JP5453003B2 - Catalyst for stereoselective asymmetric nitroaldol reaction - Google Patents
Catalyst for stereoselective asymmetric nitroaldol reaction Download PDFInfo
- Publication number
- JP5453003B2 JP5453003B2 JP2009165211A JP2009165211A JP5453003B2 JP 5453003 B2 JP5453003 B2 JP 5453003B2 JP 2009165211 A JP2009165211 A JP 2009165211A JP 2009165211 A JP2009165211 A JP 2009165211A JP 5453003 B2 JP5453003 B2 JP 5453003B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- compound
- reaction
- complex
- catalyst
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 *[C@@](C(Nc1ccccc1O)=O)NC(c1cccc(*)c1)=O Chemical compound *[C@@](C(Nc1ccccc1O)=O)NC(c1cccc(*)c1)=O 0.000 description 2
- ZMGQMTWUDWKCRL-HTAPYJJXSA-N CC(C)[C@H](C1)[C@]1(C(Nc1ccccc1O)=O)N(CCc(c1c2)ccc2O)C1=O Chemical compound CC(C)[C@H](C1)[C@]1(C(Nc1ccccc1O)=O)N(CCc(c1c2)ccc2O)C1=O ZMGQMTWUDWKCRL-HTAPYJJXSA-N 0.000 description 1
- KURSDAWWCUEEME-CQSZACIVSA-N CC(C)[C@H](CCNC(c1cc(O)ccc1)=O)CC(Nc(cc(cc1)F)c1O)=O Chemical compound CC(C)[C@H](CCNC(c1cc(O)ccc1)=O)CC(Nc(cc(cc1)F)c1O)=O KURSDAWWCUEEME-CQSZACIVSA-N 0.000 description 1
- KKCDKGPDUMXQEF-CQSZACIVSA-N CC(C)[C@H](CCNC(c1cc(O)ccc1F)=O)CC(Nc1ccccc1O)=O Chemical compound CC(C)[C@H](CCNC(c1cc(O)ccc1F)=O)CC(Nc1ccccc1O)=O KKCDKGPDUMXQEF-CQSZACIVSA-N 0.000 description 1
- DLNKOYKMWOXYQA-CBAPKCEASA-N C[C@@H]([C@@H](c1ccccc1)O)N Chemical compound C[C@@H]([C@@H](c1ccccc1)O)N DLNKOYKMWOXYQA-CBAPKCEASA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B53/00—Asymmetric syntheses
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J31/00—Catalysts comprising hydrides, coordination complexes or organic compounds
- B01J31/16—Catalysts comprising hydrides, coordination complexes or organic compounds containing coordination complexes
- B01J31/22—Organic complexes
- B01J31/2204—Organic complexes the ligands containing oxygen or sulfur as complexing atoms
- B01J31/2208—Oxygen, e.g. acetylacetonates
- B01J31/2217—At least one oxygen and one nitrogen atom present as complexing atoms in an at least bidentate or bridging ligand
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/13—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by hydroxy groups
- C07C205/14—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by hydroxy groups having nitro groups and hydroxy groups bound to acyclic carbon atoms
- C07C205/15—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by hydroxy groups having nitro groups and hydroxy groups bound to acyclic carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/13—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by hydroxy groups
- C07C205/14—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by hydroxy groups having nitro groups and hydroxy groups bound to acyclic carbon atoms
- C07C205/16—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by hydroxy groups having nitro groups and hydroxy groups bound to acyclic carbon atoms of a carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/13—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by hydroxy groups
- C07C205/26—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by hydroxy groups and being further substituted by halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/27—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by etherified hydroxy groups
- C07C205/32—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by etherified hydroxy groups having nitro groups bound to acyclic carbon atoms and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/22—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton having nitrogen atoms of amino groups bound to the carbon skeleton of the acid part, further acylated
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/02—Compositional aspects of complexes used, e.g. polynuclearity
- B01J2531/0202—Polynuclearity
- B01J2531/0205—Bi- or polynuclear complexes, i.e. comprising two or more metal coordination centres, without metal-metal bonds, e.g. Cp(Lx)Zr-imidazole-Zr(Lx)Cp
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/10—Complexes comprising metals of Group I (IA or IB) as the central metal
- B01J2531/12—Sodium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/30—Complexes comprising metals of Group III (IIIA or IIIB) as the central metal
- B01J2531/37—Lanthanum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2531/00—Additional information regarding catalytic systems classified in B01J31/00
- B01J2531/30—Complexes comprising metals of Group III (IIIA or IIIB) as the central metal
- B01J2531/38—Lanthanides other than lanthanum
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
Description
本発明は立体選択的な触媒的不斉ニトロアルドール反応、及び該反応用の触媒に関する。 The present invention relates to a stereoselective catalytic asymmetric nitroaldol reaction and a catalyst for the reaction.
有機合成化学、特に医薬品合成化学の分野において光学活性アンチ-1,2-アミノアルコール化合物は極めて有用性の高いキラルビルディングブロックとして汎用されている。例えば、β-アゴニストなどの医薬品や多くの天然生理活性化合物に光学活性アンチ-1,2-アミノアルコールが基本ユニットとして含まれており、光学活性アンチ-1,2-アミノアルコール化合物を原料化合物や反応試薬として用いることにより、これらの化合物を効率的かつ安価に製造することができる。このような理由から、光学活性アンチ-1,2-アミノアルコール化合物を効率的に製造する方法の開発が求められている。 In the field of organic synthetic chemistry, especially pharmaceutical synthetic chemistry, optically active anti-1,2-aminoalcohol compounds are widely used as chiral building blocks with extremely high utility. For example, optically active anti-1,2-aminoalcohol is included as a basic unit in pharmaceuticals such as β-agonists and many natural physiologically active compounds, and optically active anti-1,2-aminoalcohol compounds are used as starting compounds and By using it as a reaction reagent, these compounds can be produced efficiently and inexpensively. For these reasons, development of a method for efficiently producing an optically active anti-1,2-aminoalcohol compound is required.
光学活性アンチ-1,2-アミノアルコール化合物の前駆体として有用な光学活性アンチ-1,2-ニトロアルカノール化合物を触媒的不斉反応によりアンチ選択的に製造する方法として、各種アルデヒド化合物とニトロアルカンとを光学活性テトラアミノホスホニウム塩の存在下で反応させる方法が知られている(Uraguchi, D., et al., J. Am. Chem. Soc.,129, pp.12392, 2007)。しかしながら、この方法は-78℃の極低温下に行なう必要があり、工業的な製造方法として応用できないという問題があった。 As a method for producing an optically active anti-1,2-nitroalkanol compound useful as a precursor of an optically active anti-1,2-aminoalcohol compound by selective catalytic asymmetric reaction, various aldehyde compounds and nitroalkanes are used. Is known in the presence of an optically active tetraaminophosphonium salt (Uraguchi, D., et al., J. Am. Chem. Soc., 129, pp. 12392, 2007). However, this method needs to be carried out at an extremely low temperature of -78 ° C., and has a problem that it cannot be applied as an industrial production method.
一方、下記のアミド化合物:
本発明の課題は立体選択的な触媒的不斉ニトロアルドール反応、及び該反応用の触媒を提供することにある。より具体的には、光学活性アンチ-1,2-アミノアルコール化合物の前駆体として有用な光学活性アンチ-1,2-ニトロアルカノール化合物を触媒的不斉反応によりアンチ選択的に製造する方法及び該反応に用いる触媒を提供することが本発明の課題である。 An object of the present invention is to provide a stereoselective catalytic asymmetric nitroaldol reaction and a catalyst for the reaction. More specifically, a method for anti-selectively producing an optically active anti-1,2-nitroalkanol compound useful as a precursor of an optically active anti-1,2-aminoalcohol compound by catalytic asymmetric reaction, and the method It is an object of the present invention to provide a catalyst for use in the reaction.
本発明者らは上記の課題を解決すべく鋭意研究を行った結果、上記の非特許文献3に記載されたアミド化合物において芳香環上にハロゲン原子などの官能基を導入した化合物を製造し、この化合物を配位子としてネオジムなどのランタノイド及びナトリウムなどのアルカリ金属に配位させた異種金属複合型の錯体を反応触媒として用いて各種アルデヒド化合物とニトロアルカンとを用いたニトロアルドール反応を行なうことにより、光学活性アンチ-1,2-アミノアルコール化合物の前駆体である光学活性アンチ-1,2-ニトロアルカノール化合物を効率よく製造できること、及びこの反応が-40℃程度の冷却下においても速やかに進行し、高アンチ選択的に、かつ極めて高い不斉収率で目的物を製造できることを見出した。本発明は上記の知見に基づいて完成されたものである。 As a result of diligent research to solve the above problems, the inventors of the present invention produced a compound in which a functional group such as a halogen atom was introduced on the aromatic ring in the amide compound described in Non-Patent Document 3, Carry out nitroaldol reaction using various aldehyde compounds and nitroalkanes using a heterogeneous metal complex complex coordinated with lanthanoids such as neodymium and alkali metals such as sodium as ligands with this compound as a ligand. Makes it possible to efficiently produce an optically active anti-1,2-nitroalkanol compound, which is a precursor of an optically active anti-1,2-aminoalcohol compound, and this reaction can be carried out rapidly even at about -40 ° C cooling. It has been found that the target product can be produced with high anti-selectivity and extremely high asymmetric yield. The present invention has been completed based on the above findings.
すなわち、本発明により、光学活性アンチ-1,2-ニトロアルカノール化合物の製造方法であって、アルデヒド化合物と炭素数2以上のニトロアルカン化合物とを下記の一般式(I):
本発明の好ましい方法によれば、上記錯体が上記一般式(I)で表される化合物においてR1がイソブチル基である化合物、ランタン(La)、ネオジム(Nd)、サマリウム(Sm)、プラセオジム(Pr)、ユウロピウム(Eu)、ガドリニウム(Gd)、ジスプロシウム(Dy)、及びイッテルビウム(Yb)からなる群から選ばれる金属、及びアルカリ金属を含む異種金属複合型錯体である上記の方法;上記錯体が上記一般式(I)で表される化合物においてR1がイソブチル基である化合物、ネオジム(Nd)及びナトリウムを含む異種金属複合型錯体である上記の方法;
サマリウム(Sm)及びナトリウムを含む異種金属複合型錯体である上記の方法;プラセオジム(Pr)及びナトリウムを含む異種金属複合型錯体である上記の方法;ニトロアルカンがR11-CH2-NO2(式中、R11はC1-C20アルキル基を示し、該アルキル基は置換基を有していてもよい)で表されるニトロアルカンである上記の方法;反応温度を-50〜-30℃の範囲で行なう上記の方法;調製された錯体を単離して再懸濁する工程を含む上記の方法;及びランタノイドとほぼ等量の水の存在下で行なう上記の方法が提供される。
According to a preferred method of the present invention, the compound represented by the above general formula (I) wherein R 1 is an isobutyl group, lanthanum (La), neodymium (Nd), samarium (Sm), praseodymium ( Pr), europium (Eu), gadolinium (Gd), dysprosium (Dy), and a metal selected from the group consisting of ytterbium (Yb) and the above method, which is a heterogeneous metal complex complex containing an alkali metal; The above method, wherein the compound represented by the general formula (I) is a compound in which R 1 is an isobutyl group, a heterogeneous metal complex complex containing neodymium (Nd) and sodium;
The above method which is a heterometallic complex containing samarium (Sm) and sodium; the above method which is a heterometallic complex containing praseodymium (Pr) and sodium; the nitroalkane is R 11 —CH 2 —NO 2 ( Wherein R 11 represents a C 1 -C 20 alkyl group, and the alkyl group may have a substituent, and the reaction temperature is -50 to -30 There is provided the above method carried out in the range of ° C .; the above method comprising the step of isolating and resuspending the prepared complex; and the above method carried out in the presence of approximately the same amount of water as the lanthanoid.
別の観点からは、上記一般式(I)で表される化合物を配位子として含む上記の異種金属複合型錯体が本発明により提供される。また、アルデヒド化合物と炭素数2以上のニトロアルカン化合物とを反応させて光学活性アンチ-1,2-ニトロアルカノール化合物を製造するための反応触媒であって、上記錯体を含む反応触媒;光学活性アンチ-1,2-アミノアルコール化合物の製造方法であって、(A)アルデヒド化合物とニトロアルカン化合物とを上記の錯体の存在下で反応させて光学活性アンチ-1,2-ニトロアルカノール化合物を製造する工程、及び(B)上記工程(A)で得られた光学活性アンチ-1,2-ニトロアルカノール化合物を還元して光学活性アンチ-1,2-アミノアルコール化合物を製造する工程を含む方法も本発明により提供される。
さらに別の観点からは、上記一般式(I)で表される化合物も本発明により提供される。
From another viewpoint, the present invention provides the above heterogeneous metal complex complex containing the compound represented by the general formula (I) as a ligand. A reaction catalyst for producing an optically active anti-1,2-nitroalkanol compound by reacting an aldehyde compound with a nitroalkane compound having 2 or more carbon atoms, the reaction catalyst comprising the complex; A method for producing a 1,2-aminoalcohol compound, wherein (A) an aldehyde compound and a nitroalkane compound are reacted in the presence of the above complex to produce an optically active anti-1,2-nitroalkanol compound The present invention also includes a method comprising the steps of: (B) reducing the optically active anti-1,2-nitroalkanol compound obtained in the above step (A) to produce an optically active anti-1,2-aminoalcohol compound. Provided by the invention.
From another point of view, the present invention also provides a compound represented by the above general formula (I).
本発明の方法により、医薬品や天然生理活性化合物の製造のために有用な光学活性アンチ-1,2-アミノアルコール化合物の前駆体として有用な光学活性アンチ-1,2-ニトロアルカノール化合物を緩和な条件下で効率的に製造することができる。本発明の方法は-40℃程度の低温化で行なうことができるので、工業的製造方法として利用することができる。また、本発明の方法では、錯体調製において生成する沈殿を単離した後に再懸濁して触媒懸濁液として用いることができ、高純度な触媒を簡便に調製して使用することにより高アンチ選択的に、かつ極めて高い不斉収率で目的物を製造できるという優れた特徴がある。 According to the method of the present invention, an optically active anti-1,2-nitroalkanol compound useful as a precursor of an optically active anti-1,2-aminoalcohol compound useful for the production of pharmaceuticals and natural physiologically active compounds can be relaxed. It can be produced efficiently under conditions. Since the method of the present invention can be performed at a low temperature of about −40 ° C., it can be used as an industrial production method. In the method of the present invention, the precipitate produced in the complex preparation can be isolated and then resuspended to be used as a catalyst suspension. By simply preparing and using a high-purity catalyst, high anti-selection can be achieved. In particular, it has an excellent feature that the target product can be produced with an extremely high asymmetric yield.
本発明の方法はアルデヒド化合物と炭素数2以上のニトロアルカン化合物とを用いてニトロアルドール反応により光学活性アンチ-1,2-ニトロアルカノール化合物を製造する方法であり、上記反応において上記一般式(I)で表される化合物を配位子として含む異種金属複合型錯体を反応触媒として用いることを特徴としている。 The method of the present invention is a method for producing an optically active anti-1,2-nitroalkanol compound by a nitroaldol reaction using an aldehyde compound and a nitroalkane compound having 2 or more carbon atoms. In the above reaction, the above general formula (I ) Is used as a reaction catalyst.
アルデヒド化合物の種類は特に限定されず、芳香族アルデヒド化合物又は脂肪族アルデヒド化合物のいずれを用いてもよい。アルデヒド化合物は、例えばアルキル基、アルコキシ基、カルボキシル基、水酸基、ハロゲン原子など任意の置換基を1個又は2個以上有していてもよい。アルデヒド化合物を本発明の方法における原料として用いる際に、必要に応じてアルデヒド以外の反応性官能基を保護基で保護することにより、本発明の方法を好適に行なうことができる。保護基については、例えば、Greenら、Protective Groups in Organic Synthesis, 3rd Edition, 1999, John Wiley & Sons, Inc.などの成書を参照することができる。アルデヒド化合物として、例えば、ベンズアルデヒド、ハロゲノベンズアルデヒド(クロルベンズアルデヒド、ブロムベンズアルデヒドなど)、アルコキシベンズアルデヒド(メトキシベンズアルデヒド、エトキシベンズアルデヒドなど)、アルキルベンズアルデヒド(メチルベンズアルデヒド、エチルベンズアルデヒドなど)、ナフチルアルデヒドなどの芳香族アルデヒド化合物、アルキルアルデヒド(ブチルアルデヒド、シクロプロピルアルデヒドなど)などの脂肪族アルデヒド、又はアラルキルアルデヒド(フェネチルアルデヒド、ベンジルアルデヒドなど)などの芳香族置換脂肪族アルデヒドなどを用いることができるが、これらに限定されることはない。 The kind of aldehyde compound is not particularly limited, and either an aromatic aldehyde compound or an aliphatic aldehyde compound may be used. The aldehyde compound may have one or more arbitrary substituents such as an alkyl group, an alkoxy group, a carboxyl group, a hydroxyl group, and a halogen atom. When using an aldehyde compound as a raw material in the method of the present invention, the method of the present invention can be suitably carried out by protecting a reactive functional group other than the aldehyde with a protecting group as necessary. For protecting groups, reference can be made to, for example, Green et al., Protective Groups in Organic Synthesis, 3rd Edition, 1999, John Wiley & Sons, Inc. Examples of the aldehyde compound include aromatic aldehyde compounds such as benzaldehyde, halogenobenzaldehyde (chlorbenzaldehyde, bromobenzaldehyde, etc.), alkoxybenzaldehyde (methoxybenzaldehyde, ethoxybenzaldehyde, etc.), alkylbenzaldehyde (methylbenzaldehyde, ethylbenzaldehyde, etc.), and naphthylaldehyde. Aliphatic aldehydes such as alkyl aldehydes (butyraldehyde, cyclopropyl aldehyde, etc.) or aromatic substituted aliphatic aldehydes such as aralkyl aldehydes (phenethyl aldehyde, benzyl aldehyde, etc.) can be used, but are not limited thereto. There is no.
ニトロアルカン化合物の種類は炭素数2以上であれば特に限定されないが、例えば、アルキル鎖の炭素数が2〜20個程度のニトロアルカン化合物を用いることができる。ニトロアルカン化合物の主鎖を構成するアルキル基は、例えばアルキル基、アルコキシ基、カルボキシル基、水酸基、ハロゲン原子など任意の置換基を1個又は2個以上有していてもよく、アルキル鎖中に二重結合又は三重結合を任意の個数含んでいてもよい。必要に応じてニトロ基以外の反応性官能基を保護基で保護することにより、本発明の方法を好適に行なうことができる。保護基については上掲のGreenらの成書を参照することができる。ニトロアルカンとして、好ましくはニトロエタン、ニトロプロパン、ニトロブタンなどを用いることができるが、これらに限定されることはない。 The type of the nitroalkane compound is not particularly limited as long as it has 2 or more carbon atoms. For example, a nitroalkane compound having an alkyl chain with about 2 to 20 carbon atoms can be used. The alkyl group constituting the main chain of the nitroalkane compound may have one or more arbitrary substituents such as an alkyl group, an alkoxy group, a carboxyl group, a hydroxyl group, and a halogen atom. Any number of double bonds or triple bonds may be included. The method of the present invention can be suitably performed by protecting reactive functional groups other than the nitro group with a protecting group as necessary. For the protecting group, refer to the above-mentioned book by Green et al. As the nitroalkane, nitroethane, nitropropane, nitrobutane and the like can be preferably used, but are not limited thereto.
上記一般式(I)で表される錯体において、R1はC3-8アルキル基又はアラルキル基を示す。R1が示すC3-8アルキル基としては、直鎖状、分枝鎖状、環状、又はそれらの組合せからなるアルキル基を用いることができるが、分枝鎖状、環状、又はそれらの組合せからなるアルキル基が好ましく、分枝鎖状又は環状のアルキル基がより好ましい。例えば、イソプロピル基、sec-ブチル基、イソブチル基(-CH2-CH(CH3)2)、若しくはtert-ブチル基などの分枝鎖状C3-4アルキル基、又はシクロペンチル基、シクロヘキシル基、若しくはシクロヘプチル基などの環状C5-7アルキル基が好ましい。これらのうち、イソプロピル基、イソブチル基、又はtert-ブチル基がさらに好ましく、特に好ましいのはイソブチル基である。 In the complex represented by the general formula (I), R 1 represents a C 3-8 alkyl group or an aralkyl group. As the C 3-8 alkyl group represented by R 1 , an alkyl group consisting of a linear, branched, cyclic, or combination thereof can be used, but a branched, cyclic, or a combination thereof can be used. An alkyl group consisting of is preferable, and a branched or cyclic alkyl group is more preferable. For example, a branched C 3-4 alkyl group such as isopropyl group, sec-butyl group, isobutyl group (—CH 2 —CH (CH 3 ) 2 ), or tert-butyl group, or cyclopentyl group, cyclohexyl group, Alternatively, a cyclic C 5-7 alkyl group such as a cycloheptyl group is preferable. Of these, an isopropyl group, an isobutyl group, or a tert-butyl group is more preferable, and an isobutyl group is particularly preferable.
R1が示すアラルキル基としては、例えば、1個又は数個、好ましくは1個のアリール基又はヘテロアリール基により置換された直鎖状又は分枝鎖状のC1-4アルキル基を用いることができる。R1が示すアラルキル基を構成するアリール基としては単環式又は縮合多環式の芳香族炭化水素基を用いることができ、例えば、フェニル基、1-ナフチル基、2-ナフチル基、アントラニル基、又はフェナンスリル基等が挙げられるが、これらに限定されることはない。ヘテロアリール基としては、単環式又は縮合多環式の芳香族ヘテロ環基を用いることができる。環構成ヘテロ原子の個数は特に限定されないが、1個ないし数個、好ましくは1個ないし5個程度である。2個以上の環構成ヘテロ原子を含む場合にはそれらは同一でも異なっていてもよい。ヘテロ原子としては、例えば、酸素原子、窒素原子、又はイオウ原子等を挙げることができるが、これらに限定されることはない。 As the aralkyl group represented by R 1 , for example, a linear or branched C 1-4 alkyl group substituted by one or several, preferably one aryl group or heteroaryl group is used. Can do. As the aryl group constituting the aralkyl group represented by R 1 , a monocyclic or condensed polycyclic aromatic hydrocarbon group can be used, for example, a phenyl group, a 1-naphthyl group, a 2-naphthyl group, an anthranyl group Or a phenanthryl group and the like, but is not limited thereto. As the heteroaryl group, a monocyclic or condensed polycyclic aromatic heterocyclic group can be used. The number of ring-constituting heteroatoms is not particularly limited, but is about 1 to several, preferably about 1 to 5. When two or more ring heteroatoms are contained, they may be the same or different. Examples of the hetero atom include, but are not limited to, an oxygen atom, a nitrogen atom, or a sulfur atom.
R1が示すアラルキル基を構成する単環式ヘテロアリール基としては、例えば、2-フリル基、3-フリル基、2-チエニル基、3-チエニル基、1-ピロリル基、2-ピロリル基、3-ピロリル基、2-オキサゾリル基、4-オキサゾリル基、5-オキサゾリル基、3-イソオキサゾリル基、4-イソオキサゾリル基、5-イソオキサゾリル基、2-チアゾリル基、4-チアゾリル基、5-チアゾリル基、3-イソチアゾリル基、4-イソチアゾリル基、5-イソチアゾリル基、1-イミダゾリル基、2-イミダゾリル基、4-イミダゾリル基、5-イミダゾリル基、1-ピラゾリル基、3-ピラゾリル基、4-ピラゾリル基、5-ピラゾリル基、(1,2,3-オキサジアゾール)-4-イル基、(1,2,3-オキサジアゾール)-5-イル基、(1,2,4-オキサジアゾール)-3-イル基、(1,2,4-オキサジアゾール)-5-イル基、(1,2,5-オキサジアゾール)-3-イル基、(1,2,5-オキサジアゾール)-4-イル基、(1,3,4-オキサジアゾール)-2-イル基、(1,3,4-オキサジアゾール)-5-イル基、フラザニル基、(1,2,3-チアジアゾール)-4-イル基、(1,2,3-チアジアゾール)-5-イル基、(1,2,4-チアジアゾール)-3-イル基、(1,2,4-チアジアゾール)-5-イル基、(1,2,5-チアジアゾール)-3-イル基、(1,2,5-チアジアゾール)-4-イル基、(1,3,4-チアジアゾリル)-2-イル基、(1,3,4-チアジアゾリル)-5-イル基、(1H-1,2,3-トリアゾール)-1-イル基、(1H-1,2,3-トリアゾール)-4-イル基、(1H-1,2,3-トリアゾール)-5-イル基、(2H-1,2,3-トリアゾール)-2-イル基、(2H-1,2,3-トリアゾール)-4-イル基、(1H-1,2,4-トリアゾール)-1-イル基、(1H-1,2,4-トリアゾール)-3-イル基、(1H-1,2,4-トリアゾール)-5-イル基、(4H-1,2,4-トリアゾール)-3-イル基、(4H-1,2,4-トリアゾール)-4-イル基、(1H-テトラゾール)-1-イル基、(1H-テトラゾール)-5-イル基、(2H-テトラゾール)-2-イル基、(2H-テトラゾール)-5-イル基、2-ピリジル基、3-ピリジル基、4-ピリジル基、3-ピリダジニル基、4-ピリダジニル基、2-ピリミジニル基、4-ピリミジニル基、5-ピリミジニル基、2-ピラジニル基、(1,2,3-トリアジン)-4-イル基、(1,2,3-トリアジン)-5-イル基、(1,2,4-トリアジン)-3-イル基、(1,2,4-トリアジン)-5-イル基、(1,2,4-トリアジン)-6-イル基、(1,3,5-トリアジン)-2-イル基、1-アゼピニル基、1-アゼピニル基、2-アゼピニル基、3-アゼピニル基、4-アゼピニル基、(1,4-オキサゼピン)-2-イル基、(1,4-オキサゼピン)-3-イル基、(1,4-オキサゼピン)-5-イル基、(1,4-オキサゼピン)-6-イル基、(1,4-オキサゼピン)-7-イル基、(1,4-チアゼピン)-2-イル基、(1,4-チアゼピン)-3-イル基、(1,4-チアゼピン)-5-イル基、(1,4-チアゼピン)-6-イル基、(1,4-チアゼピン)-7-イル基等の5ないし7員の単環式ヘテロアリール基が挙げられるが、これらに限定されることはない。 Examples of the monocyclic heteroaryl group constituting the aralkyl group represented by R 1 include, for example, 2-furyl group, 3-furyl group, 2-thienyl group, 3-thienyl group, 1-pyrrolyl group, 2-pyrrolyl group, 3-pyrrolyl group, 2-oxazolyl group, 4-oxazolyl group, 5-oxazolyl group, 3-isoxazolyl group, 4-isoxazolyl group, 5-isoxazolyl group, 2-thiazolyl group, 4-thiazolyl group, 5-thiazolyl group, 3-isothiazolyl group, 4-isothiazolyl group, 5-isothiazolyl group, 1-imidazolyl group, 2-imidazolyl group, 4-imidazolyl group, 5-imidazolyl group, 1-pyrazolyl group, 3-pyrazolyl group, 4-pyrazolyl group, 5-pyrazolyl group, (1,2,3-oxadiazol) -4-yl group, (1,2,3-oxadiazol) -5-yl group, (1,2,4-oxadiazole) -3-yl group, (1,2,4-oxadiazol) -5-yl group, (1,2,5-oxadiazol) -3-yl group, (1,2,5-oxadiazol) -4-yl group, (1,3,4-oxadiazol) -2-yl group, (1,3,4-oxadiazol) -5-yl group , Furazanyl group, (1,2,3-thiadiazol) -4-yl group, (1,2,3-thiadiazol) -5-yl group, (1,2,4-thiadiazol) -3-yl group, 1,2,4-thiadiazol) -5-yl group, (1,2,5-thiadiazol) -3-yl group, (1,2,5-thiadiazol) -4-yl group, (1,3,4 -Thiadiazolyl) -2-yl group, (1,3,4-thiadiazolyl) -5-yl group, (1H-1,2,3-triazol) -1-yl group, (1H-1,2,3- (Triazol) -4-yl group, (1H-1,2,3-triazol) -5-yl group, (2H-1,2,3-triazol) -2-yl group, (2H-1,2,3 -Triazol) -4-yl group, (1H-1,2,4-triazol) -1-yl group, (1H-1,2,4-triazol) -3-yl group, (1H-1,2, 4-triazol) -5-yl group, (4H-1,2,4-triazol) -3-yl group, (4H-1,2,4-triazol) -4-yl group, (1H-tetrazole)- 1-yl group, (1H-te Razol) -5-yl group, (2H-tetrazol) -2-yl group, (2H-tetrazol) -5-yl group, 2-pyridyl group, 3-pyridyl group, 4-pyridyl group, 3-pyridazinyl group, 4-pyridazinyl group, 2-pyrimidinyl group, 4-pyrimidinyl group, 5-pyrimidinyl group, 2-pyrazinyl group, (1,2,3-triazin) -4-yl group, (1,2,3-triazine)- 5-yl group, (1,2,4-triazine) -3-yl group, (1,2,4-triazine) -5-yl group, (1,2,4-triazine) -6-yl group, (1,3,5-triazine) -2-yl group, 1-azepinyl group, 1-azepinyl group, 2-azepinyl group, 3-azepinyl group, 4-azepinyl group, (1,4-oxazepine) -2- Yl group, (1,4-oxazepine) -3-yl group, (1,4-oxazepine) -5-yl group, (1,4-oxazepine) -6-yl group, (1,4-oxazepine)- 7-yl group, (1,4-thiazepine) -2-yl group, (1,4-thiazepine) -3-yl group, (1,4-thiazepine) -5-yl group, (1,4-thiazepine) ) -6-yl group (1,4-thiazepine) -7-yl, but 5 to 7-membered monocyclic heteroaryl group such groups are not limited thereto.
R1が示すアラルキル基を構成する縮合多環式ヘテロアリール基としては、例えば、2-ベンゾフラニル基、3-ベンゾフラニル基、4-ベンゾフラニル基、5-ベンゾフラニル基、6-ベンゾフラニル基、7-ベンゾフラニル基、1-イソベンゾフラニル基、4-イソベンゾフラニル基、5-イソベンゾフラニル基、2-ベンゾ[b]チエニル基、3-ベンゾ[b]チエニル基、4-ベンゾ[b]チエニル基、5-ベンゾ[b]チエニル基、6-ベンゾ[b]チエニル基、7-ベンゾ[b]チエニル基、1-ベンゾ[c]チエニル基、4-ベンゾ[c]チエニル基、5-ベンゾ[c]チエニル基、1-インドリル基、1-インドリル基、2-インドリル基、3-インドリル基、4-インドリル基、5-インドリル基、6-インドリル基、7-インドリル基、(2H-イソインドール)-1-イル基、(2H-イソインドール)-2-イル基、(2H-イソインドール)-4-イル基、(2H-イソインドール)-5-イル基、(1H-インダゾール)-1-イル基、(1H-インダゾール)-3-イル基、(1H-インダゾール)-4-イル基、(1H-インダゾール)-5-イル基、(1H-インダゾール)-6-イル基、(1H-インダゾール)-7-イル基、(2H-インダゾール)-1-イル基、(2H-インダゾール)-2-イル基、(2H-インダゾール)-4-イル基、(2H-インダゾール)-5-イル基、2-ベンゾオキサゾリル基、2-ベンゾオキサゾリル基、4-ベンゾオキサゾリル基、5-ベンゾオキサゾリル基、6-ベンゾオキサゾリル基、7-ベンゾオキサゾリル基、(1,2-ベンゾイソオキサゾール)-3-イル基、(1,2-ベンゾイソオキサゾール)-4-イル基、(1,2-ベンゾイソオキサゾール)-5-イル基、(1,2-ベンゾイソオキサゾール)-6-イル基、(1,2-ベンゾイソオキサゾール)-7-イル基、(2,1-ベンゾイソオキサゾール)-3-イル基、(2,1-ベンゾイソオキサゾール)-4-イル基、(2,1-ベンゾイソオキサゾール)-5-イル基、(2,1-ベンゾイソオキサゾール)-6-イル基、(2,1-ベンゾイソオキサゾール)-7-イル基、2-ベンゾチアゾリル基、4-ベンゾチアゾリル基、5-ベンゾチアゾリル基、6-ベンゾチアゾリル基、7-ベンゾチアゾリル基、(1,2-ベンゾイソチアゾール)-3-イル基、(1,2-ベンゾイソチアゾール)-4-イル基、(1,2-ベンゾイソチアゾール)-5-イル基、(1,2-ベンゾイソチアゾール)-6-イル基、(1,2-ベンゾイソチアゾール)-7-イル基、(2,1-ベンゾイソチアゾール)-3-イル基、(2,1-ベンゾイソチアゾール)-4-イル基、(2,1-ベンゾイソチアゾール)-5-イル基、(2,1-ベンゾイソチアゾール)-6-イル基、(2,1-ベンゾイソチアゾール)-7-イル基、(1,2,3-ベンゾオキサジアゾール)-4-イル基、(1,2,3-ベンゾオキサジアゾール)-5-イル基、(1,2,3-ベンゾオキサジアゾール)-6-イル基、(1,2,3-ベンゾオキサジアゾール)-7-イル基、(2,1,3-ベンゾオキサジアゾール)-4-イル基、(2,1,3-ベンゾオキサジアゾール)-5-イル基、(1,2,3-ベンゾチアジアゾール)-4-イル基、(1,2,3-ベンゾチアジアゾール)-5-イル基、(1,2,3-ベンゾチアジアゾール)-6-イル基、(1,2,3-ベンゾチアジアゾール)-7-イル基、(2,1,3-ベンゾチアジアゾール)-4-イル基、(2,1,3-ベンゾチアジアゾール)-5-イル基、(1H-ベンゾトリアゾール)-1-イル基、(1H-ベンゾトリアゾール)-4-イル基、(1H-ベンゾトリアゾール)-5-イル基、(1H-ベンゾトリアゾール)-6-イル基、(1H-ベンゾトリアゾール)-7-イル基、(2H-ベンゾトリアゾール)-2-イル基、(2H-ベンゾトリアゾール)-4-イル基、(2H-ベンゾトリアゾール)-5-イル基、2-キノリル基、3-キノリル基、4-キノリル基、5-キノリル基、6-キノリル基、7-キノリル基、8-キノリル基、1-イソキノリル基、3-イソキノリル基、4-イソキノリル基、5-イソキノリル基、6-イソキノリル基、7-イソキノリル基、8-イソキノリル基、3-シンノリニル基、4-シンノリニル基、5-シンノリニル基、6-シンノリニル基、7-シンノリニル基、8-シンノリニル基、2-キナゾリニル基、4-キナゾリニル基、5-キナゾリニル基、6-キナゾリニル基、7-キナゾリニル基、8-キナゾリニル基、2-キノキサリニル基、5-キノキサリニル基、6-キノキサリニル基、1-フタラジニル基、5-フタラジニル基、6-フタラジニル基、2-ナフチリジニル基、3-ナフチリジニル基、4-ナフチリジニル基、2-プリニル基、6-プリニル基、7-プリニル基、8-プリニル基、2-プテリジニル基、4-プテリジニル基、6-プテリジニル基、7-プテリジニル基、1-カルバゾリル基、2-カルバゾリル基、3-カルバゾリル基、4-カルバゾリル基、9-カルバゾリル基、2-(α-カルボリニル)基、3-(α-カルボリニル)基、4-(α-カルボリニル)基、5-(α-カルボリニル)基、6-(α-カルボリニル)基、7-(α-カルボリニル)基、8-(α-カルボリニル)基、9-(α-カルボリニル)基、1-(β-カルボニリル)基、3-(β-カルボニリル)基、4-(β-カルボニリル)基、5-(β-カルボニリル)基、6-(β-カルボニリル)基、7-(β-カルボニリル)基、8-(β-カルボニリル)基、9-(β-カルボニリル)基、1-(γ-カルボリニル)基、2-(γ-カルボリニル)基、4-(γ-カルボリニル)基、5-(γ-カルボリニル)基、6-(γ-カルボリニル)基、7-(γ-カルボリニル)基、8-(γ-カルボリニル)基、9-(γ-カルボリニル)基、1-アクリジニル基、2-アクリジニル基、3-アクリジニル基、4-アクリジニル基、9-アクリジニル基、1-フェノキサジニル基、2-フェノキサジニル基、3-フェノキサジニル基、4-フェノキサジニル基、10-フェノキサジニル基、1-フェノチアジニル基、2-フェノチアジニル基、3-フェノチアジニル基、4-フェノチアジニル基、10-フェノチアジニル基、1-フェナジニル基、2-フェナジニル基、1-フェナントリジニル基、2-フェナントリジニル基、3-フェナントリジニル基、4-フェナントリジニル基、6-フェナントリジニル基、7-フェナントリジニル基、8-フェナントリジニル基、9-フェナントリジニル基、10-フェナントリジニル基、2-フェナントロリニル基、3-フェナントロリニル基、4-フェナントロリニル基、5-フェナントロリニル基、6-フェナントロリニル基、7-フェナントロリニル基、8-フェナントロリニル基、9-フェナントロリニル基、10-フェナントロリニル基、1-チアントレニル基、2-チアントレニル基、1-インドリジニル基、2-インドリジニル基、3-インドリジニル基、5-インドリジニル基、6-インドリジニル基、7-インドリジニル基、8-インドリジニル基、1-フェノキサチイニル基、2-フェノキサチイニル基、3-フェノキサチイニル基、4-フェノキサチイニル基、チエノ[2,3-b]フリル基、ピロロ[1,2-b]ピリダジニル基、ピラゾロ[1,5-a]ピリジル基、イミダゾ[11,2-a]ピリジル基、イミダゾ[1,5-a]ピリジル基、イミダゾ[1,2-b]ピリダジニル基、イミダゾ[1,2-a]ピリミジニル基、1,2,4-トリアゾロ[4,3-a]ピリジル基、1,2,4-トリアゾロ[4,3-a]ピリダジニル基等の8ないし14員の縮合多環式ヘテロアリール基が挙げられるが、これらに限定されることはない。
R1が示すアラルキル基としては、例えば、ベンジル基又は3-インドリルメチル基などを用いることができるが、これらに限定されることはない。
Examples of the condensed polycyclic heteroaryl group constituting the aralkyl group represented by R 1 include a 2-benzofuranyl group, a 3-benzofuranyl group, a 4-benzofuranyl group, a 5-benzofuranyl group, a 6-benzofuranyl group, and a 7-benzofuranyl group. 1-isobenzofuranyl group, 4-isobenzofuranyl group, 5-isobenzofuranyl group, 2-benzo [b] thienyl group, 3-benzo [b] thienyl group, 4-benzo [b] thienyl Group, 5-benzo [b] thienyl group, 6-benzo [b] thienyl group, 7-benzo [b] thienyl group, 1-benzo [c] thienyl group, 4-benzo [c] thienyl group, 5-benzo [c] thienyl group, 1-indolyl group, 1-indolyl group, 2-indolyl group, 3-indolyl group, 4-indolyl group, 5-indolyl group, 6-indolyl group, 7-indolyl group, (2H-iso Indol) -1-yl group, (2H-isoindole) -2-yl group, (2H-isoindole) -4-yl group, (2H-isoi group) Dole) -5-yl group, (1H-indazol) -1-yl group, (1H-indazol) -3-yl group, (1H-indazol) -4-yl group, (1H-indazol) -5-yl Group, (1H-indazol) -6-yl group, (1H-indazol) -7-yl group, (2H-indazol) -1-yl group, (2H-indazol) -2-yl group, (2H-indazole) ) -4-yl group, (2H-indazol) -5-yl group, 2-benzoxazolyl group, 2-benzoxazolyl group, 4-benzoxazolyl group, 5-benzoxazolyl group, 6-benzoxazolyl group, 7-benzoxazolyl group, (1,2-benzisoxazol) -3-yl group, (1,2-benzisoxazol) -4-yl group, (1,2 -Benzoisoxazol) -5-yl group, (1,2-benzisoxazol) -6-yl group, (1,2-benzisoxazol) -7-yl group, (2,1-benzisoxazole) -3-yl group, (2,1-benzoisoxazol) -4-yl group, (2,1- Nzoisoxazol) -5-yl group, (2,1-benzisoxazol) -6-yl group, (2,1-benzisoxazol) -7-yl group, 2-benzothiazolyl group, 4-benzothiazolyl group , 5-benzothiazolyl group, 6-benzothiazolyl group, 7-benzothiazolyl group, (1,2-benzisothiazol) -3-yl group, (1,2-benzisothiazol) -4-yl group, (1,2 -Benzoisothiazol) -5-yl group, (1,2-benzisothiazol) -6-yl group, (1,2-benzisothiazol) -7-yl group, (2,1-benzoisothiazole) -3-yl group, (2,1-benzoisothiazol) -4-yl group, (2,1-benzoisothiazol) -5-yl group, (2,1-benzisothiazol) -6-yl group , (2,1-Benzisothiazol) -7-yl group, (1,2,3-Benzoxadiazol) -4-yl group, (1,2,3-Benzoxadiazol) -5-yl Group, (1,2,3-benzooxadiazole) -6-yl group, (1,2,3 -Benzoxadiazol) -7-yl group, (2,1,3-benzooxadiazol) -4-yl group, (2,1,3-benzooxadiazol) -5-yl group, (1 , 2,3-benzothiadiazol) -4-yl group, (1,2,3-benzothiadiazole) -5-yl group, (1,2,3-benzothiadiazol) -6-yl group, (1,2 , 3-Benzothiadiazole) -7-yl group, (2,1,3-benzothiadiazole) -4-yl group, (2,1,3-benzothiadiazole) -5-yl group, (1H-benzotriazole) -1-yl group, (1H-benzotriazol) -4-yl group, (1H-benzotriazol) -5-yl group, (1H-benzotriazol) -6-yl group, (1H-benzotriazole) -7 -Yl group, (2H-benzotriazol) -2-yl group, (2H-benzotriazol) -4-yl group, (2H-benzotriazol) -5-yl group, 2-quinolyl group, 3-quinolyl group, 4-quinolyl group, 5-quinolyl group, 6-quinolyl group, 7-quinolyl group, 8-quinolyl group, 1- Soquinolyl group, 3-isoquinolyl group, 4-isoquinolyl group, 5-isoquinolyl group, 6-isoquinolyl group, 7-isoquinolyl group, 8-isoquinolyl group, 3-cinnolinyl group, 4-cinnolinyl group, 5-cinnolinyl group, 6- Cinnolinyl group, 7-cinnolinyl group, 8-cinnolinyl group, 2-quinazolinyl group, 4-quinazolinyl group, 5-quinazolinyl group, 6-quinazolinyl group, 7-quinazolinyl group, 8-quinazolinyl group, 2-quinoxalinyl group, 5- Quinoxalinyl group, 6-quinoxalinyl group, 1-phthalazinyl group, 5-phthalazinyl group, 6-phthalazinyl group, 2-naphthyridinyl group, 3-naphthyridinyl group, 4-naphthyridinyl group, 2-purinyl group, 6-purinyl group, 7- Purinyl group, 8-prinyl group, 2-pteridinyl group, 4-pteridinyl group, 6-pteridinyl group, 7-pteridinyl group, 1-carbazolyl group, 2-carbazolyl group, 3-carbazolyl group , 4-carbazolyl group, 9-carbazolyl group, 2- (α-carbolinyl) group, 3- (α-carbolinyl) group, 4- (α-carbolinyl) group, 5- (α-carbolinyl) group, 6- ( α-Carboninyl) group, 7- (α-Carborinyl) group, 8- (α-Carborinyl) group, 9- (α-Carborinyl) group, 1- (β-Carbonylyl) group, 3- (β-Carbonylyl) group 4- (β-Carbonylyl) group, 5- (β-Carbonylyl) group, 6- (β-Carbonylyl) group, 7- (β-Carbonylyl) group, 8- (β-Carbonylyl) group, 9- (β -Carbonylyl) group, 1- (γ-Carborinyl) group, 2- (γ-Carborinyl) group, 4- (γ-Carborinyl) group, 5- (γ-Carborinyl) group, 6- (γ-Carborinyl) group, 7- (γ-Carborinyl) group, 8- (γ-Carborinyl) group, 9- (γ-Carborinyl) group, 1-Acridinyl group, 2-Acridinyl group, 3-Acridinyl group, 4-Acridinyl group, 9-Acridinyl group Group, 1-phenoxazinyl group, 2-phenoxazinyl group, 3-phenyl Noxazinyl group, 4-phenoxazinyl group, 10-phenoxazinyl group, 1-phenothiazinyl group, 2-phenothiazinyl group, 3-phenothiazinyl group, 4-phenothiazinyl group, 10-phenothiazinyl group, 1-phenazinyl group, 2- Phenazinyl group, 1-phenanthridinyl group, 2-phenanthridinyl group, 3-phenanthridinyl group, 4-phenanthridinyl group, 6-phenanthridinyl group, 7-phenanthridinyl group 8-phenanthridinyl group, 9-phenanthridinyl group, 10-phenanthridinyl group, 2-phenanthrolinyl group, 3-phenanthrolinyl group, 4-phenanthrolinyl group, 5 -Phenanthrolinyl group, 6-phenanthrolinyl group, 7-phenanthrolinyl group, 8-phenanthrolinyl group, 9-phenanthrolinyl group, 10-phenanthrolinyl group, 1-thianthrenyl group Group, 2-thianthenyl group, 1-i Doridinyl group, 2-indolidinyl group, 3-indolidinyl group, 5-indolidinyl group, 6-indolidinyl group, 7-indolidinyl group, 8-indolidinyl group, 1-phenoxathinyl group, 2-phenoxathinyl group, 3 -Phenoxathiinyl group, 4-phenoxathiinyl group, thieno [2,3-b] furyl group, pyrrolo [1,2-b] pyridazinyl group, pyrazolo [1,5-a] pyridyl group, imidazo [ 11,2-a] pyridyl group, imidazo [1,5-a] pyridyl group, imidazo [1,2-b] pyridazinyl group, imidazo [1,2-a] pyrimidinyl group, 1,2,4-triazolo [ Examples include, but are not limited to, 8- to 14-membered fused polycyclic heteroaryl groups such as 4,3-a] pyridyl group and 1,2,4-triazolo [4,3-a] pyridazinyl group. There is no.
Examples of the aralkyl group represented by R 1 include, but are not limited to, a benzyl group or a 3-indolylmethyl group.
X1及びX2はそれぞれ独立にハロゲン原子、水酸基、アミノ基、ニトロ基、又はカルボキシル基を示す。本明細書においてハロゲン原子とはフッ素原子、塩素原子、臭素原子、又はヨウ素原子を意味するが、X1及びX2が示すハロゲン原子としては、例えば、フッ素原子、塩素原子、又は臭素原子が好ましく、フッ素原子又は塩素原子がより好ましく、フッ素原子が特に好ましい。アミノ基は1又は2個の置換基を有していてもよい。例えば、モノアルキルアミノ基、ジアルキルアミノ基、アラルキルアミノ基、又はアシルアミノ基などであってもよい。X1及びX2がともにハロゲン原子であることが好ましく、X1及びX2がともにフッ素原子であることが特に好ましい。 X 1 and X 2 each independently represent a halogen atom, a hydroxyl group, an amino group, a nitro group, or a carboxyl group. In the present specification, the halogen atom means a fluorine atom, a chlorine atom, a bromine atom, or an iodine atom. As the halogen atom represented by X 1 and X 2 , for example, a fluorine atom, a chlorine atom, or a bromine atom is preferable. , A fluorine atom or a chlorine atom is more preferable, and a fluorine atom is particularly preferable. The amino group may have 1 or 2 substituents. For example, it may be a monoalkylamino group, a dialkylamino group, an aralkylamino group, or an acylamino group. X 1 and X 2 are preferably both halogen atoms, and both X 1 and X 2 are particularly preferably fluorine atoms.
X1及びX2の置換位置は特に限定されない。例えば、X1はベンゼン環上に置換する水酸基に対してパラ位であることが好ましい。また、X2はベンゼン環上に置換する水酸基に対してメタ位であることが好ましい。X1が水酸基に対してパラ位であり、X2がアミノ基に対してパラ位であることがより好ましい。さらに好ましくはX1としてハロゲン原子が水酸基に対してパラ位に位置しており、X2としてハロゲン原子がアミノ基に対してパラ位に位置していることがさらに好ましい。特に好ましくはX1としてフッ素原子が水酸基に対してパラ位に位置しており、X2としてフッ素原子がアミノ基に対してパラ位に位置していることがさらに好ましい。 The substitution positions of X 1 and X 2 are not particularly limited. For example, X 1 is preferably para to the hydroxyl group substituted on the benzene ring. X 2 is preferably in the meta position relative to the hydroxyl group substituted on the benzene ring. More preferably, X 1 is para to the hydroxyl group and X 2 is para to the amino group. More preferably, as X 1 , the halogen atom is located in the para position with respect to the hydroxyl group, and as X 2 , it is further preferred that the halogen atom is located in the para position with respect to the amino group. Particularly preferably, as X 1 , the fluorine atom is located in the para position with respect to the hydroxyl group, and as X 2 , it is further preferred that the fluorine atom is located in the para position with respect to the amino group.
一般式(I)で表される化合物を配位子として含む異種金属複合型錯体において、金属の1種はランタノイドから選択することができる。ランタノイドはランタン系列とも呼ばれ、原子番号57〜71の15元素を含む。より具体的には、ランタン(La)、セリウム(Ce)、プラセオジム(Pr)、ネオジム(Nd)、プロメチウム(Pm)、サマリウム(Sm)、ユウロピウム(Eu)、ガドリニウム(Gd)、テルビウム(Tb)、ジスプロシウム(Dy)、ホルミウム(Ho)、エルビウム(Er)、ツリウム(Tm)、イッテルビウム(Yb)、及びルテチウム(Lu)を含む。これらの金属のうち、ランタン、ネオジム、サマリウム、プラセオジム、ユウロピウム、ガドリニウム、ジスプロシウム、及びイッテルビウムからなる群から選ばれる金属が好ましく、さらに好ましいのはネオジム、サマリウム、又はプラセオジムであり、特に好ましくはネオジム又はサマリウムである。異種金属複合型錯体において、もう1種の金属としてアルカリ金属、例えばナトリウム、カリウム、又はリチウムが用いられる。アルカリ金属としてはナトリウムが好ましい。ナトリウムとネオジム、及びナトリウムとサマリウムの組合せはジアステレオ選択性及びエナンチオ選択性の観点から最も好ましい。 In the heterogeneous metal complex complex containing the compound represented by the general formula (I) as a ligand, one type of metal can be selected from lanthanoids. Lanthanoids are also called lanthanum series and contain 15 elements with atomic numbers 57-71. More specifically, lanthanum (La), cerium (Ce), praseodymium (Pr), neodymium (Nd), promethium (Pm), samarium (Sm), europium (Eu), gadolinium (Gd), terbium (Tb) , Dysprosium (Dy), holmium (Ho), erbium (Er), thulium (Tm), ytterbium (Yb), and lutetium (Lu). Of these metals, a metal selected from the group consisting of lanthanum, neodymium, samarium, praseodymium, europium, gadolinium, dysprosium, and ytterbium is preferred, more preferred is neodymium, samarium, or praseodymium, and particularly preferred is neodymium or Samarium. In the heterogeneous metal complex, an alkali metal such as sodium, potassium, or lithium is used as the other metal. Sodium is preferred as the alkali metal. The combination of sodium and neodymium and sodium and samarium is most preferred from the viewpoint of diastereoselectivity and enantioselectivity.
上記一般式(I)で表される化合物は、側鎖としてR1を有する光学活性L又はD-アミノ酸を原料とし、該アミノ酸のカルボキシル基とX2を置換基として有する2-アミノフェノール誘導体のアミノ基とを縮合して得られる化合物に対してX1を置換基として有するサリチル酸誘導体を縮合させることにより製造することができる。必要に応じて該アミノ酸のアミノ基又は2-アミノフェノールの水酸基を適宜の保護基で保護しておくことができる。保護基については、Greenら、Protective Groups in Organic Synthesis, 3rd Edition, 1999, John Wiley & Sons, Inc.などの成書を参照することができる。アミノ基の保護基としては、例えば、アルコキシカルボニル基、アリールオキシカルボニル基、アルカノイル基、又はアリールカルボニル基などを挙げることができる(上記のアリールオキシカルボニル基又はアリールカルボニル基においてアリール環は置換又は無置換であってもよく、置換基を有する場合にはハロゲン原子やアルコキシ基などが挙げられる)。これらのうち、アルコキシカルボニル基が好ましい。アルコキシカルボニル基としては、直鎖又は分枝鎖状のC1-6アルコキシ基で構成されるアルコキシカルボニル基が好ましく、より好ましいのはメトキシカルボニル基、エトキシカルボニル基、又はtert-ブトキシカルボニル基であり、特に好ましいのはtert-ブトキシカルボニル基である。水酸基の保護基としては、ベンジル基などのアラルキル基、ベンゾイル基、トリアルキルシリル基などを用いることができるが、ベンジル基が好ましい。 The compound represented by the general formula (I) is a 2-aminophenol derivative having an optically active L or D-amino acid having R 1 as a side chain as a raw material and having a carboxyl group of the amino acid and X 2 as a substituent. It can be produced by condensing a salicylic acid derivative having X 1 as a substituent to a compound obtained by condensing with an amino group. If necessary, the amino group of the amino acid or the hydroxyl group of 2-aminophenol can be protected with an appropriate protecting group. For protecting groups, reference can be made to books such as Green et al., Protective Groups in Organic Synthesis, 3rd Edition, 1999, John Wiley & Sons, Inc. Examples of the amino-protecting group include an alkoxycarbonyl group, an aryloxycarbonyl group, an alkanoyl group, and an arylcarbonyl group (in the above aryloxycarbonyl group or arylcarbonyl group, the aryl ring is substituted or unsubstituted). It may be substituted, and when it has a substituent, examples thereof include a halogen atom and an alkoxy group). Of these, alkoxycarbonyl groups are preferred. The alkoxycarbonyl group is preferably an alkoxycarbonyl group composed of a linear or branched C 1-6 alkoxy group, more preferably a methoxycarbonyl group, an ethoxycarbonyl group, or a tert-butoxycarbonyl group. Particularly preferred is a tert-butoxycarbonyl group. As the hydroxyl-protecting group, an aralkyl group such as a benzyl group, a benzoyl group, a trialkylsilyl group, and the like can be used, and a benzyl group is preferable.
この縮合反応はアミノ基とカルボキシル基とを反応させる通常のアミド形成反応により行うことができ、例えば、活性エステル化法、混合酸無水物法、又は縮合法など適宜の方法により行われる。活性エステル化法は、溶媒中、カルボン酸を活性エステル化剤と反応させ、活性エステルを製造した後、アミノ基と反応させることによって行われる。使用される溶媒としては、不活性であれば特に限定はないが、例えば、メチレンクロリド、クロロホルムのようなハロゲン化炭化水素類、エーテル、テトラヒドロフランのようなエーテル類、ジメチルホルムアミド、ジメチルアセトアミドのようなアミド類、ベンゼン、トルエン、キシレンのような芳香族炭化水素類、酢酸エチルのようなエステル類、又はこれらの混合溶媒が好適である。活性エステル化剤としては、例えば、N-ヒドロキシサクシイミド、1-ヒドロキシベンゾトリアゾール、N-ヒドロキシ-5-ノルボルネン-2,3- ジカルボキシイミドのようなN-ヒドロキシ化合物類;1,1'-オキザリルジイミダゾール、N,N'- カルボニルジイミダゾールのようなジイミダゾール化合物類;2,2'-ジピリジルジサルファイドのようなジサルファイド化合物類;N,N'-ジサクシンイミジルカーボネートのようなコハク酸化合物類;N,N'-ビス(2- オキソ-3- オキサゾリジニル)ホスフィニッククロライドのようなホスフィニッククロライド化合物類;N,N'-ジサクシンイミジルオキザレート(DSO)、N,N'-ジフタルイミドオキザレート(DPO)、N,N'-ビス(ノルボルネニルサクシンイミジル)オキザレート(BNO)、1,1'- ビス(ベンゾトリアゾリル)オキザレート(BBTO)、1,1'- ビス(6- クロロベンゾトリアゾリル)オキザレート(BCTO)、1,1'- ビス(6-トリフルオロメチルベンゾトリアゾリル)オキザレート(BTBO)のようなオキザレート化合物類を挙げることができ、好適には、N,N'-カルボニルジイミダゾールのようなジイミダゾール化合物類を挙げることができる。 This condensation reaction can be performed by a normal amide formation reaction in which an amino group and a carboxyl group are reacted. For example, the condensation reaction is performed by an appropriate method such as an active esterification method, a mixed acid anhydride method, or a condensation method. The active esterification method is carried out by reacting a carboxylic acid with an active esterifying agent in a solvent to produce an active ester and then reacting with an amino group. The solvent used is not particularly limited as long as it is inert. For example, halogenated hydrocarbons such as methylene chloride and chloroform, ethers such as ether and tetrahydrofuran, dimethylformamide, dimethylacetamide and the like. Preference is given to amides, aromatic hydrocarbons such as benzene, toluene, xylene, esters such as ethyl acetate, or mixed solvents thereof. Examples of the active esterifying agent include N-hydroxy compounds such as N-hydroxysuccinimide, 1-hydroxybenzotriazole, N-hydroxy-5-norbornene-2,3-dicarboximide; 1,1′- Diimidazole compounds such as oxalyldiimidazole, N, N'-carbonyldiimidazole; disulfide compounds such as 2,2'-dipyridyl disulfide; such as N, N'-disuccinimidyl carbonate Succinic acid compounds; phosphinic chloride compounds such as N, N′-bis (2-oxo-3-oxazolidinyl) phosphinic chloride; N, N′-disuccinimidyl oxalate (DSO), N , N'-Diphthalimide oxalate (DPO), N, N'-bis (norbornenylsuccinimidyl) oxalate (BNO), 1,1'-bis (benzotriazolyl) oxalate (BBTO), 1 , 1'- screw ( Examples include oxalate compounds such as 6-chlorobenzotriazolyl) oxalate (BCTO) and 1,1′-bis (6-trifluoromethylbenzotriazolyl) oxalate (BTBO). Mention may be made of diimidazole compounds such as N, N′-carbonyldiimidazole.
活性エステルとアミノ基との反応は、例えば、アゾジカルボン酸ジエチル−トリフェニルホスフィンのようなアゾジカルボン酸ジ低級アルキル-トリフェニルホスフィン類、N-エチル-5-フェニルイソオキサゾリウム-3'-スルホナートのようなN-低級アルキル-5- アリールイソオキサゾリウム-3'-スルホナート類、ジエチルオキシジフォルメート(DEPC)のようなオキシジフォルメート類、N',N'-ジシクロヘキシルカルボジイミド(DCC) のようなN',N'-ジシクロアルキルカルボジイミド類、ジ-2-ピリジルジセレニドのようなジヘテロアリールジセレニド類、トリフェニルホスフィンのようなトリアリールホスフィン類、p-ニトロベンゼンスルホニルトリアゾリドのようなアリールスルホニルトリアゾリド類、2-クロル-1- メチルピリジニウム ヨーダイドのような2-ハロ-1- 低級アルキルピリジニウムハライド及びジフェニルホスホリルアジド(DPPA)のようなジアリールホスホリルアジド類、N,N'-カルボジイミダゾール(CDI) のようなイミダゾール誘導体、1-ヒドロキシベンゾトリアゾール(HOBT)のようなベンゾトリアゾール誘導体、N-ヒドロキシ-5- ノールボルネン-2,3- ジカルボキシイミド(HONB)のようなジカルボキシイミド誘導体、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド(WSC) のようなカルボジイミド誘導体、1-プロパンホスホン酸環状無水物(T3P)のようなホスホン酸環状無水物のような縮合剤の存在下に好適に行われる。好適には1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド(WSC) のようなカルボジイミド誘導体、又は1-プロパンホスホン酸環状無水物(T3P)のようなホスホン酸環状無水物を用いることができる。活性エステルの調製のための反応温度は-10℃ないし室温であり、活性エステル化合物とアミノ基との反応は室温付近であり、反応時間は30分ないし10時間程度である。 The reaction of an active ester with an amino group is, for example, azodicarboxylic acid di-lower alkyl-triphenylphosphine such as diethyl azodicarboxylate-triphenylphosphine, N-ethyl-5-phenylisoxazolium-3′- N-lower alkyl-5-arylisoxazolium-3'-sulfonates such as sulfonate, oxydiformates such as diethyloxydiformate (DEPC), N ', N'-dicyclohexylcarbodiimide (DCC ) N ', N'-dicycloalkylcarbodiimides, diheteroaryl diselenides such as di-2-pyridyl diselenide, triaryl phosphines such as triphenylphosphine, p-nitrobenzenesulfonyl Arylsulfonyl triazolides such as triazolide, 2-chloro-1-methylpyridinium 2- B-1-Lower alkyl pyridinium halides and diarylphosphoryl azides such as diphenylphosphoryl azide (DPPA), imidazole derivatives such as N, N'-carbodiimidazole (CDI), and 1-hydroxybenzotriazole (HOBT) Benzotriazole derivatives, dicarboximide derivatives such as N-hydroxy-5-norbornene-2,3-dicarboximide (HONB), 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (WSC) In the presence of a condensing agent such as a phosphonic acid cyclic anhydride such as 1-propanephosphonic acid cyclic anhydride (T3P). Preferably, a carbodiimide derivative such as 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (WSC) or a phosphonic acid cyclic anhydride such as 1-propanephosphonic acid cyclic anhydride (T3P) is used. it can. The reaction temperature for the preparation of the active ester is −10 ° C. to room temperature, the reaction between the active ester compound and the amino group is around room temperature, and the reaction time is about 30 minutes to 10 hours.
混合酸無水物法は、カルボン酸の混合酸無水物を製造した後、アミノ基を反応させることにより行われる。縮合法はカルボン酸とアミンとを前記の縮合剤の存在下で直接反応させることによって行なうことができ、上記の活性エステルを製造する反応と同様にして行われる。また、縮合は不活性溶媒中でカルボキシル基を酸ハライドに変換した後、得られた酸ハライドとアミンとを反応させることによって行うこともできる。 The mixed acid anhydride method is carried out by reacting an amino group after producing a mixed acid anhydride of a carboxylic acid. The condensation method can be carried out by directly reacting a carboxylic acid and an amine in the presence of the condensing agent, and is carried out in the same manner as in the reaction for producing the active ester. Condensation can also be carried out by converting the carboxyl group to an acid halide in an inert solvent and then reacting the resulting acid halide with an amine.
本発明の方法の一態様を具体的に反応式で示す。下記の反応において、一般式(I)で表される化合物を配位子として含む上記異種金属複合型錯体、好ましくは一般式(I)においてR1がイソプロピル基であり、X1及びX2がハロゲン原子である化合物を配位子として含むネオジム/ナトリウム錯体を反応触媒として用いることができる。特に好ましくは一般式(I)においてR1がイソブチル基であり、X1としてフッ素原子が水酸基に対してパラ位に位置しており、X2としてフッ素原子がアミノ基に対してパラ位に位置している化合物を配位子として含むネオジム/ナトリウム錯体を反応触媒として用いることができる。 One embodiment of the method of the present invention is specifically shown by a reaction formula. In the following reaction, the heterogeneous metal complex complex containing a compound represented by the general formula (I) as a ligand, preferably R 1 in the general formula (I) is an isopropyl group, and X 1 and X 2 are A neodymium / sodium complex containing a halogen atom compound as a ligand can be used as a reaction catalyst. Particularly preferably, in general formula (I), R 1 is an isobutyl group, X 1 is a fluorine atom located in the para position relative to the hydroxyl group, and X 2 is a fluorine atom located in the para position relative to the amino group. A neodymium / sodium complex containing the compound as a ligand can be used as a reaction catalyst.
本明細書において「アンチ」配置とは、一般式(II)で表される1,2-ニトロアルカノール化合物において、水酸基とニトロ基とがアンチ配置であることを意味している。本発明の方法においては上記の異種金属複合型錯体を反応触媒として用いることにより、目的物である1,2-ニトロアルカノール化合物が光学活性化合物として得られる。以下の化学式中に示される立体表記は相対配置を示しており、本明細書において示されるその他の化学式においても同様である。 In the present specification, the “anti” configuration means that in the 1,2-nitroalkanol compound represented by the general formula (II), a hydroxyl group and a nitro group are in an anti configuration. In the method of the present invention, the target 1,2-nitroalkanol compound can be obtained as an optically active compound by using the heterogeneous metal complex complex as a reaction catalyst. The steric notation shown in the following chemical formulas indicates a relative configuration, and the same applies to other chemical formulas shown in this specification.
本発明の方法は、一般的には-50℃〜-30℃程度の低温下、好ましくは-40℃程度の低温下において、テトラヒドロフラン、トルエン、キシレン、又はそれらの混合溶媒などの不活性溶媒中で行なうことができる。一般的にはランタノイドアルコキシド又はランタノイドアラルキルオキシド/アルカリ金属/一般式(I)で表される化合物を1/2/2の混合比で調製した錯体を触媒として用い、反応系内における触媒量を5〜20 mol%、好ましくは8〜10 mol%程度、特に好ましくは9 mol%程度として、ニトロアルカン化合物とアルデヒド化合物とを順次添加することにより反応を行なうことができるが、試薬の調製や反応原料の添加順序は適宜選択することが可能である。本発明の方法において、ランタノイドアルコキシド又はランタノイドアラルキルオキシド/アルカリ金属/一般式(I)で表される化合物を1/2/2の混合比で混合して得られる錯体を沈殿物として単離し、必要に応じて洗浄した後に再懸濁して触媒懸濁液として使用することができるが、この手段により高純度の錯体を簡便に調製して触媒として使用することができ、高純度の触媒を使用することにより極めて高いアンチ選択性及び不斉収率を達成できるので、この態様は本発明の方法の好ましい態様である。 The method of the present invention is generally carried out in an inert solvent such as tetrahydrofuran, toluene, xylene, or a mixed solvent thereof at a low temperature of about -50 ° C to -30 ° C, preferably at a low temperature of about -40 ° C. Can be done. Generally, a lanthanoid alkoxide or a complex prepared by mixing a compound represented by the lanthanoid aralkyl oxide / alkali metal / general formula (I) with a mixing ratio of 1/2/2 is used as a catalyst, and the amount of catalyst in the reaction system is 5 The reaction can be carried out by sequentially adding a nitroalkane compound and an aldehyde compound at about 20 mol%, preferably about 8-10 mol%, particularly preferably about 9 mol%. The order of addition can be selected as appropriate. In the method of the present invention, a complex obtained by mixing a lanthanoid alkoxide or a lanthanoid aralkyl oxide / alkali metal / a compound represented by the general formula (I) at a mixing ratio of 1/2/2 is isolated as a precipitate. Can be resuspended and used as a catalyst suspension depending on the conditions, but a high-purity complex can be easily prepared and used as a catalyst by this means, and a high-purity catalyst is used. This embodiment is a preferred embodiment of the process of the present invention because very high anti-selectivity and asymmetric yield can be achieved.
また、上記反応をランタノイドとほぼ等量、例えば、5〜20 mol%、好ましくは8〜10 mol%程度、特に好ましくは9 mol%程度の水の存在下で行なうことにより、効率的に反応を進行させることができ、単離収率、ジアステレオ選択性、及びエナンチオ選択性を改善できる場合がある。もっとも、原料化合物であるアルデヒド化合物及びニトロアルカン化合物の比率、及び反応触媒としての錯体の添加量は特に限定されることはなく、適宜選択可能である。また、上記に説明した反応条件が適宜変更可能であることは当業者に容易に理解されることであり、本明細書の実施例に具体的に示した典型的な方法を参照しつつ、反応条件を適宜選択ないし改変又は修飾することにより、本発明の方法を好ましく行なうことができる。 In addition, the above reaction is carried out in the presence of approximately the same amount of lanthanoid, for example, 5 to 20 mol%, preferably about 8 to 10 mol%, particularly preferably about 9 mol%, for efficient reaction. In some cases, and may improve isolation yield, diastereoselectivity, and enantioselectivity. But the ratio of the aldehyde compound and nitroalkane compound which are raw material compounds, and the addition amount of the complex as a reaction catalyst are not specifically limited, It can select suitably. In addition, it is easily understood by those skilled in the art that the reaction conditions described above can be appropriately changed, and the reaction can be performed while referring to typical methods specifically shown in the examples of the present specification. The method of the present invention can be preferably carried out by appropriately selecting, changing or modifying the conditions.
本発明の方法で得られた光学活性アンチ-1,2-ニトロアルカノール化合物を還元することにより医薬品や天然生理活性物質の製造に有用な光学活性アンチ-1,2-アミノアルコール化合物を得ることができる。還元反応の種類は特に限定されず、存在する官能基の種類などに応じて当業者が適宜選択できることは言うまでもない。例えば、パラジウム炭素触媒を用いた接触還元法などを例示することができるが、これに限定されることはない。 By reducing the optically active anti-1,2-nitroalkanol compound obtained by the method of the present invention, an optically active anti-1,2-aminoalcohol compound useful for the production of pharmaceuticals and natural physiologically active substances can be obtained. it can. It goes without saying that the type of the reduction reaction is not particularly limited and can be appropriately selected by those skilled in the art depending on the type of the functional group present. For example, a catalytic reduction method using a palladium carbon catalyst can be exemplified, but the method is not limited thereto.
以下、本発明を実施例によりさらに具体的に説明するが、本発明の範囲は下記の実施例に限定されることはない。
例1:配位子の合成
Example 1: Ligand synthesis
4-フルオロアニソール(3.40 mL, 30 mmol)をテトラヒドロフラン(THF, 30 mL)に溶かし、-70℃に冷却した。この溶液に、リチウムテトラメチルピペリジン(33 mmol)のTHF溶液(24 mL)を5分以上かけて滴下した後、-70℃で13時間攪拌した。続いて、破砕したドライアイス(140 g)を加え、気体の発生が収まるまで室温で攪拌した。THFを減圧留去し、1N塩酸を加え、酢酸エチルで2度抽出した。合わせた酢酸エチル層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過・濃縮後に得られる残渣を、ヘキサン/酢酸エチルから再結晶し、2-フルオロ-5-メトキシ安息香酸(化合物1, 2.74 g, 収率54%)を白色固体として得た。
化合物1
1H NMR (CDCl3) : δ 3.82 (s, 3H), 7.06-7.11 (m, 2H), 7.46-7.48 (m, 1H)
4-fluoroanisole (3.40 mL, 30 mmol) was dissolved in tetrahydrofuran (THF, 30 mL) and cooled to -70 ° C. To this solution, a THF solution (24 mL) of lithium tetramethylpiperidine (33 mmol) was added dropwise over 5 minutes and then stirred at -70 ° C for 13 hours. Subsequently, crushed dry ice (140 g) was added and stirred at room temperature until gas evolution ceased. THF was distilled off under reduced pressure, 1N hydrochloric acid was added, and the mixture was extracted twice with ethyl acetate. The combined ethyl acetate layers were washed with saturated brine and dried over anhydrous sodium sulfate. The residue obtained after filtration and concentration was recrystallized from hexane / ethyl acetate to obtain 2-fluoro-5-methoxybenzoic acid (Compound 1, 2.74 g, yield 54%) as a white solid.
Compound 1
1 H NMR (CDCl 3 ): δ 3.82 (s, 3H), 7.06-7.11 (m, 2H), 7.46-7.48 (m, 1H)
得られた化合物1(1.70 g, 10 mmol)を塩化メチレン(30 mL)に懸濁させ、N,N-ジメチルホルムアミド(3滴)を加えて0℃に冷却し、この溶液に塩化オキサリル(1.29 mL, 15 mmol)を加えて室温にて2時間攪拌した。溶媒と塩化オキサリルを減圧留去し、塩化メチレン(10 mL)に溶解させ、2-フルオロ-5-メトキシベンゾイルクロリド(10 mL, 10 mmol, 1.0 M塩化メチレン溶液)を黄色液体として得た。 The obtained compound 1 (1.70 g, 10 mmol) was suspended in methylene chloride (30 mL), N, N-dimethylformamide (3 drops) was added and cooled to 0 ° C., and oxalyl chloride (1.29) was added to this solution. mL, 15 mmol) was added, and the mixture was stirred at room temperature for 2 hours. The solvent and oxalyl chloride were distilled off under reduced pressure and dissolved in methylene chloride (10 mL) to obtain 2-fluoro-5-methoxybenzoyl chloride (10 mL, 10 mmol, 1.0 M methylene chloride solution) as a yellow liquid.
N-tert-ブチルオキシカルボニル-L-ロイシン(2.99 g, 12 mmol)をトルエン(60 mL)に懸濁液させ、氷冷下にてトリエチルアミン(1.99 mL, 14 mmol)、ピバロイルクロリド(1.61 mL, 13 mmol)を順次加え、室温にて15分攪拌した。セライト濾過により得られた濾液を0℃に冷却し、2-ベンジルオキシ-4-フルオロアニリン(2.17 g, 10 mmol)を加えた後、室温にて20分攪拌した。1N塩酸を加え、ジエチルエーテルで抽出し、得られた有機層を飽和重曹水、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過・濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル = 4/1)で精製して化合物2を得た。
化合物2
1H NMR (CDCl3) : δ 0.91 (d, J = 6.4 Hz, 6H), 1.38 (s, 9H), 1.46-1.54 (m, 1H), 1.64-1.76 (m, 2H), 4.20 (brs, 1H), 4.87 (brs, 1H), 5.09 (s, 2H), 6.63-6.67 (m, 2H), 7.32-7.40 (m, 5H), 8.27 (brs, 1H), 8.29 (dd, J = 1.4, 8.6 Hz, 1H)
N-tert-butyloxycarbonyl-L-leucine (2.99 g, 12 mmol) was suspended in toluene (60 mL), and triethylamine (1.99 mL, 14 mmol) and pivaloyl chloride (1.61) were added under ice cooling. mL, 13 mmol) was sequentially added, and the mixture was stirred at room temperature for 15 minutes. The filtrate obtained by Celite filtration was cooled to 0 ° C., 2-benzyloxy-4-fluoroaniline (2.17 g, 10 mmol) was added, and the mixture was stirred at room temperature for 20 minutes. 1N Hydrochloric acid was added, and the mixture was extracted with diethyl ether. The obtained organic layer was washed with saturated aqueous sodium hydrogen carbonate and saturated brine, and dried over anhydrous sodium sulfate. The residue obtained after filtration and concentration was purified by flash column chromatography (hexane / ethyl acetate = 4/1) to give compound 2.
Compound 2
1 H NMR (CDCl 3 ): δ 0.91 (d, J = 6.4 Hz, 6H), 1.38 (s, 9H), 1.46-1.54 (m, 1H), 1.64-1.76 (m, 2H), 4.20 (brs, 1H), 4.87 (brs, 1H), 5.09 (s, 2H), 6.63-6.67 (m, 2H), 7.32-7.40 (m, 5H), 8.27 (brs, 1H), 8.29 (dd, J = 1.4, (8.6 Hz, 1H)
得られた化合物2を塩化メチレン(10 mL)に溶かし、0℃にて4N塩化水素シクロペンチルメチルエーテル溶液(40 mL)を加え、室温で30分攪拌した。反応溶液を減圧濃縮し、得られた固形残渣をメタノールに溶解し、ジエチルエーテルを加えた後に得られた不溶性成分を濾取してジエチルエーテルで十分に洗浄した。減圧乾燥後、(2S)-2-アミノ-N-(2-ベンジルオキシ-4-フルオロ)フェニル-2-イソブチルアセタミド 塩酸塩(3.34 g, 2段階収率91%)を無色粉末として得た。 The obtained compound 2 was dissolved in methylene chloride (10 mL), 4N hydrogen chloride cyclopentyl methyl ether solution (40 mL) was added at 0 ° C., and the mixture was stirred at room temperature for 30 min. The reaction solution was concentrated under reduced pressure, and the resulting solid residue was dissolved in methanol. After adding diethyl ether, the insoluble component obtained was collected by filtration and washed thoroughly with diethyl ether. After drying under reduced pressure, (2S) -2-amino-N- (2-benzyloxy-4-fluoro) phenyl-2-isobutylacetamide hydrochloride (3.34 g, 2-step yield 91%) was obtained as a colorless powder. It was.
得られた塩酸塩(1.65 g, 4.5 mmol)の塩化メチレン(30 mL)懸濁液を0 ℃に冷却し、2-フルオロ-5-メトキシベンゾイルクロリド(5.0 mL, 5.0 mmol, 1.0 M塩化メチレン溶液)、トリエチルアミン(1.39 mL, 10 mmol)を順次加えた。室温で1時間攪拌した後、反応溶液に1N塩酸を加え塩化メチレンで2度抽出し、合わせた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過・濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル = 4/1)で精製して化合物3(2.0 g, 収率92%)を得た。
化合物3
1H NMR (CDCl3) : δ 0.93 (d, J = 6.4 Hz, 3H), 0.94 (d, J = 6.4 Hz, 3H), 1.66-1.74 (m, 2H), 1.83-1.88 (m, 1H), 3.77 (s, 3H), 4.75-4.78 (m, 1H), 5.07 (s, 2H), 6.64-6.68 (m, 2H), 6.97-7.04 (m, 2H), 7.16 (dd, J = 8.0, 14.3 Hz, 1H), 7.29-7.33 (m, 3H), 7.38 (dd, J = 1.8, 7.9 Hz, 2H), 7.51 (dd, J = 3.4, 6.1 Hz, 1H), 8.28 (dd, J = 6.1, 8.9 HZ, 1H), 8.32 (s, 1H)
A suspension of the obtained hydrochloride (1.65 g, 4.5 mmol) in methylene chloride (30 mL) was cooled to 0 ° C., and 2-fluoro-5-methoxybenzoyl chloride (5.0 mL, 5.0 mmol, 1.0 M in methylene chloride) ) And triethylamine (1.39 mL, 10 mmol) were sequentially added. After stirring at room temperature for 1 hour, 1N hydrochloric acid was added to the reaction solution, and the mixture was extracted twice with methylene chloride. The combined organic layers were washed with saturated brine, and dried over anhydrous sodium sulfate. The residue obtained after filtration and concentration was purified by flash column chromatography (hexane / ethyl acetate = 4/1) to obtain compound 3 (2.0 g, yield 92%).
Compound 3
1 H NMR (CDCl 3 ): δ 0.93 (d, J = 6.4 Hz, 3H), 0.94 (d, J = 6.4 Hz, 3H), 1.66-1.74 (m, 2H), 1.83-1.88 (m, 1H) , 3.77 (s, 3H), 4.75-4.78 (m, 1H), 5.07 (s, 2H), 6.64-6.68 (m, 2H), 6.97-7.04 (m, 2H), 7.16 (dd, J = 8.0, 14.3 Hz, 1H), 7.29-7.33 (m, 3H), 7.38 (dd, J = 1.8, 7.9 Hz, 2H), 7.51 (dd, J = 3.4, 6.1 Hz, 1H), 8.28 (dd, J = 6.1 , 8.9 HZ, 1H), 8.32 (s, 1H)
得られた化合物3(2.0 g, 4.1 mmol)を塩化メチレン(10 mL)に溶解し、0 ℃で三臭化ホウ素(23 mL, 23 mmol, 1.0 M塩化メチレン溶液)を加えた。0 ℃で3時間攪拌した後、水(70 mL)を加え、室温で30分攪拌し、混合溶液を酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過・濃縮後に得られる残渣をフラッシュカラムクロマトグラフィー(ヘキサン/酢酸エチル = 1/1)で精製し、ヘキサン/クロロホルムから再結晶し、化合物4(1.30 g, 収率84%)を無色無定型晶質体として得た。
化合物4
1H NMR (CD3OD) : δ 1.00 (d, J = 6.5 Hz, 3H), 1.02 (d, J = 6.5 Hz, 3H), 1.77-1.83 (m, 3H), 4.75-4.78 (m, 1H), 6.54 (dd, J = 8.8, 8.9 Hz, 1H), 6.58 (dd, J = 2.8, 10.1 Hz, 1H), 6.88-6.92 (m, 1H), 7.04 (dd, J = 9.2, 10.1 Hz, 1H), 7.11 (dd, J = 3.1, 5.5 Hz, 1H), 7.75 (dd, J = 6.4, 8.9 Hz, 1H)
The obtained compound 3 (2.0 g, 4.1 mmol) was dissolved in methylene chloride (10 mL), and boron tribromide (23 mL, 23 mmol, 1.0 M methylene chloride solution) was added at 0 ° C. After stirring at 0 ° C. for 3 hours, water (70 mL) was added, the mixture was stirred at room temperature for 30 minutes, and the mixed solution was extracted with ethyl acetate. The organic layer was washed with water and saturated brine, and dried over anhydrous sodium sulfate. The residue obtained after filtration and concentration was purified by flash column chromatography (hexane / ethyl acetate = 1/1) and recrystallized from hexane / chloroform to give Compound 4 (1.30 g, 84% yield) as colorless amorphous crystals. Obtained as a mass.
Compound 4
1 H NMR (CD 3 OD): δ 1.00 (d, J = 6.5 Hz, 3H), 1.02 (d, J = 6.5 Hz, 3H), 1.77-1.83 (m, 3H), 4.75-4.78 (m, 1H ), 6.54 (dd, J = 8.8, 8.9 Hz, 1H), 6.58 (dd, J = 2.8, 10.1 Hz, 1H), 6.88-6.92 (m, 1H), 7.04 (dd, J = 9.2, 10.1 Hz, 1H), 7.11 (dd, J = 3.1, 5.5 Hz, 1H), 7.75 (dd, J = 6.4, 8.9 Hz, 1H)
また、配位子の比較のため、以下の化合物を製造した。
化合物5
1H NMR (CDCl3) : δ 0.96 (d, J = 6.4 Hz, 3H), 0.98 (d, J = 6.4 Hz, 3H), 1.52-1.58 (m, 1H), 1.82-1.88 (m, 2H), 2.87-2.91 (m, 2H), 3.56 (t, J = 6.4 Hz, 2H), 5.49 (t, J = 8.6 Hz, 1H), 6.81 (dd, J = 7.9, 7.9 Hz, 1H), 6.93-6.97 (m, 2H), 7.04-7.09 (m, 3H), 7.60 (brs, 1H), 8.79 (s, 1H)
化合物6
1H NMR (CDCl3) : δ 0.97 (d, J = 6.3 Hz, 3H), 1.00 (d, J = 6.3 Hz), 1.71-1.79 (m, 2H), 1.87-1.93 (m, 1H), 4.81-4.86 (m, 1H), 6.57 (d, J = 7.5 Hz, 1H), 6.83 (dd, J = 7.5, 7.5 Hz, 1H), 6.98 (d, J = 8.6 Hz, 1H), 7.08-7.12 (m, 2H), 7.21-7.26 (m, 1H), 7.44 (d, J = 8.0 Hz, 1H), 8.84 (brs, 1H)
Compound 5
1 H NMR (CDCl 3 ): δ 0.96 (d, J = 6.4 Hz, 3H), 0.98 (d, J = 6.4 Hz, 3H), 1.52-1.58 (m, 1H), 1.82-1.88 (m, 2H) , 2.87-2.91 (m, 2H), 3.56 (t, J = 6.4 Hz, 2H), 5.49 (t, J = 8.6 Hz, 1H), 6.81 (dd, J = 7.9, 7.9 Hz, 1H), 6.93- 6.97 (m, 2H), 7.04-7.09 (m, 3H), 7.60 (brs, 1H), 8.79 (s, 1H)
Compound 6
1 H NMR (CDCl 3 ): δ 0.97 (d, J = 6.3 Hz, 3H), 1.00 (d, J = 6.3 Hz), 1.71-1.79 (m, 2H), 1.87-1.93 (m, 1H), 4.81 -4.86 (m, 1H), 6.57 (d, J = 7.5 Hz, 1H), 6.83 (dd, J = 7.5, 7.5 Hz, 1H), 6.98 (d, J = 8.6 Hz, 1H), 7.08-7.12 ( m, 2H), 7.21-7.26 (m, 1H), 7.44 (d, J = 8.0 Hz, 1H), 8.84 (brs, 1H)
化合物7
1H NMR (CD3OD) : δ 0.99 (d, J = 5.5 Hz, 3H), 1.02 (d, J = 5.8 Hz, 3H), 1.76-1.86 (m, 3H), 4.74-4.77 (m, 1H), 6.66 (ddd, J = 3.1, 8.6, 8.6 Hz, 1H), 6.76 (dd, J = 5.2, 8.6 Hz, 1H), 6.96 (d, J = 8.2 Hz, 1H), 7.27-7.33 (m, 3H), 7.83 (dd, J = 3.1, 10.7 Hz, 1H)
化合物8
1H NMR (CD3OD) : δ 1.00 (d, J = 6.4 Hz, 3H), 1.02 (d, J = 6.4 Hz, 3H), 1.79-1.83 (m, 3H), 4.76-4.79 (m, 1H), 6.80 (ddd, J = 1.5, 7.9, 7.9 Hz, 1H), 6.84 (dd, J = 1.2, 7.9 Hz, 1H), 6.89-6.92 (m, 1H), 6.97 (ddd, J = 1.5, 7.9, 7.9 Hz, 1H), 7.04 (dd, J = 8.9, 10.4 Hz, 1H), 7.10-7.12 (m, 1H), 7.80 (dd, J = 1.5, 7.9 Hz, 1H)
Compound 7
1 H NMR (CD 3 OD): δ 0.99 (d, J = 5.5 Hz, 3H), 1.02 (d, J = 5.8 Hz, 3H), 1.76-1.86 (m, 3H), 4.74-4.77 (m, 1H ), 6.66 (ddd, J = 3.1, 8.6, 8.6 Hz, 1H), 6.76 (dd, J = 5.2, 8.6 Hz, 1H), 6.96 (d, J = 8.2 Hz, 1H), 7.27-7.33 (m, 3H), 7.83 (dd, J = 3.1, 10.7 Hz, 1H)
Compound 8
1 H NMR (CD 3 OD): δ 1.00 (d, J = 6.4 Hz, 3H), 1.02 (d, J = 6.4 Hz, 3H), 1.79-1.83 (m, 3H), 4.76-4.79 (m, 1H ), 6.80 (ddd, J = 1.5, 7.9, 7.9 Hz, 1H), 6.84 (dd, J = 1.2, 7.9 Hz, 1H), 6.89-6.92 (m, 1H), 6.97 (ddd, J = 1.5, 7.9 , 7.9 Hz, 1H), 7.04 (dd, J = 8.9, 10.4 Hz, 1H), 7.10-7.12 (m, 1H), 7.80 (dd, J = 1.5, 7.9 Hz, 1H)
例2
例3
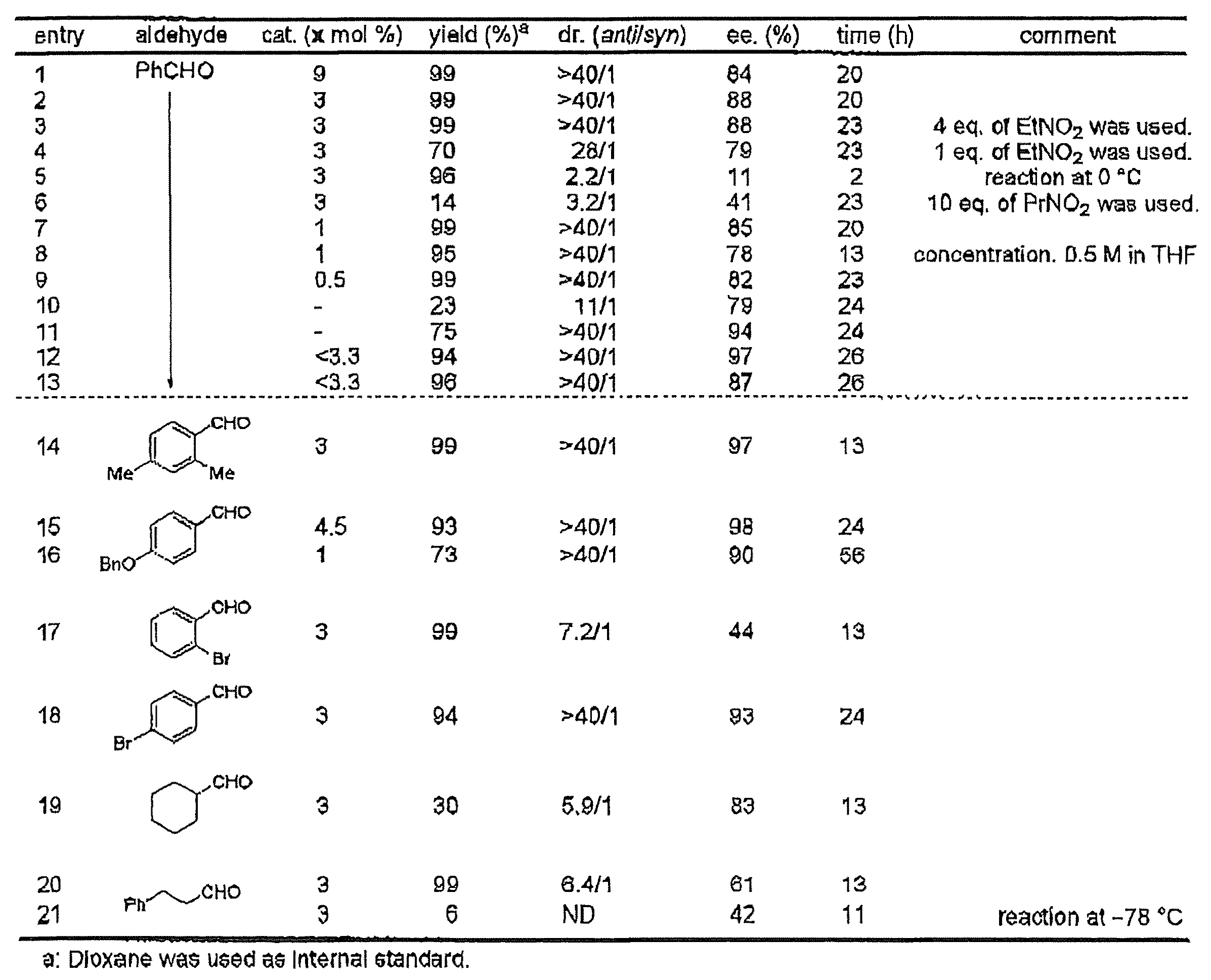
単離された触媒を用いて調製した触媒懸濁液を用いて例2と同様の反応を行った。加熱真空乾燥したガラス管に化合物4(6.8mg, 0.018 mmol)を加え、10分間真空乾燥した後、アルゴン雰囲気下にてTHF(0.30 mL)、Nd(OiPr)3(0.2 M/THF溶液, 45 μL, 0.009 mmol)を加えてボルテックスミキサーで激しく撹拌した。この懸濁液に氷冷下NaHMDS(1.0 M/THF溶液, 18 μL, 0.018 mmol)、室温にてニトロエタン(40 μL, 0.57 mmol)、水(0.2 M/THF溶液, 45 μL, 0.009 mmol)を順次加えてボルテックスミキサーで激しく撹拌した。30分後、この溶液をエッペンドルフチューブに移し、1分間遠心分離(13750 rpm)し、上清を捨てて錯体を単離した。ここにTHF(1.0 mL)を加えてボルテックスミキサーで激しく撹拌し、再度1分間遠心分離した(13750 rpm)。先と同様に上清を捨てて錯体を単離し、THF(1.3 mL)を加えて触媒懸濁液とした。別途用意した加熱真空乾燥した試験管にニトロエタン(0.21 mL, 3.0 mmol)及び調製した触媒懸濁液をアルゴン雰囲気下にて加え、試験管を-40 ℃の恒温槽に移し、ベンズアルデヒド(30 μL, 0.30 mmol)を加えた。-40 ℃で7時間攪拌した後、1N塩酸(3 mL)を加え、その混合溶液をジエチルエーテル(30 mL)にて抽出し、飽和重曹水、飽和食塩水で洗い、硫酸ナトリウムで乾燥した。化学収率99%(1H NMR, 内部標準1,4-dioxane)、anti/syn = >40/1、93% ee。この反応で得られたanti/syn比及び光学収率(ee.%)は、フッ素原子が導入されていない対応の化合物を配位子として用いた場合に比べて顕著に高いものであった(Tetrahedron Letters, 49, pp.272-276, 2008のTable 2のEntry 5を参照のこと)。
Example 3
The same reaction as in Example 2 was performed using a catalyst suspension prepared using the isolated catalyst. Compound 4 (6.8 mg, 0.018 mmol) was added to a heated and vacuum-dried glass tube, vacuum-dried for 10 minutes, then THF (0.30 mL), Nd (O i Pr) 3 (0.2 M / THF solution) under an argon atmosphere. , 45 μL, 0.009 mmol), and vigorously stirred with a vortex mixer. NaHMDS (1.0 M / THF solution, 18 μL, 0.018 mmol), nitroethane (40 μL, 0.57 mmol) and water (0.2 M / THF solution, 45 μL, 0.009 mmol) at room temperature were added to this suspension under ice cooling. Sequentially added and vigorously stirred with a vortex mixer. After 30 minutes, the solution was transferred to an Eppendorf tube, centrifuged for 1 minute (13750 rpm), and the supernatant was discarded to isolate the complex. THF (1.0 mL) was added thereto, and the mixture was vigorously stirred with a vortex mixer and centrifuged again for 1 minute (13750 rpm). As above, the supernatant was discarded to isolate the complex, and THF (1.3 mL) was added to form a catalyst suspension. Add nitroethane (0.21 mL, 3.0 mmol) and the prepared catalyst suspension to a separately prepared heated and vacuum-dried test tube in an argon atmosphere, transfer the test tube to a -40 ° C thermostatic bath, and add benzaldehyde (30 μL, 0.30 mmol) was added. After stirring at −40 ° C. for 7 hours, 1N hydrochloric acid (3 mL) was added, and the mixed solution was extracted with diethyl ether (30 mL), washed with saturated aqueous sodium hydrogen carbonate and saturated brine, and dried over sodium sulfate. Chemical yield 99% ( 1 H NMR, internal standard 1,4-dioxane), anti / syn => 40/1, 93% ee. The anti / syn ratio and optical yield (ee.%) Obtained by this reaction were significantly higher than when the corresponding compound having no fluorine atom introduced was used as the ligand ( (See Entry 5 in Table 2 of Tetrahedron Letters, 49, pp.272-276, 2008).
例4
反応条件、アルデヒド化合物、及び/又はニトロアルカンを変更して例2又は例3と同様にして反応を行った。結果を表1に示す。エントリー番号1は上記例3の結果を示し、エントリー番号3及び4はニトロエタンの量(通常は10当量)を4又は1当量に変更した結果を示し、エントリー番号5は反応温度を0℃とした場合の結果を示し、エントリー番号6はニトロエタンに代えてニトロプロパンを用いた場合の結果を示し、エントリー番号8は基質の濃度を0.2Mから0.5Mに変化させた場合の結果を示し、エントリー10及び11は、それぞれ触媒調製時の上清及び沈殿を用いた場合の結果を示し、エントリー番号12及び13は触媒調製時に得られる沈殿を一度洗って乾燥させた場合(エントリー番号12)及び同様に調製した触媒を未乾燥で用いた場合(エントリー番号13)の結果を示す。
Example 4
The reaction was carried out in the same manner as in Example 2 or Example 3 by changing the reaction conditions, the aldehyde compound, and / or the nitroalkane. The results are shown in Table 1. Entry number 1 shows the result of Example 3 above, entry numbers 3 and 4 show the result of changing the amount of nitroethane (usually 10 equivalents) to 4 or 1 equivalent, and entry number 5 sets the reaction temperature to 0 ° C. Entry number 6 shows the result when nitropropane was used instead of nitroethane, entry number 8 shows the result when the substrate concentration was changed from 0.2 M to 0.5 M, and entry 10 And 11 show the results when using the supernatant and the precipitate at the time of catalyst preparation, respectively, and entry numbers 12 and 13 are when the precipitate obtained at the time of catalyst preparation is washed once and dried (entry number 12) and similarly The results when the prepared catalyst was used undried (entry number 13) are shown.
例5
比較のため、化合物5〜8を用いて例2と同様に触媒を調製し、触媒量9 mol%、反応温度-40℃で同様のニトロアルドール反応を行った。結果を表2に示す。
For comparison, a catalyst was prepared using compounds 5 to 8 in the same manner as in Example 2, and the same nitroaldol reaction was performed at a catalyst amount of 9 mol% and a reaction temperature of -40 ° C. The results are shown in Table 2.
例6
化合物4とNd以外の希土類金属を用いて例2あるいは例3と同様に触媒を調製し、触媒量3 mol%、反応温度-40℃で同様のニトロアルドール反応を行った。結果を表3に示す。
A catalyst was prepared in the same manner as in Example 2 or Example 3 using a rare earth metal other than Compound 4 and Nd, and a similar nitroaldol reaction was performed at a catalyst amount of 3 mol% and a reaction temperature of -40 ° C. The results are shown in Table 3.
Claims (9)
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009165211A JP5453003B2 (en) | 2009-01-26 | 2009-07-14 | Catalyst for stereoselective asymmetric nitroaldol reaction |
| PCT/JP2010/000387 WO2010084772A1 (en) | 2009-01-26 | 2010-01-25 | Catalyst for stereoselective asymmetric nitroaldol reaction |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009013984 | 2009-01-26 | ||
| JP2009013984 | 2009-01-26 | ||
| JP2009165211A JP5453003B2 (en) | 2009-01-26 | 2009-07-14 | Catalyst for stereoselective asymmetric nitroaldol reaction |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010189374A JP2010189374A (en) | 2010-09-02 |
| JP5453003B2 true JP5453003B2 (en) | 2014-03-26 |
Family
ID=42355827
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009165211A Expired - Fee Related JP5453003B2 (en) | 2009-01-26 | 2009-07-14 | Catalyst for stereoselective asymmetric nitroaldol reaction |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP5453003B2 (en) |
| WO (1) | WO2010084772A1 (en) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014118799A1 (en) | 2013-02-01 | 2014-08-07 | Council Of Scientific & Industrial Research | Recyclable chiral catalyst for asymmetric nitroaldol reaction and process for the preparation thereof |
| JP6027910B2 (en) * | 2013-02-14 | 2016-11-16 | 公益財団法人微生物化学研究会 | Method for producing catalyst and method for producing optically active anti-1,2-nitroalkanol compound |
| JP6623042B2 (en) * | 2015-11-24 | 2019-12-18 | 公益財団法人微生物化学研究会 | Catalyst, method for producing the same, and method for producing optically active anti-1,2-nitroalkanol compound |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009114071A (en) * | 2007-11-01 | 2009-05-28 | Univ Of Tokyo | Catalyst for stereoselective asymmetric nitroaldol reaction |
-
2009
- 2009-07-14 JP JP2009165211A patent/JP5453003B2/en not_active Expired - Fee Related
-
2010
- 2010-01-25 WO PCT/JP2010/000387 patent/WO2010084772A1/en active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010084772A1 (en) | 2010-07-29 |
| JP2010189374A (en) | 2010-09-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2003031403A2 (en) | Process to produce derivatives from uk-2a derivatives | |
| Xu et al. | Solvent-free iodine-promoted synthesis of 3, 2′-pyrrolinyl spirooxindoles from alkylidene oxindoles and enamino esters under ball-milling conditions | |
| JP2007022932A (en) | Manufacturing method of amino acid-n-carboxylic anhydride | |
| JP5453003B2 (en) | Catalyst for stereoselective asymmetric nitroaldol reaction | |
| Huang et al. | Regioselective synthesis of 2, 3′-biindoles mediated by an NBS-induced homo-coupling of indoles | |
| Li et al. | Cu-catalyzed tandem reactions of fluorinated alkynes with sulfonyl azides en route to 2-trifluoromethylquinolines | |
| Borah et al. | Ir (iii)-Catalyzed [4+ 2] cyclization of azobenzene and diazotized Meldrum's acid for the synthesis of cinnolin-3 (2 H)-one | |
| Ikejiri et al. | Synthesis and fluorescence properties of 4-diarylmethylene analogues of the green fluorescent protein chromophore | |
| Franco et al. | Synthesis of 2-(pyrimidin-4-yl) indoles | |
| Kumar Saini et al. | I2‐DMSO‐Mediated Cascade Cyclization of β‐Ketosulfoxonium Ylides and β‐Enaminones: Synthesis of Quaternary‐Carbon‐Centered 2‐Hydroxy‐pyrrol‐3 (2H)‐ones | |
| García et al. | Synthesis, crystal structure and biological activity of β-carboline based selective CDK4-cyclin D1 inhibitors | |
| TWI290554B (en) | Process for preparation 5-(1-piperazinyl)-benzofuran-2-carboxamide by transition metal-catalyzed amination | |
| JP2009114071A (en) | Catalyst for stereoselective asymmetric nitroaldol reaction | |
| Napolitano et al. | Synthesis, kinase activity and molecular modeling of a resorcylic acid lactone incorporating an amide and a trans-enone in the macrocycle | |
| JP2022505626A (en) | N-nitrosaccharins | |
| CN113582946B (en) | 3-aryl-5-thio-1,3,4-thiadiazole-2-thioketone derivative and preparation method and application thereof | |
| JP5268052B2 (en) | Catalyst for asymmetric amination reaction | |
| WO2024031753A1 (en) | Indoline compound and preparation method therefor | |
| RU2620379C2 (en) | Method for prepairing derivatives of 2-phenyl [1,2,4] triazolo [1,5-a] pyridine | |
| JP2008169167A (en) | METHOD FOR PRODUCING beta-AMINO ESTER | |
| JP2926482B2 (en) | Method for producing hexaazaisowurtzitane having acyl group | |
| JP6507172B2 (en) | Process for producing optically active α-trifluoromethyl-β-amino acid derivative | |
| WO2006129781A1 (en) | Process for production of dibenzoxepin derivative | |
| Wang et al. | An Effective Synthesis of Indazolo [2, 1‐a] indazole‐6, 12‐diones by Regioselective Copper‐Catalyzed Cascade Acylation/Coupling Cyclization Process | |
| Dinh et al. | Control of ditryptophan cross‐linking: dihydrotryptophan as a tryptophan precursor in peptide synthesis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20120713 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20120713 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20120713 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120906 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20131015 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131119 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20131224 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140106 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5453003 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |