JP5517407B2 - Method for producing multi-metal oxide material - Google Patents
Method for producing multi-metal oxide material Download PDFInfo
- Publication number
- JP5517407B2 JP5517407B2 JP2007526256A JP2007526256A JP5517407B2 JP 5517407 B2 JP5517407 B2 JP 5517407B2 JP 2007526256 A JP2007526256 A JP 2007526256A JP 2007526256 A JP2007526256 A JP 2007526256A JP 5517407 B2 JP5517407 B2 JP 5517407B2
- Authority
- JP
- Japan
- Prior art keywords
- oxide material
- elemental
- metal oxide
- group
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000000463 material Substances 0.000 title claims description 101
- 229910044991 metal oxide Inorganic materials 0.000 title claims description 64
- 238000004519 manufacturing process Methods 0.000 title claims description 29
- 238000000034 method Methods 0.000 claims description 76
- 239000000203 mixture Substances 0.000 claims description 75
- 230000003647 oxidation Effects 0.000 claims description 75
- 238000007254 oxidation reaction Methods 0.000 claims description 75
- 230000008569 process Effects 0.000 claims description 36
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 35
- 239000002994 raw material Substances 0.000 claims description 34
- 229910052760 oxygen Inorganic materials 0.000 claims description 26
- 239000000470 constituent Substances 0.000 claims description 25
- 239000001301 oxygen Substances 0.000 claims description 23
- 150000001875 compounds Chemical class 0.000 claims description 21
- 238000010335 hydrothermal treatment Methods 0.000 claims description 21
- 239000007864 aqueous solution Substances 0.000 claims description 11
- 229910052758 niobium Inorganic materials 0.000 claims description 10
- 239000007787 solid Substances 0.000 claims description 9
- 229910052742 iron Inorganic materials 0.000 claims description 8
- 229910052759 nickel Inorganic materials 0.000 claims description 8
- 229910052763 palladium Inorganic materials 0.000 claims description 8
- 229910052697 platinum Inorganic materials 0.000 claims description 8
- 229910052720 vanadium Inorganic materials 0.000 claims description 8
- 239000012298 atmosphere Substances 0.000 claims description 7
- 229910052804 chromium Inorganic materials 0.000 claims description 7
- 239000011261 inert gas Substances 0.000 claims description 7
- 229910052714 tellurium Inorganic materials 0.000 claims description 7
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims description 6
- 229910052719 titanium Inorganic materials 0.000 claims description 6
- 229910052721 tungsten Inorganic materials 0.000 claims description 6
- 229910052725 zinc Inorganic materials 0.000 claims description 6
- 229910052726 zirconium Inorganic materials 0.000 claims description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 5
- 229910052787 antimony Inorganic materials 0.000 claims description 5
- 238000004140 cleaning Methods 0.000 claims description 5
- 238000005406 washing Methods 0.000 claims description 5
- 229910052702 rhenium Inorganic materials 0.000 claims description 4
- 229910052715 tantalum Inorganic materials 0.000 claims description 4
- 229910052737 gold Inorganic materials 0.000 claims description 3
- 229910052747 lanthanoid Inorganic materials 0.000 claims description 3
- 150000002602 lanthanoids Chemical class 0.000 claims description 3
- 150000007522 mineralic acids Chemical class 0.000 claims description 3
- 150000007524 organic acids Chemical class 0.000 claims description 3
- 229910052701 rubidium Inorganic materials 0.000 claims description 3
- 229910052712 strontium Inorganic materials 0.000 claims description 3
- 229910052782 aluminium Inorganic materials 0.000 claims description 2
- 229910052785 arsenic Inorganic materials 0.000 claims description 2
- 229910052788 barium Inorganic materials 0.000 claims description 2
- 229910052790 beryllium Inorganic materials 0.000 claims description 2
- 229910052797 bismuth Inorganic materials 0.000 claims description 2
- 229910052793 cadmium Inorganic materials 0.000 claims description 2
- 229910052792 caesium Inorganic materials 0.000 claims description 2
- 229910052791 calcium Inorganic materials 0.000 claims description 2
- 229910052802 copper Inorganic materials 0.000 claims description 2
- 229910052733 gallium Inorganic materials 0.000 claims description 2
- 229910052732 germanium Inorganic materials 0.000 claims description 2
- 229910052735 hafnium Inorganic materials 0.000 claims description 2
- 229910052738 indium Inorganic materials 0.000 claims description 2
- 229910052741 iridium Inorganic materials 0.000 claims description 2
- 229910052746 lanthanum Inorganic materials 0.000 claims description 2
- 229910052745 lead Inorganic materials 0.000 claims description 2
- 229910052744 lithium Inorganic materials 0.000 claims description 2
- 229910052749 magnesium Inorganic materials 0.000 claims description 2
- 229910052748 manganese Inorganic materials 0.000 claims description 2
- 229910052762 osmium Inorganic materials 0.000 claims description 2
- 229910052700 potassium Inorganic materials 0.000 claims description 2
- 229910052703 rhodium Inorganic materials 0.000 claims description 2
- 229910052707 ruthenium Inorganic materials 0.000 claims description 2
- 229910052706 scandium Inorganic materials 0.000 claims description 2
- 229910052709 silver Inorganic materials 0.000 claims description 2
- 229910052708 sodium Inorganic materials 0.000 claims description 2
- 229910052718 tin Inorganic materials 0.000 claims description 2
- 229910052727 yttrium Inorganic materials 0.000 claims description 2
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 claims 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims 1
- 229910052711 selenium Inorganic materials 0.000 claims 1
- 229910052716 thallium Inorganic materials 0.000 claims 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N Propene Chemical compound CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 111
- 239000012071 phase Substances 0.000 description 73
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 70
- 239000007789 gas Substances 0.000 description 51
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 39
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 38
- 239000003054 catalyst Substances 0.000 description 37
- 239000001294 propane Substances 0.000 description 35
- 230000036961 partial effect Effects 0.000 description 34
- 238000006243 chemical reaction Methods 0.000 description 33
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 30
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 26
- 239000013543 active substance Substances 0.000 description 22
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 21
- 238000000576 coating method Methods 0.000 description 20
- 239000000843 powder Substances 0.000 description 20
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 19
- 239000010955 niobium Substances 0.000 description 17
- 239000011248 coating agent Substances 0.000 description 16
- 239000007788 liquid Substances 0.000 description 16
- 239000000126 substance Substances 0.000 description 16
- 239000000047 product Substances 0.000 description 15
- 238000002441 X-ray diffraction Methods 0.000 description 14
- 239000012495 reaction gas Substances 0.000 description 14
- 238000011017 operating method Methods 0.000 description 13
- 238000010438 heat treatment Methods 0.000 description 12
- 239000011230 binding agent Substances 0.000 description 11
- 239000003085 diluting agent Substances 0.000 description 11
- 229930195733 hydrocarbon Natural products 0.000 description 10
- 150000002430 hydrocarbons Chemical class 0.000 description 10
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 10
- 239000007858 starting material Substances 0.000 description 10
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- 238000009396 hybridization Methods 0.000 description 9
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- 150000001298 alcohols Chemical class 0.000 description 8
- 239000013078 crystal Substances 0.000 description 8
- 238000001035 drying Methods 0.000 description 8
- 239000002245 particle Substances 0.000 description 8
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 7
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 7
- 235000011054 acetic acid Nutrition 0.000 description 7
- 150000001299 aldehydes Chemical class 0.000 description 7
- -1 atom ion Chemical class 0.000 description 7
- 230000000052 comparative effect Effects 0.000 description 7
- 238000010586 diagram Methods 0.000 description 7
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- 239000004215 Carbon black (E152) Substances 0.000 description 6
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 6
- 239000005977 Ethylene Substances 0.000 description 6
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical compound CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 6
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 6
- 238000002425 crystallisation Methods 0.000 description 6
- 230000008025 crystallization Effects 0.000 description 6
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 238000001027 hydrothermal synthesis Methods 0.000 description 6
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 6
- 229930195734 saturated hydrocarbon Natural products 0.000 description 6
- 238000001878 scanning electron micrograph Methods 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- LEONUFNNVUYDNQ-UHFFFAOYSA-N vanadium atom Chemical compound [V] LEONUFNNVUYDNQ-UHFFFAOYSA-N 0.000 description 6
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 239000001282 iso-butane Substances 0.000 description 5
- 230000001590 oxidative effect Effects 0.000 description 5
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 239000007921 spray Substances 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 239000010936 titanium Substances 0.000 description 5
- 239000011701 zinc Substances 0.000 description 5
- 229910015902 Bi 2 O 3 Inorganic materials 0.000 description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 4
- NIQCNGHVCWTJSM-UHFFFAOYSA-N Dimethyl phthalate Chemical compound COC(=O)C1=CC=CC=C1C(=O)OC NIQCNGHVCWTJSM-UHFFFAOYSA-N 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- 238000006555 catalytic reaction Methods 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical compound C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 238000000605 extraction Methods 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 230000002209 hydrophobic effect Effects 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 229910052756 noble gas Inorganic materials 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 238000000634 powder X-ray diffraction Methods 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- 239000011593 sulfur Substances 0.000 description 4
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 4
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical class [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- GYCMBHHDWRMZGG-UHFFFAOYSA-N Methylacrylonitrile Chemical compound CC(=C)C#N GYCMBHHDWRMZGG-UHFFFAOYSA-N 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 239000011149 active material Substances 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 238000009835 boiling Methods 0.000 description 3
- 229910002090 carbon oxide Inorganic materials 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 239000010931 gold Substances 0.000 description 3
- 239000003701 inert diluent Substances 0.000 description 3
- 238000002955 isolation Methods 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 150000002835 noble gases Chemical class 0.000 description 3
- FXADMRZICBQPQY-UHFFFAOYSA-N orthotelluric acid Chemical compound O[Te](O)(O)(O)(O)O FXADMRZICBQPQY-UHFFFAOYSA-N 0.000 description 3
- 235000019260 propionic acid Nutrition 0.000 description 3
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 3
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical compound [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 description 3
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical compound CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 2
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- STNJBCKSHOAVAJ-UHFFFAOYSA-N Methacrolein Chemical compound CC(=C)C=O STNJBCKSHOAVAJ-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- 239000002250 absorbent Substances 0.000 description 2
- 230000002745 absorbent Effects 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 229910000287 alkaline earth metal oxide Inorganic materials 0.000 description 2
- IYABWNGZIDDRAK-UHFFFAOYSA-N allene Chemical compound C=C=C IYABWNGZIDDRAK-UHFFFAOYSA-N 0.000 description 2
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- QGAVSDVURUSLQK-UHFFFAOYSA-N ammonium heptamolybdate Chemical compound N.N.N.N.N.N.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.O.[Mo].[Mo].[Mo].[Mo].[Mo].[Mo].[Mo] QGAVSDVURUSLQK-UHFFFAOYSA-N 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- UNTBPXHCXVWYOI-UHFFFAOYSA-O azanium;oxido(dioxo)vanadium Chemical compound [NH4+].[O-][V](=O)=O UNTBPXHCXVWYOI-UHFFFAOYSA-O 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- 238000001354 calcination Methods 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 229950011148 cyclopropane Drugs 0.000 description 2
- 238000006356 dehydrogenation reaction Methods 0.000 description 2
- 238000003795 desorption Methods 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- FBSAITBEAPNWJG-UHFFFAOYSA-N dimethyl phthalate Natural products CC(=O)OC1=CC=CC=C1OC(C)=O FBSAITBEAPNWJG-UHFFFAOYSA-N 0.000 description 2
- 229960001826 dimethylphthalate Drugs 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 2
- 238000011010 flushing procedure Methods 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 229910052734 helium Inorganic materials 0.000 description 2
- 150000004679 hydroxides Chemical class 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 239000011872 intimate mixture Substances 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 2
- 229910000484 niobium oxide Inorganic materials 0.000 description 2
- URLJKFSTXLNXLG-UHFFFAOYSA-N niobium(5+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[Nb+5].[Nb+5] URLJKFSTXLNXLG-UHFFFAOYSA-N 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 235000006408 oxalic acid Nutrition 0.000 description 2
- DKCWBFMZNUOFEM-UHFFFAOYSA-L oxovanadium(2+);sulfate;hydrate Chemical compound O.[V+2]=O.[O-]S([O-])(=O)=O DKCWBFMZNUOFEM-UHFFFAOYSA-L 0.000 description 2
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- MWWATHDPGQKSAR-UHFFFAOYSA-N propyne Chemical compound CC#C MWWATHDPGQKSAR-UHFFFAOYSA-N 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- NDVLTYZPCACLMA-UHFFFAOYSA-N silver oxide Chemical compound [O-2].[Ag+].[Ag+] NDVLTYZPCACLMA-UHFFFAOYSA-N 0.000 description 2
- 238000000859 sublimation Methods 0.000 description 2
- 230000008022 sublimation Effects 0.000 description 2
- 238000004448 titration Methods 0.000 description 2
- 238000009736 wetting Methods 0.000 description 2
- 229910003208 (NH4)6Mo7O24·4H2O Inorganic materials 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 238000004438 BET method Methods 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- JJLJMEJHUUYSSY-UHFFFAOYSA-L Copper hydroxide Chemical compound [OH-].[OH-].[Cu+2] JJLJMEJHUUYSSY-UHFFFAOYSA-L 0.000 description 1
- 239000005750 Copper hydroxide Substances 0.000 description 1
- QPLDLSVMHZLSFG-UHFFFAOYSA-N Copper oxide Chemical compound [Cu]=O QPLDLSVMHZLSFG-UHFFFAOYSA-N 0.000 description 1
- 239000005751 Copper oxide Substances 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- PGNKBEARDDELNB-UHFFFAOYSA-N Diethylcarbamazine citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.CCN(CC)C(=O)N1CCN(C)CC1 PGNKBEARDDELNB-UHFFFAOYSA-N 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 229910005191 Ga 2 O 3 Inorganic materials 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229910052765 Lutetium Inorganic materials 0.000 description 1
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- MCMNRKCIXSYSNV-UHFFFAOYSA-N ZrO2 Inorganic materials O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 1
- YKTSYUJCYHOUJP-UHFFFAOYSA-N [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] Chemical compound [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] YKTSYUJCYHOUJP-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000003570 air Substances 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N anhydrous glutaric acid Natural products OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- AQTIRDJOWSATJB-UHFFFAOYSA-K antimonic acid Chemical compound O[Sb](O)(O)=O AQTIRDJOWSATJB-UHFFFAOYSA-K 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- 229910000410 antimony oxide Inorganic materials 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 1
- 239000007767 bonding agent Substances 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- IAQRGUVFOMOMEM-UHFFFAOYSA-N butene Natural products CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 229910052570 clay Inorganic materials 0.000 description 1
- 230000001427 coherent effect Effects 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910001956 copper hydroxide Inorganic materials 0.000 description 1
- 229910000431 copper oxide Inorganic materials 0.000 description 1
- RKTYLMNFRDHKIL-UHFFFAOYSA-N copper;5,10,15,20-tetraphenylporphyrin-22,24-diide Chemical compound [Cu+2].C1=CC(C(=C2C=CC([N-]2)=C(C=2C=CC=CC=2)C=2C=CC(N=2)=C(C=2C=CC=CC=2)C2=CC=C3[N-]2)C=2C=CC=CC=2)=NC1=C3C1=CC=CC=C1 RKTYLMNFRDHKIL-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- HTXDPTMKBJXEOW-UHFFFAOYSA-N dioxoiridium Chemical compound O=[Ir]=O HTXDPTMKBJXEOW-UHFFFAOYSA-N 0.000 description 1
- 229910001882 dioxygen Inorganic materials 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000010304 firing Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910021505 gold(III) hydroxide Inorganic materials 0.000 description 1
- WDZVNNYQBQRJRX-UHFFFAOYSA-K gold(iii) hydroxide Chemical group O[Au](O)O WDZVNNYQBQRJRX-UHFFFAOYSA-K 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- XXMIOPMDWAUFGU-UHFFFAOYSA-N hexane-1,6-diol Chemical compound OCCCCCCO XXMIOPMDWAUFGU-UHFFFAOYSA-N 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229910000457 iridium oxide Inorganic materials 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- HCWCAKKEBCNQJP-UHFFFAOYSA-N magnesium orthosilicate Chemical compound [Mg+2].[Mg+2].[O-][Si]([O-])([O-])[O-] HCWCAKKEBCNQJP-UHFFFAOYSA-N 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 229910052919 magnesium silicate Inorganic materials 0.000 description 1
- 235000019792 magnesium silicate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000011733 molybdenum Substances 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- VTRUBDSFZJNXHI-UHFFFAOYSA-N oxoantimony Chemical class [Sb]=O VTRUBDSFZJNXHI-UHFFFAOYSA-N 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 238000005453 pelletization Methods 0.000 description 1
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 1
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 239000008262 pumice Substances 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000033458 reproduction Effects 0.000 description 1
- HYXGAEYDKFCVMU-UHFFFAOYSA-N scandium oxide Chemical compound O=[Sc]O[Sc]=O HYXGAEYDKFCVMU-UHFFFAOYSA-N 0.000 description 1
- 229930000044 secondary metabolite Natural products 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- HBMJWWWQQXIZIP-UHFFFAOYSA-N silicon carbide Chemical compound [Si+]#[C-] HBMJWWWQQXIZIP-UHFFFAOYSA-N 0.000 description 1
- 229910010271 silicon carbide Inorganic materials 0.000 description 1
- 239000010944 silver (metal) Substances 0.000 description 1
- 229910001923 silver oxide Inorganic materials 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000006104 solid solution Substances 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000003746 surface roughness Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- ZCUFMDLYAMJYST-UHFFFAOYSA-N thorium dioxide Chemical compound O=[Th]=O ZCUFMDLYAMJYST-UHFFFAOYSA-N 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 229910001928 zirconium oxide Inorganic materials 0.000 description 1
Images



Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/28—Molybdenum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/31—Chromium, molybdenum or tungsten combined with bismuth
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/64—Platinum group metals with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/652—Chromium, molybdenum or tungsten
- B01J23/6525—Molybdenum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/88—Molybdenum
- B01J23/887—Molybdenum containing in addition other metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/8877—Vanadium, tantalum, niobium or polonium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/02—Sulfur, selenium or tellurium; Compounds thereof
- B01J27/057—Selenium or tellurium; Compounds thereof
- B01J27/0576—Tellurium; Compounds thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/70—Catalysts, in general, characterised by their form or physical properties characterised by their crystalline properties, e.g. semi-crystalline
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/0215—Coating
- B01J37/0221—Coating of particles
- B01J37/0223—Coating of particles by rotation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
- B01J37/10—Heat treatment in the presence of water, e.g. steam
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B13/00—Oxygen; Ozone; Oxides or hydroxides in general
- C01B13/14—Methods for preparing oxides or hydroxides in general
- C01B13/36—Methods for preparing oxides or hydroxides in general by precipitation reactions in aqueous solutions
- C01B13/366—Methods for preparing oxides or hydroxides in general by precipitation reactions in aqueous solutions by hydrothermal processing
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B19/00—Selenium; Tellurium; Compounds thereof
- C01B19/002—Compounds containing, besides selenium or tellurium, more than one other element, with -O- and -OH not being considered as anions
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B19/00—Selenium; Tellurium; Compounds thereof
- C01B19/008—Salts of oxyacids of selenium or tellurium
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G39/00—Compounds of molybdenum
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G39/00—Compounds of molybdenum
- C01G39/006—Compounds containing, besides molybdenum, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G45/00—Compounds of manganese
- C01G45/006—Compounds containing, besides manganese, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G47/00—Compounds of rhenium
- C01G47/006—Compounds containing, besides rhenium, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G49/00—Compounds of iron
- C01G49/009—Compounds containing, besides iron, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G51/00—Compounds of cobalt
- C01G51/006—Compounds containing, besides cobalt, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G53/00—Compounds of nickel
- C01G53/006—Compounds containing, besides nickel, two or more other elements, with the exception of oxygen or hydrogen
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G55/00—Compounds of ruthenium, rhodium, palladium, osmium, iridium, or platinum
- C01G55/002—Compounds containing, besides ruthenium, rhodium, palladium, osmium, iridium, or platinum, two or more other elements, with the exception of oxygen or hydrogen
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2235/00—Indexing scheme associated with group B01J35/00, related to the analysis techniques used to determine the catalysts form or properties
- B01J2235/15—X-ray diffraction
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2235/00—Indexing scheme associated with group B01J35/00, related to the analysis techniques used to determine the catalysts form or properties
- B01J2235/30—Scanning electron microscopy; Transmission electron microscopy
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/50—Solid solutions
- C01P2002/52—Solid solutions containing elements as dopants
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/50—Solid solutions
- C01P2002/52—Solid solutions containing elements as dopants
- C01P2002/54—Solid solutions containing elements as dopants one element only
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/72—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by d-values or two theta-values, e.g. as X-ray diagram
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/01—Particle morphology depicted by an image
- C01P2004/03—Particle morphology depicted by an image obtained by SEM
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2004/00—Particle morphology
- C01P2004/10—Particle morphology extending in one dimension, e.g. needle-like
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2006/00—Physical properties of inorganic compounds
- C01P2006/12—Surface area
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Catalysts (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
本発明は以下の一般化学量論I:
Mo1VaM1 bM2 cM3 dM4 eOn (I)
[式中、M1=Al、Ga、In、Ge、Sn、Pb、As、Bi、Se、Te及びSbよりなる群から選択される少なくとも1つの元素;
M2=Sc、Y、La、Ti、Zr、Hf、Nb、Ta、Cr、W、Mn、Fe、Co、Ni、Zn、Cd及びランタノイド類よりなる群から選択される少なくとも1つの元素;
M3=Re、Ru、Rh、Pd、Os、Ir、Pt、Cu、Ag、Auよりなる群から選択される少なくとも1つの元素;
M4=Li、Na、K、Rb、Cr、Be、Mg、Ca、Sr、Ba、NH4及びTlよりなる群から選択される少なくとも1つの元素;
a=0.01〜1;
b=≧0〜1;
c=≧0〜1;
d=≧0〜0.5;
e=≧0〜0.5及び
n=(I)中の酸素以外の元素の原子価及び出現頻度によって決定される数]の一般化学量論Iの多金属酸化物材料Mの製造方法であって、
ここで、多金属酸化物材料Mの元素構成成分の原料の混合物Gは水熱処理に付され、これにより新たに形成される固体が分離される方法に関する。
The present invention provides the following general stoichiometry I:
Mo 1 V a M 1 b M 2 c M 3 d M 4 e O n (I)
[Wherein M 1 = at least one element selected from the group consisting of Al, Ga, In, Ge, Sn, Pb, As, Bi, Se, Te and Sb;
M 2 = at least one element selected from the group consisting of Sc, Y, La, Ti, Zr, Hf, Nb, Ta, Cr, W, Mn, Fe, Co, Ni, Zn, Cd and lanthanoids;
M 3 = At least one element selected from the group consisting of Re, Ru, Rh, Pd, Os, Ir, Pt, Cu, Ag, Au;
M 4 = at least one element selected from the group consisting of Li, Na, K, Rb, Cr, Be, Mg, Ca, Sr, Ba, NH 4 and Tl;
a = 0.01-1;
b = ≧ 0-1;
c = ≧ 0-1;
d = ≧ 0 to 0.5;
e = ≧ 0 to 0.5 and n = the number determined by the valence and frequency of appearance of elements other than oxygen in (I)]. And
Here, the mixture G of the raw material of the elemental component of the multimetal oxide material M is subjected to a hydrothermal treatment, whereby a newly formed solid is separated.
一般化学量論Iの多金属酸化物材料M及びその製造方法は既知である(EP−A318295、EP−A512846、EP−A767164、EP−A865809、EP−A529853、EP−A608838、EP−A962253、DE−A10248584、DE−A10119933、DE−A10118814、DE−A10029338、DE−A10359027、DE−A10321398、EP−A1407819、Applied Catalysis A:General194−195(2000)479〜485ページ;Applied Catalysis A:General200(2000)135〜143ページ;Chem.Commun.1999、517〜518ページ;Res.Chem.Intermed.26(2)(2000)、137〜144ページ;Topics Catal.15(2001)153〜160ページ;Catalysis Surveys from Japan6(1/2)(2992)33〜44及びAppl.Catal.A:General251(2003)411〜424ページを参照)。 Multi-metal oxide materials M of general stoichiometry I and processes for their preparation are known (EP-A 318295, EP-A 512846, EP-A 767164, EP-A 865809, EP-A 529853, EP-A 608838, EP-A 962533, DE -A10248584, DE-A101199933, DE-A10118814, DE-A10029338, DE-A10359027, DE-A10321398, EP-A1407819, Applied Catalysis A: General 194-195 (2000) pages 479-485; Pages 135-143; Chem. Commun. 1999, pages 517-518; termed.26 (2) (2000), pages 137-144; Topics Cat.15 (2001) pages 153-160; Catalysis Surveys from Japan6 (1/2) (2992) 33-44 and Appl. Catal.A: General 251 (2003) See pages 411-424).
また、上記先行技術からこのような多金属酸化物材料Mは、飽和及び不飽和の炭化水素、(例えば、3〜8、特に2又は3及び/又は4の炭素原子を有する)アルコール及びアルデヒドの混成に触媒された部分ガス相酸化のための、そして(追加のアンモニアの存在を通して、直接的な部分ガス相酸化とは実質的に異なっている)混成に触媒された部分ガス相アンモ酸化のための触媒用の活性物質として適当であることは既知である。これによる部分酸化生成物は、特に、α,β−モノエチレン性不飽和アルデヒド(例えば、アクロレイン及びメタアクロレイン)及びα,β−モノエチレン性不飽和カルボン酸(例えば、アクリル酸及びメタアクリル酸)及びそのニトリル(例えば、アクリロニトリル及びメタアクリロニトリル)である。目的化合物のアクロレイン、アクリル酸及び/又はアクリロニトリルは、例えば、炭化水素のプロパン及び/又はプロペンから記載の方法で得ることができる。アクロレインはまた、それ自体後者の2つの化合物の製造のための出発物質であってもよい。これらの目的化合物は例えば、接着剤として利用することができる、例えば、ポリマーの製造のために使用される重要な中間体を形成する。 Also, from the above prior art, such multi-metal oxide materials M are of saturated and unsaturated hydrocarbons, such as alcohols and aldehydes (for example having 3 to 8, in particular having 2 or 3 and / or 4 carbon atoms). For hybrid catalyzed partial gas phase oxidation and for hybrid catalyzed partial gas phase ammoxidation (which is substantially different from direct partial gas phase oxidation through the presence of additional ammonia) It is known to be suitable as an active substance for the catalyst. The resulting partial oxidation products are in particular α, β-monoethylenically unsaturated aldehydes (eg acrolein and metaacrolein) and α, β-monoethylenically unsaturated carboxylic acids (eg acrylic acid and methacrylic acid). And its nitriles (eg, acrylonitrile and methacrylonitrile). The target compounds acrolein, acrylic acid and / or acrylonitrile can be obtained, for example, from the hydrocarbon propane and / or propene by the method described. Acrolein may also itself be a starting material for the production of the latter two compounds. These target compounds form, for example, important intermediates that can be used as adhesives, for example used for the production of polymers.
相当する方法で、メタアクロレイン及びメタアクリル酸はイソブタン及びイソブテンから得ることができる。メタアクロレインはまた、メタアクリル酸の製造のための出発物質であってもよい。 In a corresponding manner, methacrolein and methacrylic acid can be obtained from isobutane and isobutene. Methacrolein may also be a starting material for the production of methacrylic acid.
更に、多金属酸化物材料Mは、互いに異なっていて、「i−相」及び「k−相」として著述中に参照されている2つの結晶相に主に現れることは評価された先行技術から既知である。 Furthermore, it is recognized from the prior art that the multi-metal oxide material M is different from each other and appears mainly in the two crystalline phases referred to in the writing as “i-phase” and “k-phase”. Known.
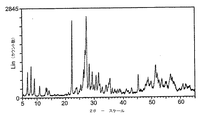
その特徴及びその結晶性構造の特徴のため、関連するX線回折図がそれぞれの結晶相の指紋として使用される。 Because of its characteristics and its crystalline structure characteristics, the associated X-ray diffractogram is used as a fingerprint for each crystalline phase.
結晶性i−相のX線回折図は、使用したX線の波長と独立に、格子面間隔d[Å]の形状で再現される以下のX線回折パターンRMiを含んでいる。 The X-ray diffraction diagram of the crystalline i-phase includes the following X-ray diffraction pattern RMi reproduced in the shape of the lattice spacing d [Å] independently of the wavelength of the X-ray used.
d[Å]
3.06±0.2
3.17±0.2
3.28±0.2
3.99±0.2
9.82±0.4
11.24±0.4
13.28±0.5
結晶性k−相のX線回折図は、使用したX線の波長と独立に、格子面間隔d[Å]の形状で再現される以下のX線回折パターンRMiを含んでいる。
d [Å]
3.06 ± 0.2
3.17 ± 0.2
3.28 ± 0.2
3.99 ± 0.2
9.82 ± 0.4
11.24 ± 0.4
13.28 ± 0.5
The X-ray diffraction diagram of the crystalline k-phase includes the following X-ray diffraction pattern RMi reproduced in the shape of the lattice spacing d [Å] independently of the X-ray wavelength used.
d[Å]
4.02±0.2
3.16±0.2
2.48±0.2
2.01±0.2
1.82±0.1
i−相及びk−相は互いに似ているが、特にk−相の回折図は通常d≧4.2Åの反射がないことが異なっている。通常、k−相はまた、3.8Å≧d≧3.35Åの範囲の反射を含んでいない。更に、k−相は原則として2.95Å≧d≧2.68Åの範囲の反射を含んでいない。
d [Å]
4.02 ± 0.2
3.16 ± 0.2
2.48 ± 0.2
2.01 ± 0.2
1.82 ± 0.1
The i-phase and the k-phase are similar to each other, except that the k-phase diffractogram usually has no reflection of d ≧ 4.24. Usually, the k-phase also does not contain reflections in the range of 3.8Å ≧ d ≧ 3.35Å. Furthermore, the k-phase in principle does not contain reflections in the range 2.95Å ≧ d ≧ 2.68Å.
更に、i−相構造を有する多金属酸化物材料の触媒的効果(活性、目標生成物の構造の選択性)は、原則として他の(例えばk−相)構造のものより優れていることが上記先行技術から既知である。 Furthermore, the catalytic effect (activity, selectivity of the target product structure) of the multi-metal oxide material having an i-phase structure should in principle be superior to that of other (eg k-phase) structures. Known from the above prior art.
しかしながら、i−相構造の多金属酸化物材料Mを生成することは非常に難しいことが上記先行技術から既知である。 However, it is known from the prior art that it is very difficult to produce an i-phase structured multi-metal oxide material M.
従って、原則として、固体溶液系が得られ、これは(例えば、k−相に加えて)ある程度のi−相部分だけを有し、最適の触媒性能の観点から見て、ここから適当な液体で他の相(例えばk−相)を洗い流すことによりi−相部分が単離される。(例えば、WO02/06199、JP−A7−232071、DE−A10254279及びEP−A1407819)。 Thus, in principle, a solid solution system is obtained, which has only a certain i-phase part (in addition to the k-phase, for example), from which a suitable liquid is obtained in view of optimum catalyst performance. The i-phase portion is isolated by washing away the other phases (eg k-phase). (For example, WO02 / 06199, JP-A7-232011, DE-A10254279 and EP-A1407819).
一般化学量論Iの多金属酸化物材料の製造は、緊密な乾燥混合物がその元素構成成分を含む出発物質(原料)から製造され、上記乾燥混合物は高温で熱処理に付される方法によって一般的に達成される。 The production of a multi-metal oxide material of general stoichiometry I is generally achieved by a process in which an intimate dry mixture is produced from a starting material (raw material) containing its elemental components and the dry mixture is subjected to a heat treatment at high temperature. To be achieved.
多金属酸化物材料Mの元素構成成分の原料混合物を水熱処理に付し、新たに形成される固体を分離する際に、原則として排他的なi−相までの高いi−相部分が得られる(US−A2003/0187299A1、DE−A10029338、EP−A1270068、EP−A1346766及びEP−A1407819を参照)。 When a raw material mixture of elemental constituents of the multimetal oxide material M is subjected to hydrothermal treatment to separate a newly formed solid, a high i-phase portion up to an exclusive i-phase is obtained in principle. (See US-A2003 / 0187299A1, DE-A10029338, EP-A1270068, EP-A1346766 and EP-A1407819).
先行技術の叙述に従い、元素構成成分の適当な原料は、化学的に結合した形状で元素構成成分を含むほぼ全ての可能な化合物である。 In accordance with the prior art statement, suitable raw materials for elemental components are almost all possible compounds containing elemental components in chemically bonded form.
しかしながら、本方法で実施する水熱製造法の欠点は、特に工業量の生産の場合、再現性が完全には満足されないことである。例えば、このような生産の中で得られるi−相部分は、反復される生産上、平均値について比較的広範囲内で変動する。しばしば、この平均値は更に比較的低いi−相部分においてである。更に、直接的に水熱で得られる生成物の触媒性能は、しばしば不満足なものであり、原則としてその改善のために次の熱処理を必要とする。
従って、本発明の目的は上述の欠点に関して、方法を改善した一般化学量論Iの多金属酸化物材料Mの水熱製造方法を供給することであった。
However, a disadvantage of the hydrothermal production process carried out in the present method is that the reproducibility is not completely satisfied, especially in the case of industrial production. For example, the i-phase portion obtained in such production varies within a relatively wide range of average values over repeated production. Often this average value is in the relatively lower i-phase part. Furthermore, the catalytic performance of products obtained directly with hydrothermal heat is often unsatisfactory and, in principle, requires a subsequent heat treatment to improve it.
Accordingly, it was an object of the present invention to provide a method for hydrothermal production of multi-metal oxide material M of general stoichiometry I which has an improved method with respect to the above mentioned drawbacks.
従って、以下の一般化学量論I:
Mo1VaM1 bM2 cM3 dM4 eOn (I)
[式中、M1=Al(+3)、Ga(+3)、In(+3)、Ge(+4)、Sn(+4)、Pb(+4)、As(+5)、Bi(+5)、Se(+6)、Te(+6)及びSb(+5)よりなる群から選択される少なくとも1つの元素;
M2=Sc(+3)、Y(+3)、La(+3)、Ti(+4)、Zr(+4)、Hf(+4)、Nb(+5)、Ta(+5)、Cr(+5)、W(+6)、Mn(+7)、Fe(+3)、Co(+3)、Ni(+3)、Zn(+2)、Cd(+2)及びランタノイド類(Ce(+4)、Pr(+3)、Nd(+3)、Pm(+3)、Sm(+3)、Eu(+3)、Gd(+3)、Tb(+4)、Dy(+3)、Ho(+3)、Er(+3)、Tm(+3)、Yb(+3)及びLu(+3))よりなる群から選択される少なくとも1つの元素;
M3=Re(+7)、Ru(+8)、Rh(+8)、Pd(+8)、Os(+8)、Ir(+8+)、Pt(+8)、Cu(+2)、Ag(+1)、Au(+1)よりなる群から選択される少なくとも1つの元素;
M4=Li(+1)、Na(+1)、K(+1)、Rb(+1)、Cs(+1)、Be(+2)、Mg(+2)、Ca(+2)、Ir(+2)、Ba(+2)、NH4(+1)及びTl(+1)よりなる群から選択される少なくとも1つの元素;
a=0.01〜1(好ましくは0.01〜0.5);
b=≧0〜1(好ましくは>0〜0.5);
c=≧0〜1(好ましくは>0〜0.5);
d=≧0〜0.5(好ましくは≧0〜0.1);
e=≧0〜0.5(好ましくは≧0〜0.1又は0)及び
n=(I)中の酸素以外の元素の原子価及び出現頻度によって決定される数]の
一般化学量論Iの多金属酸化物材料Mの製造方法が発明され、ここで、多金属酸化物材料Mの元素構成成分の原料の混合物Gは水熱処理に付され、新たに形成される固体が分離され、ここで多金属酸化物材料Mの元素構成成分及び水の元素構成成分(O、H、OH)、その元素形状の多金属酸化物材料M自体の元素構成成分及び多金属酸化物材料Mの元素構成成分を含むアンモニウム塩(例えば、元素構成成分の酸化物、水和酸化物、酸素酸、水酸化物、酸化水酸化物、元素構成成分の金属元素、及びアンモニウムメタバナデート又はアンモニウムヘプタモリブデートのような化合物)のみよりなる化合物よりなる群から排他的に選択される原料が、混合物Gに含まれるNH4及び混合物Gに含まれるMoのNH4/Moモル比MVが≦0.5及び少なくとも元素構成成分の原料の一部が、それぞれの元素構成成分の最大酸化数より低い酸化数(これは、独立に一覧される元素M1、M2、M3及びM4の後に括弧内で表示され、正の符号を有する数である)を有するこれらの原料に含まれる元素構成成分を含むという条件で、多金属酸化物材料Mの元素構成成分の原料として使用される。
Thus, the following general stoichiometry I:
Mo 1 V a M 1 b M 2 c M 3 d M 4 e O n (I)
[Wherein M 1 = Al (+3), Ga (+3), In (+3), Ge (+4), Sn (+4), Pb (+4), As (+5), Bi (+5), Se (+6 ), Te (+6) and Sb (+5) at least one element selected from the group consisting of;
M 2 = Sc (+3), Y (+3), La (+3), Ti (+4), Zr (+4), Hf (+4), Nb (+5), Ta (+5), Cr (+5), W ( +6), Mn (+7), Fe (+3), Co (+3), Ni (+3), Zn (+2), Cd (+2) and lanthanoids (Ce (+4), Pr (+3), Nd (+3) , Pm (+3), Sm (+3), Eu (+3), Gd (+3), Tb (+4), Dy (+3), Ho (+3), Er (+3), Tm (+3), Yb (+3) And at least one element selected from the group consisting of Lu (+3));
M 3 = Re (+7), Ru (+8), Rh (+8), Pd (+8), Os (+8), Ir (+8+), Pt (+8), Cu (+2), Ag (+1), Au ( +1) at least one element selected from the group consisting of;
M 4 = Li (+1), Na (+1), K (+1), Rb (+1), Cs (+1), Be (+2), Mg (+2), Ca (+2), Ir (+2), Ba ( +2), at least one element selected from the group consisting of NH 4 (+1) and Tl (+1);
a = 0.01-1 (preferably 0.01-0.5);
b = ≧ 0 to 1 (preferably> 0 to 0.5);
c = ≧ 0 to 1 (preferably> 0 to 0.5);
d = ≧ 0 to 0.5 (preferably ≧ 0 to 0.1);
e = ≧ 0 to 0.5 (preferably ≧ 0 to 0.1 or 0) and n = number determined by the valence and frequency of occurrence of elements other than oxygen in (I)] A method for producing a multi-metal oxide material M was invented. Here, a mixture G of raw materials of elemental constituents of the multi-metal oxide material M was subjected to hydrothermal treatment, and a newly formed solid was separated, The elemental component of the multimetal oxide material M and the elemental component of water (O, H, OH), the elemental component of the multimetal oxide material M itself of the element shape, and the elemental composition of the multimetal oxide material M Ammonium salts containing components (eg, elemental component oxides, hydrated oxides, oxyacids, hydroxides, hydroxide oxides, elemental component metal elements, and ammonium metavanadate or ammonium heptamolybdate) Compound) only Raw materials exclusively selected from the group consisting more is a mixture NH 4 and mixture NH 4 / Mo molar ratio part of the raw material of the MV ≦ 0.5 and at least the elemental constituents of Mo contained in the G contained in G Is an oxidation number lower than the maximum oxidation number of each elemental component (this is a number that is displayed in parentheses after the independently listed elements M 1 , M 2 , M 3 and M 4 and has a positive sign) Is used as a raw material for the elemental constituents of the multi-metal oxide material M on the condition that the constituent elements contained in these raw materials are included.
元素構成成分Vの原料の少なくとも一部(混合物G中に存在)が酸化数<+5(例えば、+4又は+3又は+2又は0)を有するバナジウムを含んでいた場合、本発明に従って好ましい。 It is preferred according to the invention if at least part of the raw material of the elemental constituent V (present in the mixture G) contains vanadium with an oxidation number <+5 (for example +4 or +3 or +2 or 0).
多金属酸化物活性物質の水熱製造は、当業者が知る通りである(例えば、Applied Catalysis A:194〜195(2000)479〜485;Kinetics and Catalysis;Vol.40、No.3、1999、pp401〜404;Chem.Commun.、1999、517〜518;JP−A6/227819及びJP−A2000/26123もまた参照)。 Hydrothermal production of multimetal oxide actives is known to those skilled in the art (eg, Applied Catalysis A: 194-195 (2000) 479-485; Kinetics and Catalysis; Vol. 40, No. 3, 1999, pp 401-404; Chem. Commun., 1999, 517-518; see also JP-A6 / 227819 and JP-A2000 / 26123).
本明細書において、本発明に従った操作法は、特に、通常>100℃〜600℃の温度で、超大気圧で水蒸気の存在下に、超大気圧(オートクレーブ)下に、容器中に、所望の多金属酸化物材料Mの元素構成成分の原料の好ましく緊密な混合物Gの熱処理という意味で理解されている。圧力範囲は典型的には(>1バール)500バールまで、好ましくは250まで拡張される。本発明に従って、600℃を超える温度及び500バールを超える水蒸気圧もまた当然ながら使用することができるが、応用技術の目的ではあまり適切ではない。 In the present description, the operating method according to the invention is preferably carried out at a temperature of> 100 ° C. to 600 ° C., in the presence of water vapor at superatmospheric pressure, in a vessel under superatmospheric pressure (autoclave), in the desired manner. It is understood in the sense of a heat treatment of a preferably intimate mixture G of the raw materials of the elemental constituents of the multimetal oxide material M. The pressure range is typically extended to (> 1 bar) up to 500 bar, preferably up to 250. According to the invention, temperatures above 600 ° C. and water vapor pressures above 500 bar can of course also be used, but are not very suitable for the purpose of application technology.
特に好都合には、本発明に従った水熱処理は水蒸気及び液体水性相が共存する条件下で達成される。相当する圧力を使用し、>100〜374.15℃(水の臨界温度)の温度範囲でこれは可能である。水の量は、液体水性相が懸濁液及び/又は溶液中の元素構成成分の出発化合物の総量を溶解できるようなものが適切である。 Particularly advantageously, the hydrothermal treatment according to the invention is achieved under conditions in which water vapor and a liquid aqueous phase coexist. This is possible in the temperature range> 100-374.15 ° C. (critical temperature of water) using corresponding pressures. Suitably the amount of water is such that the liquid aqueous phase can dissolve the total amount of elemental constituent starting compounds in the suspension and / or solution.
しかしながら、本発明に従って、元素構成成分の出発化合物の好ましく緊密な混合物Gが水蒸気と平衡に存在する液体水の量を完全に吸収する水熱操作法もまた可能である。 However, according to the invention, a hydrothermal process is also possible in which a preferably intimate mixture G of elemental constituent starting compounds completely absorbs the amount of liquid water that is in equilibrium with water vapor.
本発明に従って、本発明に従った水熱処理は>100℃〜300℃の温度、好ましくは120℃又は150℃〜250℃の温度(例えば160℃〜180℃)で好都合に達成される。 According to the invention, the hydrothermal treatment according to the invention is conveniently achieved at temperatures> 100 ° C. to 300 ° C., preferably 120 ° C. or 150 ° C. to 250 ° C. (eg 160 ° C. to 180 ° C.).
水及び混合物G(又はその中に含まれる多金属酸化物材料Mの元素構成成分の原料)の合計量に基づき、オートクレーブ中の後者の重量比率は、本発明に従って、原則として少なくとも1重量%である。通常、上述の重量比率は90重量%を超えてはならない。好ましくは重量比率は5〜60又は10〜50重量%、特に好ましくは20〜又は30〜50重量%である。 Based on the total amount of water and the mixture G (or the raw material of the elemental constituents of the multimetal oxide material M contained therein), the latter weight ratio in the autoclave is in principle at least 1% by weight according to the invention. is there. In general, the above-mentioned weight ratio should not exceed 90% by weight. The weight ratio is preferably 5 to 60 or 10 to 50% by weight, particularly preferably 20 to or 30 to 50% by weight.
多金属酸化物材料M及び水の元素構成成分の本発明による原料以外には、好ましくはそれ以上の物質は本発明に従った水熱操作法には必要としない。それ以上の物質の重量比率は混合物Gに基づいて通常≦10重量%、好都合には≦5重量%及び更に好ましくは≦3重量%であるべきである。例えば、DE−A10029338及びEP−A1407819に記述された全ての物質でH2O2以外(好ましい酸化還元条件を考慮)は、この形式の可能な外来物質として適当である。 Apart from the raw materials according to the invention of the multi-metal oxide material M and the elemental constituents of water, preferably no further substances are required for the hydrothermal operating method according to the invention. The weight ratio of further substances should usually be ≦ 10% by weight, conveniently ≦ 5% by weight and more preferably ≦ 3% by weight, based on the mixture G. For example, all substances described in DE-A1003338 and EP-A1407819 other than H 2 O 2 (taking into account preferred redox conditions) are suitable as possible foreign substances of this type.
本発明に従った水熱処理は攪拌しながら(好ましい)でも攪拌なしでも実施できる。 The hydrothermal treatment according to the invention can be carried out with (preferred) or without stirring.
好都合には、本発明に従った方法でモル比MVは≦0.3、特に好ましくは≦0.1そして非常に好ましくは0である。 Conveniently, in the process according to the invention, the molar ratio MV is ≦ 0.3, particularly preferably ≦ 0.1 and very particularly preferably 0.
混合物Gに含まれる元素構成成分の原料の総量及びこれらの原料に含まれるVの総モル量に基づいて、本発明に従った方法でこれらの原料に含まれる、好ましくは少なくとも5モル%又は少なくとも10モル%、好ましくは少なくとも20モル%又は少なくとも30モル%、非常に特に好ましくは少なくとも40モル%又は50モル%そして最も好ましくは少なくとも60モル%又は少なくとも70モル%又は80モル%又は少なくとも90モル%又はそれ以上(特に好都合には総量)のVが酸化数が<+5であるバナジウムとして含まれている。 Based on the total amount of raw materials of the elemental constituents contained in the mixture G and the total molar amount of V contained in these raw materials, it is preferably contained in these raw materials in the process according to the invention, preferably at least 5 mol% or at least 10 mol%, preferably at least 20 mol% or at least 30 mol%, very particularly preferably at least 40 mol% or 50 mol% and most preferably at least 60 mol% or at least 70 mol% or 80 mol% or at least 90 mol% % Or more (particularly advantageously total) V is included as vanadium with an oxidation number <+5.
混合物Gに含まれるバナジウム原料のVの総モル量を平均したVの数学的平均酸化数は好ましくは+3.5〜+4.5、特に好ましくは+3.8〜+4.2そして特に好ましくは+4である。 The mathematical average oxidation number of V obtained by averaging the total molar amount of V of the vanadium raw material contained in the mixture G is preferably +3.5 to +4.5, particularly preferably +3.8 to +4.2 and particularly preferably +4. is there.
非常に一般的に(特にVがその原料の少なくとも一部の中に、その最大酸化数で含まれる場合)、本発明に従った方法の混合物Gの組成は、好ましくは混合物Gのための原料に含まれる元素構成成分の酸化数に関して選択され、そのため、(V(<+5)を含む)混合物G原料中のその最大酸化数に含まれないV(+5)以外の全元素構成成分をそれぞれ最大酸化数(Vの場合、その2番目に最高の酸化数+4)に変更すると、これにより利用可能な還元能力は混合物Gの原料中に総計で含まれるVの平均酸化数を3.5〜4.5、特に好ましくは3.8〜4.2、非常に特に好ましくは4の値にするのに正に十分である。 Very generally (especially when V is included in at least part of its raw material at its maximum oxidation number), the composition of the mixture G of the process according to the invention is preferably the raw material for the mixture G Is selected with respect to the oxidation number of the elemental constituents contained in the mixture G, so that all elemental constituents other than V (+5) not included in the maximum oxidation number in the mixture G raw material (including V (<+ 5)) are maximized respectively. When changed to the oxidation number (in the case of V, the second highest oxidation number +4), the available reduction capacity is that the average oxidation number of V contained in the raw material of the mixture G is 3.5-4. .5, particularly preferably from 3.8 to 4.2, very particularly preferably to a value of 4.
本発明に従った水熱処理が酸素分子を含む酸化雰囲気下(例えば、静止し封印された空気下)で達成された場合、上述の還元能力は相当してより高くなってよい。 If the hydrothermal treatment according to the present invention is achieved under an oxidizing atmosphere containing oxygen molecules (for example under static and sealed air), the above-mentioned reducing ability may be considerably higher.
概して、本発明に従った水熱処理の条件は、好ましくは上記還元能力が実際に水熱処理中に記載された方法で履行されるように選択される。 In general, the conditions of the hydrothermal treatment according to the present invention are preferably selected so that the reducing capacity is actually implemented in the manner described during hydrothermal treatment.
V又は原料中の別の元素構成成分の酸化数の決定のために、化学化合物中のある元素の酸化数が以下の通り得られることに従って、本質的に既知の法則が適用される。
1.遊離元素中の原子の酸化数は0。
2.単一原子イオンの酸化数はその電荷と等しい。
3.既知構造の共有結合化合物において、結合電子対がより多くの電気陰性原子に完全に分配されている場合、酸化数は各原子が含む電荷に相当する。2つの同一原子間の電子対の場合、各原子は1つの電子を受け取る。
(Grundlagen der allgemeinen und organischen Chemie[Principles of general and organic chemistry]、Verlag Sauerlander、Aarau、Diesterweg Salle、Frankfurt am Main、4th Edition、1973参照)
従って、本発明に従った方法のための元素Vのために適当な原料は特にVO2、V2O3、V6O13、V3O7、V4O9及びVOのようなバナジウムの酸化物、元素バナジウム及び、本発明に従った量制限を考慮して、V2O5及びアンモニウムメタバナデートのような化合物も同様である。
For the determination of the oxidation number of V or another elemental component in the raw material, essentially known laws are applied according to the fact that the oxidation number of an element in a chemical compound is obtained as follows.
1. The oxidation number of atoms in free elements is zero.
2. The oxidation number of a single atom ion is equal to its charge.
3. In a covalently bonded compound of known structure, when the bonding electron pair is completely distributed to more electronegative atoms, the oxidation number corresponds to the charge that each atom contains. In the case of an electron pair between two identical atoms, each atom receives one electron.
(Grundgen der allgemeinen und organischen Chemie [Principles of general and organic chemistry], Verlag Sauerlander, Aarau, Diesterweig.
Accordingly, suitable raw materials for element V for the process according to the invention are in particular vanadium such as VO 2 , V 2 O 3 , V 6 O 13 , V 3 O 7 , V 4 O 9 and VO. The same applies to oxides, elemental vanadium and compounds such as V 2 O 5 and ammonium metavanadate, taking into account the quantity limits according to the invention.
本発明に従って適当な元素Moのための原料は例えば、MoO3及びMoO2のようなモリブデンの酸化物、元素Mo及び本発明に従った量制限を考慮して、アンモニウムヘプタモリブデート及びその水和物のような化合物も同様である。 Ingredients for elemental Mo suitable according to the present invention include, for example, ammonium heptamolybdate and its hydration in view of oxides of molybdenum such as MoO 3 and MoO 2 , elemental Mo and the amount restrictions according to the present invention. The same applies to compounds such as products.
本発明に従って、元素テルルの適当な原料は、例えば、TeO2のようなテルルの酸化物、金属テルル及びオルトテルル酸H6TeO6のようなテルル酸も同様である。 According to the present invention, suitable raw materials for elemental tellurium are, for example, tellurium oxides such as TeO 2 , metal tellurium and telluric acid such as orthotelluric acid H 6 TeO 6 .
本発明に従って好都合なアンチモン出発化合物は、例えば、Sb2O3のようなアンチモンの酸化物、元素Sb、及びHSb(OH)6のようなアンチモン酸も同様である。 Antimony starting compounds that are advantageous according to the invention are, for example, antimony oxides such as Sb 2 O 3 , element Sb and antimonic acid such as HSb (OH) 6 .
本発明に従って適当なニオブ原料は、例えば、Nb2O5のようなニオブの酸化物又は元素Nbである。 A suitable niobium source in accordance with the present invention is a niobium oxide such as Nb 2 O 5 or the element Nb.
本発明に従って好都合なビスマス原料はBi2O3である。好都合な金原料は水酸化金である。更に本発明に従った方法のために好都合な出発化合物は、例えば、酸化銀、水酸化銅、酸化銅、アルカリ金属及びアルカリ土金属の酸化物及び水酸化物、酸化スカンジウム、酸化イリジウム、酸化亜鉛、Ga2O、Ga2O3等である。当然ながら、1つ以上の元素構成成分が含まれ、適宜、水熱法、例えば本発明に従った水熱法により得られた混合酸化物もまた、元素構成成分の原料として適当である。更に、本発明に従って適当な元素構成成分の原料は引用された先行技術の出版物に記述されている。 A convenient bismuth source in accordance with the present invention is Bi 2 O 3 . A convenient gold source is gold hydroxide. Further convenient starting compounds for the process according to the invention are, for example, silver oxide, copper hydroxide, copper oxide, alkali and alkaline earth metal oxides and hydroxides, scandium oxide, iridium oxide, zinc oxide. , Ga 2 O, Ga 2 O 3 and the like. Of course, one or more elemental constituents are included and, if appropriate, mixed oxides obtained by hydrothermal methods, for example hydrothermal methods according to the invention, are also suitable as raw materials for elemental constituents. Furthermore, suitable elemental constituent materials in accordance with the present invention are described in the cited prior art publications.
本発明に従った水熱処理自体は、一般的に数分又は数時間から数日間持続する。48時間の期間が典型的である。水熱処理に使用されるオートクレーブがテフロンで内部コーティングされている場合、応用技術の目的で適切である。水熱処理の前に、オートクレーブは適宜含有する水性混合物を含み、好都合に排出することができる。その後、温度が上昇する前に上記オートクレーブは好ましくは不活性ガス(N2、He、Ne及び/又はアルゴンのような希ガス)で充填することができる。両方の手段を除外できるが、これはあまり好都合ではない。好都合な不活性条件を作るため、本発明に従った水熱処理の前に、当然ながら水性混合物を追加又は代わりに不活性ガスでフラッシュできる。水熱処理の前であっても、オートクレーブ中の超大気圧を確立するため、上述の不活性ガスもまた応用技術の目的で適切に使用することができる。 The hydrothermal treatment itself according to the present invention generally lasts minutes or hours to days. A period of 48 hours is typical. If the autoclave used for hydrothermal treatment is internally coated with Teflon, it is suitable for the purpose of applied technology. Prior to the hydrothermal treatment, the autoclave contains an aqueous mixture optionally contained and can be expelled conveniently. Thereafter, the autoclave before the temperature rises can preferably be filled with an inert gas (N 2, the He, noble gases such as Ne and / or argon). Both measures can be excluded, but this is not very convenient. In order to create convenient inert conditions, the aqueous mixture can of course be flushed with an inert gas instead of or before the hydrothermal treatment according to the invention. Even before the hydrothermal treatment, the above-mentioned inert gas can also be used appropriately for the purpose of applied technology in order to establish a superatmospheric pressure in the autoclave.
水熱処理終了後、オートクレーブは室温でクエンチングされるか、例えば相対的に長期間かけて(例えば放置して)ゆっくり室温に戻すことができる。 After completion of the hydrothermal treatment, the autoclave can be quenched at room temperature or can be slowly returned to room temperature, for example over a relatively long period of time (eg, left to stand).
本発明に従って重要な側面は、本発明に従った水熱処理の中で新たに形成され、水熱処理の終了後分離された固体が通常、高いi−相部分を有するか排他的にi−相を含む多金属酸化物Mであり、本発明に従って、改善された再現性で得られることである。 An important aspect according to the present invention is that the solid that is newly formed in the hydrothermal treatment according to the present invention and separated after completion of the hydrothermal treatment usually has a high i-phase part or exclusively i-phase. A multi-metal oxide M that is obtained with improved reproducibility according to the present invention.
また、本発明に従った重要性は本発明に従った方法により得られる多金属酸化物Mは水熱処理の後の熱処理を用いない場合であっても、所望の触媒活性を示すことである。 Further, the importance according to the present invention is that the multimetal oxide M obtained by the method according to the present invention exhibits a desired catalytic activity even when the heat treatment after the hydrothermal treatment is not used.
当然ながら、本発明に従って得られる多金属酸化物Mは、初めに記載された混成に触媒された方法のための活性物質として使用される前に、好都合に追加の熱後処理に付すことができる。この熱後処理は200〜1200℃、好ましくは350〜700℃、しばしば400〜650℃、たびたび400〜600℃の温度で実施することができる。 Of course, the multimetal oxide M obtained according to the invention can be conveniently subjected to an additional thermal workup before it is used as an active material for the hybrid catalyzed process described at the outset. . This thermal aftertreatment can be carried out at a temperature of 200 to 1200 ° C., preferably 350 to 700 ° C., often 400 to 650 ° C., often 400 to 600 ° C.
原則として、酸化、還元あるいは不活性(本発明に従って好ましい)雰囲気下で達成することができる。適当な酸化雰囲気は、例えば空気、酸素分子の豊富な空気又は酸素が枯渇した空気である。 In principle, it can be achieved in an oxidizing, reducing or inert (preferred according to the invention) atmosphere. Suitable oxidizing atmospheres are, for example, air, oxygen molecule rich air or oxygen depleted air.
好ましくは熱処理は不活性雰囲気下、例えば窒素分子及び/又は希ガス(He、Ar及び/又はNe)で実施される(本明細中、不活性雰囲気は常に酸素分子の含有量が通常≦5容量%、好ましくは≦3容量%、特に好ましくは≦1容量%又は≦0.1容量%、最も好ましくは0容量%であることを意味する)。当然ながら、熱後処理もまた減圧下で達成することができる。 Preferably, the heat treatment is performed under an inert atmosphere, for example, with nitrogen molecules and / or noble gases (He, Ar and / or Ne) (in this specification, the inert atmosphere always has a content of oxygen molecules usually ≦ 5 volumes). %, Preferably ≦ 3% by volume, particularly preferably ≦ 1% by volume or ≦ 0.1% by volume, most preferably 0% by volume). Of course, thermal aftertreatment can also be achieved under reduced pressure.
概して、熱後処理は24時間又はそれ以上続けてよい。熱後処理は好ましくは酸化下(酸素含有雰囲気)(例えば空気下)で150℃〜400℃又は250℃〜350℃の温度で最初に達成される。その後、熱処理は350℃〜700℃又は400℃〜650℃又は400℃〜600℃の温度で不活性ガス下で適切に継続される。当然ながら、水熱的に生成された多金属酸化物Mの熱後処理もまた、水熱的に生成された多金属酸化物Mが最初にペレットされ、次に熱後処理に付され、続いてチップに転換されるような方法で達成することができる。 In general, the thermal aftertreatment may last for 24 hours or longer. Thermal post-treatment is preferably first achieved at temperatures of 150 ° C. to 400 ° C. or 250 ° C. to 350 ° C. under oxidation (oxygen-containing atmosphere) (eg, under air). Thereafter, the heat treatment is suitably continued under inert gas at a temperature of 350 ° C. to 700 ° C. or 400 ° C. to 650 ° C. or 400 ° C. to 600 ° C. Of course, the thermal post-treatment of the hydrothermally generated multimetal oxide M is also the same as the hydrothermally generated multimetal oxide M is first pelletized and then subjected to thermal post-treatment, followed by Can be achieved in such a way as to be converted into a chip.
しかしながら、本発明に従った水熱法の中で得られる多金属酸化物Mがその熱後処理のためにチップに転換された場合、応用技術の目的では適切である。 However, if the multi-metal oxide M obtained in the hydrothermal method according to the invention is converted into chips for its thermal aftertreatment, it is appropriate for the purpose of applied technology.
更に、水熱法により本発明に従って得られる多金属酸化物材料M及び記載の通りに熱後処理されたそれに続く又はその後継の多金属酸化物材料の両方は例えば、DE−A10254279及びEP−A1407819に記載の通り適当な液体でそれらを洗浄することにより好都合な方法で更に処理することができる。適当なこのような液体は、例えば、有機酸及びその水溶液(例えば、シュウ酸、ギ酸、酢酸、クエン酸及び酒石酸)及び無機酸及びその水溶液(硫酸、過塩素酸、塩酸、硝酸、ホウ酸及び/又はテルル酸)のみならず、アルコール、上述の酸のアルコール性溶液又は過酸化水素及びその水溶液である。当然ながら、上述の洗浄液の混合物もまた洗浄のために使用することができる。更に、JP−A7−232071又はDE−A10321398もまた、適当な洗浄法を開示している。このような洗浄の中で、純粋なi−相(又は増加したi−相部分)が通常は洗浄状態で残存し、通常はまた、更に改善された触媒性能を有する。 Furthermore, both the multi-metal oxide material M obtained according to the invention by the hydrothermal method and the subsequent or successor multi-metal oxide material which has been heat-treated as described are for example DE-A 10254279 and EP-A 140 7819. Can be further processed in a convenient manner by washing them with a suitable liquid as described in. Suitable such liquids include, for example, organic acids and aqueous solutions thereof (eg, oxalic acid, formic acid, acetic acid, citric acid and tartaric acid) and inorganic acids and aqueous solutions thereof (sulfuric acid, perchloric acid, hydrochloric acid, nitric acid, boric acid and (Or telluric acid), alcohols, alcoholic solutions of the above-mentioned acids or hydrogen peroxide and aqueous solutions thereof. Of course, mixtures of the above-mentioned cleaning liquids can also be used for cleaning. Furthermore, JP-A7-231201 or DE-A10321398 also discloses suitable cleaning methods. In such washing, pure i-phase (or increased i-phase portion) usually remains in the washed state and usually also has further improved catalytic performance.
混合物Gを水熱的に処理させるための元素構成成分の出発化合物の緊密な混合は、乾燥又は湿潤形状で達成することができる。乾燥状態で達成される場合、出発化合物は微細に分離された粉末の形状で適切に使用される。しかしながら、緊密な混合は、好ましくは湿潤の水性形状で達成される。出発化合物は好ましくは互いに水溶液(適宜、少量の錯化剤を付随的に使用して)及び/又は微細に分割された懸濁液の形状で混合される。 Intimate mixing of the starting components of the elemental constituents for the hydrothermal treatment of the mixture G can be achieved in dry or wet form. If achieved in the dry state, the starting compound is suitably used in the form of a finely divided powder. However, intimate mixing is preferably achieved in wet aqueous form. The starting compounds are preferably mixed with one another in the form of an aqueous solution (optionally accompanied by a small amount of complexing agent) and / or a finely divided suspension.
初めに既に記述した反射の上及び上方に、本発明に従って水熱的に得られる多金属酸化物材料M又は、記述の通りに実施される熱後処理及び又は洗浄により得られるそれに続く物質(又は後継物質)のX線回折パターンRMiは、しばしば(含有する元素及び結晶幾何学構造(例えば、針状形状又はテーブル形状)に依存して)追加の特徴的な反射強度を有する。 Above and above the reflection already described at the beginning, the multi-metal oxide material M obtained hydrothermally according to the invention or the subsequent substance obtained by thermal post-treatment and / or cleaning carried out as described (or X-ray diffraction patterns RMi of (subsequent materials) often have additional characteristic reflection intensities (depending on the elements contained and the crystal geometry (eg, acicular or table shape)).
格子面間隔d[Å]=3.99±0.2で表される反射の強度に基づき、これらの(相対的な)反射強度I(%)は、以下の通り:
d[Å] I(%)
3.06±0.2(有利に±0.1) 5〜65
3.17±0.2(有利に±0.1) 5〜65
3.28±0.2(有利に±0.1) 15〜130、屡々15〜95
3.99±0.2(有利に±0.1) 100
9.82±0.4(有利に±0.2) 1〜50、屡々1〜30
11.24±0.4(有利に±0.2) 1〜45、屡々1〜30
13.28±0.5(有利に±0.3) 1〜35、屡々1〜15
上述に加え、使用するX線の波長に独立に格子面間隔d[Å]の形状で更に再現される特に特徴的な反射である以下の反射はまた、しばしば上述のX線回折パターンRMiで検出可能である。
Based on the reflection intensity represented by the lattice spacing d [Å] = 3.99 ± 0.2, these (relative) reflection intensities I (%) are as follows:
d [Å] I (%)
3.06 ± 0.2 (preferably ± 0.1) 5 to 65
3.17 ± 0.2 (preferably ± 0.1) 5 to 65
3.28 ± 0.2 (preferably ± 0.1) 15-130, often 15-95
3.99 ± 0.2 (preferably ± 0.1) 100
9.82 ± 0.4 (preferably ± 0.2) 1-50, often 1-30
11.24 ± 0.4 (preferably ± 0.2) 1-45, often 1-30
13.28 ± 0.5 (preferably ± 0.3) 1-35, often 1-15
In addition to the above, the following reflection, which is a particularly characteristic reflection that is further reproduced in the shape of the lattice spacing d [Å] independently of the X-ray wavelength used, is also often detected by the X-ray diffraction pattern RMi described above. Is possible.
d[Å]
8.19±0.3(好ましくは±0.15)
3.51±0.2(好ましくは±0.1)
3.42±0.2(好ましくは±0.1)
3.34±0.2(好ましくは±0.1)
2.94±0.2(好ましくは±0.1)
2.86±0.2(好ましくは±0.1)
格子面間隔d[Å]=3.99±0.2で表される反射の強度に基づき、上記の反射の(相対的な)強度I(%)は、しばしば以下の通り:
d[Å] I(%)
8.19±0.3(又は±0.15) 0〜25
3.51±0.2(又は±0.1) 2〜50
3.42±0.2(又は±0.1) 5〜75
3.34±0.2(又は±0.1) 5〜80
2.94±0.2(又は±0.1) 5〜55
2.86±0.2(又は±0.1) 5〜60
しばしば、以下の反射もまた上述のX線回折パターンRMiを補う:
d[Å]
2.54±0.2(好ましくは±0.1)
2.01±0.2(好ましくは±0.1)
これらの反射の場合、上記と同一の基準を有する(相対的な)反射強度はしばしば以下の通り:
d[Å] I(%)
2.54±0.2(又は±0.1) 0.5〜40
2.01±0.2(又は±0.1) 5〜60
本発明に従って、上述のX線回折パターンRMi(又は、上記パターンに関係する多金属酸化物材料又はその後継物質)の間で好ましいX線回折パターンは、格子面間隔d[Å]=3.99±0.2(又は±0.1)で表される反射又は格子面間隔d[Å]=3.28±0.2(又は±0.1)で表されるものが最も強い反射(最強の強度を有する)であるものである。
d [Å]
8.19 ± 0.3 (preferably ± 0.15)
3.51 ± 0.2 (preferably ± 0.1)
3.42 ± 0.2 (preferably ± 0.1)
3.34 ± 0.2 (preferably ± 0.1)
2.94 ± 0.2 (preferably ± 0.1)
2.86 ± 0.2 (preferably ± 0.1)
Based on the intensity of the reflection expressed as the lattice spacing d [Å] = 3.99 ± 0.2, the (relative) intensity I (%) of the reflection is often as follows:
d [Å] I (%)
8.19 ± 0.3 (or ± 0.15) 0-25
3.51 ± 0.2 (or ± 0.1) 2-50
3.42 ± 0.2 (or ± 0.1) 5 to 75
3.34 ± 0.2 (or ± 0.1) 5-80
2.94 ± 0.2 (or ± 0.1) 5-55
2.86 ± 0.2 (or ± 0.1) 5-60
Often, the following reflections also supplement the X-ray diffraction pattern RMi described above:
d [Å]
2.54 ± 0.2 (preferably ± 0.1)
2.01 ± 0.2 (preferably ± 0.1)
For these reflections, the (relative) reflection intensity with the same criteria as above is often as follows:
d [Å] I (%)
2.54 ± 0.2 (or ± 0.1) 0.5-40
2.01 ± 0.2 (or ± 0.1) 5-60
In accordance with the present invention, a preferred X-ray diffraction pattern among the above-described X-ray diffraction patterns RMi (or multi-metal oxide materials or successors related to the patterns) has a lattice spacing d [Å] = 3.99 Reflection represented by ± 0.2 (or ± 0.1) or lattice spacing d [Å] = 3.28 ± 0.2 (or ± 0.1) is the strongest reflection (strongest) It has a strength of
更に、好ましいX線回折パターンRMi(又は、上記パターンに関係する多金属酸化物材料又はその後継物質)は、反射d[Å]=3.99±0.2(又は±0.1)の半分の高さでの2θ全幅が≦1°、好ましくは≦0.5°であるものである。記述された他の反射の半分の高さでの2θ全幅は通常≦3°、好ましくは≦1.5°、特に好ましくは≦1°である。 Furthermore, the preferred X-ray diffraction pattern RMi (or a multi-metal oxide material or successor related to the above pattern) is half the reflection d [Å] = 3.99 ± 0.2 (or ± 0.1). The total 2θ width at a height of ≦ 1 °, preferably ≦ 0.5 °. The 2θ full width at half height of the other reflections described is usually ≦ 3 °, preferably ≦ 1.5 °, particularly preferably ≦ 1 °.
X線回折図に関する本明細の全データは、X線としてCuKα放射線(λ=1.54178Å)を使用して生成されたX線回折図に基づいている。(Siemens回折計Theta−Theta D−5000、チューブ電圧:40kV、チューブ電流:40mA、絞り径V20(可変)、コリメーターV20(可変)、第2モノクロメーター絞り径(0.1mm)、検知器絞り径(0.6mm)、測定間隔(2θ):0.02°、ステップあたりの測定時間:2.4s、検知器:シンチレーションカウンター;本明細中、X線回折図の反射強度の定義はDE−A19835247、DE−A10122027及びDE−A10051419及びDE−A10046672中に述べられた定義に基づき、本出願の参照によってここに組み入れられる;半分の高さの2θ全幅の定義も同様に適用される)
回折及び回折角θ(本明細中、2θプロットの反射の頂点が反射の位置として使用される)に使用されるX線の波長λはに以下の通りBraggの関係を介して互いに関連づいている。
All data in this specification relating to X-ray diffractograms is based on X-ray diffractograms generated using CuKα radiation (λ = 1.54178Å) as X-rays. (Siemens diffractometer Theta-Theta D-5000, tube voltage: 40 kV, tube current: 40 mA, aperture diameter V20 (variable), collimator V20 (variable), second monochromator aperture diameter (0.1 mm), detector aperture Diameter (0.6 mm), measurement interval (2θ): 0.02 °, measurement time per step: 2.4 s, detector: scintillation counter; in this specification, the definition of the reflection intensity of the X-ray diffraction diagram is DE− A19835247, DE-A10122027 and DE-A10051419 and DE-A10046672 are hereby incorporated by reference in this application; the definition of half height 2θ full width applies as well)
The X-ray wavelength λ used for diffraction and diffraction angle θ (here, the vertex of reflection in the 2θ plot is used as the position of reflection) is related to each other via the Bragg relationship as follows: .
2sinθ=λ/d
ここで、dはそれぞれの反射に関係する三次元原子配列の格子面間隔である。
2 sin θ = λ / d
Here, d is a lattice plane interval of a three-dimensional atomic arrangement related to each reflection.
本発明に従って得られる一般化学量論Iの好ましい多金属酸化物材料(及びそれらに関係する後継物質)は、以下が適用されるものである。
M1=Sb、Bi、Se、及びTeよりなる群から選択される少なくとも1つの元素;
M2=Ti、Zr、Nb、Cr、W、Fe、Co、Ni及びZnよりなる群から選択される少なくとも1つの元素;
M3=Re、Pd及びPtよりなる群から選択される少なくとも1つの元素;
M4=Rb、Cs、Sr及びBaよりなる群から選択される少なくとも1つの元素;
a=0.01〜1(好ましくは0.01〜0.5);
b=≧0〜1(好ましくは>0〜0.5);
c=≧0〜1(好ましくは>0〜0.5);
d=≧0〜0.5(好ましくは≧0〜0.1);
e=≧0〜0.5(好ましくは≧0〜0.1又は0)及び
n=(I)中の酸素以外の元素の原子価及び出現頻度によって決定される数。
Preferred multi-metal oxide materials of general stoichiometry I obtained according to the present invention (and their successors) are those to which:
M 1 = at least one element selected from the group consisting of Sb, Bi, Se, and Te;
M 2 = at least one element selected from the group consisting of Ti, Zr, Nb, Cr, W, Fe, Co, Ni and Zn;
At least one element selected from the group consisting of M 3 = Re, Pd and Pt;
M 4 = at least one element selected from the group consisting of Rb, Cs, Sr and Ba;
a = 0.01-1 (preferably 0.01-0.5);
b = ≧ 0 to 1 (preferably> 0 to 0.5);
c = ≧ 0 to 1 (preferably> 0 to 0.5);
d = ≧ 0 to 0.5 (preferably ≧ 0 to 0.1);
e = ≧ 0 to 0.5 (preferably ≧ 0 to 0.1 or 0) and n = a number determined by the valence and appearance frequency of elements other than oxygen in (I).
多金属酸化物材料M及び選ばれた元素組成の他の化学量論的係数のために好ましい範囲とは独立に、本発明に従って得られる多金属酸化物材料M(及びそれに関係する後継物質)の化学量論的係数aが0.05〜0.5、特に好ましくは0.1〜0.5である場合、本発明に従って好ましい。 Independent of the preferred range for the multi-metal oxide material M and other stoichiometric factors of the selected elemental composition, the multi-metal oxide material M (and its related successors) obtained according to the present invention A stoichiometric coefficient a of 0.05 to 0.5, particularly preferably 0.1 to 0.5 is preferred according to the invention.
多金属酸化物材料M及び選ばれた元素組成の他の化学量論的係数ために好ましい範囲とは独立に、化学量論的係数bは好ましくは>0又は0.01〜0.5、特に0.1〜0.5又は0.4である。 Independent of the preferred range for the multi-metal oxide material M and other stoichiometric coefficients of the selected elemental composition, the stoichiometric coefficient b is preferably> 0 or 0.01 to 0.5, in particular 0.1 to 0.5 or 0.4.
多金属酸化物材料M及び選ばれた元素組成の他の化学量論的係数のために好ましい範囲とは独立に、本発明に従って得られる多金属酸化物材料Mの化学量論的係数cは好都合には0.01〜0.5そして特に好ましくは0.1〜0.5又は0.4である。本発明に従って得られる多金属酸化物材料Mの他の化学量論的係数のために好ましい範囲とは独立に、本明細及び全ての選ばれた元素組成の全ての他の好ましい範囲で結合できる化学量論的係数cのために非常に特に好ましい範囲は0.05〜0.2の範囲である。 Independent of the preferred range for the multi-metal oxide material M and other stoichiometric coefficients of the selected elemental composition, the stoichiometric coefficient c of the multi-metal oxide material M obtained according to the present invention is advantageous. Is from 0.01 to 0.5 and particularly preferably from 0.1 to 0.5 or 0.4. Independent of the preferred range due to other stoichiometric coefficients of the multi-metal oxide material M obtained according to the present invention, the chemistry that can be combined in this specification and all other preferred ranges of all selected elemental compositions. A very particularly preferred range for the stoichiometric coefficient c is in the range of 0.05 to 0.2.
多金属酸化物材料M及び選ばれた元素組成の他の化学量論的係数のために好ましい範囲とは独立に、本発明に従って得られる多金属酸化物材料Mの化学量論的係数dは好ましくは>0又は0.00005又は0.0005〜0.5、特に好ましくは0.001〜0.5、しばしば0.002〜0.3及びたびたび0.005又は0.01〜0.1である。 Independent of the preferred range for the multi-metal oxide material M and other stoichiometric coefficients of the selected elemental composition, the stoichiometric coefficient d of the multi-metal oxide material M obtained according to the invention is preferably > 0 or 0.00005 or 0.0005 to 0.5, particularly preferably 0.001 to 0.5, often 0.002 to 0.3 and often 0.005 or 0.01 to 0.1 .
多金属酸化物材料M及び選ばれた元素組成の他の化学量論的係数のために好ましい範囲とは独立に、係数eもまた≧0〜0.1、好都合には0であってよい。 Independent of the preferred range for the multi-metal oxide material M and other stoichiometric coefficients of the selected elemental composition, the coefficient e can also be ≧ 0 to 0.1, conveniently 0.
本発明に従って得られ、その化学量論的係数a、b、c及びdが同時に以下の範囲内である多金属酸化物材料Mは特に好都合である:
a=0.05〜0.5;
b=0.01〜1(又は0.01〜0.5);
c=0.01〜1(又は0.01〜0.5);
d=0.0005〜0.5;及び
e=≧0〜0.5
本発明に従って得られ、その化学量論的係数a、b、c及びdが同時に以下の範囲内である多金属酸化物材料Mは特に好都合である:
a=0.1〜0.5;
b=0.1〜0.5;
c=0.1〜0.5;
d=0.001〜0.5又は0.001〜0.3又は0.001〜0.1;及び
e=≧0〜0.2又は0.1
M1は好ましくはBi、Se、Te及び/又はSb、非常に特に好ましくはTeである。
Multi-metal oxide materials M obtained according to the invention and whose stoichiometric coefficients a, b, c and d are simultaneously in the following range are particularly advantageous:
a = 0.05-0.5;
b = 0.01-1 (or 0.01-0.5);
c = 0.01-1 (or 0.01-0.5);
d = 0.005-0.5; and e = ≧ 0-0.5
Multi-metal oxide materials M obtained according to the invention and whose stoichiometric coefficients a, b, c and d are simultaneously in the following range are particularly advantageous:
a = 0.1-0.5;
b = 0.1-0.5;
c = 0.1-0.5;
d = 0.001 to 0.5 or 0.001 to 0.3 or 0.001 to 0.1; and e = ≧ 0 to 0.2 or 0.1.
M 1 is preferably Bi, Se, Te and / or Sb, very particularly preferably Te.
特に、M2の総量の少なくとも50モル%がNb、Ti、Zr、Cr、W、Fe、Co、Ni、Zn及び/又はTaであり、非常に特に好ましくはM2の総量の少なくとも50モル%又は少なくとも75モル%又はM2の総量の100モル%がNbであり、そして元素の少なくとも1つが、Ti、Zr、Cr、Ta、W、Fe、Co、Ni及びZn又はNb及び/又はTaである場合、上述の全てが適用される。 In particular, at least 50 mol% of the total amount of M 2 is Nb, Ti, Zr, Cr, W, Fe, Co, Ni, Zn and / or Ta, very particularly preferably at least 50 mol% of the total amount of M 2 or at least 75 mole%, or 100 mole% of the total amount of M 2 is Nb, and at least one element, Ti, Zr, Cr, Ta , W, Fe, Co, in Ni and Zn or Nb and / or Ta In some cases, all of the above apply.
しかしながら、M2の意味とは独立に、M3が少なくともRe、Pd又はPtよりなる群から選ばれた少なくとも1つの元素である場合もまた、適用される。 However, independently of the meaning of M 2, even if M 3 is at least one element selected from at least Re, the group consisting of Pd or Pt also apply.
しかしながら、特にM2の総量の少なくとも50モル%、又は75モル%又は100モル%がNbであり、M3がRe、Pd及びPtよりなる群から選ばれた少なくとも1つの元素である場合、上述の全てがまた適用される。 However, in particular at least 50 mol% of the total amount of M 2, or 75 mol% or 100 mol% is Nb, if at least one element M 3 is selected Re, from the group consisting of Pd and Pt, above All of this also applies.
しかしながら、特にM2の総量の少なくとも50モル%又は少なくとも75モル%又は100モル%がNbであり、そして元素の少なくとも1つがCo、Ni、Ta、W、Feであり、そしてM3がRe、Pd及びPtよりなる群から選ばれた少なくとも1つの元素である場合、上述の全てが適用される。 However, in particular at least 50 mol% or at least 75 mole%, or 100 mol% Nb of the total amount of M 2, and at least one element but is Co, Ni, Ta, W, Fe, and M 3 is Re, All of the above apply when it is at least one element selected from the group consisting of Pd and Pt.
非常に特に好ましくは、M1がTeであり、M2がNbであり、そして元素の少なくとも1つがNi、Co、Feであり、M3がPd、Re及びPtよりなる群から選ばれた少なくとも1つの元素である場合、化学量論的係数に関する全記載が適用される。 Very particularly preferably, M 1 is Te, M 2 is Nb, and at least one of the elements is Ni, Co, Fe, and M 3 is at least selected from the group consisting of Pd, Re and Pt In the case of one element, all statements regarding stoichiometric coefficients apply.
本発明に従って得られる好都合な多金属酸化物材料Mはe=0のもの(そして特に全て上述のもの)である。e>0なら、M4は好ましくはCsである。 Convenient multimetal oxide materials M obtained according to the invention are those with e = 0 (and in particular all those mentioned above). If e> 0, M 4 is preferably Cs.
更に、本発明に従った適当な化学量論Iは、引用された先行技術の化学量論(I)の多金属酸化物材料のために開示された本明細中のものである。 Further, suitable stoichiometry I according to the present invention is that disclosed herein for the cited prior art stoichiometric (I) multi-metal oxide materials.
記載の通りの水熱法により本発明に従って得られた一般化学量論Iの多金属酸化物材料又はその多金属酸化物材料に続く物質(原則として、それらは更に化学量論Iを有する)はそれ自体(たとえば粉末又はチップとして)、又は型に成形された後、例えば、飽和及び不飽和炭化水素又は低アルデヒド及び/又はアルコールの全ての部分的ガス相酸化及び又はアンモ酸化のための触媒的活性物質として使用でき、この酸化又はアンモ酸化は、本発明の序文に記載されている。触媒床は固定床、移動床又は流体床であってよい。DE−A10118814又はPCT/EP/02/04073又はDE−A10051419に記載のとおり、例えば、非支持体触媒の場合成型又はペレット化により、又は支持体への適用(コーティングされた触媒の製造)により成形を達成できる。 The general stoichiometric I multi-metal oxide material obtained according to the invention by the hydrothermal method as described or the substances following the multi-metal oxide material (in principle they have a further stoichiometry I) are Catalytically for all partial gas phase oxidation and / or ammoxidation of itself (for example as powder or chips) or after being molded into, for example, saturated and unsaturated hydrocarbons or low aldehydes and / or alcohols It can be used as an active substance and this oxidation or ammoxidation is described in the introduction of the present invention. The catalyst bed may be a fixed bed, a moving bed or a fluid bed. As described in DE-A 10118814 or PCT / EP / 02/04073 or DE-A 10051419, for example by molding or pelletizing in the case of unsupported catalysts or by application to the support (production of coated catalysts) Can be achieved.
本発明に従って得られる多金属酸化物材料M及びその後継物質のためのコーティングされた触媒の場合に使用される支持体は、好ましくは化学的に不活性、例えば本発明に従って得られる多金属酸化物材料M及びその後継物質により触媒された炭化水素(例えばプロパン及び/又はプロペンからアクリル酸)、アルコール又はアルデヒドの部分触媒ガス相酸化又はアンモ酸化の中に本質的に介在しないものである。 The support used in the case of the coated catalyst for the multimetal oxide material M obtained according to the invention and its successors is preferably chemically inert, for example the multimetal oxide obtained according to the invention Essentially absent from the partially catalyzed gas phase oxidation or ammoxidation of hydrocarbons (eg propane and / or propene to acrylic acid), alcohols or aldehydes catalyzed by material M and its successors.
本発明に従って、支持体として適当な物質は、特にアルミナ、シリカ、粘土、カオリン、ステアタイト(好ましくはC−220型のCeramTec(ドイツ)からのステアタイトで、好ましくは低水溶性アルカリ成分を有する)、軽石、ケイ酸アルミニウム及びケイ酸マグネシウムのようなケイ酸塩、炭化ケイ素、二酸化ジルコニウム及び二酸化トリウムである。 In accordance with the present invention, suitable materials for the support are in particular alumina, silica, clay, kaolin, steatite (preferably steatite from C-220 type CeramTec (Germany), preferably having a low water-soluble alkaline component. ), Silicates such as pumice, aluminum silicate and magnesium silicate, silicon carbide, zirconium dioxide and thorium dioxide.
支持体の表面は平滑でも粗くてもよい。一般的に表面の粗さは適用した活性物質コーティングの粘着強度をより強くするので、好都合には支持体の表面は粗い。 The surface of the support may be smooth or rough. In general, the surface of the support is rough because the roughness of the surface generally increases the adhesion strength of the applied active substance coating.
しばしば、支持体の表面粗さRzは5〜200μmの範囲、しばしば20〜100μmの範囲である(ドイツのHommelwerkeからの”Hommel Tester for DIN−ISO surface parameters”を使用したDIN4768シート1に従って決定)。
Often the surface roughness R z of the support is in the range of 5-200 μm, often in the range of 20-100 μm (determined according to DIN 4768
更に、支持物質は多孔質又は非多孔質であってよい。適切には、支持物質は非多孔質である(支持体の容量に基づいて、≦1容量%の総容量)。 Furthermore, the support material may be porous or non-porous. Suitably the support material is non-porous (total volume of ≦ 1% by volume, based on the volume of the support).
本発明に従ったコーティングされた触媒に存在する活性酸化物物質コーティングの厚さは、通常10〜1000μmである。しかしながら、50〜700μm、100〜600μm又は150〜400μmであってもよい。他の可能なコーティング厚は、10〜500μm、100〜500μm又は150〜300μmである。 The thickness of the active oxide material coating present in the coated catalyst according to the invention is usually between 10 and 1000 μm. However, it may be 50 to 700 μm, 100 to 600 μm, or 150 to 400 μm. Other possible coating thicknesses are 10-500 μm, 100-500 μm or 150-300 μm.
原則的に、支持体のいかなる所望の幾何学構造も本発明に従った方法のために適当である。その最長の大きさは、原則として1〜10mmである。しかしながら、球体又は円柱、特に中空円柱が支持体として好ましく使用される。球状支持体の好都合な直径は1.5〜4mmである。支持体として円柱が使用された場合、この長さは好ましくは2〜10mmであり、その直径は好ましくは4〜10mmである。環状の場合、壁厚は更に通常1〜4mmである。本発明に従って適当な環状の支持体はまた3〜6mmの長さ、4〜8mmの外径及び1〜2mmの壁厚を有してもよい。しかしながら、環状支持体はまた、7mmx3mmx4mm又は5mmx3mmx2mm(外径x長さx内径)寸法であってよい。
In principle, any desired geometric structure of the support is suitable for the method according to the invention. The longest size is in
コーティングされた触媒の製造は本発明に従って、多金属酸化物材料M又はその後の物質を前形成し、微細に分割した形状に転換し、最後にそれらを液体接合剤の助力で支持体の表面に適用することにより、最も容易に達成できる。この目的のため、支持体の表面は液体接合剤で最も簡単に潤され、活性物質のコーティングは、本発明に従って得られる微細に分割された活性多金属酸化物材料M又は微細に分割された続く物質と接触させることにより、潤された表面に粘着させられる。最後にコーティングされた支持体は乾燥される。当然ながらより厚いコーティング厚を達成するため、操作法は定期的に繰り返される。この場合、コーティングされた基礎体が新しい「支持体」等になる。 The production of the coated catalyst, according to the invention, pre-forms the multi-metal oxide material M or subsequent substance, converts it into a finely divided shape, and finally converts them to the surface of the support with the aid of a liquid binder. By applying, it can be achieved most easily. For this purpose, the surface of the support is most easily moistened with a liquid binder, and the active substance coating is followed by a finely divided active multimetal oxide material M obtained according to the invention or finely divided. By contacting with a substance, it is adhered to a moist surface. Finally, the coated substrate is dried. Of course, the procedure is repeated periodically to achieve a thicker coating thickness. In this case, the coated basic body becomes a new “support” or the like.
支持体の表面に適用される一般化学式(I)の触媒的に活性な多金属酸化物材料M又はその後継物質の微細度は、当然ながら所望のコーティング厚に適合する。100〜500μmの範囲のコーティング厚のために、例えば、粉末粒子の総数の少なくとも50%が1〜20μmのメッシュサイズのふるいを通過し、50μmを超える最長の大きさを有する粒子の数的割合が10%未満である活性物質粉末が適当である。原則として、粉末粒子の最長の大きさの分布は、生成物の結果としてガウス分布に相当する。しばしば、粒子サイズ分布は以下の通り: The fineness of the catalytically active multi-metal oxide material M of general formula (I) or its successor applied to the surface of the support is of course adapted to the desired coating thickness. For coating thicknesses in the range of 100-500 μm, for example, at least 50% of the total number of powder particles pass through a mesh size screen of 1-20 μm and the numerical proportion of particles with the longest size exceeding 50 μm An active substance powder of less than 10% is suitable. In principle, the longest size distribution of the powder particles corresponds to a Gaussian distribution as a result of the product. Often the particle size distribution is as follows:

ここで:
D=粒子径;
x=直径が≧Dの粒子の百分率;及び
y=直径が<Dの粒子の百分率。
here:
D = particle diameter;
x = percentage of particles with diameter ≧ D; and y = percentage of particles with diameter <D.
工業規模で記述されるコーティング法を実施するために、例えば、DE−A2909671に開示された操作法原理及びDE−A10051419に記載されたものを使用することが賢明であり、例えば、コーティングされる支持体は、好ましくは傾いた(傾斜角は原則として≧0°及び≦90°、一般的に≧30°及び≦90°、傾斜角は回転容器の中心軸と水平面の間の角)回転容器(例えば、回転パン又はコーティングドラム)に最初に入れられる。連続するある距離に配置された2つの計量装置の下で、回転容器は例えば球状又は円柱の支持体を輸送する。2つの計量装置の1番目は適切にはノズル(例えば、圧縮空気を処理する噴霧ノズル)に相当し、それによって回転パン中で回転する支持体は液体接合剤を噴霧され、制御された方法で湿潤させられる。2番目の計量装置は噴霧される液体接合剤の噴霧円錐外に存在し、(例えば、振とうトラフ又は粉末スクリューを介して)微細に分割された酸化活性物質を供給する役割を果たす。制御された方法で湿潤された球状支持体は、供給された活性物質粉末に溶かされ、例えば円柱又は球状である支持体の外面上の回転移動によって圧縮され、凝集性コーティングが得られる。 In order to carry out the coating process described on an industrial scale, it is advisable to use, for example, the operating principle disclosed in DE-A 2909671 and the one described in DE-A 10051419, for example the support to be coated The body is preferably tilted (tilt angles are in principle ≧ 0 ° and ≦ 90 °, generally ≧ 30 ° and ≦ 90 °, the tilt angle is the angle between the central axis of the rotating container and the horizontal plane) For example, first put in a rotating pan or coating drum). Under two metering devices arranged at a distance in succession, the rotating container transports, for example, a spherical or cylindrical support. The first of the two metering devices suitably corresponds to a nozzle (eg a spray nozzle for treating compressed air), whereby the support rotating in the rotating pan is sprayed with a liquid binder and in a controlled manner. Moistened. The second metering device is located outside the spray cone of the liquid binder to be sprayed and serves to supply finely divided oxidizing actives (for example via a shaking trough or powder screw). A spherical support wetted in a controlled manner is dissolved in the supplied active substance powder and compressed by a rotational movement on the outer surface of the support, for example cylindrical or spherical, to obtain a coherent coating.
必要なら、基礎コーティングとともにこの方法で再び供給された支持体は続く回転の間に噴霧ノズルを通過、制御された方法でこれにより湿潤され、そして従って、追加の移動等の間に微細に分割された酸化活性物質の追加のコーティングを溶かすことができる(原則として中間の乾燥は必要ない)。微細に分割された酸化活性物質及び液体接合剤は原則として連続的にかつ同時に供給される。 If necessary, the support supplied again in this way with the base coating passes through the spray nozzle during the subsequent rotation, is wetted thereby in a controlled manner, and is therefore finely divided during additional movements etc. Additional coatings of oxidatively active substances can be dissolved (in principle no intermediate drying is necessary). The finely divided oxidizing active substance and the liquid binder are in principle supplied continuously and simultaneously.
コーティング終了後、液体接合剤は、例えばN2又は空気のような熱ガスの作用によって除去することができる。意外にも、記載したコーティング操作法は互いに対する連続的コーティング及び支持体の表面に対する基礎コーティングの両方の完全に満足な粘着性が得られる。 After the coating is finished, the liquid bonding agent can be removed by the action of a hot gas such as N 2 or air. Surprisingly, the described coating operation method gives fully satisfactory adhesion of both the continuous coating to each other and the base coating to the surface of the support.
上述のコーティング法の重要な側面は、コーティングされた支持体表面の湿潤が制御された方法で実施されることである。即ち、これは、吸収された液体接合剤を有するが、液体相自体は支持体表面に見えない方法で、支持体表面が適切に湿潤されるということを意味している。支持体表面が湿潤しすぎると、微細に分割された触媒的活性酸化物物質は、表面上に吸収される或いは、固まって個々の塊を形成する。本内容の詳細な情報はDE−A2909671及びDE−A10051419に記載されている。 An important aspect of the coating method described above is that the wetting of the coated support surface is performed in a controlled manner. That is, this means that the support surface is adequately wetted in such a way that it has absorbed liquid binder but the liquid phase itself is not visible on the support surface. If the support surface is too wet, the finely divided catalytically active oxide material is absorbed on the surface or solidifies to form individual lumps. Detailed information on this content is described in DE-A 2909671 and DE-A 10051419.
使用された液体接合剤の上述された最後の除去は、例えば、蒸発及び/又は昇華によって制御した方法で実施することができる。最も単純な場合、相当する温度(しばしば、50〜300℃、たびたび150℃)の熱ガスの作用によって達成することができる。しかしながら、予備乾燥だけは、熱ガスの作用によって達成することができる。その後、最終乾燥は例えば、どのような形式(例えばベルト乾燥機)の乾燥オーブン中又は反応器中でも達成することができる。適用する温度は酸化活性物質の製造に使用する、か焼温度を超えてはいけない。乾燥は当然ながら、乾燥オーブン中で排他的に達成することができる。 The above-mentioned final removal of the used liquid binder can be carried out in a controlled manner, for example by evaporation and / or sublimation. In the simplest case, it can be achieved by the action of hot gas at the corresponding temperature (often 50-300 ° C, often 150 ° C). However, only pre-drying can be achieved by the action of hot gas. Thereafter, final drying can be accomplished, for example, in any type of drying oven (eg, belt dryer) or in a reactor. The applied temperature must not exceed the calcination temperature used for the production of the oxidation active substance. Drying can of course be achieved exclusively in a drying oven.
支持体の形式及び幾何学構造と独立に、コーティング操作法に対する結合剤として以下が使用されてよい:水、エタノール、メタノール、プロパノール及びブタノールのような1価アルコール、エチレングリコール、1,4−ブタンジオール、1,6−ヘキサンジオール又はグリセロールのような多価アルコール、プロピオン酸、シュウ酸、マロン酸、グルタル酸又はマレイン酸のような1価塩基性又は多価塩基性有機カルボン酸、エタノールアミン又はジエタノールアミンのようなアミノアルコール、及びホルムアミドのような1官能又は多官能有機アミド。好都合な結合剤はまた、水20〜90重量%及び水に溶解し、雰囲気圧(1気圧)で沸点又は昇華温度が>100℃、好ましくは>250℃である有機化合物10〜80重量%よりなる溶液である。好都合にも、有機化合物は可能な有機接合剤の上記一覧から選択される。好ましくは、上述の水性結合剤溶液の有機部分は10〜50、特に好ましくは20〜30重量%である。これにより、適当な有機成分はまた、ブドウ糖、果糖、ショ糖又は乳糖のような単糖及びオリゴ糖及びポリエチレン酸化物及びポリアクリレートである。 Independent of the type and geometry of the support, the following may be used as binders for the coating process: water, monohydric alcohols such as ethanol, methanol, propanol and butanol, ethylene glycol, 1,4-butane. Polyhydric alcohols such as diol, 1,6-hexanediol or glycerol, monovalent or polybasic organic carboxylic acids such as propionic acid, oxalic acid, malonic acid, glutaric acid or maleic acid, ethanolamine or Amino alcohols such as diethanolamine, and mono- or polyfunctional organic amides such as formamide. Convenient binders are also from 10 to 80% by weight of organic compounds which are soluble in 20 to 90% by weight of water and water and have a boiling point or sublimation temperature of> 100 ° C., preferably> 250 ° C. at atmospheric pressure (1 atm). Is a solution. Conveniently, the organic compound is selected from the above list of possible organic binders. Preferably, the organic part of the aqueous binder solution described above is 10-50, particularly preferably 20-30% by weight. Thereby, suitable organic components are also mono- and oligosaccharides such as glucose, fructose, sucrose or lactose and polyethylene oxides and polyacrylates.
適当な幾何学構造(非支持体触媒及びコーティングされた触媒の両方)は、球体、非中空円柱及び中空円柱(環状)である。上述の幾何学構造の最長の大きさは原則として1〜10mmである。円柱の場合、その長さは好ましくは2〜10mmであり、その外径は好ましくは4〜10mmである。環状の場合、壁厚は、更に通常、1〜4mmである。適当な環状の非支持体触媒はまた、3〜6mmの長さ、4〜8mmの外径及び1〜2mmの壁厚を有してよい。しかしながら、7mmx3mmx4mm寸法又は5mmx3mmx2mm寸法(外径x長さx内径)の環状の非支持体触媒もまた可能である。本発明に従って得られる多金属酸化物活性物質M及びその後継物質の適当な幾何学構造はまた、当然ながら、DE−A10101695中の全てのものである。
Suitable geometries (both non-supported and coated catalysts) are spheres, non-hollow cylinders and hollow cylinders (annular). The longest size of the above-described geometric structure is in
本発明に従って得られる多金属酸化物材料M(及びその後継物質)の特別な表面積は、しばしば1〜80m2/g又は40m2/g、しばしば11又は12〜40m2/g、たびたび15又は20〜40又は30m2/gである(BET法、窒素により決定)。 Special surface area of the multimetal oxide material M obtained according to the invention (and its successor materials) often 1~80m 2 / g or 40 m 2 / g, often 11 or 12~40m 2 / g, often 15 or 20 it is 40 or 30m 2 / g (BET method, determined by nitrogen).
上記以外の場合、DE−A10303526中に作成された陳述は本発明に従って得られた多金属酸化物材料M及びその後続物質に適用できる。 In all other cases, the statements made in DE-A 10303526 are applicable to the multi-metal oxide material M obtained according to the invention and its subsequent substances.
これは、本発明に従って得られた多金属酸化物材料M及びその後続物質もまた、当然ながら、シリカ、二酸化チタン、アルミナ、酸化ジルコニウム及び酸化ニオブのような希釈剤効果のみを本質的に有する、微細に分割された、例えばコロイド状の、物質に希釈された形状で触媒的活性物質として使用できるということを意味する。 This means that the multi-metal oxide material M obtained according to the invention and its subsequent substances also have, of course, essentially only diluent effects such as silica, titanium dioxide, alumina, zirconium oxide and niobium oxide, It means that it can be used as catalytically active material in a finely divided, eg colloidal, diluted form.
希釈質量比は9(希釈剤):1(活性物質)までであってよく、例えば、可能な希釈質量比は例えば、6(希釈剤):1(活性物質)及び3(希釈物質):1(活性物質)である。希釈剤の混合は熱処理の前及び/又は後で達成できる。 The dilution mass ratio may be up to 9 (diluent): 1 (active substance), for example possible dilution mass ratios are for example 6 (diluent): 1 (active substance) and 3 (diluent substance): 1. (Active substance). Diluent mixing can be accomplished before and / or after heat treatment.
本発明に従って得られる多金属酸化物材料M及びその後継物質は、それ自体又は上記(既に記載の通り)のように希釈された形状で、飽和及び/又は不飽和炭化水素及びアルコール及びアルデヒドの混成に触媒された部分ガス相酸化(酸素脱水素反応を含む)及び/又はアンモ酸化(例えば、DE−A10316465に従った操作法中)のための活性物質として適当である。 The multi-metal oxide material M and its successors obtained according to the invention are a mixture of saturated and / or unsaturated hydrocarbons and alcohols and aldehydes, either as such or in diluted form as described above (as already described). Suitable as active substances for partially catalyzed partial gas phase oxidation (including oxygen dehydrogenation) and / or ammoxidation (for example during the process according to DE-A 10316465).
このような飽和及び/又は不飽和炭化水素は特に、エタン、エチレン、プロパン、プロピレン、n−ブタン、イソブタン又はイソブテンである。これによる目的生成物は特にアクロレイン、アクリル酸、メタアクロレイン、メタアクリル酸、アクリロニトリル及びメタアクリロニトリルである。しかしながら、これらはまた、アクロレイン及びメタアクロレインのような化合物の混成に触媒された部分ガス相酸化及び/又はアンモ酸化に適当である。 Such saturated and / or unsaturated hydrocarbons are in particular ethane, ethylene, propane, propylene, n-butane, isobutane or isobutene. The end products thus obtained are in particular acrolein, acrylic acid, methacryloline, methacrylic acid, acrylonitrile and methacrylonitrile. However, they are also suitable for partial gas phase oxidation and / or ammoxidation catalyzed by the hybridisation of compounds such as acrolein and metaacrolein.
しかしながら、エチレン、プロピレン及び酢酸もまた目的生成物であることができる。 However, ethylene, propylene and acetic acid can also be target products.
本明細中、炭化水素、アルコール及び/又はアルデヒドの完全な酸化とは、炭化水素、アルコール及び/又はアルデヒドに含まれる炭素全てが炭素の酸化物(CO、CO2)に転換されることを意味すると理解されている。 In this specification, complete oxidation of hydrocarbons, alcohols and / or aldehydes means that all the carbon contained in the hydrocarbons, alcohols and / or aldehydes is converted to carbon oxides (CO, CO 2 ). It is understood.
反応中に酸素分子を加える炭化水素の全ての他の反応は、部分酸化という用語のもとに本明細中に組み込まれる。反応中のアンモニアの追加の添加は部分アンモ酸化とみなす。 All other reactions of hydrocarbons that add oxygen molecules during the reaction are incorporated herein under the term partial oxidation. Additional addition of ammonia during the reaction is considered partial ammoxidation.
本明細に従って得られる多金属酸化物材料M及びその後継物質は好ましくはプロパンのアクロレイン及び/又はアクリル酸への、プロパンのアクリル酸及び/又はアクリロニトリルへの、プロピレンのアクロレイン及び/又はアクリル酸への、プロピレンのアクリルニトリルへの、イソブタンのメタアクリロレイン及び/又はメタアクリル酸への、イソブタンのメタアクリル酸及び/又はメタアクリロニトリルへの、エタンのエチレンへの、エタンの酢酸への、及びエチレンの酢酸への転換のための触媒的活性物質として適当である。 The multi-metal oxide material M and its successors obtained according to the present specification are preferably propane to acrolein and / or acrylic acid, propane to acrylic acid and / or acrylonitrile, propylene to acrolein and / or acrylic acid. , Propylene to acrylonitrile, isobutane to methacrylolein and / or methacrylic acid, isobutane to methacrylic acid and / or methacrylonitrile, ethane to ethylene, ethane to acetic acid, and ethylene Suitable as catalytically active material for conversion to acetic acid.
このような部分酸化及び/又はアンモ酸化(それ自体既知の方法で制御される反応混合ガス中のアンモニア含有量の選択により、反応は排他的に部分酸化として、又は排他的に部分アンモ酸化として又は、2つの反応の重ね合わせとして本質的に設計することができる;例えばWO98/22421を参照)は、先行技術の一般化学量論Iの多金属酸化物材料からそれ自体既知であり、完全に相当する方法で実施することができる。 Such partial oxidation and / or ammoxidation (depending on the choice of the ammonia content in the reaction gas mixture controlled in a manner known per se, the reaction is either exclusively as partial oxidation or exclusively as partial ammoxidation or Can be designed essentially as a superposition of two reactions; see eg WO 98/22421), which is known per se from prior art general stoichiometric I multi-metal oxide materials and is quite comparable It can be implemented by the method.
使用された炭化水素が粗製のプロパン又は粗製のプロペンの場合、これは好ましくはDE−A10246119又はDE−A10118814又はPCT/EP/02/04073に記載された通りの組成を有する。操作法もまた、好ましくはこれらに記載の通りである。 If the hydrocarbon used is crude propane or crude propene, this preferably has a composition as described in DE-A 10246119 or DE-A 10118814 or PCT / EP / 02/04073. The operating methods are also preferably as described therein.
多金属酸化物M活性物質(又は後継活性物質)を含む触媒を使用して実施されるプロパンのアクリル酸への部分酸化は、例えば、EP−A608838、EP−A1407819、WO00/29106、JP−A10−36311及びEP−A1192987に記載された通り実施することができる。 The partial oxidation of propane to acrylic acid carried out using a catalyst comprising a multimetal oxide M active substance (or successor active substance) is, for example, EP-A 608838, EP-A 1407819, WO 00/29106, JP-A10. -36311 and EP-A 1192987.
例えば、空気、酸素が豊富な空気、酸素が涸渇した空気、又は純粋な酸素は所望の酸素分子の原料として使用することができる。 For example, air, oxygen-rich air, oxygen-depleted air, or pure oxygen can be used as a source of the desired oxygen molecules.
反応ガス出発混合物が不活性希釈ガスとして希ガス、特にヘリウムを含まない場合でさえ、このような方法は好都合である。さもなければ、反応ガス出発混合物は、当然ながら、プロパン及び酸素分子に加えて不活性希釈ガス、例えばN2、CO及びCO2を含むことができる。反応ガス混合物の成分として、水蒸気は本発明に従って好都合である。 Such a process is advantageous even when the reaction gas starting mixture does not contain a noble gas, in particular helium, as an inert diluent gas. Otherwise, the reaction gas starting mixture can of course contain inert diluent gases such as N 2 , CO and CO 2 in addition to propane and oxygen molecules. As a component of the reaction gas mixture, water vapor is advantageous according to the invention.
これは、本発明に従って得られる多金属酸化物活性物質M又はその後継物質が例えば200〜550℃又は230〜480℃又は300〜440℃の反応温度で、1〜10バール又は2〜5バールで充填されている反応ガス出発混合物は、例えば以下の組成を有してよいということを意味する。 This is because the multimetal oxide active substance M obtained according to the invention or its successor is at a reaction temperature of, for example, 200 to 550 ° C. or 230 to 480 ° C. or 300 to 440 ° C. and 1 to 10 bar or 2 to 5 bar. It means that the charged reaction gas starting mixture may have, for example, the following composition:
プロパン1〜15、好ましくは1〜7容量%、
空気44〜99容量%及び
水蒸気0〜55容量%。
Propane 1-15, preferably 1-7% by volume,
44-99% by volume of air and 0-55% by volume of water vapor.
水蒸気含有の反応ガス出発混合物は好ましい。 A reaction gas starting mixture containing water vapor is preferred.
反応ガス出発混合物の他の可能な組成として以下が適当である。 The following are suitable as other possible compositions of the reaction gas starting mixture.
プロパン70〜95容量%、
酸素分子5〜30容量%及び
水蒸気0〜25容量%。
70-95% by volume of propane,
5-30% by volume of oxygen molecules and 0-25% by volume of water vapor.
排他的にアクリル酸からなっていない生成物ガス混合物も、当然ながら、このような方法で得られる。それどころか、未変換のプロパンに加えて、生成物ガス混合物はアクリル酸から分離されるべきプロペン、アクロレイン、CO2、CO、H2O、酢酸、プロピオン酸等の副次的成分を含んでいる。 Naturally, product gas mixtures which are not exclusively composed of acrylic acid are also obtained in this way. Rather, in addition to propane unconverted, the product gas mixture contains propene to be separated from acrylic acid, acrolein, CO 2, CO, H 2 O, acetic acid, a secondary component such as propionic acid.
これは、プロペンのアクリル酸への混成に触媒されたガス相酸化から既知の方法で達成することができる。 This can be achieved in a known manner from the gas phase oxidation catalyzed by the hybridisation of propene to acrylic acid.
これは、含有するアクリル酸が水での吸収又は高沸点不活性疎水有機溶媒(例えば、ジフェニルエーテル及びジフィルの混合物で、適宜、ジメチルフタレートのような添加物もまた含有してよいもの)での吸収により生成物ガス混合物から取り出さすことができるということを意味する。次に得られた吸収剤及びアクリル酸の混合物は、それ自体既知の方法で精留、抽出及び/又は結晶化により後処理され、純粋なアクリル酸が得ることができる。或いは、生成物ガス混合物からのアクリル酸の最初の単離はまた、例えば、DE−A19924532に記載の通り、分別凝縮によって達成することができる。 This is because the acrylic acid contained is absorbed by water or absorbed by a high boiling inert hydrophobic organic solvent (for example, a mixture of diphenyl ether and difil, which may optionally contain additives such as dimethyl phthalate). Means that it can be removed from the product gas mixture. The resulting mixture of absorbent and acrylic acid can then be worked up by rectification, extraction and / or crystallization in a manner known per se to obtain pure acrylic acid. Alternatively, the initial isolation of acrylic acid from the product gas mixture can also be achieved by fractional condensation, for example as described in DE-A 19924532.
次に、得られた水性のアクリル酸凝縮物は、例えば分別結晶化(例えば、懸濁液結晶化及び/又は階層結晶化)により更に精製することができる。 The resulting aqueous acrylic acid condensate can then be further purified, for example, by fractional crystallization (eg, suspension crystallization and / or hierarchical crystallization).
アクリル酸の最初の単離で残存する残存ガス混合物は特に未転換のプロパンを含み、好ましくはガス相酸化に再利用される。この目的のため、例えば圧力下に分別精留により残存ガスから部分的又は完全に分離され、次にガス相酸化に再利用されてよい。しかしながら、残存ガスを、抽出装置中の選択的にプロパンを吸収できる疎水性有機溶媒と接触(例えば、上記ガスを通過させることにより)させることは、より好都合である。 The residual gas mixture remaining from the initial isolation of acrylic acid contains in particular unconverted propane and is preferably recycled for gas phase oxidation. For this purpose, it may be partly or completely separated from the residual gas, for example by fractional rectification under pressure, and then reused for gas phase oxidation. However, it is more convenient to contact the residual gas with a hydrophobic organic solvent that can selectively absorb propane in the extraction device (eg, by passing the gas through).
続く空気の脱離及び又は除去により、吸収されたプロパンは再び解放されてよく、本発明に従った方法に再利用することができる。この方法で、安価な全プロパン転換が遂行できる。他の分離方法と同様に、副次的成分として形成されたプロペンはプロパンから原則的に分離できない、もしくは完全には分離できず、それらとともに循環する。これは、他の同族の飽和及びオレフィン系炭化水素の場合にも適用される。特に、本発明に従って飽和炭化水素の混成に触媒された部分酸化及び/又はアンモ酸化に対して非常に一般的に事実である。 By subsequent desorption and / or removal of air, the absorbed propane may be released again and can be reused in the process according to the invention. In this way, inexpensive total propane conversion can be accomplished. As with other separation methods, propene formed as a secondary component cannot in principle be separated from propane or cannot be separated completely and circulates with them. This also applies in the case of other homologous saturated and olefinic hydrocarbons. In particular, it is very generally true for the partial oxidation and / or ammoxidation catalyzed by the hybridisation of saturated hydrocarbons according to the invention.
本発明に従って得られる多金属酸化物材料M及びその後継物質はまた、同族のオレフィン系炭化水素の同様の目的生成物への部分酸化及び/又はアンモ酸化を混成に触媒できる。従って、本発明に従って得られる多金属酸化物材料M及びその後継物質を活性物質として使用して、アクリル酸はDE−A10118814又はPCT/EP/02/04073又はJP−A7−53448に記載の通り、酸素分子としてプロペンの混成に触媒された部分ガス相酸化により製造できる。 The multi-metal oxide material M obtained in accordance with the present invention and its successors can also catalyze the partial oxidation and / or ammoxidation of homologous olefinic hydrocarbons to similar target products. Thus, using the multi-metal oxide material M obtained according to the invention and its successors as active substances, acrylic acid is as described in DE-A 10118814 or PCT / EP / 02/04073 or JP-A 7-53448, It can be produced by partial gas phase oxidation catalyzed by the hybrid of propene as oxygen molecules.
これは、単一の反応区域Aが方法を実施するのに十分であるということを意味している。この反応区域において、排他的に本発明に従って得られる多金属酸化物材料Aを含む、又は後継物質を含む触媒が触媒的に活性な物質として存在する。 This means that a single reaction zone A is sufficient to carry out the process. In this reaction zone, a catalyst containing the multimetal oxide material A obtained according to the invention exclusively or containing a successor substance is present as a catalytically active substance.
プロペンのアクリル酸への混成に触媒されたガス相酸化は非常に一般的に、連続する時間で2段階で行われるので、これは普通ではない。最初の段階で、通常プロペンは本質的にアクロレインに酸化され、そして2番目の段階で通常は最初の段階で形成されたアクロレインがアクリル酸に酸化される。 This is unusual because the gas phase oxidation catalyzed by the hybridization of propene to acrylic acid is very commonly performed in two steps in continuous time. In the first stage, usually propene is essentially oxidized to acrolein, and in the second stage, usually acrolein formed in the first stage is oxidized to acrylic acid.
従って、プロペンのアクリル酸への混成に触媒されたガス相酸化の従来の方法は、通常、2つの上述の酸化段階の各々の酸化段階に仕立てられた特別な触媒形式を使用する。 Thus, conventional methods of gas phase oxidation catalyzed by hybridization of propene to acrylic acid typically use a special catalyst type tailored to the oxidation stage of each of the two above mentioned oxidation stages.
これは、プロペンのアクリル酸への混成に触媒されたガス相酸化の従来の方法は、本発明に従った方法と対比して、2つの反応区域で処理されることを意味する。 This means that the conventional method of gas phase oxidation catalyzed by the hybridisation of propene to acrylic acid is treated in two reaction zones, in contrast to the method according to the invention.
プロペンの部分酸化の方法において、当然ながら本発明に従って得られる多金属酸化物質Mを含む、又は後継物質を含むたった1つ又は1つ以上の触媒が1つの反応地域Aに存在することは可能である。当然ながら、使用される触媒は推奨のように、例えば本明細の支持体物質として、不活性物質で希釈されてよい。 In the process of partial oxidation of propene, it is of course possible that only one or more catalysts comprising a multi-metal oxide M obtained according to the invention or containing a successor are present in one reaction zone A. is there. Of course, the catalyst used may be diluted with an inert material as recommended, for example, as a support material herein.
プロペンの部分酸化の方法において、反応地域Aを熱するための熱媒体のたった1つの温度又は反応地域Aに沿って変化する1つの温度を、1つの反応区域Aに沿って行き渡らせることができる。この温度変化は増加であっても減少であってもよい。 In the method of partial oxidation of propene, only one temperature of the heating medium for heating the reaction zone A or one temperature changing along the reaction zone A can be distributed along one reaction zone A . This temperature change may be an increase or a decrease.
本発明に従ったプロペン部分酸化方法が固定床酸化として実施された場合、その触媒チューブが触媒で充填されているチューブ内蔵反応器の中で適切に達成される。通常、液体、原則として塩浴は、触媒チューブのまわりの熱媒体として通っている。 When the propene partial oxidation process according to the present invention is carried out as fixed bed oxidation, it is suitably achieved in a tube built-in reactor whose catalyst tubes are packed with catalyst. Usually a liquid, in principle a salt bath, is passed as a heating medium around the catalyst tube.
次に、反応地域Aに沿った多数の温度地域は、触媒チューブに沿って区間ごとに触媒チューブの周囲に1つ以上の塩浴を通すことにより単純な方法で実現できる。 Next, multiple temperature zones along the reaction zone A can be realized in a simple manner by passing one or more salt baths around the catalyst tube for each section along the catalyst tube.
反応器を考慮して、反応ガス混合物は塩浴に対し順流又は逆流のいずれかで触媒チューブ内を通過する。塩浴自体は、触媒チューブに相対的に純粋に並行な流れを遂行できる。当然ながら、横断の流れをまたこれに重ね合わせてもよい。概して、塩浴はまた、反応器を考慮してだけ反応ガス混合物に対し順流又は逆流である、触媒チューブの周囲の蛇行した流動を行ってもよい。 In view of the reactor, the reaction gas mixture passes through the catalyst tube in either forward or reverse flow with respect to the salt bath. The salt bath itself can perform a purely parallel flow to the catalyst tubes. Of course, the transverse flow may also be superimposed on it. In general, the salt bath may also provide a serpentine flow around the catalyst tube that is forward or reverse flow to the reaction gas mixture only in view of the reactor.
プロペン部分酸化方法において、全体の反応地域Aに沿った反応温度は200〜500℃であってよい。通常250〜450℃である。反応温度は好ましくは330〜420℃、特に好ましくは350〜400℃である。 In the propene partial oxidation method, the reaction temperature along the entire reaction area A may be 200-500 ° C. Usually 250 to 450 ° C. The reaction temperature is preferably 330 to 420 ° C, particularly preferably 350 to 400 ° C.
プロペン部分酸化方法において、処理圧力は1バール、1バール未満又は1バールより以上であってよい。本発明に従って、典型的な処理圧力は1.5〜10バール、しばしば1.5〜5バールである。 In the propene partial oxidation process, the treatment pressure may be 1 bar, less than 1 bar, or more than 1 bar. In accordance with the present invention, typical process pressures are 1.5 to 10 bar, often 1.5 to 5 bar.
プロペン部分酸化方法に使用するプロペンはその純度に関していかなる特別な高度の要求にも合致する必要がない。 The propene used in the propene partial oxidation process need not meet any special high requirements regarding its purity.
既に述べた通り、プロペンのアクロレイン及び/又はアクリル酸への混成に触媒されたガス相酸化の全一段階又は二段階の方法は非常に一般的なので、例えば、以下の2つの仕様を有するプロペン(粗製のプロペンとしても参照)はこのような方法でプロペンとして問題なく完全に使用できる:
a)ポリマー等級のプロピレン
≧99.6重量% プロペン
≦0.4重量% プロパン
≦300重量ppm エタン及び/又はメタン
≦5重量ppm C4−炭化水素
≦1重量ppm アセチレン
≦7重量ppm エチレン
≦5重量ppm 水
≦2重量ppm O2
≦2重量ppm イオウ含有化合物(イオウ換算)
≦1重量ppm 塩素含有化合物(塩素換算)
≦5重量ppm CO2
≦5重量ppm CO
≦10重量ppm シクロプロパン
≦5重量ppm プロパジエン及び/又はプロピン
≦10重量ppm C25−炭化水素及び
≦10重量ppm カルボニル含有化合物(Ni(CO)4換算)
b)化学等級プロピレン
≧94重量% プロペン
≦6重量% プロパン
≦0.2重量% メタン及び/又はエタン
≦5重量ppm エチレン
≦1重量ppm アセチレン
≦20重量ppm プロパジエン及び/又はプロピン
≦100重量ppm シクロプロパン
≦50重量ppm ブテン
≦50重量ppm ブタジエン
≦200重量ppm C4−炭化水素
≦10重量ppm C25−炭化水素
≦2重量ppm イオウ含有化合物(イオウ換算)
≦0.1重量ppm 硫化物含有化合物(H2S換算)
≦1重量ppm 塩素含有化合物(塩素換算)
≦0.1重量ppm 塩化物含有化合物(Clθ換算)及び
≦30重量ppm 水
しかしながら、1段階又は2段階のプロペンのアクロレイン及び/又はアクリル酸への混成した触媒されたガス相酸化のための方法又は既知の方法に対する粗製のプロペンの有用性が非常に一般的に不都合に影響することなく、プロペンの全ての上述した可能な不純物は、当然ながら、粗製のプロペン中に記載された個々の量の2〜10倍それぞれ含むこともまたできる。
As already mentioned, the full one-stage or two-stage method of gas phase oxidation catalyzed by the hybridization of propene to acrolein and / or acrylic acid is very common, for example, propene having the following two specifications ( Can also be used perfectly as a propene in this way without problems:
a) Polymer grade propylene ≧ 99.6 wt% propene ≦ 0.4 wt% propane ≦ 300 wt ppm ethane and / or methane ≦ 5 wt ppm C 4 -hydrocarbon ≦ 1 wt ppm acetylene ≦ 7 wt ppm ethylene ≦ 5 Weight ppm water ≦ 2 weight ppm O 2
≦ 2 ppm by weight Sulfur-containing compounds (in terms of sulfur)
≦ 1 ppm by weight Chlorine-containing compound (chlorine conversion)
≦ 5 wt ppm CO 2
≦ 5 ppm by weight CO
≦ 10 wt ppm cyclopropane ≦ 5 wt ppm propadiene and / or propyne ≦ 10 wt ppm C 25 -hydrocarbon and ≦ 10 wt ppm carbonyl-containing compound (Ni (CO) 4 conversion)
b) Chemical grade propylene ≧ 94 wt% propene ≦ 6 wt% propane ≦ 0.2 wt% methane and / or ethane ≦ 5 wt ppm ethylene ≦ 1 wt ppm acetylene ≦ 20 wt ppm propadiene and / or propyne ≦ 100 wt ppm cyclo Propane ≦ 50 ppm by weight Butene ≦ 50 ppm by weight Butadiene ≦ 200 ppm by weight C 4 -hydrocarbon ≦ 10 ppm by weight C 25 -hydrocarbon ≦ 2 ppm by weight Sulfur-containing compound (in terms of sulfur)
≦ 0.1 ppm by weight Sulfide-containing compound (H 2 S conversion)
≦ 1 ppm by weight Chlorine-containing compound (chlorine conversion)
≦ 0.1 wt ppm Chloride-containing compound (Clθ conversion) and ≦ 30 wt ppm water However, a process for hybrid catalyzed gas phase oxidation of one or two stages of propene to acrolein and / or acrylic acid Or, the usefulness of crude propene to known methods does not very generally adversely affect all the above-mentioned possible impurities of propene, of course, in the individual amounts described in the crude propene. It can also be included 2 to 10 times each.
特に、飽和炭化水素、水蒸気、炭素の酸化物及び酸素分子の場合、これらがいかなる場合でも、上述の方法中、反応に不活性希釈ガス又は大量の反応物質のいずれかとして加える化合物である場合、これが適用される。通常、粗製のプロペン自体は、プロペンのアクロレイン及び/又はアクリル酸への混成に触媒されたガス相酸化の方法のために、再生ガス、空気及び/又は酸素分子及び/又は希釈空気及び/又は不活性ガスとともに混合物として使用される。 In particular, in the case of saturated hydrocarbons, water vapor, carbon oxides and oxygen molecules, in any case these are compounds that are added to the reaction as either an inert diluent gas or a large amount of reactants in the above-described method, This applies. Typically, the crude propene itself is regenerated gas, air and / or oxygen molecules and / or diluted air and / or non-reacted due to the process of gas phase oxidation catalyzed by the hybridization of propene to acrolein and / or acrylic acid. Used as a mixture with active gas.
しかしながら、本発明に従った方法のための別の適当なプロペン原料は本発明に従った方法とは異なる方法の中で副産物として形成され、例えば、40重量%までのプロパンを含むプロペンである。更に、このプロペンは本質的に本発明に従った方法を妨げない他の不純物を伴っていてもよい。 However, another suitable propene feedstock for the process according to the invention is formed as a by-product in a process different from the process according to the invention, for example propene containing up to 40% by weight of propane. Furthermore, the propene may be accompanied by other impurities that do not essentially interfere with the process according to the invention.
純粋な酸素及び空気又は酸素が豊富な空気又は酸素が涸渇した空気の両方とも、プロペン部分酸化方法のための酸素原料として使用してよい。 Both pure oxygen and air or oxygen rich air or oxygen depleted air may be used as oxygen feedstock for the propene partial oxidation process.
酸素分子及びプロペンに加えて、プロペン部分酸化方法に使用する反応ガス出発混合物もまた通常、少なくとも1つの希釈剤ガスを含んでいる。窒素、炭素の酸化物、希ガス及びメタン、エタン又はプロパンのような低級炭化水素はそれ自体適当である(高級炭化水素、例えばC4−炭化水素は避けるべきである)。しばしば、水蒸気もまた希釈剤ガスとして使用される。上述のガスの混合物は、しばしば、部分プロペン酸化方法のための希釈剤ガスを形成する。 In addition to molecular oxygen and propene, the reaction gas starting mixture used in the propene partial oxidation process also typically contains at least one diluent gas. Nitrogen, carbon oxides, noble gases and lower hydrocarbons such as methane, ethane or propane are suitable per se (higher hydrocarbons such as C 4 -hydrocarbons should be avoided). Often, water vapor is also used as the diluent gas. The gas mixture described above often forms a diluent gas for the partial propene oxidation process.
プロペンの混成に触媒された部分酸化は、プロパンの存在下に好都合に達成される。 The partial oxidation catalyzed by the hybrid of propene is advantageously achieved in the presence of propane.
典型的には、プロペン酸化方法のための反応ガス出発混合物は以下の組成(モル比)を有している。
プロペン:酸素:H2O:他の希釈剤ガス
=1:(0.1〜1):(0〜70):(0:20)
好ましくは、上述の比は、1:(1〜5):(1〜40):(0〜10)である。
Typically, the reaction gas starting mixture for the propene oxidation process has the following composition (molar ratio):
Propene: oxygen: H 2 O: other diluent gases = 1: (0.1-1) :( 0-70) :( 0:20)
Preferably, the above ratio is 1: (1-5) :( 1-40) :( 0-10).
プロパンが希釈剤ガスとして使用される場合、記載の通り、更に好都合にアクリル酸への部分酸化ができる。 When propane is used as the diluent gas, it can be more conveniently partially oxidized to acrylic acid as described.
本発明に従って、反応ガス出発混合物は、希釈剤ガスとして好都合に窒素分子、CO、CO2、水蒸気及びプロパンを含んでいる。 According to the invention, the reaction gas starting mixture advantageously contains molecular nitrogen, CO, CO 2 , water vapor and propane as diluent gas.
プロペン酸化方法中のプロパン:プロペンのモル比は、以下の値と想定されてよい:0〜15、しばしば0〜10、たびたび0〜5、適切には0.01〜3。 The propane: propene molar ratio in the propene oxidation process may be assumed to be the following values: 0-15, often 0-10, often 0-5, suitably 0.01-3.
部分プロペン酸化方法中のプロペンを伴う触媒床の充填は、例えば40〜250l(S.T.P.)/l/h又はそれ以上であってよい。反応ガス出発混合物を伴う充填は、しばしば500〜15000l(S.T.P.)/l/hの範囲、たびたび600〜10000l(S.T.P.)/l/hの範囲、しばしば700〜5000l(S.T.P.)/l/hである。 The packing of the catalyst bed with propene during the partial propene oxidation process may be, for example, 40-250 l (STP) / l / h or more. The charge with the reaction gas starting mixture is often in the range of 500 to 15000 l (STP) / l / h, often in the range of 600 to 10000 l (STP) / l / h, often 700 to 5000 l (STP) / l / h.
当然ながら、プロペンのアクリル酸への部分酸化の方法中、排他的にアクリル酸からなっていない生成物ガス混合物が得られる。それどころか、生成物ガス混合物は、未転換のプロペンに加えて、プロパン、アクロレイン、CO2、CO、H2O、酢酸、プロピオン酸等のようなアクリル酸から分離されなければならない副次的化合物を含んでいる。 Of course, during the process of partial oxidation of propene to acrylic acid, a product gas mixture is obtained which is not exclusively composed of acrylic acid. Rather, the product gas mixture, in addition to unconverted propene, propane, acrolein, CO 2, CO, H 2 O, acetic acid, a secondary compound that must be separated from acrylic acid such as propionic acid Contains.
これは、プロパンのアクリル酸への混成に触媒された2段階ガス相酸化(2つの反応区域で実施)から一般的に既知の方法で達成できる。 This can be achieved in a generally known manner from a two-stage gas phase oxidation (performed in two reaction zones) catalyzed by the hybridisation of propane to acrylic acid.
これは、含有するアクリル酸が水で吸収されることにより、あるいは高沸点不活性疎水有機溶媒(例えば、ジフェニルエーテル及び適宜ジメチルフタレートのような添加物を含んでもよいジフェニルの混合物)で吸収されることにより、生成物ガス混合物から取り出すことができることを意味している。吸収剤及びアクリル酸の得られた混合物は、次に精留、抽出及び/又は結晶化によりそれ自体既知の方法で後処理され、純粋なアクリル酸を得ることができる。或いは、生成物ガス混合物からのアクリル酸の最初の分離はまた、例えばDE−A19924532に記載の通り分別凝縮により達成することができる。 This is because the acrylic acid contained is absorbed by water or by a high boiling inert hydrophobic organic solvent (for example, a mixture of diphenyl which may optionally contain additives such as diphenyl ether and dimethyl phthalate). Means that the product gas mixture can be removed. The resulting mixture of absorbent and acrylic acid can then be worked up in a manner known per se by rectification, extraction and / or crystallization to obtain pure acrylic acid. Alternatively, the initial separation of acrylic acid from the product gas mixture can also be achieved by fractional condensation, for example as described in DE-A 19924532.
得られた水性のアクリル酸凝縮物は、次に、例えば分別結晶化(例えば、懸濁液結晶化及び/又は階層結晶化)により更に精製することができる。 The resulting aqueous acrylic acid condensate can then be further purified, for example, by fractional crystallization (eg, suspension crystallization and / or hierarchical crystallization).
アクリル酸の最初の単離中に残存する残存ガス混合物は特に、未転換プロペン(及びことによるとプロパン)を含んでいる。これは、例えば圧力下に分別精留により残存ガス混合物から分離し、本発明に従ったガス相酸化に再利用することができる。しかしながら、残存ガスを抽出装置内で、プロペン(及び適宜プロパン)を選択的に吸収できる疎水有機溶媒に接触させる(例えば、上記ガスを通過させることにより)ことは、より好都合である。 The residual gas mixture remaining during the initial isolation of acrylic acid contains in particular unconverted propene (and possibly propane). This can be separated from the residual gas mixture, for example by fractional rectification under pressure, and reused for gas phase oxidation according to the invention. However, it is more convenient to contact the residual gas in the extraction apparatus with a hydrophobic organic solvent that can selectively absorb propene (and optionally propane) (eg, by passing the gas).
続く空気の脱離及び又は除去により、吸収されたプロペン(及び適宜プロパン)を再び解放し、本発明に従った方法に再利用することができる。この方法で、安価な全プロペン転換が遂行できる。プロペンがプロパンの存在下に部分酸化に付された場合、プロペン及びプロパンは好ましくはともに分離され、再利用される。 Subsequent desorption and / or removal of air allows the absorbed propene (and optionally propane) to be released again and reused in the process according to the invention. In this way, an inexpensive total propene conversion can be accomplished. When propene is subjected to partial oxidation in the presence of propane, propene and propane are preferably separated and reused.
完全に相当する方法で、本発明に従って得られる多金属酸化物M及びその後継物質は、イソブタン及び/又はイソブテンのメタアクリル酸への部分酸化のための触媒として使用できる。 In a completely corresponding manner, the multimetal oxide M and its successors obtained according to the invention can be used as catalysts for the partial oxidation of isobutane and / or isobutene to methacrylic acid.
プロパン及び/又はプロペンのアンモ酸化のための使用は、例えばEP−A529853、DE−A2351151、JP−A6−166668及びJP−A7−232071に記載の通り達成できる。
n−ブタン及び/又はn−ブテンのアンモ酸化のための使用は、例えばJP−A6−211767に記載の通り達成できる。
Use for the ammoxidation of propane and / or propene can be achieved, for example, as described in EP-A 529853, DE-A 2351151, JP-A 6-166668 and JP-A 7-232011.
Use for the ammoxidation of n-butane and / or n-butene can be achieved, for example, as described in JP-A6-211767.
エタン又はエチレンの酸素脱水素反応又は酢酸への追加の反応のための使用は、US−A4,250,346又はEP−B261264に記載の通り達成できる。 Use of ethane or ethylene for oxygen dehydrogenation or additional reaction to acetic acid can be achieved as described in US-A 4,250,346 or EP-B 261264.
アクロレインのアクリル酸への部分酸化のための使用は、DE−A10261186に記載の通り達成できる。 The use for the partial oxidation of acrolein to acrylic acid can be achieved as described in DE-A 10261186.
本発明に従って得られる多金属酸化物材料M及びその後継物質は、しかしながら、他の多金属酸化物材料に合体することもできる(例えば、適宜、その微細に分割された物質を混合、圧縮及び焼成、又はスラリー(好ましくは水性)として混合、乾燥及び焼成(例えばEP−A529853に記載の通り))。焼成は好ましくは不活性ガス下にもう一度達成される。 The multi-metal oxide material M and its successors obtained according to the present invention can, however, also be merged with other multi-metal oxide materials (for example mixing, compressing and firing the finely divided substances as appropriate) Or mixed, dried and fired as a slurry (preferably aqueous) (eg as described in EP-A 529853)). Calcination is preferably accomplished once more under inert gas.
得られた多金属酸化物混合物(総物質として以下に参照)は、好ましくは本発明に従って得られる多金属酸化物材料M又はその後継物質を≧50重量%、特に好ましくは≧75重量%、非常に特に好ましくは≧90重量%又は≧95重量%含んでおり、本明細で論議されている部分酸化及び/又は部分アンモ酸化に更に適当である。 The resulting multi-metal oxide mixture (see below for the total material) is preferably ≧ 50% by weight, particularly preferably ≧ 75% by weight of the multi-metal oxide material M or its successor material obtained according to the invention. Particularly preferably ≧ 90 wt% or ≧ 95 wt%, which is more suitable for the partial oxidation and / or partial ammoxidation discussed herein.
総物質の場合、幾何学的な成形は、本発明に従って得られる多金属酸化物材料M及びその後継物質に対して記載された通り、適切に達成される。 In the case of total materials, the geometric shaping is suitably achieved as described for the multi-metal oxide material M obtained according to the invention and its successors.
プロパンのアクリル酸への混成に触媒された部分ガス相酸化のために、本発明に従って得られる多金属酸化物材料M、その後継物質及びこのような物質を含む多金属酸化物材料又は触媒は、好ましくはDE−A10122027に記載の通り処理に投入される。 Due to the hybrid gas-catalyzed partial gas phase oxidation of propane to acrylic acid, the multimetal oxide material M obtained in accordance with the present invention, its successor material and the multimetal oxide material or catalyst comprising such a material are: Preferably, it is put into the process as described in DE-A 10122027.
最後に、本発明に従った方法を使用した優れた再現性は所望の多金属酸化物Mが本質的に水及びその構成物質H、O及びOHのみを含んだ環境で得られるという事実のためであると言明すべきである。この方法で、多金属酸化物Mは、いわゆるその自然なpH下で得られ、これは明らかに操作法を特に頑健なものとしている。
実施例及び比較実施例
A)多金属酸化物材料の製造
実施例1 計量投入される化学量論Mo1V0.32Te0.027Oxの多金属酸化物材料の製造
以下のもの、内蔵攪拌器及び2.5lの内部容量を有するオートクレーブに微細に分割された形状で導入された:
MoO3 123.27g(Riedel−de−Haenブランド、30926Seelze;MoO3含有>99.9%;Mo0.86モル);
VO2 23.06g(Alfa Aesar、76057Karlsruheから;VO2含有=99.5%;VO2 0.28モル、V酸化状態=4.03);
TeO2 3.69g(Sigma Aldrich、82018Taufkirchenから;TeO2>99%;TeO2 0.023モル);
及び水1500ml。
Finally, the excellent reproducibility using the method according to the invention is due to the fact that the desired multi-metal oxide M is obtained in an environment essentially containing only water and its constituents H, O and OH. Should be stated. In this way, the multimetal oxide M is obtained under the so-called natural pH, which clearly makes the operating procedure particularly robust.
Examples and Comparative Example A) Manufacture of multi-metal oxide material Example 1 Manufacture of multi-metal oxide material with stoichiometric Mo 1 V 0.32 Te 0.027 O x Introduced in finely divided form into an agitator and an autoclave with an internal volume of 2.5 l:
123.27 g of MoO 3 (Riedel-de-Haen brand, 30926 Seelze; containing MoO 3 >99.9%; Mo 0.86 mol);
23.06 g VO 2 (from Alfa Aesar, 76057 Karlsruhe; VO 2 content = 99.5%; VO 2 0.28 mol, V oxidation state = 4.03);
3.69 g of TeO 2 (from Sigma Aldrich, 82018 Taufkirchen; TeO 2 >99%; TeO 2 0.023 mol);
And 1500 ml of water.
オートクレーブの蓋を閉じ、オートクレーブ中の水相の上部に存在する空気部分を窒素でフラッシュすることにより窒素に交換する。その後、オートクレーブを自己圧力下に10時間連続的に攪拌(700rpm)しながら連続的(直線的)に175℃まで加熱し、48時間更に攪拌しながらこの温度を保持した。次に室温(25℃)に冷却した。オートクレーブを開け、形成した黒色粉末を濾取し、25℃の温度で水200mlで3回洗浄し、次に減圧下に乾燥オーブン中に12時間80℃で乾燥した。
記載された方法で実施された実験を10回繰り返した。全ての場合において、下記のように同一の結果が得られた。
The autoclave lid is closed and the air portion present at the top of the aqueous phase in the autoclave is exchanged for nitrogen by flushing with nitrogen. Thereafter, the autoclave was continuously heated (linearly) to 175 ° C. while continuously stirring (700 rpm) for 10 hours under a self-pressure, and this temperature was maintained while further stirring for 48 hours. Next, it was cooled to room temperature (25 ° C.). The autoclave was opened and the black powder formed was collected by filtration, washed 3 times with 200 ml of water at a temperature of 25 ° C., and then dried at 80 ° C. for 12 hours in a drying oven under reduced pressure.
Experiments performed in the manner described were repeated 10 times. In all cases, the same results were obtained as follows.
図1に示す粉末X線回折図(XRD)によって示されるように、排他的にi−相が得られる。関連した走査型電子顕微鏡写真(SEM)(図2、3つの異なる倍率)は、約50〜100の高い長さ・厚さ比を有する針状結晶を示している。 The i-phase is obtained exclusively as shown by the powder X-ray diffraction diagram (XRD) shown in FIG. Related scanning electron micrographs (SEM) (FIG. 2, three different magnifications) show needle-like crystals having a high length-thickness ratio of about 50-100.
「針状」又は「繊維状」の利点は、他の(追加の)結晶の表面が等方性の物質に比べて触媒により接近可能であるという事実が見出されそうなことである。 The advantage of “needle” or “fibrous” is that the fact that the surface of other (additional) crystals is more accessible to the catalyst than isotropic materials is likely to be found.
滴定分析は、形成されたi−相中のVが4.02〜4.05の酸化状態を有していることを示している。特定の表面積は45m2/gであった。化学分析は計量投入される化学量論について満足な同意を与えた。 Titration analysis shows that V in the formed i-phase has an oxidation state of 4.02 to 4.05. The specific surface area was 45 m 2 / g. Chemical analysis gave satisfactory agreement on the stoichiometry to be metered.
比較実施例1 計量投入される化学量論Mo1V0.32Te0.027Oxの多金属酸化物材料の製造
以下のものが、実施例1から、微細に分割された形状でオートクレーブ中に導入された:
(NH4)6Mo7O244H2O 151.87g(H.C.Starck、38642Goslarから;MoO3含有81.5%;Mo0.86モル);
硫酸バナジル水和物VOSO4(H2O)x66.64g(Alfa Aesar、76057Karlsruheから;V含有=21.4重量%;V0.28モル)
TeO2 3.69g(実施例1の通り)
及び水1500ml。
Comparative Example 1 Production of multi-metal oxide material of stoichiometric Mo 1 V 0.32 Te 0.027 O x to be metered in The following from Example 1 in an autoclave in a finely divided shape Introduced in:
151,87 g of (NH 4 ) 6 Mo 7 O 24 4H 2 O (from HC Starck, 38642 Goslar; MoO 3 containing 81.5%; Mo 0.86 mol);
Vanadyl sulfate hydrate VOSO 4 (H 2 O) x 66.64 g (from Alfa Aesar, 76057 Karlsruhe; V content = 21.4 wt%; V 0.28 mol)
TeO 2 3.69 g (as in Example 1)
And 1500 ml of water.
次に、実施例1の通り(水熱的に)操作法を実施した。記載の方法で実施した実験を10回繰り返した。2回の場合について、形成した多金属酸化物粉末中に増加したi−相部分が生じた。一方、他の8回の実験においては、少ない部分のi−相しか含まない相混合物が得られた。 Next, the operation method was carried out as in Example 1 (hydrothermally). The experiment carried out in the manner described was repeated 10 times. For the two cases, an increased i-phase portion occurred in the formed multi-metal oxide powder. On the other hand, in the other 8 experiments, a phase mixture containing only a small portion of i-phase was obtained.
以下のものが他の相の間に同定された:JPDS索引81−2414(c)相[(V0.12Mo0.88)O2.94、六方晶]、JPDS索引05−0508相[MoO3、斜方晶]、JPDS索引47−0872相[NMo5.35O15.75(OH)1.6・1.7H2O]及びJPDS索引77−0649(c)相[(V0.95Mo0.97)O5、三斜晶]。 The following were identified among other phases: JPDS index 81-2414 (c) phase [(V 0.12 Mo 0.88 ) O 2.94 , hexagonal], JPDS index 05-0508 phase [ MoO 3 , orthorhombic], JPDS index 47-0872 phase [NMo 5.35 O 15.75 (OH) 1.6 · 1.7 H 2 O] and JPDS index 77-0649 (c) phase [(V 0 .95 Mo 0.97 ) O 5 , triclinic crystal].
実施例2 計量投入される化学量論Mo1V0.32 Bi 0.027Oxの多金属酸化物材料の製造
以下のものが、実施例1から、微細に分割された形状でオートクレーブ中に導入された:
MoO3123.27g(実施例1の通り)
VO223.06g(実施例1の通り)
Bi2O35.36g(Riedel−de Haenブランド、30926Seelze;Bi2O3≧99.5%;Bi0.023モル)
及び水1500ml。
Example 2 Production of a multi-metal oxide material of stoichiometric Mo 1 V 0.32 Bi 0.027 O x to be metered in The following from Example 1 in an autoclave in a finely divided shape: Introduced:
123.27 g of MoO 3 (as in Example 1)
VO 2 23.06 g (as in Example 1)
Bi 2 O 3 5.36 g (Riedel-de Haen brand, 30926 Seelze; Bi 2 O 3 ≧ 99.5%; Bi 0.023 mol)
And 1500 ml of water.
次に、実施例1の通り(水熱的に)操作法を実施した。 Next, the operation method was carried out as in Example 1 (hydrothermally).
記載された方法で実施された実験を10回繰り返した。全ての場合において、下記のように同一の結果を得た。 Experiments performed in the manner described were repeated 10 times. In all cases, the same results were obtained as follows.
黒色粉末に対し全ての場合で排他的にi−相が含まれる粉末X線回折図(図3参照)が得られた。関連した走査型電子顕微鏡写真(図4、3つの異なる倍率)は、約30〜150の高い長さ・厚さ比を有する針状結晶を示している。 A powder X-ray diffraction diagram (see FIG. 3) containing the i-phase exclusively in all cases was obtained for the black powder. The associated scanning electron micrographs (FIG. 4, three different magnifications) show needle-like crystals having a high length / thickness ratio of about 30-150.
化学分析は計量投入される化学量論について満足な同意を与えた。滴定分析に従って、形成されたi−相中のVは4.02〜4.07の酸化状態を有していた。特定の表面積は38m2/gであった。 Chemical analysis gave satisfactory agreement on the stoichiometry to be metered. According to titration analysis, V in the formed i-phase had an oxidation state of 4.02 to 4.07. The specific surface area was 38 m 2 / g.
比較実施例2 計量投入される化学量論Mo1V0.32 Bi 0.027Oxの多金属酸化物材料の製造
操作法は実施例2の通りであった。しかしながら、(NH4)6Mo7O24・4H2O 151.87g(比較実施例1の通り)がMoO3の代わりに使用され、硫酸バナジル水和物VOSO4(H2O)x66.64g(比較実施例1の通り)がVO2の代わりに使用された。実験は10回繰り返した。2回の場合について、製造された黒色粉末中に増加した比率のi−相が得られた。
Comparative Example 2 Production of Multi-Metal Oxide Material with Stoichiometric Mo 1 V 0.32 Bi 0.027 O x The operating method was as in Example 2. However, 151.87 g of (NH 4 ) 6 Mo 7 O 24 · 4H 2 O (as in Comparative Example 1) was used instead of MoO 3 and vanadyl sulfate hydrate VOSO 4 (H 2 O) x 66. 64 g (as Comparative example 1) was used instead of VO 2. The experiment was repeated 10 times. For the two cases, an increased proportion of i-phase was obtained in the black powder produced.
一方、他の8つの再現においては、少ない部分のi−相しか含まない相混合物が得られた。残存する相は、JPDS索引05−0508相[MoO3、斜方晶]として、JPDS索引47−0872相[HMo5.35O15.75(OH)1.6・1.7H2O]として、JPDS索引48−0744相(BiVO4、正方晶)として、JPDS索引85−0630相(Bi0.88Mo0.37V0.63O4)として、JPDS索引70−2321(c)相[Sb4Mo10O31、斜方晶]の結晶性構造として、JPDS索引33−0104相(Sb4Mo10O31、六方晶)の結晶性構造及び/又はJPDS索引77−0649(c)相[(V0.95Mo0.97O5、三斜晶)として同定された。加えて、追加の相がある場合に存在したが、XRD反射に基づいて追加に同定できなかった。 On the other hand, in the other 8 reproductions, a phase mixture containing only a small portion of i-phase was obtained. The remaining phases are as JPDS index 05-0508 phase [MoO 3 , orthorhombic], and as JPDS index 47-0772 phase [HMo 5.35 O 15.75 (OH) 1.6 · 1.7 H 2 O]. JPDS index 48-0744 phase (BiVO 4 , tetragonal), JPDS index 85-0630 phase (Bi 0.88 Mo 0.37 V 0.63 O 4 ), JPDS index 70-2321 (c) phase [ As the crystalline structure of Sb 4 Mo 10 O 31 , orthorhombic], the crystalline structure of JPDS index 33-0104 phase (Sb 4 Mo 10 O 31 , hexagonal crystal) and / or JPDS index 77-0649 (c) phase [(V 0.95 Mo 0.97 O 5 , triclinic). In addition, it was present when there was an additional phase, but could not be additionally identified based on XRD reflection.
実施例3 計量投入される化学量論Mo1V0.32Bi0.027Oxの多金属酸化物材料の製造
操作法は実施例2の通りであった。しかしながら、バナジウム原料はV粉末2.85g(Chempur、762304Karlsruheから;V含有>99.5%;V0.056モル)及びV2O520.36g(Gesellschaft fur Elektrometallurgie(GfE)、90431Nurnbergから;V2O5含有=99.97%;V0.224モル)の混合物を使用した。
Example 3 Production of a multi-metal oxide material of stoichiometric Mo 1 V 0.32 Bi 0.027 O x to be metered in. The operating procedure was as in Example 2. However, the vanadium raw materials were 2.85 g V powder (from Chempur, 762304 Karlsruhe; V containing>99.5%; V 0.056 mol) and 20.36 g V 2 O 5 (Gesellschaff fur Elektrometallurgie (GfE), 90431 Nurnberg 2 ; A mixture containing O 5 = 99.97%; V 0.224 mol) was used.
オートクレーブを閉じ、オートクレーブ中の水溶液の上方に存在する空気の部分を窒素でフラッシュすることにより窒素と交換した。 The autoclave was closed and replaced with nitrogen by flushing with nitrogen the portion of air present above the aqueous solution in the autoclave.
その後、オートクレーブを自己圧力下に3時間かけて連続的に攪拌(700rpm)しながら連続的(直線的)に90℃まで加熱し、この温度で10時間攪拌した。 Thereafter, the autoclave was continuously heated (linearly) to 90 ° C. while continuously stirring (700 rpm) over 3 hours under self-pressure, and stirred at this temperature for 10 hours.
その後、自己圧力下に8時間連続的に攪拌(700rpm)しながら連続的(直線的)に175℃まで加熱し、24時間攪拌しながらこの温度を保持した。その後、室温(25℃)に冷却し、実施例1の通り、形成した黒色粉末を濾取し、水で洗浄し、乾燥した。 Thereafter, the mixture was continuously heated (linearly) to 175 ° C. with continuous stirring (700 rpm) under self-pressure for 8 hours, and this temperature was maintained while stirring for 24 hours. Thereafter, the mixture was cooled to room temperature (25 ° C.), and the formed black powder was collected by filtration, washed with water and dried as in Example 1.
記載した方法で実施した実験を10回繰り返した。10回の操作法のうちの6回において、実施例2と同様の結果が得られた。 The experiment carried out in the manner described was repeated 10 times. The result similar to Example 2 was obtained in 6 times out of 10 operation methods.
10回の操作法のうちの4回において、比較実施例2からのものに相当する固体が結晶化した。 In 4 of the 10 operating procedures, a solid corresponding to that from Comparative Example 2 crystallized.
実施例4 計量投入される化学量論Mo1V0.25Oxの多金属酸化物材料の製造
操作法は実施例1の通りであった。ただし、VO218.0gのみをVO223.06gの代わりに使用し、Te化合物を除外した。
Example 4 Production of multi-metal oxide material with stoichiometric Mo 1 V 0.25 O x to be metered in The operating procedure was as in Example 1. However, using only VO 2 18.0 g instead of VO 2 23.06 g, were excluded Te compound.
実施例5 計量投入される化学量論Mo1V0.25Nb0.098Bi0.042Oxの多金属酸化物材料の製造
操作法は実施例1の通り、即ち、もう一度MoO3123.27g、及び所望の化学量論に従って、この点において必要なVO2、Nb2O5及びBi2O3の量を計量投入した。
Example 5 Preparation of a stoichiometric Mo 1 V 0.25 Nb 0.098 Bi 0.042 O x multimetal oxide material The procedure is as in Example 1, ie once again MoO 3 123. In accordance with 27 g and the desired stoichiometry, the required amounts of VO 2 , Nb 2 O 5 and Bi 2 O 3 at this point were metered in.
実施例6 計量投入される化学量論Mo1V0.3Sb0.25Nb0.12Oxの多金属酸化物材料の製造
操作法は実施例1の通り、即ち、もう一度MoO3123.27g、及び所望の化学量論に従って、この点において必要なVO2、Sb2O3及びNb2O5の量を計量投入した。
Example 6 Preparation of a stoichiometric Mo 1 V 0.3 Sb 0.25 Nb 0.12 O x multimetal oxide material The operating procedure is as in Example 1, ie once again MoO 3 123. In accordance with 27 g and the desired stoichiometry, the required amounts of VO 2 , Sb 2 O 3 and Nb 2 O 5 at this point were metered in.
実施例7 計量投入される化学量論Mo1V0.3Nb0.13Sb0.13O4.125の多金属酸化物材料の製造
操作法は実施例1の通り、即ち、もう一度MoO3123.27g、及び所望の化学量論に従って、この点において必要なVO2、Nb2O5及びSb2O3の量を計量投入した。
Example 7 Production of a stoichiometric Mo 1 V 0.3 Nb 0.13 Sb 0.13 O 4.125 multimetal oxide material The procedure is as in Example 1, ie once again MoO 3 According to 123.27 g and the desired stoichiometry, the required amounts of VO 2 , Nb 2 O 5 and Sb 2 O 3 at this point were metered in.
実施例8 計量投入される化学量論Mo1V0.3Nb0.13Ni0.13Oxの多金属酸化物材料の製造
操作法は実施例1の通り、即ち、もう一度MoO3123.27g、及び所望の化学量論に従って、この点において必要なVO2、Nb2O5及びNiOの量を計量投入した。
Example 8 Production of multi-metal oxide material of stoichiometric Mo 1 V 0.3 Nb 0.13 Ni 0.13 O x to be metered in The procedure is as in Example 1, ie once again MoO 3 123. In accordance with 27 g and the desired stoichiometry, the required amounts of VO 2 , Nb 2 O 5 and NiO at this point were metered in.
実施例9 計量投入される化学量論Mo1V0.3Nb0.13Co0.13Oxの多金属酸化物材料の製造
操作法は実施例1の通り、即ち、もう一度MoO3123.27g、及び所望の化学量論に従って、この点において必要なVO2、Nb2O5及びCoOの量を計量投入した。
Example 9 Preparation of a stoichiometric Mo 1 V 0.3 Nb 0.13 Co 0.13 O x multimetal oxide material The operating procedure is as in Example 1, ie once again MoO 3 123. In accordance with 27 g and the desired stoichiometry, the required amounts of VO 2 , Nb 2 O 5 and CoO at this point were metered in.
実施例10 計量投入される化学量論Mo1V0.3Nb0.13Cr0.031Oxの多金属酸化物材料の製造
操作法は実施例1の通り、即ち、もう一度MoO3123.27g、及び所望の化学量論に従って、この点において必要なVO2、Nb2O5及びCr2O3の量を計量投入した。
Example 10 Production of a multi-metal oxide material of stoichiometric Mo 1 V 0.3 Nb 0.13 Cr 0.031 O x to be metered in The operating procedure is as in Example 1, ie once again MoO 3 123. In accordance with 27 g and the desired stoichiometry, the required amounts of VO 2 , Nb 2 O 5 and Cr 2 O 3 at this point were metered in.
実施例11 計量投入される化学量論Mo1V0.32Bi0.027Oxの多金属酸化物材料の製造
操作法は実施例2の通りであった。得られて乾燥した多金属酸化物材料100gを水性硝酸10重量%濃度500gに添加した。得られた水性懸濁液を5時間80℃で還流下に攪拌した。次に、25℃に冷却した。黒色懸濁液中に存在する固体を濾過により水相から分離し、水で無硝酸塩に洗浄し、次に一夜120℃で透気式乾燥オーブン中に乾燥した。
Example 11 Production of multi-metal oxide material of stoichiometric Mo 1 V 0.32 Bi 0.027 O x to be metered in The operating procedure was as in Example 2. 100 g of the resulting dried multi-metal oxide material was added to 500 g of 10% aqueous nitric acid concentration. The resulting aqueous suspension was stirred under reflux at 80 ° C. for 5 hours. Next, it was cooled to 25 ° C. The solid present in the black suspension was separated from the aqueous phase by filtration, washed to the nitrate free with water and then dried overnight at 120 ° C. in an air-permeable drying oven.
実施例12
操作法は実施例2の通りであった。得られて乾燥した多金属酸化物材料100gをDE−A10029338の図1に従った回転球状炉(内部容量1リットル)中に窒素分子の気流下に(10l(S.T.P.)/l)2℃/分の加熱割合で室温から500℃まで加熱し、この温度を窒素の気流を維持しながら6時間維持した。次に、窒素の気流を維持しながら放置することにより25℃への冷却を達成した。
Example 12
The operating method was as in Example 2. 100 g of the resulting dried multimetal oxide material are placed in a rotating spherical furnace (
実施例13
熱後処理を実施例12の通り達成する以外は、実施例5の通りである。
Example 13
As in Example 5, except that the thermal post-treatment is accomplished as in Example 12.
実施例14
熱後処理を実施例12の通り達成する以外は、実施例6の通りである。
Example 14
As in Example 6, except that the thermal post-treatment is accomplished as in Example 12.
実施例15
熱後処理を実施例12の通り達成する以外は、実施例7の通りである。
Example 15
As in Example 7, except that thermal post-treatment is achieved as in Example 12.
実施例16
熱後処理を実施例12の通り達成する以外は、実施例10の通りである。
B)A)で製造された多金属酸化物材料からのコーティングされた触媒の製造
それぞれの活性物質粉末は、Retsch粉砕器(Retsch、ドイツからの遠心粉砕器、形式ZM100)中に粉砕された(粒子サイズ≦0.12mm)。
Example 16
As in Example 10, except that the thermal post-treatment is accomplished as in Example 12.
B) Production of coated catalyst from multimetal oxide material produced in A) Each active substance powder was ground in a Retsch grinder (Retsch, centrifugal grinder from Germany, type ZM100) ( Particle size ≦ 0.12 mm).
各場合において、粉砕後に存在する粉末38gを2.2〜3.2mmの直径を有する球状支持体150g(Rz=45μm、支持体物質=Ceramtec、ドイツからのステアタイトC220、支持体の総細孔容量は総支持体容量に基づいて≦1容量%)に適用した。この目的のため、支持体は最初に2lの内部容量を有するドラム(水平面に相対したドラムの中心軸の傾斜角=30°)に入れられた。ドラムは分あたり25回転で回転させられた。グリセロールと水の混合物(グリセロール:水の重量比=1:3)約25mlを圧縮空気300l(S.T.P.)/hで処理した噴霧器ノズルを介して支持体上に噴霧した。回転軌道の上半分の傾斜したドラムの最上位置で、金属ドライバープレートによりドラム内を輸送する支持体を噴霧円錐が湿潤させるような方法でノズルが取り付けられた。微細に分割された活性物質粉末が粉末スクリューを介してドラムに導入され、粉末追加の位置は回転軌道内又は噴霧円錐の下であった。湿潤及び粉末計量を定期的に繰り返すことにより、基礎コーティング自体を供給された支持体は、次の間に支持体になった。 In each case, 38 g of powder present after grinding, 150 g of spherical support having a diameter of 2.2 to 3.2 mm (Rz = 45 μm, support material = Ceramtec, steatite C220 from Germany, total pores of the support The volume was applied to ≦ 1% by volume based on the total support volume. For this purpose, the support was initially placed in a drum having an internal volume of 2 l (tilt angle of the central axis of the drum relative to the horizontal plane = 30 °). The drum was rotated at 25 revolutions per minute. About 25 ml of a mixture of glycerol and water (glycerol: water weight ratio = 1: 3) was sprayed onto the support through a sprayer nozzle treated with 300 l of compressed air (STP) / h. At the top of the inclined drum in the upper half of the rotating track, the nozzle was mounted in such a way that the spray cone wets the support transported in the drum by a metal driver plate. Finely divided active substance powder was introduced into the drum via a powder screw and the position of powder addition was in the rotating track or under the spray cone. By periodically repeating wetting and powder metering, the support supplied with the base coating itself became the support during the next.
コーティング終了後、コーティングされた支持体はマッフル炉中に150℃で16時間、空気下に乾燥された。 After coating was complete, the coated support was dried in air in a muffle furnace at 150 ° C. for 16 hours.
それぞれ活性物質20重量%を含んでいる、コーティングされた触媒SB1〜SB16及びSVB1及びSVB2が得られた。(B1はA)からの実施例1に従った多金属酸化物が使用されたことを意味する;SVB1は従って、A)からの比較実施例1に従った多金属酸化物が使用されたことを意味する)
C)B)で製造されたコーティングされた触媒の試験
鋼鉄から生産された管状の反応器(内径:8.5mm、長さ:140cm、壁厚:2.5cm)をそれぞれの場合において、B)からのそれぞれコーティングされた触媒35.0gで充填した(全ての場合において触媒床長は約53cm)。管状の反応器の残存する長さに渡って、ステアタイトビーズの予備の30cm床(直径:2.2〜3.2mm、製造者:Ceramtec、スタアタイトC220)を触媒床の前に設置し、続く同じステアサイトビーズの床を触媒床の後に設置した。
Coated catalysts SB1-SB16 and SVB1 and SVB2 were obtained, each containing 20% by weight of active substance. (B1 means that a multimetal oxide according to Example 1 from A) was used; SVB1 was therefore used as a multimetal oxide according to Comparative Example 1 from A) Means)
C) Tubular reactors (inner diameter: 8.5 mm, length: 140 cm, wall thickness: 2.5 cm) produced from the coated catalyst test steel produced in B) in each case B) Each with 35.0 g of each coated catalyst (in all cases the catalyst bed length was about 53 cm). Over the remaining length of the tubular reactor, a spare 30 cm bed of steatite beads (diameter: 2.2-3.2 mm, manufacturer: Ceramtec, Staatite C220) is placed in front of the catalyst bed and continues The same steasite bead bed was placed after the catalyst bed.
充填した反応管の外部温度Tは、電気的に加熱された加熱マットによって全長に渡り、外部的に所望の値にされた。 The external temperature T of the filled reaction tube was set to a desired value externally over the entire length by an electrically heated heating mat.
次に、反応管はモル組成プロパン:空気:H2O=1:15:14を有する反応ガス出発物質を供給される(導入する側は次の床の側である)。滞在時間(触媒床容量に基づき)は、2.4秒にされた。導入圧力は2バール絶対値であった。 The reaction tube is then fed with a reaction gas starting material having a molar composition of propane: air: H 2 O = 1: 15: 14 (the inlet side is the next bed side). The residence time (based on the catalyst bed capacity) was 2.4 seconds. The introduction pressure was 2 bar absolute.
反応管床は、本外部温度がそのそれぞれの値にされる前に24時間かけて、充填された反応管の外部温度T=350℃で、各場合において、最初に実行された。 The reaction tube bed was first run in each case at the external temperature T = 350 ° C. of the filled reaction tube for 24 hours before the external temperature was brought to its respective value.
以下の表は、使用したコーティングされた触媒及び設定された外部温度T(℃)の機能として、得られたプロパン転換(UPAN(モル%))、得られたアクリル酸構造の選択性(SACS(モル%))及びプロペン副産物の構造の選択性(SPEN(モル%))を示している。 The table below shows the propane conversion obtained (U PAN (mol%)), the selectivity of the resulting acrylic acid structure (S) as a function of the coated catalyst used and the set external temperature T (° C.). 2 shows the structural selectivity (S PEN (mol%)) of ACS (mol%)) and propene byproduct.
2004年6月9日出願の米国暫定特許出願番号60/577,929は、著述を参照することにより本出願を包含している。上述の叙述の見地から、本発明からの数の修正及び逸脱も可能である。従って、付属の請求項の範囲で、発明が特にここに記載されたものと異なる方法で実施されることができることは、想定されてよい。 US Provisional Patent Application No. 60 / 577,929, filed June 9, 2004, incorporates this application by reference to the writing. Numerous modifications and departures from the present invention are possible in light of the foregoing description. Thus, within the scope of the appended claims, it may be envisaged that the invention may be practiced otherwise than as specifically described herein.
Claims (18)
Mo1VaM1 bM2 cM3 dM4 eOn(I)
[式中、
M1=Al、Ga、In、Ge、Sn、Pb、As、Bi、Se、Te及びSbよりなる群から選択される少なくとも1つの元素;
M2=Sc、Y、La、Ti、Zr、Hf、Nb、Ta、Cr、W、Mn、Fe、Co、Ni、Zn、Cd及びランタノイド類よりなる群から選択される少なくとも1つの元素;
M3=Re、Ru、Rh、Pd、Os、Ir、Pt、Cu、Ag、Auよりなる群から選択される少なくとも1つの元素;
M4=Li、Na、K、Rb、Cs、Be、Mg、Ca、Sr、Ba、及びTlよりなる群から選択される少なくとも1つの元素;
a=0.01〜1;
b=≧0〜1;
c=≧0〜1;
d=≧0〜0.5;
e=≧0〜0.5及び
n=(I)中の酸素以外の元素の原子価及び出現頻度によって決定される数]で示される多金属酸化物材料Mを製造するにあたり、多金属酸化物材料Mの元素構成成分の原料の混合物Gを水熱処理に付し、これにより新たに形成された固体を分離する製造方法において、
多金属酸化物材料Mの元素構成成分の原料として、多金属酸化物材料Mの元素構成成分及び水の元素構成成分のみからなる化合物、及び多金属酸化物材料Mの元素構成成分自体のそれらの元素形よりなる群から選択される原料を使用し、かつ少なくとも原料の一部が、相当する元素構成成分の最大酸化数より低い酸化数を有するこれらの原料に含まれる元素構成成分を含むことを特徴とする方法。 General stoichiometry I:
Mo 1 V a M 1 b M 2 c M 3 d M 4 e O n (I)
[Where:
M 1 = at least one element selected from the group consisting of Al, Ga, In, Ge, Sn, Pb, As, Bi, Se, Te and Sb;
M 2 = at least one element selected from the group consisting of Sc, Y, La, Ti, Zr, Hf, Nb, Ta, Cr, W, Mn, Fe, Co, Ni, Zn, Cd and lanthanoids;
M 3 = At least one element selected from the group consisting of Re, Ru, Rh, Pd, Os, Ir, Pt, Cu, Ag, Au;
M 4 = at least one element selected from the group consisting of Li, Na, K, Rb, Cs, Be, Mg, Ca, Sr, Ba, and Tl;
a = 0.01-1;
b = ≧ 0-1;
c = ≧ 0-1;
d = ≧ 0 to 0.5;
e = ≧ 0 to 0.5 and n = the number determined by the valence and the frequency of appearance of elements other than oxygen in (I)] In the manufacturing method of subjecting a mixture G of raw materials of elemental constituents of material M to hydrothermal treatment, thereby separating a newly formed solid,
As a raw material of the element constituent component of the multimetal oxide material M, a compound composed of only the element constituent component of the multimetal oxide material M and the element constituent component of water, and those of the element constituent component itself of the multimetal oxide material M Using a raw material selected from the group consisting of elemental forms, and at least a portion of the raw material contains an elemental component contained in these raw materials having an oxidation number lower than the maximum oxidation number of the corresponding elemental component Feature method.
a=0.05〜0.5
b=0.01〜0.5
c=0.01〜0.5
d=0.0005〜0.5及び
e=≧0〜0.5
である、請求項1〜7の何れかに記載の方法。 The stoichiometric coefficients a, b, c and d are simultaneously in the following range:
a = 0.05-0.5
b = 0.01-0.5
c = 0.01-0.5
d = 0.005-0.5 and e = ≧ 0-0.5
The method according to any one of claims 1 to 7 , wherein
a=0.1〜0.5
b=0.1〜0.5
c=0.1〜0.5
d=0.001〜0.1及び
e=≧0〜0.1
である、請求項1〜7の何れかに記載の方法。 The stoichiometric coefficients a, b, c and d are simultaneously in the following range:
a = 0.1-0.5
b = 0.1-0.5
c = 0.1-0.5
d = 0.001 to 0.1 and e = ≧ 0 to 0.1
The method according to any one of claims 1 to 7 , wherein
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US57792904P | 2004-06-09 | 2004-06-09 | |
| DE102004027999A DE102004027999A1 (en) | 2004-06-09 | 2004-06-09 | Production of multimetal oxide material, useful as an oxidation or ammoxidation catalyst comprises subjecting a mixture of metal oxide sources to hydrothermal treatment |
| US60/577,929 | 2004-06-09 | ||
| DE102004027999.3 | 2004-06-09 | ||
| PCT/EP2005/005962 WO2005120702A1 (en) | 2004-06-09 | 2005-06-03 | Method for the production of multi-metal oxide masses |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2008501515A JP2008501515A (en) | 2008-01-24 |
| JP2008501515A5 JP2008501515A5 (en) | 2011-02-03 |
| JP5517407B2 true JP5517407B2 (en) | 2014-06-11 |
Family
ID=34968651
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007526256A Expired - Fee Related JP5517407B2 (en) | 2004-06-09 | 2005-06-03 | Method for producing multi-metal oxide material |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP1755779A1 (en) |
| JP (1) | JP5517407B2 (en) |
| WO (1) | WO2005120702A1 (en) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4727506B2 (en) * | 2006-06-07 | 2011-07-20 | 旭化成ケミカルズ株式会社 | Method for producing oxide having bronze structure |
| EP2179793A1 (en) | 2008-10-21 | 2010-04-28 | Sued-Chemie AG | Phosphorous-containing mixed oxide catalysts |
| EP2179790A1 (en) | 2008-10-21 | 2010-04-28 | Sued-Chemie AG | Bismuth-containing mixed oxide catalysts |
| CN101862661B (en) * | 2010-06-04 | 2012-01-04 | 浙江大学 | Method for preparing V-S co-doped titanium dioxide photocatalyst |
| DE102012207811A1 (en) * | 2012-05-10 | 2012-07-12 | Basf Se | Heterogeneously catalyzed gas phase partial oxidation of (meth)acrolein to (meth)acrylic acid using a catalytically active multimetal oxide mass |
| US9409156B2 (en) * | 2012-10-19 | 2016-08-09 | Instituto Mexicano Del Petroleo | Oxidative dehydrogenation of ethane to ethylene and preparation of multimetallic mixed oxide catalyst for such process |
| DE102013202048A1 (en) | 2013-02-07 | 2013-04-18 | Basf Se | Preparing catalytically active composition useful for preparing a catalyst, comprises e.g. thermally treating geometrical precursor bodies formed by a mixture obtained by uniformly mixing e.g. a spray-dried powder and molybdenum oxide |
| EP3219386A1 (en) * | 2016-03-14 | 2017-09-20 | Evonik Degussa GmbH | Method for the hydrothermal preparation of molybdenum-bismuth-cobalt-iron-based mixed oxide catalysts |
| CA2936448C (en) * | 2016-07-19 | 2024-02-20 | Nova Chemicals Corporation | Controlled pressure hydrothermal treatment of odh catalyst |
| DE102017000862A1 (en) | 2017-01-31 | 2018-08-02 | Clariant Produkte (Deutschland) Gmbh | Synthesis of a MoVNbTe catalyst with reduced content of niobium and tellurium and higher activity for the oxidative dehydrogenation of ethane |
| DE102017000865A1 (en) | 2017-01-31 | 2018-08-02 | Clariant Produkte (Deutschland) Gmbh | Synthesis of a MoVNbTe catalyst with increased specific surface area and higher activity for the oxidative dehydrogenation of ethane to ethylene |
| DE102017121709A1 (en) | 2017-09-19 | 2019-03-21 | Clariant International Ltd | Synthesis of a MoVNbTe coated catalyst for the oxidative dehydrogenation of ethane to ethylene |
| JP7520043B2 (en) | 2019-05-02 | 2024-07-22 | ダウ グローバル テクノロジーズ エルエルシー | Process for reducing aldehyde by-products in acrylic acid production using highly active and selective catalysts |
| JP7375639B2 (en) * | 2020-03-18 | 2023-11-08 | 三菱ケミカル株式会社 | Method for producing catalyst for unsaturated carboxylic acid synthesis |
| JP7375638B2 (en) * | 2020-03-18 | 2023-11-08 | 三菱ケミカル株式会社 | Method for producing catalyst for unsaturated carboxylic acid synthesis |
| CN115443189A (en) | 2020-04-21 | 2022-12-06 | 巴斯夫欧洲公司 | Method for producing catalytically active multielement oxides comprising the elements Mo, W, V and Cu |
| CN116490275A (en) | 2020-10-29 | 2023-07-25 | 巴斯夫欧洲公司 | Method for preparing core-shell catalyst |
| EP4392180A1 (en) * | 2021-08-23 | 2024-07-03 | Dow Global Technologies LLC | Catalyst and process for the dehydrogenation of alkanes to olefins |
| WO2024120861A1 (en) | 2022-12-07 | 2024-06-13 | Basf Se | Process for producing a catalytically active multi-element oxide containing the elements mo, w, v, cu and sb |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3500682B2 (en) * | 1994-02-23 | 2004-02-23 | 三菱化学株式会社 | Catalyst for the production of nitriles from alkanes |
| DE19542755A1 (en) * | 1995-11-16 | 1997-05-22 | Basf Ag | Multimetal oxides |
| JP4212154B2 (en) * | 1997-12-24 | 2009-01-21 | 旭化成ケミカルズ株式会社 | Catalyst and method for producing unsaturated nitrile using the same |
| DE10029338A1 (en) * | 2000-06-20 | 2002-01-03 | Basf Ag | Production of acrolein or acrylic acid involves absorption of propane and propene from a gas mixture followed by desorption and oxidation, with no catalytic dehydrogenation of propane and no added oxygen |
| DE10046672A1 (en) * | 2000-09-20 | 2002-03-28 | Basf Ag | Production of acrolein or acrylic acid involves absorption of propane and propene from a gas mixture followed by desorption and oxidation, with no catalytic dehydrogenation of propane and no added oxygen |
| DE10119933A1 (en) * | 2001-04-23 | 2002-10-24 | Basf Ag | Production of acrolein or acrylic acid involves absorption of propane and propene from a gas mixture followed by desorption and oxidation, with no catalytic dehydrogenation of propane and no added oxygen |
| AU2001287622A1 (en) * | 2000-07-18 | 2002-01-30 | Basf Aktiengesellschaft | Method for producing acrylic acid by the heterogeneously catalysed gas-phase oxidation of propane |
| US6642173B2 (en) * | 2001-04-25 | 2003-11-04 | Rohm And Haas Company | Catalyst |
| JP4002437B2 (en) * | 2001-12-28 | 2007-10-31 | 株式会社日本触媒 | Exhaust gas treatment catalyst and exhaust gas treatment method |
| DE10261186A1 (en) * | 2002-12-20 | 2004-07-08 | Basf Ag | Heterogeneously catalyzed gas-phase partial oxidation of acrolein to acrylic acid, used to produce polymers for adhesives, involves using active multimetal oxide material containing e.g. molybdenum and vanadium |
| US7038082B2 (en) * | 2002-10-17 | 2006-05-02 | Basf Aktiengesellschaft | Preparation of a multimetal oxide material |
| JP4155087B2 (en) * | 2003-04-16 | 2008-09-24 | 東亞合成株式会社 | Method for producing metal oxide catalyst |
| DE10321398A1 (en) * | 2003-05-12 | 2004-05-27 | Basf Ag | Multi-metal oxides, useful as catalysts for gas phase oxidation or ammoxidation, contain molybdenum and vanadium along with other elements and have an i-phase structure |
| FR2855516B1 (en) * | 2003-05-27 | 2005-07-08 | Atofina | OXIDATION OF ACRYLIC ACID PROPANE BY USING CATALYSTS MIXED WITH CRYSTALLINE PHASES |
| US20050054869A1 (en) * | 2003-06-06 | 2005-03-10 | Lugmair Claus G. | Mixed metal oxide catalysts for propane and isobutane oxidation and ammoxidation, and methods of preparing same |
-
2005
- 2005-06-03 EP EP05745626A patent/EP1755779A1/en not_active Withdrawn
- 2005-06-03 WO PCT/EP2005/005962 patent/WO2005120702A1/en not_active Application Discontinuation
- 2005-06-03 JP JP2007526256A patent/JP5517407B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| WO2005120702A1 (en) | 2005-12-22 |
| JP2008501515A (en) | 2008-01-24 |
| EP1755779A1 (en) | 2007-02-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5517407B2 (en) | Method for producing multi-metal oxide material | |
| JP4465275B2 (en) | Multi-metal oxide composition | |
| JP4197254B2 (en) | Production of acrylic acid by heterogeneous catalytic partial oxidation of propane | |
| US6867328B2 (en) | Method for producing acrylic acid by the heterogeneously catalysed gas-phase oxidation of propane | |
| JP4204327B2 (en) | Method for producing acrylic acid by vapor phase oxidation of propene with heterogeneous catalyst using molecular oxygen in reaction zone | |
| EP1871522B8 (en) | Process for preparing improved catalysts for selective oxidation of propane into acrylic acid | |
| JP2006502950A5 (en) | ||
| US6461996B2 (en) | Halogen promoted multi-metal oxide catalyst | |
| JP2004148302A (en) | Hydrothermally synthesized mo-v-m-nb-x oxide catalyst selectively oxidizing hydrocarbon | |
| WO2018148240A1 (en) | Process for the preparation of propylene ammoxidation catalysts | |
| TW201002420A (en) | Method for producing catalyst for use in production of unsaturated aldehyde and/or unsaturated carboxylic acid, and method for producing unsaturated aldehyde and/or unsaturated carboxylic acid | |
| ZA200503901B (en) | Multimetallic oxide composition | |
| US20050277546A1 (en) | Process for the preparation of a multimetal oxide material | |
| JP2002510591A (en) | Composite metal oxide material with two-layer structure | |
| US7495121B2 (en) | Mo- and V-containing multimetal oxide materials | |
| JP4868519B2 (en) | Multi-metal oxide material containing Mo, V and alkali metals present in pure phase I | |
| US7019169B2 (en) | Preparation of (meth)acrylic acid | |
| RU2352390C2 (en) | Mass of metal oxides | |
| US7253310B2 (en) | Preparation of (meth)acrylic acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100603 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100901 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100908 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20101001 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20101013 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20101101 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20101109 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20101203 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20101228 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111222 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120309 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130111 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130409 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130416 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130509 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130516 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130610 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130617 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130709 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130805 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20131105 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20131112 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20131203 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20131210 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20131227 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140114 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140303 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140401 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5517407 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |