JPWO2013150902A1 - Method for producing 5'-terminal phosphorylated oligodeoxynucleotide by cleaving oligodeoxynucleotide containing non-natural base - Google Patents
Method for producing 5'-terminal phosphorylated oligodeoxynucleotide by cleaving oligodeoxynucleotide containing non-natural base Download PDFInfo
- Publication number
- JPWO2013150902A1 JPWO2013150902A1 JP2014509104A JP2014509104A JPWO2013150902A1 JP WO2013150902 A1 JPWO2013150902 A1 JP WO2013150902A1 JP 2014509104 A JP2014509104 A JP 2014509104A JP 2014509104 A JP2014509104 A JP 2014509104A JP WO2013150902 A1 JPWO2013150902 A1 JP WO2013150902A1
- Authority
- JP
- Japan
- Prior art keywords
- dna
- reaction
- oligodeoxynucleotide
- substituted
- unsubstituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 COP(O)(OC(C1)C(COP(O)(O*)=O)OC1N(C=C(C(N1)=O)C#Cc2ccccc2)C1=O)=O Chemical compound COP(O)(OC(C1)C(COP(O)(O*)=O)OC1N(C=C(C(N1)=O)C#Cc2ccccc2)C1=O)=O 0.000 description 5
- IIEFLCXENFHLBX-STBIYBPSSA-N CCCCN(CCCC)/C=N/c1c2nc(-c3c(C=O)cccc3)[n](C(C3)OC(CO)C3O)c2ncn1 Chemical compound CCCCN(CCCC)/C=N/c1c2nc(-c3c(C=O)cccc3)[n](C(C3)OC(CO)C3O)c2ncn1 IIEFLCXENFHLBX-STBIYBPSSA-N 0.000 description 1
- PHQKUDUQIGSISC-UHFFFAOYSA-N Nc1c2nc(-c3c(C=O)cccc3)[n](C(C3)OC(CO)C3O)c2ncn1 Chemical compound Nc1c2nc(-c3c(C=O)cccc3)[n](C(C3)OC(CO)C3O)c2ncn1 PHQKUDUQIGSISC-UHFFFAOYSA-N 0.000 description 1
Images





































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/073—Pyrimidine radicals with 2-deoxyribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/173—Purine radicals with 2-deoxyribosyl as the saccharide radical
Abstract
非天然型塩基を含むデオキシヌクレオチドをDNA鎖に導入することにより、化学反応によって低コスト且つ簡単な実験操作でDNA鎖の切断を行い、結果として生じた末端がリン酸化されたオリゴデオキシヌクレオチドを提供する。本発明は、求核剤と式Iで表されるオリゴデオキシヌクレオチドとを接触させることによって、式Iで表されるオリゴデオキシヌクレオチドに含まれる非天然型塩基を含むデオキシヌクレオシドBの3’位とCの5’末端との間のホスホジエステル結合が、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断されて、5’末端がリン酸化されたオリゴデオキシヌクレオチドCを得る、5’末端リン酸化オリゴデオキシヌクレオチド形成工程を含む、オリゴデオキシヌクレオチドの製造方法に関する。By introducing deoxynucleotides containing non-natural bases into the DNA strand, the resulting DNA strand is cleaved by chemical reaction at low cost and with simple experimental operations, and the resulting end is phosphorylated. To do. The present invention comprises contacting the nucleophile with the oligodeoxynucleotide represented by formula I to form the 3′-position of deoxynucleoside B containing an unnatural base contained in the oligodeoxynucleotide represented by formula I. The phosphodiester bond between the 5 ′ end of C is cleaved by elimination of the oligodeoxynucleotide moiety phosphorylated at the 5 ′ end of C from the 3 ′ position of B, and the 5 ′ end is phosphorylated. The present invention relates to a method for producing an oligodeoxynucleotide, which comprises a 5′-terminal phosphorylated oligodeoxynucleotide forming step for obtaining the resulting oligodeoxynucleotide C.
Description
本発明は、非天然型塩基を含むオリゴデオキシヌクレオチドの切断による5’末端リン酸化オリゴデオキシヌクレオチドの製造方法に関する。 The present invention relates to a method for producing a 5'-terminal phosphorylated oligodeoxynucleotide by cleaving an oligodeoxynucleotide containing a non-natural base.
分子生物学分野の研究においては、デオキシリボ核酸(以下「DNA」とも記載する)を複製又は増幅、切断、及び連結する技術が使用される。このうち、DNAを切断する技術としては、制限酵素を用いる方法が、DNAを連結する技術としては、DNA連結酵素(DNAリガーゼ)を用いる方法が、それぞれ一般的である。例えば、タイプIIの制限酵素を用いて突出末端を有するDNA断片を調製し(非特許文献1及び2)、当該突出末端を有するDNA断片と、当該突出末端と相補的な突出末端を有する別のDNA断片とをDNA連結酵素を用いて連結することができる。
In research in the field of molecular biology, techniques for replicating or amplifying, cleaving, and ligating deoxyribonucleic acid (hereinafter also referred to as “DNA”) are used. Among them, a method using a restriction enzyme is generally used as a technique for cleaving DNA, and a method using a DNA ligase (DNA ligase) is generally used as a technique for ligating DNA. For example, a DNA fragment having a protruding end is prepared using a type II restriction enzyme (Non-Patent
しかしながら、上記の方法には、いくつかの問題点がある。第一に、タイプIIの制限酵素を用いて作製された突出末端は突出部分が短い。第二に、突出末端の突出部分の塩基配列が回文配列となる。第三に、制限酵素の認識配列でしか突出末端を作製することができない。これらの問題点のうち、第一及び第二の問題点は、DNA連結酵素によるDNA断片同士の連結効率の低下の原因となり、第三の問題点は、DNA断片を連結した結果形成される新たなDNAにおける塩基配列の大きな制約となる。 However, the above method has several problems. First, overhanging ends made using type II restriction enzymes have short overhangs. Second, the base sequence of the protruding portion at the protruding end is a palindromic sequence. Thirdly, overhanging ends can only be created with restriction enzyme recognition sequences. Among these problems, the first and second problems cause a decrease in the efficiency of ligation between DNA fragments by the DNA ligation enzyme, and the third problem is a new problem formed as a result of ligating DNA fragments. It becomes a big restriction of the base sequence in DNA.
このような問題点を解決するため、非天然型塩基を含むデオキシ又はリボヌクレオチドを予めDNA鎖に導入しておき、非天然型塩基を含むデオキシ又はリボヌクレオチドの部位でDNA鎖を切断する技術が開発された。特許文献1及び2、並びに非特許文献3〜5は、非天然型塩基としてウリジン(リボヌクレオチド)を導入したDNAプライマーを用いてポリメラーゼ連鎖反応(PCR)等でDNAを増幅した後、増幅されたDNAをウリジン(リボヌクレオチド)の位置で切断する方法を記載する。また、非特許文献6〜12は、非天然型塩基として2’-デオキシウリジンを導入したDNAプライマーを用いてPCR等でDNAを増幅した後、増幅されたDNAを2’-デオキシウリジンの位置で切断する方法を記載する。上記文献に記載の方法は、DNAの切断にRNA分解酵素又はウラシル除去酵素のような酵素を用いる。それ故、コスト面で不利である。また、一般に酵素による切断反応は、pH及び/又は金属イオン濃度などに関する特別な反応条件を満たす必要がある。
In order to solve such problems, there is a technique in which deoxy or ribonucleotide containing a non-natural base is introduced into a DNA strand in advance and the DNA strand is cleaved at the site of deoxy or ribonucleotide containing a non-natural base. It has been developed.
非特許文献13は、デプリネーションとして知られるDNA塩基の脱離反応とそれによって生じるアベーシック部位を経由する、DNAの切断方法を記載する。デプリネーションは、酸性条件下で容易に引き起こされるが、塩基性条件下では非常に起きにくいことが知られている(非特許文献14)。非特許文献15は、天然型塩基ではデプリネーションが起こりにくい塩基性条件下で、デプリネーションと類似の反応を引き起こす非天然型塩基を含むデオキシヌクレオチドを記載する。しかしながら、当該文献記載のデオキシヌクレオシドを用いる場合、DNA切断のために酸素バブリングという煩雑な操作を必要とする。また、DNA自動合成機によるDNAの合成条件下及び/又はPCRのようなDNAの複製条件下で安定であり、且つ天然型塩基と同様にDNAの複製反応で鋳型として機能する非天然型塩基を含むデオキシヌクレオチドは報告されていない。
Non-Patent
オリゴデオキシヌクレオチド(以下「DNAオリゴマー」又は「オリゴDNA」とも記載する)を人工的に合成する場合、DNA自動合成機が使用される。DNA自動合成機を用いて合成されたDNAオリゴマーは、通常、5’末端がリン酸化されていない。このため、DNA連結酵素を用いて当該合成DNAオリゴマーを他のDNA(DNA誘導体)と連結する場合、予め5’末端をリン酸化する必要がある。 When an oligodeoxynucleotide (hereinafter also referred to as “DNA oligomer” or “oligo DNA”) is artificially synthesized, an automatic DNA synthesizer is used. A DNA oligomer synthesized using an automatic DNA synthesizer is usually not phosphorylated at the 5 'end. For this reason, when the synthetic DNA oligomer is linked to other DNA (DNA derivative) using a DNA ligation enzyme, it is necessary to phosphorylate the 5 'end in advance.
DNAの5’及び/又は3’末端をリン酸化する方法としては、DNA断片をポリヌクレオチドキナーゼ処理する方法(5’末端リン酸化の場合)、並びにリン酸化アミダイトを用いる合成DNAの末端リン酸化法を挙げることができる(特許文献3並びに非特許文献16及び17)。このうち、リン酸化アミダイトを用いる方法の場合、リン酸化アミダイトのカップリング効率が100%ではないため、リン酸化されたDNA及びリン酸化されなかったDNAが生成する。ここで、5’末端のリン酸化の場合、リン酸化されたDNAとリン酸化されなかったDNAとは、逆相HPLCでは分離が困難という問題点が存在する。逆相HPLCにおいてリン酸化されたDNAとリン酸化されなかったDNAとの分離を改善する方法として、DNAの末端リン酸部分をDMTr基のような保護基で保護した形態でDNAオリゴマーを精製し、その後、酸処理によってDMTr基を脱保護するという方法がある。しかしながら、この方法の場合、脱保護に酸を使用するため、デプリネーション反応が起こってDNAが破壊されるという問題が存在する(非特許文献18)。
Methods for phosphorylating the 5 ′ and / or 3 ′ end of DNA include a method of treating a DNA fragment with a polynucleotide kinase (in the case of 5 ′ end phosphorylation), and a method for terminal phosphorylation of synthetic DNA using phosphorylated amidite (
DNAを切断する方法として公知の特許文献1及び2、並びに非特許文献3〜12に記載の方法は、上記のようにDNAの切断にRNA分解酵素又はウラシル除去酵素のような酵素を用いる。それ故、コスト面で不利である。また、一般に酵素による切断反応は、pH及び/又は金属イオン濃度などに関する特別な反応条件を満たす必要がある。非特許文献15に記載の方法は、酵素を使用せず化学反応によってDNAの切断を行うため、コスト及び反応効率の点で有利である。しかしながら、当該文献に記載の非天然型塩基を含むデオキシヌクレオチドは、対応する天然型塩基(A、T、G又はC)を含むデオキシヌクレオチドと比較して、DNA自動合成機によるDNAの合成条件下及び/又はPCRのようなDNAの複製条件下における安定性が低く、且つDNA複製反応のようなin vivo及びin vitroの反応において使用した場合にフィデリティが低くなる。また、当該文献記載のデオキシヌクレオシドを用いる場合、DNA切断のために酸素バブリングという煩雑な操作を必要とする。
The methods described in
DNAの5’及び/又は3’末端をリン酸化する方法として公知の特許文献3並びに非特許文献16及び17に記載の方法は、上記のようにリン酸化アミダイトのカップリング効率が100%ではなく、且つリン酸化されたDNAとリン酸化されなかったDNAとの分離が困難という問題点が存在する。
The methods described in
所望の部位でのDNA鎖の切断、及び切断された末端のリン酸化を行うためには、化学反応によってDNAを二つの断片に切断し、それによって生じるDNA断片の5’末端をリン酸末端とすることが可能である、非天然型塩基を含むデオキシヌクレオチドを切断部位としてDNA鎖に導入する方法が、低コストで、反応条件の制約が少なく、且つ実験操作が簡便である点で有利である。それ故、本発明は、非天然型塩基を含むデオキシヌクレオチドをDNA鎖に導入することにより、化学反応によって低コスト且つ簡単な実験操作でDNA鎖の切断を行い、結果として生じた末端がリン酸化されたオリゴデオキシヌクレオチドを提供することを目的とする。 In order to cleave the DNA strand at the desired site and phosphorylate the cleaved end, the DNA is cleaved into two fragments by a chemical reaction, and the 5 ′ end of the resulting DNA fragment becomes the phosphate end. The method of introducing a deoxynucleotide containing a non-natural base into a DNA strand as a cleavage site is advantageous in that it is low-cost, has few restrictions on reaction conditions, and is easy to carry out experimental operations. . Therefore, the present invention introduces deoxynucleotides containing unnatural bases into the DNA strand, thereby cleaving the DNA strand with a low-cost and simple experimental operation by a chemical reaction, and the resulting end is phosphorylated. It is an object of the present invention to provide an oligodeoxynucleotide.
本発明者らは、前記課題を解決するための手段を種々検討した結果、求核剤によって脱離可能な部分を有する、非天然型塩基を含むデオキシヌクレオチド又はデオキシヌクレオシドを用いることにより、上記目的を達成できることを見出し、本発明を完成した。 As a result of various studies on means for solving the above-mentioned problems, the present inventors have used the above object by using a deoxynucleotide or deoxynucleoside containing a non-natural base having a moiety removable by a nucleophile. The present invention has been completed.
すなわち、本発明の要旨は以下の通りである。 That is, the gist of the present invention is as follows.
(1) 求核剤と、式I:
(A)n-B-C
[式中、
Aは、オリゴデオキシヌクレオチドであり、
Bは、求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドであり、
Cはオリゴデオキシヌクレオチドであり、
nは0又は1であり、
nが1のとき、Aの3’末端とBの5’位とは、ホスホジエステル結合で連結されており、
Bの3’位とCの5’末端とは、ホスホジエステル結合で連結されており、
BとCとの間のホスホジエステル結合は、求核剤の攻撃によって誘発されるEの脱離に伴って、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断される]
で表されるオリゴデオキシヌクレオチドとを接触させることによって、BとCとの間のホスホジエステル結合が、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断されて、前記Cの5’末端がリン酸化されたオリゴデオキシヌクレオチドと、式II:
(A)n-B’
[式中、
A及びnは、式Iにおいて定義したのと同様の意味であり、
B’は、求核剤の攻撃によるEの脱離に伴って形成されるBの残基部分である]
で表されるオリゴデオキシヌクレオチドとを得る、5’末端リン酸化オリゴデオキシヌクレオチド形成工程を含む、オリゴデオキシヌクレオチドの製造方法。(1) Nucleophiles and formula I:
(A) n -BC
[Where:
A is an oligodeoxynucleotide,
B is a deoxynucleoside containing a non-natural base having a moiety E removable by a nucleophile;
C is an oligodeoxynucleotide,
n is 0 or 1,
When n is 1, the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond,
The 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond,
The phosphodiester bond between B and C is such that the oligodeoxynucleotide part phosphorylated at the 5 'end of C from the 3' position of B with E elimination induced by nucleophile attack. Disconnected by detachment]
By contacting the oligodeoxynucleotide represented by the following formula, the phosphodiester bond between B and C is released from the 3 'position of B by phosphorylation of the 5' end of C. An oligodeoxynucleotide which has been cleaved to phosphorylate the 5 ′ end of C, and a compound of formula II:
(A) n -B '
[Where:
A and n have the same meaning as defined in Formula I,
B ′ is the residue part of B that is formed with the elimination of E by nucleophile attack]
A method for producing an oligodeoxynucleotide, comprising a 5′-terminal phosphorylated oligodeoxynucleotide forming step, which obtains an oligodeoxynucleotide represented by the formula:
(2) 式II:
(A)n-B’
[式中、
A及びB’は、前記(1)と同様の意味であり、
nは1である]
で表されるオリゴデオキシヌクレオチドを反応させることによって、B’とAとの間のホスホジエステル結合が、Aの3’末端がリン酸化されたオリゴデオキシヌクレオチド部分がB’から脱離することによって切断されて、式IIa:
A-PO3H2
で表される3’末端がリン酸化されたオリゴデオキシヌクレオチドを得る、3’末端リン酸化オリゴデオキシヌクレオチド形成工程をさらに含む、前記(1)の方法。(2) Formula II:
(A) n -B '
[Where:
A and B ′ have the same meaning as in the above (1),
n is 1]
By reacting with oligodeoxynucleotide represented by Formula IIa:
A-PO 3 H 2
The method according to (1), further comprising a 3′-terminal phosphorylated oligodeoxynucleotide forming step of obtaining an oligodeoxynucleotide phosphorylated at the 3 ′ end represented by:
(3) 前記非天然型塩基を含むデオキシヌクレオシドBが、以下:

[式中、
Rは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロアリールアルキル又はカルボニルであり;
R’は、水素、又は置換若しくは非置換のアルキル、アルケニル若しくはアルキニルであり;
rは、0〜4の整数であり;
R’’は、ハロゲン、又は置換若しくは非置換のアルキル、アルケニル若しくはアルキニルであり;
R’’’は、-COZ、-C≡C-Z又は-CNであり;
Zは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、又は置換若しくは非置換のヘテロアリールアルキルである]
で表されるデオキシヌクレオシドから選択される、前記(1)又は(2)の方法。[Where:
R is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, Substituted or unsubstituted heteroarylalkyl or carbonyl;
R ′ is hydrogen or substituted or unsubstituted alkyl, alkenyl or alkynyl;
r is an integer from 0 to 4;
R ″ is halogen or substituted or unsubstituted alkyl, alkenyl or alkynyl;
R ′ ″ is —COZ, —C≡CZ or —CN;
Z is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, Or substituted or unsubstituted heteroarylalkyl]
The method according to (1) or (2), wherein the method is selected from deoxynucleosides represented by:
(4) 前記5’末端リン酸化オリゴデオキシヌクレオチド形成工程の前に、求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドB又はその誘導体と、天然型塩基を含む1個以上のデオキシヌクレオシド又はその誘導体とを含む基質を用いて、式Iで表されるオリゴデオキシヌクレオチドを合成する、オリゴデオキシヌクレオチド合成工程をさらに含む、前記(1)〜(3)のいずれかの方法。 (4) Before the 5′-terminal phosphorylated oligodeoxynucleotide forming step, a deoxynucleoside B containing a non-natural base having a moiety E removable by a nucleophile or a derivative thereof, and a natural base Any of the above (1) to (3), further comprising an oligodeoxynucleotide synthesis step of synthesizing an oligodeoxynucleotide represented by formula I using a substrate containing one or more deoxynucleosides or derivatives thereof the method of.
(5) 式I:
(A)n-B-C
[式中、
Aは、オリゴデオキシヌクレオチドであり、
Bは、求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドであり、
Cはオリゴデオキシヌクレオチドであり、
nは0又は1であり、
nが1のとき、Aの3’末端とBの5’位とは、ホスホジエステル結合で連結されており、
Bの3’位とCの5’末端とは、ホスホジエステル結合で連結されており、
BとCとの間のホスホジエステル結合は、求核剤の攻撃によって誘発されるEの脱離に伴って、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断される]
で表されるオリゴデオキシヌクレオチドをプライマーとして用いて、直鎖状二本鎖DNA断片を増幅するDNA断片増幅工程;
求核剤と、増幅された直鎖状二本鎖DNA断片とを接触させることによって、BとCとの間のホスホジエステル結合が、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断されて、直鎖状二本鎖DNA断片に含まれる前記Cの5’末端がリン酸化されており、且つ式Iで表されるプライマー部分の少なくとも一部と相補的な配列を含む3’突出末端を有する直鎖状二本鎖DNA断片を形成させる、3’突出末端形成工程;並びに
前記3’突出末端を有する直鎖状二本鎖DNA断片と、別の二本鎖DNA断片とを連結する連結工程;
を含む、二本鎖DNAの連結方法。(5) Formula I:
(A) n -BC
[Where:
A is an oligodeoxynucleotide,
B is a deoxynucleoside containing a non-natural base having a moiety E removable by a nucleophile;
C is an oligodeoxynucleotide,
n is 0 or 1,
When n is 1, the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond,
The 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond,
The phosphodiester bond between B and C is such that the oligodeoxynucleotide part phosphorylated at the 5 'end of C from the 3' position of B with E elimination induced by nucleophile attack. Disconnected by detachment]
A DNA fragment amplification step of amplifying a linear double-stranded DNA fragment using the oligodeoxynucleotide represented by
By contacting the nucleophile with the amplified linear double-stranded DNA fragment, the phosphodiester bond between B and C becomes an oligodeoxynucleotide moiety phosphorylated at the 5 'end of C. It is cleaved by detaching from the 3 ′ position of B, the 5 ′ end of C contained in the linear double-stranded DNA fragment is phosphorylated, and at least the primer part represented by Formula I Forming a linear double-stranded DNA fragment having a 3 ′ protruding end containing a sequence complementary to a part thereof; and a linear double-stranded DNA fragment having the 3 ′ protruding end; And a ligation step of ligating another double-stranded DNA fragment;
A method for ligating double-stranded DNA.
(6) 前記(1)〜(4)のいずれかのオリゴデオキシヌクレオチドの製造方法又は前記(5)の二本鎖DNAの連結方法に使用するためのキットであって、式I:
(A)n-B-C
[式中、
Aは、オリゴデオキシヌクレオチドであり、
Bは、求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドであり、
Cはオリゴデオキシヌクレオチドであり、
nは0又は1であり、
nが1のとき、Aの3’末端とBの5’位とは、ホスホジエステル結合で連結されており、
Bの3’位とCの5’末端とは、ホスホジエステル結合で連結されており、
BとCとの間のホスホジエステル結合は、求核剤の攻撃によって誘発されるEの脱離に伴って、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断される]
で表されるオリゴデオキシヌクレオチド、又は求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドB若しくはその誘導体を少なくとも含む、前記キット。(6) A kit for use in the method for producing an oligodeoxynucleotide according to any one of (1) to (4) or the method for ligating double-stranded DNA according to (5), wherein the formula I:
(A) n -BC
[Where:
A is an oligodeoxynucleotide,
B is a deoxynucleoside containing a non-natural base having a moiety E removable by a nucleophile;
C is an oligodeoxynucleotide,
n is 0 or 1,
When n is 1, the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond,
The 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond,
The phosphodiester bond between B and C is such that the oligodeoxynucleotide part phosphorylated at the 5 'end of C from the 3' position of B with E elimination induced by nucleophile attack. Disconnected by detachment]
Or at least a deoxynucleoside B containing a non-natural base or a derivative thereof having a moiety E removable by a nucleophile.
(7) 以下の式:
[式中、
Rは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロアリールアルキル又はカルボニルであり;
R'は、メチルであり;
rは、0〜4の整数であり;
R’’は、ハロゲン、又は置換若しくは非置換のアルキル、アルケニル若しくはアルキニルであり;
R’’’は、-COZ、-C≡C-Z又は-CNであり;
Zは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、又は置換若しくは非置換のヘテロアリールアルキルである]
で表される非天然型塩基を含むデオキシヌクレオシド。[Where:
R is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, Substituted or unsubstituted heteroarylalkyl or carbonyl;
R ′ is methyl;
r is an integer from 0 to 4;
R ″ is halogen or substituted or unsubstituted alkyl, alkenyl or alkynyl;
R ′ ″ is —COZ, —C≡CZ or —CN;
Z is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, Or substituted or unsubstituted heteroarylalkyl]
A deoxynucleoside comprising an unnatural base represented by
本発明により、化学反応によって低コスト且つ簡単な実験操作でDNA鎖の切断を行い、結果として生じた末端がリン酸化されたオリゴデオキシヌクレオチドを提供することが可能となる。 According to the present invention, it is possible to cleave a DNA strand by a chemical reaction at a low cost and with a simple experimental operation, and to provide an oligodeoxynucleotide whose resulting end is phosphorylated.
本明細書は、本願の優先権の基礎である日本国特許出願第2012-084249号の明細書及び/又は図面に記載される内容を包含する。 This specification includes the contents described in the specification and / or drawings of Japanese Patent Application No. 2012-084249, which is the basis of the priority of the present application.
以下、本発明の好ましい実施形態について詳細に説明する。 Hereinafter, preferred embodiments of the present invention will be described in detail.
<1. 定義>
本明細書において、「オリゴデオキシヌクレオチド」は、デオキシリボ核酸(DNA)、一本鎖若しくは二本鎖のオリゴデオキシヌクレオチド若しくはポリデオキシヌクレオチド、又はそれらの誘導体を意味する。オリゴデオキシヌクレオチド(DNAオリゴマー又はオリゴDNA)は、天然のものでも合成のものでもよく、ポリメラーゼ連鎖反応(PCR)産物であってもよい。好適なオリゴデオキシヌクレオチド誘導体としては、限定するものではないが、例えば、オリゴデオキシヌクレオチド中のホスホジエステル結合が、ホスホロチオエート結合、N3’-P5’ホスホロアミダイト結合又はペプチド結合に変換されたオリゴデオキシヌクレオチド誘導体、オリゴデオキシヌクレオチド中のデオキシリボースが、モルホリン、2’-O-プロピルリボース、2’-O-メチルリボース、2’-フルオロ-2’-デオキシリボース、トレオフラノース(TNA)又は2’位のOと4’位のCとをメチレンで架橋したリボース(ロックされた核酸(LNA)若しくは架橋された核酸(BNA))に変換されたオリゴデオキシヌクレオチド誘導体、並びにペプチド核酸(PNA)を挙げることができる。<1. Definition>
As used herein, “oligodeoxynucleotide” means deoxyribonucleic acid (DNA), single-stranded or double-stranded oligodeoxynucleotide or polydeoxynucleotide, or a derivative thereof. The oligodeoxynucleotide (DNA oligomer or oligo DNA) may be natural or synthetic and may be a polymerase chain reaction (PCR) product. Suitable oligodeoxynucleotide derivatives include, but are not limited to, for example, oligodeoxynucleotides in which the phosphodiester bond in the oligodeoxynucleotide is converted to a phosphorothioate bond, an N3′-P5 ′ phosphoramidite bond, or a peptide bond Deoxyribose in the derivative, oligodeoxynucleotide is morpholine, 2'-O-propylribose, 2'-O-methylribose, 2'-fluoro-2'-deoxyribose, treofuranose (TNA) or 2'-position Examples include oligodeoxynucleotide derivatives converted to ribose (locked nucleic acid (LNA) or cross-linked nucleic acid (BNA)) obtained by cross-linking O and C at the 4 'position with methylene, and peptide nucleic acids (PNA). it can.
オリゴデオキシヌクレオチドの5’末端及び/又は3’末端は、特に記載する場合を除き、リン酸化されていてもよく、リン酸化されていなくてもよい。或いは、DNA自動合成機を用いるDNAオリゴマー合成において、最後のデオキシヌクレオチドの5’位ヒドロキシル基の保護基を脱保護しない条件で合成を行う場合のように、5’位のヒドロキシル基が保護基で保護された形態であってもよい。いずれの場合も、オリゴデオキシヌクレオチドに包含されるものとする。 The oligodeoxynucleotide 5 'end and / or 3' end may be phosphorylated or not phosphorylated unless otherwise specified. Alternatively, in the synthesis of a DNA oligomer using an automatic DNA synthesizer, the 5′-position hydroxyl group is a protecting group as in the case of synthesis under the condition that the 5′-position hydroxyl group protecting group of the last deoxynucleotide is not deprotected. It may be in a protected form. In either case, the oligodeoxynucleotide shall be included.
本明細書において、「保護基」は、末端ヒドロキシル基と結合して望ましくない副反応を実質的に抑制又は防止する基を意味し、限定するものではないが、例えば、トリチル基、モノ置換若しくはジ置換トリチル基、o-ニトロベンジル基及びレブリニル基を挙げることができる。ジメトキシトリチル(DMTr)であることが好ましい。上記の保護基は、所定の脱保護処理をすることによって除去される。保護基がトリチル基又はモノ置換若しくはジ置換トリチル基の場合、脱保護処理を行うために用いる脱保護剤としては、トリクロロ酢酸及びトリフルオロ酢酸のような酸を挙げることができる。保護基がレブリニル基の場合、脱保護処理を行うために用いる脱保護剤としては、ヒドラジンを挙げることができる。保護基がo-ニトロベンジル基の場合、脱保護処理としては、UV光照射処理を挙げることができる。 As used herein, “protecting group” means a group that binds to a terminal hydroxyl group to substantially suppress or prevent unwanted side reactions, and includes, but is not limited to, for example, a trityl group, a mono-substituted group, or Mention may be made of disubstituted trityl groups, o-nitrobenzyl groups and levulinyl groups. Dimethoxytrityl (DMTr) is preferred. The protecting group is removed by performing a predetermined deprotection treatment. When the protecting group is a trityl group or a mono-substituted or di-substituted trityl group, examples of the deprotecting agent used for the deprotection treatment include acids such as trichloroacetic acid and trifluoroacetic acid. When the protecting group is a levulinyl group, examples of the deprotecting agent used for performing the deprotecting treatment include hydrazine. When the protecting group is an o-nitrobenzyl group, examples of the deprotection treatment include UV light irradiation treatment.
オリゴデオキシヌクレオチドは、5’末端及び/若しくは3’末端のヒドロキシル基、並びに/又は塩基がプローブ分子で標識された形態であってもよい。いずれの場合も、オリゴデオキシヌクレオチドに包含されるものとする。本明細書において、「プローブ(標識)分子」は、オリゴデオキシヌクレオチドの標識のために当該技術分野で慣用される分子を意味し、ビオチン、ポリエチレングリコール(PEG)、蛍光色素、ジコキシゲニン、タンパク質(例えば、細胞透過性ペプチド又は細胞内局在シグナルペプチドを含む)、及び標識分子を付与するためのリンカー分子を挙げることができる。フルオレセイン誘導体及びシアニン色素からなる群より選択される蛍光色素が好ましい。当該プローブ分子は、そのままの形態であってもよく、活性エステル(例えばスクシンイミジルエステル)、アジド基、アミノ基、チオ―ル基、マレインイミド基又はアルキニル基のような、他の官能基との間で共有結合を形成し得る基が導入された形態でもよい。いずれの形態も、プローブ分子に包含されるものとする。 The oligodeoxynucleotide may be in a form in which the hydroxyl group at the 5 'end and / or the 3' end and / or the base is labeled with a probe molecule. In either case, the oligodeoxynucleotide shall be included. As used herein, “probe (label) molecule” refers to a molecule conventionally used in the art for labeling oligodeoxynucleotides, such as biotin, polyethylene glycol (PEG), fluorescent dye, dicoxygenin, protein (eg, And a linker molecule for providing a labeling molecule). Fluorescent dyes selected from the group consisting of fluorescein derivatives and cyanine dyes are preferred. The probe molecule may be in its native form, with other functional groups such as active esters (eg succinimidyl esters), azide groups, amino groups, thiol groups, maleimide groups or alkynyl groups. It may be in a form in which a group capable of forming a covalent bond is introduced. Any form shall be included in the probe molecule.
本明細書において、「アルキル」は、特定の数の炭素原子を含む、直鎖又は分枝鎖の脂肪族炭化水素基を意味する。例えば、「員数1〜20のアルキル」及び「C1〜C20アルキル」は、少なくとも1個且つ多くても20個の炭素原子を含む、直鎖又は分枝鎖の炭化水素鎖を意味する。好適なアルキルは、限定するものではないが、例えばメチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチル、イソブチル、tert-ブチル、n-ペンチル、n-ヘキシル、n-ヘプチル及びn-オクチル等を挙げることができる。As used herein, “alkyl” means a straight or branched chain aliphatic hydrocarbon group containing the specified number of carbon atoms. For example, “1-20 alkyl” and “C 1 -C 20 alkyl” mean a straight or branched hydrocarbon chain containing at least 1 and at most 20 carbon atoms. Suitable alkyls include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl, n-pentyl, n-hexyl, n-heptyl and n -Octyl and the like can be mentioned.
本明細書において、「アルケニル」は、前記アルキルの1個以上のC-C単結合が二重結合に置換された基を意味する。好適なアルケニルは、限定するものではないが、例えばビニル、1-プロペニル、アリル、1-メチルエテニル(イソプロペニル)、1-ブテニル、2-ブテニル、3-ブテニル、1-メチル-2-プロペニル、2-メチル-2-プロペニル、1-メチル-1-プロペニル、2-メチル-1-プロペニル、1-ペンテニル、1-ヘキセニル、n-ヘプテニル及び1-オクテニル等を挙げることができる。 In the present specification, “alkenyl” means a group in which one or more C—C single bonds of the alkyl are substituted with double bonds. Suitable alkenyls include, but are not limited to, vinyl, 1-propenyl, allyl, 1-methylethenyl (isopropenyl), 1-butenyl, 2-butenyl, 3-butenyl, 1-methyl-2-propenyl, 2 Examples include -methyl-2-propenyl, 1-methyl-1-propenyl, 2-methyl-1-propenyl, 1-pentenyl, 1-hexenyl, n-heptenyl and 1-octenyl.
本明細書において、「アルキニル」は、前記アルキルの1個以上のC-C単結合が三重結合に置換された基を意味する。好適なアルキニルは、限定するものではないが、例えばエチニル、1-プロピニル、2-プロピニル、1-ブチニル、2-ブチニル、3-ブチニル、1-メチル-2-プロピニル、1-ペンチニル、1-ヘキシニル、1-ヘプチニル及び1-オクチニル等を挙げることができる。 In the present specification, “alkynyl” means a group in which one or more C—C single bonds of the alkyl are substituted with triple bonds. Suitable alkynyls include but are not limited to ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1-methyl-2-propynyl, 1-pentynyl, 1-hexynyl 1-heptynyl, 1-octynyl and the like.
本明細書において、「シクロアルキル」は、特定の数の炭素原子を含む、脂環式アルキルを意味する。例えば、「(環の)員数3〜20のシクロアルキル」及び「C3〜C20シクロアルキル」は、少なくとも3個且つ多くても20個の炭素原子を含む、環式の炭化水素基を意味する。好適なシクロアルキルは、限定するものではないが、例えばシクロプロピル、シクロブチル、シクロペンチル及びシクロヘキシル等を挙げることができる。As used herein, “cycloalkyl” means an alicyclic alkyl containing the specified number of carbon atoms. For example, "(ring) cycloalkyl Number 3-20" and "C 3 -C 20 cycloalkyl" refers to at least three and at most containing 20 carbon atoms, a cyclic hydrocarbon group To do. Suitable cycloalkyls include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and the like.
本明細書において、「シクロアルケニル」は、前記シクロアルキルの1個以上のC-C単結合が二重結合に置換された基を意味する。好適なシクロアルケニルは、限定するものではないが、例えばシクロプロペニル、シクロブテニル、シクロペンテニル及びシクロヘキセニル等を挙げることができる。 In the present specification, “cycloalkenyl” means a group in which one or more C—C single bonds of the cycloalkyl are substituted with double bonds. Suitable cycloalkenyl includes, but is not limited to, cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, and the like.
本明細書において、「シクロアルキニル」は、前記シクロアルキルの1個以上のC-C単結合が三重結合に置換された基を意味する。好適なシクロアルキニルは、限定するものではないが、例えばシクロブチニル、シクロペンチニル及びシクロヘキシニル等を挙げることができる。 In the present specification, “cycloalkynyl” means a group in which one or more C—C single bonds of the cycloalkyl are substituted with triple bonds. Suitable cycloalkynyl includes, but is not limited to, cyclobutynyl, cyclopentynyl, cyclohexynyl, and the like.
本明細書において、「ヘテロシクリル」は、前記シクロアルキル、シクロアルケニル又はシクロアルキニルの1個以上の炭素原子が、それぞれ独立して窒素原子(N)、硫黄原子(S)及び酸素原子(O)から選択される複素原子に置換された基を意味する。例えば、「(環の)員数3〜20のヘテロシクリル」及び「C3〜C20ヘテロシクリル」は、少なくとも3個且つ多くても20個の炭素原子を含む環式の炭化水素基の1個以上の炭素原子が、それぞれ独立して上記の複素原子に置換された基を意味する。この場合において、N又はSによる置換は、それぞれN-オキシド又はSのオキシド若しくはジオキシドによる置換を包含する。好適なヘテロシクリルは、限定するものではないが、例えばピロリジニル、テトラヒドロフラニル、ジヒドロフラニル、テトラヒドロチエニル、テトラヒドロピラニル、ジヒドロピラニル、テトラヒドロチオピラニル、ピペリジニル、モルホリニル、チオモルホリニル及びピペラジニル等を挙げることができる。In the present specification, “heterocyclyl” means that one or more carbon atoms of the cycloalkyl, cycloalkenyl or cycloalkynyl are each independently a nitrogen atom (N), a sulfur atom (S) and an oxygen atom (O). It means a group substituted by a selected hetero atom. For example, “(cyclic) 3-20 membered heterocyclyl” and “C 3 -C 20 heterocyclyl” include one or more cyclic hydrocarbon groups containing at least 3 and at most 20 carbon atoms. It means a group in which carbon atoms are independently substituted with the above heteroatoms. In this case, substitution with N or S includes substitution with N-oxide or S oxide or dioxide, respectively. Suitable heterocyclyls include, but are not limited to, pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidinyl, morpholinyl, thiomorpholinyl, and piperazinyl. it can.
本明細書において、「アリール」は、5〜20の炭素原子数を有する芳香族基を意味する。例えば、「(環の)員数5〜20のアリール」及び「C5〜C20アリール」は、少なくとも5個且つ多くても20個の炭素原子を含む芳香族基を意味する。好適なアリールは、限定するものではないが、例えばフェニル、ナフチル及びアントリル(アントラセニル)等を挙げることができる。In the present specification, “aryl” means an aromatic group having 5 to 20 carbon atoms. For example, “aryl of (ring) 5-20” and “C 5 -C 20 aryl” mean an aromatic group containing at least 5 and at most 20 carbon atoms. Suitable aryls include, but are not limited to, phenyl, naphthyl, anthryl (anthracenyl), and the like.
本明細書において、「アリールアルキル」は、前記アルキルの水素原子の1個が前記アリールに置換された基を意味する。好適なアリールアルキルは、限定するものではないが、例えばベンジル、1-フェネチル及び2-フェネチル等を挙げることができる。 In the present specification, “arylalkyl” means a group in which one of hydrogen atoms of the alkyl is substituted with the aryl. Suitable arylalkyls include, but are not limited to, benzyl, 1-phenethyl, 2-phenethyl, and the like.
本明細書において、「アリールアルケニル」は、前記アルケニルの水素原子の1個が前記アリールに置換された基を意味する。好適なアリールアルケニルは、限定するものではないが、例えばスチリル等を挙げることができる。 In the present specification, “arylalkenyl” means a group in which one of the hydrogen atoms of the alkenyl is substituted with the aryl. Suitable arylalkenyls include, but are not limited to, styryl and the like.
本明細書において、「ヘテロアリール」は、前記アリールの1個以上の炭素原子が、それぞれ独立して窒素原子(N)、硫黄原子(S)及び酸素原子(O)から選択される複素原子に置換された基を意味する。例えば、「(環の)員数5〜20のヘテロアリール」及び「C5〜C20ヘテロアリール」は、少なくとも5個且つ多くても20個の炭素原子を含む芳香族基の1個以上の炭素原子が、それぞれ独立して上記の複素原子に置換された基を意味する。この場合において、N又はSによる置換は、それぞれN-オキシド又はSのオキシド若しくはジオキシドによる置換を包含する。好適なヘテロアリールは、限定するものではないが、例えばフラニル、チエニル、ピロリル、イミダゾリル、ピラゾリル、トリアゾリル、テトラゾリル、チアゾリル、オキサゾリル、イソオキサゾリル、オキサジアゾリル、チアジアゾリル、イソチアゾリル、ピリジル、ピリダジニル、ピラジニル、ピリミジニル、キノリニル、イソキノリニル及びインドリル等を挙げることができる。As used herein, “heteroaryl” refers to a heteroatom in which one or more carbon atoms of the aryl are each independently selected from a nitrogen atom (N), a sulfur atom (S), and an oxygen atom (O). Means a substituted group. For example, and "(ring) heteroaryl Number 5-20""C 5 -C 20 heteroaryl", one or more carbons of the aromatic group containing at least five and at most 20 carbon atoms It means a group in which atoms are each independently substituted with the above heteroatoms. In this case, substitution with N or S includes substitution with N-oxide or S oxide or dioxide, respectively. Suitable heteroaryl include, but are not limited to, furanyl, thienyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, thiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl, isothiazolyl, pyridyl, pyridazinyl, pyrazinyl, pyrimidinyl, quinolinyl, Examples thereof include isoquinolinyl and indolyl.
本明細書において、「ヘテロアリールアルキル」は、前記アルキルの水素原子の1個が前記ヘテロアリールに置換された基を意味する。 In the present specification, “heteroarylalkyl” means a group in which one of the hydrogen atoms of the alkyl is substituted with the heteroaryl.
本明細書において、「カルボニル」は、カルボニル基を有する一価の基を意味する。好適なカルボニルは、限定するものではないが、例えばホルミル、アセチル、ベンゾイル、カルボキシ、メトキシカルボニル、ハロカルボニル及びカルバモイル(アミノカルボニル)等を挙げることができる。 In the present specification, “carbonyl” means a monovalent group having a carbonyl group. Suitable carbonyls include, but are not limited to, formyl, acetyl, benzoyl, carboxy, methoxycarbonyl, halocarbonyl, carbamoyl (aminocarbonyl), and the like.
上記で説明した基は、それぞれ独立して、酸素原子(O)、窒素原子(N)、硫黄原子(S)、ケイ素原子(Si)及びリン原子(P)からなる群より選択される1個以上の複素原子を含んでいてもよい。酸素原子、窒素原子又は硫黄原子であることが好ましい。この場合、上記の基を構成する1個以上の炭素原子が、それぞれ独立してこれらの複素原子に置換される。それ故、上記で説明した基は、それ自体の形態だけでなく、該基において一部の炭素原子が複素原子と置き換えられた形態も含むものとする。 Each of the groups described above is independently selected from the group consisting of an oxygen atom (O), a nitrogen atom (N), a sulfur atom (S), a silicon atom (Si), and a phosphorus atom (P). The above heteroatoms may be included. It is preferably an oxygen atom, a nitrogen atom or a sulfur atom. In this case, one or more carbon atoms constituting the above group are each independently substituted with these heteroatoms. Thus, the groups described above are intended to include not only the form itself, but also the form in which some of the carbon atoms are replaced with heteroatoms.
上記で説明した基は、それぞれ独立して、非置換であるか、又は1個以上のハロゲン、ヒドロキシル基、置換若しくは非置換のアミノ基、ニトロ基、シアノ基、メルカプト基、カルボニル基、アミド基又は複素原子を含んでいてもよい置換若しくは非置換の一価の炭化水素基(例えば置換若しくは非置換のC1〜C20アルキル、C2〜C20アルケニル、C2〜C20アルキニル、C1〜C20アルコキシル、C3〜C20シクロアルキル、C3〜C20シクロアルキルオキシ、C3〜C20シクロアルケニル、C3〜C20シクロアルケニルオキシ、C4〜C20シクロアルキニル、C4〜C20シクロアルキニルオキシ、C3〜C20ヘテロシクリル、C3〜C20ヘテロシクリルオキシ、C5〜C20アリール、C5〜C20アリールオキシ、C6〜C20アリールアルキル、C6〜C20アリールアルケニル、C5〜C20ヘテロアリール、C5〜C20ヘテロアリールオキシ又はC6〜C20ヘテロアリールアルキルであって、これらの基において一部の炭素原子が複素原子と置き換えられていてもよい)によって置換することもできる。この場合、上記の基は、それぞれ独立して、1個以上の上記の基で更に置換されていてもよい。The groups described above are each independently unsubstituted or one or more halogen, hydroxyl group, substituted or unsubstituted amino group, nitro group, cyano group, mercapto group, carbonyl group, amide group or substituted or also contain hetero atoms unsubstituted monovalent hydrocarbon group (e.g., a substituted or unsubstituted C 1 -C 20 alkyl, C 2 -C 20 alkenyl, C 2 -C 20 alkynyl, C 1 -C 20 alkoxyl, C 3 -C 20 cycloalkyl, C 3 -C 20 cycloalkyloxy, C 3 -C 20 cycloalkenyl, C 3 -C 20 cycloalkenyloxy, C 4 -C 20 cycloalkynyl, C 4 ~ C 20 cycloalkylene alkynyloxy, C 3 -C 20 heterocyclyl, C 3 -C 20 heterocyclyloxy, C 5 -C 20 aryl, C 5 -C 20 aryloxy, C 6 -C 20 arylalkyl, C 6 -C 20 aryl alkenyl, C 5 ~ C 20 heteroaryl, a C 5 -C 20 heteroaryloxy or C 6 -C 20 heteroarylalkyl, a part of carbon atoms are replaced by may) be replaced with hetero atoms in these groups You can also. In this case, each of the above groups may independently be further substituted with one or more of the above groups.
なお、本明細書において、「ハロゲン」又は「ハロ」は、フッ素、塩素、臭素又はヨウ素を意味する。 In the present specification, “halogen” or “halo” means fluorine, chlorine, bromine or iodine.
<2. オリゴデオキシヌクレオチド>
[2-1. オリゴデオキシヌクレオチド]
本発明者らは、化学反応によって低コスト且つ簡単な実験操作でDNA鎖の切断及び切断された末端のリン酸化を行う手段を検討したところ、以下において説明する非天然型塩基を含むデオキシヌクレオシドを導入したDNAオリゴマーと一級アミンのような求核剤とを反応させると、DNAオリゴマーが非天然型塩基を含むデオキシヌクレオシドの部位で切断されて、5’末端がリン酸化されたオリゴデオキシヌクレオチドが形成されることを見出した。当該反応は、前記DNAオリゴマーを求核剤存在下(塩基性条件下)で反応させることによって進行するため、デプリネーション反応のような酸性条件下で進行するDNAの分解反応を実質的に抑制又は防止することが可能となる。また、非天然型塩基を含むデオキシヌクレオシドは、DNA自動合成機によるDNA合成条件下で安定であるため、当該非天然型塩基を含むデオキシヌクレオシドをモノマーユニットとして、DNA自動合成機を用いてDNAを高収率で化学合成することが可能となる。加えて、当該非天然型塩基は、PCRのようなDNA複製条件下で天然型塩基と実質的に同一又は略同一の相補性を示すため、非天然型塩基を含むDNAは、in vivo及びin vitroにおける反応において、天然型塩基からなるDNAと実質的に同一又は略同一の鋳型としての機能を発揮することが可能となる。<2. Oligodeoxynucleotide>
[2-1. Oligodeoxynucleotide]
The inventors of the present invention have studied means for performing DNA strand cleavage and phosphorylation of a cleaved end by chemical reaction at low cost and simple experimental operation. As a result, deoxynucleosides containing unnatural bases described below are obtained. When the introduced DNA oligomer is reacted with a nucleophile such as a primary amine, the DNA oligomer is cleaved at a deoxynucleoside site containing a non-natural base to form an oligodeoxynucleotide phosphorylated at the 5 'end. I found out that Since the reaction proceeds by reacting the DNA oligomer in the presence of a nucleophile (under basic conditions), the DNA degradation reaction that proceeds under acidic conditions such as depreciation reaction is substantially suppressed or It becomes possible to prevent. In addition, deoxynucleosides containing unnatural bases are stable under the conditions of DNA synthesis using an automated DNA synthesizer. Chemical synthesis can be achieved with high yield. In addition, since the non-natural base exhibits substantially the same or substantially the same complementarity as the natural base under DNA replication conditions such as PCR, DNA containing the non-natural base can be used in vivo and in vivo. In the in vitro reaction, it is possible to exhibit a function as a template that is substantially the same or substantially the same as DNA comprising a natural base.
本発明の方法において、式I:
(A)n-B-C
で表されるオリゴデオキシヌクレオチドを使用する。In the process of the invention, the formula I:
(A) n -BC
The oligodeoxynucleotide represented by these is used.
ここで、式I中、
Aは、オリゴデオキシヌクレオチドであり、
Bは、求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドであり、
Cはオリゴデオキシヌクレオチドであり、
nは0又は1であり、
nが1のとき、Aの3’末端とBの5’位とは、ホスホジエステル結合で連結されており、
Bの3’位とCの5’末端とは、ホスホジエステル結合で連結されており、
BとCとの間のホスホジエステル結合は、求核剤の攻撃によって誘発されるEの脱離に伴って、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断される。Where in formula I
A is an oligodeoxynucleotide,
B is a deoxynucleoside containing a non-natural base having a moiety E removable by a nucleophile;
C is an oligodeoxynucleotide,
n is 0 or 1,
When n is 1, the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond,
The 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond,
The phosphodiester bond between B and C is such that the oligodeoxynucleotide part phosphorylated at the 5 'end of C from the 3' position of B with E elimination induced by nucleophile attack. It is cut by detaching.
式Iにおいて、オリゴデオキシヌクレオチドA及びCは、上記で説明した特徴を有するオリゴデオキシヌクレオチドであることが好ましい。オリゴデオキシヌクレオチドA及びCの塩基数は特に限定されないが、オリゴデオキシヌクレオチドAの場合、通常、0〜100,000の範囲であり、0〜20,000の範囲であることが好ましく、0〜10,000の範囲であることがより好ましい。オリゴデオキシヌクレオチドCの場合、通常、1〜100,000の範囲であり、1〜20,000の範囲であることが好ましく、1〜10,000の範囲であることがより好ましい。 In formula I, oligodeoxynucleotides A and C are preferably oligodeoxynucleotides having the characteristics described above. The number of bases of oligodeoxynucleotides A and C is not particularly limited, but in the case of oligodeoxynucleotide A, it is usually in the range of 0 to 100,000, preferably in the range of 0 to 20,000, and in the range of 0 to 100,000. It is more preferable. In the case of oligodeoxynucleotide C, it is usually in the range of 1 to 100,000, preferably in the range of 1 to 20,000, and more preferably in the range of 1 to 10,000.
求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドBは、以下で説明する特徴を有する非天然型塩基を含むデオキシヌクレオシドであることが好ましい。 The deoxynucleoside B containing a non-natural base having a moiety E removable by a nucleophile is preferably a deoxynucleoside containing a non-natural base having the characteristics described below.
式Iにおいて、nが0の場合、非天然型塩基を含むデオキシヌクレオシドBの5’末端のヒドロキシル基は、無保護の形態であってもよく、トリチル基又はモノ置換若しくはジ置換トリチル基、好ましくはDMTr基のような上記で説明した保護基で保護された形態であってもよい。いずれの形態も式Iで表されるオリゴヌクレオチドに包含されるものとする。 In Formula I, when n is 0, the hydroxyl group at the 5 ′ end of deoxynucleoside B containing an unnatural base may be in an unprotected form, and is preferably a trityl group or a mono- or di-substituted trityl group, preferably May be in a form protected with a protecting group as described above, such as a DMTr group. Any form shall be encompassed by the oligonucleotide represented by Formula I.
上記の特徴を有する式Iで表されるオリゴデオキシヌクレオチドを用いることにより、化学反応によって低コスト且つ簡単な実験操作でDNA鎖の切断を行い、結果として生じた末端がリン酸化されたオリゴデオキシヌクレオチドを得ることが可能となる。 By using the oligodeoxynucleotide represented by the formula I having the above characteristics, the DNA strand is cleaved by a chemical reaction at a low cost and with a simple experimental operation, and the resulting end is phosphorylated at the end. Can be obtained.
[2-2. 非天然型塩基を含むデオキシヌクレオシド]
本発明の方法で使用される非天然型塩基を含むデオキシヌクレオシドBは、求核剤によって脱離可能な部分を有する。具体的には、以下の特徴を有する。[2-2. Deoxynucleosides containing unnatural bases]
Deoxynucleoside B containing an unnatural base used in the method of the present invention has a moiety that can be eliminated by a nucleophile. Specifically, it has the following characteristics.
(i)DNA合成条件下において安定に存在する。 (I) It exists stably under DNA synthesis conditions.
(ii)in vivo及び/又はin vitroにおける反応(例えばPCR反応)のようなDNA複製条件下において安定に存在する。 (Ii) It exists stably under DNA replication conditions such as in vivo and / or in vitro reactions (eg, PCR reactions).
(iii) 求核剤存在下(塩基性条件下)で反応させることによって脱離可能な部分Eを有する。 (Iii) having a moiety E that can be eliminated by reacting in the presence of a nucleophile (under basic conditions).
(iv) in vivo及び/又はin vitroにおける反応(例えばPCR反応)のようなDNA複製条件下において、フィデリティが高く、天然型塩基と実質的に同一又は略同一の相補性を示す。 (Iv) It has high fidelity under DNA replication conditions such as in vivo and / or in vitro reaction (for example, PCR reaction) and exhibits substantially the same or substantially the same complementarity as a natural base.
(v) 3’位のヒドロキシル基において、別のオリゴデオキシヌクレオチドCの5’末端のリン酸基とホスホジエステル結合を形成している場合、Bの3’位とCの5’末端との間のホスホジエステル結合は、求核剤の攻撃によって誘発されるEの脱離に伴って、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断される。これにより、5’末端がリン酸化された別のオリゴヌクレオチドCが形成される。 (V) When forming a phosphodiester bond with a phosphate group at the 5 ′ end of another oligodeoxynucleotide C in the hydroxyl group at the 3 ′ position, between the 3 ′ position of B and the 5 ′ end of C The phosphodiester bond is cleaved by elimination of the oligodeoxynucleotide moiety phosphorylated at the 5 'end of C from the 3' position of B upon E elimination induced by nucleophile attack. Is done. This forms another oligonucleotide C phosphorylated at the 5 'end.
本明細書において、「DNA合成条件」は、当該技術分野において通常使用されるDNA自動合成機によってDNA合成を行う際に適用される、ホスホロアミダイト法のような一般的な合成条件を意味する。前記DNA自動合成機としては、限定するものではないが、例えば、ABI 392 DNA/RNAシンセサイザー(アプライドバイオシステムズ社)、及びNTS H-6 DNA/RNAシンセサイザー(日本テクノサービス社)を挙げることができる。DNA合成条件は、式Iで表されるオリゴデオキシヌクレオチドを合成する際の基質として使用される、非天然型塩基を含むデオキシヌクレオシドB及び天然型塩基を含む1個以上のデオキシヌクレオシドの組み合わせに基づいて、例えばDNA自動合成機に添付される取扱説明書を参照することによって適宜設定することができる。 In the present specification, “DNA synthesis conditions” means general synthesis conditions such as phosphoramidite method applied when DNA synthesis is performed by an automatic DNA synthesizer usually used in the art. . Examples of the automatic DNA synthesizer include, but are not limited to, ABI 392 DNA / RNA synthesizer (Applied Biosystems) and NTS H-6 DNA / RNA synthesizer (Nippon Techno Service). . DNA synthesis conditions are based on a combination of deoxynucleoside B containing a non-natural base and one or more deoxynucleosides containing a natural base used as a substrate when synthesizing an oligodeoxynucleotide represented by Formula I. For example, it can be set as appropriate by referring to the instruction manual attached to the DNA automatic synthesizer.
特徴(i)「DNA合成条件下において安定に存在する」とは、具体的には以下の特徴を満足することを意味する。 Feature (i) “Stablely present under DNA synthesis conditions” specifically means that the following features are satisfied.
(i-1)カップリング反応生成物の亜リン酸エステル部分の酸化に用いられるヨウ素存在下において、ヨウ素と実質的に反応しないこと。 (I-1) It does not substantially react with iodine in the presence of iodine used for oxidation of the phosphite part of the coupling reaction product.
(i-2)DMTr基の脱保護に用いられるトリクロロ酢酸のような酸存在下(酸性条件下)において、酸と実質的に反応しないこと。 (I-2) It should not substantially react with an acid in the presence of an acid such as trichloroacetic acid (acidic conditions) used for deprotection of the DMTr group.
(i-3)合成されたDNAの切り出し及び脱保護に用いられるアンモニア水のようなアルカリ存在下(アルカリ性条件下)において、アルカリと実質的に反応しないこと。 (I-3) It should not substantially react with an alkali in the presence of an alkali such as aqueous ammonia used for excision and deprotection of the synthesized DNA (alkaline conditions).
また、本明細書において、「DNA複製条件」は、当該技術分野において通常使用されるPCR反応によってDNAを複製する際に適用される、一般的な反応条件を意味する。DNA複製条件は、式Iで表されるオリゴデオキシヌクレオチドを複製する際の鋳型として使用される当該オリゴデオキシヌクレオチドの塩基配列に基づいて、例えばKramer M. F.及びCoen D. M., “Enzymatic amplification of DNA by PCR: standard procedures and optimization.”, Curr Protoc Cytom. 2006 Aug;Appendix 3:Appendix 3Kを参照することによって適宜設定することができる。 Moreover, in this specification, "DNA replication conditions" means general reaction conditions applied when replicating DNA by a PCR reaction usually used in the art. DNA replication conditions are based on the base sequence of the oligodeoxynucleotide used as a template when replicating the oligodeoxynucleotide represented by Formula I, for example, Kramer MF and Coen DM, “Enzymatic amplification of DNA by PCR: "standard procedures and optimization.", Curr Protoc Cytom. 2006 Aug; Appendix 3: Appendix 3K.
特徴(ii)「in vivo及び/又はin vitroにおける反応(例えばPCR反応)のようなDNA複製条件下において安定に存在する」とは、具体的には以下の特徴を満足することを意味する。 Characteristic (ii) “is stably present under DNA replication conditions such as in vivo and / or in vitro reaction (eg, PCR reaction)” specifically means that the following characteristics are satisfied.
(ii-1)PCRで通常使用される緩衝溶液(例えば塩化マグネシウム又は塩化カリウムのような塩を含む、常温でpH8付近のトリスバッファー(場合により界面活性剤又はBSAを含んでもよい))中、PCRサイクルにおいて95℃で加熱しても実質的に分解されない。 (Ii-1) In a buffer solution usually used in PCR (for example, a tris buffer containing a salt such as magnesium chloride or potassium chloride and having a pH of around 8 at room temperature (optionally containing a surfactant or BSA)), Heating at 95 ° C in the PCR cycle does not substantially decompose.
非天然型塩基を含むデオキシヌクレオシドBは、より具体的には、以下の特徴(vi)及び(vii)を有することが好ましい。 More specifically, deoxynucleoside B containing an unnatural base preferably has the following characteristics (vi) and (vii).
(vi) 非天然型塩基が、置換若しくは非置換のアルケニル又はアルキニルで置換されたプリン型若しくはピリミジン型塩基の構造を有する。 (Vi) The unnatural base has a structure of a purine or pyrimidine base substituted with a substituted or unsubstituted alkenyl or alkynyl.
(vii) 求核剤存在下(塩基性条件下)で反応させることによって、デオキシリボースの1’位の炭素原子が求核剤の攻撃を受け、グリコシド結合が切断されるとともに、グリコシド結合を形成していた電子対の流れがアルケニル又はアルキニルの炭素原子をさらに攻撃することにより、非天然型塩基部分で分子内環化反応を生じながら非天然型塩基に由来する部分Eが脱離する。 (Vii) By reacting in the presence of a nucleophile (under basic conditions), the carbon atom at the 1 'position of deoxyribose is attacked by the nucleophile, and the glycosidic bond is cleaved to form a glycosidic bond. When the flow of the electron pair further attacks the carbon atom of alkenyl or alkynyl, the moiety E derived from the non-natural base is eliminated while causing an intramolecular cyclization reaction at the non-natural base moiety.
或いは、非天然型塩基を含むデオキシヌクレオシドBは、以下の特徴(vi’)及び(vii’)を有することが好ましい。 Alternatively, deoxynucleoside B containing an unnatural base preferably has the following characteristics (vi ′) and (vii ′).
(vi’) 非天然型塩基が、置換若しくは非置換のアルケニル又はアルキニルで置換されたプリン型若しくはピリミジン型塩基の水付加体若しくは求核剤付加体、又はそれらの互変異性体の構造を有する。 (Vi ') The unnatural base has a structure of a water adduct or a nucleophile adduct of a purine or pyrimidine base substituted with a substituted or unsubstituted alkenyl or alkynyl, or a tautomer thereof. .
(vii’) 求核剤存在下(塩基性条件下)で反応させることによって、置換若しくは非置換のアルケニル又はアルキニルで置換されたプリン型若しくはピリミジン型塩基に水若しくは求核剤が付加して該デオキシヌクレオシドの水付加体若しくは求核剤付加体を形成するか、又はそれらの互変異性体を形成する。そして、前記デオキシヌクレオシドの水付加体若しくは求核剤付加体、又はそれらの互変異性体のデオキシリボースの1’位の炭素原子が求核剤の攻撃を受け、グリコシド結合が切断されるとともに、非天然型塩基に由来する部分Eが脱離する。 (Vii ') By reacting in the presence of a nucleophile (under basic conditions), water or a nucleophile is added to a purine or pyrimidine type base substituted with a substituted or unsubstituted alkenyl or alkynyl, Form water adducts or nucleophile adducts of deoxynucleosides or form their tautomers. And, the deoxyribose 1'-position carbon atom of the deoxynucleoside water adduct or nucleophile adduct, or their tautomers is attacked by the nucleophile, and the glycosidic bond is cleaved, Part E derived from a non-natural base is eliminated.
或いは、非天然型塩基を含むデオキシヌクレオシドBは、以下の特徴(vi’’)及び(vii’’)を有することが好ましい。 Alternatively, deoxynucleoside B containing an unnatural base preferably has the following characteristics (vi ″) and (vii ″).
(vi’’) 非天然型塩基が、カルボニル、置換若しくは非置換のアルキニル若しくはシアノを有する置換若しくは非置換のアリールで置換されたプリン型塩基の構造を有する。 (Vi ″) A non-natural base has a structure of a purine base substituted with a substituted or unsubstituted aryl having carbonyl, substituted or unsubstituted alkynyl or cyano.
(vii’’) 求核剤存在下(塩基性条件下)で反応させることによって、デオキシリボースの1’位の炭素原子が求核剤の攻撃を受け、グリコシド結合が切断されるとともに、グリコシド結合を形成していた電子対の流れがカルボニル、置換若しくは非置換のアルキニル又はシアノの炭素原子をさらに攻撃することにより、非天然型塩基に由来する部分Eが脱離する。 (Vii '') By reacting in the presence of a nucleophile (under basic conditions), the 1'-position carbon atom of deoxyribose is attacked by the nucleophile and the glycosidic bond is cleaved. When the electron pair stream that formed further attacks the carbon atom of carbonyl, substituted or unsubstituted alkynyl or cyano, the moiety E derived from the unnatural base is eliminated.
本明細書において、「非天然型塩基を含むデオキシヌクレオシドB」は、化合物B自体だけでなく、該化合物の水付加体若しくは求核剤付加体又はそれらの互変異性体も意味するものとする。 In the present specification, “deoxynucleoside B containing a non-natural base” means not only compound B itself but also a water adduct or a nucleophile adduct of the compound or a tautomer thereof. .
例えば、非天然型塩基を含むデオキシヌクレオシドBが上記の特徴(vi)及び(vii)を有する下記の式:
[式中、R及びR’は、下記で定義するものと同様の基を意味する。]
で表される化合物である場合、Bを含むオリゴデオキシヌクレオチドを、求核剤存在下(塩基性条件下)で反応させることにより、以下の反応が進行すると考えられる。なお、本発明の作用効果は、下記の反応機構に限定されるものではない。
In the case of a compound represented by the formula, it is considered that the following reaction proceeds when an oligodeoxynucleotide containing B is reacted in the presence of a nucleophile (under basic conditions). In addition, the effect of this invention is not limited to the following reaction mechanism.
上記の反応式において、R及びR’は下記で定義するものと同様の基を意味し、DNAはオリゴデオキシヌクレオチドを、Nuは求核剤を意味する。 In the above reaction formula, R and R 'are the same groups as defined below, DNA is an oligodeoxynucleotide, and Nu is a nucleophile.
例えば、非天然型塩基を含むデオキシヌクレオシドBが上記の特徴(vi’)及び(vii’)を有する下記の式:
[式中、Rは、下記で定義するものと同様の基を意味する。]
で表される化合物である場合、Bを含むオリゴデオキシヌクレオチドを、求核剤存在下(塩基性条件下)で反応させることにより、該化合物の互変異性化を伴う以下の反応が進行すると考えられる。なお、本発明の作用効果は、下記の反応機構に限定されるものではない。
In the case of a compound represented by the formula, it is considered that the following reaction involving tautomerization of the compound proceeds by reacting an oligodeoxynucleotide containing B in the presence of a nucleophile (under basic conditions). It is done. In addition, the effect of this invention is not limited to the following reaction mechanism.
上記の反応式において、Rは下記で定義するものと同様の基を意味し、DNAはオリゴデオキシヌクレオチドを、Nuは求核剤を意味する。 In the above reaction formula, R means the same group as defined below, DNA means an oligodeoxynucleotide, and Nu means a nucleophile.
例えば、非天然型塩基を含むデオキシヌクレオシドBが上記の特徴(vi’)及び(vii’)を有する下記の式:
[式中、Rは、下記で定義するものと同様の基を意味する。]
で表される化合物である場合、Bを含むオリゴデオキシヌクレオチドを、求核剤存在下(塩基性条件下)で反応させることにより、該化合物の水付加反応又は求核剤付加反応が進行する可能性がある。
In the case of a compound represented by the formula, by reacting an oligodeoxynucleotide containing B in the presence of a nucleophile (under basic conditions), the water addition reaction or nucleophile addition reaction of the compound can proceed. There is sex.
前記反応によって生じたBの水付加体又は求核剤付加体を含むオリゴデオキシヌクレオチドを、求核剤存在下(塩基性条件下)でさらに反応させることにより、以下の反応が進行すると考えられる。なお、本発明の作用効果は、下記の反応機構に限定されるものではない。
上記の反応式において、Rは下記で定義するものと同様の基を意味し、DNAはオリゴデオキシヌクレオチドを、Nuは求核剤を、RNuは求核剤付加に伴って生じる基を意味する。In the above reaction formula, R means the same group as defined below, DNA means an oligodeoxynucleotide, Nu means a nucleophile, and R Nu means a group generated upon addition of a nucleophile. .
例えば、非天然型塩基を含むデオキシヌクレオシドBが上記の特徴(vi’’)及び(vii’’)を有する下記の式:
[式中、r、R’’、R’’’及びZは、下記で定義するものと同様の基を意味する。]
で表される化合物である場合、Bを含むオリゴデオキシヌクレオチドを、求核剤存在下(塩基性条件下)で反応させることにより、以下の反応が進行すると考えられる。以下の反応式では、R’’’がホルミル基(-COZ, Z=H)の場合を例示したが、R’’’が他の基の場合であっても同様の反応が進行すると考えられる。なお、本発明の作用効果は、下記の反応機構に限定されるものではない。
In the case of a compound represented by the formula, it is considered that the following reaction proceeds when an oligodeoxynucleotide containing B is reacted in the presence of a nucleophile (under basic conditions). In the following reaction formula, the case where R ′ ″ is a formyl group (—COZ, Z = H) is exemplified, but it is considered that the same reaction proceeds even when R ′ ″ is another group. . In addition, the effect of this invention is not limited to the following reaction mechanism.
上記の反応式において、r及びR’’は下記で定義するものと同様の基を意味し、DNAはオリゴデオキシヌクレオチドを、Nuは求核剤を意味する。 In the above reaction formula, r and R ″ represent the same groups as defined below, DNA represents an oligodeoxynucleotide, and Nu represents a nucleophile.
上記の特徴を有する非天然型塩基を含むデオキシヌクレオシドBを本発明の方法に用いることにより、求核剤存在下(塩基性条件下)で反応させることによって低コスト且つ簡単な実験操作でDNA鎖の切断を行い、結果として生じた末端がリン酸化されたオリゴデオキシヌクレオチドを得ることが可能となる。また、本発明の方法によるDNAの切断は、求核剤の攻撃によって誘発される、非天然型塩基を含むデオキシヌクレオシドからのEの脱離に伴って生じることから、当該一塩基のデオキシヌクレオシドBの挿入又は置換によってDNA鎖の任意の位置に切断部位を導入することが可能となる。 By using deoxynucleoside B containing an unnatural base having the above-mentioned characteristics in the method of the present invention, a DNA strand can be reacted at a low cost and with a simple experimental operation by reacting in the presence of a nucleophile (under basic conditions). As a result, it is possible to obtain an oligodeoxynucleotide whose resulting end is phosphorylated. Further, since the cleavage of DNA by the method of the present invention is accompanied by the elimination of E from the deoxynucleoside containing a non-natural base induced by the attack of a nucleophile, the single-base deoxynucleoside B It becomes possible to introduce a cleavage site at an arbitrary position of the DNA strand by insertion or substitution.
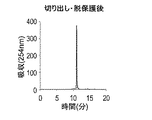
DNA自動合成機を用いて式Iで表されるオリゴデオキシヌクレオチドを調製する場合、モノマーユニットとして準備される非天然型塩基を含むデオキシヌクレオシドBの5’末端のヒドロキシル基は、トリチル基又はモノ置換若しくはジ置換トリチル基、好ましくはDMTr基のような上記で説明した保護基で保護された形態であることが好ましい。また、式Iで表されるオリゴデオキシヌクレオチドにおいて、nが0である場合、非天然型塩基を含むデオキシヌクレオシドBの5’末端のヒドロキシル基は、無保護、或いはトリチル基又はモノ置換若しくはジ置換トリチル基、好ましくはDMTr基のような上記で説明した保護基で保護された形態であることが好ましい。 When preparing an oligodeoxynucleotide represented by Formula I using an automatic DNA synthesizer, the hydroxyl group at the 5 ′ end of deoxynucleoside B containing a non-natural base prepared as a monomer unit is a trityl group or mono-substituted. Alternatively, it is preferably in a form protected with a protecting group as described above, such as a disubstituted trityl group, preferably a DMTr group. In addition, in the oligodeoxynucleotide represented by formula I, when n is 0, the hydroxyl group at the 5 ′ end of deoxynucleoside B containing an unnatural base is unprotected, or is a trityl group, monosubstituted or disubstituted It is preferably in a form protected with a protecting group as described above, such as a trityl group, preferably a DMTr group.
本明細書において、「非天然型塩基を含むデオキシヌクレオチドB」は、非天然型塩基を含むデオキシヌクレオシドBの5’末端のヒドロキシル基がリン酸化された形態を意味する。 In the present specification, “deoxynucleotide B containing an unnatural base” means a form in which the hydroxyl group at the 5 ′ end of deoxynucleoside B containing an unnatural base is phosphorylated.
非天然型塩基を含むデオキシヌクレオシドBは、以下:
で表されるデオキシヌクレオシドであることが好ましく、式B-1、B-2、B-3、B-5、B-6、B-7、B-21又はB-22で表されるデオキシヌクレオシドであることがより好ましい。前記の化合物のうち、式B-3、B-7、B-21及び22(式中、Rは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロアリールアルキル又はカルボニルであり;R'は、メチルであり;rは、0〜4の整数であり;R’’は、ハロゲン、又は置換若しくは非置換のアルキル、アルケニル若しくはアルキニルであり;R’’’は、-COZ、-C≡C-Z又は-CNであり;Zは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、又は置換若しくは非置換のヘテロアリールアルキルである)で表される化合物は、本発明者らが見出した新規化合物である。 Is preferably a deoxynucleoside represented by the formula: B-1, B-2, B-3, B-5, B-6, B-7, B-21 or B-22 It is more preferable that Of the above compounds, formulas B-3, B-7, B-21 and 22 wherein R is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl , Substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl or carbonyl; R ′ is methyl; Is an integer of 4; R ″ is halogen or substituted or unsubstituted alkyl, alkenyl, or alkynyl; R ′ ″ is —COZ, —C≡CZ, or —CN; Z is hydrogen Substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted aryl Le, a substituted or unsubstituted heteroaryl, or substituted or a compound represented by unsubstituted heteroaryl alkyl) is a novel compound discovered by the present inventors.
式B-1〜B-7、B-11〜B-14、B-21及びB-22において、Rは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロアリールアルキル又はカルボニルである。具体的には、Rは、水素、置換若しくは非置換のC1〜C10アルキル、置換若しくは非置換のC3〜C20シクロアルキル、置換若しくは非置換のC3〜C20ヘテロシクリル、置換若しくは非置換のC5〜C20アリール、置換若しくは非置換のC6〜C20アリールアルキル、置換若しくは非置換のC5〜C20ヘテロアリール、置換若しくは非置換のC5〜C20ヘテロアリールアルキル又はカルボニルであることが好ましい。特に好適なRは、水素、メチル、エチル、フェニル、ヒドロキシメチル、アミノメチル、メルカプトメチル、ハロメチル、ホルミル、カルボキシル、カルバモイル及びアセチルを挙げることができる。水素、メチル、フェニル又はカルバモイルであることが特に好ましい。In formulas B-1 to B-7, B-11 to B-14, B-21 and B-22, R is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted A heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl or carbonyl. Specifically, R, hydrogen, a substituted or unsubstituted C 1 -C 10 alkyl, substituted or unsubstituted C 3 -C 20 cycloalkyl, substituted or unsubstituted C 3 -C 20 heterocyclyl, substituted or unsubstituted substituted C 5 -C 20 aryl, substituted or unsubstituted C 6 -C 20 arylalkyl, substituted or unsubstituted C 5 -C 20 heteroaryl, substituted or unsubstituted C 5 -C 20 heteroarylalkyl or carbonyl It is preferable that Particularly suitable R can include hydrogen, methyl, ethyl, phenyl, hydroxymethyl, aminomethyl, mercaptomethyl, halomethyl, formyl, carboxyl, carbamoyl and acetyl. Particular preference is given to hydrogen, methyl, phenyl or carbamoyl.
式B-1〜B-7、B-11〜B-14、B-21及びB-22において、R’が存在する場合、R'は、水素、又は置換若しくは非置換のアルキル、アルケニル若しくはアルキニルである。具体的には、R'は、水素原子、又は置換若しくは非置換のC1〜C10アルキル、C2〜C10アルケニル若しくはC2〜C10アルキニルであることが好ましく、メチルであることが特に好ましい。In formulas B-1 to B-7, B-11 to B-14, B-21 and B-22, when R ′ is present, R ′ is hydrogen or substituted or unsubstituted alkyl, alkenyl or alkynyl. It is. Specifically, R ′ is preferably a hydrogen atom or a substituted or unsubstituted C 1 -C 10 alkyl, C 2 -C 10 alkenyl or C 2 -C 10 alkynyl, particularly methyl. preferable.
式B-21及びB-22において、rは、0〜4の整数であり、R’’は、ハロゲン、又は置換若しくは非置換のアルキル、アルケニル若しくはアルキニルである。具体的には、rは、0〜4の整数であり、R’’は、ハロゲン、又は置換若しくは非置換のC1〜C10アルキル、C2〜C10アルケニル若しくはC2〜C10アルキニルであることが好ましく、rは、0であることが特に好ましい。In formulas B-21 and B-22, r is an integer from 0 to 4 and R ″ is halogen or substituted or unsubstituted alkyl, alkenyl or alkynyl. Specifically, r is an integer from 0 to 4, R '' is halogen, or substituted or unsubstituted C 1 -C 10 alkyl, with C 2 -C 10 alkenyl or C 2 -C 10 alkynyl Preferably, r is particularly preferably 0.
式B-21及びB-22において、R’’’は、-COZ、-C≡C-Z又は-CNである。具体的には、R’’’は、-COZであることが好ましい。 In formulas B-21 and B-22, R ″ ″ is —COZ, —C≡C—Z or —CN. Specifically, R ″ ″ is preferably —COZ.
式B-21及びB-22において、Zは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、又は置換若しくは非置換のヘテロアリールアルキルである。具体的には、Zは、水素、置換若しくは非置換のC1〜C10アルキル、置換若しくは非置換のC3〜C20シクロアルキル、置換若しくは非置換のC3〜C20ヘテロシクリル、置換若しくは非置換のC5〜C20アリール、置換若しくは非置換のC6〜C20アリールアルキル、置換若しくは非置換のC5〜C20ヘテロアリール、又は置換若しくは非置換のC5〜C20ヘテロアリールアルキルであることが好ましい。特に好適なRは、水素、メチル、エチル、フェニル、ヒドロキシメチル、アミノメチル、メルカプトメチル又はハロメチルを挙げることができる。水素、メチル又はフェニルであることが好ましく、水素であることがより好ましい。In formulas B-21 and B-22, Z is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted aryl Alkyl, substituted or unsubstituted heteroaryl, or substituted or unsubstituted heteroarylalkyl. Specifically, Z is hydrogen, substituted or unsubstituted C 1 -C 10 alkyl, substituted or unsubstituted C 3 -C 20 cycloalkyl, substituted or unsubstituted C 3 -C 20 heterocyclyl, substituted or unsubstituted C 5 -C 20 aryl substituted, substituted or unsubstituted C 6 -C 20 arylalkyl, substituted or unsubstituted C 5 -C 20 heteroaryl, or substituted or unsubstituted of C 5 -C 20 heteroarylalkyl Preferably there is. Particularly suitable R may include hydrogen, methyl, ethyl, phenyl, hydroxymethyl, aminomethyl, mercaptomethyl or halomethyl. Hydrogen, methyl or phenyl is preferable, and hydrogen is more preferable.
上記の基は、一部の炭素原子が、1又は複数、好ましくは1〜5個、より好ましくは1〜3個の複素原子と置き換えられていてもよい。前記複素原子は、酸素原子、窒素原子、硫黄原子、ケイ素原子及びリン原子からなる群より選択される複素原子であることが好ましい。 In the above groups, some of the carbon atoms may be replaced with one or more, preferably 1 to 5, more preferably 1 to 3 heteroatoms. The hetero atom is preferably a hetero atom selected from the group consisting of an oxygen atom, a nitrogen atom, a sulfur atom, a silicon atom and a phosphorus atom.
また、上記の基が置換されている場合、当該基は、1個以上のハロゲン、ヒドロキシル基、置換若しくは非置換のアミノ基、ニトロ基、シアノ基、メルカプト基、カルボニル基、アミド基又は複素原子を含んでいてもよい置換若しくは非置換の一価の炭化水素基(例えば置換若しくは非置換のC1〜C20アルキル、C1〜C20アルコキシル、C3〜C20シクロアルキル、C3〜C20シクロアルキルオキシ、C3〜C20ヘテロシクリル、C3〜C20ヘテロシクリルオキシ、C5〜C20アリール、C5〜C20アリールオキシ、C6〜C20アリールアルキル、C5〜C20ヘテロアリール、C5〜C20ヘテロアリールオキシ又はC6〜C20ヘテロアリールアルキルであって、これらの基において一部の炭素原子が複素原子と置き換えられていてもよい)によって置換されていることが好ましい。When the above group is substituted, the group is one or more halogen, hydroxyl group, substituted or unsubstituted amino group, nitro group, cyano group, mercapto group, carbonyl group, amide group, or hetero atom. the comprise also be substituted or unsubstituted optionally monovalent hydrocarbon group (e.g., a substituted or unsubstituted C 1 -C 20 alkyl, C 1 -C 20 alkoxyl, C 3 -C 20 cycloalkyl, C 3 -C 20 cycloalkyloxy, C 3 -C 20 heterocyclyl, C 3 -C 20 heterocyclyloxy, C 5 -C 20 aryl, C 5 -C 20 aryloxy, C 6 -C 20 arylalkyl, C 5 -C 20 heteroaryl , C 5 -C 20 heteroaryloxy or C 6 -C 20 heteroarylalkyl, in which some of the carbon atoms may be replaced by heteroatoms). That's right.
非天然型塩基を含むデオキシヌクレオシドBは、該化合物自体だけでなく、その塩も包含する。非天然型塩基を含むデオキシヌクレオシドBの塩の対イオンとしては、限定するものではないが、例えば、ナトリウムイオン、カリウムイオン、カルシウムイオン若しくはマグネシウムイオンのようなカチオン、又は塩化物イオン、臭化物イオン、ギ酸イオン、酢酸イオン、マレイン酸イオン、フマル酸イオン、安息香酸イオン、アスコルビン酸イオン、パモ酸イオン、コハク酸イオン、ビスメチレンサリチル酸イオン、メタンスルホン酸イオン、エタンジスルホン酸イオン、プロピオン酸イオン、酒石酸イオン、サリチル酸イオン、クエン酸イオン、グルコン酸イオン、アスパラギン酸イオン、ステアリン酸イオン、パルミチン酸イオン、イタコン酸イオン、グリコール酸イオン、p-アミノ安息香酸イオン、グルタミン酸イオン、ベンゼンスルホン酸イオン、シクロヘキシルスルファミン酸イオン、メタンスルホン酸イオン、エタンスルホン酸イオン、イセチオン酸イオン、ベンゼンスルホン酸イオン、p-トルエンスルホン酸イオン、ナフタレンスルホン酸イオン、リン酸イオン、硝酸イオン、硫酸イオン、炭酸イオン、炭酸水素イオン又は過塩素酸イオンのようなアニオンが好ましい。 Deoxynucleoside B containing a non-natural base includes not only the compound itself but also a salt thereof. The counter ion of the salt of deoxynucleoside B containing a non-natural base includes, but is not limited to, for example, a cation such as sodium ion, potassium ion, calcium ion or magnesium ion, or chloride ion, bromide ion, Formate, acetate, maleate, fumarate, benzoate, ascorbate, pamoate, succinate, bismethylenesalicylate, methanesulfonate, ethanedisulfonate, propionate, tartaric acid Ion, salicylate ion, citrate ion, gluconate ion, aspartate ion, stearate ion, palmitate ion, itaconic acid ion, glycolate ion, p-aminobenzoate ion, glutamate ion, benzene Phosphonate ion, cyclohexylsulfamate ion, methanesulfonate ion, ethanesulfonate ion, isethionate ion, benzenesulfonate ion, p-toluenesulfonate ion, naphthalenesulfonate ion, phosphate ion, nitrate ion, sulfate ion, Anions such as carbonate ions, bicarbonate ions or perchlorate ions are preferred.
非天然型塩基を含むデオキシヌクレオシドBは、該化合物自体だけでなく、その溶媒和物も包含する。非天然型塩基を含むデオキシヌクレオシドBと溶媒和物を形成し得る溶媒としては、限定するものではないが、例えば、メタノール、エタノール、2-プロパノール(イソプロピルアルコール)、ジメチルスルホキシド(DMSO)、酢酸、エタノールアミン若しくは酢酸エチルのような有機溶媒、又は水が好ましい。 The deoxynucleoside B containing an unnatural base includes not only the compound itself but also a solvate thereof. Solvents that can form solvates with deoxynucleoside B containing an unnatural base include, but are not limited to, for example, methanol, ethanol, 2-propanol (isopropyl alcohol), dimethyl sulfoxide (DMSO), acetic acid, Organic solvents such as ethanolamine or ethyl acetate, or water are preferred.
また、非天然型塩基を含むデオキシヌクレオシドBは、該化合物自体だけでなく、その保護形態も包含する。本明細書において、「保護形態」は、1個又は複数の官能基に保護基が導入された形態を意味する。 In addition, deoxynucleoside B containing a non-natural base includes not only the compound itself but also a protected form thereof. In the present specification, “protected form” means a form in which a protecting group is introduced into one or more functional groups.
上記の特徴を有する非天然型塩基を含むデオキシヌクレオシドBを本発明の方法に用いることにより、求核剤存在下(塩基性条件下)で反応させることによって低コスト且つ簡単な実験操作でDNA鎖の切断を行い、結果として生じた末端がリン酸化されたオリゴデオキシヌクレオチドを得ることが可能となる。 By using deoxynucleoside B containing an unnatural base having the above-mentioned characteristics in the method of the present invention, a DNA strand can be reacted at a low cost and with a simple experimental operation by reacting in the presence of a nucleophile (under basic conditions). As a result, it is possible to obtain an oligodeoxynucleotide whose resulting end is phosphorylated.
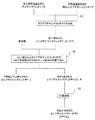
<3. オリゴデオキシヌクレオチドの製造方法>
図1は、本発明のオリゴデオキシヌクレオチドの製造方法の一実施形態を示す工程図である。以下、図1に基づき、本発明の方法の好ましい実施形態について詳細に説明する。<3. Method for producing oligodeoxynucleotide>
FIG. 1 is a process diagram showing one embodiment of a method for producing an oligodeoxynucleotide of the present invention. Hereinafter, a preferred embodiment of the method of the present invention will be described in detail with reference to FIG.
[3-1. 5’末端リン酸化オリゴヌクレオチド形成工程]
本発明の方法は、求核剤と、式I:
(A)n-B-C
で表されるオリゴデオキシヌクレオチドとを接触させることによって、前記Cの5’末端がリン酸化されたオリゴデオキシヌクレオチドと、式II:
(A)n-B’
で表されるオリゴデオキシヌクレオチドとを得る、5’末端リン酸化オリゴヌクレオチド形成工程を含む(工程S2)。本工程は、BとCとの間のホスホジエステル結合を、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分をBの3’位から脱離させることによって切断して、前記Cの5’末端がリン酸化されたオリゴデオキシヌクレオチドを得ることを目的とする。[3-1. 5′-terminal phosphorylated oligonucleotide formation process]
The method of the present invention comprises a nucleophile and a compound of formula I:
(A) n -BC
And an oligodeoxynucleotide phosphorylated at the 5 ′ end of C, by contacting with an oligodeoxynucleotide represented by formula II:
(A) n -B '
A 5′-terminal phosphorylated oligonucleotide formation step for obtaining an oligodeoxynucleotide represented by (Step S2). In this step, the phosphodiester bond between B and C is cleaved by removing the oligodeoxynucleotide moiety phosphorylated at the 5 ′ end of C from the 3 ′ position of B. 'To obtain oligodeoxynucleotides phosphorylated at the ends.
式I及びIIにおいて、A、B、C及びnは、上記で定義したのと同様の意味を表す。 In formulas I and II, A, B, C and n represent the same meaning as defined above.
式IIにおいて、B’は、求核剤の攻撃によるEの脱離に伴って形成されるBの残基部分である。 In Formula II, B 'is the residue part of B that is formed upon elimination of E by nucleophilic attack.
式IIにおいて、AとB’とは、ホスホジエステル結合で連結されている。より具体的には、B’は、Bの5’位に由来する位置において、Aの3’末端とホスホジエステル結合で連結されている。本明細書において、「B’の5’位」は、B’において、Bの5’位に由来する位置を意味する。 In Formula II, A and B 'are linked by a phosphodiester bond. More specifically, B 'is linked to the 3' end of A by a phosphodiester bond at a position derived from the 5 'position of B. In the present specification, “the 5′-position of B ′” means a position derived from the 5′-position of B in B ′.
式Iにおいて、nが0である場合、求核剤と、式I’:
B-C
で表されるオリゴデオキシヌクレオチドとを接触させることによって、BとCとの間のホスホジエステル結合を、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分をBの3’位から脱離させることによって切断して、前記Cの5’末端がリン酸化されたオリゴデオキシヌクレオチドを生じる。また、式Iにおいて、nが1である場合、求核剤と式Iで表されるオリゴデオキシヌクレオチドとを接触させることによって、BとCとの間のホスホジエステル結合を、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分をBの3’位から脱離させることによって切断して、前記Cの5’末端がリン酸化されたオリゴデオキシヌクレオチドと、式IIで表されるオリゴデオキシヌクレオチドとを生じる。それ故、DNA自動合成機を用いて式I’で表されるオリゴデオキシヌクレオチドを合成した後、本工程を実施することにより、5’末端がリン酸化されたオリゴデオキシヌクレオチドCを製造することが可能となる。In formula I, when n is 0, a nucleophile and formula I ′:
BC
The phosphodiester bond between B and C is removed from the 3 'position of B by contacting the phosphodiester bond between B and C by contacting with the oligodeoxynucleotide represented by Resulting in oligodeoxynucleotides phosphorylated at the 5 ′ end of the C. In Formula I, when n is 1, the phosphodiester bond between B and C is brought into contact with the oligodeoxynucleotide represented by Formula I by contacting the nucleophile with the 5 ′ end of C. An oligodeoxynucleotide that is cleaved by removing the phosphorylated oligodeoxynucleotide moiety from the 3 ′ position of B and phosphorylated at the 5 ′ end of C and an oligodeoxynucleotide represented by Formula II And produce. Therefore, after synthesizing the oligodeoxynucleotide represented by formula I ′ using an automatic DNA synthesizer, this step can be performed to produce oligodeoxynucleotide C phosphorylated at the 5 ′ end. It becomes possible.
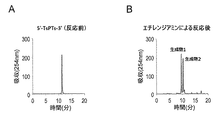
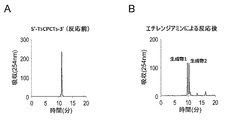
本工程は、酵素処理を必要とせず、化学反応(求核剤による求核反応)によって式I又は式I’で表されるオリゴデオキシヌクレオチドの切断と、5’末端のリン酸化が行われる。それ故、ポリヌクレオチドキナーゼのような酵素を用いる従来技術の方法と比較して、低コスト且つ簡単な実験操作で5’末端がリン酸化されたオリゴデオキシヌクレオチドを製造することが可能となる。 In this step, enzymatic treatment is not required, and the oligodeoxynucleotide represented by Formula I or Formula I ′ is cleaved and phosphorylated at the 5 ′ end by a chemical reaction (nucleophilic reaction by a nucleophile). Therefore, it is possible to produce oligodeoxynucleotides phosphorylated at the 5 'end with a low-cost and simple experimental operation as compared with the prior art method using an enzyme such as polynucleotide kinase.
例えば、非天然型塩基を含むデオキシヌクレオシドBが式B-1で表される化合物である場合、本工程における反応は以下のように進行すると考えられる。なお、本発明の作用効果は、以下の原理に限定されるものではない。
前記の反応式において、A、C及びEは、上記で定義された式Iと同様の意味であり、RNuは、分解したヌクレオチドに求核剤(Nu)が一分子付加して非天然型塩基が除去されてできたものを意味する。In the above reaction formula, A, C, and E have the same meaning as in formula I defined above, and R Nu is a non-natural type obtained by adding one molecule of a nucleophile (Nu) to a degraded nucleotide. This means that the base has been removed.
本工程において、非天然型塩基を含むデオキシヌクレオシドB-1を含む式Iで表されるオリゴデオキシヌクレオチドを求核剤存在下で反応させることにより、非天然型塩基を含むデオキシヌクレオシドB-1から部分Eが脱離すると考えられる(特徴(iii))。より具体的には、デオキシリボースの1’位の炭素原子が求核剤Nuの攻撃を受け、グリコシド結合が切断されるとともに、グリコシド結合を形成していた電子対の流れがアルキニルの炭素原子をさらに攻撃することにより、非天然型塩基部分で分子内環化反応を生じながら非天然型塩基に由来する部分Eが脱離すると考えられる(特徴(vii))。上記式では、1’位の炭素原子が求核剤の攻撃を受けた場合の反応機構を示したが、4’位の炭素原子が求核剤の攻撃を受けた場合にも同様の反応が進行すると考えられる。なお、ここでは、特徴(vi)及び(vii)を有するチミンに対応する非天然型塩基を含むデオキシヌクレオシドB-1を例示して説明した。前記の反応機構は、チミン及びシトシンのようなピリミジン型塩基に対応する非天然型塩基だけでなく、アデニン及びグアニンのようなプリン型塩基に対応する非天然型塩基を含むデオキシヌクレオシドBについても適用される。また、前記の反応機構は、特徴(vi)及び(vii)を有する非天然型塩基を含むデオキシヌクレオシドBだけでなく、特徴(vi’)及び(vii’)又は特徴(vi’’)及び(vii’’)を有する非天然型塩基を含むデオキシヌクレオシドBについても、これらの非天然型塩基を含むデオキシヌクレオシドBに関する前記の説明を考慮することにより、同様に適用することができる。 In this step, deoxynucleoside B-1 containing unnatural base is reacted with an oligodeoxynucleotide represented by formula I containing deoxynucleoside B-1 containing unnatural base in the presence of a nucleophile. Part E is considered to be eliminated (feature (iii)). More specifically, the 1'-position carbon atom of deoxyribose was attacked by the nucleophile Nu, the glycosidic bond was cleaved, and the flow of electron pairs that had formed the glycosidic bond replaced the alkynyl carbon atom. By further attack, it is considered that the portion E derived from the non-natural base is eliminated while causing an intramolecular cyclization reaction at the non-natural base portion (feature (vii)). In the above formula, the reaction mechanism is shown when the carbon atom at the 1 'position is attacked by a nucleophile, but the same reaction occurs when the carbon atom at the 4' position is attacked by a nucleophile. It is thought to progress. Here, deoxynucleoside B-1 containing an unnatural base corresponding to thymine having characteristics (vi) and (vii) has been described as an example. The above reaction mechanism applies not only to non-natural bases corresponding to pyrimidine-type bases such as thymine and cytosine, but also to deoxynucleoside B containing non-natural bases corresponding to purine-type bases such as adenine and guanine. Is done. In addition, the reaction mechanism includes not only deoxynucleoside B containing unnatural bases having characteristics (vi) and (vii), but also characteristics (vi ′) and (vii ′) or characteristics (vi ″) and ( The same applies to deoxynucleoside B containing non-natural bases having vii ″) in consideration of the above explanation regarding deoxynucleoside B containing these non-natural bases.
この反応過程は、以下に示すデプリネーション反応で生じるアベーシック部位の分解反応と類似していると考えられる(非特許文献13)。
デプリネーション反応は、DNAに含まれるプリン塩基であるアデニン及び/又はグアニンがヌクレオシドより脱離する反応である。ここで、アデニン及びグアニンは、プロトン化することによって脱離しやすくなるので、デプリネーション反応は塩基性条件下では起こりにくく酸性条件下で起こりやすい。また、ピリミジン塩基であるシトシン及びチミンは、プリン塩基よりも脱離しにくく、塩基性条件及び中性条件だけでなく、酸性条件でもDNAからのピリミジン塩基の脱離は起きにくい。 The depurination reaction is a reaction in which adenine and / or guanine, which are purine bases contained in DNA, are eliminated from the nucleoside. Here, since adenine and guanine are easily released by protonation, the depurination reaction hardly occurs under basic conditions, and easily occurs under acidic conditions. In addition, the pyrimidine bases cytosine and thymine are less likely to desorb than purine bases, and the pyrimidine base is not likely to be desorbed from DNA not only under basic and neutral conditions but also under acidic conditions.
また、以下に示すように、塩基部分の3位の水素が脱離する環化反応は公知である(非特許文献26〜29)。しかしながら、上記のようにデオキシヌクレオシドBに含まれる非天然型塩基が環化すると同時にヌクレオシドから脱離すると考えられるメカニズムによりDNAが切断される反応はこれまでに報告されておらず、本発明者らが見出した新規な反応である。
DNAポリメラーゼを用いるDNAの複製反応において、チミジンとして働くチミジン誘導体の非天然型塩基を含むDNAは公知である(特許文献4及び5並びに非特許文献30〜34)。しかしながら、本工程の如く、塩基性条件下(求核剤存在下)で反応させた場合、DNAに含まれる天然型塩基は損傷を受けず、非天然型塩基を含むデオキシヌクレオチドの部位でのみ特異的にDNAが切断され、且つ切断で生じる5’末端がリン酸化される反応はこれまでに報告されておらず、本発明者らが見出した新規な反応である。
DNAs containing unnatural bases of thymidine derivatives that act as thymidine in DNA replication reactions using DNA polymerase are known (
本工程において使用される式Iで表されるオリゴデオキシヌクレオチドは、予め合成されたものを購入等することによって準備してもよく、以下で説明するオリゴデオキシヌクレオチド合成工程を実施することによって準備してもよい。いずれの場合も、本工程の実施形態に包含されるものとする。 The oligodeoxynucleotide represented by the formula I used in this step may be prepared by purchasing a pre-synthesized one or by performing the oligodeoxynucleotide synthesis step described below. May be. In either case, it is included in the embodiment of this step.
本工程において使用される求核剤は、式Iで表されるオリゴデオキシヌクレオチドに含まれる非天然型塩基を含むデオキシヌクレオシドBの部位を攻撃して、Bから脱離可能な部分Eを脱離させ、且つEの脱離に伴って、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分をBの3’位から脱離させることによって切断することができる化合物である。求核剤は、限定するものではないが、例えば、一級アミンを挙げることができる。メチルアミン、エチレンジアミン、2-アミノエタノール、エチルアミン、プロピルアミン、2-アミノエタンチオール、1,3-ジアミノプロパン及びトリス(2-アミノエチル)アミンからなる群より選択される一級アミンであることが好ましい。上記の求核剤を用いることにより、式Iで表されるオリゴデオキシヌクレオチドに含まれるBとCとの間のホスホジエステル結合を、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分をBの3’位から脱離させることによって切断して、5’末端がリン酸化されたオリゴデオキシヌクレオチドCを得ることが可能となる。 The nucleophilic agent used in this step attacks the deoxynucleoside B site containing the unnatural base contained in the oligodeoxynucleotide represented by Formula I, and removes the detachable moiety E from B. And an oligodeoxynucleotide moiety phosphorylated at the 5 ′ end of C along with the elimination of E, can be cleaved by elimination from the 3 ′ position of B. Although a nucleophile is not limited, For example, a primary amine can be mentioned. A primary amine selected from the group consisting of methylamine, ethylenediamine, 2-aminoethanol, ethylamine, propylamine, 2-aminoethanethiol, 1,3-diaminopropane and tris (2-aminoethyl) amine is preferred. . By using the above nucleophile, the phosphodiester bond between B and C contained in the oligodeoxynucleotide represented by formula I is converted to the oligodeoxynucleotide moiety phosphorylated at the 5 ′ end of C. It is possible to obtain oligodeoxynucleotide C in which the 5 ′ end is phosphorylated by cleaving it from the 3 ′ position.
本工程は、通常、求核剤と式Iで表されるオリゴデオキシヌクレオチドとを所定の反応条件下で接触させることにより実施される。前記反応条件は、本工程で使用される求核剤及び非天然型塩基を含むデオキシヌクレオシドBの組み合わせに基づき適宜設定すればよい。具体的には、反応温度は、0〜90℃の範囲の温度であることが好ましく、4〜70℃の範囲の温度であることがより好ましい。また、前記反応条件が加熱条件の場合、反応温度は、60〜90℃の範囲の温度であることが好ましく、70〜80℃の範囲の温度であることがより好ましい。反応温度が90℃以下、特に4〜70℃の範囲の場合、低温であればあるほど長鎖のオリゴヌクレオチドのアルカリ変性を抑制することができる。反応時間は、0.1〜168時間の範囲の時間であることが好ましく、0.1〜48時間の範囲の時間であることがより好ましく、0.1〜24時間の範囲の時間であることがさらに好ましく、0.1〜12時間の範囲の時間であることが特に好ましい。上記の反応条件で本工程を実施することにより、求核剤と式Iで表されるオリゴデオキシヌクレオチドとの反応を促進することが可能となる。 This step is usually carried out by bringing a nucleophile and an oligodeoxynucleotide represented by formula I into contact under predetermined reaction conditions. The reaction conditions may be set as appropriate based on the combination of the nucleophile used in this step and deoxynucleoside B containing an unnatural base. Specifically, the reaction temperature is preferably in the range of 0 to 90 ° C, and more preferably in the range of 4 to 70 ° C. When the reaction conditions are heating conditions, the reaction temperature is preferably in the range of 60 to 90 ° C, more preferably in the range of 70 to 80 ° C. When the reaction temperature is 90 ° C. or lower, particularly in the range of 4 to 70 ° C., the lower the temperature, the more alkali denaturation of the long-chain oligonucleotide can be suppressed. The reaction time is preferably a time in the range of 0.1 to 168 hours, more preferably a time in the range of 0.1 to 48 hours, further preferably a time in the range of 0.1 to 24 hours, A time in the range of 12 hours is particularly preferred. By carrying out this step under the above reaction conditions, the reaction between the nucleophile and the oligodeoxynucleotide represented by formula I can be promoted.
本工程は、通常、求核剤と式Iで表されるオリゴデオキシヌクレオチドとを媒体中で接触させることにより実施される。媒体は、本工程で使用される求核剤及び非天然型塩基を含むデオキシヌクレオシドBの組み合わせに基づき適宜設定すればよい。具体的には、水であることが好ましい。上記の媒体中で本工程を実施することにより、望ましくない副反応を抑制し、且つ求核剤と式Iで表されるオリゴデオキシヌクレオチドとの反応を促進することが可能となる。 This step is usually performed by bringing a nucleophile and an oligodeoxynucleotide represented by formula I into contact in a medium. The medium may be appropriately set based on the combination of the nucleophile used in this step and deoxynucleoside B containing an unnatural base. Specifically, water is preferable. By carrying out this step in the above medium, it is possible to suppress undesirable side reactions and promote the reaction between the nucleophile and the oligodeoxynucleotide represented by formula I.
本工程は、脱保護された形態の式Iで表されるオリゴデオキシヌクレオチドを用いて実施してもよく、例えば5’末端がトリチル基又はモノ置換若しくはジ置換トリチル基、好ましくはDMTr基のような上記で説明した保護基で保護された形態の式Iで表されるオリゴデオキシヌクレオチドを用いて実施してもよい。後者の場合、本工程で得られる式IIで表されるオリゴデオキシヌクレオチドは、5’末端が保護された形態となる。この場合、所望により、以下で説明する3’末端リン酸化オリゴデオキシヌクレオチド形成工程において、上記で説明した脱保護処理を行うことによって脱保護すればよい。 This step may be carried out using a deprotected oligodeoxynucleotide represented by Formula I, for example, a 5 ′ end is a trityl group or a mono- or di-substituted trityl group, preferably a DMTr group. Alternatively, the oligodeoxynucleotide represented by the formula I protected with the protecting group described above may be used. In the latter case, the oligodeoxynucleotide represented by Formula II obtained in this step is in a form in which the 5 'end is protected. In this case, if desired, deprotection may be performed by performing the deprotection treatment described above in the 3′-terminal phosphorylated oligodeoxynucleotide forming step described below.
本工程において得られた5’末端がリン酸化されたオリゴデオキシヌクレオチドC、及び場合により式IIで表されるオリゴデオキシヌクレオチドは、当該技術分野で通常使用される精製手段により、互いに並びに原料及び副生成物から分離されることによって、精製及び単離することができる。それ故、本工程は、精製単離工程をさらに含んでもよい。精製手段としては、限定するものではないが、例えば、中圧液体クロマトグラフィー(MPLC)、高速液体クロマトグラフィー(HPLC)、減圧濃縮(留去)、凍結乾燥、再結晶、アガロースゲル電気泳動、ポリアクリルアミドゲル電気泳動、逆相若しくはイオン交換担体を充填した簡易カートリッジによる処理及びエタノール沈殿、並びにそれらの組み合わせを挙げることができる。或いは、本工程では精製単離工程を実施せず、以下において説明する3’末端リン酸化オリゴデオキシヌクレオチド形成工程において、5’末端がリン酸化されたオリゴデオキシヌクレオチドC及び3’末端がリン酸化されたオリゴデオキシヌクレオチドの精製及び単離を一緒に実施してもよい。上記の精製単離工程を実施することにより、高純度の5’末端がリン酸化されたオリゴデオキシヌクレオチドCを製造することが可能となる。 The oligodeoxynucleotide C phosphorylated at the 5 ′ end obtained in this step and optionally the oligodeoxynucleotide represented by the formula II are separated from each other, the raw material and the auxiliary by a purification means usually used in the art. By being separated from the product, it can be purified and isolated. Therefore, this step may further include a purification and isolation step. Examples of purification means include, but are not limited to, medium pressure liquid chromatography (MPLC), high performance liquid chromatography (HPLC), vacuum concentration (evaporation), lyophilization, recrystallization, agarose gel electrophoresis, poly Mention may be made of acrylamide gel electrophoresis, reverse phase or treatment with a simple cartridge filled with an ion exchange carrier and ethanol precipitation, and combinations thereof. Alternatively, the purification and isolation step is not performed in this step, and in the 3′-terminal phosphorylated oligodeoxynucleotide forming step described below, oligodeoxynucleotide C having a phosphorylated 5 ′ end and the 3 ′ end are phosphorylated. The purification and isolation of the oligodeoxynucleotides may be performed together. By performing the above-described purification and isolation step, it is possible to produce oligodeoxynucleotide C having a highly purified 5 'end phosphorylated.
[3-2. 3’末端リン酸化オリゴデオキシヌクレオチド形成工程]
本発明の方法は、場合により式II:
(A)n-B’
で表されるオリゴデオキシヌクレオチドを反応させて、式IIa:
A-PO3H2
で表される3’末端がリン酸化されたオリゴデオキシヌクレオチドを得る、3’末端リン酸化オリゴデオキシヌクレオチド形成工程をさらに含んでもよい(工程S3)。本工程は、B’とAとの間のホスホジエステル結合を、Aの3’末端がリン酸化されたオリゴデオキシヌクレオチド部分をBから脱離させることによって切断して、式IIaで表される3’末端がリン酸化されたオリゴデオキシヌクレオチドを得ることを目的とする。[3-2. 3'-terminal phosphorylated oligodeoxynucleotide forming step]
The method of the present invention optionally comprises Formula II:
(A) n -B '
Is reacted with an oligodeoxynucleotide of formula IIa:
A-PO 3 H 2
A 3′-terminal phosphorylated oligodeoxynucleotide forming step of obtaining an oligodeoxynucleotide phosphorylated at the 3 ′ end represented by the formula (3) may be further included (step S3). This step cleaves the phosphodiester bond between B ′ and A by removing the oligodeoxynucleotide moiety phosphorylated at the 3 ′ end of A from B 3 'To obtain oligodeoxynucleotides phosphorylated at the ends.
式II及びIIaにおいて、A及びB’は、上記と同様の意味であり、
nは1である。In Formulas II and IIa, A and B ′ have the same meaning as described above,
n is 1.
本工程は、化学反応(脱離反応)によって式IIで表されるオリゴデオキシヌクレオチドに含まれるB’とAとの間のホスホジエステル結合を切断することにより、Aの3’末端がリン酸化されたオリゴデオキシヌクレオチドを生じる。それ故、リン酸化アミダイトを用いる合成DNAの末端リン酸化法と比較して、低コスト且つ簡単な実験操作で3’末端がリン酸化されたオリゴデオキシヌクレオチドを製造することが可能となる。 In this step, the 3 'end of A is phosphorylated by cleaving the phosphodiester bond between B' and A contained in the oligodeoxynucleotide represented by Formula II by a chemical reaction (elimination reaction). Resulting in oligodeoxynucleotides. Therefore, it is possible to produce an oligodeoxynucleotide phosphorylated at the 3 'end with a low-cost and simple experimental operation, compared to the terminal phosphorylation method of synthetic DNA using phosphorylated amidite.
非天然型塩基を含むデオキシヌクレオシドBが式B-1で表される化合物である場合、本工程における反応は以下のように進行すると考えられる。なお、本発明の作用効果は、以下の原理に限定されるものではない。
前記の反応式において、A、C及びEは、上記で定義された式Iと同様の意味であり、RNuは、分解したヌクレオチドに求核剤(Nu)が一分子付加して非天然型塩基が除去されてできたものを意味する。In the above reaction formula, A, C, and E have the same meaning as in formula I defined above, and R Nu is a non-natural type obtained by adding one molecule of a nucleophile (Nu) to a degraded nucleotide. This means that the base has been removed.
前工程における求核剤の攻撃によるEの脱離に伴って形成された式IIで表されるオリゴデオキシヌクレオチドA-(B-1’)は、本工程において加熱処理することにより、B-1’とAとの間のホスホジエステル結合が、Aの3’末端がリン酸化されたオリゴデオキシヌクレオチド部分がB-1’から脱離することによって切断されて、Aの3’末端がリン酸化されたオリゴデオキシヌクレオチドを生じる。 The oligodeoxynucleotide A- (B-1 ′) represented by the formula II formed along with the elimination of E due to the attack of the nucleophile in the previous step is subjected to heat treatment in this step to obtain B-1 The phosphodiester bond between 'and A is cleaved by elimination of the oligodeoxynucleotide moiety phosphorylated at the 3' end of A from B-1 'and phosphorylated at the 3' end of A Resulting in oligodeoxynucleotides.
本工程は、通常、式IIで表されるオリゴデオキシヌクレオチドを所定の反応条件下で反応させることにより実施される。前記反応条件は、使用される非天然型塩基を含むデオキシヌクレオシドB及びBの切断により形成されたB’に基づき適宜設定すればよい。具体的には、反応温度は、0〜120℃の範囲の温度であることが好ましく、4〜70℃の範囲の温度であることがより好ましい。また、前記反応条件が加熱条件の場合、反応温度は、90〜120℃の範囲の温度であることが好ましく、110〜120℃の範囲の温度であることがより好ましい。反応時間は、0.1〜168時間の範囲の時間であることが好ましく、0.1〜48時間の範囲の時間であることがより好ましく、0.1〜24時間の範囲の時間であることがさらに好ましく、0.1〜1時間の範囲の時間であることが特に好ましい。また、反応系のpHは、通常は中性条件であり、具体的には、6〜8の範囲の値であることが好ましく、6.8〜7.2の範囲の値であることがより好ましい。上記の条件で本工程を実施することにより、Aの3’末端がリン酸化されたオリゴデオキシヌクレオチド部分をB’から脱離させて、B’とAとの間のホスホジエステル結合を切断することが可能となる。 This step is usually performed by reacting the oligodeoxynucleotide represented by formula II under predetermined reaction conditions. The reaction conditions may be set as appropriate based on deoxynucleosides B and B 'formed by cleavage of the unnatural base used. Specifically, the reaction temperature is preferably in the range of 0 to 120 ° C, and more preferably in the range of 4 to 70 ° C. When the reaction condition is a heating condition, the reaction temperature is preferably in the range of 90 to 120 ° C, more preferably in the range of 110 to 120 ° C. The reaction time is preferably a time in the range of 0.1 to 168 hours, more preferably a time in the range of 0.1 to 48 hours, further preferably a time in the range of 0.1 to 24 hours, A time in the range of 1 hour is particularly preferred. Further, the pH of the reaction system is usually neutral conditions, and specifically, it is preferably a value in the range of 6 to 8, more preferably a value in the range of 6.8 to 7.2. By carrying out this step under the above conditions, the oligodeoxynucleotide part phosphorylated at the 3 'end of A is eliminated from B' and the phosphodiester bond between B 'and A is cleaved. Is possible.
本工程は、通常、式IIで表されるオリゴデオキシヌクレオチドを媒体中で反応させることにより実施される。媒体は、本工程で使用される式IIで表されるオリゴデオキシヌクレオチドに基づき適宜設定すればよい。具体的には、水であることが好ましい。また、媒体は、反応系のpHを上記で説明した好ましいpH範囲に保持するために、1以上の緩衝成分を含んでもよい。前記緩衝成分は、限定するものではないが、例えば、リン酸塩及びカコジル酸塩、並びにそれらの組み合わせを挙げることができる。リン酸塩であることが好ましい。緩衝成分の濃度は、10〜1000 mMの範囲であることが好ましく、10〜100 mMの範囲であることがより好ましい。上記の媒体中で本工程を実施することにより、望ましくない副反応を抑制し、且つAの3’末端がリン酸化されたオリゴデオキシヌクレオチド部分をB’から脱離させて、B’とAとの間のホスホジエステル結合を切断することが可能となる。 This step is usually performed by reacting an oligodeoxynucleotide represented by formula II in a medium. The medium may be appropriately set based on the oligodeoxynucleotide represented by formula II used in this step. Specifically, water is preferable. The medium may also contain one or more buffer components in order to maintain the pH of the reaction system in the preferred pH range described above. Examples of the buffer component include, but are not limited to, phosphate and cacodylate, and combinations thereof. A phosphate is preferred. The concentration of the buffer component is preferably in the range of 10 to 1000 mM, more preferably in the range of 10 to 100 mM. By carrying out this step in the above medium, an undesirable side reaction is suppressed, and the oligodeoxynucleotide moiety phosphorylated at the 3 ′ end of A is eliminated from B ′, and B ′ and A It becomes possible to cleave the phosphodiester bond between.
本工程において得られた式IIaで表される3’末端がリン酸化されたオリゴデオキシヌクレオチドは、当該技術分野で通常使用される精製手段により、原料及び副生成物から分離されることによって、精製及び単離することができる。それ故、本工程は、精製単離工程をさらに含んでもよい。精製手段としては、限定するものではないが、例えば、中圧液体クロマトグラフィー(MPLC)、高速液体クロマトグラフィー(HPLC)、減圧濃縮(留去)、凍結乾燥及び再結晶、アガロースゲル電気泳動、ポリアクリルアミドゲル電気泳動、逆相若しくはイオン交換担体を充填した簡易カートリッジによる処理及びエタノール沈殿、並びにそれらの組み合わせを挙げることができる。上記の精製単離工程を実施することにより、高純度の式IIaで表される3’末端がリン酸化されたオリゴデオキシヌクレオチドを製造することが可能となる。 The oligodeoxynucleotide phosphorylated at the 3 ′ end represented by Formula IIa obtained in this step is purified by being separated from the raw materials and by-products by purification means usually used in the art. And can be isolated. Therefore, this step may further include a purification and isolation step. Examples of purification means include, but are not limited to, medium pressure liquid chromatography (MPLC), high performance liquid chromatography (HPLC), vacuum concentration (evaporation), lyophilization and recrystallization, agarose gel electrophoresis, poly Mention may be made of acrylamide gel electrophoresis, reverse phase or treatment with a simple cartridge filled with an ion exchange carrier and ethanol precipitation, and combinations thereof. By performing the above-described purification and isolation step, it is possible to produce a highly pure oligodeoxynucleotide having a phosphorylated 3 ′ end represented by Formula IIa.
本工程は、脱保護された形態の式IIで表されるオリゴデオキシヌクレオチドを用いて実施してもよく、例えば5’末端がトリチル基又はモノ置換若しくはジ置換トリチル基、好ましくはDMTr基のような上記で説明した保護基で保護された形態の式IIで表されるオリゴデオキシヌクレオチドを用いて実施してもよい。後者の場合、本工程で得られる式IIaで表される3’末端がリン酸化されたオリゴデオキシヌクレオチドは、5’末端が保護された形態となる。この場合、所望により、上記で説明した脱保護処理を行うことによって脱保護することにより、脱保護された形態の式IIaで表される3’末端がリン酸化されたオリゴデオキシヌクレオチドを得ることが可能となる。 This step may be performed using a deprotected oligodeoxynucleotide represented by Formula II, for example, a 5 ′ end is a trityl group or a mono- or di-substituted trityl group, preferably a DMTr group. Alternatively, the oligodeoxynucleotide represented by the formula II protected with the protecting group described above may be used. In the latter case, the oligodeoxynucleotide phosphorylated at the 3 'end represented by Formula IIa obtained in this step is in a form in which the 5' end is protected. In this case, if desired, the deprotection treatment described above may be performed to obtain a deprotected oligodeoxynucleotide having a phosphorylated 3 ′ end represented by Formula IIa. It becomes possible.
[3-3. オリゴデオキシヌクレオチド合成工程]
本発明の方法は、予め合成された式Iで表されるオリゴデオキシヌクレオチドを購入等することによって実施してもよい。しかしながら、式Iで表されるオリゴデオキシヌクレオチドを合成した後に、その場で又は逐次的に5’末端リン酸化オリゴデオキシヌクレオチド形成工程を実施してもよい。それ故、本発明の方法は、場合により前記5’末端リン酸化オリゴデオキシヌクレオチド形成工程の前に、求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドB又はその誘導体と、天然型塩基を含む1個以上のデオキシヌクレオシド又はその誘導体とを含む基質を用いて、式I:
(A)n-B-C
で表されるオリゴデオキシヌクレオチドを合成する、オリゴデオキシヌクレオチド合成工程をさらに含んでもよい(工程S1)。本工程は、前記5’末端リン酸化オリゴデオキシヌクレオチド形成工程の原料となる、式Iで表されるオリゴデオキシヌクレオチドを得ることを目的とする。[3-3. Oligodeoxynucleotide synthesis process]
You may implement the method of this invention by purchasing the oligodeoxynucleotide represented by Formula I previously synthesized. However, after synthesizing the oligodeoxynucleotide represented by Formula I, a 5′-terminal phosphorylated oligodeoxynucleotide forming step may be performed in situ or sequentially. Therefore, in the method of the present invention, deoxynucleoside B containing a non-natural base or a portion thereof having a moiety E removable by a nucleophile, optionally before the 5′-terminal phosphorylated oligodeoxynucleotide forming step. Using a substrate comprising a derivative and one or more deoxynucleosides or derivatives thereof comprising a natural base, the formula I:
(A) n -BC
An oligodeoxynucleotide synthesis step for synthesizing the oligodeoxynucleotide represented by the formula (1) may be further included (step S1). The purpose of this step is to obtain an oligodeoxynucleotide represented by the formula I, which is a raw material for the 5′-terminal phosphorylated oligodeoxynucleotide forming step.
式Iにおいて、A、B及びCは、上記と同様の意味であり、
nは0又は1である。In the formula I, A, B and C have the same meaning as above,
n is 0 or 1.
本工程において、求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドBは、上記で説明した非天然型塩基を含むデオキシヌクレオシドBを使用することができる。また、天然型塩基を含む1個以上のデオキシヌクレオシドは、式Iで表されるオリゴデオキシヌクレオチドに含まれるデオキシヌクレオチドのうち、非天然型塩基を含むデオキシヌクレオチドB以外の、アデニン、チミン、グアニン及びシトシンからなる群より選択される天然型塩基を含むデオキシヌクレオチドに対応するものを意味し、当該技術分野で通常使用される純度の天然型塩基を含むデオキシヌクレオシドを使用することができる。 In this step, as the deoxynucleoside B containing a non-natural base having a moiety E that can be eliminated by a nucleophile, the deoxynucleoside B containing a non-natural base described above can be used. In addition, one or more deoxynucleosides containing a natural base are adenine, thymine, guanine and other than deoxynucleotide B containing an unnatural base among the deoxynucleotides contained in the oligodeoxynucleotide represented by Formula I. It means the one corresponding to a deoxynucleotide containing a natural base selected from the group consisting of cytosine, and a deoxynucleoside containing a natural base having a purity usually used in the art can be used.
求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドBの誘導体、及び天然型塩基を含むデオキシヌクレオシドの誘導体は、限定するものではないが、例えば、3’位にホスホロアミダイト基を有する当該デオキシヌクレオシドの誘導体を挙げることができる。上記の誘導体を用いることにより、当該技術分野において通常使用されるDNA自動合成機によって式Iで表されるオリゴデオキシヌクレオチドを得ることが可能となる。 A derivative of deoxynucleoside B containing a non-natural base and a derivative of deoxynucleoside containing a natural base having a moiety E that can be eliminated by a nucleophile is not limited, for example, at the 3 ′ position. Mention may be made of derivatives of the deoxynucleoside having a phosphoramidite group. By using the above derivative, it is possible to obtain the oligodeoxynucleotide represented by the formula I by an automatic DNA synthesizer usually used in the art.
本工程において使用される基質は、上記で説明した求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドB又はその誘導体と、天然型塩基を含む1個以上のデオキシヌクレオシド又はその誘導体とを含む。前記基質は、所望により、他の非天然型塩基を含むデオキシヌクレオシド若しくはその誘導体、又は蛍光色素、ビオチン若しくはジコギシゲニンなどの標識分子のような他の成分を含んでもよい。 The substrate used in this step is a deoxynucleoside B containing a non-natural base having a moiety E that can be eliminated by a nucleophile described above or a derivative thereof, and one or more deoxy containing a natural base. A nucleoside or a derivative thereof. The substrate may optionally include other components such as deoxynucleosides or derivatives thereof containing other unnatural bases, or labeling molecules such as fluorescent dyes, biotin or dichogigenin.
本工程は、前記基質と、所望により、アセトニトリル又はN,N-ジメチルホルムアミドのような媒体と、テトラゾール、テトラゾール誘導体又は4,5-ジシアノイミダゾールのような活性化剤とを接触させることにより、実施することができる。この場合、本工程は、上記で説明したDNA合成条件下で実施すればよい。 This step is carried out by contacting the substrate with a medium such as acetonitrile or N, N-dimethylformamide and an activator such as tetrazole, a tetrazole derivative or 4,5-dicyanoimidazole, if desired. can do. In this case, this step may be performed under the DNA synthesis conditions described above.
上記の条件で本工程を実施することにより、式Iで表されるオリゴデオキシヌクレオチドを高収率で得ることが可能となる。 By carrying out this step under the above conditions, the oligodeoxynucleotide represented by formula I can be obtained in high yield.
<4. 二本鎖DNAの連結方法>
本発明の方法により、低コスト且つ簡単な実験操作でDNA鎖の切断を行い、結果として生じた末端がリン酸化されたオリゴデオキシヌクレオチドを得ることができる。それ故、本発明はまた、式I:
(A)n-B-C
[式中、A、B、C及びnは、上記で定義したのと同様の意味を表し、
nが1のとき、Aの3’末端とBの5’位とは、ホスホジエステル結合で連結されており、
Bの3’位とCの5’末端とは、ホスホジエステル結合で連結されており、
BとCとの間のホスホジエステル結合は、求核剤の攻撃によって誘発されるEの脱離に伴って、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断される]
で表されるオリゴデオキシヌクレオチドをプライマーとして用いて、直鎖状二本鎖DNA断片を増幅するDNA断片増幅工程;
求核剤と、増幅された直鎖状二本鎖DNA断片とを接触させることによって、BとCとの間のホスホジエステル結合が、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断されて、直鎖状二本鎖DNA断片に含まれる前記Cの5’末端がリン酸化されており、且つ式Iで表されるプライマー部分の少なくとも一部と相補的な配列を含む3’突出末端を有する直鎖状二本鎖DNA断片を形成させる、3’突出末端形成工程;並びに
前記3’突出末端を有する直鎖状二本鎖DNA断片と、別の二本鎖DNA断片とを連結する連結工程;
を含む、二本鎖DNAの連結方法に関する。<4. Method of linking double-stranded DNA>
According to the method of the present invention, a DNA strand can be cleaved by a low-cost and simple experimental operation, and the resulting oligodeoxynucleotide whose end is phosphorylated can be obtained. Therefore, the present invention also provides formula I:
(A) n -BC
[Wherein A, B, C and n represent the same meaning as defined above,
When n is 1, the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond,
The 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond,
The phosphodiester bond between B and C is such that the oligodeoxynucleotide part phosphorylated at the 5 'end of C from the 3' position of B with E elimination induced by nucleophile attack. Disconnected by detachment]
A DNA fragment amplification step of amplifying a linear double-stranded DNA fragment using the oligodeoxynucleotide represented by
By contacting the nucleophile with the amplified linear double-stranded DNA fragment, the phosphodiester bond between B and C becomes an oligodeoxynucleotide moiety phosphorylated at the 5 'end of C. It is cleaved by detaching from the 3 ′ position of B, the 5 ′ end of C contained in the linear double-stranded DNA fragment is phosphorylated, and at least the primer part represented by Formula I Forming a linear double-stranded DNA fragment having a 3 ′ protruding end containing a sequence complementary to a part thereof; and a linear double-stranded DNA fragment having the 3 ′ protruding end; And a ligation step of ligating another double-stranded DNA fragment;
And a method for ligating double-stranded DNA.
DNA断片増幅工程において、直鎖状二本鎖DNA断片を増幅する手段としては、限定するものではないが、例えば、ポリメラーゼ連鎖反応(PCR)を挙げることができる。この場合、本工程は、上記で説明したDNA複製条件下で実施すればよい。また、式Iで表されるオリゴデオキシヌクレオチドにおいて、オリゴデオキシヌクレオチドCは、ポリメラーゼ連鎖反応の標的となる鋳型DNAの少なくとも一部と相補的な塩基配列を有すればよい。 In the DNA fragment amplification step, means for amplifying a linear double-stranded DNA fragment is not limited, and examples thereof include a polymerase chain reaction (PCR). In this case, this step may be performed under the DNA replication conditions described above. In the oligodeoxynucleotide represented by the formula I, the oligodeoxynucleotide C may have a base sequence complementary to at least a part of the template DNA that is a target of the polymerase chain reaction.
3’突出末端形成工程において使用される求核剤、及び本工程の具体的な反応条件としては、5’末端リン酸化オリゴデオキシヌクレオチド形成工程に関して上記で説明した条件を適用することが好ましい。 As the nucleophile used in the 3 'protruding end forming step and the specific reaction conditions of this step, it is preferable to apply the conditions described above with respect to the 5' end phosphorylated oligodeoxynucleotide forming step.
本発明の方法は、タイプIIの制限酵素を用いる方法と比較して、切断するDNA断片中の制限酵素認識配列の有無を考慮せずにDNAを切断でき、且つ突出末端の突出部分の長さ及び配列を任意に設定できるという利点を有する。それ故、本発明の方法で製造された3’突出末端を有する直鎖状二本鎖DNA断片は、DNA連結酵素を用いる連結の際に、タイプIIの制限酵素を用いて製造されたDNA断片と比較して高い連結効率を発揮することが可能となる。 Compared with a method using a type II restriction enzyme, the method of the present invention can cleave DNA without considering the presence or absence of a restriction enzyme recognition sequence in the DNA fragment to be cleaved, and the length of the protruding portion at the protruding end. And it has the advantage that the arrangement can be set arbitrarily. Therefore, a linear double-stranded DNA fragment having a 3 ′ overhang produced by the method of the present invention is a DNA fragment produced using a type II restriction enzyme during ligation using a DNA ligation enzyme. Compared to the above, it is possible to exhibit high connection efficiency.
<5. オリゴデオキシヌクレオチドの製造方法に使用するためのキット>
本発明の方法により、5’末端がリン酸化されたオリゴデオキシヌクレオチドC及び/又は式IIaで表される3’末端がリン酸化されたオリゴデオキシヌクレオチドを得ることができる。それ故、本発明はまた、上記で説明した本発明のオリゴデオキシヌクレオチドの製造方法又は二本鎖DNAの連結方法に使用するためのキットに関する。<5. Kit for use in the production method of oligodeoxynucleotide>
By the method of the present invention, oligodeoxynucleotide C phosphorylated at the 5 ′ end and / or oligodeoxynucleotide phosphorylated at the 3 ′ end represented by Formula IIa can be obtained. Therefore, the present invention also relates to a kit for use in the above-described method for producing the oligodeoxynucleotide of the present invention or the method for ligating double-stranded DNA.
前記キットは、式I:
(A)n-B-C
で表されるオリゴデオキシヌクレオチド、又は求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドB若しくはその誘導体を少なくとも含む。The kit has the formula I:
(A) n -BC
Or a deoxynucleoside B containing a non-natural base or a derivative thereof having a moiety E removable by a nucleophile.
式Iにおいて、A、B及びCは、上記と同様の意味であり、
nは0又は1である。In the formula I, A, B and C have the same meaning as above,
n is 0 or 1.
また、前記キットは、式Iで表されるオリゴデオキシヌクレオチド又は非天然型塩基を含むデオキシヌクレオシドB若しくはその誘導体に加えて、アデニン、チミン、グアニン及びシトシンからなる群より選択される天然型塩基を含む1個以上のデオキシヌクレオシド又はその誘導体、求核剤、媒体、触媒、DNA連結酵素(リガーゼ)及び使用説明書からなる群より選択される1個以上のさらなる要素を含んでもよい。ここで、前記求核剤、媒体、触媒及びDNA連結酵素は、上記で説明した本発明の方法に使用し得るものの中から適宜選択することができる。上記の要素を含むことにより、前記キットを用いて本発明のオリゴデオキシヌクレオチドの製造方法を実施することが可能となる。 The kit further comprises a natural base selected from the group consisting of adenine, thymine, guanine and cytosine in addition to the oligodeoxynucleotide represented by formula I or deoxynucleoside B containing a non-natural base or a derivative thereof. It may contain one or more additional elements selected from the group consisting of one or more deoxynucleosides or derivatives thereof, nucleophiles, media, catalysts, DNA-ligating enzymes (ligases) and instructions for use. Here, the nucleophile, the medium, the catalyst, and the DNA ligating enzyme can be appropriately selected from those that can be used in the method of the present invention described above. By including the above-described elements, the method for producing an oligodeoxynucleotide of the present invention can be carried out using the kit.
本発明のキットは、必ずしも上記の要素が一体化された形態で提供される必要はなく、該要素が別々に包装された形態で提供されてもよい。いずれの形態も前記キットに包含されるものとする。 The kit of the present invention does not necessarily need to be provided in an integrated form, and may be provided in a form in which the elements are packaged separately. Any form shall be included in the kit.
上記の特徴を有するキットを用いることにより、本発明のオリゴデオキシヌクレオチドの製造方法を実施して、低コスト且つ簡単な実験操作で5’末端がリン酸化されたオリゴデオキシヌクレオチドC及び/又は式IIaで表される3’末端がリン酸化されたオリゴデオキシヌクレオチドを得ることが可能となる。 By using the kit having the above characteristics, the oligodeoxynucleotide production method of the present invention is carried out, and the oligodeoxynucleotide C and / or formula IIa phosphorylated at the 5 ′ end by a low-cost and simple experimental operation It is possible to obtain an oligodeoxynucleotide phosphorylated at the 3 ′ end represented by
以上詳細に説明したように、本発明により、化学反応によって低コスト且つ簡単な実験操作でDNA鎖の切断を行い、さらにポリヌクレオチドキナーゼのような酵素法又はリン酸化アミダイトを用いる合成DNAの末端リン酸化法と比較してより高収率で末端のリン酸化を行うことが可能となる。 As described above in detail, according to the present invention, a DNA strand is cleaved by a chemical reaction at a low cost and with a simple experimental operation, and further, the terminal phosphorylation of a synthetic DNA using an enzymatic method such as polynucleotide kinase or a phosphorylated amidite. It becomes possible to perform terminal phosphorylation with higher yield compared to the oxidation method.
以下、実施例を用いて本発明をさらに具体的に説明する。但し、本発明の技術的範囲はこれら実施例に限定されるものではない。 Hereinafter, the present invention will be described more specifically with reference to examples. However, the technical scope of the present invention is not limited to these examples.
<1. 有機合成及びDNA合成の試薬並びに機器>
有機合成及び/又はDNA合成のための試薬は、Sigma-Aldrich、和光純薬、東京化成、ナカライテスク又はグレンリサーチより購入した。合成反応に用いた溶媒は無水のものを使用した。合成反応は窒素雰囲気下で行った。合成した化合物の精製で使用したシリカゲルはワコーゲルC-200(和光純薬)を使用した。DNAの精製にはギルソン社製のHPLCシステムを使用した。逆相カラムはケムコ社のケムコボンド5-ODS-H (10×150 mm)を使用した。NMRスペクトルは日本電子社製のJNM-ECS400を使用して測定した。1H-NMRのケミカルシフトは、DMSO-d6の残留プロトンのケミカルシフト(2.49 ppm)を基準としてppm単位で記載した。HRMSスペクトルは、ウォータース社製のSYNAPT G2 MSを使用して測定した。MALDI TOF-MSスペクトルは、島津製作所社製のAXIMA Assuranceを使用して測定した。<1. Organic synthesis and DNA synthesis reagents and equipment>
Reagents for organic synthesis and / or DNA synthesis were purchased from Sigma-Aldrich, Wako Pure Chemicals, Tokyo Kasei, Nacalai Tesque or Glen Research. The solvent used for the synthesis reaction was anhydrous. The synthesis reaction was performed under a nitrogen atmosphere. Wako Gel C-200 (Wako Pure Chemical Industries) was used as the silica gel used for purification of the synthesized compound. An HPLC system manufactured by Gilson was used for DNA purification. The reverse phase column used was Chemco Bond 5-ODS-H (10 × 150 mm) manufactured by Chemco. The NMR spectrum was measured using JNM-ECS400 manufactured by JEOL. The chemical shift of 1 H-NMR was described in ppm based on the chemical shift (2.49 ppm) of the residual proton in DMSO-d6. The HRMS spectrum was measured using SYNAPT G2 MS manufactured by Waters. The MALDI TOF-MS spectrum was measured using AXIMA Assurance manufactured by Shimadzu Corporation.
<2. 合成例1:5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマー>
[合成例1-1:5-フェニルエチニル-2’-デオキシウリジンの合成]
3.54 g(10.0 mmol)の5-ヨード-2’-デオキシウリジン(分子量354.10)と1.16 g(2.00 mmol)のPd(PPh3)4(分子量1155.56)と190 mg(1.00 mmol)のCuI(分子量190.45)とを撹拌子とともにナスフラスコに入れ、そこへ20 mLのDMFと2.78 mL(20.0 mmol)のEt3N(分子量101.19, 比重0.73 g/mL)と2.52 mL(23.0 mmol)のエチニルベンゼン(分子量102.13, 比重0.93 g/mL)とを加え、窒素雰囲気下、80℃で加熱しながら1時間撹拌した。溶媒を減圧留去し、シリカゲルカラムで目的物質を精製した(展開溶媒は2〜10% MeOH/AcOEt)。目的のフラクションの溶媒を減圧留去し、少量のメタノールに溶かし、約200 mLの酢酸エチルを加え、減圧下で溶液を濃縮し、生じた白色沈殿をろ過、真空ポンプで乾燥することにより2.02 g(6.15 mmol, 収率61.5%)の目的物質(分子量328.32)を白色粉末として得た。<2. Synthesis Example 1: DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine>
[Synthesis Example 1-1: Synthesis of 5-phenylethynyl-2′-deoxyuridine]
3.54 g (10.0 mmol) 5-iodo-2'-deoxyuridine (molecular weight 354.10), 1.16 g (2.00 mmol) Pd (PPh 3 ) 4 (molecular weight 1155.56) and 190 mg (1.00 mmol) CuI (molecular weight 190.45) ) With a stir bar, 20 mL of DMF, 2.78 mL (20.0 mmol) Et 3 N (molecular weight 101.19, specific gravity 0.73 g / mL) and 2.52 mL (23.0 mmol) ethynylbenzene (molecular weight). 102.13, specific gravity 0.93 g / mL) was added, and the mixture was stirred for 1 hour while heating at 80 ° C. in a nitrogen atmosphere. The solvent was distilled off under reduced pressure, and the target substance was purified with a silica gel column (developing solvent was 2 to 10% MeOH / AcOEt). The solvent of the desired fraction was distilled off under reduced pressure, dissolved in a small amount of methanol, about 200 mL of ethyl acetate was added, the solution was concentrated under reduced pressure, and the resulting white precipitate was filtered and dried with a vacuum pump to obtain 2.02 g. The target substance (molecular weight 328.32) (6.15 mmol, yield 61.5%) was obtained as a white powder.
得られた物質の1H NMRスペクトルを測定したところ、非特許文献19(Cho, J. H.ら, Eur. J. Org. Chem. 2010年, p. 3678-3683)に記載の1H NMRスペクトルデータとほぼ一致した。The 1 H NMR spectrum of the obtained material was measured,
1H NMR(400 MHz, DMSO-d6) δ11.67 (s, 1H), 8.37 (s, 1H), 7.47-7.38 (m, 5H), 6.12 (t, J = 6.6 Hz, 1H), 5.25 (d, J = 4.1 Hz, 1H), 5.15 (t, J = 4.8 Hz, 1H), 4.26-4.24 (m, 1H), 3.81-3.79 (m, 1H), 3.64-3.58 (m, 2H), 2.18-2.14 (m, 2H).
[合成例1-2:5-フェニルエチニル-5’-O-ジメトキシトリチル-2’-デオキシウリジンの合成]
656 mg(2.00 mmol)の5-フェニルエチニル-2’-デオキシウリジン(分子量328.32)と813 mg(4.00 mmol)のDMTrCl(分子量338.83)とをナスフラスコに入れ、そこへ10 mLのピリジンを加え、室温で1時間撹拌した。約2 mLのメタノールを加え、反応を停止させ、溶媒を減圧留去した。シリカゲルカラムで目的物質を精製した(展開溶媒は2〜5% MeOH, 2% Et3N/CH2Cl2)。目的のフラクションの溶媒を減圧留去し、真空ポンプで乾燥した。少量の塩化メチレンに溶かし、約400 mLのヘキサンを加え、生じた白色沈殿をろ過、真空ポンプで乾燥することにより1.20 g(1.64 mmol, 収率82.0%)の目的物質(分子量731.88(トリエチルアミン一分子を含む))を白色粉末として得た。 1 H NMR (400 MHz, DMSO-d6) δ11.67 (s, 1H), 8.37 (s, 1H), 7.47-7.38 (m, 5H), 6.12 (t, J = 6.6 Hz, 1H), 5.25 ( d, J = 4.1 Hz, 1H), 5.15 (t, J = 4.8 Hz, 1H), 4.26-4.24 (m, 1H), 3.81-3.79 (m, 1H), 3.64-3.58 (m, 2H), 2.18 -2.14 (m, 2H).
[Synthesis Example 1-2: Synthesis of 5-phenylethynyl-5′-O-dimethoxytrityl-2′-deoxyuridine]
656 mg (2.00 mmol) 5-phenylethynyl-2′-deoxyuridine (molecular weight 328.32) and 813 mg (4.00 mmol) DMTrCl (molecular weight 338.83) were placed in an eggplant flask, and 10 mL of pyridine was added thereto, Stir at room temperature for 1 hour. About 2 mL of methanol was added to stop the reaction, and the solvent was distilled off under reduced pressure. The target substance was purified by a silica gel column (developing solvent was 2-5% MeOH, 2% Et 3 N / CH 2 Cl 2 ). The solvent of the objective fraction was distilled off under reduced pressure and dried with a vacuum pump. Dissolve in a small amount of methylene chloride, add about 400 mL of hexane, filter the resulting white precipitate, and dry with a vacuum pump to obtain 1.20 g (1.64 mmol, 82.0% yield) of the target substance (molecular weight 731.88 (one triethylamine molecule) Was obtained as a white powder.
得られた物質の1H NMRスペクトルを測定したところ、一分子のトリエチルアミンが残った点を除いて、非特許文献24(Rospigliosi, A.ら, Langmuir 2007年, 23, 15, p. 8264-8271)に記載の1H NMRスペクトルデータとほぼ一致した。When the 1 H NMR spectrum of the obtained substance was measured, it was found that non-patent document 24 (Rospigliosi, A. et al.,
1H NMR(400 MHz, DMSO-d6) δ11.75 (s, 1H), 8.06 (s, 1H), 7.41-7.39 (m, 2H), 7.33-7.26 (m, 9H), 7.16-7.10 (m, 3H), 6.84-6.80 (m, 4H), 6.14 (t, J= 6.9 Hz, 1H), 5.40 (d, J = 4.1 Hz, 1H), 4.34-4.28 (m, 1H), 3.98-3.95 (m, 1H), 3.644 (s, 3H), 3.640 (s, 3H), 3.24-3.14 (m, 2H), 3.04-3.03 (m, 6H), 2.32-2.21 (m, 2H), 1.19 (t, J = 7.3 Hz, 9H).
[合成例1-3:5-フェニルエチニル-5’-O-ジメトキシトリチル-2’-デオキシウリジン-3’-N,N-ジイソプロピル(シアノエチル)ホスホロアミダイトの合成]
366 mg(0.500 mmol)の5-フェニルエチニル-5’-O-ジメトキシトリチル-2’-デオキシウリジン(分子量731.88(トリエチルアミン一分子を含む))をナスフラスコに入れ、5 mLのCH3CNと261μL(1.50 mmol)のN,N-ジイソプロピルエチルアミン(分子量129.25, 比重0.742 g/mL)を加え、撹拌し、さらに223μL(1.00 mmol)の2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロアミダイト(分子量236.68, 比重1.061 g/mL)を加え、室温で30分間撹拌した。25 mLのAcOEtと25 mLの飽和食塩水とを加え、分液し、有機層をさらに2回飽和食塩水で洗った。有機層を無水硫酸マグネシウムで乾燥した後、フィルターでろ過し、ろ液の溶媒を減圧留去した。アセトニトリルで3回共沸した後、10 mLのアセトニトリルを加えた。溶液を0.45μmのフィルターに通し、少量のモレキュラーシーブを加えた。DNA自動合成機に取り付けてDNA合成に使用した。 1 H NMR (400 MHz, DMSO-d6) δ11.75 (s, 1H), 8.06 (s, 1H), 7.41-7.39 (m, 2H), 7.33-7.26 (m, 9H), 7.16-7.10 (m , 3H), 6.84-6.80 (m, 4H), 6.14 (t, J = 6.9 Hz, 1H), 5.40 (d, J = 4.1 Hz, 1H), 4.34-4.28 (m, 1H), 3.98-3.95 ( m, 1H), 3.644 (s, 3H), 3.640 (s, 3H), 3.24-3.14 (m, 2H), 3.04-3.03 (m, 6H), 2.32-2.21 (m, 2H), 1.19 (t, J = 7.3 Hz, 9H).
[Synthesis Example 1-3: Synthesis of 5-phenylethynyl-5′-O-dimethoxytrityl-2′-deoxyuridine-3′-N, N-diisopropyl (cyanoethyl) phosphoramidite]
366 mg (0.500 mmol) of 5-phenylethynyl-5′-O-dimethoxytrityl-2′-deoxyuridine (molecular weight 731.88 (including one molecule of triethylamine)) was placed in an eggplant flask, 5 mL of CH 3 CN and 261 μL (1.50 mmol) N, N-diisopropylethylamine (molecular weight 129.25, specific gravity 0.742 g / mL) was added, stirred, and 223 μL (1.00 mmol) 2-cyanoethyl N, N, N ', N'-tetraisopropylphospho Loamidite (molecular weight 236.68, specific gravity 1.061 g / mL) was added, and the mixture was stirred at room temperature for 30 minutes. 25 mL of AcOEt and 25 mL of saturated saline were added and separated, and the organic layer was further washed twice with saturated saline. The organic layer was dried over anhydrous magnesium sulfate and filtered through a filter, and the solvent of the filtrate was distilled off under reduced pressure. After azeotroping with acetonitrile three times, 10 mL of acetonitrile was added. The solution was passed through a 0.45 μm filter and a small amount of molecular sieve was added. Attached to an automatic DNA synthesizer and used for DNA synthesis
[合成例1-4:5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーの合成]
チミジン誘導体のカップリング時間を30分に伸ばした以外は通常のDNA自動合成機のDNA合成のプロトコールで、1μmolスケールでホスホロアミダイト法により合成した(Synthetic strategies and parameters involved in the synthesis of oligodeoxyribonucleotides according to the phosphoramidite method. Beaucage, S. L.及びCaruthers, M. H. Current Protocols in Nucleic Acid Chemistry UNIT 3.3)。担体からの切り出しは28%アンモニア水で行った。脱保護はDNAオリゴマーを切り出した後、室温で16時間静置することにより行った。脱保護後、アンモニアを遠心濃縮装置により減圧留去し、DNAを含む溶液を0.45μmのフィルターに通した後、逆相HPLCにより10分付近の保持時間に溶出されたメインピークを精製し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置と凍結乾燥によりギ酸アンモニウムと溶媒とを留去した。得られたDNAオリゴマーはMALDI TOF-MS測定により同定した。合成したDNAオリゴマーは、約500μLの蒸留水を加えて水溶液とした。[Synthesis Example 1-4: Synthesis of DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine]
A synthetic strategy and parameters involved in the synthesis of oligodeoxyribonucleotides according to the phosphoramidite method. Beaucage, SL and Caruthers, MH Current Protocols in Nucleic Acid Chemistry UNIT 3.3). Cutting out from the carrier was performed with 28% aqueous ammonia. Deprotection was performed by cutting out the DNA oligomer and then allowing to stand at room temperature for 16 hours. After deprotection, the ammonia was distilled off under reduced pressure using a centrifugal concentrator, and the DNA-containing solution was passed through a 0.45 μm filter. -50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and the solvent were distilled off by centrifugal concentrator and lyophilization. The obtained DNA oligomer was identified by MALDI TOF-MS measurement. The synthesized DNA oligomer was made into an aqueous solution by adding about 500 μL of distilled water.
以下において、5-フェニルエチニル-2’-デオキシウリジン:
を「P」で示す。 Is indicated by “P”.
合成したDNAオリゴマーの逆相HPLCクロマトグラムを図2〜7に示す。 The reverse phase HPLC chromatogram of the synthesized DNA oligomer is shown in FIGS.
合成したDNAオリゴマーの上記逆相HPLCにおける保持時間は以下の通りである。 The retention time in the reverse phase HPLC of the synthesized DNA oligomer is as follows.
5’-TTTTCTTPTCCTTTT-3’について10.9分(配列番号1)
5’-TTTTTTPTTTTTT-3’について11.0分(配列番号2)
5’-TTTTTAPATTTTT-3’について10.9分(配列番号3)
5’-TTTTTCPCTTTTT-3’について10.8分(配列番号4)
5’-TTTTTGPGTTTTT-3’について10.6分(配列番号5)
5’-CGCAATPTAACGC-3’について10.0分(配列番号6)
合成したDNAオリゴマーのMALDI TOF-MSのデータは以下の通りである。10.9 minutes for 5'-TTTTCTTPTCCTTTT-3 '(SEQ ID NO: 1)
11.0 minutes for 5'-TTTTTTPTTTTTT-3 '(SEQ ID NO: 2)
10.9 minutes for 5'-TTTTTAPATTTTT-3 '(SEQ ID NO: 3)
10.8 minutes for 5'-TTTTTCPCTTTTT-3 '(SEQ ID NO: 4)
10.6 minutes for 5'-TTTTTGPGTTTTT-3 '(SEQ ID NO: 5)
10.0 minutes for 5'-CGCAATPTAACGC-3 '(SEQ ID NO: 6)
The MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
5’-TTTTCTTPTCCTTTT-3’について分子式C154H195N33O100P14、[M-H]-の計算値 4541.0、実測値 4541.9(配列番号1)
5’-TTTTTTPTTTTTT-3’ について分子式C137H172N26O89P12、[M-H]-の計算値 3977.6、実測値 3978.0(配列番号2)
5’-TTTTTAPATTTTT-3’ について分子式C137H170N32O85P12、[M-H]-の計算値 3995.7、実測値 3996.3(配列番号3)
5’-TTTTTCPCTTTTT-3’ について分子式C135H170N28O87P12、[M-H]-の計算値 3947.6、実測値 3948.4(配列番号4)
5’-TTTTTGPGTTTTT-3’ について分子式C137H170N32O87P12、[M-H]-の計算値 4027.7、実測値 4028.2(配列番号5)
5’-CGCAATPTAACGC-3’ について分子式C133H162N48O75P12、[M-H]-の計算値 4003.7、実測値 4004.7(配列番号6)
<3. 使用例1: 5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマー>
[使用例1-1:DNAオリゴマーの濃度決定]
DNAの濃度決定は、5’-TTTTCTTPTCCTTTT-3’について、DNAの水溶液10μLを10μLの酵素分解溶液(子ウシ腸由来アルカリホスファターゼ (100 U/mL)とP1ヌクレアーゼ (100 U/mL)とを含む水溶液)と混合して、室温で12時間放置してデオキシヌクレオシドに分解したものを、逆相HPLC(3-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)により定量することによって行った。この結果より、5’-TTTTCTTPTCCTTTT-3’の260 nmのモル吸光定数は119,000と計算された。この値は、5’-TTTTCTTTTCCTTTT-3’の260 nmにおけるモル吸光定数の計算値である119,400と非常に近い値であったので(Predicting ultraviolet spectrum of single and double stranded deoxyribonucleic acids. Tataurov, A. V., You, Y.及びOwczarzy, R. Biophys. Chem. 2008, 133, 66-70)、Pを一つ含むDNAオリゴマーの260 nmにおけるモル吸光係数は、PをTに置換して計算したDNAのモル吸光係数に近い値であることが示唆された。よって、本実施例では、DNAオリゴマーを希釈して260 nmの吸光度を測定し、PをTに置換して計算した260 nmにおけるモル吸光係数を用いて(Predicting ultraviolet spectrum of single and double stranded deoxyribonucleic acids. Tataurov, A. V., You, Y.及びOwczarzy, R. Biophys. Chem. 2008, 133, 66-70)、元のDNAオリゴマーの濃度を決定した。For 5′-TTTTCTTPTCCTTTT-3 ′, the molecular formula C 154 H 195 N 33 O 100 P 14 , [MH] − calculated value 4541.0, measured value 4541.9 (SEQ ID NO: 1)
5'-TTTTTTPTTTTTT-3 'molecular formula C 137 H 172 N 26 O 89 P 12 about, [MH] - Calculated 3977.6, Found 3978.0 (SEQ ID NO: 2)
For 5′-TTTTTAPATTTTT-3 ′, the molecular formula C 137 H 170 N 32 O 85 P 12 , [MH] − calculated value 3995.7, measured value 3996.3 (SEQ ID NO: 3)
For 5′-TTTTTCPCTTTTT-3 ′, the molecular formula C 135 H 170 N 28 O 87 P 12 , calculated value of [MH] − 3947.6, measured value 3948.4 (SEQ ID NO: 4)
For 5′-TTTTTGPGTTTTT-3 ′, the molecular formula C 137 H 170 N 32 O 87 P 12 , calculated for [MH] − 4027.7, measured value 4028.2 (SEQ ID NO: 5)
For 5′-CGCAATPTAACGC-3 ′, the molecular formula C 133 H 162 N 48 O 75 P 12 , [MH] − calculated value 4003.7, measured value 4004.7 (SEQ ID NO: 6)
<3. Use example 1: DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine>
[Use Example 1-1: Determination of DNA oligomer concentration]
Determine the concentration of DNA. For 5'-TTTTCTTPTCCTTTT-3 ', 10μL of DNA aqueous solution contains 10μL of enzymatic degradation solution (calf intestinal alkaline phosphatase (100 U / mL) and P1 nuclease (100 U / mL) And then decomposed to deoxynucleoside by standing at room temperature for 12 hours by reverse-phase HPLC (3-20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm) Went by. From this result, the molar absorption constant at 260 nm of 5′-TTTTCTTPTCCTTTT-3 ′ was calculated to be 119,000. This value was very close to the calculated value of 119,400 for the molar absorption constant at 260 nm of 5'-TTTTCTTTTCCTTTT-3 '(Predicting ultraviolet spectrum of single and double stranded deoxyribonucleic acids. Tataurov, AV, You , Y. and Owczarzy, R. Biophys. Chem. 2008, 133, 66-70), the molar extinction coefficient at 260 nm of a DNA oligomer containing one P is the molar extinction of DNA calculated by substituting P for T. It was suggested that the value is close to the coefficient. Therefore, in this example, the DNA oligomer was diluted and the absorbance at 260 nm was measured, and the molar extinction coefficient at 260 nm calculated by substituting P for T (Predicting ultraviolet spectrum of single and double stranded deoxyribonucleic acids) Tataurov, AV, You, Y. and Owczarzy, R. Biophys. Chem. 2008, 133, 66-70), the concentration of the original DNA oligomer was determined.
PをTに置換した場合の260 nmにおける各DNAオリゴマーのモル吸光定数εは以下の通りである。 The molar extinction constant ε of each DNA oligomer at 260 nm when P is replaced with T is as follows.
5’-TTTTCTTPTCCTTTT-3’についてε= 119,400
5’-TTTTTTPTTTTTT-3’についてε= 105,900
5’-TTTTTAPATTTTT-3’についてε= 117,700
5’-TTTTTCPCTTTTT-3’についてε= 104,100
5’-TTTTTGPGTTTTT-3’についてε= 111,000
5’-CGCAATPTAACGC-3’についてε= 122,700
[使用例1-2:5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーの切断反応]
一般的な方法では、DNA水溶液に等体積の一級アミンを加え、密閉状態で70℃に加熱すると、DNAオリゴマーは切断される。ここで、使用される一級アミンが異なると、切断反応の速度及び切断で生じる一方のDNA断片が異なる。切断で生じる2つのDNA断片のうち、3’-末端を新たに生じるDNA断片には使用されるアミンが付加するので、使用される一級アミンにより生成するDNA断片は異なる。このDNA断片がさらに分解されて3’-リン酸末端になる場合があるが、その場合、使用される一級アミンに依らず、同一のDNA断片が生じる。切断で生じる2つのDNA断片のうち、5’-リン酸末端を新たに生じるDNA断片については、原料DNAオリゴマーが同じ場合、使用される一級アミンに依らず同じDNA断片が生じる。具体的な一級アミンの反応については以下に示した。For 5'-TTTTCTTPTCCTTTT-3 'ε = 119,400
For 5'-TTTTTTPTTTTTT-3 ', ε = 105,900
Ε = 117,700 for 5'-TTTTTAPATTTTT-3 '
For 5'-TTTTTCPCTTTTT-3 'ε = 104,100
For 5'-TTTTTGPGTTTTT-3 ', ε = 111,000
For 5'-CGCAATPTAACGC-3 ', ε = 122,700
[Use Example 1-2: Cleavage reaction of DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine]
In a general method, when an equal volume of primary amine is added to an aqueous DNA solution and heated to 70 ° C. in a sealed state, the DNA oligomer is cleaved. Here, when the primary amine used is different, the rate of the cleavage reaction and the one DNA fragment produced by the cleavage are different. Of the two DNA fragments generated by cleavage, the DNA fragment generated by the primary amine used is different because the amine used is added to the newly generated DNA fragment at the 3′-end. This DNA fragment may be further degraded to the 3′-phosphate end, in which case the same DNA fragment is generated regardless of the primary amine used. Of the two DNA fragments generated by cleavage, a DNA fragment that newly generates a 5′-phosphate terminus, the same DNA fragment is generated regardless of the primary amine used when the source DNA oligomer is the same. Specific reaction of the primary amine is shown below.
[使用例1-3:メチルアミンによる5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーの切断反応及び生成物の同定]
スクリューチューブに30μLの5’-TTTTCTTPTCCTTTT-3’の水溶液(ストランド濃度 500μM)と30μLのメチルアミン(40% 水溶液)とを加え、水浴中で70℃、12時間加熱した。反応後は、メチルアミンを遠心濃縮装置で減圧留去し、逆相HPLCにより分析して生成物を分取した(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。分取した生成物は、MALDI TOF-MSにより分子量を測定した。さらに生成物の一部について、酵素分解して得られたデオキシヌクレオシドの比を求めた。pTCCTTTTに関しては、アルカリホスファターゼ処理した生成物の逆相HPLCにおける保持時間が、別途合成したTCCTTTTと一致し、MALDI TOF-MS測定による分子量も一致することを確認した。DNAオリゴマーの脱塩が不十分でギ酸アンモニウムがDNAオリゴマーに含まれている場合は、生成物2の一部がさらに分解して3’-リン酸末端となる場合がある。[Use Example 1-3: Cleavage reaction of DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine with methylamine and identification of product]
30 μL of 5′-TTTTCTTPTCCTTTT-3 ′ aqueous solution (
合成したDNAオリゴマーの反応前後の逆相HPLCクロマトグラムを図8に示す。 FIG. 8 shows reverse phase HPLC chromatograms of the synthesized DNA oligomer before and after the reaction.
生成物1及び2のMALDI TOF-MSのデータは以下の通りである。以下において、「RNu」は、切断反応に伴って生成するP由来の部分を意味する。The MALDI TOF-MS data for
生成物1:pTCCTTTT 分子式C68H91N16O48P7 FW 2117.3 [M-H]- 2116.3 実測値 2116.4
生成物2:TTTTCTTpRNu 分子式C75H103N16O51P7 FW 2261.5 [M-H]- 2260.5 実測値 2262.2
DNAオリゴマーを分解して生じるデオキシヌクレオシドの比は以下の通りである。Product 1: pTCCTTTT molecular formula C 68 H 91 N 16 O 48 P 7 FW 2117.3 [MH] - 2116.3 Found 2116.4
Product 2: TTTTCTTpR Nu molecular formula C 75 H 103 N 16 O 51 P 7 FW 2261.5 [MH] - 2260.5 Found 2262.2
The ratio of deoxynucleosides generated by decomposing DNA oligomers is as follows.
生成物1:dC:dT = 2:5
生成物2:dC:dT = 1:6
メチルアミンによるDNAオリゴマーの分解反応を下記に示す。下記反応式において、左側の生成物が生成物2に、右側の生成物が生成物1に、それぞれ対応する。
Product 2: dC: dT = 1: 6
Degradation reaction of DNA oligomer by methylamine is shown below. In the following reaction formula, the product on the left corresponds to the
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
[使用例1-4:メチルアミン、エチレンジアミン又は2-アミノエタノールによる5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーの切断反応速度の比較]
スクリューチューブに30μLの5’-TTTTCTTPTCCTTTT-3’の水溶液(ストランド濃度 500μM)と30μLの一級アミン(メチルアミン、エチレンジアミン又は2-アミノエタノール)を加え、水浴中で70℃、1時間加熱した。反応混合物を酢酸で中和し、逆相HPLCにより分析した(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。各反応混合物のHPLCクロマトグラムを図9に示す。[Use Example 1-4: Comparison of cleavage reaction rate of DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine with methylamine, ethylenediamine or 2-aminoethanol]
30 μL of 5′-TTTTCTTPTCCTTTT-3 ′ aqueous solution (
5’-TCCTTTT-3’の260 nmにおけるモル吸光係数ε=55,500、5’-TTTTCTTTTCCTTTT-3’の260 nmにおけるモル吸光係数ε=119,400を利用して、HPLCクロマトグラム上のピーク面積に基づき定量したところ、メチルアミンを用いて得られた反応混合物における原料消失は24%、エチレンジアミンを用いて得られた反応混合物における原料消失は76%、そして2-アミノエタノールを用いて得られた反応混合物における原料消失は31%と計算された。この結果より、メチルアミン、エチレンジアミン又は2-アミノエタノールの一級アミンを用いるDNAオリゴマーの切断反応において、最も反応速度の速いものはエチレンジアミンを用いる切断反応であることが示唆された。 Using 5'-TCCTTTT-3 'molar extinction coefficient at 260 nm ε = 55,500, 5'-TTTTCTTTTCCTTTT-3' molar extinction coefficient at 260 nm ε = 119,400, based on the peak area on the HPLC chromatogram As a result, the disappearance of the raw material in the reaction mixture obtained using methylamine was 24%, the disappearance of the raw material in the reaction mixture obtained using ethylenediamine was 76%, and in the reaction mixture obtained using 2-aminoethanol. The disappearance of raw material was calculated as 31%. From these results, it was suggested that in the cleavage reaction of DNA oligomer using primary amine of methylamine, ethylenediamine or 2-aminoethanol, the one having the fastest reaction rate is the cleavage reaction using ethylenediamine.
[使用例1-5:エチレンジアミンによる5-フェニルエチニル-2’-デオキシウリジンを含むさまざまな配列を有するDNAオリゴマーの切断反応及び生成物の同定]
スクリューチューブに30μLのDNAオリゴマー水溶液(ストランド濃度 500μM)と30μLのエチレンジアミン(無水)とを加え、水浴中で70℃、3時間加熱した。反応混合物を酢酸で中和し、逆相HPLCにより分析し、生成物を分取した(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。分取した生成物は、MALDI TOF-MSにより分子量を測定した。[Use Example 1-5: Cleavage reaction of DNA oligomers having various sequences including 5-phenylethynyl-2′-deoxyuridine with ethylenediamine and identification of products]
30 μL of DNA oligomer aqueous solution (
合成した各DNAオリゴマーの反応前後の逆相HPLCクロマトグラムを図10〜15に示す。 The reverse phase HPLC chromatogram before and behind reaction of each synthetic | combination DNA oligomer is shown to FIGS.
各DNAオリゴマーにおける生成物1及び2のMALDI TOF-MSのデータは以下の通りである。以下において、「RNu」は、切断反応に伴って生成するP由来の部分を意味する。The MALDI TOF-MS data of
5’-TTTTCTTPTCCTTTT-3’について
生成物1:pTCCTTTT 分子式C68H91N16O48P7 FW 2117.3 [M-H]- 2116.3 実測値 2116.4
生成物2:TTTTCTTpRNu 分子式C76H106N17O51P7 FW 2290.6 [M-H]- 2289.5 実測値 2290.4
5’-TTTTTTPTTTTTT-3’について
生成物1:pTTTTTT 分子式C60H80N12O43P6 FW 1843.2 [M-H]- 1842.2 実測値 1842.3
生成物2:TTTTTTpRNu 分子式C67H94N14O45P6 FW 2001.4 [M-H]- 2000.4 実測値 2000.3
5’-TTTTTAPATTTTT-3’について
生成物1:pATTTTT 分子式C60H79N15O41P6 FW 1852.2 [M-H]- 1851.2 実測値 1851.0
生成物2:TTTTTApRNu 分子式C67H93N17O43P6 FW 2010.4 [M-H]- 2009.4 実測値 2009.2
5’-TTTTTCPCTTTTT-3’について
生成物1:pCTTTTT 分子式C59H79N13O42P6 FW 1828.2 [M-H]- 1827.2 実測値 1826.9
生成物2:TTTTTCpRNu 分子式C66H93N15O44P6 FW 1986.4 [M-H]- 1985.4 実測値 1985.5
5’-TTTTTGPGTTTTT-3’について
生成物1:pGTTTTT 分子式C60H79N15O42P6 FW 1868.2 [M-H]- 1867.2 実測値 1867.2
生成物2:TTTTTGpRNu 分子式C67H93N17O44P6 FW 2026.4 [M-H]- 2025.4 実測値 2025.2
5’-CGCAATPTAACGC-3’について
生成物1:pTAACGC 分子式C58H75N23O36P6 FW 1856.2 [M-H]- 1855.2 実測値 1855.2
生成物2:CGCAATpRNu 分子式C65H89N25O38P6 FW 2014.4 [M-H]- 2013.4 実測値 2013.2
エチレンジアミンによるDNAオリゴマーの分解反応を下記に示す。下記反応式において、左側の生成物が生成物2に、右側の生成物が生成物1に、それぞれ対応する。
Product 2: TTTTCTTpR Nu molecular formula C 76 H 106 N 17 O 51 P 7 FW 2290.6 [MH] - 2289.5 Found 2290.4
5'-TTTTTTPTTTTTT-3 'product for 1: pTTTTTT molecular formula C 60 H 80 N 12 O 43 P 6 FW 1843.2 [MH] - 1842.2 Found 1842.3
Product 2: TTTTTTpR Nu molecular formula C 67 H 94 N 14 O 45 P 6 FW 2001.4 [MH] - 2000.4 Found 2000.3
5'-TTTTTAPATTTTT-3 'product for 1: pATTTTT molecular formula C 60 H 79 N 15 O 41 P 6 FW 1852.2 [MH] - 1851.2 Found 1851.0
Product 2: TTTTTApR Nu molecular formula C 67 H 93 N 17 O 43 P 6 FW 2010.4 [MH] - 2009.4 Found 2009.2
5'-TTTTTCPCTTTTT-3 'product for 1: pCTTTTT molecular formula C 59 H 79 N 13 O 42 P 6 FW 1828.2 [MH] - 1827.2 Found 1826.9
Product 2: TTTTTCpR Nu molecular formula C 66 H 93 N 15 O 44 P 6 FW 1986.4 [MH] - 1985.4 Found 1985.5
5'-TTTTTGPGTTTTT-3 'product for 1: pGTTTTT molecular formula C 60 H 79 N 15 O 42 P 6 FW 1868.2 [MH] - 1867.2 Found 1867.2
Product 2: TTTTTGpR Nu molecular formula C 67 H 93 N 17 O 44 P 6 FW 2026.4 [MH] - 2025.4 Found 2025.2
5'-CGCAATPTAACGC-3 'product for 1: pTAACGC molecular formula C 58 H 75 N 23 O 36 P 6 FW 1856.2 [MH] - 1855.2 Found 1855.2
Product 2: CGCAATpR Nu molecular formula C 65 H 89 N 25 O 38 P 6 FW 2014.4 [MH] - 2013.4 Found 2013.2
Degradation reaction of DNA oligomer by ethylenediamine is shown below. In the following reaction formula, the product on the left corresponds to the
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
[使用例1-6:2-アミノエタノールによる5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーの切断反応及び生成物の同定]
スクリューチューブに30μLの5’-TTTTCTTPTCCTTTT-3’の水溶液(ストランド濃度 500μM)と30μLの2-アミノエタノール(無水)とを加え、水浴中で70℃、12時間加熱した。反応混合物を酢酸で中和し、逆相HPLCにより分析し、生成物を分取した(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。分取した生成物は、MALDI TOF-MSにより分子量を測定した。[Use Example 1-6: Cleavage reaction of DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine with 2-aminoethanol and identification of the product]
30 μL of 5′-TTTTCTTPTCCTTTT-3 ′ aqueous solution (strand concentration: 500 μM) and 30 μL of 2-aminoethanol (anhydrous) were added to the screw tube and heated in a water bath at 70 ° C. for 12 hours. The reaction mixture was neutralized with acetic acid and analyzed by reverse phase HPLC to fractionate the product (0-50% CH 3 CN / 50 mM aqueous ammonium formate, 20 min, detected at 254 nm). The molecular weight of the fractionated product was measured by MALDI TOF-MS.
合成したDNAオリゴマーの反応前後の逆相HPLCクロマトグラムを図16に示す。 FIG. 16 shows reverse phase HPLC chromatograms of the synthesized DNA oligomer before and after the reaction.
生成物1、2及び3のMALDI TOF-MSのデータは以下の通りである。以下において、「RNu」は、切断反応に伴って生成するP由来の部分を意味する。MALDI TOF-MS data for
生成物1:pTCCTTTT 分子式C68H91N16O48P7 FW 2117.3 [M-H]- 2116.3 実測値 2116.2
生成物2:TTTTCTTp 分子式C69H92N15O49P7 FW2132.4 [M-H]- 2131.3 実測値 2131.1
生成物3:TTTTCTTpRNu 分子式C76H105N16O52P7 FW 2291.5 [M-H]- 2290.5 実測値 2290.5
2-アミノエタノールによるDNAオリゴマーの分解反応を下記に示す。2-アミノエタノールの場合、反応混合物にギ酸アンモニウムが含まれていなくても、切断で生じる5'-末端側の生成物の一部がさらに分解して、3’-リン酸末端を形成する。下記反応式において、左側の生成物が生成物3に、右側の生成物が生成物1に、それぞれ対応する。生成物2は、生成物3から糖由来の残基部分が脱離して3’-リン酸末端を形成した化合物に対応する。
Product 2: TTTTCTTp molecular formula C 69 H 92 N 15 O 49 P 7 FW2132.4 [MH] - 2131.3 Found 2131.1
Product 3: TTTTCTTpR Nu molecular formula C 76 H 105 N 16 O 52 P 7 FW 2291.5 [MH] - 2290.5 Found 2290.5
The degradation reaction of DNA oligomer with 2-aminoethanol is shown below. In the case of 2-aminoethanol, even if ammonium formate is not contained in the reaction mixture, a part of the product on the 5′-terminal side generated by cleavage is further decomposed to form a 3′-phosphate terminal. In the following reaction formula, the product on the left corresponds to the
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
[使用例1-7:5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーを用いる5’-リン酸化(1)]
DNAオリゴマーの脱保護とチミジン誘導体の位置でのDNA切断反応とを同時に行った。[Use Example 1-7: 5′-Phosphorylation Using DNA Oligomers Containing 5-Phenylethynyl-2′-Deoxyuridine (1)]
The deprotection of the DNA oligomer and the DNA cleavage reaction at the position of the thymidine derivative were performed simultaneously.
DNA自動合成機を用いて、5’-PTTATTGTT-3’の配列を有するDNAオリゴマーを上記と同じ方法で合成した。担体からの切り出しを28%アンモニア水と40%メチルアミン水溶液との1:1混合溶液で行った。切り出したDNAオリゴマー溶液をスクリューチューブに入れ、水浴中で70℃、16時間加熱することによって脱保護を行った。遠心濃縮装置を用いてアンモニア及びメチルアミンを減圧留去し、0.45μmのフィルターに通した。その後、逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。MALDI TOF-MS測定により、得られたDNAオリゴマーは5’-末端がリン酸化された5’-TTATTGTT-3’の分子量と一致することを確認した。得られたDNAオリゴマーの水溶液10μLを、10μLのアルカリホスファターゼ(子ウシ腸由来アルカリホスファターゼ (100 U/mL))と混合し、40℃で2時間加熱することにより、末端リン酸部分を除去した。生成物と5’-TTATTGTT-3’とを混合して逆相HPLCで分析したところ、両化合物は同一の保持時間を示すことが確認された。また、この生成物は、5’-TTATTGTT-3’と同一の分子量であることをMALDI TOF-MSによって確認した。A DNA oligomer having a 5′-PTTATTGTT-3 ′ sequence was synthesized by the same method as described above using an automatic DNA synthesizer. Cutting out from the carrier was performed with a 1: 1 mixed solution of 28% aqueous ammonia and 40% aqueous methylamine. The DNA oligomer solution thus cut out was put in a screw tube and deprotected by heating in a water bath at 70 ° C. for 16 hours. Ammonia and methylamine were removed under reduced pressure using a centrifugal concentrator and passed through a 0.45 μm filter. Thereafter, the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off. By MALDI TOF-MS measurement, it was confirmed that the obtained DNA oligomer coincided with the molecular weight of 5′-TTATTGTT-3 ′ phosphorylated at the 5′-end. 10 μL of the obtained aqueous solution of DNA oligomer was mixed with 10 μL of alkaline phosphatase (calf intestine-derived alkaline phosphatase (100 U / mL)) and heated at 40 ° C. for 2 hours to remove the terminal phosphate portion. When the product and 5′-TTATTGTT-3 ′ were mixed and analyzed by reverse phase HPLC, it was confirmed that both compounds showed the same retention time. Further, it was confirmed by MALDI TOF-MS that this product had the same molecular weight as 5′-TTATTGTT-3 ′.
切り出し及び脱保護後のDNAオリゴマーの逆相HPLCクロマトグラムを図17に示す。 FIG. 17 shows a reverse phase HPLC chromatogram of the DNA oligomer after excision and deprotection.
メインピーク:5’-pTTATTGTT-3’ 分子式C80H104N22O54P8 FW 2485.6 [M-H]- 2484.6 実測値 2484.7
反応スキームを下記に示す。
The reaction scheme is shown below.
[使用例1-8:5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーを用いる5’-リン酸化(2)]
DNAオリゴマーの切り出し及び脱保護とチミジン誘導体の位置でのDNA切断反応とを別々に行った。[Use Example 1-8: 5′-Phosphorylation (2) using DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine]
The excision and deprotection of the DNA oligomer and the DNA cleavage reaction at the position of the thymidine derivative were performed separately.
DNA自動合成機を用いて、5’-PTTATTGTT-3’の配列を有するDNAオリゴマーを上記と同じ方法で合成した。担体からの切り出しを28%アンモニア水で行い、脱保護は室温(約25℃)で16時間静置することにより行った。遠心濃縮装置を用いてアンモニアを減圧留去し、0.45μmのフィルターに通した。その後、逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。得られた凍結乾燥物に30μLの蒸留水及び30μLのエチレンジアミンを加え、水浴中で70℃、3時間加熱した。反応混合物を酢酸で中和した後、上記使用例1-7と同様の方法によりDNAオリゴマーを精製した。得られたDNAオリゴマーはMALDI TOF-MSにより同定した。得られたDNAオリゴマーの水溶液10μLを、10μLのアルカリホスファターゼ(子ウシ腸由来アルカリホスファターゼ (100 U/mL))と混合し、40℃で2時間加熱することにより、末端リン酸部分を除去した。生成物と5’-TTATTGTT-3’とを混合して逆相HPLCで分析したところ、両化合物は同一の保持時間を示すことが確認された。また、この生成物は、5’-TTATTGTT-3’と同一の分子量であることをMALDI TOF-MSによって確認した。A DNA oligomer having a 5′-PTTATTGTT-3 ′ sequence was synthesized by the same method as described above using an automatic DNA synthesizer. Cleavage from the carrier was performed with 28% aqueous ammonia, and deprotection was performed by standing at room temperature (about 25 ° C.) for 16 hours. Ammonia was distilled off under reduced pressure using a centrifugal concentrator and passed through a 0.45 μm filter. Thereafter, the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off. 30 μL of distilled water and 30 μL of ethylenediamine were added to the resulting lyophilized product and heated in a water bath at 70 ° C. for 3 hours. After neutralizing the reaction mixture with acetic acid, the DNA oligomer was purified by the same method as in Use Example 1-7. The obtained DNA oligomer was identified by MALDI TOF-MS. 10 μL of the obtained aqueous solution of DNA oligomer was mixed with 10 μL of alkaline phosphatase (calf intestine-derived alkaline phosphatase (100 U / mL)) and heated at 40 ° C. for 2 hours to remove the terminal phosphate portion. When the product and 5′-TTATTGTT-3 ′ were mixed and analyzed by reverse phase HPLC, it was confirmed that both compounds showed the same retention time. Further, it was confirmed by MALDI TOF-MS that this product had the same molecular weight as 5′-TTATTGTT-3 ′.
切り出し及び脱保護後のDNAオリゴマーの逆相HPLCクロマトグラムを図18に、エチレンジアミンによる反応後のDNAオリゴマーの逆相HPLCクロマトグラムを図19に、それぞれ示す。 FIG. 18 shows a reverse phase HPLC chromatogram of the DNA oligomer after excision and deprotection, and FIG. 19 shows a reverse phase HPLC chromatogram of the DNA oligomer after the reaction with ethylenediamine.
切り出し及び脱保護後のDNAオリゴマー
メインピーク:5’-PTTATTGTT-3’ 分子式C97H118N24O58P8 FW 2795.9 [M-H]- 2794.9 実測値 2795.1
エチレンジアミンによる反応後のDNAオリゴマー
メインピーク:5’-PTTATTGTT-3’ 分子式C97H118N24O58P8 FW 2795.9 [M-H]- 2794.9 実測値 2795.1
反応スキームを下記に示す。
DNA oligomers main peak after reaction with ethylene diamine: 5'-PTTATTGTT-3 'molecular formula C 97 H 118 N 24 O 58 P 8 FW 2795.9 [MH] - 2794.9 Found 2795.1
The reaction scheme is shown below.
[使用例1-9:5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーを用いる3’-リン酸化(1)]
DNAオリゴマーの脱保護とチミジン誘導体の位置でのDNA切断反応とを同時に行った。[Use Example 1-9: 3′-Phosphorylation (1) using a DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine]
The deprotection of the DNA oligomer and the DNA cleavage reaction at the position of the thymidine derivative were performed simultaneously.
DNA自動合成機を用いて、5’-TTATTGTTPT-3’の配列を有するDNAオリゴマーを上記と同じ方法で合成した。担体からの切り出しを28%アンモニア水と40%メチルアミン水溶液との1:1混合溶液で行った。切り出したDNAオリゴマー溶液をスクリューチューブに入れ、水浴中で70℃、16時間加熱することによって脱保護を行った。遠心濃縮装置を用いてアンモニア及びメチルアミンを減圧留去し、0.45μmのフィルターに通した。その後、逆相HPLCにより保持時間10分付近のピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。分取HPLCクロマトグラム上において、メインピークに近い小さなピークは、メインピークの生成物から糖に由来する部分が脱離して3’-リン酸末端になったものであった。得られたDNAオリゴマーに100 mMリン酸バッファー500μL(pH 7)を加え、スクリューチューブに移し、120℃に設定した乾燥器中で一時間静置した。逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。分取HPLCクロマトグラム上において、メインピークより少し後に溶出されたピークは、3’-末端のリン酸が脱離したものであった。MALDI TOF-MS測定により、得られたDNAオリゴマーは3’-末端がリン酸化された5’-TTATTGTT-3’の分子量と一致することを確認した。得られたDNAオリゴマーの水溶液10μLを、10μLのアルカリホスファターゼ(子ウシ腸由来アルカリホスファターゼ (100 U/mL))と混合し、40℃で2時間加熱することにより、末端リン酸部分を除去した。生成物と5’-TTATTGTT-3’とを混合して逆相HPLCで分析したところ、両化合物は同一の保持時間を示すことが確認された。また、この生成物は、5’-TTATTGTT-3’と同一の分子量であることをMALDI TOF-MSによって確認した。Using an automatic DNA synthesizer, a DNA oligomer having the
切り出し及び脱保護後のDNAオリゴマーの逆相HPLCクロマトグラムを図20に、100 mMリン酸バッファー中で120℃処理後のDNAオリゴマーの逆相HPLCクロマトグラムを図21に、それぞれ示す。 FIG. 20 shows a reverse phase HPLC chromatogram of the DNA oligomer after excision and deprotection, and FIG. 21 shows a reverse phase HPLC chromatogram of the DNA oligomer after treatment at 120 ° C. in 100 mM phosphate buffer.
切り出し及び脱保護後のDNAオリゴマー
メインピーク:5’-TTATTGTTpRNu-3’ 分子式C86H115N23O56P8 FW 2614.7 [M-H]- 2613.7 実測値 2613.4
リン酸バッファー中で120℃処理後のDNAオリゴマー
メインピーク:5’-TTATTGTTp-3’ 分子式C80H104N22O54P8 FW 2485.6 [M-H]- 2484.6 実測値 2484.6
反応スキームを下記に示す。
DNA oligomers main peak of 120 ° C. after treatment in phosphoric acid buffer: 5'-TTATTGTTp-3 'molecular formula C 80 H 104 N 22 O 54 P 8 FW 2485.6 [MH] - 2484.6 Found 2484.6
The reaction scheme is shown below.
生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
[使用例1-10:5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーを用いる3’-リン酸化(2)]
DNAオリゴマーの切り出し及び脱保護とチミジン誘導体の位置でのDNA切断反応とを別々に行った。[Use Example 1-10: 3′-Phosphorylation (2) using a DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine]
The excision and deprotection of the DNA oligomer and the DNA cleavage reaction at the position of the thymidine derivative were performed separately.
DNA自動合成機を用いて、5’-TTATTGTTPT-3’の配列を有するDNAオリゴマーを上記と同じ方法で合成した。担体からの切り出しを28 %アンモニア水で行い、脱保護は室温(約25℃)で16時間静置することにより行った。遠心濃縮装置を用いてアンモニアを減圧留去し、0.45μmのフィルターに通した。その後、逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。得られた凍結乾燥物に30μLの蒸留水及び30μLのエチレンジアミンを加え、水浴中で70℃、3時間加熱した。反応混合物を酢酸で中和した後、0.45μmのフィルターに通した。その後、逆相HPLCにより保持時間10分付近のピークを精製し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。得られた凍結乾燥物に500μLの100 mMリン酸バッファー(pH 7)を加えた後、スクリューチューブに移し、120℃に設定した乾燥器中で一時間静置した。逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。得られたDNAオリゴマーはMALDI TOF-MSにより同定した。得られたDNAオリゴマーの水溶液10μLを、10μLのアルカリホスファターゼ(子ウシ腸由来アルカリホスファターゼ (100 U/mL))と混合し、40℃で2時間加熱することにより、末端リン酸部分を除去した。生成物と5’-TTATTGTT-3’とを混合して逆相HPLCで分析したところ、両化合物は同一の保持時間を示すことが確認された。また、この生成物は、5’-TTATTGTT-3’と同一の分子量であることをMALDI TOF-MSによって確認した。Using an automatic DNA synthesizer, a DNA oligomer having the
切り出し及び脱保護後のDNAオリゴマーの逆相HPLCクロマトグラムを図22に、エチレンジアミンによる反応後のDNAオリゴマーの逆相HPLCクロマトグラムを図23に、100 mMリン酸バッファー中で120℃処理後のDNAオリゴマーの逆相HPLCクロマトグラムを図24に、それぞれ示す。 Fig. 22 shows the reverse-phase HPLC chromatogram of the DNA oligomer after excision and deprotection, and Fig. 23 shows the reverse-phase HPLC chromatogram of the DNA oligomer after reaction with ethylenediamine. DNA after treatment at 120 ° C in 100 mM phosphate buffer. The reverse phase HPLC chromatogram of the oligomer is shown in FIG. 24, respectively.
切り出し及び脱保護後のDNAオリゴマー
メインピーク:5’-TTATTGTTPT-3’ 分子式C107H131N26O65P9 FW 3100.1 [M-H]- 3099.1 実測値 3099.4
エチレンジアミンによる反応後のDNAオリゴマー
メインピーク:5’-TTATTGTTpRNu-3’ 分子式C87H118N24O56P8 FW 2643.8 [M-H]- 2642.8 実測値 2642.9
リン酸バッファー中で120℃処理後のDNAオリゴマー
メインピーク:5’-TTATTGTTp-3’ 分子式C80H104N22O54P8 FW 2485.6 [M-H]- 2484.6 実測値 2484.7
反応スキームを下記に示す。
DNA oligomers main peak after reaction with ethylene diamine: 5'-TTATTGTTpR Nu -3 'molecular formula C 87 H 118 N 24 O 56 P 8 FW 2643.8 [MH] - 2642.8 Found 2642.9
DNA oligomers main peak of 120 ° C. after treatment in phosphoric acid buffer: 5'-TTATTGTTp-3 'molecular formula C 80 H 104 N 22 O 54 P 8 FW 2485.6 [MH] - 2484.6 Found 2484.7
The reaction scheme is shown below.
生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
[使用例1-11:5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーを用いるDMTr ON合成]
DNA自動合成機を用いるDNAオリゴマー合成において、最後のデオキシヌクレオシドの5’-末端のDMTr基を脱保護しない条件(以下「DMTr ON」とも記載する)で合成を行った。[Use Example 1-11: DMTr ON synthesis using a DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine]
In DNA oligomer synthesis using an automatic DNA synthesizer, synthesis was carried out under conditions that do not deprotect the DMTr group at the 5′-end of the last deoxynucleoside (hereinafter also referred to as “DMTr ON”).
DNA自動合成機を用いて、5’-PTTATTGTT-3’の配列を有するDNAオリゴマーを上記と同じ方法で合成した。但し、最後のデオキシヌクレオシドの5’-末端のDMTr基は脱保護せず、「DMTr ON」の条件で合成を行った。担体からの切り出しを28%アンモニア水で行い、脱保護は室温(約25℃)で16時間静置することにより行った。遠心濃縮装置を用いてアンモニアを減圧留去し、0.45μmのフィルターに通した。その後、逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。得られた凍結乾燥物に100μLの蒸留水及び100μLのエチレンジアミンを加え、水浴中で70℃、8時間加熱した。反応混合物を水で1 mLに希釈し、酢酸で中和した後、逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。得られたDNAオリゴマーは、5’-末端がリン酸化された5’-TTATTGTT-3’と同一の分子量であることをMALDI TOF-MSによって確認した。A DNA oligomer having a 5′-PTTATTGTT-3 ′ sequence was synthesized by the same method as described above using an automatic DNA synthesizer. However, the DMTr group at the 5′-end of the last deoxynucleoside was not deprotected and was synthesized under the conditions of “DMTr ON”. Cleavage from the carrier was performed with 28% aqueous ammonia, and deprotection was performed by standing at room temperature (about 25 ° C.) for 16 hours. Ammonia was distilled off under reduced pressure using a centrifugal concentrator and passed through a 0.45 μm filter. Thereafter, the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off. 100 μL of distilled water and 100 μL of ethylenediamine were added to the resulting lyophilized product and heated in a water bath at 70 ° C. for 8 hours. The reaction mixture was diluted to 1 mL with water and was neutralized with acetic acid, it was collected fractions containing the main peak by
切り出し及び脱保護後の5’-末端がDMTr保護されたDNAオリゴマーの逆相HPLCクロマトグラムを図25に、エチレンジアミンによる反応後の5’-末端がリン酸化されたDNAオリゴマーの逆相HPLCクロマトグラムを図26に、それぞれ示す。 Figure 25 shows the reverse-phase HPLC chromatogram of the DNA oligomer with DMTr protection at the 5'-end after excision and deprotection. Figure 25 shows the reverse-phase HPLC chromatogram of the DNA oligomer phosphorylated at the 5'-end after reaction with ethylenediamine. Are shown in FIG.
切り出し及び脱保護後の5’-末端がDMTr保護されたDNAオリゴマー
メインピーク:5’-DMTr-PTTATTGTT-3’ 分子式C118H137N24O60P8 FW 3099.3 [M-H]- 3098.3 実測値 3097.7
エチレンジアミンによる反応後の5’-末端がリン酸化されたDNAオリゴマー
メインピーク:5’-pTTATTGTT-3’ 分子式C80H104N22O54P8 FW 2485.6 [M-H]- 2484.6 実測値 2484.6
反応スキームを下記に示す。
DNA oligomers 5'-end of the post-reaction with ethylenediamine was phosphorylated main peak: 5'-pTTATTGTT-3 'molecular formula C 80 H 104 N 22 O 54 P 8 FW 2485.6 [MH] - 2484.6 Found 2484.6
The reaction scheme is shown below.
[使用例1-12:5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーより調製された5’末端がリン酸化されたDNAオリゴマーと別のDNAオリゴマーとの連結反応]
DNA自動合成機を用いて、下記の配列を有するDNAオリゴマーを上記と同じ方法で合成した。但し、TS3は、最後のデオキシヌクレオシドの5’末端のDMTr基は脱保護せず、「DMTr ON」の条件で合成を行った。5’末端及び3’末端のFAM修飾には、それぞれグレンリサーチ社の5’-フルオレセインホスホロアミダイト (カタログ番号10-5901)及び3’-(6-FAM)-CPG (カタログ番号20-2961)を使用した。TF3の5'末端へのアミノ基の導入は、非特許文献35に記載の方法に従って行った。[Use Example 1-12: Ligation reaction between a DNA oligomer prepared from a DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine and phosphorylated at the 5 ′ end and another DNA oligomer]
Using an automatic DNA synthesizer, a DNA oligomer having the following sequence was synthesized in the same manner as described above. However, TS3 was synthesized under the conditions of “DMTr ON” without deprotecting the DMTr group at the 5 ′ end of the last deoxynucleoside. FAM modifications at the 5 'and 3' ends include 5'-fluorescein phosphoramidite (Catalog No. 10-5901) and 3 '-(6-FAM) -CPG (Catalog No. 20-2961) from Glen Research, respectively. It was used. The amino group was introduced into the 5 ′ end of TF3 according to the method described in Non-Patent Document 35.
TS1:5’-FAM-ATAGTGAGTCGTATTAGGCCCGATCCAGGCCATCGCGCATGCTCAG-3’(配列番号7)
TS3:5’-ATAGTGAGTCGTATTAGGCCCGATCCAGGCCATCGCGCATGCTCAG-3’(配列番号8)
TF3:5’-NH2-TTAACTGAGCATGCGCGATGGCCTG-3’(配列番号9)
TT1:5’-pGATCGGGCCTAATACGACTCACTAT-FAM-3’(配列番号10)
TT2:5’-pATCGGGCCTAATACGACTCACTAT-FAM-3’(配列番号11)
5’末端がリン酸化されたDNAオリゴマーであるTT1及びTT2は、以下の方法で調製した。5-フェニルエチニル-2’-デオキシウリジンを用いて、5’-PGATCGGGCCTAATACGACTCACTAT-FAM-3’(配列番号12)及び5’-PATCGGGCCTAATACGACTCACTAT-FAM-3’(配列番号13)の配列を有するDNAオリゴマーを上記使用例1-7と同じ方法で合成した。担体からの切り出しを28%アンモニア水と40%メチルアミン水溶液との1:1混合溶液で行った。切り出したDNAオリゴマー溶液をスクリューチューブに入れ、水浴中で70℃、16時間加熱することによって脱保護を行った。遠心濃縮装置を用いてアンモニア及びメチルアミンを減圧留去し、0.45μmのフィルターに通した。その後、逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。MALDI TOF-MS測定により、得られたDNAオリゴマーは5’末端がリン酸化されたDNAオリゴマーであるTT1及びTT2の分子量とそれぞれ一致することを確認した。TS1: 5'-FAM-ATAGTGAGTCGTATTAGGCCCGATCCAGGCCATCGCGCATGCTCAG-3 '(SEQ ID NO: 7)
TS3: 5'-ATAGTGAGTCGTATTAGGCCCGATCCAGGCCATCGCGCATGCTCAG-3 '(SEQ ID NO: 8)
TF3: 5'-NH 2 -TTAACTGAGCATGCGCGATGGCCTG-3 '(SEQ ID NO: 9)
TT1: 5'-pGATCGGGCCTAATACGACTCACTAT-FAM-3 '(SEQ ID NO: 10)
TT2: 5'-pATCGGGCCTAATACGACTCACTAT-FAM-3 '(SEQ ID NO: 11)
TT1 and TT2, which are DNA oligomers phosphorylated at the 5 ′ end, were prepared by the following method. Using 5-phenylethynyl-2′-deoxyuridine, a DNA oligomer having sequences of 5′-PGATCGGGCCTAATACGACTCACTAT-FAM-3 ′ (SEQ ID NO: 12) and 5′-PATCGGGCCTAATACGACTCACTAT-FAM-3 ′ (SEQ ID NO: 13) The compound was synthesized in the same manner as in the above Use Example 1-7. Cutting out from the carrier was performed with a 1: 1 mixed solution of 28% aqueous ammonia and 40% aqueous methylamine. The DNA oligomer solution thus cut out was put in a screw tube and deprotected by heating in a water bath at 70 ° C. for 16 hours. Ammonia and methylamine were removed under reduced pressure using a centrifugal concentrator and passed through a 0.45 μm filter. Thereafter, the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off. It was confirmed by MALDI TOF-MS measurement that the obtained DNA oligomers were in agreement with the molecular weights of TT1 and TT2, which are DNA oligomers phosphorylated at the 5 ′ end.
切り出し及び脱保護後のTT1及びTT2の逆相HPLCクロマトグラムを図27A及びBにそれぞれ示す。 The reverse phase HPLC chromatograms of TT1 and TT2 after excision and deprotection are shown in FIGS. 27A and B, respectively.
TT1:
メインピーク:5’-pGATCGGGCCTAATACGACTCACTAT-FAM-3’ 分子式C270H332N94O161P26 FW 8275.4 [M-H]-8274.4 実測値 8273.7
TT2:
メインピーク:5’-pATCGGGCCTAATACGACTCACTAT-FAM-3’ 分子式C260H320N89O155P25 FW 7946.2 [M-H]-7945.2 実測値 7946.1
5’末端がFAM修飾されたDNAオリゴマー(以下「5’-FAM」とも称する)の構造を下記に示す。
Main peak: 5'-pGATCGGGCCTAATACGACTCACTAT-FAM-3 ' molecular formula C 270 H 332 N 94 O 161 P 26 FW 8275.4 [MH] - 8274.4 Found 8273.7
TT2:
Main peak: 5'-pATCGGGCCTAATACGACTCACTAT-FAM-3 ' molecular formula C 260 H 320 N 89 O 155 P 25 FW 7946.2 [MH] - 7945.2 Found 7946.1
The structure of a DNA oligomer (hereinafter also referred to as “5′-FAM”) in which the 5 ′ end is FAM-modified is shown below.
5’末端がアミノ基で修飾されたDNAオリゴマー(以下「5’-NH2」とも称する)の構造を下記に示す。
3’末端がFAM修飾されたDNAオリゴマー(以下「FAM-3’」とも称する)の構造を下記に示す。
DNAオリゴマーの連結反応は、40μMのTF3及びTS3と、33μMのTT1(比較のためTT2)との混合溶液15μLに対して、15μLのLigation high(TOYOBO社製 カタログ番号LGK-101)を混合し、16℃において1時間から3時間振盪することによって行った。対照区として、TT1をTT2に変更した試料を上記と同様に反応させた。得られた反応混合物を、7 M 尿素を含む15% ポリアクリルアミドゲル電気泳動により分析して、連結反応の確認を行った。各DNAオリゴマーの塩基配列及びポリアクリルアミドゲル電気泳動による分析結果を図28に示す。 For the DNA oligomer ligation reaction, 15 μL of Ligation high (TOYOBO catalog number LGK-101) is mixed with 15 μL of a mixed solution of 40 μM TF3 and TS3 and 33 μM TT1 (TT2 for comparison) This was done by shaking at 16 ° C. for 1 to 3 hours. As a control, a sample in which TT1 was changed to TT2 was reacted in the same manner as described above. The obtained reaction mixture was analyzed by 15% polyacrylamide gel electrophoresis containing 7 M urea to confirm the ligation reaction. FIG. 28 shows the base sequence of each DNA oligomer and the analysis results by polyacrylamide gel electrophoresis.
図28Bに示すように、ポリアクリルアミドゲルに紫外線を照射すると、蛍光色素FAMが蛍光を発するため、FAM修飾されたDNAオリゴマーはゲル上で白いバンドとして検出される。左から一番目のレーンのバンドは、46塩基からなるDNAオリゴマーであるTS1に、左から二番目及び六番目のレーンのバンドは、それぞれ25塩基及び24塩基からなるDNAオリゴマーであるTT1及びTT2に、それぞれ対応する。左から三番目〜五番目のレーンは、TS3を鋳型としてTT1及びTF3を1、2又は3時間連結反応させて得られたDNAオリゴマーのうち、FAM修飾されたTT1由来のDNAオリゴマーのゲル上の位置を示す。これらのレーンで検出されたメインバンドの位置は、46塩基からなるTS1の位置と近いことから、連結反応混合物中ではTT1及びTF3が連結されて、50塩基からなるDNAオリゴマーが形成されたことが示唆される。 As shown in FIG. 28B, when the polyacrylamide gel is irradiated with ultraviolet rays, the fluorescent dye FAM emits fluorescence, so that the FAM-modified DNA oligomer is detected as a white band on the gel. The first lane band from the left is TS1 which is a DNA oligomer consisting of 46 bases, and the second and sixth lane bands from the left are the DNA oligomers consisting of 25 bases and 24 bases, TT1 and TT2, respectively. , Respectively. The third to fifth lanes from the left are the DNA oligomers obtained by ligation reaction of TT1 and TF3 for 1, 2 or 3 hours using TS3 as a template on the gel of FAM-modified TT1-derived DNA oligomers. Indicates the position. Since the position of the main band detected in these lanes is close to the position of TS1 consisting of 46 bases, TT1 and TF3 were connected in the ligation reaction mixture, and a DNA oligomer consisting of 50 bases was formed. It is suggested.
右から一番目のレーンは、TS3を鋳型としてTT2及びTF3を3時間連結反応させて得られた対照区のDNAオリゴマーのゲル上の位置を示す。このレーンで検出されたメインバンドの位置は、24塩基からなる連結前のDNAオリゴマーであるTT2の位置と近いことから、TT2は、鋳型であるTS3とアニールするもののTF3とは連結されなかったことが示唆される。TT2は、TT1の5’末端が1塩基欠失した塩基配列からなるDNAオリゴマーであるため、TS3を鋳型としてTT2及びTF3をアニールさせると、TT2とTF3との間には1塩基のギャップが生じる(図28A)。このため、対照区の試料では連結反応が進行しなかったと考えられる。 The first lane from the right shows the position on the gel of the DNA oligomer in the control group obtained by ligating TT2 and TF3 for 3 hours using TS3 as a template. Since the position of the main band detected in this lane is close to the position of TT2, which is a DNA oligomer before ligation consisting of 24 bases, TT2 annealed with the template TS3 but was not ligated with TF3. Is suggested. TT2 is a DNA oligomer consisting of a base sequence with one base deleted from the 5 'end of TT1, so when TT2 and TF3 are annealed using TS3 as a template, a 1 base gap is generated between TT2 and TF3. (Figure 28A). For this reason, it is considered that the ligation reaction did not proceed in the control sample.
<4. 合成例2:5-(1-プロピニル)-2’-デオキシウリジンを含むDNAオリゴマー>
非天然型塩基を含むデオキシヌクレオシド(5-(1-プロピニル)-2’-デオキシウリジン)のモノマーユニットであるホスホロアミダイト(非特許文献36〜41)は、グレンリサーチ社より購入した(非特許文献49)。DNA自動合成機を用いて、5-(1-プロピニル)-2’-デオキシウリジンを含むDNAオリゴマーを上記と同じ方法で合成した(非特許文献42〜49)。担体からの切り出しを28%アンモニア水で行い、脱保護は室温(約25℃)で16時間静置することにより行った。遠心濃縮装置を用いてアンモニアを減圧留去し、0.45μmのフィルターに通した。その後、逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。得られたDNAオリゴマーはMALDI TOF-MS測定により同定した。<4. Synthesis Example 2: DNA oligomer containing 5- (1-propynyl) -2′-deoxyuridine>
Phosphoramidite (non-patent documents 36 to 41), which is a monomer unit of deoxynucleoside (5- (1-propynyl) -2'-deoxyuridine) containing an unnatural base, was purchased from Glen Research (non-patent document). Reference 49). Using an automatic DNA synthesizer, a DNA oligomer containing 5- (1-propynyl) -2'-deoxyuridine was synthesized by the same method as described above (
以下において、5-(1-プロピニル)-2’-デオキシウリジン:
を「Pr」で示す。 Is indicated by “Pr”.
合成したDNAオリゴマーの逆相HPLCクロマトグラムを図29〜30に示す。 The reverse phase HPLC chromatogram of the synthesized DNA oligomer is shown in FIGS.
合成したDNAオリゴマーの上記逆相HPLCにおける保持時間は以下の通りである。 The retention time in the reverse phase HPLC of the synthesized DNA oligomer is as follows.
5’-TTTTCTT(Pr)TCCTTTT-3’について10.5分(配列番号14)
5’-TTTTTT(Pr)TTTTTT-3’について10.7分(配列番号15)
合成したDNAオリゴマーのMALDI TOF-MSのデータは以下の通りである。10.5 minutes for 5'-TTTTCTT (Pr) TCCTTTT-3 '(SEQ ID NO: 14)
10.7 minutes for 5'-TTTTTT (Pr) TTTTTT-3 '(SEQ ID NO: 15)
The MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
5’-TTTTCTT(Pr)TCCTTTT-3’について分子式C149H193N33O100P14、[M-H]-の計算値 4478.8、実測値 4479.4(配列番号14)
5’-TTTTTT(Pr)TTTTTT-3’について分子式C132H170N26O89P12、[M-H]-の計算値 3915.6、実測値 3915.8(配列番号15)
<5. 使用例2:5-(1-プロピニル)-2’-デオキシウリジンを含むDNAオリゴマー>
[使用例2-1:5-(1-プロピニル)-2’-デオキシウリジンを含むDNAオリゴマーの切断反応]
スクリューチューブに30μLのDNAオリゴマー水溶液(ストランド濃度 500μM)と30μLのエチレンジアミン(無水)とを加え、水浴中で70℃、3時間又は16時間加熱した。反応混合物を酢酸で中和し、逆相HPLCにより分析し、生成物を分取した(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。分取した生成物は、MALDI TOF-MSにより分子量を測定した(16時間反応後の試料の生成物)。For 5′-TTTTCTT (Pr) TCCTTTT-3 ′, the molecular formula C 149 H 193 N 33 O 100 P 14 , [MH] − calculated value 4478.8, measured value 4479.4 (SEQ ID NO: 14)
For 5′-TTTTTT (Pr) TTTTTT-3 ′, the molecular formula C 132 H 170 N 26 O 89 P 12 , [MH] − calculated value 3915.6, measured value 3915.8 (SEQ ID NO: 15)
<5. Use Example 2: DNA oligomer containing 5- (1-propynyl) -2′-deoxyuridine>
[Use Example 2-1: Cleavage reaction of DNA oligomer containing 5- (1-propynyl) -2′-deoxyuridine]
30 μL of DNA oligomer aqueous solution (
合成した各DNAオリゴマーの反応前後の逆相HPLCクロマトグラムを図31及び33に、生成物1及び他の副生成物のMALDI TOF-MSスペクトルを図32及び34に、それぞれ示す。
Reverse phase HPLC chromatograms of the synthesized DNA oligomers before and after the reaction are shown in FIGS. 31 and 33, and MALDI TOF-MS spectra of the
各DNAオリゴマーにおける生成物1のMALDI TOF-MSのデータは以下の通りである。
The MALDI TOF-MS data of
5’-TTTTCTT(Pr)TCCTTTT-3’について
生成物1:pTCCTTTT 分子式C68H91N16O48P7 FW 2117.3 [M-H]- 2116.3 実測値 2116.4
5’-TTTTTT(Pr)TTTTTT-3’について
生成物1:pTTTTTT 分子式C60H80N12O43P6 FW 1843.2 [M-H]- 1842.2 実測値 1841.9
エチレンジアミンによるDNAオリゴマーの分解反応を下記に示す。下記反応式において、右側の生成物が生成物1に対応する。左側の生成物は、反応時間が長くなったことに起因して、糖残基の脱離又は分子量が64増す反応が生じてさらに別の副生成物を形成したことがMALDI TOF-MSスペクトルより予想される。
5'-TTTTTT (Pr) TTTTTT- 3 ' product for 1: pTTTTTT molecular formula C 60 H 80 N 12 O 43 P 6 FW 1843.2 [MH] - 1842.2 Found 1841.9
Degradation reaction of DNA oligomer by ethylenediamine is shown below. In the following reaction formula, the product on the right side corresponds to
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
<6. 合成例3:5-エチニル-2’-デオキシウリジンを含むDNAオリゴマー>
非天然型塩基を含むデオキシヌクレオシドである5-エチニル-2’-デオキシウリジンは、抗ウイルス活性及び抗癌活性を有する公知の化合物である(非特許文献50〜53)。また、当該デオキシヌクレオシドは、細胞中のDNA標識法にも用いられている(非特許文献54〜63)。細胞培地に前記デオキシヌクレオシドを加えると、該デオキシヌクレオシドは、細胞のDNA中にチミジンと間違われて取り込まれる。このデオキシヌクレオシドは、エチニル基を有するため、クリックケミストリーによってアジド基を有する別の標識分子(例えば蛍光色素など)と容易にカップリングさせることができる。上記の機構により、細胞のDNA中に取り込まれた前記デオキシヌクレオシドを、蛍光色素などの標識により検出することが可能となる。或いは、ラマン分光を利用することにより、上記のような標識反応を行うことなく前記デオキシヌクレオシドを検出することもできる。それ故、生細胞中でも当該デオキシヌクレオシドをモニターすることが可能となる(非特許文献64)。<6. Synthesis Example 3: DNA oligomer containing 5-ethynyl-2′-deoxyuridine>
5-Ethynyl-2′-deoxyuridine, which is a deoxynucleoside containing an unnatural base, is a known compound having antiviral activity and anticancer activity (
5-エチニル-2’-デオキシウリジンの合成は、これまでに多くの方法が報告されている(例えば、非特許文献65〜67)。また、DNA自動合成機を用いて、5-エチニル-2’-デオキシウリジンを含むDNAオリゴマーを合成する方法も、すでに報告されている(非特許文献68及び69)。しかしながら、前記デオキシヌクレオシドを、DNA鎖の切断及び切断された末端のリン酸化に適用した例は存在しない。 Many methods have so far been reported for the synthesis of 5-ethynyl-2'-deoxyuridine (for example, non-patent documents 65 to 67). In addition, a method of synthesizing a DNA oligomer containing 5-ethynyl-2'-deoxyuridine using an automatic DNA synthesizer has already been reported (Non-patent Documents 68 and 69). However, there is no example in which the deoxynucleoside is applied to DNA strand breaks and phosphorylated phosphorylated ends.
5-エチニル-2’-デオキシウリジンのモノマーユニットであるホスホロアミダイトは、非特許文献66及び68に記載の方法に従い合成した。DNA自動合成機を用いて、5-エチニル-2’-デオキシウリジンを含むDNAオリゴマーを合成例1-4と同じ方法で合成した(非特許文献69)。担体からの切り出し及び脱保護を、50 mM K2CO3/MeOH中、室温(約25℃)で16時間静置することにより行った。DNAオリゴマーがアデニン及び/又はグアニンを含む場合、DNAオリゴマーに水を500μL加えた後、遠心濃縮装置を用いてメタノールを減圧留去し、さらに一体積分の28%アンモニア水を加えて撹拌して密閉し、24時間静置することによって、塩基部分を完全に脱保護することが可能である。遠心濃縮装置を用いてメタノールを減圧留去し、1 mLの蒸留水を加えた後に0.45μmのフィルターに通した。その後、逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。得られたDNAオリゴマーはMALDI TOF-MS測定により同定した。The phosphoramidite, which is a monomer unit of 5-ethynyl-2′-deoxyuridine, was synthesized according to the methods described in Non-Patent Documents 66 and 68. Using an automatic DNA synthesizer, a DNA oligomer containing 5-ethynyl-2′-deoxyuridine was synthesized by the same method as in Synthesis Example 1-4 (Non-patent Document 69). Cleavage from the carrier and deprotection were performed by standing in 50 mM K 2 CO 3 / MeOH at room temperature (about 25 ° C.) for 16 hours. If the DNA oligomer contains adenine and / or guanine, 500 μL of water is added to the DNA oligomer, and then methanol is distilled off under reduced pressure using a centrifugal concentrator. However, it is possible to completely deprotect the base moiety by leaving it to stand for 24 hours. Methanol was distilled off under reduced pressure using a centrifugal concentrator, 1 mL of distilled water was added, and then passed through a 0.45 μm filter. Thereafter, the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off. The obtained DNA oligomer was identified by MALDI TOF-MS measurement.
以下において、5-エチニル-2’-デオキシウリジン:
を「Pa」で示す。 Is indicated by “Pa”.
合成したDNAオリゴマーの逆相HPLCクロマトグラムを図35及び36に示す。 The reverse phase HPLC chromatogram of the synthesized DNA oligomer is shown in FIGS.
合成したDNAオリゴマーの上記逆相HPLCにおける保持時間は以下の通りである。 The retention time in the reverse phase HPLC of the synthesized DNA oligomer is as follows.
5’-TTTTCTT(Pa)TCCTTTT-3’について10.4分(配列番号16)
5’-TTTTTT(Pa)TTTTTT-3’について10.7分(配列番号17)
合成したDNAオリゴマーのMALDI TOF-MSのデータは以下の通りである。10.4 minutes for 5'-TTTTCTT (Pa) TCCTTTT-3 '(SEQ ID NO: 16)
10.7 minutes for 5'-TTTTTT (Pa) TTTTTT-3 '(SEQ ID NO: 17)
The MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
5’-TTTTCTT(Pa)TCCTTTT-3’について分子式C148H191N33O100P14、[M-H]-の計算値 4464.9、実測値 4465.3(配列番号16)
5’-TTTTTT(Pa)TTTTTT-3’について分子式C131H168N26O89P12、[M-H]-の計算値 3901.5、実測値 3901.8(配列番号17)
<7. 使用例3:5-エチニル-2’-デオキシウリジンを含むDNAオリゴマー>
[使用例3-1:5-エチニル-2’-デオキシウリジンを含むDNAオリゴマーの切断反応]
スクリューチューブに30μLのDNAオリゴマー水溶液(ストランド濃度 500μM)と30μLのメチルアミン(40% 水溶液)とを加え、水浴中で70℃、10分間又は30分間加熱した。遠心濃縮装置を用いて反応混合物からメチルアミンを減圧留去し、該反応混合物を逆相HPLCにより分析し、生成物を分取した(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。分取した生成物は、MALDI TOF-MSにより分子量を測定した(30分間反応後の試料の生成物)。For 5′-TTTTCTT (Pa) TCCTTTT-3 ′, the molecular formula C 148 H 191 N 33 O 100 P 14 , [MH] − calculated value 4464.9, measured value 4465.3 (SEQ ID NO: 16)
5'-TTTTTT (Pa) molecular formula for TTTTTT-3 'C 131 H 168 N 26 O 89
<7. Use Example 3: DNA oligomer containing 5-ethynyl-2′-deoxyuridine>
[Use Example 3-1: Cleavage reaction of DNA oligomer containing 5-ethynyl-2'-deoxyuridine]
30 μL of DNA oligomer aqueous solution (
合成した各DNAオリゴマーの反応前後の逆相HPLCクロマトグラムを図37及び38に、それぞれ示す。 The reverse phase HPLC chromatogram before and after the reaction of each synthesized DNA oligomer is shown in FIGS. 37 and 38, respectively.
各DNAオリゴマーにおける生成物1のMALDI TOF-MSのデータは以下の通りである。
The MALDI TOF-MS data of
5’-TTTTCTT(Pa)TCCTTTT-3’について
生成物1:pTCCTTTT 分子式C68H91N16O48P7 FW 2117.3 [M-H]- 2116.3 実測値 2116.2
生成物2:TTTTCTTRNu 分子式C75H103N16O51P7 FW 2261.5 [M-H]- 2260.5 実測値 2260.6
5’-TTTTTT(Pa)TTTTTT-3’ について
生成物1:pTTTTTT 分子式C60H80N12O43P6 FW 1843.2 [M-H]- 1842.2 実測値 1841.8
生成物2:TTTTTTRNu 分子式C66H91N13O45P6 FW 1972.3 [M-H]- 1971.3 実測値 1971.3
メチルアミンによるDNAオリゴマーの分解反応を下記に示す。下記反応式において、右側の生成物が生成物1に、左側の生成物が生成物2にそれぞれ対応することがMALDI TOF-MSスペクトルより示された。
Product 2: TTTTCTTR Nu molecular formula C 75 H 103 N 16 O 51 P 7 FW 2261.5 [MH] - 2260.5 Found 2260.6
5'-TTTTTT (Pa) TTTTTT- 3 ' product for 1: pTTTTTT molecular formula C 60 H 80 N 12 O 43 P 6 FW 1843.2 [MH] - 1842.2 Found 1841.8
Product 2: TTTTTTR Nu molecular formula C 66 H 91 N 13 O 45 P 6 FW 1972.3 [MH] - 1971.3 Found 1971.3
Degradation reaction of DNA oligomer by methylamine is shown below. In the following reaction formula, it was shown from the MALDI TOF-MS spectrum that the product on the right side corresponds to
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
上記のように、5-エチニル-2’-デオキシウリジンを含むDNAオリゴマーは、70℃で30分間加熱することにより切断反応が略完結した。この結果から、5-エチニル-2’-デオキシウリジンを含むDNAオリゴマーは、5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマー(使用例1)及び5-(1-プロピニル)-2’-デオキシウリジンを含むDNAオリゴマー(使用例2)と比較して極めて迅速に切断反応が進行することが明らかとなった。 As described above, the cleavage reaction of the DNA oligomer containing 5-ethynyl-2'-deoxyuridine was almost completed by heating at 70 ° C for 30 minutes. From this result, a DNA oligomer containing 5-ethynyl-2′-deoxyuridine is a DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine (Use Example 1) and 5- (1-propynyl) -2′- It was revealed that the cleavage reaction proceeded very rapidly as compared with a DNA oligomer containing deoxyuridine (Use Example 2).
[使用例3-2:メチルアミン又はエチレンジアミンによる5-エチニル-2’-デオキシウリジン(Pa)を含むDNAオリゴマーの25℃における切断反応速度の比較]
スクリューチューブに、200μLの5’-T6(Pa)T6-3’の水溶液(ストランド濃度 100μM)と200μLのメチルアミン又はエチレンジアミン(40%水溶液)とを加え、よく撹拌し、25℃で40時間静置した。メチルアミンによる反応の場合、反応後、反応溶液を加熱せずに遠心濃縮機で濃縮し、該反応溶液からメチルアミンを減圧留去した。エチレンジアミンによる反応の場合、酢酸で反応溶液を中和した。得られたそれぞれの反応混合物を、逆相HPLCにより分析した(8-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。反応溶液のHPLCチャートを図39に示す。[Use Example 3-2: Comparison of cleavage reaction rates of DNA oligomers containing 5-ethynyl-2′-deoxyuridine (Pa) with methylamine or ethylenediamine at 25 ° C.]
Add 200 μL of 5′-T 6 (Pa) T 6 -3 ′ aqueous solution (
図39に示すように、メチルアミンによる反応の場合、保持時間8.2分及び10.3分付近に(図39A)、エチレンジアミンによる反応の場合、保持時間8.1分及び10.3分付近に(図39B)、それぞれピークが検出された。前記使用例3-1と同様のMALDI TOF-MSスペクトル測定により、これらのピークが5’-T6(Pa)T6-3’の切断に由来する生成物に対応することが確認された。以上の結果から、25℃においてもDNAオリゴマーの切断が進行することが確認された。As shown in FIG. 39, in the case of reaction with methylamine, the retention time was around 8.2 minutes and 10.3 minutes (FIG. 39A), and in the case of reaction with ethylenediamine, the retention time was around 8.1 minutes and 10.3 minutes (FIG. 39B), respectively. Was detected. The same MALDI TOF-MS spectrum measurement as in Use Example 3-1 confirmed that these peaks correspond to products derived from cleavage of 5′-T 6 (Pa) T 6 -3 ′. From the above results, it was confirmed that the cleavage of the DNA oligomer proceeds even at 25 ° C.
[使用例3-3:炭酸カリウム水溶液中におけるPaを含むDNAオリゴマーの切断反応と生成物の同定]
スクリューチューブに、100μLの5’-T6PaT6-3’の水溶液(ストランド濃度 400μM)と100μLの炭酸カリウム水溶液(1 M)とを加え、乾燥機で120℃、4時間加熱した。反応溶液を酢酸で中和した。得られた反応混合物を、逆相HPLCにより分析し、生成物を分取した(5-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。DNAオリゴマーの切断反応前後のHPLCチャートを図40に示す。[Use Example 3-3: Cleavage reaction of Pa-containing DNA oligomer in potassium carbonate aqueous solution and product identification]
100 μL of 5′-T 6 PaT 6 -3 ′ aqueous solution (
図40に示すように、前記の切断反応により、生成物1及び2が生成した。分取した生成物1及び2は、MALDI TOF-MSにより分子量を測定した。生成物1及び2のMALDI TOF-MSのデータは以下の通りである。
As shown in FIG. 40,
生成物1:pTTTTTT 分子式C60H80N12O43P6 FW 1843.2 [M-H]- 1842.2 実測値 1842.2
HPLCの保持時間 12.1分
生成物2:TTTTTTp 分子式C60H80N12O43P6 FW 1843.2 [M-H]- 1842.2 実測値 1842.0
HPLCの保持時間 12.8分
炭酸カリウムによるDNAオリゴマーの分解反応を下記に示す。下記反応式において、右側の生成物が生成物1に、左側の生成物が生成物2にそれぞれ対応することがMALDI TOF-MSスペクトルより示された。
HPLC retention time 12.1 min product 2: TTTTTTp molecular formula C 60 H 80 N 12 O 43 P 6 FW 1843.2 [MH] - 1842.2 Found 1842.0
HPLC retention time 12.8 minutes Degradation reaction of DNA oligomer with potassium carbonate is shown below. In the following reaction formula, it was shown from the MALDI TOF-MS spectrum that the product on the right side corresponds to
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
生成物1は、メチルアミンによる分解生成物との重ね打ちによるHPLC分析により、同一の保持時間を有することが確認された。本反応の求核種は、水酸化物イオンと考えられる。本反応では、非特許文献13に示されたアベーシックサイトを有するDNAの水中における分解反応と、同様の反応が起きたものと予想される。シトシンを含むDNAオリゴマーの反応では、生成物のピークが2つよりも多くなった。この結果から、アルカリ条件下で加熱することにより、シトシンのデアミネーションが起きることが示唆される。
<8. 合成例4:5-[(E)-2-カルバモイルビニル]-2’-デオキシウリジンを含むDNAオリゴマー>
[合成例4-1:5’-O-ジメトキシトリチル-5-[(E)-2-カルボメトキシビニル]-2’-デオキシウリジンの合成]
非特許文献73に従って合成した767 mg(2.46 mmol)の5-[(E)-2-カルボメトキシビニル]-2’-デオキシウリジン(分子量312.28)と1.05 g(3.10 mmol)のDMTrCl(分子量338.83)とをナスフラスコに入れ、そこへ20 mLのピリジンを加え、室温で1時間撹拌した。約2 mLのメタノールを加え、反応を停止させた後、溶媒を減圧留去した。シリカゲルカラムで目的物質を精製した(展開溶媒は2〜5% MeOH, 2% Et3N/CH2Cl2)。目的のフラクションの溶媒を減圧留去し、真空ポンプで乾燥した。少量の塩化メチレンに溶かし、ヘキサンを加え、溶媒を減圧留去した後、真空ポンプで乾燥することにより、1.49 g(1.94 mmol, 収率79%)の目的物質(分子量766.43トリエチルアミン1.5分子を含む)を白色粉末として得た。<8. Synthesis Example 4: DNA oligomer containing 5-[(E) -2-carbamoylvinyl] -2'-deoxyuridine>
[Synthesis Example 4-1: Synthesis of 5′-O-dimethoxytrityl-5-[(E) -2-carbomethoxyvinyl] -2′-deoxyuridine]
767 mg (2.46 mmol) 5-[(E) -2-carbomethoxyvinyl] -2'-deoxyuridine (molecular weight 312.28) and 1.05 g (3.10 mmol) DMTrCl (molecular weight 338.83) synthesized according to Non-Patent Document 73 Were placed in an eggplant flask, 20 mL of pyridine was added thereto, and the mixture was stirred at room temperature for 1 hour. About 2 mL of methanol was added to stop the reaction, and the solvent was distilled off under reduced pressure. The target substance was purified by a silica gel column (developing solvent was 2-5% MeOH, 2% Et 3 N / CH 2 Cl 2 ). The solvent of the objective fraction was distilled off under reduced pressure and dried with a vacuum pump. Dissolve in a small amount of methylene chloride, add hexane, distill off the solvent under reduced pressure, and dry with a vacuum pump to obtain 1.49 g (1.94 mmol, yield 79%) of the target substance (including 1.5 molecules of molecular weight 766.43 triethylamine) Was obtained as a white powder.
1H NMR(400 MHz, DMSO-d6) δ11.70 (s, 1H), 8.01 (s, 1H), 7.39-7.36 (m, 2H),7.29-7.15 (m, 8H), 6.86-6.81 (m, 5H), 6.15 (t, J = 6.4 Hz, 1H), 5.34 (d, J= 5.0Hz, 1H), 4.23-4.20 (m, 1H), 3.93-3.88 (m, 1H),3.70 (s, 1H), 3.69 (s, 1H), 3.63 (s, 1H), 3.26-3.12 (m, 2H),3.07-2.98 (m, 9H), 2.36-2.28 (m, 1H), 2.24-2.16 (m, 1H), 1.20-1.15 (m, 13.5 H); HRMS (ESI): C34H34N2O9Naに対する計算値: 637.2157 [M+Na]+; 実測値: 637.2162.
[合成例4-2:5’-O-ジメトキシトリチル-5-[(E)-2-カルボメトキシビニル]-2’-デオキシウリジン-3’-N,N-ジイソプロピル(シアノエチル) ホスホロアミダイトの合成]
300 mg(0.391 mmol)の5’-O-ジメトキシトリチル-5-[(E)-2-カルボメトキシビニル]-2’-デオキシウリジン(分子量766.43;トリエチルアミン1.5分子を含む)をナスフラスコに入れ、5 mLのCH3CNと261μL(1.50 mmol)のN,N-ジイソプロピルエチルアミン(分子量129.25, 比重0.742 g/mL)とを加え、撹拌し、さらに223μL(1.00 mmol)の2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロアミダイト(分子量236.68, 比重1.061 g/mL)を加え、室温で30分間撹拌した。25 mLのAcOEtと25 mLの飽和食塩水とを加え、分液し、有機層をさらに2回飽和食塩水で洗った。有機層を無水硫酸マグネシウムで乾燥した後、フィルターでろ過し、ろ液の溶媒を減圧留去した。アセトニトリルで3回共沸した後、10 mLのアセトニトリルを加えた。溶液を0.45μmのフィルターに通し、少量のモレキュラーシーブを加えた。DNA自動合成機に取り付けてDNA合成に使用した。 1 H NMR (400 MHz, DMSO-d6) δ11.70 (s, 1H), 8.01 (s, 1H), 7.39-7.36 (m, 2H), 7.29-7.15 (m, 8H), 6.86-6.81 (m , 5H), 6.15 (t, J = 6.4 Hz, 1H), 5.34 (d, J = 5.0Hz, 1H), 4.23-4.20 (m, 1H), 3.93-3.88 (m, 1H), 3.70 (s, 1H), 3.69 (s, 1H), 3.63 (s, 1H), 3.26-3.12 (m, 2H), 3.07-2.98 (m, 9H), 2.36-2.28 (m, 1H), 2.24-2.16 (m, 1H), 1.20-1.15 (m, 13.5 H); HRMS (ESI): Calculated for C 34 H 34 N 2 O 9 Na: 637.2157 [M + Na] + ; Found: 637.2162.
[Synthesis Example 4-2: 5′-O-dimethoxytrityl-5-[(E) -2-carbomethoxyvinyl] -2′-deoxyuridine-3′-N, N-diisopropyl (cyanoethyl) phosphoramidite Composition]
300 mg (0.391 mmol) of 5′-O-dimethoxytrityl-5-[(E) -2-carbomethoxyvinyl] -2′-deoxyuridine (molecular weight 766.43; containing 1.5 molecules of triethylamine) was placed in an eggplant flask, 5 mL of CH 3 CN and 261 μL (1.50 mmol) of N, N-diisopropylethylamine (molecular weight 129.25, specific gravity 0.742 g / mL) were added, stirred, and further 223 μL (1.00 mmol) of 2-cyanoethyl N, N, N ′, N′-tetraisopropyl phosphoramidite (molecular weight 236.68, specific gravity 1.061 g / mL) was added, and the mixture was stirred at room temperature for 30 minutes. 25 mL of AcOEt and 25 mL of saturated saline were added and separated, and the organic layer was further washed twice with saturated saline. The organic layer was dried over anhydrous magnesium sulfate and filtered through a filter, and the solvent of the filtrate was distilled off under reduced pressure. After azeotroping with acetonitrile three times, 10 mL of acetonitrile was added. The solution was passed through a 0.45 μm filter and a small amount of molecular sieve was added. Attached to an automatic DNA synthesizer and used for DNA synthesis
[合成例4-3:5-[(E)-2-カルバモイルビニル]-2’-デオキシウリジンを含むDNAオリゴマーの合成]
DNA自動合成機を用いて、5-[(E)-2-カルバモイルビニル]-2’-デオキシウリジンを含むDNAオリゴマーを上記と同じ方法で合成した(非特許文献70〜72)。担体からの切り出しを28%アンモニア水で行い、脱保護は室温(約25℃)で16時間静置することにより行った。塩基部分のカルボメトキシビニル基は脱保護条件でカルバモイルビニル基に変換される。遠心濃縮装置を用いてアンモニアを減圧留去し、0.45μmのフィルターに通した。その後、逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。得られたDNAオリゴマーはMALDI TOF-MS測定により同定した。[Synthesis Example 4-3: Synthesis of DNA oligomer containing 5-[(E) -2-carbamoylvinyl] -2'-deoxyuridine]
Using an automatic DNA synthesizer, a DNA oligomer containing 5-[(E) -2-carbamoylvinyl] -2′-deoxyuridine was synthesized by the same method as described above (Non-patent Documents 70 to 72). Cleavage from the carrier was performed with 28% aqueous ammonia, and deprotection was performed by standing at room temperature (about 25 ° C.) for 16 hours. The carbomethoxyvinyl group in the base moiety is converted to a carbamoylvinyl group under deprotection conditions. Ammonia was distilled off under reduced pressure using a centrifugal concentrator and passed through a 0.45 μm filter. Thereafter, the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off. The obtained DNA oligomer was identified by MALDI TOF-MS measurement.
以下において、5-[(E)-2-カルバモイルビニル]-2’-デオキシウリジン:
を「CvU」で示す。 Is indicated by “CvU”.
合成したDNAオリゴマーの逆相HPLCクロマトグラムを図41に示す。 FIG. 41 shows a reverse phase HPLC chromatogram of the synthesized DNA oligomer.
合成したDNAオリゴマーの上記逆相HPLCにおける保持時間は以下の通りである。 The retention time in the reverse phase HPLC of the synthesized DNA oligomer is as follows.
5’-TTTTCTT(CvU)TCCTTTT-3’について10.5分(配列番号18)
5’-TTTTTT(CvU)TTTTTT-3’について10.6分(配列番号19)
合成したDNAオリゴマーのMALDI TOF-MSのデータは以下の通りである。10.5 minutes for 5'-TTTTCTT (CvU) TCCTTTT-3 '(SEQ ID NO: 18)
10.6 minutes for 5'-TTTTTT (CvU) TTTTTT-3 '(SEQ ID NO: 19)
The MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
5’-TTTTCTT(CvU)TCCTTTT-3’について分子式C149H194N34O101P14、[M-H]-の計算値 4509.9、実測値 4510.0(配列番号18)
5’-TTTTTT(CvU)TTTTTT-3’について分子式C132H171N27O90P12、[M-H]-の計算値 3946.6、実測値 3946.6(配列番号19)
<9. 使用例4:5-[(E)-2-カルバモイルビニル]-2’-デオキシウリジンを含むDNAオリゴマー>
[使用例4-1:5-[(E)-2-カルバモイルビニル]-2’-デオキシウリジンを含むDNAオリゴマーの切断反応]
スクリューチューブに30μLのDNAオリゴマー水溶液(ストランド濃度 500μM)と30μLのエチレンジアミン(無水)とを加え、水浴中で70℃、3時間又は16時間加熱した。反応混合物を酢酸で中和し、逆相HPLCにより分析し、生成物を分取した(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。分取した生成物は、MALDI TOF-MSにより分子量を測定した(8時間反応後の試料の生成物)。For 5′-TTTTCTT (CvU) TCCTTTT-3 ′, the molecular formula C 149 H 194 N 34 O 101 P 14 , [MH] − calculated value 4509.9, actual value 4510.0 (SEQ ID NO: 18)
For 5′-TTTTTT (CvU) TTTTTT-3 ′, the molecular formula C 132 H 171 N 27 O 90 P 12 , [MH] − calculated value 3946.6, measured value 3946.6 (SEQ ID NO: 19)
<9. Use Example 4: DNA oligomer containing 5-[(E) -2-carbamoylvinyl] -2'-deoxyuridine>
[Use Example 4-1: Cleavage reaction of DNA oligomer containing 5-[(E) -2-carbamoylvinyl] -2'-deoxyuridine]
30 μL of DNA oligomer aqueous solution (
合成した各DNAオリゴマーの反応前後の逆相HPLCクロマトグラムを図42及び44に、生成物1及び他の副生成物のMALDI TOF-MSスペクトルを図43及び45に、それぞれ示す。
42 and 44 show reverse phase HPLC chromatograms of the synthesized DNA oligomers before and after the reaction, and FIGS. 43 and 45 show MALDI TOF-MS spectra of the
各DNAオリゴマーにおける生成物1のMALDI TOF-MSのデータは以下の通りである。
The MALDI TOF-MS data of
5’-TTTTCTT(CvU)TCCTTTT-3’について
生成物1:pTCCTTTT 分子式C68H91N16O48P7 FW 2117.3 [M-H]- 2116.3 実測値 2116.2
5’-TTTTTT(CvU)TTTTTT-3’について
生成物1:pTTTTTT 分子式C60H80N12O43P6 FW 1843.2 [M-H]- 1842.2 実測値 1841.9
エチレンジアミンによるDNAオリゴマーの分解反応を下記に示す。下記反応式において、右側の生成物がHPLCクロマトグラム上の小さなピークである生成物1に対応する。反応後の分取HPLCにおいて原料のDNAオリゴマーと同じリテンションタイムに溶出されるピークを含むフラクションをMALDI TOF-MS測定すると、MALDI TOF-MSスペクトル上に原料+60に相当するピークが検出された。この結果から、反応後の前記フラクションには、原料のDNAオリゴマーにエチレンジアミンが一分子付加した化合物が多く含まれていることが示唆された。それ故、DNAオリゴマーの分解反応とは別の反応が同時に起きたことが予想される。
5'-TTTTTT (CvU) TTTTTT- 3 ' product for 1: pTTTTTT molecular formula C 60 H 80 N 12 O 43 P 6 FW 1843.2 [MH] - 1842.2 Found 1841.9
Degradation reaction of DNA oligomer by ethylenediamine is shown below. In the following reaction formula, the product on the right side corresponds to
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
<10. 合成例5:N6-(3-プロピニル)-5-メチル-2’-デオキシシチジンを含むDNAオリゴマー>
[合成例5-1:N6-(3-プロピニル)-5-メチル-2’-デオキシシチジンの合成]
1.21 g(5.00 mmol)のチミジン(分子量242.23)を20 mLのアセトニトリルに懸濁し、そこへ5.58 mL(40.0 mmol)のトリエチルアミン(分子量101.19, 比重0.73 g/mL)と1.89 mL(20.0 mmol)の無水酢酸(分子量102.09, 比重1.08 g/mL)とを加え、25℃で21時間撹拌した。溶媒を減圧留去し、100 mLの酢酸エチルと100 mLの飽和食塩水とを加えて分液した。有機層を100 mLの飽和食塩水で2回洗浄した後、有機層を硫酸マグネシウムで乾燥した。乾燥後の有機層をろ過し、ろ液の溶媒を減圧留去して、真空ポンプにより乾燥した。3.45 g(50.0 mmol)の1,2,4-トリアゾール(分子量69.07)を加え、30 mLのアセトニトリルを加え、7.67 mL(55.0 mmol)のトリエチルアミン(分子量101.19, 比重0.73 g/mL)を加えた。混合物をよく撹拌しながら932μL(10.0 mmol)の塩化ホスホリル(分子量153.33, 比重1.645 g/mL)を加えた。25℃で1時間撹拌した。溶媒を減圧留去し、100 mLの酢酸エチルと100 mLの飽和食塩水とを加えて分液した。有機層を100 mLの飽和食塩水で2回洗浄した後、有機層を硫酸マグネシウムで乾燥した。乾燥後の有機層をろ過し、ろ液の溶媒を減圧留去して、真空ポンプにより乾燥した。約30 mLの1,4-ジオキサンを加え、溶解させた後、961μL(15.0 mmol)のプロパルギルアミン(分子量55.08, 比重0.86)を加え、25℃で2時間撹拌した。溶媒を減圧留去し、30 mLのメタノールと30 mLの28%アンモニア水とを加えて25℃で24時間撹拌した。溶媒を減圧留去し、シリカゲルカラムで目的生成物を精製した(10-15% MeOH/CH2Cl2)。873 mg(3.13 mmol, 収率63%)の目的物質(分子量279.29)を白色粉末として得た。<10. Synthesis Example 5: DNA oligomer containing N6- (3-propynyl) -5-methyl-2'-deoxycytidine>
[Synthesis Example 5-1: Synthesis of N6- (3-propynyl) -5-methyl-2′-deoxycytidine]
1.21 g (5.00 mmol) thymidine (molecular weight 242.23) was suspended in 20 mL acetonitrile, and 5.58 mL (40.0 mmol) triethylamine (molecular weight 101.19, specific gravity 0.73 g / mL) and 1.89 mL (20.0 mmol) anhydrous Acetic acid (molecular weight 102.09, specific gravity 1.08 g / mL) was added, and the mixture was stirred at 25 ° C. for 21 hours. The solvent was distilled off under reduced pressure, and 100 mL of ethyl acetate and 100 mL of saturated brine were added for liquid separation. The organic layer was washed twice with 100 mL of saturated brine, and then the organic layer was dried over magnesium sulfate. The organic layer after drying was filtered, and the solvent of the filtrate was distilled off under reduced pressure, followed by drying with a vacuum pump. 3.45 g (50.0 mmol) of 1,2,4-triazole (molecular weight 69.07) was added, 30 mL of acetonitrile was added, and 7.67 mL (55.0 mmol) of triethylamine (molecular weight 101.19, specific gravity 0.73 g / mL) was added. 932 μL (10.0 mmol) of phosphoryl chloride (molecular weight 153.33, specific gravity 1.645 g / mL) was added while stirring the mixture well. Stir at 25 ° C. for 1 hour. The solvent was distilled off under reduced pressure, and 100 mL of ethyl acetate and 100 mL of saturated brine were added for liquid separation. The organic layer was washed twice with 100 mL of saturated brine, and then the organic layer was dried over magnesium sulfate. The organic layer after drying was filtered, and the solvent of the filtrate was distilled off under reduced pressure, followed by drying with a vacuum pump. About 30 mL of 1,4-dioxane was added and dissolved, and then 961 μL (15.0 mmol) of propargylamine (molecular weight 55.08, specific gravity 0.86) was added, and the mixture was stirred at 25 ° C. for 2 hours. The solvent was distilled off under reduced pressure, 30 mL of methanol and 30 mL of 28% aqueous ammonia were added, and the mixture was stirred at 25 ° C. for 24 hours. The solvent was removed under reduced pressure and the desired product was purified on a silica gel column (10-15% MeOH / CH 2 Cl 2 ). 873 mg (3.13 mmol, yield 63%) of the target substance (molecular weight 279.29) was obtained as a white powder.
1H NMR(400 MHz, DMSO-d6) δ7.65 (d, J= 0.9Hz, 1H), 7.57 (t, J= 5.7Hz, 1H), 5.19 (d, J= 4.6Hz, 1H), 5.01 (t, J= 5.3Hz, 1H), 4.23-4.18 (m, 1H), 4.14-4.01 (m, 2H),3.76-3.74 (m, 1H),3.62-3.50 (m, 2H), 3.04 (t, J = 2.5 Hz, 1H),2.12-2.05 (m, 1H),1.99-1.92 (m, 1H),1.84 (s, 3H); HRMS (ESI): C13H17N3O4Naに対する計算値: 302.1116 [M+Na]+; 実測値: 302.1117.
[合成例5-2:5’-O-ジメトキシトリチル- N6-(3-プロピニル)-5-メチル-2’-デオキシシチジンの合成]
312 mg(1.12 mmol)のN6-(3-プロピニル)-5-メチル-2’-デオキシシチジン(分子量279.29)と454 mg(1.34 mmol)のDMTrCl(分子量338.83)とをナスフラスコに入れ、そこへ10 mLのピリジンを加え、室温で1時間撹拌した。約2 mLのメタノールを加え、反応を停止させ、溶媒を減圧留去した。シリカゲルカラムで目的物質を精製した(展開溶媒は2〜5% MeOH, 2% Et3N/CH2Cl2)。目的のフラクションの溶媒を減圧留去し、真空ポンプで乾燥した。少量の塩化メチレンに溶かし、ヘキサンを加え、溶媒を減圧留去し、真空ポンプで乾燥することにより、631 mg(0.924 mmol, 収率83%)の目的物質(分子量682.85;トリエチルアミン一分子を含む)を白色粉末として得た。 1 H NMR (400 MHz, DMSO-d6) δ7.65 (d, J = 0.9Hz, 1H), 7.57 (t, J = 5.7Hz, 1H), 5.19 (d, J = 4.6Hz, 1H), 5.01 (t, J = 5.3Hz, 1H), 4.23-4.18 (m, 1H), 4.14-4.01 (m, 2H), 3.76-3.74 (m, 1H), 3.62-3.50 (m, 2H), 3.04 (t , J = 2.5 Hz, 1H), 2.12-2.05 (m, 1H), 1.99-1.92 (m, 1H), 1.84 (s, 3H); HRMS (ESI): calculation for C 13 H 17 N 3 O 4 Na Value: 302.1116 [M + Na] + ; Found: 302.1117.
[Synthesis Example 5-2: Synthesis of 5′-O-dimethoxytrityl-N6- (3-propynyl) -5-methyl-2′-deoxycytidine]
312 mg (1.12 mmol) N6- (3-propynyl) -5-methyl-2'-deoxycytidine (molecular weight 279.29) and 454 mg (1.34 mmol) DMTrCl (molecular weight 338.83) were placed in an eggplant flask. 10 mL of pyridine was added and stirred at room temperature for 1 hour. About 2 mL of methanol was added to stop the reaction, and the solvent was distilled off under reduced pressure. The target substance was purified by a silica gel column (developing solvent was 2-5% MeOH, 2% Et 3 N / CH 2 Cl 2 ). The solvent of the objective fraction was distilled off under reduced pressure and dried with a vacuum pump. Dissolve in a small amount of methylene chloride, add hexane, evaporate the solvent under reduced pressure, and dry with a vacuum pump to obtain 631 mg (0.924 mmol, 83% yield) of the target substance (molecular weight 682.85; including one triethylamine molecule) Was obtained as a white powder.
1H NMR(400 MHz, DMSO-d6) δ7.63 (t, J= 5.7Hz, 1H), 7.51 (s, 1H), 7.39-7.35 (m, 2H),7.32-7.19 (m, 7H),6.90-6.86 (m, 4H),6.21 (t, J = 6.6 Hz, 1H), 5.34 (d, J= 4.1Hz, 1H), 4.31-4.25 (m, 1H), 4.14-4.02 (m, 2H),3.90-3.86 (m, 1H),3.72 (s, 6H), 3.22-3.16 (m, 2H),3.09-3.00 (m, 6H),2.21-2.06 (m, 2H),1.50 (s, 3H), 1.23-1.15 (m, 9H); HRMS (ESI): C34H35N3O6Naに対する計算値: 604.2525 [M+Na]+;実測値: 604.2424.
[合成例5-3:5’-O-ジメトキシトリチル-N6-(3-プロピニル)-5-メチル-2’-デオキシシチジン-3’-N,N-ジイソプロピル(シアノエチル)ホスホロアミダイトの合成]
291 mg(0.426 mmol)の5’-O-ジメトキシトリチル-N6-(3-プロピニル)-5-メチル-2’-デオキシシチジン (分子量682.85;トリエチルアミン一分子を含む)をナスフラスコに入れ、5 mLのCH3CNと261μL(1.50 mmol)のN,N-ジイソプロピルエチルアミン(分子量129.25, 比重0.742 g/mL)とを加え、撹拌し、さらに223μL(1.00 mmol)の2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロアミダイト(分子量236.68, 比重1.061 g/mL)を加え、室温で30分間撹拌した。25 mLのAcOEtと25 mLの飽和食塩水とを加え、分液し、有機層をさらに2回飽和食塩水で洗った。有機層を無水硫酸マグネシウムで乾燥した後、フィルターでろ過し、ろ液の溶媒を減圧留去した。アセトニトリルで3回共沸した後、10 mLのアセトニトリルを加えた。溶液を0.45μmのフィルターに通し、少量のモレキュラーシーブを加えた。DNA自動合成機に取り付けてDNA合成に使用した。 1 H NMR (400 MHz, DMSO-d6) δ7.63 (t, J = 5.7Hz, 1H), 7.51 (s, 1H), 7.39-7.35 (m, 2H), 7.32-7.19 (m, 7H), 6.90-6.86 (m, 4H), 6.21 (t, J = 6.6 Hz, 1H), 5.34 (d, J = 4.1Hz, 1H), 4.31-4.25 (m, 1H), 4.14-4.02 (m, 2H) , 3.90-3.86 (m, 1H), 3.72 (s, 6H), 3.22-3.16 (m, 2H), 3.09-3.00 (m, 6H), 2.21-2.06 (m, 2H), 1.50 (s, 3H) , 1.23-1.15 (m, 9H); HRMS (ESI): Calculated for C 34 H 35 N 3 O 6 Na: 604.2525 [M + Na] + ; Found: 604.2424.
[Synthesis Example 5-3: Synthesis of 5′-O-dimethoxytrityl-N6- (3-propynyl) -5-methyl-2′-deoxycytidine-3′-N, N-diisopropyl (cyanoethyl) phosphoramidite]
291 mg (0.426 mmol) of 5′-O-dimethoxytrityl-N6- (3-propynyl) -5-methyl-2′-deoxycytidine (molecular weight 682.85; containing one molecule of triethylamine) was placed in an eggplant flask and 5 mL Of CH 3 CN and 261 μL (1.50 mmol) of N, N-diisopropylethylamine (molecular weight: 129.25, specific gravity: 0.742 g / mL), stirred, and further 223 μL (1.00 mmol) of 2-cyanoethyl N, N, N ′ , N'-tetraisopropyl phosphoramidite (molecular weight 236.68, specific gravity 1.061 g / mL) was added, and the mixture was stirred at room temperature for 30 minutes. 25 mL of AcOEt and 25 mL of saturated saline were added and separated, and the organic layer was further washed twice with saturated saline. The organic layer was dried over anhydrous magnesium sulfate and filtered through a filter, and the solvent of the filtrate was distilled off under reduced pressure. After azeotroping with acetonitrile three times, 10 mL of acetonitrile was added. The solution was passed through a 0.45 μm filter and a small amount of molecular sieve was added. Attached to an automatic DNA synthesizer and used for DNA synthesis
[合成例5-4:N6-(3-プロピニル)-5-メチル-2’-デオキシシチジンを含むDNAオリゴマーの合成]
DNA自動合成機を用いて、N6-(3-プロピニル)-5-メチル-2’-デオキシシチジンを含むDNAオリゴマーを上記と同じ方法で合成した。担体からの切り出しを28 %アンモニア水で行い、脱保護は室温(約25℃)で16時間静置することにより行った。遠心濃縮装置を用いてアンモニアを減圧留去し、0.45μmのフィルターに通した。その後、逆相HPLCによりメインピークを含むフラクションを分取し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置及び凍結乾燥機を用いてギ酸アンモニウム及び溶媒を留去した。得られたDNAオリゴマーはMALDI TOF-MS測定により同定した。[Synthesis Example 5-4: Synthesis of DNA oligomer containing N6- (3-propynyl) -5-methyl-2′-deoxycytidine]
Using an automatic DNA synthesizer, a DNA oligomer containing N6- (3-propynyl) -5-methyl-2'-deoxycytidine was synthesized by the same method as described above. Cutting out from the carrier was performed with 28% aqueous ammonia, and deprotection was performed by standing at room temperature (about 25 ° C.) for 16 hours. Ammonia was distilled off under reduced pressure using a centrifugal concentrator and passed through a 0.45 μm filter. Thereafter, the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off. The obtained DNA oligomer was identified by MALDI TOF-MS measurement.
以下において、N6-(3-プロピニル)-5-メチル-2’-デオキシシチジン:
を「PC」で示す。 Is indicated by “PC”.
合成したDNAオリゴマーの逆相HPLCクロマトグラムを図46に示す。 FIG. 46 shows a reverse phase HPLC chromatogram of the synthesized DNA oligomer.
合成したDNAオリゴマーの上記逆相HPLCにおける保持時間は以下の通りである。 The retention time in the reverse phase HPLC of the synthesized DNA oligomer is as follows.
5’-TTTTCTT(PC)TCCTTTT-3’について10.6分(配列番号20)
5’-TTTTTT(PC)TTTTTT-3’について10.9分(配列番号21)
合成したDNAオリゴマーのMALDI TOF-MSのデータは以下の通りである。10.6 minutes for 5'-TTTTCTT (PC) TCCTTTT-3 '(SEQ ID NO: 20)
10.9 minutes for 5'-TTTTTT (PC) TTTTTT-3 '(SEQ ID NO: 21)
The MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
5’-TTTTCTT(PC)TCCTTTT-3’について分子式C150H196N34O99P14、[M-H]-の計算値 4492.0、実測値 4491.9(配列番号20)
5’-TTTTTT(PC)TTTTTT-3’について分子式C133H173N27O88P12、[M-H]-の計算値 3928.6、実測値 3928.8(配列番号21)
<11. 使用例5:N6-(3-プロピニル)-5-メチル-2’-デオキシシチジンを含むDNAオリゴマー>
[使用例5-1:N6-(3-プロピニル)-5-メチル-2’-デオキシシチジンを含むDNAオリゴマーの切断反応]
スクリューチューブに30μLのDNAオリゴマー水溶液(ストランド濃度 500μM)と30μLのエチレンジアミン(無水)とを加え、水浴中で70℃、3時間又は16時間加熱した。反応混合物を酢酸で中和し、逆相HPLCにより分析し、生成物を分取した(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。分取した生成物は、MALDI TOF-MSにより分子量を測定した(16時間反応後の試料の生成物)。5'-TTTTCTT (PC) TCCTTTT- 3 ' molecular formula C 150 H 196 N 34 O 99 P 14 about, [MH] - calcd 4492.0, found 4491.9 (SEQ ID NO: 20)
5'-TTTTTT (PC) TTTTTT- 3 molecular formula C 133 for 'H 173 N 27 O 88 P 12, [MH] - Calculated 3928.6, Found 3928.8 (SEQ ID NO: 21)
<11. Use Example 5: DNA oligomer containing N6- (3-propynyl) -5-methyl-2'-deoxycytidine>
[Use Example 5-1: Cleavage reaction of a DNA oligomer containing N6- (3-propynyl) -5-methyl-2'-deoxycytidine]
30 μL of DNA oligomer aqueous solution (
合成した各DNAオリゴマーの反応前後の逆相HPLCクロマトグラムを図47及び49に、生成物1及び他の副生成物のMALDI TOF-MSスペクトルを図48及び50に、それぞれ示す。
47 and 49 show the reverse phase HPLC chromatograms of the synthesized DNA oligomers before and after the reaction, and FIGS. 48 and 50 show the MALDI TOF-MS spectra of the
各DNAオリゴマーにおける生成物1のMALDI TOF-MSのデータは以下の通りである。
The MALDI TOF-MS data of
5’-TTTTCTT(PC)TCCTTTT-3’について
生成物1:pTCCTTTT 分子式C68H91N16O48P7 FW 2117.3 [M-H]- 2116.3 実測値 2116.5
5’-TTTTTT(PC)TTTTTT-3’について
生成物1:pTTTTTT 分子式C60H80N12O43P6 FW 1843.2 [M-H]- 1842.2 実測値 1841.8
エチレンジアミンによるDNAオリゴマーの分解反応を下記に示す。下記反応式において、右側の生成物がHPLCクロマトグラム上の小さなピークである生成物1に対応する。反応後の分取HPLCにおいて原料のDNAオリゴマーと同じリテンションタイムに溶出されるピークを含むフラクションをMALDI TOF-MS測定したところ、原料のDNAオリゴマーが多く含まれていることが示唆された。それ故、当該DNAオリゴマーの分解反応は、使用例1の5-フェニルエチニル-2’-デオキシウリジンを含むDNAオリゴマーの場合と比較してかなり遅いことが示唆された。
5'-TTTTTT (PC) TTTTTT- 3 ' product for 1: pTTTTTT molecular formula C 60 H 80 N 12 O 43 P 6 FW 1843.2 [MH] - 1842.2 Found 1841.8
Degradation reaction of DNA oligomer by ethylenediamine is shown below. In the following reaction formula, the product on the right side corresponds to
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
<12. 使用例6:非天然塩基を含むDNAオリゴマーを用いたポリメラーゼ連鎖反応によるDNAの増幅と増幅産物の連結反応>
[使用例6-1:非天然塩基を含むDNAオリゴマーを用いたポリメラーゼ連鎖反応によるDNAの増幅]
鋳型DNAとして、λDNA(New England BioLabs社製 カタログ番号 N3011)を、酵素としてKOD FX Neo(TOYOBO 社製 カタログ番号 KFX-201)を用い、5-フェニルエチニル-2’-デオキシウリジン(P)又は5-エチニル-2’-デオキシウリジン(Pa)を含むDNAオリゴマーをプライマーとして用いて、ポリメラーゼ連鎖反応を行った。ポリメラーゼ連鎖反応後の増幅産物に対して本発明のDNA鎖の切断反応を実施することにより、リガーゼによる連結が可能な突出末端が生成するように非天然塩基を含むDNAオリゴマーの配列設計を行い、2つの標的DNAを鋳型としてポリメラーゼ連鎖反応を実施した。<12. Use Example 6: DNA amplification by polymerase chain reaction using DNA oligomers containing unnatural bases and ligation of amplified products>
[Example of use 6-1: Amplification of DNA by polymerase chain reaction using a DNA oligomer containing an unnatural base]
Using λDNA (catalog number N3011 manufactured by New England BioLabs) as template DNA and KOD FX Neo (catalog number KFX-201 manufactured by TOYOBO) as an enzyme, 5-phenylethynyl-2′-deoxyuridine (P) or 5 Polymerase chain reaction was performed using a DNA oligomer containing -ethynyl-2′-deoxyuridine (Pa) as a primer. By performing the DNA chain cleavage reaction of the present invention on the amplified product after the polymerase chain reaction, the sequence design of the DNA oligomer containing an unnatural base is performed so that a protruding end that can be ligated by ligase is generated, Polymerase chain reaction was performed using two target DNAs as templates.
標的DNAの塩基配列は、λDNA中において5’末端から28634-28770番目に相当する遺伝子1(136塩基対)と、28743-28932番目に相当する遺伝子2(179塩基対)とからなる。遺伝子1のフォワードプライマー鎖として5’-GTAGCGATCAAGCCATGAATGTAACG-3’(配列番号22)、及びリバースプライマー鎖として5’-ACAGGCGAATCGCAAXCACTG-3’(X = 5-フェニルエチニル-2’-デオキシウリジン(P)又は5-エチニル-2’-デオキシウリジン(Pa))(配列番号23)を、遺伝子2のフォワードプライマー鎖として5’-ATTGCGATTCGCCTGXCTCTGC-3’(5-フェニルエチニル-2’-デオキシウリジン(P)又は5-エチニル-2’-デオキシウリジン(Pa))(配列番号24)、及びリバースプライマー鎖として5’-GGTTGACTTAAATCGACCAGTAACAGG-3’(配列番号25)を設計した。
The base sequence of the target DNA consists of gene 1 (136 base pairs) corresponding to the 28634-28770th position from the 5 'end and gene 2 (179 base pairs) corresponding to the 28743-28932 position from the 5' end in the lambda DNA. 5′-GTAGCGATCAAGCCATGAATGTAACG-3 ′ (SEQ ID NO: 22) as the forward primer strand of
ポリメラーゼ連鎖反応は、終濃度0.3μM プライマー、1 ng λDNA、0.4 mM dNTPs、1 U KOD Fx neo、及び製品添付のバッファーを用いて、TaKaRa PCR Thermal Cycler Dice(登録商標) Gradient(Takara社製サーマルサイクラ―、カタログ番号 TP600)により、アニーリング:94℃, 2分、変性:98℃, 10秒、伸長:68℃, 10秒(変性→伸長を35回繰り返し)の条件下において実施した。増幅産物を、10%非変性ポリアクリルアミドゲルに供することによって、ポリメラーゼ連鎖反応による増幅産物の確認を行った。結果を図51に示す。 Polymerase chain reaction was performed using TaKaRa PCR Thermal Cycler Dice (registered trademark) Gradient (Takara thermal cycler) using a final concentration of 0.3 μM primer, 1 ng λDNA, 0.4 mM dNTPs, 1 U KOD Fx neo, and a buffer attached to the product. -, Catalog number TP600), annealing: 94 ° C., 2 minutes, denaturation: 98 ° C., 10 seconds, elongation: 68 ° C., 10 seconds (denaturation → extension repeated 35 times). The amplified product was subjected to 10% non-denaturing polyacrylamide gel to confirm the amplified product by polymerase chain reaction. The results are shown in FIG.
図51では、電気泳動後のゲルを、30分間室温にてエチジウムブロマイド溶液に浸潤させることで、二本鎖DNA選択的な染色操作を行い、紫外線照射して発する蛍光を検出することにより、DNA鎖を白いバンドとして検出している。レーン1のバンドは、遺伝子マーカーとして100塩基対毎にバンドが検出されるTrackIt(商標) 100 bp DNAラダー(Invitrogen社製 カタログ番号 10488-058)を用いた。レーン2及び3のバンドは、それぞれX = 5-フェニルエチニル-2’-デオキシウリジン(P)又は5-エチニル-2’-デオキシウリジン(Pa)を含むDNAオリゴマーをプライマー鎖として用いた場合の遺伝子1の増幅産物を示しており、レーン4及び5のバンドは、それぞれX = 5-フェニルエチニル-2’-デオキシウリジン(P)又は5-エチニル-2’-デオキシウリジン(Pa)を含むDNAオリゴマーをプライマー鎖として用いた場合の遺伝子2の増幅産物を示している。マーカー由来の100塩基対と200塩基対の泳動度から、目的産物の増幅が確認された。
In FIG. 51, the gel after electrophoresis is infiltrated into an ethidium bromide solution at room temperature for 30 minutes, thereby performing a double-stranded DNA selective staining operation and detecting fluorescence emitted by ultraviolet irradiation to detect DNA. The strand is detected as a white band. The band in
[使用例6-2:非天然塩基を含むDNA増幅産物の切断反応及び得られた5’末端リン酸化DNA断片の連結反応]
PCRチューブに、50μLの使用例5-1で調製されたDNA増幅産物及び50μLのエチレンジアミンを加え、TaKaRa PCR Thermal Cycler Dice(登録商標) Gradientにより、70℃、3時間加熱した。反応後、50μLの蒸留水を加え、さらに150μLのPCI(フェノール:クロロホルム:イソアミルアルコール=25:24:1)混合溶液を加えた後、ボルテックスミキサーを用いて前記溶液を激しく混ぜ、2層に均一に層分離するまで遠心分離した。上清を回収し、再び150μLのPCI混合溶液を加えた後、ボルテックスミキサーを用いて前記溶液を激しく混ぜ、2層に均一に層分離するまで遠心分離した。この操作を合計5回繰り返した。上記の操作によって得られた上清を、Bio-Spin 6カラム(BIO-RAD社製 カタログ番号 732-6227)に供した後、エタノール沈殿により精製を行った。遠心濃縮装置を用いて5’末端リン酸化DNA断片を乾燥させ、5μLのTEバッファーに再溶解させた。[Use Example 6-2: Cleavage reaction of DNA amplification product containing unnatural base and ligation reaction of the obtained 5′-terminal phosphorylated DNA fragment]
50 μL of the DNA amplification product prepared in Use Example 5-1 and 50 μL of ethylenediamine were added to the PCR tube, and heated by TaKaRa PCR Thermal Cycler Dice (registered trademark) Gradient at 70 ° C. for 3 hours. After the reaction, add 50 μL of distilled water, and then add 150 μL of PCI (phenol: chloroform: isoamyl alcohol = 25: 24: 1) mixed solution, then vigorously mix the solution using a vortex mixer to make two layers even Centrifugation until layer separation. The supernatant was collected, 150 μL of PCI mixed solution was added again, and then the solution was vigorously mixed using a vortex mixer and centrifuged until the two layers were uniformly separated. This operation was repeated a total of 5 times. The supernatant obtained by the above operation was applied to a Bio-Spin 6 column (catalog number 732-6227 manufactured by BIO-RAD), and then purified by ethanol precipitation. The 5′-end phosphorylated DNA fragment was dried using a centrifugal concentrator and redissolved in 5 μL of TE buffer.
それぞれ5μLの遺伝子1及び遺伝子2の5’末端リン酸化DNA断片を含む溶液を混合し、1.5μLの1 M NaCl及び3.5μLの蒸留水を加え、TaKaRa PCR Thermal Cycler Dice(登録商標) Gradientにより、98℃、15分間加熱した後に室温まで徐冷した。15μLのLigation High (TOYOBO社製 カタログ番号 LGK-101)を加え、16℃、40時間振盪させた。リガーゼ反応後のサンプルを、PCI抽出操作及びエタノール沈殿により精製し、10%非変性ポリアクリルアミドゲルに供し、連結反応の解析を行った。結果を図52に示す。
A solution containing 5 μL of the
レーン1のバンドは、遺伝子マーカーとして100塩基対毎にバンドが検出されるTrackIt(商標)100 bp DNAラダーを用いた。レーン2及び3のバンドは、それぞれX = 5-フェニルエチニル-2’-デオキシウリジン(P)を含む遺伝子1又は遺伝子2の増幅産物を本発明の切断反応で切断し、得られた5’末端リン酸化DNA断片を示しており、レーン4及び5のバンドは、それぞれX = 5-フェニルエチニル-2’-デオキシウリジン(P)又は5-エチニル-2’-デオキシウリジン(Pa)を含む遺伝子1及び2のDNA増幅産物を本発明の切断反応で切断し、得られた遺伝子1及び2の5’末端リン酸化DNA断片をリガーゼと反応させた連結反応産物を示している。レーン6のバンドは、λDNA中において5’末端から28634-28932番目に相当する塩基配列を有するDNA断片、すなわち遺伝子1及び2が連結した連結反応産物と同一の塩基配列を有する遺伝子3(299塩基対)の増幅産物を示している。X = P又はPaを含む遺伝子1及び2のDNA増幅産物を本発明の切断反応で切断し、得られた5’末端リン酸化DNA断片をリガーゼと反応させた連結反応産物(レーン4及び5)は、遺伝子3(レーン6)の泳動度と一致することから、連結反応が高効率に進行していることが示された。
The
<13. 合成例7:2’-デオキシ-4-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-5-フェニルエチニルシチジンを含むDNAオリゴマー>
[合成例7-1:2’-デオキシ-4-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-5-フェニルエチニルシチジンの合成]
2’-デオキシ-5-フェニルエチニルシチジンは、非特許文献41に従って合成した。708 mg(2.16 mmol)の2’-デオキシ-5-フェニルエチニルシチジン(分子量327.33)を10 mLのメタノールに溶解した。得られた溶液に、2.0 mL(8.6 mmol)のN,N-ジ(n-ブチル)ホルムアミジンジメチルアセタール(分子量203.32、密度0.87 g/mL)(Dialkylformamidines: depurination resistant N6-protecting group for deoxyadenosine. Froehler, B. C.及びMatteucci, M. D. Tetrahedron Letters 1983年, 11, p. 8031-8036)を加え、25℃で2時間撹拌した。反応液の溶媒を減圧留去し、シリカゲルカラムで目的物質を精製した(展開溶媒はCH2Cl2のみ〜5〜10% MeOH/CH2Cl2)。目的のフラクションの溶媒を減圧留去し、700 mg(1.50 mmol, 収率69 %)の目的物質(分子量466.57)を、うす褐色のオイル状物質として得た。<13. Synthesis Example 7: DNA oligomer containing 2′-deoxy-4-N- {N ′, N′-di (n-butyl) formamidine} -5-phenylethynylcytidine>
[Synthesis Example 7-1: Synthesis of 2′-deoxy-4-N- {N ′, N′-di (n-butyl) formamidine} -5-phenylethynylcytidine]
2′-Deoxy-5-phenylethynylcytidine was synthesized according to Non-Patent Document 41. 708 mg (2.16 mmol) of 2′-deoxy-5-phenylethynylcytidine (molecular weight of 327.33) was dissolved in 10 mL of methanol. To the resulting solution, 2.0 mL (8.6 mmol) of N, N-di (n-butyl) formamidine dimethyl acetal (molecular weight 203.32, density 0.87 g / mL) (Dialkylformamidines: depurination resistant N6-protecting group for deoxyadenosine. , BC and Matteucci,
1H NMR(400 MHz, DMSO-d6) δ8.64 (s, 1H), 8.44 (s, 1H), 7.43-7.34 (m, 5H), 6.12 (t, J = 6.2 Hz, 1H), 5.23 (d, J = 4.1 Hz, 1H), 5.13 (t, J = 4.8 Hz, 1H), 4.26-4.21 (m, 1H), 3.84-3.81 (m, 1H), 3.70-3.44 (m, 6H), 2.26-2.20 (m, 1H), 2.08-2.01 (m, 1H), 1.66-1.52 (m, 4H), 1.32-1.22 (m, 4H), 0.90 (t, J = 7.3 Hz, 3H), 0.82 (t, J = 7.3 Hz, 3H); 13C NMR(100 MHz, DMSO-d6) δ169.7, 157.4, 153.4, 145.3, 130.7, 128.6, 128.1, 123.2, 97.5, 91.6, 87.6, 85.8, 84.5, 69.7, 60.7, 54.9, 51.4, 45.4, 41.0, 30.4, 28.5, 19.6, 19.1, 13.5; HRMS (ESI): C26H35N4O4に対する計算値: 467.2658 [M+H]+; 実測値: 467.2653.
[合成例7-2:4-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-5-フェニルエチニル-5’-O-ジメトキシトリチル-2’-デオキシシチジンの合成]
700 mg(1.50 mmol)の2’-デオキシ-4-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-5-フェニルエチニル-シチジン (分子量466.57)を、10 mLのピリジンに溶解した。得られた溶液に、763 mg(2.25 mmol)の4,4’-ジメトキシトリチルクロリド(分子量338.83)を加え、25℃で1時間撹拌した。反応液に約10 mLのメタノールを加え、溶媒を減圧留去した。得られた残渣に、少量の塩化メチレン及びヘキサンを加えて混合した。前記混合物を3回共沸した後、シリカゲルカラムで目的物質を精製した(展開溶媒は2% Et3N、0〜2〜5% MeOH/CH2Cl2)。目的のフラクションの溶媒を減圧留去し、少量の塩化メチレン及びヘキサンを加え共沸した。924 mg(1.20 mmol, 収率80%)の目的生成物を、白色粉末として得た。 1 H NMR (400 MHz, DMSO-d6) δ8.64 (s, 1H), 8.44 (s, 1H), 7.43-7.34 (m, 5H), 6.12 (t, J = 6.2 Hz, 1H), 5.23 ( d, J = 4.1 Hz, 1H), 5.13 (t, J = 4.8 Hz, 1H), 4.26-4.21 (m, 1H), 3.84-3.81 (m, 1H), 3.70-3.44 (m, 6H), 2.26 -2.20 (m, 1H), 2.08-2.01 (m, 1H), 1.66-1.52 (m, 4H), 1.32-1.22 (m, 4H), 0.90 (t, J = 7.3 Hz, 3H), 0.82 (t , J = 7.3 Hz, 3H); 13 C NMR (100 MHz, DMSO-d6) δ169.7, 157.4, 153.4, 145.3, 130.7, 128.6, 128.1, 123.2, 97.5, 91.6, 87.6, 85.8, 84.5, 69.7, 60.7, 54.9, 51.4, 45.4, 41.0, 30.4, 28.5, 19.6, 19.1, 13.5; HRMS (ESI): Calculated for C 26 H 35 N 4 O 4 : 467.2658 [M + H] + ; Found: 467.2653.
[Synthesis Example 7-2: Synthesis of 4-N- {N ′, N′-di (n-butyl) formamidine} -5-phenylethynyl-5′-O-dimethoxytrityl-2′-deoxycytidine]
700 mg (1.50 mmol) 2'-deoxy-4-N- {N ', N'-di (n-butyl) formamidine} -5-phenylethynyl-cytidine (molecular weight 466.57) in 10 mL pyridine Dissolved. To the obtained solution, 763 mg (2.25 mmol) of 4,4′-dimethoxytrityl chloride (molecular weight: 338.83) was added and stirred at 25 ° C. for 1 hour. About 10 mL of methanol was added to the reaction solution, and the solvent was distilled off under reduced pressure. A small amount of methylene chloride and hexane were added to the resulting residue and mixed. After the mixture was azeotroped three times, the target substance was purified with a silica gel column (developing solvent was 2% Et 3 N, 0 to 2 to 5% MeOH / CH 2 Cl 2 ). The solvent of the objective fraction was distilled off under reduced pressure, and a small amount of methylene chloride and hexane were added and azeotroped. 924 mg (1.20 mmol, 80% yield) of the desired product was obtained as a white powder.
1H NMR(400 MHz, DMSO-d6) δ8.65 (s, 1H), 8.17 (s, 1H), 7.44-6.81 (m, 18H), 6.14 (t, J = 6.6 Hz, 1H), 5.32 (d, J = 4.6 Hz, 1H), 4.31-4.26 (m, 1H), 4.02-3.97 (m, 1H), 3.644 (s, 3H), 3.637 (s, 3H), 3.57-3.49 (m, 4H), 3.23-3.16 (m, 2H), 2.36-2.30 (m, 1H), 2.17-2.10 (m, 1H), 1.66-1.53 (m, 4H), 1.32-1.22 (m, 4H), 0.91 (t, J = 7.3 Hz, 3H), 0.82 (t, J = 7.3 Hz, 3H); 13C NMR(100 MHz, DMSO-d6) δ169.7, 158.03, 158.01, 157.5, 153.4, 144.7, 144.3, 135.6, 135.4, 130.6, 129.7, 129.6, 128.3, 127.93, 127.86, 127.6, 126.6, 123.0, 113.2, 97.9, 91.7, 86.2, 85.9, 83.9, 70.6, 63.5, 54.9, 54.5, 45.4, 41.2, 30.4, 28.5, 19.6, 19.1, 13.5; HRMS (ESI): C47H53N4O6に対する計算値: 769.3965 [M+H]+; 実測値: 769.3985.
[合成例7-3:4-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-5-フェニルエチニル-5’-O-ジメトキシトリチル-2’-デオキシシチジン-3’-N,N-ジイソプロピル(シアノエチル)ホスホロアミダイトの合成]
109 mg(0.142 mmol)の4-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-5-フェニルエチニル-5’-O-ジメトキシトリチル-2’-デオキシシチジン(分子量768.94)をナスフラスコに入れ、5 mLのCH3CN及び157μL(1.13 mmol)のトリエチルアミン(分子量101.19, 比重0.726 g/mL)を加え、撹拌し、さらに126μL(0.56 mmol)の2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロジアミダイト(分子量236.68, 比重1.061 g/mL)を加え、室温で30分間撹拌した。得られた混合物に、25 mLのAcOEtと25 mLの飽和食塩水とを加え、分液し、有機層をさらに2回飽和食塩水で洗った。得られた有機層を、無水硫酸マグネシウムで乾燥した後、フィルターでろ過し、ろ液の溶媒を減圧留去した。アセトニトリルで3回共沸した後、4.7 mLのアセトニトリルを加えた。前記溶液に、少量のモレキュラーシーブを加えた。DNA自動合成機に取り付けてDNA合成に使用した。 1 H NMR (400 MHz, DMSO-d6) δ8.65 (s, 1H), 8.17 (s, 1H), 7.44-6.81 (m, 18H), 6.14 (t, J = 6.6 Hz, 1H), 5.32 ( d, J = 4.6 Hz, 1H), 4.31-4.26 (m, 1H), 4.02-3.97 (m, 1H), 3.644 (s, 3H), 3.637 (s, 3H), 3.57-3.49 (m, 4H) , 3.23-3.16 (m, 2H), 2.36-2.30 (m, 1H), 2.17-2.10 (m, 1H), 1.66-1.53 (m, 4H), 1.32-1.22 (m, 4H), 0.91 (t, J = 7.3 Hz, 3H), 0.82 (t, J = 7.3 Hz, 3H); 13 C NMR (100 MHz, DMSO-d6) δ169.7, 158.03, 158.01, 157.5, 153.4, 144.7, 144.3, 135.6, 135.4 , 130.6, 129.7, 129.6, 128.3, 127.93, 127.86, 127.6, 126.6, 123.0, 113.2, 97.9, 91.7, 86.2, 85.9, 83.9, 70.6, 63.5, 54.9, 54.5, 45.4, 41.2, 30.4, 28.5, 19.6, 19.1 , 13.5; HRMS (ESI): Calculated for C 47 H 53 N 4 O 6 : 769.3965 [M + H] + ; Found: 769.3985.
[Synthesis Example 7-3: 4-N- {N ′, N′-di (n-butyl) formamidine} -5-phenylethynyl-5′-O-dimethoxytrityl-2′-deoxycytidine-3′- Synthesis of N, N-diisopropyl (cyanoethyl) phosphoramidite]
109 mg (0.142 mmol) 4-N- {N ', N'-di (n-butyl) formamidine} -5-phenylethynyl-5'-O-dimethoxytrityl-2'-deoxycytidine (molecular weight 768.94) Was added to an eggplant flask, 5 mL of CH 3 CN and 157 μL (1.13 mmol) of triethylamine (molecular weight 101.19, specific gravity 0.726 g / mL) were added, stirred, and 126 μL (0.56 mmol) of 2-cyanoethyl N, N, N ′, N′-tetraisopropyl phosphorodiamidite (molecular weight 236.68, specific gravity 1.061 g / mL) was added, and the mixture was stirred at room temperature for 30 minutes. To the obtained mixture, 25 mL of AcOEt and 25 mL of saturated brine were added and separated, and the organic layer was further washed twice with saturated brine. The obtained organic layer was dried over anhydrous magnesium sulfate and filtered through a filter, and the solvent of the filtrate was distilled off under reduced pressure. After azeotroping with acetonitrile three times, 4.7 mL of acetonitrile was added. A small amount of molecular sieve was added to the solution. Attached to an automatic DNA synthesizer and used for DNA synthesis
[合成例7-4:5-フェニルエチニル-2’-デオキシシチジン(Q)を含むDNAオリゴマーの合成]
通常のDNA自動合成機のDNA合成のプロトコールで、1μmolスケール、DMTr ONでホスホロアミダイト法により合成した。担体からの切り出しは28%アンモニア水で行い、脱保護は25℃で16時間静置することにより行った。脱保護後、アンモニアを遠心濃縮装置により減圧留去し、DNAを含む溶液を0.45μmのフィルターに通した後、逆相HPLCによりメインピークを精製し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置と凍結乾燥によりギ酸アンモニウムと溶媒とを留去した。得られた残渣に100μLの80%酢酸水溶液を加え、よく混合し、25℃で30分間静置した。その後、前記混合物に蒸留水を約1 mLとなるように加えて、0.45μmのフィルターに通した後、逆相HPLCにより13〜14分の保持時間に溶出されたメインピークを精製し(8-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置と凍結乾燥によりギ酸アンモニウムと溶媒とを留去した。得られたDNAオリゴマーはMALDI TOF-MS測定により同定した。合成したDNAオリゴマーは、約500μLの蒸留水を加えて水溶液とした。[Synthesis Example 7-4: Synthesis of DNA oligomer containing 5-phenylethynyl-2′-deoxycytidine (Q)]
The DNA was synthesized by the phosphoramidite method with 1 μmol scale and DMTr ON according to the protocol for DNA synthesis of a normal automatic DNA synthesizer. Cutting out from the carrier was performed with 28% aqueous ammonia, and deprotection was performed by standing at 25 ° C. for 16 hours. After deprotection, the ammonia was distilled off under reduced pressure using a centrifugal concentrator, the solution containing DNA was passed through a 0.45 μm filter, and the main peak was purified by reverse-phase HPLC (0-50% CH 3 CN / 50 mM formic acid. Ammonium aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and the solvent were distilled off by centrifugal concentrator and lyophilization. To the obtained residue, 100 μL of 80% aqueous acetic acid solution was added, mixed well, and allowed to stand at 25 ° C. for 30 minutes. Thereafter, distilled water was added to the mixture so as to be about 1 mL, passed through a 0.45 μm filter, and the main peak eluted at a retention time of 13 to 14 minutes was purified by reverse phase HPLC (8- 20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and the solvent were distilled off by centrifugal concentrator and lyophilization. The obtained DNA oligomer was identified by MALDI TOF-MS measurement. The synthesized DNA oligomer was made into an aqueous solution by adding about 500 μL of distilled water.
以下において、5-フェニルエチニル-2’-デオキシシチジン:
を「Q」で示す。 Is indicated by “Q”.
DNAの濃度決定は、QをCと近似して260 nmのモル吸光係数を計算し、260 nmのUV吸収より濃度を計算した。 The concentration of DNA was determined by calculating the molar extinction coefficient at 260 nm by approximating Q to C and calculating the concentration from the UV absorption at 260 nm.
合成したDNAオリゴマーの逆相HPLCクロマトグラムを図53及び54に示す。 53 and 54 show reverse phase HPLC chromatograms of the synthesized DNA oligomers.
合成したDNAオリゴマーの上記逆相HPLCにおける保持時間は以下の通りである。 The retention time in the reverse phase HPLC of the synthesized DNA oligomer is as follows.
5’-TTTTTTQTTTTTT-3’について13.6分(配列番号26)
5’-TTTTCTTQTCCTTTT-3’について13.1分(配列番号27)
合成したDNAオリゴマーのMALDI TOF-MSのデータは以下の通りである。13.6 minutes for 5'-TTTTTTQTTTTTT-3 '(SEQ ID NO: 26)
13.1 minutes for 5'-TTTTCTTQTCCTTTT-3 '(SEQ ID NO: 27)
The MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
5’-TTTTTTQTTTTTT-3’について分子式C137H173N27O88P12、[M-H]-の計算値 3976.6、実測値 3977.1(配列番号26)
5’-TTTTCTTQTCCTTTT-3’について分子式C154H196N34O99P14、[M-H]-の計算値 4540.0、実測値 4540.0(配列番号27)
<14. 使用例7: 5-フェニルエチニル-2’-デオキシシチジンを含むDNAオリゴマー>
[使用例7-1:メチルアミンによる5-フェニルエチニル-2’-デオキシシチジンを含むDNAオリゴマーの切断反応及び生成物の同定]
スクリューチューブに300μLの5’-T6QT6-3’又は5’-TTTTCTTQTCCTTTT-3’の水溶液(ストランド濃度 100μM)と300μLのメチルアミン(40% 水溶液)とを加え、水浴中で70℃、12時間加熱した。反応後は、メチルアミンを遠心濃縮装置で減圧留去し、逆相HPLCにより分析して生成物を分取した(8-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。分取した生成物は、MALDI TOF-MSにより分子量を測定した。For 5′-TTTTTTQTTTTTT-3 ′, the molecular formula C 137 H 173 N 27 O 88 P 12 , calculated value of [MH] − 3976.6, measured value 3977.1 (SEQ ID NO: 26)
5'-TTTTCTTQTCCTTTT-3 molecular formula C 154 for 'H 196 N 34 O 99 P 14, [MH] - Calculated 4540.0, Found 4540.0 (SEQ ID NO: 27)
<14. Use Example 7: DNA oligomer containing 5-phenylethynyl-2′-deoxycytidine>
[Use Example 7-1: Cleavage reaction of DNA oligomer containing 5-phenylethynyl-2′-deoxycytidine with methylamine and identification of the product]
Add 300 μL of 5′-T 6 QT 6 -3 ′ or 5′-TTTTCTTQTCCTTTT-3 ′ aqueous solution (strand concentration: 100 μM) and 300 μL of methylamine (40% aqueous solution) to the screw tube. Heated for 12 hours. After the reaction, methylamine was distilled off under reduced pressure with a centrifugal concentrator and analyzed by reverse phase HPLC to fractionate the product (8-20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, at 254 nm. detection). The molecular weight of the fractionated product was measured by MALDI TOF-MS.
合成した各DNAオリゴマーの反応後の逆相HPLCクロマトグラムを図55及び56に示す。 The reverse phase HPLC chromatogram after reaction of each synthesized DNA oligomer is shown in FIGS.
各DNAオリゴマーにおける生成物1及び2のMALDI TOF-MSのデータは以下の通りである。以下において、「RNu」は、切断反応に伴って生成するQ由来の部分を意味する。The MALDI TOF-MS data of
5’-TTTTTTQTTTTTT-3’について
生成物1:pTTTTTT 分子式C60H80N12O43P6 FW 1843.2 [M-H]- 1842.2 実測値 1842.0
HPLCの保持時間 8.5分
生成物2:TTTTTTpRNu 分子式C66H91N13O45P6 FW 1972.3 [M-H]- 1971.3 実測値 1971.1
HPLCの保持時間 10.6分
5’-TTTTCTTQTCCTTTT-3’について
生成物1:pTCCTTTT 分子式C68H91N16O48P7 FW 2117.3 [M-H]- 2116.3 実測値 2116.3
HPLCの保持時間7.1分
生成物2:TTTTCTTpRNu 分子式C75H103N16O51P7 FW 2261.5 [M-H]- 2260.5 実測値 2260.5
HPLCの保持時間 9.7分
メチルアミンによるDNAオリゴマーの分解反応を下記に示す。下記反応式において、左側の生成物が生成物2に、右側の生成物が生成物1に、それぞれ対応する。
HPLC retention time 8.5 min product 2: TTTTTTpR Nu molecular formula C 66 H 91 N 13 O 45 P 6 FW 1972.3 [MH] - 1971.3 Found 1971.1
HPLC retention time 10.6 minutes
5'-TTTTCTTQTCCTTTT-3 'product for 1: pTCCTTTT molecular formula C 68 H 91 N 16 O 48 P 7 FW 2117.3 [MH] - 2116.3 Found 2116.3
HPLC retention time 7.1 min product 2: TTTTCTTpR Nu molecular formula C 75 H 103 N 16 O 51 P 7 FW 2261.5 [MH] - 2260.5 Found 2260.5
HPLC retention time 9.7 minutes Degradation reaction of DNA oligomer by methylamine is shown below. In the following reaction formula, the product on the left corresponds to the
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
[使用例7-2:メチルアミン又はエチレンジアミンによる5-フェニルエチニル-2’-デオキシシチジンを含むDNAオリゴマーの切断反応速度の比較]
スクリューチューブに30μLの5’-T6(Q)T6-3’又は5’-TTTTCTT(Q)TCCTTTT-3’の水溶液(ストランド濃度 100μM)と、30μLのメチルアミン40%水溶液又はエチレンジアミンを加え、水浴中で70℃、2時間加熱した。反応混合物を酢酸で中和し、逆相HPLCにより分析した(8-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。5’-T6(Q)T6-3’の反応混合物のHPLCチャートを図57に、5’-TTTTCTT(Q)TCCTTTT-3’の反応混合物のHPLCチャートを図58に、それぞれ示す。[Use Example 7-2: Comparison of cleavage reaction rate of DNA oligomer containing 5-phenylethynyl-2′-deoxycytidine with methylamine or ethylenediamine]
Add 30 μL of 5′-T 6 (Q) T 6 -3 ′ or 5′-TTTTCTT (Q) TCCTTTT-3 ′ aqueous solution (
5’-T6QT6-3’の5’-側の切断生成物である5’-T6p-3’のピーク面積に基づき定量したところ、5’-T6QT6-3’の5’-側の切断産物の量は、メチルアミン処理の方がエチレンジアミン処理よりも3.6倍多いことが計算された。また、5’-TTTTCTTQTCCTTTT-3’の5’-側の切断生成物である5’-TTTTCTTp-3’のピーク面積に基づき定量したところ、5’-TTTTCTTQTCCTTTT-3’の5’-側の切断産物の量は、メチルアミン処理の方がエチレンジアミン処理よりも2.6倍多いことが計算された。この結果より、メチルアミン又はエチレンジアミンの一級アミンを用いるDNAオリゴマーの切断反応において、最も反応速度の速いものはメチルアミンを用いる切断反応であることが示唆された。5'-T 6 QT 6 -3 was quantified based on the peak area of 'which is the 5'-cleavage product of 5'-T 6 p-3' , the 5'-T 6 QT 6 -3 ' The amount of cleavage product on the 5′-side was calculated to be 3.6 times higher with methylamine treatment than with ethylenediamine treatment. In addition, when quantified based on the peak area of 5'-TTTTCTTp-3 'which is the 5'-side cleavage product of 5'-TTTTCTTQTCCTTTT-3', 5'-TTTTCTTQTCCTTTT-3 '5'-side cleavage The amount of product was calculated to be 2.6 times higher with methylamine treatment than with ethylenediamine treatment. From this result, it was suggested that in the cleavage reaction of DNA oligomer using methylamine or primary amine of ethylenediamine, the one having the highest reaction rate is the cleavage reaction using methylamine.
合成例7で得られたQには、下記に示すように複数の互変異性体が存在する。それ故、Qを含むオリゴデオキシヌクレオチドにおいて、Qの互変異性体が求核剤と反応することにより、Eの脱離反応及びオリゴデオキシヌクレオチドの切断反応が進行すると考えられる。
上記の反応式において、DNAはオリゴデオキシヌクレオチドを、Nuは求核剤を意味する。 In the above reaction formula, DNA means oligodeoxynucleotide and Nu means nucleophile.
<15. 合成例8:5-(2-オキシ-2-フェニルエチル)-2’-デオキシウリジン(HP)を含むDNAオリゴマー>
[合成例8-1:5-(2-オキシ-2-フェニルエチル)-2’-デオキシウリジン(HP)を含むDNAオリゴマーの合成]
ヌクレオシドレベルで公知の反応に基づき、合成例1で得られたPを含むDNAオリゴマーにおいて、Pの塩基部分を銀イオンで環化させて(A facile synthesis of fluorophores based on 5-phenylethynyluracils. Hudson, H. E. R.及びMoszybski, J. M. Synlett 2006年, 18, p. 2997-3000)、その後、水酸化ナトリウム処理することにより、Pを5-(2-オキシ-2-フェニルエチル)-2’-デオキシウリジンに変換した(非特許文献39)。
[Synthesis Example 8-1: Synthesis of DNA oligomer containing 5- (2-oxy-2-phenylethyl) -2′-deoxyuridine (HP)]
Based on a known reaction at the nucleoside level, in the DNA oligomer containing P obtained in Synthesis Example 1, the base portion of P was cyclized with silver ions (A facile synthesis of fluorophores based on 5-phenylethynyluracils.Hudson, HER And Moszybski, JM Synlett 2006, 18, p. 2997-3000), followed by sodium hydroxide treatment to convert P to 5- (2-oxy-2-phenylethyl) -2'-deoxyuridine (Non-patent document 39).
合成例1で得られた5’-T6PT6-3’水溶液(1 mMストランド濃度)200μLに、硝酸銀水溶液(100 mM)200μLを加え、よく混合し、25℃で16時間静置した。前記混合物に、NaOH水溶液(10規定)20μLを加え、よく混合した。得られた混合物を、25℃で2時間静置した。その後、前記混合物に水を加えて約1 mLとし、酢酸20μLを加えた。得られた溶液を0.45μmのフィルターに通した後、逆相HPLCで精製 (8-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)し、脱塩した。メインピークとして得られた生成物は、MALDI TOF-MS測定により同定した。To 200 μL of the 5′-T 6 PT 6 -3 ′ aqueous solution (1 mM strand concentration) obtained in Synthesis Example 1, 200 μL of an aqueous silver nitrate solution (100 mM) was added, mixed well, and allowed to stand at 25 ° C. for 16 hours. To the mixture, 20 μL of NaOH aqueous solution (10N) was added and mixed well. The resulting mixture was allowed to stand at 25 ° C. for 2 hours. Thereafter, water was added to the mixture to make about 1 mL, and 20 μL of acetic acid was added. The obtained solution was passed through a 0.45 μm filter, and purified by reverse phase HPLC (8-20% CH 3 CN / 50 mM aqueous ammonium formate solution, 20 minutes, detected at 254 nm) and desalted. The product obtained as the main peak was identified by MALDI TOF-MS measurement.
以下において、5-(2-オキシ-2-フェニルエチル)-2’-デオキシウリジン:
を「HP」で示す。 Is indicated by “HP”.
DNAの濃度決定は、HPをTと近似して260 nmのモル吸光係数を計算し、260 nmのUV吸収より濃度を計算した。 The concentration of DNA was determined by approximating HP with T and calculating the molar extinction coefficient at 260 nm, and calculating the concentration from UV absorption at 260 nm.
合成したDNAオリゴマーの逆相HPLCクロマトグラムを図59及び60に示す。 The reverse phase HPLC chromatogram of the synthesized DNA oligomer is shown in FIGS.
合成したDNAオリゴマーの上記逆相HPLCにおける保持時間は以下の通りである。 The retention time in the reverse phase HPLC of the synthesized DNA oligomer is as follows.
5’-TTTTTT(HP)TTTTTT-3’について12.5分(配列番号28)
5’-TTTTCTT(HP)TCCTTTT-3’について11.4分(配列番号29)
合成したDNAオリゴマーのMALDI TOF-MSのデータは以下の通りである。12.5 minutes for 5'-TTTTTT (HP) TTTTTT-3 '(SEQ ID NO: 28)
11.4 minutes for 5'-TTTTCTT (HP) TCCTTTT-3 '(SEQ ID NO: 29)
The MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
5’-TTTTTT(HP)TTTTTT-3’について分子式C137H174N26O90P12、[M-H]-の計算値 3995.6、実測値 3995.6(配列番号28)
5’-TTTTCTT(HP)TCCTTTT-3’について分子式C154H197N33O101P14、[M-H]-の計算値 4559.0、実測値 4559.4(配列番号29)
<16. 使用例8: 5-(2-オキシ-2-フェニルエチル)-2’-デオキシウリジンを含むDNAオリゴマー>
[使用例8-1:メチルアミンによる5-(2-オキシ-2-フェニルエチル)-2’-デオキシウリジンを含むDNAオリゴマーの切断反応及び生成物の同定]
スクリューチューブに300μLの5’-T6(HP)T6-3’又は5’-TTTTCTT(HP)TCCTTTT-3’の水溶液(ストランド濃度100μM)と300μLのメチルアミン(40% 水溶液)を加え、水浴中で70℃、12時間加熱した。反応後は、メチルアミンを遠心濃縮装置で減圧留去し、逆相HPLCにより分析して生成物を分取した(8-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。分取した生成物は、MALDI TOF-MSにより分子量を測定した。5'-TTTTTT (HP) molecular formula for TTTTTT-3 'C 137 H 174 N 26 O 90
For 5′-TTTTCTT (HP) TCCTTTT-3 ′, the molecular formula C 154 H 197 N 33 O 101 P 14 , [MH] − calculated value 4559.0, measured value 4559.4 (SEQ ID NO: 29)
<16. Use Example 8: DNA oligomer containing 5- (2-oxy-2-phenylethyl) -2'-deoxyuridine>
[Use Example 8-1: Cleavage reaction of DNA oligomer containing 5- (2-oxy-2-phenylethyl) -2'-deoxyuridine with methylamine and identification of the product]
Add 300 μL of 5′-T 6 (HP) T 6 -3 ′ or 5′-TTTTCTT (HP) TCCTTTT-3 ′ aqueous solution (
合成した各DNAオリゴマーの反応後の逆相HPLCクロマトグラムを図61及び62に示す。 61 and 62 show reverse phase HPLC chromatograms after the reaction of each synthesized DNA oligomer.
各DNAオリゴマーにおける生成物1及び2のMALDI TOF-MSのデータは以下の通りである。以下において、「RNu」は、切断反応に伴って生成するHP由来の部分を意味する。The MALDI TOF-MS data of
5’-TTTTTT(HP)TTTTTT-3’について
生成物1:pTTTTTT 分子式C60H80N12O43P6 FW 1843.2 [M-H]- 1842.2 実測値 1841.6
HPLCの保持時間 8.5分
生成物2:TTTTTTpRNu 分子式C66H91N13O45P6 FW1972.3 [M-H]- 1971.3 実測値 1971.0
HPLCの保持時間 10.6分
5’-TTTTCTT(HP)TCCTTTT-3’について
生成物1:pTCCTTTT 分子式C68H91N16O48P7 FW 2117.3 [M-H]- 2116.3 実測値 2116.2
HPLCの保持時間 7.3分
生成物2:TTTTCTTpRNu 分子式C75H103N16O51P7 FW 2261.5 [M-H]- 2260.5 実測値 2260.5
HPLCの保持時間 9.9分
メチルアミンによるDNAオリゴマーの分解反応を下記に示す。下記反応式において、左側の生成物が生成物2に、右側の生成物が生成物1に、それぞれ対応する。
HPLC retention time 8.5 min product 2: TTTTTTpR Nu molecular formula C 66 H 91 N 13 O 45 P 6 FW1972.3 [MH] - 1971.3 Found 1971.0
HPLC retention time 10.6 minutes
5'-TTTTCTT (HP) TCCTTTT- 3 ' product for 1: pTCCTTTT molecular formula C 68 H 91 N 16 O 48 P 7 FW 2117.3 [MH] - 2116.3 Found 2116.2
HPLC retention time 7.3 min product 2: TTTTCTTpR Nu molecular formula C 75 H 103 N 16 O 51 P 7 FW 2261.5 [MH] - 2260.5 Found 2260.5
HPLC retention time 9.9 minutes Degradation reaction of DNA oligomer by methylamine is shown below. In the following reaction formula, the product on the left corresponds to the
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
[使用例8-2:メチルアミン又はエチレンジアミンによる5-(2-オキシ-2-フェニルエチル)-2’-デオキシウリジンを含むDNAオリゴマーの切断反応速度の比較]
スクリューチューブに30μLの5’-T6(HP)T6-3’又は5’-TTTTCTT(HP)TCCTTTT-3’の水溶液(ストランド濃度100μM)と、30μLのメチルアミン40%水溶液又はエチレンジアミンを加え、水浴中で70℃、2時間加熱した。反応混合物を酢酸で中和し、逆相HPLCにより分析した(8-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。5’-T6(HP)T6-3’の反応混合物のHPLCチャートを図63に、5’-TTTTCTT(HP)TCCTTTT-3’の反応混合物のHPLCチャートを図64に、それぞれ示す。[Use Example 8-2: Comparison of cleavage reaction rate of DNA oligomer containing 5- (2-oxy-2-phenylethyl) -2'-deoxyuridine with methylamine or ethylenediamine]
Add 30 μL of 5′-T 6 (HP) T 6 -3 ′ or 5′-TTTTCTT (HP) TCCTTTT-3 ′ aqueous solution (
5’-T6(HP)T6-3’の5’-側の切断生成物である5’-T6p-3’のピーク面積に基づき定量したところ、5’-T6(HP)T6-3’の5’-側の切断産物の量は、メチルアミン処理の方がエチレンジアミン処理よりも2.4倍多いことが計算された。また、5’-TTTTCTT(HP)TCCTTTT-3’の5’-側の切断生成物である5’-TTTTCTTp-3’のピーク面積に基づき定量したところ、5’-TTTTCTT(HP)TCCTTTT-3’の5’-側の切断産物の量は、メチルアミン処理の方がエチレンジアミン処理よりも2.1倍多いことが計算された。この結果より、メチルアミン又はエチレンジアミンの一級アミンを用いるDNAオリゴマーの切断反応において、最も反応速度の速いものはメチルアミンを用いる切断反応であることが示唆された。Was quantified based on the peak area of the 5'-T 6 (HP) T 6 -3 '5'a cleavage product of the side 5'-T 6 p-3' , 5'-T 6 (HP) The amount of cleavage product on the 5′-side of T 6 -3 ′ was calculated to be 2.4 times greater for the methylamine treatment than for the ethylenediamine treatment. In addition, when quantified based on the peak area of 5'-TTTTCTTp-3 'which is a 5'-side cleavage product of 5'-TTTTCTT (HP) TCCTTTT-3', 5'-TTTTCTT (HP) TCCTTTT-3 The amount of cleavage product on the 5'-side of 'was calculated to be 2.1 times higher with methylamine treatment than with ethylenediamine treatment. From this result, it was suggested that in the cleavage reaction of DNA oligomer using methylamine or primary amine of ethylenediamine, the one having the highest reaction rate is the cleavage reaction using methylamine.
合成例8で得られたHPは、合成例1で得られたPの三重結合に水が付加した構造を有する。Pは、求核剤として用いるアミン存在下で加熱することにより、三重結合にアミンが付加したアミン付加体を形成し、さらに互変異性及びアミンの脱離を経由して、HPを形成する可能性がある。
上記の反応式において、RNuは、求核剤付加に伴って生じる基を意味する。上記のオリゴデオキシヌクレオチドは、下記のような分子内環化反応を生じながら非天然塩基に由来する部分が脱離して、切断されると考えられる。
或いは、アミン付加体を含むオリゴデオキシヌクレオチドは、下記のような分子内環化反応を生じた後、切断されるとも考えられる。
<17. 合成例9:8-(2-ホルミルフェニル)-2’-デオキシグアノシンンを含むDNAオリゴマー>
[合成例9-1:8-(2-ホルミルフェニル)-2’-デオキシグアノシンンの合成]
[Synthesis Example 9-1: Synthesis of 8- (2-formylphenyl) -2′-deoxyguanosine]
8-ブロモ-2’-デオキシグアノシンンは、公知文献(Cation-Mediated Energy Transfer in G-Quadruplexes Revealed by an Internal Fluorescent Probe. Dumas A.及びLuedtke, N. W. J. Am. Chem. Soc. 2010年, 132, p. 18004-18007)に従って合成した。3.46 g(10.0 mmol)の8-ブロモ-2’-デオキシグアノシンン(分子量346.14)と3.00 g(20.0 mmol)の2-ホルミルベンゼンボロン酸(分子量149.94)と224 mg(1.00 mmol)の酢酸パラジウム(分子量224.51)と787 mg(3.00 mmol)のトリフェニルホスフィン(分子量262.29)と2.76 g(20.0 mmol)の炭酸カリウムをナスフラスコに加え、そこへ80 mLのアセトニトリルと40 mLの水を加え、80℃で4時間撹拌した。反応液の溶媒を減圧留去し、メタノールを200 mL加え、ろ過した。残渣をメタノールで洗浄した。ろ液を回収し、溶媒を減圧留去し、シリカゲルカラムで目的物質を精製した(展開溶媒は10〜20% MeOH/CH2Cl2)。目的のフラクションの溶媒を減圧留去し、少量のメタノールとアセトニトリルで生じる沈殿をろ過し、減圧下で乾燥した。ろ液を濃縮して少量のメタノールとアセトニトリルで生じる沈殿をろ過し、減圧下で乾燥する操作を計3回繰り返し、2.97 g(8.00 mmol, 収率80%)の目的物質(分子量371.35)を、薄黄色粉末として得た。8-Bromo-2'-deoxyguanosine is known in the literature (Cation-Mediated Energy Transfer in G-Quadruplexes Revealed by an Internal Fluorescent Probe. Dumas A. and Luedtke, NWJ Am. Chem. Soc. 2010, 132, p. 18004-18007). 3.46 g (10.0 mmol) 8-bromo-2'-deoxyguanosine (molecular weight 346.14), 3.00 g (20.0 mmol) 2-formylbenzeneboronic acid (molecular weight 149.94) and 224 mg (1.00 mmol) palladium acetate ( (Molecular weight 224.51), 787 mg (3.00 mmol) of triphenylphosphine (molecular weight 262.29) and 2.76 g (20.0 mmol) of potassium carbonate were added to an eggplant flask, and 80 mL of acetonitrile and 40 mL of water were added thereto, For 4 hours. The solvent of the reaction solution was distilled off under reduced pressure, 200 mL of methanol was added and filtered. The residue was washed with methanol. The filtrate was collected, the solvent was distilled off under reduced pressure, and the target substance was purified with a silica gel column (developing solvent was 10 to 20% MeOH / CH 2 Cl 2 ). The solvent of the objective fraction was distilled off under reduced pressure, and the precipitate formed with a small amount of methanol and acetonitrile was filtered and dried under reduced pressure. Concentrate the filtrate, filter the precipitate formed with a small amount of methanol and acetonitrile, and repeat the operation of drying under reduced pressure three times in total.2.97 g (8.00 mmol, 80% yield) of the target substance (molecular weight 371.35) Obtained as a pale yellow powder.
1H NMR(400 MHz, DMSO-d6) δ10.80 (br, 1H), 9.85 (s, 1H), 7.98 (dd, J = 7.8, 1.4 Hz, 1H),7.84-7.80 (m, 1H), 7.75-7.67 (m, 2H), 6.47 (br, 2H), 5.92 (dd, J = 8.2, 6.4 Hz, 1H), 5.10 (d, J = 4.1 Hz, 1H), 4.89-4.86 (m, 1H), 4.21-4.18 (m, 1H), 3.72-3.69 (m, 1H), 3.49-3.38 (m, 2H), 2.98 (ddd, J = 13.2, 8.2, 5.9 Hz, 1H), 4.20 (ddd, J = 13.3, 6.4, 2.3 Hz, 1H); 13C NMR(100 MHz, DMSO-d6) δ191.7, 156.5, 153.3, 151.8, 143.4, 135.5, 133.5, 132.5, 131.4, 130.1, 128.4, 117.5, 87.8, 84.6, 71.0, 61.9, 36.8; HRMS (ESI): C17H17N5O5Naに対する計算値: 394.1127 [M+Na]+; 実測値: 394.1115.
[合成例9-2:2-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-8-(2-ホルミルフェニル)-2’-デオキシグアノシンンの合成]
[Synthesis Example 9-2: Synthesis of 2-N- {N ′, N′-di (n-butyl) formamidine} -8- (2-formylphenyl) -2′-deoxyguanosine]
2.32 g(6.25 mmol)の8-(2-ホルミルフェニル)-2’-デオキシグアノシンン(分子量371.35)を20 mLのメタノールに溶解した。得られた溶液に11.7 mL(50.0 mmol)のN,N-ジ(n-ブチル)ホルムアミドジメチルアセタール(分子量203.32、密度0.87 g/mL) (Dialkylformamidines: depurination resistant N6-protecting group for deoxyadenosine. Froehler, B. C.及びMatteucci, M. D. Tetrahedron Letters 1983年, 11, p. 8031-8036)を加え、25℃で2時間撹拌した。溶媒を減圧留去し、シリカゲルカラムで目的物質を精製した(展開溶媒は5〜10% MeOH/CH2Cl2)。目的のフラクションの溶媒を減圧留去し、少量の酢酸エチルとヘキサンで生じる沈殿をろ過し、減圧下で乾燥した。2.10 g(4.11 mmol, 収率66%)の目的物質(分子量510.59)を、薄黄色粉末として得た。2.32 g (6.25 mmol) of 8- (2-formylphenyl) -2′-deoxyguanosine (molecular weight 371.35) was dissolved in 20 mL of methanol. 11.7 mL (50.0 mmol) N, N-di (n-butyl) formamide dimethyl acetal (molecular weight 203.32, density 0.87 g / mL) (Dialkylformamidines: depurination resistant N6-protecting group for deoxyadenosine.Froehler, BC And Matteucci,
1H NMR(400 MHz, DMSO-d6) δ11.51 (s, 1H), 9.89 (s, 1H), 8.53 (s, 1H), 8.02-8.00 (m, 1H), 7.87-7.83 (m, 1H), 7.77-7.73 (m, 1H), 7.69-7.67 (m, 1H), 5.92 (dd, J = 7.8, 6.9 Hz, 1H), 5.16 (d, J = 4.2 Hz, 1H), 4.85-4.82 (m, 1H), 4.33-4.29 (m, 1H), 3.76-3.73 (m, 1H), 3.58-3.35 (m, 6H), 3.14 (ddd, J = 13.7, 7.3, 6.4 Hz, 1H), 2.05 (ddd, J = 13.3, 6.4, 2.8 Hz, 1H), 1.63-1.54 (m, 4H), 1.35-1.23 (m, 4H), 0.94-0.89 (m, 6H); 13C NMR(100 MHz, DMSO-d6) δ191.7, 157.8, 157.4, 157.3, 150.5, 144.6, 135.5, 133.6, 132.1, 131.3, 130.2, 128.7, 120.5, 87.8, 85.0, 71.0, 62.0, 51.2, 44.9, 37.3, 30.3, 28.5, 19.6, 19.1, 13.7, 13.5; HRMS (ESI): C26H34N6O5Naに対する計算値: 533.2488 [M+Na]+; 実測値: 533.2481.
[合成例9-3:2-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-8-(2-ホルミルフェニル)-5’-O-ジメトキシトリチル-2’-デオキシグアノシンンの合成]
[Synthesis Example 9-3: 2-N- {N ′, N′-di (n-butyl) formamidine} -8- (2-formylphenyl) -5′-O-dimethoxytrityl-2′-deoxyguanosine Synthesis]
2.10 g(4.11 mmol)の2-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-8-(2-ホルミルフェニル)-2’-デオキシグアノシンン (分子量510.59)を16 mLのピリジンに溶解した。得られた溶液に1.53 g(4.52 mmol)の4,4’-ジメトキシトリチルクロリド(分子量338.83)を加え、25℃で1時間撹拌した。約10 mLのメタノールを加え、溶媒を減圧留去した。残渣に少量の塩化メチレンとヘキサンを加え、3回共沸した後、シリカゲルカラムで目的物質を精製した(展開溶媒は2% Et3N, AcOEt:n-ヘキサン=8:2〜AcOEt)。目的のフラクションの溶媒を減圧留去し、少量の塩化メチレンとヘキサンを加え共沸した。2.20 g(2.71 mmol, 収率66%)の目的生成物(分子量812.95)を、薄黄色粉末として得た。16 mL of 2.10 g (4.11 mmol) 2-N- {N ', N'-di (n-butyl) formamidine} -8- (2-formylphenyl) -2'-deoxyguanosine (molecular weight 510.59) In pyridine. 1.53 g (4.52 mmol) of 4,4′-dimethoxytrityl chloride (molecular weight 338.83) was added to the resulting solution, and the mixture was stirred at 25 ° C. for 1 hour. About 10 mL of methanol was added, and the solvent was distilled off under reduced pressure. A small amount of methylene chloride and hexane were added to the residue and azeotroped three times, and then the target substance was purified with a silica gel column (developing solvent was 2% Et 3 N, AcOEt: n-hexane = 8: 2 to AcOEt). The solvent of the objective fraction was distilled off under reduced pressure, and a small amount of methylene chloride and hexane were added and azeotroped. 2.20 g (2.71 mmol, 66% yield) of the desired product (molecular weight 812.95) was obtained as a pale yellow powder.
1H NMR(400 MHz, DMSO-d6) δ11.52 (s, 1H), 9.88 (s, 1H), 8.38 (s, 1H), 7.98 (d, J = 7.8 Hz, 1H), 7.80-7.79 (m, 2H), 7.74-7.70 (m, 1H), 7.31-7.29 (m, 2H), 7.20-7.15 (m, 7H), 6.79-6.72 (m, 4H), 6.00 (dd, J = 8.0, 4.8 Hz, 1H), 5.19 (d, J = 5.1 Hz, 1H), 4.54-4.48 (m, 1H), 3.84-3.80 (m, 1H), 3.70 (s, 3H), 3.69 (s, 3H), 3.47-3.43 (m, 2H), 3.31-3.26 (m, 1H), 3.21-3.13 (m, 3H), 3.07-3.04 (m, 1H), 2.17 (ddd, J = 13.7, 8.0, 5.8 Hz, 1H), 1.59-1.51 (m, 2H), 1.45-1.36 (m, 2H), 1.33-1.24 (m, 2H), 1.15-1.09 (m, 2H), 0.93-0.89 (m, 3H), 0.79-0.75 (m, 3H); 13C NMR(100 MHz, DMSO-d6) δ191.6, 158.0, 157.9, 157.5, 157.3, 156.9, 150.3, 144.9, 144.8, 135.7, 135.6, 133.4, 132.5, 131.2, 130.1, 129.6, 129.4, 128.3, 127.6, 126.5, 120.4, 112.9, 85.4, 85.2, 84.0, 70.6, 63.6, 55.0, 54.9, 51.2, 45.0, 37.6, 30.2, 28.5, 19.6, 19.0, 13.7, 13.5; HRMS (ESI): C47H53N6O7に対する計算値: 813.3976 [M+H]+; 実測値: 813.3976.
[合成例9-4:2-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-8-(2-ホルミルフェニル)-5’-O-ジメトキシトリチル-2’-デオキシグアノシンン-3’-N,N-ジイソプロピル(シアノエチル)ホスホロアミダイトの合成]
[Synthesis Example 9-4: 2-N- {N ′, N′-di (n-butyl) formamidine} -8- (2-formylphenyl) -5′-O-dimethoxytrityl-2′-deoxyguanosine Of 3'-N, N-diisopropyl (cyanoethyl) phosphoramidite]
163 mg(0.200 mmol)の2-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-8-(2-ホルミルフェニル)-5’-O-ジメトキシトリチル-2’-デオキシグアノシンン(分子量812.95)をナスフラスコに入れ5 mLのCH3CNと223μL(1.600 mmol)のトリエチルアミン(分子量101.19, 比重0.726 g/mL)を加え、撹拌し、さらに178μL(0.800 mmol)の2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロアミダイト(分子量236.68, 比重1.061 g/mL)を加え、室温で30分間撹拌した。25 mLのAcOEtと25 mLの飽和食塩水を加え、分液し、有機層をさらに2回飽和食塩水で洗った。得られた有機層を、無水硫酸マグネシウムで乾燥した後、フィルターでろ過し、ろ液の溶媒を減圧留去した。アセトニトリルで3回共沸した後、10 mLのアセトニトリルを加えて溶解し、少量のモレキュラーシーブを加えた。DNA自動合成機に取り付けてDNA合成に使用した。163 mg (0.200 mmol) 2-N- {N ', N'-di (n-butyl) formamidine} -8- (2-formylphenyl) -5'-O-dimethoxytrityl-2'-deoxyguanosine (Molecular weight 812.95) was added to an eggplant flask, 5 mL of CH 3 CN and 223 μL (1.600 mmol) of triethylamine (molecular weight 101.19, specific gravity 0.726 g / mL) were added, stirred, and further 178 μL (0.800 mmol) of 2-cyanoethyl. N, N, N ′, N′-tetraisopropyl phosphoramidite (molecular weight 236.68, specific gravity 1.061 g / mL) was added, and the mixture was stirred at room temperature for 30 minutes. 25 mL of AcOEt and 25 mL of saturated saline were added, and the mixture was separated. The organic layer was further washed twice with saturated brine. The obtained organic layer was dried over anhydrous magnesium sulfate and filtered through a filter, and the solvent of the filtrate was distilled off under reduced pressure. After azeotroping with acetonitrile three times, 10 mL of acetonitrile was added and dissolved, and a small amount of molecular sieve was added. Attached to an automatic DNA synthesizer and used for DNA synthesis
[合成例9-5:8-(2-ホルミルフェニル)-2’-デオキシグアノシン(fPG)を含むDNAオリゴマーの合成]
通常のDNA自動合成機のDNA合成のプロトコールで1μmolスケール、DMTr ONでホスホロアミダイト法により合成した。担体からの切り出しは28%アンモニア水で行い、脱保護は25℃で16時間静置することにより行った。脱保護後、アンモニアを遠心濃縮装置により減圧留去し、DNAを含む溶液を0.45μmのフィルターに通した後、逆相HPLCによりメインピークを精製し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置と凍結乾燥によりギ酸アンモニウムと溶媒を留去した。得られた残渣に100μLの80%酢酸水溶液を加え、よく混合し、25℃で30分間静置した。その後、前記混合物に蒸留水を約1 mLとなるように加えて、0.45μmのフィルターに通した後、逆相HPLCにより13〜14分の保持時間に溶出されたメインピークを精製し(5-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置と凍結乾燥によりギ酸アンモニウムと溶媒とを留去した。得られたDNAオリゴマーはMALDI TOF-MS測定により同定した。合成したDNAオリゴマーは、約500μLの蒸留水を加えて水溶液とした。The DNA was synthesized by the phosphoramidite method with DMTr ON using a DNA synthesis protocol of a normal automatic DNA synthesizer with 1 μmol scale. Cutting out from the carrier was performed with 28% aqueous ammonia, and deprotection was performed by standing at 25 ° C. for 16 hours. After deprotection, the ammonia was distilled off under reduced pressure using a centrifugal concentrator, the solution containing DNA was passed through a 0.45 μm filter, and the main peak was purified by reverse-phase HPLC (0-50% CH 3 CN / 50 mM formic acid. Ammonium aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and solvent were distilled off by centrifugal concentrator and lyophilization. To the obtained residue, 100 μL of 80% aqueous acetic acid was added, mixed well, and allowed to stand at 25 ° C. for 30 minutes. Thereafter, distilled water was added to the mixture to make about 1 mL, passed through a 0.45 μm filter, and then the main peak eluted at a retention time of 13 to 14 minutes was purified by reverse phase HPLC (5- 20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and the solvent were distilled off by centrifugal concentrator and lyophilization. The obtained DNA oligomer was identified by MALDI TOF-MS measurement. The synthesized DNA oligomer was made into an aqueous solution by adding about 500 μL of distilled water.
以下において、8-(2-ホルミルフェニル)-2’-デオキシグアノシンン:
を「fPG」で示す。 Is indicated by “fPG”.
DNAの濃度決定は、fPGをCと近似して260 nmのモル吸光係数を計算し、260 nmのUV吸収より濃度を計算した。 For determination of DNA concentration, the molar extinction coefficient at 260 nm was calculated by approximating fPG to C, and the concentration was calculated from UV absorption at 260 nm.
合成したDNAオリゴマーの逆相HPLCクロマトグラムを図65及び66に示す。 65 and 66 show reverse phase HPLC chromatograms of the synthesized DNA oligomers.
合成したDNAオリゴマーの上記逆相HPLCにおける保持時間は以下の通りである。 The retention time in the reverse phase HPLC of the synthesized DNA oligomer is as follows.
5’-TTTTTT(fPG)TTTTTT-3’について14.6分(配列番号30)
5’-TTTTCTT(fPG)TCCTTTT-3’について14.0分(配列番号31)
合成したDNAオリゴマーのMALDI TOF-MSのデータは以下の通りである。14.6 minutes for 5'-TTTTTT (fPG) TTTTTT-3 '(SEQ ID NO: 30)
14.0 minutes for 5'-TTTTCTT (fPG) TCCTTTT-3 '(SEQ ID NO: 31)
The MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
5’-TTTTTT(fPG)TTTTTT-3’について分子式C137H173N29O89P12、[M-H]-の計算値 4020.7、実測値 4022.0(配列番号30)
5’-TTTTCTT(fPG)TCCTTTT-3’について分子式C154H196N36O100P14、[M-H]-の計算値 4584.0、実測値 4584.3(配列番号31)
<18. 使用例9: 8-(2-ホルミルフェニル)-2’-デオキシグアノシンン(fPG)を含むDNAオリゴマー>
[使用例9-1:8-(2-ホルミルフェニル)-2’-デオキシグアノシンン(fPG)を含むDNAオリゴマーのメチルアミンによる切断反応と生成物の同定]
スクリューチューブに300μLの5’-T6(fPG)T6-3’又は5’-TTTTCTT(fPG)TCCTTTT-3’の水溶液(ストランド濃度100μM)と300μLのメチルアミン(40% 水溶液)とを加え、水浴中で70℃、6時間加熱した。反応後は、メチルアミンを遠心濃縮装置で減圧留去し、逆相HPLCにより分析して生成物を分取した(5-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。分取した生成物は、MALDI TOF-MSにより分子量を測定した。5'-TTTTTT (fPG) TTTTTT- 3 ' molecular formula C 137 H 173 N 29 O 89 P 12 about, [MH] - calcd 4020.7, found 4022.0 (SEQ ID NO: 30) of
For 5′-TTTTCTT (fPG) TCCTTTT-3 ′, the molecular formula C 154 H 196 N 36 O 100 P 14 , calculated value of [MH] − 4584.0, measured value 4584.3 (SEQ ID NO: 31)
<18. Use Example 9: DNA oligomer containing 8- (2-formylphenyl) -2'-deoxyguanosine (fPG)>
[Use Example 9-1: Cleavage reaction of methyl oligomer containing 8- (2-formylphenyl) -2'-deoxyguanosine (fPG) with methylamine and identification of the product]
Add 300 μL of 5′-T 6 (fPG) T 6 -3 ′ or 5′-TTTTCTT (fPG) TCCTTTT-3 ′ aqueous solution (
合成した各DNAオリゴマーの反応後の逆相HPLCクロマトグラムを図67及び68に示す。 67 and 68 show reverse phase HPLC chromatograms after the reaction of each synthesized DNA oligomer.
各DNAオリゴマーにおける生成物1及び2のMALDI TOF-MSのデータは以下の通りである。以下において、「RNu」は、切断反応に伴って生成するfPG由来の部分を意味する。The MALDI TOF-MS data of
5’-TTTTTT(fPG)TTTTTT-3’について
生成物1:pTTTTTT 分子式C60H80N12O43P6 FW 1843.2 [M-H]- 1842.2 実測値 1842.2
HPLCの保持時間 12.0分
生成物2:TTTTTTpRNu 分子式C66H91N13O45P6 FW 1972.3 [M-H]- 1971.3 実測値 1971.0
HPLCの保持時間 13.9分
5’-TTTTCTT(fPG)TCCTTTT-3’について
生成物1:pTCCTTTT 分子式C68H91N16O48P7 FW 2117.3 [M-H]- 2116.3 実測値 2116.2
HPLCの保持時間11.1分
生成物2:TTTTCTTpRNu 分子式C75H103N16O51P7 FW 2261.5 [M-H]- 2260.5 実測値 2260.5
HPLCの保持時間 13.3分
メチルアミンによるDNAオリゴマーの分解反応を下記に示す。下記反応式において、左側の生成物が生成物2に、右側の生成物が生成物1に、それぞれ対応する。
HPLC retention time 12.0 min product 2: TTTTTTpR Nu molecular formula C 66 H 91 N 13 O 45 P 6 FW 1972.3 [MH] - 1971.3 Found 1971.0
HPLC retention time 13.9 minutes
5'-TTTTCTT (fPG) TCCTTTT- 3 ' product for 1: pTCCTTTT molecular formula C 68 H 91 N 16 O 48 P 7 FW 2117.3 [MH] - 2116.3 Found 2116.2
HPLC retention time 11.1 min product 2: TTTTCTTpR Nu molecular formula C 75 H 103 N 16 O 51 P 7 FW 2261.5 [MH] - 2260.5 Found 2260.5
HPLC retention time 13.3 minutes Degradation reaction of DNA oligomer by methylamine is shown below. In the following reaction formula, the product on the left corresponds to the
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
<19. 合成例10:8-(2-ホルミルフェニル)-2’-デオキシアデノシンを含むDNAオリゴマー>
[合成例10-1:8-(2-ホルミルフェニル)-2’-デオキシアデノシンの合成]
[Synthesis Example 10-1: Synthesis of 8- (2-formylphenyl) -2′-deoxyadenosine]
8-ブロモ-2’-デオキシアデノシンは、公知文献(特開2011-37796号公報)に従って合成した。2.20 g(6.66 mmol)の8-ブロモ-2’-デオキシアデノシン(分子量330.14)と2.00g(13.3 mmol)の2-ホルミルベンゼンボロン酸(分子量149.94)と150 mg(0.666 mmol)の酢酸パラジウム(分子量224.51)と525 mg(2.00 mmol)のトリフェニルホスフィン(分子量262.29)と1.84 g(13.3 mmol)の炭酸カリウムをナスフラスコに加え、そこへ40 mLのアセトニトリルと20 mLの水を加え、80℃で4時間撹拌した。反応液の溶媒を減圧留去し、メタノールを200 mL加え、ろ過した。残渣をメタノールで洗浄した。ろ液を回収し、溶媒を減圧留去し、シリカゲルカラムで目的物質を精製した(展開溶媒は10% MeOH/CH2Cl2)。目的のフラクションの溶媒を減圧留去し、少量のアセトニトリルに溶かし、塩化メチレンを加え、さらにヘキサンを加えることで生じる沈殿をろ過し、減圧下で乾燥した。1.81 g(5.09 mmol, 収率76%)の目的物質(分子量355.35)を、白色粉末として得た。8-Bromo-2'-deoxyadenosine was synthesized according to a known document (Japanese Patent Laid-Open No. 2011-37796). 2.20 g (6.66 mmol) 8-bromo-2'-deoxyadenosine (molecular weight 330.14), 2.00 g (13.3 mmol) 2-formylbenzeneboronic acid (molecular weight 149.94) and 150 mg (0.666 mmol) palladium acetate (molecular weight) 224.51) and 525 mg (2.00 mmol) of triphenylphosphine (molecular weight 262.29) and 1.84 g (13.3 mmol) of potassium carbonate were added to an eggplant flask, and 40 mL of acetonitrile and 20 mL of water were added thereto at 80 ° C. Stir for 4 hours. The solvent of the reaction solution was distilled off under reduced pressure, 200 mL of methanol was added and filtered. The residue was washed with methanol. The filtrate was collected, the solvent was distilled off under reduced pressure, and the target substance was purified with a silica gel column (developing solvent was 10% MeOH / CH 2 Cl 2 ). The solvent of the objective fraction was distilled off under reduced pressure, dissolved in a small amount of acetonitrile, methylene chloride was added, and the precipitate formed by adding hexane was filtered and dried under reduced pressure. 1.81 g (5.09 mmol, yield 76%) of the target substance (molecular weight 355.35) was obtained as a white powder.
1H NMR(400 MHz, DMSO-d6) δ9.93 (s, 1H), 8.18 (s, 1H), 8.09-8.07 (m, 1H), 7.90-7.80 (m, 2H), 7.74-7.72 (m, 1H), 7.46 (br, 2H), 5.90 (dd, J = 8.5, 6.2 Hz, 1H), 5.49 (dd, J = 8.3, 4.1 Hz, 1H), 5.17 (d, J = 4.1 Hz, 1H), 4.39-4.35 (m, 1H), 3.82-3.79 (m, 1H), 3.65-3.59 (m, 1H), 3.50-3.44 (m, 1H), 3.19 (ddd, J = 12.8, 8.7, 6.0 Hz, 1H), 2.06 (ddd, J = 13.0, 6.2, 1.8 Hz, 1H); 13C NMR(100 MHz, DMSO-d6) δ191.7, 156.2, 152.3, 149.6, 147.4, 135.4, 133.8, 131.5, 131.4, 130.8, 129.5, 119.3, 88.3, 85.7, 71.3, 62.2, 37.4; HRMS (ESI): C17H18N5O4に対する計算値: 356.1359 [M+H]+; 実測値: 356.1364.
[合成例10-2:6-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-8-(2-ホルミルフェニル)-2’-デオキシアデノシンの合成]
[Synthesis Example 10-2: Synthesis of 6-N- {N ′, N′-di (n-butyl) formamidine} -8- (2-formylphenyl) -2′-deoxyadenosine]
1.55 g(4.36 mmol)の8-(2-ホルミルフェニル)-2’-デオキシアデノシン(分子量355.35)を20 mLのメタノールに溶解し、得られた溶液に8.14 mL(34.8 mmol)のN,N-ジ(n-ブチル)ホルムアミドジメチルアセタール(分子量203.32、密度0.87 g/mL)(Dialkylformamidines: depurination resistant N6-protecting group for deoxyadenosine. Froehler, B. C.及びMatteucci, M. D. Tetrahedron Letters 1983年, 11, p. 8031-8036)を加え、25℃で2時間撹拌した。溶媒を減圧留去し、シリカゲルカラムで目的物質を精製した(展開溶媒は2〜5% MeOH/CH2Cl2)。目的のフラクションの溶媒を減圧留去し、少量の塩化メチレンを加え、ジエチルエーテルを加え、さらにヘキサンを加えることで生じる沈殿をろ過し、減圧下で乾燥した。1.40 g(2.83 mmol, 収率65%)の目的物質(分子量494.59)を、白色粉末として得た。1.55 g (4.36 mmol) of 8- (2-formylphenyl) -2'-deoxyadenosine (molecular weight 355.35) was dissolved in 20 mL of methanol, and 8.14 mL (34.8 mmol) of N, N- Dialkylformamidines: depurination resistant N6-protecting group for deoxyadenosine. Froehler, BC and Matteucci,
1H NMR(400 MHz, DMSO-d6) δ9.92 (s, 1H), 8.91 (s, 1H), 8.45 (s, 1H), 8.09 (d, J = 7.8 Hz, 1H), 7.91-7.82 (m, 2H), 7.74 (d, J = 7.3 Hz, 1H), 5.94 (dd, J = 7.8, 6.8 Hz, 1H), 5.28 (dd, J = 7.6, 4.4 Hz, 1H), 5.18 (d, J = 4.1 Hz, 1H), 4.40-4.36 (m, 1H), 3.81-3.77 (m, 1H), 3.64-3.41 (m, 6H), 3.25-3.18(m, 1H), 2.09-2.04 (m, 1H), 1.62-1.52 (m, 4H), 1.33-1.22 (m, 4H), 0.94-0.83 (m, 6H); 13C NMR(100 MHz, DMSO-d6) δ191.8, 159.5, 157.9, 151.7, 151.6, 149.3, 135.5, 133.8, 131.5, 131.3, 130.9, 129.7, 125.8, 88.2, 85.5, 71.2, 62.1, 50.9, 44.4, 37.2, 30.4, 28.7, 19.6, 19.1,13.7, 13.5; HRMS (ESI): C26H35N6O4に対する計算値: 495.2720 [M+H]+; 実測値: 495.2718.
[合成例10-3:6-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-8-(2-ホルミルフェニル)-5’-O-ジメトキシトリチル-2’-デオキシアデノシンの合成]
[Synthesis Example 10-3: 6-N- {N ′, N′-di (n-butyl) formamidine} -8- (2-formylphenyl) -5′-O-dimethoxytrityl-2′-deoxyadenosine Synthesis]
1.31 g(2.64 mmol)の6-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-8-(2-ホルミルフェニル)-2’-デオキシグアノシン (分子量494.59)を10 mLのピリジンに溶解した。得られた溶液に991 mg(2.92 mmol)の4,4’-ジメトキシトリチルクロリド(分子量338.83)を加え、25℃で1時間撹拌した。約10 mLのメタノールを加え、溶媒を減圧留去した。残渣に少量の塩化メチレンとヘキサンを加え、3回共沸した後、シリカゲルカラムで目的物質を精製した(展開溶媒は2% Et3N, 2% MeOH/CH2Cl2)。目的のフラクションの溶媒を減圧留去し、少量の塩化メチレンとヘキサンを加え共沸した。1.24 g(1.56 mmol, 収率59%)の目的生成物(分子量796.95)を、白色粉末として得た。1.31 g (2.64 mmol) 6-N- {N ', N'-di (n-butyl) formamidine} -8- (2-formylphenyl) -2'-deoxyguanosine (molecular weight 494.59) in 10 mL Dissolved in pyridine. To the obtained solution, 991 mg (2.92 mmol) of 4,4′-dimethoxytrityl chloride (molecular weight: 338.83) was added and stirred at 25 ° C. for 1 hour. About 10 mL of methanol was added, and the solvent was distilled off under reduced pressure. A small amount of methylene chloride and hexane were added to the residue, and the mixture was azeotroped three times. Then, the target substance was purified with a silica gel column (developing solvent was 2% Et 3 N, 2% MeOH / CH 2 Cl 2 ). The solvent of the objective fraction was distilled off under reduced pressure, and a small amount of methylene chloride and hexane were added and azeotroped. 1.24 g (1.56 mmol, 59% yield) of the desired product (molecular weight 796.95) was obtained as a white powder.
1H NMR(400 MHz, DMSO-d6) δ9.89 (s, 1H), 8.88 (s, 1H), 8.25 (s, 1H), 8.06-8.04 (m, 1H), 7.88-7.78 (m, 3H), 7.33-7.31 (m, 2H), 7.21-7.15 (m, 7H), 6.81-6.73 (m, 4H), 5.98 (t, J = 6.9 Hz, 1H), 5.25 (d, J = 4.6 Hz, 1H), 4.57-4.53 (m, 1H), 3.94-3.90 (m, 1H), 3.71 (s, 3H), 3.69 (s, 3H), 3.59-3.53 (m, 2H), 3.44-3.32 (m, 3H), 3.20-3.12 (m, 2H), 2.11 (ddd, J = 13.0, 7.3, 4.3 Hz, 1H), 1.62-1.52 (m, 4H), 1.34-1.23 (m, 4H), 0.90-0.87 (m, 6H); 13C NMR(100 MHz, DMSO-d6) δ191.6, 159.2, 157.9, 157.9, 157.7, 151.8, 149.5, 145.0, 135.7, 135.60, 135.55, 133.6, 131.7, 131.4, 130.7, 129.7, 129.5, 129.1, 127.6, 126.5, 125.6, 113.0, 112.9, 85.8, 85.2, 84.8, 71.0, 63.6, 54.94, 54.90, 50.9, 44.4, 36.3, 30.4, 28.7, 19.6, 19.1, 13.7, 13.5; HRMS (ESI): C47H53N6O6に対する計算値: 797.4027 [M+H]+; 実測値: 797.4046.
[合成例10-4:6-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-8-(2-ホルミルフェニル)-5’-O-ジメトキシトリチル-2’-デオキシアデノシン-3’-N,N-ジイソプロピル(シアノエチル)ホスホロアミダイトの合成]
[Synthesis Example 10-4: 6-N- {N ′, N′-di (n-butyl) formamidine} -8- (2-formylphenyl) -5′-O-dimethoxytrityl-2′-deoxyadenosine Synthesis of -3'-N, N-diisopropyl (cyanoethyl) phosphoramidite]
159 mg(0.200 mmol)の6-N-{N’,N’-ジ(n-ブチル)ホルムアミジン}-8-(2-ホルミルフェニル)-5’-O-ジメトキシトリチル-2’-デオキシアデノシン(分子量796.95)をナスフラスコに入れ5 mLのCH3CNと223μL(1.600 mmol)のトリエチルアミン(分子量101.19, 比重0.726 g/mL)を加え、撹拌し、さらに178μL(0.800 mmol)の2-シアノエチル N,N,N’,N’-テトライソプロピルホスホロジアミダイト(分子量236.68, 比重1.061 g/mL)を加え、室温で30分間撹拌した。25 mLのAcOEtと25 mLの飽和食塩水を加え、分液し、有機層をさらに2回飽和食塩水で洗った。有機層を無水硫酸マグネシウムで乾燥した後、フィルターでろ過し、ろ液の溶媒を減圧留去した。アセトニトリルで3回共沸した後、10 mLのアセトニトリルを加えて溶解し、少量のモレキュラーシーブを加えた。DNA自動合成機に取り付けてDNA合成に使用した。159 mg (0.200 mmol) 6-N- {N ', N'-di (n-butyl) formamidine} -8- (2-formylphenyl) -5'-O-dimethoxytrityl-2'-deoxyadenosine (Molecular weight 796.95) was placed in an eggplant flask, 5 mL of CH 3 CN and 223 μL (1.600 mmol) of triethylamine (molecular weight 101.19, specific gravity 0.726 g / mL) were added, stirred, and 178 μL (0.800 mmol) of 2-cyanoethyl N , N, N ′, N′-Tetraisopropylphosphorodiamidite (molecular weight 236.68, specific gravity 1.061 g / mL) was added, and the mixture was stirred at room temperature for 30 minutes. 25 mL of AcOEt and 25 mL of saturated saline were added, and the mixture was separated. The organic layer was further washed twice with saturated brine. The organic layer was dried over anhydrous magnesium sulfate and filtered through a filter, and the solvent of the filtrate was distilled off under reduced pressure. After azeotroping with acetonitrile three times, 10 mL of acetonitrile was added and dissolved, and a small amount of molecular sieve was added. Attached to an automatic DNA synthesizer and used for DNA synthesis
[合成例10-5:8-(2-ホルミルフェニル)-2’-デオキシアデノシン(fPA)を含むDNAオリゴマーの合成]
通常のDNA自動合成機のDNA合成のプロトコールで1μmolスケール、DMTr ONでホスホロアミダイト法により合成した。切り出しは28%アンモニア水で行い、脱保護は25℃で16時間静置することにより行った。脱保護後、アンモニアを遠心濃縮装置により減圧留去し、DNAを含む溶液を0.45μmのフィルターに通した後、逆相HPLCによりメインピークを精製し(0-50% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置と凍結乾燥によりギ酸アンモニウムと溶媒を留去した。得られた残渣に100μLの80%酢酸水溶液を加え、よく混合し、25℃で30分間静置した。その後、前記混合物に蒸留水を約1 mLとなるように加えて、0.45μmのフィルターに通した後、逆相HPLCで13-14分の保持時間に溶出されたメインピークを精製し(5-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)、遠心濃縮装置と凍結乾燥によりギ酸アンモニウムと溶媒とを留去した。得られたDNAオリゴマーはMALDI TOF-MS測定により同定した。合成したDNAオリゴマーは、約500μLの蒸留水を加えて水溶液とした。The DNA was synthesized by the phosphoramidite method with DMTr ON using a DNA synthesis protocol of a normal automatic DNA synthesizer with 1 μmol scale. Cutout was performed with 28% aqueous ammonia, and deprotection was performed by standing at 25 ° C. for 16 hours. After deprotection, the ammonia was distilled off under reduced pressure using a centrifugal concentrator, the solution containing DNA was passed through a 0.45 μm filter, and the main peak was purified by reverse-phase HPLC (0-50% CH 3 CN / 50 mM formic acid. Ammonium aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and solvent were distilled off by centrifugal concentrator and lyophilization. To the obtained residue, 100 μL of 80% aqueous acetic acid was added, mixed well, and allowed to stand at 25 ° C. for 30 minutes. Thereafter, distilled water was added to the mixture so as to be about 1 mL, passed through a 0.45 μm filter, and then the main peak eluted at a retention time of 13-14 minutes was purified by reverse phase HPLC (5- 20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and the solvent were distilled off by centrifugal concentrator and lyophilization. The obtained DNA oligomer was identified by MALDI TOF-MS measurement. The synthesized DNA oligomer was made into an aqueous solution by adding about 500 μL of distilled water.
以下において、8-(2-ホルミルフェニル)-2’-デオキシアデノシン:
を「fPA」で示す。 Is indicated by “fPA”.
DNAの濃度決定は、fPAをCと近似して260 nmのモル吸光係数を計算し、260 nmのUV吸収より濃度を計算した。 The concentration of DNA was determined by calculating the molar extinction coefficient at 260 nm by approximating fPA to C, and calculating the concentration from the UV absorption at 260 nm.
合成したDNAオリゴマーの逆相HPLCクロマトグラムを図69及び70に示す。 69 and 70 show reverse phase HPLC chromatograms of the synthesized DNA oligomers.
合成したDNAオリゴマーの上記逆相HPLCにおける保持時間は以下の通りである。 The retention time in the reverse phase HPLC of the synthesized DNA oligomer is as follows.
5’-TTTTTT(fPA)TTTTTT-3’について14.9分(配列番号32)
5’-TTTTCTT(fPA)TCCTTTT-3’について13.8分(配列番号33)
合成したDNAオリゴマーのMALDI TOF-MSのデータは以下の通りである。14.9 minutes for 5'-TTTTTT (fPA) TTTTTT-3 '(SEQ ID NO: 32)
13.8 minutes for 5'-TTTTCTT (fPA) TCCTTTT-3 '(SEQ ID NO: 33)
The MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
5’-TTTTTT(fPA)TTTTTT-3’について分子式C137H173N29O88P12、[M-H]-の計算値 4004.7、実測値 4005.2(配列番号32)
5’-TTTTCTT(fPA)TCCTTTT-3’について分子式C154H196N36O99P14、[M-H]-の計算値 4568.0、実測値 4568.5(配列番号33)
<20. 使用例10: 8-(2-ホルミルフェニル)-2’-デオキシアデノシン(fPA)を含むDNAオリゴマー>
[使用例10-1:8-(2-ホルミルフェニル)-2’-デオキシアデノシン(fPA)を含むDNAオリゴマーのメチルアミンによる切断反応と生成物の同定]
スクリューチューブに300μLの5’-T6(fPA)T6-3’又は5’-TTTTCTT(fPA)TCCTTTT-3’の水溶液(ストランド濃度 100μM)と300μLのメチルアミン(40% 水溶液)とを加え、水浴中で70℃、6時間加熱した。反応後は、メチルアミンを遠心濃縮装置で減圧留去し、逆相HPLCにより分析して生成物を分取した(5-20% CH3CN/50 mM ギ酸アンモニウム水溶液, 20分, 254 nmで検出)。分取した生成物は、MALDI TOF-MSにより分子量を測定した。5'-TTTTTT (fPA) TTTTTT- 3 ' molecular formula C 137 H 173 N 29 O 88 P 12 about, [MH] - Calculated 4004.7, Found 4005.2 (SEQ ID NO: 32)
For 5′-TTTTCTT (fPA) TCCTTTT-3 ′, the molecular formula C 154 H 196 N 36 O 99 P 14 , calculated value of [MH] − 4568.0, measured value 4568.5 (SEQ ID NO: 33)
<20. Use Example 10: DNA oligomer containing 8- (2-formylphenyl) -2'-deoxyadenosine (fPA)>
[Use Example 10-1: Cleavage reaction of methyl oligomer containing 8- (2-formylphenyl) -2'-deoxyadenosine (fPA) with methylamine and identification of the product]
Add 300 µL of 5'-T 6 (fPA) T 6 -3 'or 5'-TTTTCTT (fPA) TCCTTTT-3' aqueous solution (
合成した各DNAオリゴマーの反応後の逆相HPLCクロマトグラムを図71及び72に示す。 71 and 72 show reverse phase HPLC chromatograms after the reaction of each synthesized DNA oligomer.
各DNAオリゴマーにおける生成物1及び2のMALDI TOF-MSのデータは以下の通りである。以下において、「RNu」は、切断反応に伴って生成するfPA由来の部分を意味する。The MALDI TOF-MS data of
5’-TTTTTT(fPA)TTTTTT-3’について
生成物1:pTTTTTT 分子式C60H80N12O43P6 FW 1843.2 [M-H]- 1842.2 実測値 1842.0と4019.5(後者の分子量ピークの方が大きい)
HPLCの保持時間 12.1分
生成物2:TTTTTTpRNu 分子式C66H91N13O45P6 FW 1972.3 [M-H]- 1971.3 実測値 1971.2
HPLCの保持時間 13.9分
5’-TTTTCTT(fPA)TCCTTTT-3’について
生成物1:pTCCTTTT 分子式C68H91N16O48P7 FW 2117.3 [M-H]- 2116.3 実測値 2116.2
HPLCの保持時間11.1分
生成物2:TTTTCTTpRNu 分子式C75H103N16O51P7 FW 2261.5 [M-H]- 2260.5 実測値 2260.6
HPLCの保持時間 13.2分
生成物の分子量より、5’-T6(fPA)T6-3’に関しては、生成物1と同じ保持時間に別の生成物が同時に溶出されたことが予想される。メチルアミンとアデノシン類似体上のアルデヒドにより、安定なイミン(分子式C138H176N30O87P12、分子量4018.7)の生成が予想される。5'-TTTTTT (fPA) TTTTTT- 3 ' product for 1: pTTTTTT molecular formula C 60 H 80 N 12 O 43 P 6 FW 1843.2 [MH] - 1842.2 Found 1842.0 and 4019.5 (the larger the latter molecular weight peak)
HPLC retention time 12.1 min product 2: TTTTTTpR Nu molecular formula C 66 H 91 N 13 O 45 P 6 FW 1972.3 [MH] - 1971.3 Found 1971.2
HPLC retention time 13.9 minutes
5'-TTTTCTT (fPA) TCCTTTT- 3 ' product for 1: pTCCTTTT molecular formula C 68 H 91 N 16 O 48 P 7 FW 2117.3 [MH] - 2116.3 Found 2116.2
HPLC retention time 11.1 min product 2: TTTTCTTpR Nu molecular formula C 75 H 103 N 16 O 51 P 7 FW 2261.5 [MH] - 2260.5 Found 2260.6
HPLC retention time 13.2 minutes From the molecular weight of the product, for 5'-T 6 (fPA) T 6 -3 ', it is expected that another product was eluted at the same retention time as
メチルアミンによるDNAオリゴマーの分解反応を下記に示す。下記反応式において、左側の生成物が生成物2に、右側の生成物が生成物1に、それぞれ対応する。
前記の反応式において、A及びCは、上記で定義された式Iと同様の意味である。生成物の分子量より、RNuは、分解したヌクレオチドに求核剤が一分子付加して非天然型塩基が除去されてできたものと予想される。In the above reaction formula, A and C have the same meaning as in formula I defined above. Based on the molecular weight of the product, R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
fPG又はfPAを含むオリゴデオキシヌクレオチドは、下記のような分子内環化反応を生じながら非天然塩基に由来する部分が脱離して、切断されると考えられる。下記反応式は、fPGを含むオリゴデオキシヌクレオチドの場合であるが、fPAを含むオリゴデオキシヌクレオチドの場合も同様の反応が進行すると考えられる。
<21. 使用例11: 5-エチニル-2’-デオキシウリジン(Pa)を含むDNAオリゴマーを用いたポリメラーゼ連鎖反応による比較的長いDNAの増幅及び切断と増幅産物の連結反応>
[使用例11-1:5-エチニル-2’-デオキシウリジン(Pa)を含むDNAオリゴマーを用いたポリメラーゼ連鎖反応によるDNAの増幅]
合成例3と同様の手順で、下記の塩基配列を有する5-エチニル-2’-デオキシウリジン(Pa)を含むDNAオリゴマーを合成した。<21. Use Example 11: Amplification and cleavage of relatively long DNA by polymerase chain reaction using a DNA oligomer containing 5-ethynyl-2′-deoxyuridine (Pa) and ligation of amplification products>
[Use Example 11-1: Amplification of DNA by polymerase chain reaction using a DNA oligomer containing 5-ethynyl-2′-deoxyuridine (Pa)]
A DNA oligomer containing 5-ethynyl-2′-deoxyuridine (Pa) having the following base sequence was synthesized in the same procedure as in Synthesis Example 3.
LL1: 5’-AGG(Pa)GCTTTATGACTCTGCCG-3’(配列番号34)
LL2: 5’-AGGTGCTTTA(Pa)GACTCTGCCG-3’(配列番号35)
LL3: 5’-ACCGAAGCG(Pa)GGTGTTTACGAAGG-3’(配列番号36)
LL4: 5’-AGCG(Pa)GGTGTTTACGAAGG-3’(配列番号37)
LL5: 5’-ACGC(Pa)TCGGTATCGCTCTG-3’(配列番号38)
LL6: 5’-ACGCTTCGG(Pa)ATCGCTCTG-3’(配列番号39)
LL7: 5’-ATAAAGCACC(Pa)CGGTGGTGAAAGG-3’(配列番号40)
LL8: 5’-ACC(Pa)CGGTGGTGAAAGGGCAG-3’(配列番号41)
鋳型DNAとして、λDNA(New England BioLabs社製 カタログ番号 N3011)を、酵素としてKOD FX Neo(TOYOBO 社製 カタログ番号 KFX-201)を用い、前記DNAオリゴマーをプライマーとして用いて、ポリメラーゼ連鎖反応を行った。ストランド濃度10μM プライマーを2μL×2、1 ng/μL λDNAを1μL、各2 mM dNTPsを10μL、 KOD Fx neo(東洋紡社製 KFX-201)を1 μL(1 U)、及び製品添付のバッファー(×2)を25μL、ポリメラーゼ連鎖反応用チューブに加えた。前記反応用チューブを、TaKaRa PCR Thermal Cycler Dice(登録商標) Gradient(Takara社製サーマルサイクラ―、カタログ番号 TP600)に装着し、94℃, 2分→[98℃, 10秒→60℃, 30秒→68℃, 2分]×30→4℃の温度サイクルで反応させて、約1.5 kb及び約2.3 kbのDNAを増幅した。LL1又はLL2とLL3又はLL4とのプライマーセットを用いることにより、約1.5 kbのDNAが、LL5又はLL6とLL7又はLL8とのプライマーセットを用いることにより、約2.3 kbのDNAが、それぞれ増幅された。ポリメラーゼ連鎖反応後の反応溶液(0.5μL)を、1%アガロースゲル電気泳動で分析した。電気泳動後のゲルをエチジウムブロマイド染色して、DNAを蛍光検出した。エチジウムブロマイド染色後のゲルを図73に示す。なお、図中の写真は、白黒を反転させている。LL1: 5'-AGG (Pa) GCTTTATGACTCTGCCG-3 '(SEQ ID NO: 34)
LL2: 5'-AGGTGCTTTA (Pa) GACTCTGCCG-3 '(SEQ ID NO: 35)
LL3: 5'-ACCGAAGCG (Pa) GGTGTTTACGAAGG-3 '(SEQ ID NO: 36)
LL4: 5'-AGCG (Pa) GGTGTTTACGAAGG-3 '(SEQ ID NO: 37)
LL5: 5'-ACGC (Pa) TCGGTATCGCTCTG-3 '(SEQ ID NO: 38)
LL6: 5'-ACGCTTCGG (Pa) ATCGCTCTG-3 '(SEQ ID NO: 39)
LL7: 5'-ATAAAGCACC (Pa) CGGTGGTGAAAGG-3 '(SEQ ID NO: 40)
LL8: 5'-ACC (Pa) CGGTGGTGAAAGGGCAG-3 '(SEQ ID NO: 41)
Polymerase chain reaction was performed using λDNA (catalog number N3011 manufactured by New England BioLabs) as a template DNA, KOD FX Neo (catalog number KFX-201 manufactured by TOYOBO) as an enzyme, and the DNA oligomer as a primer. . Strand concentration: 10
図73において、レーン1のバンドは、DNAマーカー(100、200、300、400、500、600、700、800、900、1000、1100、1200、1300、1400、1500及び2072 bp、強く現れたバンドは、600、1500及び2072 bpのDNAである)を示す。レーン2のバンドは、LL1及びLL3をプライマーセットとして用いた場合の増幅産物のDNAを示しており、レーン3のバンドは、LL2及びLL4をプライマーセットとして用いた場合の増幅産物のDNAを示しており、レーン4のバンドは、LL5及びLL8をプライマーセットとして用いた場合の増幅産物のDNAを示しており、レーン5のバンドは、LL6及びLL7をプライマーセットとして用いた場合の増幅産物のDNAを示している。
In FIG. 73, the bands in
[使用例11-2:5-エチニル-2’-デオキシウリジン(Pa)を含むDNA増幅産物の切断反応及び得られた5’末端リン酸化DNA断片の連結反応(1)]
使用例11-1で調製された2種類のDNA増幅産物を当量混合した。得られた混合物に、1体積分の40%メチルアミン水溶液を加えて、よく混合した。得られた混合物を、25℃で2日間(48時間)静置した。遠心濃縮装置を用いて、加熱せずにメチルアミンを前記溶液中から除去した。その後、水を加えて、元の反応溶液と同じ体積に調製した。この溶液を2μL、水を6.5μL、Taq DNAリガーゼ(New England BioLabs社製 カタログ番号 M0208S)を0.5μL、及び製品添付のバッファー(×10)を1μL、ポリメラーゼ連鎖反応用チューブに加えた。前記反応用チューブを、TaKaRa PCR Thermal Cycler Dice(登録商標) Gradient(Takara社製サーマルサイクラ―、カタログ番号 TP600)に装着し、[80℃, 1分→45℃, 5分]×10、20又は30→4℃の温度サイクルで反応させて、DNAを連結した。連結反応後の反応溶液(4μL)を、1%アガロースゲル電気泳動で分析した。電気泳動後のゲルをエチジウムブロマイド染色して、DNAを蛍光検出した。エチジウムブロマイド染色後のゲルを図74に示す。なお、図中の写真は、白黒を反転させている。[Use Example 11-2: Cleavage reaction of DNA amplification product containing 5-ethynyl-2′-deoxyuridine (Pa) and ligation reaction of the obtained 5 ′ terminal phosphorylated DNA fragment (1)]
Two kinds of DNA amplification products prepared in Use Example 11-1 were mixed in an equivalent amount. One volume of 40% methylamine aqueous solution was added to the resulting mixture and mixed well. The resulting mixture was allowed to stand at 25 ° C. for 2 days (48 hours). Using a centrifugal concentrator, methylamine was removed from the solution without heating. Thereafter, water was added to prepare the same volume as the original reaction solution. 2 μL of this solution, 6.5 μL of water, 0.5 μL of Taq DNA ligase (New England BioLabs catalog number M0208S), and 1 μL of the buffer (× 10) attached to the product were added to the polymerase chain reaction tube. The reaction tube is attached to a TaKaRa PCR Thermal Cycler Dice (registered trademark) Gradient (Takara thermal cycler, catalog number TP600), and [80 ° C., 1 minute → 45 ° C., 5 minutes] × 10, 20 or The DNA was ligated by reaction at a temperature cycle of 30 → 4 ° C. The reaction solution (4 μL) after the ligation reaction was analyzed by 1% agarose gel electrophoresis. The gel after electrophoresis was stained with ethidium bromide to detect the fluorescence of DNA. FIG. 74 shows the gel after ethidium bromide staining. In the photo in the figure, black and white are reversed.
図74において、レーン1のバンドは、DNAマーカー(0.5, 1.0, 1.5, 2, 3, 4, 6, 8, 10及び12 kb)を示す。レーン2及び3のバンドは、10回の温度サイクルで連結反応を行った場合の連結反応産物のDNAを示しており、レーン4及び5のバンドは、20回の温度サイクルで連結反応を行った場合の連結反応産物のDNAを示しており、レーン6及び7のバンドは、30回の温度サイクルで連結反応を行った場合の連結反応産物のDNAを示している。また、レーン2、4及び6のバンドは、LL1及びLL3をプライマーセットとして用いた場合の増幅産物のDNAと、LL6及びLL7をプライマーセットとして用いた場合の増幅産物のDNAとを連結反応させた場合の連結反応産物のDNAを示しており、レーン3、5及び7のバンドは、LL2及びLL4をプライマーセットとして用いた場合の増幅産物のDNAと、LL6及びLL7をプライマーセットとして用いた場合の増幅産物のDNAとを連結反応させた場合の連結反応産物のDNAを示している。
In FIG. 74, the band in
図74に示すように、1.5 kb及び2.3 kbの原料DNAを連結反応させることにより、3.8 kbのDNAのバンドが確認された。これは、メチルアミン処理によりDNAが切断されて、1ヌクレオチドギャップ(欠損部位)を有する二つのDNA鎖のハイブリダイゼーションがライゲーションのための加熱及び冷却のサイクル中におきたことを示唆している。リガーゼにより、二つのDNA間に共有結合が生じていると考えられる。しかしながら、どの程度の割合で、リガーゼにより二つのDNAが共有結合で結ばれたかは、アガロースゲル電気泳動による分析の結果のみからは不明である。ハイブリダイズしたDNAを大腸菌などに導入すると、連結されていない場合のニック(切れ目)は一般に修復される。 As shown in FIG. 74, a 3.8 kb DNA band was confirmed by ligation reaction of the 1.5 kb and 2.3 kb source DNA. This suggests that the DNA was cleaved by methylamine treatment, and hybridization of two DNA strands having one nucleotide gap (deletion site) occurred during the heating and cooling cycles for ligation. Ligase is thought to cause a covalent bond between the two DNAs. However, it is unclear only from the results of analysis by agarose gel electrophoresis how often the two DNAs were covalently linked by ligase. When hybridized DNA is introduced into E. coli or the like, nicks (cuts) that are not linked are generally repaired.
連結前のDNAと連結後のDNAとを比較するために、これらのDNAを一緒に1%アガロースゲル電気泳動で分析した。電気泳動後のゲルをエチジウムブロマイド染色して、DNAを蛍光検出した。エチジウムブロマイド染色後のゲルを図75に示す。なお、図中の写真は、白黒を反転させている。 In order to compare the DNA before ligation with the DNA after ligation, these DNAs were analyzed together by 1% agarose gel electrophoresis. The gel after electrophoresis was stained with ethidium bromide to detect the fluorescence of DNA. The gel after ethidium bromide staining is shown in FIG. In the photo in the figure, black and white are reversed.
図75において、レーン1のバンドは、DNAマーカー(0.5, 1.0, 1.5, 2, 3, 4, 6, 8, 10及び12 kb)を示す。レーン2及び3のバンドは、メチルアミン処理を9時間行ってDNAの切断反応を行った場合の切断反応産物のDNAを示しており、レーン4及び5のバンドは、メチルアミン処理を24時間行ってDNAの切断反応を行った場合の切断反応産物のDNAを示しており、レーン6及び7のバンドは、メチルアミン処理を48時間行ってDNAの切断反応を行った場合の切断反応産物のDNAを示している。レーン8及び9のバンドは、メチルアミン処理を48時間行ってDNAの切断反応を行った後、30回の温度サイクルで連結反応を行った場合の連結反応産物のDNAを示している。また、レーン2、4、6及び8のバンドは、LL1及びLL3をプライマーセットとして用いた場合の増幅産物のDNAと、LL6及びLL7をプライマーセットとして用いた場合の増幅産物のDNAとを切断反応させた場合の切断反応産物のDNA又は切断反応及び連結反応させた場合の連結反応産物のDNAを示しており、レーン3、5、7及び9のバンドは、LL2及びLL4をプライマーセットとして用いた場合の増幅産物のDNAと、LL6及びLL7をプライマーセットとして用いた場合の増幅産物のDNAとを切断反応させた場合の切断反応産物のDNA又は切断反応及び連結反応させた場合の連結反応産物のDNAを示している。
In FIG. 75, the band in
DNA増幅産物の切断反応において、DNA増幅産物にメチルアミンを加えて加熱した場合、DNAがアルカリ変性し、鮮明なバンドが検出されなかった。これに対し、図74及び75に示すように、DNA増幅産物をメチルアミン存在下、25℃で反応させた場合、DNAのアルカリ変性が抑えられたことが確認された。また、図75に示すように、連結反応前の状態では、原料DNAを混合しても、連結反応産物に相当する3.8 kbのDNAバンドは生じないことが確認された。これは、切断後のDNAの末端部分には、切断で生じた短いDNA断片が結合しており、切断後のDNAをただ混合したのみではDNAの末端部分同士がハイブリダイゼーションしにくい状態にあることを示唆している。 In the cleavage reaction of the DNA amplification product, when methylamine was added to the DNA amplification product and heated, the DNA was alkali-denatured and no clear band was detected. In contrast, as shown in FIGS. 74 and 75, it was confirmed that when the DNA amplification product was reacted at 25 ° C. in the presence of methylamine, the alkaline denaturation of DNA was suppressed. Further, as shown in FIG. 75, it was confirmed that in the state before the ligation reaction, even when the raw material DNA was mixed, a 3.8 kb DNA band corresponding to the ligation product was not generated. This is because the short DNA fragments produced by the cleavage are bound to the terminal portions of the DNA after cleavage, and it is difficult for the DNA end portions to hybridize with each other simply by mixing the DNA after the cleavage. It suggests.
5位置換2'-デオキシウリジン誘導体は、DNAの複製反応の基質又は鋳型として適していることが知られており、5位置換2’-デオキシウリジン三リン酸を利用したDNAの複製反応の報告は数多い(例えば非特許文献74〜76)。Paと類似の構造の5-(1-プロピニル)-2’-デオキシウリジン三リン酸を利用するDNAの複製及び増幅反応も報告されており、比較的高い忠誠度で複製反応が効率よく進行することが知られている(非特許文献77)。これまで、Paを鋳型とするDNA複製反応の報告は存在しなかった。本使用例で、DNAの連結が確認されたので、Paを鋳型としてDNAの伸長が起きたことが示唆される。 5 position substituted 2'-deoxyuridine derivatives are known to be suitable as substrates or templates for DNA replication reactions, and report of DNA replication reactions using 5 position substituted 2'-deoxyuridine triphosphates. Are many (for example, non-patent documents 74 to 76). A DNA replication and amplification reaction using 5- (1-propynyl) -2'-deoxyuridine triphosphate having a structure similar to Pa has also been reported, and the replication reaction proceeds efficiently with a relatively high degree of loyalty. It is known (Non-patent Document 77). Until now, there has been no report of DNA replication reaction using Pa as a template. In this use example, DNA ligation was confirmed, suggesting that DNA elongation occurred using Pa as a template.
[使用例11-3:5-エチニル-2’-デオキシウリジン(Pa)を含むDNA増幅産物の切断反応及び得られた5’末端リン酸化DNA断片の連結反応(2)]
使用例11-2では、2種類のDNA増幅産物の片側の末端のみが完全に相補的で連結されるように、DNA増幅産物の組み合わせを選択した。本使用例では、2種類のDNA増幅産物の両側の末端が相補的で連結されるように、DNA増幅産物の組み合わせを選択した。使用例11-2と同様の手順でメチルアミン処理したDNAの混合溶液を2又は4μL、水を6.5又は4.5μL、Taq DNAリガーゼ(New England BioLabs社製 カタログ番号 M0208S)又はT4 DNAリガーゼ(New England BioLabs社製 カタログ番号 M0202S)を0.5μL、及び製品添付のバッファー(×10)を1μL、ポリメラーゼ連鎖反応用チューブに加えた。Taq DNAリガーゼを用いる場合、前記反応用チューブを、TaKaRa PCR Thermal Cycler Dice(登録商標) Gradient(Takara社製サーマルサイクラ―、カタログ番号 TP600)に装着し、[80℃, 1分→45℃, 5分]×30→4℃の温度サイクルで反応させて、DNAを連結した。T4 DNAリガーゼを用いる場合、前記反応用チューブを、25℃で18時間静置することにより、DNAを連結した。連結反応後の反応溶液(4又は2μL)を、1%アガロースゲル電気泳動で分析した。電気泳動後のゲルをエチジウムブロマイド染色して、DNAを蛍光検出した。エチジウムブロマイド染色後のゲルを図76に示す。なお、図中の写真は、白黒を反転させている。[Use Example 11-3: Cleavage reaction of a DNA amplification product containing 5-ethynyl-2′-deoxyuridine (Pa) and ligation reaction of the obtained 5′-terminal phosphorylated DNA fragment (2)]
In Use Example 11-2, a combination of DNA amplification products was selected so that only one end of two types of DNA amplification products was completely complementary and ligated. In this use example, the combination of DNA amplification products was selected so that the ends on both sides of the two types of DNA amplification products were complementary and ligated. 2 or 4 μL of methylamine-treated DNA mixed solution in the same procedure as in Use Example 11-2, 6.5 or 4.5 μL of water, Taq DNA ligase (New England BioLabs, catalog number M0208S) or T4 DNA ligase (New England 0.5 μL of BioLabs catalog number M0202S) and 1 μL of the buffer (× 10) attached to the product were added to the polymerase chain reaction tube. When Taq DNA ligase is used, the reaction tube is attached to TaKaRa PCR Thermal Cycler Dice (registered trademark) Gradient (Takara Thermal Cycler, Catalog No. TP600), [80 ° C, 1 minute → 45 ° C, 5 ° C] The reaction was carried out at a temperature cycle of [minute] × 30 → 4 ° C. to ligate the DNA. When T4 DNA ligase was used, the reaction tubes were allowed to stand at 25 ° C. for 18 hours to ligate DNA. The reaction solution (4 or 2 μL) after the ligation reaction was analyzed by 1% agarose gel electrophoresis. The gel after electrophoresis was stained with ethidium bromide to detect the fluorescence of DNA. The gel after ethidium bromide staining is shown in FIG. In the photo in the figure, black and white are reversed.
図76において、レーン1のバンドは、DNAマーカー(0.5, 1.0, 1.5, 2, 3, 4, 6, 8, 10及び12 kb)を示す。レーン2、3、6及び7のバンドは、LL1及びLL4をプライマーセットとして用いた場合の増幅産物のDNAと、LL5及びLL8をプライマーセットとして用いた場合の増幅産物のDNAとを連結反応させた場合の連結反応産物のDNAを示しており、レーン4、5、8及び9のバンドは、LL2及びLL3をプライマーセットとして用いた場合の増幅産物のDNAと、LL6及びLL7をプライマーセットとして用いた場合の増幅産物のDNAとを連結反応させた場合の連結反応産物のDNAを示している。レーン2〜5のバンドは、T4 DNAリガーゼを用いてDNAを連結反応させた場合の連結反応産物のDNAを示しており、レーン6〜9のバンドは、Taq DNAリガーゼを用いてDNAを連結反応させた場合の連結反応産物のDNAを示している。また、レーン2、4、6及び8のバンドは、2μLの前記増幅産物のDNAの混合溶液を用いてDNAを連結反応させた場合の連結反応産物のDNAを示しており、レーン3、5、7及び9のバンドは、4μLの前記増幅産物のDNAの混合溶液を用いてDNAを連結反応させた場合の連結反応産物のDNAを示している。
In FIG. 76, the band in
図76に示すように、本発明の切断反応によって長い突出末端を形成させたDNAを、Taq DNAリガーゼを用いて連結反応させた場合(レーン8及び9)に、最も効率よくDNAがハイブリダイゼーション又は連結されて、非常に長いDNAを生成したことが確認された。
As shown in FIG. 76, when DNA having a long protruding end formed by the cleavage reaction of the present invention was ligated using Taq DNA ligase (
本発明の方法により、化学反応によって低コスト且つ簡単な実験操作でDNA鎖の切断を行い、さらにポリヌクレオチドキナーゼのような酵素法又はリン酸化アミダイトを用いる合成DNAの末端リン酸化法と比較してより高収率で末端がリン酸化されたオリゴデオキシヌクレオチドを得ることが可能となる。 By the method of the present invention, a DNA strand is cleaved by a chemical reaction at a low cost and with a simple experimental operation, and compared with an enzymatic method such as polynucleotide kinase or a terminal phosphorylation method of synthetic DNA using phosphorylated amidite. It is possible to obtain oligodeoxynucleotides whose ends are phosphorylated with higher yield.
本明細書で引用した全ての刊行物、特許及び特許出願をそのまま参考として本明細書にとり入れるものとする。 All publications, patents and patent applications cited herein are incorporated herein by reference in their entirety.
Claims (7)
(A)n-B-C
[式中、
Aは、オリゴデオキシヌクレオチドであり、
Bは、求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドであり、
Cはオリゴデオキシヌクレオチドであり、
nは0又は1であり、
nが1のとき、Aの3’末端とBの5’位とは、ホスホジエステル結合で連結されており、
Bの3’位とCの5’末端とは、ホスホジエステル結合で連結されており、
BとCとの間のホスホジエステル結合は、求核剤の攻撃によって誘発されるEの脱離に伴って、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断される]
で表されるオリゴデオキシヌクレオチドとを接触させることによって、BとCとの間のホスホジエステル結合が、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断されて、前記Cの5’末端がリン酸化されたオリゴデオキシヌクレオチドと、式II:
(A)n-B’
[式中、
A及びnは、式Iにおいて定義したのと同様の意味であり、
B’は、求核剤の攻撃によるEの脱離に伴って形成されるBの残基部分である]
で表されるオリゴデオキシヌクレオチドとを得る、5’末端リン酸化オリゴデオキシヌクレオチド形成工程を含む、オリゴデオキシヌクレオチドの製造方法。Nucleophiles and formula I:
(A) n -BC
[Where:
A is an oligodeoxynucleotide,
B is a deoxynucleoside containing a non-natural base having a moiety E removable by a nucleophile;
C is an oligodeoxynucleotide,
n is 0 or 1,
When n is 1, the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond,
The 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond,
The phosphodiester bond between B and C is such that the oligodeoxynucleotide part phosphorylated at the 5 'end of C from the 3' position of B with E elimination induced by nucleophile attack. Disconnected by detachment]
By contacting the oligodeoxynucleotide represented by the following formula, the phosphodiester bond between B and C is released from the 3 'position of B by phosphorylation of the 5' end of C. An oligodeoxynucleotide which has been cleaved to phosphorylate the 5 ′ end of C, and a compound of formula II:
(A) n -B '
[Where:
A and n have the same meaning as defined in Formula I,
B ′ is the residue part of B that is formed with the elimination of E by nucleophile attack]
A method for producing an oligodeoxynucleotide, comprising a 5′-terminal phosphorylated oligodeoxynucleotide forming step, which obtains an oligodeoxynucleotide represented by the formula:
(A)n-B’
[式中、
A及びB’は、請求項1と同様の意味であり、
nは1である]
で表されるオリゴデオキシヌクレオチドを反応させることによって、B’とAとの間のホスホジエステル結合が、Aの3’末端がリン酸化されたオリゴデオキシヌクレオチド部分がB’から脱離することによって切断されて、式IIa:
A-PO3H2
で表される3’末端がリン酸化されたオリゴデオキシヌクレオチドを得る、3’末端リン酸化オリゴデオキシヌクレオチド形成工程をさらに含む、請求項1の方法。Formula II:
(A) n -B '
[Where:
A and B ′ have the same meaning as in claim 1,
n is 1]
By reacting with oligodeoxynucleotide represented by Formula IIa:
A-PO 3 H 2
The method according to claim 1, further comprising a step of forming a 3'-terminal phosphorylated oligodeoxynucleotide, wherein an oligodeoxynucleotide phosphorylated at the 3 'end represented by:
Rは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロアリールアルキル又はカルボニルであり;
R’は、水素、又は置換若しくは非置換のアルキル、アルケニル若しくはアルキニルであり;
rは、0〜4の整数であり;
R’’は、ハロゲン、又は置換若しくは非置換のアルキル、アルケニル若しくはアルキニルであり;
R’’’は、-COZ、-C≡C-Z又は-CNであり;
Zは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、又は置換若しくは非置換のヘテロアリールアルキルである]
で表されるデオキシヌクレオシドから選択される、請求項1又は2の方法。The deoxynucleoside B containing the unnatural base is:
R is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, Substituted or unsubstituted heteroarylalkyl or carbonyl;
R ′ is hydrogen or substituted or unsubstituted alkyl, alkenyl or alkynyl;
r is an integer from 0 to 4;
R ″ is halogen or substituted or unsubstituted alkyl, alkenyl or alkynyl;
R ′ ″ is —COZ, —C≡CZ or —CN;
Z is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, Or substituted or unsubstituted heteroarylalkyl]
The method of Claim 1 or 2 selected from the deoxynucleoside represented by these.
(A)n-B-C
[式中、
Aは、オリゴデオキシヌクレオチドであり、
Bは、求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドであり、
Cはオリゴデオキシヌクレオチドであり、
nは0又は1であり、
nが1のとき、Aの3’末端とBの5’位とは、ホスホジエステル結合で連結されており、
Bの3’位とCの5’末端とは、ホスホジエステル結合で連結されており、
BとCとの間のホスホジエステル結合は、求核剤の攻撃によって誘発されるEの脱離に伴って、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断される]
で表されるオリゴデオキシヌクレオチドをプライマーとして用いて、直鎖状二本鎖DNA断片を増幅するDNA断片増幅工程;
求核剤と、増幅された直鎖状二本鎖DNA断片とを接触させることによって、BとCとの間のホスホジエステル結合が、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断されて、直鎖状二本鎖DNA断片に含まれる前記Cの5’末端がリン酸化されており、且つ式Iで表されるプライマー部分の少なくとも一部と相補的な配列を含む3’突出末端を有する直鎖状二本鎖DNA断片を形成させる、3’突出末端形成工程;並びに
前記3’突出末端を有する直鎖状二本鎖DNA断片と、別の二本鎖DNA断片とを連結する連結工程;
を含む、二本鎖DNAの連結方法。Formula I:
(A) n -BC
[Where:
A is an oligodeoxynucleotide,
B is a deoxynucleoside containing a non-natural base having a moiety E removable by a nucleophile;
C is an oligodeoxynucleotide,
n is 0 or 1,
When n is 1, the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond,
The 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond,
The phosphodiester bond between B and C is such that the oligodeoxynucleotide part phosphorylated at the 5 'end of C from the 3' position of B with E elimination induced by nucleophile attack. Disconnected by detachment]
A DNA fragment amplification step of amplifying a linear double-stranded DNA fragment using the oligodeoxynucleotide represented by
By contacting the nucleophile with the amplified linear double-stranded DNA fragment, the phosphodiester bond between B and C becomes an oligodeoxynucleotide moiety phosphorylated at the 5 'end of C. It is cleaved by detaching from the 3 ′ position of B, the 5 ′ end of C contained in the linear double-stranded DNA fragment is phosphorylated, and at least the primer part represented by Formula I Forming a linear double-stranded DNA fragment having a 3 ′ protruding end containing a sequence complementary to a part thereof; and a linear double-stranded DNA fragment having the 3 ′ protruding end; And a ligation step of ligating another double-stranded DNA fragment;
A method for ligating double-stranded DNA.
(A)n-B-C
[式中、
Aは、オリゴデオキシヌクレオチドであり、
Bは、求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドであり、
Cはオリゴデオキシヌクレオチドであり、
nは0又は1であり、
nが1のとき、Aの3’末端とBの5’位とは、ホスホジエステル結合で連結されており、
Bの3’位とCの5’末端とは、ホスホジエステル結合で連結されており、
BとCとの間のホスホジエステル結合は、求核剤の攻撃によって誘発されるEの脱離に伴って、Cの5’末端がリン酸化されたオリゴデオキシヌクレオチド部分がBの3’位から脱離することによって切断される]
で表されるオリゴデオキシヌクレオチド、又は求核剤によって脱離可能な部分Eを有する、非天然型塩基を含むデオキシヌクレオシドB若しくはその誘導体を少なくとも含む、前記キット。A kit for use in the method for producing an oligodeoxynucleotide according to any one of claims 1 to 4 or the method for ligating double-stranded DNA according to claim 5, comprising the formula I:
(A) n -BC
[Where:
A is an oligodeoxynucleotide,
B is a deoxynucleoside containing a non-natural base having a moiety E removable by a nucleophile;
C is an oligodeoxynucleotide,
n is 0 or 1,
When n is 1, the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond,
The 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond,
The phosphodiester bond between B and C is such that the oligodeoxynucleotide part phosphorylated at the 5 'end of C from the 3' position of B with E elimination induced by nucleophile attack. Disconnected by detachment]
Or at least a deoxynucleoside B containing a non-natural base or a derivative thereof having a moiety E removable by a nucleophile.
Rは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、置換若しくは非置換のヘテロアリールアルキル又はカルボニルであり;
R'は、メチルであり;
rは、0〜4の整数であり;
R’’は、ハロゲン、又は置換若しくは非置換のアルキル、アルケニル若しくはアルキニルであり;
R’’’は、-COZ、-C≡C-Z又は-CNであり;
Zは、水素、置換若しくは非置換のアルキル、置換若しくは非置換のシクロアルキル、置換若しくは非置換のヘテロシクリル、置換若しくは非置換のアリール、置換若しくは非置換のアリールアルキル、置換若しくは非置換のヘテロアリール、又は置換若しくは非置換のヘテロアリールアルキルである]
で表される非天然型塩基を含むデオキシヌクレオシド。The following formula:
R is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, Substituted or unsubstituted heteroarylalkyl or carbonyl;
R ′ is methyl;
r is an integer from 0 to 4;
R ″ is halogen or substituted or unsubstituted alkyl, alkenyl or alkynyl;
R ′ ″ is —COZ, —C≡CZ or —CN;
Z is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, Or substituted or unsubstituted heteroarylalkyl]
A deoxynucleoside comprising an unnatural base represented by
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014509104A JPWO2013150902A1 (en) | 2012-04-02 | 2013-03-22 | Method for producing 5'-terminal phosphorylated oligodeoxynucleotide by cleaving oligodeoxynucleotide containing non-natural base |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012084249 | 2012-04-02 | ||
| JP2012084249 | 2012-04-02 | ||
| JP2014509104A JPWO2013150902A1 (en) | 2012-04-02 | 2013-03-22 | Method for producing 5'-terminal phosphorylated oligodeoxynucleotide by cleaving oligodeoxynucleotide containing non-natural base |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JPWO2013150902A1 true JPWO2013150902A1 (en) | 2015-12-17 |
Family
ID=49300395
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014509104A Pending JPWO2013150902A1 (en) | 2012-04-02 | 2013-03-22 | Method for producing 5'-terminal phosphorylated oligodeoxynucleotide by cleaving oligodeoxynucleotide containing non-natural base |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2013150902A1 (en) |
| WO (1) | WO2013150902A1 (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016094845A2 (en) | 2014-12-12 | 2016-06-16 | Woolf Tod M | Compositions and methods for editing nucleic acids in cells utilizing oligonucleotides |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0991779A1 (en) * | 1997-08-28 | 2000-04-12 | The Perkin-Elmer Corporation | Improved detection of mutations in nucleic acids by chemical cleavage |
| US7057026B2 (en) * | 2001-12-04 | 2006-06-06 | Solexa Limited | Labelled nucleotides |
| EP1573056A4 (en) * | 2002-05-17 | 2007-11-28 | Nugen Technologies Inc | Methods for fragmentation, labeling and immobilization of nucleic acids |
-
2013
- 2013-03-22 JP JP2014509104A patent/JPWO2013150902A1/en active Pending
- 2013-03-22 WO PCT/JP2013/058349 patent/WO2013150902A1/en active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| WO2013150902A1 (en) | 2013-10-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2459347C (en) | Locked nucleic acid (lna) compositions and uses thereof | |
| EP1483280B1 (en) | Fluorescent nitrogenous base and nucleosides incorporating same | |
| JP5689054B2 (en) | RNA chemical synthesis method | |
| JP2004505988A (en) | Nucleic acid binding compounds containing pyrazolo [3,4-d] pyrimidine analogs of purine-2,6-diamine and uses thereof | |
| JP2004500330A (en) | Guanidinium-functionalized oligomers and their preparation | |
| EP3137478B1 (en) | Phosphorous protecting groups and methods of preparation and use thereof | |
| CN102443030A (en) | Photocleavable labeled nucleotides and nucleosides and labeled nucleotides and nucleosides and methods for their use in DNA sequencing | |
| Boháčová et al. | Protected 5-(hydroxymethyl) uracil nucleotides bearing visible-light photocleavable groups as building blocks for polymerase synthesis of photocaged DNA | |
| CN101663313A (en) | nucleotide with an alpha-phosphate mimetic | |
| JP4903163B2 (en) | Phosphoramidite activators for oligonucleotide synthesis | |
| US20130052690A1 (en) | Pcr primer and method for reducing non-specific nucleic acid amplification using a photolabile protecting group | |
| Matyašovský et al. | 2-Substituted 2′-deoxyinosine 5′-triphosphates as substrates for polymerase synthesis of minor-groove-modified DNA and effects on restriction endonuclease cleavage | |
| WO2013150902A1 (en) | Method for producing 5'-terminal-phosphorylated oligodeoxynucleotide by cleaving oligodeoxynucleotide containing non-naturally-occurring nucleotide | |
| Lauritsen et al. | Oligodeoxynucleotides containing amide-linked LNA-type dinucleotides: synthesis and high-affinity nucleic acid hybridization | |
| Meher et al. | Nucleobase Protection of Deoxyribo‐and Ribonucleosides | |
| EP3137477A1 (en) | Compounds compositions and methods including thermally labile moieties | |
| Seela et al. | 2′‐Deoxyuridine and 2′‐Deoxyisocytidine as Constituents of DNA with Parallel Chain Orientation: The Stabilization of the iCd⋅ Gd Base Pair by the 5‐Methyl Group | |
| Moriguchi et al. | Novel method of the synthesis and hybridization properties of an oligonucleotide containing non-ionic diisopropylsilyl internucleotide linkage | |
| Eritja | Nucleic Acids Chemistry: Modifications and Conjugates for Biomedicine and Nanotechnology | |
| US11912734B2 (en) | Solid-phase synthesis of oligonucleotides containing N6-(2-deoxy-alpha,beta-derythropentofuranosyl)-2,6-diamino-4-hydroxy-5-formamidopyrimidine (Fapy⋅dG) | |
| EP1700922B1 (en) | 3-Substituted 5-Nitroindole derivatives and labeled oligonucleotide probes containing them | |
| JP4180681B2 (en) | Antisense oligonucleotide | |
| WO2022175685A1 (en) | Modified guanines | |
| Vaníková | Construction of modified DNAs with selected reactive or protective groups | |
| Ohkubo et al. | Non-protected Synthesis of Oligonucleotides |