RU2575249C2 - Novel palladium catalyst, method for production thereof and use thereof - Google Patents
Novel palladium catalyst, method for production thereof and use thereof Download PDFInfo
- Publication number
- RU2575249C2 RU2575249C2 RU2013132388/04A RU2013132388A RU2575249C2 RU 2575249 C2 RU2575249 C2 RU 2575249C2 RU 2013132388/04 A RU2013132388/04 A RU 2013132388/04A RU 2013132388 A RU2013132388 A RU 2013132388A RU 2575249 C2 RU2575249 C2 RU 2575249C2
- Authority
- RU
- Russia
- Prior art keywords
- catalyst
- palladium
- reaction
- nmr
- mhz
- Prior art date
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 111
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 title claims abstract description 69
- 229910052763 palladium Inorganic materials 0.000 title claims abstract description 21
- 238000004519 manufacturing process Methods 0.000 title description 3
- ITJHLZVYLDBFOJ-UHFFFAOYSA-N tris[3,5-bis(trifluoromethyl)phenyl]phosphane Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(P(C=2C=C(C=C(C=2)C(F)(F)F)C(F)(F)F)C=2C=C(C=C(C=2)C(F)(F)F)C(F)(F)F)=C1 ITJHLZVYLDBFOJ-UHFFFAOYSA-N 0.000 claims abstract description 11
- 150000003839 salts Chemical class 0.000 claims abstract description 10
- 239000011780 sodium chloride Substances 0.000 claims abstract description 10
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 7
- 238000005984 hydrogenation reaction Methods 0.000 claims abstract description 7
- 238000005859 coupling reaction Methods 0.000 claims abstract description 6
- 238000006243 chemical reaction Methods 0.000 claims description 70
- 238000000354 decomposition reaction Methods 0.000 claims description 30
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 30
- 239000000758 substrate Substances 0.000 claims description 22
- 150000001875 compounds Chemical class 0.000 claims description 19
- 238000006880 cross-coupling reaction Methods 0.000 claims description 12
- 238000006069 Suzuki reaction reaction Methods 0.000 claims description 8
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 claims description 6
- 238000005160 1H NMR spectroscopy Methods 0.000 claims description 5
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 claims description 4
- 239000012298 atmosphere Substances 0.000 claims description 4
- 238000002844 melting Methods 0.000 claims description 4
- 238000007341 Heck reaction Methods 0.000 claims description 3
- 238000000113 differential scanning calorimetry Methods 0.000 claims description 3
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 claims description 3
- 238000002955 isolation Methods 0.000 claims description 2
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 claims description 2
- 238000011084 recovery Methods 0.000 claims description 2
- 238000003477 Sonogashira cross-coupling reaction Methods 0.000 claims 1
- 230000001808 coupling Effects 0.000 claims 1
- 238000010168 coupling process Methods 0.000 claims 1
- 230000037029 cross reaction Effects 0.000 claims 1
- 230000003595 spectral Effects 0.000 claims 1
- 239000000126 substance Substances 0.000 abstract description 23
- 230000000694 effects Effects 0.000 abstract 1
- 239000000047 product Substances 0.000 description 40
- 239000011541 reaction mixture Substances 0.000 description 37
- 235000005979 Citrus limon Nutrition 0.000 description 29
- 240000002268 Citrus limon Species 0.000 description 29
- OKKJLVBELUTLKV-UHFFFAOYSA-N methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 26
- 239000000203 mixture Substances 0.000 description 26
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- 239000002904 solvent Substances 0.000 description 18
- 230000015572 biosynthetic process Effects 0.000 description 13
- PPBRXRYQALVLMV-UHFFFAOYSA-N styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 13
- 238000005755 formation reaction Methods 0.000 description 12
- ZKHQWZAMYRWXGA-KQYNXXCUSA-N Adenosine triphosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-N 0.000 description 11
- 239000003153 chemical reaction reagent Substances 0.000 description 11
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- -1 tri- [3,5-bis (fluoromethyl) phenyl] phosphine Chemical compound 0.000 description 10
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 9
- 230000035484 reaction time Effects 0.000 description 9
- ZWRMSOMIVOVMQX-UHFFFAOYSA-M 2-(2-aminoethyldisulfanyl)ethanamine;palladium(2+);chloride Chemical compound [Cl-].[Pd+2].NCCSSCCN ZWRMSOMIVOVMQX-UHFFFAOYSA-M 0.000 description 8
- 150000001499 aryl bromides Chemical class 0.000 description 8
- HEDRZPFGACZZDS-UHFFFAOYSA-N chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- VLKZOEOYAKHREP-UHFFFAOYSA-N hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 7
- 229910052751 metal Inorganic materials 0.000 description 7
- 239000002184 metal Substances 0.000 description 7
- IMRWILPUOVGIMU-UHFFFAOYSA-N 2-bromopyridine Chemical compound BrC1=CC=CC=N1 IMRWILPUOVGIMU-UHFFFAOYSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N acetic acid ethyl ester Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 239000012300 argon atmosphere Substances 0.000 description 6
- 230000000875 corresponding Effects 0.000 description 6
- 230000003197 catalytic Effects 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- 239000012535 impurity Substances 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 238000005259 measurement Methods 0.000 description 5
- 239000001184 potassium carbonate Substances 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 238000005070 sampling Methods 0.000 description 5
- UEXCJVNBTNXOEH-UHFFFAOYSA-N Phenylacetylene Chemical group C#CC1=CC=CC=C1 UEXCJVNBTNXOEH-UHFFFAOYSA-N 0.000 description 4
- 150000001298 alcohols Chemical class 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- UHOVQNZJYSORNB-UHFFFAOYSA-N benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 4
- 230000027455 binding Effects 0.000 description 4
- 238000004090 dissolution Methods 0.000 description 4
- 238000004817 gas chromatography Methods 0.000 description 4
- 239000008079 hexane Substances 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N n-butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- 230000000717 retained Effects 0.000 description 4
- PJANXHGTPQOBST-VAWYXSNFSA-N (E)-Stilbene Chemical class C=1C=CC=CC=1/C=C/C1=CC=CC=C1 PJANXHGTPQOBST-VAWYXSNFSA-N 0.000 description 3
- KNNUFFUGRZXENC-UHFFFAOYSA-N O=C1BCCC1=O Chemical compound O=C1BCCC1=O KNNUFFUGRZXENC-UHFFFAOYSA-N 0.000 description 3
- 230000024881 catalytic activity Effects 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 239000012429 reaction media Substances 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- ZBTMRBYMKUEVEU-UHFFFAOYSA-N 1-bromo-4-methylbenzene Chemical compound CC1=CC=C(Br)C=C1 ZBTMRBYMKUEVEU-UHFFFAOYSA-N 0.000 description 2
- OPCXBVHARFWOOU-UHFFFAOYSA-N 2-(4-ethoxy-3-methylphenyl)-1,3,2-dioxaborolane Chemical compound C1=C(C)C(OCC)=CC=C1B1OCCO1 OPCXBVHARFWOOU-UHFFFAOYSA-N 0.000 description 2
- ZTLKPHPUXGOLHV-UHFFFAOYSA-N 2-(4-ethoxy-3-methylphenyl)pyridine Chemical compound C1=C(C)C(OCC)=CC=C1C1=CC=CC=N1 ZTLKPHPUXGOLHV-UHFFFAOYSA-N 0.000 description 2
- VXWVFZFZYXOBTA-UHFFFAOYSA-N 5-bromo-1H-indole Chemical compound BrC1=CC=C2NC=CC2=C1 VXWVFZFZYXOBTA-UHFFFAOYSA-N 0.000 description 2
- CYJZJGYYTFQQBY-UHFFFAOYSA-N 5-bromoisoquinoline Chemical compound N1=CC=C2C(Br)=CC=CC2=C1 CYJZJGYYTFQQBY-UHFFFAOYSA-N 0.000 description 2
- QARVLSVVCXYDNA-UHFFFAOYSA-N Bromobenzene Chemical compound BrC1=CC=CC=C1 QARVLSVVCXYDNA-UHFFFAOYSA-N 0.000 description 2
- 238000007125 Buchwald synthesis reaction Methods 0.000 description 2
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 125000004432 carbon atoms Chemical group C* 0.000 description 2
- 238000006555 catalytic reaction Methods 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 150000002475 indoles Chemical class 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N iso-propanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 239000005373 porous glass Substances 0.000 description 2
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N t-BuOH Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- YXFVVABEGXRONW-UHFFFAOYSA-N toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 2
- ZISCOWXWCHUSMH-VOTSOKGWSA-N (E)-4-nitrostilbene Chemical compound C1=CC([N+](=O)[O-])=CC=C1\C=C\C1=CC=CC=C1 ZISCOWXWCHUSMH-VOTSOKGWSA-N 0.000 description 1
- IPLYZSMSIDWHMI-VAWYXSNFSA-N 1,3-dimethyl-2-[(E)-2-phenylethenyl]benzene Chemical compound CC1=CC=CC(C)=C1\C=C\C1=CC=CC=C1 IPLYZSMSIDWHMI-VAWYXSNFSA-N 0.000 description 1
- QYMXXJHRSKCEHY-CMDGGOBGSA-N 1,3-dimethyl-5-[(E)-2-phenylethenyl]benzene Chemical compound CC1=CC(C)=CC(\C=C\C=2C=CC=CC=2)=C1 QYMXXJHRSKCEHY-CMDGGOBGSA-N 0.000 description 1
- NUAGVTUKHLNXKJ-MDZDMXLPSA-N 1-fluoro-3-[(E)-2-phenylethenyl]benzene Chemical compound FC1=CC=CC(\C=C\C=2C=CC=CC=2)=C1 NUAGVTUKHLNXKJ-MDZDMXLPSA-N 0.000 description 1
- MDRVHDXASYPUCB-VAWYXSNFSA-N 1-methyl-4-[(E)-2-phenylethenyl]benzene Chemical compound C1=CC(C)=CC=C1\C=C\C1=CC=CC=C1 MDRVHDXASYPUCB-VAWYXSNFSA-N 0.000 description 1
- AVFZOVWCLRSYKC-UHFFFAOYSA-N 1-methylpyrrolidine Chemical compound CN1CCCC1 AVFZOVWCLRSYKC-UHFFFAOYSA-N 0.000 description 1
- JWCIMYGAABSRQD-UHFFFAOYSA-N 1-nitro-4-(2-phenylethynyl)benzene Chemical compound C1=CC([N+](=O)[O-])=CC=C1C#CC1=CC=CC=C1 JWCIMYGAABSRQD-UHFFFAOYSA-N 0.000 description 1
- LLSKXGRDUPMXLC-UHFFFAOYSA-N 1-phenylpiperidine Chemical compound C1CCCCN1C1=CC=CC=C1 LLSKXGRDUPMXLC-UHFFFAOYSA-N 0.000 description 1
- FQUPCEHJCVWBRZ-CMDGGOBGSA-N 2,4-dimethoxy-1-[(E)-2-phenylethenyl]benzene Chemical compound COC1=CC(OC)=CC=C1\C=C\C1=CC=CC=C1 FQUPCEHJCVWBRZ-CMDGGOBGSA-N 0.000 description 1
- AEWIQMNETZVDBM-UHFFFAOYSA-N 2-(2-phenylethynyl)-1,3,5-tri(propan-2-yl)benzene Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C#CC1=CC=CC=C1 AEWIQMNETZVDBM-UHFFFAOYSA-N 0.000 description 1
- SZVSCIGDTZNNPO-UHFFFAOYSA-N 2-(4-ethoxy-3-methoxyphenyl)pyridine Chemical compound C1=C(OC)C(OCC)=CC=C1C1=CC=CC=N1 SZVSCIGDTZNNPO-UHFFFAOYSA-N 0.000 description 1
- DBELOSOZLGEZBM-UHFFFAOYSA-N 2-bromo-3-methylbut-2-ene Chemical compound CC(C)=C(C)Br DBELOSOZLGEZBM-UHFFFAOYSA-N 0.000 description 1
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- WBHPBFALEAUFHE-UHFFFAOYSA-N 5-(3,4-dimethoxyphenyl)-1H-indole Chemical compound C1=C(OC)C(OC)=CC=C1C1=CC=C(NC=C2)C2=C1 WBHPBFALEAUFHE-UHFFFAOYSA-N 0.000 description 1
- UPEZZOOZPQQCTL-UHFFFAOYSA-N 5-(4-methylphenyl)-1H-indole Chemical compound C1=CC(C)=CC=C1C1=CC=C(NC=C2)C2=C1 UPEZZOOZPQQCTL-UHFFFAOYSA-N 0.000 description 1
- QGUXCJZUDOLSPJ-OUKQBFOZSA-N CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1\C=C\C1=CC=CC=C1 Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1\C=C\C1=CC=CC=C1 QGUXCJZUDOLSPJ-OUKQBFOZSA-N 0.000 description 1
- 206010065929 Cardiovascular insufficiency Diseases 0.000 description 1
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N Potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 238000006161 Suzuki-Miyaura coupling reaction Methods 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N Trifluoromethanesulfonic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- 229940054051 antipsychotic Indole derivatives Drugs 0.000 description 1
- 125000004429 atoms Chemical group 0.000 description 1
- 235000020127 ayran Nutrition 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N boron Chemical group [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 239000002026 chloroform extract Substances 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 201000002574 conversion disease Diseases 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N deuterated chloroform Substances [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical class C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 1
- 239000012156 elution solvent Substances 0.000 description 1
- SQYOVAAJHMOZIX-UHFFFAOYSA-N ethanol;2-methylpropan-2-ol;propan-2-ol Chemical compound CCO.CC(C)O.CC(C)(C)O SQYOVAAJHMOZIX-UHFFFAOYSA-N 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 238000000806 fluorine-19 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 229940074076 glycerol formal Drugs 0.000 description 1
- 125000005842 heteroatoms Chemical group 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 150000002537 isoquinolines Chemical class 0.000 description 1
- 238000002386 leaching Methods 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 238000000034 method Methods 0.000 description 1
- 238000003541 multi-stage reaction Methods 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L na2so4 Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 125000000962 organic group Chemical group 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 238000005020 pharmaceutical industry Methods 0.000 description 1
- 125000004437 phosphorous atoms Chemical group 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 238000001394 phosphorus-31 nuclear magnetic resonance spectrum Methods 0.000 description 1
- NQRYJNQNLNOLGT-UHFFFAOYSA-N piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 1
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Inorganic materials [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- 239000003638 reducing agent Substances 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000002194 synthesizing Effects 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N tin hydride Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-M toluene-4-sulfonate Chemical compound CC1=CC=C(S([O-])(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-M 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 125000001680 trimethoxyphenyl group Chemical group 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Images
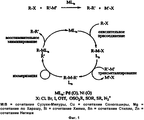
Abstract
Description
Изобретение относится к новому палладиевому катализатору, представляющему собой комплекс три-[3,5-бис(фторметил)-фенил]-фосфина и палладия(0).The invention relates to a new palladium catalyst, which is a complex of tri- [3,5-bis (fluoromethyl) phenyl] phosphine and palladium (0).
Изобретение дополнительно относится к способу получения нового катализатора. Изобретение также относится к применению нового катализатора в реакциях, требующих такие катализаторы, в частности в реакциях с образованием С-С связи (реакции кросс-сочетания, такие как реакция Сузуки, Хека, Стилле и др. сочетания), в реакциях с образованием связи С-гетероатом (C-N, С-О, C-S, С-Р, главным образом C-N) (например, реакция Бухвальда), а также при реакциях гидрогенизации.The invention further relates to a method for producing a new catalyst. The invention also relates to the use of a new catalyst in reactions requiring such catalysts, in particular in reactions with the formation of a C — C bond (cross-coupling reactions, such as the Suzuki, Heck, Stille, and other combinations), in reactions with the formation of a C bond a heteroatom (CN, CO, CS, CP, mainly CN) (for example, the Buchwald reaction), as well as in hydrogenation reactions.
Так как в настоящее время реакции кросс-сочетания, которые катализируются комплексами переходных металлов (наиболее часто комплексами Pd и Ni), играют исключительную роль в формировании С-С связи и такие реакции привели к радикальным изменениям в путях синтеза, изобретение будет обсуждаться ниже в первую очередь в связи с реакциями кросс-сочетания, однако без ограничения его объема данным способом применения.Since at present cross-coupling reactions, which are catalyzed by transition metal complexes (most often Pd and Ni complexes), play an exceptional role in the formation of CC bonds and such reactions have led to radical changes in the synthesis pathways, the invention will be discussed below in the first queue in connection with cross-coupling reactions, but without limiting its scope by this method of application.
Общий процесс реакций кросс-сочетания можно описать какThe general process of cross-coupling reactions can be described as
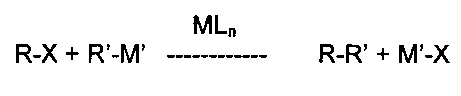
гдеWhere
R и R′ представляют собой органические группы, подлежащие связыванию С-С связью,R and R ′ are organic groups to be bound by a C — C bond,
М представляет сбой металлический компонент каталитического комплекса,M represents a failure of the metal component of the catalytic complex,
L представляет собой лиганды, присутствующие в каталитическом комплексе,L represents ligands present in the catalytic complex,
n представляет собой число присутствующих лигандов,n represents the number of ligands present,
X представляет собой замещаемый атом или группу (например, Cl, Br, I, трифлат, мезилат, тозилат), иX represents a substituted atom or group (e.g., Cl, Br, I, triflate, mesylate, tosylate), and
М′ представляет собой металл или группу, содержащую металл, соответствующий типу рассматриваемой реакции кросс-сочетания (например, данным металлическим компонентом является бор для сочетания Сузуки-Мияуры, медь для сочетания Соногаширы, магний для сочетания по Харашу, кремний для сочетания Хияма, олово для сочетания Стилле, цинк для сочетания Негиши и т.д.).M ′ is a metal or group containing a metal corresponding to the type of cross-coupling reaction under consideration (for example, this metal component is boron for the Suzuki-Miyaura combination, copper for the Sonogashira combination, magnesium for the Harash combination, silicon for the Hiyama combination, tin for Stille combinations, zinc for Negishi combinations, etc.).
Общий механизм реакций кросс-сочетания изображен на Фиг. 1.The general mechanism of cross-coupling reactions is depicted in FIG. one.
Однако в аспектах практического применения данные способы имеют некоторые недостатки, которые особенно выражены в области фармацевтической промышленности. Одним из них является то, что требуются довольно большие количества катализатора (1-5 мол. % относительно субстрата), помимо этого, металлические примеси, источником которых является катализатор, как правило, можно удалить из конечного продукта в основном только утомительными и дорогими способами. Последнее является особенно справедливым для палладиевых катализаторов, которые, кроме того, в значительной степени подвержены разложению. В качестве примера, когда тетракис(трифенилфосфин)палладий (0) формулы (II)However, in aspects of practical application of these methods have some disadvantages, which are especially pronounced in the field of pharmaceutical industry. One of them is that rather large amounts of catalyst are required (1-5 mol.% Relative to the substrate), in addition, metal impurities, the source of which is the catalyst, can usually be removed from the final product only by tedious and expensive methods. The latter is especially true for palladium catalysts, which, in addition, are highly susceptible to decomposition. As an example, when tetrakis (triphenylphosphine) palladium (0) of the formula (II)

который до сих пор является катализатором, часто применяемым в промышленном масштабе, хранят на воздухе при комнатной температуре, значительное количество палладиевой черни отделяется в течение короткого времени, таким образом, его желательно хранить в холодильнике в атмосфере аргона. Несмотря на то что реакции кросс-сочетания с применением катализатора формулы (II) проводят в инертной атмосфере, отделение палладиевой черни все еще широко распространено, что приводит не только к значительной потере катализатора, но и к тому, что следует вводить утомительные трудоемкие и дорогостоящие стадии очистки.which is still a catalyst that is often used on an industrial scale, is stored in air at room temperature, a significant amount of palladium black is separated in a short time, so it is advisable to store it in a refrigerator in an argon atmosphere. Although cross-coupling reactions using a catalyst of formula (II) are carried out in an inert atmosphere, the separation of palladium black is still widespread, which leads not only to a significant loss of catalyst, but also to the introduction of tedious laborious and expensive stages cleaning up.
Целью данного изобретения было получение нового катализатора на основе комплекса палладия (0), который является значительно более стабильным, чем катализаторы на основе комплекса палладия (0), которые применяли раньше для реакций кросс-сочетания, а также делает возможным значительно уменьшить количество катализатора, требуемое на 1 моль субстрата. В рамках данной области нашей основной целью являлось исключить образование палладиевой черни, так как палладиевая чернь, образующаяся из комплексов Pd(0), является конечным состоянием, причем разложение катализатора заметно снижает общую каталитическую активность. Кроме того, неконтролируемое разложение катализатора часто приводит к тому, что неконтролируемые количества P выщелачиваются в продукт.The aim of this invention was to obtain a new catalyst based on a palladium complex (0), which is significantly more stable than catalysts based on a palladium complex (0), which were used earlier for cross-coupling reactions, and also makes it possible to significantly reduce the amount of catalyst required per 1 mol of substrate. Within the framework of this field, our main goal was to exclude the formation of palladium black, since palladium black formed from Pd (0) complexes is the final state, and decomposition of the catalyst noticeably reduces the total catalytic activity. In addition, uncontrolled decomposition of the catalyst often leads to uncontrolled amounts of P leaching into the product.
На данный момент обнаружили, что катализатор на основе комплекса палладия (0) согласно изобретению полностью удовлетворяет указанным выше требованиям, а также имеет дополнительные преимущества.It has now been discovered that the catalyst based on the palladium complex (0) according to the invention fully satisfies the above requirements, and also has additional advantages.
Таким образом, в одном аспекте настоящее изобретение относится к комплексу палладия (0).Thus, in one aspect, the present invention relates to a palladium complex (0).
Данное соединение представляет собой твердое вещество яркого лимонно-желтого цвета с превосходной стабильностью: образование палладиевой черни не наблюдалось даже в образцах, которые хранили на воздухе при комнатной температуре в течение более 20 месяцев.This compound is a bright lemon yellow solid with excellent stability: the formation of palladium black was not observed even in samples that were stored in air at room temperature for more than 20 months.
Данное соединение хранили на воздухе при различных значениях T температуры и влажности. Образцы периодически отбирали из хранящегося продукта и разложение продукта анализировали на основе 31P, 19F, 13C и 1H спектров ЯМР (ядерно-магнитный резонанс). Результаты приведены в следующей таблице.This compound was stored in air at various T values of temperature and humidity. Samples were periodically taken from the stored product and product decomposition was analyzed based on 31 P, 19 F, 13 C and 1 H NMR spectra (nuclear magnetic resonance). The results are shown in the following table.
При испытании соединения согласно изобретению ДСК (дифференциальная сканирующая калориметрия) разложение наблюдали при 169,5°C на воздухе при атмосферном давлении. При проведении испытания в инертной атмосфере, обнаружили, что точка плавления соединения составляет 220°C. Просто для сравнения, нефторированный катализатор формулы (II) начинал разлагаться при 98°C.When testing the compound according to the invention, DSC (differential scanning calorimetry) decomposition was observed at 169.5 ° C. in air at atmospheric pressure. When tested in an inert atmosphere, it was found that the melting point of the compound is 220 ° C. Just for comparison, the non-fluorinated catalyst of formula (II) began to decompose at 98 ° C.
Проводили испытания на стабильность на катализаторах на основе комплекса[три-[замещенный фенил]-фосфина и Pd(0), где две из трех 3,5-(трифторметил)-фенильных групп, присоединенных к атому фосфора в лиганде, сохранены, а третья группа замещена моно-, ди- или триметоксифенильной, три-изопропилфенильной или 2-пиридильной группой. Ни одно из таких соединений не могло даже приблизиться к соединению согласно изобретению по стабильности при хранении. Таким образом, превосходная стабильность данного соединения при хранении является очень удивительной характеристикой, которая не проявляется даже у очень близких структурных аналогов.The stability tests were carried out on catalysts based on the complex of [tri- [substituted phenyl] phosphine and Pd (0), where two of the three 3,5- (trifluoromethyl) phenyl groups attached to the phosphorus atom in the ligand were retained, and the third the group is substituted by a mono-, di- or trimethoxyphenyl, tri-isopropylphenyl or 2-pyridyl group. None of these compounds could even come close to the compound according to the invention for storage stability. Thus, the excellent storage stability of this compound is a very surprising characteristic that does not occur even in very close structural analogues.
При исследовании стабильности катализатора в условиях протекания реакции кросс-сочетания обнаружили, что катализатор не чувствителен к увеличению температуры; он сохраняет свою стабильность при любой температуре ниже своей точки плавления. Аналогичным образом, увеличение давления не оказывало влияния на стабильность катализатора.When studying the stability of the catalyst under the conditions of the cross-coupling reaction, it was found that the catalyst is not sensitive to temperature increase; it retains its stability at any temperature below its melting point. Similarly, an increase in pressure did not affect the stability of the catalyst.
При исследовании стабильности катализатора согласно изобретению было обнаружено следующее.When studying the stability of the catalyst according to the invention, the following was discovered.
Катализатор не растворим в воде при промышленных температурах; в то же самое время он остается неограниченно стабильным при хранении в воде.The catalyst is insoluble in water at industrial temperatures; at the same time, it remains unlimited when stored in water.
Растворимость катализатора в спиртах при комнатной температуре увеличивается с увеличением числа атомов углерода в спирте; однако при исследованных температурных интервалах каталитических реакций (110-130°C) его стабильность в спиртах снижается параллельно с увеличением числа атомов углерода спирта. Однако стабильность катализатора можно повысить или даже полностью восстановить при добавлении воды к реакционной смеси. В водных спиртах растворение катализатора начинается при температуре около 90°C и, в зависимости от рассматриваемого спирта, заканчивается при 110-130°C, где каталитическая активность достигает своего максимума. Однако даже при температурах, приводящих к полному растворению, отделения палладиевой черни не наблюдалось. Иногда происходило незначительное допустимое разложение, на которое указывало несильное потемнение цвета реакционной смеси (от лимонно-желтого до желтовато-коричневого). Особенно примечательным является то, что даже в таких условиях можно было достигнуть полного (100%) химического превращения. Для сравнения: когда соединение формулы (II) применяли в качестве катализатора при значительно более мягких условиях, чем те, которые обсуждались выше (атмосферное давление; точка кипения реакционной смеси), было невозможно избежать образования палладиевой черни, что ясно указывает на значительное разложение катализатора.The solubility of the catalyst in alcohols at room temperature increases with an increase in the number of carbon atoms in alcohol; however, at the studied temperature ranges of catalytic reactions (110-130 ° C), its stability in alcohols decreases in parallel with an increase in the number of carbon atoms of the alcohol. However, the stability of the catalyst can be improved or even completely restored by adding water to the reaction mixture. In aqueous alcohols, the dissolution of the catalyst begins at a temperature of about 90 ° C and, depending on the alcohol in question, ends at 110-130 ° C, where the catalytic activity reaches its maximum. However, even at temperatures leading to complete dissolution, separation of palladium black was not observed. Sometimes a slight permissible decomposition occurred, indicated by a slight darkening of the color of the reaction mixture (from lemon yellow to tan). Particularly noteworthy is that, even under such conditions, a complete (100%) chemical conversion could be achieved. For comparison: when the compound of formula (II) was used as a catalyst under much milder conditions than those discussed above (atmospheric pressure; boiling point of the reaction mixture), it was impossible to avoid the formation of palladium black, which clearly indicates a significant decomposition of the catalyst.
Для того чтобы избежать применения давлений выше атмосферного, что нежелательно в промышленных аспектах, стабильность катализатора согласно изобретению также испытывали в важных в промышленном отношении полярных апротонных и неполярных апротонных органических растворителях (например, диметилсульфоксид, диметилформамид, этилметилкетон, метилизобутилкетон, N-метилпирролидин и тетрагидрофуран), в которых катализатор полностью растворяется при более низких температурах. В данных растворителях также не наблюдалось образования палладиевой черни, хотя иногда цвет реакционной смеси становился в некоторой степени темнее во время каталитической реакции (наблюдали изменения цвета от лимонно-желтого до розового, оранжевого, красного или коричневатого). Как и в случае спиртов, обсуждаемых выше, в некоторых из данных растворителей небольшое снижение стабильности катализатора может быть значительно остановлено путем добавления воды к реакционной смеси.In order to avoid the use of pressures above atmospheric, which is undesirable in industrial aspects, the catalyst according to the invention was also tested in industrially important polar aprotic and nonpolar aprotic organic solvents (e.g. dimethyl sulfoxide, dimethylformamide, ethyl methyl ketone, methyl isobutyl ketone, N-methylpyrrolidine and tetrahydrofuran) in which the catalyst is completely soluble at lower temperatures. In these solvents, the formation of palladium black was also not observed, although sometimes the color of the reaction mixture became somewhat darker during the catalytic reaction (color changes from lemon yellow to pink, orange, red or brownish were observed). As with the alcohols discussed above, in some of these solvents, a slight decrease in catalyst stability can be significantly stopped by adding water to the reaction mixture.
При исследовании каталитической активности данного соединения в реакциях кросс-сочетания обнаружили, что при одном и том же субстрате и при прочих равных условиях реакции требуемое количество нового катализатора может быть снижено до части количества похожих известных катализаторов (с 1-5 мол.% относительно субстрата до 0,1-0,3 мол.% относительно субстрата) без какого-либо значимого уменьшения выхода и химического превращения, достигнутых за то же время реакции. Хотя выход и химическое превращение, которые достигаются при тех же условиях реакции, за данное время реакции снижаются, когда количество катализатора дополнительно уменьшают ниже данного уровня, это может быть достаточно скомпенсировано повышением температуры и/или времени реакции. В качестве примера: в реакции сочетания Сузуки 2-бромпиридина и 2-(4-этокси-3-метилфенил)-1,3,2-диоксаборолана, проводимой в 10:1 об./об. смеси метанола и воды, в присутствии K2CO3 при 110°C под давлением, 100% химического превращения достигали в течение 1 часа при применении 0,25 мол.% катализатора согласно изобретению. При уменьшении количества катализатора до 0,05 мол.% (что составляет 20% от прежней величины) достигнутое за один час химическое превращение еще оставалось довольно высоким (81%), а при применении всего 0,005 мол.% катализатора (что составляет 2% от прежней величины и 1-5 тысячных обычных промышленных значений) химическое превращение на 50% даже могло быть достигнуто за 1 час.In the study of the catalytic activity of this compound in cross-coupling reactions, it was found that under the same substrate and ceteris paribus the required amount of new catalyst can be reduced to a fraction of the number of similar known catalysts (from 1-5 mol.% Relative to the substrate to 0.1-0.3 mol% relative to the substrate) without any significant decrease in yield and chemical conversion achieved during the same reaction time. Although the yield and chemical conversion, which are achieved under the same reaction conditions, during a given reaction time decrease when the amount of catalyst is further reduced below this level, this can be sufficiently compensated by an increase in temperature and / or reaction time. As an example: in the reaction of the combination of Suzuki 2-bromopyridine and 2- (4-ethoxy-3-methylphenyl) -1,3,2-dioxaborolan, carried out in 10: 1 vol./about. mixtures of methanol and water, in the presence of K 2 CO 3 at 110 ° C under pressure, 100% chemical conversion was achieved within 1 hour using 0.25 mol.% of the catalyst according to the invention. With a decrease in the amount of catalyst to 0.05 mol% (which is 20% of the previous value), the chemical conversion achieved in one hour still remained quite high (81%), and when using only 0.005 mol% of the catalyst (which is 2% of the previous value and 1-5 thousandths of ordinary industrial values) a chemical conversion of 50% could even be achieved in 1 hour.
В большинстве случаев удаление палладия из продукта не требуется по той причине, что благодаря малому количеству и высокой стабильности нового катализатора, в продукте не остается палладия либо количество остаточного палладия ниже допустимого уровня. Если остаточный палладий все-таки следует удалить, дорогостоящие способы очистки [определенные операции для связывания Pd(0)], обычно применяющиеся для данной цели, могут быть полностью опущены. Остаточный, еще находящийся в комплексах палладий можно удалить путем простых операций (хроматография; фильтрация через недорогой угольный фильтр и т.д.), обычно применяющихся в промышленности, и, как правило, не требуется более одной стадии очистки.In most cases, the removal of palladium from the product is not required because, due to the small amount and high stability of the new catalyst, the product does not have palladium or the amount of residual palladium is below the permissible level. If residual palladium should still be removed, costly purification methods [certain Pd (0) binding operations] commonly used for this purpose may be completely omitted. Residual palladium still in the complexes can be removed by simple operations (chromatography; filtration through an inexpensive carbon filter, etc.), commonly used in industry, and as a rule, more than one purification step is not required.
Изобретение дополнительно относится к способу получения данного катализатора.The invention further relates to a method for producing this catalyst.
Катализатор согласно изобретению можно легко получить путем взаимодействия соли палладия (II) с три-[3,5-бис(трифторметил)-фенил]-фосфином, предпочтительно, с по меньшей мере четырехкратным молярным избытком указанного соединения, и восстановления палладия (II) до палладия (0) в полученной в результате комплексной соли в одно- или многостадийной реакции, предпочтительно проводящейся в одном реакционном сосуде без выделения промежуточных соединений. В качестве соли палладия (II) можно предпочтительно применять дихлорид палладия; предпочтительным восстанавливающим агентом является гидразин-гидрат.The catalyst according to the invention can be easily obtained by reacting a palladium (II) salt with tri- [3,5-bis (trifluoromethyl) phenyl] phosphine, preferably with at least a four-fold molar excess of said compound, and reducing palladium (II) to palladium (0) in the resulting complex salt in a one- or multi-stage reaction, preferably carried out in a single reaction vessel without isolation of intermediate compounds. As the palladium (II) salt, palladium dichloride can preferably be used; a preferred reducing agent is hydrazine hydrate.
Три-[3,5-бис(трифторметил)-фенил]-фосфин, используемый в качестве комплексообразующего агента, является известным веществом [см., например, Н.G. Alt, R. Baumgaertner, Н.A. Brune: Chemische Berichte 119(5), 1694-1703 (1986)].Tri- [3,5-bis (trifluoromethyl) phenyl] phosphine, used as a complexing agent, is a known substance [see, for example, H. G. Alt, R. Baumgaertner, N.A. Brune: Chemische Berichte 119 (5), 1694-1703 (1986)].
Изобретение также относится к применению соединения согласно изобретению в качестве катализатора в реакциях сочетания C-C и C-гетероатом, а также при гидрогенизации.The invention also relates to the use of a compound according to the invention as a catalyst in C-C and C-heteroatom coupling reactions, as well as in hydrogenation.
Обнаружили, что соединение согласно изобретению можно применять в любом типе данных реакций. Условия таких реакций могут быть такими же, как применялись при использовании других катализаторов на основе комплексов Pd(0), с той разницей, что при применении соединения согласно изобретению в качестве катализатора обычно меньших, иногда гораздо меньших количеств катализатора все еще достаточно для проведения реакции. На основе своих общих знаний и на основе информации, представленной в данном описании, специалист в данной области может легко определить оптимальные параметры для реакций с применением катализатора, применяя стандартные способы или иногда простые испытания и принимая во внимание характеристики растворения катализатора. Здесь следует отметить, что идея применения катализатора согласно изобретению, приготовленного in situ (например, Pd2(dba)3 с PPh3(CF3)6), не является целесообразной, так как неконтролируемое образование комплекса и практически мгновенное появление Pd-черни приводит к низким выходам.It was found that the compound according to the invention can be used in any type of these reactions. The conditions of such reactions may be the same as those used with other catalysts based on Pd (0) complexes, with the difference that when using the compounds of the invention as a catalyst, usually smaller, sometimes much smaller amounts of catalyst are still sufficient to carry out the reaction. Based on his general knowledge and on the basis of the information presented in this description, a person skilled in the art can easily determine the optimal parameters for reactions using a catalyst using standard methods or sometimes simple tests and taking into account the dissolution characteristics of the catalyst. It should be noted that the idea of using the catalyst according to the invention, prepared in situ (for example, Pd 2 (dba) 3 with PPh 3 (CF 3 ) 6 ), is not feasible, since the uncontrolled complexation and almost instantaneous appearance of Pd-black leads to low outputs.
Следующие Примеры служат для иллюстрации дополнительных подробностей изобретенияThe following Examples serve to illustrate further details of the invention.
Пример 1Example 1
Получение катализатора согласно изобретениюObtaining a catalyst according to the invention
Аргон барботировали через 30 мл диметилсульфоксида при комнатной температуре, и затем добавляли 6,7 г (0,01 моль) три-[3,5-бис(трифторметил)-фенил]-фосфина и 0,355 г (0,002 моль) хлорида палладия (II). После этого смесь нагревали до 110-130°C. После того как достигали абсолютной прозрачности раствора, что свидетельствовало о том, что комплекс образовался, к смеси добавляли 0,5 г (0,01 моль) гидразин-гидрата. Затем колбу погружали в ледяную воду. Отделившийся продукт фильтровали через пористый стеклянный фильтр и три раза промывали хлороформом. Получали твердое кристаллическое вещество яркого лимонно-желтого цвета с выходом 90%.Argon was bubbled through 30 ml of dimethyl sulfoxide at room temperature, and then 6.7 g (0.01 mol) of tri- [3,5-bis (trifluoromethyl) phenyl] phosphine and 0.355 g (0.002 mol) of palladium (II) chloride were added. ) After that, the mixture was heated to 110-130 ° C. After the solution was completely transparent, indicating that the complex had formed, 0.5 g (0.01 mol) of hydrazine hydrate was added to the mixture. Then the flask was immersed in ice water. The separated product was filtered through a porous glass filter and washed three times with chloroform. A solid crystalline solid of bright lemon yellow color was obtained with a yield of 90%.
Характеристические данные спектра ЯМР: 1Н-ЯМР (300 МГц, ТГФ-d8 (тетрагидрофуран), δ=3,58 млн-1) 8.17 (s, 12H), 7.84 (s, 24H); 13C-ЯМР (75 МГц, ТГФ-d8, δ=67,3 млн-1) 138.1 (C), 133.7 (q, J=38,7 Гц, C), 133.4 (CH), 126.3 (CH), 123.4 (q, J=271,57 Гц, CF3); 31P-ЯМР (300 МГц, ТГФ-d8) 28.77; 19F-ЯМР (300 МГц, ТГФ-d8) -62.94.Characteristic data of the NMR spectrum: 1 H-NMR (300 MHz, THF-d 8 (tetrahydrofuran), δ = 3.58 ppm -1 ) 8.17 (s, 12H), 7.84 (s, 24H); 13 C-NMR (75 MHz, THF-d 8, δ = 67,3 million -1) 138.1 (C), 133.7 (q, J = 38,7 Hz, C), 133.4 (CH) , 126.3 (CH) 123.4 (q, J = 271.57 Hz, CF 3 ); 31 P-NMR (300 MHz, THF-d 8 ) 28.77; 19 F-NMR (300 MHz, THF-d 8 ) -62.94.
Пример 2Example 2
Получение 2-(4-этокси-3-метилфенил)пиридина с помощью реакции сочетания Сузуки с применением 10/1 об./об. смеси метанола и воды в качестве растворителя и катализатора согласно изобретениюObtaining 2- (4-ethoxy-3-methylphenyl) pyridine using the Suzuki coupling reaction using 10/1 vol./about. mixtures of methanol and water as solvent and catalyst according to the invention
Общий протоколGeneral protocol
Количество катализатора, которое будет приведено ниже, 618 мг (3 ммоля) 2-(4-этокси-3-метилфенил)-1,3,2-диоксаборолана и 553 мг (4 ммоля) карбоната калия взвешивали в колбу. Затем колбу помещали в атмосферу аргона и добавляли 10 мл метанола и 1 мл воды. В конце вводили 316 мг (190 мкл, 2 ммоля) 2-бромпиридина (субстрат) с использованием автоматической пипетки. Колбу закрывали и реакционную смесь перемешивали при температуре и в течение времени, приведенных ниже, необязательно при давлении выше атмосферного.The amount of catalyst that will be given below, 618 mg (3 mmol) of 2- (4-ethoxy-3-methylphenyl) -1,3,2-dioxaborolan and 553 mg (4 mmol) of potassium carbonate was weighed into a flask. Then the flask was placed in an argon atmosphere and 10 ml of methanol and 1 ml of water were added. At the end, 316 mg (190 μl, 2 mmol) of 2-bromopyridine (substrate) was introduced using an automatic pipette. The flask was closed and the reaction mixture was stirred at a temperature and for the time given below, optionally at a pressure above atmospheric.
В целях обработки, охлажденную реакционную смесь экстрагировали четыре раза 5 мл хлороформа каждый раз; таким образом, практически все количество катализатора удаляли из продукта. Так как хлороформный экстракт все еще содержал примесь диоксоборолана, отделенное таким образом вещество дополнительно очищали колоночной хроматографией с силикагелем, применяя 3/1 об./об. смесь гексана и этилацетата в качестве элюирующего агента.For processing purposes, the cooled reaction mixture was extracted four times with 5 ml of chloroform each time; thus, almost the entire amount of catalyst was removed from the product. Since the chloroform extract still contained an dioxoborolan impurity, the substance thus separated was further purified by silica gel column chromatography using 3/1 v / v. a mixture of hexane and ethyl acetate as an eluting agent.
Серия испытаний (А)Test Series (A)
В данной серии испытаний реакции проводили при температуре 110°C и при давлении выше атмосферного в течение 1 часа. Количество соединения согласно изобретению было различным, и проверяли, как это его изменение влияет на достигнутое химическое превращение.In this series of tests, the reactions were carried out at a temperature of 110 ° C and at a pressure above atmospheric for 1 hour. The amount of the compound according to the invention was different, and it was checked how its change affects the achieved chemical conversion.
Во всех случаях, представленных в данном описании, величины химического превращения определяли на основе спектров 1H ЯМР или с помощью газовой хроматографии. Результаты представлены в Таблице 1. Несмотря на эти довольно маломасштабные испытываемые реакции, обработка смеси влияет на практический выход, эти данные также приведены для справки.In all cases presented in this description, the chemical conversion values were determined on the basis of 1 H NMR spectra or by gas chromatography. The results are presented in Table 1. Despite these rather small-scale test reactions, the processing of the mixture affects the practical yield; these data are also provided for reference.
Отделения палладиевой черни не наблюдалось ни в одном случае; цвет реакционной смеси сохранялся лимонно-желтым во всех реакциях. Особенно примечательным является то, что 50% химическое превращение еще могло быть достигнуто в течение 1 часа, когда количество катализатора согласно изобретению составляло только 0,005 мол.%. Согласно опыту, приобретенному в других испытаниях, данное снижение эффективности химического превращения может быть скомпенсировано увеличением времени реакции и/или температуры реакции.No separation of palladium mobile was observed in any case; the color of the reaction mixture remained lemon yellow in all reactions. Particularly noteworthy is that 50% chemical conversion could still be achieved within 1 hour, when the amount of catalyst according to the invention was only 0.005 mol%. According to the experience gained in other tests, this decrease in the efficiency of the chemical transformation can be compensated by an increase in the reaction time and / or reaction temperature.
В испытаниях, проводимых в целях проверки, вышеприведенную реакцию повторяли таким образом, что катализатор не добавляли к реакционной смеси. Таким образом, намеревались удостовериться в том, что образование продукта действительно может объясняться тем, что катализатор вводится в очень малом количестве, а не действием каких-либо металлических примесей, которые могли бы присутствовать в растворителях или в колбах. В данных условиях эффективность химического превращения была равна нулю, таким образом, можно утверждать с полной уверенностью, что катализатор согласно изобретению является активным даже в количестве 0,005 мол.%.In tests conducted for verification purposes, the above reaction was repeated so that the catalyst was not added to the reaction mixture. Thus, they intended to make sure that the formation of the product can really be explained by the fact that the catalyst is introduced in a very small amount, and not by the action of any metal impurities that might be present in solvents or in flasks. Under these conditions, the chemical conversion efficiency was zero, thus, it can be stated with full confidence that the catalyst according to the invention is active even in an amount of 0.005 mol%.
Серия испытаний (Б)Test Series (B)
В данной серии испытаний применяли 0,25 мол.% катализатора согласно изобретению для 1 моля 2-бромпиридина в качестве субстрата и реакции проводили в течение 1 часа при температурах, приведенных в Таблице 2, при давлении выше атмосферного, при необходимости. Проверяли, каким образом изменения температуры влияют на достигнутое химическое превращение. Результаты приведены в Таблице 2; практические выходы также приведены для справки.In this series of tests, 0.25 mol% of the catalyst according to the invention was used for 1 mol of 2-bromopyridine as a substrate and the reaction was carried out for 1 hour at temperatures shown in Table 2, above atmospheric pressure, if necessary. Checked how temperature changes affect the achieved chemical conversion. The results are shown in Table 2; practical outputs are also provided for reference.
Наблюдаемые результаты показывают, что при применении 10/1 об./об. смеси метанола и воды в качестве реакционной среды, рекомендуется проводить реакцию сочетания при температуре выше 90°C и при давлении выше атмосферного, что позволяет реакционной смеси оставаться в жидком состоянии. Это можно объяснить тем, что замечательное растворение катализатора происходит при таких температурах. Образования палладиевой черни или любого другого признака разложения катализатора не могли наблюдать ни в одной из реакций. Для сравнения; когда в реакции, которую проводили при температуре 110°C, катализатор согласно изобретению заменяли тем же количеством катализатора формулы (II), реакционная смесь чернела в течение нескольких минут. После завершения реакции очень трудно удалить металлические примеси. Продукт, полученный в данной последней реакции, оставался оранжево-желтым/темно-оранжево-желтым даже после полного удаления палладиевой черни, в то время как при применении катализатора согласно изобретению, получали белоснежный продукт.The observed results show that when applying 10/1 vol./about. mixtures of methanol and water as a reaction medium, it is recommended to carry out the coupling reaction at a temperature above 90 ° C and at a pressure above atmospheric, which allows the reaction mixture to remain in a liquid state. This can be explained by the fact that a remarkable dissolution of the catalyst occurs at such temperatures. The formation of palladium black or any other sign of decomposition of the catalyst could not be observed in any of the reactions. For comparison; when in the reaction that was carried out at a temperature of 110 ° C, the catalyst according to the invention was replaced with the same amount of the catalyst of formula (II), the reaction mixture blackened for several minutes. After completion of the reaction, it is very difficult to remove metallic impurities. The product obtained in this last reaction remained orange-yellow / dark orange-yellow even after the complete removal of palladium black, while using the catalyst according to the invention, a snow-white product was obtained.
Физические константы всех образцов продукта, полученные в Примере 2, в пределах точности измерений хорошо согласовывались друг с другом и с соответствующими параметрами образца аутентичного продукта. Для справки ниже приведены физические константы, измеренные для образца 2-(4-этокси-3-метоксифенил)пиридина, приготовленного в 10/1 об./об. смеси метанола и воды при 110°C в течение 1 часа с применением 0,25 мол.% катализатора согласно изобретению:The physical constants of all product samples obtained in Example 2, within the measurement accuracy, were in good agreement with each other and with the corresponding parameters of the sample of the authentic product. For reference, the physical constants measured for a sample of 2- (4-ethoxy-3-methoxyphenyl) pyridine prepared in 10/1 v / v are given below. mixtures of methanol and water at 110 ° C for 1 hour using 0.25 mol.% of the catalyst according to the invention:
1H ЯМР (300 МГц, CDCl3, δTMS=0 млн-1): 8.65 (d, J=4,8 Гц, 1H), 7.75 (m, 4H), 7.16 (m, 1H), 6.90 (d, J=8,4 Гц, 1H), 4.10 (q, J=6,9 Гц, 2H), 2.31 (s, 3H), 1.45 (t, J=7,2 Гц). 1 H NMR (300 MHz, CDCl 3, δ TMS = 0 million -1): 8.65 (d, J = 4,8 Hz, 1H), 7.75 (m, 4H), 7.16 (m, 1H), 6.90 (d , J = 8.4 Hz, 1H), 4.10 (q, J = 6.9 Hz, 2H), 2.31 (s, 3H), 1.45 (t, J = 7.2 Hz).
13C-ЯМР (75 МГц, CDCl3, δCDCl3=77,00 млн-1): 158.2 (C), 157.3 (C), 149.3 (CH), 136.7 (CH), 131.1 (C), 129 (CH), 127.1 (C), 125.5 (CH), 121.2 (CH), 119.9 (CH), 111.0 (CH), 63.6 (CH2), 16.4 (CH3), 14.9 (CH3). 13 C-NMR (75 MHz, CDCl 3, δ CDCl3 = 77,00 mn -1): 158.2 (C), 157.3 (C), 149.3 (CH), 136.7 (CH), 131.1 (C), 129 (CH ), 127.1 (C), 125.5 (CH), 121.2 (CH), 119.9 (CH), 111.0 (CH), 63.6 (CH 2 ), 16.4 (CH 3 ), 14.9 (CH 3 ).
ИК (KBr, ν см-1): 1604, 1587, 1561, 1467, 1433, 1394, 1309, 1281, 1247, 1181, 1151, 1131, 1109, 1042, 926, 884, 777, 742, 618.IR (KBr, ν cm -1 ): 1604, 1587, 1561, 1467, 1433, 1394, 1309, 1281, 1247, 1181, 1151, 1131, 1109, 1042, 926, 884, 777, 742, 618.
Пример 3Example 3
Получение 2-(4-этокси-3-метилфенил)пиридина с помощью реакции сочетания Сузуки в реакционной среде, отличающейся от 10/1 об./об. смеси метанола и воды, и с применением соединения согласно изобретению в качестве катализатораObtaining 2- (4-ethoxy-3-methylphenyl) pyridine using the Suzuki coupling reaction in a reaction medium other than 10/1 v / v. mixtures of methanol and water, and using the compound of the invention as a catalyst
Реакцию сочетания Сузуки, описанную в Примере 2, повторяли с применением 316 мг (190 мкл, 2 ммоля) 2-бромпиридина в качестве субстрата и общим количеством реакционной среды, составляющим 11 мл, однако условия реакции (состав реакционной смеси; количество катализатора; количество реагента диоксоборолана; время реакции; температура) различались, как показано в Таблице 3. Химическое превращение оценивали, как описано в Примере 2. Результаты приведены в Таблице 3.The Suzuki coupling reaction described in Example 2 was repeated using 316 mg (190 μl, 2 mmol) of 2-bromopyridine as a substrate and a total amount of the reaction medium of 11 ml, however, the reaction conditions (composition of the reaction mixture; amount of catalyst; amount of reagent dioxoborolan; reaction time; temperature) varied as shown in Table 3. Chemical conversion was evaluated as described in Example 2. The results are shown in Table 3.
Когда применяли водный этанол, водный изопропанол и водный трет-бутанол, при времени реакции 1 час, цвет реакционной смеси постепенно становился насыщеннее и становился коричневым; порядок насыщения цвета был следующим: этанол-изопропанол-трет-бутанол. Однако палладиевая чернь не отделялась ни в одном из примеров и степень химического превращения оставалась 100%, отражая то, что катализатор сохранял свою активность. При проведении реакции в смесях гексан/вода, диметоксиэтан/вода и тетрагидрофуран/вода, обнаружили, что качество органического растворителя как компонента реакционной смеси в высокой степени влияет на степень химического превращения, достигаемую за заданный период времени. Это обычное явление для реакций кросс-сочетания. И в этом случае с данными растворителями образования палладиевой черни не могли наблюдать, хотя иногда цвет реакционной смеси становился насыщеннее в течение реакции. Результаты испытаний, которые проводили в смеси тетрагидрофуран/вода, являются особенно примечательными. Испытание также проводили с чрезвычайно малым количеством катализатора (0,002 мол.%, около одной тысячной количества, необходимого для известных катализаторов). Как и в Примере 2, данное чрезвычайно малое количество катализатора вводили в смесь в виде стокового раствора в тетрагидрофуране. Данные ясно показывают, что снижение степени химического превращения может быть достаточно скомпенсировано увеличением времени реакции и/или температуры реакции: при повышении температуры до 130°C и времени реакции до 19 часов можно было достигнуть 100% химического превращения даже с этим чрезвычайно малым количеством катализатора. После проведения контрольного испытания, описанного в Примере 2 (реакция без катализатора), снова убедились в том, что образование продукта может объясняться исключительно наличием катализатора, а не действием каких-либо возможных примесей металлов, которые могли бы присутствовать в растворителях или в колбах. Превосходную стабильность катализатора согласно изобретению хорошо иллюстрирует тот факт, что никакого признака разложения катализатора не могли наблюдать даже после того, как реакцию проводили при температуре 130°C в течение 19 часов, что является очень жестким условием.When aqueous ethanol, aqueous isopropanol and aqueous tert-butanol were used, with a reaction time of 1 hour, the color of the reaction mixture gradually became more saturated and turned brown; the order of color saturation was as follows: ethanol-isopropanol-tert-butanol. However, palladium black did not separate in any of the examples and the degree of chemical conversion remained 100%, reflecting that the catalyst remained active. When carrying out the reaction in hexane / water, dimethoxyethane / water and tetrahydrofuran / water mixtures, it was found that the quality of the organic solvent as a component of the reaction mixture highly affects the degree of chemical conversion achieved over a given period of time. This is common for cross-coupling reactions. In this case, the formation of palladium black could not be observed with these solvents, although sometimes the color of the reaction mixture became more saturated during the reaction. The test results that were carried out in a tetrahydrofuran / water mixture are particularly noteworthy. The test was also carried out with an extremely small amount of catalyst (0.002 mol%, about one thousandth of the amount required for known catalysts). As in Example 2, this extremely small amount of catalyst was introduced into the mixture as a stock solution in tetrahydrofuran. The data clearly show that a decrease in the degree of chemical conversion can be sufficiently compensated by an increase in the reaction time and / or reaction temperature: by increasing the temperature to 130 ° C and the reaction time to 19 hours, 100% chemical conversion could be achieved even with this extremely small amount of catalyst. After the control test described in Example 2 (reaction without a catalyst), they were again convinced that the formation of the product can be explained solely by the presence of a catalyst, and not by the action of any possible metal impurities that could be present in solvents or in flasks. The excellent stability of the catalyst according to the invention is well illustrated by the fact that no sign of decomposition of the catalyst could be observed even after the reaction was carried out at a temperature of 130 ° C for 19 hours, which is a very stringent condition.
Пример 4Example 4
Получение производных пиридина с помощью реакции сочетания Сузуки с применением катализатора согласно изобретениюObtaining pyridine derivatives using the Suzuki coupling reaction using a catalyst according to the invention
Общий протоколGeneral protocol
14 мг (0,25 мол.% относительно субстрата 2-бромпиридин) катализатора, 3 ммоля реагента диоксаборолан и 553 мг (4 ммоля) карбоната калия взвешивали в колбу. Затем колбу помещали в атмосферу аргона и добавляли 10 мл метанола и 1 мл воды. В конце вводили 316 мг (190 мкл, 2 ммоля) 2-бромпиридина (субстрат) автоматической пипеткой. Затем колбу закрывали и реакционную смесь перемешивали в течение 1 часа при температуре 110°C при давлении, необходимом для поддержания жидкого состояния реакционной смеси. Затем реакционную смесь обрабатывали, как описано в Примере 2.14 mg (0.25 mol% relative to the substrate 2-bromopyridine) catalyst, 3 mmol of dioxaborolan reagent and 553 mg (4 mmol) of potassium carbonate were weighed into a flask. Then the flask was placed in an argon atmosphere and 10 ml of methanol and 1 ml of water were added. At the end, 316 mg (190 μl, 2 mmol) of 2-bromopyridine (substrate) was introduced using an automatic pipette. Then the flask was closed and the reaction mixture was stirred for 1 hour at a temperature of 110 ° C at a pressure necessary to maintain the liquid state of the reaction mixture. Then the reaction mixture was processed as described in Example 2.
Применявшиеся реагенты, полученные продукты и их физические константы, а также практические выходы (%) приведены в Таблице 4.The reagents used, the products obtained and their physical constants, as well as practical yields (%) are shown in Table 4.
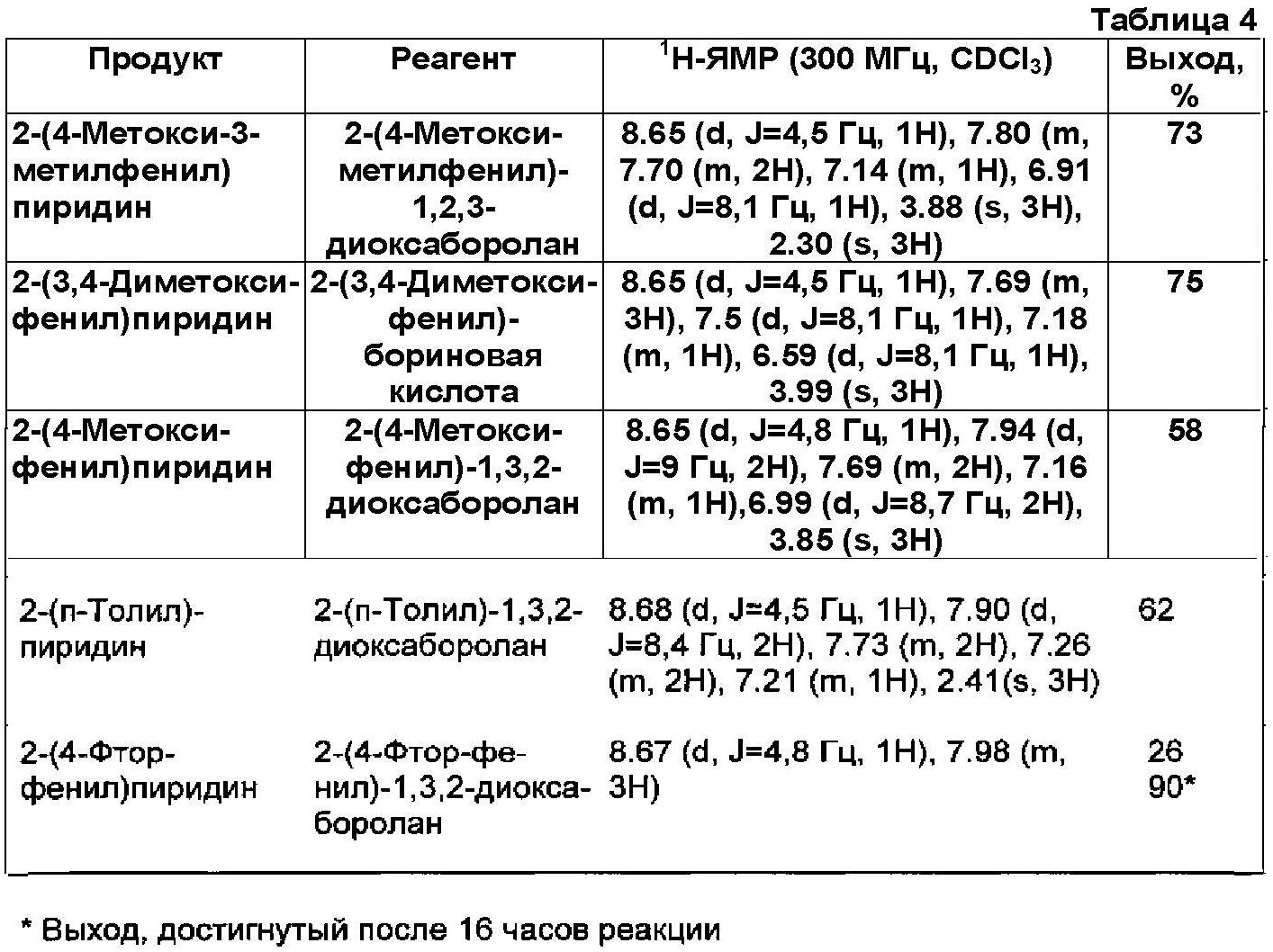
Физические константы всех полученных продуктов, в пределах точности измерений, хорошо согласовывались с соответствующими параметрами образцов аутентичного продукта. Реакционные смеси всегда сохраняли лимонно-желтый цвет, даже после времени реакции, составляющего 16 часов. Никакого признака, относящегося к дополнительному разложению катализатора, не обнаруживали.The physical constants of all the products obtained, within the limits of measurement accuracy, were in good agreement with the corresponding parameters of the samples of the authentic product. The reaction mixtures always maintained a lemon yellow color, even after a reaction time of 16 hours. No evidence of further decomposition of the catalyst was detected.
Пример 5Example 5
Получение производных индола с помощью реакции сочетания Сузуки с применением катализатора согласно изобретениюObtaining indole derivatives using the Suzuki coupling reaction using a catalyst according to the invention
Общий протоколGeneral protocol
14 мг (0,25 мол. % относительно субстрата 5-броминдол) катализатора согласно изобретению, 3 ммоля реагента диоксаборолана, 553 мг (4 ммоля) карбоната калия и 390 мг (2 ммоля) 5-броминдола взвешивали в колбу. Затем колбу помещали в атмосферу аргона и добавляли 10 мл метанола и 1 мл воды. Затем колбу закрывали и реакционную смесь перемешивали в течение 1 ч при 110°C под давлением, необходимым для поддержания жидкого состояния реакционной смеси.14 mg (0.25 mol% relative to the substrate 5-bromoindole) of the catalyst according to the invention, 3 mmol of dioxaborolan reagent, 553 mg (4 mmol) of potassium carbonate and 390 mg (2 mmol) of 5-bromindole were weighed into a flask. Then the flask was placed in an argon atmosphere and 10 ml of methanol and 1 ml of water were added. Then the flask was closed and the reaction mixture was stirred for 1 h at 110 ° C under the pressure necessary to maintain the liquid state of the reaction mixture.
Из полученных конечных продуктов только 5-(п-толил)-1Н-индол растворим в воде. При получении данного соединения реакционную смесь обрабатывали, как описано в Примере 2.Of the final products obtained, only 5- (p-tolyl) -1H-indole is soluble in water. Upon receipt of this compound, the reaction mixture was processed as described in Example 2.
Реакционные смеси, содержащие другие (нерастворимые в воде) индольные соединения, обрабатывали следующим образом.Reaction mixtures containing other (water-insoluble) indole compounds were treated as follows.
К реакционной смеси добавляли 9 мл воды и отделенную твердую фазу, которая содержит катализатор и продукт, отфильтровывали через пористый стеклянный фильтр. Для того чтобы удалить катализатор, полученную в результате твердую фазу растворяли в хлороформе, нерастворимый в хлороформе катализатор отфильтровывали, фильтрат сушили над сульфатом натрия и затем упаривали в вакууме.9 ml of water was added to the reaction mixture, and the separated solid phase, which contains the catalyst and the product, was filtered through a porous glass filter. In order to remove the catalyst, the resulting solid phase was dissolved in chloroform, the chloroform insoluble catalyst was filtered off, the filtrate was dried over sodium sulfate and then evaporated in vacuo.
Используемые реагенты, полученные продукты и их физические константы, а также практические выходы (%) приведены в Таблице 5.The reagents used, the products obtained and their physical constants, as well as practical yields (%) are shown in Table 5.
Физические константы всех полученных продуктов, в пределах точности измерений, хорошо согласовывались с соответствующими параметрами образцов аутентичного продукта. В реакционных смесях не наблюдали образования палладиевой черни; отделенный катализатор во всех случаях сохранял лимонно-желтый цвет.The physical constants of all the products obtained, within the limits of measurement accuracy, were in good agreement with the corresponding parameters of the samples of the authentic product. In the reaction mixtures, the formation of palladium black was not observed; the separated catalyst in all cases retained a lemon yellow color.
Пример 6Example 6
Получение производных изохинолина с помощью реакции сочетания Сузуки с применением катализатора согласно изобретениюObtaining isoquinoline derivatives using the Suzuki coupling reaction using a catalyst according to the invention
Общий протоколGeneral protocol
14 мг (0,25 мол. % относительно субстрата 5-бром-изохинолин) катализатора согласно изобретению, 3 ммоля реагента диоксаборолана, 553 мг (4 ммоля) карбоната калия и 416 мг (2 ммоля) 5-бром-изохинолина взвешивали в колбу. Затем колбу помещали в атмосферу аргона и добавляли 10 мл метанола и 1 мл воды. Затем колбу закрывали и реакционную смесь перемешивали в течение 1 ч при 110°C под давлением, необходимым для поддержания жидкого состояния реакционной смеси. Полученные в результате реакционные смеси обрабатывали, как описано в Примере 2.14 mg (0.25 mol% relative to the substrate 5-bromo-isoquinoline) of the catalyst according to the invention, 3 mmol of dioxaborolan reagent, 553 mg (4 mmol) of potassium carbonate and 416 mg (2 mmol) of 5-bromo-isoquinoline were weighed into a flask. Then the flask was placed in an argon atmosphere and 10 ml of methanol and 1 ml of water were added. Then the flask was closed and the reaction mixture was stirred for 1 h at 110 ° C under the pressure necessary to maintain the liquid state of the reaction mixture. The resulting reaction mixtures were processed as described in Example 2.
Используемые реагенты, полученные продукты и их физические константы, а также практические выходы (%) приведены в Таблице 6.The reagents used, the products obtained and their physical constants, as well as practical yields (%) are shown in Table 6.
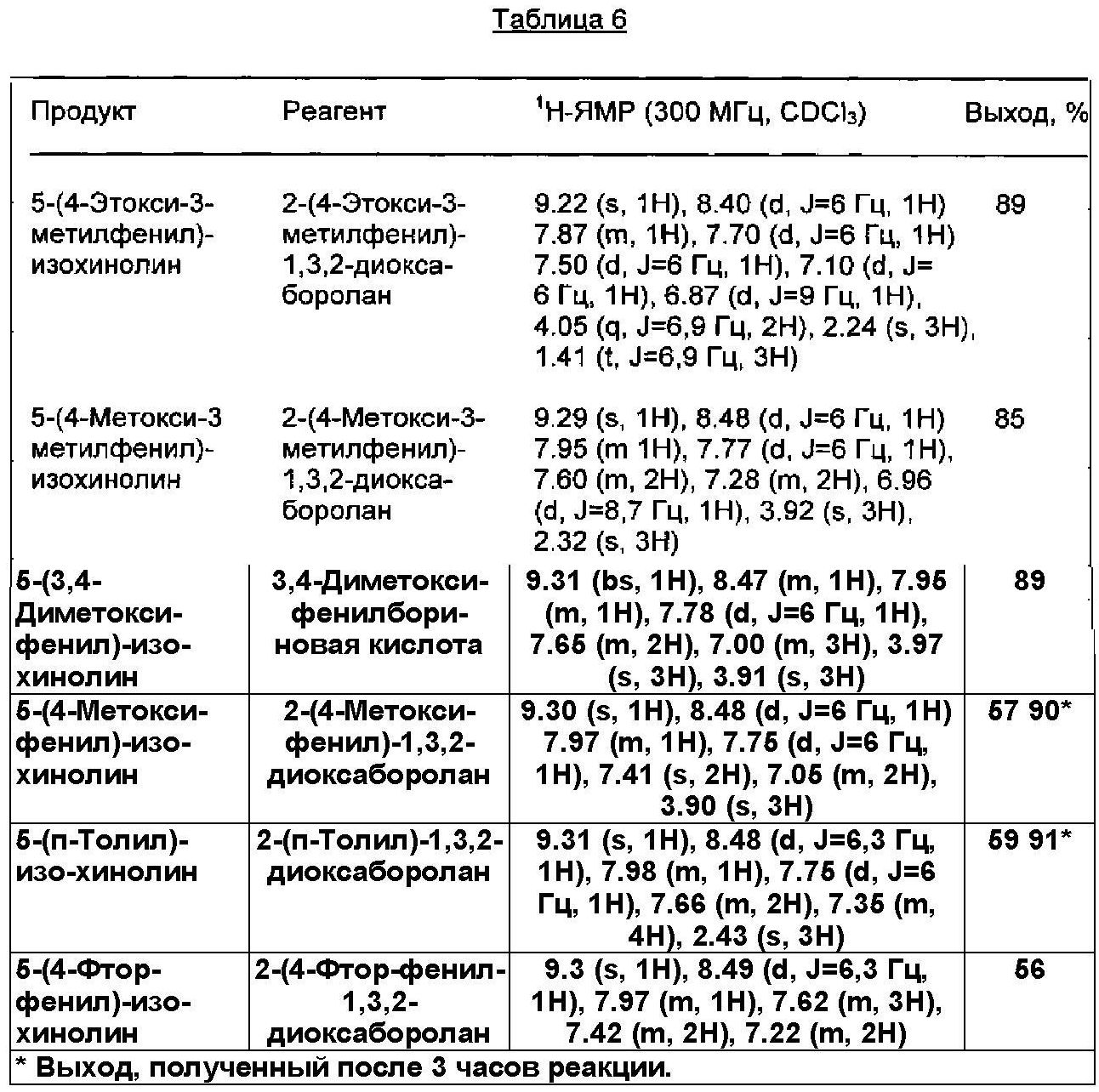
Физические константы всех полученных продуктов, в пределах точности измерений, хорошо согласовывались с соответствующими параметрами образцов аутентичного продукта. Реакционные смеси во всех случаях сохраняли лимонно-желтый цвет, и никакого признака, относящегося к дополнительному разложению катализатора, не могли обнаружить.The physical constants of all the products obtained, within the limits of measurement accuracy, were in good agreement with the corresponding parameters of the samples of the authentic product. The reaction mixtures in all cases retained a lemon yellow color, and no sign of additional decomposition of the catalyst could be detected.
Пример 7Example 7
Получение производных бифенила с помощью реакции сочетания Сузуки с применением катализатора согласно изобретениюObtaining biphenyl derivatives using the Suzuki coupling reaction using a catalyst according to the invention
Общий протоколGeneral protocol
14 мг (0,25 мол.% относительно субстрата п-бромтолуол) катализатора согласно изобретению, 3 ммоля реагента диоксаборолана, 553 мг (4 ммоля) карбоната калия и 342 мг (2 ммоля) п-бромтолуола взвешивали в колбу. Затем колбу помещали в атмосферу аргона и добавляли 10 мл метанола и 1 мл воды. Затем колбу закрывали и реакционную смесь перемешивали в течение 1 ч при 110°C под давлением, необходимым для поддержания жидкого состояния реакционной смеси. Полученные в результате реакционные смеси обрабатывали, как описано в Примере 5.14 mg (0.25 mol% relative to the substrate p-bromtoluene) of the catalyst according to the invention, 3 mmol of dioxaborolan reagent, 553 mg (4 mmol) of potassium carbonate and 342 mg (2 mmol) of p-bromtoluene were weighed into a flask. Then the flask was placed in an argon atmosphere and 10 ml of methanol and 1 ml of water were added. Then the flask was closed and the reaction mixture was stirred for 1 h at 110 ° C under the pressure necessary to maintain the liquid state of the reaction mixture. The resulting reaction mixtures were processed as described in Example 5.
Используемые реагенты, полученные продукты и их физические константы, а также практические выходы (%) приведены в Таблице 7.The reagents used, the products obtained and their physical constants, as well as practical yields (%) are shown in Table 7.
Физические константы всех полученных продуктов, в пределах точности измерений, хорошо согласовывались с соответствующими параметрами образцов аутентичного продукта. Реакционные смеси во всех случаях сохраняли лимонно-желтый цвет, и никакого признака, относящегося к дополнительному разложению катализатора, не могли обнаружить.The physical constants of all the products obtained, within the limits of measurement accuracy, were in good agreement with the corresponding parameters of the samples of the authentic product. The reaction mixtures in all cases retained a lemon yellow color, and no sign of additional decomposition of the catalyst could be detected.
Пример 8Example 8
Получение производных стильбена с помощью реакции сочетания Хека с применением катализатора согласно изобретениюObtaining stilbene derivatives using the Heck coupling reaction using a catalyst according to the invention
Производные стильбена получали с помощью реакции стирола с различными арилбромидами, как показано на следующей схеме реакции:Derivatives of stilbene were obtained by the reaction of styrene with various aryl bromides, as shown in the following reaction scheme:
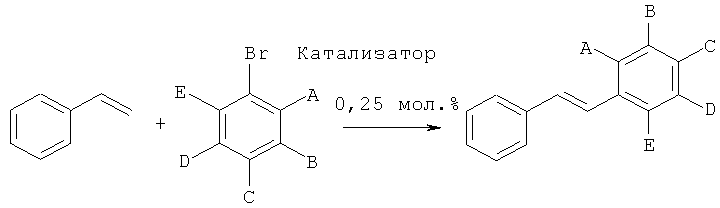
Общий протоколGeneral protocol
552 мг (4 ммоля, 2 экв.) K2CO3, 14 мг (0,25 мол.%, рассчитанных на субстрат арилбромид) катализатора согласно изобретению, 312 мг (0,343 мл, 3 ммоля, 1,5 экв.) стирола, 2 ммоля (1 экв.) субстрата арилбромид и 10 мл 10:1 смеси метанола и воды загружали в высушенный в сушильной печи сосуд Шленка. Реакцию проводили при 110°C в течение 3 часов или 20 часов, как показано в Таблице 8. Степень химического превращения определяли, подвергая реакционные смеси ГХ (газовая хроматография), и затем продукт выделяли. Для испытаний №1, 2, 3 и 5 продукты осаждались из смеси при охлаждении, таким образом, они могли быть выделены путем простой фильтрации; тогда как для испытаний №4, 6 и 7 продукты выделяли с помощью флеш-хроматографии.552 mg (4 mmol, 2 equiv.) K 2 CO 3 , 14 mg (0.25 mol%, calculated on the substrate aryl bromide) of the catalyst according to the invention, 312 mg (0.343 ml, 3 mmol, 1.5 equiv.) Styrene , 2 mmol (1 equiv.) Of aryl bromide substrate and 10 ml of a 10: 1 mixture of methanol and water were loaded into a Schlenk vessel dried in an oven. The reaction was carried out at 110 ° C for 3 hours or 20 hours, as shown in Table 8. The degree of chemical conversion was determined by subjecting the reaction mixture to GC (gas chromatography), and then the product was isolated. For tests No. 1, 2, 3, and 5, the products precipitated from the mixture upon cooling, so that they could be isolated by simple filtration; while for tests No. 4, 6 and 7, the products were isolated using flash chromatography.
Результаты представлены в Таблице 8.The results are presented in Table 8.
Данные ЯМР для полученных в результате производных стильбена являются следующими:NMR data for the resulting stilbene derivatives are as follows:
(E)-3-Фторстильбен: 1H ЯМР (300 МГц, CDCl3) δ 7.53 (d, J=7,5 Гц, 2H), 7.39 (t, J=7,5 Гц, 2H), 7.41-7.22 (m, 4H), 7.11 (s, 1H), 7.10 (s, 1H), 6.99-6.94 (m, 1H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 163.5 (C, d, J=244 Гц), 139.9 (C, d, J=7,65 Гц), 137.1 (C), 130.3 (CH, d, J=8,18 Гц), 129.0 (CH), 128.2 (CH), 127.7 (CH, d, J=2.70 Гц), 126.9 (CH), 122.7 (CH, d, J=2,78 Гц), 114.62 (CH, d, J=21,5 Гц), 113.0 (CH, d, J=21,5 Гц).(E) -3-Fluorostilbene: 1 H NMR (300 MHz, CDCl 3 ) δ 7.53 (d, J = 7.5 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.41-7.22 (m, 4H), 7.11 (s, 1H), 7.10 (s, 1H), 6.99-6.94 (m, 1H); 13 C NMR (ATP) (75 MHz, CDCl 3 ) δ 163.5 (C, d, J = 244 Hz), 139.9 (C, d, J = 7.65 Hz), 137.1 (C), 130.3 (CH, d , J = 8.18 Hz), 129.0 (CH), 128.2 (CH), 127.7 (CH, d, J = 2.70 Hz), 126.9 (CH), 122.7 (CH, d, J = 2.78 Hz), 114.62 (CH, d, J = 21.5 Hz), 113.0 (CH, d, J = 21.5 Hz).
(E)-4-Нитростильбен: 1H ЯМР (300 МГц, CDCl3) δ 8.23-8.21 (m, 2H), 7.63 (d, J=8,7 Гц, 2H), 7.55 (d, J=7,5 Гц, 2H), 7.43-7.25 (m, 4H), 7.14 (d, J=16,5 Гц, 1H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 147.0 (C), 136.4 (C), 133.6 (CH), 129.1 (CH), 127.3 (CH), 127.1 (CH), 126.5 (CH), 124.4 (CH).(E) -4-Nitrostilbene: 1 H NMR (300 MHz, CDCl 3 ) δ 8.23-8.21 (m, 2H), 7.63 (d, J = 8.7 Hz, 2H), 7.55 (d, J = 7, 5 Hz, 2H), 7.43-7.25 (m, 4H), 7.14 (d, J = 16.5 Hz, 1H); 13 C NMR (ATP) (75 MHz, CDCl 3 ) δ 147.0 (C), 136.4 (C), 133.6 (CH), 129.1 (CH), 127.3 (CH), 127.1 (CH), 126.5 (CH), 124.4 (CH).
(E)-4-Метилстильбен: 1H ЯМР (300 МГц, CDCl3) δ 7.54 (d, J=7,8 Гц, 2H), 7.46 (d, J=7,8 Гц, 2H), 7.41-7.36 (m, 2H), 7.31-7.26 (m, 1H), 7.21 (d, J=7,8 Гц, 2H), 7.12 (s, 2H), 2.40 (s, 3H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 137.8 (C), 137.8 (C), 134.8 (C), 129.7 (CH), 128,9 (CH), 128.0 (CH), 127.7 (CH), 126.7 (CH), 21.5 (CH3).(E) -4-Methylstilbene: 1 H NMR (300 MHz, CDCl 3 ) δ 7.54 (d, J = 7.8 Hz, 2H), 7.46 (d, J = 7.8 Hz, 2H), 7.41-7.36 (m, 2H), 7.31-7.26 (m, 1H), 7.21 (d, J = 7.8 Hz, 2H), 7.12 (s, 2H), 2.40 (s, 3H); 13 C NMR (ATP) (75 MHz, CDCl 3 ) δ 137.8 (C), 137.8 (C), 134.8 (C), 129.7 (CH), 128.9 (CH), 128.0 (CH), 127.7 (CH) , 126.7 (CH), 21.5 ( CH 3).
(E)-2,4-Диметоксистильбен: 1H ЯМР (300 МГц, CDCl3) δ 7.53 (d, J=8,4 Гц, 3H), 7.42 (d, J=16,5 Гц, 1H), 7.35 (t, J=7,5 Гц, 2H), 7.24 (dd, J=4,9 Гц, 12,1 Гц, 1H), 7.02 (d, J=16,5 Гц, 1H), 6.53 (dd, J=2,2 Гц, 9,9 Гц, 1H), 6.49 (d, J=2,4 Гц, 1H), 3.88 (s, 1H), 3.84 (s, 1H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 160.5 (C), 138.3 (C), 128.5 (CH), 127.2 (CH), 127.0 (CH), 126.9 (CH), 126.3 (CH), 123.3 (CH), 119.5 (C), 105.0 (CH), 98.5 (CH), 55.5 (CH3), 55.4 (CH3).(E) -2,4-Dimethoxystilbene: 1 H NMR (300 MHz, CDCl 3 ) δ 7.53 (d, J = 8.4 Hz, 3H), 7.42 (d, J = 16.5 Hz, 1H), 7.35 (t, J = 7.5 Hz, 2H), 7.24 (dd, J = 4.9 Hz, 12.1 Hz, 1H), 7.02 (d, J = 16.5 Hz, 1H), 6.53 (dd, J = 2.2 Hz, 9.9 Hz, 1H), 6.49 (d, J = 2.4 Hz, 1H), 3.88 (s, 1H), 3.84 (s, 1H); 13 C NMR (ATP) (75 MHz, CDCl 3 ) δ 160.5 (C), 138.3 (C), 128.5 (CH), 127.2 (CH), 127.0 (CH), 126.9 (CH), 126.3 (CH), 123.3 (CH), 119.5 (C), 105.0 (CH), 98.5 (CH), 55.5 (CH 3 ), 55.4 (CH 3 ).
(E)-3,5-Диметилстильбен: 1H ЯМР (300 МГц, CDCl3) δ 7.55-7.53 (m, 2H), 7.41-7.36 (m, 2H), 7.31-7.26 (m, 1H), 7.18 (s, 2H), 7.11 (d, J=2,4 Гц, 2H), 6.95 (s, 1H), 2.38 (s, 6H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 138.3 (C), 137.5 (C), 129.7 (CH), 129.1 (CH), 128.9 (CH), 128.5 (CH), 127.7 (CH), 126.7 (CH), 124.7 (CH), 21.5 (CH3).(E) -3,5-Dimethylstilbene: 1 H NMR (300 MHz, CDCl 3 ) δ 7.55-7.53 (m, 2H), 7.41-7.36 (m, 2H), 7.31-7.26 (m, 1H), 7.18 ( s, 2H), 7.11 (d, J = 2.4 Hz, 2H), 6.95 (s, 1H), 2.38 (s, 6H); 13 C NMR (ATP) (75 MHz, CDCl 3 ) δ 138.3 (C), 137.5 (C), 129.7 (CH), 129.1 (CH), 128.9 (CH), 128.5 (CH), 127.7 (CH), 126.7 (CH), 124.7 (CH), 21.5 (CH 3 ).
(E)-2,6-Диметилстильбен: 1H ЯМР (300 МГц, CDCl3) δ 7.54-7.52 (m, 2H), 7.42-7.37 (m, 2H), 7.32-7.26 (m, 1H), 7.13 (d, J=16,8 Гц, 1H), 7.1 (m, 3H), (m, 3H), 6.63 (d, J=16,8 Гц, 1H), 2.39 (s, 6H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 137.6 (C), 137.0 (C), 136.2 (C), 134.0 (CH), 128.7 (CH), 127.9 (CH), 127.6 (CH), 126.9 (CH), 126.7 (CH), 126.3 (CH), 21.0 (CH3).(E) -2,6-Dimethylstilbene: 1 H NMR (300 MHz, CDCl 3 ) δ 7.54-7.52 (m, 2H), 7.42-7.37 (m, 2H), 7.32-7.26 (m, 1H), 7.13 ( d, J = 16.8 Hz, 1H), 7.1 (m, 3H), (m, 3H), 6.63 (d, J = 16.8 Hz, 1H), 2.39 (s, 6H); 13 C NMR (ATP) (75 MHz, CDCl 3 ) δ 137.6 (C), 137.0 (C), 136.2 (C), 134.0 (CH), 128.7 (CH), 127.9 (CH), 127.6 (CH), 126.9 (CH), 126.7 (CH), 126.3 (CH), 21.0 (CH 3 ).
(E)-2,4,6-Триизопропилстильбен: 1H ЯМР (300 М Гц, CDCl3) δ 7.54-7.52 (m, 2H), 7.43-7.38 (m, 2H), 7.33-7.26 (m, 1H), 7.22 (d, J=16,5 Гц, 1H), 7.07 (s, 2H), 6.52 (d, J=16,8 Гц, 1H), 3.31 (h, J=6,9 Гц, 2H), 2.94 (h, J=6,9 Гц, 1H), 1.33-1.23 (m, 18H); 13C ЯМР (АТФ) (75 М Гц, CDCl3) δ 142.4 (C), 141.4 (C), 132.3 (C), 128.6 (CH), 127.7 (C), 123.4 (CH), 122.1 (CH), 121.7 (CH), 121.0 (CH), 115.3 (CH), 29.0 (CH), 24.9 (CH), 18.7 (CH3), 18.5 (CH3).(E) -2,4,6-Triisopropylstilbene: 1 H NMR (300 M Hz, CDCl 3 ) δ 7.54-7.52 (m, 2H), 7.43-7.38 (m, 2H), 7.33-7.26 (m, 1H) , 7.22 (d, J = 16.5 Hz, 1H), 7.07 (s, 2H), 6.52 (d, 16.8 Hz, 1H), 3.31 (h, 6.9 Hz, 2H), 2.94 (h, J = 6.9 Hz, 1H); 1.33-1.23 (m, 18H); 13 C NMR (ATP) (75 M Hz, CDCl 3 ) δ 142.4 (C), 141.4 (C), 132.3 (C), 128.6 (CH), 127.7 (C), 123.4 (CH), 122.1 (CH), 121.7 (CH), 121.0 (CH ), 115.3 (CH), 29.0 (CH), 24.9 (CH), 18.7 (CH 3), 18.5 (CH 3).
Пример 9Example 9
Получение производных фенилаиетилена с помощью реакции сочетания Соногаширы с применением катализатора согласно изобретениюPreparation of Phenyl-Ethylene Derivatives Using the Sonogashira Combination Reaction Using the Catalyst of the Invention
Производные фенилацетилена получали с помощью реакции фенилацетилена с различными арилбромидами, как показано на следующей схеме реакции:Derivatives of phenylacetylene were obtained by the reaction of phenylacetylene with various aryl bromides, as shown in the following reaction scheme:
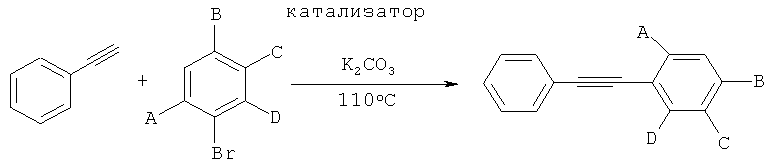
Общий протокол:General Protocol:
276 мг (2 ммоля, 1 экв.) K2CO3, 7 мг (0,25 мол.%, рассчитанных на субстрат арилбромид) катализатора согласно изобретению, 0,165 мл (1,5 ммоля, 1,5 экв.) фенилацетилена, 1 ммоль (1 экв.) субстрата арилбромида и 5 мл растворителя [растворитель (а): 5:1 смесь метанола и воды; растворитель (б): н-бутанол; растворитель (в): глицерол формаль] загружали в высушенный в сушильной печи сосуд Шленка. Реакцию проводили при 110°C в течение 3 часов или 24 часов, как показано в Таблице 9. Количества продуктов определяли, подвергая реакционную смесь ГХ.276 mg (2 mmol, 1 equiv.) K 2 CO 3 , 7 mg (0.25 mol%, calculated on the substrate aryl bromide) of the catalyst according to the invention, 0.165 ml (1.5 mmol, 1.5 equiv.) Of phenylacetylene, 1 mmol (1 equiv.) Of aryl bromide substrate and 5 ml of solvent [solvent (a): 5: 1 mixture of methanol and water; solvent (b): n-butanol; solvent (c): glycerol formal] was loaded into a Schlenk vessel dried in an oven. The reaction was carried out at 110 ° C for 3 hours or 24 hours, as shown in Table 9. The quantities of the products were determined by subjecting the reaction mixture to GC.
Результаты представлены в Таблице 9.The results are presented in Table 9.
Данные ЯМР для полученных в результате производных фенилацетилена выглядят следующим образом:NMR data for the resulting phenylacetylene derivatives are as follows:
1-Метил-4-(фенилэтинил)-бензол: 1H ЯМР (300 МГц, CDCl3) δ 7.43 (d, J=8,0 Гц, 2H), 7.26-7.22 (m, 5H), 6.70 (d, J=8,3 Гц, 1H), 3.75 (s, 3H), 2.13 (s, 3H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 158.2 (C), 133.9 (CH), 131.5 (CH), 130.6 (CH), 128.5 (CH), 128.3 (CH), 127.9 (CH), 123.8 (C), 109.9 (CH), 89.9 (C), 55.4 (CH), 16.1 (CH).1-Methyl-4- (phenylethinyl) benzene: 1 H NMR (300 MHz, CDCl 3 ) δ 7.43 (d, J = 8.0 Hz, 2H), 7.26-7.22 (m, 5H), 6.70 (d, J = 8.3 Hz, 1H), 3.75 (s, 3H), 2.13 (s, 3H); 13 C NMR (ATP) (75 MHz, CDCl 3 ) δ 158.2 (C), 133.9 (CH), 131.5 (CH), 130.6 (CH), 128.5 (CH), 128.3 (CH), 127.9 (CH), 123.8 (C), 109.9 (CH), 89.9 (C), 55.4 (CH), 16.1 (CH).
1-Нитро-4-(фенилэтинил)-бензол: 1H ЯМР (300 МГц, CDCl3) δ 8.17 (d, J=9,0 Гц, 2H), 7.62 (d, J=9,0 Гц, 2H), 7.58-7.55 (m, 2H), 7.41-7.38 (m, 3H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 147.0 (C), 132.3 (CH), 131.9 (CH), 130.3 (C), 129.4 (CH), 129.0 (CH), 123.7 (CH), 122.2 (C), 94.8 (C), 87.7 (C).1-Nitro-4- (phenylethynyl) benzene: 1 H NMR (300 MHz, CDCl 3 ) δ 8.17 (d, J = 9.0 Hz, 2H), 7.62 (d, J = 9.0 Hz, 2H) 7.58-7.55 (m, 2H); 7.41-7.38 (m, 3H); 13 C NMR (ATP) (75 MHz, CDCl 3 ) δ 147.0 (C), 132.3 (CH), 131.9 (CH), 130.3 (C), 129.4 (CH), 129.0 (CH), 123.7 (CH), 122.2 (C), 94.8 (C), 87.7 (C).
1-Метил-4-(фенилэтинил)-бензол: 1H ЯМР (300 МГц, CDCl3) δ 7.60-7.57 (m, 2H), 7.49 (d, J=8,1 Гц, 2H), 7.40-7.35 (m, 3H), 7.20 (d, J=7,9 Гц, 2H), 2.41 (s, 3H); 13C ЯМР (АТФ) (75 МГц, CDCl3) δ 138.6 (C), 131.8 (CH), 131.7 (CH), 129.3 (CH), 128.5 (CH), 128.3 (CH), 123.7 (C), 120.4 (C), 89.8 (C), 89.0 (CH), 21.7 (CH3).1-Methyl-4- (phenylethinyl) benzene: 1 H NMR (300 MHz, CDCl 3 ) δ 7.60-7.57 (m, 2H), 7.49 (d, J = 8.1 Hz, 2H), 7.40-7.35 ( m, 3H), 7.20 (d, J = 7.9 Hz, 2H), 2.41 (s, 3H); 13 C NMR (ATP) (75 MHz, CDCl 3 ) δ 138.6 (C), 131.8 (CH), 131.7 (CH), 129.3 (CH), 128.5 (CH), 128.3 (CH), 123.7 (C), 120.4 (C), 89.8 (C), 89.0 (CH), 21.7 (CH 3 ).
Фенил-(2,4,6-триизопропил-фенил)-ацетилен: 1H ЯМР (300 МГц, CDCl3) δ 7.58 (dd, J=8,0 Гц, 1,4 Гц, 2H), 7.43-7.35 (m, 3H), 7.07 (s, 2H), 3.65 (sept, J=6,9 Гц, 2H), 2.96 (sept, J=6,9 Гц, 1H), 1.36 (d, J=6,9 Гц, 12H), 1.32 (d, J=6,9 Гц, 6H);Phenyl- (2,4,6-triisopropyl-phenyl) -acetylene: 1 H NMR (300 MHz, CDCl 3 ) δ 7.58 (dd, J = 8.0 Hz, 1.4 Hz, 2H), 7.43-7.35 ( m, 3H), 7.07 (s, 2H), 3.65 (sept, J = 6.9 Hz, 2H), 2.96 (sept, J = 6.9 Hz, 1H), 1.36 (d, J = 6.9 Hz , 12H), 1.32 (d, J = 6.9 Hz, 6H);
13C ЯМР (АТФ) (75 МГц, CDCl3) δ 150.9 (C), 149.5 (C), 131.5 (CH), 128.6 (CH), 128.1 (CH), 124.6 (CH), 120.7 (CH), 118.7 (C), 97.0 (C), 87.3 (C), 4.9 (CH3), 32.2 (CH3), 24.2 (CH3), 23.6 (CH3). 13 C NMR (ATP) (75 MHz, CDCl 3 ) δ 150.9 (C), 149.5 (C), 131.5 (CH), 128.6 (CH), 128.1 (CH), 124.6 (CH), 120.7 (CH), 118.7 (C), 97.0 (C), 87.3 (C), 4.9 (CH 3 ), 32.2 (CH 3 ), 24.2 (CH 3 ), 23.6 (CH 3 ).
Описанную выше реакцию повторяли с применением 2-бром-3-метил-бут-2-ена в качестве субстрата. Полученные результаты приведены в Таблице 10.The above reaction was repeated using 2-bromo-3-methyl-but-2-ene as a substrate. The results are shown in Table 10.
Данные ЯМР для полученного в результате продукта выглядят следующим образом: 1H ЯМР (300 МГц, CDCl3) δ 7.45-7.39 (m, 2H), 7.34-7.22 (m, 3H), 2.02 (s, 3H), 1.89 (s, 3H), 1.79 (s, 3H).NMR data for the resulting product are as follows: 1 H NMR (300 MHz, CDCl 3 ) δ 7.45-7.39 (m, 2H), 7.34-7.22 (m, 3H), 2.02 (s, 3H), 1.89 (s , 3H), 1.79 (s, 3H).
Пример 10Example 10
Получение N-фенилпиперидина с помощью реакции Бухвальда с применением соединения согласно изобретению в качестве катализатораObtaining N-phenylpiperidine using the Buchwald reaction using the compound according to the invention as a catalyst
224 мг (2 ммоля) трет-бутоксида калия, 70 мг (0,25 мол.%, рассчитанных на субстрат бромбензол) катализатора согласно изобретению, 105 мкл (1 ммоль) бромбензола, 198 мкл (2 ммоля) пиперидина и 5 мл растворителя загружали в высушенный в сушильной печи сосуд Шленка. Реакционную смесь нагревали на масляной бане при температуре 110°C в течение 24 часов, затем позволяли ей охладиться до комнатной температуры, и выпаривали при пониженном давлении. Для получения желаемого продукта остаток очищали колоночной хроматографией на силикагеле (растворитель для элюирования: гексан/этилацетат); 1H ЯМР (300 МГц, CDCl3) δ 7.31-7.26 (m, 2H), 7.00-6.97 (m, 2H), 6.89-6.84 (m, 1H), 3.19 (t, J=5,6 Гц, 4H), 1.79-1.72 (m, 4H), 1.64-1.59 (m, 2H).224 mg (2 mmol) of potassium tert-butoxide, 70 mg (0.25 mol%, calculated on the bromobenzene substrate) of the catalyst according to the invention, 105 μl (1 mmol) of bromobenzene, 198 μl (2 mmol) of piperidine and 5 ml of solvent were charged into a Schlenk vessel dried in an oven. The reaction mixture was heated in an oil bath at 110 ° C for 24 hours, then allowed to cool to room temperature and evaporated under reduced pressure. To obtain the desired product, the residue was purified by silica gel column chromatography (elution solvent: hexane / ethyl acetate); 1 H NMR (300 MHz, CDCl 3 ) δ 7.31-7.26 (m, 2H), 7.00-6.97 (m, 2H), 6.89-6.84 (m, 1H), 3.19 (t, J = 5.6 Hz, 4H ), 1.79-1.72 (m, 4H), 1.64-1.59 (m, 2H).
Результаты испытаний, проведенных в различных растворителях, приведены в Таблице 11.The results of tests carried out in various solvents are shown in Table 11.
Пример 11Example 11
В тестовых реакциях в качестве растворителей использовали метанол и н-бутанол. Тестировали восстановление стирола, применяя во всех случаях давление водорода 6 бар. В реактор помещали 21 мг катализатора формулы (I) (1 моль % по отношению к субстрату, представляющему собой стирол), 1 ммоль стирола и 10 мл растворителя. Сначала колбу промывали газообразным аргоном, потом заполняли ее газообразным Н2 (6 бар). Реакционную смесь перемешивали 6 часов при 110°C при температуре 110°C. Реакционное превращение мониторили с помощью газовой хроматоргафииIn the test reactions, methanol and n-butanol were used as solvents. Styrene recovery was tested using, in all cases, a hydrogen pressure of 6 bar. 21 mg of the catalyst of formula (I) (1 mol% with respect to the styrene substrate), 1 mmol of styrene and 10 ml of solvent were placed in the reactor. First, the flask was washed with gaseous argon, then it was filled with gaseous H2 (6 bar). The reaction mixture was stirred for 6 hours at 110 ° C at a temperature of 110 ° C. Reaction conversion was monitored by gas chromatography

Скрининг суперстабильного катализатора Pd(0) в отношении гидрогенгизации стриролаScreening for the Superstable Pd (0) Catalyst for Hydrogenation of Styrene
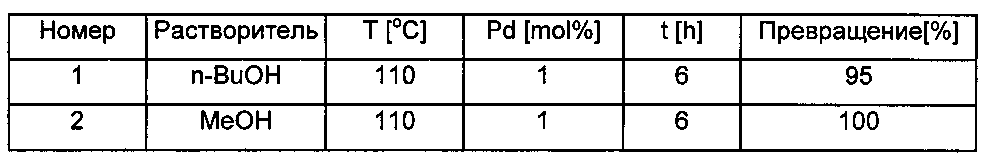
Claims (18)
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HUP-1000668 | 2010-12-16 | ||
| HU1000668A HU230438B1 (en) | 2010-12-16 | 2010-12-16 | New palladium catalyst, process for preparation and use thereof |
| PCT/HU2011/000122 WO2012093271A1 (en) | 2010-12-16 | 2011-12-13 | A new palladium catalyst, method for its preparation and its use |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2013132388A RU2013132388A (en) | 2015-01-27 |
| RU2575249C2 true RU2575249C2 (en) | 2016-02-20 |
Family
ID=
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SU408647A1 (en) * | 1971-04-17 | 1973-11-30 | Т. М. Белослюдова , Л. А. Ильина Всесоюзный научно исследовательский витаминный институт | METHOD FOR PRODUCING A CATALYST FOR HYDROGENATION OF ORGANIC COMPOUNDS |
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SU408647A1 (en) * | 1971-04-17 | 1973-11-30 | Т. М. Белослюдова , Л. А. Ильина Всесоюзный научно исследовательский витаминный институт | METHOD FOR PRODUCING A CATALYST FOR HYDROGENATION OF ORGANIC COMPOUNDS |
Non-Patent Citations (1)
| Title |
|---|
| SOBERATS B. et al, Conventional Tetrakis(triphenylphosphine)palladium-Copper(I) Iodide-Catalyzed Sonogashira Coupling of Free and BOC-Protected Propargylic Amines "On Water", Advanced Synthesis & Catalysis, 2009, v. 351, no. 11-12, p. 1727-1731. CLARKE M.L. et al, The electron-poor phosphines P{ C6H3(CF3)2-3,5} 3 and P(C6F5)3 do not mimic phosphites as ligands for hydroformylation. A comparison of the coordination chemistry of P{ C6H3(CF3)2-3,5} 3 and P(C6F5)3 and the unexpectedly low hydroformylation activity of their rhodium complexes, Dalton Trans., 2005, p. 1294-1300. TORTOSA ESTORACH C. et al, Hydrocarboxylation of terminal alkenes in supercritical carbon dioxide, Eur. J. Inorg. Chem., 2008, p. 3524-3531. HAI-CHEN WU et al, Possible Origin of Electronic Effects in Rh(I)-Catalyzed Enantioselective Hydrogenation, J. Am. Chem. Soc., 2009, v. 131, no. 28, p. 9604-9605. ALTINEL H. et al, Fluorinated rhodium-phosphine complexes as efficient homogeneous catalysts for the hydrogenation of * |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Narr et al. | Selective measurements of a nitroxide–nitroxide distance of 5nm and a nitroxide–copper distance of 2.5 nm in a terpyridine-based copper (II) complex by pulse EPR | |
| Huynh et al. | Pincer-type di (1, 2, 4-triazolin-5-ylidene) Pd (II) complexes and their catalytic activities towards Cu-and amine-free Sonogashira reaction | |
| US9056314B2 (en) | Palladium catalyst, method for its preparation and its use | |
| Borah et al. | A cyclometalated Ir (III)–NHC complex as a recyclable catalyst for acceptorless dehydrogenation of alcohols to carboxylic acids | |
| Muthumari et al. | Highly efficient palladium (II) hydrazone based catalysts for the Suzuki coupling reaction in aqueous medium | |
| JP2014511339A5 (en) | ||
| RU2575249C2 (en) | Novel palladium catalyst, method for production thereof and use thereof | |
| Zong et al. | Ruthenium carbonyl complexes supported by pyridine-alkoxide ligands: synthesis, structure and catalytic oxidation of secondary alcohols | |
| Dey et al. | Polymer-incarcerated palladium-catalyzed facile in situ carbonylation for the synthesis of aryl aldehydes and diaryl ketones using CO surrogates under ambient conditions | |
| JP4906711B2 (en) | Method for producing 3-cyclopentyloxy-4-methoxybenzaldehyde | |
| Kiss et al. | Synthesis and characterization of [4‐{(CH2O) 2CH} C6H4] 2Hg,[4‐(O= CH) C6H4] 2Hg and [(E)‐4‐(RN= CH) C6H4] 2Hg (R= 2′‐py, 4′‐py, 2′‐pyCH2, 4′‐pyCH2) | |
| Sýkora et al. | Two structural types of 1, 3-alternate tetrapropoxycalix [4] arene derivatives in the solid state | |
| JP6904508B2 (en) | Selective reduction of aldehydes and ketones | |
| Roman | Selected Michael additions to thiophene-containing analogues of chalcone | |
| Mohite et al. | Nickel–PNN catalysed sustainable synthesis of polysubstituted quinolines under microwave irradiation | |
| JPS6031824B2 (en) | Method for producing hydroxyphenylpiperidinyl methanol derivative | |
| Potapov et al. | Synthesis of monomeric and oligomeric 1, 1′-methylenebis-(1 H-pyrazoles) contaning ethynyl fragments | |
| Deng et al. | Unusual C–O bond cleavage of aromatic ethers in ruthenium complexes bearing a 2-alkoxypyridyl fragment | |
| EP3679033B1 (en) | Method of synthesis of 5,6-bis(5-alkoxythiophen-2-yl)pyrazine-2,3-dicarbodinitriles, derivatives of dicyanopyrazine and use thereof | |
| RU2626403C1 (en) | Method of preparation (2-hydroxynaphthalene-1-yl) azin | |
| Mohite et al. | Catalytic utility of PNN-based Mn I pincer complexes in the synthesis of quinolines and transfer hydrogenation of carbonyl derivatives | |
| Zhang et al. | Green synthesis and antitumor activity of (E)-diethyl 2-styrylquinoline-3, 4-dicarboxylates | |
| Liu et al. | Synthesis of silica-supported selenide palladium (0) complex and its catalytic properties for phenylation of acid chlorides and aryl iodides or bromides | |
| KR880001850B1 (en) | Preparation process for derivertives of 5-fluoro pyridone | |
| CN110746278A (en) | Nonmetal-catalyzed method for preparing 1, 3-diketone compound based on alkynone |