboratory strain of Saccharomyces cerevisiae using the Nucleon MiY DNA extraction kit (Amersham Biosciences, USA) according to the supplier's instructions. Amplification of the partial yeast gene was performed using standard PCR using yeast genomic DNA as template. In the first step of amplification, a forward primer containing a restriction enzyme site and a reverse primer containing a universal linker sequence were used. In this step, 20 bp was added to the 3′-end of the amplicon, next to the stop codon. In the second step of amplification, the reverse primer was exchanged with a nested primer containing a poly-T 20 tail and a restriction enzyme site. The SWI5 amplicon contains 730 bp of the SWI5 ORF plus 20 bp universal linker sequence and a poly-A 20 tail.
- PCR primers used were; YDR146C-For-EcoRI: acgtgaattcaaatacagacaatgaaggagatga (SEQ ID NO:) YDR146C-Rev-Uni: gatccccgggaattgccatgttacctttgattag (SEQ ID NO:) ttttcattggc Uni-polyT-BamHI: acgtggatcctttttttttttttttttttttttttgatc (SEQ ID NO:) cccgggaattgccatg,
- the PCR amplicon was cut with the restriction enzymes, EcoRI +BamHI.
- the DNA fragment was ligated into the pTRIamp 18 vector (Ambion, USA) using the Quick Ligation Kit (New England Biolabs, USA) according to the supplier's instructions and transformed into E. coli DH-5 ⁇ by standard methods.
- the Arabidopsis thaliana Rubisco small subunit ssu2b gene fragment (gil7064721) was amplified from genomic DNA by primers named DJ305 5′-ACTATGATGGACGATACTGGAC-3′ (SEQ ID NO:) and DJ306 5′-ATTGGATCGATCCGATGATCCTAATGAAGGC-3′ (SEQ ID NO:), containing ClaI restriction site linkers.
- the purified PCR fragment was digested with ClaI and then cloned into the swi5 (gI:7839148) vector at the unique ClaI site (atcgat) giving each insert a flanking sequence from the original yeast SWI5 insert (named exon01and exon 03, FIG. 11).
- the product was inserted in the reverse orientation, so that the insert sequence is: atcgatCCGATGATCCTAATGAAGGCGCCCGGGTACT (SEQ ID NO:) CCTTCTTGCATTCTTCAACTTCCTTCAACACTTGAGC GGAGTCGGTGCATCCGAACAATGGAAGCTTCCACATT GTCCAGTATCGTCCATCATAGTatcgat
- the Arabidopsis thaliana Lea gene (gil 526423) was amplified from genomic DNA with primers named DJ307 5′-GGAATTATCGATGTGTGATAGGATCAGTGTTCAG-3′ (SEQ ID NO:) and DJ308 5′-AATTGGATCGATATTAGCAGTCTCCTTCGCC-3′ (SEQ ID NO:) including the ClaI linker sites as above.
- the PCR fragment was digested with ClaI cloned into the yeast SWI5 IVT construct as above at the unique ClaI site.
- the fragment was inserted in the forward orientation, resulting in the following insert sequence: atcgatGTGTGATAGGTTCAGTGTTCAGGGCTGTCCA (SEQ ID NO:) AGGAACGTATGAGCATGCGAGAGACGCTGTAGTTGGA AAAACCCACGAAGCGGCTGAGTCTACCAAAGAAGGAG CTCAGATAGCTTCAGAGAAAGCGGTTGGAGCAAAGGA CGCAACCGTCGAGAAAGCTAAGGAAACCGCTGATTAT ACTGCGGAGAAGGTGGGTGAGTATAAAGACTATACGG TTGATAAAGCTAAAGAGGCTAAGGACACAACTGCAGA GAAGGCGAAGGAGACTGCTAATatcgat.
- RNA Preparation from Splice Variant Vectors In vitro RNA from the splice variants were made using the MEGAscriptTM high yield transcription kit according to the manufacturer's instructions (Ambion, USA). The yield of IVT RNA was quantified at a Nanodrop spectrophotometer (Nanodrop Technologies, USA).
- C. elegans wild-type strain (Bristol-N2) was maintained on nematode growth medium (NG) plates seeded with Escherichia coli strain OP50 at 20° C., and the mixed stages of the nematode were prepared as described in Hope, I. A. (ed.) “ C. elegans -A Practical Approach”, Oxford University Press 1999. The samples were immediately flash frozen in liquid N 2 and stored at ⁇ 80° C. until RNA isolation.
- NG nematode growth medium
- a 100 ⁇ l aliquot of packed C. elegans worms from a mixed stage population was homogenized using the FastPrep Biol 101 from Kem-En-Tec for 1 minute, speed 6 followed by isolation of total RNA from the extracts using the FastPrep Bio 101 kit (Kem-En-Tec) according to the manufacturer's instructions.
- the eluted total RNA was ethanol precipitated for 24 hours at ⁇ 20° C. by addition of 2.5 volumes of 96% EtOH and 0.1 volume of 3M Na-acetate, pH 5.2 (Ambion, USA), followed by centrifugation of the total RNA sample for 30 minutes at 13200 rpm.
- the total RNA pellet was air-dried and redissolved in 10 ⁇ l of diethylpyrocarbonate (DEPC)-treated water (Ambion, USA) and stored at ⁇ 80° C.
- DEPC diethylpyrocarbonate
- RNAs corresponding to the splice variant #1 (exon01-INS3-exon03 (1-INS3-3) and splice variant #2 (exon01-INS4-exon03 (1-INS3-3) were spiked into 10 ⁇ g of C. elegans reference total RNA sample in two different ratios.
- the first target pool (KU007) contained 10 ng of splice variant #1 (1-INS3-3) transcript and 2 ng of variant #2 (1-INS4-3) transcript, a ratio of 5:1.
- the second target pool (KU008) contained 2 ng variant #1 (1-INS3-3) transcript and 10 ng of splice variant #2 (1-INS4-3) transcript, a ratio of 1:5. Both mRNA pools were combined in separate labeling reactions with 5 ⁇ g anchored oligo(dT 2 O) primer and DEPC-treated water to a final volume of 8 ⁇ l. The mixture was heated at 70° C.
- elegans reference RNA alone labeled with Cy3-dCTP according to the labeling method described above for the splice variant spikes. All four cDNA syntheses were carried out at 42° C. for 2 hours, and the reaction was stopped by incubation at 70° C. for 5 minutes, followed by incubation on ice for 5 minutes.
- Unincorporated dNTPs were removed by gel filtration using MicroSpin S-400 HRcolumns as described below.
- the column was pre-spun for 1 minute at 1500 ⁇ g in a 1.5 ml tube, and the column was placed in a new 1.5 ml tube.
- the cDNA sample was slowly to the top center of the resin, spun 1500-xg for 2 minutes, and the eluate was collected.
- the RNA was hydrolyzed by adding 3 ⁇ l of 0.5 M NaOH, mixing, and incubating at 70° C. for 15 minutes.
- the samples were neutralized by adding 3 ⁇ l of 0.5 M HCl and mixing, followed by addition of 450 ⁇ l 1xTE, pH 7.5 to the neutralized sample and transfer onto a Microcon-30 concentrator (prior to use, 500 ⁇ l 1 ⁇ TE was spun through the column to remove residual glycerol).
- the samples were centrifuged at 14000-xg in a microcentrifuge for 12 minutes. Spinning was continued until volume was reduced to 511.
- the labelled cDNA probes were eluted by inverting the Microcon-30 tube and spinning at 1000-xg for 3 minutes.
- the labelled cDNA target samples were filtered in Millipore 0.22 ⁇ filter spin column (Ultrafree-MC, Millipore, USA) according to the manufacturer's instructions, followed by incubation of the reaction mixture at 100° C. for 2-5 minutes.
- the cDNA probes were cooled at room temp for 2-5 minutes by spinning at maximum speed in a microcentrifuge.
- a LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on ImmobilizerTM MicroArray Slide, and the hybridization mixture was applied to the array from the side.
- An aliquot of 30 ⁇ L of 3 ⁇ SSC was added to both ends of the hybridization chamber, and the ImmobilizerTM MicroArray Slide was placed in the hybridization chamber (DieTech, USA).
- the chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol. The slides were washed sequentially by plunging gently in 2 ⁇ SSC/0.1% SDS at room temperature until the cover slip falls off into the washing solution, then in 1 ⁇ SSC pH 7.0 (150 mM NaCl, 15 mM Sodium Citrate) at room temperature for 1 minute, then in 0.2 ⁇ SSC, pH 7.0 (30 mM NaCl, 3 mM Sodium Citrate) at room temperature for 1 minute, and finally in 0.05 ⁇ SSC (7.5 mM NaCl, 0.75 mM Sodium Citrate) for 5 seconds, followed by drying of the slides by spinning at 1000 ⁇ g for 2 minutes. The slides were stored in a slide box in the dark until scanning.
- the splice variant microarray was scanned in a ScanArray 4000XL confocal laser scanner (Packard Instruments, USA). The hybridization data were analysed using the GenePix Pro 4.01 microarray analysis software (Axon, USA). Only the Cy5 (650 nm) data were examined as both hybridizations produced comparable, and acceptably low, signal from the C. elegans reference RNA alone (Cy3 channel).
- the ratio of signal in hybridization KU007/KU008 should be 5 for probes designed to exon junctions of the INS3 splice variant #1 and 0.2 for probes corresponding to 1-INS4 splice variant #2.
- the capture probes were designed to a 602-nucleotide sequence in the 3′-region of the Yeast ( S. cerevisiae ) 70 kDa heat shock protein (SSA4) gene.
- the 602-base pair sequence is shown in Table 16.
- For the LNA-spiked oligonucleotide capture probes every third DNA nucleotide was substituted with a LNA nucleotide. All capture probes are shown in Table 17.
- TABLE 16 Six hundred and two (602) base pair sequence stretch of the S. cerevisiae ssa4 gene. The underlined segments indicate the position of the capture probes. First underline is equal to capture probe YER103W-554, second underline is equal to capture probe YER103W-492 and so forth. (SEQ ID NO:)
- the SSA4 capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by a hexaethyleneglycol-2 (HEG2) linker.
- the capture probes (Table 17) were first diluted to a 20 ⁇ M final concentration in 100 mM Na-phosphate buffer pH 7.0, and spotted on the Immobilizer microarray slides (Exiqon, Denmark) using the Biochip Arrayer One (Packard Biochip Technologies) with a spot volume of 2 ⁇ 300 pl and 400 ⁇ m between the spots.
- the capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker with 2300 ⁇ joules (Stratagene, USA).
- Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides two times 15 minutes in 1 ⁇ SSC. After washing, the slides were dried by centrifugation at 1000xg for 2 minutes, and stored in a slide box until microarray hybridization.
- Saccharomyces cerevisiae wild-type (BY4741, MATa; his3 ⁇ 1; leu2 ⁇ 0; met15 ⁇ 0 ; ura3 ⁇ O) and ⁇ ssa4 (MATa; his3 ⁇ 1; leu2 ⁇ 0; met15 ⁇ 0; ura3 ⁇ O; YER103w::kanMX4) mutant strains (EUROSCARF) were grown in YPD at 30° C. until the A 600 density of the cultures reached 0.8. Half of the cultures were collected by centrifugation and resuspended in one volume of 40° C. preheated YPD. Incubation was continued for an additional 30 minutes at 30° C. or 40° C. for the standard and heat-shocked cultures, respectively. Cells were harvested by centrifugation and stored at ⁇ 80° C.
- a total of seven cDNA assay mixtures were produced; each with ten (10) ⁇ g total RNA from wtand combined with 5 ⁇ g anchored oligo(dT 2 O) primer and DEPC-treated water to a final volume of 8 ⁇ l. The mixtures were heated at 70° C.
- FIG. 29 shows the gel electrophoresis of fragmented cDNA from the yeast wild-type strain.
- the labelled cDNA target samples were filtered in Millipore 0.22 ⁇ filter spin column (Ultrafree-MC, Millipore, USA) according to the manufacturer's instructions, followed by incubation of the reaction mixture at 100° C. for 2-5 minutes.
- the cDNA probes were cooled at room temp for 2-5 minutes by spinning at maximum speed in a microcentrifuge.
- a LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the SSA4 microarrays spotted on ImmobilizerTM MicroArray Slide, and the hybridization mixture was applied to the array from the side.
- An aliquot of 30 ⁇ L of 3 ⁇ SSC was added to both ends of the hybridization chamber, and the slide was placed in the hybridization chamber (DieTech, USA).
- the chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol. The slides were washed sequentially by plunging gently in 2 ⁇ SSC/0.1% SDS at room temperature until the cover slip falls of into the washing solution, then in 1 ⁇ SSC pH 7.0 (150 mM NaCl, 15 mM Sodium Citrate) at room temperature for 1 minute, then in 0.2 ⁇ SSC, pH 7.0 (30 mM NaCl, 3 mM Sodium Citrate) at room temperature for 1 minute, and finally in 0.05 ⁇ SSC (7.5 mM NaCl, 0.75 mM Sodium Citrate) for 5 seconds, followed by drying of the slides by spinning at 1000 ⁇ g for 2 minutes. The slides were stored in a slide box in the dark until scanning.
- the eukaryotic pre-mRNA is the subject of Splicing and Alternative Splicing, hence sequences refer to RNA sequences, Original sequence refers to pre-mRNA, and splice forms refer to mRNA sequences.
- the splicing is conducted by a cellular machinery named the spliceosome.
- exons and introns can be used to refer to regions of pre-RNA sequences (or more specifically a single splice form). It is noted that a part of the corresponding DNA/pre-mRNA sequence that is an exon (not excised) in one splice form can potentially be absent in another splice form (e.g., partly absent in exon truncation and completely absent in exon skipping).
- the terms “constant regions” and “variable regions” are useful for characterizing the process of identifying different splice forms.
- Splicing can be defined as the production of a new sequence via the excision of part(s) of an original sequence (FIG. 31).
- Alternative splicing can be defined as the production of more than one novel sequence via the excision of different parts of the original sequence.
- When comparing two different splice forms they can be divided into a constant region that is shared by both sequences and a variable region by which the two splice forms differ (FIG. 32).
- Alternative splicing can be categorized in terms of (i) whether or not the variable region is flanked by a single constant region or surrounded by two constant regions, (ii) the size of the variable region (e.g., exon skipping/intron retention vs. extension and truncation) [(intron/exon) 5′ and 3′], and (iii) the number of variable regions (and hence the number of splice forms).
- Capture Probe design can be divided into 3 distinct types according to their position: Merged Probes (MP) or Junction Probes, Unique Internal Probes (UIP), and Shared Internal Probes (SIP) (FIG. 33). Considering the case of a single variable region surrounded by constant regions, there are several different possible capture probe positions for each type (FIG. 34).
- the aim of the analyses can be to determine (i) whether a given original sequence is subject to alternative splicing (i.e., whether there is more than one splice form present), and (ii) whether there is a difference in alternative splicing of the original sequence between two biological samples (i.e., whether the proportions between the two splice forms differ between biological samples).
- the analysis can also be used for data validation.
- Possible biases in the microarray platform include (a) noise in terms of non-specific binding and subsequent false signal, (b) differences in dye labeling efficiency, (c) differences in capture probe affinity, (d) differences in sample conditions (e.g., number of cells, and amount of RNA), and (e) differences in reverse transcriptase efficiency of different splice forms. Biases can be corrected for by various means of normalization and/or standardization.
- 0 denotes the relation between the proportions of the signals (capture probes a and b) between the labeled extracts from biological samples (labeled with Cy5 & Cy3). That is,
- ⁇ can be compared between different transcripts to determine whether they have correlated expression, and ⁇ 's from sets of capture probes from the same transcript (different probes) can be averaged.
- the nucleic acid arrays of the invention can be generated by standard methods for either synthesis of nucleic acid probes that are then bonded to a solid support or synthesis of the nucleic acid probes on a solid support (e.g., by sequential addition of nucleotides to a reactive group on the solid support).
- photogenerated acids are produced in light-irradiate sites of the chip and used to deprotect the 5′-OH group of nucleic acid monomers and oligomers (e.g., to remove an acid-labile protecting group such as 5′—O—DMT) to which a nucleotide is to be added (Gao et al., Nucleic Acid Research 29:4744-4750, 2001).
- Standard methods can also be used to label the nucleic acids in a test sample with, e.g., a fluorescent label, incubate the labeled nucleic acid sample with the array, and remove any unbound or weakly bound test nucleic acids from the array.
- capture probes were immobilized using AQ technology with a HEG5 linker (U.S.P.N. 6,033,784) onto an ImmobilizerTM slide.
- An exemplary chip consists of 288 spots in four replicates (i.e., 1152 spots) with a pitch of 250 ⁇ m, and an exemplary hybridization buffer is 5 ⁇ SSCT (i.e., 750 mM NaCl, 75 mM Sodium Citrate, pH 7.2, 0.05% Tween) and 10 mM MgCl 2 .
- An exemplary target is a 45-mer oligonucleotide with Cy5 at the 5′ end and with a final concentration in the hybridization solution of 1 ⁇ M.
- Hybridization was performed with 200 1L hybridization solution in a hybridization chamber created by attaching a CoverWell gasket to the Immobilizer slide. The incubation was conducted overnight at 4° C. After hybridization, the hybridization solution was removed, and the chamber was flushed with 3 ⁇ 1.0 mL hybridization buffer described above without any target nucleic acid. A coverWellTM chamber was then filled with 200 ⁇ L hybridization solution without target. The slide was observed with a Zeiss Axioplan 2 epifluorescence microscope with a 5 ⁇ Fluar objective and a Cy5 filterset from OMEGA. The temperature of the microscope stage was controlled with a Peltier element. Thirty-five images at each temperature were acquired automatically with a Photometrics camera, automated shutter, and motorized microscope stage. The images were acquired, stitched together, calibrated and stored in stack by the software package “MetaVue”
- Arrays can be generated using capture probes of any desired length (e.g., arrays of pentamers, hexamers, or heptamers.)
- 1, 2, 3, 4, 5, 6, 7, 8, or more nucleotides of the probes are LNA nucleotides.
- at least 1, 2, 3, 5, 7, 9, or all of the A and T nucleotides in the probes are LNA A and LNA T nucleotides.
- LNA nucleotides can be placed in any position of the capture probe, such as at the 5′ terminus, between the 5′ and 3′ termini, or at the 3′ terminus. LNA nucleotides may be consecutive or may be separated by one or more other nucleotides.
- the microarrays can be used to analyze target nucleic acids of any “AT” or “GC” content, and are especially useful for analyzing nucleic acids with high “AT” content because of the increased affinity of the microarrays of the present invention for such nucleic acids compared to traditional microarrays.
- the array has at least 100, 200, 300, 400, 500, 600, 800, 1000, 2000, 5000, 8000, 10000, 15000, 20000, or more different probes.
- nucleotides with a universal base can be included in the capture probes to increase the T m of the capture probes (e.g., capture probes of less than 7, 6, 5, or 4 nucleotides).
- non-discriminatory nucleotides include inosine, random nucleotides, 5 nitro-indole, LNA, inosine, and LNA 2-aminopurine.
- 1, 2, 3, 4, 5, or more nucleotides with a universal base are located at the 5′ and/or 3′ termini of the capture probes.
- An exemplary application of these methods includes comparing hybridization patterns of cDNA or cRNA from a patient sample to classify early-tumors or detect an infection or a diseased state.
- the microarrays of the invention may also be used as a general tool to analyze the PCR products generated by amplification of a test sample with PCR primers for one or more nucleic acids of interest.
- PCR primers can be used to amplify nucleic acids with a particular exon or exon-exon combination, and then the PCR products can be identified and/or quantified using a microarray of the invention.
- PCR primers to specific exons can be used to amplify nucleic acids that are then applied to a microarray for detection and/or quantification as described herein.
- species-specific PCR primers e.g., primers specific for an exon whose sequence differs among species
- the hybridization pattern of the PCR products to the array can be used to distinguish between different bacteria, viruses, or yeast and even between different strains of the same pathogenic species.
- the array is used to determine whether a patient sample contains a bacteria strain that is known to be resistant or susceptible to particular antibiotics or contains a virus or yeast strain known to be resistant or susceptible to certain drugs. Changes in product composition or raw material origin can also be detected using a microarray. The arrays can also be used to determine the composition of mRNA cocktails.
- Exemplary environmental microbiology applications of these arrays include identification of major rRNA types in contaminated soil samples and classification of microbial isolates. These rRNA amplificates are formed from rRNA by rtPCR or from the rDNA gene by conventional PCR. Numerous general and selective primers for different groups of organisms have been published. Most frequently an almost full length amplificate of the 16S rDNA gene is used (e.g., the primers 26F and 1492R). For purifying rRNA from a soil sample, standard methods such as one or more commercial extraction kits from companies such as QIAGEN (“Rneasy”, Q-biogene “RNA PLUS,” or “Total RNA safe” can be used.
- any simultaneous use of more than one primer or probe is made difficult because the involved primers or probes must work under the same conditions.
- An indication of whether or not two or more primers or probes will work under the same conditions is the relative T ms at which the hybridized oligonucleotides dissociate.
- the ⁇ T m is of importance.
- ATM expresses the difference between T m of the match and the T m of the mismatch hybridizations. Generally, the larger ⁇ T m obtained, the more specific detection of the sequence of interest.
- a large ⁇ T m facilitates more probes to be used simultaneously and in this way a higher degree of multiplexity can be applied.
- High affinity nucleotide analogs such a LNA can be also be used universally to equalize the melting properties of oligonucleotides with different AT and CG content.
- the increased affinity of LNA adenosine and LNA thymidine corresponds approximately to the normal affinity of DNA guanine and DNA cytosine.
- An overall substitution of all DNA-A and DNA-T with LNA-A and LNA-T results in melting properties that are nearly sequence independent but only depend on the length of the oligonucleotide. This may be important for design of oligonucleotide probes used in large multiplex analysis.
- the effect of LNA A and T substitutions has been evaluated by predicting the T m value of all possible 9-mer oligonucleotides with different universal substitutions.
- the distribution of the 262,000 T m -values exhibits a very homogeneous T m value for universally LNA A and T substituted oligonucleotides.
- the standard deviation of the melting temperature for all 9-mers drops from 7.7° C. for pure DNA to only 2.2° C. for LNA A and T substituted oligonucleotides. This equalizing effect may also be utilized for photomediated on-chip synthesis of oligonucleotides.
- LNA can be mixed with DNA during standard oligonucleotide synthesis
- LNA can be placed at optimal positions in probes in order to adjust T m .
- the specificity of PCR may also be enhanced by the use of LNA in primers, or probes, and this facilitates a higher degree of multiplexity.
- the T m of the primers or probes can be adjusted to work at the same temperature. Amplification or hybridization is more specific when LNA is included in primers or probes. This is due to the LNA increased ATM, which relates to higher specificity. Once ⁇ T m of the primers or probes is high, more primers or probes can potentially be brought to work together.
- LNA can be used to enhance any experiment that is based on hybridization.
- the series of algorithms described herein have been developed to predict the optimal use of LNA. Melting properties of 129 different LNA substituted capture probes hybridized described herein to their corresponding DNA targets were measured in solution using UV-spectrophotometry.
- the data set was divided into a training set with 90 oligonucleotides and a test set with 39 oligonucleotides.
- the training set was used for training of both linear regression models and neural networks. Neural networks trained with nearest neighbour information, length, and DNA/LNA neighbour effect are efficient for prediction of T m with the given set of data.
- All assays in which DNA/RNA hybridization is conducted may benefit from the use of LNA in terms of increased specificity and quality.
- Exemplary uses include sequencing, primer extension assays, PCR amplification, such as multiplex PCR, allele specific PRamplification, molecular beacons, (e.g., nucleic acids be multiplexed with one colour based on multiple T m 's), Taq-man probes, in situ hybridization probes (e.g., chromosomal and bacterial 16S rRNA probes), capture probes to the mRNA poly-A tail, capture probes for microarray detection of SNPs, capture probes for expression microarrays (sensitivity increased 5-8 times), and capture probes for assessment of alternative mRNA splicing.
- LNA units have different melting properties than DNA and RNA nucleotides.
- thermodynamical models for melting temperature prediction have existed for DNA and RNA only, but not for LNA.
- T m prediction model for LNA/DNA mixed oligonucleotides has been developed (John SantaLucia, Jr. (1998) Proc. Natl. Acad. Sci. 95 1460-1465 and T ⁇ stensen et al. “Prediction of Melting Temperature for LNA (Locked Nucleic Acid) Modified Oligonucleotides” Bioinformatics 2002, Bergen, Norway April 4-7 2002).
- the T m prediction tool is available on-line at the Exiqon website (www.LNA-T m .com and http://www.exiqon.com/Poster/T m pred-ET-view.pdf).
- T m is usually computed using a two-state thermodynamical model (Breslauer, Meth. Enzymol., 259:221-242, 1995).
- T m is usually computed using a two-state thermodynamical model (Breslauer, Meth. Enzymol., 259:221-242, 1995).
- Several different groups have estimated model parameters for nearest neighbors in the sequence based on experimental data (for a review see SantaLucia, Proc. Natl. Acad. Sci., 95:1460-1465, 1998).
- the model described herein predicts the T m of duplexes of mixed LNA/DNA oligonucleotides hybridized to their complementary DNA strands.
- DNA monomers are denoted with lowercase letters
- LNA monomers are denoted with uppercase letters, e.g., there are eight types of monomers in the mixed strand: a, c, g, t, A, C, G and T.
- the model is based on the formula (SantaLucia, 1998, supra; Allawi et al., Biochemistry 36:10581-10594, 1997).
- T m ⁇ ⁇ ⁇ H ⁇ ⁇ ⁇ S + R ⁇ ln ⁇ ( C - C m / 2 ) + 0.368 ⁇ ( L - 1 ) ⁇ ln ⁇ [ Na + ] ,
- C and C m are the concentrations of the two strands where C ⁇ C m , and L is the length of the strands.
- C ⁇ C m /2 is replaced by the total strand concentration C T and a symmetry correction of ⁇ 1.4 cal/k mol is added to ⁇ S (SantaLucia, 1998, supra).
- the LNA model differs from SantaLucia's DNA model in the way the changes in enthalpy ⁇ H and entropy ⁇ S are calculated. As in SantaLucia's model, they depend on nearest neighbor sequence information and special contributions for the terminal base-pairs in the two ends of the duplex. However, with eight types of monomers (LNA and DNA) the increased number of nearest neighbor combinations requires more model parameters to be determined and hence more data.
- ⁇ H and ⁇ s are calculated as a sum of contributions from all nearest neighbor pairs in the sequence.
- the inclusion of LNA doubles the number of monomer types and quadruples the number of possible nearest neighbor pairs.
- Parameter reduction strategies are used for matching the model complexity to limited data sets.
- a strategy for reducing model complexity is to sum ⁇ H from single base-pair contributions, which do not take the influence of adjacent nucleotides into account.
- nearest neighbor contributions are added as a correction term to the single base-pair contributions.
- Another strategy is to use hierarchically reduced monomer alphabets.
- similar monomers are identified with the same letter.
- the smallest alphabet, ⁇ D,L ⁇ simply identifies the monomer type: DNA or LNA.
- the sequence GcTAAcTt can be written as SsWWWsWw or as LDLLLDLD.
- Model parameters were determined using data from melting experiments on hundreds of oligonucleotides.
- thermodynamic quantities The aim of this work has been to estimate T m values as accurately as possible.
- a machine learning approach has been adopted in which the prediction of the physical ⁇ H and ⁇ S quantities is less important.
- the parameters of this model may be inaccurate as thermodynamic quantities.
- the gradient descent algorithm produces a broad ensemble of models in which the ⁇ H and ⁇ S parameters can vary substantially, while maintaining an accuracy in the predicted T m .
- the thermodynamic meaning of ⁇ H and ⁇ S is based on a two-state assumption, which may not be realistic in every case. Even short oligonucleotides can form different secondary structures or melt through multiple-state transitions (T ⁇ stensen et al., J. Phys. Chem. B.
- the T m prediction model has been tested on two data sets that were not used during the training process.
- One set consisted of pure DNA oligonucleotides without LNA monomers and had a standard deviation of the residuals (SEP) of 1.57 degrees.
- the other set consisted of mixed oligonucleotides with both LNA and DNA and had a SEP of 5.25 degrees.
- SEP standard deviation of the residuals
- the difference in prediction accuracy between the two types of oligonucleotides suggests that T m prediction of mixed strands is a more complex task than T m prediction of pure DNA. This is possibly due to irregularities in the duplex helical structure induced by the LNA monomers (Nielsen et al., Bioconjug. Chem. 11:228-238, 2000).
- High affinity nucleotides such as LNA and other nucleotides that are conformationally restricted to prefer the C3′-endo conformation or nucleotides with a modified backbone and/or nucleobase stabilize a double helix configuration.
- the most stable duplex between a high affinity capture oligonucleotide and an unmodified target oligonucleotide should generally arise when all nucleotides in the capture probe or primer are replaced by their high affinity analogue.
- the most stable duplex should thus be formed between a fully modified LNA capture probe and the corresponding DNA/RNA target molecule.
- Such a fully modified capture probe should be more efficient in capturing target molecules, and the resulting duplex is more thermally stable.
- a fully modified capture probe may thus form duplexes with itself, or if it is long enough, internal hairpins that are even more stable than duplexes with the desired target molecule.
- Probes with even a small inverse repeat segment where all constituent positions are substituted with high affinity nucleotides may bind to itself and be unable to bind the target.
- a sequence dependent substitution pattern is desirably used to avoid substitutions in positions that may form self-complementary base-pairs.
- a computer algorithm can be used to automatically determine the optimal substitution pattern for any given capture probe sequence according to the following two criteria.
- the capture probe should contain as many substitutions as possible in order to bind as much target as possible at any given temperature and to increase the thermal stability of the formed duplex.
- the second criterion is substituted with the following alternative criterion to obtain capture probes with similar thermal stability. The number and position of capture probe substitutions should be adjusted so that all the duplexes between capture probes and targets have a similar thermal stability (i.e., T m equalization).
- the second criterion for increasing thermal stability is more desirable that the alternative second criterion for T m equalization.
- the second alternative criterion is desirably used since T m equalization is desirable for these probes and primers.
- An exemplary algorithm works as follows. For each nucleotide sequence in an array of length n, all possible substitution patterns, i.e., 2 ′ different sequences are evaluated. Each evaluation consist of estimating the energetic stability of the duplex between the substituted capture sequence and a perfect match unmodified target (“target duplex”) and the energetic stability of the most stable duplex that can be formed between two substituted capture probes themselves (“self duplex”).
- target duplex perfect match unmodified target
- self duplex the energetic stability of the most stable duplex that can be formed between two substituted capture probes themselves
- the energetic stability estimate for a duplex may be calculated, e.g., using a Smith-Waterman algorithm with the following scoring matrix.
- Gap continuation penalty ⁇ 50 a c g t A C G T a ⁇ 2 c ⁇ 2 ⁇ 2 g ⁇ 2 3 ⁇ 2 t 2 ⁇ 2 1 ⁇ 2 A ⁇ 3 ⁇ 3 ⁇ 3 4 ⁇ 3 C ⁇ 3 ⁇ 3 6 ⁇ 3 ⁇ 3 ⁇ 3 G ⁇ 3 6 ⁇ 3 2 ⁇ 3 9 ⁇ 3 T 4 ⁇ 3 2 ⁇ 3 6 ⁇ 3 3 ⁇ 3
- This scoring matrix was partly based on the best parameter fit to a large (over 1000) number of melting curves of different DNA and LNA containing duplexes and partly by visual scoring of test capture probe efficiency. If desired, this scoring matrix may be optimized by optimizing the parameter fit as well as increasing or optimizing the dataset used to obtain these parameters.
- the heptamer sequence ATGCAGA in which each position can be either an LNA or a DNA nucleotide is used.
- the target duplex formed between a fully modified capture probes with this sequence and its unmodified target receive a score of 34 as illustrated below.
- the difference between the stability of the desired target duplex and the undesired self duplex can be further increased by using the capture sequence AtgcaGA where the target duplex has a score of 24.
- the additional destabilization of the self duplex is generally not required if the difference in stability between the target duplex and self duplex is above a threshold of 25% of the target duplex stability, as illustrated below.
- ATCcaGA is the substitution pattern with the highest degree of substitution for which the stability of the target duplex is adequately more stable than the stability of the best self duplex (e.g., above 25%).
- This algorithm can be used to determine desirable substitution patterns for any size capture probe or any given probe sequence.
- the following simple design rules may also be applied for probe design, especially for short probes.
- the best self alignment for the corresponding DNA capture probe in the sequence is determined using a simple Smith-Waterman scoring matrix of: a c g t a ⁇ 2 c ⁇ 2 ⁇ 2 g ⁇ 2 3 ⁇ 2 t 2 ⁇ 2 1 ⁇ 2
- Exemplary programs are provided in the Computer Program Listing Appendix submitted on compact disc herewith.
- the oligod program (50287.007002_oligod.txt) takes a gene sequence as input and returns sequences for LNA spiked oligonucleotides.
- the dyp program (50287.007002_dyp.txt) is used by oligod to predict the secondary structure and self annealing properties of the oligonucleotides.
- the expression_array_param file (50287.007002_expression_array_param.txt) contains parameters used by the oligod program.
- 50287.007002_T m prediction.txt contains code for a T m prediction program
- 50287.007002_T m thermodynamic.txt contains code for a T m thermodynamic model.
- FIG. 35 shows such an exemplary computer system.
- Computer system 2 includes internal and external components.
- the internal components include a processor 4 coupled to a memory 6.
- the external components include a mass-storage device 8, e.g., a hard disk drive, user input devices 10, e.g., a keyboard and a mouse, a display 12, e.g., a monitor, and usually, a network link 14 capable of connecting the computer system to other computers to allow sharing of data and processing tasks. Programs are loaded into the memory 6 of this system 2 during operation.
- These programs include an operating system 16, e.g., Microsoft Windows, which manages the computer system, software 18 that encodes common languages and functions to assist programs that implement the methods of this invention, and software 20 that encodes the methods of the invention in a procedural language or symbolic package.
- Languages that can be used to program the methods include, without limitation, Visual C/C ++ from Microsoft.
- the methods of the invention are programmed in mathematical software packages that allow symbolic entry of equations and high-level specification of processing, including algorithms used in the execution of the programs, thereby freeing a user of the need to program procedurally individual equations or algorithms.
- An exemplary mathematical software package useful for this purpose is Matlab from Mathworks (Natick, Mass.).
- PVM Parallel Virtual Machine
- MPI Message Passing Interface
- LNA Locked Nucleic Acids
- LNA are DNA analogues that form DNA- or RNA-heteroduplexes with exceptionally high thermal stability.
- LNA units include bicyclic compounds as shown immediately below where ENA refers to 2′O,4′C.-ethylene-bridged nucleic acids:
- Desirable LNA monomers and oligomers share some chemical properties of DNA and RNA; they are water soluble, can be separated by agarose gel electrophoresis, and can be ethanol precipitated.
- Desirable LNA monomers and oligonucleotide units include nucleoside units having a 2′-4′ cyclic linkage, as described in the International Patent Application WO 99/14226 and WO 0056746, WO 0056748, and WO 0066604. Desirable LNA monomers structures are exemplified in the formulae Ia and Ib below. In formula Ia the configuration of the furanose is denoted D- ⁇ and in formula Ib the configuration is denoted L- ⁇ . Configurations which are composed of mixtures of the two, e.g. D- ⁇ and L- ⁇ are also included.
- X is oxygen, sulfur and carbon
- B is a universal or modified base (particularly non-natural occurring base) e.g. pyrene and pyridyloxazole derivatives, pyrenyl, pyrenylmethylglycerol moieties, all of which may be optionally substituted.
- Other desirable universal bases include, pyrrole, diazole or triazole moieties, all of which may be optionally substituted, and other groups e.g.
- R 1 , R 2 or R 2 , R 2′ or R 3 , R 5 and R 5 are hydrogen, methyl, ethyl, propyl, propynyl, aminoalkyl, methoxy, propoxy, methoxy-ethoxy, fluoro, or chloro.
- P designates the radical position for an internucleoside linkage to a succeeding monomer, or a 5′-terminal group
- R 3 or R 3′ is an internucleoside linkage to a preceding monomer, or a 3′-terminal group.
- the internucleotide linkage may be a phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, or methyl phosphonate.
- the internucleotide linkage may also contain non-phosphorous linkers, hydroxylamine derivatives (e.g.—CH 2 —NCH 3 —O—CH 2 —), hydrazine derivatives, e.g.—CH 2 —NCH 3 —NCH 3 —CH 2 , amid derivatives, e.g.—CH 2 —CO—NH—CH 2 —, CH 2 —NH—CO—CH 2 —.
- hydroxylamine derivatives e.g.—CH 2 —NCH 3 —O—CH 2 —
- hydrazine derivatives e.g.—CH 2 —NCH 3 —NCH 3 —CH 2
- amid derivatives e.g.—CH 2 —CO—NH—CH 2 —, CH 2 —NH—CO—CH 2 —.
- R 4 ′ and R 2 ′ together designate —CH 2 —O—, —CH 2 —S—, —CH 2 —NH—, —CH 2 —NMe-, —CH 2 —CH 2 —O—, —CH 2 —CH 2 —S—, —CH 2 —CH 2 —NH—, or —CH 2 —CH 2 —NMe—where the oxygen, sulfuir or nitrogen, respectively, is attached to the 2′-position.
- R 4 ′ and R 2 together designate —CH 2 —O—, —CH 2 —S—, —CH 2 —NH—, —CH 2 —NMe-, —CH 2 —CH 2 —O—, —CH 2 —CH 2 —S—, —CH 2 —CH 2 —NH—, or —CH 2 —CH 2 —NMe- where the oxygen, sulphur or nitrogen, respectively, is attached to the 2-position (R 2 configuration).
- Desirable LNA monomer structures are structures in which X is oxygen (Formula Ia and Ib); B is a universal base such as pyrene; R 1 , R 2 or R 2 , R 3 or R 3 , R 5 and R 5′ are hydrogen; P is a phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, and methyl phosphornates; R 3 or R 3′ is an internucleoside linkage to a preceding monomer, or a 3′-terminal group.
- R 4 and R 2 together designate —CH 2 —O—, —CH 2 —S—, —CH 2 —NH—, —CH 2 —NMe—, —CH 2 —CH 2 —O—, —CH 2 —CH 2 —S—, —CH 2 —CH 2 —NH—, or —CH 2 —CH 2 —NMe—where the oxygen, sulphur or nitrogen, respectively, is attached to the 2′-position
- R 4 ′ and R 2 together designate —CH 2 —O—, —CH 2 —S—, —CH 2 —NH—, —CH 2 —NMe—, —CH 2 —CH 2 —O—, —CH 2 —CH 2 —S—, —CH 2 —CH 2 —NH—, or —CH 2 —CH 2 —NMe—where the oxygen, sulphur or nitrogen, respectively, is attached to the 2′-position in the R
- LNA monomer for incorporation into an oligonucleotide of the invention include those of the following formula Ia
- B is a modified base as discussed above e.g. an optionally substituted carbocyclic aryl such as optionally substituted pyrene or optionally substituted pyrenylmethylglycerol, or an optionally substituted heteroalicylic or optionally substituted heteroaromatic such as optionally substituted pyridyloxazole.
- R 1* , R 2 , R 3 , R 5 and R 5* are hydrogen;
- P designates the radical position for an internucleoside linkage to a succeeding monomer, or a 5′-terminal group,
- R 3* is an internucleoside linkage to a preceding monomer, or a 3′-terminal group;
- R 2* and R 4* together designate —O—CH 2 — or —CH 2 —CH 2 —O—, where the oxygen is attached in the 2′-position, or a linkage of —(CH 2 ) n — where n is 2, 3 or 4, desirably 2, or a linkage of —S—CH 2 — or —NH—CH 2 —.
- LNA units of formula Ia where R 2* and R 4* contain oxygen are sometimes referred to herein as “oxy-LNA”
- units of formula IIa where R 2* and R 4* contain sulfur are sometimes referred to herein as “thio-LNA”
- units of formula Ia where R 2* and R 4* contain nitrogen are sometimes referred to herein as “amino-LNA”.
- oxy-LNA units are desirable modified nucleic acid units of oligonucleotides of the invention.
- LNA monomers for use in oligonucleotides of the invention are 2′-deoxyribonucleotides, ribonucleotides, and analogues thereof that are modified at the 2′-position in the ribose, such as 2′—O—methyl, 2′-fluoro, 2′-trifluoromethyl, 2′—O—(2-methoxyethyl), 2′—O—aminopropyl, 2′—O—dimethylamino-oxyethyl, 2′—O—fluoroethyl or 2′—O—propenyl, and analogues wherein the modification involves both the 2 ′ and 3′ position, desirably such analogues wherein the modifications links the 2′- and 3′-position in the ribose, such as those described in Nielsen et al., J.
- ⁇ -L-ribo Of particular use are ⁇ -L-ribo, the ⁇ -D-xylo and the ⁇ -L-xylo configurations (see Beier et al., Science, 1999, 283, 699 and Eschenmoser, Science, 1999, 284, 2118), in particular those having a 2′-4′-CH 2 —S—, —CH 2 —NH—, —CH 2 —O— or —CH 2 —NMe- bridge.
- LNA modified oligonucleotides used in this invention comprises oligonucleotides containing at least one LNA monomeric unit of the general scheme A above, wherein X, B, P are defined as above.
- One of the substituents R 2 , R 2* , R 3 , and R 3* is a group P* which designates an internucleoside linkage to a preceding monomer, or a 2′/3′-terminal group.
- R 1* , R 2 , R 2* , R 3 , R 4* , R 5 , R 5* , R 6 , R 6* , R 7 , and R 7* when taken together designate a biradical structure selected from —(CR * R * ) r —M—(CR* R*)s—, —(CR * R * ) r —M—(CR * R * ) s —M—, —M—(CR * R * ) r+s —M—, —M—(CR * R * ) r —M—(CR * R * ) s —, —(CR * R * ) r+s —, —M—, —M—M—M—M—, wherein each M is independently selected from —O—, —S—, —Si(R * ) 2 —, —N(R * )—, >C ⁇ O, —C( ⁇ O)—N(
- R * and R 1(1*) —R 7(7*) which are not involved in the biradical, are independently selected from hydrogen, halogen, azido, cyano, nitro, hydroxy, mercapto, amino, mono- or di(C 1-6 -alkyl)amino, optionally substituted C 1-6 -alkoxy, optionally substituted C 1-6 -alkyl-alkyl, DNA intercalators, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands, and/or two adjacent (non-geminal) R* may together designate a double bond, and each of r and s is 0-4 with the proviso that the sum r+s is 1-5.
- P designates the radical position for an internucleoside linkage to a succeeding monomer, nucleoside such as an L-nucleoside, or a 5′-terminal group, such internucleoside linkage or 5′-terminal group optionally including the substituent R 5 .
- R 1 , R 2* , R 3 , and R 3* is a group P* which designates an internucleoside linkage to a preceding monomer, or a 2′/3′-terminal group.
- Desirable nucleosides are L-nucleosides such as for example, derived dinucleoside monophosphates.
- the nucleoside can be comprised of either a beta-D, a beta-L or an alpha-L nucleoside.
- Desirable nucleosides may be linked as dimers wherein at least one of the nucleosides is a beta-L or alpha-L.
- B may also designate the pyrimidine bases cytosine, 5-methyl-cytosine, thymine, uracil, or 5-fluorouridine (5-FUdR) other 5-halo compounds, or the purine bases, adenosine, guanosine or inosine.
- LNA units may be employed in the monomers and oligomers of the invention including bicyclic and tricyclic DNA or RNA having a 2′-4′ or 2′-3′ sugar linkages; 2′-O,4′-C-methylene- ⁇ -D-ribofuranosyl moiety, known to adopt a locked C3′-endo RNA-like furanose conformation.
- Illustrative modified structures that may be included in oligonucleotides of the invention are shown in FIG. 1.
- nucleic acid units that may be included in an oligonucleotide of the invention may comprise 2′-deoxy-2′-fluoro ribonucleotides; 2′—O—methyl ribonucleotides; 2′—O—methoxyethyl ribonucleotides; peptide nucleic acids; 5-propynyl pyrimidine ribonucleotides; 7-deazapurine ribonucleotides; 2,6-diaminopurine ribonucleotides; and 2-thio-pyrimidine ribonucleotides, and nucleotides with other sugar groups (e.g. xylose).
- Oligonucleotides containing LNA are readily synthesized by standard phosphoramidite chemistry.
- the flexibility of the phosphoramidite synthesis approach further facilitates the easy production of LNA oligos carrying all types of standard linkers, fluorophores and reporter groups.
- SBC Selective Binding Complementary
- SBC nucleotides are unable to form stable hybrids with each other, yet are able to form stable, sequence-specific hybrids with complementary unmodified strands of nucleic acids.
- SBC oligonucleotides to form intramolecular hydrogen bond base-pairs between regions of substantially complementary sequence causes a reduced level of secondary structure.
- Self-complementarity is an important issue in nucleic acid technologies as reported for DNA, PNA and LNA, and in different biological applications especially in the field of homogeneous assays.
- LNA:LNA duplexes are the most thermally stable nucleic acid type duplex system known, making the reduction of self-complementarity even more important.
- Exemplary SBC oligonucleotides contain 2-amino-A (D) and 2 sT incorporated in the same oligonucleotide as replacements of A and T, respectively.
- the SBC name refers to the fact that D and 2 ST form a destabilised base-pair compared to the A-T base-pair, but D-T and 2 ST-A base-pairs are normally more stable than the original A-T base-pair.
- Exemplary SBC-G nucleotides include inosine or LNA-inosine
- exemplary SBC-C nucleotides includee PyrroloPyr, LNA- PyrroloPyr, 2sC, and LNA-2SC (FIG. 3). Other exemplary SBC nucleotides are shown in FIGS. 2 and 3. If desired, SBC nucleotides may be incorporated into the nucleic acids and arrays of the invention, using standard methods.
- the systems disclosed herein can provide significant nucleic acid probes for universal hybridization.
- universal hybridization can be accomplished with a conformationally restricted monomer, including a desirable pyrene LNA monomer.
- Universal hybridization behavior also can be accomplished in an RNA context.
- the binding affinity of probes for universal hybridization can be increased by the introduction of high affinity monomers without compromising the base-pairing selectivity of bases neighboring the universal base.
- LNA oligonucleotides can often self-hybridize, rather than hybridize to another oligonucleotide.
- Use of one or more modified bases with the LNA units can modulate the propensity of the oligonucleotide to form double stranded structures with other oligonucleotides containing modified nucleobases including internal duplex formation, thereby inhibiting undesired self-hybridization.
- 2-Thiopyrimidine nucleosides can be prepared in several ways as described below.
- the 2-thiouridine-nucleosides (IV) can be synthesized from a substituted uridine nucleoside (VIII) as described in the scheme below.
- By protection of the 04-position (IX) on the nucleobase thionation can be performed, O 2 position, which results in the 2-thio-uridine nucleoside (IV).
- Performing sulphurisation on both O 2 and 04 results in 2,4-dithio-uridine nucleoside (X) which may be transformed into the 2-thio-uridine nucleoside (IV) (Saladino, et.
- lewis acid-catalyzed.condensation of a properly substituted sugar (I) and a substituted 2-thio-uracil (II) can result in a substituted 2-thio-uridine nucleoside of the structure (III) which by further synthetic manipulations can be transformed into the LNA 2-thiouridine nucleoside (IV) (Hamamura et. al., Moffatt, J. Med. Chem., 1972, 15, 1061; Bretner et. al., J. Med. Chem., 1993, 36, 3611).
- a 2-thio-uridine nucleoside can be synthesized through ring-synthesis of the nucleobase by reaction of the amino sugar (V) and an substituted isothiocyanate(VI), yielding the substituted LNA 2-thio-uracil nucleoside (VI) (Shaw and Warrener, J. Chem. Soc. 1957, 153; Cusack et al., J. Chem. Soc. Perkin 1, 1973, 1721).
- Strategy A involves coupling a glycosyl-donor and a nucleobase, using standard methodology for synthesis of existing LNA monomers.
- Strategy B involves ring synthesis of the nucleobase. This strategy is desirable because the availability of 1-amino-LNA enables introduction of a variety of new nucleobases.
- Strategy C includes modification of T-LNA; the easy synthesis of LNA-T diol makes this an attractive pathway.
- 2 sT-LNA is synthesized as illustrated in the scheme below.
- the known coupling sugar 1,2-di—O—acetyl-3, 5 di—O—benzyl, 4-C- mesyloxymethyl, ⁇ , 62 -D-ribofuranose 1 was coupled with the nucleobase 2-thio-thymidine in a Vorbruggen type reaction.
- the nucleobase was silyilated and condensed with the sugar using SnCl 4 as catalyst to promote the reaction affording nucleoside 2.
- Mass spectrometry and NMR subsequently identified the isolated product as the desired one. NMR data were compared with published data of a 2-thio-thymindine derivative (Kuimelis and Nambiar, Nucleic Acid Res., 1994, 22, 1429-1436) in order to validate the correct attachment point of the nucleobase.
- the LNA derivative was protected at the nucleobase with the toluoyl protective group to give 4.
- This group is well known for the protection of 2-thio-thymidine derivatives, (Kuimelis and Nambiar, Nucleic Acid Res., 1994, 22, 1429-1436).
- the protection of the nucleobase occurs at both the N-3 and the 0-4 position and hence the compound is isolated as a mixture of two compounds. NMR shows that the ratio of the two isomers in the isolated mixture is 2:1.
- LNA-I hypoxanthine (or inosine)
- LNA-D 2,6-diaminopurine
- LNA-2AP 2-aminopurine
- hypoxantine the base found in the nucleotides inosine and deoxyinosine, is considered as a guanine analogue in nucleic acids.
- Oligonucleotides containing 2,6-diaminopurine replacements for adenines are expected to bind more strongly to their complementary sequences especially as part of A-type helixes due to the potential formation of three hydrogen bounds with thymine or uracil.
- the reported effect of 2,6-diaminopurine deoxyriboside (D) on the stability of polynucleotide duplexes reaches, on average, about 1.5° C. per modification. Higher stabilization effects for mismatches were observed for D nucleosides involved in formation of duplexes prone to form A-type helixes.
- LNA D and LNA 2′-OMe-D are expected to have increased stabilization and mismatch discrimination.
- LNA can be used in combination with 2-thio-T for construction of selectively binding complementary oligonucleotides. Taking into consideration the extremely high stability of LNA:LNA duplexes, this approach might be very useful for constructing of LNA containing capture probes and antisense reagents including drugs.
- 2-Aminopurine (2-AP) is a fluorescent nucleobase (emission at 363 nm), which is useful for probing nucleic acids structure and dynamics and for hybridizing with thymine in Watson-crick geometry.
- LNA-I, LNA-D, and/or LNA-2AP may be used in the nucleic acids of the present invention, e.g., to increase the priming efficiency of DNA oligonucleotides in PCR experiments and to construct selectively binding complementary agents.
- Deprotection of the 5′-hydroxy group of 6 was accomplished via two-step procedure developed for the syntheses of other LNA nucleosides ( Koshkin et al., supra).
- 5′—O—mesyl group was displaced by sodium benzoate to produce nucleoside 7.
- the latter was converted into 5′-hydroxy derivative 8 after saponification of the 5′-benzoate.
- Direct removal of the 3′—O—benzyl group from compound 8 was unsuccessful under the conditions tested due to a solubility problem. Therefore, compound 8 was converted to DMT-protected nucleoside 9 prior to catalytic debenzylation of the 3′—O—hydroxy group.
- the phosphoramidite 11 was finally afforded via standard phosphitylation (McBride et al., Tetrahedron Lett. 24:245, 1983; Sinha et al., Tetrahedron Lett. 24:5843, 1983; and Sinha et al., Nucleic Acids Res. 12:4539, 1984) of the nucleoside 10.
- compound 7 was successfully converted into the known LNA-A derivative 13 (Koshkin et al., supra) (Scheme 2).
- a treatment of 7 with phosphoryl chloride according to the procedure reported by Martin (Helv. Chim. Acta 78:486, 1995) resulted in a high yield formation of 6-chloropurine derivative 12.
- the adenosine derivative 13 was derived from 12 after reaction with ammonia.
- Data for compound 8 includes the following: mp 302-305° C. (dec).
- 1 H NMR (DMSO-d 6 ): 88.16, (s, 1H), 8.06 (s, 1H), 7.30-7.20 (m, 5H), 5.95 (s, 1H), 4.69 (s, 1H), 4.63 (s, 2H), 4.28 (s, 1H), 3.95 (d, J 7.7, 1H), 3.83 (m, 3H).
- Data for compound 23 includes the following: crystallized from MeOH. mp. 227.5-229° C. (dec).
- the 31 P NMR (DMSO-d 6 ) spectrum has a signal at 148.93 and 148.85.
- the intermediate derivative 16 was also used for the synthesis of LNA-2AP nucleoside.
- the 5′—O—benzoyl group of 16 was hydrolyzed by aqueous sodium hydroxide to give the nucleoside derivative 22 in 72% yield.
- the conditions of catalytic transfer hydrogenation usually used for removal of the 3′—O—benzyl group turned out to be suitable for complete dechlorination of the nucleobase of 22.
- totally deprotected LNA-2AP nucleoside 23 was afforded in high yield after refluxing of the methanolic solution of 22 in the presence of paladium hydroxide and ammonium formate.
- the 2-amine of 23 was selectively protected with an amidine group after treatment with N,N-dimethylformamide dimethyl acetal.
- the resulting diol 24 was then 5′—O—DMT protected and 3′—O—phosphitylated to yield the desired phosphoramidite LNA-2AP monomer 25 (McBride et al., J. Am. Chem. Soc. 108:2040, 1986).
- oligonucleotides were performed by treatment with concentrated ammonium hydroxide for five hours at 60° C. After that, the LNA-D containing oligonucleotides were additionally treated with AMA (concentrated ammonium hydroxide/40% aqueous MeNH 2 ; 1/1 v/v) for one hour at 60° C. All the synthesized oligonucleotides were purified by RP-HPLC, and their structures were verified by MALDI-TOF mass spectra.
- AMA concentrated ammonium hydroxide/40% aqueous MeNH 2 ; 1/1 v/v
- the average melting temperatures of DNA and LNA 8-mers against coplementary DNAs typically differ by about 40° C.
- the replacement of one internal LNA-A nucleotide by LNA-D resulted in the further stabilization of the complementary duplex (i.e., compare entries 8 and 11) by 6.2° C.
- the analogous replacement made in an DNA octamer destabilized the corresponding duplex by 0.5° C. (i.e., entries 1 and 4).
- D-nucleosides may facilitate a B to A helix transition, because the A-type structure of an LNA:DNA duplex is more suitable for effective D:t pairing.
- LNAs are CAPITALIZED
- s 2-thio-deoxythymidine
- d diamino- purineriboside
- the furanopyrimidine phosphoramidite 6 pC used for incorporation of the pyrroloC analogue can be synthesized from LNA-U through a series of reactions as illustrated below and in FIG. 6.
- LNA-U lpC iodine can be introduced on the 5 position on the nucleobase (Chang and Welch, J. Med. Chem. 1963, 6, 428).
- This compound can be used in a Sonogashira type palladium coupling reaction (Sonogashira, Tohda and Hagihara, Tetrahedron Lett. 1975, 4467) resulting in the 5-ethynyl-LNA-U 3 pC.
- the 5-ethynyl-LNA-U 3 pC can be transformed to the furanopyrimidie LNA analogue 4 pC when reacted with CuI, and then transformed into the DMT-protected phosphoramidite 6 pC (Woo, Meyer, and Gamper, Nucleic Acids Res., 1996, 24, 2470).
- LNA-PyrroloPyr-SBC-C is formed when 6 pC or an oligonucleotide containing 6 pC is deprotected with ammonia.
- Desirable modified bases are covalently linked to the 1′-position of a furanosyl ring, particularly to the 1′-position of a 2′,4′-linked furanosyl ring, especially to the 1′-position of a 2′-0,4′-C-methylene-beta-D-ribofuranosyl ring.
- modified bases contain one or more carbon alicyclic or carbocyclic aryl units, i.e. non-aromatic or aromatic cyclic units that contain only carbon atoms as ring members.
- Modified bases that contain carbocyclic aryl groups are generally desirable, particularly a moiety that contains multiple linked aromatic groups, particularly groups that contain fused rings.
- optionally substituted polynuclear aromatic groups are especially desirable such as optionally substituted naphthyl, optionally substituted anthracenyl, optionally substituted phenanthrenyl, optionally substituted pyrenyl, optionally substituted chrysenyl, optionally substituted benzanthracenyl, optionally substituted dibenzanthracenyl, optionally substituted benzopyrenyl, with substituted or unsubstituted pyrenyl being particularly desirable.
- Modified bases that contain one or more heteroalicyclic or heteroaromatic groups also are suitable for use in LNA units, particularly such non-aromatic and aromatic groups that contains one or more N, O or S atoms as ring members, particularly at least one sulfur atom, and from 5 to about 8 ring members. Also desirable is a nucleo base that contains two or more fused rings, where at least one of the rings is a heteroalicyclic or heteroaromatic group containing 1, 2, or 3 N, O, or S atoms as ring members.
- modified bases that contain 2, 3, 4, 5, 6, 7 or 8 fused rings, which may be carbon alicyclic, heteroalicyclic, carbocyclic aryl and/or heteroaromatic; more desirably modified bases that contain 3, 4, 5, or 6 fused rings, which may be carbon alicyclic, heteroalicyclic, carbocyclic aryl and/or heteroaromatic, and desirably the fused rings are each aromatic, particularly carbocyclic aryl.
- the base is not an optionally substituted oxazole, optionally substituted imidazole, or optionally substituted isoxazole modified base.
- Suitable modified bases for use in LNA units in accordance with the invention include optionally substituted pyridyloxazole, optionally substituted pyrenylmethylglycerol, optionally substituted pyrrole, optionally substituted diazole and optionally substituted triazole groups.
- Desirable modified bases of the present invention when incorporated into an oligonucleotide containing all LNA units or a mixture of LNA and DNA or RNA units will exhibit substantially constant T m values upon hybridization with a complementary oligonucleotide, irrespective of the bases present on the complementary oligonucleotide.
- one or more of the common RNA or commonly used derivatives thereof such as 2′—O—methyl, 2′-fluoro, 2′-allyl, and 2′—O—methoxyethoxy derivatives are combined with at least one nucleotide with a universal base to generate an oligonucleotide having between five to 100 nucleotides.
- Modified nucleic acid compounds may comprise a variety of nucleic acid units e.g. nucleoside and/or nucleotide units.
- an LNA nucleic acid unit has a carbon or hetero alicyclic ring with four to six ring members, e.g. a furanose ring, or other alicyclic ring structures such as a cyclopentyl, cycloheptyl, tetrahydropyranyl, oxepanyl, tetrahydrothiophenyl, pyrrolidinyl, thianyl, thiepanyl, piperidinyl, and the like.
- At least one ring atom of the carbon or hetero alicyclic group is taken to form a further cyclic linkage to thereby provide a multi-cyclic group.
- the cyclic linkage may include one or more, typically two atoms, of the carbon or hetero alicyclic group.
- the cyclic linkage also may include one or more atoms that are substituents, but not ring members, of the carbon or hetero alicyclic group.
- an alicyclic group as referred to herein is inclusive of group having all carbon ring members as well as groups having one or more hetero atom (e.g. N, O, S or Se) ring members.
- the disclosure of the group as a “carbon or hetero alicyclic group” further indicates that the alicyclic group may contain all carbon ring members (i.e. a carbon alicyclic) or may contain one or more hetero atom ring members (i.e. a hetero alicyclic).
- Alicyclic groups are understood not to be aromatic, and typically are fully saturated within the ring (i.e. no endocyclic multiple bonds).
- the alicyclic ring is a hetero alicyclic, i.e. the alicyclic group has one or more hetero atoms ring members, typically one or two hetero atom ring members such as O, N, S or Se, with oxygen being often desirable.
- the one or more cyclic linkages of an alicyclic group may be comprised completely of carbon atoms, or generally more desirable, one or more hetero atoms such as O, S, N or Se, desirably oxygen for at least some embodiments.
- the cyclic linkage will typically contain one or two or three hetero atoms, more typically one or two hetero atoms in a single cyclic linkage.
- the one or more cyclic linkages of a nucleic acid compound of the invention can have a number of alternative configurations and/or configurations.
- cyclic linkages of nucleic acid compounds of the invention will include at least one alicyclic ring atom.
- the cyclic linkage may be disubstituted to a single alicyclic atom, or two adjacent or non-adjacent alicyclic ring atoms may be included in a cyclic linkage.
- a cyclic linkage may include a single alicyclic ring atom, and a further atom that is a substituent but not a ring member of the alicyclic group.
- desirable cyclic linkages include the following: C-1′, C-2′; C-2′, C-3′; C-2′, C-4′; or a C-2′, C-5′ linkage.
- a cyclic linkage will typically comprise, in addition to the one or more alicyclic group ring atoms, 2 to 6 atoms in addition to the alicyclic ring members, more typically 3 or 4 atoms in addition to the alicyclic ring member(s).
- the alicyclic group atoms that are incorporated into a cyclic linkage are typically carbon atoms, but hetero atoms such as nitrogen of the alicyclic group also may be incorporated into a cyclic linkage.
- Specifically desirable modified nucleic acids for use oligonucleotides of the invention include locked nucleic acids as disclosed in WO99/14226 (which include bicyclic and tricyclic DNA or RNA having a 2′-4′ or 2′-3′ sugar linkages); 2′-deoxy-2′-fluoro ribonucleotides; 2′—O—methyl ribonucleotides; 2′—O—methoxyethyl ribonucleotides; peptide nucleic acids; 5-propynyl pyrimidine ribonucleotides; 7-deazapurine ribonucleotides; 2,6-diaminopurine ribonucleotides; and 2-thio-pyrimidine ribonucleotides.
- LNA units as disclosed in WO 99/14226 are in general particularly desirable modified nucleic acids for incorporation into an oligonucleotide of the invention. Additionally, the nucleic acids may be modified at either the 3′ and/or 5′ end by any type of modification known in the art. For example, either or both ends may be capped with a protecting group, attached to a flexible linking group, attached to a reactive group to aid in attachment to the substrate surface, etc. Desirable LNA units also are disclosed in WO 0056746, WO 0056748, and WO 0066604.
- modified nucleic acids may be employed, including those that have 2′—modification of hydroxyl, 2′-O-methyl, 2′-fluoro, 2′-trifluoromethyl, 2′—O—(2-methoxyethyl), 2′O-aminopropyl, 2′-O- dimethylamino-oxyethyl, 2′—O—fluoroethyl or 2′—O—propenyl.
- the nucleic acid may further include 3′modification, desirably where the 2′- and 3′-position of the ribose group is linked.
- the nucleic acid also may contain a modification at the 4′-position, desirably where the 2′-and 4′positions of the ribose group are linked such as by a 2′-4′ link of —CH 2 —S—, -CH 2 —NH—, or —CH 2 —NMe—bridge.
- the nucleotide also may have a variety of configurations such as ⁇ -D-ribo, ⁇ -D-xylo, or ⁇ -L-xylo configuration.
- the internucleoside linkages of the units of oligos of the invention may be natural phosphorodiester linkages, or other linkages such as —O—P(O) 2 —O—, —O—P(O,S)—O—, —O—P(S) 2 —O—, -NR H —P(O) 2 —O—P(O,NR H )—O—, —O—PO(R′′)—O—, —O—PO(CH 3 )—O—, and —O—PO(NHR N )—O—, where R H is selected from hydrogen and C 1-4 -alkyl, and R′′ is selected from C 1-6 -alkyl and phenyl.
- a further desirable group of modified nucleic acids for incorporation into oligomers of the invention include those of the following formula:
- X is —O—;
- B is a modified base as discussed above e.g. an optionally substituted carbocyclic aryl such as optionally substituted pyrene or optionally substituted pyrenylmethylglycerol, or an optionally substituted heteroalicylic or optionally substituted heteroaromatic such as optionally substituted pyridyloxazole.
- Other desirable universal bases include, pyrrole, diazole or triazole moieties, all of which may be optionally substituted.
- R 1* is hydrogen.
- P designates the radical position for an internucleoside linkage to a succeeding monomer, or a 5′-terminal group, such internucleoside linkage or 5′-terminal group optionally including the substituent R 5 , R 5 being hydrogen or included in an internucleoside linkage.
- R 3* is a group P* which designates an internucleoside linkage to a preceding monomer, or a 3′-terminal group.
- Z is selected from —O—, —S—, and —N(R a )-, and R a and R b each is independently selected from hydrogen, optionally substituted C 1-6 -alkyl -alkyl, optionally substituted C 2-6 -alkenyl, hydroxy, C 1-6 -alkyl, C 2-6 -alkenyloxy, carboxy, C 1-6 -alkoxycarbonyl, C 1-6 -alkylcarbonyl, formyl, amino, mono- and di(C 1-6 -alkyl)amino, carbamoyl, mono- and di(C 1-6 -alkyl)-amino-carbonyl, amino-C 1-6 -alkyl-aminocarbonyl, mono- and di(C 1-6 -alkyl)amino-C 1-6 -alkyl-aminocarbonyl, C 1-6 -alkyl-carbonylamino, carbamido, C 1-6 -al
- Modified nucleobases and nucleosidic bases may comprise a cyclic unit (e.g. a carbocyclic unit such as pyrenyl) that is joined to a nucleic unit, such as a 1′-position of furasonyl ring through a linker, such as a straight of branched chain alkylene or alkenylene group.
- Alkylene groups suitably having from 1 (i.e.—CH 2 —) to about 12 carbon atoms, more typically 1 to about 8 carbon atoms, still more typically 1 to about 6 carbon atoms.
- Alkenylene groups suitably have one, two or three carbon-carbon double bounds and from 2 to 12 carbon atoms, more typically 2 to 8 carbon atoms, still more typically 2 to 6 carbon atoms.
- Desirable LNA units include those that contain a furanosyl-type ring and one or more of the following linkages: C-1′, C-2′; C-2′, C-3′; C-2′, C-4′; or a C-2′, C-5′ linkage.
- a C-2′, C-4′ is particularly desirable.
- desirable LNA units are compounds having a substituent on the 2′-position of the central sugar moiety (e.g., ribose or xylose), or derivatives thereof, which favors the C3′-endo conformation, commonly referred to as the North (or simply N for short) conformation.
- the oligonucleotide has at least one LNA unit with a modified base as disclosed herein.
- Suitable oligonucleotides also may contain natural DNA or RNA units (e.g., nucleotides) with natural bases, as well as LNA units that contain natural bases.
- the oligonucleotides of the invention also may contain modified DNA or RNA, such as 2′—O—methyl RNA, with natural or modified nucleobases (e.g., pyrene).
- Desirable oligonucleotides contain at least one of and desirably both of 1) one or more DNA or RNA units (e.g., nucleotides) with natural bases, and 2) one or more LNA units with natural bases, in addition to LNA units with a modified base.
- the nucleic acid does not contain a modified base.
- Oligonucleotides of the invention desirably contain at least 50 percent or more, more desirably 55, 60, 65, or 70 percent or more of non-modified or natural DNA or RNA units (e.g., nucleotides) or units other than LNA units based on the total number of units or residues of the oligo.
- a non-modified nucleic acid as referred to herein means that the nucleic acid upon incorporation into a 1 0-mer oligomer will not increase the T m of the oligomer in excess of 1° C. or 2° C.
- the non-modified nucleic acid unit is a substantially or completely “natural” nucleic acid, i.e. containing a non-modified base of uracil, cytosine, 5-methyl-cytosine, thymine, adenine or guanine and a non-modified pentose sugar unit of ⁇ -D-ribose (in the case of RNA) or ⁇ -D-2-deoxyribose (in the case of DNA).
- Oligonucleotides of the invention suitably may contain only a single modified (i.e. LNA) nucleic acid unit, but desirably an oligonucleotide will contain 2, 3, 4 or 5 or more modified nucleic acid units.
- an oligonucleotide contains from about 5 to about 40 or 45 percent modified (LNA) nucleic acid units, based on total units of the oligo, more desirably where the oligonucleotide contains from about 5 or 10 percent to about 20, 25, 30 or 35 percent modified nucleic acid units, based on total units of the oligo.
- Typical oligonucleotides that contain one or more LNA units with a modified base as disclosed herein suitably contain from 3 or 4 to about 200 nucleic acid repeat units, with at least one unit being an LNA unit with a modified base, more typically from about 3 or 4 to about 5, 6, 7, 8, 9, 10, 15, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140 or 150 nucleic acid units, with 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 LNA units with a modified base being present.
- particularly desirable oligonucleotides contain a non- modified DNA or RNA unit at the 3′ terminus and a modified DNA or RNA unit at one position upstream from (generally referred to hereing as the ⁇ 1 or penultimate position) the 3′ terminal non-modified nucleic acid unit.
- the modified base is at the 3′ terminal position of a nucleic acid primer, such as a primer for the detection of a single nucleotide polymorphism.
- Other particularly desirable nucleic acids have an LNA unit with or without a modified base in the 5′ and/or 3′ terminal position.
- oligonucleotides that do not have an extended stretches of modified DNA or RNA units, e.g. greater than about 4, 5 or 6 consecutive modified DNA or RNA units. That is, desirably one or more non-modified DNA or RNA will be present after a consecutive stretch of about 3, 4 or 5 modified nucleic acids.
- oligonucleotides that contain a mixture of LNA units that have non-modified or natural nucleobases (i.e., adenine, guanine, cytosine, 5-methyl-cytosine, uracil, or thymine) and LNA units that have modified bases as disclosed herein.
- non-modified or natural nucleobases i.e., adenine, guanine, cytosine, 5-methyl-cytosine, uracil, or thymine
- Particularly desirable oligonucleotides of the invention include those where an LNA unit with a modified base is interposed between two LNA units each having non-modified or natural bases (adenine, guanine, cytosine, 5-methyl-cytosine, uracil, or thymine.
- the LNA “flanking” units with natural base moieties may be directly adjacent to the LNA with modified base moiety, or desirably is within 2, 3, 4 or 5 nucleic acid units of the LNA unit with modified base.
- Nucleic acid units that may be spaced between an LNA unit with a modified base and an LNA unit with natural nucleobasis suitably are DNA and/or RNA and/or alkyl-modified RNA/DNA units, typically with natural base moieties, although the DNA and or RNA units also may contain modified base moieties.
- oligonucleotides of the present invention are comprised of at least about one universal base. Oligonucleotides of the present can also be comprised, for exmple, of between about one to six 2′-Ome-RNA unit, at least about two LNA units and at least about one LNA pyrene unit.
- target genes may be suitably single-stranded or double-stranded DNA or RNA; however, single-stranded DNA or RNA targets are desirable.
- target to which the nucleic acids of the invention are directed include allelic forms of the targeted gene and the corresponding mRNAs including splice variants.
- Desirable mRNA targets include the 5′ cap site, tRNA primer binding site, the initiation codon site, the mRNA donor splice site, and the mRNA acceptor splice site, e.g., Goodchild et al., U.S. Pat. No. 4,806,463.
- the chimeric oligos of the present invention are highly suitable for a variety of diagnostic purposes such as for the isolation, purification, amplification, detection, identification, quantification, or capture of nucleic acids such as DNA, mRNA or non-protein coding cellular RNAs, such as tRNA, rRNA, snRNA and scRNA, or synthetic nucleic acids, in vivo or in vitro.
- nucleic acids such as DNA, mRNA or non-protein coding cellular RNAs, such as tRNA, rRNA, snRNA and scRNA, or synthetic nucleic acids, in vivo or in vitro.
- the oligomer can comprise a photochemically active group, a thermochemically active group, a chelating group, a reporter group, or a ligand that facilitates the direct or indirect detection of the oligomer or the immobilization of the oligomer onto a solid support.
- a photochemically active group typically attached to the oligo when it is intended as a probe for in situ hybridization, in Southern hybridization, Dot blot hybridization, reverse Dot blot hybridization, or in Northern hybridization.
- the spacer may suitably comprise a chemically cleavable group.
- An additional object of the present invention is to provide oligonucleotides which combines an increased ability to discriminate between complementary and mismatched targets with the ability to act as substrates for nucleic acid active enzymes such as for example DNA and RNA polymerases, ligases, phosphatases.
- nucleic acid active enzymes such as for example DNA and RNA polymerases, ligases, phosphatases.
- Such oligonucleotides may be used for instance as primers for sequencing nucleic acids and as primers in any of the several well known amplification reactions, such as the PCR reaction.
- LNA monomers with natural bases into either DNA, RNA, or pure LNA oligonucleotides can result in extremely high thermal stability of duplexes with complementary DNA or RNA, while at the same time obeying the Watson-Crick base-pairing rules. In general, the thermal stability of heteroduplexes is increased 3-8° C. per LNA monomer in the duplex.
- Oligonucleotides containing LNA can be designed to be substrates for polymerases (e.g., Taq polymerase), and PCR based on LNA primers is more discriminatory towards single base mutations in the template DNA compared to normal DNA-primers (e.g., allele specific PCR).
- very short LNA oligos e.g. 5-mers or 8-mers which have high T m 's when compared to similar DNA oligos can be used as highly specific catching probes with outstanding discriminatory power towards single base mutations (e.g., SNP detection).
- LNA oligonucleotides are capable of hybridizing with double-stranded DNA target molecules as well as RNA secondary structures by strand invasion as well as of specifically blocking a wide selection of enzymatic reactions such as, digestion of double-stranded DNA by restriction endonucleases; and digestion of DNA and RNA with deoxyribonucleases and ribonucleases, respectively.
- oligonucleotides of the invention may be used to construct new affinity pairs with exhibit enhanced specificity towards each other.
- the affinity constants can easily be adjusted over a wide range and a vast number of affinity pairs can be designed and synthesized.
- One part of the affinity pair can be attached to the molecule of interest (e.g. proteins, amplicons, enzymes, polysaccharides, antibodies, haptens, peptides, etc.) by standard methods, while the other part of the affinity pair can be attached to e.g. a solid support such as beads, membranes, micro-titer plates, sticks, tubes, etc.
- the solid support may be chosen from a wide range of polymer materials such as for instance polypropylene, polystyrene, polycarbonate or polyethylene.
- the affinity pairs may be used in selective isolation, purification, capture and detection of a diversity of the target molecules.
- Oligonucleotides of the invention also may be employed as probes in the purification, isolation and detection of for instance pathogenic organisms such as viral, bacteria and fungi etc. Oligonucleotides of the invention also may be used as generic tools for the purification, isolation, amplification and detection of nucleic acids from groups of related species such as for instance rRNA from gram-positive or gram negative bacteria, fungi, mammalian cells etc.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
The invention features improved nucleic acids and methods for expression profiling of mRNAs, identifying and profiling of particular mRNA splice variants, and detecting mutations, deletions, or duplications of particular exons or other splice variants, e.g., alterations associated with a disease such as cancer, in a nucleic acid sample, e.g., a biological sample or a patient sample.
Description
- This application claims priority to U.S. Provisional Application No. 60/420,278, filed on Oct. 21, 2002 and Danish Application No. PA 2003 00752, filed on May 19, 2003, each of which is hereby incorporated by reference.
- A compact disc containing the files: 50287.007002_oligod.txt, 60 kB; 50287.007002_dyp.txt, 52 kB; 50287.007002_expression_array_param.txt, 3 kB; 50287.007002_T m prediction.txt, 3 kB; 50287.007002_Tm thermodynamic.txt, 47 kB, all created on Oct. 19, 2003, has been submitted in duplicate and is hereby incorporated by reference.
- The invention relates to nucleic acids and methods for expression profiling of mRNAs, identifying and profiling of particular mRNA splice variants, and detecting mutations, deletions, or duplications of particular exons, e.g., alterations associated with a disease such as cancer, in a nucleic acid sample, e.g., a patient sample. The invention furthermore relates to methods for detecting nucleic acids by fluorescence in situ hybridization.
- The field of the invention is oligonucleotides (e.g., oligonucleotide arrays) that are useful for detecting nucleic acids of interest and for detecting differences between nucleic acid samples (e.g, such as samples from a cancer patient and a healthy patient).
- DNA chip technology utilizes minituarized arrays of DNA molecules immobilized on solid surfaces for biochemical analyses. The power of DNA microarrays as experimental tools relies on the specific molecular recognition via complementary base-pairing, which makes them highly useful for massive parallel analyses. In the post-genomic era, microarray technology has thus become the method of choice for many hybridization-based assays, such as expression profiling, SNP detection, DNA re-sequencing, and genotyping on a genomic scale.
- Expression microarrays are capable of profiling gene expression patterns of tens of thousands of genes in a single experiment. Hence, this technology provides a powerful tool for deciphering complex biological systems, and thereby greatly facilitates research in basic biology and living processes, as well as disease diagnostics, theranostics, and drug development. In a typical cell, the mRNAs are distributed in three frequency classes: (i) superprevalent (10-20% of the total mRNA mass); (ii) intermediate (40-45%); and (iii) low-abundant mRNAs (40-45%). It is therefore of utmost importance that the dynamic range and sensitivity of the expression arrays are optimal, especially when analyzing expression levels of messages or mRNA splice variants belonging to the low-abundant class.
- The recent explosion of interest in DNA microarray technology has been sparked by two key innovations. The first was the use of non-porous solid support, such as glass or polymer as opposed to nylon or nitrocellulose filters, which has facilitated miniaturization and fluorescence-based detection. Roughly 20,000 cDNAs can be robotically spotted onto a microscope slide and hybridized with a double-labeled probe. The second was the development of methods for high-density spatial synthesis of oligonucleotides. The two key array technologies are outlined in the following.
- Oligonucleotide arrays
- An efficient strategy for oligonucleotide microarray manufacturing involves DNA synthesis on solid surfaces using combinatorial chemistry. Most of the current technology is developed by Affymetrix and Rosetta Inpharmatics. Glass is currently preferred as the synthesis support because of its inert chemical properties and low level of intrinsic fluorescence as well as the ability to chemically derivatize the surface. Of the three approaches currently used to manufacture oligonucleotide arrays, the light-directed deprotection method is the most effective one in generating high density microchips. A single round of synthesis involves light-directed deprotection, followed by nucleotide coupling. Photolithographic masking is used to control the regions of the chip designated for illumination. Affynietrix uses a combination of photolithography and combinatorial chemistry to manufacture its GeneChip Arrays. Using technologies adapted from the semiconductor industry, GeneChip manufacturing begins with a 5-inch square quartz wafer. Initially the quartz is washed to ensure uniform hydroxylation across its surface. The wafer is placed in a bath of silane, which reacts with the hydroxyl groups of the quartz and forms a matrix of covalently linked molecules. The distance between these silane molecules determines the probes' packing density, allowing arrays to hold over 500,000 features within 1.28 square centimeters. The principal disadvantage of this method is that a significant amount of chip design work and cost is associated with the mask design. Once a set of masks has been made, a large number of chips can be produced at a reasonable cost. The current pricing of oligonucleotide arrays available from Affymetrix are in the range of 5-10 fold more expensive than cDNA microarrays.
- DNA-DNA hybridization using oligonucleotide chips is clearly different from that of cDNA microarrays. Hybridizations involving oligos are much more sensitive to the GC content of individual heteroduplexes. In addition, single base mismatches have a pronounced effect on the hybridization reassociation of short oligos, and point mutations can thus be readily detected using oligo chips.
- cDNA Microarrays
- cDNA microarrays containing large DNA segments such as cDNAs are generated by physically depositing small amounts of each DNA of interest onto known locations on glass surfaces. Two technologies for printing microarrays are (1) mechanical microspotting, and (2) ink-jetting. Mechanical microspotting has been extensively used at, e.g., Stanford University, and it utilizes pins or capillaries to deposit small quantities of DNA onto known addresses using motion control systems. Recent advances in microspotting technology using modem arraying robots allow for the preparation of 100 microarrays containing over 10,000 features in less than 12 hours. A DNA arrayer is relatively easy to set up, and the cost is usually low compared to on-chip oligoarrayers. cDNA microarrays are capable of profiling gene expression patterns of tens of thousands of genes in a single experiment. To compare the relative abundance of the arrayed gene sequences in two DNA or RNA samples, e.g., the total mRNA isolated from two different cell populations, the two samples are first labeled using two different fluorescent dyes such as Cy-3 and Cy-5. The labeled samples are mixed and hybridized to the clones on the array slide. After the hybridization, laser excitation of the incorporated, fluorescent target molecules yields an emission with a characteristic spectra, which is measured with a confocal laser scanner. The monochrome images from the scanner are imported to the software in which the images are pseudo-colored and merged. Data from a single hybridization is viewed as a normalized ratio in which significant deviations from the ratio are indicative of either increased or decreased expression levels relative to the reference sample. Data from multiple experiments can be examined using any number of data mining tools.
- Current status of array technology
- It has now become clear that cDNA microarrays, originally developed by Pat Brown and co-workers at the Stanford University, are sensitive, but may not be sufficiently specific with respect to, e.g., discrimination of homologous transcripts in gene families and alternatively spliced isoforms. On the other hand, the Affymetrix GeneChip system is specific, but may not be sensitive enough. This lack of sensitivity may explain why Affymetrix uses 1 6×26-mer perfect match capture probes together with 1 6×25-mer mismatch probes per transcript in its expression profiling chips resulting in enormous data sets in genome-wide arrays. Therefore, the functional genomics field is in the process of switching, as they run out of samples, from existing PCR-amplified cDNA fragment libraries for microarraying to custom longmer oligonucleotide arrays comprising transcript-specific oligonucleotide capture probes typically in the range of 30-mers to 80-mers, thus addressing both specificity and sensitivity.
- Alternative splicing
- As the field of genomics research is shifting from the acquisition of genome sequences to high-throughput functional genomics, there is an increasing need to understand the dynamics within the genetic regulation as well as RNA and protein sequences in order to elucidate gene expression in all its complexity. A common feature for eukaryotic genes is that they are composed of protein-encoding exons and introns. Introns (intra-genic-regions) are non-coding DNA which interrupt the exons. Introns are characterized by being excised from the pre-mRNA molecule in RNA splicing, as the sequences on each side of the intron are spliced together. RNA splicing not only provides functional mRNA, but is also responsible for generating additional diversity. This phenomenon is called alternative splicing, which results in the production of different mRNAs from the same gene. The mRNAs that represent isoforms arising from a single gene can differ by the use of alternative exons or retention of an intron that disrupts two exons. This process often leads to different protein products that may have related or drastically different, even antagonistic, cellular functions. There is increasing evidence indicating that alternative splicing is very widespread (Croft et al. Nature Genetics, 2000). Recent studies have revealed that at least 60% of the roughly 35,000 genes in the human genome are alternatively spliced. Clearly, by combining different types of modifications and thus generating different possible combinations of transcripts of different genes, alternative splicing is a potent mechanism for generating protein diversity. Analysis of the spliceome, in turn, represents a novel approach to both functional genomics and pharmacogenomics.
- Antisense Transcription in Eukaryotes
- RNA-mediated gene regulation is widespread in higher eukaryotes and complex genetic phenomena like RNA interference, co-suppression, transgene silencing, imprinting, methylation, and possibly position-effect variegation and transvection, all involve intersecting pathways based on or connected to RNA signalling (Mattick 2001; EMBO reports 2, 11: 986-991). Recent studies indicate that antisense transcription is a very common phenomenon in the mouse and human genomes (Okazaki et al. 2002; Nature 420: 563-573; Yelin et al. 2003, Nature Biotechnol.). Thus, antisense modulation of gene expression in e.g. human cells may be a common regulatory mechanism. In light of this, the present invention provides novel tools, in which non-naturally occurring nucleic acids, such as LNA oligonucleotides, can be designed to silence or modulate the regulation of a given mRNA by non-coding antisense RNA, by designing a complementary sense LNA oligonucleotide for the regulatory antisense RNA. This has a high potential in target identification, target validation and therapeutic use of LNA oligonucleotides as modulating and silencing sense nucleic acid agents. Misplaced control of alternative splicing can cause disease
- The detection of the detailed structure of all transcripts is an important goal for molecular characterization of a cell or tissue. Without the ability to detect and quantify the splice variants present in one tissue, the transcript content or the protein content cannot be described accurately. Molecular medical research shows that many cancers result in altered levels of splice variants, so an accurate method to detect and quantify these transcripts is required. Mutations that produce an aberrant splice form can also be the primary cause of such severe diseases such as spinal muscular dystrophy and cystic fibrosis.
- Much of the study of human disease, indeed much of genetics is based upon the study of a few model organisms. The evolutionary stability of alternative splicing patterns and the degree to which splicing changes according to mutations and environmental and cellular conditions influence the relevance of these model systems. At present, there is little understanding of the rates at which alternative splicing patterns change, and the factors influencing these rates. Table 1 shows a set of genes that are known to be alternatively spliced and that are orthologs of known human disease genes.
TABLE 1 C. elegans disease orthologs that are known to be differentially spliced in C. elegans. Disease C. elegans gene BLAST E value brABL1 M79.1A 1.00E−162 X-Linked Lymphoprol.-SH2D1A M79.1A 2.00E−58 Cyclin Dep. Kinase 4-CDK4 F18H3.5A 1.00E−124 HNPCC*-PMS2 H12C20.2A 1.00E−123 Neurofibromatosis 2-NF2 C01G8.5A 5.00E−163 Duchenne MD+-DMD F32B4.3A 0.00E+00 Coffin-Lowry-RPS6KA3 T01H8.1A 2.00E−13 Septooptic Dysplasia-HESX1 Y113G7A.6A 1.00E−152 Non-Insulin Dep. Diabet.-PCSK1 F11A6.1A 1.00E−166 Bartter's-SLC12A1 Y37A1C.1A 1.00E−167 Gitelmans-SLC12A3 Y37A1C.1A 0.00E+00 Hered. Spherocytosis-ANK1 B0350.2A 1.00E−09 Darier-White-SERCA K11D9.2A 0.00E+00 Spondyloepip.Dysp.-COL2A1 F01G12.5A/let-2 9.00E−20 - Previously, other microarray analyses have been performed with the aim of detecting either splicing of RNA transcripts per se in yeast, or of detecting putative exon skipping splicing events in rat tissues, but neither of these approaches had sufficient resolution to estimate quantities of splice variants, a factor that could be essential to an understanding of the changes in cell life cycle and disease.
- Thus, improved methods are needed for nucleic acid amplification, hybridization, and classification. Desirable methods can distinguish between mRNA splice variants and quantitate the amount of each variant in a sample. Other desirable methods can detect differences in expressions patterns between patient nucleic acid samples and nucleic acid standards.
- The present invention demonstrates the usefulness of LNA-modified oligonucleotides in the construction of highly specific and sensitive microarrays for expression profiling (e.g., mRNA splice variant detection) and comparative genomic hybridization. The invention provides novel technology platforms based on nucleic acids with LNA or other high affinity nucleotides for sensitive and specific assessment of alternative splicing using microarray technology. As opposed to high-density cDNA or DNA oligonucleotide microarrays, LNA microarrays are able to discriminate between highly homologous as well as differentially spliced transcripts. The invention furthermore provides methods for highly sensitive and specific nucleic acid detection by fluorescence in situ hybridization using LNA-modified oligonucleotides. The present methods greatly facilitate the analysis of gene expression patterns from a particular species, tissue, cell type. The analysis of the human spliceome provides important information for pharmacogenetics. Thus, the present methods are highly valuable in medical research and diagnostics as well as in drug development and toxicological studies.
- In general, the invention features populations of high affinity nucleic acids that have duplex stabilizing properties and thus are useful for a variety of nucleic acid detection, amplification, and hybridization methods (e.g., expression or mRNA splice variant profiling). Some of these oligonucleotides contain novel nucleotides created by combining specialized synthetic nucleobases with an LNA backbone, thus creating high affinity oligonucleotides with specialized properties such as reduced sequence discrimination for the complementary strand or reduced ability to form intramolecular double stranded structures. The invention also provides improved methods for identifying nucleic acids in a sample and for classifying a nucleic acid sample by comparing its pattern of hybridization to an array to the corresponding pattern of hybridization of one or more standards to the array (e.g., comparative genomic hybridization).
- Other desirable modified bases have decreased ability to self-anneal or to form duplexes with oligonucleotides containing one or more modified bases. The invention also provides arrays of nucleic acids containing these modified bases that have a decreased variance in melting temperature and/or an increased capture efficiency compared to naturally-occurring nucleic acids. These arrays as well as the oligonucleotides in solution can be used in a variety of applications for the detection, characterization, identification, and/or amplification of one or more target nucleic acids. These oligonucleotides and oligonucleotides of the invention in general can also be used for solution assays, such as homogeneous assays.
- Merged Probes
- In one aspect, the invention features a non-naturally-occurring nucleic acid with a melting temperature that is at least 3, 5, 8, 10, 12, 15, 20, 25, 30, 35, or 40° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides. The nucleic acid hybridizes to a first region within a first exon of a target nucleic acid and to a second region within a second exon of the target nucleic acid that is adjacent to the first exon.
- In a related aspect, the invention provides a non-naturally-occurring nucleic acid with a melting temperature that is at least 3, 5, 8, 10, 12, 15, 20, 25, 30, 35, or 4 0° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides. The nucleic acid hybridizes to a first region within an exon of a target nucleic acid and to a second region within an intron of the target nucleic acid that is adjacent to the exon.
- In another aspect, the invention features a non-naturally-occurring nucleic acid with a melting temperature that is at least 3, 5, 8, 10, 12, 15, 20, 25, 30, 35, or 40° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides. The nucleic acid hybridizes to a first region within a first intron of a target nucleic acid and to a second region within a second intron of the target nucleic acid that is adjacent to the first intron.
- In yet another aspect, the invention provides a nucleic acid that is a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10, 25, 50, 100, 150, 200, 500, 800, 1000, or 1200% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of the nucleic acid. The nucleic acid hybridizes to a first region within a first exon of a target nucleic acid and to a second region within a second exon of the target nucleic acid that is adjacent to the first exon.
- In a related aspect, the invention features a nucleic acid that is a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10, 25, 50, 100, 150, 200, 500, 800, 1000, or 1200% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of the nucleic acid. The nucleic acid hybridizes to a first region within an exon of a target nucleic acid and to a second region within an intron of the target nucleic acid that is adjacent to the exon.
- In yet another aspect, the invention provides a nucleic acid that is a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10, 25, 50, 100, 150, 200, 500, 800, 1000, or 1200% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of the nucleic acid. The nucleic acid hybridizes to a first region within a first intron of a target nucleic acid and to a second region within a second intron of the target nucleic acid that is adjacent to the first intron.
- In desirable embodiments, the nucleic acids of the invention featuring a non-naturally occurring nucleic acid exhibit increased duplex stability due to slower rates of dissociation of the nucleic acid complexes (the off-rate) (Christensen et al. 2001, Biochem. J. 354: 481-484).
- In one aspect of the invention the structure of desirable adenosine, thymine, guanine and cytosine analogs are those disclosed in PCT Publication No. WO 97/12896,
Formula - In still another aspect, the invention features a nucleic acid that is an LNA (i.e., a nucleic acids with one or more LNA units) and that hybridizes to a first region within a first exon of a target nucleic acid and to a second region within a second exon of the target nucleic acid that is adjacent to the first exon.
- In another aspect, the invention features a nucleic acid that is an LNA and that hybridizes to a first region within an exon of a target nucleic acid and to a second region within an intron of the target nucleic acid that is adjacent to the exon.
- In one aspect, the invention provides nucleic acid that is an LNA and that hybridizes to a first region within a first intron of a target nucleic acid and to a second region within a second intron of the target nucleic acid that is adjacent to the first intron.
- In desirable embodiments of any of the above aspects, the length of the segment of the nucleic acid hybridizing to the first region and the length of the segment of the nucleic acid hybridizing to the second region are between 3 and 50 nucleotides, 10 and 40 nucleotides, or 20 and 30 nucleotides, inclusive. The length of the segment of the nucleic acid hybridizing to the first region and the length of the segment of the nucleic acid hybridizing to the second region may be the same length or different lengths. Desirably, the nucleic acid containing LNA units are symmetrically spaced on both sides of a junction between either two exons, an exon and an intron, or two introns, or alternatively, the nucleic acid containing LNA units are spaced on both sides of a junction based on equalized duplex melting temperatures of the segments. Desirably, the nucleic acid has one or more LNA units within 5, 4, 3, 2, or 1 nucleotides of a junction between either two exons, an exon and an intron, or two introns.
- In another aspect, the invention features a population of nucleic acids that includes one or more nucleic acids of any one of the above aspects.
- Internal Probes
- In another aspect, the invention features a non-naturally-occurring nucleic acid with a melting temperature that is at least 3, 5, 8, 10, 12, 15, 20, 25, 30, 35, or 40° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides. The nucleic acid hybridizes to only one exon or to only one intron of a target nucleic acid.
- In a related aspect, the invention features a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10, 25, 50, 100, 150, 200, 500, 800, 1000, or 1200% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of the nucleic acid. The nucleic acid hybridizes to only one exon or to only one intron of a target nucleic acid.
- In another aspect, the invention features a nucleic acid that is an LNA and that hybridizes to only one exon or to only one intron of a target nucleic acid.
- In desirable embodiments of the above aspects for nucleic acids that hybridizes to only one exon or only one intron, the nucleic acid does not hybridize to both an exon and an intron.
- In another aspect, the invention features a population of nucleic acids that includes one or more nucleic acids of any one of the above aspects.
- Pharmaceutical Compositions and Nucleic Acid Populations
- In another aspect, the invention features a pharmaceutical composition that includes one or more of the nucleic acids of the invention and a pharmaceutically acceptable carrier, such as one of the carriers described herein.
- In another aspect, the invention features a population of two or more nucleic acids of the invention. The populations of nucleic acids of the invention may contain any number of unique molecules. For example, the population may contain as few as 10, 10 2, 10 4, or 105 unique molecules or as many as 107, 108, 109 or more unique molecules. In desirable embodiments, at least 1, 5, 10, 50, 100 or more of the polynucleotide sequences are a non-naturally-occurring sequence. Desirably, at least 20, 40, or 60% of the unique polynucleotide sequences are non-naturally-occurring sequences. Desirably, the nucleic acids are all the same length; however, some of the molecules may differ in length.
- Desirable Embodiments of Any of the Above Aspects
- In desirable embodiments of any of the above aspects, the length of one or more nucleic acids (e.g., nucleic acids in a nucleic acid population of the invention) is between 15 and 150 nucleotides, 5 and 100 nucleotides, 20 and 80 nucleotides, or 30 and 60 nucleotides in length, inclusive. In particular embodiments, the nucleic acid is 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 30, 40, 50 nucleotides or at least 60, 70, 80, 90, 100, 120, or 130 nucleotides in length. In additional embodiments, the nucleic acid is between 8 and 40 nucleotides, such as between 9 and 30, or 12 and 25, or 15 and 20 nucleotides. Desirably, at least 5, 10, 15, 20, 30, 40, 50, 60, or 70% of the nucleotides in the nucleic acid are LNA units. In desirable embodiments, every second nucleotide, every third, every fourth nucleotide, every fifth nucleotide, or every sixth nucleotide in the nucleic acid is an LNA unit. In various embodiments, (i) every second and every third nucleotide, (ii) every second and every fourth nucleotide, (iii) every second and every fifth nucleotide, (iv) every second and every sixth nucleotide, (v) every third and every fourth nucleotide, (vi) every third and every fifth nucleotide, (vii) every third and every sixth nucleotide, (viii) every fourth and every fifth nucleotide, (ix) every fourth and every sixth nucleotide, and/or (x) every fifth and every sixth nucleotide in the nucleic acid is an LNA unit. Desirably, every second, every third, and every fourth nucleotide in the nucleic acid is an LNA unit. In desirable embodiments, the nucleic acids of the invention have one or more of the following substitution patterns which is repeated throughout the nucleic acids: XxXx, xXxX, XxxXxx, xXxxXx, xxXxxX, XxxxXxxx, xXxxxXxx, xxXxxxXx, or xxxXxxxX in which “X” denotes an LNA unit and “x” denotes a DNA or RNA unit. In some embodiments, the nucleotides that are not LNA units are naturally-occuring DNA or RNA nucleotides.
- In various embodiments, the nucleic acid comprises two or more contiguous LNA units. Desirably, the nucleic acid comprises at least 2, 3, 4, 5, 6, 7, or 8 contiguous LNA units. In desirable embodiments, the number of contiguous LNA units is between 5 and 20% or 10 and 15% of the total length of the nucleic acid. In a particular embodiment, 5 contiguous nucleotides of a 50-mer merged probe are LNA units. In one embodiment, the nucleic acid does not have greatly extended stretches of modified DNA or RNA residues, e.g. greater than about 4, 5, 6, 7, or 8 consecutive modified DNA or RNA residues. According to this embodiment, one or more non-modified DNA or RNA units are present after a consecutive stretch of about 3, 4, 5, 6, 7, or 8 modified nucleic acids.
- Other desirable nucleic acids have an LNA substitution pattern that results in the formation of negligible secondary structure by the nucleic acids with itself. In one such embodiment, the nucleic acids do not form hairpins or do not form other secondary structures that would otherwise inhibit or prevent their binding to a target nucleic acid. Desirably, opposing nucleotides in a palindrome pair or opposing nucleotides in inverted repeats or in reverse complements are not both LNA units. In various embodiments, the nucleic acids in the first population form less than 3, 2, or 1 intramolecular base-pairs or base-pairs between two identical molecules. In desirable embodiments, the nucleic acid does not have LNA-5-nitroindole: LNA-5-nitroindole intramolecular base-pairs.
- In other desirable embodiments, at least one LNA unit (e.g., at least 2, 3, 4, 5, 6, 7, 8, 9, or 10 LNA units) in the nucleic acid hybridizes to a first region within a first exon of a target nucleic acid and at least one LNA unit (e.g., at least 2, 3, 4, 5, 6, 7, 8, 9, or 10 LNA units) in the nucleic acid hybridizes to a second region within a second exon of the target nucleic acid that is adjacent to the first exon. The number of LNA units that bind to each region can be the same or different. In some embodiments, the 5′ terminal nucleotide of the nucleic acid is or is not an LNA unit. Desirably, the 3′ terminal nucleotide of the nucleic acid is not an LNA unit (e.g., the nucleic acid may contain a 3′ terminal naturally-occurring nucleotide).
- Desirably, the nucleic acid can distinguish between different nucleic acids (e.g., mRNA splice variants) that cannot be distinguished using a naturally-occurring control nucleic acid (e.g., a control nucleic acid that consists of only 2′-deoxynucleotides such as a control nucleic acid of the same length as the nucleic acid of the invention). Desirably, the hybridization intensity of the nucleic acid for an exon of interest is at least 2, 3, 4, 5, 6, or 10 fold greater than the hybridization intensity of the nucleic acid for another exon in the same target nucleic acid (e.g., mRNA) or in another nucleic acid. Desirably, the hybridization intensity of the nucleic acid for target nucleic acid is at least 2, 3, 4, 5, 6, or 10 fold greater than the hybridization intensity for a non-target nucleic acid with less than 99, 95, 90, 80, 70, or 60% sequence identity to the target nucleic acid.
- Desirably, all of the nucleic acids of the population or all of the nucleic acids of a subpopulation of the population are the same length. In some embodiments, the population includes one or more nucleic acids of a different length. In some embodiments, longer nucleic acids contain one or more nucleotides with universal bases. For example, nucleotides with universal bases can be used to increase the thermal stability of nucleic acids that would otherwise have a thermal stability lower than some or all of the nucleic acids in the population. In some embodiments, one or more nucleic acids have a universal base located at the 5′ or 3′ terminus of the nucleic acid. In desirable embodiments, one or more (e.g., 2, 3, 4, 5, 6, or more) universal bases are located at the 5′ and 3′ termini of the nucleic acid. Desirably, all of the nucleic acids in the population have the same number of universal bases. Desirable universal bases include inosine, pyrene, 3-nitropyrrole, and 5-nitroindole.
- In desirable embodiments, the nucleic acid has at least one LNA A or LNA T. In some embodiments, each nucleic acid has at least one LNA A or LNA T. Desirably, all of the adenine and thymine-containing nucleotides in the LNA are LNA A and LNA T, respectively. In some embodiments, a nucleic acid with a increased capture efficiency or melting temperature compared to a control nucleic acid has at least one LNA T or LNA C. In some embodiments, all of the thymine and cytosine-containing nucleotides in the LNA are LNA T and LNA C, respectively. In some embodiments, a nucleic acid with an increased specificity or decreased self-complementarity compared to a control nucleic acid has at least one LNA A or LNA C. In some embodiments, all of the adenine and cytosine-containing nucleotides in the LNA are LNA A and LNA C, respectively.
- Desirably, at least 10, 20, 25, 30, 40, 50, 60, 70, 80, 90, or 100% of the nucleic acids in the population have one ore more LNA units.
- In desirable embodiments, the LNA has at least one 2,6-diaminopurine, 2-aminopurine, 2-thio-thymine, 2-thio-uracil, inosine, or hypoxanthine base. Desirably, the LNA has a nucleotide with a 2′0, 4° C.-methylene linkage between the 2′ and 4′ position of a sugar moiety. In some embodiments, one or more nucleic acids in the first population are LNA/DNA, LNA/RNA, or LNA/DNA/RNA chimeras.
- In desirable embodiments of any of the above aspects, the variance in the melting temperature of the population is at least 10, 20, 30, 40, 50, 60, or 70% less than the variance in the melting temperature of the corresponding control population of nucleic acids of the same length with 2′-deoxynucleotides (e.g., DNA nucleotides) instead of LNA units or other modified or non-naturally-occurring units. In desirable embodiments, the standard deviation in melting temperature is less than 10, 9.5, 9, 8.5, 8, 7.5, 7, 6.5, or 6. In certain embodiment, the range in melting temperatures for nucleic acids in the population is less than 70, 60, 50, 40, 30, or 20° C. Desirably, the variance in the melting temperature of the population is less than 59, 50, 40, 30, 25, 20, 15, 10, or 5° C.
- In still other embodiments, the nucleic acids are covalently bonded to a solid support. Desirably, the nucleic acids are in a predefined arrangement. In various embodiments, the first population has at least 10; 100; 1,000; 5,000; 10,000; 100,000; or 1,000,000 different nucleic acids. Desirably, the nucleic acids in the population together hybridize to at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, or 100% of the exons of a target nucleic acid. In desirable embodiments, the population includes nucleic acids that together hybridize to at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, or 100% of the nucleic acids expressed by a particular cell or tissue. In some embodiments, the population includes nucleic acids that together hybridize to at least one exon from at least 1,5, 10, 20, 25, 30, 40, 50, 60, 70, 80, 90, or 100% of the nucleic acid sequences expressed by a particular cell or tissue at a given point in time (e.g., an expression array with sequences corresponding to the sequences of mRNA molecules expressed by a particular cell type or a cell under a particular set of conditions). In some embodiments, the plurality of nucleic acids are used as PCR primers or FISH probes.
- Desirable modified bases of the present invention when incorporated into the central position of a 9-mer oligonucleotide (all other eight residues or units being natural DNA or RNA units with natural bases) exhibit a T m difference equal to or less than about 15, 12, 10, 9, 8, 7, 6, 5, 4, 3 or 2° C. upon hybridizing to the four complementary oligonucleotide variants that are identical except for the unit corresponding to the LNA unit, where each variant has one of the natural bases uracil, cytosine, thymine, adenine or guanine. That is, the highest and the lowest Tm (referred to herein as the Tm differential) obtained with such four complementary sequences is 15,12, 10,9,8,7,6,5,4, 3 or 2° C. or less.
- Modified nucleic acid oligomers of the invention desirably contain at least one LNA unit, such as an LNA unit with a modified nucleobase. Modified nucleobases or nucleosidic bases desirably base-pair with adenine, guanine, cytosine, uracil, or thymine. Exemplary oligomers contain 2 to 100, 5 to 100, 4 to 50, 5 to 50, 5 to 30, or 8 to 15 nucleic acid units. In some embodiments, one or more LNA units with natural nucleobases are incorporated into the oligonucleotide at a distance from the LNA unit having a modified base of 1 to 6 (e.g., 1 to 4) bases. In certain embodiments, at least two LNA units with natural nucleobases are flanking an LNA unit having a modified base. Desirably, at least two LNA units independently are positioned at a distance from the LNA unit having the modified base of 1 to 6 (e.g., 1 to 4 bases).
- Desirable modified nucleobases or nucleosidic bases for use in nucleic acid compositions of the invention include optionally substituted carbon alicyclic or carbocyclic aryl groups (i.e., only carbon ring members), particularly multi-ring carbocyclic aryl groups such as groups having 2, 3, 4, 5, 6, 7, or 8 linked, particularly fused carbocyclic aryl moieties. Optionally substituted pyrene is also desirable. Such nucleobases or nucleosidic bases can provide significant performance results, as demonstrated in the examples which follow. Heteroalicyclic and heteroaromatic nucleobases or nucleosidic bases also are suitable. In some embodiments, the carbocyclic moiety is linked to the 1′-position of the LNA unit through a linker (e.g., a branched or straight alkylene or alkenylene).
- Desirable LNA units have a carbon or hetero alicyclic ring with four to six ring members, e.g. a furanose ring, or other alicyclic ring structures such as a cyclopentyl, cycloheptyl, tetrahydropyranyl, oxepanyl, tetrahydrothiophenyl, pyrrolidinyl, thianyl, thiepanyl, piperidinyl, and the like. In one aspect, at least one ring atom of the carbon or hetero alicyclic group is taken to form a further cyclic linkage to thereby provide a multi-cyclic group. The cyclic linkage may include one or more, typically two atoms, of the carbon or hetero alicyclic group. The cyclic linkage also may include one or more atoms that are substituents, but not ring members, of the carbon or hetero alicyclic group. Other desirable LNA units are compounds having a substituent on the 2′-position of the central sugar moiety (e.g., ribose or xylose), or derivatives thereof, which favors the C3′-endo conformation, commonly referred to as the North (or simply N for short) conformation. These LNA units include ENA (2′-O,4′-C-ethylene-bridged nucleic acids such as those disclosed in WO 00/47599) units as well as non-bridged riboses such as 2′-F or 2′—O—methyl.
- For any of the above aspects, an exemplary control nucleic acid has β-D-2-deoxyribose instead of one or more bicyclic or sugar groups of a LNA unit or other modified or non-naturally-occurring units in a nucleic acid of the first population. In some embodiments, the nucleic acid or population of the invention and the control nucleic acid or population only have naturally-occurring nucleobases. If a nucleic acid in the nucleic acid or population of the invention has one or more non-naturally-occurring nucleobases, the capture efficiency of the corresponding control nucleic acid is calculated as the average capture efficiency for all of the nucleic acids that have either A, T, C, G or mC (methyl Cytosin) in each position corresponding to a non-naturally-occurring nucleobase in the nucleic acid in the first population.
- Complex of Target Nucleic Acids and Nucleic Acid Probes
- In one aspect, the invention features a complex of one or more target nucleic acids and nucleic acid of the invention (e.g., nucleic acid probes) in which one or more target nucleic acids are hybridized to a plurality of nucleic acids of the invention. Desirably, at least 2, 3, 4, 5, 6, 7, 10, 15, 20, 30, or 40 different target nucleic acids are hybridized. In some embodiments, the target nucleic acids are cDNA molecules reverse transcribed from a patient sample or cRNA molecules amplified from a patient sample using a T7 RNA polymerase-based linear amplification system or the like. The target nucleic acids are labeled prior to hybridization to the nucleic acids of invention.
- Methods for Detecting or Amplifying Target Nucleic Acids
- In one aspect, the invention features a method for detecting the presence of one or more target nucleic acids in a sample. This method involves incubating a nucleic acid sample with one or more nucleic acids of the invention under conditions that allow at least one target nucleic acid to hybridize to at least one of the nucleic acids of the invention. Desirably, hybridization is detected for at least 2, 3, 4, 5, 6, 8, 10, or 12 target nucleic acids. In some embodiments, the method further includes contacting the target nucleic acid with a second nucleic acid or a population of second nucleic acids that binds to a different region of the target molecule than the first nucleic acid. Desirably, the method further involves identifying one or more hybridized target nucleic acids and/or determining the amount of one or more hybridized target nucleic acids. In desirable embodiments, the method further includes determining the presence or absence of an mRNA splice variant of interest in the sample and/or determining the presence or absence of a mutation, deletion, and/or duplication of an exon of interest. In some embodiments, the mutation, deletion, and/or duplication is indicative of a disease, disorder, or condition, such as cancer.
- In desirable embodiments of any of the above detection methods, at least 5, 10, 15, 20, 30, 40, 50, 80, 100, 150, 200, or more target nucleic acids hybridize to the nucleic acids of the invention. Desirably, the method is repeated under one or more different incubation conditions. In particular embodiments, the method is repeated at 1, 3, 5, 8, 10, 15, 20, 30, 40 or more different temperatures, cation concentrations (e.g., concentrations of monovalent cations such as Na+ and K + or divalent cations such as Mg 2+ and Ca2+), denaturants (e.g., hydrogen bond donors or acceptors that interfere with the hydrogen bonds keeping the base-pairs together such as formamide or urea). Desirably, the method also includes identifying the target nucleic acid hybridized to the nucleic acids of the invention and/or determining the amount of the target nucleic acid hybridized to the nucleic acids of the invention. In particular embodiments, the target nucleic acids are labeled with a fluorescent group. In certain embodiments, the labeling is repeated using different fluorescent groups (e.g., labelling for so-called dye-swap labeling experiments). In desirable embodiments, the determination of the amount of bound target nucleic acid involves one or more of the following: (i) adjusting for the varying intensity of the excitation light source used for detection of the hybridization, (ii) adjusting for photobleaching of the fluorescent group, and/or (iii) comparing the fluorescent intensity of the target nucleic acid(s) hybridized to the nucleic acids of the invention of nucleic acids to the fluorescent intensity of adifferent sample of nucleic acids hybridized to the nucleic acids of the invention (e.g., a different sample hybridized to the same population of nucleic acids of the invention on the same or a different solid support such as the same chip or a different chip). Desirably, this comparison in fluorescent intensity involves adjusting for a difference in the amount of the nucleic acids of the invention used for hybridization to each sample and/or adjusting for a difference in the buffer (e.g., a difference in Mg2+ concentration) used for hybridization to each sample or scaling for different labeling efficiencies with different fluorochromes.
- Desirably, the target nucleic acids are cDNA molecules reverse transcribed from a patient sample or cRNA molecules amplified using a T7 RNA polymerase-based linear amplification system or the like from a patient sample. In particular embodiments, the sample has nucleic acids that are amplified using one or more primers specific for an exon of a target nucleic acid, and the method involves determining the presence or absence of an mRNA splice variant with the exon in the sample. Desirably, one or more of the primers are specific for an exon or exon-exon junction of a pathogen of interest, and the method involves determining the presence or absence of a nucleic acid with the exon in the sample.
- In a desirable embodiment, the nucleic acids of the invention are covalently bonded to a solid support by reaction of a nucleoside phosphoramidite with an activated solid support, and subsequent reaction of a nucleoside phosphoramide with an activated nucleotide or nucleic acid bound to the solid support. In some embodiments, the solid support or the growing nucleic acid bound to the solid support is activated by illumination, a photogenerated acid, or electric current.
- In another aspect, the invention features a method for amplifying a target nucleic acid molecule. The method involves (a) incubating a first nucleic acid of the invention with a target nucleic acid under conditions that allow the first nucleic acid to bind the target nucleic acid; and (b) extending the first nucleic acid with the target nucleic acid as a template. Desirably, the method further involves contacting the target nucleic acid with a second nucleic acid (e.g., a second nucleic acid of the invention) that binds to a different region of the target nucleic acid than the first nucleic acid. In various embodiments, the sequence of the target nucleic acid is known or unknown.
- In one aspect, the invention features a method of detecting a nucleic acid of a pathogen (e.g., a nucleic acid in a sample such as a blood or urine sample from a mammal). This method involves contacting a nucleic acid probe of the invention (e.g., a probe specific for an exon or a mRNA from a particular pathogen or family of pathogens) with a nucleic acid sample under conditions that allow the probe to hybridize to at least one nucleic acid in the sample. The probe is desirably at least 60, 70, 80, 90, 95, or 100% complementary to a nucleic acid of a pathogen (e.g., a bacteria, virus, or yeast such as any of the pathogens described herein). Hybridization between the probe and a nucleic acid in the sample is detected, indicating that the sample contains the corresponding nucleic acid from a pathogen. In some embodiments, the method is used to determine what strain of a pathogen has infected a mammal (e.g., a human) by determining whether a particular nucleic acid is present in the sample. In other embodiments, the probe has a universal base in a position corresponding to a nucleotide that varies among-different strains of a pathogen, and thus the probe detects the presence of a nucleic acid from any of a multiple of pathogenic strains.
- Methods for Classifying Nucleic Acids Samples
- In one aspect, the invention features a method for classifying a test nucleic acid sample including target nucleic acids. This method involves (a) incubating a test nucleic acid sample with a one or more nucleic acids of the invention under conditions that allow at least one of the nucleic acids in the test sample to hybridize to at least one nucleic acid of the invention, (b) detecting a hybridization pattern of the test nucleic acid sample, and (c) comparing the hybridization pattern to a hybridization pattern of a first nucleic acid standard, whereby the comparison indicates whether or not the test sample has the same classification as the first standard. Desirably, the method also includes comparing a hybridization pattern of the test nucleic acid sample to a hybridization pattern of a second standard. In various embodiments, a hybridization pattern of the test nucleic acid sample is compared to at least 3, 4, 5, 8, 10, 15, 20, 30, 40, or more standards.
- Desirably, the method also includes identifying the hybridized target nucleic acid and/or determining the amount of hybridized target nucleic acid. In particular embodiments, the target nucleic acids are labeled with a fluorescent group. Desirably, the first nucleic acid standard is labeled with a different fluorescent group. The fluorescence of the target nucleic acids and the first nucleic acid standard can be detected simultaneously or sequentially.
- In desirable embodiments, the method further includes determining the presence or absence of an mRNA splice variant of interest in the sample and/or determining the presence or absence of a mutation, deletion, and/or duplication of an exon of interest. In some embodiments, the mutation, deletion, and/or duplication is indicative of a disease, disorder, or condition, such as cancer.
- In desirable embodiments, the determination of the amount of bound target nucleic acid involves one or more of the following: (i) adjusting for the varying intensity of the excitation light source used for detection of the hybridization, (ii) adjusting for photobleaching of the fluorescent group, and/or (iii) comparing the fluorescent intensity of the target nucleic acid(s) hybridized to the nucleic acids of the invention to the fluorescent intensity of a different sample of nucleic acids hybridized to the nucleic acids of the invention (e.g., a different sample hybridized to same set of nucleic acids of the invention on the same or a different solid support such as the same chip or a different chip). Desirably, this comparison in fluorescent intensity involves adjusting for a difference in the amount of the plurality used for hybridization to each sample and/or adjusting for a difference in the buffer (e.g., a difference in Mg 2+ concentration) used for hybridization to each sample.
- Desirably, the nucleic acids in the population together hybridize to at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, or 100% of the exons of a target nucleic acid. In desirable embodiments, the population includes nucleic acids that together hybridize to at least 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, or 100% of the nucleic acids expressed by a particular cell or tissue. In some embodiments, the population includes nucleic acids that together hybridize to at least one exon from at least 1, 5, 10, 20, 25, 30, 40, 50, 60, 70, 80, 90, or 100% of the nucleic acid sequences expressed by a particular cell or tissue at a given point in time (e.g., an expression array with sequences corresponding to the sequences of mRNA molecules expressed by a particular cell type or a cell under a particular set of conditions). Desirably, the method further includes using a nucleic acid or a region of a nucleic acid that is present in a first test sample but not present in a first standard or not present in a second test sample as a probe or primer for the detection, amplification, or characterization of the nucleic acid.
- In desirable embodiments of any of the above methods, at least 5, 10, 15, 20, 30, 40, 50, 80, 100, 150, 200, or more target nucleic acids hybridize to the nucleic acids of the invention. Desirably, the method is repeated under one or more different incubation or hybridization conditions. In particular embodiments, the method is repeated at 1, 3, 5, 8, 10, 15, 20, 30, 40 or more different temperatures, cation concentrations (e.g., concentration of monovalent cations such as Na+ and K + or divalent cations such as Mg2+and Ca2),′ denaturants (e.g., hydrogen bond donors or acceptors that interfere with the hydrogen bonds keeping the base-pairs together such as formamide or urea).
- In particular embodiments, the sample has nucleic acids that are amplified using one or more primers specific for an exon of a target nucleic acid, and the method involves determining the presence or absence of an mRNA splice variant with the exon in the sample. Desirably, one or more of the primers are specific for an exon or exon-exon junction of a pathogen of interest, and the method involves determining the presence or absence of a nucleic acid with the exon in the sample.
- Desirably, the comparison of the hybridization pattern of a patient nucleic acid sample to that of one or more standards is used to determine whether or not a patient has a particular disease, disorder, condition, or infection or an increased risk for a particular disease, disorder, condition, or infection. In some embodiments, the comparison is used to determine what pathogen has infected a patient and to select a therapeutic for the treatment of the patient. Desirably, the comparison is used to select a therapeutic for the treatment or prevention of a disease or disorder in the patient. In yet other embodiments, the comparison is used to include or exclude the patient from a group in a clinical trial. Desirably, the comparison is used to compare the expression of nucleic acids (e.g., mRNA splice forms associated with toxicity) in the presence and absence of a candidate compound (e.g., a lead compound for drug development). In other embodiments, the comparison is used to determine differences in expression of nucleic acids (e.g., mRNA splice variants) under particular conditions (e.g., under different environmental stress conditions) or at different developmental time points. In particular embodiments, the expression of one or more members from a particular enzyme class (e.g., protein kinase splice variants) is measured.
- In a desirable embodiment, the nucleic acids of the invention are covalently bonded to a solid support by reaction of a nucleoside phosphoramidite with an activated solid support, and subsequent reaction of a nucleoside phosphoramide with an activated nucleotide or nucleic acid bound to the solid support. In some embodiments, the solid support or the growing nucleic acid bound to the solid support is activated by illumination, a photogenerated acid, or electric current.
- The use of a variety of different monomers in the nucleic acids of the invention offers a means to “fine tune” the chemical, physical, biological, pharmacokinetic, and pharmacological properties of the nucleic acids thereby facilitating improvement in their safety and efficacy profiles when used as a therapeutic drug.
- Applications for the Nucleic Acids of the Invention
- In another aspect, the invention features the use of one ore more nucleic acids of the invention for the detection, amplification, or classification of a nucleic acid of interest or a population of nucleic acids of interest.
- In another aspect, the invention features the use of one or more nucleic acids of the invention for alternative mRNA splice variant detection, expression profiling, comparative genomic hybridization, or real-time PCR. In exemplary real-time PCR applications, the nucleic acids are used to determine the amount of one or more target nucleic acids (e.g., mRNA splice variants) in a sample. In particular embodiments, fluorescently labeled RT-PCR products from the amplification of a test nucleic acid sample are hybridized to a population of nucleic acids of the invention. Desirably, the amount of one or more RT-PCR products is measured to determine the amount of the corresponding nucleic acid in the initial sample.
- In yet another aspect, the invention features the use of a nucleic of the invention as a PCR primer or FISH probe.
- Methods for Selecting a Population of Nucleic Acid
- In one aspect, the invention features a method of selecting a nucleic acid for a population of nucleic acids. This method involves (a) determining the melting temperature of a nucleic acid of the invention, determining the ability of the nucleic acid to self-anneal, determining the ability of the nucleic acid to hybridize to one or more exons or introns of a target nucleic acid, and/or determining the ability of the nucleic acid to hybridize to a non-target nucleic acid, and (b) selecting the nucleic acid for inclusion or exclusion from the population based on the determination in step (a). In desirable embodiments, step (a) is performed for at least 2, 3, 4, 5, 6, 10, 20, 50, 100, 200, 500, 1,000, 5,000 or more nucleic acids, and a subset of the nucleic acids are selected for inclusion in the population based on the determination in step (a). Desirably, the nucleic acids with the highest melting temperatures and/or ability to hybridize to one or more exons or introns of a target nucleic acid are selected. Desirably, the nucleic acids with the lowest ability to self-anneal and/or hybridize to a non-target nucleic acid are selected.
- Databases with Hybridization Patterns of Nucleic Acids Samples and/or Standards
- The invention also features a variety of databases. These databases are useful for storing the information obtained in any of the methods of the invention. These databases may also be used in the diagnosis of disease or an increased risk for a disease or in the selection of a desirable therapeutic for a particular patient or class of patients.
- Accordingly, in one such aspect, the invention provides an electronic database including at least 1, 10, 10 2, 10 3, 5×10 3, 104, 10 5, 10 6, 107, 10 8, or 109 records of a nucleic acid of interest or a population of nucleic acids of interest (e.g., one or more nucleic acids in a standard or in a test nucleic acid sample) correlated to records of its hybridization pattern to a plurality of nucleic acids of the invention under one or more incubation conditions (e.g., one or more temperatures, denaturant concentrations, or salt concentrations).
- In another aspect, the invention features computer including the database of the above aspect and a user interface (i) capable of displaying a hybridization pattern for a nucleic acid of interest or a population of nucleic acids of interest whose record is stored in the computer or (ii) capable of displaying a nucleic acid of interest (e.g., displaying the polynucleotide sequence or another identifying characteristic of the nucleic acid of interest) or a population of nucleic acids of interest that produces a hybridization pattern whose record is stored in the computer.
- Methods for Silencing a Target Nucleic Acid in a Cell or Animal
- One method for inhibiting specific gene expression involves the use of antisense or double stranded oligonucleotides, which are complementary to a specific target messenger RNA (mRNA) sequence, such as a specific mRNA splice variant. Of special interest are oligonucleotides with a modified backbone (such as LNA or phosphorothioate) that are not readily degraded by endonucleases in the target cells.
- In one aspect, the invention features the use of a nucleic acid of the invention for the manufacture of a pharmaceutical composition for treatment of a disease curable by an antisense or RNAi technology.
- In one aspect, the invention provides a method for inhibiting the expression of a target nucleic acid in a cell. The method involves introducing into the cell a nucleic acid of the invention in an amount sufficient to specifically attenuate expression of the target nucleic acid. The introduced nucleic acid has a nucleotide sequence that is essentially complementary to a region of desirably at least 20 nucleotides of the target nucleic acid. Desirably, the cell is in a mammal.
- In a related aspect, the invention provides a method for preventing, stabilizing, or treating a disease, disorder, or condition associated with a target nucleic acid in a mammal. This method involves introducing into the mammal a nucleic acid of the invention in an amount sufficient to specifically attenuate expression of the target nucleic acid, wherein the introduced nucleic acid has a nucleotide sequence that is essentially complementary to a region of desirably at least 20 nucleotides of the target nucleic acid.
- In another aspect, the invention provides a method for preventing, stabilizing, or treating a pathogenic infection in a mammal by introducing into the mammal a nucleic acid of the invention in an amount sufficient to specifically attenuate expression of a target nucleic acid of a pathogen. The introduced nucleic acid has a nucleotide sequence that is essentially complementary to a region of desirably at least 20 nucleotides of the target nucleic acid.
- In desirable embodiments of the therapeutic methods of the above aspects, the mammal is a human. In some embodiments, the introduced nucleic acid is single stranded or double stranded.
- With respect to the therapeutic methods of the invention, it is not intended that the administration of nucleic acids to a mammal be limited to a particular mode of administration, dosage, or frequency of dosing; the present invention contemplates all modes of administration, including oral, intraperitoneal, intramuscular, intravenous, intraarticular, intralesional, subcutaneous, or any other route sufficient to provide a dose adequate to prevent or treat a disease (e.g., a disease associated with the expression of a target nucleic acid that is silenced with a nucleic acid of the invention). One or more nucleic acids may be administered to the mammal in a single dose or multiple doses. When multiple doses are administered, the doses may be separated from one another by, for example, one week, one month, one year, or ten years. It is to be understood that, for any particular subject, specific dosage regimes should be adjusted over time according to the individual need and the professional judgment of the person administering or supervising the administration of the compositions.
- Exemplary mammals that can be treated using the methods of the invention include humans, primates such as monkeys, animals of veterinary interest (e.g., cows, sheep, goats, buffalos, and horses), and domestic pets (e.g., dogs and cats). Exemplary cells in which one or more target genes can be silenced using the methods of the invention include invertebrate, plant, bacteria, yeast, and vertebrate (e.g., mammalian or human) cells.
- Optimum dosages for gene silencing applications may vary depending on the relative potency of individual oligonucleotides, and can generally be estimated based on EC 50 values found to be effective in in vitro and in vivo animal models. In general, dosage is from 0.001 μg to 100 g per kg of body weight (e.g., 0.001 μg/kg to 1 g/kg), and may be given once or more daily, weekly, monthly or yearly, or even once every 2 to 20 years (U.S.P.N. 6,440,739). Persons of ordinary skill in the art can easily estimate repetition rates for dosing based on measured residence times and concentrations of the drug in bodily fluids or tissues. Following successful treatment, it may be desirable to have the patient undergo maintenance therapy to prevent the recurrence of the disease state, wherein the oligonucleotide is administered in maintenance doses, ranging from 0.001 ug to 100 g per kg of body weight (e.g., 0.001 1 g/kg to 1 g/kg), once or more daily, to once every 20 years. If desired, conventional treatments may be used in combination with the nucleic acids of the present invention.
- Suitable carriers include, but are not limited to, saline, buffered saline, dextrose, water, glycerol, ethanol, and combinations thereof. The composition can be adapted for the mode of administration and can be in the form of, for example, a pill, tablet, capsule, spray, powder, or liquid. In some embodiments, the pharmaceutical composition contains one or more pharmaceutically acceptable additives suitable for the selected route and mode of administration. These compositions may be administered by, without limitation, any parenteral route including intravenous, intra-arterial, intramuscular, subcutaneous, intradermal, intraperitoneal, intrathecal, as well as topically, orally, and by mucosal routes of delivery such as intranasal, inhalation, rectal, vaginal, buccal, and sublingual. In some embodiments, the pharmaceutical compositions of the invention are prepared for administration to vertebrate (e.g., mammalian) subjects in the form of liquids, including sterile, non-pyrogenic liquids for injection, emulsions, powders, aerosols, tablets, capsules, enteric coated tablets, or suppositories.
- Exemplary Oligomers of the Invention and Methods for Synthesizing Them
- In desirable embodiments, the invention features a method of synthesizing a nucleic acid. This method involves synthesizing a 2-thio-uridine nucleoside or nucleotide of formula IV using a compound of formula VIII, IX, X, XI, or XII as shown below. The nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.

- In a particular embodiment, nucleobase thiolation is performed on the O 2 position of compound XI to form compound IV. In another embodiment, sulphurization on both O2 and 04 in compound VIII generates a 2,4-dithio-uridine nucleoside or nucleotide of formula X which is converted into compound IV. In yet another embodiment, a cyclic ether of formula XI is transferred into compound IV or a 2—O—alkyl-uridine nucleoside or nucleotide of formula XII through reaction with the 5′ position. In other embodiments, a 2—O—alkyl-uridine nucleoside or nucleotide of formula XII is generated by direct alkylation of a uridine nucleoside or nucleotide of formula VIII.
- In desirable embodiments R 4 and R2 are each independently alkyl (e.g., methyl or ethyl), acyl (e.g., acetyl or benzoyl), or any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl). R5” is any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, trityl(triphenylmethyl), acetyl, benzoyl, or benzyl. In desirable embodiments, R5 is hydrogen, alkyl (e.g., methyl or ethyl), 1-propynyl, thiazol-2-yl, pyridine-2-yl, thien-2-yl, imidazol-2-yl, (4/5-methyl)-thiazol-2-yl, 3-(iodoacetamido)propyl, 4-[N,N-bis(3-aminopropyl)amino]butyl), or halo (e.g., chloro, bromo, iodo, fluoro).
- The group —OR 3′in the formulas IV, VIII, IX, X, XI, and XII is any of the groups listed for R3 or R3′ in formula IIa or formula Ib or listed for R3 or R3* in formula Ia, Scheme A, or Scheme B, or the group —OR3 or R3′ in the formulas IV, VIII, IX, X, XI, and XII is selected from the group consisting of H, —OH, P(Q(CH2)2CN)N(iPr)2, P(O(CH2)2CN)N(iPr)2, phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g., acetyl or benzoyl), aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl)), linkers (e.g., a linker containing an amine, ethylene glycol, quinone such as anthraquinone), detectable labels (e.g., radiolabels or fluorescent labels), and biotin.
- The group —OR 5′ in the formulas IV, and VIII, IX, X, and XII is any of the groups listed for R5 or R5′ in formula Ia or formula Ib or listed for R5 or R5*in formula Ia, Scheme A, or Scheme B, or the group —OR5′ or R5′0 in the formulas IV, and VIII, IX, X, and XII is selected from the group consisting of H, —OH, P(O(CH2)2CN)N(iPr)2, P(O(CH2)2CN)N(iPr)2, phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g. acetyl or benzoyl), aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl)), linkers (e.g., a linker containing an amine, ethylene glycol, quinone such as anthraquinone), detectable labels (e.g., radiolabels or fluorescent labels), and biotin.
- In yet another aspect, the invention features a method of synthesizing a nucleic acid. This method involves synthesizing a 2-thiopyrimidine nucleoside or nucleotide of formula IV using a compound of formula III or compounds of the formula I, II, and III as shown below. The nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.

- In some embodiments, lewis acid-catalyzed condensation of a substituted sugar of formula I and a substituted 2-thio-uracil of formula II results in a substituted 2-thio-uridine nucleoside or nucleotide of the formula III. In some embodiments, a compound of formula III is converted into a LNA 2-thiouridine nucleoside or nucleotide of formula IV.
- In desirable embodiments R 4′ and R5′ are, e.g., methanesulfonyloxy, p-toluenesulfonyloxy, or any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, trityl(triphenylmethyl), acetyl, benzoyl, or benzyl, R″ is, e.g., acetyl, benzoyl, alkoxy (e.g., methoxy). R2 is, e.g.,acetyl or benzoyl, and R3 is any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, trityl(triphenylmethyl), acetyl, or benzoyl. In desirable embodiments, R5 is hydrogen, alkyl (e.g. methyl or ethyl), 1-propynyl, thiazol-2-yl, pyridine-2-yl, thien-2-yl, imidazol-2-yl, (4/5-methyl)-thiazol-2-yl, 3-(iodoacetamido)propyl, 4-[N,N-bis(3-aminopropyl)amino]butyl), or halo (e.g., chloro, bromo, iodo, fluoro).
- The group —OR 3′ in the formulas I, III, and IV is any of the groups listed for R3 or R3′ in formula Ia or formula Ib or listed for R3 or R3* in formula Ia, Scheme A, or Scheme B, or the group —OR3′ or R3′ in the formulas I, III, and IV is selected from the group consisting of H, —OH, P(O(CH2)2CN)N(iPr)2, phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g. acetyl or benzoyl), aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl)), linkers (e.g., a linker containing an amine, ethylene glycol, quinone such as anthraquinone), detectable labels (e.g., radiolabels or fluorescent labels), and biotin.
- The group R 5′ in the formulas I, III, and IV is any of the groups listed for R5 or R5′ in formula Ia or formula Ib or listed for R5 or R5* in formula Ia, Scheme A, or Scheme B, or R5′ in the formulas I, III, and IV is selected from the group consisting of H, —OH, P(O(CH2)2CN)N(iPr)2, phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g. acetyl or benzoyl), aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl)), linkers (e.g., a linker containing an amine, ethylene glycol, quinone such as anthraquinone), detectable labels (e.g., radiolabels or fluorescent labels), and biotin.
- In still another aspect, the invention features a method of synthesizing a nucleic acid. This method involves synthesizing a 2-thiopyrimidine nucleoside or nucleotide of formula IV using a compound of formula VII, compounds of the formula V, VI, and VII, or compounds of the formula I, V, VI, and VII as shown below. The nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.

- In some embodiments, a 2-thio-uridine nucleoside or nucleotide of the formula IV is synthesized through ring-synthesis of the nucleobase by reaction of an amino sugar of the formula V and a substituted isothiocyanate of the formula VI.
- In desirable embodiments, R 4′ and R5′ are each idenpendently, e.g., methanesulfonyloxy, p-toluenesulfonyloxy, or any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, trityl(triphenylmethyl), acetyl, benzoyl, or benzyl. R″ is, e.g., acetyl or benzoyl or alkoxy (e.g., methoxy), and Ris, e.g., acetyl or benzoyl, R3′ is any appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, trityl(triphenylmethyl), acetyl, or benzoyl. R5 are R6 each idenpendently, e.g., hydrogen or alkyl (e.g. methyl or ethyl). R6 can also be, e.g., an appropriate protecting group such as silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl). In desirable embodiments, R5 is hydrogen or methyl, and R6 is methyl or ethyl.
- The group —OR 3 in the formulas I, V, VII, and IV is any of the groups listed for R3 or R3′ in formula Ia or formula Ib or listed for R3 or R3* in formula Ia, Scheme A, or Scheme B, or the group —ORor R3 in the formulas I, V, VII, and IV is selected from the group consisting of H, —OH, P(O(CH2)2CN)N(iPr)2, phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g. acetyl or benzoyl), aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl)), linkers (e.g., a linker containing an amine, ethylene glycol, quinone such as anthraquinone), detectable labels (e.g., radiolabels or fluorescent labels), and biotin.
- R 5′ in the formulas I, V, VII, and IV is any of the groups listed for R5 or R5 in formula Ia or formula Ib or listed for R5 or R5* in formula Ia, Scheme A, or Scheme B, or R5 in the formulas I, V, VII, and IV is selected from the group consisting of H, —OH, P(O(CH2)2CN)N(iPr)2, phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g. acetyl or benzoyl), aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl)), linkers (e.g., a linker containing an amine, ethylene glycol, quinone such as anthraquinone), detectable labels (e.g., radiolabels or fluorescent labels), and biotin.
- In another aspect, the invention features a method of synthesizing a nucleic acid. This method involves synthesizing a 2-thiopyrimidine nucleoside as shown below. In desirable embodiments, the method further comprises reacting one or both compounds of the
formula 4 with a phosphodiamidite (e.g., 2-cyanoethyl tetraisopropylphosphorodiamidite) to produce the corresponding nucleoside phosphoramidite. The nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention.
- In some embodiments, a glycosyl-donor is coupled to a nucleobase as shown in pathway A. In other embodiments, ring synthesis of the nucleobase is performed as show in pathway B. In still other embodiments, LNA-T diol is modified as shown in pathway C.
- In desirable embodiments, R is hydrogen, methyl, 1-propynyl, thiazol-2-yl, pyridine-2-yl, thien-2-yl, imidazol-2-yl, (4/5-methyl)-thiazol-2-yl, 3-(iodoacetamido)propyl, 4-[N,N-bis(3-aminopropyl)amino]butyl, or halo (e.g., chloro, bromo, iodo, fluoro). Desirably, R 1, R2, and R3 are each any appropriate protecting group such as acetyl, benzyl, silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl).
- In another aspect, the invention features a method of synthesizing a nucleic acid. This method involves synthesizing a 2-thiopyrimidine nucleoside or nucleotide of
formula 4 using a compound offormula 3, compounds of theformula formula 
- In desirable embodiments, the method further comprises reacting one or both compounds of the
formula 4 with a phosphodiamidite (e.g., 2-cyanoethyl tetraisopropylphosphorodiamidite) to produce the corresponding nucleoside phosphoramidite. - In another aspect, the invention features a method of synthesizing a nucleic acid. This method involves synthesizing a nucleoside or nucleotide of
formula formula 5 and any one of the formulas 6-9, or compounds of theformula 
- In some embodiments, a compound of
formula 4 is used as a glycosyl donor in a coupling reaction with silylated hypoxantine to form a compound of theformula 5. In certain embodiments, a compound of theformula 5 is used in a ring closing reaction to form a compound of theformula 6. Desirably, deprotection of the 5′-hydroxy group ofcompound 6 is performed by displacing the 5′—O—mesyl group with sodium benzoate to produce a compound of theformula 7 that is converted into a compound of theformula 8 after saponification of the 5′-benzoate. In some embodiments,compound 8 is converted to a DMT-protectedcompound 9 prior to debenzylation of the 3′—O—hydroxy group. In desirable embodiments, a phosphoramidite of theformula 11 is generated by phosphitylation of a nucleoside of theformula 10. - In desirable embodiments, the R 1 is H or P(O(CH2)2CN)N(iPr)2. In other embodiments, the group R1 or —OR1 is any of the groups listed for R3 or R3 in formula Ia or formula Ib or listed for R3 or R3* in formula Ia, Scheme A, or Scheme B, or the group —OR1 or R1 is selected from the group consisting of-OH, P(O(CH2)2CN)N(iPr)2, phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g. acetyl or benzoyl), aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl)), linkers (e.g., a linker containing an amine, ethylene glycol, quinone such as anthraquinone), detectable labels (e.g., radiolabels or fluorescent labels), and biotin.
- In another aspect, the invention features a method of synthesizing a nucleic acid. This method involves synthesizing a nucleoside or nucleotide of
formula compound 4 is the same sugar shown in the above aspect. The nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention. This method can also be performed using any other appropriate protecting groups instead of DMT, Bn, Bz (benzoyl), Ac, or Ms. Additionally, the method can be performed with any other halogen (e.g., fluoro or bromo) instead of chloro.
- In desirable embodiments to promote the ring closing reaction, a solution of
compound 14 inaqueous 1,4-dioxane is treated with sodium hydroxide to give abicyclic compound 15. In some embodiments, sodium benzoate is used for displacement of 5′-mesylate ofcompound 15 to givecompound 16. In some embodiments,compound 17 is formed by reaction ofcompound 16 with sodium azide. In some embodiments,compound 18 is produced by saponification of the 5′-benzoate ofcompound 17. In certain embodiments, hydrogenation ofcompound 18 producescompound 19. In certain embodiments, the peracelation method is used to benzolylate the 2- and 6-amino groups ofcompound 19, yielding 20, which is desirably converted into thephosphoramidite compound 21. - In a related aspect, the invention features a derivative of a compound of the
formula 
- In yet another aspect, the invention features a method of synthesizing a nucleic acid. This method involves synthesizing a nucleoside or nucleotide of
formula - In some embodiments,
compound 17 is formed by reaction ofcompound 7 withl,3-dichloro-1,1,3,3-tetraisopropyldisiloxane. Desirably,compound 18 is formed by reaction ofcompound 17 with phenoxyacetic anhydride. In some embodiments,compound 19 is generated by reaction ofcompound 18 with acid. Desirably,compound 20 is produced by reactingcompound 19 with DMT-Cl. In desirably embodiments,compound 20 is reacted with 2-cyanoethyl tetraisopropylphosphorodiamidite to give thephosphoramidite 21. - In desirable embodiments, the Ris H or P(O(CH 2)2CN)N(iPr)2. In other embodiments, the Ror —ORis any of the groups listed for R3 or R3 in formula Ia or formula Ib or listed for R3 or R3* in formula IIa, Scheme A, or Scheme B, or the group —ORor Ris selected from the group consisting of-OH, P(O(CH2)2CN)N(iPr)2, phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g. acetyl or benzoyl), aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl)), linkers (e.g., a linker containing an amine, ethylene glycol, quinone such as anthraquinone), detectable labels (e.g., radiolabels or fluorescent labels), and biotin.
- In yet another aspect, the invention features a method of synthesizing a nucleic acid. This method involves synthesizing a nucleoside or nucleotide of
formula 24 or 25 as shown below. The nucleoside, nucleoside phosphoramidite, or nucleotide is incorporated into a nucleic acid of the invention. This method can also be performed using any other appropriate protecting groups instead of Bz, Bn, and DMT. Additionally, the method can be performed with any other halogen (e.g., fluoro or bromo) instead of chloro.
- In some embodiments, the
compound 16 is formed fromcompounds compound 16 is hydrolyzed by aqueous sodium hydroxyde to givecompound 22.Compound 23 is desirably produced by incubation ofcompound 22 in the presence of paladium hydroxide and ammonium formate. Desirably, the 2-amine ofcompound 23 is selectively protected with an amidine group after treatment with N,N-dimethylformamide dimethyl acetal to yieldcompound 24. In some embodiments, thediol 24 is 5′—O—DMT protected and 3′—O—phosphitylated produce the phosphoramidite LNA-2AP compound 25. - In some embodiments, compound 25 has one of the following groups instead of the P(O(CH 2)2CN)N(iPr)2 group: any of the groups listed for R3 or R3′ in formula Ia or formula Ib or listed for R3 or R3* in formula Ia, Scheme A, or Scheme B, or a group selected from the group consisting of-OH, phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, methyl phosphonate, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g. acetyl or benzoyl), aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl)), linkers (e.g., a linker containing an amine, ethylene glycol, quinone such as anthraquinone), detectable labels (e.g., radiolabels or fluorescent labels), and biotin.
- In another aspect, the invention features a nucleic acid of the invention that includes a compound of the
formula 6 pCor the product of a compound of theformula 6 pC treated with ammonia as described herein. In a related aspect, the invention features a method of synthesizing a nucleic acid that involves performing one or more of the steps described herein for the synthesis of a compound of theformula 6 pCor the product of a compound oftheformula 6 pC treated with ammonia. - In yet another aspect, the invention features a method of synthesizing a nucleic acid. This method involves one or more of any of the nucleosides or nucleotides of the invention with (i) any other nucleoside or nucleotide of the invention, (ii) any other nucleoside or nucleotide of formula Ia, formula Ib, formula Ia, Scheme A, or Scheme B, and/or (iii) any naturally-occurring nucleoside or nucleotide. Desirably, the method involves reacting one or more nucleoside phosphoramidites of any of the above aspects with a nucleotide or nucleic acid.
- Methods for Synthesis of Nucleic Acids on a Solid Support
- In another aspect, the invention provides a method for the synthesis of a population of nucleic acids (e.g., a population of nucleic acids of the invention) on a solid support. This method involves the reaction of a plurality of nucleoside phosphoramidites with an activated solid support (e.g., a solid support with an activated linker) and the subsequent reaction of a plurality of nucleoside phosphoramidites with activated nucleotides or nucleic acids bound to the solid support.
- In some embodiments of any of the above aspects, the solid support or the growing nucleic acid bound to the solid support is activated by illumination, a photogenerated acid, or electric current. In desirable embodiments, one or more spots or regions (e.g., a region with an area of less than 1
cm 2, 0.1 cm2, 0.01 cm2, 1 mm2, or 0.1 mm that desirably contains one particular nucleic acid monomer or oligomer) on the solid support are irradiated to produce a photogenerated acid that removes the 5′-OH protecting group of one or more nucleic acid monomers or oligomers to which a nucleotide is subsequently added. In other embodiments, an electric current is applied to one or more spots or regions (e.g., a region with an area of less than 1cm 2,0.1 cm2,0.01 cm2, 1 mm2, or 0. mm2 that desirably contains one particular nucleic acid monomer or oligomer) on the solid support to remove an electrochemically sensitive protecting group of one or more nucleic acid monomers or oligomers to which a nucleotide is subsequently added. In still other embodiments, one or more spots or regions (e.g., a region with an area of less than 1 cm2, 0.1 cm2, 0.01 cm2, 1 mm2, or 0.1 mm2 that desirably contains one particular nucleic acid monomer or oligomer) on the solid support are irradiated to remove a photosensitive protecting group of one or more nucleic acid monomers or oligomers to which a nucleotide is subsequently added. In various embodiments, the solid support (e.g., chip, coverslip, microscope glass slide, quartz, or silicon) is less than 1, 0.5, 0.1. or 0.05 mm thick. - Methods for the Synthesis of Nucleic Acids
- In another aspect, the invention features a method of reacting a population of nucleic acids of the invention with one or more nucleic acids. This method involves incubating an immobilized population of nucleic acids of the invention with a solution that includes one or more probes (e.g., at least 2, 3, 4, 5, 10, 15, 20, 30, 40, 50, 60, 80, 100, or 150 different nucleic acids) and one or more target nucleic acids (e.g., at least 2, 3, 4, 5, 10, 15, 20, 30, 40, 50, 60, 80, 100, or 150 different target nucleic acids). The incubation is performed in the presence of a ligase under conditions that allow the ligase to covalently react one or more immobilized nucleic acids with one or more nucleic acid probes in solution that hybridize to the same target nucleic acid. Desirably, at least 2, 5, 10, 15, 20, 30, 40, 50, 80, or 100 pairs of immobilized nucleic acids and nucleic acid probes are ligated. In various embodiments, the incubation occurs between 15 and 45° C., such as between 20 and 40° C. or between 25 and 35° C.
- Desirable Embodiments of Any of the Aspects of the Invention
- In other embodiments of any of various aspects of the invention, a nucleic acid probe or primer specifically hybridizes to a target nucleic acid but does not substantially hybridize to non-target molecules, which include other nucleic acids in a cell or biological sample having a sequence that is less than 99, 95, 90, 80, or 70% identical or complementary to that of the target nucleic acid. Desirably, the amount of the these non-target molecules hybridized to, or associated with, the nucleic acid probe or primer, as measured using standard assays, is 2-fold, desirably 5-fold, more desirably 10-fold, and most desirably 50-fold lower than the amount of the target nucleic acid hybridized to, or associated with, the nucleic acid probe or primer. In other embodiments, the amount of a target nucleic acid hybridized to, or associated with, the nucleic acid probe or primer, as measured using standard assays, is 2-fold, desirably 5-fold, more desirably 10-fold, and most desirably 50-fold greater than the amount of a control nucleic acid hybridized to, or associated with, the nucleic acid probe or primer. In certain embodiments, the nucleic acid probe or primer is substantially complementary (e.g., at least 80, 90, 95, 98, or 100% complementary) to a target nucleic acid or a group of target nucleic acids from a cell. In other embodiments, the probe or primer is homologous to multiple RNA or DNA molecules, such as RNA or DNA molecules from the same gene family. In other embodiments, the probe or primer is homologous to a large number of RNA or DNA molecules. In desirable embodiments, the probe or primer binds to nucleic acids which have polynucleotide sequences that differ in sequence at a position that corresponds to the position of a universal base in the probe or primer. Examples of control nucleic acids include nucleic acids with a random sequence or nucleic acids known to have little, if any, affinity for the nucleic acid probe or primer. In some embodiments, the target nucleic acid is an RNA, DNA, or cDNA molecule.
- Desirably, the association constant (Ka) of the nucleic acid toward a complementary target molecule is higher than the association constant of the complementary strands of the double stranded target molecule. In some desirable embodiments, the melting temperature of a duplex between the nucleic acid and a complementary target molecule is higher than the melting temperature of the complementary strands of the double stranded target molecule.
- In some embodiments, the LNA-pyrene is in a position corresponding to the position of a non-base (e.g., a unit without a base) in another nucleic acid, such as a target nucleic acid. Incorporation of pyrene in a DNA strand that is hybridized against the four natural bases decreases the T m by −4.5° C. to −6.8° C.; however, incorporation of pyrene in a DNA strand in a position opposite a non-base only decreases the Tm by −2.3° C. to −4.6° C., most likely due to the better accommodation of the pyrene in the B-type duplex (Matray and Kool, J. Am. Chem. Soc. 120, 6191, 1998). Thus, incorporation on LNA-pyrene into a nucleic acid in a position opposite a non-base (e.g., a unit without a base or a unit with a small group such as a noncyclic group instead of a base) in a target nucleic acid may also minimize any potential decrease in Tm due to the pyrene substitution
- In various embodiments, the number of molecules in the plurality of nucleic acids of the invention is at least 2, 4, 5, 6, 7, 8, or 10-fold greater than the number of molecules in the test nucleic acid sample. In some embodiments, a LNA is a triplex-forming oligonucleotide.
- In desirable embodiments of any of the aspects of the invention, the target nucleic acids (e.g., cDNA molecules reverse transcribed from a patient sample or cRNA molecules amplified from a patient sample using a T7 RNA polymerase-based amplification system or the like) are fragmented using an enzyme such as a uracil-DNA glycosylase (e.g., E. coli uracil-DNA glycosylase) or using chemical hydrolysis such as alkaline hydrolysis. In various embodiments, the average size of the fragmented nucleic acids is between 300 and 50 nucleic acids, such as approximately 300, 200, 100, or 50 nucleotides.
- Advantages
- The present invention has a variety of advantages related to nucleic acid analysis methods. The ability to equalize melting temperatures of a series of nucleic acids is generally applicable and desirable in all situations where more than one sequence is used simultaneously (e.g. DNA arrays with more than one capture probe, PCR and especially multiplex PCR, homogeneous assays such as Taqman and Molecular beacon). Sample preparation of specific sequences (e.g., DNA or RNA extraction using capture probes on filters or magnetic beads) is another area where melting temperature equalization of specific probe sequences is useful.
- For example, the invention provides high affinity nucleotides (e.g., LNA and other high affinity nucleotides with a modified base and/or backbone) that can be used, e.g., arraysof the invention. In particular, the nucleic acids of the invention containing LNA units exhibited a suprising ability to discriminate between different mRNA splice variants compared to naturally-occurring nucleic acids. If desired, universal bases can be added as part of flanking regions in capture probes (e.g., probes of an array) to stabilize hybridization with high affinity nucleotides in the capture probes. Replacement of one or more DNA-t nucleotides with LNA-T and/or replacement of one or more DNA-a nucleotides with LNA-A reduces the variability of melting temperatures for capture probes of similar length but different GC and AT content by desirably at least 10, 20, 30, 40 or 50%. Additionally, replacement of one or more DNA-t nucleotides with LNA-T and/or replacement of one or more DNA-c with LNA-C increases the stability of a large number of capture probes, while desirably avoiding self-complementary sequences with LNA:LNA base-pairs within a capture probe that would otherwise reduce or eliminate the binding of target molecules to the probe. Although a general T and C substitution may not reduce the variability of melting temperatures of the probes, this substitution increases the melting temperature and binding efficiency of many capture probes that contain these two nucleotides.
- The invention also provides a general substitution algorithm for enhancement of the hybridization signal of a test nucleic acid sample by inclusion of high affinity monomers (e.g., LNA and other high affinity nucleotides with a modified base and/or backbone) in the array. This method increases the stability and binding affinity of capture probes while avoiding substitutions in positions that may form self-complementary base-pairs which may otherwise inhibit binding to a target molecule. The substitution algorithm is broadly useful for specialized arrays, as well as for PCR primers and FISH probes.
- Other features and advantages of the invention will be apparent from the following detailed description.
- Definitions
- When used herein, the term “LNA” (Locked Nucleoside Analogues) refers to nucleoside analogues (e.g., bicyclic nucleoside analogues, e.g., as disclosed in WO 9914226) either incorporated in an oligonucleotide or as a discrete chemical species (e.g., LNA nucleoside and LNA nucleotide). Furthermore, the term “LNA” includes the compounds as described in the present specificatiion including the compounds described in Example 17.
- The term “monomeric LNA” may, e.g., refer to the monomers LNA A, LNA T, LNA C, or any other LNA monomers.
- By “LNA unit” is meant an individual LNA monomer (e.g., an LNA nucleoside or LNA nucleotide).or an oligomer (e.g., an oligonucleotide or nucleic acid) that includes at least one LNA monomer. LNA units as disclosed in WO 99/14226 are in general particularly desirable modified nucleic acids for incorporation into an oligonucleotide of the invention. Additionally, the nucleic acids may be modified at either the 3′ and/or 5′ end by any type of modification known in the art. For example, either or both ends may be capped with a protecting group, attached to a flexible linking group, attached to a reactive group to aid in attachment to the substrate surface, etc. Desirable LNA units and their method of synthesis also are disclosed in WO 0056746, WO 0056748, WO 0066604, Morita et al., Bioorg. Med. Chem. Lett. 12(1):73-76, 2002; Hakansson et al., Bioorg. Med. Chem. Lett. 11 (7):935-938, 2001; Koshkin et al., J. Org. Chem. 66(25):8504-8512, 2001; Kvaerno et al., J. Org. Chem. 66(16):5498-5503, 2001; Hakansson et al., J. Org. Chem. 65(17):5161-5166, 2000; Kvaerno et al., J. Org. Chem. 65(17):5167-5176, 2000; Pfundheller et al., Nucleosides Nucleotides 18(9):2017-2030, 1999; and Kumar et al., Bioorg. Med. Chem. Lett. 8(16):2219-2222, 1998.
- By “LNA modified oligonucleotide” is meant a oligonucleotide comprising at least one LNA monomeric unit of the general scheme A, described infra, having the below described illustrative examples of modifications:

- wherein X is selected from —O—, —S—, —N(R N)—, —C(R6R6*)—, —O—C(R7R7*)—, —C(R6R6*)—O—, —S—C(R7R7*)—, —C(R1R6*)—S—, —N(RN*)-C(R7R7*)—, —C(R6R6*)—N(RN*)—, and —C(R6R6*)—C(R7R7*).
- B is selected from a modified base as discussed above e.g. an optionally substituted carbocyclic aryl such as optionally substituted pyrene or optionally substituted pyrenylmethylglycerol, or an optionally substituted heteroalicylic or optionally substituted heteroaromatic such as optionally substituted pyridyloxazole, optionally substituted pyrrole, optionally substituted diazole or optionally substituted triazole moieties; hydrogen, hydroxy, optionally substituted C 1-4-alkoxy, optionally substituted C1-4-alkyl, optionally substituted C1-4-acyloxy, nucleobases, DNA intercalators, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands.
- P designates the radical position for an internucleoside linkage to a succeeding monomer, or a 5′-terminal group, such internucleoside linkage or 5′-terminal group optionally including the substituent R 5. One of the substituents R2, R2*, R3, and R3* is a group P* which designates an internucleoside linkage to a preceding monomer, or a 2′/3′-terminal group. The substituents of R1*, R4*, R5, R5*, R6, R6*, R7, R7*, RN, and the ones of R2, R2*, R3, and R3*not designating P* each designates a biradical comprising about 1-8 groups/atoms selected from —C(RaRb)—, —C(Ra)=C(Ra)—, —C(Ra)=N—, —C(Ra)—O—, —O—, —Si(Ra)2—, —C(Ra)—S, —S—, —SO2—, —C(Ra)—N(Rb)—, N(Ra)—, and >C=Q, wherein Q is selected from —O—, —S—, and —N(Ra)—, and Ra and Rb each is independently selected from hydrogen, optionally substituted C1-12-alkyl, optionally substituted C2-12-alkenyl, optionally substituted C2-12-alkynyl, hydroxy, C1-12-alkoxy, C2-12-alkenyloxy, carboxy, C1-12-alkoxycarbonyl, C1-12-alkylcarbonyl, formyl, aryl, aryloxy-carbonyl, aryloxy, arylcarbonyl, heteroaryl, hetero-aryloxy-carbonyl, heteroaryloxy, heteroarylcarbonyl, amino, mono- and di(C1-6-alkyl)amino, carbamoyl, mono- and di(C1-6-alkyl)-amino-carbonyl, amino-C1-6-alkyl-aminocarbonyl, mono- and di(C1-6-alkyl)amino-C1-6-alkyl-aminocarbonyl, C1-6-alkyl-carbonylamino, carbamido, C1-6-alkanoyloxy, sulphono, C1-6-alkylsulphonyloxy, nitro, azido, sulphanyl, C1-6-alkylthio, halogen, DNA intercalators, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands, where aryl and heteroaryl may be optionally substituted, and where two geminal substituents Ra and Rb together may designate optionally substituted methylene (═CH2), and wherein two non-geminal or geminal substituents selected from Ra, Rb, and any of the substituents R1*, R2, R2, R3, R3*, R4*, R5, R5*, R6 and R6*, R7, and R7*which are present and not involved in P, P* or the biradical(s) together may form an associated biradical selected from biradicals of the same kind as defined before; the pair(s) of non-geminal substituents thereby forming a mono- or bicyclic entity together with (i) the atoms to which said non-geminal substituents are bound and (ii) any intervening atoms.
- Each of the substituents R 1*, R2, R2*, R3, R4*, R5, R5*, R6* and R6, R7, and R7* which are present and not involved in P, P* or the biradical(s), is independently selected from hydrogen, optionally substituted C1-12-alkyl, optionally substituted C2-12-alkenyl, optionally substituted C2-12-alkynyl, hydroxy, C1-12-alkoxy, C2-12-alkenyloxy, carboxy, C1-12-alkoxycarbonyl, C1-12-alkylcarbonyl, formyl, aryl, aryloxy-carbonyl, aryloxy, arylcarbonyl, heteroaryl, heteroaryloxy-carbonyl, heteroaryloxy, heteroarylcarbonyl, amino, mono- and di(C1-6-alkyl)amino, carbamoyl, mono- and di(C1-6-alkyl)-amino-carbonyl, amino-C1-6-alkyl-aminocarbonyl, mono- and di(C1-6-alkyl)amino-C1-6-alkyl-aminocarbonyl, C1-6-alkyl-carbonylamino, carbamido, C1-6-alkanoyloxy, sulphono, C1-6-alkylsulphonyloxy, nitro, azido, sulphanyl, C1-6-alkylthio, halogen, DNA intercalators, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands, where aryl and heteroaryl may be optionally substituted, and where two geminal substituents together may designate oxo, thioxo, imino, or optionally substituted methylene, or together may form a spiro biradical consisting of a 1-5 carbon atom(s) alkylene chain which is optionally interrupted and/or terminated by one or more heteroatoms/groups selected from —O—, —S—, and —(NRN)— where RN is selected from hydrogen and C1-4-alkyl, and where two adjacent (non-geminal) substituents may designate an additional bond resulting in a double bond; and RN, when present and not involved in a biradical, is selected from hydrogen and C1-4-alkyl; and basic salts and acid addition salts thereof.
- Exemplary 5′, 3′, and/or 2′ terminal groups include —H, —OH, halo (e.g., chloro, fluoro, iodo, or bromo), optionally substituted aryl, (e.g., phenyl or benzyl), alkyl (e.g, methyl or ethyl), alkoxy (e.g., methoxy), acyl (e.g. acetyl or benzoyl), aroyl, aralkyl, hydroxy, hydroxyalkyl, alkoxy, aryloxy, aralkoxy, nitro, cyano, carboxy, alkoxycarbonyl, aryloxycarbonyl, aralkoxycarbonyl, acylamino, aroylamine, alkylsulfonyl, arylsulfonyl, heteroarylsulfonyl, alkylsulfinyl, arylsulfinyl, heteroarylsulfinyl, alkylthio, arylthio, heteroarylthio, aralkylthio, heteroaralkylthio,amidino, amino, carbamoyl, sulfamoyl, alkene, alkyne, protecting groups (e.g., silyl, 4,4′-dimethoxytrityl, monomethoxytrityl, or trityl(triphenylmethyl)), linkers (e.g., a linker containing an amine, ethylene glycol, quinone such as anthraquinone), detectable labels (e.g., radiolabels or fluorescent labels), and biotin.
- It is understood that references herein to a nucleic acid unit, nucleic acid residue, LNA unit, or similar term are inclusive of both individual nucleoside units and nucleotide units and nucleoside units and nucleotide units within an oligonucleotide.
- A “modified base” or other similar term refers to a composition (e.g., a non-naturally occuring nucleobase or nucleosidic base) which can pair with a natural base (e.g., adenine, guanine, cytosine, uracil, and/or thymine) and/or can pair with a non-naturally occurring nucleobase or nucleosidic base. Desirably, the modified base provides a T m differential of 15, 12, 10, 8, 6, 4, or 2° C. or less as described herein. Exemplary modified bases are described in
EP 1 072 679 and WO 97/12896. - By “nucleobase” is meant the naturally occurring nucleobases adenine (A), guanine (G), cytosine (C), thymine (T) and uracil (U) as well as non-naturally occurring nucleobases such as xanthine, diaminopurine, 8-oxo-N 6-methyladenine, 7-deazaxanthine, 7-deazaguanine, N4,N4-ethanocytosin, N′,N′-ethano-2,6-diaminopurine, 5-methylcytosine (mC), 5-(C3—C6)-alkynyl-cytosine, 5-fluorouracil, 5-bromouracil, pseudoisocytosine, 2-hydroxy-5-methyl-4-triazolopyridin, isocytosine, isoguanine, inosine and the “non-naturally occurring” nucleobases described in Benner et al., U.S. Pat. No. 5,432,272 and Susan M. Freier and Karl-Heinz Altmann, Nucleic Acids Research, 1997, vol. 25, pp 4429-4443. The term “nucleobase” thus includes not only the known purine and pyrimidine heterocycles, but also heterocyclic analogues and tautomers thereof. Further naturally and non-naturally occurring nucleobases include those disclosed in U.S. Pat. No. 3,687,808 (Merigan, et al.), in
Chapter 15 by Sanghvi, in Antisense Research and Application, Ed. S. T. Crooke and B. Lebleu, CRC Press, 1993, in Englisch et al., Angewandte Chemie, International Edition, 1991, 30, 613-722 (see especially pages 622 and 623, and in the Concise Encyclopedia of Polymer Science and Engineering, J. I. Kroschwitz Ed., John Wiley & Sons, 1990, pages 858-859, Cook,Anti-Cancer Drug Design 1991, 6, 585-607, each of which are hereby incorporated by reference in their entirety). The term “nucleosidic base” or “base unit” is further intended to include compounds such as heterocyclic compounds that can serve like nucleobases including certain “universal bases” that are not nucleosidic b.ases in the most classical sense but serve as nucleosidic bases. Especially mentioned as universal bases are 3-nitropyrrole, optionally substituted indoles (e.g., 5-nitroindole), and optionally substituted hypoxanthine. Other desirable universal bases include, pyrrole, diazole or triazole derivatives, including those universal bases known in the art. - As described herein, various groups of an LNA unit may be optionally substituted. A “substituted” group such as a nucleobase or nucleosidic base and the like may be substituted by other than hydrogen at one or more available positions, typically 1 to 3 or 4 positions, by one or more suitable groups such as those disclosed herein. Suitable groups that may be present on a “substituted” group include e.g. halogen such as fluoro, chloro, bromo and iodo; cyano; hydroxyl; nitro; azido; alkanoyl such as a C 1-6 alkanoyl group such as acyl and the like; carboxamido; alkyl groups including those groups having 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5, or 6 carbon atoms; alkenyl and alkynyl groups including groups having one or more unsaturated linkages and from 2 to 12 carbon, or 2, 3, 4, 5 or 6 carbon atoms; alkoxy groups including those having one or more oxygen linkages and from 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5 or 6 carbon atoms; aryloxy such as phenoxy; alkylthio groups including those moieties having one or more thioether linkages and from 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5 or 6 carbon atoms; alkylsulfinyl groups including those moieties having one or more sulfinyl linkages and from 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5, or 6 carbon atoms; alkylsulfonyl groups including those moieties having one or more sulfonyl linkages and from 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5, or 6 carbon atoms; aminoalkyl groups such as groups having one or more N atoms and from 1 to about 12 carbon atoms, or 1, 2, 3, 4, 5 or 6 carbon atoms; carbocyclic aryl having 6 or more carbons; aralkyl having 1 to 3 separate or fused rings and from 6 to about 18 carbon ring atoms, with benzyl being a desirable group; aralkoxy having 1 to 3 separate or fused rings and from 6 to about 18 carbon ring atoms, with O-benzyl being a desirable group; or a heteroaromatic or heteroalicyclic group having 1 to 3 separate or fused rings with 3 to about 8 members per ring and one or more N, O or S atoms, e.g. coumarinyl, quinolinyl, pyridyl, pyrazinyl, pyrimidyl, furyl, pyrrolyl, thienyl, thiazolyl, oxazolyl, imidazolyl, indolyl, benzofuranyl, benzothiazolyl, tetrahydrofuranyl, tetrahydropyranyl, piperidinyl, morpholino and pyrrolidinyl.
- By “oxy-LNA monomer or unit” is meant any nucleoside or nucleotide which contains an oxygen atom in a 2′-4′ linkage.
- A “non-oxy-LNA” monomer or unit is broadly defined as any nucleoside or nucleotide which does not contain an oxygen atom in a 2′-4′-linkage. Examples of non-oxy-LNA monomers include 2′-deoxynucleotides (DNA) or nucleotides (RNA) or any analogues of these monomers which are not oxy-LNA, such as for example the thio-LNA and amino-LNA described herein with respect to formula Ia and in Singh et al. J. Org. Chem. 1998, 6, 6078-9, and the derivatives described in Susan M. Freier and Karl-Heinz Altmann, Nucleic Acids Research, 1997, vol 25, pp 4429-4443.
- By “universal base” is meant a naturally-occurring or desirably a non-naturally occurring compound or moiety that can pair with a natural base (e.g., adenine, guanine, cytosine, uracil, and/or thymine), and that has a T m differential of 15, 12, 10, 8, 6, 4, or 2° C. or less as described herein.
- By “oligonucleotide,” “oligomer,” or “oligo” is meant a successive chain of monomers (e.g., glycosides of heterocyclic bases) connected via internucleoside linkages. The linkage between two successive monomers in the oligo consist of 2 to 4, desirably 3, groups/atoms selected from —CH 2—, —O—, —S—, —NRH_, >C═O, >C═NRH, >C═S, —Si(R″)2—, —SO—, —S(O)2—, —P(O)2—, —PO(BH3)—, —P(O,S)—, —P(S)2—, —PO(R″)—, —PO(OCH3)—, and —PO(NHRH)—, where RH is selected from hydrogen and C1-4-alkyl, and R″ is selected from C1-6-alkyl and phenyl. Illustrative examples of such linkages are —CH2—CH2—CH2—, —CH2—CO—CH2—, —CH2—CHOH—CH2—, —O—CH2—O—, —O—CH2—CH2—, —O—CH2—CH=(including R5 when used as a linkage to a succeeding monomer), —CH2—CH2—O—, —NRH—CH2—CH2—, —CH2—CH2—NRH—, —CH2—NRH—CH2—, —O—CH2—CH2—NRH—, —NRH—CO—O—, —NRH—CO—NRH—, —NRH—CS—NRH—, —NRH—C(═NRH)—NRH—, —NRH—CO—CH2—NRH—O—CO—O—, —O—CO—CH2—O—, —O—CH2—CO—O—, —CH2—CO—NRH—, —O—CO—NRH—, —NRH—CO—CH2—, —O—CH2—CO—NRH—, —O—CH2—CH2—NRH—, —CH═N—O—, —CH2—NRH—O—, —CH2—O—N═(including R5 when used as a linkage to a succeeding monomer), —CH2—O—NRH—, —CO—NRH—CH2—, —CH2—NRH—O—, —CH2—NRH—CO—, —O—NRH—CH2—, —O—NRH, —O—CH2—S—, —S—CH2—O—, —CH2—CH2—S—, —O—CH2—CH2—S—, —S—CH2—CH=(including R5 when used as a linkage to a succeeding monomer), —S—CH2—CH2—, —S—CH2—CH2—O—, —S—CH2—CH2—S—, —CH2—S—CH2—, —CH2—SO—CH2—, —CH2—SO2—CH2—, —O—SO—O—, —O—S(O)2—O—, —O—S(O)2—CH2—, —O—S(O)2—NRH—, —NRH—S(O)2—CH2—; —O—S(O)2—CH2—, —O—P(O)2—O—, —O—P(O,S)—O—, —O—P(S)2—O—, —S—P(O)2—O—, —S—P(O,S)—O—, —S—P(S)2—O—, —O—P(O)2—S—, —O—P(O,S)—S—, —O—P(S)2—S—, —S—P(O)2—S—, —S—P(O,S)—S—, —S—P(S)2—S—, —O—PO(R″)—O—, —O—PO(OCH3)—O—, —O—PO(OCH2CH3)—O—, —O—PO(OCH2CH2S—R)—O—, —O—PO(BH3)—O—, —O—PO(NHRN)—O—, —O—P(O)2—NRH H—, —NRH—P(O)2—O—, —O—P(O,NRH)—O—, —CH2—P(O)2—O—, —O—P(O)2—CH2—, and —O—Si(R″)2—O—; among which —CH2—CO—NRH—, —CH2—NRH—O—, —S—CH2—O—, —O—P(O)2—O—-O—P(O,S)—O—, —O—P(S)2—O—, —NRH P(O)2—O—, —O—P(O,NRH)—O—, —O—PO(R″)—O—, —O—PO(CH3)—O—, and —O—PO(NHRN)—O—, where RH is selected form hydrogen and C1-4-alkyl, and R″ is selected from C1-6-alkyl and phenyl, are especially desirable. Further illustrative examples are given in Mesmaeker et. al., Current Opinion in
Structural Biology 1995, 5, 343-355 and Susan M. Freier and Karl-Heinz Altmann, Nucleic Acids Research, 1997, vol 25, pp 4429-4443. The left-hand side of the internucleoside linkage is bound to the 5-membered ring as substituent P at the 3′-position, whereas the right-hand side is bound to the 5′-position of a preceding monomer. - By “succeeding monomer” is meant the neighboring monomer in the 5′-terminal direction, and by “preceding monomer” is meant the neighboring monomer in the 3′-terminal direction.
- By “LNA spiked oligo” is meant an oligonucleotide, such as a DNA oligonucleotide, wherein at least one unit (and preferably not all units) has been substituted by the corresponding LNA nucleoside monomer.
- The term “T m ” is used in reference to the “melting temperature.” The melting temperature is the temperature at which 50% of a population of double-stranded nucleic acid molecules becomes dissociated into single strands. The equation for calculating the Tm of nucleic acids is well-known in the art. The Tm of a hybrid nucleic acid is often estimated using a formula adopted from hybridization assays in 1 M salt, and commonly used for calculating Tm for PCR primers: Tm=[(number of A+T)×2° C.+(number of G+C)×4° C.]. C. R. Newton et al. PCR, 2nd Ed., Springer-Verlag (New York: 1997), p. 24. This formula was found to be inaccurate for primers longer that 20 nucleotides. Id. Other more sophisticated computations exist in the art which take structural as well as sequence characteristics into account for the calculation of Tm. A calculated Tm is merely an estimate; the optimum temperature is commonly determined empirically.
- A nucleic acid compound that has a T m differential of a specified amount (e.g., less than 15, 12, 10, 8, 6, 4, 2, or 1° C.) means the nucleic acid exhibits that specified Tm differential when incorporated into a specified 9-mer oligonucleotide with respect to the four complementary variants, as defined immediately below.
- Unless otherwise indicated, a T m differential provided by a particular modified base is calculated by the following protocol (steps a) through d)):
- a) incorporating the modified base of interest into the following
oligonucleotide 5′-d(GTGAMATGC), wherein M is the modified base; - b) mixing 1.5×10 −6M of the oligonucleotide having incorporated therein the modified base with each of 1.5xl 0−6M of the four oligonucleotides having the
sequence 3′-d(CACTYTACG), wherein Y is A, C, G, T, respectively, in a buffer of 10 mM sodium phosphate, 100 mM sodium chloride, 0.1 mM EDTA, pH 7.0; - c) allowing the oligonucleotides to hybridize; and
- d) detecting the T m for each of the four hybridized nucleotides by heating the hybridized nucleotides and observing the temperature at which the maximum of the first derivative of the melting curve recorded at a wavelength of 260 nm is:obtained.
- Unless otherwise indicated, a T m differential for a particular modified base is determined by subtracting the highest Tm value determined in steps a) through d) immediately above from the lowest Tm value determined by steps a) through d) immediately above.
- By “variance in T m is meant the variance in the values of the melting temperatures for a population of nucleic acids. The Tm for each nucleic acid is determined by experimentally measuring or computationally predicting the temperature at which 50% of a population double-stranded molecules with the sequence of the nucleic acid becomes dissociated into single strands. For a nucleic acid with only A, T, C, G, and/or U bases, the Tm is the temperature at which 50% of a population of 100% complementary double-stranded molecules with the sequence of the nucleic acid becomes dissociated into single strands. For determining the Tm variance when a nucleic acid has one or more nucleobases other than A, T, C, G, or U, the Tm of this “modified” nucleic acid is approximated by determining the Tm for each possible double stranded molecule in which one strand is the modified nucleic acid and the other strand has either A, T, C, or G in each position corresponding to a nucleobase other than A, T, C, G, or U in the modified nucleic acid. For example, if the modified nucleic acid has the sequence XMX in which X is 0, 1, or more A, T, C, G or U bases and M is any other nucleobase or nucleosidic base, the Tm is calculated for each possible double stranded molecule in which one strand is XMX and the other strand is X′YX′ in which X′ is the base complementary to the corresponding X base and Y is either A, T, C, or G. The average is then calculated for the Tm values for each possible double stranded molecule (i.e., four possible duplexes per modified nucleobase or nucleoside base in the modified nucleic acid) and used as the approximate Tm value for the modified nucleic acid.
- By “capture efficiency” is meant the amount of target nucleic acid(s) bound to a particular nucleic acid or a population of nucleic acids. Standard methods can be used to calculate the capture efficiency by measuring the amount of bound target nucleic acid(s) and/or measuring the amount of unbound target nucleic acid(s). The capture efficiency of a nucleic acid or nucleic acid population of the invention is typically compared to the capture efficiency of a control nucleic acid or nucleic acid population under the same incubation conditions (e.g., using same buffer and temperature).
- For example, a control nucleic acid may have β-D-2-deoxyribose instead of one or more bicyclic or sugar groups of a LNA unit or other modified or non-naturally-occurring units in a nucleic acid of the invention. In some embodiments, the nucleic acid of the invention and the control nucleic acid only have naturally-occurring nucleobases. If a nucleic acid of the invention has one or more non-naturally-occurring nucleobases, the capture efficiency of the corresponding control nucleic acid is calculated as the average capture efficiency for all of the nucleic acids that have either A, T, C, or G in each position corresponding to a non-naturally-occurring nucleobase in the nucleic acid of the invention.
- Monomers are referred to as being “complementary” if they contain nucleobases that can form hydrogen bonds according to Watson-Crick base-pairing rules (e.g., G with C, A with T, or A with U) or other hydrogen bonding motifs such as for example diaminopurine with T, inosine with C, and pseudoisocytosine with G.
- By “substantially complementarity” is meant having a sequence that is at least 60, 70, 80, 90, 95, or 100% complementary to that of another sequence. Sequence complementarity is typically measured using sequence analysis software with the default parameters specified therein (e.g., Sequence Analysis Software Package of the Genetics Computer Group, University of Wisconsin Biotechnology Center, 1710 University Avenue, Madison, Wis. 53705). This software program matches similar sequences by assigning degrees of homology to various substitutions, deletions, and other modifications.
- The term “homology” refers to a degree of complementarity. There can be partial homology or complete homology (i.e., identity). A partially complementary sequence that at least partially inhibits a completely complementary sequence from hybridizing to a target nucleic acid is referred to using the functional term “substantially homologous.”
- When used in reference to a double-stranded nucleic acid sequence such as a cDNA or genomic clone, the term “substantially homologous” refers to a probe that can hybridize to a strand of the double-stranded nucleic acid sequence under conditions of low stringency, e.g. using a hybridization buffer comprising 20% formamide in 0.8M saline/0.08M sodium citrate (SSC) buffer at a temperature of 37° C. and remaining bound when subject to washing once with that SSC buffer at 37° C.
- When used in reference to a single-stranded nucleic acid sequence, the term “substantially homologous” refers to a probe that can hybridize to (i.e., is the complement of) the single-stranded nucleic acid template sequence under conditions of low stringency, e.g. using a hybridization buffer comprising 20% formamide in 0.8M saline/0.08M sodium citrate (SSC) buffer at a temperature of 37° C. and remaining bound when subject to washing once with that SSC buffer at 37° C.
- By “internal probe” is meant a nucleic acid (e.g., a probe or primer) that hybridizes to either only one exon or only one intron of a nucleic acid (e.g., mRNA). The internal probe may hybridize to the 5′ end of the exon or intron, the 3′ end of the exon or intron, or between the 5′ end and the 3′ end of the exon or intron. Desirably, the internal probe is at least 90, 95, 96, 97, 98, 99, or 100% identical to the corresponding region of a target nucleic acid.
- By “merged probe” is meant a nucleic acid (e.g., a probe or primer) that hybridizes to more than one exon and/or intron of a nucleic acid (e.g., mRNA). Desirably, the merged probe hybridizes to two consecutive exons (e.g., exons in a mature mRNA transcript that may or may not be consecutive in the corresponding DNA molecule). In another desirable embodiment, the merged probe hybridizes to an exon and the consecutive intron. In desirable embodiments, the merged probe hybridizes to the same number of nucleotides in each exon or to the same number of nucleotides in the exon and intron. In various embodiments, the length of the region of the merged probe that hybridizes to one exon differs by less than 60, 40, 20, 10, or 5% from the length of the region of the merged probe that hybridizes to the other exon or to the intron. Desirably, the merged probe is at least 90, 95, 96, 97, 98, 99, or 100% identical to the corresponding region of a target nucleic acid.
- By “poly-T 20 tail “is meant a DNA polymer consisting of 20 DNA-t units added by polymerase chain reaction as a tail to a nucleic acid sequence, which is subsequently cloned in a plasmid vector allowing in vitro synthesis of poly(A)20 polyadenylated RNA.
- By “mixmer” or “mixmer probe” is meant a nucleic acid (e.g., a probe or primer) that contains at least one LNA unit and at least one RNA or DNA unit (e.g., a naturally-occurring RNA or DNA unit).
- By “corresponding unmodified reference nucleobase” is meant a nucleobase that is not part of an LNA unit and is in the same orientation as the nucleobase in an LNA unit.
- By “mutation” is meant an alteration in a naturally-occurring or reference nucleic acid sequence, such as an insertion, deletion, frameshift mutation, silent mutation, nonsense mutation, or missense mutation. Desirably, the amino acid sequence encoded by the nucleic acid sequence has at least one amino acid alteration from a naturally-occurring sequence.
- By “selecting” is meant substantially partitioning a molecule from other molecules in a population. Desirably, the partitioning provides at least a 2-fold, desirably, a 30-fold, more desirably, a 100-fold, and most desirably, a 1,000-fold enrichment of a desired molecule relative to undesired molecules in a population following the selection step. The selection step may be repeated a number of times, and different types of selection steps may be combined in a given approach. The population desirably contains at least 109 molecules, more desirably at least 101, 1013, or 1014 molecules and, most desirably, at least 1015 molecules.
- By a “population” is meant more than one nucleic acid. A “population” according to the invention desirably means more than 101, 102, 103, or 104 different molecules.
- By “photochemically active groups” is meant compounds which are able to undergo chemical reactions upon irradiation with light. Illustrative examples of functional groups are quinones, especially 6-methyl-1,4-naphtoquinone, anthraquinone, naphtoquinone, and 1,4-dimethyl-anthraquinone, diazirines, aromatic azides, benzophenones, psoralens, diazo compounds, and diazirino compounds.
- By “thermochemically reactive group” is meant a functional group which is able to undergo thermochemically-induced covalent bond formation with other groups. Illustrative examples of functional parts of thermochemically reactive groups are carboxylic acids, carboxylic acid esters such as activated esters, carboxylic acid halides such as acid fluorides, acid chlorides, acid bromide, acid iodides, carboxylic acid azides, carboxylic acid hydrazides, sulfonic acids, sulfonic acid esters, sulfonic acid halides, semicarbazides, thiosemicarbazides, aldehydes, ketones, primary alcohols, secondary alcohols, tertiary alcohols, phenols, alkyl halides, thiols, disulphides, primary amines, secondary amines, tertiary amines, hydrazines, epoxides, maleimides, and boronic acid derivatives.
- By “chelating group” is meant a molecule that contains more than one binding site and frequently binds to another molecule, atom, or ion through more than one binding site at the same time. Examples of functional parts of chelating groups are iminodiacetic acid, nitrilotriacetic acid, ethylenediamine tetraacetic acid (EDTA), and aminophosphonic acid.
- By “reporter group” is meant a group which is detectable either by itself or as a part of an detection series. Examples of functional parts of reporter groups are biotin, digoxigenin, fluorescent groups (e.g., groups which are able to absorb electromagnetic radiation, e.g. light or X-rays, of a certain wavelength, and which subsequently reemit the energy absorbed as radiation of longer wavelength; such as dansyl (5-dimethylamino)-1-naphthalenesulfonyl), DOXYL (N-oxyl-4,4-dimethyloxazolidine), PROXYL (N-oxyl-2,2,5,5-tetramethylpyrrolidine), TEMPO(N-oxyl-2,2,6,6-tetramethylpiperidine), dinitrophenyl, acridines, coumarins, Cy3 and Cy5 (trademarks for Biological Detection Systems, Inc.), erythrosine, coumaric acid, umbelliferone, Texas red, rhodamine, tetramethyl rhodamine, Rox, 7-nitrobenzo-2-oxa- 1-diazole (NBD), pyrene, fluorescein, Europium, Ruthenium, Samarium, and other rare earth metals), radioisotopic labels, chemiluminescence labels (i.e., labels that are detectable via the emission of light during a chemical reaction), spin labels (a free radical e.g., substituted organic nitroxides) or other paramagnetic probes (e.g., Cu2+ or Mg 2+) bound to a biological molecule being detectable by the use of electron spin resonance spectroscopy), enzymes (such as peroxidases, alkaline phosphatases, β-galactosidases, and glycose oxidases), antigens, antibodies, haptens (e.g., groups which are able to combine with an antibody, but which cannot initiate an immune response by itself, such as peptides and steroid hormones), carrier systems for cell membrane penetration, fatty acid units, steroid moieties (cholesteryl), vitamin A, vitamin D, vitamin E, folic acid peptides for specific receptors, groups for mediating endocytose, epidermal growth factor (EGF), bradykinin, and platelet derived growth factor (PDGF). Especially desirable groups are biotin, fluorescein, Texas Red, rhodamine, dinitrophenyl, digoxigenin, Ruthenium, Europium, Cy5, and Cy3.
- By “ligand” is meant a compound which binds. Ligands can comprise functional groups such as aromatic groups (such as benzene, pyridine, naphthalene, anthracene, and phenanthrene), heteroaromatic groups (such as thiophene, furan, tetrahydrofuran, pyridine, dioxane, and pyrimidine), carboxylic acids, carboxylic acid esters, carboxylic acid halides, carboxylic acid azides, carboxylic acid hydrazides, sulfonic acids, sulfohic acid esters, sulfonic acid halides, semicarbazides, thiosemicarbazides, aldehydes, ketones, primary alcohols, secondary alcohols, tertiary alcohols, phenols, alkyl halides, thiols, disulphides, primary amines, secondary amines, tertiary amines, hydrazines, epoxides, maleimides, C 1-C20 alkyl groups optionally interrupted or terminated with one or more heteroatoms such as oxygen atoms, nitrogen atoms, and/or sulphur atoms, optionally containing aromatic or mono/polyunsaturated hydrocarbons, polyoxyethylene such as polyethylene glycol, oligo/polyamides such as poly-α-alanine, polyglycine, polylysine, peptides, oligo/polysaccharides, oligo/polyphosphates, toxins, antibiotics, cell poisons, and steroids. “Affinity ligands” include functional groups or biomolecules that have a specific affinity for sites on particular proteins, antibodies, poly- and oligosaccharides, and other biomolecules.
- It should be understood that the above-mentioned specific examples under DNA intercalators, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands correspond to the “active/functional” part of the groups in question. For the person skilled in the art it is furthermore clear that DNA intercalators, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands are typically represented in the form M-K- where M is the “active/functional” part of the group in question and where K is a spacer through which the “active/functional” part is attached to the 5- or 6-membered ring. Thus, it should be understood that the group B, in the case where B is selected from DNA intercalators, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands, has the form M-K-, where M is the “active/functional” part of the DNA intercalator, photochemically active group, thermochemically active group, chelating group, reporter group, and ligand, respectively, and where K is an optional spacer comprising 1-50 atoms, desirably 1-30 atoms, in particular 1-15 atoms, between the 5- or 6-membered ring and the “active/functional” part.
- By “spacer” is meant a thermochemically and photochemically non-active distance-making group and is used to join two or more different moieties of the types defined above. Spacers are selected on the basis of a variety of characteristics including their hydrophobicity, hydrophilicity, molecular flexibility and length (e.g., Hermanson et. al., “Immobilized Affinity Ligand Techniques,” Academic Press, San Diego, Calif. (1992). Generally, the length of the spacers is less than or about 400 A, in some applications desirably less than 100 A. The spacer, thus, comprises a chain of carbon atoms optionally interrupted or terminated with one or more heteroatoms, such as oxygen atoms, nitrogen atoms, and/or sulphur atoms. Thus, the spacer K may comprise one or more amide, ester, amino, ether, and/or thioether functionalities, and optionally aromatic or mono/polyunsaturated hydrocarbons, polyoxyethylene such as polyethylene glycol, oligo/polyamides such as poly-α-alanine, polyglycine, polylysine, peptides, oligosaccharides, or oligo/polyphosphates. Moreover the spacer may consist of combined units thereof. The length of the spacer may vary, taking into consideration the desired or necessary positioning and spatial orientation of the “active/functional” part of the group in question in relation to the 5- or 6-membered ring. In particularl embodiments, the spacer includes a chemically cleavable group. Examples of such chemically cleavable groups include disulphide groups cleavable under reductive conditions and peptide fragments cleavable by peptidases.
- By “target nucleic acid” or “nucleic acid target” is meant a particular nucleic acid sequence of interest. Thus, the “target” can exist in the presence of other nucleic acid molecules or within a larger nucleic acid molecule.
- By “solid support” is meant any rigid or semi-rigid material to which a nucleic acid binds or is directly or indirectly attached. The support can be any porous or non-porous water insoluble material, including without limitation, membranes, filters, chips, slides, wafers, fibers, magnetic or nonmagnetic beads, gels, tubing, strips, plates, rods, polymers, particles, microparticles, capillaries, and the like. The support can have a variety of surface forms, such as wells, trenches, pins, channels and pores.
- By an “array” is meant a fixed pattern of at least two different immobilized nucleic acids on a solid support. Desirably, the array includes at least 102, more desirably, at least 103, and, most desirably, at least 1 different nucleic acids.
- By “antisense nucleic acid” is meant a nucleic acid, regardless of length, that is complementary to a coding strand or mRNA of interest. In some embodiments, the antisense molecule inhibits the expression of only one nucleic acid, and in other embodiments, the antisense molecule inhibits the expression of more than one nucleic acid. Desirably, the antisense nucleic acid decreases the expression or biological activity of a nucleic and or encoded protein by at least 20, 40, 50, 60, 70, 80, 90, 95, or 100%. An antisense molecule can be introduced, e.g., to an individual cell or to whole animals, for example, it may be introduced systemically via the bloodstream. Desirably, a region of the antisense nucleic acid or the entire antisense nucleic acid is at least 70, 80, 90, 95, 98, or 100% complementary to a coding sequence, regulatory region (5′ or 3′ untranslated region), or an mRNA of interest. Desirably, the region of complementarity includes at least 5, 10, 20, 30, 50, 75, 100, 200, 500, 1000, 2000 or 5000 nucleotides or includes all of the nucleotides in the antisense nucleic acid.
- In some embodiments, the antisense molecule is less than 200, 150, 100, 75, 50, or 25 nucleotides in length. In other embodiments, the antisense molecule is less than 50,000; 10,000; 5,000; or 2,000 nucleotides in length. In certain embodiments, the antisense molecule is at least 200, 300, 500, 1000, or 5000 nucleotides in length. In some embodiments, the number of nucleotides in the antisense molecule is contained in one of the following ranges: 5-15 nucleotides, 16-20 nucleotides, 21-25 nucleotides, 26-35 nucleotides, 36-45 nucleotides, 46-60 nucleotides, 61-80 nucleotides, 81-100 nucleotides, 101-150 nucleotides, or 151-200 nucleotides, inclusive. In addition, the antisense molecule may contain a sequence that is less than a full-length sequence or may contain a full-length sequence.
- By “double stranded nucleic acid” is meant a nucleic acid containing a region of two or more nucleotides that are in a double stranded conformation. In various embodiments, the double stranded nucleic acids consist entirely of LNA units or a mixture of LNA units, ribonucleotides, and/or deoxynucleotides. The double stranded nucleic acid may be a single molecule with a region of self-complementarity such that nucleotides in one segment of the molecule base-pair with nucleotides in another segment of the molecule. Alternatively, the double stranded nucleic acid may include two different strands that have a region of complementarity to each other. Desirably, the regions of complementarity are at least 70, 80, 90, 95, 98, or 100% identical. Desirably, the region of the double stranded nucleic acid that is present in a double stranded conformation includes at least 5, 10, 20, 30, 50, 75, 100, 200, 500, 1000, 2000 or 5000 nucleotides or includes all of the nucleotides in the double stranded nucleic acid. Desirable double stranded nucleic acid molecules have a strand or region that is at least 70, 80, 90, 95, 98, or 100% identical to a coding region or a regulatory sequence (e.g., a transcription factor binding site, a promoter, or a 5′ or 3′ untranslated region) of a nucleic acid of interest. In some embodiments, the double stranded nucleic acid is less than 200, 150, 100, 75, 50, or 25 nucleotides in length. In other embodiments, the double stranded nucleic acid is less than 50,000; 10,000; 5,000; or 2,000 nucleotides in length. In certain embodiments, the double stranded nucleic acid is at least 200, 300, 500, 1000, or 5000 nucleotides in length. In some embodiments, the number of nucleotides in the double stranded nucleic acid is contained in one of the following ranges: 5-15 nucleotides, 16-20 nucleotides, 21-25 nucleotides, 26-35 nucleotides, 36-45 nucleotides, 46-60 nucleotides, 61-80 nucleotides, 81-100 nucleotides, 101-150 nucleotides, or 151-200 nucleotides, inclusive. In addition, the double stranded nucleic acid may contain a sequence that is less than a full-length sequence or may contain a full-length sequence.
- In some embodiments, the double stranded nucleic acid inhibits the expression of only one nucleic acid, and in other embodiments, the double stranded nucleic acid molecule inhibits the expression of more than one nucleic acid. Desirably, the nucleic acid decreases the expression or biological activity of a nucleic acid of interest or a protein encoded by a nucleic acid of interest by at least 20, 40, 50, 60, 70, 80, 90, 95, or 100%. A double stranded nucleic acid can be introduced, e.g., to an individual cell or to whole animals, for example, it may be introduced systemically via the bloodstream.
- In various embodiments, the double stranded nucleic acid or antisense molecule includes one or more LNA nucleotides, one or more universal bases, and/or one or more modified nucleotides in which the 2′ position in the sugar (e.g., ribose or xylose) contains a halogen (such as fluorine group) or contains an alkoxy group (such as a methoxy group) which increases the half-life of the double stranded nucleic acid or antisense molecule in vitro or in vivo compared to the corresponding double stranded nucleic acid or antisense molecule in which the corresponding 2′ position contains a hydrogen or an hydroxyl group. In yet other embodiments, the double stranded nucleic acid or antisense molecule includes one or more linkages between adjacent nucleotides other than a naturally-occurring phosphodiester linkage. Examples of such linkages include phosphoramide, phosphorothioate, and phosphorodithioate linkages. Desirably, the double stranded or antisense molecule is purified.
- By “purified” is meant separated from other components that naturally accompany it. Typically, a factor is substantially pure when it is at least 50%, by weight, free from proteins, antibodies, and naturally-occurring organic molecules with which it is naturally associated. Desirably, the factor is at least 75%, more desirably, at least 90%, and most desirably, at least 99%, by weight, pure. A substantially pure factor may be obtained by chemical synthesis, separation of the factor from natural sources, or production of the factor in a recombinant host cell that does not naturally produce the factor. Nucleic acids and proteins may be purified by one skilled in the art using standard techniques such as those described by Ausubel et al. (Current Protocols in Molecular Biology, John Wiley & Sons, New York, 2000). The factor is desirably at least 2, 5, or 10 times as pure as the starting material, as measured using polyacrylamide gel electrophoresis, column chromatography, optical density, HPLC analysis, or western analysis (Ausubel et al., supra). Desirable methods of purification include immunoprecipitation, column chromatography such as immunoaffinity chromatography, magnetic bead immunoaffinity purification, and panning with a plate-bound antibody.
- By “treating, stabilizing, or preventing a disease, disorder, or condition” is meant preventing or delaying an initial or subsequent occurrence of a disease, disorder, or condition; increasing the disease-free survival time between the disappearance of a condition and its reoccurrence; stabilizing or reducing an adverse symptom associated with a condition; or inhibiting or stabilizing the progression of a condition. Desirably, at least 20, 40, 60, 80, 90, or 95% of the treated subjects have a complete remission in which all evidence of the disease disappears. In another desirable embodiment, the length of time a patient survives after being diagnosed with a condition and treated with a nucleic acid of the invention is at least 20, 40, 60, 80, 100, 200, or even 500% greater than (i) the average amount of time an untreated patient survives or (ii) the average amount of time a patient treated with another therapy survives.
- By “treating, stabilizing, or preventing cancer” is meant causing a reduction in the size of a tumor, slowing or preventing an increase in the size of a tumor, increasing the disease-free survival time between the disappearance of a tumor and its reappearance, preventing an initial or subsequent occurrence of a tumor, or reducing an adverse symptom associated with a tumor. In one desirable embodiment, the number of cancerous cells surviving the treatment is at least 20, 40, 60, 80, or 100% lower than the initial number of cancerous cells, as measured using any standard assay. Desirably, the decrease in the number of cancerous cells induced by administration of a nucleic acid of the invention (e.g., a nucleic acid with substantial complementarily to a nucleic acid associated with cancer such as an oncogene) is at least 2, 5, 10, 20, or 50-fold greater than the decrease in the number of non-cancerous cells. In yet another desirable embodiment, the number of cancerous cells present after administration of a nucleic acid of the invention is at least 2, 5, 10, 20, or 50-fold lower than the number of cancerous cells present prior to the administration of the compound or after administration of a buffer control.
- Desirably, the methods of the present invention result in a decrease of 20, 40, 60, 80, or 100% in the size of a tumor as determined using standard methods. Desirably, at least 20, 40, 60, 80, 90, or 95% of the treated subjects have a complete remission in which all evidence of the cancer disappears. Desirably, the cancer does not reappear or reappears after at least 5, 10, 15, or 20 years.
- Exemplary cancers that can be treated, stabilized, or prevented using the above methods include prostate cancers, breast cancers, ovarian cancers, pancreatic cancers, gastric cancers, bladder cancers, salivary gland carcinomas, gastrointestinal cancers, lung cancers, colon cancers, melanomas, brain tumors, leukemias, lymphomas, and carcinomas. Benign tumors may also be treated or prevented using the methods and nucleic acids of the present invention.
- By “infection” is meant the invasion of a host animal by a pathogen (e.g., a bacteria, yeast, or virus). For example, the infection may include the excessive growth of a pathogen that is normally present in or on the body of an animal or growth of a pathogen that is not normally present in or on the animal. More generally, an infection can be any situation in which the presence of a pathogen population(s) is damaging to a host. Thus, an animal is “suffering” from an infection when an excessive amount of a pathogen population is present in or on the animal's body, or when the presence of a pathogen population(s) is damaging the cells or other tissue of the animal. In one embodiment, the number of a particular genus or species of pathogen is at least 2, 4, 6, or 8 times the number normally found in the animal.
- A bacterial infection may be due to gram positive and/or gram negative bacteria. In desirable embodiments, the bacterial infection is due to one or more of the following bacteria: Chlamydophila pneumoniae, C. psittaci, C. abortus, Chlamydia trachomatis, Simkania negevensis, Parachlamydia acanthamoebae, Pseudomonas aeruginosa, P. alcaligenes, P. chlororaphis, P. fluorescens, P. luteola, P. mendocina, P. monteilii, P. oryzihabitans, P. pertocinogena, P. pseudalcaligenes, P. putida, P. stutzeri, Burkholderia cepacia, Aeromonas hydrophilia, Escherichia coli, Citrobacter freundii, Salmonella typhimurium, S. typhi, S. paratyphi, S. enteritidis, Shigella dysenteriae, S. flexneri, S. sonnei, Enterobacter cloacae, E. aerogenes, Klebsiella pneumoniae, K. oxytoca, Serratia marcescens, Francisella tularensis, Morganella morganii, Proteus mirabilis, Proteus vulgaris, Providencia alcalifaciens, P. rettgeri, P. stuartii, Acinetobacter calcoaceticus, A. haemolyticus, Yersinia enterocolitica, Y. pestis, Y. pseudotuberculosis, Y. intermedia, Bordetella pertussis, B. parapertussis, B. bronchiseptica, Haemophilus influenzae, H. parainfluenzae, H. haemolyticus, H. parahaemolyticus, H. ducreyi, Pasteurella multocida, P. haemolytica, Branhamella catarrhalis, Helicobacter pylori, Campylobacter fetus, C. jejuni, C. coli, Borrelia burgdorferi, V. cholerae, V parahaemolyticus, Legionella pneumophila, Listeria monocytogenes, Neisseria gonorrhea, N. meningitidis, Kingella dentrificans, K. kingae, K. oralis, Moraxella catarrhalis, M atlantae, M lacunata, M. nonliquefaciens, M. osloensis, M phenylpyruvica, Gardnerella vaginalis, Bacteroidesfragilis, Bacteroides distasonis, Bacteroides 3452A homology group, Bactero ides vulgatus, B. ovalus, B. thetaiotaomicron, B. uniformis, B. eggerthii, B. splanchnicus, Clostridium difficile, Mycobacterium tuberculosis, M. avium, M intracellulare, M leprae, C. diphtheriae, C. ulcerans, C. accolens, C. afermentans, C. amycolatum, C. argentorense, C. auris, C. bovis, C. confusum, C. coyleae, C. durum, C. falsenii, C. glucuronolyticum, C. imitans, C. jeikeium, C. kutscheri, C. kroppenstedtii, C. lipophilum, C. macginleyi, C. matruchoti, C. mucifaciens, C. pilosum, C. propinquum, C. renale, C. riegelii, C. sanguinis, C. singulare, C. striatum, C. sundsvallense, C. thomssenii, C. urealyticum, C. xerosis, Streptococcus pneumoniae, S. agalactiae, S. pyogenes, Enterococcus avium, E. casseliflavus, E. cecorum, E. dispar, E. durans, E. faecalis, E. faecium, E. flavescens, E. gallinarum, E. hirae, E. malodoratus, E. mundtii, E. pseudoavium, E. raffinosus, E. solitarius, Staphylococcus aureus, S. epidermidis, S. saprophyticus, S. intermedius, S. hyicus, S. haemolyticus, S. hominis, and/or S. saccharolyticus. Desirably, a nucleic acid is administered in an amount sufficient to prevent, stabilize, or inhibit the growth of a pathogenic bacteria or to kill the bacteria.
- In various embodiments, the viral infection relevant to the methods of the invention is an infection by one or more of the following viruses: West Nile virus (e.g., Samuel, “Host genetic variability and West Nile virus susceptibility,” Proc. Natl. Acad. Sci. USA Aug. 21, 2002; Beasley, Virology 296:17-23, 2002), Hepatitis, picornarirus, polio, HIV, coxsacchie, herpes (e.g., zoster, simplex, EBV, or CMV), adenovirus, retrovius, falvi, pox, rhabdovirus, picorna virus (e.g., coxsachie, entero, hoof and mouth, polio, or rhinovirus), St. Louis encephalitis, Epstein-Barr, myxovirus, JC, coxsakievirus B, togavirus, measles, paramyxovirus, echovirus, bunyavirus, cytomegalovirus, varicella-zoster, mumps, equine encephalitis, lymphocytic choriomeningitis, rabies,
simian virus 40, polyoma virus, parvovirus, papilloma virus, primate adenovirus, and/or BK. - By “mammal in need of treatment” is meant a mammal in which a disease, disorder, or condition is treated, stabilized, or prevented by the administration of a nucleic acid of the invention.
- Other aspects and embodiments of the invention are in the detailed description and claims below. Additionally, other nucleic acids and methods described in U.S. Ser. No. 10/105,639 (Jakobsen et al., “Modified Oligonucleotides and Uses Thereof”) or U.S. S. No. 60/410,061 (Ramsing et al., “Populations of Oligonucleotides with Duplex Stabilizing Properties and Uses Thereof”) which are hereby incorporated by reference, can be used in the present invention.
- FIG. 1 shows the structures of selected nucleotide monomers: DNA (T), LNA (TL), pyrene DNA (Py), 2′-OMe-RNA [2′-OMe(T)], abasic LNA (AbL), phenyl LNA (17a), and pyrenyl LNA (17d).
- FIG. 2 illustrates the chemical structures of Selective Binding Complementary (SBC) nucleotides.
- FIG. 3 shows the base-pairing between modified bases and naturally-occurring nucleotides. These modified bases may be incorporated as part of an LNA, DNA, or RNA unit and used any of the oligomers of the invention.
- FIGS. 4A and 4B show the sensitivity of 50-mer LNA capture probes compared to 50-mer DNA capture probes. SWI 5-specific 50-mer DNA oligonucleotides (green bars) and 50-mer capture probes with an LNA nucleotide incorporated at every third nucleotide position (red bars) were printed at the oligo concentration indicated below. The slides were hybridized at 65° C. in 3×SSC (FIG. 4A) and at 70° C. in 3×SSC (FIG. 4B).
- FIGS. 5A and 5B show the specificity of 40-mer LNA capture probes (red bars) compared to DNA capture probes (green bars). The hybridizations were carried out at 65° C. in 3×SSC.
Bars - FIG. 6 shows the detection principle for alternative exon skipping in the C. elegans let-2 gene using LNA oligonucleotide capture probes and comparative expression profiling.
- FIG. 7 shows the detection of alternative splicing of C. elegans Let-2
exon - FIGS. 8A and 8B show the comparison of DNA and LNA-modified oligonucleotide capture probes in the specific capture of the C. elegans T01 D3.3 mRNA,
exon 4. - FIG. 9 illustrates the LNA exon-exon junction (merged) probe concept.
- FIGS. 10A and 10B show the capture probe specificity for the C. elegans T10D3.3 mRNA, exon 4 (FIG. 10A) and exon 5 (FIG. 10B) as validated by short complementary target oligonucleotides.
- FIG. 11 shows the construction of the recombinant splice variants in the in vitro transcription vector. The small bars show the location of the hybridization for the oligonucleotide capture probes used in this example. The sequences of the capture probes are described herein.
- FIGS. 12A (LNA probes) and 12B (DNA control probes) show the detection of
splice variant # 1 and #2, respectively using merged capture probes in a comparative, two-color hybridization. - FIG. 13 shows the sensitivity of 50-mer LNA capture probes compared to 50-mer DNA capture probes. SWI5-specific 50-mer DNA oligonucleotides and 50-mer capture probes with an LNA nucleotide incorporated at every second (LNA2) or third (LNA3) nucleotide position. The slides were hybridized at 65° C. in 3×SSC.
- FIG. 14 is a bar graph of the signal intensities of a patient DNA sample hybridized to an array of the invention. The names of the probes in FIGS. 14 and 17 match, although the numbers used in FIG. 14 are abbreviated, e.g., probe No. 10580 Menkes.14 50NH2C 6-2.LNA in FIG. 17 corresponds to the second probe counted from the left “14.2” LNA in the lower graph of FIG. 14.
- FIG. 15 is a graph comparing the spot intensity for probes of the invention with different LNA substitution patterns.
- FIG. 16 is a bar graph of the spot intensity for LNA probes for different exons.
- FIG. 17 is a table of comparative genome hybridization (CGH) capture probe sequences.
- FIG. 18 is a flow chart of the steps of oligo design software of the invention. The OligoDesign software features LNA modified oligonucleotide secondary structure prediction, LNA spiked oligonucleotide melting temperature prediction, genome wide cross hybridization prediction, secondary structure prediction of the target, and recognition and filtering of the target in the genome. These features are determined for each possible probe of the query gene and presented to an artificial neural network. The probes are then ranked according to the neural network prediction and the top scoring probes are returned.
- FIGS. 19A-19F are a schematic illustration of the OligoDesign software of the invention.
- FIG. 20 illustrates photo-activated immobilization of nucleic acids of the invention, which enables polarized coupling of anthraquinone (AQ)-linked LNA oligonucleotides onto the polymer surface. No pretreatment of the slide is needed. A covalent bond is formed between the oligonucleotide and the polymer using a UV source, e.g. Stratalinker.
- FIG. 21 illustrates an injection-molded polymer slide. Finger indents ease slide handling. The slide has a well-defined printing and hybridization window, frosted surface for identification and orientation, and space for barcodes.
- FIG. 22 illustrates spot quality on different slides that can be used to immobilize nucleic acids of the invention. The hydrophobic slide surface ensures that extremely homogenous spots are generated when hydrophilic spotting solution is applied to the surface. A high spot quality is obtained on the Immobilizer™ polymer slide compared to a glass slide when using a spot-to-spot distance of 150 μM. The high-quality arrays simplify downstream image analyses.
- FIG. 23 is a schematic illustration of a method of the invention.
- FIG. 24 is a table of exemplary target nucleic acids (Holstege et al. (1998)(Cell 95, 717-728, and Causton et al. (2001)
Mol. Biol. Cell 12, 323-337). - FIG. 25 is a graph of Cy5 intensity. Yeast actin 1-specific 50-mer capture probes were synthesized as DNA and DNA/LNA mixmer oligonucleotides. LNA-substituted mixmer capture probes contain an LNA at every 4 th, 5th, and 6th nucleotide position (
LNA —4,LNA —5, LNA—6). On-chip melting profiles demonstrate a 8-10° C. increase in Tm obtained with LNA capture probes. - FIG. 26A illustrates the heat-shock response in yeast. The array was hybridized with Cy3-labeled standard and Cy5-labelled heat-shock yeast cDNA. FIG. 26B also illustrates the heat-shock response in yeast. The microarray data were normalized using
yeast actin 1. The ssa4 gene encoding heat shock protein HSP70 is up-regulated over 2-fold. Expression of the gual gene is down-regulated. - FIG. 27A compares expression of wild-type and ssa4 mutant yeast. The array was hybridized with Cy3-labeled wild-type and Cy5-labelled ssa4 mutant yeast cDNA. FIG. 27B also compares wild-type and ssa4 yeast. The hybridization data were normalized using
yeast actin 1. ssa4 is detected in the wild-type yeast strain, but not in the ssa4 knock-out strain. - FIG. 28 illustrates mRNA splicing.
- FIG. 29 is a picture showing gel electrophoresis of fragmented cDNA from the yeast wild-type strain. The molecular marker (
lane 1 and 9) is from Life technologies, USA. Lanes 2-8 represents the UDG-fragmented cDNA 1-7 according to the different dUTP/dTTP ratios in Table 18. - FIG. 30 is a graph of the log ratios of the normalized fluorescence intensities from the wild-type yeast strain (signal) and those from the Δssa4 yeast strain (noise) as a function of capture probe position in the 3′ region of the SSA4 mRNA.
- FIG. 31 is a schematic illustration of mRNA splicing.
- FIG. 32 is a schematic illustration of alternative mRNA splicing.
- FIG. 33 is a schematic illustration of probes of the invention.
- FIG. 34 is a schematic illustration of probes of the invention.
- FIG. 35 illustrates an exemplary computer for use in the methods of the invention.
- FIGS. 36 a-36 d show the sensitivity and specificity of LNA oligonucleotide capture probes (black solid bars) compared to DNA capture probes (white, open bars) on expression microarrays. Fluorescence intensity is shown in arbitrary units (relative measurements). The arrays comprising 50-mer and 40-mer perfect match and 1-5 mismatch capture probes were hybridized at 65° C. in 3×SSC with Cy3-labelled cDNA from 10 μg C. elegans total RNA spiked with yeast a) SWI5 RNA and c) THI4 RNA. FIGS. 36b and 36 d demonstrate the improved mismatch discrimination with the 50-mer LNA probes by increasing the hybridization temperature from 65° C. to 70° C. hybridized with Cy3-labelled cDNA from 10 μg C. elegans total RNA spiked with yeast b) SWI5 RNA and d) THI4 RNA.
- FIG. 37 shows the expected (black, solid bars) and observed (white, open bars) fold-of-change in the expression levels of the Cy3-ULS-labelled yeast HSP78 spike RNA as measured by on-chip capture using three different 25-mer oligonucleotide capture probes (DNA control, LNA-T substituted,
LNA —3 substituted in which every third nucleotide was substituted with an LNA monomer). In the hybridization experiment, one ng of HSP78 in vitro spike RNA or 200 pg HSP78 in vitro spike RNA was used, respectively. Thus, the fold change of the HSP78 RNA in the two hybridizations in the comparison is 5-fold. Fourteen additional synthetic in vitro mRNA spike controls were included in the hybridisation solution as a semi-complex background RNA mixture. Seven of these spikes were used as normalization controls, the remaining seven were used as negative controls. Hybridization temperature was 65° C. for 16 hours, and post-hybridization washes as described. Both LNA_T andLNA —3 substituted 25-mer probes are capable of providing highly accurate measurements for fold-of-changes in gene expression levels, as depicted in FIG. 37. Under these conditions the DNA capture probes did not hybridize. - FIG. 38 shows the measured intensity levels by on-chip capture using three different 25-mer oligonucleotide capture probe designs (DNA control, LNA_T substituted and LNA C and T substituted probes). One (1) ng biotin-labeled HSP78 target was used in the hybridization experiments, followed by staining with Streptavidin Phycoerythrin. The LNA_T and LNA_TC substituted 25-mer capture probes show a significantly enhanced on-chip capture of the HSP78 RNA target, compared to the DNA 25-mer control probes under four different hybridization stringency conditions in dicated on the graph.
- FIG. 39 shows the detection of alternatively spliced mRNAs using LNA-substituted 50-mer oligonucleotide capture probes. Parts per million (ppm) calculations indicate spike transcripts per total transcripts in the hybridisation mix. Calculations are based on an average C. elegans RNA being 1000 nucleotides as in Hill et al. (2000) Science 290:809-812. The 50-mer LNA-DNA mixmer capture probes, substituted with an LNA nucleotide at every third nucleotide position, are able to provide highly accurate measurements for fold-changes in the expression of three homologous, alternatively spliced mRNA variants in the concentration range of 1000 ppm to 10 ppm. The quantification of the splice isoforms was carried out using a set of both internal, exon-specific probes and merged, splice junction specific probes, printed onto microarrays and hybridized with complex cDNA target pools spiked with different cloned artificial splice isoforms in which the middle exon was either alternatively skipped or excluded completely resulting in the three different splice isoforms; 01-INS3-03, 01-INS4-03 and 01-03.
- FIG. 40 shows the detection of alternatively spliced mRNAs using LNA-substituted 40-mer oligonucleotide capture probes. Parts per million (ppm) calculations indicate spike transcripts per total transcripts in the hybridisation mix. Calculations are based on an average C. elegans RNA being 1000 nucleotides as in Hill et al. (2000) Science 290:809-812. The 40-mer LNA-DNA mixmer capture probes, substituted with an LNA nucleotide at every third nucleotide position, are able to provide highly accurate measurements for fold-changes in the expression of three homologous, alternatively spliced mRNA variants in the concentration range of 1000 ppm to 10 ppm. The quantification of the splice isoforms was carried out using a set of both internal, exon-specific probes and merged, splice junction specific probes, printed onto microarrays and hybridized with complex cDNA target pools spiked with different cloned artificial splice isoforms in which the middle exon was either alternatively skipped or excluded completely resulting in the three different splice isoforms; 01-INS3-03, 01-INS4-03 and 01-03.
- FIGS. 41A-41E show the comparison of different LNA/DNA mixmer oligonucleotide probes in the detection of human satellite-2 repeats by fluorescence in situ hybridization. Experiment conditions: 6.4 pmoles of Cy3 labeled probe was hybridized for 30 minutes at 37° C., after simultaneous denaturation of the target and the probe at 75° C. for 5 minutes. A. LNA-2 giving signals on
chromosomes chromosomes chromosomes chromosome 1, E. DNA control oligonucleotide FISH probe giving no signals on any of the chromosomes. - FIG. 42. Illustrates the hybridisation of the Cy3-labelled human telomere repeat specific, LNA-2 substituted oligonucleotide probe on human metaphase chromosomes resulted in prominent signals on the telomeres.
- Detection and Analysis of mRNA Splice Variants
- Alternative splicing is the process by which different mature messenger RNAs are produced from the same pre-mRNA. Because the mRNA composition of a given cell determines the proteins present in a cell, this process is an important aspect of a cells gene expression profile. Current investigations of transcriptomes (i.e., the total complexity of RNA transcripts produced by an organism) indicate that at least 50-60% of the genes of complex eukaryotes produce more than one splice variant. The present invention provides a novel method for detecting and quantifying the levels of splice variants in complex mRNA pools using LNA discriminating probes and high-throughput LNA oligonucleotide microarray technology. The detection concept which uses internal LNA exon probes and/or splice-variant specific exon-exon junction or exon-intron or intron-exon (so-called merged) probes is depicted in FIG. 9.
- Internal, exon-specific (or intron-specific) LNA oligonucleotide probes are designed and used to detect the relative levels of a given exon (or intron) in complex mRNA pools using oligonucleotide microarray technology or similar techniques. Exon-exon LNA junction probes are designed for multiple or all possible exon-exon combinations or exon-intron combinations. The LNA discriminating probes are highly specific and superior compared to DNA oligonucleotides due to the higher ΔT m of LNA probes. These probes can be used to determine the sequential order of each sub-element (i.e., exon structure or exon-intron structure) in a given alternatively spliced mRNA isoform, thus giving the exact composition of the mRNA. Subsequently, the ratios of each splice variant can be quantified using the combined readouts from both internal and merged LNA probes and control probes. The invention is applicable both in single fluor (single channel) or comparative two-fluor (two channel) microarray hybridizations.
- Several “artificial,”alternatively spliced mRNA molecules may be constructed in an in vitro transcription vector for the production of clean IVT RNA. Both internal and junction-specific LNA oligonucleotide capture probes are designed, synthesized, and spotted onto, e.g., Exiqon's polymer microarray platform. The resulting splice-specific microarray is used to validate the LNA discriminating probe concept by spiking the in vitro RNAs individually as well as in different ratios into a complex RNA background for fluorochrome-labelling and array hybridization.
- The internal and merged probes of the invention can also be used in any standard method for the analysis of mRNA splice variants (see, for example, Yeakley et al., Nature Biotechnology 20:353-358, 2002; Clark et al., Science 296:907-910, 2002; Mutch et al., Genome Biology 2(12):preprint00009.1-0009.31, 2001).
- Exemplary Applications of Internal and/or Merged Probes
- The internal and/or merged probes of the invention can also be used for gene expression profiling of alternative splice variants, oligonucleotide expression microarrays, real-time PCR, and profiling of alternatively spliced mRNAs using microtiterplate assays or fiber-optic arrays.
- Detection and characterization of alternative splicing is particularly useful for the study and treatment of human disease (exonhit website, “Inaugural Splicing 2002 Concludes: Alternative Splicing May Make All the Difference in Discovering the Origin of Disease). In particular, RNA splicing is now widely recognized as a means to generate protein diversity. Alternative splicing is a key mechanism for regulating gene expression, and any mutation or defect in its regulation can impact considerably cell functions. Therefore, it is likely to be an important source of novel gene and protein targets implicated in human pathology. Industry has long recognized the need for innovative discovery technologies that focuses on the origin of disease for the development of novel diagnostics and therapeutics.
- In particular, there are many examples of human pathologies caused by alterations in normal patterns of alternative RNA splicing. Because a large number of human genes undergo alternative splicing, the protein isoforms that result from this process represent a major source of targets for commercial development of therapies and diagnostics. In particular, splicing processes play a significant role in the onset and development of cardiovascular, muscular, CNS diseases, and cancer. Early evidence indicates the origin of many diseases can be identified by examining alternative splicing—which leads to the point of intervention for discovering future generations of drugs. The study of splicing enables the discovery of new mechanisms underlying disease progression.
- Comparative Genomic Hybridization
- Comparative Genomic Hybridization (CGH) is a powerful technology for detection of unbalanced chromosome rearrangements and holds much promise for screening and identification of interstitial submicroscopic rearrangements that otherwise cannot be detected using classical cytogenetic or FISH technologies. The adaptation of CGH onto an oligo microarray platform allows detection of small single exon deletion/duplications on a genome wide scale. There is a strong need for developing microarrays that can detect, e.g., single exon aberrations. This detection can be achieved by employing LNA mixmer oligos as capture probes for individual exons in selected genes. A model system for these methods is the Menkes loci. Menkes disease is a lethal-X linked recessive disorder associated with copper metabolism disturbance leading to death in early childhood. The Menkes locus has been mapped to Xq13. The gene spans about 150 kb genomic region, contains 23 exons, and encodes a 8.5 kb gene transcript. The gene for Menkes disease (now designated as A TP7A) encodes a 1500 amino acid membrane-bound Cu-binding P-type ATPase (ATP7A). The 8.5 kb transcript is expressed in all tissues from normal individuals (though only trace amounts are present in liver), but is diminished or absent in Menkes disease patients. Several different kinds of mutations, like chromosome aberrations, point mutations and partial gene deletions affecting A TP7A have been identified in MD patients. 50-mer capture probes with LNA spiked in every second, third, and fourth position have been designed for every exon (23 exons) representing ATP7A, using the OligoDesign software tool, described herein. The C6-amino-linked capture probes were spotted onto Immobilizer slides and hybridized with patient samples with Cy3 fluorescent dye and a known reference genomic sample with Cy5. After mixing equal amounts of the labelled DNA, the probe is hybridized it to array. The ratio of Cy5 signal to Cy3 for each clone indicates differences in chromosome/DNA material. For example, the Cy5 signal is higher than Cy3 if the patient genome has a deletion, and is lower if there is duplication. In regions that are unchanged, the Cy5:Cy3 ratio is 1:1. These methods can be used to analyze a number of well-characterized Menkes patients with a range of partial deletions of A TP7A.
- LNA oligonucleotide-based CGH makes it possible to assess a large number of chromosomal aberrations that are being screen for in the cytogenetic clinic. In contrast, standard FISH analysis typically only detects large chromosomal rearrangements. In desirable embodiments, an array that contains a series of overlapping probes is used to detect a chromosomal deletion in a nucleic acid sample, such as a patient sample.
- Clinical diagnostics
- Clinical diagnosis is a key element in healthcare management and point-of-care. A large number of analyses in the hospitals are based on the use of robust, cost efficient, sensitive and highly specific diagnostic tests. Thus, the diagnosis of various diseases is performed with a high selectivity and reliability, resulting in confirmation of medical diagnosis, choice of therapy and follow-up treatment as well as prevention. In addition to its importance in the quality of healthcare provided to patients, clinical diagnosis also contributes to the control of healthcare costs. The field of clinical diagnostics involves analyzing biological fluid samples (blood, urine, etc.) or biopsies collected from patients in order to establish the diagnosis of diseases, whether of infectious, metabolic, endocrine or cancerous origin. Medical analysis of infectious diseases involves testing and identifying the micro-organisms causing the infection e.g. testing for and identifying a micro-organism in blood and determining its susceptibility to antibiotics or detecting an antigen-antibody reaction produced as a response to an attack by a micro-organism in the human body, e.g. testing for antibodies for the diagnosis of hepatitis. The accurate diagnosis of metabolic and endocrine diseases and cancers, resulting in a disease phenotype with a bodily imbalance, involves the measurement of diagnostic substances or elements present in the biological fluids or biopsies. These substances are examined and results are interpreted with reference to known normal values.
- Use of Diagnostic Kits in Microbiological Control
- The pharmaceutical, cosmetics and agri-food industries are being confronted with increasingly strict quality standards. Thus, the purpose of industrial microbiological control testing is to detect and measure the presence of potentially pathogenic microbial contaminants throughout the manufacturing process from raw materials to the finished products, as well as in the production environment. The obtained results are subsequently compared to the current regulatory guidelines and industry standards.
- Application of Molecular Biological Techniques to In Vitro Diagnostics RECENTLY, several different molecular biological techniques have been used successfully in accurate quantification of RNA levels in clinical diagnosis as well as in microbiological control. The applications are wide-ranging and include methods for quantification of the regulation and expression of drug resistance markers in tumour cells, monitoring of the responses to chemotherapy, measuring the biodistribution and transcription of gene-encoded therapeutics, molecular assessment of the tumor stage in a given cancer, detecting circulating tumor cells in cancer patients and detection of bacterial and viral pathogens. The reverse transcription polymerase chain reaction (RT-PCR) is the most sensitive method for the detection of mRNA, including low abundant mRNAs, often obtained from limited tissue samples in clinical diagnostics. The application of fluorescence techniques to RT-PCR combined with suitable instrumentation has led to development of quantitative RT-PCR methods, combining amplification, detection and quantification in a closed system avoid from contamination and with minimized hands-on time. The two most commonly used quantitative RT-PCR techniques are the Taqman RT-PCR assay (ABI, Foster City, USA) and the Lightcycler assay (Roche, USA). A third method applied to detection and quantification of RNA levels is real-time nucleic acid sequence based amplification (NASBA) combined with molecular beacon detection molecules. NASBA is a singe-step isothermal RNA-specific amplification method that amplifies mRNA in a double stranded DNA environment, and this method has recently proven useful in the detection of various mRNAs and in the detection of both viral and bacterial RNA in clinical samples. Finally, the recent explosion in microarray technology holds the promise of using microarrays in clinical diagnostics. For example van't Veer et al. (Nature 2002: 415, 31) describe the successful use of microarrays in obtaining digital mRNA signatures from breast tumors and the use of these signatures in the precise prediction of the clinical outcome of breast cancer in patients.
- The success of exploiting molecular biological techniques in diagnostics and diagnostic kits depends on continuous optimization of the technologies and the development of new robust and cost-effective technology platforms for producing accurate, reproducible and valid clinical data. Locked nucleic acid (LNA) oligonucleotides constitute a novel class of bicyclic RNA analogs having an exceptionally high affinity and specificity toward their complementary DNA and RNA target molecules. Besides increased thermal stability, LNA-containing oligonucleotides show significantly increased mismatch discrimination, and allow full control of the melting temperature across microarray hybridizations. The LNA chemistry is completely compatible with conventional DNA phosphoramidite chemistry and thus LNA substituted oligonucleotides can be designed to optimize performance. LNA oligonucleotides would be well-suited for large-scale clinical studies providing highly accurate genotyping by direct competitive hybridization of two allele-specific LNA probes to e.g. microarrays of immobilized patient amplicons. In addition, the use of LNA substituted oligonucleotides would increase both sensitivity and specificity in detection and quantification of mRNA levels in clinical samples, either by quantitative RT-PCR, quantitative NASBA or oligonucleotide microarrays, compared with DNA probes. Application of LNA oligonucleotides into diagnostic kits would thus significantly enhance their performance. Finally, the use of LNA substituted oligonucleotides would increase the sensititity and specificity in the detection of alternatively spliced mRNA isoforms and non-coding RNAs either by homogeneous assays (Taqman assay, Lightcycler assay, NASBA) or by oligonucleotide microarrays in a massive parallel analysis setup.
- Optimized Nucleic Acids of the Invention
- Decreasing the variation in melting temperatures (T m) of a population of nucleic acids allows the nucleic acids to hybridize to target molecules under similar binding conditions, thereby simplifying the simultaneous hybridization of multiple nucleic acids. Similar melting temperatures also allow the same hybridization conditions to be used for multiple experiments, which is particularly useful for assays involving hybridization to nucleic acids of varying “AT” content. For example, current methods often require less stringent conditions for hybridization of nucleic acids with high “AT” content compared to nucleic acids with low “AT” content. Due to this variation in hybridization stringency, current methods may require significant trial and error to optimize the hybridization conditions for each experiment.
- To overcome limitations in current nucleic acid hybridization and/or amplification techniques, we have developed populations of nucleic acid probes or primers with minimal variation in melting temperature (U.S. S. No. 60/410,061). For example, the unique properties of LNA nucleotide analogs increase their binding affinity for DNA and RNA. The stability of duplexes can generally be ranked as follows: DNA:DNA <DNA:RNA <RNA:RNA <LNA:DNA <LNA:RNA <LNA:LNA. The DNA:DNA duplex is thus the least stable and the LNA:LNA duplex the most stable. The affinity of the LNA nucleotides A and T corresponds approximately to the affinity of DNA G and C to their complementary bases. General substitution of one or more A and T nucleotides with LNA A and LNA T in DNA oligonucleotides is therefore a simple way of equalizing differences in T m. Furthermore, the mean melting temperature is increased significantly, which is often important for shorter oligonucleotides. For example, predictions of melting temperature of all possible 9-mer oligonucleotides have shown that the mean temperature increases from 39.7° C. to 59.3° C. by substituting all DNA A and T nucleotides with LNA A and T nucleotides. The variance in Tm of all 9-mers furthermore decreases from 59.6° C. for DNA oligonucleotides to only 4.7° C. for the LNA substituted oligonucleotides. The estimations are based on the latest LNA Tm prediction algorithms such as those disclosed herein, which have a variance of 6-7° C.
- If desired, the capture efficiency of one or more nucleic acids can be increased by including any of the high affinity nucleotides (e.g., LNA units) described herein within the nucleic acids. The examples herein also provide algorithms for optimizing the substitution patterns of the nucleic acids to minimize self-complementarity that may otherwise inhibit the binding of the nucleic acids to target molecules.
- For various applications of the nucleic acids and arrays of the invention, LNA A and LNA T substitutions are made to equalize the melting temperatures of the nucleic acids. In other embodiments, LNA A and LNA C substitutions are made to minimize self-complementarity and to increase specificity. LNA C and LNA T substitutions also minimize self-complementarity. Additionally, oligonucleotides containing LNA C and LNA T are desirable because these modified nucleotides are easy to synthesis and are especially useful for applications such as antisense technology in which minimizing cost is especially desirable.
- The following non-limiting examples are illustrative of the invention. All documents mentioned herein are incorporated herein by reference in their entirety. In the following Examples, compound reference numbers designate the compound as shown in
Scheme - This example demonstrates the advantages of using LNA oligonucleotide microarrays in gene expression profiling experiments. Capture probes for the Saccharomyces cerevisiae genes SWI5 (YDR146C) and THI4 (YGR144W) were designed as 50-mer standard DNA and different LNA/DNA “mixmer” oligonucleotides (i.e., oligonucleotides containing both LNA and DNA nucleotides) respectively, for comparison (Table 2). In addition, 40-mer oligonucleotides were designed as truncated versions of the 50-mer capture probes (Table 2). The specificity of the LNA oligoarrays was addressed by introducing 1-5 consecutive mismatches positioned in the middle of 40-mer LNA/DNA mixmer capture probes with LNA in every fourth position. To assess the sensitivity of DNA versus LNA capture probes in complex hybridization mixtures, in vitro synthesized yeast RNA for either SWI5 or THI4 was spiked into Caenorhabditis elegans total RNA for cDNA target synthesis. These experiments are described further below. Cultivation of Caenorhabditis elegans worms
- Mixed stage C. elegans cultures were grown according to standard methods. Samples were harvested by centrifugation at 3,000×g, suspended in RNA Later storage buffer (Ambion, USA), and immediately frozen in liquid nitrogen.
- RNA Extraction
- RNA was extracted from the worm samples using the FastRNA® Kit, GREEN (Q-BIO) essentially according to the suppliers' instructions.
- In Vitro RNA Synthesis
- Amplification of the yeast genes was performed using standard PCR with yeast genomic DNA as the template. In the first step, a forward primer containing a restriction enzyme site and a reverse primer containing a universal linker sequence were used. In the second PCR reaction, the reverse primer was exchanged with a nested primer containing a poly-T20 tail and a restriction enzyme site. The DNA fragments were ligated into the
pTRIamp 18 vector (Ambion, USA) using the Quick Ligation Kit (New England Biolabs, USA) according to the supplier's instructions and transformed into E. coli DH-5α by standard methods. The PCR clones were sequenced using M13 forward and M13 reverse primers on an ABI 377 (Applied Biosystems, USA). Synthesis of in vitro RNA was carried out using the MEGAscript™ T7 Kit (Ambion, USA) according to the manufacturer's instructions. - Design and Synthesis of the LNA Capture Probes
- To design the capture probes, regions with unique mRNA sequence of the selected target genes were identified. The optimal 50-mer oligonucleotide sequences with respect to T m, self-complementarity, and secondary structure were selected. LNA modifications were incorporated to increase affinity and specificity.
- Printing of the LNA Microarrays
- The microarrays were printed on Immobilizerm MicroArray Slides (Exiqon, Denmark) using the Biochip One Arrayer from Packard Biochip technologies (Packard, USA). The arrays were printed with a spot volume of 2×300 pl of a 10 μM capture probe solution. Four replicas of the capture probes were printed on each slide.
- Synthesis of Fluorochrome Labelled First Strand cDNA from Total RNA
- Ten ng of S. cerevisiae in vitro synthesized RNA (either SWI5 or THI4) was combined with 10 μg of C. elegans total RNA and 5 μg oligo dT primer (T20VN) in an RNase free, pre-siliconized 1.5 mL tube, and the final volume was adjusted with DEPC-water to 8 μL. The reaction mixture was heated at +70° C. for 10 minutes, quenched on ice for 5 minutes, and spun for 20 seconds, followed by addition of 1 μl SUPERase-In™ (20U/μL, RNAse inhibitor, Ambion, USA), 4 μL SxRTase buffer (Invitrogen, USA), 2 μL 0.1 M DTT (Invitrogen, USA), 1 μL dNTP (20 mM dATP, dGTP, dTTP; 0.4 mM dCTP in DEPC-water, Amersham Pharmacia Biotech, USA), and 3 μL Cy3™-dCTP or Cy5™-dCTP (Amersham Pharmacia Biotech, USA). First strand cDNA synthesis was carried out by adding 1 μL of Superscript™ II (Invitrogen, 200 U/mL), mixing, and incubating the reaction mixture for one hour at 42° C. An additional 1 μL of Superscript™ II was added, and the cDNA synthesis reaction mixture was incubated for an additional one hour at 42° C.; the reaction was stopped by heating at 70° C. for 5 minutes, and quenching on ice for 2 minutes. The RNA was hydrolyzed by adding 3 μL of 0.5 M NaOH, and incubating at 70° C. for 15 minutes. The samples were neutralized by adding 3 μL of 0.5 M HCl, and purified by adding 450
μL 1×TE buffer, pH 7.5 to the neutralized sample and transferring the samples onto a Microcon-30 concentrator. The samples were centrifuged at 14000×g in a microcentrifuge for 8 minutes, the flow-through was discarded and the washing step was repeated twice by refilling the filter with 450μl 1×TE buffer and by spinning for ˜12 minutes. Centrifugation was continued until the volume was reduced to 5 μL, and finally the labelled cDNA probe was eluted by inverting the Microcon-30 tube and spinning at 1000×g for 3 minutes. - Hybridization with Fluorochrome-Labelled cDNA
- The arrays were hybridized overnight using the following protocol. The Cy3™ or Cy5™-labelled cDNA samples were combined in one tube followed by addition of 3 μL 21×SSC (3×SSC final), 0.5 μL 1 M HEPES, pH 7.0 (25 mM final), 25 μg yeast tRNA (1.25 μg/tL final), 10 μg PolyA blocker (0.5 μg/mL final), 0.6 μL 10% SDS (0.3% final), and DEPC-treated water to 20 μL final volume. The labelled cDNA target sample was filtered in a Millipore 0.22 micron spin column according to the manufacturer's instructions (Millipore, USA), and the probe was denatured by incubating the reaction at 100° C. for 2 minutes. The sample was cooled at 20-25° C. for 5 minutes by spinning at maximum speed in a microcentrifuge. A LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on Immobilizer™ MicroArray Slide, and the hybridization mixture was applied to the array from the side. An aliquot of 30 μL of 3×SSC was added to both ends of the hybridization chamber, and the Immobilizer™ MicroArray Slide was placed in the hybridization chamber. The chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol. The Lifterslip coverslip was washed off in 2×SSC, pH 7.0 containing 0.1% SDS at room temperature for one minute, followed by washing of the microarrays subsequently in 1.0×SSC, pH 7.0 at room temperature for one minute, and then in 0.2×SSC, pH 7.0 at room temperature for one minute. Finally, the slides were washed for 5 seconds in 0.05×SSC, pH 7.0. The slides were then dried by centrifugation in a swinging bucket rotor at approximately 600 rpm for 5 minutes.
- Data analysis
- Following washing and drying, the slides were scanned using a ScanArray 4000XL scanner (Perkin-Elmer Life Sciences, USA), and the array data were processed using the GenePix™ Pro 4.0 software package (Axon, USA).
- Results
- Incorporation of LNA nucleotides at every third nucleotide position in standard 50-mer expression array oligonucleoitde capture probes resulted in a 3-fold increase in fluorescence intensity levels, when hybridized under standard stringency conditions (FIGS. 4A and 4B). When the hybridization temperature is increased from 65° C. to 70° C., the capture of the SWI5 spike mRNA by LNA 50-mer oligos is increased by 8-fold relative to the DNA controls. Thus, it can clearly be concluded that oligonucleotides containing LNA units are more sensitive in expression profiling compared to oligonucleotides containing only DNA units. The specificity of 40-mer LNA/DNA mixmer capture probes in the discrimination of highly homologous target sequences was addressed by introducing 1-5 consecutive mismatches in the middle of SWI5 and THI4 capture oligos together with the corresponding DNA controls. As demonstrated in FIGS. 5A and 5B, the LNA-spiked (LNA modification at every fourth nucleotide position) 40-mer triple mismatch oligos showed a 3-fold signal intensity decrease relative to the perfectly matched duplexes, whereas the corresponding 40-mer standard DNA capture probes did not form duplexes under standard hybridization stringency. Further, the 40-mer perfect match LNA capture probes showed a 5-fold to 14-fold increase in the intensity levels compared to DNA oligonucleotides under standard hybridization conditions. Capture probes of other lengths and/or with other LNA substitution patterns can be used similarly.
TABLE 2 DNA and LNA-modified SWI5 (YDR146C) and THI4 (YGR144W) oligonucleotide capture probes. LNA modifications are depicted by uppercase letters in the sequence, mt denotes the number of mismatches (bolded) in the center of the oligonucleotide with respect to its target cDNA (mRNA), and “mC” denotes LNA methyl cytosine. (SEQ ID NOs:) Oligo Name Sequence YDR146C-40 acggggattatggtttcgccaatgaaaactaatcaaaggt YDR146C-40_mt1 acggggattatggtttcgcctatgaaaactaatcaaaggt YDR146C-40_mt2 acggggattatggtttcgcgtatgaaaactaatcaaaggt YDR146C-40_mt3 acggggattatggtttcgggtatgaaaactaatcaaaggt YDR146C-40_mt4 acggggattatggtttcgggtttgaaaactaatcaaaggt YDR146C-40_mt5 acggggattatggtttcgggttagaaaactaatcaaaggt YDR146C-40_LNA4 acGgggAttaTggtTtcgmCcaaTgaaAactAatcAaagGt YDR146C-40_LNA4_mt1 acGgggAttaTggtTtcgmCctaTgaaAactAatcAaagGt YDR146C-40_LNA4_mt2 acGgggAttaTggtTtcgmCgtaTgaaAactAatcAaagGt YDR146C-40_LNA4_mt3 acGgggAttaTggtTtcgGgtaTgaaAactAatcAaagGt YDR146C-40_LNA4_mt4 acGgggAttaTggtTtcgGgttTgaaAactAatcAaagGt YDR146C-40_LNA4_mt5 acGgggAttaTggtTtcgGgttAgaaAactAatcAaagGt YDR146C-50 tgggaatggaacggggattatggtttcgccaatgaaaactaatcaaaggt YDR146C-50_mt1 tgggaatggaacggggattatggtatcgccaatgaaaactaatcaaaggt YDR146C-50_mt2 tgggaatggaacggggattatggtaacgccaatgaaaactaatcaaaggt YDR146C-50_mt3 tgggaatggaacggggattatggtaaggccaatgaaaactaatcaaaggt YDR146C-50_mt4 tgggaatggaacggggattatggaaaggccaatgaaaactaatcaaaggt YDR146C-50_mt5 tgggaatggaacggggattatggaaagcccaatgaaaactaatcaaaggt YDR146C-50_LNA2 TgGgAaTgGaAcGgGgAtTaTgGtTtmCgmCcAaTgAaAamCtAaTcAaAgGt YDR146C-50_LNA3 TggGaaTggAacGggGatTatGgtTtcGccAatGaaAacTaaTcaAagGt YDR146C-50_LNA4 TgggAatgGaacGgggAttaTggtTtcgmCcaaTgaaAactAatcAaagGt YDR146C-50_LNA5 TgggaAtggaAcgggGattaTggttTcgccAatgaAaactAatcaAaggt YDR146C-50_LNA6 TgggaaTggaacGgggatTatggtTtcgccAatgaaAactaaTcaaagGt YDR146C-50_LNA3_mt1 TggGaaTggAacGggGatTatGgtAtcGccAatGaaAacTaaTcaAagGt YDR146C-50_LNA3_mt2 TggGaaTggAacGggGatTatGgtAacGccAatGaaAacTaaTcaAagGt YDR146C-50_LNA3_mt3 TggGaaTggAacGggGatTatGgtAagGccAatGaaAacTaaTcaAagGt YDR146C-50_LNA3_mt4 TggGaaTggAacGggGatTatGgaAagGccAatGaaAacTaaTcaAagGt YDR146C-50_LNA3_mt5 TggGaaTggAacGggGatTatGgaAagmCccAatGaaAacTaaTcaAagGt YGR144W-40 ttgctgaactggatggattaaaccgtatgggtccaacttt YGR144W-40_mt1 ttgctgaactggatggatttaaccgtatgggtccaacttt YGR144W-40_mt2 ttgctgaactggatggatataaccgtatgggtccaacttt YGR144W-40_mt3 ttgctgaactggatggatattaccgtatgggtccaacttt YGR144W-40_mt4 ttgctgaactggatggatatttccgtatgggtccaacttt YGR144W-40_mt5 ttgctgaactggatggatatttgcgtatgggtccaacttt YGR144W-40_LNA4 ttGctgAactGgatGgatTaaamCcgtAtggGtccAactTt YGR144W-40_LNA4_mt1 ttGctgAactGgatGgatTtaamCcgtAtggGtccAactTt YGR144W-40_LNA4_mt2 ttGctgAactGgatGgatAtaamCcgtAtggGtccAactTt YGR144W-40_LNA4_mt3 ttGctgAactGgatGgatAttamCcgtAtggGtccAactTt YGR144W-40_LNA4_mt4 ttGctgAactGgatGgatAtttmCcgtAtggGtccAactTt YGR144W-40_LNA4_mt5 ttGctgAactGgatGgatAtttGcgtAtggGtccAactTt YGR144W-50 ggtatggaagttgctgaactggatggattaaaccgtatgggtccaacttt YGR144W-50_mt1 ggtatggaagttgctgaactggatcgattaaaccgtatgggtccaacttt YGR144W-50_mt2 ggtatggaagttgctgaactggatccattaaaccgtatgggtccaacttt YGR144W-50_mt3 ggtatggaagttgctgaactggaaccattaaaccgtatgggtccaacttt YGR144W-50_mt4 ggtatggaagttgctgaactggaacctttaaaccgtatgggtccaacttt YGR144W-50_mt5 ggtatggaagttgctgaactggaacctataaaccgtatgggtccaacttt YGR144W-50_LNA3 GgtAtgGaaGttGctGaamCtgGatGgaTtaAacmCgtAtgGgtmCcaActTt YGR144W-50_LNA3_mt1 GgtAtgGaaGttGctGaamCtgGatmCgaTtaAacmCgtAtgGgtmCcaActTt YGR144W-50_LNA3_mt2 GgtAtgGaaGttGctGaamCtgGatmCcaTtaAacmCgtAtgGgtmCcaActTt YGR144W-50_LNA3_mt3 GgtAtgGaaGttGctGaamCtgGaamCcaTtaAacmCgtAtgGgtmCcaActTt YGR144W-50_LNA3_mt4 GgtAtgGaaGttGctGaamCtgGaamCctTtaAacmCgtAtgGgtmCcaActTt YGR144W-50_LNA3_mt5 GgtAtgGaaGttGctGaamCtgGaamCctAtaAacmCgtAtgGgtmCcaActTt - Capture Probe Design
- Finding the Regions of Interest
- From the database “intronerator” (W. Jim Kent and Al M Zahler, “The Intronerator: exploring introns and alternative splicing in C. elegans,” Nucleic Acids Research Jan. 1, 2000; 28(1):91-93 and “Conservation, Regulation, Synteny, and Introns in a Large-scale C. briggsae-C. elegans Genomic Alignment” in Genome Research August, 2000; 10(8):1115-1125) as well as scientific literature, the C. elegans Let-2 gene encoding type IV collagen was found according to the following criteria. The generation of mature mRNA desirably involves either complete exon or intron skipping. ESTs (expressed sequence tags) desirably indicate different isoforms. If ESTs were only different from the gene annotation(s), this could simply mean that the prediction is wrong, and nothing more. Desirably, there are different EST splice indications in different developmental stages. Two gene prediction algorithms (e.g., GeneFinder and Genie) desirably agree upon the number of genes in a coding segment. Exons of interest (e.g., exons being skipped and their flanking exons) in the C. elegans gene TOID3.3 desirably exceed 70 base-pairs. Other genes of interest may be selected using one or more of the above criteria or using other criteria, such as the medical relevance of the gene.
- Determining Melting Temperatures and Palindromic Properties of the C. elegans let-2 Gene/
Exons - The script PICK70 (which was kindly provided by Jingchun Zhu from the Joe DeRisi Laboratory and which is publicly available) was used to run a sliding 50 base-pair window across the regions in which an oligonucleotide capture probe should be designed. The output data were saved for later.
- Determining the Uniqueness of the Regions
- All regions were compared using a publicly available BLAST program to the complete set of annotated transcripts from the C. elegans genome downloaded from NCBI. For each region a list with the location of all BLAST hits was made.
- Choice of Desirable T m for Capture Probes
- From the PICK70 output, the distribution of melting temperatures for all possible oligonucleotides was collected. As these centered around approximately 80° C., this temperature was chosen as the desirable temperature.
- For each region, an oligonucleotide with a palindromic value below 100 (default value in PICK70, value based on Smith-Waterman algorithm) and with a melting temperature closest to 80° C. was picked. The location of the oligonucleotide within the region was then compared to the list made using the above BLAST search. If the oligonucleotide did not coincide with a BLAST hit exceeding around 25 (consecutive) base-pairs, this oligonucleotide sequence was chosen as a 50-mer capture probe. Otherwise, a new oligo sequence was picked from the PICK70 output.
- Checking Probe Sequences
- The selected 50-mer oligonucleotide sequences were “BLASTed” against the C. elegans transcripts again, as described above.
- Accounting for the Introns.
- The oligonucleotide sequences were “BLASTed” against the complete C. elegans genome. The matches were run against a list made from the GenBank reports of the complete genome, indexing whether positions in the genome were genic or intergenic.
- It was checked to determine whether new hits to genic regions appeared (compared to the initial BLAST search using the PICK70 output). If this was not case, the oligonucleotide sequences were selected for capture probe synthesis.
- Design of the LNA-Modified Capture Probes
- For the LNA-modified oligonucleotide capture probes, every fourth DNA nucleotide was substituted with an LNA nucleotide, as shown in Table 3. The oligonucleotides were synthesized with an anthraquinone (AQ) moiety at the 5′-end of each oligonucleotide (e.g., as described in allowed U.S. Ser. No. 09/611,833), followed by a hexaethyleneglycol tetramer linker region and the LNA/DNA mixmer capture oligonucleotide sequence.
TABLE 3 C. elegans let-2 gene/ exons probes (SEQ ID NOs:) Sequence (LNA = uppercase, Capture probe DNA = lowercase letters) CE42.08-0HEG4 GgctGgatmCcccAggaAaccmCaggAatcGgaaGcatTggamCcaaAaggAg CE42.09-0HEG4 mCaccGgatmCcggmCtcaAttgTcggAcctmCgcgGaaamCcctGgagAaaaGg CE42.10-0HEG4 TccgmCcagGcccAatcGcctmCcacmCatgTccaAgggAaccAttaTcggTc CE42.11-0HEG4 GagcmCaggAgagGgagGtcaAcgcGgttAcccAggaAatgGaggActcTc - Strains and Growth Conditions
- C. elegans wild-type strain (Bristol-N2) was maintained on nematode growth medium (NG) plates seeded with Escherichia coli strain OP50 at 20° C., and the eggs and L1 larvae were prepared as described in Hope, I. A. (ed.) “C. elegans —A Practical Approach “, Oxford University Press 1999. The samples were immediately flash frozen in liquid N2 and stored at −80° C. until RNA isolation.
- Isolation of Total RNA
- A 100 1 aliquot of packed C. elegans worms from a L1 larvae population was homogenized using the FastPrep Bio101 from Kem-En-Tec for 1 minute at
speed 6 followed by isolation of total RNA from the extracts using the FastPrep Bio101 kit (Kem-En-Tec) according to the manufacturer's instructions. A 50 μl aliquot of packed C. elegans eggs was homogenized in lysis buffer (RNeasy total RNA purification kit, QIAGEN) containing quartz sand for 3 minutes using a Pellet Pestle Motor followed by isolation of total RNA according to the manufacturer's manual. - The eluted total RNA from worms (L1 larvae) as well as eggs was ethanol precipitated for 24 hours at −20° C. by addition of 2.5 volumes of 96% EtOH and 0.1 volume of 3M Na-acetate, pH 5.2 (Ambion, USA), followed by centrifugation of the total RNA sample for 30 minutes at 13200 rpm. The total RNA pellet was air-dried and redissolved in 6 μl (worms) or 2.5 μl (eggs) of diethylpyrocarbonate (DEPC)-treated water (Ambion, USA) and stored at −80° C. Reverse transcription (RT)-PCR
- Total RNA (1.5 μg) from eggs or 1 μg total RNA from worms (L1 larvae) were mixed with 5 μg oligo(dT)1 2-18 primer (Amersham Pharmacia Biotech, USA) and 0.5 μg of random hexamers, pd(N) 6 (Amersham Pharmacia Biotech, USA) and DEPC-treated water to a final volume of 7 μl. The mixture was heated at 70° C. for 10 minutes, quenched on ice for 5 minutes, followed by addition of 20 units of Superasin RNase inhibitor (Ambion, USA), 4 μl of 5×Superscript buffer (Life Technologies, USA), 2 μl of 100 mM DTT, 1 μlof dNTP solution (20 mM each dATP, dGTP, dTTP and dCTP, Amersham Pharmacia Biotech, USA), and 3 μl of DEPC-treated water.
- The primers were pre-annealed at 37° C. for 5 minutes, followed by addition of 400 units of Superscipt II reverse transcriptase (Invitrogen, USA). First strand cDNA synthesis was carried out at 37° C. for 30 minutes, followed by 2 hours at 42° C., and the reaction was stopped by incubation at 70° C. for 5 minutes, followed by incubation on ice for 5 minutes.
- Unincorporated dNTPs were removed by gel filtration using MicroSpin S-400 HR columns as described below. The column was pre-spun for 1 minute at 735×g in a 1.5 ml tube, and the column was placed in a new 1.5 ml tube. The cDNA sample was slowly applied to the top center of the resin and spun at 735×g for 2 minutes. The eluate was collected. The volume of the eluate was adjusted to 50 μl with TE-buffer pH 7.0 before being used as the template for linear PCR. Four μl template (RT from eggs or worms) was combined with 1 μl dNTP solution (10 mM each dATP, dGTP, dTTP and dCTP, Amersham Phamacia Biotech, USA), 1 μl of each primer (20 μM CE42.07 sense: gatcgaattcctccaggagagaagggagatg (SEQ ID NO:)), and CE42.12 antisense: 5′gatcaagcttatctcttcctgggtatccagctt (SEQ ID NO:)), 5
μl 10×AmpliTaq Gold Polymerase buffer, 5 μl 25 mM MgCl2, 0.5 μl AmpliTaq Gold DNA polymerase (5μl, Applied Biosystems), 2 μl Cy3-dCTP (Amersham Phamacia Biotech, USA) (eggs) or 2 μl Cy5-dCTP (Amersham Pharmacia Biotech, USA) (worms), and 31.5 μl DEPC-treated water to a final volume of 50 μl. The PCR reactions were carried out using the following program: 95° C. for 5 minutes followed by 30 cycles of PCR using the following cycling program (denaturation at 95° C. for 45 seconds, annealing at 60° C. for 30 seconds, and extension at 72° C. for 1 minute) followed by a final extension step at 72° C. for 10 minutes and incubation on ice for 5 minutes. - Purification of the PCR amplicons from eggs as well as worms was performed using a Qiaquick PCR purification kit (QIAGEN) according to the manufacturer's instructions.
- Fluorochrome-Labeling of the let-2 cDNA Fragments Using Primer Extension
- Four (4) μl template (RT from eggs or worms) was combined with 1 μl dNTP solution (10 mM each dATP, dGTP, dTTP and dCTP, Amersham Phamacia Biotech, USA), 1 μl of each primer (20μM CE42.12
antisense 5′gatcaagcttatctcttcctgggtatccagctt (SEQ ID NO:)), 5μl 10×AmpliTaq Gold Polymerase buffer, 5 μl 25 mM MgCl2, 0.5 μl AmpliTaq Gold DNA polymerase (5U/μl, Applied Biosystems), 2 μl Cy3-dCTP (Amersham Phamacia Biotech, USA) (eggs) or 2 μl Cy5-dCTP (Amersham Phamacia Biotech, USA) (worms), and 31.5 μl DEPC-treated water to a final volume of 50 μl. The PCR reactions were carried out using the following program: 95° C. for 5 minutes followed by 30 cycles of PCR using the following cycling program (deriaturation at 95° C. for 45 seconds annealing at 60° C. for 30 seconds extension at 72° C. for 1 minute) followed by a final extension step at 72° C. for 10 minutes and incubation on ice for 5 minutes. - Purification of the PCR amplicons from eggs as well as worms were performed using a Qiaquick PCR purification kit (QIAGEN) according to the manufacturer's instructions. Unincorporated dNTP nucleotides were removed by gel filtration using MicroSpin S-400 HRcolumns as described above before the eluted, fluorochrome-labelled DNA fragments were stored at −20° C. in the dark until microarray hybridization.
- Printing and Coupling of the C. elegans Let-2 Exon 8-11 Microarrays
- The C. elegans gene Let-2/exon 8-11 capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by a hexaethyleneglycol-4 (HEG4) linker (Table 3).
- The capture probes were first diluted to a 10 μM final concentration in 100 mM Na-phosphate buffer pH 7.0 and spotted on Euray COP microarray slides using the Biochip Arrayer One (Packard Biochip Technologies) with a spot volume of 300 μl and 300 μm between the spots.
- The capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker for 90 seconds at full power (Stratagene, USA). Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides for ½ hour in 30% acetone before rising in milli-Q H2O. After washing, the slides were centrifuged at 800 rpm for 2 minutes and stored in a slide box until microarray hybridization.
- Comparative Hybridization of the C. elegans microarrays and Post-Hybridization Washes
- The slides were hybridized with 2.5 μl of the Cy3-labelled and 2.5 μl of the Cy5-labelled target preparation from eggs and worms, respectively, as described above (see “Reverse transcription (RT)-PCR″ section) in 25 μl of hybridization solution, containing 25 mM HEPES, pH 7.0, 3×SSC, 0.3% SDS, and 25 μg of yeast tRNA. The target probe was filtered in a Millipore 0.22 micron spin column (Ultrafree-MC, Millipore, USA), denatured by incubation at 100° C. for 5 minutes, cooled at room temperature for 5 minutes, and then carefully applied onto the prepared microarray. One-third of a cover slip was laid over the microarray, and the hybridization was performed for 16-18 hours at 65° C. in a hybridization chamber (DieTech, model Joe deRisi, USA).
- Following hybridization, the slides were washed sequentially by plunging gently in 2×SSC/0.1% SDS at room temperature until the cover slip falls off into the washing solution, then in 1×SSC pH 7.0 (150 mM NaCl, 15 mM Sodium Citrate) at room temperature for 1 minute, then in 0.2×SSC, pH 7.0 (30 mM NaCl, 3 mM Sodium Citrate) at room temperature for 1 minute, and finally in 0.05×SSC (7.5 mM NaCl, 0.75 mM Sodium Citrate) for 5 seconds, followed by drying of the slides by spinning at 500 rpm for 5 minutes. The slides were stored in a slide box in the dark until scanning.
- Microarray Data Analysis
- The C. elegans let-2 gene microarray was scanned in an ArrayWoRx Scanner (Applied Precision, USA) using an exposure time of 5 seconds, resolution of 5.0, and high (high level) sensitivity. The hybridization data were analyzed using the ArrayVision image analysis software package 5.1 (IMAGING Research Inc., USA). The detection principle for alternative exon skipping the C. elegans let-2 gene is shown in FIG. 6. As demonstrated in FIG. 7, analysis of the comparative hybridization data from the C. elegans Let-2 exon 8-1 1 array demonstrates detection of alternative exon skipping of the let-2 exon 9 (eggs) and exon 10 (L1 larvae) using LNA-modified 50-mer capture probes. Capture probes of other lengths and/or with other LNA substitution patterns can be used similarly.
- Capture Probe Design: The design method of exon-specific capture probes for the C. elegans gene T01D3.3
exon 4 has been described in example 2. - Design of the LNA-modified Capture Probes: For the LNA-spiked oligonucleotide capture probes, every fourth DNA nucleotide was substituted with an LNA nucleotide, as shown in Table 4.
TABLE 4 C. elegans gene T01D3.3/exon 4-specific capture probes. (SEQ ID NOs:) Capture probes Sequence (LNA = uppercase, DNA = lowercase letters) CEgene26.04-70 ggctggaacagaagtttgttggtgcgtgacaaggtatggaagaagattatccggaaaagaaagcaa agac CEgene26.04-50 ggctggaacagaagtttgttggtgcgtgacaaggtatggaagaagattat CEgene26.04-40 ggctggaacagaagtttgttggtgcgtgacaaggtatgga CEgene26.04-30 gaacagaagtttgttggtgcgtgacaaggt CEgene26.04-50HEG2 GgctGgaamCagaAgttTgttGgtgmCgtgAcaaGgtaTggaAgaaGattAt CEgene26.04-50HEG4 GgctGgaamCagaAgttTgttGgtgmCgtgAcaaGgtaTggaAgaaGattAt CEgene26.04-40HEG2 GgctGgaamCagaAgttTgttGgtgmCgtgAcaaGgtaTgga CEgene26.04-40HEG4 GgctGgaamCagaAgttTgttGgtgmCgtgAcaaGgtaTgga CEgene26.04-30HEG2 GaacAgaaGtttGttgGtgcGtgamCaagGt CEgene26.04-30HEG4 GaacAgaaGtttGttgGtgcGtgamCaagGt - Cultivation of Caenorhabditis elegans Worms
- Mixed stage C. elegans cultures were grown according to standard methods. Samples were harvested by centrifugation at 3000×g, suspended in RNA Later (Ambion, USA), and immediately frozen in liquid nitrogen.
- mRNA Isolation from C. elegans Mixed Stages Worms
- Poly(A) + RNA was isolated from the worm samples using the Pick-Pen (Bio-Nobile, Finland) Starter kit combined with the KingFisher mRNA purification kit (Thermol.absystems, Finland) according to the manufacturer's instructions. The yield was 1-2 μg poly(A)+ RNA from approximately 50 mg of C. elegans worms.
- Synthesis Of Fluorochrome Labelled First Strand cDNA from C. elegans mRNA
- One μg of C. elegans poly(A)++ RNA was combined with 2 μg oligo dT primer (T20VN) in an RNase free, pre-siliconized 1.5 mL tube, and the final volume was adjusted with DEPC-water to 8 μL. The reaction mixture was heated at +70° C. for 10 minutes, quenched on
ice 5 minutes, spun for 20 seconds, followed by addition of 1 μL SUPERase-In™(20U/μL, RNAse inhibitor, Ambion, USA), 4 μL 5xRTase buffer (Invitrogen, USA), 2 μL 0.1 M DTT (Invitrogen, USA), 1 μL dNTP (20 mM dATP, dGTP, dTTP; 4 mM dCTP in DEPC-water, Amersham Pharmacia Biotech, USA), and 3 μL Cy3™-dCTP (Amersham Pharmacia Biotech, USA). First strand cDNA synthesis was carried out by adding 1 μL of Superscript™ II (Invitrogen, 200 U/mL), mixing, and incubating the reaction mixture for one hour at 42° C. An additional 1 μL of Superscript™ II was added and the cDNA synthesis reaction mixture was incubated for an additional one hour at 42° C.; the reaction was stopped by heating at 70° C. for 5 minutes, and quenching on ice for 2 minutes. The RNA was hydrolyzed by adding 3 μL of 0.5 M NaOH and incubating at 70° C. for 15 minutes. The samples were neutralized by adding 3 μL of 0.5 M HCl and purified by adding 450μL 1×TE buffer, pH 7.5 to the neutralized sample and transferring the samples onto a Microcon-30 concentrator. The samples were centrifuged at 14000×g in a microcentrifuge for ˜8 minutes, the flow-through was discarded, and the washing step was repeated twice by refilling the filter with 450μl 1×TE buffer and by spinning for ˜12 minutes. Centrifugation was continued until the volume was reduced to 5 μL, and finally the labelled cDNA probe was eluted by inverting the Microcon-30 tube and spinning at 1000×g for 3 minutes. - Printing and Coupling of the C. elegans Microarrays
- The C. elegans gene T01D3.3/
exon 4 capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by either a hexaethyleneglycol-2 or a hexaethyleneglycol-4 (HEG2/HEG4) linker (Table 4). The capture probes were first diluted to a 10 μM final concentration in 100 mM Na-phosphate buffer pH 7.0, followed by a two-fold dilution series (10 μM, 5 μM, 2.5 uM, 1.25 μM, 0.625 μM, 0.31 μM, and 0.155 μM) and spotted on Exiqon's polycarbonate microarray slides using the Biochip Arrayer One (Packard Biochip Technologies, USA) with a spot volume of 3×300 pl and 400 μm between the spots. The capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker for 90 seconds at full power (Stratagene, USA). Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides for 24 hours in milli-Q H2O. After washing, the slides were dried in an oven at 37° C. for 30 minutes and stored in a slide box until microarray hybridization. - Hybridization with Cy3-Labelled cDNA
- The arrays were hybridized overnight using the following protocol. The Cy3™-labelled cDNA sample was combined with 3
1L 21×SSC (3×SSC final), 0.5 μL 1 M HEPES, pH 7.0 (25 mM final), 25 μg yeast tRNA (1.25 μg/mL final), 10 μg PolyA blocker (0.5 μg/μL final), 0.6 μL 10% SDS (0.3% final), and DEPC-treated water to 20 μL final volume. The labelled cDNA target sample was filtered in a Millipore 0.22 micron spin column according to the manufacturer's instructions (Millipore, USA), and the probe was denatured by incubating the reaction at 100° C. for 2 minutes. The sample was cooled at 20-25° C. for 5 minutes by spinning at maxium speed in a microcentrifuge, and then carefully applied on top of the microarray. A cover slip was laid over the microarray and the hybridization was performed for 16 hours at 63° C. in a hybridization chamber (Coming, USA) submerged in a water bath, with an aliquot of 30 μL of 3×SSC added to both ends of the hybridization chamber to prevent evaporation. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol. The coverslip was washed off in 2×SSC, pH 7.0 containing 0.1% SDS at room temperature for one minute, followed by washing of the microarrays subsequently in 1.0 x SSC, pH 7.0 at room temperature for one minute, and then in 0.2×SSC, pH 7.0 at room temperature for one minute. Finally, the slides were washed for 5 seconds in 0.05×SSC, pH 7.0. The slides were then dried by centrifugation in a swinging bucket rotor at approximately 600 rpm for 5 minutes. - Microarray Data Analysis
- The C. elegans gene TOl D3.3
exon 4 array was scanned in an ArrayWoRx Scanner (Applied Precision, USA) using an exposure time of 5 seconds, resolution of 5.0, and high (high level) sensitivity. The hybridization data were analyzed using the ArrayVision image analysis software package 5.1 (IMAGING Research Inc., USA). As shown in FIGS. 8A and 8B, analysis of the hybridization data from the C. elegans gene 26/T01D3.3exon 4 array demonstrates that the use of LNA-modified capture probes for the C. elegans T10D3.3 exon results in 5-fold increased sensitivity inexon 4 capture compared to the corresponding DNA oligonucleotide capture probe controls printed on the same microarray. Capture probes of other lengths and/or with other LNA substitution patterns can be used similarly. - Capture probe design: Exon-specific capture probes for the C. elegans gene T01D3.3
exons - Design of the LNA-modified capture probes: For the LNA-spiked oligonucleotide capture probes, every fourth DNA nucleotide was substituted with an LNA nucleotide, as shown in Table 5: C. elegans gene TOID3.3/
exons 4 and 5-specific capture probes and synthetic target oligonucleotides.TABLE 5 (SEQ ID NOs:) Sequence (LNA = uppercase, DNA = lowercase letters) Capture probes CEgene26.04-70 ggctggaacagaagtttgttggtgcgtgacaaggtatggaagaagattatccggaaaagaa agcaaagac CEgene26.05-70 tatgtggcgcgaatgagcaatattcagcatgtttctcctcttgtcaaccatcatgtcaagatcctt caac CEgene26.04-50 ggctggaacagaagtttgttggtgcgtgacaaggtatggaagaagattat CEgene26.05-50 tatgtggcgcgaatgagcaatattcagcatgtttctcctcttgtcaacca CEgene26.04-40 ggctggaacagaagtttgttggtgcgtgacaaggtatgga CEgene26.05-40 tatgtggcgcgaatgagcaatattcagcatgtttctcctc CEgene26.04-30 gaacagaagtttgttggtgcgtgacaaggt CEgene26.05-30 tatgtggcgcgaatgagcaatattcagcat CEgene26.04-50HEG2 GgctGgaamCagaAgttTgttGgtgmCgtgAcaaGgtaTggaAgaaGattAt CEgene26.04-50HEG4 GgctGgaamCagaAgttTgttGgtgmCgtgAcaaGgtaTggaAgaaGattAt CEgene26.05-50HEG2 TatgTggcGcgaAtgaGcaaTattmCagcAtgtTtctmCctcTtgtmCaacmCa CEgene26.05-50HEG4 TatgTggcGcgaAtgaGcaaTattmCagcAtgtTtctmCctcTtgtmCaacmCa CEgene26.04-40HEG2 GgctGgaamCagaAgttTgttGgtgmCgtgAcaaGgtaTgga CEgene26.04-40HEG4 GgctGgaamCagaAgttTgttGgtgmCgtgAcaaGgtaTgga CEgene26.05-40HEG2 TatgTggcGcgaAtgaGcaaTattmCagcAtgtTtctmCctc CEgene26.05-40HEG4 TatgTggcGcgaAtgaGcaaTattmCagcAtgtTtctmCctc CEgene26.04-30HEG2 GaacAgaaGtttGttgGtgcGtgamCaagGt CEgene26.04-30HEG4 GaacAgaaGtttGttgGtgcGtgamCaagGt CEgene26.05-30HEG2 TatgTggcGcgaAtgaGcaaTattmCagcAt CEgene26.05-30HEG4 TatgTggcGcgaAtgaGcaaTattmCagcAt Target oligos CEgene26.04-biotarget accttgtcacgcaccaacaaacttctgttc CEgene26.05-biotarget atgctgaatattgctcattcgcgccacata - Printing and Coupling of the C. elegans Gene T01D3.3/exon 4-5 Microarrays
- The C. elegans gene T01D3.3/exon 4-5 capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by either a hexaethyleneglycol-2 or a hexaethyleneglycol-4 (HEG2/HEG4) linker (Table 5). The capture probes were first diluted to a 10 μM final concentration in 100 mM Na-phosphate buffer pH 7.0, followed by a two-fold dilution series (10 μM, 5 μM, 2.5 μM, 1.25 μM, 0.625 1M, 0.31, μM, and 0.155 μM) and spotted on Euray polycarbonate microarray slides using the Biochip Arrayer One (Packard Biochip Technologies) with a spot volume of 3×300 pl and 400 μm between the spots. The capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker for 90 seconds at full power (Stratagene, USA). Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides for 24 hours in milli-Q H2O. After washing, the slides were dried in an oven at 37° C. for 30 minutes, and stored in a slide box until microarray hybridization.
- Hybridization of the C. elegans microarrays and post-hybridization washes
- The slides were hybridized with a high (saturated) concentration of 1 μM of each gene T10D3.3,
exon - Following hybridization, the slides were washed sequentially by plunging gently in 1×SSCT (150 mM NaCl, 15 mM Sodium Citrate+Tween 20) at room temperature for one minute, then in 0.2×SSCT (30 mM NaCl, 3 mM Sodium Citrate+Tween 20) at room temperature for one minute, and finally in Milli Q water, followed by drying of the slides in an oven at 37° C. for 30 minutes. The slides were Cy5 labelled using a Cy5-straptavidin target. Thirty μl of a Cy5-streptavidin (2 μg/ml in 1×SSCT) were carefully applied onto the hybridized microarray and incubated one hour at room temperature before an additional washing step were performed in 1×SSCT (150 mM NaCl, 15 mM Sodium Citrate+Tween 20) at room temperature for one minute, then in 0.2×SSCT (30 mM NaCl, 3 mM Sodium Citrate+Tween 20) at room temperature for one minute, and finally in Milli Q water. Following washing, the slides were drying in an oven at 37° C. for 30 minutes and stored in a slide box in the dark until scanning.
- Microarray data analysis
- The C. elegans gene T01D3.3 exon 4-5 microarray was scanned in an ArrayWoRx Scanner (Applied Precision, USA) using an exposure time of 5 seconds, resolution of 5.0, and high (high level) sensitivity. The hybridization data were analyzed using the ArrayVision image analysis software package 5.1 (IMAGING Research Inc., USA). As shown in FIGS. 10A and 10B, analysis of the hybridization data from the C. elegans gene 26/TOI D3.3 array demonstrates that both the DNA as well as the LNA capture probes for the C. elegans TOID3.3 exons 4 (FIG. 10A) and exon 5 (FIG. 10B), respectively are highly specific with a very low level of cross-hybridization between their respective target oligonucleotides. The exon-specific design of the oligonucleotide capture probes is thus validated. Capture probes of other lengths and/or with other LNA substitution patterns can be used similarly.
- Oligonucleotide Design for Microarrays.
- Methods for designing exon-specific internal oligonucleotide capture probes has been described in Example 2.
- Design of the LNA-Modified Capture Probes
- For the LNA-modified oligonucleotide capture probes, every third DNA nucleotide was substituted with an LNA nucleotide. The probes designed to capture the junction of the recombinant splice variants were designed with LNA modifications in a block of five consecutive LNAs nucleotides, two on the 5′ side of the splice junction and three on the 3′ side of the splice junction. All capture probes are shown in Table 6.
TABLE 6 Internal, exon-specific and merged, exon—exon junction specific oligonucleotide capture probes. (SEQ ID NOs:) Sequence (LNA = uppercase, Capture probes DNA lowercase letters) gene78.01a cctgaaagtagatttgttatttccgaaacgccttctcccgttcttaagtc gene78.01b catataccacaaatagtccctcaaaaatcacaagaaaactcacaacactg gene78.03a gatttgcagcggtggtaaaaagtatgaaaacgtggtaattaaaaggtctc gene78.03b ccaatgaaaactaatcaaaggtaaacgtggatcccatggcaattcccggg gene78.m01INS3 caacactgcccagaggttcaatcgatccgatgatcctaatgaaggcgccc gene78.mINS303 gtccagtatcgtccatcatagtatcgataaatatgtgaaggaaatgcctg gene78.m01INS4 caacactgcccagaggttcaatcgatgtgtgataggatcagtgttcaggg gene78.mINS403 gaaggcgaaggagactgctaatatcgataaatatgtgaaggaaatgcctg gene78.01a_50_LNA3 mCctGaaAgtAgaTttGttAttlccGaaAcgmCctTctmCccGttmCttAagTc gene78.01b_50_LNA3 mCatAtamCcamCaaAtaGtcmCctmCaaAaaTcamCaaGaaAacTcamCaamCacTg gene78.03a_50_LNA3 GatTtgmCagmCggTggTaaAaaGtaTgaAaamCgtGgtAatTaaAagGtcTc gene78.03b_50_LNA3 mCcaAtgAaaActAatmCaaAggTaaAcgTggAtcmCcaTggmCaaTtcmCcgGg gene78.m01INS3_50_block caacactgcccagaggttcaatcGATmCmCgatgatcctaatgaaggcgccc gene78.mINS303_50_block gtccagtatcgtccatcatAGTATcgataaatatgtgaaggaaatgcctg gene78.m01INS4_50_block caacactgcccagaggttcaatcGATGTgtgataggatcagtgttcaggg gene78.mINS403_50_block gaaggcgaaggagactgctAATATcgataaatatgtgaaggaaatgcctg - Printing and Coupling of the Splice Isoform-Specific Microarrays
- The splice variant capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by a hexaethyleneglycol-2 (HEG2) linker. The capture probes were first diluted to a 20 μM final concentration in 100 mM Na-phosphate buffer pH 7.0, and spotted on the Immobilizer polymer microarray slides (Exiqon, Denmark) using the Biochip Arrayer One (Packard Biochip Technologies, USA) with a spot volume of 2×300 pl and 300 μm between the spots. The capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker with 2300 μjoules (Stratagene, USA). Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides two
times 15 minutes in 1×SSC. After washing, the slides were dried by centrifugation at 1000×g for 2 minutes, and stored in a slide box until microarray hybridization. - Construction of Splice Variant Vectors
- The recombinant splice variant constructs were cloned into the
Triampl 8 vector (Ambion, USA). The constructs were sequenced to confirm their construction. The plasmid clones were transformed into E. coli XLI O-Gold (Stratagene, USA). Triamp18/SWI5 vector construct - Genomic DNA was prepared from a wild-type standard laboratory strain of Saccharomyces cerevisiae using the Nucleon MiY DNA extraction kit (Amersham Biosciences, USA) according to the supplier's instructions. Amplification of the partial yeast gene was performed using standard PCR with yeast genomic DNA as the template. In the first step of amplification, a forward primer containing a restriction enzyme site and a reverse primer containing a universal linker sequence were used. In this step, 20 base-pairs were added to the 3′-end of the amplicon, next to the stop codon. In the second step of amplification, the reverse primer was exchanged with a nested primer containing a poly-T20 tail and a restriction enzyme site. The SWI5 amplicon contains 730 bp of the SWI5 ORF plus a 20 bp universal linker sequence and a poly-A20 tail. The PCR primers used were YDR146C-For-EcoRI (acgtgaattcaaatacagacaatgaaggagatga) (SEQ ID NO:), YDR146C-Rev-Uni (gatccccgggaattgccatgttacctttgattagttttcattggc (SEQ ID NO:)), and Uni-polyT-BamHI (acgtggatccttttttttttttttttttttgatccccgggaattgccatg (SEQ ID NO:)).
- The PCR amplicon was cleaved with the restriction enzymes EcoRI and BamHI. The DNA fragment was ligated into the
pTRIampl 8 vector (Ambion, USA) using the Quick Ligation Kit (New England Biolabs, USA) according to the supplier's instructions and transformed into E. coli DH-5α(by standard methods. - Construction of the Recombinant Splice Variant #1 (Triamp18/swi5-rubisco)
- The Arabidopsis thaliana Rubisco small subunit ssu2b gene fragment (gil 7064721) was amplified from genomic DNA using primers named DJ305 (5′-ACTATGATGGACGATACTGGAC-3′ (SEQ ID NO:)) and DJ306 (5′-ATTGGATCGATCCGATGATCCTAATGAAGGC-3′ (SEQ ID NO:)), containing ClaI restriction site linkers. The purified PCR fragment was digested with ClaI and then cloned into the swi5 (gI:7839148) vector at the unique ClaI site (atcgat) giving each insert a flanking sequence from the original yeast SWI5 insert (named exon01 and
exon 03, FIG. 11). The product was inserted in the reverse orientation, so that the insert sequence is as follows:AtcgatCCGATGATCCTAATGAAGGCGCCCGGGTACTCCTTCTTGCATTCTTCAACTTCC (SEQ ID NO:) TTCAACACTTGAGCGGAGTCGGTGCATCCGAACAATGGAAGCTTCCACATTGTCCAG TATCGTCCATCATAGTatcgat. - Nucleotide sequence analysis revealed a difference between the sequence of A. thaliana rubisco expected from the GenBank database and that obtained from all sequenced constructs and PCR products.
Position 30 in the Rubisco insert is “C” rather than the expected “A.” This SNP was probably created by PCR. None of the oligonucleotide capture probes used in the example cover this region. The Rubisco sequence in Genbank is TCCTAATGAAGGCGCCA (SEQ ID NO:), and the sequence obtained from the plasmid contruct is TCCTAATGAAGGCGCCC (SEQ ID NO:). - Construction of the Recombinant Splice Variant # 2 (Triamp18/swi5-lea)
- The Arabidopsis thaliana Lea gene (gil 526423) was amplified from genomic DNA with primers named DJ307 (5′-GGAATTATCGATGTGTGATAGGATCAGTGTTCAG-3′ (SEQ ID NO:)), and DJ308 (5′-AATTGGATCGATATTAGCAGTCTCCTTCGCC-3′ (SEQ ID NO:)), including the ClaI linker sites as above. The PCR fragment was digested with ClaI cloned into the yeast SWI5 IVT construct as above at the unique ClaI site.
- The fragment was inserted in the forward orientation, resulting in the following insert sequence:
atcgatGTGTGATAGGTTCAGTGTTCAGGGCTGTCCAAGGAACGTATGAGCATGCGAGA (SEQ ID NO:) GACGCTGTAGTTGGAAAAACCCACGAAGCGGCTGAGTCTACCAAAGAAGGAGCTCA GATAGCTTCAGAGAAAGCGGTTGGAGCAAAGGACGCAACCGTCGAGAAAGCTAAG GAAACCGCTGATTATACTGCGGAGAAGGTGGGTGAGTATAAAGACTATACGGTTGA TAAAGCTAAAGAGGCTAAGGACACAACTGCAGAGAAGGCGAAGGAGACTGCTAATa tcgat. - In Vitro RNA PREPARATION from Splice Variant Vectors
- In vitro RNA from the splice variants were made using the MEGAscript™ high yield transcription kit according to the manufacturer's instructions (Ambion, USA). The yield of IVT RNA was quantified at a Nanodrop spectrophotometer (Nanodrop Technologies, USA, FIG. 11). Isolation of total RNA from C. elegans C. elegans wild-type strain (Bristol-N2) was maintained on nematode growth medium (NG) plates seeded with Escherichia coli strain OP50 at 20° C., and the mixed stages of the nematode were prepared as described by Hope (ed.) (“C. elegans —A Practical Approach”, Oxford University Press 1999). The samples were immediately flash frozen in liquid N2 and stored at −80° C. until RNA isolation.
- A 100 μl aliquot of packed C. elegans worms from a mixed stage population was homogenized using the FastPrep Bio101 from Kem-En-Tec for one minute,
speed 6 followed by isolation of total RNA from the extracts using the FastPrep BiolΦkit (Kem-En-Tec) according to the manufacturer's instructions. - The eluted total RNA was ethanol precipitated for 24 hours at −20° C. by addition of 2.5 volumes of 96% EtOH and 0.1 volume of 3M Na-acetate, pH 5.2 (Ambion, USA), followed by centrifugation of the total RNA sample for 30 minutes at 13200 rpm. The total RNA pellet was air-dried and redissolved in 10 μl of diethylpyrocarbonate (DEPC)-treated water (Ambion, USA) and stored at −80° C.
- Fluorochrome-Labelling of the Target
- Ten (10) μg total RNA from C. elegans and 1 ng of in vitro RNA from Splice variant #were combined with 5 μg anchored oligo(dT2O) primer and DEPC-treated water to a final volume of 8 μl. The mixture was heated at 70° C. for 10 minutes, quenched on ice for 5 minutes, followed by addition of 20 units of Superasin RNase inhibitor (Ambion, USA), 1 μl dNTP solution (10 mM each dATP, dGTP, dTTP and 0.4 mM dCTP, and 3 μl Cy5-dCTP, Amersham Biosciensces, USA), 4±5×RTase buffer (Invitrogen), 2 μl 0.1 mM DTT (Invitrogen), 400 units of Superscript II reverse transcriptase (Invitrogen, USA), and DEPC-treated water to 20 μl final volume.
- A parallel set-up was made with 10 μg total RNA from C. elegans and 1 ng of in vitro RNA from
Splice variant # 2, labelling with Cy3-dCTP. Both cDNA syntheses were carried out at 42° C. for 2 hours, and the reactions were stopped by incubation at 70° C. for 5 minutes, followed by incubation on ice for 5 minutes. - Unincorporated dNTPs were removed by gel filtration using MicroSpin S-400 HR columns as described below. The column was pre-spun for one minute at 1500×g in a 1.5 ml tube, and the column was placed in a new 1.5 ml tube. The cDNA sample was slowly applied to the top center of the resin and spun 1500-x g for 2 minutes. The eluate was collected. RNA was degraded by adding 3 μl of 0.5 M NaOH. The solution was mixed well and incubated at 70° C. for 15 minutes. The solution was neutralized by adding 3 μl of 0.5 M HCl and mixed well.
- Then, 450
μl 1×TE, pH 7.5 was added to the neutralized sample, and the sample was transferred onto a Microcon-30 concentrator (prior to use, 500μl 1×TE was spun through the column to remove residual glycerol). The samples were spun at 14000×g in a micro centrifuge for 12 minutes, and the volume was checked. Spinning was continued until the volume was reduced to μl. The labelled cDNA probe was eluted by inverting the Microcon-30 tube and spinning at 1000×g for 3 minutes. The Microcon filter was checked for proper elution. - Comparative Hybridization of the Splice Variant Microarrays and Post-hybridization washes
- The Cy3 and Cy5-labelled cDNA samples, respectively, were combined in one tube. The following was added: 3.75 μl 20×SSC (3×SSC final, pass through 0.22 μ filter prior to use to remove particulates), yeast tRNA (1 μg/μl final), 0.625 μl 1 M HEPES, pH 7.0 (25 mM final, pass through 0.22 μ filter prior to use to remove particulates), 0.75 μl 10% SDS (0.3% final), and DEPC-water to 25 μl final volume. The labelled cDNA target sample was filtered in Millipore 0.22 μl filter spin column (Ultrafree-MC, Millipore, USA) according to the manufacturer's instructions, followed by incubation of the reaction mixture at 100° C. for 2-5 minutes. The cDNA probes were cooled at room temperature for 2-5 minutes by spinning at maxium speed in a microcentrifuge. A LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on Immobilizerm MicroArray Slide, and the hybridization mixture was applied to the array from the side. An aliquot of 30 μL of 3×SSC was added to both ends of the hybridization chamber, and the Immobilizer™ MicroArray Slide was placed in the hybridization chamber (DieTech, USA). The chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol.
- The slides were washed sequentially by plunging gently in 2×SSC/0.1% SDS at room temperature until the cover slip falls of into the washing solution, then in 1×SSC pH 7.0 (150 mM NaCl, 15 mM Sodium Citrate) at room temperature for one minute, then in 0.2×SSC, pH 7.0 (30 mM NaCl, 3 mM Sodium Citrate) at room temperature for one minute, and finally in 0.05×SSC (7.5 mM NaCl, 0.75 mM Sodium Citrate) for 5 seconds, followed by drying of the slides by spinning at 1000×g for 2 minutes. The slides were stored in a slide box in the dark until scanning.
- Microarray data analysis
- The splice variant microarray was scanned in a ScanArray 4000XL confocal laser scanner (Packard Instruments, USA). The hybridization data were analyzed using the GenePix Pro 4.01 microarray analysis software (Axon, USA).
- In the data analysis, the experimental variation in the labelling efficiency between the two fluorescent dyes was normalized (scaled) as follows. The average signal intensities from the “exonI” and “exon3” internal capture probes (Table 6), were used to calculate normalization factor of 2.75. This factor was multiplied to the signal intensity values from the Cy-3 target.
- Analysis of the data from the specific detection of the two recombinant splice variants in a complex RNA pool demonstrates that the merged capture probes containing a LNA block have significantly higher signals and a very low level of cross-hybridization, compared to the DNA capture probes (FIGS. 12A and 1 2B). In addition, the specific detection of the two artificial
splice variants # 1 and #2 is validated with the results from LNA-modified oligonucleotide capture probes. Capture probes of other lengths and/or with other LNA substitution patterns can be used similarly. In contrast, the corresponding DNA oligonucleotide capture probes fail to detect splice variant #1 (FIG. 12B). - This example demonstrates the advantages of using LNA oligonucleotide microarrays in gene expression profiling experiments. Capture probes for the Saccharomyces cerevisiae gene SWI5 (YDR146C) were designed as 50-mer standard DNA and two different LNA-modified oligonucleotides with LNA substitutions at every second or every third nucleotide position, respectively, for comparison (Table 7). To assess the sensitivity of DNA versus LNA capture probes, hybridizations with different amounts of biotin-labelled antisense oligonucleotides in a 10-fold dilution series were performed.
- Design and Synthesis of the LNA Capture Probes
- To design capture probes, regions with unique mRNA sequence of the selected target genes were identified. Optimized 50-mer oligonucleotide sequences with respect to T m, self-complementarity, and secondary structure were selected. LNA modifications were incorporated to increase affinity and specificity. The biotin-labelled antisense DNA target oligonucleotide corresponds to the reverse complement sequence.
- Printing of the LNA Microarrays
- The microarrays were printed on Immobilizer™ MicroArray Slides (Exiqon, Denmark) using the Biochip One Arrayer from Packard Biochip technologies (Packard, USA). The arrays were printed with a spot volume of 2×300 pl of a 10 μM (final concentration) capture probe dilution. Four replicas of the capture probes were printed on each slide
- Hybridization with Biotin-labelled Antisense Oligonucleotide
- The arrays were hybridized overnight using the following protocol. The desired amount of biotin-labelled oligonucleotide was combined in one tube followed by addition of 3 μL 21×SSC (3×SSC final), 0.5 1L 1 M HEPES, pH 7.0 (25 mM final), 25 μg yeast tRNA (1.25 μg/μL final), 0.6 μL 10% SDS (0.3% final), and DEPC-treated water to 20 μL final volume. The biotin-labelled target sample was filtered in a Millipore 0.22 micron spin column according to the manufacturer's instructions (Millipore, USA), and the probe was denatured by incubating the reaction at 100° C. for 2 minutes. The sample was cooled at 20-25° C. for 5 minutes by spinning at maxium speed in a microcentrifuge. A LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on Immobilizer™ MicroArray Slide, and the hybridization mixture was applied to the array from the side. An aliquot of 30 μL of 3×SSC was added to both ends of the hybridization chamber, and the Immobilizer™ MicroArray Slide was placed in the hybridization chamber. The chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol. The Lifterslip coverslip was washed off in 2×SSC, pH 7.0 containing 0.1% SDS at room temperature for 1 minute, followed by washing of the microarrays subsequently in 10.1×SSC, pH 7.0 at room temperature for 1 minute, and then in 0.2×SSC, pH 7.0 at room temperature for 1 minute. Finally, the slides were washed for 5 seconds in 0.05×SSC, pH 7.0. The slides were then dried by centrifugation in a swinging bucket rotor at approximately 200 G for 2 minutes. To visualize the biotin containing duplexes, an aliquot of 40 μL of the 2 μg/ml streptavidin-Cy3 in 1×SSC+0.05% Tween solution was applied to the slide as described for the hybridization mixture above. The slide was incubated in a humidified chamber for 1 hour at room temperature. The coverslip was washed off in 1×SSC+0.05% Tween for 1 minute, followed by wash in 0.2×SSC+0.05% Tween for 1 minute and then 10 seconds in MilliQ-water. The slide was dried by centrifugation in a swinging bucket rotor for 2 minutes at 200 G.
- Data Analysis
- Following washing and drying, the slides were scanned using a ScanArray 4000XL scanner (Perkin-Elmer Life Sciences, USA), and the array data were processed using the GenePix™ Pro 4.0 software package (Axon, USA).
- Results
- Incorporation of LNA nucleotides at every second or third nucleotide position in standard 50-mer expression array oligonucleotide capture probes results in a 2-7-fold increase in fluorescence intensity levels using an unsaturated target concentration and hybridizing under standard stringency conditions (FIG. 13). Thus, it can clearly be concluded that the LNA oligonucleotides are more sensitive in expression profiling compared to DNA oligonucleotides.
TABLE 7 DNA and LNA-modified SWI5 (YDR146C) oligonucleotide capture probes. LNA modifications are depicted by uppercase letters in the sequence; “mC” denotes LNA methyl cytosine. (SEQ ID NO:) Oligo Name Sequence YDR146C-50 tgggaatggaacggggattatggtttcgccaatgaaaactaatcaaaggt YDR146C-50_LNA2 TgGgAaTgGaAcGgGgAtTaTgGtTtmCgmCcAaTgAaAamCtAaTcAaAgGt YDR146C-50_LNA3 TggGaaTggAacGggGatTatGgtTtcGccAatGaaAacTaaTcaAagGt - This example demonstrates the advantages of using LNA oligonucleotide microarrays in Comparative Genome Hybridization (CGH) experiments. Capture probes for all 23 exons of the Menkes gene (A TP7A) were designed as 50-mer standard DNA and different LNA/DNA mixmer oligonucleotides, respectively, for comparison (FIG. 17). The C6-amino-linked capture probes were applied to Immobilizer slides and hybridized with patient DNA samples labelled with a Cy3 fluorescent dye.
- Design and Synthesis of the LNA Capture Probes
- To design the capture probes, regions comprising individual exons of the Menkes gene were identified. The optimal 50-mer oligonucleotide sequences with respect to T m, self-complementarity, and secondary structure were selected for each exon. LNA modifications were incorporated to increase affinity and specificity. A software tool “OligoDesign”, which automatically designs capture probes that are optimized for sequence specificity, Tm, self-complementarity, secondary structure, and LNA modifications was used for oligonucleotide design.
- Results
- Fluorescent Cy3 labelled patient genomic DNA was hybridized to microarrays spotted with the CGH capture probes listed in FIG. 17. Compared to DNA capture probes, capture probes with LNA in every second position (LNA-2) had a significantly better capture rate of non-amplified labelled genomic patient DNA as shown in FIGS. 14-16. Capture probes of other lengths and/or with other LNA substitution patterns can be used similarly.
- This example demonstrates the use of the C. elegans LNA tox oligoarray in gene expression profiling experiments in the nematode Caenorhabditis elegans. The C. elegans tox oligoarray monitors the expression of a selection of 1 10 genes relevant for general stress response and for the metabolism of toxic compounds. Two different capture probes for each of these target genes were designed and included in the LNA tox array. In addition, the C. elegans LNA tox oligoarray contained capture probes providing control for cDNA synthesis efficiency and the developmental stage of the nematode. Capture probes for constitutively expressed genes for data set normalization were also included on the C. elegans LNA tox oligoarray.
- Cultivation of C. elegans Worms
- For all cultures, the sample was divided into two, and one half of the sample was used as the control, the other was used as the treated sample. Worm samples were harvested and sucrose cleaned by standard methods. For heat shock treatment, the heat shock sample was added to S-media preheated to 33° C. in a 1 L flask suspended in a water bath at 33° C., the other sample was added to a 1 L flask with S-media at 25° C. Both samples were shaken at approximately 100 rpm for an hour. For Lansoprazole treatment, 0.5 mL of 10 mg/mL Lansoprazole (Sigma) in DMSO was added to each 500 mL volume of S-media culture after 28 hours of growth from LI. At the same time, 0.5 mL of DMSO was added to the control. Incubation was for 24 hours. Samples were then harvested by centrifugation at 3000×g suspended in RNALater™(Ambion) and immediately frozen in liquid nitrogen.
- RNA Extraction
- RNA was extracted from the worrn samples using the FastRNA® Kit, GREEN (Q-BIO) essentially according to the suppliers' instructions.
- Design and Synthesis of the LNA Capture Probes
- To design the capture probes, regions with unique mRNA sequence of the selected target genes were identified. The optimal 50-mer oligonucleotide sequences with respect to T m, self-complementarity, and secondary structure were selected. LNA modifications were incorporated to increase affinity and specificity.
- Printing of the LNA Microarrays
- The microarrays were printed on ImmobilizerM MicroArray Slides (Exiqon, Denmark) using the Biochip One Arrayer from Packard Biochip technologies (Packard, USA). The arrays were printed with a spot volume of 2×300 pl of a 10 1M capture probe solution. Four replicas of the capture probes were printed on each slide.
- Synthesis of Fluorochrome Labelled First Strand cDNA from Total RNA
- 15 μg of C. elegans total RNA was combined with 5 μl oligo dT primer (T20VN) in an RNase free, pre-siliconized 1.5 mL tube, and the final volume was adjusted with DEPC-water to 8 μL. The reaction mixture was heated at +70° C. for 10 minutes, quenched on
ice 5 minutes, spin 20 seconds, followed by addition of 1 μL SUPERase-In™ (20U/μL, Ambion, USA), 4 μL 5xRTase buffer (Invitrogen, USA), 2 μL 0.1 M DTT (Invitrogen, USA), 1 μL dNTP (20 mM dATP, dGTP, dTTP; 0.4 mM dCTP in DEPC-water, Amersham Pharmacia Biotech, USA), and 3 μL Cy3™-dCTP or Cy5™-dCTP (Amersham Pharmacia Biotech, USA). First strand cDNA synthesis was carried out by adding μL of Superscript™ II (Invitrogen, 200 U/mL), mixing, and incubating the reaction mixture for 1 hour at 42° C. An additional 1 μL of Superscript™ II was added, and the cDNA synthesis reaction mixture was incubated for an additional 1 hour at 42° C.; the reaction was stopped by heating at 70° C. for 5 minutes, and quenching on ice for 2 minutes. The RNA was hydrolyzed by adding 3 μL of 0.5 M NaOH, and incubating at 70° C. for 15 minutes. The samples were neutralized by adding 3 μL of 0.5 M HCl, and purified by adding 450μL 1×TE buffer, pH 7.5 to the neutralized sample and transferring the samples onto a Microcon-30 concentrator. The samples were centrifuged at 14000×g in a microcentrifuge for 8 minutes, the flow-through was discarded, and the washing step was repeated twice by refilling the filter with 450μl 1×TE buffer and by spinning for ˜12 minutes. Centrifugation was continued until the volume was reduced to 5 μL, and finally the labelled cDNA probe was eluted by inverting the Microcon-30 tube and spinning at 1 000xg for 3 minutes. - Hybridization with Fluorochrome-Labelled cDNA
- The arrays were hybridized overnight using the following protocol. The Cy3™ and Cy5™-labelled cDNA samples were combined in one tube followed by addition of 3 μL 21×SSC (3×SSC final), 0.5 μL 1 M HEPES, pH 7.0 (25 mM final), 25 μg yeast tRNA (1.25 μg/μL final), 0.6 μL 10% SDS (0.3% final), and DEPC-treated water to 20 μL final volume. The labelled cDNA target sample was filtered in a Millipore 0.22 micron spin column according to the manufacturer's instructions (Millipore, USA), and the probe was denatured by incubating the reaction at 100° C. for 2 minutes. The sample was cooled at 20-25° C. for 5 minutes by spinning at maximum speed in a microcentrifuge. A LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on Immobilizer™ MicroArray Slide, and the hybridization mixture was applied to the array from the side. An aliquot of 30 μL of 3×SSC was added to both ends of the hybridization chamber, and the Immobilizer™ MicroArray Slide was placed in the hybridization chamber. The chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridisation, the slide was removed carefully from the hybridization chamber and washed using the following protocol. The Lifterslip coverslip was washed off in 2×S SC, pH 7.0 containing 0.1% SDS at room temperature for 1 minute, followed by washing of the microarrays subsequently in 10.1×SSC, pH 7.0 at room temperature for 1 minute, and then in 0.2×SSC, pH 7.0 at room temperature for 1 minute. Finally, the slides were washed for 5 seconds in 0.05×SSC, pH 7.0. The slides were then dried by centrifugation in a swinging bucket rotor at approximately 200 G for 2 minutes. The slide was then ready for scanning.
- Data analysis.
- Following washing and drying, the slides were scanned using a ScanArray 4000XL scanner (Perkin-Elmer Life Sciences, USA), and the array data were processed using the GenePix™ Pro 4.0 software package (Axon, USA). The data in each image was normalized so that the ratio of means of all of the features is equal to 1.
- Results
- Use of LNA-modified oligonucleotide capture probes in the C. elegans LNA tox oligoarray clearly allows the identification of distinct expression profiles for C. elegans genes relevant for general stress response and for the metabolism of toxic compounds.
TABLE 12 Expression profiling using LNA Oligonucleotide Microarrays. Log2 transformed fold of changes for five selected genes in the two expression profiling experiments. Protein name (clone name) Heat shock Lansoprazole HSP70 (F44E5.4/5) 4.11 nd CYP37A (F01D5.9) nd 0.98 Ubiquitin (M7.1) 0.16 −0.12 Histone 1Q (C01B10.5) −1.49 nd HSP90 (C47E8.5) nd −1.17 -
TABLE 13 LNA-modified oligonucleotide capture probes. LNA modifications are depicted by uppercase letters in the sequence; “mC” denotes LNA methyl cytosine. (SEQ ID NOs:) Oligo Name Sequence CEABC_C34G6.4_u293_LNA3 TgcmCatTgcAcgGgcActTgtTcgAtcTccTtcTgtTttActTttGgaTg CEABC_C34G6.4_u375_LNA3 TcaTtcTagGatTgcmCagAtgGttAlgAtamCtcAtgTcgGagAgaAagGa CEABC_F57C12.4_u15_LNA3 mCcaAtgTtgTttAalTggTtgTaaTgtmdllGatGacmCtgmCatAatmCatAt CEABC_F57C12.4_u480_LNA3 mCacAagAtcmCtgTgtTgtTctmCcgGaamCaaTgaAaaTgaActTagAtcmCa CEABC_F57C12.5_u111_LNA3 TacTtgTtcTcgAcaAagGttGtgTagmCcgAgtTtgAcamCtcmCgaAgaAa CEABC_F57C12.5_u444_LNA3 TgaActTggAtcmCclTctTtgmCatTtaGcgAtgAtcAaaTttGggAagmCg CEABC_K08E7.9_d8_LNA3 TcaTtaAttTtgTgtAgcTttmCttTctmCgaTttTtgmCacGatmCttTccmCc CEABC_K08E7.9_u51_LNA3 AggGtgmCctActAcaAacTgamCccAaaAgcAgaTgamCcgAgaAgaAatAa CEABC_Y39D8C.1_u37_LNA3 AttGaaAgcGacGcgGaaAgtGccAtgTatTtcTaaTttTgtTttmCttTa CEABC_Y39D8C.1_u422_LNA3 TtgTcaGcaTatmCaaGagTagAtaTggAagTggAtamCacTctGctAatmCc CEADH_H24K24.3a_d3_LNA3 mCacmCttAttGcgTtcAatrttTgtTtcmCacmCtamCtamCtamCgaAtamCgtTg CEADH_H24K24.3a_u50_LNA3 TcamCaaGggAgaGagTctGcgGtcGgtGctGgcGttmCgaGaaAatAtaAc CEAPEX_R09B3.1_u191_LNA3 mCatGcaTccmCgamCgaGaaGaaGtamCtcAttTtgGagrtaTctGgcGaaTt CEAPEX_R09B3.1_u37_LNA3 GacmCatGctmCcgGtcGtcAtgmCaaAtcGacTtcTaaAttGctTctGatTa CEAPO_C35D10.9_u15_LNA3 TtgmCatGctGttAaaAccTatmCgtGtamCaaTatTgcmCtgTatAttmCccmCt CEAPO_C35D10.9_u609_LNA3 TggmCacAgcTtaAtaAcaAatTggAaaGtcGagGatTagTcgGtgTtgAa CEAPO_C48D1.2_u176_LNA3 GacAcamCgcAaaGgaTatGgaTgtTgtTgaGctGctGacTgaAgtmCaaTa CEAPO_C48D1.2_u23_LNA3 AgcAcgAaamCtcTgcmCgtmCtaAaaTtcActmCgtGatTcaTtgmCccAatTg CEAPO_F20C5.1_u453_LNA3 AtgGtcAtamCtcTaaAatGggmCagAacTtcAacmCaaAtcAttmCtcGtcAg CEAPO_F20C5.1_u96_LNA3 AacmCcgAgcTtgmCcgmCaaAgtGcaAgaAaaTtaTagAacGaaTgaAacAg CEATPase_B0365.3_u31_LNA3 GgaTggGtcGagmCgtGagAccTacTacTaaAgaAcaGctTgtGaaTctTt mCaamCgtTctmCgaTtcmCtamCggAcaAgaAtgGacmCtaTgcmCaamCagAaa CEATPase_B0365.3_u386_LNA3 Ga CEATPase_C17H12.14_u356_LNA3 TgcTcgTtaTccAgcTatTttGaaGggActTgtmCatGcaAggActTctTc CEATPase_C17H12.14_u89_LNA3 mCcgTttAgaGctTatTgcTaamCcaGatTgtmCccAcaAgtmCagAacAgcTc CEATPase_F55F3.3_u215_LNA3 TgamCggAcgmCtamCtamCccAtaTgtAttTgtTccAtcTtamCcaGcaAccAa CEATPase_F55F3.3_u275_LNA3 AgcTacTtcAttmCgamCaaGgaAcaTctmCggAaaAgtmCaaGtamCatmCccGg CEATPase_Y49A3A.2_u103_LNA3 AaaTtcAagGatmCcaGttGccGatGgtGaaGccAagAttmCgcAagGatTa CEATPase_Y49A3A.2_u272_LNA3 mCgaTcgTttmCtgmCccAttmCtamCaaGacTgtmCggTatGctmCaaGaaTatGa CECALR_Y38A10A.5_u238_LNA3 TcaGgaAcgAtcTttGacAacAttAtcAtcAccGacTctGttGagGagGc CECALR_Y38A10A.5_u296_LNA3 TgaActmCtamCtcTtaTgaAagmCtgGggAgcmCatmCggAttmCgaTttGtgGc CECAT_Y54G11A.5b_u137_LNA3 GaamCttTgcAggGccGctmCggGgaAtgTcaTgaTttmCatTatTaaGggAa CECAT_Y54G11A.5b_u189_LNA3 GtcAatTctGggAgaAggTgtTggAtamCcgGggmCtcGggAgaGaaTgtGc CECC_C03D6.3_u275_LNA3 AtgTaaAgaAggAatGctTccmCgaAtgGatTggAtaTttAttTgtmCcaGa CECC_C03D6.3_u430_LNA3 GgamCcgAaaTttGtgmCagmCatGtcGgamCacGaaAttGatGgtmCtcAttTt CECC_C07G2.3_d9_LNA3 mCagAcamCgaAggTtamCgaTagAtaAccAtcTctmCaaAgtmCtaTcgAccTc CECC_C07G2.3_u44_LNA3 mCgamCgaTgtGcgTgtTccTgamCgaTgaAagAatGggAtaTtaAgaAaamCc CECC_Y46G5A.2_u331_LNA3 TtgTgcTccAtcGctGctmCcgmCttAcaGacTtgAcaAcgmCtcAccTttGc CECC_Y46G5A.2_u385_LNA3 AatGagmCggTtgTgcmCgtGtgAcgTcamCttmCgtmCacAgtGttGctmCtamCt CECoA_C29F3.1_u316_LNA3 AaaTtgAcamCcaAtcAaaTctGtcTcaTctmCctGagGacmCgtmCaamCttmCg CECoA_C29F3.1_u392_LNA3 AatmCttTgtGtamCggAgaTggGgcAaaAggmCagmCaaGaaAgtAaamCcaAg CECoA_F08A8.4_u1094_LNA3 AggAcaAggGgcActActGgcAcaGgcTttGatTatTgcAgtGagAtaTt CECoA_F08A8.4_u1260_LNA3 TtaAtgGagGtgAcaAtgGgtTccTtgGatTcgAtaAatTccGagTgcmCc CECoA_F59F4.1_u109_LNA3 GctmCttmCtcmCagTggGctmCaaAatAgtmCaamCtcAacAgaTcgGaaGttmCt CECoA_F59F4.1_u424_LNA3 AaaGctTcgAgaTggmCacGttmCgtmCtgTatmCtcGtgAagAacTtaTtgmCa CECoA_Y25C1A.13_u115_LNA3 GatTcgmCtgAacTttAtcAagAcgTggAatAtgAgcmCagmCtcmCtgTcgAc CECoA_Y25C1A.13_u451_LNA3 GatmCttAtcAccGcgTgcGatAttmCgaGtaGctTcamCagGatGcgAttTt CECOL_C27H5.5_u493_LNA3 GgaAagGaaGgaTccAttmCtcAgcTctGcamCttmCcamCcaTcaGagmCcaTg CECOL_C27H5.5_u680_LNA3 TggAtamCaaGgaGggAtcTggmCagTggTggAtcTggAagTggTggAtaTg CECOQ_ZC395.2_u199_LNA3 TtgAaaGaamCtcmCttGccGacGatmCctGaaAcamCacAaaGaaTtgmCtgAa CECOQ_ZC395.2_u400_LNA3 AtgTggGatGagGagAaaGaamCatTtaGatAcaAtgGaaAgaTtaGctGc CECRYZ_F39B2.3_u171_LNA3 AggmCtgAgcTctTggActTtgGcaTcaAcaTtgTctmCatTctTgaAggAa CECRYZ_F39B2.3_u222_LNA3 TtaTggTtamCagAagGagmCtgTttAcgGtgTagmCatTggGaaTgtmCttmCc mCacTtcAacmCaamCtcmCgtGttAatmCaaGcaAgcmCgcmCacmCatmCtaAtg CECyclin_R02F2.1a_u24_LNA3 Ag CECyclin_R02F2.1a_u312_LNA3 TctmCatTgcTcgTcgAggmCtamCcaAcaAacActGgcAatAccmCaaTtaAt CECyclin_ZC168.4_u203_LNA3 TaaGaaAgtmCatTgaGgaTgcTgtmCgcTttGctmCgcmCgaAgtmCtcGtaTa CECyclin_ZC168.4_u273_LNA3 AagTtcAtcmCtgTtgAcgGaaTcgAggmCggAgaAtgmCtgTatmCggTcaTt CECYP_B0213.15_u133_LNA3 AcaGgaAatAtgAttTtgGatTtcGatTttGaaTcgGttGgtGctGccmCc CECYP_B0213.15_u202_LNA3 GctGagmCtgTatTtgGctAgtGaaAtgTgtGttTttGatActTtaAatGa CECYP_B0304.3_u38_LNA3 AcgAggTttGgaTcamCaaTcaGaaTtcTgtGaaAtaAgcGttTttTggGa CECYP_B0304.3_u89_LNA3 AgtTctmCggTctAacAgtGtcTccmCgtTgaAtaTtcTtgTaaAatmCacAc CECYP_C03G6.14_u706_LNA3 AtgAccActmCaaAatActGctAaaAgaTttGcaGcgGcaGaaGccGttAa CECYP_C03G6.14_u768_LNA3 TtgAtaTggmCtgTacmCtgTatGgtTttTgaGgamCgtTttTtaGgaGtcGa CECYP_C03G6.15_d9_LNA3 AttTatTcaTtcAtcmCatGtaAacTgtAtaTttTgaAttTgtGttGtaAa CECYP_C03G6.15_u148_LNA3 GccAaaGcaGaaTtgTatTtgAtcTtcGgtAacmCttmCtcmCttmCgcTacAa CECYP_C06B3.3_u102_LNA3 AttTtgAatmCttmCtgGgaAaaTgcmCatmCcamCtcGagAaamCcgTtcmCgtTt CECYP_C06B3.3_u474_LNA3 mCtaAcgGagGatmCtcGccAatTatmCttTgaGagAcaAaamCtgAaamCtcmCt CECYP_C12D5.7_u399_LNA3 AtcTagTccmCaaTgaAtcTccmCacAtgmCtgTtamCtcGtgAtgTtcAacTc CECYP_C12D5.7_u65_LNA3 TttTgcTttmCatmCgcAaaAgcTcaAgaTtamCacAtgTcaGgtmCaaGccAa CECYP_C45H4.17_u27_LNA3 mCcgmCgamCttTaaAgaGaaGatmCatAaaTttGcaTtgTttTttGttTgtAt CECYP_C45H4.17_u598_LNA3 mCgaGggTgaTtcGgaGacTttmCagTaaTgtmCcaActTtcAaaTgtTtgmCa CECYP_C45H4.2_u110_LNA3 TagAtamCaaGatAcaTccmCtcAaaAgaAggmCctAccGtcAatGgcmCaaAg CECYP_C45H4.2_u429_LNA3 TcaAcgmCgtmCtaTaaAtgAatmCacAacGagGtaTcaAcaTtcTccmCccTg CECYP_C49C8.4_u363_LNA3 AtgmCtgAtgTtgAaaTtgmCtgGctAccGtaTtcmCaaAagAtamCtgTaaTc CECYP_C49C8.4_u883_LNA3 AtgAatmCcaTggmCttGgamCatmCtcmCcgTttTtcAagGgaTatAaaAatGt CECYP_C49G7.8_d6_LNA3 AtgmCaamCgaAttAgtGaaAaaTtcAtcmCtgGaaTaaAaaAtaAttmCtaAa CECYP_C49G7.8_u795_LNA3 AtcGctAcgAcaAtcmmCcgAtgmCctTcgAagTttmCgaAagmCttTctmCt CECYP_F01D5.9_u374_LNA3 GagGtcGgtGgaGgaGgaAgtGgaAatTgamCggmCaaAatmCctGccmCaaGg CECYP_F01D5.9_u46_LNA3 mCccTctTtgGgaTttmCcamCtcAagTttActGttmCggmCagmCagTgaTatAa CECYP_F08F3.7_u25_LNA3 GagTtgGttmCcamCagAatGctTagGacGttTaaAttmCgtmCacAaamCttTt CECYP_F08F3.7_u401_LNA3 mCaaTatGgtTccmCatTttAgcAacTcaTatGaamCacAgaAgaTgtmCctTg CECYP_F14F7.2_u397_LNA3 GaaAaaGgcGtcGacAttTtaTgtGacAcgTggAcamCttmCacTatGacAa CECYP_F14F7.2_u68_LNA3 TaaTtgAatTacGggTctTttGtamCatAttAatTttAgtAtamCttTgtGa CECYP_F42A9.5_u435_LNA3 AtaTcaAtgmCaamCtaTtaAtgAatmCacAacGtcTtgmCcaAtcTtcTccmCg CECYP_F42A9.5_u55_LNA3 GgaGtgActAtgAaaGcaAagAgtTacmCgaTtgAaamCtgAaaGacAgamCa CECYP_K07C6.3_u3_LNA3 AatmCttTaaTgaTaaTttAtgGgaTctGtaTttmCtcTttmCtgTcaAtaAa CECYP_K07C6.3_u354_LNA3 AtgAgcmCcamCaaAtgTaaAagGatAcgAgaTtgAttmCggGaamCagTcaTg CECYP_K07C6.4_u118_LNA3 AtcmCtgmCgaTatGacAttAagmCcamCatGgtTctGaamCctTcaAcaGaaGa mCtgAacmCttmCaamCagAagAtaAacTtcmCgtAtaGcgmCtgGaaAaamCtcmC CECYP_K07C6.4_u87_LNA3 t CECYP_K07C6.5_u7_LNA3 AttTaaAggAatTcamCagmCtcAaaAaaTaaTaamCtamCcgGttmCagAgaTt CECYP_K07C6.5_u99_LNA3 AatTtgAgcmCacAtgGcaAgtTatmCaamCagAggAgamCaaTgcmCgtAcaGt CECYP_K09A11.3_u362_LNA3 TgamCatTctActTaaAggGaaGaaAatAccAacTggTacmCctTgtAttTg CECYP_K09A11.3_u48_LNA3 TcamCcamCaaAgcmCatAcaTatGcgAgcTagTtcmCtcAggmCtgmCttAaamCc CECYP_K09A11.4_u238_LNA3 TtcGacAaaActAttTtgGaaAgaAcaAtcmCcaTtcAgtGtcGgcAaamCg CECYP_K09A11.4_u68_LNA3 TctGacAacAaaGccAtamCacGtgmCcgActAatTccAcaAtcAgcTagAa CECYP_K09D9.2_u151_LNA3 TtgGcaAaaGcaGaaTtgTatTtaAtcTttGgaAacmCtcmCttmCttmCgcTa CECYP_K09D9.2_u866_LNA3 TgaAtcTttmCaaActTatmCacTccTttTaaTacTacmCgtTccTgtTtgGa CECYP_T10B9.10_u410_LNA AttGagAttGtaTccAttGgcGtcTctTgtTcamCaaTcgAaaAtgTctmCa CECYP_T10B9.10_u56_LNA AacTgcTacTatTgcGccAtcAagTgtGctGctmCaaActTaaAtcmCagGt CECYP_T10B9.7_u102_LNA3 TtgAgamCagGaaAtaAgamCtaGaaTtcmCttTgaAacTggTggGaaGtgmCt CECYP_T10B9.7_u267_LNA3 AagAtgTcaAagAatTcaAgcmCagAacGatGgtmCcamCcgAcgAgcmCatTa CECYP_T19B10.1_u100_LNA3 AttGaamCcaActmCtgAaaTatAatGacAcaAaamCcaTgtmCtgGaaGtgGt CECYP_T19B10.1_u319_LNA3 GgcAatGtgAcaAtaTctmCcaAtgGttmCttmCacAgcAatmCatmCacGtgTt CECYP_Y49C4A.9_u121_LNA3 mCtaTtcAatmCgaTatTttAtcAcamCcaTccAgtGctGgamCctmCcaTcaTt CECYP_Y49C4A.9_u413_LNA3 GtcTcaGagAtgTgtAaaTttActTccmCtgmCaaTttGttTcamCgcAacTa CECYP_ZK177.5_u394_LNA3 TtcmCgaAtgTttmCcaAttGggActGaaGttTcaAgaGtcAccmCagAaaAa CECYP_ZK177.5_u445_LNA3 GatmCcaGcaTctTccAagmCttAcaTtcmCtcmCgtGctTgtAtcAagGaaAc CEDAO_C47A10.5_d9_LNA3 TttGaaAacmCtgTttTatTatTaaAatAgaTaaTtgAttAgtTctGtamCg CEDAO_C47A10.5_u269_LNA3 AtamCgtTgcActGcaTccGgcTatGagGgaGccAaaAatmCttAggGgaGt CEDC_C01A2.3_u373_LNA3 GcamCttmCcaTtcAtcTctGcaGctActAtgGctTtgGtgAcaAaaGttGg CEDC_C01A2.3_u96_LNA4 mCcgTccAaaAgaAtgmCcaTctmCacAagTctTgaAatmCttAtaAagGtaGt CEDC_C34F6.1_u301_LNA3 GagGgaTcaAcaGtaAccTcgTgcGgtAttGacAagGgaTgtmCcgGaaGg CEDC_C34F6.1_u450_LNA3 GatGgtTctTcgAtcGcaAacAaaAcaGatGtgmCtcmCatTtamCatAcgGa CEDC_F33D11.3_u126_LNA3 AtgGagAaaAtgGatmCtgAtgGagTtgmCagGaaGtgAtgGagmCtcmCagGa CEDC_F33D11.3_u14_LNA3 TgaAtcTccAtaAatTatTcaAtgTttmCcaAatAttTaaTttAtcAatTg GctmCaamCacGgtAggAtcmCtaTggAacmCgtmCggAggAgcAggmCctmCggA CEDC_F46E10.2_u392_LNA3 g CEDC_F46E10.2_u54_LNA3 mCgtGacAacmCtcTtaTttAttTctGtaAaamCtgAttmCgcmCaaActTttGt CEDC_F56G4.2_u382_LNA3 GaaGctTtcAaamCcaAatGagTtcmCttmCccGgaAtcmCcaAagAatAccAa CEDC_F56G4.2_u82_LNA3 AcaAtgAaaAgaGagGatGgaAagGaaAtcGaaGtcTctGttmCttGacGa CEDC_M162.2_u103_LNA3 GatGagGtamCatAacTttGtgTgcAgtTatAggmCcaTctAcaGtamCctGc CEDC_M162.2_u480_LNA3 TtcmCatmCatmCacTaamCcgAttGtcmCtgAcaTtgAtgGccAaamCcaGggAa CEDC_R10E4.11_u274_LNA3 TcamCatTatmCgaAcaAgtActAgtAagmCatGctGtgAtgGagTgcmCgcTa CEDC_R10E4.11_u397_LNA3 mCacGgaGatmCacGacAtcAaaGcgGatTgcTtaGagTgtGgaAacmCgtmCt CEDC_T04C9.1_u321_LNA3 ActAtcTacGtgGcamCgtTggActmCatmCatmCgaTggGaamCgamCgtAtaAg CEDC_T04C9.1_u64_LNA3 TctmCtgGccAgtTcamCttTgtGatmCaaTctmCagAttmCgtmCcamCacAagAt mCtamCttmCcgmCaaGaaGgcmCcgTcgTttmCtaAtcGatmCgaAcaTctmCacA CEDC_W02A2.3_u32_LNA3 c AtgGatGatmCgamCccActTgcmCacTgamCccAcaAtcmCcgmCacTcamCtam CEDC_W02A2.3_u374_LNA3 Cc CEDC_W05G11.3_u153_LNA3 AagAcgGagAggmCtgGagAgaAcgGtamCcgAtgGagAgcmCagGaamCtgAt CEDC_W05G11.3_u51_LNA3 mCcamCccAggAggAggGatAcaAgaGaaGaaAgtAcaGatTctmCcaActAa CEDC_ZK863.5_u256_LNA3 AgtTtcAcamCttmCttTttGccGttTtgGttmCccGttAtcAatmCcaTtgAt CEDC_ZK863.5_u324_LNA3 mCttTtaTatTctmCatmCaaTttGttTccTacTtgGtcAgcTgaGgaTcgTt CEEPHX_Y55B1BR.4_u161_LNA3 TtcGgcAcaAatGgaGcaAaaGtaTcgTggTtaTtgTgaTgcGatTatTc CEEPHX_Y55B1BR.4_u93_LNA3 mCtamCtaTgaAtgAgcTcamCtgGacTcaTttAtcAacTcgAgtmCaaAagmCc CEER_18S_u388_LNA3 GttGgcGaaTctTcgGgtTcgTatAacTtcTtaGagGgaTaaGcgGtgTt CEER_18S_u82_LNA3 GaamCtgAttmCgaGaaGagTggGgamCtgTcgmCttmCgaGgtTtaAcgActTc CEER_26S_u342_LNA3 TgtTatTgcGaaAgtAatmCctGctTagTacGagAggAacAgcGggTtcAa CEER_26S_u38_LNA3 TgcAtamCgamCttGgtmCtcTtgGtcAagGtgTtgTatTcaGtaGagmCagTc CEFOXO_R13H8.1b_u331_LNA3 TgtGctmCagAatmCcamCttmCttmCgaAatmCcaAttGtgmCcaAgcActAacTt CEFOXO_R13H8.1b_u393_LNA3 TtaAgamCggAacmCaaTtgmCtcmCacmCacmCatmCatAccAcgAgtTgaAcaGt CEGAPDH_K10B3.7_u21_LNA3 AcaTtgmCtamCcaAggmCctAagmCcgmCttmCaaAttmCtcTaaGtcTgaAatGa CEGAPDH_K10B3.7_u727_LNA3 GttGagTccAccGgaGtcTtcAccAccAtcGagAagGccAatGctmCacTt CEGBA_F11E6.1a_u232_LNA3 AgtAaaTtcmCttmCcamCgtGgaTctActmCgtGtgTtcAcaAagAtcGagGg CEGBA_F11E6.1a_u451_LNA3 GgtmCcaAtaAtgGgaGacTggTtcmCgcGcaGaaAgtTatGcaGatGatAt CEGLU_C02A12.1_u264_LNA3 AgaAaamCttmCgtTggAccmCtgmCtaAggAgaAgtAttTcaAgcTtcTgaGc CEGLU_C02A12.1_u55_LNA3 GagmCacmCcgAagmCtcAagmCcaTatTtgGaaAcaAgamCcaTacTctTcaAa CEGLU_C46F11.2_u271_LNA3 GttAccmCtcTacAaaTctmCgcTtcAatmCcaAtgTtgTtcGcaGtcAccAa CEGLU_C46F11.2_u45_LNA3 mCcgAagAgcTcgTtamCtaTgcGagGagGtgTgaAgcmCggAatAatTttTt CEGLU_F26E4.12_u109_LNA3 AagTtcTtgGttGgamCgcGatGggAaaAttAtcAagAgaTttGgamCcaAc CEGLU_F26E4.12_u480_LNA3 AcgAttTcaAcgTcaAaaAtgmCtaAtgGtgAtgAcgTgtmCacTttmCggAt CEGLU_R07B1.4_u166_LNA3 AccTggGttGatGttTttGcgGctGaaAgtTtcTccAagmCtcAttGatTa CEGLU_R07B1.4_u38_LNA3 GaaGtamCgtmCtcmCcaAagAaaAgcTacmCccAgcTtaAggmCatTgcAcaAt CEGLU_T09A12.2_u220_LNA3 GcgmCcaGatAtgTatTcaAagAtcGagGtaAatGgtmCagAacActmCatmCc CEGLU_T09A12.2_u335_LNA3 AatmCtamCagGgaAaaAggAttTcgAgtTgcmCgcGttTccAtgmCaaTcaAt CEGLU_T28A11.11_u299_LNA3 AgaTggmCaaAgaAgcAtamCatAacTgaAacTctTccmCggGgaGctActAc CEGLU_T28A11.11_u54_LNA3 TgaAtaAacGggmCcgAacTaaAtcmCatTcgTcaGtgGaaAtgGgaAacAa CEGPD_B0035.5_u256_LNA3 GtcmCgtmCttmCctGatGctTatGaamCgcmCtaTttmCtcGaaGtaTtcAtgGg CEGPD_B0035.5_u478_LNA3 TgtGgaAaaGctmCtcAacGagAagAaaGcaGaaGttmCgtAtamCaaTtcAa CEHSP_C09B8.6_d8_LNA3 AtaTcgmCcgmCctGctTccTcamCcaAccmCgaAtaAcgmCaamCaaAaamCttTa CEHSP_C09B8.6_u286_LNA3 AagAgcmCcamCtcAtcAagGatGaaAgtGatGgaAagActmCttmCgtmCtcAg CEHSP_C12C8.1_u127_LNA3 mCaaGatAttTtaAcaAaaAtgmCatmCaamCaaGaaGccmCaaTcaGgtTccGg CEHSP_C12C8.1_u1531_LNA3 mCttGggmCatTctGtamCggGatGctGtcAttActGtgmCctGcaTatTttAa CEHSP_C47E8.5_u310_LNA3 AagAagmCatmCtcGaaAtcAacmCcaGacmCacGctAtcAtgAagAcamCttmCg CEHSP_C47E8.5_u361_LNA3 AtgAaaGctmCaaGctmCttmCgtGatTccTctActAtgGgaTacAtgGccGc CEHSP_F26D10.3_u276_LNA3 TtaAgcAgamCcaTtgAggAcgAgaAgcTcaAggAtaAgaTcaGccmCagAa CEHSP_F26D10.3_u397_LNA3 mCgtmCttTccAagGatGacAttGaamCgcAtgGtcAacGaaGctGagAaaTa CEHSP_F43D9.4_u169_LNA3 GtcGacTtgGctmCacAtcmCacAccGtcAtcAacAagGaaGgamCagAtgAc mCaaTctTgaGggAcamCgtTctmCacmCatTgaGggAcamCcamCgaGgtmCaa CEHSP_F43D9.4_u275_LNA3 Ga CEHSP_F44E5.4/5_u123_LNA3 TcamCtaAaaTgcAccAatmCtgGacAatmCttmCtgmCttmCtgmCtgGatGcgmCt CEHSP_F44E5.4/5_u380_LNA3 TcaTgaAgcTaaAcaAttmCgaAaaGgaAgaTggTgaAcaAcgGgaAcgTg CEHSP_F52E1.7_u175_LNA3 AagTatAacmCttmCcaAcaGggGtcmCgtmCcaGaamCaaAtcAagTccGaaTt CEHSP_F52E1.7_u448_LNA3 TttAacmCatGgcmCgcAgaTtcTtcGatGacGtcGacTttGatmCgcmCacAt CEHSP_F54D5.8_u252_LNA3 GcgTcgAaaAgaTctmCccTgaAgtmCtgmCatTgamCtgGccTtgAtaTtaTg CEHSP_F54D5.8_u318_LNA3 AcaTagTctTcgTcaTcaAggAtaAgcmCacAccmCgaAatTcaAgcGagAg CEHUS_H26D21.1_u117_LNA3 TcgmCcaAcamCtcGgamCacGtgmCcaAaaTgaAtaTcaTctmCaaAtcGaaTg CEHUS_H26D21.1_u478_LNA3 GtcGaaGttAgaAatmCcaGaaGccGatAttGttTctmCatmCaaAttmCcaAt CEMRE_ZC302.1_u169_LNA3 ActActmCgtGgaAgaTccAatAaaGttGttTcaAcgmCgamCaaAtcGatTc CEMRE_ZC302.1_u292_LNA3 GgcAgtGaaGatGaaGtgGcaAatTctGatGaaGaaAtgGgaAgcAgtAt CEMTL_T08G5.10_d127_LNA3 TtgTcaAcgAccAgaAgcAaaAatTatGggAatmCgcGatAaaAttmCaaGg CEMTL_T08G5.10_u45_LNA3 GatGcaAgtGtgmCcaActGcgAatGtgmCtcAggmCtgmCtcAttAatTtgAa CENAP_D2096.8_u356_LNA3 GacGatAtgTtcGatTtcmCcaGgaGagGacGgtGatGatGtgTcaGacTt CENAP_D2096.8_u70_LNA3 GacGatAtgTtcGatTtcmCcaGgaGagGacGgtGatGatGtgTcaGacTt CEPAI_F56D12.5_u241_LNA3 GagGtcGtcGtaAtcmCacAagGctmCcaAgaAagmCaaGtgmCtcGacAttTc CEPAI_F56D12.5_u301_LNA3 GatActTttGgcAagmCtcGttmCcaAtcAagAagGagGtcAtcmCcaGatmCg CEPDI_C07A12.4_u28_LNA3 GatGagGagGgamCacAccGagmCtcTaaAtcmCacAttmCcaAtamCagTtcAa CEPDI_C07A12.4_u433_LNA3 mCttAtgTccGaaGatAtcmCcaGagGatTggGacAagAacmCcaGtcAagAt TacmCccAgtmCgamCtaTgaTggAgamCagAaamCctmCgaGaaGttmCgaAgaA CEPDI_C14B1.1_u119_LNA3 t CEPDI_C14B1.1_u358_LNA3 mCtcGtcGccTccAacTtcAacGaaAttGccmCttGatGaaAccAagActGt CEPGK_T03F1.3_d9_LNA3 TtcTatTgtTtaTtcmCttGccmCaaTagTgtAttTgtAttTatTctTtcTc CEPGK_T03F1.3_u424_LNA3 mCaaAtcmCatmCtcmCcaGtgGatTtcGtcAttGclGacAagTtcGccGagGa CEPON_E01A2.7_u223_LNA3 GttTctGatTcgAcamCttTatGgamCcaTctmCaaGttmCtgmCgaGttTctTt CEPON_E01A2.7_u79_LNA3 GggAaamCaaAtgAttGttGgtAcaGtaGccmCgcmCctGctAttmCacTgtGa mCgaGcamCatmCatmCcaAtcGttmCctGttmCaamCaaGgcmCttmCtaAtcGttA CEPPGB_F13D12.6_u44_LNA3 g CEPPGB_F13D12.6_u440_LNA3 TgaTgaGagmCccAgtAacmCaaTtaTttGaamCcgTcaGgaTgtGcgTaaGg CEPPS_T14G10.1_d2_LNA3 mCgtmCtaAtcGaaGaaGggGatmCgtGggmCaaTcaTaamCtaAttAacmCttmCa CEPPS_T14G10.1_u240_LNA3 mCaaTggmCtcmCagGtcTttmCtgmCtcTtcAtaTacTtcmCatTccGagTtgmCt CEPRDX_R07E5.2_u405_LNA3 GttmCtcTtgGagmCtgAagTtgTcgmCgtGctmCgtGtgAttmCtcActTctmCt CEPRDX_R07E5.2_u42_LNA3 TcgmCtamCcaGcaAggAatActTcaAcaAggTcaAcaAgtGatmCacAcaGa CEPYC_D2023.2_u256_LNA3 AagGaaAttGtaActmCgcmCcaAgaGctmCtcmCcaGgtGtcmCgtGgamCatAt CEPYC_D2023.2_u427_LNA3 TtgActGgaTtgGagAttGcgGaaGaaGttGatGttGaaAtcGagAgtGg CERAD_F10G7.4_u169_LNA3 GccAagTctmCaaGcaAtaAgtGttGatmCaaTcaGagmCcaTacGgaGagAt AtaTtgAgamCttmCggGacAagmCggActTctmCatmCtgTcamCagmCaamCtgm CERAD_F10G7.4_u267_LNA3 Cc CERAD_F32A11.2_u250_LNA3 GatmCcgmCagAgaAtcGagTatTtcmCtcTcgAgamCccAtgGatAtcAacTg CERAD_F32A11.2_u380_LNA3 TccGttAagAagmCtcActGgaAaaAcamCacGgcTcgAacGaaAttGgaAt CERAD_T04H1.4_u274_LNA3 AatTtgGatGagAgcAaaGtgGaaGgaAtgGctAtcGttTtgGcaGatAt CERAD_T04H1.4_u375_LNA3 GtgmCtgGtcAaaAaaTgcTtgmCttmCgtTgcTtaTtcGcaTtgmCacTcgmCa CERAD_W06D4.6_u325_LNA3 mCttmCgaGaamCtcTtcAagTtgGaaTcaAcaGtgGcaTcgGatAcamCatGa CERAD_W06D4.6_u34_LNA3 GtgmCctTctGaaGccGaaGaaAacGacGatTagTtaAatGttTccAagTt CERAD_Y116A8C.13_u289_LNA3 GatAaaAtcGatAgcGacGacGatGagGaaGccGatGatGagGagmCtcGa CERAD_Y116A8C.13_u59_LNA3 GcaGgtGgaTacGgaTgtGgaGctGacTttTgcGttTtaTcaAgaAtcTc CERAD_Y39A1A.23_u221_LNA3 TccmCgtAgaAgtAgaAatGctAgaAgaAccTgaAcaAgaAgaTcaAgaAa CERAD_Y39A1A.23_u276_LNA3 TgcAagAtgTcaGtaTtgAaamCaaTtcmCtgTagAgamCccmCcgAagAaaAt CERAD_Y41C4A.14_u509_LNA3 AgtmCtcGtaTccGggAatGttTcaGccTgtGaaAatGctTgtTgaAgamCg CERAD_Y41C4A.14_u731_LNA3 mCttmCaaAacmCgtmCgcTttTaaGgaTacAggAacGtgGcamCgcTtcmCgaGg CERAD_Y43C5A.6_u131_LNA3 mCagAttGtamCctTcgAaaAggAaaAggAgaGaaTcgmCgtmCgcAaaAatGg CERAD_Y43C5A.6_u429_LNA3 TgaTggmCttTgaTtaTtcGagmCagGagmCaaTgaTgtmCcgAgaGtcGttAt CERFC_F31E3.3_u128_LNA3 mCaaTgamCgaGaaTatTggAgtAatGggGaaActGgtTgcGacTtgmCgaAa CERFC_F31E3.3_u55_LNA3 TtgGaaAacAatmCtcmCtcGacTttmCtgmCtcActmCttmCgtGaaActAtcmCa CERPL_K11H12.2_d1_LNA3 TctTgtTatTttAttTtgTttTggGctTgtTccGaaAatGaaAtgGttGt mCaaTggAtcAccAagmCcaGttmCacAagmCacmCgtGagmCaaAgaGgamCtc CERPL_K11H12.2_u172_LNA3 Ac CERT_F36A4.7_u1396_LNA3 mCttTgtGatGtgAtgActGcgAagGgamCacTtgAtgGctAttAcgAgamCa CERT_F36A4.7_u2302_LNA3 GagmCcaGctActmCagAtgAcamCtcAacAcgTtcmCatTatGcaGgaGttTc CERT_F36A4.7_u289_LNA3 TacActmCcaTccTcgmCcgAcaTacAatmCcaAcaTctmCcamCgcGgaTtcTc CERT_F36A4.7_u2919_LNA3 AtgGagAagAtgGttTggAtgGaaTgtGggTtgAgaAtcAgaAtaTgcmCg CERT_F36A4.7_u4269_LNA3 AacmCggGatAccGtgTcgAacGtcAcaTgaAagAtgGcgAtaTaaTcgTc CERT_F36A4.7_u5485_LNA3 GagGagAttAaamCgcAtgTcaGtgGctmCatGtcGagTttmCcaGaaGtcTa CESLC_F52F12.1a_u249_LNA3 AgaTatTgcmCtcTacTtaTcaTggGccTgaTggmCttTgtmCtgmCcgGtaTt GaaTctmCaamCcamCttmCtgGaamCccmCatAcamCcaAtgGatAgaAgamCgg CESLC_F52F12.1a_u76_LNA3 Ag CESLC_K11G9.5_u400_LNA3 GttGttmCttTttTccGtgAtcTttTcaTgtTtaTgtmCtgAacGtgGcaGg CESLC_K11G9.5_u462_LNA3 GacTcgTtgGtgTctTgcTagGatGtcTtgGgtTcaTtcmCtcAatmCgtTg CESLC_Y32F6B.1_u179_LNA3 GtamCtgGgcTcgAggGctGaaActAatmCgaAgaAgaAacTccAgaAgaTa CESLC_Y32F6B.1_u280_LNA3 GgaTcaTgcTctGttTacGacActGatGagTtaAgaGtcAgamCtgmCacGt CESLC_Y37A1C.1a_u104_LNA3 mCgaTggTtcTtcTcgTctAtcAtaTcgGggTagTtgmCcgAagTgtTgaAa CESLC_Y37A1C.1a_u404_LNA3 mCaaAtcGaamCtgGtaTaaAggAggAccGacGgaGacGaaTttGaamCgaGa CESLC_Y70G10A.3_u383_LNA3 AttmCgaTcaAagAacTctGgcTctmCggmCgtTaamCtgGacAttTgtTcgTc CESLC_Y70G10A.3_u46_LNA3 mCtcmCccGagmCagGcgAttAttmCacGctAgtTatGctmCaaAtgTgaTctGt CESOD_C15F1.7_u435_LNA3 mCcgGtamCtaTctGgaTcamCacAgaAgtmCcgAaaAtgAccAggmCagTtaTt CESOD_C15F1.7_u9_LNA3 mCccAgtGacTacmCtgAatmCgcGtcTctGaaTctmCcamCacAatTccTacTa CESOD_F10D11.1_u326_LNA3 GgaGttGctmCacmCgcAatTaaGagmCgamCttmCggAtcTctGgaTaaTctTc CESOD_F10D11.1_u477_LNA3 AaaTtgAggAaaAgcTtcAcgAggmCggTctmCcaAagGaaAcgTcaAagAa CESULT_EEED8.2_u316_LNA3 mCaaTcgTacmCatGaaAgaAgtTggAagmCcamCgtGcaAgaGaaGaaAtcmCa CESULT_EEED8.2_u82_LNA3 AagAagAttmCctGacmCagAgaGacTcamCgtGctTacmCcaAgaAgcAtcTa CESULT_Y113G7A.11_u252_LNA3 AgcAttGgtGgaAatAcgAaaTggmCatGggAagAgaAacmCccTctmCaaTt CESULT_Y113G7A.11_u96_LNA3 mCtgGttAcgGtaGtgTatGgtmCccTgtmCctmCtcAgaAtgmCaaAtaTgtmCg CESULT_Y67A10A.4_u108_LNA3 TctAcgTcgAtgGaaAagmCcgAttTaamCaaTcaAagmCcaAcaAcgmCagTt CESULT_Y67A10A.4_u327_LNA3 GgaAagGtgmCcaAaaAgtTgamCagmCaaTtgGagGatmCttAttmCatTgcmCa CETOPO_K12D12.1_u398_LNA3 AgaTgaTgaTgaAgtTccTgcAaaGaaGccTgcTccAgcGaaGaaAgcTg CETOPO_K12D12.1_u449_LNA3 AaaAccTcgTacTggAaaAggAgcTgcGaaAgcGgaAgtTatmCgaTttGt CETOPO_M01E5.5b_u256_LNA3 GagAagGccmCagAagAagTacGacAgamCtgAagGagmCagTtgAaaAagTt CETOPO_M01E5.5b_u429_LNA3 TtcTgtmCatAcaAtcGtgmCtaAtcGgcAggTtgmCgaTccTttGtaAccAt CEUbi_F25B5.4_u186_LNA3 AagmCttmCggAcamCcaTtgAgaAtgTcaAagmCcaAaaTccAggAtaAggAg CEUbi_F25B5.4_u2_LNA3 AatmCgaAccmCatmCaaTtcActmCgtTatTccTccTcgAtcTccGttmCaaGt CEUbi_F29B9.6_u145_LNA3 mCtgAacmCatmCcaAatAttGaaGatmCcaGctmCagGctGaaGccTatmCagAt CEUbi_F29B9.6_u230_LNA3 mCgtGtgmCttAtcTctTctGgaTgaAaamCaaGgaTtgGaaGccGtcAatmCt CEUbi_M7.1_u239_LNA3 mCggAagmCatmCtgmCctTgamCatTctmCcgTtcGcaGtgGtcGccGgcTctG CEUbi_M7.1_u53_LNA3 AaaGtamCgcTatGtgAggAggmCtaAcamCcaTtcAtaTaaGaamCgcAgcmCa CEUGT_F39G3.1_u40_LNA3 TgtTgcmCgtAgaAgaGagActAaaActAagAacGatTgaTtgAagGtcTg CEUGT_F39G3.1_u466_LNA3 TacAatTctTtgmCagGaaGcaAtaTccGccGgaGtcmCccmCttAtcActAt CEUGT_M88.1_u480_LNA3 mCtcAcgGagGttAtaAttmCtaTgcAggAggmCaaTttmCtgmCtgGagTtcmCa CEUGT_M88.1_u72_LNA3 AccGttTcaTgaGagmCtgTaaTcaGgtGttGttTctGtaAaaAgtGtgAa YAL009W_u145_LNA3 GtgGatGtgAaaTtaGtcmCtcAacmCccAgaGcaTttAgtGcaGagAttAg YAL009W_u341_LNA3 GcaGttTaaTgtGaaGctAgtTaaAgtAcaGtcTacGtgGgamCgaGaaAt YAL059W_u262_LNA3 AttGccAagTccAttTctmCgtGccAagTacAttmCaaAatAcaAgaAagGc YAL059W_u51_LNA3 AgamCtcmCtamCaaAtaGatTcgGtgTccTgcmCagAcgAtgTtgAagAatAg YER109C_u109_LNA3 TtgAagTttGggAatAttGgtAtgGttGaaGacmCaaGgamCcgGatTacGa YER109C_u436_LNA3 GagGcgmCaaGtaGgcAatGatTcaAgaAgtAgtAaaGgcAatmCgtAacAc YHR152W_u128_LNA3 TgaGcamCaaAgtTaaGatGttmCggAaaGaaAaaGaaAgtmCaaTccTatGa mCaaGtgAccAatmCagmCacGcamCggmCttmCcaTccTcaAgamCtgAtaTtam YHR152W_u510_LNA3 Cc YKL130C_u211_LNA3 AttAaaTgcGcaGatGagGacGgaAcgAatAtcGgaGaaActGatAatAt YKL130C_u85_LNA3 GatGgtAagmCtgAgcGccTtgGacGaaGaaTttGatGttGtcGctActAa YKL178C_u199_LNA3 TacGtcAcgmCaaGgamCagAgcTttGacGacGaaAtaTcamCttGgaGgaTt YKL178C_u367_LNA3 TctmCccTgtGtaGgtAcamCcaAtaTcamCaaGcgmCatTtcTatGtcGacTa YLR443W_u179_LNA3 TgcTaamCacmCagTttAgamCcaTggAaaTccmCacmCgcAaaTatAagmCaaTg YLR443W_u86_LNA3 GcaGgamCatAagAttmCcgGtcAagmCaamCgamCagTgaAgaAagTatGcaAa YOR092W_u251_LNA3 mCcgTctAgtGaaAgcGggAtgGctAaaTtgGgaAaamCgamCaaGatGttAt YOR092W_u82_LNA3 GatGctTcaAtaTccTttGatGgtmCgtTagTttAccAttTttGgtGtcTt YPL263C_u132_LNA3 tCatTtgAgtTatGtgAagAccGttGgtGggAaaGaaGagAtcAggTg YPL263C_u257_LNA3 GtcTtgGctAccAcamCccAaaAccGttmCgaAacTttAagAgcAttmCtamCt - Oligonucleotide Design for Microarrays.
- The methods for designing exon-specific internal oligonucleotide capture probes are described above.
- Design of the LNA-Modified Capture Probes
- For the internal LNA-modified oligonucleotide capture probes, every third DNA nucleotide was substituted with an LNA nucleotide. The probes designed to capture the junction of the recombinant splice variants were designed with LNA modifications in a block of five consecutive LNAs nucleotides, two on the 5′ side of the splice junction and three on the 3′ side of the splice junction. All capture probes are shown in Table 14.
TABLE 14 Internal, exon-specific and merged, exon—exon junction specific oligonucleotide capture probes used in this example. (SEQ ID NOs:) Sequence (LNA = uppercase, DNA Capture probes lowercase letters) gene78.01a_50_LNA3 mCctGaaAgtAgaTttGttAttTccGaaAcgmCctTctmCccGttmCttAagTc gene78.01b_50_LNA3 mCatAtamCcamCaaAtaGtcmCctmCaaAaaTcamCaaGaaAacTcamcaamCacTg gene78.03a_50_LNA3 GatTtgmCagmCggTggTaaAaaGtaTgaAaamCgtGgtAatTaaAagGtcTc gene78.03b_50_LNA3 mCcaAtgAaaActAatmCaaAggTaaAcgTggAtcmCcaTggmCaaTtcmCcgGg gene78.m01INS3_50_block caacactgcccagaggttcaatcGATmCmCgatgatcctaatgaaggcgccc gene78.mINS303_50_block gtccagtatcgtccatcatAGTATcgataaatatgtgaaggaaatgcctg gene78.m01INS4_50_block caacactgcccagaggttcaatcGATGTgtgataggatcagtgttcaggg gene78.mINS403_50_block gaaggcgaaggagactgctAATATcgataaatatgtgaaggaaatgcctg - Printing and Coupling of the Splice Isoform-specific Microarrays
- The splice variant capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by a hexaethyleneglycol-2 (HEG2) linker. The capture probes were first diluted to a 20 uM final concentration in 100 mM Na-phosphate buffer pH 7.0, and spotted on the Immobilizer polymer microarray slides (Exiqon, Denmark) using the Biochip Arrayer One (Packard Biochip Technologies, USA) with a spot volume of 2×300 pl and 300 μm between the spots. The capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker with 2300 μjoules (Stratagene, USA). Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides two
times 15 minutes in 1×SSC. After washing, the slides were dried by centrifugation at 1000×g for 2 minutes, and stored in a slide box until microarray hybridization. - Construction of Splice Variant Vectors
- The recombinant splice variant constructs were cloned into the
Triamp 18 vector (Ambion, USA). The constructs were sequenced to confirm their construction. The plasmid clones were transformed into E. coli XLI0-Gold (Stratagene, USA). Triamp] 8/SWI5 Vector Construct - Genomic DNA was prepared from a wild type standard laboratory strain of Saccharomyces cerevisiae using the Nucleon MiY DNA extraction kit (Amersham Biosciences, USA) according to the supplier's instructions. Amplification of the partial yeast gene was performed using standard PCR using yeast genomic DNA as template. In the first step of amplification, a forward primer containing a restriction enzyme site and a reverse primer containing a universal linker sequence were used. In this step, 20 bp was added to the 3′-end of the amplicon, next to the stop codon. In the second step of amplification, the reverse primer was exchanged with a nested primer containing a poly-T20 tail and a restriction enzyme site. The SWI5 amplicon contains 730 bp of the SWI5 ORF plus 20 bp universal linker sequence and a poly-A20 tail.
- The PCR primers used were;
YDR146C-For-EcoRI: acgtgaattcaaatacagacaatgaaggagatga (SEQ ID NO:) YDR146C-Rev-Uni: gatccccgggaattgccatgttacctttgattag (SEQ ID NO:) ttttcattggc Uni-polyT-BamHI: acgtggatccttttttttttttttttttttgatc (SEQ ID NO:) cccgggaattgccatg, - The PCR amplicon was cut with the restriction enzymes, EcoRI +BamHI. The DNA fragment was ligated into the
pTRIamp 18 vector (Ambion, USA) using the Quick Ligation Kit (New England Biolabs, USA) according to the supplier's instructions and transformed into E. coli DH-5α by standard methods. - Construction of the Recombinant Splice Variant #1 (Triamp18/swi5-rubisco)
- The Arabidopsis thaliana Rubisco small subunit ssu2b gene fragment (gil7064721) was amplified from genomic DNA by primers named
DJ305 5′-ACTATGATGGACGATACTGGAC-3′ (SEQ ID NO:) andDJ306 5′-ATTGGATCGATCCGATGATCCTAATGAAGGC-3′ (SEQ ID NO:), containing ClaI restriction site linkers. The purified PCR fragment was digested with ClaI and then cloned into the swi5 (gI:7839148) vector at the unique ClaI site (atcgat) giving each insert a flanking sequence from the original yeast SWI5 insert (namedexon01and exon 03, FIG. 11). The product was inserted in the reverse orientation, so that the insert sequence is:atcgatCCGATGATCCTAATGAAGGCGCCCGGGTACT (SEQ ID NO:) CCTTCTTGCATTCTTCAACTTCCTTCAACACTTGAGC GGAGTCGGTGCATCCGAACAATGGAAGCTTCCACATT GTCCAGTATCGTCCATCATAGTatcgat - Nucleotide sequence analysis revealed a difference between the sequence of A. thaliana rubisco expected from the GenBank database and that obtained from all sequenced constructs and PCR products.
Position 30 in the Rubisco insert is “C” rather than the expected “A”. This SNP was probably created by PCR. None of the oligonucleotide capture probes used in the example cover this region. Rubisco sequence in genbank is TCCTAATGAAGGCGCCA (SEQ ID NO:). The sequence obtained from the plasmid contruct is TCCTAATGAAGGCGCCC (SEQ ID NO:). - Construction of the Recombinant Splice variant # 2 (
Triampl 8/swi5-lea) - The Arabidopsis thaliana Lea gene (gil 526423) was amplified from genomic DNA with primers named
DJ307 5′-GGAATTATCGATGTGTGATAGGATCAGTGTTCAG-3′ (SEQ ID NO:) andDJ308 5′-AATTGGATCGATATTAGCAGTCTCCTTCGCC-3′ (SEQ ID NO:) including the ClaI linker sites as above. The PCR fragment was digested with ClaI cloned into the yeast SWI5 IVT construct as above at the unique ClaI site. The fragment was inserted in the forward orientation, resulting in the following insert sequence:atcgatGTGTGATAGGTTCAGTGTTCAGGGCTGTCCA (SEQ ID NO:) AGGAACGTATGAGCATGCGAGAGACGCTGTAGTTGGA AAAACCCACGAAGCGGCTGAGTCTACCAAAGAAGGAG CTCAGATAGCTTCAGAGAAAGCGGTTGGAGCAAAGGA CGCAACCGTCGAGAAAGCTAAGGAAACCGCTGATTAT ACTGCGGAGAAGGTGGGTGAGTATAAAGACTATACGG TTGATAAAGCTAAAGAGGCTAAGGACACAACTGCAGA GAAGGCGAAGGAGACTGCTAATatcgat. - Preparation of Target
- In vitro RNA Preparation from Splice Variant Vectors In vitro RNA from the splice variants were made using the MEGAscript™ high yield transcription kit according to the manufacturer's instructions (Ambion, USA). The yield of IVT RNA was quantified at a Nanodrop spectrophotometer (Nanodrop Technologies, USA).
- Isolation of Total RNA from C. elegans
- C. elegans wild-type strain (Bristol-N2) was maintained on nematode growth medium (NG) plates seeded with Escherichia coli strain OP50 at 20° C., and the mixed stages of the nematode were prepared as described in Hope, I. A. (ed.) “C. elegans-A Practical Approach”, Oxford University Press 1999. The samples were immediately flash frozen in liquid N2 and stored at −80° C. until RNA isolation.
- A 100 μl aliquot of packed C. elegans worms from a mixed stage population was homogenized using the
FastPrep Biol 101 from Kem-En-Tec for 1 minute,speed 6 followed by isolation of total RNA from the extracts using theFastPrep Bio 101 kit (Kem-En-Tec) according to the manufacturer's instructions. The eluted total RNA was ethanol precipitated for 24 hours at −20° C. by addition of 2.5 volumes of 96% EtOH and 0.1 volume of 3M Na-acetate, pH 5.2 (Ambion, USA), followed by centrifugation of the total RNA sample for 30 minutes at 13200 rpm. The total RNA pellet was air-dried and redissolved in 10 μl of diethylpyrocarbonate (DEPC)-treated water (Ambion, USA) and stored at −80° C. - Fluorochrome-Labelling of the Target
- The following fluorochrome-labelled cDNA targets were synthesized to test the performance of ‘merged’ probes that span exon borders. Synthetic RNAs corresponding to the splice variant #1 (exon01-INS3-exon03 (1-INS3-3) and splice variant #2 (exon01-INS4-exon03 (1-INS3-3) were spiked into 10 μg of C. elegans reference total RNA sample in two different ratios. The first target pool (KU007) contained 10 ng of splice variant #1 (1-INS3-3) transcript and 2 ng of variant #2 (1-INS4-3) transcript, a ratio of 5:1. The second target pool (KU008) contained 2 ng variant #1 (1-INS3-3) transcript and 10 ng of splice variant #2 (1-INS4-3) transcript, a ratio of 1:5. Both mRNA pools were combined in separate labeling reactions with 5 μg anchored oligo(dT2O) primer and DEPC-treated water to a final volume of 8 μl. The mixture was heated at 70° C. for 10 minutes, quenched on ice for 5 minutes, followed by addition of 20 units of Superasin RNase inhibitor (Ambion, USA), 1 μldNTP solution (10 mM each dATP, dGTP, dTTP and 0.4 mM dCTP, and 3 μl Cy5-dCTP, Amersham Biosciensces, USA), 4
μl 5×RTase buffer (Invitrogen), 2 μ0.1 mM DTT (Invitrogen), 400 units of Superscript II reverse transcriptase (Invitrogen, USA) and DEPC-treated water to 20 μl final volume. Background hybridization to merged capture probes was monitored in both hybridizations using the other fuor channel with 10 μg of C. elegans reference RNA alone labeled with Cy3-dCTP, according to the labeling method described above for the splice variant spikes. All four cDNA syntheses were carried out at 42° C. for 2 hours, and the reaction was stopped by incubation at 70° C. for 5 minutes, followed by incubation on ice for 5 minutes. - Unincorporated dNTPs were removed by gel filtration using MicroSpin S-400 HRcolumns as described below. The column was pre-spun for 1 minute at 1500×g in a 1.5 ml tube, and the column was placed in a new 1.5 ml tube. The cDNA sample was slowly to the top center of the resin, spun 1500-xg for 2 minutes, and the eluate was collected. The RNA was hydrolyzed by adding 3 μl of 0.5 M NaOH, mixing, and incubating at 70° C. for 15 minutes. The samples were neutralized by adding 3 μl of 0.5 M HCl and mixing, followed by addition of 450 μl 1xTE, pH 7.5 to the neutralized sample and transfer onto a Microcon-30 concentrator (prior to use, 500
μl 1×TE was spun through the column to remove residual glycerol). The samples were centrifuged at 14000-xg in a microcentrifuge for 12 minutes. Spinning was continued until volume was reduced to 511. The labelled cDNA probes were eluted by inverting the Microcon-30 tube and spinning at 1000-xg for 3 minutes. - Microarray Hybridization
- The fluorochrome-labelled cDNA samples, respectively, were combined (the two different ratios separately). The following were added: 3.75 μl 20×SSC (3×SSC final, which was passed through a 0.22 filter prior to use to remove particulates) yeast tRNA (1 μg/μl final) 0.625 μl 1 M HEPES, pH 7.0 (25 mM final, which was passed through 0.22 μ filter prior to use to remove particulates) 0.75
μl 10% SDS (0.3% final) and DEPC-water to 25 μl final volume. The labelled cDNA target samples were filtered in Millipore 0.22 μ filter spin column (Ultrafree-MC, Millipore, USA) according to the manufacturer's instructions, followed by incubation of the reaction mixture at 100° C. for 2-5 minutes. The cDNA probes were cooled at room temp for 2-5 minutes by spinning at maximum speed in a microcentrifuge. A LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on Immobilizer™ MicroArray Slide, and the hybridization mixture was applied to the array from the side. An aliquot of 30 μL of 3×SSC was added to both ends of the hybridization chamber, and the Immobilizer™ MicroArray Slide was placed in the hybridization chamber (DieTech, USA). The chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol. The slides were washed sequentially by plunging gently in 2×SSC/0.1% SDS at room temperature until the cover slip falls off into the washing solution, then in 1×SSC pH 7.0 (150 mM NaCl, 15 mM Sodium Citrate) at room temperature for 1 minute, then in 0.2×SSC, pH 7.0 (30 mM NaCl, 3 mM Sodium Citrate) at room temperature for 1 minute, and finally in 0.05×SSC (7.5 mM NaCl, 0.75 mM Sodium Citrate) for 5 seconds, followed by drying of the slides by spinning at 1000×g for 2 minutes. The slides were stored in a slide box in the dark until scanning. - Microarray data analysis
- The splice variant microarray was scanned in a ScanArray 4000XL confocal laser scanner (Packard Instruments, USA). The hybridization data were analysed using the GenePix Pro 4.01 microarray analysis software (Axon, USA). Only the Cy5 (650 nm) data were examined as both hybridizations produced comparable, and acceptably low, signal from the C. elegans reference RNA alone (Cy3 channel).
- Normalization
- Data was normalized so that it could be compared between hybridizations. Both hybridizations contained the same amount of RNA from
synthetic exons 01 and exons 03 (10+2 ng), so signal from the capture probes designed to internal regions of these exons is expected to be equal. The ratio of raw Cy5 signal between the two different labeled cDNA target pools, designated as KU007 and KU008 hybridizations, for each probe corresponding to either of these exons was calculated, that is for each probe iwe calculated the ratio probeiKU007/probeiKU008). The average of all of these ratios was used as the normalization ratio. - Expectations of Normalized Data.
- To reflect the proportions of RNA spiked into the hybridization, the ratio of signal in hybridization KU007/KU008 should be 5 for probes designed to exon junctions of the INS3
splice variant # 1 and 0.2 for probes corresponding to 1-INS4splice variant # 2. Data was 10g2 transformed: 10g2(5)=+2.32, log2(0.2)=-2.32. The merged probe corresponding to the exon 01-exon 03 border desirably produces a consistently low value that is desirably independent of which transcript was more abundant, i.e., log2(ratio)=0. - Array Results
- Results are summarized in Table 15.50-mer capture probes containing LNA in a block spanning exon-exon junctions were consistent in producing the expected ratios.
TABLE 15 LNA 50-mer block probes are most consistent in producing overall data closest to expected ratios. Observed ratio (log2) with merged Capture probe Expected ratio (log2) LNA block capture probes gene78.m0103 0.00 −0.24 gene78.m01INS3 2.32 2.93 gene78.m01INS4 −2.32 −2.39 gene78.mINS303 2.32 3.11 gene78.mINS403 −2.32 −0.86 - Capture Probe Design
- The capture probes were designed to a 602-nucleotide sequence in the 3′-region of the Yeast ( S. cerevisiae) 70 kDa heat shock protein (SSA4) gene. The 602-base pair sequence is shown in Table 16. For the LNA-spiked oligonucleotide capture probes, every third DNA nucleotide was substituted with a LNA nucleotide. All capture probes are shown in Table 17.
TABLE 16 Six hundred and two (602) base pair sequence stretch of the S. cerevisiae ssa4 gene. The underlined segments indicate the position of the capture probes. First underline is equal to capture probe YER103W-554, second underline is equal to capture probe YER103W-492 and so forth. (SEQ ID NO:) 
-
TABLE 17 Capture probes for the SSA4 tile array. (SEQ ID NOs:) Oligo Name Sequence YER103W-1-DNA gccccactggagcaccagacaacggcccaacggt tgaagaggttgattag YER103W-38-DNA gccccaggagcaggcccagttccgggtgctggag caggccccactggagc YER103W-73-DNA ccattatgagtaaattttacggagctgcaggtgg tgccccaggagcaggc YER103W-92-DNA ctagaaggtgttgcaaaccccattatgagtaaat tttacggagctgcagg YER103W-127-DNA cctccaccgaggaatacaaggaaaggcaaaagga actagaaggtgttgca YER103W-200-DNA ggtgaagaggatgccaggaaattggaagccgccg cccaagatgctataaa YER103W-245-DNA actttgaaaaattctgtgagcgaaaataacttca aggagaaggtgggtga YER103W-272-DNA aagaatcagctagaatcgtacgcgtttactttga aaaattctgtgagcga YER103W-336-DNA aatggttgctgaggcagaaaagttcaaggccgaa gatgaacaagaagctc YER103W-393-DNA taacaagattacaattactaacgataagggaaga ttatcgaaggaagata YER103W-447-DNA cgatgcaaatggtattctgaacgtatctgccgtt gaaaaaggtactggta YER103W-492-DNA acccgctccaagaggcgtaccacaaattgaagtt acatttgatatcgatg YER103W-554-DNA ggtgaaaggacaaggacaaaagacaacaatctac tgggtaaatttgagtt YER103W-1-LNA1 GccmCcamCtgGagmCacmCagAcaAcgGccmCa amCggTtgAagAggTtgAttAg YER103W-38-LNA1 GccmCcaGgaGcaGgcmCcaGttmCcgGgtGctG gaGcaGgcmCccActGgaGc YER103W-73-LNA1 mCcaTtaTgaGtaAatTttAcgGagmCtgmCagG tgGtgmCccmCagGagmCagGc YER103W-92-LNA1 mCtaGaaGgtGttGcaAacmCccAttAtgAgtAa aTttTacGgaGctGcaGg YER103W-127-LNA1 mCctmCcamCcgAggAatAcaAggAaaGgcAaaA ggAacTagAagGtgTtgmCa YER103W-200-LNA1 GgtGaaGagGatGccAggAaaTtgGaaGccGccG ccmCaaGatGctAtaAa YER103W-245-LNA1 ActTtgAaaAatTctGtgAgcGaaAatAacTtcA agGagAagGtgGgtGa YER103W-272-LNA1 AagAatmCagmCtaGaaTcgTacGcgTttActTt gAaaAatTctGtgAgcGa YER103W-336-LNA1 AatGgtTgcTgaGgcAgaAaaGttmCaaGgcmCg aAgaTgaAcaAgaAgcTc YER103W-393-LNA1 TaamCaaGatTacAatTacTaamCgaTaaGggAa gAttAtcGaaGgaAgaTa YER103W-447-LNA1 mCgaTgcAaaTggTatTctGaamCgtAtcTgcmC gtTgaAaaAggTacTggTa YER103W-492-LNA1 AccmCgcTccAagAggmCgtAccAcaAatTgaAg tTacAttTgaTatmCgaTg YER103W-554-LNA1 GgtGaaAggAcaAggAcaAaaGacAacAatmCta mCtgGgtAaaTttGagTt Control capture probes YFL039C-50 acaagaatacgacgaaagtggtccatctatcgtt caccacaagtgtttct YFL039C-50_LNA3 AcaAgaAtamCgamCgaAagTggTccAtcTatmC gtTcamCcamCaaGtgTttmCt YDR146C-50 tgggaatggaacggggattatggtttcgccaatg aaaactaatcaaaggt YDR146C-50_LNA3 TggGaaTggAacGggGatTatGgtTtcGccAatG aaAacTaaTcaAagGt - Printing and Coupling of the Yeast SSA4 Tile Microarrays
- The SSA4 capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by a hexaethyleneglycol-2 (HEG2) linker. The capture probes (Table 17) were first diluted to a 20 μM final concentration in 100 mM Na-phosphate buffer pH 7.0, and spotted on the Immobilizer microarray slides (Exiqon, Denmark) using the Biochip Arrayer One (Packard Biochip Technologies) with a spot volume of 2×300 pl and 400 μm between the spots. The capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker with 2300 μjoules (Stratagene, USA). Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides two
times 15 minutes in 1×SSC. After washing, the slides were dried by centrifugation at 1000xg for 2 minutes, and stored in a slide box until microarray hybridization. - Yeast Cultures
- Saccharomyces cerevisiae wild-type (BY4741, MATa; his3Δ1; leu2Δ0; met15Δ0; ura3ΔO) and Δssa4 (MATa; his3 Δ1; leu2 Δ0; met15Δ0; ura3ΔO; YER103w::kanMX4) mutant strains (EUROSCARF) were grown in YPD at 30° C. until the A600 density of the cultures reached 0.8. Half of the cultures were collected by centrifugation and resuspended in one volume of 40° C. preheated YPD. Incubation was continued for an additional 30 minutes at 30° C. or 40° C. for the standard and heat-shocked cultures, respectively. Cells were harvested by centrifugation and stored at −80° C.
- RNA Extraction
- Total RNA was extracted using the FastRNA Kit-RED (BIO 101) according to suppliers' instructions. The quantity and quality of the RNA preparations were examined by standard spectrophotometry on a NanoDrop ND-1000 (USA) and by gel electrophoresis. Only high quality RNA preparations were used for microarray analyses.
- Fluorochrome-labelling of the Target
- A total of seven cDNA assay mixtures were produced; each with ten (10) μg total RNA from wtand combined with 5 μg anchored oligo(dT 2O) primer and DEPC-treated water to a final volume of 8 μl. The mixtures were heated at 70° C. for 10 minutes, quenched on ice for 5 minutes, followed by addition of 20 units of Superasin RNase inhibitor (Ambion, USA), 3 μl Cy3-dCTP (Amersham Biosciences), 10 mM final concentration of dATP and dGTP, 4
μl 5×RTase buffer (Invitrogen), 2[0.1 mM DTT (Invitrogen), 400 units of Superscript II reverse transcriptase (Invitrogen, USA), dUTP and dTTP accordingly to Table 18, and DEPC-treated water to 20 μl final volume. A parallel set-up was made with 10 μg total RNA from Δssa4 for target cDNA labelling with Cy5-dCTP. All cDNA syntheses were carried out at 42° C. for 2 hours, and the reaction was stopped by incubation at 70° C. for 5 minutes, followed by incubation on ice for 5 minutes. Each cDNA pool (except the unfragmented control pool) was incubated at 37° C. for 2 hours with 2 units of Uracil-DNA Glycosylase (UDG, New England Biolabs, USA) and by addition of 2.4 μl (1×final concentration in the reaction mixture) of UDG reaction buffer. The enzyme was heat-inactivated at 95° C. for 10 minutes. Unincorporated dNTPs were removed by gel filtration using MicroSpin S-400 HR columns as described in Example 9.TABLE 18 dUTP and dTTP ratios in cDNA target labelling. Assay # Final conc. dUTP Final conc. dTTP 1 0.5 0 2 0.25 0.25 3 0.125 0.375 4 0.05 0.45 5 0.025 0.475 6 0.0125 0.4875 7 0 0.5 - Gel Electrophoresis of the cDNA Target Pools
- 0.5 μl of each of the seven fragmented cDNA pools were analysed on a 2% agarose-gel. The data show that the cDNA is fragmented linearly with respect to the concentration of dUTP used in the synthesis. FIG. 29 shows the gel electrophoresis of fragmented cDNA from the yeast wild-type strain.
- Comparative Hybridization of the SSA4 Tile Array with Fluorochrome-Labelled Wild-Type and Δssa4 cDNA Target Pools and Post-hybridization Washes
- The fluorochrome-labelled cDNA samples, respectively, were combined (the different UDG-fragmented samples separately). The following were added: 3.75 μl 20×SSC (3×SSC final, pass through 0.22 μ filter prior to use to remove particulates) yeast tRNA (1 μg/μl final) 0.625 μl 1 M HEPES, pH 7.0 (25 mM final, pass through 0.22 It filter prior to use to remove particulates) 0.75
μl 10% SDS (0.3% final) and DEPC-water to 25 μl final volume. The labelled cDNA target samples were filtered in Millipore 0.22 μ filter spin column (Ultrafree-MC, Millipore, USA) according to the manufacturer's instructions, followed by incubation of the reaction mixture at 100° C. for 2-5 minutes. The cDNA probes were cooled at room temp for 2-5 minutes by spinning at maximum speed in a microcentrifuge. A LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the SSA4 microarrays spotted on Immobilizer™ MicroArray Slide, and the hybridization mixture was applied to the array from the side. An aliquot of 30 μL of 3×SSC was added to both ends of the hybridization chamber, and the slide was placed in the hybridization chamber (DieTech, USA). The chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridization, the slide was removed carefully from the hybridization chamber and washed using the following protocol. The slides were washed sequentially by plunging gently in 2×SSC/0.1% SDS at room temperature until the cover slip falls of into the washing solution, then in 1×SSC pH 7.0 (150 mM NaCl, 15 mM Sodium Citrate) at room temperature for 1 minute, then in 0.2×SSC, pH 7.0 (30 mM NaCl, 3 mM Sodium Citrate) at room temperature for 1 minute, and finally in 0.05×SSC (7.5 mM NaCl, 0.75 mM Sodium Citrate) for 5 seconds, followed by drying of the slides by spinning at 1000×g for 2 minutes. The slides were stored in a slide box in the dark until scanning. - Microarray Data Analysis
- The slides were scanned in a ScanArray 4000XL confocal laser scanner (Packard Instruments, USA). The hybridization data were analysed using the GenePix Pro 4.01 microarray analysis software (Axon, USA).
- In the data analysis, the differences in labelling efficiency between the two fluorescent dyes were scaled by using an internal normalization approach. The average signal intensities from the control capture probes (Table 17) were used to calculate the normalization factor. This factor was multiplied to the signal intensity values from the Cy-3 target. Analysis of the data demonstrates that capture probes with LNA in every third position have up to 5.2 fold higher signal-to-noise ratios, compared to the DNA capture probes (FIG. 30).
- This example illustrates the interpretation of microarray analysis of alternative mRNA splicing. Different LNA capture probe design types are formalized, and the expression constant Ø is introduced as a measurement of alternative splicing.
- Introduction
- The eukaryotic pre-mRNA is the subject of Splicing and Alternative Splicing, hence sequences refer to RNA sequences, Original sequence refers to pre-mRNA, and splice forms refer to mRNA sequences. The splicing is conducted by a cellular machinery named the spliceosome. The terms exons and introns can be used to refer to regions of pre-RNA sequences (or more specifically a single splice form). It is noted that a part of the corresponding DNA/pre-mRNA sequence that is an exon (not excised) in one splice form can potentially be absent in another splice form (e.g., partly absent in exon truncation and completely absent in exon skipping). Thus, the terms “constant regions” and “variable regions” (see below) are useful for characterizing the process of identifying different splice forms.
- Splicing can be defined as the production of a new sequence via the excision of part(s) of an original sequence (FIG. 31). Alternative splicing can be defined as the production of more than one novel sequence via the excision of different parts of the original sequence. When comparing two different splice forms, they can be divided into a constant region that is shared by both sequences and a variable region by which the two splice forms differ (FIG. 32).
- Alternative splicing can be categorized in terms of (i) whether or not the variable region is flanked by a single constant region or surrounded by two constant regions, (ii) the size of the variable region (e.g., exon skipping/intron retention vs. extension and truncation) [(intron/exon) 5′ and 3′], and (iii) the number of variable regions (and hence the number of splice forms).
- Capture Probe Design
- Capture Probe design can be divided into 3 distinct types according to their position: Merged Probes (MP) or Junction Probes, Unique Internal Probes (UIP), and Shared Internal Probes (SIP) (FIG. 33). Considering the case of a single variable region surrounded by constant regions, there are several different possible capture probe positions for each type (FIG. 34).
- Data Interpretation
- The aim of the analyses can be to determine (i) whether a given original sequence is subject to alternative splicing (i.e., whether there is more than one splice form present), and (ii) whether there is a difference in alternative splicing of the original sequence between two biological samples (i.e., whether the proportions between the two splice forms differ between biological samples). The analysis can also be used for data validation.
- Possible biases in the microarray platform include (a) noise in terms of non-specific binding and subsequent false signal, (b) differences in dye labeling efficiency, (c) differences in capture probe affinity, (d) differences in sample conditions (e.g., number of cells, and amount of RNA), and (e) differences in reverse transcriptase efficiency of different splice forms. Biases can be corrected for by various means of normalization and/or standardization.
- Data Analysis
- In order to analyze the expression of the different splice forms, the expression constant 0 is introduced. 0 denotes the relation between the proportions of the signals (capture probes a and b) between the labeled extracts from biological samples (labeled with Cy5 & Cy3). That is,
- (Cy5a/Cy3a)=(Cy5b/Cy3b)* 0 or,

- Considering normalization due to different biases and given a sample normalization factor S due to differences between the samples in terms of amounts of RNA, RT-efficiency, dye properties, etc. and a probe normalization factor P due to differences in probes in terms of affinity, position in target sequence, etc., the following equations apply.
- For two probes: a and b, a*P=b
- For two samples Cy5 & Cy3, Cy5*S=Cy3,
- Thus, considering two probes from two samples the signals are:
- Cy5a*P* S
- Cy5b* S
- Cy3a* P
- Cy3b
- With respect to Ø:

- Note that the calculation of Ø is not affected by the normalization factors S and P, hence it is not necessary to normalize the array data when interpreting alternative splice arrays with the use of the Expression constant Ø.
- Properties of Ø
- If Ø= 1, there is no difference in the proportions of the targets of capture probes a and b in the two samples. Even in the case of alternative splicing, it is not possible to determine whether there is more than a single splice form present using this particular method. If Ø≠1, there is a difference in the proportions of the targets of capture probes a and b in the two samples, thus there is a difference in splice pattern and therefore there must be more than one splice form present.
- Comparing Ø's
- Ø can be compared between different transcripts to determine whether they have correlated expression, and Ø's from sets of capture probes from the same transcript (different probes) can be averaged.
- Example
- Considering a simple example of a single large variable region surrounded by constant regions using a combination of a Merged Probe and a Shared Internal Probe. Calculating Ø of a single splice form can be performed using the following equation:
- Ø=(Cy5 MP*Cy3SIP)/ (Cy3 MP*Cy5SIP)
- If Ø=1, there is no difference in the proportions of the targets of capture probes a and b in the two samples, and it may not be possible to determine whether multiple splice forms are present using this particular method. If Ø≠1, there is a difference in the proportions of the targets of capture probes a and b, thus there is a difference in splice pattern and therefore there must be more than one splice form present.
- Conclusions
- It is possible to infer difference in expression level of two capture probe targets from two tissues when one is comparing the proportions of signals from one capture probe with the proportion of signals from the other probe. In contrast, single signals may be subject to biases from normalizations and standardizations for each probe and sample.
- The nucleic acid arrays of the invention can be generated by standard methods for either synthesis of nucleic acid probes that are then bonded to a solid support or synthesis of the nucleic acid probes on a solid support (e.g., by sequential addition of nucleotides to a reactive group on the solid support). In desirable methods for on-chip synthesis of the capture probes, photogenerated acids are produced in light-irradiate sites of the chip and used to deprotect the 5′-OH group of nucleic acid monomers and oligomers (e.g., to remove an acid-labile protecting group such as 5′—O—DMT) to which a nucleotide is to be added (Gao et al., Nucleic Acid Research 29:4744-4750, 2001). Standard methods can also be used to label the nucleic acids in a test sample with, e.g., a fluorescent label, incubate the labeled nucleic acid sample with the array, and remove any unbound or weakly bound test nucleic acids from the array. Exemplary methods are described, for example, in U.S.P.N. 6,410,229; 6,406,844; 6,403,957; 6,403,320; 6,403,317; 6,346,413; 6,344,316; 6,329,143; 6,310,189; 6,309,831; 6,309,823; 6,261,776; 6,239,273; 6,238,862; 6,156,501; 5,945,334; 5,919,523; 5,889,165; 5,885,837; 5,744,305; 5,445,934; 5,800,9927; and 5,874,219.
- In an exemplary method for synthesis of an array, capture probes were immobilized using AQ technology with a HEG5 linker (U.S.P.N. 6,033,784) onto an Immobilizer™ slide. An exemplary chip consists of 288 spots in four replicates (i.e., 1152 spots) with a pitch of 250 μm, and an exemplary hybridization buffer is 5×SSCT (i.e., 750 mM NaCl, 75 mM Sodium Citrate, pH 7.2, 0.05% Tween) and 10 mM MgCl 2. An exemplary target is a 45-mer oligonucleotide with Cy5 at the 5′ end and with a final concentration in the hybridization solution of 1 μM.
- Hybridization was performed with 200 1L hybridization solution in a hybridization chamber created by attaching a CoverWell gasket to the Immobilizer slide. The incubation was conducted overnight at 4° C. After hybridization, the hybridization solution was removed, and the chamber was flushed with 3×1.0 mL hybridization buffer described above without any target nucleic acid. A coverWell™ chamber was then filled with 200 μL hybridization solution without target. The slide was observed with a
Zeiss Axioplan 2 epifluorescence microscope with a 5×Fluar objective and a Cy5 filterset from OMEGA. The temperature of the microscope stage was controlled with a Peltier element. Thirty-five images at each temperature were acquired automatically with a Photometrics camera, automated shutter, and motorized microscope stage. The images were acquired, stitched together, calibrated and stored in stack by the software package “MetaVue” - Arrays can be generated using capture probes of any desired length (e.g., arrays of pentamers, hexamers, or heptamers.) In various embodiments, 1, 2, 3, 4, 5, 6, 7, 8, or more nucleotides of the probes are LNA nucleotides. Desirably, at least 1, 2, 3, 5, 7, 9, or all of the A and T nucleotides in the probes are LNA A and LNA T nucleotides. LNA nucleotides can be placed in any position of the capture probe, such as at the 5′ terminus, between the 5′ and 3′ termini, or at the 3′ terminus. LNA nucleotides may be consecutive or may be separated by one or more other nucleotides. The microarrays can be used to analyze target nucleic acids of any “AT” or “GC” content, and are especially useful for analyzing nucleic acids with high “AT” content because of the increased affinity of the microarrays of the present invention for such nucleic acids compared to traditional microarrays. Desirably, the array has at least 100, 200, 300, 400, 500, 600, 800, 1000, 2000, 5000, 8000, 10000, 15000, 20000, or more different probes. If desired, nucleotides with a universal base can be included in the capture probes to increase the T m of the capture probes (e.g., capture probes of less than 7, 6, 5, or 4 nucleotides). Exemplary “non-discriminatory” nucleotides include inosine, random nucleotides, 5 nitro-indole, LNA, inosine, and LNA 2-aminopurine. In desirable embodiments, 1, 2, 3, 4, 5, or more nucleotides with a universal base are located at the 5′ and/or 3′ termini of the capture probes.
- An exemplary application of these methods includes comparing hybridization patterns of cDNA or cRNA from a patient sample to classify early-tumors or detect an infection or a diseased state. The microarrays of the invention may also be used as a general tool to analyze the PCR products generated by amplification of a test sample with PCR primers for one or more nucleic acids of interest. For example, PCR primers can be used to amplify nucleic acids with a particular exon or exon-exon combination, and then the PCR products can be identified and/or quantified using a microarray of the invention. For identification of splice variants, PCR primers to specific exons can be used to amplify nucleic acids that are then applied to a microarray for detection and/or quantification as described herein. To detect microbial pathogens, species-specific PCR primers (e.g., primers specific for an exon whose sequence differs among species) can be used to amplify nucleic acids in a sample for subsequent analysis using a microarray. For example, the hybridization pattern of the PCR products to the array can be used to distinguish between different bacteria, viruses, or yeast and even between different strains of the same pathogenic species. In particular embodiments, the array is used to determine whether a patient sample contains a bacteria strain that is known to be resistant or susceptible to particular antibiotics or contains a virus or yeast strain known to be resistant or susceptible to certain drugs. Changes in product composition or raw material origin can also be detected using a microarray. The arrays can also be used to determine the composition of mRNA cocktails.
- Exemplary environmental microbiology applications of these arrays include identification of major rRNA types in contaminated soil samples and classification of microbial isolates. These rRNA amplificates are formed from rRNA by rtPCR or from the rDNA gene by conventional PCR. Numerous general and selective primers for different groups of organisms have been published. Most frequently an almost full length amplificate of the 16S rDNA gene is used (e.g., the primers 26F and 1492R). For purifying rRNA from a soil sample, standard methods such as one or more commercial extraction kits from companies such as QIAGEN (“Rneasy”, Q-biogene “RNA PLUS,” or “Total RNA safe” can be used.
- Any simultaneous use of more than one primer or probe is made difficult because the involved primers or probes must work under the same conditions. An indication of whether or not two or more primers or probes will work under the same conditions is the relative T ms at which the hybridized oligonucleotides dissociate. In cases where probes are applied for specific detection of homologous sequences such as splice variants, the ΔTm is of importance. A™ expresses the difference between Tm of the match and the Tm of the mismatch hybridizations. Generally, the larger ΔTm obtained, the more specific detection of the sequence of interest. In addition, a large ΔTm facilitates more probes to be used simultaneously and in this way a higher degree of multiplexity can be applied.
- High affinity nucleotide analogs such a LNA can be also be used universally to equalize the melting properties of oligonucleotides with different AT and CG content. The increased affinity of LNA adenosine and LNA thymidine corresponds approximately to the normal affinity of DNA guanine and DNA cytosine. An overall substitution of all DNA-A and DNA-T with LNA-A and LNA-T results in melting properties that are nearly sequence independent but only depend on the length of the oligonucleotide. This may be important for design of oligonucleotide probes used in large multiplex analysis. The effect of LNA A and T substitutions has been evaluated by predicting the T m value of all possible 9-mer oligonucleotides with different universal substitutions. The distribution of the 262,000 Tm-values exhibits a very homogeneous Tm value for universally LNA A and T substituted oligonucleotides. The standard deviation of the melting temperature for all 9-mers drops from 7.7° C. for pure DNA to only 2.2° C. for LNA A and T substituted oligonucleotides. This equalizing effect may also be utilized for photomediated on-chip synthesis of oligonucleotides.
- It is often difficult to design probes and primers with the same range of melting temperature due to the variance in A/T and G/C content of the probing sites. Highly A/T rich regions typically give lower T m values. Furthermore, if single mismatches are to be resolved, G/T mismatches are known to contribute little to A™. As discussed above, the use of LNA is a desirable way to solve problems related to multiplex use of primers and probes. LNA offers the possibility to adjust Tm and increase the ΔTm at the same time. LNA increases Tm with 4-8° C./substitution and increases ΔTm in many cases (Table 9).
TABLE 9 Demonstration of LNA controlled increase of Tm and Δ Tm. (SEQ ID NOs:) Perfect Single Tm of LNA: match mismatch DNA Duplexes 3′-ACGACCAC-5′ 3′-ACGGCCAC-5′ ΔTm LNA 8-mer 71° C. 45° C. 26° C. 5′-TGCTGGTG-3′ DNA 8- mer 35° C. 25° C. 10° C. 5′-TGCTGGTG-3′ - As LNA can be mixed with DNA during standard oligonucleotide synthesis, LNA can be placed at optimal positions in probes in order to adjust T m. The specificity of PCR may also be enhanced by the use of LNA in primers, or probes, and this facilitates a higher degree of multiplexity. By incorporation of LNA, the Tm of the primers or probes can be adjusted to work at the same temperature. Amplification or hybridization is more specific when LNA is included in primers or probes. This is due to the LNA increased A™, which relates to higher specificity. Once ΔTm of the primers or probes is high, more primers or probes can potentially be brought to work together.
- Prediction of T m
- LNA can be used to enhance any experiment that is based on hybridization. The series of algorithms described herein have been developed to predict the optimal use of LNA. Melting properties of 129 different LNA substituted capture probes hybridized described herein to their corresponding DNA targets were measured in solution using UV-spectrophotometry. The data set was divided into a training set with 90 oligonucleotides and a test set with 39 oligonucleotides. The training set was used for training of both linear regression models and neural networks. Neural networks trained with nearest neighbour information, length, and DNA/LNA neighbour effect are efficient for prediction of T m with the given set of data.
- Applications of the Normalization of Thermal Stability by LNA A and T Nucleotide Substitutions
- All assays in which DNA/RNA hybridization is conducted may benefit from the use of LNA in terms of increased specificity and quality. Exemplary uses include sequencing, primer extension assays, PCR amplification, such as multiplex PCR, allele specific PRamplification, molecular beacons, (e.g., nucleic acids be multiplexed with one colour based on multiple T m 's), Taq-man probes, in situ hybridization probes (e.g., chromosomal and bacterial 16S rRNA probes), capture probes to the mRNA poly-A tail, capture probes for microarray detection of SNPs, capture probes for expression microarrays (sensitivity increased 5-8 times), and capture probes for assessment of alternative mRNA splicing.
- LNA units have different melting properties than DNA and RNA nucleotides. Until recently, thermodynamical models for melting temperature prediction have existed for DNA and RNA only, but not for LNA. Now a T m prediction model for LNA/DNA mixed oligonucleotides has been developed (John SantaLucia, Jr. (1998) Proc. Natl. Acad. Sci. 95 1460-1465 and Tøstensen et al. “Prediction of Melting Temperature for LNA (Locked Nucleic Acid) Modified Oligonucleotides” Bioinformatics 2002, Bergen, Norway April 4-7 2002). The Tm prediction tool is available on-line at the Exiqon website (www.LNA-Tm.com and http://www.exiqon.com/Poster/Tm pred-ET-view.pdf).
- Numerous applications in molecular biology are based on the ability of DNA and RNA to hybridize in a temperature dependent manner (e.g. the microarray techniques, PCR reactions and blotting techniques). The melting properties of nucleic acid duplexes, in particular the melting temperature T m, are crucial for optimal design of such experiments. Tm is usually computed using a two-state thermodynamical model (Breslauer, Meth. Enzymol., 259:221-242, 1995). Several different groups have estimated model parameters for nearest neighbors in the sequence based on experimental data (for a review see SantaLucia, Proc. Natl. Acad. Sci., 95:1460-1465, 1998).
- The model described herein predicts the T m of duplexes of mixed LNA/DNA oligonucleotides hybridized to their complementary DNA strands. DNA monomers are denoted with lowercase letters, and LNA monomers are denoted with uppercase letters, e.g., there are eight types of monomers in the mixed strand: a, c, g, t, A, C, G and T. The model is based on the formula (SantaLucia, 1998, supra; Allawi et al., Biochemistry 36:10581-10594, 1997).
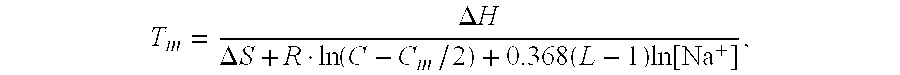
- in which the salt concentration [Na+] enters as an entropic correction together with the oligonucleotide concentrations. Ris the gas constant, C and C m are the concentrations of the two strands where C≧Cm, and L is the length of the strands. For self-complementary sequences, C−Cm/2 is replaced by the total strand concentration CT and a symmetry correction of −1.4 cal/k mol is added to ΔS (SantaLucia, 1998, supra).
- The LNA model differs from SantaLucia's DNA model in the way the changes in enthalpy ΔH and entropy ΔS are calculated. As in SantaLucia's model, they depend on nearest neighbor sequence information and special contributions for the terminal base-pairs in the two ends of the duplex. However, with eight types of monomers (LNA and DNA) the increased number of nearest neighbor combinations requires more model parameters to be determined and hence more data.
- Parameter Reduction
- Usually ΔH and Δs are calculated as a sum of contributions from all nearest neighbor pairs in the sequence. The inclusion of LNA doubles the number of monomer types and quadruples the number of possible nearest neighbor pairs. Parameter reduction strategies are used for matching the model complexity to limited data sets. A strategy for reducing model complexity is to sum ΔH from single base-pair contributions, which do not take the influence of adjacent nucleotides into account. However, nearest neighbor contributions are added as a correction term to the single base-pair contributions.
- Another strategy is to use hierarchically reduced monomer alphabets. Here, similar monomers are identified with the same letter. A four-letter alphabet, {w,s,W,S}, defines classes according to binding strength: w={a,t}, s={c,g}, W={A,T} and S={C,G}. The smallest alphabet, {D,L}, simply identifies the monomer type: DNA or LNA. As an example, the sequence GcTAAcTt can be written as SsWWWsWw or as LDLLLDLD.
- The principle is to split ΔH and ΔS into contributions that depend on different levels of detail of the sequence. The fine levels of detail require many parameters to be determined, while the coarse levels need fewer parameters. The more detailed contributions can then be treated as minor corrections, thus effectively reducing the total number of model parameters.
- Training
- Model parameters were determined using data from melting experiments on hundreds of oligonucleotides. The oligonucleotides were random sequences with lengths between 8 and 20 and a percentage of LNA between 20 and 70. Melting curves were obtained using a Perkin-Elmer UV λ-40 spectrophotometer, but only the T m values were used for modeling. Model parameters were adjusted using a gradient descent algorithm that minimizes the error function

- i.e., the distance between predicted and experimental T m values. Many different models were trained in this way and their performance was evaluated on test sets distinct from the training data. Seven reliable models were chosen and combined to form the committee model implemented at the Exiqon website (www.LNA-Tm.com.)
- Machine Learning And Thermodynamics
- The aim of this work has been to estimate T m values as accurately as possible. To this end, a machine learning approach has been adopted in which the prediction of the physical ΔH and ΔS quantities is less important. The parameters of this model may be inaccurate as thermodynamic quantities. First, the gradient descent algorithm produces a broad ensemble of models in which the ΔH and ΔS parameters can vary substantially, while maintaining an accuracy in the predicted Tm. Second, the thermodynamic meaning of ΔH and ΔS is based on a two-state assumption, which may not be realistic in every case. Even short oligonucleotides can form different secondary structures or melt through multiple-state transitions (Tøstensen et al., J. Phys. Chem. B. 105:1618-1630, 2001). Third, the use of an optical instrument instead of a calorimetric instrument (DSC) introduces an error in the measured ΔH and ΔS. Nevertheless, the uncertain thermodynamic interpretation of the ΔH and ΔS model parameters does not imply that the Tm prediction model is unreliable.
- Results
- The T m prediction model has been tested on two data sets that were not used during the training process. One set consisted of pure DNA oligonucleotides without LNA monomers and had a standard deviation of the residuals (SEP) of 1.57 degrees. The other set consisted of mixed oligonucleotides with both LNA and DNA and had a SEP of 5.25 degrees. The difference in prediction accuracy between the two types of oligonucleotides suggests that Tm prediction of mixed strands is a more complex task than Tm prediction of pure DNA. This is possibly due to irregularities in the duplex helical structure induced by the LNA monomers (Nielsen et al., Bioconjug. Chem. 11:228-238, 2000). The obtained prediction accuracy is in both cases adequate for most biological applications. In conclusion, the reduced nearest neighbor model implemented at the Exiqon website (www.LNA-Tm.com) can predict Tm surprisingly well for both types of oligonucleotides. This indicates that the parameter reduction strategy is applicable for other types of modified oligonucleotides.
- High affinity nucleotides such as LNA and other nucleotides that are conformationally restricted to prefer the C3′-endo conformation or nucleotides with a modified backbone and/or nucleobase stabilize a double helix configuration. As these effects are generally additive, the most stable duplex between a high affinity capture oligonucleotide and an unmodified target oligonucleotide should generally arise when all nucleotides in the capture probe or primer are replaced by their high affinity analogue. The most stable duplex should thus be formed between a fully modified LNA capture probe and the corresponding DNA/RNA target molecule. Such a fully modified capture probe should be more efficient in capturing target molecules, and the resulting duplex is more thermally stable.
- However, many high affinity nucleotides (e.g., as LNA) have an even higher affinity for other high affinity nucleotides (e.g., as LNA) than for DNA/RNA. A fully modified capture probe may thus form duplexes with itself, or if it is long enough, internal hairpins that are even more stable than duplexes with the desired target molecule. Probes with even a small inverse repeat segment where all constituent positions are substituted with high affinity nucleotides may bind to itself and be unable to bind the target. Thus, a sequence dependent substitution pattern is desirably used to avoid substitutions in positions that may form self-complementary base-pairs.
- For example, a computer algorithm can be used to automatically determine the optimal substitution pattern for any given capture probe sequence according to the following two criteria. First, the difference between the stability of (i) the duplex formed between the capture probe and the target molecule and (ii) the best possible duplex between two capture probes should be above a certain threshold. If this is not possible, then the substitution pattern with the largest possible difference is chosen. Second, the capture probe should contain as many substitutions as possible in order to bind as much target as possible at any given temperature and to increase the thermal stability of the formed duplex. Alternatively, the second criterion is substituted with the following alternative criterion to obtain capture probes with similar thermal stability. The number and position of capture probe substitutions should be adjusted so that all the duplexes between capture probes and targets have a similar thermal stability (i.e., T m equalization).
- For oligonucleotide capture probes such, incomplete matches between target and capture probe are likely to be a reproducible feature of the recorded biosignatures. For short probes, the second criterion for increasing thermal stability is more desirable that the alternative second criterion for T m equalization. For long capture probes and PCR primers, the second alternative criterion is desirably used since Tm equalization is desirable for these probes and primers.
- An exemplary algorithm works as follows. For each nucleotide sequence in an array of length n, all possible substitution patterns, i.e., 2′ different sequences are evaluated. Each evaluation consist of estimating the energetic stability of the duplex between the substituted capture sequence and a perfect match unmodified target (“target duplex”) and the energetic stability of the most stable duplex that can be formed between two substituted capture probes themselves (“self duplex”).
- The energetic stability estimate for a duplex may be calculated, e.g., using a Smith-Waterman algorithm with the following scoring matrix.
- Gap initiation penalty: −8
- Gap continuation penalty: −50
a c g t A C G T a −2 c −2 −2 g −2 3 −2 t 2 −2 1 −2 A −3 −3 −3 4 −3 C −3 −3 6 −3 −3 −3 G −3 6 −3 2 −3 9 −3 T 4 −3 2 −3 6 −3 3 −3 - This scoring matrix was partly based on the best parameter fit to a large (over 1000) number of melting curves of different DNA and LNA containing duplexes and partly by visual scoring of test capture probe efficiency. If desired, this scoring matrix may be optimized by optimizing the parameter fit as well as increasing or optimizing the dataset used to obtain these parameters.
- As an example of these calculations, the heptamer sequence ATGCAGA in which each position can be either an LNA or a DNA nucleotide is used. The target duplex formed between a fully modified capture probes with this sequence and its unmodified target receive a score of 34 as illustrated below.
Capture sequence: A-T-G-C-A-G-A | | | | | | | Target sequence: t-a-c-g-t-c-t Score: 4+4+6+6+4+6+4 = 34 - The most stable self duplex that can be formed between two modified capture probes has an almost equivalent energetic stability with a score of 30 as illustrated below.
Capture sequence: A-T-G-C-A-G-A | | | | Target sequence: A-G-A-C-G-T-A Score: +6+9+9+6 = 30 - Thus, the capture probe efficiency of a fully modified probe is likely reduced by its propensity to form a stable duplex with itself. In contrast, by choosing a slightly different substitution pattern, ATGcaGA in which capital letters represent LNA nucleotides, the stability of the target duplex is reduced slightly from 34 to 29.
Capture sequence: A-T-G-c-a-G-A | | | | | | | Target sequence: t-a-c-g-t-c-t Score: 4+4+6+3+2+6+4 = 29 - However, the most stable self complementary duplex that can be formed is reduced much more from 30 to 20, as illustrated below.
Capture sequence: A-T-G-c-a-G-A | | | | Target sequence: A-G-a-c-G-T-A Score: +4+6+6+4 = 20 - The difference between the stability of the desired target duplex and the undesired self duplex can be further increased by using the capture sequence AtgcaGA where the target duplex has a score of 24.
Capture sequence: A-t-g-c-a-G-A | | | | | | | Target sequence: t-a-c-g-t-c-t Score: 4+2+3+3+2+6+4 = 24 - Whereas the score of the self duplex is only 10, as shown below.
Capture sequence: A-t-g-c-a-G-A | | | | Target sequence: A-G-a-c-g-t-A Score: +2+3+3+2 = 10 - The additional destabilization of the self duplex is generally not required if the difference in stability between the target duplex and self duplex is above a threshold of 25% of the target duplex stability, as illustrated below.
- Discrimination for ATGCAGA=(34-30)/34=12%<threshold (25%)
- Discrimination for ATGcaGA =(29-20)/29=31%>threshold (25%)
- Discrimination for ATGCAGA=(24-10)/24=58%>threshold (25%)
- Thus, ATCcaGA is the substitution pattern with the highest degree of substitution for which the stability of the target duplex is adequately more stable than the stability of the best self duplex (e.g., above 25%).
- This algorithm can be used to determine desirable substitution patterns for any size capture probe or any given probe sequence. The following simple design rules may also be applied for probe design, especially for short probes. The best self alignment for the corresponding DNA capture probe in the sequence is determined using a simple Smith-Waterman scoring matrix of:
a c g t a −2 c −2 −2 g −2 3 −2 t 2 −2 1 −2 - Additionally, all possible positions in the sequence are substituted, with the exception of desirably avoiding the substitution of both bases of a self-complementary base-pair. The most stable self duplex thus does not contain any LNA:LNA base-pairs but only LNA:DNA basepairs.
- Exemplary programs are provided in the Computer Program Listing Appendix submitted on compact disc herewith. The oligod program (50287.007002_oligod.txt) takes a gene sequence as input and returns sequences for LNA spiked oligonucleotides. The dyp program (50287.007002_dyp.txt) is used by oligod to predict the secondary structure and self annealing properties of the oligonucleotides. The expression_array_param file (50287.007002_expression_array_param.txt) contains parameters used by the oligod program. 50287.007002_T m prediction.txt contains code for a Tm prediction program, and 50287.007002_Tm thermodynamic.txt contains code for a Tm thermodynamic model.
- Exemplary Computer
- Any of the methods described herein may be implemented using virtually any computer. FIG. 35 shows such an exemplary computer system.
Computer system 2 includes internal and external components. The internal components include aprocessor 4 coupled to amemory 6. The external components include a mass-storage device 8, e.g., a hard disk drive,user input devices 10, e.g., a keyboard and a mouse, adisplay 12, e.g., a monitor, and usually, anetwork link 14 capable of connecting the computer system to other computers to allow sharing of data and processing tasks. Programs are loaded into thememory 6 of thissystem 2 during operation. These programs include anoperating system 16, e.g., Microsoft Windows, which manages the computer system,software 18 that encodes common languages and functions to assist programs that implement the methods of this invention, andsoftware 20 that encodes the methods of the invention in a procedural language or symbolic package. Languages that can be used to program the methods include, without limitation, Visual C/C++ from Microsoft. In preferred applications, the methods of the invention are programmed in mathematical software packages that allow symbolic entry of equations and high-level specification of processing, including algorithms used in the execution of the programs, thereby freeing a user of the need to program procedurally individual equations or algorithms. An exemplary mathematical software package useful for this purpose is Matlab from Mathworks (Natick, Mass.). Using the Matlab software, one can also apply the Parallel Virtual Machine (PVM) module and Message Passing Interface (MPI), which supports processing on multiple processors. This implementation of PVM and MPI with the methods herein is accomplished using methods known in the art. Alternatively, the software or a portion thereof is encoded in dedicated circuitry by methods known in the art. - As disclosed in WO 99/14226, LNA are DNA analogues that form DNA- or RNA-heteroduplexes with exceptionally high thermal stability. LNA units include bicyclic compounds as shown immediately below where ENA refers to 2′O,4′C.-ethylene-bridged nucleic acids:

- References herein to Locked Nucleoside Analogues, LNA units, LNA monomers, or similar terms are inclusive of such compounds as disclosed in WO 99/14226, WO 00/56746, WO 00/56748, and WO 00/66604.
- Desirable LNA monomers and oligomers share some chemical properties of DNA and RNA; they are water soluble, can be separated by agarose gel electrophoresis, and can be ethanol precipitated.
- Desirable LNA monomers and oligonucleotide units include nucleoside units having a 2′-4′ cyclic linkage, as described in the International Patent Application WO 99/14226 and WO 0056746, WO 0056748, and WO 0066604. Desirable LNA monomers structures are exemplified in the formulae Ia and Ib below. In formula Ia the configuration of the furanose is denoted D-α and in formula Ib the configuration is denoted L-α. Configurations which are composed of mixtures of the two, e.g. D-β and L-α are also included.

- In Ia and Ib, X is oxygen, sulfur and carbon; B is a universal or modified base (particularly non-natural occurring base) e.g. pyrene and pyridyloxazole derivatives, pyrenyl, pyrenylmethylglycerol moieties, all of which may be optionally substituted. Other desirable universal bases include, pyrrole, diazole or triazole moieties, all of which may be optionally substituted, and other groups e.g. modified adenine, cytosine, 5-methylcytosine, isocytosine, pseudoisocytosine, guanine, thymine, uracil, 5-bromouracil, 5-propynyluracil, 5-propyny-6-fluoroluracil, 5-methylthiazoleuracil, 6-aminopurine, 2-aminopurine, inosine, diaminopurine, 7-propyne-7-deazaadenine, 7-propyne-7-deazaguanine. R 1, R2 or R2, R2′or R3, R5 and R5, are hydrogen, methyl, ethyl, propyl, propynyl, aminoalkyl, methoxy, propoxy, methoxy-ethoxy, fluoro, or chloro.
- P designates the radical position for an internucleoside linkage to a succeeding monomer, or a 5′-terminal group, R 3 or R3′ is an internucleoside linkage to a preceding monomer, or a 3′-terminal group. The internucleotide linkage may be a phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, phosphoroselenoate, phosphorodiselenoate, alkylphosphotriester, or methyl phosphonate. The internucleotide linkage may also contain non-phosphorous linkers, hydroxylamine derivatives (e.g.—CH2—NCH3—O—CH2—), hydrazine derivatives, e.g.—CH2—NCH3—NCH3—CH2, amid derivatives, e.g.—CH2—CO—NH—CH2—, CH2—NH—CO—CH2—. In Ia, R4′ and R2′ together designate —CH2—O—, —CH2—S—, —CH2—NH—, —CH2—NMe-, —CH2—CH2—O—, —CH2—CH2—S—, —CH2—CH2—NH—, or —CH2—CH2—NMe—where the oxygen, sulfuir or nitrogen, respectively, is attached to the 2′-position. In Formula Ib, R4′ and R2 together designate —CH2—O—, —CH2—S—, —CH2—NH—, —CH2—NMe-, —CH2—CH2—O—, —CH2—CH2—S—, —CH2—CH2—NH—, or —CH2—CH2—NMe- where the oxygen, sulphur or nitrogen, respectively, is attached to the 2-position (R2 configuration).
- Desirable LNA monomer structures are structures in which X is oxygen (Formula Ia and Ib); B is a universal base such as pyrene; R 1, R2 or R2, R3 or R3, R5 and R5′ are hydrogen; P is a phosphate, phosphorothioate, phosphorodithioate, phosphoramidate, and methyl phosphornates; R3 or R3′ is an internucleoside linkage to a preceding monomer, or a 3′-terminal group. In Formula 1a, R4 and R2 together designate —CH2—O—, —CH2—S—, —CH2—NH—, —CH2—NMe—, —CH2—CH2—O—, —CH2—CH2—S—, —CH2—CH2—NH—, or —CH2—CH2—NMe—where the oxygen, sulphur or nitrogen, respectively, is attached to the 2′-position, and in Formula Ib, R4′ and R2 together designate —CH2—O—, —CH2—S—, —CH2—NH—, —CH2—NMe—, —CH2—CH2—O—, —CH2—CH2—S—, —CH2—CH2—NH—, or —CH2—CH2—NMe—where the oxygen, sulphur or nitrogen, respectively, is attached to the 2′-position in the R2 configuration.
- Particularly desirable LNA monomer for incorporation into an oligonucleotide of the invention include those of the following formula Ia

- wherein X oxygen, sulfur, nitrogen, substituted nitrogen, carbon and substituted carbon, and desirably is oxygen; B is a modified base as discussed above e.g. an optionally substituted carbocyclic aryl such as optionally substituted pyrene or optionally substituted pyrenylmethylglycerol, or an optionally substituted heteroalicylic or optionally substituted heteroaromatic such as optionally substituted pyridyloxazole. Other desirable universal bases include, pyrrole, diazole or triazole moieties, all of which may be optionally substituted; R 1*, R2, R3, R5 and R5*are hydrogen; P designates the radical position for an internucleoside linkage to a succeeding monomer, or a 5′-terminal group, R3* is an internucleoside linkage to a preceding monomer, or a 3′-terminal group; and R2*and R4* together designate —O—CH2— or —CH2—CH2—O—, where the oxygen is attached in the 2′-position, or a linkage of —(CH2)n— where n is 2, 3 or 4, desirably 2, or a linkage of —S—CH2— or —NH—CH2—.
- LNA units of formula Ia where R 2* and R4* contain oxygen are sometimes referred to herein as “oxy-LNA”; units of formula IIa where R2* and R4* contain sulfur are sometimes referred to herein as “thio-LNA”; and units of formula Ia where R2* and R4* contain nitrogen are sometimes referred to herein as “amino-LNA”. For many applications, oxy-LNA units are desirable modified nucleic acid units of oligonucleotides of the invention.
- Particularly desirable LNA monomers for use in oligonucleotides of the invention are 2′-deoxyribonucleotides, ribonucleotides, and analogues thereof that are modified at the 2′-position in the ribose, such as 2′—O—methyl, 2′-fluoro, 2′-trifluoromethyl, 2′—O—(2-methoxyethyl), 2′—O—aminopropyl, 2′—O—dimethylamino-oxyethyl, 2′—O—fluoroethyl or 2′—O—propenyl, and analogues wherein the modification involves both the 2 ′ and 3′ position, desirably such analogues wherein the modifications links the 2′- and 3′-position in the ribose, such as those described in Nielsen et al., J. Chem. Soc., Perkin Trans. 1, 1997, 3423-33, and in WO 99/14226, and analogues wherein the modification involves both the 2′- and 4′-position, desirably such analogues wherein the modifications links the 2′- and 4′-position in the ribose, such as analogues having a —CH 2—S— or a —CH2—NH— or a —CH2—NMe- bridge (see Singh et al. J. Org. Chem. 1998, 6, 6078-9). Although LNA monomers having the α-D-ribo configuration are often the most applicable, other configurations also are suitable for purposes of the invention. Of particular use are α-L-ribo, the β-D-xylo and the α-L-xylo configurations (see Beier et al., Science, 1999, 283, 699 and Eschenmoser, Science, 1999, 284, 2118), in particular those having a 2′-4′-CH2—S—, —CH2—NH—, —CH2—O— or —CH2—NMe- bridge.
- In another desirable embodiment, LNA modified oligonucleotides used in this invention comprises oligonucleotides containing at least one LNA monomeric unit of the general scheme A above, wherein X, B, P are defined as above. One of the substituents R 2, R2*, R3, and R3* is a group P* which designates an internucleoside linkage to a preceding monomer, or a 2′/3′-terminal group. Two of the substituents of R1*, R2, R2*, R3, R4*, R5, R5*, R6, R6*, R7, and R7* when taken together designate a biradical structure selected from —(CR*R*)r—M—(CR* R*)s—, —(CR*R*)r—M—(CR*R*)s—M—, —M—(CR*R*)r+s—M—, —M—(CR*R*)r—M—(CR*R*)s—, —(CR*R*)r+s—, —M—, —M—M—, wherein each M is independently selected from —O—, —S—, —Si(R*)2—, —N(R*)—, >C═O, —C(═O)—N(R*)—, and —N(R*)—C(═O)—. Each R* and R1(1*)—R7(7*), which are not involved in the biradical, are independently selected from hydrogen, halogen, azido, cyano, nitro, hydroxy, mercapto, amino, mono- or di(C1-6-alkyl)amino, optionally substituted C1-6-alkoxy, optionally substituted C1-6-alkyl-alkyl, DNA intercalators, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands, and/or two adjacent (non-geminal) R* may together designate a double bond, and each of r and s is 0-4 with the proviso that the sum r+s is 1-5.
- Examples of LNA units are shown scheme B:

- wherein the groups, X and B are defined as above. P designates the radical position for an internucleoside linkage to a succeeding monomer, nucleoside such as an L-nucleoside, or a 5′-terminal group, such internucleoside linkage or 5′-terminal group optionally including the substituent R 5. One of the substituents R1, R2*, R3, and R3* is a group P* which designates an internucleoside linkage to a preceding monomer, or a 2′/3′-terminal group.
- Desirable nucleosides are L-nucleosides such as for example, derived dinucleoside monophosphates. The nucleoside can be comprised of either a beta-D, a beta-L or an alpha-L nucleoside. Desirable nucleosides may be linked as dimers wherein at least one of the nucleosides is a beta-L or alpha-L. B may also designate the pyrimidine bases cytosine, 5-methyl-cytosine, thymine, uracil, or 5-fluorouridine (5-FUdR) other 5-halo compounds, or the purine bases, adenosine, guanosine or inosine.
- As discussed above, a variety of LNA units may be employed in the monomers and oligomers of the invention including bicyclic and tricyclic DNA or RNA having a 2′-4′ or 2′-3′ sugar linkages; 2′-O,4′-C-methylene-β-D-ribofuranosyl moiety, known to adopt a locked C3′-endo RNA-like furanose conformation. Illustrative modified structures that may be included in oligonucleotides of the invention are shown in FIG. 1. Other nucleic acid units that may be included in an oligonucleotide of the invention may comprise 2′-deoxy-2′-fluoro ribonucleotides; 2′—O—methyl ribonucleotides; 2′—O—methoxyethyl ribonucleotides; peptide nucleic acids; 5-propynyl pyrimidine ribonucleotides; 7-deazapurine ribonucleotides; 2,6-diaminopurine ribonucleotides; and 2-thio-pyrimidine ribonucleotides, and nucleotides with other sugar groups (e.g. xylose).
- Oligonucleotides containing LNA are readily synthesized by standard phosphoramidite chemistry. The flexibility of the phosphoramidite synthesis approach further facilitates the easy production of LNA oligos carrying all types of standard linkers, fluorophores and reporter groups.
- Selective Binding Complementary (SBC) nucleotides are unable to form stable hybrids with each other, yet are able to form stable, sequence-specific hybrids with complementary unmodified strands of nucleic acids. Thus, the reduced ability of SBC oligonucleotides to form intramolecular hydrogen bond base-pairs between regions of substantially complementary sequence causes a reduced level of secondary structure. Self-complementarity is an important issue in nucleic acid technologies as reported for DNA, PNA and LNA, and in different biological applications especially in the field of homogeneous assays. LNA:LNA duplexes are the most thermally stable nucleic acid type duplex system known, making the reduction of self-complementarity even more important.
- Exemplary SBC oligonucleotides contain 2-amino-A (D) and 2sT incorporated in the same oligonucleotide as replacements of A and T, respectively. The SBC name refers to the fact that D and 2ST form a destabilised base-pair compared to the A-T base-pair, but D-T and 2ST-A base-pairs are normally more stable than the original A-T base-pair. Exemplary SBC-G nucleotides include inosine or LNA-inosine, and exemplary SBC-C nucleotides inclue PyrroloPyr, LNA- PyrroloPyr, 2sC, and LNA-2SC (FIG. 3). Other exemplary SBC nucleotides are shown in FIGS. 2 and 3. If desired, SBC nucleotides may be incorporated into the nucleic acids and arrays of the invention, using standard methods.
- The systems disclosed herein can provide significant nucleic acid probes for universal hybridization. In particular, universal hybridization can be accomplished with a conformationally restricted monomer, including a desirable pyrene LNA monomer. Universal hybridization behavior also can be accomplished in an RNA context. Additionally, the binding affinity of probes for universal hybridization can be increased by the introduction of high affinity monomers without compromising the base-pairing selectivity of bases neighboring the universal base.
- Incorporation of one or more modified nucleobases or nucleosidic bases into an oligonucleotide can provide significant advantages. Among other things, LNA oligonucleotides can often self-hybridize, rather than hybridize to another oligonucleotide. Use of one or more modified bases with the LNA units can modulate the propensity of the oligonucleotide to form double stranded structures with other oligonucleotides containing modified nucleobases including internal duplex formation, thereby inhibiting undesired self-hybridization.
- 2-Thiopyrimidine nucleosides can be prepared in several ways as described below. For example, the 2-thiouridine-nucleosides (IV) can be synthesized from a substituted uridine nucleoside (VIII) as described in the scheme below. By protection of the 04-position (IX) on the nucleobase thionation can be performed, O 2 position, which results in the 2-thio-uridine nucleoside (IV). Performing sulphurisation on both O2 and 04 results in 2,4-dithio-uridine nucleoside (X) which may be transformed into the 2-thio-uridine nucleoside (IV) (Saladino, et. al., Tetrahedron, 1996, 52, 6759). Another way is to generate a cyclic ether (XI) through reaction with the 5′ position this product can then be transformed to the 2-thio-uridine nucleoside (IV) or the 2—O—alkyl-uridine nucleoside (XII). The 2—O—alkyl-uridine nucleoside (XII) can also be generated by direct alkylation of the uridine nucleoside (VIII). Treatment of the 2—O—alkyl-uridine nucleoside (XII) can also be transformed into the 2-thio-uridine nucleoside (Brown et. al., J. Chem. Soc. 1957, 868; Singer, et. al., Proc. Natl. Acad. Sci. USA, 1983, 80, 4884; Rajur and McLaughlin, Tetrahedron Lett., 1992, 33, 6081).

- In another method, lewis acid-catalyzed.condensation of a properly substituted sugar (I) and a substituted 2-thio-uracil (II) can result in a substituted 2-thio-uridine nucleoside of the structure (III) which by further synthetic manipulations can be transformed into the LNA 2-thiouridine nucleoside (IV) (Hamamura et. al., Moffatt, J. Med. Chem., 1972, 15, 1061; Bretner et. al., J. Med. Chem., 1993, 36, 3611).

- Using a properly substituted amino-sugar (V), a 2-thio-uridine nucleoside can be synthesized through ring-synthesis of the nucleobase by reaction of the amino sugar (V) and an substituted isothiocyanate(VI), yielding the substituted LNA 2-thio-uracil nucleoside (VI) (Shaw and Warrener, J. Chem. Soc. 1957, 153; Cusack et al., J. Chem. Soc.
Perkin 1, 1973, 1721).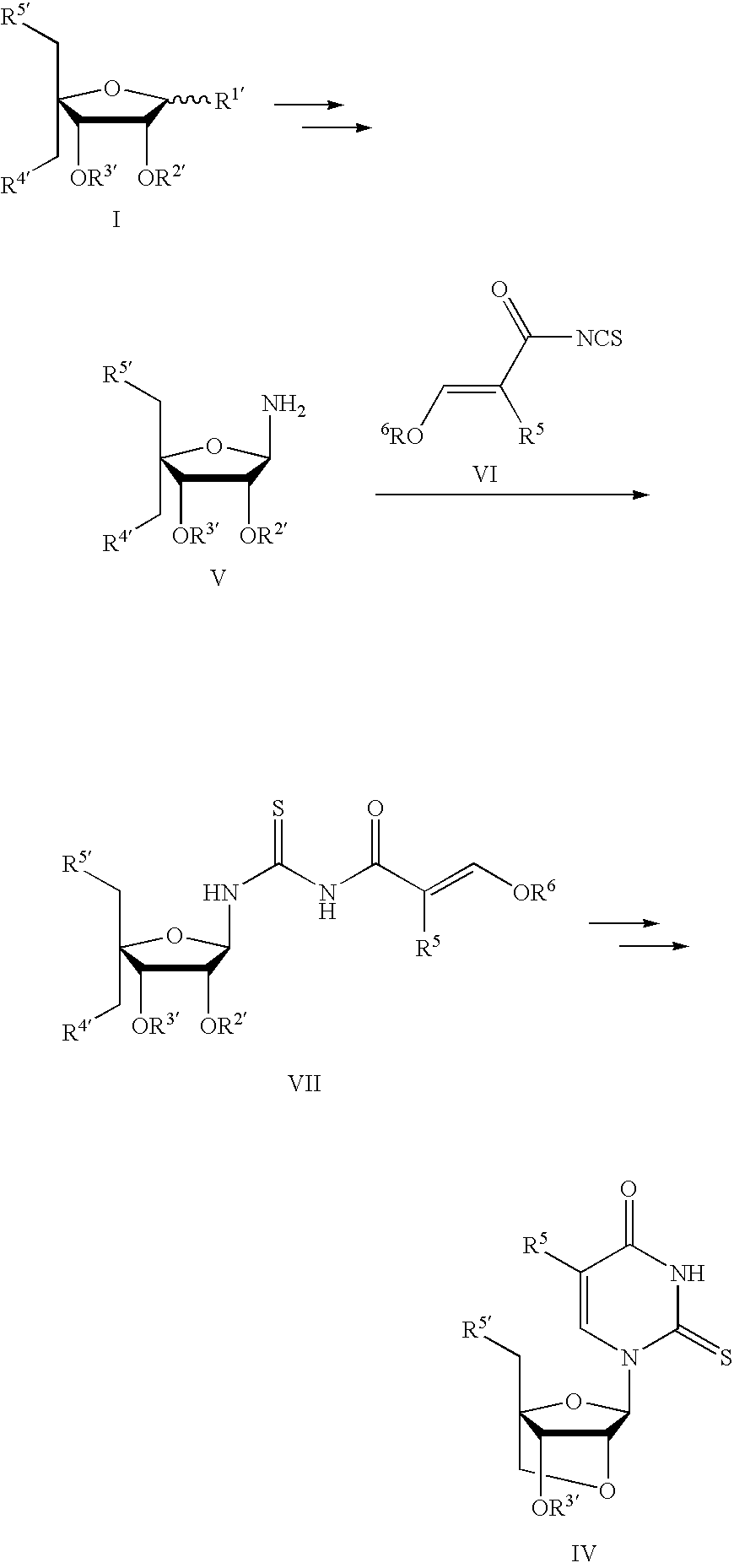
- Three different strategies for synthesis of 2sT-LNA are outlined in the Summary of the Invention section. Strategy A involves coupling a glycosyl-donor and a nucleobase, using standard methodology for synthesis of existing LNA monomers. Strategy B involves ring synthesis of the nucleobase. This strategy is desirable because the availability of 1-amino-LNA enables introduction of a variety of new nucleobases. Strategy C includes modification of T-LNA; the easy synthesis of LNA-T diol makes this an attractive pathway.
- In a desirable embodiment, 2sT-LNA is synthesized as illustrated in the scheme below.

- In particular, the known
coupling sugar 1,2-di—O—acetyl-3, 5 di—O—benzyl, 4-C- mesyloxymethyl, α,62-D-ribofuranose 1 was coupled with the nucleobase 2-thio-thymidine in a Vorbruggen type reaction. Thus, the nucleobase was silyilated and condensed with the sugar using SnCl4 as catalyst to promote thereaction affording nucleoside 2. Mass spectrometry and NMR subsequently identified the isolated product as the desired one. NMR data were compared with published data of a 2-thio-thymindine derivative (Kuimelis and Nambiar, Nucleic Acid Res., 1994, 22, 1429-1436) in order to validate the correct attachment point of the nucleobase. - Subsequently, a base mediated ring-closing reaction afforded the di-
benzylated LNA derivative 3 in 77% yield. The signals in the 1H-NMR spectrum of the compound appeared as singlets, thus proving that the cyclization had occurred to give the LNA skeleton, in which the 1′-H and 2′-H are perpendicular to each other causing the 3J1′,2′ to be 0 Hz. MALDI mass spectrometry was likewise used for the identification of the compound. - The LNA derivative was protected at the nucleobase with the toluoyl protective group to give 4. This group is well known for the protection of 2-thio-thymidine derivatives, (Kuimelis and Nambiar, Nucleic Acid Res., 1994, 22, 1429-1436). The protection of the nucleobase occurs at both the N-3 and the 0-4 position and hence the compound is isolated as a mixture of two compounds. NMR shows that the ratio of the two isomers in the isolated mixture is 2:1.
- These methods are described further below.
- 1-(2—O—acetyl-3-O, 5—O—dibenzyl, 4-C-mesyloxymethyl-β-D-ribofuranosyl)-2-thio-thymine (2)
- 1,2-di—O—acetyl-3, 5 di—O—dibenzyl, 4-C-mesyloxymethyl, α,β-D-ribofuranose (1, 2.0g, 3.83 mmol) and 2-thio-thymine (552 mg, 3.89 mmol) were co-evaporated with anhydrous acetonitrile (100 ml) and redissolved in anhydrous acetonitrile (80 ml), N,O-bistrimethylsilylacetamide (1.5, 5.85 mmol) was added, and the reaction was stirred at 80° C. for one hour. The mixture was cooled to 0° C., SnCl 4 (0.9 ml, 7.66 mmol) was added, and the reaction was left to stir for 24 hours. The reaction mixture was diluted with EtOAc and washed with NaHCO3 and subsequently with water. The organic phase was dried (Na2SO4) and evaporated to dryness. The product was purified using column chromatography, giving the thio-thymidine derivative 2 (1.1 g, 1.82 mmol, 40%) as a white foam. Rf(10% THF/dichloromethane): 0.75. MALDI-MS: 627 (M+Na) 13C-NMR (CDCl3): δ=174.40, 169.29, 159.89, 136.13, 136.51, 136.05, 128.62, 128.56, 128.41, 128.29, 128.07, 127.89, 12767, 116.18, 91.41, 86.21, 75.59, 75.31, 74.46, 74.22, 73.61, 69.25, 69.04, 37.52, 20.62, 11.91
- (1R,3R,4R, 7S)-7-(benzyloxy)-1-(benzyloxymethyl)-3-(2-thiothymidine)-2,5-dioxabicyclo[2.2.1]heptane (3)
- 1-(2—O—acetyl-3-0,5—O—dibenzyl, 4-C-mesyloxymethyl-β-D-ribofuranosyl)-2-thio-thymine (2, 630 mg, 1.04 mmol) was dissolved in dioxane (15 ml) and water (8 ml), and aqueous NaOH (2M, 5 ml) was added, and the reaction was left to stir at room temperature for one hour. The yellow solution was neutralized with HCl (IM, 6 ml) affording a precipitation. The mixture was diluted with dichloromethane and ethyl acetate causing an emulsion. After separation, the aqueous phase extracted with ethyl acetate, and the combined organic phase was dried (Na 2SO4) and evaporated to dryness. The compound was purified by column chromatography (0-2, then 5% THF/dichloromethane), giving the ring
closed compound 3 as a white foam (370 mg, 0.79 mmol, 77%). Rf (2% MeOH/dichloromethane): 0.23. - MALDI-MS: 488 (M+Na) 13C-NMR (CDCl3): δ=173.14, 160.39, 137.20, 136.63, 136.00, 128.46, 128.34, 128.02, 127.66, 115.52, 90.29, 87.77, 77.39,75.26, 73.77, 72.07, 71.70, 64.15, 30.17, 12.33
- 1H-NMR (CDCl3): δ=9.87 (s, 1H), 7.69 (d, 1.1 Hz, 1H), 7.26-7.37 (m, 10H), 6.13 (s, 1H), 4.84 (s, 1H), 4.66 (d, J=11.3 Hz, 1H), 4.61 (s, 2H), 4.52 (d, 11.5 Hz, 1H), 4.04 (d, J=7.7 Hz, 1H), 3.93 (s, 1H), 3.88 (d, J=11.0 Hz, 1H), 3.82 (d, J=7.7 Hz, 1H), 3.82 (d, J=10.8 Hz, 1H), 1.59 (d, J=1.1 Hz, 3H)
- (1R,3R,4R, 7S)-7-(benzyloxy)-1-(benzyloxymethyl)-3-(2-thio-N3/04-toluoyl- thymidine)-2,5-dioxabicyclo[2.2.1]heptane (4)
- (1R, 3R, 4R, 7S)-7-(benzyloxy)-1-(benzyloxymethyl)-3-(2-thiothymidine)-2,5-dioxabicyclo[2.2.1]heptane (3, 290 mg, 0.62 mmol) was dissolved in anhydrous pyridine and diisopropylethylamine (0.2 ml, 1.1 5 mmol), toluoyl chloride (0.25 ml, 1.89 mmol) was added, and the reaction mixture was stirred at room temperature for three hours. After completion, the reaction mixture was diluted with dichloromethane, and the reaction was quenched by addition of water. The phases were separated, and the organic phase was dried (Na 2SO4) and evaporate to dryness. The residue was co-evaporated with toluene. The product was purified by column chromatography (0-1% MeOH/dichloromethane) to give
nucleoside 4 as a white foam (320 mg, 0.55 mmol, 89%). Rf (2% MeOH/dichloromethane): 0.78. - MALDI-MS: 606 (M+Na) 13C-NMR (CDCl3): δ=171.98, 168.30, 160.30, 145.92, 145.82, 137.22, 136.65, 135.98, 130.39, 130.27, 129.85, 129.50, 128.51, 128.41, 128.08, 127.73, 115.11, 90.10, 87.81, 76.01, 75.80, 75.39, 75.01, 73.83, 72.19, 72.09, 71.74, 64.15, 21.75, 12.40.
- 2′-O, 4′-C-methylene linked (LNA) nucleosides containing hypoxanthine (or inosine) (LNA-I), 2,6-diaminopurine (LNA-D), and 2-aminopurine (LNA-2AP) nucleobases were efficiently prepared via convergent syntheses. The nucleosides were converted into phosphoramidite monomers and incorporated into LNA oligonucleotides using an automated phosphoramidite method. The complexing properties of oligonucleotides containing these LNA nucleosides were assessed against perfect and singly mismatch DNA.

- Hypoxantine, the base found in the nucleotides inosine and deoxyinosine, is considered as a guanine analogue in nucleic acids.
- Oligonucleotides containing 2,6-diaminopurine replacements for adenines are expected to bind more strongly to their complementary sequences especially as part of A-type helixes due to the potential formation of three hydrogen bounds with thymine or uracil. The reported effect of 2,6-diaminopurine deoxyriboside (D) on the stability of polynucleotide duplexes reaches, on average, about 1.5° C. per modification. Higher stabilization effects for mismatches were observed for D nucleosides involved in formation of duplexes prone to form A-type helixes. LNA D and
LNA 2′-OMe-D are expected to have increased stabilization and mismatch discrimination. LNA can be used in combination with 2-thio-T for construction of selectively binding complementary oligonucleotides. Taking into consideration the extremely high stability of LNA:LNA duplexes, this approach might be very useful for constructing of LNA containing capture probes and antisense reagents including drugs. - 2-Aminopurine (2-AP) is a fluorescent nucleobase (emission at 363 nm), which is useful for probing nucleic acids structure and dynamics and for hybridizing with thymine in Watson-crick geometry. LNA-I, LNA-D, and/or LNA-2AP may be used in the nucleic acids of the present invention, e.g., to increase the priming efficiency of DNA oligonucleotides in PCR experiments and to construct selectively binding complementary agents.
- Synthesis of LNA-I
- The convergent method adopted for preparation of LNA monomers (Koshkin et al., J. Org. Chem. 66:8504, 2001) was successfully applied for syntheses of the modified LNA nucleotides 1-3. The synthetic route to LNA-I phosphoramidite 11 is depicted in the scheme below. The previously described 4-C-branched furanose 4 (Koshkin et al., supra) was used as a glycosyl donor in coupling reaction with silylated hypoxantine by the method of Vorbriiggen et al. (Vorbrüggen et al., Chem. Ber. 114:1234, 1981; Vorbrüggen et al., Chem. Ber. 114:1256, 1981; and Vorbrigen, Acta Biochim. Pol., 43:25, 1996). The reaction resulted in high yield formation of desired β-
configurated nucleoside derivative 5. However, analogous to the coupling reaction of 4 with protected guanines, the formation of undesired N-7 isomer (ratio of N-91N-7=4:1) was also detected. The mixture of the isomers was used for the ring closing reaction and protectedLNA nucleoside 6 was isolated in 68% yield as a crystalline compound. The correct structure of the isolated isomer was confirmed later by chemical conversion of LNA-I into LNA-A nucleoside (vide infra). Deprotection of the 5′-hydroxy group of 6 was accomplished via two-step procedure developed for the syntheses of other LNA nucleosides ( Koshkin et al., supra). First, 5′—O—mesyl group was displaced by sodium benzoate to producenucleoside 7. The latter was converted into 5′-hydroxy derivative 8 after saponification of the 5′-benzoate. Direct removal of the 3′—O—benzyl group fromcompound 8 was unsuccessful under the conditions tested due to a solubility problem. Therefore,compound 8 was converted to DMT-protectednucleoside 9 prior to catalytic debenzylation of the 3′—O—hydroxy group. Thephosphoramidite 11 was finally afforded via standard phosphitylation (McBride et al., Tetrahedron Lett. 24:245, 1983; Sinha et al., Tetrahedron Lett. 24:5843, 1983; and Sinha et al., Nucleic Acids Res. 12:4539, 1984) of thenucleoside 10. In order to verify the correct orientation of the glycoside bond (N-9 isomer) in synthesized LNA-I nucleoside,compound 7 was successfully converted into the known LNA-A derivative 13 (Koshkin et al., supra) (Scheme 2). Thus, a treatment of 7 with phosphoryl chloride according to the procedure reported by Martin (Helv. Chim. Acta 78:486, 1995) resulted in a high yield formation of 6-chloropurine derivative 12. Theadenosine derivative 13 was derived from 12 after reaction with ammonia. - Exemplary Analytical Data
- Data for
compound 8 includes the following: mp 302-305° C. (dec). 1H NMR (DMSO-d6): 88.16, (s, 1H), 8.06 (s, 1H), 7.30-7.20 (m, 5H), 5.95 (s, 1H), 4.69 (s, 1H), 4.63 (s, 2H), 4.28 (s, 1H), 3.95 (d, J=7.7, 1H), 3.83 (m, 3H). 13C NMR (DMSO-d6): δ 156.6, 147.3, 146.1, 137.9, 137.3, 128.3, 127.6, 127.5, 124.5, 88.2, 85.4, 77.0, 72.1, 71.3, 56.7. MALDI-MS m/z: (M+H)+. - Anal. Calcd for C 18H18N4O5.{fraction (5/12)}H2O: C, 57,21; H, 5.02; N, 14.82. Found: C, 57,47; H, 4.95; N, 14.17.
- Analysis of
compound 11 indicated that 31p NMR (DMSO-d6): δ 148.90.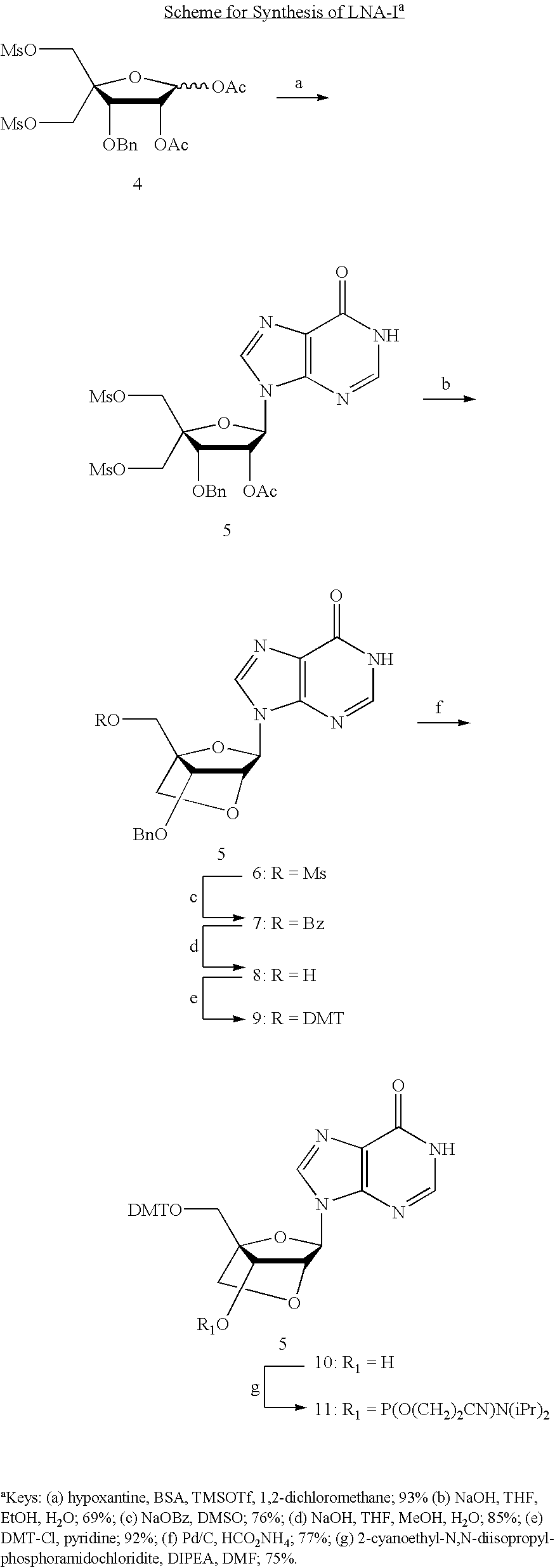
- Exemplary Experimental Conditions
- (1R,3R,4R, 7S)-7-(2-Cyanoethoxy(diisopropylamino)phosphinoxy)-1-(4,4′-dimethoxytrityloxymethyl)-3-(hyroxanthin-9-yl)-dioxabicyclo [2.2.1]heptane (11)
- Compound 10 (530 mg, 0.90 mmol, described previously, (see for example, WO 00/56746) was dissolved in anhydrous EtOAc (5 mL) and cooled in an ice-bath. DIPEA (0.47 mL, 2.7 mmol) and (250 μL, 1.1 mmol) were added under intensive stirring. Formation of insoluble material was observed, and CH 2Cl2 (3 mL) was added to produce a clear solution. More 2-cyanoethyl-N,N-diisopropylphosphoramidochloridite (200 μL, 0.88 mmol) was added after one hour, and the mixture was stirred overnight. EtOAc (30 mL) was added, the mixture was washed with sat. NaHCO3 (2×50 mL), brine (50 mL), dried (Na2SO4), and concentrated to a solid residue. Purification by silica gel HPLC (1-5% MeOH/CH2Cl2 v/v, containing 0.1% of pyridine) gave compound 11 (495 mg, 75%) as a white solid material. 31p NMR (DMSO-d6): δ 148.90.
- Synthesis of LNA-D
- Taking advantage of a high availability of the natural deoxy- and riboguanosines, a number of effective methods were developed for their conversion into 2,6-diaminopurine (D) nucleosides (Fathi et al., Tetrahedron Lett. 31:319, 1990; Gryaznov et al., Tetrahedron Lett., 35:2489, 1994; and Lakshman et al., Org. Lett., 2:927, 2000). However, the production of LNA-G nucleoside is a multi-step synthetic procedure.

- For the synthesis of LNA-D nucleoside, a novel synthesis method was developed that employed a common convergent scheme, related to the strategy used earlier for the synthesis of its anhydrohexitol counterpart (Boudou et al., Nucleic Acids Res. 27:1450, 1999). In particular, a properly protected carbohydride unit was conjugated with 6-chloro-2-aminopurine to give a stable 6-chloro intermediate derivative (scheme below) which was further converted into desired diaminopurine nucleoside.

- Thus, it was shown that glycosylation of 2-chloro-6-aminopurine with
compound 4 resulted in highly stereoselective formation of thenucleoside derivative 14. To promote the ring closing reaction, a solution of 14 inaqueous 1,4-dioxane was treated with 1 0-fold excess of sodium

- hydroxide to give
bicyclic compound 15 in 87% yield. The standard reaction with sodium benzoate in hot DMF was then successfully applied for displacement of 5′-mesylate of 15. Notably, this reaction proceeded in very selective manner and no side products originating from the modification of the nucleobase were detected. The desiredcompound 16 was precipitated from the reaction mixture after addition of water. In order to introduce the 6-amino group into nucleobase structure, intermediate 6-azido derivative 17 was synthesized via reaction of 16 with sodium azide. Thenucleoside derivative 18 was isolated as a crystalline compound after saponification of the 5′-benzoate of 17. Subsequent catalytic hydrogenation of 18 on palladium hydroxide resulted in simultaneous reduction of 6-azido and 3′-benzyl groups to give LNA-D diol 19 after crystallization from water. By the use of peracelation method, 2- and 6-amino groups of 19 were benzoylated at the next step to give the nucleobase protected derivative 20, which was in the standard way further converted intophosphoramidite monomer 21. This phosphoramidite has been produced in a quantity of 0.5 grams. - Exemplary Analytical Data
- Data for
compound 19 includes the following: 1H NMR (DMSO-d6): δ 7.81 (s, 1H), 6.78 (br s, 2H), 5.91 (br s, 2H), 5.71 (s, 1H), 5.66 (br s, 1H), 5.04 (br s, 1H), 4.31 (s, 1H), 4.20 (s, 1H), 3.90 (d, J=7.7 Hz, 1H), 3.77 (m, 2H), 3.73 (d, J=7.7 Hz, 1H). 13C NMR(DMSO-d6): δ 160.5, 156.2, 150.9, 134.2, 113.4, 88.3, 85.0, 79.3, 71.5, 70.0, 56.8. MALDI-MS m/z: 295.0 (M+H)+. Anal. Calcd for C11H14N6O4.1.5H2O: C, 41,12; H, 5.33; N, 26.15. Found: C, 41.24; H, 5.19; N, 25.80. - The 31P NMR (DMSO-d6) spectrum for
compound 24 contained signals at δ 149.19 and 148.98. - Data for
compound 23 includes the following: crystallized from MeOH. mp. 227.5-229° C. (dec). 1H NMR (DMSO-d6): δ 8.60 (s, 1H), 8.15 (s, 1H), 6.64 (br s, 2H), 5.82 (s, 1H), 5.71 (br s, 1H), 5.04 (br s, 1H), 4.40 (s, 1H), 4.21 (s, 1H), 3.92 (d, J=7.7 Hz, 1H), 3.79 (m, 2H), 3.75 (d, J=7.7 Hz, 1H). 13C NMR(DMSO-d6): δ 160.6, 152.0, 149.4, 139.3, 127.1, 88.6, 84.8, 79.1, 71.6, 70.2, 56.8. MALDI-MS m/z: 334.7 (M+H)+. - For protected
compound 23, the 31 P NMR (DMSO-d6) spectrum has a signal at 148.93 and 148.85. - Exemplary Experimental Conditions
- (1S,3R,4R, 7S)-3-(2-amino-6-chloropurin-9-yl)-7-benzyloxy-1-methanesulfonoxymethyl-2,5-dioxabicyclo[2.2.11heptane (15)
- To a solution of compound 14 (40 g, 64.5 mmol) in 1,4-dioxane (300 mL) was added 1 M NaOH (350 mL). The mixture was stirred for one hour at 0° C., neutralized with AcOH (40 mL), and washed with CH 2Cl2 (2×200 mL). The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The solid residue was purified by silica gel flash chromatography to give compound 15 (27.1 g, 87%) as a white solid material. 1H NMR (CDCl3): δ 7.84 (s, 1H), 7.32-7.26 (m, 5H), 5.91 (s, 1H), 4.73 (s, 1H), 4.66 (d, J=11.7 Hz, 1H), 4.61 (d, J=11.7 Hz, 1H), 4.59 (s, 2H), 4.31 (s, 1H), 4.18 (d, J=8.0 Hz, 2H), 3.99 (d, J=7.9 Hz, 1H), 3.05 (s, 3H). 13C NMR (CDCl3) δ 158.9, 152.2, 151.4, 139.1, 136.4, 128.4, 128.2, 127.7, 125.3, 86.5, 85.2, 77.2, 76.8, 72.4, 72.1, 64.0, 37.7. MALDI-MS m/z 482.1 [M+H]+.
- (1S,3R,4R, 7S)-3-(2-amino-6-chloropurin-9-yl)-1-benzoyloxymethyl-7-benzyloxy-2,5-dioxabicyclo[2.2.1]heptane (16)
- A mixture of sodium benzoate (7.78 g, 54 mmol) and compound 1513g, 27 mmol) was suspended in anhydrous DMF (150 mL) and stirred for two hours at 105° C. Ice-cold water (500 mL) was added to the solution under intensive stirring. The precipitate was filtered off, washed with water, and dried in vacuo. The intermediate product 16 (8 g) was used for ext step without further purification. Analytical sample was additionally purified by silica gel HPLC (0-2% MeOH/CH 2Cl2 v/v). 1H NMR (CDCl3) δ 7.98-7.95 (m, 2H), 7.79 (s, 1H), 7.62-7.58 (m, 1H), 7.48-7.44 (m, 2H), 7.24 (m, 5H), 5.93 (s, 1H), 4.80 (d, J=12.6 Hz, 1H), 4.77 (s, 1H), 4.67 (d, J=11.9 Hz, 1H), 4.65 (d, J=12.6 Hz, 1H), 4.56 (d, J=11.9 Hz, 1H), 4.27 (d, J=8.0 Hz, 1H), 4.25 (s, 1H), 4.08 (d, J=7.9 Hz, 1H). 3C NMR (CDCl3) δ 165.7, 158.8, 152.1, 151.3, 138.9, 136.4, 133.4, 129.4, 129.0, 128.5, 128.4, 128.2, 127.6, 125.4, 86.4, 85.7, 77.2, 76.7, 72.5, 72.3, 59.5. MALDI-MS m/z 508.0 [M+H]+. (1s,3R,4R, 75)-3-(2-amino-6-azidopurin-9-yl)-7-benzyloxy-1-hydroxymethyl-2,5-dioxabicyclo[2.2.1jheptane (18)
- All the amount of
compound 16 from the previous experiment was dissolved in anhydrous DMSO (100 mL) and NaN3 (5.4 g, 83 mmol) was added. The mixture was stirred for two hours at 100° C. and cooled to room temperature. Water (400 ml) was added, and the mixture was stirred for 30 minutes at 0° C. (ice-bath) to give a yellowish precipitate 17. The precipitate was filtered off, washed with water, and dissolved in THF (25 mL). 2M NaOH (30 mL) was then added to the solution, and after 15 minutes of stirring the mixture was neutralized with AcOH (4 mL). The mixture was concentrated to approximately ½ of its volume and cooled in an ice-bath. The titel compound was collected by filtration, washed with cold water, and dried in vacuo. - Yield: 8.8 g (79% from 15). 1H NMR (DMSO-d6) δ 8.53 (br s, 2H), 8.23 (s, 1H), 7.31-7.26 (m, 5H), 6.00 (s, 1H), 5.26 (t, J=5.7 Hz,1H), 4.76 (s,1H), 4.64 (s, 1H), 4.31 (s, 1H), 3.99 (d, J=7.9 Hz, 1H), 3.88-3.85 (m, 3H). 13C NMR (DMSO-d6) δ 146.0, 144.0, 143.8, 137.9, 137.0, 128.3, 127.7, 127.6, 112.3, 88.3, 85.6, 77.1, 77.0, 72.2, 71.4, 56.8. MALDI-MS m/z 384.7 [M+H]+for 2,6-diaminopurine product, 410.5 [M+H]+. Anal. Calcd for C18H18 N8O4: C, 52.68; H, 4.42; N. 27.30. Found: C, 52.62; H, 4.36; N. 26.94.
- (1S,3,4, 7s)-3-(2,6-Diaminopurin-9-yl)-7-hydroxy-1-hydroxymethyl-2,5-dioxabicyclo[2.2.1]heptane (19)
- To a suspension of compound 18 (8 g, 19.5 mmol) in MeOH (100 mL) were added Pd(OH) 2/C (20%, 5.5 g) and HCO2NH4 (3g). The mixture was refluxed for 30 minutes and more HCO2NH4 (3g) was added. After refluxing for further 30 minutes, the catalyst was filtered off and washed with boiling MeOH/H2O (1/1 v/v, 200 mL). The combined filtrates were concentrated to approximately 100 mL and cooled in an ice-bath. The precipitate was filtered off, washed with ice-cold H2O and dried in vacuo to give compound 19 (5.4 g, 94%) as a white solid material. 1H NMR (DMSO-d6): δ 7.81 (s, 1H), 6.78 (br s, 2H), 5.91 (br s, 2H), 5.71 (s, 1H), 5.66 (br s, 1H), 5.04 (br s, 1H), 4.31 (s, 1H), 4.20 (s, 1H), 3.90 (d, J=7.7 Hz, 1H), 3.77 (m, 2H), 3.73 (d, J=7.7 Hz, 1H). 13C NMR(DMSO-d6) δ 160.5, 156.2, 150.9, 134.2, 113.4, 88.3, 85.0, 79.3, 71.5, 70.0, 56.8. MALDI-MS m/z: 295.0 (M+H)+. Anal. Calcd for C11H14N6O4.1.5H2O: C, 41,12; H, 5.33; N, 26.15. Found: C, 41.24; H, 5.19; N, 25.80.
- (1S,3R,4R, 7S)-3-(2,6-Di-(N-benzoylamino)purin-9-yl)-7-hydroxy-1-hydroxymethyl-2,5-dioxabicyclo[2.2.1]heptane (20)
- A solution of compound 19 (0.5 g, 1.7 mmol) in anhydrous pyridine (20 mL) was cooled in an ice-bath and benzoyl chloride (1.5 mL, 12.9 mmol) was added under intensive stirring. The mixture was allowed to warm to room temperature and was stirred overnight. Ethanol (20 mL) and 2 M NaOH (20 mL) were added, and the mixture was stirred for an additional hour. EtOAc (75 mL) was added and the solution was washed with water (2×50 mL). The combined aqueous layers were washed with CH 2Cl2 (2×50 mL). The combined organic phases were dried (Na2SO4) and concentrated under reduced pressure to a solid residue. The residue was suspended in Et2O (75 mL, under refluxing for 30 minutes) and cooled in an ice-bath. The product was collected by filtration, washed with cold Et2O, and dried in vacuo to give compound 20 (530 mg, 62%) as a slightly yellow solid material.
- (1R,3R,4R, 7S)-3-(2,6-Di-(N-benzoylamino)purin-9-yl)-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-2,5-dioxabicyclo[2.2.1]heptane (21)
- Compound 20 (530 mg, 1.06 mmol) was co-evaporated with anhydrous pyridine (2×20 mL) and dissolved in anhydrous piridine (10 mL). DMT-Cl (600 mg, 1.77 mmol) was added, and the solution was stirred overnight at rt. The mixture was diluted with EtOAc (100 mL), washed with saturated NaHCO 3 (100 mL) and brine (50 mL). Organic layer was dried over Na2SO4 and concentrated under reduced pressure. Purification by silica gel HPLC (20-100% EtOAc/hexane v/v, containing 0.1% of pyridine) gave compound 21 (670 mg, 79%) as a white solid material.
- 1H NMR (CD3OD): δ 8.41 (s, 1H), 8.15-8.03 (m, 4H), 7.71-7.22 (m, 15H), 6.92-6.86 (m, 4H), 6.23 (s, 1H), 4.77 (s, 1H), 4.62 (s, 1H), 4.03 (d, J=7.9 Hz, 1H), 3.99 (d, J=7.9 Hz, 1H), 3.79 (s, 6H), 3.67 (d, J=10.9 Hz, 1H), 3.54 (d, J=10.8 Hz, 1H),. MALDI-MS m/z: 826 (M+Na)+. Anal. Calcd for C46H4 0N608-H2O: C, 67.14; H, 5.14; N, 10.21. Found: C, 67.24; H, 4.97; N, 10.11.
- (1R,3R,4R, 7S)-7-(2-Cyanoethoxy(diisopropylamino)phosphinoxy)-3-(2,6-di-(N-benzoylamino)purin-9-yl)-1-(4,4′-dimethoxytrityloxymethyl)-2,5-dioxabicyclo[2.2]heptane (21).
- To a stirred solution of compound 20 (640 mg, 0.8 mmol) in anhydrous DMF (5 mL) were added DIPEA (420 L, 2.4 mmol) and 2-cyanoethyl-N,N-diisopropylphosphoramidochloridite (300 μL, 1.2 mmol). The mixture was stirred for 1.5 hours at room temperature, diluted with EtOAc (100 mL), and washed with saturated NaHCO 3 (2×100 mL) and brine (50 mL). Organic layer was dried (Na2SO4) and concentrated under reduced pressure to give a yellow solid residue. Purification by silica gel HPLC (20-100% EtOAc/hexene containing 0.1% of pyridine) gave compound 21 (590 mg, 74%) as a white solid material. 31p NMR (DMSO-d6) δ 149.19, 148.98.
- Synthesis ofpac-protected LNA-D amidite
- The following scheme illustrates a method for synthesizing a Pac-protected version of LNA-D amidite.

-
Compound 17 - Compound 7 (1 g, 3.39 mmol) was co-evaporated with anhydrous DMF (2×10 mL) and dissolved in DMF (10 mL). Imidazole (0.69 g, 10.17 mmol) and 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (1.4 mL, 4.37 mmol) were added, and the mixture was stirred overnight. H 2O (100 mL) was added under intensive stirring to precipitate nucleoside material. The precipitate was filtered off, washed with H2O, and dried in vacuo. Crystallization from ethanol gave compound 17 (1.15 g, 63%) as a white solid material. MALDI-MS: m/z 537.3 (M+H)+.
-
Compound 18 - To a solution of compound 17 (1.15 g, 2.14 mmol) in anhydrous pyridine (5 mL) was added phenoxyacetic anhydride (2 g, 7.0 mmol) and the mixture was stirred for four hours. EtOAc (100 mL) was added, and the solution was washed with sat. NaHCO 3 (2×100 mL), brine (50 mL), dried (Na2SO4), and concentrated to a solid residue. Purification by silica gel HPLC (50-100% v/v EtOAc/hexane) gave compound 18 (1.65 g, 95%) as a white solid material. MALDI-MS: m/z 827.3 (M+Na)+.
- (1S,3R,4R, 7S)-3-(2,6-Di-(N-phenoxyacetylamino)purin-9-yl)-7-hydroxy-1-hydroxymethyl-2,5-dioxabicyclo[2.2.1]heptane (19)
- To a solution of compound 18 (0.96 g, 1.19 mmol) in anhydrous THF (10 mL) was added Et 3N 3HF (0.2 mL) and the mixture was stirred overnight at room temperature. The formed precipitate was collected by filtration and washed with THF (5 mL) and pentane (5 mL) to give after drying compound 19 (650 mg, 97%) as a white solid material. MALDI-MS: m/z 563.0 (M+H)+.
- (1R,3R,4R, 7S)-3-(2,6-Di-(N-phenoxyacetylamino)-purin-9-yl)-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-2,5-dioxabicyclo[2.2.1]heptane (20)
- To a solution of compound 19 (650 mg, 1.15 mmol) was added DMT-CI (500 mg, 1.48 mmol). The mixture was stirred for five hours, diluted with EtOAc (100 mL), and washed with sat. NaHCO 3 (2×100 mL). The organic layer was dried and concentrated to a solid residue. Crystallization from EtOAc gave compound 20 (810 mg, 81%) as a white solid material.
- (1R,3R,4R, 7S)-7-(2-Cyanoethoxy(diisopropylamino)phosphinoxy)-3-(2,6-di-(N-phenoxyacetylamino)-purin-9-yl)-1-(4,4′-dimethoxytrityloxymethyl)-2,5-dioxabicyclo[2.2.1]heptane (21)
- To a solution of compound 20 (800 mg, 0.92 mmol) in anhydrous DMF (10 mL) were added 0.75 M solution of DCI in EtOAc (0.7 mL) and 2-cyanoethyl tetraisopropylphosphorodiamidite (0.32 mL, 1.01 mmol). The mixture was stirred at room temperature overnight and EtOAc (75 mL) was added. The resulting solution was washed with sat. NaHCO 3 and brine, dried and concentrated to a solid residue. Purification by silica gel HPLC (30-100% v/v EtOAc/hexane, containing 0.1% of pyridine) gave phosphoramidite 21 (550 mg, 56%) as a white solid material.
- 31P NMR (DMSO-d6): δ 149.08, 148.8.
- Synthesis of LNA-2AP
- The
intermediate derivative 16 was also used for the synthesis of LNA-2AP nucleoside. First, the 5′—O—benzoyl group of 16 was hydrolyzed by aqueous sodium hydroxide to give thenucleoside derivative 22 in 72% yield. The conditions of catalytic transfer hydrogenation usually used for removal of the 3′—O—benzyl group turned out to be suitable for complete dechlorination of the nucleobase of 22. Thus, totally deprotected LNA-2AP nucleoside 23 was afforded in high yield after refluxing of the methanolic solution of 22 in the presence of paladium hydroxide and ammonium formate. The 2-amine of 23 was selectively protected with an amidine group after treatment with N,N-dimethylformamide dimethyl acetal. The resultingdiol 24 was then 5′—O—DMT protected and 3′—O—phosphitylated to yield the desired phosphoramidite LNA-2AP monomer 25 (McBride et al., J. Am. Chem. Soc. 108:2040, 1986).
- Exemplary Experimental Conditions
- (1S,3,4R, 7s)-3-(2-amino-6-chloropurin-9-yl)-7-benzyloxy-1-hydroxymethyl-2,5-dioxabicyclo[2.2.1]heptane (22)
- To a solution of compound 16 (3 g, 5.92 mmol) in 1,4-dioxane (20 mL) was added 2 M NaOH (20 mL) and the mixture was stirred for one hour. AcOH (3 mL) was added, and the solvents were removed under reduced pressure. The solid residue was re-dissolved in 20% MeOH/EtAc (50 mL), washed with NaHCO 3 (2×50 mL), dried (Na2SO4) and concentrated to a solid residue. The residue was purified by silica gel column chromatography (1-2% MeOH/EtAc v/v) to give compound 22 (1.72 g, 72%) as a white solid material.
- (1S,3R,4R, 7S)-3-(2-aminopurin-9-yl)-7-hydroxy-1-hydroxymethyl-2,5-dioxabicyclo[2.2.1] lheptane (23)
- To a solution of compound 22 (0.72 g, 1.79 mmol) in MeOH/dioxane (1/1 v/v) were added Pd(OH) 2/C (20%, 0.5 g) and HCO2NH4 (1.5 g, 23.8 mmol). The mixture was stirred under refluxing for 30 minutes and cooled to room temperature. The catalyst was filtered off and washed with MeOH. The combined filtrates were concentrated under reduced pressure to yield compound 23 (0.44 g, 89%) as a white solid material. Analytical sample was crystallized from MeOH. mp. 227.5-229° C. (dec). 1H NMR (DMSO-d6): δ 8.60 (s, 1H), 8.15 (s, 1H), 6.64 (br s, 2H), 5.82 (s, 1H), 5.71 (br s, 1H), 5.04 (br s, 1H), 4.40 (s, 1H), 4.21 (s, 1H), 3.92 (d, J=7.7 Hz, 1H), 3.79 (m, 2H), 3.75 (d, J=7.7 Hz,1H). 13C NMR (DMSO-d6): δ 160.6, 152.0, 149.4, 139.3, 127.1, 88.6, 84.8, 79.1, 71.6, 70.2, 56.8.
- (1R,3,4R, 7S)-1-(4,4′-di meth oxytrityloxymethyl)-3-(2-N-(dimethylaminomethylidene)aminopurin-9-yl)-7-hydroxy-2,5-dioxabicyclo[2.2.1]heptane (5′ DMT protected version of 24)
- Compound 23 (0.4 g, 1.43 mmol) was co-evaporated with anhydrous DMF (10 mL) and dissolved in DMF (15 mL). N,N-Dimethylformamide dimethylacetal (0.8 mL) was added and the solution was stirred for three days at room temperature. Water (5 mL) was added, and the solvents were removed under reduced pressure. The solid residue was co-evaporated with anhydrous pyridine (2×10 mL) and dissolved in anhydrous pyridine (5 mL). DMT-Cl (0.7 g, 2.1 mmol) was added, the solution was stirred for four hours, diluted with EtOAc (50 mL), and washed with NaHCO 3 (2×50 mL) and brine (50 mL). Organic layer was dried (Na2SO4) and concentrated to a yellow solid residue. Purification by silica gel HPLC (1-6% MeOH/CH2Cl2 v/v, containing 0.1% of pyridine) gave the 5′ DMT protected version of compound 24 (0.87 g, 87%) as a white solid material.
- (1R,3R,4R, 7S)-7-(2-Cya noethoxy(diisopropylamino)phosphinoxy)-1-(4,4′-dimethoxytrityloxymethyl)-3-(2-N-(dimethylaminomethylidene)aminopurin-9-yl)-2,5-dioxabicyclo[2.2]heptane (25)
- The 5′ DMT protected version of compound 24 (0.5 g, 0.79 mmol) was dissolved in anhydrous DMF (10 mL) and DIPEA (350 μL) and 2-cyanoethyl-N,N-diisopropylphosphoramidochloridite (250 μL) were added. The mixture was stirred for one hour, diluted with EtOAc (50 mL), washed with saturated NaHCO 3 (2×100 mL) and brine (50 mL), dried (Na2SO4), and concentrated to a solid residue. Purification by silica gel HPLC (0-3% MeOH/CH2Cl2 v/v, containing 0.1% of pyridine) gave compound 25 (0.42 g, 64%) as a white solid material.31P NMR (DMSO-d6) δ 148.93, 148.85.
- Synthesis of Oligomers
- Along with previously described LNA phosphoramidites (Koshkin et al., supra; and Pedersen et al., Synthesis p. 802, 2002), the
phosphoramidite monomers - The complexing properties of oligonucleotides containing new LNA monomers 1-3 were assessed. Comparative binding data from an 8-mer LNA sequence is shown in Table 9 as the melting temperatures against complementary single stranded DNA. An exemplary sequence for this comparison is GACATAGG, which is the central part of a capture probe used for SNP detection in Gluc1VS7-7asA (A:a mismatch position). The thermal stabilities of reference DNA duplexes (entries 1-7, Table 9) can be directly compared with their LNA counterparts (entries 8-14). The hybridizing ability of all LNA 8-mers is superior to that of isosequencial DNA ogonucleotides. The average melting temperatures of DNA and LNA 8-mers against coplementary DNAs typically differ by about 40° C. The replacement of one internal LNA-A nucleotide by LNA-D resulted in the further stabilization of the complementary duplex (i.e., compare
entries 8 and 11) by 6.2° C. Interestingly, the analogous replacement made in an DNA octamer destabilized the corresponding duplex by 0.5° C. (i.e.,entries 1 and 4). D-nucleosides may facilitate a B to A helix transition, because the A-type structure of an LNA:DNA duplex is more suitable for effective D:t pairing. This stabilizing effect is expected to be even more pronounced for LNA:RNA duplexes, which can be very useful for construction of antisense or other gene-silencing reagents. The mismatch discrimination ability of the D-nucleoside was also studied (entry 11). In comparison to LNA-A (entry 8) D-nucleoside demonstrated remarkable increased mismatch discrimination against DNA-g nucleoside.TABLE 9 Melting temperatures (Tm) of the complementary DNA—DNA and LNA-DNA duplexes.a Modified monomers (LNA are in CAPITALs): I = inosine; D = 2,6-diaminopurine; X = 2-aminopurine. Tm (± 0.5° C.) of the duplexes Oligonucleotide with complementary deoxynucleotide Entry structure 3′- ctgtatcc 3′- ctgaatcc 3′- ctggatcc 3′- ctgcatcc 1 5′-gacatagg 23.8 <10 <10 <10 2 5′-gacttagg <10 22.6 <10 <10 3 5′-gacgtagg <10 <10 <10 25.0 4 5′-gacdtagg 23.3 <10 <10 <10 5 5′-gdcdtdgg 33.4 <10 <10 17.7 6 5′-gacitagg <10 <10 <10 20.9 7 5′-gacxtagg <10 <10 <10 <10 8 5′-GACATAGG 61.6 38.2 43.4 40.6 9 5′-GACTTAGG 28.0 60.7 36.4 23.5 10 5′-GACGTAGG 55.0 32b 41b 70.9 11 5′-GACDTAGG 67.8 42.2 41.4 52.4 12 5′-GDCDTDGG 78.3 55.9 54.7 63.8 13 5′-GACITAGG 53.1 48.2 43.0 59.9 14 5′-GACXTAGG 60.8 45.5 44.0 53.9 -
TABLE 10 The mismatch discrimination effect of the chimeric LNA-DNA 12-mers containing LNA-A or LNA-D nucleo- sides against the point of mutation (SEQ ID NOs:). Tm (±0.5° C.) of the complementary duplexes with The structure of DNA oligonucleotides (Δ Tm LNA-DNA between singly mismatched oligonucleotide and perfect duplexes) HNFas128A-2 caacatcccaca caacaacccaca tGtggGATGttg 61.0 45.9 (−15.1) tGtggGDTGttg 65.5 49.7 (−15.8) Gluc53as-A aagagtccagtg aagaggccagtg cAmCtgGAmCtctt 61.5 50.6 (−10.9) cAmCtgGDmCtctt 65.3 45.4 (−19.9) -
TABLE 11 Melting temperatures of the LNA and DNA duplexes (LNAs are CAPITALIZED) containing 2-thio-deoxythymidine (s) and diamino- purineriboside (d). See Table 9 for experimental conditions. oligo Tm (±0.5° C.) of complementary duplexes with structure 3′- ctgtatcc 3′- ctgsatcc 3′- CTGsATCC 3′- CTGtATCC 3′- CTGTATCC 5′-gacatagg 23.8 27 54.4 49.4 54.6 5′-gacdtagg 23.3 <6 45.4 55.2 60.5 5′-GACATAGG 61.6 64.6 87* 88 88 5′-GACDTAGG 67.8 59.4 80 >90 >90 - The
furanopyrimidine phosphoramidite 6 pC used for incorporation of the pyrroloC analogue can be synthesized from LNA-U through a series of reactions as illustrated below and in FIG. 6. Starting from LNA-U lpC iodine can be introduced on the 5 position on the nucleobase (Chang and Welch, J. Med. Chem. 1963, 6, 428). This compound can be used in a Sonogashira type palladium coupling reaction (Sonogashira, Tohda and Hagihara, Tetrahedron Lett. 1975, 4467) resulting in the 5-ethynyl-LNA-U 3 pC. The 5-ethynyl-LNA-U 3 pC can be transformed to thefuranopyrimidie LNA analogue 4 pC when reacted with CuI, and then transformed into the DMT-protectedphosphoramidite 6 pC (Woo, Meyer, and Gamper, Nucleic Acids Res., 1996, 24, 2470). LNA-PyrroloPyr-SBC-C is formed when 6 pC or an oligonucleotide containing 6 pC is deprotected with ammonia.
- Desirable modified bases are covalently linked to the 1′-position of a furanosyl ring, particularly to the 1′-position of a 2′,4′-linked furanosyl ring, especially to the 1′-position of a 2′-0,4′-C-methylene-beta-D-ribofuranosyl ring.
- As discussed above, other desirable modified bases contain one or more carbon alicyclic or carbocyclic aryl units, i.e. non-aromatic or aromatic cyclic units that contain only carbon atoms as ring members. Modified bases that contain carbocyclic aryl groups are generally desirable, particularly a moiety that contains multiple linked aromatic groups, particularly groups that contain fused rings. That is, optionally substituted polynuclear aromatic groups are especially desirable such as optionally substituted naphthyl, optionally substituted anthracenyl, optionally substituted phenanthrenyl, optionally substituted pyrenyl, optionally substituted chrysenyl, optionally substituted benzanthracenyl, optionally substituted dibenzanthracenyl, optionally substituted benzopyrenyl, with substituted or unsubstituted pyrenyl being particularly desirable.
- Without being bound by any theory, it is believed that such carbon alicyclic and/or carbocyclic aryl modified bases can increase hydrophobic interaction with neighboring bases of an oligonucleotide. Those interactions can enhance the stability of a hybridized oligo pair, without necessity of interactions between bases of the distinct oligos of the hybridized pair.
- Again without being bound by any theory, it is further believed that such hydrophobic interactions can be particularly favored by platelike stacking of neighboring bases, i.e. intercalation. Such intercalation will be promoted if the base comprises a moiety with a relatively planar extended structure, such as provided by an aromatic group, particularly a carbocyclic aryl group having multiple fused rings. This is indicated by the increases in T m values exhibited by oligos having LNA units with pyrenyl nucleobases relative to comparable oligos having LNA units with naphthyl nucleobases.
- Modified bases that contain one or more heteroalicyclic or heteroaromatic groups also are suitable for use in LNA units, particularly such non-aromatic and aromatic groups that contains one or more N, O or S atoms as ring members, particularly at least one sulfur atom, and from 5 to about 8 ring members. Also desirable is a nucleo base that contains two or more fused rings, where at least one of the rings is a heteroalicyclic or heteroaromatic group containing 1, 2, or 3 N, O, or S atoms as ring members.
- In general, desirable are modified bases that contain 2, 3, 4, 5, 6, 7 or 8 fused rings, which may be carbon alicyclic, heteroalicyclic, carbocyclic aryl and/or heteroaromatic; more desirably modified bases that contain 3, 4, 5, or 6 fused rings, which may be carbon alicyclic, heteroalicyclic, carbocyclic aryl and/or heteroaromatic, and desirably the fused rings are each aromatic, particularly carbocyclic aryl.
- In some embodiments, the base is not an optionally substituted oxazole, optionally substituted imidazole, or optionally substituted isoxazole modified base.
- Other suitable modified bases for use in LNA units in accordance with the invention include optionally substituted pyridyloxazole, optionally substituted pyrenylmethylglycerol, optionally substituted pyrrole, optionally substituted diazole and optionally substituted triazole groups.
- Desirable modified bases of the present invention when incorporated into an oligonucleotide containing all LNA units or a mixture of LNA and DNA or RNA units will exhibit substantially constant T m values upon hybridization with a complementary oligonucleotide, irrespective of the bases present on the complementary oligonucleotide.
- In some embodiments, one or more of the common RNA or commonly used derivatives thereof, such as 2′—O—methyl, 2′-fluoro, 2′-allyl, and 2′—O—methoxyethoxy derivatives are combined with at least one nucleotide with a universal base to generate an oligonucleotide having between five to 100 nucleotides.
- Modified nucleic acid compounds may comprise a variety of nucleic acid units e.g. nucleoside and/or nucleotide units. As discussed above, an LNA nucleic acid unit has a carbon or hetero alicyclic ring with four to six ring members, e.g. a furanose ring, or other alicyclic ring structures such as a cyclopentyl, cycloheptyl, tetrahydropyranyl, oxepanyl, tetrahydrothiophenyl, pyrrolidinyl, thianyl, thiepanyl, piperidinyl, and the like.
- In an aspect of the invention, at least one ring atom of the carbon or hetero alicyclic group is taken to form a further cyclic linkage to thereby provide a multi-cyclic group. The cyclic linkage may include one or more, typically two atoms, of the carbon or hetero alicyclic group. The cyclic linkage also may include one or more atoms that are substituents, but not ring members, of the carbon or hetero alicyclic group.
- Unless indicated otherwise, an alicyclic group as referred to herein is inclusive of group having all carbon ring members as well as groups having one or more hetero atom (e.g. N, O, S or Se) ring members. The disclosure of the group as a “carbon or hetero alicyclic group” further indicates that the alicyclic group may contain all carbon ring members (i.e. a carbon alicyclic) or may contain one or more hetero atom ring members (i.e. a hetero alicyclic). Alicyclic groups are understood not to be aromatic, and typically are fully saturated within the ring (i.e. no endocyclic multiple bonds).
- Desirably, the alicyclic ring is a hetero alicyclic, i.e. the alicyclic group has one or more hetero atoms ring members, typically one or two hetero atom ring members such as O, N, S or Se, with oxygen being often desirable.
- The one or more cyclic linkages of an alicyclic group may be comprised completely of carbon atoms, or generally more desirable, one or more hetero atoms such as O, S, N or Se, desirably oxygen for at least some embodiments. The cyclic linkage will typically contain one or two or three hetero atoms, more typically one or two hetero atoms in a single cyclic linkage.
- The one or more cyclic linkages of a nucleic acid compound of the invention can have a number of alternative configurations and/or configurations. For instance, cyclic linkages of nucleic acid compounds of the invention will include at least one alicyclic ring atom. The cyclic linkage may be disubstituted to a single alicyclic atom, or two adjacent or non-adjacent alicyclic ring atoms may be included in a cyclic linkage. Still further, a cyclic linkage may include a single alicyclic ring atom, and a further atom that is a substituent but not a ring member of the alicyclic group.
- For instance, as discussed above, if the alicyclic group is a furanosyl-type ring, desirable cyclic linkages include the following: C-1′, C-2′; C-2′, C-3′; C-2′, C-4′; or a C-2′, C-5′ linkage.
- A cyclic linkage will typically comprise, in addition to the one or more alicyclic group ring atoms, 2 to 6 atoms in addition to the alicyclic ring members, more typically 3 or 4 atoms in addition to the alicyclic ring member(s).
- The alicyclic group atoms that are incorporated into a cyclic linkage are typically carbon atoms, but hetero atoms such as nitrogen of the alicyclic group also may be incorporated into a cyclic linkage.
- Specifically desirable modified nucleic acids for use oligonucleotides of the invention include locked nucleic acids as disclosed in WO99/14226 (which include bicyclic and tricyclic DNA or RNA having a 2′-4′ or 2′-3′ sugar linkages); 2′-deoxy-2′-fluoro ribonucleotides; 2′—O—methyl ribonucleotides; 2′—O—methoxyethyl ribonucleotides; peptide nucleic acids; 5-propynyl pyrimidine ribonucleotides; 7-deazapurine ribonucleotides; 2,6-diaminopurine ribonucleotides; and 2-thio-pyrimidine ribonucleotides.
- LNA units as disclosed in WO 99/14226 are in general particularly desirable modified nucleic acids for incorporation into an oligonucleotide of the invention. Additionally, the nucleic acids may be modified at either the 3′ and/or 5′ end by any type of modification known in the art. For example, either or both ends may be capped with a protecting group, attached to a flexible linking group, attached to a reactive group to aid in attachment to the substrate surface, etc. Desirable LNA units also are disclosed in WO 0056746, WO 0056748, and WO 0066604.
- Desirable syntheses of pyrene-LNA monomers is shown in the following
Schemes below Schemes 

- A wide variety of modified nucleic acids may be employed, including those that have 2′—modification of hydroxyl, 2′-O-methyl, 2′-fluoro, 2′-trifluoromethyl, 2′—O—(2-methoxyethyl), 2′O-aminopropyl, 2′-O- dimethylamino-oxyethyl, 2′—O—fluoroethyl or 2′—O—propenyl. The nucleic acid may further include 3′modification, desirably where the 2′- and 3′-position of the ribose group is linked. The nucleic acid also may contain a modification at the 4′-position, desirably where the 2′-and 4′positions of the ribose group are linked such as by a 2′-4′ link of —CH 2—S—, -CH2—NH—, or —CH2—NMe—bridge.
- The nucleotide also may have a variety of configurations such as α-D-ribo, β-D-xylo, or α-L-xylo configuration.
- The internucleoside linkages of the units of oligos of the invention may be natural phosphorodiester linkages, or other linkages such as —O—P(O) 2—O—, —O—P(O,S)—O—, —O—P(S)2—O—, -NRH—P(O)2—O—P(O,NRH)—O—, —O—PO(R″)—O—, —O—PO(CH3)—O—, and —O—PO(NHRN)—O—, where RH is selected from hydrogen and C1-4-alkyl, and R″ is selected from C1-6-alkyl and phenyl.
- A further desirable group of modified nucleic acids for incorporation into oligomers of the invention include those of the following formula:

- wherein X is —O—; B is a modified base as discussed above e.g. an optionally substituted carbocyclic aryl such as optionally substituted pyrene or optionally substituted pyrenylmethylglycerol, or an optionally substituted heteroalicylic or optionally substituted heteroaromatic such as optionally substituted pyridyloxazole. Other desirable universal bases include, pyrrole, diazole or triazole moieties, all of which may be optionally substituted. R 1* is hydrogen.
- P designates the radical position for an internucleoside linkage to a succeeding monomer, or a 5′-terminal group, such internucleoside linkage or 5′-terminal group optionally including the substituent R 5, R5 being hydrogen or included in an internucleoside linkage. R3* is a group P* which designates an internucleoside linkage to a preceding monomer, or a 3′-terminal group. One or two pairs of non-geminal substituents selected from the present substituents of R2, R2, R3, R4*, may designate a biradical consisting of 1-4 groups/atoms selected from —C(RaRb)-, —C(Ra)=C(Ra)—, —C(Ra)=N—, —O—, —S—, —SO2—, —N(Ra)—, and >C=Z. Z is selected from —O—, —S—, and —N(Ra)-, and Ra and Rbeach is independently selected from hydrogen, optionally substituted C1-6-alkyl -alkyl, optionally substituted C2-6-alkenyl, hydroxy, C1-6-alkyl, C2-6-alkenyloxy, carboxy, C1-6-alkoxycarbonyl, C1-6-alkylcarbonyl, formyl, amino, mono- and di(C1-6-alkyl)amino, carbamoyl, mono- and di(C1-6-alkyl)-amino-carbonyl, amino-C1-6-alkyl-aminocarbonyl, mono- and di(C1-6-alkyl)amino-C1-6-alkyl-aminocarbonyl, C1-6-alkyl-carbonylamino, carbamido, C1-6-alkanoyloxy, sulphono, C1-6-alkylsulphonyloxy, nitro, azido, sulphanyl, C1-6-alkylthio, halogen, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands, the possible pair of non-geminal substituents thereby forming a monocyclic entity together with (i) the atoms to which the non-geminal substituents are bound and (ii) any intervening atoms; and each of the substituents R2, R2, R3, R4 which are present and not involved in the possible biradical is independently selected from hydrogen, optionally substituted C1-6-alkyl, optionally substituted C2-6-alkenyl, hydroxy, C1-6-alkyl, C2-6-alkenyloxy, carboxy, C1-6-alkoxycarbonyl, C1-6-alkylcarbonyl, formyl, amino, mono- and di(C1-6-alkyl)amino, carbamoyl, mono- and di(C1-6-alkyl)-amino-carbonyl, amino-C1-6-alkyl-aminocarbonyl, mono- and di(C1-6-alkyl)amino-C1-6-alkyl-aminocarbonyl, C1-6-alkyl-carbonylamino, carbamido, C1-6-alkanoyloxy, sulphono, C1-6-alkylsulphonyloxy, nitro, azido, sulphanyl, C1-6-alkylthio, halogen, photochemically active groups, thermochemically active groups, chelating groups, reporter groups, and ligands; and basic salts and acid addition salts thereof.
- Modified nucleobases and nucleosidic bases may comprise a cyclic unit (e.g. a carbocyclic unit such as pyrenyl) that is joined to a nucleic unit, such as a 1′-position of furasonyl ring through a linker, such as a straight of branched chain alkylene or alkenylene group. Alkylene groups suitably having from 1 (i.e.—CH 2—) to about 12 carbon atoms, more typically 1 to about 8 carbon atoms, still more typically 1 to about 6 carbon atoms. Alkenylene groups suitably have one, two or three carbon-carbon double bounds and from 2 to 12 carbon atoms, more typically 2 to 8 carbon atoms, still more typically 2 to 6 carbon atoms.
- Desirable LNA units include those that contain a furanosyl-type ring and one or more of the following linkages: C-1′, C-2′; C-2′, C-3′; C-2′, C-4′; or a C-2′, C-5′ linkage. A C-2′, C-4′ is particularly desirable. In another aspect of the invention, desirable LNA units are compounds having a substituent on the 2′-position of the central sugar moiety (e.g., ribose or xylose), or derivatives thereof, which favors the C3′-endo conformation, commonly referred to as the North (or simply N for short) conformation. Desirable LNA In various embodiments, the oligonucleotide has at least one LNA unit with a modified base as disclosed herein. Suitable oligonucleotides also may contain natural DNA or RNA units (e.g., nucleotides) with natural bases, as well as LNA units that contain natural bases. Furthermore, the oligonucleotides of the invention also may contain modified DNA or RNA, such as 2′—O—methyl RNA, with natural or modified nucleobases (e.g., pyrene). Desirable oligonucleotides contain at least one of and desirably both of 1) one or more DNA or RNA units (e.g., nucleotides) with natural bases, and 2) one or more LNA units with natural bases, in addition to LNA units with a modified base. In other embodiments, the nucleic acid does not contain a modified base.
- Oligonucleotides of the invention desirably contain at least 50 percent or more, more desirably 55, 60, 65, or 70 percent or more of non-modified or natural DNA or RNA units (e.g., nucleotides) or units other than LNA units based on the total number of units or residues of the oligo. A non-modified nucleic acid as referred to herein means that the nucleic acid upon incorporation into a 1 0-mer oligomer will not increase the T m of the oligomer in excess of 1° C. or 2° C. More desirably, the non-modified nucleic acid unit (e.g., nucleotide) is a substantially or completely “natural” nucleic acid, i.e. containing a non-modified base of uracil, cytosine, 5-methyl-cytosine, thymine, adenine or guanine and a non-modified pentose sugar unit of β-D-ribose (in the case of RNA) or β-D-2-deoxyribose (in the case of DNA).
- Oligonucleotides of the invention suitably may contain only a single modified (i.e. LNA) nucleic acid unit, but desirably an oligonucleotide will contain 2, 3, 4 or 5 or more modified nucleic acid units. Typically desirable is where an oligonucleotide contains from about 5 to about 40 or 45 percent modified (LNA) nucleic acid units, based on total units of the oligo, more desirably where the oligonucleotide contains from about 5 or 10 percent to about 20, 25, 30 or 35 percent modified nucleic acid units, based on total units of the oligo.
- Typical oligonucleotides that contain one or more LNA units with a modified base as disclosed herein suitably contain from 3 or 4 to about 200 nucleic acid repeat units, with at least one unit being an LNA unit with a modified base, more typically from about 3 or 4 to about 5, 6, 7, 8, 9, 10, 15, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 130, 140 or 150 nucleic acid units, with 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 LNA units with a modified base being present.
- As discussed above, particularly desirable oligonucleotides contain a non- modified DNA or RNA unit at the 3′ terminus and a modified DNA or RNA unit at one position upstream from (generally referred to hereing as the −1 or penultimate position) the 3′ terminal non-modified nucleic acid unit. In some embodiments, the modified base is at the 3′ terminal position of a nucleic acid primer, such as a primer for the detection of a single nucleotide polymorphism. Other particularly desirable nucleic acids have an LNA unit with or without a modified base in the 5′ and/or 3′ terminal position.
- Also desirable are oligonucleotides that do not have an extended stretches of modified DNA or RNA units, e.g. greater than about 4, 5 or 6 consecutive modified DNA or RNA units. That is, desirably one or more non-modified DNA or RNA will be present after a consecutive stretch of about 3, 4 or 5 modified nucleic acids.
- Generally desirable are oligonucleotides that contain a mixture of LNA units that have non-modified or natural nucleobases (i.e., adenine, guanine, cytosine, 5-methyl-cytosine, uracil, or thymine) and LNA units that have modified bases as disclosed herein.
- Particularly desirable oligonucleotides of the invention include those where an LNA unit with a modified base is interposed between two LNA units each having non-modified or natural bases (adenine, guanine, cytosine, 5-methyl-cytosine, uracil, or thymine. The LNA “flanking” units with natural base moieties may be directly adjacent to the LNA with modified base moiety, or desirably is within 2, 3, 4 or 5 nucleic acid units of the LNA unit with modified base. Nucleic acid units that may be spaced between an LNA unit with a modified base and an LNA unit with natural nucleobasis suitably are DNA and/or RNA and/or alkyl-modified RNA/DNA units, typically with natural base moieties, although the DNA and or RNA units also may contain modified base moieties.
- The oligonucleotides of the present invention are comprised of at least about one universal base. Oligonucleotides of the present can also be comprised, for exmple, of between about one to six 2′-Ome-RNA unit, at least about two LNA units and at least about one LNA pyrene unit.
- In the practice of the present invention, target genes may be suitably single-stranded or double-stranded DNA or RNA; however, single-stranded DNA or RNA targets are desirable. It is understood that the target to which the nucleic acids of the invention are directed include allelic forms of the targeted gene and the corresponding mRNAs including splice variants. There is substantial guidance in the literature for selecting particular sequences for nucleic acids with LNA or other high affinity nucleotides given a knowledge of the sequence of the target polynucleotide, e.g., Peyman and Ulmann, Chemical Reviews, 90:543-584, 1990; Crooke, Ann. Rev. Pharmacol. Toxicol., 32:329-376 (1992); and Zamecnik and Stephenson, Proc. Natl. Acad. Sci., 75:280-284 (1974). Desirable mRNA targets include the 5′ cap site, tRNA primer binding site, the initiation codon site, the mRNA donor splice site, and the mRNA acceptor splice site, e.g., Goodchild et al., U.S. Pat. No. 4,806,463.
- The chimeric oligos of the present invention are highly suitable for a variety of diagnostic purposes such as for the isolation, purification, amplification, detection, identification, quantification, or capture of nucleic acids such as DNA, mRNA or non-protein coding cellular RNAs, such as tRNA, rRNA, snRNA and scRNA, or synthetic nucleic acids, in vivo or in vitro.
- The oligomer can comprise a photochemically active group, a thermochemically active group, a chelating group, a reporter group, or a ligand that facilitates the direct or indirect detection of the oligomer or the immobilization of the oligomer onto a solid support. Such group are typically attached to the oligo when it is intended as a probe for in situ hybridization, in Southern hybridization, Dot blot hybridization, reverse Dot blot hybridization, or in Northern hybridization.
- When the photochemically active group, the thermochemically active group, the chelating group, the reporter group, or the ligand includes a spacer (K), the spacer may suitably comprise a chemically cleavable group.
- An additional object of the present invention is to provide oligonucleotides which combines an increased ability to discriminate between complementary and mismatched targets with the ability to act as substrates for nucleic acid active enzymes such as for example DNA and RNA polymerases, ligases, phosphatases. Such oligonucleotides may be used for instance as primers for sequencing nucleic acids and as primers in any of the several well known amplification reactions, such as the PCR reaction.
- Introduction of LNA monomers with natural bases into either DNA, RNA, or pure LNA oligonucleotides can result in extremely high thermal stability of duplexes with complementary DNA or RNA, while at the same time obeying the Watson-Crick base-pairing rules. In general, the thermal stability of heteroduplexes is increased 3-8° C. per LNA monomer in the duplex. Oligonucleotides containing LNA can be designed to be substrates for polymerases (e.g., Taq polymerase), and PCR based on LNA primers is more discriminatory towards single base mutations in the template DNA compared to normal DNA-primers (e.g., allele specific PCR). Furthermore, very short LNA oligos (e.g. 5-mers or 8-mers) which have high T m 's when compared to similar DNA oligos can be used as highly specific catching probes with outstanding discriminatory power towards single base mutations (e.g., SNP detection).
- LNA oligonucleotides are capable of hybridizing with double-stranded DNA target molecules as well as RNA secondary structures by strand invasion as well as of specifically blocking a wide selection of enzymatic reactions such as, digestion of double-stranded DNA by restriction endonucleases; and digestion of DNA and RNA with deoxyribonucleases and ribonucleases, respectively.
- In a further aspect, oligonucleotides of the invention may be used to construct new affinity pairs with exhibit enhanced specificity towards each other. The affinity constants can easily be adjusted over a wide range and a vast number of affinity pairs can be designed and synthesized. One part of the affinity pair can be attached to the molecule of interest (e.g. proteins, amplicons, enzymes, polysaccharides, antibodies, haptens, peptides, etc.) by standard methods, while the other part of the affinity pair can be attached to e.g. a solid support such as beads, membranes, micro-titer plates, sticks, tubes, etc. The solid support may be chosen from a wide range of polymer materials such as for instance polypropylene, polystyrene, polycarbonate or polyethylene. The affinity pairs may be used in selective isolation, purification, capture and detection of a diversity of the target molecules.
- Oligonucleotides of the invention also may be employed as probes in the purification, isolation and detection of for instance pathogenic organisms such as viral, bacteria and fungi etc. Oligonucleotides of the invention also may be used as generic tools for the purification, isolation, amplification and detection of nucleic acids from groups of related species such as for instance rRNA from gram-positive or gram negative bacteria, fungi, mammalian cells etc.
- Oligonucleotides of the invention also may be employed as an aptamer in molecular diagnostics, e.g. in RNA mediated catalytic processes, in specific binding of antibiotics, drugs, amino acids, peptides, structural proteins, protein receptors, protein enzymes, saccharides, polysaccharides, biological cofactors, nucleic acids, or triphosphates or in the separation of enantiomers from racemic mixtures by stereospecific binding.
- Oligonucleotides of the invention also may be used for labeling of cells, e.g. in methods wherein the label allows the cells to be separated from unlabelled cells.
- Oligonucleotides also may be conjugated to a compound selected from proteins, amplicons, enzymes, polysaccharides, antibodies, haptens, and peptides.
- Kits are also provided containing one or more oligonucleotides of the invention for the isolation, purification, amplification, detection, identification, quantification, or capture of natural or synthetic nucleic acids. The kit typically will contain a reaction body, e.g. a slide or biochip. One or more oligonucleotides of the invention may be suitably immobilized on such a reaction body.
- The invention also provides methods for using kits of the invention for carrying out a variety of bioassays, e.g. for diagnostic purposes. Any type of assay wherein one component is immobilized may be carried out using the substrate platforms of the invention. Bioassays utilizing an immobilized component are well known in the art. Examples of assays utilizing an immobilized component include for example, immunoassays, analysis of protein-protein interactions, analysis of protein-nucleic acid interactions, analysis of nucleic acid-nucleic acid interactions, receptor binding assays, enzyme assays, phosphorylation assays, diagnostic assays for determination of disease state, genetic profiling for drug compatibility analysis, and SNP detection (U.S.P.N. 6,316,198; 6,303,315).
- Identification of a nucleic acid sequence capable of binding to a biomolecule of interest can be achieved by immobilizing a library of nucleic acids onto the substrate surface so that each unique nucleic acid was located at a defined position to form an array. The array would then be exposed to the biomolecule under conditions which favored binding of the biomolecule to the nucleic acids. Non-specifically binding biomolecules could be washed away using mild to stringent buffer conditions depending on the level of specificity of binding desired. The nucleic acid array would then be analyzed to determine which nucleic acid sequences bound to the biomolecule. Desirably the biomolecules would carry a fluorescent tag for use in detection of the location of the bound nucleic acids.
- Oligonucleotides of the invention can be employed in a wide range of applications, particularly those applications involving a hybridization reaction. Oligonucleotides also may be used in DNA sequencing aiming at improved throughput in large-scale, shotgun genome sequencing projects, improved throughput in capillary DNA sequencing (e.g. ABI prism 3700) as well as at an improved method for 1) sequencing large, tandemly repeated genomic regions, 2) closing gaps in genome sequencing projects and 3) sequencing of GC-rich templates. In DNA sequencing, oligonucleotide sequencing primers are combined with LNA enhancer elements for the read-through of GC-rich and/or tandemly repeated genomic regions, which often present many challenges for genome sequencing projects. LNA may increase the specificity of certain sequencing primers and thus facilitate selection of a particular version of a repeated sequence and possibly also use strand invasion to open up recalcitrant GC rich sequences.
- The incorporation of one or more universal nucleosides into the oligomer makes bonding to unknown bases possible and allows the oligonucleotide to match ambiguous or unknown nucleic acid sequences.
- As discussed above, oligonucleotides of the invention may be used for therapeutic applications, e.g. as an antisense, antigene or ribozyme or double stranded nucleic acid therapeutic agents. In these therapeutic methods, one or more oligonucleotides of the invention is administered as desired to a patient suffering from or susceptible the targeted disease or disorder, e.g. a viral infection.
- In an exemplary in vitro method for measuring the ability of a nucleic acid of the invention to silence a target gene, cells are cultured in standard medium supplemented with 1% fetal calf serum as previously described (Lykkesfeld et al., Int. J. Cancer 61:529-534, 1995). At the start of the experiment cells are approximately 40% confluent. The serum containing medium is removed and replaced with serum-free medium. Transfection is performed using, e.g., Lipofectin (GibcoBRL cat. No 18292-011) diluted 40×in medium without serum and combined with the oligo to a concentration of 750 nM oligo, 0.8 ug/ml Lipofectin. Then, the medium is removed from the cells and replaced with the medium containing oligo-Lipofectin complex. The cells are incubated at 37° C. for 6 hours, rinsed once with medium without serum and incubated for a further 18 hours in DME/
F 12 with 1% FCS at 3 7° C. Standard methods are used to measure the level of mRNA or protein encoded by the target gene to measure the level of gene silencing. - It is also contemplated that information on the structures assumed by a target nucleic acid may be used in the design of the probes, such that regions that are known or suspected to be involved in folding may be chosen as hybridization sites. Such an approach will reduce the number of probes that are likely to be needed to distinguish between targets of interest.
- There are many methods used to obtain structural information involving nucleic acids, including the use of chemicals that are sensitive to the nucleic acid structure, such as phenanthroline/copper, EDTA-Fe 2+, cisplatin, ethylnitrosourea, dimethylpyrocarbonate, hydrazine, dimethyl sulfate, and bisulfite. Enzymatic probing using structure-specific nucleases from a variety of sources, such as the Cleavase™ enzymes (Third Wave Technologies, Inc., Madison, Wis.), Taq DNA polymerase, E. coli DNA polymerase I, and eukaryotic structure-specific endonucleases (e.g., human, murine and Xenopus XPG enzymes, yeast RAD2 enzymes), murine FEN-1 endonucleases (Harrington and Lieber, Genes and Develop., 3:1344 [1994]) and
calfthymus 5′ to 3′ exonuclease (Murante et al., J. Biol. Chem., 269:1191 [1994]). In addition, enzymes having 3′ nuclease activity such as members of the family of DNA repair endonucleases (e.g., the RrpI enzyme from Drosophila melanogaster, the yeast RAD1/RAD10 complex and E. coli Exo III), are also suitable for examining the structures of nucleic acids. - If analysis of structure as a step in probe selection is to be used for a segment of nucleic acid for which no information is available concerning regions likely to form secondary structures, the sites of structure-induced modification or cleavage must be identified. It is most convenient if the modification or cleavage can be done under partially reactive conditions (i.e., such that in the population of molecules in a test sample, each individual will receive only one or a few cuts or modifications). When the sample is analyzed as a whole, each reactive site should be represented, and all the sites may be thus identified. Using a Cleavase Fragment Length Polymorphism™ cleavage reaction as an example, when the partial cleavage products of an end labeled nucleic acid fragment are resolved by size (e.g., by electrophoresis), the result is a ladder of bands indicating the site of each cleavage, measured from the labeled end. Similar analysis can be done for chemical modifications that block DNA synthesis; extension of a primer on molecules that have been partially modified will yield a nested set of termination products. Determining the sites of cleavage/modification may be done with some degree of accuracy by comparing the products to size markers (e.g., commercially available fragments of DNA for size comparison) but a more accurate measure is to create a DNA sequencing ladder for the same segment of nucleic acid to resolve alongside the test sample. This allows rapid identification of the precise site of cleavage or modification.
- Reactions were conducted under an atmosphere of nitrogen when anhydrous solvents were used. All reactions were monitored by thin-layer chromatography (TLC) using EM reagent plates with fluorescence indicator (SiO 2-60, F-254). The compounds were visualized under UV light and by spraying with a mixture of 5% aqueous sulfuric acid and ethanol followed by heating. Silica gel 60 (particle size 0.040-0.063 mm, Merck) was used for flash column chromatography. NMR spectra were recorded at 300 MHz for 1H NMR, 75.5 MHz for 13C NMR and 121.5 MHz for 31P NMR on a
Varian Unity 300 spectrometer. δ-Values are in ppm relative to tetramethyl silane as internal standard (1H and 13C NMR) and relative to 85% H3PO4 as external standard (31P NMR). Coupling constants are given in Hertz. The assignments, when given, are tentative, and the assignments of methylene protons, when given, may be interchanged. Bicyclic compounds are named according to the Von Bayer nomenclature. Fast atom bombardment mass spectra (FAB-MS) were recorded in positive ion mode on a Kratos MS50TC spectrometer. The composition of the oligonucleotides were verified by MALDI-MS on a Micromass Tof Spec E mass spectrometer using a matrix of diammonium citrate and 2,6-dihydroxyacetophenone. - Mesyl chloride (8.6 g, 7.5 mmol) was dropwise added to a stirred solution of 4-C-hydroxymethyl-1,2—O—isopropylidene-3—O—p-methoxybenzyl- α-D-ribofuranose [R. Yamaguchi, T. Imanishi, S. Kohgo, H. Horie and H. Ohrui, Biosci. Biotechnol. Biochem., 1999, 63, 736] (1, 10.0 g, 29.4 mmol) in anhydrous pyridine (30 cm 3) and the reaction mixture was stirred overnight at room temperature. The mixture was evaporated to dryness under reduced pressure to give a residue which was co-evaporated with toluene (2×25 cm3), dissolved in CH2Cl2 (200 cm3) and washed successively with saturated aqueous NaHCO3 (2×100 cm3) and brine (50 cm3). The organic phase was dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The colorless viscous oil obtained was purified by column chromatography [0.5-1% (v/v) MeOH in CH2Cl2 as eluent], followed by crystallization from MeOH to give furanose 2 as a white solid material (13.6 g, 93%); Rf 0.57 (CH2Cl2/MeOH 95:5, v/v); & (CDCl3) 7.30 (2H, d, J 8.7), 6.90 (2H, d, J 8.5), 5.78 (1H, d, J3.7), 4.86 (1H, d, J 12.0), 4.70 (1H, d, J 11.4), 4.62 (1H, dd, J5.0 and 3.8), 4.50 (1H, d, J 11.1), 4.39 (1H, d, J 12.3), 4.31 (1H, d, J 11.0), 4.17 (1H, d, J5.1), 4.11(1H, d, J 11.0), 3.81 (3H, s), 3.07 (3H, s), 2.99(3H, s), 1.68 (3H, s), 1.34(3H, s); δc (CDCl3) 159.8, 129.9, 128.8, 114.1, 114.0, 104.5, 83.2, 78.0, 77.9, 72.6, 69.6, 68.8, 55.4, 38.1, 37.5, 26.3, 25.7.
- A suspension of furanoside 2 (13.5 g, 27.2 mmol) in a mixture of H 2O (45 cm3) and 15% HCl in MeOH (450 cm3, w/w) was stirred at room temperature for 72 h. The mixture was carefully neutralized by addition of saturated aqueous NaHCO3 (100 cm3) followed by NaHCO3 (s) whereupon the mixture was evaporated to dryness under reduced pressure. H2O (100 cm 3) was added, and extraction was performed with EtOAc (3×100 cm3). The combined organic phase was washed with brine (100 cm3), dried (Na2SO4), filtered and then evaporated to dryness under reduced pressure. The residue was coevaporated with toluene (2×25 cm3) and purified by column chromatography [1-2% (v/v) MeOH in CH2Cl2] to give
furanoside 3 as an anomeric mixture (clear oil, 11.0 g, 86%, ratio between anomers ca. 6:1); Rf 0.39, 0.33 (CH2Cl2/MeOH 95:5, v/v); δH (CDCl3, major anomer only) 7.28 (2H, d, J8.4), 6.91 (2H, d, J8.9), 4.87 (1H, s), 4.62 (1H, d, J 11.4), 4.53 (1H, d, J 11.2), 4.41 (2H, s), 4.31 (1H, d, J9.8), 4.24 (1H, d, J4.6), 4.06 (1H, d, J 10.0), 3.98 (1H, br s), 3.81 (3H, s), 3.33 (3H, s), 3.06 (3H, s), 3.03 (3H,s); δc (CDCl3, major anomer only) 160.0, 130.1, 128.5, 114.3, 107.8, 81.7, 81.2, 73.8, 73.6, 69.7, 69.6, 55.5, 55.4, 37.5, 37.4. - To a stirred solution of the anomeric mixture of Compound 3 (10.9 g, 23.2 mmol) in anhydrous DMF (50 cm 3) at 0° C. was during 10 min added sodium hydride (2.28 g of a 60% suspension in mineral oil (w/w), 95.2 mmol) and the mixture was stirred for 12 h at room temperature. Ice-cold H2O (200 cm3) was slowly added and extraction was performed using EtOAc (3×200 cm3). The combined organic phase was washed successively with saturated aqueous NaHCO3 (2×100 cm3) and brine (50 cm3), dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The residue was purified by column chromatography [0.5-1% (v/v) MeOH in CH2Cl2] to give first the major isomer (6.42 g, 74%) and then [1.5% (v/v) MeOH in CH2Cl2] the minor isomer (1.13 g, 13%), both as clear oils; Rf 0.56, 0.45 (CH2Cl2/MeOH 95:5, v/v); δH (CDCl3, major isomer) 7.16 (2H, d, J8.8), 6.74 (2H, d, J 8.4), 4.65 (1H, s), 4.42-4.32 (4H, m), 3.95-3.94 (2H, m), 3.84 (1H, d, J 7.4), 3.66 (3H, s), 3.54 (1H, d,J7.4), 3.21 (3H, s), 2.90 (3H, s); δc (CDCl3, major isomer) 159.6, 129.5, 129.3, 114.0, 105.3, 83.2, 78.6, 77.2, 72.1, 71.8, 66.3, 55.6, 55.4, 37.8; & (CDCl3, minor isomer) 7.27 (2H, d, J8.9), 6.89(2H, d, J8.6), 4.99(1H, s), 4.63-4.39(4H, m), 4.19(1H, s), 4.10-3.94(2H, m), 3.91 (1H, s), 3.81 (3H, s), 3.47 (3H, s), 3.05 (3H, s); δc (CDCl3, minor isomer) 159.7, 129.6, 129.5, 114.1, 104.4, 86.4, 79.3, 77.1, 72.3, 71.9, 66.2, 56.4, 55.4, 37.7.
- To a stirred solution of furanoside 4 (major isomer, 6.36 g, 17.0 mmol) in dioxane (25 cm 3) was added 18-crown-6 (9.0 g, 34.1 mmol) and KOAc (8.4 g, 85.6 mmol). The stirred mixture was heated under refluxed for 12 h and subsequently evaporated to dryness under reduced pressure. The residue was dissolved in CH2Cl2 (100 cm3) and washing was performed, successively, with saturated aqueous NaHCO3 (2×5O cm3) and brine (50 cm3). The separated organic phase was dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The residue was purified by column chromatography [1% (v/v) MeOH in CH2Cl2] to give
furanoside 5 as a white solid material (one anomer, 5.23 g, 91%); Rf 0.63 (CH2Cl2/MeOH 95:5, v/v); δH (CDCl3) 7.27-7.24 (2H, m), 6.90-6.87 (2H, m), 4.79 (1H, s), 4.61 (1H, d, J 11.0), 4.49 (2H, m), 4.28 (1H, d, J 11.0), 4.04 (3H, m), 3.80 (3H, s), 3.68 (1H, m), 3.36 (3H, s), 2.06 (3H, s); δC (CDCl3) 170.7, 159.5, 129.5, 129.4, 113.9, 105.1, 83.3, 78.9, 77.2, 72.0, 71.9, 61.0, 55.4, 55.3, 20.8. - A solution of furanoside 5 (one anomer, 5.16 g, 15.3 mmol) in saturated methanolic ammonia (200 cm 3) was stirred at room temperature for 48 h. The reaction mixture was evaporated to dryness under reduced pressure, coevaporated with toluene (2×50 cm3), and the residue purified by column chromatography [2-3% (v/v) MeOH in CH2Cl2] to give
furanoside 6 as a white solid material (one anomer, 3.98 g, 88%); Rf 0.43 (CH2Cl2/MeOH 95:5, v/v); δH (CDCl3) 7.27(2H, d, J8.6), 6.88 (2H, d, J8.9), 4.79(1H, s), 4.59 (1H, d, J 11.3), 4.53 (1H, d, J 11.4), 4.09 (2H, s), 3.97 (1H, d, J7.5), 3.86 (2H, br s), 3.80 (3H, s), 3.75-3.62 (2H, m), 3.37 (3H, s); δC (CDCl3) 159.4, 129.7, 129.3, 113.9, 105.2, 85.6, 78.3, 77.4, 71.9, 71.8, 58.8, 55.5, 55.3. - To a stirred solution of furanoside 6 (one anomer, 3.94 g, 13.3 mmol) in anhydrous DMF (50 cm 3) at 0° C. was added a suspension of NaH [60% in mineral oil (w/w), 1.46 g, 60.8 mmol] followed by dropwise addition ofp-methoxybenzyl chloride (2.74 g, 17.5 mmol). The mixture was allowed to warm to room temperature and stirring was continued for another 4 h whereupon ice-cold H2O (50 cm3) was dropwise added. The mixture was extracted with CH2Cl2 (3×100 cm3) and the combined organic phase was washed with brine (100 cm3), dried (Na2SO4), filtered, evaporated to dryness under reduced pressure and coevaporated with toluene (3×50 cm3). The residue (4.71g) tentatively assigned as a mixture of 7 and
aldehyde 11 was used in the preparation of 11 (see below) without further purification. - 4-C-Hydroxymethyl-3,5-di—O—(p-methoxybenzyl)-1,2—O—isopropylidene-α-D-ribofuranose [R. Yamaguchi, T. Imanishi, S. Kohgo, H. Horie and H. Ohrui, Biosci. Biotechnol. Biochem., 1999, 63, 736] (8, 3.2 g, 6.95 mmol) was mesylated using MsCl (2.00 g, 17.5 mmol) and pyridine (10 cm3) following the procedure described for 2. After work-up, the colorless viscous oil was purified by column chromatography [1% (v/v) MeOH in CH2Cl2] to give derivative 9 in 89% yield (3.17 g) as a clear oil; Rf 0.45 (CH2Cl2/MeOH 98:2, v/v); /H (CDCl3) 7.22 (2H, d, J8.9), 7.18 (2H, d, J8.7), 6.86 (4H, d, J 8.3), 5.76 (1H, d, J3.8), 4.83 (1H, d, J 12.0), 4.64 (1H, d, J 11.6), 4.59 (1H, m), 4.49-4.35 (4H, m), 4.24 (1H, d, J5.3), 3.80 (6H, s), 3.56 (1H, d, J 10.5), 3.45 (1H, d, J 10.5), 3.06 (3H, s), 1.67 (3H, s), 1.33 (3H, s); δC (CDCl3) 159.6, 159.4, 129.9, 129.8, 129.7, 129.5, 129.4, 129.3, 114.0, 113.9, 113.8, 113.7, 113.6, 104.5, 84.9, 78.6, 78.1, 73.4, 72.4, 71.0, 69.9, 55.3, 38.0, 26.4, 25.9.
- Methanolysis of furanoside 9 (3.1 g, 5.76 mmol) was performed using a mixture of a solution of 15% HCl in MeOH (w/w, 120 cm 3) and H2O (12 cm3) following the procedure described for the synthesis of 3. After work-up, the crude product was purified by column chromatography [0.5-1% (v/v) MeOH in CH2Cl2] to give the major anomer of 10 (1.71 g, 58%) and [1-1.5% (v/v) MeOH in CH2Cl2] the minor anomer of 10 (0.47 g, 16%), both as clear oils; Rf 0.31, 0.24 (CH2Cl2/MeOH 98:2, v/v); δC (major anomer, CDCl3) 159.8, 159.5, 129.9, 129.8, 129.6, 129.5, 129.0, 114.2, 114.1, 114.0, 113.9, 107.9, 84.7, 79.9, 74.2, 73.5, 73.5, 70.2, 64.4, 55.6, 55.4, 37.4.
- Ring closure of furanoside 10 (major anomer, 1.68 g, 3.28 mmol) was achieved using NaH (60% suspension in mineral oil (w/w), 0.32 g, 13.1 mmol) in anhydrous DMF (10 cm 3) following the procedure described for the synthesis of 4 to give a crude product tentatively assigned as a mixture of
furanoside 7 and aldehyde 11 (see below) (1.13 g). - A solution of crude furanoside 7 (as a mixture with 11 as prepared as described above, 5.80 g) in 80% glacial acetic acid (100 cm 3) was stirred at 50° C. for 4 h. The solvent was distilled off under reduced pressure and the residue was successively coevaporated with absolute ethanol (3×25 cm3) and toluene (2×25 cm3) and purified by column chromatography [4-5% (v/v) MeOH in CH2Cl2] to give
aldehyde 11 as a colorless oil (4.60 g); Rf 0.37 (CH2Cl2/MeOH 95:5, v/v); δH (CDCl3) 9.64 (1H, br s), 7.27-7.17 (4H, m), 6.87-6.84 (4H, m), 4.59 (1H, d, J 11.6), 4.51-4.41 (2H, m), 4.35 (1H, s), 3.92-3.90 (2H, m), 3.79 (6H, s), 3.77-3.68 (3H, m), 3.55 (2H, br s); δC (CDCl3) 203.6, 159.5, 159.4, 129.7, 129.6, 129.5, 129.2, 114.0, 113.9, 113.8, 87.3,86.7, 81.0, 75.1, 73.4, 71.6, 67.6, 55.3. - A solution of aldehyde 11 (Scheme 2) in anhydrous THF (10 cm 3) was added dropwise during 5 min to a stirred solution of the aryl magnesium bromide dissolved in anhydrous THF at 0° C. The mixture was allowed to heat to room temperature and stirred for 12 h. The mixture was evaporated to dryness under reduced pressure and the residue diluted with CH2Cl2 and washed several times with saturated aqueous NH4Cl. The organic phase was dried (Na2SO4), filtered, and evaporated to dryness under reduced pressure. Column chromatography of the crude product obtained afforded the compounds 12a-e as shown in
Scheme 2. - Grignard reaction of phenylmagnesium bromide (1.0 M solution in THF, 14.2 cm 3, 14.2 mmol) with aldehyde 11 (515 mg, 1.28 mmol) afforded 12a as shown in
Scheme 2. The crude product was purified by column chromatography [4% (v/v) MeOH in CH2Cl2] to give tetrahydrofuran 12a (540 mg, 88%) as a colorless oil; Rf 0.34 (CH2Cl2/MeOH 95:5, v/v); δC (CDCl3) 7.40-7.19 (7H, m), 6.91-6.73 (6H, m), 4.73 (1H, d, J6.4), 4.48 (2H, s), 4.08 (2H, s), 3.88 (1H, d, J 9.4), 3.79 (1H, m), 3.78 (3H, s), 3.76 (3H, s), 3.75-3.69 (2H, m), 3.50 (1H, d, J 9.4), 3.45 (1H, s), 3.42 (1H, br s), 3.26 (1H, br s); δC, (CDCl3) 159.5, 159.3, 140.7, 129.7, 129.6, 129.5, 129.2, 128.5, 128.0, 127.3, 113.9, 113.8, 113.7, 89.4, 84.6, 81.8, 75.3, 74.7, 73.5, 71.6, 69.3, 55.3; m/z (FAB) 503 [M+Na]+, 479 [M−H]+, 461 [M−H—H2O]+. - Grignard reaction of 4-fluoro-3-methylphenylmagnesium bromide (1.0 M solution in THF, 15.0 cm 3, 15.0 mmol) with aldehyde 11 (603 mg, 1.5 mmol) afforded 12b as shown in
Scheme 2. The crude product was purified by column chromatography [4-5% (v/v) MeOH in CH2Cl2] to give tetrahydrofuran 12b (611 mg, 85%) as a colorless oil; Rf 0.34 (CH2Cl2/MeOH 95:5, v/v); & (CDCl3) 7.24-7.12 (5H, m), 6.98-6.84 (5H, m), 6.77 (1H, d, J8.5), 4.65 (1H, dd, J2.8 and 6.4), 4.49 (2H, s), 4.15 (2H, s), 4.01 (1H, dd, J2.3 and 6.5), 3.87 (1H, d, J9.3), 3.79 (3H, s), 3.78 (3H, s), 3.76-3.68 (2H, m), 3.52 (1H, s), 3.47 (1H, d, J 10.3), 3.42 (1H, d, J2.9), 3.22 (1H, s), 2.24 (3H, d, J0.8); δC (CDCl3) 162.7, 159.5, 159.4, 136.2, 136.1, 130.3, 130.2, 129.7, 129.6, 129.5, 129.4, 129.1, 126.1, 126.0, 115.1, 114.8, 114.0, 113.9, 113.8, 89.3, 84.5, 81.8, 75.3, 74.0, 73.5, 71.7, 69.2, 55.4, 55.3, 14.7 (d, J 3.9); m/z (FAB) 535 [M+Na]+, 511 [M-H]+, 493 [M−H—H2O]+. - 1-Bromonaphthalene (1.55 g, 7.5 mmol) was added to a stirred mixture of magnesium turnings (182 mg, 7.5 mmol) and iodine (10 mg) in THF (10 cm3). The mixture was stirred at 40° C. for 1 h whereupon it was allowed to cool to room temperature. A solution of aldehyde 11 (603 mg, 1.5 mmol) in THF (10 cm3) was added slowly and the reaction was stirred for 12 h. The crude product was purified by column chromatography [4-5% (v/v) MeOH in CH2Cl21 to give tetrahydrofuran 12c (756 mg, 95%) as a colorless oil; Rf 0.35 (CH2Cl2MeOH 95:5, v/v); & (CDCl3) 8.08 (1H, m), 7.86 (1H, m), 7.79 (1H, d, J 8.2), 7.72 (1H, d, J 7.2), 7.49-7.44 (3H, m), 7.18 (2H, d, J 8.4), 6.84 (2H, d, J 8.6), 6.74 (2H, d, J 8.7), 6.68 (2H, d, J 8.8), 5.52 (1H, dd, J3.7 and 5.6), 4.45 (2H, s), 4.34 (1H, dd, J2.5 and 5.9), 4.03 (1H, d, J 11.0), 3.96 (1H, d, J 11.0), 3.93 (1H, d, J9.5), 3.80 (1H, d, J9.3), 3.77 (3H, s), 3.75 (1H, d, J2.6), 3.72 (3H, s), 3.68 (1H, d, J9.3), 3.56 (1H, d, J3.7), 3.49 (1H, d, J 9.3), 3.34 (1H, s); δC (CDCl3) 159.5, 159.3, 136.3, 134.0, 131.0, 129.7, 129.6, 129.5, 129.4, 129.0, 128.6, 128.2, 125.6, 125.5, 123.5, 114.0, 113.8, 113.7, 88.7, 84.7, 81.9, 75.5, 73.5, 71.7, 71.3, 69.3, 55.4, 55.3; m/z (FAB) 553 [M+Na]+, 529 [M−H]+, 511 [M−H—H2O]+.
- Tetrahydrofuran 12d was synthesized from aldehyde 11(515 mg, 1.28 mmol), 1-bromopyrene (1.0 g, 3.56 mmol), magnesium turnings (155 mg, 6.4 mmol), iodine (10 mg) and THF (20 cm 3) following the procedure described for synthesis of compound 12c. The crude product was purified by column chromatography [3-4% (v/v) MeOH in CH2Cl2] to give tetrahydrofuran 12d (690 mg, 89%) as a pale yellow solid; Rf 0.35 (CH2Cl2MeOH 95:5, v/v); δH (CDCl3) 8.23 (2H, d, J8.4 and 9.2), 8.19-8.13 (3H, m), 8.05-7.99 (4H, m), 7.14 (2H, d, J8.8), 6.82 (2H, d, J9.0), 6.30 (2H, d, J 8.7), 6.20 (2H, d, J 8.6), 5.87 (1H, d, J 7.2), 4.43 (2H, s), 4.41 (1H, m), 4.01 (1H, d, J9.4), 3.91 (1H, d, J 11.8), 3.86 (1H, d, J9.2), 3.77 (1H, d, J 1.9), 3.76 (3H, s), 3.70-3.64 (3H, m), 3.52-3.45 (1H, m), 3.44 (3H, s); δc (CDCl3) 159.5, 158.9, 133.9, 131.4, 131.1, 130.7, 129.7, 129.5, 129.2, 128.9, 128.5, 127.8, 127.7, 127.5, 126.0, 125.5, 125.3, 125.2, 125.1, 125.0, 124.9, 122.9, 113.9, 113.3, 89.5, 83.5, 82.0, 75.7, 73.4, 71.3, 71.0, 69.3, 55.3, 55.0; m/z (MALDI) 627 [M+Na]+, 609 [M++Na-H2O]+.
- Tetrahydrofuran 12e was synthesized from aldehyde 11 (515 mg, 1.28 mmol), 1-bromo-2,4,5-trimethylbenzene (1.28 g, 6.4 mmol), magnesium turnings (155 mg, 6.4 mmol), iodine (10 mg) and THF (20 cm 3) following the procedure described for synthesis of compound 12c. The crude product was purified by column chromatography [3-4% (v/v) MeOH in CH2Cl2] to give tetrahydrofuran 12e (589 mg, 88%) as a colorless oil; Rf 0.34 (CH2Cl2/MeOH 95:5, v/v); & (CDCl3) 7.25 (2H, d, J 8.7), 7.21 (2H, d, J 8.9), 6.90 (1H, s), 6.87 (1H, s), 6.85 (2H, d, J 8.9), 6.76 (2H, d, J 8.7), 4.95 (1H, dd, J 3.6 and 5.9), 4.48 (2H, s), 4.18-4.08 (3H, m), 3.89 (1H, d, J9.6), 3.80 (1H, m), 3.79 (3H, s), 3.77 (3H, s), 3.71 (1H, d, J9.2), 3.64 (1H, d, J2.6), 3.51 (1H, d, J9.4), 3.24 (1H, s), 3.18 (1H, d, J3.4), 2.25 (3H,s), 2.22 (3H,s), 2.21 (3H, s); δc (CDCl3) 159.5, 159.3, 136.0, 135.8, 134.2, 132.5, 132.0, 129.8, 129.7, 129.6, 129.5, 128.5, 113.9, 113.8, 88.6, 84.7, 81.7, 75.4, 73.5, 71.7, 70.9, 69.4, 55.3, 19.5, 19.4, 19.0; m/z (FAB) 545 [M+Na]+, 521 [M−H]+, 503 [M−H—H2O]+.
- N,N,N′,N′-Tetramethylazodicarboxamide (TMAD) was added in one portion to a stirred solution of the compounds 12a-e as shown in
Scheme 2 and tributylphosphine in benzene at 0° C. The mixture was stirred for 12 h at room temperature whereupon it was diluted with diethyl ether (50 cm3). The organic phase was washed successively with saturated aqueous NH4Cl (2×20 cm3) and brine (25 cm3), dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The crude product obtained was purified by column chromatography [1.5-2% (v/v) MeOH in CH2Cl2] to give compounds 13a-e as shown inScheme 2. - Cyclization of compound 12a (540 mg, 1.13 mmol) in the presence of TMAD (310 mg, 1.8 mmol), PBu 3 (364 mg, 1.8 mmol) and benzene (10 cm3) followed by the general work-up procedure and column chromatography afforded compound 13a as a colorless oil (400 mg, 77%); Rf 0.51 (CH2Cl2/MeOH 98:2, v/v); & (CDCl3) 7.36-7.33 (7H, m), 7.10 (2H, d, J8.3), 6.88 (2H, d, J8.7), 6.78(2H, d, J8.7), 5.17 (1H, s, H-3), 4.59 (2H, br s, —CH2(MPM)), 4.43 (1H, d, J 11.3, —CH2(MPM)), 4.34 (1H, d, J 11.3, —CH2(MPM)),4.19 (1H, s, H-4), 4.09 (1H, d, J 7.7, H-6), 4.06 (1H, d, J 7.7, H-6), 4.01 (1H, s, H-7), 3.82-3.77 (5H, m, —C1-CH2—O—, OCH3), 3.76 (3H, s, —OCH3);δc, (CDCl3) 159.4, 159.3, 139.4 (C-1′), 130.3, 129.7, 129.5, 129.3, 128.5, 127.5, 125.4, 113.9, 113.8, 85.9 (C-1), 84.1 (C-3), 81.1 (C-4), 77.4 (C-7), 73.7 (—CH2(MPM)), 73.4 (C-6), 71.8 (—CH2(MPM)), 66.3 (—C1-CH2—O—), 55.4 (—OCH3), 55.3 (—OCH3); m/z (FAB) 467 [M+Na-H2O]+, 461 [M−H]+.
- Cyclization of compound 12b (550 mg, 1.08 mmol) in the presence of TMAD (275 mg, 1.6 mmol), PBu 3 (325 mg, 1.6 mmol) and benzene (10 cm3) followed by the general work-up procedure and column chromatography afforded compound 13b as a colorless oil (445 mg, 84%); Rf 0.52 (CH2Cl2/MeOH 98:2, v/v); δh(CDCl3) 7.28 (2H, d, J 8.7), 7.11 (2H, d, J 8.6), 7.08-7.09 (2H, m, H-2′ and H-6′), 6.94 (1H, dd, J 8.5 and 9.2, H-5′), 6.88 (2H, d, J 8.6), 6.79 (2H, d, J 8.4), 5.08(1H, s, H-3), 4.62-4.55 (2H, m, —CH2(MPM)), 4.45 (1H, d, J 11.1, —CH2(MPM)), 4.36 (1H, d, J 11.6, —CH2(MPM)), 4.13 (1H, s, H-4), 4.07, 4.03 (1H each, 2d, J 7.6 each, H-6), 3.99 (1H, s, H-7), 3.81 (2H, m, —C1-CH2—O—), 3.80 (3H, s, —OCH3), 3.77 (3H, s, —OCH3), 2.23 (3H, d, J 1.6, Ar—CH3); δc (CDCl3) 162.3 (C-4′), 159.4, 159.3, 134.8, 134.7, 130.3, 129.6, 129.5, 129.2, 128.5, 128.4, 128.3, 124.2, 115.1, 114.8, 113.9, 113.8, 85.9 (C-1), 83.5 (C-3), 81.0 (C-4), 77.1 (C-7), 73.6 (—CH2(MPM)), 73.4 (C-6), 71.8 (—CH2(MPM)), 66.2 (—C1-CH2—O—), 55.4 (—OCH3), 55.3 (—OCH3), 14.7 (d,J3.3, Ar—CH3); m/z (FAB) 494 [M]+, 493 [M−H]+.
- Cyclization of compound 12c (700 mg, 1.32 mmol) in the presence of TMAD (345 mg, 2.0 mmol), PBu 3 (405 mg, 2.0 mmol) and benzene (15 cm3) followed by the general work-up procedure and column chromatography afforded compound 13c as a colorless oil (526 mg, 78%); Rf 0.53 (CH2Cl2/MeOH 98:2, v/v); δH (CDCl3) 7.91-7.86 (2H, m), 7.78 (1H, d, J 8.2), 7.73 (1H, d, J 7.1), 7.53-7.46 (3H, m), 7.32 (2H, d, J 8.7), 7.04 (2H, d, J 8.7), 6.90 (2H, d, J 8.3), 6.71 (2H, d,J8.6), 5.79 (1H, s, H-3), 4.67-4.61 (2H, m, —CH2(MPM)), 4.43 (1H, s, H-4), 4.38 (1H, d, J 11.2, —CH2(MPM)), 4.27 (1H, d, J 10.9, —CH2(MPM)), 4.16 (2H, br s, H-6), 4.08 (1H, s, H-7), 3.91, 3.87 (1H each, 2d, J 11.0 each, —C1-CH2—O—), 3.81 (3H, s, —OCH3), 3.72 (3H, s, —OCH3); δc (CDCl3) 159.3, 134.6 (C-1′), 133.5, 130.3, 129.8, 129.7, 129.4, 129.3, 128.9, 128.1, 126.4, 125.8, 125.6, 123.8, 122.7, 113.9, 113.7, 85.7 (C-1), 82.3 (C-3), 79.9 (C-4), 78.2 (C-7), 73.7 (—OCH2(MPM)), 73.5 (C-6), 71.8 (—OCH2(MPM)), 66.3 (—C1-CH2—O—), 55.4 (—OCH3), 55.3 (—OCH3); m/z (FAB) 512 [M]+, 511 [M−H]+.
- Cyclization of compound 12d (650 mg, 1.08 mmol) in the presence of TMAD (275 mg, 1.6 mmol), PBu 3 (325 mg, 1.6 mmol) and benzene (10 cm3) followed by the general work-up procedure and column chromatography afforded compound 13d as a pale yellow solid (496 mg, 79%); Rf 0.53 (CH2Cl2/MeOH 98:2, v/v); δH (CDCl3) 8.29 (1H, d, J8.2), 8.18-8.12 (5H, m), 8.08-8.01 (2H, m), 7.96 (1H, d, J 7.5), 7.35 (2H, d, J 8.5), 6.97 (2H, d, J 8.9), 6.92 (2H, d, J 8.8), 6.60 (2H, d, J8.8), 6.09 (1H, s, H-3), 4.71-4.65 (2H, m, —CH2(MPM)), 4.49 (1H, s, H-4), 4.34 (1H, d, J 1 1.4, —CH2(MPM)), 4.23 (1H, d, J 11.1, —CH2(MPM)), 4.25 (1H, d, J 7.6, H-6), 4.21 (1H, d, J7.8, H-6), 4.16 (1H, s, H-7), 3.95-3.94 (2H, m, —C1-CH2—O—), 3.81 (3H, s, —OCH3), 3.59 (3H, s, —OCH3); δC (CDCl3) 159.4, 159.3, 132.2 (C-1′), 131.4, 130.8, 130.7, 130.4, 129.5, 129.4, 128.0, 127.5, 127.4, 126.9, 126.1, 125.6, 125.4, 124.9, 124.8, 124.7, 123.6, 122.0, 113.9, 113.7, 85.9 (C-1), 82.7 (C-3), 80.6 (C-4), 77.9 (C-7), 73.9 (—OCH2(MPM)), 73.5 (C-6), 71.8 (—OCH2(MPM)), 66.3 (—C]-CH2—O—), 55.4 (—OCH3), 55.2 (—OCH3); m/z (FAB) 587 [M+H]+, 586 [M]+
- Cyclization of compound 12e (550 mg, 1.05 mmol) in the presence of TMAD (275 mg, 1.6 mmol), PBu 3 (325 mg, 1.6 mmol) and benzene (10 cm3) followed by the general work-up procedure and column chromatography afforded compound 13e as a colorless oil (425 mg, 80%); Rf 0.52 (CH2Cl2/MeOH 98:2, v/v); δH (CDCl3) 7.30 (2H, d, J9.0), 7.24 (1H, s, H-6′), 7.13 (2H, d, J 8.9), 6.89 (1H, s, H-3′), 6.88 (2H, d, J 8.8), 6.79 (2H, d, J 8.6), 5.18 (1H, s, H-3), 4.64-4.57 (2H, m, —CH2(MPM)), 4.46 (1H, d, J 11.2, —CH2(MPM)), 4.36 (1H, d, J 11.5, —CH2(MPM)), 4.18 (1H, s, H-4), 4.14 (1H, s, H-7), 4.09 (1H, d, J7.9, H-6), 4.04 (1H, d, J7.7, H-6), 3.86 (2H, s, —C1-CH2—O—), 3.80 (3H, s, —OCH3), 3.76 (3H, s, —OCH3), 2.21 (6H, s, 2×Ar—CH3), 2.17 (3H, s, Ar—CH3);δc. (CDCl3) 159.4, 159.3, 135.5 (C-1′), 134.4, 134.0, 131.7, 131.3, 130.5, 129.9, 129.4, 129.2, 127.2, 113.9, 113.8, 85.6 (C-1), 82.4 (C-3), 79.4 (C-4), 77.6 (C-7), 73.5 (—OCH2(MPM)), 73.4 (C-6), 71.8 (—OCH2(MPM)), 66.3 (—C1-CH2—O—), 55.4 (—OCH3), 55.3 (—OCH3), 19.5 (—CH3), 19.3 (—CH3), 18.4 (—CH3); m/z (FAB) 504 [M]+, 503 [M−H]+.
- To a stirred solution of Compound 13a-e in CH 2Cl2 (containing a small amount of H2O) at room temperature, was added 2,3-dichloro-5,6-dicyanoquinone (DDQ) which resulted in an immediate appearance of a deep greenish-black color which slowly faded into pale brownish-yellow. The reaction mixture was vigorously stirred at room temperature for 4 h. The precipitate was removed by filtration through a short pad of silica gel and washed with EtOAc. The combined filtrate was washed, successively, with saturated aqueous NaHCO3 (2×25 cm3) and brine (25 cm3). The separated organic phase was dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The crude product obtained was purified by column chromatography [4-5% (v/v) MeOH in CH2Cl2] to give compounds 14a-e.
- Compound 13a (400 mg, 0.86 mmol) was treated with DDQ (600 mg, 2.63 mmol) in a mixture of CH 2Cl2 (10 cm3) and H2O (0.5 cm3). After the general work-up procedure and column chromatography, compound 14a was obtained as a white solid material (128 mg, 66%); Rf 0.30 (CH2Cl2/MeOH 9:1, v/v); δH (CD3)2CO/CD3OD; (CD3)2CO was added to the compound followed by addition of CD3OD until a clear solution appeared) 7.40-7.22 (5H, m), 4.99 (1H, s), 4.09 (1H, s), 4.04 (1H, s), 4.01 (1H, d, J7.7), 3.86 (1H, d, J7.7), 3.90 (2H, br s), 3.77 (2H, br s); δc ((CD3)2CO/CD3OD; (CD3)2CO was added to the compound followed by addition of CD3OD until a clear solution appeared) 140.0, 128.2, 127.2, 125.4, 87.2, 83.7, 83.5, 72.3, 70.2, 58.4; m/z (FAB) 223 [M+H]+.
- Compound 13b (400 mg, 0.81 mmol) was treated with DDQ (570 mg, 2.5 mmol) in a mixture of CH 2Cl2 (10 cm3) and H2O (0.5 cm3). After the general work-up procedure and column chromatography, compound 14b was obtained as a white solid material (137 mg, 67%); Rf 0.31 (CH2Cl2/MeOH 9: 1, v/v); & (CD3OD) 7.23 (1H, d, J 8.1), 7.19 (1H, m), 6.99 (1H, dd, J 8.5 and 9.3), 4.99 (1H, s), 4.09 (1H, s), 4.06 (1H, s), 4.03 (1H, d, J 7.6), 3.93-3.91 (3H, m), 2.25 (3H, d, J 1.4); δC, (CD3OD) 161.9 (d, J 243.3), 136.4 (d, J 3.4), 129.6 (d, J 5.0), 126.1 (d, J 22.8), 125.5 (d, J8.0), 115.7 (d, J22.9), 88.5, 85.0, 84.3, 73.5, 71.3, 59.4, 14.5 (d, J3.7); m/z (FAB) 255 [M+H]+.
- Compound 13c (475 mg, 0.93 mmol) was treated with DDQ (600 mg, 2.63 mmol) in a mixture of CH 2Cl2 (10 cm3) and H2O (0.5 cm3). After the general work-up procedure and column chromatography, compound 14c was obtained as a white solid material (170 mg, 67%); Rf 0.31 (CH2Cl2/MeOH 9:1, v/v); δH (CDCl3/CD3OD; CD3OD was added to the compound followed by addition of CDCl3 until a clear solution appeared) 7.94-7.86 (2H, m), 7.80-7.74 (2H, m), 7.55-7.46 (3H, m), 5.74 (1H, s), 4.56 (2H, br s), 4.37 (1H, s), 4.24 (1H, s), 4.17-4.11 (2H, m), 4.04 (2H, br s); δC (CDCl3/CD3OD; CD3OD was added to the compound followed by addition of CDCl3 until a clear solution appeared 134.7, 134.0, 130.2, 129.3, 128.6, 126.8, 126.2, 125.8, 123.8, 122.8, 87.4, 83.1, 82.2, 73.1, 71.5, 59.0; m/z (FAB) 273 [M+H]+, 272 [M]+.
- Compound 13d (411 mg, 0.7 mmol) was treated with DDQ (570 mg, 2.5 mmol) in a mixture of CH 2Cl2 (10 cm3) and H2O (0.5 cm3). After the general work-up procedure and column chromatography, compound 14d was obtained as a white solid material (182 mg, 75%); Rf 0.32 (CH2Cl2/MeOH 9: 1, v/v); & (CDCl3/CD3OD; CD3OD was added to the compound followed by addition of CDCl3 until a clear solution appeared) 8.32 (1H, d, J 7.8), 8.23-8.18 (5H, m), 8.06 (2H, br s), 8.01 (1H, d, J7.6), 6.06 (1H, s), 4.47 (1H, s), 4.36 (1H, s), 4.27-4.18 (2H, m), 4.10 (2H, br s); δc (CDCl3/CD3OD) 132.2, 131.0, 128.5, 127.8, 127.3, 126.5, 125.9, 125.7, 125.1, 123.6, 122.1, 87.7, 83.7, 82.6, 73.1, 71.4, 58.9; m/z (FAB) 347 [M+H]+, 346 [M]+.
- Compound 13e (355 mg, 0.7 mmol) was treated with DDQ (570 mg, 2.5 mmol) in a mixture of CH 2Cl2 (10 cm3) and H2O (0.5 cm3). After the general usual work-up procedure and column chromatography, compound 14e was obtained as a white solid material (120 mg, 65%); Rf 0.31 (CH2Cl2/MeOH 9:1, v/v); δH (CDCl3/CD3OD; CD3OD was added to the compound followed by addition of CDCl3 until a clear solution appeared) 7.23 (1H, s), 6.92 (1H, s), 5.14 (1H, s), 4.26 (1H, s), 4.10 (1H, s), 4.08, (1H, d,J7.7), 4.00-3.95 (3H, m), 2.23 (6H, s), 2.21 (1H, s); δC(CDCl3/CD3OD; CD3OD was added to the compound followed by addition of CDCl3 until a clear solution appeared) 135.6, 133.9, 133.8, 131.7, 131.2, 126.6, 86.6, 82.1, 81.9, 72.3, 70.6, 58.5, 19.2, 19.0, 18.1; m/z (FAB) 265 [M+H]+, 264 [M]+.
- 4,4′-Dimethoxytrityl chloride (DMTCI) was added in one portion to a stirred solution of compound 14a-e in anhydrous pyridine. After stirring the mixture at room temperature for 4 h, methanol (0.2 cm 3) was added and the resulting mixture was evaporated to dryness under reduced pressure. The residue was coevaporated with anhydrous CH3CN (2×5 cm3) and anhydrous toluene (2×5 cm3) and then dissolved in CH2Cl2 (20 cm3, traces of acid removed by filtration through a short pad of basic alumina). The resulting solution was washed, successively, with saturated aqueous NaHCO3 (2×10 cm3) and brine (10 cm3). The separated organic phase was dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The crude product obtained was purified by column chromatography [0.25-0.50% (v/v) MeOH in CH2Cl2, containing 0.5% Et3N] affording compounds 15a-e.
- Dimethoxytritylation of compound 14a (108 mg, 0.49 mmol) using DMTCI (214 mg, 0.63 mmol) in anhydrous pyridine (2 cm 3) followed by the general work-up procedure and column chromatography afforded compound 15a as a white solid material (180 mg, 71%); Rf 0.31 (CH2Cl2/MeOH 98:2, v/v); δH (CDCl3) 7.66-7.21 (14H, m), 6.84 (4H, d, J8.8), 5.19 (1H, s), 4.29 (1H, s), 4.13 (1H, s), 4.07 (1H, d, J8.4), 4.01 (1H, d, J8.3), 3.78 (6H, s), 3.55 (1H, d, J 10.2), 3.50 (1H, d, J 10.7), 2.73 (1H, br s); δC (CDCl3) 158.6, 149.8, 144.9, 139.4, 136.2, 135.9, 135.8, 130.3, 130.2, 128.5, 128.3, 128.0, 127.6, 126.9, 125.4, 123.9, 113.3, 86.4, 86.0, 83.8, 83.4, 73.0, 71.6, 60.2, 55.3; m/z (FAB) 525 [M+H]+, 524 [M]+.
- Dimethoxytritylation of compound 14b (95 mg, 0.38 mmol) using DMTCI (129 mg, 0.42 mmol) in anhydrous pyridine (2 cm 3) followed by the general work-up procedure and column chromatography afforded compound 15b as a white solid material (126 mg, 61%); Rf 0.32 (CH2Cl2/MeOH 98:2, v/v); δh (CDCl3) 7.53-7.15 (11H, m), 6.97 (1H, dd, J 8.7 and 8.9), 6.84 (4H, d, J8.8), 5.11 (1H, s), 4.26 (1H, d, J3.9), 4.08 (1H, s), 4.03 (1H, d, J8.0), 3.95 (1H, d, J 8.0), 3.78 (6H, s), 3.54 (1H, d, J 10.5), 3.47 (1H, d, J 10.1), 2.26 (3H, d, J 1.5), 2.08 (1H, br s); δc (CDCl3) 160.8 (d, J244.1), 158.7, 144.9, 135.9, 134.7, 134.6, 130.3, 130.2, 130.1, 128.5, 128.4, 128.3, 128.0, 127.0, 125.2, 124.9, 124.4, 124.3, 115.2, 114.9, 113.4, 86.5, 86.0, 83.7, 83.0, 72.9, 71.7, 60.1, 55.3, 14.8 (d, J3.1); m/z (FAB) 556 [M]+.
- Dimethoxytritylation of compound 14c (125 mg, 0.46 mmol) using DMTCI (170 mg, 0.5 mmol) in anhydrous pyridine (2 cm 3) followed by the general work-up procedure and column chromatography afforded compound 15c as a white solid material (158 mg, 60%); Rf 0.35 (CH2Cl2MeOH 98:2, v/v); δH (CDCl3) 7.95-7.86 (3H, m), 7.79 (1H, d, J 8.3), 7.58-7.41 (9H, m), 7.35-7.23 (3H, m), 6.86 (4H, d, J 8.8), 5.80 (1H, s), 4.36 (1H, s), 4.32 (1H, d, J 6.5), 4.17 (1H, d, J8.3), 4.06 (1H, d, J8.0), 3.78 (6H, s), 3.62-3.56 (2H, m), 2.00 (1H, d, J6.6); δH (CDCl3) 158.7, 144.9, 136.0, 135.9, 134.5, 133.6, 130.3, 129.8, 129.0, 128.3, 128.2, 128.1, 127.0, 126.5, 125.9, 125.6, 123.9, 122.6, 113.4, 86.6, 85.7, 82.5, 81.7, 73.1, 72.6, 60.2, 55.3; m/z (FAB) 575 [M+H]+, 574 [M]+.
- Dimethoxytritylation of the compound 14d (130 mg, 0.38 mmol) using DMTCI (140 mg, 0.42 mmol) in anhydrous pyridine (2 cm 3) followed by the general work-up procedure and column chromatography afforded compound 15d as a white solid material (147 mg, 61%); Rf 0.37 (CH2Cl2/MeOH 98:2, v/v); & (CDCl3) 8.46 (1H, d, J 8.0), 8.19-8.00 (7H, m), 7.61 (2H, dd, J 1.6 and 7.4), 7.48 (4H, d, J8.3), 7.35 (2H, dd, J7.2 and 7.5), 7.25 (1H, m), 7.15 (1H, m), 6.88 (4H, d, J9.0), 6.10 (1H, s), 4.46 (1H, s), 4.43 (1H, br s), 4.25 (1H, d, J8.1), 4.12 (1H, d, J8.1), 3.79 (6H, s), 3.71-3.63 (2H, m), 2.22 (1H, br s); δC (CDCl3) 158.7, 149.8, 144.9, 136.1, 136.0, 135.9, 132.1, 131.4, 130.9, 130.6, 130.3, 130.2, 129.2, 129.1, 128.4, 128.3, 128.2, 128.1, 127.5, 127.4, 127.0, 126.9, 126.2, 125.5, 125.4, 124.9, 124.8, 124.7, 123.8, 123.7, 121.9, 113.4, 86.6, 86.1, 83.2, 82.2, 73.2, 72.4, 60.3, 55.3; m/z (FAB) 649 [M+H]+, 648 [M]+.
- Dimethoxytritylation of compound 14e (80 mg, 0.3 mmol) using DMTCI (113 mg, 0.33 mmol) in anhydrous pyridine (2 cm 3) followed by the general work-up procedure and column chromatography afforded compound 15e as a white solid material (134 mg, 78%); Rf 0.32 (CH2Cl2/MeOH 98:2, v/v); δH (CDCl3) 7.55 (2H, d, J 7.9), 7.45-7.42 (4H, m), 7.32-7.21 (4H, m), 6.93 (1H, s), 6.84 (4H, d, J 8.2), 5.20 (1H, s), 4.40 (1H, s), 4.08 (1H, s), 4.04 (1H, d, J 8.3), 3.95 (1H, d, J 8.2), 3.78 (6H, s), 3.56 (1H, d, J 10.5), 3.47 (1H, d, J 10.2), 2.24 (3H, s), 2.22 (3H, s), 2.19 (3H, s); δC (CDCl3) 158.6, 145.0, 136.0, 135.7, 134.4, 134.2, 131.8, 131.3, 130.3, 130.2, 128.3, 128.0, 127.2, 126.9, 113.3, 86.4, 85.7, 82.1, 81.8, 73.0, 71.8, 60.2, 55.3, 19.6, 19.3, 18.4; m/z (FAB) 567 [M+H]+, 566 [M]+.
- 2-Cyanoethyl N,N′-diisopropylphosphoramidochloridite was added dropwise to a stirred solution of nucleoside 15a-e and N,N-diisopropylethylamine (DIPEA) in anhydrous CH 2Cl2 at room temperature. After stirring the mixture at room temperature for 6 h, methanol (0.2 cm3) was added and the resulting mixture diluted with EtOAc (20 cm3, containing 0.5% Et3N, v/v). The organic phase was washed, successively, with saturated a. NaHCO3 (2×10 cm3) and brine (10 cm3). The separated organic phase was dried (Na2SO4), filtered and evaporated to dryness under reduced pressure. The residue obtained was purified by column chromatography [25-30% (v/v) EtOAc in n-hexane containing 0.5% Et3N] to give the amidites 16a-e.
- Treatment of compound 15a (170 mg, 0.32 mmol) with 2-cyanoethyl N,N′-diisopropylphosphoramidochloridite (85 mg, 0.36 mmol) in the presence of DIPEA (0.4 cm 3) and anhydrous CH2Cl2 (2.0 cm3) followed by the general work-up procedure and column chromatography afforded phosphoramidite 16a as a white solid material (155 mg, 66%); Rf 0.45, 0.41 (CH2Cl2/MeOH 98:2, v/v); δp (CDCl3) 149.3, 148.9.
- Treatment of compound 15b (95 mg, 0.17 mmol) with 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (53 mg, 0.22 mmol) in the presence of DIPEA (0.3 cm 3) and anhydrous CH2Cl2 (2.0 cm3) followed by the general work-up procedure and column chromatography afforded phosphoramidite 16b as a white solid material (85 mg, 66%); Rf 0.45, 0.41 (CH2Cl2/MeOH 98:2, v/v); δP (CDCl3) 149.3, 148.8.
- Treatment of compound 5c (158 mg, 0.28 mmol) with 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (75.7 mg, 0.32 mmol) in the presence of DIPEA (0.4 cm 3) and anhydrous CH2Cl2 (2.0 cm3) followed by the general work-up procedure and column chromatography afforded phosphoramidite 16c as a white solid material (127 mg, 60%); Rf 0.47, 0.44 (CH2Cl2/MeOH 98:2, v/v); δp (CDCl3) 149.2, 149.1.
- Treatment of compound 15d (140 mg, 0.22 mmol) with 2-cyanoethyl N,N′-diisopropylphosphoramidochloridite (64 mg, 0.27 mmol) in the presence of DIPEA (0.3 cm 3) and anhydrous CH2Cl2 (2.0 cm3) followed by the general work-up procedure and column chromatography afforded phosphoramidite 16d as a white solid material (124 mg, 68%); Rf 0.51, 0.47 (CH2Cl2/MeOH 98:2, v/v); δp (CDCl3) 149.4, 149.1.
- Treatment of compound 15e (130 mg, 0.23 mmol) with 2-cyanoethyl N,N-diisopropylphosphoramidochloridite (64 mg, 0.27 mmol) in the presence of DIPEA (0.3 cm 3) and anhydrous CH2Cl2 (2.0 cm3) followed by the general work-up procedure and column chromatography afforded phosphoramidite 16e as a white solid material (111 mg, 63%); Rf 0.44, 0.42 (CH2Cl2/MeOH 98:2, v/v); bp (CDCl3) 149.0.
- All oligomers were prepared using the phosphoramidite approach on a Biosearch 8750 DNA synthesizer in 0.2 μmol scale on CPG solid supports (BioGenex). The stepwise coupling efficiencies for phosphoramidites 16a-c (10 min coupling time) and phosphoramidites 16d and 16e (20 min coupling time) were >96% and for unmodified deoxynucleoside and ribonucleoside phosphoramidites (with standard coupling time) generally >99%, in all cases using 1H-tetrazole as activator. After standard deprotection and cleavage from the solid support using 32% aqueous ammonia (12 h, 55° C.), the oligomers were purified by precipitation from ethanol. The composition of the oligomers were verified by MALDI-MS analysis and the purity (>80%) by capillary gel electrophoresis. Selected MALDI-MS data ([M−H] -; found/calcd.: ON3 2731/2733; ON4 2857/2857; ON6 3094/3093).
- The thermal denaturation experiments were performed on a Perkin-Elmer UVVIS spectrometer fitted with a PTP-6 Peltier temperature-programming element using a medium salt buffer solution (10 mM sodium phosphate, 100 mM sodium chloride, 0.1 mM EDTA, pH 7.0). Concentrations of 1.5 mM of the two complementary strands were used assuming identical extinction coefficients for modified and unmodified oligonucleotides. The absorbance was monitored at 260 nm while raising the temperature at a rate of 1° C. per min. The melting temperatures (T m values) of the duplexes were determined as the maximum of the first derivatives of the melting curves obtained.
- LNA containing the derivatives 17a-17e (FIG. 1, Schemel, Scheme 2), were synthesized, all based on the LNA-
type 2′-O,4′-C-methylene-, BD-ribofuranosyl moiety which is known to adopt a locked C3′-endo RNA-like furanose conformation [S. Obika, D. Nanbu, Y. Hari, K. Morio, Y. In, T. Ishida, and T. Imanishi, Tetrahedron Lett., 1997, 38, 8735; S. K. Singh, P. Nielsen, A. A. Koshkin and J. Wengel, Chem. Commun., 1998, 455; A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, Tetrahedron, 1998, 54, 3607; S. Obika, D. Nanbu, Y. Hari, J. Andoh, K. Morio, T. Doi and T. Imanishi, Tetrahedron Lett., 1998, 39, 5401]. The syntheses of the phosphoramidite building blocks 16a-16e suitable for incorporation of the LNA-type aryl C-glycosides 17a-17e are shown inScheme 1 andScheme 2 and described in details in the experimental section. In the design of an appropriate synthetic route, it was decided to utilize a reaction similar to one described recently in the literature. Thus, stereoselective attack of Grignard reagents of various heterocycles on a carbonyl group of an aldehyde corresponding to aldehyde 11 (Scheme 2) but with two O-benzyl groups instead of the two p-methoxybenzyl groups of aldehyde 11 (Scheme 2) has been reported to furnish locked-C-nucleosides [S. Obika, Y. Hari, K. Morio and T. Imanishi, Tetrahedron Lett., 2000, 41, 215; S. Obika, Y. Hari, K. Morio and T. Imanishi, Tetrahedron Lett., 2000, 41, 221]. The key intermediate in the synthetic route selected herein, namely the novel aldehyde 1.1 was synthesized from the known furanoside 1 [R. Yamaguchi, T. Imanishi, S. Kohgo, H. Horie and H. Ohrui, Biosci. Biotechnol. Biochem.,1999, 63, 736] following two different routes. In general, O-(p-Methoxy)benzyl protection was desirable instead of O-benzyl protection as removal of the benzyl protection at a later stage (i.e. 13->14) could also likely result in the cleavage of the benzylic O—C1, bond present, e.g., incompounds 13 and 14 (Scheme 2). In one route to givealdehyde 11, regioselectivep-methoxybenzylation of thefuranoside 1, followed by mesylation and methanolysis yielded the anomeric mixture of themethyl furanosides 9. Base induced cyclization followed by acetyl hydrolysis afforded thealdehyde 11 in approximately 24% overall yield from 1 (Scheme 1 and Scheme 2). This yield was improved to following a different strategy. Thus, di—O—mesylation of 1 followed by methanolysis and base induced intramolecular nucleophilic attack from the 2-OH group afforded the cyclized anomeric mixture ofmethyl furanoside 4. Substitution of the remaining mesyloxy group of 4 with an acetate group, followed by deacetylation, p-methoxybenzylation and then acetyl hydrolysis afforded the required aldehyde 11 (Scheme 1). - Coupling of the
aldehyde 11 with different aryl Grignard reagents yielded selectively one epimer of each of the compounds 12a-e in good yields (see experimental section for further details on this and other synthetic steps). Each of the diols 12a-e was cyclized under Mitsunobu conditions (TMAD, PBu3) to afford the bicyclic, C-nucleoside derivatives 13a-e. Oxidative removal of the p-methoxybenzyl protections was achieved in satisfactory yields using DDQ. Subsequent, selective 4,4′-dimethoxytritylation (to give compounds 15a-e) followed by phosphorylation afforded the phosphoramidite building blocks 16a-e in satisfactory yields. The configuration ofcompounds 13, and thus also compounds 11, 12 and 14-17 were assigned based on 1H NMR spectroscopy, including NOE experiments. - All oligomers were prepared in the 0.2 μmol scale using the phosphoramidite approach. The stepwise coupling efficiencies for phosphoramidites 16a-c (10 min coupling time) and phosphoramidites 16d and 16e (20 min coupling time) were >96% and for unmodified deoxynucleoside and ribonucleoside phosphoramidites (with standard coupling time) generally >99%, in all cases using 1H-tetrazole as activator. After standard deprotection and cleavage from the solid support using 32% aqueous ammonia (12 h, 55° C.), the oligomers were purified by precipitation from ethanol. The composition of the oligomers were verified by MALDI-MS analysis and the purity (>80%) by capillary gel electrophoresis.
- The hybridization of the oligonucleotides ON1—ON11 (Table 8 below) toward four 9-mer DNA targets with the central base being each of four natural bases were studied by thermal denaturation experiments (T m measurements; see the experimental section for details). Compared to the DNA reference ON1, introduction of one abasic LNA monomer AbL (ON2) has earlier been reported to prevent the formation of a stable duplex above 0° C. (only evaluated with adenine as the opposite base) [L. KvaernØ and J. Wengel, Chem. Commun., 1999, 657]. With the phenyl monomer 17a (ON3), Tm values in the range of 5-12° C. was observed. Thus, the phenyl moiety stabilizes the duplexes compared to AbL, but universal hybridization is not achieved as a preference for a central adenine base in the complementary target strand is indicated (Table 8). In addition, significant destabilization compared to the ON1:DNA reference duplex was observed. Results similar to those obtained for ON3 were obtained for oligomers isosequential with ON3 but containing 17b, 17c or 17e instead of 17a as the central monomer (Table 8, ON7, ON8 and ON9, respectively).
TABLE 8 Thermal denaturation experiments (Tm values shown) for ON1-ON11 towards DNA complements with each of the four natural bases in the central positiona Y: DNA target: 3′-d(CACTYTACG) A C G T ON1 5′-d(GTGATATGC) 28 11 12 19 ON2 5′-d(GTGAAb LATGC)<3 n.d. n.d. n.d. ON3 5′-d(GTGA17aATGC)12 5 6 7 ON4 5′-d(GTGA17dATGC)18 17 18 19 ON5 5′-d[2′-OMe(GTGATATGC)]35 14 19 21 ON6 5′-d[2′-OMe(GT LGA17dAT LGC)]39 38 37 40 ON7 5′-d(GTGA17bATGC)15 7 6 8 ON8 5′-d(GTGA17cATGC)15 7 6 9 ON9 5′-d(GTGA17eATGC)13 6 6 7 ON10 5′-d[2′-OMe(GT LGA17bAT LGC)]31 25 26 27 ON11 5′-d[2′-OMe(GT LGA17cAT LGC)]34 27 27 32 # 17a-17e; DNA sequences are shown as d(sequence) and 2′-OMe-RNA sequences as 2′-OMe(sequence); “n.d.” denotes “not determined”. The data reported for ON1 have been reported earlier [A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, Tetrahedron, 1998, 54, 3607]. The data reported for ON2 has been reported earlier [L. Kværnø and J. Wengel, Chem. Commun., 1999, 657]. - The
pyrene LNA nucleotide 17d (in ON4) displays more encouraging properties (Table 8). Firstly, the binding affinity towards all four complements is increased compared to ON3 (containing 17a). Secondly, universal hybridization is observed as shown by the four Tm values all being within 17-19° C. With respect to universal hybridization, 17d thus parallels the pyrene DNA derivative Py [T. J. Matray and E. T. Kool, J. Am. Chem. Soc., 1998, 120, 6191], but the decrease in thermal stability compared to the ON1:DNA reference is more pronounced for 17d (˜10° C.) than reported for Py (˜5° C. in a 12-mer polypyrimidine DNA sequence) [T. J. Matray and E. T. Kool, J. Am. Chem. Soc., 1998, 120, 6191]. It therefore appears that stacking (or intercalation) by the0 pyrene moiety is not favored by the conformational restriction of the furanose ring of 17d, although comparison of the thermal stabilities of ON2, ON3 and ON4 strongly indicate interaction of the pyrene moiety within the helix. - When measured against an RNA target [3′-r(CACUAUACG)], the T m values (using identical experimental conditions as for the experiments descried above) of ON3 was 11.9° C. and of ON4 was 12.7° C. For oligomers ON7, ON8 and ON9 (Table 8), the corresponding Tm values were 11.7, 8.8 and 10.2° C., respectively.
- ON5, ON6, ON10 and ON11 (see Table 8 above), were synthesized. The former being composed entirely of 2′-OMe-RNA monomers and the latter three of six 2′-OMe-RNA monomers (see FIG. 1), two LNA thymine monomers TL (see FIG. 1), and one central
LNA pyrene monomer 17d (oligomer ON6), or one central monomer 17b (ON10) or 17c (ON11). A sequence corresponding to ON6 but with three TL monomers has earlier been shown to form a duplex with complementary DNA of very high thermal stability. ON6 is therefore suitable for evaluation of the effect of introducing high-affinity monomers around a universal base. As seen in Table 8, the 2′-OMe-RNA reference ON5 binds to the DNA complement with slightly increased thermal stability and conserved Watson-Crick discrimination (compared to the DNA reference ON1). Indeed, the LNA/2′-OMe-RNA chimera ON6 displays universal hybridization behavior as revealed from the four Tm values (37, 38, 39 and 40° C.). All four Tm values obtained for ON6 are higher than the Tm values obtained for the two fully complementary reference duplexes ON1:DNA (Tm=28° C.) and ON5:DNA (Tm=35° C.). - These novel data demonstrate that the
pyrene LNA monomer 17d display universal hybridization behavior both in a DNA context (ON4) and in an RNA-like context (ON6), and that the problem of decreased affinity of universal hybridization probes can be solved by the introduction of high-affinity monomers, e.g. 2′-OMe-RNA and/or LNA monomers. Increased affinities compared to ON7 and ON8 were obtained for ON10 and ON11, respectively, but universal hybridization behavior was not obtained as a preference for a central adenine base in the complementary target strand is indicated (Table 8 above). - A systematic thermal denaturation study with ON6 (Table 11) was performed to determine base-pairing selectivity. For each of the four DNA complements (DNA target strands; monomer Y=A, C, G or T) used in the study shown in Table 8 above, ON6, containing a central
pyrene LNA monomer 17d, was hybridized with all four base combinations in the neighboring position towards the 3′-end of ON6 (DNA target strands; monomer Z=A, C, G or T, monomer X=T) and the same towards the 5′-end of ON6 (DNA target strands; monomer X=A, C, G or T, monomer Z=T). In all eight subsets of four data points, satisfactory to excellent Watson-Crick discrimination was observed between the match and the three mismatches (Table 11 below, ΔTm values in the range of 5-25° C.).TABLE 11 Thermal denaturation experiments (Tm values shown) to evaluate the base-pairing selectivity of the bases neighboring the universal pyrene LNA monomer 17d in the2′-OMe-RNA/LNA chimera ON6. In the target strand [3′-d(CAC-XYZ-ACG)], the central three bases XYZ are varied among each of the four natural bases a5′[2′-OMe(GT LG-A17dA-T LGC)] 3′-d(CAC-X Y Z-ACG) XYZ Tm/° C. XYZ T m/° C. XYZ Tm/° C. XYZ Tm/° C. TAA 26 TCA 22 TGA 22 TTA 29 TAC 26 TCC 29 TGC 26 TTG 31 TAG 24 TCG 24 TGG 30 TTC 32 TAT 39 TCT 38 TGT 37 TTT 40 AAT 18 ACT 27 AGT 22 ATT 28 CAT 30 CCT 31 CGT 27 CTT 35 GAT 14 GCT 28 GGT 16 GTT 27 TAT 39 TCT 38 TGT 37 TTT 40 - The results reported herein have several important implications for the design of probes for universal hybridization: (I) Universal hybridization is possible with a conformationally restricted monomer as demonstrated for the pyrene LNA monomer; (2) Universal hybridization behavior is feasible in an RNA context; (3) The binding affinity of probes for universal hybridization can be increased by the introduction of high-affinity monomers without compromising the universal hybridization and the base-pairing selectivity of bases neighboring the universal base.
- Based on the results reported herein, that chimeric oligonucleotides comprising pyrene and other known universal bases attached at various backbones (e.g. LNA-type monomers, ribofuranose monomers or deoxyribose monomers in 2′-OMe-RNA/LNA chimeric oligos) likewise will display attractive properties with respect to universal hybridization behavior. For example, an oligomer identical with the 2′-OMe-RNA/LNA oligo ON6 but with the 17d monomer substituted by a pyrenyl-2′-OMe-ribonucleotide monomer.
- These chimeric oligonucleotides are comprised of pyrene and other known universal bases attached at various backbones (e.g. LNA-type monomers, ribofuranose monomers or deoxyribose monomers in 2′-OMe-RNA/LNA oligos). Experimentation with these chimeric oligonucleotides are for evaluating the possibility of obtaining similar results to the 2′-OMe-RNA/LNA oligo ON6 at a lower cost, for example, by substituting Py L with a pyrenyl-2′-OMe-ribonucleotide monomer.
- A. In vitro Synthesis of the Yeast Spike RNAs
- Amplification of the yeast genes was carried out by standard two-step PCR using yeast genomic DNA as template (21). In the second PCR, a poly-T 20 tail was inserted in the amplicon. The DNA fragments were ligated into the
pTRlamp 18 vector (Ambion, USA) using the Quick Ligation Kit (New England Biolabs, USA) according to the manufacturers instructions and transformed into E. coli DH-5α (21). Synthesis of in vitro RNA was done using the MEGAscript™ T7 Kit (Ambion, USA) according to the manufacturers instructions. - B. Design of the LNA Expression Arrays
- Capture probes were designed using the OligoDesign™ software as described in the previous examples.
- C. Printing and hybridisation of the LNA expression microarrays
- The LNA oligonucleotide microarrays were printed onto Immobilizer™ MicroArray Slides (Exiqon, Denmark) using the Packard Biochip I Arrayer (Packard, USA), with a spot volume of 2×300 pl of a 10 μM capture probe solution. Four replicas of each capture probe were printed on each slide. Mixed staged Caenorhabditis elegans worm cultures were cultivated according to standard protocols. RNA was extracted from worm samples using the FastRNA Kit, GREEN (Q-BIO, USA) according to the supplier's instructions. Fluorochrome-labelled first strand cDNA was synthesized from worm total RNA or in vitro synthesized RNA as described (22) followed by purification of the cDNA target, hybridisation of the microarrays overnight at 65° C., washing of the slides and drying of the arrays (22). The slides were scanned using a
ScanArray 4000 XL scanner (Perkin-Elmer, USA), and the array data were processed using the GenePix™ Pro 4.0 software package (Axon, USA). - D. Assessment of Sensitivity and Specificity in LNA Expression Microarrays
- To enable direct comparisons between LNA and DNA capture probes in measuring gene expression levels, specific oligonucleotide capture probes for the Saccharomyces cerevisiae genes SWI5 and TH14 were designed in the 3′-end of the two ORFs. The capture probes were synthesized as 50-mer DNA and corresponding LNA-modified oligonucleotides, respectively, with an LNA substitution at every 3rd nucleotide position. In addition, 40-mer DNA and LNA oligonucleotides were designed as truncated versions of the 50-mer capture probes, along with oligonucleotides with 1 to 5 consecutive mismatches positioned centrally in the 50-mer and 40-mer capture probes. All capture probes were synthesized with an anthraquinone group at the 5′-end and a hexaethyleneglycol dimer linker region (HEG2 spacer arm) enabling photocoupling onto polymer microarray slides as described in U.S. Pat. No. 6,531,591.
- To assess the sensitivity and specificity of the oligonucleotide microarrays, in vitro synthesized yeast RNA for either SWI5 or THI4 was spiked into C. elegans total RNA for cDNA target synthesis followed by hybridization of the microarrays with fluorochrome-labelled cDNA target pools. The incorporation of LNA nucleotides into 50-mer DNA oligonucleotide capture probes results in a 3 to 4-fold increase in fluorescence intensity levels, when hybridized with the spiked, complex cDNA target pools under standard stringency conditions (FIGS. 36a and 36 c). The sensitivity increase is even more pronounced, 5 to 12-fold, when 40-mer LNA capture probes are employed. None of the yeast capture probes showed cross-hybridization to C. elegans cDNA target control without yeast spike RNA under the same conditions.
- The specificity of the oligonucleotide capture probes was examined using a panel of LNA mismatch oligonucleotides together with the DNA controls. As demonstrated in FIGS. 36 a and 36c, the fluorescence intensities obtained with the LNA-modified 40-mer triple mismatch oligonucleotides show a 3-fold intensity decrease relative to the perfectly matched duplexes. In contrast, the corresponding 40-mer standard DNA capture probes are neither capable of forming stable duplexes nor discriminating between the perfect match and mismatched targets under standard hybridization stringency conditions, resulting in low intensity values from all 40-mer DNA capture probes (FIGS. 36a and 36 c). Interestingly, mismatch discrimination with the 50-mer LNA probes could be significantly improved by increasing the hybridization temperature from 65° C. to 70° C. (FIGS. 36b and 36 d), without compromising their capture sensitivity. By comparison, the signal intensities from all 50-mer DNA capture probes including the perfect match oligonucleotides were reduced under the same conditions (FIGS. 36b and 36 d). Considered together, our results strongly support the contention that LNA oligonucleotide capture probes are significantly more sensitive and specific than DNA probes, being able to discriminate between highly homologous (90%) mRNAs with a 5 to 10-fold increase in sensitivity.
- In a typical cell, mRNAs can be subdivided into three kinetic classes: (i) highly abundant (30-90% of the total mRNA mass, 0.1% of the sequence complexity); (ii) medium abundant (50% mass, 2-5% of complexity); and (iii) low-abundant mRNAs (<1% mass, >90% of complexity). In addition, alternative splicing has been shown to be prevalent in higher eukaryotes, where at least 50% of the genes appear to be alternatively spliced, thereby generating additional diversity within the transcriptome. It is thus of utmost importance that the dynamic range, sensitivity and specificity of the expression profiling technology used are optimal, especially when analyzing expression levels of messages and mRNA splice variants belonging to the low-abundant class of high mRNA sequence complexity. A common problem for all DNA oligonucleotide microarrays is the need for an adequate compromise with respect to the sensitivity and specificity of the platform. In the present example LNA oligonucleotide microarrays perform better in expression profiling than microarrays with corresponding DNA probes. Our results clearly demonstrate that both the specificity and sensitivity in target molecule capture can be improved using LNA oligonucleotide microarrays, enabling discrimination between highly homologous mRNAs and alternative splice variants with a simultaneous increase in sensitivity.
- FIGS. 36 a-36 d shows the sensitivity and specificity of LNA oligonucleotide capture probes (black bars) compared to DNA capture probes (white bars) on expression microarrays. Fluorescence intensity is shown in arbitrary units (relative measurements). The arrays comprising 50-mer and 40-mer perfect match and 1-5 mismatch capture probes were hybridized at 65° C. in 3×SSC with Cy3-labelled cDNA from 10 μg C. elegans total RNA spiked with yeast a) SWI5 RNA and c) THI4 RNA. Demonstration of improved mismatch discrimination with the 50-mer LNA probes by increasing the hybridization temperature from 65° C. to 70° C. hybridized with Cy3-labelled cDNA from 10 μg C. elegans total RNA spiked with yeast b) SWI5 RNA and d) THI4 RNA.
- A. Capture Probe Design
- Unique capture probes for the yeast HSP78 gene were designed using the OligoDesign software, described in FIGS. 19A-19F. The design options used were: (i) length of each oligonucleotide probe was 25 nucleotides; (ii)
Blast word length 7; (iii) Blast expectation cut-off 1000; other options were as default; 24 DNA capture probes were selected. Furthermore, three different LNA-substituted probes were designed based on the sequences of the 24 DNA capture probes, selected by OligoDesign: optimal LNA_T, optimal LNA_TC andLNA —3. In the LNA_T design the DNA t nucleotides were substituted with LNA T. For the LNA_TC design, LNA T and C nucleotides were used to substitute DNA t and c. In theLNA —3 design every third DNA nucleotide was substituted with the corresponding LNA nucleotide. For the LNA_T and LNA_TC design, no blocks of LNAs were allowed; in addition the LNAs were substituted in a pattern providing a more narrow duplex melting temperature range compared to the DNA Tm range. In addition, an equivalent set of capture probes with a single mismatch in the central nucleotide position was designed. Altogether, 192 capture probes were designed including a anthraquinone (AQ29) 5′-modifier and a hexaethyleneglycol dimer (HEG2) at the 5′ end of each probe —as shown in Table 13. - B. Determination of the Duplex Melting Temperatures (T m).
- The duplex melting temperatures of the DNA, LNA T and LNA TC designed probes were measured using the
Perkin Elmer Lambda 40 Spectrophotometer and according to Wahlestedt et al. PNAS 97/10 2000. All oligos were measured twice and if the replica values deviated more than 1° C., then a third or a fourth measurement was carried out. The average Tm for each oligonucleotide duplex is presented in Table 14. - C. In Vitro Synthesis of Fluorochrome-Labeled Yeast HSP78 RNA
- C1. Genomic DNA was prepared from a wild type standard laboratory strain of Saccharomyces cerevisiae using the Nucleon MiY DNA extraction kit (Amersham Biosciences) according to supplier's instructions.
- C2. PCR amplification of the partial yeast gene was done by standard PCR using yeast genomic DNA as template. In the first step of amplification, a forward primer containing a restriction enzyme site and a reverse primer containing a universal linker sequence were used. In this
step 20 bp was added to the 3′-end of the amplicon, next to the stop codon. In the second step of amplification, the reverse primer was exchanged with a nested primer containing a poly-T20 tail and a restriction enzyme site. The SWI5 amplicon contains 730 bp of the SWI5 ORF plus 20 bp universal linker sequence and a poly-A20 tail. - The PCR primers used were:
YDR258C-For-SacI: acgtgagctcttttgacatgtcagaatttcaag (SEQ ID NO:) YDR258C-Rev-Uni: gatccccgggaattgccatgttacttttcagct (SEQ ID NO:) tcctcttcaac Uni-polyT-BamHI: acgtggatccttttttttttttttttttttgat (SEQ ID NO:) ccccgggaattgccatg, - C3. Plasmid DNA Constructs
- The PCR amplicon was cut with the restriction enzymes, EcoRI +BamHI. The DNA fragment was ligated into the pTRIampl8 vector (Ambion) using the Quick Ligation Kit (New England Biolabs) according to the supplier's instructions and transformed into E. coli DH-5(x by standard methods.
- C4. DNA Sequencing
- To verify the cloning of the PCR amplicon, plasmid DNA was sequenced using M13 forward and M13 reverse primers and analysed on an ABI 377.
- C5. Biotin Labeling of cRNA
- One μg of plasmid containing the HSP78 sequence was linearized with restriction enzyme BamHI (Amersham Pharmacia Biotech, USA) for 2 hours at 37° C. The RNA was labeled with biotin-CTP and biotin-UTP using the Message AMP aRNA kit from Ambion (USA) according to the manufacturer's instructions. Following hybridisation, the slides were stained with Streptavidin Phycoerythrin (Molecular Probes, S-866, USA) according to the GeneChip Expression Analysis Technical Manual (Affymetrix, USA)
- C6. Fluorochrome Labeling of Spike RNA
- In vitro synthesized spike RNA from the HSP78 plasmid construct was labeled with either Cy3-ULS or Cy5-ULS (Amersham Biosciences, USA) according to the manufacturer's instructions, followed by filtration through a ProbeQuant G50 Micro Column and Microcon30 (Millipore, USA). The labeling efficiency was monitored using the Nanodrop spectrophotometer (Nanodrop Technologies, USA)
- D. Microarray Fabrication
- The microarrays were printed on Immobilizer™ MicroArray Slides (Exiqon, Denmark) using the MicroGrid II from Biorobotics (UK) using a 20 pM capture probe solution for each oligonucleotide probe. Four replicas of each capture probe were printed on the slides.
- E. Hybridization with Fluorochrome-Labelled cRNA
- The arrays were hybridized for 16 hours using the following protocol. The labelled RNA samples were hybridized in a hybridization solution (20 μL final volume) containing 3×SSC (final concentration), 25 mM HEPES, pH 7.0 (final concentration), 1.25 μg/μL yeast tRNA, 0.3% SDS. The labeled RNA target sample was filtered in a Millipore 0.22 micron spin column according to the manufacturer's instructions (Millipore, USA), and the probe was denatured by incubating the reaction at 100° C. for 2 min. The sample was cooled at 20-25° C. for 5 min. by spinning at max speed in a microcentrifuge. A LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on Immobilizerm MicroArray Slide and the hybridization mixture was applied to the array from the side. An aliquot of 30 μL of 3×SSC was added to both ends of the hybridization chamber, and the Immobilizer™ MicroArray Slide was placed in the hybridization chamber. The chamber was sealed watertight and incubated at 45° C., 55° C. or 65° C. for 16-18 hours submerged in a water bath. After hybridisation, the slide was removed carefully from the hybridization chamber and washed using the following protocol. The Lifterslip coverslip was washed off in 6×SSC, pH 7.0 containing 0.1% Tween20 at 50° C. for 15 min., followed by washing of the microarrays in 0.4×SSC, pH 7.0 at 50° C. for 30 min. Finally the slides were washed for 5 seconds in 0.05×SSC, pH 7.0. The slides were then dried by centrifugation in a swinging bucket rotor at approximately 200 G for 2 min.
- F. Data Analysis.
- Following washing and drying, the slides were scanned using a ScanArray 4000XL scanner (Perkin-Elmer Life Sciences, USA), and the array data were processed using the GenePix™ Pro 4.0 software package (Axon, USA).
- Results
- G1. Duplex Melting Temperatures
- The Tm data clearly shows that LNA-substituted oligonucleotide capture probes have a significantly increased average duplex melting temperature compared to the corresponding DNA probes. Furthermore, the difference in melting temperature between the perfectly matched (PM) and single mismatched (MM) probes, designated as A™, is significantly higher than the corresponding ΔTm for DNA probes (Table 12).
TABLE 12 The average difference in melting temperature between the perfectly matched (PM) and single mismatched (MM) probes in different capture probe designs. The observed difference between the DNA and LNA substituted probes is statistically significant as revealed by a t-Test; Two-Sample Assuming Unequal Variances. Average Max Min Δ Tm St. dev. T-Test MM-DNA 60.4 68.5 54.3 MM-LNA T 67.6 74 58.1 MM-LNA TC 72.0 79.3 61.6 PM-DNA 66.3 72.8 61 5.9 1.52 PM-LNA T 74.2 80.8 66.3 6.6 1.32 0.047 PM-LNA TC 80.0 86.8 71.4 8.0 2.65 0.001/0.017 - G2. Microarray Hybridization Results
- Both LNA_T and
LNA —3 substituted 25-mer probes are capable of providing highly accurate measurements for fold-of-changes in gene expression levels, as depicted in FIG. 37. The DNA capture probes did not provide any hybridisation signals under the given microarray hybridisation conditions (FIG. 37). FIG. 37 shows the expected (black bars) and observed (white bars) fold-of-change in the expression levels of the Cy3-ULS labelled HSP78 spike RNA as measured by on-chip capture using different oligonucleotide capture probes. In the hybridization experiment, 1 ng HSP78 in vitro spike RNA or 200 pg HSP78 in vitro spike RNA was used, respectively. Thus, the fold change of the HSP78 RNA in the two hybridizations in the comparison is 5-fold. Fourteen additional synthetic in vitro mRNA spike controls were included in the hybridisation solution as a semi-complex background RNA mixture. Seven of these spikes were used as normalization controls, the other seven were used as negative controls. Hybridization temperature was 65° C. for 16 hours, and post-hybridization washes as described above. Under these conditions the DNA capture probes did not produce hybridization signals. FIG. 38 shows measured intensity levels by on-chip capture using three different 25-mer oligonucleotide capture probe designs. One (1) ng biotin-labeled HSP78 target was used in the hybridization experiments, followed by staining with Streptavidin Phycoerythrin. The LNA_T and LNA_TC substituted 25-mer capture probes show a significantly enhanced on-chip capture of the HSP78 RNA target, compared to the DNA 25-mer control probes under four different hybridization stringency conditions.TABLE 13 Design of the yeast HSP78 capture probes. YDR258C denotes the ORF name of the S. cerevisiae HSP78 gene. The numbers refer to the nucleotide position from the 3′-end of the HSP78 mRNA sequence. PM = perfectly matched probe, MM = single mismatch probe, LNA substitutions are depicted by capital letters, mC denotes LNA methyl-C (SEQ ID NOs:) Oligo name Sequence Oligo name Sequence YDR258C_PM_043 tttggtagcacgacaagcttagtat YDR258C_PM_043T TTTggTagcacgacaagcTTagTat YDR258C_PM_078 cactctaacagtttcgccgtttcta YDR258C_PM_078T cacTcTaacagtTtcgccgTTTcTa YDR258C_PM_124 gttgccatggagttcaaaatctgtc YDR258C_PM_1241 gTTgccaTggagTtcaaaaTcTgTc YDR258C_PM_164 tcaatggccttgcaccatataattg YDR258C_PM_1641 TcaaTggccTTgcaccaTaTaaTTg YDR258C_PM_201 tcagttagccaatccttcgcttcat YDR258C_PM_2011 TcagTTagccaaTccTTcgcTTcat YDR258C_PM_249 tttttcggccaaacgatcttgaatt YDR258C_PM_2491 TtTTTcggccaaacgaTcTTgaaTt YDR258C_PM_295 attgacctcaaaactttcttggata YDR258C_PM_295T aTTgaccTcaaaacTTTcTTggaTa YDR258C_PM_356 gatgaactcaggtggataggatctt YDR258C_PM_3561 gaTgaacTcaggTggaTaggaTcTt YDR258C_PM_424 accatcatcacccaactttgtgtcg YDR258C_PM_4241 accaTcaTcacccaacTTTgTgTcg YDR258C_PM_433 cccaactttgtgtcgtttaataaaa YDR258C_PM_4331 cccaacTTTgTgTcgTTTaaTaaaa YDR258C_PM_486 caatgatcgtgttacggaaatcaac YDR258C_PM_4861 caaTgaTcgTgtTacggaaaTcaac YDR258C_PM_515 tggcctagggaatcggtcagcttac YDR258C_PM_5151 TggccTagggaaTcggTcagcTTac YDR258C_PM_566 ttggaaacatcggggtgcgcttttt YDR258C_PM_5661 TTggaaacaTcggggTgcgcTTTTt YDR258C_PM_569 gaaacatcggggtgcgctttttcaa YDR258C_PM_5691 gaaacaTcggggTgcgcTTTTTcaa YDR258C_PM_604 aaaacgacagcataaggctttcttc YDR258C_PM_6041 aaaacgacagcaTaaggcTTTcTTc YDR258C_PM_631 gacagcctcagttaattggccacca YDR258C_PM_6311 gacagccTcagtTaaTTggccacca YDR258C_PM_686 ccgattaaacgagagacagtatgct YDR258C_PM_6861 ccgaTTaaacgagagacagTaTgct YDR258C_PM_757 atcaaataggaattcagctaaagcc YDR258C_PM_7571 aTcaaaTaggaaTTcagcTaaagcc YDR258C_PM_813 gacctaagaacataaagctggcaat YDR258C_PM_8131 gaccTaagaacaTaaagcTggcaat YDR258C_PM_823 ataaagctggcaataggtctctttt YDR258C_PM_823T aTaaagcTggcaaTaggTcTcTTTt YDR258C_PM_870 agacgtacagcatcagaaatagcag YDR258C_PM_8701 agacgTacagcaTcagaaaTagcag YDR258C_PM_888 aaatagcagcaatggcctcgtcttg YDR258C_PM_8881 aaaTagcagcaaTggccTcgTcTTg YDR258C_PM_890 atagcagcaatggcctcgtcttggc YDR258C_PM_8901 aTagcagcaaTggccTcgTcTTggc YDR258C_PM_896 gcaatggcctcgtcttggccaacga YDR258C_PM_896T gcaaTggccTcgTcTTggccaacga YDR258C_PM_043TC TtTggTagmCamCgamCaagmCtTagTat YDR258C_PM_LNA3_043 TttGgtAgcAcgAcaAgcTtaGtat YDR258C_PM_078TC mCamCtmCtaamCagtTtcgmCcgTtTmCta YDR258C_PM_LNA3_078 mCacTctAacAgtTtcGccGttTcta YDR258C_PM_124TC gTtgmCmCaTggagTtmCaaaaTmCtgTc YDR258C_PM_LNA3_124 GttGccAtgGagTtcAaaAtcTgtc YDR258C_PM_164TC TmCaaTggmCcTTgmCacmCaTaTaaTTg YDR258C_PM_LNA3_164 TcaAtgGccTtgmCacmCatAtaAttg YDR258C_PM_201TC TmCagTtagcmCaaTcmCtTmCgmCttmCat YDR258C_PM_LNA3_424 AccAtcAtcAccmCaamCttTgtGtcg YDR258C_PM_249TC TtTtTmCggmCmCaaamCgatmCtTgaaTt YDR258C_PM_LNA3_486 mCaaTgaTcgTgtTacGgaAatmCaac YDR258C_PM_295TC aTTgamCmCTmCaaaamCTTtmGTTggaTa YDR258C_PM_LNA3_515 TggmCctAggGaaTcgGtcAgcTtac YDR258C_PM_356TC gaTgaamCTmCaggTggaTaggaTmCTt YDR258C_PM_LNA3_566 TtgGaaAcaTcgGggTgcGctTttt YDR258C_PM_424TC amCcaTcaTcacmCmCaaccTtgTgtmCg YDR258C_PM_LNA3_569 GaaAcaTcgGggTgcGctTttTcaa YDR258C_PM_433TC mCmCmCaacTTtgTgTcgTTTaaTaaaa YDR258C_PM_LNA3_604 AaaAcgAcaGcaTaaGgcTttmGttc YDR258C_PM_486TC mCaaTgaTmCgTgtTamCggaaaTmCaac YDR258C_PM_LNA3_757 AtcAaaTagGaaTtcAgcTaaAgcc YDR258C_PM_515TC TggmCcTagggaaTmCggTcagmCtTacTC YDR258C_PM_LNA3_813 GacmCtaAgaAcaTaaAgcTggmCaat YDR258G_PM_566TC TTggaaamCatmCggggTgmCgctTtTt YDR258C_PM_LNA3_823 AtaAagmCtgGcaAtaGgtmCtcTttt YDR258C_PM_569TC gaaamCaTmCggggTgmCgctttTtmCaa YDR258C_PM_LNA3_870 AgamCgtAcaGcaTcaGaaAtaGcag YDR258C_PM_604TC aaaamCgamCagmCaTaaggmCTtTmCtTc YDR258C_PM_LNA3_888 AaaTagmCagmCaaTggmCctmCgtmCttg YDR258C_PM_631TC gamCagmCcTcagtTaaTTggcmCacmCa YDR258C_PM_LNA3_890 AtaGcaGcaAtgGccTcgTctTggc YDR258C_PM_686TC mCmCgaTTaaamCgagagamCagTaTgmCt YDR258C_PM_LNA3_896 GcaAtgGccTcgTctTggmCcaAcga YDR258C_PM_757TC aTmCaaaTaggaaTtmCagmCTaaagmCc YDR258C_PM_LNA3_631 GacAgcmCtcAgtTaaTtgGccAcca YDR258C_PM_813TC gamCmCTaagaamCaTaaagmCTggmCaat YDR258C_PM_LNA3_686 mCcgAttAaamCgaGagAcaGtaTgct YDR258C_PM_823TC aTaaagmCTggmCaaTaggTmCTcTTTt YDR258C_PM_LNA3_356 GatGaamCtcAggTggAtaGgaTctt YDR258C_PM_870TC agamCgTamCagmCaTmCagaaaTagmCag YDR258C_PM_LNA3_201 TcaGttAgcmCaaTccTtcGctTcat YDR258G_PM_888TC aaaTagmCagmCaaTggmCcTmCgTctTg YDR258C_PM_LNA3_249 TttTtcGgcmCaaAcgAtcTtgAatt YDR258G_PM_890TC aTagmCagcaaTggmCcTcgtmCtTggc YDR258C_PM_LNA3_295 AttGacmCtcAaaActTtcTtgGata YDR258C_PM_896TC gcaatggcctmCgTmCttggccaacga YDR258C_PM_LNA3_433 mCccAacTttGtgTcgTttAatAaaa YDR258C_MM_043 tttggtagcacgtcaagcttagtat YDR258C_MM_043T TTTggTagcacgtcaagcTTagTat YDR258C_MM_078 cactctaacagtatcgccgtttcta YDR258C_MM_078T cacTcTaacagtatcgccgTTTcTa YDR258C_MM_124 gttgccatggagatcaaaatctgtc YDR258C_MM_124T gTTgccaTggagatcaaaaTcTgTc YDR258C_MM_164 tcaatggccttggaccatataattg YDR258C_MM_164T TcaaTggccTTggaccaTaTaaTTg YDR258C_MM_201 tcagttagccaaaccttcgcttcat YDR258C_MM_201T TcagTTagccaaaccTTcgcTTcat YDR258C_MM_249 tttttcggccaatcgatcttgaatt YDR258C_MM_249T TtTTTcggccaatcgaTcTTgaaTt YDR258C_MM_295 attgacctcaaatctttcttggata YDR258C_MM_295T aTTgaccTcaaatcTTTcTTggaTa YDR258C_MM_356 gatgaactcaggaggataggatctt YDR258C_MM_356T gaTgaacTcaggaggaTaggaTcTt YDR258C_MM_424 accatcatcaccgaactttgtgtcg YDR258C_MM_424T accaTcaTcaccgaacTTTgTgTcg YDR258C_MM_433 cccaactttgtgacgtttaataaaa YDR258C_MM_433T cccaacTTTgTgacgTTTaaTaaaa YDR258C_MM_486 caatgatcgtgtaacggaaatcaac YDR258C_MM_486T caaTgaTcgTgtaacggaaaTcaac YDR258C_MM_515 tggcctagggaaacggtcagcttac YDR258C_MM_51ST TggccTagggaaacggTcagcTTac YDR258C_MM_566 ttggaaacatcgcggtgcgcttttt YDR258C_MM_566T TTggaaacaTcgcggTgcgcTTTTt YDR258C_MM_569 gaaacatcggggagcgctttttcaa YDR258C_MM_569T gaaacaTcggggagcgcTTTTTcaa YDR258C_MM_604 aaaacgacagcaaaaggctttcttc YDR258C_MM_604T aaaacgacagcaaaaggcTTTcTTc YDR258C_MM_631 gacagcctcagtaaattggccacca YDR258C_MM_631T gacagccTcagtaaaTTggccacca YDR258C_MM_686 ccgattaaacgacagacagtatgct YDR258C_MM_686T ccgaTTaaacgacagacagTaTgct YDR258C_MM_757 atcaaataggaaatcagctaaagcc YDR258C_MM_757T aTcaaaTaggaaatcagcTaaagcc YDR258C_MM_813 gacctaagaacaaaaagctggcaat YDR258C_MM_813T gaccTaagaacaaaaagcTggcaat YDR258C_MM_823 ataaagctggcattaggtctctttt YDR258C_MM_823T aTaaagcTggcaaaaggTcTcTTTt YDR258C_MM_870 agacgtacagcaacagaaatagcag YDR258C_MM_870T agacgTacagcaacagaaaTagcag YDR258C_MM_888 aaatagcagcaaaggcctcgtcttg YDR258C_MM_888T aaaTagcagcaaaggccTcgTcTTg YDR258C_MM_890 atagcagcaatgccctcgtcttggc YDR258C_MM_890T aTagcagcaaTgcccTcgTcTTggc YDR258C MM 896 gcaatggcctcgacttggccaacga YDR258C_MM_896T gcaaTggccTcgacTTggccaacga YDR258C_MM_043TC TtTggTagmCamCgtcaagmCtTagTat YDR258C_MM_LNA3_043 TttGgtAgcAcgTcaAgcTtaGtat YDR258C_MM_078TC mCamCtmCtaamCagtatcgmCcgTtTmCTa YDR258C_MM_LNA3_078 mCacTctAacAgtAtcGccGttTcta YDR258C_MM_124TC gTtgmCmCaTggagatmCaaaaTmCtgTc YDR258C_MM_LNA3_124 GttGccAtgGagAtcAaaAtcTgtc YDR258C_MM_164TC TmCaaTggmCcTTggacmCaTaTaaTTg YDR258C_MM_LNA3_164 TcaAtgGccTtgGacmCatAtaAttg YDR258C_MM_201TC tmCagTtagcmCaaacctTcgmCttmCat YDR258C_MM_LNA3_201 TcaGttAgcmCaaAccTtcGctTcat YDR258C_MM_249TC TtTtTmCggmCmCaatcgatmCtTgaaTt YDR258C_MM_LNA3_249 TttTtcGgcmCaaTcgAtcTtgAatt YDR258C_MM_295TC aTTgamCmCTmCaaatcTTtmCTTggaTa YDR258C_MM_LNA3_295 AttGacmCtcAaaTctTtcTtgGata YDR258G_MM_356TC gaTgaamCTmCaggaggaTaggaTmCTt YDR258C_MM_LNA3_356 GatGaamCtcAggAggAtaGgaTctt YDR258C_MM_424TC amCcaTcaTcacccaaccTtgTgtmGg YDR258C_MM_LNA3_424 AccAtcAtcAccGaamCttTgtGtcg YDR258C_MM_433TC mCmCmCaacTTtgTgacgTTTaaTaaaa YDR258C_MM_LNA3_433 mCccAacTttGtgAcgTttAatAaaa YDR258C_MM_486TC mCaaTgaTmCgTgttamCggaaaTmCaac YDR258C_MM_LNA3_486 mCaaTgaTcgTgtAacGgaAatmCaac YDR258C_MM_515TC TggmCcTagggaaacggTcagmCtTac YDR258C_MM_LNA3_515 TggmCctAggGaaAcgGtcAgcTtac YDR258C_MM_566TC TTggaaamCatmCgcggTgmCgctTtTt YDR258C_MM_LNA3_566 TtgGaaAcaTcgmCggTgcGctTttt YDR258C_MM_569TC gaaamCaTmCggggagmCgctuTtmCaa YDR258C_MM_LNA3_569 GaaAcaTcgGggAgcGctTttTcaa YDR258C_MM_604TC aaaamCgamCagmCaaaaggmCTtTmCtTc YDR258C_MM_LNA3_604 AaaAcgAcaGcaAaaGgcTttmCttc YDR258C_MM_631TC gamCagmCcTcagtaaaTTggcmCacmCa YDR258C_MM_LNA3_631 GacAgcmCtcAgtAaaTtgGccAcca YDR258C_MM_686TC mCmCgaTTaaamCgacagamCagTaTgmCt YDR258C_MM_LNA3_686 mCcgAttAaamCgamCagAcaGtaTgct YDR258C_MM_757TC aTmCaaaTaggaaatmCagmCTaaagmCc YDR258C_MM_LNA3_757 AtcAaaTagGaaAtcAgcTaaAgcc YDR258C_MM_813TC gamCmCTaagaamCaaaaagmCTggmCaat YDR258C_MM_LNA3_813 GacmCtaAgaAcaAaaAgcTggmCaat YDR258C_MM_823TC aTaaagmCTggmCattaggTmCTcTTTt YDR258C_MM_LNA3_823 AtaAagmCtgGcaTtaGgtmCtcTttt YDR258C_MM_870TC agamCgTamCagmCaacagaaaTagmCag YDR258C_MM_LNA3_870 AgamCgtAcaGcaAcaGaaAtaGcag YDR258C_MM_888TC aaaTagmCagmGaaaggmCcTmCgTctTg YDR258C_MM_LNA3_888 AaaTagmCagmCaaAggmCctmCgtmCttg YDR258C_MM_890TC aTagmCagcaaTgcccTcgtmCtTggc YDR258C_MM_LNA3_890 AtaGcaGcaAtgmCccTcgTctTggc YDR258C_MM_896TC gmCaaTggcctmCgactggccaamCga YDR258C_MM LNA3_896 GcaAtgGccTcgActTggmGcaAcga -
TABLE 14!Duplex melting temperatures (Tm) for the 144 different 25-mer oligonucleotide? ? !capture probes. The design column denotes the sequence design of the probe.? !PM = perfectly matched probe, MM = single mismatch probe, LNA substitutions are? !depicted by capital letters, mC denotes LNA methyl-C (SEQ ID NOs:)? !? ? ? ? Average? ? !Probe? ? ? ? duplex melting? !number? Oligonucleotide target name? Complementary target sequence? Design? temp. (° C.) 12696 YDR258C_Tm_predic_043 tttggtagcacgtcaagcttagtat MM-DNA 59.2 12699 YDR258C_Tm_predic_078 cactctaacagtatcgccgtttcta MM-DNA 61.1 12694 YDR258C_Tm_predic_124 gttgccatggagatcaaaatctgtc MM-DNA 60 12700 YDR258C_Tm_predic_164 tcaatggccttggaccatataattg MM-DNA 59.5 12693 YDR258C_Tm_predic_201 tcagttagccaaaccttcgcttcat MM-DNA 61.4 12698 YDR258C_Tm_predic_249 tttttcggccaatcgatcttgaatt MM-DNA 58.8 12702 YDR258C_Tm_predic_295 attgacctcaaatctttcttggata MM-DNA 54.5 12692 YDR258C_Tm_predic_356 gatgaactcaggaggataggatctt MM-DNA 58.2 12680 YDR258C_Tm_predic_424 accatcatcaccgaactttgtgtcg MM-DNA 63.6 12703 YDR258C_Tm_predic_433 cccaactttgtgacgtttaataaaa MM-DNA 56.2 12681 YDR258C_Tm_predic_486 caatgatcgtgtaacggaaatcaac MM-DNA 59.3 12682 YDR258C_Tm_predic_515 tggcctagggaaacggtcagcttac MM-DNA 65.2 12683 YDR258C_Tm_predic_566 ttggaaacatcgcggtgcgcttttt MM-DNA 61.5 12684 YDR258C_Tm_predic_569 gaaacatcggggagcgctttttcaa MM-DNA 63.8 12701 YDR258C_Tm_predic_604 aaaacgacagcaaaaggctttcttc MM-DNA 58.8 12685 YDR258C_Tm_predic_631 gacagcctcagtaaattggccacca MM-DNA 64.8 12686 YDR258C_Tm_predic_686 ccgattaaacgacagacagtatgct MM-DNA 57.1 12687 YDR258C_Tm_predic_757 atcaaataggaaatcagctaaagcc MM-DNA 54.3 12688 YDR258C_Tm_predic_813 gacctaagaacaaaaagctggcaat MM-DNA 58.2 12697 YDR258C_Tm_predic_823 ataaagctggcattaggtctctttt MM-DNA 58.4 12695 YDR258G_Tm_predic_870 agacgtacagcaacagaaatagcag MM-DNA 61.2 12689 YDR258C_Tm_predic_888 aaatagcagcaaaggcctcgtcttg MM-DNA 64 12690 YDR258C_Tm_predic_890 atagcagcaatgccctcgtcttggc MM-DNA 62.9 12691 YDR258C_Tm_predic 896 gcaatggcctcgacrtggccaacga MM-DNA 68.5 12720 YDR258C_Tm_predic_043T TTTggTagcacgtcaagcTTagTat MM-T 68.6 12723 YDR258C_Tm_predic_078T cacTcTaacagtatcgccgTTTcTa MM-T 69.7 12718 YDR258C_Tm_predic_124T gTTgccaTggagatcaaaaTcTgTc MM-T 69.2 12724 YDR258C_Tm_predic_164T TcaaTggccTTggaccaTaTaaTTg MM-T 69.7 12717 YDR258C_Tm_predic_201T TcagTTagccaaaccTTcgcTTcat MM-T 69.4 12722 YDR258C_Tm_predic_249T TtTTTcggccaatcgaTcTTgaaTt MM-T 65.9 12726 YDR258C_Tm_predic_295T aTTgaccTcaaatcTTTcTTggaTa MM-T 65.1 12716 YDR258C_Tm_predic_356T gaTgaacTcaggaggaTaggaTcTt MM-T 64.9 12704 YDR258C_Tm_predic_424T accaTcaTcaccgaacTTrgTgTcg MM-T 74 12727 YDR258C_Tm_predic_433T cccaacTTTgTgacgTTTaaTaaaa MM-T 66.5 12705 YDR258C_Tm_predic_486T caaTgaTcgTgtaacggaaaTcaac MM-T 65.2 12706 YDR258C_Tm_predic_515T TggccTagggaaacggTcagcTTac MM-T 71.6 12707 YDR258C_Tm_predic_566T TTggaaacaTcgcggTgcgcTTTTt MM-T 68.6 12708 YDR258C_Tm_predic_569T gaaacaTcggggagcgcTTTTTcaa MM-T 69.9 12725 YDR258C_Tm_predic_604T aaaacgacagcaaaaggcTTTcTTc MM-T 65.9 12709 YDR258C_Tm_predic 631T gacagccTcagtaaaTTggccacca MM-T 68.8 12710 YDR258C_Tm_predic_686T ccgaTTaaacgacagacagTaTgct MM-T 63.5 12711 YDR258C_Tm_predic_757T aTcaaaTaggaaatcagcTaaagcc MM-T 58.1 12712 YDR258C_Tm_predic_813T gaccTaagaacaaaaagcTggcaat MM-T 61.1 12721 YDR258C_Tm_predic_823T aTaaagcTggcaaaaggTcTcTTTt MM-T 67.2 13 + 14 12719 YDR258C_Tm_predic_870T agacgTacagcaacagaaaTagcag MM-T 65 12713 YDR258C_Tm_predic_888T aaaTagcagcaaaggccTcgTcTTg MM-T 70.4 12714 YDR258C_Tm_predic_890T aTagcagcaaTgcccTcgTcTTggc MM-T 71.3 12715 YDR258C_Tm_predic_896T gcaaTggccTcgacTTggccaacga MM-T 73.7 12744 YDR258C_Tm_predic_043TC TtTggTagmCamCgtcaagmCtTagTat MM-TC 73.3 12747 YDR258C_Tm_predic_078TC mCamCtmCtaamCagtatcgmCcgTtTmCTa MM-TC 75.2 12742 YDR258C_Tm_predic_124TC gTtgmCmCaTggagatmCaaaaTmCtgTc MM-TC 61.6 12748 YDR258C_Tm_predic_164TC TmCaaTggmCcTTggacmCaTaTaaTTg MM-TC 74.8 12741 YDR258C_Tm_predic_201TC tmCagTtagcmCaaacctTcgmCttmCat MM-TC 70.6 12746 YDR258C_Tm_predic_249TC TtTtTmCggmCmCaatcgatmCtTgaaTt MM-TC 71 12750 YDR258C_Tm_predic_295TC aTTgamCmCTmCaaatcTTtmCTTggaTa MM-TC 72.2 12740 YDR258C_Tm_predic_356TC gaTgaamCTmCaggaggaTaggaTmCTt MM-TC 70.4 12728 YDR258C_Tm_predic_424TC amCcaTcaTcacccaaccTtgTgtmCg MM-TC 70.2 13 + 16 12751 YDR258C_Tm_predic_433TC mCmCmCaacTTtgTgacgTTTaaTaaaa MM-TC 67.6 12730 YDR258C_Tm_predic_515TC TggmCcTagggaaacggTcagmCtTac MM-TC 75.5 12731 YDR258C_Tm_predic_566TC TTggaaamCatmCgcggTgmCgctTtTt MM-TC 72 12732 YDR258C_Tm_predic_569TC gaaamCaTmCggggagmCgctttTtmCaa MM-TC 74.8 12749 YDR258C_Tm_predic_604TC aaaamCgamCagmCaaaaggmCTtTmCtTc MM-TC 72 12733 YDR258C_Tm_predic_631TC gamCagmCcTcagtaaaTTggcmCacmCa MM-TC 77.4 12734 YDR258C_Tm_predic_686TC mCmCgaTTaaamCgacagamCagTaTgmCt MM-TC 70.2 12735 YDR258C_Tm_predic_757TC aTmCaaaTaggaaatmCagmCTaaagmCc MM-TC 64.6 12736 YDR258C_Tm_predic_813TC gamCmCTaagaamCaaaaagmCTggmCaat MM-TC 71.4 12745 YDR258C_Tm_predic_823TC aTaaagmCTggmCattaggTmCTcTTTt MM-TC 74 12743 YDR258C_Tm_predic_870TC agamCgTamCagmCaacagaaaTagmCag MM-TC 73.6 12737 YDR258C_Tm_predic_888TC aaaTagmCagmCaaaggmCcTmCgTctTg MM-TC 79.3 12738 YDR258C_Tm_predic_890TC aTagmCagcaaTgcccTcgtmCtTggc MM-TC 71.5 12739 YDR258C_Tm_predic_896TC gcaatggcctmCgamCttggccaacga MM-TC 73.1 12768 YDR258C_Tm_predic_043_PM tttggtagcacgacaagcttagtat PM-DNA 65.7 12771 YDR258C_Tm_predic_078_PM cactctaacagtttcgccgtttcta PM-DNA 66.3 12766 YDR258C_Tm_predic_124 PM gttgccatggagttcaaaatctgtc PM-DNA 65.8 12772 YDR258C_Tm_predic_164_PM tcaatggccttgcaccatataattg PM-DNA 64 12765 YDR258C_Tm_predic_201_PM tcagttagccaatccttcgcttcat PM-DNA 66.1 12770 YDR258C_Tm_predic_249_PM tttttcggccaaacgatcttgaatt PM-DNA 65 12774 YDR258C_Tm_predic_295_PM attgacctcaaaactttcttggata PM-DNA 61.8 12764 YDR258C_Tm_predic_356_PM gatgaactcaggtggataggatctt PM-DNA 64 12752 YDR258C_Tm_predic_424_PM accatcatcacccaactttgtgtcg PM-DNA 67.5 12775 YDR258C_Tm_predic_433_PM cccaactttgtgtcgtttaataaaa PM-DNA 61 12753 YDR258C_Tm_predic_486_PM caatgatcgtgttacggaaatcaac PM-DNA 64.2 12754 YDR258C_Tm_predic_515 PM tggcctagggaatcggtcagcttac PM-DNA 70.1 12755 YDR258C_Tm_predic_566_PM ttggaaacatcggggtgcgcttttt PM-DNA 70.8 12756 YDR258C_Tm_predic_569_PM gaaacatcggggtgcgctttttcaa PM-DNA 68.7 12773 YDR258C_Tm_predic_604_PM aaaacgacagcataaggctttcttc PM-DNA 64.5 12757 YDR258C_Tm_predic_631_PM gacagcctcagttaattggccacca PM-DNA 70.2 12758 YDR258C_Tm_predic_686_PM ccgattaaacgagagacagtatgct PM-DNA 65.4 12759 YDR258C_Tm_predic_757_PM atcaaataggaattcagctaaagcc PM-DNA 61.5 12760 YDR258C_Tm_predic_813_PM gacctaagaacataaagctggcaat PM-DNA 63.5 12769 YDR258C_Tm_predic_823_PM ataaagctggcaataggtctctttt PM-DNA 63.6 12767 YDR258C_Tm_predic_870 PM agacgtacagcatcagaaatagcag PM-DNA 66.6 12761 YDR258C_Tm_predic_888_PM aaatagcagcaatggcctcgtcttg PM-DNA 69.2 12762 YDR258C_Tm_predic_890_PM atagcagcaatggcctcgtcttggc PM-DNA 72.7 12763 YDR258C_Tm_predic_896_PM gcaatggcctcgtcttggccaacga PM-DNA 72.8 12792 YDR258C_Tm_predic_043T_PM TTTggTagcacgacaagcTTagTat PM-T 74.2 12795 YDR258C_Tm_predic_078T_PM cacTcTaacagtTtcgccgTTTcTa PM-T 75.5 12790 YDR258C_Tm_predic_124T_PM gTTgccaTggagTtcaaaaTcTgTc PM-T 74.7 12796 YDR258C_Tm_predic_164T_PM TcaaTggccTTgcaccaTaTaaTTg PM-T 74 12789 YDR258C_Tm_predic_201T_PM TcagTTagccaaTccTTcgcTTcat PM-T 75.7 12794 YDR258C_Tm_predic_249T_PM TtTTTcggccaaacgaTcTTgaaTt PM-T 71 12798 YDR258C_Tm_predic_295T_PM TtTTgaccTcaaaacTTTcTTggaTa PM-T 70.2 12788 YDR258C_Tm_predic_356T_PM gaTgaacTcaggTggaTaggaTcTt PM-T 71 12776 YDR258C_Tm_predic_424T_PM accaTcaTcacccaacTTTgTgTcg PM-T 79.2 12799 YDR258C_Tm_predic_433T_PM cccaacTrTgTgTcgTTTaaTaaaa PM-T 75.1 12777 YDR258C_Tm_predic_486T_PM caaTgaTcgTgtTacggaaaTcaac PM-T 72.2 12778 YDR258C_Tm_predic_515T_PM TggccTagggaaTcggTcagcTTac PM-T 76.9 12779 YDR258C_Tm_predic_566T_PM TTggaaacaTcggggTgcgcTTTTt PM-T 76.7 12780 YDR258C_Tm_predic_569T_PM gaaacaTcggggTgcgcTTTTTcaa PM-T 78 12797 YDR258C_Tm_predic_604T_PM aaaacgacagcaTaaggcTTTcTTc PM-T 71.7 12781 YDR258C_Tm_predic_631T_PM gacagccTcagtTaaTTggccacca PM-T 75.4 12782 YDR258C_Tm_predic_686T_PM ccgaTTaaacgagagacagTaTgct PM-T 72.2 12783 YDR258C_Tm_predic_757T_PM aTcaaaTaggaaTTcagcTaaagcc PM-T 66.3 12784 YDR258C_Tm_predic_813T_PM gaccTaagaacaTaaagcTggcaat PM-T 67.5 12793 YDR258C_Tm_predic_823T_PM aTaaagcTggcaaTaggTcTcTTTt PM-T 74.3 12791 YDR258C_Tm_predic_870T_PM agacgTacagcaTcagaaaTagcag PM-T 71.4 12785 YDR258C_Tm_predic_888T_PM aaaTagcagcaaTggccTcgTcTTg PM-T 77.2 12786 YDR258C_Tm_predic_890T_PM aTagcagcaaTggccTcgTcTTggc PM-T 80.1 12787 YDR258C_Tm_predic_896T_PM gcaaTggccTcgTcTTggccaacga PM-T 80.8 12816 YDR258C_Tm_predic_043TC_PM TtTggTagmCamCgamCaagmCtTagTat PM-TC 79 12819 YDR258C_Tm_predic_078TC_PM mCamCtmCtaamCagtTtcgmCcgTtTmCTa PM-TC 81.9 12814 YDR258C_Tm_predic_T24TC_PM gTtgmCmCaTggagTtmCaaaaTmCtgTc PM-TC 78.3 12820 YDR258C_Tm_predic_164TC_PM TmCaaTggrnCcTTgmCacmCaTaTaaTTg PM-TC 83.5 12813 YDR258C_Tm_predic_201TC_PM TmCagTtagcmCaaTcmCtTmCgmCttmCat PM-TC 81.8 12818 YDR258C_Tm_predic_249TC_PM TtTtTmCggmCmCaaamCgatmCtTgaaTt PM-TC 77.8 12822 YDR258C_Tm_predic_295TC_PM aTTgamCmCTmCaaaamCTTtmCTTggaTa PM-TC 75.6 12812 YDR258C_Tm_predic_356TC_PM gaTgaamCTmCaggTggaTaggaTmCTt PM-TC 77.3 12800 YDR258C_Tm_predic_424TC_PM amCcaTcaTcacmCmCaaccTtgTgtmCg PM-TC 74.2 12823 YDR258C_Tm_predic_433TC_PM mCmCmCaacTTtgTgTcgTTTaaTaaaa PM-TC 74.8 12729 YDR258C_Tm_predic_486TC_PM mCaaTgaTmCgTgttamCggaaaTmCaac PM-TC 75.5 12802 YDR258C_Tm_predic_515TC_PM TggmCcTagggaaTmCggTcagmCtTac PM-TC 83.6 12803 YDR258C_Tm_predic_566TC_PM TTggaaamCatmCggggTgmCgctTtTt PM-TG 80.8 12804 YDR258C_Tm_predic_569TC_PM gaaamCaTmGggggTgmCgctttTtmCaa PM-TG 83.2 12821 YDR258C_Tm_predic_604TG PM aaaamCgamCagmCaTaaggmCTtTmCtTc PM-TC 79 12805 YDR258C_Tm_predic_631TC_PM gamCagmCcTcagtTaarrggcmCacmCa PM-TC 82.5 12806 YDR258C_Tm_predic_686TC_PM mCmCgaTTaaamCgagagamCagTaTgmGt PM-TC 79.4 12807 YDR258C_Tm_predic_757TC_PM aTmCaaaTaggaaTtmCagmCTaaagmCc PM-TC 71.4 12808 YDR258C_Tm_predic_813TC_PM gamCmCTaagaamCaTaaagmCTggmCaat PM-TC 78.9 12817 YDR258C_Tm_predic_823TC_PM aTaaagmCTggmCaaTaggTmCTcTTTt PM-TC 81.2 12815 YDR258C_Tm_predic_870TC_PM agamCgTamCagmCaTmCagaaaTagmGag PM-TC 81.9 12809 YDR258C_Tm_predic_888TC_PM aaaTagmCagmCaaTggmCcTmCgTctTg PM-TC 86.8 12810 YDR258C_Tm_predic_890TC_PM aTagmCagcaaTggmCcTcgtmCtTggc PM-TC 83 12811 YDR258C_Tm_predic_896TC_PM gcaatggcctmCgTmCttggccaacga PM-TC 79.3 - Example 52. Performance Analysis of LNA Substituted Oligonucleotide Capture Probes Designed to Detect Splice Variants in Complex RNA Pools.
- A. Oliconucleotide design for microarrays. The methods for designing exon-specific internal oligonucleotide capture probes has been described in example 2.
- A1 Design of the LNA-Modified Capture Probes
- For the internal LNA-modified oligonucleotide capture probes, every third DNA nucleotide was substituted with an LNA nucleotide. The probes designed to capture the splice junction of the recombinant splice variants were designed with LNA substitutions at every third nucleotide position. All capture probes are shown in Table 15.
TABLE 15 Internal, exon-specific and merged, exon—exon splice junction specific oligonucleotide capture probes used in the example. Capital letters denote LNA nucleotides and mC LNA methyl-cytosine (SEQ ID NOs:) EQ No Oligo Name Sequence 10716 >gene78.01a cctgaaagtagatttgttatttccgaaacgccttctcccgttcttaagtc 10717 >gene78.01b catataccacaaatagtccctcaaaaatcacaagaaaactcacaacactg 10718 >gene78.03a gatttgcagcggtggtaaaaagtatgaaaacgtggtaattaaaaggtctc 10719 >gene78.03b ccaatgaaaactaatcaaaggtaaacgtggatcccatggcaattcccggg 10720 >gene78.m0103 cacaacactgcccagaggttcaatcgataaatatgtgaaggaaatgcctg 10721 >gene78.m01INS3 caacactgcccagaggttcaatcgatccgatgatcctaatgaaggcgccc 10722 >gene78.mINS303 gtccagtatcgtccatcatagtatcgataaatatgtgaaggaaatgcctg 10723 >gene78.INS3 ctccttcttgcattcttcaacttccttcaacacttgagcggagtcggtgc 10724 >gene78.m01INS4 caacactgcccagaggttcaatcgatgtgtgataggatcagtgttcaggg 10725 >gene78.mINS403 gaaggcgaaggagactgctaatatcgataaatatgTgaaggaaatgcctg 10726 >gene78.INS4a gaacgtatgagcatgcgagagacgctgtagttggaaaaacccacgaagcg 10727 >gene78.INS4b gaaaccgctgattatactgcggagaaggtgggtgagtataaagactatac 11345 >gene78.01a_40 aagtagatttgttatttccgaaacgccttctcccgttctt 11346 >gene78.01b_40 accacaaatagtccctcaaaaatcacaagaaaactcacaa 11347 >gene78.03a_40 gcagcggtggtaaaaagtatgaaaacgtggtaattaaaag 11348 >gene78.03b_40 gaaaactaatcaaaggtaaacgtggatcccatggcaattc 11349 >gene78.m0103_40 cactgcccagaggttcaatcgataaatatgtgaaggaaat 11350 >gene78.m01INS3_40 ctgcccagaggttcaatcgatccgatgatcctaatgaagg 11351 >gene78.mINS303_40 gtatcgtccatcatagtatcgataaatatgtgaaggaaat 11352 >gene78.INS3_40 tcttgcattcttcaacttccttcaacacttgagcggagtc 11353 >gene78.m01INS4_40 ctgcccagaggttcaatcgatgtgtgataggatcagtgtt 11354 >gene78.mINS403_40 cgaaggagactgctaatatcgataaatatgtgaaggaaat 11355 >gene78.INS4a_40 tatgagcatgcgagagacgctgtagttggaaaaacccacg 11356 >gene78.INS4b_40 cgctgattatactgcggagaaggtgggtgagtataaagac 11357 >gene78.01a_50_LNA3 mCctGaaAgtAgaTttGttAttTccGaaAcgmCctTctmCccGttmCttAagTc 11358 >gene78.01b_50_LNA3 mCatAtamCcamCaaAtaGtcmCctmCaaAaaTcamCaaGaaAacTcamCaamCacTg 11359 >gene78.03a_50_LNA3 GatTtgmCagmCggTggTaaAaaGtaTgaAaamCgtGgtAatTaaAagGtcTc 11360 >gene78.03b_50_LNA3 mCcaAtgAaaActAatmCaaAggTaaAcgTggAtcmCcaTggmCaaTtcmCcgGg 11361 >gene78.m0103_50_LNA3 mCacAacActGccmCagAggTtcAatmCgaTaaAtaTgtGaaGgaAatGccTg 11362 >gene78.m01INS3_50_LNA3 mCaamCacTgcmCcaGagGttmCaaTcgAtcmCgaTgaTccTaaTgaAggmCgcmCc 11363 >gene78.mINS303_50_LNA3 GtcmCagTatmCgtmCcaTcaTagTatmCgaTaaAtaTgtGaaGgaAatGccTg 11364 >gene78.INS3_50_LNA3 mCtcmCttmCttGcaTtcTtcAacTtcmCttmCaamCacTtgAgcGgaGtcGgtGc 11365 >gene78.m01INS4_50_LNA3 mCaamCacTgcmCcaGagGttmCaaTcgAtgTgtGatAggAtcAgtGttmCagGg 11366 >gene78.mINS403_50_LNA3 GaaGgcGaaGgaGacTgcTaaTatmCgaTaaAtaTgtGaaGgaAatGccTg 11367 >gene78.INS4a_50_LNA3 GaamCgtAtgAgcAtgmCgaGagAcgmCtgTagTtgGaaAaamCccAcgAagmCg 11368 >gene78.INS4b_50_LNA3 GaaAccGctGatTatActGcgGagAagGtgGgtGagTatAaaGacTatAc 11369 >gene78.01a_40_LNA3 aAgtAgaTttGttAttTccGaaAcgmCctTctmCccGttmCtt 11370 >gene78.01b_40_LNA3 amCcamCaaAtaGtcmCctmCaaAaaTcamCaaGaaAacTcamCaa 11371 >gene78.03a_40_LNA3 gmCagmCggTggTaaAaaGtaTgaAaamCgtGgtAatTaaAag 11372 >gene78.03b_40_LNA3 gAaaActAatmCaaAggTaaAcgTggAtcinCcaTggmCaaTtc 11373 >gene78.m0103_40_LNA3 cActGccmCagAggTtcAatmCgaTaaAtaTgtGaaGgaAat 11374 >gene78.m01INS3_40_LNA3 cTgcmCcaGagGttmCaaTcgAtcmCgaTgaTccTaaTgaAgg 11375 >gene78.mINS303_40_LNA3 gTatmCgtmCcaTcaTagTatmCgaTaaAtaTgtGaaGgaAat 11376 >gene78.INS3_40_LNA3 tmCttGcaTtcTtcAacTtcmCttmCaamCacTtgAgcGgaGtc 11377 >gene78.m01INS4_40_LNA3 cTgcmCcaGagGttmCaaTcgAtgTgtGatAggAtcAgtGtt 11378 >gene78.mINS403_40_LNA3 cGaaGgaGacTgcTaaTatinCgaTaaAtaTgtGaaGgaAat 11379 >gene78.INS4a_40_LNA3 tAtgAgcAtgmCgaGagAcgmCtgTagTtgGaaAaamCccAc~ 11380 >gene78.INS4b_40_LNA3 cGctGatTatActGcgGagAagGtgGgtGagTatAaaGac - B. Printing and Coupling of the Splice Isoform-Specific Microarrays
- The splice variant capture probes were synthesized with a 5′ anthraquinone (AQ)-modification, followed by a hexaethyleneglycol-2 (HEG2) linker. The capture probes were first diluted to a 20 μM final concentration in 100 mM Na-phosphate buffer pH 7.0, and spotted on the Immobilizer polymer microarray slides (Exiqon, Denmark) using the Biochip Arrayer One (Packard Biochip Technologies, USA) with a spot volume of 2×300 pl and 300 μm between the spots. The capture probes were immobilized onto the microarray slide by UV irradiation in a Stratalinker with 2300 μjoules (Stratagene, USA). Non-immobilized capture probe oligonucleotides were removed from the slides by washing the slides two
times 15 min. in 1×SSC. After washing, the slides were dried by centrifugation at 1000×g for 2 min., and stored in a slide box until microarray hybridization. - C. Construction of the Splice Variant Clones
- The recombinant splice variant constructs were cloned into the
Triamp 18 vector (Ambion, USA). The constructs were sequenced to confirm their construction. The plasmid clones were transformed into E. coli XL1 0-Gold (Stratagene, USA). - Genomic DNA was prepared from a wild type standard laboratory strain of Saccharomyces cerevisiae using the Nucleon MiY DNA extraction kit (Amersham Biosciences, USA) according to the supplier's instructions. Amplification of the partial yeast gene was done by standard PCR using yeast genomic DNA as template. In the first step of amplification, a forward primer containing a restriction enzyme site and a reverse primer containing a universal linker sequence were used. In this
step 20 bp was added to the 3′-end of the amplicon, next to the stop codon. In the second step of amplification, the reverse primer was exchanged with a nested primer containing a poly-T20 tail and a restriction enzyme site. The SWI5 amplicon contains 730 bp of the SWI5 ORF plus 20 bp universal linker sequence and a poly-A20 tail. The PCR primers used were; - YDR146C-For-EcoRI: acgtgaattcaaatacagacaatgaaggagatga (SEQ ID NO:)
- YDR146C-Rev-Uni: gatccccgggaattgccatgttacctttgattagttttcattggc (SEQ ID NO:)
- Uni-polyT-BamI: acgtggatccttttttttttttttttttttgatccccgggaattgccatg (SEQ ID NO:),
- The PCR amplicon was cut with the restriction enzymes, EcoRI +BamHI. The DNA fragment was ligated into the
pTRIamp 18 vector (Ambion, USA) using the Quick Ligation Kit (New England Biolabs, USA) according to the supplier's instructions and transformed into E. coli DH-5 αby standard methods. - C1. Construction of the recombinant splice variant #1 (Triamp18/swi5-rubisco) The Arabidopsis thaliana Rubisco small subunit ssu2b gene fragment (gil 7064721) was amplified from genomic DNA by primers named
DJ305 5′-ACTATGATGGACGATACTGGAC-3′ (SEQ ID NO:) andDJ306 5′-ATTGGATCGATCCGATGATCCTAATGAAGGC-3′ (SEQ ID NO:), containing ClaI restriction site linkers. The purified PCR fragment was digested with ClaI and then cloned into the swi5 (gI:7839148) vector at the unique ClaI site (atcgat) giving each insert a flanking sequence from the original yeast SWI5 insert (namedexon01and exon 03, see FIG. 19). The product was inserted in the reverse orientation, so that the insert sequence is:atcgatCCGATGATCCTAATGAAGGCGCCCGGGTACT (SEQ ID NO:) CCTTCTTGCATTCTTCAACTTCCTTCAACACTTGAGC GGAGTCGGTGCATCCGAACAATGGAAGCTTCCACATT GTCCAGTATCGTCCATCATAGTatcgat - Nucleotide sequence analysis revealed a difference between the sequence of A. thaliana Rubisco expected from the GenBank database and that obtained from all sequenced constructs and PCR products.
Position 30 in the Rubisco insert is C rather than the expected A. This SNP was probably created by PCR. None of the oligonucleotide capture probes used in the example cover this region.Rubisco seq. in genbank TCCTAATGAAGGCGCCA (SEQ ID NO:) The sequence obtained from the plasmid contruct TCCTAATGAAGGCGCCC (SEQ ID NO:) - C2. Construction of the recombinant splice variant #2 (
Triamp 18/swi5-lea) The Arabidopsis thaliana Lea gene (gil 526423) was amplified from genomic DNA with primers namedDJ307 5′-GGAATTATCGATGTGTGATAGGATCAGTGTTCAG-3′ (SEQ ID NO:), andDJ308 5′-AATTGGATCGATATTAGCAGTCTCCTTCGCC-3′ (SEQ ID NO:), including the ClaI linker sites as above. The PCR fragment was digested with ClaI cloned into the yeast SWI5 IVT construct as above at the unique ClaI site. - The fragment was inserted in the forward orientation, resulting in the following insert sequence:
atcgatGTGTGATAGGTTCAGTGTTCAGGGCTGTCCA (SEQ ID NO:) AGGAACGTATGAGCATGCGAGAGACGCTGTAGTTGGA AAAACCCACGAAGCGGCTGAGTCTACCAAAGAAGGAG CTCAGATAGCTTCAGAGAAAGCGGTTGGAGCAAAGGA CGCAACCGTCGAGAAAGCTAAGGAAACCGCTGATTAT ACTGCGGAGAAGGTGGGTGAGTATAAAGACTATACGG TTGATAAAGCTAAAGAGGCTAAGGACACAACTGCAGA AGGCGAAGGAGACTGCTAATatcgat. - FIG. 11 shows the construction of the recombinant splice variants in the in vitro transcription vector. The small bars show the location of the oligonucleotide capture probes used in this example. The sequences of the capture probes are shown in Table 15.
- D. Preparation of Target D1. In vitro RNA preparation from splice variant vectors
- In vitro RNA from the splice variants were made using the MEGAscript™ high yield transcription kit according to the manufacturer's instructions (Ambion, USA). The yield of IVT RNA was quantified at a Nanodrop spectrophotometer (Nanodrop Technologies, USA)
- D2. Isolation of Total RNA from C. elegans
- Strains and growth conditions: C. elegans wild-type strain (Bristol-N2) was maintained on nematode growth medium (NG) plates seeded with Escherichia coli strain OP50 at 20° C., and the mixed stages of the nematode were prepared as described in Hope, I. A. (ed.) “C. elegans —A Practical Approach “, Oxford University Press 1999. The samples were immediately flash frozen in liquid N2 and stored at −80° C. until RNA isolation.
- A 100 μl aliquot of packed C. elegans worms from a mixed stage population was homogenized using the
FastPrep Bio 101 from Kem-En-Tec for 1 min,speed 6 followed by isolation of total RNA from the extracts using the FastPrep Bio101 kit (Kem-En-Tec) according to the manufacturer's instructions. The eluted total RNA was ethanol precipitated for 24 hours at −20° C. by addition of 2.5 volumes of 96% EtOH and 0.1 volume of 3M Na-acetate, pH 5.2 (Ambion, USA), followed by centrifugation of the total RNA sample for 30 min at 13200 rpm. The total RNA pellet was air-dried and redissolved in 10 μl of diethylpyrocarbonate (DEPC)-treated water (Ambion, USA) and stored at −80° C. - E. Fluorochrome-labelling of the target
- The following fluorochrome-labelled cDNA targets were synthesized to test the performance of ‘merged’ splice junction probes that encompass exon borders. Synthetic RNAs corresponding to three artificial splice variants; #1 (exon01-INS3-exon03 (1-INS3-3), #2 (exon01-INS4-exon03) (01-INS3-3) and #3 without the middle exon (01-03) were spiked into 10 μg of C. elegans reference total RNA samples in various combinations and concentrations prior to fluorochrome-labelling with either Cy3 or Cy5 as indicated in Table 16. At the
same time 10 μg of C. elegans reference total RNA was labeled with Cy3 for control experiments. Hybridizations were performed with Cy3- and Cy5 labeled C. elegans RNA+spike RNA mix. The details of RNA samples and synthetic RNA spikes are shown in Table 16. The RNA samples were combined in individual labeling reactions with 5 μg anchored oligo(dT20) primer and DEPC-treated water to a final volume of 8 μl. The mixture was heated at 70° C. for 10 min, quenched on ice for 5 min, followed by addition of 20 units of Superasin RNase inhibitor (Ambion, USA), 1 μl dNTP solution (10 mM each dATP, dGTP, dTTP and 0.4 mM dCTP, and 3 μl of Cy3-dCTP or Cy5-dCTP, Amersham Biosciensces, USA), 4 μl ×RTase buffer (Invitrogen), 2 μl 0.1 mM DTT (Invitrogen), 400 units of Superscript II reverse transcriptase (Invitrogen, USA) and DEPC-treated water to 20 μl final volume. Background hybridization to merged capture probes was monitored with 10 μg of C. elegans reference RNA alone labeled with Cy3-dCTP; according to the labeling method described above for the splice variant spikes. All cDNA syntheses were carried out at 42° C. for 2 hours, and the reaction was stopped by incubation at 70° C. for 5 min., followed by incubation on ice for 5 min. - Unincorporated dNTPs were removed by gel filtration using MicroSpin S-400 HRcolumns as described in the following: Pre-spin the
column 1 min at 1500×g in a 1.5-ml tube and place the column in a new 1.5 ml tube. Slowly apply the cDNA sample to the top centre of the resin, spin 1500-x g for 2 min and collect the eluate. The RNA was hydrolyzed by adding 3 μl of 0.5 M NaOH, mixing and incubating at 70° C. for 15 min. The samples were neutralized by adding 3 μl of 0.5 M HCl and mixing, followed by addition of 450μl 1×TE, pH 7.5 to the neutralized sample and transfer onto a Microcon-30 concentrator (prior to use, spin 500μl 1×TE through the column to remove residual glycerol). The samples were centrifuged at 14000-x g in a microcentrifuge for 12 min. Spinning was continued until volume was reduced to 5 μl. The labelled cDNA probes were eluted by inverting the Microcon-30 tube and spinning at 1000-x g for 3 minTABLE 16 Synthetic splice variant RNAs spiked into C. elegans samples*. Spike RNA Observed ratio STDEV Observed ratio STDEV concentration Ratio Splice variant RNAs LNA 50 mer LNA 50 mer LNA 40 mer LNA 40 mer Expected ratio 1000 ppm 5 Cy3: spike 01-INS3-03 0.76 0.05 0.61 0.19 0.83 1 Cy3: spike 01-03 0.24 0.05 0.39 0.19 0.17 1 Cy5: spike 01-INS3-03 0.16 0.04 0.06 0.03 0.17 5 Cy5: spike 01-03 0.84 0.04 0.94 0.03 0.83 1000 ppm 5 Cy3: spike 01-INS3-03 0.77 0.11 0.68 0.22 0.83 1 Cy3: spike 01-INS4-03 0.23 0.11 0.32 0.22 0.17 1 Cy5: spike 01-INS3-03 0.12 0.04 0.11 0.15 0.17 5 Cy5: spike 01-INS4-03 0.88 0.04 0.89 0.15 0.83 1000 ppm 5 Cy3: spike 01-INS3-03 0.88 0.08 0.87 0.10 0.83 1 Cy3: spike 01-INS4-03 0.12 0.08 0.13 0.10 0.17 1 Cy5: spike 01-INS3-03 0.22 0.12 0.15 0.12 0.17 5 Cy5: spike 01-INS4-03 0.78 0.12 0.85 0.12 0.83 100 ppm 5 Cy3: spike 01-INS3-03 0.89 0.15 0.11 0.08 0.83 1 Cy3: spike 01-INS4-03 0.11 0.15 0.89 0.08 0.17 1 Cy5: spike 01-INS3-03 0.48 0.2 0.57 0.31 0.17 5 Cy5: spike 01-INS4-03 0.52 0.2 0.43 0.31 0.83 10 ppm 5 Cy3: spike 01-INS3-03 0.61 0.2 0.09 0.14 0.83 1 Cy3: spike 01-INS4-03 0.39 0.2 0.91 0.14 0.17 1 Cy5: spike 01-INS3-03 0.34 0.18 0.19 0.22 0.17 5 Cy5: spike 01-INS4-03 0.66 0.18 0.81 0.22 0.83 - F. Microarray Hybridization
- The fluorochrome-labelled cDNA samples, respectively, were combined. The following was added: 3.75 μl 20×SSC (3×SSC final, pass through 0.22 μ filter prior to use to remove particulates) yeast tRNA (1 μg/μl final) 0.625 μl 1 M HEPES, pH 7.0 (25 mM final, pass through 0.22 μfilter prior to use to remove particulates) 0.75
μl 10% SDS (0.3% final) and DEPC- water to 25 μl final volume. The labelled cDNA target samples were filtered in Millipore 0.22 μ filter spin column (Ultrafree-MC, Millipore, USA) according to the manufacturer's instructions, followed by incubation of the reaction mixture at 100° C. for 2-5 min. The cDNA probes were cooled at room temp for 2-5 min by spinning at max speed in a microcentrifuge. A LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on Immobilizer™ MicroArray Slide and the hybridization mixture was applied to the array from the side. An aliquot of 30 μL of 3×SSC was added to both ends of the hybridization chamber, and the Immobilizerm MicroArray Slide was placed in the hybridization chamber (DieTech, USA). The chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridisation, the slide was removed carefully from the hybridization chamber and washed using the following protocol. The slides were washed sequentially by plunging gently in 2×SSC/0.1% SDS at room temperature until the cover slip falls of into the washing solution, then in 1×SSC pH 7.0 (150 mM NaCl, 15 mM Sodium Citrate) at room temperature for 1 min, then in 0.2×SSC, pH 7.0 (30 mM NaCl, 3 mM Sodium Citrate) at room temperature for 1 min, and finally in 0.05×SSC (7.5 mM NaCl, 0.75 mM Sodium Citrate) for 5 sec, followed by drying of the slides by spinning at 1000×g for 2 min. The slides were stored in a slide box in the dark until scanning. G. Microarray data analysis. - The splice variant microarray was scanned in a ScanArray 4000XL confocal laser scanner (Packard Instruments, USA). The hybridisation data were analysed using the GenePix Pro 4.01 microarray analysis software (Axon, USA). The C. elegans reference RNA alone converted to first strand cDNA and labelled with Cy3-dCTP did not produce significant fluorescence intensity signals from the LNA substituted spike RNA specific capture probes.
- G1. A mathematical formula for analysis of the microarray data for alternative splicing
- One of the major limitations to comparative microarray hybridisation assays is that only identical probes can be compared between samples. Different alternative splice forms are detected using different probes, and this will tell directly if one splice form is more abundant in a given tissue compared to another. However, the estimation of the ratios between splice forms in a single tissue is not directly accessible. Given an example similar to that described below we employ the following calculations to calculate quantities of splice variants from array data. The theoretical justification is shown. To our knowledge this justification has not been used by any previous analysis.
- The above scenario is tested in a comparative hybridisation, with two channels: I & II (signal from probe2 in channel I is called probe2(I), and so forth).
Probe 1 hybridises to both splice forms, Probe2 hybridises to A only, Probe3 hybridises to B only. - Since every transcript will hybridise to probe 1, and every transcript will hybridise to either probe2 or probe3, there exists some relationship between the following:
- probe I (I)˜{probe2(I) and probe3(I)}.
- Probe 1 (II)˜{probe2(II) and probe3(II)}.
- For simplicity we assume that systematic differences between channels have already been eliminated through normalisation, although this is not essential. We now imagine a factor (x) that will transform the signal of probe2 into a value directly comparable to
probe 1. Likewise we imagine factor (y) for probe3. As long as we are not facing saturation in the hybridisations, the assumption of a linear relationship between absolute probe signals is reasonable. - The introduction of variables x & y will give the following equations:
- Probe 1 (I)=(x)probe2(I)+(y)probe3(I).
- probe 1(II)=(x)probe2(II)+(y)probe3(II).
- Since all signals are measurable, the above is two linear equations with two unknown variables, that can easily be solved. Further the ratio between (x)probe2(I) & (y)probe3(I) will provide the ratio between splice form A and B in channel I. Similarly, the ratio of (x)probe2(II) to (y)probe3(II) is used for channel II.
- Data normalization is not required for this method.
- In the above equations,
Probe 1 denotes all probes that will hybridize to both spliceforms, probe2 denotes probes that specifically will hybridize to spliceform A but not B, and probe3 denotes probes that will specifically hybridize to spliceform B but not A. - In the case where two spliceforms consist of gene78 with two different inserts middle exons (INS3& INS4), probes can be grouped as in Table 20 (only LNA 40mers are considered here):
TABLE 20 Probes that will hybridize to Probes that will hybridize to Probes that will hybridize to both constructs INS3 constructs only INS4 constructs only Gene78.01a_40_LNA3 Gene78.INS3_40_LNA3 Gene78.INS4_40_LNA3 Gene78.01b_40_LNA3 Gene78.m01INS3_40_LNA3 Gene78.m01INS4_40_LNA3 Gene78.03a_40_LNA3 Gene78.mINS303_40_LNA3 Gene78.m1NS403_40_LNA3 Gene78.03b_40_LNA3 - The equations can be solved with any combinations of one representative from each probe group. This gives a total of 48 (4×3×3) possible ways of solving the equations. The estimated quantities of the constructs are given as the average of all possible solutions (equations yielding non-positive solutions are ignored). This was done for all comparative hybridizations. Note that when comparing with gene78 with no insert, only 12 equations are possible (The, since the artificial splice variant construct with no insert has only one specific probe). The results from analysis of the microarray hybridization data from the RNA pools spiked with different splice isoforms at different ratios and concentrations are shown in FIGS. 39 and 40.
- RESULTS
- FIG. 39 shows detection of alternatively spliced mRNAs using LNA-substituted 50-mer oligonucleotide capture probes in a bar diagram format. FIG. 40 shows detection of alternatively spliced mRNAs using LNA-substituted 40-mer oligonucleotide capture probes. Both 50-mer and 40-mer LNA-DNA mixmer substituted oligonucleotide capture probes, substituted with an LNA nucleotide at every third nucleotide position, were able to provide highly accurate measurements for fold-changes in the expression of three homologous, alternatively spliced mRNA variants in the concentration range of 1000 ppm to 10 ppm. The quantification of the splice isoforms was carried out using a set of both internal, exon-specific probes and merged, splice junction specific probes, printed onto microarrays and hybridized with complex cDNA target pools spiked with different cloned artificial splice isoforms in which the middle exon was either alternatively skipped or excluded completely resulting in the three different splice isoforms; 01-INS3-03, 01-INS4-03 and 01-03.
- The present patent example demonstrates the use of the Exiqon C. elegans LNA tox oligoarray in gene expression profiling experiments in the nematode Caenorhabditis elegans. The C. elegans tox oligoarray will monitor the expression of a selection of 110 genes relevant for general stress response and for the metabolism of toxic compounds. Two different capture probes for each of these target genes have been designed, and included on the LNA tox array. In addition, the C. elegans LNA tox oligoarray contains capture probes providing control for cDNA synthesis efficiency and the developmental stage of the nematode. Capture probes for constitutively expressed genes for data set normalization is also included on the C. elegans LNA tox oligoarray.
- A. Cultivation of C. elegans worms
- For all cultures the sample is divided into two, and one half of the sample is used as the control, the other as the treated sample. Worm samples are harvested and sucrose cleaned by standard methods. For heat shock treatment: the heat shock sample is added to S-media preheated to 33° C. in a 1 L flask suspended in a water bath at 33° C., the other sample is added to a 1 L flask with S-media at 25° C. Both samples are shaken at approximately 100 rpm. for an hour. For β-naphthoflavone and primaquine treatment: 0.5 mL of 5 mg/mL β-naphthoflavone in DMSO or 0.5 mL of 20 mg/mL primaquine in DMSO were added to each 500 mL volume of S-media culture after 28 hours of growth from LI. At the same time 0.5 mL of DMSO was added to the control. Incubation was for 24 hours. Samples are then harvested by centrifugation at 3000×g suspended in RNALaterM (Ambion) and immediately frozen in liquid nitrogen.
- B. RNA Extraction
- RNA was extracted from the worm samples using the FastRNA™ Kit, GREEN (Q-BIO) essentially according to the suppliers' instructions.
- C. Design and Synthesis of the LNA Capture Probes
- Capture probes were designed using an in-house developed software. Regions with unique mRNA sequence of the selected target genes were identified. The optimal 50-mer oligonucleotide sequences with respect to T m, self-complementarity and secondary structure were selected. LNA modifications were incorporated to increase affinity and specificity.
- D. Printing of the LNA Microarrays
- The microarrays were printed on Immobilizer™ MicroArray Slides (Exiqon, Denmark) using the MicroGrid II from Biorobotics (UK). The arrays were printed with a 10 μM capture probe solution. Two replicas of the capture probes were printed on each slide. E. Synthesis of fluorochrome labelled first strand cDNA from total RNA
- 15 μg of C. elegans total RNA was combined with 5 μg oligo dT primer (T20VN) in an RNase free, pre-siliconized 1.5 mL tube, and the final volume was adjusted with DEPC-water to 14.5 μL. The reaction mixture was heated at +70° C. for 10 min., quenched on
ice 5 min., spin 20 seconds, followed by addition of 1 μL SUPERase-In™(20U/μL, Ambion, USA), 6 μL 5xRTase buffer (Invitrogen, USA), 3 μL 0.1 M DTT (Invitrogen, USA), 1.5 1L dNTP (20 mM dATP, dGTP, dTTP; 4 mM dCTP in DEPC-water, Amersham Biosciences, USA), and 3 μL Cy3™-dCTP or Cy5™-dCTP (Amersham Biosciences, USA). First strand cDNA synthesis was carried out by adding 1 μL of Superscript™ II (Invitrogen, 200 U/mL), mixing and incubating the reaction mixture for 1 hour at 42° C. An additional 1 μL of Superscript™ II was added and the cDNA synthesis reaction mixture was incubated for an additional 1 hour at 42° C.; the reaction was stopped by heating at 70° C. for 5 min., and quenching on ice for 2 min. The RNA was hydrolyzed by adding 5 1L of 1 M NaOH, and incubating at 70° C. for 15 min. The samples were neutralized by adding 5 μL of 1 M HCl, and purified by adding 450μL 1×TE buffer, pH 7.5 to the neutralized sample and transferring the samples onto a Microcon-30 concentrator. The samples were centrifuged at 14000×g in a microcentrifuge for 8 min, the flow-through was discarded and the washing step was repeated twice by refilling the filter with 450μl 1×TE buffer and by spinning for ˜12 min. centrifugation was continued until the volume was reduced to about 5 μL, and finally the labelled cDNA probe was eluted by inverting the Microcon-30 tube and spinning at 1 000xg for 3 min. - F. Synthesis of Fluorochrome Labelled cRNA from Total RNA
- First and second strand cDNA syntheses were made using the MessageAmp™ aRNA Kit (Ambion) according to suppliers' instructions. Five microgram of C. elegans total RNA was used as template for cDNA syntheses. Syntheses of fluorescent cRNA were made according to the MessageAmp™ aRNA Kit (Ambion) protocol with minor modifications. Cy3™-UTP or Cy5™-UTP (611 of a 5 mM solution Amersham Biosciences, USA) replaced biotin-CTP. The final concentration of ATP, CTP, and GTP was 7.5 mM whereas the concentration of UTP was reduced to 4.9 mM.
- G. Hybridization with Fluorochrome-Labelled cDNA or cRNA
- The arrays were hybridized overnight using the following protocol. The Cy3™ and Cy5™-labelled cDNA or cRNA samples were combined in one tube followed by addition of 3 μL 21×SSC (3×SSC final), 0.5 μL 1 M HEPES, pH 7.0 (25 mM final), 25 μg yeast tRNA (1.25 μg/μL final), 0.6 μL 10% SDS (0.3% final), and DEPC-treated water to 20 μL final volume. The labelled cDNA target sample was filtered in a Millipore 0.22 micron spin column according to the manufacturer's instructions (Millipore, USA), and the probe was denatured by incubating the reaction at 1 00° C. for 2 min. The sample was cooled at 20-25° C. for 5 min. by spinning at max speed in a microcentrifuge. A LifterSlip (Erie Scientific Company, USA) was carefully placed on top of the microarray spotted on Immobilizer™ MicroArray Slide and the hybridization mixture was applied to the array from the side. An aliquot of 30 μL of 3×SSC was added to both ends of the hybridization chamber, and the Immobilizer™ MicroArray Slide was placed in the hybridization chamber. The chamber was sealed watertight and incubated at 65° C. for 16-18 hours submerged in a water bath. After hybridisation, the slide was removed carefully from the hybridization chamber and washed using the following protocol. The Lifterslip coverslip was washed off in 2×SSC, pH 7.0 containing 0.1% SDS at room temperature for 1 min., followed by washing of the microarrays subsequently in 10.1×SSC, pH 7.0 at room temperature for 1 min, and then in 0.2×SSC, pH 7.0 at room temperature for 1 min. Finally the slides were washed for 5 seconds in 0.05×SSC, pH 7.0. The slides were then dried by centrifugation in a swinging bucket rotor at approximately 200 G for 2 min. The slide is now ready for scanning.
- H. Data Analysis.
- Following washing and drying, the slides were scanned using a ScanArray 400XL scanner (Perkin-Elmer Life Sciences, USA), and the array data were processed using the GenePix™ Pro 4.0 software package (Axon, USA). The data in each image was normalized so that the ratio of means of all of the features is equal to 1.
- RESULTS
- Use of LNA-modified oligonucleotide capture probes in Exiqons C. elegans LNA tox oligoarray clearly allows the identification of distinct expression profiles for C. elegans genes relevant for general stress response and for the metabolism of toxic compounds.
TABLE 17 Expression profiling using LNA Oligonucleotide Microarrays. Log2 transformed fold of changes for selected genes in the two expression profiling experiments hybridised with cRNA target. Tox compound Gene name Primaquine beta-naphthoflavone ABC_C34G6.4 1.01 ABC_F57C12.4 −1.11 CYP_C03G6.15 2.35 CYP_C06B3.3 2.47 CYP_C49G7.8 2.40 CYP_F14F7.2 −1.24 −1.03 CYP_K07C6.5 2.68 CYP_K09D9.2 2.14 DC_W05G11.3 1.16 ER_26S −1.09 −1.01 HSP_C47E8.5 1.17 HSP_F26D10.3 1.05 HSP_F43D9.4 1.27 NAP_D2096.8 1.14 PPGB_F13D12.6 1.08 1.21 RAD_Y116A8C.133 1.13 RPL_K11H12.2 1.15 1.42 Ubi_F25B5.4 1.37 -
TABLE 18 LNA-modified oligonucleotide capture probes. LNA modifications are depicted by uppercase letters in the sequence, mC denotes LNA methyl cytosine. (SEQ ID NOs:) Oligo Name Sequence CEABC_C34G6.4_u293_LNA3 TgcmCatTgcAcgGgcActTgtTcgAtcTccTtcTgtTttActTttGgaTg CEABC_C34G6.4_u375_LNA3 TcaTtcTagGatTgcmCagAtgGttAtgAtamCtcAtgTcgGagAgaAagGa CEABC_F57C12.4_u15_LNA3 mCcaAtgTtgTttAatTggTtgTaaTgtmCttGatGacmCtgmCatAatmCatAt CEABC_F57C12.4_u480_LNA3 mCacAagAtcmCtgTgtTgtTctmCcgGaamCaaTgaAaaTgaActTagAtcmCa CEABC_F57C12.5_u111_LNA3 TacTtgTtcTcgAcaAagGttGtgTagmCcgAgtTtgAcamCtcmCgaAgaAa CEABC_F57C12.5_u444_LNA3 TgaActTggAtcmCctTctTtgmCatTtaGcgAtgAtcAaaTttGggAagmCg CEABC_K08E7.9_d8_LNA3 TcaTtaAttTtgTgtAgcTttmCttTctmCgaTttTtgmCacGatmCttTccmCc CEABC_K08E7.9_u51_LNA3 AggGtgmCctActAcaAacTgamCccAaaAgcAgaTgamCcgAgaAgaAatAa CEABC_Y39D8C.1_u37_LNA3 AttGaaAgcGacGcgGaaAgtGccAtgTatTtcTaaTttTgtTttmCttTa CEABC_Y39D8C.1_u422_LNA3 TtgTcaGcaTatmCaaGagTagAtaTggAagTggAtamCacTctGctAatmCc CEADH_H24K24.3a_d3_LNA3 mCacmCttAttGcgTtcAatTttTgtTtcmCacmCtamCtamCtamCgaAtamCgtTg CEADH_H24K24.3a_u50_LNA3 TcamCaaGggAgaGagTctGcgGtcGgtGctGgcGttmCgaGaaAatAtaAc CEAPEX_R09B3.1_u191_LNA3 mCatGcaTccmCgamCgaGaaGaaGtamCtcAttTtgGagTtaTctGgcGaaTt CEAPEX_R09B3.1_u37_LNA3 GacmCatGctmCcgGtcGtcAtgmCaaAtcGacTtcTaaAttGctTctGatTa CEAPO_C35D10.9_u15_LNA3 TtgmCatGctGttAaaAccTatmCgtGtamCaaTatTgcmCtgTatAttmCccmCt CEAPO_C35D10.9_u609_LNA3 TggmCacAgcTtaAtaAcaAatTggAaaGtcGagGatTagTcgGtgTtgAa CEAPO_C48D1.2_u176_LNA3 GacAcamCgcAaaGgaTatGgaTgtTgtTgaGctGctGacTgaAgtmCaaTa CEAPO_C48D1.2_u23_LNA3 AgcAcgAaamCtcTgcmCgtmCtaAaaTtcActmCgtGatTcaTtgmCccAatTg CEAPO_F20C5.1_u453_LNA3 AtgGtcAtamCtcTaaAatGggmCagAacTtcAacmCaaAtcAttmCtcGtcAg CEAPO_F20C5.1_u96_LNA3 AacmCcgAgcTtgmCcgmCaaAgtGcaAgaAaaTtaTagAacGaaTgaAacAg CEATPase_B0365.3_u31_LNA3 GgaTggGtcGagmCgtGagAccTacTacTaaAgaAcaGctTgtGaaTctTt CEATPase_B0365.3_u386_LNA3 mCaamCgtTctmCgaTtcmCtamCggAcaAgaAtgGacmCtaTgcmCaamCagAaaGa CEATPase_C17H12.14_u356_LNA3 TgcTcgTtaTccAgcTatTttGaaGggActTgtmCatGcaAggActTctTc CEATPase_C17H12.14_u89_LNA3 mCcgTttAgaGctTatTgcTaamCcaGatTgtmCccAcaAgtmCagAacAgcTc CEATPase_F55F3.3_u215_LNA3 TgamCggAcgmCtamCtamCccAtaTgtAttTgtTccAtcTtamCcaGcaAccAa CEATPase_F55F3.3_u275_LNA3 AgcTacTtcAttmCgamCaaGgaAcaTctmCggAaaAgtmCaaGtamCatmCccGg CEATPase_Y49A3A.2_u103_LNA3 AaaTtcAagGatmCcaGttGccGatGgtGaaGccAagAttmCgcAagGatTa CEATPase_Y49A3A.2_u272_LNA3 mCgaTcgTttmCtgmCccAttmCtamCaaGacTgtmCggTatGctmCaaGaaTatGa CECALR_Y38A10A.5_u238_LNA3 TcaGgaAcgAtcTttGacAacAttAtcAtcAccGacTctGttGagGagGc CECALR_Y38A10A.5_u296_LNA3 TgaActmCtamCtcTtaTgaAagmCtgGggAgcmCatmCggAttmCgaTttGtgGc CECAT_Y54G11A.5b_u137_LNA3 GaamCttTgcAggGccGctmCggGgaAtgTcaTgaTttmCatTatTaaGggAa CECAT_Y54G11A.5b_u189_LNA3 GtcAatTctGggAgaAggTgtTggAtamCcgGggmCtcGggAgaGaaTgtGc CECC_C03D6.3_u275_LNA3 AtgTaaAgaAggAatGctTccmCgaAtgGatTggAtaTttAttTgtmCcaGa CECC_C03D6.3_u430_LNA3 GgamCcgAaaTttGtgmCagmCatGtcGgamCacGaaAttGatGgtmCtcAttTt CECC_C07G2.3_d9_LNA3 mCagAcamCgaAggTtamCgaTagAtaAccAtcTctmCaaAgtmCtaTcgAccTc CECC_C07G2.3_u44_LNA3 mCgamCgaTgtGcgTgtTccTgamCgaTgaAagAatGggAtaTtaAgaAaamCc CECC_Y46G5A.2_u331_LNA3 TtgTgcTccAtcGctGctmCcgmCttAcaGacTtgAcaAcgmCtcAccTttGc CECC_Y46G5A.2_u385_LNA3 AatGagmCggTtgTgcmCgtGtgAcgTcamCttmCgtmCacAgtGttGctmCtamCt CECoA_C29F3.1_u316_LNA3 AaaTtgAcamCcaAtcAaaTctGtcTcaTctmCctGagGacmCgtmCaamCttmCg CECoA_C29F3.1_u392_LNA3 AatmCttTgtGtamCggAgaTggGgcAaaAggmCagmCaaGaaAgtAaamCcaAg CECoA_F08A8.4_u1094_LNA3 AggAcaAggGgcActActGgcAcaGgcTttGatTatTgcAgtGagAtaTt CECoA_F08A8.4_u1260_LNA3 TtaAtgGagGtgAcaAtgGgtTccTtgGatTcgAtaAatTccGagTgcmCc CECoA_F59F4.1_u109_LNA3 GctmCttmCtcmCagTggGctmCaaAatAgtmCaamCtcAacAgaTcgGaaGttmCt CECoA_F59F4.1_u424_LNA3 AaaGctTcgAgaTggmCacGttmCgtmCtgTatmCtcGtgAagAacTtaTtgmCa CECoA_Y25C1A.13_u115_LNA3 GatTcgmCtgAacTttAtcAagAcgTggAatAtgAgcmCagmCtcmCtgTcgAc CECoA_Y25C1A.13_u451_LNA3 GatmCttAtcAccGcgTgcGatAttmCgaGtaGctTcamCagGatGcgAttTt CECOL_C27H5.5_u493_LNA3 GgaAagGaaGgaTccAttmCtcAgcTctGcamCttmCcamCcaTcaGagmCcaTg CECOL_C27H5.5_u680_LNA3 TggAtamCaaGgaGggAtcTggmCagTggTggAtcTggAagTggTggAtaTg CECOQ_ZC395.2_u199_LNA3 TtgAaaGaamCtcmCttGccGacGatmCctGaaAcamCacAaaGaaTtgmCtgAa CECOQ_ZC395.2_u400_LNA3 AtgTggGatGagGagAaaGaamCatTtaGatAcaAtgGaaAgaTtaGctGc CECRYZ_F39B2.3_u171_LNA3 AggmCtgAgcTctTggActTtgGcaTcaAcaTtgTctmCatTctTgaAggAa CECRYZ_F3982.3_u222_LNA3 TtaTggTtamCagAagGagmCtgTttAcgGtgTagmCatTggGaaTgtmCttmCc CECyclin_R02F2.1a_u24_LNA3 mCacTtcAacmCaamCtcmCgtGttAatmCaaGcaAgcmCgcmCacmCatmCtaAtgAg CECyclin_R02F2.1a_u312_LNA3 TctmCatTgcTcgTcgAggmCtamCcaAcaAacActGgcAatAccmCaaTtaAt CECyclin_ZC168.4_u203_LNA3 TaaGaaAgtmCatTgaGgaTgcTgtmCgcTttGctmCgcmCgaAgtmCtcGtaTa CECyclin_ZC168.4_u273_LNA3 AagTtcAtcmCtgTtgAcgGaaTcgAggmCggAgaAtgmCtgTatmCggTcaTt CECYP_B0213.15_u133_LNA3 AcaGgaAatAtgAttTtgGatTtcGatTttGaaTcgGttGgtGctGccmCc CECYP_B0213.15_u202_LNA3 GctGagmCtgTatTtgGctAgtGaaAtgTgtGttTttGatActTtaAatGa CECYP_B0304.3_u38_LNA3 AcgAggTttGgaTcamCaaTcaGaaTtcTgtGaaAtaAgcGttTttTggGa CECYP_B0304.3_u89_LNA3 AgtTctmCggTctAacAgtGtcTccmCgtTgaAtaTtcTtgTaaAatmCacAc CECYP_C03G6.14_u706_LNA3 AtgAccActmCaaAatActGctAaaAgaTttGcaGcgGcaGaaGccGttAa CECYP_C03G6.14_u768_LNA3 TtgAtaTggmCtgTacmCtgTatGgtTttTgaGgamCgtTttTtaGgaGtcGa CECYP_C03G6.15_d9_LNA3 AttTatTcaTtcAtcmCatGtaAacTgtAtaTttTgaAttTgtGttGtaAa CECYP_C03G6.15_u148_LNA3 GccAaaGcaGaaTtgTatTtgAtcTtcGgtAacmCttmCtcmCttmCgcTacAa CECYP_C06B3.3_u102_LNA3 AttTtgAatmCttmCtgGgaAaaTgcmCatmCcamCtcGagAaamCcgTtcmCgtTt CECYP_C06B3.3_u474_LNA3 mCtaAcgGagGatmCtcGccAatTatmCttTgaGagAcaAaamCtgAaamCtcmCt CECYP_C12D5.7_u399_LNA3 AtcTagTccmCaaTgaAtcTccmCacAtgmCtgTtamCtcGtgAtgTtcAacTc CECYP_C12D5.7_u65_LNA3 TttTgcTttmCatmCgcAaaAgcTcaAgaTtamCacAtgTcaGgtmCaaGccAa CECYP_C45H4.17_u27_LNA3 mCcgmCgamCttTaaAgaGaaGatmCatAaaTttGcaTtgTttTttGttTgtAt CECYP_C45H4.17_u598_LNA3 mCgaGggTgaTtcGgaGacTttmCagTaaTgtmCcaActTtcAaaTgtTtgmCa CECYP_C45H4.2_u110_LNA3 TagAtamCaaGatAcaTccmCtcAaaAgaAggmCctAccGtcAatGgcmCaaAg CECYP_C45H4.2_u429_LNA3 TcaAcgmCgtmCtaTaaAtgAatmCacAacGagGtaTcaAcaTtcTccmCccTg CECYP_C49C8.4_u363_LNA3 AtgmCtgAtgTtgAaaTtgmCtgGctAccGtaTtcmCaaAagAtamCtgTaaTc CECYP_C49C8.4_u883_LNA3 AtgAatmCcaTggmCttGgamCatmCtcmCcgTttTtcAagGgaTatAaaAatGt CECYP_C49G7.8_d6_LNA3 AtgmCaamCgaAttAgtGaaAaaTtcAtcmCtgGaaTaaAaaAtaAttmCtaAa CECYP_C49G7.8_u795_LNA3 AtcGctAcgAcaAtcTttmCcgAtgmCctTcgAagTttmCgaAagmCttTctmCt CECYP_F01D5.9_u374_LNA3 GagGtcGgtGgaGgaGgaAgtGgaAatTgamCggmCaaAatmCctGccmCaaGg CECYP_F01D5.9_u46_LNA3 mCccTctTtgGgaTttmCcamCtcAagTttActGttmCggmCagmCagTgaTatAa CECYP_F08F3.7_u25_LNA3 GagTtgGttmCcamCagAatGctTagGacGttTaaAttmCgtmCacAaamCttTt CECYP_F08F3.7_u401_LNA3 mCaaTatGgtTccmCatTttAgcAacTcaTatGaamCacAgaAgaTgtmCctTg CECYP_F14F7.2_u397_LNA3 GaaAaaGgcGtcGacAttTtaTgtGacAcgTggAcamCttmCacTatGacAa CECYP_F14F7.2_u68_LNA3 TaaTtgAatTacGggTctTttGtamCatAttAatTttAgtAtamCttTgtGa CECYP_F42A9.5_u435_LNA3 AtaTcaAtgmCaamCtaTtaAtgAatmCacAacGtcTtgmCcaAtcTtcTccmCg CECYP_F42A9.5_u55_LNA3 GgaGtgActAtgAaaGcaAagAgtTacmCgaTtgAaamCtgAaaGacAgamCa CECYP_K07C6.3_u3_LNA3 AatmCttTaaTgaTaaTttAtgGgaTctGtaTttmCtcTttmCtgTcaAtaAa CECYP_K07C6.3_u354_LNA3 AtgAgcmCcamCaaAtgTaaAagGatAcgAgaTtgAttmCggGaamCagTcaTg CECYP_K07C6.4_u118_LNA3 AtcmCtgmCgaTatGacAttAagmCcamCatGgtTctGaamCctTcaAcaGaaGa CECYP_K07C6.4_u87_LNA3 mCtgAacmCttmCaamCagAagAtaAacTtcmCgtAtaGcgmCtgGaaAaamCtcmCt CECYP_K07C6.5_u7_LNA3 AttTaaAggAatTcamCagmCtcAaaAaaTaaTaamCtamCcgGttmCagAgaTt CECYP_K07C6.5_u99_LNA3 AatTtgAgcmCacAtgGcaAgtTatmCaamCagAggAgamCaaTgcmCgtAcaGt CECYP_K09A11.3_u362_LNA3 TgamCatTctActTaaAggGaaGaaAatAccAacTggTacmCctTgtAttTg CECYP_K09A11.3_u48_LNA3 TcamCcamCaaAgcmCatAcaTatGcgAgcTagTtcmCtcAggmCtgmCttAaamCc CECYP_K09A11.4_u238_LNA3 TtcGacAaaActAttTtgGaaAgaAcaAtcmCcaTtcAgtGtcGgcAaamCg CECYP_K09A11.4_u68_LNA3 TctGacAacAaaGccAtamCacGtgmCcgActAatTccAcaAtcAgcTagAa CECYP_K09D9.2_u151_LNA3 TtgGcaAaaGcaGaaTtgTatTtaAtcTttGgaAacmCtcmCttmCttmCgcTa CECYP_K09D9.2_u866_LNA3 TgaAtcTttmCaaActTatmCacTccTttTaaTacTacmCgtTccTgtTtgGa CECYP_T1089.10_u410_LNA AttGagAttGtaTccAttGgcGtcTctTgtTcamCaaTcgAaaAtgTctmCa CECYP_T1089.10_u56_LNA AacTgcTacTatTgcGccAtcAagTgtGctGctmCaaActTaaAtcmCagGt CECYP_T1089.7_u102_LNA3 TtgAgamCagGaaAtaAgamCtaGaaTtcmCttTgaAacTggTggGaaGtgmCt CECYP_T1089.7_u267_LNA3 AagAtgTcaAagAatTcaAgcmCagAacGatGgtmCcamCcgAcgAgcmCatTa CECYP_T19810.1_u100_LNA3 AttGaamCcaActmCtgAaaTatAatGacAcaAaamCcaTgtmCtgGaaGtgGt CECYP_T19810.1_u319_LNA3 GgcAatGtgAcaAtaTctmCcaAtgGttmCttmCacAgcAatmCatmCacGtgTt CECYP_Y49C4A.9_u121_LNA3 mCtaTtcAatmCgaTatTttAtcAcamCcaTccAgtGctGgamCctmCcaTcaTt CECYP_Y49C4A.9_u413_LNA3 GtcTcaGagAtgTgtAaaTttActTccmCtgmCaaTttGttTcamCgcAacTa CECYP_ZK177.5_u394_LNA3 TtcmCgaAtgTttmCcaAttGggActGaaGttTcaAgaGtcAccmCagAaaAa CECYP_ZK177.5_u445_LNA3 GatmCcaGcaTctTccAagmCttAcaTtcmCtcmCgtGctTgtAtcAagGaaAc CEDAO_C47A10.5_d9_LNA3 TttGaaAacmCtgTttTatTatTaaAatAgaTaaTtgAttAgtTctGtamCg CEDAO_C47A10.5_u269_LNA3 AtamCgtTgcActGcaTccGgcTatGagGgaGccAaaAatmCttAggGgaGt CEDC_C01A2.3_u373_LNA3 GcamCttmCcaTtcAtcTctGcaGctActAtgGctTtgGtgAcaAaaGttGg CEDC_C01A2.3_u96_LNA4 mCcgTccAaaAgaAtgmCcaTctmCacAagTctTgaAatmCttAtaAagGtaGt CEDC_C34F6.1_u301_LNA3 GagGgaTcaAcaGtaAccTcgTgcGgtAttGacAagGgaTgtmCcgGaaGg CEDC_C34F6.1_u450_LNA3 GatGgtTctTcgAtcGcaAacAaaAcaGatGtgmCtcmCatTtamCatAcgGa CEDC_F33D11.3_u126_LNA3 AtgGagAaaAtgGatmCtgAtgGagTtgmCagGaaGtgAtgGagmCtcmCagGa CEDC_F33D11.3_u14_LNA3 TgaAtcTccAtaAatTatTcaAtgTttmCcaAatAttTaaTttAtcAatTg CEDC_F46E10.2_u392_LNA3 GctmCaamCacGgtAggAtcmCtaTggAacmCgtmCggAggAgcAggmCctmCggAg CEDC_F46E10.2_u54_LNA3 mCgtGacAacmCtcTtaTttAttTctGtaAaamCtgAttmCgcmCaaActTttGt CEDC_F56G4.2_u382_LNA3 GaaGctTtcAaamCcaAatGagTtcmCttmCccGgaAtcmCcaAagAatAccAa CEDC_F56G4.2_u82_LNA3 AcaAtgAaaAgaGagGatGgaAagGaaAtcGaaGtcTctGttmCttGacGa CEDC_M162.2_u103_LNA3 GatGagGtamCatAacTttGtgTgcAgtTatAggmCcaTctAcaGtamCctGc CEDC_M162.2_u480_LNA3 TtcmCatmCatmCacTaamCcgAttGtcmCtgAcaTtgAtgGccAaamCcaGggAa CEDC_R10E4.11_u274_LNA3 TcamCatTatmCgaAcaAgtActAgtAagmCatGctGtgAtgGagTgcmCgcTa CEDC_R10E4.11_u397_LNA3 mCacGgaGatmCacGacAtcAaaGcgGatTgcTtaGagTgtGgaAacmCgtmCt CEDC_T04C9.1_u321_LNA3 ActAtcTacGtgGcamCgtTggActmCatmCatmCgaTggGaamCgamCgtAtaAg CEDC_T04C9.1_u64_LNA3 TctmCtgGccAgtTcamCttTgtGatmCaaTctmCagAttmCgtmCcamCacAagAt CEDC_W02A2.3_u32_LNA3 mCtamCttmCcgmCaaGaaGgcmCcgTcgTttmCtaAtcGatmCgaAcaTctmCacAc CEDC_W02A2.3_u374_LNA3 AtgGatGatmCgamCccActTgcmCacTgamCccAcaAtcmCcgmCacTcamCtamCc CEDC_W05G11.3_u153_LNA3 AagAcgGagAggmCtgGagAgaAcgGtamCcgAtgGagAgcmCagGaamCtgAt CEDC_W05G11.3_u51_LNA3 mCcamCccAggAggAggGatAcaAgaGaaGaaAgtAcaGatTctmCcaActAa CEDC_ZK863.5_u256_LNA3 AgtTtcAcamCttmCttTttGccGttTtgGttmCccGttAtcAatmCcaTtgAt CEDC_ZK863.5_u324_LNA3 mCttTtaTatTctmCatmCaaTttGttTccTacTtgGtcAgcTgaGgaTcgTt CEEPHX_Y55B1BR.4_u161_LNA3 TtcGgcAcaAatGgaGcaAaaGtaTcgTggTtaTtgTgaTgcGatTatTc CEEPHX_Y55B1BR.4_u93_LNA3 mCtamCtaTgaAtgAgcTcamCtgGacTcaTttAtcAacTcgAgtmCaaAagmCc CEER_18S_u388_LNA3 GttGgcGaaTctTcgGgtTcgTatAacTtcTtaGagGgaTaaGcgGtgTt CEER_18S_u82_LNA3 GaamCtgAttmCgaGaaGagTggGgamCtgTcgmCttmCgaGgtTtaAcgActTc CEER_26S_u342_LNA3 TgtTatTgcGaaAgtAatmCctGctTagTacGagAggAacAgcGggTtcAa CEER_26S_u38_LNA3 TgcAtamCgamCttGgtmCtcTtgGtcAagGtgTtgTatTcaGtaGagmCagTc CEFOXO_R13H8.1b_u331_LNA3 TgtGctmCagAatmCcamCttmCttmCgaAatmCcaAttGtgmCcaAgcActAacTt CEFOXO_R13H8.1b_u393_LNA3 TtaAgamCggAacmCaaTtgmCtcmCacmCacmCatmCatAccAcgAgtTgaAcaGt CEGAPDH_K10B3.7_u21_LNA3 AcaTtgmCtamCcaAggmCctAagmCcgmCttmCaaAttmCtcTaaGtcTgaAatGa CEGAPDH_K10B3.7_u727_LNA3 GttGagTccAccGgaGtcTtcAccAccAtcGagAagGccAatGctmCacTt CEGBA_F11E6.1a_u232_LNA3 AgtAaaTtcmCttmCcamCgtGgaTctActmCgtGtgTtcAcaAagAtcGagGg CEGBA_F11E6.1a_u451_LNA3 GgtmCcaAtaAtgGgaGacTggTtcmCgcGcaGaaAgtTatGcaGatGatAt CEGLU_C02A12.1_u264_LNA3 AgaAaamCttmCgtTggAccmCtgmCtaAggAgaAgtAttTcaAgcTtcTgaGc CEGLU_C02A12.1_u55_LNA3 GagmcacmCcgAagmCtcAagmCcaTatTtgGaaAcaAgamCcaTacTctTcaAa CEGLU_C46F11.2_u271_LNA3 GttAccmCtcTacAaaTctmCgcTtcAatmCcaAtgTtgTtcGcaGtcAccAa CEGLU_C46F11.2_u45_LNA3 mCcgAagAgcTcgTtamCtaTgcGagGagGtgTgaAgcmCggAatAatTttTt CEGLU_F26E4.12_u109_LNA3 AagTtcTtgGttGgamCgcGatGggAaaAttAtcAagAgaTttGgamCcaAc CEGLU_F26E4.12_u480_LNA3 AcgAttTcaAcgTcaAaaAtgmCtaAtgGtgAtgAcgTgtmCacTttmCggAt CEGLU_R07B1.4_u166_LNA3 AccTggGttGatGttTttGcgGctGaaAgtTtcTccAagmCtcAttGatTa CEGLU_R07B1.4_u38_LNA3 GaaGtamCgtmCtcmCcaAagAaaAgcTacmCccAgcTtaAggmCatTgcAcaAt CEGLU_T09A12.2_u220_LNA3 GcgmCcaGatAtgTatTcaAagAtcGagGtaAatGgtmCagAacActmCatmCc CEGLU_T09A12.2_u335_LNA3 AatmCtamCagGgaAaaAggAttTcgAgtTgcmCgcGttTccAtgmCaaTcaAt CEGLU_T28A11.11_u299_LNA3 AgaTggmCaaAgaAgcAtamCatAacTgaAacTctTccmCggGgaGctActAc CEGLU_T28A11.11_u54_LNA3 TgaAtaAacGggmCcgAacTaaAtcmCatTcgTcaGtgGaaAtgGgaAacAa CEGPD_B0035.5_u256_LNA3 GtcmCgtmCttmCctGatGctTatGaamCgcmCtaTttmCtcGaaGtaTtcAtgGg CEGPD_B0035.5_u478_LNA3 TgtGgaAaaGctmCtcAacGagAagAaaGcaGaaGttmCgtAtamCaaTtcAa CEHSP_C09B8.6_d8_LNA3 AtaTcgmCcgmCctGctTccTcamCcaAccmCgaAtaAcgmCaamCaaAaamCttTa CEHSP_C0988.6_u286_LNA3 AagAgcmCcamCtcAtcAagGatGaaAgtGatGgaAagActmCttmCgtmCtcAg CEHSP_C12C8.1_u127_LNA3 mCaaGatAttTtaAcaAaaAtgmCatmCaamCaaGaaGccmCaaTcaGgtTccGg CEHSP_C12C8.1_u1531_LNA3 mCttGggmCatTctGtamCggGatGctGtcAttActGtgmCctGcaTatTttAa CEHSP_C47E8.5_u310_LNA3 AagAagmCatmCtcGaaAtcAacmCcaGacmCacGctAtcAtgAagAcamCttmCg CEHSP_C47E8.5_u361_LNA3 AtgAaaGctmCaaGctmCttmCgtGatTccTctActAtgGgaTacAtgGccGc CEHSP_F26D10.3_u276_LNA3 TtaAgcAgamCcaTtgAggAcgAgaAgcTcaAggAtaAgaTcaGccmCagAa CEHSP_F26D10.3_u397_LNA3 mCgtmCttTccAagGatGacAttGaamCgcAtgGtcAacGaaGctGagAaaTa CEHSP_F43D9.4_u169_LNA3 GtcGacTtgGctmCacAtcmCacAccGtcAtcAacAagGaaGgamCagAtgAc CEHSP_F43D9.4_u275_LNA3 mCaaTctTgaGggAcamCgtTctmCacmCatTgaGggAcamCcamCgaGgtmCaaGa CEHSP_F44E5.4/5_u123_LNA3 TcamCtaAaaTgcAccAatmCtgGacAatmCttmCtgmCttmCtgmCtgGatGcgmCt CEHSP_F44E5.4/5_u380_LNA3 TcaTgaAgcTaaAcaAttmCgaAaaGgaAgaTggTgaAcaAcgGgaAcgTg CEHSP_F52E1.7_u175_LNA3 AagTatAacmCttmCcaAcaGggGtcmCgtmCcaGaamCaaAtcAagTccGaaTt CEHSP_F52E1.7_u448_LNA3 TttAacmCatGgcmCgcAgaTtcTtcGatGacGtcGacTttGatmCgcmCacAt CEHSP_F54D5.8_u252_LNA3 GcgTcgAaaAgaTctmCccTgaAgtmCtgmCatTgamCtgGccTtgAtaTtaTg CEHSP_F54D5.8_u318_LNA3 AcaTagTctTcgTcaTcaAggAtaAgcmCacAccmCgaAatTcaAgcGagAg CEHUS_H26D21.1_u117_LNA3 TcgmCcaAcamCtcGgamCacGtgmCcaAaaTgaAtaTcaTctmCaaAtcGaaTg CEHUS_H26D21.1_u478_LNA3 GtcGaaGttAgaAatmCcaGaaGccGatAttGttTctmCatmCaaAttmCcaAt CEMRE_ZC302.1_u169_LNA3 ActActmCgtGgaAgaTccAatAaaGttGttTcaAcgmCgamCaaAtcGatTc CEMRE_ZC302.1_u292_LNA3 GgcAgtGaaGatGaaGtgGcaAatTctGatGaaGaaAtgGgaAgcAgtAt CEMTL_T08G5.10_d127_LNA3 TtgTcaAcgAccAgaAgcAaaAatTatGggAatmCgcGatAaaAttmCaaGg CEMTL_T08G5.10_u45_LNA3 GatGcaAgtGtgmCcaActGcgAatGtgmCtcAggmCtgmCtcAttAatTtgAa CENAP_D2096.8_u356_LNA3 GacGatAtgTtcGatTtcmCcaGgaGagGacGgtGatGatGtgTcaGacTt CENAP_D2096.8_u70_LNA3 GacGatAtgTtcGatTtcmCcaGgaGagGacGgtGatGatGtgTcaGacTt CEPAI_F56D12.5_u241_LNA3 GagGtcGtcGtaAtcmCacAagGctmCcaAgaAagmCaaGtgmCtcGacAttTc CEPAI_F56D12.5_u301_LNA3 GatActTttGgcAagmCtcGttmCcaAtcAagAagGagGtcAtcmCcaGatmCg CEPDI_C07A12.4_u28_LNA3 GatGagGagGgamCacAccGagmCtcTaaAtcmCacAttmCcaAtamCagTtcAa CEPDI_C07A12.4_u433_LNA3 mCttAtgTccGaaGatAtcmCcaGagGatTggGacAagAacmCcaGtcAagAt CEPDI_C1481.1_u119_LNA3 TacmCccAgtmCgamCtaTgaTggAgamCagAaamCctmCgaGaaGttmCgaAgaAt CEPDI_01481.1_u358_LNA3 mCtcGtcGccTccAacTtcAacGaaAttGccmCttGatGaaAccAagActGt CEPGK_T03F1.3_d9_LNA3 TtcTatTgtTtaTtcmCttGccmCaaTagTgtAttTgtAttTatTctTtcTc CEPGK_T03F1.3_u424_LNA3 mCaaAtcmCatmCtomCcaGtgGatTtcGtcAttGctGacAagTtcGccGagGa CEPON_E01A2.7_u223_LNA3 GttTctGatTcgAcamCttTatGgamCcaTctmCaaGttmCtgmCgaGttTctTt CEPON_E01A2.7_u79_LNA3 GggAaamCaaAtgAttGttGgtAcaGtaGccmCgcmCctGctAttmCacTgtGa CEPPGB_F13D12.6_u44_LNA3 mCgaGcamCatmCatmCcaAtcGttmCctGttmCaamCaaGgcmCttmCtaAtcGttAg CEPPGB_F13D12.6_u440_LNA3 TgaTgaGagmCccAgtAacmCaaTtaTttGaamCcgTcaGgaTgtGcgTaaGg CEPPS_T14G10.1_d2_LNA3 mCgtmCtaAtcGaaGaaGggGatmCgtGggmCaaTcaTaamCtaAttAacmCttmCa CEPPS_T14G10.1_u240_LNA3 mCaaTggmCtcmCagGtcTttmCtgmCtcTtcAtaTacTtcmCatTccGagTtgmCt CEPRDX_R07E5.2_u405_LNA3 GttmCtcTtgGagmCtgAagTtgTcgmCgtGctmCgtGtgAttmCtcActTctmCt CEPRDX_R07E5.2_u42_LNA3 TcgmCtamCcaGcaAggAatActTcaAcaAggTcaAcaAgtGatmCacAcaGa CEPYC_D2023.2_u256_LNA3 AagGaaAttGtaActmCgcmCcaAgaGctmCtcmCcaGgtGtcmCgtGgamCatAt CEPYC_D2023.2_u427_LNA3 TtgActGgaTtgGagAttGcgGaaGaaGttGatGttGaaAtcGagAgtGg CERAD_F10G7.4_u169_LNA3 GccAagTctmCaaGcaAtaAgtGttGatmCaaTcaGagmCcaTacGgaGagAt CERAD_F10G7.4_u267_LNA3 AtaTtgAgamCttmCggGacAagmCggActTctmCatmCtgTcamCagmCaamCtgmCc CERAD_F32A11.2_u250_LNA3 GatmCcgmCagAgaAtcGagTatTtcmCtcTcgAgamCccAtgGatAtcAacTg CERAD_F32A11.2_u380_LNA3 TccGttAagAagmCtcActGgaAaaAcamCacGgcTcgAacGaaAttGgaAt CERAD_T04H1.4_u274_LNA3 AatTtgGatGagAgcAaaGtgGaaGgaAtgGctAtcGttTtgGcaGatAt CERAD_T04H1.4_u375_LNA3 GtgmCtgGtcAaaAaaTgcTtgmCttmCgtTgcTtaTtcGcaTtgmCacTcgmCa CERAD_W06D4.6_u325_LNA3 mCttmCgaGaamCtcTtcAagTtgGaaTcaAcaGtgGcaTcgGatAcamCatGa CERAD_W06D4.6_u34_LNA3 GtgmCctTctGaaGccGaaGaaAacGacGatTagTtaAatGttTccAagTt CERAD_Y116A8C.13_u289_LNA3 GatAaaAtcGatAgcGacGacGatGagGaaGccGatGatGagGagmCtcGa CERAD_Y116ABC.13_u59_LNA3 GcaGgtGgaTacGgaTgtGgaGctGacTttTgcGttTtaTcaAgaAtcTc CERAD_Y39A1A.23_u221_LNA3 TccmCgtAgaAgtAgaAatGctAgaAgaAccTgaAcaAgaAgaTcaAgaAa CERAD_Y39A1A.23_u276_LNA3 TgcAagAtgTcaGtaTtgAaamCaaTtcmCtgTagAgamCccmCcgAagAaaAt CERAD_Y41C4A.14_u509_LNA3 AgtmCtcGtaTccGggAatGttTcaGccTgtGaaAatGctTgtTgaAgamCg CERAD_Y41C4A.14_u731_LNA3 mCttmCaaAacmCgtmCgcTttTaaGgaTacAggAacGtgGcamCgcTtcmCgaGg CERAD_Y43C5A.6_u131_LNA3 mCagAttGtamCctTcgAaaAggAaaAggAgaGaaTcgmCgtmCgcAaaAatGg CERAD_Y43C5A.6_u429_LNA3 TgaTggmCttTgaTtaTtcGagmCagGagmCaaTgaTgtmCcgAgaGtcGttAt CERFC_F31E3.3_u128_LNA3 mCaaTgamCgaGaaTatTggAgtAatGggGaaActGgtTgcGacTtgmCgaAa CERFC_F31E3.3_u55_LNA3 TtgGaaAacAatmCtcmCtcGacTttmCtgmCtcActmCttmCgtGaaActAtcmCa CERPL_K11H12.2_d1_LNA3 TctTgtTatTttAttTtgTttTggGctTgtTccGaaAatGaaAtgGttGt CERPL_K11H12.2_u172_LNA3 mCaaTggAtcAccAagmCcaGttmCacAagmCacmCgtGagmCaaAgaGgamCtcAc CERT_F36A4.7_u1396_LNA3 mCttTgtGatGtgAtgActGcgAagGgamCacTtgAtgGctAttAcgAgamCa CERT_F36A4.7_u2302_LNA3 GagmCcaGctActmCagAtgAcamCtcAacAcgTtcmCatTatGcaGgaGttTc CERT_F36A4.7_u289_LNA3 TacActmCcaTccTcgmCcgAcaTacAatmCcaAcaTctmCcamCgcGgaTtcTc CERT_F36A4.7_u2919_LNA3 AtgGagAagAtgGttTggAtgGaaTgtGggTtgAgaAtcAgaAtaTgcmCg CERT_F36A4.7_u4269_LNA3 AacmCggGatAccGtgTcgAacGtcAcaTgaAagAtgGcgAtaTaaTcgTc CERT_F36A4.7_u5485_LNA3 GagGagAttAaamCgcAtgTcaGtgGctmCatGtcGagTttmCcaGaaGtcTa CESLC_F52F12.1a_u249_LNA3 AgaTatTgcmCtcTacTtaTcaTggGccTgaTggmCttTgtmCtgmCcgGtaTt CESLC_F52F12.1a_u76_LNA3 GaaTctmCaamCcamCttmCtgGaamCccmCatAcamCcaAtgGatAgaAgamCggAg CESLC_K11G9.5_u400_LNA3 GttGttmCttTttTccGtgAtcTttTcaTgtTtaTgtmCtgAacGtgGcaGg CESLC_K11G9.5_u462_LNA3 GacTcgTtgGtgTctTgcTagGatGtcTtgGgtTcaTtcmCtcAatmCgtTg CESLC_Y32F6B.1_u179_LNA3 GtamCtgGgcTcgAggGctGaaActAatmCgaAgaAgaAacTccAgaAgaTa CESLC_Y32F6B.1_u280_LNA3 GgaTcaTgcTctGttTacGacActGatGagTtaAgaGtcAgamCtgmCacGt CESLC_Y37A1C.1a_u104_LNA3 mCgaTggTtcTtcTcgTctAtcAtaTcgGggTagTtgmCcgAagTgtTgaAa CESLC_Y37A1C.1a_u404_LNA3 mCaaAtcGaamCtgGtaTaaAggAggAccGacGgaGacGaaTttGaamCgaGa CESLC_Y70G10A.3_u383_LNA3 AttmCgaTcaAagAacTctGgcTctmCggmCgtTaamCtgGacAttTgtTcgTc CESLC_Y70G10A.3_u46_LNA3 mCtcmCccGagmCagGcgAttAttmCacGctAgtTatGctmCaaAtgTgaTctGt CESOD_C15F1.7_u435_LNA3 mCcgGtamCtaTctGgaTcamCacAgaAgtmCcgAaaAtgAccAggmCagTtaTt CESOD_C15F1.7_u9_LNA3 mCccAgtGacTacmCtgAatmCgcGtcTctGaaTctmCcamCacAatTccTacTa CESOD_F10011.1_u326_LNA3 GgaGttGctmCacmCgcAatTaaGagmCgamCttmCggAtcTctGgaTaaTctTc CESOD_F10011.1_u477_LNA3 AaaTtgAggAaaAgcTtcAcgAggmCggTctmCcaAagGaaAcgTcaAagAa CESULT_EEED8.2_u316_LNA3 mCaaTcgTacmCatGaaAgaAgtTggAagmCcamCgtGcaAgaGaaGaaAtcmCa CESULT_EEED8.2_u82_LNA3 AagAagAttmCctGacmCagAgaGacTcamCgtGctTacmCcaAgaAgcAtcTa CESULT_Y113G7A.11_u252_LNA3 AgcAttGgtGgaAatAcgAaaTggmCatGggAagAgaAacmCccTctmCaaTt CESULT_Y113G7A.11_u96_LNA3 mCtgGttAcgGtaGtgTatGgtmCccTgtmCctmCtcAgaAtgmCaaAtaTgtmCg CESULT_Y67A10A.4_u108_LNA3 TctAcgTcgAtgGaaAagmCcgAttTaamCaaTcaAagmCcaAcaAcgmCagTt CESULT_Y67A10A.4_u327_LNA3 GgaAagGtgmCcaAaaAgtTgamCagmCaaTtgGagGatmCttAttmCatTgcmCa CETOPO_K12D12.1_u398_LNA3 AgaTgaTgaTgaAgtTccTgcAaaGaaGccTgcTccAgcGaaGaaAgcTg CETOPO_K12D12.1_u449_LNA3 AaaAccTcgTacTggAaaAggAgcTgcGaaAgcGgaAgtTatmCgaTttGt CETOPO_M01E5.5b_u256_LNA3 GagAagGccmCagAagAagTacGacAgamCtgAagGagmCagTtgAaaAagTt CETOPO_M01E5.5b_u429_LNA3 TtcTgtmCatAcaAtcGtgmCtaAtcGgcAggTtgmCgaTccTttGtaAccAt CEUbi_F25B5.4_u186_LNA3 AagmCttmCggAcamCcaTtgAgaAtgTcaAagmCcaAaaTccAggAtaAggAg CEUbi_F25B5.4_u2_LNA3 AatmCgaAccmCatmCaaTtcActmCgtTatTccTccTcgAtcTccGttmCaaGt CEUbi_F29B9.6_u145_LNA3 mCtgAacmCatmCcaAatAttGaaGatmCcaGctmCagGctGaaGccTatmCagAt CEUbi_F29B9.6_u230_LNA3 mCgtGtgmCttAtcTctTctGgaTgaAaamCaaGgaTtgGaaGccGtcAatmCt CEUbi_M7.1_u239_LNA3 mCggAagmCatmCtgmCctTgamCatTctmCcgTtcGcaGtgGtcGccGgcTctG CEUbi_M7.1_u53_LNA3 AaaGtamCgcTatGtgAggAggmCtaAcamCcaTtcAtaTaaGaamCgcAgcmCa CEUGT_F39G3.1_u40_LNA3 TgtTgcmCgtAgaAgaGagActAaaActAagAacGatTgaTtgAagGtcTg CEUGT_F39G3.1_u466_LNA3 TacAatTctTtgmCagGaaGcaAtaTccGccGgaGtcmCccmCttAtcActAt CEUGT_M88.1_u480_LNA3 mCtcAcgGagGttAtaAttmCtaTgcAggAggmCaaTttmCtgmCtgGagTtcmCa CEUGT_M88.1_u72_LNA3 AccGttTcaTgaGagmCtgTaaTcaGgtGttGttTctGtaAaaAgtGtgAa YAL009W_u145_LNA3 GtgGatGtgAaaTtaGtcmCtcAacmCccAgaGcaTttAgtGcaGagAttAg YAL009W_u341_LNA3 GcaGttTaaTgtGaaGctAgtTaaAgtAcaGtcTacGtgGgamCgaGaaAt YAL059W_u262_LNA3 AttGccAagTccAttTctmCgtGccAagTacAttmCaaAatAcaAgaAagGc YAL059W_u51_LNA3 AgamCtcmCtamCaaAtaGatTcgGtgTccTgcmCagAcgAtgTtgAagAatAg YER109C_u109_LNA3 TtgAagTttGggAatAttGgtAtgGttGaaGacmCaaGgamCcgGatTacGa YER109C_u436_LNA3 GagGcgmCaaGtaGgcAatGatTcaAgaAgtAgtAaaGgcAatmCgtAacAc YHR152W_u128_LNA3 TgaGcamCaaAgtTaaGatGttmCggAaaGaaAaaGaaAgtmCaaTccTatGa YHR152W_uS10_LNA3 mCaaGtgAccAatmCagmCacGcamCggmCttmCcaTccTcaAgamCtgAtaTtamCc YKL130C_u211_LNA3 AttAaaTgcGcaGatGagGacGgaAcgAatAtcGgaGaaActGatAatAt YKL130C_u85_LNA3 GatGgtAagmCtgAgcGccTtgGacGaaGaaTttGatGttGtcGctActAa YKL178C_u199_LNA3 TacGtcAcgmCaaGgamCagAgcTttGacGacGaaAtaTcamCttGgaGgaTt YKL178C_u367_LNA3 TctmCccTgtGtaGgtAcamCcaAtaTcamCaaGcgmCatTtcTatGtcGacTa YLR443W_u179_LNA3 TgcTaamCacmCagTttAgamCcaTggAaaTccmCacmCgcAaaTatAagmCaaTg YLR443W_u86_LNA3 GcaGgamCatAagAttmCcgGtcAagmCaamCgamCagTgaAgaAagTatGcaAa YOR092W_u251_LNA3 mCcgTctAgtGaaAgcGggAtgGctAaaTtgGgaAaamCgamCaaGatGttAt YOR092W_u82_LNA3 GatGctTcaAtaTccTttGatGgtmCgtTagTttAccAttTttGgtGtcTt YPL263C_u132_LNA3 AgtmCatTtgAgtTatGtgAagAccGttGgtGggAaaGaaGagAtcAggTg YPL263C_u257_LNA3 GtcTtgGctAccAcamCccAaaAccGttmCgaAacTttAagAgcAttmCtamCt - Locked Nucleic Acids (LNA) constitute a novel class of DNA analogues that have an exceptionally high affinity towards complementary DNA and RNA. Using human classical satellite-2 repeat sequence clusters as targets, we demonstrate that LNA/DNA mixmer oligonucleotides are excellent probes for FISH combining high binding affinity with short hybridization time and even with the ability to hybridize without prior termal denaturation of the template. The development of molecular probes and image analysis has made fluorescence in situ hybridization (FISH) a powerful investigative tool. Although FISH has proved to be a useful technique in many areas, it is a fairly time-consuming procedure with limitations in sensitivity. Probes with higher DNA affinity may potentially reduce the time needed for hybridization and the sensitivity of the technique. Thus, improvement in hybridization characteristics has been reported for the DNA mimic peptide nucleic acid (PNA). This example describes the development of LNA substituted oligonucleotides as probes for fluorescence in situ hybridization on metaphase chromosomes and interphase nuclei. In each experiment a different LNA substituted oligonucleotide of the same 23-bp human satellite-2 repeat sequence (attccattcgattccattcgatc) have been used, cf. Jeanpierre, M. (1994).
Human satellites - A1. Chromosome Preparations
- Chromosome preparations were made by standard methods from peripheral lymphocyte cultures of normal males. Slides were prepared 1-4 days prior to an experiment and treated with RNAse (10 μg/μl) at 37° C. for one hour before hybridization.
- A2. Probe Preparation
- The 23 bp human satellite-2 repeat sequence, attccattcgattccattcgatc, was used to prepare the LNA/DNA mixmers with different content and sequence order of LNA modifications (Table 19). All mixmers were labeled in the 5′ end with either Cy3 or biotin. Biotin amidite was purchased from Applied Biosystems and Cy3 amidite was purchased from Amersham Bioscience. A DNA oligonucleotide of the same sequence without any LNA modifications was used as a control in each experiment.
- A3. Fluorescence in Situ Hybridization
- FISH was carried out as described by Silahtaroglu A N, Hacihanefioglu S, Guven G S, Cenani A, Wirth J, Tommerup N, Tumer Z.(1998) Not para-, not peri-, but centric inversion of
chromosome 12. J Med Genet. 35(8):682-4. (1); with the following modifications including: The amounts of probe were 6.4, 10, 13.4 and 20 pmoles. Denaturation of the target DNA and the probe were performed at 75° C. for 5 minutes either separately using 70% formamide or simultaneously under the coverslip in the presence of the hybridization mixture containing 50% formamide. In addition the effect of denaturation was also tested. Two alternative hybridization mixtures were used: 50% formamide/2×SSC (pH 7.0)/10% dextran sulphate or 2×SSC (pH 7.0) /10% dextran sulphate. Hybridization times included 30 min, 1 hr, 2 hrs, 3 hrs and overnight. Hybridization temperatures included: 37° C., 55° C., 60° C. and 72° C. Post washing was either as for standard FISH (1), or with 50% formamide/2×SSC at 60° C., or without formamide. Hybridization signals with biotin labeled LNA substituted oligonucleotide probes were visualized indirectly using two layers of fluorescein-labeled avidin (Vector Labs) linked by a biotinylated anti-avidin molecule, which amplified the signal 8-64 times. The hybridization of Cy3 labeled molecules however, was visualized directly after a short washing procedure. Slides were mounted in Vectashield (Vector Laboratories) containing 4′-6′-diamidino-2-phenylindole (DAPI). The whole procedure was carried out in the dark. The signals were visualized using a Leica DMRB epifluorescence microscope equipped with a SenSys charge-coupled device camera (Photometrics, Tucson, Ariz.), and IPLAB Spectrum Quips FISH software (Applied Imaging international Ltd, Newcastle, UK) within two days after hybridization. - Satellite-II DNA, composed of multiple repeats of a 23 bp and a 26 bp sequence, is especially concentrated in the large heterochromatic regions of
human chromosomes chromosomes 9, Y, 15 and in other minor sites like the short arms/satellites of the acrocentric chromosomes and some centromeric regions. Classical satellite DNA can be visualised by FISH with traditional genomic and DNA oligonucleotide probes (see Kokalj-Vokac N, Alemeida A, Gerbault-Seureau M, Malfoy B, Dutrillaux B. (1993) Two-color FISH characterization of i(1 q) and der(1; 16) in human breast cancer cells. Genes Chromosomes Cancer. 7, 8-14; and Tagarro I, Femandez-Peralta A M, Gonzales-Aguilera J J. (1994) Chromosomal localization ofhuman satellites chromosome 1 was always stronger and appeared earlier, followed by signals onchromosomes - B1. Effect of LNA Content of the LNA Substituted Oligonucleotides (LNA/DNA Mixmers)
- The LNA-2 molecule which had every other nucleotide modified as LNA. (aTtCcAtTcGaTtCcAtTcGaTc (SEQ ID NO:)) gave the best results in all the experiment performed. The LNA-3 molecule, with every third oligonucleotide modified as LNA, (aTtcCatTcgAtTccAttCgaTc (SEQ ID NO:)) also gave hybridization signals, but with less efficiency than the LNA-2 probes. Preferably, an LNA-2 oligonucleotide molecule has an LNA unit at every other nucleotide position in the sequence and an LNA-3 oligonuclotide molecule has an LNA unit at every third position of the sequence. However, minor deviations, e.g. in one position or less than 5-10 percent of the nucleotide positions in the sequence may still provide the general features of an LNA-2 or an LNA-3 molecule.
- The Dispersed LNA (aTtccatTcgaTtccAttcgaTc (SEQ ID NO:)), which had 5 dispersed LNA modifications, was less efficient in short term hybridization, but gave signals on both
chromosomes - B2. Effect of Amount of the LNA/DNA Mixmers
- The initial experiments performed with 20 pmol of LNA/DNA mixmer resulted in bright and large signals, but with an extremely high background. Thus, lower concentrations were tested (13.4 pmol, 10 pmol and 6.4 pmol). The concentration giving the optimal signal to noise ratio was found to be 6.4 pmol.
- B3. Effect of Denaturation
- The signals on the major sites of hybridization (1q, 16q) were equally bright after both types of denaturation. However, smaller and weaker signals were observed on the minor sites with the simultaneous denaturation protocol. To check the potential “strand invasion” property of LNA, some of the experiments were performed without a denaturation step. As expected, no signals were obtained by the control DNA oligonucleotide probe. In contrast, hybridization signals on
chromosomes - B4. Effect of Hybridization Time, Temperature and Post-Hybridization Washes
- Although signals could be observed after only 30 min of hybridization, the optimal hybridization time and temperature for LNA-2, which gave the best signals, was 1 hr at 37° C. A 3×5 min wash with 0.1×SSC/60° C. and 4×SSC/0.05% Tween/37° C., respectively, followed by a 5 min PBS wash was found to be sufficient for washing the slides after hybridization with DNA-LNA mixmers. There was no specific difference between a wash with 50% formamide at 42° C. or 60° C.
- The signals faded away in most of the slides within two days. When hybridized with directly labeled LNA, the whole slide was stained with Cy3 after three days. Thus, slides had to be analyzed within 48 hours after hybridization.
- The experiments have demonstrated, that LNA substituted oligonucleotides are very efficient FISH probes. LNA substituted oligonucleotide probes gave strong signals after only 1 hr of hybridization, and it was possible to omit the use of formamide both from the denaturation and from the post hybridization washing steps and still obtain a very good signal to noise ratio. The ability of LNA to hybridize without prior denaturation could be due to a strand invasion property of LNA and this warrants further investigation with other LNA probes and at different conditions.Based on the combined results of these experiments, the optimal LNA-FISH procedure was defined as follows: 6.4 pmoles of Cy-3 labeled LNA-2 probe was denatured together with the target at 75° C. for 5 minutes, and hybridized for one hour then followed by a short post wash without any formamide (3×5 minutes 0.1×SSC at 60° C.; 2×5
minutes 4×SSC/0.05% Tween at 37° C.; 5 minute PBS). The FISH experiments indicate that LNA containing probes would be valuable for the detection of a variety of other repetitive elements, such as centromeric α-repeats and telomeric repeats. In addition, the superior hybridization characteristics of LNA containing oligonucleotides could lead to detection of single base pair differences between repetitive sequences as well as single copy sequences. - C1. FIG. 41 shows a comparison of different LNA/DNA mixmer oligonucleotides. Experiment conditions: 6.4 pmoles of Cy3 labeled probe was hybridized for 30 minutes at 37° C., after simultaneous denaturation of the target and the probe at 75° C. for 5 minutes. A. LNA-2 giving signals on
chromosomes chromosomes chromosomes chromosome 1, E. DNA oligo giving no signals on any of the chromosomes.TABLE 19 DNA/LNA mixmers for human satellite 2repeat sequence used in this study. (SEQ ID NOs:) LNA Name FISH probe sequences monomers Tm* DNA oligo attccattcgattccattcgatc 0 60 Dispersed LNA aTtccatTcgaTtccAttcgaTc 5 71 LNA-3 aTtcCatTcgAtTccAttCgaTc 8 77 LNA Blocks aTTCcattcgATTccattcGATc 9 73 LNA-2 ATtCcAtTcGaTtCcAtTcGaTc 11 84 - LNA modifications are depicted in capital letters and *Tm values for each molecule have been calculated using Exiqon's Tm Prediction program accessible at http://lna-tm.com/ and as appears in FIGS. 19A-19F herein.
- 1. Chromosome Preparations
- Chromosome preparations were made by standard methods from peripheral lymphocyte cultures of two normal males. Slides were prepared 1-6 days prior to an experiment and treated with (10 μg/μl) at 3 7° C. for one hour before hybridization.
- 2. FISH probe preparation
- A Cy3-labelled, LNA-2 design of the 24-bp telomere sequence (ttagggttagggttagggttaggg) representing 4 blocks of 6-bp telomere repeat (ttaggg) was used as a probe. A DNA oligomer of the same sequence without any LNA modifications was used as a control in each experiment.
- 3. Fluorescence in situ hybridization
- FISH was carried out as described previously (Silahtaroglu et al, 1998) with the following modifications. The amount of probe was 5 pmoles. Denaturation of the target DNA and the probe were performed at 75° C. for 5 minutes simultaneously under the coverslip in the presence of hybridization mix containing 50% formamide. Slides were washed after 30 min. hybridization at 37° C. Post washing steps included a 2×5 min 0.1×SSC at 60° C.; 5
min 2×SSC at 37° C.; 3min 4×SSC/0.05% Tween20 at 37° C. and 5 min PBS. Slides were mounted in Vectashield (Vector Laboratories) containing 4′-6′-diamidino-2-phenylindole (DAPI). The whole procedure was carried out in the dark. The signals were visualized using a Leica DMRB epifluorescence microscope equipped with a SenSys charge-coupled device camera (Photometrics, Tucson, Ariz.), and IPLAB Spectrum Quips FISH software (Applied Imaging international Ltd., Newcastle, UK). - 4. Results
- The human telomere repeat specific LNA oligonucleotide probe gave prominent signals on the telomeres, when used as a FISH probe (FIG. 42), whereas no signals could be detected with the corresponding DNA control probe when using the hybridization conditions specified above. Thus, the experiments described here for human telomere repeat, demonstrates that LNA substituted oligonucleotides are highly efficient as FISH probes.
- 5. References
- Silahtaroglu, A.N., Hacihanefioglu, S., Guven, G.S., Cenani, A., Wirth, J., Tommerup, N., Tumer, Z.(1998) Not para-, not peri-, but centric inversion of
chromosome 12. Journal of Medical Genetics 35(8), 682-684. - 1. Chromosome Preparations
- Chromosome preparations were made by standard methods from peripheral lymphocyte cultures of a normal female. Slides were prepared 5 days prior to use. Before use slides were treated with RNAse A (10 μg/μl) at 37° C. for one hour and proteinase K for 10 minutes washed, with 2×
SSC 3times 3 min, before dehydration through a cold ethanol series. - 2. Probe Preparation
- A 5′ biotin-labelled, LNA substituted 15-mer FISH probe (aCcCaGcCaAaGgAg (SEQ ID NO:), LNA uppercase, DNA lowercase) and a 5′ biotin-labelled LNA substituted 24-mer FISH probe (TgTgTaCcCaGcCaAaGgAgTtGa (SEQ ID NO:), LNA uppercase, DNA lowercase) specific for the centromeric
human chromosome 21 alphaRI(680) locus alpha-satellite repeat were used as probes. Biotin-labelled DNA probes of the same sequence without any LNA modifications were used as controls in each experiment. - 3. Fluorescence in situ hybridization
- FISH was carried out as described previously (Silahtaroglu et al, 1998) with the following modifications. The amount of probe was 11 M for 1 the 15-
mer chromocsome 21 FISH probe and 1.4 μM for the 24.mer FISH probe. Denaturation of the target DNA and the probe were performed simultaneously at 79° C. for 4 minutes under the coverslip in the presence of hybridization mix containing 50% formamide. Slides were washed after 40 min. hybridization at RT. Post washing steps included a 2×5 min 0.1×SSC at 65° C.;Smin 3min 4×SSC/0.05% Tween20 at 37° C. Slides are then incubated 10 min with 1% blocking reagent and a layer of Flourescein conjugated Avidin (Vector Labs) has been applied for 20 minutes at 37° C. After a 3times 3 minute wash with 4×SSC/0.05% Tween20, slides are dehydrated and mounted in Vectashield (Vector Laboratories) containing 4′-6′-diamidino-2-phenylindole (DAPI). The whole procedure was carried out in the dark. The signals were visualized using a Leica DMRB epifluorescence microscope equipped with a SenSys charge-coupled device camera (Photometrics, Tucson, Ariz.), and IPLAB Spectrum Quips FISH software (Applied Imaging international Ltd., Newcastle, UK). - 4. Results
- The LNA substituted 15-mer oligonucleotide probe specific for the centromeric
human chromosome 21 alphaRI(680) locus alpha-satellite repeat gave prominent signals onchromosome 21, when used as a FISH probe, whereas no signals could be detected with the corresponding DNA control probe when using the hybridization conditions specified above. The LNA substituted 24-mer oligonucleotide probe specific for the centromeric alphaRI(680) locus alpha-satellite repeat gave prominent signals both onchromosomes chromosomes human chromosome 21, demonstrates that LNA substituted oligonucleotides are highly efficient as FISH probes and can be used in diagnosis ofchromosome 21 trisomy. - Other Embodiments
- From the foregoing description, it will be apparent that variations and modifications may be made to the invention described herein to adopt it to various usages and conditions. The foregoing description of the invention is merely illustrative thereof, and it understood that variations and modifications can be effected without departing from the scope or spirit of the invention.
- All publications, patent applications, and patents mentioned in this specification are herein incorporated by reference to the same extent as if each independent publication, patent, or patent application was specifically and individually indicated to be incorporated by reference.
- Other embodiments are in the claims.
-
1 852 1 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 1 acggggatta tggtttcgcc aatgaaaact aatcaaaggt 40 2 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 2 acggggatta tggtttcgcc tatgaaaact aatcaaaggt 40 3 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 3 acggggatta tggtttcgcg tatgaaaact aatcaaaggt 40 4 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 4 acggggatta tggtttcggg tatgaaaact aatcaaaggt 40 5 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 5 acggggatta tggtttcggg tttgaaaact aatcaaaggt 40 6 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 6 acggggatta tggtttcggg ttagaaaact aatcaaaggt 40 7 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 7 acggggatta tggtttcgnc aatgaaaact aatcaaaggt 40 8 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 8 acggggatta tggtttcgnc tatgaaaact aatcaaaggt 40 9 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 9 acggggatta tggtttcgng tatgaaaact aatcaaaggt 40 10 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 10 acggggatta tggtttcggg tatgaaaact aatcaaaggt 40 11 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 11 acggggatta tggtttcggg tttgaaaact aatcaaaggt 40 12 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 12 acggggatta tggtttcggg ttagaaaact aatcaaaggt 40 13 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 13 tgggaatgga acggggatta tggtttcgcc aatgaaaact aatcaaaggt 50 14 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 14 tgggaatgga acggggatta tggtatcgcc aatgaaaact aatcaaaggt 50 15 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 15 tgggaatgga acggggatta tggtaacgcc aatgaaaact aatcaaaggt 50 16 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 16 tgggaatgga acggggatta tggtaaggcc aatgaaaact aatcaaaggt 50 17 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 17 tgggaatgga acggggatta tggaaaggcc aatgaaaact aatcaaaggt 50 18 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 18 tgggaatgga acggggatta tggaaagccc aatgaaaact aatcaaaggt 50 19 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 19 tgggaatgga acggggatta tggtttngnc aatgaaaant aatcaaaggt 50 20 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 20 tgggaatgga acggggatta tggtttcgcc aatgaaaact aatcaaaggt 50 21 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 21 tgggaatgga acggggatta tggtttcgnc aatgaaaact aatcaaaggt 50 22 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 22 tgggaatgga acggggatta tggtttcgcc aatgaaaact aatcaaaggt 50 23 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 23 tgggaatgga acggggatta tggtttcgcc aatgaaaact aatcaaaggt 50 24 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 24 tgggaatgga acggggatta tggtatcgcc aatgaaaact aatcaaaggt 50 25 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 25 tgggaatgga acggggatta tggtaacgcc aatgaaaact aatcaaaggt 50 26 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 26 tgggaatgga acggggatta tggtaaggcc aatgaaaact aatcaaaggt 50 27 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 27 tgggaatgga acggggatta tggaaaggcc aatgaaaact aatcaaaggt 50 28 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 28 tgggaatgga acggggatta tggaaagncc aatgaaaact aatcaaaggt 50 29 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 29 ttgctgaact ggatggatta aaccgtatgg gtccaacttt 40 30 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 30 ttgctgaact ggatggattt aaccgtatgg gtccaacttt 40 31 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 31 ttgctgaact ggatggatat aaccgtatgg gtccaacttt 40 32 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 32 ttgctgaact ggatggatat taccgtatgg gtccaacttt 40 33 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 33 ttgctgaact ggatggatat ttccgtatgg gtccaacttt 40 34 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 34 ttgctgaact ggatggatat ttgcgtatgg gtccaacttt 40 35 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 35 ttgctgaact ggatggatta aancgtatgg gtccaacttt 40 36 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 36 ttgctgaact ggatggattt aancgtatgg gtccaacttt 40 37 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 37 ttgctgaact ggatggatat aancgtatgg gtccaacttt 40 38 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 38 ttgctgaact ggatggatat tancgtatgg gtccaacttt 40 39 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 39 ttgctgaact ggatggatat ttncgtatgg gtccaacttt 40 40 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 40 ttgctgaact ggatggatat ttgcgtatgg gtccaacttt 40 41 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 41 ggtatggaag ttgctgaact ggatggatta aaccgtatgg gtccaacttt 50 42 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 42 ggtatggaag ttgctgaact ggatcgatta aaccgtatgg gtccaacttt 50 43 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 43 ggtatggaag ttgctgaact ggatccatta aaccgtatgg gtccaacttt 50 44 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 44 ggtatggaag ttgctgaact ggaaccatta aaccgtatgg gtccaacttt 50 45 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 45 ggtatggaag ttgctgaact ggaaccttta aaccgtatgg gtccaacttt 50 46 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 46 ggtatggaag ttgctgaact ggaacctata aaccgtatgg gtccaacttt 50 47 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 47 ggtatggaag ttgctgaant ggatggatta aacngtatgg gtncaacttt 50 48 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 48 ggtatggaag ttgctgaant ggatngatta aacngtatgg gtncaacttt 50 49 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 49 ggtatggaag ttgctgaant ggatncatta aacngtatgg gtncaacttt 50 50 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 50 ggtatggaag ttgctgaant ggaancatta aacngtatgg gtncaacttt 50 51 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 51 ggtatggaag ttgctgaant ggaancttta aacngtatgg gtncaacttt 50 52 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 52 ggtatggaag ttgctgaant ggaanctata aacngtatgg gtncaacttt 50 53 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 53 ggctggatnc ccaggaaacc naggaatcgg aagcattgga ncaaaaggag 50 54 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 54 naccggatnc ggntcaattg tcggacctng cggaaancct ggagaaaagg 50 55 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 55 tccgncaggc ccaatcgcct ncacnatgtc caagggaacc attatcggtc 50 56 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 56 gagcnaggag agggaggtca acgcggttac ccaggaaatg gaggactctc 50 57 31 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 57 gatcgaattc ctccaggaga gaagggagat g 31 58 33 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 58 gatcaagctt atctcttcct gggtatccag ctt 33 59 69 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 59 gctggaacag aagtttgttg gtgcgtgaca aggtatggaa gaagattatc cggaaaagaa 60 agcaaagac 69 60 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 60 ggctggaaca gaagtttgtt ggtgcgtgac aaggtatgga agaagattat 50 61 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 61 ggctggaaca gaagtttgtt ggtgcgtgac aaggtatgga 40 62 30 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 62 gaacagaagt ttgttggtgc gtgacaaggt 30 63 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 63 ggctggaana gaagtttgtt ggtgngtgac aaggtatgga agaagattat 50 64 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 64 ggctggaana gaagtttgtt ggtgngtgac aaggtatgga agaagattat 50 65 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 65 ggctggaana gaagtttgtt ggtgngtgac aaggtatgga 40 66 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 66 ggctggaana gaagtttgtt ggtgngtgac aaggtatgga 40 67 30 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 67 gaacagaagt ttgttggtgc gtganaaggt 30 68 30 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 68 gaacagaagt ttgttggtgc gtganaaggt 30 69 70 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 69 tatgtggcgc gaatgagcaa tattcagcat gtttctcctc ttgtcaacca tcatgtcaag 60 atccttcaac 70 70 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 70 tatgtggcgc gaatgagcaa tattcagcat gtttctcctc ttgtcaacca 50 71 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 71 tatgtggcgc gaatgagcaa tattcagcat gtttctcctc 40 72 30 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 72 tatgtggcgc gaatgagcaa tattcagcat 30 73 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 73 tatgtggcgc gaatgagcaa tattnagcat gtttctnctc ttgtnaacna 50 74 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 74 tatgtggcgc gaatgagcaa tattnagcat gtttctnctc ttgtnaacna 50 75 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 75 tatgtggcgc gaatgagcaa tattnagcat gtttctnctc 40 76 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 76 tatgtggcgc gaatgagcaa tattnagcat gtttctnctc 40 77 30 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 77 tatgtggcgc gaatgagcaa tattnagcat 30 78 30 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 78 tatgtggcgc gaatgagcaa tattnagcat 30 79 30 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 79 accttgtcac gcaccaacaa acttctgttc 30 80 30 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 80 atgctgaata ttgctcattc gcgccacata 30 81 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 81 cctgaaagta gatttgttat ttccgaaacg ccttctcccg ttcttaagtc 50 82 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 82 catataccac aaatagtccc tcaaaaatca caagaaaact cacaacactg 50 83 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 83 gatttgcagc ggtggtaaaa agtatgaaaa cgtggtaatt aaaaggtctc 50 84 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 84 ccaatgaaaa ctaatcaaag gtaaacgtgg atcccatggc aattcccggg 50 85 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 85 caacactgcc cagaggttca atcgatccga tgatcctaat gaaggcgccc 50 86 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 86 gtccagtatc gtccatcata gtatcgataa atatgtgaag gaaatgcctg 50 87 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 87 caacactgcc cagaggttca atcgatgtgt gataggatca gtgttcaggg 50 88 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 88 gaaggcgaag gagactgcta atatcgataa atatgtgaag gaaatgcctg 50 89 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 89 nctgaaagta gatttgttat ttccgaaacg ncttctnccg ttnttaagtc 50 90 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 90 natatancan aaatagtcnc tnaaaaatca naagaaaact canaanactg 50 91 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 91 gatttgnagn ggtggtaaaa agtatgaaaa ngtggtaatt aaaaggtctc 50 92 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 92 ncaatgaaaa ctaatnaaag gtaaacgtgg atcncatggn aattcncggg 50 93 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 93 caacactgcc cagaggttca atcgatnnga tgatcctaat gaaggcgccc 50 94 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 94 gtccagtatc gtccatcata gtatcgataa atatgtgaag gaaatgcctg 50 95 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 95 caacactgcc cagaggttca atcgatgtgt gataggatca gtgttcaggg 50 96 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 96 gaaggcgaag gagactgcta atatcgataa atatgtgaag gaaatgcctg 50 97 34 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 97 acgtgaattc aaatacagac aatgaaggag atga 34 98 45 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 98 gatccccggg aattgccatg ttacctttga ttagttttca ttggc 45 99 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 99 acgtggatcc tttttttttt tttttttttt gatccccggg aattgccatg 50 100 22 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 100 actatgatgg acgatactgg ac 22 101 31 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 101 attggatcga tccgatgatc ctaatgaagg c 31 102 139 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 102 atcgatccga tgatcctaat gaaggcgccc gggtactcct tcttgcattc ttcaacttcc 60 ttcaacactt gagcggagtc ggtgcatccg aacaatggaa gcttccacat tgtccagtat 120 cgtccatcat agtatcgat 139 103 17 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 103 tcctaatgaa ggcgcca 17 104 17 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 104 tcctaatgaa ggcgccc 17 105 34 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 105 ggaattatcg atgtgtgata ggatcagtgt tcag 34 106 31 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 106 aattggatcg atattagcag tctccttcgc c 31 107 287 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 107 atcgatgtgt gataggttca gtgttcaggg ctgtccaagg aacgtatgag catgcgagag 60 acgctgtagt tggaaaaacc cacgaagcgg ctgagtctac caaagaagga gctcagatag 120 cttcagagaa agcggttgga gcaaaggacg caaccgtcga gaaagctaag gaaaccgctg 180 attatactgc ggagaaggtg ggtgagtata aagactatac ggttgataaa gctaaagagg 240 ctaaggacac aactgcagag aaggcgaagg agactgctaa tatcgat 287 108 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 108 tctgttgagg gtatgacttg caattcctgt gtttggacca ttgagcagca 50 109 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 109 tctgttgagg gtatganttg naattcctgt gtttggacca ttgagcagna 50 110 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 110 tctgttgagg gtatgacttg caattcctgt gtttggacca ttgagnagna 50 111 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 111 tctgttgagg gtatganttg naattcntgt gtttggacna ttgagcagna 50 112 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 112 agaaaagcaa tagaggctgt atcaccgggg ctatatagag ttagtatcac 50 113 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 113 agaaaagcaa tagaggntgt atcancgggg ctatatagag ttagtatcac 50 114 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 114 agaaaagcaa tagaggctgt atcancgggg ntatatagag ttagtatcac 50 115 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 115 agaaaagcaa tagaggntgt atnancgggg ntatatagag ttagtatcac 50 116 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 116 gctgttatac aacccccaat gatagcagag ttcatccgag aacttggatt 50 117 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 117 gctgttatac aancccnaat gatagcagag ttnatcngag aacttggatt 50 118 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 118 gctgttatan aanccncaat gatagcagag ttcatcngag aanttggatt 50 119 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 119 gctgttatac aancncnaat gatagcagag ttnatcngag aanttggatt 50 120 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 120 tctttggtca agaaggatcg gtcagcaagt cacttagatc ataaacgaga 50 121 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 121 tctttggtna agaaggatcg gtcagcaagt canttagatc ataaacgaga 50 122 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 122 tctttggtca agaaggatng gtcagcaagt nacttagatn ataaangaga 50 123 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 123 tctttggtna agaaggatng gtnagcaagt nanttagatc ataaacgaga 50 124 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 124 ttataaagca ctgaagcata agacagcaaa tatggacgta ctgattgtgc 50 125 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 125 ttataaagna ctgaagnata agacagcaaa tatggangta ntgattgtgc 50 126 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 126 ttataaagca ctgaagcata agacagcaaa tatggangta ctgattgtgc 50 127 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 127 ttataaagna ntgaagnata agacagnaaa tatggangta ntgattgtgc 50 128 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 128 aacaagtgga tgtggaactt gtacaacgtg gagatatcat taaagtagtt 50 129 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 129 aacaagtgga tgtggaactt gtacaacgtg gagatatcat taaagtagtt 50 130 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 130 aacaagtgga tgtggaactt gtacaacgtg gagatatcat taaagtagtt 50 131 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 131 aanaagtgga tgtggaactt gtacaangtg gagatatcat taaagtagtt 50 132 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 132 ccattgccac cctcttggta tggattgtaa ttggatttct gaattttgaa 50 133 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 133 ncattgccac cctcttggta tggattgtaa ttggatttct gaattttgaa 50 134 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 134 ncattgncan cctcttggta tggattgtaa ttggatttct gaattttgaa 50 135 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 135 ncattgncac nctcttggta tggattgtaa ttggatttnt gaattttgaa 50 136 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 136 ggtatttgat aagactggaa ccattactca cggaacccca gtggtgaatc 50 137 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 137 ggtatttgat aagactggaa ncattactna cggaacncca gtggtgaatc 50 138 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 138 ggtatttgat aagactggaa cnattactca nggaacncca gtggtgaatc 50 139 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 139 ggtatttgat aagantggaa ncattantna nggaacncna gtggtgaatc 50 140 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 140 attggtaacc gggagtggat gattagaaat ggtcttgtca ttaataacga 50 141 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 141 attggtaanc gggagtggat gattagaaat ggtcttgtca ttaataacga 50 142 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 142 attggtaacn gggagtggat gattagaaat ggtnttgtca ttaataacga 50 143 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 143 attggtaanc gggagtggat gattagaaat ggtcttgtna ttaataacga 50 144 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 144 tggcacaggc acagatgtag ccattgaagc agctgatgtg gttttgataa 50 145 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 145 tggcacaggc acagatgtag ncattgaagc agntgatgtg gttttgataa 50 146 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 146 tggnacaggn acagatgtag cnattgaagc agctgatgtg gttttgataa 50 147 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 147 tggcacaggc acagatgtag ncattgaagc agntgatgtg gttttgataa 50 148 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 148 gtganttctc ngattgtgtg agntttgttg gagcntgcgt acgtggattt 50 149 49 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 149 tttaantgan ancttgtttn tgantgttan ggcgtnantg antttgcna 49 150 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 150 natanaggtc actggcatga nttgngnttc ntgtgtagna aanattgaac 50 151 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 151 tgaggggaat gangtgtgnc tcntgcgtac ataaaataga gtntagtctc 50 152 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 152 tgtattnctg taatggggnt gatgacatat atgatggtta tggancacna 50 153 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 153 acatnagagg ntnttgnaaa gttaatttna ntacaagcta nagaagnaac 50 154 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 154 ttncattaac nagaacgggt nantgcttat ntgcgcaana natgttggag 50 155 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 155 ncattgncac nctcttggta tggattgtaa ttggatttnt gaattttgaa 50 156 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 156 gaaangataa tangatttgc tttcnaagnc tctatcacag ttntgttgna 50 157 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 157 atgaacagtc atnaacttng tcttncatga ttattgatgc ncagatntna 50 158 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 158 gttctgatga ntggagacaa nagtaaaana gctagatcta ttgcttntna 50 159 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 159 tggcaagtat tgacttatna agaaagacag tcaagaggat tcggataaat 50 160 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 160 gcntntataa actcactant gtntgataaa ngntncntaa acagtgttgt 50 161 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 161 ntggatggga tctgnagcaa tggctgnttc atntgtttnt gtagtanttt 50 162 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 162 tgcnattgca cgggcacttg ttcgatctcc ttctgtttta cttttggatg 50 163 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 163 tcattctagg attgcnagat ggttatgata ntcatgtcgg agagaaagga 50 164 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 164 ncaatgttgt ttaattggtt gtaatgtntt gatgacntgn ataatnatat 50 165 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 165 nacaagatcn tgtgttgttc tncggaanaa tgaaaatgaa cttagatcna 50 166 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 166 tacttgttct cgacaaaggt tgtgtagncg agtttgacan tcngaagaaa 50 167 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 167 tgaacttgga tcncttcttt gnatttagcg atgatcaaat ttgggaagng 50 168 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 168 tcattaattt tgtgtagctt tntttctnga tttttgnacg atntttccnc 50 169 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 169 agggtgncta ctacaaactg anccaaaagc agatgancga gaagaaataa 50 170 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 170 attgaaagcg acgcggaaag tgccatgtat ttctaatttt gttttnttta 50 171 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 171 ttgtcagcat atnaagagta gatatggaag tggatanact ctgctaatnc 50 172 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 172 nacnttattg cgttcaattt ttgtttcnac ntantantan gaatangttg 50 173 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 173 tcanaaggga gagagtctgc ggtcggtgct ggcgttngag aaaatataac 50 174 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 174 natgcatccn gangagaaga agtantcatt ttggagttat ctggcgaatt 50 175 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 175 gacnatgctn cggtcgtcat gnaaatcgac ttctaaattg cttctgatta 50 176 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 176 ttgnatgctg ttaaaaccta tngtgtanaa tattgcntgt atattnccnt 50 177 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 177 tggnacagct taataacaaa ttggaaagtc gaggattagt cggtgttgaa 50 178 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 178 gacacangca aaggatatgg atgttgttga gctgctgact gaagtnaata 50 179 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 179 agcacgaaan tctgcngtnt aaaattcact ngtgattcat tgnccaattg 50 180 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 180 atggtcatan tctaaaatgg gnagaacttc aacnaaatca ttntcgtcag 50 181 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 181 aacncgagct tgncgnaaag tgcaagaaaa ttatagaacg aatgaaacag 50 182 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 182 ggatgggtcg agngtgagac ctactactaa agaacagctt gtgaatcttt 50 183 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 183 naangttctn gattcntang gacaagaatg gacntatgcn aanagaaaga 50 184 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 184 tgctcgttat ccagctattt tgaagggact tgtnatgcaa ggacttcttc 50 185 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 185 ncgtttagag cttattgcta ancagattgt nccacaagtn agaacagctc 50 186 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 186 tganggacgn tantanccat atgtatttgt tccatcttan cagcaaccaa 50 187 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 187 agctacttca ttnganaagg aacatctngg aaaagtnaag tanatnccgg 50 188 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 188 aaattcaagg atncagttgc cgatggtgaa gccaagattn gcaaggatta 50 189 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 189 ngatcgtttn tgnccattnt anaagactgt nggtatgctn aagaatatga 50 190 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 190 tcaggaacga tctttgacaa cattatcatc accgactctg ttgaggaggc 50 191 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 191 tgaactntan tcttatgaaa gntggggagc natnggattn gatttgtggc 50 192 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 192 gaantttgca gggccgctng gggaatgtca tgatttnatt attaagggaa 50 193 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 193 gtcaattctg ggagaaggtg ttggatancg gggntcggga gagaatgtgc 50 194 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 194 atgtaaagaa ggaatgcttc cngaatggat tggatattta tttgtncaga 50 195 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 195 ggancgaaat ttgtgnagna tgtcgganac gaaattgatg gtntcatttt 50 196 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 196 nagacangaa ggttangata gataaccatc tctnaaagtn tatcgacctc 50 197 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 197 ngangatgtg cgtgttcctg angatgaaag aatgggatat taagaaaanc 50 198 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 198 ttgtgctcca tcgctgctnc gnttacagac ttgacaacgn tcacctttgc 50 199 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 199 aatgagnggt tgtgcngtgt gacgtcantt ngtnacagtg ttgctntant 50 200 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 200 aaattgacan caatcaaatc tgtctcatct nctgaggacn gtnaanttng 50 201 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 201 aatntttgtg tanggagatg gggcaaaagg nagnaagaaa gtaaancaag 50 202 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 202 aggacaaggg gcactactgg cacaggcttt gattattgca gtgagatatt 50 203 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 203 ttaatggagg tgacaatggg ttccttggat tcgataaatt ccgagtgcnc 50 204 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 204 gctnttntcn agtgggctna aaatagtnaa ntcaacagat cggaagttnt 50 205 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 205 aaagcttcga gatggnacgt tngtntgtat ntcgtgaaga acttattgna 50 206 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 206 gattcgntga actttatcaa gacgtggaat atgagcnagn tcntgtcgac 50 207 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 207 gatnttatca ccgcgtgcga tattngagta gcttcanagg atgcgatttt 50 208 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 208 ggaaaggaag gatccattnt cagctctgca nttncancat cagagncatg 50 209 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 209 tggatanaag gagggatctg gnagtggtgg atctggaagt ggtggatatg 50 210 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 210 ttgaaagaan tcnttgccga cgatnctgaa acanacaaag aattgntgaa 50 211 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 211 atgtgggatg aggagaaaga anatttagat acaatggaaa gattagctgc 50 212 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 212 aggntgagct cttggacttt ggcatcaaca ttgtctnatt cttgaaggaa 50 213 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 213 ttatggttan agaaggagnt gtttacggtg tagnattggg aatgtnttnc 50 214 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 214 nacttcaacn aantcngtgt taatnaagca agcngcnacn atntaatgag 50 215 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 215 tctnattgct cgtcgaggnt ancaacaaac actggcaata ccnaattaat 50 216 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 216 taagaaagtn attgaggatg ctgtngcttt gctngcngaa gtntcgtata 50 217 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 217 aagttcatcn tgttgacgga atcgaggngg agaatgntgt atnggtcatt 50 218 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 218 acaggaaata tgattttgga tttcgatttt gaatcggttg gtgctgccnc 50 219 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 219 gctgagntgt atttggctag tgaaatgtgt gtttttgata ctttaaatga 50 220 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 220 acgaggtttg gatcanaatc agaattctgt gaaataagcg ttttttggga 50 221 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 221 agttctnggt ctaacagtgt ctccngttga atattcttgt aaaatnacac 50 222 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 222 atgaccactn aaaatactgc taaaagattt gcagcggcag aagccgttaa 50 223 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 223 ttgatatggn tgtacntgta tggtttttga ggangttttt taggagtcga 50 224 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 224 atttattcat tcatcnatgt aaactgtata ttttgaattt gtgttgtaaa 50 225 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 225 gccaaagcag aattgtattt gatcttcggt aacnttntcn ttngctacaa 50 226 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 226 attttgaatn ttntgggaaa atgcnatnca ntcgagaaan cgttcngttt 50 227 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 227 ntaacggagg atntcgccaa ttatntttga gagacaaaan tgaaantcnt 50 228 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 228 atctagtccn aatgaatctc cnacatgntg ttantcgtga tgttcaactc 50 229 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 229 ttttgctttn atngcaaaag ctcaagatta nacatgtcag gtnaagccaa 50 230 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 230 ncgnganttt aaagagaaga tnataaattt gcattgtttt ttgtttgtat 50 231 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 231 ngagggtgat tcggagactt tnagtaatgt ncaactttca aatgtttgna 50 232 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 232 tagatanaag atacatccnt caaaagaagg nctaccgtca atggcnaaag 50 233 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 233 tcaacgngtn tataaatgaa tnacaacgag gtatcaacat tctccncctg 50 234 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 234 atgntgatgt tgaaattgnt ggctaccgta ttcnaaaaga tantgtaatc 50 235 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 235 atgaatncat ggnttggana tntcncgttt ttcaagggat ataaaaatgt 50 236 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 236 atgnaangaa ttagtgaaaa attcatcntg gaataaaaaa taattntaaa 50 237 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 237 atcgctacga caatctttnc gatgncttcg aagtttngaa agntttctnt 50 238 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 238 gaggtcggtg gaggaggaag tggaaattga nggnaaaatn ctgccnaagg 50 239 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 239 ncctctttgg gatttncant caagtttact gttnggnagn agtgatataa 50 240 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 240 gagttggttn canagaatgc ttaggacgtt taaattngtn acaaantttt 50 241 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 241 naatatggtt ccnattttag caactcatat gaanacagaa gatgtncttg 50 242 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 242 gaaaaaggcg tcgacatttt atgtgacacg tggacanttn actatgacaa 50 243 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 243 taattgaatt acgggtcttt tgtanatatt aattttagta tantttgtga 50 244 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 244 atatcaatgn aantattaat gaatnacaac gtcttgncaa tcttctccng 50 245 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 245 ggagtgacta tgaaagcaaa gagttacnga ttgaaantga aagacagana 50 246 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 246 aatntttaat gataatttat gggatctgta tttntctttn tgtcaataaa 50 247 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 247 atgagcncan aaatgtaaaa ggatacgaga ttgattnggg aanagtcatg 50 248 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 248 atcntgngat atgacattaa gncanatggt tctgaanctt caacagaaga 50 249 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 249 ntgaacnttn aanagaagat aaacttcngt atagcgntgg aaaaantcnt 50 250 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 250 atttaaagga attcanagnt caaaaaataa taantancgg ttnagagatt 50 251 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 251 aatttgagcn acatggcaag ttatnaanag aggaganaat gcngtacagt 50 252 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 252 tganattcta cttaaaggga agaaaatacc aactggtacn cttgtatttg 50 253 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 253 tcancanaaa gcnatacata tgcgagctag ttcntcaggn tgnttaaanc 50 254 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 254 ttcgacaaaa ctattttgga aagaacaatc ncattcagtg tcggcaaang 50 255 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 255 tctgacaaca aagccatana cgtgncgact aattccacaa tcagctagaa 50 256 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 256 ttggcaaaag cagaattgta tttaatcttt ggaaacntcn ttnttngcta 50 257 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 257 tgaatctttn aaacttatna ctccttttaa tactacngtt cctgtttgga 50 258 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 258 attgagattg tatccattgg cgtctcttgt tcanaatcga aaatgtctna 50 259 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 259 aactgctact attgcgccat caagtgtgct gctnaaactt aaatcnaggt 50 260 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 260 ttgaganagg aaataagant agaattcntt tgaaactggt gggaagtgnt 50 261 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 261 aagatgtcaa agaattcaag cnagaacgat ggtncancga cgagcnatta 50 262 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 262 attgaancaa ctntgaaata taatgacaca aaancatgtn tggaagtggt 50 263 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 263 ggcaatgtga caatatctnc aatggttntt nacagcaatn atnacgtgtt 50 264 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 264 ntattcaatn gatattttat cacancatcc agtgctggan ctncatcatt 50 265 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 265 gtctcagaga tgtgtaaatt tacttccntg naatttgttt cangcaacta 50 266 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 266 ttcngaatgt ttncaattgg gactgaagtt tcaagagtca ccnagaaaaa 50 267 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 267 gatncagcat cttccaagnt tacattcntc ngtgcttgta tcaaggaaac 50 268 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 268 tttgaaaacn tgttttatta ttaaaataga taattgatta gttctgtang 50 269 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 269 atangttgca ctgcatccgg ctatgaggga gccaaaaatn ttaggggagt 50 270 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 270 gcanttncat tcatctctgc agctactatg gctttggtga caaaagttgg 50 271 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 271 ncgtccaaaa gaatgncatc tnacaagtct tgaaatntta taaaggtagt 50 272 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 272 gagggatcaa cagtaacctc gtgcggtatt gacaagggat gtncggaagg 50 273 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 273 gatggttctt cgatcgcaaa caaaacagat gtgntcnatt tanatacgga 50 274 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 274 atggagaaaa tggatntgat ggagttgnag gaagtgatgg agntcnagga 50 275 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 275 tgaatctcca taaattattc aatgtttnca aatatttaat ttatcaattg 50 276 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 276 gctnaanacg gtaggatcnt atggaacngt nggaggagca ggnctnggag 50 277 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 277 ngtgacaacn tcttatttat ttctgtaaaa ntgattngcn aaacttttgt 50 278 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 278 gaagctttca aancaaatga gttcnttncc ggaatcncaa agaataccaa 50 279 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 279 acaatgaaaa gagaggatgg aaaggaaatc gaagtctctg ttnttgacga 50 280 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 280 gatgaggtan ataactttgt gtgcagttat aggncatcta cagtanctgc 50 281 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 281 ttcnatnatn actaancgat tgtcntgaca ttgatggcca aancagggaa 50 282 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 282 tcanattatn gaacaagtac tagtaagnat gctgtgatgg agtgcngcta 50 283 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 283 nacggagatn acgacatcaa agcggattgc ttagagtgtg gaaacngtnt 50 284 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 284 actatctacg tggcangttg gactnatnat ngatgggaan gangtataag 50 285 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 285 tctntggcca gttcantttg tgatnaatct nagattngtn canacaagat 50 286 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 286 ntanttncgn aagaaggcnc gtcgtttnta atcgatngaa catctnacac 50 287 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 287 atggatgatn ganccacttg cnactgancc acaatcncgn actcantanc 50 288 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 288 aagacggaga ggntggagag aacggtancg atggagagcn aggaantgat 50 289 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 289 ncanccagga ggagggatac aagagaagaa agtacagatt ctncaactaa 50 290 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 290 agtttcacan ttntttttgc cgttttggtt nccgttatca atncattgat 50 291 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 291 nttttatatt ctnatnaatt tgtttcctac ttggtcagct gaggatcgtt 50 292 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 292 ttcggcacaa atggagcaaa agtatcgtgg ttattgtgat gcgattattc 50 293 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 293 ntantatgaa tgagctcant ggactcattt atcaactcga gtnaaaagnc 50 294 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 294 gttggcgaat cttcgggttc gtataacttc ttagagggat aagcggtgtt 50 295 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 295 gaantgattn gagaagagtg gggantgtcg nttngaggtt taacgacttc 50 296 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 296 tgttattgcg aaagtaatnc tgcttagtac gagaggaaca gcgggttcaa 50 297 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 297 tgcatangan ttggtntctt ggtcaaggtg ttgtattcag tagagnagtc 50 298 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 298 tgtgctnaga atncanttnt tngaaatnca attgtgncaa gcactaactt 50 299 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 299 ttaagangga acnaattgnt cnacnacnat nataccacga gttgaacagt 50 300 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 300 acattgntan caaggnctaa gncgnttnaa attntctaag tctgaaatga 50 301 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 301 gttgagtcca ccggagtctt caccaccatc gagaaggcca atgctnactt 50 302 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 302 agtaaattcn ttncangtgg atctactngt gtgttcacaa agatcgaggg 50 303 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 303 ggtncaataa tgggagactg gttcngcgca gaaagttatg cagatgatat 50 304 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 304 agaaaanttn gttggaccnt gntaaggaga agtatttcaa gcttctgagc 50 305 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 305 gagnacncga agntcaagnc atatttggaa acaagancat actcttcaaa 50 306 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 306 gttaccntct acaaatctng cttcaatnca atgttgttcg cagtcaccaa 50 307 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 307 ncgaagagct cgttantatg cgaggaggtg tgaagcngga ataatttttt 50 308 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 308 aagttcttgg ttggangcga tgggaaaatt atcaagagat ttggancaac 50 309 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 309 acgatttcaa cgtcaaaaat gntaatggtg atgacgtgtn actttnggat 50 310 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 310 acctgggttg atgtttttgc ggctgaaagt ttctccaagn tcattgatta 50 311 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 311 gaagtangtn tcncaaagaa aagctacncc agcttaaggn attgcacaat 50 312 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 312 gcgncagata tgtattcaaa gatcgaggta aatggtnaga acactnatnc 50 313 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 313 aatntanagg gaaaaaggat ttcgagttgc ngcgtttcca tgnaatcaat 50 314 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 314 agatggnaaa gaagcatana taactgaaac tcttccnggg gagctactac 50 315 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 315 tgaataaacg ggncgaacta aatcnattcg tcagtggaaa tgggaaacaa 50 316 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 316 gtcngtnttn ctgatgctta tgaangcnta tttntcgaag tattcatggg 50 317 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 317 tgtggaaaag ctntcaacga gaagaaagca gaagttngta tanaattcaa 50 318 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 318 atatcgncgn ctgcttcctc ancaaccnga ataacgnaan aaaaanttta 50 319 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 319 aagagcncan tcatcaagga tgaaagtgat ggaaagactn ttngtntcag 50 320 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 320 naagatattt taacaaaaat gnatnaanaa gaagccnaat caggttccgg 50 321 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 321 nttgggnatt ctgtanggga tgctgtcatt actgtgnctg catattttaa 50 322 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 322 aagaagnatn tcgaaatcaa cncagacnac gctatcatga agacanttng 50 323 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 323 atgaaagctn aagctnttng tgattcctct actatgggat acatggccgc 50 324 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 324 ttaagcagan cattgaggac gagaagctca aggataagat cagccnagaa 50 325 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 325 ngtntttcca aggatgacat tgaangcatg gtcaacgaag ctgagaaata 50 326 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 326 gtcgacttgg ctnacatcna caccgtcatc aacaaggaag ganagatgac 50 327 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 327 naatcttgag ggacangttc tnacnattga gggacancan gaggtnaaga 50 328 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 328 tcantaaaat gcaccaatnt ggacaatntt ntgnttntgn tggatgcgnt 50 329 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 329 tcatgaagct aaacaattng aaaaggaaga tggtgaacaa cgggaacgtg 50 330 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 330 aagtataacn ttncaacagg ggtcngtnca gaanaaatca agtccgaatt 50 331 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 331 tttaacnatg gcngcagatt cttcgatgac gtcgactttg atngcnacat 50 332 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 332 gcgtcgaaaa gatctncctg aagtntgnat tgantggcct tgatattatg 50 333 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 333 acatagtctt cgtcatcaag gataagcnac accngaaatt caagcgagag 50 334 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 334 tcgncaacan tcgganacgt gncaaaatga atatcatctn aaatcgaatg 50 335 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 335 gtcgaagtta gaaatncaga agccgatatt gtttctnatn aaattncaat 50 336 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 336 actactngtg gaagatccaa taaagttgtt tcaacgngan aaatcgattc 50 337 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 337 ggcagtgaag atgaagtggc aaattctgat gaagaaatgg gaagcagtat 50 338 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 338 ttgtcaacga ccagaagcaa aaattatggg aatngcgata aaattnaagg 50 339 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 339 gatgcaagtg tgncaactgc gaatgtgntc aggntgntca ttaatttgaa 50 340 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 340 gacgatatgt tcgatttcnc aggagaggac ggtgatgatg tgtcagactt 50 341 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 341 gacgatatgt tcgatttcnc aggagaggac ggtgatgatg tgtcagactt 50 342 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 342 gaggtcgtcg taatcnacaa ggctncaaga aagnaagtgn tcgacatttc 50 343 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 343 gatacttttg gcaagntcgt tncaatcaag aaggaggtca tcncagatng 50 344 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 344 gatgaggagg ganacaccga gntctaaatc nacattncaa tanagttcaa 50 345 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 345 nttatgtccg aagatatcnc agaggattgg gacaagaacn cagtcaagat 50 346 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 346 tacnccagtn gantatgatg gaganagaaa nctngagaag ttngaagaat 50 347 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 347 ntcgtcgcct ccaacttcaa cgaaattgcc nttgatgaaa ccaagactgt 50 348 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 348 ttctattgtt tattcnttgc cnaatagtgt atttgtattt attctttctc 50 349 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 349 naaatcnatn tcncagtgga tttcgtcatt gctgacaagt tcgccgagga 50 350 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 350 gtttctgatt cgacanttta tggancatct naagttntgn gagtttcttt 50 351 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 351 gggaaanaaa tgattgttgg tacagtagcc ngcnctgcta ttnactgtga 50 352 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 352 ngagcanatn atncaatcgt tnctgttnaa naaggcnttn taatcgttag 50 353 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 353 tgatgagagn ccagtaacna attatttgaa ncgtcaggat gtgcgtaagg 50 354 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 354 ngtntaatcg aagaagggga tngtgggnaa tcataantaa ttaacnttna 50 355 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 355 naatggntcn aggtctttnt gntcttcata tacttcnatt ccgagttgnt 50 356 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 356 gttntcttgg agntgaagtt gtcgngtgct ngtgtgattn tcacttctnt 50 357 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 357 tcgntancag caaggaatac ttcaacaagg tcaacaagtg atnacacaga 50 358 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 358 aaggaaattg taactngcnc aagagctntc ncaggtgtcn gtgganatat 50 359 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 359 ttgactggat tggagattgc ggaagaagtt gatgttgaaa tcgagagtgg 50 360 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 360 gccaagtctn aagcaataag tgttgatnaa tcagagncat acggagagat 50 361 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 361 atattgagan ttngggacaa gnggacttct natntgtcan agnaantgnc 50 362 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 362 gatncgnaga gaatcgagta tttcntctcg aganccatgg atatcaactg 50 363 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 363 tccgttaaga agntcactgg aaaaacanac ggctcgaacg aaattggaat 50 364 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 364 aatttggatg agagcaaagt ggaaggaatg gctatcgttt tggcagatat 50 365 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 365 gtgntggtca aaaaatgctt gnttngttgc ttattcgcat tgnactcgna 50 366 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 366 nttngagaan tcttcaagtt ggaatcaaca gtggcatcgg atacanatga 50 367 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 367 gtgncttctg aagccgaaga aaacgacgat tagttaaatg tttccaagtt 50 368 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 368 gataaaatcg atagcgacga cgatgaggaa gccgatgatg aggagntcga 50 369 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 369 gcaggtggat acggatgtgg agctgacttt tgcgttttat caagaatctc 50 370 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 370 tccngtagaa gtagaaatgc tagaagaacc tgaacaagaa gatcaagaaa 50 371 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 371 tgcaagatgt cagtattgaa anaattcntg tagaganccn cgaagaaaat 50 372 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 372 agtntcgtat ccgggaatgt ttcagcctgt gaaaatgctt gttgaagang 50 373 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 373 nttnaaaacn gtngctttta aggatacagg aacgtggcan gcttcngagg 50 374 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 374 nagattgtan cttcgaaaag gaaaaggaga gaatcgngtn gcaaaaatgg 50 375 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 375 tgatggnttt gattattcga gnaggagnaa tgatgtncga gagtcgttat 50 376 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 376 naatgangag aatattggag taatggggaa actggttgcg acttgngaaa 50 377 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 377 ttggaaaaca atntcntcga ctttntgntc actnttngtg aaactatcna 50 378 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 378 tcttgttatt ttattttgtt ttgggcttgt tccgaaaatg aaatggttgt 50 379 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 379 naatggatca ccaagncagt tnacaagnac ngtgagnaaa gaggantcac 50 380 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 380 ntttgtgatg tgatgactgc gaaggganac ttgatggcta ttacgagana 50 381 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 381 gagncagcta ctnagatgac antcaacacg ttcnattatg caggagtttc 50 382 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 382 tacactncat cctcgncgac atacaatnca acatctncan gcggattctc 50 383 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 383 atggagaaga tggtttggat ggaatgtggg ttgagaatca gaatatgcng 50 384 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 384 aacngggata ccgtgtcgaa cgtcacatga aagatggcga tataatcgtc 50 385 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 385 gaggagatta aangcatgtc agtggctnat gtcgagtttn cagaagtcta 50 386 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 386 agatattgcn tctacttatc atgggcctga tggntttgtn tgncggtatt 50 387 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 387 gaatctnaan canttntgga anccnataca ncaatggata gaaganggag 50 388 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 388 gttgttnttt tttccgtgat cttttcatgt ttatgtntga acgtggcagg 50 389 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 389 gactcgttgg tgtcttgcta ggatgtcttg ggttcattcn tcaatngttg 50 390 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 390 gtantgggct cgagggctga aactaatnga agaagaaact ccagaagata 50 391 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 391 ggatcatgct ctgtttacga cactgatgag ttaagagtca gantgnacgt 50 392 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 392 ngatggttct tctcgtctat catatcgggg tagttgncga agtgttgaaa 50 393 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 393 naaatcgaan tggtataaag gaggaccgac ggagacgaat ttgaangaga 50 394 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 394 attngatcaa agaactctgg ctctnggngt taantggaca tttgttcgtc 50 395 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 395 ntcnccgagn aggcgattat tnacgctagt tatgctnaaa tgtgatctgt 50 396 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 396 ncggtantat ctggatcana cagaagtncg aaaatgacca ggnagttatt 50 397 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 397 nccagtgact acntgaatng cgtctctgaa tctncanaca attcctacta 50 398 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 398 ggagttgctn acngcaatta agagngantt nggatctctg gataatcttc 50 399 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 399 aaattgagga aaagcttcac gaggnggtct ncaaaggaaa cgtcaaagaa 50 400 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 400 naatcgtacn atgaaagaag ttggaagnca ngtgcaagag aagaaatcna 50 401 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 401 aagaagattn ctgacnagag agactcangt gcttacncaa gaagcatcta 50 402 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 402 agcattggtg gaaatacgaa atggnatggg aagagaaacn cctctnaatt 50 403 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 403 ntggttacgg tagtgtatgg tncctgtnct ntcagaatgn aaatatgtng 50 404 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 404 tctacgtcga tggaaaagnc gatttaanaa tcaaagncaa caacgnagtt 50 405 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 405 ggaaaggtgn caaaaagttg anagnaattg gaggatntta ttnattgcna 50 406 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 406 agatgatgat gaagttcctg caaagaagcc tgctccagcg aagaaagctg 50 407 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 407 aaaacctcgt actggaaaag gagctgcgaa agcggaagtt atngatttgt 50 408 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 408 gagaaggccn agaagaagta cgacagantg aaggagnagt tgaaaaagtt 50 409 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 409 ttctgtnata caatcgtgnt aatcggcagg ttgngatcct ttgtaaccat 50 410 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 410 aagnttngga cancattgag aatgtcaaag ncaaaatcca ggataaggag 50 411 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 411 aatngaaccn atnaattcac tngttattcc tcctcgatct ccgttnaagt 50 412 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 412 ntgaacnatn caaatattga agatncagct naggctgaag cctatnagat 50 413 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 413 ngtgtgntta tctcttctgg atgaaaanaa ggattggaag ccgtcaatnt 50 414 49 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 414 nggaagnatn tgncttgana ttctncgttc gcagtggtcg ccggctctg 49 415 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 415 aaagtangct atgtgaggag gntaacanca ttcatataag aangcagcna 50 416 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 416 tgttgcngta gaagagagac taaaactaag aacgattgat tgaaggtctg 50 417 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 417 tacaattctt tgnaggaagc aatatccgcc ggagtcnccn ttatcactat 50 418 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 418 ntcacggagg ttataattnt atgcaggagg naatttntgn tggagttcna 50 419 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 419 accgtttcat gagagntgta atcaggtgtt gtttctgtaa aaagtgtgaa 50 420 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 420 gtggatgtga aattagtcnt caacnccaga gcatttagtg cagagattag 50 421 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 421 gcagtttaat gtgaagctag ttaaagtaca gtctacgtgg gangagaaat 50 422 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 422 attgccaagt ccatttctng tgccaagtac attnaaaata caagaaaggc 50 423 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 423 agantcntan aaatagattc ggtgtcctgc nagacgatgt tgaagaatag 50 424 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 424 ttgaagtttg ggaatattgg tatggttgaa gacnaaggan cggattacga 50 425 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 425 gaggcgnaag taggcaatga ttcaagaagt agtaaaggca atngtaacac 50 426 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 426 tgagcanaaa gttaagatgt tnggaaagaa aaagaaagtn aatcctatga 50 427 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 427 naagtgacca atnagnacgc anggnttnca tcctcaagan tgatattanc 50 428 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 428 attaaatgcg cagatgagga cggaacgaat atcggagaaa ctgataatat 50 429 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 429 gatggtaagn tgagcgcctt ggacgaagaa tttgatgttg tcgctactaa 50 430 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 430 tacgtcacgn aagganagag ctttgacgac gaaatatcan ttggaggatt 50 431 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 431 tctncctgtg taggtacanc aatatcanaa gcgnatttct atgtcgacta 50 432 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 432 tgctaanacn agtttaganc atggaaatcc nacngcaaat ataagnaatg 50 433 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 433 gcagganata agattncggt caagnaanga nagtgaagaa agtatgcaaa 50 434 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 434 ncgtctagtg aaagcgggat ggctaaattg ggaaaangan aagatgttat 50 435 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 435 gatgcttcaa tatcctttga tggtngttag tttaccattt ttggtgtctt 50 436 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 436 agtnatttga gttatgtgaa gaccgttggt gggaaagaag agatcaggtg 50 437 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 437 gtcttggcta ccacanccaa aaccgttnga aactttaaga gcattntant 50 438 603 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 438 ggtgaaagga caaggacaaa agacaacaat ctactgggta aatttgagtt gagcggtatt 60 ccacccgctc caagaggcgt accacaaatt gaagttacat ttgatatcga tgcaaatggt 120 attctgaacg tatctgccgt tgaaaaaggt actggtaaat ctaacaagat tacaattact 180 aacgataagg gaagattatc gaaggaagat atcgataaaa tggttgctga ggcagaaaag 240 ttcaaggccg aagatgaaca agaagctcaa cgtgttcaag ctaagaatca gctagaatcg 300 tacgcgttta ctttgaaaaa ttctgtgagc gaaaataact tcaaggagaa ggtgggtgaa 360 gaggatgcca ggaaattgga agccgccgcc caagatgcta taaattggtt agatgcttcg 420 caagcggcct ccaccgagga atacaaggaa aggcaaaagg aactagaagg tgttgcaaac 480 cccattatga gtaaatttta cggagctgca ggtggtgccc caggagcagg cccagttccg 540 ggtgctggag caggccccac tggagcacca gacaacggcc caacggttga agaggttgat 600 tag 603 439 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 439 gccccactgg agcaccagac aacggcccaa cggttgaaga ggttgattag 50 440 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 440 gccccaggag caggcccagt tccgggtgct ggagcaggcc ccactggagc 50 441 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 441 ccattatgag taaattttac ggagctgcag gtggtgcccc aggagcaggc 50 442 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 442 ctagaaggtg ttgcaaaccc cattatgagt aaattttacg gagctgcagg 50 443 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 443 cctccaccga ggaatacaag gaaaggcaaa aggaactaga aggtgttgca 50 444 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 444 ggtgaagagg atgccaggaa attggaagcc gccgcccaag atgctataaa 50 445 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 445 actttgaaaa attctgtgag cgaaaataac ttcaaggaga aggtgggtga 50 446 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 446 aagaatcagc tagaatcgta cgcgtttact ttgaaaaatt ctgtgagcga 50 447 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 447 aatggttgct gaggcagaaa agttcaaggc cgaagatgaa caagaagctc 50 448 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 448 taacaagatt acaattacta acgataaggg aagattatcg aaggaagata 50 449 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 449 cgatgcaaat ggtattctga acgtatctgc cgttgaaaaa ggtactggta 50 450 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 450 acccgctcca agaggcgtac cacaaattga agttacattt gatatcgatg 50 451 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 451 ggtgaaagga caaggacaaa agacaacaat ctactgggta aatttgagtt 50 452 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 452 gccncantgg agnacnagac aacggccnaa nggttgaaga ggttgattag 50 453 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 453 gccncaggag caggcncagt tncgggtgct ggagcaggcn ccactggagc 50 454 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 454 ncattatgag taaattttac ggagntgnag gtggtgnccn aggagnaggc 50 455 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 455 ntagaaggtg ttgcaaacnc cattatgagt aaattttacg gagctgcagg 50 456 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 456 nctncancga ggaatacaag gaaaggcaaa aggaactaga aggtgttgna 50 457 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 457 ggtgaagagg atgccaggaa attggaagcc gccgccnaag atgctataaa 50 458 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 458 actttgaaaa attctgtgag cgaaaataac ttcaaggaga aggtgggtga 50 459 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 459 aagaatnagn tagaatcgta cgcgtttact ttgaaaaatt ctgtgagcga 50 460 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 460 aatggttgct gaggcagaaa agttnaaggc ngaagatgaa caagaagctc 50 461 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 461 taanaagatt acaattacta angataaggg aagattatcg aaggaagata 50 462 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 462 ngatgcaaat ggtattctga angtatctgc ngttgaaaaa ggtactggta 50 463 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 463 accngctcca agaggngtac cacaaattga agttacattt gatatngatg 50 464 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 464 ggtgaaagga caaggacaaa agacaacaat ntantgggta aatttgagtt 50 465 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 465 acaagaatac gacgaaagtg gtccatctat cgttcaccac aagtgtttct 50 466 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 466 acaagaatan gangaaagtg gtccatctat ngttcancan aagtgtttnt 50 467 12 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 467 caacatccca ca 12 468 12 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 468 caacaaccca ca 12 469 12 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 469 tgtgggatgt tg 12 470 12 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 470 tgtgggdtgt tg 12 471 12 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 471 aagagtccag tg 12 472 12 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 472 aagaggccag tg 12473 12 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 473 cantggantc tt 12474 12 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 474 cantggdntc tt 12475 33 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 475 acgtgagctc ttttgacatg tcagaatttc aag 33476 44 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 476 gatccccggg aattgccatg ttacttttca gcttcctctt caac 44 477 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 477 tttggtagca cgacaagctt agtat 25 478 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 478 tttggtagca cgacaagctt agtat 25 479 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 479 cactctaaca gtttcgccgt ttcta 25 480 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 480 cactctaaca gtttcgccgt ttcta 25 481 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 481 gttgccatgg agttcaaaat ctgtc 25 482 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 482 gttgccatgg agttcaaaat ctgtc 25 483 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 483 tcaatggcct tgcaccatat aattg 25 484 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 484 tcaatggcct tgcaccatat aattg 25 485 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 485 tcagttagcc aatccttcgc ttcat 25 486 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 486 tcagttagcc aatccttcgc ttcat 25 487 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 487 tttttcggcc aaacgatctt gaatt 25 488 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 488 tttttcggcc aaacgatctt gaatt 25 489 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 489 attgacctca aaactttctt ggata 25 490 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 490 attgacctca aaactttctt ggata 25 491 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 491 gatgaactca ggtggatagg atctt 25 492 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 492 gatgaactca ggtggatagg atctt 25 493 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 493 accatcatca cccaactttg tgtcg 25 494 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 494 accatcatca cccaactttg tgtcg 25 495 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 495 cccaactttg tgtcgtttaa taaaa 25 496 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 496 cccaactttg tgtcgtttaa taaaa 25 497 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 497 caatgatcgt gttacggaaa tcaac 25 498 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 498 caatgatcgt gttacggaaa tcaac 25 499 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 499 tggcctaggg aatcggtcag cttac 25 500 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 500 tggcctaggg aatcggtcag cttac 25 501 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 501 ttggaaacat cggggtgcgc ttttt 25 502 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 502 ttggaaacat cggggtgcgc ttttt 25 503 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 503 gaaacatcgg ggtgcgcttt ttcaa 25 504 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 504 gaaacatcgg ggtgcgcttt ttcaa 25 505 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 505 aaaacgacag cataaggctt tcttc 25 506 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 506 aaaacgacag cataaggctt tcttc 25 507 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 507 gacagcctca gttaattggc cacca 25 508 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 508 gacagcctca gttaattggc cacca 25 509 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 509 ccgattaaac gagagacagt atgct 25 510 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 510 ccgattaaac gagagacagt atgct 25 511 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 511 atcaaatagg aattcagcta aagcc 25 512 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 512 atcaaatagg aattcagcta aagcc 25 513 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 513 gacctaagaa cataaagctg gcaat 25 514 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 514 gacctaagaa cataaagctg gcaat 25 515 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 515 ataaagctgg caataggtct ctttt 25 516 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 516 ataaagctgg caataggtct ctttt 25 517 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 517 agacgtacag catcagaaat agcag 25 518 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 518 agacgtacag catcagaaat agcag 25 519 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 519 aaatagcagc aatggcctcg tcttg 25 520 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 520 aaatagcagc aatggcctcg tcttg 25 521 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 521 atagcagcaa tggcctcgtc ttggc 25 522 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 522 atagcagcaa tggcctcgtc ttggc 25 523 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 523 gcaatggcct cgtcttggcc aacga 25 524 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 524 gcaatggcct cgtcttggcc aacga 25 525 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 525 tttggtagna nganaagntt agtat 25 526 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 526 tttggtagca cgacaagctt agtat 25 527 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 527 nantntaana gtttcgncgt ttnta 25 528 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 528 nactctaaca gtttcgccgt ttcta 25 529 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 529 gttgnnatgg agttnaaaat ntgtc 25 530 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 530 gttgccatgg agttcaaaat ctgtc 25 531 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 531 tnaatggnct tgnacnatat aattg 25 532 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 532 tcaatggcct tgnacnatat aattg 25 533 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 533 tnagttagcn aatcnttngn ttnat 25 534 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 534 accatcatca ccnaantttg tgtcg 25 535 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 535 tttttnggnn aaangatntt gaatt 25 536 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 536 naatgatcgt gttacggaaa tnaac 25 537 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 537 attganntna aaantttntt ggata 25 538 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 538 tggnctaggg aatcggtcag cttac 25 539 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 539 gatgaantna ggtggatagg atntt 25 540 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 540 ttggaaacat cggggtgcgc ttttt 25 541 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 541 ancatcatca cnnaaccttg tgtng 25 542 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 542 gaaacatcgg ggtgcgcttt ttcaa 25 543 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 543 nnnaactttg tgtcgtttaa taaaa 25 544 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 544 aaaacgacag cataaggctt tnttc 25 545 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 545 naatgatngt gttanggaaa tnaac 25 546 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 546 atcaaatagg aattcagcta aagcc 25 547 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 547 tggnctaggg aatnggtcag nttac 25 548 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 548 gacntaagaa cataaagctg gnaat 25 549 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 549 ttggaaanat nggggtgngc ttttt 25 550 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 550 ataaagntgg caataggtnt ctttt 25 551 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 551 gaaanatngg ggtgngcttt ttnaa 25 552 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 552 agangtacag catcagaaat agcag 25 553 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 553 aaaanganag nataaggntt tnttc 25 554 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 554 aaatagnagn aatggnctng tnttg 25 555 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 555 ganagnctca gttaattggc nacna 25 556 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 556 atagcagcaa tggcctcgtc ttggc 25 557 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 557 nngattaaan gagaganagt atgnt 25 558 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 558 gcaatggcct cgtcttggnc aacga 25 559 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 559 atnaaatagg aattnagnta aagnc 25 560 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 560 gacagcntca gttaattggc cacca 25 561 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 561 ganntaagaa nataaagntg gnaat 25 562 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 562 ncgattaaan gagagacagt atgct 25 563 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 563 ataaagntgg naataggtnt ctttt 25 564 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 564 gatgaantca ggtggatagg atctt 25 565 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 565 agangtanag natnagaaat agnag 25 566 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 566 tcagttagcn aatccttcgc ttcat 25 567 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 567 aaatagnagn aatggnctng tcttg 25 568 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 568 tttttcggcn aaacgatctt gaatt 25 569 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 569 atagnagcaa tggnctcgtn ttggc 25 570 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 570 attgacntca aaactttctt ggata 25 571 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 571 gcaatggcct ngtnttggcc aacga 25 572 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 572 nccaactttg tgtcgtttaa taaaa 25 573 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 573 tttggtagca cgtcaagctt agtat 25 574 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 574 tttggtagca cgtcaagctt agtat 25 575 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 575 cactctaaca gtatcgccgt ttcta 25 576 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 576 cactctaaca gtatcgccgt ttcta 25 577 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 577 gttgccatgg agatcaaaat ctgtc 25 578 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 578 gttgccatgg agatcaaaat ctgtc 25 579 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 579 tcaatggcct tggaccatat aattg 25 580 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 580 tcaatggcct tggaccatat aattg 25 581 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 581 tcagttagcc aaaccttcgc ttcat 25 582 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 582 tcagttagcc aaaccttcgc ttcat 25 583 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 583 tttttcggcc aatcgatctt gaatt 25 584 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 584 tttttcggcc aatcgatctt gaatt 25 585 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 585 attgacctca aatctttctt ggata 25 586 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 586 attgacctca aatctttctt ggata 25 587 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 587 gatgaactca ggaggatagg atctt 25 588 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 588 gatgaactca ggaggatagg atctt 25 589 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 589 accatcatca ccgaactttg tgtcg 25 590 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 590 accatcatca ccgaactttg tgtcg 25 591 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 591 cccaactttg tgacgtttaa taaaa 25 592 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 592 cccaactttg tgacgtttaa taaaa 25 593 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 593 caatgatcgt gtaacggaaa tcaac 25 594 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 594 caatgatcgt gtaacggaaa tcaac 25 595 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 595 tggcctaggg aaacggtcag cttac 25 596 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 596 tggcctaggg aaacggtcag cttac 25 597 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 597 ttggaaacat cgcggtgcgc ttttt 25 598 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 598 ttggaaacat cgcggtgcgc ttttt 25 599 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 599 gaaacatcgg ggagcgcttt ttcaa 25 600 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 600 gaaacatcgg ggagcgcttt ttcaa 25 601 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 601 aaaacgacag caaaaggctt tcttc 25 602 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 602 aaaacgacag caaaaggctt tcttc 25 603 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 603 gacagcctca gtaaattggc cacca 25 604 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 604 gacagcctca gtaaattggc cacca 25 605 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 605 ccgattaaac gacagacagt atgct 25 606 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 606 ccgattaaac gacagacagt atgct 25 607 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 607 atcaaatagg aaatcagcta aagcc 25 608 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 608 atcaaatagg aaatcagcta aagcc 25 609 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 609 gacctaagaa caaaaagctg gcaat 25 610 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 610 gacctaagaa caaaaagctg gcaat 25 611 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 611 ataaagctgg cattaggtct ctttt 25 612 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 612 ataaagctgg caaaaggtct ctttt 25 613 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 613 agacgtacag caacagaaat agcag 25 614 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 614 agacgtacag caacagaaat agcag 25 615 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 615 aaatagcagc aaaggcctcg tcttg 25 616 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 616 aaatagcagc aaaggcctcg tcttg 25 617 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 617 atagcagcaa tgccctcgtc ttggc 25 618 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 618 atagcagcaa tgccctcgtc ttggc 25 619 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 619 gcaatggcct cgacttggcc aacga 25 620 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 620 gcaatggcct cgacttggcc aacga 25 621 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 621 tttggtagna ngtcaagntt agtat 25 622 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 622 tttggtagca cgtcaagctt agtat 25 623 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 623 nantntaana gtatcgncgt ttnta 25 624 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 624 nactctaaca gtatcgccgt ttcta 25 625 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 625 gttgnnatgg agatnaaaat ntgtc 25 626 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 626 gttgccatgg agatcaaaat ctgtc 25 627 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 627 tnaatggnct tggacnatat aattg 25 628 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 628 tcaatggcct tggacnatat aattg 25 629 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 629 tnagttagcn aaaccttcgn ttnat 25 630 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 630 tcagttagcn aaaccttcgc ttcat 25 631 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 631 tttttnggnn aatcgatntt gaatt 25 632 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 632 tttttcggcn aatcgatctt gaatt 25 633 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 633 attganntna aatctttntt ggata 25 634 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 634 attgacntca aatctttctt ggata 25 635 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 635 gatgaantna ggaggatagg atntt 25 636 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 636 gatgaantca ggaggatagg atctt 25 637 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 637 ancatcatca cccaaccttg tgtng 25 638 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 638 accatcatca ccgaantttg tgtcg 25 639 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 639 nnnaactttg tgacgtttaa taaaa 25 640 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 640 nccaactttg tgacgtttaa taaaa 25 641 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 641 naatgatngt gttanggaaa tnaac 25 642 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 642 naatgatcgt gtaacggaaa tnaac 25 643 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 643 tggnctaggg aaacggtcag nttac 25 644 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 644 tggnctaggg aaacggtcag cttac 25 645 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 645 ttggaaanat ngcggtgngc ttttt 25 646 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 646 ttggaaacat cgnggtgcgc ttttt 25 647 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 647 gaaanatngg ggagngcttt ttnaa 25 648 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 648 gaaacatcgg ggagcgcttt ttcaa 25 649 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 649 aaaanganag naaaaggntt tnttc 25 650 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 650 aaaacgacag caaaaggctt tnttc 25 651 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 651 ganagnctca gtaaattggc nacna 25 652 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 652 gacagcntca gtaaattggc cacca 25 653 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 653 nngattaaan gacaganagt atgnt 25 654 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 654 ncgattaaan ganagacagt atgct 25 655 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 655 atnaaatagg aaatnagnta aagnc 25 656 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 656 atcaaatagg aaatcagcta aagcc 25 657 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 657 ganntaagaa naaaaagntg gnaat 25 658 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 658 gacntaagaa caaaaagctg gnaat 25 659 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 659 ataaagntgg nattaggtnt ctttt 25 660 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 660 ataaagntgg cattaggtnt ctttt 25 661 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 661 agangtanag naacagaaat agnag 25 662 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 662 agangtacag caacagaaat agcag 25 663 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 663 aaatagnagn aaaggnctng tcttg 25 664 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 664 aaatagnagn aaaggnctng tnttg 25 665 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 665 atagnagcaa tgccctcgtn ttggc 25 666 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 666 atagcagcaa tgncctcgtc ttggc 25 667 24 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 667 gnaatggcct ngactggcca anga 24668 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 668 gcaatggcct cgacttggnc aacga 25 669 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 669 tttggtagca cgtcaagctt agtat 25 670 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 670 cactctaaca gtatcgccgt ttcta 25 671 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 671 gttgccatgg agatcaaaat ctgtc 25 672 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 672 tcaatggcct tggaccatat aattg 25 673 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 673 tcagttagcc aaaccttcgc ttcat 25 674 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 674 tttttcggcc aatcgatctt gaatt 25 675 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 675 attgacctca aatctttctt ggata 25 676 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 676 gatgaactca ggaggatagg atctt 25 677 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 677 accatcatca ccgaactttg tgtcg 25 678 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 678 cccaactttg tgacgtttaa taaaa 25 679 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 679 caatgatcgt gtaacggaaa tcaac 25 680 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 680 tggcctaggg aaacggtcag cttac 25 681 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 681 ttggaaacat cgcggtgcgc ttttt 25 682 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 682 gaaacatcgg ggagcgcttt ttcaa 25 683 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 683 aaaacgacag caaaaggctt tcttc 25 684 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 684 gacagcctca gtaaattggc cacca 25 685 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 685 ccgattaaac gacagacagt atgct 25 686 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 686 atcaaatagg aaatcagcta aagcc 25 687 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 687 gacctaagaa caaaaagctg gcaat 25 688 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 688 ataaagctgg cattaggtct ctttt 25 689 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 689 agacgtacag caacagaaat agcag 25 690 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 690 aaatagcagc aaaggcctcg tcttg 25 691 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 691 atagcagcaa tgccctcgtc ttggc 25 692 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 692 gcaatggcct cgacttggcc aacga 25 693 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 693 tttggtagca cgtcaagctt agtat 25 694 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 694 cactctaaca gtatcgccgt ttcta 25 695 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 695 gttgccatgg agatcaaaat ctgtc 25 696 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 696 tcaatggcct tggaccatat aattg 25 697 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 697 tcagttagcc aaaccttcgc ttcat 25 698 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 698 tttttcggcc aatcgatctt gaatt 25 699 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 699 attgacctca aatctttctt ggata 25 700 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 700 gatgaactca ggaggatagg atctt 25 701 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 701 accatcatca ccgaactttg tgtcg 25 702 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 702 cccaactttg tgacgtttaa taaaa 25 703 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 703 caatgatcgt gtaacggaaa tcaac 25 704 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 704 tggcctaggg aaacggtcag cttac 25 705 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 705 ttggaaacat cgcggtgcgc ttttt 25 706 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 706 gaaacatcgg ggagcgcttt ttcaa 25 707 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 707 aaaacgacag caaaaggctt tcttc 25 708 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 708 gacagcctca gtaaattggc cacca 25 709 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 709 ccgattaaac gacagacagt atgct 25 710 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 710 atcaaatagg aaatcagcta aagcc 25 711 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 711 gacctaagaa caaaaagctg gcaat 25 712 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 712 ataaagctgg caaaaggtct ctttt 25 713 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 713 agacgtacag caacagaaat agcag 25 714 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 714 aaatagcagc aaaggcctcg tcttg 25 715 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 715 atagcagcaa tgccctcgtc ttggc 25 716 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 716 gcaatggcct cgacttggcc aacga 25 717 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 717 tttggtagna ngtcaagntt agtat 25 718 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 718 nantntaana gtatcgncgt ttnta 25 719 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 719 gttgnnatgg agatnaaaat ntgtc 25 720 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 720 tnaatggnct tggacnatat aattg 25 721 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 721 tnagttagcn aaaccttcgn ttnat 25 722 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 722 tttttnggnn aatcgatntt gaatt 25 723 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 723 attganntna aatctttntt ggata 25 724 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 724 gatgaantna ggaggatagg atntt 25 725 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 725 ancatcatca cccaaccttg tgtng 25 726 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 726 nnnaactttg tgacgtttaa taaaa 25 727 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 727 tggnctaggg aaacggtcag nttac 25 728 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 728 ttggaaanat ngcggtgngc ttttt 25 729 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 729 gaaanatngg ggagngcttt ttnaa 25 730 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 730 aaaanganag naaaaggntt tnttc 25 731 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 731 ganagnctca gtaaattggc nacna 25 732 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 732 nngattaaan gacaganagt atgnt 25 733 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 733 atnaaatagg aaatnagnta aagnc 25 734 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 734 ganntaagaa naaaaagntg gnaat 25 735 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 735 ataaagntgg nattaggtnt ctttt 25 736 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 736 agangtanag naacagaaat agnag 25 737 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 737 aaatagnagn aaaggnctng tcttg 25 738 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 738 atagnagcaa tgccctcgtn ttggc 25 739 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 739 gcaatggcct nganttggcc aacga 25 740 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 740 tttggtagca cgacaagctt agtat 25 741 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 741 cactctaaca gtttcgccgt ttcta 25 742 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 742 gttgccatgg agttcaaaat ctgtc 25 743 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 743 tcaatggcct tgcaccatat aattg 25 744 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 744 tcagttagcc aatccttcgc ttcat 25 745 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 745 tttttcggcc aaacgatctt gaatt 25 746 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 746 attgacctca aaactttctt ggata 25 747 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 747 gatgaactca ggtggatagg atctt 25 748 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 748 accatcatca cccaactttg tgtcg 25 749 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 749 cccaactttg tgtcgtttaa taaaa 25 750 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 750 caatgatcgt gttacggaaa tcaac 25 751 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 751 tggcctaggg aatcggtcag cttac 25 752 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 752 ttggaaacat cggggtgcgc ttttt 25 753 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 753 gaaacatcgg ggtgcgcttt ttcaa 25 754 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 754 aaaacgacag cataaggctt tcttc 25 755 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 755 gacagcctca gttaattggc cacca 25 756 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 756 ccgattaaac gagagacagt atgct 25 757 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 757 atcaaatagg aattcagcta aagcc 25 758 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 758 gacctaagaa cataaagctg gcaat 25 759 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 759 ataaagctgg caataggtct ctttt 25 760 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 760 agacgtacag catcagaaat agcag 25 761 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 761 aaatagcagc aatggcctcg tcttg 25 762 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 762 atagcagcaa tggcctcgtc ttggc 25 763 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 763 gcaatggcct cgtcttggcc aacga 25 764 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 764 tttggtagca cgacaagctt agtat 25 765 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 765 cactctaaca gtttcgccgt ttcta 25 766 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 766 gttgccatgg agttcaaaat ctgtc 25 767 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 767 tcaatggcct tgcaccatat aattg 25 768 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 768 tcagttagcc aatccttcgc ttcat 25 769 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 769 tttttcggcc aaacgatctt gaatt 25 770 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 770 attgacctca aaactttctt ggata 25 771 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 771 gatgaactca ggtggatagg atctt 25 772 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 772 accatcatca cccaactttg tgtcg 25 773 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 773 cccaactttg tgtcgtttaa taaaa 25 774 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 774 caatgatcgt gttacggaaa tcaac 25 775 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 775 tggcctaggg aatcggtcag cttac 25 776 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 776 ttggaaacat cggggtgcgc ttttt 25 777 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 777 gaaacatcgg ggtgcgcttt ttcaa 25 778 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 778 aaaacgacag cataaggctt tcttc 25 779 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 779 gacagcctca gttaattggc cacca 25 780 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 780 ccgattaaac gagagacagt atgct 25 781 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 781 atcaaatagg aattcagcta aagcc 25 782 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 782 gacctaagaa cataaagctg gcaat 25 783 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 783 ataaagctgg caataggtct ctttt 25 784 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 784 agacgtacag catcagaaat agcag 25 785 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 785 aaatagcagc aatggcctcg tcttg 25 786 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 786 atagcagcaa tggcctcgtc ttggc 25 787 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 787 gcaatggcct cgtcttggcc aacga 25 788 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 788 tttggtagna nganaagntt agtat 25 789 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 789 nantntaana gtttcgncgt ttnta 25 790 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 790 gttgnnatgg agttnaaaat ntgtc 25 791 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 791 tnaatggnct tgnacnatat aattg 25 792 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 792 tnagttagcn aatcnttngn ttnat 25 793 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 793 tttttnggnn aaangatntt gaatt 25 794 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 794 attganntna aaantttntt ggata 25 795 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 795 gatgaantna ggtggatagg atntt 25 796 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 796 ancatcatca cnnaaccttg tgtng 25 797 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 797 nnnaactttg tgtcgtttaa taaaa 25 798 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 798 naatgatngt gttanggaaa tnaac 25 799 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 799 tggnctaggg aatnggtcag nttac 25 800 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 800 ttggaaanat nggggtgngc ttttt 25 801 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 801 gaaanatngg ggtgngcttt ttnaa 25 802 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 802 aaaanganag nataaggntt tnttc 25 803 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 803 ganagnctca gttaattggc nacna 25 804 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 804 nngattaaan gagaganagt atgnt 25 805 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 805 atnaaatagg aattnagnta aagnc 25 806 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 806 ganntaagaa nataaagntg gnaat 25 807 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 807 ataaagntgg naataggtnt ctttt 25 808 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 808 agangtanag natnagaaat agnag 25 809 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 809 aaatagnagn aatggnctng tcttg 25 810 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 810 atagnagcaa tggnctcgtn ttggc 25 811 25 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 811 gcaatggcct ngtnttggcc aacga 25 812 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 812 cacaacactg cccagaggtt caatcgataa atatgtgaag gaaatgcctg 50813 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 813 ctccttcttg cattcttcaa cttccttcaa cacttgagcg gagtcggtgc 50814 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 814 gaacgtatga gcatgcgaga gacgctgtag ttggaaaaac ccacgaagcg 50815 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 815 gaaaccgctg attatactgc ggagaaggtg ggtgagtata aagactatac 50816 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 816 aagtagattt gttatttccg aaacgccttc tcccgttctt 40817 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 817 accacaaata gtccctcaaa aatcacaaga aaactcacaa 40818 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 818 gcagcggtgg taaaaagtat gaaaacgtgg taattaaaag 40819 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 819 gaaaactaat caaaggtaaa cgtggatccc atggcaattc 40820 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 820 cactgcccag aggttcaatc gataaatatg tgaaggaaat 40821 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 821 ctgcccagag gttcaatcga tccgatgatc ctaatgaagg 40822 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 822 gtatcgtcca tcatagtatc gataaatatg tgaaggaaat 40 823 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 823 tcttgcattc ttcaacttcc ttcaacactt gagcggagtc 40824 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 824 ctgcccagag gttcaatcga tgtgtgatag gatcagtgtt 40 825 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 825 cgaaggagac tgctaatatc gataaatatg tgaaggaaat 40 826 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 826 tatgagcatg cgagagacgc tgtagttgga aaaacccacg 40827 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 827 cgctgattat actgcggaga aggtgggtga gtataaagac 40828 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 828 nacaacactg ccnagaggtt caatngataa atatgtgaag gaaatgcctg 50 829 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 829 naanactgcn cagaggttna atcgatcnga tgatcctaat gaaggngcnc 50 830 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 830 gtcnagtatn gtncatcata gtatngataa atatgtgaag gaaatgcctg 50831 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 831 ntcnttnttg cattcttcaa cttcnttnaa nacttgagcg gagtcggtgc 50832 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 832 naanactgcn cagaggttna atcgatgtgt gataggatca gtgttnaggg 50 833 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 833 gaaggcgaag gagactgcta atatngataa atatgtgaag gaaatgcctg 50834 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 834 gaangtatga gcatgngaga gacgntgtag ttggaaaaan ccacgaagng 50 835 50 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 835 gaaaccgctg attatactgc ggagaaggtg ggtgagtata aagactatac 50836 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 836 aagtagattt gttatttccg aaacgncttc tnccgttntt 40 837 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 837 ancanaaata gtcnctnaaa aatcanaaga aaactcanaa 40 838 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 838 gnagnggtgg taaaaagtat gaaaangtgg taattaaaag 40839 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 839 gaaaactaat naaaggtaaa cgtggatcnc atggnaattc 40840 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 840 cactgccnag aggttcaatn gataaatatg tgaaggaaat 40841 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 841 ctgcncagag gttnaatcga tcngatgatc ctaatgaagg 40842 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 842 gtatngtnca tcatagtatn gataaatatg tgaaggaaat 40843 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 843 tnttgcattc ttcaacttcn ttnaanactt gagcggagtc 40844 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 844 ctgcncagag gttnaatcga tgtgtgatag gatcagtgtt 40845 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 845 cgaaggagac tgctaatatn gataaatatg tgaaggaaat 40846 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 846 tatgagcatg ngagagacgn tgtagttgga aaaanccacg 40847 40 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 847 cgctgattat actgcggaga aggtgggtga gtataaagac 40848 23 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 848 attccattcg attccattcg atc 23849 24 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 849 ttagggttag ggttagggtt aggg 24850 15 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 850 acccagccaa aggag 15 851 24 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 851 tgtgtaccca gccaaaggag ttga 24852 32 DNA Artificial Sequence Synthetic Oligonucleotide Sequence 852 ccagctctaa gagcggcgcg cttactgcgt gt 32
Claims (184)
1. A nucleic acid that is a non-naturally-occurring nucleic acid with a melting temperature that is at least 3° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides; wherein said nucleic acid is capable of hybridizing to a first region within a first exon of a target nucleic acid and to a second region within a second exon of said target nucleic acid that is adjacent to said first exon.
2. A nucleic acid that is a non-naturally-occurring nucleic acid with a melting temperature that is at least 3° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides; wherein said nucleic acid is capable of hybridizing to a first region within an exon of a target nucleic acid and to a second region within an intron of said target nucleic acid that is adjacent to said exon.
3. A nucleic acid that is a non-naturally-occurring nucleic acid with a melting temperature that is at least 3° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides; wherein said nucleic acid is capable of hybridizing to a first region within a first intron of a target nucleic acid and to a second region within a second intron of said target nucleic acid that is adjacent to said first intron.
4. A nucleic acid that is a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of said nucleic acid; wherein said nucleic acid is capable of hybridizing to a first region within a first exon of a target nucleic acid and to a second region within a second exon of said target nucleic acid that is adjacent to said first exon.
5. A nucleic acid that is a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of said nucleic acid; wherein said nucleic acid is capable of hybridizing to a first region within an exon of a target nucleic acid and to a second region within an intron of said target nucleic acid that is adjacent to said exon.
6. A nucleic acid that is a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of said nucleic acid; wherein said nucleic acid is capable of hybridizing to a first region within a first intron of a target nucleic acid and to a second region within a second intron of said target nucleic acid that is adjacent to said first intron.
7. A nucleic acid that is an LNA capable of hybridizing to a first region within a first exon of a target nucleic acid and to a second region within a second exon of said target nucleic acid that is adjacent to said first exon.
8. A nucleic acid that is an LNA capable of hybridizing to a first region within an exon of a target nucleic acid and to a second region within an intron of said target nucleic acid that is adjacent to said exon.
9. A nucleic acid that is an LNA capable of hybridizing to a first region within a first intron of a target nucleic acid and to a second region within a second intron of said target nucleic acid that is adjacent to said first intron.
10. The nucleic acid of any one of claims 1-9, wherein the length of said first region and the length of said second region are between 3 and 50 nucleotides, inclusive.
11. The nucleic acid of claim 10 , wherein the length of said first region and the length of said second region are between 10 and 40 nucleotides, inclusive.
12. The nucleic acid of claim 11 , wherein the length of said first region and the length of said second region are between 20 and 30 nucleotides, inclusive.
13. The nucleic acid of any one of claims 1-9, wherein said first region and said second region are the same length.
14. The nucleic acid of any one of claims 1-9, wherein said first region and said second region are a different length.
15. A population of nucleic acids that includes a nucleic acid of any one of claims 1-1 4.
16. A population of nucleic acids that includes a non-naturally-occurring nucleic acid with a melting temperature that is at least 3° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides; wherein said nucleic acid is capable of hybridizing to only one exon or to only one intron of a target nucleic acid.
17. A population of nucleic acids that includes a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of said nucleic acid; wherein said nucleic acid is capable of hybridizing to only one exon or to only one intron of a target nucleic acid.
18. A population of nucleic acids that includes a nucleic acid that is an LNA capable of hybridizing to only one exon or to only one intron of a target nucleic acid.
19. A nucleic acid of any one of claims 1-9 that is a non-naturally occurring nucleic acid with a melting temperature that is at least 3° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides; wherein said nucleic acid is capable of hybridizing to only one exon or to only one intron of a target nucleic acid.
20. A nucleic acid of any one of claims 1-9 that is a non-naturally-occurring nucleic acid with a capture efficiency that is at least 10% greater than that of a corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of said nucleic acid; wherein said nucleic acid is capable of hybridizing to only one exon or to only one intron of a target nucleic acid.
21. A nucleic acid of any one of claims 1-9 that comprises an LNA capable of hybridizing to only one exon or to only one intron of a target nucleic acid.
22. The population of any one of claims 15-18, wherein said nucleic acid is between 15 and 150 nucleotides in length, inclusive.
23. The population of claim 22 , wherein said nucleic acid is between 5 and 100 nucleotides in length, inclusive.
24. The population of claim 23 , wherein said nucleic acid is between 20 and 80 nucleotides in length, inclusive.
25. The population of claim 24 , wherein said nucleic acid is between 30 and 60 nucleotides in length, inclusive.
26. The population of claim 25 , wherein said nucleic acid is 40 nucleotides in length.
27. The population of claim 25 , wherein said nucleic acid is 50 nucleotides in length.
28. The nucleic acid of any one of claims 1-9 and 19-21 wherein said nucleic acid is between 8 and 70 nucleotides in length.
29. The nucleic acid of any one of claims 1-9 and 19-21 wherein said nucleic acid is between 9 and 50 nucleotides in length.
30. The nucleic acid of any one of claims 1-9 and 19-21 wherein said nucleic acid is between 12 and 40 nucleotides in length.
31. The nucleic acid of any one of claims 1-9 and 19-21 wherein said nucleic acid is between 15 and 35 nucleotides in length.
32. The population of any one of claims 15-18, wherein at least 5% of the nucleotides in said nucleic acid are LNA units.
33. The population of claim 32 , wherein at least 10% of the nucleotides in said nucleic acid are LNA units.
34. The population of claim 32 , wherein at least 20% of the nucleotides in said nucleic acid are LNA units.
35. The population of any one of claims 15-18, wherein every second nucleotide in said nucleic acid is an LNA unit.
36. The population of any one of claims 15-18, wherein every third nucleotide in said nucleic acid is an LNA unit.
37. The population of any one of claims 15-18, wherein every fourth nucleotide in said nucleic acid is an LNA unit.
38. The population of any one of claims 15-18, wherein every fifth nucleotide in said nucleic acid is an LNA unit.
39. The population of any one of claims 15-18, wherein every sixth nucleotide in said nucleic acid is an LNA unit.
40. The population of any one of claims 15-18, wherein (i) every second and every third nucleotide, (ii) every second and every fourth nucleotide, (iii) every second and every fifth nucleotide, (iv) every second and every sixth nucleotide, (v) every third and every fourth nucleotide, (vi) every third and every fifth nucleotide, (vii) every third and every sixth nucleotide, (viii) every fourth and every fifth nucleotide, (ix) every fourth and every sixth nucleotide, and/or (x) every fifth and every sixth nucleotide in said nucleic acid is an LNA unit.
41. The population of claim 40 , wherein every second, every third, and every fourth nucleotide in said nucleic acid is an LNA unit.
42. The population of any one of claims 15-18, wherein said nucleic acid comprises two or more contiguous LNA units.
43. The population of claim 42 , wherein said nucleic acid comprises at least 4 contiguous LNA units.
44. The population of claim 42 , wherein said nucleic acid comprises at least 5 contiguous LNA units.
45. The population of claim 42 , wherein the number of contiguous LNA units is between 5 and 20% of the total length of said nucleic acid.
46. The population of claim 45 , wherein the number of contiguous LNA units is between 10 and 15% of the total length of said nucleic acid.
47. The population of claim 42 , wherein at least one LNA unit in said nucleic acid is capable of hybridizing to a first region within a first exon of a target nucleic acid and at least one LNA unit in said nucleic acid is capable of hybridizing to a second region within a second exon of said target nucleic acid that is adjacent to said first exon.
48. The population of claim 47 , wherein at least two LNA units in said nucleic acid hybridize to a first region within a first exon of a target nucleic acid and at least two LNA units in said nucleic acid hybridize to a second region within a second exon of said target nucleic acid that is adjacent to said first exon.
49. The population of claim 48 , wherein at least three LNA units in said nucleic acid hybridize to a first region within a first exon of a target nucleic acid and at least three LNA units in said nucleic acid hybridize to a second region within a second exon of said target nucleic acid that is adjacent to said first exon.
50. The population of any one of claims 15-18, wherein the 5′ terminal nucleotide of said nucleic acid is not an LNA unit.
51. The population of any one of claims 15-18, wherein the 5′ terminal nucleotide of said nucleic acid is an LNA unit.
52. The population of any one of claims 15-18, wherein the 3′ terminal nucleotide of said nucleic acid is not an LNA unit.
53. The population of any one of claims 15-18, wherein said nucleic acid can distinguish between different nucleic acids that cannot be distinguished using a naturally-occurring control nucleic acid.
54. The population of claim 53 , wherein said control nucleic acid consists of only 2′-deoxynucleotides.
55. The population of claim 53 , wherein said different nucleic acids are mRNA splice variants.
56. The population of any one of claims 15-18, wherein said nucleic acid comprises one or more universal bases.
57. The population of claim 56 , wherein said universal base is located at the 5′ or 3′ terminus of said nucleic acid.
58. The population of claim 56 , wherein one or more universal base are located at the 5′ and 3′ termini of said nucleic acid.
59. The population of claim 56 , wherein all of said nucleic acids of said population have the same number of universal bases.
60. The population of claim 56 , wherein said universal base is inosine, pyrene, 3-nitropyrrole, or 5-nitroindole.
61. The population of any one of claims 15-18, wherein said nucleic acid comprises at least one LNA A or LNA T.
62. The population of any one of claims 15-18, wherein each nucleic acid in said population comprises at least one LNA A or LNA T.
63. The population of claim 61 , wherein all of the adenine and thymine-containing nucleotides in.said LNA are LNA A and LNA T, respectively.
64. The population of any one of claims 15-18, wherein said nucleic acid comprises a at least one 2,6,-diaminopurine or 2-thio-thymine base.
65. The population of any one of claims 15-18, wherein at least 5% of the nucleic acids in said population are LNA.
66. The population of any one of claims 15-18, wherein at least 10% of the nucleic acid in said population are LNA.
67. The population of any one of claims 15-18, that includes nucleic acids that together hybridize to at least 10% of the nucleic acids expressed by a particular cell or tissue.
68. The population of claim 67 , that includes nucleic acids that together hybridize to at least 50% of the exons of a target nucleic acid.
69. The population of any one of claims 15-18, wherein said nucleic acid does not form a hairpin that would otherwise inhibit its binding to a target nucleic acid.
70. The population of any one of claims 15-18, wherein opposing nucleotides in a palindrome pair or opposing nucleotides in inverted repeats of said nucleic acid are not both LNA units.
71. The population of any one of claims 15-18, wherein said nucleic acid forms less than 3 intramolecular base-pairs.
72. The population of any one of claims 15-18, wherein said nucleic acid does not have LNA-5-nitroindole: LNA-5-nitroindole intramolecular base-pairs.
73. The population of any one of claims 15-18, wherein said nucleic acid has a LNA unit with a 2,6,-diaminopurine, 2-aminopurine, 2-thio-thymine, 2-thio-uracil, inosine, or hypoxanthine base.
74. The population of any one of claims 15-18, wherein said nucleic acid has a 2′O, 4° C.-methylene linkage.
75. The population of any one of claims 15-18, wherein one or more nucleic acids are LNA/DNA, LNA/RNA, or LNA/DNA/RNA chimeras.
76. The population of any one of claims 15-18, wherein said nucleic acid has a melting temperature that is at least 110C higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides.
77. The population of claim 76 , wherein said nucleic acid has a melting temperature that is at least 20° C. higher than that of the corresponding control nucleic acid with 2′-deoxynucleotides.
78. The population of any one of claims 15-18, wherein said nucleic acid has a capture efficiency that is at least 100% greater than that of the corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of said nucleic acid.
79. The population of claim 78 , wherein said nucleic acid has a capture efficiency that is at least 400% greater than that of the corresponding control nucleic acid with 2′-deoxynucleotides at the temperature equal to the melting temperature of said nucleic acid.
80. The population of any one of claims 15-18, wherein said nucleic acids are covalently bonded to a solid support.
81. The population of claim 80 , wherein said nucleic acids are in a predefined arrangement.
82. The population of any one of claims 15-18, comprising at least 10 different nucleic acids.
83. The population of claims 82, comprising at least 100 different nucleic acids.
84. The population of claim 83 , comprising at least 500 different nucleic acids.
85. The population of claim 84 , comprising at least 1,000 different nucleic acids.
86. The population of claim 85 , comprising at least 5,000 different nucleic acids.
87. A complex of one or more target nucleic acids and one or more nucleic acids or populations of any one of claims 1-9, 15-18 or 19-21 in which one or more target nucleic acids are hybridized to one or more said nucleic acids or populations.
88. The complex of claim 87 , wherein at least 10 different target nucleic acids are hybridized.
89. The complex of claim 87 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample.
90. The complex of claim 87 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase.
91. The complex of claim 87 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase to an average size of 300 nucleotides.
92. The complex of claim 87 , wherein the target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. Coli Uracil-DNA Glycosylase to an average size of 200 nucleotides.
93. The complex of claim 87 , wherein the target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase to an average size of 100 nucleotides.
94. The complex of claim 87 , wherein the target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase to an average size of 50 nucleotides.
95. The complex of claim 87 , wherein said target nucleic acids are cRNA molecules amplified from a sample.
96. The complex of claim 87 , wherein said target nucleic acids are cRNA molecules amplified from a sample and fragmented using alkaline hydrolysis.
97. The complex of claim 87 , wherein at least 10 different target nucleic acids are labeled and hybridized.
98. The complex of claim 87 , wherein said target nucleic acids are RNA molecules isolated from a sample and labeled chemically directly, e.g. using platinum-linked fluorescent dyes.
99. The complex of claim 87 , wherein said target nucleic acids are RNA molecules isolated from a sample and labeled chemically directly, e.g. using platinum-linked fluorescent dyes and fragmented using alkaline hydrolysis.
100. A method for detecting the presence of one or more target nucleic acids in a sample, said method comprising incubating a labeled nucleic acid sample with one or more first nucleic acids of any one of claims 1-9 or 19-21 or one or more populations of first nucleic acids of any one of claims 15-18 under conditions that allow at least one target nucleic acid to hybridize to at least one of said first nucleic acids.
101. The method of claim 100 , wherein hybridization is detected between at least 5 target nucleic acids and said first nucleic acids.
102. The method of claim 100 , further comprising identifying one or more hybridized target nucleic acids.
103. The method of claim 100 , further comprising determining the amount of one or more hybridized target nucleic acids.
104. The method of claim 100 , wherein one or more target nucleic acids are labeled with a fluorescent group, and wherein the determination of the amount of said hybridized target nucleic acid involves one or more of the following: (i) adjusting for the varying intensity of an excitation light source used for detection of said hybridization, (ii) adjusting for photobleaching of said fluorescent group, and/or (iii) comparing the fluorescent intensity of said hybridized target nucleic acid(s) to the fluorescent intensity of a different sample of hybridized nucleic acids.
105. The method of claim 100 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample.
106. The method of claim 105 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase.
107. The method of claim 105 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase to an average size of 300 nucleotides.
108. The method of claim 105 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase to an average size of 200 nucleotides.
109. The method of claim 105 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase to an average size of 100 nucleotides.
110. The method of claim 105 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase to a n average size of 50 nucleotides.
111. The method of claim 105 , wherein said target nucleic acids are cRNA molecules amplified from a sample that is optionally fragmented uing alkaline hydrolysis.
112. The method of claim 100 , further comprising determining the presence or absence of an mRNA splice variant of interest in said sample or determining the presence or absence of a mutation, deletion, and/or duplication of an exon of interest.
113. The method of claim 100 , wherein the sample has nucleic acids that are amplified using one or more primers specific for an exon of a target nucleic acid, and wherein said method involves determining the presence or absence of an mRNA splice variant with said exon in said sample.
114. The method of claim 113 , wherein one or more of said primers are specific for an exon or exon-exon junction of interest, and said method involves determining the presence or absence of a nucleic acid with said exon in said sample.
115. The method of claim 100 , wherein said first nucleic acids are covalently bonded to a solid support by reaction of a nucleoside phosphoramidite with an activated solid support, and subsequent reaction of a nucleoside phosphoramide with an activated nucleotide or nucleic acid bound to said solid support.
116. The method of claim 100 , further comprising contacting said target nucleic acid with a second nucleic acid or a population of second nucleic acids that binds to a different region of the target molecule than said first nucleic acid.
117. A method for amplifying a target nucleic acid, said method comprising the steps of: (a) incubating one or more first nucleic acids of any one of claims 1-9 or 19-21 or one or more populations of first nucleic acids of any one of claims 15-18 with a target nucleic acid under conditions that allow said first nucleic acid to bind said target nucleic acid; and (b) extending said first nucleic acid with said target nucleic acid as a template.
118. The method of claim 117 , further comprising contacting said target nucleic acid with a second nucleic acid or a population of second nucleic acids that binds to a different region of the target molecule than said first nucleic acid.
119. A method for classifying a test nucleic acid sample comprising a target nucleic acid, said method comprising the steps of: (a) incubating a test nucleic acid sample with one or more nucleic acids probes of any one of claims 1-9 or 19-21 or one or more populations of nucleic acids probes of any one of claims 15-18 under conditions that allow at least one of the nucleic acids in said test sample to hybridize to at least one nucleic acid probe; (b) detecting a hybridization pattern of said test nucleic acid sample; and (c) comparing said hybridization pattern to a hybridization pattern of a first nucleic acid standard, whereby said comparison indicates whether or not said test sample has the same classification as said first standard.
120. The method of claim 119 , further comprising comparing a hybridization pattern of said test nucleic acid sample to a hybridization pattern of a second standard.
121. The method of claim 119 , wherein at least 5 target nucleic acids hybridize to said nucleic acid probes.
122. The method of claim 119 , further comprising identifying a hybridized target nucleic acid.
123. The method of claim 119 , further comprising determining the amount of said hybridized target nucleic acid.
124. The method of claim 119 , wherein said target nucleic acids are labeled with fluorescent groups.
125. The method of claim 124 , wherein said determination comprises scaling for the varying labeling efficiency for the different fluorescent groups used for detection of said hybridization.
126. The method of claim 124 , wherein said determination comprises adjusting for the varying intensity of the excitation light source used for detection of said hybridization.
127. The method of claim 124 , wherein said determination comprises adjusting for photobleaching of said fluorescent group.
128. The method of claim 124 , wherein said comparison comprises adjusting for a difference in the amount of said nucleic acid probes used for hybridization to said test sample and said first standard.
129. The method of claim 124 , wherein said comparison comprises adjusting for a difference in the buffer used for hybridization to said test sample and said first standard.
130. The method of claim 129 , wherein said difference is a difference in Na+ concentration.
131. The method of claim 124 , wherein said first nucleic acid standard is labeled with a different fluorescent group.
132. The method of claim 119 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and optionally fragmented using E. coli Uracil-DNA Glycosylase.
133. The method of claim 119 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase to an average size of 300 nucleotides.
134. The method of claim 119 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase to an average size of 200 nucleotides.
135. The method of claim 119 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. coli Uracil-DNA Glycosylase to an average size of 100 nucleotides.
136. The method of claim 119 , wherein said target nucleic acids are cDNA molecules reverse transcribed from a sample and fragmented using E. Coli Uracil-DNA Glycosylase to an average size of 50 nucleotides.
137. The method of claim 119 , wherein said target nucleic acids are cRNA molecules amplified from a sample.
138. The method of claim 119 , wherein said target nucleic acids are cRNA molecules amplified from a sample and fragmented using alkaline hydrolysis.
139. The method of claim 119 , further comprising determining the presence or absence of an mRNA splice variant of interest in said sample.
140. The method of claim 119 , further comprising determining the presence or absence of a mutation, deletion, and/or duplication of an exon of interest.
141. The method of claim 140 , wherein said mutation, deletion, and/or duplication is indicative of a disease, disorder, or condition.
142. The method of claim 141 , wherein said disease is cancer.
143. The method of claim 119 , wherein the sample has nucleic acids that are amplified using one or more primers specific for an exon of a target nucleic acid, and wherein said method involves determining the presence or absence of an mRNA splice variant with said exon in said sample.
144. The method of claim 143 , wherein one or more of said primers are specific for an exon or exon-exon junction of interest, and said method involves determining the presence or absence of a nucleic acid with said exon in said sample.
145. The method of claim 119 , wherein said first nucleic acids are covalently bonded to a solid support by reaction of a nucleoside phosphoramidite with an activated solid support, and subsequent reaction of a nucleoside phosphoramide with an activated nucleotide or nucleic acid bound to said solid support.
146. A method of selecting a nucleic acid for a population of nucleic acids, said method comprising the steps of: (a) determining the melting temperature of a nucleic acid, determining the ability of said nucleic acid to self-anneal, determining the ability of said nucleic acid to hybridize to one or more exons or introns of a target nucleic acid, and/or determining the ability of said nucleic acid to hybridize to a non-target nucleic acid, and (b) selecting said nucleic acid for inclusion or exclusion from said population based on the determination in step (a), wherein said nucleic acid is a nucleic acid of any one of claims 1-9 or 19-21 or a nucleic acid that has least one LNA unit and that is capable of hybridizing to only one exon or to only one intron of a target nucleic acid.
147. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 for the detection, amplification, or classification of a nucleic acid of interest or a population of nucleic acids of interest.
148. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 for alternative mRNA splice variant detection, expression profiling, comparative genomic hybridization, or real-time PCR.
149. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 as a PCR primer or FISH probe.
150. Use according to claim 149 wherein said nucleic acid or said population of nucleic acids comprise LNA in about every other or about every third position of the nucleotide sequence.
151. Use according to claim 149 wherein said nucleic acid or said population of nucleic acids are used for the detection of a repetitive element.
152. Use according to claim 151 wherein said repetitive element is a centromeric alpha-repeat or a telomeric repeat.
153. Use according to claim 149 wherein said nucleic acid or said population of nucleic acids are used for the detection of a single base pair difference between repetitive sequences or for the detection of single copy sequences.
154. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 as a probe in fluorescent in situ hybridisation of a target DNA without a denaturation step.
155. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 in a homogeneous assay such as quantitative real-time RT-PCR assay, e.g. Taqman real-time RT-PCR, Lightcycler real-time RT-PCR, or real-time NASBA (nucleic acid sequence-based amplification) assay with a Molecular Beacon-based detection.
156. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 in a diagnostic kit based on a homogeneous assay such as quantitative real-time RT-PCR assay, e.g. Taqman real-time RT-PCR, Lightcycler real-time RT-PCR, or real-time NASBA (nucleic acid sequence-based amplification) assay with a Molecular Beacon-based detection.
157. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 as an antisense, antigene or ribozyme or double stranded nucleic acid therapeutic agent, or in therapeutic use as modulating and silencing sense nucleic acid agents.
158. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 as an antisense, antigene or ribozyme or double stranded nucleic acid therapeutic agent, in which the said nucleic acid or population of nucleic acids hybridize to specific splice isofoms or isoforms.
159. Use of a nucleic acid or a population of nucleic acids of any one of claims 1-9 or 19-21 or of claims 15-18 as an antisense, antigene or ribozyme or double stranded nucleic acid therapeutic agent, in which the said nucleic acid or population of nucleic acids hybridize to a non-coding antisense RNA or RNAs.
160. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 in a diagnostic kit, based on detection of diagnostic splice isoforms or splice patterns, or diagnostic nucleic acids, such as viral or bacterial mRNAs.
161. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 in a diagnostic microarray based on detection of mRNA signatures or splice isoform signatures in the concentration range of 10000 ppm to 1000 ppm.
162. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 in a diagnostic microarray based on detection of mRNA signatures or splice isoform signatures in the concentration range of 1000 ppm to 100 ppm.
163. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 in a diagnostic microarray based on detection of mRNA signatures or splice isoform signatures in the concentration range of 100 ppm to 10 ppm.
164. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 in a diagnostic microarray based on detection of mRNA signatures or splice isoform signatures in the concentration range of 10 ppm to 1 ppm.
165. Use of a nucleic acid of any one of claims 1-9 or 19-21 or a population of nucleic acids of any one of claims 15-18 in a diagnostic microarray based on detection of mRNA signatures or splice isoform signatures.
166. Use of a non-naturally occurring nucleic acid of any one of claims 1-9 or 19-21 or use of a population of non-naturally occurring nucleic acids of any one of claims 15-18 in a method for generating an array of said non-naturally occurring nucleic acids of chosen lengths within discrete locations of a support material.
167. Use according to claim 166 , where said non-naturally occurring nucleic acids are LNA oligonucleotides.
168. Use according to any one of claims 166-167, where said non-naturally occurring nucleic acids are LNA oligonucleotides containing a photochemically active group at the 5′ end or the 3′ end, separated by a linker from the LNA oligonucleotides.
169. Use according to any one of claims 166-168, wherein said linker is a hexaethylene monomer, dimer, trimer, tetramer, pentamer or hexamer, or a poly-T sequence of 10-50 nucleotides in length, or a poly-C sequence of 10-50 nucleotides in length.
170. Use according to any one of claims 168-169 for generating an array comprising the steps of a) selecting said nucleic acid or population of nucleic acids according to the method of claim 146 , b) synthesis of the said nucleic acids using phoshoramidite chemistry and DNA and LNA phosphoramidites; c) purification of the said nucleic acids; d) printing of the said nucleic acids of chosen lengths and sequence onto a polymer surface; and e) coupling of said nucleic acids via excitation of the photochemically active group at the 5′ end or the 3′ end covalently onto the polymer surface using UV light.
171. The method of claims 166-170 wherein a computer-controlled microarray printing robot delivers the said nucleic acids onto discrete locations on the polymer surface.
172. The method of claims 166-171, wherein the size of each discrete location is between an average size of 10 and 150 microns.
173. The method of claims 166-172, wherein said nucleic acids are printed at a density of at least 50 nucleic acids per square cm.
174. The method of claims 166-172, wherein said nucleic acids are printed at a density of at least 100 nucleic acids per square cm.
175. The method of claims 166-172, wherein said nucleic acids are printed at a density of at least 500 nucleic acids per square cm.
176. The method of claims 166-172, wherein said nucleic acids are printed at a density of at least 1000 nucleic acids per square cm.
177. The method of claims 166-172, wherein said nucleic acids are printed at a density of at least 2500 nucleic acids per square cm.
178. The method of claims 166-172, wherein said nucleic acids are printed at a density of at least 4000 nucleic acids per square cm.
179. The method of claims 166-172, wherein said nucleic acids are printed at a density of at least 10000 nucleic acids per square cm.
180. The method of claim 139 , wherein said mRNA splice variant is indicative of a disease, disorder, or condition.
181. The method of claim 180 , wherein said disease is cancer.
182. Use according to claim 149 wherein said nucleic acid or said population of nucleic acids are used for the detection of a repetitive element in human chromosomes 13 and 21.
183. Use according to claim 153 wherein said nucleic acid or said population of nucleic acids are used for the detection of a single base pair difference between repetitive sequences of human chromosomes 13 and 21.
184. Use according to claim 183 wherein said repetitive element is a centromeric alpha-repeat.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/690,487 US20040219565A1 (en) | 2002-10-21 | 2003-10-21 | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
| US11/643,615 US20070117144A1 (en) | 2002-10-21 | 2006-12-21 | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
| US13/295,615 US9464106B2 (en) | 2002-10-21 | 2011-11-14 | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US42027802P | 2002-10-21 | 2002-10-21 | |
| DKPA200300752 | 2003-05-19 | ||
| DKPA200300752 | 2003-05-19 | ||
| US10/690,487 US20040219565A1 (en) | 2002-10-21 | 2003-10-21 | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/643,615 Division US20070117144A1 (en) | 2002-10-21 | 2006-12-21 | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20040219565A1 true US20040219565A1 (en) | 2004-11-04 |
Family
ID=43216788
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/690,487 Abandoned US20040219565A1 (en) | 2002-10-21 | 2003-10-21 | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
| US11/643,615 Abandoned US20070117144A1 (en) | 2002-10-21 | 2006-12-21 | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
| US13/295,615 Expired - Lifetime US9464106B2 (en) | 2002-10-21 | 2011-11-14 | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/643,615 Abandoned US20070117144A1 (en) | 2002-10-21 | 2006-12-21 | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
| US13/295,615 Expired - Lifetime US9464106B2 (en) | 2002-10-21 | 2011-11-14 | Oligonucleotides useful for detecting and analyzing nucleic acids of interest |
Country Status (1)
| Country | Link |
|---|---|
| US (3) | US20040219565A1 (en) |
Cited By (122)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060142228A1 (en) * | 2004-12-23 | 2006-06-29 | Ambion, Inc. | Methods and compositions concerning siRNA's as mediators of RNA interference |
| WO2007097876A2 (en) * | 2006-02-15 | 2007-08-30 | Agilent Technologies, Inc. | Normalization probes for comparative genome hybridization arrays |
| US20070259363A1 (en) * | 2006-04-28 | 2007-11-08 | Hakima Amri | Phylogenetic Analysis of Mass Spectrometry or Gene Array Data for the Diagnosis of Physiological Conditions |
| WO2007146511A2 (en) | 2006-05-05 | 2007-12-21 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating gene expression |
| US20080003573A1 (en) * | 2006-06-28 | 2008-01-03 | Bio-Id Diagnostic Inc. | Determination of variants produced upon replication or transcription of nucleic acid sequences |
| US20080014579A1 (en) * | 2003-02-11 | 2008-01-17 | Affymetrix, Inc. | Gene expression profiling in colon cancers |
| US20080188005A1 (en) * | 2007-01-10 | 2008-08-07 | Chunnian Shi | Inhibition of mismatch hybridization by a universal competitor dna |
| WO2008098248A2 (en) | 2007-02-09 | 2008-08-14 | Northwestern University | Particles for detecting intracellular targets |
| WO2008150729A2 (en) | 2007-05-30 | 2008-12-11 | Isis Pharmaceuticals, Inc. | N-substituted-aminomethylene bridged bicyclic nucleic acid analogs |
| US20080311669A1 (en) * | 2007-06-04 | 2008-12-18 | Northwestern University | Screening sequence selectivity of oligonucleotide-binding molecules using nanoparticle based colorimetric assay |
| WO2009149182A1 (en) | 2008-06-04 | 2009-12-10 | The Board Of Regents Of The University Of Texas System | Modulation of gene expression through endogenous small rna targeting of gene promoters |
| US20100015604A1 (en) * | 2005-08-17 | 2010-01-21 | Evriklia Lianidou | Composition and method for determination of ck19 expression |
| WO2010014592A1 (en) | 2008-07-29 | 2010-02-04 | The Board Of Regents Of The University Of Texas Sytem | Selective inhibition of polyglutamine protein expression |
| US7666854B2 (en) | 2006-05-11 | 2010-02-23 | Isis Pharmaceuticals, Inc. | Bis-modified bicyclic nucleic acid analogs |
| US20100144834A1 (en) * | 2006-11-27 | 2010-06-10 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US7741457B2 (en) | 2006-01-27 | 2010-06-22 | Isis Pharmaceuticals, Inc. | 6-modified bicyclic nucleic acid analogs |
| US7750131B2 (en) | 2006-05-11 | 2010-07-06 | Isis Pharmaceuticals, Inc. | 5′-modified bicyclic nucleic acid analogs |
| US20100222414A1 (en) * | 2007-09-19 | 2010-09-02 | Applied Biosystems, Llc | SiRNA Sequence-Independent Modification Formats for Reducing Off-Target Phenotypic Effects in RNAi, and Stabilized Forms Thereof |
| WO2010120420A1 (en) | 2009-04-15 | 2010-10-21 | Northwestern University | Delivery of oligonucleotide-functionalized nanoparticles |
| WO2010124231A2 (en) | 2009-04-24 | 2010-10-28 | The Board Of Regents Of The University Of Texas System | Modulation of gene expression using oligomers that target gene regions downstream of 3' untranslated regions |
| US20100279305A1 (en) * | 2008-01-14 | 2010-11-04 | Applied Biosystems, Llc | Compositions, methods, and kits for detecting ribonucleic acid |
| US20100331538A1 (en) * | 2007-11-21 | 2010-12-30 | Seth Punit P | Carbocyclic alpha-l-bicyclic nucleic acid analogs |
| WO2011017521A2 (en) | 2009-08-06 | 2011-02-10 | Isis Pharmaceuticals, Inc. | Bicyclic cyclohexose nucleic acid analogs |
| WO2011053994A1 (en) | 2009-11-02 | 2011-05-05 | Alnylam Pharmaceuticals, Inc. | Modulation of ldl receptor gene expression with double-stranded rnas targeting the ldl receptor gene promoter |
| WO2011056215A1 (en) | 2009-11-03 | 2011-05-12 | Landers James P | Versatile, visible method for detecting polymeric analytes |
| WO2011056687A2 (en) | 2009-10-27 | 2011-05-12 | Swift Biosciences, Inc. | Polynucleotide primers and probes |
| WO2011063403A1 (en) | 2009-11-23 | 2011-05-26 | Swift Biosciences, Inc. | Devices to extend single stranded target molecules |
| WO2011085102A1 (en) | 2010-01-11 | 2011-07-14 | Isis Pharmaceuticals, Inc. | Base modified bicyclic nucleosides and oligomeric compounds prepared therefrom |
| JP2011522556A (en) * | 2008-06-13 | 2011-08-04 | ナショナル ユニバーシティー オブ アイルランド, ゴールウェイ | SWI5 gene as a diagnostic target for the identification of fungal and yeast species |
| WO2011097388A1 (en) | 2010-02-03 | 2011-08-11 | Alnylam Pharmaceuticals, Inc. | Selective inhibition of polyglutamine protein expression |
| US8008010B1 (en) | 2007-06-27 | 2011-08-30 | Applied Biosystems, Llc | Chimeric oligonucleotides for ligation-enhanced nucleic acid detection, methods and compositions therefor |
| WO2011113054A2 (en) | 2010-03-12 | 2011-09-15 | Aurasense Llc | Crosslinked polynucleotide structure |
| US8022046B2 (en) | 2008-04-18 | 2011-09-20 | Baxter International, Inc. | Microsphere-based composition for preventing and/or reversing new-onset autoimmune diabetes |
| WO2011115818A1 (en) | 2010-03-17 | 2011-09-22 | Isis Pharmaceuticals, Inc. | 5'-substituted bicyclic nucleosides and oligomeric compounds prepared therefrom |
| WO2011133695A2 (en) | 2010-04-20 | 2011-10-27 | Swift Biosciences, Inc. | Materials and methods for nucleic acid fractionation by solid phase entrapment and enzyme-mediated detachment |
| WO2011139702A2 (en) | 2010-04-28 | 2011-11-10 | Isis Pharmaceuticals, Inc. | Modified nucleosides and oligomeric compounds prepared therefrom |
| WO2011150226A1 (en) | 2010-05-26 | 2011-12-01 | Landers James P | Method for detecting nucleic acids based on aggregate formation |
| WO2011156278A1 (en) | 2010-06-07 | 2011-12-15 | Isis Pharmaceuticals, Inc. | Bicyclic nucleosides and oligomeric compounds prepared therefrom |
| US8093222B2 (en) | 2006-11-27 | 2012-01-10 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| EP2410054A1 (en) | 2006-10-18 | 2012-01-25 | Isis Pharmaceuticals, Inc. | Antisense compounds |
| US20120029891A1 (en) * | 2010-08-02 | 2012-02-02 | Integrated Dna Technologies, Inc. | Methods for Predicting Stability and Melting Temperatures of Nucleic Acid Duplexes |
| WO2012106509A1 (en) | 2011-02-02 | 2012-08-09 | The Trustees Of Princeton University | Sirtuin modulators as virus production modulators |
| US8252756B2 (en) | 2005-06-14 | 2012-08-28 | Northwestern University | Nucleic acid functionalized nanoparticles for therapeutic applications |
| US8278426B2 (en) | 2007-06-08 | 2012-10-02 | Isis Pharmaceuticals, Inc. | Carbocyclic bicyclic nucleic acid analogs |
| US8278283B2 (en) | 2007-07-05 | 2012-10-02 | Isis Pharmaceuticals, Inc. | 6-disubstituted or unsaturated bicyclic nucleic acid analogs |
| EP2505647A1 (en) | 2006-05-05 | 2012-10-03 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of DGAT2 |
| WO2012149154A1 (en) | 2011-04-26 | 2012-11-01 | Swift Biosciences, Inc. | Polynucleotide primers and probes |
| WO2012151268A1 (en) | 2011-05-02 | 2012-11-08 | University Of Virginia Patent Foundation | Method and system for high throughput optical and label free detection of analytes |
| WO2012151289A2 (en) | 2011-05-02 | 2012-11-08 | University Of Virginia Patent Foundation | Method and system to detect aggregate formation on a substrate |
| US20120303283A1 (en) * | 2011-01-19 | 2012-11-29 | Jian Han | Polymerase Preference Index |
| WO2013040499A1 (en) | 2011-09-14 | 2013-03-21 | Northwestern University | Nanoconjugates able to cross the blood-brain barrier |
| US8501805B2 (en) | 2008-09-24 | 2013-08-06 | Isis Pharmaceuticals, Inc. | Substituted alpha-L-bicyclic nucleosides |
| US8518908B2 (en) | 2009-09-10 | 2013-08-27 | University Of Idaho | Nucleobase-functionalized conformationally restricted nucleotides and oligonucleotides for targeting of nucleic acids |
| US8524680B2 (en) | 2002-02-01 | 2013-09-03 | Applied Biosystems, Llc | High potency siRNAS for reducing the expression of target genes |
| US8530640B2 (en) | 2008-02-07 | 2013-09-10 | Isis Pharmaceuticals, Inc. | Bicyclic cyclohexitol nucleic acid analogs |
| WO2013138536A1 (en) | 2012-03-13 | 2013-09-19 | Swift Biosciences, Inc. | Methods and compositions for size-controlled homopolymer tailing of substrate polynucleotides by a nucleic acid polymerase |
| WO2013154798A1 (en) | 2012-04-09 | 2013-10-17 | Isis Pharmaceuticals, Inc. | Tricyclic nucleic acid analogs |
| WO2013154799A1 (en) | 2012-04-09 | 2013-10-17 | Isis Pharmaceuticals, Inc. | Tricyclic nucleosides and oligomeric compounds prepared therefrom |
| WO2014045126A2 (en) | 2012-09-18 | 2014-03-27 | Uti Limited Partnership | Treatment of pain by inhibition of usp5 de-ubiquitinase |
| US20140087962A1 (en) * | 2011-03-22 | 2014-03-27 | Life Technologies Corporation | Identification of Linkage Using Multiplex Digital PCR |
| US20140120540A1 (en) * | 2011-07-01 | 2014-05-01 | Htg Molecular Diagnostics, Inc. | Methods of detecting gene fusions |
| US8815821B2 (en) | 2002-02-01 | 2014-08-26 | Life Technologies Corporation | Double-stranded oligonucleotides |
| WO2014134179A1 (en) | 2013-02-28 | 2014-09-04 | The Board Of Regents Of The University Of Texas System | Methods for classifying a cancer as susceptible to tmepai-directed therapies and treating such cancers |
| US8889350B2 (en) | 2010-03-26 | 2014-11-18 | Swift Biosciences, Inc. | Methods and compositions for isolating polynucleotides |
| EP2826863A1 (en) | 2007-05-30 | 2015-01-21 | Northwestern University | Nucleic acid functionalized nanoparticles for therapeutic applications |
| US9029335B2 (en) | 2012-10-16 | 2015-05-12 | Isis Pharmaceuticals, Inc. | Substituted 2′-thio-bicyclic nucleosides and oligomeric compounds prepared therefrom |
| US9139827B2 (en) | 2008-11-24 | 2015-09-22 | Northwestern University | Polyvalent RNA-nanoparticle compositions |
| WO2015168172A1 (en) | 2014-04-28 | 2015-11-05 | Isis Pharmaceuticals, Inc. | Linkage modified oligomeric compounds |
| WO2016028940A1 (en) | 2014-08-19 | 2016-02-25 | Northwestern University | Protein/oligonucleotide core-shell nanoparticle therapeutics |
| US9273349B2 (en) | 2013-03-14 | 2016-03-01 | Affymetrix, Inc. | Detection of nucleic acids |
| US9290534B2 (en) | 2008-04-04 | 2016-03-22 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds having at least one neutrally linked terminal bicyclic nucleoside |
| WO2016081911A2 (en) | 2014-11-21 | 2016-05-26 | Northwestern University | The sequence-specific cellular uptake of spherical nucleic acid nanoparticle conjugates |
| WO2016100716A1 (en) | 2014-12-18 | 2016-06-23 | Vasant Jadhav | Reversirtm compounds |
| US9376690B2 (en) | 2009-10-30 | 2016-06-28 | Northwestern University | Templated nanoconjugates |
| WO2017015109A1 (en) | 2015-07-17 | 2017-01-26 | Alnylam Pharmaceuticals, Inc. | Multi-targeted single entity conjugates |
| US20170029818A1 (en) * | 2012-01-27 | 2017-02-02 | Biomarin Technologies B.V. | RNA Modulating Oligonucleotides with Improved Characteristics for the Treatment of Duchenne and Becker Muscular Dystrophy |
| US9688707B2 (en) | 2014-12-30 | 2017-06-27 | Ionis Pharmaceuticals, Inc. | Bicyclic morpholino compounds and oligomeric compounds prepared therefrom |
| US9695418B2 (en) | 2012-10-11 | 2017-07-04 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleosides and uses thereof |
| US9777275B2 (en) | 2002-02-01 | 2017-10-03 | Life Technologies Corporation | Oligonucleotide compositions with enhanced efficiency |
| US20170298415A1 (en) * | 2014-09-30 | 2017-10-19 | Ge Healthcare Bio-Sciences Corp. | Method for nucleic acid analysis directly from an unpurified biological sample |
| US9879222B2 (en) | 2007-12-14 | 2018-01-30 | Mofa Group Llc | Gender-specific separation of sperm cells and embryos |
| US9885082B2 (en) | 2011-07-19 | 2018-02-06 | University Of Idaho | Embodiments of a probe and method for targeting nucleic acids |
| US9914922B2 (en) | 2012-04-20 | 2018-03-13 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleotides and uses thereof |
| US10017764B2 (en) | 2011-02-08 | 2018-07-10 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleotides and uses thereof |
| US10098958B2 (en) | 2009-01-08 | 2018-10-16 | Northwestern University | Delivery of oligonucleotide functionalized nanoparticles |
| CN108754010A (en) * | 2018-06-14 | 2018-11-06 | 中国农业科学院蔬菜花卉研究所 | It is a kind of quickly to detect the remaining method of genomic DNA in total serum IgE sample |
| US10182988B2 (en) | 2013-12-03 | 2019-01-22 | Northwestern University | Liposomal particles, methods of making same and uses thereof |
| US10301622B2 (en) | 2013-11-04 | 2019-05-28 | Northwestern University | Quantification and spatio-temporal tracking of a target using a spherical nucleic acid (SNA) |
| US10385388B2 (en) | 2013-12-06 | 2019-08-20 | Swift Biosciences, Inc. | Cleavable competitor polynucleotides |
| WO2019217459A1 (en) | 2018-05-07 | 2019-11-14 | Alnylam Pharmaceuticals, Inc. | Extrahepatic delivery |
| WO2020056341A2 (en) | 2018-09-14 | 2020-03-19 | Northwestern University | Programming protein polymerization with dna |
| WO2020069055A1 (en) | 2018-09-28 | 2020-04-02 | Alnylam Pharmaceuticals, Inc. | Transthyretin (ttr) irna compositions and methods of use thereof for treating or preventing ttr-associated ocular diseases |
| WO2020118259A1 (en) | 2018-12-06 | 2020-06-11 | Northwestern University | Protein crystal engineering through dna hybridization interactions |
| WO2020226960A1 (en) | 2019-05-03 | 2020-11-12 | Dicerna Pharmaceuticals, Inc. | Double-stranded nucleic acid inhibitor molecules with shortened sense strands |
| WO2020236600A1 (en) | 2019-05-17 | 2020-11-26 | Alnylam Pharmaceuticals, Inc. | Oral delivery of oligonucleotides |
| US10876114B2 (en) | 2007-10-26 | 2020-12-29 | Biomarin Technologies B.V. | Methods and means for efficient skipping of at least one of the following exons of the human Duchenne muscular dystrophy gene: 43, 46, 50-53 |
| USRE48468E1 (en) | 2007-10-26 | 2021-03-16 | Biomarin Technologies B.V. | Means and methods for counteracting muscle disorders |
| WO2021092371A2 (en) | 2019-11-06 | 2021-05-14 | Alnylam Pharmaceuticals, Inc. | Extrahepatic delivery |
| WO2021092145A1 (en) | 2019-11-06 | 2021-05-14 | Alnylam Pharmaceuticals, Inc. | Transthyretin (ttr) irna composition and methods of use thereof for treating or preventing ttr-associated ocular diseases |
| CN114585749A (en) * | 2019-10-16 | 2022-06-03 | 斯蒂拉科技公司 | Determination of the concentration of nucleic acid sequences |
| US11364304B2 (en) | 2016-08-25 | 2022-06-21 | Northwestern University | Crosslinked micellar spherical nucleic acids |
| WO2022147223A2 (en) | 2020-12-31 | 2022-07-07 | Alnylam Pharmaceuticals, Inc. | 2'-modified nucleoside based oligonucleotide prodrugs |
| WO2022147214A2 (en) | 2020-12-31 | 2022-07-07 | Alnylam Pharmaceuticals, Inc. | Cyclic-disulfide modified phosphate based oligonucleotide prodrugs |
| WO2022192038A1 (en) | 2021-03-12 | 2022-09-15 | Northwestern University | Antiviral vaccines using spherical nucleic acids |
| WO2022213118A1 (en) | 2021-03-31 | 2022-10-06 | Entrada Therapeutics, Inc. | Cyclic cell penetrating peptides |
| US11466311B2 (en) * | 2014-10-08 | 2022-10-11 | Cornell University | Method for identification and quantification of nucleic acid expression, splice variant, translocation, copy number, or methylation changes |
| WO2022240721A1 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | Compositions and methods for modulating interferon regulatory factor-5 (irf-5) activity |
| WO2022241408A1 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | Compositions and methods for modulating tissue distribution of intracellular therapeutics |
| WO2022240760A2 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | COMPOSITIONS AND METHODS FOR MODULATING mRNA SPLICING |
| WO2022271818A1 (en) | 2021-06-23 | 2022-12-29 | Entrada Therapeutics, Inc. | Antisense compounds and methods for targeting cug repeats |
| WO2023283403A2 (en) | 2021-07-09 | 2023-01-12 | Alnylam Pharmaceuticals, Inc. | Bis-rnai compounds for cns delivery |
| WO2023003922A1 (en) | 2021-07-21 | 2023-01-26 | Alnylam Pharmaceuticals, Inc. | Metabolic disorder-associated target gene irna compositions and methods of use thereof |
| WO2023034817A1 (en) | 2021-09-01 | 2023-03-09 | Entrada Therapeutics, Inc. | Compounds and methods for skipping exon 44 in duchenne muscular dystrophy |
| WO2023064530A1 (en) | 2021-10-15 | 2023-04-20 | Alnylam Pharmaceuticals, Inc. | Extra-hepatic delivery irna compositions and methods of use thereof |
| US11690920B2 (en) | 2017-07-13 | 2023-07-04 | Northwestern University | General and direct method for preparing oligonucleotide-functionalized metal-organic framework nanoparticles |
| US11732261B2 (en) | 2011-08-11 | 2023-08-22 | Ionis Pharmaceuticals, Inc. | Selective antisense compounds and uses thereof |
| WO2023220744A2 (en) | 2022-05-13 | 2023-11-16 | Alnylam Pharmaceuticals, Inc. | Single-stranded loop oligonucleotides |
| WO2024006999A2 (en) | 2022-06-30 | 2024-01-04 | Alnylam Pharmaceuticals, Inc. | Cyclic-disulfide modified phosphate based oligonucleotide prodrugs |
| WO2024040041A1 (en) | 2022-08-15 | 2024-02-22 | Dicerna Pharmaceuticals, Inc. | Regulation of activity of rnai molecules |
| WO2024039776A2 (en) | 2022-08-18 | 2024-02-22 | Alnylam Pharmaceuticals, Inc. | Universal non-targeting sirna compositions and methods of use thereof |
| WO2024073732A1 (en) | 2022-09-30 | 2024-04-04 | Alnylam Pharmaceuticals, Inc. | Modified double-stranded rna agents |
| WO2024168010A2 (en) | 2023-02-09 | 2024-08-15 | Alnylam Pharmaceuticals, Inc. | Reversir molecules and methods of use thereof |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2385828T3 (en) | 2003-06-20 | 2012-08-01 | Exiqon A/S | Probes, libraries and kits for analysis of nucleic acid mixtures and procedures for building them |
| WO2010039275A1 (en) * | 2008-10-03 | 2010-04-08 | Oligonix, Inc. | Method, array and system for detecting intergenic fusions |
| NO2576783T3 (en) * | 2010-05-26 | 2018-04-28 | ||
| EP3702460A1 (en) | 2010-11-12 | 2020-09-02 | The General Hospital Corporation | Polycomb-associated non-coding rnas |
| US9920317B2 (en) | 2010-11-12 | 2018-03-20 | The General Hospital Corporation | Polycomb-associated non-coding RNAs |
| DK2850186T3 (en) | 2012-05-16 | 2019-04-08 | Translate Bio Ma Inc | COMPOSITIONS AND PROCEDURES FOR MODULATING SMN GENFAMILY EXPRESSION |
| WO2013173598A1 (en) | 2012-05-16 | 2013-11-21 | Rana Therapeutics, Inc. | Compositions and methods for modulating atp2a2 expression |
| WO2013173645A1 (en) | 2012-05-16 | 2013-11-21 | Rana Therapeutics, Inc. | Compositions and methods for modulating utrn expression |
| US10174315B2 (en) | 2012-05-16 | 2019-01-08 | The General Hospital Corporation | Compositions and methods for modulating hemoglobin gene family expression |
| US10837014B2 (en) | 2012-05-16 | 2020-11-17 | Translate Bio Ma, Inc. | Compositions and methods for modulating SMN gene family expression |
| EP2850190B1 (en) | 2012-05-16 | 2020-07-08 | Translate Bio MA, Inc. | Compositions and methods for modulating mecp2 expression |
| KR102324117B1 (en) | 2013-11-22 | 2021-11-10 | 오리온 디아그노스티카 오와이 | Detection of nucleic acids by strand invasion based amplification |
| GB201410022D0 (en) | 2014-06-05 | 2014-07-16 | Orion Diagnostica Oy | Method |
| CA2966044A1 (en) | 2014-10-30 | 2016-05-06 | The General Hospital Corporation | Methods for modulating atrx-dependent gene repression |
| EP3256591A4 (en) | 2015-02-13 | 2018-08-08 | Translate Bio Ma, Inc. | Hybrid oligonucleotides and uses thereof |
| WO2016149455A2 (en) | 2015-03-17 | 2016-09-22 | The General Hospital Corporation | The rna interactome of polycomb repressive complex 1 (prc1) |
| WO2017004203A1 (en) * | 2015-06-29 | 2017-01-05 | Fluidigm Canada Inc. | Systems, methods and compositions for simultaneous detection of rna and protein by mass spectrometry |
| US11497758B2 (en) | 2018-11-15 | 2022-11-15 | Samuel L. Shepherd | Method of treating oxidative stress due to radiation exposure |
| CA3103750A1 (en) * | 2018-12-17 | 2020-06-25 | Illumina Cambridge Limited | Primer oligonucleotide for sequencing |
| AU2022204064A1 (en) | 2021-07-16 | 2023-02-02 | Samuel L. Shepherd | Method of treating oxidative stress due to radiation exposure |
Citations (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) * | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4806463A (en) * | 1986-05-23 | 1989-02-21 | Worcester Foundation For Experimental Biology | Inhibition of HTLV-III by exogenous oligonucleotides |
| US5432272A (en) * | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5445934A (en) * | 1989-06-07 | 1995-08-29 | Affymax Technologies N.V. | Array of oligonucleotides on a solid substrate |
| US5744305A (en) * | 1989-06-07 | 1998-04-28 | Affymetrix, Inc. | Arrays of materials attached to a substrate |
| US5800992A (en) * | 1989-06-07 | 1998-09-01 | Fodor; Stephen P.A. | Method of detecting nucleic acids |
| US5874219A (en) * | 1995-06-07 | 1999-02-23 | Affymetrix, Inc. | Methods for concurrently processing multiple biological chip assays |
| US5885837A (en) * | 1991-11-22 | 1999-03-23 | Affymetrix, Inc. | Very large scale immobilized polymer synthesis using mechanically directed flow paths |
| US5919523A (en) * | 1995-04-27 | 1999-07-06 | Affymetrix, Inc. | Derivatization of solid supports and methods for oligomer synthesis |
| US5945334A (en) * | 1994-06-08 | 1999-08-31 | Affymetrix, Inc. | Apparatus for packaging a chip |
| US6033784A (en) * | 1995-04-07 | 2000-03-07 | Jacobsen; Mogens Havsteen | Method of photochemical immobilization of ligands using quinones |
| US6156501A (en) * | 1993-10-26 | 2000-12-05 | Affymetrix, Inc. | Arrays of modified nucleic acid probes and methods of use |
| US6238862B1 (en) * | 1995-09-18 | 2001-05-29 | Affymetrix, Inc. | Methods for testing oligonucleotide arrays |
| US6239273B1 (en) * | 1995-02-27 | 2001-05-29 | Affymetrix, Inc. | Printing molecular library arrays |
| US6303315B1 (en) * | 1999-03-18 | 2001-10-16 | Exiqon A/S | One step sample preparation and detection of nucleic acids in complex biological samples |
| US6309831B1 (en) * | 1998-02-06 | 2001-10-30 | Affymetrix, Inc. | Method of manufacturing biological chips |
| US6309823B1 (en) * | 1993-10-26 | 2001-10-30 | Affymetrix, Inc. | Arrays of nucleic acid probes for analyzing biotransformation genes and methods of using the same |
| US6310189B1 (en) * | 1989-06-07 | 2001-10-30 | Affymetrix, Inc. | Nucleotides and analogs having photoremoveable protecting groups |
| US6316198B1 (en) * | 1999-03-18 | 2001-11-13 | Exiqon A/S | Detection of mutations in genes by specific LNA primers |
| US6344316B1 (en) * | 1996-01-23 | 2002-02-05 | Affymetrix, Inc. | Nucleic acid analysis techniques |
| US20020068709A1 (en) * | 1999-12-23 | 2002-06-06 | Henrik Orum | Therapeutic uses of LNA-modified oligonucleotides |
| US6403320B1 (en) * | 1989-06-07 | 2002-06-11 | Affymetrix, Inc. | Support bound probes and methods of analysis using the same |
| US6403317B1 (en) * | 1999-03-26 | 2002-06-11 | Affymetrix, Inc. | Electronic detection of hybridization on nucleic acid arrays |
| US6406844B1 (en) * | 1989-06-07 | 2002-06-18 | Affymetrix, Inc. | Very large scale immobilized polymer synthesis |
| US6410229B1 (en) * | 1995-09-15 | 2002-06-25 | Affymetrix, Inc. | Expression monitoring by hybridization to high density nucleic acid arrays |
| US6440739B1 (en) * | 2001-07-17 | 2002-08-27 | Isis Pharmaceuticals, Inc. | Antisense modulation of glioma-associated oncogene-2 expression |
| US20030077609A1 (en) * | 2001-03-25 | 2003-04-24 | Jakobsen Mogens Havsteen | Modified oligonucleotides and uses thereof |
| US20060147924A1 (en) * | 2002-09-11 | 2006-07-06 | Ramsing Neils B | Population of nucleic acids including a subpopulation of lna oligomers |
Family Cites Families (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4889818A (en) | 1986-08-22 | 1989-12-26 | Cetus Corporation | Purified thermostable enzyme |
| CA1340807C (en) | 1988-02-24 | 1999-11-02 | Lawrence T. Malek | Nucleic acid amplification process |
| US5162209A (en) | 1991-01-18 | 1992-11-10 | Beth Israel Hospital Association | Synthesis of full-length, double-stranded dna from a single-stranded linear dna template |
| US5719262A (en) | 1993-11-22 | 1998-02-17 | Buchardt, Deceased; Ole | Peptide nucleic acids having amino acid side chains |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US5714331A (en) | 1991-05-24 | 1998-02-03 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| JP3236295B2 (en) | 1991-09-24 | 2001-12-10 | ケイヘーネ・エヌ・ベー | Selective restriction fragment amplification: General DNA fingerprinting |
| US5525470A (en) | 1994-01-26 | 1996-06-11 | Hybridon, Inc. | Method of sequencing [short] oligonucleotides |
| DE69516325T2 (en) * | 1994-02-22 | 2001-01-18 | Mitsubishi Chemical Corp., Tokio/Tokyo | Oligonucleotide and method for analyzing the base sequence of the nucleic acid |
| US5874229A (en) | 1995-08-07 | 1999-02-23 | Kyoto Daiichi Kagaku Co., Ltd. | Method for avoiding influence of hemoglobin |
| US5912340A (en) | 1995-10-04 | 1999-06-15 | Epoch Pharmaceuticals, Inc. | Selective binding complementary oligonucleotides |
| US6117635A (en) | 1996-07-16 | 2000-09-12 | Intergen Company | Nucleic acid amplification oligonucleotides with molecular energy transfer labels and methods based thereon |
| US5989165A (en) | 1996-09-30 | 1999-11-23 | Cybex International, Inc. | Incline press apparatus for exercising regions of the upper body |
| US6043060A (en) | 1996-11-18 | 2000-03-28 | Imanishi; Takeshi | Nucleotide analogues |
| JP3756313B2 (en) | 1997-03-07 | 2006-03-15 | 武 今西 | Novel bicyclonucleosides and oligonucleotide analogues |
| US6310183B1 (en) | 1997-09-10 | 2001-10-30 | Novo Nordisk A/S | Coagulation factor VIIa composition |
| US6794499B2 (en) | 1997-09-12 | 2004-09-21 | Exiqon A/S | Oligonucleotide analogues |
| ES2242291T5 (en) | 1997-09-12 | 2016-03-11 | Exiqon A/S | Bicyclic and tricyclic nucleoside analogs, nucleotides and oligonucleotides |
| EP0950683A4 (en) | 1997-09-17 | 2001-01-31 | Japan Polychem Corp | Resin material for foam molding, foamed sheet obtained therefrom, and process for producing the same |
| CZ296576B6 (en) | 1999-02-12 | 2006-04-12 | Sankyo Company Limited | Nucleoside analogue, oligonucleotide analogue, pharmaceutical composition, probe and a primer containing thereof |
| DE60044241D1 (en) | 1999-03-18 | 2010-06-02 | Exiqon As | XYLO-LNA ANALOG |
| DK1163250T3 (en) | 1999-03-24 | 2006-11-13 | Exiqon As | Improved synthesis of [2.2.1] bicyclonucleosides |
| GB9909801D0 (en) | 1999-04-28 | 1999-06-23 | Btg Int Ltd | Ultrasound detectable instrument |
| ATE356824T1 (en) | 1999-05-04 | 2007-04-15 | Santaris Pharma As | L-RIBO-LNA ANALOGUE |
| AU5429000A (en) | 1999-06-25 | 2001-01-31 | Sankyo Company Limited | Novel bicyclonucleoside derivatives |
| CA2378070A1 (en) | 1999-07-19 | 2001-01-25 | Cambridge University Technical Services Limited | A method for amplifying low abundance nucleic acid sequences and means for performing said method |
| EP1072679A3 (en) | 1999-07-20 | 2002-07-31 | Agilent Technologies, Inc. (a Delaware corporation) | Method of producing nucleic acid molecules with reduced secondary structure |
| JP4151751B2 (en) | 1999-07-22 | 2008-09-17 | 第一三共株式会社 | New bicyclonucleoside analogues |
| US6617442B1 (en) | 1999-09-30 | 2003-09-09 | Isis Pharmaceuticals, Inc. | Human Rnase H1 and oligonucleotide compositions thereof |
| JP2003511016A (en) | 1999-10-04 | 2003-03-25 | エクシコン エ/エス | Design of High Affinity RNase H to Replenish Oligonucleotides |
| JP2003530894A (en) | 2000-04-25 | 2003-10-21 | アフィメトリックス インコーポレイテッド | Methods for monitoring the expression of alternatively spliced genes |
| US6706476B1 (en) | 2000-08-22 | 2004-03-16 | Azign Bioscience A/S | Process for amplifying and labeling single stranded cDNA by 5′ ligated adaptor mediated amplification |
| AU2001297830A1 (en) | 2000-10-25 | 2002-12-09 | Exiqon A/S | Closed substrate platforms suitable for analysis of biomolecules |
| JP2004532615A (en) | 2000-12-27 | 2004-10-28 | インヴィトロジェン コーポレーション | Primers and methods for nucleic acid detection and identification |
| US6558907B2 (en) | 2001-05-16 | 2003-05-06 | Corning Incorporated | Methods and compositions for arraying nucleic acids onto a solid support |
| AU2002317437A1 (en) | 2001-05-18 | 2002-12-03 | Cureon A/S | Therapeutic uses of lna-modified oligonucleotides in infectious diseases |
| CA2459347C (en) | 2001-09-04 | 2012-10-09 | Exiqon A/S | Locked nucleic acid (lna) compositions and uses thereof |
| EP1430128B1 (en) | 2001-09-28 | 2018-04-25 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Micro-rna molecules |
| EP1546344A4 (en) | 2002-09-18 | 2007-10-03 | Isis Pharmaceuticals Inc | Efficient reduction of target rna's by single- and double-stranded oligomeric compounds |
| US20040235005A1 (en) | 2002-10-23 | 2004-11-25 | Ernest Friedlander | Methods and composition for detecting targets |
| US20040175732A1 (en) | 2002-11-15 | 2004-09-09 | Rana Tariq M. | Identification of micrornas and their targets |
| ES2607471T3 (en) | 2002-11-18 | 2017-03-31 | Roche Innovation Center Copenhagen A/S | Antisense design |
| US7217807B2 (en) | 2002-11-26 | 2007-05-15 | Rosetta Genomics Ltd | Bioinformatically detectable group of novel HIV regulatory genes and uses thereof |
| MXPA05006633A (en) | 2002-12-18 | 2006-05-25 | Third Wave Tech Inc | Detection of small nucleic acids. |
| WO2004081564A1 (en) | 2003-03-14 | 2004-09-23 | Peter Maccallum Cancer Institute | Expression profiling of tumours |
| WO2005018534A2 (en) | 2003-05-16 | 2005-03-03 | Rosetta Inpharmatics, Llc | Methods and compositions for rna interference |
| CA2531123A1 (en) | 2003-07-02 | 2005-01-13 | Perkinelmer Las, Inc. | Assay and process for labeling and detection of micro rna and small interfering rna sequences |
| WO2005013901A2 (en) | 2003-07-31 | 2005-02-17 | Isis Pharmaceuticals, Inc. | Oligomeric compounds and compositions for use in modulation of small non-coding rnas |
| CA2539654A1 (en) | 2003-10-14 | 2005-05-06 | Novartis Ag | Oligonucleotide microarray |
| ATE498022T1 (en) | 2003-12-23 | 2011-02-15 | Genomic Health Inc | UNIVERSAL DUPLICATION OF FRAGMENTED RNA |
| JP2007529758A (en) | 2004-03-19 | 2007-10-25 | ユー.エス. ジェノミクス, インコーポレイテッド | Compositions and methods for single molecule detection |
| WO2005098029A2 (en) | 2004-04-07 | 2005-10-20 | Exiqon A/S | Methods for quantification of micrornas and small interfering rnas |
| WO2006076025A2 (en) | 2004-05-14 | 2006-07-20 | Amaox, Inc. | Immune cell biosensors and methods of using same |
| EP1784501B1 (en) | 2004-05-14 | 2015-11-18 | Rosetta Genomics Ltd | VIRAL AND VIRUS ASSOCIATED MicroRNAS AND USES THEREOF |
| CN101031658A (en) | 2004-05-26 | 2007-09-05 | 罗塞塔金诺米克斯有限公司 | Viral and viral associated mirnas and uses thereof |
| US7575863B2 (en) | 2004-05-28 | 2009-08-18 | Applied Biosystems, Llc | Methods, compositions, and kits comprising linker probes for quantifying polynucleotides |
| KR20070057761A (en) | 2004-06-04 | 2007-06-07 | 아비아라디엑스, 인코포레이티드 | Identification of tumors |
| WO2006016978A1 (en) | 2004-06-30 | 2006-02-16 | Applera Corporation | Analog probe complexes |
| AU2005327199A1 (en) | 2004-07-09 | 2006-08-17 | Amaox, Inc. | Immune cell biosensors and methods of using same |
| US20060185027A1 (en) | 2004-12-23 | 2006-08-17 | David Bartel | Systems and methods for identifying miRNA targets and for altering miRNA and target expression |
| EP1838870A2 (en) | 2004-12-29 | 2007-10-03 | Exiqon A/S | NOVEL OLIGONUCLEOTIDE COMPOSITIONS AND PROBE SEQUENCES USEFUL FOR DETECTION AND ANALYSIS OF MICRORNAS AND THEIR TARGET MRNAs |
| EP1851336B1 (en) | 2005-01-25 | 2010-09-08 | Rosetta Inpharmatics LLC | Methods for quantitating small rna molecules |
| US8071306B2 (en) | 2005-01-25 | 2011-12-06 | Merck Sharp & Dohme Corp. | Methods for quantitating small RNA molecules |
| US20060211000A1 (en) | 2005-03-21 | 2006-09-21 | Sorge Joseph A | Methods, compositions, and kits for detection of microRNA |
| US20070065840A1 (en) | 2005-03-23 | 2007-03-22 | Irena Naguibneva | Novel oligonucleotide compositions and probe sequences useful for detection and analysis of microRNAS and their target mRNAS |
| AU2006242225B2 (en) | 2005-04-29 | 2012-01-12 | Rockefeller University | Human microRNAs and methods for inhibiting same |
| AU2006300864A1 (en) | 2005-10-10 | 2007-04-19 | Council Of Scientific And Industrial Research | Human microrna targets in HIV genome and a method of identification thereof |
| CA2629323A1 (en) | 2005-11-10 | 2007-05-24 | The University Of North Carolina At Chapel Hill | Splice switching oligomers for tnf superfamily receptors and their use in treatment of disease |
| WO2008061537A2 (en) | 2006-11-23 | 2008-05-29 | Querdenker Aps | Oligonucleotides for modulating target rna activity |
| WO2008074328A2 (en) | 2006-12-21 | 2008-06-26 | Exiqon A/S | Microrna target site blocking oligos and uses thereof |
| WO2008111908A1 (en) | 2007-03-15 | 2008-09-18 | Jyoti Chattopadhyaya | Five- and six-membered conformationally locked 2',4'- carbocyclic ribo-thymidines for the treatment of infections and cancer |
| CN101054580A (en) | 2007-04-06 | 2007-10-17 | 哈尔滨医科大学 | Imitation miRNA sequence and preparation method thereof |
| CN101054576A (en) | 2007-04-06 | 2007-10-17 | 哈尔滨医科大学 | A miRNA barrier technique |
| CN101821390A (en) | 2007-06-14 | 2010-09-01 | 米尔克斯治疗有限责任公司 | Oligonucleotides for modulation of target RNA activity |
-
2003
- 2003-10-21 US US10/690,487 patent/US20040219565A1/en not_active Abandoned
-
2006
- 2006-12-21 US US11/643,615 patent/US20070117144A1/en not_active Abandoned
-
2011
- 2011-11-14 US US13/295,615 patent/US9464106B2/en not_active Expired - Lifetime
Patent Citations (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) * | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4806463A (en) * | 1986-05-23 | 1989-02-21 | Worcester Foundation For Experimental Biology | Inhibition of HTLV-III by exogenous oligonucleotides |
| US6403320B1 (en) * | 1989-06-07 | 2002-06-11 | Affymetrix, Inc. | Support bound probes and methods of analysis using the same |
| US5445934A (en) * | 1989-06-07 | 1995-08-29 | Affymax Technologies N.V. | Array of oligonucleotides on a solid substrate |
| US5744305A (en) * | 1989-06-07 | 1998-04-28 | Affymetrix, Inc. | Arrays of materials attached to a substrate |
| US5800992A (en) * | 1989-06-07 | 1998-09-01 | Fodor; Stephen P.A. | Method of detecting nucleic acids |
| US6261776B1 (en) * | 1989-06-07 | 2001-07-17 | Affymetrix, Inc. | Nucleic acid arrays |
| US5889165A (en) * | 1989-06-07 | 1999-03-30 | Affymetrix, Inc. | Photolabile nucleoside protecting groups |
| US6406844B1 (en) * | 1989-06-07 | 2002-06-18 | Affymetrix, Inc. | Very large scale immobilized polymer synthesis |
| US6346413B1 (en) * | 1989-06-07 | 2002-02-12 | Affymetrix, Inc. | Polymer arrays |
| US6329143B1 (en) * | 1989-06-07 | 2001-12-11 | Affymetrix, Inc. | Very large scale immobilized polymer synthesis |
| US6310189B1 (en) * | 1989-06-07 | 2001-10-30 | Affymetrix, Inc. | Nucleotides and analogs having photoremoveable protecting groups |
| US6403957B1 (en) * | 1989-06-07 | 2002-06-11 | Affymetrix, Inc. | Nucleic acid reading and analysis system |
| US5432272A (en) * | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| US5885837A (en) * | 1991-11-22 | 1999-03-23 | Affymetrix, Inc. | Very large scale immobilized polymer synthesis using mechanically directed flow paths |
| US6309823B1 (en) * | 1993-10-26 | 2001-10-30 | Affymetrix, Inc. | Arrays of nucleic acid probes for analyzing biotransformation genes and methods of using the same |
| US6156501A (en) * | 1993-10-26 | 2000-12-05 | Affymetrix, Inc. | Arrays of modified nucleic acid probes and methods of use |
| US5945334A (en) * | 1994-06-08 | 1999-08-31 | Affymetrix, Inc. | Apparatus for packaging a chip |
| US6239273B1 (en) * | 1995-02-27 | 2001-05-29 | Affymetrix, Inc. | Printing molecular library arrays |
| US6033784A (en) * | 1995-04-07 | 2000-03-07 | Jacobsen; Mogens Havsteen | Method of photochemical immobilization of ligands using quinones |
| US5919523A (en) * | 1995-04-27 | 1999-07-06 | Affymetrix, Inc. | Derivatization of solid supports and methods for oligomer synthesis |
| US5874219A (en) * | 1995-06-07 | 1999-02-23 | Affymetrix, Inc. | Methods for concurrently processing multiple biological chip assays |
| US6410229B1 (en) * | 1995-09-15 | 2002-06-25 | Affymetrix, Inc. | Expression monitoring by hybridization to high density nucleic acid arrays |
| US6238862B1 (en) * | 1995-09-18 | 2001-05-29 | Affymetrix, Inc. | Methods for testing oligonucleotide arrays |
| US6344316B1 (en) * | 1996-01-23 | 2002-02-05 | Affymetrix, Inc. | Nucleic acid analysis techniques |
| US6309831B1 (en) * | 1998-02-06 | 2001-10-30 | Affymetrix, Inc. | Method of manufacturing biological chips |
| US6303315B1 (en) * | 1999-03-18 | 2001-10-16 | Exiqon A/S | One step sample preparation and detection of nucleic acids in complex biological samples |
| US6316198B1 (en) * | 1999-03-18 | 2001-11-13 | Exiqon A/S | Detection of mutations in genes by specific LNA primers |
| US6403317B1 (en) * | 1999-03-26 | 2002-06-11 | Affymetrix, Inc. | Electronic detection of hybridization on nucleic acid arrays |
| US20020068709A1 (en) * | 1999-12-23 | 2002-06-06 | Henrik Orum | Therapeutic uses of LNA-modified oligonucleotides |
| US20030077609A1 (en) * | 2001-03-25 | 2003-04-24 | Jakobsen Mogens Havsteen | Modified oligonucleotides and uses thereof |
| US6440739B1 (en) * | 2001-07-17 | 2002-08-27 | Isis Pharmaceuticals, Inc. | Antisense modulation of glioma-associated oncogene-2 expression |
| US20060147924A1 (en) * | 2002-09-11 | 2006-07-06 | Ramsing Neils B | Population of nucleic acids including a subpopulation of lna oligomers |
Cited By (229)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10196640B1 (en) | 2002-02-01 | 2019-02-05 | Life Technologies Corporation | Oligonucleotide compositions with enhanced efficiency |
| US10036025B2 (en) | 2002-02-01 | 2018-07-31 | Life Technologies Corporation | Oligonucleotide compositions with enhanced efficiency |
| US8815821B2 (en) | 2002-02-01 | 2014-08-26 | Life Technologies Corporation | Double-stranded oligonucleotides |
| US9592250B2 (en) | 2002-02-01 | 2017-03-14 | Life Technologies Corporation | Double-stranded oligonucleotides |
| US8524680B2 (en) | 2002-02-01 | 2013-09-03 | Applied Biosystems, Llc | High potency siRNAS for reducing the expression of target genes |
| US9796978B1 (en) | 2002-02-01 | 2017-10-24 | Life Technologies Corporation | Oligonucleotide compositions with enhanced efficiency |
| US10106793B2 (en) | 2002-02-01 | 2018-10-23 | Life Technologies Corporation | Double-stranded oligonucleotides |
| US9777275B2 (en) | 2002-02-01 | 2017-10-03 | Life Technologies Corporation | Oligonucleotide compositions with enhanced efficiency |
| US10626398B2 (en) | 2002-02-01 | 2020-04-21 | Life Technologies Corporation | Oligonucleotide compositions with enhanced efficiency |
| US20080014579A1 (en) * | 2003-02-11 | 2008-01-17 | Affymetrix, Inc. | Gene expression profiling in colon cancers |
| US20060142228A1 (en) * | 2004-12-23 | 2006-06-29 | Ambion, Inc. | Methods and compositions concerning siRNA's as mediators of RNA interference |
| US8058255B2 (en) | 2004-12-23 | 2011-11-15 | Applied Biosystems, Llc | Methods and compositions concerning siRNA's as mediators of RNA interference |
| US9719089B2 (en) | 2005-06-14 | 2017-08-01 | Northwestern University | Nucleic acid functionalized nonoparticles for therapeutic applications |
| US10370661B2 (en) | 2005-06-14 | 2019-08-06 | Northwestern University | Nucleic acid functionalized nanoparticles for therapeutic applications |
| US8252756B2 (en) | 2005-06-14 | 2012-08-28 | Northwestern University | Nucleic acid functionalized nanoparticles for therapeutic applications |
| US8999947B2 (en) | 2005-06-14 | 2015-04-07 | Northwestern University | Nucleic acid functionalized nanoparticles for therapeutic applications |
| US20100015604A1 (en) * | 2005-08-17 | 2010-01-21 | Evriklia Lianidou | Composition and method for determination of ck19 expression |
| US8022193B2 (en) | 2006-01-27 | 2011-09-20 | Isis Pharmaceuticals, Inc. | 6-modified bicyclic nucleic acid analogs |
| EP2332951A2 (en) | 2006-01-27 | 2011-06-15 | ISIS Pharmaceuticals, Inc. | 6-modified bicyclic nucleic acid analogs |
| EP2314594A1 (en) | 2006-01-27 | 2011-04-27 | Isis Pharmaceuticals, Inc. | 6-modified bicyclic nucleic acid analogs |
| US7741457B2 (en) | 2006-01-27 | 2010-06-22 | Isis Pharmaceuticals, Inc. | 6-modified bicyclic nucleic acid analogs |
| WO2007097876A3 (en) * | 2006-02-15 | 2008-04-10 | Agilent Technologies Inc | Normalization probes for comparative genome hybridization arrays |
| US20090275482A1 (en) * | 2006-02-15 | 2009-11-05 | Jing Gao | Normalization Probes for Comparative Genome Hybridization Arrays |
| GB2449048A (en) * | 2006-02-15 | 2008-11-05 | Agilent Technologies Inc | Normalization probes for comparative genome hybridization arrays |
| US8221978B2 (en) | 2006-02-15 | 2012-07-17 | Agilent Technologies, Inc. | Normalization probes for comparative genome hybridization arrays |
| WO2007097876A2 (en) * | 2006-02-15 | 2007-08-30 | Agilent Technologies, Inc. | Normalization probes for comparative genome hybridization arrays |
| US8849576B2 (en) * | 2006-04-28 | 2014-09-30 | Hakima Amri | Phylogenetic analysis of mass spectrometry or gene array data for the diagnosis of physiological conditions |
| US20070259363A1 (en) * | 2006-04-28 | 2007-11-08 | Hakima Amri | Phylogenetic Analysis of Mass Spectrometry or Gene Array Data for the Diagnosis of Physiological Conditions |
| US9045754B2 (en) | 2006-05-05 | 2015-06-02 | Isis Pharmaceuticals, Inc. | Short antisense compounds with gapmer configuration |
| EP2505649A1 (en) | 2006-05-05 | 2012-10-03 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of GCGR |
| US8143230B2 (en) | 2006-05-05 | 2012-03-27 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of PCSK9 |
| US8969316B2 (en) | 2006-05-05 | 2015-03-03 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of DGAT2 |
| US8188059B2 (en) | 2006-05-05 | 2012-05-29 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of GCGR |
| EP2458006A1 (en) | 2006-05-05 | 2012-05-30 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression APOB |
| US8673871B2 (en) | 2006-05-05 | 2014-03-18 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression ApoB |
| EP2397551A1 (en) | 2006-05-05 | 2011-12-21 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of PCSK9 |
| US8586554B2 (en) | 2006-05-05 | 2013-11-19 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of PTP1B |
| US20090318532A1 (en) * | 2006-05-05 | 2009-12-24 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of ptp1b |
| US8372967B2 (en) | 2006-05-05 | 2013-02-12 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of GCCR |
| US8362232B2 (en) | 2006-05-05 | 2013-01-29 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of SGLT2 |
| EP2505650A1 (en) | 2006-05-05 | 2012-10-03 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of PCSK9 |
| EP2363482A1 (en) | 2006-05-05 | 2011-09-07 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating gene expression |
| EP2363481A1 (en) | 2006-05-05 | 2011-09-07 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating gene expression |
| WO2007146511A2 (en) | 2006-05-05 | 2007-12-21 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating gene expression |
| EP2505648A1 (en) | 2006-05-05 | 2012-10-03 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of PTP1B |
| US20080015162A1 (en) * | 2006-05-05 | 2008-01-17 | Sanjay Bhanot | Compounds and methods for modulating gene expression |
| EP2505647A1 (en) | 2006-05-05 | 2012-10-03 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of DGAT2 |
| EP2505646A1 (en) | 2006-05-05 | 2012-10-03 | Isis Pharmaceuticals, Inc. | Compounds and methods for modulating expression of CRP |
| US8030467B2 (en) | 2006-05-11 | 2011-10-04 | Isis Pharmaceuticals, Inc. | 5′-modified bicyclic nucleic acid analogs |
| US8268980B2 (en) | 2006-05-11 | 2012-09-18 | Isis Pharmaceuticals, Inc. | 5′-modified bicyclic nucleic acid analogs |
| US7666854B2 (en) | 2006-05-11 | 2010-02-23 | Isis Pharmaceuticals, Inc. | Bis-modified bicyclic nucleic acid analogs |
| US8088746B2 (en) | 2006-05-11 | 2012-01-03 | Isis Pharmaceuticals, Inc. | Bis-modified bicyclic nucleic acid analogs |
| US7750131B2 (en) | 2006-05-11 | 2010-07-06 | Isis Pharmaceuticals, Inc. | 5′-modified bicyclic nucleic acid analogs |
| US10370656B2 (en) | 2006-06-08 | 2019-08-06 | Northwestern University | Nucleic acid functionalized nanoparticles for therapeutic applications |
| US9506056B2 (en) | 2006-06-08 | 2016-11-29 | Northwestern University | Nucleic acid functionalized nanoparticles for therapeutic applications |
| US20080003573A1 (en) * | 2006-06-28 | 2008-01-03 | Bio-Id Diagnostic Inc. | Determination of variants produced upon replication or transcription of nucleic acid sequences |
| US9150906B2 (en) * | 2006-06-28 | 2015-10-06 | Bio-Id Diagnostic Inc. | Determination of variants produced upon replication or transcription of nucleic acid sequences |
| US10036053B2 (en) | 2006-06-28 | 2018-07-31 | Bio-ID Diagnostics Inc. | Determination of variants produced upon replication or transcription of nucleic acid sequences |
| EP2410053A1 (en) | 2006-10-18 | 2012-01-25 | Isis Pharmaceuticals, Inc. | Antisense compounds |
| EP2410054A1 (en) | 2006-10-18 | 2012-01-25 | Isis Pharmaceuticals, Inc. | Antisense compounds |
| US9550988B2 (en) | 2006-10-18 | 2017-01-24 | Ionis Pharmaceuticals, Inc. | Antisense compounds |
| EP3916095A1 (en) | 2006-10-18 | 2021-12-01 | Ionis Pharmaceuticals, Inc. | Antisense compounds |
| EP3202905A1 (en) | 2006-10-18 | 2017-08-09 | Ionis Pharmaceuticals, Inc. | Antisense compounds |
| US20100144834A1 (en) * | 2006-11-27 | 2010-06-10 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US8093222B2 (en) | 2006-11-27 | 2012-01-10 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US8084437B2 (en) | 2006-11-27 | 2011-12-27 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US9650636B2 (en) | 2006-11-27 | 2017-05-16 | Ionis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US11530410B2 (en) | 2006-11-27 | 2022-12-20 | Ionis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US8912160B2 (en) | 2006-11-27 | 2014-12-16 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US8664190B2 (en) | 2006-11-27 | 2014-03-04 | Isis Pharmaceuticals, Inc. | Methods for treating hypercholesterolemia |
| US20080188005A1 (en) * | 2007-01-10 | 2008-08-07 | Chunnian Shi | Inhibition of mismatch hybridization by a universal competitor dna |
| US7820389B2 (en) * | 2007-01-10 | 2010-10-26 | Geneohm Sciences, Inc. | Inhibition of mismatch hybridization by a universal competitor DNA |
| US9890427B2 (en) | 2007-02-09 | 2018-02-13 | Northwestern University | Particles for detecting intracellular targets |
| WO2008098248A2 (en) | 2007-02-09 | 2008-08-14 | Northwestern University | Particles for detecting intracellular targets |
| US8507200B2 (en) | 2007-02-09 | 2013-08-13 | Northwestern University | Particles for detecting intracellular targets |
| EP2826863A1 (en) | 2007-05-30 | 2015-01-21 | Northwestern University | Nucleic acid functionalized nanoparticles for therapeutic applications |
| US8278425B2 (en) | 2007-05-30 | 2012-10-02 | Isis Pharmaceuticals, Inc. | N-substituted-aminomethylene bridged bicyclic nucleic acid analogs |
| WO2008150729A2 (en) | 2007-05-30 | 2008-12-11 | Isis Pharmaceuticals, Inc. | N-substituted-aminomethylene bridged bicyclic nucleic acid analogs |
| US7807372B2 (en) | 2007-06-04 | 2010-10-05 | Northwestern University | Screening sequence selectivity of oligonucleotide-binding molecules using nanoparticle based colorimetric assay |
| US20080311669A1 (en) * | 2007-06-04 | 2008-12-18 | Northwestern University | Screening sequence selectivity of oligonucleotide-binding molecules using nanoparticle based colorimetric assay |
| US8278426B2 (en) | 2007-06-08 | 2012-10-02 | Isis Pharmaceuticals, Inc. | Carbocyclic bicyclic nucleic acid analogs |
| US8889355B2 (en) | 2007-06-27 | 2014-11-18 | Applied Biosystems, Llc | Chimeric oligonucleotides for ligation-enhanced nucleic acid detection, methods and compositions therefor |
| US8008010B1 (en) | 2007-06-27 | 2011-08-30 | Applied Biosystems, Llc | Chimeric oligonucleotides for ligation-enhanced nucleic acid detection, methods and compositions therefor |
| US8278283B2 (en) | 2007-07-05 | 2012-10-02 | Isis Pharmaceuticals, Inc. | 6-disubstituted or unsaturated bicyclic nucleic acid analogs |
| US9284551B2 (en) | 2007-09-19 | 2016-03-15 | Applied Biosystems, Llc | RNAi sequence-independent modification formats, and stabilized forms thereof |
| US9771583B2 (en) | 2007-09-19 | 2017-09-26 | Applied Biosystems, Llc | siRNA sequence-independent modification formats for reducing off-target phenotypic effects in RNAI, and stabilized forms thereof |
| EP2548962A1 (en) | 2007-09-19 | 2013-01-23 | Applied Biosystems, LLC | Sirna sequence-independent modification formats for reducing off-target phenotypic effects in rnai, and stabilized forms thereof |
| US8524681B2 (en) | 2007-09-19 | 2013-09-03 | Applied Biosystems, Llc | siRNA sequence-independent modification formats for reducing off-target phenotypic effects in RNAi, and stabilized forms thereof |
| US20100222414A1 (en) * | 2007-09-19 | 2010-09-02 | Applied Biosystems, Llc | SiRNA Sequence-Independent Modification Formats for Reducing Off-Target Phenotypic Effects in RNAi, and Stabilized Forms Thereof |
| US9273312B2 (en) | 2007-09-19 | 2016-03-01 | Applied Biosystems, Llc | SiRNA sequence-independent modification formats for reducing off-target phenotypic effects in RNAi, and stabilized forms thereof |
| US10329564B2 (en) | 2007-09-19 | 2019-06-25 | Applied Biosystems, Llc | siRNA sequence-independent modification formats for reducing off-target phenotypic effects in RNAi, and stabilized forms thereof |
| US10900038B2 (en) | 2007-09-19 | 2021-01-26 | Applied Biosystems, Llc | siRNA sequence-independent modification formats for reducing off-target phenotypic effects in RNAI, and stabilized forms thereof |
| USRE48468E1 (en) | 2007-10-26 | 2021-03-16 | Biomarin Technologies B.V. | Means and methods for counteracting muscle disorders |
| US11427820B2 (en) | 2007-10-26 | 2022-08-30 | Biomarin Technologies B.V. | Methods and means for efficient skipping of exon 45 in Duchenne muscular dystrophy pre-mRNA |
| US10876114B2 (en) | 2007-10-26 | 2020-12-29 | Biomarin Technologies B.V. | Methods and means for efficient skipping of at least one of the following exons of the human Duchenne muscular dystrophy gene: 43, 46, 50-53 |
| US8546556B2 (en) | 2007-11-21 | 2013-10-01 | Isis Pharmaceuticals, Inc | Carbocyclic alpha-L-bicyclic nucleic acid analogs |
| US20100331538A1 (en) * | 2007-11-21 | 2010-12-30 | Seth Punit P | Carbocyclic alpha-l-bicyclic nucleic acid analogs |
| US9879222B2 (en) | 2007-12-14 | 2018-01-30 | Mofa Group Llc | Gender-specific separation of sperm cells and embryos |
| US20100279305A1 (en) * | 2008-01-14 | 2010-11-04 | Applied Biosystems, Llc | Compositions, methods, and kits for detecting ribonucleic acid |
| US10829808B2 (en) | 2008-01-14 | 2020-11-10 | Applied Biosystems, Llc | Amplification and detection of ribonucleic acids |
| US9834816B2 (en) | 2008-01-14 | 2017-12-05 | Applied Biosystems, Llc | Amplification and detection of ribonucleic acids |
| US8192941B2 (en) | 2008-01-14 | 2012-06-05 | Applied Biosystems, Llc | Amplification and detection of ribonucleic acid |
| US10240191B2 (en) | 2008-01-14 | 2019-03-26 | Applied Biosystems, Llc | Amplification and detection of ribonucleic acids |
| US9416406B2 (en) | 2008-01-14 | 2016-08-16 | Applied Biosystems, Llc | Amplification and detection of ribonucleic acids |
| US9624534B2 (en) | 2008-01-14 | 2017-04-18 | Applied Biosystems, Llc | Amplification and detection of ribonucleic acids |
| US8932816B2 (en) | 2008-01-14 | 2015-01-13 | Applied Biosystems, Llc | Amplification and detection of ribonucleic acids |
| US8530640B2 (en) | 2008-02-07 | 2013-09-10 | Isis Pharmaceuticals, Inc. | Bicyclic cyclohexitol nucleic acid analogs |
| US9290534B2 (en) | 2008-04-04 | 2016-03-22 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds having at least one neutrally linked terminal bicyclic nucleoside |
| US8022046B2 (en) | 2008-04-18 | 2011-09-20 | Baxter International, Inc. | Microsphere-based composition for preventing and/or reversing new-onset autoimmune diabetes |
| EP2982753A1 (en) | 2008-04-18 | 2016-02-10 | Baxter International Inc | Microsphere-based composition for preventing and/or reversing new-onset autoimmune diabetes |
| WO2009149182A1 (en) | 2008-06-04 | 2009-12-10 | The Board Of Regents Of The University Of Texas System | Modulation of gene expression through endogenous small rna targeting of gene promoters |
| US9745636B2 (en) * | 2008-06-13 | 2017-08-29 | National University Of Ireland, Galway | SWI5 gene as a diagnostic target for the identification of fungal and yeast species |
| US20110212445A1 (en) * | 2008-06-13 | 2011-09-01 | National University Of Ireland, Galway | Swi5 gene as a diagnostic target for the identification of fungal and yeast species |
| AU2009256588B2 (en) * | 2008-06-13 | 2015-03-19 | National University Of Ireland, Galway | SWI5 gene as a diagnostic target for the identification of fungal and yeast species |
| JP2011522556A (en) * | 2008-06-13 | 2011-08-04 | ナショナル ユニバーシティー オブ アイルランド, ゴールウェイ | SWI5 gene as a diagnostic target for the identification of fungal and yeast species |
| WO2010014592A1 (en) | 2008-07-29 | 2010-02-04 | The Board Of Regents Of The University Of Texas Sytem | Selective inhibition of polyglutamine protein expression |
| US8501805B2 (en) | 2008-09-24 | 2013-08-06 | Isis Pharmaceuticals, Inc. | Substituted alpha-L-bicyclic nucleosides |
| US10391116B2 (en) | 2008-11-24 | 2019-08-27 | Northwestern University | Polyvalent RNA-nanoparticle compositions |
| US9844562B2 (en) | 2008-11-24 | 2017-12-19 | Northwestern University | Polyvalent RNA-nanoparticle compositions |
| US9139827B2 (en) | 2008-11-24 | 2015-09-22 | Northwestern University | Polyvalent RNA-nanoparticle compositions |
| EP3335705A1 (en) | 2008-11-24 | 2018-06-20 | Northwestern University | Polyvalent rna-nanoparticle compositions |
| US11633503B2 (en) | 2009-01-08 | 2023-04-25 | Northwestern University | Delivery of oligonucleotide-functionalized nanoparticles |
| US10098958B2 (en) | 2009-01-08 | 2018-10-16 | Northwestern University | Delivery of oligonucleotide functionalized nanoparticles |
| WO2010120420A1 (en) | 2009-04-15 | 2010-10-21 | Northwestern University | Delivery of oligonucleotide-functionalized nanoparticles |
| WO2010124231A2 (en) | 2009-04-24 | 2010-10-28 | The Board Of Regents Of The University Of Texas System | Modulation of gene expression using oligomers that target gene regions downstream of 3' untranslated regions |
| WO2011017521A2 (en) | 2009-08-06 | 2011-02-10 | Isis Pharmaceuticals, Inc. | Bicyclic cyclohexose nucleic acid analogs |
| US8912318B2 (en) | 2009-09-10 | 2014-12-16 | University Of Idaho | Nucleobase-functionalized conformationally restricted nucleotides and oligonucleotides for targeting nucleic acids |
| US8518908B2 (en) | 2009-09-10 | 2013-08-27 | University Of Idaho | Nucleobase-functionalized conformationally restricted nucleotides and oligonucleotides for targeting of nucleic acids |
| US10480030B2 (en) | 2009-10-27 | 2019-11-19 | Swift Biosciences, Inc. | Polynucleotide primers and probes |
| WO2011056687A2 (en) | 2009-10-27 | 2011-05-12 | Swift Biosciences, Inc. | Polynucleotide primers and probes |
| US9757475B2 (en) | 2009-10-30 | 2017-09-12 | Northwestern University | Templated nanoconjugates |
| US9376690B2 (en) | 2009-10-30 | 2016-06-28 | Northwestern University | Templated nanoconjugates |
| WO2011053994A1 (en) | 2009-11-02 | 2011-05-05 | Alnylam Pharmaceuticals, Inc. | Modulation of ldl receptor gene expression with double-stranded rnas targeting the ldl receptor gene promoter |
| WO2011056215A1 (en) | 2009-11-03 | 2011-05-12 | Landers James P | Versatile, visible method for detecting polymeric analytes |
| WO2011063403A1 (en) | 2009-11-23 | 2011-05-26 | Swift Biosciences, Inc. | Devices to extend single stranded target molecules |
| US8779118B2 (en) | 2010-01-11 | 2014-07-15 | Isis Pharmaceuticals, Inc. | Base modified bicyclic nucleosides and oligomeric compounds prepared therefrom |
| WO2011085102A1 (en) | 2010-01-11 | 2011-07-14 | Isis Pharmaceuticals, Inc. | Base modified bicyclic nucleosides and oligomeric compounds prepared therefrom |
| WO2011097388A1 (en) | 2010-02-03 | 2011-08-11 | Alnylam Pharmaceuticals, Inc. | Selective inhibition of polyglutamine protein expression |
| WO2011113054A2 (en) | 2010-03-12 | 2011-09-15 | Aurasense Llc | Crosslinked polynucleotide structure |
| WO2011115818A1 (en) | 2010-03-17 | 2011-09-22 | Isis Pharmaceuticals, Inc. | 5'-substituted bicyclic nucleosides and oligomeric compounds prepared therefrom |
| US8889350B2 (en) | 2010-03-26 | 2014-11-18 | Swift Biosciences, Inc. | Methods and compositions for isolating polynucleotides |
| WO2011133695A2 (en) | 2010-04-20 | 2011-10-27 | Swift Biosciences, Inc. | Materials and methods for nucleic acid fractionation by solid phase entrapment and enzyme-mediated detachment |
| WO2011139702A2 (en) | 2010-04-28 | 2011-11-10 | Isis Pharmaceuticals, Inc. | Modified nucleosides and oligomeric compounds prepared therefrom |
| EP3173419A1 (en) | 2010-04-28 | 2017-05-31 | Ionis Pharmaceuticals, Inc. | Modified nucleosides, analogs thereof and oligomeric compounds prepared therefrom |
| WO2011150226A1 (en) | 2010-05-26 | 2011-12-01 | Landers James P | Method for detecting nucleic acids based on aggregate formation |
| US8957200B2 (en) | 2010-06-07 | 2015-02-17 | Isis Pharmaceuticals, Inc. | Bicyclic nucleosides and oligomeric compounds prepared therefrom |
| WO2011156278A1 (en) | 2010-06-07 | 2011-12-15 | Isis Pharmaceuticals, Inc. | Bicyclic nucleosides and oligomeric compounds prepared therefrom |
| US9081737B2 (en) * | 2010-08-02 | 2015-07-14 | Integrated Dna Technologies, Inc. | Methods for predicting stability and melting temperatures of nucleic acid duplexes |
| US20120029891A1 (en) * | 2010-08-02 | 2012-02-02 | Integrated Dna Technologies, Inc. | Methods for Predicting Stability and Melting Temperatures of Nucleic Acid Duplexes |
| US20120303283A1 (en) * | 2011-01-19 | 2012-11-29 | Jian Han | Polymerase Preference Index |
| US10860684B2 (en) * | 2011-01-19 | 2020-12-08 | iRepertoire, Inc. | Polymerase preference index |
| WO2012106509A1 (en) | 2011-02-02 | 2012-08-09 | The Trustees Of Princeton University | Sirtuin modulators as virus production modulators |
| US10813934B2 (en) | 2011-02-02 | 2020-10-27 | The Trustees Of Princeton University | Sirtuin modulators as inhibitors of cytomegalovirus |
| US10017764B2 (en) | 2011-02-08 | 2018-07-10 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleotides and uses thereof |
| US20140087962A1 (en) * | 2011-03-22 | 2014-03-27 | Life Technologies Corporation | Identification of Linkage Using Multiplex Digital PCR |
| WO2012149154A1 (en) | 2011-04-26 | 2012-11-01 | Swift Biosciences, Inc. | Polynucleotide primers and probes |
| WO2012151289A2 (en) | 2011-05-02 | 2012-11-08 | University Of Virginia Patent Foundation | Method and system to detect aggregate formation on a substrate |
| WO2012151268A1 (en) | 2011-05-02 | 2012-11-08 | University Of Virginia Patent Foundation | Method and system for high throughput optical and label free detection of analytes |
| US20140120540A1 (en) * | 2011-07-01 | 2014-05-01 | Htg Molecular Diagnostics, Inc. | Methods of detecting gene fusions |
| US10294515B2 (en) * | 2011-07-01 | 2019-05-21 | Htg Molecular Diagnostics, Inc. | Methods of detecting gene fusions |
| US11268133B2 (en) * | 2011-07-01 | 2022-03-08 | Htg Molecular Diagnostics, Inc. | Methods of detecting gene fusions |
| US9885082B2 (en) | 2011-07-19 | 2018-02-06 | University Of Idaho | Embodiments of a probe and method for targeting nucleic acids |
| US11732261B2 (en) | 2011-08-11 | 2023-08-22 | Ionis Pharmaceuticals, Inc. | Selective antisense compounds and uses thereof |
| US9889209B2 (en) | 2011-09-14 | 2018-02-13 | Northwestern University | Nanoconjugates able to cross the blood-brain barrier |
| US10398784B2 (en) | 2011-09-14 | 2019-09-03 | Northwestern Univerity | Nanoconjugates able to cross the blood-brain barrier |
| WO2013040499A1 (en) | 2011-09-14 | 2013-03-21 | Northwestern University | Nanoconjugates able to cross the blood-brain barrier |
| US20170029818A1 (en) * | 2012-01-27 | 2017-02-02 | Biomarin Technologies B.V. | RNA Modulating Oligonucleotides with Improved Characteristics for the Treatment of Duchenne and Becker Muscular Dystrophy |
| US10179912B2 (en) * | 2012-01-27 | 2019-01-15 | Biomarin Technologies B.V. | RNA modulating oligonucleotides with improved characteristics for the treatment of duchenne and becker muscular dystrophy |
| US10913946B2 (en) | 2012-01-27 | 2021-02-09 | Biomarin Technologies B.V. | RNA modulating oligonucleotides with improved characteristics for the treatment of Duchenne and Becker muscular dystrophy |
| WO2013138536A1 (en) | 2012-03-13 | 2013-09-19 | Swift Biosciences, Inc. | Methods and compositions for size-controlled homopolymer tailing of substrate polynucleotides by a nucleic acid polymerase |
| US9221864B2 (en) | 2012-04-09 | 2015-12-29 | Isis Pharmaceuticals, Inc. | Tricyclic nucleic acid analogs |
| WO2013154798A1 (en) | 2012-04-09 | 2013-10-17 | Isis Pharmaceuticals, Inc. | Tricyclic nucleic acid analogs |
| WO2013154799A1 (en) | 2012-04-09 | 2013-10-17 | Isis Pharmaceuticals, Inc. | Tricyclic nucleosides and oligomeric compounds prepared therefrom |
| US9914922B2 (en) | 2012-04-20 | 2018-03-13 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleotides and uses thereof |
| US11566245B2 (en) | 2012-04-20 | 2023-01-31 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleotides and uses thereof |
| WO2014045126A2 (en) | 2012-09-18 | 2014-03-27 | Uti Limited Partnership | Treatment of pain by inhibition of usp5 de-ubiquitinase |
| US9695418B2 (en) | 2012-10-11 | 2017-07-04 | Ionis Pharmaceuticals, Inc. | Oligomeric compounds comprising bicyclic nucleosides and uses thereof |
| US9029335B2 (en) | 2012-10-16 | 2015-05-12 | Isis Pharmaceuticals, Inc. | Substituted 2′-thio-bicyclic nucleosides and oligomeric compounds prepared therefrom |
| WO2014134179A1 (en) | 2013-02-28 | 2014-09-04 | The Board Of Regents Of The University Of Texas System | Methods for classifying a cancer as susceptible to tmepai-directed therapies and treating such cancers |
| US9273349B2 (en) | 2013-03-14 | 2016-03-01 | Affymetrix, Inc. | Detection of nucleic acids |
| US10266879B2 (en) | 2013-03-14 | 2019-04-23 | Affymetrix, Inc. | Detection of nucleic acids |
| US10301622B2 (en) | 2013-11-04 | 2019-05-28 | Northwestern University | Quantification and spatio-temporal tracking of a target using a spherical nucleic acid (SNA) |
| US10182988B2 (en) | 2013-12-03 | 2019-01-22 | Northwestern University | Liposomal particles, methods of making same and uses thereof |
| US10792251B2 (en) | 2013-12-03 | 2020-10-06 | Northwestern University | Liposomal particles, methods of making same and uses thereof |
| US11883535B2 (en) | 2013-12-03 | 2024-01-30 | Northwestern University | Liposomal particles, methods of making same and uses thereof |
| US10385388B2 (en) | 2013-12-06 | 2019-08-20 | Swift Biosciences, Inc. | Cleavable competitor polynucleotides |
| EP3647318A1 (en) | 2014-04-28 | 2020-05-06 | Ionis Pharmaceuticals, Inc. | Linkage modified oligomeric compounds |
| WO2015168172A1 (en) | 2014-04-28 | 2015-11-05 | Isis Pharmaceuticals, Inc. | Linkage modified oligomeric compounds |
| US9926556B2 (en) | 2014-04-28 | 2018-03-27 | Ionis Pharmaceuticals, Inc. | Linkage modified oligomeric compounds |
| WO2016028940A1 (en) | 2014-08-19 | 2016-02-25 | Northwestern University | Protein/oligonucleotide core-shell nanoparticle therapeutics |
| US11987836B2 (en) * | 2014-09-30 | 2024-05-21 | Global Life Sciences Solutions Usa Llc | Method for nucleic acid analysis directly from an unpurified biological sample |
| US20170298415A1 (en) * | 2014-09-30 | 2017-10-19 | Ge Healthcare Bio-Sciences Corp. | Method for nucleic acid analysis directly from an unpurified biological sample |
| US11466311B2 (en) * | 2014-10-08 | 2022-10-11 | Cornell University | Method for identification and quantification of nucleic acid expression, splice variant, translocation, copy number, or methylation changes |
| US11213593B2 (en) | 2014-11-21 | 2022-01-04 | Northwestern University | Sequence-specific cellular uptake of spherical nucleic acid nanoparticle conjugates |
| WO2016081911A2 (en) | 2014-11-21 | 2016-05-26 | Northwestern University | The sequence-specific cellular uptake of spherical nucleic acid nanoparticle conjugates |
| WO2016100716A1 (en) | 2014-12-18 | 2016-06-23 | Vasant Jadhav | Reversirtm compounds |
| US9688707B2 (en) | 2014-12-30 | 2017-06-27 | Ionis Pharmaceuticals, Inc. | Bicyclic morpholino compounds and oligomeric compounds prepared therefrom |
| EP3919619A1 (en) | 2015-07-17 | 2021-12-08 | Alnylam Pharmaceuticals, Inc. | Multi-targeted single entity conjugates |
| WO2017015109A1 (en) | 2015-07-17 | 2017-01-26 | Alnylam Pharmaceuticals, Inc. | Multi-targeted single entity conjugates |
| US11364304B2 (en) | 2016-08-25 | 2022-06-21 | Northwestern University | Crosslinked micellar spherical nucleic acids |
| US11690920B2 (en) | 2017-07-13 | 2023-07-04 | Northwestern University | General and direct method for preparing oligonucleotide-functionalized metal-organic framework nanoparticles |
| WO2019217459A1 (en) | 2018-05-07 | 2019-11-14 | Alnylam Pharmaceuticals, Inc. | Extrahepatic delivery |
| CN108754010A (en) * | 2018-06-14 | 2018-11-06 | 中国农业科学院蔬菜花卉研究所 | It is a kind of quickly to detect the remaining method of genomic DNA in total serum IgE sample |
| WO2020056341A2 (en) | 2018-09-14 | 2020-03-19 | Northwestern University | Programming protein polymerization with dna |
| WO2020069055A1 (en) | 2018-09-28 | 2020-04-02 | Alnylam Pharmaceuticals, Inc. | Transthyretin (ttr) irna compositions and methods of use thereof for treating or preventing ttr-associated ocular diseases |
| WO2020118259A1 (en) | 2018-12-06 | 2020-06-11 | Northwestern University | Protein crystal engineering through dna hybridization interactions |
| WO2020226960A1 (en) | 2019-05-03 | 2020-11-12 | Dicerna Pharmaceuticals, Inc. | Double-stranded nucleic acid inhibitor molecules with shortened sense strands |
| WO2020236600A1 (en) | 2019-05-17 | 2020-11-26 | Alnylam Pharmaceuticals, Inc. | Oral delivery of oligonucleotides |
| CN114585749A (en) * | 2019-10-16 | 2022-06-03 | 斯蒂拉科技公司 | Determination of the concentration of nucleic acid sequences |
| WO2021092371A2 (en) | 2019-11-06 | 2021-05-14 | Alnylam Pharmaceuticals, Inc. | Extrahepatic delivery |
| WO2021092145A1 (en) | 2019-11-06 | 2021-05-14 | Alnylam Pharmaceuticals, Inc. | Transthyretin (ttr) irna composition and methods of use thereof for treating or preventing ttr-associated ocular diseases |
| WO2022147214A2 (en) | 2020-12-31 | 2022-07-07 | Alnylam Pharmaceuticals, Inc. | Cyclic-disulfide modified phosphate based oligonucleotide prodrugs |
| WO2022147223A2 (en) | 2020-12-31 | 2022-07-07 | Alnylam Pharmaceuticals, Inc. | 2'-modified nucleoside based oligonucleotide prodrugs |
| WO2022192038A1 (en) | 2021-03-12 | 2022-09-15 | Northwestern University | Antiviral vaccines using spherical nucleic acids |
| WO2022213118A1 (en) | 2021-03-31 | 2022-10-06 | Entrada Therapeutics, Inc. | Cyclic cell penetrating peptides |
| WO2022240721A1 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | Compositions and methods for modulating interferon regulatory factor-5 (irf-5) activity |
| WO2022240760A2 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | COMPOSITIONS AND METHODS FOR MODULATING mRNA SPLICING |
| WO2022241408A1 (en) | 2021-05-10 | 2022-11-17 | Entrada Therapeutics, Inc. | Compositions and methods for modulating tissue distribution of intracellular therapeutics |
| WO2022271818A1 (en) | 2021-06-23 | 2022-12-29 | Entrada Therapeutics, Inc. | Antisense compounds and methods for targeting cug repeats |
| WO2023283403A2 (en) | 2021-07-09 | 2023-01-12 | Alnylam Pharmaceuticals, Inc. | Bis-rnai compounds for cns delivery |
| WO2023003922A1 (en) | 2021-07-21 | 2023-01-26 | Alnylam Pharmaceuticals, Inc. | Metabolic disorder-associated target gene irna compositions and methods of use thereof |
| WO2023034817A1 (en) | 2021-09-01 | 2023-03-09 | Entrada Therapeutics, Inc. | Compounds and methods for skipping exon 44 in duchenne muscular dystrophy |
| WO2023064530A1 (en) | 2021-10-15 | 2023-04-20 | Alnylam Pharmaceuticals, Inc. | Extra-hepatic delivery irna compositions and methods of use thereof |
| WO2023220744A2 (en) | 2022-05-13 | 2023-11-16 | Alnylam Pharmaceuticals, Inc. | Single-stranded loop oligonucleotides |
| WO2024006999A2 (en) | 2022-06-30 | 2024-01-04 | Alnylam Pharmaceuticals, Inc. | Cyclic-disulfide modified phosphate based oligonucleotide prodrugs |
| WO2024040041A1 (en) | 2022-08-15 | 2024-02-22 | Dicerna Pharmaceuticals, Inc. | Regulation of activity of rnai molecules |
| WO2024039776A2 (en) | 2022-08-18 | 2024-02-22 | Alnylam Pharmaceuticals, Inc. | Universal non-targeting sirna compositions and methods of use thereof |
| WO2024073732A1 (en) | 2022-09-30 | 2024-04-04 | Alnylam Pharmaceuticals, Inc. | Modified double-stranded rna agents |
| WO2024168010A2 (en) | 2023-02-09 | 2024-08-15 | Alnylam Pharmaceuticals, Inc. | Reversir molecules and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US9464106B2 (en) | 2016-10-11 |
| US20070117144A1 (en) | 2007-05-24 |
| US20120157333A1 (en) | 2012-06-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9464106B2 (en) | Oligonucleotides useful for detecting and analyzing nucleic acids of interest | |
| US7060809B2 (en) | LNA compositions and uses thereof | |
| EP0628051B1 (en) | Applications of fluorescent n-nucleosides and fluorescent structural analogs of n-nucleosides | |
| US8057997B2 (en) | Nucleic acid binding compounds containing pyrazolo[3,4-D]pyrimidine analogues of purin-2,6-diamine and their uses | |
| El-Sagheer et al. | Click chemistry with DNA | |
| US8383344B2 (en) | Methods for quantification of microRNAs and small interfering RNAs | |
| EP1735459B1 (en) | Methods for quantification of micrornas and small interfering rnas | |
| US20060147924A1 (en) | Population of nucleic acids including a subpopulation of lna oligomers | |
| JP2005532618A (en) | System and method for predicting melting point (Tm) of oligonucleotide | |
| EP1975256B1 (en) | Modified oligonucleotides for mismatch discrimination | |
| EP2891714B1 (en) | Method for analyzing target nucleic acid, kit, and analyzer | |
| JP2003525292A5 (en) | ||
| US6063571A (en) | Process for amplifying nucleic acids using DNA/PNA primers | |
| WO2004035819A2 (en) | Oligonucleotide analogues for detecting and analyzing nucleic acids | |
| EP0669928A1 (en) | Applications of fluorescent n-nucleosides and fluorescent structural analogs of n-nucleosides | |
| EP1882748A2 (en) | A population of nucleic acids including a subpopulation of LNA oligomers | |
| Ghosh | Modified Nucleotides and Nucleic Acids as Molecular Probes | |
| Booth | Propargylamino modified bases for DNA duplex stabilisation | |
| Dempcy et al. | T m leveling compositions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: EXIQON A/S, DENMARK Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNORS:KAUPPINEN, SAKARI;ALSBO, CARSTEN;NIELSEN, PETER S.;AND OTHERS;REEL/FRAME:015635/0868;SIGNING DATES FROM 20041025 TO 20041217 |
|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |