RU2578604C1 - Chimeric antibiotics based on azithromycin and glycopeptide antibiotics, having antibacterial activity and synthesis method thereof - Google Patents
Chimeric antibiotics based on azithromycin and glycopeptide antibiotics, having antibacterial activity and synthesis method thereof Download PDFInfo
- Publication number
- RU2578604C1 RU2578604C1 RU2014151833/04A RU2014151833A RU2578604C1 RU 2578604 C1 RU2578604 C1 RU 2578604C1 RU 2014151833/04 A RU2014151833/04 A RU 2014151833/04A RU 2014151833 A RU2014151833 A RU 2014151833A RU 2578604 C1 RU2578604 C1 RU 2578604C1
- Authority
- RU
- Russia
- Prior art keywords
- azithromycin
- antibiotics
- vancomycin
- glycopeptide
- eremomycin
- Prior art date
Links
- YHVUVJYEERGYNU-UHFFFAOYSA-N CC(C(C(C)(C1)OC)O)OC1O Chemical compound CC(C(C(C)(C1)OC)O)OC1O YHVUVJYEERGYNU-UHFFFAOYSA-N 0.000 description 1
- 0 CCC[C@]([C@](C[C@@](*)NCCC[C@@](CCCC1C)O)N=O)OC1=O Chemical compound CCC[C@]([C@](C[C@@](*)NCCC[C@@](CCCC1C)O)N=O)OC1=O 0.000 description 1
Images




Landscapes
- Peptides Or Proteins (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract

Description
Изобретение относится к фармацевтической промышленности и касается новых производных на основе антибиотика азитромицина и гликопептидных антибиотиков, и способа их получения.The invention relates to the pharmaceutical industry and relates to new derivatives based on the antibiotic azithromycin and glycopeptide antibiotics, and a method for their preparation.
Азитромицин (1) (Фигура 1) является полусинтетическим антибиотиком, первым представителем подкласса азалидов, несколько отличающихся по структуре от классических макролидов, основу химической структуры которых составляет макроциклическое лактонное кольцо. Получен модификацией 14-членных макролидов путем включения атома азота в лактонное кольцо между 9 и 10 атомами углерода, кольцо при этом превращается в 15-членное. Данная структурная перестройка обусловливает значительное повышение кислотоустойчивости препарата - в 300 раз по сравнению с эритромицином. Антибиотик широкого спектра действия действует бактериостатически. Связываясь с 50S-субъединицей рибосом, угнетает пептидтранслоказу на стадии трансляции, подавляет синтез белка, замедляет рост и размножение бактерий, в высоких концентрациях оказывает бактерицидный эффект. Действует на вне- и внутриклеточных возбудителей. Неактивен в отношении грамположительных бактерий, устойчивых к эритромицину.Azithromycin (1) (Figure 1) is a semi-synthetic antibiotic, the first representative of a subclass of azalides that are slightly different in structure from classical macrolides, the basis of the chemical structure of which is a macrocyclic lactone ring. Obtained by the modification of 14-membered macrolides by the inclusion of a nitrogen atom in the lactone ring between 9 and 10 carbon atoms, while the ring turns into a 15-membered one. This structural adjustment leads to a significant increase in the acid resistance of the drug - 300 times compared with erythromycin. A broad-spectrum antibiotic acts bacteriostatically. By binding to the 50S subunit of ribosomes, it inhibits the peptide translocase at the translation stage, inhibits protein synthesis, slows the growth and reproduction of bacteria, and has a bactericidal effect in high concentrations. It acts on extra- and intracellular pathogens. Inactive against gram-positive bacteria resistant to erythromycin.
Обладает уникальной по сравнению с другими макролидами способностью накапливаться в органах и тканях. Азитромицин активно поглощается разными клетками, включая лейкоциты, фибробласты, макрофаги и фагоциты, и вместе с ними транспортируется к месту инфекции (воспаления) [Lalak N.J., Morris D.L., Azithromycin Clinical Pharmacokinetics, Clinical Pharmacokinetics, 1993, V. 25, P. 370-374; Gladue R.P, Bright G.M., Isaacson R.E., Newborg M.F. In vitro and in vivo uptake of azithromycin (CP-62,993) by phagocytic cells: possible mechanism of delivery and release at sites of infection. Antimicrobial Agents Chemotherapy, 1990, V. 34, P. 1056-1060]. Азитромицин относится к тканевым антибиотикам, так как его концентрации в сыворотке крови значительно ниже тканевых. Он хорошо распределяется в организме, создавая высокие концентрации в различных тканях и органах (в том числе в предстательной железе), особенно при воспалении. При этом азитромицин проникает внутрь клеток и создает высокие внутриклеточные концентрации. Метаболизируется в печени при участии микросомальной системы цитохрома Р-450, метаболиты выводятся преимущественно с желчью. Период полувыведения азитромицина 55 ч. При почечной недостаточности этот параметр не изменяется [Практическое руководство по антиинфекционной химиотерапии. НИИАХ СГМА. 2007. Под редакцией Л.С. Страчунского, Ю.Б. Белоусова, C.H. Козлова].It has a unique ability to accumulate in organs and tissues in comparison with other macrolides. Azithromycin is actively absorbed by various cells, including leukocytes, fibroblasts, macrophages and phagocytes, and is transported with them to the site of infection (inflammation) [Lalak NJ, Morris DL, Azithromycin Clinical Pharmacokinetics, Clinical Pharmacokinetics, 1993, V. 25, P. 370- P. 370- 374; Gladue R.P., Bright G.M., Isaacson R.E., Newborg M.F. In vitro and in vivo uptake of azithromycin (CP-62,993) by phagocytic cells: possible mechanism of delivery and release at sites of infection. Antimicrobial Agents Chemotherapy, 1990, V. 34, P. 1056-1060]. Azithromycin refers to tissue antibiotics, since its concentration in the blood serum is much lower than tissue. It is well distributed in the body, creating high concentrations in various tissues and organs (including the prostate gland), especially with inflammation. In this case, azithromycin penetrates into the cells and creates high intracellular concentrations. It is metabolized in the liver with the participation of the microsomal system of cytochrome P-450, metabolites are excreted mainly with bile. The half-life of azithromycin is 55 hours. With renal failure, this parameter does not change [Practical Guide to Anti-Infectious Chemotherapy. NIIAH SGMA. 2007. Edited by L.S. Strachunsky, Yu.B. Belousova, C.H. Kozlova].
Ванкомицин (2) представляет собой трициклический гептапептид, к которому присоединен дисахарид, состоящий из аминодезоксисахара (ванкозамина) и D-глюкозы (формула 1) [Sztaricskai F., Pelyva′s-Ferenczik I.. Chemistry of carbohydrate components. In Glycopeptide Antibiotics, 1st edn (Ed. Nagarajan R.), 1994, Marcel Dekker, New York, NY, USA.].Vancomycin (2) is a tricyclic heptapeptide to which a disaccharide consisting of aminodeoxysugar (vancosamine) and D-glucose (Formula 1) is attached [Sztaricskai F., Pelyva′s-Ferenczik I. Chemistry of carbohydrate components. In Glycopeptide Antibiotics, 1st edn (Ed. Nagarajan R.), 1994, Marcel Dekker, New York, NY, USA.].
Эремомицин (3) - оригинальный отечественный антибиотик, агликон которого отличается от агликона ванкомицина отсутствием атома хлора в боковом ароматическом радикале аминокислоты 6, структурой дисахаридной цепи (2-(O-(α-L-эремозаминил)-β-D-глюкопиранозил) и наличием третьего углеводного остатка (L-эремозаминил) в боковом радикале аминокислоты 6 (формула 1) [Gause G.F., Brazhnikova M.G., Lomakina N.N., Berdnikova T.F., Fedorova G.B., Tokareva N.L., Borisova V.N., Batta G. Eremomycin - new glycopeptide antibiotics. Chemical properties and structure. J. Antibiotics, 1989, V. 42, P. 1790-1799].Eremomycin (3) is an original domestic antibiotic whose aglycon differs from vancomycin aglycon by the absence of a chlorine atom in the lateral aromatic radical of
Агликон тейкопланина (4) представляет собой гептапептид, в котором в отличие от агликонов ваномицина и эремомицина, боковые радикалы аминокислот 1 и 3 являются ароматическими и соединены между собой эфирной связью (формула 1).Teicoplanin aglycone (4) is a heptapeptide in which, unlike vancomycin and eremomycin aglycones, the side radicals of
Ванкомицин применяется в клинической практике с 1958 г., тейкопланин - с середины 80-х годов. В последнее время интерес к гликопептидам возрос в связи с увеличением частоты нозокомиальных инфекций грамположительными микроорганизмами. Гликопептиды активны в отношении грамположительных аэробных и анаэробных микроорганизмов, включая метициллин-устойчивые Staphylococcus aureus (MSRA), метициллин-устойчивые Staphylococcus epidermidis (MRSE), стрептококков, пневмококков (включая АРП), энтерококков, включая резистентных к ампицилину и аминогликозидам, пептострептококков, листерий, коринебактерий, клостридий (включая С. difficile). Грамотрицательные микроорганизмы устойчивы к гликопептидам. Vancomycin has been used in clinical practice since 1958, teicoplanin - since the mid-80s. Recently, interest in glycopeptides has increased due to an increase in the frequency of nosocomial infections with gram-positive microorganisms. Glycopeptides are active against gram-positive aerobic and anaerobic microorganisms, including methicillin-resistant Staphylococcus aureus (MSRA), methicillin-resistant Staphylococcus epidermidis (MRSE), streptococci, pneumococci (including ARP), enterococci, ampecoccus tuberculosis, pepticaria, resistant to corynebacteria, clostridia (including C. difficile). Gram-negative microorganisms are resistant to glycopeptides.
Гликопептиды нарушают синтез клеточной стенки бактерий. Оказывают бактерицидное действие, однако в отношении энтерококков, некоторых стрептококков и коагулазонегативных стафилококков действуют бактериостатически. Гликопептиды практически не всасываются при приеме внутрь. Биодоступность тейкопланина при внутримышечном введении составляет около 90%. Гликопептиды не метаболизируются, выводятся почками в неизмененном виде, поэтому при почечной недостаточности требуется коррекция доз. Препараты не удаляются при гемодиализе. Период полувыведения ванкомицина при нормальной функции почек составляет 6-8 ч, тейкопланина - от 40 ч до 70 ч. Длительный период полувыведения тейкопланина дает возможность назначать его один раз в сутки [Практическое руководство по антиинфекционной химиотерапии. НИИАХ СГМА. 2007. Под редакцией Л.С. Страчунского, Ю.Б. Белоусова, C.H. Козлова].Glycopeptides disrupt the synthesis of the bacterial cell wall. They have a bactericidal effect, however, they act bacteriostatically against enterococci, some streptococci and coagulase-negative staphylococci. Glycopeptides are practically not absorbed when taken orally. The bioavailability of teicoplanin with intramuscular injection is about 90%. Glycopeptides are not metabolized, excreted by the kidneys unchanged, so dose adjustment is required for renal failure. Drugs are not removed during hemodialysis. The half-life of vancomycin with normal kidney function is 6-8 hours, teicoplanin is from 40 hours to 70 hours. The long half-life of teicoplanin makes it possible to prescribe it once a day [Practical Guide to Anti-Infectious Chemotherapy. NIIAH SGMA. 2007. Edited by L.S. Strachunsky, Yu.B. Belousova, C.H. Kozlova].
По спектру антимикробной активности ванкомицин и тейкопланин сходны, однако имеются некоторые различия в уровне природной активности и приобретенной резистентности. Их успешное применение в клинике поставлено под угрозу распространением устойчивых к ванкомицину грамположительных бактерий: энтерококков (VRE) и ванкомицин-резистентных Staphilococcus aureus (VRSA) [Mendez-Alvarez S., Perez-Hernandez X., Claverie-Martin F. Glycopeptide resistance in enterococci // Internat. Microbiol. - 2000. - V. 3. - P. 71-80; Cetinkaya Y., Falk P., Mayhall C.G. Vancomycin-resistant Enterococci // Clin. Microboil. Rev. - 2000. - V. 13. - P. 686-707]. Штаммы VRE, и в особенности VRSA, устойчивы к подавляющему большинству применяемых антибиотиков, и поэтому распространение таких микроорганизмов представляет очень серьезную проблему.Vancomycin and teicoplanin are similar in the spectrum of antimicrobial activity, however, there are some differences in the level of natural activity and acquired resistance. Their successful use in the clinic is jeopardized by the spread of vancomycin-resistant gram-positive bacteria: enterococci (VRE) and vancomycin-resistant Staphilococcus aureus (VRSA) [Mendez-Alvarez S., Perez-Hernandez X., Claverie-Martin F. Glycopeptide resistance in enterocococ // Internat. Microbiol. - 2000. - V. 3. - P. 71-80; Cetinkaya Y., Falk P., Mayhall C.G. Vancomycin-resistant Enterococci // Clin. Microboil. Rev. - 2000. - V. 13. - P. 686-707]. VRE strains, and in particular VRSA, are resistant to the vast majority of antibiotics used, and therefore the spread of such microorganisms is a very serious problem.
Широкое распространение резистентности к антибиотикам среди возбудителей заболеваний привело к утрате клинической значимости ряда лекарственных препаратов и послужило стимулом для поиска новых эффективных антимикробных агентов. Одной из перспективных стратегий, направленной на создание препаратов, активных в отношении резистентных микроорганизмов, является создание антибиотиков двойного действия - «химерных» антибиотиков. Они состоят из молекул разных антибиотиков, связанных между собой различными способами через так называемые «спейсеры». Они обладают расширенным спектром действия по сравнению с исходными антибиотиками и замедляют развитие антибиотикорезистентности.The wide spread of antibiotic resistance among pathogens has led to the loss of the clinical significance of a number of drugs and served as an incentive for the search for new effective antimicrobial agents. One of the promising strategies aimed at creating drugs that are active against resistant microorganisms is the creation of double-acting antibiotics - “chimeric” antibiotics. They consist of molecules of different antibiotics, interconnected in various ways through the so-called "spacers". They have an extended spectrum of action compared to the original antibiotics and slow down the development of antibiotic resistance.
Настоящее изобретение призвано получить антибиотики двойного действия на основе макролидного антибиотика азитромицина и гликопептидных антибиотиков, обладающие антибактериальной активностью.The present invention is intended to obtain dual-action antibiotics based on the macrolide antibiotic azithromycin and glycopeptide antibiotics with antibacterial activity.
Объединение в одной молекуле азитромицина с другим антибактериальным агентом потенциально может расширить спектр действия полученного химерного антибиотика по сравнению со спектрами действия исходных антибиотиков. Фармакокинетические свойства такого химерного антибиотика могут превосходить фармакокинетические свойства второго антибактериального агента, использованного для его синтеза.The combination of azithromycin in one molecule with another antibacterial agent can potentially expand the spectrum of action of the obtained chimeric antibiotic in comparison with the spectrum of action of the original antibiotics. The pharmacokinetic properties of such a chimeric antibiotic may be superior to the pharmacokinetic properties of the second antibacterial agent used for its synthesis.
Гликопептидные антибиотики выбраны в качестве второго антибактериального агента при синтезе химерных антибиотиков на основе азитромицина, поскольку они активны в отношении грамположительных аэробных и анаэробных микроорганизмов и являются препаратами выбора при инфекциях, вызванных широко распространенными в клиниках резистентными грамположительными бактериями.Glycopeptide antibiotics were chosen as the second antibacterial agent in the synthesis of chimeric azithromycin-based antibiotics, since they are active against gram-positive aerobic and anaerobic microorganisms and are the drugs of choice for infections caused by resistant gram-positive bacteria that are widespread in clinics.
Изобретение включает соединения, соответствующие структурной формуле, изображенной на фигуре 2, где n=1-10, a R представляет собой остаток гликопептидного антибиотика, такого как ванкомицин, или эремомицин, или агликон тейкопланина.The invention includes compounds corresponding to the structural formula depicted in Figure 2, where n = 1-10, and R is the remainder of a glycopeptide antibiotic such as vancomycin, or eremomycin, or teicoplanin aglycon.
Изобретение также включает в себя способ получения антибиотиков на основе азитромицина и гликопептидных антибиотиков (Фигура 1), заключающийся в проведении реакции ацилирования 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) гликопептидным антибиотиком (эремомицином (2), или ванкомицином (3), или агликоном тейкопланина (4)) в присутствии конденсирующего агента (Фигура 3).The invention also includes a method for producing antibiotics based on azithromycin and glycopeptide antibiotics (Figure 1), which consists in carrying out the acylation reaction of 11-O- (ω-aminoalkylcarbamoyl) azithromycin (6) with a glycopeptide antibiotic (eremomycin (2), or vancomycin (3) , or teicoplanin aglycon (4)) in the presence of a condensing agent (Figure 3).
11-O-(ω-Аминоалкилкарбамоил)азитромицин (6) получен из 2′-O-ацетил-11,12-циклического карбоната азитромицина (5) методом, аналогичным описанному в литературе [X. Li, S. Ma, M. Yan, Y. Wang, S Ma. Synthesis and antibacterial evaluation of novel 11,4′′-disubstituted azithromycin analogs with greatly improved activity against erythromycin-resistant bacteria. European Journal of Medicinal Chemistry, 2013, V. 59, pp 209-217].11-O- (ω-aminoalkylcarbamoyl) azithromycin (6) was obtained from 2′-O-acetyl-11,12-cyclic azithromycin carbonate (5) by a method similar to that described in the literature [X. Li, S. Ma, M. Yan, Y. Wang, S Ma. Synthesis and antibacterial evaluation of novel 11.4 ′ ′ - disubstituted azithromycin analogs with greatly improved activity against erythromycin-resistant bacteria. European Journal of Medicinal Chemistry, 2013, V. 59, pp 209-217].
В этой работе описано получение 11,4′′-дизамещенных производных азитромицина - производных 11-O-(аминофенилкарбамоил)-4′′-O-(амино)карбамоилазитромицина, которые показали высокую активность в отношении эритромицин-чувствительных штаммов S. pneumoniae и значительно улучшенную активность в отношении эритромицин-резистентных штаммов S. pneumonia (МПК 2÷16 мкг/мл против 256 мкг/мл).This work describes the preparation of 11,4 ″ disubstituted derivatives of azithromycin, derivatives of 11-O- (aminophenylcarbamoyl) -4 ″ O O- (amino) carbamoylazitromycin, which showed high activity against erythromycin-sensitive S. pneumoniae strains and significantly improved activity against erythromycin-resistant strains of S. pneumonia (
Реакцию ацилирования 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) (Фиг. 3, Схема 1) гликопептидным антибиотиком с получением соединения формулы 2 проводят в присутствии конденсирующих агентов, известных из уровня техники и применяемых для образования амидной связи, например бензотриазол-1-ил-окси-триспирролидинофосфоний гексафторфосфата (РуВОР) или O-(бензотриазол-1-ил)-N,N,N′,N′-бис(тетраметилен)) гексафторфосфат мочевины (HBPyU). Реакцию ацилирования 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) (Фигура 3) гликопептидным антибиотиком с получением соединения структурной формулы, изображенной на фигуре 2, и показанного на фигуре 4, проводят в растворителе, выбираемом из метанола, этанола, Ν,Ν-диметилформамида или диметилсульфоксида.The acylation reaction of 11-O- (ω-aminoalkylcarbamoyl) azithromycin (6) (Fig. 3, Scheme 1) with a glycopeptide antibiotic to produce a compound of
Соединения структурной формулы, изображенной на фигуре 2, обладают выраженной антибактериальной активностью, в том числе в отношении штаммов, устойчивых к ванкомицину (см. Пример 2) и могут быть использованы для лечения инфекционных заболеваний.Compounds of the structural formula depicted in figure 2 have pronounced antibacterial activity, including against strains resistant to vancomycin (see Example 2) and can be used to treat infectious diseases.
Вспомогательные средстваAids
Эремомицин сульфат получен на опытной установке НИИ по изысканию новых антибиотиков им. Г.Ф. Гаузе РАМН. Ванкомицин гидрохлорид был коммерческим продуктом фирмы Aldrich (США). Агликон тейкопланина был получен от фирмы Lepetit Research Center (Gerenzano (Varese), Италия). Бензотриазол-1-ил-окси-триспирролидинофосфоний гексафторфосфат (РуВОР), O-(бензотриазол-1-ил)-N,N,N′,N′-бис(тетраметилен)) гексафторфосфат мочевины (HBPyU) были коммерческими продуктами фирмы Acros. Все растворы высушивали над сульфатом натрия и упаривали при температуре не выше 40°C.Eremomycin sulfate was obtained at a pilot plant of the Research Institute for the search for new antibiotics named after G.F. Gause RAMS. Vancomycin hydrochloride was a commercial product of Aldrich (USA). Teicoplanin aglycon was obtained from Lepetit Research Center (Gerenzano (Varese), Italy). Benzotriazol-1-yl-hydroxy-trispyrrolidinophosphonium hexafluorophosphate (RuBOP), O- (benzotriazol-1-yl) -N, N, N ′, N′-bis (tetramethylene)) urea hexafluorophosphate (HBPyU) were commercial products of Acros. All solutions were dried over sodium sulfate and evaporated at a temperature not exceeding 40 ° C.
Тонкослойную хроматографию осуществляли на пластинках с силикагелем G60 (Merck) в смеси растворителей: система (A) AcOEt-nPrOH-NH4OH, 1:1:2. Для препаративной очистки использовали колоночную хроматографию на силанизированном силикагеле Merck с размером частиц 0.040-0.063 µм.Thin layer chromatography was performed on G60 silica gel plates (Merck) in a solvent mixture: system (A) AcOEt-nPrOH-NH 4 OH, 1: 1: 2. For preparative purification, column chromatography on Merck silanized silica gel with a particle size of 0.040-0.063 μm was used.
Аналитическую ВЭЖХ осуществляли на хроматографе LC-10 (Shimadzu, Япония) с использованием УФ-детектора и колонки Kromasil 100-С18 4×250 мм, размер частиц 6 мкм (АО БиоХимМак СТ, РФ). Подвижной фазой служили системы, состоящие из двух компонентов А и Б:Analytical HPLC was carried out on an LC-10 chromatograph (Shimadzu, Japan) using a UV detector and a Kromasil 100-
Система (В): А (0.2% HCOONH4 рН 4.5) и Б (MeCN), изократический режим 8% ацетонитрила от 0 до 5 минут, затем линейный градиент концентрации ацетонитрила 8→70% от 5 до 40 мин, скорость потока 1.0 мл/мин.System (C): A (0.2% HCOONH 4 pH 4.5) and B (MeCN), isocratic regime of 8% acetonitrile from 0 to 5 minutes, then a linear gradient of the concentration of
ИК-спектры снимали в таблетке KBr на спектрофотометре DTGS. Температуры плавления получены на приборе Buchi SMP-20.IR spectra were recorded in a KBr tablet on a DTGS spectrophotometer. Melting points were obtained on a Buchi SMP-20 device.
Масс-спектры при ионизации электрораспылением (ESI) получали на приборе Finnigan MAT 900S (Германия).Mass spectra during electrospray ionization (ESI) were obtained on a Finnigan MAT 900S instrument (Germany).
Примеры получения производных на основе азитромицина и гликопептидных антибиотиков по настоящему изобретению и изучение их антибактериальной активности:Examples of the preparation of derivatives of azithromycin and glycopeptide antibiotics of the present invention and the study of their antibacterial activity:
Пример 1. Общая методика получения химерных антибиотиков на основе остатков азитромицина и гликопептидного антибиотика.Example 1. General method for producing chimeric antibiotics based on residues of azithromycin and a glycopeptide antibiotic.
2′-O-ацетил-11,12-циклического карбоната азитромицина (5).2′-O-acetyl-11,12-cyclic azithromycin carbonate (5).
К раствору азитромицина (5 г, 3.27 ммоль) в 24 мл этилацетата добавляли K2CO3 (0.64 г, 4.63 ммоль), нагревали смесь до кипения и затем медленно, в течение 20 мин при кипячении добавляли 1.6 г (18.2 ммоль) этиленкарбоната. Далее смесь кипятили 24 ч, затем этилацетат упаривали. Остаток растворяли в дихлорметане (30 мл) при комнатной температуре, затем добавляли уксусный ангидрид (0.61 мл, 4.37 ммоль) и триэтиламин (1.8 мл, 13 ммоль), реакционную массу премешивали 24 ч при комнатной температуре. К реакционной смеси добавляли 5% водный раствор NaHCO3 (30 мл), и водный раствор экстагировали дихлорметаном (3×10 мл). Объединенные слои дихлорметана высушивали безводным Na2SO4 и высушивали в вакууме. Далее остаток растворяли в хлороформе и наносили на хроматографическую колонку (45×1.5 см) с силикагелем Merck, уравновешенную хлороформом, затем элюировали хлороформом (40 мл), затем смесью хлороформ-метанол (10:1). Фракции, содержащие целевое вещество, упаривали и высушивали в вакууме.To a solution of azithromycin (5 g, 3.27 mmol) in 24 ml of ethyl acetate was added K 2 CO 3 (0.64 g, 4.63 mmol), the mixture was heated to boiling, and then 1.6 g (18.2 mmol) of ethylene carbonate was added slowly over 20 minutes while boiling. The mixture was boiled for 24 hours, then ethyl acetate was evaporated. The residue was dissolved in dichloromethane (30 ml) at room temperature, then acetic anhydride (0.61 ml, 4.37 mmol) and triethylamine (1.8 ml, 13 mmol) were added, and the reaction mixture was stirred for 24 h at room temperature. A 5% aqueous solution of NaHCO 3 (30 ml) was added to the reaction mixture, and the aqueous solution was extracted with dichloromethane (3 × 10 ml). The combined dichloromethane layers were dried with anhydrous Na 2 SO 4 and dried in vacuo. Next, the residue was dissolved in chloroform and applied onto a chromatographic column (45 × 1.5 cm) with Merck silica gel equilibrated with chloroform, then eluted with chloroform (40 ml), then with a mixture of chloroform-methanol (10: 1). Fractions containing the target substance were evaporated and dried in vacuo.
11-O-(ω-Аминоалкилкарбамоил)азитромицин (6).11-O- (ω-aminoalkylcarbamoyl) azithromycin (6).
2′-O-ацетил-11,12-циклического карбоната азитромицина (5) растворяли в минимальном объеме Να,Νω-диаминоалкана и перемешивали при комнатной температуре 48 ч. Далее в реакционную смесь добавляли CHCl3 (20 мл) и воду (20 мл), смесь встряхивали. Органический слой отделяли и промывали водой 6 раз. Органические слои объединяли и далее добавляли воду и 0.5 n HCl, встряхивая слои так, чтобы рН водного слоя составил 8. Органический слой отделяли, добавляли Na2SO4, выдерживали 1 ч, осадок Na2SO4 отфильтровывали, промывая хлороформом. Органический слой упаривали и высушивали в вакууме.2′-O-acetyl-11,12-cyclic azithromycin carbonate (5) was dissolved in a minimal volume of Να, Νω-diaminoalkane and stirred at room temperature for 48 hours. Then, CHCl 3 (20 ml) and water (20 ml) were added to the reaction mixture. ), the mixture was shaken. The organic layer was separated and washed with
Общая методика проведения реакции ацилирования 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) гликопептидными антибиотиками (ванкомицином или эремомицином или аглконом тейкопланина), отщепления ацетильной группы и очистки.The general procedure for carrying out the acylation reaction of 11-O- (ω-aminoalkylcarbamoyl) azithromycin (6) with glycopeptide antibiotics (vancomycin or eremomycin or teicoplanin agglone), acetyl group cleavage and purification.
К раствору гликопептидного антибиотика (0.47 ммоль) в ДМСО (7 мл) добавляли 11-O-(ω-аминоалкилкарбамоил)азитромицина (6) (0.5 экв., 0.235 ммоль), значение рН реакционной смеси доводили до ~7.5 добавлением Et3N. Порциями в течение 1 ч добавляли РуВОР (1.1 экв., 0.26 ммоль), поддерживая рН реакционной смеси ~7.5 добавлением Et3N. Реакционную смесь перемешивали в течение 20 часов при комнатной температуре, затем добавляли пятикратный объем диэтилового эфира. Полученную смесь интенсивно перемешивали, затем эфирный слой удаляли. Процедуру повторяли дважды, до получения вязкого масла, затем добавляли метанол (0.5 мл), ацетон (2 мл) и избыток диэтилового эфира, выпавший осадок отфильтровывали, промывали диэтиловым эфиром и высушивали. Продукт далее очищали методом колоночной хроматографии на силинизированном силикагеле. Вещество растворяли в 30% водном растворе МеОН, добавляли 1 см3 силанизированного силикагеля и высушивали эту смесь в вакууме, далее наносили ее на колонку с силанизированным силикагелем, уравновешенную водой. Элюцию осуществляли сначала водой (100 мл), затем 0.05 M раствором CH3COOH, затем системой МеОН-0.05 M CH3COOH (30:70) (100 мл) для соединений 7-9 или системой МеОН-0.05 M CH3COOH (30:70) (100 мл) для соединений 10-11. Фракции, содержащее целевое вещество, объединяли, упаривали в роторном испарителе с добавлением n-BuOH, к остатку добавляли ацетон и диэтиловый эфир. Выпавший осадок отфильтровывали, промывали ацетоном и высушивали в вакууме. Физико-химические данные для производных 7-11 (Фигура 4) представлены в Таблице 1.To a solution of the glycopeptide antibiotic (0.47 mmol) in DMSO (7 ml) was added 11-O- (ω-aminoalkylcarbamoyl) azithromycin (6) (0.5 eq., 0.235 mmol), the pH of the reaction mixture was adjusted to ~ 7.5 by adding Et 3 N. RuBOP (1.1 eq., 0.26 mmol) was added in portions over 1 h, maintaining the reaction mixture at a pH of ~ 7.5 by adding Et 3 N. The reaction mixture was stirred for 20 hours at room temperature, then a five-fold volume of diethyl ether was added. The resulting mixture was stirred vigorously, then the ether layer was removed. The procedure was repeated twice until a viscous oil was obtained, then methanol (0.5 ml), acetone (2 ml) and an excess of diethyl ether were added, the precipitate formed was filtered off, washed with diethyl ether and dried. The product was further purified by column chromatography on silicinized silica gel. The substance was dissolved in a 30% aqueous solution of MeOH, 1 cm 3 of silanized silica gel was added and the mixture was dried in vacuo, then it was applied to a silanized silica gel column equilibrated with water. The elution was carried out first with water (100 ml), then with a 0.05 M solution of CH 3 COOH, then with a MeOH-0.05 M CH 3 COOH system (30:70) (100 ml) for compounds 7-9 or with a MeOH-0.05 M CH 3 COOH system ( 30:70) (100 ml) for compounds 10-11. The fractions containing the target substance were combined, evaporated in a rotary evaporator with the addition of n-BuOH, acetone and diethyl ether were added to the residue. The precipitate was filtered off, washed with acetone and dried in vacuo. Physico-chemical data for derivatives 7-11 (Figure 4) are presented in Table 1.


Пример 2. Изучение антибактериальной активности антибиотиков на основе азитромицина и гликопептидных антибиотиковExample 2. The study of the antibacterial activity of antibiotics based on azithromycin and glycopeptide antibiotics
Антибактериальная активность производных 7-11 в сравнении с азитромицином (1) и ванкомицином (2) изучена на панели штаммов грамположительных и грамотрицательных бактерий (Таблица 2).The antibacterial activity of derivatives 7-11 in comparison with azithromycin (1) and vancomycin (2) was studied on the panel of strains of gram-positive and gram-negative bacteria (Table 2).
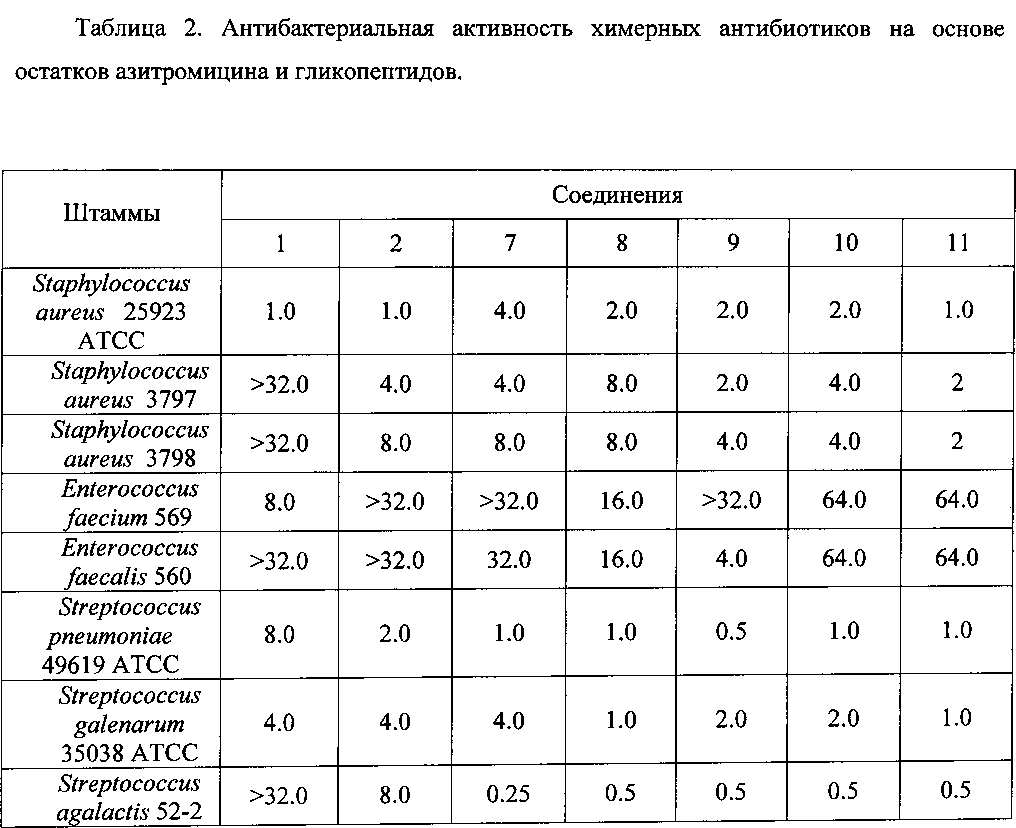
Полученные данные свидетельствуют, что все синтезированные химерные антибиотики на основе азитромицина и гликопептидов 7-11 не уступали или превосходили азитромицин и ванкомицин по антибактериальной активности в отношении всех изученных штаммов грамположительных бактерий. Ценной является высокая активность производных (выше, чем у азитромицина и ванкомицина) в отношении пневмокков, являющихся причиной большинства случаев менингитов, внебольничных пневмоний и ряда гнойно-септических инфекций. Производные на основе эремомицина и азитромицина оказались активны в отношении штаммов Enterococcus faecium и Enterococcus faecalis, устойчивых к ванкомицину.The data obtained indicate that all synthesized chimeric antibiotics based on azithromycin and glycopeptides 7-11 were not inferior or superior to azithromycin and vancomycin in antibacterial activity against all studied strains of gram-positive bacteria. The high activity of derivatives (higher than that of azithromycin and vancomycin) against pneumococci, which are the cause of most cases of meningitis, community-acquired pneumonia, and a number of purulent-septic infections, is valuable. Derivatives based on eremomycin and azithromycin were active against vancomycin resistant strains of Enterococcus faecium and Enterococcus faecalis.
Таким образом, предложенный способ получения антибиотиков на основе азитромицина и гликопептидных антибиотиков позволяет получать новые соединения, обладающие высокой антибактериальной активностью, в том числе, в отношении ряда штаммов Enterococcus, устойчивых к ванкомицину.Thus, the proposed method for producing antibiotics based on azithromycin and glycopeptide antibiotics allows to obtain new compounds with high antibacterial activity, including against a number of Enterococcus strains resistant to vancomycin.
Claims (2)
где n=1-10, a R представляет собой остаток гликопептидного антибиотика, такого как ванкомицин, или эремомицин, или агликон тейкопланина.1. Compounds corresponding to formula 2
where n = 1-10, a R represents the remainder of a glycopeptide antibiotic, such as vancomycin, or eremomycin, or teicoplanin aglycon.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2014151833/04A RU2578604C1 (en) | 2014-12-22 | 2014-12-22 | Chimeric antibiotics based on azithromycin and glycopeptide antibiotics, having antibacterial activity and synthesis method thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2014151833/04A RU2578604C1 (en) | 2014-12-22 | 2014-12-22 | Chimeric antibiotics based on azithromycin and glycopeptide antibiotics, having antibacterial activity and synthesis method thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| RU2578604C1 true RU2578604C1 (en) | 2016-03-27 |
Family
ID=55656751
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2014151833/04A RU2578604C1 (en) | 2014-12-22 | 2014-12-22 | Chimeric antibiotics based on azithromycin and glycopeptide antibiotics, having antibacterial activity and synthesis method thereof |
Country Status (1)
| Country | Link |
|---|---|
| RU (1) | RU2578604C1 (en) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6437119B1 (en) * | 1998-05-07 | 2002-08-20 | William Lawrence Truett | Compounds formed from two or three antibiotics and their processes of preparation |
| WO2004112713A2 (en) * | 2003-06-20 | 2004-12-29 | Royer Biomedical, Inc. | Drug polymer complexes |
| RU2278675C1 (en) * | 2005-01-17 | 2006-06-27 | Юрий Борисович Иванов | Antibacterial agent and pharmaceutical composition comprising effective amount of antibacterial agent |
| WO2011132171A1 (en) * | 2010-04-23 | 2011-10-27 | Piramal Life Sciences Limited | Nitric oxide releasing prodrugs of therapeutic agents |
| WO2014080251A1 (en) * | 2012-11-24 | 2014-05-30 | Hangzhou Dac Biotech Co., Ltd. | Hydrophilic linkers and their uses for conjugation of drugs to cell binding molecules |
-
2014
- 2014-12-22 RU RU2014151833/04A patent/RU2578604C1/en not_active IP Right Cessation
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6437119B1 (en) * | 1998-05-07 | 2002-08-20 | William Lawrence Truett | Compounds formed from two or three antibiotics and their processes of preparation |
| WO2004112713A2 (en) * | 2003-06-20 | 2004-12-29 | Royer Biomedical, Inc. | Drug polymer complexes |
| RU2278675C1 (en) * | 2005-01-17 | 2006-06-27 | Юрий Борисович Иванов | Antibacterial agent and pharmaceutical composition comprising effective amount of antibacterial agent |
| WO2011132171A1 (en) * | 2010-04-23 | 2011-10-27 | Piramal Life Sciences Limited | Nitric oxide releasing prodrugs of therapeutic agents |
| WO2014080251A1 (en) * | 2012-11-24 | 2014-05-30 | Hangzhou Dac Biotech Co., Ltd. | Hydrophilic linkers and their uses for conjugation of drugs to cell binding molecules |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DK2125850T3 (en) | MACROCYCLIC POLYMORPHS, COMPOSITIONS INCLUDING SUCH POLYMORPHS, PROCEDURES FOR PREPARING AND USING THEREOF | |
| CA2327775C (en) | 15-membered lactams ketolides with antibacterial activity | |
| JP4843614B2 (en) | Acylated nonadepsipeptide II | |
| Kirst et al. | Synthesis and evaluation of tylosin-related macrolides modified at the aldehyde function: a new series of orally effective antibiotics | |
| Szűcs et al. | N-Terminal guanidine derivatives of teicoplanin antibiotics strongly active against glycopeptide resistant Enterococcus faecium | |
| US20190211003A1 (en) | Novel 16-member triamilide derivatives and uses thereof | |
| He | Mannopeptimycins, a novel class of glycopeptide antibiotics active against Gram-positive bacteria | |
| Li et al. | Synthesis and structure-bactericidal activity relationships of non-ketolides: 9-Oxime clarithromycin 11, 12-cyclic carbonate featured with three-to eight-atom-length spacers at 3-OH | |
| RU2578604C1 (en) | Chimeric antibiotics based on azithromycin and glycopeptide antibiotics, having antibacterial activity and synthesis method thereof | |
| Hunziker et al. | Novel ketolide antibiotics with a fused five-membered lactone ring––synthesis, physicochemical and antimicrobial properties | |
| WO2000042067A1 (en) | Saccharides linked to compounds that bind cell-surface peptides or proteins | |
| CN111533771A (en) | Gamithromycin related substance and synthesis and separation method thereof | |
| Minowa et al. | Synthesis and antibacterial activity of novel neamine derivatives | |
| He et al. | Mannopeptimycin esters and carbonates, potent antibiotic agents against drug-resistant bacteria | |
| Sum et al. | Novel ether derivatives of mannopeptimycin glycopeptide antibiotic | |
| Bauer et al. | Impact of stereochemistry on the biological activity of novel oleandomycin derivatives | |
| HRP990116A2 (en) | NOVEL 8a AND 9a- 15-MEMBERED LACTAMES | |
| Gebhardt et al. | Semisynthetic preparation of leucomycin derivatives: Introduction of aromatic side chains by reductive amination | |
| RU2570425C1 (en) | Chimeric antibiotics based on glycopeptides and 11,12-cyclic carbonate azithromycin and method for production thereof | |
| Malabarba et al. | Synthesis and biological evaluation of de (acetylglucosaminyl) didehydrodeoxy derivatives of teicoplanin antibiotics | |
| Chu et al. | A novel everninomicin antibiotic active against multidrug-resistant bacteria | |
| Shao et al. | Synthesis and antibacterial activity of N4-mono alkyl derivatives of novel glycopeptide LYV07ww01 | |
| CA2256017C (en) | .beta.,.beta.-disubstituted derivatives of 9-deoxo-9a-n-ethenyl-9a-aza-9a-homoerythromycin a | |
| CN104788519B (en) | Sixteen-ring triamine lactone derivatives and its application | |
| US20040266997A1 (en) | Degradation products of azithromycin, and methods for their indentification |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20181223 |