US20030191086A1 - Substituted nitrogen heterocyclic derivatives and pharmaceutical use thereof - Google Patents
Substituted nitrogen heterocyclic derivatives and pharmaceutical use thereof Download PDFInfo
- Publication number
- US20030191086A1 US20030191086A1 US10/358,674 US35867403A US2003191086A1 US 20030191086 A1 US20030191086 A1 US 20030191086A1 US 35867403 A US35867403 A US 35867403A US 2003191086 A1 US2003191086 A1 US 2003191086A1
- Authority
- US
- United States
- Prior art keywords
- group
- alkyl
- substituted
- formula
- carbon atoms
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
- 0 *.[2*]C1=CC=CC([6*])=C1 Chemical compound *.[2*]C1=CC=CC([6*])=C1 0.000 description 75
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/16—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/40—Heterocyclic compounds containing purine ring systems with halogen atoms or perhalogeno-alkyl radicals directly attached in position 2 or 6
Definitions
- the present invention relates generally to new purine and pyrazolopyrimidine derivatives and their deaza analogues and to their use in suitable utilities, especially diagnostic and therapeutic methods.
- the present invention relates, in particular, to purine derivatives and their inhibitory effect with respect to cyclin-dependent kinase proteins, abbreviated cdks and also with an inhibitory effect with respect to viruses and immunostimulation.
- Purine analogues as cdk inhibitors are disclosed for example in WO 97/16452, WO 98/05335 and WO97/20842. The teaching of these patents includes 2,6,9-trisubstituted and less substituted purine derivatives only.
- Tetrasubstituted purines are disclosed in WO 98/01448 in which substituents are short hydrocarbonyl chains, usually represented by hydrogen.
- Substituents at C6 represents hydrogen or amine optionally substituted by one or two hydrocarbon groups; substituents at C8 are hydroxy, mercapto, acyloxy or oxycarbonyl substituted by aliphatic alkyl only.
- Nucleotide analogues containing phosphonate groups are disclosed for example in U.S. Pat. Nos.
- Typical purine base is adenine, 2,6-diaminopurine and guanine.
- the purine bases may include the deaza analogues thereof, 6,9-substituted and 2,6,9-trisubstituted purines and related analogues are disclosed in WO 96/33200. 2,8,9-, 6,8,9-trisubstituted and 2,6,8,9-tetrasubstituted purines and tri- and tetrasubstituted pyrazolopyrimidines and their deaza analogues have not yet been described.
- the invention concerns substituted nitrogen heterocyclic derivatives of the formula I—
- A is a divalent group selected from the ensemble consisting of
- Z is N or CH, provided that at least one Z being N;
- R2 and R6 are independent of one another, represent H, halogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, cycloalkyl alkyl, arylalkyl, heteroalkyl, heteroarylalkyl, heterocycloalkyl alkyl or R6′-X wherein
- X is an —NH—, —N(C 1 -C 6 -alkyl)-, —O— or —S— moiety;
- R6′ is H, alkyl, substituted alkyl, acyl, amido, sulpho, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycle, heteroaryl, substituted heteroaryl, arylalkyl, heterocycloalkyl, substituted heterocycloalkyl, heteroarylalkyl, heteroalkyl, cycloalkyl alkyl and heterocycloalkyl alkyl;
- R8 is halogen, hydroxyl, amino, carboxyl, cyano, nitro, amido, sulpho, sulphamino, carbamino, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, arylalkyl, heteroalkyl, heteroarylalkyl, cycloalkyl alkyl, heterocycloalkyl alkyl or R8′-X wherein
- X is —NH—, —N(alkyl)-, —O— or —S— moiety
- R8′ is according to any one of the substituents defined above for R2′ or R6′.
- R9 is alkyl, substituted alkyl, acyl, carboxyl, amido, sulphamino, cycloalkyl, substituted cycloalkyl, cycloalkyl alkyl, heteroalkylcycloalkyl alkyl, heterocycloalkyl, substituted heterocycloalkyl, aryl, substituted aryl, heterocycle, heteroaryl, substituted heteroaryl, arylalkyl, heteroarylalkyl, heteroalkyl or —B-R9′ wherein
- B is —CH 2 —, —(CH 2 ) 2 —, —CH(CH 3 )CH 2 —, —CH(CH 2 F)CH 2 —, —CH(CH 2 OH)CH 2 , or the groups of the following structure,
- R4 and R5 that are independent of one another, represent hydrogen, hydroxyl, halogen, amino, acyloxy substituent having 1-5 carbon atoms, alkoxy substituent having 1-5 carbon atoms, alkylmercapto substituent having 1-5 carbon atoms, alkylamino substituent having 1-5 carbon atoms and dialkylamino in which each alkyl substituent has 1-5 carbon atoms;
- R7 and R10 that are independent of one another, represent H or alkyl substituent having 1-10 carbon atoms;
- R9′ is -X(CH 2 ) m Y wherein
- X is —O—, —S—, —NH— or —N (alkyl)- substituent having 1-6 carbon atoms;
- Y is carboxyl, amido, sulpho, sulphamino, hydroxyl, carboxyl, mercapto, carbylmercapto, amino, alkylamino, carbamino —PO(OH) 2 , —PO(O-C 1 -C 6 -alkyl) 2 , —PO(NH-C 1 -C 6 -alkyl) —PO(O-C 1 -C 6 -alkyl) (NH-C 1 -C 6 -alkyl), —PO(OH) (O-C 1 -C 6 -alkyl);
- R9′, X, m and Y are as defined above and
- D is alkyl, substituted alkyl, —PO(OH) 2 , —PO (OH) (O-C 1 -C 6 -alkyl), —PO(OH)(NH-C 1 -C 6 -alkyl).
- Halogen refers to fluorine, bromine, chlorine, and iodine atoms.
- Alkyl refers to
- Substituted alkyl refers to a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and having substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl.
- substituent generic groups having the meanings being identical with the definitions of the corresponding groups as defined in this legend.
- Carbyloxy denotes the group —OR a , where R a is alkyl, substituted alkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl, cycloalkyl, substituted cycloalkyl, heterocycloalkyl or substituted heterocycloalkyl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Carbylmercapto denotes the group -SR b , where R b is alkyl, substituted alkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl, cycloalkyl, substituted cycloalkyl, heterocycloalkyl or substituted heterocycloalkyl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Acyl denotes the group —C(O)R e , where R e is hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl, cycloalkyl, substituted cycloalkyl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Aryloxy denotes the group —OAr, where Ar is an aryl, substituted aryl, heteroaryl or substituted heteroaryl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Alkylamino denotes the group —NR f R g , where R f and R g , that are independent of one another, represent hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, heteroaryl or substituted heteroaryl, provided that R f and R g are not both hydrogens, whereas these generic substituents have meanings which are identical with definitions of the corresponding groups defined herein.
- “Amido” denotes the group - c (O)NR h R i ′, where R h and R i ′ may independently be hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Carboxyl denotes the group —C(O)OR j , where R j is hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, heteroaryl or substituted heteroaryl, whereas these generic substituents have meanings which are identical with definitions of the corresponding groups defined herein.
- “Carbamino” denotes the group —NHCOR k , where R k may be hydrogen, alkyl, substituted alkyl, heterocycle, aryl, substituted aryl, heteroaryl and substituted heteroaryl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Aryl refers to an aromatic carbocyclic group having from 6 to 18 carbon atoms and being composed of at least one aromatic or multiple condensed rings in which at least one of which being aromatic.
- Substituted aryl refers to an aromatic carbocyclic group having from 6 to 18 carbon atoms and being composed of at least one aromatic ring or of multiple condensed rings at least one of which being aromatic.
- the ring(s) are optionally substituted with one or more substituents selected from the group consisting of halogen, alkyl, hydroxyl, carbylmercapto, alkylamino, carbyloxy, amino, amido, carboxyl, nitro, mercapto or sulpho, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Heterocycle refers to a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S.
- Heteroaryl refers to a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S with at least one ring of this group being aromatic.
- Substituted heteroaryl refers to a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S with at least one ring of this group being aromatic and this group being substituted with one or more substituents selected from the group consisting of halogen, alkyl, carbyloxy, carbylmercapto, alkylamino, amido, carboxyl, hydroxyl, nitro, mercapto or sulpho, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Arylalkyl denotes the group -R 1 —Ar where R i is
- Ar is an aromatic carbocyclic group having from 6 to 18 carbon atoms and being composed of at least one aromatic ring or of multiple condensed rings at least one of which being aromatic and the group being optionally substituted with one or more substituents selected from the group consisting of halogen, alkyl, hydroxyl, carbylmercapto, alkylamino, carbyloxy, amino, amido, carboxyl, nitro, mercapto or sulpho, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Heteroalkyl denotes the group -R m -L where R m is
- L is a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S and the group being unsubstituted or substituted with one or more substituents selected from the group consisting of halogen, alkyl, alkoxy, alkylmercapto, alkylamino, amido, carboxyl, hydroxy, nitro, mercapto or sulpho, whereas these generic substituents have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Heteroarylalkyl denotes the group -R n -G where R n is
- a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S with at least one ring of which being aromatic and the group being optionally substituted with one or more substituents selected from the group consisting of halogen, alkyl, carbyloxy, carbylmercapto, alkylamino, amido, carboxyl, hydroxyl, nitro, mercapto or sulpho, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Cycloalkyl refers to a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms.
- Substituted cycloalkyl refers to a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms and being substituted by one or more substituents selected from the group consisting of halogen, alkyl, substituted alkyl, carbyloxy, carbylmercapto, aryl, nitro, mercapto or sulpho, whereas these generic substituent groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Heterocycloalkyl refers to a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms which at least one ring carbon atom of its cyclic structure being replaced with a heteroatom selected from the group consisting of N, O, S or P.
- Substituted heterocycloalkyl refers to a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms which at least one ring carbon atom of its cyclic structure being replaced with a heteroatom selected from the group consisting of N, O, S or P and the group is containing one or more substituents selected from the group consisting of halogen, alkyl, substituted alkyl, carbyloxy, carbylmercapto, aryl, nitro, mercapto or sulpho, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Cycloalkyl alkyl denotes the group -R o -J where R o is
- a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms which contains one or more substituents selected from the group consisting of halogen, alkyl, substituted alkyl, carbyloxy, carbylmercapto, aryl, nitro, mercapto or sulpho, whereas these generic substituent groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- Heterocycloalkylalkyl denotes the group -R p V where R p is
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula I, wherein R6 ⁇ H and R2, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula I, wherein R2 ⁇ H and R6, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ia,
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ia, wherein R6 ⁇ H and R2, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ia, wherein R2 ⁇ H and R6, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ib,
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ib,
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ib, wherein R2 ⁇ H and R6, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ic,
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ic, wherein R6 ⁇ H and R2, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ic, wherein R2 ⁇ H and R6, R8 and R9 have above mentioned meanings.
- the inventions concerns further substituted nitrogen heterocyclic derivatives of the formula Id, wherein R6 ⁇ H and R2, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Id, wherein R2 ⁇ H and R6, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ie,
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ie, wherein R6 ⁇ H and R2, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ie, wherein R2 ⁇ H and R6, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula If, wherein R6 ⁇ H and R2, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula If, wherein R2 ⁇ H and R6, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ig,
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ig, wherein R6 ⁇ H and R2, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ig, wherein R2 ⁇ H and R6, R8 and R9 have above mentioned meanings.
- the invention concerns further substituted nitrogen heterocyclic derivatives of the formula I selected from the group consisting of 2-(1-hydroxymethylpropylamino)-6-benzylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ⁇ -hydroxyalkylamino, ⁇ -hydroxyalkyloxy, ⁇ -carboxyalkylamino, ⁇ -aminoalkylamino, ⁇ -fosfonoalkylamino, ⁇ -fosfonoalkyloxy, propinyl)-9-isopropylpurine, 2-(2-aminopropylamino)-6-benzylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ⁇ -hydroxyalkylamino, ⁇ -
- the invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6, R8 and R9 have above mentioned meanings, A is a group of formula
- R2, R6 and R9 have above mentioned meanings, is brominated with using a brominating system selected from a group consisting of bromine/acetic acid, bromine/chloroform, bromine/acetate buffer, bromine/water, N-bromosuccinimide/dimethylformamide and bromoacetamide/dimethylformamide to obtain a derivative of formula XIa,
- the invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6, R8 and R9 have above mentioned meanings, A is a group of formula
- R6 and R9 have above mentioned meanings, is brominated with using a brominating system selected from a group consisting of bromine/acetic acid, bromine/chloroform, bromine/acetate buffer, bromine/water, N-bromosuccinimide/dimethylformamide and bromoacetamide/dimethylformamide to obtain a derivative of formula XIc,
- the invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6 and R8 have above mentioned meanings, R9 is alkyl as defined above, A is a group of formula
- R6 is alkylated in position 9 with using an appropriate alkylating agent in a system selected from a group consisting of K 2 CO 3 /dimethylformamide, Cs 2 CO 3 /dimethylformamide, t-BuOK/dimethylformamide, t-BuOK/dimethylsulphoxide and NaH/dimethylformamide or under conditions of Mitsunobu reaction to obtain a derivative of formula XIIb
- R6 and alkyl are as defined above, and the chlorine atoms in positions 2 and 8 of the derivative of formula XIIb are optionally, either progressively or simultaneously, subjected to a substitution in order to replace them by another substituents R2 and R8, that have above mentioned meanings.
- the invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives, wherein R2, R6 and R8 have above mentioned meanings, R9 is alkyl as defined above, A is a group of formula
- R6 has above mentioned meanings
- a group R6 and/or the chlorine atom in position 2 and/or the chlorine atom in position 8 are optionally converted to another substituents R2, R6 and R8, that have above mentioned meanings and in such optionally modificated product of formula XIIc
- the 2-tetrahydropyranyl group is split off to obtain a corresponding derivative of formula XIIc wherein R9 is hydrogen
- so obtained product is then alkylated in position 9 with using an appropriate alkylating agent in a system selected from a group consisting of K 2 CO 3 /dimethylformamide, Cs 2 CO 3 /dimethylformamide, t-BuOK/dimethylformamide, t-BuOK/dimethylsulphoxide and NaH/dimethylformamide or under conditions of Mitsunobu reaction to obtain a tetrasubstituted derivative of formula XIIc, wherein R2,R6,R8 and R9 have above mentioned meanings
- the invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6 and R8 have above mentioned meanings, R9 is alkyl as defined above, A is a group
- Z is N, characterized in that, 2,6,8-trichloropurine is subjected to a nucleophilic substitution in position 6 in order to replace the chlorine atom in position 6 by another substituent R6 as defined above to obtain a derivative of formula XIIa,
- the invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2 is hydrogen, R6 and R8 have above mentioned meanings, R9 is alkyl as defined above, A is a group of formula
- R6 has above mentioned meanings
- so obtained derivative is alkylated in position 9 with using an appropriate alkylating agent in a system selected from a group consisting of K 2 CO 3 /dimethylformamide, Cs 2 CO 3 /dimethylformamide, t-BuOK/dimethylformamide, t-BuOK/dimethylsulphoxide and NaH/dimethylformamide or under conditions of Mitsunobu reaction to obtain a derivative of formula XVIa,
- the invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6 and R8 have above mentioned meanings, R9 is alkyl as defined above, A is a group of formula
- R6 is as defined above, is alkylated in position 9 with using either methyl acrylate or acrylonitrile or oxirane to obtain a derivative of formula XVIIa
- the invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R6 is a halogen or hydrogen, R2, R8 and R9 have above mentioned meanings, A is a group of formula
- R2, R8 and R9 have above mentioned meanings, is halogenated via diazotation with using, for example, amylnitrite/CH 2 Br 2 or amylnitrite/CHI 3 , and so obtained derivative wherein R6 is halogen is optionally hydrogenolyzed with using H 2 /Pd catalyst to obtain a corresponding derivative wherein R6 is hydrogen.
- the invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R9 has above mentioned meanings and one substituent of R2, R6 or R8 is alkinyl or alkenyl containing 2 to 6 carbon atoms whereas two other substituents of R2, R6 and R8 have above mentioned meanings, A is a group of formula I, wherein R9 has above mentioned meanings and one substituent of R2, R6 or R8 is alkinyl or alkenyl containing 2 to 6 carbon atoms whereas two other substituents of R2, R6 and R8 have above mentioned meanings, A is a group of formula
- Z is N or CH, characterized in that a derivative of formula I, wherein R9 has above mentioned meanings and one substituent of R2, R6 and R8 is a halogen whereas two other substituents of R2, R6 and R8 have above mentioned meanings, is alkinylated in the position of the halogen with using alkine/triphenylphosphinePdCl 2 /CuI/triethylamine system and so obtained 2-, 6- or 8-alkinyl derivative is optionally converted to 2-, 6- or 8-alkenyl derivative with using a Lindlar catalyst.
- the invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6, R8 and R9 have above mentioned meanings, a is a group of formula
- Z is N, characterized in that 2,5-dialkyl-3-alkoxycarbonyl-4-aminopyrazole, wherein alkyls and alkoxy have above mentioned meanings, is reacted with formamidine acetate/triethylamine system to obtain 1,3-dialkyl-7-hydroxypyrazolo[4,3-d]pyrimidine and so obtained 7-hydroxy derivative is then optionally transferred to a corresponding 7-chloro derivative by reaction with thionylchloride, which product is optionally substituted to obtain a corresponding R6-derivative, wherein R6 has above mentioned meanings.
- the invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6, R8 and R9 have above mentioned meanings, A is a group of formula
- [0169] and Z is N, characterized in that 3,7-dialkylpyrazolo [4,3-d]pyrimidine, wherein alkyls have above mentioned meanings, is alkylated with using an appropriate alkylating agent in a system selected from a group consisting of K 2 CO 3 /dimethylformamide and Cs 2 CO 3 /dimethylformamide to obtain a mixture of trialkylated and tetraalkylated products and the trialkylated product is separated by means of the column chromatography.
- an appropriate alkylating agent in a system selected from a group consisting of K 2 CO 3 /dimethylformamide and Cs 2 CO 3 /dimethylformamide to obtain a mixture of trialkylated and tetraalkylated products and the trialkylated product is separated by means of the column chromatography.
- the invention also relates to substituted nitrogen heterocyclic derivatives of formula I and pharmaceutically acceptable salts thereof for use as medicaments.
- the invention also relates to substituted nitrogen heterocyclic derivatives of formula I for use as means for preparing affinity adsorption matrices, immobilized enzymes for process control, immunoassay reagents, diagnostic samples, 14 C-, 3 H-, avidin- or biotin-labelled compounds, oligonucleotides and diagnostic samples.
- the invention also relates to substituted nitrogen heterocyclic derivatives of formula I for use as antimitotic drugs, in particular drugs for elimination or reduction of viral spread or growth in tissue culture systems during the production of biopharmaceutical or other products such as proteins and vaccines, drugs for elimination or reduction of viral spread and growth in clinical samples such as blood, and drugs for stopping of growth of tissue culture cells while leaving the cells to carry on with protein production.
- the invention also relates to a pharmaceutical composition with cytostatic, anticancer, antimitotic, antineurodegenerative, immunosuppressive and antimicrobial activity, characterised in that it comprises, in addition to auxiliary pharmaceutical matters, at least one substituted nitrogen heterocyclic derivative of formula I.
- the invention also relates to a combined pharmaceutical composition with cytostatic effect, characterised in that it comprises, in addition to auxiliary pharmaceutical matters, a combination of at least one substituted nitrogen heterocyclic derivative of formula I and a cytostatic agent selected from a group consisting of mitoxantron, cis- and carbo-platin, methotrexate, taxol and doxorubicin.
- a cytostatic agent selected from a group consisting of mitoxantron, cis- and carbo-platin, methotrexate, taxol and doxorubicin.
- the invention also relates to a use of substituted nitrogen heterocyclic derivatives of formula I for preparing medicaments destined for treating tumours, cancers, psoriasis, rheumatoid arthritis, lupus, type I diabetes, multiple sclerosis, restenosis, polycyclic kidney disease, host graft disease and gout, parasitoses, such as those caused by fungi or protists, or Alzheimer's disease, or for preparing antineurodegenerative drugs and suppress immunostimulation agents.
- novel compounds of this invention have a wide variety of diagnostic, therapeutic and industrial utilities.
- the compounds of this invention are suitable as intermediates for use in the preparation of affinity absorption matrices.
- the phosphonate groups in matrix bound form are useful in the chromatographic separation of positively charged molecules.
- Other immobilised examples of the compounds herein are useful in purifying proteins, e.g., cell cycle enzymes (cdk's), enzymes involved in recognition of the compound of this invention, e.g. transport proteins.
- cdk's cell cycle enzymes
- Suitable methods of incorporation of the compounds of this invention into polymeric resins will be readily apparent to the skilled artisan, for instance the compounds are incorporated by cross-linking hydroxyl groups of the phosphonate or hydroxymethyl substituents using cross-linking agents heretofore known. Linking through a group other than the heterocyclic base will produce a resin useful in hydrophobic affinity chromatography.
- Other suitable linking methods are described in Cihlar (supra).
- the compounds of the formula I and their pharmaceutically acceptable salts inhibit selectively the enzyme p34 cdc2 /cyclin B kinase and related cdks (cdk2, cdk5, cdk7, erk1, erk2).
- this invention is a method for inhibiting cdks and cell proliferation in mammals comprising administering a therapeutically effective amount of the composition of claim 1 to the mammal.
- the cdk inhibiting molecules are useful for treating cell proliferation disorders such as rheumatoid arthritis, lupus, type I diabetes, multiple sclerosis, cancer, restenosis, Alzheimer's disease, growth of parasites (animal, protists), host graft disease, and gout.
- this invention is a composition useful for treating fungal infections (fungi) in humans, animal, and in plants.
- this kinase controls certain steps of cell division cycles, in particular the transition from GI phase into the S phase and in particular the transition from the G2 phase into the M phase.
- hypoproliferative diseases such as cancer, restenosis and Alzheimer's disease.
- they are capable of inhibiting cell cycle transitions (G1/S, G2/M, M-phase/metaphase) carried out on the different animal bodies and embryos.
- the compounds are useful in treating autoimune diseases, e.g.
- rheumatoidal arthritis in treating of cancer, cardiovascular disease such as restenosis, host vs graft disease, gout, polycystic kidney disease and other proliferative diseases whose pathogenesis involves abnormal cell proliferation.
- This invention also concerns novel compounds that have been discovered to be potent and specific inhibitors of I ⁇ B- ⁇ kinase which prevents signal induced NF- ⁇ B activation and cytokine synthesis in vitro and in vivo.
- Such inhibitors are expected to inhibit synthesis of cytokines and adhesion proteins whose synthesis is transcriptionally regulated by NF- ⁇ B.
- Pro-inflammatory cytokines such as IL-1, IL-6, TNF and adhesion proteins (e.g. ICAM, VCAM and selections) belong to this class of molecules and have implicated in the pathogenesis of inflammatory diseases.
- a potent inhibitor of I ⁇ B- ⁇ kinase is useful in the clinical management of diseases where the NF- ⁇ B activation is required for disease induction.
- p53 the mammal cell's own natural brake gene for stopping uncontrolled cell proliferation (cancer), thus being able to switch off the cancer.
- p53 as well as retinoblastoma (Rb) are two well-characterised tumour suppressors whose inactivation may led to uncontrolled cell proliferation and malignancy. Phosphorylation of these two proteins, which are involved in the cell cycle regulatory mechanisms, is known to modulate their function.
- a potent cdk inhibitor represent a good toll for treatment of cancers due to induction of wild type p53 protein in cancers expressing mutant p53.
- the interest is therefore assessed by application of the derivatives of the invention for stimulating on of p53-independent apoptosis in the cells, which have stopped at stage G2 through damage to the DNA using agents such as mitoxantrone or cis-platinum.
- the cdk inhibitors of this invention can thus increase the therapeutic potential of the anti-tumour agents currently used.
- the compounds of this invention will generally be terminally incorporated into the oligonucleotide. If they do not contain phosphonyl group attached to the hydroxyl group, they optionally are incorporated internally into the sequence of the oligonucleotide. Terminally incorporated diphosphonyl compounds of this invention which contain no free hydroxyl capable of participating in chain elongation also are useful in DNA sequencing in essentially the same manner as deoxyNTPs have been used in the past (see example 8 of U.S. Pat. No. 5,276,143).
- the nucleotide analogue is included in a kit with other reagents (such as Klenow polymerase or T4 polymerase, dNTPs, etc) needed for DNA sequencing (Otvos, et al. “Nucl. Acids. Res.” 15:1763-1777 (1987).
- the oligonucleotide-incorporated compound of this invention is bindingcompetent for its complementary sequence, i.e., if it is capable of base pairing, then this nucleotide monomer will participate in hybridisation. It is not necessary, however, that the incorporated nucleotide analogue of this invention base pair or otherwise participate in hybridisation. If it is located at the terminus of the oligonucleotide it will be useful as an immunological recognition site, or haptenic recognition site, to facilitate detection of the oligonucleotide by an antibody capable of binding the compound of this invention.
- the compounds of this invention also are useful as linkers or spacers in preparation affinity absorption matrices (as opposed to functioning as affinity moieties per se as noted above), immobilised enzymes for process control, or immunoassay reagents.
- the compounds herein contain a multiplicity of functional groups that are suitable as sites for cross-linking desired substances. For example, it is conventional to link affinity reagents such as hormones, peptides, antibodies, drugs, and the like to insoluble substrates. These insolubilised reagents are employed in known fashion to absorb binding partners for the affinity reagents from manufactured preparations, diagnostic samples and other impure mixture. Similarly, immobilised enzymes are used to perform catalytic conversions with facile recovery of enzyme. Bifunctional compounds are commonly used to link analytes to detectable groups in preparing diagnostic reagents.
- the phosphonic acid is used to form esters with alcohols or amides with amines.
- the R groups substituted with OH, azido (which is reduced to amino if desired before cross-linking) or vinyl are exemplary suitable sites.
- the amino, halo, acyl and other reactive sites found on group B are suitable. Suitable protection of reactive groups will be used where necessary while assembling the cross-linked reagent.
- the compounds here are used by linking them through phosphonic acid or amino group to the hydroxyl or amino groups of the linking partner in the same fashion as shown herein, and covalently bonded to the other binding partner through an R group.
- a first binding partner such as a steroid hormone is esterified and then this conjugate is cross-linked through hydroxymethyl R to cyanogen bromide activated Sepharose, whereby immobilised steroid is obtained.
- Other chemistries for conjugation are well known. See for example Maggio, “EnzymeImmunoassay” (CRC, 1988, pp 71-135) and references cited therein.
- the oligonucleotides of this invention are labeled with any conventional detectable label, e.g. a fluorescent moiety such a fluorescein, radioisotopes such as 14 C or 3 H, stable free radicals, avidin, biotin and the like all of which previously have been used as labels for immunoassays or diagnostic probes.
- a fluorescent moiety such as fluorescein, radioisotopes such as 14 C or 3 H, stable free radicals, avidin, biotin and the like all of which previously have been used as labels for immunoassays or diagnostic probes.
- the label will be present on the oligonucleotide or on the residue of an analogue of this invention.
- Suitable labelling methods are well known and are readily used with reactive groups such as hydroxyl, allyl and the like.
- a simple method is to label the compound of this invention with H 3 by proton exchange.
- the compounds also are biotinylated using conventional methods. See for instance U.S
- the compounds of this invention also are useful directly in diagnostic probe assays without an exogenous detectable label.
- antibodies are raised against the compounds of this invention. Such antibodies (which in turn are labelled or used in a double antibody configuration) bind to the analogue of this invention and thereby are useful in detecting its presence as label for a protein or oligonucleotide.
- the compounds of the invention are useful for treatment of microbial infections, for treatment of tumours or for other indications described below.
- Microbial infections treatable by the compounds of this invention include viruses, parasites, yeast and fungi, but it is believed that the compounds are most effective against viruses, which constitutes the preferred utility.
- Exemplary viral infections include infections caused by DNA or RNA viruses including herpes viruses (CMV, HSV 1, HSV 2, EBV, varicella zoster virus (VZV), bovid herpesvirus type 1, equid herpesvirus type 1, HVV-6, papillomaviruses (HPV types 1-55 including carcinogenic HPV), flaviviruses (including yellow fever virus, African swine fever virus and Japanese encephalitis virus), togaviruses (including Venezuelan equine encephalomyelitis virus), influenza viruses (types A-C), retroviruses (HIV-1, HIV-2, HTLV-I, HTLV-II, SIV, FeLV, FIV, MoMSV), adenoviruses (types 1-8), poxviruses (vaccinia virus), enteroviruses (poliovirus types 1-3, Coxsackie, hepatitis A virus, and ECHO virus), gastroenteritis viruses (Norwalk viruses, rot
- the antiviral activity of individual compounds is determined by routine assay of antiviral (or other antimicrobial) activity using enzyme inhibition assays, tissue culture assays, animal model assays and the like as will be understood by those skilled in the art.
- protozoa include those members of the subphyla Sarcomastigophora and Sporozoa of the phylum Protozoa. More particularly, the term protozoa as used herein include genera of parasitic protozoa, which are important to man, because they either cause disease in man or in his domestic animals. These genera for the most part are classified in the superclass Mastigophora of the subphylum Sarcomastigophora and the class Telesporea of the subphylum Sporozoa in the classification according to Baker (1969).
- Illustrative genera of these parasitic protozoa include Histomonas, Pneumocystis, Trypanosoma, Giardia, Trichomonas, Eimeria, Isopora, Leishmania, Entamoeba, Toxoplasma and Plasmodium.
- Parasitic protozoans include Plasmodium falciparum, Plasmodium berghei, Plasmodium malariae, Plasmodium vivax, Leishmania braziliensis, Leishmania donovani, Trypanosoma cruzi, Trypanosoma brucei, Trypanosoma rhodesiense, Pneumocystis carinii, Entamoeba histolytica, Trichomonas vaginalis and the like (de Vries, E., et al, “Mol. Biochem. Parasitol” 47:43-50 (1991) and trypanosomes (Kaminsky et al. “J.Parasitol.” 80(6): 1026-30 (1994).
- the compounds in which R is CH 2 OH and B is 3-deazaadenine are particularly interesting in the treatment of malarial parasites.
- Compounds of the invention are used to treat yeast or fungal infections caused by Candida glabrata, Candida ropicalis, Candida albicans, and other Candida species, Cryptococcus species including Cryptococcus neoformans, Blastomyces species including Blastomyces dermatidis, Torulopsis species including Torulopsis glabrata, Coccidioides species including Coccidioides immitis, Aspergillus species and the like.
- the compounds of the invention can also be (1) applied to tissue culture systems to eliminate or reduce viral spread or growth during the production of biopharmaceutical or other products (such as proteins or vaccines), (2) used to eliminate or reduce viral spread or growth in clinical sample (such as blood), and (3) used to stop growth of tissue culture cells while leaving the cells to carry on with protein production.
- biopharmaceutical or other products such as proteins or vaccines
- clinical sample such as blood
- the compounds herein have been found to suppress immunostimulation. Accordingly, they can suppress metabolic activities of T-lymphocytes stimulated by diverse agents, e.g. concavalin A, they principally will find application in the treatment of autoimmune diseases, e.g. arthritis, or in suppression of transplant rejection. Their therapeutically active concentrations are in the range of 1 mg/kg to 50 mg/kg of body weight.
- Suitable routes for administration include oral, rectal, vassal, topical (including ocular, buccal and sublingual), vaginal and parental (including subcutaneous, intramuscular, intravitreous, intravenous, intradermal, intrathecal and epidural).
- the preferred route of administration will depend upon the condition of the patient, the toxicity of the compound and the site of infection, among other considerations known to the clinician.
- the therapeutical composition comprise about 1% to about 95% of the active ingredient, single-dose forms of administration preferably comprising about 20% to about 90% of the active ingredient and administration forms which are not single-dose preferably comprising about 5% to about 20% of the active ingredient.
- Unit dose forms are, for example, coated tablets, tablets, ampoules, vials, suppositories or capsules.
- Other forms of administration are, for example, ointments, creams, pastes, foams, tinctures, lipsticks, drops, sprays, dispersions-and the like. Examples are capsules containing from about 0.05 g to about 1.0 g of the active ingredient.
- compositions of the present invention are prepared in a manner known per se, for example by means of convential mixing, granulating, coating, dissolving or lyophilising processes.
- solutions of the active ingredient, and in addition also suspensions or dispersions, especially isotonic aqueous solutions, dispersions or suspensions are used, it being possible for these to be prepared before use, for example in the case of lyophilised compositions which comprise the active substance by itself or together with a carrier, for example mannitol.
- the pharmaceutical compositions can be sterilised and/or comprise excipients, for example preservatives, stabilisers, wetting agents and/or emulsifiers, solubilizing agents, salts for regulating the osmotic pressure and/or buffers, and they are prepared in a manner known per se, for example by means of convential dissolving or lyophilising processes.
- the solutions or suspensions mentioned can comprise viscosity-increasing substances, such as sodium carboxymethylcellulose, carboxymethylcellulose, dextran, polyvinylpyrrolidone or gelatin.
- Suspensions in oil comprise, as the oily component, the vegetable, synthetic or semisynthetic oils customary for injection purposes.
- Oils which may be mentioned are, in particular, liquid fatty acid esters which contain, as the acid component, a long-chain fatty acid having 8-22, in particular 12-22, carbon atoms, for example lauric acid, tridecylic acid, myristic acid, pentadecylic acid, palmitic acid, margaric acid, stearic acid, arachidinic acid, behenic acid or corresponding unsaturated acids, for example oleic acid, elaidic acid, euric acid, brasidic acid or linoleic acid, if appropriate with the addition of antioxidants, for example vitamin E, ( ⁇ -carotene or 3,5-di-tert-butyl-4 hydroxytoluene.
- the alcohol component of these fatty acid esters has not more than 6 carbon atoms and is mono- or polyhydric, for example mono-, di- or trihydric alcohol, for example methanol, ethanol, propanol, butanol, or pentanol, or isomers thereof, but in particular glycol and glycerol.
- Fatty acid esters are therefore, for example: ethyl oleate, isopropyl myristate, isopropyl palmitate, “Labrafil M 2375” (polyoxyethylene glycerol trioleate from Gattefosee, Paris), “Labrafil M 1944 CS” (unsaturated polyglycolated glycerides prepared by an alcoholysis of apricot kernel oil and made up of glycerides and polyethylene glycol esters; from Gattefosee, Paris), “Labrasol” (saturated polyglycolated glycerides prepared by an alcoholysis of TCM and made up of glycerides and polyethylene glycol esters; from Gattefosee, Paris) and/or “Miglyol 812” (triglyceride of saturated fatty acids of chain length C 8 to C 12 from Hills AG, Germany), and in particular vegetable oils, such as cottonseed oil, almond oil, olive oil, castor oil, sesame oil,
- the preparation of the injection compositions is carried out in the customary manner under sterile conditions, as are bottling, for example in ampoules or vials, and closing of the containers.
- compositions for oral use can be obtained by combining the active ingredient with one or more solid carriers, if appropriate granulating the resulting mixture, and, if desired, processing the mixture or granules to tablets or coated tablet cores, if appropriate by addition of additional excipients.
- Suitable carriers are, in particular, fillers, such as sugars, for example lactose, sucrose, mannitol or sorbitol, cellulose preparations and/or calcium phosphates, for example tricalcium phosphate, or calcium hydrogen phosphate, and furthermore binders, such as starches, for example maize, wheat, rice or potato starch, methylcellulose, hydroxypropyl-methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidine, and/or, if desired, desintegrators, such as the above mentioned starches, and furthermore carboxymethyl-starch, crosslinked polyvinylpyrrolidone, alginic acid or a salt thereof, such as sodium alginate.
- fillers such as sugars, for example lactose, sucrose, mannitol or sorbitol
- cellulose preparations and/or calcium phosphates for example tricalcium phosphate, or calcium hydrogen phosphate
- binders such as starches, for example maize
- Additional excipients are, in particular, flow regulators and lubricants, for example salicylic acid, talc, stearic acid or salts thereof, such as magnesium stearate or calcium stearate, and/or polyethylene glycol, or derivatives thereof.
- flow regulators and lubricants for example salicylic acid, talc, stearic acid or salts thereof, such as magnesium stearate or calcium stearate, and/or polyethylene glycol, or derivatives thereof.
- Coated tablet cores ca be provided with suitable coatings which, if appropriate, are resistant to gastric juice, the coatings used being, inter alia, concentrated sugar solutions, which, if appropriate, comprise gum arabic, talc, polyvinylpyrrolidine, polyethylene glycol and/or titanium dioxide, coating solutions in suitable organic solvents or solvent mixtures or, for the preparation of coatings which are resistant to gastric juice, solutions of suitable cellulose preparations, such as acetylcellulose phthalate or hydroxypropylmethylcellulose phthalate. Dyes or pigments can be admixed to the tablets or coated tablet coatings, for example for identification or characterisation of different doses of active ingredient.
- suitable coatings which, if appropriate, are resistant to gastric juice
- the coatings used being, inter alia, concentrated sugar solutions, which, if appropriate, comprise gum arabic, talc, polyvinylpyrrolidine, polyethylene glycol and/or titanium dioxide, coating solutions in suitable organic solvents or solvent mixtures or, for the preparation of coatings which are resistant
- compositions which can be used orally, are also hard capsules of gelatin and soft, closed capsules of gelatin and a plasticiser, such as glycerol or sorbitol.
- the hard capsules can contain the active ingredient in the form of granules, mixed for example with fillers, such as maize starch, binders and/or lubricants, such as talc or magnesium stearate, and stabilisers if appropriate.
- the active ingredient is preferably dissolved or suspended in suitable liquid excipients, such as greasy oils, parrafin oil or liquid polyethylene glycols or fatty acid esters of ethylene glycol or propylene glycol, it being likewise possible to add stabilisers and detergents, for example of the polyethylene sorbitan fatty acid ester type.
- suitable liquid excipients such as greasy oils, parrafin oil or liquid polyethylene glycols or fatty acid esters of ethylene glycol or propylene glycol, it being likewise possible to add stabilisers and detergents, for example of the polyethylene sorbitan fatty acid ester type.
- oral forms of administration are, for example, syrups prepared in the customary manner, which comprise the active ingredient, for example, in suspended form and in a concentration of about 5% to 20%, preferably about 10% or in a similar concentration which results in a suitable individual dose, for example, when 5 or 10 ml are measured out.
- Other forms are, for example, also pulverulent or liquid concentrates for preparing of shakes, for example in milk. Such concentrates can also be packed in unit dose quantities.
- compositions which can be used rectally, are, for example, suppositories that comprise a combination of the active ingredient with a suppository base.
- Suitable suppository bases are, for example, naturally occurring or synthetic triglycerides, paraffin hydrocarbons, polyethylene glycols or higher alkanols.
- compositions which are suitable for parenteral administration are aqueous solutions of an active ingredient in water-soluble form, for example of water-soluble salt, or aqueous injection suspensions, which comprise viscosity-increasing substances, for example sodium carboxymethylcellulose, sorbitol and/or dextran, and if appropriate stabilisers.
- the active ingredient can also be present here in the form of a lyophilisate, if appropriate together with excipients, and be dissolved before parenteral administration by addition of suitable solvents. Solutions such as are used, for example, for parental administration can also be used as infusion solutions. Preferred preservatives are, for example.
- Antioxidants such as ascorbic acid, or microbicides, such as sorbic or benzoic acid.
- Ointments are oil-in-water emulsions, which comprise not more than 70%, but preferably 20-50% of water or aqueous phase.
- the fatty phase consists, in particular, hydrocarbons, for example vaseline, paraffin oil or hard paraffin's, which preferably comprise suitable hydroxy compounds, such as fatty alcohol's or esters thereof, for example cetyl alcohol or wool wax alcohols, such as wool wax, to improve the waterbinding capacity.
- Emulsifiers are corresponding lipophilic substances, such as sorbitan fatty acid esters (Spans), for example sorbitan oleate and/or sorbitan isostearate.
- Additives to the aqueous phase are, for example, humectants, such as polyalcohols, for example glycerol, propylene glycol, sorbitol and/or polyethylene glycol, or preservatives and odoriferous substances.
- humectants such as polyalcohols, for example glycerol, propylene glycol, sorbitol and/or polyethylene glycol, or preservatives and odoriferous substances.
- Fatty ointments are anhydrous and comprise, as the base, in particular, hydrocarbons, for example paraffin, vaseline or paraffin oil, and furthermore naturally occurring or semisynthetic fats, for example hydrogenated coconut-fatty acid triglycerides, or, preferably, hydrogenated oils, for example hydrogenated groundnut or castor oil, and furthermore fatty acid partial esters of glycerol, for example glycerol mono- and/or distearate, and for example, the fatty alcohols. They also contain emulsifiers and/or additives mentioned in connection with the ointments which increase uptake of water.
- hydrocarbons for example paraffin, vaseline or paraffin oil
- furthermore naturally occurring or semisynthetic fats for example hydrogenated coconut-fatty acid triglycerides, or, preferably, hydrogenated oils, for example hydrogenated groundnut or castor oil, and furthermore fatty acid partial esters of glycerol, for example glycerol mono- and/or di
- Creams are oil-in-water emulsions, which comprise more than 50% of water.
- Oily bases used are, in particular, fatty alcohols, for example lauryl, cetyl or stearyl alcohols, fatty acids, for example palmitic or stearic acid, liquid to solid waxes, for example isopropyl myristate, wool wax or beeswax, and/or hydrocarbons, for example vaseline (petrolatum) or paraffin oil.
- Emulsifiers are surface-active substances with predominantly hydrophilic properties, such as corresponding nonionic emulsifiers, for example fatty acid esters of polyalcohols or ethyleneoxy adducts thereof, such as polyglyceric acid fatty acid esters or polyethylene sorbitan fatty esters (Tweens), and furthermore polyoxyethylene fatty alcohol ethers or polyoxyethylene fatty acid esters, or corresponding ionic emulsifiers, such as alkali metal salts of fatty alcohol sulfates, for example sodium lauryl sulfate, sodium cetyl sulfate or sodium stearyl sulfate, which are usually used in the presence of fatty alcohols, for example cetyl stearyl alcohol or stearyl alcohol.
- corresponding nonionic emulsifiers for example fatty acid esters of polyalcohols or ethyleneoxy adducts thereof, such as polyglyceric acid fatty acid esters or polyethylene
- Additives to the aqueous phase are, inter alia, agents which prevent the creams from drying out, for example polyalcohols, such as glycerol, sorbitol, propylene glycol and/or polyethylene glycols, and furthermore preservatives and odoriferous substances.
- polyalcohols such as glycerol, sorbitol, propylene glycol and/or polyethylene glycols, and furthermore preservatives and odoriferous substances.
- Pastes are creams and ointments having secretion-absorbing powder constituents, such as metal oxides, for example titanium oxide or zinc oxide, and furthermore talc and/or aluminium silicates, which have the task of binding the moisture or secretions present.
- secretion-absorbing powder constituents such as metal oxides, for example titanium oxide or zinc oxide, and furthermore talc and/or aluminium silicates, which have the task of binding the moisture or secretions present.
- Foams are administered from pressurised containers and they are liquid oil-inwater emulsions present in aerosol for.
- halogenated hydrocarbons such as chlorofluoro-lower alkanes, for example dichlorofluoromethane and dichlorotetrafluoroethane, or, preferably, non-halogenated gaseous hydrocarbons, air, N 2 O, or carbon dioxide are used.
- the oily phases used are, inter alia, those mentioned above for ointments and creams, and the additives mentioned there are likewise used.
- Tinctures and solutions usually comprise an aqueous-ethanolic base to which, humectants for reducing evaporation, such as polyalcohols, for example glycerol, glycols and/or polyethylene glycol, and re-oiling substances, such as fatty acid esters with lower polyethylene glycols, i.e. lipophilic substances soluble in the aqueous mixture to substitute the fatty substances removed from the skin with the ethanol, and, if necessary, other excipients and additives, are admixed.
- humectants for reducing evaporation such as polyalcohols, for example glycerol, glycols and/or polyethylene glycol
- re-oiling substances such as fatty acid esters with lower polyethylene glycols, i.e. lipophilic substances soluble in the aqueous mixture to substitute the fatty substances removed from the skin with the ethanol, and, if necessary, other excipients and additives, are
- the present invention further provides veterinary compositions comprising at least one active ingredient as above defined together with a veterinary carrier therefor.
- Veterinary carriers are materials for administering the composition and may be solid, liquid or gaseous materials, which are inert or acceptable in the veterinary art and are compatible with the active ingredient. These veterinary compositions may be administered orally, parenterally or by any other desired route.
- the invention also relates to a process or method for treatment of the disease states mentioned above.
- the compounds can be administered prophylactically or therapeutically as such or in the form of pharmaceutical compositions, preferably in an amount, which is effective against the diseases mentioned.
- a warm-blooded animal for example a human, requiring such treatment, the compounds are used, in particular, in the form of pharmaceutical composition.
- a daily dose of about 0.1 to about 5 g, preferably 0.5 g to about 2 g, of a compound of the present invention is administered here for a body weight of about 70 kg.
- 6-Amino-2-(3-hydroxypropylamino)-9-methyl-8-(3-hydroxypropyloxy)purine was brominated in the position 6 with amylnitrite/CH 2 Br 2 .
- the 6-bromo derivative was then hydrogenolyzed with PdO/BaSO 4 in strong alkaline solution to give 2,8,9-trisubstituted purine.
- the crude product was purified by column chromatography (silica gel, chloroform-MeOH-conc. NH 4 OH (8:2:0.2)). The total yield was 35%.
- 6-(Benzylamino)-2,8-dichloropurine prepared from 2,6,8-trichloropurine, m.p.220-223° C., 50% yield, was dissolved in 2-aminobutanol and heated to 150° C. for 12 hours. After evaporation of the solvent in vacuo the product, 6-benzylamino-2-[(1-hydroxymethyl) propylamino]-8-chloropurine was purified by column chromatography (silica gel, chloroform—MeOH—conc. NH 4 OH 9:1:0.1) with total yield 36%. MS-ESI(+) m/z 348 [M+H] + .
- Freeze-dried epoxy activated Sepharose 6B (Pharmacia LKB, Piscataway, N.J.) was chosen for the coupling reaction due to its ability to form an ether bond between a hydroxyl-containing ligand and the epoxide group on the Sepharose.
- the resin was activated with three alternating cycles of twenty column volumes each of pH 4.0 (0.1 M acetate, 0.5 M NaCl) and pH 8.0 (0.1 M tris-HCI, 0.5 M NaCl) buffers followed by twenty column volumes of reaction buffer (20 mM HEPES, pH 7.3, 10 mM MgCl 2 , 15 mM glycerophosphate, 0.5 mM sodium orthovanadate, 0.5 mM EGTA).
- the column was stored at 4° C in the reaction buffer containing 0.1% sodium azide and regenerated prior to each use with alternating cycles of low and high pH as described above.
- the Sf9 insect cell lysate (500 ⁇ g protein in 1-ml reaction buffer) was passed over the affinity column matrix sequentially five times and the flow through was saved, (unbound material). The matrix was then washed three times with 1 ml reaction buffer (wash 1-3) then three times each reaction buffer containing 0.5M NaCl (eluate 1-3). The coupled proteins were eluted at low pH (pH 4.0, 0.1M acetate, 0.5M NaCl) as described above and aliquots (20 ⁇ from 1 ml) of each sample were assayed for their ability to phosphorylate histone H1 and other substrate proteins as described in Example 12. The presence of CDK complexes was also determined by SDS-PAGE.
- Cyclin-dependent kinases (p34 cdc2 , p33 cdk2 , p33 cdk4 ) and cyclins (cyclin B, E and D1) are produced in Sf9 insect cells coinfected with appropriate baculoviral constructs.
- the cells are harvested 68-72 hrs post infection in lysis buffer for 30 min on ice and the soluble fraction is recovered by centrifugation at 14.000 g for 10 min.
- the protein extract is stored at ⁇ 80° C.
- Rb-GST is produced using an E. coli expression system, containing sequence encoding C terminus of retinoblastoma protein (aminoacids 773-928), which is known to be phosphorylated by p33 cdk4 kinase.
- the fusion protein is purified on glutathioneagarose beads.
- Lysis buffer 50 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 20 mM NaF, 1% Tween, 1 mM DTT, 0.1 mM PMSF, leupeptine, aprotonine.
- the p34 cdc2 and p33 cdk2 kinase inhibition determination involves the use of 1 mg/ml histone H1 (Sigma, type III-S) in the presence of 15 ⁇ M [ ⁇ - 32 P]ATP (500-100 cpm/pmol) (Amersham) in a final volume of 20 ⁇ l, inhibition of p33 cdk4 kinase is determined with Rb-GST (0.2 mg/ml) as a substrate.
- Kinase activity is determined at 30° C. in the kinase buffer.
- Tested compounds are usually dissolved to 100 mM solutions in DMSO, final concentration of DMSO in reaction mixture never exceeds 1%.
- the controls contain suitable dilutions of DMSO.
- kinase activity is expressed as a percentage of maximum activity. The apparent inhibition constants are determined by graphic analysis.
- Kinase buffer 50 mM Hepes pH 7.4. 10 mM MgCl 2 , 5 mM EGTA.
- Table 4 shows the results of inhibitory activity of novel compounds against CDC2 and I ⁇ B- ⁇ in comparison with the data on the prototype compounds. Most of the 2,6,8,9-tetrasubstituted purine derivatives showed marked inhibitory activity in in vitro kinase assays. Modification of 2,6,8-trisubstituted purines by small substituent in R8 position led usually to increase in cdk inhibitory activity of the tested compound.
- Metaphase-arrested Xenopus egg extracts were prepared as described previously by Blow “J.Cell Biol.” 122:993 (1993) and stored in liquid nitrogen. Demembranated Xenopus sperm nuclei were prepared as described by Blow & Laskey “Cell” 47:577 (1986). After thawing, extracts were supplemented with 25 mM phosphocreatine, 5 ⁇ g/ml creatine phosphokinase, 250 ⁇ g/ml cycloheximide, [ ⁇ - 32 P]dATP (for DNA synthesis assays).
- Demembranated sperm nuclei were added to a final sperm concentration of 3 ng/ ⁇ l DNA extract and CDK inhibitor tested was then added at different concentrations. M-phase promoting factor inhibition by different CDK inhibitors was monitored 1.5 h after addition by assessing the amount of sperm nuclei that had been assembled into interphase nuclei, possessing a complete phase-dense nuclear envelope. DNA synthesis was assessed by releasing extract into interphase by the addition of 0.3 mM CaCl 2 and measuring the total amount of [ ⁇ - 32 P]dATP incorporation after 3 h by TCA co-precipitation.
- One of the parameters used, as the basis for colorimetric assays, is the metabolic activity of viable cells.
- a microtiter assay which uses the tetrazolium salt MTT, is now widely used to quantitate cell proliferation and cytotoxicity.
- this assay is used in drug screening programs and in chemosensitivity testing. Because only metabolically active cells cleave tetrazolium salts, these assays detect viable cells exclusively.
- MTT assay yellow soluble tetrazolium salt is reduced to coloured water-insoluble formazan salt. After it is solubilized, the formazan formed can easily and rapidly be quantified in a conventional ELISA plate reader at 570 nm (maximum absorbance). The quantity of reduced formazan corresponds to number of vital cells in the culture.
- Human T-lymphoblastic leukemia cell line CEM promyelocytic HL-60 and monocytic U937 leukemias; breast carcinoma cell lines MCF-7, BT549, MDA-MB-231; glioblastoma U87MG cells; cervical carcinoma cells HELA; sarcoma cells U20S and Saos2; hepatocellular carcinoma HepG2; mouse fibroblasts NIH3T3; mouse immortalized bone marrow macrophages B2.4 and B10A.4; P388D1 and L1210 leukemia; B16 and B16F10 melanomas were used for routine screening of compounds.
- the cells were maintained in Nunc/Corning 80 cm2 plastic tissue culture flasks and cultured in cell culture medium (DMEM with 5 g/l glucose, 2 mM glutamine, 100 U/ml penicillin, 100 ⁇ g/ml streptomycin, 10% fetal calf serum and sodium bicarbonate).
- DMEM cell culture medium
- TCS tumour cell survival
- Novel Compounds Induce Apoptosis in Tumour Cells.
- apoptosis produces little or no inflammation, since the neighbouring cells, especially macrophages, rather than being released into the extracellular fluid, engulf shrunken portions of the cell. In contract, in necrosis, cellular contents are released into the extracellular fluid, and thus have an irritant affect on the nearby cells, causing inflammation.
- apoptotic cells exhibit shrinkage and blebbing of the cytoplasm, preservation of structure of cellular organelles including the mitochondria, condensation and margination of chromatin, fragmentation of nuclei, and formation of apoptotic bodies, thought not all of these are seen in all cell types.
- HL-60 cell line was cultured in 6-well culture plates with or without 70 ⁇ M concentration of novel derivatives at 37° C. and 5% CO 2 for 3-24 hours. Following the incubation, cells were pelleted, washed in Hank's buffered salt solution and processed as described below.
- lymphocytes One of the most important parameters of specific cellular immunity is proliferative response of lymphocytes to antigens or polyclonal mitogens.
- Majority of normal mammalian peripheral lymphocytes is resting cells.
- Antigens or nonspecific—polyclonal mitogens have capacity to activate lymphoid cells and it is accompanied with dramatic changes of intracellular metabolism (mitochondrial activity, protein synthesis, nucleic acids synthesis, formation of blastic cells and cellular proliferation).
- Compounds with ability to selectively inhibit lymphocyte proliferation are potent immunosuppressants. Variety of in vitro assays were developed to measure proliferative response of lymphocytes. The most commonly used is 3 H-thymidine incorporation method.
- the cells were diluted at target density of 1.100.000 cells/ml were added by pipet (180 ⁇ l) into 96/well microtiter plates. Four-fold dilutions of the intended test concentration were added at time zero in 20 ⁇ l aliquots to the microtiter plate wells. Usually, test compound was evaluated at six 4-fold dilutions. In routine testing, the highest well concentration was 266.7 ⁇ M. All drug concentrations were examined in duplicates. All wells with exception of unstimulated controls were activated with 50 ⁇ l of concanavalin A (25 ⁇ g/ml). Incubations of cells with test compounds lasted for 72 hours at 37° C., in 5% CO2 atmosphere and 100% humidity. At the end of incubation period, the cells were assayed by using the [ 3 H]-TdR:
- ED immunosuppressant
- CCID 50 is a virus quantity which causes cytopathicity effect in 50% of the cells under the experimental conditions
- the infected cell cultures were incubated at 37° C. for 5 days in a humidified CO 2 incubator.
- the cytopathicity of the virus was examined by determination of MT-4 cell viability by trypan blue dye staining. The results are summarised in Table 8 with comparison on the prototype compounds.
- Table 8 also shows the results of activity testing of novel compounds against MSV-induced transformation in murine embryo fibroblast C3H/3T3 cells.
- the cells were seeded in 1 ml wells of 48-well plates and exposed to 80 PFU (plague forming units) for 60-90 min. The virus was subsequently removed and culture medium containing appropriate concentrations of the tested compounds was added (1 ml per well). At day 6-post infection, MSV-induced transformation of the cell culture was examined microscopically.
- Preparation process The powdered substances mentioned are pressed through a sieve of mesh width 0.6 mm. Portions of 0.33 g of the mixture are transferred to gelatine capsules with the aid of a capsule-filling machine.
- Preparation process The powdered active ingredient is suspended in Lauroglycol® (propylene glycol laurate, Gattefosse S. A., Saint Priest, France) and ground in a wet-pulveriser to a particle size of about 1 to 3 ⁇ m. Portions of in each case 0.419 g of the mixture are then transferred to soft gelatine capsules by means of a capsule-filling machine.
- Lauroglycol® propylene glycol laurate, Gattefosse S. A., Saint Priest, France
- Preparation process The powdered active ingredient is suspended in PEG 400 (polyethylene glycol of Mr between 380 and about 420, Sigma, Fluka, Aldrich, USA) and Tween® 80 (polyoxyethylene sorbitan monolaurate, Atlas Chem. Inc., USA, supplied by Sigma, Fluka, Aldrich) and ground in a wet-pulveriser to a particle size of about 1 to 3 mm. Portions of in each case 0.43 g. of the mixture are then transferred to soft gelatine capsules by means of a capsule-filling machine.
- PEG 400 polyethylene glycol of Mr between 380 and about 420, Sigma, Fluka, Aldrich, USA
- Tween® 80 polyoxyethylene sorbitan monolaurate, Atlas Chem. Inc., USA, supplied by Sigma, Fluka, Aldrich
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Transplantation (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Various substituted nitrogen heterocyclic derivatives and their pharmaceutically acceptable salt derivatives are provided for use as medicaments, and particularly, as antimitotic, anti-viral, anti-cancer, anti-degenerative, immunosuppressive, and anti-microbial drugs or, vaccines. These heterocyclic derivatives can be used as an active agent in a pharmaceutical, as well as a diagnostic utility. To this end, several families of heterocyclic derivatives are provided including pyrrolopyrimidines, pyrazolopyrimidines, purines, and imidazopyridines. In particular, certain tri-substituted and tetra-substituted purines and pyrazolopyrimidines and their deaza analogues are provided for inhibiting cyclin-dependent kinase (“cdk”) proteins, viruses, and immunostimulation.
Description
- The present application is a divisional application of U.S. application No. 09/889,176 filed on Jul. 12, 2001, from which priority is claimed, the entirety of which is hereby incorporated by reference.
- The present invention relates generally to new purine and pyrazolopyrimidine derivatives and their deaza analogues and to their use in suitable utilities, especially diagnostic and therapeutic methods.
- The present invention relates, in particular, to purine derivatives and their inhibitory effect with respect to cyclin-dependent kinase proteins, abbreviated cdks and also with an inhibitory effect with respect to viruses and immunostimulation. Purine analogues as cdk inhibitors are disclosed for example in WO 97/16452, WO 98/05335 and WO97/20842. The teaching of these patents includes 2,6,9-trisubstituted and less substituted purine derivatives only.
- Tetrasubstituted purines are disclosed in WO 98/01448 in which substituents are short hydrocarbonyl chains, usually represented by hydrogen. Substituents at C6 represents hydrogen or amine optionally substituted by one or two hydrocarbon groups; substituents at C8 are hydroxy, mercapto, acyloxy or oxycarbonyl substituted by aliphatic alkyl only. Nucleotide analogues containing phosphonate groups are disclosed for example in U.S. Pat. Nos. 4,659,825; 4,724,233; 5,124,051; 5,302,585; 5,208,221; 5,352,786; 5,356,886; 5,142,051; in EP publication numbers 269,947; 481,214; 630,381; 369,409; 454,427; 618,214; 398,231; 454,427; 468,119; 481,119; 481,214; 434,450 and in WO 95/07920; WO 94/03467; and WO96/33200. Typical purine base is adenine, 2,6-diaminopurine and guanine. The purine bases may include the deaza analogues thereof, 6,9-substituted and 2,6,9-trisubstituted purines and related analogues are disclosed in WO 96/33200. 2,8,9-, 6,8,9-trisubstituted and 2,6,8,9-tetrasubstituted purines and tri- and tetrasubstituted pyrazolopyrimidines and their deaza analogues have not yet been described.
- It is an object of this invention to provide anticancer, antiviral, neurodepressive and immunosuppressive compounds with improved selectivity and efficiency index, i.e. that are less toxic yet more efficacious than analogues known heretofore.
- The various features of novelty which characterize the invention are pointed out with particularity in the claims annexed to and forming a part of this disclosure. For a better understanding of the invention, its operating advantages and specific objects attained by its uses, reference is made to the accompanying descriptive matter in which a preferred embodiment of the invention is illustrated.
- The invention concerns substituted nitrogen heterocyclic derivatives of the formula I—

- wherein,
- A is a divalent group selected from the ensemble consisting of

- Z is N or CH, provided that at least one Z being N;
- R2 and R6 are independent of one another, represent H, halogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, cycloalkyl alkyl, arylalkyl, heteroalkyl, heteroarylalkyl, heterocycloalkyl alkyl or R6′-X wherein
- X is an —NH—, —N(C 1-C6-alkyl)-, —O— or —S— moiety;
- R6′ is H, alkyl, substituted alkyl, acyl, amido, sulpho, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycle, heteroaryl, substituted heteroaryl, arylalkyl, heterocycloalkyl, substituted heterocycloalkyl, heteroarylalkyl, heteroalkyl, cycloalkyl alkyl and heterocycloalkyl alkyl;
- R8 is halogen, hydroxyl, amino, carboxyl, cyano, nitro, amido, sulpho, sulphamino, carbamino, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, arylalkyl, heteroalkyl, heteroarylalkyl, cycloalkyl alkyl, heterocycloalkyl alkyl or R8′-X wherein
- X is —NH—, —N(alkyl)-, —O— or —S— moiety, and
- R8′ is according to any one of the substituents defined above for R2′ or R6′.
- R9 is alkyl, substituted alkyl, acyl, carboxyl, amido, sulphamino, cycloalkyl, substituted cycloalkyl, cycloalkyl alkyl, heteroalkylcycloalkyl alkyl, heterocycloalkyl, substituted heterocycloalkyl, aryl, substituted aryl, heterocycle, heteroaryl, substituted heteroaryl, arylalkyl, heteroarylalkyl, heteroalkyl or —B-R9′ wherein
- B is —CH 2—, —(CH2)2—, —CH(CH3)CH2—, —CH(CH2F)CH2—, —CH(CH2OH)CH2, or the groups of the following structure,

- wherein the left hand bond is linked to nitrogen of 5-membered ring of compounds of the formula I;
- R4 and R5, that are independent of one another, represent hydrogen, hydroxyl, halogen, amino, acyloxy substituent having 1-5 carbon atoms, alkoxy substituent having 1-5 carbon atoms, alkylmercapto substituent having 1-5 carbon atoms, alkylamino substituent having 1-5 carbon atoms and dialkylamino in which each alkyl substituent has 1-5 carbon atoms;
- R7 and R10, that are independent of one another, represent H or alkyl substituent having 1-10 carbon atoms;
- or R9 is —(CH 2)n-R9′, wherein n=1-2 and the
- R9′ is -X(CH 2)mY wherein
- X is —O—, —S—, —NH— or —N (alkyl)- substituent having 1-6 carbon atoms;
- m=1-2;
- Y is carboxyl, amido, sulpho, sulphamino, hydroxyl, carboxyl, mercapto, carbylmercapto, amino, alkylamino, carbamino —PO(OH) 2, —PO(O-C1-C6-alkyl)2, —PO(NH-C1-C6-alkyl) —PO(O-C1-C6-alkyl) (NH-C1-C6-alkyl), —PO(OH) (O-C1-C6-alkyl);
- —PO(OH) (NH-C 1-C6-alkyl) or —(CH2CHD)-R9′, wherein
- R9′, X, m and Y are as defined above and
- D is alkyl, substituted alkyl, —PO(OH) 2, —PO (OH) (O-C1-C6-alkyl), —PO(OH)(NH-C1-C6-alkyl).
- The above not yet defined generic groups having meanings as introduced in the following legend.
- “Halogen” refers to fluorine, bromine, chlorine, and iodine atoms.
- “Alkyl” refers to
- a branched or unbranched alkyl group having 1-6 carbon atoms,
- a branched or unbranched alkenyl group having 2-6 carbon atoms,
- a branched or unbranched alkinyl group having 2-6 carbon atoms.
- “Substituted alkyl” refers to a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and having substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl. These substituent generic groups having the meanings being identical with the definitions of the corresponding groups as defined in this legend.
- “Carbyloxy” denotes the group —OR a, where Ra is alkyl, substituted alkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl, cycloalkyl, substituted cycloalkyl, heterocycloalkyl or substituted heterocycloalkyl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Carbylmercapto” denotes the group -SR b, where Rb is alkyl, substituted alkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl, cycloalkyl, substituted cycloalkyl, heterocycloalkyl or substituted heterocycloalkyl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Sulpho” denotes the group —SO 3Rc, where Rc is
- hydrogen,
- a branched or unbranched alkyl group having 1-6 carbon atoms,
- a branched or unbranched alkenyl group having 2-6 carbon atoms,
- a branched or unbranched alkinyl group having 2-6 carbon atoms,
- a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl, whereas these generic groups have meanings which are identical with the definitions of the corresponding groups as defined in this legend.
- “Sulphamino” denotes the group —NHSO 3Rd, wherein Rd is
- hydrogen,
- a branched or unbranched alkyl group having 1-6 carbon atoms,
- a branched or unbranched alkenyl group having 2-6 carbon atoms,
- a branched or unbranched alkinyl group having 2-6 carbon atoms,
- a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl, whereas these generic substituents have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Acyl” denotes the group —C(O)R e, where Re is hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl, cycloalkyl, substituted cycloalkyl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Aryloxy” denotes the group —OAr, where Ar is an aryl, substituted aryl, heteroaryl or substituted heteroaryl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Alkylamino” denotes the group —NR fRg, where Rf and Rg, that are independent of one another, represent hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, heteroaryl or substituted heteroaryl, provided that Rf and Rg are not both hydrogens, whereas these generic substituents have meanings which are identical with definitions of the corresponding groups defined herein.
- “Amido” denotes the group - c(O)NRhRi′, where Rh and Ri′ may independently be hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Carboxyl” denotes the group —C(O)OR j, where Rj is hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, heteroaryl or substituted heteroaryl, whereas these generic substituents have meanings which are identical with definitions of the corresponding groups defined herein.
- “Carbamino” denotes the group —NHCOR k, where Rk may be hydrogen, alkyl, substituted alkyl, heterocycle, aryl, substituted aryl, heteroaryl and substituted heteroaryl whereas these generic groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Aryl” refers to an aromatic carbocyclic group having from 6 to 18 carbon atoms and being composed of at least one aromatic or multiple condensed rings in which at least one of which being aromatic.
- “Substituted aryl” refers to an aromatic carbocyclic group having from 6 to 18 carbon atoms and being composed of at least one aromatic ring or of multiple condensed rings at least one of which being aromatic. The ring(s) are optionally substituted with one or more substituents selected from the group consisting of halogen, alkyl, hydroxyl, carbylmercapto, alkylamino, carbyloxy, amino, amido, carboxyl, nitro, mercapto or sulpho, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Heterocycle” refers to a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S.
- “Heteroaryl” refers to a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S with at least one ring of this group being aromatic.
- “Substituted heteroaryl” refers to a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S with at least one ring of this group being aromatic and this group being substituted with one or more substituents selected from the group consisting of halogen, alkyl, carbyloxy, carbylmercapto, alkylamino, amido, carboxyl, hydroxyl, nitro, mercapto or sulpho, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Arylalkyl” denotes the group -R 1—Ar where Ri is
- a branched or unbranched alkyl group having 1-6 carbon atoms,
- a branched or unbranched alkenyl group having 2-6 carbon atoms,
- a branched or unbranched alkinyl group having 2-6 carbon atoms,
- and Ar is an aromatic carbocyclic group having from 6 to 18 carbon atoms and being composed of at least one aromatic ring or of multiple condensed rings at least one of which being aromatic and the group being optionally substituted with one or more substituents selected from the group consisting of halogen, alkyl, hydroxyl, carbylmercapto, alkylamino, carbyloxy, amino, amido, carboxyl, nitro, mercapto or sulpho, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Heteroalkyl” denotes the group -R m-L where Rm is
- a branched or unbranched alkyl group having 1-6 carbon atoms,
- a branched or unbranched alkenyl group having 2-6 carbon atoms,
- a branched or unbranched alkinyl group having 2-6 carbon atoms,
- a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend;
- and L is a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S and the group being unsubstituted or substituted with one or more substituents selected from the group consisting of halogen, alkyl, alkoxy, alkylmercapto, alkylamino, amido, carboxyl, hydroxy, nitro, mercapto or sulpho, whereas these generic substituents have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Heteroarylalkyl” denotes the group -R n-G where Rn is
- a branched or unbranched alkyl group having 1-6 carbon atoms,
- a branched or unbranched alkenyl group having 2-6 carbon atoms,
- a branched or unbranched alkinyl group having 2-6 carbon atoms,
- a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend;
- and G is
- a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S with at least one ring of which being aromatic and the group being optionally substituted with one or more substituents selected from the group consisting of halogen, alkyl, carbyloxy, carbylmercapto, alkylamino, amido, carboxyl, hydroxyl, nitro, mercapto or sulpho, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Cycloalkyl” refers to a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms.
- “Substituted cycloalkyl” refers to a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms and being substituted by one or more substituents selected from the group consisting of halogen, alkyl, substituted alkyl, carbyloxy, carbylmercapto, aryl, nitro, mercapto or sulpho, whereas these generic substituent groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Heterocycloalkyl” refers to a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms which at least one ring carbon atom of its cyclic structure being replaced with a heteroatom selected from the group consisting of N, O, S or P.
- “Substituted heterocycloalkyl” refers to a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms which at least one ring carbon atom of its cyclic structure being replaced with a heteroatom selected from the group consisting of N, O, S or P and the group is containing one or more substituents selected from the group consisting of halogen, alkyl, substituted alkyl, carbyloxy, carbylmercapto, aryl, nitro, mercapto or sulpho, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Cycloalkyl alkyl” denotes the group -R o-J where Ro is
- a branched or unbranched alkyl group having 1-6 carbon atoms,
- a branched or unbranched alkenyl group having 2-6 carbon atoms,
- a branched or unbranched alkinyl group having 2-6 carbon atoms,
- a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl, whereas these generic substituent group have meanings which are identical with definitions of the corresponding groups as defined in this legend;
- and J is
- a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms;
- a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms which contains one or more substituents selected from the group consisting of halogen, alkyl, substituted alkyl, carbyloxy, carbylmercapto, aryl, nitro, mercapto or sulpho, whereas these generic substituent groups have meanings which are identical with definitions of the corresponding groups as defined in this legend.
- “Heterocycloalkylalkyl” denotes the group -R pV where Rp is
- a branched or unbranched alkyl group having 1-6 carbon atoms,
- a branched or unbranched alkenyl group having 2-6 carbon atoms,
- a branched or unbranched alkinyl group having 2-6 carbon atoms,
- a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl, whereas these generic substituent groups have meanings which are identical with definitions of the corresponding groups as defined in this legend;
- and V is
- a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms with at least one being replaced with a heteroatom selected from the group consisting of N, O, S or P;
- a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms with at least one being replaced with a heteroatom selected from the group consisting of N, O, S or P and the group contains one or more substituents selected from the group consisting of halogen, alkyl, substituted alkyl, carbyloxy, carbylmercapto, aryl, nitro, mercapto or sulpho, whereas these generic substituent groups have meanings which are identical with definitions of the corresponding groups as defined in this legend, and the pharmaceutically acceptable acid salts, racemates and optical isomers thereof.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula I, wherein R6═H and R2, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula I, wherein R2═H and R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ia,

- wherein R2, R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ia, wherein R6═H and R2, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ia, wherein R2═H and R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ib,

- wherein R2, R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ib,
- wherein R6═H and R2, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ib, wherein R2═H and R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ic,
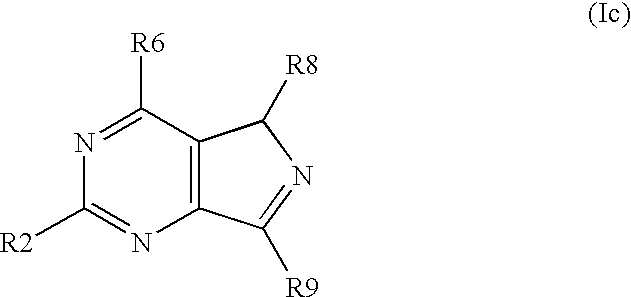
- wherein R2, R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ic, wherein R6═H and R2, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ic, wherein R2═H and R6, R8 and R9 have above mentioned meanings.
- The inventions concerns further substituted nitrogen heterocyclic derivatives of the formula Id
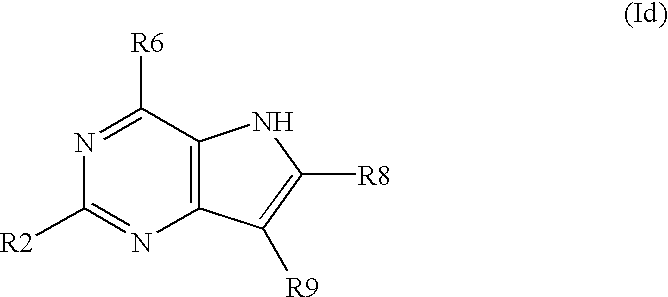
- wherein R2, R6, R8 and R9 have above mentioned meanings.
- The inventions concerns further substituted nitrogen heterocyclic derivatives of the formula Id, wherein R6═H and R2, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Id, wherein R2═H and R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ie,
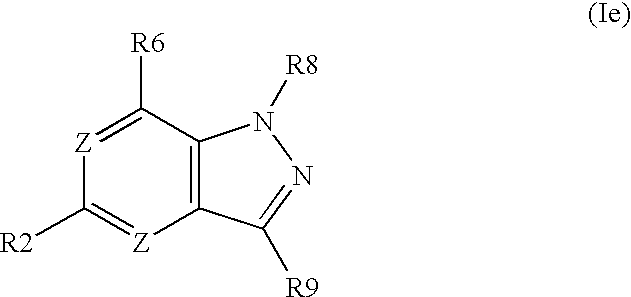
- wherein R2, R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ie, wherein R6═H and R2, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ie, wherein R2═H and R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula If,

- wherein R2, R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula If, wherein R6═H and R2, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula If, wherein R2═H and R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ig,
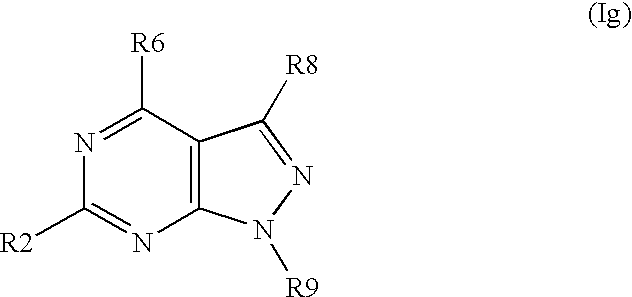
- wherein R2, R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ig, wherein R6═H and R2, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula Ig, wherein R2═H and R6, R8 and R9 have above mentioned meanings.
- The invention concerns further substituted nitrogen heterocyclic derivatives of the formula I selected from the group consisting of 2-(1-hydroxymethylpropylamino)-6-benzylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-isopropylpurine, 2-(2-aminopropylamino)-6-benzylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-isopropylpurine, 2-(2-hydroxypropylamino)-6-benzylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-isopropylpurine, 2-diethylamino-6-(4-methoxybenzylamino)-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-isopropylpurine, 2-(2-hydroxypropylamino)-6-(3-chloroanilino)-8-chloro (or hydroxy, bromo, amino, C 1-C6 alkyl, methyl, ethyl, propyl, isopropyl, vinyl, allyl, propargyl)-9-isopropylpurine, 2-(2-hydroxypropylamino)-6-(3-chloro-4-carboxyanilino)-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-isopropylpurine, 2-(R)-(2-hydroxypyrrolidin-1-yl) -6-benzylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-isopropylpurine, 2-(R)-(1-isopropyl-2-hydroxyethylamino)-6(3-chloro-4-carboxyanilino)-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-isopropylpurine, 2-(R)-(1-isopropyl-2-hydroxyethylamino)-6-benzylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-isopropylpurine, 2-(R)-(1-isopropyl-2-hydroxyethylamino)-6-(3-chloroanilino)-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-isopropylpurine, 2-alkylamino-6-dimethylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-(R)-(2-phosphonomethoxypropyl) purine, 2-alkylamino-6-diethylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9(R)-(2-phosphonomethoxypropyl)purine, 2-alkylamino-6-butylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-(R)-(2-phosphonomethoxypropyl)purine, 2-alkylamino-6- (2-butylamino)-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-(R)-(2-phosphonomethoxypropyl)purine, 2-alkylamino-6-cyclopropylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-(R)-(2-phosphonomethoxypropyl)purine, 2-amino-6-cyclohexylamino-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy propinyl)-9-(R)-(2-phosphonomethoxypropyl)purine, 2-alkylamino-6-(pyrrolidin-1-yl)-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-(R)-(2-phosphonomethoxypropyl) purine, 2-alkylamino-6-(morpholin-1-yl)-8-chloro (or hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, propinyl)-9-(R)-(2-phosphonomethoxypropyl) purine.
- The invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6, R8 and R9 have above mentioned meanings, A is a group of formula

- and Z is N, characterized in that a trisubstituted derivative of formula XI,

- wherein R2, R6 and R9 have above mentioned meanings, is brominated with using a brominating system selected from a group consisting of bromine/acetic acid, bromine/chloroform, bromine/acetate buffer, bromine/water, N-bromosuccinimide/dimethylformamide and bromoacetamide/dimethylformamide to obtain a derivative of formula XIa,
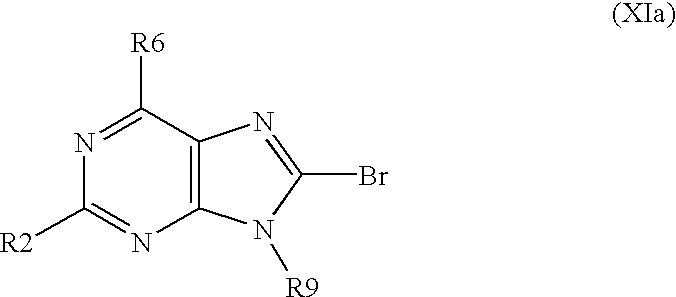
- wherein R2, R6 and R9 have above mentioned meanings, and the bromine atom in position 8 of the derivative of formula XIa is then optionally subjected to a substitution in order to replace it by another substituent R8, which has above mentioned meanings.
- The invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6, R8 and R9 have above mentioned meanings, A is a group of formula
- and Z is N, characterized in that a trisubstituted derivative of formula XIb,
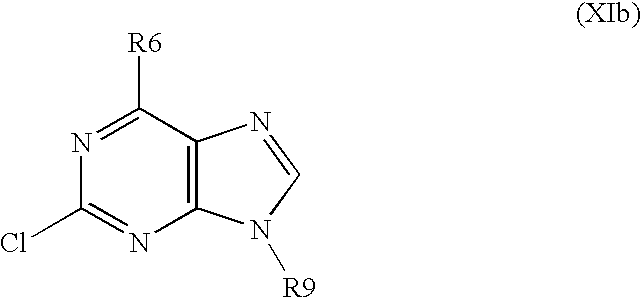
- wherein R6 and R9 have above mentioned meanings, is brominated with using a brominating system selected from a group consisting of bromine/acetic acid, bromine/chloroform, bromine/acetate buffer, bromine/water, N-bromosuccinimide/dimethylformamide and bromoacetamide/dimethylformamide to obtain a derivative of formula XIc,

- wherein R6 and R9 have above mentioned meanings, and the chlorine atom in position 2 and the bromine atom in position 8 of the derivative of formula XIc are optionally, either progressively or simultaneously, subjected to a nucleophilic substitution in order to replace them by other substituents R2 and R8, that have above mentioned meanings.
- The invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6 and R8 have above mentioned meanings, R9 is alkyl as defined above, A is a group of formula

- and Z is N, characterized in that, a trisubstituted derivative of formula XIIa,

- wherein R6 is as defined above, is alkylated in position 9 with using an appropriate alkylating agent in a system selected from a group consisting of K 2CO3/dimethylformamide, Cs2CO3/dimethylformamide, t-BuOK/dimethylformamide, t-BuOK/dimethylsulphoxide and NaH/dimethylformamide or under conditions of Mitsunobu reaction to obtain a derivative of formula XIIb

- wherein R6 and alkyl are as defined above, and the chlorine atoms in positions 2 and 8 of the derivative of formula XIIb are optionally, either progressively or simultaneously, subjected to a substitution in order to replace them by another substituents R2 and R8, that have above mentioned meanings.
- The invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives, wherein R2, R6 and R8 have above mentioned meanings, R9 is alkyl as defined above, A is a group of formula

- and Z is N, characterized in that a trisubstituted derivative of formula XIIa,

- wherein R6 has above mentioned meanings, is protected by reacting, for example, with 2-dihydropyrane/H + to obtain a derivative of formula XIIc,

- wherein R6 has above mentioned meanings, and a group R6 and/or the chlorine atom in position 2 and/or the chlorine atom in position 8 are optionally converted to another substituents R2, R6 and R8, that have above mentioned meanings and in such optionally modificated product of formula XIIc, the 2-tetrahydropyranyl group is split off to obtain a corresponding derivative of formula XIIc wherein R9 is hydrogen, and so obtained product is then alkylated in position 9 with using an appropriate alkylating agent in a system selected from a group consisting of K 2CO3/dimethylformamide, Cs2CO3/dimethylformamide, t-BuOK/dimethylformamide, t-BuOK/dimethylsulphoxide and NaH/dimethylformamide or under conditions of Mitsunobu reaction to obtain a tetrasubstituted derivative of formula XIIc, wherein R2,R6,R8 and R9 have above mentioned meanings.
- The invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6 and R8 have above mentioned meanings, R9 is alkyl as defined above, A is a group

- and Z is N, characterized in that, 2,6,8-trichloropurine is subjected to a nucleophilic substitution in position 6 in order to replace the chlorine atom in position 6 by another substituent R6 as defined above to obtain a derivative of formula XIIa,

- wherein R6 has above mentioned meanings, and so obtained product is then alkylated in position 9 with using an appropriate alkylating agent in a system selected from a group consisting of K 2CO3/dimethylformamide, Cs2CO3/dimethylformamide, t-BuOK/dimethylformamide, t-BuOK/dimethylsulphoxide and NaH/dimethylformamide or under conditions of Mitsunobu reaction to obtain a derivative of formula XIIb,

- wherein R6 and alkyl have above mentioned meanings, and the chlorine atoms in positions 2 and 8 of the derivative of formula XIIb are optionally, either progressively or simultaneously, subjected to a nucleophilic substitution in order to replace them by another substituents R2 and R8 that have above mentioned meanings.
- The invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2 is hydrogen, R6 and R8 have above mentioned meanings, R9 is alkyl as defined above, A is a group of formula

- and Z is N, characterized in that 6,8-dichloropurine is subjected to a substitution in position 6 in order to replace the chlorine atom in position 6 by another substituent R6 which has above mentioned meanings to obtain a derivative of formula XVI,

- wherein R6 has above mentioned meanings, and so obtained derivative is alkylated in position 9 with using an appropriate alkylating agent in a system selected from a group consisting of K 2CO3/dimethylformamide, Cs2CO3/dimethylformamide, t-BuOK/dimethylformamide, t-BuOK/dimethylsulphoxide and NaH/dimethylformamide or under conditions of Mitsunobu reaction to obtain a derivative of formula XVIa,

- wherein R6 and alkyl have above mentioned meanings, and the chlorine atom in position 8 of the derivative of formula XVIa is optionally subjected to a substitution in order to replace it by another substituent R8 which has above mentioned meanings.
- The invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6 and R8 have above mentioned meanings, R9 is alkyl as defined above, A is a group of formula

- and Z is N, characterized in that 2,6-dichloropurine is subjected to a nucleophilic substitution in position 6 in order to replace the chlorine atom in position 6 by another substituent R6 as defined above, and so obtained derivative of formula XVII,

- wherein R6 is as defined above, is alkylated in position 9 with using either methyl acrylate or acrylonitrile or oxirane to obtain a derivative of formula XVIIa
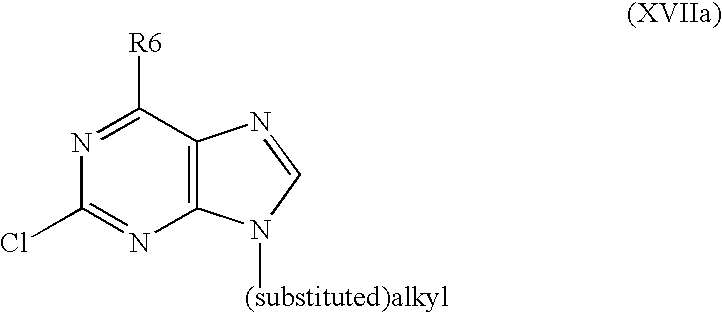
- wherein R6 and alkyl have above mentioned meanings, which product is then brominated in position 8 with using a brominating system selected from a group consisting of bromine/acetic acid, bromine/chloroform, bromine/acetate buffer, bromine/water, N-bromosuccinimide/dimethylformamide and bromoacetamide/dimethylformamide to obtain a derivative of formula XVIIb,

- wherein R6 and alkyl have above mentioned meanings, and so obtained product is then optionally, either progressively or simultaneously, subjected to a nucleophilic substitution in positions 2 and 8 in order to replace the chlorine atom in position 2 and the bromine atom in position 8 by another substituents R2 and R8 that have above mentioned meanings.
- The invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R6 is a halogen or hydrogen, R2, R8 and R9 have above mentioned meanings, A is a group of formula

- and Z is N, characterized in that a derivative of formula XVIII,

- wherein R2, R8 and R9 have above mentioned meanings, is halogenated via diazotation with using, for example, amylnitrite/CH 2Br2 or amylnitrite/CHI3, and so obtained derivative wherein R6 is halogen is optionally hydrogenolyzed with using H2/Pd catalyst to obtain a corresponding derivative wherein R6 is hydrogen.
- The invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R9 has above mentioned meanings and one substituent of R2, R6 or R8 is alkinyl or alkenyl containing 2 to 6 carbon atoms whereas two other substituents of R2, R6 and R8 have above mentioned meanings, A is a group of formula

- and Z is N or CH, characterized in that a derivative of formula I, wherein R9 has above mentioned meanings and one substituent of R2, R6 and R8 is a halogen whereas two other substituents of R2, R6 and R8 have above mentioned meanings, is alkinylated in the position of the halogen with using alkine/triphenylphosphinePdCl 2/CuI/triethylamine system and so obtained 2-, 6- or 8-alkinyl derivative is optionally converted to 2-, 6- or 8-alkenyl derivative with using a Lindlar catalyst.
- The invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6, R8 and R9 have above mentioned meanings, a is a group of formula

- and Z is N, characterized in that 2,5-dialkyl-3-alkoxycarbonyl-4-aminopyrazole, wherein alkyls and alkoxy have above mentioned meanings, is reacted with formamidine acetate/triethylamine system to obtain 1,3-dialkyl-7-hydroxypyrazolo[4,3-d]pyrimidine and so obtained 7-hydroxy derivative is then optionally transferred to a corresponding 7-chloro derivative by reaction with thionylchloride, which product is optionally substituted to obtain a corresponding R6-derivative, wherein R6 has above mentioned meanings.
- The invention also relates to a method for preparing substituted nitrogen heterocyclic derivatives of formula I, wherein R2, R6, R8 and R9 have above mentioned meanings, A is a group of formula

- and Z is N, characterized in that 3,7-dialkylpyrazolo [4,3-d]pyrimidine, wherein alkyls have above mentioned meanings, is alkylated with using an appropriate alkylating agent in a system selected from a group consisting of K 2CO3/dimethylformamide and Cs2CO3/dimethylformamide to obtain a mixture of trialkylated and tetraalkylated products and the trialkylated product is separated by means of the column chromatography.
- The invention also relates to substituted nitrogen heterocyclic derivatives of formula I and pharmaceutically acceptable salts thereof for use as medicaments.
- The invention also relates to substituted nitrogen heterocyclic derivatives of formula I for use as means for preparing affinity adsorption matrices, immobilized enzymes for process control, immunoassay reagents, diagnostic samples, 14C-, 3H-, avidin- or biotin-labelled compounds, oligonucleotides and diagnostic samples.
- The invention also relates to substituted nitrogen heterocyclic derivatives of formula I for use as antimitotic drugs, in particular drugs for elimination or reduction of viral spread or growth in tissue culture systems during the production of biopharmaceutical or other products such as proteins and vaccines, drugs for elimination or reduction of viral spread and growth in clinical samples such as blood, and drugs for stopping of growth of tissue culture cells while leaving the cells to carry on with protein production.
- The invention also relates to a pharmaceutical composition with cytostatic, anticancer, antimitotic, antineurodegenerative, immunosuppressive and antimicrobial activity, characterised in that it comprises, in addition to auxiliary pharmaceutical matters, at least one substituted nitrogen heterocyclic derivative of formula I.
- The invention also relates to a combined pharmaceutical composition with cytostatic effect, characterised in that it comprises, in addition to auxiliary pharmaceutical matters, a combination of at least one substituted nitrogen heterocyclic derivative of formula I and a cytostatic agent selected from a group consisting of mitoxantron, cis- and carbo-platin, methotrexate, taxol and doxorubicin.
- The invention also relates to a use of substituted nitrogen heterocyclic derivatives of formula I for preparing medicaments destined for treating tumours, cancers, psoriasis, rheumatoid arthritis, lupus, type I diabetes, multiple sclerosis, restenosis, polycyclic kidney disease, host graft disease and gout, parasitoses, such as those caused by fungi or protists, or Alzheimer's disease, or for preparing antineurodegenerative drugs and suppress immunostimulation agents.
- The novel compounds of this invention have a wide variety of diagnostic, therapeutic and industrial utilities.
- The compounds of this invention are suitable as intermediates for use in the preparation of affinity absorption matrices. For example, the phosphonate groups in matrix bound form are useful in the chromatographic separation of positively charged molecules. Other immobilised examples of the compounds herein are useful in purifying proteins, e.g., cell cycle enzymes (cdk's), enzymes involved in recognition of the compound of this invention, e.g. transport proteins. Suitable methods of incorporation of the compounds of this invention into polymeric resins will be readily apparent to the skilled artisan, for instance the compounds are incorporated by cross-linking hydroxyl groups of the phosphonate or hydroxymethyl substituents using cross-linking agents heretofore known. Linking through a group other than the heterocyclic base will produce a resin useful in hydrophobic affinity chromatography. Other suitable linking methods are described in Cihlar (supra).
- The compounds of the formula I and their pharmaceutically acceptable salts inhibit selectively the enzyme p34 cdc2/cyclin B kinase and related cdks (cdk2, cdk5, cdk7, erk1, erk2).
- In another embodiment, this invention is a method for inhibiting cdks and cell proliferation in mammals comprising administering a therapeutically effective amount of the composition of claim 1 to the mammal. The cdk inhibiting molecules are useful for treating cell proliferation disorders such as rheumatoid arthritis, lupus, type I diabetes, multiple sclerosis, cancer, restenosis, Alzheimer's disease, growth of parasites (animal, protists), host graft disease, and gout.
- In another embodiment, this invention is a composition useful for treating fungal infections (fungi) in humans, animal, and in plants.
- 2,6,8,9-tetrasubstituted and 6,8,9- or 2,8,9-trisubstituted adenine derivatives exhibit extremely high potency against DNA viruses on the part of the defined compounds. Such compounds otherwise have been considered to have little or no activity against DNA viruses. Moreover, surprisingly the chirally enriched or pure (S)-enantiomer is antivirally active. Heretofore, only the (R)-enantiomer was notably antivirally active, and then only against retroviruses.
- In addition to other cdc2-related kinases, this kinase controls certain steps of cell division cycles, in particular the transition from GI phase into the S phase and in particular the transition from the G2 phase into the M phase. Out the basis of this findings, it can be expected that the compounds of the formula I, II and their pharmaceutically acceptable salts can be used as antimitotic compounds or for treatment of hypoproliferative diseases, such as cancer, restenosis and Alzheimer's disease. Thus in very low concentration (micromolar and lower), they are capable of inhibiting cell cycle transitions (G1/S, G2/M, M-phase/metaphase) carried out on the different animal bodies and embryos. Furthermore, the compounds are useful in treating autoimune diseases, e.g. rheumatoidal arthritis, lupus, type I diabetes, multiple sclerosis, etc., in treating of cancer, cardiovascular disease such as restenosis, host vs graft disease, gout, polycystic kidney disease and other proliferative diseases whose pathogenesis involves abnormal cell proliferation.
- This invention also concerns novel compounds that have been discovered to be potent and specific inhibitors of IκB-α kinase which prevents signal induced NF-κB activation and cytokine synthesis in vitro and in vivo. Such inhibitors are expected to inhibit synthesis of cytokines and adhesion proteins whose synthesis is transcriptionally regulated by NF-κB. Pro-inflammatory cytokines such as IL-1, IL-6, TNF and adhesion proteins (e.g. ICAM, VCAM and selections) belong to this class of molecules and have implicated in the pathogenesis of inflammatory diseases. Thus a potent inhibitor of IκB-α kinase is useful in the clinical management of diseases where the NF-κB activation is required for disease induction.
- It also relates to novel compounds activating p53, the mammal cell's own natural brake gene for stopping uncontrolled cell proliferation (cancer), thus being able to switch off the cancer. p53 as well as retinoblastoma (Rb) are two well-characterised tumour suppressors whose inactivation may led to uncontrolled cell proliferation and malignancy. Phosphorylation of these two proteins, which are involved in the cell cycle regulatory mechanisms, is known to modulate their function. Thus a potent cdk inhibitor represent a good toll for treatment of cancers due to induction of wild type p53 protein in cancers expressing mutant p53.
- In addition, studies carried out on the derivatives of the invention have demonstrated strong effect on apoptosis of many cancer cell lines. It has been seen that apoptosis can be induced at stage G1 or G2 and following damage of the DNA, some cells stop at stage G1 and p53-dependent apoptotic pathway is then induced. In other situations, it seems that cells stop at G2/M stage in response to damage caused to the DNA, and activation of an independent p53 apoptotic path is observed. This path has proved to be particularly significant in the therapy of tumours in which a less of active p53 is observed. The interest is therefore assessed by application of the derivatives of the invention for stimulating on of p53-independent apoptosis in the cells, which have stopped at stage G2 through damage to the DNA using agents such as mitoxantrone or cis-platinum. The cdk inhibitors of this invention can thus increase the therapeutic potential of the anti-tumour agents currently used.
- The compounds of this invention will generally be terminally incorporated into the oligonucleotide. If they do not contain phosphonyl group attached to the hydroxyl group, they optionally are incorporated internally into the sequence of the oligonucleotide. Terminally incorporated diphosphonyl compounds of this invention which contain no free hydroxyl capable of participating in chain elongation also are useful in DNA sequencing in essentially the same manner as deoxyNTPs have been used in the past (see example 8 of U.S. Pat. No. 5,276,143). The nucleotide analogues of the invention (when diphosphorylated) are useful as chain terminators for dideoxynucleotide-type DNA sequencing protocols, provided that the nucleotide analogue lacks a free hydroxyl group suitable for polymerase mediated chain elongation. These compounds will not have R=hydroxymethyl and do not possess a cyclic structure incorporating the phosphorus atom (although compounds having such excluded structures can be intermediates). The nucleotide analogue is included in a kit with other reagents (such as Klenow polymerase or T4 polymerase, dNTPs, etc) needed for DNA sequencing (Otvos, et al. “Nucl. Acids. Res.” 15:1763-1777 (1987).
- If the oligonucleotide-incorporated compound of this invention is bindingcompetent for its complementary sequence, i.e., if it is capable of base pairing, then this nucleotide monomer will participate in hybridisation. It is not necessary, however, that the incorporated nucleotide analogue of this invention base pair or otherwise participate in hybridisation. If it is located at the terminus of the oligonucleotide it will be useful as an immunological recognition site, or haptenic recognition site, to facilitate detection of the oligonucleotide by an antibody capable of binding the compound of this invention.
- The compounds of this invention also are useful as linkers or spacers in preparation affinity absorption matrices (as opposed to functioning as affinity moieties per se as noted above), immobilised enzymes for process control, or immunoassay reagents. The compounds herein contain a multiplicity of functional groups that are suitable as sites for cross-linking desired substances. For example, it is conventional to link affinity reagents such as hormones, peptides, antibodies, drugs, and the like to insoluble substrates. These insolubilised reagents are employed in known fashion to absorb binding partners for the affinity reagents from manufactured preparations, diagnostic samples and other impure mixture. Similarly, immobilised enzymes are used to perform catalytic conversions with facile recovery of enzyme. Bifunctional compounds are commonly used to link analytes to detectable groups in preparing diagnostic reagents.
- Many functional groups present in the compounds of this invention are suitable for use in cross-linking. For example, the phosphonic acid is used to form esters with alcohols or amides with amines. The R groups substituted with OH, azido (which is reduced to amino if desired before cross-linking) or vinyl are exemplary suitable sites. Similarly, the amino, halo, acyl and other reactive sites found on group B are suitable. Suitable protection of reactive groups will be used where necessary while assembling the cross-linked reagent. In general, the compounds here are used by linking them through phosphonic acid or amino group to the hydroxyl or amino groups of the linking partner in the same fashion as shown herein, and covalently bonded to the other binding partner through an R group. For example a first binding partner such as a steroid hormone is esterified and then this conjugate is cross-linked through hydroxymethyl R to cyanogen bromide activated Sepharose, whereby immobilised steroid is obtained. Other chemistries for conjugation are well known. See for example Maggio, “EnzymeImmunoassay” (CRC, 1988, pp 71-135) and references cited therein.
- The oligonucleotides of this invention are labeled with any conventional detectable label, e.g. a fluorescent moiety such a fluorescein, radioisotopes such as 14C or 3H, stable free radicals, avidin, biotin and the like all of which previously have been used as labels for immunoassays or diagnostic probes. The label will be present on the oligonucleotide or on the residue of an analogue of this invention. Suitable labelling methods are well known and are readily used with reactive groups such as hydroxyl, allyl and the like. A simple method is to label the compound of this invention with H3 by proton exchange. The compounds also are biotinylated using conventional methods. See for instance U.S. Pat. No. 5,276,143 for analogous structures. However, the compounds of this invention also are useful directly in diagnostic probe assays without an exogenous detectable label. In one embodiment of this alternative, antibodies are raised against the compounds of this invention. Such antibodies (which in turn are labelled or used in a double antibody configuration) bind to the analogue of this invention and thereby are useful in detecting its presence as label for a protein or oligonucleotide.
- The compounds of the invention are useful for treatment of microbial infections, for treatment of tumours or for other indications described below. Microbial infections treatable by the compounds of this invention include viruses, parasites, yeast and fungi, but it is believed that the compounds are most effective against viruses, which constitutes the preferred utility. Exemplary viral infections include infections caused by DNA or RNA viruses including herpes viruses (CMV, HSV 1, HSV 2, EBV, varicella zoster virus (VZV), bovid herpesvirus type 1, equid herpesvirus type 1, HVV-6, papillomaviruses (HPV types 1-55 including carcinogenic HPV), flaviviruses (including yellow fever virus, African swine fever virus and Japanese encephalitis virus), togaviruses (including Venezuelan equine encephalomyelitis virus), influenza viruses (types A-C), retroviruses (HIV-1, HIV-2, HTLV-I, HTLV-II, SIV, FeLV, FIV, MoMSV), adenoviruses (types 1-8), poxviruses (vaccinia virus), enteroviruses (poliovirus types 1-3, Coxsackie, hepatitis A virus, and ECHO virus), gastroenteritis viruses (Norwalk viruses, rotaviruses), hantaviruses (Hantaan virus), polyomavirus, papovaviruses, rhinoviruses, parainfluenza virus types 1-4, rabies virus, respiratory synctial virus (RSV), hepatitis viruses A, B, C and E, and the like.
- The antiviral activity of individual compounds is determined by routine assay of antiviral (or other antimicrobial) activity using enzyme inhibition assays, tissue culture assays, animal model assays and the like as will be understood by those skilled in the art.
- Protozoan parasite infections are treated using the compounds of the invention. The term protozoa include those members of the subphyla Sarcomastigophora and Sporozoa of the phylum Protozoa. More particularly, the term protozoa as used herein include genera of parasitic protozoa, which are important to man, because they either cause disease in man or in his domestic animals. These genera for the most part are classified in the superclass Mastigophora of the subphylum Sarcomastigophora and the class Telesporea of the subphylum Sporozoa in the classification according to Baker (1969). Illustrative genera of these parasitic protozoa include Histomonas, Pneumocystis, Trypanosoma, Giardia, Trichomonas, Eimeria, Isopora, Leishmania, Entamoeba, Toxoplasma and Plasmodium. Parasitic protozoans include Plasmodium falciparum, Plasmodium berghei, Plasmodium malariae, Plasmodium vivax, Leishmania braziliensis, Leishmania donovani, Trypanosoma cruzi, Trypanosoma brucei, Trypanosoma rhodesiense, Pneumocystis carinii, Entamoeba histolytica, Trichomonas vaginalis and the like (de Vries, E., et al, “Mol. Biochem. Parasitol” 47:43-50 (1991) and trypanosomes (Kaminsky et al. “J.Parasitol.” 80(6): 1026-30 (1994). The compounds in which R is CH2OH and B is 3-deazaadenine are particularly interesting in the treatment of malarial parasites.
- Compounds of the invention are used to treat yeast or fungal infections caused by Candida glabrata, Candida ropicalis, Candida albicans, and other Candida species, Cryptococcus species including Cryptococcus neoformans, Blastomyces species including Blastomyces dermatidis, Torulopsis species including Torulopsis glabrata, Coccidioides species including Coccidioides immitis, Aspergillus species and the like.
- The compounds of the invention can also be (1) applied to tissue culture systems to eliminate or reduce viral spread or growth during the production of biopharmaceutical or other products (such as proteins or vaccines), (2) used to eliminate or reduce viral spread or growth in clinical sample (such as blood), and (3) used to stop growth of tissue culture cells while leaving the cells to carry on with protein production.
- The compounds herein have been found to suppress immunostimulation. Accordingly, they can suppress metabolic activities of T-lymphocytes stimulated by diverse agents, e.g. concavalin A, they principally will find application in the treatment of autoimmune diseases, e.g. arthritis, or in suppression of transplant rejection. Their therapeutically active concentrations are in the range of 1 mg/kg to 50 mg/kg of body weight.
- Therapeutic administration
- Suitable routes for administration include oral, rectal, vassal, topical (including ocular, buccal and sublingual), vaginal and parental (including subcutaneous, intramuscular, intravitreous, intravenous, intradermal, intrathecal and epidural). The preferred route of administration will depend upon the condition of the patient, the toxicity of the compound and the site of infection, among other considerations known to the clinician.
- The therapeutical composition comprise about 1% to about 95% of the active ingredient, single-dose forms of administration preferably comprising about 20% to about 90% of the active ingredient and administration forms which are not single-dose preferably comprising about 5% to about 20% of the active ingredient. Unit dose forms are, for example, coated tablets, tablets, ampoules, vials, suppositories or capsules. Other forms of administration are, for example, ointments, creams, pastes, foams, tinctures, lipsticks, drops, sprays, dispersions-and the like. Examples are capsules containing from about 0.05 g to about 1.0 g of the active ingredient.
- The pharmaceutical compositions of the present invention are prepared in a manner known per se, for example by means of convential mixing, granulating, coating, dissolving or lyophilising processes.
- Preferably, solutions of the active ingredient, and in addition also suspensions or dispersions, especially isotonic aqueous solutions, dispersions or suspensions, are used, it being possible for these to be prepared before use, for example in the case of lyophilised compositions which comprise the active substance by itself or together with a carrier, for example mannitol. The pharmaceutical compositions can be sterilised and/or comprise excipients, for example preservatives, stabilisers, wetting agents and/or emulsifiers, solubilizing agents, salts for regulating the osmotic pressure and/or buffers, and they are prepared in a manner known per se, for example by means of convential dissolving or lyophilising processes. The solutions or suspensions mentioned can comprise viscosity-increasing substances, such as sodium carboxymethylcellulose, carboxymethylcellulose, dextran, polyvinylpyrrolidone or gelatin.
- Suspensions in oil comprise, as the oily component, the vegetable, synthetic or semisynthetic oils customary for injection purposes. Oils which may be mentioned are, in particular, liquid fatty acid esters which contain, as the acid component, a long-chain fatty acid having 8-22, in particular 12-22, carbon atoms, for example lauric acid, tridecylic acid, myristic acid, pentadecylic acid, palmitic acid, margaric acid, stearic acid, arachidinic acid, behenic acid or corresponding unsaturated acids, for example oleic acid, elaidic acid, euric acid, brasidic acid or linoleic acid, if appropriate with the addition of antioxidants, for example vitamin E, (β-carotene or 3,5-di-tert-butyl-4 hydroxytoluene. The alcohol component of these fatty acid esters has not more than 6 carbon atoms and is mono- or polyhydric, for example mono-, di- or trihydric alcohol, for example methanol, ethanol, propanol, butanol, or pentanol, or isomers thereof, but in particular glycol and glycerol. Fatty acid esters are therefore, for example: ethyl oleate, isopropyl myristate, isopropyl palmitate, “Labrafil M 2375” (polyoxyethylene glycerol trioleate from Gattefosee, Paris), “Labrafil M 1944 CS” (unsaturated polyglycolated glycerides prepared by an alcoholysis of apricot kernel oil and made up of glycerides and polyethylene glycol esters; from Gattefosee, Paris), “Labrasol” (saturated polyglycolated glycerides prepared by an alcoholysis of TCM and made up of glycerides and polyethylene glycol esters; from Gattefosee, Paris) and/or “Miglyol 812” (triglyceride of saturated fatty acids of chain length C 8 to C12 from Hills AG, Germany), and in particular vegetable oils, such as cottonseed oil, almond oil, olive oil, castor oil, sesame oil, soybean oil and, in particular, groundnut oil.
- The preparation of the injection compositions is carried out in the customary manner under sterile conditions, as are bottling, for example in ampoules or vials, and closing of the containers.
- For example, pharmaceutical compositions for oral use can be obtained by combining the active ingredient with one or more solid carriers, if appropriate granulating the resulting mixture, and, if desired, processing the mixture or granules to tablets or coated tablet cores, if appropriate by addition of additional excipients.
- Suitable carriers are, in particular, fillers, such as sugars, for example lactose, sucrose, mannitol or sorbitol, cellulose preparations and/or calcium phosphates, for example tricalcium phosphate, or calcium hydrogen phosphate, and furthermore binders, such as starches, for example maize, wheat, rice or potato starch, methylcellulose, hydroxypropyl-methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidine, and/or, if desired, desintegrators, such as the above mentioned starches, and furthermore carboxymethyl-starch, crosslinked polyvinylpyrrolidone, alginic acid or a salt thereof, such as sodium alginate. Additional excipients are, in particular, flow regulators and lubricants, for example salicylic acid, talc, stearic acid or salts thereof, such as magnesium stearate or calcium stearate, and/or polyethylene glycol, or derivatives thereof.
- Coated tablet cores ca be provided with suitable coatings which, if appropriate, are resistant to gastric juice, the coatings used being, inter alia, concentrated sugar solutions, which, if appropriate, comprise gum arabic, talc, polyvinylpyrrolidine, polyethylene glycol and/or titanium dioxide, coating solutions in suitable organic solvents or solvent mixtures or, for the preparation of coatings which are resistant to gastric juice, solutions of suitable cellulose preparations, such as acetylcellulose phthalate or hydroxypropylmethylcellulose phthalate. Dyes or pigments can be admixed to the tablets or coated tablet coatings, for example for identification or characterisation of different doses of active ingredient.
- Pharmaceutical compositions, which can be used orally, are also hard capsules of gelatin and soft, closed capsules of gelatin and a plasticiser, such as glycerol or sorbitol. The hard capsules can contain the active ingredient in the form of granules, mixed for example with fillers, such as maize starch, binders and/or lubricants, such as talc or magnesium stearate, and stabilisers if appropriate. In soft capsules, the active ingredient is preferably dissolved or suspended in suitable liquid excipients, such as greasy oils, parrafin oil or liquid polyethylene glycols or fatty acid esters of ethylene glycol or propylene glycol, it being likewise possible to add stabilisers and detergents, for example of the polyethylene sorbitan fatty acid ester type.
- Other oral forms of administration are, for example, syrups prepared in the customary manner, which comprise the active ingredient, for example, in suspended form and in a concentration of about 5% to 20%, preferably about 10% or in a similar concentration which results in a suitable individual dose, for example, when 5 or 10 ml are measured out. Other forms are, for example, also pulverulent or liquid concentrates for preparing of shakes, for example in milk. Such concentrates can also be packed in unit dose quantities.
- Pharmaceutical compositions, which can be used rectally, are, for example, suppositories that comprise a combination of the active ingredient with a suppository base. Suitable suppository bases are, for example, naturally occurring or synthetic triglycerides, paraffin hydrocarbons, polyethylene glycols or higher alkanols.
- Compositions which are suitable for parenteral administration are aqueous solutions of an active ingredient in water-soluble form, for example of water-soluble salt, or aqueous injection suspensions, which comprise viscosity-increasing substances, for example sodium carboxymethylcellulose, sorbitol and/or dextran, and if appropriate stabilisers. The active ingredient can also be present here in the form of a lyophilisate, if appropriate together with excipients, and be dissolved before parenteral administration by addition of suitable solvents. Solutions such as are used, for example, for parental administration can also be used as infusion solutions. Preferred preservatives are, for example. Antioxidants, such as ascorbic acid, or microbicides, such as sorbic or benzoic acid.
- Ointments are oil-in-water emulsions, which comprise not more than 70%, but preferably 20-50% of water or aqueous phase. The fatty phase consists, in particular, hydrocarbons, for example vaseline, paraffin oil or hard paraffin's, which preferably comprise suitable hydroxy compounds, such as fatty alcohol's or esters thereof, for example cetyl alcohol or wool wax alcohols, such as wool wax, to improve the waterbinding capacity. Emulsifiers are corresponding lipophilic substances, such as sorbitan fatty acid esters (Spans), for example sorbitan oleate and/or sorbitan isostearate. Additives to the aqueous phase are, for example, humectants, such as polyalcohols, for example glycerol, propylene glycol, sorbitol and/or polyethylene glycol, or preservatives and odoriferous substances.
- Fatty ointments are anhydrous and comprise, as the base, in particular, hydrocarbons, for example paraffin, vaseline or paraffin oil, and furthermore naturally occurring or semisynthetic fats, for example hydrogenated coconut-fatty acid triglycerides, or, preferably, hydrogenated oils, for example hydrogenated groundnut or castor oil, and furthermore fatty acid partial esters of glycerol, for example glycerol mono- and/or distearate, and for example, the fatty alcohols. They also contain emulsifiers and/or additives mentioned in connection with the ointments which increase uptake of water.
- Creams are oil-in-water emulsions, which comprise more than 50% of water. Oily bases used are, in particular, fatty alcohols, for example lauryl, cetyl or stearyl alcohols, fatty acids, for example palmitic or stearic acid, liquid to solid waxes, for example isopropyl myristate, wool wax or beeswax, and/or hydrocarbons, for example vaseline (petrolatum) or paraffin oil. Emulsifiers are surface-active substances with predominantly hydrophilic properties, such as corresponding nonionic emulsifiers, for example fatty acid esters of polyalcohols or ethyleneoxy adducts thereof, such as polyglyceric acid fatty acid esters or polyethylene sorbitan fatty esters (Tweens), and furthermore polyoxyethylene fatty alcohol ethers or polyoxyethylene fatty acid esters, or corresponding ionic emulsifiers, such as alkali metal salts of fatty alcohol sulfates, for example sodium lauryl sulfate, sodium cetyl sulfate or sodium stearyl sulfate, which are usually used in the presence of fatty alcohols, for example cetyl stearyl alcohol or stearyl alcohol. Additives to the aqueous phase are, inter alia, agents which prevent the creams from drying out, for example polyalcohols, such as glycerol, sorbitol, propylene glycol and/or polyethylene glycols, and furthermore preservatives and odoriferous substances.
- Pastes are creams and ointments having secretion-absorbing powder constituents, such as metal oxides, for example titanium oxide or zinc oxide, and furthermore talc and/or aluminium silicates, which have the task of binding the moisture or secretions present.
- Foams are administered from pressurised containers and they are liquid oil-inwater emulsions present in aerosol for. As the propellant gases, halogenated hydrocarbons, such as chlorofluoro-lower alkanes, for example dichlorofluoromethane and dichlorotetrafluoroethane, or, preferably, non-halogenated gaseous hydrocarbons, air, N 2O, or carbon dioxide are used. The oily phases used are, inter alia, those mentioned above for ointments and creams, and the additives mentioned there are likewise used.
- Tinctures and solutions usually comprise an aqueous-ethanolic base to which, humectants for reducing evaporation, such as polyalcohols, for example glycerol, glycols and/or polyethylene glycol, and re-oiling substances, such as fatty acid esters with lower polyethylene glycols, i.e. lipophilic substances soluble in the aqueous mixture to substitute the fatty substances removed from the skin with the ethanol, and, if necessary, other excipients and additives, are admixed.
- The present invention further provides veterinary compositions comprising at least one active ingredient as above defined together with a veterinary carrier therefor. Veterinary carriers are materials for administering the composition and may be solid, liquid or gaseous materials, which are inert or acceptable in the veterinary art and are compatible with the active ingredient. These veterinary compositions may be administered orally, parenterally or by any other desired route.
- The invention also relates to a process or method for treatment of the disease states mentioned above. The compounds can be administered prophylactically or therapeutically as such or in the form of pharmaceutical compositions, preferably in an amount, which is effective against the diseases mentioned. With a warm-blooded animal, for example a human, requiring such treatment, the compounds are used, in particular, in the form of pharmaceutical composition. A daily dose of about 0.1 to about 5 g, preferably 0.5 g to about 2 g, of a compound of the present invention is administered here for a body weight of about 70 kg.
- 1 mmol of 2-(3-hydroxypropylamino)-6-benzylamino-9-isopropylpurine was dissolved in chloroform and 1.1 mmol of bromine was added. The 8-bromo derivative hydrobromide was removed by filtration and crystallized from n-propanol—ether. M.p. 156-161° C. Yield 90%. TLC: chloroform-methanol (96:4), single spot. MS-ESI(+) m/z: 420, 422 [M+H] +
TABLE 1 Compounds Prepared by the Method of Example 1 SUBSTITUENT C2 N6 C8 N9 2-hydroxyethylamino benzylamino bromo methyl 3-hydroxypropylamino benzylamino bromo isopropyl (R)-1-(hydroxymethyl) propylamino benzylamino bromo isopropyl (R)-1-(hydroxymethyl) propylamino 3-iodobenzylamino bromo isopropyl diethanolamino 3-chloroanilino bromo isopropyl (R)-1-(hydroxymethyl) isobutylamino 4-methoxybenzylamino bromo isopropyl (R)-1-(hydroxymethyl) isobutylamino 3-chloroanilino bromo isopropyl - 2-(3-Hydroxypropylamino)-6-benzylamino-8-bromo-9-isopropylpurine (0.15 mmol), prepared as described in Example 1, was dissolved in 2 mL dimethylformamide, 0.9 mmol of CH 3SNa was added and the mixture stirred at 40° C. for 1 hour. The solvent was removed in vacuo and the rest partitioned between water-EtOAc. The organic layer was dried, evaporated and the rest purified by column chromatography (silica gel, chloroform-heptane (8:2)). Yield 75%. M.p. 64-67° C. 1H NMR (300 MHz, CDCl3): 7.28-7.40 m, 5.55 t, 4.85 t, 4.75 d, 4.65 m, 3.62 m, 2.62 s, 1.74 dd, 1.60 d. MS-ESI(+) m/z: 387 [M+H]+
TABLE 2 Compounds Prepared by the Method of Example 2 SUBSTITUENT C2 N6 C8 N9 2-hydroxyethylamino benzylamino methyl methyl 2-hydroxyethylamino benzylamino mercapto methyl 2-hydroxyethylamino benzylamino hydroxy methyl 2-hydroxyethylamino benzylamino amino methyl 2-hydroxyethylamino benzylamino 2-hydroxyethylamino methyl 2-hydroxyethylamino benzylamino aminomethylamino methyl 3-hydroxypropylamino benzylamino methyl isopropyl 3-hydroxypropylamino benzylamino mercapto isopropyl 3-hydroxypropylamino benzylamino hydroxy isopropyl 3-hydroxypropylamino benzylamino amino isopropyl 3-hydroxypropylamino benzylamino 2-hydroxyethylamino isopropyl 3-hydroxypropylamino benzylamino aminomethylamino isopropyl (R)-1-(hydroxymethyl) benzylamino methyl isopropyl propylamino (R)-1-(hydroxymethyl) benzylamino mercapto isopropyl propylamino (R)-1-(hydroxymethyl) benzylamino hydroxy isopropyl propylamino (R)-1-(hydroxymethyl) benzylamino amino isopropyl propylamino (R)-I-(hydroxymethyl) benzylamino 2-hydroxyethylamino isopropyl propylamino (R)-1-(hydroxymethyl) benzylamino aminomethylamino isopropyl propylamino (R)-1-(hydroxymethyl) 3-iodobenzylamino methyl isopropyl propylamino (R)-1-(hydroxymethyl) 3-iodobenzylamino mercapto isopropyl propylamino (R)-1-(hydroxymethyl) 3-iodobenzylamino hydroxy isopropyl propylamino (R)-1-(hydroxymethyl) 3-iodobenzylamino amino isopropyl propylamino (R)-1-(hydroxymethyl) 3-iodobenzylamino 2-hydroxyethylamino isopropyl propylamino (R)-1-(hydroxymethyl) 3-iodobenzylamino aminomethylamino isopropyl propylamino diethanolamino 3-chloroanilino methyl isopropyl diethanolamino 3-chloroanilino mercapto isopropyl diethanolamino 3-chloroanilino hydroxy isopropyl diethanolamino 3-chloroanilino amino isopropyl diethanolamino 3-chloroanilino 2-hydroxyethylamino isopropyl diethanolamino 3-chloroanilino aminomethylamino isopropyl (R)-1-(hydroxymethyl) 4-methoxybenzylamino mercapto isopropyl isobutylamino (R)-1-(hydroxymethyl) 4-methoxybenzylamino hydroxy isopropyl isobutylamino (R)-1-(hydroxymethyl) 4-methoxybenzylamino amino isopropyl isobutylamino (R)-1-(hydroxymethyl) 4-methoxybenzylamino 2-hydroxyethylamino isopropyl isobutylamino (R)-1-(hydroxymethyl) 4-methoxybenzylamino aminomethylamino isopropyl isobutylamino (R)-1-(hydroxymethyl) 3-chloroanilino methyl isopropyl isobutylamino (R)-1-(hydroxymethyl) 3-chloroanilino mercapto isopropyl isobutylamino (R)-1-(hydroxymethyl) 3-chloroanilino hydroxy isopropyl isobutylamino (R)-1-(hydroxymethyl) 3-chloroanilino amino isopropyl isobutylamino (R)-1-(hydroxymethyl) 3-chloroanilino 2-hydroxyethylamino isopropyl isobutylamino (R)-1-(hydroxymethyl) 3-chloroanilino aminomethylamino isopropyl isobutylamino - 1 mmol of 6-(3-methoxybenzylamino)-2-chloro-9-isopropylpurine was brominated with 1.1 mmol of bromine as described in Example 1. The crude 2,8-dihalogeno derivative prepared was alkalinized with NH 3/MeOH and extracted between water—EtOAc. The product crystallizes after partial evaporation of ethylacetate. M.p.123-125° C., yield 88%. MS-ESI (+) m/z 409.5, 411.4, 413.3 [M+H]+
- 6-Phenylamino-2-(3-aminopropylamino)-9-isopropyl-9-benzylaminopurine, prepared from appropriate 8-bromo derivative, was dissolved in glacial acetic acid. To this solution, 50 mg Pd/BaSO 4 (10%) was added and the mixture hydrogenated to constant consumption of hydrogen (2 hours). The catalyst was removed by centrifugation and acetic acid was evaporated in vacuo. The rest was purified by column chromatography (silica gel, chloroform-MeOH-conc. NH4OH (95:5:1). Yield 75%. MS-ESI (+) m/z 340.5 [M+H]+
- 6-(4-Methoxybenzylamino)-2,8-bis(3-hydroxypropylamino)purine (0.07 mmol) was stirred in 2 mL dimethylformamide with 0.14 mmol of allylbromide and with 0.36 mmol of dry potassium carbonate at ambient temperature (6 hours). The solvent was evaporated to dryness and the rest extracted between ethylacetate—water. The organic layer was dried, ethylacetate was evaporated and the rest purified by column chromatography (silica gel, chloroform). Yield 55%. MS-ESI(+) m/z 441.4 [M+H] +
TABLE 3 Compounds Prepared by the Method of Example 5 SUBSTITUENT C2 N6 C8 N9 2-hydroxyethylamino benzylamino 2-hydroxyethylamino allyl 3-hydroxypropylamino 4-methoxybenzylamino 3-hydroxypropylamino allyl 2-aminoethylamino benzylamino 2-aminoethylamino isopropyl 3-aminopropylamino benzylamino 3-aminopropylamino isopropyl aminomethylamino benzylamino aminomethylamino isopropyl diethanolamino benzylamino diethanolamino isopropyl hydroxymethylamino benzylamino hydroxymethylamino isopropyl 3-aminopropylamino benzylamino 3-aminopropylamino isopropyl 2-hydroxyethylamino 3-chloroanilino 2-hydroxyethylamino isopropyl 3-hydroxypropylamino 3-chloroanilino 3-hydroxypropylamino isopropyl 2-aminoethylamino 3-chloroanilino 2-aminoethylamino isopropyl 3-aminopropylamino 3-chloroanilino 3-aminopropylamino isopropyl diethanolamino benzylamino diethanolamino isopropyl hydroxymethylamino benzylamino hydroxymethylamino isopropyl (R)-1-(hydroxymethyl) 3-chloroanilino (R)-1-(hydroxymethyl)- isopropyl isobutylamino isobutylamino - 1 mmol of 6-chloropurine was alkylated with isopropylbromide in DMF as described in Example 5. The product, 6-chloro-9-isopropylpurine was brominated in acetic acid similarly as described in Example 1. After purification by column chromatography, 6-benzylamino-9-isopropyl-8-bromopurine was treated with 2-aminoethanol to give 6-benzylamino-8-hydroxyethylamino-9-isopropylpurine. Yield 40%. MS-ESI(+) m/z 326 [M+H] +
- 6-Amino-2-(3-hydroxypropylamino)-9-methyl-8-(3-hydroxypropyloxy)purine was brominated in the position 6 with amylnitrite/CH 2Br2. The 6-bromo derivative was then hydrogenolyzed with PdO/BaSO4 in strong alkaline solution to give 2,8,9-trisubstituted purine. The crude product was purified by column chromatography (silica gel, chloroform-MeOH-conc. NH4OH (8:2:0.2)). The total yield was 35%. MS-ESI(+) 281[M+H]+
- 0.02 mmol of 2-Methyl-4-amino-3-alkoxycarbonyl-5-isopropylpyrazole (prepared from appropriate 4-nitro derivative by catalytic hydrogenation) was dissolved in 30 mL of 2-ethoxyethanol and 0.02 mmol of formamidin acetate and 5.2 mL of triethylamine were added. The mixture was heated to 90° C. for 2 hours. The solution was concentrated in vacuo, dissolved in chloroform to give crystalline product. After recrystallization from ethanol m.p. 295-298° C. MS-ESI(+) m/z 193 [M+H] +
- 1.1 mmol of 7-Hydroxy-l-methyl-3-isopropyl pyrazolo[4,3]pyrimidine (Example 8) was dissolved in the mixture of 0.12 mL of dimethylformamide and 5 mL dry chloroform. 11 mmol of thionylchloride (0.81 mL) was added and the solution heated to 80° C. for 1 hour. A new portion of 0.8 mL of thionylchloride and more chloroformdimethylformamide were then added and heating continued for 3 hours. The solvents were evaporated in vacuo and the rest partitioned between water-chloroform. The organic extract was dried and used in next reaction without purification. The chloroform solution was heated with excess of 2-hydroxybenzylamine and N-ethyldiisopropylamine for 1 hour. The product was purified by column chromatography (silica gel, chloroform MeOH-AcOH (20:0.4:0.1)). M.p.205-210° C. Yield 40%. MS-ESI(+) m/z 297 [M+H] +
- 6-(Benzylamino)-2,8-dichloropurine, prepared from 2,6,8-trichloropurine, m.p.220-223° C., 50% yield, was dissolved in 2-aminobutanol and heated to 150° C. for 12 hours. After evaporation of the solvent in vacuo the product, 6-benzylamino-2-[(1-hydroxymethyl) propylamino]-8-chloropurine was purified by column chromatography (silica gel, chloroform—MeOH—conc. NH 4OH 9:1:0.1) with total yield 36%. MS-ESI(+) m/z 348 [M+H]+. The product was subsequently alkylated with isopropyliodide/K2CO3/dimethylformamide to give, after preparative column chromatography (silica gel, chloroform) 70% yield of the desired compound. MS (ESI+): 388.4, 390.6 (M+H)+.
- Freeze-dried epoxy activated Sepharose 6B (Pharmacia LKB, Piscataway, N.J.) was chosen for the coupling reaction due to its ability to form an ether bond between a hydroxyl-containing ligand and the epoxide group on the Sepharose. The gel was swollen according to the manufacturer's instructions, (100 mg) of any one of the compound defined by Formula I (preferably with R6=aminooctylamino, 3- or 4-benzylamino, etc.) was dissolved in 1 ml coupling solution (1.2:1, v/v, DMF, 0.1N NaOH) and mixed with 0.5 ml of swollen gel at pH 10-11 for 72 h at room temperature with gentle agitation. Excess reactive groups were blocked with 1M ethanolamine for 4 hours at 50° C. and the gel slurry was poured into 1-ml syringe column. The resin was activated with three alternating cycles of twenty column volumes each of pH 4.0 (0.1 M acetate, 0.5 M NaCl) and pH 8.0 (0.1 M tris-HCI, 0.5 M NaCl) buffers followed by twenty column volumes of reaction buffer (20 mM HEPES, pH 7.3, 10 mM MgCl 2, 15 mM glycerophosphate, 0.5 mM sodium orthovanadate, 0.5 mM EGTA). The column was stored at 4° C in the reaction buffer containing 0.1% sodium azide and regenerated prior to each use with alternating cycles of low and high pH as described above.
- The Sf9 insect cell lysate (500 μg protein in 1-ml reaction buffer) was passed over the affinity column matrix sequentially five times and the flow through was saved, (unbound material). The matrix was then washed three times with 1 ml reaction buffer (wash 1-3) then three times each reaction buffer containing 0.5M NaCl (eluate 1-3). The coupled proteins were eluted at low pH (pH 4.0, 0.1M acetate, 0.5M NaCl) as described above and aliquots (20μ from 1 ml) of each sample were assayed for their ability to phosphorylate histone H1 and other substrate proteins as described in Example 12. The presence of CDK complexes was also determined by SDS-PAGE.
- Proteins
- Cyclin-dependent kinases (p34 cdc2, p33cdk2, p33cdk4) and cyclins (cyclin B, E and D1) are produced in Sf9 insect cells coinfected with appropriate baculoviral constructs. The cells are harvested 68-72 hrs post infection in lysis buffer for 30 min on ice and the soluble fraction is recovered by centrifugation at 14.000 g for 10 min. The protein extract is stored at −80° C.
- Rb-GST is produced using an E. coli expression system, containing sequence encoding C terminus of retinoblastoma protein (aminoacids 773-928), which is known to be phosphorylated by p33cdk4 kinase. The fusion protein is purified on glutathioneagarose beads.
- Lysis buffer: 50 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 20 mM NaF, 1% Tween, 1 mM DTT, 0.1 mM PMSF, leupeptine, aprotonine.
- Enzyme inhibition assays
- To carry out experiments on kinetics under linear conditions, the final point test system for kinase activity measurement is used. The kinase is added to reaction mixture in such a way as to obtain linear activity with respect to the concentration of enzyme and with respect to time.
- The p34 cdc2 and p33cdk2 kinase inhibition determination involves the use of 1 mg/ml histone H1 (Sigma, type III-S) in the presence of 15 μM [γ-32P]ATP (500-100 cpm/pmol) (Amersham) in a final volume of 20 μl, inhibition of p33cdk4 kinase is determined with Rb-GST (0.2 mg/ml) as a substrate. Kinase activity is determined at 30° C. in the kinase buffer.
- Tested compounds are usually dissolved to 100 mM solutions in DMSO, final concentration of DMSO in reaction mixture never exceeds 1%. The controls contain suitable dilutions of DMSO.
- After 10 min, addition 3× SDS sample buffer stops the incubations. Phosphorylated proteins are separated electrophoretically using 12.5% SDS polyacrylamide gel. The measurement of kinase activity is done using digital image analysis.
- The kinase activity is expressed as a percentage of maximum activity. The apparent inhibition constants are determined by graphic analysis. Kinase buffer: 50 mM Hepes pH 7.4. 10 mM MgCl 2, 5 mM EGTA. 10 mM 2-glycerolphosphate, 1 mM NaF, 1 mM DTT
TABLE 4 Kinase Inhibitory Activity of 2,6,8,9-Tetrasubstituted Purine Derivatives CDC2 IKB-α SUBSTITUENT IC50 IC50 C2 N6 C8 N9 (μM) (μM) 2-hydroxyethylamino benzylamino — methyl 7 15.4 OLOMOUCINE 2-hydroxyethylamino benzylamino fluoro methyl 4.2 7.8 2-hydroxyethylamino benzylamino bromo methyl 16.2 22.8 2-hydroxyethylamino benzylamino mercapto methyl 22.4 35.7 2-hydroxyethylamino benzylamino hydroxy methyl 13.5 16.2 2-hydroxyethylamino benzylamino amino methyl 14.2 17.1 2-hydroxyethylamino benzylamino 2-hydroxy- methyl 125.2 153.8 ethylamino 2-hydroxyethylamino benzylamino aminomethyl methyl 114.3 126.5 amino 3-hydroxypropylamino benzylamino — isopropyl 1 3.2 BOHEMINE 3-hydroxypropylamino benzylamino bromo isopropyl 12.93 24.5 3-hydroxypropylamino benzylamino mercapto isopropyl 13.1 20.9 3-hydroxypropylamino benzylamino hydroxy isopropyl 6.48 12.6 3-hydroxypropylamino benzylamino amino isopropyl 8.52 12.8 3-hydroxypropylamino benzylamino 2-hydroxy- isopropyl 143.8 152.6 ethylamino 3-hydroxypropylamino benzylamino aminomethyl isopropyl 123.5 138.7 amino (R)-1-(hydroxymethyl) benzylamino isopropyl 0.45 1.4 propylamino ROSCOVITINE (R)-1-(hydroxymethyl) benzylamino fluoro isopropyl 0.38 1.2 propylamino (R)-1-(hydroxymethyl) benzylamino bromo isopropyl 10.38 11.2 propylamino (R)-1-(hydroxymethyl) benzylamino mercapto isopropyl 41.11 43.8 propylamino (R)-1-(hydroxylethyl) benzylamino hydroxy isopropyl 10.15 10.94 propylamino (R)-1-(hydroxylethyl) benzylamino amino isopropyl 20.17 20.85 propylamino (R)-1-(hydroxymethyl)- benzylamino 2-hydroxy- isopropyl 151.93 164.95 propylamino ethylamino (R)-1-(hydroxymethyl)- benzylamino aminomethyl- isopropyl 181.05 193.28 propylamino amino (R)-1-(hydroxymethyl) 3-chloroanilino isopropyl 4 nM 11.2 nM isobutylamino PURVALANOL A (R)-1-(hydroxymethyl) 3-chloroanilino fluoro isopropyl 3.8 nM 11 nM isobutylamino (R)-1-(hydroxymethyl) 3-chloroanilino bromo isopropyl 43.8 51 nM isobutylamino nM (R)-1-(hydroxymethyl) 3-chloroanilino mercapto isopropyl 83.7 105.6 isobutylamino nM nM (R)-1-(hydroxymethyl) 3-chloroanilino hydroxy isopropyl 72.1 89.5 nM isobutylamino nM (R)-1-(hydroxymethyl) 3-chloroanilino amino isopropyl 91.5 92.8 nM isobutylamino nM (R)-1-(hydroxymethyl)- 3-chloroanilino 2-hydroxy- isopropyl 1529.6 1726.8 isobutylamino ethylamino nM nM (R)-1-(hydroxymethyl)- 3-chloroanilino aminomethyl- isopropyl 2521.3 3287.2 isobutylamino amino nM nM - Table 4 shows the results of inhibitory activity of novel compounds against CDC2 and IκB-α in comparison with the data on the prototype compounds. Most of the 2,6,8,9-tetrasubstituted purine derivatives showed marked inhibitory activity in in vitro kinase assays. Modification of 2,6,8-trisubstituted purines by small substituent in R8 position led usually to increase in cdk inhibitory activity of the tested compound.
- Metaphase-arrested Xenopus egg extracts were prepared as described previously by Blow “J.Cell Biol.” 122:993 (1993) and stored in liquid nitrogen. Demembranated Xenopus sperm nuclei were prepared as described by Blow & Laskey “Cell” 47:577 (1986). After thawing, extracts were supplemented with 25 mM phosphocreatine, 5 μg/ml creatine phosphokinase, 250 μg/ml cycloheximide, [α- 32P]dATP (for DNA synthesis assays). Demembranated sperm nuclei were added to a final sperm concentration of 3 ng/μl DNA extract and CDK inhibitor tested was then added at different concentrations. M-phase promoting factor inhibition by different CDK inhibitors was monitored 1.5 h after addition by assessing the amount of sperm nuclei that had been assembled into interphase nuclei, possessing a complete phase-dense nuclear envelope. DNA synthesis was assessed by releasing extract into interphase by the addition of 0.3 mM CaCl2 and measuring the total amount of [α-32P]dATP incorporation after 3 h by TCA co-precipitation.
- At concentrations of CDK inhibitors (see Table 5) ranging from 0.1-2 μM, chromosomes remained highly condensed and no nuclear envelope was visible. At 4-6 μM and higher concentrations, interphase nuclei appeared with partially decondensed chromatin and an intact nuclear envelope. Replication was significantly inhibited at 1-5 μM CDK inhibitors tested. For the inhibition effect to become detectable, the first 15-min incubation of the interphase extract is probably sufficient.
TABLE 5 Antimitotic Activities of 2,6,8,9-Tetrasubstituted Purine Derivatives Inhibition of MPF Inhibition activity of DNA SUBSTITUENT IC50 synthesis C2 N6 C8 N9 (μM) IC50 (μM) hydroxyethylamino benzylamino — methyl 12 15 OLOMOUCINE hydroxypropylamino benzylamino fluoro isopropyl 2.6 3.2 (R)-1-(hydroxyethyl) benzylamino fluoro isopropyl 1.5 2.4 propylamino (R)-1-(hydroxyethyl) 3-chloroanilino fluoro isopropyl 0.8 1.5 propylamino (R)-1-(hydroxyethyl) 3-chloro-4-carboxy- fluoro isopropyl 0.1 0.25 propylamino anilino (R)-1-(hydroxymethyl) 3-chloroanilino amino isopropyl 3.12 3.3 propylamino (R)-1-(hydroxymethyl) 3-chloroanilino hydroxy isopropyl 4.03 4.2 propylamino 2-hydroxyethylamino 3-chloroanilino 2- isopropyl 41.7 63.5 hydroxy ethyl- amino (R)-1-(hydroxymethyl)- 3-chloroanilino bromo isopropyl 31.6 52.8 propylamino - One of the parameters used, as the basis for colorimetric assays, is the metabolic activity of viable cells. For example, a microtiter assay, which uses the tetrazolium salt MTT, is now widely used to quantitate cell proliferation and cytotoxicity. For instance, this assay is used in drug screening programs and in chemosensitivity testing. Because only metabolically active cells cleave tetrazolium salts, these assays detect viable cells exclusively. In the case of MTT assay, yellow soluble tetrazolium salt is reduced to coloured water-insoluble formazan salt. After it is solubilized, the formazan formed can easily and rapidly be quantified in a conventional ELISA plate reader at 570 nm (maximum absorbance). The quantity of reduced formazan corresponds to number of vital cells in the culture.
- Human T-lymphoblastic leukemia cell line CEM; promyelocytic HL-60 and monocytic U937 leukemias; breast carcinoma cell lines MCF-7, BT549, MDA-MB-231; glioblastoma U87MG cells; cervical carcinoma cells HELA; sarcoma cells U20S and Saos2; hepatocellular carcinoma HepG2; mouse fibroblasts NIH3T3; mouse immortalized bone marrow macrophages B2.4 and B10A.4; P388D1 and L1210 leukemia; B16 and B16F10 melanomas were used for routine screening of compounds. The cells were maintained in Nunc/Corning 80 cm2 plastic tissue culture flasks and cultured in cell culture medium (DMEM with 5 g/l glucose, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10% fetal calf serum and sodium bicarbonate).
- The cell suspensions that were prepared and diluted according to the particular cell type and the expected target cell density (2.500-30.000 cells per well based on cell growth characteristics) were added by pipette (80 μl) into 96/well microtiter plates. Inoculates were allowed a pre-incubation period of 24 hours at 37° C. and 5% CO 2 for stabilisation. Four-fold dilutions of the intended test concentration were added at time zero in 20 μl aliquots to the microtiter plate wells. Usually, test compound was evaluated at six 4-fold dilution. In routine testing, the highest well concentration was 266.7 μM, but it can be the matter of change dependent on the agent. All drug concentrations were examined in duplicates. Incubations of cells with the test compounds lasted for 72 hours at 37° C., in 5% CO2 atmosphere and 100% humidity. At the end of incubation period, the cells were assayed by using the MTT. Ten microliters of the MTT stock solution were pipetted into each well and incubated further for 1-4 hours. After this incubation period, formazan was solubilized by addition of 100 μl/well of 10% SDS in water (pH=5.5) followed by further incubation at 37° C. overnight. The optical density (OD) was measured at 540 nm with the Labsystem iEMS Reader MF (UK). The tumour cell survival (TCS) was calculated using the following equitation: TCS=(ODdrug exposed well/mean ODcontrol wells)×100%. The TCS50 value, the drug concentration lethal to 50% of the tumour cells, was calculated from the obtained dose response curves.
- Cytoxicity of novel compounds was tested on panel of cell lines of different histogenetic and species origin (Table 6). We show here that equal activities were found in all tumour cell lines tested, however, the non-malignant cells, e.g. NIH3T3 fibroblasts and normal human lymphocytes, were resistant to synthetic CDK inhibitors induced cytotoxicity. As demonstrated in Table 6, IC 50 for NIH3T3 fibroblasts and normal human lymphocytes was always higher than 250 μM. Effective novel derivatives killed tumour cells in concentrations close to 1-5 μM. Notably, the identical effectiveness of purine derivatives was also found in cell lines bearing various mutations or deletions in cell cycle associated proteins, e.g. HL-60, BT549, Hela, U20S, MDA-MB231, and Saos2 (Table 6). It indicates that these substances should be equally effective in tumours with various alterations of tumour suppresser genes, namely p53, Rb, etc. Importantly, this observation distinguishes novel compounds from flavopiridol and related compounds, as their biological activity is dependent on p53 status.
TABLE 6 Cytotoxicity of Novel Compounds for Different Cancer Cells CEM B16 SUBSTITUENT IC50 IC50 C2 N6 C8 N9 (μM) (μM) 2-hydroxyethylamino benzylamino — methyl 70 85.4 OLOMOUCINE 2-hydroxyethylamino benzylamino fluoro methyl 45.2 62.8 2-hydroxyethylamino benzylamino bromo methyl 165.2 172.8 2-hydroxyethylamino benzylamino mercapto methyl 92.4 85.7 2-hydroxyethylamino benzylamino hydroxy methyl 45.5 56.2 2-hydroxyethylamino benzylamino amino methyl 84.2 67.1 2-hydroxyethylamino benzylamino 2-hydroxy- methyl >166.7 >166.7 ethylamino 2-hydroxyethylamino benzylamino aminomethyl- methyl 114.3 126.5 amino 3-hydroxypropylamino benzylamino — isopropyl 4.1 7.2 BOHEMINE 3-hydroxypropylamino benzylamino fluoro isopropyl 3.93 5.5 3-hydroxypropylamino benzylamino bromo isopropyl 116.9 76.4 3-hydroxypropylamino benzylamino methylthio isopropyl 68.4 60.6 3-hydroxypropylamino benzylamino hydroxy isopropyl 7.48 5.6 3-hydroxypropylamino benzylamino amino isopropyl 8.52 6.8 3-hydroxypropylamino benzylamino 2-hydroxy- isopropyl >166.7 >166.7 ethylamino 3-hydroxypropylamino benzylamino 2-aminoethyl isopropyl 29.9 2.5 amino (R)-1-(hydroxymethyl)- benzylamino isopropyl 3.45 4.4 propylamino ROSCOVITINE (R)-1-(hydroxymethyl)- benzylamino fluoro isopropyl 3.38 3.2 propylamino (R)-1-(hydroxymethyl)- — bromo isopropyl 48.4 59.2 propylamino benzylamino (R)-1-(hydroxymethyl)- benzylamino mercapto isopropyl 30.7 125.6 propylamino (R)-1-(hydroxymethyl)- benzylamino hydroxy isopropyl 12.15 12.94 propylamino (R)-1-(hydroxymethyl)- benzylamino amino isopropyl 12.17 12.85 propylamino (R)-1-(hydroxymethyl)- benzylamino 2-hydroxy- isopropyl 140.2 124.95 propylamino ethylethoxy (R)-1-(hydroxymethyl)- benzylamino 1,2- isopropyl 128.2 147.5 propylamino dihydroxy- propylamino (R)-1-(hydroxymethyl)- benzylamino 2-aminoethyl isopropyl 96.1 >166.7 propylamino amino (R)-1-(hydroxymethyl)- benzylamino hydrazido isopropyl 58 61.5 propylamino (R)-1-(hydroxymethyl)- 3-chloroanilino isopropyl 2.4 3.5 isobutylamino PURVALANOL A (R)-1-(hydroxymethyl)- 3-chloroanilino fluoro isopropyl 2.5 3.5 isobutylamino (R)-1-(hydroxymethyl)- 3-chloroanilino bromo isopropyl 22.5 23.5 isobutylamino (R)-1-(hydroxymethyl)- 3-chloroanilino mercapto isopropyl 25.8 47.6 isobutylamino (R)-1-(hydroxymethyl)- 3-chloroanilino hydroxy isopropyl 11.8 11.3 isobutylamino (R)-1-(hydroxymethyl)- 3-chloroanilino amino isopropyl 11.5 11.8 isobutylamino (R)-1-(hydroxymethyl)- 3-chloroanilino 2- isopropyl 78 150 isobutylamino hydroxyethyl amino (R)-1-(hydroxymethyl)- 3-chloroanilino 2- isopropyl 56 130 isobutylamino aminomethyl- amino - To analyse the mechanisms of induced cytotoxicity by novel compounds, it is important to distinguish apoptosis from the other major form of cell death, necrosis. First, at the tissue level, apoptosis produces little or no inflammation, since the neighbouring cells, especially macrophages, rather than being released into the extracellular fluid, engulf shrunken portions of the cell. In contract, in necrosis, cellular contents are released into the extracellular fluid, and thus have an irritant affect on the nearby cells, causing inflammation. Second, at the cellular level, apoptotic cells exhibit shrinkage and blebbing of the cytoplasm, preservation of structure of cellular organelles including the mitochondria, condensation and margination of chromatin, fragmentation of nuclei, and formation of apoptotic bodies, thought not all of these are seen in all cell types. Third, at the molecular level, a number of biochemical processes take an important role in induction of apoptosis. However, majority of them is not well understood, and they result in activation of proteases and nucleases, which finally destruct key biological macromolecules-proteins and DNA. For detection of apoptotic versus necrotic mode of cell death, two independent methods were employed: assessment of morphology by electron microscopy and analysis of DNA fragmentation by flow-cytometry.
- HL-60 cell line was cultured in 6-well culture plates with or without 70 μM concentration of novel derivatives at 37° C. and 5% CO 2 for 3-24 hours. Following the incubation, cells were pelleted, washed in Hank's buffered salt solution and processed as described below.
- Cells were suspended in 2% glutaraldehyde/PBS, fixed overnight at 4° C., pelleted and embedded into 1% agar (Agar Noble, Difco) thereafter. Agar block containing fixed cells was epoxide polymerised, ultrathin sectioned, osmium tetraoxide postfixed, uranium acetate contrasted and examined under electron microscope.
- Initial phase contrast microscopy examinations indicated that treated HL-60 line exhibit typical morphological features of apoptotic cells, and it was later confirmed by electron microscopy. Typical morphological criteria of apoptosis were identified in cells treated with tetrasubstituted purine derivatives: chromatin condensation, nuclear fragmentation, cytoplasmatic blebbing, and formation of apoptotic bodies.
- One of the most important parameters of specific cellular immunity is proliferative response of lymphocytes to antigens or polyclonal mitogens. Majority of normal mammalian peripheral lymphocytes is resting cells. Antigens or nonspecific—polyclonal mitogens have capacity to activate lymphoid cells and it is accompanied with dramatic changes of intracellular metabolism (mitochondrial activity, protein synthesis, nucleic acids synthesis, formation of blastic cells and cellular proliferation). Compounds with ability to selectively inhibit lymphocyte proliferation are potent immunosuppressants. Variety of in vitro assays were developed to measure proliferative response of lymphocytes. The most commonly used is 3H-thymidine incorporation method.
- During cell proliferation, the DNA has to be replicated before the cell is divided into two daughter cells. This close association between cell doublings and DNA synthesis is very attractive for assessing cell proliferation. If labeled DNA precursors are added to the cell culture, cells that are about to divide incorporate the labeled nucleotide into their DNA. Traditionally, those assays usually involve the use of radiolabeled nucleosides, particularly tritiated thymidine ([ 3H]-TdR). The amount of [3H]-TdR incorporated into the cellular DNA is quantified by liquid scintillation counting.
- Human heparinized peripheral blood was obtained from healthy volunteers by cubital vein punction. The blood was diluted in PBS (1:3) and mononuclear cells were separated by centrifugation in Ficoll-Hypaque density gradient (Pharmacia, 1.077 g/ml) at 2200 g for 30 minutes. Following centrifugation, lymphocytes were washed in PBS and resuspended in cell culture medium (RMPI 1640, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10% fetal calf serum and sodium bicarbonate).
- The cells were diluted at target density of 1.100.000 cells/ml were added by pipet (180 μl) into 96/well microtiter plates. Four-fold dilutions of the intended test concentration were added at time zero in 20 μl aliquots to the microtiter plate wells. Usually, test compound was evaluated at six 4-fold dilutions. In routine testing, the highest well concentration was 266.7 μM. All drug concentrations were examined in duplicates. All wells with exception of unstimulated controls were activated with 50 μl of concanavalin A (25 μg/ml). Incubations of cells with test compounds lasted for 72 hours at 37° C., in 5% CO2 atmosphere and 100% humidity. At the end of incubation period, the cells were assayed by using the [ 3H]-TdR:
- Cell cultures were incubated with 0.5 μCi (20 μl of stock solution 500 μCi/ml) per well for 6 hours at 37° C. and 5% CO 2. The automated cell harvester was used to lyse cells in water and adsorb the DNA onto glass-fiber filters in the format of microtiter plate. The DNA incorporated [3H]-TdR was retained on the filter while unincorporated material passes through. The filters were dried at room temperature overnight, sealed into a sample bag with 10-12 ml of scintillant. The amount of [3H]-TdR present in each filter (in CCPM) was determined by scintillation counting in a Betaplate liquid scintillation counter. The effective dose of immunosuppressant (ED) was calculated using the following equotation: ED=(CCPMdrug exposed well/mean CCPMcontrolwells)×100%. The ED50 value, the drug concentration inhibiting proliferation of 50% of lymphocytes, was calculated from the obtained dose response curves.
- To evaluate immunosuppressive activity of tetrasubstituted adenines, their ability to inhibit polyclonal mitogen induced proliferation of normal human lymphocytes was analyzed (Table 7). Our data demonstrate that these compounds have only marginal activity on 3H-thymidine incorporation, nonetheless, they efficiently inhibit proliferation of activated lymphocytes. Effective immunosuppressive dose of tetrasubstituted derivatives under in vitro conditions was close to 1-20 μM.
TABLE 7 Immunosuppressive effects of compounds on spontaneous and mitogen activated proliferation of lymphocytes spont- mitogen SUBSTITUENT aneous activated C2 N6 C8 N9 IC50 (μM) IC50 (μM) hydroxyethylamino benzylamino — methyl 245 72.1 OLOMOUCIN 2-hydroxyethylamino benzylamino bromo methyl 228 68.3 2-hydroxyethylamino benzylamino mercapto methyl 250 83.5 2-hydroxyethylamino benzylamino hydroxy methyl 201 48.6 2-hydroxyethylamino benzylamino amino methyl 170.3 26.7 2-hydroxyethylamino benzylamino 2-hydroxy- methyl 250 169.3 ethylamino 2-hydroxyethylamino benzylamino aminomethyl- methyl 250 113.5 amino 3-hydroxypropylamino benzylamino — isopropyl 172 5.7 BOHEMIN 3-hydroxypropylamino benzylamino bromo isopropyl 178 5.9 3-hydroxypropylamino benzylamino mercapto isopropyl 197 18.4 3-hydroxypropylamino benzylamino hydroxy isopropyl 163 2.1 3-hydroxypropylamino benzylamino amino isopropyl 143 1.59 3-hydroxypropylamino benzylamino 2-hydroxy- isopropyl 181 29.3 ethylamino 3-hydroxypropylamino benzylamino aminomethyl- isopropyl 174 26.8 amino (R)-1-(hydroxymethyl)- benzylamino isopropyl 164 6.4 propylamino ROSCOVITIN (R)-1-(hydroxymethyl)- benzylamino bromo isopropyl 168 17.1 propylamino (R)-1-(hydroxymethyl)- benzylamino mercapto isopropyl 174 18.5 propylamino (R)-1-(hydroxymethyl)- benzylamino hydroxy isopropyl 153 12.8 propylamino (R)-1-(hydroxymethyl)- benzylamino amino isopropyl 149 13.3 propylamino (R)-1-(hydroxymethyl)- benzylamino 2-hydroxy- isopropyl 182 31.5 propylamino ethylamino (R)-1-(hydroxymethyl)- benzylamino aminomethyl- isopropyl 177 25.8 propylamino amino (R)-1-(hydroxymethyl)- 3-chloroanilino isopropyl 158 5.7 isobutylamino PURVALANOL A (R)-1-(hydroxymethyl)- 3-chloroanilino bromo isopropyl 142 13.7 isobutylamino (R)-1-(hydroxymethyl)- 3-chloroanilino mercapto isopropyl 164 26.9 isobutylamino (R)-1-(hydroxymethyl)- 3-chloroanilino hydroxy isopropyl 126 2.98 isobutylamino (R)1-(hydroxymethyl)- 3-chloroanilino amino isopropyl 103 3.1 isobutylamino (R)-1-(hydroxymethyl)- 3-chloroanilino 2-hydroxy- isopropyl 205 150 isobutylamino ethylamino (R)-1-(hydroxymethyl)- 3-chloroanilino aminomethyl- isopropyl 230 138 isobutylamino amino - The activity of the compounds against HIV-1 and HIV-2 induced cytopathicity was examined in human lymphocyte MT-4 cells. The cells (300 000 cells/ml) were infected with 100 CCID 50 (1 CCID50 is a virus quantity which causes cytopathicity effect in 50% of the cells under the experimental conditions) of HIV-1 or HIV-2 and added to 200 μl wells of a microtiter plate containing different dilutions of the tested compounds. The infected cell cultures were incubated at 37° C. for 5 days in a humidified CO2 incubator. The cytopathicity of the virus was examined by determination of MT-4 cell viability by trypan blue dye staining. The results are summarised in Table 8 with comparison on the prototype compounds.
- Table 8 also shows the results of activity testing of novel compounds against MSV-induced transformation in murine embryo fibroblast C3H/3T3 cells. The cells were seeded in 1 ml wells of 48-well plates and exposed to 80 PFU (plague forming units) for 60-90 min. The virus was subsequently removed and culture medium containing appropriate concentrations of the tested compounds was added (1 ml per well). At day 6-post infection, MSV-induced transformation of the cell culture was examined microscopically.
TABLE 8 Antiretroviral Activity of Purine Compounds, wherein R9 is either PMP (2-phosphonomethoxypropyl)group or PME (2-phosphonomethoxyethyl)group. (μg/ml), (R2 = NH2). HIV-1 HIV-2 R6 R8 MSV MT-4 CEM MT-4 CEM PME-derivatives amino 0.6 2.67 6.9 ND ND amino fluoro 0.57 3.12 7.2 ND ND amino mercapto 1.23 5.87 10.8 ND ND amino hydroxy 0.42 1.95 5.2 ND ND amino amino 0.58 1.98 4.8 ND ND amino 2-hydroxyethyl- 1.54 8.33 10.8 ND ND amino amino aminomethyl- 132 7.54 9.3 ND ND amino cyclohexylamino 0.26 5.7 >20 4.8 >20 cyclohexylamino fluoro 0.24 6.3 >20 4.6 >20 cyclohexylamino mercapto 0.35 8.9 >20 8.5 >20 cyclohexylamino hydroxy 0.21 4.3 >20 2.1 >20 cyclohexylamino amino 0.19 3.5 >20 1.6 >20 cyclohexylamino 2-hydroxyethyl- 0.45 12.7 >20 12.7 >20 amino cyclohexylamino aminomethyl- 0.43 10.8 >20 9.5 >20 amino benzylamino 1.5 50 >20 49 >20 benzylamino fluoro 1.3 47 >20 45 >20 benzylamino mercapto 1.8 56 >20 57 >20 benzylamino hydroxy 0.9 45 >20 32 >20 benzylamino amino 0.8 48 >20 31 >20 benzylamino 2-hydroxyethyl- 1.7 67 >20 48 >20 amino benzylamino aminomethyl- 1.4- 55 >20 52 >20 amino PMP-derivatives amino 0.07 0.29 10 0.24 10 amino fluoro 0.06 3.12 3.54 ND amino mercapto 0.15 4.18 ND 4.15 ND amino hydroxy 0.05 0.25 ND 0.21 ND amino amino 0.06 0.19 ND 0.20 ND amino 2-hydroxyethyl- 0.35 5.16 ND 4.87 ND amino amino aminomethyl- 0.42 4.58 ND 4.65 ND amino cyclohexylamino 3.78 3.4 4.5 5.8 8.5 cyclohexylamino fluoro 2.54 3.2 4.1 4.6 8.3 cyclohexylamino mercapto 6.32 10.1 >20 11.2 >20 cyclohexylamino hydroxy 1.37 2.1 5.2 3.2 7.8 cyclohexylamino amino 1.25 1.8 4.7 2.8 8.1 cyclohexylamino 2-hydroxyethyl- 5.42 12.7 >20 25.1 >20 amino cyclohexylamino aminomethyl- 4.98 9.8 >20 18.6 >20 amino benzylamino 0.3 10.3 11.6 8.3 12.5 benzylamino fluoro 0.27 8.5 12.5 6.4 11.1 benzylamino mercapto 1.35 21.8 >20 >20 >20 benzylamino hydroxy 0.21 3.7 10.3 4.3 11.1 benzylamino amino 0.18 2.9 10.7 3.8 11.9 benzylamino 2-hydroxyethyl- 2.17 19.5 >20 >20 >20 amino benzylamino aminomethyl- 1.89 16.8 >20 >20 >20 amino - Most of the PMP (9-(2-phosphonomethoxypropyl) derivative) and PME (9-(2phosphonomethoxyethyl) derivative) compounds of the formula I showed marked antiHIV activity in vitro. HIV-1 and HIV-2 did not differ in their sensitivity to the test compounds. (R)-PMP compounds were markedly inhibitory to retroviruses at 2-3 μg/ml and not toxic to the cells at 100 μg/ml. Its selectivity index (ratio cytotoxic dose/antivirally active dose) proved superior over that of the prototype compound PME. The (S)-enantiomer of PME was devoid of marked antiretroviral activity. (R)-PMPD were exquisitely inhibitory to retrovirus replication (EC 50 0.01-0.1 μg/ml) and not toxic to the cells at 100 μg/ml. It proved superior over PMEA and other prototype compounds in terms of both antiviral activity and lack of toxicity. Its selectivity index was higher than 2000 for HIV-1 and HIV-2.
- 5000 capsules, each of which contain 0.25 g of one of the compounds of the formula I mentioned in the preceding Examples as active ingredient, are prepared as follows:
- Composition:
- Active ingredient 1250 g
- Talc 180 g
- Wheat starch 120 g
- Magnesium stearate 80 g
- Lactose 20 g
- Preparation process: The powdered substances mentioned are pressed through a sieve of mesh width 0.6 mm. Portions of 0.33 g of the mixture are transferred to gelatine capsules with the aid of a capsule-filling machine.
- 5000 soft gelatine capsules, each of which contain 0.05 g of one of the compounds of the formula I mentioned in the preceding Examples as active ingredient, are prepared as follows:
- Composition:
- Active ingredient 250 g
- Lauroglycol 2 liters
- Preparation process: The powdered active ingredient is suspended in Lauroglycol® (propylene glycol laurate, Gattefosse S. A., Saint Priest, France) and ground in a wet-pulveriser to a particle size of about 1 to 3 μm. Portions of in each case 0.419 g of the mixture are then transferred to soft gelatine capsules by means of a capsule-filling machine.
- 5000 soft gelatine capsules, each of which contain 0.05 g of one of the compounds of the formula I mentioned in the preceding or following Examples as active ingredient, are prepared as follows:
- Composition
- Active ingredient 250 g
- PEG 400 1 liter
- Tween 80 1 liter
- Preparation process: The powdered active ingredient is suspended in PEG 400 (polyethylene glycol of Mr between 380 and about 420, Sigma, Fluka, Aldrich, USA) and Tween® 80 (polyoxyethylene sorbitan monolaurate, Atlas Chem. Inc., USA, supplied by Sigma, Fluka, Aldrich) and ground in a wet-pulveriser to a particle size of about 1 to 3 mm. Portions of in each case 0.43 g. of the mixture are then transferred to soft gelatine capsules by means of a capsule-filling machine.
- While a specific embodiment of the invention has been shown and described in detail to illustrate the application of the principles of the invention, it will be understood that the invention may be embodied otherwise without departing from such principles.
Claims (16)
1. Substituted nitrogen heterocyclic derivatives of the formula I—

wherein,


A is a divalent group
Z is N;
R2 and R6 are independent of one another, represent H, halogen, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, cycloalkyl alkyl, arylalkyl, heteroalkyl, heteroarylalkyl, heterocycloalkyl alkyl or R6′-X wherein
X is an —NH—, —N(C1-C6-alkyl)-, —O— or —S— moiety;
R6′ is H, alkyl, substituted alkyl, acyl, amido, sulpho, cycloalkyl, substituted cycloalkyl, aryl, substituted aryl, heterocycle, heteroaryl, substituted heteroaryl, arylalkyl, heterocycloalkyl, substituted heterocycloalkyl, heteroarylalkyl, heteroalkyl, cycloalkyl alkyl and heterocycloalkyl alkyl;
R8 is halogen, hydroxyl, amino, carboxyl, cyano, nitro, amido, sulpho, sulphamino, carbamino, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, arylalkyl, heteroalkyl, heteroarylalkyl, cycloalkyl alkyl, heterocycloalkyl alkyl or
R8′-X wherein
X is —NH—, —N(alkyl)—, —O— or —S— moiety, and
R8′ is according to any one of the substituents defined above for R2′ or R6′;
R9 is alkyl, substituted alkyl, acyl, carboxyl, amido, sulphamino, cycloalkyl, substituted cycloalkyl, cycloalkyl alkyl, heteroalkylcycloalkyl alkyl, heterocycloalkyl, substituted heterocycloalkyl, aryl, substituted aryl, heterocycle, heteroaryl, substituted heteroaryl, arylalkyl, heteroarylalkyl, heteroalkyl or —B-R9′ wherein
B is —CH2—, —(CH2)2—, —CH(CH3)CH2—, —CH(CH2F)CH2—, —CH(CH2OH)CH2, or the groups of the following structure,
wherein the left hand bond is linked to nitrogen of 5-membered ring of compounds of the formula I;
R4 and R5, that are independent of one another, represent hydrogen, hydroxyl, halogen, amino, acyloxy substituent having 1-5 carbon atoms, alkoxy substituent having 1-5 carbon atoms, alkylmercapto substituent having 1-5 carbon atoms, alkylamino substituent having 1-5 carbon atoms and dialkylamino in which each alkyl substituent has 1-5 carbon atoms;
R7 and R10, that are independent of one another, represent H or alkyl substituent having 1-10 carbon atoms;
or R9 is —(CH2)n-R9′, wherein n=1-2 and the
R9′ is -X(CH2)mY wherein
X is —O—, —S—, —NH— or —N (alkyl)- substituent having 1-6 carbon atoms;
m=1-2;
Y is carboxyl, amido, sulpho, sulphamino, hydroxyl, carboxyl, mercapto, carbylmercapto, amino, alkylamino, carbamino —PO(OH)2, —PO(O-C1-C6-alkyl)2, —PO(NH-C1-C6-alkyl)2, —PO(O-C1-C6-alkyl) (NH-C1-C6-alkyl) —PO(OH) (O-C1-C6-alkyl), —PO(OH)(NH-C1-C6-alkyl) or —(CH2CHD)-R9′, wherein
D is alkyl, substituted alkyl, —PO(OH)2,
—PO(OH) (O-C1-C6-alkyl), —PO(OH) (NH-C1-C6-alkyl)
wherein:
“halogen” is fluorine, bromine, chlorine, and iodine atoms;
“alkyl” is:
a) a branched or unbranched alkyl group having 1-6 carbon atoms,
b) a branched or unbranched alkenyl group having 2-6 carbon atoms, and
c) a branched or unbranched alkinyl group having 2-6 carbon atoms;
“substituted alkyl” is a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and having substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl;
“carbyloxy” is the group —ORa, where Ra is alkyl, substituted alkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl, cycloalkyl, substituted cycloalkyl, heterocycloalkyl or substituted heterocycloalkyl whereas these generic groups have meanings being identical with definitions of the corresponding groups as defined in this legend;
“carbylmercapto” is the group -SRb where Rb is alkyl, substituted alkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl, cycloalkyl, substituted cycloalkyl, heterocycloalkyl or substituted heterocycloalkyl whereas these general groups have meanings being identical with definitions of the corresponding groups as defined in this legend;
“sulpho” is the group —SO3Rc where Rc is:
a) hydrogen,
b) a branched or unbranched alkyl group having 1-6 carbon atoms,
c) a branched or unbranched alkenyl group having 2-6 carbon atoms,
d) a branched or unbranched alkinyl group having 2-6 carbon atoms, and
e) a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl, whereas these generic groups have meanings being identical with the definitions of the corresponding groups as defined in this legend;
“sulphamino” is the group —NHSO3Rd wherein Rd is:
a) hydrogen,
b) a branched or unbranched alkyl group having 1-6 carbon atoms,
c) a branched or unbranched alkenyl group having 2-6 carbon atoms,
d) a branched or unbranched alkinyl group having 2-6 carbon atoms, and
e) a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl, whereas these generic substituents have meanings being identical with definitions of the corresponding groups as defined in this legend;
“acyl” is the group —C(O)Re where Re is hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl, cycloalkyl, substituted cycloalkyl whereas these generic groups have meanings being identical with definitions of the corresponding groups as defined in this legend;
“aryloxy” is the group —OAr, where Ar is an aryl, substituted aryl, heteroaryl or substituted heteroaryl whereas these generic groups have meanings being identical with definitions of the corresponding groups as defined in this legend;
“alkylamino” is the group —NRfRg where Rf and Rg, that are independent of one another, represent hydrogen, alkyl, substituted alkyl, provided that Rf and Rg are not both hydrogens;
“amido” is the group —C(O)NRhRi′, where Rh and Ri′ may independently be hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl whereas these generic groups have meanings being identical with definitions of the corresponding groups as defined in this legend;
“carboxyl” is the group —C(O)ORj where Rj is hydrogen, alkyl, substituted alkyl, aryl, substituted aryl, heteroaryl or substituted heteroaryl;
“carbamino” is the group —NHCORk where Rk may be hydrogen, alkyl, substituted alkyl, heterocycle, aryl, substituted aryl, heteroaryl and substituted heteroaryl whereas these generic groups have meanings being identical with definitions of the corresponding groups as defined in this legend;
“aryl” is an aromatic carbocyclic group having from 6 to 18 carbon atoms and being composed of at least one aromatic or multiple condensed rings in which at least one of which being aromatic;
“substituted aryl” is an aromatic carbocyclic group having from 6 to 18 carbon atoms and being composed of at least one aromatic ring or of multiple condensed rings at least one of which being aromatic; The ring(s) are optionally substituted with one or more substituents selected from the group consisting of halogen, alkyl, hydroxyl, carbylmercapto, alkylamino, carbyloxy, amino, amido, carboxyl, nitro, mercapto or sulpho;
“heterocycle” is a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S;
“heteroaryl” is a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S with at least one ring of this group being aromatic;
“substituted heteroaryl” is a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S with at least one ring of this group being aromatic and this group being substituted with one or more substituents selected from the group consisting of halogen, alkyl, carbyloxy, carbylmercapto, alkylamino, amido, carboxyl, hydroxyl, nitro, mercapto or sulpho;
“arylalkyl” is the group —R1—Ar wherein R1 is:
a) a branched or unbranched alkyl group having 1-6 carbon atoms,
b) a branched or unbranched alkenyl group having 2-6 carbon atoms,
c) a branched or unbranched alkinyl group having 2-6 carbon atoms;
“Ar” is an aromatic carbocyclic group having from 6-18 carbon atoms and being composed of at least one aromatic ring or of multiple condensed rings at least one of which being aromatic and the group being optionally substituted with one or more substituents selected from the group consisting of halogen, alkyl, hydroxyl, carbylmercapto, alkylamino, carbyloxy, amino, amido, carboxyl, nitro, mercapto or sulpho;
“heteroalkyl” is the group -Rm-L wherein Rm is:
a) a branched or unbranched alkyl group having 1-6 carbon atoms,
b) a branched or unbranched alkenyl group having 2-6 carbon atoms,
c) a branched or unbranched alkinyl group having 2-6 carbon atoms,
d) a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl; and
L is a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S and the group being unsubstituted or substituted with one or more substituents selected from the group consisting of halogen, alkyl, alkoxy, alkylmercapto, alkylamino, amido, carboxyl, hydroxy, nitro, mercapto or sulpho;
“heteroarylalkyl” is the group -Rn-G wherein Rn is
a) a branched or unbranched alkyl group having 1-6 carbon atoms,
b) a branched or unbranched alkenyl group having 2-6 carbon atoms,
c) a branched or unbranched alkinyl group having 2-6 carbon atoms,
d) a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl; and
G is a heterocyclic group having from 4 to 9 carbon atoms and at least one heteroatom selected from the group consisting of N, O or S with at least one ring of which being aromatic and the group being optionally substituted with one or more substituents selected from the group consisting of halogen, alkyl, carbyloxy, carbylmercapto, alkylamino, amido, carboxyl, hydroxyl, nitro, mercapto or sulpho;
“cycloalkyl” is a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms;
“substituted cycloalkyl” is a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms and being substituted by one or more substituents selected from the group consisting of halogen, alkyl, substituted alkyl, carbyloxy, carbylmercapto, aryl, nitro, mercapto or sulpho;
“heterocycloalkyl” is a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms which at least one ring carbon atom of its cyclic structure being replaced with a heteroatom selected from the group consisting of N, O, S or P;
“substituted heterocycloalkyl” is a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms which at least one ring carbon atom of its cyclic structure being replaced with a heteroatom selected from the group consisting of N, O, S or P and the group is containing one or more substituents selected from the group consisting of halogen, alkyl, substituted alkyl, carbyloxy, carbylmercapto, aryl, nitro, mercapto or sulpho;
“cycloalkyl alkyl” is the group -Ro-J where Ro is:
a) a branched or unbranched alkyl group having 1-6 carbon atoms,
b) a branched or unbranched alkenyl group having 2-6 carbon atoms,
c) a branched or unbranched alkinyl group having 2-6 carbon atoms,
d) a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl, whereas these generic substituent group have meanings being identical with definitions of the corresponding groups as defined in this legend; and
J is:
a) a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms; and
b) a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms which contains one or more substituents selected from the group consisting of halogen, alkyl, substituted alkyl, carbyloxy, carbylmercapto, aryl, nitro, mercapto or sulpho;
“heterocycloalkylalkyl” is the group -RpV where Rp is:
a) a branched or unbranched alkyl group having 1-6 carbon atoms,
b) a branched or unbranched alkenyl group having 2-6 carbon atoms,
c) a branched or unbranched alkinyl group having 2-6 carbon atoms, and
d) a branched or unbranched alkyl, alkenyl or alkinyl group having 1-6 carbon atoms and being substituted by one or more substituents selected from the group consisting of hydroxyl, mercapto, carbylmercapto, halogen, carbyloxy, amino, amido, carboxyl, cycloalkyl, sulpho or acyl; and
V is:
a) a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms with at least one being replaced with a heteroatom selected from the group consisting of N, O, S or P; and
b) a monocyclic or polycyclic alkyl group containing 3 to 15 carbon atoms with at least one being replaced with a heteroatom selected from the group consisting of N, O, S or P and the group contains one or more substituents selected from the group consisting of halogen, alkyl, substituted alkyl, carbyloxy, carbylmercapto, aryl, nitro, mercapto or sulpho.
2. Substituted nitrogen heterocyclic derivatives of the formula I according to claim 1 , wherein R6═H.
3. Substituted nitrogen heterocyclic derivatives of the formula I according to claim 1 , wherein R2═H and R6, R8 and R9 are as defined in claim 1 .
4. Substituted nitrogen heterocyclic derivatives according to claim 1 , of the formula Ib,

wherein R2, R6, R8 and R9 are as defined in claim 1 .
5. Substituted nitrogen heterocyclic derivatives of the formula Ib according to claim 4 , wherein R6═H and R2, R8 and R9 are defined as in claim 1 .
6. Substituted nitrogen heterocyclic derivatives of the formula Ib according to claim 4 , wherein R2═H and R6, R8 and R9 are as defined in claim 1 .
7. Substituted nitrogen heterocyclic derivatives of the formula I according to claim 1 , selected from the group consisting of
a) 2-(l-hydroxymethylpropylamino)-6-benzylamino-8-Q-9-isopropylpurine;
b) 2-(2-aminopropylamino)-6-benzylamino-8-Q-9-isopropylpurine;
c) 2-(2-hydroxypropylamino)-6-benzylamino-8-Q-9-isopropylpurine;
d) 2-diethylamino-6-(4-methoxybenzylamino)-8-Q-9-isopropylpurine;
e) 2-(2-hydroxypropylamino)-6-(3-chloro-4-carboxyanilino)-8-Q-9-isopropylpurine;
f) 2-(R)-(2-hydroxypyrrolidin-1-yl)-6-benzylamino-8-Q-9-isopropylpurine;
g) 2-(R)-(l-isopropyl-2-hydroxyethylamino)-6(3-chloro-4-carboxyanilino)-8-Q-9-isopropylpurine;
h) 2-(R)-(1-isopropyl-2-hydroxyethylamino)-6-benzylamino-8-Q-9-isopropylpurine;
i) 2-(R)-(1-isopropyl-2-hydroxyethylamino)-6-(3-chloroanilino)-8-Q-9-isopropylpurine;
j) 2-alkylamino-6-dimethylamino-8-Q -9-(R)-(2-phosphonomethoxypropyl)purine;
k) 2-alkylamino-6-diethylamino-8-Q-9(R)-(2-phosphonomethoxypropyl)purine;
l) 2-alkylamino-6 -butylamino-8-Q-9-(R)-(2-phosphonomethoxypropyl) purine;
m) 2-alkylamino-6-(2-butylamino)-8-Q-9-(R)-(2-phosphonomethoxypropyl)purine;
n) 2-alkylamino6-cyclopropylamino-8-Q-9-(R)-(2-phosphonomethoxypropyl)purine;
o) 2-amino-6-cyclohexylamino-8-Q-9-(R)-(2-phosphonomethoxypropyl)purine;
p) 2-alkylamino-6-(pyrrolidin-1-yl)-8-Q-9-(R)-(2-phosphonomethoxypropyl)purine;
q) 2-alkylamino-6-(morpholin-1-yl)-8-Q-9-(R)-(2-phosphonomethoxypropyl)purine;
wherein Q is chloro, hydroxy, bromo, fluoro, amino, amido, carboxy, cyano, methylamino, thio, methylthio, ω-hydroxyalkylamino, ω-hydroxyalkyloxy, ω-carboxyalkylamino, ω-aminoalkylamino, ω-fosfonoalkylamino, ω-fosfonoalkyloxy, or propinyl.
8. Substituted nitrogen heterocyclic derivatives of the formula I according to claim 1 , selected from the group consisting of:
2-(2-hydroxypropylamino)-6-(3-chloroanilino)-8-S-9-isopropylpurine;
wherein S is chloro, hydroxy, bromo, amino, C1-C6 alkyl, methyl, ethyl, propyl, isopropyl, vinyl, allyl, propargyl.
9. A method for preparing substituted nitrogen heterocyclic derivatives of formula I according to claim 1 , wherein R2, R6, R8 and R9 are as defined in claim 1 , A is a group of formula

and Z is N, characterized in that a trisubstituted derivative of formula XI,

wherein R2, R6 and R9 are as defined in claim 1 , is brominated by using a brominating system selected from a group consisting of bromine/acetic acid, bromine/chloroform, bromine/acetate buffer, bromine/water, N-bromosuccinimide/dimethylformamide and bromoacetamide/dimethylformamide to obtain a derivative of formula XIa,
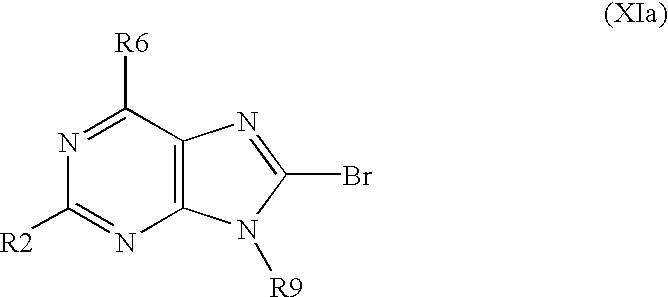
wherein R2, R6 and R9 are as defined in claim 1 , and a bromine atom in position 8 of the derivative of formula XIa is then optionally subjected to a substitution in order to replace said bromine atom by another substituent R8 as defined in claim 1 .
10. A method for preparing substituted nitrogen heterocyclic derivatives of the formula I according to claim 1 , wherein R2, R6, R8 and R9 are as defined in claim 1 , A is a group of formula

and Z is N, characterized in that a trisubstituted derivative of formula XIb,

wherein R6 and R9 are as defined in claim 1 , is brominated by using a brominating system selected from a group consisting of bromine/acetic acid, bromine/chloroform, bromine/acetate buffer, bromine/water, N-bromosuccinimide/dimethylformamide and bromoacetamide/dimethylformamide to obtain a derivative of formula XIc,

wherein R6 and R9 are as defined in claim 1 , and a chlorine atom in position 2 and a bromine atom in position 8 of the derivative of formula XIc are optionally, either progressively or simultaneously, subjected to a nucleophilic substitution in order to replace said chlorine and bromine atoms by other substituents R2 and R8 as defined in claim 1 .
11. A method for preparing substituted nitrogen heterocyclic derivatives of the formula I according to claim 1 , wherein R2, R6 and R8 are as defined in claim 1 , R9 is alkyl as defined in claim 1 , A is a group of formula

and Z is N, characterized in that, a trisubstituted derivative of formula XIIa,

wherein R6 is as defined in claim 1 ,
and said derivative of formula XIIa is alkylated in position 9 by using an alkylating agent in a system selected from a group consisting of K2CO3/ dimethylformamide,
Cs2CO3/dimethylformamide, t-BuOK/dimethylformamide, t-BuOK/dimethylsulphoxide and NaH/dimethylformamide or under conditions of Mitsunobu reaction to obtain a derivative of formula XIIb

wherein R6 and alkyl are as defined in claim 1 , and chlorine atoms in positions 2 and 8 of the derivative of formula XIIb are optionally, either progressively or simultaneously, subjected to a substitution in order to replace said chlorine atoms by other substituents R2 and R8, as defined in claim 1 .
12. A method for preparing substituted nitrogen heterocyclic derivatives of the formula I according to claim 1 , wherein R2, R6 and R8 are as defined in claim 1 , R9 is alkyl as defined in claim 1 , A is a group of formula

and Z is N, characterized in that a trisubstituted derivative of formula XIIa,
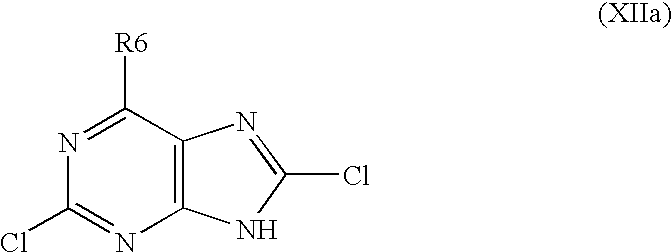
wherein R6 is as defined in claim 1 , is protected by reacting with 2-dihydropyrane/H+ to obtain a derivative of formula XIIc,

wherein R6 is as defined in claim 1 , and the group R6 and/or a chlorine atom in position 2 and/or a chlorine atom in position 8 are optionally converted to other substituents R2, R6 and R8, as defined in claim 1 , and in such optionally modified derivative of formula XIIc, the 2-tetrahydropyranyl group is split off to obtain a corresponding derivative of formula XIIc wherein R9 is hydrogen, and so obtained derivative of formula XIIc is then alkylated in position 9 by using an alkylating agent in a system selected from a group consisting of K2CO3/dimethylformamide, Cs2CO3/dimethylformamide, t-BuOK/dimethylformamide, t-BuOK/dimethylsulphoxide and NaH/dimethylformamide or under conditions of Mitsunobu reaction to obtain a tetrasubstituted derivative of formula XIIc, wherein R2,R6,R8 and R9 are as defined in claim 1 .
13. A method for preparing substituted nitrogen heterocyclic derivatives of the formula I according to claim 1 , wherein R2, R6 and R8 are as defined in claim 1 , R9 is alkyl as defined in claim 1 , A is a group

and Z is N, characterized in that, 2,6,8-trichloropurine is subjected to a nucleophilic substitution in position 6 in order to replace a chlorine atom in position 6 by another substituent R6 as defined in claim 1 to obtain a derivative of formula XIIa,

wherein R6 is as defined in claim 1 ,
and said derivative of formula XIIa is then alkylated in position 9 by using an alkylating agent in a system selected from a group consisting of K2CO3/dimethylformamide, Cs2CO3/dimethylformamide, t-BuOK/dimethylformamide, t-BuOK/dimethylsulphoxide and NaH/dimethylformamide or under conditions of Mitsunobu reaction to obtain a derivative of formula XIIb,

wherein R6 and alkyl are as defined in claim 1 , and chlorine atoms in positions 2 and 8 of the derivative of formula XIIb are optionally, either progressively or simultaneously, subjected to a nucleophilic substitution in order to replace said chlorine atoms by other substituents R2 and R8 as defined in claim 1 .
14. A method for preparing substituted nitrogen heterocyclic derivatives of the formula I according to claim 1 , wherein R2 is hydrogen, R6 and R8 are as defined in claim 1 , R9 is alkyl as defined in claim 1 , A is a group of formula
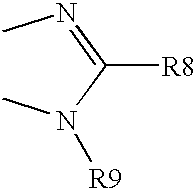
and Z is N, characterized in that 6,8-dichloropurine is subjected to a nucleophilic substitution in position 6 in order to replace a chlorine atom in position 6 by another substituent R6 to obtain a derivative of formula XVI,

wherein R6 is as defined in claim 1 ,
and said derivative of formula XVI is alkylated in position 9 by using an alkylating agent in a system selected from a group consisting of K2CO3/dimethylformamide,
Cs2CO3/dimethylformamide, t-BuOK/dimethylformamide, t-BuOK/dimethylsulphoxide and NaH/dimethylformamide or under conditions of Mitsunobu reaction to obtain a derivative of formula XVIa,

wherein R6 and alkyl are as defined in claim 1 , and a chlorine atom in position 8 of the derivative of formula XVIa is optionally subjected to a substitution in order to replace said chlorine atom by another substituent R8 as defined in claim 1 .
15. A method for preparing substituted nitrogen heterocyclic derivatives of the formula I according to claim 1 , wherein R2, R6 and R8 are as defined in claim 1 , R9 is alkyl as defined in claim 1 , A is a group of formula
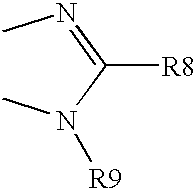
and Z is N, characterized in that 2,6-dichloropurine is subjected to a nucleophilic substitution in position 6 in order to replace a chlorine atom in position 6 by another substituent R6, to obtain a derivative of formula XVII,

wherein R6 is as defined in claim 1 ,
and said derivative of formula XVII is alkylated in position 9 by using either methyl acrylate or acrylonitrile or oxirane to obtain a derivative of formula XVIIa

wherein R6 and alkyl are as defined in claim 1 , and said derivative of formula XVIIa is then brominated in position 8 by using a brominating system selected from a group consisting of bromine/acetic acid, bromine/chloroform, bromine/acetate buffer, bromine/water, N-bromosuccinimide/dimethylformamide and bromoacetamide/dimethylformamide to obtain a derivative of formula XVIIb,
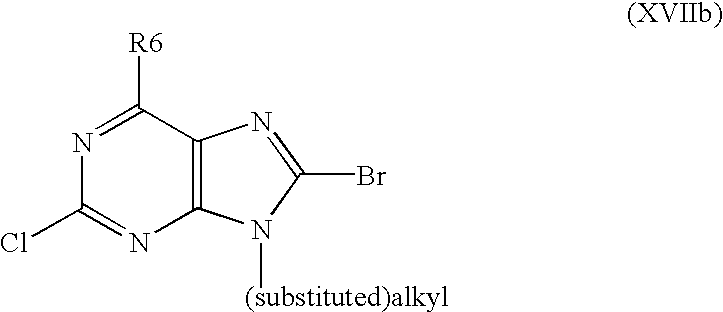
wherein R6 and alkyl are as defined in claim 1;
and said derivative of formula XVIIb is then optionally, either progressively or simultaneously, subjected to a nucleophilic substitution in positions 2 and 8 in order to replace a chlorine atom in position 2 and a bromine atom in position 8 by other substituents R2 and R8 as defined in claim 1 .
16. A method for preparing substituted nitrogen heterocyclic derivatives of the formula I according to claim 1 , wherein R6 is a halogen or hydrogen, R2, R8 and R9 are as defined in claim 1 , A is a group of formula

and Z is N, characterized in that a derivative of formula XVIII,

wherein R2, R8 and R9 are defined in claim 1 , is halogenated via diazotation by using amylnitrite/CH2Br2 or amylnitratite/CHI3, and so an obtained derivative where R6 is halogen is optionally hydrogenolyzed by using H2/Pd catalyst to obtain a corresponding derivative where R6 is hydrogen.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/358,674 US20030191086A1 (en) | 1999-01-26 | 2003-02-05 | Substituted nitrogen heterocyclic derivatives and pharmaceutical use thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CZ1999273A CZ27399A3 (en) | 1999-01-26 | 1999-01-26 | Substituted nitrogen heterocyclic derivatives process of their preparation, the derivatives employed as medicaments, pharmaceutical composition and a compound pharmaceutical preparation in which these derivatives are comprised as well as use of these derivatives for preparing medicaments |
| CZPV273-99 | 1999-01-26 | ||
| US09/889,176 US6552192B1 (en) | 1999-01-26 | 2000-01-25 | Substituted nitrogen heterocyclic derivatives and pharmaceutical use thereof |
| US10/358,674 US20030191086A1 (en) | 1999-01-26 | 2003-02-05 | Substituted nitrogen heterocyclic derivatives and pharmaceutical use thereof |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/889,176 Division US6552192B1 (en) | 1999-01-26 | 2000-01-25 | Substituted nitrogen heterocyclic derivatives and pharmaceutical use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| US20030191086A1 true US20030191086A1 (en) | 2003-10-09 |
Family
ID=5461478
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/889,176 Expired - Fee Related US6552192B1 (en) | 1999-01-26 | 2000-01-25 | Substituted nitrogen heterocyclic derivatives and pharmaceutical use thereof |
| US10/358,674 Abandoned US20030191086A1 (en) | 1999-01-26 | 2003-02-05 | Substituted nitrogen heterocyclic derivatives and pharmaceutical use thereof |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/889,176 Expired - Fee Related US6552192B1 (en) | 1999-01-26 | 2000-01-25 | Substituted nitrogen heterocyclic derivatives and pharmaceutical use thereof |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US6552192B1 (en) |
| EP (1) | EP1147108B1 (en) |
| AT (1) | ATE247115T1 (en) |
| AU (1) | AU2276100A (en) |
| CZ (1) | CZ27399A3 (en) |
| DE (1) | DE60004475D1 (en) |
| WO (1) | WO2000043394A1 (en) |
Cited By (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060052403A1 (en) * | 2002-09-27 | 2006-03-09 | Yoshiaki Isobe | Novel adenine compound and use thereof |
| US20060116376A1 (en) * | 2004-11-29 | 2006-06-01 | Warner-Lambert Company Llc | Therapeutic pyrazolo[3,4-B]pyridines and indazoles |
| US7157465B2 (en) | 2001-04-17 | 2007-01-02 | Dainippon Simitomo Pharma Co., Ltd. | Adenine derivatives |
| US20070225275A1 (en) * | 2006-03-21 | 2007-09-27 | Allison Brett D | Tetrahydro-pyrimidoazepines as modulators of TRPV1 |
| US20070225303A1 (en) * | 2004-03-26 | 2007-09-27 | Haruhisa Ogita | 8-Oxoadenine Compound |
| US20080269240A1 (en) * | 2005-09-22 | 2008-10-30 | Dainippon Sumitomo Pharma Co., Ltd. a corporation of Japan | Novel Adenine Compound |
| US20080300244A1 (en) * | 2006-12-14 | 2008-12-04 | Astrazeneca Ab | Novel compounds |
| US20090082332A1 (en) * | 2005-09-22 | 2009-03-26 | Philip Abbot | Purine derivatives for the treatment of viral or allergic diseases and cancers |
| US20090099216A1 (en) * | 2005-09-22 | 2009-04-16 | Astrazeneca Aktiebolag A Corporation Of Sweden | Novel adenine compound |
| US20090118263A1 (en) * | 2005-09-22 | 2009-05-07 | Dainippon Sumitomo Pharma Co., Ltd. | Novel Adenine Compound |
| US20090143400A1 (en) * | 2005-09-16 | 2009-06-04 | Mcinally Thomas | Purine derivatives having immuno-modulating properties |
| US20090156598A1 (en) * | 2007-12-17 | 2009-06-18 | Lebsack Alec D | Imidazolopyrimidine modulators of TRPV1 |
| US20090156599A1 (en) * | 2007-12-17 | 2009-06-18 | Bryan James Branstetter | Imidazolo-, oxazolo-, and thiazolopyrimidine modulators of TRPV1 |
| US20090192153A1 (en) * | 2005-09-22 | 2009-07-30 | Dainippon Sumitomo Pharma Co., Ltd. a corporation of Japan | Novel adenine compound |
| US20090202626A1 (en) * | 2008-02-07 | 2009-08-13 | Carson Dennis A | Treatment of bladder diseases with a tlr7 activator |
| US20090209524A1 (en) * | 2007-11-22 | 2009-08-20 | Astrazeneca Ab | Novel Compounds |
| US20090324551A1 (en) * | 2005-08-22 | 2009-12-31 | The Regents Of The University Of California Office Of Technology Transfer | Tlr agonists |
| US20100087443A1 (en) * | 2007-03-19 | 2010-04-08 | Roger Victor Bonnert | 9-substituted-8-oxo-adenine compounds as toll-like receptor (tlr7) modulators |
| US20100093998A1 (en) * | 2007-03-20 | 2010-04-15 | Dainippon Sumitomo Pharma Co., Ltd. | Novel adenine compound |
| US20100099870A1 (en) * | 2007-03-20 | 2010-04-22 | Dainippon Sumitomo Phama Co., Ltd | Novel adenine compound |
| US20100130491A1 (en) * | 2007-03-19 | 2010-05-27 | Roger Victor Bonnert | 9-substituted-8-oxo-adenine compounds as toll-like receptor (tlr7 ) modulators |
| US20100210598A1 (en) * | 2009-02-11 | 2010-08-19 | Regents Of The University Of California, San Diego | Toll-like receptor modulators and treatment of diseases |
| US20100240623A1 (en) * | 2006-07-05 | 2010-09-23 | Anthony Cook | 8-oxoadenine derivatives acting as modulators of tlr7 |
| US20100280001A1 (en) * | 2007-05-08 | 2010-11-04 | Roger Victor Bonnert | Imidazoquinolines with immuno-modulating properties |
| US20100298364A1 (en) * | 2009-05-21 | 2010-11-25 | Astrazeneca Ab | salts 756 |
| US20110054168A1 (en) * | 2008-01-17 | 2011-03-03 | Ayumu Kurimoto | Method for preparing adenine compound |
| US20110098294A1 (en) * | 2006-05-31 | 2011-04-28 | Carson Dennis A | Purine analogs |
| US20110136801A1 (en) * | 2009-12-03 | 2011-06-09 | Dainippon Sumitomo Pharma Co. Ltd. | Novel Compounds |
| US8012964B2 (en) | 2004-03-26 | 2011-09-06 | Dainippon Sumitomo Pharma Co., Ltd. | 9-substituted 8-oxoadenine compound |
| RU2461559C2 (en) * | 2007-04-04 | 2012-09-20 | Сайкласел Лимитед | Compounds |
| US8357374B2 (en) | 2007-02-07 | 2013-01-22 | The Regents Of The University Of California | Conjugates of synthetic TLR agonists and uses therefor |
| US8563575B2 (en) | 2010-06-24 | 2013-10-22 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds |
| US8673907B2 (en) | 2007-12-17 | 2014-03-18 | Astrazeneca Ab | Pharmaceutically acceptable salts of methyl (3-{ [[3-(6-amino- 2-butoxy-8-oxo-7,8-dihydro-9H-purin-9-yl) propyl] (3-morpholin-4-ylpropyl) amino] methyl }phenyl) acetate and their use in therapy |
| US8865896B2 (en) | 2008-01-17 | 2014-10-21 | Astrazeneca Aktiebolag | Method for preparing adenine compound |
| US8895570B2 (en) | 2010-12-17 | 2014-11-25 | Astrazeneca Ab | Purine derivatives |
| US9045472B2 (en) | 2010-12-16 | 2015-06-02 | Astrazeneca Ab | Imidazoquinoline compounds |
| US9050319B2 (en) | 2010-04-30 | 2015-06-09 | Telormedix, Sa | Phospholipid drug analogs |
| US9066940B2 (en) | 2009-02-06 | 2015-06-30 | Telormedix, Sa | Pharmaceutical compositions comprising imidazoquinolin(amines) and derivatives thereof suitable for local administration |
| US9120788B2 (en) | 2013-02-19 | 2015-09-01 | Pfizer Inc. | Azabenzimidazole compounds |
| US9173936B2 (en) | 2010-04-30 | 2015-11-03 | Telormedix Sa | Phospholipid drug analogs |
| WO2016057370A1 (en) * | 2014-10-06 | 2016-04-14 | Signal Pharmaceuticals, Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| US9376398B2 (en) | 2012-05-18 | 2016-06-28 | Sumitomo Dainippon Pharma Co., Ltd | Carboxylic acid compounds |
| US9428512B2 (en) | 2012-11-20 | 2016-08-30 | Glaxosmithkline Llc | Compounds |
| US9533978B2 (en) | 2009-05-21 | 2017-01-03 | Sumitomo Dainippon Pharma Co., Ltd | Pyrimidine derivatives and their use in the treatment of cancer and further diseases |
| US9540383B2 (en) | 2012-11-20 | 2017-01-10 | Glaxosmithkline Llc | Pyrrolopyrimidines as therapeutic agents for the treatment of diseases |
| US9550785B2 (en) | 2012-11-20 | 2017-01-24 | Glaxosmithkline Llc | Pyrrolopyrimidines as therapeutic agents for the treatment of diseases |
| US9555036B2 (en) | 2012-08-24 | 2017-01-31 | Glaxosmithkline Llc | Pyrazolopyrimidine compounds |
| US9598421B2 (en) | 2014-08-06 | 2017-03-21 | Pfizer Inc. | Imidazopyridazine compounds |
| US9877968B2 (en) | 2008-08-11 | 2018-01-30 | Glaxosmithkline Llc | 6-amino-purin-8-one compounds |
| US10112946B2 (en) | 2011-07-22 | 2018-10-30 | Glaxosmithkline Llc | Composition |
| US10131669B2 (en) | 2014-07-24 | 2018-11-20 | Pfizer Inc. | Pyrazolopyrimidine compounds |
| US10543214B2 (en) | 2017-10-04 | 2020-01-28 | Celgene Corporation | Compositions and methods of use of cis-4-[2-{[(3S,4R)-3-fluorooxan-4-yl]amino}-8-(2,4,6-trichloroanilino)-9H-purin-9-yl]-1-methylcyclohexane-1-carboxamide |
| US10590136B2 (en) | 2017-10-04 | 2020-03-17 | Celgene Corporation | Processes for the preparation of cis-4-[2-{[(3S,4R)-3-fluorooxan-4-yl]amino}-8-(2,4,6-trichloroanilino)-9H-purin-9-yl]-1-methylcyclohexane-1-carboxamide |
| US11697851B2 (en) | 2016-05-24 | 2023-07-11 | The Regents Of The University Of California | Early ovarian cancer detection diagnostic test based on mRNA isoforms |
Families Citing this family (138)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6992188B1 (en) * | 1995-12-08 | 2006-01-31 | Pfizer, Inc. | Substituted heterocyclic derivatives |
| US6255485B1 (en) * | 1997-08-07 | 2001-07-03 | The Regents Of The University Of California | Purine inhibitors of protein kinases, G proteins and polymerases |
| DE60008988T2 (en) * | 1999-09-17 | 2005-04-07 | Pna Diagnostics Aps | Synthesis of peptide nucleic acids |
| ATE423120T1 (en) * | 2000-06-26 | 2009-03-15 | Pfizer Prod Inc | PYRROLOÄ2,3-DÜPYRIMIDINE COMPOUNDS AS IMMUNOSUPPRESSIVE ACTIVES |
| MXPA03009925A (en) * | 2001-04-30 | 2004-06-30 | Bayer Pharmaceuticals Corp | Novel 4-amino-5,6-substituted thiopheno[2,3-d]pyrimidines. |
| DE60205376T2 (en) * | 2001-06-27 | 2006-04-06 | Cyclacel Ltd. | 2,6,9-SUBSTITUTED PURINE DERIVATIVES AND THEIR USE IN THE TREATMENT OF PROLIFERATIVE DISEASES |
| WO2003037860A2 (en) | 2001-10-30 | 2003-05-08 | Conforma Therapeutics Corporation | Purine analogs having hsp90-inhibiting activity |
| ES2314224T3 (en) | 2002-03-07 | 2009-03-16 | F. Hoffmann-La Roche Ag | BIRCINAL PYRIMIDINE AND PYRIDINE INHIBITORS OF P38 QUINASA. |
| MXPA04008797A (en) | 2002-03-13 | 2004-11-26 | Janssen Pharmaceutica Nv | Inhibitors of histone deacetylase. |
| GB0219052D0 (en) * | 2002-08-15 | 2002-09-25 | Cyclacel Ltd | New puring derivatives |
| DE10238722A1 (en) | 2002-08-23 | 2004-03-11 | Bayer Ag | Improving attention, concentration, cognition, learning and/or memory performance, using selective phosphodiesterase 9A inhibitors, preferably 4H-pyrazolo-(3,4-d)-pyrimidin-4-one derivatives |
| GB0225873D0 (en) * | 2002-11-06 | 2002-12-11 | Cyclacel Ltd | Combination |
| MXPA05007485A (en) | 2003-01-14 | 2006-01-30 | Arena Pharm Inc | 1,2,3-trisubstituted aryl and heteroaryl derivatives as modulators of metabolism and the prpphylaxis and treatment of disorders related thereto such as diabetes and hyperglycemia. |
| US20060128956A1 (en) * | 2003-04-21 | 2006-06-15 | Ustav Organicke Chemie A Biochemie Akademie Ved Ceske Republiky | (Purin-6-yl) amino acid and production method thereof |
| ES2351624T3 (en) * | 2003-05-06 | 2011-02-08 | Ústav Experimentálni Botaniky Av Cr, V.V.I. (Institute Of Experimental Botany Academy Of Sciences Of The Czech Republic, Pro) | PIRAZOLO [4,3-D] PYRIMIDINS, PROCEDURE FOR PREPARATION AND USE. |
| US7429596B2 (en) * | 2003-06-20 | 2008-09-30 | The Regents Of The University Of California | 1H-pyrrolo [2,3-D] pyrimidine derivatives and methods of use thereof |
| RS20060018A (en) * | 2003-07-14 | 2007-12-31 | Arena Pharmaceuticals Inc., | Fused-aryl and heteroaryl derivatives as modulators of metabolism and the prophylaxis and treatment of disorders related thereto |
| MXPA06002997A (en) | 2003-09-18 | 2007-02-08 | Conforma Therapeutics Corp | Novel heterocyclic compounds as hsp90-inhibitors. |
| CN100475815C (en) * | 2003-09-25 | 2009-04-08 | 詹森药业有限公司 | HIV replication inhibiting purine derivatives |
| CA2569016C (en) * | 2004-06-02 | 2012-11-27 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compound |
| KR101261305B1 (en) | 2004-07-28 | 2013-05-08 | 얀센 파마슈티카 엔.브이. | Substituted indolyl alkyl amino derivatives as novel inhibitors of histone deacetylase |
| US20080125404A1 (en) * | 2004-08-27 | 2008-05-29 | Cyclacel Limited | Purine and Pyrimidine Cdk Inhitbitors and Their use for The Treatment of Autoimmune Diseases |
| JPWO2006051951A1 (en) * | 2004-11-12 | 2008-05-29 | 一仁 富澤 | Diabetes therapeutic agent containing Cdk5 inhibitor |
| EP1831225A2 (en) * | 2004-11-19 | 2007-09-12 | The Regents of the University of California | Anti-inflammatory pyrazolopyrimidines |
| US7759342B2 (en) * | 2005-01-13 | 2010-07-20 | Signal Pharmaceuticals, Llc | Methods of treatment and prevention using haloaryl substituted aminopurines |
| US7723340B2 (en) * | 2005-01-13 | 2010-05-25 | Signal Pharmaceuticals, Llc | Haloaryl substituted aminopurines, compositions thereof, and methods of treatment therewith |
| US7521446B2 (en) | 2005-01-13 | 2009-04-21 | Signal Pharmaceuticals, Llc | Haloaryl substituted aminopurines, compositions thereof, and methods of treatment therewith |
| EP1846408B1 (en) | 2005-01-14 | 2013-03-20 | Janssen Pharmaceutica NV | 5-membered annelated heterocyclic pyrimidines as kinase inhibitors |
| WO2006074984A1 (en) * | 2005-01-14 | 2006-07-20 | Janssen Pharmaceutica N.V. | Pyrazolopyrimidines as cell cycle kinase inhibitors |
| AU2006244195B2 (en) | 2005-05-05 | 2012-03-15 | Ardea Biosciences, Inc. | Diaryl-purine, azapurines and -deazapurines as non-nucleoside reverse transcriptase inhibitors for treatment of HIV |
| WO2007002587A2 (en) * | 2005-06-24 | 2007-01-04 | The Brigham And Women's Hospital, Inc. | Inhibitors of epstein barr virus nuclear antigen 1 |
| US7932257B2 (en) | 2005-07-22 | 2011-04-26 | Sunesis Pharmaceuticals, Inc. | Substituted pyrazolo[4,3-d]pyrimidines as aurora kinase inhibitors |
| WO2007059219A1 (en) | 2005-11-15 | 2007-05-24 | Vertex Pharmaceuticals Incorporated | Azaindazoles useful as inhibitors of kinases |
| UA95788C2 (en) | 2005-12-15 | 2011-09-12 | Ф. Хоффманн-Ля Рош Аг | Fused pyrrole derivatives |
| WO2007082874A1 (en) | 2006-01-19 | 2007-07-26 | Janssen Pharmaceutica N.V. | Pyridine and pyrimidine derivatives as inhibitors of histone deacetylase |
| JP2009528989A (en) | 2006-02-17 | 2009-08-13 | ファイザー・リミテッド | 3-Deazapurine derivatives as TLR7 modulators |
| WO2007114926A2 (en) | 2006-04-04 | 2007-10-11 | The Regents Of The University Of California | Kinase antagonists |
| PE20080695A1 (en) | 2006-04-27 | 2008-06-28 | Banyu Pharma Co Ltd | DIHYDROPIRAZOLOPYRIMIDINONE DERIVATIVES AS KINASE WEEL INHIBITORS |
| US7851468B2 (en) * | 2006-05-15 | 2010-12-14 | Cephalon, Inc. | Substituted pyrazolo[3,4-d]pyrimidines |
| DE102006029074A1 (en) * | 2006-06-22 | 2007-12-27 | Friedrich-Schiller-Universität Jena | 4-Amino-3-arylamino-6-arylpyrazolo [3,4-d] pyrimidine derivatives, process for their preparation and their use as antiviral agents |
| EP2617423A1 (en) * | 2006-10-19 | 2013-07-24 | Genzyme Corporation | Purine derivatives for the treatment of cystic diseases |
| AR065372A1 (en) * | 2007-02-19 | 2009-06-03 | Smithkline Beecham Corp | PURINA DERIVATIVES |
| GB0708507D0 (en) * | 2007-05-02 | 2007-06-13 | Queen Mary & Westfield College | Substituted phosphonates and their use |
| GB2467670B (en) * | 2007-10-04 | 2012-08-01 | Intellikine Inc | Chemical entities and therapeutic uses thereof |
| JP5498392B2 (en) | 2007-11-30 | 2014-05-21 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | 1,5-Dihydro-pyrazolo [3,4-D] pyrimidin-4-one derivatives and their use as PDE9A modulators for the treatment of CNS disorders |
| ES2389752T3 (en) | 2007-12-14 | 2012-10-31 | Ardea Biosciences, Inc. | Reverse Transcriptase Inhibitors |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| MX2010007419A (en) | 2008-01-04 | 2010-11-12 | Intellikine Inc | CERTAIN CHEMICAL ENTITIES, COMPOSITIONS AND METHODS. |
| EP2252293B1 (en) * | 2008-03-14 | 2018-06-27 | Intellikine, LLC | Kinase inhibitors and methods of use |
| US8993580B2 (en) * | 2008-03-14 | 2015-03-31 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| UA105362C2 (en) | 2008-04-02 | 2014-05-12 | Бьорингер Ингельхайм Интернациональ Гмбх | 1-heterocyclyl-1, 5-dihydro-pyrazolo [3, 4-d] pyrimidin-4-one derivatives and their use as pde9a modulators |
| US20110224223A1 (en) | 2008-07-08 | 2011-09-15 | The Regents Of The University Of California, A California Corporation | MTOR Modulators and Uses Thereof |
| BRPI0915231A2 (en) * | 2008-07-08 | 2018-06-12 | Intellikine Inc | kinase inhibitor compounds and methods of use |
| BRPI0917013A2 (en) | 2008-08-11 | 2016-02-16 | Glaxosmithkline Llc | methods for treating allergic diseases and other inflammatory conditions, and for treating or preventing disease, compound, pharmaceutical composition, and use of a compound |
| US8802684B2 (en) | 2008-08-11 | 2014-08-12 | Glaxosmithkline Llc | Adenine derivatives |
| EP2326646B1 (en) | 2008-08-11 | 2013-07-31 | GlaxoSmithKline LLC | Purine derivatives for use in the treatment of allergic, inflammatory and infectious diseases |
| KR101335843B1 (en) | 2008-08-20 | 2013-12-02 | 조에티스 엘엘씨 | Pyrrolo [2,3-D] pyrimidine compound |
| KR20110063447A (en) | 2008-09-08 | 2011-06-10 | 베링거 인겔하임 인터내셔날 게엠베하 | Pyrazolopyrimidines and Their Uses for the Treatment of CNS Disorders |
| CA2738429C (en) | 2008-09-26 | 2016-10-25 | Intellikine, Inc. | Heterocyclic kinase inhibitors |
| EP2358720B1 (en) | 2008-10-16 | 2016-03-02 | The Regents of The University of California | Fused ring heteroaryl kinase inhibitors |
| US8476431B2 (en) | 2008-11-03 | 2013-07-02 | Itellikine LLC | Benzoxazole kinase inhibitors and methods of use |
| RS53347B (en) | 2008-12-09 | 2014-10-31 | Gilead Sciences, Inc. | TOLL-SIMILAR RECEPTOR MODULATORS |
| PT2414363E (en) | 2009-03-31 | 2014-02-26 | Boehringer Ingelheim Int | 1-heterocyclyl-1,5-dihydro-pyrazolo[3,4-d]pyrimidin-4-one derivatives and their use as pde9a modulators |
| JP5789252B2 (en) | 2009-05-07 | 2015-10-07 | インテリカイン, エルエルシー | Heterocyclic compounds and uses thereof |
| WO2011014239A1 (en) * | 2009-07-27 | 2011-02-03 | Virostatics Srl | Anti-proliferative substituted pyrazolo[3,4-d]pyrimidines derivatives (spp) to inhibit immune activation, virus replication and tumor growth |
| US8846673B2 (en) | 2009-08-11 | 2014-09-30 | Bristol-Myers Squibb Company | Azaindazoles as kinase inhibitors and use thereof |
| CZ302618B6 (en) | 2009-09-10 | 2011-08-03 | Univerzita Palackého | Platinum cyclobutane-1,1-dicarboxylate complexes with N6-benzyladenine derivatives, process of their preparation and use of these complexes as medicaments in antitumor therapy |
| WO2011047384A2 (en) | 2009-10-16 | 2011-04-21 | The Regents Of The University Of California | Methods of inhibiting ire1 |
| JP5694345B2 (en) | 2009-10-22 | 2015-04-01 | ギリアード サイエンシーズ, インコーポレイテッド | Regulators of TOLL-like receptors |
| JP5856980B2 (en) | 2010-01-27 | 2016-02-10 | アリーナ ファーマシューティカルズ, インコーポレイテッド | (R) -2- (7- (4-Cyclopentyl-3- (trifluoromethyl) benzyloxy) -1,2,3,4-tetrahydrocyclopenta [b] indol-3-yl) acetic acid and its salts Process for preparation |
| WO2011098451A1 (en) | 2010-02-10 | 2011-08-18 | Glaxosmithkline Llc | Purine derivatives and their pharmaceutical uses |
| CA2799579A1 (en) | 2010-05-21 | 2011-11-24 | Intellikine, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| EP2603511B1 (en) | 2010-08-12 | 2017-03-15 | Boehringer Ingelheim International GmbH | 6-cycloalkyl-1, 5-dihydro-pyrazolo [3, 4-d] pyrimidin-4-one derivatives and their use as pde9a inhibitors |
| EP3323818A1 (en) | 2010-09-22 | 2018-05-23 | Arena Pharmaceuticals, Inc. | Modulators of the gpr119 receptor and the treatment of disorders related thereto |
| JP2013545749A (en) | 2010-11-10 | 2013-12-26 | インフィニティー ファーマシューティカルズ, インコーポレイテッド | Heterocyclic compounds and uses thereof |
| ES2637113T3 (en) | 2011-01-10 | 2017-10-10 | Infinity Pharmaceuticals, Inc. | Procedures for preparing isoquinolinones and solid forms of isoquinolinones |
| US8809345B2 (en) | 2011-02-15 | 2014-08-19 | Boehringer Ingelheim International Gmbh | 6-cycloalkyl-pyrazolopyrimidinones for the treatment of CNS disorders |
| US9295673B2 (en) | 2011-02-23 | 2016-03-29 | Intellikine Llc | Combination of mTOR inhibitors and P13-kinase inhibitors, and uses thereof |
| AR088218A1 (en) | 2011-07-19 | 2014-05-21 | Infinity Pharmaceuticals Inc | USEFUL HETEROCICLICAL COMPOUNDS AS PI3K INHIBITORS |
| HK1198443A1 (en) | 2011-07-19 | 2015-04-24 | 无限药品股份有限公司 | Heterocyclic compounds and uses thereof |
| MX2014002542A (en) | 2011-08-29 | 2014-07-09 | Infinity Pharmaceuticals Inc | Heterocyclic compounds and uses thereof. |
| CA2846496C (en) | 2011-09-02 | 2020-07-14 | The Regents Of The University Of California | Substituted pyrazolo[3,4-d]pyrimidines and uses thereof |
| PL2766367T3 (en) * | 2011-10-14 | 2022-08-22 | Scandion Oncology A/S | 4-Amino-3-phenylamino-6-phenylpyrazolo[3,4-d]pyrimidine derivatives for the treatment of viral infections, in particular picornavirus infections |
| DE102012004736A1 (en) | 2011-10-20 | 2013-04-25 | Dritte Patentportfolio Beteiligungsgesellschaft Mbh & Co.Kg | New 4-amino-3-phenylamino-6-phenylpyrazolo(3,4-d)pyrimidine derivatives useful as a medicament for prophylactic- or therapeutic treatment of viral infections, preferably picornavirus infections and rhinovirus infections |
| EP3578563B1 (en) | 2011-12-22 | 2021-04-14 | Geron Corporation | Guanine analogs as telomerase substrates and telomere length affectors |
| US8889730B2 (en) | 2012-04-10 | 2014-11-18 | Pfizer Inc. | Indole and indazole compounds that activate AMPK |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| JP2015532287A (en) | 2012-09-26 | 2015-11-09 | ザ・リージエンツ・オブ・ザ・ユニバーシテイー・オブ・カリフオルニア | IRE1 regulation |
| AU2013337717B2 (en) | 2012-11-01 | 2018-10-25 | Infinity Pharmaceuticals, Inc. | Treatment of cancers using PI3 kinase isoform modulators |
| GEP201606600B (en) | 2013-02-22 | 2017-01-10 | Pfizer | Pyrrolo [2, 3 -d]pyrimidine derivatives as inhibitors of janus- related kinases (jak) |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| EP2970177A1 (en) | 2013-03-15 | 2016-01-20 | Pfizer Inc. | Indole compounds that activate ampk |
| WO2014153509A1 (en) | 2013-03-22 | 2014-09-25 | Millennium Pharmaceuticals, Inc. | Combination of catalytic mtorc 1/2 inhibitors and selective inhibitors of aurora a kinase |
| PT3052485T (en) | 2013-10-04 | 2021-10-22 | Infinity Pharmaceuticals Inc | HETEROCYCLIC COMPOUNDS AND THEIR USES |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| MX382033B (en) | 2014-03-19 | 2025-03-13 | Infinity Pharmaceuticals Inc | HETEROCYCLIC COMPOUNDS FOR USE IN THE TREATMENT OF PI3K-GAMMA-MEDIATED DISORDERS. |
| WO2015160975A2 (en) | 2014-04-16 | 2015-10-22 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| JP6522732B2 (en) | 2014-07-11 | 2019-05-29 | ギリアード サイエンシーズ, インコーポレイテッド | Modulators of Toll-like receptors for treating HIV |
| WO2016024185A1 (en) | 2014-08-12 | 2016-02-18 | Pfizer Inc. | Pyrrolo[2,3-d]pyrimidine derivatives useful for inhibiting janus kinase |
| SG11201701520TA (en) | 2014-09-16 | 2017-04-27 | Gilead Sciences Inc | Solid forms of a toll-like receptor modulator |
| WO2016054491A1 (en) | 2014-10-03 | 2016-04-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| CN107454899B (en) | 2014-10-27 | 2020-05-29 | 大学健康网络 | RIPK2 inhibitors and methods of treating cancer therewith |
| CZ306984B6 (en) | 2014-12-15 | 2017-11-01 | Ústav experimentální botaniky AV ČR, v. v. i. | 6,8-Disubstituted-9-(heterocyclyl)purines, preparations containing these derivatives and their use in cosmetic and medical applications |
| PL3242666T3 (en) | 2015-01-06 | 2025-02-17 | Arena Pharmaceuticals, Inc. | Compound for use in treating conditions related to the s1p1 receptor |
| ES2662822T3 (en) * | 2015-03-04 | 2018-04-09 | Scandion Oncology A/S | 4-Amino-3-phenylamino-6-phenylpyrazol [3,4-d] pyrimidine derivatives for use as BCRP inhibitors in therapeutic treatments |
| BR112017027656B1 (en) | 2015-06-22 | 2023-12-05 | Arena Pharmaceuticals, Inc. | CRYSTALLINE HABIT OF SALT-FREE PLATE OF ACID L-ARGININE (R)-2-(7-(4- CYCLOPENTYL-3-(TRIFLUOROMETHYL)BENZYLOXY)- 1,2,3,4-TETRA-HYDROCYCLO-PENTA[B ]INDOL-3- IL)ACETIC, PHARMACEUTICAL COMPOSITION THAT COMPRISES IT, ITS USES AND METHOD OF PREPARATION THEREOF |
| WO2017048702A1 (en) | 2015-09-14 | 2017-03-23 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinone derivatives, process of making, compositions comprising, and methods of using the same |
| CN108473495B (en) | 2015-11-20 | 2022-04-12 | 福马治疗有限公司 | Purinones as ubiquitin-specific protease 1 inhibitors |
| WO2017161116A1 (en) | 2016-03-17 | 2017-09-21 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as pi3k kinase inhibitors |
| WO2017214269A1 (en) | 2016-06-08 | 2017-12-14 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| SG11201811237WA (en) | 2016-06-24 | 2019-01-30 | Infinity Pharmaceuticals Inc | Combination therapies |
| CN115819417A (en) | 2016-09-09 | 2023-03-21 | 因赛特公司 | Pyrazolopyridine derivatives as HPK1 modulators and their use for the treatment of cancer |
| WO2018049214A1 (en) | 2016-09-09 | 2018-03-15 | Incyte Corporation | Pyrazolopyridine derivatives as hpk1 modulators and uses thereof for the treatment of cancer |
| US10280164B2 (en) | 2016-09-09 | 2019-05-07 | Incyte Corporation | Pyrazolopyridone compounds and uses thereof |
| US20180072741A1 (en) | 2016-09-09 | 2018-03-15 | Incyte Corporation | Pyrazolopyrimidine compounds and uses thereof |
| GB201700587D0 (en) | 2017-01-13 | 2017-03-01 | Redag Crop Prot Ltd | Agricultural chemicals |
| WO2018152220A1 (en) | 2017-02-15 | 2018-08-23 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| MX2019009841A (en) | 2017-02-16 | 2020-01-30 | Arena Pharm Inc | Compounds and methods for treatment of primary biliary cholangitis. |
| AU2018281131B2 (en) | 2017-06-08 | 2022-01-20 | Merck Sharp & Dohme Corp. | Pyrazolopyrimidine PDE9 inhibitors |
| US10722495B2 (en) | 2017-09-08 | 2020-07-28 | Incyte Corporation | Cyanoindazole compounds and uses thereof |
| US10752635B2 (en) | 2018-02-20 | 2020-08-25 | Incyte Corporation | Indazole compounds and uses thereof |
| EP3755703B1 (en) | 2018-02-20 | 2022-05-04 | Incyte Corporation | N-(phenyl)-2-(phenyl)pyrimidine-4-carboxamide derivatives and related compounds as hpk1 inhibitors for treating cancer |
| US10745388B2 (en) | 2018-02-20 | 2020-08-18 | Incyte Corporation | Indazole compounds and uses thereof |
| US11299473B2 (en) | 2018-04-13 | 2022-04-12 | Incyte Corporation | Benzimidazole and indole compounds and uses thereof |
| CA3102136A1 (en) | 2018-06-06 | 2019-12-12 | Arena Pharmaceuticals, Inc. | Methods of treating conditions related to the s1p1 receptor |
| US10899755B2 (en) | 2018-08-08 | 2021-01-26 | Incyte Corporation | Benzothiazole compounds and uses thereof |
| WO2020068729A1 (en) | 2018-09-25 | 2020-04-02 | Incyte Corporation | Pyrazolo[4,3-d]pyrimidine compounds as alk2 abd/or fgfr modulators |
| US12522583B2 (en) | 2018-11-07 | 2026-01-13 | Dana-Farber Cancer Institute, Inc. | Benzimidazole derivatives and aza-benzimidazole derivatives as Janus kinase 2 inhibitors and uses thereof |
| EP3877371A4 (en) | 2018-11-07 | 2022-07-27 | Dana-Farber Cancer Institute, Inc. | IMIDAZOPYRIDINE DERIVATIVES AND AZA-IMIDAZOPYRIDINE DERIVATIVES USED AS JANUS KINASE 2 INHIBITORS AND ASSOCIATED USES |
| EP3876939A4 (en) | 2018-11-07 | 2022-08-10 | Dana-Farber Cancer Institute, Inc. | BENZOTHIAZOLE DERIVATIVES AND 7-AZA-BENZOTHIAZOLE DERIVATIVES AS JANUS KINASE-2 INHIBITORS AND USES THEREOF |
| NL2022471B1 (en) | 2019-01-29 | 2020-08-18 | Vationpharma B V | Solid state forms of oclacitinib |
| TWI810456B (en) | 2019-05-22 | 2023-08-01 | 美商基利科學股份有限公司 | Combination of a tlr7 modulating compound and an hiv vaccine |
| BR112022002059A2 (en) | 2019-08-06 | 2022-06-07 | Incyte Corp | Solid forms of an hpk1 inhibitor |
| US11691963B2 (en) | 2020-05-06 | 2023-07-04 | Ajax Therapeutics, Inc. | 6-heteroaryloxy benzimidazoles and azabenzimidazoles as JAK2 inhibitors |
| US12043632B2 (en) | 2020-12-23 | 2024-07-23 | Ajax Therapeutics, Inc. | 6-heteroaryloxy benzimidazoles and azabenzimidazoles as JAK2 inhibitors |
| WO2023086319A1 (en) | 2021-11-09 | 2023-05-19 | Ajax Therapeutics, Inc. | 6-he tero aryloxy benzimidazoles and azabenzimidazoles as jak2 inhibitors |
| US12162881B2 (en) | 2021-11-09 | 2024-12-10 | Ajax Therapeutics, Inc. | Forms and compositions of inhibitors of JAK2 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030187261A1 (en) * | 2000-01-07 | 2003-10-02 | Libor Havlicek | Purine derivatives, process for their preparation and use thereof |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4923872A (en) * | 1986-08-26 | 1990-05-08 | Warner-Lambert Co. | Analogues of pyrrolo[3,2d]pyrimidin-4-ones |
| EP0374096B1 (en) * | 1988-12-14 | 1992-12-02 | Ciba-Geigy Ag | Combination therapy involving 2',3'-dideoxypurine nucleoside and an inhibitor of purine nucleoside phosphorylase, and its compositions |
| US5514688A (en) * | 1990-09-14 | 1996-05-07 | Merrell Dow Pharmaceuticals Inc. | Carbocyclic adenosine analogs useful as immunosuppressants |
| US5236926A (en) * | 1992-02-03 | 1993-08-17 | Warner-Lambert Company | 9-substituted-8-halo or -8-hydroxy-9-deazaguanines as inhibitors or PNP for pharmaceutical compositions |
| US5977061A (en) * | 1995-04-21 | 1999-11-02 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | N6 - substituted nucleotide analagues and their use |
| EP0770080B1 (en) * | 1995-05-12 | 1999-07-14 | Neurogen Corporation | Novel deazapurine derivatives; a new class of crf1 specific ligands |
| AU7296896A (en) * | 1995-11-01 | 1997-05-22 | Novartis Ag | Purine derivatives and processes for their preparation |
| FR2741881B1 (en) * | 1995-12-01 | 1999-07-30 | Centre Nat Rech Scient | NOVEL PURINE DERIVATIVES HAVING IN PARTICULAR ANTI-PROLIFERATIVE PRORIETES AND THEIR BIOLOGICAL APPLICATIONS |
| US5650511A (en) * | 1995-12-12 | 1997-07-22 | Biocryst Pharmaceuticals, Inc. | Process for the preparation of 9-deazaguanine derivatives |
| ES2232871T3 (en) * | 1996-07-03 | 2005-06-01 | Sumitomo Pharmaceuticals Company, Limited | NEW DERIVATIVES OF PURINA. |
| US5866702A (en) * | 1996-08-02 | 1999-02-02 | Cv Therapeutics, Incorporation | Purine inhibitors of cyclin dependent kinase 2 |
| US6255485B1 (en) * | 1997-08-07 | 2001-07-03 | The Regents Of The University Of California | Purine inhibitors of protein kinases, G proteins and polymerases |
| CN1130363C (en) * | 1997-11-12 | 2003-12-10 | 三菱化学株式会社 | Purine derivatives and medicaments containing them as active ingredients |
| DE69817393T2 (en) * | 1997-11-28 | 2004-06-17 | Sumitomo Pharmaceuticals Co., Ltd. | NEW HETEROCYCLIC CONNECTIONS |
| AU1688599A (en) * | 1998-01-05 | 1999-07-26 | Eisai Co. Ltd. | Purine derivatives and adenosine a2 receptor antagonists serving as preventives/remedies for diabetes |
| AU753015B2 (en) * | 1998-03-04 | 2002-10-03 | Cyclacel Limited | Cyclin dependent kinase inhibitor |
| US6630476B2 (en) * | 2000-07-07 | 2003-10-07 | Bristol-Myers Squibb Pharma Company | Pyrrolo [3,4-d] pyrimidines as corticotropin releasing factor (CRF) antagonists |
-
1999
- 1999-01-26 CZ CZ1999273A patent/CZ27399A3/en unknown
-
2000
- 2000-01-25 DE DE60004475T patent/DE60004475D1/en not_active Expired - Lifetime
- 2000-01-25 US US09/889,176 patent/US6552192B1/en not_active Expired - Fee Related
- 2000-01-25 AT AT00901478T patent/ATE247115T1/en not_active IP Right Cessation
- 2000-01-25 EP EP00901478A patent/EP1147108B1/en not_active Expired - Lifetime
- 2000-01-25 AU AU22761/00A patent/AU2276100A/en not_active Abandoned
- 2000-01-25 WO PCT/CZ2000/000002 patent/WO2000043394A1/en not_active Ceased
-
2003
- 2003-02-05 US US10/358,674 patent/US20030191086A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030187261A1 (en) * | 2000-01-07 | 2003-10-02 | Libor Havlicek | Purine derivatives, process for their preparation and use thereof |
Cited By (116)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7157465B2 (en) | 2001-04-17 | 2007-01-02 | Dainippon Simitomo Pharma Co., Ltd. | Adenine derivatives |
| US20070037832A1 (en) * | 2001-04-17 | 2007-02-15 | Dainippon Sumitomo Pharma Co., Ltd. | Novel adenine derivatives |
| US7521454B2 (en) | 2001-04-17 | 2009-04-21 | Dainippon Sumitomo Pharma Co., Ltd. | Adenine derivatives |
| US20100256118A1 (en) * | 2002-09-27 | 2010-10-07 | Dainippon Sumitomo Pharma Co.,Ltd. | Novel adenine compound and use thereof |
| US20060052403A1 (en) * | 2002-09-27 | 2006-03-09 | Yoshiaki Isobe | Novel adenine compound and use thereof |
| US7754728B2 (en) | 2002-09-27 | 2010-07-13 | Dainippon Sumitomo Pharma Co., Ltd. | Adenine compound and use thereof |
| US8148371B2 (en) | 2002-09-27 | 2012-04-03 | Dainippon Sumitomo Pharma Co., Ltd. | Adenine compound and use thereof |
| US8012964B2 (en) | 2004-03-26 | 2011-09-06 | Dainippon Sumitomo Pharma Co., Ltd. | 9-substituted 8-oxoadenine compound |
| US8575180B2 (en) | 2004-03-26 | 2013-11-05 | Astrazeneca Aktiebolag | 9-substituted 8-oxoadenine compound |
| US8969362B2 (en) | 2004-03-26 | 2015-03-03 | Astrazeneca Aktiebolag | 9-substituted 8-oxoadenine compound |
| US20070225303A1 (en) * | 2004-03-26 | 2007-09-27 | Haruhisa Ogita | 8-Oxoadenine Compound |
| US20080293715A1 (en) * | 2004-11-29 | 2008-11-27 | Warner Lambert Company Llc | Therapeutic Pyrazolo[3,4-b]Pyridines and Indazoles |
| US7485636B2 (en) | 2004-11-29 | 2009-02-03 | Pfizer Inc. | Therapeutic pyrazolo[3,4-b]pyridines and indazoles |
| US7423054B2 (en) | 2004-11-29 | 2008-09-09 | Warner Lambert Company Llc | Therapeutic pyrazolo[3,4-b]pyridines and indazoles |
| US20060116376A1 (en) * | 2004-11-29 | 2006-06-01 | Warner-Lambert Company Llc | Therapeutic pyrazolo[3,4-B]pyridines and indazoles |
| US9359360B2 (en) | 2005-08-22 | 2016-06-07 | The Regents Of The University Of California | TLR agonists |
| US20090324551A1 (en) * | 2005-08-22 | 2009-12-31 | The Regents Of The University Of California Office Of Technology Transfer | Tlr agonists |
| US20090143400A1 (en) * | 2005-09-16 | 2009-06-04 | Mcinally Thomas | Purine derivatives having immuno-modulating properties |
| US20080269240A1 (en) * | 2005-09-22 | 2008-10-30 | Dainippon Sumitomo Pharma Co., Ltd. a corporation of Japan | Novel Adenine Compound |
| US20090192153A1 (en) * | 2005-09-22 | 2009-07-30 | Dainippon Sumitomo Pharma Co., Ltd. a corporation of Japan | Novel adenine compound |
| US20090118263A1 (en) * | 2005-09-22 | 2009-05-07 | Dainippon Sumitomo Pharma Co., Ltd. | Novel Adenine Compound |
| US20090099216A1 (en) * | 2005-09-22 | 2009-04-16 | Astrazeneca Aktiebolag A Corporation Of Sweden | Novel adenine compound |
| US20090082332A1 (en) * | 2005-09-22 | 2009-03-26 | Philip Abbot | Purine derivatives for the treatment of viral or allergic diseases and cancers |
| US9738649B2 (en) | 2006-03-21 | 2017-08-22 | Janssen Pharmaceutica N.V. | Tetrahydro-pyrimidoazepines as modulators of TRPV1 |
| US8673895B2 (en) | 2006-03-21 | 2014-03-18 | Janssen Pharmaceutica Nv | Tetrahydro-pyrimidoazepines as modulators of TRPV1 |
| US20070225275A1 (en) * | 2006-03-21 | 2007-09-27 | Allison Brett D | Tetrahydro-pyrimidoazepines as modulators of TRPV1 |
| US9422293B2 (en) | 2006-03-21 | 2016-08-23 | Janssen Pharmaceutica Nv | Tetrahydro-pyrimidoazepines as modulators of TRPV1 |
| US20110098294A1 (en) * | 2006-05-31 | 2011-04-28 | Carson Dennis A | Purine analogs |
| US8846697B2 (en) | 2006-05-31 | 2014-09-30 | The Regents Of The University Of California | Purine analogs |
| US20100240623A1 (en) * | 2006-07-05 | 2010-09-23 | Anthony Cook | 8-oxoadenine derivatives acting as modulators of tlr7 |
| US8138172B2 (en) | 2006-07-05 | 2012-03-20 | Astrazeneca Ab | 8-oxoadenine derivatives acting as modulators of TLR7 |
| US8067411B2 (en) | 2006-12-14 | 2011-11-29 | Astrazeneca Ab | Compounds |
| US20080300244A1 (en) * | 2006-12-14 | 2008-12-04 | Astrazeneca Ab | Novel compounds |
| US8790655B2 (en) | 2007-02-07 | 2014-07-29 | The Regents Of The University Of California | Conjugates of synthetic TLR agonists and uses therefor |
| US9050376B2 (en) | 2007-02-07 | 2015-06-09 | The Regents Of The University Of California | Conjugates of synthetic TLR agonists and uses therefor |
| US8357374B2 (en) | 2007-02-07 | 2013-01-22 | The Regents Of The University Of California | Conjugates of synthetic TLR agonists and uses therefor |
| US20100087443A1 (en) * | 2007-03-19 | 2010-04-08 | Roger Victor Bonnert | 9-substituted-8-oxo-adenine compounds as toll-like receptor (tlr7) modulators |
| US20100130491A1 (en) * | 2007-03-19 | 2010-05-27 | Roger Victor Bonnert | 9-substituted-8-oxo-adenine compounds as toll-like receptor (tlr7 ) modulators |
| US8063051B2 (en) | 2007-03-19 | 2011-11-22 | Astrazeneca Ab | 9-substituted-8-oxo-adenine compounds as toll-like receptor (TLR7) modulators |
| US8067413B2 (en) | 2007-03-19 | 2011-11-29 | Astrazeneca Ab | 9-substituted-8-oxo-adenine compounds as toll-like receptor (TLR7 ) modulators |
| US20100099870A1 (en) * | 2007-03-20 | 2010-04-22 | Dainippon Sumitomo Phama Co., Ltd | Novel adenine compound |
| US8044056B2 (en) | 2007-03-20 | 2011-10-25 | Dainippon Sumitomo Pharma Co., Ltd. | Adenine compound |
| US20100093998A1 (en) * | 2007-03-20 | 2010-04-15 | Dainippon Sumitomo Pharma Co., Ltd. | Novel adenine compound |
| US20110028715A1 (en) * | 2007-03-20 | 2011-02-03 | Dainippon Sumitomo Pharma Co., Ltd. | Novel adenine compound |
| RU2461559C2 (en) * | 2007-04-04 | 2012-09-20 | Сайкласел Лимитед | Compounds |
| US8436178B2 (en) | 2007-05-08 | 2013-05-07 | Astrazeneca Ab | Imidazoquinolines with immuno-modulating properties |
| US20100280001A1 (en) * | 2007-05-08 | 2010-11-04 | Roger Victor Bonnert | Imidazoquinolines with immuno-modulating properties |
| US20090209524A1 (en) * | 2007-11-22 | 2009-08-20 | Astrazeneca Ab | Novel Compounds |
| US8268990B2 (en) | 2007-11-22 | 2012-09-18 | Astrazeneca Ab | Compounds |
| US8765939B2 (en) | 2007-11-22 | 2014-07-01 | Astrazeneca Ab | Pyrimidline derivatives having immune modulating properties that act via TLR7 for the treatment of viral or allergic diseases and cancers |
| US8288397B2 (en) | 2007-12-17 | 2012-10-16 | Janssen Pharmaceutica Nv | Imidazolo-, oxazolo-, and thiazolopyrimidine modulators of TRPV1 |
| US9440978B2 (en) | 2007-12-17 | 2016-09-13 | Janssen Pharmaceutica Nv | Imidazolo-, oxazolo-, and thiazolopyrimidine modulators of TRPV1 |
| US20090156599A1 (en) * | 2007-12-17 | 2009-06-18 | Bryan James Branstetter | Imidazolo-, oxazolo-, and thiazolopyrimidine modulators of TRPV1 |
| US8637527B2 (en) | 2007-12-17 | 2014-01-28 | Janssen Pharmaceutica Nv | Imidazolo-, oxazolo-, and thiazolopyrimidine modulators of TRPV1 |
| US20090156598A1 (en) * | 2007-12-17 | 2009-06-18 | Lebsack Alec D | Imidazolopyrimidine modulators of TRPV1 |
| US8673907B2 (en) | 2007-12-17 | 2014-03-18 | Astrazeneca Ab | Pharmaceutically acceptable salts of methyl (3-{ [[3-(6-amino- 2-butoxy-8-oxo-7,8-dihydro-9H-purin-9-yl) propyl] (3-morpholin-4-ylpropyl) amino] methyl }phenyl) acetate and their use in therapy |
| US8865896B2 (en) | 2008-01-17 | 2014-10-21 | Astrazeneca Aktiebolag | Method for preparing adenine compound |
| US20110054168A1 (en) * | 2008-01-17 | 2011-03-03 | Ayumu Kurimoto | Method for preparing adenine compound |
| US20090202626A1 (en) * | 2008-02-07 | 2009-08-13 | Carson Dennis A | Treatment of bladder diseases with a tlr7 activator |
| US10117873B2 (en) | 2008-08-11 | 2018-11-06 | Glaxosmithkline Llc | 6-amino-purin-8-one compounds |
| US9877968B2 (en) | 2008-08-11 | 2018-01-30 | Glaxosmithkline Llc | 6-amino-purin-8-one compounds |
| US9066940B2 (en) | 2009-02-06 | 2015-06-30 | Telormedix, Sa | Pharmaceutical compositions comprising imidazoquinolin(amines) and derivatives thereof suitable for local administration |
| US9107919B2 (en) | 2009-02-06 | 2015-08-18 | Telormedix Sa | Pharmaceutical compositions comprising imidazoquinolin(amines) and derivatives thereof suitable for local administration |
| US20100210598A1 (en) * | 2009-02-11 | 2010-08-19 | Regents Of The University Of California, San Diego | Toll-like receptor modulators and treatment of diseases |
| US8729088B2 (en) | 2009-02-11 | 2014-05-20 | The Regents Of The University Of California | Toll-like receptor modulators and treatment of diseases |
| US8476288B2 (en) | 2009-05-21 | 2013-07-02 | Astrazeneca Ab | Salts 756 |
| US9533978B2 (en) | 2009-05-21 | 2017-01-03 | Sumitomo Dainippon Pharma Co., Ltd | Pyrimidine derivatives and their use in the treatment of cancer and further diseases |
| US20100298364A1 (en) * | 2009-05-21 | 2010-11-25 | Astrazeneca Ab | salts 756 |
| US20110136801A1 (en) * | 2009-12-03 | 2011-06-09 | Dainippon Sumitomo Pharma Co. Ltd. | Novel Compounds |
| US9173936B2 (en) | 2010-04-30 | 2015-11-03 | Telormedix Sa | Phospholipid drug analogs |
| US9173935B2 (en) | 2010-04-30 | 2015-11-03 | Telormedix Sa | Phospholipid drug analogs |
| US9180183B2 (en) | 2010-04-30 | 2015-11-10 | Telormedix Sa | Phospholipid drug analogs |
| US9050319B2 (en) | 2010-04-30 | 2015-06-09 | Telormedix, Sa | Phospholipid drug analogs |
| US8846713B2 (en) | 2010-06-24 | 2014-09-30 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds as phosphodiesterases (PDEs) inhibitors |
| US8563575B2 (en) | 2010-06-24 | 2013-10-22 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds |
| US9226921B2 (en) | 2010-06-24 | 2016-01-05 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds as phosphodiesterases (PDES) inhibitors |
| US8940758B2 (en) | 2010-06-24 | 2015-01-27 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds |
| US9045472B2 (en) | 2010-12-16 | 2015-06-02 | Astrazeneca Ab | Imidazoquinoline compounds |
| US8895570B2 (en) | 2010-12-17 | 2014-11-25 | Astrazeneca Ab | Purine derivatives |
| US10112946B2 (en) | 2011-07-22 | 2018-10-30 | Glaxosmithkline Llc | Composition |
| US11299465B2 (en) | 2012-05-18 | 2022-04-12 | Sumitomo Dainippon Pharma Co., Ltd. | Carboxylic acid compounds |
| US10150743B2 (en) | 2012-05-18 | 2018-12-11 | Sumitomo Dainippon Pharma Co., Ltd. | Carboxylic acid compounds |
| US9376398B2 (en) | 2012-05-18 | 2016-06-28 | Sumitomo Dainippon Pharma Co., Ltd | Carboxylic acid compounds |
| US10562861B2 (en) | 2012-05-18 | 2020-02-18 | Sumitomo Dainippon Pharma Co., Ltd. | Carboxylic acid compounds |
| US12077510B2 (en) | 2012-05-18 | 2024-09-03 | Sumitomo Pharma Co., Ltd. | Carboxylic acid compounds |
| US9662336B2 (en) | 2012-08-24 | 2017-05-30 | Glaxosmithkline Llc | Pyrazolopyrimidine compounds |
| US9555036B2 (en) | 2012-08-24 | 2017-01-31 | Glaxosmithkline Llc | Pyrazolopyrimidine compounds |
| US10022442B2 (en) | 2012-08-24 | 2018-07-17 | Glaxosmithkline Llc | Pyrazolopyrimidine compounds |
| US9540383B2 (en) | 2012-11-20 | 2017-01-10 | Glaxosmithkline Llc | Pyrrolopyrimidines as therapeutic agents for the treatment of diseases |
| US9428512B2 (en) | 2012-11-20 | 2016-08-30 | Glaxosmithkline Llc | Compounds |
| US9907847B2 (en) | 2012-11-20 | 2018-03-06 | Glaxosmithkline Llc | Pyrrolopyrimidines as therapeutic agents for the treatment of diseases |
| US9550785B2 (en) | 2012-11-20 | 2017-01-24 | Glaxosmithkline Llc | Pyrrolopyrimidines as therapeutic agents for the treatment of diseases |
| US9120788B2 (en) | 2013-02-19 | 2015-09-01 | Pfizer Inc. | Azabenzimidazole compounds |
| US9815832B2 (en) | 2013-02-19 | 2017-11-14 | Pfizer Inc. | Azabenzimidazole compounds |
| US10131669B2 (en) | 2014-07-24 | 2018-11-20 | Pfizer Inc. | Pyrazolopyrimidine compounds |
| US9598421B2 (en) | 2014-08-06 | 2017-03-21 | Pfizer Inc. | Imidazopyridazine compounds |
| US10669279B2 (en) | 2014-08-06 | 2020-06-02 | Pfizer Inc. | Imidazopyridazine compounds |
| US10077269B2 (en) | 2014-08-06 | 2018-09-18 | Pfizer Inc. | Imidazopyridazine compounds |
| US11590139B2 (en) | 2014-10-06 | 2023-02-28 | Signal Pharmaceuticals, Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| US9737541B2 (en) | 2014-10-06 | 2017-08-22 | Signal Pharmaceuticals, Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| US10398700B2 (en) | 2014-10-06 | 2019-09-03 | Signal Pharmaceuticals, Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| WO2016057370A1 (en) * | 2014-10-06 | 2016-04-14 | Signal Pharmaceuticals, Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| KR20170063740A (en) * | 2014-10-06 | 2017-06-08 | 시그날 파마소티칼 엘엘씨 | Substituted Aminopurine Compounds, Compositions Thereof, and Methods of Treatment Therewith |
| CN107001372A (en) * | 2014-10-06 | 2017-08-01 | 西格诺药品有限公司 | Substituted aminopurine compounds, compositions and methods of treatment thereof |
| US10646493B2 (en) | 2014-10-06 | 2020-05-12 | Signal Pharmaceuticals, Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| US10149849B2 (en) | 2014-10-06 | 2018-12-11 | Signal Pharmaceuticals, Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| KR102504849B1 (en) | 2014-10-06 | 2023-03-02 | 시그날 파마소티칼 엘엘씨 | Substituted Aminopurine Compounds, Compositions Thereof, and Methods of Treatment Therewith |
| US10940152B2 (en) | 2014-10-06 | 2021-03-09 | Signal Pharmaceuticals, Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| CN113248506A (en) * | 2014-10-06 | 2021-08-13 | 西格诺药品有限公司 | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| US9512124B2 (en) | 2014-10-06 | 2016-12-06 | Signal Pharmaceuticals, Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| US11697851B2 (en) | 2016-05-24 | 2023-07-11 | The Regents Of The University Of California | Early ovarian cancer detection diagnostic test based on mRNA isoforms |
| US10590136B2 (en) | 2017-10-04 | 2020-03-17 | Celgene Corporation | Processes for the preparation of cis-4-[2-{[(3S,4R)-3-fluorooxan-4-yl]amino}-8-(2,4,6-trichloroanilino)-9H-purin-9-yl]-1-methylcyclohexane-1-carboxamide |
| US10898488B2 (en) | 2017-10-04 | 2021-01-26 | Celgene Corporation | Compositions and methods of use of cis-4-[2-{[(3S,4R)-3-fluorooxan-4-yl]amino}-8-(2,4,6-trichloroanilino)-9H-purin-9-yl]-1-methylcyclohexane-1-carboxamide |
| US10653697B2 (en) | 2017-10-04 | 2020-05-19 | Celgene Corporation | Compositions and methods of use of cis-4-[2-{[3S,4R)-3-fluorooxan-4-yl]amino}8-(2,4,6-trichloroanilino)-9H-purin-9-yl]-1-methylcyclohexane-1-carboxamide |
| US11701362B2 (en) | 2017-10-04 | 2023-07-18 | Celgene Corporation | Compositions and methods of use of cis-4-[2-{[(3S,4R)-3-fluorooxan-4-yl]amino}-8-(2,4,6-trichloroanilino)-9H-purin-9-yl]-1- methylcyclohexane-1-carboxamide |
| US10543214B2 (en) | 2017-10-04 | 2020-01-28 | Celgene Corporation | Compositions and methods of use of cis-4-[2-{[(3S,4R)-3-fluorooxan-4-yl]amino}-8-(2,4,6-trichloroanilino)-9H-purin-9-yl]-1-methylcyclohexane-1-carboxamide |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2000043394A1 (en) | 2000-07-27 |
| DE60004475D1 (en) | 2003-09-18 |
| AU2276100A (en) | 2000-08-07 |
| EP1147108B1 (en) | 2003-08-13 |
| ATE247115T1 (en) | 2003-08-15 |
| EP1147108A1 (en) | 2001-10-24 |
| CZ27399A3 (en) | 2000-08-16 |
| US6552192B1 (en) | 2003-04-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6552192B1 (en) | Substituted nitrogen heterocyclic derivatives and pharmaceutical use thereof | |
| EP1244668B1 (en) | Purine derivatives, process for their preparation and use thereof | |
| US7816350B2 (en) | Substituted [1,2,3] triazolo[4,5-D]pyrimidines as cdk inhibitors | |
| EP1348707B1 (en) | Pyrazolo[4,3-d]pyrimidines, processes for their preparation and methods for therapy | |
| EP1575973B1 (en) | Substitution derivatives of n sp 6 sp -benzyladenosine, methods of their preparation, their use for preparation of drugs, cosmetic preparations and growth regulators, pharmaceutical preparations, cosmetic preparations and growth regulators containing these compounds | |
| US6072053A (en) | Dideoxycarbocyclic nucleosides | |
| US4975434A (en) | Antiviral and anticancer cyclopentenyl cytosine | |
| EP2430036B1 (en) | Substituted 6-(benzylamino) purine riboside derivatives, use thereof and compositions containing these derivatives | |
| Michael et al. | Alkylpurines as immunopotentiating agents. Synthesis and antiviral activity of certain alkylguanines | |
| EP0351215B1 (en) | Ester of 9-(2-hydroxyethoxymethyl) guanine | |
| Swayze et al. | Synthesis, antiproliferative, and antiviral evaluation of certain acyclic 6-substituted pyrrolo [2, 3-d]-pyrimidine nucleoside analogs related to sangivamycin and toyocamycin | |
| US5567703A (en) | Method for treating HIV infections with dideoxycarbocyclic nucleosides | |
| DE69718831T2 (en) | 5H-PYRANO [2,3-D: 6,5-D '] DIPYRIMIDINE DERIVATIVES WITH ANTIBACTERIAL, ANTIVIRAL AND IMMUNO-MODULATIVE EFFECT | |
| US9073961B2 (en) | Substitution derivatives of N6 -benzyladenosine-5′-monophosphate, methods of preparation thereof, use thereof as medicaments, and therapeutic preparations containing these compounds | |
| AU618982B2 (en) | Pyrimidine and purine 1,2-butadiene-4-ols as anti-retroviral agents | |
| RU2188201C2 (en) | Derivatives of 5h-pyrano[2,3-d:6,5-d]dipyrimidine showing antibacterial, antiviral and immunomodulating effect | |
| US5175292A (en) | Intermediates for the preparation of dideoxycarbocyclic nucleosides | |
| PL162956B1 (en) | Method for the production of a new esterified PL PL 5-propynylpyrimidine nucleoside | |
| WO1993017035A1 (en) | 2'ISODIDEOXY-β-D-NUCLEOSIDES AS STABLE ANTIVIRAL AGENTS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| STCB | Information on status: application discontinuation |
Free format text: ABANDONED -- FAILURE TO RESPOND TO AN OFFICE ACTION |